User login
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
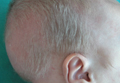
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
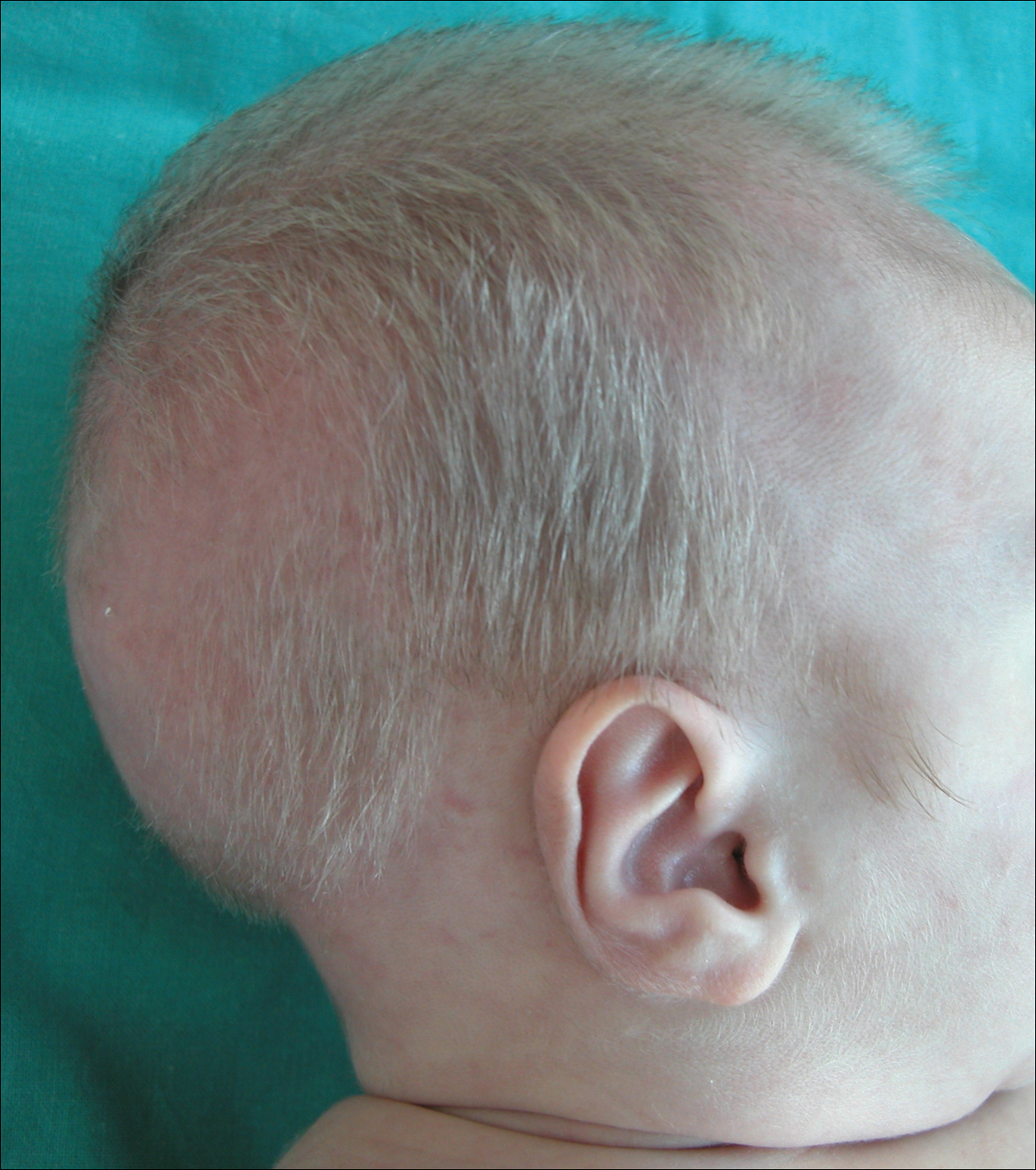
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.


Comment
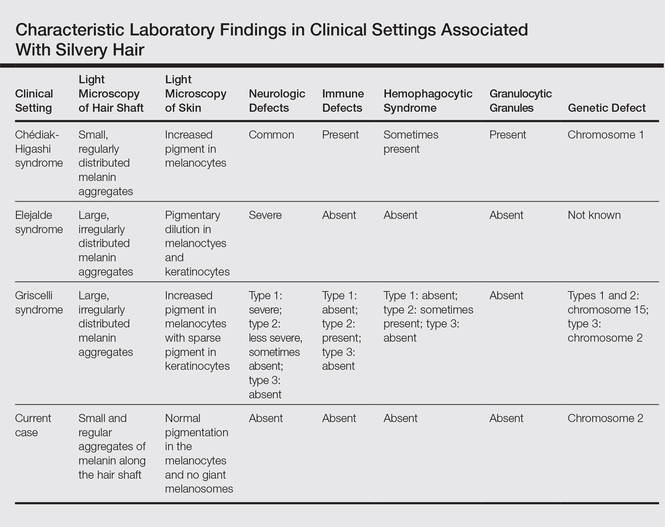
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
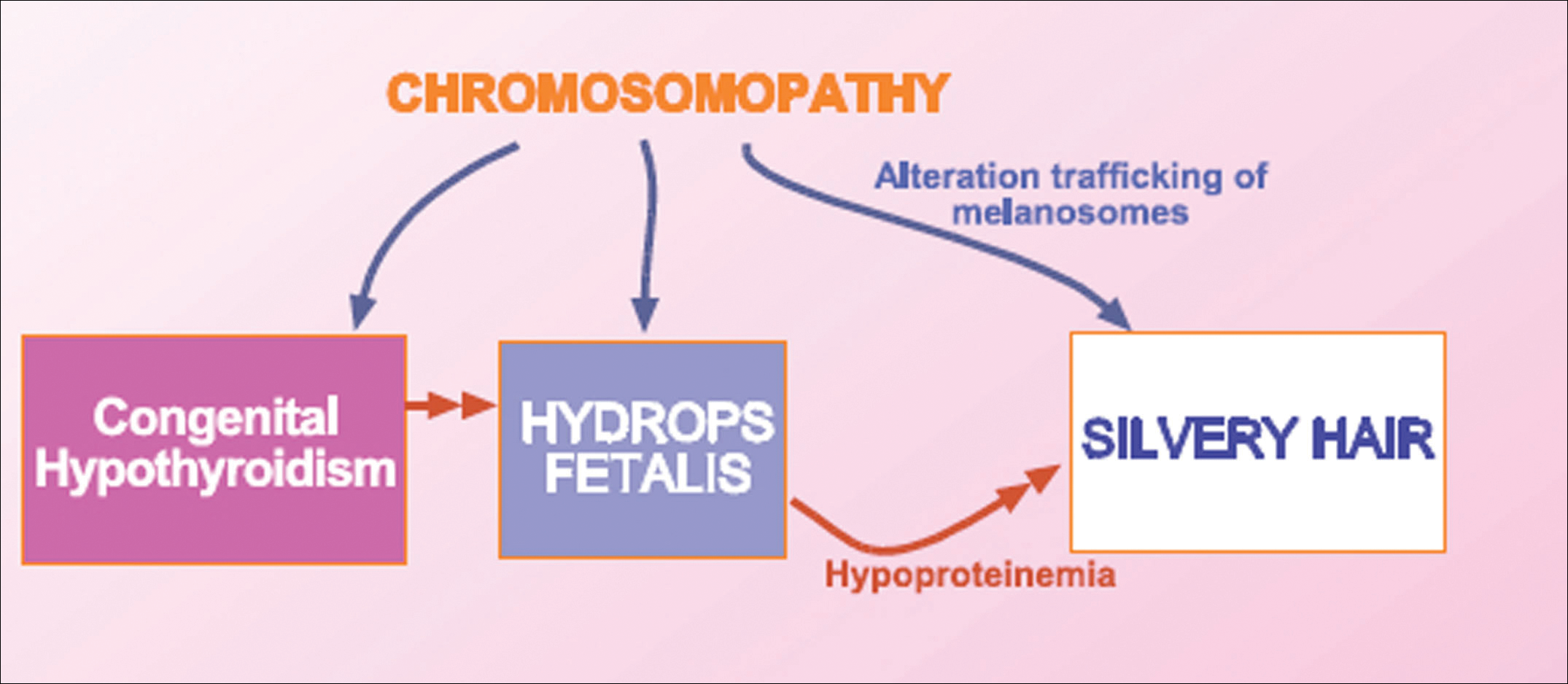
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.
Comment
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.
Comment
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
Practice Points
- Silvery hair is characteristic of 3 rare autosomal-recessive disorders: Chédiak-Higashi syndrome, Elejalde syndrome, and Griscelli syndrome.
- Hypopigmentation is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes.
- Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.