User login
The balance between dietary intake and excretion of phosphorus can be impaired in patients with decreased renal function, leading to hyperphosphatemia. Many patients with end-stage renal disease on dialysis require phosphorus-binding drugs to control their serum phosphorus levels.
See related editorial and article
In this review, we discuss the pathophysiology of hyperphosphatemia in kidney disease, its consequences, and how to control it, focusing on the different classes of phosphorus binders.
ROLE OF THE INTERNIST
With kidney disease common and on the increase,1 nephrologists and internists need to work together to provide optimal care.
Further, many internists in managed care plans and accountable care organizations now handle many tasks previously left to specialists—including prescribing and managing phosphorus binders in patients with kidney disease.
PATHOPHYSIOLOGY OF HYPERPHOSPHATEMIA
The pathophysiology of bone mineral disorders in kidney disease is complex. To simplify the discussion, we will address it in 3 parts:
- Phosphorus balance
- The interplay of hormones, including fibroblast growth factor 23 (FGF23)
- The mechanism of hyperphosphatemia in kidney disease.
Phosphorus balance
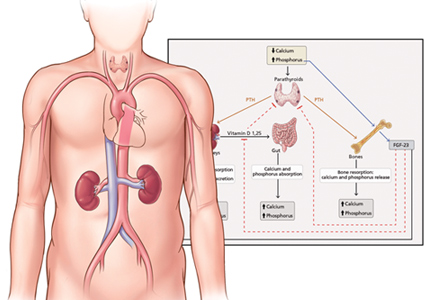
Phosphorus is a macronutrient essential for a range of cellular functions that include structure, energy production, metabolism, and cell signaling. It exists primarily in the form of inorganic phosphate.

An average Western diet provides 20 mg of phosphorus per kilogram of body weight per day. Of this, 13 mg/kg is absorbed, and the rest is excreted in the feces.2
Absorption of dietary phosphorus occurs mainly in the jejunum. It is mediated by both a paracellular sodium-independent pathway (driven by high intraluminal phosphorus content) and by active sodium-dependent cotransporters. It is also influenced by diet and promoted by active vitamin D (1,25 dihydroxyvitamin D3, also called calcitriol).3
Absorbed phosphorus enters the extracellular fluid and shifts in and out of the skeleton under the influence of parathyroid hormone.
Phosphorus excretion is handled almost entirely by the kidneys. Phosphorus is freely filtered at the glomerulus and reabsorbed mainly in the proximal tubule by sodium-phosphate cotransporters.
Normally, when phosphorus intake is adequate, most of the filtered phosphorus is reabsorbed and only 10% to 20% is excreted in the urine. However, the threshold for phosphorus reabsorption in the proximal tubule is influenced by parathyroid hormone, FGF23, and dietary phosphorus intake: low serum phosphate levels lead to an increase in the synthesis of sodium-phosphorus cotransporters, resulting in increased (nearly complete) proximal reabsorption and an increase in the serum phosphorus concentration.4 Conversely, both parathyroid hormone and FGF23 are phosphaturic and decrease the number of phosphorus transporters, which in turn leads to increased phosphorus excretion and a decrease in serum phosphorus concentration.5
Interplay of hormones
FGF23 is a phosphaturic glycoprotein secreted by osteoblasts and osteocytes. It acts by binding to fibroblastic growth receptor 1 in the presence of its coreceptor, the Klotho protein.6
FGF23 is regulated by serum phosphorus levels and plays a major role in the response to elevated serum phosphorus. It causes a direct increase in urinary phosphorus excretion, a decrease in intestinal phosphorus absorption (indirectly via inhibition of calcitriol), and decreased bone resorption via a decrease in parathyroid hormone production.7


Mechanism of hyperphosphatemia in kidney disease
In chronic kidney disease, phosphorus retention can trigger secondary hyperparathyroidism, as rising phosphorus levels stimulate FGF23. In the early stages of chronic kidney disease, this response can correct the phosphorus levels, but with several consequences:
- Decreased calcitriol due to its inhibition by FGF239
- Hypocalcemia due to decreased calcitriol (leading to decreased intestinal calcium absorption) and calcium binding of retained phosphorus
- Elevated parathyroid hormone due to low calcitriol levels (lack of inhibitory feedback by calcitriol), hyperphosphatemia, and hypocalcemia (direct parathyroid hormone stimulation).

As the elevated phosphorus level is likely to be the triggering event behind secondary renal hyperparathyroidism, it needs to be controlled. This is accomplished by restricting dietary phosphorus and using phosphorus binders.
HYPERPHOSPHATEMIA MAY LEAD TO VASCULAR CALCIFICATION
Elevated serum phosphorus levels (normal range 2.48–4.65 mg/dL in adults11) are associated with cardiovascular calcification and subsequent increases in mortality and morbidity rates. Elevations in serum phosphorus and calcium levels are associated with progression in vascular calcification12 and likely account for the accelerated vascular calcification that is seen in kidney disease.13
Hyperphosphatemia has been identified as an independent risk factor for death in patients with end-stage renal disease,14 but that relationship is less clear in patients with chronic kidney disease. A study in patients with chronic kidney disease and not on dialysis found a lower mortality rate in those who were prescribed phosphorus binders,15 but the study was criticized for limitations in its design.
Hyperphosphatemia can also lead to adverse effects on bone health due to complications such as renal osteodystrophy.
However, in its 2017 update, the Kidney Disease: Improving Global Outcomes (KDIGO) program only “suggests” lowering elevated phosphorus levels “toward” the normal range in patients with chronic kidney disease stages G3a through G5D, ie, those with glomerular filtration rates less than 60 mL/min/1.73 m2, including those on dialysis. The recommendation is graded 2C, ie, weak, based on low-quality evidence (https://kdigo.org/guidelines/ckd-mbd).
DIETARY RESTRICTION OF PHOSPHORUS
Diet is the major source of phosphorus intake. The average daily phosphorus consumption is 20 mg/kg, or 1,400 mg, and protein is the major source of dietary phosphorus.
In patients with stage 4 or 5 chronic kidney disease, the Kidney Disease Outcomes Quality Initiative recommends limiting protein intake to 0.6 mg/kg/day.16 However, in patients on hemodialysis, they recommend increasing protein intake to 1.1 mg/kg/day while limiting phosphorus intake to about 800 to 1,000 mg/day. This poses a challenge, as limiting phosphorus intake can reduce protein intake.

Sources of protein can be broadly classified as plant-based or animal-based. Animal protein contains organic phosphorus, which is easily absorbed.18 Plant protein may not be absorbed as easily.
Moe et al19 studied the importance of the protein source of phosphorus after 7 days of controlled diets. Despite equivalent protein and phosphorus concentrations in the vegetarian and meat-based diets, participants on the vegetarian diet had lower serum phosphorus levels, a trend toward lower 24-hour urinary phosphorus excretion, and significantly lower FGF23 levels than those on the meat-based diet. This suggests that a vegetarian diet may have advantages in terms of preventing hyperphosphatemia.
Another measure to reduce phosphorus absorption from meat is to boil it, which reduces the phosphorus content by 50%.20
Processed foods containing additives and preservatives are very high in phosphorus21 and should be avoided, particularly as there is no mandate to label phosphorus content in food.
PHOSPHORUS AND DIALYSIS
Although hemodialysis removes phosphorus, it does not remove enough to keep levels within normal limits. Indeed, even when patients adhere to a daily phosphorus limit of 1,000 mg, phosphorus accumulates. If 70% of the phosphorus in the diet is absorbed, this is 4,500 to 5,000 mg in a week. A 4-hour hemodialysis session will remove only 1,000 mg of phosphorus, which equals about 3,000 mg for patients undergoing dialysis 3 times a week,22 far less than phosphorus absorption.
In patients on continuous ambulatory peritoneal dialysis, a daily regimen of 4 exchanges of 2 L per exchange removes about 200 mg of phosphorus per day. In a 2012 study, patients on nocturnal dialysis or home dialysis involving longer session length had greater lowering of phosphorus levels than patients undergoing routine hemodialysis.23
Hence, phosphorus binders are often necessary in patients on routine hemodialysis or peritoneal dialysis.
PHOSPHORUS BINDERS
Phosphorus binders reduce serum phosphorus levels by binding with ingested phosphorus in the gastrointestinal tract and forming insoluble complexes that are not absorbed. For this reason they are much more effective when taken with meals. Phosphorus binders come in different formulations: pills, capsules, chewable tablets, liquids, and even powders that can be sprinkled on food.
The potency of each binder is quantified by its “phosphorus binder equivalent dose,” ie, its binding capacity compared with that of calcium carbonate as a reference.24
Phosphorus binders are broadly divided into those that contain calcium and those that do not.
Calcium-containing binders
The 2 most commonly used preparations are calcium carbonate (eg, Tums) and calcium acetate (eg, Phoslo). While these are relatively safe, some studies suggest that their use can lead to accelerated vascular calcification.25
According to KDIGO,26 calcium-containing binders should be avoided in hypercalcemia and adynamic bone disease. Additionally, the daily elemental calcium intake from binders should be limited to 1,500 mg, with a total daily intake that does not exceed 2,000 mg.
The elemental calcium content of calcium carbonate is about 40% of its weight (eg, 200 mg of elemental calcium in a 500-mg tablet of Tums), while the elemental calcium content of calcium acetate is about 25%. Therefore, a patient who needs 6 g of calcium carbonate for efficacy will be ingesting 2.4 g of elemental calcium per day, and that exceeds the recommended daily maximum. The main advantage of calcium carbonate is its low cost and easy availability. Commonly reported side effects include nausea and constipation.
A less commonly used calcium-based binder is calcium citrate (eg, Calcitrate). It should, however, be avoided in chronic kidney disease because of the risk of aluminum accumulation. Calcium citrate can enhance intestinal absorption of aluminum from dietary sources, as aluminum can form complexes with citrate.27
Calcium-free binders
There are several calcium-free binders. Some are based on metals such as aluminum, magnesium, iron, and lanthanum; others, such as sevelamer, are resin-based.
Aluminum- and magnesium-based binders are generally not used long-term in kidney disease because of the toxicity associated with aluminum and magnesium accumulation. However, aluminum hydroxide has an off-label use as a phosphorus binder in the acute setting, particularly when serum phosphorus levels are above 7 mg/dL.28 The dose is 300 to 600 mg 3 times daily with meals for a maximum of 4 weeks.
Sevelamer. Approved by the US Food and Drug Administration (FDA) in 1998, sevelamer acts by trapping phosphorus through ion exchange and hydrogen binding. It has the advantage of being calcium-free, which makes it particularly desirable in patients with hypercalcemia.
The Renagel in New Dialysis25 and Treat-To-Goal29 studies were randomized controlled trials that looked at the effects of sevelamer vs calcium-based binders on the risk of vascular calcification. The primary end points were serum phosphorus and calcium levels, while the secondary end points were coronary artery calcification on computed tomography and thoracic vertebral bone density. Both studies demonstrated a higher risk of vascular calcification with the calcium-based binders.
Another possible benefit of sevelamer is an improvement in lipid profile. Sevelamer lowers total cholesterol and low-density lipoprotein cholesterol levels without affecting high-density lipoprotein cholesterol or triglyceride levels.30 This is likely due to its bile acid-binding effect.31 Sevelamer has also been shown to lower C-reactive protein levels.32 While the cardiovascular profile appears to be improved with the treatment, there are no convincing data to confirm that those properties translate to a proven independent survival benefit.
The Calcium Acetate Renagel Evaluation33 was a randomized controlled study comparing sevelamer and calcium acetate. The authors attempted to control for the lipid-lowering effects of sevelamer by giving atorvastatin to all patients in both groups who had a low-density lipoprotein level greater than 70 mg/dL. The study found sevelamer to be not inferior to calcium acetate in terms of mortality and coronary calcification.
Further studies such as the Brazilian Renagel and Calcium trial34 and the Dialysis Clinical Outcomes Revisited trial failed to show a significant long-term benefit of sevelamer over calcium-based binders. However, a secondary statistical analysis of the latter study showed possible benefit of sevelamer over calcium acetate among those age 65 and older.35
To understand how sevelamer could affect vascular calcification, Yilmaz et al36 compared the effects of sevelamer vs calcium acetate on FGF23 and fetuin A levels. Fetuin A is an important inhibitor of vascular calcification and is progressively diminished in kidney disease, leading to accelerated calcification.37 Patients on sevelamer had higher levels of fetuin A than their counterparts on calcium acetate.37 The authors proposed increased fetuin A levels as a mechanism for decreased vascular calcification.
In summary, some studies suggest that sevelamer may offer the advantage of decreasing vascular calcification, but the data are mixed and do not provide a solid answer. The main disadvantages of sevelamer are a high pill burden and side effects of nausea and dyspepsia.
Lanthanum, a metallic element, was approved as a phosphorus binder by the FDA in 2008. It comes as a chewable tablet and offers the advantage of requiring the patient to take fewer pills than sevelamer and calcium-based binders.

Sucroferric oxyhydroxide comes as a chewable tablet. It was approved by the FDA in 2013. Although each tablet contains 500 mg of iron, it has not been shown to improve iron markers. In terms of phosphorus-lowering ability, it has been shown to be noninferior to sevelamer.39 Advantages include a significantly lower pill burden. Disadvantages include gastrointestinal side effects such as diarrhea and nausea and the drug’s high cost.
Ferric citrate was approved by the FDA in 2014, and 1 g delivers 210 mg of elemental iron. The main advantage of ferric citrate is its ability to increase iron markers. The phase 3 trial that demonstrated its efficacy as a binder showed an increase in ferritin compared with the active control.40 The study also showed a decrease in the need to use intravenous iron and erythropoesis-stimulating agents. This was thought to be due to improved iron stores, leading to decreased erythropoietin resistance.41
The mean number of ferric citrate tablets needed to achieve the desired phosphorus-lowering effect was 8 per day, containing 1,680 mg of iron. In comparison, oral ferrous sulfate typically provides 210 mg of iron per day.42
Disadvantages of ferric citrate include high pill burden, high cost, and gastrointestinal side effects such as nausea and constipation.
Chitosan binds salivary phosphorus. It can potentially be used, but it is not approved, and its efficacy in lowering serum phosphorus remains unclear.43
CHOOSING THE APPROPRIATE PHOSPHORUS BINDER
The choice of phosphorus binder is based on the patient’s serum calcium level and iron stores and on the drug’s side effect profile, iron pill burden, and cost. Involving patients in the choice after discussing potential side effects, pill burden, and cost is important for shared decision-making and could play a role in improving adherence.
Phosphorus binders are a major portion of the pill burden in patients with end-stage renal disease, possibly affecting patient adherence. The cost of phosphorus binders is estimated at half a billion dollars annually, underlining the significant economic impact of phosphorus control.11
Calcium-based binders should be the first choice when there is secondary hyperparathyroidism without hypercalcemia. There is no clear evidence regarding the benefit of correcting hypocalcemia, but KDIGO recommends keeping the serum calcium level within the reference range. KDIGO also recommends restricting calcium-based binders in persistent hypercalcemia, arterial calcification, and adynamic bone disease. This recommendation is largely based on expert opinion.
Noncalcium-based binders, which in theory might prevent vascular calcification, should be considered for patients with at least 1 of the following44:
- Complicated diabetes mellitus
- Vascular or valvular calcification
- Persistent inflammation.
Noncalcium-based binders are also preferred in low bone-turnover states such as adynamic bone disease, as elevated calcium can inhibit parathyroid hormone.
However, the advantage of noncalcium-based binders regarding vascular calcification is largely theoretical and has not been proven clinically. Indeed, there are data comparing long-term outcomes of the different classes of phosphorus binders, but studies were limited by short follow-up, and individual studies have lacked power to detect statistical significance between two classes of binders on long-term outcomes. Meta-analyses have provided conflicting data, with some suggesting better outcomes with sevelamer than with calcium-based binders, and with others failing to show any difference.45
Because iron deficiency is common in kidney disease, ferric citrate, which can improve iron markers, may be a suitable option, provided its cost is covered by insurance.
SPECIAL CIRCUMSTANCES FOR THE USE OF PHOSPHORUS BINDERS
Tumor lysis syndrome
Tumor lysis syndrome occurs when tumor cells release their contents into the bloodstream, either spontaneously or in response to therapy, leading to the characteristic findings of hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia.46 Phosphorus binders in conjunction with intravenous hydration are used to treat hyperphosphatemia, but evidence about their efficacy in this setting is limited.
Hypocalcemia in tumor lysis syndrome is usually not treated unless symptomatic, as the calcium-phosphorus product can increase, leading to calcium phosphate crystallization. When the calcium-phosphorus product is greater than 60, there is a higher risk of calcium phosphate deposition in the renal tubules that can lead to acute renal failure in tumor lysis syndrome.47 To lower the risk of calcium phosphate crystallization, calcium-based binders should be avoided in tumor lysis syndrome.
Total parenteral nutrition
Since patients on total parenteral nutrition do not eat, phosphorus binders are considered ineffective; there are no concrete data showing that phosphorus binders are effective in these patients.48 In patients with kidney disease, the phosphorus content in the parenteral nutrition formulation must be reduced.
Pregnancy
Data on phosphorus binders in pregnancy are limited. Calcium can cross the placenta. Calcium carbonate can be used in pregnancy, and fetal harm is not expected if calcium concentrations are within normal limits.49 Calcium acetate, sevelamer, and lanthanum are considered pregnancy category C drugs. Patients with advanced chronic kidney disease and end-stage renal disease who become pregnant must receive specialized obstetric care for high-risk pregnancy.
FUTURE DIRECTIONS
Future therapies may target FGF23 and other inflammatory markers that are up-regulated in renal hyperparathyroidism. However, trials studying these markers are needed to provide a better understanding of their role in bone mineral and cardiovascular health and in overall long-term outcomes. Additionally, randomized controlled trials are needed to study long-term nonsurrogate outcomes such as reduction in cardiovascular disease and rates of overall mortality.
- Collins AJ, Foley RN, Herzog C, et al. US renal data system 2012 annual data report. Am J Kidney Dis 2013; 61(1 suppl 1):A7,e1–476. doi:10.1053/j.ajkd.2012.11.031
- Tenenhouse HS. Regulation of phosphorus homeostasis by the type iia Na/phosphate cotransporter. Annu Rev Nutr 2005; 25:197–214. doi:10.1146/annurev.nutr.25.050304.092642
- Lederer E. Regulation of serum phosphate. J Physiol 2014; 592(18):3985–3995. doi:10.1113/jphysiol.2014.273979
- Lederer E. Renal phosphate transporters. Curr Opin Nephrol Hypertens 2014; 23(5):502–506. doi:10.1097/MNH.0000000000000053
- Weinman EJ, Lederer ED. NHERF-1 and the regulation of renal phosphate reabsoption: a tale of three hormones. Am J Physiol Renal Physiol 2012; 303(3):F321–F327. doi:10.1152/ajprenal.00093.2012
- Block GA, Ix JH, Ketteler M, et al. Phosphate homeostasis in CKD: report of a scientific symposium sponsored by the National Kidney Foundation. Am J Kidney Dis 2013; 62(3):457–473. doi:10.1053/j.ajkd.2013.03.042
- Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev 2012; 92(1):131–155. doi:10.1152/physrev.00002.2011
- Nissenson RA, Juppner H. Parathyroid hormone. In: Rosen CJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 8th ed. Ames, IA: Wiley-Blackwell; 2013:208–214.
- Chauhan V, Kelepouris E, Chauhan N, Vaid M. Current concepts and management strategies in chronic kidney disease-mineral and bone disorder. South Med J 2012; 105(9):479–485. doi:10.1097/SMJ.0b013e318261f7fe
- Slatopolsky E, Robson AM, Elkan I, Bricker NS. Control of phosphate excretion in uremic man. J Clin Invest 1968; 47(8):1865–1874. doi:10.1172/JCI105877
- Ritter CS, Slatopolsky E. Phosphate toxicity in CKD: the killer among us. Clin J Am Soc Nephrol 2016; 11(6):1088–1100. doi:10.2215/CJN.11901115
- Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 2004; 15(8):2208–2218. doi:10.1097/01.ASN.0000133041.27682.A2
- Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
- Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 1998; 31(4):607–617. pmid:9531176
- Bhandari SK, Liu IA, Kujubu DA, et al. Use of phosphorus binders among non-dialysis chronic kidney disease patients and mortality outcomes. Am J Nephrol 2017; 45(5):431–441. doi:10.1159/000474959
- Clinical practice guidelines for nutrition in chronic renal failure. K/DOQI, National Kidney Foundation. Am J Kidney Dis 2000; 35(6 suppl 2):S1–S140. pmid:10895784
- Streja E, Lau WL, Goldstein L, et al. Hyperphosphatemia is a combined function of high serum PTH and high dietary protein intake in dialysis patients. Kidney Int Suppl (2011) 2013; 3(5):462–468. doi:10.1038/kisup.2013.96
- Kalantar-Zadeh K, Gutekunst L, Mehrotra R, et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin J Am Soc Nephrol 2010; 5(3):519–530. doi:10.2215/CJN.06080809
- Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
- Cupisti A, Comar F, Benini O, et al. Effect of boiling on dietary phosphate and nitrogen intake. J Ren Nutr 2006; 16(1):36–40. doi:10.1053/j.jrn.2005.10.005
- Uribarri J, Calvo MS. Hidden sources of phosphorus in the typical American diet: does it matter in nephrology? Semin Dial 2003; 16(3):186–188. pmid:12753675
- Hou SH, Zhao J, Ellman CF, et al. Calcium and phosphorus fluxes during hemodialysis with low calcium dialysate. Am J Kidney Dis 1991; 18(2):217–224. pmid:1867178
- Daugirdas JT, Chertow GM, Larive B, et al; Frequent Hemodialysis Network (FHN) Trial Group. Effects of frequent hemodialysis on measures of CKD mineral and bone disorder. J Am Soc Nephrol 2012; 23(4):727–738. doi:10.1681/ASN.2011070688
- Daugirdas JT, Finn WF, Emmett M, Chertow GM; Frequent Hemodialysis Network Trial Group. The phosphate binder equivalent dose. Semin Dial 2011; 24(1):41–49. doi:10.1111/j.1525-139X.2011.00849.x
- Block GA, Spiegel DM, Ehrlich J, et al. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int 2005; 68(4):1815–1824. doi:10.1111/j.1523-1755.2005.00600.x
- National Kidney Foundation. KDOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(4 suppl 3):S1–S201. pmid:14520607
- Nolan CR, Califano JR, Butzin CA. Influence of calcium acetate or calcium citrate on intestinal aluminum absorption. Kidney Int 1990; 38(5):937–941. pmid:2266679
- Schucker JJ, Ward KE. Hyperphosphatemia and phosphate binders. Am J Health Syst Pharm 2005; 62(22):2355–2361. doi:10.2146/ajhp050198
- Chertow GM, Burke SK, Raggi P; Treat to Goal Working Group. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int 2002; 62(1):245–252. doi:10.1046/j.1523-1755.2002.00434.x
- Chertow GM, Burke SK, Dillon MA, Slatopolsky E. Long-term effects of sevelamer hydrochloride on the calcium x phosphate product and lipid profile of haemodialysis patients. Nephrol Dial Transplant 1999; 14(12):2907–2914. pmid:10570096
- Braunlin W, Zhorov E, Guo A, et al. Bile acid binding to sevelamer HCl. Kidney Int 2002; 62(2):611–619. doi:10.1046/j.1523-1755.2002.00459.x
- Yamada K, Fujimoto S, Tokura T, et al. Effect of sevelamer on dyslipidemia and chronic inflammation in maintenance hemodialysis patients. Ren Fail 2005; 27(4):361–365. pmid:16060120
- Qunibi W, Moustafa M, Muenz LR, et al; CARE-2 Investigators. A 1-year randomized trial of calcium acetate versus sevelamer on progression of coronary artery calcification in hemodialysis patients with comparable lipid control: the Calcium Acetate Renagel Evaluation-2 (CARE-2) study. Am J Kidney Dis 2008; 51(6):952–965. doi:10.1053/j.ajkd.2008.02.298
- Barreto DV, Barreto Fde C, de Carvalho AB, et al. Phosphate binder impact on bone remodeling and coronary calcification—results from the BRIC study. Nephron Clin Pract 2008; 110(4):c273–c283. doi:10.1159/000170783
- Cozzolino M, Mazzaferro S, Brandenburg V. The treatment of hyperphosphataemia in CKD: calcium-based or calcium-free phosphate binders? Nephrol Dial Transplant 2011; 26(2):402–407. doi:10.1093/ndt/gfq691
- Yilmaz MI, Sonmez A, Saglam M, et al. Comparison of calcium acetate and sevelamer on vascular function and fibroblast growth factor 23 in CKD patients: a randomized clinical trial. Am J Kidney Dis 2012; 59(2):177–185. doi:10.1053/j.ajkd.2011.11.007
- Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
- Hutchison AJ, Wilson RJ, Garafola S, Copley JB. Lanthanum carbonate: safety data after 10 years. Nephrology (Carlton) 2016; 21(12):987–994. doi:10.1111/nep.12864
- Floege J, Covic AC, Ketteler M, et al; PA21 Study Group. A phase III study of the efficacy and safety of a novel iron-based phosphate binder in dialysis patients. Kidney Int 2014; 86(3):638–647. doi:10.1038/ki.2014.58
- Lewis JB, Sika M, Koury MJ, et al; Collaborative Study Group. Ferric citrate controls phosphorus and delivers iron in patients on dialysis. J Am Soc Nephrol 2015; 26(2):493–503. doi:10.1681/ASN.2014020212
- Liu K, Kaffes AJ. Iron deficiency anemia: a review of diagnosis, investigation and management. Eur J Gastroenterol Hepatol 2012; 24(2):109–116. doi:10.1097/MEG.0b013e32834f3140
- Shah HH, Hazzan AD, Fishbane S. Novel iron-based phosphate binders in patients with chronic kidney disease. Curr Opin Nephrol Hypertens 2015; 24(4):330–335. doi:10.1097/MNH.0000000000000128
- Eknoyan G. Salivary phosphorus binding: a novel approach to control hyperphosphatemia. J Am Soc Nephrol 2009; 20(3):460–462. doi:10.1681/ASN.2009010067
- Raggi P, Vukicevic S, Moysés RM, Wesseling K, Spiegel DM. Ten-year experience with sevelamer and calcium salts as phosphate binders. Clin J Am Soc Nephrol 2010; 5(suppl 1):S31–S40. doi:10.2215/CJN.05880809
- Airy M, Winkelmayer WC, Navaneethan SD. Phosphate binders: the evidence gap persists. Am J Kidney Dis 2016; 68(5):667–670. doi:10.1053/j.ajkd.2016.08.008
- Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med 2011; 364(19):1844–1854. doi:10.1056/NEJMra0904569
- Van den Berg H, Reintsema AM. Renal tubular damage in rasburicase: risks of alkalinisation. Ann Oncol 2004; 15(1):175–176. pmid:14679140
- Suzuki NT. Hyperphosphatemia in nondialyzed TPN patients. JPEN J Parenter Enteral Nutr 1987; 11(5):512. doi:10.1177/0148607187011005512
- Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab 2011; 96(1):53–58. doi:10.1210/jc.2010-2704
The balance between dietary intake and excretion of phosphorus can be impaired in patients with decreased renal function, leading to hyperphosphatemia. Many patients with end-stage renal disease on dialysis require phosphorus-binding drugs to control their serum phosphorus levels.
See related editorial and article
In this review, we discuss the pathophysiology of hyperphosphatemia in kidney disease, its consequences, and how to control it, focusing on the different classes of phosphorus binders.
ROLE OF THE INTERNIST
With kidney disease common and on the increase,1 nephrologists and internists need to work together to provide optimal care.
Further, many internists in managed care plans and accountable care organizations now handle many tasks previously left to specialists—including prescribing and managing phosphorus binders in patients with kidney disease.
PATHOPHYSIOLOGY OF HYPERPHOSPHATEMIA
The pathophysiology of bone mineral disorders in kidney disease is complex. To simplify the discussion, we will address it in 3 parts:
- Phosphorus balance
- The interplay of hormones, including fibroblast growth factor 23 (FGF23)
- The mechanism of hyperphosphatemia in kidney disease.
Phosphorus balance
Phosphorus is a macronutrient essential for a range of cellular functions that include structure, energy production, metabolism, and cell signaling. It exists primarily in the form of inorganic phosphate.
An average Western diet provides 20 mg of phosphorus per kilogram of body weight per day. Of this, 13 mg/kg is absorbed, and the rest is excreted in the feces.2
Absorption of dietary phosphorus occurs mainly in the jejunum. It is mediated by both a paracellular sodium-independent pathway (driven by high intraluminal phosphorus content) and by active sodium-dependent cotransporters. It is also influenced by diet and promoted by active vitamin D (1,25 dihydroxyvitamin D3, also called calcitriol).3
Absorbed phosphorus enters the extracellular fluid and shifts in and out of the skeleton under the influence of parathyroid hormone.
Phosphorus excretion is handled almost entirely by the kidneys. Phosphorus is freely filtered at the glomerulus and reabsorbed mainly in the proximal tubule by sodium-phosphate cotransporters.
Normally, when phosphorus intake is adequate, most of the filtered phosphorus is reabsorbed and only 10% to 20% is excreted in the urine. However, the threshold for phosphorus reabsorption in the proximal tubule is influenced by parathyroid hormone, FGF23, and dietary phosphorus intake: low serum phosphate levels lead to an increase in the synthesis of sodium-phosphorus cotransporters, resulting in increased (nearly complete) proximal reabsorption and an increase in the serum phosphorus concentration.4 Conversely, both parathyroid hormone and FGF23 are phosphaturic and decrease the number of phosphorus transporters, which in turn leads to increased phosphorus excretion and a decrease in serum phosphorus concentration.5
Interplay of hormones
FGF23 is a phosphaturic glycoprotein secreted by osteoblasts and osteocytes. It acts by binding to fibroblastic growth receptor 1 in the presence of its coreceptor, the Klotho protein.6
FGF23 is regulated by serum phosphorus levels and plays a major role in the response to elevated serum phosphorus. It causes a direct increase in urinary phosphorus excretion, a decrease in intestinal phosphorus absorption (indirectly via inhibition of calcitriol), and decreased bone resorption via a decrease in parathyroid hormone production.7
Mechanism of hyperphosphatemia in kidney disease
In chronic kidney disease, phosphorus retention can trigger secondary hyperparathyroidism, as rising phosphorus levels stimulate FGF23. In the early stages of chronic kidney disease, this response can correct the phosphorus levels, but with several consequences:
- Decreased calcitriol due to its inhibition by FGF239
- Hypocalcemia due to decreased calcitriol (leading to decreased intestinal calcium absorption) and calcium binding of retained phosphorus
- Elevated parathyroid hormone due to low calcitriol levels (lack of inhibitory feedback by calcitriol), hyperphosphatemia, and hypocalcemia (direct parathyroid hormone stimulation).
As the elevated phosphorus level is likely to be the triggering event behind secondary renal hyperparathyroidism, it needs to be controlled. This is accomplished by restricting dietary phosphorus and using phosphorus binders.
HYPERPHOSPHATEMIA MAY LEAD TO VASCULAR CALCIFICATION
Elevated serum phosphorus levels (normal range 2.48–4.65 mg/dL in adults11) are associated with cardiovascular calcification and subsequent increases in mortality and morbidity rates. Elevations in serum phosphorus and calcium levels are associated with progression in vascular calcification12 and likely account for the accelerated vascular calcification that is seen in kidney disease.13
Hyperphosphatemia has been identified as an independent risk factor for death in patients with end-stage renal disease,14 but that relationship is less clear in patients with chronic kidney disease. A study in patients with chronic kidney disease and not on dialysis found a lower mortality rate in those who were prescribed phosphorus binders,15 but the study was criticized for limitations in its design.
Hyperphosphatemia can also lead to adverse effects on bone health due to complications such as renal osteodystrophy.
However, in its 2017 update, the Kidney Disease: Improving Global Outcomes (KDIGO) program only “suggests” lowering elevated phosphorus levels “toward” the normal range in patients with chronic kidney disease stages G3a through G5D, ie, those with glomerular filtration rates less than 60 mL/min/1.73 m2, including those on dialysis. The recommendation is graded 2C, ie, weak, based on low-quality evidence (https://kdigo.org/guidelines/ckd-mbd).
DIETARY RESTRICTION OF PHOSPHORUS
Diet is the major source of phosphorus intake. The average daily phosphorus consumption is 20 mg/kg, or 1,400 mg, and protein is the major source of dietary phosphorus.
In patients with stage 4 or 5 chronic kidney disease, the Kidney Disease Outcomes Quality Initiative recommends limiting protein intake to 0.6 mg/kg/day.16 However, in patients on hemodialysis, they recommend increasing protein intake to 1.1 mg/kg/day while limiting phosphorus intake to about 800 to 1,000 mg/day. This poses a challenge, as limiting phosphorus intake can reduce protein intake.
Sources of protein can be broadly classified as plant-based or animal-based. Animal protein contains organic phosphorus, which is easily absorbed.18 Plant protein may not be absorbed as easily.
Moe et al19 studied the importance of the protein source of phosphorus after 7 days of controlled diets. Despite equivalent protein and phosphorus concentrations in the vegetarian and meat-based diets, participants on the vegetarian diet had lower serum phosphorus levels, a trend toward lower 24-hour urinary phosphorus excretion, and significantly lower FGF23 levels than those on the meat-based diet. This suggests that a vegetarian diet may have advantages in terms of preventing hyperphosphatemia.
Another measure to reduce phosphorus absorption from meat is to boil it, which reduces the phosphorus content by 50%.20
Processed foods containing additives and preservatives are very high in phosphorus21 and should be avoided, particularly as there is no mandate to label phosphorus content in food.
PHOSPHORUS AND DIALYSIS
Although hemodialysis removes phosphorus, it does not remove enough to keep levels within normal limits. Indeed, even when patients adhere to a daily phosphorus limit of 1,000 mg, phosphorus accumulates. If 70% of the phosphorus in the diet is absorbed, this is 4,500 to 5,000 mg in a week. A 4-hour hemodialysis session will remove only 1,000 mg of phosphorus, which equals about 3,000 mg for patients undergoing dialysis 3 times a week,22 far less than phosphorus absorption.
In patients on continuous ambulatory peritoneal dialysis, a daily regimen of 4 exchanges of 2 L per exchange removes about 200 mg of phosphorus per day. In a 2012 study, patients on nocturnal dialysis or home dialysis involving longer session length had greater lowering of phosphorus levels than patients undergoing routine hemodialysis.23
Hence, phosphorus binders are often necessary in patients on routine hemodialysis or peritoneal dialysis.
PHOSPHORUS BINDERS
Phosphorus binders reduce serum phosphorus levels by binding with ingested phosphorus in the gastrointestinal tract and forming insoluble complexes that are not absorbed. For this reason they are much more effective when taken with meals. Phosphorus binders come in different formulations: pills, capsules, chewable tablets, liquids, and even powders that can be sprinkled on food.
The potency of each binder is quantified by its “phosphorus binder equivalent dose,” ie, its binding capacity compared with that of calcium carbonate as a reference.24
Phosphorus binders are broadly divided into those that contain calcium and those that do not.
Calcium-containing binders
The 2 most commonly used preparations are calcium carbonate (eg, Tums) and calcium acetate (eg, Phoslo). While these are relatively safe, some studies suggest that their use can lead to accelerated vascular calcification.25
According to KDIGO,26 calcium-containing binders should be avoided in hypercalcemia and adynamic bone disease. Additionally, the daily elemental calcium intake from binders should be limited to 1,500 mg, with a total daily intake that does not exceed 2,000 mg.
The elemental calcium content of calcium carbonate is about 40% of its weight (eg, 200 mg of elemental calcium in a 500-mg tablet of Tums), while the elemental calcium content of calcium acetate is about 25%. Therefore, a patient who needs 6 g of calcium carbonate for efficacy will be ingesting 2.4 g of elemental calcium per day, and that exceeds the recommended daily maximum. The main advantage of calcium carbonate is its low cost and easy availability. Commonly reported side effects include nausea and constipation.
A less commonly used calcium-based binder is calcium citrate (eg, Calcitrate). It should, however, be avoided in chronic kidney disease because of the risk of aluminum accumulation. Calcium citrate can enhance intestinal absorption of aluminum from dietary sources, as aluminum can form complexes with citrate.27
Calcium-free binders
There are several calcium-free binders. Some are based on metals such as aluminum, magnesium, iron, and lanthanum; others, such as sevelamer, are resin-based.
Aluminum- and magnesium-based binders are generally not used long-term in kidney disease because of the toxicity associated with aluminum and magnesium accumulation. However, aluminum hydroxide has an off-label use as a phosphorus binder in the acute setting, particularly when serum phosphorus levels are above 7 mg/dL.28 The dose is 300 to 600 mg 3 times daily with meals for a maximum of 4 weeks.
Sevelamer. Approved by the US Food and Drug Administration (FDA) in 1998, sevelamer acts by trapping phosphorus through ion exchange and hydrogen binding. It has the advantage of being calcium-free, which makes it particularly desirable in patients with hypercalcemia.
The Renagel in New Dialysis25 and Treat-To-Goal29 studies were randomized controlled trials that looked at the effects of sevelamer vs calcium-based binders on the risk of vascular calcification. The primary end points were serum phosphorus and calcium levels, while the secondary end points were coronary artery calcification on computed tomography and thoracic vertebral bone density. Both studies demonstrated a higher risk of vascular calcification with the calcium-based binders.
Another possible benefit of sevelamer is an improvement in lipid profile. Sevelamer lowers total cholesterol and low-density lipoprotein cholesterol levels without affecting high-density lipoprotein cholesterol or triglyceride levels.30 This is likely due to its bile acid-binding effect.31 Sevelamer has also been shown to lower C-reactive protein levels.32 While the cardiovascular profile appears to be improved with the treatment, there are no convincing data to confirm that those properties translate to a proven independent survival benefit.
The Calcium Acetate Renagel Evaluation33 was a randomized controlled study comparing sevelamer and calcium acetate. The authors attempted to control for the lipid-lowering effects of sevelamer by giving atorvastatin to all patients in both groups who had a low-density lipoprotein level greater than 70 mg/dL. The study found sevelamer to be not inferior to calcium acetate in terms of mortality and coronary calcification.
Further studies such as the Brazilian Renagel and Calcium trial34 and the Dialysis Clinical Outcomes Revisited trial failed to show a significant long-term benefit of sevelamer over calcium-based binders. However, a secondary statistical analysis of the latter study showed possible benefit of sevelamer over calcium acetate among those age 65 and older.35
To understand how sevelamer could affect vascular calcification, Yilmaz et al36 compared the effects of sevelamer vs calcium acetate on FGF23 and fetuin A levels. Fetuin A is an important inhibitor of vascular calcification and is progressively diminished in kidney disease, leading to accelerated calcification.37 Patients on sevelamer had higher levels of fetuin A than their counterparts on calcium acetate.37 The authors proposed increased fetuin A levels as a mechanism for decreased vascular calcification.
In summary, some studies suggest that sevelamer may offer the advantage of decreasing vascular calcification, but the data are mixed and do not provide a solid answer. The main disadvantages of sevelamer are a high pill burden and side effects of nausea and dyspepsia.
Lanthanum, a metallic element, was approved as a phosphorus binder by the FDA in 2008. It comes as a chewable tablet and offers the advantage of requiring the patient to take fewer pills than sevelamer and calcium-based binders.
Sucroferric oxyhydroxide comes as a chewable tablet. It was approved by the FDA in 2013. Although each tablet contains 500 mg of iron, it has not been shown to improve iron markers. In terms of phosphorus-lowering ability, it has been shown to be noninferior to sevelamer.39 Advantages include a significantly lower pill burden. Disadvantages include gastrointestinal side effects such as diarrhea and nausea and the drug’s high cost.
Ferric citrate was approved by the FDA in 2014, and 1 g delivers 210 mg of elemental iron. The main advantage of ferric citrate is its ability to increase iron markers. The phase 3 trial that demonstrated its efficacy as a binder showed an increase in ferritin compared with the active control.40 The study also showed a decrease in the need to use intravenous iron and erythropoesis-stimulating agents. This was thought to be due to improved iron stores, leading to decreased erythropoietin resistance.41
The mean number of ferric citrate tablets needed to achieve the desired phosphorus-lowering effect was 8 per day, containing 1,680 mg of iron. In comparison, oral ferrous sulfate typically provides 210 mg of iron per day.42
Disadvantages of ferric citrate include high pill burden, high cost, and gastrointestinal side effects such as nausea and constipation.
Chitosan binds salivary phosphorus. It can potentially be used, but it is not approved, and its efficacy in lowering serum phosphorus remains unclear.43
CHOOSING THE APPROPRIATE PHOSPHORUS BINDER
The choice of phosphorus binder is based on the patient’s serum calcium level and iron stores and on the drug’s side effect profile, iron pill burden, and cost. Involving patients in the choice after discussing potential side effects, pill burden, and cost is important for shared decision-making and could play a role in improving adherence.
Phosphorus binders are a major portion of the pill burden in patients with end-stage renal disease, possibly affecting patient adherence. The cost of phosphorus binders is estimated at half a billion dollars annually, underlining the significant economic impact of phosphorus control.11
Calcium-based binders should be the first choice when there is secondary hyperparathyroidism without hypercalcemia. There is no clear evidence regarding the benefit of correcting hypocalcemia, but KDIGO recommends keeping the serum calcium level within the reference range. KDIGO also recommends restricting calcium-based binders in persistent hypercalcemia, arterial calcification, and adynamic bone disease. This recommendation is largely based on expert opinion.
Noncalcium-based binders, which in theory might prevent vascular calcification, should be considered for patients with at least 1 of the following44:
- Complicated diabetes mellitus
- Vascular or valvular calcification
- Persistent inflammation.
Noncalcium-based binders are also preferred in low bone-turnover states such as adynamic bone disease, as elevated calcium can inhibit parathyroid hormone.
However, the advantage of noncalcium-based binders regarding vascular calcification is largely theoretical and has not been proven clinically. Indeed, there are data comparing long-term outcomes of the different classes of phosphorus binders, but studies were limited by short follow-up, and individual studies have lacked power to detect statistical significance between two classes of binders on long-term outcomes. Meta-analyses have provided conflicting data, with some suggesting better outcomes with sevelamer than with calcium-based binders, and with others failing to show any difference.45
Because iron deficiency is common in kidney disease, ferric citrate, which can improve iron markers, may be a suitable option, provided its cost is covered by insurance.
SPECIAL CIRCUMSTANCES FOR THE USE OF PHOSPHORUS BINDERS
Tumor lysis syndrome
Tumor lysis syndrome occurs when tumor cells release their contents into the bloodstream, either spontaneously or in response to therapy, leading to the characteristic findings of hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia.46 Phosphorus binders in conjunction with intravenous hydration are used to treat hyperphosphatemia, but evidence about their efficacy in this setting is limited.
Hypocalcemia in tumor lysis syndrome is usually not treated unless symptomatic, as the calcium-phosphorus product can increase, leading to calcium phosphate crystallization. When the calcium-phosphorus product is greater than 60, there is a higher risk of calcium phosphate deposition in the renal tubules that can lead to acute renal failure in tumor lysis syndrome.47 To lower the risk of calcium phosphate crystallization, calcium-based binders should be avoided in tumor lysis syndrome.
Total parenteral nutrition
Since patients on total parenteral nutrition do not eat, phosphorus binders are considered ineffective; there are no concrete data showing that phosphorus binders are effective in these patients.48 In patients with kidney disease, the phosphorus content in the parenteral nutrition formulation must be reduced.
Pregnancy
Data on phosphorus binders in pregnancy are limited. Calcium can cross the placenta. Calcium carbonate can be used in pregnancy, and fetal harm is not expected if calcium concentrations are within normal limits.49 Calcium acetate, sevelamer, and lanthanum are considered pregnancy category C drugs. Patients with advanced chronic kidney disease and end-stage renal disease who become pregnant must receive specialized obstetric care for high-risk pregnancy.
FUTURE DIRECTIONS
Future therapies may target FGF23 and other inflammatory markers that are up-regulated in renal hyperparathyroidism. However, trials studying these markers are needed to provide a better understanding of their role in bone mineral and cardiovascular health and in overall long-term outcomes. Additionally, randomized controlled trials are needed to study long-term nonsurrogate outcomes such as reduction in cardiovascular disease and rates of overall mortality.
The balance between dietary intake and excretion of phosphorus can be impaired in patients with decreased renal function, leading to hyperphosphatemia. Many patients with end-stage renal disease on dialysis require phosphorus-binding drugs to control their serum phosphorus levels.
See related editorial and article
In this review, we discuss the pathophysiology of hyperphosphatemia in kidney disease, its consequences, and how to control it, focusing on the different classes of phosphorus binders.
ROLE OF THE INTERNIST
With kidney disease common and on the increase,1 nephrologists and internists need to work together to provide optimal care.
Further, many internists in managed care plans and accountable care organizations now handle many tasks previously left to specialists—including prescribing and managing phosphorus binders in patients with kidney disease.
PATHOPHYSIOLOGY OF HYPERPHOSPHATEMIA
The pathophysiology of bone mineral disorders in kidney disease is complex. To simplify the discussion, we will address it in 3 parts:
- Phosphorus balance
- The interplay of hormones, including fibroblast growth factor 23 (FGF23)
- The mechanism of hyperphosphatemia in kidney disease.
Phosphorus balance
Phosphorus is a macronutrient essential for a range of cellular functions that include structure, energy production, metabolism, and cell signaling. It exists primarily in the form of inorganic phosphate.
An average Western diet provides 20 mg of phosphorus per kilogram of body weight per day. Of this, 13 mg/kg is absorbed, and the rest is excreted in the feces.2
Absorption of dietary phosphorus occurs mainly in the jejunum. It is mediated by both a paracellular sodium-independent pathway (driven by high intraluminal phosphorus content) and by active sodium-dependent cotransporters. It is also influenced by diet and promoted by active vitamin D (1,25 dihydroxyvitamin D3, also called calcitriol).3
Absorbed phosphorus enters the extracellular fluid and shifts in and out of the skeleton under the influence of parathyroid hormone.
Phosphorus excretion is handled almost entirely by the kidneys. Phosphorus is freely filtered at the glomerulus and reabsorbed mainly in the proximal tubule by sodium-phosphate cotransporters.
Normally, when phosphorus intake is adequate, most of the filtered phosphorus is reabsorbed and only 10% to 20% is excreted in the urine. However, the threshold for phosphorus reabsorption in the proximal tubule is influenced by parathyroid hormone, FGF23, and dietary phosphorus intake: low serum phosphate levels lead to an increase in the synthesis of sodium-phosphorus cotransporters, resulting in increased (nearly complete) proximal reabsorption and an increase in the serum phosphorus concentration.4 Conversely, both parathyroid hormone and FGF23 are phosphaturic and decrease the number of phosphorus transporters, which in turn leads to increased phosphorus excretion and a decrease in serum phosphorus concentration.5
Interplay of hormones
FGF23 is a phosphaturic glycoprotein secreted by osteoblasts and osteocytes. It acts by binding to fibroblastic growth receptor 1 in the presence of its coreceptor, the Klotho protein.6
FGF23 is regulated by serum phosphorus levels and plays a major role in the response to elevated serum phosphorus. It causes a direct increase in urinary phosphorus excretion, a decrease in intestinal phosphorus absorption (indirectly via inhibition of calcitriol), and decreased bone resorption via a decrease in parathyroid hormone production.7
Mechanism of hyperphosphatemia in kidney disease
In chronic kidney disease, phosphorus retention can trigger secondary hyperparathyroidism, as rising phosphorus levels stimulate FGF23. In the early stages of chronic kidney disease, this response can correct the phosphorus levels, but with several consequences:
- Decreased calcitriol due to its inhibition by FGF239
- Hypocalcemia due to decreased calcitriol (leading to decreased intestinal calcium absorption) and calcium binding of retained phosphorus
- Elevated parathyroid hormone due to low calcitriol levels (lack of inhibitory feedback by calcitriol), hyperphosphatemia, and hypocalcemia (direct parathyroid hormone stimulation).
As the elevated phosphorus level is likely to be the triggering event behind secondary renal hyperparathyroidism, it needs to be controlled. This is accomplished by restricting dietary phosphorus and using phosphorus binders.
HYPERPHOSPHATEMIA MAY LEAD TO VASCULAR CALCIFICATION
Elevated serum phosphorus levels (normal range 2.48–4.65 mg/dL in adults11) are associated with cardiovascular calcification and subsequent increases in mortality and morbidity rates. Elevations in serum phosphorus and calcium levels are associated with progression in vascular calcification12 and likely account for the accelerated vascular calcification that is seen in kidney disease.13
Hyperphosphatemia has been identified as an independent risk factor for death in patients with end-stage renal disease,14 but that relationship is less clear in patients with chronic kidney disease. A study in patients with chronic kidney disease and not on dialysis found a lower mortality rate in those who were prescribed phosphorus binders,15 but the study was criticized for limitations in its design.
Hyperphosphatemia can also lead to adverse effects on bone health due to complications such as renal osteodystrophy.
However, in its 2017 update, the Kidney Disease: Improving Global Outcomes (KDIGO) program only “suggests” lowering elevated phosphorus levels “toward” the normal range in patients with chronic kidney disease stages G3a through G5D, ie, those with glomerular filtration rates less than 60 mL/min/1.73 m2, including those on dialysis. The recommendation is graded 2C, ie, weak, based on low-quality evidence (https://kdigo.org/guidelines/ckd-mbd).
DIETARY RESTRICTION OF PHOSPHORUS
Diet is the major source of phosphorus intake. The average daily phosphorus consumption is 20 mg/kg, or 1,400 mg, and protein is the major source of dietary phosphorus.
In patients with stage 4 or 5 chronic kidney disease, the Kidney Disease Outcomes Quality Initiative recommends limiting protein intake to 0.6 mg/kg/day.16 However, in patients on hemodialysis, they recommend increasing protein intake to 1.1 mg/kg/day while limiting phosphorus intake to about 800 to 1,000 mg/day. This poses a challenge, as limiting phosphorus intake can reduce protein intake.
Sources of protein can be broadly classified as plant-based or animal-based. Animal protein contains organic phosphorus, which is easily absorbed.18 Plant protein may not be absorbed as easily.
Moe et al19 studied the importance of the protein source of phosphorus after 7 days of controlled diets. Despite equivalent protein and phosphorus concentrations in the vegetarian and meat-based diets, participants on the vegetarian diet had lower serum phosphorus levels, a trend toward lower 24-hour urinary phosphorus excretion, and significantly lower FGF23 levels than those on the meat-based diet. This suggests that a vegetarian diet may have advantages in terms of preventing hyperphosphatemia.
Another measure to reduce phosphorus absorption from meat is to boil it, which reduces the phosphorus content by 50%.20
Processed foods containing additives and preservatives are very high in phosphorus21 and should be avoided, particularly as there is no mandate to label phosphorus content in food.
PHOSPHORUS AND DIALYSIS
Although hemodialysis removes phosphorus, it does not remove enough to keep levels within normal limits. Indeed, even when patients adhere to a daily phosphorus limit of 1,000 mg, phosphorus accumulates. If 70% of the phosphorus in the diet is absorbed, this is 4,500 to 5,000 mg in a week. A 4-hour hemodialysis session will remove only 1,000 mg of phosphorus, which equals about 3,000 mg for patients undergoing dialysis 3 times a week,22 far less than phosphorus absorption.
In patients on continuous ambulatory peritoneal dialysis, a daily regimen of 4 exchanges of 2 L per exchange removes about 200 mg of phosphorus per day. In a 2012 study, patients on nocturnal dialysis or home dialysis involving longer session length had greater lowering of phosphorus levels than patients undergoing routine hemodialysis.23
Hence, phosphorus binders are often necessary in patients on routine hemodialysis or peritoneal dialysis.
PHOSPHORUS BINDERS
Phosphorus binders reduce serum phosphorus levels by binding with ingested phosphorus in the gastrointestinal tract and forming insoluble complexes that are not absorbed. For this reason they are much more effective when taken with meals. Phosphorus binders come in different formulations: pills, capsules, chewable tablets, liquids, and even powders that can be sprinkled on food.
The potency of each binder is quantified by its “phosphorus binder equivalent dose,” ie, its binding capacity compared with that of calcium carbonate as a reference.24
Phosphorus binders are broadly divided into those that contain calcium and those that do not.
Calcium-containing binders
The 2 most commonly used preparations are calcium carbonate (eg, Tums) and calcium acetate (eg, Phoslo). While these are relatively safe, some studies suggest that their use can lead to accelerated vascular calcification.25
According to KDIGO,26 calcium-containing binders should be avoided in hypercalcemia and adynamic bone disease. Additionally, the daily elemental calcium intake from binders should be limited to 1,500 mg, with a total daily intake that does not exceed 2,000 mg.
The elemental calcium content of calcium carbonate is about 40% of its weight (eg, 200 mg of elemental calcium in a 500-mg tablet of Tums), while the elemental calcium content of calcium acetate is about 25%. Therefore, a patient who needs 6 g of calcium carbonate for efficacy will be ingesting 2.4 g of elemental calcium per day, and that exceeds the recommended daily maximum. The main advantage of calcium carbonate is its low cost and easy availability. Commonly reported side effects include nausea and constipation.
A less commonly used calcium-based binder is calcium citrate (eg, Calcitrate). It should, however, be avoided in chronic kidney disease because of the risk of aluminum accumulation. Calcium citrate can enhance intestinal absorption of aluminum from dietary sources, as aluminum can form complexes with citrate.27
Calcium-free binders
There are several calcium-free binders. Some are based on metals such as aluminum, magnesium, iron, and lanthanum; others, such as sevelamer, are resin-based.
Aluminum- and magnesium-based binders are generally not used long-term in kidney disease because of the toxicity associated with aluminum and magnesium accumulation. However, aluminum hydroxide has an off-label use as a phosphorus binder in the acute setting, particularly when serum phosphorus levels are above 7 mg/dL.28 The dose is 300 to 600 mg 3 times daily with meals for a maximum of 4 weeks.
Sevelamer. Approved by the US Food and Drug Administration (FDA) in 1998, sevelamer acts by trapping phosphorus through ion exchange and hydrogen binding. It has the advantage of being calcium-free, which makes it particularly desirable in patients with hypercalcemia.
The Renagel in New Dialysis25 and Treat-To-Goal29 studies were randomized controlled trials that looked at the effects of sevelamer vs calcium-based binders on the risk of vascular calcification. The primary end points were serum phosphorus and calcium levels, while the secondary end points were coronary artery calcification on computed tomography and thoracic vertebral bone density. Both studies demonstrated a higher risk of vascular calcification with the calcium-based binders.
Another possible benefit of sevelamer is an improvement in lipid profile. Sevelamer lowers total cholesterol and low-density lipoprotein cholesterol levels without affecting high-density lipoprotein cholesterol or triglyceride levels.30 This is likely due to its bile acid-binding effect.31 Sevelamer has also been shown to lower C-reactive protein levels.32 While the cardiovascular profile appears to be improved with the treatment, there are no convincing data to confirm that those properties translate to a proven independent survival benefit.
The Calcium Acetate Renagel Evaluation33 was a randomized controlled study comparing sevelamer and calcium acetate. The authors attempted to control for the lipid-lowering effects of sevelamer by giving atorvastatin to all patients in both groups who had a low-density lipoprotein level greater than 70 mg/dL. The study found sevelamer to be not inferior to calcium acetate in terms of mortality and coronary calcification.
Further studies such as the Brazilian Renagel and Calcium trial34 and the Dialysis Clinical Outcomes Revisited trial failed to show a significant long-term benefit of sevelamer over calcium-based binders. However, a secondary statistical analysis of the latter study showed possible benefit of sevelamer over calcium acetate among those age 65 and older.35
To understand how sevelamer could affect vascular calcification, Yilmaz et al36 compared the effects of sevelamer vs calcium acetate on FGF23 and fetuin A levels. Fetuin A is an important inhibitor of vascular calcification and is progressively diminished in kidney disease, leading to accelerated calcification.37 Patients on sevelamer had higher levels of fetuin A than their counterparts on calcium acetate.37 The authors proposed increased fetuin A levels as a mechanism for decreased vascular calcification.
In summary, some studies suggest that sevelamer may offer the advantage of decreasing vascular calcification, but the data are mixed and do not provide a solid answer. The main disadvantages of sevelamer are a high pill burden and side effects of nausea and dyspepsia.
Lanthanum, a metallic element, was approved as a phosphorus binder by the FDA in 2008. It comes as a chewable tablet and offers the advantage of requiring the patient to take fewer pills than sevelamer and calcium-based binders.
Sucroferric oxyhydroxide comes as a chewable tablet. It was approved by the FDA in 2013. Although each tablet contains 500 mg of iron, it has not been shown to improve iron markers. In terms of phosphorus-lowering ability, it has been shown to be noninferior to sevelamer.39 Advantages include a significantly lower pill burden. Disadvantages include gastrointestinal side effects such as diarrhea and nausea and the drug’s high cost.
Ferric citrate was approved by the FDA in 2014, and 1 g delivers 210 mg of elemental iron. The main advantage of ferric citrate is its ability to increase iron markers. The phase 3 trial that demonstrated its efficacy as a binder showed an increase in ferritin compared with the active control.40 The study also showed a decrease in the need to use intravenous iron and erythropoesis-stimulating agents. This was thought to be due to improved iron stores, leading to decreased erythropoietin resistance.41
The mean number of ferric citrate tablets needed to achieve the desired phosphorus-lowering effect was 8 per day, containing 1,680 mg of iron. In comparison, oral ferrous sulfate typically provides 210 mg of iron per day.42
Disadvantages of ferric citrate include high pill burden, high cost, and gastrointestinal side effects such as nausea and constipation.
Chitosan binds salivary phosphorus. It can potentially be used, but it is not approved, and its efficacy in lowering serum phosphorus remains unclear.43
CHOOSING THE APPROPRIATE PHOSPHORUS BINDER
The choice of phosphorus binder is based on the patient’s serum calcium level and iron stores and on the drug’s side effect profile, iron pill burden, and cost. Involving patients in the choice after discussing potential side effects, pill burden, and cost is important for shared decision-making and could play a role in improving adherence.
Phosphorus binders are a major portion of the pill burden in patients with end-stage renal disease, possibly affecting patient adherence. The cost of phosphorus binders is estimated at half a billion dollars annually, underlining the significant economic impact of phosphorus control.11
Calcium-based binders should be the first choice when there is secondary hyperparathyroidism without hypercalcemia. There is no clear evidence regarding the benefit of correcting hypocalcemia, but KDIGO recommends keeping the serum calcium level within the reference range. KDIGO also recommends restricting calcium-based binders in persistent hypercalcemia, arterial calcification, and adynamic bone disease. This recommendation is largely based on expert opinion.
Noncalcium-based binders, which in theory might prevent vascular calcification, should be considered for patients with at least 1 of the following44:
- Complicated diabetes mellitus
- Vascular or valvular calcification
- Persistent inflammation.
Noncalcium-based binders are also preferred in low bone-turnover states such as adynamic bone disease, as elevated calcium can inhibit parathyroid hormone.
However, the advantage of noncalcium-based binders regarding vascular calcification is largely theoretical and has not been proven clinically. Indeed, there are data comparing long-term outcomes of the different classes of phosphorus binders, but studies were limited by short follow-up, and individual studies have lacked power to detect statistical significance between two classes of binders on long-term outcomes. Meta-analyses have provided conflicting data, with some suggesting better outcomes with sevelamer than with calcium-based binders, and with others failing to show any difference.45
Because iron deficiency is common in kidney disease, ferric citrate, which can improve iron markers, may be a suitable option, provided its cost is covered by insurance.
SPECIAL CIRCUMSTANCES FOR THE USE OF PHOSPHORUS BINDERS
Tumor lysis syndrome
Tumor lysis syndrome occurs when tumor cells release their contents into the bloodstream, either spontaneously or in response to therapy, leading to the characteristic findings of hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia.46 Phosphorus binders in conjunction with intravenous hydration are used to treat hyperphosphatemia, but evidence about their efficacy in this setting is limited.
Hypocalcemia in tumor lysis syndrome is usually not treated unless symptomatic, as the calcium-phosphorus product can increase, leading to calcium phosphate crystallization. When the calcium-phosphorus product is greater than 60, there is a higher risk of calcium phosphate deposition in the renal tubules that can lead to acute renal failure in tumor lysis syndrome.47 To lower the risk of calcium phosphate crystallization, calcium-based binders should be avoided in tumor lysis syndrome.
Total parenteral nutrition
Since patients on total parenteral nutrition do not eat, phosphorus binders are considered ineffective; there are no concrete data showing that phosphorus binders are effective in these patients.48 In patients with kidney disease, the phosphorus content in the parenteral nutrition formulation must be reduced.
Pregnancy
Data on phosphorus binders in pregnancy are limited. Calcium can cross the placenta. Calcium carbonate can be used in pregnancy, and fetal harm is not expected if calcium concentrations are within normal limits.49 Calcium acetate, sevelamer, and lanthanum are considered pregnancy category C drugs. Patients with advanced chronic kidney disease and end-stage renal disease who become pregnant must receive specialized obstetric care for high-risk pregnancy.
FUTURE DIRECTIONS
Future therapies may target FGF23 and other inflammatory markers that are up-regulated in renal hyperparathyroidism. However, trials studying these markers are needed to provide a better understanding of their role in bone mineral and cardiovascular health and in overall long-term outcomes. Additionally, randomized controlled trials are needed to study long-term nonsurrogate outcomes such as reduction in cardiovascular disease and rates of overall mortality.
- Collins AJ, Foley RN, Herzog C, et al. US renal data system 2012 annual data report. Am J Kidney Dis 2013; 61(1 suppl 1):A7,e1–476. doi:10.1053/j.ajkd.2012.11.031
- Tenenhouse HS. Regulation of phosphorus homeostasis by the type iia Na/phosphate cotransporter. Annu Rev Nutr 2005; 25:197–214. doi:10.1146/annurev.nutr.25.050304.092642
- Lederer E. Regulation of serum phosphate. J Physiol 2014; 592(18):3985–3995. doi:10.1113/jphysiol.2014.273979
- Lederer E. Renal phosphate transporters. Curr Opin Nephrol Hypertens 2014; 23(5):502–506. doi:10.1097/MNH.0000000000000053
- Weinman EJ, Lederer ED. NHERF-1 and the regulation of renal phosphate reabsoption: a tale of three hormones. Am J Physiol Renal Physiol 2012; 303(3):F321–F327. doi:10.1152/ajprenal.00093.2012
- Block GA, Ix JH, Ketteler M, et al. Phosphate homeostasis in CKD: report of a scientific symposium sponsored by the National Kidney Foundation. Am J Kidney Dis 2013; 62(3):457–473. doi:10.1053/j.ajkd.2013.03.042
- Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev 2012; 92(1):131–155. doi:10.1152/physrev.00002.2011
- Nissenson RA, Juppner H. Parathyroid hormone. In: Rosen CJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 8th ed. Ames, IA: Wiley-Blackwell; 2013:208–214.
- Chauhan V, Kelepouris E, Chauhan N, Vaid M. Current concepts and management strategies in chronic kidney disease-mineral and bone disorder. South Med J 2012; 105(9):479–485. doi:10.1097/SMJ.0b013e318261f7fe
- Slatopolsky E, Robson AM, Elkan I, Bricker NS. Control of phosphate excretion in uremic man. J Clin Invest 1968; 47(8):1865–1874. doi:10.1172/JCI105877
- Ritter CS, Slatopolsky E. Phosphate toxicity in CKD: the killer among us. Clin J Am Soc Nephrol 2016; 11(6):1088–1100. doi:10.2215/CJN.11901115
- Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 2004; 15(8):2208–2218. doi:10.1097/01.ASN.0000133041.27682.A2
- Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
- Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 1998; 31(4):607–617. pmid:9531176
- Bhandari SK, Liu IA, Kujubu DA, et al. Use of phosphorus binders among non-dialysis chronic kidney disease patients and mortality outcomes. Am J Nephrol 2017; 45(5):431–441. doi:10.1159/000474959
- Clinical practice guidelines for nutrition in chronic renal failure. K/DOQI, National Kidney Foundation. Am J Kidney Dis 2000; 35(6 suppl 2):S1–S140. pmid:10895784
- Streja E, Lau WL, Goldstein L, et al. Hyperphosphatemia is a combined function of high serum PTH and high dietary protein intake in dialysis patients. Kidney Int Suppl (2011) 2013; 3(5):462–468. doi:10.1038/kisup.2013.96
- Kalantar-Zadeh K, Gutekunst L, Mehrotra R, et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin J Am Soc Nephrol 2010; 5(3):519–530. doi:10.2215/CJN.06080809
- Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
- Cupisti A, Comar F, Benini O, et al. Effect of boiling on dietary phosphate and nitrogen intake. J Ren Nutr 2006; 16(1):36–40. doi:10.1053/j.jrn.2005.10.005
- Uribarri J, Calvo MS. Hidden sources of phosphorus in the typical American diet: does it matter in nephrology? Semin Dial 2003; 16(3):186–188. pmid:12753675
- Hou SH, Zhao J, Ellman CF, et al. Calcium and phosphorus fluxes during hemodialysis with low calcium dialysate. Am J Kidney Dis 1991; 18(2):217–224. pmid:1867178
- Daugirdas JT, Chertow GM, Larive B, et al; Frequent Hemodialysis Network (FHN) Trial Group. Effects of frequent hemodialysis on measures of CKD mineral and bone disorder. J Am Soc Nephrol 2012; 23(4):727–738. doi:10.1681/ASN.2011070688
- Daugirdas JT, Finn WF, Emmett M, Chertow GM; Frequent Hemodialysis Network Trial Group. The phosphate binder equivalent dose. Semin Dial 2011; 24(1):41–49. doi:10.1111/j.1525-139X.2011.00849.x
- Block GA, Spiegel DM, Ehrlich J, et al. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int 2005; 68(4):1815–1824. doi:10.1111/j.1523-1755.2005.00600.x
- National Kidney Foundation. KDOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(4 suppl 3):S1–S201. pmid:14520607
- Nolan CR, Califano JR, Butzin CA. Influence of calcium acetate or calcium citrate on intestinal aluminum absorption. Kidney Int 1990; 38(5):937–941. pmid:2266679
- Schucker JJ, Ward KE. Hyperphosphatemia and phosphate binders. Am J Health Syst Pharm 2005; 62(22):2355–2361. doi:10.2146/ajhp050198
- Chertow GM, Burke SK, Raggi P; Treat to Goal Working Group. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int 2002; 62(1):245–252. doi:10.1046/j.1523-1755.2002.00434.x
- Chertow GM, Burke SK, Dillon MA, Slatopolsky E. Long-term effects of sevelamer hydrochloride on the calcium x phosphate product and lipid profile of haemodialysis patients. Nephrol Dial Transplant 1999; 14(12):2907–2914. pmid:10570096
- Braunlin W, Zhorov E, Guo A, et al. Bile acid binding to sevelamer HCl. Kidney Int 2002; 62(2):611–619. doi:10.1046/j.1523-1755.2002.00459.x
- Yamada K, Fujimoto S, Tokura T, et al. Effect of sevelamer on dyslipidemia and chronic inflammation in maintenance hemodialysis patients. Ren Fail 2005; 27(4):361–365. pmid:16060120
- Qunibi W, Moustafa M, Muenz LR, et al; CARE-2 Investigators. A 1-year randomized trial of calcium acetate versus sevelamer on progression of coronary artery calcification in hemodialysis patients with comparable lipid control: the Calcium Acetate Renagel Evaluation-2 (CARE-2) study. Am J Kidney Dis 2008; 51(6):952–965. doi:10.1053/j.ajkd.2008.02.298
- Barreto DV, Barreto Fde C, de Carvalho AB, et al. Phosphate binder impact on bone remodeling and coronary calcification—results from the BRIC study. Nephron Clin Pract 2008; 110(4):c273–c283. doi:10.1159/000170783
- Cozzolino M, Mazzaferro S, Brandenburg V. The treatment of hyperphosphataemia in CKD: calcium-based or calcium-free phosphate binders? Nephrol Dial Transplant 2011; 26(2):402–407. doi:10.1093/ndt/gfq691
- Yilmaz MI, Sonmez A, Saglam M, et al. Comparison of calcium acetate and sevelamer on vascular function and fibroblast growth factor 23 in CKD patients: a randomized clinical trial. Am J Kidney Dis 2012; 59(2):177–185. doi:10.1053/j.ajkd.2011.11.007
- Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
- Hutchison AJ, Wilson RJ, Garafola S, Copley JB. Lanthanum carbonate: safety data after 10 years. Nephrology (Carlton) 2016; 21(12):987–994. doi:10.1111/nep.12864
- Floege J, Covic AC, Ketteler M, et al; PA21 Study Group. A phase III study of the efficacy and safety of a novel iron-based phosphate binder in dialysis patients. Kidney Int 2014; 86(3):638–647. doi:10.1038/ki.2014.58
- Lewis JB, Sika M, Koury MJ, et al; Collaborative Study Group. Ferric citrate controls phosphorus and delivers iron in patients on dialysis. J Am Soc Nephrol 2015; 26(2):493–503. doi:10.1681/ASN.2014020212
- Liu K, Kaffes AJ. Iron deficiency anemia: a review of diagnosis, investigation and management. Eur J Gastroenterol Hepatol 2012; 24(2):109–116. doi:10.1097/MEG.0b013e32834f3140
- Shah HH, Hazzan AD, Fishbane S. Novel iron-based phosphate binders in patients with chronic kidney disease. Curr Opin Nephrol Hypertens 2015; 24(4):330–335. doi:10.1097/MNH.0000000000000128
- Eknoyan G. Salivary phosphorus binding: a novel approach to control hyperphosphatemia. J Am Soc Nephrol 2009; 20(3):460–462. doi:10.1681/ASN.2009010067
- Raggi P, Vukicevic S, Moysés RM, Wesseling K, Spiegel DM. Ten-year experience with sevelamer and calcium salts as phosphate binders. Clin J Am Soc Nephrol 2010; 5(suppl 1):S31–S40. doi:10.2215/CJN.05880809
- Airy M, Winkelmayer WC, Navaneethan SD. Phosphate binders: the evidence gap persists. Am J Kidney Dis 2016; 68(5):667–670. doi:10.1053/j.ajkd.2016.08.008
- Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med 2011; 364(19):1844–1854. doi:10.1056/NEJMra0904569
- Van den Berg H, Reintsema AM. Renal tubular damage in rasburicase: risks of alkalinisation. Ann Oncol 2004; 15(1):175–176. pmid:14679140
- Suzuki NT. Hyperphosphatemia in nondialyzed TPN patients. JPEN J Parenter Enteral Nutr 1987; 11(5):512. doi:10.1177/0148607187011005512
- Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab 2011; 96(1):53–58. doi:10.1210/jc.2010-2704
- Collins AJ, Foley RN, Herzog C, et al. US renal data system 2012 annual data report. Am J Kidney Dis 2013; 61(1 suppl 1):A7,e1–476. doi:10.1053/j.ajkd.2012.11.031
- Tenenhouse HS. Regulation of phosphorus homeostasis by the type iia Na/phosphate cotransporter. Annu Rev Nutr 2005; 25:197–214. doi:10.1146/annurev.nutr.25.050304.092642
- Lederer E. Regulation of serum phosphate. J Physiol 2014; 592(18):3985–3995. doi:10.1113/jphysiol.2014.273979
- Lederer E. Renal phosphate transporters. Curr Opin Nephrol Hypertens 2014; 23(5):502–506. doi:10.1097/MNH.0000000000000053
- Weinman EJ, Lederer ED. NHERF-1 and the regulation of renal phosphate reabsoption: a tale of three hormones. Am J Physiol Renal Physiol 2012; 303(3):F321–F327. doi:10.1152/ajprenal.00093.2012
- Block GA, Ix JH, Ketteler M, et al. Phosphate homeostasis in CKD: report of a scientific symposium sponsored by the National Kidney Foundation. Am J Kidney Dis 2013; 62(3):457–473. doi:10.1053/j.ajkd.2013.03.042
- Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev 2012; 92(1):131–155. doi:10.1152/physrev.00002.2011
- Nissenson RA, Juppner H. Parathyroid hormone. In: Rosen CJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 8th ed. Ames, IA: Wiley-Blackwell; 2013:208–214.
- Chauhan V, Kelepouris E, Chauhan N, Vaid M. Current concepts and management strategies in chronic kidney disease-mineral and bone disorder. South Med J 2012; 105(9):479–485. doi:10.1097/SMJ.0b013e318261f7fe
- Slatopolsky E, Robson AM, Elkan I, Bricker NS. Control of phosphate excretion in uremic man. J Clin Invest 1968; 47(8):1865–1874. doi:10.1172/JCI105877
- Ritter CS, Slatopolsky E. Phosphate toxicity in CKD: the killer among us. Clin J Am Soc Nephrol 2016; 11(6):1088–1100. doi:10.2215/CJN.11901115
- Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 2004; 15(8):2208–2218. doi:10.1097/01.ASN.0000133041.27682.A2
- Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
- Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 1998; 31(4):607–617. pmid:9531176
- Bhandari SK, Liu IA, Kujubu DA, et al. Use of phosphorus binders among non-dialysis chronic kidney disease patients and mortality outcomes. Am J Nephrol 2017; 45(5):431–441. doi:10.1159/000474959
- Clinical practice guidelines for nutrition in chronic renal failure. K/DOQI, National Kidney Foundation. Am J Kidney Dis 2000; 35(6 suppl 2):S1–S140. pmid:10895784
- Streja E, Lau WL, Goldstein L, et al. Hyperphosphatemia is a combined function of high serum PTH and high dietary protein intake in dialysis patients. Kidney Int Suppl (2011) 2013; 3(5):462–468. doi:10.1038/kisup.2013.96
- Kalantar-Zadeh K, Gutekunst L, Mehrotra R, et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin J Am Soc Nephrol 2010; 5(3):519–530. doi:10.2215/CJN.06080809
- Moe SM, Zidehsarai MP, Chambers MA, et al. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 2011; 6(2):257–264. doi:10.2215/CJN.05040610
- Cupisti A, Comar F, Benini O, et al. Effect of boiling on dietary phosphate and nitrogen intake. J Ren Nutr 2006; 16(1):36–40. doi:10.1053/j.jrn.2005.10.005
- Uribarri J, Calvo MS. Hidden sources of phosphorus in the typical American diet: does it matter in nephrology? Semin Dial 2003; 16(3):186–188. pmid:12753675
- Hou SH, Zhao J, Ellman CF, et al. Calcium and phosphorus fluxes during hemodialysis with low calcium dialysate. Am J Kidney Dis 1991; 18(2):217–224. pmid:1867178
- Daugirdas JT, Chertow GM, Larive B, et al; Frequent Hemodialysis Network (FHN) Trial Group. Effects of frequent hemodialysis on measures of CKD mineral and bone disorder. J Am Soc Nephrol 2012; 23(4):727–738. doi:10.1681/ASN.2011070688
- Daugirdas JT, Finn WF, Emmett M, Chertow GM; Frequent Hemodialysis Network Trial Group. The phosphate binder equivalent dose. Semin Dial 2011; 24(1):41–49. doi:10.1111/j.1525-139X.2011.00849.x
- Block GA, Spiegel DM, Ehrlich J, et al. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int 2005; 68(4):1815–1824. doi:10.1111/j.1523-1755.2005.00600.x
- National Kidney Foundation. KDOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(4 suppl 3):S1–S201. pmid:14520607
- Nolan CR, Califano JR, Butzin CA. Influence of calcium acetate or calcium citrate on intestinal aluminum absorption. Kidney Int 1990; 38(5):937–941. pmid:2266679
- Schucker JJ, Ward KE. Hyperphosphatemia and phosphate binders. Am J Health Syst Pharm 2005; 62(22):2355–2361. doi:10.2146/ajhp050198
- Chertow GM, Burke SK, Raggi P; Treat to Goal Working Group. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int 2002; 62(1):245–252. doi:10.1046/j.1523-1755.2002.00434.x
- Chertow GM, Burke SK, Dillon MA, Slatopolsky E. Long-term effects of sevelamer hydrochloride on the calcium x phosphate product and lipid profile of haemodialysis patients. Nephrol Dial Transplant 1999; 14(12):2907–2914. pmid:10570096
- Braunlin W, Zhorov E, Guo A, et al. Bile acid binding to sevelamer HCl. Kidney Int 2002; 62(2):611–619. doi:10.1046/j.1523-1755.2002.00459.x
- Yamada K, Fujimoto S, Tokura T, et al. Effect of sevelamer on dyslipidemia and chronic inflammation in maintenance hemodialysis patients. Ren Fail 2005; 27(4):361–365. pmid:16060120
- Qunibi W, Moustafa M, Muenz LR, et al; CARE-2 Investigators. A 1-year randomized trial of calcium acetate versus sevelamer on progression of coronary artery calcification in hemodialysis patients with comparable lipid control: the Calcium Acetate Renagel Evaluation-2 (CARE-2) study. Am J Kidney Dis 2008; 51(6):952–965. doi:10.1053/j.ajkd.2008.02.298
- Barreto DV, Barreto Fde C, de Carvalho AB, et al. Phosphate binder impact on bone remodeling and coronary calcification—results from the BRIC study. Nephron Clin Pract 2008; 110(4):c273–c283. doi:10.1159/000170783
- Cozzolino M, Mazzaferro S, Brandenburg V. The treatment of hyperphosphataemia in CKD: calcium-based or calcium-free phosphate binders? Nephrol Dial Transplant 2011; 26(2):402–407. doi:10.1093/ndt/gfq691
- Yilmaz MI, Sonmez A, Saglam M, et al. Comparison of calcium acetate and sevelamer on vascular function and fibroblast growth factor 23 in CKD patients: a randomized clinical trial. Am J Kidney Dis 2012; 59(2):177–185. doi:10.1053/j.ajkd.2011.11.007
- Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21(1):103–112. doi:10.1681/ASN.2009060640
- Hutchison AJ, Wilson RJ, Garafola S, Copley JB. Lanthanum carbonate: safety data after 10 years. Nephrology (Carlton) 2016; 21(12):987–994. doi:10.1111/nep.12864
- Floege J, Covic AC, Ketteler M, et al; PA21 Study Group. A phase III study of the efficacy and safety of a novel iron-based phosphate binder in dialysis patients. Kidney Int 2014; 86(3):638–647. doi:10.1038/ki.2014.58
- Lewis JB, Sika M, Koury MJ, et al; Collaborative Study Group. Ferric citrate controls phosphorus and delivers iron in patients on dialysis. J Am Soc Nephrol 2015; 26(2):493–503. doi:10.1681/ASN.2014020212
- Liu K, Kaffes AJ. Iron deficiency anemia: a review of diagnosis, investigation and management. Eur J Gastroenterol Hepatol 2012; 24(2):109–116. doi:10.1097/MEG.0b013e32834f3140
- Shah HH, Hazzan AD, Fishbane S. Novel iron-based phosphate binders in patients with chronic kidney disease. Curr Opin Nephrol Hypertens 2015; 24(4):330–335. doi:10.1097/MNH.0000000000000128
- Eknoyan G. Salivary phosphorus binding: a novel approach to control hyperphosphatemia. J Am Soc Nephrol 2009; 20(3):460–462. doi:10.1681/ASN.2009010067
- Raggi P, Vukicevic S, Moysés RM, Wesseling K, Spiegel DM. Ten-year experience with sevelamer and calcium salts as phosphate binders. Clin J Am Soc Nephrol 2010; 5(suppl 1):S31–S40. doi:10.2215/CJN.05880809
- Airy M, Winkelmayer WC, Navaneethan SD. Phosphate binders: the evidence gap persists. Am J Kidney Dis 2016; 68(5):667–670. doi:10.1053/j.ajkd.2016.08.008
- Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med 2011; 364(19):1844–1854. doi:10.1056/NEJMra0904569
- Van den Berg H, Reintsema AM. Renal tubular damage in rasburicase: risks of alkalinisation. Ann Oncol 2004; 15(1):175–176. pmid:14679140
- Suzuki NT. Hyperphosphatemia in nondialyzed TPN patients. JPEN J Parenter Enteral Nutr 1987; 11(5):512. doi:10.1177/0148607187011005512
- Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab 2011; 96(1):53–58. doi:10.1210/jc.2010-2704
KEY POINTS
- Serum phosphorus is maintained within normal levels in a tightly regulated system involving interplay between organs, hormones, diet, and other factors.
- Dietary phosphorus comes mainly from protein, so restricting phosphorus without introducing protein deficiency is difficult. Food with a low phosphorus-to-protein ratio and plant-based sources of protein may be preferable.
- Although dialysis removes phosphorus, it usually does not remove enough, and many patients require phosphorus-binding drugs.
- Selection of an appropriate binder should consider serum calcium levels, pill burden, serum iron stores, and cost.