User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Quo vadis, psychopharmacology?
The psychopharmacology era that began 6 decades ago has had a momentous and transformative effect on the identity and practice of psychiatry. It enabled community-based treatment and follow-up to supplant institutional warehousing of persons with serious mental disorders. The discovery of neurotransmitter pathways and receptors involved in the mechanism of action of antipsychotic, antidepressant, and anxiolytic agents sparked the neuroscience revolution that has become 1 of the fastest-moving frontiers in medicine.
Over the past few years, the shine seems to have worn off and psychopharmacology appears to be in limbo between the serendipitous but aging discoveries of the past and the exciting but unfulfilled promise of future breakthroughs. Psychopharmacology is in urgent need of a renaissance to propel it into new directions that will maintain its credibility as the core of psychiatric therapeutics. The following are some issues and challenges that may influence how psychopharmacology can surge forward and restore its “mojo.”
Scientific challenges. Psychopharmacology must decisively move from serendipity and its corollaries to rational, pathophysiology-based drug development. We need a translational “Marshall Plan” to exploit genetic and molecular neurobiology advances to develop radically new pharmacologic biotherapies for psychiatric brain disorders.
Conceptual challenges. As long as psychiatric diagnoses are based on clusters of symptoms assembled by committees, it makes little sense for the FDA to mandate that a drug must work for a DSM diagnosis instead of specific symptoms. Psychiatric disorders share many symptoms such as depressed mood, anxiety, agitation, hallucinations, delusions, insomnia, impulsivity, etc. Approving new drugs for target symptoms rather than a DSM diagnosis might eliminate the often deplored—yet necessary—practice referred to as “off-label” pharmacotherapy. Frankly, it is silly that an antipsychotic must be approved separately for schizophrenia, schizoaffective disorder, psychotic mania, delusional disorder, or brief reactive psychosis when these disorders all share delusions or hallucinations that respond to that same agent. The high cost of conducting redundant clinical trials for all antipsychotic drugs in each psychotic disorder is far better invested in discovering agents with new mechanisms of action.
Disease heterogeneity. Research strongly points to a substantial heterogeneity in practically every psychiatric disorder, with multiple genotypes and phenotypes that share common features. Therefore, there will always be full responders, partial responders, and refractory patients in any psychiatric illness. Rational psychopharmacology must develop strategies to prospectively identify these subgroups by using pharmacogenetic markers that should become a vital component of guiding treatment selection.
Big picture issues
Who will spearhead the psychopharmacology of the future? There is a tremendous unmet need, with >80% of psychiatric disorders having no FDA-approved medication, and a substantial proportion of patients who do receive an approved drug often remain disabled even after symptomatic improvement. This unmet need is not just for new drugs, but more effective drugs.
Funding. It is expensive to develop new mediations. Only the private sector (pharmaceutical industry) develops drugs for psychiatry. Unless the government decides to allocate a trillion dollars to take over that role, it should provide incentives to attract the private sector to invest in psychiatry instead of abandoning it, as some companies recently have done. One possibility is to substantially extend the patent life for a medication with a new mechanism of action. This will spur innovation and benefit millions of sick individuals.
Medico-legal liability. The antidote to innovation is a class-action lawsuit. All drugs will inevitably cause side effects. When millions of people receive a vaccine during an epidemic, a couple hundred may die or suffer serious side effects. Imagine if vaccine development stops and many millions die as a consequence. The FDA currently approves a drug after careful study. Therefore, shouldn’t the FDA share liability for unexpected serious adverse effects or waive such liability altogether if these effects were completely unforeseen during the clinical trials?
So quo vadis, psychopharmacology? This temporary lull is worrisome but there is reason to believe brighter days lie ahead. However, it is obvious that innovative advances in psychopharmacology are not only dependent on scientific breakthroughs but also on completely new paradigms of clinical diagnosis, less rigid regulatory policies, creative financing, and a change in liability laws that stifle drug development. All these are feasible and achievable. This is a time to stop dithering and to start envisioning new directions. Millions of patients are eagerly awaiting the psychopharmacology of the future.

Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, visit http://www.facebook.com/CurrentPsychiatry, or click on the “Send Letters” link below.
Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, visit http://www.facebook.com/CurrentPsychiatry, or click on the “Send Letters” link below.
Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, visit http://www.facebook.com/CurrentPsychiatry, or click on the “Send Letters” link below.
The psychopharmacology era that began 6 decades ago has had a momentous and transformative effect on the identity and practice of psychiatry. It enabled community-based treatment and follow-up to supplant institutional warehousing of persons with serious mental disorders. The discovery of neurotransmitter pathways and receptors involved in the mechanism of action of antipsychotic, antidepressant, and anxiolytic agents sparked the neuroscience revolution that has become 1 of the fastest-moving frontiers in medicine.
Over the past few years, the shine seems to have worn off and psychopharmacology appears to be in limbo between the serendipitous but aging discoveries of the past and the exciting but unfulfilled promise of future breakthroughs. Psychopharmacology is in urgent need of a renaissance to propel it into new directions that will maintain its credibility as the core of psychiatric therapeutics. The following are some issues and challenges that may influence how psychopharmacology can surge forward and restore its “mojo.”
Scientific challenges. Psychopharmacology must decisively move from serendipity and its corollaries to rational, pathophysiology-based drug development. We need a translational “Marshall Plan” to exploit genetic and molecular neurobiology advances to develop radically new pharmacologic biotherapies for psychiatric brain disorders.
Conceptual challenges. As long as psychiatric diagnoses are based on clusters of symptoms assembled by committees, it makes little sense for the FDA to mandate that a drug must work for a DSM diagnosis instead of specific symptoms. Psychiatric disorders share many symptoms such as depressed mood, anxiety, agitation, hallucinations, delusions, insomnia, impulsivity, etc. Approving new drugs for target symptoms rather than a DSM diagnosis might eliminate the often deplored—yet necessary—practice referred to as “off-label” pharmacotherapy. Frankly, it is silly that an antipsychotic must be approved separately for schizophrenia, schizoaffective disorder, psychotic mania, delusional disorder, or brief reactive psychosis when these disorders all share delusions or hallucinations that respond to that same agent. The high cost of conducting redundant clinical trials for all antipsychotic drugs in each psychotic disorder is far better invested in discovering agents with new mechanisms of action.
Disease heterogeneity. Research strongly points to a substantial heterogeneity in practically every psychiatric disorder, with multiple genotypes and phenotypes that share common features. Therefore, there will always be full responders, partial responders, and refractory patients in any psychiatric illness. Rational psychopharmacology must develop strategies to prospectively identify these subgroups by using pharmacogenetic markers that should become a vital component of guiding treatment selection.
Big picture issues
Who will spearhead the psychopharmacology of the future? There is a tremendous unmet need, with >80% of psychiatric disorders having no FDA-approved medication, and a substantial proportion of patients who do receive an approved drug often remain disabled even after symptomatic improvement. This unmet need is not just for new drugs, but more effective drugs.
Funding. It is expensive to develop new mediations. Only the private sector (pharmaceutical industry) develops drugs for psychiatry. Unless the government decides to allocate a trillion dollars to take over that role, it should provide incentives to attract the private sector to invest in psychiatry instead of abandoning it, as some companies recently have done. One possibility is to substantially extend the patent life for a medication with a new mechanism of action. This will spur innovation and benefit millions of sick individuals.
Medico-legal liability. The antidote to innovation is a class-action lawsuit. All drugs will inevitably cause side effects. When millions of people receive a vaccine during an epidemic, a couple hundred may die or suffer serious side effects. Imagine if vaccine development stops and many millions die as a consequence. The FDA currently approves a drug after careful study. Therefore, shouldn’t the FDA share liability for unexpected serious adverse effects or waive such liability altogether if these effects were completely unforeseen during the clinical trials?
So quo vadis, psychopharmacology? This temporary lull is worrisome but there is reason to believe brighter days lie ahead. However, it is obvious that innovative advances in psychopharmacology are not only dependent on scientific breakthroughs but also on completely new paradigms of clinical diagnosis, less rigid regulatory policies, creative financing, and a change in liability laws that stifle drug development. All these are feasible and achievable. This is a time to stop dithering and to start envisioning new directions. Millions of patients are eagerly awaiting the psychopharmacology of the future.
The psychopharmacology era that began 6 decades ago has had a momentous and transformative effect on the identity and practice of psychiatry. It enabled community-based treatment and follow-up to supplant institutional warehousing of persons with serious mental disorders. The discovery of neurotransmitter pathways and receptors involved in the mechanism of action of antipsychotic, antidepressant, and anxiolytic agents sparked the neuroscience revolution that has become 1 of the fastest-moving frontiers in medicine.
Over the past few years, the shine seems to have worn off and psychopharmacology appears to be in limbo between the serendipitous but aging discoveries of the past and the exciting but unfulfilled promise of future breakthroughs. Psychopharmacology is in urgent need of a renaissance to propel it into new directions that will maintain its credibility as the core of psychiatric therapeutics. The following are some issues and challenges that may influence how psychopharmacology can surge forward and restore its “mojo.”
Scientific challenges. Psychopharmacology must decisively move from serendipity and its corollaries to rational, pathophysiology-based drug development. We need a translational “Marshall Plan” to exploit genetic and molecular neurobiology advances to develop radically new pharmacologic biotherapies for psychiatric brain disorders.
Conceptual challenges. As long as psychiatric diagnoses are based on clusters of symptoms assembled by committees, it makes little sense for the FDA to mandate that a drug must work for a DSM diagnosis instead of specific symptoms. Psychiatric disorders share many symptoms such as depressed mood, anxiety, agitation, hallucinations, delusions, insomnia, impulsivity, etc. Approving new drugs for target symptoms rather than a DSM diagnosis might eliminate the often deplored—yet necessary—practice referred to as “off-label” pharmacotherapy. Frankly, it is silly that an antipsychotic must be approved separately for schizophrenia, schizoaffective disorder, psychotic mania, delusional disorder, or brief reactive psychosis when these disorders all share delusions or hallucinations that respond to that same agent. The high cost of conducting redundant clinical trials for all antipsychotic drugs in each psychotic disorder is far better invested in discovering agents with new mechanisms of action.
Disease heterogeneity. Research strongly points to a substantial heterogeneity in practically every psychiatric disorder, with multiple genotypes and phenotypes that share common features. Therefore, there will always be full responders, partial responders, and refractory patients in any psychiatric illness. Rational psychopharmacology must develop strategies to prospectively identify these subgroups by using pharmacogenetic markers that should become a vital component of guiding treatment selection.
Big picture issues
Who will spearhead the psychopharmacology of the future? There is a tremendous unmet need, with >80% of psychiatric disorders having no FDA-approved medication, and a substantial proportion of patients who do receive an approved drug often remain disabled even after symptomatic improvement. This unmet need is not just for new drugs, but more effective drugs.
Funding. It is expensive to develop new mediations. Only the private sector (pharmaceutical industry) develops drugs for psychiatry. Unless the government decides to allocate a trillion dollars to take over that role, it should provide incentives to attract the private sector to invest in psychiatry instead of abandoning it, as some companies recently have done. One possibility is to substantially extend the patent life for a medication with a new mechanism of action. This will spur innovation and benefit millions of sick individuals.
Medico-legal liability. The antidote to innovation is a class-action lawsuit. All drugs will inevitably cause side effects. When millions of people receive a vaccine during an epidemic, a couple hundred may die or suffer serious side effects. Imagine if vaccine development stops and many millions die as a consequence. The FDA currently approves a drug after careful study. Therefore, shouldn’t the FDA share liability for unexpected serious adverse effects or waive such liability altogether if these effects were completely unforeseen during the clinical trials?
So quo vadis, psychopharmacology? This temporary lull is worrisome but there is reason to believe brighter days lie ahead. However, it is obvious that innovative advances in psychopharmacology are not only dependent on scientific breakthroughs but also on completely new paradigms of clinical diagnosis, less rigid regulatory policies, creative financing, and a change in liability laws that stifle drug development. All these are feasible and achievable. This is a time to stop dithering and to start envisioning new directions. Millions of patients are eagerly awaiting the psychopharmacology of the future.
Is there a rational management strategy for tardive dyskinesia?
Introduced into clinical practice more than a half century ago, antipsychotics are still the mainstay of schizophrenia treatment. However, from the earliest reports, antipsychotic efficacy was seemingly inseparable from extrapyramidal side effects (EPS) that manifested as acute and chronic involuntary movement disorders. Although acute extrapyramidal side effects could be prevented and treated, the late-arising symptoms of tardive dyskinesia (TD) seemed irreversible in most cases.
Concerns over TD stimulated extensive research and fueled efforts to develop new antipsychotics that spared the extrapyramidal motor system. Numerous industry-sponsored trials found a reduced risk of EPS—including TD—with newer, second-generation antipsychotics (SGAs), although this advantage diminished when modest doses of low- or mid-potency first-generation antipsychotics (FGAs) were used as the comparator.1-3 Nevertheless, in addition to the continued potential risk of introducing new cases of TD—even with SGAs—several other factors underscore the need to develop a rational strategy for clinical management of TD, including:
- thousands of patients are left with TD as a legacy of past treatment
- the neurophysiologic mechanisms underlying TD are not well understood
- there is no uniformly effective treatment to reverse TD
- TD may be irreversible in most cases.
Prevention
Because there is no “gold standard” treatment for TD, it is important to minimize the risk of TD by taking preventive measures and detecting incipient signs of the disorder. Preventive principles include:
- confirming and documenting the indication for antipsychotics
- using conservative maintenance doses and opting for lower potency or newer agents
- informing patients and caregivers of risk
- assessing for incipient signs of TD using the Abnormal Involuntary Movement Scale (AIMS),4 which should be administered at least every 3 to 6 months.
Confirming the diagnosis
TD presents as a polymorphous involuntary movement disorder,5-8 most often with nonrhythmic, repetitive, purposeless hyperkinetic symptoms. It usually affects orofacial and lingual musculature (“buccolinguomasticatory syndrome”) with chewing; bruxism; protrusion, curling, or twisting of the tongue; lip smacking, puckering, sucking, and pursing; retraction, grimacing or bridling of the mouth; bulging of the cheeks; or eye blinking and blepharospasm. Choreoathetoid movements of the fingers, hands, or upper or lower extremities also are common. Patients may experience axial symptoms affecting the neck, shoulders, spine, or pelvis. When severe, dyskinesias can affect breathing, swallowing, or speech, and interfere with walking and activities of daily living.
TD may present with nonchoreoathetoid symptoms that can be difficult to distinguish from acute EPS. These may co-exist with classic TD symptoms, but may represent separate subtypes with increased risk of progression, persistence, and severe disability. For example, tardive dystonia, which is estimated to occur in 1% to 4% of patients treated with antipsychotics,9 may be more generalized and disabling than TD, and may respond to anticholinergic agents. Akathisia and other movement disorders also occur as tardive variants.10
Multiple diagnostic schemes for TD have been proposed; criteria proposed by Schooler and Kane have been widely accepted (Table 1).11 TD onset occurs insidiously over ≥3 months of antipsychotic treatment and may begin with tic-like movements or increased eye blinking. TD often is suppressed or masked by ongoing antipsychotic treatment and becomes apparent only when the drug is reduced, switched, or discontinued. Dyskinesias increase with emotional arousal, activation, or distraction, and diminish with relaxation, sleep, or volitional effort. As a result, TD symptoms fluctuate over time; therefore, repeated measurements are necessary for reliable assessment of severity and persistence.
The differential diagnosis of TD necessitates conducting a careful medical and neurologic evaluation of all patients with new-onset movement disorders. Clues to neurologic causes include a family history of movement disorders, sudden onset or progressive course, associated medical or neurologic abnormalities, and asymmetry of symptoms. Some of the medical, neurologic, and psychiatric conditions to consider are listed in Table 2.12
Table 1
Schooler-Kane diagnostic criteria for TD
|
| Probable TD: meets criteria 1 through 3 Masked TD: meets criteria 1 through 3 but movements suppressed within 2 weeks by antipsychotic drugs Transient TD: movements not observed on subsequent examination within 3 months Withdrawal TD: movements observed within 2 weeks of antipsychotic drug discontinuation Persistent TD: movements persist for 3 months |
| TD: tardive dyskinesia Source: Reference 11 |
Table 2
Differential diagnosis of tardive dyskinesia
| Primary movement disorders |
|
| Secondary movement disorders |
|
| Source: Reference 12 |
Treatment decisions
If a patient develops TD, clinicians need to make several decisions (Algorithm). First, consider tapering any anticholinergic drugs unless acute EPS are prominent or tardive dystonia is present. Anticholinergic agents can worsen TD but not tardive dystonia; 60% of TD cases improve after discontinuing anticholinergics.13 Second, decide whether antipsychotics could be safely tapered or discontinued. If antipsychotics cannot be safely tapered, decide whether to maintain the patient’s present antipsychotic or switch to a more or less potent agent. Finally, decide whether a trial of an adjunctive antidyskinetic drug is warranted. All of these decisions require thorough discussion with patients and their families, accompanied by careful documentation.
Discontinuing, continuing, or switching antipsychotics. Discontinuing antipsychotics once TD becomes apparent is an option. However, the natural course of TD after drug withdrawal is unclear. Although drug withdrawal had been recommended to increase the odds of TD resolution, early studies showed withdrawing antipsychotics may lead to an initial worsening of TD in 33% to 53% of patients (unmasking or withdrawal dyskinesia).14 With long-term follow-up, 36% to 55% of patients eventually improved, which supports recommendations for drug reduction or withdrawal.14 However, complete and permanent reversibility beyond the withdrawal period is rare; Glazer et al found only 2% of patients showed complete reversal of TD after drug discontinuation.15,16 In a meta-analysis, Soares and McGrath17 reported 37% of patients assigned to placebo across studies showed at least some improvement in TD, but concluded insufficient evidence existed to support drug cessation or reduction as effective treatments for TD, especially when contrasted with robust evidence for the risk of psychotic relapse after drug withdrawal in patients with schizophrenia (53% within 9 months).18
A second option for a stable patient with good control of psychotic symptoms but established or long-term TD is to continue the antipsychotic, try to gradually reduce the dose, inform patients and caregivers of risks, document the decision, and monitor carefully. In most cases, TD may not progress even with continued antipsychotic treatment, although symptoms may worsen in some cases. However, in a patient with new-onset or early signs of TD, the clinician may be obligated to switch to a lower-potency antipsychotic or newer SGA to improve the chance of resolution; switching is discussed below.
Data on the change in prevalence of TD within a population during continued antipsychotic treatment have been inconsistent. Some studies show an increase, while others show a decrease or no change at all.19 However, prevalence rates obscure the dynamics of TD in individual patients. Roughly 50% of patients with TD have persistent symptoms, 10% to 30% have reduced symptoms, and 10% to 30% experience increased symptoms during treatment.13 Long-term studies estimated that up to 23% of patients may show loss of observable TD symptoms during treatment with FGAs in 1 year.19,20 Similarly, studies of SGAs have shown reduction of TD ratings; some found greater reductions, some found less reductions, and some no difference compared with FGAs.19,20 In some studies, improved TD outcomes were correlated with younger age, lower antipsychotic doses, reduced duration of drug treatment and dyskinesia, and increased length of follow-up.
In the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study, there was a significant decline in TD severity ratings among 200 patients with TD at baseline who were randomized to receive 1 of 4 SGAs, but there were no significant differences among these SGAs in decline in AIMS scores (Figure).19 Fifty-five percent of these patients met criteria for TD at 2 consecutive post-baseline visits, 76% met criteria at some or all post-baseline visits, and 24% did not meet criteria at any subsequent visit. In addition, 32% showed ≥50% decrease and 7% showed ≥50% increase in AIMS score. Thus, similar to past evidence on the course of TD during treatment with FGAs or SGAs, most patients in this trial showed either persistence or fluctuation in observable TD symptoms.
Another alternative is to switch antipsychotics, keeping in mind the risk of destabilizing a patient and precipitating psychotic relapse. More potent antipsychotics—such as haloperidol—suppress TD in approximately 67% of patients and may be necessary to consider in patients with severe, disabling symptoms, although the safety of these drugs in relation to their impact on long-term TD outcome is unclear.13,21,22 On the other hand, lower-potency drugs and SGAs also have been associated with reduced TD symptoms23,24; this was confirmed by results of the CATIE trial cited above in which SGAs were associated with a significant reduction in TD severity ratings.19 Clozapine in particular has been recommended for suppressing TD, especially in cases of tardive dystonia.20 Surprisingly, data are limited and inconsistent in addressing whether high-potency FGAs suppress TD symptoms more than low-potency drugs or SGAs, and whether SGAs may suppress TD by mechanisms other than dopamine receptor blockade, which would enhance symptom remission.19,25,26
Apart from short-term suppression of TD symptoms, the advantage of switching to lower-potency antipsychotics or other SGAs would be to increase the odds of eventual TD resolution. Although there has been speculation that in contrast to high-potency FGAs, SGAs may increase the possibility of remission by actively reversing TD or by passively allowing time for TD to resolve, existing data are inconclusive as to whether treatment with SGAs or FGAs results in true recovery rather than symptom suppression. To distinguish remission from suppression, a few studies discontinued SGAs. Some reported continued absence of TD,27,28 but others found unmasking and reappearance of TD.29-31
Adjunctive antidyskinetic drugs. Agents that have been tested off-label for antidyskinetic effects could be considered if symptoms of TD remain problematic despite optimization of antipsychotic treatment, although none have been confirmed as uniformly effective in randomized controlled trials replicated by different investigators.13,17,22 These include dopamine-depleting agents, dopamine agonists, noradrenergic agonists and antagonists, GABAergic drugs (benzodiazepines, valproate, levetiracetam), lithium, calcium channel blockers, serotonergic drugs, antioxidants (vitamin E and B6), branched-chain amino acids, neuropeptides, cholinergic precursors, and cholinesterase inhibitors. Electroconvulsive therapy and botulinum toxin or surgical intervention (for tardive dystonia) also may be considered.
Hypotheses proposed to explain TD pathophysiology and thereby justify trials of specific antidyskinetic agents include dopamine receptor hypersensitivity, GABA insufficiency, and structural damage resulting from increased catecholamine metabolism and oxidative free radical production.32 Another hypothesis proposes that TD results from damage to striatal cholinergic interneurons due to loss of dopamine-mediated inhibition.33 If correct, this implies that cholinesterase inhibitors or cholinergic agonists may suppress TD by directly enhancing post-synaptic cholinergic activity, thereby compensating for the loss of pre-synaptic cholinergic neurons. Several preliminary trials that explored the use of cholinesterase inhibitors had mixed results.34-36 However, this hypothesis is supported by evidence from animal and human studies that correlated antipsychotic-induced changes in cholinergic activity with the delay in onset, irreversibility, and age-related risk of TD, the worsening of symptoms due to anticholinergic drugs, and the reduced liability of SGAs for causing TD. These findings suggest that further investigation of cholinergic mechanisms underlying TD may be worthwhile.35
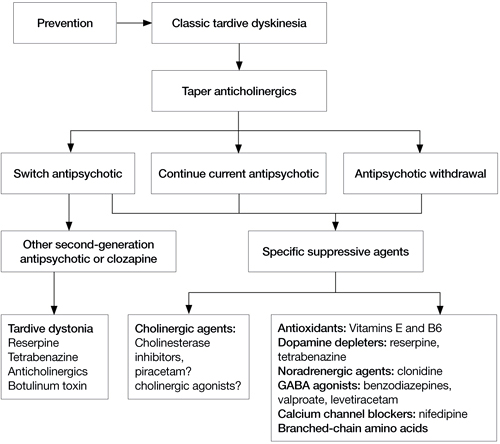
Algorithm: Proposed treatment algorithm for tardive dyskinesia
Source: Reprinted from Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE Schizophrenia Trial. Neurol Clin. 2011;29:127-148 with permission from Elsevier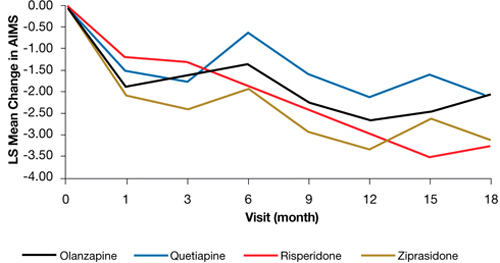
Figure: Adjusteda repeated measures model of change in total AIMS scores for patients with tardive dyskinesia at baseline in CATIE
a Model adjusted for baseline AIMS, baseline PANSS, and duration of illness. Adjusted P value for reduction in total AIMS score from baseline for all patients: P < .001. Treatment differences between the second-generation antipsychotics during the trial: P = .811
AIMS: Abnormal Involuntary Movement Scale; CATIE: Clinical Antipsychotic Trials of Intervention Effectiveness; PANSS: Positive and Negative Syndrome Scale
Source: Reprinted from Caroff SN, Davis VG, Miller DD, et al; for the CATIE Investigators. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303 with permission from Physician Postgraduate Press, Inc.Related Resources
- National Institute of Neurological Disorders and Stroke. NINDS Tardive Dyskinesia Information Page. www.ninds.nih.gov/disorders/tardive/tardive.htm.
- WE MOVE (Worldwide Education and Awareness for Movement Disorders). www.wemove.org.
Drug Brand Names
- Botulinum toxin • Botox, Dysport, others
- Clonidine • Catapres
- Clozapine • Clozaril
- Haloperidol • Haldol
- Levetiracetam • Keppra
- Levodopa • Dopar, Larodopa
- Lithium • Lithobid, Eskalith, others
- Nifedipine • Adalat, Afeditab CR, others
- Olanzapine • Zyprexa
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Reserpine • Serpasil
- Risperidone • Risperdal
- Tetrabenazine • Xenazine
- Valproate • Depakote
- Ziprasidone • Geodon
Disclosures
Drs. Caroff, Dhopesh, and Campbell report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Miller receives research/grant support from AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Ortho-McNeil-Janssen, and Pfizer Inc. and is a consultant to GlaxoSmithKline and Otsuka.
1. Miller DD, Caroff SN, Davis SM, et al. Extrapyramidal side-effects of antipsychotics in a randomised trial. Br J Psychiatry. 2008;193(4):279-288.
2. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223.
3. Leucht S, Wahlbeck K, Hamann J, et al. New generation antipsychotics versus low-potency conventional antipsychotics: a systematic review and meta-analysis. Lancet. 2003;361(9369):1581-1589.
4. Guy W. Abnormal involuntary movement scale (AIMS). In: Guy W ed. ECDEU assessment manual for psychopharmacology. Rockville, MD: U.S. Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute of Mental Health, Psychopharmacology Research Branch, Division of Extramural Research Programs; 1976:534–537.
5. Tarsy D. Neuroleptic-induced extrapyramidal reactions: classification description, and diagnosis. Clin Neuropharmacol. 1983;6(1):9-26.
6. Kane JM. Tardive dyskinesia: epidemiological and clinical presentation. In: Bloom FE Kupfer DJ, eds. Psychopharmacology: the fourth generation of progress. New York, NY: Raven Press; 1995:1485–1495.
7. Casey DE. Neuroleptic drug-induced extrapyramidal syndromes and tardive dyskinesia. Schizophr Res. 1991;4(2):109-120.
8. Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011;29(1):127-148.
9. Dayalu P, Chou KL. Antipsychotic-induced extrapyramidal symptoms and their management. Expert Opin Pharmacother. 2008;9(9):1451-1462.
10. Burke RE, Kang UJ, Jankovic J, et al. Tardive akathisia: an analysis of clinical features and response to open therapeutic trials. Mov Disord. 1989;4(2):157-175.
11. Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486-487.
12. American Psychiatric Association. Tardive dyskinesia: a task force report of the American Psychiatric Association. Washington DC: American Psychiatric Press, Inc; 1992.
13. Egan MF, Apud J, Wyatt RJ. Treatment of tardive dyskinesia. Schizophr Bull. 1997;23(4):583-609.
14. Casey DE, Gerlach J. Tardive dyskinesia: what is the long-term outcome? In: Casey DE Gardos G, eds. Tardive dyskinesia and neuroleptics: from dogma to reason. Washington, DC: American Psychiatric Press, Inc; 1986:76–97.
15. Glazer WM, Moore DC, Schooler NR, et al. Tardive dyskinesia. A discontinuation study. Arch Gen Psychiatry. 1984;41(6):623-627.
16. Glazer WM, Morgenstern H, Schooler N, et al. Predictors of improvement in tardive dyskinesia following discontinuation of neuroleptic medication. Br J Psychiatry. 1990;157:585-592.
17. Soares KV, McGrath JJ. The treatment of tardive dyskinesia—a systematic review and meta-analysis. Schizophr Res. 1999;39(1):1-16.
18. Gilbert PL, Harris MJ, McAdams LA, et al. Neuroleptic withdrawal in schizophrenic patients. A review of the literature. Arch Gen Psychiatry. 1995;52(3):173-188.
19. Caroff SN, Davis VG, Miller DD, et al. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303.
20. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
21. Jeste DV, Wyatt RJ. In search of treatment for tardive dyskinesia: review of the literature. Schizophr Bull. 1979;5(2):251-293.
22. Jeste DV, Lohr JB, Clark K, et al. Pharmacological treatments of tardive dyskinesia in the 1980s. J Clin Psychopharmacol. 1988;8(4 suppl):38S-48S.
23. Caroff SN, Mann SC, Campbell EC, et al. Movement disorders associated with atypical antipsychotic drugs. J Clin Psychiatry. 2002;63(suppl 4):12-19.
24. Tarsy D, Baldessarini RJ, Tarazi FI. Effects of newer antipsychotics on extrapyramidal function. CNS Drugs. 2002;16(1):23-45.
25. Emsley R, Turner HJ, Schronen J, et al. A single-blind, randomized trial comparing quetiapine and haloperidol in the treatment of tardive dyskinesia. J Clin Psychiatry. 2004;65(5):696-701.
26. Glazer WM, Hafez H. A comparison of masking effects of haloperidol versus molindone in tardive dyskinesia. Schizophr Res. 1990;3(5-6):315-320.
27. Kinon BJ, Jeste DV, Kollack-Walker S, et al. Olanzapine treatment for tardive dyskinesia in schizophrenia patients: a prospective clinical trial with patients randomized to blinded dose reduction periods. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(6):985-996.
28. Tamminga CA, Thaker GK, Moran M, et al. Clozapine in tardive dyskinesia: observations from human and animal model studies. J Clin Psychiatry. 1994;55(suppl B):102-106.
29. Simpson GM, Lee JH, Shrivastava RK. Clozapine in tardive dyskinesia. Psychopharmacology (Berl). 1978;56(1):75-80.
30. Ahmed S, Chengappa KN, Naidu VR, et al. Clozapine withdrawal-emergent dystonias and dyskinesias: a case series. J Clin Psychiatry. 1998;59(9):472-477.
31. Small JG, Milstein V, Marhenke JD, et al. Treatment outcome with clozapine in tardive dyskinesia, neuroleptic sensitivity, and treatment-resistant psychosis. J Clin Psychiatry. 1987;48(7):263-267.
32. Casey DE. Tardive dyskinesia: pathophysiology and animal models. J Clin Psychiatry. 2000;61(suppl 4):5-9.
33. Miller R, Chouinard G. Loss of striatal cholinergic neurons as a basis for tardive and L-dopa-induced dyskinesias neuroleptic-induced supersensitivity psychosis and refractory schizophrenia. Biol Psychiatry. 1993;34(10):713-738.
34. Caroff SN, Campbell EC, Havey J, et al. Treatment of tardive dyskinesia with donepezil: a pilot study. J Clin Psychiatry. 2001;62(10):772-775.
35. Caroff SN, Walker P, Campbell C, et al. Treatment of tardive dyskinesia with galantamine: a randomized controlled crossover trial. J Clin Psychiatry. 2007;68(3):410-415.
36. Caroff SN, Martine R, Kleiner-Fisman G, et al. Treatment of levodopa-induced dyskinesias with donepezil. Parkinsonism Relat Disord. 2006;12(4):261-263.
Introduced into clinical practice more than a half century ago, antipsychotics are still the mainstay of schizophrenia treatment. However, from the earliest reports, antipsychotic efficacy was seemingly inseparable from extrapyramidal side effects (EPS) that manifested as acute and chronic involuntary movement disorders. Although acute extrapyramidal side effects could be prevented and treated, the late-arising symptoms of tardive dyskinesia (TD) seemed irreversible in most cases.
Concerns over TD stimulated extensive research and fueled efforts to develop new antipsychotics that spared the extrapyramidal motor system. Numerous industry-sponsored trials found a reduced risk of EPS—including TD—with newer, second-generation antipsychotics (SGAs), although this advantage diminished when modest doses of low- or mid-potency first-generation antipsychotics (FGAs) were used as the comparator.1-3 Nevertheless, in addition to the continued potential risk of introducing new cases of TD—even with SGAs—several other factors underscore the need to develop a rational strategy for clinical management of TD, including:
- thousands of patients are left with TD as a legacy of past treatment
- the neurophysiologic mechanisms underlying TD are not well understood
- there is no uniformly effective treatment to reverse TD
- TD may be irreversible in most cases.
Prevention
Because there is no “gold standard” treatment for TD, it is important to minimize the risk of TD by taking preventive measures and detecting incipient signs of the disorder. Preventive principles include:
- confirming and documenting the indication for antipsychotics
- using conservative maintenance doses and opting for lower potency or newer agents
- informing patients and caregivers of risk
- assessing for incipient signs of TD using the Abnormal Involuntary Movement Scale (AIMS),4 which should be administered at least every 3 to 6 months.
Confirming the diagnosis
TD presents as a polymorphous involuntary movement disorder,5-8 most often with nonrhythmic, repetitive, purposeless hyperkinetic symptoms. It usually affects orofacial and lingual musculature (“buccolinguomasticatory syndrome”) with chewing; bruxism; protrusion, curling, or twisting of the tongue; lip smacking, puckering, sucking, and pursing; retraction, grimacing or bridling of the mouth; bulging of the cheeks; or eye blinking and blepharospasm. Choreoathetoid movements of the fingers, hands, or upper or lower extremities also are common. Patients may experience axial symptoms affecting the neck, shoulders, spine, or pelvis. When severe, dyskinesias can affect breathing, swallowing, or speech, and interfere with walking and activities of daily living.
TD may present with nonchoreoathetoid symptoms that can be difficult to distinguish from acute EPS. These may co-exist with classic TD symptoms, but may represent separate subtypes with increased risk of progression, persistence, and severe disability. For example, tardive dystonia, which is estimated to occur in 1% to 4% of patients treated with antipsychotics,9 may be more generalized and disabling than TD, and may respond to anticholinergic agents. Akathisia and other movement disorders also occur as tardive variants.10
Multiple diagnostic schemes for TD have been proposed; criteria proposed by Schooler and Kane have been widely accepted (Table 1).11 TD onset occurs insidiously over ≥3 months of antipsychotic treatment and may begin with tic-like movements or increased eye blinking. TD often is suppressed or masked by ongoing antipsychotic treatment and becomes apparent only when the drug is reduced, switched, or discontinued. Dyskinesias increase with emotional arousal, activation, or distraction, and diminish with relaxation, sleep, or volitional effort. As a result, TD symptoms fluctuate over time; therefore, repeated measurements are necessary for reliable assessment of severity and persistence.
The differential diagnosis of TD necessitates conducting a careful medical and neurologic evaluation of all patients with new-onset movement disorders. Clues to neurologic causes include a family history of movement disorders, sudden onset or progressive course, associated medical or neurologic abnormalities, and asymmetry of symptoms. Some of the medical, neurologic, and psychiatric conditions to consider are listed in Table 2.12
Table 1
Schooler-Kane diagnostic criteria for TD
|
| Probable TD: meets criteria 1 through 3 Masked TD: meets criteria 1 through 3 but movements suppressed within 2 weeks by antipsychotic drugs Transient TD: movements not observed on subsequent examination within 3 months Withdrawal TD: movements observed within 2 weeks of antipsychotic drug discontinuation Persistent TD: movements persist for 3 months |
| TD: tardive dyskinesia Source: Reference 11 |
Table 2
Differential diagnosis of tardive dyskinesia
| Primary movement disorders |
|
| Secondary movement disorders |
|
| Source: Reference 12 |
Treatment decisions
If a patient develops TD, clinicians need to make several decisions (Algorithm). First, consider tapering any anticholinergic drugs unless acute EPS are prominent or tardive dystonia is present. Anticholinergic agents can worsen TD but not tardive dystonia; 60% of TD cases improve after discontinuing anticholinergics.13 Second, decide whether antipsychotics could be safely tapered or discontinued. If antipsychotics cannot be safely tapered, decide whether to maintain the patient’s present antipsychotic or switch to a more or less potent agent. Finally, decide whether a trial of an adjunctive antidyskinetic drug is warranted. All of these decisions require thorough discussion with patients and their families, accompanied by careful documentation.
Discontinuing, continuing, or switching antipsychotics. Discontinuing antipsychotics once TD becomes apparent is an option. However, the natural course of TD after drug withdrawal is unclear. Although drug withdrawal had been recommended to increase the odds of TD resolution, early studies showed withdrawing antipsychotics may lead to an initial worsening of TD in 33% to 53% of patients (unmasking or withdrawal dyskinesia).14 With long-term follow-up, 36% to 55% of patients eventually improved, which supports recommendations for drug reduction or withdrawal.14 However, complete and permanent reversibility beyond the withdrawal period is rare; Glazer et al found only 2% of patients showed complete reversal of TD after drug discontinuation.15,16 In a meta-analysis, Soares and McGrath17 reported 37% of patients assigned to placebo across studies showed at least some improvement in TD, but concluded insufficient evidence existed to support drug cessation or reduction as effective treatments for TD, especially when contrasted with robust evidence for the risk of psychotic relapse after drug withdrawal in patients with schizophrenia (53% within 9 months).18
A second option for a stable patient with good control of psychotic symptoms but established or long-term TD is to continue the antipsychotic, try to gradually reduce the dose, inform patients and caregivers of risks, document the decision, and monitor carefully. In most cases, TD may not progress even with continued antipsychotic treatment, although symptoms may worsen in some cases. However, in a patient with new-onset or early signs of TD, the clinician may be obligated to switch to a lower-potency antipsychotic or newer SGA to improve the chance of resolution; switching is discussed below.
Data on the change in prevalence of TD within a population during continued antipsychotic treatment have been inconsistent. Some studies show an increase, while others show a decrease or no change at all.19 However, prevalence rates obscure the dynamics of TD in individual patients. Roughly 50% of patients with TD have persistent symptoms, 10% to 30% have reduced symptoms, and 10% to 30% experience increased symptoms during treatment.13 Long-term studies estimated that up to 23% of patients may show loss of observable TD symptoms during treatment with FGAs in 1 year.19,20 Similarly, studies of SGAs have shown reduction of TD ratings; some found greater reductions, some found less reductions, and some no difference compared with FGAs.19,20 In some studies, improved TD outcomes were correlated with younger age, lower antipsychotic doses, reduced duration of drug treatment and dyskinesia, and increased length of follow-up.
In the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study, there was a significant decline in TD severity ratings among 200 patients with TD at baseline who were randomized to receive 1 of 4 SGAs, but there were no significant differences among these SGAs in decline in AIMS scores (Figure).19 Fifty-five percent of these patients met criteria for TD at 2 consecutive post-baseline visits, 76% met criteria at some or all post-baseline visits, and 24% did not meet criteria at any subsequent visit. In addition, 32% showed ≥50% decrease and 7% showed ≥50% increase in AIMS score. Thus, similar to past evidence on the course of TD during treatment with FGAs or SGAs, most patients in this trial showed either persistence or fluctuation in observable TD symptoms.
Another alternative is to switch antipsychotics, keeping in mind the risk of destabilizing a patient and precipitating psychotic relapse. More potent antipsychotics—such as haloperidol—suppress TD in approximately 67% of patients and may be necessary to consider in patients with severe, disabling symptoms, although the safety of these drugs in relation to their impact on long-term TD outcome is unclear.13,21,22 On the other hand, lower-potency drugs and SGAs also have been associated with reduced TD symptoms23,24; this was confirmed by results of the CATIE trial cited above in which SGAs were associated with a significant reduction in TD severity ratings.19 Clozapine in particular has been recommended for suppressing TD, especially in cases of tardive dystonia.20 Surprisingly, data are limited and inconsistent in addressing whether high-potency FGAs suppress TD symptoms more than low-potency drugs or SGAs, and whether SGAs may suppress TD by mechanisms other than dopamine receptor blockade, which would enhance symptom remission.19,25,26
Apart from short-term suppression of TD symptoms, the advantage of switching to lower-potency antipsychotics or other SGAs would be to increase the odds of eventual TD resolution. Although there has been speculation that in contrast to high-potency FGAs, SGAs may increase the possibility of remission by actively reversing TD or by passively allowing time for TD to resolve, existing data are inconclusive as to whether treatment with SGAs or FGAs results in true recovery rather than symptom suppression. To distinguish remission from suppression, a few studies discontinued SGAs. Some reported continued absence of TD,27,28 but others found unmasking and reappearance of TD.29-31
Adjunctive antidyskinetic drugs. Agents that have been tested off-label for antidyskinetic effects could be considered if symptoms of TD remain problematic despite optimization of antipsychotic treatment, although none have been confirmed as uniformly effective in randomized controlled trials replicated by different investigators.13,17,22 These include dopamine-depleting agents, dopamine agonists, noradrenergic agonists and antagonists, GABAergic drugs (benzodiazepines, valproate, levetiracetam), lithium, calcium channel blockers, serotonergic drugs, antioxidants (vitamin E and B6), branched-chain amino acids, neuropeptides, cholinergic precursors, and cholinesterase inhibitors. Electroconvulsive therapy and botulinum toxin or surgical intervention (for tardive dystonia) also may be considered.
Hypotheses proposed to explain TD pathophysiology and thereby justify trials of specific antidyskinetic agents include dopamine receptor hypersensitivity, GABA insufficiency, and structural damage resulting from increased catecholamine metabolism and oxidative free radical production.32 Another hypothesis proposes that TD results from damage to striatal cholinergic interneurons due to loss of dopamine-mediated inhibition.33 If correct, this implies that cholinesterase inhibitors or cholinergic agonists may suppress TD by directly enhancing post-synaptic cholinergic activity, thereby compensating for the loss of pre-synaptic cholinergic neurons. Several preliminary trials that explored the use of cholinesterase inhibitors had mixed results.34-36 However, this hypothesis is supported by evidence from animal and human studies that correlated antipsychotic-induced changes in cholinergic activity with the delay in onset, irreversibility, and age-related risk of TD, the worsening of symptoms due to anticholinergic drugs, and the reduced liability of SGAs for causing TD. These findings suggest that further investigation of cholinergic mechanisms underlying TD may be worthwhile.35
Algorithm: Proposed treatment algorithm for tardive dyskinesia
Source: Reprinted from Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE Schizophrenia Trial. Neurol Clin. 2011;29:127-148 with permission from Elsevier
Figure: Adjusteda repeated measures model of change in total AIMS scores for patients with tardive dyskinesia at baseline in CATIE
a Model adjusted for baseline AIMS, baseline PANSS, and duration of illness. Adjusted P value for reduction in total AIMS score from baseline for all patients: P < .001. Treatment differences between the second-generation antipsychotics during the trial: P = .811
AIMS: Abnormal Involuntary Movement Scale; CATIE: Clinical Antipsychotic Trials of Intervention Effectiveness; PANSS: Positive and Negative Syndrome Scale
Source: Reprinted from Caroff SN, Davis VG, Miller DD, et al; for the CATIE Investigators. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303 with permission from Physician Postgraduate Press, Inc.Related Resources
- National Institute of Neurological Disorders and Stroke. NINDS Tardive Dyskinesia Information Page. www.ninds.nih.gov/disorders/tardive/tardive.htm.
- WE MOVE (Worldwide Education and Awareness for Movement Disorders). www.wemove.org.
Drug Brand Names
- Botulinum toxin • Botox, Dysport, others
- Clonidine • Catapres
- Clozapine • Clozaril
- Haloperidol • Haldol
- Levetiracetam • Keppra
- Levodopa • Dopar, Larodopa
- Lithium • Lithobid, Eskalith, others
- Nifedipine • Adalat, Afeditab CR, others
- Olanzapine • Zyprexa
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Reserpine • Serpasil
- Risperidone • Risperdal
- Tetrabenazine • Xenazine
- Valproate • Depakote
- Ziprasidone • Geodon
Disclosures
Drs. Caroff, Dhopesh, and Campbell report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Miller receives research/grant support from AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Ortho-McNeil-Janssen, and Pfizer Inc. and is a consultant to GlaxoSmithKline and Otsuka.
Introduced into clinical practice more than a half century ago, antipsychotics are still the mainstay of schizophrenia treatment. However, from the earliest reports, antipsychotic efficacy was seemingly inseparable from extrapyramidal side effects (EPS) that manifested as acute and chronic involuntary movement disorders. Although acute extrapyramidal side effects could be prevented and treated, the late-arising symptoms of tardive dyskinesia (TD) seemed irreversible in most cases.
Concerns over TD stimulated extensive research and fueled efforts to develop new antipsychotics that spared the extrapyramidal motor system. Numerous industry-sponsored trials found a reduced risk of EPS—including TD—with newer, second-generation antipsychotics (SGAs), although this advantage diminished when modest doses of low- or mid-potency first-generation antipsychotics (FGAs) were used as the comparator.1-3 Nevertheless, in addition to the continued potential risk of introducing new cases of TD—even with SGAs—several other factors underscore the need to develop a rational strategy for clinical management of TD, including:
- thousands of patients are left with TD as a legacy of past treatment
- the neurophysiologic mechanisms underlying TD are not well understood
- there is no uniformly effective treatment to reverse TD
- TD may be irreversible in most cases.
Prevention
Because there is no “gold standard” treatment for TD, it is important to minimize the risk of TD by taking preventive measures and detecting incipient signs of the disorder. Preventive principles include:
- confirming and documenting the indication for antipsychotics
- using conservative maintenance doses and opting for lower potency or newer agents
- informing patients and caregivers of risk
- assessing for incipient signs of TD using the Abnormal Involuntary Movement Scale (AIMS),4 which should be administered at least every 3 to 6 months.
Confirming the diagnosis
TD presents as a polymorphous involuntary movement disorder,5-8 most often with nonrhythmic, repetitive, purposeless hyperkinetic symptoms. It usually affects orofacial and lingual musculature (“buccolinguomasticatory syndrome”) with chewing; bruxism; protrusion, curling, or twisting of the tongue; lip smacking, puckering, sucking, and pursing; retraction, grimacing or bridling of the mouth; bulging of the cheeks; or eye blinking and blepharospasm. Choreoathetoid movements of the fingers, hands, or upper or lower extremities also are common. Patients may experience axial symptoms affecting the neck, shoulders, spine, or pelvis. When severe, dyskinesias can affect breathing, swallowing, or speech, and interfere with walking and activities of daily living.
TD may present with nonchoreoathetoid symptoms that can be difficult to distinguish from acute EPS. These may co-exist with classic TD symptoms, but may represent separate subtypes with increased risk of progression, persistence, and severe disability. For example, tardive dystonia, which is estimated to occur in 1% to 4% of patients treated with antipsychotics,9 may be more generalized and disabling than TD, and may respond to anticholinergic agents. Akathisia and other movement disorders also occur as tardive variants.10
Multiple diagnostic schemes for TD have been proposed; criteria proposed by Schooler and Kane have been widely accepted (Table 1).11 TD onset occurs insidiously over ≥3 months of antipsychotic treatment and may begin with tic-like movements or increased eye blinking. TD often is suppressed or masked by ongoing antipsychotic treatment and becomes apparent only when the drug is reduced, switched, or discontinued. Dyskinesias increase with emotional arousal, activation, or distraction, and diminish with relaxation, sleep, or volitional effort. As a result, TD symptoms fluctuate over time; therefore, repeated measurements are necessary for reliable assessment of severity and persistence.
The differential diagnosis of TD necessitates conducting a careful medical and neurologic evaluation of all patients with new-onset movement disorders. Clues to neurologic causes include a family history of movement disorders, sudden onset or progressive course, associated medical or neurologic abnormalities, and asymmetry of symptoms. Some of the medical, neurologic, and psychiatric conditions to consider are listed in Table 2.12
Table 1
Schooler-Kane diagnostic criteria for TD
|
| Probable TD: meets criteria 1 through 3 Masked TD: meets criteria 1 through 3 but movements suppressed within 2 weeks by antipsychotic drugs Transient TD: movements not observed on subsequent examination within 3 months Withdrawal TD: movements observed within 2 weeks of antipsychotic drug discontinuation Persistent TD: movements persist for 3 months |
| TD: tardive dyskinesia Source: Reference 11 |
Table 2
Differential diagnosis of tardive dyskinesia
| Primary movement disorders |
|
| Secondary movement disorders |
|
| Source: Reference 12 |
Treatment decisions
If a patient develops TD, clinicians need to make several decisions (Algorithm). First, consider tapering any anticholinergic drugs unless acute EPS are prominent or tardive dystonia is present. Anticholinergic agents can worsen TD but not tardive dystonia; 60% of TD cases improve after discontinuing anticholinergics.13 Second, decide whether antipsychotics could be safely tapered or discontinued. If antipsychotics cannot be safely tapered, decide whether to maintain the patient’s present antipsychotic or switch to a more or less potent agent. Finally, decide whether a trial of an adjunctive antidyskinetic drug is warranted. All of these decisions require thorough discussion with patients and their families, accompanied by careful documentation.
Discontinuing, continuing, or switching antipsychotics. Discontinuing antipsychotics once TD becomes apparent is an option. However, the natural course of TD after drug withdrawal is unclear. Although drug withdrawal had been recommended to increase the odds of TD resolution, early studies showed withdrawing antipsychotics may lead to an initial worsening of TD in 33% to 53% of patients (unmasking or withdrawal dyskinesia).14 With long-term follow-up, 36% to 55% of patients eventually improved, which supports recommendations for drug reduction or withdrawal.14 However, complete and permanent reversibility beyond the withdrawal period is rare; Glazer et al found only 2% of patients showed complete reversal of TD after drug discontinuation.15,16 In a meta-analysis, Soares and McGrath17 reported 37% of patients assigned to placebo across studies showed at least some improvement in TD, but concluded insufficient evidence existed to support drug cessation or reduction as effective treatments for TD, especially when contrasted with robust evidence for the risk of psychotic relapse after drug withdrawal in patients with schizophrenia (53% within 9 months).18
A second option for a stable patient with good control of psychotic symptoms but established or long-term TD is to continue the antipsychotic, try to gradually reduce the dose, inform patients and caregivers of risks, document the decision, and monitor carefully. In most cases, TD may not progress even with continued antipsychotic treatment, although symptoms may worsen in some cases. However, in a patient with new-onset or early signs of TD, the clinician may be obligated to switch to a lower-potency antipsychotic or newer SGA to improve the chance of resolution; switching is discussed below.
Data on the change in prevalence of TD within a population during continued antipsychotic treatment have been inconsistent. Some studies show an increase, while others show a decrease or no change at all.19 However, prevalence rates obscure the dynamics of TD in individual patients. Roughly 50% of patients with TD have persistent symptoms, 10% to 30% have reduced symptoms, and 10% to 30% experience increased symptoms during treatment.13 Long-term studies estimated that up to 23% of patients may show loss of observable TD symptoms during treatment with FGAs in 1 year.19,20 Similarly, studies of SGAs have shown reduction of TD ratings; some found greater reductions, some found less reductions, and some no difference compared with FGAs.19,20 In some studies, improved TD outcomes were correlated with younger age, lower antipsychotic doses, reduced duration of drug treatment and dyskinesia, and increased length of follow-up.
In the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study, there was a significant decline in TD severity ratings among 200 patients with TD at baseline who were randomized to receive 1 of 4 SGAs, but there were no significant differences among these SGAs in decline in AIMS scores (Figure).19 Fifty-five percent of these patients met criteria for TD at 2 consecutive post-baseline visits, 76% met criteria at some or all post-baseline visits, and 24% did not meet criteria at any subsequent visit. In addition, 32% showed ≥50% decrease and 7% showed ≥50% increase in AIMS score. Thus, similar to past evidence on the course of TD during treatment with FGAs or SGAs, most patients in this trial showed either persistence or fluctuation in observable TD symptoms.
Another alternative is to switch antipsychotics, keeping in mind the risk of destabilizing a patient and precipitating psychotic relapse. More potent antipsychotics—such as haloperidol—suppress TD in approximately 67% of patients and may be necessary to consider in patients with severe, disabling symptoms, although the safety of these drugs in relation to their impact on long-term TD outcome is unclear.13,21,22 On the other hand, lower-potency drugs and SGAs also have been associated with reduced TD symptoms23,24; this was confirmed by results of the CATIE trial cited above in which SGAs were associated with a significant reduction in TD severity ratings.19 Clozapine in particular has been recommended for suppressing TD, especially in cases of tardive dystonia.20 Surprisingly, data are limited and inconsistent in addressing whether high-potency FGAs suppress TD symptoms more than low-potency drugs or SGAs, and whether SGAs may suppress TD by mechanisms other than dopamine receptor blockade, which would enhance symptom remission.19,25,26
Apart from short-term suppression of TD symptoms, the advantage of switching to lower-potency antipsychotics or other SGAs would be to increase the odds of eventual TD resolution. Although there has been speculation that in contrast to high-potency FGAs, SGAs may increase the possibility of remission by actively reversing TD or by passively allowing time for TD to resolve, existing data are inconclusive as to whether treatment with SGAs or FGAs results in true recovery rather than symptom suppression. To distinguish remission from suppression, a few studies discontinued SGAs. Some reported continued absence of TD,27,28 but others found unmasking and reappearance of TD.29-31
Adjunctive antidyskinetic drugs. Agents that have been tested off-label for antidyskinetic effects could be considered if symptoms of TD remain problematic despite optimization of antipsychotic treatment, although none have been confirmed as uniformly effective in randomized controlled trials replicated by different investigators.13,17,22 These include dopamine-depleting agents, dopamine agonists, noradrenergic agonists and antagonists, GABAergic drugs (benzodiazepines, valproate, levetiracetam), lithium, calcium channel blockers, serotonergic drugs, antioxidants (vitamin E and B6), branched-chain amino acids, neuropeptides, cholinergic precursors, and cholinesterase inhibitors. Electroconvulsive therapy and botulinum toxin or surgical intervention (for tardive dystonia) also may be considered.
Hypotheses proposed to explain TD pathophysiology and thereby justify trials of specific antidyskinetic agents include dopamine receptor hypersensitivity, GABA insufficiency, and structural damage resulting from increased catecholamine metabolism and oxidative free radical production.32 Another hypothesis proposes that TD results from damage to striatal cholinergic interneurons due to loss of dopamine-mediated inhibition.33 If correct, this implies that cholinesterase inhibitors or cholinergic agonists may suppress TD by directly enhancing post-synaptic cholinergic activity, thereby compensating for the loss of pre-synaptic cholinergic neurons. Several preliminary trials that explored the use of cholinesterase inhibitors had mixed results.34-36 However, this hypothesis is supported by evidence from animal and human studies that correlated antipsychotic-induced changes in cholinergic activity with the delay in onset, irreversibility, and age-related risk of TD, the worsening of symptoms due to anticholinergic drugs, and the reduced liability of SGAs for causing TD. These findings suggest that further investigation of cholinergic mechanisms underlying TD may be worthwhile.35
Algorithm: Proposed treatment algorithm for tardive dyskinesia
Source: Reprinted from Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE Schizophrenia Trial. Neurol Clin. 2011;29:127-148 with permission from Elsevier
Figure: Adjusteda repeated measures model of change in total AIMS scores for patients with tardive dyskinesia at baseline in CATIE
a Model adjusted for baseline AIMS, baseline PANSS, and duration of illness. Adjusted P value for reduction in total AIMS score from baseline for all patients: P < .001. Treatment differences between the second-generation antipsychotics during the trial: P = .811
AIMS: Abnormal Involuntary Movement Scale; CATIE: Clinical Antipsychotic Trials of Intervention Effectiveness; PANSS: Positive and Negative Syndrome Scale
Source: Reprinted from Caroff SN, Davis VG, Miller DD, et al; for the CATIE Investigators. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303 with permission from Physician Postgraduate Press, Inc.Related Resources
- National Institute of Neurological Disorders and Stroke. NINDS Tardive Dyskinesia Information Page. www.ninds.nih.gov/disorders/tardive/tardive.htm.
- WE MOVE (Worldwide Education and Awareness for Movement Disorders). www.wemove.org.
Drug Brand Names
- Botulinum toxin • Botox, Dysport, others
- Clonidine • Catapres
- Clozapine • Clozaril
- Haloperidol • Haldol
- Levetiracetam • Keppra
- Levodopa • Dopar, Larodopa
- Lithium • Lithobid, Eskalith, others
- Nifedipine • Adalat, Afeditab CR, others
- Olanzapine • Zyprexa
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Reserpine • Serpasil
- Risperidone • Risperdal
- Tetrabenazine • Xenazine
- Valproate • Depakote
- Ziprasidone • Geodon
Disclosures
Drs. Caroff, Dhopesh, and Campbell report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Miller receives research/grant support from AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Ortho-McNeil-Janssen, and Pfizer Inc. and is a consultant to GlaxoSmithKline and Otsuka.
1. Miller DD, Caroff SN, Davis SM, et al. Extrapyramidal side-effects of antipsychotics in a randomised trial. Br J Psychiatry. 2008;193(4):279-288.
2. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223.
3. Leucht S, Wahlbeck K, Hamann J, et al. New generation antipsychotics versus low-potency conventional antipsychotics: a systematic review and meta-analysis. Lancet. 2003;361(9369):1581-1589.
4. Guy W. Abnormal involuntary movement scale (AIMS). In: Guy W ed. ECDEU assessment manual for psychopharmacology. Rockville, MD: U.S. Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute of Mental Health, Psychopharmacology Research Branch, Division of Extramural Research Programs; 1976:534–537.
5. Tarsy D. Neuroleptic-induced extrapyramidal reactions: classification description, and diagnosis. Clin Neuropharmacol. 1983;6(1):9-26.
6. Kane JM. Tardive dyskinesia: epidemiological and clinical presentation. In: Bloom FE Kupfer DJ, eds. Psychopharmacology: the fourth generation of progress. New York, NY: Raven Press; 1995:1485–1495.
7. Casey DE. Neuroleptic drug-induced extrapyramidal syndromes and tardive dyskinesia. Schizophr Res. 1991;4(2):109-120.
8. Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011;29(1):127-148.
9. Dayalu P, Chou KL. Antipsychotic-induced extrapyramidal symptoms and their management. Expert Opin Pharmacother. 2008;9(9):1451-1462.
10. Burke RE, Kang UJ, Jankovic J, et al. Tardive akathisia: an analysis of clinical features and response to open therapeutic trials. Mov Disord. 1989;4(2):157-175.
11. Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486-487.
12. American Psychiatric Association. Tardive dyskinesia: a task force report of the American Psychiatric Association. Washington DC: American Psychiatric Press, Inc; 1992.
13. Egan MF, Apud J, Wyatt RJ. Treatment of tardive dyskinesia. Schizophr Bull. 1997;23(4):583-609.
14. Casey DE, Gerlach J. Tardive dyskinesia: what is the long-term outcome? In: Casey DE Gardos G, eds. Tardive dyskinesia and neuroleptics: from dogma to reason. Washington, DC: American Psychiatric Press, Inc; 1986:76–97.
15. Glazer WM, Moore DC, Schooler NR, et al. Tardive dyskinesia. A discontinuation study. Arch Gen Psychiatry. 1984;41(6):623-627.
16. Glazer WM, Morgenstern H, Schooler N, et al. Predictors of improvement in tardive dyskinesia following discontinuation of neuroleptic medication. Br J Psychiatry. 1990;157:585-592.
17. Soares KV, McGrath JJ. The treatment of tardive dyskinesia—a systematic review and meta-analysis. Schizophr Res. 1999;39(1):1-16.
18. Gilbert PL, Harris MJ, McAdams LA, et al. Neuroleptic withdrawal in schizophrenic patients. A review of the literature. Arch Gen Psychiatry. 1995;52(3):173-188.
19. Caroff SN, Davis VG, Miller DD, et al. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303.
20. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
21. Jeste DV, Wyatt RJ. In search of treatment for tardive dyskinesia: review of the literature. Schizophr Bull. 1979;5(2):251-293.
22. Jeste DV, Lohr JB, Clark K, et al. Pharmacological treatments of tardive dyskinesia in the 1980s. J Clin Psychopharmacol. 1988;8(4 suppl):38S-48S.
23. Caroff SN, Mann SC, Campbell EC, et al. Movement disorders associated with atypical antipsychotic drugs. J Clin Psychiatry. 2002;63(suppl 4):12-19.
24. Tarsy D, Baldessarini RJ, Tarazi FI. Effects of newer antipsychotics on extrapyramidal function. CNS Drugs. 2002;16(1):23-45.
25. Emsley R, Turner HJ, Schronen J, et al. A single-blind, randomized trial comparing quetiapine and haloperidol in the treatment of tardive dyskinesia. J Clin Psychiatry. 2004;65(5):696-701.
26. Glazer WM, Hafez H. A comparison of masking effects of haloperidol versus molindone in tardive dyskinesia. Schizophr Res. 1990;3(5-6):315-320.
27. Kinon BJ, Jeste DV, Kollack-Walker S, et al. Olanzapine treatment for tardive dyskinesia in schizophrenia patients: a prospective clinical trial with patients randomized to blinded dose reduction periods. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(6):985-996.
28. Tamminga CA, Thaker GK, Moran M, et al. Clozapine in tardive dyskinesia: observations from human and animal model studies. J Clin Psychiatry. 1994;55(suppl B):102-106.
29. Simpson GM, Lee JH, Shrivastava RK. Clozapine in tardive dyskinesia. Psychopharmacology (Berl). 1978;56(1):75-80.
30. Ahmed S, Chengappa KN, Naidu VR, et al. Clozapine withdrawal-emergent dystonias and dyskinesias: a case series. J Clin Psychiatry. 1998;59(9):472-477.
31. Small JG, Milstein V, Marhenke JD, et al. Treatment outcome with clozapine in tardive dyskinesia, neuroleptic sensitivity, and treatment-resistant psychosis. J Clin Psychiatry. 1987;48(7):263-267.
32. Casey DE. Tardive dyskinesia: pathophysiology and animal models. J Clin Psychiatry. 2000;61(suppl 4):5-9.
33. Miller R, Chouinard G. Loss of striatal cholinergic neurons as a basis for tardive and L-dopa-induced dyskinesias neuroleptic-induced supersensitivity psychosis and refractory schizophrenia. Biol Psychiatry. 1993;34(10):713-738.
34. Caroff SN, Campbell EC, Havey J, et al. Treatment of tardive dyskinesia with donepezil: a pilot study. J Clin Psychiatry. 2001;62(10):772-775.
35. Caroff SN, Walker P, Campbell C, et al. Treatment of tardive dyskinesia with galantamine: a randomized controlled crossover trial. J Clin Psychiatry. 2007;68(3):410-415.
36. Caroff SN, Martine R, Kleiner-Fisman G, et al. Treatment of levodopa-induced dyskinesias with donepezil. Parkinsonism Relat Disord. 2006;12(4):261-263.
1. Miller DD, Caroff SN, Davis SM, et al. Extrapyramidal side-effects of antipsychotics in a randomised trial. Br J Psychiatry. 2008;193(4):279-288.
2. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223.
3. Leucht S, Wahlbeck K, Hamann J, et al. New generation antipsychotics versus low-potency conventional antipsychotics: a systematic review and meta-analysis. Lancet. 2003;361(9369):1581-1589.
4. Guy W. Abnormal involuntary movement scale (AIMS). In: Guy W ed. ECDEU assessment manual for psychopharmacology. Rockville, MD: U.S. Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute of Mental Health, Psychopharmacology Research Branch, Division of Extramural Research Programs; 1976:534–537.
5. Tarsy D. Neuroleptic-induced extrapyramidal reactions: classification description, and diagnosis. Clin Neuropharmacol. 1983;6(1):9-26.
6. Kane JM. Tardive dyskinesia: epidemiological and clinical presentation. In: Bloom FE Kupfer DJ, eds. Psychopharmacology: the fourth generation of progress. New York, NY: Raven Press; 1995:1485–1495.
7. Casey DE. Neuroleptic drug-induced extrapyramidal syndromes and tardive dyskinesia. Schizophr Res. 1991;4(2):109-120.
8. Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011;29(1):127-148.
9. Dayalu P, Chou KL. Antipsychotic-induced extrapyramidal symptoms and their management. Expert Opin Pharmacother. 2008;9(9):1451-1462.
10. Burke RE, Kang UJ, Jankovic J, et al. Tardive akathisia: an analysis of clinical features and response to open therapeutic trials. Mov Disord. 1989;4(2):157-175.
11. Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486-487.
12. American Psychiatric Association. Tardive dyskinesia: a task force report of the American Psychiatric Association. Washington DC: American Psychiatric Press, Inc; 1992.
13. Egan MF, Apud J, Wyatt RJ. Treatment of tardive dyskinesia. Schizophr Bull. 1997;23(4):583-609.
14. Casey DE, Gerlach J. Tardive dyskinesia: what is the long-term outcome? In: Casey DE Gardos G, eds. Tardive dyskinesia and neuroleptics: from dogma to reason. Washington, DC: American Psychiatric Press, Inc; 1986:76–97.
15. Glazer WM, Moore DC, Schooler NR, et al. Tardive dyskinesia. A discontinuation study. Arch Gen Psychiatry. 1984;41(6):623-627.
16. Glazer WM, Morgenstern H, Schooler N, et al. Predictors of improvement in tardive dyskinesia following discontinuation of neuroleptic medication. Br J Psychiatry. 1990;157:585-592.
17. Soares KV, McGrath JJ. The treatment of tardive dyskinesia—a systematic review and meta-analysis. Schizophr Res. 1999;39(1):1-16.
18. Gilbert PL, Harris MJ, McAdams LA, et al. Neuroleptic withdrawal in schizophrenic patients. A review of the literature. Arch Gen Psychiatry. 1995;52(3):173-188.
19. Caroff SN, Davis VG, Miller DD, et al. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303.
20. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
21. Jeste DV, Wyatt RJ. In search of treatment for tardive dyskinesia: review of the literature. Schizophr Bull. 1979;5(2):251-293.
22. Jeste DV, Lohr JB, Clark K, et al. Pharmacological treatments of tardive dyskinesia in the 1980s. J Clin Psychopharmacol. 1988;8(4 suppl):38S-48S.
23. Caroff SN, Mann SC, Campbell EC, et al. Movement disorders associated with atypical antipsychotic drugs. J Clin Psychiatry. 2002;63(suppl 4):12-19.
24. Tarsy D, Baldessarini RJ, Tarazi FI. Effects of newer antipsychotics on extrapyramidal function. CNS Drugs. 2002;16(1):23-45.
25. Emsley R, Turner HJ, Schronen J, et al. A single-blind, randomized trial comparing quetiapine and haloperidol in the treatment of tardive dyskinesia. J Clin Psychiatry. 2004;65(5):696-701.
26. Glazer WM, Hafez H. A comparison of masking effects of haloperidol versus molindone in tardive dyskinesia. Schizophr Res. 1990;3(5-6):315-320.
27. Kinon BJ, Jeste DV, Kollack-Walker S, et al. Olanzapine treatment for tardive dyskinesia in schizophrenia patients: a prospective clinical trial with patients randomized to blinded dose reduction periods. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(6):985-996.
28. Tamminga CA, Thaker GK, Moran M, et al. Clozapine in tardive dyskinesia: observations from human and animal model studies. J Clin Psychiatry. 1994;55(suppl B):102-106.
29. Simpson GM, Lee JH, Shrivastava RK. Clozapine in tardive dyskinesia. Psychopharmacology (Berl). 1978;56(1):75-80.
30. Ahmed S, Chengappa KN, Naidu VR, et al. Clozapine withdrawal-emergent dystonias and dyskinesias: a case series. J Clin Psychiatry. 1998;59(9):472-477.
31. Small JG, Milstein V, Marhenke JD, et al. Treatment outcome with clozapine in tardive dyskinesia, neuroleptic sensitivity, and treatment-resistant psychosis. J Clin Psychiatry. 1987;48(7):263-267.
32. Casey DE. Tardive dyskinesia: pathophysiology and animal models. J Clin Psychiatry. 2000;61(suppl 4):5-9.
33. Miller R, Chouinard G. Loss of striatal cholinergic neurons as a basis for tardive and L-dopa-induced dyskinesias neuroleptic-induced supersensitivity psychosis and refractory schizophrenia. Biol Psychiatry. 1993;34(10):713-738.
34. Caroff SN, Campbell EC, Havey J, et al. Treatment of tardive dyskinesia with donepezil: a pilot study. J Clin Psychiatry. 2001;62(10):772-775.
35. Caroff SN, Walker P, Campbell C, et al. Treatment of tardive dyskinesia with galantamine: a randomized controlled crossover trial. J Clin Psychiatry. 2007;68(3):410-415.
36. Caroff SN, Martine R, Kleiner-Fisman G, et al. Treatment of levodopa-induced dyskinesias with donepezil. Parkinsonism Relat Disord. 2006;12(4):261-263.
Opioid use disorder during pregnancy
For 3 years, your mental health clinic has been treating Ms. J, age 23, for bipolar disorder. She is single, unemployed, lives alone, and receives Social Security disability assistance and financial support from her parents. She has been successfully maintained on aripiprazole, 15 mg/d, and citalopram, 20 mg/d, for 18 months. Six months ago she began to miss therapy sessions and physician visits.
Her parents inform Ms. J’s therapist that she is “snorting oxycontin” with her new boyfriend. At her next visit Ms. J confirms she has been struggling to manage an opioid use disorder for more than 1 year, and requests help.
After you educate her about the diagnosis, pathophysiology, and treatment of opioid addiction, she chooses to include pharmacotherapy as part of her treatment. After informed consent, Ms. J agrees to take buprenorphine and naloxone, meet with her therapist weekly, and attend twice-weekly Narcotics Anonymous (NA) meetings. Over the ensuing months she is gradually inducted onto buprenorphine and naloxone, 12 mg, shows improved insight and motivation, provides negative urine drug screens, and demonstrates increased ability to manage her recovery. Two weeks later Ms. J tells you she may be pregnant but wants to continue buprenorphine and naloxone.
Opioid use disorder (OUD) during pregnancy is among the most difficult clinical scenarios to manage. The prevalence of OUD during pregnancy is largely unknown. However, stigma against pregnant patients with OUD is substantial.1 This article briefly summarizes identification, assessment, and treatment of OUD during pregnancy. To avoid confusion with the term “physical dependence, “ we will use “opioid use disorder” instead of “opioid dependence. “ The DSM-5 Substance Use Disorders Workgroup recommends combining abuse and dependence into a single disorder of graded clinical severity; however, this has not been finalized.2
Early identification is crucial
Early identification of OUD in pregnant women can be challenging. Self-reports underestimate use3 and shame, fear of prosecution or involvement of child welfare services, and guilt can further erode self-report. Women with OUD may have irregular menses and might not be aware of their pregnancy until several months after conception.4 Also, women with OUD who are maintained on opioid agonist therapies may misinterpret early signs of pregnancy—such as fatigue, nausea, vomiting, headaches, and cramps—as withdrawal symptoms and may respond by increasing their opioid dosing, thus exposing their fetus to increased drug levels. Finally, many women with OUD experience amenorrhea as a result of their stressful, unhealthy lifestyle, which may preclude pregnancy despite sexual activity. When these women later enroll in an opioid maintenance program, their endocrine function may return to normal, leading to unexpected pregnancy.5
Screening for OUD in pregnant patients has not been well studied. An interviewer’s nonjudgmental, empathic attitude may be more important than the specific questions he or she asks. It may be best to begin with less threatening questions and proceed to more specific questions after developing a therapeutic alliance.6
Chasnoff et al7 studied >2, 000 Medicaid-eligible pregnant patients from 9 prenatal clinics to identify risk factors for substance use during pregnancy. Alcohol or tobacco use in the month before pregnancy most differentiated current drug or alcohol use from nonuse while pregnant; however, a wide variation in use rates among patients in this study limits the generalizability of these findings. Consider OUD in women with:
-
physical examination findings or history that suggests substance use or withdrawal symptoms
-
positive drug test results for illicit or nonprescribed opioids
-
aberrant medication-taking behaviors in those receiving prescribed opioids
-
nicotine or alcohol use in the month before they knew they were pregnant
-
a history of addiction-related disorders
-
evidence of diseases associated with drug use, such as human immunodeficiency virus or hepatitis C
-
poor prenatal care attendance
-
unexplained fetal growth abnormalities.
Chasnoff et al demonstrated the reliability and effectiveness of a 1-minute, 5-item instrument (the “4 P’s Plus”) to screen for substance use, including heroin, during pregnancy (Table 1)8 In a study of 228 pregnant women, the overall internal consistency of this instrument was low but acceptable. More than three-quarters of patients (78%) were correctly classified as positive or negative, sensitivity was 87%, specificity was 76%, negative predictive validity was extremely high (97%), and positive predictive validity was low (36%). This low positive predictive validity may be acceptable in this population because over-identification of women at risk may be preferred to under-identification. The 4 P’s Plus identifies light and infrequent substance users who otherwise would go undetected, although it may place undue burden on providers to follow up on what later may be revealed to be a false positive screen.9 OUD-specific screening approaches are lacking; screening for general substance use is discussed elsewhere in the literature.10
A combination of interviewing and biologic drug screening may be more effective than either approach alone.11 Drug screening should include opioids typically screened for (morphine, codeine, heroin metabolite) and those for which additional tests may be required (eg, semi-synthetics such as oxycodone and synthetics such as fentanyl). Learn your state’s civil mandates regarding drug-using pregnant women, guidelines for addiction treatment, and confidentiality provisions, especially as they relate to drug testing and mandatory reporting. Ideally, patients should be informed of these issues before they undergo drug testing or other procedures. These requirements may vary according to physician specialty or role in providing care.
Diagnosis of opioid dependence is based on DSM-IV-TR criteria; however, the proposed DSM-5 criteria for OUD may better emphasize cautions about including tolerance or withdrawal when diagnosing OUD in the setting of medically supervised and appropriate opioid use.2
Stigma against pregnant women with OUD easily can erode therapeutic efforts. Perhaps the most important element of assessment is maximizing the therapeutic alliance to ensure that the patient complies with prenatal obstetric care and maternal addiction services. Pregnancy may be an opportune time to motivate women with OUD to make a change because they may be more open to receiving help.12 Motivational interventions are helpful for many but not all patients; the best approach to such interventions is still uncertain.13 Regardless of the mother’s motivation, prenatal care is fundamental.
Table 1 The ‘4P’s Plus’ screen for substance use during pregnancy
| Parents: Did either of your parents ever have a problem with alcohol or drugs? |
| Partner: Does your partner have a problem with alcohol or drugs? |
| Past: Have you ever drunk beer, wine, or liquor? |
| Pregnancy: In the month before you knew you were pregnant, how many cigarettes did you smoke? |
| In the month before you knew you were pregnant, how many beers/how much wine/ how much liquor did you drink? |
| A positive screen results when a patient answers either of the 2 questions relating to pregnancy, indicating any alcohol or tobacco use in the month before she knew she was pregnant |
| Source:Reference 8 |
Office management
OUD-specific treatment decreases opioid use and improves birth outcomes14; however, retaining these patients in treatment can be difficult. Addressing social issues— including financial burdens, unstable living conditions, intimate partner violence, transportation difficulties, and limited access to medical and child care—can facilitate treatment.5 The Addiction Severity Index version tailored to women and pregnancy15 examines 7 domains of functioning (drugs, alcohol, psychological, social, medical, legal, and employment), informs treatment planning, quantifies treatment progress, and has predictive validity.16 Services are more likely to be effective if started during pregnancy as opposed to after delivery. Although detoxification is possible under carefully monitored conditions, many women relapse after detoxifying, and neonatal abstinence syndrome (NAS)—a disorder in which an addicted newborn experiences drug withdrawal—is common. Therefore, the risks of detoxification often outweigh benefits.5,17,18
Rehabilitation services for the mother can be provided at various levels of care, including outpatient, intensive outpatient, day hospital, residential, and inpatient. Although pregnancy-specific OUD treatment is ideal, it may not be available. Clinicians should attempt to locate services that can incorporate resources for pregnant women. Providing a means for child care during treatment is paramount to compliance. Develop a plan for nonconfrontational counseling, job skills training/education, and ongoing care after delivery (including child care and transportation resources) at the onset of treatment. The length of time maintained in treatment is one of the strongest predictors of abstinence.5
Pregnant women with OUD should be screened for comorbid medical, obstetric, and psychiatric complications and referred accordingly (Table 2 and Table 3).6 Coordination among the patient’s psychiatrist, primary care provider, and obstetrician/gynecologist is essential. Programs that integrate these approaches into a single treatment team may be ideal. Although pregnancy per se may not be associated with higher risk of mental disorders, the risk of major depressive disorder may be increased during the postpartum period.19 Young, unmarried women with recent stressful life events, complicated pregnancies, and poor overall health may face a significantly increased risk of psychiatric illness during pregnancy.19 Patients whose opioid use has caused pregnancy complications may experience guilt and grief.
Increased education and screening for substance use as the pregnancy approaches term is necessary because patients may mistake early labor for symptoms of opioid withdrawal or worry that delivery room pain management will be inadequate and therefore relapse. Among pregnant women with addiction, preterm labor may be most common in those with OUD.12
Table 2 Medical complications common to pregnancy and substance abuse
| Anemia |
| Bacteremia/sepsis |
| Endocarditis |
| Cellulitis |
| Depression/anxiety |
| Gestational diabetes |
| Hepatitis (chronic and acute) |
| Hypertension/tachycardia |
| Phlebitis |
| Pneumonia |
| Gingivitis/poor oral hygiene |
| Sexually transmitted diseases |
|
|
| Tetanus |
| Cystitis |
| Pyelonephritis |
| AIDS: acquired immune deficiency syndrome; HIV: human immunodeficiency virus |
| Source:Reference 6 |
Table 3 Obstetric complications in women with addiction disorders
| Placental abruption |
| Chorioamnionitis |
| Placental insufficiency |
| Intrauterine growth restriction |
| Hypoxic/ischemic brain injury |
| Meconium passage |
| Neonatal abstinence syndrome |
| Spontaneous abortion |
| Intrauterine fetal death |
| Premature labor and delivery |
| Preterm, premature rupture of membranes |
| Postpartum hemorrhage |
| Hypertensive emergencies/preeclampsia |
| Source:Reference 6 |
Opioid agonist therapy
Obstetric complications in women with OUD may be related to rapid, frequent fluctuations of opioid blood levels during intoxication and withdrawal. Therefore, the first goal of pharmacotherapy is to reduce physical stress associated with cycling opioid blood levels. Opioid agonist medications can be extremely effective. Opioid agonist treatment for pregnant patients is similar to that of nonpregnant patients but includes pregnancy-specific objectives (Table 4).20
Few anti-relapse medications have been studied in pregnant patients. Pharmacotherapies for OUD include methadone and buprenorphine. In our experience, opioid antagonists such as naltrexone typically would not be considered for pregnant patients because:
-
their expected efficacy in reducing relapse in pregnant patients is lower than that of other medications
-
their expected risk for inducing withdrawal is higher compared with methadone or buprenorphine
-
research on the use of naltrexone during pregnancy is lacking.
Methadone has been used to treat OUD during pregnancy since the late 1970s.5 It requires adherence to strict federal regulations and is FDA pregnancy class C (animal reproduction studies have shown an adverse effect on the fetus and there are no adequate well-controlled studies in humans, but potential benefits may warrant use in pregnant women despite potential risks). Pregnant women have been safely maintained on methadone without adverse long-term maternal or fetal effects, and the National Institutes of Health recommends it as the standard of care for pregnant women with OUD. A woman steadily maintained on methadone is more likely to have a healthy pregnancy and infant than a woman who uses alcohol or other drugs.21 Further, the structure and services of methadone maintenance treatment can improve compliance with prenatal care and help prepare patients for parental responsibilities.
Fluctuating blood opioid levels are minimized when methadone dosage is individually determined. Dosages should be based on a woman’s stage of pregnancy, relapse risk, pre-pregnancy methadone dose, experience with methadone, and clinical history. Some women experience lowered methadone blood levels during pregnancy because of increased fluid space, a larger tissue reservoir that can store methadone, and increased drug metabolism by both placenta and fetus. As a result, increased or split (twice daily) dosing may be indicated.22-24
Buprenorphine is FDA pregnancy class C. Although not approved for use during pregnancy, it has been used successfully for pregnant patients with OUD.12,25 It is a partial agonist of the mu opioid receptor and an antagonist of the kappa opioid receptor, which may reduce its abuse liability and NAS severity.
The few randomized clinical trials comparing methadone with buprenorphine during pregnancy suggest that buprenorphine is not inferior to methadone in safety and discomfort of induction from a short-acting opioid, nor in outcome measures assessing NAS and maternal and neonatal safety.26,27 Results from the recent Maternal Opioid Treatment: Human Experimental Research project suggest that buprenorphine may have some advantages over methadone in pregnancy. Buprenorphine-maintained neonates may need less morphine, have shorter hospital stays, and require shorter treatment for NAS.28 However, treatment retention may be lower for buprenorphine-maintained mothers; any resultant long-term consequences on maternal and child health are as yet unexplored. These findings require replication.
Methadone and buprenorphine are not interchangeable. Many patients maintained on methadone do not respond optimally to buprenorphine. Clinics that dispense maintenance methadone are required to provide counseling services and random drug testing; these requirements do not apply to physicians who prescribe buprenorphine. Moreover, in our experience buprenorphine at times has been prescribed without close regard to psychosocial issues, adequate random drug testing, or coordination of care with other providers.
In pregnant patients, buprenorphine is preferred over buprenorphine and naloxone to avoid fetal exposure to naloxone, which may cause intrauterine withdrawal and maternal-fetal hormonal changes. To reduce abuse or diversion, patients should undergo drug testing to ensure buprenorphine is present, smaller prescriptions may be provided, and tablets can be counted. Limited data suggests buprenorphine is not teratogenic. Some data show low placental transfer of buprenorphine, thereby limiting fetal exposure and lowering risk for intrauterine growth restriction.29
Table 4 Opioid agonist treatment objectives for addicted patients who are pregnant
| General objectives |
| Prevent opioid withdrawal signs and symptoms |
| Provide a comfortable induction onto the medication |
| Block the euphoric and reinforcing effects of illicit opioids while also attenuating the motivation (craving, social interactions) to use illicit opioids and other drugs |
| Enhance treatment retention |
| Create a more optimal environment for behavioral and psychosocial interventions |
| Pregnancy-specific objectives |
| Eliminate or reduce fetal exposure to illicit opioids and other illicit drugs |
| Stabilize the intrauterine environment |
| Enhance involvement in prenatal care |
| Create an optimal environment to address pregnancy-specific problems |
| Source:Reference 20 |
Delivery and postnatal care
Compared with those not in treatment, women who are engaged in a multidisciplinary treatment program at the time of delivery demonstrated higher gestational age, increased birth weights, and lower rates of neonatal ICU admissions. They also realized a cost savings of $4, 644 per mother-infant pair.30
During delivery, pain medication should not be withheld solely because a pregnant woman has a history of addiction-related disorders; these women are subject to pain during delivery as much as other women. Avoid using mixed agonists/antagonists such as nalbuphine or butorphanol in women receiving opioid maintenance medication. Labor and delivery pain management for a pregnant patient maintained on opioid agonist therapies is discussed elsewhere in the literature.31 Every effort should be made to ensure that the mother remains in treatment through delivery and beyond.
To read about advising women with OUD on the benefits and risks of breastfeeding while receiving opioid agonist maintenance treatment, see the Box below.
CASE CONTINUED: Medication change
Ms. J’s boyfriend has left her and her parents have not readily accepted her pregnancy and need for support. She continues to attend NA meetings and weekly therapy. After educating her about the differences between buprenorphine and buprenorphine and naloxone in relation to risk, benefits, and side effects, you switch Ms. J to buprenorphine, 12 mg/d, while maintaining her on aripiprazole and citalopram. She consents to exchanging information about her medical, mental health, and addiction-related treatment with her primary care provider, who helps locate an obstetrician/gynecologist comfortable with her OUD and buprenorphine. Ms. J’s therapist helps link her with social services agencies to ensure prenatal care, assist with removing barriers to care, and plan for her needs as a parent.
After checking your state’s mandates, you determine you are not required to report Ms. J’s drug testing results. Ms. J’s ongoing drug testing shows the presence of buprenorphine and the absence of other opioids and all drugs of abuse.
Ms. J’s delivery is uncomplicated medically; however, family, financial, and parental role issues remain problematic. Encouraging her involvement in therapy and social services as part of her continued buprenorphine prescribing proves beneficial.
Related Resources
-
Jones HE, Martin PR, Heil SH, et al. Treatment of opioid dependent pregnant women: clinical and research issues. J Subst Abuse Treat. 2008; 35(3): 245-259.
-
Johnson RE, Jones HE, Fischer G. Use of buprenorphine in pregnancy: patient management and effects on the neonate. Drug Alcohol Depend. 2003; 70(suppl 1 ): S87-S101.
-
Velez M, Jansson LM. The opioid dependent mother and the newborn dyad: nonpharmacologic care. J Addict Med. 2008; 2(3): 113-120.
Drug Brand Names
Aripiprazole • Abilify
Buprenorphine and naloxone •Suboxone
Buprenorphine • Subutex
Butorphanol • Stadol
Citalopram • Celexa
Fentanyl • Duragesic, Sublimaze, others
Methadone • Dolophine
Naloxone • Narcan
Naltrexone • ReVia
Nalbuphine • Nubain
Oxycodone • Oxycontin
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Fernandez’ time toward this project was funded by the University Hospital/University of Cincinnati Addiction Psychiatry Fellowship Training Program operated by the Center for Treatment, Research, and Education in Addictive Disorders (CeTREAD), Department of Psychiatry and Behavioral Neuroscience, University of Cincinnati and by the Veterans Affairs Medical Center, Cincinnati, OH.
The statements in this publication do not necessarily reflect the views or opinions of the Department of Veterans Affairs, the United States Government, or Opiate Addiction Recovery Services.
For 3 years, your mental health clinic has been treating Ms. J, age 23, for bipolar disorder. She is single, unemployed, lives alone, and receives Social Security disability assistance and financial support from her parents. She has been successfully maintained on aripiprazole, 15 mg/d, and citalopram, 20 mg/d, for 18 months. Six months ago she began to miss therapy sessions and physician visits.
Her parents inform Ms. J’s therapist that she is “snorting oxycontin” with her new boyfriend. At her next visit Ms. J confirms she has been struggling to manage an opioid use disorder for more than 1 year, and requests help.
After you educate her about the diagnosis, pathophysiology, and treatment of opioid addiction, she chooses to include pharmacotherapy as part of her treatment. After informed consent, Ms. J agrees to take buprenorphine and naloxone, meet with her therapist weekly, and attend twice-weekly Narcotics Anonymous (NA) meetings. Over the ensuing months she is gradually inducted onto buprenorphine and naloxone, 12 mg, shows improved insight and motivation, provides negative urine drug screens, and demonstrates increased ability to manage her recovery. Two weeks later Ms. J tells you she may be pregnant but wants to continue buprenorphine and naloxone.
Opioid use disorder (OUD) during pregnancy is among the most difficult clinical scenarios to manage. The prevalence of OUD during pregnancy is largely unknown. However, stigma against pregnant patients with OUD is substantial.1 This article briefly summarizes identification, assessment, and treatment of OUD during pregnancy. To avoid confusion with the term “physical dependence, “ we will use “opioid use disorder” instead of “opioid dependence. “ The DSM-5 Substance Use Disorders Workgroup recommends combining abuse and dependence into a single disorder of graded clinical severity; however, this has not been finalized.2
Early identification is crucial
Early identification of OUD in pregnant women can be challenging. Self-reports underestimate use3 and shame, fear of prosecution or involvement of child welfare services, and guilt can further erode self-report. Women with OUD may have irregular menses and might not be aware of their pregnancy until several months after conception.4 Also, women with OUD who are maintained on opioid agonist therapies may misinterpret early signs of pregnancy—such as fatigue, nausea, vomiting, headaches, and cramps—as withdrawal symptoms and may respond by increasing their opioid dosing, thus exposing their fetus to increased drug levels. Finally, many women with OUD experience amenorrhea as a result of their stressful, unhealthy lifestyle, which may preclude pregnancy despite sexual activity. When these women later enroll in an opioid maintenance program, their endocrine function may return to normal, leading to unexpected pregnancy.5
Screening for OUD in pregnant patients has not been well studied. An interviewer’s nonjudgmental, empathic attitude may be more important than the specific questions he or she asks. It may be best to begin with less threatening questions and proceed to more specific questions after developing a therapeutic alliance.6
Chasnoff et al7 studied >2, 000 Medicaid-eligible pregnant patients from 9 prenatal clinics to identify risk factors for substance use during pregnancy. Alcohol or tobacco use in the month before pregnancy most differentiated current drug or alcohol use from nonuse while pregnant; however, a wide variation in use rates among patients in this study limits the generalizability of these findings. Consider OUD in women with:
-
physical examination findings or history that suggests substance use or withdrawal symptoms
-
positive drug test results for illicit or nonprescribed opioids
-
aberrant medication-taking behaviors in those receiving prescribed opioids
-
nicotine or alcohol use in the month before they knew they were pregnant
-
a history of addiction-related disorders
-
evidence of diseases associated with drug use, such as human immunodeficiency virus or hepatitis C
-
poor prenatal care attendance
-
unexplained fetal growth abnormalities.
Chasnoff et al demonstrated the reliability and effectiveness of a 1-minute, 5-item instrument (the “4 P’s Plus”) to screen for substance use, including heroin, during pregnancy (Table 1)8 In a study of 228 pregnant women, the overall internal consistency of this instrument was low but acceptable. More than three-quarters of patients (78%) were correctly classified as positive or negative, sensitivity was 87%, specificity was 76%, negative predictive validity was extremely high (97%), and positive predictive validity was low (36%). This low positive predictive validity may be acceptable in this population because over-identification of women at risk may be preferred to under-identification. The 4 P’s Plus identifies light and infrequent substance users who otherwise would go undetected, although it may place undue burden on providers to follow up on what later may be revealed to be a false positive screen.9 OUD-specific screening approaches are lacking; screening for general substance use is discussed elsewhere in the literature.10
A combination of interviewing and biologic drug screening may be more effective than either approach alone.11 Drug screening should include opioids typically screened for (morphine, codeine, heroin metabolite) and those for which additional tests may be required (eg, semi-synthetics such as oxycodone and synthetics such as fentanyl). Learn your state’s civil mandates regarding drug-using pregnant women, guidelines for addiction treatment, and confidentiality provisions, especially as they relate to drug testing and mandatory reporting. Ideally, patients should be informed of these issues before they undergo drug testing or other procedures. These requirements may vary according to physician specialty or role in providing care.
Diagnosis of opioid dependence is based on DSM-IV-TR criteria; however, the proposed DSM-5 criteria for OUD may better emphasize cautions about including tolerance or withdrawal when diagnosing OUD in the setting of medically supervised and appropriate opioid use.2
Stigma against pregnant women with OUD easily can erode therapeutic efforts. Perhaps the most important element of assessment is maximizing the therapeutic alliance to ensure that the patient complies with prenatal obstetric care and maternal addiction services. Pregnancy may be an opportune time to motivate women with OUD to make a change because they may be more open to receiving help.12 Motivational interventions are helpful for many but not all patients; the best approach to such interventions is still uncertain.13 Regardless of the mother’s motivation, prenatal care is fundamental.
Table 1 The ‘4P’s Plus’ screen for substance use during pregnancy
| Parents: Did either of your parents ever have a problem with alcohol or drugs? |
| Partner: Does your partner have a problem with alcohol or drugs? |
| Past: Have you ever drunk beer, wine, or liquor? |
| Pregnancy: In the month before you knew you were pregnant, how many cigarettes did you smoke? |
| In the month before you knew you were pregnant, how many beers/how much wine/ how much liquor did you drink? |
| A positive screen results when a patient answers either of the 2 questions relating to pregnancy, indicating any alcohol or tobacco use in the month before she knew she was pregnant |
| Source:Reference 8 |
Office management
OUD-specific treatment decreases opioid use and improves birth outcomes14; however, retaining these patients in treatment can be difficult. Addressing social issues— including financial burdens, unstable living conditions, intimate partner violence, transportation difficulties, and limited access to medical and child care—can facilitate treatment.5 The Addiction Severity Index version tailored to women and pregnancy15 examines 7 domains of functioning (drugs, alcohol, psychological, social, medical, legal, and employment), informs treatment planning, quantifies treatment progress, and has predictive validity.16 Services are more likely to be effective if started during pregnancy as opposed to after delivery. Although detoxification is possible under carefully monitored conditions, many women relapse after detoxifying, and neonatal abstinence syndrome (NAS)—a disorder in which an addicted newborn experiences drug withdrawal—is common. Therefore, the risks of detoxification often outweigh benefits.5,17,18
Rehabilitation services for the mother can be provided at various levels of care, including outpatient, intensive outpatient, day hospital, residential, and inpatient. Although pregnancy-specific OUD treatment is ideal, it may not be available. Clinicians should attempt to locate services that can incorporate resources for pregnant women. Providing a means for child care during treatment is paramount to compliance. Develop a plan for nonconfrontational counseling, job skills training/education, and ongoing care after delivery (including child care and transportation resources) at the onset of treatment. The length of time maintained in treatment is one of the strongest predictors of abstinence.5
Pregnant women with OUD should be screened for comorbid medical, obstetric, and psychiatric complications and referred accordingly (Table 2 and Table 3).6 Coordination among the patient’s psychiatrist, primary care provider, and obstetrician/gynecologist is essential. Programs that integrate these approaches into a single treatment team may be ideal. Although pregnancy per se may not be associated with higher risk of mental disorders, the risk of major depressive disorder may be increased during the postpartum period.19 Young, unmarried women with recent stressful life events, complicated pregnancies, and poor overall health may face a significantly increased risk of psychiatric illness during pregnancy.19 Patients whose opioid use has caused pregnancy complications may experience guilt and grief.
Increased education and screening for substance use as the pregnancy approaches term is necessary because patients may mistake early labor for symptoms of opioid withdrawal or worry that delivery room pain management will be inadequate and therefore relapse. Among pregnant women with addiction, preterm labor may be most common in those with OUD.12
Table 2 Medical complications common to pregnancy and substance abuse
| Anemia |
| Bacteremia/sepsis |
| Endocarditis |
| Cellulitis |
| Depression/anxiety |
| Gestational diabetes |
| Hepatitis (chronic and acute) |
| Hypertension/tachycardia |
| Phlebitis |
| Pneumonia |
| Gingivitis/poor oral hygiene |
| Sexually transmitted diseases |
|
|
| Tetanus |
| Cystitis |
| Pyelonephritis |
| AIDS: acquired immune deficiency syndrome; HIV: human immunodeficiency virus |
| Source:Reference 6 |
Table 3 Obstetric complications in women with addiction disorders
| Placental abruption |
| Chorioamnionitis |
| Placental insufficiency |
| Intrauterine growth restriction |
| Hypoxic/ischemic brain injury |
| Meconium passage |
| Neonatal abstinence syndrome |
| Spontaneous abortion |
| Intrauterine fetal death |
| Premature labor and delivery |
| Preterm, premature rupture of membranes |
| Postpartum hemorrhage |
| Hypertensive emergencies/preeclampsia |
| Source:Reference 6 |
Opioid agonist therapy
Obstetric complications in women with OUD may be related to rapid, frequent fluctuations of opioid blood levels during intoxication and withdrawal. Therefore, the first goal of pharmacotherapy is to reduce physical stress associated with cycling opioid blood levels. Opioid agonist medications can be extremely effective. Opioid agonist treatment for pregnant patients is similar to that of nonpregnant patients but includes pregnancy-specific objectives (Table 4).20
Few anti-relapse medications have been studied in pregnant patients. Pharmacotherapies for OUD include methadone and buprenorphine. In our experience, opioid antagonists such as naltrexone typically would not be considered for pregnant patients because:
-
their expected efficacy in reducing relapse in pregnant patients is lower than that of other medications
-
their expected risk for inducing withdrawal is higher compared with methadone or buprenorphine
-
research on the use of naltrexone during pregnancy is lacking.
Methadone has been used to treat OUD during pregnancy since the late 1970s.5 It requires adherence to strict federal regulations and is FDA pregnancy class C (animal reproduction studies have shown an adverse effect on the fetus and there are no adequate well-controlled studies in humans, but potential benefits may warrant use in pregnant women despite potential risks). Pregnant women have been safely maintained on methadone without adverse long-term maternal or fetal effects, and the National Institutes of Health recommends it as the standard of care for pregnant women with OUD. A woman steadily maintained on methadone is more likely to have a healthy pregnancy and infant than a woman who uses alcohol or other drugs.21 Further, the structure and services of methadone maintenance treatment can improve compliance with prenatal care and help prepare patients for parental responsibilities.
Fluctuating blood opioid levels are minimized when methadone dosage is individually determined. Dosages should be based on a woman’s stage of pregnancy, relapse risk, pre-pregnancy methadone dose, experience with methadone, and clinical history. Some women experience lowered methadone blood levels during pregnancy because of increased fluid space, a larger tissue reservoir that can store methadone, and increased drug metabolism by both placenta and fetus. As a result, increased or split (twice daily) dosing may be indicated.22-24
Buprenorphine is FDA pregnancy class C. Although not approved for use during pregnancy, it has been used successfully for pregnant patients with OUD.12,25 It is a partial agonist of the mu opioid receptor and an antagonist of the kappa opioid receptor, which may reduce its abuse liability and NAS severity.
The few randomized clinical trials comparing methadone with buprenorphine during pregnancy suggest that buprenorphine is not inferior to methadone in safety and discomfort of induction from a short-acting opioid, nor in outcome measures assessing NAS and maternal and neonatal safety.26,27 Results from the recent Maternal Opioid Treatment: Human Experimental Research project suggest that buprenorphine may have some advantages over methadone in pregnancy. Buprenorphine-maintained neonates may need less morphine, have shorter hospital stays, and require shorter treatment for NAS.28 However, treatment retention may be lower for buprenorphine-maintained mothers; any resultant long-term consequences on maternal and child health are as yet unexplored. These findings require replication.
Methadone and buprenorphine are not interchangeable. Many patients maintained on methadone do not respond optimally to buprenorphine. Clinics that dispense maintenance methadone are required to provide counseling services and random drug testing; these requirements do not apply to physicians who prescribe buprenorphine. Moreover, in our experience buprenorphine at times has been prescribed without close regard to psychosocial issues, adequate random drug testing, or coordination of care with other providers.
In pregnant patients, buprenorphine is preferred over buprenorphine and naloxone to avoid fetal exposure to naloxone, which may cause intrauterine withdrawal and maternal-fetal hormonal changes. To reduce abuse or diversion, patients should undergo drug testing to ensure buprenorphine is present, smaller prescriptions may be provided, and tablets can be counted. Limited data suggests buprenorphine is not teratogenic. Some data show low placental transfer of buprenorphine, thereby limiting fetal exposure and lowering risk for intrauterine growth restriction.29
Table 4 Opioid agonist treatment objectives for addicted patients who are pregnant
| General objectives |
| Prevent opioid withdrawal signs and symptoms |
| Provide a comfortable induction onto the medication |
| Block the euphoric and reinforcing effects of illicit opioids while also attenuating the motivation (craving, social interactions) to use illicit opioids and other drugs |
| Enhance treatment retention |
| Create a more optimal environment for behavioral and psychosocial interventions |
| Pregnancy-specific objectives |
| Eliminate or reduce fetal exposure to illicit opioids and other illicit drugs |
| Stabilize the intrauterine environment |
| Enhance involvement in prenatal care |
| Create an optimal environment to address pregnancy-specific problems |
| Source:Reference 20 |
Delivery and postnatal care
Compared with those not in treatment, women who are engaged in a multidisciplinary treatment program at the time of delivery demonstrated higher gestational age, increased birth weights, and lower rates of neonatal ICU admissions. They also realized a cost savings of $4, 644 per mother-infant pair.30
During delivery, pain medication should not be withheld solely because a pregnant woman has a history of addiction-related disorders; these women are subject to pain during delivery as much as other women. Avoid using mixed agonists/antagonists such as nalbuphine or butorphanol in women receiving opioid maintenance medication. Labor and delivery pain management for a pregnant patient maintained on opioid agonist therapies is discussed elsewhere in the literature.31 Every effort should be made to ensure that the mother remains in treatment through delivery and beyond.
To read about advising women with OUD on the benefits and risks of breastfeeding while receiving opioid agonist maintenance treatment, see the Box below.
CASE CONTINUED: Medication change
Ms. J’s boyfriend has left her and her parents have not readily accepted her pregnancy and need for support. She continues to attend NA meetings and weekly therapy. After educating her about the differences between buprenorphine and buprenorphine and naloxone in relation to risk, benefits, and side effects, you switch Ms. J to buprenorphine, 12 mg/d, while maintaining her on aripiprazole and citalopram. She consents to exchanging information about her medical, mental health, and addiction-related treatment with her primary care provider, who helps locate an obstetrician/gynecologist comfortable with her OUD and buprenorphine. Ms. J’s therapist helps link her with social services agencies to ensure prenatal care, assist with removing barriers to care, and plan for her needs as a parent.
After checking your state’s mandates, you determine you are not required to report Ms. J’s drug testing results. Ms. J’s ongoing drug testing shows the presence of buprenorphine and the absence of other opioids and all drugs of abuse.
Ms. J’s delivery is uncomplicated medically; however, family, financial, and parental role issues remain problematic. Encouraging her involvement in therapy and social services as part of her continued buprenorphine prescribing proves beneficial.
Related Resources
-
Jones HE, Martin PR, Heil SH, et al. Treatment of opioid dependent pregnant women: clinical and research issues. J Subst Abuse Treat. 2008; 35(3): 245-259.
-
Johnson RE, Jones HE, Fischer G. Use of buprenorphine in pregnancy: patient management and effects on the neonate. Drug Alcohol Depend. 2003; 70(suppl 1 ): S87-S101.
-
Velez M, Jansson LM. The opioid dependent mother and the newborn dyad: nonpharmacologic care. J Addict Med. 2008; 2(3): 113-120.
Drug Brand Names
Aripiprazole • Abilify
Buprenorphine and naloxone •Suboxone
Buprenorphine • Subutex
Butorphanol • Stadol
Citalopram • Celexa
Fentanyl • Duragesic, Sublimaze, others
Methadone • Dolophine
Naloxone • Narcan
Naltrexone • ReVia
Nalbuphine • Nubain
Oxycodone • Oxycontin
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Fernandez’ time toward this project was funded by the University Hospital/University of Cincinnati Addiction Psychiatry Fellowship Training Program operated by the Center for Treatment, Research, and Education in Addictive Disorders (CeTREAD), Department of Psychiatry and Behavioral Neuroscience, University of Cincinnati and by the Veterans Affairs Medical Center, Cincinnati, OH.
The statements in this publication do not necessarily reflect the views or opinions of the Department of Veterans Affairs, the United States Government, or Opiate Addiction Recovery Services.
For 3 years, your mental health clinic has been treating Ms. J, age 23, for bipolar disorder. She is single, unemployed, lives alone, and receives Social Security disability assistance and financial support from her parents. She has been successfully maintained on aripiprazole, 15 mg/d, and citalopram, 20 mg/d, for 18 months. Six months ago she began to miss therapy sessions and physician visits.
Her parents inform Ms. J’s therapist that she is “snorting oxycontin” with her new boyfriend. At her next visit Ms. J confirms she has been struggling to manage an opioid use disorder for more than 1 year, and requests help.
After you educate her about the diagnosis, pathophysiology, and treatment of opioid addiction, she chooses to include pharmacotherapy as part of her treatment. After informed consent, Ms. J agrees to take buprenorphine and naloxone, meet with her therapist weekly, and attend twice-weekly Narcotics Anonymous (NA) meetings. Over the ensuing months she is gradually inducted onto buprenorphine and naloxone, 12 mg, shows improved insight and motivation, provides negative urine drug screens, and demonstrates increased ability to manage her recovery. Two weeks later Ms. J tells you she may be pregnant but wants to continue buprenorphine and naloxone.
Opioid use disorder (OUD) during pregnancy is among the most difficult clinical scenarios to manage. The prevalence of OUD during pregnancy is largely unknown. However, stigma against pregnant patients with OUD is substantial.1 This article briefly summarizes identification, assessment, and treatment of OUD during pregnancy. To avoid confusion with the term “physical dependence, “ we will use “opioid use disorder” instead of “opioid dependence. “ The DSM-5 Substance Use Disorders Workgroup recommends combining abuse and dependence into a single disorder of graded clinical severity; however, this has not been finalized.2
Early identification is crucial
Early identification of OUD in pregnant women can be challenging. Self-reports underestimate use3 and shame, fear of prosecution or involvement of child welfare services, and guilt can further erode self-report. Women with OUD may have irregular menses and might not be aware of their pregnancy until several months after conception.4 Also, women with OUD who are maintained on opioid agonist therapies may misinterpret early signs of pregnancy—such as fatigue, nausea, vomiting, headaches, and cramps—as withdrawal symptoms and may respond by increasing their opioid dosing, thus exposing their fetus to increased drug levels. Finally, many women with OUD experience amenorrhea as a result of their stressful, unhealthy lifestyle, which may preclude pregnancy despite sexual activity. When these women later enroll in an opioid maintenance program, their endocrine function may return to normal, leading to unexpected pregnancy.5
Screening for OUD in pregnant patients has not been well studied. An interviewer’s nonjudgmental, empathic attitude may be more important than the specific questions he or she asks. It may be best to begin with less threatening questions and proceed to more specific questions after developing a therapeutic alliance.6
Chasnoff et al7 studied >2, 000 Medicaid-eligible pregnant patients from 9 prenatal clinics to identify risk factors for substance use during pregnancy. Alcohol or tobacco use in the month before pregnancy most differentiated current drug or alcohol use from nonuse while pregnant; however, a wide variation in use rates among patients in this study limits the generalizability of these findings. Consider OUD in women with:
-
physical examination findings or history that suggests substance use or withdrawal symptoms
-
positive drug test results for illicit or nonprescribed opioids
-
aberrant medication-taking behaviors in those receiving prescribed opioids
-
nicotine or alcohol use in the month before they knew they were pregnant
-
a history of addiction-related disorders
-
evidence of diseases associated with drug use, such as human immunodeficiency virus or hepatitis C
-
poor prenatal care attendance
-
unexplained fetal growth abnormalities.
Chasnoff et al demonstrated the reliability and effectiveness of a 1-minute, 5-item instrument (the “4 P’s Plus”) to screen for substance use, including heroin, during pregnancy (Table 1)8 In a study of 228 pregnant women, the overall internal consistency of this instrument was low but acceptable. More than three-quarters of patients (78%) were correctly classified as positive or negative, sensitivity was 87%, specificity was 76%, negative predictive validity was extremely high (97%), and positive predictive validity was low (36%). This low positive predictive validity may be acceptable in this population because over-identification of women at risk may be preferred to under-identification. The 4 P’s Plus identifies light and infrequent substance users who otherwise would go undetected, although it may place undue burden on providers to follow up on what later may be revealed to be a false positive screen.9 OUD-specific screening approaches are lacking; screening for general substance use is discussed elsewhere in the literature.10
A combination of interviewing and biologic drug screening may be more effective than either approach alone.11 Drug screening should include opioids typically screened for (morphine, codeine, heroin metabolite) and those for which additional tests may be required (eg, semi-synthetics such as oxycodone and synthetics such as fentanyl). Learn your state’s civil mandates regarding drug-using pregnant women, guidelines for addiction treatment, and confidentiality provisions, especially as they relate to drug testing and mandatory reporting. Ideally, patients should be informed of these issues before they undergo drug testing or other procedures. These requirements may vary according to physician specialty or role in providing care.
Diagnosis of opioid dependence is based on DSM-IV-TR criteria; however, the proposed DSM-5 criteria for OUD may better emphasize cautions about including tolerance or withdrawal when diagnosing OUD in the setting of medically supervised and appropriate opioid use.2
Stigma against pregnant women with OUD easily can erode therapeutic efforts. Perhaps the most important element of assessment is maximizing the therapeutic alliance to ensure that the patient complies with prenatal obstetric care and maternal addiction services. Pregnancy may be an opportune time to motivate women with OUD to make a change because they may be more open to receiving help.12 Motivational interventions are helpful for many but not all patients; the best approach to such interventions is still uncertain.13 Regardless of the mother’s motivation, prenatal care is fundamental.
Table 1 The ‘4P’s Plus’ screen for substance use during pregnancy
| Parents: Did either of your parents ever have a problem with alcohol or drugs? |
| Partner: Does your partner have a problem with alcohol or drugs? |
| Past: Have you ever drunk beer, wine, or liquor? |
| Pregnancy: In the month before you knew you were pregnant, how many cigarettes did you smoke? |
| In the month before you knew you were pregnant, how many beers/how much wine/ how much liquor did you drink? |
| A positive screen results when a patient answers either of the 2 questions relating to pregnancy, indicating any alcohol or tobacco use in the month before she knew she was pregnant |
| Source:Reference 8 |
Office management
OUD-specific treatment decreases opioid use and improves birth outcomes14; however, retaining these patients in treatment can be difficult. Addressing social issues— including financial burdens, unstable living conditions, intimate partner violence, transportation difficulties, and limited access to medical and child care—can facilitate treatment.5 The Addiction Severity Index version tailored to women and pregnancy15 examines 7 domains of functioning (drugs, alcohol, psychological, social, medical, legal, and employment), informs treatment planning, quantifies treatment progress, and has predictive validity.16 Services are more likely to be effective if started during pregnancy as opposed to after delivery. Although detoxification is possible under carefully monitored conditions, many women relapse after detoxifying, and neonatal abstinence syndrome (NAS)—a disorder in which an addicted newborn experiences drug withdrawal—is common. Therefore, the risks of detoxification often outweigh benefits.5,17,18
Rehabilitation services for the mother can be provided at various levels of care, including outpatient, intensive outpatient, day hospital, residential, and inpatient. Although pregnancy-specific OUD treatment is ideal, it may not be available. Clinicians should attempt to locate services that can incorporate resources for pregnant women. Providing a means for child care during treatment is paramount to compliance. Develop a plan for nonconfrontational counseling, job skills training/education, and ongoing care after delivery (including child care and transportation resources) at the onset of treatment. The length of time maintained in treatment is one of the strongest predictors of abstinence.5
Pregnant women with OUD should be screened for comorbid medical, obstetric, and psychiatric complications and referred accordingly (Table 2 and Table 3).6 Coordination among the patient’s psychiatrist, primary care provider, and obstetrician/gynecologist is essential. Programs that integrate these approaches into a single treatment team may be ideal. Although pregnancy per se may not be associated with higher risk of mental disorders, the risk of major depressive disorder may be increased during the postpartum period.19 Young, unmarried women with recent stressful life events, complicated pregnancies, and poor overall health may face a significantly increased risk of psychiatric illness during pregnancy.19 Patients whose opioid use has caused pregnancy complications may experience guilt and grief.
Increased education and screening for substance use as the pregnancy approaches term is necessary because patients may mistake early labor for symptoms of opioid withdrawal or worry that delivery room pain management will be inadequate and therefore relapse. Among pregnant women with addiction, preterm labor may be most common in those with OUD.12
Table 2 Medical complications common to pregnancy and substance abuse
| Anemia |
| Bacteremia/sepsis |
| Endocarditis |
| Cellulitis |
| Depression/anxiety |
| Gestational diabetes |
| Hepatitis (chronic and acute) |
| Hypertension/tachycardia |
| Phlebitis |
| Pneumonia |
| Gingivitis/poor oral hygiene |
| Sexually transmitted diseases |
|
|
| Tetanus |
| Cystitis |
| Pyelonephritis |
| AIDS: acquired immune deficiency syndrome; HIV: human immunodeficiency virus |
| Source:Reference 6 |
Table 3 Obstetric complications in women with addiction disorders
| Placental abruption |
| Chorioamnionitis |
| Placental insufficiency |
| Intrauterine growth restriction |
| Hypoxic/ischemic brain injury |
| Meconium passage |
| Neonatal abstinence syndrome |
| Spontaneous abortion |
| Intrauterine fetal death |
| Premature labor and delivery |
| Preterm, premature rupture of membranes |
| Postpartum hemorrhage |
| Hypertensive emergencies/preeclampsia |
| Source:Reference 6 |
Opioid agonist therapy
Obstetric complications in women with OUD may be related to rapid, frequent fluctuations of opioid blood levels during intoxication and withdrawal. Therefore, the first goal of pharmacotherapy is to reduce physical stress associated with cycling opioid blood levels. Opioid agonist medications can be extremely effective. Opioid agonist treatment for pregnant patients is similar to that of nonpregnant patients but includes pregnancy-specific objectives (Table 4).20
Few anti-relapse medications have been studied in pregnant patients. Pharmacotherapies for OUD include methadone and buprenorphine. In our experience, opioid antagonists such as naltrexone typically would not be considered for pregnant patients because:
-
their expected efficacy in reducing relapse in pregnant patients is lower than that of other medications
-
their expected risk for inducing withdrawal is higher compared with methadone or buprenorphine
-
research on the use of naltrexone during pregnancy is lacking.
Methadone has been used to treat OUD during pregnancy since the late 1970s.5 It requires adherence to strict federal regulations and is FDA pregnancy class C (animal reproduction studies have shown an adverse effect on the fetus and there are no adequate well-controlled studies in humans, but potential benefits may warrant use in pregnant women despite potential risks). Pregnant women have been safely maintained on methadone without adverse long-term maternal or fetal effects, and the National Institutes of Health recommends it as the standard of care for pregnant women with OUD. A woman steadily maintained on methadone is more likely to have a healthy pregnancy and infant than a woman who uses alcohol or other drugs.21 Further, the structure and services of methadone maintenance treatment can improve compliance with prenatal care and help prepare patients for parental responsibilities.
Fluctuating blood opioid levels are minimized when methadone dosage is individually determined. Dosages should be based on a woman’s stage of pregnancy, relapse risk, pre-pregnancy methadone dose, experience with methadone, and clinical history. Some women experience lowered methadone blood levels during pregnancy because of increased fluid space, a larger tissue reservoir that can store methadone, and increased drug metabolism by both placenta and fetus. As a result, increased or split (twice daily) dosing may be indicated.22-24
Buprenorphine is FDA pregnancy class C. Although not approved for use during pregnancy, it has been used successfully for pregnant patients with OUD.12,25 It is a partial agonist of the mu opioid receptor and an antagonist of the kappa opioid receptor, which may reduce its abuse liability and NAS severity.
The few randomized clinical trials comparing methadone with buprenorphine during pregnancy suggest that buprenorphine is not inferior to methadone in safety and discomfort of induction from a short-acting opioid, nor in outcome measures assessing NAS and maternal and neonatal safety.26,27 Results from the recent Maternal Opioid Treatment: Human Experimental Research project suggest that buprenorphine may have some advantages over methadone in pregnancy. Buprenorphine-maintained neonates may need less morphine, have shorter hospital stays, and require shorter treatment for NAS.28 However, treatment retention may be lower for buprenorphine-maintained mothers; any resultant long-term consequences on maternal and child health are as yet unexplored. These findings require replication.
Methadone and buprenorphine are not interchangeable. Many patients maintained on methadone do not respond optimally to buprenorphine. Clinics that dispense maintenance methadone are required to provide counseling services and random drug testing; these requirements do not apply to physicians who prescribe buprenorphine. Moreover, in our experience buprenorphine at times has been prescribed without close regard to psychosocial issues, adequate random drug testing, or coordination of care with other providers.
In pregnant patients, buprenorphine is preferred over buprenorphine and naloxone to avoid fetal exposure to naloxone, which may cause intrauterine withdrawal and maternal-fetal hormonal changes. To reduce abuse or diversion, patients should undergo drug testing to ensure buprenorphine is present, smaller prescriptions may be provided, and tablets can be counted. Limited data suggests buprenorphine is not teratogenic. Some data show low placental transfer of buprenorphine, thereby limiting fetal exposure and lowering risk for intrauterine growth restriction.29
Table 4 Opioid agonist treatment objectives for addicted patients who are pregnant
| General objectives |
| Prevent opioid withdrawal signs and symptoms |
| Provide a comfortable induction onto the medication |
| Block the euphoric and reinforcing effects of illicit opioids while also attenuating the motivation (craving, social interactions) to use illicit opioids and other drugs |
| Enhance treatment retention |
| Create a more optimal environment for behavioral and psychosocial interventions |
| Pregnancy-specific objectives |
| Eliminate or reduce fetal exposure to illicit opioids and other illicit drugs |
| Stabilize the intrauterine environment |
| Enhance involvement in prenatal care |
| Create an optimal environment to address pregnancy-specific problems |
| Source:Reference 20 |
Delivery and postnatal care
Compared with those not in treatment, women who are engaged in a multidisciplinary treatment program at the time of delivery demonstrated higher gestational age, increased birth weights, and lower rates of neonatal ICU admissions. They also realized a cost savings of $4, 644 per mother-infant pair.30
During delivery, pain medication should not be withheld solely because a pregnant woman has a history of addiction-related disorders; these women are subject to pain during delivery as much as other women. Avoid using mixed agonists/antagonists such as nalbuphine or butorphanol in women receiving opioid maintenance medication. Labor and delivery pain management for a pregnant patient maintained on opioid agonist therapies is discussed elsewhere in the literature.31 Every effort should be made to ensure that the mother remains in treatment through delivery and beyond.
To read about advising women with OUD on the benefits and risks of breastfeeding while receiving opioid agonist maintenance treatment, see the Box below.
CASE CONTINUED: Medication change
Ms. J’s boyfriend has left her and her parents have not readily accepted her pregnancy and need for support. She continues to attend NA meetings and weekly therapy. After educating her about the differences between buprenorphine and buprenorphine and naloxone in relation to risk, benefits, and side effects, you switch Ms. J to buprenorphine, 12 mg/d, while maintaining her on aripiprazole and citalopram. She consents to exchanging information about her medical, mental health, and addiction-related treatment with her primary care provider, who helps locate an obstetrician/gynecologist comfortable with her OUD and buprenorphine. Ms. J’s therapist helps link her with social services agencies to ensure prenatal care, assist with removing barriers to care, and plan for her needs as a parent.
After checking your state’s mandates, you determine you are not required to report Ms. J’s drug testing results. Ms. J’s ongoing drug testing shows the presence of buprenorphine and the absence of other opioids and all drugs of abuse.
Ms. J’s delivery is uncomplicated medically; however, family, financial, and parental role issues remain problematic. Encouraging her involvement in therapy and social services as part of her continued buprenorphine prescribing proves beneficial.
Related Resources
-
Jones HE, Martin PR, Heil SH, et al. Treatment of opioid dependent pregnant women: clinical and research issues. J Subst Abuse Treat. 2008; 35(3): 245-259.
-
Johnson RE, Jones HE, Fischer G. Use of buprenorphine in pregnancy: patient management and effects on the neonate. Drug Alcohol Depend. 2003; 70(suppl 1 ): S87-S101.
-
Velez M, Jansson LM. The opioid dependent mother and the newborn dyad: nonpharmacologic care. J Addict Med. 2008; 2(3): 113-120.
Drug Brand Names
Aripiprazole • Abilify
Buprenorphine and naloxone •Suboxone
Buprenorphine • Subutex
Butorphanol • Stadol
Citalopram • Celexa
Fentanyl • Duragesic, Sublimaze, others
Methadone • Dolophine
Naloxone • Narcan
Naltrexone • ReVia
Nalbuphine • Nubain
Oxycodone • Oxycontin
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Fernandez’ time toward this project was funded by the University Hospital/University of Cincinnati Addiction Psychiatry Fellowship Training Program operated by the Center for Treatment, Research, and Education in Addictive Disorders (CeTREAD), Department of Psychiatry and Behavioral Neuroscience, University of Cincinnati and by the Veterans Affairs Medical Center, Cincinnati, OH.
The statements in this publication do not necessarily reflect the views or opinions of the Department of Veterans Affairs, the United States Government, or Opiate Addiction Recovery Services.
How proposed changes to personality disorders in DSM-5 will affect researchers
‘Scared’ and short of breath
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Paranoid and scared
Police bring Mr. C, age 42, to a local crisis center after he is found masturbating in public the same day he was released from jail after serving time for the same behavior. Previously, Mr. C was diagnosed with schizophrenia, paranoid type, and alcohol dependence. He is single, unemployed, and lives with his parents. He has had 3 previous admissions to a psychiatric hospital, but no preexisting medical illness. A judge involuntarily commits Mr. C to our psychiatric facility.
Mr. C looks older than his age and has poor hygiene. He appears bizarre, makes poor eye contact, and speaks slowly but with normal volume. His speech is not coherent, relevant, or goal-directed. He is not able to answer questions properly, chanting “it’s eternity, eternity, eternity.” He shows no tremors, repetitive motor behavior, or muscle rigidity. His affect is flat and he has no suicidal or homicidal ideations. Based on Mr. C’s history, we diagnose him with schizophrenia, paranoid type and alcohol dependence.
Over the next 9 days, Mr. C receives trials of haloperidol, lorazepam, diphenhydramine, ziprasidone, olanzapine, hydroxyzine, trazodone, and benztropine to treat his schizophrenia. From days 1 to 3, all medications are given on an as-needed basis. On day 1, Mr. C receives haloperidol, 20 mg, lorazepam, 9 mg, diphenhydramine, 150 mg, and ziprasidone, 20 mg. On day 2, he receives haloperidol, 15 mg, lorazepam, 10 mg, olanzapine, 20 mg, hydroxyzine, 100 mg, and trazodone, 50 mg. On day 3, he receives haloperidol, 20 mg, lorazepam, 6 mg, and trazodone, 100 mg. On days 4 to 8, in addition to scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d, he receives haloperidol, 5 mg, and lorazepam, 2 mg, as needed. On day 9, he receives the scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d.
During his stay, Mr. C is incoherent and disorganized. On day 9, he eats all of his lunch, none of his dinner, but sips milk and juice and eats snacks. He drinks 2 small cups of water with medication and 2 small cups of water during oral care. His mucosa and tongue are dry. At 11:30 pm, while lying in bed mumbling “scared, scared,” he experiences shortness of breath. His temperature is 99.6°F, blood pressure is 151/93 mm Hg, pulse is 125 beats per minute, respiratory rate is 40 breaths per minute, and oxygen saturation is 91% on ambient air. Twenty minutes later, his blood pressure increases to 180/120 mm Hg. On physical examination, he has “lead pipe” rigidity of both arms. He is awake, confused, and not able to communicate, still mumbling “scared, scared.” Changes in his blood pressure, pulse, and temperature during his stay in the psychiatric hospital are depicted in Figures 1 and 2, respectively.
Figure 1: Mr. C’s blood pressure and pulse changes from day 4 to day 9 in the psychiatric hospital
BP: blood pressure
Figure 2: Mr. C’s temperature changes from day 4 to day 9 in the psychiatric hospital
The authors’ observations
NMS is a life-threatening, iatrogenic neurologic emergency associated with antipsychotic use. Early incidence rate estimates ran as high as 3% of patients treated with antipsychotics; however, more recent data suggest an incidence of 0.01% to 0.02%.1 This decrease in frequency likely reflects increased awareness of the disorder, more conservative prescribing patterns, and a shift to using atypical antipsychotics.2 In the mid 1980s and early 1990s the mortality rate was 25% to 30% if NMS was not promptly recognized and treated3; however, progression to more fulminant, lethal NMS episodes now occurs less often and the mortality rate ranges from 10% to 20%.4
If NMS is suspected, immediate transfer to an emergency department (ED) is necessary. Even with early diagnosis, however, complications of NMS are still likely, including:
- rhabdomyolysis
- renal failure
- seizures
- respiratory failure
- aspiration pneumonia
- disseminated intravascular coagulation
- venous thromboembolism.5-9
Caroff et al reported observing a residual catatonic state after acute NMS symptoms subsided.10
Although the pathophysiology of NMS is complex—involving a cascade of dysregulation in multiple neurochemical and neuroendocrine systems—dopamine blockade likely plays a pivotal role in triggering the condition.2 In addition, evidence supports the hypothesis that dysregulated sympathetic nervous system hyperactivity is responsible for most NMS features.11
TREATMENT: Arrival in the ED
Based on his elevated blood pressure (151/93 mm Hg), “lead pipe” rigidity, and increased body temperature associated with Mr. C’s history of haloperidol use for 9 days, the treatment team suspects NMS. Labile blood pressure, which changed from 151/93 to 180/120 mm Hg in 20 minutes, reinforces the NMS diagnosis. Approximately 30 minutes after Mr. C shows signs of NMS, he is transferred to a local ED. He is awake, alert, and communicative after he arrives in the ED, but becomes confused and noncommunicative the next morning. When he arrives in the ED, he is found to have tachycardia (114 beats per minute), tachypnea (26 breaths per minute), blood pressure of 132/84 mm Hg, and temperature of 102°F. In the ED, he is given IV normal saline, diphenhydramine, 25 mg, and IV lorazepam, 1 mg. His rigidity slightly improves.
Early the next morning, his blood pressure is 182/89 mm Hg, respirations are 30 to 40 breaths per minute, and heart rate is 120 beats per minute. He then receives IV lorazepam, 2 mg, after which his tachypnea, tachycardia, and elevated blood pressure improve.
The authors’ observations
A case-control study by Keck et al12 comparing 18 patients with NMS and 36 matched neuroleptic-treated patients with no history of the syndrome identified greater psychomotor agitation, significantly higher doses of neuroleptics, greater rates of dosage increase, and a greater number of IM injections as potential risk factors. Other potential risk factors include use of restraints, pre-existing CNS dopamine activity or receptor function abnormalities, and iron deficiency.2 Agitation, dehydration, and exhaustion were found to be the most consistent systemic factors predisposing patients taking antipsychotics to NMS in small case-control studies.13,14 Well-supported risk factors also include use of high-potency antipsychotics, prior episodes of NMS, age <40, male sex, malnutrition, organic brain syndromes, and lithium use.3,5,15
There is no way to predict the risk of NMS for an individual patient. Usually, symptoms develop within 4 weeks of starting an antipsychotic, but can occur after taking the same dose for many months. The onset may be within hours, but on average it is 4 to 14 days after initiating therapy. Among patients who develop NMS, 90% do so within 10 days.3,5
Mr. C’s risk factors include high-potency antipsychotic use, male sex, relatively high dose (haloperidol, 30 to 35 mg/d), agitation, dehydration, and exhaustion.
Managing NMS
The standard approaches for managing patients with NMS include discontinuing suspected triggering drugs and providing supportive care. Beyond supportive care, oral or IV benzodiazepines may relieve symptoms and speed recovery.2 Dopaminergic drugs, such as bromocriptine or amantadine, used alone or with other treatments, can reduce parkinsonism and disease duration and mortality.2 Dantrolene may be useful only for NMS patients who exhibit extreme temperature elevations, rigidity, and true hypermetabolism.16 Electroconvulsive therapy may be effective for NMS patients whose symptoms do not respond to supportive care and drug therapy or those with residual catatonic or parkinsonian symptoms.2
OUTCOME: Improvement, discharge
Mr. C is admitted to the hospital with the diagnosis of NMS and transferred to the intensive care unit (ICU) for treatment. After Mr. C is admitted to the ICU, apart from continuing the medication given in the ED, he also receives dantrolene, 2 mg/kg, then 1 mg/kg, 4 times a day, as well as IV lorazepam, 1 mg every 6 hours. His other medications include IV pantoprazole, 40 mg/d, for prophylaxis of stress ulcer. Diphenhydramine administration is changed to as needed. On the second day in the ICU, he has only mild upper extremity rigidity but no lower extremity rigidity. However, he suffers 1 seizure, which is treated with IV fosphenytoin at the loading dose, 18 mg/kg, then a maintaining dose of 5 mg phenytoin equivalent/kg/d.
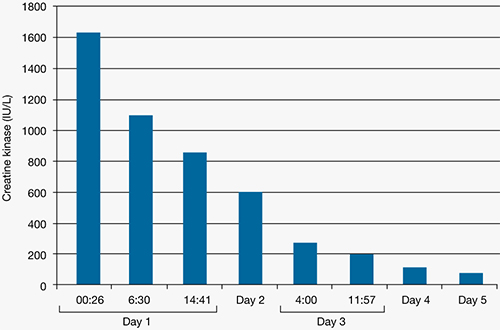
Figure 3: Mr. C’s creatine kinase level (IU/L) during the first 5 days in the intensive care unit
Figure 4: Mr. C’s blood pressure before and after admission
Figure 5: Mr. C’s temperature before and after admissionMr. C remains in the ICU for 7 days. There he receives valproic acid, titrated to 500 mg in the morning and 1,000 mg at bedtime, for agitation. He also receives olanzapine, 5 mg/d, for psychotic symptoms. He develops deep vein thrombosis in the right cephalic vein, which is treated with subcutaneous enoxaparin, 1 mg/kg, and warfarin, 5 mg/d.
He is discharged from the hospital after 2 weeks and returns to the psychiatric facility. He continues to be treated for paranoid schizophrenia with olanzapine, 5 mg/d.
The authors’ observations
High-potency, typical antipsychotics can cause NMS, as shown in Mr. C’s case. It also can be caused by typical low-potency antipsychotics,3 atypical antipsychotics,17 antiemetic drugs,18 and lithium,19,20 and can occur after the withdrawal of levodopa and similar dopaminergic agents during Parkinson’s disease treatment.21 Atypical antipsychotics reported to be associated with NMS include clozapine, risperidone, olanzapine, quetiapine, aripiprazole, ziprasidone, and paliperidone.22-27 Atypical antipsychotic-induced NMS also has been reported in children and adolescents.22,28-30
With the broad application of atypical antipsychotics, physicians should be aware of atypical NMS presentation. Although NMS diagnosis commonly requires core symptoms of hyperthermia and muscle rigidity (Table 1 and 2),31 atypical presentations may not demonstrate temperature changes and/or muscle rigidity or may progress slowly over several days, leading to a delay in diagnosis and treatment.28,30,32,33 Therefore, clinicians should evaluate any patient taking antipsychotics for features of NMS and not prematurely exclude a NMS diagnosis in cases where severe rigidity or hyperthermia is not initially apparent.33
Table 1
DSM-IV-TR criteria for neuroleptic malignant syndrome
| A. The development of severe muscle rigidity and elevated temperature associated with the use of neuroleptic medication |
| B. 2 (or more) of the following: |
|
| Source: Reference 31 |
Table 2
Diagnostic features of neuroleptic malignant syndrome
| Essential features: severe muscle rigidity and elevated temperature in an individual using neuroleptic medication |
| Elevated temperature: from mild (eg, 99º to 100ºF) to markedly hyperthermic states (eg, 106ºF) |
| Creatine kinase: typically elevated, ranging from minor elevations to extremely high levels (exceeding 16,000 IU) |
| Other features: mental status changes, unstable blood pressure, diaphoresis, other signs of autonomic dysfunction |
| Source: Reference 31 |
Related Resource
- Neuroleptic Malignant Syndrome Information Service. www.nmsis.org.
Drug Brand Names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Benztropine • Cogentin
- Bromocriptine • Parlodel
- Clozapine • Clozaril
- Dantrolene • Dantrium
- Diphenhydramine • Benadryl
- Enoxaparin • Lovenox
- Fosphenytoin • Cerebyx
- Haloperidol • Haldol
- Hydroxyzine • Vistaril
- Levodopa • Sinemet
- Lithium • Eskalith, Lithobid, others
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Paliperidone • Invega
- Pantoprazole • Protonix
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Trazodone • Desyrel, Oleptro
- Valproic acid • Depakote
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgements
The authors are very grateful for the critical reviews by James R. Allen, MD, MPH, professor of Child and Adolescent Psychiatry Fellowship Program at the University of Oklahoma and Lori Hake, DO, director of Psychiatry Residency Training Program at Griffin Memorial Hospital in Norman, OK.
1. Stubner S, Rustenbeck E, Grohmann R, et al. Severe and uncommon involuntary movement disorders due to psychotropic drugs. Pharmacopsychiatry. 2004;37:S54-S64.
2. Strawn JR, Keck PE, Jr, Caroff SN. Neuroleptic malignant syndrome. Am J Psychiatry. 2007;164:870-876.
3. Ropper AH, Brown RH. Adams and Victor’s principles of neurology. 8th ed. New York, NY: McGraw Hill; 2005;1025-1026.
4. Sheil AT, Collins KA, Schandl CA, et al. Fetal neurotoxic response to neuroleptic medications: case report and review of the literature. Am J Forensic Med Pathol. 2007;28:116-120.
5. Balzan MV. The neuroleptic malignant syndrome: a logical approach to the patient with temperature and rigidity. Postgrad Med J. 1998;74:72-76.
6. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77:185-202.
7. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the critical care unit. Crit Care Med. 2002;30:2609-2610.
8. Caroff SN, Campbell EC, Sullivan KA. Neuroleptic malignant syndrome in elderly patients. Expert Rev Neurother. 2007;7:423-431.
9. Gurrera RJ, Simpson JC, Tsuang MT. Meta-analytic evidence of systematic bias in estimates of neuroleptic malignant syndrome incidence. Compr Psychiatry. 2007;48:205-211.
10. Caroff SN, Mann SC, Keck PE, Jr, et al. Residual catatonic state following neuroleptic malignant syndrome. J Clin Psychopharmacol. 2001;21:121-122.
11. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
12. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome. A case-control study. Arch Gen Psychiatry. 1989;46:914-918.
13. Berardi D, Amore M, Keck PE, Jr, et al. Clinical and pharmacologic risk factors for neuroleptic malignant syndrome: a case-control study. Biol Psychiatry. 1998;44:748-754.
14. Rosebush PI, Stewart TD. A prospective analysis of 24 episodes of neuroleptic malignant syndrome. Am J Psychiatry. 1989;146:717-725.
15. Martinez M, Marangell LB, Martinez JM. Psychopharmacology. In: Hales RE, Yudofsky SC, Gabbard GO, eds. American Psychiatric Publishing textbook of psychiatry. Arlington, VA: American Psychiatric Publishing, Inc.; 2008:1059-1132.
16. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, et al, eds. Neuroleptic malignant syndrome and related conditions. Washington, DC: American Psychiatric Publishing; 2003:1-44.
17. Hammerman S, Lam C, Caroff SN. Neuroleptic malignant syndrome and aripiprazole. J Am Acad Child Adolesc Psychiatry. 2006;45:639-641.
18. Stein MH, Sorscher M, Caroff SN. Neuroleptic malignant syndrome induced by metoclopramide in an infant with Freeman-Sheldon syndrome. Anesth Analg. 2006;103:786-787.
19. Borovicka MC, Bond LC, Gaughan KM. Ziprasidone- and lithium-induced neuroleptic malignant syndrome. Ann Pharmacother. 2006;40:139-142.
20. Gill J, Singh H, Nugent K. Acute lithium intoxication and neuroleptic malignant syndrome. Pharmacotherapy. 2003;23:811-815.
21. Ward C. Neuroleptic malignant syndrome in a patient with Parkinson’s disease: a case study. J Neurosci Nurs. 2005;37:160-162.
22. Leibold J, Patel V, Hasan RA. Neuroleptic malignant syndrome associated with ziprasidone in an adolescent. Clin Ther. 2004;26:1105-1108.
23. Corallo CE, Ernest D. Atypical neuroleptic malignant syndrome with long-term clozapine. Crit Care Resusc. 2007;9:338-340.
24. Molina D, Tingle LE, Lu X. Aripiprazole as the causative agent of neuroleptic malignant syndrome: a case report. Prim Care Companion J Clin Psychiatry. 2007;9:148-150.
25. Trollor JN, Chen X, Sachdev PS. Neuroleptic malignant syndrome associated with atypical antipsychotic drugs. CNS Drugs. 2009;23:477-492.
26. Gortney JS, Fagan A, Kissack JC. Neuroleptic malignant syndrome secondary to quetiapine. Ann Pharmacother. 2009;43:785-791.
27. Han C, Lee SJ, Pae CU. Paliperidone-associated atypical neuroleptic malignant syndrome: a case report. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:650-651.
28. Hanft A, Eggleston CF, Bourgeois JA. Neuroleptic malignant syndrome in an adolescent after brief exposure to olanzapine. J Child Adolesc Psychopharmacol. 2004;14:481-487.
29. Abu-Kishk I, Toledano M, Reis A, et al. Neuroleptic malignant syndrome in a child treated with an atypical antipsychotic. J Toxicol Clin Toxicol. 2004;42:921-925.
30. Neuhut R, Lindenmayer JP, Silva R. Neuroleptic malignant syndrome in children and adolescents on atypical antipsychotic medication: a review. J Child Adolesc Psychopharmacol. 2009;19:415-422.
31. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association: 2000.
32. Carroll BT, Surber SA. The problem of atypical neuroleptic malignant syndrome: a case report. Psychiatry (Edgmont). 2009;6:45-47.
33. Picard LS, Lindsay S, Strawn JR, et al. Atypical neuroleptic malignant syndrome: diagnostic controversies and considerations. Pharmacotherapy. 2008;28:530-535.
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Paranoid and scared
Police bring Mr. C, age 42, to a local crisis center after he is found masturbating in public the same day he was released from jail after serving time for the same behavior. Previously, Mr. C was diagnosed with schizophrenia, paranoid type, and alcohol dependence. He is single, unemployed, and lives with his parents. He has had 3 previous admissions to a psychiatric hospital, but no preexisting medical illness. A judge involuntarily commits Mr. C to our psychiatric facility.
Mr. C looks older than his age and has poor hygiene. He appears bizarre, makes poor eye contact, and speaks slowly but with normal volume. His speech is not coherent, relevant, or goal-directed. He is not able to answer questions properly, chanting “it’s eternity, eternity, eternity.” He shows no tremors, repetitive motor behavior, or muscle rigidity. His affect is flat and he has no suicidal or homicidal ideations. Based on Mr. C’s history, we diagnose him with schizophrenia, paranoid type and alcohol dependence.
Over the next 9 days, Mr. C receives trials of haloperidol, lorazepam, diphenhydramine, ziprasidone, olanzapine, hydroxyzine, trazodone, and benztropine to treat his schizophrenia. From days 1 to 3, all medications are given on an as-needed basis. On day 1, Mr. C receives haloperidol, 20 mg, lorazepam, 9 mg, diphenhydramine, 150 mg, and ziprasidone, 20 mg. On day 2, he receives haloperidol, 15 mg, lorazepam, 10 mg, olanzapine, 20 mg, hydroxyzine, 100 mg, and trazodone, 50 mg. On day 3, he receives haloperidol, 20 mg, lorazepam, 6 mg, and trazodone, 100 mg. On days 4 to 8, in addition to scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d, he receives haloperidol, 5 mg, and lorazepam, 2 mg, as needed. On day 9, he receives the scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d.
During his stay, Mr. C is incoherent and disorganized. On day 9, he eats all of his lunch, none of his dinner, but sips milk and juice and eats snacks. He drinks 2 small cups of water with medication and 2 small cups of water during oral care. His mucosa and tongue are dry. At 11:30 pm, while lying in bed mumbling “scared, scared,” he experiences shortness of breath. His temperature is 99.6°F, blood pressure is 151/93 mm Hg, pulse is 125 beats per minute, respiratory rate is 40 breaths per minute, and oxygen saturation is 91% on ambient air. Twenty minutes later, his blood pressure increases to 180/120 mm Hg. On physical examination, he has “lead pipe” rigidity of both arms. He is awake, confused, and not able to communicate, still mumbling “scared, scared.” Changes in his blood pressure, pulse, and temperature during his stay in the psychiatric hospital are depicted in Figures 1 and 2, respectively.
Figure 1: Mr. C’s blood pressure and pulse changes from day 4 to day 9 in the psychiatric hospital
BP: blood pressure
Figure 2: Mr. C’s temperature changes from day 4 to day 9 in the psychiatric hospital
The authors’ observations
NMS is a life-threatening, iatrogenic neurologic emergency associated with antipsychotic use. Early incidence rate estimates ran as high as 3% of patients treated with antipsychotics; however, more recent data suggest an incidence of 0.01% to 0.02%.1 This decrease in frequency likely reflects increased awareness of the disorder, more conservative prescribing patterns, and a shift to using atypical antipsychotics.2 In the mid 1980s and early 1990s the mortality rate was 25% to 30% if NMS was not promptly recognized and treated3; however, progression to more fulminant, lethal NMS episodes now occurs less often and the mortality rate ranges from 10% to 20%.4
If NMS is suspected, immediate transfer to an emergency department (ED) is necessary. Even with early diagnosis, however, complications of NMS are still likely, including:
- rhabdomyolysis
- renal failure
- seizures
- respiratory failure
- aspiration pneumonia
- disseminated intravascular coagulation
- venous thromboembolism.5-9
Caroff et al reported observing a residual catatonic state after acute NMS symptoms subsided.10
Although the pathophysiology of NMS is complex—involving a cascade of dysregulation in multiple neurochemical and neuroendocrine systems—dopamine blockade likely plays a pivotal role in triggering the condition.2 In addition, evidence supports the hypothesis that dysregulated sympathetic nervous system hyperactivity is responsible for most NMS features.11
TREATMENT: Arrival in the ED
Based on his elevated blood pressure (151/93 mm Hg), “lead pipe” rigidity, and increased body temperature associated with Mr. C’s history of haloperidol use for 9 days, the treatment team suspects NMS. Labile blood pressure, which changed from 151/93 to 180/120 mm Hg in 20 minutes, reinforces the NMS diagnosis. Approximately 30 minutes after Mr. C shows signs of NMS, he is transferred to a local ED. He is awake, alert, and communicative after he arrives in the ED, but becomes confused and noncommunicative the next morning. When he arrives in the ED, he is found to have tachycardia (114 beats per minute), tachypnea (26 breaths per minute), blood pressure of 132/84 mm Hg, and temperature of 102°F. In the ED, he is given IV normal saline, diphenhydramine, 25 mg, and IV lorazepam, 1 mg. His rigidity slightly improves.
Early the next morning, his blood pressure is 182/89 mm Hg, respirations are 30 to 40 breaths per minute, and heart rate is 120 beats per minute. He then receives IV lorazepam, 2 mg, after which his tachypnea, tachycardia, and elevated blood pressure improve.
The authors’ observations
A case-control study by Keck et al12 comparing 18 patients with NMS and 36 matched neuroleptic-treated patients with no history of the syndrome identified greater psychomotor agitation, significantly higher doses of neuroleptics, greater rates of dosage increase, and a greater number of IM injections as potential risk factors. Other potential risk factors include use of restraints, pre-existing CNS dopamine activity or receptor function abnormalities, and iron deficiency.2 Agitation, dehydration, and exhaustion were found to be the most consistent systemic factors predisposing patients taking antipsychotics to NMS in small case-control studies.13,14 Well-supported risk factors also include use of high-potency antipsychotics, prior episodes of NMS, age <40, male sex, malnutrition, organic brain syndromes, and lithium use.3,5,15
There is no way to predict the risk of NMS for an individual patient. Usually, symptoms develop within 4 weeks of starting an antipsychotic, but can occur after taking the same dose for many months. The onset may be within hours, but on average it is 4 to 14 days after initiating therapy. Among patients who develop NMS, 90% do so within 10 days.3,5
Mr. C’s risk factors include high-potency antipsychotic use, male sex, relatively high dose (haloperidol, 30 to 35 mg/d), agitation, dehydration, and exhaustion.
Managing NMS
The standard approaches for managing patients with NMS include discontinuing suspected triggering drugs and providing supportive care. Beyond supportive care, oral or IV benzodiazepines may relieve symptoms and speed recovery.2 Dopaminergic drugs, such as bromocriptine or amantadine, used alone or with other treatments, can reduce parkinsonism and disease duration and mortality.2 Dantrolene may be useful only for NMS patients who exhibit extreme temperature elevations, rigidity, and true hypermetabolism.16 Electroconvulsive therapy may be effective for NMS patients whose symptoms do not respond to supportive care and drug therapy or those with residual catatonic or parkinsonian symptoms.2
OUTCOME: Improvement, discharge
Mr. C is admitted to the hospital with the diagnosis of NMS and transferred to the intensive care unit (ICU) for treatment. After Mr. C is admitted to the ICU, apart from continuing the medication given in the ED, he also receives dantrolene, 2 mg/kg, then 1 mg/kg, 4 times a day, as well as IV lorazepam, 1 mg every 6 hours. His other medications include IV pantoprazole, 40 mg/d, for prophylaxis of stress ulcer. Diphenhydramine administration is changed to as needed. On the second day in the ICU, he has only mild upper extremity rigidity but no lower extremity rigidity. However, he suffers 1 seizure, which is treated with IV fosphenytoin at the loading dose, 18 mg/kg, then a maintaining dose of 5 mg phenytoin equivalent/kg/d.
Figure 3: Mr. C’s creatine kinase level (IU/L) during the first 5 days in the intensive care unit
Figure 4: Mr. C’s blood pressure before and after admission
Figure 5: Mr. C’s temperature before and after admissionMr. C remains in the ICU for 7 days. There he receives valproic acid, titrated to 500 mg in the morning and 1,000 mg at bedtime, for agitation. He also receives olanzapine, 5 mg/d, for psychotic symptoms. He develops deep vein thrombosis in the right cephalic vein, which is treated with subcutaneous enoxaparin, 1 mg/kg, and warfarin, 5 mg/d.
He is discharged from the hospital after 2 weeks and returns to the psychiatric facility. He continues to be treated for paranoid schizophrenia with olanzapine, 5 mg/d.
The authors’ observations
High-potency, typical antipsychotics can cause NMS, as shown in Mr. C’s case. It also can be caused by typical low-potency antipsychotics,3 atypical antipsychotics,17 antiemetic drugs,18 and lithium,19,20 and can occur after the withdrawal of levodopa and similar dopaminergic agents during Parkinson’s disease treatment.21 Atypical antipsychotics reported to be associated with NMS include clozapine, risperidone, olanzapine, quetiapine, aripiprazole, ziprasidone, and paliperidone.22-27 Atypical antipsychotic-induced NMS also has been reported in children and adolescents.22,28-30
With the broad application of atypical antipsychotics, physicians should be aware of atypical NMS presentation. Although NMS diagnosis commonly requires core symptoms of hyperthermia and muscle rigidity (Table 1 and 2),31 atypical presentations may not demonstrate temperature changes and/or muscle rigidity or may progress slowly over several days, leading to a delay in diagnosis and treatment.28,30,32,33 Therefore, clinicians should evaluate any patient taking antipsychotics for features of NMS and not prematurely exclude a NMS diagnosis in cases where severe rigidity or hyperthermia is not initially apparent.33
Table 1
DSM-IV-TR criteria for neuroleptic malignant syndrome
| A. The development of severe muscle rigidity and elevated temperature associated with the use of neuroleptic medication |
| B. 2 (or more) of the following: |
|
| Source: Reference 31 |
Table 2
Diagnostic features of neuroleptic malignant syndrome
| Essential features: severe muscle rigidity and elevated temperature in an individual using neuroleptic medication |
| Elevated temperature: from mild (eg, 99º to 100ºF) to markedly hyperthermic states (eg, 106ºF) |
| Creatine kinase: typically elevated, ranging from minor elevations to extremely high levels (exceeding 16,000 IU) |
| Other features: mental status changes, unstable blood pressure, diaphoresis, other signs of autonomic dysfunction |
| Source: Reference 31 |
Related Resource
- Neuroleptic Malignant Syndrome Information Service. www.nmsis.org.
Drug Brand Names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Benztropine • Cogentin
- Bromocriptine • Parlodel
- Clozapine • Clozaril
- Dantrolene • Dantrium
- Diphenhydramine • Benadryl
- Enoxaparin • Lovenox
- Fosphenytoin • Cerebyx
- Haloperidol • Haldol
- Hydroxyzine • Vistaril
- Levodopa • Sinemet
- Lithium • Eskalith, Lithobid, others
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Paliperidone • Invega
- Pantoprazole • Protonix
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Trazodone • Desyrel, Oleptro
- Valproic acid • Depakote
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgements
The authors are very grateful for the critical reviews by James R. Allen, MD, MPH, professor of Child and Adolescent Psychiatry Fellowship Program at the University of Oklahoma and Lori Hake, DO, director of Psychiatry Residency Training Program at Griffin Memorial Hospital in Norman, OK.
Discuss this article at www.facebook.com/CurrentPsychiatry
CASE: Paranoid and scared
Police bring Mr. C, age 42, to a local crisis center after he is found masturbating in public the same day he was released from jail after serving time for the same behavior. Previously, Mr. C was diagnosed with schizophrenia, paranoid type, and alcohol dependence. He is single, unemployed, and lives with his parents. He has had 3 previous admissions to a psychiatric hospital, but no preexisting medical illness. A judge involuntarily commits Mr. C to our psychiatric facility.
Mr. C looks older than his age and has poor hygiene. He appears bizarre, makes poor eye contact, and speaks slowly but with normal volume. His speech is not coherent, relevant, or goal-directed. He is not able to answer questions properly, chanting “it’s eternity, eternity, eternity.” He shows no tremors, repetitive motor behavior, or muscle rigidity. His affect is flat and he has no suicidal or homicidal ideations. Based on Mr. C’s history, we diagnose him with schizophrenia, paranoid type and alcohol dependence.
Over the next 9 days, Mr. C receives trials of haloperidol, lorazepam, diphenhydramine, ziprasidone, olanzapine, hydroxyzine, trazodone, and benztropine to treat his schizophrenia. From days 1 to 3, all medications are given on an as-needed basis. On day 1, Mr. C receives haloperidol, 20 mg, lorazepam, 9 mg, diphenhydramine, 150 mg, and ziprasidone, 20 mg. On day 2, he receives haloperidol, 15 mg, lorazepam, 10 mg, olanzapine, 20 mg, hydroxyzine, 100 mg, and trazodone, 50 mg. On day 3, he receives haloperidol, 20 mg, lorazepam, 6 mg, and trazodone, 100 mg. On days 4 to 8, in addition to scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d, he receives haloperidol, 5 mg, and lorazepam, 2 mg, as needed. On day 9, he receives the scheduled haloperidol, 30 mg/d, benztropine, 1 mg/d, and trazodone, 100 mg/d.
During his stay, Mr. C is incoherent and disorganized. On day 9, he eats all of his lunch, none of his dinner, but sips milk and juice and eats snacks. He drinks 2 small cups of water with medication and 2 small cups of water during oral care. His mucosa and tongue are dry. At 11:30 pm, while lying in bed mumbling “scared, scared,” he experiences shortness of breath. His temperature is 99.6°F, blood pressure is 151/93 mm Hg, pulse is 125 beats per minute, respiratory rate is 40 breaths per minute, and oxygen saturation is 91% on ambient air. Twenty minutes later, his blood pressure increases to 180/120 mm Hg. On physical examination, he has “lead pipe” rigidity of both arms. He is awake, confused, and not able to communicate, still mumbling “scared, scared.” Changes in his blood pressure, pulse, and temperature during his stay in the psychiatric hospital are depicted in Figures 1 and 2, respectively.
Figure 1: Mr. C’s blood pressure and pulse changes from day 4 to day 9 in the psychiatric hospital
BP: blood pressure
Figure 2: Mr. C’s temperature changes from day 4 to day 9 in the psychiatric hospital
The authors’ observations
NMS is a life-threatening, iatrogenic neurologic emergency associated with antipsychotic use. Early incidence rate estimates ran as high as 3% of patients treated with antipsychotics; however, more recent data suggest an incidence of 0.01% to 0.02%.1 This decrease in frequency likely reflects increased awareness of the disorder, more conservative prescribing patterns, and a shift to using atypical antipsychotics.2 In the mid 1980s and early 1990s the mortality rate was 25% to 30% if NMS was not promptly recognized and treated3; however, progression to more fulminant, lethal NMS episodes now occurs less often and the mortality rate ranges from 10% to 20%.4
If NMS is suspected, immediate transfer to an emergency department (ED) is necessary. Even with early diagnosis, however, complications of NMS are still likely, including:
- rhabdomyolysis
- renal failure
- seizures
- respiratory failure
- aspiration pneumonia
- disseminated intravascular coagulation
- venous thromboembolism.5-9
Caroff et al reported observing a residual catatonic state after acute NMS symptoms subsided.10
Although the pathophysiology of NMS is complex—involving a cascade of dysregulation in multiple neurochemical and neuroendocrine systems—dopamine blockade likely plays a pivotal role in triggering the condition.2 In addition, evidence supports the hypothesis that dysregulated sympathetic nervous system hyperactivity is responsible for most NMS features.11
TREATMENT: Arrival in the ED
Based on his elevated blood pressure (151/93 mm Hg), “lead pipe” rigidity, and increased body temperature associated with Mr. C’s history of haloperidol use for 9 days, the treatment team suspects NMS. Labile blood pressure, which changed from 151/93 to 180/120 mm Hg in 20 minutes, reinforces the NMS diagnosis. Approximately 30 minutes after Mr. C shows signs of NMS, he is transferred to a local ED. He is awake, alert, and communicative after he arrives in the ED, but becomes confused and noncommunicative the next morning. When he arrives in the ED, he is found to have tachycardia (114 beats per minute), tachypnea (26 breaths per minute), blood pressure of 132/84 mm Hg, and temperature of 102°F. In the ED, he is given IV normal saline, diphenhydramine, 25 mg, and IV lorazepam, 1 mg. His rigidity slightly improves.
Early the next morning, his blood pressure is 182/89 mm Hg, respirations are 30 to 40 breaths per minute, and heart rate is 120 beats per minute. He then receives IV lorazepam, 2 mg, after which his tachypnea, tachycardia, and elevated blood pressure improve.
The authors’ observations
A case-control study by Keck et al12 comparing 18 patients with NMS and 36 matched neuroleptic-treated patients with no history of the syndrome identified greater psychomotor agitation, significantly higher doses of neuroleptics, greater rates of dosage increase, and a greater number of IM injections as potential risk factors. Other potential risk factors include use of restraints, pre-existing CNS dopamine activity or receptor function abnormalities, and iron deficiency.2 Agitation, dehydration, and exhaustion were found to be the most consistent systemic factors predisposing patients taking antipsychotics to NMS in small case-control studies.13,14 Well-supported risk factors also include use of high-potency antipsychotics, prior episodes of NMS, age <40, male sex, malnutrition, organic brain syndromes, and lithium use.3,5,15
There is no way to predict the risk of NMS for an individual patient. Usually, symptoms develop within 4 weeks of starting an antipsychotic, but can occur after taking the same dose for many months. The onset may be within hours, but on average it is 4 to 14 days after initiating therapy. Among patients who develop NMS, 90% do so within 10 days.3,5
Mr. C’s risk factors include high-potency antipsychotic use, male sex, relatively high dose (haloperidol, 30 to 35 mg/d), agitation, dehydration, and exhaustion.
Managing NMS
The standard approaches for managing patients with NMS include discontinuing suspected triggering drugs and providing supportive care. Beyond supportive care, oral or IV benzodiazepines may relieve symptoms and speed recovery.2 Dopaminergic drugs, such as bromocriptine or amantadine, used alone or with other treatments, can reduce parkinsonism and disease duration and mortality.2 Dantrolene may be useful only for NMS patients who exhibit extreme temperature elevations, rigidity, and true hypermetabolism.16 Electroconvulsive therapy may be effective for NMS patients whose symptoms do not respond to supportive care and drug therapy or those with residual catatonic or parkinsonian symptoms.2
OUTCOME: Improvement, discharge
Mr. C is admitted to the hospital with the diagnosis of NMS and transferred to the intensive care unit (ICU) for treatment. After Mr. C is admitted to the ICU, apart from continuing the medication given in the ED, he also receives dantrolene, 2 mg/kg, then 1 mg/kg, 4 times a day, as well as IV lorazepam, 1 mg every 6 hours. His other medications include IV pantoprazole, 40 mg/d, for prophylaxis of stress ulcer. Diphenhydramine administration is changed to as needed. On the second day in the ICU, he has only mild upper extremity rigidity but no lower extremity rigidity. However, he suffers 1 seizure, which is treated with IV fosphenytoin at the loading dose, 18 mg/kg, then a maintaining dose of 5 mg phenytoin equivalent/kg/d.
Figure 3: Mr. C’s creatine kinase level (IU/L) during the first 5 days in the intensive care unit
Figure 4: Mr. C’s blood pressure before and after admission
Figure 5: Mr. C’s temperature before and after admissionMr. C remains in the ICU for 7 days. There he receives valproic acid, titrated to 500 mg in the morning and 1,000 mg at bedtime, for agitation. He also receives olanzapine, 5 mg/d, for psychotic symptoms. He develops deep vein thrombosis in the right cephalic vein, which is treated with subcutaneous enoxaparin, 1 mg/kg, and warfarin, 5 mg/d.
He is discharged from the hospital after 2 weeks and returns to the psychiatric facility. He continues to be treated for paranoid schizophrenia with olanzapine, 5 mg/d.
The authors’ observations
High-potency, typical antipsychotics can cause NMS, as shown in Mr. C’s case. It also can be caused by typical low-potency antipsychotics,3 atypical antipsychotics,17 antiemetic drugs,18 and lithium,19,20 and can occur after the withdrawal of levodopa and similar dopaminergic agents during Parkinson’s disease treatment.21 Atypical antipsychotics reported to be associated with NMS include clozapine, risperidone, olanzapine, quetiapine, aripiprazole, ziprasidone, and paliperidone.22-27 Atypical antipsychotic-induced NMS also has been reported in children and adolescents.22,28-30
With the broad application of atypical antipsychotics, physicians should be aware of atypical NMS presentation. Although NMS diagnosis commonly requires core symptoms of hyperthermia and muscle rigidity (Table 1 and 2),31 atypical presentations may not demonstrate temperature changes and/or muscle rigidity or may progress slowly over several days, leading to a delay in diagnosis and treatment.28,30,32,33 Therefore, clinicians should evaluate any patient taking antipsychotics for features of NMS and not prematurely exclude a NMS diagnosis in cases where severe rigidity or hyperthermia is not initially apparent.33
Table 1
DSM-IV-TR criteria for neuroleptic malignant syndrome
| A. The development of severe muscle rigidity and elevated temperature associated with the use of neuroleptic medication |
| B. 2 (or more) of the following: |
|
| Source: Reference 31 |
Table 2
Diagnostic features of neuroleptic malignant syndrome
| Essential features: severe muscle rigidity and elevated temperature in an individual using neuroleptic medication |
| Elevated temperature: from mild (eg, 99º to 100ºF) to markedly hyperthermic states (eg, 106ºF) |
| Creatine kinase: typically elevated, ranging from minor elevations to extremely high levels (exceeding 16,000 IU) |
| Other features: mental status changes, unstable blood pressure, diaphoresis, other signs of autonomic dysfunction |
| Source: Reference 31 |
Related Resource
- Neuroleptic Malignant Syndrome Information Service. www.nmsis.org.
Drug Brand Names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Benztropine • Cogentin
- Bromocriptine • Parlodel
- Clozapine • Clozaril
- Dantrolene • Dantrium
- Diphenhydramine • Benadryl
- Enoxaparin • Lovenox
- Fosphenytoin • Cerebyx
- Haloperidol • Haldol
- Hydroxyzine • Vistaril
- Levodopa • Sinemet
- Lithium • Eskalith, Lithobid, others
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Paliperidone • Invega
- Pantoprazole • Protonix
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Trazodone • Desyrel, Oleptro
- Valproic acid • Depakote
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgements
The authors are very grateful for the critical reviews by James R. Allen, MD, MPH, professor of Child and Adolescent Psychiatry Fellowship Program at the University of Oklahoma and Lori Hake, DO, director of Psychiatry Residency Training Program at Griffin Memorial Hospital in Norman, OK.
1. Stubner S, Rustenbeck E, Grohmann R, et al. Severe and uncommon involuntary movement disorders due to psychotropic drugs. Pharmacopsychiatry. 2004;37:S54-S64.
2. Strawn JR, Keck PE, Jr, Caroff SN. Neuroleptic malignant syndrome. Am J Psychiatry. 2007;164:870-876.
3. Ropper AH, Brown RH. Adams and Victor’s principles of neurology. 8th ed. New York, NY: McGraw Hill; 2005;1025-1026.
4. Sheil AT, Collins KA, Schandl CA, et al. Fetal neurotoxic response to neuroleptic medications: case report and review of the literature. Am J Forensic Med Pathol. 2007;28:116-120.
5. Balzan MV. The neuroleptic malignant syndrome: a logical approach to the patient with temperature and rigidity. Postgrad Med J. 1998;74:72-76.
6. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77:185-202.
7. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the critical care unit. Crit Care Med. 2002;30:2609-2610.
8. Caroff SN, Campbell EC, Sullivan KA. Neuroleptic malignant syndrome in elderly patients. Expert Rev Neurother. 2007;7:423-431.
9. Gurrera RJ, Simpson JC, Tsuang MT. Meta-analytic evidence of systematic bias in estimates of neuroleptic malignant syndrome incidence. Compr Psychiatry. 2007;48:205-211.
10. Caroff SN, Mann SC, Keck PE, Jr, et al. Residual catatonic state following neuroleptic malignant syndrome. J Clin Psychopharmacol. 2001;21:121-122.
11. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
12. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome. A case-control study. Arch Gen Psychiatry. 1989;46:914-918.
13. Berardi D, Amore M, Keck PE, Jr, et al. Clinical and pharmacologic risk factors for neuroleptic malignant syndrome: a case-control study. Biol Psychiatry. 1998;44:748-754.
14. Rosebush PI, Stewart TD. A prospective analysis of 24 episodes of neuroleptic malignant syndrome. Am J Psychiatry. 1989;146:717-725.
15. Martinez M, Marangell LB, Martinez JM. Psychopharmacology. In: Hales RE, Yudofsky SC, Gabbard GO, eds. American Psychiatric Publishing textbook of psychiatry. Arlington, VA: American Psychiatric Publishing, Inc.; 2008:1059-1132.
16. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, et al, eds. Neuroleptic malignant syndrome and related conditions. Washington, DC: American Psychiatric Publishing; 2003:1-44.
17. Hammerman S, Lam C, Caroff SN. Neuroleptic malignant syndrome and aripiprazole. J Am Acad Child Adolesc Psychiatry. 2006;45:639-641.
18. Stein MH, Sorscher M, Caroff SN. Neuroleptic malignant syndrome induced by metoclopramide in an infant with Freeman-Sheldon syndrome. Anesth Analg. 2006;103:786-787.
19. Borovicka MC, Bond LC, Gaughan KM. Ziprasidone- and lithium-induced neuroleptic malignant syndrome. Ann Pharmacother. 2006;40:139-142.
20. Gill J, Singh H, Nugent K. Acute lithium intoxication and neuroleptic malignant syndrome. Pharmacotherapy. 2003;23:811-815.
21. Ward C. Neuroleptic malignant syndrome in a patient with Parkinson’s disease: a case study. J Neurosci Nurs. 2005;37:160-162.
22. Leibold J, Patel V, Hasan RA. Neuroleptic malignant syndrome associated with ziprasidone in an adolescent. Clin Ther. 2004;26:1105-1108.
23. Corallo CE, Ernest D. Atypical neuroleptic malignant syndrome with long-term clozapine. Crit Care Resusc. 2007;9:338-340.
24. Molina D, Tingle LE, Lu X. Aripiprazole as the causative agent of neuroleptic malignant syndrome: a case report. Prim Care Companion J Clin Psychiatry. 2007;9:148-150.
25. Trollor JN, Chen X, Sachdev PS. Neuroleptic malignant syndrome associated with atypical antipsychotic drugs. CNS Drugs. 2009;23:477-492.
26. Gortney JS, Fagan A, Kissack JC. Neuroleptic malignant syndrome secondary to quetiapine. Ann Pharmacother. 2009;43:785-791.
27. Han C, Lee SJ, Pae CU. Paliperidone-associated atypical neuroleptic malignant syndrome: a case report. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:650-651.
28. Hanft A, Eggleston CF, Bourgeois JA. Neuroleptic malignant syndrome in an adolescent after brief exposure to olanzapine. J Child Adolesc Psychopharmacol. 2004;14:481-487.
29. Abu-Kishk I, Toledano M, Reis A, et al. Neuroleptic malignant syndrome in a child treated with an atypical antipsychotic. J Toxicol Clin Toxicol. 2004;42:921-925.
30. Neuhut R, Lindenmayer JP, Silva R. Neuroleptic malignant syndrome in children and adolescents on atypical antipsychotic medication: a review. J Child Adolesc Psychopharmacol. 2009;19:415-422.
31. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association: 2000.
32. Carroll BT, Surber SA. The problem of atypical neuroleptic malignant syndrome: a case report. Psychiatry (Edgmont). 2009;6:45-47.
33. Picard LS, Lindsay S, Strawn JR, et al. Atypical neuroleptic malignant syndrome: diagnostic controversies and considerations. Pharmacotherapy. 2008;28:530-535.
1. Stubner S, Rustenbeck E, Grohmann R, et al. Severe and uncommon involuntary movement disorders due to psychotropic drugs. Pharmacopsychiatry. 2004;37:S54-S64.
2. Strawn JR, Keck PE, Jr, Caroff SN. Neuroleptic malignant syndrome. Am J Psychiatry. 2007;164:870-876.
3. Ropper AH, Brown RH. Adams and Victor’s principles of neurology. 8th ed. New York, NY: McGraw Hill; 2005;1025-1026.
4. Sheil AT, Collins KA, Schandl CA, et al. Fetal neurotoxic response to neuroleptic medications: case report and review of the literature. Am J Forensic Med Pathol. 2007;28:116-120.
5. Balzan MV. The neuroleptic malignant syndrome: a logical approach to the patient with temperature and rigidity. Postgrad Med J. 1998;74:72-76.
6. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993;77:185-202.
7. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the critical care unit. Crit Care Med. 2002;30:2609-2610.
8. Caroff SN, Campbell EC, Sullivan KA. Neuroleptic malignant syndrome in elderly patients. Expert Rev Neurother. 2007;7:423-431.
9. Gurrera RJ, Simpson JC, Tsuang MT. Meta-analytic evidence of systematic bias in estimates of neuroleptic malignant syndrome incidence. Compr Psychiatry. 2007;48:205-211.
10. Caroff SN, Mann SC, Keck PE, Jr, et al. Residual catatonic state following neuroleptic malignant syndrome. J Clin Psychopharmacol. 2001;21:121-122.
11. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry. 1999;156:169-180.
12. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome. A case-control study. Arch Gen Psychiatry. 1989;46:914-918.
13. Berardi D, Amore M, Keck PE, Jr, et al. Clinical and pharmacologic risk factors for neuroleptic malignant syndrome: a case-control study. Biol Psychiatry. 1998;44:748-754.
14. Rosebush PI, Stewart TD. A prospective analysis of 24 episodes of neuroleptic malignant syndrome. Am J Psychiatry. 1989;146:717-725.
15. Martinez M, Marangell LB, Martinez JM. Psychopharmacology. In: Hales RE, Yudofsky SC, Gabbard GO, eds. American Psychiatric Publishing textbook of psychiatry. Arlington, VA: American Psychiatric Publishing, Inc.; 2008:1059-1132.
16. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, et al, eds. Neuroleptic malignant syndrome and related conditions. Washington, DC: American Psychiatric Publishing; 2003:1-44.
17. Hammerman S, Lam C, Caroff SN. Neuroleptic malignant syndrome and aripiprazole. J Am Acad Child Adolesc Psychiatry. 2006;45:639-641.
18. Stein MH, Sorscher M, Caroff SN. Neuroleptic malignant syndrome induced by metoclopramide in an infant with Freeman-Sheldon syndrome. Anesth Analg. 2006;103:786-787.
19. Borovicka MC, Bond LC, Gaughan KM. Ziprasidone- and lithium-induced neuroleptic malignant syndrome. Ann Pharmacother. 2006;40:139-142.
20. Gill J, Singh H, Nugent K. Acute lithium intoxication and neuroleptic malignant syndrome. Pharmacotherapy. 2003;23:811-815.
21. Ward C. Neuroleptic malignant syndrome in a patient with Parkinson’s disease: a case study. J Neurosci Nurs. 2005;37:160-162.
22. Leibold J, Patel V, Hasan RA. Neuroleptic malignant syndrome associated with ziprasidone in an adolescent. Clin Ther. 2004;26:1105-1108.
23. Corallo CE, Ernest D. Atypical neuroleptic malignant syndrome with long-term clozapine. Crit Care Resusc. 2007;9:338-340.
24. Molina D, Tingle LE, Lu X. Aripiprazole as the causative agent of neuroleptic malignant syndrome: a case report. Prim Care Companion J Clin Psychiatry. 2007;9:148-150.
25. Trollor JN, Chen X, Sachdev PS. Neuroleptic malignant syndrome associated with atypical antipsychotic drugs. CNS Drugs. 2009;23:477-492.
26. Gortney JS, Fagan A, Kissack JC. Neuroleptic malignant syndrome secondary to quetiapine. Ann Pharmacother. 2009;43:785-791.
27. Han C, Lee SJ, Pae CU. Paliperidone-associated atypical neuroleptic malignant syndrome: a case report. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:650-651.
28. Hanft A, Eggleston CF, Bourgeois JA. Neuroleptic malignant syndrome in an adolescent after brief exposure to olanzapine. J Child Adolesc Psychopharmacol. 2004;14:481-487.
29. Abu-Kishk I, Toledano M, Reis A, et al. Neuroleptic malignant syndrome in a child treated with an atypical antipsychotic. J Toxicol Clin Toxicol. 2004;42:921-925.
30. Neuhut R, Lindenmayer JP, Silva R. Neuroleptic malignant syndrome in children and adolescents on atypical antipsychotic medication: a review. J Child Adolesc Psychopharmacol. 2009;19:415-422.
31. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association: 2000.
32. Carroll BT, Surber SA. The problem of atypical neuroleptic malignant syndrome: a case report. Psychiatry (Edgmont). 2009;6:45-47.
33. Picard LS, Lindsay S, Strawn JR, et al. Atypical neuroleptic malignant syndrome: diagnostic controversies and considerations. Pharmacotherapy. 2008;28:530-535.
A complex subject
Thank you for your excellent article “Pharmacologic treatment of borderline personality disorder” (Current Psychiatry, August 2011, p. 30-40). This subject is very complex and poorly understood, both in primary diagnosis and the implications it has on other axis I conditions. Your attempts to educate and suggest possible treatment options are appreciated.
Bruce Miller, PA-C
Department of Corrections
VA Medical Center
Sartell, MN
Thank you for your excellent article “Pharmacologic treatment of borderline personality disorder” (Current Psychiatry, August 2011, p. 30-40). This subject is very complex and poorly understood, both in primary diagnosis and the implications it has on other axis I conditions. Your attempts to educate and suggest possible treatment options are appreciated.
Bruce Miller, PA-C
Department of Corrections
VA Medical Center
Sartell, MN
Thank you for your excellent article “Pharmacologic treatment of borderline personality disorder” (Current Psychiatry, August 2011, p. 30-40). This subject is very complex and poorly understood, both in primary diagnosis and the implications it has on other axis I conditions. Your attempts to educate and suggest possible treatment options are appreciated.
Bruce Miller, PA-C
Department of Corrections
VA Medical Center
Sartell, MN
Treating family members
I enjoyed the article by Drs. Mossman, Farrell, and Gilday addressing the practice of physicians prescribing medications for relatives or friends (“Should you prescribe medications for family and friends?” Current Psychiatry, June 2011, p. 41-51). I would like to add additional reasons why physicians may accept or decline a relative’s request for treatment.
Some doctors treat relatives out of a desire to reciprocate for financial or emotional support, or to compensate for deficiencies in other parts of their relationship.1 Other reasons to treat include convenience for the relative by not having to wait for an appointment, or for the relative’s physician by not wanting to bother them with a minor problem and associated paperwork.2 A belief that the physician-relative can offer better care than what is available or currently provided has been given as a motivation for treatment.3 The fear of misdiagnosis and a subsequent guilty conscience have been offered as reasons to refrain from treating family members.1 Other doctors refrain from treating because of role conflict or the possibility of treatment noncompliance; a relative may not follow the treatment plan seriously because of familiarity with the physician.2,3 Interestingly, a desire for confidentiality has been given as a reason to either treat or refrain from treating family members.1
Because most physicians will be solicited for treatment from family members or friends during their careers, I applaud the authors for addressing this topic. Increasing awareness of guidelines, reasons for and against treatment, and recommendations for treatment should be a priority in medical school curricula and graduate medical education.
Jonathan R. Scarff, MD
Resident in General Psychiatry
University of Louisville
Louisville, KY
1. Boiko PE, Schuman SH, Rust PF. Physicians treating their own spouses: relationship of physicians to their own family’s health care. J Fam Pract. 1984;18(6):891-896.
2. Reagan B, Reagan P, Sinclair A. ‘Common sense and a thick hide’. Physicians providing care to their own family members. Arch Fam Med. 1994;3(7):599-604.
3. La Puma J, Stocking CB, La Voie D, et al. When physicians treat members of their own families. Practices in a community hospital. N Engl J Med. 1991;325(18):1290-1294.
I enjoyed the article by Drs. Mossman, Farrell, and Gilday addressing the practice of physicians prescribing medications for relatives or friends (“Should you prescribe medications for family and friends?” Current Psychiatry, June 2011, p. 41-51). I would like to add additional reasons why physicians may accept or decline a relative’s request for treatment.
Some doctors treat relatives out of a desire to reciprocate for financial or emotional support, or to compensate for deficiencies in other parts of their relationship.1 Other reasons to treat include convenience for the relative by not having to wait for an appointment, or for the relative’s physician by not wanting to bother them with a minor problem and associated paperwork.2 A belief that the physician-relative can offer better care than what is available or currently provided has been given as a motivation for treatment.3 The fear of misdiagnosis and a subsequent guilty conscience have been offered as reasons to refrain from treating family members.1 Other doctors refrain from treating because of role conflict or the possibility of treatment noncompliance; a relative may not follow the treatment plan seriously because of familiarity with the physician.2,3 Interestingly, a desire for confidentiality has been given as a reason to either treat or refrain from treating family members.1
Because most physicians will be solicited for treatment from family members or friends during their careers, I applaud the authors for addressing this topic. Increasing awareness of guidelines, reasons for and against treatment, and recommendations for treatment should be a priority in medical school curricula and graduate medical education.
Jonathan R. Scarff, MD
Resident in General Psychiatry
University of Louisville
Louisville, KY
I enjoyed the article by Drs. Mossman, Farrell, and Gilday addressing the practice of physicians prescribing medications for relatives or friends (“Should you prescribe medications for family and friends?” Current Psychiatry, June 2011, p. 41-51). I would like to add additional reasons why physicians may accept or decline a relative’s request for treatment.
Some doctors treat relatives out of a desire to reciprocate for financial or emotional support, or to compensate for deficiencies in other parts of their relationship.1 Other reasons to treat include convenience for the relative by not having to wait for an appointment, or for the relative’s physician by not wanting to bother them with a minor problem and associated paperwork.2 A belief that the physician-relative can offer better care than what is available or currently provided has been given as a motivation for treatment.3 The fear of misdiagnosis and a subsequent guilty conscience have been offered as reasons to refrain from treating family members.1 Other doctors refrain from treating because of role conflict or the possibility of treatment noncompliance; a relative may not follow the treatment plan seriously because of familiarity with the physician.2,3 Interestingly, a desire for confidentiality has been given as a reason to either treat or refrain from treating family members.1
Because most physicians will be solicited for treatment from family members or friends during their careers, I applaud the authors for addressing this topic. Increasing awareness of guidelines, reasons for and against treatment, and recommendations for treatment should be a priority in medical school curricula and graduate medical education.
Jonathan R. Scarff, MD
Resident in General Psychiatry
University of Louisville
Louisville, KY
1. Boiko PE, Schuman SH, Rust PF. Physicians treating their own spouses: relationship of physicians to their own family’s health care. J Fam Pract. 1984;18(6):891-896.
2. Reagan B, Reagan P, Sinclair A. ‘Common sense and a thick hide’. Physicians providing care to their own family members. Arch Fam Med. 1994;3(7):599-604.
3. La Puma J, Stocking CB, La Voie D, et al. When physicians treat members of their own families. Practices in a community hospital. N Engl J Med. 1991;325(18):1290-1294.
1. Boiko PE, Schuman SH, Rust PF. Physicians treating their own spouses: relationship of physicians to their own family’s health care. J Fam Pract. 1984;18(6):891-896.
2. Reagan B, Reagan P, Sinclair A. ‘Common sense and a thick hide’. Physicians providing care to their own family members. Arch Fam Med. 1994;3(7):599-604.
3. La Puma J, Stocking CB, La Voie D, et al. When physicians treat members of their own families. Practices in a community hospital. N Engl J Med. 1991;325(18):1290-1294.
Reserpine for schizophrenia
It seems unfortunate “Treatment-resistant schizophrenia: What can we do about it?” (Current Psychiatry, June 2011, p. 52-59) made no mention of reserpine, an alternative antipsychotic with a completely different mechanism of action. Many articles have reported augmentation when reserpine is combined with D2-blocking antipsychotics. Several articles also document a lack of convincing evidence for the common opinion that reserpine causes or worsens depression. Many psychiatrists may not know it is still available for prescription.
Dale Simpson, MD
Psychiatrist, Private Practice
Sanford, NC
Dr. Citrome responds
Thank you for bringing up an agent that has been forgotten but played a pivotal role in the early history of the psychopharmacology of schizophrenia. I could not locate a report of a randomized controlled study of reserpine specifically for treatment-resistant or refractory schizophrenia; therefore, I did not include this agent in my review. Christison et al1 published a succinct summary of the use of reserpine in schizophrenia in which they reviewed the extant controlled studies for reserpine for psychosis. All were conducted more than 50 years ago using the standards for such trials as they existed at that time. The authors comment there is anecdotal and uncontrolled evidence that some patients who respond poorly to “neuroleptics” improve with reserpine. They suggest trials of reserpine may be warranted in some neuroleptic-resistant patients but effects such as severe depression, significant hypotension, exacerbation of asthma, peptic ulceration and hemorrhage, and extrapyramidal side effects can be problematic. There is evidence that a gradual increase to a full dose reduces some side effects.
Leslie Citrome, MD, MPH
Professor of Psychiatry
New York University School of Medicine
New York, NY
Reference
1. Christison GW, Kirch DG, Wyatt RJ. When symptoms persist: choosing among alternative somatic treatments for schizophrenia. Schizophr Bull. 1991;17(2):217-245.
It seems unfortunate “Treatment-resistant schizophrenia: What can we do about it?” (Current Psychiatry, June 2011, p. 52-59) made no mention of reserpine, an alternative antipsychotic with a completely different mechanism of action. Many articles have reported augmentation when reserpine is combined with D2-blocking antipsychotics. Several articles also document a lack of convincing evidence for the common opinion that reserpine causes or worsens depression. Many psychiatrists may not know it is still available for prescription.
Dale Simpson, MD
Psychiatrist, Private Practice
Sanford, NC
Dr. Citrome responds
Thank you for bringing up an agent that has been forgotten but played a pivotal role in the early history of the psychopharmacology of schizophrenia. I could not locate a report of a randomized controlled study of reserpine specifically for treatment-resistant or refractory schizophrenia; therefore, I did not include this agent in my review. Christison et al1 published a succinct summary of the use of reserpine in schizophrenia in which they reviewed the extant controlled studies for reserpine for psychosis. All were conducted more than 50 years ago using the standards for such trials as they existed at that time. The authors comment there is anecdotal and uncontrolled evidence that some patients who respond poorly to “neuroleptics” improve with reserpine. They suggest trials of reserpine may be warranted in some neuroleptic-resistant patients but effects such as severe depression, significant hypotension, exacerbation of asthma, peptic ulceration and hemorrhage, and extrapyramidal side effects can be problematic. There is evidence that a gradual increase to a full dose reduces some side effects.
Leslie Citrome, MD, MPH
Professor of Psychiatry
New York University School of Medicine
New York, NY
It seems unfortunate “Treatment-resistant schizophrenia: What can we do about it?” (Current Psychiatry, June 2011, p. 52-59) made no mention of reserpine, an alternative antipsychotic with a completely different mechanism of action. Many articles have reported augmentation when reserpine is combined with D2-blocking antipsychotics. Several articles also document a lack of convincing evidence for the common opinion that reserpine causes or worsens depression. Many psychiatrists may not know it is still available for prescription.
Dale Simpson, MD
Psychiatrist, Private Practice
Sanford, NC
Dr. Citrome responds
Thank you for bringing up an agent that has been forgotten but played a pivotal role in the early history of the psychopharmacology of schizophrenia. I could not locate a report of a randomized controlled study of reserpine specifically for treatment-resistant or refractory schizophrenia; therefore, I did not include this agent in my review. Christison et al1 published a succinct summary of the use of reserpine in schizophrenia in which they reviewed the extant controlled studies for reserpine for psychosis. All were conducted more than 50 years ago using the standards for such trials as they existed at that time. The authors comment there is anecdotal and uncontrolled evidence that some patients who respond poorly to “neuroleptics” improve with reserpine. They suggest trials of reserpine may be warranted in some neuroleptic-resistant patients but effects such as severe depression, significant hypotension, exacerbation of asthma, peptic ulceration and hemorrhage, and extrapyramidal side effects can be problematic. There is evidence that a gradual increase to a full dose reduces some side effects.
Leslie Citrome, MD, MPH
Professor of Psychiatry
New York University School of Medicine
New York, NY
Reference
1. Christison GW, Kirch DG, Wyatt RJ. When symptoms persist: choosing among alternative somatic treatments for schizophrenia. Schizophr Bull. 1991;17(2):217-245.
Reference
1. Christison GW, Kirch DG, Wyatt RJ. When symptoms persist: choosing among alternative somatic treatments for schizophrenia. Schizophr Bull. 1991;17(2):217-245.
T3 in depression
Thanks to Drs. Gih, Bostwick, and Casher for their valuable reminder about the use of triiodothyronine (T3) in treatment-resistant depression (“Augmenting antidepressants with triiodothyronine: An underutilized strategy,” Current Psychiatry, July 2011, p. 43-44).
The authors note “If TSH [thyroid-stimulating hormone] is elevated, a free thyroxine (T4) level should be ordered to detect clinical hypothyroidism.” I have found a significant number of patients with normal TSH levels but low free T4 levels, which is perhaps because of the number of traumatic brain injuries (TBI) in the veteran population I see. I realize a TSH screening alone is more cost-effective than both a TSH and free T4, but I wonder if this is the best course in evaluating treatment-resistant depression, particularly in patient populations where subtle brain damage may confound a normal TSH value. How many cases of hypothyroidism might be missed in the interest of economy?
Scott D. Mendelson, MD, PhD
Consultation Liaison Psychiatrist
Roseburg VA Medical Center
Roseburg, OR
The authors respond
Thank you for your interest in our article. Our opinion is TSH alone is a sufficient screening laboratory in “routine” depressed populations. Although a case could be made for adding free T4 (with TSH), economics and clinical guidelines would argue otherwise. In patients who are asymptomatic but have abnormal thyroid values, referral to an endocrinologist is highly advised.
Although we do not routinely see brain traumatized patients, a 2007 article by Rothman et al1 affirms your astute clinical observations about TBI and associated neuroendocrine findings.
Many clinicians are checking and supplementing vitamin D levels, in addition to including omega-3 fatty acids in their psychotropic regimens. Although the jury remains out on these interventions, preliminary data suggest these may be helpful adjuncts for depressed patients and have a low burden of side effects. Let us not forget that evidence-based psychotherapies also should be included in the armamentarium for treating “resistant” depressed patients. Psychosocial interventions should be given the same “adequate dose and duration” guidance as psychotropics.
Daniel E. Gih, MD
Clinical Assistant Professor of Child
and Adolescent Psychiatry
Department of Psychiatry
University of Michigan Health System
Ann Arbor, MI
Jolene Bostwick, PharmD, BCPS, BCPP
Clinical Assistant Professor of Pharmacy
Clinical Pharmacist
University of Michigan Health System
Ann Arbor, MI
Michael I. Casher, MD
Clinical Assistant Professor of Psychiatry
Director, Adult Inpatient Unit
University of Michigan Health System
Ann Arbor, MI
Reference
1. Rothman MS, Arciniegas DB, Filley CM, et al. The neuroendocrine effects of traumatic brain injury. J Neuropsychiatry Clin Neurosci. 2007;19(4):363-372
Thanks to Drs. Gih, Bostwick, and Casher for their valuable reminder about the use of triiodothyronine (T3) in treatment-resistant depression (“Augmenting antidepressants with triiodothyronine: An underutilized strategy,” Current Psychiatry, July 2011, p. 43-44).
The authors note “If TSH [thyroid-stimulating hormone] is elevated, a free thyroxine (T4) level should be ordered to detect clinical hypothyroidism.” I have found a significant number of patients with normal TSH levels but low free T4 levels, which is perhaps because of the number of traumatic brain injuries (TBI) in the veteran population I see. I realize a TSH screening alone is more cost-effective than both a TSH and free T4, but I wonder if this is the best course in evaluating treatment-resistant depression, particularly in patient populations where subtle brain damage may confound a normal TSH value. How many cases of hypothyroidism might be missed in the interest of economy?
Scott D. Mendelson, MD, PhD
Consultation Liaison Psychiatrist
Roseburg VA Medical Center
Roseburg, OR
The authors respond
Thank you for your interest in our article. Our opinion is TSH alone is a sufficient screening laboratory in “routine” depressed populations. Although a case could be made for adding free T4 (with TSH), economics and clinical guidelines would argue otherwise. In patients who are asymptomatic but have abnormal thyroid values, referral to an endocrinologist is highly advised.
Although we do not routinely see brain traumatized patients, a 2007 article by Rothman et al1 affirms your astute clinical observations about TBI and associated neuroendocrine findings.
Many clinicians are checking and supplementing vitamin D levels, in addition to including omega-3 fatty acids in their psychotropic regimens. Although the jury remains out on these interventions, preliminary data suggest these may be helpful adjuncts for depressed patients and have a low burden of side effects. Let us not forget that evidence-based psychotherapies also should be included in the armamentarium for treating “resistant” depressed patients. Psychosocial interventions should be given the same “adequate dose and duration” guidance as psychotropics.
Daniel E. Gih, MD
Clinical Assistant Professor of Child
and Adolescent Psychiatry
Department of Psychiatry
University of Michigan Health System
Ann Arbor, MI
Jolene Bostwick, PharmD, BCPS, BCPP
Clinical Assistant Professor of Pharmacy
Clinical Pharmacist
University of Michigan Health System
Ann Arbor, MI
Michael I. Casher, MD
Clinical Assistant Professor of Psychiatry
Director, Adult Inpatient Unit
University of Michigan Health System
Ann Arbor, MI
Thanks to Drs. Gih, Bostwick, and Casher for their valuable reminder about the use of triiodothyronine (T3) in treatment-resistant depression (“Augmenting antidepressants with triiodothyronine: An underutilized strategy,” Current Psychiatry, July 2011, p. 43-44).
The authors note “If TSH [thyroid-stimulating hormone] is elevated, a free thyroxine (T4) level should be ordered to detect clinical hypothyroidism.” I have found a significant number of patients with normal TSH levels but low free T4 levels, which is perhaps because of the number of traumatic brain injuries (TBI) in the veteran population I see. I realize a TSH screening alone is more cost-effective than both a TSH and free T4, but I wonder if this is the best course in evaluating treatment-resistant depression, particularly in patient populations where subtle brain damage may confound a normal TSH value. How many cases of hypothyroidism might be missed in the interest of economy?
Scott D. Mendelson, MD, PhD
Consultation Liaison Psychiatrist
Roseburg VA Medical Center
Roseburg, OR
The authors respond
Thank you for your interest in our article. Our opinion is TSH alone is a sufficient screening laboratory in “routine” depressed populations. Although a case could be made for adding free T4 (with TSH), economics and clinical guidelines would argue otherwise. In patients who are asymptomatic but have abnormal thyroid values, referral to an endocrinologist is highly advised.
Although we do not routinely see brain traumatized patients, a 2007 article by Rothman et al1 affirms your astute clinical observations about TBI and associated neuroendocrine findings.
Many clinicians are checking and supplementing vitamin D levels, in addition to including omega-3 fatty acids in their psychotropic regimens. Although the jury remains out on these interventions, preliminary data suggest these may be helpful adjuncts for depressed patients and have a low burden of side effects. Let us not forget that evidence-based psychotherapies also should be included in the armamentarium for treating “resistant” depressed patients. Psychosocial interventions should be given the same “adequate dose and duration” guidance as psychotropics.
Daniel E. Gih, MD
Clinical Assistant Professor of Child
and Adolescent Psychiatry
Department of Psychiatry
University of Michigan Health System
Ann Arbor, MI
Jolene Bostwick, PharmD, BCPS, BCPP
Clinical Assistant Professor of Pharmacy
Clinical Pharmacist
University of Michigan Health System
Ann Arbor, MI
Michael I. Casher, MD
Clinical Assistant Professor of Psychiatry
Director, Adult Inpatient Unit
University of Michigan Health System
Ann Arbor, MI
Reference
1. Rothman MS, Arciniegas DB, Filley CM, et al. The neuroendocrine effects of traumatic brain injury. J Neuropsychiatry Clin Neurosci. 2007;19(4):363-372
Reference
1. Rothman MS, Arciniegas DB, Filley CM, et al. The neuroendocrine effects of traumatic brain injury. J Neuropsychiatry Clin Neurosci. 2007;19(4):363-372
Comments & Controversies
Discuss this article at www.facebook.com/CurrentPsychiatry
T3 in depression
Thanks to Drs. Gih, Bostwick, and Casher for their valuable reminder about the use of triiodothyronine (T3) in treatment-resistant depression (“Augmenting antidepressants with triiodothyronine: An underutilized strategy,” Current Psychiatry, July 2011, p. 43-44).
The authors note “If TSH [thyroid-stimulating hormone] is elevated, a free thyroxine (T4) level should be ordered to detect clinical hypothyroidism.” I have found a significant number of patients with normal TSH levels but low free T4 levels, which is perhaps because of the number of traumatic brain injuries (TBI) in the veteran population I see. I realize a TSH screening alone is more cost-effective than both a TSH and free T4, but I wonder if this is the best course in evaluating treatment-resistant depression, particularly in patient populations where subtle brain damage may confound a normal TSH value. How many cases of hypothyroidism might be missed in the interest of economy?
Scott D. Mendelson, MD, PhD
Consultation Liaison Psychiatrist
Roseburg VA Medical Center
Roseburg, OR
The authors respond
Thank you for your interest in our article. Our opinion is TSH alone is a sufficient screening laboratory in “routine” depressed populations. Although a case could be made for adding free T4 (with TSH), economics and clinical guidelines would argue otherwise. In patients who are asymptomatic but have abnormal thyroid values, referral to an endocrinologist is highly advised.
Although we do not routinely see brain traumatized patients, a 2007 article by Rothman et al1 affirms your astute clinical observations about TBI and associated neuroendocrine findings.
Many clinicians are checking and supplementing vitamin D levels, in addition to including omega-3 fatty acids in their psychotropic regimens. Although the jury remains out on these interventions, preliminary data suggest these may be helpful adjuncts for depressed patients and have a low burden of side effects. Let us not forget that evidence-based psychotherapies also should be included in the armamentarium for treating “resistant” depressed patients. Psychosocial interventions should be given the same “adequate dose and duration” guidance as psychotropics.
Daniel E. Gih, MD
Clinical Assistant Professor of Child
and Adolescent Psychiatry
Department of Psychiatry
University of Michigan Health System
Ann Arbor, MI
Jolene Bostwick, PharmD, BCPS, BCPP
Clinical Assistant Professor of Pharmacy
Clinical Pharmacist
University of Michigan Health System
Ann Arbor, MI
Michael I. Casher, MD
Clinical Assistant Professor of Psychiatry
Director, Adult Inpatient Unit
University of Michigan Health System
Ann Arbor, MI
Reserpine for schizophrenia
It seems unfortunate “Treatment-resistant schizophrenia: What can we do about it?” (Current Psychiatry, June 2011, p. 52-59) made no mention of reserpine, an alternative antipsychotic with a completely different mechanism of action. Many articles have reported augmentation when reserpine is combined with D2-blocking antipsychotics. Several articles also document a lack of convincing evidence for the common opinion that reserpine causes or worsens depression. Many psychiatrists may not know it is still available for prescription.
Dale Simpson, MD
Psychiatrist, Private Practice
Sanford, NC
Dr. Citrome responds
Thank you for bringing up an agent that has been forgotten but played a pivotal role in the early history of the psychopharmacology of schizophrenia. I could not locate a report of a randomized controlled study of reserpine specifically for treatment-resistant or refractory schizophrenia; therefore, I did not include this agent in my review. Christison et al1 published a succinct summary of the use of reserpine in schizophrenia in which they reviewed the extant controlled studies for reserpine for psychosis. All were conducted more than 50 years ago using the standards for such trials as they existed at that time. The authors comment there is anecdotal and uncontrolled evidence that some patients who respond poorly to “neuroleptics” improve with reserpine. They suggest trials of reserpine may be warranted in some neuroleptic-resistant patients but effects such as severe depression, significant hypotension, exacerbation of asthma, peptic ulceration and hemorrhage, and extrapyramidal side effects can be problematic. There is evidence that a gradual increase to a full dose reduces some side effects.
Leslie Citrome, MD, MPH
Professor of Psychiatry
New York University School of Medicine
New York, NY
Treating family members
I enjoyed the article by Drs. Mossman, Farrell, and Gilday addressing the practice of physicians prescribing medications for relatives or friends (“Should you prescribe medications for family and friends?” Current Psychiatry, June 2011, p. 41-51). I would like to add additional reasons why physicians may accept or decline a relative’s request for treatment.
Some doctors treat relatives out of a desire to reciprocate for financial or emotional support, or to compensate for deficiencies in other parts of their relationship.1 Other reasons to treat include convenience for the relative by not having to wait for an appointment, or for the relative’s physician by not wanting to bother them with a minor problem and associated paperwork.2 A belief that the physician-relative can offer better care than what is available or currently provided has been given as a motivation for treatment.3 The fear of misdiagnosis and a subsequent guilty conscience have been offered as reasons to refrain from treating family members.1 Other doctors refrain from treating because of role conflict or the possibility of treatment noncompliance; a relative may not follow the treatment plan seriously because of familiarity with the physician.2,3 Interestingly, a desire for confidentiality has been given as a reason to either treat or refrain from treating family members.1
Because most physicians will be solicited for treatment from family members or friends during their careers, I applaud the authors for addressing this topic. Increasing awareness of guidelines, reasons for and against treatment, and recommendations for treatment should be a priority in medical school curricula and graduate medical education.
Jonathan R. Scarff, MD
Resident in General Psychiatry
University of Louisville
Louisville, KY
A complex subject
Thank you for your excellent article “Pharmacologic treatment of borderline personality disorder” (Current Psychiatry, August 2011, p. 30-40). This subject is very complex and poorly understood, both in primary diagnosis and the implications it has on other axis I conditions. Your attempts to educate and suggest possible treatment options are appreciated.
Bruce Miller, PA-C
Department of Corrections
VA Medical Center
Sartell, MN
Discuss this article at www.facebook.com/CurrentPsychiatry
T3 in depression
Thanks to Drs. Gih, Bostwick, and Casher for their valuable reminder about the use of triiodothyronine (T3) in treatment-resistant depression (“Augmenting antidepressants with triiodothyronine: An underutilized strategy,” Current Psychiatry, July 2011, p. 43-44).
The authors note “If TSH [thyroid-stimulating hormone] is elevated, a free thyroxine (T4) level should be ordered to detect clinical hypothyroidism.” I have found a significant number of patients with normal TSH levels but low free T4 levels, which is perhaps because of the number of traumatic brain injuries (TBI) in the veteran population I see. I realize a TSH screening alone is more cost-effective than both a TSH and free T4, but I wonder if this is the best course in evaluating treatment-resistant depression, particularly in patient populations where subtle brain damage may confound a normal TSH value. How many cases of hypothyroidism might be missed in the interest of economy?
Scott D. Mendelson, MD, PhD
Consultation Liaison Psychiatrist
Roseburg VA Medical Center
Roseburg, OR
The authors respond
Thank you for your interest in our article. Our opinion is TSH alone is a sufficient screening laboratory in “routine” depressed populations. Although a case could be made for adding free T4 (with TSH), economics and clinical guidelines would argue otherwise. In patients who are asymptomatic but have abnormal thyroid values, referral to an endocrinologist is highly advised.
Although we do not routinely see brain traumatized patients, a 2007 article by Rothman et al1 affirms your astute clinical observations about TBI and associated neuroendocrine findings.
Many clinicians are checking and supplementing vitamin D levels, in addition to including omega-3 fatty acids in their psychotropic regimens. Although the jury remains out on these interventions, preliminary data suggest these may be helpful adjuncts for depressed patients and have a low burden of side effects. Let us not forget that evidence-based psychotherapies also should be included in the armamentarium for treating “resistant” depressed patients. Psychosocial interventions should be given the same “adequate dose and duration” guidance as psychotropics.
Daniel E. Gih, MD
Clinical Assistant Professor of Child
and Adolescent Psychiatry
Department of Psychiatry
University of Michigan Health System
Ann Arbor, MI
Jolene Bostwick, PharmD, BCPS, BCPP
Clinical Assistant Professor of Pharmacy
Clinical Pharmacist
University of Michigan Health System
Ann Arbor, MI
Michael I. Casher, MD
Clinical Assistant Professor of Psychiatry
Director, Adult Inpatient Unit
University of Michigan Health System
Ann Arbor, MI
Reserpine for schizophrenia
It seems unfortunate “Treatment-resistant schizophrenia: What can we do about it?” (Current Psychiatry, June 2011, p. 52-59) made no mention of reserpine, an alternative antipsychotic with a completely different mechanism of action. Many articles have reported augmentation when reserpine is combined with D2-blocking antipsychotics. Several articles also document a lack of convincing evidence for the common opinion that reserpine causes or worsens depression. Many psychiatrists may not know it is still available for prescription.
Dale Simpson, MD
Psychiatrist, Private Practice
Sanford, NC
Dr. Citrome responds
Thank you for bringing up an agent that has been forgotten but played a pivotal role in the early history of the psychopharmacology of schizophrenia. I could not locate a report of a randomized controlled study of reserpine specifically for treatment-resistant or refractory schizophrenia; therefore, I did not include this agent in my review. Christison et al1 published a succinct summary of the use of reserpine in schizophrenia in which they reviewed the extant controlled studies for reserpine for psychosis. All were conducted more than 50 years ago using the standards for such trials as they existed at that time. The authors comment there is anecdotal and uncontrolled evidence that some patients who respond poorly to “neuroleptics” improve with reserpine. They suggest trials of reserpine may be warranted in some neuroleptic-resistant patients but effects such as severe depression, significant hypotension, exacerbation of asthma, peptic ulceration and hemorrhage, and extrapyramidal side effects can be problematic. There is evidence that a gradual increase to a full dose reduces some side effects.
Leslie Citrome, MD, MPH
Professor of Psychiatry
New York University School of Medicine
New York, NY
Treating family members
I enjoyed the article by Drs. Mossman, Farrell, and Gilday addressing the practice of physicians prescribing medications for relatives or friends (“Should you prescribe medications for family and friends?” Current Psychiatry, June 2011, p. 41-51). I would like to add additional reasons why physicians may accept or decline a relative’s request for treatment.
Some doctors treat relatives out of a desire to reciprocate for financial or emotional support, or to compensate for deficiencies in other parts of their relationship.1 Other reasons to treat include convenience for the relative by not having to wait for an appointment, or for the relative’s physician by not wanting to bother them with a minor problem and associated paperwork.2 A belief that the physician-relative can offer better care than what is available or currently provided has been given as a motivation for treatment.3 The fear of misdiagnosis and a subsequent guilty conscience have been offered as reasons to refrain from treating family members.1 Other doctors refrain from treating because of role conflict or the possibility of treatment noncompliance; a relative may not follow the treatment plan seriously because of familiarity with the physician.2,3 Interestingly, a desire for confidentiality has been given as a reason to either treat or refrain from treating family members.1
Because most physicians will be solicited for treatment from family members or friends during their careers, I applaud the authors for addressing this topic. Increasing awareness of guidelines, reasons for and against treatment, and recommendations for treatment should be a priority in medical school curricula and graduate medical education.
Jonathan R. Scarff, MD
Resident in General Psychiatry
University of Louisville
Louisville, KY
A complex subject
Thank you for your excellent article “Pharmacologic treatment of borderline personality disorder” (Current Psychiatry, August 2011, p. 30-40). This subject is very complex and poorly understood, both in primary diagnosis and the implications it has on other axis I conditions. Your attempts to educate and suggest possible treatment options are appreciated.
Bruce Miller, PA-C
Department of Corrections
VA Medical Center
Sartell, MN
Discuss this article at www.facebook.com/CurrentPsychiatry
T3 in depression
Thanks to Drs. Gih, Bostwick, and Casher for their valuable reminder about the use of triiodothyronine (T3) in treatment-resistant depression (“Augmenting antidepressants with triiodothyronine: An underutilized strategy,” Current Psychiatry, July 2011, p. 43-44).
The authors note “If TSH [thyroid-stimulating hormone] is elevated, a free thyroxine (T4) level should be ordered to detect clinical hypothyroidism.” I have found a significant number of patients with normal TSH levels but low free T4 levels, which is perhaps because of the number of traumatic brain injuries (TBI) in the veteran population I see. I realize a TSH screening alone is more cost-effective than both a TSH and free T4, but I wonder if this is the best course in evaluating treatment-resistant depression, particularly in patient populations where subtle brain damage may confound a normal TSH value. How many cases of hypothyroidism might be missed in the interest of economy?
Scott D. Mendelson, MD, PhD
Consultation Liaison Psychiatrist
Roseburg VA Medical Center
Roseburg, OR
The authors respond
Thank you for your interest in our article. Our opinion is TSH alone is a sufficient screening laboratory in “routine” depressed populations. Although a case could be made for adding free T4 (with TSH), economics and clinical guidelines would argue otherwise. In patients who are asymptomatic but have abnormal thyroid values, referral to an endocrinologist is highly advised.
Although we do not routinely see brain traumatized patients, a 2007 article by Rothman et al1 affirms your astute clinical observations about TBI and associated neuroendocrine findings.
Many clinicians are checking and supplementing vitamin D levels, in addition to including omega-3 fatty acids in their psychotropic regimens. Although the jury remains out on these interventions, preliminary data suggest these may be helpful adjuncts for depressed patients and have a low burden of side effects. Let us not forget that evidence-based psychotherapies also should be included in the armamentarium for treating “resistant” depressed patients. Psychosocial interventions should be given the same “adequate dose and duration” guidance as psychotropics.
Daniel E. Gih, MD
Clinical Assistant Professor of Child
and Adolescent Psychiatry
Department of Psychiatry
University of Michigan Health System
Ann Arbor, MI
Jolene Bostwick, PharmD, BCPS, BCPP
Clinical Assistant Professor of Pharmacy
Clinical Pharmacist
University of Michigan Health System
Ann Arbor, MI
Michael I. Casher, MD
Clinical Assistant Professor of Psychiatry
Director, Adult Inpatient Unit
University of Michigan Health System
Ann Arbor, MI
Reserpine for schizophrenia
It seems unfortunate “Treatment-resistant schizophrenia: What can we do about it?” (Current Psychiatry, June 2011, p. 52-59) made no mention of reserpine, an alternative antipsychotic with a completely different mechanism of action. Many articles have reported augmentation when reserpine is combined with D2-blocking antipsychotics. Several articles also document a lack of convincing evidence for the common opinion that reserpine causes or worsens depression. Many psychiatrists may not know it is still available for prescription.
Dale Simpson, MD
Psychiatrist, Private Practice
Sanford, NC
Dr. Citrome responds
Thank you for bringing up an agent that has been forgotten but played a pivotal role in the early history of the psychopharmacology of schizophrenia. I could not locate a report of a randomized controlled study of reserpine specifically for treatment-resistant or refractory schizophrenia; therefore, I did not include this agent in my review. Christison et al1 published a succinct summary of the use of reserpine in schizophrenia in which they reviewed the extant controlled studies for reserpine for psychosis. All were conducted more than 50 years ago using the standards for such trials as they existed at that time. The authors comment there is anecdotal and uncontrolled evidence that some patients who respond poorly to “neuroleptics” improve with reserpine. They suggest trials of reserpine may be warranted in some neuroleptic-resistant patients but effects such as severe depression, significant hypotension, exacerbation of asthma, peptic ulceration and hemorrhage, and extrapyramidal side effects can be problematic. There is evidence that a gradual increase to a full dose reduces some side effects.
Leslie Citrome, MD, MPH
Professor of Psychiatry
New York University School of Medicine
New York, NY
Treating family members
I enjoyed the article by Drs. Mossman, Farrell, and Gilday addressing the practice of physicians prescribing medications for relatives or friends (“Should you prescribe medications for family and friends?” Current Psychiatry, June 2011, p. 41-51). I would like to add additional reasons why physicians may accept or decline a relative’s request for treatment.
Some doctors treat relatives out of a desire to reciprocate for financial or emotional support, or to compensate for deficiencies in other parts of their relationship.1 Other reasons to treat include convenience for the relative by not having to wait for an appointment, or for the relative’s physician by not wanting to bother them with a minor problem and associated paperwork.2 A belief that the physician-relative can offer better care than what is available or currently provided has been given as a motivation for treatment.3 The fear of misdiagnosis and a subsequent guilty conscience have been offered as reasons to refrain from treating family members.1 Other doctors refrain from treating because of role conflict or the possibility of treatment noncompliance; a relative may not follow the treatment plan seriously because of familiarity with the physician.2,3 Interestingly, a desire for confidentiality has been given as a reason to either treat or refrain from treating family members.1
Because most physicians will be solicited for treatment from family members or friends during their careers, I applaud the authors for addressing this topic. Increasing awareness of guidelines, reasons for and against treatment, and recommendations for treatment should be a priority in medical school curricula and graduate medical education.
Jonathan R. Scarff, MD
Resident in General Psychiatry
University of Louisville
Louisville, KY
A complex subject
Thank you for your excellent article “Pharmacologic treatment of borderline personality disorder” (Current Psychiatry, August 2011, p. 30-40). This subject is very complex and poorly understood, both in primary diagnosis and the implications it has on other axis I conditions. Your attempts to educate and suggest possible treatment options are appreciated.
Bruce Miller, PA-C
Department of Corrections
VA Medical Center
Sartell, MN