User login
Dermoscopy in family medicine: A primer
Dermoscopy, the use of a handheld instrument to magnify the skin 10-fold while providing a light source, is a quick, useful, cost-effective tool for detecting melanoma in family medicine.1-4 The device, which allows the physician to visualize structures below the stratum corneum that are not routinely discernible with the naked eye, can be attached to a smartphone so that photos can be taken and reviewed with the patient. The photo can also be reviewed after a biopsy result is obtained.
Its use among non-dermatologist US physicians appears to be relatively low, but rising. One small study of physicians working in family medicine, internal medicine, and plastic surgery found that only 15% had ever used a dermatoscope and 6% were currently using one.5
As a family physician, you can expand your diagnostic abilities in dermatology with the acquisition of a dermatoscope (FIGURE 1) and some time invested in learning to interpret visible patterns. With that in mind, this review focuses on the diagnosis of skin cancers and benign growths using dermoscopy. We begin with a brief look at the research on dermoscopy and how it is performed. From there, we’ll detail an algorithm to guide dermoscopic analysis. And to round things out, we provide guidance that will help you to get started. (See “Choosing a dermatoscope—and making the most of it,” and “To learn more about dermoscopy …”.)

SIDEBAR
Choosing a dermatoscope—and making the most of it
1. Consider acquiring a hybrid dermatoscope.
Nonpolarized dermatoscopes (NPDs) and polarized dermatoscopes (PDs) provide different but complementary information. PDs enable users to identify features such as vessels and shiny white structures that are highly indicative of skin cancer. Because PDs are highly sensitive for detecting skin cancer and do not require a liquid interface or direct skin contact, they are the ideal dermatoscopes to use for skin cancer screening.
However, maintaining the highest specificity requires the complementary use of NPDs, which are better at identifying surface structures seen in seborrheic keratoses and other benign lesions. Thus, if the aim is to maintain the highest diagnostic accuracy for all types of lesions, then the preferred dermatoscope is a hybrid that permits the user to toggle between polarized and nonpolarized features in one device.
2. Choose a dermatoscope that attaches to your smartphone and/or camera.
This helps you capture digital dermoscopic images that can be analyzed on a larger screen, which permits:
- enlarging certain areas for in-depth analysis of structures and patterns
- sharing the image with the patient to explain why a biopsy is, or isn’t, needed
- sharing the image with a colleague for the purpose of a consult or a referral, or using the images for teaching purposes
- saving the images in order to follow lesions over time when monitoring is indicated
- ongoing learning. After each biopsy result comes back, we recommend correlating the dermoscopic images with the biopsy report. If your suspected diagnosis was correct, this reinforces your knowledge. If the pathology diagnosis is unexpected, you can learn by revisiting the original images to look for structures or patterns you may have missed upon first examination. You may even question the pathology report based on the dermoscopy, prompting a call to the pathologist.
- keeping a safe distance from the patient when looking for scabies mites.
SIDEBAR
To learn more about dermoscopy…
FREE APPS:
Dermoscopy 2-Step Algorithm. Available for free on iTunes, Google Play, and at https://usatinemedia.com/app/dermoscopy-two-step-algorithm/, this free app (developed by 3 of the 4 authors) is intended to help you interpret the dermoscopic patterns seen with your dermatoscope. It asks a series of questions that lead you to the most probable diagnosis. The app also contains more than 80 photos and charts to help you with your diagnosis. No Internet connection is needed to view the full app. There are 50 interactive cases to solve.
YOUdermoscopy Training (Available for free on iTunes, Google Play, and at https://www.youdermoscopytraining.org/) offers a fun game interface to test and expand your dermoscopy skills.
OTHER INTERNET RESOURCES:
- Dermoscopedia provides state-of-the-art information on dermoscopy. It’s available at: https://dermoscopedia.org.
- A free dermoscopy tutorial is available at: http://www.dermoscopy.org/
- The International Dermoscopy Society’s Web site, which offers various tutorials and other information, can be found at: http://www.dermoscopy-ids.org/.
COURSES:
Dermoscopy courses are a great way to get started and/or to advance your skills. The following courses are taught by the authors of this article:
- The American Dermoscopy Meeting is held yearly in the summer in a national park. See http://www.americandermoscopy.com/.
- Memorial Sloan Kettering Cancer Center holds a yearly dermoscopy workshop each fall in New York City. See http://www.mskcc.org/events/.
- The yearly American Academy of Family Physicians' FMX meeting offers dermoscopy workshops. See https://www.aafp.org/events/fmx.html.
Continue to: What the research says
What the research says
Dermoscopy improves sensitivity for detecting melanoma over the naked eye alone; it also allows for the detection of melanoma at earlier stages, which improves prognosis.6
A meta-analysis of dermoscopy use in clinical settings showed that, following training, dermoscopy increases the average sensitivity of melanoma diagnosis from 71% to more than 90% without a significant decrease in specificity.7 In a study of 74 primary care physicians, there was an improvement in both clinical and dermoscopic diagnosis of melanoma among those who received training in dermoscopy, compared with a control group.8 Another study found that primary care physicians can reduce their baseline benign-to-melanoma ratio (the number of suspicious benign lesions biopsied to find 1 melanoma) from 9.5:1 with naked eye examination to 3.5:1 with dermoscopy.9
The exam begins by choosing 1 of 3 modes of dermoscopy
Dermatoscopes can have a polarized or nonpolarized light source. Some dermatoscopes combine both types of light (hybrid dermatoscopes; see “Choosing a dermatoscope—and making the most of it.”)
There are 3 modes of dermoscopy:
- nonpolarized contact dermoscopy
- polarized contact dermoscopy
- polarized non-contact dermoscopy.
Dermatoscopes with nonpolarized light require direct skin contact and a liquid interface (eg, alcohol, gel, mineral oil) between the scope’s glass plate and the skin for the visualization of subsurface structures. In contrast, dermatoscopes with polarized light do not require direct skin contact or a liquid interface; however, contacting the skin and using a liquid interface will provide a sharper image.
Continue to: Two major algorithms guide dermoscopic analysis
Two major algorithms guide dermoscopic analysis
The first of 2 major algorithms that can be used to guide dermoscopic analysis is a modified pattern analysis put forth by Kittler.10 This descriptive system based on geometric elements, patterns, colors, and clues guides the observer to a specific diagnosis without categorizing lesions as being either melanocytic or nonmelanocytic. Because this is not the preferred method of the authors, we will move on to Method 2.
The second method, a 2-step algorithm, is a qualitative system that guides the observer through differentiating melanocytic from nonmelanocytic lesions in order to differentiate nevi from melanoma (FIGURE 2). At the same time, it serves as an aid to correctly diagnose non-melanocytic lesions. The 2-step algorithm forms the foundation for the dermoscopic evaluation of skin lesions in this article.

Not all expert dermoscopists employ structured analytical systems or methods to reach a diagnosis. Because of their vast experience, many rely purely on pattern recognition. But algorithms can facilitate non-experts in dermoscopy in the differentiation of nevi from melanoma or, simply, in differentiating the benign from the malignant.
Although each algorithm has its unique criteria, all of them require training and practice and familiarity with the terms used to describe morphologic structures. The International Dermoscopy Society recently published a consensus paper designating some terms as preferred over others.11
Continue to: Step 1...
Step 1: Melanocytic vs non-melanocytic
Step 1 of the 2-step algorithm requires the observer to determine whether the lesion is melanocytic (ie, originates from melanocytes and, therefore, could be a melanoma) or nonmelanocytic in origin.
A melanocytic lesion usually will display at least 1 of the following structures:
- pigment network (FIGURE 3A) (This can include angulated lines.)
- negative network (FIGURE 3B) (hypopigmented lines connecting pigmented structures in a serpiginous fashion)
- streaks (FIGURE 3C)
- homogeneous blue pigmentation (FIGURE 3D)
- globules (aggregated or as a peripheral rim) (FIGURE 3E)
- pseudonetwork (facial skin) (FIGURE 3F)
- parallel pigment pattern (acral lesions) (FIGURE 3G).

Exceptions. Sometimes, nonmelanocytic lesions will present with pigment network. Dermatofibromas, for example, are one exception in which the pattern trumps the network. Two other exceptions are solar lentigo and supernumerary or accessory nipple.
If the lesion does not display any structure, it is considered structureless. In these cases, proceed to the second step to rule out a melanoma.
Doesn’t meet criteria for a melanocytic lesion?
If the lesion does not reveal any of the criteria for a melanocytic lesion, then look for structures seen in nonmelanocytic lesions: dermatofibromas; seborrheic keratosis; angiomas and angiokeratomas; sebaceous hyperplasia; clear-cell acanthomas; basal cell carcinomas (BCCs); and squamous cell carcinomas (SCCs).
Continue to: Benign nonmelanocytic lesions
Benign nonmelanocytic lesions
Dermatofibromas are benign symmetric lesions that feel firm and may dimple upon application of lateral pressure. They are fibrotic scar-like lesions that present with 1 or more of the following dermoscopic features (FIGURE 4):
- peripheral pigment network, due to increased melanin in keratinocytes
- homogeneous brown pigmented areas
- central scar-like area
- shiny white lines
- vascular structures (ie, dotted, polymorphous vessels), usually seen within the scar-like area
- ring-like globules, usually seen in the zone between the scar-like depigmentation and the peripheral network. They correspond to widened hyperpigmented rete ridges.

Seborrheic keratosis (SK) is a benign skin growth that often has a stuck-on appearance (FIGURE 5). Features often include:
- multiple (>2) milia-like cysts
- comedo-like openings
- a network-like structure that corresponds to gyri and sulci and which in some cases can create a cerebriform pattern
- fingerprint-like structures
- moth-eaten borders
- jelly sign. This consists of semicircular u-shaped structures that have a smudged appearance and are aligned in the same direction. The appearance resembles jelly as it is spread on a piece of bread.
- hairpin (looped or twisted-looped) vessels surrounded by a white halo.

Other clues include a sharp demarcation and a negative wobble sign (which we’ll describe in a moment). The presence or absence of a wobble sign is determined by using a dermatoscope that touches the skin. Mild vertical pressure is applied to the lesion while moving the scope back and forth horizontally. If the lesion slides across the skin surface, the diagnosis of an epidermal keratinocytic tumor (ie, SK) is favored. If, on the other hand, the lesion wobbles (rolls back and forth), then the diagnosis of a neoplasm with a dermal component (ie, intradermal or compound nevus) is more likely.
Angiomas and angiokeratomas. Angiomas demonstrate lacunae that are often separated by septae (FIGURE 6). Lacunae can vary in size and color. They can be red, red-white, red-blue, maroon, blue, blue-black, or even black (when thrombosis is present).

Angiokeratomas (FIGURE 7) can reveal lacunae of varying colors including black, red, purple, and maroon. In addition, a blue-whitish veil, erythema, and hemorrhagic crusts can be present.

Continue to: Sebaceous hyperplasia...
Sebaceous hyperplasia is the overgrowth of sebaceous glands. It can mimic BCC on the face. Sebaceous hyperplasia presents with multiple vessels in a crown-like arrangement that do not cross the center of the lesion. The sebaceous glands resemble popcorn (FIGURE 8).

Clear-cell acanthoma is a benign erythematous epidermal tumor usually found on the leg with a string-of-pearls pattern. This pattern is vascular so the pearls are red in color (FIGURE 9).

Malignant nonmelanocytic lesions
BCC is the most common type of skin cancer. Features often include:
- spoke-wheel-like structures or concentric structures (FIGURE 10A)
- leaf-like areas (FIGURE 10B)
- arborizing vessels (FIGURE 10b and 10C)large blue-gray ovoid nest (FIGURE 10A)
- multiple blue-gray non-aggregated globules
- ulceration or multiple small erosions
- shiny white structures and strands (FIGURE 10C).

Additional dermoscopic clues include short, fine, superficial telangiectasias and multiple in-focus dots in a buck-shot scatter distribution.
Squamous cell carcinomas (SCCs) of the skin are keratinizing malignant tumors. Each SCC generally has some of the following features (FIGURE 11):
- dotted and/or glomerular vessels, commonly distributed focally at the periphery. They can also be diffuse or aligned linearly within the lesion.
- scale (yellow or white)
- rosettes (seen with polarized light)
- white circles or keratin pearls
- brown circles
- ulcerations
- brown dots or globules arranged in a linear configuration.

Continue to: Step 2...
Step 2: It’s melanocytic, but is it a nevus or a melanoma?
If, by following Step 1 of the algorithm, the lesion is determined to be of melanocytic origin, then one proceeds to Step 2 to decide whether the growth is a nevus, a suspicious lesion, or a melanoma. For this purpose, several additional algorithms are available.12-17

Benign nevi tend to manifest with 1 of the following 10 patterns: (FIGURE 12)
- diffuse reticular
- patchy reticular
- peripheral reticular with central hypopigmentation
- peripheral reticular with central hyperpigmentation
- homogeneous
- peripheral globules/starburst. It has been suggested that lesions that show starburst morphology on dermoscopy require complete excision and follow-up since 13% of Spitzoid-looking symmetric lesions in patients older than 12 years were found to be melanoma in one study.18
- peripheral reticular with central globules
- globular
- 2-component
- symmetric multicomponent (this pattern should be interpreted with caution, and a biopsy is probably warranted for dermoscopic novices).

Melanomas tend to deviate from the benign patterns described earlier. Structures in melanomas are often distributed in an asymmetric fashion (which is the basis for diagnosis in many of the other algorithms), and most of them will reveal 1 or more of the melanoma-specific structures (FIGURE 13). The melanomas in FIGURES 14 A-H each show at least 2 melanoma-specific structures. On the face or sun-damaged skin, melanoma may present with grey color, a circle-in-circle pattern, and/or polygonal lines (FIGURE 15). Note that melanoma on the soles or palms may present with a parallel ridge pattern (FIGURE 16).

How to proceed after the evaluation of melanocytic lesions
After evaluating the lesion for benign patterns and melanoma-specific structures, there are 3 possible pathways:
1. The lesion adheres to one of the nevi patterns and does not display a melanoma-specific structure. You can reassure the patient that the lesion is benign.
2. The lesion:
A. Adheres to one nevus pattern, but also displays a melanoma-specific structure.
B. Does not adhere to any of the benign patterns and does not have any melanoma-specific structures.

This is considered a suspicious lesion, and the choices of action include performing a biopsy or short-term monitoring by comparing dermoscopic images over a 3-month interval. (Caveat: Never monitor raised lesions because nodular melanomas can grow quickly and develop a worsened prognosis in a short time. Instead you’ll want to biopsy the lesion that day or very soon thereafter.)
3. The lesion deviates from the benign patterns and has at least 1 melanoma-specific structure. Biopsy the lesion to rule out melanoma.

Continue to: A bonus...
A bonus: Diagnosing scabies
Increasingly, dermoscopy is being used in the diagnosis of many other skin, nail, and hair problems. In fact, one great bonus to owning a dermatoscope is the accurate diagnosis of scabies. Dermoscopy can be helpful in detecting the scabies mite without having to scrape and use the microscope. Moreover, the sensitivity and specificity of a dermoscopic diagnosis is higher than for scraping and microscopy.19
What you’ll see
The anterior legs and mouth parts of the mite resemble a triangle (arrowhead, delta-wing jet) (FIGURE 17). Look for a burrow, and the mite can be seen at the end of the burrow as a faint circle with a leading darker triangle. The burrow itself has a distinctive pattern that has more morphology than an excoriation and has been described as the contrail of a jet plane. Using a dermatoscope attached to your smartphone allows you to magnify the image even further while maintaining a safe distance from the mite.

CORRESPONDENCE
Richard P. Usatine, MD, 903 W. Martin, Skin Clinic – Historic Building, San Antonio, TX 78207; [email protected].
1. Herschorn A. Dermoscopy for melanoma detection in family practice. Can Fam Physician. 2012;58:740-745.
2. Buckley D, McMonagle C. Melanoma in primary care. The role of the general practitioner. Ir J Med Sci. 2014;183:363-368.
3. Mayer JE, Swetter SM, Fu T, et al. Screening, early detection, education, and trends for melanoma: current status (2007-2013) and future directions: Part I Epidemiology, high-risk groups, clinical strategies, and diagnostic technology. J Am Acad Dermatol. 2014;71:599.e1-599.e12.
4. Mayer JE, Swetter SM, Fu T, et al. Screening, early detection, education, and trends for melanoma: current status (2007-2013) and future directions: Part II Screening, education, and future directions. J Am Acad Dermatol. 2014;71:611.e1-611.e10.
5. Morris JB, Alfonso SV, Hernandez N, et al. Use of and intentions to use dermoscopy among physicians in the United States. Dermatol Pract Concept. 2017;7:2.
6. Salerni G, Terán T, Alonso C, et al. The role of dermoscopy and digital dermoscopy follow-up in the clinical diagnosis of melanoma: clinical and dermoscopic features of 99 consecutive primary melanomas. Dermatol Pract Concept. 2014;4:39-46.
7. Vestergaard ME, Macaskill P, Holt PE, et al. Dermoscopy compared with naked eye examination for the diagnosis of primary melanoma: a meta-analysis of studies performed in a clinical setting. Br J Dermatol. 2008;159:669-676.
8. Westerhoff K, McCarthy WH, Menzies SW. Increase in the sensitivity for melanoma diagnosis by primary care physicians using skin surface microscopy. Br J Dermatol. 2000;143:1016-1020.
9. Menzies SW, Emery J, Staples M, et al. Impact of dermoscopy and short-term sequential digital dermoscopy imaging for the management of pigmented lesions in primary care: a sequential intervention trial. Br J Dermatol. 2009;161:1270-1277.
10. Kittler H. Dermatoscopy: introduction of a new algorithmic method based on pattern analysis for diagnosis of pigmented skin lesions. Dermatopathology: Practical & Conceptual. 2007;13:3.
11. Kittler H, Marghoob AA, Argenziano G, et al. Standardization of terminology in dermoscopy/dermatoscopy: results of the third consensus conference of the International Society of Dermoscopy. J Am Acad Dermatol. 2016;74:1093-1106.
12. Stolz W, Riemann A, Cognetta AB, et al. ABCD rule of dermoscopy: a new practical method for early recognition of malignant melanoma. Eur J Dermatol. 1994;4:521-527.
13. Pehamberger H, Steiner A, Wolff K. In vivo epiluminescence microscopy of pigmented skin lesions I Pattern analysis of pigmented skin lesions. J Am Acad Dermatol. 1987;17:571-583.
14. Menzies SW, Ingvar C, McCarthy WH. A sensitivity and specificity analysis of the surface microscopy features of invasive melanoma. Melanoma Res. 1996;6:55-62.
15. Argenziano G, Fabbrocini G, Carli P, et al. Epiluminescence microscopy for the diagnosis of doubtful melanocytic skin lesions. Comparison of the ABCD rule of dermatoscopy and a new 7-point checklist based on pattern analysis. Arch Dermatol. 1998;134:1563-1570.
16. Henning JS, Dusza SW, Wang SQ, et al. The CASH (color, architecture, symmetry, and homogeneity) algorithm for dermoscopy. J Am Acad Dermatol. 2007;56:45-52.
17. Soyer HP, Argenziano G, Zalaudek I, et al. Three-point checklist of dermoscopy. A new screening method for early detection of melanoma. Dermatology. 2004;208:27-31.
18. Lallas A, Moscarella E, Longo C, et al. Likelihood of finding melanoma when removing a Spitzoid-looking lesion in patients aged 12 years or older. J Am Acad Dermatol. 2015;72:47-53.
19. Dupuy A, Dehen L, Bourrat E, et al. Accuracy of standard dermoscopy for diagnosing scabies. J Am Acad Dermatol. 2007;56:53-62.
Dermoscopy, the use of a handheld instrument to magnify the skin 10-fold while providing a light source, is a quick, useful, cost-effective tool for detecting melanoma in family medicine.1-4 The device, which allows the physician to visualize structures below the stratum corneum that are not routinely discernible with the naked eye, can be attached to a smartphone so that photos can be taken and reviewed with the patient. The photo can also be reviewed after a biopsy result is obtained.
Its use among non-dermatologist US physicians appears to be relatively low, but rising. One small study of physicians working in family medicine, internal medicine, and plastic surgery found that only 15% had ever used a dermatoscope and 6% were currently using one.5
As a family physician, you can expand your diagnostic abilities in dermatology with the acquisition of a dermatoscope (FIGURE 1) and some time invested in learning to interpret visible patterns. With that in mind, this review focuses on the diagnosis of skin cancers and benign growths using dermoscopy. We begin with a brief look at the research on dermoscopy and how it is performed. From there, we’ll detail an algorithm to guide dermoscopic analysis. And to round things out, we provide guidance that will help you to get started. (See “Choosing a dermatoscope—and making the most of it,” and “To learn more about dermoscopy …”.)
SIDEBAR
Choosing a dermatoscope—and making the most of it
1. Consider acquiring a hybrid dermatoscope.
Nonpolarized dermatoscopes (NPDs) and polarized dermatoscopes (PDs) provide different but complementary information. PDs enable users to identify features such as vessels and shiny white structures that are highly indicative of skin cancer. Because PDs are highly sensitive for detecting skin cancer and do not require a liquid interface or direct skin contact, they are the ideal dermatoscopes to use for skin cancer screening.
However, maintaining the highest specificity requires the complementary use of NPDs, which are better at identifying surface structures seen in seborrheic keratoses and other benign lesions. Thus, if the aim is to maintain the highest diagnostic accuracy for all types of lesions, then the preferred dermatoscope is a hybrid that permits the user to toggle between polarized and nonpolarized features in one device.
2. Choose a dermatoscope that attaches to your smartphone and/or camera.
This helps you capture digital dermoscopic images that can be analyzed on a larger screen, which permits:
- enlarging certain areas for in-depth analysis of structures and patterns
- sharing the image with the patient to explain why a biopsy is, or isn’t, needed
- sharing the image with a colleague for the purpose of a consult or a referral, or using the images for teaching purposes
- saving the images in order to follow lesions over time when monitoring is indicated
- ongoing learning. After each biopsy result comes back, we recommend correlating the dermoscopic images with the biopsy report. If your suspected diagnosis was correct, this reinforces your knowledge. If the pathology diagnosis is unexpected, you can learn by revisiting the original images to look for structures or patterns you may have missed upon first examination. You may even question the pathology report based on the dermoscopy, prompting a call to the pathologist.
- keeping a safe distance from the patient when looking for scabies mites.
SIDEBAR
To learn more about dermoscopy…
FREE APPS:
Dermoscopy 2-Step Algorithm. Available for free on iTunes, Google Play, and at https://usatinemedia.com/app/dermoscopy-two-step-algorithm/, this free app (developed by 3 of the 4 authors) is intended to help you interpret the dermoscopic patterns seen with your dermatoscope. It asks a series of questions that lead you to the most probable diagnosis. The app also contains more than 80 photos and charts to help you with your diagnosis. No Internet connection is needed to view the full app. There are 50 interactive cases to solve.
YOUdermoscopy Training (Available for free on iTunes, Google Play, and at https://www.youdermoscopytraining.org/) offers a fun game interface to test and expand your dermoscopy skills.
OTHER INTERNET RESOURCES:
- Dermoscopedia provides state-of-the-art information on dermoscopy. It’s available at: https://dermoscopedia.org.
- A free dermoscopy tutorial is available at: http://www.dermoscopy.org/
- The International Dermoscopy Society’s Web site, which offers various tutorials and other information, can be found at: http://www.dermoscopy-ids.org/.
COURSES:
Dermoscopy courses are a great way to get started and/or to advance your skills. The following courses are taught by the authors of this article:
- The American Dermoscopy Meeting is held yearly in the summer in a national park. See http://www.americandermoscopy.com/.
- Memorial Sloan Kettering Cancer Center holds a yearly dermoscopy workshop each fall in New York City. See http://www.mskcc.org/events/.
- The yearly American Academy of Family Physicians' FMX meeting offers dermoscopy workshops. See https://www.aafp.org/events/fmx.html.
Continue to: What the research says
What the research says
Dermoscopy improves sensitivity for detecting melanoma over the naked eye alone; it also allows for the detection of melanoma at earlier stages, which improves prognosis.6
A meta-analysis of dermoscopy use in clinical settings showed that, following training, dermoscopy increases the average sensitivity of melanoma diagnosis from 71% to more than 90% without a significant decrease in specificity.7 In a study of 74 primary care physicians, there was an improvement in both clinical and dermoscopic diagnosis of melanoma among those who received training in dermoscopy, compared with a control group.8 Another study found that primary care physicians can reduce their baseline benign-to-melanoma ratio (the number of suspicious benign lesions biopsied to find 1 melanoma) from 9.5:1 with naked eye examination to 3.5:1 with dermoscopy.9
The exam begins by choosing 1 of 3 modes of dermoscopy
Dermatoscopes can have a polarized or nonpolarized light source. Some dermatoscopes combine both types of light (hybrid dermatoscopes; see “Choosing a dermatoscope—and making the most of it.”)
There are 3 modes of dermoscopy:
- nonpolarized contact dermoscopy
- polarized contact dermoscopy
- polarized non-contact dermoscopy.
Dermatoscopes with nonpolarized light require direct skin contact and a liquid interface (eg, alcohol, gel, mineral oil) between the scope’s glass plate and the skin for the visualization of subsurface structures. In contrast, dermatoscopes with polarized light do not require direct skin contact or a liquid interface; however, contacting the skin and using a liquid interface will provide a sharper image.
Continue to: Two major algorithms guide dermoscopic analysis
Two major algorithms guide dermoscopic analysis
The first of 2 major algorithms that can be used to guide dermoscopic analysis is a modified pattern analysis put forth by Kittler.10 This descriptive system based on geometric elements, patterns, colors, and clues guides the observer to a specific diagnosis without categorizing lesions as being either melanocytic or nonmelanocytic. Because this is not the preferred method of the authors, we will move on to Method 2.
The second method, a 2-step algorithm, is a qualitative system that guides the observer through differentiating melanocytic from nonmelanocytic lesions in order to differentiate nevi from melanoma (FIGURE 2). At the same time, it serves as an aid to correctly diagnose non-melanocytic lesions. The 2-step algorithm forms the foundation for the dermoscopic evaluation of skin lesions in this article.
Not all expert dermoscopists employ structured analytical systems or methods to reach a diagnosis. Because of their vast experience, many rely purely on pattern recognition. But algorithms can facilitate non-experts in dermoscopy in the differentiation of nevi from melanoma or, simply, in differentiating the benign from the malignant.
Although each algorithm has its unique criteria, all of them require training and practice and familiarity with the terms used to describe morphologic structures. The International Dermoscopy Society recently published a consensus paper designating some terms as preferred over others.11
Continue to: Step 1...
Step 1: Melanocytic vs non-melanocytic
Step 1 of the 2-step algorithm requires the observer to determine whether the lesion is melanocytic (ie, originates from melanocytes and, therefore, could be a melanoma) or nonmelanocytic in origin.
A melanocytic lesion usually will display at least 1 of the following structures:
- pigment network (FIGURE 3A) (This can include angulated lines.)
- negative network (FIGURE 3B) (hypopigmented lines connecting pigmented structures in a serpiginous fashion)
- streaks (FIGURE 3C)
- homogeneous blue pigmentation (FIGURE 3D)
- globules (aggregated or as a peripheral rim) (FIGURE 3E)
- pseudonetwork (facial skin) (FIGURE 3F)
- parallel pigment pattern (acral lesions) (FIGURE 3G).
Exceptions. Sometimes, nonmelanocytic lesions will present with pigment network. Dermatofibromas, for example, are one exception in which the pattern trumps the network. Two other exceptions are solar lentigo and supernumerary or accessory nipple.
If the lesion does not display any structure, it is considered structureless. In these cases, proceed to the second step to rule out a melanoma.
Doesn’t meet criteria for a melanocytic lesion?
If the lesion does not reveal any of the criteria for a melanocytic lesion, then look for structures seen in nonmelanocytic lesions: dermatofibromas; seborrheic keratosis; angiomas and angiokeratomas; sebaceous hyperplasia; clear-cell acanthomas; basal cell carcinomas (BCCs); and squamous cell carcinomas (SCCs).
Continue to: Benign nonmelanocytic lesions
Benign nonmelanocytic lesions
Dermatofibromas are benign symmetric lesions that feel firm and may dimple upon application of lateral pressure. They are fibrotic scar-like lesions that present with 1 or more of the following dermoscopic features (FIGURE 4):
- peripheral pigment network, due to increased melanin in keratinocytes
- homogeneous brown pigmented areas
- central scar-like area
- shiny white lines
- vascular structures (ie, dotted, polymorphous vessels), usually seen within the scar-like area
- ring-like globules, usually seen in the zone between the scar-like depigmentation and the peripheral network. They correspond to widened hyperpigmented rete ridges.
Seborrheic keratosis (SK) is a benign skin growth that often has a stuck-on appearance (FIGURE 5). Features often include:
- multiple (>2) milia-like cysts
- comedo-like openings
- a network-like structure that corresponds to gyri and sulci and which in some cases can create a cerebriform pattern
- fingerprint-like structures
- moth-eaten borders
- jelly sign. This consists of semicircular u-shaped structures that have a smudged appearance and are aligned in the same direction. The appearance resembles jelly as it is spread on a piece of bread.
- hairpin (looped or twisted-looped) vessels surrounded by a white halo.
Other clues include a sharp demarcation and a negative wobble sign (which we’ll describe in a moment). The presence or absence of a wobble sign is determined by using a dermatoscope that touches the skin. Mild vertical pressure is applied to the lesion while moving the scope back and forth horizontally. If the lesion slides across the skin surface, the diagnosis of an epidermal keratinocytic tumor (ie, SK) is favored. If, on the other hand, the lesion wobbles (rolls back and forth), then the diagnosis of a neoplasm with a dermal component (ie, intradermal or compound nevus) is more likely.
Angiomas and angiokeratomas. Angiomas demonstrate lacunae that are often separated by septae (FIGURE 6). Lacunae can vary in size and color. They can be red, red-white, red-blue, maroon, blue, blue-black, or even black (when thrombosis is present).
Angiokeratomas (FIGURE 7) can reveal lacunae of varying colors including black, red, purple, and maroon. In addition, a blue-whitish veil, erythema, and hemorrhagic crusts can be present.
Continue to: Sebaceous hyperplasia...
Sebaceous hyperplasia is the overgrowth of sebaceous glands. It can mimic BCC on the face. Sebaceous hyperplasia presents with multiple vessels in a crown-like arrangement that do not cross the center of the lesion. The sebaceous glands resemble popcorn (FIGURE 8).
Clear-cell acanthoma is a benign erythematous epidermal tumor usually found on the leg with a string-of-pearls pattern. This pattern is vascular so the pearls are red in color (FIGURE 9).
Malignant nonmelanocytic lesions
BCC is the most common type of skin cancer. Features often include:
- spoke-wheel-like structures or concentric structures (FIGURE 10A)
- leaf-like areas (FIGURE 10B)
- arborizing vessels (FIGURE 10b and 10C)large blue-gray ovoid nest (FIGURE 10A)
- multiple blue-gray non-aggregated globules
- ulceration or multiple small erosions
- shiny white structures and strands (FIGURE 10C).
Additional dermoscopic clues include short, fine, superficial telangiectasias and multiple in-focus dots in a buck-shot scatter distribution.
Squamous cell carcinomas (SCCs) of the skin are keratinizing malignant tumors. Each SCC generally has some of the following features (FIGURE 11):
- dotted and/or glomerular vessels, commonly distributed focally at the periphery. They can also be diffuse or aligned linearly within the lesion.
- scale (yellow or white)
- rosettes (seen with polarized light)
- white circles or keratin pearls
- brown circles
- ulcerations
- brown dots or globules arranged in a linear configuration.
Continue to: Step 2...
Step 2: It’s melanocytic, but is it a nevus or a melanoma?
If, by following Step 1 of the algorithm, the lesion is determined to be of melanocytic origin, then one proceeds to Step 2 to decide whether the growth is a nevus, a suspicious lesion, or a melanoma. For this purpose, several additional algorithms are available.12-17
Benign nevi tend to manifest with 1 of the following 10 patterns: (FIGURE 12)
- diffuse reticular
- patchy reticular
- peripheral reticular with central hypopigmentation
- peripheral reticular with central hyperpigmentation
- homogeneous
- peripheral globules/starburst. It has been suggested that lesions that show starburst morphology on dermoscopy require complete excision and follow-up since 13% of Spitzoid-looking symmetric lesions in patients older than 12 years were found to be melanoma in one study.18
- peripheral reticular with central globules
- globular
- 2-component
- symmetric multicomponent (this pattern should be interpreted with caution, and a biopsy is probably warranted for dermoscopic novices).
Melanomas tend to deviate from the benign patterns described earlier. Structures in melanomas are often distributed in an asymmetric fashion (which is the basis for diagnosis in many of the other algorithms), and most of them will reveal 1 or more of the melanoma-specific structures (FIGURE 13). The melanomas in FIGURES 14 A-H each show at least 2 melanoma-specific structures. On the face or sun-damaged skin, melanoma may present with grey color, a circle-in-circle pattern, and/or polygonal lines (FIGURE 15). Note that melanoma on the soles or palms may present with a parallel ridge pattern (FIGURE 16).
How to proceed after the evaluation of melanocytic lesions
After evaluating the lesion for benign patterns and melanoma-specific structures, there are 3 possible pathways:
1. The lesion adheres to one of the nevi patterns and does not display a melanoma-specific structure. You can reassure the patient that the lesion is benign.
2. The lesion:
A. Adheres to one nevus pattern, but also displays a melanoma-specific structure.
B. Does not adhere to any of the benign patterns and does not have any melanoma-specific structures.
This is considered a suspicious lesion, and the choices of action include performing a biopsy or short-term monitoring by comparing dermoscopic images over a 3-month interval. (Caveat: Never monitor raised lesions because nodular melanomas can grow quickly and develop a worsened prognosis in a short time. Instead you’ll want to biopsy the lesion that day or very soon thereafter.)
3. The lesion deviates from the benign patterns and has at least 1 melanoma-specific structure. Biopsy the lesion to rule out melanoma.
Continue to: A bonus...
A bonus: Diagnosing scabies
Increasingly, dermoscopy is being used in the diagnosis of many other skin, nail, and hair problems. In fact, one great bonus to owning a dermatoscope is the accurate diagnosis of scabies. Dermoscopy can be helpful in detecting the scabies mite without having to scrape and use the microscope. Moreover, the sensitivity and specificity of a dermoscopic diagnosis is higher than for scraping and microscopy.19
What you’ll see
The anterior legs and mouth parts of the mite resemble a triangle (arrowhead, delta-wing jet) (FIGURE 17). Look for a burrow, and the mite can be seen at the end of the burrow as a faint circle with a leading darker triangle. The burrow itself has a distinctive pattern that has more morphology than an excoriation and has been described as the contrail of a jet plane. Using a dermatoscope attached to your smartphone allows you to magnify the image even further while maintaining a safe distance from the mite.
CORRESPONDENCE
Richard P. Usatine, MD, 903 W. Martin, Skin Clinic – Historic Building, San Antonio, TX 78207; [email protected].
Dermoscopy, the use of a handheld instrument to magnify the skin 10-fold while providing a light source, is a quick, useful, cost-effective tool for detecting melanoma in family medicine.1-4 The device, which allows the physician to visualize structures below the stratum corneum that are not routinely discernible with the naked eye, can be attached to a smartphone so that photos can be taken and reviewed with the patient. The photo can also be reviewed after a biopsy result is obtained.
Its use among non-dermatologist US physicians appears to be relatively low, but rising. One small study of physicians working in family medicine, internal medicine, and plastic surgery found that only 15% had ever used a dermatoscope and 6% were currently using one.5
As a family physician, you can expand your diagnostic abilities in dermatology with the acquisition of a dermatoscope (FIGURE 1) and some time invested in learning to interpret visible patterns. With that in mind, this review focuses on the diagnosis of skin cancers and benign growths using dermoscopy. We begin with a brief look at the research on dermoscopy and how it is performed. From there, we’ll detail an algorithm to guide dermoscopic analysis. And to round things out, we provide guidance that will help you to get started. (See “Choosing a dermatoscope—and making the most of it,” and “To learn more about dermoscopy …”.)
SIDEBAR
Choosing a dermatoscope—and making the most of it
1. Consider acquiring a hybrid dermatoscope.
Nonpolarized dermatoscopes (NPDs) and polarized dermatoscopes (PDs) provide different but complementary information. PDs enable users to identify features such as vessels and shiny white structures that are highly indicative of skin cancer. Because PDs are highly sensitive for detecting skin cancer and do not require a liquid interface or direct skin contact, they are the ideal dermatoscopes to use for skin cancer screening.
However, maintaining the highest specificity requires the complementary use of NPDs, which are better at identifying surface structures seen in seborrheic keratoses and other benign lesions. Thus, if the aim is to maintain the highest diagnostic accuracy for all types of lesions, then the preferred dermatoscope is a hybrid that permits the user to toggle between polarized and nonpolarized features in one device.
2. Choose a dermatoscope that attaches to your smartphone and/or camera.
This helps you capture digital dermoscopic images that can be analyzed on a larger screen, which permits:
- enlarging certain areas for in-depth analysis of structures and patterns
- sharing the image with the patient to explain why a biopsy is, or isn’t, needed
- sharing the image with a colleague for the purpose of a consult or a referral, or using the images for teaching purposes
- saving the images in order to follow lesions over time when monitoring is indicated
- ongoing learning. After each biopsy result comes back, we recommend correlating the dermoscopic images with the biopsy report. If your suspected diagnosis was correct, this reinforces your knowledge. If the pathology diagnosis is unexpected, you can learn by revisiting the original images to look for structures or patterns you may have missed upon first examination. You may even question the pathology report based on the dermoscopy, prompting a call to the pathologist.
- keeping a safe distance from the patient when looking for scabies mites.
SIDEBAR
To learn more about dermoscopy…
FREE APPS:
Dermoscopy 2-Step Algorithm. Available for free on iTunes, Google Play, and at https://usatinemedia.com/app/dermoscopy-two-step-algorithm/, this free app (developed by 3 of the 4 authors) is intended to help you interpret the dermoscopic patterns seen with your dermatoscope. It asks a series of questions that lead you to the most probable diagnosis. The app also contains more than 80 photos and charts to help you with your diagnosis. No Internet connection is needed to view the full app. There are 50 interactive cases to solve.
YOUdermoscopy Training (Available for free on iTunes, Google Play, and at https://www.youdermoscopytraining.org/) offers a fun game interface to test and expand your dermoscopy skills.
OTHER INTERNET RESOURCES:
- Dermoscopedia provides state-of-the-art information on dermoscopy. It’s available at: https://dermoscopedia.org.
- A free dermoscopy tutorial is available at: http://www.dermoscopy.org/
- The International Dermoscopy Society’s Web site, which offers various tutorials and other information, can be found at: http://www.dermoscopy-ids.org/.
COURSES:
Dermoscopy courses are a great way to get started and/or to advance your skills. The following courses are taught by the authors of this article:
- The American Dermoscopy Meeting is held yearly in the summer in a national park. See http://www.americandermoscopy.com/.
- Memorial Sloan Kettering Cancer Center holds a yearly dermoscopy workshop each fall in New York City. See http://www.mskcc.org/events/.
- The yearly American Academy of Family Physicians' FMX meeting offers dermoscopy workshops. See https://www.aafp.org/events/fmx.html.
Continue to: What the research says
What the research says
Dermoscopy improves sensitivity for detecting melanoma over the naked eye alone; it also allows for the detection of melanoma at earlier stages, which improves prognosis.6
A meta-analysis of dermoscopy use in clinical settings showed that, following training, dermoscopy increases the average sensitivity of melanoma diagnosis from 71% to more than 90% without a significant decrease in specificity.7 In a study of 74 primary care physicians, there was an improvement in both clinical and dermoscopic diagnosis of melanoma among those who received training in dermoscopy, compared with a control group.8 Another study found that primary care physicians can reduce their baseline benign-to-melanoma ratio (the number of suspicious benign lesions biopsied to find 1 melanoma) from 9.5:1 with naked eye examination to 3.5:1 with dermoscopy.9
The exam begins by choosing 1 of 3 modes of dermoscopy
Dermatoscopes can have a polarized or nonpolarized light source. Some dermatoscopes combine both types of light (hybrid dermatoscopes; see “Choosing a dermatoscope—and making the most of it.”)
There are 3 modes of dermoscopy:
- nonpolarized contact dermoscopy
- polarized contact dermoscopy
- polarized non-contact dermoscopy.
Dermatoscopes with nonpolarized light require direct skin contact and a liquid interface (eg, alcohol, gel, mineral oil) between the scope’s glass plate and the skin for the visualization of subsurface structures. In contrast, dermatoscopes with polarized light do not require direct skin contact or a liquid interface; however, contacting the skin and using a liquid interface will provide a sharper image.
Continue to: Two major algorithms guide dermoscopic analysis
Two major algorithms guide dermoscopic analysis
The first of 2 major algorithms that can be used to guide dermoscopic analysis is a modified pattern analysis put forth by Kittler.10 This descriptive system based on geometric elements, patterns, colors, and clues guides the observer to a specific diagnosis without categorizing lesions as being either melanocytic or nonmelanocytic. Because this is not the preferred method of the authors, we will move on to Method 2.
The second method, a 2-step algorithm, is a qualitative system that guides the observer through differentiating melanocytic from nonmelanocytic lesions in order to differentiate nevi from melanoma (FIGURE 2). At the same time, it serves as an aid to correctly diagnose non-melanocytic lesions. The 2-step algorithm forms the foundation for the dermoscopic evaluation of skin lesions in this article.
Not all expert dermoscopists employ structured analytical systems or methods to reach a diagnosis. Because of their vast experience, many rely purely on pattern recognition. But algorithms can facilitate non-experts in dermoscopy in the differentiation of nevi from melanoma or, simply, in differentiating the benign from the malignant.
Although each algorithm has its unique criteria, all of them require training and practice and familiarity with the terms used to describe morphologic structures. The International Dermoscopy Society recently published a consensus paper designating some terms as preferred over others.11
Continue to: Step 1...
Step 1: Melanocytic vs non-melanocytic
Step 1 of the 2-step algorithm requires the observer to determine whether the lesion is melanocytic (ie, originates from melanocytes and, therefore, could be a melanoma) or nonmelanocytic in origin.
A melanocytic lesion usually will display at least 1 of the following structures:
- pigment network (FIGURE 3A) (This can include angulated lines.)
- negative network (FIGURE 3B) (hypopigmented lines connecting pigmented structures in a serpiginous fashion)
- streaks (FIGURE 3C)
- homogeneous blue pigmentation (FIGURE 3D)
- globules (aggregated or as a peripheral rim) (FIGURE 3E)
- pseudonetwork (facial skin) (FIGURE 3F)
- parallel pigment pattern (acral lesions) (FIGURE 3G).
Exceptions. Sometimes, nonmelanocytic lesions will present with pigment network. Dermatofibromas, for example, are one exception in which the pattern trumps the network. Two other exceptions are solar lentigo and supernumerary or accessory nipple.
If the lesion does not display any structure, it is considered structureless. In these cases, proceed to the second step to rule out a melanoma.
Doesn’t meet criteria for a melanocytic lesion?
If the lesion does not reveal any of the criteria for a melanocytic lesion, then look for structures seen in nonmelanocytic lesions: dermatofibromas; seborrheic keratosis; angiomas and angiokeratomas; sebaceous hyperplasia; clear-cell acanthomas; basal cell carcinomas (BCCs); and squamous cell carcinomas (SCCs).
Continue to: Benign nonmelanocytic lesions
Benign nonmelanocytic lesions
Dermatofibromas are benign symmetric lesions that feel firm and may dimple upon application of lateral pressure. They are fibrotic scar-like lesions that present with 1 or more of the following dermoscopic features (FIGURE 4):
- peripheral pigment network, due to increased melanin in keratinocytes
- homogeneous brown pigmented areas
- central scar-like area
- shiny white lines
- vascular structures (ie, dotted, polymorphous vessels), usually seen within the scar-like area
- ring-like globules, usually seen in the zone between the scar-like depigmentation and the peripheral network. They correspond to widened hyperpigmented rete ridges.
Seborrheic keratosis (SK) is a benign skin growth that often has a stuck-on appearance (FIGURE 5). Features often include:
- multiple (>2) milia-like cysts
- comedo-like openings
- a network-like structure that corresponds to gyri and sulci and which in some cases can create a cerebriform pattern
- fingerprint-like structures
- moth-eaten borders
- jelly sign. This consists of semicircular u-shaped structures that have a smudged appearance and are aligned in the same direction. The appearance resembles jelly as it is spread on a piece of bread.
- hairpin (looped or twisted-looped) vessels surrounded by a white halo.
Other clues include a sharp demarcation and a negative wobble sign (which we’ll describe in a moment). The presence or absence of a wobble sign is determined by using a dermatoscope that touches the skin. Mild vertical pressure is applied to the lesion while moving the scope back and forth horizontally. If the lesion slides across the skin surface, the diagnosis of an epidermal keratinocytic tumor (ie, SK) is favored. If, on the other hand, the lesion wobbles (rolls back and forth), then the diagnosis of a neoplasm with a dermal component (ie, intradermal or compound nevus) is more likely.
Angiomas and angiokeratomas. Angiomas demonstrate lacunae that are often separated by septae (FIGURE 6). Lacunae can vary in size and color. They can be red, red-white, red-blue, maroon, blue, blue-black, or even black (when thrombosis is present).
Angiokeratomas (FIGURE 7) can reveal lacunae of varying colors including black, red, purple, and maroon. In addition, a blue-whitish veil, erythema, and hemorrhagic crusts can be present.
Continue to: Sebaceous hyperplasia...
Sebaceous hyperplasia is the overgrowth of sebaceous glands. It can mimic BCC on the face. Sebaceous hyperplasia presents with multiple vessels in a crown-like arrangement that do not cross the center of the lesion. The sebaceous glands resemble popcorn (FIGURE 8).
Clear-cell acanthoma is a benign erythematous epidermal tumor usually found on the leg with a string-of-pearls pattern. This pattern is vascular so the pearls are red in color (FIGURE 9).
Malignant nonmelanocytic lesions
BCC is the most common type of skin cancer. Features often include:
- spoke-wheel-like structures or concentric structures (FIGURE 10A)
- leaf-like areas (FIGURE 10B)
- arborizing vessels (FIGURE 10b and 10C)large blue-gray ovoid nest (FIGURE 10A)
- multiple blue-gray non-aggregated globules
- ulceration or multiple small erosions
- shiny white structures and strands (FIGURE 10C).
Additional dermoscopic clues include short, fine, superficial telangiectasias and multiple in-focus dots in a buck-shot scatter distribution.
Squamous cell carcinomas (SCCs) of the skin are keratinizing malignant tumors. Each SCC generally has some of the following features (FIGURE 11):
- dotted and/or glomerular vessels, commonly distributed focally at the periphery. They can also be diffuse or aligned linearly within the lesion.
- scale (yellow or white)
- rosettes (seen with polarized light)
- white circles or keratin pearls
- brown circles
- ulcerations
- brown dots or globules arranged in a linear configuration.
Continue to: Step 2...
Step 2: It’s melanocytic, but is it a nevus or a melanoma?
If, by following Step 1 of the algorithm, the lesion is determined to be of melanocytic origin, then one proceeds to Step 2 to decide whether the growth is a nevus, a suspicious lesion, or a melanoma. For this purpose, several additional algorithms are available.12-17
Benign nevi tend to manifest with 1 of the following 10 patterns: (FIGURE 12)
- diffuse reticular
- patchy reticular
- peripheral reticular with central hypopigmentation
- peripheral reticular with central hyperpigmentation
- homogeneous
- peripheral globules/starburst. It has been suggested that lesions that show starburst morphology on dermoscopy require complete excision and follow-up since 13% of Spitzoid-looking symmetric lesions in patients older than 12 years were found to be melanoma in one study.18
- peripheral reticular with central globules
- globular
- 2-component
- symmetric multicomponent (this pattern should be interpreted with caution, and a biopsy is probably warranted for dermoscopic novices).
Melanomas tend to deviate from the benign patterns described earlier. Structures in melanomas are often distributed in an asymmetric fashion (which is the basis for diagnosis in many of the other algorithms), and most of them will reveal 1 or more of the melanoma-specific structures (FIGURE 13). The melanomas in FIGURES 14 A-H each show at least 2 melanoma-specific structures. On the face or sun-damaged skin, melanoma may present with grey color, a circle-in-circle pattern, and/or polygonal lines (FIGURE 15). Note that melanoma on the soles or palms may present with a parallel ridge pattern (FIGURE 16).
How to proceed after the evaluation of melanocytic lesions
After evaluating the lesion for benign patterns and melanoma-specific structures, there are 3 possible pathways:
1. The lesion adheres to one of the nevi patterns and does not display a melanoma-specific structure. You can reassure the patient that the lesion is benign.
2. The lesion:
A. Adheres to one nevus pattern, but also displays a melanoma-specific structure.
B. Does not adhere to any of the benign patterns and does not have any melanoma-specific structures.
This is considered a suspicious lesion, and the choices of action include performing a biopsy or short-term monitoring by comparing dermoscopic images over a 3-month interval. (Caveat: Never monitor raised lesions because nodular melanomas can grow quickly and develop a worsened prognosis in a short time. Instead you’ll want to biopsy the lesion that day or very soon thereafter.)
3. The lesion deviates from the benign patterns and has at least 1 melanoma-specific structure. Biopsy the lesion to rule out melanoma.
Continue to: A bonus...
A bonus: Diagnosing scabies
Increasingly, dermoscopy is being used in the diagnosis of many other skin, nail, and hair problems. In fact, one great bonus to owning a dermatoscope is the accurate diagnosis of scabies. Dermoscopy can be helpful in detecting the scabies mite without having to scrape and use the microscope. Moreover, the sensitivity and specificity of a dermoscopic diagnosis is higher than for scraping and microscopy.19
What you’ll see
The anterior legs and mouth parts of the mite resemble a triangle (arrowhead, delta-wing jet) (FIGURE 17). Look for a burrow, and the mite can be seen at the end of the burrow as a faint circle with a leading darker triangle. The burrow itself has a distinctive pattern that has more morphology than an excoriation and has been described as the contrail of a jet plane. Using a dermatoscope attached to your smartphone allows you to magnify the image even further while maintaining a safe distance from the mite.
CORRESPONDENCE
Richard P. Usatine, MD, 903 W. Martin, Skin Clinic – Historic Building, San Antonio, TX 78207; [email protected].
1. Herschorn A. Dermoscopy for melanoma detection in family practice. Can Fam Physician. 2012;58:740-745.
2. Buckley D, McMonagle C. Melanoma in primary care. The role of the general practitioner. Ir J Med Sci. 2014;183:363-368.
3. Mayer JE, Swetter SM, Fu T, et al. Screening, early detection, education, and trends for melanoma: current status (2007-2013) and future directions: Part I Epidemiology, high-risk groups, clinical strategies, and diagnostic technology. J Am Acad Dermatol. 2014;71:599.e1-599.e12.
4. Mayer JE, Swetter SM, Fu T, et al. Screening, early detection, education, and trends for melanoma: current status (2007-2013) and future directions: Part II Screening, education, and future directions. J Am Acad Dermatol. 2014;71:611.e1-611.e10.
5. Morris JB, Alfonso SV, Hernandez N, et al. Use of and intentions to use dermoscopy among physicians in the United States. Dermatol Pract Concept. 2017;7:2.
6. Salerni G, Terán T, Alonso C, et al. The role of dermoscopy and digital dermoscopy follow-up in the clinical diagnosis of melanoma: clinical and dermoscopic features of 99 consecutive primary melanomas. Dermatol Pract Concept. 2014;4:39-46.
7. Vestergaard ME, Macaskill P, Holt PE, et al. Dermoscopy compared with naked eye examination for the diagnosis of primary melanoma: a meta-analysis of studies performed in a clinical setting. Br J Dermatol. 2008;159:669-676.
8. Westerhoff K, McCarthy WH, Menzies SW. Increase in the sensitivity for melanoma diagnosis by primary care physicians using skin surface microscopy. Br J Dermatol. 2000;143:1016-1020.
9. Menzies SW, Emery J, Staples M, et al. Impact of dermoscopy and short-term sequential digital dermoscopy imaging for the management of pigmented lesions in primary care: a sequential intervention trial. Br J Dermatol. 2009;161:1270-1277.
10. Kittler H. Dermatoscopy: introduction of a new algorithmic method based on pattern analysis for diagnosis of pigmented skin lesions. Dermatopathology: Practical & Conceptual. 2007;13:3.
11. Kittler H, Marghoob AA, Argenziano G, et al. Standardization of terminology in dermoscopy/dermatoscopy: results of the third consensus conference of the International Society of Dermoscopy. J Am Acad Dermatol. 2016;74:1093-1106.
12. Stolz W, Riemann A, Cognetta AB, et al. ABCD rule of dermoscopy: a new practical method for early recognition of malignant melanoma. Eur J Dermatol. 1994;4:521-527.
13. Pehamberger H, Steiner A, Wolff K. In vivo epiluminescence microscopy of pigmented skin lesions I Pattern analysis of pigmented skin lesions. J Am Acad Dermatol. 1987;17:571-583.
14. Menzies SW, Ingvar C, McCarthy WH. A sensitivity and specificity analysis of the surface microscopy features of invasive melanoma. Melanoma Res. 1996;6:55-62.
15. Argenziano G, Fabbrocini G, Carli P, et al. Epiluminescence microscopy for the diagnosis of doubtful melanocytic skin lesions. Comparison of the ABCD rule of dermatoscopy and a new 7-point checklist based on pattern analysis. Arch Dermatol. 1998;134:1563-1570.
16. Henning JS, Dusza SW, Wang SQ, et al. The CASH (color, architecture, symmetry, and homogeneity) algorithm for dermoscopy. J Am Acad Dermatol. 2007;56:45-52.
17. Soyer HP, Argenziano G, Zalaudek I, et al. Three-point checklist of dermoscopy. A new screening method for early detection of melanoma. Dermatology. 2004;208:27-31.
18. Lallas A, Moscarella E, Longo C, et al. Likelihood of finding melanoma when removing a Spitzoid-looking lesion in patients aged 12 years or older. J Am Acad Dermatol. 2015;72:47-53.
19. Dupuy A, Dehen L, Bourrat E, et al. Accuracy of standard dermoscopy for diagnosing scabies. J Am Acad Dermatol. 2007;56:53-62.
1. Herschorn A. Dermoscopy for melanoma detection in family practice. Can Fam Physician. 2012;58:740-745.
2. Buckley D, McMonagle C. Melanoma in primary care. The role of the general practitioner. Ir J Med Sci. 2014;183:363-368.
3. Mayer JE, Swetter SM, Fu T, et al. Screening, early detection, education, and trends for melanoma: current status (2007-2013) and future directions: Part I Epidemiology, high-risk groups, clinical strategies, and diagnostic technology. J Am Acad Dermatol. 2014;71:599.e1-599.e12.
4. Mayer JE, Swetter SM, Fu T, et al. Screening, early detection, education, and trends for melanoma: current status (2007-2013) and future directions: Part II Screening, education, and future directions. J Am Acad Dermatol. 2014;71:611.e1-611.e10.
5. Morris JB, Alfonso SV, Hernandez N, et al. Use of and intentions to use dermoscopy among physicians in the United States. Dermatol Pract Concept. 2017;7:2.
6. Salerni G, Terán T, Alonso C, et al. The role of dermoscopy and digital dermoscopy follow-up in the clinical diagnosis of melanoma: clinical and dermoscopic features of 99 consecutive primary melanomas. Dermatol Pract Concept. 2014;4:39-46.
7. Vestergaard ME, Macaskill P, Holt PE, et al. Dermoscopy compared with naked eye examination for the diagnosis of primary melanoma: a meta-analysis of studies performed in a clinical setting. Br J Dermatol. 2008;159:669-676.
8. Westerhoff K, McCarthy WH, Menzies SW. Increase in the sensitivity for melanoma diagnosis by primary care physicians using skin surface microscopy. Br J Dermatol. 2000;143:1016-1020.
9. Menzies SW, Emery J, Staples M, et al. Impact of dermoscopy and short-term sequential digital dermoscopy imaging for the management of pigmented lesions in primary care: a sequential intervention trial. Br J Dermatol. 2009;161:1270-1277.
10. Kittler H. Dermatoscopy: introduction of a new algorithmic method based on pattern analysis for diagnosis of pigmented skin lesions. Dermatopathology: Practical & Conceptual. 2007;13:3.
11. Kittler H, Marghoob AA, Argenziano G, et al. Standardization of terminology in dermoscopy/dermatoscopy: results of the third consensus conference of the International Society of Dermoscopy. J Am Acad Dermatol. 2016;74:1093-1106.
12. Stolz W, Riemann A, Cognetta AB, et al. ABCD rule of dermoscopy: a new practical method for early recognition of malignant melanoma. Eur J Dermatol. 1994;4:521-527.
13. Pehamberger H, Steiner A, Wolff K. In vivo epiluminescence microscopy of pigmented skin lesions I Pattern analysis of pigmented skin lesions. J Am Acad Dermatol. 1987;17:571-583.
14. Menzies SW, Ingvar C, McCarthy WH. A sensitivity and specificity analysis of the surface microscopy features of invasive melanoma. Melanoma Res. 1996;6:55-62.
15. Argenziano G, Fabbrocini G, Carli P, et al. Epiluminescence microscopy for the diagnosis of doubtful melanocytic skin lesions. Comparison of the ABCD rule of dermatoscopy and a new 7-point checklist based on pattern analysis. Arch Dermatol. 1998;134:1563-1570.
16. Henning JS, Dusza SW, Wang SQ, et al. The CASH (color, architecture, symmetry, and homogeneity) algorithm for dermoscopy. J Am Acad Dermatol. 2007;56:45-52.
17. Soyer HP, Argenziano G, Zalaudek I, et al. Three-point checklist of dermoscopy. A new screening method for early detection of melanoma. Dermatology. 2004;208:27-31.
18. Lallas A, Moscarella E, Longo C, et al. Likelihood of finding melanoma when removing a Spitzoid-looking lesion in patients aged 12 years or older. J Am Acad Dermatol. 2015;72:47-53.
19. Dupuy A, Dehen L, Bourrat E, et al. Accuracy of standard dermoscopy for diagnosing scabies. J Am Acad Dermatol. 2007;56:53-62.

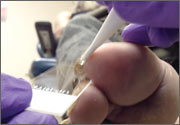
Persistent rash on feet
A 49-year-old Hispanic woman presented with a 4-month history of scaling and a macerated rash localized between her toes (FIGURE 1). The rash was malodorous, mildly erythematous, and sometimes associated with pruritus. The patient had no relevant medical history. Potassium hydroxide (KOH) testing was performed and found to be negative. So a Wood’s lamp was used to examine the patient’s toes—and it revealed the diagnosis.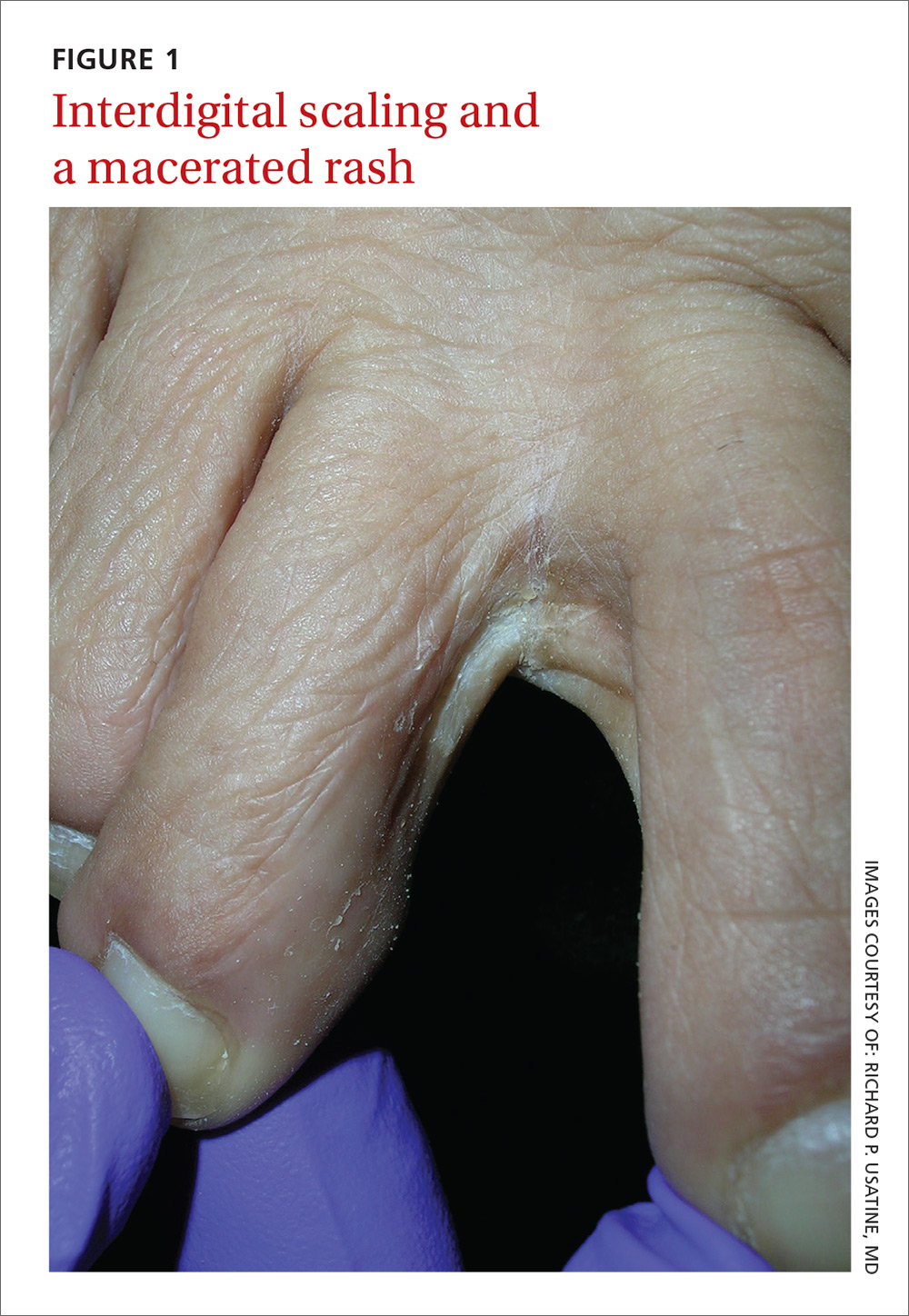
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Erythrasma
The Wood’s lamp revealed a coral-red fluorescence in the interdigital spaces (FIGURE 2), which led us to a diagnosis of erythrasma.
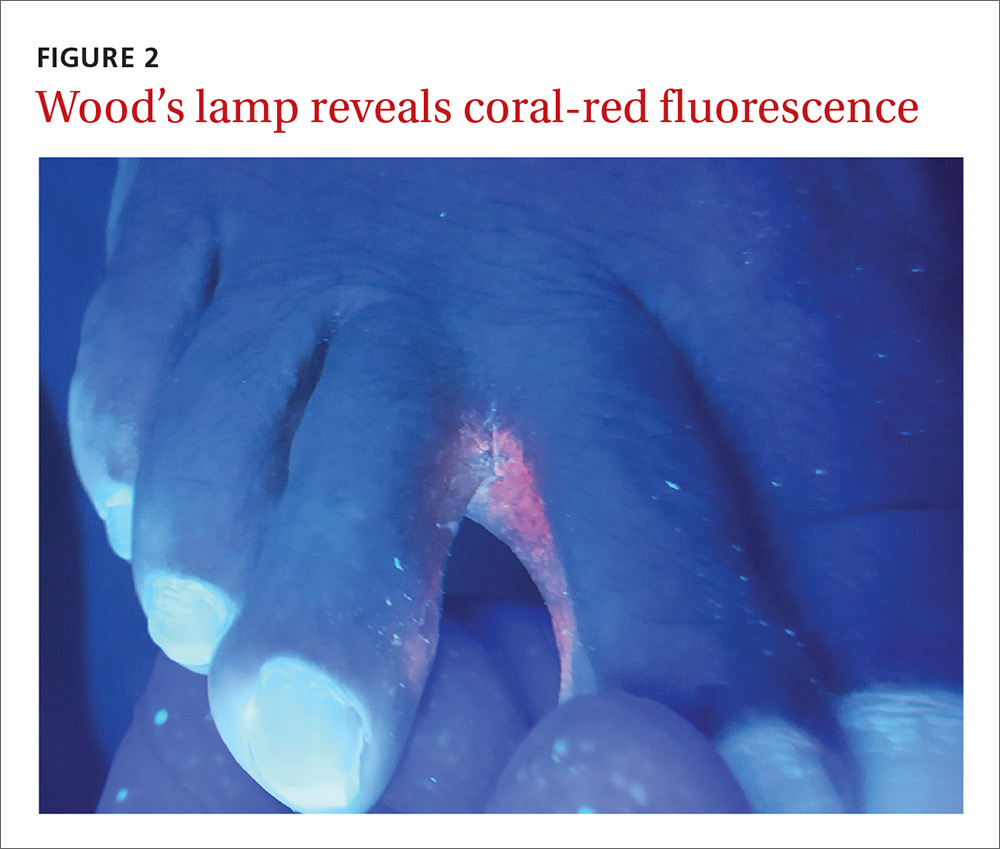
The coral-red fluorescence seen under the Wood’s lamp is due to porphyrins produced by Corynebacterium minutissimum. The organism invades the stratum corneum where it proliferates and causes erythrasma. Erythrasma typically appears as delineated, dry, red-brown patches in intertriginous areas, such as the axilla, groin, interdigital spaces, intergluteal cleft, perianal skin, and inframammary area.1,2
Interdigital erythrasma is more common than previously thought; in one study of 151 patients with erythrasma, the most common site was the toe webs (64.9%), followed by the inguinal region (17.9%), the axillary region (14.6%), and the inframammary region (2.6%).2 Erythrasma affects 4% of the population; risk factors include poor hygiene, hyperhidrosis, obesity, warm climate, diabetes, and an immunocompromised state.3
Differential includes “athlete’s foot”
The differential diagnosis for a pruritic rash between the toes includes:
Tinea pedis. Erythrasma is often mistaken for tinea pedis, because both conditions cause scaling between the toes. A Wood’s lamp exam can quickly differentiate between the 2,1 as tinea pedis does not fluoresce under ultraviolet light.
Contact dermatitis mimics many conditions, but a negative Wood’s lamp exam and history of worsening with contact to specific substances helps to make this diagnosis.
Prevention and Tx hinge on good hygiene, topical agents
First-line management of erythrasma includes both nonpharmacologic and pharmacologic modalities. Good hygiene and, depending on the area affected, loose-fitting cotton undergarments can help treat and prevent erythrasma.
Topical 2% miconazole bid for 2 weeks has resulted in clearance rates as high as 88%.4 Its affordable price, over-the-counter availability, and lack of adverse effects make miconazole a reasonable choice.4,5 It is also a smart treatment choice when erythrasma is coexisting with tinea, because it can treat both conditions. This is not uncommon in the interdigital spaces between the toes and in the groin.
Topical 1% clindamycin or 2% erythromycin solution or gel bid for 2 weeks can also be used to treat the condition.3,6 However, given that topical antibiotics are more expensive than single-dose oral treatment and are no better than the oral formulations of these antibiotics,6 clarithromycin 1 g taken once orally may be preferred.2,6
Our patient was treated with a single dose of clarithromycin 1 g. At follow-up, her erythrasma was clear.
CORRESPONDENCE
Richard P. Usatine, MD, University of Texas Health at San Antonio, 7703 Floyd Curl Dr., San Antonio, TX 78229; [email protected].
1. Polat M, lhan MN. The prevalence of interdigital erythrasma: a prospective study from an outpatient clinic in Turkey. J Am Podiatr Med Assoc. 2015;105:121-124.
2. Avci O, Tanyildizi T, Kusku E. A comparison between the effectiveness of erythromycin, single-dose clarithromycin and topical fusidic acid in the treatment of erythrasma. J Dermatolog Treat. 2013;24:70-74.
3. Kibbi AG, Sleiman M. Erythrasma. Available at: http://emedicine.medscape.com/article/1052532-overview#a0199. Accessed December 10, 2016.
4. Pitcher DG, Noble WC, Seville RH. Treatment of erythrasma with miconazole. Clin Exp Dermatol. 1979;4:453-456.
5. Clayton YM, Knight AG. A clinical double-blind trial of topical miconazole and clotrimazole against superficial fungal infections and erythrasma. Clin Exp Dermatol. 1976;1:225-232.
6. Holdiness MR. Management of cutaneous erythrasma. Drugs. 2002;62:1131-1141.
A 49-year-old Hispanic woman presented with a 4-month history of scaling and a macerated rash localized between her toes (FIGURE 1). The rash was malodorous, mildly erythematous, and sometimes associated with pruritus. The patient had no relevant medical history. Potassium hydroxide (KOH) testing was performed and found to be negative. So a Wood’s lamp was used to examine the patient’s toes—and it revealed the diagnosis.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Erythrasma
The Wood’s lamp revealed a coral-red fluorescence in the interdigital spaces (FIGURE 2), which led us to a diagnosis of erythrasma.
The coral-red fluorescence seen under the Wood’s lamp is due to porphyrins produced by Corynebacterium minutissimum. The organism invades the stratum corneum where it proliferates and causes erythrasma. Erythrasma typically appears as delineated, dry, red-brown patches in intertriginous areas, such as the axilla, groin, interdigital spaces, intergluteal cleft, perianal skin, and inframammary area.1,2
Interdigital erythrasma is more common than previously thought; in one study of 151 patients with erythrasma, the most common site was the toe webs (64.9%), followed by the inguinal region (17.9%), the axillary region (14.6%), and the inframammary region (2.6%).2 Erythrasma affects 4% of the population; risk factors include poor hygiene, hyperhidrosis, obesity, warm climate, diabetes, and an immunocompromised state.3
Differential includes “athlete’s foot”
The differential diagnosis for a pruritic rash between the toes includes:
Tinea pedis. Erythrasma is often mistaken for tinea pedis, because both conditions cause scaling between the toes. A Wood’s lamp exam can quickly differentiate between the 2,1 as tinea pedis does not fluoresce under ultraviolet light.
Contact dermatitis mimics many conditions, but a negative Wood’s lamp exam and history of worsening with contact to specific substances helps to make this diagnosis.
Prevention and Tx hinge on good hygiene, topical agents
First-line management of erythrasma includes both nonpharmacologic and pharmacologic modalities. Good hygiene and, depending on the area affected, loose-fitting cotton undergarments can help treat and prevent erythrasma.
Topical 2% miconazole bid for 2 weeks has resulted in clearance rates as high as 88%.4 Its affordable price, over-the-counter availability, and lack of adverse effects make miconazole a reasonable choice.4,5 It is also a smart treatment choice when erythrasma is coexisting with tinea, because it can treat both conditions. This is not uncommon in the interdigital spaces between the toes and in the groin.
Topical 1% clindamycin or 2% erythromycin solution or gel bid for 2 weeks can also be used to treat the condition.3,6 However, given that topical antibiotics are more expensive than single-dose oral treatment and are no better than the oral formulations of these antibiotics,6 clarithromycin 1 g taken once orally may be preferred.2,6
Our patient was treated with a single dose of clarithromycin 1 g. At follow-up, her erythrasma was clear.
CORRESPONDENCE
Richard P. Usatine, MD, University of Texas Health at San Antonio, 7703 Floyd Curl Dr., San Antonio, TX 78229; [email protected].
A 49-year-old Hispanic woman presented with a 4-month history of scaling and a macerated rash localized between her toes (FIGURE 1). The rash was malodorous, mildly erythematous, and sometimes associated with pruritus. The patient had no relevant medical history. Potassium hydroxide (KOH) testing was performed and found to be negative. So a Wood’s lamp was used to examine the patient’s toes—and it revealed the diagnosis.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Erythrasma
The Wood’s lamp revealed a coral-red fluorescence in the interdigital spaces (FIGURE 2), which led us to a diagnosis of erythrasma.
The coral-red fluorescence seen under the Wood’s lamp is due to porphyrins produced by Corynebacterium minutissimum. The organism invades the stratum corneum where it proliferates and causes erythrasma. Erythrasma typically appears as delineated, dry, red-brown patches in intertriginous areas, such as the axilla, groin, interdigital spaces, intergluteal cleft, perianal skin, and inframammary area.1,2
Interdigital erythrasma is more common than previously thought; in one study of 151 patients with erythrasma, the most common site was the toe webs (64.9%), followed by the inguinal region (17.9%), the axillary region (14.6%), and the inframammary region (2.6%).2 Erythrasma affects 4% of the population; risk factors include poor hygiene, hyperhidrosis, obesity, warm climate, diabetes, and an immunocompromised state.3
Differential includes “athlete’s foot”
The differential diagnosis for a pruritic rash between the toes includes:
Tinea pedis. Erythrasma is often mistaken for tinea pedis, because both conditions cause scaling between the toes. A Wood’s lamp exam can quickly differentiate between the 2,1 as tinea pedis does not fluoresce under ultraviolet light.
Contact dermatitis mimics many conditions, but a negative Wood’s lamp exam and history of worsening with contact to specific substances helps to make this diagnosis.
Prevention and Tx hinge on good hygiene, topical agents
First-line management of erythrasma includes both nonpharmacologic and pharmacologic modalities. Good hygiene and, depending on the area affected, loose-fitting cotton undergarments can help treat and prevent erythrasma.
Topical 2% miconazole bid for 2 weeks has resulted in clearance rates as high as 88%.4 Its affordable price, over-the-counter availability, and lack of adverse effects make miconazole a reasonable choice.4,5 It is also a smart treatment choice when erythrasma is coexisting with tinea, because it can treat both conditions. This is not uncommon in the interdigital spaces between the toes and in the groin.
Topical 1% clindamycin or 2% erythromycin solution or gel bid for 2 weeks can also be used to treat the condition.3,6 However, given that topical antibiotics are more expensive than single-dose oral treatment and are no better than the oral formulations of these antibiotics,6 clarithromycin 1 g taken once orally may be preferred.2,6
Our patient was treated with a single dose of clarithromycin 1 g. At follow-up, her erythrasma was clear.
CORRESPONDENCE
Richard P. Usatine, MD, University of Texas Health at San Antonio, 7703 Floyd Curl Dr., San Antonio, TX 78229; [email protected].
1. Polat M, lhan MN. The prevalence of interdigital erythrasma: a prospective study from an outpatient clinic in Turkey. J Am Podiatr Med Assoc. 2015;105:121-124.
2. Avci O, Tanyildizi T, Kusku E. A comparison between the effectiveness of erythromycin, single-dose clarithromycin and topical fusidic acid in the treatment of erythrasma. J Dermatolog Treat. 2013;24:70-74.
3. Kibbi AG, Sleiman M. Erythrasma. Available at: http://emedicine.medscape.com/article/1052532-overview#a0199. Accessed December 10, 2016.
4. Pitcher DG, Noble WC, Seville RH. Treatment of erythrasma with miconazole. Clin Exp Dermatol. 1979;4:453-456.
5. Clayton YM, Knight AG. A clinical double-blind trial of topical miconazole and clotrimazole against superficial fungal infections and erythrasma. Clin Exp Dermatol. 1976;1:225-232.
6. Holdiness MR. Management of cutaneous erythrasma. Drugs. 2002;62:1131-1141.
1. Polat M, lhan MN. The prevalence of interdigital erythrasma: a prospective study from an outpatient clinic in Turkey. J Am Podiatr Med Assoc. 2015;105:121-124.
2. Avci O, Tanyildizi T, Kusku E. A comparison between the effectiveness of erythromycin, single-dose clarithromycin and topical fusidic acid in the treatment of erythrasma. J Dermatolog Treat. 2013;24:70-74.
3. Kibbi AG, Sleiman M. Erythrasma. Available at: http://emedicine.medscape.com/article/1052532-overview#a0199. Accessed December 10, 2016.
4. Pitcher DG, Noble WC, Seville RH. Treatment of erythrasma with miconazole. Clin Exp Dermatol. 1979;4:453-456.
5. Clayton YM, Knight AG. A clinical double-blind trial of topical miconazole and clotrimazole against superficial fungal infections and erythrasma. Clin Exp Dermatol. 1976;1:225-232.
6. Holdiness MR. Management of cutaneous erythrasma. Drugs. 2002;62:1131-1141.
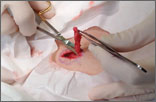
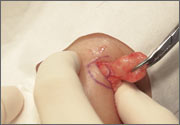
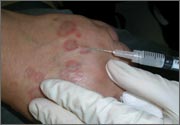
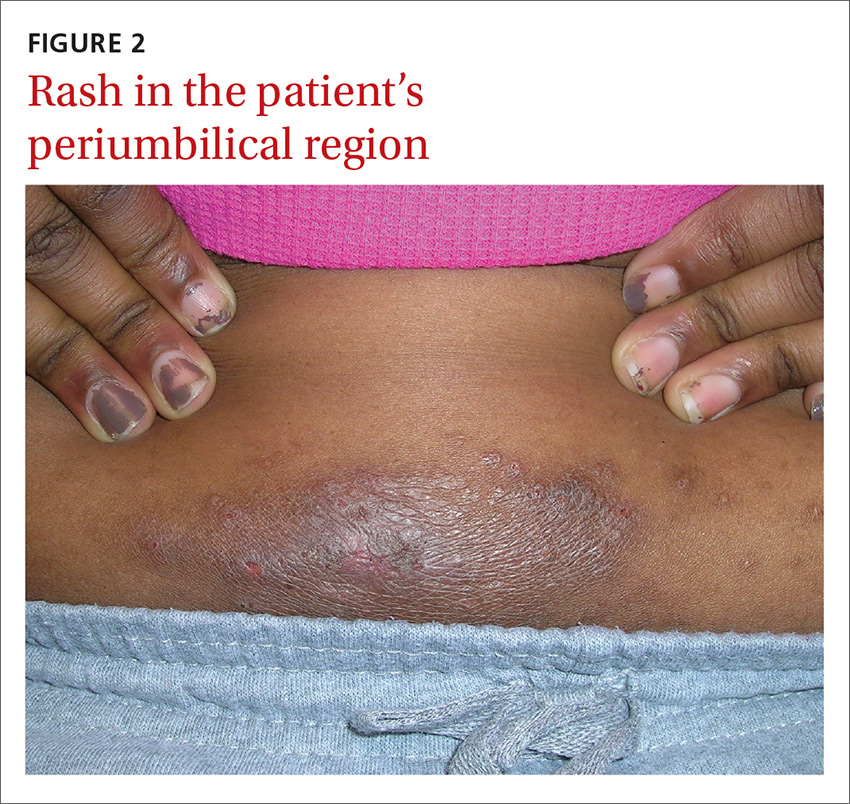
Rash on eyebrows and periumbilical region
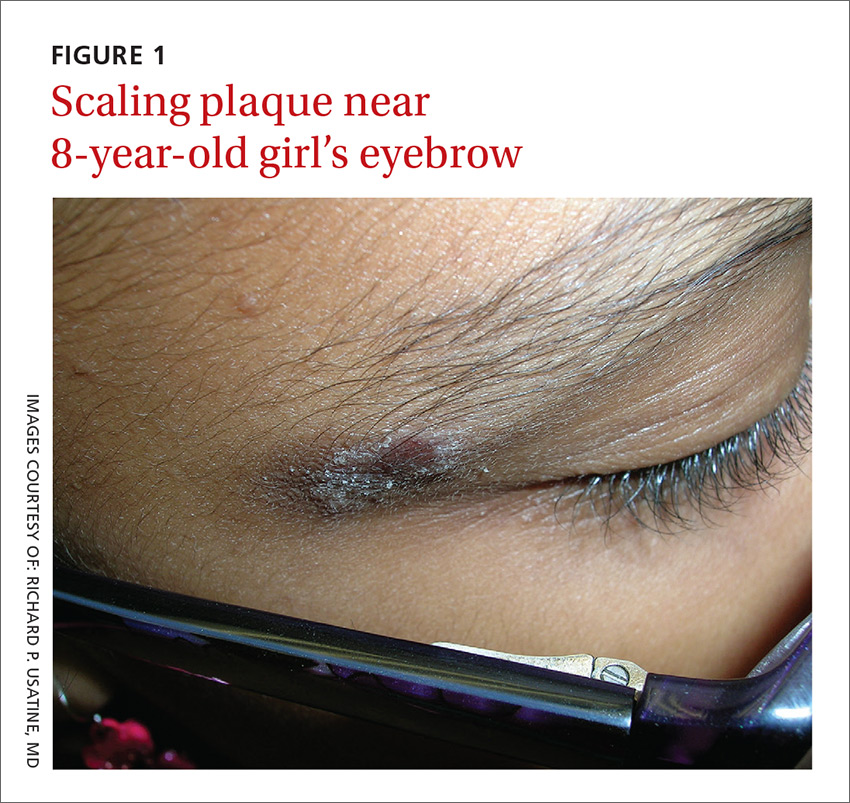
An 8-year-old girl was brought to her family physician’s office (RU) because of a persistent rash on her lateral eyebrows and periumbilical region. The family indicated that she’d had the rash for more than 6 months. They also mentioned that the child had received a new pair of eyeglasses 8 months earlier. The child was otherwise in good health. The physical examination revealed erythematous scaling plaques near both lateral eyebrows and around the belly button (FIGURES 1 AND 2).
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Allergic contact dermatitis
We recognized that this was a case of allergic contact dermatitis (ACD), based on the clinical presentation. The distribution of the erythema, scale, and postinflammatory hyperpigmentation was highly suggestive of an ACD to nickel. In this case, the nickel in the patient’s eyeglasses and the snaps found on her pants were the culprits.
The lichenification of the plaque near the umbilicus suggested that the dermatitis was not acute and that the patient had likely been scratching the area due to itching. The plaque near the patient’s eye was actually in the shape of the metal on the inside of her glasses.
Most prevalent contact allergens. Patch testing data indicate that the 5 most prevalent contact allergens out of more than 3700 that are known are: nickel (14.3% of patients tested), fragrance mix (14%), the topical antibiotic neomycin (11.6%), balsam of Peru (used in some perfumes, toiletries, and pharmaceutical items) (10.4%), and the mercury-based vaccine preservative thimerosal (10.4%).1
ACD is a delayed-type hypersensitivity reaction in which a foreign substance comes into contact with the skin and is linked to skin proteins forming an antigen complex that leads to sensitization. When the epidermis is re-exposed to the antigen, the sensitized T cells initiate an inflammatory cascade, leading to the skin changes seen in ACD.2
Silverberg et al reported that in 30 children with a personal history of umbilical or wrist dermatitis or a family history of nickel ACD, 100% demonstrated a positive reaction to nickel sulfate.3 Nickel continues to be used (and unregulated) in a wide range of products, including costume jewelry, piercing posts, belt buckles, eyeglasses, and personal electronics (eg, tablets, cell phones, and laptop computers).
Making the diagnosis. Contact dermatitis can sometimes be diagnosed clinically with a good history and physical exam. However, there are many cases in which patch testing is needed to find the offending allergens or confirm the suspicion regarding a specific allergen. The only convenient and ready-to-use patch test in the United States is the T.R.U.E. test.
The differential includes other superficial skin infections
ACD characteristically presents with eczematoid plaques that are primarily in the area(s) of cutaneous contact with an allergen. The condition typically appears within a few days of exposure.
The differential diagnosis for ACD includes cutaneous candidiasis, impetigo, plaque psoriasis, and seborrheic dermatitis.
Cutaneous candidiasis is a superficial infection of the skin with a candida species. It can present as beefy red erythematous plaques on the buttocks, lower abdomen, thighs, or in intertriginous areas or oral commissures. A hallmark sign is pinpoint pustulovesicular satellite lesions.
Impetigo is a superficial bacterial skin infection that presents with edema, erythema, tenderness on palpation, and possible purulent drainage. It appears as honey-colored crusts with erythema and occurs most often on the face—especially around a child’s nose and mouth—but can occur anywhere on the head and body.
Plaque psoriasis presents as erythematous silver-scaled plaques on extensor surfaces, including the elbows and knees. Inverse psoriasis may present as erythema and maceration in intertriginous areas.
Seborrheic dermatitis appears as well-circumscribed greasy scale overlying erythematous skin. It is commonly found on the scalp, eyebrows, nasolabial folds, chest, face, and in the ear canals. It is thought to be an inflammatory reaction to Malassezia furfur.
Cool compresses, topical steroids can relieve symptoms
Patients with ACD should avoid the allergen that is causing the reaction. In cases of nickel ACD, the patient may cover the metal tab of their jeans with an iron-on patch or a few coats of clear nail polish. A better option is to buy jeans and pants that do not have nickel in the metal tab. (Levi’s has removed nickel from their pants.) Cool compresses can soothe the symptoms of acute cases of ACD.4 Calamine and colloidal oatmeal baths may help to dry and soothe acute, oozing lesions. Localized acute ACD lesions respond best to mid-potency (eg, 0.1% triamcinolone) to high-potency (eg, 0.05% clobetasol) topical steroids.4
On areas of thinner skin (eg, flexural surfaces, eyelids, face, anogenital region), lower-potency steroids such as desonide ointment can minimize the risk of skin atrophy.3,4 Be aware that some patients are actually allergic to topical steroids. This unfortunate situation can be diagnosed with patch testing.
We recommended that our patient get different glasses that were nickel-free. Fortunately, there are many frames for glasses that have no nickel in them. We also gave her advice on how to avoid the nickel that still exists in some pants. We gave her desonide 0.05% cream to apply to the affected area for symptomatic relief.
CORRESPONDENCE
Richard P. Usatine, MD, Skin clinic, University of Texas Health Science Center at San Antonio, 903 W. Martin, San Antonio, TX 78207; [email protected].
1. Krob HA, Fleischer AB Jr, D’Agostino R Jr, et al. Prevalence and relevance of contact dermatitis allergens: a meta-analysis of 15 years of published T.R.U.E. test data. J Am Acad Dermatol. 2004;51:349-353.
2. Usatine RP, Riojas M. Diagnosis and management of contact dermatitis. Am Fam Physician. 2010;82:249-255.
3. Silverberg NB, Licht J, Friedler S, et al. Nickel contact hypersensitivity in children. Pediatr Dermatol. 2002;19:110-113.
4. Beltrani VS, Bernstein IL, Cohen DE, et al. Contact dermatitis: a practice parameter. Ann Allergy Asthma Immunol. 2006;97:S1-S38.
An 8-year-old girl was brought to her family physician’s office (RU) because of a persistent rash on her lateral eyebrows and periumbilical region. The family indicated that she’d had the rash for more than 6 months. They also mentioned that the child had received a new pair of eyeglasses 8 months earlier. The child was otherwise in good health. The physical examination revealed erythematous scaling plaques near both lateral eyebrows and around the belly button (FIGURES 1 AND 2).
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Allergic contact dermatitis
We recognized that this was a case of allergic contact dermatitis (ACD), based on the clinical presentation. The distribution of the erythema, scale, and postinflammatory hyperpigmentation was highly suggestive of an ACD to nickel. In this case, the nickel in the patient’s eyeglasses and the snaps found on her pants were the culprits.
The lichenification of the plaque near the umbilicus suggested that the dermatitis was not acute and that the patient had likely been scratching the area due to itching. The plaque near the patient’s eye was actually in the shape of the metal on the inside of her glasses.
Most prevalent contact allergens. Patch testing data indicate that the 5 most prevalent contact allergens out of more than 3700 that are known are: nickel (14.3% of patients tested), fragrance mix (14%), the topical antibiotic neomycin (11.6%), balsam of Peru (used in some perfumes, toiletries, and pharmaceutical items) (10.4%), and the mercury-based vaccine preservative thimerosal (10.4%).1
ACD is a delayed-type hypersensitivity reaction in which a foreign substance comes into contact with the skin and is linked to skin proteins forming an antigen complex that leads to sensitization. When the epidermis is re-exposed to the antigen, the sensitized T cells initiate an inflammatory cascade, leading to the skin changes seen in ACD.2
Silverberg et al reported that in 30 children with a personal history of umbilical or wrist dermatitis or a family history of nickel ACD, 100% demonstrated a positive reaction to nickel sulfate.3 Nickel continues to be used (and unregulated) in a wide range of products, including costume jewelry, piercing posts, belt buckles, eyeglasses, and personal electronics (eg, tablets, cell phones, and laptop computers).
Making the diagnosis. Contact dermatitis can sometimes be diagnosed clinically with a good history and physical exam. However, there are many cases in which patch testing is needed to find the offending allergens or confirm the suspicion regarding a specific allergen. The only convenient and ready-to-use patch test in the United States is the T.R.U.E. test.
The differential includes other superficial skin infections
ACD characteristically presents with eczematoid plaques that are primarily in the area(s) of cutaneous contact with an allergen. The condition typically appears within a few days of exposure.
The differential diagnosis for ACD includes cutaneous candidiasis, impetigo, plaque psoriasis, and seborrheic dermatitis.
Cutaneous candidiasis is a superficial infection of the skin with a candida species. It can present as beefy red erythematous plaques on the buttocks, lower abdomen, thighs, or in intertriginous areas or oral commissures. A hallmark sign is pinpoint pustulovesicular satellite lesions.
Impetigo is a superficial bacterial skin infection that presents with edema, erythema, tenderness on palpation, and possible purulent drainage. It appears as honey-colored crusts with erythema and occurs most often on the face—especially around a child’s nose and mouth—but can occur anywhere on the head and body.
Plaque psoriasis presents as erythematous silver-scaled plaques on extensor surfaces, including the elbows and knees. Inverse psoriasis may present as erythema and maceration in intertriginous areas.
Seborrheic dermatitis appears as well-circumscribed greasy scale overlying erythematous skin. It is commonly found on the scalp, eyebrows, nasolabial folds, chest, face, and in the ear canals. It is thought to be an inflammatory reaction to Malassezia furfur.
Cool compresses, topical steroids can relieve symptoms
Patients with ACD should avoid the allergen that is causing the reaction. In cases of nickel ACD, the patient may cover the metal tab of their jeans with an iron-on patch or a few coats of clear nail polish. A better option is to buy jeans and pants that do not have nickel in the metal tab. (Levi’s has removed nickel from their pants.) Cool compresses can soothe the symptoms of acute cases of ACD.4 Calamine and colloidal oatmeal baths may help to dry and soothe acute, oozing lesions. Localized acute ACD lesions respond best to mid-potency (eg, 0.1% triamcinolone) to high-potency (eg, 0.05% clobetasol) topical steroids.4
On areas of thinner skin (eg, flexural surfaces, eyelids, face, anogenital region), lower-potency steroids such as desonide ointment can minimize the risk of skin atrophy.3,4 Be aware that some patients are actually allergic to topical steroids. This unfortunate situation can be diagnosed with patch testing.
We recommended that our patient get different glasses that were nickel-free. Fortunately, there are many frames for glasses that have no nickel in them. We also gave her advice on how to avoid the nickel that still exists in some pants. We gave her desonide 0.05% cream to apply to the affected area for symptomatic relief.
CORRESPONDENCE
Richard P. Usatine, MD, Skin clinic, University of Texas Health Science Center at San Antonio, 903 W. Martin, San Antonio, TX 78207; [email protected].
An 8-year-old girl was brought to her family physician’s office (RU) because of a persistent rash on her lateral eyebrows and periumbilical region. The family indicated that she’d had the rash for more than 6 months. They also mentioned that the child had received a new pair of eyeglasses 8 months earlier. The child was otherwise in good health. The physical examination revealed erythematous scaling plaques near both lateral eyebrows and around the belly button (FIGURES 1 AND 2).
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Allergic contact dermatitis
We recognized that this was a case of allergic contact dermatitis (ACD), based on the clinical presentation. The distribution of the erythema, scale, and postinflammatory hyperpigmentation was highly suggestive of an ACD to nickel. In this case, the nickel in the patient’s eyeglasses and the snaps found on her pants were the culprits.
The lichenification of the plaque near the umbilicus suggested that the dermatitis was not acute and that the patient had likely been scratching the area due to itching. The plaque near the patient’s eye was actually in the shape of the metal on the inside of her glasses.
Most prevalent contact allergens. Patch testing data indicate that the 5 most prevalent contact allergens out of more than 3700 that are known are: nickel (14.3% of patients tested), fragrance mix (14%), the topical antibiotic neomycin (11.6%), balsam of Peru (used in some perfumes, toiletries, and pharmaceutical items) (10.4%), and the mercury-based vaccine preservative thimerosal (10.4%).1
ACD is a delayed-type hypersensitivity reaction in which a foreign substance comes into contact with the skin and is linked to skin proteins forming an antigen complex that leads to sensitization. When the epidermis is re-exposed to the antigen, the sensitized T cells initiate an inflammatory cascade, leading to the skin changes seen in ACD.2
Silverberg et al reported that in 30 children with a personal history of umbilical or wrist dermatitis or a family history of nickel ACD, 100% demonstrated a positive reaction to nickel sulfate.3 Nickel continues to be used (and unregulated) in a wide range of products, including costume jewelry, piercing posts, belt buckles, eyeglasses, and personal electronics (eg, tablets, cell phones, and laptop computers).
Making the diagnosis. Contact dermatitis can sometimes be diagnosed clinically with a good history and physical exam. However, there are many cases in which patch testing is needed to find the offending allergens or confirm the suspicion regarding a specific allergen. The only convenient and ready-to-use patch test in the United States is the T.R.U.E. test.
The differential includes other superficial skin infections
ACD characteristically presents with eczematoid plaques that are primarily in the area(s) of cutaneous contact with an allergen. The condition typically appears within a few days of exposure.
The differential diagnosis for ACD includes cutaneous candidiasis, impetigo, plaque psoriasis, and seborrheic dermatitis.
Cutaneous candidiasis is a superficial infection of the skin with a candida species. It can present as beefy red erythematous plaques on the buttocks, lower abdomen, thighs, or in intertriginous areas or oral commissures. A hallmark sign is pinpoint pustulovesicular satellite lesions.
Impetigo is a superficial bacterial skin infection that presents with edema, erythema, tenderness on palpation, and possible purulent drainage. It appears as honey-colored crusts with erythema and occurs most often on the face—especially around a child’s nose and mouth—but can occur anywhere on the head and body.
Plaque psoriasis presents as erythematous silver-scaled plaques on extensor surfaces, including the elbows and knees. Inverse psoriasis may present as erythema and maceration in intertriginous areas.
Seborrheic dermatitis appears as well-circumscribed greasy scale overlying erythematous skin. It is commonly found on the scalp, eyebrows, nasolabial folds, chest, face, and in the ear canals. It is thought to be an inflammatory reaction to Malassezia furfur.
Cool compresses, topical steroids can relieve symptoms
Patients with ACD should avoid the allergen that is causing the reaction. In cases of nickel ACD, the patient may cover the metal tab of their jeans with an iron-on patch or a few coats of clear nail polish. A better option is to buy jeans and pants that do not have nickel in the metal tab. (Levi’s has removed nickel from their pants.) Cool compresses can soothe the symptoms of acute cases of ACD.4 Calamine and colloidal oatmeal baths may help to dry and soothe acute, oozing lesions. Localized acute ACD lesions respond best to mid-potency (eg, 0.1% triamcinolone) to high-potency (eg, 0.05% clobetasol) topical steroids.4
On areas of thinner skin (eg, flexural surfaces, eyelids, face, anogenital region), lower-potency steroids such as desonide ointment can minimize the risk of skin atrophy.3,4 Be aware that some patients are actually allergic to topical steroids. This unfortunate situation can be diagnosed with patch testing.
We recommended that our patient get different glasses that were nickel-free. Fortunately, there are many frames for glasses that have no nickel in them. We also gave her advice on how to avoid the nickel that still exists in some pants. We gave her desonide 0.05% cream to apply to the affected area for symptomatic relief.
CORRESPONDENCE
Richard P. Usatine, MD, Skin clinic, University of Texas Health Science Center at San Antonio, 903 W. Martin, San Antonio, TX 78207; [email protected].
1. Krob HA, Fleischer AB Jr, D’Agostino R Jr, et al. Prevalence and relevance of contact dermatitis allergens: a meta-analysis of 15 years of published T.R.U.E. test data. J Am Acad Dermatol. 2004;51:349-353.
2. Usatine RP, Riojas M. Diagnosis and management of contact dermatitis. Am Fam Physician. 2010;82:249-255.
3. Silverberg NB, Licht J, Friedler S, et al. Nickel contact hypersensitivity in children. Pediatr Dermatol. 2002;19:110-113.
4. Beltrani VS, Bernstein IL, Cohen DE, et al. Contact dermatitis: a practice parameter. Ann Allergy Asthma Immunol. 2006;97:S1-S38.
1. Krob HA, Fleischer AB Jr, D’Agostino R Jr, et al. Prevalence and relevance of contact dermatitis allergens: a meta-analysis of 15 years of published T.R.U.E. test data. J Am Acad Dermatol. 2004;51:349-353.
2. Usatine RP, Riojas M. Diagnosis and management of contact dermatitis. Am Fam Physician. 2010;82:249-255.
3. Silverberg NB, Licht J, Friedler S, et al. Nickel contact hypersensitivity in children. Pediatr Dermatol. 2002;19:110-113.
4. Beltrani VS, Bernstein IL, Cohen DE, et al. Contact dermatitis: a practice parameter. Ann Allergy Asthma Immunol. 2006;97:S1-S38.
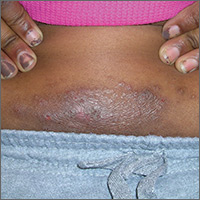
Pruritus since childhood
A 48-year-old woman experiencing homelessness presented to our clinic with a 4-week history of an intensely pruritic rash on her upper back and bilateral upper extremities. She reported that she had experienced exacerbations and remissions of the rash in similar locations for the past several years and during childhood. Factors that exacerbated the rash included being outdoors and being exposed to heat. Her pruritus was intensified by scratching the skin and was significantly worse at night. Previous doctors had diagnosed her with psoriasis and prescribed a short trial of hydrocortisone cream and oral antihistamines, but they provided minimal relief.
The patient indicated that the itching interrupted her sleep and her skin’s appearance made it difficult to get a job. The physical exam revealed excoriated and erythematous papules and patches on her upper back, the extensor and flexor aspects of her bilateral forearms, and the dorsal surface of her bilateral wrists, hands, and fingers (FIGURE 1). Her skin was dry and scaly with pigmentary changes and skin thickening (FIGURE 2). She denied any other systemic symptoms. Her hair and nails were normal, she had no palpable lymph nodes, and she was afebrile. She reported suffering from seasonal allergies, but wasn’t aware of a family history of skin disorders.


WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Chronic atopic dermatitis
Although the patient was told she had psoriasis by previous doctors, we diagnosed her condition as atopic dermatitis based on its clinical appearance. There is no single test that can establish a diagnosis of atopic dermatitis. While serum total IgE levels are often elevated, testing is not currently recommended.
The United Kingdom working group on atopic dermatitis published diagnostic criteria based on clinical history and physical exam that include pruritic skin in addition to the presence of 3 or more of the following: skin crease involvement, chronically dry skin, symptom onset before 2 years of age, and visible evidence of dermatitis involving flexural surfaces.1 Our patient fulfilled all but one condition, as she wasn’t sure if her symptoms began before age 2.
Atopic dermatitis is a chronic and inflammatory cutaneous disease that affects approximately 10% to 12% of children and less than 1% of adults in the United States.2 Approximately 90% of cases present before the age of 5 and the literature demonstrates a slight female predominance.3,4
Disease severity is classified as mild, moderate, or severe.5 Mild disease is characterized by dry skin and minimal itching with little impairment of the patient’s physical and psychological wellbeing. Moderate disease includes frequent pruritus and erythema with or without secondary skin changes and a moderate impact on physical and mental health. In severe disease, extensive secondary skin changes exist and the patient’s daily activities, sleep, and mental health may be severely impaired.
Etiology is multifactorial. Causes of atopic dermatitis include abnormalities in the epidermal stratum corneum and tight junctions, a heightened type-2 helper T-cell response to environmental antigens, innate immunity defects, and altered microbial skin flora.6,7
Genetic influences appear to play a substantial role in disease development. Approximately 70% of patients have a positive family history of an atopic disease such as eczema, asthma, or allergic rhinitis.8 Genetic defects are believed to be related to defective proteins and lipids in the epidermis that lead to disruption of the epidermal barrier and subsequent cutaneous inflammation.6,7
Clinical presentation: Lesion distribution varies with age
Intense pruritus and dry scaly skin occur in both children and adults, although the distribution of lesions may vary with age. Children typically exhibit erythematous patches with papules and crusting on the face, scalp, extremities, or trunk. In adults, lesions are primarily located on the hands and feet, but may also present on the face, wrists, forearms, and flexural areas.3
Adults also frequently present with secondary skin changes such as thickened skin, pigmentation changes, lichenification, and excoriated papules due to chronic rubbing or scratching.3 Our patient presented with significant lichenification and hyperpigmentation of the skin that was most prominent on the wrists and forearms.
Additional clinical features consistent with atopic dermatitis include a personal history of allergic conditions and a disease course characterized by exacerbations and remissions. Exacerbations may be caused by heat exposure, dry climates, anxiety, rapid temperature variations, contact with certain chemical substances, or microbial infections.8
Differential Dx includes psoriasis and scabies
The differential diagnosis of chronic atopic dermatitis consists of allergic or irritant contact dermatitis, plaque psoriasis, seborrheic dermatitis, scabies, and drug eruptions. Early diagnosis of atopic dermatitis is imperative to prevent sleep disturbances, chronic secondary skin changes, scarring, and the development of skin infections.
Allergic or irritant contact dermatitis is a cutaneous inflammation occurring after contact with an allergen or irritant. The lesions include erythematous, scaling areas with marked borders that are commonly pruritic. Acute cases often present with vesicles and bullae, while lichenification with cracks and fissures are common among chronic cases. Patch testing may be performed if the diagnosis is suspected.
Plaque psoriasis is characterized by areas of dry, erythematous, and well-demarcated plaques with silver scales that are most commonly found on the knees, elbows, scalp, and lower back. Systemic manifestations can include joint pain and joint swelling. Nail pitting and onycholysis are also common. While our patient had skin thickening, it was from the lichenification that is common in atopic dermatitis.
Psoriasis and atopic dermatitis are often confused. Psoriasis has discrete plaques on extensor surfaces and is often associated with nail changes, while the thickening of the skin that comes with chronic itch and scratching of atopic dermatitis is often less well circumscribed and found on flexor surfaces. Family physicians are frequently the first to encounter patients with atopic dermatitis and psoriasis and must be able to distinguish these conditions, as their treatments differ.
Seborrheic dermatitis is a chronic, relapsing inflammatory condition characterized by pruritic, erythematous, greasy, scaly patches on sebum-rich skin such as the scalp, face, and upper trunk. Seborrheic dermatitis is a clinical diagnosis.
Scabies is a pruritic skin condition caused by Sarcoptes scabiei var hominis. Characteristic linear burrows often appear as serpiginous, gray, threadlike elevations in the webbed spaces of the fingers, scrotum, areolae, elbows, axillae, feet, and wrist flexors. Secondary lesions from scratching or inflammation include excoriations, erythema, and hyperpigmentation. The diagnosis is made clinically and confirmed by dermoscopy, when available. Alternatively, mites or eggs may be observed on skin scrapings using light microscopy.
Drug eruptions should be considered in individuals taking medications who develop acute, symmetric cutaneous eruptions that may be morbilliform, urticarial, papulosquamous, pustular, or bullous in nature.
Treatment depends on severity, area of involvement, and patient’s age
Components of atopic dermatitis treatment include skin hydration, negative stimuli avoidance, pharmacologic modalities, and patient education. Improved skin hydration can be achieved by applying thick emollients containing little to no water at least twice daily and after bathing.
Topical corticosteroids are added when emollient use alone fails. Potency selection is based upon the patient’s age, involved body region, and the severity of skin inflammation.8 In order to reduce cutaneous atrophy, only low-potency corticosteroids should be applied to the face, groin, and axillae. Patients with mild disease may apply desonide 0.05% or hydrocortisone 2.5% cream or ointment once or twice daily for 2 to 4 weeks.8 Patients without improvement or with moderate disease may need medium- to high-potency steroids such as fluocinolone 0.025% or triamcinolone 0.1%.
Patients with atopic dermatitis on the face, eyelids, neck, and skin folds or those who do not obtain relief from combined emollients and topical corticosteroids may benefit from topical calcineurin inhibitors such as pimecrolimus or tacrolimus.9 However, these agents should be utilized only for short periods of time and avoided in immunocompromised patients and those younger than 2 years of age.9
Patients with uncontrolled moderate to severe refractory disease may consider a trial of phototherapy or cyclosporine if phototherapy is ineffective or unavailable.10 A meta-analysis has shown that once remission is achieved, intermittent therapy with moderate- to high-potency corticosteroids or tacrolimus may be effective in reducing subsequent flares.11
In all cases, sedating antihistamines such as diphenhydramine or hydroxyzine can be utilized for pruritic relief, particularly at night. Additionally, signs suggestive of infection should prompt antibiotic treatment with agents that provide coverage for Staphylococcus and Streptococcus species. Lastly, patients must be adequately educated on stimuli avoidance (eg, hot water, wool) and counseled on the medical and psychological issues that often accompany atopic dermatitis.
Our patient was placed on triamcinolone 0.1% for 4 weeks and her condition improved.
CORRESPONDENCE
Andrea Richardson, MD, MPH, 7414 Carriage Bay, San Antonio, TX 78249; [email protected].
1. Williams HC, Burney PG, Pembroke AC, et al. The U.K. Working Party’s Diagnostic Criteria for Atopic Dermatitis. III. Independent hospital validation. Br J Dermatol. 1994;131:406-416.
2. Horii KA, Simon SD, Liu DY, et al. Atopic dermatitis in children in the United States, 1997-2004: visit trends, patient and provider characteristics, and prescribing patterns. Pediatrics. 2007;120:e527-e534.
3. Rudikoff D, Lebwohl M. Atopic dermatitis. Lancet. 1998;351:1715-1721.
4. Kang K, Polster AM, Nedorost ST, et al. Atopic dermatitis. In: Dermatology. Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Mosby, New York;2003:199.
5. Lewis-Jones S, Mugglestone MA; Guideline Development Group. Management of atopic eczema in children aged up to 12 years: summary of NICE guidance. BMJ. 2007;335:1263-1264.
6. Kuo IH, Yoshida T, De Benedetto A, et al. The cutaneous innate immune response in patients with atopic dermatitis. J Allergy Clin Immunol. 2013;131:266-278.
7. Boguniewicz M, Leung DY. Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunol Rev. 2011;242:233-246.
8. Eichenfield LF, Tom WL, Chamlin SL, et al. Guidelines of care for the management of atopic dermatitis: section 1. Diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70:338-351.
9. Ashcroft DM, Dimmock P, Garside R, et al. Efficacy and tolerability of topical pimecrolimus and tacrolimus in the treatment of atopic dermatitis: meta-analysis of randomised controlled trials. BMJ. 2005;330:516.
10. Garritsen FM, Brouwer MW, Limpens J, et al. Photo(chemo)therapy in the management of atopic dermatitis: an updated systematic review with implications for practice and research. Br J Dermatol. 2014;170:501-513.
11. Schmitt J, von Kobyletzki L, Svensson A, et al. Efficacy and tolerability of proactive treatment with topical corticosteroids and calcineurin inhibitors for atopic eczema: systematic review and meta-analysis of randomized controlled trials. Br J Dermatol. 2011;164:415-428.
A 48-year-old woman experiencing homelessness presented to our clinic with a 4-week history of an intensely pruritic rash on her upper back and bilateral upper extremities. She reported that she had experienced exacerbations and remissions of the rash in similar locations for the past several years and during childhood. Factors that exacerbated the rash included being outdoors and being exposed to heat. Her pruritus was intensified by scratching the skin and was significantly worse at night. Previous doctors had diagnosed her with psoriasis and prescribed a short trial of hydrocortisone cream and oral antihistamines, but they provided minimal relief.
The patient indicated that the itching interrupted her sleep and her skin’s appearance made it difficult to get a job. The physical exam revealed excoriated and erythematous papules and patches on her upper back, the extensor and flexor aspects of her bilateral forearms, and the dorsal surface of her bilateral wrists, hands, and fingers (FIGURE 1). Her skin was dry and scaly with pigmentary changes and skin thickening (FIGURE 2). She denied any other systemic symptoms. Her hair and nails were normal, she had no palpable lymph nodes, and she was afebrile. She reported suffering from seasonal allergies, but wasn’t aware of a family history of skin disorders.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Chronic atopic dermatitis
Although the patient was told she had psoriasis by previous doctors, we diagnosed her condition as atopic dermatitis based on its clinical appearance. There is no single test that can establish a diagnosis of atopic dermatitis. While serum total IgE levels are often elevated, testing is not currently recommended.
The United Kingdom working group on atopic dermatitis published diagnostic criteria based on clinical history and physical exam that include pruritic skin in addition to the presence of 3 or more of the following: skin crease involvement, chronically dry skin, symptom onset before 2 years of age, and visible evidence of dermatitis involving flexural surfaces.1 Our patient fulfilled all but one condition, as she wasn’t sure if her symptoms began before age 2.
Atopic dermatitis is a chronic and inflammatory cutaneous disease that affects approximately 10% to 12% of children and less than 1% of adults in the United States.2 Approximately 90% of cases present before the age of 5 and the literature demonstrates a slight female predominance.3,4
Disease severity is classified as mild, moderate, or severe.5 Mild disease is characterized by dry skin and minimal itching with little impairment of the patient’s physical and psychological wellbeing. Moderate disease includes frequent pruritus and erythema with or without secondary skin changes and a moderate impact on physical and mental health. In severe disease, extensive secondary skin changes exist and the patient’s daily activities, sleep, and mental health may be severely impaired.
Etiology is multifactorial. Causes of atopic dermatitis include abnormalities in the epidermal stratum corneum and tight junctions, a heightened type-2 helper T-cell response to environmental antigens, innate immunity defects, and altered microbial skin flora.6,7
Genetic influences appear to play a substantial role in disease development. Approximately 70% of patients have a positive family history of an atopic disease such as eczema, asthma, or allergic rhinitis.8 Genetic defects are believed to be related to defective proteins and lipids in the epidermis that lead to disruption of the epidermal barrier and subsequent cutaneous inflammation.6,7
Clinical presentation: Lesion distribution varies with age
Intense pruritus and dry scaly skin occur in both children and adults, although the distribution of lesions may vary with age. Children typically exhibit erythematous patches with papules and crusting on the face, scalp, extremities, or trunk. In adults, lesions are primarily located on the hands and feet, but may also present on the face, wrists, forearms, and flexural areas.3
Adults also frequently present with secondary skin changes such as thickened skin, pigmentation changes, lichenification, and excoriated papules due to chronic rubbing or scratching.3 Our patient presented with significant lichenification and hyperpigmentation of the skin that was most prominent on the wrists and forearms.
Additional clinical features consistent with atopic dermatitis include a personal history of allergic conditions and a disease course characterized by exacerbations and remissions. Exacerbations may be caused by heat exposure, dry climates, anxiety, rapid temperature variations, contact with certain chemical substances, or microbial infections.8
Differential Dx includes psoriasis and scabies
The differential diagnosis of chronic atopic dermatitis consists of allergic or irritant contact dermatitis, plaque psoriasis, seborrheic dermatitis, scabies, and drug eruptions. Early diagnosis of atopic dermatitis is imperative to prevent sleep disturbances, chronic secondary skin changes, scarring, and the development of skin infections.
Allergic or irritant contact dermatitis is a cutaneous inflammation occurring after contact with an allergen or irritant. The lesions include erythematous, scaling areas with marked borders that are commonly pruritic. Acute cases often present with vesicles and bullae, while lichenification with cracks and fissures are common among chronic cases. Patch testing may be performed if the diagnosis is suspected.
Plaque psoriasis is characterized by areas of dry, erythematous, and well-demarcated plaques with silver scales that are most commonly found on the knees, elbows, scalp, and lower back. Systemic manifestations can include joint pain and joint swelling. Nail pitting and onycholysis are also common. While our patient had skin thickening, it was from the lichenification that is common in atopic dermatitis.
Psoriasis and atopic dermatitis are often confused. Psoriasis has discrete plaques on extensor surfaces and is often associated with nail changes, while the thickening of the skin that comes with chronic itch and scratching of atopic dermatitis is often less well circumscribed and found on flexor surfaces. Family physicians are frequently the first to encounter patients with atopic dermatitis and psoriasis and must be able to distinguish these conditions, as their treatments differ.
Seborrheic dermatitis is a chronic, relapsing inflammatory condition characterized by pruritic, erythematous, greasy, scaly patches on sebum-rich skin such as the scalp, face, and upper trunk. Seborrheic dermatitis is a clinical diagnosis.
Scabies is a pruritic skin condition caused by Sarcoptes scabiei var hominis. Characteristic linear burrows often appear as serpiginous, gray, threadlike elevations in the webbed spaces of the fingers, scrotum, areolae, elbows, axillae, feet, and wrist flexors. Secondary lesions from scratching or inflammation include excoriations, erythema, and hyperpigmentation. The diagnosis is made clinically and confirmed by dermoscopy, when available. Alternatively, mites or eggs may be observed on skin scrapings using light microscopy.
Drug eruptions should be considered in individuals taking medications who develop acute, symmetric cutaneous eruptions that may be morbilliform, urticarial, papulosquamous, pustular, or bullous in nature.
Treatment depends on severity, area of involvement, and patient’s age
Components of atopic dermatitis treatment include skin hydration, negative stimuli avoidance, pharmacologic modalities, and patient education. Improved skin hydration can be achieved by applying thick emollients containing little to no water at least twice daily and after bathing.
Topical corticosteroids are added when emollient use alone fails. Potency selection is based upon the patient’s age, involved body region, and the severity of skin inflammation.8 In order to reduce cutaneous atrophy, only low-potency corticosteroids should be applied to the face, groin, and axillae. Patients with mild disease may apply desonide 0.05% or hydrocortisone 2.5% cream or ointment once or twice daily for 2 to 4 weeks.8 Patients without improvement or with moderate disease may need medium- to high-potency steroids such as fluocinolone 0.025% or triamcinolone 0.1%.
Patients with atopic dermatitis on the face, eyelids, neck, and skin folds or those who do not obtain relief from combined emollients and topical corticosteroids may benefit from topical calcineurin inhibitors such as pimecrolimus or tacrolimus.9 However, these agents should be utilized only for short periods of time and avoided in immunocompromised patients and those younger than 2 years of age.9
Patients with uncontrolled moderate to severe refractory disease may consider a trial of phototherapy or cyclosporine if phototherapy is ineffective or unavailable.10 A meta-analysis has shown that once remission is achieved, intermittent therapy with moderate- to high-potency corticosteroids or tacrolimus may be effective in reducing subsequent flares.11
In all cases, sedating antihistamines such as diphenhydramine or hydroxyzine can be utilized for pruritic relief, particularly at night. Additionally, signs suggestive of infection should prompt antibiotic treatment with agents that provide coverage for Staphylococcus and Streptococcus species. Lastly, patients must be adequately educated on stimuli avoidance (eg, hot water, wool) and counseled on the medical and psychological issues that often accompany atopic dermatitis.
Our patient was placed on triamcinolone 0.1% for 4 weeks and her condition improved.
CORRESPONDENCE
Andrea Richardson, MD, MPH, 7414 Carriage Bay, San Antonio, TX 78249; [email protected].
A 48-year-old woman experiencing homelessness presented to our clinic with a 4-week history of an intensely pruritic rash on her upper back and bilateral upper extremities. She reported that she had experienced exacerbations and remissions of the rash in similar locations for the past several years and during childhood. Factors that exacerbated the rash included being outdoors and being exposed to heat. Her pruritus was intensified by scratching the skin and was significantly worse at night. Previous doctors had diagnosed her with psoriasis and prescribed a short trial of hydrocortisone cream and oral antihistamines, but they provided minimal relief.
The patient indicated that the itching interrupted her sleep and her skin’s appearance made it difficult to get a job. The physical exam revealed excoriated and erythematous papules and patches on her upper back, the extensor and flexor aspects of her bilateral forearms, and the dorsal surface of her bilateral wrists, hands, and fingers (FIGURE 1). Her skin was dry and scaly with pigmentary changes and skin thickening (FIGURE 2). She denied any other systemic symptoms. Her hair and nails were normal, she had no palpable lymph nodes, and she was afebrile. She reported suffering from seasonal allergies, but wasn’t aware of a family history of skin disorders.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Chronic atopic dermatitis
Although the patient was told she had psoriasis by previous doctors, we diagnosed her condition as atopic dermatitis based on its clinical appearance. There is no single test that can establish a diagnosis of atopic dermatitis. While serum total IgE levels are often elevated, testing is not currently recommended.
The United Kingdom working group on atopic dermatitis published diagnostic criteria based on clinical history and physical exam that include pruritic skin in addition to the presence of 3 or more of the following: skin crease involvement, chronically dry skin, symptom onset before 2 years of age, and visible evidence of dermatitis involving flexural surfaces.1 Our patient fulfilled all but one condition, as she wasn’t sure if her symptoms began before age 2.
Atopic dermatitis is a chronic and inflammatory cutaneous disease that affects approximately 10% to 12% of children and less than 1% of adults in the United States.2 Approximately 90% of cases present before the age of 5 and the literature demonstrates a slight female predominance.3,4
Disease severity is classified as mild, moderate, or severe.5 Mild disease is characterized by dry skin and minimal itching with little impairment of the patient’s physical and psychological wellbeing. Moderate disease includes frequent pruritus and erythema with or without secondary skin changes and a moderate impact on physical and mental health. In severe disease, extensive secondary skin changes exist and the patient’s daily activities, sleep, and mental health may be severely impaired.
Etiology is multifactorial. Causes of atopic dermatitis include abnormalities in the epidermal stratum corneum and tight junctions, a heightened type-2 helper T-cell response to environmental antigens, innate immunity defects, and altered microbial skin flora.6,7
Genetic influences appear to play a substantial role in disease development. Approximately 70% of patients have a positive family history of an atopic disease such as eczema, asthma, or allergic rhinitis.8 Genetic defects are believed to be related to defective proteins and lipids in the epidermis that lead to disruption of the epidermal barrier and subsequent cutaneous inflammation.6,7
Clinical presentation: Lesion distribution varies with age
Intense pruritus and dry scaly skin occur in both children and adults, although the distribution of lesions may vary with age. Children typically exhibit erythematous patches with papules and crusting on the face, scalp, extremities, or trunk. In adults, lesions are primarily located on the hands and feet, but may also present on the face, wrists, forearms, and flexural areas.3
Adults also frequently present with secondary skin changes such as thickened skin, pigmentation changes, lichenification, and excoriated papules due to chronic rubbing or scratching.3 Our patient presented with significant lichenification and hyperpigmentation of the skin that was most prominent on the wrists and forearms.
Additional clinical features consistent with atopic dermatitis include a personal history of allergic conditions and a disease course characterized by exacerbations and remissions. Exacerbations may be caused by heat exposure, dry climates, anxiety, rapid temperature variations, contact with certain chemical substances, or microbial infections.8
Differential Dx includes psoriasis and scabies
The differential diagnosis of chronic atopic dermatitis consists of allergic or irritant contact dermatitis, plaque psoriasis, seborrheic dermatitis, scabies, and drug eruptions. Early diagnosis of atopic dermatitis is imperative to prevent sleep disturbances, chronic secondary skin changes, scarring, and the development of skin infections.
Allergic or irritant contact dermatitis is a cutaneous inflammation occurring after contact with an allergen or irritant. The lesions include erythematous, scaling areas with marked borders that are commonly pruritic. Acute cases often present with vesicles and bullae, while lichenification with cracks and fissures are common among chronic cases. Patch testing may be performed if the diagnosis is suspected.
Plaque psoriasis is characterized by areas of dry, erythematous, and well-demarcated plaques with silver scales that are most commonly found on the knees, elbows, scalp, and lower back. Systemic manifestations can include joint pain and joint swelling. Nail pitting and onycholysis are also common. While our patient had skin thickening, it was from the lichenification that is common in atopic dermatitis.
Psoriasis and atopic dermatitis are often confused. Psoriasis has discrete plaques on extensor surfaces and is often associated with nail changes, while the thickening of the skin that comes with chronic itch and scratching of atopic dermatitis is often less well circumscribed and found on flexor surfaces. Family physicians are frequently the first to encounter patients with atopic dermatitis and psoriasis and must be able to distinguish these conditions, as their treatments differ.
Seborrheic dermatitis is a chronic, relapsing inflammatory condition characterized by pruritic, erythematous, greasy, scaly patches on sebum-rich skin such as the scalp, face, and upper trunk. Seborrheic dermatitis is a clinical diagnosis.
Scabies is a pruritic skin condition caused by Sarcoptes scabiei var hominis. Characteristic linear burrows often appear as serpiginous, gray, threadlike elevations in the webbed spaces of the fingers, scrotum, areolae, elbows, axillae, feet, and wrist flexors. Secondary lesions from scratching or inflammation include excoriations, erythema, and hyperpigmentation. The diagnosis is made clinically and confirmed by dermoscopy, when available. Alternatively, mites or eggs may be observed on skin scrapings using light microscopy.
Drug eruptions should be considered in individuals taking medications who develop acute, symmetric cutaneous eruptions that may be morbilliform, urticarial, papulosquamous, pustular, or bullous in nature.
Treatment depends on severity, area of involvement, and patient’s age
Components of atopic dermatitis treatment include skin hydration, negative stimuli avoidance, pharmacologic modalities, and patient education. Improved skin hydration can be achieved by applying thick emollients containing little to no water at least twice daily and after bathing.
Topical corticosteroids are added when emollient use alone fails. Potency selection is based upon the patient’s age, involved body region, and the severity of skin inflammation.8 In order to reduce cutaneous atrophy, only low-potency corticosteroids should be applied to the face, groin, and axillae. Patients with mild disease may apply desonide 0.05% or hydrocortisone 2.5% cream or ointment once or twice daily for 2 to 4 weeks.8 Patients without improvement or with moderate disease may need medium- to high-potency steroids such as fluocinolone 0.025% or triamcinolone 0.1%.
Patients with atopic dermatitis on the face, eyelids, neck, and skin folds or those who do not obtain relief from combined emollients and topical corticosteroids may benefit from topical calcineurin inhibitors such as pimecrolimus or tacrolimus.9 However, these agents should be utilized only for short periods of time and avoided in immunocompromised patients and those younger than 2 years of age.9
Patients with uncontrolled moderate to severe refractory disease may consider a trial of phototherapy or cyclosporine if phototherapy is ineffective or unavailable.10 A meta-analysis has shown that once remission is achieved, intermittent therapy with moderate- to high-potency corticosteroids or tacrolimus may be effective in reducing subsequent flares.11
In all cases, sedating antihistamines such as diphenhydramine or hydroxyzine can be utilized for pruritic relief, particularly at night. Additionally, signs suggestive of infection should prompt antibiotic treatment with agents that provide coverage for Staphylococcus and Streptococcus species. Lastly, patients must be adequately educated on stimuli avoidance (eg, hot water, wool) and counseled on the medical and psychological issues that often accompany atopic dermatitis.
Our patient was placed on triamcinolone 0.1% for 4 weeks and her condition improved.
CORRESPONDENCE
Andrea Richardson, MD, MPH, 7414 Carriage Bay, San Antonio, TX 78249; [email protected].
1. Williams HC, Burney PG, Pembroke AC, et al. The U.K. Working Party’s Diagnostic Criteria for Atopic Dermatitis. III. Independent hospital validation. Br J Dermatol. 1994;131:406-416.
2. Horii KA, Simon SD, Liu DY, et al. Atopic dermatitis in children in the United States, 1997-2004: visit trends, patient and provider characteristics, and prescribing patterns. Pediatrics. 2007;120:e527-e534.
3. Rudikoff D, Lebwohl M. Atopic dermatitis. Lancet. 1998;351:1715-1721.
4. Kang K, Polster AM, Nedorost ST, et al. Atopic dermatitis. In: Dermatology. Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Mosby, New York;2003:199.
5. Lewis-Jones S, Mugglestone MA; Guideline Development Group. Management of atopic eczema in children aged up to 12 years: summary of NICE guidance. BMJ. 2007;335:1263-1264.
6. Kuo IH, Yoshida T, De Benedetto A, et al. The cutaneous innate immune response in patients with atopic dermatitis. J Allergy Clin Immunol. 2013;131:266-278.
7. Boguniewicz M, Leung DY. Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunol Rev. 2011;242:233-246.
8. Eichenfield LF, Tom WL, Chamlin SL, et al. Guidelines of care for the management of atopic dermatitis: section 1. Diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70:338-351.
9. Ashcroft DM, Dimmock P, Garside R, et al. Efficacy and tolerability of topical pimecrolimus and tacrolimus in the treatment of atopic dermatitis: meta-analysis of randomised controlled trials. BMJ. 2005;330:516.
10. Garritsen FM, Brouwer MW, Limpens J, et al. Photo(chemo)therapy in the management of atopic dermatitis: an updated systematic review with implications for practice and research. Br J Dermatol. 2014;170:501-513.
11. Schmitt J, von Kobyletzki L, Svensson A, et al. Efficacy and tolerability of proactive treatment with topical corticosteroids and calcineurin inhibitors for atopic eczema: systematic review and meta-analysis of randomized controlled trials. Br J Dermatol. 2011;164:415-428.
1. Williams HC, Burney PG, Pembroke AC, et al. The U.K. Working Party’s Diagnostic Criteria for Atopic Dermatitis. III. Independent hospital validation. Br J Dermatol. 1994;131:406-416.
2. Horii KA, Simon SD, Liu DY, et al. Atopic dermatitis in children in the United States, 1997-2004: visit trends, patient and provider characteristics, and prescribing patterns. Pediatrics. 2007;120:e527-e534.
3. Rudikoff D, Lebwohl M. Atopic dermatitis. Lancet. 1998;351:1715-1721.
4. Kang K, Polster AM, Nedorost ST, et al. Atopic dermatitis. In: Dermatology. Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Mosby, New York;2003:199.
5. Lewis-Jones S, Mugglestone MA; Guideline Development Group. Management of atopic eczema in children aged up to 12 years: summary of NICE guidance. BMJ. 2007;335:1263-1264.
6. Kuo IH, Yoshida T, De Benedetto A, et al. The cutaneous innate immune response in patients with atopic dermatitis. J Allergy Clin Immunol. 2013;131:266-278.
7. Boguniewicz M, Leung DY. Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunol Rev. 2011;242:233-246.
8. Eichenfield LF, Tom WL, Chamlin SL, et al. Guidelines of care for the management of atopic dermatitis: section 1. Diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70:338-351.
9. Ashcroft DM, Dimmock P, Garside R, et al. Efficacy and tolerability of topical pimecrolimus and tacrolimus in the treatment of atopic dermatitis: meta-analysis of randomised controlled trials. BMJ. 2005;330:516.
10. Garritsen FM, Brouwer MW, Limpens J, et al. Photo(chemo)therapy in the management of atopic dermatitis: an updated systematic review with implications for practice and research. Br J Dermatol. 2014;170:501-513.
11. Schmitt J, von Kobyletzki L, Svensson A, et al. Efficacy and tolerability of proactive treatment with topical corticosteroids and calcineurin inhibitors for atopic eczema: systematic review and meta-analysis of randomized controlled trials. Br J Dermatol. 2011;164:415-428.
Dermoscopy

Vulvar pain in pregnancy
A 30-year-old pregnant woman presented to a rural Panamanian hospital with new onset genital pain, vaginal itching, and dysuria that she’d had for 48 hours. The patient was in the first trimester of her pregnancy and indicated that she’d had recent unprotected sex with a new partner who wasn’t the father of the developing fetus. The patient had never experienced symptoms like these before and denied ever having a sexually transmitted infection (STI). On physical exam, the physician noted numerous pustules covering tender, swollen labia (FIGURE). A small amount of white discharge was noted at the introitus.
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Herpes simplex virus
The physician on-call diagnosed candida vaginitis along with a bacterial skin infection, and admitted the patient to the hospital for intravenous (IV) antibiotics. Fortunately, we were there on a medical mission and were consulted on the case.
We diagnosed a primary herpes simplex virus type 2 (HSV-2) infection in this patient, based on the classic presentation of grouped pustules and vesicles on erythematous and swollen labia, and the patient’s complaint of dysuria.
Herpes cultures weren’t available in the hospital, but the clinical picture was unmistakable for HSV infection. Since multiple STIs may occur simultaneously, we ordered a serum rapid plasma reagin (RPR) test for syphilis, and tested her urine for gonorrhea and chlamydia. The tests were negative.
Differential Dx includes other STIs and a fixed drug eruption
Herpes is a common STI and most people don’t have symptoms. In 2012, an estimated 417 million people worldwide were living with genital herpes caused by HSV-2.1
The differential diagnosis for HSV infection includes primary syphilis, chancroid, folliculitis, and fixed drug eruptions.
Primary syphilis (Treponema pallidum) commonly presents with a painless, ulcerated, clean-based ulcer. While the chancre of primary syphilis can sometimes be painful, this patient did not have ulcers at the time of her presentation. Her pustules would likely ulcerate over time, but would not resemble the chancre of syphilis.
Chancroid (Haemophilus ducreyi) is a less common STI than syphilis and HSV infection. It presents with deep, sharply defined, purulent ulcers that are often associated with painful adenopathy. The ulcers can appear grey or yellowish in color.
Folliculitis presents with pustules surrounding hair follicles. Some of the pustules were surrounding hair follicles in this patient’s case, but others were independent of the hair. The patient’s marked swelling and tenderness along with dysuria also did not fit the characteristics of folliculitis.
Fixed drug eruptions can occur in the genital region, but the patient had neither bullous nor ulcerated eruptions (as one would expect with this condition). Fixed drug eruptions are usually hyperpigmented and require a history of taking medication, such as an antibiotic or a nonsteroidal anti-inflammatory drug.
Questions that help narrow the differential. Zeroing in on the cause of a patient’s genital lesions requires that you ask whether the lesions are painful, if the patient has dysuria, if there are any constitutional symptoms, and if this has happened before. Other distinguishing factors include enlarged lymph nodes and the presence of multiple (vs single) lesions.
Viral cell cultures are the preferred lab test
Common laboratory tests to make the diagnosis include viral culture, direct fluorescence antibody (DFA), polymerase chain reaction (PCR), and type-specific serologic tests.
Viral cell culture is the preferred test for suspected HSV of the skin and mucous membranes.2 PCR is the preferred test for suspected herpes meningitis or encephalitis when cerebrospinal fluid has been obtained through lumbar puncture.3 DFA and herpes culture can be ordered simultaneously. DFA can provide a quick result, and herpes culture can provide a more sensitive result (this may take 5-7 days before results are available).
No evidence that antivirals pose risk during pregnancy
Treatment with antivirals (acyclovir, famciclovir, or valacyclovir) may help to reduce the length of the outbreak. Oral antivirals are usually sufficient for uncomplicated HSV; IV antivirals may be needed in complicated cases. The current recommendation for acyclovir (the most commonly prescribed drug for HSV infection) is 400 mg 3 times daily or 200 mg 5 times daily for 7 to 10 days in a primary outbreak.3
Antiviral therapy is most effective if begun within 72 hours of symptom onset in primary herpes genitalis.4 Analgesics can help with pain control and sitz baths are helpful for women with severe dysuria.
Maternal–fetal transmission of HSV is associated with significant morbidity and mortality in children.5 The Centers for Disease Control and Prevention and the American College of Obstetricians and Gynecologists recommend that cesarean delivery be offered as soon as possible to women who have active HSV lesions or, in those with a history of genital herpes, symptoms of vulvar pain or burning at the time of delivery.3
There is no evidence that the use of antiviral agents in women who are pregnant and have a history of genital herpes prevents perinatal transmission of HSV to neonates.6 However, antenatal antiviral prophylaxis has been shown to reduce viral shedding, recurrences at delivery, and the need for cesarean delivery.7
Our patient was treated with oral acyclovir 400 mg 3 times a day for 10 days. One day after seeking care, she had less pain, swelling, and tenderness and was discharged. (Based on the severity of the outbreak and lack of sanitary living conditions, hospitalization was the safest and most reliable option.) The patient was counseled on the ramifications of HSV infection in pregnancy, including the fact that she might need a cesarean section. She was told that she must get prenatal care and that she needed to tell her primary care physician about her HSV infection. She was also warned about the risk of disease transmission to sexual partners and the importance of using barrier contraception to minimize the risk of future transmission.
CORRESPONDENCE
Luke Wallis, BS, 6410 Rambling Trail Drive, San Antonio, TX 78240; [email protected].
1. World Health Organization. Herpes simplex virus. World Health Organization Web site. Available at: http://www.who.int/mediacentre/factsheets/fs400/en/. Accessed February 8, 2016.
2. Ramaswamy M, McDonald C, Smith M, et al. Diagnosis of genital herpes by real time PCR in routine clinical practice. Sex Transm Infect. 2004;80:406-410.
3. Workowski KA, Berman S; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2010. MMWR Recomm Rep. 2010;59:1-110.
4. Cernik C, Gallina K, Brodell RT. The treatment of herpes simplex infections: an evidence-based review. Arch Intern Med. 2008;168:1137-1144.
5. Flagg EW, Weinstock H. Incidence of neonatal herpes simplex virus infections in the United States, 2006. Pediatrics. 2011;127:e1-e8.
6. Wenner C, Nashelsky J. Antiviral agents for pregnant women with genital herpes. Am Fam Physician. 2005;72:1807-1808.
7. Hollier LM, Wendel GD. Third trimester antiviral prophylaxis for preventing maternal genital herpes simplex virus (HSV) recurrences and neonatal infection. Cochrane Database Syst Rev. 2008;CD004946.
A 30-year-old pregnant woman presented to a rural Panamanian hospital with new onset genital pain, vaginal itching, and dysuria that she’d had for 48 hours. The patient was in the first trimester of her pregnancy and indicated that she’d had recent unprotected sex with a new partner who wasn’t the father of the developing fetus. The patient had never experienced symptoms like these before and denied ever having a sexually transmitted infection (STI). On physical exam, the physician noted numerous pustules covering tender, swollen labia (FIGURE). A small amount of white discharge was noted at the introitus.
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Herpes simplex virus
The physician on-call diagnosed candida vaginitis along with a bacterial skin infection, and admitted the patient to the hospital for intravenous (IV) antibiotics. Fortunately, we were there on a medical mission and were consulted on the case.
We diagnosed a primary herpes simplex virus type 2 (HSV-2) infection in this patient, based on the classic presentation of grouped pustules and vesicles on erythematous and swollen labia, and the patient’s complaint of dysuria.
Herpes cultures weren’t available in the hospital, but the clinical picture was unmistakable for HSV infection. Since multiple STIs may occur simultaneously, we ordered a serum rapid plasma reagin (RPR) test for syphilis, and tested her urine for gonorrhea and chlamydia. The tests were negative.
Differential Dx includes other STIs and a fixed drug eruption
Herpes is a common STI and most people don’t have symptoms. In 2012, an estimated 417 million people worldwide were living with genital herpes caused by HSV-2.1
The differential diagnosis for HSV infection includes primary syphilis, chancroid, folliculitis, and fixed drug eruptions.
Primary syphilis (Treponema pallidum) commonly presents with a painless, ulcerated, clean-based ulcer. While the chancre of primary syphilis can sometimes be painful, this patient did not have ulcers at the time of her presentation. Her pustules would likely ulcerate over time, but would not resemble the chancre of syphilis.
Chancroid (Haemophilus ducreyi) is a less common STI than syphilis and HSV infection. It presents with deep, sharply defined, purulent ulcers that are often associated with painful adenopathy. The ulcers can appear grey or yellowish in color.
Folliculitis presents with pustules surrounding hair follicles. Some of the pustules were surrounding hair follicles in this patient’s case, but others were independent of the hair. The patient’s marked swelling and tenderness along with dysuria also did not fit the characteristics of folliculitis.
Fixed drug eruptions can occur in the genital region, but the patient had neither bullous nor ulcerated eruptions (as one would expect with this condition). Fixed drug eruptions are usually hyperpigmented and require a history of taking medication, such as an antibiotic or a nonsteroidal anti-inflammatory drug.
Questions that help narrow the differential. Zeroing in on the cause of a patient’s genital lesions requires that you ask whether the lesions are painful, if the patient has dysuria, if there are any constitutional symptoms, and if this has happened before. Other distinguishing factors include enlarged lymph nodes and the presence of multiple (vs single) lesions.
Viral cell cultures are the preferred lab test
Common laboratory tests to make the diagnosis include viral culture, direct fluorescence antibody (DFA), polymerase chain reaction (PCR), and type-specific serologic tests.
Viral cell culture is the preferred test for suspected HSV of the skin and mucous membranes.2 PCR is the preferred test for suspected herpes meningitis or encephalitis when cerebrospinal fluid has been obtained through lumbar puncture.3 DFA and herpes culture can be ordered simultaneously. DFA can provide a quick result, and herpes culture can provide a more sensitive result (this may take 5-7 days before results are available).
No evidence that antivirals pose risk during pregnancy
Treatment with antivirals (acyclovir, famciclovir, or valacyclovir) may help to reduce the length of the outbreak. Oral antivirals are usually sufficient for uncomplicated HSV; IV antivirals may be needed in complicated cases. The current recommendation for acyclovir (the most commonly prescribed drug for HSV infection) is 400 mg 3 times daily or 200 mg 5 times daily for 7 to 10 days in a primary outbreak.3
Antiviral therapy is most effective if begun within 72 hours of symptom onset in primary herpes genitalis.4 Analgesics can help with pain control and sitz baths are helpful for women with severe dysuria.
Maternal–fetal transmission of HSV is associated with significant morbidity and mortality in children.5 The Centers for Disease Control and Prevention and the American College of Obstetricians and Gynecologists recommend that cesarean delivery be offered as soon as possible to women who have active HSV lesions or, in those with a history of genital herpes, symptoms of vulvar pain or burning at the time of delivery.3
There is no evidence that the use of antiviral agents in women who are pregnant and have a history of genital herpes prevents perinatal transmission of HSV to neonates.6 However, antenatal antiviral prophylaxis has been shown to reduce viral shedding, recurrences at delivery, and the need for cesarean delivery.7
Our patient was treated with oral acyclovir 400 mg 3 times a day for 10 days. One day after seeking care, she had less pain, swelling, and tenderness and was discharged. (Based on the severity of the outbreak and lack of sanitary living conditions, hospitalization was the safest and most reliable option.) The patient was counseled on the ramifications of HSV infection in pregnancy, including the fact that she might need a cesarean section. She was told that she must get prenatal care and that she needed to tell her primary care physician about her HSV infection. She was also warned about the risk of disease transmission to sexual partners and the importance of using barrier contraception to minimize the risk of future transmission.
CORRESPONDENCE
Luke Wallis, BS, 6410 Rambling Trail Drive, San Antonio, TX 78240; [email protected].
A 30-year-old pregnant woman presented to a rural Panamanian hospital with new onset genital pain, vaginal itching, and dysuria that she’d had for 48 hours. The patient was in the first trimester of her pregnancy and indicated that she’d had recent unprotected sex with a new partner who wasn’t the father of the developing fetus. The patient had never experienced symptoms like these before and denied ever having a sexually transmitted infection (STI). On physical exam, the physician noted numerous pustules covering tender, swollen labia (FIGURE). A small amount of white discharge was noted at the introitus.
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Herpes simplex virus
The physician on-call diagnosed candida vaginitis along with a bacterial skin infection, and admitted the patient to the hospital for intravenous (IV) antibiotics. Fortunately, we were there on a medical mission and were consulted on the case.
We diagnosed a primary herpes simplex virus type 2 (HSV-2) infection in this patient, based on the classic presentation of grouped pustules and vesicles on erythematous and swollen labia, and the patient’s complaint of dysuria.
Herpes cultures weren’t available in the hospital, but the clinical picture was unmistakable for HSV infection. Since multiple STIs may occur simultaneously, we ordered a serum rapid plasma reagin (RPR) test for syphilis, and tested her urine for gonorrhea and chlamydia. The tests were negative.
Differential Dx includes other STIs and a fixed drug eruption
Herpes is a common STI and most people don’t have symptoms. In 2012, an estimated 417 million people worldwide were living with genital herpes caused by HSV-2.1
The differential diagnosis for HSV infection includes primary syphilis, chancroid, folliculitis, and fixed drug eruptions.
Primary syphilis (Treponema pallidum) commonly presents with a painless, ulcerated, clean-based ulcer. While the chancre of primary syphilis can sometimes be painful, this patient did not have ulcers at the time of her presentation. Her pustules would likely ulcerate over time, but would not resemble the chancre of syphilis.
Chancroid (Haemophilus ducreyi) is a less common STI than syphilis and HSV infection. It presents with deep, sharply defined, purulent ulcers that are often associated with painful adenopathy. The ulcers can appear grey or yellowish in color.
Folliculitis presents with pustules surrounding hair follicles. Some of the pustules were surrounding hair follicles in this patient’s case, but others were independent of the hair. The patient’s marked swelling and tenderness along with dysuria also did not fit the characteristics of folliculitis.
Fixed drug eruptions can occur in the genital region, but the patient had neither bullous nor ulcerated eruptions (as one would expect with this condition). Fixed drug eruptions are usually hyperpigmented and require a history of taking medication, such as an antibiotic or a nonsteroidal anti-inflammatory drug.
Questions that help narrow the differential. Zeroing in on the cause of a patient’s genital lesions requires that you ask whether the lesions are painful, if the patient has dysuria, if there are any constitutional symptoms, and if this has happened before. Other distinguishing factors include enlarged lymph nodes and the presence of multiple (vs single) lesions.
Viral cell cultures are the preferred lab test
Common laboratory tests to make the diagnosis include viral culture, direct fluorescence antibody (DFA), polymerase chain reaction (PCR), and type-specific serologic tests.
Viral cell culture is the preferred test for suspected HSV of the skin and mucous membranes.2 PCR is the preferred test for suspected herpes meningitis or encephalitis when cerebrospinal fluid has been obtained through lumbar puncture.3 DFA and herpes culture can be ordered simultaneously. DFA can provide a quick result, and herpes culture can provide a more sensitive result (this may take 5-7 days before results are available).
No evidence that antivirals pose risk during pregnancy
Treatment with antivirals (acyclovir, famciclovir, or valacyclovir) may help to reduce the length of the outbreak. Oral antivirals are usually sufficient for uncomplicated HSV; IV antivirals may be needed in complicated cases. The current recommendation for acyclovir (the most commonly prescribed drug for HSV infection) is 400 mg 3 times daily or 200 mg 5 times daily for 7 to 10 days in a primary outbreak.3
Antiviral therapy is most effective if begun within 72 hours of symptom onset in primary herpes genitalis.4 Analgesics can help with pain control and sitz baths are helpful for women with severe dysuria.
Maternal–fetal transmission of HSV is associated with significant morbidity and mortality in children.5 The Centers for Disease Control and Prevention and the American College of Obstetricians and Gynecologists recommend that cesarean delivery be offered as soon as possible to women who have active HSV lesions or, in those with a history of genital herpes, symptoms of vulvar pain or burning at the time of delivery.3
There is no evidence that the use of antiviral agents in women who are pregnant and have a history of genital herpes prevents perinatal transmission of HSV to neonates.6 However, antenatal antiviral prophylaxis has been shown to reduce viral shedding, recurrences at delivery, and the need for cesarean delivery.7
Our patient was treated with oral acyclovir 400 mg 3 times a day for 10 days. One day after seeking care, she had less pain, swelling, and tenderness and was discharged. (Based on the severity of the outbreak and lack of sanitary living conditions, hospitalization was the safest and most reliable option.) The patient was counseled on the ramifications of HSV infection in pregnancy, including the fact that she might need a cesarean section. She was told that she must get prenatal care and that she needed to tell her primary care physician about her HSV infection. She was also warned about the risk of disease transmission to sexual partners and the importance of using barrier contraception to minimize the risk of future transmission.
CORRESPONDENCE
Luke Wallis, BS, 6410 Rambling Trail Drive, San Antonio, TX 78240; [email protected].
1. World Health Organization. Herpes simplex virus. World Health Organization Web site. Available at: http://www.who.int/mediacentre/factsheets/fs400/en/. Accessed February 8, 2016.
2. Ramaswamy M, McDonald C, Smith M, et al. Diagnosis of genital herpes by real time PCR in routine clinical practice. Sex Transm Infect. 2004;80:406-410.
3. Workowski KA, Berman S; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2010. MMWR Recomm Rep. 2010;59:1-110.
4. Cernik C, Gallina K, Brodell RT. The treatment of herpes simplex infections: an evidence-based review. Arch Intern Med. 2008;168:1137-1144.
5. Flagg EW, Weinstock H. Incidence of neonatal herpes simplex virus infections in the United States, 2006. Pediatrics. 2011;127:e1-e8.
6. Wenner C, Nashelsky J. Antiviral agents for pregnant women with genital herpes. Am Fam Physician. 2005;72:1807-1808.
7. Hollier LM, Wendel GD. Third trimester antiviral prophylaxis for preventing maternal genital herpes simplex virus (HSV) recurrences and neonatal infection. Cochrane Database Syst Rev. 2008;CD004946.
1. World Health Organization. Herpes simplex virus. World Health Organization Web site. Available at: http://www.who.int/mediacentre/factsheets/fs400/en/. Accessed February 8, 2016.
2. Ramaswamy M, McDonald C, Smith M, et al. Diagnosis of genital herpes by real time PCR in routine clinical practice. Sex Transm Infect. 2004;80:406-410.
3. Workowski KA, Berman S; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2010. MMWR Recomm Rep. 2010;59:1-110.
4. Cernik C, Gallina K, Brodell RT. The treatment of herpes simplex infections: an evidence-based review. Arch Intern Med. 2008;168:1137-1144.
5. Flagg EW, Weinstock H. Incidence of neonatal herpes simplex virus infections in the United States, 2006. Pediatrics. 2011;127:e1-e8.
6. Wenner C, Nashelsky J. Antiviral agents for pregnant women with genital herpes. Am Fam Physician. 2005;72:1807-1808.
7. Hollier LM, Wendel GD. Third trimester antiviral prophylaxis for preventing maternal genital herpes simplex virus (HSV) recurrences and neonatal infection. Cochrane Database Syst Rev. 2008;CD004946.
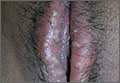
KOH preparation