User login
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 76-YEAR-OLD MAN sought care for a rash that had gotten progressively worse over the previous 3 weeks. He indicated that the rash was initially red, itchy, and located over his abdomen, but as time went by, new blisters developed in the axillae and groin, and they were painful. The patient did not have any arthralgias or systemic symptoms. The medications he was taking included simvastatin, albuterol, and finasteride.
On physical examination, the patient was in mild distress due to the pain and anxiety, and his temperature was 36.5°C (97.7°F). He had confluent areas of erythematous, denuded skin spanning his trunk, back, and proximal upper and lower extremities (FIGURE 1). Tense, fluid-filled blisters were most prominent in the groin and in the axillae, bilaterally.
FIGURE 1
Diffuse rash on the trunk and in the axillae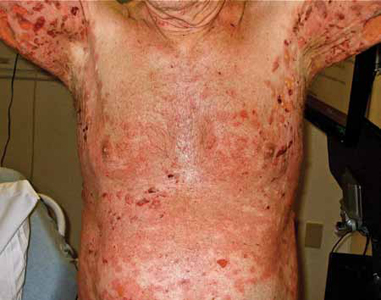
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Bullous pemphigoid
This patient had a severe and refractory case of bullous pemphigoid (BP), which was confirmed with a biopsy of the lesions.
BP is a rare autoimmune, blistering skin disease that typically occurs after age 60.1 The incidence rises with age, and is higher among women than men.1 The pathogenesis of BP involves development of autoantibodies against the subepidermal basement membrane. Deposition of immunoglobulin G (IgG) occurs, leading to immune-mediated destruction and subepidermal blistering.2
Patients will present with new-onset, widespread eruptions of bullous lesions and urticarial plaques (FIGURE 2).2 Bullae are frequent on flexural surfaces such as the groin and axillae. Urticarial plaques are often pruritic. Oral involvement occurs in a minority of cases.2 Nikolsky’s sign—exfoliation of the outermost layer of skin upon slight rubbing—is absent in BP.
FIGURE 2
Fluid-filled vesicles and bullae on right anterior thigh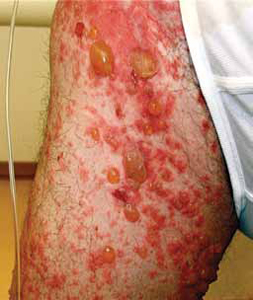
Differential: Other autoimmune and blistering skin conditions
Two additional pemphigoid subtypes are part of the differential when a patient presents with a blistering skin condition: pemphigoid gestationis and mucous membrane pemphigoid.2
Pemphigoid gestationis occurs exclusively during pregnancy and the puerperium, and is self-limited.
Mucous membrane pemphigoid is pathophysiologically similar to BP, but distributes preferentially on mucosal surfaces.
Pemphigus vulgaris, another autoimmune blistering skin disease, is characterized by sparse intact bullae. The mucous membrane is frequently involved, and there is a positive Nikolsky’s sign.
Additional conditions to keep in mind include epidermolysis bullosa acquisita, dermatitis herpetiformis, bullous erythema multiforme, and bullous lupus erythematosus.
Biopsy confirms the Dx
A biopsy of a lesion confirms the diagnosis of BP and will help differentiate it from the conditions mentioned above.
Light microscopy shows eosinophil-rich subepidermal inflammatory infiltrate.2 Direct immunofluorescence displays the characteristic linear deposits of IgG and complement C3 along the basement membrane. Immunofluorescent testing on human salt-split skin may also be performed.
Drug induced? There is a subset of BP, called drug-induced BP, in which the onset of the disease is associated with the initiation of a medication. Furosemide is the most common culprit,3 although many additional medications have been described.
The pathophysiology of drug-induced BP is poorly understood.3 In some cases, discontinuation of the offending medication may halt progression and prevent recurrence. In other cases, the disease will progress to a chronic form regardless of medication discontinuation. It is reasonable to attempt medication discontinuation trials in cases where drug-induced BP is suspected.
Treat with corticosteroids
The traditional treatment of BP is high-dose oral corticosteroids. However, long-term use of systemic corticosteroids can cause significant morbidity and has been linked to an increased mortality rate in the elderly population.4 A potent topical corticosteroid, such as clobetasol propionate cream 10 to 30 g/d tapering over 4 months, or 40 g/d tapering over 12 months,5,6 is an effective alternative (strength of recommendation [SOR]: A).
Other options include methotrexate, mycophenolate, azathioprine, niacinamide, doxycycline, intravenous (IV) immunoglobulin, and plasma exchange. These therapies are typically used in combination with corticosteroids, or after initial treatment failure. Evidence regarding their effectiveness is limited7 (SOR: B).
Although the disease is occasionally self-limited after the initial episode, most patients with BP will achieve clinical remission with medical intervention. Patients often experience recurrent outbreaks and require chronic use of immunosuppressive agents.
Our patient required ongoing care
Our patient was prescribed prednisone 80 mg/d PO in combination with topical clobetasol cream. Despite these treatments, the disease progressed. One week later, approximately 80% of his body surface was involved. He was admitted for fluid replacement and monitoring for infection.
Subsequent initiation of methotrexate, niacinamide, doxycycline, and topical clobetasol led to clinical remission. Unfortunately, the patient relapsed approximately 3 months later and required a second hospital stay.
In the ensuing months, the patient’s course was marked by frequent relapses and significant morbidity. Further treatment trials have included IV immunoglobulin, mycophenolate, and azathioprine.
CORRESPONDENCE
Casey Z. MacVane, MD, MPH, Department of Emergency Medicine, Maine Medical Center, 47 Bramhall Street, Portland, ME 04102; [email protected]
1. Langan SM, Smeeth L, Hubbard R, et al. Bullous pemphigoid and pemphigus vulgaris—incidence and mortality in the UK: population based cohort study. BMJ. 2008;337:a180.-
2. Yancey KB, Egan CA. Pemphigoid: clinical, histological, immunopathologic, and therapeutic considerations. JAMA. 2000;248:350-356.
3. Lee JJ, Downham TF, 2nd. Furosemide-induced bullous pemphigoid: case report and review of literature. J Drugs Dermatol. 2006;5:562-564.
4. Rzany B, Partscht K, Jung M, et al. Risk factors for lethal outcome in patients with bullous pemphigoid. Arch Dermatol. 2002;138:903-908.
5. Joly P, Roujeau JC, Benichou J, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med. 2002;346:321-327.
6. Joly P, Roujeau JC, Benichou J, et al. A comparison of two regimens of topical corticosteroids in the treatment of patients with bullous pemphigoid: a multicenter randomized study. J Invest Dermatol. 2009;129:1681-1687.
7. Kirtschig G, Middleton P, Bennett C, et al. Interventions for bullous pemphigoid. Cochrane Database Syst Rev. 2010;(10):CD002292.
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 76-YEAR-OLD MAN sought care for a rash that had gotten progressively worse over the previous 3 weeks. He indicated that the rash was initially red, itchy, and located over his abdomen, but as time went by, new blisters developed in the axillae and groin, and they were painful. The patient did not have any arthralgias or systemic symptoms. The medications he was taking included simvastatin, albuterol, and finasteride.
On physical examination, the patient was in mild distress due to the pain and anxiety, and his temperature was 36.5°C (97.7°F). He had confluent areas of erythematous, denuded skin spanning his trunk, back, and proximal upper and lower extremities (FIGURE 1). Tense, fluid-filled blisters were most prominent in the groin and in the axillae, bilaterally.
FIGURE 1
Diffuse rash on the trunk and in the axillae
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Bullous pemphigoid
This patient had a severe and refractory case of bullous pemphigoid (BP), which was confirmed with a biopsy of the lesions.
BP is a rare autoimmune, blistering skin disease that typically occurs after age 60.1 The incidence rises with age, and is higher among women than men.1 The pathogenesis of BP involves development of autoantibodies against the subepidermal basement membrane. Deposition of immunoglobulin G (IgG) occurs, leading to immune-mediated destruction and subepidermal blistering.2
Patients will present with new-onset, widespread eruptions of bullous lesions and urticarial plaques (FIGURE 2).2 Bullae are frequent on flexural surfaces such as the groin and axillae. Urticarial plaques are often pruritic. Oral involvement occurs in a minority of cases.2 Nikolsky’s sign—exfoliation of the outermost layer of skin upon slight rubbing—is absent in BP.
FIGURE 2
Fluid-filled vesicles and bullae on right anterior thigh
Differential: Other autoimmune and blistering skin conditions
Two additional pemphigoid subtypes are part of the differential when a patient presents with a blistering skin condition: pemphigoid gestationis and mucous membrane pemphigoid.2
Pemphigoid gestationis occurs exclusively during pregnancy and the puerperium, and is self-limited.
Mucous membrane pemphigoid is pathophysiologically similar to BP, but distributes preferentially on mucosal surfaces.
Pemphigus vulgaris, another autoimmune blistering skin disease, is characterized by sparse intact bullae. The mucous membrane is frequently involved, and there is a positive Nikolsky’s sign.
Additional conditions to keep in mind include epidermolysis bullosa acquisita, dermatitis herpetiformis, bullous erythema multiforme, and bullous lupus erythematosus.
Biopsy confirms the Dx
A biopsy of a lesion confirms the diagnosis of BP and will help differentiate it from the conditions mentioned above.
Light microscopy shows eosinophil-rich subepidermal inflammatory infiltrate.2 Direct immunofluorescence displays the characteristic linear deposits of IgG and complement C3 along the basement membrane. Immunofluorescent testing on human salt-split skin may also be performed.
Drug induced? There is a subset of BP, called drug-induced BP, in which the onset of the disease is associated with the initiation of a medication. Furosemide is the most common culprit,3 although many additional medications have been described.
The pathophysiology of drug-induced BP is poorly understood.3 In some cases, discontinuation of the offending medication may halt progression and prevent recurrence. In other cases, the disease will progress to a chronic form regardless of medication discontinuation. It is reasonable to attempt medication discontinuation trials in cases where drug-induced BP is suspected.
Treat with corticosteroids
The traditional treatment of BP is high-dose oral corticosteroids. However, long-term use of systemic corticosteroids can cause significant morbidity and has been linked to an increased mortality rate in the elderly population.4 A potent topical corticosteroid, such as clobetasol propionate cream 10 to 30 g/d tapering over 4 months, or 40 g/d tapering over 12 months,5,6 is an effective alternative (strength of recommendation [SOR]: A).
Other options include methotrexate, mycophenolate, azathioprine, niacinamide, doxycycline, intravenous (IV) immunoglobulin, and plasma exchange. These therapies are typically used in combination with corticosteroids, or after initial treatment failure. Evidence regarding their effectiveness is limited7 (SOR: B).
Although the disease is occasionally self-limited after the initial episode, most patients with BP will achieve clinical remission with medical intervention. Patients often experience recurrent outbreaks and require chronic use of immunosuppressive agents.
Our patient required ongoing care
Our patient was prescribed prednisone 80 mg/d PO in combination with topical clobetasol cream. Despite these treatments, the disease progressed. One week later, approximately 80% of his body surface was involved. He was admitted for fluid replacement and monitoring for infection.
Subsequent initiation of methotrexate, niacinamide, doxycycline, and topical clobetasol led to clinical remission. Unfortunately, the patient relapsed approximately 3 months later and required a second hospital stay.
In the ensuing months, the patient’s course was marked by frequent relapses and significant morbidity. Further treatment trials have included IV immunoglobulin, mycophenolate, and azathioprine.
CORRESPONDENCE
Casey Z. MacVane, MD, MPH, Department of Emergency Medicine, Maine Medical Center, 47 Bramhall Street, Portland, ME 04102; [email protected]
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 76-YEAR-OLD MAN sought care for a rash that had gotten progressively worse over the previous 3 weeks. He indicated that the rash was initially red, itchy, and located over his abdomen, but as time went by, new blisters developed in the axillae and groin, and they were painful. The patient did not have any arthralgias or systemic symptoms. The medications he was taking included simvastatin, albuterol, and finasteride.
On physical examination, the patient was in mild distress due to the pain and anxiety, and his temperature was 36.5°C (97.7°F). He had confluent areas of erythematous, denuded skin spanning his trunk, back, and proximal upper and lower extremities (FIGURE 1). Tense, fluid-filled blisters were most prominent in the groin and in the axillae, bilaterally.
FIGURE 1
Diffuse rash on the trunk and in the axillae
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Bullous pemphigoid
This patient had a severe and refractory case of bullous pemphigoid (BP), which was confirmed with a biopsy of the lesions.
BP is a rare autoimmune, blistering skin disease that typically occurs after age 60.1 The incidence rises with age, and is higher among women than men.1 The pathogenesis of BP involves development of autoantibodies against the subepidermal basement membrane. Deposition of immunoglobulin G (IgG) occurs, leading to immune-mediated destruction and subepidermal blistering.2
Patients will present with new-onset, widespread eruptions of bullous lesions and urticarial plaques (FIGURE 2).2 Bullae are frequent on flexural surfaces such as the groin and axillae. Urticarial plaques are often pruritic. Oral involvement occurs in a minority of cases.2 Nikolsky’s sign—exfoliation of the outermost layer of skin upon slight rubbing—is absent in BP.
FIGURE 2
Fluid-filled vesicles and bullae on right anterior thigh
Differential: Other autoimmune and blistering skin conditions
Two additional pemphigoid subtypes are part of the differential when a patient presents with a blistering skin condition: pemphigoid gestationis and mucous membrane pemphigoid.2
Pemphigoid gestationis occurs exclusively during pregnancy and the puerperium, and is self-limited.
Mucous membrane pemphigoid is pathophysiologically similar to BP, but distributes preferentially on mucosal surfaces.
Pemphigus vulgaris, another autoimmune blistering skin disease, is characterized by sparse intact bullae. The mucous membrane is frequently involved, and there is a positive Nikolsky’s sign.
Additional conditions to keep in mind include epidermolysis bullosa acquisita, dermatitis herpetiformis, bullous erythema multiforme, and bullous lupus erythematosus.
Biopsy confirms the Dx
A biopsy of a lesion confirms the diagnosis of BP and will help differentiate it from the conditions mentioned above.
Light microscopy shows eosinophil-rich subepidermal inflammatory infiltrate.2 Direct immunofluorescence displays the characteristic linear deposits of IgG and complement C3 along the basement membrane. Immunofluorescent testing on human salt-split skin may also be performed.
Drug induced? There is a subset of BP, called drug-induced BP, in which the onset of the disease is associated with the initiation of a medication. Furosemide is the most common culprit,3 although many additional medications have been described.
The pathophysiology of drug-induced BP is poorly understood.3 In some cases, discontinuation of the offending medication may halt progression and prevent recurrence. In other cases, the disease will progress to a chronic form regardless of medication discontinuation. It is reasonable to attempt medication discontinuation trials in cases where drug-induced BP is suspected.
Treat with corticosteroids
The traditional treatment of BP is high-dose oral corticosteroids. However, long-term use of systemic corticosteroids can cause significant morbidity and has been linked to an increased mortality rate in the elderly population.4 A potent topical corticosteroid, such as clobetasol propionate cream 10 to 30 g/d tapering over 4 months, or 40 g/d tapering over 12 months,5,6 is an effective alternative (strength of recommendation [SOR]: A).
Other options include methotrexate, mycophenolate, azathioprine, niacinamide, doxycycline, intravenous (IV) immunoglobulin, and plasma exchange. These therapies are typically used in combination with corticosteroids, or after initial treatment failure. Evidence regarding their effectiveness is limited7 (SOR: B).
Although the disease is occasionally self-limited after the initial episode, most patients with BP will achieve clinical remission with medical intervention. Patients often experience recurrent outbreaks and require chronic use of immunosuppressive agents.
Our patient required ongoing care
Our patient was prescribed prednisone 80 mg/d PO in combination with topical clobetasol cream. Despite these treatments, the disease progressed. One week later, approximately 80% of his body surface was involved. He was admitted for fluid replacement and monitoring for infection.
Subsequent initiation of methotrexate, niacinamide, doxycycline, and topical clobetasol led to clinical remission. Unfortunately, the patient relapsed approximately 3 months later and required a second hospital stay.
In the ensuing months, the patient’s course was marked by frequent relapses and significant morbidity. Further treatment trials have included IV immunoglobulin, mycophenolate, and azathioprine.
CORRESPONDENCE
Casey Z. MacVane, MD, MPH, Department of Emergency Medicine, Maine Medical Center, 47 Bramhall Street, Portland, ME 04102; [email protected]
1. Langan SM, Smeeth L, Hubbard R, et al. Bullous pemphigoid and pemphigus vulgaris—incidence and mortality in the UK: population based cohort study. BMJ. 2008;337:a180.-
2. Yancey KB, Egan CA. Pemphigoid: clinical, histological, immunopathologic, and therapeutic considerations. JAMA. 2000;248:350-356.
3. Lee JJ, Downham TF, 2nd. Furosemide-induced bullous pemphigoid: case report and review of literature. J Drugs Dermatol. 2006;5:562-564.
4. Rzany B, Partscht K, Jung M, et al. Risk factors for lethal outcome in patients with bullous pemphigoid. Arch Dermatol. 2002;138:903-908.
5. Joly P, Roujeau JC, Benichou J, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med. 2002;346:321-327.
6. Joly P, Roujeau JC, Benichou J, et al. A comparison of two regimens of topical corticosteroids in the treatment of patients with bullous pemphigoid: a multicenter randomized study. J Invest Dermatol. 2009;129:1681-1687.
7. Kirtschig G, Middleton P, Bennett C, et al. Interventions for bullous pemphigoid. Cochrane Database Syst Rev. 2010;(10):CD002292.
1. Langan SM, Smeeth L, Hubbard R, et al. Bullous pemphigoid and pemphigus vulgaris—incidence and mortality in the UK: population based cohort study. BMJ. 2008;337:a180.-
2. Yancey KB, Egan CA. Pemphigoid: clinical, histological, immunopathologic, and therapeutic considerations. JAMA. 2000;248:350-356.
3. Lee JJ, Downham TF, 2nd. Furosemide-induced bullous pemphigoid: case report and review of literature. J Drugs Dermatol. 2006;5:562-564.
4. Rzany B, Partscht K, Jung M, et al. Risk factors for lethal outcome in patients with bullous pemphigoid. Arch Dermatol. 2002;138:903-908.
5. Joly P, Roujeau JC, Benichou J, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med. 2002;346:321-327.
6. Joly P, Roujeau JC, Benichou J, et al. A comparison of two regimens of topical corticosteroids in the treatment of patients with bullous pemphigoid: a multicenter randomized study. J Invest Dermatol. 2009;129:1681-1687.
7. Kirtschig G, Middleton P, Bennett C, et al. Interventions for bullous pemphigoid. Cochrane Database Syst Rev. 2010;(10):CD002292.