User login
To the Editor:
Acrokeratosis paraneoplastica (AP) of Bazex is a rare but distinctive acral psoriasiform dermatosis associated with internal malignancy, usually squamous cell carcinoma (SCC), of the upper aerodigestive tract.1,2 Recognizing this paraneoplastic condition is paramount because cutaneous findings often precede the onset of symptoms associated with an occult malignancy.3
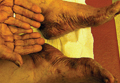
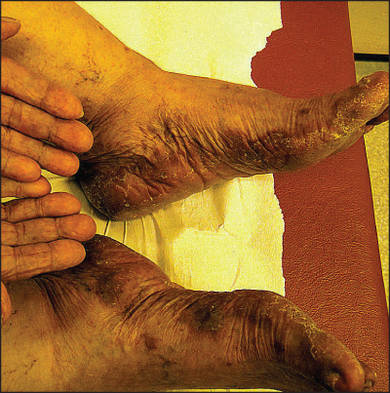
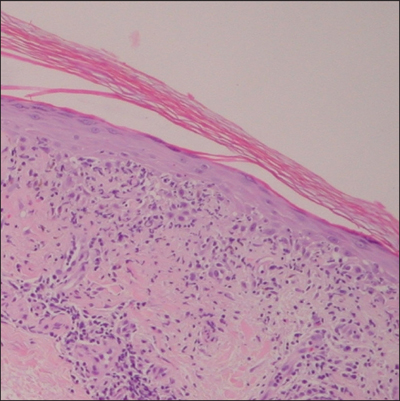
A 76-year-old woman with adenocarcinoma of the transverse colon of 3 years’ duration was referred to the dermatology department. She had a hemicolectomy and was doing well until tumor recurrence with peritoneal metastasis was detected following an exploratory laparotomy 1 year prior to presentation to us. She underwent a total hysterectomy and bilateral salpingo-oophorectomy. Palliative chemotherapy was initiated, and she completed 5 cycles of capecitabine and oxaliplatin. Long-term medications for diabetes mellitus, hypertension, and hyperlipidemia included glibenclamide, nifedipine, hydrochlorothiazide, and losartan. The patient had a progressive pruritic rash of 6 months’ duration that started on the hands and forearms and spread to involve the feet and lower limbs as well as the ears and face. She also experienced progressive thickening of the palms and soles. She had been treated with topical steroids and emollients with no improvement. Clinical examination revealed erythematous scaly patches on all of her limbs, especially on the elbows and knees, and on the ear helices and nose. There also was notable palmoplantar keratoderma with central sparing (Figure 1), onycholysis, and subungual hyperkeratosis. A skin biopsy of the forearm was performed, and histology revealed orthokeratosis, hypergranulosis and basal vacuolar alteration, and superficial perivascular lymphohistiocytic infiltrate with melanophages (Figure 2). Direct immunofluorescence studies and fungal cultures were negative. The cutaneous features of a treatment-resistant acral dermatosis supported the clinical diagnosis of AP, especially in the setting of an internal malignancy. The patient was started on palliative radiotherapy with no notable resolution of the cutaneous lesions. She was lost to follow-up.
First described by Bazex et al1 in 1965, AP is an uncommon but well-recognized paraneoplastic dermatosis associated with an underlying neoplasm. Fewer than 160 cases have been reported, and the majority of cases have been men older than 40 years. The most commonly associated malignancy was SCC (at least 50% of reported cases) involving mainly the oropharynx and larynx, with lung and esophageal SCC also described.2-4 Only a few cases of adenocarcinoma-associated AP have been described, such as adenocarcinoma of the lung, prostate, and stomach.3-5 In a reported case of AP associated with early colon adenocarcinoma, the patient had remarkable cutaneous resolution following successful tumor resection.6 Other reported rare hematologic associations included Hodgkin disease, peripheral T-cell lymphoma, and multiple myeloma.3-5 Karabulut et al4 described a case associated with cholangiocarcinoma and studied the primary sites of malignancies in another 133 patients with AP (118 patients with documented cell type): oropharynx and larynx in 55 (41%); lung in 23 (17%); unknown location in 20 (15%); esophagus in 14 (11%); prostate in 3 (2%); stomach in 3 (2%); and isolated cases involving the liver, thymus, uterus, vulva, breast, urinary bladder, lymph nodes, and bone marrow.
A review of 113 cases of AP showed that only 15% developed cutaneous lesions after the malignancy had been discovered; in the majority of cases (67%), cutaneous lesions were present for an average of 1 year preceding the diagnosis of malignancy.3 In our patient, the late-onset cutaneous involvement corresponded to the progression of the underlying colon adenocarcinoma. In the absence of initial cutaneous involvement or when successful treatment of a tumor results in cutaneous resolution, subsequent emergence of cutaneous lesions of AP signifies tumor progression. The evolution of cutaneous features in AP was well described by Bazex et al.1 Stage 1 of the disease shows initial erythema and psoriasiform scaling of the fingers and toes, spreading to the nose and ear helices (hence acrokeratosis); the tumor frequently remains asymptomatic or undetected. Stage 2 shows palmoplantar keratoderma with central sparing and more extensive facial lesions; progression to stage 3 occurs if the tumor remains undetected or untreated, with further spread of psoriasiform lesions to the elbows, knees, trunk, and scalp.1 Nail involvement occurs in nearly 75% of cases5; typical changes include subungual hyperkeratosis, onycholysis, longitudinal streaks, yellow pigmentation, and rarely onychomadesis. A high index of suspicion of AP is paramount when evaluating any recalcitrant acral dermatosis that fails to respond to appropriate therapy, especially in the presence of constitutional symptoms, typical bulbous enlargement of distal phalanges, or isolated involvement of helices. These findings should prompt physicians to perform an extensive search for an underlying malignancy with complete physical examination, particularly of the head and neck region, with appropriate endoscopic assessment and imaging studies.
A myriad of nonspecific histologic features of AP commonly reported include hyperkeratosis, parakeratosis, acanthosis, and dermal perivascular lymphohistiocytic infiltrate.7 Less common features include dyskeratotic keratinocytes, vacuolar degeneration, bandlike infiltrate, and melanin incontinence.7 The pathogenesis of AP remains elusive. A postulated immunologic mechanism is based on reports of immunoglobulins (IgG, IgA, IgM) and complement (C3) deposition along the basement membrane zone.8 Association with autoimmune disorders such as alopecia areata and vitiligo also has been reported.9 Another possible mechanism is cross-reactivity between antigens found in the tumor and skin, resulting in a T-cell–mediated immune response to tumorlike antigens in the epidermis, or secretion of tumor-originating growth factors responsible for the hyperkeratotic skin changes, such as epidermal growth factor, transforming growth factor a, or insulinlike growth factor 1.3,7,10,11
Spontaneous remission of cutaneous lesions in untreated underlying malignancy is rare. Isolated reports of treatment using topical and systemic steroids, salicylic acid, topical vitamin D analogues, etretinate, and psoralen plus UVA showed minimal improvement.5,7 The mainstay in attaining cutaneous resolution is to detect and eradicate the underlying neoplasm with surgery, chemotherapy, or radiotherapy, or combination therapy.
Our case is noteworthy because of the patient’s gender (female), underlying malignancy (adenocarcinoma of the colon), and late onset of cutaneous involvement, which are all uncommon associations related to paraneoplastic syndrome. The clinical features of AP should be recognized early to facilitate an extensive search for an occult malignancy, and late-onset cutaneous involvement also should be recognized as a marker of tumor relapse or progression.
1. Bazex A, Salvador R, Dupré A, et al. Late symptomatic hepatic porphyria developing to the picture of lipoidoproteinsosis [in French]. Bull Soc Fr Dermatol Syphiligr. 1965;72:182.
2. Witkowski JA, Parish LC. Bazex’s syndrome. paraneoplastic acrokeratosis. JAMA. 1982;248:2883-2884.
3. Bolognia JL. Bazex syndrome: acrokeratosis paraneoplastica. Semin Dermatol. 1995;14:84-89.
4. Karabulut AA, Sahin S, Sahin M, et al. Paraneoplastic acrokeratosis of Bazex (Bazex’s syndrome): report of a female case associated with cholangiocarcinoma and review of the published work. J Dermatol. 2006;33:850-854.
5. Valdivielso M, Longo I, Suárez R. Acrokeratosis paraneoplastica: Bazex syndrome. J Eur Acad Dermatol Venereol. 2005;19:340-344.
6. Hsu YS, Lien GS, Lai HH, et al. Acrokeratosis paraneoplastica (Bazex syndrome) with adenocarcinoma of the colon: report of a case and review of the literature. J Gastroenterol. 2000;35:460-464.
7. Bolognia JL, Brewer YP, Cooper DL. Bazex syndrome (acrokeratosis paraneoplastica). an analytic review. Medicine (Baltimore). 1991;70:269-280.
8. Mutasim DF, Meiri G. Bazex syndrome mimicking a primary autoimmune bullous disorder. J Am Acad Dermatol. 1999;40(5, pt 2):822-825.
9. Hara M, Hunayama M, Aiba S, et al. Acrokeratosis paraneoplastica (Bazex syndrome) associated with primary cutaneous squamous cell carcinoma of the lower leg, vitiligo and alopecia areata. Br J Dermatol. 1995;133:121-124.
10. Stone SP, Buescher LS. Life-threatening paraneoplastic cutaneous syndromes. Clin Dermatol. 2005;23:301-306.
11. Politi Y, Ophir J, Brenner S. Cutaneous paraneoplastic syndromes. Acta Derm Venereol. 1993;73:161-170.
To the Editor:
Acrokeratosis paraneoplastica (AP) of Bazex is a rare but distinctive acral psoriasiform dermatosis associated with internal malignancy, usually squamous cell carcinoma (SCC), of the upper aerodigestive tract.1,2 Recognizing this paraneoplastic condition is paramount because cutaneous findings often precede the onset of symptoms associated with an occult malignancy.3
A 76-year-old woman with adenocarcinoma of the transverse colon of 3 years’ duration was referred to the dermatology department. She had a hemicolectomy and was doing well until tumor recurrence with peritoneal metastasis was detected following an exploratory laparotomy 1 year prior to presentation to us. She underwent a total hysterectomy and bilateral salpingo-oophorectomy. Palliative chemotherapy was initiated, and she completed 5 cycles of capecitabine and oxaliplatin. Long-term medications for diabetes mellitus, hypertension, and hyperlipidemia included glibenclamide, nifedipine, hydrochlorothiazide, and losartan. The patient had a progressive pruritic rash of 6 months’ duration that started on the hands and forearms and spread to involve the feet and lower limbs as well as the ears and face. She also experienced progressive thickening of the palms and soles. She had been treated with topical steroids and emollients with no improvement. Clinical examination revealed erythematous scaly patches on all of her limbs, especially on the elbows and knees, and on the ear helices and nose. There also was notable palmoplantar keratoderma with central sparing (Figure 1), onycholysis, and subungual hyperkeratosis. A skin biopsy of the forearm was performed, and histology revealed orthokeratosis, hypergranulosis and basal vacuolar alteration, and superficial perivascular lymphohistiocytic infiltrate with melanophages (Figure 2). Direct immunofluorescence studies and fungal cultures were negative. The cutaneous features of a treatment-resistant acral dermatosis supported the clinical diagnosis of AP, especially in the setting of an internal malignancy. The patient was started on palliative radiotherapy with no notable resolution of the cutaneous lesions. She was lost to follow-up.
First described by Bazex et al1 in 1965, AP is an uncommon but well-recognized paraneoplastic dermatosis associated with an underlying neoplasm. Fewer than 160 cases have been reported, and the majority of cases have been men older than 40 years. The most commonly associated malignancy was SCC (at least 50% of reported cases) involving mainly the oropharynx and larynx, with lung and esophageal SCC also described.2-4 Only a few cases of adenocarcinoma-associated AP have been described, such as adenocarcinoma of the lung, prostate, and stomach.3-5 In a reported case of AP associated with early colon adenocarcinoma, the patient had remarkable cutaneous resolution following successful tumor resection.6 Other reported rare hematologic associations included Hodgkin disease, peripheral T-cell lymphoma, and multiple myeloma.3-5 Karabulut et al4 described a case associated with cholangiocarcinoma and studied the primary sites of malignancies in another 133 patients with AP (118 patients with documented cell type): oropharynx and larynx in 55 (41%); lung in 23 (17%); unknown location in 20 (15%); esophagus in 14 (11%); prostate in 3 (2%); stomach in 3 (2%); and isolated cases involving the liver, thymus, uterus, vulva, breast, urinary bladder, lymph nodes, and bone marrow.
A review of 113 cases of AP showed that only 15% developed cutaneous lesions after the malignancy had been discovered; in the majority of cases (67%), cutaneous lesions were present for an average of 1 year preceding the diagnosis of malignancy.3 In our patient, the late-onset cutaneous involvement corresponded to the progression of the underlying colon adenocarcinoma. In the absence of initial cutaneous involvement or when successful treatment of a tumor results in cutaneous resolution, subsequent emergence of cutaneous lesions of AP signifies tumor progression. The evolution of cutaneous features in AP was well described by Bazex et al.1 Stage 1 of the disease shows initial erythema and psoriasiform scaling of the fingers and toes, spreading to the nose and ear helices (hence acrokeratosis); the tumor frequently remains asymptomatic or undetected. Stage 2 shows palmoplantar keratoderma with central sparing and more extensive facial lesions; progression to stage 3 occurs if the tumor remains undetected or untreated, with further spread of psoriasiform lesions to the elbows, knees, trunk, and scalp.1 Nail involvement occurs in nearly 75% of cases5; typical changes include subungual hyperkeratosis, onycholysis, longitudinal streaks, yellow pigmentation, and rarely onychomadesis. A high index of suspicion of AP is paramount when evaluating any recalcitrant acral dermatosis that fails to respond to appropriate therapy, especially in the presence of constitutional symptoms, typical bulbous enlargement of distal phalanges, or isolated involvement of helices. These findings should prompt physicians to perform an extensive search for an underlying malignancy with complete physical examination, particularly of the head and neck region, with appropriate endoscopic assessment and imaging studies.
A myriad of nonspecific histologic features of AP commonly reported include hyperkeratosis, parakeratosis, acanthosis, and dermal perivascular lymphohistiocytic infiltrate.7 Less common features include dyskeratotic keratinocytes, vacuolar degeneration, bandlike infiltrate, and melanin incontinence.7 The pathogenesis of AP remains elusive. A postulated immunologic mechanism is based on reports of immunoglobulins (IgG, IgA, IgM) and complement (C3) deposition along the basement membrane zone.8 Association with autoimmune disorders such as alopecia areata and vitiligo also has been reported.9 Another possible mechanism is cross-reactivity between antigens found in the tumor and skin, resulting in a T-cell–mediated immune response to tumorlike antigens in the epidermis, or secretion of tumor-originating growth factors responsible for the hyperkeratotic skin changes, such as epidermal growth factor, transforming growth factor a, or insulinlike growth factor 1.3,7,10,11
Spontaneous remission of cutaneous lesions in untreated underlying malignancy is rare. Isolated reports of treatment using topical and systemic steroids, salicylic acid, topical vitamin D analogues, etretinate, and psoralen plus UVA showed minimal improvement.5,7 The mainstay in attaining cutaneous resolution is to detect and eradicate the underlying neoplasm with surgery, chemotherapy, or radiotherapy, or combination therapy.
Our case is noteworthy because of the patient’s gender (female), underlying malignancy (adenocarcinoma of the colon), and late onset of cutaneous involvement, which are all uncommon associations related to paraneoplastic syndrome. The clinical features of AP should be recognized early to facilitate an extensive search for an occult malignancy, and late-onset cutaneous involvement also should be recognized as a marker of tumor relapse or progression.
To the Editor:
Acrokeratosis paraneoplastica (AP) of Bazex is a rare but distinctive acral psoriasiform dermatosis associated with internal malignancy, usually squamous cell carcinoma (SCC), of the upper aerodigestive tract.1,2 Recognizing this paraneoplastic condition is paramount because cutaneous findings often precede the onset of symptoms associated with an occult malignancy.3
A 76-year-old woman with adenocarcinoma of the transverse colon of 3 years’ duration was referred to the dermatology department. She had a hemicolectomy and was doing well until tumor recurrence with peritoneal metastasis was detected following an exploratory laparotomy 1 year prior to presentation to us. She underwent a total hysterectomy and bilateral salpingo-oophorectomy. Palliative chemotherapy was initiated, and she completed 5 cycles of capecitabine and oxaliplatin. Long-term medications for diabetes mellitus, hypertension, and hyperlipidemia included glibenclamide, nifedipine, hydrochlorothiazide, and losartan. The patient had a progressive pruritic rash of 6 months’ duration that started on the hands and forearms and spread to involve the feet and lower limbs as well as the ears and face. She also experienced progressive thickening of the palms and soles. She had been treated with topical steroids and emollients with no improvement. Clinical examination revealed erythematous scaly patches on all of her limbs, especially on the elbows and knees, and on the ear helices and nose. There also was notable palmoplantar keratoderma with central sparing (Figure 1), onycholysis, and subungual hyperkeratosis. A skin biopsy of the forearm was performed, and histology revealed orthokeratosis, hypergranulosis and basal vacuolar alteration, and superficial perivascular lymphohistiocytic infiltrate with melanophages (Figure 2). Direct immunofluorescence studies and fungal cultures were negative. The cutaneous features of a treatment-resistant acral dermatosis supported the clinical diagnosis of AP, especially in the setting of an internal malignancy. The patient was started on palliative radiotherapy with no notable resolution of the cutaneous lesions. She was lost to follow-up.
First described by Bazex et al1 in 1965, AP is an uncommon but well-recognized paraneoplastic dermatosis associated with an underlying neoplasm. Fewer than 160 cases have been reported, and the majority of cases have been men older than 40 years. The most commonly associated malignancy was SCC (at least 50% of reported cases) involving mainly the oropharynx and larynx, with lung and esophageal SCC also described.2-4 Only a few cases of adenocarcinoma-associated AP have been described, such as adenocarcinoma of the lung, prostate, and stomach.3-5 In a reported case of AP associated with early colon adenocarcinoma, the patient had remarkable cutaneous resolution following successful tumor resection.6 Other reported rare hematologic associations included Hodgkin disease, peripheral T-cell lymphoma, and multiple myeloma.3-5 Karabulut et al4 described a case associated with cholangiocarcinoma and studied the primary sites of malignancies in another 133 patients with AP (118 patients with documented cell type): oropharynx and larynx in 55 (41%); lung in 23 (17%); unknown location in 20 (15%); esophagus in 14 (11%); prostate in 3 (2%); stomach in 3 (2%); and isolated cases involving the liver, thymus, uterus, vulva, breast, urinary bladder, lymph nodes, and bone marrow.
A review of 113 cases of AP showed that only 15% developed cutaneous lesions after the malignancy had been discovered; in the majority of cases (67%), cutaneous lesions were present for an average of 1 year preceding the diagnosis of malignancy.3 In our patient, the late-onset cutaneous involvement corresponded to the progression of the underlying colon adenocarcinoma. In the absence of initial cutaneous involvement or when successful treatment of a tumor results in cutaneous resolution, subsequent emergence of cutaneous lesions of AP signifies tumor progression. The evolution of cutaneous features in AP was well described by Bazex et al.1 Stage 1 of the disease shows initial erythema and psoriasiform scaling of the fingers and toes, spreading to the nose and ear helices (hence acrokeratosis); the tumor frequently remains asymptomatic or undetected. Stage 2 shows palmoplantar keratoderma with central sparing and more extensive facial lesions; progression to stage 3 occurs if the tumor remains undetected or untreated, with further spread of psoriasiform lesions to the elbows, knees, trunk, and scalp.1 Nail involvement occurs in nearly 75% of cases5; typical changes include subungual hyperkeratosis, onycholysis, longitudinal streaks, yellow pigmentation, and rarely onychomadesis. A high index of suspicion of AP is paramount when evaluating any recalcitrant acral dermatosis that fails to respond to appropriate therapy, especially in the presence of constitutional symptoms, typical bulbous enlargement of distal phalanges, or isolated involvement of helices. These findings should prompt physicians to perform an extensive search for an underlying malignancy with complete physical examination, particularly of the head and neck region, with appropriate endoscopic assessment and imaging studies.
A myriad of nonspecific histologic features of AP commonly reported include hyperkeratosis, parakeratosis, acanthosis, and dermal perivascular lymphohistiocytic infiltrate.7 Less common features include dyskeratotic keratinocytes, vacuolar degeneration, bandlike infiltrate, and melanin incontinence.7 The pathogenesis of AP remains elusive. A postulated immunologic mechanism is based on reports of immunoglobulins (IgG, IgA, IgM) and complement (C3) deposition along the basement membrane zone.8 Association with autoimmune disorders such as alopecia areata and vitiligo also has been reported.9 Another possible mechanism is cross-reactivity between antigens found in the tumor and skin, resulting in a T-cell–mediated immune response to tumorlike antigens in the epidermis, or secretion of tumor-originating growth factors responsible for the hyperkeratotic skin changes, such as epidermal growth factor, transforming growth factor a, or insulinlike growth factor 1.3,7,10,11
Spontaneous remission of cutaneous lesions in untreated underlying malignancy is rare. Isolated reports of treatment using topical and systemic steroids, salicylic acid, topical vitamin D analogues, etretinate, and psoralen plus UVA showed minimal improvement.5,7 The mainstay in attaining cutaneous resolution is to detect and eradicate the underlying neoplasm with surgery, chemotherapy, or radiotherapy, or combination therapy.
Our case is noteworthy because of the patient’s gender (female), underlying malignancy (adenocarcinoma of the colon), and late onset of cutaneous involvement, which are all uncommon associations related to paraneoplastic syndrome. The clinical features of AP should be recognized early to facilitate an extensive search for an occult malignancy, and late-onset cutaneous involvement also should be recognized as a marker of tumor relapse or progression.
1. Bazex A, Salvador R, Dupré A, et al. Late symptomatic hepatic porphyria developing to the picture of lipoidoproteinsosis [in French]. Bull Soc Fr Dermatol Syphiligr. 1965;72:182.
2. Witkowski JA, Parish LC. Bazex’s syndrome. paraneoplastic acrokeratosis. JAMA. 1982;248:2883-2884.
3. Bolognia JL. Bazex syndrome: acrokeratosis paraneoplastica. Semin Dermatol. 1995;14:84-89.
4. Karabulut AA, Sahin S, Sahin M, et al. Paraneoplastic acrokeratosis of Bazex (Bazex’s syndrome): report of a female case associated with cholangiocarcinoma and review of the published work. J Dermatol. 2006;33:850-854.
5. Valdivielso M, Longo I, Suárez R. Acrokeratosis paraneoplastica: Bazex syndrome. J Eur Acad Dermatol Venereol. 2005;19:340-344.
6. Hsu YS, Lien GS, Lai HH, et al. Acrokeratosis paraneoplastica (Bazex syndrome) with adenocarcinoma of the colon: report of a case and review of the literature. J Gastroenterol. 2000;35:460-464.
7. Bolognia JL, Brewer YP, Cooper DL. Bazex syndrome (acrokeratosis paraneoplastica). an analytic review. Medicine (Baltimore). 1991;70:269-280.
8. Mutasim DF, Meiri G. Bazex syndrome mimicking a primary autoimmune bullous disorder. J Am Acad Dermatol. 1999;40(5, pt 2):822-825.
9. Hara M, Hunayama M, Aiba S, et al. Acrokeratosis paraneoplastica (Bazex syndrome) associated with primary cutaneous squamous cell carcinoma of the lower leg, vitiligo and alopecia areata. Br J Dermatol. 1995;133:121-124.
10. Stone SP, Buescher LS. Life-threatening paraneoplastic cutaneous syndromes. Clin Dermatol. 2005;23:301-306.
11. Politi Y, Ophir J, Brenner S. Cutaneous paraneoplastic syndromes. Acta Derm Venereol. 1993;73:161-170.
1. Bazex A, Salvador R, Dupré A, et al. Late symptomatic hepatic porphyria developing to the picture of lipoidoproteinsosis [in French]. Bull Soc Fr Dermatol Syphiligr. 1965;72:182.
2. Witkowski JA, Parish LC. Bazex’s syndrome. paraneoplastic acrokeratosis. JAMA. 1982;248:2883-2884.
3. Bolognia JL. Bazex syndrome: acrokeratosis paraneoplastica. Semin Dermatol. 1995;14:84-89.
4. Karabulut AA, Sahin S, Sahin M, et al. Paraneoplastic acrokeratosis of Bazex (Bazex’s syndrome): report of a female case associated with cholangiocarcinoma and review of the published work. J Dermatol. 2006;33:850-854.
5. Valdivielso M, Longo I, Suárez R. Acrokeratosis paraneoplastica: Bazex syndrome. J Eur Acad Dermatol Venereol. 2005;19:340-344.
6. Hsu YS, Lien GS, Lai HH, et al. Acrokeratosis paraneoplastica (Bazex syndrome) with adenocarcinoma of the colon: report of a case and review of the literature. J Gastroenterol. 2000;35:460-464.
7. Bolognia JL, Brewer YP, Cooper DL. Bazex syndrome (acrokeratosis paraneoplastica). an analytic review. Medicine (Baltimore). 1991;70:269-280.
8. Mutasim DF, Meiri G. Bazex syndrome mimicking a primary autoimmune bullous disorder. J Am Acad Dermatol. 1999;40(5, pt 2):822-825.
9. Hara M, Hunayama M, Aiba S, et al. Acrokeratosis paraneoplastica (Bazex syndrome) associated with primary cutaneous squamous cell carcinoma of the lower leg, vitiligo and alopecia areata. Br J Dermatol. 1995;133:121-124.
10. Stone SP, Buescher LS. Life-threatening paraneoplastic cutaneous syndromes. Clin Dermatol. 2005;23:301-306.
11. Politi Y, Ophir J, Brenner S. Cutaneous paraneoplastic syndromes. Acta Derm Venereol. 1993;73:161-170.