User login
The Diagnosis: Granular Cell Tumor
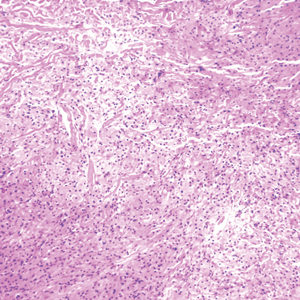
Histopathologic analysis from the axillary excision demonstrated cords and sheets of large polygonal cells in the dermis with uniform, oval, hyperchromatic nuclei and ample pink granular-staining cytoplasm (quiz images). An infiltrative growth pattern was noted; however, there was no evidence of conspicuous mitoses, nuclear pleomorphism, or necrosis. These results in conjunction with the immunohistochemistry findings were consistent with a benign granular cell tumor (GCT), a rare neoplasm considered to have neural/Schwann cell origin.1-3
Our case demonstrates the difficulty in clinically diagnosing cutaneous GCTs. The tumor often presents as a solitary, 0.5- to 3-cm, asymptomatic, firm nodule4,5; however, GCTs also can appear verrucous, eroded, or with other variable morphologies, which can create diagnostic challenges.5,6 Accordingly, a 1980 study of 110 patients with GCTs found that the preoperative clinical diagnosis was incorrect in all but 3 cases,7 emphasizing the need for histologic evaluation. Benign GCTs tend to exhibit sheets of polygonal tumor cells with eosinophilic granular cytoplasm and small central nuclei.3,5 The cytoplasmic granules are periodic acid-Schiff positive and diastase resistant.6 Many cases feature pseudoepitheliomatous hyperplasia, which can misleadingly resemble squamous cell carcinoma.3,5,6 Of note, invasive growth patterns on histology can occur with benign GCTs, as in our patient's case, and do not impact prognosis.3,4 On immunohistochemistry, benign, atypical, and malignant GCTs often stain positive for S-100 protein, vimentin, neuron-specific enolase, SOX10, and CD68.1,3
Although our patient's GCTs were benign, an estimated 1% to 2% are malignant.1,4 In 1998, Fanburg-Smith et al1 defined 6 histologic criteria that characterize malignant GCTs: necrosis, tumor cell spindling, vesicular nuclei with large nucleoli, high nuclear to cytoplasmic ratio, increased mitosis, and pleomorphism. Neoplasms with 3 or more of these features are classified as malignant, those with 1 or 2 are considered atypical, and those with only pleomorphism or no other criteria met are diagnosed as benign.1
Multiple GCTs have been reported in 10% to 25% of cases and, as highlighted in our case, can occur in both a metachronous and synchronous manner.2-4,6 Our patient developed a solitary GCT on the inferior lip 3 years prior to the appearance of 2 additional GCTs within 6 months of each other. The presence of multiple GCTs has been associated with genetic syndromes, such as neurofibromatosis type 1 and Noonan syndrome with multiple lentigines3,8; however, as our case demonstrates, multiple GCTs can occur in nonsyndromic patients as well. When multiple GCTs develop at distant sites, they can resemble metastasis.3 To differentiate these clinical scenarios, Machado et al3 proposed utilizing histology and anatomic location. Multiple tumors with benign characteristics on histology likely represent multiple GCTs, whereas tumors arising at sites common to GCT metastasis, such as lymph node, bone, or viscera, are more concerning for metastatic disease. It has been suggested that patients with multiple GCTs should be monitored with physical examination and repeat magnetic resonance imaging or computed tomography every 6 to 12 months.2 Given our patient's presentation with new tumors arising within 6 months of one another, we recommended a 6-month follow-up interval rather than 1 year. Due to the rarity of GCTs, clinical trials to define treatment guidelines and recommendations have not been performed.3 However, the most commonly utilized treatment modality is wide local excision, as performed in our patient.2,4
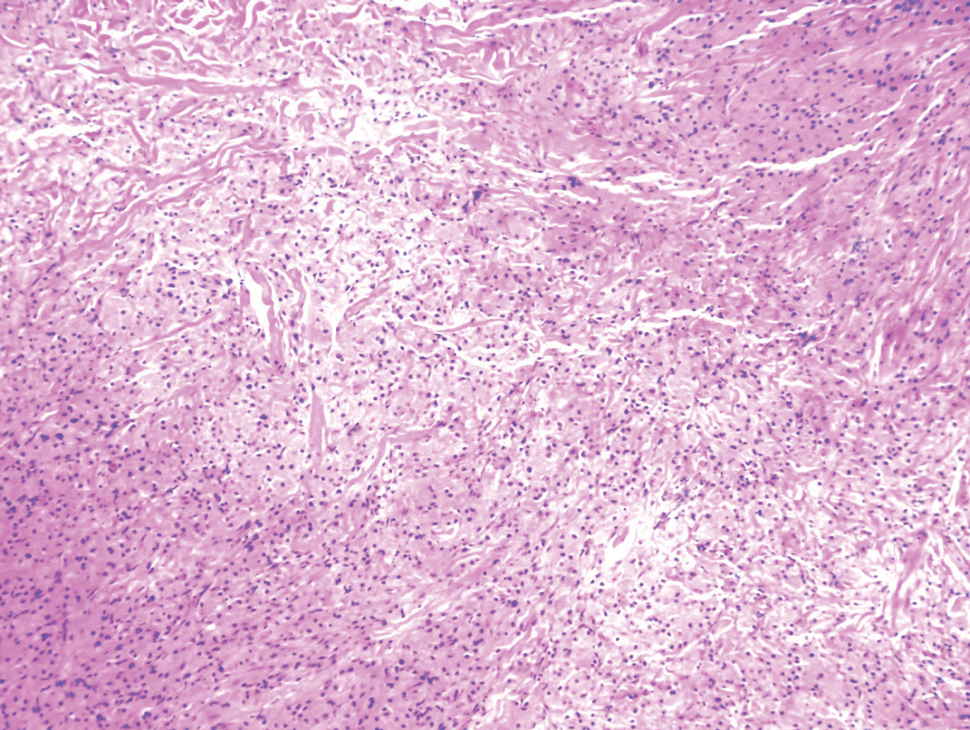
Melanoma, atypical fibroxanthoma (AFX), xanthoma, and leiomyosarcoma may be difficult to distinguish from GCT.1,3,4 Melanoma incidence has increased dramatically over the last several decades, with rates in the United States rising from 6.8 cases per 100,000 individuals in the 1970s to 20.1 in the early 2000s. Risk factors for its development include UV radiation exposure and particularly severe sunburns during childhood, along with a number of host risk factors such as total number of melanocytic nevi, family history, and fair complexion.9 Histologically, it often demonstrates irregularly distributed, poorly defined melanocytes with pagetoid spread and dyscohesive nests (Figure 1).10 Melanoma metastasis occasionally can present as a soft-tissue mass and often stains positive for S-100 and vimentin, thus resembling GCT1,4; however, unlike melanoma, GCTs lack melanosomes and stain negative for more specific melanocyte markers, such as melanoma antigen recognized by T cells 1 (MART-1).1,3,4
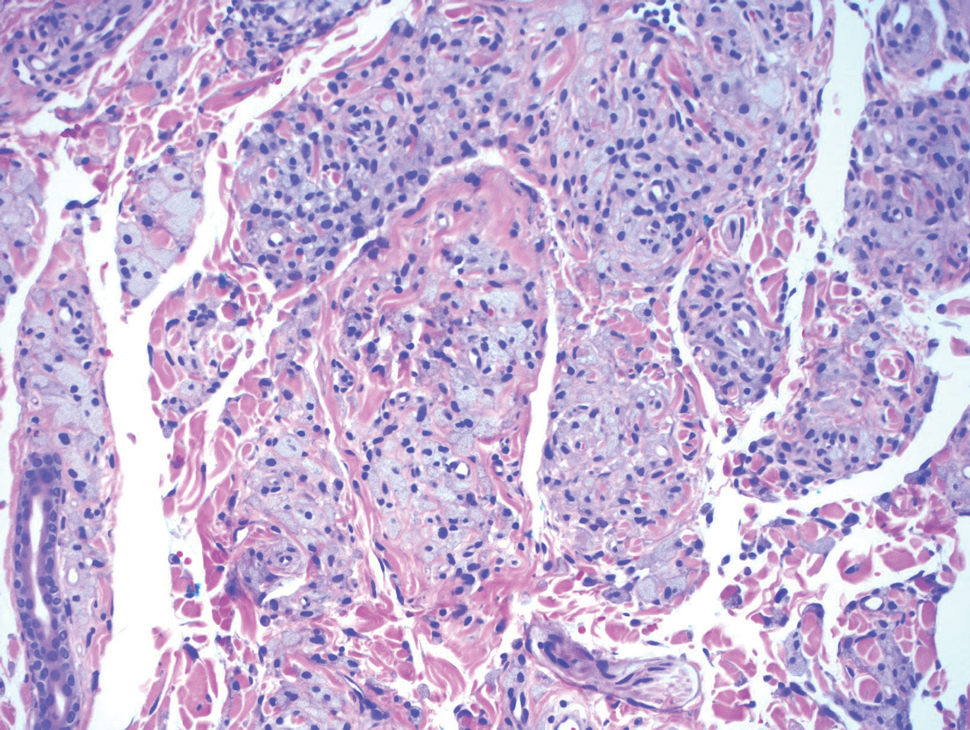
Atypical fibroxanthoma is a cutaneous neoplasm with fibrohistiocytic mesenchymal origin.11 These tumors typically arise on the head and neck in elderly individuals, particularly men with sun-damaged skin. They often present as superficial, rapidly growing nodules with the potential to ulcerate and bleed.11,12 Histologic features include pleomorphic spindle and epithelioid cells, whose nuclei appear hyperchromatic with atypical mitoses (Figure 2).12 Granular cell changes occur infrequently with AFXs, but in such cases immunohistochemistry can readily distinguish AFX from GCT. Although both tend to stain positive for CD68 and vimentin, AFXs lack S-100 protein and SOX10 expression that frequently is observed in GCTs.3,12

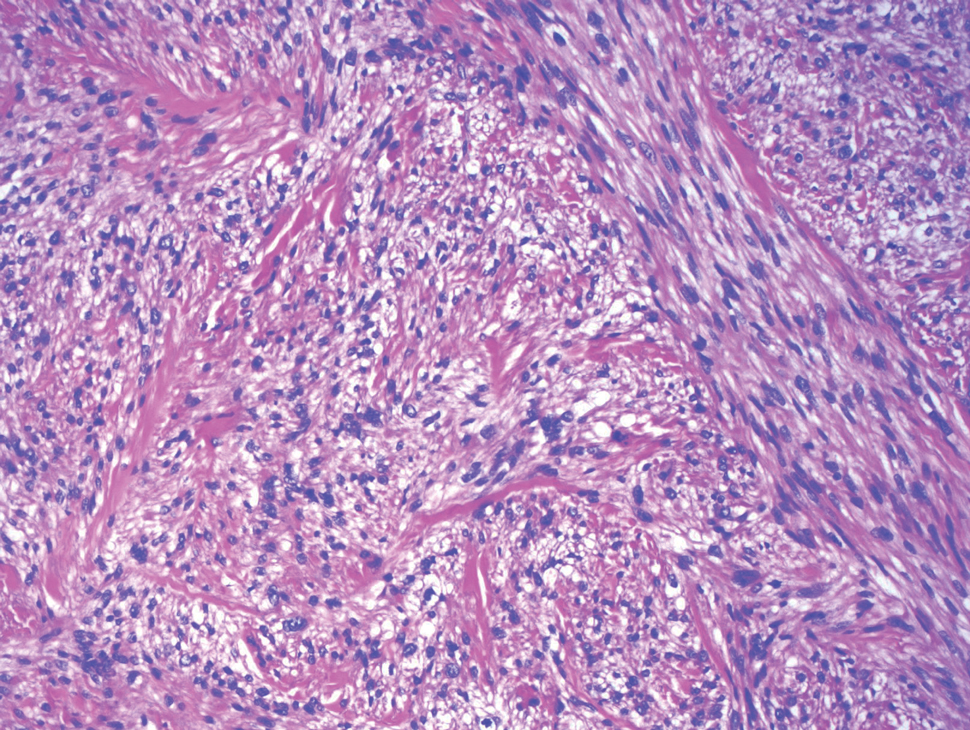
Xanthomas are localized lipid deposits in the connective tissue of the skin that often arise in association with dyslipidemia.13 They typically present as soft to semisolid yellow papules, plaques, or nodules. Their clinical appearance can resemble GCTs; however, histologic analysis enables differentiation with ease, as xanthomas demonstrate characteristic foam cells, consisting of lipid-laden macrophages (Figure 3).13
Cutaneous leiomyosarcoma is a rare dermal neoplasm, accounting for 2% to 3% of all sarcomas.14 They typically occur in White males during the fifth to seventh decades of life and often present as asymptomatic lesions on the lower extremities. They frequently arise from pilar smooth muscle. Unlike uterine and soft-tissue leiomyosarcoma, cutaneous leiomyosarcoma tends to follow an indolent course and rarely metastasizes.14 Histologically, these tumors display intersecting, well-defined, spindle-cell fascicles with abundant eosinophilic cytoplasm and cigar-shaped, blunt-ended nuclei (Figure 4).15 Occasionally, leiomyosarcomas can demonstrate cytoplasmic granularity due to lysosome accumulation4; nevertheless, the diagnosis usually can be elucidated by examining more typical histologic areas and utilizing immunohistochemistry, which often stains positive for α-smooth muscle actin, desmin, and h-caldesmon.4,15
- Fanburg-Smith JC, Meis-Kindblom JM, Fante R, et al. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol. 1998;22:779-794.
- Moten AS, Movva S, von Mehren M, et al. Granular cell tumor experience at a comprehensive cancer center. J Surg Res. 2018;226:1-7.
- Machado I, Cruz J, Lavernia J, et al. Solitary, multiple, benign, atypical, or malignant: the "granular cell tumor" puzzle. Virchows Arch. 2016;468:527-538.
- Ordóñez NG. Granular cell tumor: a review and update. Adv Anat Pathol. 1999;6:186-203.
- Vaughan V, Ferringer T. Granular cell tumor. Cutis. 2014;94:275, 279-280.
- Van L, Parker SR. Multiple morphologically distinct cutaneous granular cell tumors occurring in a single patient. Cutis. 2016;97:E26-E29.
- Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
- Bamps S, Oyen T, Legius E, et al. Multiple granular cell tumors in a child with Noonan syndrome. Eur J Pediatr Surg. 2013;23:257-259.
- Rastrelli M, Tropea S, Rossi CR, et al. Melanoma: epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo. 2014;28:1005-1011.
- Smoller BR. Histologic criteria for diagnosing primary cutaneousmalignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Cardis MA, Ni J, Bhawan J. Granular cell differentiation: a review of the published work. J Dermatol. 2017;44:251-258.
- Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations [published online April 29, 2014]. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188.
- Sandhu N, Sauvageau AP, Groman A, et al. Cutaneous leiomyosarcoma: a SEER database analysis. Dermatol Surg. 2020;46:159-164.
- George S, Serrano C, Hensley ML, et al. Soft tissue and uterine leiomyosarcoma. J Clin Oncol. 2018;36:144-150.
The Diagnosis: Granular Cell Tumor
Histopathologic analysis from the axillary excision demonstrated cords and sheets of large polygonal cells in the dermis with uniform, oval, hyperchromatic nuclei and ample pink granular-staining cytoplasm (quiz images). An infiltrative growth pattern was noted; however, there was no evidence of conspicuous mitoses, nuclear pleomorphism, or necrosis. These results in conjunction with the immunohistochemistry findings were consistent with a benign granular cell tumor (GCT), a rare neoplasm considered to have neural/Schwann cell origin.1-3
Our case demonstrates the difficulty in clinically diagnosing cutaneous GCTs. The tumor often presents as a solitary, 0.5- to 3-cm, asymptomatic, firm nodule4,5; however, GCTs also can appear verrucous, eroded, or with other variable morphologies, which can create diagnostic challenges.5,6 Accordingly, a 1980 study of 110 patients with GCTs found that the preoperative clinical diagnosis was incorrect in all but 3 cases,7 emphasizing the need for histologic evaluation. Benign GCTs tend to exhibit sheets of polygonal tumor cells with eosinophilic granular cytoplasm and small central nuclei.3,5 The cytoplasmic granules are periodic acid-Schiff positive and diastase resistant.6 Many cases feature pseudoepitheliomatous hyperplasia, which can misleadingly resemble squamous cell carcinoma.3,5,6 Of note, invasive growth patterns on histology can occur with benign GCTs, as in our patient's case, and do not impact prognosis.3,4 On immunohistochemistry, benign, atypical, and malignant GCTs often stain positive for S-100 protein, vimentin, neuron-specific enolase, SOX10, and CD68.1,3
Although our patient's GCTs were benign, an estimated 1% to 2% are malignant.1,4 In 1998, Fanburg-Smith et al1 defined 6 histologic criteria that characterize malignant GCTs: necrosis, tumor cell spindling, vesicular nuclei with large nucleoli, high nuclear to cytoplasmic ratio, increased mitosis, and pleomorphism. Neoplasms with 3 or more of these features are classified as malignant, those with 1 or 2 are considered atypical, and those with only pleomorphism or no other criteria met are diagnosed as benign.1
Multiple GCTs have been reported in 10% to 25% of cases and, as highlighted in our case, can occur in both a metachronous and synchronous manner.2-4,6 Our patient developed a solitary GCT on the inferior lip 3 years prior to the appearance of 2 additional GCTs within 6 months of each other. The presence of multiple GCTs has been associated with genetic syndromes, such as neurofibromatosis type 1 and Noonan syndrome with multiple lentigines3,8; however, as our case demonstrates, multiple GCTs can occur in nonsyndromic patients as well. When multiple GCTs develop at distant sites, they can resemble metastasis.3 To differentiate these clinical scenarios, Machado et al3 proposed utilizing histology and anatomic location. Multiple tumors with benign characteristics on histology likely represent multiple GCTs, whereas tumors arising at sites common to GCT metastasis, such as lymph node, bone, or viscera, are more concerning for metastatic disease. It has been suggested that patients with multiple GCTs should be monitored with physical examination and repeat magnetic resonance imaging or computed tomography every 6 to 12 months.2 Given our patient's presentation with new tumors arising within 6 months of one another, we recommended a 6-month follow-up interval rather than 1 year. Due to the rarity of GCTs, clinical trials to define treatment guidelines and recommendations have not been performed.3 However, the most commonly utilized treatment modality is wide local excision, as performed in our patient.2,4
Melanoma, atypical fibroxanthoma (AFX), xanthoma, and leiomyosarcoma may be difficult to distinguish from GCT.1,3,4 Melanoma incidence has increased dramatically over the last several decades, with rates in the United States rising from 6.8 cases per 100,000 individuals in the 1970s to 20.1 in the early 2000s. Risk factors for its development include UV radiation exposure and particularly severe sunburns during childhood, along with a number of host risk factors such as total number of melanocytic nevi, family history, and fair complexion.9 Histologically, it often demonstrates irregularly distributed, poorly defined melanocytes with pagetoid spread and dyscohesive nests (Figure 1).10 Melanoma metastasis occasionally can present as a soft-tissue mass and often stains positive for S-100 and vimentin, thus resembling GCT1,4; however, unlike melanoma, GCTs lack melanosomes and stain negative for more specific melanocyte markers, such as melanoma antigen recognized by T cells 1 (MART-1).1,3,4
Atypical fibroxanthoma is a cutaneous neoplasm with fibrohistiocytic mesenchymal origin.11 These tumors typically arise on the head and neck in elderly individuals, particularly men with sun-damaged skin. They often present as superficial, rapidly growing nodules with the potential to ulcerate and bleed.11,12 Histologic features include pleomorphic spindle and epithelioid cells, whose nuclei appear hyperchromatic with atypical mitoses (Figure 2).12 Granular cell changes occur infrequently with AFXs, but in such cases immunohistochemistry can readily distinguish AFX from GCT. Although both tend to stain positive for CD68 and vimentin, AFXs lack S-100 protein and SOX10 expression that frequently is observed in GCTs.3,12
Xanthomas are localized lipid deposits in the connective tissue of the skin that often arise in association with dyslipidemia.13 They typically present as soft to semisolid yellow papules, plaques, or nodules. Their clinical appearance can resemble GCTs; however, histologic analysis enables differentiation with ease, as xanthomas demonstrate characteristic foam cells, consisting of lipid-laden macrophages (Figure 3).13
Cutaneous leiomyosarcoma is a rare dermal neoplasm, accounting for 2% to 3% of all sarcomas.14 They typically occur in White males during the fifth to seventh decades of life and often present as asymptomatic lesions on the lower extremities. They frequently arise from pilar smooth muscle. Unlike uterine and soft-tissue leiomyosarcoma, cutaneous leiomyosarcoma tends to follow an indolent course and rarely metastasizes.14 Histologically, these tumors display intersecting, well-defined, spindle-cell fascicles with abundant eosinophilic cytoplasm and cigar-shaped, blunt-ended nuclei (Figure 4).15 Occasionally, leiomyosarcomas can demonstrate cytoplasmic granularity due to lysosome accumulation4; nevertheless, the diagnosis usually can be elucidated by examining more typical histologic areas and utilizing immunohistochemistry, which often stains positive for α-smooth muscle actin, desmin, and h-caldesmon.4,15
The Diagnosis: Granular Cell Tumor
Histopathologic analysis from the axillary excision demonstrated cords and sheets of large polygonal cells in the dermis with uniform, oval, hyperchromatic nuclei and ample pink granular-staining cytoplasm (quiz images). An infiltrative growth pattern was noted; however, there was no evidence of conspicuous mitoses, nuclear pleomorphism, or necrosis. These results in conjunction with the immunohistochemistry findings were consistent with a benign granular cell tumor (GCT), a rare neoplasm considered to have neural/Schwann cell origin.1-3
Our case demonstrates the difficulty in clinically diagnosing cutaneous GCTs. The tumor often presents as a solitary, 0.5- to 3-cm, asymptomatic, firm nodule4,5; however, GCTs also can appear verrucous, eroded, or with other variable morphologies, which can create diagnostic challenges.5,6 Accordingly, a 1980 study of 110 patients with GCTs found that the preoperative clinical diagnosis was incorrect in all but 3 cases,7 emphasizing the need for histologic evaluation. Benign GCTs tend to exhibit sheets of polygonal tumor cells with eosinophilic granular cytoplasm and small central nuclei.3,5 The cytoplasmic granules are periodic acid-Schiff positive and diastase resistant.6 Many cases feature pseudoepitheliomatous hyperplasia, which can misleadingly resemble squamous cell carcinoma.3,5,6 Of note, invasive growth patterns on histology can occur with benign GCTs, as in our patient's case, and do not impact prognosis.3,4 On immunohistochemistry, benign, atypical, and malignant GCTs often stain positive for S-100 protein, vimentin, neuron-specific enolase, SOX10, and CD68.1,3
Although our patient's GCTs were benign, an estimated 1% to 2% are malignant.1,4 In 1998, Fanburg-Smith et al1 defined 6 histologic criteria that characterize malignant GCTs: necrosis, tumor cell spindling, vesicular nuclei with large nucleoli, high nuclear to cytoplasmic ratio, increased mitosis, and pleomorphism. Neoplasms with 3 or more of these features are classified as malignant, those with 1 or 2 are considered atypical, and those with only pleomorphism or no other criteria met are diagnosed as benign.1
Multiple GCTs have been reported in 10% to 25% of cases and, as highlighted in our case, can occur in both a metachronous and synchronous manner.2-4,6 Our patient developed a solitary GCT on the inferior lip 3 years prior to the appearance of 2 additional GCTs within 6 months of each other. The presence of multiple GCTs has been associated with genetic syndromes, such as neurofibromatosis type 1 and Noonan syndrome with multiple lentigines3,8; however, as our case demonstrates, multiple GCTs can occur in nonsyndromic patients as well. When multiple GCTs develop at distant sites, they can resemble metastasis.3 To differentiate these clinical scenarios, Machado et al3 proposed utilizing histology and anatomic location. Multiple tumors with benign characteristics on histology likely represent multiple GCTs, whereas tumors arising at sites common to GCT metastasis, such as lymph node, bone, or viscera, are more concerning for metastatic disease. It has been suggested that patients with multiple GCTs should be monitored with physical examination and repeat magnetic resonance imaging or computed tomography every 6 to 12 months.2 Given our patient's presentation with new tumors arising within 6 months of one another, we recommended a 6-month follow-up interval rather than 1 year. Due to the rarity of GCTs, clinical trials to define treatment guidelines and recommendations have not been performed.3 However, the most commonly utilized treatment modality is wide local excision, as performed in our patient.2,4
Melanoma, atypical fibroxanthoma (AFX), xanthoma, and leiomyosarcoma may be difficult to distinguish from GCT.1,3,4 Melanoma incidence has increased dramatically over the last several decades, with rates in the United States rising from 6.8 cases per 100,000 individuals in the 1970s to 20.1 in the early 2000s. Risk factors for its development include UV radiation exposure and particularly severe sunburns during childhood, along with a number of host risk factors such as total number of melanocytic nevi, family history, and fair complexion.9 Histologically, it often demonstrates irregularly distributed, poorly defined melanocytes with pagetoid spread and dyscohesive nests (Figure 1).10 Melanoma metastasis occasionally can present as a soft-tissue mass and often stains positive for S-100 and vimentin, thus resembling GCT1,4; however, unlike melanoma, GCTs lack melanosomes and stain negative for more specific melanocyte markers, such as melanoma antigen recognized by T cells 1 (MART-1).1,3,4
Atypical fibroxanthoma is a cutaneous neoplasm with fibrohistiocytic mesenchymal origin.11 These tumors typically arise on the head and neck in elderly individuals, particularly men with sun-damaged skin. They often present as superficial, rapidly growing nodules with the potential to ulcerate and bleed.11,12 Histologic features include pleomorphic spindle and epithelioid cells, whose nuclei appear hyperchromatic with atypical mitoses (Figure 2).12 Granular cell changes occur infrequently with AFXs, but in such cases immunohistochemistry can readily distinguish AFX from GCT. Although both tend to stain positive for CD68 and vimentin, AFXs lack S-100 protein and SOX10 expression that frequently is observed in GCTs.3,12
Xanthomas are localized lipid deposits in the connective tissue of the skin that often arise in association with dyslipidemia.13 They typically present as soft to semisolid yellow papules, plaques, or nodules. Their clinical appearance can resemble GCTs; however, histologic analysis enables differentiation with ease, as xanthomas demonstrate characteristic foam cells, consisting of lipid-laden macrophages (Figure 3).13
Cutaneous leiomyosarcoma is a rare dermal neoplasm, accounting for 2% to 3% of all sarcomas.14 They typically occur in White males during the fifth to seventh decades of life and often present as asymptomatic lesions on the lower extremities. They frequently arise from pilar smooth muscle. Unlike uterine and soft-tissue leiomyosarcoma, cutaneous leiomyosarcoma tends to follow an indolent course and rarely metastasizes.14 Histologically, these tumors display intersecting, well-defined, spindle-cell fascicles with abundant eosinophilic cytoplasm and cigar-shaped, blunt-ended nuclei (Figure 4).15 Occasionally, leiomyosarcomas can demonstrate cytoplasmic granularity due to lysosome accumulation4; nevertheless, the diagnosis usually can be elucidated by examining more typical histologic areas and utilizing immunohistochemistry, which often stains positive for α-smooth muscle actin, desmin, and h-caldesmon.4,15
- Fanburg-Smith JC, Meis-Kindblom JM, Fante R, et al. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol. 1998;22:779-794.
- Moten AS, Movva S, von Mehren M, et al. Granular cell tumor experience at a comprehensive cancer center. J Surg Res. 2018;226:1-7.
- Machado I, Cruz J, Lavernia J, et al. Solitary, multiple, benign, atypical, or malignant: the "granular cell tumor" puzzle. Virchows Arch. 2016;468:527-538.
- Ordóñez NG. Granular cell tumor: a review and update. Adv Anat Pathol. 1999;6:186-203.
- Vaughan V, Ferringer T. Granular cell tumor. Cutis. 2014;94:275, 279-280.
- Van L, Parker SR. Multiple morphologically distinct cutaneous granular cell tumors occurring in a single patient. Cutis. 2016;97:E26-E29.
- Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
- Bamps S, Oyen T, Legius E, et al. Multiple granular cell tumors in a child with Noonan syndrome. Eur J Pediatr Surg. 2013;23:257-259.
- Rastrelli M, Tropea S, Rossi CR, et al. Melanoma: epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo. 2014;28:1005-1011.
- Smoller BR. Histologic criteria for diagnosing primary cutaneousmalignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Cardis MA, Ni J, Bhawan J. Granular cell differentiation: a review of the published work. J Dermatol. 2017;44:251-258.
- Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations [published online April 29, 2014]. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188.
- Sandhu N, Sauvageau AP, Groman A, et al. Cutaneous leiomyosarcoma: a SEER database analysis. Dermatol Surg. 2020;46:159-164.
- George S, Serrano C, Hensley ML, et al. Soft tissue and uterine leiomyosarcoma. J Clin Oncol. 2018;36:144-150.
- Fanburg-Smith JC, Meis-Kindblom JM, Fante R, et al. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol. 1998;22:779-794.
- Moten AS, Movva S, von Mehren M, et al. Granular cell tumor experience at a comprehensive cancer center. J Surg Res. 2018;226:1-7.
- Machado I, Cruz J, Lavernia J, et al. Solitary, multiple, benign, atypical, or malignant: the "granular cell tumor" puzzle. Virchows Arch. 2016;468:527-538.
- Ordóñez NG. Granular cell tumor: a review and update. Adv Anat Pathol. 1999;6:186-203.
- Vaughan V, Ferringer T. Granular cell tumor. Cutis. 2014;94:275, 279-280.
- Van L, Parker SR. Multiple morphologically distinct cutaneous granular cell tumors occurring in a single patient. Cutis. 2016;97:E26-E29.
- Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
- Bamps S, Oyen T, Legius E, et al. Multiple granular cell tumors in a child with Noonan syndrome. Eur J Pediatr Surg. 2013;23:257-259.
- Rastrelli M, Tropea S, Rossi CR, et al. Melanoma: epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo. 2014;28:1005-1011.
- Smoller BR. Histologic criteria for diagnosing primary cutaneousmalignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Cardis MA, Ni J, Bhawan J. Granular cell differentiation: a review of the published work. J Dermatol. 2017;44:251-258.
- Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations [published online April 29, 2014]. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188.
- Sandhu N, Sauvageau AP, Groman A, et al. Cutaneous leiomyosarcoma: a SEER database analysis. Dermatol Surg. 2020;46:159-164.
- George S, Serrano C, Hensley ML, et al. Soft tissue and uterine leiomyosarcoma. J Clin Oncol. 2018;36:144-150.

A 26-year-old woman with a history of dysplastic nevi with severe atypia presented with a growth on the lower lip of 3 years’ duration. She denied any inciting event, such as prior trauma to the area, and reported that the lesion had been asymptomatic without a notable change in size. Physical examination revealed a translucent, soft, compressible cystic papule on the left inferior vermilion lip. Wide local excision following incisional biopsy was performed. Six months later, the patient returned to our clinic with a lesion on the right lateral tongue of 6 weeks’ duration as well as a 1-cm subcutaneous cyst in the left axilla of 6 months’ duration. Excisional biopsies of both lesions were performed for histopathologic analysis. Pathology results were similar among the lip, tongue, and axillary lesions. Immunohistochemistry revealed strong positive staining with antibodies to S-100 protein, SOX10, and CD68.