User login
Lambert-Eaton Myasthenic Syndrome and Merkel Cell Carcinoma
Merkel cell carcinoma (MCC) is an aggressive neuroendocrine malignancy of the skin that is thought to arise from neural crest cells. It has an estimated annual incidence of 0.6 per 100,000 individuals, typically occurs in the elderly population, and is most common in white males.1 The tumor presents as a rapidly growing, violaceous nodule in sun-exposed areas of the skin; early in the course, it can be mistaken for a benign entity such as an epidermal cyst.2 Merkel cell carcinoma has a propensity to spread to regional lymph nodes, and in some cases, it occurs in the absence of skin findings.3 Histologically, MCC is nearly indistinguishable from small cell lung carcinoma (SCLC).4 The overall prognosis for patients with MCC is poor and largely dependent on the stage at diagnosis. Patients with regional and distant metastases have a 5-year survival rate of 26% to 42% and 18%, respectively.3
Lambert-Eaton myasthenic syndrome (LEMS) is a paraneoplastic or autoimmune disorder of the neuromuscular junction that is found in 3% of cases of SCLC.4 Reported cases of LEMS in patients with MCC are exceedingly rare.5-8 We provide a full report and longitudinal clinical follow-up of a case that was briefly discussed by Simmons et al,8 and we review the literature regarding paraneoplastic syndromes associated with MCC and other extrapulmonary small cell carcinomas (EPSCCs).
Case Report
A 63-year-old man was evaluated in the neurology clinic due to difficulty walking, climbing stairs, and performing push-ups over the last month. Prior to the onset of symptoms, he was otherwise healthy, walking 3 miles daily; however, at presentation he required use of a cane. Leg weakness worsened as the day progressed. In addition, he reported constipation, urinary urgency, dry mouth, mild dysphagia, reduced sensation below the knees, and a nasal quality in his speech. He had no ptosis, diplopia, dysarthria, muscle cramps, myalgia, or facial weakness. He denied fevers, chills, and night sweats but did admit to an unintentional 10- to 15-lb weight loss over the preceding few months.
The neurologic examination revealed mild proximal upper extremity weakness in the bilateral shoulder abductors, infraspinatus, hip extensors, and hip flexors (Medical Research Council muscle scale grade 4). All deep tendon reflexes, except the Achilles reflex, were present. Despite subjective sensory concerns, objective examination of all sensory modalities was normal. Cranial nerve examination was normal, except for a slight nasal quality to his voice.
A qualitative assay was positive for the presence of P/Q-type voltage-gated calcium channel (VGCC) antibodies. Other laboratory studies were within reference range, including acetylcholine-receptor antibodies (blocking, binding, and modulating) and muscle-specific kinase antibodies.
Lumbar and cervical spine magnetic resonance imaging revealed multilevel neuroforaminal stenosis without spinal canal stenosis or myelopathy. Computed tomography (CT) of the chest was notable for 2 pathologically enlarged lymph nodes in the left axilla and no evidence of primary pulmonary malignancy. Nerve-conduction studies (NCSs) in conjunction with other clinical findings were consistent with the diagnosis of LEMS.
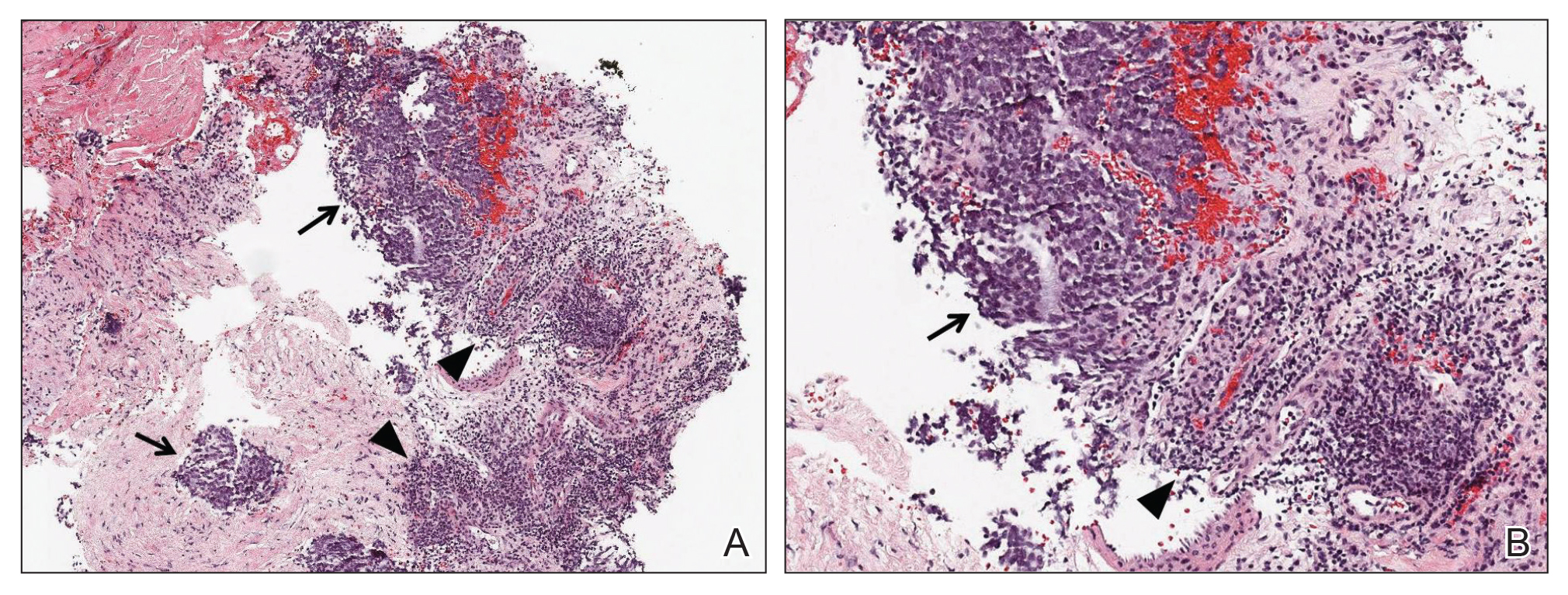
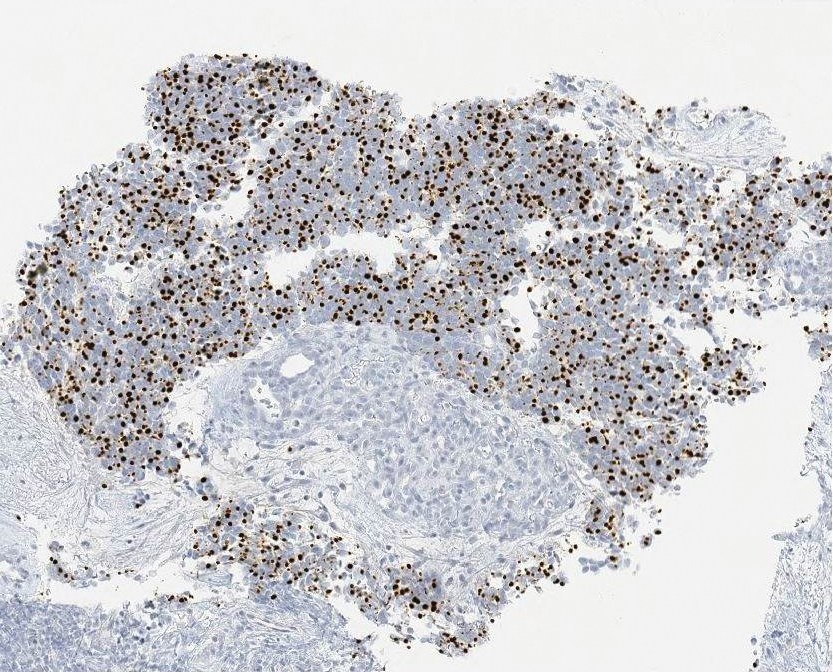
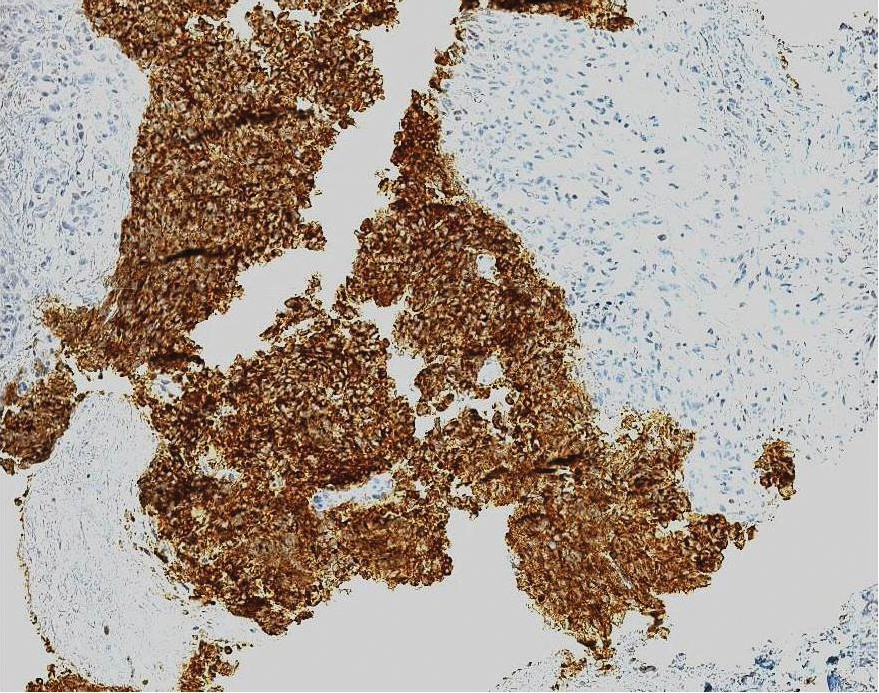
Ultrasound-guided biopsy of the enlarged axillary lymph nodes demonstrated sheets and nests of small round blue tumor cells with minimal cytoplasm, high mitotic rate, and foci of necrosis (Figure 1). The tumor cells were positive for pancytokeratin (Lu-5) and cytokeratin (CK) 20 in a perinuclear dotlike pattern (Figure 2), as well as for the neuroendocrine markers synaptophysin (Figure 3), chromogranin A, and CD56. The tumor cells showed no immunoreactivity for CK7, thyroid transcription factor 1, CD3, CD5, or CD20. Flow cytometry demonstrated low cellularity, low specimen viability, and no evidence of an abnormal B-cell population. These findings were consistent with the diagnosis of MCC.
The patient underwent surgical excision of the involved lymph nodes. Four weeks after surgery, he reported dramatic improvement in strength, with complete resolution of the nasal speech, dysphagia, dry mouth, urinary retention, and constipation. Two months after surgery, his strength had normalized, except for slight persistent weakness in the bilateral shoulder abductors, trace weakness in the hip flexors, and a slight Trendelenburg gait. He was able to rise from a chair without using his arms and no longer required a cane for ambulation.
The patient underwent adjuvant radiation therapy after 2-month surgical follow-up with 5000-cGy radiation treatment to the left axillary region. Six months following primary definitive surgery and 4 months following adjuvant radiation therapy, he reported a 95% subjective return of physical strength. The patient was able to return to near-baseline physical activity. He continued to deny symptoms of dry mouth, incontinence, or constipation. Objectively, he had no focal neurologic deficits or weakness; no evidence of new skin lesions or lymphadenopathy was noted.
Comment
MCC vs SCLC
Merkel cell carcinoma is classified as a type of EPSCC. The histologic appearance of MCC is indistinguishable from SCLC. Both tumors are composed of uniform sheets of small round cells with a high nucleus to cytoplasm ratio, and both can express neuroendocrine markers, such as neuron-specific enolase, chromogranin A, and synaptophysin.9 Immunohistochemical positivity for CK20 and neurofilaments in combination with negative staining for thyroid transcription factor 1 and CK7 effectively differentiate MCC from SCLC.9 In addition, MCC often displays CK20 positivity in a perinuclear dotlike or punctate pattern, which is characteristic of this tumor.3,9,10 Negative immunohistochemical markers for B cells (CD20) and T cells (CD3) are important in excluding lymphoma.
LEMS Diagnosis
Lambert-Eaton myasthenic syndrome is a paraneoplastic or autoimmune disorder involving the neuromuscular junction. Autoantibodies to VGCC impair calcium influx into the presynaptic terminal, resulting in marked reduction of acetylcholine release into the synaptic cleft. The reduction in acetylcholine activity impairs production of muscle fiber action potentials, resulting in clinical weakness. The diagnosis of LEMS rests on clinical presentation, positive serology, and confirmatory neurophysiologic testing by NCS. Clinically, patients present with proximal weakness, hyporeflexia or areflexia, and autonomic dysfunction. Antibodies to P/Q-type VGCCs are found in 85% to 90% of cases of LEMS and are thought to play a direct causative role in the development of weakness.11 The finding of postexercise facilitation on motor NCS is the neurophysiologic hallmark and is highly specific for the diagnosis.
Approximately 50% to 60% of patients who present with LEMS have an underlying tumor, the vast majority of which are SCLC.11 There are a few reports of LEMS associated with other malignancies, including lymphoma; thymoma; neuroblastoma; and carcinoma of the breast, stomach, prostate, bladder, kidney, and gallbladder.12 Patients with nontumor or autoimmune LEMS tend to be younger, and there is no male predominance, as there is in paraneoplastic LEMS.13 Given the risk of underlying malignancy in LEMS, Titulaer et al14 proposed a screening protocol for patients presenting with LEMS, recommending initial primary screening using CT of the chest. If the CT scan is negative, total-body fludeoxyglucose positron emission tomography should be performed to assess for fludeoxyglucose avid lesions. If both initial studies are negative, routine follow-up with CT of the chest at 6-month intervals for a minimum of 2 to 4 years after the initial diagnosis of LEMS was recommended. An exception to this protocol was suggested to allow consideration to stop screening after the first 6-month follow-up chest CT for patients younger than 45 years who have never smoked and who have an HLA 8.1 haplotype for which nontumor LEMS would be a more probable diagnosis.14
In addition to a screening protocol, a validated prediction tool, the Dutch-English LEMS Tumor Association prediction score, was developed. It uses common signs and symptoms of LEMS and risk factors for SCLC to help guide the need for further screening.15
Paraneoplastic Syndromes Associated With MCC
Other paraneoplastic syndromes have been reported in association with MCC. A patient with brainstem encephalitis associated with MCC was reported in a trial of a novel immunotherapy for paraneoplastic neurologic syndromes.16,17 A syndrome of inappropriate antidiuretic hormone (SIADH) secretion was reported in a patient with N-type calcium channel antibodies.18 Two cases of paraneoplastic cerebellar degeneration have been reported; the first was associated with a novel 70-kD antibody,19 and the second was associated with the P/Q-type VGCC antibody.20 Anti-Hu antibodies have been found in a handful of reports of neurologic deterioration in patients with MCC. Hocar et al21 reported a severe necrotizing myopathy; Greenlee et al22 described a syndrome of progressive sensorimotor and autonomic neuropathy with encephalopathy; and Lopez et al23 described a constellation of vision changes, gait imbalance, and proximal weakness. Support for a pathophysiologic connection among these 3 cases is suggested by the finding of Hu antigen expression by MCC in 2 studies.24,25 Because MCC can present with occult lymph node involvement in the absence of primary cutaneous findings,3 there are more cases of paraneoplastic neurologic syndromes that were not recognized.
Extrapulmonary small cell carcinomas such as MCC are morphologically indistinguishable from their pulmonary counterparts and have been reported in most anatomic regions of the body, including gynecologic organs (eg, ovaries, cervix), genitourinary organs (eg, bladder, prostate), the gastrointestinal tract (eg, esophagus), skin (eg, MCC), and the head and neck region. Extrapulmonary small cell carcinoma is a rare entity, with the most common form found in the gynecologic tract, representing only 2% of gynecologic malignancies.26
Paraneoplastic syndromes of EPSCC are rare given the paucity of the malignancy. Several case reports discuss findings of SIADH in EPSCC of the cervix, as well as hypercalcemia, polyneuropathy, Cushing syndrome, limbic encephalitis, and peripheral neuropathy in EPSCC of the prostate.27,28 In contrast, SCLC has long been associated with paraneoplastic syndromes. Numerous case reports have been published describing SCLC-associated paraneoplastic syndromes to include hypercalcemia, Cushing syndrome, SIADH, vasoactive peptide production, cerebellar degeneration, limbic encephalitis, visceral plexopathy, autonomic dysfunction, and LEMS.29 As more cases of EPSCC with paraneoplastic syndromes are identified and reported, we might gain a better understanding of this interesting phenomenon.
Conclusion
Merkel cell carcinoma is an aggressive neuroendocrine malignancy associated with paraneoplastic neurologic syndromes, including LEMS. A thorough search for an underlying malignancy is highly recommended in patients with diagnosed LEMS without clear cause. Early identification and treatment of the primary tumor can lead to improvement of neurologic symptoms.
We present a case of LEMS with no clearly identifiable cause on presentation with later diagnosis of metastatic MCC of unknown primary origin. After surgical excision of affected lymph nodes and adjuvant radiation therapy, the patient had near-complete resolution of LEMS symptoms at 6-month follow-up, without additional findings of lymphadenopathy or skin lesions. Although this patient is not undergoing routine surveillance imaging to monitor for recurrence of MCC, a chest CT or positron emission tomography–CT for secondary screening would be considered if the patient experienced clinical symptoms consistent with LEMS.
In cases of LEMS without pulmonary malignancy, we recommend considering MCC in the differential diagnosis during the workup of an underlying malignancy
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2010;37:20-27.
- Senchenkov A, Moran SL. Merkel cell carcinoma: diagnosis, management, and outcomes. Plast Reconstr Surg. 2013;131:E771-E778.
- Han SY, North JP, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
- Vernino S. Paraneoplastic disorders affecting the neuromuscular junction or anterior horn cell. CONTINUUM Lifelong Learning in Neurology. 2009;15:132-146.
- Eggers SD, Salomao DR, Dinapoli RP, et al. Paraneoplastic and metastatic neurologic complications of Merkel cell carcinoma. Mayo Clin Proc. 2001;76:327-330.
- Siau RT, Morris A, Karoo RO. Surgery results in complete cure of Lambert-Eaton myasthenic syndrome in a patient with metastatic Merkel cell carcinoma. J Plast Reconstr Aesthet Surg. 2014;67:e162-e164.
- Bombelli F, Lispi L, Calabrò F, et al. Lambert-Eaton myasthenic syndrome associated to Merkel cell carcinoma: report of a case. Neurol Sci. 2015;36:1491-1492.
- Simmons DB, Duginski TM, McClean JC, et al. Lambert-eaton myasthenic syndrome and merkel cell carcinoma. Muscle Nerve. 2015;53:325-326.
- Bobos M, Hytiroglou P, Kostopoulos I, et al. Immunohistochemical distinction between Merkel cell carcinoma and small cell carcinoma of the lung. Am J Dermatopathol. 2006;28:99-104.
- Jensen K, Kohler S, Rouse RV. Cytokeratin staining in Merkel cell carcinoma: an immunohistochemical study of cytokeratins 5/6, 7, 17, and 20. Appl Immunohistochem Mol Morphol. 2000;8:310-315.
- Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011;10:1098-1107.
- Sanders DB. Lambert-Eaton myasthenic syndrome. In: Daroff R, Aminoff MJ, eds. Encyclopedia of the Neurological Sciences. Vol 2. New York, NY: Elsevier; 2009:819-822.
- Wirtz PW, Smallegange TM, Wintzen AR, et al. Differences in clinical features between the Lambert-Eaton myasthenic syndrome with and without cancer: an analysis of 227 published cases. Clin Neurol Neurosurg. 2002;104:359-363.
- Titulaer MJ, Wirtz PW, Willems LN, et al. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. 2008;26:4276-4281.
- Titulaer MJ, Maddison P, Sont JK, et al. Clinical Dutch-English Lambert-Eaton Myasthenic Syndrome (LEMS) Tumor Association prediction score accurately predicts small-cell lung cancer in the LEMS. J Clin Oncol. 2011;7:902-908.
- Cher LM, Hochberg FH, Teruya J, et al. Therapy for paraneoplastic neurologic syndromes in six patients with protein A column immunoadsorption. Cancer. 1995;75:1678-1683.
- Batchelor TT, Platten M, Hochberg FH. Immunoadsorption therapy for paraneoplastic syndromes. J Neurooncol. 1998;40:131-136.
- Blondin NA, Vortmeyer AO, Harel NY. Paraneoplastic syndrome of inappropriate antidiuretic hormone mimicking limbic encephalitis. Arch Neurol. 2011;68:1591-1594.
- Balegno S, Ceroni M, Corato M, et al. Antibodies to cerebellar nerve fibres in two patients with paraneoplastic cerebellar ataxia. Anticancer Res. 2005;25:3211-3214.
- Zhang C, Emery L, Lancaster E. Paraneoplastic cerebellar degeneration associated with noncutaneous Merkel cell carcinoma. Neurol Neuroimmunol Neuroinflamm. 2014;1:e17.
- Hocar O, Poszepczynska-Guigné E, Faye O, et al. Severe necrotizing myopathy subsequent to Merkel cell carcinoma. Ann Dermatol Venereol. 2011;138:130-134.
- Greenlee JE, Steffens JD, Clawson SA, et al. Anti-Hu antibodies in Merkel cell carcinoma. Ann Neurol. 2002;52:111-115.
- Lopez MC, Pericay C, Agustí M, et al. Merkel cell carcinoma associated with a paraneoplastic neurologic syndrome. Histopathology. 2004;44:628-629.
- Dalmau J, Furneaux HM, Cordon-Cardo C, et al. The expression of the Hu (paraneoplastic encephalomyelitis/sensory neuronopathy) antigen in human normal and tumor tissues. Am J Pathol. 1992;141:881-886.
- Gultekin SH, Rosai J, Demopoulos A, et al. Hu immunolabeling as a marker of neural and neuroendocrine differentiation in normal and neoplastic human tissues: assessment using a recombinant anti-Hu Fab fragment. Int J Surg Pathol. 2000;8:109-117.
- Zheng X, Liu D, Fallon JT, et al. Distinct genetic alterations in small cell carcinoma from different anatomic sites. Exp Hematol Oncol. 2015;4:2.
- Kim D, Yun H, Lee Y, et al. Small cell neuroendocrine carcinoma of the uterine cervix presenting with syndrome of inappropriate antidiuretic hormone secretion. Obstet Gynecol Sci. 2013;56:420-425.
- Venkatesh PK, Motwani B, Sherman N, et al. Metastatic pure small-cell carcinoma of prostate. Am J Med Sci. 2004;328:286-289.
- Kaltsas G, Androulakis II, de Herder WW, et al. Paraneoplastic syndromes secondary to neuroendocrine tumours. Endocr Relat Cancer. 2010;17:R173-R193.
Merkel cell carcinoma (MCC) is an aggressive neuroendocrine malignancy of the skin that is thought to arise from neural crest cells. It has an estimated annual incidence of 0.6 per 100,000 individuals, typically occurs in the elderly population, and is most common in white males.1 The tumor presents as a rapidly growing, violaceous nodule in sun-exposed areas of the skin; early in the course, it can be mistaken for a benign entity such as an epidermal cyst.2 Merkel cell carcinoma has a propensity to spread to regional lymph nodes, and in some cases, it occurs in the absence of skin findings.3 Histologically, MCC is nearly indistinguishable from small cell lung carcinoma (SCLC).4 The overall prognosis for patients with MCC is poor and largely dependent on the stage at diagnosis. Patients with regional and distant metastases have a 5-year survival rate of 26% to 42% and 18%, respectively.3
Lambert-Eaton myasthenic syndrome (LEMS) is a paraneoplastic or autoimmune disorder of the neuromuscular junction that is found in 3% of cases of SCLC.4 Reported cases of LEMS in patients with MCC are exceedingly rare.5-8 We provide a full report and longitudinal clinical follow-up of a case that was briefly discussed by Simmons et al,8 and we review the literature regarding paraneoplastic syndromes associated with MCC and other extrapulmonary small cell carcinomas (EPSCCs).
Case Report
A 63-year-old man was evaluated in the neurology clinic due to difficulty walking, climbing stairs, and performing push-ups over the last month. Prior to the onset of symptoms, he was otherwise healthy, walking 3 miles daily; however, at presentation he required use of a cane. Leg weakness worsened as the day progressed. In addition, he reported constipation, urinary urgency, dry mouth, mild dysphagia, reduced sensation below the knees, and a nasal quality in his speech. He had no ptosis, diplopia, dysarthria, muscle cramps, myalgia, or facial weakness. He denied fevers, chills, and night sweats but did admit to an unintentional 10- to 15-lb weight loss over the preceding few months.
The neurologic examination revealed mild proximal upper extremity weakness in the bilateral shoulder abductors, infraspinatus, hip extensors, and hip flexors (Medical Research Council muscle scale grade 4). All deep tendon reflexes, except the Achilles reflex, were present. Despite subjective sensory concerns, objective examination of all sensory modalities was normal. Cranial nerve examination was normal, except for a slight nasal quality to his voice.
A qualitative assay was positive for the presence of P/Q-type voltage-gated calcium channel (VGCC) antibodies. Other laboratory studies were within reference range, including acetylcholine-receptor antibodies (blocking, binding, and modulating) and muscle-specific kinase antibodies.
Lumbar and cervical spine magnetic resonance imaging revealed multilevel neuroforaminal stenosis without spinal canal stenosis or myelopathy. Computed tomography (CT) of the chest was notable for 2 pathologically enlarged lymph nodes in the left axilla and no evidence of primary pulmonary malignancy. Nerve-conduction studies (NCSs) in conjunction with other clinical findings were consistent with the diagnosis of LEMS.
Ultrasound-guided biopsy of the enlarged axillary lymph nodes demonstrated sheets and nests of small round blue tumor cells with minimal cytoplasm, high mitotic rate, and foci of necrosis (Figure 1). The tumor cells were positive for pancytokeratin (Lu-5) and cytokeratin (CK) 20 in a perinuclear dotlike pattern (Figure 2), as well as for the neuroendocrine markers synaptophysin (Figure 3), chromogranin A, and CD56. The tumor cells showed no immunoreactivity for CK7, thyroid transcription factor 1, CD3, CD5, or CD20. Flow cytometry demonstrated low cellularity, low specimen viability, and no evidence of an abnormal B-cell population. These findings were consistent with the diagnosis of MCC.
The patient underwent surgical excision of the involved lymph nodes. Four weeks after surgery, he reported dramatic improvement in strength, with complete resolution of the nasal speech, dysphagia, dry mouth, urinary retention, and constipation. Two months after surgery, his strength had normalized, except for slight persistent weakness in the bilateral shoulder abductors, trace weakness in the hip flexors, and a slight Trendelenburg gait. He was able to rise from a chair without using his arms and no longer required a cane for ambulation.
The patient underwent adjuvant radiation therapy after 2-month surgical follow-up with 5000-cGy radiation treatment to the left axillary region. Six months following primary definitive surgery and 4 months following adjuvant radiation therapy, he reported a 95% subjective return of physical strength. The patient was able to return to near-baseline physical activity. He continued to deny symptoms of dry mouth, incontinence, or constipation. Objectively, he had no focal neurologic deficits or weakness; no evidence of new skin lesions or lymphadenopathy was noted.
Comment
MCC vs SCLC
Merkel cell carcinoma is classified as a type of EPSCC. The histologic appearance of MCC is indistinguishable from SCLC. Both tumors are composed of uniform sheets of small round cells with a high nucleus to cytoplasm ratio, and both can express neuroendocrine markers, such as neuron-specific enolase, chromogranin A, and synaptophysin.9 Immunohistochemical positivity for CK20 and neurofilaments in combination with negative staining for thyroid transcription factor 1 and CK7 effectively differentiate MCC from SCLC.9 In addition, MCC often displays CK20 positivity in a perinuclear dotlike or punctate pattern, which is characteristic of this tumor.3,9,10 Negative immunohistochemical markers for B cells (CD20) and T cells (CD3) are important in excluding lymphoma.
LEMS Diagnosis
Lambert-Eaton myasthenic syndrome is a paraneoplastic or autoimmune disorder involving the neuromuscular junction. Autoantibodies to VGCC impair calcium influx into the presynaptic terminal, resulting in marked reduction of acetylcholine release into the synaptic cleft. The reduction in acetylcholine activity impairs production of muscle fiber action potentials, resulting in clinical weakness. The diagnosis of LEMS rests on clinical presentation, positive serology, and confirmatory neurophysiologic testing by NCS. Clinically, patients present with proximal weakness, hyporeflexia or areflexia, and autonomic dysfunction. Antibodies to P/Q-type VGCCs are found in 85% to 90% of cases of LEMS and are thought to play a direct causative role in the development of weakness.11 The finding of postexercise facilitation on motor NCS is the neurophysiologic hallmark and is highly specific for the diagnosis.
Approximately 50% to 60% of patients who present with LEMS have an underlying tumor, the vast majority of which are SCLC.11 There are a few reports of LEMS associated with other malignancies, including lymphoma; thymoma; neuroblastoma; and carcinoma of the breast, stomach, prostate, bladder, kidney, and gallbladder.12 Patients with nontumor or autoimmune LEMS tend to be younger, and there is no male predominance, as there is in paraneoplastic LEMS.13 Given the risk of underlying malignancy in LEMS, Titulaer et al14 proposed a screening protocol for patients presenting with LEMS, recommending initial primary screening using CT of the chest. If the CT scan is negative, total-body fludeoxyglucose positron emission tomography should be performed to assess for fludeoxyglucose avid lesions. If both initial studies are negative, routine follow-up with CT of the chest at 6-month intervals for a minimum of 2 to 4 years after the initial diagnosis of LEMS was recommended. An exception to this protocol was suggested to allow consideration to stop screening after the first 6-month follow-up chest CT for patients younger than 45 years who have never smoked and who have an HLA 8.1 haplotype for which nontumor LEMS would be a more probable diagnosis.14
In addition to a screening protocol, a validated prediction tool, the Dutch-English LEMS Tumor Association prediction score, was developed. It uses common signs and symptoms of LEMS and risk factors for SCLC to help guide the need for further screening.15
Paraneoplastic Syndromes Associated With MCC
Other paraneoplastic syndromes have been reported in association with MCC. A patient with brainstem encephalitis associated with MCC was reported in a trial of a novel immunotherapy for paraneoplastic neurologic syndromes.16,17 A syndrome of inappropriate antidiuretic hormone (SIADH) secretion was reported in a patient with N-type calcium channel antibodies.18 Two cases of paraneoplastic cerebellar degeneration have been reported; the first was associated with a novel 70-kD antibody,19 and the second was associated with the P/Q-type VGCC antibody.20 Anti-Hu antibodies have been found in a handful of reports of neurologic deterioration in patients with MCC. Hocar et al21 reported a severe necrotizing myopathy; Greenlee et al22 described a syndrome of progressive sensorimotor and autonomic neuropathy with encephalopathy; and Lopez et al23 described a constellation of vision changes, gait imbalance, and proximal weakness. Support for a pathophysiologic connection among these 3 cases is suggested by the finding of Hu antigen expression by MCC in 2 studies.24,25 Because MCC can present with occult lymph node involvement in the absence of primary cutaneous findings,3 there are more cases of paraneoplastic neurologic syndromes that were not recognized.
Extrapulmonary small cell carcinomas such as MCC are morphologically indistinguishable from their pulmonary counterparts and have been reported in most anatomic regions of the body, including gynecologic organs (eg, ovaries, cervix), genitourinary organs (eg, bladder, prostate), the gastrointestinal tract (eg, esophagus), skin (eg, MCC), and the head and neck region. Extrapulmonary small cell carcinoma is a rare entity, with the most common form found in the gynecologic tract, representing only 2% of gynecologic malignancies.26
Paraneoplastic syndromes of EPSCC are rare given the paucity of the malignancy. Several case reports discuss findings of SIADH in EPSCC of the cervix, as well as hypercalcemia, polyneuropathy, Cushing syndrome, limbic encephalitis, and peripheral neuropathy in EPSCC of the prostate.27,28 In contrast, SCLC has long been associated with paraneoplastic syndromes. Numerous case reports have been published describing SCLC-associated paraneoplastic syndromes to include hypercalcemia, Cushing syndrome, SIADH, vasoactive peptide production, cerebellar degeneration, limbic encephalitis, visceral plexopathy, autonomic dysfunction, and LEMS.29 As more cases of EPSCC with paraneoplastic syndromes are identified and reported, we might gain a better understanding of this interesting phenomenon.
Conclusion
Merkel cell carcinoma is an aggressive neuroendocrine malignancy associated with paraneoplastic neurologic syndromes, including LEMS. A thorough search for an underlying malignancy is highly recommended in patients with diagnosed LEMS without clear cause. Early identification and treatment of the primary tumor can lead to improvement of neurologic symptoms.
We present a case of LEMS with no clearly identifiable cause on presentation with later diagnosis of metastatic MCC of unknown primary origin. After surgical excision of affected lymph nodes and adjuvant radiation therapy, the patient had near-complete resolution of LEMS symptoms at 6-month follow-up, without additional findings of lymphadenopathy or skin lesions. Although this patient is not undergoing routine surveillance imaging to monitor for recurrence of MCC, a chest CT or positron emission tomography–CT for secondary screening would be considered if the patient experienced clinical symptoms consistent with LEMS.
In cases of LEMS without pulmonary malignancy, we recommend considering MCC in the differential diagnosis during the workup of an underlying malignancy
Merkel cell carcinoma (MCC) is an aggressive neuroendocrine malignancy of the skin that is thought to arise from neural crest cells. It has an estimated annual incidence of 0.6 per 100,000 individuals, typically occurs in the elderly population, and is most common in white males.1 The tumor presents as a rapidly growing, violaceous nodule in sun-exposed areas of the skin; early in the course, it can be mistaken for a benign entity such as an epidermal cyst.2 Merkel cell carcinoma has a propensity to spread to regional lymph nodes, and in some cases, it occurs in the absence of skin findings.3 Histologically, MCC is nearly indistinguishable from small cell lung carcinoma (SCLC).4 The overall prognosis for patients with MCC is poor and largely dependent on the stage at diagnosis. Patients with regional and distant metastases have a 5-year survival rate of 26% to 42% and 18%, respectively.3
Lambert-Eaton myasthenic syndrome (LEMS) is a paraneoplastic or autoimmune disorder of the neuromuscular junction that is found in 3% of cases of SCLC.4 Reported cases of LEMS in patients with MCC are exceedingly rare.5-8 We provide a full report and longitudinal clinical follow-up of a case that was briefly discussed by Simmons et al,8 and we review the literature regarding paraneoplastic syndromes associated with MCC and other extrapulmonary small cell carcinomas (EPSCCs).
Case Report
A 63-year-old man was evaluated in the neurology clinic due to difficulty walking, climbing stairs, and performing push-ups over the last month. Prior to the onset of symptoms, he was otherwise healthy, walking 3 miles daily; however, at presentation he required use of a cane. Leg weakness worsened as the day progressed. In addition, he reported constipation, urinary urgency, dry mouth, mild dysphagia, reduced sensation below the knees, and a nasal quality in his speech. He had no ptosis, diplopia, dysarthria, muscle cramps, myalgia, or facial weakness. He denied fevers, chills, and night sweats but did admit to an unintentional 10- to 15-lb weight loss over the preceding few months.
The neurologic examination revealed mild proximal upper extremity weakness in the bilateral shoulder abductors, infraspinatus, hip extensors, and hip flexors (Medical Research Council muscle scale grade 4). All deep tendon reflexes, except the Achilles reflex, were present. Despite subjective sensory concerns, objective examination of all sensory modalities was normal. Cranial nerve examination was normal, except for a slight nasal quality to his voice.
A qualitative assay was positive for the presence of P/Q-type voltage-gated calcium channel (VGCC) antibodies. Other laboratory studies were within reference range, including acetylcholine-receptor antibodies (blocking, binding, and modulating) and muscle-specific kinase antibodies.
Lumbar and cervical spine magnetic resonance imaging revealed multilevel neuroforaminal stenosis without spinal canal stenosis or myelopathy. Computed tomography (CT) of the chest was notable for 2 pathologically enlarged lymph nodes in the left axilla and no evidence of primary pulmonary malignancy. Nerve-conduction studies (NCSs) in conjunction with other clinical findings were consistent with the diagnosis of LEMS.
Ultrasound-guided biopsy of the enlarged axillary lymph nodes demonstrated sheets and nests of small round blue tumor cells with minimal cytoplasm, high mitotic rate, and foci of necrosis (Figure 1). The tumor cells were positive for pancytokeratin (Lu-5) and cytokeratin (CK) 20 in a perinuclear dotlike pattern (Figure 2), as well as for the neuroendocrine markers synaptophysin (Figure 3), chromogranin A, and CD56. The tumor cells showed no immunoreactivity for CK7, thyroid transcription factor 1, CD3, CD5, or CD20. Flow cytometry demonstrated low cellularity, low specimen viability, and no evidence of an abnormal B-cell population. These findings were consistent with the diagnosis of MCC.
The patient underwent surgical excision of the involved lymph nodes. Four weeks after surgery, he reported dramatic improvement in strength, with complete resolution of the nasal speech, dysphagia, dry mouth, urinary retention, and constipation. Two months after surgery, his strength had normalized, except for slight persistent weakness in the bilateral shoulder abductors, trace weakness in the hip flexors, and a slight Trendelenburg gait. He was able to rise from a chair without using his arms and no longer required a cane for ambulation.
The patient underwent adjuvant radiation therapy after 2-month surgical follow-up with 5000-cGy radiation treatment to the left axillary region. Six months following primary definitive surgery and 4 months following adjuvant radiation therapy, he reported a 95% subjective return of physical strength. The patient was able to return to near-baseline physical activity. He continued to deny symptoms of dry mouth, incontinence, or constipation. Objectively, he had no focal neurologic deficits or weakness; no evidence of new skin lesions or lymphadenopathy was noted.
Comment
MCC vs SCLC
Merkel cell carcinoma is classified as a type of EPSCC. The histologic appearance of MCC is indistinguishable from SCLC. Both tumors are composed of uniform sheets of small round cells with a high nucleus to cytoplasm ratio, and both can express neuroendocrine markers, such as neuron-specific enolase, chromogranin A, and synaptophysin.9 Immunohistochemical positivity for CK20 and neurofilaments in combination with negative staining for thyroid transcription factor 1 and CK7 effectively differentiate MCC from SCLC.9 In addition, MCC often displays CK20 positivity in a perinuclear dotlike or punctate pattern, which is characteristic of this tumor.3,9,10 Negative immunohistochemical markers for B cells (CD20) and T cells (CD3) are important in excluding lymphoma.
LEMS Diagnosis
Lambert-Eaton myasthenic syndrome is a paraneoplastic or autoimmune disorder involving the neuromuscular junction. Autoantibodies to VGCC impair calcium influx into the presynaptic terminal, resulting in marked reduction of acetylcholine release into the synaptic cleft. The reduction in acetylcholine activity impairs production of muscle fiber action potentials, resulting in clinical weakness. The diagnosis of LEMS rests on clinical presentation, positive serology, and confirmatory neurophysiologic testing by NCS. Clinically, patients present with proximal weakness, hyporeflexia or areflexia, and autonomic dysfunction. Antibodies to P/Q-type VGCCs are found in 85% to 90% of cases of LEMS and are thought to play a direct causative role in the development of weakness.11 The finding of postexercise facilitation on motor NCS is the neurophysiologic hallmark and is highly specific for the diagnosis.
Approximately 50% to 60% of patients who present with LEMS have an underlying tumor, the vast majority of which are SCLC.11 There are a few reports of LEMS associated with other malignancies, including lymphoma; thymoma; neuroblastoma; and carcinoma of the breast, stomach, prostate, bladder, kidney, and gallbladder.12 Patients with nontumor or autoimmune LEMS tend to be younger, and there is no male predominance, as there is in paraneoplastic LEMS.13 Given the risk of underlying malignancy in LEMS, Titulaer et al14 proposed a screening protocol for patients presenting with LEMS, recommending initial primary screening using CT of the chest. If the CT scan is negative, total-body fludeoxyglucose positron emission tomography should be performed to assess for fludeoxyglucose avid lesions. If both initial studies are negative, routine follow-up with CT of the chest at 6-month intervals for a minimum of 2 to 4 years after the initial diagnosis of LEMS was recommended. An exception to this protocol was suggested to allow consideration to stop screening after the first 6-month follow-up chest CT for patients younger than 45 years who have never smoked and who have an HLA 8.1 haplotype for which nontumor LEMS would be a more probable diagnosis.14
In addition to a screening protocol, a validated prediction tool, the Dutch-English LEMS Tumor Association prediction score, was developed. It uses common signs and symptoms of LEMS and risk factors for SCLC to help guide the need for further screening.15
Paraneoplastic Syndromes Associated With MCC
Other paraneoplastic syndromes have been reported in association with MCC. A patient with brainstem encephalitis associated with MCC was reported in a trial of a novel immunotherapy for paraneoplastic neurologic syndromes.16,17 A syndrome of inappropriate antidiuretic hormone (SIADH) secretion was reported in a patient with N-type calcium channel antibodies.18 Two cases of paraneoplastic cerebellar degeneration have been reported; the first was associated with a novel 70-kD antibody,19 and the second was associated with the P/Q-type VGCC antibody.20 Anti-Hu antibodies have been found in a handful of reports of neurologic deterioration in patients with MCC. Hocar et al21 reported a severe necrotizing myopathy; Greenlee et al22 described a syndrome of progressive sensorimotor and autonomic neuropathy with encephalopathy; and Lopez et al23 described a constellation of vision changes, gait imbalance, and proximal weakness. Support for a pathophysiologic connection among these 3 cases is suggested by the finding of Hu antigen expression by MCC in 2 studies.24,25 Because MCC can present with occult lymph node involvement in the absence of primary cutaneous findings,3 there are more cases of paraneoplastic neurologic syndromes that were not recognized.
Extrapulmonary small cell carcinomas such as MCC are morphologically indistinguishable from their pulmonary counterparts and have been reported in most anatomic regions of the body, including gynecologic organs (eg, ovaries, cervix), genitourinary organs (eg, bladder, prostate), the gastrointestinal tract (eg, esophagus), skin (eg, MCC), and the head and neck region. Extrapulmonary small cell carcinoma is a rare entity, with the most common form found in the gynecologic tract, representing only 2% of gynecologic malignancies.26
Paraneoplastic syndromes of EPSCC are rare given the paucity of the malignancy. Several case reports discuss findings of SIADH in EPSCC of the cervix, as well as hypercalcemia, polyneuropathy, Cushing syndrome, limbic encephalitis, and peripheral neuropathy in EPSCC of the prostate.27,28 In contrast, SCLC has long been associated with paraneoplastic syndromes. Numerous case reports have been published describing SCLC-associated paraneoplastic syndromes to include hypercalcemia, Cushing syndrome, SIADH, vasoactive peptide production, cerebellar degeneration, limbic encephalitis, visceral plexopathy, autonomic dysfunction, and LEMS.29 As more cases of EPSCC with paraneoplastic syndromes are identified and reported, we might gain a better understanding of this interesting phenomenon.
Conclusion
Merkel cell carcinoma is an aggressive neuroendocrine malignancy associated with paraneoplastic neurologic syndromes, including LEMS. A thorough search for an underlying malignancy is highly recommended in patients with diagnosed LEMS without clear cause. Early identification and treatment of the primary tumor can lead to improvement of neurologic symptoms.
We present a case of LEMS with no clearly identifiable cause on presentation with later diagnosis of metastatic MCC of unknown primary origin. After surgical excision of affected lymph nodes and adjuvant radiation therapy, the patient had near-complete resolution of LEMS symptoms at 6-month follow-up, without additional findings of lymphadenopathy or skin lesions. Although this patient is not undergoing routine surveillance imaging to monitor for recurrence of MCC, a chest CT or positron emission tomography–CT for secondary screening would be considered if the patient experienced clinical symptoms consistent with LEMS.
In cases of LEMS without pulmonary malignancy, we recommend considering MCC in the differential diagnosis during the workup of an underlying malignancy
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2010;37:20-27.
- Senchenkov A, Moran SL. Merkel cell carcinoma: diagnosis, management, and outcomes. Plast Reconstr Surg. 2013;131:E771-E778.
- Han SY, North JP, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
- Vernino S. Paraneoplastic disorders affecting the neuromuscular junction or anterior horn cell. CONTINUUM Lifelong Learning in Neurology. 2009;15:132-146.
- Eggers SD, Salomao DR, Dinapoli RP, et al. Paraneoplastic and metastatic neurologic complications of Merkel cell carcinoma. Mayo Clin Proc. 2001;76:327-330.
- Siau RT, Morris A, Karoo RO. Surgery results in complete cure of Lambert-Eaton myasthenic syndrome in a patient with metastatic Merkel cell carcinoma. J Plast Reconstr Aesthet Surg. 2014;67:e162-e164.
- Bombelli F, Lispi L, Calabrò F, et al. Lambert-Eaton myasthenic syndrome associated to Merkel cell carcinoma: report of a case. Neurol Sci. 2015;36:1491-1492.
- Simmons DB, Duginski TM, McClean JC, et al. Lambert-eaton myasthenic syndrome and merkel cell carcinoma. Muscle Nerve. 2015;53:325-326.
- Bobos M, Hytiroglou P, Kostopoulos I, et al. Immunohistochemical distinction between Merkel cell carcinoma and small cell carcinoma of the lung. Am J Dermatopathol. 2006;28:99-104.
- Jensen K, Kohler S, Rouse RV. Cytokeratin staining in Merkel cell carcinoma: an immunohistochemical study of cytokeratins 5/6, 7, 17, and 20. Appl Immunohistochem Mol Morphol. 2000;8:310-315.
- Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011;10:1098-1107.
- Sanders DB. Lambert-Eaton myasthenic syndrome. In: Daroff R, Aminoff MJ, eds. Encyclopedia of the Neurological Sciences. Vol 2. New York, NY: Elsevier; 2009:819-822.
- Wirtz PW, Smallegange TM, Wintzen AR, et al. Differences in clinical features between the Lambert-Eaton myasthenic syndrome with and without cancer: an analysis of 227 published cases. Clin Neurol Neurosurg. 2002;104:359-363.
- Titulaer MJ, Wirtz PW, Willems LN, et al. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. 2008;26:4276-4281.
- Titulaer MJ, Maddison P, Sont JK, et al. Clinical Dutch-English Lambert-Eaton Myasthenic Syndrome (LEMS) Tumor Association prediction score accurately predicts small-cell lung cancer in the LEMS. J Clin Oncol. 2011;7:902-908.
- Cher LM, Hochberg FH, Teruya J, et al. Therapy for paraneoplastic neurologic syndromes in six patients with protein A column immunoadsorption. Cancer. 1995;75:1678-1683.
- Batchelor TT, Platten M, Hochberg FH. Immunoadsorption therapy for paraneoplastic syndromes. J Neurooncol. 1998;40:131-136.
- Blondin NA, Vortmeyer AO, Harel NY. Paraneoplastic syndrome of inappropriate antidiuretic hormone mimicking limbic encephalitis. Arch Neurol. 2011;68:1591-1594.
- Balegno S, Ceroni M, Corato M, et al. Antibodies to cerebellar nerve fibres in two patients with paraneoplastic cerebellar ataxia. Anticancer Res. 2005;25:3211-3214.
- Zhang C, Emery L, Lancaster E. Paraneoplastic cerebellar degeneration associated with noncutaneous Merkel cell carcinoma. Neurol Neuroimmunol Neuroinflamm. 2014;1:e17.
- Hocar O, Poszepczynska-Guigné E, Faye O, et al. Severe necrotizing myopathy subsequent to Merkel cell carcinoma. Ann Dermatol Venereol. 2011;138:130-134.
- Greenlee JE, Steffens JD, Clawson SA, et al. Anti-Hu antibodies in Merkel cell carcinoma. Ann Neurol. 2002;52:111-115.
- Lopez MC, Pericay C, Agustí M, et al. Merkel cell carcinoma associated with a paraneoplastic neurologic syndrome. Histopathology. 2004;44:628-629.
- Dalmau J, Furneaux HM, Cordon-Cardo C, et al. The expression of the Hu (paraneoplastic encephalomyelitis/sensory neuronopathy) antigen in human normal and tumor tissues. Am J Pathol. 1992;141:881-886.
- Gultekin SH, Rosai J, Demopoulos A, et al. Hu immunolabeling as a marker of neural and neuroendocrine differentiation in normal and neoplastic human tissues: assessment using a recombinant anti-Hu Fab fragment. Int J Surg Pathol. 2000;8:109-117.
- Zheng X, Liu D, Fallon JT, et al. Distinct genetic alterations in small cell carcinoma from different anatomic sites. Exp Hematol Oncol. 2015;4:2.
- Kim D, Yun H, Lee Y, et al. Small cell neuroendocrine carcinoma of the uterine cervix presenting with syndrome of inappropriate antidiuretic hormone secretion. Obstet Gynecol Sci. 2013;56:420-425.
- Venkatesh PK, Motwani B, Sherman N, et al. Metastatic pure small-cell carcinoma of prostate. Am J Med Sci. 2004;328:286-289.
- Kaltsas G, Androulakis II, de Herder WW, et al. Paraneoplastic syndromes secondary to neuroendocrine tumours. Endocr Relat Cancer. 2010;17:R173-R193.
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2010;37:20-27.
- Senchenkov A, Moran SL. Merkel cell carcinoma: diagnosis, management, and outcomes. Plast Reconstr Surg. 2013;131:E771-E778.
- Han SY, North JP, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
- Vernino S. Paraneoplastic disorders affecting the neuromuscular junction or anterior horn cell. CONTINUUM Lifelong Learning in Neurology. 2009;15:132-146.
- Eggers SD, Salomao DR, Dinapoli RP, et al. Paraneoplastic and metastatic neurologic complications of Merkel cell carcinoma. Mayo Clin Proc. 2001;76:327-330.
- Siau RT, Morris A, Karoo RO. Surgery results in complete cure of Lambert-Eaton myasthenic syndrome in a patient with metastatic Merkel cell carcinoma. J Plast Reconstr Aesthet Surg. 2014;67:e162-e164.
- Bombelli F, Lispi L, Calabrò F, et al. Lambert-Eaton myasthenic syndrome associated to Merkel cell carcinoma: report of a case. Neurol Sci. 2015;36:1491-1492.
- Simmons DB, Duginski TM, McClean JC, et al. Lambert-eaton myasthenic syndrome and merkel cell carcinoma. Muscle Nerve. 2015;53:325-326.
- Bobos M, Hytiroglou P, Kostopoulos I, et al. Immunohistochemical distinction between Merkel cell carcinoma and small cell carcinoma of the lung. Am J Dermatopathol. 2006;28:99-104.
- Jensen K, Kohler S, Rouse RV. Cytokeratin staining in Merkel cell carcinoma: an immunohistochemical study of cytokeratins 5/6, 7, 17, and 20. Appl Immunohistochem Mol Morphol. 2000;8:310-315.
- Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011;10:1098-1107.
- Sanders DB. Lambert-Eaton myasthenic syndrome. In: Daroff R, Aminoff MJ, eds. Encyclopedia of the Neurological Sciences. Vol 2. New York, NY: Elsevier; 2009:819-822.
- Wirtz PW, Smallegange TM, Wintzen AR, et al. Differences in clinical features between the Lambert-Eaton myasthenic syndrome with and without cancer: an analysis of 227 published cases. Clin Neurol Neurosurg. 2002;104:359-363.
- Titulaer MJ, Wirtz PW, Willems LN, et al. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. 2008;26:4276-4281.
- Titulaer MJ, Maddison P, Sont JK, et al. Clinical Dutch-English Lambert-Eaton Myasthenic Syndrome (LEMS) Tumor Association prediction score accurately predicts small-cell lung cancer in the LEMS. J Clin Oncol. 2011;7:902-908.
- Cher LM, Hochberg FH, Teruya J, et al. Therapy for paraneoplastic neurologic syndromes in six patients with protein A column immunoadsorption. Cancer. 1995;75:1678-1683.
- Batchelor TT, Platten M, Hochberg FH. Immunoadsorption therapy for paraneoplastic syndromes. J Neurooncol. 1998;40:131-136.
- Blondin NA, Vortmeyer AO, Harel NY. Paraneoplastic syndrome of inappropriate antidiuretic hormone mimicking limbic encephalitis. Arch Neurol. 2011;68:1591-1594.
- Balegno S, Ceroni M, Corato M, et al. Antibodies to cerebellar nerve fibres in two patients with paraneoplastic cerebellar ataxia. Anticancer Res. 2005;25:3211-3214.
- Zhang C, Emery L, Lancaster E. Paraneoplastic cerebellar degeneration associated with noncutaneous Merkel cell carcinoma. Neurol Neuroimmunol Neuroinflamm. 2014;1:e17.
- Hocar O, Poszepczynska-Guigné E, Faye O, et al. Severe necrotizing myopathy subsequent to Merkel cell carcinoma. Ann Dermatol Venereol. 2011;138:130-134.
- Greenlee JE, Steffens JD, Clawson SA, et al. Anti-Hu antibodies in Merkel cell carcinoma. Ann Neurol. 2002;52:111-115.
- Lopez MC, Pericay C, Agustí M, et al. Merkel cell carcinoma associated with a paraneoplastic neurologic syndrome. Histopathology. 2004;44:628-629.
- Dalmau J, Furneaux HM, Cordon-Cardo C, et al. The expression of the Hu (paraneoplastic encephalomyelitis/sensory neuronopathy) antigen in human normal and tumor tissues. Am J Pathol. 1992;141:881-886.
- Gultekin SH, Rosai J, Demopoulos A, et al. Hu immunolabeling as a marker of neural and neuroendocrine differentiation in normal and neoplastic human tissues: assessment using a recombinant anti-Hu Fab fragment. Int J Surg Pathol. 2000;8:109-117.
- Zheng X, Liu D, Fallon JT, et al. Distinct genetic alterations in small cell carcinoma from different anatomic sites. Exp Hematol Oncol. 2015;4:2.
- Kim D, Yun H, Lee Y, et al. Small cell neuroendocrine carcinoma of the uterine cervix presenting with syndrome of inappropriate antidiuretic hormone secretion. Obstet Gynecol Sci. 2013;56:420-425.
- Venkatesh PK, Motwani B, Sherman N, et al. Metastatic pure small-cell carcinoma of prostate. Am J Med Sci. 2004;328:286-289.
- Kaltsas G, Androulakis II, de Herder WW, et al. Paraneoplastic syndromes secondary to neuroendocrine tumours. Endocr Relat Cancer. 2010;17:R173-R193.
Practice Points
- Approximately 50% to 60% of patients with Lambert-Eaton myasthenic syndrome (LEMS) have an underlying tumor, most commonly small cell lung carcinoma.
- A thorough search for an underlying malignancy is highly recommended in patients with diagnosed LEMS without clear cause; to this end, a screening protocol comprising computed tomography and total-body fludeoxyglucose positron emission tomography has been established.
- Because Merkel cell carcinoma (MCC) can present as occult lymph node involvement with primary cutaneous findings absent, it is recommended that MCC be considered in the differential diagnosis of an underlying malignancy in a LEMS patient.
- Early identification and treatment of the primary tumor can lead to improvement of neurologic symptoms.
Novel Neuroendocrine Tumor in Multiple Endocrine Neoplasia Type 1 (FULL)
Neuroendocrine tumors (NETs) are uncommon and can occur in the context of genetic conditions. Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant disorder of the tumor suppressor gene of the same name—MEN1, which encodes for the protein menin. Multiple endocrine neoplasia type 1 is characterized clinically by the presence of 2 or more of the following NETs: parathyroid, pituitary, and pancreaticoduodenal.1 Pancreaticoduodenal NETs occur in 30% to 80% of patients with MEN1 and have malignant potential. Although the majority of pancreaticoduodenal NETs are nonfunctioning, patients may present with symptoms secondary to mass effect.
Genetic testing exists for MEN1, but not all genetic mutations that cause MEN1 have been discovered. Therefore, because negative genetic testing does not rule out MEN1, a diagnosis is based on tumor type and location. Neuroendocrine tumors of the biliary tree are rare, and there
are no well-accepted guidelines on how to stage them.2-4 The following case demonstrates an unusual initial presentation of a NET in the context of MEN1.
Case Report
A 29-year-old, active-duty African-American man deployed in Kuwait presented with icterus, flank pain, and hematuria. His past medical history was significant for nephrolithiasis, and his family history was notable for hyperparathyroidism. Laboratory results showed primary hyperparathyroidism and evidence of biliary obstruction.
A sestamibi scan demonstrated uptake in a location corresponding with the right inferior parathyroid gland. A computed tomography (CT) scan showed nephrolithiasis and hepatic biliary ductal dilatation. Magnetic resonance cholangiopancreatography (MRCP) revealed both intra- and extrahepatic ductal dilatation, focal narrowing of the proximal common bile duct, and possible adenopathy that was concerning for cholangiocarcinoma. Endoscopic retrograde cholangiopancreatography (ERCP) demonstrated a 1 cm to 2 cm focal stricture within the mid-common bile duct with intra- and extrahepatic ductal dilatation (Figure 1). An endoscopy showed no masses in the duodenum, and anendoscopic ultrasound showed no masses in the pancreas. Endoscopic brushings and endoscopic, ultrasound-guided, fine-needle aspiration
cytology were nondiagnostic. Exploratory laparotomy revealed a dilated hepatic bile duct, an inflamed porta hepatis, and a mass involving the distal hepatic bile duct. 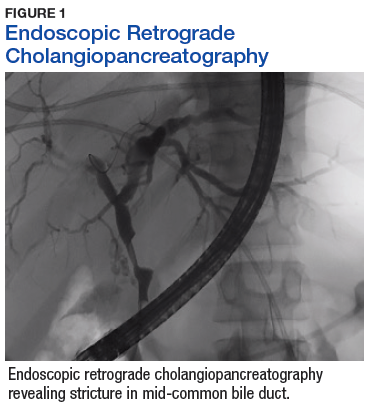
The patient underwent cholecystectomy, radical extra hepatic bile duct resection to the level of the hepatic bifurcation, and hepaticojejunostomy. Gross examination of the specimen showed a nodule centered in the distal common hepatic duct with an adjacent, 2-cm lymph node. The histologic examination revealed a neoplastic proliferation consisting of epithelioid cells with round nuclei and granular chromatin with amphophilic cytoplasm in a trabecular and nested architecture.
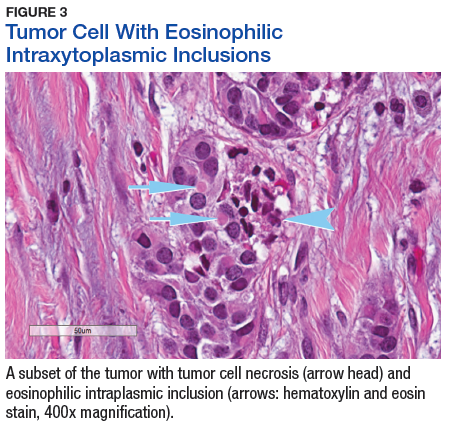
The tumor was centered in the submucosa, which is typical of gastrointestinal NETs (Figure 2). There was no evidence of direct tumor extension elsewhere. About 40% of the tumor cells contained eosinophilic, intracytoplasmic inclusions (Figure 3). The tumor did not involve the margins or lymph node. 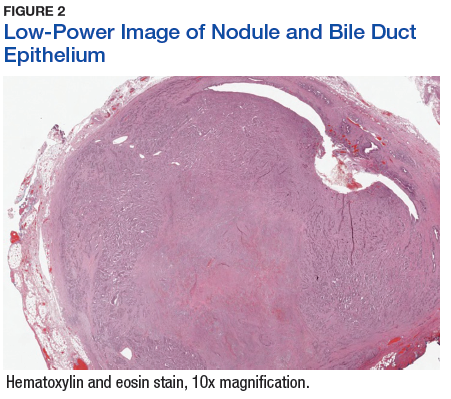
Positive staining with the neuroendocrine markers synaptophysin and chromagranin A confirmed a well-differentiated NET. The intracytoplasmic inclusions stained strongly positive for cytokeratin CAM 5.2. The tumor had higher-grade features, including tumor cell necrosis, a Ki-67 labeling index of 3%, and perineural invasion. The 2010 World Health Organization (WHO) criteria for NET of the digestive system classified this tumor as a grade 2, well-differentiated NET and as stage 1a (limited to the bile duct).4
Postoperatively, octreotide scan with single-photon emission computed tomography (SPECT)-CT did not show additional masses or lesions. Serum pancreatic polypeptide was elevated, with the remaining serum and plasma NET markers—including gastrin, glucagon, insulin, chromogranin A, and vasoactive intestinal polypeptide (VIP)—being within reference ranges. Genetic testing (GeneDx, Inc, Gaithersburg, MD) showed an E563X nonsense mutation in the MEN1 gene, confirming a MEN1 disorder. The patient then underwent a 4-gland parathyroidectomy with reimplantation; the parathyroid glands demonstrated hyperplasia in all 4 glands.
Biochemical follow-up at 14 months showed that the serum pancreatic polypeptide had normalized. There was no evidence of pituitary orpancreatic hypersecretion. The patient developed hypoparathyroidism, requiring calcium and calcitriol supplementation. Radiographic follow-up using abdominal magnetic resonance imaging at 16 months showed no evidence of disease.
Discussion
This case illustrates a genetic disease with an unusual initial presentation. Primary extrahepatic bile duct NETs are rare and have been reported previously in patients without MEN1.5-9 Neuroendocrine tumors in the hepatic bile duct in patients with MEN1 also have been reported but only after these tumors first appeared in the pancreas or duodenum.10 An extensive literature search revealed no prior reports extrahepatic bile duct NETs with MEN1 as the primary site or with biliary obstruction, which is why this patient’s presentation is particularly interesting.5,6,10-13 The table summarizes select reports of NETs. 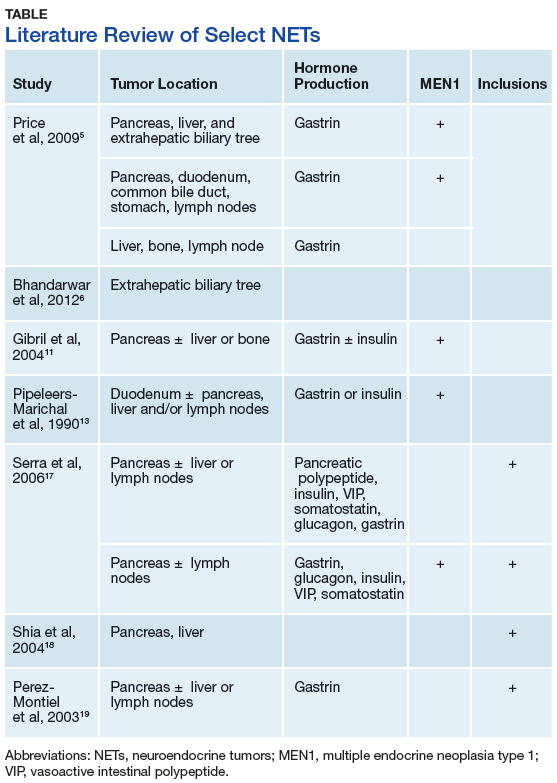
Tumor location in this patient was atypical, and genetic testing guided the management. Serum MEN1 genetic testing is indicated in patients with ≥ 2 tumors that are atypical but possibly associated with MEN1 (such as adrenal tumors, gastrinomas, and carcinoids) and in patients aged < 45 years with primary hyperparathyroidism.14,15 The patient in this study was aged 29 years and had hyperparathyroidism and an NET of the hepatic bile duct. This condition was sufficient to warrant genetic testing, the results of which affected the patient’s subsequent parathyroid surgery.15 Despite the suggestion of unifocal localization on the sestamibi scan, the patient underwent the more appropriate subtotal parathyroidectomy.14 The patient’s tumor most likely originated from a germline mutation of the MEN1 gene.
As a result of the patient’s genetic test results, his daughter also was tested. She was found to have the same mutation as her father and will undergo proper tumor surveillance for MEN1. There was no personal or family history of hemangioblastomas, renal cell carcinomas, or cystadenomas, which would have prompted testing for von Hippel-Lindau disease. Likewise, there was no personal or family history of café-au-lait macules and neurofibromas, which would have prompted testing for neurofibromatosis type 1.
Due to the paucity of cases, there are currently no well-accepted guidelines on how to stage extrahepatic biliary NETs.3-5,16 The WHO recommends staging according to adenocarcinomas of the gallbladder and bile duct.3 As such, the pathologic stage of this tumor would be stage 1a.
The significance of the intracytoplasmic inclusion in this case is unknown. Pancreatic NETs and neuroendocrine carcinomas have demonstrated intracytoplasmic inclusions that stain positively for keratin and may indicate more aggressive tumor behavior.17-19 In 1 report, electron microscopic examination demonstrated intermediate filaments with entrapped neurosecretory granules.18 In a series of 84 cases of pancreatic endocrine tumors, 14 had intracytoplasmic inclusions; of these, 5 had MEN1.17 In the present case, the patient continues to show no evidence of tumor recurrence at 16 months after resection.
Conclusion
Extrahepatic biliary neuroendocrine tumors are rare. Further investigation into biliary tree NET staging and future studies to determine the significance of intracytoplasmic inclusions may be beneficial. This case highlights the appropriate use of genetic testing and supports expanding the clinical diagnosis of MEN1 to include NETs of the extrahepatic bile duct.
Click here to read the digital edition.
1. Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, eds. Williams Textbook of Endocrinology. 12th ed. Philadelphia, PA: WB Saunders; 2011.
2. American Joint Committee on Cancer. Neuroendocrine Tumors. In: Edge S, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A, eds. American Joint Committee on Cancer Staging Handbook. 7th ed. From the AJCC Cancer Staging Manual. New York, NY: Springer-Verlag; 2010:227-236.
3. Komminoth P, Arnold R, Capella C, et al. Neuroendocrine neoplasms of the gallbladder and extrahepatic bile ducts. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, et al, eds. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon, France: IARC Press; 2010:274-276.
4. Rindi G, Arnold R, Bosman FT. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, et al, eds. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon, France: IARC Press; 2010:13.
5. Price TN, Thompson GB, Lewis JT, Lloyd RV, Young WF. Zollinger-Ellison syndrome due to primary gastrinoma of the extrahepatic biliary tree: three case reports and review of literature. Endocr Pract. 2009;15(7):737-749.
6. Bhandarwar AH, Shaikh TA, Borisa AD, et al. Primary neuroendocrine tumor of the left hepatic duct: a case report with review of the literature. Case Rep Surg. 2012:786432.
7. Bhalla P, Powle V, Shah RC, Jagannath P. Neuroendocrine tumor of common hepatic duct. Indian J Gastroenterol. 2012;31(3):144-146.
8. Khan FA, Stevens-Chase A, Chaudhry R, Hashmi A, Edelman D, Weaver D. Extrahepatic biliary obstrution secondary to neuroendocrine tumor of the common hepatic duct. Int J Surg Case Rep. 2017;30:46-49.
9. Hong N, Kim HJ, Byun JH, et al. Neuroendocrine neoplasms of the extrahepatic bile duct: radiologic and clinical characteristics. Abdom Imaging. 2015;40(1):181-191.
10. Tonelli F, Giudici F, Nesi G, Batignani G, Brandi ML. Biliary tree gastrinomas in multiple endocrine neoplasia type 1 syndrome. World J Gastroenterol. 2013;19(45):8312-8320.
11. Gibril F, Schumann M, Pace A, Jensen RT. Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (Baltimore). 2004;83(1):43-83.
12. Pieterman CRC, Conemans EB, Dreijerink KMA, et al. Thoracic and duodenopancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1: natural history and function of menin in tumorigenesis. Endocr Relat Cancer. 2014;21(3):R121-R142.
13. Pipeleers-Marichal M, Somers G, Willems G, et al. Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. N Engl J Med. 1990;322(11):723-727.
14. Thakker RV, Newey PJ, Walls GV, et al; Endocrine Society. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012;97(9):2990-3011.
15. Eastell R, Brandi ML, Costa AG, et al. Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the Fourth International Workshop. J Clin Endocrinol Metab. 2014;99(10):3570-3579.
16. Michalopoulos N, Papavramidis TS, Karayannopoulou G, Pliakos I, Papavramidis ST, Kanellos I. Neuroendocrine tumors of extrahepatic biliary tract. Pathol Oncol Res. 2014;20(4):765-775.
17. Serra S, Asa SL, Chetty R. Intracytoplasmic inclusions (including the so-called “rhabdoid” phenotype) in pancreatic endocrine tumors. Endocr Pathol. 2006;17(1):75-81.
18. Shia J, Erlandson RA, Klimstra DS. Whorls of intermediate filaments with entrapped neurosecretory granules correspond to the “rhabdoid” inclusions seen in pancreatic endocrine
neoplasms. Am J Surg Pathol. 2004;28(2):271-273.
19. Perez-Montiel MD, Frankel WL, Suster S. Neuroendocrine carcinomas of the pancreas with ‘Rhabdoid’ features. Am J Surg Pathol. 2003;27(5):642-649.
Neuroendocrine tumors (NETs) are uncommon and can occur in the context of genetic conditions. Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant disorder of the tumor suppressor gene of the same name—MEN1, which encodes for the protein menin. Multiple endocrine neoplasia type 1 is characterized clinically by the presence of 2 or more of the following NETs: parathyroid, pituitary, and pancreaticoduodenal.1 Pancreaticoduodenal NETs occur in 30% to 80% of patients with MEN1 and have malignant potential. Although the majority of pancreaticoduodenal NETs are nonfunctioning, patients may present with symptoms secondary to mass effect.
Genetic testing exists for MEN1, but not all genetic mutations that cause MEN1 have been discovered. Therefore, because negative genetic testing does not rule out MEN1, a diagnosis is based on tumor type and location. Neuroendocrine tumors of the biliary tree are rare, and there
are no well-accepted guidelines on how to stage them.2-4 The following case demonstrates an unusual initial presentation of a NET in the context of MEN1.
Case Report
A 29-year-old, active-duty African-American man deployed in Kuwait presented with icterus, flank pain, and hematuria. His past medical history was significant for nephrolithiasis, and his family history was notable for hyperparathyroidism. Laboratory results showed primary hyperparathyroidism and evidence of biliary obstruction.
A sestamibi scan demonstrated uptake in a location corresponding with the right inferior parathyroid gland. A computed tomography (CT) scan showed nephrolithiasis and hepatic biliary ductal dilatation. Magnetic resonance cholangiopancreatography (MRCP) revealed both intra- and extrahepatic ductal dilatation, focal narrowing of the proximal common bile duct, and possible adenopathy that was concerning for cholangiocarcinoma. Endoscopic retrograde cholangiopancreatography (ERCP) demonstrated a 1 cm to 2 cm focal stricture within the mid-common bile duct with intra- and extrahepatic ductal dilatation (Figure 1). An endoscopy showed no masses in the duodenum, and anendoscopic ultrasound showed no masses in the pancreas. Endoscopic brushings and endoscopic, ultrasound-guided, fine-needle aspiration
cytology were nondiagnostic. Exploratory laparotomy revealed a dilated hepatic bile duct, an inflamed porta hepatis, and a mass involving the distal hepatic bile duct.
The patient underwent cholecystectomy, radical extra hepatic bile duct resection to the level of the hepatic bifurcation, and hepaticojejunostomy. Gross examination of the specimen showed a nodule centered in the distal common hepatic duct with an adjacent, 2-cm lymph node. The histologic examination revealed a neoplastic proliferation consisting of epithelioid cells with round nuclei and granular chromatin with amphophilic cytoplasm in a trabecular and nested architecture.
The tumor was centered in the submucosa, which is typical of gastrointestinal NETs (Figure 2). There was no evidence of direct tumor extension elsewhere. About 40% of the tumor cells contained eosinophilic, intracytoplasmic inclusions (Figure 3). The tumor did not involve the margins or lymph node.
Positive staining with the neuroendocrine markers synaptophysin and chromagranin A confirmed a well-differentiated NET. The intracytoplasmic inclusions stained strongly positive for cytokeratin CAM 5.2. The tumor had higher-grade features, including tumor cell necrosis, a Ki-67 labeling index of 3%, and perineural invasion. The 2010 World Health Organization (WHO) criteria for NET of the digestive system classified this tumor as a grade 2, well-differentiated NET and as stage 1a (limited to the bile duct).4
Postoperatively, octreotide scan with single-photon emission computed tomography (SPECT)-CT did not show additional masses or lesions. Serum pancreatic polypeptide was elevated, with the remaining serum and plasma NET markers—including gastrin, glucagon, insulin, chromogranin A, and vasoactive intestinal polypeptide (VIP)—being within reference ranges. Genetic testing (GeneDx, Inc, Gaithersburg, MD) showed an E563X nonsense mutation in the MEN1 gene, confirming a MEN1 disorder. The patient then underwent a 4-gland parathyroidectomy with reimplantation; the parathyroid glands demonstrated hyperplasia in all 4 glands.
Biochemical follow-up at 14 months showed that the serum pancreatic polypeptide had normalized. There was no evidence of pituitary orpancreatic hypersecretion. The patient developed hypoparathyroidism, requiring calcium and calcitriol supplementation. Radiographic follow-up using abdominal magnetic resonance imaging at 16 months showed no evidence of disease.
Discussion
This case illustrates a genetic disease with an unusual initial presentation. Primary extrahepatic bile duct NETs are rare and have been reported previously in patients without MEN1.5-9 Neuroendocrine tumors in the hepatic bile duct in patients with MEN1 also have been reported but only after these tumors first appeared in the pancreas or duodenum.10 An extensive literature search revealed no prior reports extrahepatic bile duct NETs with MEN1 as the primary site or with biliary obstruction, which is why this patient’s presentation is particularly interesting.5,6,10-13 The table summarizes select reports of NETs.
Tumor location in this patient was atypical, and genetic testing guided the management. Serum MEN1 genetic testing is indicated in patients with ≥ 2 tumors that are atypical but possibly associated with MEN1 (such as adrenal tumors, gastrinomas, and carcinoids) and in patients aged < 45 years with primary hyperparathyroidism.14,15 The patient in this study was aged 29 years and had hyperparathyroidism and an NET of the hepatic bile duct. This condition was sufficient to warrant genetic testing, the results of which affected the patient’s subsequent parathyroid surgery.15 Despite the suggestion of unifocal localization on the sestamibi scan, the patient underwent the more appropriate subtotal parathyroidectomy.14 The patient’s tumor most likely originated from a germline mutation of the MEN1 gene.
As a result of the patient’s genetic test results, his daughter also was tested. She was found to have the same mutation as her father and will undergo proper tumor surveillance for MEN1. There was no personal or family history of hemangioblastomas, renal cell carcinomas, or cystadenomas, which would have prompted testing for von Hippel-Lindau disease. Likewise, there was no personal or family history of café-au-lait macules and neurofibromas, which would have prompted testing for neurofibromatosis type 1.
Due to the paucity of cases, there are currently no well-accepted guidelines on how to stage extrahepatic biliary NETs.3-5,16 The WHO recommends staging according to adenocarcinomas of the gallbladder and bile duct.3 As such, the pathologic stage of this tumor would be stage 1a.
The significance of the intracytoplasmic inclusion in this case is unknown. Pancreatic NETs and neuroendocrine carcinomas have demonstrated intracytoplasmic inclusions that stain positively for keratin and may indicate more aggressive tumor behavior.17-19 In 1 report, electron microscopic examination demonstrated intermediate filaments with entrapped neurosecretory granules.18 In a series of 84 cases of pancreatic endocrine tumors, 14 had intracytoplasmic inclusions; of these, 5 had MEN1.17 In the present case, the patient continues to show no evidence of tumor recurrence at 16 months after resection.
Conclusion
Extrahepatic biliary neuroendocrine tumors are rare. Further investigation into biliary tree NET staging and future studies to determine the significance of intracytoplasmic inclusions may be beneficial. This case highlights the appropriate use of genetic testing and supports expanding the clinical diagnosis of MEN1 to include NETs of the extrahepatic bile duct.
Click here to read the digital edition.
Neuroendocrine tumors (NETs) are uncommon and can occur in the context of genetic conditions. Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant disorder of the tumor suppressor gene of the same name—MEN1, which encodes for the protein menin. Multiple endocrine neoplasia type 1 is characterized clinically by the presence of 2 or more of the following NETs: parathyroid, pituitary, and pancreaticoduodenal.1 Pancreaticoduodenal NETs occur in 30% to 80% of patients with MEN1 and have malignant potential. Although the majority of pancreaticoduodenal NETs are nonfunctioning, patients may present with symptoms secondary to mass effect.
Genetic testing exists for MEN1, but not all genetic mutations that cause MEN1 have been discovered. Therefore, because negative genetic testing does not rule out MEN1, a diagnosis is based on tumor type and location. Neuroendocrine tumors of the biliary tree are rare, and there
are no well-accepted guidelines on how to stage them.2-4 The following case demonstrates an unusual initial presentation of a NET in the context of MEN1.
Case Report
A 29-year-old, active-duty African-American man deployed in Kuwait presented with icterus, flank pain, and hematuria. His past medical history was significant for nephrolithiasis, and his family history was notable for hyperparathyroidism. Laboratory results showed primary hyperparathyroidism and evidence of biliary obstruction.
A sestamibi scan demonstrated uptake in a location corresponding with the right inferior parathyroid gland. A computed tomography (CT) scan showed nephrolithiasis and hepatic biliary ductal dilatation. Magnetic resonance cholangiopancreatography (MRCP) revealed both intra- and extrahepatic ductal dilatation, focal narrowing of the proximal common bile duct, and possible adenopathy that was concerning for cholangiocarcinoma. Endoscopic retrograde cholangiopancreatography (ERCP) demonstrated a 1 cm to 2 cm focal stricture within the mid-common bile duct with intra- and extrahepatic ductal dilatation (Figure 1). An endoscopy showed no masses in the duodenum, and anendoscopic ultrasound showed no masses in the pancreas. Endoscopic brushings and endoscopic, ultrasound-guided, fine-needle aspiration
cytology were nondiagnostic. Exploratory laparotomy revealed a dilated hepatic bile duct, an inflamed porta hepatis, and a mass involving the distal hepatic bile duct.
The patient underwent cholecystectomy, radical extra hepatic bile duct resection to the level of the hepatic bifurcation, and hepaticojejunostomy. Gross examination of the specimen showed a nodule centered in the distal common hepatic duct with an adjacent, 2-cm lymph node. The histologic examination revealed a neoplastic proliferation consisting of epithelioid cells with round nuclei and granular chromatin with amphophilic cytoplasm in a trabecular and nested architecture.
The tumor was centered in the submucosa, which is typical of gastrointestinal NETs (Figure 2). There was no evidence of direct tumor extension elsewhere. About 40% of the tumor cells contained eosinophilic, intracytoplasmic inclusions (Figure 3). The tumor did not involve the margins or lymph node.
Positive staining with the neuroendocrine markers synaptophysin and chromagranin A confirmed a well-differentiated NET. The intracytoplasmic inclusions stained strongly positive for cytokeratin CAM 5.2. The tumor had higher-grade features, including tumor cell necrosis, a Ki-67 labeling index of 3%, and perineural invasion. The 2010 World Health Organization (WHO) criteria for NET of the digestive system classified this tumor as a grade 2, well-differentiated NET and as stage 1a (limited to the bile duct).4
Postoperatively, octreotide scan with single-photon emission computed tomography (SPECT)-CT did not show additional masses or lesions. Serum pancreatic polypeptide was elevated, with the remaining serum and plasma NET markers—including gastrin, glucagon, insulin, chromogranin A, and vasoactive intestinal polypeptide (VIP)—being within reference ranges. Genetic testing (GeneDx, Inc, Gaithersburg, MD) showed an E563X nonsense mutation in the MEN1 gene, confirming a MEN1 disorder. The patient then underwent a 4-gland parathyroidectomy with reimplantation; the parathyroid glands demonstrated hyperplasia in all 4 glands.
Biochemical follow-up at 14 months showed that the serum pancreatic polypeptide had normalized. There was no evidence of pituitary orpancreatic hypersecretion. The patient developed hypoparathyroidism, requiring calcium and calcitriol supplementation. Radiographic follow-up using abdominal magnetic resonance imaging at 16 months showed no evidence of disease.
Discussion
This case illustrates a genetic disease with an unusual initial presentation. Primary extrahepatic bile duct NETs are rare and have been reported previously in patients without MEN1.5-9 Neuroendocrine tumors in the hepatic bile duct in patients with MEN1 also have been reported but only after these tumors first appeared in the pancreas or duodenum.10 An extensive literature search revealed no prior reports extrahepatic bile duct NETs with MEN1 as the primary site or with biliary obstruction, which is why this patient’s presentation is particularly interesting.5,6,10-13 The table summarizes select reports of NETs.
Tumor location in this patient was atypical, and genetic testing guided the management. Serum MEN1 genetic testing is indicated in patients with ≥ 2 tumors that are atypical but possibly associated with MEN1 (such as adrenal tumors, gastrinomas, and carcinoids) and in patients aged < 45 years with primary hyperparathyroidism.14,15 The patient in this study was aged 29 years and had hyperparathyroidism and an NET of the hepatic bile duct. This condition was sufficient to warrant genetic testing, the results of which affected the patient’s subsequent parathyroid surgery.15 Despite the suggestion of unifocal localization on the sestamibi scan, the patient underwent the more appropriate subtotal parathyroidectomy.14 The patient’s tumor most likely originated from a germline mutation of the MEN1 gene.
As a result of the patient’s genetic test results, his daughter also was tested. She was found to have the same mutation as her father and will undergo proper tumor surveillance for MEN1. There was no personal or family history of hemangioblastomas, renal cell carcinomas, or cystadenomas, which would have prompted testing for von Hippel-Lindau disease. Likewise, there was no personal or family history of café-au-lait macules and neurofibromas, which would have prompted testing for neurofibromatosis type 1.
Due to the paucity of cases, there are currently no well-accepted guidelines on how to stage extrahepatic biliary NETs.3-5,16 The WHO recommends staging according to adenocarcinomas of the gallbladder and bile duct.3 As such, the pathologic stage of this tumor would be stage 1a.
The significance of the intracytoplasmic inclusion in this case is unknown. Pancreatic NETs and neuroendocrine carcinomas have demonstrated intracytoplasmic inclusions that stain positively for keratin and may indicate more aggressive tumor behavior.17-19 In 1 report, electron microscopic examination demonstrated intermediate filaments with entrapped neurosecretory granules.18 In a series of 84 cases of pancreatic endocrine tumors, 14 had intracytoplasmic inclusions; of these, 5 had MEN1.17 In the present case, the patient continues to show no evidence of tumor recurrence at 16 months after resection.
Conclusion
Extrahepatic biliary neuroendocrine tumors are rare. Further investigation into biliary tree NET staging and future studies to determine the significance of intracytoplasmic inclusions may be beneficial. This case highlights the appropriate use of genetic testing and supports expanding the clinical diagnosis of MEN1 to include NETs of the extrahepatic bile duct.
Click here to read the digital edition.
1. Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, eds. Williams Textbook of Endocrinology. 12th ed. Philadelphia, PA: WB Saunders; 2011.
2. American Joint Committee on Cancer. Neuroendocrine Tumors. In: Edge S, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A, eds. American Joint Committee on Cancer Staging Handbook. 7th ed. From the AJCC Cancer Staging Manual. New York, NY: Springer-Verlag; 2010:227-236.
3. Komminoth P, Arnold R, Capella C, et al. Neuroendocrine neoplasms of the gallbladder and extrahepatic bile ducts. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, et al, eds. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon, France: IARC Press; 2010:274-276.
4. Rindi G, Arnold R, Bosman FT. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, et al, eds. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon, France: IARC Press; 2010:13.
5. Price TN, Thompson GB, Lewis JT, Lloyd RV, Young WF. Zollinger-Ellison syndrome due to primary gastrinoma of the extrahepatic biliary tree: three case reports and review of literature. Endocr Pract. 2009;15(7):737-749.
6. Bhandarwar AH, Shaikh TA, Borisa AD, et al. Primary neuroendocrine tumor of the left hepatic duct: a case report with review of the literature. Case Rep Surg. 2012:786432.
7. Bhalla P, Powle V, Shah RC, Jagannath P. Neuroendocrine tumor of common hepatic duct. Indian J Gastroenterol. 2012;31(3):144-146.
8. Khan FA, Stevens-Chase A, Chaudhry R, Hashmi A, Edelman D, Weaver D. Extrahepatic biliary obstrution secondary to neuroendocrine tumor of the common hepatic duct. Int J Surg Case Rep. 2017;30:46-49.
9. Hong N, Kim HJ, Byun JH, et al. Neuroendocrine neoplasms of the extrahepatic bile duct: radiologic and clinical characteristics. Abdom Imaging. 2015;40(1):181-191.
10. Tonelli F, Giudici F, Nesi G, Batignani G, Brandi ML. Biliary tree gastrinomas in multiple endocrine neoplasia type 1 syndrome. World J Gastroenterol. 2013;19(45):8312-8320.
11. Gibril F, Schumann M, Pace A, Jensen RT. Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (Baltimore). 2004;83(1):43-83.
12. Pieterman CRC, Conemans EB, Dreijerink KMA, et al. Thoracic and duodenopancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1: natural history and function of menin in tumorigenesis. Endocr Relat Cancer. 2014;21(3):R121-R142.
13. Pipeleers-Marichal M, Somers G, Willems G, et al. Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. N Engl J Med. 1990;322(11):723-727.
14. Thakker RV, Newey PJ, Walls GV, et al; Endocrine Society. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012;97(9):2990-3011.
15. Eastell R, Brandi ML, Costa AG, et al. Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the Fourth International Workshop. J Clin Endocrinol Metab. 2014;99(10):3570-3579.
16. Michalopoulos N, Papavramidis TS, Karayannopoulou G, Pliakos I, Papavramidis ST, Kanellos I. Neuroendocrine tumors of extrahepatic biliary tract. Pathol Oncol Res. 2014;20(4):765-775.
17. Serra S, Asa SL, Chetty R. Intracytoplasmic inclusions (including the so-called “rhabdoid” phenotype) in pancreatic endocrine tumors. Endocr Pathol. 2006;17(1):75-81.
18. Shia J, Erlandson RA, Klimstra DS. Whorls of intermediate filaments with entrapped neurosecretory granules correspond to the “rhabdoid” inclusions seen in pancreatic endocrine
neoplasms. Am J Surg Pathol. 2004;28(2):271-273.
19. Perez-Montiel MD, Frankel WL, Suster S. Neuroendocrine carcinomas of the pancreas with ‘Rhabdoid’ features. Am J Surg Pathol. 2003;27(5):642-649.
1. Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, eds. Williams Textbook of Endocrinology. 12th ed. Philadelphia, PA: WB Saunders; 2011.
2. American Joint Committee on Cancer. Neuroendocrine Tumors. In: Edge S, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A, eds. American Joint Committee on Cancer Staging Handbook. 7th ed. From the AJCC Cancer Staging Manual. New York, NY: Springer-Verlag; 2010:227-236.
3. Komminoth P, Arnold R, Capella C, et al. Neuroendocrine neoplasms of the gallbladder and extrahepatic bile ducts. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, et al, eds. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon, France: IARC Press; 2010:274-276.
4. Rindi G, Arnold R, Bosman FT. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, et al, eds. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon, France: IARC Press; 2010:13.
5. Price TN, Thompson GB, Lewis JT, Lloyd RV, Young WF. Zollinger-Ellison syndrome due to primary gastrinoma of the extrahepatic biliary tree: three case reports and review of literature. Endocr Pract. 2009;15(7):737-749.
6. Bhandarwar AH, Shaikh TA, Borisa AD, et al. Primary neuroendocrine tumor of the left hepatic duct: a case report with review of the literature. Case Rep Surg. 2012:786432.
7. Bhalla P, Powle V, Shah RC, Jagannath P. Neuroendocrine tumor of common hepatic duct. Indian J Gastroenterol. 2012;31(3):144-146.
8. Khan FA, Stevens-Chase A, Chaudhry R, Hashmi A, Edelman D, Weaver D. Extrahepatic biliary obstrution secondary to neuroendocrine tumor of the common hepatic duct. Int J Surg Case Rep. 2017;30:46-49.
9. Hong N, Kim HJ, Byun JH, et al. Neuroendocrine neoplasms of the extrahepatic bile duct: radiologic and clinical characteristics. Abdom Imaging. 2015;40(1):181-191.
10. Tonelli F, Giudici F, Nesi G, Batignani G, Brandi ML. Biliary tree gastrinomas in multiple endocrine neoplasia type 1 syndrome. World J Gastroenterol. 2013;19(45):8312-8320.
11. Gibril F, Schumann M, Pace A, Jensen RT. Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (Baltimore). 2004;83(1):43-83.
12. Pieterman CRC, Conemans EB, Dreijerink KMA, et al. Thoracic and duodenopancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1: natural history and function of menin in tumorigenesis. Endocr Relat Cancer. 2014;21(3):R121-R142.
13. Pipeleers-Marichal M, Somers G, Willems G, et al. Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. N Engl J Med. 1990;322(11):723-727.
14. Thakker RV, Newey PJ, Walls GV, et al; Endocrine Society. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012;97(9):2990-3011.
15. Eastell R, Brandi ML, Costa AG, et al. Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the Fourth International Workshop. J Clin Endocrinol Metab. 2014;99(10):3570-3579.
16. Michalopoulos N, Papavramidis TS, Karayannopoulou G, Pliakos I, Papavramidis ST, Kanellos I. Neuroendocrine tumors of extrahepatic biliary tract. Pathol Oncol Res. 2014;20(4):765-775.
17. Serra S, Asa SL, Chetty R. Intracytoplasmic inclusions (including the so-called “rhabdoid” phenotype) in pancreatic endocrine tumors. Endocr Pathol. 2006;17(1):75-81.
18. Shia J, Erlandson RA, Klimstra DS. Whorls of intermediate filaments with entrapped neurosecretory granules correspond to the “rhabdoid” inclusions seen in pancreatic endocrine
neoplasms. Am J Surg Pathol. 2004;28(2):271-273.
19. Perez-Montiel MD, Frankel WL, Suster S. Neuroendocrine carcinomas of the pancreas with ‘Rhabdoid’ features. Am J Surg Pathol. 2003;27(5):642-649.