User login
Our knowledge of the pathogenesis of thrombotic microangiopathies has greatly advanced in the last decade, improving the diagnosis and treatment of these diseases.

Many conditions involve thrombotic microangiopathies (Table 1). This article reviews the most common ones, ie, thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, atypical hemolytic uremic syndrome, and antiphospholipid syndrome—their clinical features (focusing on the kidney), course, and management. Of note, although the diseases are similar, their pathogeneses and treatments differ.
DIFFERENT PATHWAYS TO MULTIORGAN THROMBOSIS
The thrombotic microangiopathies are multisystem disorders that can affect children and adults and often present with prominent renal and neurologic involvement. Endothelial injury is likely the inciting factor leading to thrombosis in the kidney and in many other organs. The causes variously include:
- Toxins from bacteria or drugs
- Abnormal complement activation, genetic or autoantibody-induced
- Procoagulant factors, eg, antiphospholipid antibodies
- Loss of anticoagulants, eg, from a defect of ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13); ADAMTS13 is also known as von Willebrand factor-cleaving protease
- Severe hypertension.
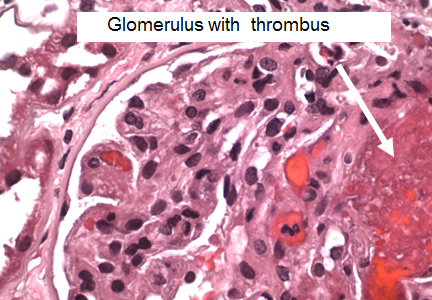
The histopathologic features are similar in all the thrombotic microangiopathies. Laboratory findings include thrombocytopenia, microangiopathic hemolytic anemia (with schistocytes on the peripheral blood smear), and high serum lactate dehydrogenase (LDH) levels; these are also markers of treatment progress. Bilirubin may be elevated and haptoglobin absent. Renal biopsy reveals thrombi in the glomeruli and arterioles.
THROMBOTIC THROMBOCYTOPENIC PURPURA
A young woman with fever, bruising, and renal failure, then blindness
A 36-year-old black woman who had been previously healthy presents to her doctor with fever and bruising.
Her hematocrit is 28% (reference range 38%–46%), platelet count 15 x 109/L (150–450), and prothrombin and partial thromboplastin times are normal. Her peripheral blood smear shows microangiopathic hemolytic anemia with schistocytes.
Over the next few days, her urine output declines and she develops sudden blindness followed by decreased mental acuity. Blood is drawn and sent for ADAMTS13 assay. Treatment is started at once with daily therapeutic plasma exchange. The assay results, when they arrive, show marked ADAMTS13 reduction (< 5%). Over the ensuing weeks, her mental acuity improves, her vision returns, and her renal function improves.
ADAMTS13 deficiency is definitive
Thrombotic thrombocytopenic purpura is characterized by:
- Neurologic abnormalities and acute renal failure
- Thrombocytopenia and microangiopathic hemolytic anemia
- Histologic evidence of thrombotic microangiopathy
- Deficiency of von Willebrand factor-cleaving protease (ADAMTS13 < 10%).
von Willebrand factor forms ultralarge multimers in the circulation that interact with platelets; these are normally cleaved by ADAMTS13. With ADAMTS13 deficiency (from either a genetic mutation or autoantibodies), the ultralarge multimers lead to coagulation as blood flows through small vessels.1
In 2003, Tsai2 evaluated 127 patients over age 10 who had thrombocytopenia and microangiopathic hemolysis with no plausible cause or features suggestive of hemolytic uremic syndrome. All were severely deficient in ADAMTS13. Subsequently, thrombotic thrombocytopenic purpura has been defined by a severe actual or effective deficiency of ADAMTS13.
Prompt plasma exchange is critical
Although the ADAMTS13 assay is important for diagnosing thrombotic thrombocytopenic purpura, in suspected cases daily plasma exchange should be started promptly, before test results return. Plasma exchange removes autoantibodies to ADAMTS13 from the blood, removes circulating ultralarge von Willebrand factor multimers, and replaces the missing ADAMTS13. Untreated, the disease is progressive, with irreversible renal failure, neurologic deterioration, and a 90% mortality rate. Plasma exchange reduces the mortality rate to less than 15%. If another diagnosis is confirmed, plasma exchange can be stopped.
Plasma exchange has been shown in clinical trials to be superior to plasma infusion in normalizing platelet counts and reducing mortality.3,4 Mortality rates were comparable with different replacement fluids vs fresh-frozen plasma, including solvent or detergent-treated plasma, and cryo-poor (cryosupernatant) plasma.4 Antiplatelet therapy, platelet transfusions, and splenectomy are ineffective.
Glucocorticoids for early treatment
An appropriate strategy is to add a glucocorticoid to plasma-exchange therapy at once (oral prednisone 1 mg/kg per day or intravenous methylprednisolone 125 mg twice daily) and withdraw it after several days if it is determined that it is not needed. Steroids for suspected thrombotic thrombocytopenic purpura can be justified for several reasons:
- The results of the ADAMTS13 assay are usually delayed, so steroids provide coverage for other diagnoses.
- They are helpful if thrombotic thrombocytopenic purpura is idiopathic (which is true for most cases) and if the patient has a poor response to initial therapy with plasma exchange.
- They are indicated for patients whose platelet counts do not increase with several days of plasma exchange or whose thrombocytopenia recurs as plasma exchange is decreased.
Rituximab improves survival
Rituximab, a chimeric (half murine) monoclonal antibody against CD19 and CD20 B cells, suppresses antibody production by knocking out the precursors of antibody-producing cells.
Anecdotal reports and small studies involving a total of 42 patients have been published on the use of rituximab for thrombotic thrombocytopenic purpura. Courses of rituximab varied greatly, from 1 to 13 weekly doses at 375 mg/m2, with 4 doses being the most common. Complete remission occurred in 90% of cases.5,6 A typical study from 2014 involved 48 patients (30 of whom received rituximab) followed by severe ADAMTS13 deficiency during remission.7 Despite the small study size, the investigators found significantly improved relapse-free survival rates with rituximab treatment.
But rituximab can cost $25,000 for 2 doses of 1,000 mg, and this will most likely prohibit its routine use. The cost and insurance coverage vary with location and policies.
Based on such studies, a reasonable strategy is to treat thrombotic thrombocytopenic purpura with:
- Daily plasma exchange
- Steroids, at least until the diagnosis is certain
- Rituximab if warranted.
New targeted therapies
Caplacizumab, a humanized immunoglobulin that inhibits the interaction between ultralarge von Willebrand factor multimers and platelets, has the potential to change this strategy when it receives US Food and Drug Administration approval, which is expected soon.
Peyvandi et al8 randomized 75 patients with acquired thrombotic thrombocytopenic purpura to either subcutaneous caplacizumab 10 mg daily for 30 days or placebo. Both groups had daily plasma exchange. The treatment group had a 39% reduction in median time to normalization of platelets vs the placebo group, and 3 of 36 patients had exacerbations, compared with 11 of 39 patients in the placebo group. Although 8 patients relapsed within the first month after stopping caplacizumab, their cases were brought under control. There were also more bleeding episodes with caplacizumab (54% vs 38%), most being mild to moderate. Two patients in the placebo group died, but none in the treatment group.
The fact that platelet normalization occurred significantly faster with caplacizumab, even in some patients who had not yet had plasma exchange therapy initiated, has enormous clinical significance. The low platelet count in thrombotic thrombocytopenic purpura is a marker of susceptibility to rapid damage to the brain and kidneys, so correcting it quickly is critical.
Other strategies for new drug development include replacing the deficient ADAMTS13 with a recombinant molecule and blocking antibody production (the same mode of action as rituximab and glucocorticoids).9 Using all 3 strategies to treat thrombotic thrombocytopenic purpura may be the future standard of care.
HEMOLYTIC UREMIC SYNDROME
A child with sudden onset of bloody diarrhea and kidney failure
A 4-year-old girl plays with baby animals at a petting zoo and does not wash her hands immediately afterwards. Three days later, she develops fever, abdominal cramps, nausea, vomiting, and bloody diarrhea. Her pediatrician gives her antibiotics. On day 6, she develops ecchymoses on the extremities and lips, thrombocytopenia, low urine output, and seizures. Her stool tests positive for Escherichia coli O157:H7
Classic presentation: Young patient with bloody diarrhea
The classic presentation of hemolytic uremic syndrome is of a young patient with bloody diarrhea typically lasting 5 to 10 days. Kidney failure may follow, requiring dialysis in about 60% of patients for a mean of 10 days. About one-fourth of patients develop neurologic symptoms, and about the same fraction are left with long-term morbidity, eg, hypertension, proteinuria, and reduced glomerular filtration rate. The mortality rate is typically 4%10,11 but varies with the outbreak.
Histologically, the kidneys look identical to those in thrombotic thrombocytopenic purpura, with thrombi in glomeruli and small vessels.
E coli is the most common culprit, but other bacteria, including Shigella dysenteriae, and viruses are sometimes the cause. Fewer than 10% of children infected with Shiga toxin-positive E coli, also known as enterohemorrhagic E coli (O157:H7, O104:H4), develop hemolytic uremic syndrome.
Lessons from outbreaks
Petting zoos are a common source of transmission of pathogenic bacteria. Disease can be extremely serious: in 15 cases linked to a Florida petting zoo, 3 children died.
Other outbreaks involving pathogenic E coli have been tied to fresh vegetables and to undercooked hamburger at fast-food chains.
In Germany in 2011, more than 3,000 people acquired Shiga toxin nonhemolytic uremic syndrome due to E coli, and 16 of them died. In addition, 845 acquired hemolytic uremic syndrome, and 36 died. This outbreak was associated with the more virulent and less common O104:H4 strain, which has acquired a Shiga toxin-encoding phage. Patients were treated with quinolone antibiotics, which actually increase toxin production in this strain.12
Unusual in the German epidemic was that more adults were affected (88%), especially women (68% of cases).13 The source of infection was eventually found to be alfalfa sprouts, the seeds of which had been contaminated by E coli. Women did not harbor any intrinsic factor making them more susceptible; rather, they were more likely to eat salads.13
Supportive management
Supportive care is most important. Transfusion with packed red blood cells is indicated for hemoglobin below 6 g/dL. Hypertension should be controlled and dialysis provided. For central nervous system involvement or severe disease, plasma exchange is sometimes used.
Eculizumab was tried for a time as therapy but did not prove to be of benefit. Shiga toxin-binding agents have been developed, but by the time they are given it is too late in the disease process to help.
Antibiotics may harm; it is possible that they kill beneficial bacteria, allowing the Shiga toxin-producing E coli to better proliferate. Antimotility agents also are contraindicated. Other agents not recommended include urokinase, heparin, dipyridamole, and vincristine. Splenectomy is not advised.
The most important way to control hemolytic uremic syndrome is to prevent it by thoroughly cooking meat, cleaning fresh produce, and having children wash their hands after petting animals.
ATYPICAL HEMOLYTIC UREMIC SYNDROME
A young man in renal failure
A 28-year-old man has a history of “thrombotic thrombocytopenic purpura-hemolytic uremic syndrome” at age 12. He slowly progresses to end-stage renal disease and receives a renal transplant from his mother at age 20 that fails after 3 months. The renal transplant biopsy report at the time reads “thrombotic microangiopathy.” The patient’s brother also requires dialysis.
The patient’s complement values are low, especially C3. His father is offering him a kidney at this time, and the patient wants to know whether to proceed.
Normal ADAMTS13, no diarrhea
Hemolytic uremic syndrome without diarrhea is now called atypical hemolytic uremic syndrome. Patients have normal levels of ADAMTS13, do not have diarrhea, and have no evidence of Shiga toxin-producing E coli.
Continuous complement pathway activation
The complement system is part of the innate immune system, which provides immediate defense against infection and does not evolve as does the adaptive immune system. The classic complement pathway is activated by the C1 antibody-antigen complex. The alternative complement pathway leads to the same pathway via C3.14 Both pathways lead to the formation of C5 through C9 membrane attack complexes, which form channels across the membranes of target cells, leading to cell lysis and death.
The alternate pathway does not require an antibody trigger so is always active at a low level. Inhibitory factors (factor H, factor I, membrane cofactor protein, factor H-related proteins) are naturally present and slow it down at various steps. People who are born with an abnormal factor or, more commonly, develop antibodies against one of the factors, have uninhibited complement activation. If this happens in the blood vessels, massive coagulation and atypical hemolytic uremic syndrome ensues. The endothelial damage and clotting in the brain, kidney, and other organs are identical to that of hemolytic uremic syndrome caused by Shiga toxin.
Treat with eculizumab
Historically, atypical hemolytic uremic syndrome was treated with plasma exchange, which replaces defective complement regulatory proteins and removes inhibitory antibodies.
Understanding the complement pathways is key to developing drugs that target atypical hemolytic uremic syndrome, and about 60 are in the pipeline. The only one currently approved in the United States for atypical hemolytic uremic syndrome is eculizumab, a humanized monoclonal antibody that binds with high affinity to C5, blocking the end of the complement cascade and preventing formation of the membrane attack complex.15–18
The effects of eculizumab on atypical hemolytic uremic syndrome were studied in 2 prospective trials.19 Platelet counts rose rapidly within weeks of starting treatment, and kidney function improved. Benefits continued throughout the 64 weeks studied. There were no deaths among the 37 patients enrolled, and although these were single-arm trials, they provide evidence of dramatic benefit considering the high mortality risk of this disease.
Eculizumab is now considered the treatment of choice. It may be used empirically for patients with hemolytic uremic syndrome who test negative for Shiga toxin and antiphospholipid antibody, and who do not have a very low level of ADAMTS13. The big drawback of eculizumab is its high price,20–22 which varies by amount used, location, and pharmacy negotiation, but can be in the hundreds of thousands of dollars.
For a patient with atypical hemolytic uremic syndrome on dialysis, treatment with eculizumab should continue for 4 to 6 months if there are no extrarenal manifestations. But many patients continue to have the defect in the complement system, so the problem may recur.
Case revisited
For our patient considering a kidney transplant, many experts feel that a transplant can be done as long as platelet counts are monitored and treatment with eculizumab is restarted if needed. One can also make the case for waiting a few years for new oral drugs to become available before offering transplant.
ANTIPHOSPHOLIPID SYNDROME
A young woman with a history of thrombosis and miscarriages
A 27-year-old woman presents with arthralgias, low-grade fever, and malaise. She has a history of 3 spontaneous abortions and Raynaud phenomenon. Two years ago, she had deep vein thrombosis of the right calf after a long automobile trip.
She now has swollen metacarpophalangeal and proximal interphalangeal joints, livedo reticularis (a mottled venous pattern of the skin best seen under fluorescent light) of the legs and arms, and ankle edema (2-cm indentation).
Her blood pressure is 152/92 mm Hg. Laboratory values:
- White blood cell count 3.6 × 109/L (reference range 4.5–11.0)
- Hematocrit 24% (36%–47%)
- Platelet count 89 × 109/L (150–450)
- Urinalysis: protein 4+, heme 3+, red blood cells 8–15 per high-power field (< 3), red blood cell casts present
- Blood urea nitrogen 43 mg/dL (10–20)
- Creatinine 2.6 mg/dL (0.5–1.1).
- Prothrombin time 14.6 s (10–14)
- Partial thromboplastin time 85 s (25–35)
- Antinuclear antibody positive at 1:160
- Anti-double-stranded DNA and serum complement normal
- Syphilis serologic screening (VDRL) positive.
The patient has leukopenia, anemia, thrombocytopenia, hematuria, proteinuria, high blood urea nitrogen, and markedly elevated partial thromboplastin time. Although she has a positive antinuclear antibody test and renal dysfunction, her anti-dsDNA and serum complement tests are normal, making the diagnosis of systemic lupus erythematosus unlikely.
Consider antiphospholipid syndrome
In any patient with multiple pregnancy losses, lupus, or a history of thrombosis, antiphospholipid syndrome should be considered.
In a series of patients with antiphospholipid antibody who underwent kidney biopsy, more than half were men, indicating that, unlike lupus, this is not primarily a disease of young women.
Diagnosis based on specific criteria
Clinical criteria require at least one of the following in the patient’s history23:
- One or more episodes of arterial, venous, or small-vessel thrombosis
- Unexplained pregnancy morbidity (death of a fetus or neonate with normal morphology or 3 or more spontaneous abortions).
Serologic criteria for any of the following antiphospholipid antibodies require that at least one of the following tests be positive at least twice and at least 12 weeks apart:
- Anticardiolipin antibodies—high-titer immunoglobulin (Ig) G or IgM
- Autoantibodies for beta 2-glycoprotein
- Lupus anticoagulant—autoantibodies that increase clotting time in vitro and target beta 2-glycoprotein I and prothrombin (despite its name and actions in vitro, lupus anticoagulant functions as a coagulant).
As with the other thrombotic microangiopathies, patients with anticardiolipin syndrome have microthrombi in the glomeruli and blood vessels that are evident on kidney biopsy.
Suspect condition in likely groups
Antiphospholipid syndrome is surprisingly common.24 In a case-control study, de Groot et al25 found that 3.1% of patients under age 70 with a first episode of venous thrombosis and no known cancer were positive for lupus anticoagulant vs 0.9% of controls. In another case-control study, Urbanus et al26 found that 17% of women under age 50 with a stroke tested positive for lupus anticoagulant compared with less than 1% of controls. Because of such studies, it has become routine to test for anticardiolipin and lupus anticoagulant in young patients presenting with a stroke.
About 1% of women trying to have children have recurrent miscarriages, and of these, 10% to 15% have antiphospholipid antibody present.27–30
Pathogenesis
Patients with antiphospholipid syndrome have a much higher proportion of plasma beta 2-glycoprotein in the oxidized form than do healthy controls. The level is also higher than in patients with a different autoimmune disease whether or not they have antibodies against beta 2-glycoprotein 1. Although about 40% of patients with lupus have an anticardiolipin antibody, only a small percentage develop antiphospholipid syndrome with clotting.
It is thought that antiphospholipid syndrome involves initial injury to the endothelium, then potentiation of thrombus formation. Oxidized beta 2-glycoprotein complexes may bind to the endothelial cell surface, causing it to become the target of antibodies. The exact relationships between the factors are not yet understood.
The risk of a thrombotic event in an asymptomatic patient positive for all 3 factors—lupus anticoagulant, anticardiolipin antibody, and anti-beta 2-glycoprotein I antibody—is more than 5% per year.31
Manage thrombosis with anticoagulation
Khamashta et al,32 in a 1995 study, retrospectively studied patients with antiphospholipid antibodies and a history of thrombosis. Of 147 patients, 66 had idiopathic primary disease, 62 had systemic lupus, and 19 had “lupus-like” disease. Almost 70% (101 patients) had a recurrence of thrombosis, totaling 186 events. The mean time to recurrence was 12 months (range 2 weeks to 12 years). Recurrence rates were 0.01 events per patient per year with high-dose warfarin, 0.23 with low-dose warfarin, and 0.18 with aspirin. But the highest bleeding rates were in the 6 months after warfarin withdrawal; 29 patients had bleeding events, one-fourth of which were severe.
Standard therapy has become anticoagulation, starting with heparin or enoxaparin, then warfarin. There is inadequate evidence for the role of newer oral anticoagulant therapy.
A very high INR is not generally better than a moderately elevated level
For a time, it was thought that the international normalized ratio (INR) should be kept on the very high side to prevent thrombosis.
Crowther et al33 conducted a randomized, double-blind trial comparing moderate warfarin therapy (INR 2.0–3.0) and high-intensity warfarin therapy (INR 3.1–4.0) in antiphospholipid syndrome. Thrombosis actually recurred more frequently in the high-intensity therapy group (10.7% vs 3.4%), with no significant difference in major bleeding events.
A reasonable strategy is to keep the INR between 2.5 and 3.0, keeping in mind that values fluctuate in any individual patient. A higher goal often leads to excessive anticoagulation and bleeding. If the goal is too low, recurrent thrombosis becomes more likely. There are fewer data on the newer oral anticoagulants, but their role is likely to increase as reversal agents are developed.
Recommendations published in 2003 for treating antiphospholipid syndrome include34:
- Warfarin (INR 2.0–3.0) after the first thrombotic event
- Warfarin (INR 3.0–4.0) if a clot develops despite warfarin
- Warfarin (INR > 3.0) for an arterial event.
For the rare but catastrophic antiphospholipid syndrome in which thrombosis occurs in multiple organs, recommendations are for heparin plus steroids, with or without intravenous immunoglobulin and plasmapheresis. This approach has not always been successful, and the mortality rate is high.
Treatment of asymptomatic carriers is uncertain
Treatment of asymptomatic carriers of the antiphospholipid antibody is controversial. Evidence for management is scarce; some experts recommend aspirin therapy, but benefit has yet to be proven in clinical trials.
Canaud et al35 documented the role of activation of the kinase mammalian target of rapamycin (mTOR) in the vascular changes characteristic of antiphospholipid nephropathy. Postkidney transplant surveillance biopsies of patients with antiphospholipid antibodies showed vascular damage occurring over time (despite patients being asymptomatic) compared with other renal transplant patients. Patients with antiphospholipid antibodies who were treated with the immunosuppressive drug sirolimus were protected from developing these changes. Twelve years after transplant, 70% of patients with antiphospholipid antibodies taking sirolimus still had a functioning graft compared with 11% of untreated patients.
- Sadler JE. Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. Blood 2008; 112:11–18.
- Tsai HM. Advances in the pathogenesis, diagnosis, and treatment of thrombotic thrombocytopenic purpura. J Am Soc Nephrol 2003; 14:1072–1081.
- Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med 1991; 325:393–397.
- Brunskill SJ, Tusold A, Benjamin S, Stanworth SJ, Murphy MF. A systematic review of randomized controlled trials for plasma exchange in the treatment of thrombotic thrombocytopenic purpura. Transfus Med 2007; 17:17–35.
- Jasti S, Coyle T, Gentile T, Rosales L, Poiesz B. Rituximab as an adjunct to plasma exchange in TTP: a report of 12 cases and review of literature. J Clin Apher 2008; 23:151–156.
- Ling HT, Field JJ, Blinder MA. Sustained response with rituximab in patients with thrombotic thrombocytopenic purpura: a report of 13 cases and review of the literature. Am J Hematol 2009; 84:418–421.
- Hie M, Gay J, Galicier L, et al; French Thrombotic Microangiopathies Reference Centre. Preemptive rituximab infusions after remission efficiently prevent relapses in acquired thrombotic thrombocytopenic purpura. Blood 2014; 124:204–210.
- Peyvandi F, Scully M, Kremer Hovinga JA, et al; TITAN Investigators. Caplacizumab for acquired thrombotic thrombocytopenic purpura. N Engl J Med 2016; 374:511–522.
- Veyradier A. Von Willebrand factor—a new target for TTP treatment? N Engl J Med 2016; 374:583–585.
- Boyce TG, Swerdlow DL, Griffin PM. Escherichia coli O157:H7 and the hemolytic-uremic syndrome. N Engl J Med 1995; 333:364–368.
- Gerber A, Karch H, Allerberger F, Verweyen HM, Zimmerhackl LB. Clinical course and the role of Shiga toxin-producing Escherichia coli infection in the hemolytic-uremic syndrome in pediatric patients, 1997–2000, in Germany and Austria: a prospective study. J Infect Dis 2002; 186:493–500.
- Rasko DA, Webster DR, Sahl JW, et al. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N Engl J Med 2011; 365:709–717.
- Frank C, Werber D, Cramer JP, et al; HUS Investigation Team. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N Engl J Med 2011; 365:1771–1780.
- Bomback AS, Appel GB. Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nat Rev Nephrol 2012; 8:634–642.
- Figueroa JE, Densen P. Infectious diseases associated with complement deficiencies. Clin Microbiol Rev 1991; 4:359–395.
- Walport MJ. Complement. First of two parts. N Engl J Med 2001; 344:1058–1066.
- Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol 2007; 25:1256–1264.
- Soliris (eculizumab). Prescribing information. Alexion Pharmaceuticals, Inc.
- Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic–uremic syndrome. N Engl J Med 2013; 368:2169–2181.
- Kim JJ, Waller SC, Reid CJ. Eculizumab in atypical haemolytic-uraemic syndrome allows cessation of plasma exchange and dialysis. Clin Kidney J 2012; 5:34–36.
- Povey H, Vundru R, Junglee N, Jibani M. Renal recovery with eculizumab in atypical hemolytic uremic syndrome following prolonged dialysis. Clin Nephrol 2014; 82:326–331.
- Gargau M, Azancot M, Ramos R, Sanchez-Corral P, Montero MA, Seron D. Early treatment with eculizumab may be beneficial in atypical haemolytic uraemic syndrome. Clin Kidney J 2012; 5:1–3.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Giannakopoulos B, Krilis SA. The pathogenesis of the antiphospholipid syndrome. N Engl J Med 2013; 368:1033–1044.
- de Groot PG, Lutters B, Derksen RH, Lisman T, Meijers JC, Rosendaal FR. Lupus anticoagulants and the risk of a first episode of deep venous thrombosis. J Thromb Haemost 2005; 3:1993–1997.
- Urbanus RT, Siegerink B, Roest M, Rosendaal FR, de Groot PG, Algra A. Antiphospholipid antibodies and risk of myocardial infarction and ischaemic stroke in young women in the RATIO study: a case-control study. Lancet Neurol 2009; 8:998–1005.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Stirrat GM. Recurrent miscarriage I: definition and epidemiology. Lancet 1990; 336:673–675.
- Rai RS, Regan L, Clifford K, et al. Antiphospholipid antibodies and beta 2-glycoprotein-I in 500 women with recurrent miscarriage: results of a comprehensive screening approach. Hum Reprod 1995; 10:2001–2005.
- Yetman DL, Kutteh WH. Antiphospholipid antibody panels and recurrent pregnancy loss: prevalence of anticardiolipin antibodies compared with other antiphospholipid antibodies. Fertil Steril 1996; 66:540–546.
- Pengo V, Ruffatti A, Legnani C, et al. Incidence of a first thromboembolic event in asymptomatic carriers of high-risk antiphospholipid antibody profile: a multicenter prospective study. Blood 2011; 118:4714–4718.
- Khamashta MA, Cuadrado MJ, Mujic F, Taub NA, Hunt BJ, Hughes GR. The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med 1995; 332:993–997.
- Crowther MA, Ginsberg JS, Julian J, et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N Engl J Med 2003; 349:1133–1138.
- Lockshin M, Tenedios F, Petri M, et al. Cardiac disease in the antiphospholipid syndrome: recommendations for treatment. Committee consensus report. Lupus 2003; 12:518–523.
- Canaud G, Bienaimé F, Tabarin F, et al. Inhibition of the mTORC pathway in the antiphospholipid syndrome. N Engl J Med 2014; 371:303–312.
Our knowledge of the pathogenesis of thrombotic microangiopathies has greatly advanced in the last decade, improving the diagnosis and treatment of these diseases.
Many conditions involve thrombotic microangiopathies (Table 1). This article reviews the most common ones, ie, thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, atypical hemolytic uremic syndrome, and antiphospholipid syndrome—their clinical features (focusing on the kidney), course, and management. Of note, although the diseases are similar, their pathogeneses and treatments differ.
DIFFERENT PATHWAYS TO MULTIORGAN THROMBOSIS
The thrombotic microangiopathies are multisystem disorders that can affect children and adults and often present with prominent renal and neurologic involvement. Endothelial injury is likely the inciting factor leading to thrombosis in the kidney and in many other organs. The causes variously include:
- Toxins from bacteria or drugs
- Abnormal complement activation, genetic or autoantibody-induced
- Procoagulant factors, eg, antiphospholipid antibodies
- Loss of anticoagulants, eg, from a defect of ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13); ADAMTS13 is also known as von Willebrand factor-cleaving protease
- Severe hypertension.
The histopathologic features are similar in all the thrombotic microangiopathies. Laboratory findings include thrombocytopenia, microangiopathic hemolytic anemia (with schistocytes on the peripheral blood smear), and high serum lactate dehydrogenase (LDH) levels; these are also markers of treatment progress. Bilirubin may be elevated and haptoglobin absent. Renal biopsy reveals thrombi in the glomeruli and arterioles.
THROMBOTIC THROMBOCYTOPENIC PURPURA
A young woman with fever, bruising, and renal failure, then blindness
A 36-year-old black woman who had been previously healthy presents to her doctor with fever and bruising.
Her hematocrit is 28% (reference range 38%–46%), platelet count 15 x 109/L (150–450), and prothrombin and partial thromboplastin times are normal. Her peripheral blood smear shows microangiopathic hemolytic anemia with schistocytes.
Over the next few days, her urine output declines and she develops sudden blindness followed by decreased mental acuity. Blood is drawn and sent for ADAMTS13 assay. Treatment is started at once with daily therapeutic plasma exchange. The assay results, when they arrive, show marked ADAMTS13 reduction (< 5%). Over the ensuing weeks, her mental acuity improves, her vision returns, and her renal function improves.
ADAMTS13 deficiency is definitive
Thrombotic thrombocytopenic purpura is characterized by:
- Neurologic abnormalities and acute renal failure
- Thrombocytopenia and microangiopathic hemolytic anemia
- Histologic evidence of thrombotic microangiopathy
- Deficiency of von Willebrand factor-cleaving protease (ADAMTS13 < 10%).
von Willebrand factor forms ultralarge multimers in the circulation that interact with platelets; these are normally cleaved by ADAMTS13. With ADAMTS13 deficiency (from either a genetic mutation or autoantibodies), the ultralarge multimers lead to coagulation as blood flows through small vessels.1
In 2003, Tsai2 evaluated 127 patients over age 10 who had thrombocytopenia and microangiopathic hemolysis with no plausible cause or features suggestive of hemolytic uremic syndrome. All were severely deficient in ADAMTS13. Subsequently, thrombotic thrombocytopenic purpura has been defined by a severe actual or effective deficiency of ADAMTS13.
Prompt plasma exchange is critical
Although the ADAMTS13 assay is important for diagnosing thrombotic thrombocytopenic purpura, in suspected cases daily plasma exchange should be started promptly, before test results return. Plasma exchange removes autoantibodies to ADAMTS13 from the blood, removes circulating ultralarge von Willebrand factor multimers, and replaces the missing ADAMTS13. Untreated, the disease is progressive, with irreversible renal failure, neurologic deterioration, and a 90% mortality rate. Plasma exchange reduces the mortality rate to less than 15%. If another diagnosis is confirmed, plasma exchange can be stopped.
Plasma exchange has been shown in clinical trials to be superior to plasma infusion in normalizing platelet counts and reducing mortality.3,4 Mortality rates were comparable with different replacement fluids vs fresh-frozen plasma, including solvent or detergent-treated plasma, and cryo-poor (cryosupernatant) plasma.4 Antiplatelet therapy, platelet transfusions, and splenectomy are ineffective.
Glucocorticoids for early treatment
An appropriate strategy is to add a glucocorticoid to plasma-exchange therapy at once (oral prednisone 1 mg/kg per day or intravenous methylprednisolone 125 mg twice daily) and withdraw it after several days if it is determined that it is not needed. Steroids for suspected thrombotic thrombocytopenic purpura can be justified for several reasons:
- The results of the ADAMTS13 assay are usually delayed, so steroids provide coverage for other diagnoses.
- They are helpful if thrombotic thrombocytopenic purpura is idiopathic (which is true for most cases) and if the patient has a poor response to initial therapy with plasma exchange.
- They are indicated for patients whose platelet counts do not increase with several days of plasma exchange or whose thrombocytopenia recurs as plasma exchange is decreased.
Rituximab improves survival
Rituximab, a chimeric (half murine) monoclonal antibody against CD19 and CD20 B cells, suppresses antibody production by knocking out the precursors of antibody-producing cells.
Anecdotal reports and small studies involving a total of 42 patients have been published on the use of rituximab for thrombotic thrombocytopenic purpura. Courses of rituximab varied greatly, from 1 to 13 weekly doses at 375 mg/m2, with 4 doses being the most common. Complete remission occurred in 90% of cases.5,6 A typical study from 2014 involved 48 patients (30 of whom received rituximab) followed by severe ADAMTS13 deficiency during remission.7 Despite the small study size, the investigators found significantly improved relapse-free survival rates with rituximab treatment.
But rituximab can cost $25,000 for 2 doses of 1,000 mg, and this will most likely prohibit its routine use. The cost and insurance coverage vary with location and policies.
Based on such studies, a reasonable strategy is to treat thrombotic thrombocytopenic purpura with:
- Daily plasma exchange
- Steroids, at least until the diagnosis is certain
- Rituximab if warranted.
New targeted therapies
Caplacizumab, a humanized immunoglobulin that inhibits the interaction between ultralarge von Willebrand factor multimers and platelets, has the potential to change this strategy when it receives US Food and Drug Administration approval, which is expected soon.
Peyvandi et al8 randomized 75 patients with acquired thrombotic thrombocytopenic purpura to either subcutaneous caplacizumab 10 mg daily for 30 days or placebo. Both groups had daily plasma exchange. The treatment group had a 39% reduction in median time to normalization of platelets vs the placebo group, and 3 of 36 patients had exacerbations, compared with 11 of 39 patients in the placebo group. Although 8 patients relapsed within the first month after stopping caplacizumab, their cases were brought under control. There were also more bleeding episodes with caplacizumab (54% vs 38%), most being mild to moderate. Two patients in the placebo group died, but none in the treatment group.
The fact that platelet normalization occurred significantly faster with caplacizumab, even in some patients who had not yet had plasma exchange therapy initiated, has enormous clinical significance. The low platelet count in thrombotic thrombocytopenic purpura is a marker of susceptibility to rapid damage to the brain and kidneys, so correcting it quickly is critical.
Other strategies for new drug development include replacing the deficient ADAMTS13 with a recombinant molecule and blocking antibody production (the same mode of action as rituximab and glucocorticoids).9 Using all 3 strategies to treat thrombotic thrombocytopenic purpura may be the future standard of care.
HEMOLYTIC UREMIC SYNDROME
A child with sudden onset of bloody diarrhea and kidney failure
A 4-year-old girl plays with baby animals at a petting zoo and does not wash her hands immediately afterwards. Three days later, she develops fever, abdominal cramps, nausea, vomiting, and bloody diarrhea. Her pediatrician gives her antibiotics. On day 6, she develops ecchymoses on the extremities and lips, thrombocytopenia, low urine output, and seizures. Her stool tests positive for Escherichia coli O157:H7
Classic presentation: Young patient with bloody diarrhea
The classic presentation of hemolytic uremic syndrome is of a young patient with bloody diarrhea typically lasting 5 to 10 days. Kidney failure may follow, requiring dialysis in about 60% of patients for a mean of 10 days. About one-fourth of patients develop neurologic symptoms, and about the same fraction are left with long-term morbidity, eg, hypertension, proteinuria, and reduced glomerular filtration rate. The mortality rate is typically 4%10,11 but varies with the outbreak.
Histologically, the kidneys look identical to those in thrombotic thrombocytopenic purpura, with thrombi in glomeruli and small vessels.
E coli is the most common culprit, but other bacteria, including Shigella dysenteriae, and viruses are sometimes the cause. Fewer than 10% of children infected with Shiga toxin-positive E coli, also known as enterohemorrhagic E coli (O157:H7, O104:H4), develop hemolytic uremic syndrome.
Lessons from outbreaks
Petting zoos are a common source of transmission of pathogenic bacteria. Disease can be extremely serious: in 15 cases linked to a Florida petting zoo, 3 children died.
Other outbreaks involving pathogenic E coli have been tied to fresh vegetables and to undercooked hamburger at fast-food chains.
In Germany in 2011, more than 3,000 people acquired Shiga toxin nonhemolytic uremic syndrome due to E coli, and 16 of them died. In addition, 845 acquired hemolytic uremic syndrome, and 36 died. This outbreak was associated with the more virulent and less common O104:H4 strain, which has acquired a Shiga toxin-encoding phage. Patients were treated with quinolone antibiotics, which actually increase toxin production in this strain.12
Unusual in the German epidemic was that more adults were affected (88%), especially women (68% of cases).13 The source of infection was eventually found to be alfalfa sprouts, the seeds of which had been contaminated by E coli. Women did not harbor any intrinsic factor making them more susceptible; rather, they were more likely to eat salads.13
Supportive management
Supportive care is most important. Transfusion with packed red blood cells is indicated for hemoglobin below 6 g/dL. Hypertension should be controlled and dialysis provided. For central nervous system involvement or severe disease, plasma exchange is sometimes used.
Eculizumab was tried for a time as therapy but did not prove to be of benefit. Shiga toxin-binding agents have been developed, but by the time they are given it is too late in the disease process to help.
Antibiotics may harm; it is possible that they kill beneficial bacteria, allowing the Shiga toxin-producing E coli to better proliferate. Antimotility agents also are contraindicated. Other agents not recommended include urokinase, heparin, dipyridamole, and vincristine. Splenectomy is not advised.
The most important way to control hemolytic uremic syndrome is to prevent it by thoroughly cooking meat, cleaning fresh produce, and having children wash their hands after petting animals.
ATYPICAL HEMOLYTIC UREMIC SYNDROME
A young man in renal failure
A 28-year-old man has a history of “thrombotic thrombocytopenic purpura-hemolytic uremic syndrome” at age 12. He slowly progresses to end-stage renal disease and receives a renal transplant from his mother at age 20 that fails after 3 months. The renal transplant biopsy report at the time reads “thrombotic microangiopathy.” The patient’s brother also requires dialysis.
The patient’s complement values are low, especially C3. His father is offering him a kidney at this time, and the patient wants to know whether to proceed.
Normal ADAMTS13, no diarrhea
Hemolytic uremic syndrome without diarrhea is now called atypical hemolytic uremic syndrome. Patients have normal levels of ADAMTS13, do not have diarrhea, and have no evidence of Shiga toxin-producing E coli.
Continuous complement pathway activation
The complement system is part of the innate immune system, which provides immediate defense against infection and does not evolve as does the adaptive immune system. The classic complement pathway is activated by the C1 antibody-antigen complex. The alternative complement pathway leads to the same pathway via C3.14 Both pathways lead to the formation of C5 through C9 membrane attack complexes, which form channels across the membranes of target cells, leading to cell lysis and death.
The alternate pathway does not require an antibody trigger so is always active at a low level. Inhibitory factors (factor H, factor I, membrane cofactor protein, factor H-related proteins) are naturally present and slow it down at various steps. People who are born with an abnormal factor or, more commonly, develop antibodies against one of the factors, have uninhibited complement activation. If this happens in the blood vessels, massive coagulation and atypical hemolytic uremic syndrome ensues. The endothelial damage and clotting in the brain, kidney, and other organs are identical to that of hemolytic uremic syndrome caused by Shiga toxin.
Treat with eculizumab
Historically, atypical hemolytic uremic syndrome was treated with plasma exchange, which replaces defective complement regulatory proteins and removes inhibitory antibodies.
Understanding the complement pathways is key to developing drugs that target atypical hemolytic uremic syndrome, and about 60 are in the pipeline. The only one currently approved in the United States for atypical hemolytic uremic syndrome is eculizumab, a humanized monoclonal antibody that binds with high affinity to C5, blocking the end of the complement cascade and preventing formation of the membrane attack complex.15–18
The effects of eculizumab on atypical hemolytic uremic syndrome were studied in 2 prospective trials.19 Platelet counts rose rapidly within weeks of starting treatment, and kidney function improved. Benefits continued throughout the 64 weeks studied. There were no deaths among the 37 patients enrolled, and although these were single-arm trials, they provide evidence of dramatic benefit considering the high mortality risk of this disease.
Eculizumab is now considered the treatment of choice. It may be used empirically for patients with hemolytic uremic syndrome who test negative for Shiga toxin and antiphospholipid antibody, and who do not have a very low level of ADAMTS13. The big drawback of eculizumab is its high price,20–22 which varies by amount used, location, and pharmacy negotiation, but can be in the hundreds of thousands of dollars.
For a patient with atypical hemolytic uremic syndrome on dialysis, treatment with eculizumab should continue for 4 to 6 months if there are no extrarenal manifestations. But many patients continue to have the defect in the complement system, so the problem may recur.
Case revisited
For our patient considering a kidney transplant, many experts feel that a transplant can be done as long as platelet counts are monitored and treatment with eculizumab is restarted if needed. One can also make the case for waiting a few years for new oral drugs to become available before offering transplant.
ANTIPHOSPHOLIPID SYNDROME
A young woman with a history of thrombosis and miscarriages
A 27-year-old woman presents with arthralgias, low-grade fever, and malaise. She has a history of 3 spontaneous abortions and Raynaud phenomenon. Two years ago, she had deep vein thrombosis of the right calf after a long automobile trip.
She now has swollen metacarpophalangeal and proximal interphalangeal joints, livedo reticularis (a mottled venous pattern of the skin best seen under fluorescent light) of the legs and arms, and ankle edema (2-cm indentation).
Her blood pressure is 152/92 mm Hg. Laboratory values:
- White blood cell count 3.6 × 109/L (reference range 4.5–11.0)
- Hematocrit 24% (36%–47%)
- Platelet count 89 × 109/L (150–450)
- Urinalysis: protein 4+, heme 3+, red blood cells 8–15 per high-power field (< 3), red blood cell casts present
- Blood urea nitrogen 43 mg/dL (10–20)
- Creatinine 2.6 mg/dL (0.5–1.1).
- Prothrombin time 14.6 s (10–14)
- Partial thromboplastin time 85 s (25–35)
- Antinuclear antibody positive at 1:160
- Anti-double-stranded DNA and serum complement normal
- Syphilis serologic screening (VDRL) positive.
The patient has leukopenia, anemia, thrombocytopenia, hematuria, proteinuria, high blood urea nitrogen, and markedly elevated partial thromboplastin time. Although she has a positive antinuclear antibody test and renal dysfunction, her anti-dsDNA and serum complement tests are normal, making the diagnosis of systemic lupus erythematosus unlikely.
Consider antiphospholipid syndrome
In any patient with multiple pregnancy losses, lupus, or a history of thrombosis, antiphospholipid syndrome should be considered.
In a series of patients with antiphospholipid antibody who underwent kidney biopsy, more than half were men, indicating that, unlike lupus, this is not primarily a disease of young women.
Diagnosis based on specific criteria
Clinical criteria require at least one of the following in the patient’s history23:
- One or more episodes of arterial, venous, or small-vessel thrombosis
- Unexplained pregnancy morbidity (death of a fetus or neonate with normal morphology or 3 or more spontaneous abortions).
Serologic criteria for any of the following antiphospholipid antibodies require that at least one of the following tests be positive at least twice and at least 12 weeks apart:
- Anticardiolipin antibodies—high-titer immunoglobulin (Ig) G or IgM
- Autoantibodies for beta 2-glycoprotein
- Lupus anticoagulant—autoantibodies that increase clotting time in vitro and target beta 2-glycoprotein I and prothrombin (despite its name and actions in vitro, lupus anticoagulant functions as a coagulant).
As with the other thrombotic microangiopathies, patients with anticardiolipin syndrome have microthrombi in the glomeruli and blood vessels that are evident on kidney biopsy.
Suspect condition in likely groups
Antiphospholipid syndrome is surprisingly common.24 In a case-control study, de Groot et al25 found that 3.1% of patients under age 70 with a first episode of venous thrombosis and no known cancer were positive for lupus anticoagulant vs 0.9% of controls. In another case-control study, Urbanus et al26 found that 17% of women under age 50 with a stroke tested positive for lupus anticoagulant compared with less than 1% of controls. Because of such studies, it has become routine to test for anticardiolipin and lupus anticoagulant in young patients presenting with a stroke.
About 1% of women trying to have children have recurrent miscarriages, and of these, 10% to 15% have antiphospholipid antibody present.27–30
Pathogenesis
Patients with antiphospholipid syndrome have a much higher proportion of plasma beta 2-glycoprotein in the oxidized form than do healthy controls. The level is also higher than in patients with a different autoimmune disease whether or not they have antibodies against beta 2-glycoprotein 1. Although about 40% of patients with lupus have an anticardiolipin antibody, only a small percentage develop antiphospholipid syndrome with clotting.
It is thought that antiphospholipid syndrome involves initial injury to the endothelium, then potentiation of thrombus formation. Oxidized beta 2-glycoprotein complexes may bind to the endothelial cell surface, causing it to become the target of antibodies. The exact relationships between the factors are not yet understood.
The risk of a thrombotic event in an asymptomatic patient positive for all 3 factors—lupus anticoagulant, anticardiolipin antibody, and anti-beta 2-glycoprotein I antibody—is more than 5% per year.31
Manage thrombosis with anticoagulation
Khamashta et al,32 in a 1995 study, retrospectively studied patients with antiphospholipid antibodies and a history of thrombosis. Of 147 patients, 66 had idiopathic primary disease, 62 had systemic lupus, and 19 had “lupus-like” disease. Almost 70% (101 patients) had a recurrence of thrombosis, totaling 186 events. The mean time to recurrence was 12 months (range 2 weeks to 12 years). Recurrence rates were 0.01 events per patient per year with high-dose warfarin, 0.23 with low-dose warfarin, and 0.18 with aspirin. But the highest bleeding rates were in the 6 months after warfarin withdrawal; 29 patients had bleeding events, one-fourth of which were severe.
Standard therapy has become anticoagulation, starting with heparin or enoxaparin, then warfarin. There is inadequate evidence for the role of newer oral anticoagulant therapy.
A very high INR is not generally better than a moderately elevated level
For a time, it was thought that the international normalized ratio (INR) should be kept on the very high side to prevent thrombosis.
Crowther et al33 conducted a randomized, double-blind trial comparing moderate warfarin therapy (INR 2.0–3.0) and high-intensity warfarin therapy (INR 3.1–4.0) in antiphospholipid syndrome. Thrombosis actually recurred more frequently in the high-intensity therapy group (10.7% vs 3.4%), with no significant difference in major bleeding events.
A reasonable strategy is to keep the INR between 2.5 and 3.0, keeping in mind that values fluctuate in any individual patient. A higher goal often leads to excessive anticoagulation and bleeding. If the goal is too low, recurrent thrombosis becomes more likely. There are fewer data on the newer oral anticoagulants, but their role is likely to increase as reversal agents are developed.
Recommendations published in 2003 for treating antiphospholipid syndrome include34:
- Warfarin (INR 2.0–3.0) after the first thrombotic event
- Warfarin (INR 3.0–4.0) if a clot develops despite warfarin
- Warfarin (INR > 3.0) for an arterial event.
For the rare but catastrophic antiphospholipid syndrome in which thrombosis occurs in multiple organs, recommendations are for heparin plus steroids, with or without intravenous immunoglobulin and plasmapheresis. This approach has not always been successful, and the mortality rate is high.
Treatment of asymptomatic carriers is uncertain
Treatment of asymptomatic carriers of the antiphospholipid antibody is controversial. Evidence for management is scarce; some experts recommend aspirin therapy, but benefit has yet to be proven in clinical trials.
Canaud et al35 documented the role of activation of the kinase mammalian target of rapamycin (mTOR) in the vascular changes characteristic of antiphospholipid nephropathy. Postkidney transplant surveillance biopsies of patients with antiphospholipid antibodies showed vascular damage occurring over time (despite patients being asymptomatic) compared with other renal transplant patients. Patients with antiphospholipid antibodies who were treated with the immunosuppressive drug sirolimus were protected from developing these changes. Twelve years after transplant, 70% of patients with antiphospholipid antibodies taking sirolimus still had a functioning graft compared with 11% of untreated patients.
Our knowledge of the pathogenesis of thrombotic microangiopathies has greatly advanced in the last decade, improving the diagnosis and treatment of these diseases.
Many conditions involve thrombotic microangiopathies (Table 1). This article reviews the most common ones, ie, thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, atypical hemolytic uremic syndrome, and antiphospholipid syndrome—their clinical features (focusing on the kidney), course, and management. Of note, although the diseases are similar, their pathogeneses and treatments differ.
DIFFERENT PATHWAYS TO MULTIORGAN THROMBOSIS
The thrombotic microangiopathies are multisystem disorders that can affect children and adults and often present with prominent renal and neurologic involvement. Endothelial injury is likely the inciting factor leading to thrombosis in the kidney and in many other organs. The causes variously include:
- Toxins from bacteria or drugs
- Abnormal complement activation, genetic or autoantibody-induced
- Procoagulant factors, eg, antiphospholipid antibodies
- Loss of anticoagulants, eg, from a defect of ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13); ADAMTS13 is also known as von Willebrand factor-cleaving protease
- Severe hypertension.
The histopathologic features are similar in all the thrombotic microangiopathies. Laboratory findings include thrombocytopenia, microangiopathic hemolytic anemia (with schistocytes on the peripheral blood smear), and high serum lactate dehydrogenase (LDH) levels; these are also markers of treatment progress. Bilirubin may be elevated and haptoglobin absent. Renal biopsy reveals thrombi in the glomeruli and arterioles.
THROMBOTIC THROMBOCYTOPENIC PURPURA
A young woman with fever, bruising, and renal failure, then blindness
A 36-year-old black woman who had been previously healthy presents to her doctor with fever and bruising.
Her hematocrit is 28% (reference range 38%–46%), platelet count 15 x 109/L (150–450), and prothrombin and partial thromboplastin times are normal. Her peripheral blood smear shows microangiopathic hemolytic anemia with schistocytes.
Over the next few days, her urine output declines and she develops sudden blindness followed by decreased mental acuity. Blood is drawn and sent for ADAMTS13 assay. Treatment is started at once with daily therapeutic plasma exchange. The assay results, when they arrive, show marked ADAMTS13 reduction (< 5%). Over the ensuing weeks, her mental acuity improves, her vision returns, and her renal function improves.
ADAMTS13 deficiency is definitive
Thrombotic thrombocytopenic purpura is characterized by:
- Neurologic abnormalities and acute renal failure
- Thrombocytopenia and microangiopathic hemolytic anemia
- Histologic evidence of thrombotic microangiopathy
- Deficiency of von Willebrand factor-cleaving protease (ADAMTS13 < 10%).
von Willebrand factor forms ultralarge multimers in the circulation that interact with platelets; these are normally cleaved by ADAMTS13. With ADAMTS13 deficiency (from either a genetic mutation or autoantibodies), the ultralarge multimers lead to coagulation as blood flows through small vessels.1
In 2003, Tsai2 evaluated 127 patients over age 10 who had thrombocytopenia and microangiopathic hemolysis with no plausible cause or features suggestive of hemolytic uremic syndrome. All were severely deficient in ADAMTS13. Subsequently, thrombotic thrombocytopenic purpura has been defined by a severe actual or effective deficiency of ADAMTS13.
Prompt plasma exchange is critical
Although the ADAMTS13 assay is important for diagnosing thrombotic thrombocytopenic purpura, in suspected cases daily plasma exchange should be started promptly, before test results return. Plasma exchange removes autoantibodies to ADAMTS13 from the blood, removes circulating ultralarge von Willebrand factor multimers, and replaces the missing ADAMTS13. Untreated, the disease is progressive, with irreversible renal failure, neurologic deterioration, and a 90% mortality rate. Plasma exchange reduces the mortality rate to less than 15%. If another diagnosis is confirmed, plasma exchange can be stopped.
Plasma exchange has been shown in clinical trials to be superior to plasma infusion in normalizing platelet counts and reducing mortality.3,4 Mortality rates were comparable with different replacement fluids vs fresh-frozen plasma, including solvent or detergent-treated plasma, and cryo-poor (cryosupernatant) plasma.4 Antiplatelet therapy, platelet transfusions, and splenectomy are ineffective.
Glucocorticoids for early treatment
An appropriate strategy is to add a glucocorticoid to plasma-exchange therapy at once (oral prednisone 1 mg/kg per day or intravenous methylprednisolone 125 mg twice daily) and withdraw it after several days if it is determined that it is not needed. Steroids for suspected thrombotic thrombocytopenic purpura can be justified for several reasons:
- The results of the ADAMTS13 assay are usually delayed, so steroids provide coverage for other diagnoses.
- They are helpful if thrombotic thrombocytopenic purpura is idiopathic (which is true for most cases) and if the patient has a poor response to initial therapy with plasma exchange.
- They are indicated for patients whose platelet counts do not increase with several days of plasma exchange or whose thrombocytopenia recurs as plasma exchange is decreased.
Rituximab improves survival
Rituximab, a chimeric (half murine) monoclonal antibody against CD19 and CD20 B cells, suppresses antibody production by knocking out the precursors of antibody-producing cells.
Anecdotal reports and small studies involving a total of 42 patients have been published on the use of rituximab for thrombotic thrombocytopenic purpura. Courses of rituximab varied greatly, from 1 to 13 weekly doses at 375 mg/m2, with 4 doses being the most common. Complete remission occurred in 90% of cases.5,6 A typical study from 2014 involved 48 patients (30 of whom received rituximab) followed by severe ADAMTS13 deficiency during remission.7 Despite the small study size, the investigators found significantly improved relapse-free survival rates with rituximab treatment.
But rituximab can cost $25,000 for 2 doses of 1,000 mg, and this will most likely prohibit its routine use. The cost and insurance coverage vary with location and policies.
Based on such studies, a reasonable strategy is to treat thrombotic thrombocytopenic purpura with:
- Daily plasma exchange
- Steroids, at least until the diagnosis is certain
- Rituximab if warranted.
New targeted therapies
Caplacizumab, a humanized immunoglobulin that inhibits the interaction between ultralarge von Willebrand factor multimers and platelets, has the potential to change this strategy when it receives US Food and Drug Administration approval, which is expected soon.
Peyvandi et al8 randomized 75 patients with acquired thrombotic thrombocytopenic purpura to either subcutaneous caplacizumab 10 mg daily for 30 days or placebo. Both groups had daily plasma exchange. The treatment group had a 39% reduction in median time to normalization of platelets vs the placebo group, and 3 of 36 patients had exacerbations, compared with 11 of 39 patients in the placebo group. Although 8 patients relapsed within the first month after stopping caplacizumab, their cases were brought under control. There were also more bleeding episodes with caplacizumab (54% vs 38%), most being mild to moderate. Two patients in the placebo group died, but none in the treatment group.
The fact that platelet normalization occurred significantly faster with caplacizumab, even in some patients who had not yet had plasma exchange therapy initiated, has enormous clinical significance. The low platelet count in thrombotic thrombocytopenic purpura is a marker of susceptibility to rapid damage to the brain and kidneys, so correcting it quickly is critical.
Other strategies for new drug development include replacing the deficient ADAMTS13 with a recombinant molecule and blocking antibody production (the same mode of action as rituximab and glucocorticoids).9 Using all 3 strategies to treat thrombotic thrombocytopenic purpura may be the future standard of care.
HEMOLYTIC UREMIC SYNDROME
A child with sudden onset of bloody diarrhea and kidney failure
A 4-year-old girl plays with baby animals at a petting zoo and does not wash her hands immediately afterwards. Three days later, she develops fever, abdominal cramps, nausea, vomiting, and bloody diarrhea. Her pediatrician gives her antibiotics. On day 6, she develops ecchymoses on the extremities and lips, thrombocytopenia, low urine output, and seizures. Her stool tests positive for Escherichia coli O157:H7
Classic presentation: Young patient with bloody diarrhea
The classic presentation of hemolytic uremic syndrome is of a young patient with bloody diarrhea typically lasting 5 to 10 days. Kidney failure may follow, requiring dialysis in about 60% of patients for a mean of 10 days. About one-fourth of patients develop neurologic symptoms, and about the same fraction are left with long-term morbidity, eg, hypertension, proteinuria, and reduced glomerular filtration rate. The mortality rate is typically 4%10,11 but varies with the outbreak.
Histologically, the kidneys look identical to those in thrombotic thrombocytopenic purpura, with thrombi in glomeruli and small vessels.
E coli is the most common culprit, but other bacteria, including Shigella dysenteriae, and viruses are sometimes the cause. Fewer than 10% of children infected with Shiga toxin-positive E coli, also known as enterohemorrhagic E coli (O157:H7, O104:H4), develop hemolytic uremic syndrome.
Lessons from outbreaks
Petting zoos are a common source of transmission of pathogenic bacteria. Disease can be extremely serious: in 15 cases linked to a Florida petting zoo, 3 children died.
Other outbreaks involving pathogenic E coli have been tied to fresh vegetables and to undercooked hamburger at fast-food chains.
In Germany in 2011, more than 3,000 people acquired Shiga toxin nonhemolytic uremic syndrome due to E coli, and 16 of them died. In addition, 845 acquired hemolytic uremic syndrome, and 36 died. This outbreak was associated with the more virulent and less common O104:H4 strain, which has acquired a Shiga toxin-encoding phage. Patients were treated with quinolone antibiotics, which actually increase toxin production in this strain.12
Unusual in the German epidemic was that more adults were affected (88%), especially women (68% of cases).13 The source of infection was eventually found to be alfalfa sprouts, the seeds of which had been contaminated by E coli. Women did not harbor any intrinsic factor making them more susceptible; rather, they were more likely to eat salads.13
Supportive management
Supportive care is most important. Transfusion with packed red blood cells is indicated for hemoglobin below 6 g/dL. Hypertension should be controlled and dialysis provided. For central nervous system involvement or severe disease, plasma exchange is sometimes used.
Eculizumab was tried for a time as therapy but did not prove to be of benefit. Shiga toxin-binding agents have been developed, but by the time they are given it is too late in the disease process to help.
Antibiotics may harm; it is possible that they kill beneficial bacteria, allowing the Shiga toxin-producing E coli to better proliferate. Antimotility agents also are contraindicated. Other agents not recommended include urokinase, heparin, dipyridamole, and vincristine. Splenectomy is not advised.
The most important way to control hemolytic uremic syndrome is to prevent it by thoroughly cooking meat, cleaning fresh produce, and having children wash their hands after petting animals.
ATYPICAL HEMOLYTIC UREMIC SYNDROME
A young man in renal failure
A 28-year-old man has a history of “thrombotic thrombocytopenic purpura-hemolytic uremic syndrome” at age 12. He slowly progresses to end-stage renal disease and receives a renal transplant from his mother at age 20 that fails after 3 months. The renal transplant biopsy report at the time reads “thrombotic microangiopathy.” The patient’s brother also requires dialysis.
The patient’s complement values are low, especially C3. His father is offering him a kidney at this time, and the patient wants to know whether to proceed.
Normal ADAMTS13, no diarrhea
Hemolytic uremic syndrome without diarrhea is now called atypical hemolytic uremic syndrome. Patients have normal levels of ADAMTS13, do not have diarrhea, and have no evidence of Shiga toxin-producing E coli.
Continuous complement pathway activation
The complement system is part of the innate immune system, which provides immediate defense against infection and does not evolve as does the adaptive immune system. The classic complement pathway is activated by the C1 antibody-antigen complex. The alternative complement pathway leads to the same pathway via C3.14 Both pathways lead to the formation of C5 through C9 membrane attack complexes, which form channels across the membranes of target cells, leading to cell lysis and death.
The alternate pathway does not require an antibody trigger so is always active at a low level. Inhibitory factors (factor H, factor I, membrane cofactor protein, factor H-related proteins) are naturally present and slow it down at various steps. People who are born with an abnormal factor or, more commonly, develop antibodies against one of the factors, have uninhibited complement activation. If this happens in the blood vessels, massive coagulation and atypical hemolytic uremic syndrome ensues. The endothelial damage and clotting in the brain, kidney, and other organs are identical to that of hemolytic uremic syndrome caused by Shiga toxin.
Treat with eculizumab
Historically, atypical hemolytic uremic syndrome was treated with plasma exchange, which replaces defective complement regulatory proteins and removes inhibitory antibodies.
Understanding the complement pathways is key to developing drugs that target atypical hemolytic uremic syndrome, and about 60 are in the pipeline. The only one currently approved in the United States for atypical hemolytic uremic syndrome is eculizumab, a humanized monoclonal antibody that binds with high affinity to C5, blocking the end of the complement cascade and preventing formation of the membrane attack complex.15–18
The effects of eculizumab on atypical hemolytic uremic syndrome were studied in 2 prospective trials.19 Platelet counts rose rapidly within weeks of starting treatment, and kidney function improved. Benefits continued throughout the 64 weeks studied. There were no deaths among the 37 patients enrolled, and although these were single-arm trials, they provide evidence of dramatic benefit considering the high mortality risk of this disease.
Eculizumab is now considered the treatment of choice. It may be used empirically for patients with hemolytic uremic syndrome who test negative for Shiga toxin and antiphospholipid antibody, and who do not have a very low level of ADAMTS13. The big drawback of eculizumab is its high price,20–22 which varies by amount used, location, and pharmacy negotiation, but can be in the hundreds of thousands of dollars.
For a patient with atypical hemolytic uremic syndrome on dialysis, treatment with eculizumab should continue for 4 to 6 months if there are no extrarenal manifestations. But many patients continue to have the defect in the complement system, so the problem may recur.
Case revisited
For our patient considering a kidney transplant, many experts feel that a transplant can be done as long as platelet counts are monitored and treatment with eculizumab is restarted if needed. One can also make the case for waiting a few years for new oral drugs to become available before offering transplant.
ANTIPHOSPHOLIPID SYNDROME
A young woman with a history of thrombosis and miscarriages
A 27-year-old woman presents with arthralgias, low-grade fever, and malaise. She has a history of 3 spontaneous abortions and Raynaud phenomenon. Two years ago, she had deep vein thrombosis of the right calf after a long automobile trip.
She now has swollen metacarpophalangeal and proximal interphalangeal joints, livedo reticularis (a mottled venous pattern of the skin best seen under fluorescent light) of the legs and arms, and ankle edema (2-cm indentation).
Her blood pressure is 152/92 mm Hg. Laboratory values:
- White blood cell count 3.6 × 109/L (reference range 4.5–11.0)
- Hematocrit 24% (36%–47%)
- Platelet count 89 × 109/L (150–450)
- Urinalysis: protein 4+, heme 3+, red blood cells 8–15 per high-power field (< 3), red blood cell casts present
- Blood urea nitrogen 43 mg/dL (10–20)
- Creatinine 2.6 mg/dL (0.5–1.1).
- Prothrombin time 14.6 s (10–14)
- Partial thromboplastin time 85 s (25–35)
- Antinuclear antibody positive at 1:160
- Anti-double-stranded DNA and serum complement normal
- Syphilis serologic screening (VDRL) positive.
The patient has leukopenia, anemia, thrombocytopenia, hematuria, proteinuria, high blood urea nitrogen, and markedly elevated partial thromboplastin time. Although she has a positive antinuclear antibody test and renal dysfunction, her anti-dsDNA and serum complement tests are normal, making the diagnosis of systemic lupus erythematosus unlikely.
Consider antiphospholipid syndrome
In any patient with multiple pregnancy losses, lupus, or a history of thrombosis, antiphospholipid syndrome should be considered.
In a series of patients with antiphospholipid antibody who underwent kidney biopsy, more than half were men, indicating that, unlike lupus, this is not primarily a disease of young women.
Diagnosis based on specific criteria
Clinical criteria require at least one of the following in the patient’s history23:
- One or more episodes of arterial, venous, or small-vessel thrombosis
- Unexplained pregnancy morbidity (death of a fetus or neonate with normal morphology or 3 or more spontaneous abortions).
Serologic criteria for any of the following antiphospholipid antibodies require that at least one of the following tests be positive at least twice and at least 12 weeks apart:
- Anticardiolipin antibodies—high-titer immunoglobulin (Ig) G or IgM
- Autoantibodies for beta 2-glycoprotein
- Lupus anticoagulant—autoantibodies that increase clotting time in vitro and target beta 2-glycoprotein I and prothrombin (despite its name and actions in vitro, lupus anticoagulant functions as a coagulant).
As with the other thrombotic microangiopathies, patients with anticardiolipin syndrome have microthrombi in the glomeruli and blood vessels that are evident on kidney biopsy.
Suspect condition in likely groups
Antiphospholipid syndrome is surprisingly common.24 In a case-control study, de Groot et al25 found that 3.1% of patients under age 70 with a first episode of venous thrombosis and no known cancer were positive for lupus anticoagulant vs 0.9% of controls. In another case-control study, Urbanus et al26 found that 17% of women under age 50 with a stroke tested positive for lupus anticoagulant compared with less than 1% of controls. Because of such studies, it has become routine to test for anticardiolipin and lupus anticoagulant in young patients presenting with a stroke.
About 1% of women trying to have children have recurrent miscarriages, and of these, 10% to 15% have antiphospholipid antibody present.27–30
Pathogenesis
Patients with antiphospholipid syndrome have a much higher proportion of plasma beta 2-glycoprotein in the oxidized form than do healthy controls. The level is also higher than in patients with a different autoimmune disease whether or not they have antibodies against beta 2-glycoprotein 1. Although about 40% of patients with lupus have an anticardiolipin antibody, only a small percentage develop antiphospholipid syndrome with clotting.
It is thought that antiphospholipid syndrome involves initial injury to the endothelium, then potentiation of thrombus formation. Oxidized beta 2-glycoprotein complexes may bind to the endothelial cell surface, causing it to become the target of antibodies. The exact relationships between the factors are not yet understood.
The risk of a thrombotic event in an asymptomatic patient positive for all 3 factors—lupus anticoagulant, anticardiolipin antibody, and anti-beta 2-glycoprotein I antibody—is more than 5% per year.31
Manage thrombosis with anticoagulation
Khamashta et al,32 in a 1995 study, retrospectively studied patients with antiphospholipid antibodies and a history of thrombosis. Of 147 patients, 66 had idiopathic primary disease, 62 had systemic lupus, and 19 had “lupus-like” disease. Almost 70% (101 patients) had a recurrence of thrombosis, totaling 186 events. The mean time to recurrence was 12 months (range 2 weeks to 12 years). Recurrence rates were 0.01 events per patient per year with high-dose warfarin, 0.23 with low-dose warfarin, and 0.18 with aspirin. But the highest bleeding rates were in the 6 months after warfarin withdrawal; 29 patients had bleeding events, one-fourth of which were severe.
Standard therapy has become anticoagulation, starting with heparin or enoxaparin, then warfarin. There is inadequate evidence for the role of newer oral anticoagulant therapy.
A very high INR is not generally better than a moderately elevated level
For a time, it was thought that the international normalized ratio (INR) should be kept on the very high side to prevent thrombosis.
Crowther et al33 conducted a randomized, double-blind trial comparing moderate warfarin therapy (INR 2.0–3.0) and high-intensity warfarin therapy (INR 3.1–4.0) in antiphospholipid syndrome. Thrombosis actually recurred more frequently in the high-intensity therapy group (10.7% vs 3.4%), with no significant difference in major bleeding events.
A reasonable strategy is to keep the INR between 2.5 and 3.0, keeping in mind that values fluctuate in any individual patient. A higher goal often leads to excessive anticoagulation and bleeding. If the goal is too low, recurrent thrombosis becomes more likely. There are fewer data on the newer oral anticoagulants, but their role is likely to increase as reversal agents are developed.
Recommendations published in 2003 for treating antiphospholipid syndrome include34:
- Warfarin (INR 2.0–3.0) after the first thrombotic event
- Warfarin (INR 3.0–4.0) if a clot develops despite warfarin
- Warfarin (INR > 3.0) for an arterial event.
For the rare but catastrophic antiphospholipid syndrome in which thrombosis occurs in multiple organs, recommendations are for heparin plus steroids, with or without intravenous immunoglobulin and plasmapheresis. This approach has not always been successful, and the mortality rate is high.
Treatment of asymptomatic carriers is uncertain
Treatment of asymptomatic carriers of the antiphospholipid antibody is controversial. Evidence for management is scarce; some experts recommend aspirin therapy, but benefit has yet to be proven in clinical trials.
Canaud et al35 documented the role of activation of the kinase mammalian target of rapamycin (mTOR) in the vascular changes characteristic of antiphospholipid nephropathy. Postkidney transplant surveillance biopsies of patients with antiphospholipid antibodies showed vascular damage occurring over time (despite patients being asymptomatic) compared with other renal transplant patients. Patients with antiphospholipid antibodies who were treated with the immunosuppressive drug sirolimus were protected from developing these changes. Twelve years after transplant, 70% of patients with antiphospholipid antibodies taking sirolimus still had a functioning graft compared with 11% of untreated patients.
- Sadler JE. Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. Blood 2008; 112:11–18.
- Tsai HM. Advances in the pathogenesis, diagnosis, and treatment of thrombotic thrombocytopenic purpura. J Am Soc Nephrol 2003; 14:1072–1081.
- Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med 1991; 325:393–397.
- Brunskill SJ, Tusold A, Benjamin S, Stanworth SJ, Murphy MF. A systematic review of randomized controlled trials for plasma exchange in the treatment of thrombotic thrombocytopenic purpura. Transfus Med 2007; 17:17–35.
- Jasti S, Coyle T, Gentile T, Rosales L, Poiesz B. Rituximab as an adjunct to plasma exchange in TTP: a report of 12 cases and review of literature. J Clin Apher 2008; 23:151–156.
- Ling HT, Field JJ, Blinder MA. Sustained response with rituximab in patients with thrombotic thrombocytopenic purpura: a report of 13 cases and review of the literature. Am J Hematol 2009; 84:418–421.
- Hie M, Gay J, Galicier L, et al; French Thrombotic Microangiopathies Reference Centre. Preemptive rituximab infusions after remission efficiently prevent relapses in acquired thrombotic thrombocytopenic purpura. Blood 2014; 124:204–210.
- Peyvandi F, Scully M, Kremer Hovinga JA, et al; TITAN Investigators. Caplacizumab for acquired thrombotic thrombocytopenic purpura. N Engl J Med 2016; 374:511–522.
- Veyradier A. Von Willebrand factor—a new target for TTP treatment? N Engl J Med 2016; 374:583–585.
- Boyce TG, Swerdlow DL, Griffin PM. Escherichia coli O157:H7 and the hemolytic-uremic syndrome. N Engl J Med 1995; 333:364–368.
- Gerber A, Karch H, Allerberger F, Verweyen HM, Zimmerhackl LB. Clinical course and the role of Shiga toxin-producing Escherichia coli infection in the hemolytic-uremic syndrome in pediatric patients, 1997–2000, in Germany and Austria: a prospective study. J Infect Dis 2002; 186:493–500.
- Rasko DA, Webster DR, Sahl JW, et al. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N Engl J Med 2011; 365:709–717.
- Frank C, Werber D, Cramer JP, et al; HUS Investigation Team. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N Engl J Med 2011; 365:1771–1780.
- Bomback AS, Appel GB. Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nat Rev Nephrol 2012; 8:634–642.
- Figueroa JE, Densen P. Infectious diseases associated with complement deficiencies. Clin Microbiol Rev 1991; 4:359–395.
- Walport MJ. Complement. First of two parts. N Engl J Med 2001; 344:1058–1066.
- Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol 2007; 25:1256–1264.
- Soliris (eculizumab). Prescribing information. Alexion Pharmaceuticals, Inc.
- Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic–uremic syndrome. N Engl J Med 2013; 368:2169–2181.
- Kim JJ, Waller SC, Reid CJ. Eculizumab in atypical haemolytic-uraemic syndrome allows cessation of plasma exchange and dialysis. Clin Kidney J 2012; 5:34–36.
- Povey H, Vundru R, Junglee N, Jibani M. Renal recovery with eculizumab in atypical hemolytic uremic syndrome following prolonged dialysis. Clin Nephrol 2014; 82:326–331.
- Gargau M, Azancot M, Ramos R, Sanchez-Corral P, Montero MA, Seron D. Early treatment with eculizumab may be beneficial in atypical haemolytic uraemic syndrome. Clin Kidney J 2012; 5:1–3.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Giannakopoulos B, Krilis SA. The pathogenesis of the antiphospholipid syndrome. N Engl J Med 2013; 368:1033–1044.
- de Groot PG, Lutters B, Derksen RH, Lisman T, Meijers JC, Rosendaal FR. Lupus anticoagulants and the risk of a first episode of deep venous thrombosis. J Thromb Haemost 2005; 3:1993–1997.
- Urbanus RT, Siegerink B, Roest M, Rosendaal FR, de Groot PG, Algra A. Antiphospholipid antibodies and risk of myocardial infarction and ischaemic stroke in young women in the RATIO study: a case-control study. Lancet Neurol 2009; 8:998–1005.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Stirrat GM. Recurrent miscarriage I: definition and epidemiology. Lancet 1990; 336:673–675.
- Rai RS, Regan L, Clifford K, et al. Antiphospholipid antibodies and beta 2-glycoprotein-I in 500 women with recurrent miscarriage: results of a comprehensive screening approach. Hum Reprod 1995; 10:2001–2005.
- Yetman DL, Kutteh WH. Antiphospholipid antibody panels and recurrent pregnancy loss: prevalence of anticardiolipin antibodies compared with other antiphospholipid antibodies. Fertil Steril 1996; 66:540–546.
- Pengo V, Ruffatti A, Legnani C, et al. Incidence of a first thromboembolic event in asymptomatic carriers of high-risk antiphospholipid antibody profile: a multicenter prospective study. Blood 2011; 118:4714–4718.
- Khamashta MA, Cuadrado MJ, Mujic F, Taub NA, Hunt BJ, Hughes GR. The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med 1995; 332:993–997.
- Crowther MA, Ginsberg JS, Julian J, et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N Engl J Med 2003; 349:1133–1138.
- Lockshin M, Tenedios F, Petri M, et al. Cardiac disease in the antiphospholipid syndrome: recommendations for treatment. Committee consensus report. Lupus 2003; 12:518–523.
- Canaud G, Bienaimé F, Tabarin F, et al. Inhibition of the mTORC pathway in the antiphospholipid syndrome. N Engl J Med 2014; 371:303–312.
- Sadler JE. Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. Blood 2008; 112:11–18.
- Tsai HM. Advances in the pathogenesis, diagnosis, and treatment of thrombotic thrombocytopenic purpura. J Am Soc Nephrol 2003; 14:1072–1081.
- Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med 1991; 325:393–397.
- Brunskill SJ, Tusold A, Benjamin S, Stanworth SJ, Murphy MF. A systematic review of randomized controlled trials for plasma exchange in the treatment of thrombotic thrombocytopenic purpura. Transfus Med 2007; 17:17–35.
- Jasti S, Coyle T, Gentile T, Rosales L, Poiesz B. Rituximab as an adjunct to plasma exchange in TTP: a report of 12 cases and review of literature. J Clin Apher 2008; 23:151–156.
- Ling HT, Field JJ, Blinder MA. Sustained response with rituximab in patients with thrombotic thrombocytopenic purpura: a report of 13 cases and review of the literature. Am J Hematol 2009; 84:418–421.
- Hie M, Gay J, Galicier L, et al; French Thrombotic Microangiopathies Reference Centre. Preemptive rituximab infusions after remission efficiently prevent relapses in acquired thrombotic thrombocytopenic purpura. Blood 2014; 124:204–210.
- Peyvandi F, Scully M, Kremer Hovinga JA, et al; TITAN Investigators. Caplacizumab for acquired thrombotic thrombocytopenic purpura. N Engl J Med 2016; 374:511–522.
- Veyradier A. Von Willebrand factor—a new target for TTP treatment? N Engl J Med 2016; 374:583–585.
- Boyce TG, Swerdlow DL, Griffin PM. Escherichia coli O157:H7 and the hemolytic-uremic syndrome. N Engl J Med 1995; 333:364–368.
- Gerber A, Karch H, Allerberger F, Verweyen HM, Zimmerhackl LB. Clinical course and the role of Shiga toxin-producing Escherichia coli infection in the hemolytic-uremic syndrome in pediatric patients, 1997–2000, in Germany and Austria: a prospective study. J Infect Dis 2002; 186:493–500.
- Rasko DA, Webster DR, Sahl JW, et al. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N Engl J Med 2011; 365:709–717.
- Frank C, Werber D, Cramer JP, et al; HUS Investigation Team. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N Engl J Med 2011; 365:1771–1780.
- Bomback AS, Appel GB. Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nat Rev Nephrol 2012; 8:634–642.
- Figueroa JE, Densen P. Infectious diseases associated with complement deficiencies. Clin Microbiol Rev 1991; 4:359–395.
- Walport MJ. Complement. First of two parts. N Engl J Med 2001; 344:1058–1066.
- Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol 2007; 25:1256–1264.
- Soliris (eculizumab). Prescribing information. Alexion Pharmaceuticals, Inc.
- Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic–uremic syndrome. N Engl J Med 2013; 368:2169–2181.
- Kim JJ, Waller SC, Reid CJ. Eculizumab in atypical haemolytic-uraemic syndrome allows cessation of plasma exchange and dialysis. Clin Kidney J 2012; 5:34–36.
- Povey H, Vundru R, Junglee N, Jibani M. Renal recovery with eculizumab in atypical hemolytic uremic syndrome following prolonged dialysis. Clin Nephrol 2014; 82:326–331.
- Gargau M, Azancot M, Ramos R, Sanchez-Corral P, Montero MA, Seron D. Early treatment with eculizumab may be beneficial in atypical haemolytic uraemic syndrome. Clin Kidney J 2012; 5:1–3.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Giannakopoulos B, Krilis SA. The pathogenesis of the antiphospholipid syndrome. N Engl J Med 2013; 368:1033–1044.
- de Groot PG, Lutters B, Derksen RH, Lisman T, Meijers JC, Rosendaal FR. Lupus anticoagulants and the risk of a first episode of deep venous thrombosis. J Thromb Haemost 2005; 3:1993–1997.
- Urbanus RT, Siegerink B, Roest M, Rosendaal FR, de Groot PG, Algra A. Antiphospholipid antibodies and risk of myocardial infarction and ischaemic stroke in young women in the RATIO study: a case-control study. Lancet Neurol 2009; 8:998–1005.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Stirrat GM. Recurrent miscarriage I: definition and epidemiology. Lancet 1990; 336:673–675.
- Rai RS, Regan L, Clifford K, et al. Antiphospholipid antibodies and beta 2-glycoprotein-I in 500 women with recurrent miscarriage: results of a comprehensive screening approach. Hum Reprod 1995; 10:2001–2005.
- Yetman DL, Kutteh WH. Antiphospholipid antibody panels and recurrent pregnancy loss: prevalence of anticardiolipin antibodies compared with other antiphospholipid antibodies. Fertil Steril 1996; 66:540–546.
- Pengo V, Ruffatti A, Legnani C, et al. Incidence of a first thromboembolic event in asymptomatic carriers of high-risk antiphospholipid antibody profile: a multicenter prospective study. Blood 2011; 118:4714–4718.
- Khamashta MA, Cuadrado MJ, Mujic F, Taub NA, Hunt BJ, Hughes GR. The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med 1995; 332:993–997.
- Crowther MA, Ginsberg JS, Julian J, et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N Engl J Med 2003; 349:1133–1138.
- Lockshin M, Tenedios F, Petri M, et al. Cardiac disease in the antiphospholipid syndrome: recommendations for treatment. Committee consensus report. Lupus 2003; 12:518–523.
- Canaud G, Bienaimé F, Tabarin F, et al. Inhibition of the mTORC pathway in the antiphospholipid syndrome. N Engl J Med 2014; 371:303–312.
KEY POINTS
- Thrombotic thrombocytopenic purpura is diagnosed with the ADAMTS13 assay. As soon as it is suspected, it should be treated with daily plasma exchange, steroids (at least until the diagnosis is certain), and, if additional treatment is needed, rituximab.
- Hemolytic uremic syndrome is seen in children who handle farm animals and in children and adults in food outbreaks. It is managed supportively with transfusion of packed red blood cells and dialysis.
- Atypical hemolytic uremic syndrome should be suspected in patients with normal ADAMTS13 and without diarrhea or evidence of Shiga toxin-producing Escherichia coli. It often responds well to eculizumab, a blocker of C5 (the fifth component of complement).
- Antiphospholipid syndrome should be investigated in women who have multiple miscarriages or thrombotic events. Symptomatic disease requires long-term anticoagulation therapy.