User login
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
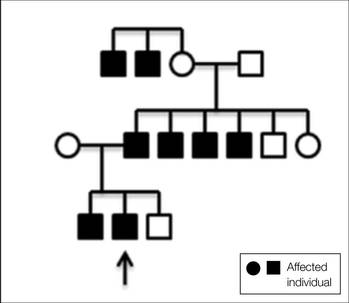
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
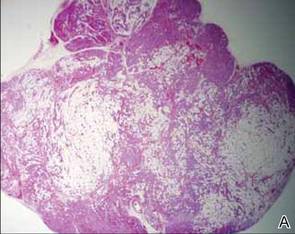
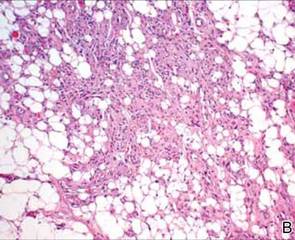
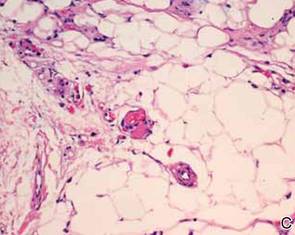
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
Practice Points
- Dermatologists should be familiar with the clinical and histological features of angiolipomas along with their potential inheritance patterns.
- Familial angiolipomatosis is a rare condition characterized by multiple angiolipomas that has been described as having an autosomal-recessive transmission pattern. Autosomal-dominant inheritance also may occur, as illustrated in the current case report.
- Awareness of the autosomal-dominant form of this entity is important to prevent its misdiagnosis as
neurofibromatosis type I, which has a similar family history and clinical presentation.