User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Diabetes screening: Which patients, what tests, and how often?
Dr. Keenan is associate professor, department of medicine, University of California, Davis.
Principal Source: U.S. Preventive Services Task Force. Screening for type 2 diabetes mellitus in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2008;148(11):846-854.—Discussant: Craig R. Keenan, MD
- Screen annually for type 2 diabetes mellitus (T2DM), prediabetes, weight gain, and lipid abnormalities in all patients taking atypical antipsychotics.
- Screen annually psychiatric patients age ≥30 who do not take atypicals for T2DM and prediabetes.
- For patients age <30, regularly review your patients’ risk factors for diabetes to determine whom to screen for T2DM or prediabetes.
- Screening is done most simply by ordering a fasting plasma glucose test.
Psychiatric patients—especially those with schizophrenia or taking atypical antipsychotics—are at risk for developing type 2 diabetes mellitus (T2DM) and prediabetes conditions. T2DM can be present for years without significant symptoms and even asymptomatic conditions increase the risk of cardiovascular, renal, retinal, and neurologic complications.
Despite a need for T2DM screening and treatment, expert guidelines disagree on who and how to screen (Table 1). Although testing patients who have diabetes symptoms—including polyuria, polydipsia, and weight loss—is indicated, some medical groups advocate screening asymptomatic persons for T2DM.
Screening recommendations
Consensus guidelines. In 2004, the American Diabetes Association (ADA), American Psychiatric Association (APA), American Association of Clinical Endocrinologists (AACE), and North American Association for the Study of Obesity (NAASO) created consensus guidelines for screening psychiatric patients receiving atypical antipsychotics. In addition to diabetes risk, psychiatric patients are at higher risk for metabolic syndrome, dyslipidemia, obesity, and hypertension.1 The ADA, APA, AACE, and NAASO recommend regularly screening for weight gain and dyslipidemia, obtaining baseline values of fasting plasma glucose (FPG), rechecking FPG after 3 months, and then screening annually for diabetes or prediabetes. For patients with risk factors for diabetes and those who develop diabetes or prediabetes while taking an atypical antipsychotic, consider an atypical with a lower risk of diabetes—specifically aripiprazole or ziprasidone.1 For psychiatric patients who do not take atypicals, there is no consensus on who and how to screen for T2DM.
The U.S. Preventive Services Task Force (USPSTF) recommends screening only adults with hypertension.2 Its review found insufficient evidence that early detection and treatment leads to improved clinical outcomes in asymptomatic adults.
The ADA recommends more liberal screening, including individuals age ≥45 or anyone age <45 who is overweight and has any other diabetes risk factors.3 The ADA admits that no trials show a benefit of screening asymptomatic patients but notes that the duration of glycemic burden predicts adverse outcomes and effective interventions for diabetes and prediabetes are available.
AACE guidelines recommend screening starting at age 30 if the patient has risk factors for T2DM. This is the only group that includes psychiatric illness as a risk factor.4
European Association for the Study of Diabetes (EASD) guidelines calculate a risk score based on common risk factors to determine who should be screened and recommend using the oral glucose tolerance test (OGTT) rather the FPG.5 The OGTT identifies more cases of diabetes and pre-diabetes but takes >2 hours to administer.
Table 1
General population screening recommendations for type 2 diabetes mellitus or prediabetes
| Organization | Year | Whom to screen | How to screen |
|---|---|---|---|
| U.S. Preventive Services Task Force (USPSTF) | 2008 | Asymptomatic adults with sustained blood pressure >135/80 mmHg (treated or untreated) | FPG or OGTT every 3 years |
| American Diabetes Association (ADA) | 2009 | All adults age ≥45 Adults of any age with BMI >25 kg/m2 and ≥1 risk factors for diabetes (Table 2) | FPG or 2-hour OGTT every 3 years or more frequently, depending on initial results and risks |
| American Association of Clinical Endocrinologists (AACE) | 2007 | All adults age ≥30 with risk factors for diabetes (Table 2) | FPG or 2-hour OGTT (frequency not specified) |
| European Association for the Study of Diabetes (EASD) and European Society of Cardiology (ESC) | 2007 | All adults with elevated risk score* | OGTT (frequency not indicated) |
| FPG: fasting plasma glucose; OGTT: oral glucose tolerance test (75 gm glucose load); BMI: body mass index | |||
| *Risk scoring tool available at www.diabetes.fi/english/risktest | |||
Discussion
Despite a lack evidence showing benefit to the screened population, treating diabetes and its comorbidities improves outcomes, and the potential risks of therapy are low. Therefore, it seems reasonable to screen more patients than the USPSTF recommends.
Using the EASD risk score is intriguing, but difficult to implement in a busy practice. Therefore, I recommend following the AACE guidelines, which recognize psychiatric illness as a risk factor, for screening psychiatric patients who are not receiving atypicals.
Annually screen psychiatric patients age ≥30, especially those with schizophrenia or affective disorders. I also follow the ADA guidelines and screen overweight adults age ≤30 if they have any of the other risk factors listed in Table 2. The most common risk factors seen in practice are being a member of a high-risk ethnic group, hypertension, lipid abnormalities, and cardiovascular disease. For overweight adults without other risk factors, I start screening at age 30.
Other practitioners can be more or less conservative and still be within accepted guidelines. The FPG—glucose level drawn from a vein after at least 8 hours of fasting—is probably the easiest screening test in practice. Any patient with a value >100mg/dL should be referred to the patient’s primary care physician. Any patient who develops diabetes symptoms—including polyuria, polydipsia, and weight loss—should be tested immediately. The hemoglobin A1C test is not recommended for screening.
Table 2
Risk factors identified for diabetes or prediabetes
| American Diabetes Association (ADA) |
|
| American Association of Clinical Endocrinologists |
|
Clinical presentation
Screening detects overt diabetes and can identify prediabetes. Prediabetes includes conditions of impaired fasting glucose (IFG) or impaired glucose tolerance (IGT). IFG is defined as a fasting glucose of 100 to 125 mg/dL, and IGT is defined as having a 2-hour glucose of 140 to 199 mg/dL on an OGTT.
Approximately one-quarter of the adult population has prediabetes, and interventions can prevent the progression of prediabetes to overt diabetes and reverse prediabetes. The Diabetes Prevention Trial found that lifestyle measures—including exercise and diet—were most effective, with a 53% reduction in the rate of progression to diabetes.6 Metformin also was effective, but less so than lifestyle measures alone.
Treatment slows the development or progression of microvascular complications, such as retinopathy, nephropathy, and neuropathy. Aggressive treatment of comorbid conditions, including hyperlipidemia and hypertension, also reduces the risk of cardiovascular events.
Drug brand names
- Aripiprazole • Abilify
- Metformin • Glucophage
- Ziprasidone • Geodon
Related resources
- American Diabetes Association. Diabetes risk calculator. www.diabetes.org/risk-test.jsp.
- Dagogo-Jack S. The role of antipsychotic agents in the development of diabetes mellitus. Nat Clin Pract Endocrinol Metab. 2009;5(1):22-23. Quick, up-to-date review of the association between atypical antipsychotics and diabetes mellitus.
Disclosure
Dr. Keenan reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
2. U.S. Preventive Services Task Force. Screening for type 2 diabetes mellitus in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2008;148(11):846-854.
3. American Diabetes Association. Standards of medical care in diabetes—2009. Diabetes Care. 2009;32(suppl 1):S13-61.
4. Rodbard HW, Blonde L, Braithwaite SS, et al. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the management of diabetes mellitus. Endocr Pract. 2007;13(suppl 1):1-68.
5. Rydén L, Standl E, Bartnik M, et al. Guidelines on diabetes, pre-diabetes and cardiovascular diseases: executive summary. The Task Force on Diabetes and Cardio-vascular Diseases of the European Society of Cardiology (ESC) and of the European Association for the Study of Diabetes (EASD). Eur Heart J. 2007;28(1):88-136.
6. Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393-403.

Robert M. McCarron, DO
Series Editor
Robert M. McCarron, DO
Series Editor
Robert M. McCarron, DO
Series Editor
Dr. Keenan is associate professor, department of medicine, University of California, Davis.
Principal Source: U.S. Preventive Services Task Force. Screening for type 2 diabetes mellitus in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2008;148(11):846-854.—Discussant: Craig R. Keenan, MD
- Screen annually for type 2 diabetes mellitus (T2DM), prediabetes, weight gain, and lipid abnormalities in all patients taking atypical antipsychotics.
- Screen annually psychiatric patients age ≥30 who do not take atypicals for T2DM and prediabetes.
- For patients age <30, regularly review your patients’ risk factors for diabetes to determine whom to screen for T2DM or prediabetes.
- Screening is done most simply by ordering a fasting plasma glucose test.
Psychiatric patients—especially those with schizophrenia or taking atypical antipsychotics—are at risk for developing type 2 diabetes mellitus (T2DM) and prediabetes conditions. T2DM can be present for years without significant symptoms and even asymptomatic conditions increase the risk of cardiovascular, renal, retinal, and neurologic complications.
Despite a need for T2DM screening and treatment, expert guidelines disagree on who and how to screen (Table 1). Although testing patients who have diabetes symptoms—including polyuria, polydipsia, and weight loss—is indicated, some medical groups advocate screening asymptomatic persons for T2DM.
Screening recommendations
Consensus guidelines. In 2004, the American Diabetes Association (ADA), American Psychiatric Association (APA), American Association of Clinical Endocrinologists (AACE), and North American Association for the Study of Obesity (NAASO) created consensus guidelines for screening psychiatric patients receiving atypical antipsychotics. In addition to diabetes risk, psychiatric patients are at higher risk for metabolic syndrome, dyslipidemia, obesity, and hypertension.1 The ADA, APA, AACE, and NAASO recommend regularly screening for weight gain and dyslipidemia, obtaining baseline values of fasting plasma glucose (FPG), rechecking FPG after 3 months, and then screening annually for diabetes or prediabetes. For patients with risk factors for diabetes and those who develop diabetes or prediabetes while taking an atypical antipsychotic, consider an atypical with a lower risk of diabetes—specifically aripiprazole or ziprasidone.1 For psychiatric patients who do not take atypicals, there is no consensus on who and how to screen for T2DM.
The U.S. Preventive Services Task Force (USPSTF) recommends screening only adults with hypertension.2 Its review found insufficient evidence that early detection and treatment leads to improved clinical outcomes in asymptomatic adults.
The ADA recommends more liberal screening, including individuals age ≥45 or anyone age <45 who is overweight and has any other diabetes risk factors.3 The ADA admits that no trials show a benefit of screening asymptomatic patients but notes that the duration of glycemic burden predicts adverse outcomes and effective interventions for diabetes and prediabetes are available.
AACE guidelines recommend screening starting at age 30 if the patient has risk factors for T2DM. This is the only group that includes psychiatric illness as a risk factor.4
European Association for the Study of Diabetes (EASD) guidelines calculate a risk score based on common risk factors to determine who should be screened and recommend using the oral glucose tolerance test (OGTT) rather the FPG.5 The OGTT identifies more cases of diabetes and pre-diabetes but takes >2 hours to administer.
Table 1
General population screening recommendations for type 2 diabetes mellitus or prediabetes
| Organization | Year | Whom to screen | How to screen |
|---|---|---|---|
| U.S. Preventive Services Task Force (USPSTF) | 2008 | Asymptomatic adults with sustained blood pressure >135/80 mmHg (treated or untreated) | FPG or OGTT every 3 years |
| American Diabetes Association (ADA) | 2009 | All adults age ≥45 Adults of any age with BMI >25 kg/m2 and ≥1 risk factors for diabetes (Table 2) | FPG or 2-hour OGTT every 3 years or more frequently, depending on initial results and risks |
| American Association of Clinical Endocrinologists (AACE) | 2007 | All adults age ≥30 with risk factors for diabetes (Table 2) | FPG or 2-hour OGTT (frequency not specified) |
| European Association for the Study of Diabetes (EASD) and European Society of Cardiology (ESC) | 2007 | All adults with elevated risk score* | OGTT (frequency not indicated) |
| FPG: fasting plasma glucose; OGTT: oral glucose tolerance test (75 gm glucose load); BMI: body mass index | |||
| *Risk scoring tool available at www.diabetes.fi/english/risktest | |||
Discussion
Despite a lack evidence showing benefit to the screened population, treating diabetes and its comorbidities improves outcomes, and the potential risks of therapy are low. Therefore, it seems reasonable to screen more patients than the USPSTF recommends.
Using the EASD risk score is intriguing, but difficult to implement in a busy practice. Therefore, I recommend following the AACE guidelines, which recognize psychiatric illness as a risk factor, for screening psychiatric patients who are not receiving atypicals.
Annually screen psychiatric patients age ≥30, especially those with schizophrenia or affective disorders. I also follow the ADA guidelines and screen overweight adults age ≤30 if they have any of the other risk factors listed in Table 2. The most common risk factors seen in practice are being a member of a high-risk ethnic group, hypertension, lipid abnormalities, and cardiovascular disease. For overweight adults without other risk factors, I start screening at age 30.
Other practitioners can be more or less conservative and still be within accepted guidelines. The FPG—glucose level drawn from a vein after at least 8 hours of fasting—is probably the easiest screening test in practice. Any patient with a value >100mg/dL should be referred to the patient’s primary care physician. Any patient who develops diabetes symptoms—including polyuria, polydipsia, and weight loss—should be tested immediately. The hemoglobin A1C test is not recommended for screening.
Table 2
Risk factors identified for diabetes or prediabetes
| American Diabetes Association (ADA) |
|
| American Association of Clinical Endocrinologists |
|
Clinical presentation
Screening detects overt diabetes and can identify prediabetes. Prediabetes includes conditions of impaired fasting glucose (IFG) or impaired glucose tolerance (IGT). IFG is defined as a fasting glucose of 100 to 125 mg/dL, and IGT is defined as having a 2-hour glucose of 140 to 199 mg/dL on an OGTT.
Approximately one-quarter of the adult population has prediabetes, and interventions can prevent the progression of prediabetes to overt diabetes and reverse prediabetes. The Diabetes Prevention Trial found that lifestyle measures—including exercise and diet—were most effective, with a 53% reduction in the rate of progression to diabetes.6 Metformin also was effective, but less so than lifestyle measures alone.
Treatment slows the development or progression of microvascular complications, such as retinopathy, nephropathy, and neuropathy. Aggressive treatment of comorbid conditions, including hyperlipidemia and hypertension, also reduces the risk of cardiovascular events.
Drug brand names
- Aripiprazole • Abilify
- Metformin • Glucophage
- Ziprasidone • Geodon
Related resources
- American Diabetes Association. Diabetes risk calculator. www.diabetes.org/risk-test.jsp.
- Dagogo-Jack S. The role of antipsychotic agents in the development of diabetes mellitus. Nat Clin Pract Endocrinol Metab. 2009;5(1):22-23. Quick, up-to-date review of the association between atypical antipsychotics and diabetes mellitus.
Disclosure
Dr. Keenan reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Keenan is associate professor, department of medicine, University of California, Davis.
Principal Source: U.S. Preventive Services Task Force. Screening for type 2 diabetes mellitus in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2008;148(11):846-854.—Discussant: Craig R. Keenan, MD
- Screen annually for type 2 diabetes mellitus (T2DM), prediabetes, weight gain, and lipid abnormalities in all patients taking atypical antipsychotics.
- Screen annually psychiatric patients age ≥30 who do not take atypicals for T2DM and prediabetes.
- For patients age <30, regularly review your patients’ risk factors for diabetes to determine whom to screen for T2DM or prediabetes.
- Screening is done most simply by ordering a fasting plasma glucose test.
Psychiatric patients—especially those with schizophrenia or taking atypical antipsychotics—are at risk for developing type 2 diabetes mellitus (T2DM) and prediabetes conditions. T2DM can be present for years without significant symptoms and even asymptomatic conditions increase the risk of cardiovascular, renal, retinal, and neurologic complications.
Despite a need for T2DM screening and treatment, expert guidelines disagree on who and how to screen (Table 1). Although testing patients who have diabetes symptoms—including polyuria, polydipsia, and weight loss—is indicated, some medical groups advocate screening asymptomatic persons for T2DM.
Screening recommendations
Consensus guidelines. In 2004, the American Diabetes Association (ADA), American Psychiatric Association (APA), American Association of Clinical Endocrinologists (AACE), and North American Association for the Study of Obesity (NAASO) created consensus guidelines for screening psychiatric patients receiving atypical antipsychotics. In addition to diabetes risk, psychiatric patients are at higher risk for metabolic syndrome, dyslipidemia, obesity, and hypertension.1 The ADA, APA, AACE, and NAASO recommend regularly screening for weight gain and dyslipidemia, obtaining baseline values of fasting plasma glucose (FPG), rechecking FPG after 3 months, and then screening annually for diabetes or prediabetes. For patients with risk factors for diabetes and those who develop diabetes or prediabetes while taking an atypical antipsychotic, consider an atypical with a lower risk of diabetes—specifically aripiprazole or ziprasidone.1 For psychiatric patients who do not take atypicals, there is no consensus on who and how to screen for T2DM.
The U.S. Preventive Services Task Force (USPSTF) recommends screening only adults with hypertension.2 Its review found insufficient evidence that early detection and treatment leads to improved clinical outcomes in asymptomatic adults.
The ADA recommends more liberal screening, including individuals age ≥45 or anyone age <45 who is overweight and has any other diabetes risk factors.3 The ADA admits that no trials show a benefit of screening asymptomatic patients but notes that the duration of glycemic burden predicts adverse outcomes and effective interventions for diabetes and prediabetes are available.
AACE guidelines recommend screening starting at age 30 if the patient has risk factors for T2DM. This is the only group that includes psychiatric illness as a risk factor.4
European Association for the Study of Diabetes (EASD) guidelines calculate a risk score based on common risk factors to determine who should be screened and recommend using the oral glucose tolerance test (OGTT) rather the FPG.5 The OGTT identifies more cases of diabetes and pre-diabetes but takes >2 hours to administer.
Table 1
General population screening recommendations for type 2 diabetes mellitus or prediabetes
| Organization | Year | Whom to screen | How to screen |
|---|---|---|---|
| U.S. Preventive Services Task Force (USPSTF) | 2008 | Asymptomatic adults with sustained blood pressure >135/80 mmHg (treated or untreated) | FPG or OGTT every 3 years |
| American Diabetes Association (ADA) | 2009 | All adults age ≥45 Adults of any age with BMI >25 kg/m2 and ≥1 risk factors for diabetes (Table 2) | FPG or 2-hour OGTT every 3 years or more frequently, depending on initial results and risks |
| American Association of Clinical Endocrinologists (AACE) | 2007 | All adults age ≥30 with risk factors for diabetes (Table 2) | FPG or 2-hour OGTT (frequency not specified) |
| European Association for the Study of Diabetes (EASD) and European Society of Cardiology (ESC) | 2007 | All adults with elevated risk score* | OGTT (frequency not indicated) |
| FPG: fasting plasma glucose; OGTT: oral glucose tolerance test (75 gm glucose load); BMI: body mass index | |||
| *Risk scoring tool available at www.diabetes.fi/english/risktest | |||
Discussion
Despite a lack evidence showing benefit to the screened population, treating diabetes and its comorbidities improves outcomes, and the potential risks of therapy are low. Therefore, it seems reasonable to screen more patients than the USPSTF recommends.
Using the EASD risk score is intriguing, but difficult to implement in a busy practice. Therefore, I recommend following the AACE guidelines, which recognize psychiatric illness as a risk factor, for screening psychiatric patients who are not receiving atypicals.
Annually screen psychiatric patients age ≥30, especially those with schizophrenia or affective disorders. I also follow the ADA guidelines and screen overweight adults age ≤30 if they have any of the other risk factors listed in Table 2. The most common risk factors seen in practice are being a member of a high-risk ethnic group, hypertension, lipid abnormalities, and cardiovascular disease. For overweight adults without other risk factors, I start screening at age 30.
Other practitioners can be more or less conservative and still be within accepted guidelines. The FPG—glucose level drawn from a vein after at least 8 hours of fasting—is probably the easiest screening test in practice. Any patient with a value >100mg/dL should be referred to the patient’s primary care physician. Any patient who develops diabetes symptoms—including polyuria, polydipsia, and weight loss—should be tested immediately. The hemoglobin A1C test is not recommended for screening.
Table 2
Risk factors identified for diabetes or prediabetes
| American Diabetes Association (ADA) |
|
| American Association of Clinical Endocrinologists |
|
Clinical presentation
Screening detects overt diabetes and can identify prediabetes. Prediabetes includes conditions of impaired fasting glucose (IFG) or impaired glucose tolerance (IGT). IFG is defined as a fasting glucose of 100 to 125 mg/dL, and IGT is defined as having a 2-hour glucose of 140 to 199 mg/dL on an OGTT.
Approximately one-quarter of the adult population has prediabetes, and interventions can prevent the progression of prediabetes to overt diabetes and reverse prediabetes. The Diabetes Prevention Trial found that lifestyle measures—including exercise and diet—were most effective, with a 53% reduction in the rate of progression to diabetes.6 Metformin also was effective, but less so than lifestyle measures alone.
Treatment slows the development or progression of microvascular complications, such as retinopathy, nephropathy, and neuropathy. Aggressive treatment of comorbid conditions, including hyperlipidemia and hypertension, also reduces the risk of cardiovascular events.
Drug brand names
- Aripiprazole • Abilify
- Metformin • Glucophage
- Ziprasidone • Geodon
Related resources
- American Diabetes Association. Diabetes risk calculator. www.diabetes.org/risk-test.jsp.
- Dagogo-Jack S. The role of antipsychotic agents in the development of diabetes mellitus. Nat Clin Pract Endocrinol Metab. 2009;5(1):22-23. Quick, up-to-date review of the association between atypical antipsychotics and diabetes mellitus.
Disclosure
Dr. Keenan reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
2. U.S. Preventive Services Task Force. Screening for type 2 diabetes mellitus in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2008;148(11):846-854.
3. American Diabetes Association. Standards of medical care in diabetes—2009. Diabetes Care. 2009;32(suppl 1):S13-61.
4. Rodbard HW, Blonde L, Braithwaite SS, et al. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the management of diabetes mellitus. Endocr Pract. 2007;13(suppl 1):1-68.
5. Rydén L, Standl E, Bartnik M, et al. Guidelines on diabetes, pre-diabetes and cardiovascular diseases: executive summary. The Task Force on Diabetes and Cardio-vascular Diseases of the European Society of Cardiology (ESC) and of the European Association for the Study of Diabetes (EASD). Eur Heart J. 2007;28(1):88-136.
6. Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393-403.
1. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
2. U.S. Preventive Services Task Force. Screening for type 2 diabetes mellitus in adults: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2008;148(11):846-854.
3. American Diabetes Association. Standards of medical care in diabetes—2009. Diabetes Care. 2009;32(suppl 1):S13-61.
4. Rodbard HW, Blonde L, Braithwaite SS, et al. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the management of diabetes mellitus. Endocr Pract. 2007;13(suppl 1):1-68.
5. Rydén L, Standl E, Bartnik M, et al. Guidelines on diabetes, pre-diabetes and cardiovascular diseases: executive summary. The Task Force on Diabetes and Cardio-vascular Diseases of the European Society of Cardiology (ESC) and of the European Association for the Study of Diabetes (EASD). Eur Heart J. 2007;28(1):88-136.
6. Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393-403.
Clozapine drug levels guide dosing
Finding the best clozapine dosage for your psychotic patient can be challenging because any given dose of the drug yields highly variable clozapine serum levels. This interindividual variability reflects clozapine’s complex metabolism.
Obtaining serum levels will help you determine if your patient remains psychotic because of insufficient dosing or if your asymptomatic patient can safely receive a lower dose to minimize side effects without risking psychotic relapse. Following these guidelines will help you make the ban use of clozapine drug levels.
Obtaining clozapine levels
Measure clozapine as steady-state trough levels. I usually draw them 12 hours after the last dose (such as in the morning after the nightly dose) and several days after treatment begins.
When you order a clozapine level, most laboratories report 3 numbers: clozapine, norclozapine, and their sum. The literature addresses only the clinical use of clozapine levels and ignores the much less active metabolite, norclozapine.
Interpreting clozapine levels
Although there is no simple relationship among clozapine levels, therapeutic efficacy, and toxicity, a randomized clinical trial of patients with chronic schizophrenia1 compared 3 non-overlapping ranges and found:
- “medium” range (200 to 300 ng/mL) is a good initial target
- low range (50 to 150 ng/mL) is not as effective as medium or high levels
- high range (350 to 450 ng/mL) can be tried if clinical response is insufficient, although the high range was no more effective than the medium range
- overy high levels (ie >1,000 ng/mL combined clozapine and norclozapine levels) have no proven benefit and increase seizure risk.
These guidelines are based on bid or tid dosing. If your patient receives clozapine only at night, take into account the higher morning level compared with the same dose administered on a split schedule.
Adjusting clozapine dose
Now that you have an accurate drug level, take advantage of clozapine’s linear pharmacokinetics. If you double the dose, you double the level; if you halve the dose, you halve the level.
For example, consider the case of a schizophrenia patient who remains psychotic despite a clozapine dose of 200 mg bid (400 mg/d). His clozapine level is 100 ng/mL (ie, low range) and his norclozapine level is 50 ng/mL. This patient would need double his dose (800 mg/d) to achieve a clozapine level at the low end of the medium range (200 ng/mL). Note that the norclozapine level is ignored for this calculation.
Reference
1. VanderZwaag C, McGee M, McEvoy JP, et al. Response of patients with treatment-refractory schizophrenia to clozapine within three serum level ranges. Am J Psychiatry. 1996;153:1579-1584.
Finding the best clozapine dosage for your psychotic patient can be challenging because any given dose of the drug yields highly variable clozapine serum levels. This interindividual variability reflects clozapine’s complex metabolism.
Obtaining serum levels will help you determine if your patient remains psychotic because of insufficient dosing or if your asymptomatic patient can safely receive a lower dose to minimize side effects without risking psychotic relapse. Following these guidelines will help you make the ban use of clozapine drug levels.
Obtaining clozapine levels
Measure clozapine as steady-state trough levels. I usually draw them 12 hours after the last dose (such as in the morning after the nightly dose) and several days after treatment begins.
When you order a clozapine level, most laboratories report 3 numbers: clozapine, norclozapine, and their sum. The literature addresses only the clinical use of clozapine levels and ignores the much less active metabolite, norclozapine.
Interpreting clozapine levels
Although there is no simple relationship among clozapine levels, therapeutic efficacy, and toxicity, a randomized clinical trial of patients with chronic schizophrenia1 compared 3 non-overlapping ranges and found:
- “medium” range (200 to 300 ng/mL) is a good initial target
- low range (50 to 150 ng/mL) is not as effective as medium or high levels
- high range (350 to 450 ng/mL) can be tried if clinical response is insufficient, although the high range was no more effective than the medium range
- overy high levels (ie >1,000 ng/mL combined clozapine and norclozapine levels) have no proven benefit and increase seizure risk.
These guidelines are based on bid or tid dosing. If your patient receives clozapine only at night, take into account the higher morning level compared with the same dose administered on a split schedule.
Adjusting clozapine dose
Now that you have an accurate drug level, take advantage of clozapine’s linear pharmacokinetics. If you double the dose, you double the level; if you halve the dose, you halve the level.
For example, consider the case of a schizophrenia patient who remains psychotic despite a clozapine dose of 200 mg bid (400 mg/d). His clozapine level is 100 ng/mL (ie, low range) and his norclozapine level is 50 ng/mL. This patient would need double his dose (800 mg/d) to achieve a clozapine level at the low end of the medium range (200 ng/mL). Note that the norclozapine level is ignored for this calculation.
Finding the best clozapine dosage for your psychotic patient can be challenging because any given dose of the drug yields highly variable clozapine serum levels. This interindividual variability reflects clozapine’s complex metabolism.
Obtaining serum levels will help you determine if your patient remains psychotic because of insufficient dosing or if your asymptomatic patient can safely receive a lower dose to minimize side effects without risking psychotic relapse. Following these guidelines will help you make the ban use of clozapine drug levels.
Obtaining clozapine levels
Measure clozapine as steady-state trough levels. I usually draw them 12 hours after the last dose (such as in the morning after the nightly dose) and several days after treatment begins.
When you order a clozapine level, most laboratories report 3 numbers: clozapine, norclozapine, and their sum. The literature addresses only the clinical use of clozapine levels and ignores the much less active metabolite, norclozapine.
Interpreting clozapine levels
Although there is no simple relationship among clozapine levels, therapeutic efficacy, and toxicity, a randomized clinical trial of patients with chronic schizophrenia1 compared 3 non-overlapping ranges and found:
- “medium” range (200 to 300 ng/mL) is a good initial target
- low range (50 to 150 ng/mL) is not as effective as medium or high levels
- high range (350 to 450 ng/mL) can be tried if clinical response is insufficient, although the high range was no more effective than the medium range
- overy high levels (ie >1,000 ng/mL combined clozapine and norclozapine levels) have no proven benefit and increase seizure risk.
These guidelines are based on bid or tid dosing. If your patient receives clozapine only at night, take into account the higher morning level compared with the same dose administered on a split schedule.
Adjusting clozapine dose
Now that you have an accurate drug level, take advantage of clozapine’s linear pharmacokinetics. If you double the dose, you double the level; if you halve the dose, you halve the level.
For example, consider the case of a schizophrenia patient who remains psychotic despite a clozapine dose of 200 mg bid (400 mg/d). His clozapine level is 100 ng/mL (ie, low range) and his norclozapine level is 50 ng/mL. This patient would need double his dose (800 mg/d) to achieve a clozapine level at the low end of the medium range (200 ng/mL). Note that the norclozapine level is ignored for this calculation.
Reference
1. VanderZwaag C, McGee M, McEvoy JP, et al. Response of patients with treatment-refractory schizophrenia to clozapine within three serum level ranges. Am J Psychiatry. 1996;153:1579-1584.
Reference
1. VanderZwaag C, McGee M, McEvoy JP, et al. Response of patients with treatment-refractory schizophrenia to clozapine within three serum level ranges. Am J Psychiatry. 1996;153:1579-1584.
Is Darwin still relevant? Advanced human brain breaks evolutionary rules
You may have noticed the buzz about Charles Darwin in the news: 2009 marks the 200th anniversary of his birth and the 150th anniversary of his monumental description of evolution in On the Origin of Species. Celebrations are scheduled around the world to honor the scientist who coined the phrase “natural selection” to explain the heritable process by which adaptive evolution occurs.
But is Darwin’s theory of evolution still relevant? The “game-changer” that is transforming evolution is the genetic mutation that led to dramatic growth in the primate cortex—especially the frontal lobe—culminating in the emergence of Homo sapiens. The overdeveloped brain that has helped our species adapt and survive may be transforming us into predators of all other species and a hazard to our planet.
Survival regardless of ‘fitness’
Humans are discovering so much about biology and treatment of disease that we are disrupting natural selection and undermining the tyranny of “survival of the fittest.” A triumph of modern medicine (the antithesis of eugenics) is salvaging those who in Darwin’s day would have died and allowing them to survive and perpetuate their genes:
- Children born with metabolic errors no longer are doomed to succumb before their childbearing years.
- Premature 1-pound infants who never would have survived before are routinely doing so now.
- Women can conceive in their 60s, well after menopause.
- Medical advances have enabled humans to parry the deadly assaults of bacteria, viruses, and parasites.
- Future scientific advances will certainly include genetic engineering, which will eliminate the sometimes grim determinism of heredity.
Conversely, humans’ intelligence has enabled us to wreak havoc on other species. Tens of thousands of animals and plants have vanished because flourishing humans have survived and wittingly or unwittingly exploited, polluted, and injured the environment. Of course, a pathologic evolution may backfire on us, despite our highly evolved brain. However, humans may still adapt and survive, albeit in a dramatically altered, even inhospitable world. Or maybe not, as war and weapons of mass destruction appear to be unique inventions of the human brain.
Darwin’s psychiatric challenges
What does this have to do with psychiatry? Disorders of thought, emotions, cognition, and volition are probably the price humans paid for a dramatically evolved brain. Evolutionary psychiatrists have linked these disorders to traits that may have survival value in carriers but cause maladaptive behaviors in some individuals.
Darwin’s own struggles with disabling mental illness1-5 are perhaps the most eloquent testimonial that classic evolutionary principles may not apply to humans. His symptoms included panic attacks, agoraphobia, social anxiety, depersonalization, obsessions and compulsions, depression, suicidal impulses, visual hallucinations, and psychosomatic complaints (eczema, dizziness, and irritable bowel syndrome). Beginning in his 20s, he confined himself to his home for decades, undergoing futile interventions (such as water therapy) and taking antiquated remedies for his afflictions.
Yet Darwin’s genius was never compromised by his lack of psychiatric or physical fitness. His explanation for evolution—which made him one of the most famous scientists of all time—emerged in the midst of and despite his overwhelming health problems.
Thus, Darwin’s achievements embody the notion that the mutation that led to the hyper-developed human brain is the nemesis of evolution as he conceived it. He also serves as an inspiration to everyone who suffers from mental illness. His landmark contribution to science is a forceful rebuttal to anyone who regards psychiatric illness as synonymous with “lack of fitness.”
Happy birthday, Darwin!
1. Barlow N, ed. The autobiography of Charles Darwin, 1809-1882. London, UK: Collins; 1958.
2. Colp R, Jr. To be an invalid: the illness of Charles Darwin. Chicago, IL: The University of Chicago Press; 1977.
3. Desmond A, Moore J. Darwin: the life of a tormented evolutionist. New York, NY: Warner Books; 1991.
4. Barloon T, Noyes R. Charles Darwin and panic disorder. JAMA. 1997;277(2):138-141.
5. Picover CA. Strange brains and genius: the secret lives of eccentric scientists and madmen. New York, NY: Quill William Morrow; 1998.

Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, contact Dr. Nasrallah at [email protected] or click here.
Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, contact Dr. Nasrallah at [email protected] or click here.
Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, contact Dr. Nasrallah at [email protected] or click here.
You may have noticed the buzz about Charles Darwin in the news: 2009 marks the 200th anniversary of his birth and the 150th anniversary of his monumental description of evolution in On the Origin of Species. Celebrations are scheduled around the world to honor the scientist who coined the phrase “natural selection” to explain the heritable process by which adaptive evolution occurs.
But is Darwin’s theory of evolution still relevant? The “game-changer” that is transforming evolution is the genetic mutation that led to dramatic growth in the primate cortex—especially the frontal lobe—culminating in the emergence of Homo sapiens. The overdeveloped brain that has helped our species adapt and survive may be transforming us into predators of all other species and a hazard to our planet.
Survival regardless of ‘fitness’
Humans are discovering so much about biology and treatment of disease that we are disrupting natural selection and undermining the tyranny of “survival of the fittest.” A triumph of modern medicine (the antithesis of eugenics) is salvaging those who in Darwin’s day would have died and allowing them to survive and perpetuate their genes:
- Children born with metabolic errors no longer are doomed to succumb before their childbearing years.
- Premature 1-pound infants who never would have survived before are routinely doing so now.
- Women can conceive in their 60s, well after menopause.
- Medical advances have enabled humans to parry the deadly assaults of bacteria, viruses, and parasites.
- Future scientific advances will certainly include genetic engineering, which will eliminate the sometimes grim determinism of heredity.
Conversely, humans’ intelligence has enabled us to wreak havoc on other species. Tens of thousands of animals and plants have vanished because flourishing humans have survived and wittingly or unwittingly exploited, polluted, and injured the environment. Of course, a pathologic evolution may backfire on us, despite our highly evolved brain. However, humans may still adapt and survive, albeit in a dramatically altered, even inhospitable world. Or maybe not, as war and weapons of mass destruction appear to be unique inventions of the human brain.
Darwin’s psychiatric challenges
What does this have to do with psychiatry? Disorders of thought, emotions, cognition, and volition are probably the price humans paid for a dramatically evolved brain. Evolutionary psychiatrists have linked these disorders to traits that may have survival value in carriers but cause maladaptive behaviors in some individuals.
Darwin’s own struggles with disabling mental illness1-5 are perhaps the most eloquent testimonial that classic evolutionary principles may not apply to humans. His symptoms included panic attacks, agoraphobia, social anxiety, depersonalization, obsessions and compulsions, depression, suicidal impulses, visual hallucinations, and psychosomatic complaints (eczema, dizziness, and irritable bowel syndrome). Beginning in his 20s, he confined himself to his home for decades, undergoing futile interventions (such as water therapy) and taking antiquated remedies for his afflictions.
Yet Darwin’s genius was never compromised by his lack of psychiatric or physical fitness. His explanation for evolution—which made him one of the most famous scientists of all time—emerged in the midst of and despite his overwhelming health problems.
Thus, Darwin’s achievements embody the notion that the mutation that led to the hyper-developed human brain is the nemesis of evolution as he conceived it. He also serves as an inspiration to everyone who suffers from mental illness. His landmark contribution to science is a forceful rebuttal to anyone who regards psychiatric illness as synonymous with “lack of fitness.”
Happy birthday, Darwin!
You may have noticed the buzz about Charles Darwin in the news: 2009 marks the 200th anniversary of his birth and the 150th anniversary of his monumental description of evolution in On the Origin of Species. Celebrations are scheduled around the world to honor the scientist who coined the phrase “natural selection” to explain the heritable process by which adaptive evolution occurs.
But is Darwin’s theory of evolution still relevant? The “game-changer” that is transforming evolution is the genetic mutation that led to dramatic growth in the primate cortex—especially the frontal lobe—culminating in the emergence of Homo sapiens. The overdeveloped brain that has helped our species adapt and survive may be transforming us into predators of all other species and a hazard to our planet.
Survival regardless of ‘fitness’
Humans are discovering so much about biology and treatment of disease that we are disrupting natural selection and undermining the tyranny of “survival of the fittest.” A triumph of modern medicine (the antithesis of eugenics) is salvaging those who in Darwin’s day would have died and allowing them to survive and perpetuate their genes:
- Children born with metabolic errors no longer are doomed to succumb before their childbearing years.
- Premature 1-pound infants who never would have survived before are routinely doing so now.
- Women can conceive in their 60s, well after menopause.
- Medical advances have enabled humans to parry the deadly assaults of bacteria, viruses, and parasites.
- Future scientific advances will certainly include genetic engineering, which will eliminate the sometimes grim determinism of heredity.
Conversely, humans’ intelligence has enabled us to wreak havoc on other species. Tens of thousands of animals and plants have vanished because flourishing humans have survived and wittingly or unwittingly exploited, polluted, and injured the environment. Of course, a pathologic evolution may backfire on us, despite our highly evolved brain. However, humans may still adapt and survive, albeit in a dramatically altered, even inhospitable world. Or maybe not, as war and weapons of mass destruction appear to be unique inventions of the human brain.
Darwin’s psychiatric challenges
What does this have to do with psychiatry? Disorders of thought, emotions, cognition, and volition are probably the price humans paid for a dramatically evolved brain. Evolutionary psychiatrists have linked these disorders to traits that may have survival value in carriers but cause maladaptive behaviors in some individuals.
Darwin’s own struggles with disabling mental illness1-5 are perhaps the most eloquent testimonial that classic evolutionary principles may not apply to humans. His symptoms included panic attacks, agoraphobia, social anxiety, depersonalization, obsessions and compulsions, depression, suicidal impulses, visual hallucinations, and psychosomatic complaints (eczema, dizziness, and irritable bowel syndrome). Beginning in his 20s, he confined himself to his home for decades, undergoing futile interventions (such as water therapy) and taking antiquated remedies for his afflictions.
Yet Darwin’s genius was never compromised by his lack of psychiatric or physical fitness. His explanation for evolution—which made him one of the most famous scientists of all time—emerged in the midst of and despite his overwhelming health problems.
Thus, Darwin’s achievements embody the notion that the mutation that led to the hyper-developed human brain is the nemesis of evolution as he conceived it. He also serves as an inspiration to everyone who suffers from mental illness. His landmark contribution to science is a forceful rebuttal to anyone who regards psychiatric illness as synonymous with “lack of fitness.”
Happy birthday, Darwin!
1. Barlow N, ed. The autobiography of Charles Darwin, 1809-1882. London, UK: Collins; 1958.
2. Colp R, Jr. To be an invalid: the illness of Charles Darwin. Chicago, IL: The University of Chicago Press; 1977.
3. Desmond A, Moore J. Darwin: the life of a tormented evolutionist. New York, NY: Warner Books; 1991.
4. Barloon T, Noyes R. Charles Darwin and panic disorder. JAMA. 1997;277(2):138-141.
5. Picover CA. Strange brains and genius: the secret lives of eccentric scientists and madmen. New York, NY: Quill William Morrow; 1998.
1. Barlow N, ed. The autobiography of Charles Darwin, 1809-1882. London, UK: Collins; 1958.
2. Colp R, Jr. To be an invalid: the illness of Charles Darwin. Chicago, IL: The University of Chicago Press; 1977.
3. Desmond A, Moore J. Darwin: the life of a tormented evolutionist. New York, NY: Warner Books; 1991.
4. Barloon T, Noyes R. Charles Darwin and panic disorder. JAMA. 1997;277(2):138-141.
5. Picover CA. Strange brains and genius: the secret lives of eccentric scientists and madmen. New York, NY: Quill William Morrow; 1998.
The treatment-resistant catatonia patient
Case: Worsening psychosis
Ms. R, age 21, is admitted to our psychiatric facility while experiencing paranoid delusions and auditory hallucinations. Upon admission, she is agitated and her mood is labile.
Ms. R has 4 previous brief psychiatric admissions and was diagnosed with schizoaffective disorder, bipolar type and moderate mental retardation. Her family history is positive for psychiatric illness, as her mother was diagnosed with schizophrenia. Prior to admission, Ms. R was taking ziprasidone, 160 mg/d, and lithium, 450 mg/d, for 11 months. Both were discontinued during the first week of admission because Ms. R was not responding.
During this admission, the treating psychiatrist assesses Ms. R using the Schedules for Clinicians’ Interview in Psychiatry (SCIP), an instrument developed by the lead author (AA) for psychiatrists to use in conjunction with their routine clinical interviews in inpatient and outpatient settings (see Related Resources). The SCIP includes a 25-question screening section and a diagnostic section that consists of 7 modules that represent major psychiatric diagnoses defined by DSM and International Classification of Diseases criteria.1
During the first week of admission, we monitor Ms. R and administer haloperidol as needed, 10 mg total. Eight days after admission, she develops severe catatonia. On the catatonia scale of the SCIP, Ms. R scores the maximum on measures of immobility, catalepsy/waxy flexibility, and mutism (Table).
How would you treat Ms. R’s catatonia?
Table
Patient’s catatonia symptoms: Response to pharmacotherapy
| Lorazepam only | Lorazepam + risperidone | Risperidone oral only | Risperidone long-acting injection only | |
|---|---|---|---|---|
| Dosage(s) | 7 mg total over 7 days | Lorazepam: 4 mg/d Risperidone: 4 mg/d | 8 mg/d | 37.5 mg every 2 weeks |
| Scores on SCIP catatonia scale:* | ||||
| Immobility | 2 | 1 | 0 | 0 |
| Catalepsy/waxy flexibility† | 2 | 1 | 0 | 0 |
| Mutism | 2 | 0 | 0 | 0 |
| Total score | 6 | 2 | 0 | 0 |
| *Scale of 0 to 2, with 0=none, 1=less than half the time, and 2=more than half the time. Symptoms are evaluated over a 1-day period | ||||
| †For this category, 0=none, 1=brief (usually 1 minute | ||||
| SCIP: Schedules for Clinicians’ Interview in Psychiatry | ||||
The authors’ observations
DSM-IV-TR recognizes catatonia as a schizophrenia subtype, as a descriptor for mania and major depression, and as being caused by various medical conditions, such as neuroleptic malignant syndrome, encephalopathy, or renal failure.2 Kahlbaum initially described catatonia in 1873 as a brain disease characterized by motor abnormalities such as akinesia, rigidity, negativism, mutism, grimacing, posturing, catalepsy, waxy flexibility, and verbigerations.3 Catatonia is characterized by hypo- and hyperkinetic features. Catalepsy, stupor, rigidity, and catatonic posturing with waxy flexibility might alternate with violent catatonic excitement.4
Catatonia can be life-threatening; patients might not be able to eat or chew food, which puts them at risk for aspiration. Those with immobility might not move to urinate or defecate. During the first half of the 20th century, catatonia was documented in up to 50% of patients with schizophrenia.5 Since then, the incidence of catatonia has decreased, possibly the result of advances in psychopharmacology.6
Two days after Ms. R develops catatonia, we transfer her to a local hospital for evaluation to rule out a medical cause of her catatonic symptoms.
EVALUATION: No medical cause
At the hospital, physical examination, electroencephalography, drug screening, and liver and thyroid function tests are within normal limits, eliminating an organic cause of Ms. R’s catatonia. MRI of the head shows a 3-mm mass at the base of the infundibulum, which is unchanged from a prior MRI. Ms. R received 7 mg total of lorazepam over 4 days without relief of her catatonia. She is transferred back to our facility.
The authors’ observations
Benzodiazepines and ECT are effective treatments for catatonia.7 Benzodiazepines are considered first-line treatment because of their efficacy and favorable side-effect profile.7 Lorazepam frequently is used to treat catatonia in the short term.8 Long-term use of benzodiazepines, however, is associated with tolerance, addiction, and rebound phenomena.8,9
Patients with catatonia who do not respond to benzodiazepines may benefit from ECT.9 ECT can cause serious side effects, however, including memory impairment, confusion, delirium, and cardiac arrhythmias.10
Atypical antipsychotics may alleviate motor symptoms of catatonia by virtue of their 5-HT2A receptor antagonistic action.9 In 2 case reports, risperidone successfully treated catatonia.4,11 Kopala et al11 found risperidone, 4 mg/d, was effective in treating severe, first-episode catatonic schizophrenia in a neuroleptic-naive young man. This efficacy was sustained over a 3.5-year outpatient follow-up.
In another report, risperidone, 6 mg/d, effectively treated catatonia and prevented further episodes in a patient with schizophrenia who developed severe catatonia after receiving adequate treatment for Lyme disease with encephalitis.4 Two relapses of catatonic syndrome occurred when risperidone was reduced to 2 mg/d, and remission occurred after risperidone was increased to 6 mg/d. Risperidone’s antagonistic activity of the 5-HT2/D2 receptors may be relevant to its anticatatonic effect.12
Other atypical antipsychotics—ziprasidone and olanzapine—also have been shown to be effective in treating catatonia. Levy et al13 reported successful treatment of a catatonic state (with catalepsy, stupor, and mutism) using intramuscular ziprasidone followed by oral ziprasidone. A data analysis by Martenyi et al14 showed olanzapine to be effective in treating nonspecific signs and symptoms of catatonia, as measured by the Positive and Negative Syndrome Scale.
TREATMENT: Trying risperidone
Based on case reports showing risperidone’s efficacy for catatonia, we start Ms. R on risperidone, 4 mg/d, and lorazepam, 4 mg/d. Eight days later, her catatonic symptoms decrease substantially—she scores 2/6 on the SCIP catatonia scale (Table)—and she starts to talk with the staff.
We continue this regimen for 30 days, then discontinue lorazepam to avoid long-term side effects—such as dependence—and titrate risperidone to 8 mg/d. Ms. R continues to improve while taking risperidone only. Twenty-three days after stopping lorazepam, she is free of catatonic symptoms, scoring 0/6 on the SCIP catatonia scale.
We discharge Ms. R on risperidone. Because she has a history of medication nonadherence, we prescribe risperidone long-acting injection, 37.5 mg every 2 weeks, while continuing oral risperidone for 3 weeks after the first injection. She does well on this medication, experiencing no catatonic symptoms or adverse effects over the next 15 months as measured by the SCIP assessment.
The authors’ observations
This is the third case report in the literature to show that risperidone is effective in short- and long-term treatment of catatonia.4,11 Although Ms. R’s initial response can be attributed at least partially to lorazepam—which is known to be effective in treating catatonia—she continued to show improvement while taking risperidone only and remained free from catatonic symptoms for 15 months, until she was readmitted for reasons unrelated to catatonia.
We recommend using risperidone to treat catatonia in patients who do not respond to a benzodiazepine, especially those with other psychotic symptoms such as delusions or hallucinations. While administering risperidone, watch for long-term side effects, such as hyperlipidemia, weight gain, and diabetes. For catatonia in patients who cannot tolerate risperidone, consider olanzapine or ziprasidone.
- Schedules for Clinicians’ Interview in Psychiatry (SCIP). Available from Ahmed Aboraya, [email protected].
- Valevski A, Loebl T, Keren T, et al. Response of catatonia to risperidone: two case reports. Clin Neuropharmacol. 2001;24(4):228-231.
- Van Den Eede F, Van Hecke J, Van Dalfsen A, et al. The use of atypical antipsychotics in the treatment of catatonia. Eur Psychiatry. 2005;20(5-6):422-429.
- Haloperidol • Haldol
- Lithium • Eskalith, Lithobid
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Risperidone • Risperdal
- Risperidone long-acting injection • Risperdal Consta
- Ziprasidone • Geodon
The authors have no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Aboraya A, Tien A. Schedules for Clinicians’ Interviews in Psychiatry (SCIP): work in progress. International Journal of Mental Health and Addiction. Available at: http://www.ijma-journal.com/pdf/c01a09.pdf. Accessed February 4, 2009.
2. Diagnostic and statistical manual of disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
3. Kahlbaum KL. In: Levi Y, Pridon T, trans. Catatonia. Baltimore, MD: Johns Hopkins University Press; 1973.
4. Hesslinger B, Walden J, Normann C. Acute and long-term treatment of catatonia with risperidone. Pharmacopsychiatry. 2001;34(1):25-26.
5. Bleuler E. Dementia praecox. New York, NY: International University Press; 1950.
6. Blumer D. Catatonia and the neuroleptics: psychobiologic significance of remote and recent findings. Compr Psychiatry. 1997;38(4):193-201.
7. Bush G, Fink M, Petrides G, et al. Catatonia. II Treatment with lorazepam and electroconvulsive therapy. Acta Psychiatr Scand. 1996;93(2):137-143.
8. Duggal HS, Gandotra G. Risperidone treatment of periodic catatonia. Can J Psychiatry. 2005;50(4):241-242.
9. Duggal HS. Risperidone treatment of febrile catatonia in first-episode psychosis. Gen Hosp Psychiatry. 2005;27(1):80-81.
10. Rudorfer M, Henry M, Sackeim H. Electroconvulsive therapy. In: Tasman A, Kay J, Lieberman JA, eds. Psychiatry: therapeutics. London, UK: John Wiley & Sons; 2003:167-203.
11. Kopala LC, Caudle C. Acute and longer-term effects of risperidone in a case of first-episode catatonic schizophrenia. J Psychopharmacol. 1998;12(3):314-317.
12. Poyurousky M, Bergman J, Weizman A. Risperidone in the treatment of catatonia in a schizophrenic patient. Isr J Psychiatry Relat Sci. 1997;34(4):323-324.
13. Levy WO, Nunez CY. Use of ziprasidone to treat bipolar-associated catatonia. Bipolar Disord. 2004;6(2):166-167.
14. Martenyi F, Metcalfe S, Schausberger B, et al. An efficacy analysis of olanzapine treatment data in schizophrenia patients with catatonic signs and symptoms. J Clin Psychiatry. 2001;62(suppl 2):225-227.
Case: Worsening psychosis
Ms. R, age 21, is admitted to our psychiatric facility while experiencing paranoid delusions and auditory hallucinations. Upon admission, she is agitated and her mood is labile.
Ms. R has 4 previous brief psychiatric admissions and was diagnosed with schizoaffective disorder, bipolar type and moderate mental retardation. Her family history is positive for psychiatric illness, as her mother was diagnosed with schizophrenia. Prior to admission, Ms. R was taking ziprasidone, 160 mg/d, and lithium, 450 mg/d, for 11 months. Both were discontinued during the first week of admission because Ms. R was not responding.
During this admission, the treating psychiatrist assesses Ms. R using the Schedules for Clinicians’ Interview in Psychiatry (SCIP), an instrument developed by the lead author (AA) for psychiatrists to use in conjunction with their routine clinical interviews in inpatient and outpatient settings (see Related Resources). The SCIP includes a 25-question screening section and a diagnostic section that consists of 7 modules that represent major psychiatric diagnoses defined by DSM and International Classification of Diseases criteria.1
During the first week of admission, we monitor Ms. R and administer haloperidol as needed, 10 mg total. Eight days after admission, she develops severe catatonia. On the catatonia scale of the SCIP, Ms. R scores the maximum on measures of immobility, catalepsy/waxy flexibility, and mutism (Table).
How would you treat Ms. R’s catatonia?
Table
Patient’s catatonia symptoms: Response to pharmacotherapy
| Lorazepam only | Lorazepam + risperidone | Risperidone oral only | Risperidone long-acting injection only | |
|---|---|---|---|---|
| Dosage(s) | 7 mg total over 7 days | Lorazepam: 4 mg/d Risperidone: 4 mg/d | 8 mg/d | 37.5 mg every 2 weeks |
| Scores on SCIP catatonia scale:* | ||||
| Immobility | 2 | 1 | 0 | 0 |
| Catalepsy/waxy flexibility† | 2 | 1 | 0 | 0 |
| Mutism | 2 | 0 | 0 | 0 |
| Total score | 6 | 2 | 0 | 0 |
| *Scale of 0 to 2, with 0=none, 1=less than half the time, and 2=more than half the time. Symptoms are evaluated over a 1-day period | ||||
| †For this category, 0=none, 1=brief (usually 1 minute | ||||
| SCIP: Schedules for Clinicians’ Interview in Psychiatry | ||||
The authors’ observations
DSM-IV-TR recognizes catatonia as a schizophrenia subtype, as a descriptor for mania and major depression, and as being caused by various medical conditions, such as neuroleptic malignant syndrome, encephalopathy, or renal failure.2 Kahlbaum initially described catatonia in 1873 as a brain disease characterized by motor abnormalities such as akinesia, rigidity, negativism, mutism, grimacing, posturing, catalepsy, waxy flexibility, and verbigerations.3 Catatonia is characterized by hypo- and hyperkinetic features. Catalepsy, stupor, rigidity, and catatonic posturing with waxy flexibility might alternate with violent catatonic excitement.4
Catatonia can be life-threatening; patients might not be able to eat or chew food, which puts them at risk for aspiration. Those with immobility might not move to urinate or defecate. During the first half of the 20th century, catatonia was documented in up to 50% of patients with schizophrenia.5 Since then, the incidence of catatonia has decreased, possibly the result of advances in psychopharmacology.6
Two days after Ms. R develops catatonia, we transfer her to a local hospital for evaluation to rule out a medical cause of her catatonic symptoms.
EVALUATION: No medical cause
At the hospital, physical examination, electroencephalography, drug screening, and liver and thyroid function tests are within normal limits, eliminating an organic cause of Ms. R’s catatonia. MRI of the head shows a 3-mm mass at the base of the infundibulum, which is unchanged from a prior MRI. Ms. R received 7 mg total of lorazepam over 4 days without relief of her catatonia. She is transferred back to our facility.
The authors’ observations
Benzodiazepines and ECT are effective treatments for catatonia.7 Benzodiazepines are considered first-line treatment because of their efficacy and favorable side-effect profile.7 Lorazepam frequently is used to treat catatonia in the short term.8 Long-term use of benzodiazepines, however, is associated with tolerance, addiction, and rebound phenomena.8,9
Patients with catatonia who do not respond to benzodiazepines may benefit from ECT.9 ECT can cause serious side effects, however, including memory impairment, confusion, delirium, and cardiac arrhythmias.10
Atypical antipsychotics may alleviate motor symptoms of catatonia by virtue of their 5-HT2A receptor antagonistic action.9 In 2 case reports, risperidone successfully treated catatonia.4,11 Kopala et al11 found risperidone, 4 mg/d, was effective in treating severe, first-episode catatonic schizophrenia in a neuroleptic-naive young man. This efficacy was sustained over a 3.5-year outpatient follow-up.
In another report, risperidone, 6 mg/d, effectively treated catatonia and prevented further episodes in a patient with schizophrenia who developed severe catatonia after receiving adequate treatment for Lyme disease with encephalitis.4 Two relapses of catatonic syndrome occurred when risperidone was reduced to 2 mg/d, and remission occurred after risperidone was increased to 6 mg/d. Risperidone’s antagonistic activity of the 5-HT2/D2 receptors may be relevant to its anticatatonic effect.12
Other atypical antipsychotics—ziprasidone and olanzapine—also have been shown to be effective in treating catatonia. Levy et al13 reported successful treatment of a catatonic state (with catalepsy, stupor, and mutism) using intramuscular ziprasidone followed by oral ziprasidone. A data analysis by Martenyi et al14 showed olanzapine to be effective in treating nonspecific signs and symptoms of catatonia, as measured by the Positive and Negative Syndrome Scale.
TREATMENT: Trying risperidone
Based on case reports showing risperidone’s efficacy for catatonia, we start Ms. R on risperidone, 4 mg/d, and lorazepam, 4 mg/d. Eight days later, her catatonic symptoms decrease substantially—she scores 2/6 on the SCIP catatonia scale (Table)—and she starts to talk with the staff.
We continue this regimen for 30 days, then discontinue lorazepam to avoid long-term side effects—such as dependence—and titrate risperidone to 8 mg/d. Ms. R continues to improve while taking risperidone only. Twenty-three days after stopping lorazepam, she is free of catatonic symptoms, scoring 0/6 on the SCIP catatonia scale.
We discharge Ms. R on risperidone. Because she has a history of medication nonadherence, we prescribe risperidone long-acting injection, 37.5 mg every 2 weeks, while continuing oral risperidone for 3 weeks after the first injection. She does well on this medication, experiencing no catatonic symptoms or adverse effects over the next 15 months as measured by the SCIP assessment.
The authors’ observations
This is the third case report in the literature to show that risperidone is effective in short- and long-term treatment of catatonia.4,11 Although Ms. R’s initial response can be attributed at least partially to lorazepam—which is known to be effective in treating catatonia—she continued to show improvement while taking risperidone only and remained free from catatonic symptoms for 15 months, until she was readmitted for reasons unrelated to catatonia.
We recommend using risperidone to treat catatonia in patients who do not respond to a benzodiazepine, especially those with other psychotic symptoms such as delusions or hallucinations. While administering risperidone, watch for long-term side effects, such as hyperlipidemia, weight gain, and diabetes. For catatonia in patients who cannot tolerate risperidone, consider olanzapine or ziprasidone.
- Schedules for Clinicians’ Interview in Psychiatry (SCIP). Available from Ahmed Aboraya, [email protected].
- Valevski A, Loebl T, Keren T, et al. Response of catatonia to risperidone: two case reports. Clin Neuropharmacol. 2001;24(4):228-231.
- Van Den Eede F, Van Hecke J, Van Dalfsen A, et al. The use of atypical antipsychotics in the treatment of catatonia. Eur Psychiatry. 2005;20(5-6):422-429.
- Haloperidol • Haldol
- Lithium • Eskalith, Lithobid
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Risperidone • Risperdal
- Risperidone long-acting injection • Risperdal Consta
- Ziprasidone • Geodon
The authors have no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Case: Worsening psychosis
Ms. R, age 21, is admitted to our psychiatric facility while experiencing paranoid delusions and auditory hallucinations. Upon admission, she is agitated and her mood is labile.
Ms. R has 4 previous brief psychiatric admissions and was diagnosed with schizoaffective disorder, bipolar type and moderate mental retardation. Her family history is positive for psychiatric illness, as her mother was diagnosed with schizophrenia. Prior to admission, Ms. R was taking ziprasidone, 160 mg/d, and lithium, 450 mg/d, for 11 months. Both were discontinued during the first week of admission because Ms. R was not responding.
During this admission, the treating psychiatrist assesses Ms. R using the Schedules for Clinicians’ Interview in Psychiatry (SCIP), an instrument developed by the lead author (AA) for psychiatrists to use in conjunction with their routine clinical interviews in inpatient and outpatient settings (see Related Resources). The SCIP includes a 25-question screening section and a diagnostic section that consists of 7 modules that represent major psychiatric diagnoses defined by DSM and International Classification of Diseases criteria.1
During the first week of admission, we monitor Ms. R and administer haloperidol as needed, 10 mg total. Eight days after admission, she develops severe catatonia. On the catatonia scale of the SCIP, Ms. R scores the maximum on measures of immobility, catalepsy/waxy flexibility, and mutism (Table).
How would you treat Ms. R’s catatonia?
Table
Patient’s catatonia symptoms: Response to pharmacotherapy
| Lorazepam only | Lorazepam + risperidone | Risperidone oral only | Risperidone long-acting injection only | |
|---|---|---|---|---|
| Dosage(s) | 7 mg total over 7 days | Lorazepam: 4 mg/d Risperidone: 4 mg/d | 8 mg/d | 37.5 mg every 2 weeks |
| Scores on SCIP catatonia scale:* | ||||
| Immobility | 2 | 1 | 0 | 0 |
| Catalepsy/waxy flexibility† | 2 | 1 | 0 | 0 |
| Mutism | 2 | 0 | 0 | 0 |
| Total score | 6 | 2 | 0 | 0 |
| *Scale of 0 to 2, with 0=none, 1=less than half the time, and 2=more than half the time. Symptoms are evaluated over a 1-day period | ||||
| †For this category, 0=none, 1=brief (usually 1 minute | ||||
| SCIP: Schedules for Clinicians’ Interview in Psychiatry | ||||
The authors’ observations
DSM-IV-TR recognizes catatonia as a schizophrenia subtype, as a descriptor for mania and major depression, and as being caused by various medical conditions, such as neuroleptic malignant syndrome, encephalopathy, or renal failure.2 Kahlbaum initially described catatonia in 1873 as a brain disease characterized by motor abnormalities such as akinesia, rigidity, negativism, mutism, grimacing, posturing, catalepsy, waxy flexibility, and verbigerations.3 Catatonia is characterized by hypo- and hyperkinetic features. Catalepsy, stupor, rigidity, and catatonic posturing with waxy flexibility might alternate with violent catatonic excitement.4
Catatonia can be life-threatening; patients might not be able to eat or chew food, which puts them at risk for aspiration. Those with immobility might not move to urinate or defecate. During the first half of the 20th century, catatonia was documented in up to 50% of patients with schizophrenia.5 Since then, the incidence of catatonia has decreased, possibly the result of advances in psychopharmacology.6
Two days after Ms. R develops catatonia, we transfer her to a local hospital for evaluation to rule out a medical cause of her catatonic symptoms.
EVALUATION: No medical cause
At the hospital, physical examination, electroencephalography, drug screening, and liver and thyroid function tests are within normal limits, eliminating an organic cause of Ms. R’s catatonia. MRI of the head shows a 3-mm mass at the base of the infundibulum, which is unchanged from a prior MRI. Ms. R received 7 mg total of lorazepam over 4 days without relief of her catatonia. She is transferred back to our facility.
The authors’ observations
Benzodiazepines and ECT are effective treatments for catatonia.7 Benzodiazepines are considered first-line treatment because of their efficacy and favorable side-effect profile.7 Lorazepam frequently is used to treat catatonia in the short term.8 Long-term use of benzodiazepines, however, is associated with tolerance, addiction, and rebound phenomena.8,9
Patients with catatonia who do not respond to benzodiazepines may benefit from ECT.9 ECT can cause serious side effects, however, including memory impairment, confusion, delirium, and cardiac arrhythmias.10
Atypical antipsychotics may alleviate motor symptoms of catatonia by virtue of their 5-HT2A receptor antagonistic action.9 In 2 case reports, risperidone successfully treated catatonia.4,11 Kopala et al11 found risperidone, 4 mg/d, was effective in treating severe, first-episode catatonic schizophrenia in a neuroleptic-naive young man. This efficacy was sustained over a 3.5-year outpatient follow-up.
In another report, risperidone, 6 mg/d, effectively treated catatonia and prevented further episodes in a patient with schizophrenia who developed severe catatonia after receiving adequate treatment for Lyme disease with encephalitis.4 Two relapses of catatonic syndrome occurred when risperidone was reduced to 2 mg/d, and remission occurred after risperidone was increased to 6 mg/d. Risperidone’s antagonistic activity of the 5-HT2/D2 receptors may be relevant to its anticatatonic effect.12
Other atypical antipsychotics—ziprasidone and olanzapine—also have been shown to be effective in treating catatonia. Levy et al13 reported successful treatment of a catatonic state (with catalepsy, stupor, and mutism) using intramuscular ziprasidone followed by oral ziprasidone. A data analysis by Martenyi et al14 showed olanzapine to be effective in treating nonspecific signs and symptoms of catatonia, as measured by the Positive and Negative Syndrome Scale.
TREATMENT: Trying risperidone
Based on case reports showing risperidone’s efficacy for catatonia, we start Ms. R on risperidone, 4 mg/d, and lorazepam, 4 mg/d. Eight days later, her catatonic symptoms decrease substantially—she scores 2/6 on the SCIP catatonia scale (Table)—and she starts to talk with the staff.
We continue this regimen for 30 days, then discontinue lorazepam to avoid long-term side effects—such as dependence—and titrate risperidone to 8 mg/d. Ms. R continues to improve while taking risperidone only. Twenty-three days after stopping lorazepam, she is free of catatonic symptoms, scoring 0/6 on the SCIP catatonia scale.
We discharge Ms. R on risperidone. Because she has a history of medication nonadherence, we prescribe risperidone long-acting injection, 37.5 mg every 2 weeks, while continuing oral risperidone for 3 weeks after the first injection. She does well on this medication, experiencing no catatonic symptoms or adverse effects over the next 15 months as measured by the SCIP assessment.
The authors’ observations
This is the third case report in the literature to show that risperidone is effective in short- and long-term treatment of catatonia.4,11 Although Ms. R’s initial response can be attributed at least partially to lorazepam—which is known to be effective in treating catatonia—she continued to show improvement while taking risperidone only and remained free from catatonic symptoms for 15 months, until she was readmitted for reasons unrelated to catatonia.
We recommend using risperidone to treat catatonia in patients who do not respond to a benzodiazepine, especially those with other psychotic symptoms such as delusions or hallucinations. While administering risperidone, watch for long-term side effects, such as hyperlipidemia, weight gain, and diabetes. For catatonia in patients who cannot tolerate risperidone, consider olanzapine or ziprasidone.
- Schedules for Clinicians’ Interview in Psychiatry (SCIP). Available from Ahmed Aboraya, [email protected].
- Valevski A, Loebl T, Keren T, et al. Response of catatonia to risperidone: two case reports. Clin Neuropharmacol. 2001;24(4):228-231.
- Van Den Eede F, Van Hecke J, Van Dalfsen A, et al. The use of atypical antipsychotics in the treatment of catatonia. Eur Psychiatry. 2005;20(5-6):422-429.
- Haloperidol • Haldol
- Lithium • Eskalith, Lithobid
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Risperidone • Risperdal
- Risperidone long-acting injection • Risperdal Consta
- Ziprasidone • Geodon
The authors have no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Aboraya A, Tien A. Schedules for Clinicians’ Interviews in Psychiatry (SCIP): work in progress. International Journal of Mental Health and Addiction. Available at: http://www.ijma-journal.com/pdf/c01a09.pdf. Accessed February 4, 2009.
2. Diagnostic and statistical manual of disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
3. Kahlbaum KL. In: Levi Y, Pridon T, trans. Catatonia. Baltimore, MD: Johns Hopkins University Press; 1973.
4. Hesslinger B, Walden J, Normann C. Acute and long-term treatment of catatonia with risperidone. Pharmacopsychiatry. 2001;34(1):25-26.
5. Bleuler E. Dementia praecox. New York, NY: International University Press; 1950.
6. Blumer D. Catatonia and the neuroleptics: psychobiologic significance of remote and recent findings. Compr Psychiatry. 1997;38(4):193-201.
7. Bush G, Fink M, Petrides G, et al. Catatonia. II Treatment with lorazepam and electroconvulsive therapy. Acta Psychiatr Scand. 1996;93(2):137-143.
8. Duggal HS, Gandotra G. Risperidone treatment of periodic catatonia. Can J Psychiatry. 2005;50(4):241-242.
9. Duggal HS. Risperidone treatment of febrile catatonia in first-episode psychosis. Gen Hosp Psychiatry. 2005;27(1):80-81.
10. Rudorfer M, Henry M, Sackeim H. Electroconvulsive therapy. In: Tasman A, Kay J, Lieberman JA, eds. Psychiatry: therapeutics. London, UK: John Wiley & Sons; 2003:167-203.
11. Kopala LC, Caudle C. Acute and longer-term effects of risperidone in a case of first-episode catatonic schizophrenia. J Psychopharmacol. 1998;12(3):314-317.
12. Poyurousky M, Bergman J, Weizman A. Risperidone in the treatment of catatonia in a schizophrenic patient. Isr J Psychiatry Relat Sci. 1997;34(4):323-324.
13. Levy WO, Nunez CY. Use of ziprasidone to treat bipolar-associated catatonia. Bipolar Disord. 2004;6(2):166-167.
14. Martenyi F, Metcalfe S, Schausberger B, et al. An efficacy analysis of olanzapine treatment data in schizophrenia patients with catatonic signs and symptoms. J Clin Psychiatry. 2001;62(suppl 2):225-227.
1. Aboraya A, Tien A. Schedules for Clinicians’ Interviews in Psychiatry (SCIP): work in progress. International Journal of Mental Health and Addiction. Available at: http://www.ijma-journal.com/pdf/c01a09.pdf. Accessed February 4, 2009.
2. Diagnostic and statistical manual of disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
3. Kahlbaum KL. In: Levi Y, Pridon T, trans. Catatonia. Baltimore, MD: Johns Hopkins University Press; 1973.
4. Hesslinger B, Walden J, Normann C. Acute and long-term treatment of catatonia with risperidone. Pharmacopsychiatry. 2001;34(1):25-26.
5. Bleuler E. Dementia praecox. New York, NY: International University Press; 1950.
6. Blumer D. Catatonia and the neuroleptics: psychobiologic significance of remote and recent findings. Compr Psychiatry. 1997;38(4):193-201.
7. Bush G, Fink M, Petrides G, et al. Catatonia. II Treatment with lorazepam and electroconvulsive therapy. Acta Psychiatr Scand. 1996;93(2):137-143.
8. Duggal HS, Gandotra G. Risperidone treatment of periodic catatonia. Can J Psychiatry. 2005;50(4):241-242.
9. Duggal HS. Risperidone treatment of febrile catatonia in first-episode psychosis. Gen Hosp Psychiatry. 2005;27(1):80-81.
10. Rudorfer M, Henry M, Sackeim H. Electroconvulsive therapy. In: Tasman A, Kay J, Lieberman JA, eds. Psychiatry: therapeutics. London, UK: John Wiley & Sons; 2003:167-203.
11. Kopala LC, Caudle C. Acute and longer-term effects of risperidone in a case of first-episode catatonic schizophrenia. J Psychopharmacol. 1998;12(3):314-317.
12. Poyurousky M, Bergman J, Weizman A. Risperidone in the treatment of catatonia in a schizophrenic patient. Isr J Psychiatry Relat Sci. 1997;34(4):323-324.
13. Levy WO, Nunez CY. Use of ziprasidone to treat bipolar-associated catatonia. Bipolar Disord. 2004;6(2):166-167.
14. Martenyi F, Metcalfe S, Schausberger B, et al. An efficacy analysis of olanzapine treatment data in schizophrenia patients with catatonic signs and symptoms. J Clin Psychiatry. 2001;62(suppl 2):225-227.
‘Night owls’: Reset the physiologic clock in delayed sleep phase disorder
Jason, age 16, has had difficulty with sleep initiation for 2 years. He describes going to bed at 10:30 PM on school nights but falling asleep no sooner than midnight and typically after 1:30 AM. He denies contributions from an “active mind” or environmental disturbances, and his bedroom contains no TV, computer, or other media devices. He does not sleep better with a change in environment. He denies pervasive low mood symptoms and believes his mood hinges predominantly on his ability to achieve sufficient sleep.
Once asleep, Jason generally enjoys good sleep consolidation until he needs to arise at 6:30 AM. His mother awakens him with difficulty, as he often sleeps through his alarm. He sleeps approximately 5 hours nightly during the school week, endorses impaired concentration, and often dozes during his first several classes. When he returns home from school, he finds it very difficult to resist napping.
On weekends he retires at 1 AM or later and typically falls asleep within 30 minutes. He usually awakens at noon but can sleep as late as 4:30 PM. He feels slightly more refreshed on weekends and describes his mood then as improved. During a recent spring break, he felt much better when allowed to sleep as much as he wanted.
Delayed sleep phase disorder (DSPD)—characterized by a pathological “night owl” circadian preference—is seen most commonly in adolescents and is associated with psychiatric morbidity, psychosocial impairment, and poor academic performance. Proper identification of the condition can be enhanced with a variety of assessment tools, and successful treatment requires an awareness of potential endogenous and exogenous contributors.
This article describes what is known about DSPD and uses the case example to illustrate diagnostic assessment and treatment choices. Intriguing data support various pathophysiologic explanations for DSPD (Box 1).1-6 Facilitating the adjustment of patients’ physiologic clocks is the overall goal in managing DSPD.
In individuals normally entrained to the light/dark cycle, circadian rhythms are:
- delayed by evening exposure to bright light (≥ 2,500 lux) prior to the core body temperature minimum (Tmin)
- advanced by morning light exposure after the Tmin.1
These opposing effects attune most people to the light/dark cycle, with sleep and wakefulness occurring on a conventional schedule. Persons with delayed sleep phase disorder (DSPD) live at a delayed phase that resists advancement and is incompatible with their personal and social obligations.
Theories have been proposed, but DSPD’s etiology has not been fully explained. Affected adolescents may exhibit an extreme in circadian preference. Case reports also describe DSPD emerging after traumatic brain injury.2
Intriguing evidence supports various pathophysiologic explanations for DSPD. An abnormally long intrinsic circadian period (>25 hours) was recently demonstrated during temporal isolation in 1 individual with DSPD.3 Both this case report and controlled studies describe deviations from expected relationships between the sleep/wake cycle and physiologic circadian markers. Most consistently described are longer intervals from Tmin4 to sleep offset (final rise time) in DSPD patients compared with controls.
Other research suggests:
Extreme ‘eveningness’
Because of their extreme seemingly innate preference to retire and arise at relatively late clock hours (an “eveningness” trait), school-aged patients with DSPD represent a high-risk population for problematic sleepiness. In a survey of 612 high school students, the 63% who felt they needed more sleep on school nights showed a strong eveningness preference (as assessed by questionnaire), compared with students who described getting sufficient sleep.7 Other studies have revealed psychiatric morbidity (including affective and personality disorders), psychosocial impairment, and poor academic performance associated with the condition.8-10
DSPD may affect 7% to 16% of patients presenting with insomnia complaints in sleep medicine clinics.11 The condition appears most common among young cohorts and has been reported to affect up to 7% of adolescents in the United States.12 Its high frequency in this age group may be a pathologic exaggeration of the normal tendency toward delayed timing of sleep and wakefulness linked with pubertal development.13
Sleep and wakefulness regulation
Conceptually, 2 processes govern sleep and wakefulness:
- The homeostatic drive to sleep (process S) is proportional to the duration of sleep restriction and becomes maximal at about 40 hours.
- Circadian regulation (process C) creates a drive for wakefulness that variably opposes process S and depends upon intrinsic rhythms.14
Neurons of the suprachiasmatic nucleus in the hypothalamus exert master coordination of this sleep/wake rhythm, along with other behavioral and physiologic variables.15 Because the typical intrinsic period is slightly longer than 24 hours, synchronization to the 24-hour day (entrainment) is accomplished by environmental inputs (zeitgebers, or “time givers”), the most important of which is exposure to light.16
Misalignment between endogenous circadian rhythms and the light/dark cycle can result in circadian rhythm sleep disorders, such as:
- delayed sleep timing (DSPD)
- advanced sleep timing (advanced sleep phase disorder)
- erratic sleep timing (irregular sleep/wake rhythm)
- complete dissociation from the light/dark cycle (circadian rhythm sleep disorder, free-running type).
These 4 conditions are thought to involve predominantly intrinsic mechanisms, but circadian dysrhythmias also can be induced by exogenous factors. Extreme work schedules or rapid travel across time zones can challenge the circadian system’s ability to acclimate and the individual’s ability to achieve a desired sleep schedule.17
Differential diagnosis
Because DSPD relates primarily to an aberration in timing of sleep, it is characterized as a disorder only if the individual’s preferred schedule interferes substantially with social or occupational functioning. The International Classification of Sleep Disorders (ICSD) provides detailed diagnostic criteria (Table).17
Table
Diagnostic criteria for delayed sleep phase disorder
A. Delay exists in the phase of the major sleep period in relation to desired sleep time and wake-up time, as evidenced by:
|
| B. When allowed to choose a preferred schedule, patients exhibit normal sleep quality and duration for age and maintain a delayed but stable phase of entrainment to the 24-hour sleep/wake pattern. |
| C. Monitoring with a sleep log or actigraphy (including sleep diary) for at least 7 days demonstrates a stable delay in the timing of the habitual sleep period. |
| D. The sleep disturbance is not better explained by another sleep disorder, medical or neurologic disorder, mental disorder, medication use, or substance use disorder. |
| Source: Adapted and reprinted with permission from International classification of sleep disorders. Diagnostic and coding manual. 2nd ed17 |
Depression and anxiety often manifest with sleep difficulties, as do inadequate sleep hygiene and other conditions associated with prolonged sleep initiation. According to ICSD criteria, primary insomnia can be differentiated from DSPD if the patient readily initiates and maintains sleep when allowed to sleep on his/her desired sleep/wake schedule. Accumulated evidence has largely debunked this notion, however, as polysomnographic studies have demonstrated both prolonged sleep latency and impaired sleep efficiency in DSPD patients versus matched controls.3
Assessment tools can complement the clinical history in diagnosing DSPD. Either a sleep log or actigraphy is required to demonstrate a stable phase delay, but actigraphy typically generates more reliable data.18 Actigraphs are compact “motion detectors” whose output while being worn by patients allows longitudinal assessment of sleep/wake parameters.
Eveningness tendencies of presumptive DSPD patients can be further verified with the Morningness-Eveningness Questionnaire (MEQ) (Box 2).19 Low scores are associated with evening types—felt to correspond to the endogenous circadian period—and can help narrow the differential diagnosis of sleep-initiation complaints.20
The Morningness-Eveningness Questionnaire (MEQ) developed by Horne and Ostberg19 can be used to verify eveningness tendencies of patients with presumptive delayed sleep phase disorder. The MEQ is a 19-item self-assessment tool with responses that are assigned values totaling up to 86 points. Examples of the questions include:
- Considering only your own ‘feeling best’ rhythm, at what time would you get up if you were entirely free to plan your day?
- Considering only your own ‘feeling best’ rhythm, at what time would you go to bed if you were entirely free to plan your day?
- How easy do you find it to get up each day?
- When you have no commitments the next day, how much later do you go to bed compared to your usual bedtime?
- One hears about ‘morning’ and ‘evening’ types of people. Which ONE of these types do you consider yourself to be?
Lower scores are associated with evening types—felt to correspond to the endogenous circadian period—and can help in narrowing the differential diagnosis of sleep-initiation complaints.20 Scores on the MEQ are interpreted as:
- 70 to 86: definite morning type
- 59 to 69: moderately morning type
- 42 to 58: neither type
- 31 to 41: moderately evening type
- 16 to 30: definite evening type
CASE CONTINUED: ‘Definite evening type’
Jason scores 28 on the MEQ, consistent with a “definite evening type.” Actigraphic monitoring is scheduled during a school holiday, when he is instructed to sleep according to his preferred schedule with the least possible restriction.
A clearly delayed sleep phase is evident, with the habitual sleep period occurring between 5 AM and 1 PM. Even on days when he was quite sleep-restricted because of an enforced wake time, sleep onset on the ensuing evening was substantially delayed, suggesting an obligate nature for the delayed sleep/wake schedule. Overall, Jason had few complaints with respect to impaired alertness while on this unrestricted schedule and experienced a much more stable mood.
Interventions
Without physiologic assessments, understanding the patient’s “natural” sleep schedule can allow for rational recommendations about using phototherapy and oral melatonin (Figure21). However, referral to a sleep specialist is required unless the general psychiatrist has experience in treating circadian rhythm sleep disorders.
Morning phototherapy. Properly timed morning bright light therapy (≥2,500 lux) has been shown to help DSPD patients achieve physiologically measured sleep phase advances, objective improvements in daytime alertness, and earlier reported bedtimes compared with controls.22 Unfortunately, the described 2-hour treatment duration make this research protocol clinically impractical, and most clinicians commence with a 30-minute duration of therapy, as described in the seasonal affective disorder literature.
Relatively new and widely available blue light boxes have been reported to exhibit at least equivalent efficacy to bright light devices (as reported in the literature pertaining to seasonal affective disorder), but with markedly decreased light intensity and fewer associated adverse effects.23 As the research addressing their use in the treatment of circadian rhythm sleep disorders is still emerging, their future role remains uncertain.
Precautions. Most psychiatrists would not perform a physiologic determination of a patient’s circadian phase, and further undesired phase delays can occur if phototherapy is administered before the core body temperature minimum (Tmin).24 Also, use caution if prescribing phototherapy to patients taking photosensitizing drugs and/or those with ocular or retinal pathology.20
Evening light avoidance. Whether or not you prescribe morning phototherapy, recommending that DSPD patients avoid evening light is essential to avoid further induction or exacerbation of phase delays. Protective eyewear is warranted in instances where these advisory precautions are insufficient (see Related Resources). Such an intervention has been shown effective in decreasing light exposure and undesired phase advances in studies involving subjects exposed to simulated shift work.25
Oral melatonin. Abundant evidence supports melatonin use in achieving phase advances in individuals with DSPD.26,27 A synergistic effect can be obtained when melatonin is combined with phototherapy.28
Proper timing of melatonin to achieve a maximal phase advance can be estimated based on the individual’s dim light melatonin onset (DLMO), which occurs approximately 14 hours after the habitual (unrestricted) wake time.29 Maximal phase advances appear to occur when melatonin is given approximately 6 hours before the DLMO.26 Thus, a rational practice is to recommend that patients take melatonin 8 hours after their natural wake time. Doses of ≤0.5 mg appear to achieve the maximal chronobiotic effect while avoiding an undesired hypnotic effect.30
Precautions. Verifying the purity of over-the-counter melatonin is difficult. A review by the National Academy of Sciences states that short-term use of melatonin, ≤10 mg/d, appears to be safe in healthy adults but recommends caution in children/adolescents and women of reproductive age. Doses recommended for circadian-based interventions are typically physiologic in nature (i.e., ≤0.5 mg), which may serve to mitigate these concerns.
Adverse effects such as headaches, somnolence, hypotension, hypertension, gastrointestinal upset, and exacerbation of alopecia areata have been reported at higher melatonin doses in healthy adults and at lower doses in persons with preexisting central nervous system, cardiovascular, gastrointestinal, or dermatologic conditions.31
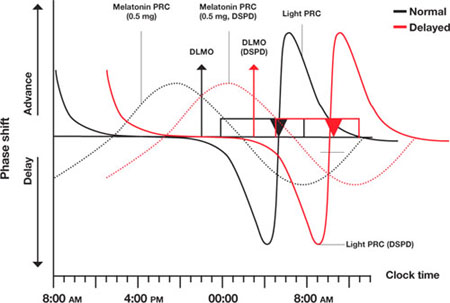
Figure Light and melatonin phase response curves: Normal vs. delayed
This schematic compares ‘normal sleep’ phase response curves (PRCs) to light and exogenous melatonin with postulated PRCs for an individual with delayed sleep phase disorder (DSPD), presumed to be 5 hours ‘out of phase.’ Y-axis shows the direction and relative magnitude of phase shifts produced by light or melatonin at times shown on the x-axis. X-axis covers >24 hours to better illustrate the PRCs.
Relationships between ‘normal sleepers’ and DSPD patients are depicted by:
- rectangles (sleep period)
- triangles (core body temperature minimum [Tmin])
- arrows (dim light melatonin onsets [DLMOs]).
‘Normal’ sleep is shown to occur from midnight to 8 AM, and the DSPD patient’s sleep from 5 AM to 1 PM; DLMO and Tmin are similarly delayed by 5 hours in the DSPD patient. This schematic assumes that phase relationships are maintained in DSPD patients, which is not a certainty.
Source: Adapted from reference 21
CASE CONTINUED: Under the bright lights
Jason starts phototherapy treatment during his winter break, administering bright light daily upon natural awakening using a 10,000 lux light box for at least 30 minutes. As instructed, he gradually advances the time of administration by approximately 30 minutes every other day, striving for a nocturnal sleep period of 11 PM to 7 AM. He also wears protective eyewear to reduce light exposure during evening hours to avoid further delays in sleep phase. To further promote a phase advance, he takes oral melatonin, 0.5 mg/d at approximately 8 PM, as determined by his self-report and results of actigraphic recording.
Other options
Hypnotics. Little evidence supports the use of hypnotics in DSPD,32 and patients may show resistance to these drugs.33 Nevertheless, hypnotics can heighten confidence in the ability to initiate sleep in individuals with a concomitant conditioned insomnia.
With chronotherapy, patients are prescribed a sleep schedule that is delayed several hours incrementally until sleep is aligned to a target bedtime. The individual then is advised to rigorously maintain a regular sleep/wake schedule, repeating the process as necessary.
Although case reports have shown positive results with chronotherapy for DSPD,34 no controlled trials have demonstrated its efficacy or safety. One study reported high relapse rates,31 and 1 patient with DSPD developed free-running circadian rhythms.35 Clinical experience suggests chronotherapy is impractical for patients who must adhere to a fixed schedule.
Behavioral approaches
For an adolescent with DSPD, consider asking the school district to allow him or her a later school start-time. This alone often can substantially increase total sleep time and mitigate associated impairments.36 In all instances pursue and address external contributors to DSPD, such as poor sleep hygiene (including excessive caffeine use) and substance misuse.
Emphasize regular wake times, as arising later on weekends can cause phase delays.37 DSPD patients may have a concomitant conditioned insomnia that responds to evidence-based behavioral treatments.38
Whatever intervention you choose, schedule a follow-up appointment in approximately 2 months to evaluate patients’ progress and compliance. Encourage them to contact you with questions or concerns in the interim. Review sleep logs or actigraphy during this visit, and adjust the timing and/or nature of interventions as needed. Adolescents can be particularly noncompliant with clinical interventions, and therapeutic goals cannot be reached without their full investment.
Because no guidelines exist on how long to treat DSPD, stop on a “trial-and-error” basis when symptoms are controlled, and resume if they recur. Another approach is to maintain a desired sleep/wake schedule with bedtime melatonin and encourage continued adherence to other measures.
CASE CONTINUED: Maintenance therapy
Jason returns to the clinic approximately 10 weeks later. After an obviously concerted effort to adhere to treatment, his progress is quite remarkable. He rarely falls asleep later than 11 PM, so he is obtaining 2.5 hours more sleep each night before arising for school at 6:30 AM. Sleepiness at school is rarely problematic, and his mood is more stable.
He nevertheless describes a persistent tendency to retire and arise later and asks to continue melatonin and phototherapy. Because no guidelines exist for long-term therapy of DSPD, he is advised to switch melatonin to bedtime dosing (with a presumed phase-neutral “maintenance” effect), and to continue phototherapy as prescribed.
- Wyatt JK. Delayed sleep phase syndrome: pathophysiology and treatment options. Sleep. 2004;27(6):1195-1203.
- Crowley SJ, Acebo C, Carskadon MA. Sleep, circadian rhythms, and delayed sleep phase in adolescence. Sleep Med. 2007;8(6):602-612.
- National Sleep Foundation. Adolescent sleep needs and patterns: research report and resource guide. Washington, DC; 2000:1-30.
- Products designed to assist in the avoidance of light at improper times. www.lowbluelights.com.
Disclosure
Dr. Auger reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Khalsa SB, Jewett ME, Cajochen C, et al. A phase response curve to single bright light pulses in human subjects. J Physiol. 2003;549(Pt 3):945-952.
2. Quinto C, Gellido C, Chokroverty S, et al. Posttraumatic delayed sleep phase syndrome. Neurology. 2000;54(1):250-252.
3. Campbell SS, Murphy PJ. Delayed sleep phase disorder in temporal isolation. Sleep. 2007;30(9):1225-1228.
4. Uchiyama M, Okawa M, Shibui K, et al. Altered phase relation between sleep timing and core body temperature rhythm in delayed sleep phase syndrome and non-24-hour sleep-wake syndrome in humans. Neurosci Lett. 2000;294(2):101-104.
5. Aoki H, Ozeki Y, Yamada N. Hypersensitivity of melatonin suppression in response to light in patients with delayed sleep phase syndrome. Chronobiol Int. 2001;18(2):263-271.
6. Uchiyama M, Okawa M, Shibui K, et al. Poor compensatory function for sleep loss as a pathogenic factor in patients with delayed sleep phase syndrome. Sleep. 2000;23(4):553-558.
7. Mercer PW, Merritt SL, Cowell JM. Differences in reported sleep need among adolescents. J Adolesc Health. 1998;23(5):259-263.
8. Krahn LE, Pankratz VS, Harris AM, et al. Long-term outcome of adolescents with delayed sleep phase disorder [abstract]. Sleep. 2003;26:A115.-
9. Dagan Y, Stein D, Steinbock M, et al. Frequency of delayed sleep phase syndrome among hospitalized adolescent psychiatric patients. J Psychosom Res. 1998;45(1):15-20.
10. Thorpy MJ, Korman E, Spielman AJ, et al. Delayed sleep phase syndrome in adolescents. J Adolesc Health Care. 1998;9(1):22-27.
11. Regestein QR, Monk TH. Delayed sleep phase syndrome: a review of the clinical aspects. Am J Psychiatry. 1995;152(4):602-608.
12. Pelayo RP, Thorpy MJ, Glovinsky P. Prevalence of delayed sleep phase syndrome among adolescents [abstract]. Sleep Res. 1988;17:391.-
13. Gau SF, Soong WT. The transition of sleep-wake patterns in early adolescence. Sleep. 2003;26(4):449-454.
14. Beersma DG, Gordijn MC. Circadian control of the sleep-wake cycle. Physiol Behav. 2007;90(2-3):190-195.
15. Ralph MR, Foster RG, Davis FC, et al. Transplanted suprachiasmatic nucleus determines circadian period. Science. 1990;247(4945):975-978.
16. Waterhouse J, DeCoursey PJ. Chronobiology: biological timekeeping. Sunderland, MA: Sinauer Associates, Inc. Publishers; 2004:291-323.
17. American Academy of Sleep Medicine. International classification of sleep disorders. Diagnostic and coding manual. 2nd ed. Westchester, IL: American Academy of Sleep Medicine; 2005.
18. Bradshaw DA, Yanagi MA, Pak ES, et al. Nightly sleep duration in the 2-week period preceding multiple sleep latency testing. J Clin Sleep Med. 2007;3(6):613-619.
19. Horne JA, Ostberg O. A self-assessment questionnaire to determine morningness-eveningness in human circadian rhythms. Int J Chronobiol. 1976;4(2):97-110.
20. Sack RL, Auckley D, Auger RR, et al. Circadian rhythm sleep disorders: Part I. basic principles, shift work and jet lag disorders. Sleep. 2007;30(11):1460-1483.
21. Burgess HJ, Sharkey KM, Eastman CI. Bright light, dark and melatonin can promote circadian adaptation in night shift workers. Sleep Med Rev. 2002;6(5):407-420.
22. Rosenthal NE, Joseph-Vanderpool JR, Levendosky AA, et al. Phase-shifting effects of bright morning light as treatment for delayed sleep phase syndrome. Sleep. 1990;13(4):354-361.
23. Glickman G, Byrne B, Pineda C, et al. Light therapy for seasonal affective disorder with blue narrow-band light-emitting diodes (LEDs). Biol Psychiatry. 2006;59:502-507.
24. Czeisler C, Wright K, Jr. Influence of light on circadian rhythmicity in humans. New York, NY: Marcel Dekker; 1999:149-180.
25. Crowley SJ, Lee C, Tseng CY, et al. Combinations of bright light, scheduled dark, sunglasses, and melatonin to facilitate circadian entrainment to night shift work. J Biol Rhythms. 2003;18(6):513-523.
26. Mundey K, Benloucif S, Harsanyi K, et al. Phase-dependent treatment of delayed sleep phase syndrome with melatonin. Sleep. 2005;28(10):1271-1278.
27. Sack RL, Auckley D, Auger RR, et al. Circadian rhythm sleep disorders: Part II, advanced sleep phase disorder, delayed sleep phase disorder, free-running disorder, and irregular sleep-wake rhythm. Sleep. 2007;30(11):1484-1501.
28. Revell VL, Burgess HJ, Gazda CJ, et al. Advancing human circadian rhythms with afternoon melatonin and morning intermittent bright light. J Clin Endocrinol Metab. 2006;91(1):54-59.
29. Burgess HJ, Eastman CI. The dim light melatonin onset following fixed and free sleep schedules. J Sleep Res. 2005;14(3):229-237.
30. Lewy AJ. Clinical applications of melatonin in circadian disorders. Dialog Clin Neurosci. 2003;5:399-413.
31. Committee on the Framework for Evaluating the Safety of Dietary Supplements FaNB, Board on Life Sciences, Institute of Medicine and National Research Council of the National Academies. Dietary supplements: a framework for evaluating safety. Washington, DC: The National Academies Press; 2005.
32. Ito A, Ando K, Hayakawa T, et al. Long-term course of adult patients with delayed sleep phase syndrome. Jpn J Psychiatry Neurol. 1993;47(3):563-567.
33. Auger RR. Circadian rhythm sleep disorder, delayed sleep phase type (pediatric case). In: Winkelman JW (chair), Henderson JH, Kotagal S, et al, eds. Case book of sleep medicine. Westchester, IL: American Academy of Sleep Medicine; 2008:195-199.
34. Czeisler C, Weitzman E, Moore, et al. Chronotherapy: resetting the circadian clocks of patients with delayed sleep phase insomnia. Sleep. 1981;4:1-21.
35. Oren DA, Wehr TA. Hypernyctohemeral syndrome after chronotherapy for delayed sleep phase syndrome. N Engl J Med. 1992;327(24):1762.-
36. Wahlstrom K. Changing times: findings from the first longitudinal study of later high school start times. NASSP Bulletin. 2002;86(633):3-21.
37. Burgess HJ, Eastman CI. A late wake time phase delays the human dim light melatonin rhythm. Neurosci Lett. 2006;395(3):191-195.
38. Morgenthaler T, Kramer M, Alessi C, et al. Practice parameters for the psychological and behavioral treatment of insomnia: an update. An American Academy of Sleep Medicine report. Sleep. 2006;29(11):1415-1419.
Jason, age 16, has had difficulty with sleep initiation for 2 years. He describes going to bed at 10:30 PM on school nights but falling asleep no sooner than midnight and typically after 1:30 AM. He denies contributions from an “active mind” or environmental disturbances, and his bedroom contains no TV, computer, or other media devices. He does not sleep better with a change in environment. He denies pervasive low mood symptoms and believes his mood hinges predominantly on his ability to achieve sufficient sleep.
Once asleep, Jason generally enjoys good sleep consolidation until he needs to arise at 6:30 AM. His mother awakens him with difficulty, as he often sleeps through his alarm. He sleeps approximately 5 hours nightly during the school week, endorses impaired concentration, and often dozes during his first several classes. When he returns home from school, he finds it very difficult to resist napping.
On weekends he retires at 1 AM or later and typically falls asleep within 30 minutes. He usually awakens at noon but can sleep as late as 4:30 PM. He feels slightly more refreshed on weekends and describes his mood then as improved. During a recent spring break, he felt much better when allowed to sleep as much as he wanted.
Delayed sleep phase disorder (DSPD)—characterized by a pathological “night owl” circadian preference—is seen most commonly in adolescents and is associated with psychiatric morbidity, psychosocial impairment, and poor academic performance. Proper identification of the condition can be enhanced with a variety of assessment tools, and successful treatment requires an awareness of potential endogenous and exogenous contributors.
This article describes what is known about DSPD and uses the case example to illustrate diagnostic assessment and treatment choices. Intriguing data support various pathophysiologic explanations for DSPD (Box 1).1-6 Facilitating the adjustment of patients’ physiologic clocks is the overall goal in managing DSPD.
In individuals normally entrained to the light/dark cycle, circadian rhythms are:
- delayed by evening exposure to bright light (≥ 2,500 lux) prior to the core body temperature minimum (Tmin)
- advanced by morning light exposure after the Tmin.1
These opposing effects attune most people to the light/dark cycle, with sleep and wakefulness occurring on a conventional schedule. Persons with delayed sleep phase disorder (DSPD) live at a delayed phase that resists advancement and is incompatible with their personal and social obligations.
Theories have been proposed, but DSPD’s etiology has not been fully explained. Affected adolescents may exhibit an extreme in circadian preference. Case reports also describe DSPD emerging after traumatic brain injury.2
Intriguing evidence supports various pathophysiologic explanations for DSPD. An abnormally long intrinsic circadian period (>25 hours) was recently demonstrated during temporal isolation in 1 individual with DSPD.3 Both this case report and controlled studies describe deviations from expected relationships between the sleep/wake cycle and physiologic circadian markers. Most consistently described are longer intervals from Tmin4 to sleep offset (final rise time) in DSPD patients compared with controls.
Other research suggests:
Extreme ‘eveningness’
Because of their extreme seemingly innate preference to retire and arise at relatively late clock hours (an “eveningness” trait), school-aged patients with DSPD represent a high-risk population for problematic sleepiness. In a survey of 612 high school students, the 63% who felt they needed more sleep on school nights showed a strong eveningness preference (as assessed by questionnaire), compared with students who described getting sufficient sleep.7 Other studies have revealed psychiatric morbidity (including affective and personality disorders), psychosocial impairment, and poor academic performance associated with the condition.8-10
DSPD may affect 7% to 16% of patients presenting with insomnia complaints in sleep medicine clinics.11 The condition appears most common among young cohorts and has been reported to affect up to 7% of adolescents in the United States.12 Its high frequency in this age group may be a pathologic exaggeration of the normal tendency toward delayed timing of sleep and wakefulness linked with pubertal development.13
Sleep and wakefulness regulation
Conceptually, 2 processes govern sleep and wakefulness:
- The homeostatic drive to sleep (process S) is proportional to the duration of sleep restriction and becomes maximal at about 40 hours.
- Circadian regulation (process C) creates a drive for wakefulness that variably opposes process S and depends upon intrinsic rhythms.14
Neurons of the suprachiasmatic nucleus in the hypothalamus exert master coordination of this sleep/wake rhythm, along with other behavioral and physiologic variables.15 Because the typical intrinsic period is slightly longer than 24 hours, synchronization to the 24-hour day (entrainment) is accomplished by environmental inputs (zeitgebers, or “time givers”), the most important of which is exposure to light.16
Misalignment between endogenous circadian rhythms and the light/dark cycle can result in circadian rhythm sleep disorders, such as:
- delayed sleep timing (DSPD)
- advanced sleep timing (advanced sleep phase disorder)
- erratic sleep timing (irregular sleep/wake rhythm)
- complete dissociation from the light/dark cycle (circadian rhythm sleep disorder, free-running type).
These 4 conditions are thought to involve predominantly intrinsic mechanisms, but circadian dysrhythmias also can be induced by exogenous factors. Extreme work schedules or rapid travel across time zones can challenge the circadian system’s ability to acclimate and the individual’s ability to achieve a desired sleep schedule.17
Differential diagnosis
Because DSPD relates primarily to an aberration in timing of sleep, it is characterized as a disorder only if the individual’s preferred schedule interferes substantially with social or occupational functioning. The International Classification of Sleep Disorders (ICSD) provides detailed diagnostic criteria (Table).17
Table
Diagnostic criteria for delayed sleep phase disorder
A. Delay exists in the phase of the major sleep period in relation to desired sleep time and wake-up time, as evidenced by:
|
| B. When allowed to choose a preferred schedule, patients exhibit normal sleep quality and duration for age and maintain a delayed but stable phase of entrainment to the 24-hour sleep/wake pattern. |
| C. Monitoring with a sleep log or actigraphy (including sleep diary) for at least 7 days demonstrates a stable delay in the timing of the habitual sleep period. |
| D. The sleep disturbance is not better explained by another sleep disorder, medical or neurologic disorder, mental disorder, medication use, or substance use disorder. |
| Source: Adapted and reprinted with permission from International classification of sleep disorders. Diagnostic and coding manual. 2nd ed17 |
Depression and anxiety often manifest with sleep difficulties, as do inadequate sleep hygiene and other conditions associated with prolonged sleep initiation. According to ICSD criteria, primary insomnia can be differentiated from DSPD if the patient readily initiates and maintains sleep when allowed to sleep on his/her desired sleep/wake schedule. Accumulated evidence has largely debunked this notion, however, as polysomnographic studies have demonstrated both prolonged sleep latency and impaired sleep efficiency in DSPD patients versus matched controls.3
Assessment tools can complement the clinical history in diagnosing DSPD. Either a sleep log or actigraphy is required to demonstrate a stable phase delay, but actigraphy typically generates more reliable data.18 Actigraphs are compact “motion detectors” whose output while being worn by patients allows longitudinal assessment of sleep/wake parameters.
Eveningness tendencies of presumptive DSPD patients can be further verified with the Morningness-Eveningness Questionnaire (MEQ) (Box 2).19 Low scores are associated with evening types—felt to correspond to the endogenous circadian period—and can help narrow the differential diagnosis of sleep-initiation complaints.20
The Morningness-Eveningness Questionnaire (MEQ) developed by Horne and Ostberg19 can be used to verify eveningness tendencies of patients with presumptive delayed sleep phase disorder. The MEQ is a 19-item self-assessment tool with responses that are assigned values totaling up to 86 points. Examples of the questions include:
- Considering only your own ‘feeling best’ rhythm, at what time would you get up if you were entirely free to plan your day?
- Considering only your own ‘feeling best’ rhythm, at what time would you go to bed if you were entirely free to plan your day?
- How easy do you find it to get up each day?
- When you have no commitments the next day, how much later do you go to bed compared to your usual bedtime?
- One hears about ‘morning’ and ‘evening’ types of people. Which ONE of these types do you consider yourself to be?
Lower scores are associated with evening types—felt to correspond to the endogenous circadian period—and can help in narrowing the differential diagnosis of sleep-initiation complaints.20 Scores on the MEQ are interpreted as:
- 70 to 86: definite morning type
- 59 to 69: moderately morning type
- 42 to 58: neither type
- 31 to 41: moderately evening type
- 16 to 30: definite evening type
CASE CONTINUED: ‘Definite evening type’
Jason scores 28 on the MEQ, consistent with a “definite evening type.” Actigraphic monitoring is scheduled during a school holiday, when he is instructed to sleep according to his preferred schedule with the least possible restriction.
A clearly delayed sleep phase is evident, with the habitual sleep period occurring between 5 AM and 1 PM. Even on days when he was quite sleep-restricted because of an enforced wake time, sleep onset on the ensuing evening was substantially delayed, suggesting an obligate nature for the delayed sleep/wake schedule. Overall, Jason had few complaints with respect to impaired alertness while on this unrestricted schedule and experienced a much more stable mood.
Interventions
Without physiologic assessments, understanding the patient’s “natural” sleep schedule can allow for rational recommendations about using phototherapy and oral melatonin (Figure21). However, referral to a sleep specialist is required unless the general psychiatrist has experience in treating circadian rhythm sleep disorders.
Morning phototherapy. Properly timed morning bright light therapy (≥2,500 lux) has been shown to help DSPD patients achieve physiologically measured sleep phase advances, objective improvements in daytime alertness, and earlier reported bedtimes compared with controls.22 Unfortunately, the described 2-hour treatment duration make this research protocol clinically impractical, and most clinicians commence with a 30-minute duration of therapy, as described in the seasonal affective disorder literature.
Relatively new and widely available blue light boxes have been reported to exhibit at least equivalent efficacy to bright light devices (as reported in the literature pertaining to seasonal affective disorder), but with markedly decreased light intensity and fewer associated adverse effects.23 As the research addressing their use in the treatment of circadian rhythm sleep disorders is still emerging, their future role remains uncertain.
Precautions. Most psychiatrists would not perform a physiologic determination of a patient’s circadian phase, and further undesired phase delays can occur if phototherapy is administered before the core body temperature minimum (Tmin).24 Also, use caution if prescribing phototherapy to patients taking photosensitizing drugs and/or those with ocular or retinal pathology.20
Evening light avoidance. Whether or not you prescribe morning phototherapy, recommending that DSPD patients avoid evening light is essential to avoid further induction or exacerbation of phase delays. Protective eyewear is warranted in instances where these advisory precautions are insufficient (see Related Resources). Such an intervention has been shown effective in decreasing light exposure and undesired phase advances in studies involving subjects exposed to simulated shift work.25
Oral melatonin. Abundant evidence supports melatonin use in achieving phase advances in individuals with DSPD.26,27 A synergistic effect can be obtained when melatonin is combined with phototherapy.28
Proper timing of melatonin to achieve a maximal phase advance can be estimated based on the individual’s dim light melatonin onset (DLMO), which occurs approximately 14 hours after the habitual (unrestricted) wake time.29 Maximal phase advances appear to occur when melatonin is given approximately 6 hours before the DLMO.26 Thus, a rational practice is to recommend that patients take melatonin 8 hours after their natural wake time. Doses of ≤0.5 mg appear to achieve the maximal chronobiotic effect while avoiding an undesired hypnotic effect.30
Precautions. Verifying the purity of over-the-counter melatonin is difficult. A review by the National Academy of Sciences states that short-term use of melatonin, ≤10 mg/d, appears to be safe in healthy adults but recommends caution in children/adolescents and women of reproductive age. Doses recommended for circadian-based interventions are typically physiologic in nature (i.e., ≤0.5 mg), which may serve to mitigate these concerns.
Adverse effects such as headaches, somnolence, hypotension, hypertension, gastrointestinal upset, and exacerbation of alopecia areata have been reported at higher melatonin doses in healthy adults and at lower doses in persons with preexisting central nervous system, cardiovascular, gastrointestinal, or dermatologic conditions.31
Figure Light and melatonin phase response curves: Normal vs. delayed
This schematic compares ‘normal sleep’ phase response curves (PRCs) to light and exogenous melatonin with postulated PRCs for an individual with delayed sleep phase disorder (DSPD), presumed to be 5 hours ‘out of phase.’ Y-axis shows the direction and relative magnitude of phase shifts produced by light or melatonin at times shown on the x-axis. X-axis covers >24 hours to better illustrate the PRCs.
Relationships between ‘normal sleepers’ and DSPD patients are depicted by:
- rectangles (sleep period)
- triangles (core body temperature minimum [Tmin])
- arrows (dim light melatonin onsets [DLMOs]).
‘Normal’ sleep is shown to occur from midnight to 8 AM, and the DSPD patient’s sleep from 5 AM to 1 PM; DLMO and Tmin are similarly delayed by 5 hours in the DSPD patient. This schematic assumes that phase relationships are maintained in DSPD patients, which is not a certainty.
Source: Adapted from reference 21
CASE CONTINUED: Under the bright lights
Jason starts phototherapy treatment during his winter break, administering bright light daily upon natural awakening using a 10,000 lux light box for at least 30 minutes. As instructed, he gradually advances the time of administration by approximately 30 minutes every other day, striving for a nocturnal sleep period of 11 PM to 7 AM. He also wears protective eyewear to reduce light exposure during evening hours to avoid further delays in sleep phase. To further promote a phase advance, he takes oral melatonin, 0.5 mg/d at approximately 8 PM, as determined by his self-report and results of actigraphic recording.
Other options
Hypnotics. Little evidence supports the use of hypnotics in DSPD,32 and patients may show resistance to these drugs.33 Nevertheless, hypnotics can heighten confidence in the ability to initiate sleep in individuals with a concomitant conditioned insomnia.
With chronotherapy, patients are prescribed a sleep schedule that is delayed several hours incrementally until sleep is aligned to a target bedtime. The individual then is advised to rigorously maintain a regular sleep/wake schedule, repeating the process as necessary.
Although case reports have shown positive results with chronotherapy for DSPD,34 no controlled trials have demonstrated its efficacy or safety. One study reported high relapse rates,31 and 1 patient with DSPD developed free-running circadian rhythms.35 Clinical experience suggests chronotherapy is impractical for patients who must adhere to a fixed schedule.
Behavioral approaches
For an adolescent with DSPD, consider asking the school district to allow him or her a later school start-time. This alone often can substantially increase total sleep time and mitigate associated impairments.36 In all instances pursue and address external contributors to DSPD, such as poor sleep hygiene (including excessive caffeine use) and substance misuse.
Emphasize regular wake times, as arising later on weekends can cause phase delays.37 DSPD patients may have a concomitant conditioned insomnia that responds to evidence-based behavioral treatments.38
Whatever intervention you choose, schedule a follow-up appointment in approximately 2 months to evaluate patients’ progress and compliance. Encourage them to contact you with questions or concerns in the interim. Review sleep logs or actigraphy during this visit, and adjust the timing and/or nature of interventions as needed. Adolescents can be particularly noncompliant with clinical interventions, and therapeutic goals cannot be reached without their full investment.
Because no guidelines exist on how long to treat DSPD, stop on a “trial-and-error” basis when symptoms are controlled, and resume if they recur. Another approach is to maintain a desired sleep/wake schedule with bedtime melatonin and encourage continued adherence to other measures.
CASE CONTINUED: Maintenance therapy
Jason returns to the clinic approximately 10 weeks later. After an obviously concerted effort to adhere to treatment, his progress is quite remarkable. He rarely falls asleep later than 11 PM, so he is obtaining 2.5 hours more sleep each night before arising for school at 6:30 AM. Sleepiness at school is rarely problematic, and his mood is more stable.
He nevertheless describes a persistent tendency to retire and arise later and asks to continue melatonin and phototherapy. Because no guidelines exist for long-term therapy of DSPD, he is advised to switch melatonin to bedtime dosing (with a presumed phase-neutral “maintenance” effect), and to continue phototherapy as prescribed.
- Wyatt JK. Delayed sleep phase syndrome: pathophysiology and treatment options. Sleep. 2004;27(6):1195-1203.
- Crowley SJ, Acebo C, Carskadon MA. Sleep, circadian rhythms, and delayed sleep phase in adolescence. Sleep Med. 2007;8(6):602-612.
- National Sleep Foundation. Adolescent sleep needs and patterns: research report and resource guide. Washington, DC; 2000:1-30.
- Products designed to assist in the avoidance of light at improper times. www.lowbluelights.com.
Disclosure
Dr. Auger reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Jason, age 16, has had difficulty with sleep initiation for 2 years. He describes going to bed at 10:30 PM on school nights but falling asleep no sooner than midnight and typically after 1:30 AM. He denies contributions from an “active mind” or environmental disturbances, and his bedroom contains no TV, computer, or other media devices. He does not sleep better with a change in environment. He denies pervasive low mood symptoms and believes his mood hinges predominantly on his ability to achieve sufficient sleep.
Once asleep, Jason generally enjoys good sleep consolidation until he needs to arise at 6:30 AM. His mother awakens him with difficulty, as he often sleeps through his alarm. He sleeps approximately 5 hours nightly during the school week, endorses impaired concentration, and often dozes during his first several classes. When he returns home from school, he finds it very difficult to resist napping.
On weekends he retires at 1 AM or later and typically falls asleep within 30 minutes. He usually awakens at noon but can sleep as late as 4:30 PM. He feels slightly more refreshed on weekends and describes his mood then as improved. During a recent spring break, he felt much better when allowed to sleep as much as he wanted.
Delayed sleep phase disorder (DSPD)—characterized by a pathological “night owl” circadian preference—is seen most commonly in adolescents and is associated with psychiatric morbidity, psychosocial impairment, and poor academic performance. Proper identification of the condition can be enhanced with a variety of assessment tools, and successful treatment requires an awareness of potential endogenous and exogenous contributors.
This article describes what is known about DSPD and uses the case example to illustrate diagnostic assessment and treatment choices. Intriguing data support various pathophysiologic explanations for DSPD (Box 1).1-6 Facilitating the adjustment of patients’ physiologic clocks is the overall goal in managing DSPD.
In individuals normally entrained to the light/dark cycle, circadian rhythms are:
- delayed by evening exposure to bright light (≥ 2,500 lux) prior to the core body temperature minimum (Tmin)
- advanced by morning light exposure after the Tmin.1
These opposing effects attune most people to the light/dark cycle, with sleep and wakefulness occurring on a conventional schedule. Persons with delayed sleep phase disorder (DSPD) live at a delayed phase that resists advancement and is incompatible with their personal and social obligations.
Theories have been proposed, but DSPD’s etiology has not been fully explained. Affected adolescents may exhibit an extreme in circadian preference. Case reports also describe DSPD emerging after traumatic brain injury.2
Intriguing evidence supports various pathophysiologic explanations for DSPD. An abnormally long intrinsic circadian period (>25 hours) was recently demonstrated during temporal isolation in 1 individual with DSPD.3 Both this case report and controlled studies describe deviations from expected relationships between the sleep/wake cycle and physiologic circadian markers. Most consistently described are longer intervals from Tmin4 to sleep offset (final rise time) in DSPD patients compared with controls.
Other research suggests:
Extreme ‘eveningness’
Because of their extreme seemingly innate preference to retire and arise at relatively late clock hours (an “eveningness” trait), school-aged patients with DSPD represent a high-risk population for problematic sleepiness. In a survey of 612 high school students, the 63% who felt they needed more sleep on school nights showed a strong eveningness preference (as assessed by questionnaire), compared with students who described getting sufficient sleep.7 Other studies have revealed psychiatric morbidity (including affective and personality disorders), psychosocial impairment, and poor academic performance associated with the condition.8-10
DSPD may affect 7% to 16% of patients presenting with insomnia complaints in sleep medicine clinics.11 The condition appears most common among young cohorts and has been reported to affect up to 7% of adolescents in the United States.12 Its high frequency in this age group may be a pathologic exaggeration of the normal tendency toward delayed timing of sleep and wakefulness linked with pubertal development.13
Sleep and wakefulness regulation
Conceptually, 2 processes govern sleep and wakefulness:
- The homeostatic drive to sleep (process S) is proportional to the duration of sleep restriction and becomes maximal at about 40 hours.
- Circadian regulation (process C) creates a drive for wakefulness that variably opposes process S and depends upon intrinsic rhythms.14
Neurons of the suprachiasmatic nucleus in the hypothalamus exert master coordination of this sleep/wake rhythm, along with other behavioral and physiologic variables.15 Because the typical intrinsic period is slightly longer than 24 hours, synchronization to the 24-hour day (entrainment) is accomplished by environmental inputs (zeitgebers, or “time givers”), the most important of which is exposure to light.16
Misalignment between endogenous circadian rhythms and the light/dark cycle can result in circadian rhythm sleep disorders, such as:
- delayed sleep timing (DSPD)
- advanced sleep timing (advanced sleep phase disorder)
- erratic sleep timing (irregular sleep/wake rhythm)
- complete dissociation from the light/dark cycle (circadian rhythm sleep disorder, free-running type).
These 4 conditions are thought to involve predominantly intrinsic mechanisms, but circadian dysrhythmias also can be induced by exogenous factors. Extreme work schedules or rapid travel across time zones can challenge the circadian system’s ability to acclimate and the individual’s ability to achieve a desired sleep schedule.17
Differential diagnosis
Because DSPD relates primarily to an aberration in timing of sleep, it is characterized as a disorder only if the individual’s preferred schedule interferes substantially with social or occupational functioning. The International Classification of Sleep Disorders (ICSD) provides detailed diagnostic criteria (Table).17
Table
Diagnostic criteria for delayed sleep phase disorder
A. Delay exists in the phase of the major sleep period in relation to desired sleep time and wake-up time, as evidenced by:
|
| B. When allowed to choose a preferred schedule, patients exhibit normal sleep quality and duration for age and maintain a delayed but stable phase of entrainment to the 24-hour sleep/wake pattern. |
| C. Monitoring with a sleep log or actigraphy (including sleep diary) for at least 7 days demonstrates a stable delay in the timing of the habitual sleep period. |
| D. The sleep disturbance is not better explained by another sleep disorder, medical or neurologic disorder, mental disorder, medication use, or substance use disorder. |
| Source: Adapted and reprinted with permission from International classification of sleep disorders. Diagnostic and coding manual. 2nd ed17 |
Depression and anxiety often manifest with sleep difficulties, as do inadequate sleep hygiene and other conditions associated with prolonged sleep initiation. According to ICSD criteria, primary insomnia can be differentiated from DSPD if the patient readily initiates and maintains sleep when allowed to sleep on his/her desired sleep/wake schedule. Accumulated evidence has largely debunked this notion, however, as polysomnographic studies have demonstrated both prolonged sleep latency and impaired sleep efficiency in DSPD patients versus matched controls.3
Assessment tools can complement the clinical history in diagnosing DSPD. Either a sleep log or actigraphy is required to demonstrate a stable phase delay, but actigraphy typically generates more reliable data.18 Actigraphs are compact “motion detectors” whose output while being worn by patients allows longitudinal assessment of sleep/wake parameters.
Eveningness tendencies of presumptive DSPD patients can be further verified with the Morningness-Eveningness Questionnaire (MEQ) (Box 2).19 Low scores are associated with evening types—felt to correspond to the endogenous circadian period—and can help narrow the differential diagnosis of sleep-initiation complaints.20
The Morningness-Eveningness Questionnaire (MEQ) developed by Horne and Ostberg19 can be used to verify eveningness tendencies of patients with presumptive delayed sleep phase disorder. The MEQ is a 19-item self-assessment tool with responses that are assigned values totaling up to 86 points. Examples of the questions include:
- Considering only your own ‘feeling best’ rhythm, at what time would you get up if you were entirely free to plan your day?
- Considering only your own ‘feeling best’ rhythm, at what time would you go to bed if you were entirely free to plan your day?
- How easy do you find it to get up each day?
- When you have no commitments the next day, how much later do you go to bed compared to your usual bedtime?
- One hears about ‘morning’ and ‘evening’ types of people. Which ONE of these types do you consider yourself to be?
Lower scores are associated with evening types—felt to correspond to the endogenous circadian period—and can help in narrowing the differential diagnosis of sleep-initiation complaints.20 Scores on the MEQ are interpreted as:
- 70 to 86: definite morning type
- 59 to 69: moderately morning type
- 42 to 58: neither type
- 31 to 41: moderately evening type
- 16 to 30: definite evening type
CASE CONTINUED: ‘Definite evening type’
Jason scores 28 on the MEQ, consistent with a “definite evening type.” Actigraphic monitoring is scheduled during a school holiday, when he is instructed to sleep according to his preferred schedule with the least possible restriction.
A clearly delayed sleep phase is evident, with the habitual sleep period occurring between 5 AM and 1 PM. Even on days when he was quite sleep-restricted because of an enforced wake time, sleep onset on the ensuing evening was substantially delayed, suggesting an obligate nature for the delayed sleep/wake schedule. Overall, Jason had few complaints with respect to impaired alertness while on this unrestricted schedule and experienced a much more stable mood.
Interventions
Without physiologic assessments, understanding the patient’s “natural” sleep schedule can allow for rational recommendations about using phototherapy and oral melatonin (Figure21). However, referral to a sleep specialist is required unless the general psychiatrist has experience in treating circadian rhythm sleep disorders.
Morning phototherapy. Properly timed morning bright light therapy (≥2,500 lux) has been shown to help DSPD patients achieve physiologically measured sleep phase advances, objective improvements in daytime alertness, and earlier reported bedtimes compared with controls.22 Unfortunately, the described 2-hour treatment duration make this research protocol clinically impractical, and most clinicians commence with a 30-minute duration of therapy, as described in the seasonal affective disorder literature.
Relatively new and widely available blue light boxes have been reported to exhibit at least equivalent efficacy to bright light devices (as reported in the literature pertaining to seasonal affective disorder), but with markedly decreased light intensity and fewer associated adverse effects.23 As the research addressing their use in the treatment of circadian rhythm sleep disorders is still emerging, their future role remains uncertain.
Precautions. Most psychiatrists would not perform a physiologic determination of a patient’s circadian phase, and further undesired phase delays can occur if phototherapy is administered before the core body temperature minimum (Tmin).24 Also, use caution if prescribing phototherapy to patients taking photosensitizing drugs and/or those with ocular or retinal pathology.20
Evening light avoidance. Whether or not you prescribe morning phototherapy, recommending that DSPD patients avoid evening light is essential to avoid further induction or exacerbation of phase delays. Protective eyewear is warranted in instances where these advisory precautions are insufficient (see Related Resources). Such an intervention has been shown effective in decreasing light exposure and undesired phase advances in studies involving subjects exposed to simulated shift work.25
Oral melatonin. Abundant evidence supports melatonin use in achieving phase advances in individuals with DSPD.26,27 A synergistic effect can be obtained when melatonin is combined with phototherapy.28
Proper timing of melatonin to achieve a maximal phase advance can be estimated based on the individual’s dim light melatonin onset (DLMO), which occurs approximately 14 hours after the habitual (unrestricted) wake time.29 Maximal phase advances appear to occur when melatonin is given approximately 6 hours before the DLMO.26 Thus, a rational practice is to recommend that patients take melatonin 8 hours after their natural wake time. Doses of ≤0.5 mg appear to achieve the maximal chronobiotic effect while avoiding an undesired hypnotic effect.30
Precautions. Verifying the purity of over-the-counter melatonin is difficult. A review by the National Academy of Sciences states that short-term use of melatonin, ≤10 mg/d, appears to be safe in healthy adults but recommends caution in children/adolescents and women of reproductive age. Doses recommended for circadian-based interventions are typically physiologic in nature (i.e., ≤0.5 mg), which may serve to mitigate these concerns.
Adverse effects such as headaches, somnolence, hypotension, hypertension, gastrointestinal upset, and exacerbation of alopecia areata have been reported at higher melatonin doses in healthy adults and at lower doses in persons with preexisting central nervous system, cardiovascular, gastrointestinal, or dermatologic conditions.31
Figure Light and melatonin phase response curves: Normal vs. delayed
This schematic compares ‘normal sleep’ phase response curves (PRCs) to light and exogenous melatonin with postulated PRCs for an individual with delayed sleep phase disorder (DSPD), presumed to be 5 hours ‘out of phase.’ Y-axis shows the direction and relative magnitude of phase shifts produced by light or melatonin at times shown on the x-axis. X-axis covers >24 hours to better illustrate the PRCs.
Relationships between ‘normal sleepers’ and DSPD patients are depicted by:
- rectangles (sleep period)
- triangles (core body temperature minimum [Tmin])
- arrows (dim light melatonin onsets [DLMOs]).
‘Normal’ sleep is shown to occur from midnight to 8 AM, and the DSPD patient’s sleep from 5 AM to 1 PM; DLMO and Tmin are similarly delayed by 5 hours in the DSPD patient. This schematic assumes that phase relationships are maintained in DSPD patients, which is not a certainty.
Source: Adapted from reference 21
CASE CONTINUED: Under the bright lights
Jason starts phototherapy treatment during his winter break, administering bright light daily upon natural awakening using a 10,000 lux light box for at least 30 minutes. As instructed, he gradually advances the time of administration by approximately 30 minutes every other day, striving for a nocturnal sleep period of 11 PM to 7 AM. He also wears protective eyewear to reduce light exposure during evening hours to avoid further delays in sleep phase. To further promote a phase advance, he takes oral melatonin, 0.5 mg/d at approximately 8 PM, as determined by his self-report and results of actigraphic recording.
Other options
Hypnotics. Little evidence supports the use of hypnotics in DSPD,32 and patients may show resistance to these drugs.33 Nevertheless, hypnotics can heighten confidence in the ability to initiate sleep in individuals with a concomitant conditioned insomnia.
With chronotherapy, patients are prescribed a sleep schedule that is delayed several hours incrementally until sleep is aligned to a target bedtime. The individual then is advised to rigorously maintain a regular sleep/wake schedule, repeating the process as necessary.
Although case reports have shown positive results with chronotherapy for DSPD,34 no controlled trials have demonstrated its efficacy or safety. One study reported high relapse rates,31 and 1 patient with DSPD developed free-running circadian rhythms.35 Clinical experience suggests chronotherapy is impractical for patients who must adhere to a fixed schedule.
Behavioral approaches
For an adolescent with DSPD, consider asking the school district to allow him or her a later school start-time. This alone often can substantially increase total sleep time and mitigate associated impairments.36 In all instances pursue and address external contributors to DSPD, such as poor sleep hygiene (including excessive caffeine use) and substance misuse.
Emphasize regular wake times, as arising later on weekends can cause phase delays.37 DSPD patients may have a concomitant conditioned insomnia that responds to evidence-based behavioral treatments.38
Whatever intervention you choose, schedule a follow-up appointment in approximately 2 months to evaluate patients’ progress and compliance. Encourage them to contact you with questions or concerns in the interim. Review sleep logs or actigraphy during this visit, and adjust the timing and/or nature of interventions as needed. Adolescents can be particularly noncompliant with clinical interventions, and therapeutic goals cannot be reached without their full investment.
Because no guidelines exist on how long to treat DSPD, stop on a “trial-and-error” basis when symptoms are controlled, and resume if they recur. Another approach is to maintain a desired sleep/wake schedule with bedtime melatonin and encourage continued adherence to other measures.
CASE CONTINUED: Maintenance therapy
Jason returns to the clinic approximately 10 weeks later. After an obviously concerted effort to adhere to treatment, his progress is quite remarkable. He rarely falls asleep later than 11 PM, so he is obtaining 2.5 hours more sleep each night before arising for school at 6:30 AM. Sleepiness at school is rarely problematic, and his mood is more stable.
He nevertheless describes a persistent tendency to retire and arise later and asks to continue melatonin and phototherapy. Because no guidelines exist for long-term therapy of DSPD, he is advised to switch melatonin to bedtime dosing (with a presumed phase-neutral “maintenance” effect), and to continue phototherapy as prescribed.
- Wyatt JK. Delayed sleep phase syndrome: pathophysiology and treatment options. Sleep. 2004;27(6):1195-1203.
- Crowley SJ, Acebo C, Carskadon MA. Sleep, circadian rhythms, and delayed sleep phase in adolescence. Sleep Med. 2007;8(6):602-612.
- National Sleep Foundation. Adolescent sleep needs and patterns: research report and resource guide. Washington, DC; 2000:1-30.
- Products designed to assist in the avoidance of light at improper times. www.lowbluelights.com.
Disclosure
Dr. Auger reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Khalsa SB, Jewett ME, Cajochen C, et al. A phase response curve to single bright light pulses in human subjects. J Physiol. 2003;549(Pt 3):945-952.
2. Quinto C, Gellido C, Chokroverty S, et al. Posttraumatic delayed sleep phase syndrome. Neurology. 2000;54(1):250-252.
3. Campbell SS, Murphy PJ. Delayed sleep phase disorder in temporal isolation. Sleep. 2007;30(9):1225-1228.
4. Uchiyama M, Okawa M, Shibui K, et al. Altered phase relation between sleep timing and core body temperature rhythm in delayed sleep phase syndrome and non-24-hour sleep-wake syndrome in humans. Neurosci Lett. 2000;294(2):101-104.
5. Aoki H, Ozeki Y, Yamada N. Hypersensitivity of melatonin suppression in response to light in patients with delayed sleep phase syndrome. Chronobiol Int. 2001;18(2):263-271.
6. Uchiyama M, Okawa M, Shibui K, et al. Poor compensatory function for sleep loss as a pathogenic factor in patients with delayed sleep phase syndrome. Sleep. 2000;23(4):553-558.
7. Mercer PW, Merritt SL, Cowell JM. Differences in reported sleep need among adolescents. J Adolesc Health. 1998;23(5):259-263.
8. Krahn LE, Pankratz VS, Harris AM, et al. Long-term outcome of adolescents with delayed sleep phase disorder [abstract]. Sleep. 2003;26:A115.-
9. Dagan Y, Stein D, Steinbock M, et al. Frequency of delayed sleep phase syndrome among hospitalized adolescent psychiatric patients. J Psychosom Res. 1998;45(1):15-20.
10. Thorpy MJ, Korman E, Spielman AJ, et al. Delayed sleep phase syndrome in adolescents. J Adolesc Health Care. 1998;9(1):22-27.
11. Regestein QR, Monk TH. Delayed sleep phase syndrome: a review of the clinical aspects. Am J Psychiatry. 1995;152(4):602-608.
12. Pelayo RP, Thorpy MJ, Glovinsky P. Prevalence of delayed sleep phase syndrome among adolescents [abstract]. Sleep Res. 1988;17:391.-
13. Gau SF, Soong WT. The transition of sleep-wake patterns in early adolescence. Sleep. 2003;26(4):449-454.
14. Beersma DG, Gordijn MC. Circadian control of the sleep-wake cycle. Physiol Behav. 2007;90(2-3):190-195.
15. Ralph MR, Foster RG, Davis FC, et al. Transplanted suprachiasmatic nucleus determines circadian period. Science. 1990;247(4945):975-978.
16. Waterhouse J, DeCoursey PJ. Chronobiology: biological timekeeping. Sunderland, MA: Sinauer Associates, Inc. Publishers; 2004:291-323.
17. American Academy of Sleep Medicine. International classification of sleep disorders. Diagnostic and coding manual. 2nd ed. Westchester, IL: American Academy of Sleep Medicine; 2005.
18. Bradshaw DA, Yanagi MA, Pak ES, et al. Nightly sleep duration in the 2-week period preceding multiple sleep latency testing. J Clin Sleep Med. 2007;3(6):613-619.
19. Horne JA, Ostberg O. A self-assessment questionnaire to determine morningness-eveningness in human circadian rhythms. Int J Chronobiol. 1976;4(2):97-110.
20. Sack RL, Auckley D, Auger RR, et al. Circadian rhythm sleep disorders: Part I. basic principles, shift work and jet lag disorders. Sleep. 2007;30(11):1460-1483.
21. Burgess HJ, Sharkey KM, Eastman CI. Bright light, dark and melatonin can promote circadian adaptation in night shift workers. Sleep Med Rev. 2002;6(5):407-420.
22. Rosenthal NE, Joseph-Vanderpool JR, Levendosky AA, et al. Phase-shifting effects of bright morning light as treatment for delayed sleep phase syndrome. Sleep. 1990;13(4):354-361.
23. Glickman G, Byrne B, Pineda C, et al. Light therapy for seasonal affective disorder with blue narrow-band light-emitting diodes (LEDs). Biol Psychiatry. 2006;59:502-507.
24. Czeisler C, Wright K, Jr. Influence of light on circadian rhythmicity in humans. New York, NY: Marcel Dekker; 1999:149-180.
25. Crowley SJ, Lee C, Tseng CY, et al. Combinations of bright light, scheduled dark, sunglasses, and melatonin to facilitate circadian entrainment to night shift work. J Biol Rhythms. 2003;18(6):513-523.
26. Mundey K, Benloucif S, Harsanyi K, et al. Phase-dependent treatment of delayed sleep phase syndrome with melatonin. Sleep. 2005;28(10):1271-1278.
27. Sack RL, Auckley D, Auger RR, et al. Circadian rhythm sleep disorders: Part II, advanced sleep phase disorder, delayed sleep phase disorder, free-running disorder, and irregular sleep-wake rhythm. Sleep. 2007;30(11):1484-1501.
28. Revell VL, Burgess HJ, Gazda CJ, et al. Advancing human circadian rhythms with afternoon melatonin and morning intermittent bright light. J Clin Endocrinol Metab. 2006;91(1):54-59.
29. Burgess HJ, Eastman CI. The dim light melatonin onset following fixed and free sleep schedules. J Sleep Res. 2005;14(3):229-237.
30. Lewy AJ. Clinical applications of melatonin in circadian disorders. Dialog Clin Neurosci. 2003;5:399-413.
31. Committee on the Framework for Evaluating the Safety of Dietary Supplements FaNB, Board on Life Sciences, Institute of Medicine and National Research Council of the National Academies. Dietary supplements: a framework for evaluating safety. Washington, DC: The National Academies Press; 2005.
32. Ito A, Ando K, Hayakawa T, et al. Long-term course of adult patients with delayed sleep phase syndrome. Jpn J Psychiatry Neurol. 1993;47(3):563-567.
33. Auger RR. Circadian rhythm sleep disorder, delayed sleep phase type (pediatric case). In: Winkelman JW (chair), Henderson JH, Kotagal S, et al, eds. Case book of sleep medicine. Westchester, IL: American Academy of Sleep Medicine; 2008:195-199.
34. Czeisler C, Weitzman E, Moore, et al. Chronotherapy: resetting the circadian clocks of patients with delayed sleep phase insomnia. Sleep. 1981;4:1-21.
35. Oren DA, Wehr TA. Hypernyctohemeral syndrome after chronotherapy for delayed sleep phase syndrome. N Engl J Med. 1992;327(24):1762.-
36. Wahlstrom K. Changing times: findings from the first longitudinal study of later high school start times. NASSP Bulletin. 2002;86(633):3-21.
37. Burgess HJ, Eastman CI. A late wake time phase delays the human dim light melatonin rhythm. Neurosci Lett. 2006;395(3):191-195.
38. Morgenthaler T, Kramer M, Alessi C, et al. Practice parameters for the psychological and behavioral treatment of insomnia: an update. An American Academy of Sleep Medicine report. Sleep. 2006;29(11):1415-1419.
1. Khalsa SB, Jewett ME, Cajochen C, et al. A phase response curve to single bright light pulses in human subjects. J Physiol. 2003;549(Pt 3):945-952.
2. Quinto C, Gellido C, Chokroverty S, et al. Posttraumatic delayed sleep phase syndrome. Neurology. 2000;54(1):250-252.
3. Campbell SS, Murphy PJ. Delayed sleep phase disorder in temporal isolation. Sleep. 2007;30(9):1225-1228.
4. Uchiyama M, Okawa M, Shibui K, et al. Altered phase relation between sleep timing and core body temperature rhythm in delayed sleep phase syndrome and non-24-hour sleep-wake syndrome in humans. Neurosci Lett. 2000;294(2):101-104.
5. Aoki H, Ozeki Y, Yamada N. Hypersensitivity of melatonin suppression in response to light in patients with delayed sleep phase syndrome. Chronobiol Int. 2001;18(2):263-271.
6. Uchiyama M, Okawa M, Shibui K, et al. Poor compensatory function for sleep loss as a pathogenic factor in patients with delayed sleep phase syndrome. Sleep. 2000;23(4):553-558.
7. Mercer PW, Merritt SL, Cowell JM. Differences in reported sleep need among adolescents. J Adolesc Health. 1998;23(5):259-263.
8. Krahn LE, Pankratz VS, Harris AM, et al. Long-term outcome of adolescents with delayed sleep phase disorder [abstract]. Sleep. 2003;26:A115.-
9. Dagan Y, Stein D, Steinbock M, et al. Frequency of delayed sleep phase syndrome among hospitalized adolescent psychiatric patients. J Psychosom Res. 1998;45(1):15-20.
10. Thorpy MJ, Korman E, Spielman AJ, et al. Delayed sleep phase syndrome in adolescents. J Adolesc Health Care. 1998;9(1):22-27.
11. Regestein QR, Monk TH. Delayed sleep phase syndrome: a review of the clinical aspects. Am J Psychiatry. 1995;152(4):602-608.
12. Pelayo RP, Thorpy MJ, Glovinsky P. Prevalence of delayed sleep phase syndrome among adolescents [abstract]. Sleep Res. 1988;17:391.-
13. Gau SF, Soong WT. The transition of sleep-wake patterns in early adolescence. Sleep. 2003;26(4):449-454.
14. Beersma DG, Gordijn MC. Circadian control of the sleep-wake cycle. Physiol Behav. 2007;90(2-3):190-195.
15. Ralph MR, Foster RG, Davis FC, et al. Transplanted suprachiasmatic nucleus determines circadian period. Science. 1990;247(4945):975-978.
16. Waterhouse J, DeCoursey PJ. Chronobiology: biological timekeeping. Sunderland, MA: Sinauer Associates, Inc. Publishers; 2004:291-323.
17. American Academy of Sleep Medicine. International classification of sleep disorders. Diagnostic and coding manual. 2nd ed. Westchester, IL: American Academy of Sleep Medicine; 2005.
18. Bradshaw DA, Yanagi MA, Pak ES, et al. Nightly sleep duration in the 2-week period preceding multiple sleep latency testing. J Clin Sleep Med. 2007;3(6):613-619.
19. Horne JA, Ostberg O. A self-assessment questionnaire to determine morningness-eveningness in human circadian rhythms. Int J Chronobiol. 1976;4(2):97-110.
20. Sack RL, Auckley D, Auger RR, et al. Circadian rhythm sleep disorders: Part I. basic principles, shift work and jet lag disorders. Sleep. 2007;30(11):1460-1483.
21. Burgess HJ, Sharkey KM, Eastman CI. Bright light, dark and melatonin can promote circadian adaptation in night shift workers. Sleep Med Rev. 2002;6(5):407-420.
22. Rosenthal NE, Joseph-Vanderpool JR, Levendosky AA, et al. Phase-shifting effects of bright morning light as treatment for delayed sleep phase syndrome. Sleep. 1990;13(4):354-361.
23. Glickman G, Byrne B, Pineda C, et al. Light therapy for seasonal affective disorder with blue narrow-band light-emitting diodes (LEDs). Biol Psychiatry. 2006;59:502-507.
24. Czeisler C, Wright K, Jr. Influence of light on circadian rhythmicity in humans. New York, NY: Marcel Dekker; 1999:149-180.
25. Crowley SJ, Lee C, Tseng CY, et al. Combinations of bright light, scheduled dark, sunglasses, and melatonin to facilitate circadian entrainment to night shift work. J Biol Rhythms. 2003;18(6):513-523.
26. Mundey K, Benloucif S, Harsanyi K, et al. Phase-dependent treatment of delayed sleep phase syndrome with melatonin. Sleep. 2005;28(10):1271-1278.
27. Sack RL, Auckley D, Auger RR, et al. Circadian rhythm sleep disorders: Part II, advanced sleep phase disorder, delayed sleep phase disorder, free-running disorder, and irregular sleep-wake rhythm. Sleep. 2007;30(11):1484-1501.
28. Revell VL, Burgess HJ, Gazda CJ, et al. Advancing human circadian rhythms with afternoon melatonin and morning intermittent bright light. J Clin Endocrinol Metab. 2006;91(1):54-59.
29. Burgess HJ, Eastman CI. The dim light melatonin onset following fixed and free sleep schedules. J Sleep Res. 2005;14(3):229-237.
30. Lewy AJ. Clinical applications of melatonin in circadian disorders. Dialog Clin Neurosci. 2003;5:399-413.
31. Committee on the Framework for Evaluating the Safety of Dietary Supplements FaNB, Board on Life Sciences, Institute of Medicine and National Research Council of the National Academies. Dietary supplements: a framework for evaluating safety. Washington, DC: The National Academies Press; 2005.
32. Ito A, Ando K, Hayakawa T, et al. Long-term course of adult patients with delayed sleep phase syndrome. Jpn J Psychiatry Neurol. 1993;47(3):563-567.
33. Auger RR. Circadian rhythm sleep disorder, delayed sleep phase type (pediatric case). In: Winkelman JW (chair), Henderson JH, Kotagal S, et al, eds. Case book of sleep medicine. Westchester, IL: American Academy of Sleep Medicine; 2008:195-199.
34. Czeisler C, Weitzman E, Moore, et al. Chronotherapy: resetting the circadian clocks of patients with delayed sleep phase insomnia. Sleep. 1981;4:1-21.
35. Oren DA, Wehr TA. Hypernyctohemeral syndrome after chronotherapy for delayed sleep phase syndrome. N Engl J Med. 1992;327(24):1762.-
36. Wahlstrom K. Changing times: findings from the first longitudinal study of later high school start times. NASSP Bulletin. 2002;86(633):3-21.
37. Burgess HJ, Eastman CI. A late wake time phase delays the human dim light melatonin rhythm. Neurosci Lett. 2006;395(3):191-195.
38. Morgenthaler T, Kramer M, Alessi C, et al. Practice parameters for the psychological and behavioral treatment of insomnia: an update. An American Academy of Sleep Medicine report. Sleep. 2006;29(11):1415-1419.
Fibromyalgia: Psychiatric drugs target CNS-linked symptoms
WEB AUDIO
Listen to Dr. Stanford discuss,
"Is fibromyalgia a somatoform disorder?
Patients with fibromyalgia are a heterogeneous group, yet many describe a common experience: seeing multiple physicians who seem unable or unwilling to provide a diagnosis or treat their symptoms. This situation may be changing with the recent FDA approval of an anticonvulsant and 2 antidepressants for managing fibromyalgia symptoms.
These medications—pregabalin, duloxetine, and milnacipran—reflect a revised understanding of fibromyalgia as a CNS condition, rather than an inflammatory process in the muscles or connective tissue. As a result, psychiatrists—because of our experience with CNS phenomena and managing antidepressant and anticonvulsant medications—are likely to play a larger role in treating fibromyalgia.
CASE REPORT: ‘Just too tired’
Ms. D, age 50, has a history of migraine headaches and is referred by her primary physician for evaluation of depression and anxiety. She reports deteriorating mood over 6 months, beginning when a minor car accident left her “very sore the next day.”
Ms. D reports she is depressed because she feels “just too tired” after work to keep up with social activities or housework. Her physician’s referral notes a normal physical exam except for tenderness over her upper back and hips. Laboratory testing is negative.
Making the diagnosis
American College of Rheumatology (ACR) criteria for fibromyalgia require widespread pain for at least 3 months. “Widespread” is defined as pain in the axial skeleton, left and right sides of the body, and above and below the waist. Pain must be found in at least 11 of 18 tender point sites on digital palpation using a force of approximately 4 kg/cm2.1 For many fibromyalgia patients, however, musculoskeletal pain is not their most problematic symptom (Table 1). They may suffer:
- migraine and tension headaches (10% to 80% of patients)
- irritable bowel syndrome (32% to 80%)2
- mood disorders (major depressive disorder [62%], bipolar disorder [11%])
- anxiety disorders (panic disorder [29%], posttraumatic stress disorder [21%], social phobia [19%]).3
Because patients with fibromyalgia often meet criteria for somatization or somatoform disorders, how to classify them—as medically or psychiatrically ill—is controversial. Some patients believe their mood or anxiety problem stems from the difficulty they experience dealing with their physical symptoms, and if they could feel better physically they would not be depressed or anxious. Others believe their psychiatric symptoms impede their ability to help themselves feel better.
Consider fibromyalgia in any patient with widespread pain of unknown cause. Before making the diagnosis, rule out other illnesses that present with similar symptoms (Table 3). Because many patients newly diagnosed with fibromyalgia worry that something “more serious” may be going on, confirm the diagnosis with appropriate testing and physical examination, usually by a rheumatologist or primary care physician.
Table 1
Medical and cognitive symptoms related to fibromyalgia
| Neurologic Tension/migraine headache |
| Psychiatric Memory and cognitive difficulties Mood disturbance Anxiety disorders |
| Ear, nose, throat Sicca symptoms Vasomotor rhinitis Vestibular complaints |
| Cardiovascular Neurally mediated hypotension Mitral valve prolapse Noncardiac chest pain |
| Gastrointestinal Esophageal dysmotility Irritable bowel syndrome |
| Urological Interstitial cystitis |
| Gynecological Vulvodynia Chronic pelvic pain |
| Oral/dental Temporomandibular joint syndrome |
| Other (general) Chronic fatigue syndrome Sleep disturbances Idiopathic low back pain Multiple chemical sensitivity |
Fibromyalgia: Structured interview for diagnosis
| A. Generalized pain affecting the axial, plus upper and lower segments, plus left and rights sides of the body |
| Either B or C: |
| B. At least 11 of 18 reproducible tender points |
C. At least 4 of the following symptoms:
|
| D. It cannot be established that disturbance was due to another systematic condition |
| Source: Reference 4 |
Differentiating fibromyalgia from illnesses with similar symptoms
| Illness | Tests to differentiate from primary fibromyalgia |
|---|---|
| Rheumatic diseases Osteoarthritis Spondyloarthropathies, rheumatoid arthritis Systemic lupus erythematosus, polymyalgia rheumatica Osteomalacia Myopathy | Radiographs Rheumatic markers (antinuclear antibody, rheumatoid factor, antibodies) Inflammatory markers (ESR, C-reactive protein) Vitamin D level CPK |
| Neurologic Multiple sclerosis, Chiari’s malformation, spinal stenosis, radiculopathy Neuropathy | MRI EMG |
| Endocrine Hypothyroidism Diabetes | TSH Basic chemistry panel with fasting glucose |
| Other Infectious Lyme disease Hepatitis Anemia Cancers | CBC Lyme titer Hepatitis antibody panel, liver function tests Hemoglobin/hematocrit Routine cancer screening tests, bone scan, blood chemistries specific for suspected primary cancer |
| ESR: erythrocyte sedimentation rate; CPK: creatine phosphokinase; EMG: electromyography; TSH: thyroid-stimulating hormone; CBC: complete blood count | |
CASE CONTINUED: Central pain sensitization
As you elicit more details about Ms. D’s mood, she continues to focus on her physical symptoms. She states that some days she wishes to die because her pain gets so bad, but she denies any plan or intent to harm herself. She worries that her symptoms will worsen and that she will become completely disabled.
Her primary physician attempted to relieve Ms. D’s pain with multiple trials of nonsteroidal anti-inflammatory drugs (NSAIDs) and cyclobenzaprine. She says she gained no benefit from the NSAIDs and discontinued the muscle relaxant because it made her too sleepy. Fibromyalgia affects 3.5% of women and 0.5% of men.5 It runs in families with histories of fibromyalgia and major mood and anxiety disorders, suggesting genetic links.6 Defects in genes controlling serotonin and norepinephrine have been implicated.7-9
Fibromyalgia patients show lower levels of serotonin, norepinephrine, and dopamine metabolites in cerebrospinal fluid (CSF), compared with controls.10 These neurotransmitters may inhibit descending pain pathways in the CNS, and low levels in the brain and spinal cord may inhibit CNS regulation of pain impulses from the periphery.11
Although many patients describe muscle pain, evidence suggests central pain augmentation rather than an abnormality of muscle or connective tissue.12 Some studies have found evidence of “windup,” in which second-order neurons in the spinal cord become sensitized by repeated signals from first-order neurons in the periphery, resulting in amplified and prolonged pain signals traveling to the brain.13
Levels of substance P—a primary transmitter of pain impulses—are significantly higher in CSF of fibromyalgia patients compared with controls.14 This finding, in addition to low levels of serotonin and norepinephrine, indicates that pain signals are ascending unchecked to be processed by the brain.
- Twice as much pressure was required before controls rated their pain at a level similar to that of fibromyalgia patients.
- When controls and fibromyalgia patients reported similar pain, a very high degree of overlap was seen in brain areas responsible for pain processing. This indicates that fibromyalgia patients and controls were experiencing the pain they reported in the same way.15
Treating the whole patient
As a clinician who specializes in fibromyalgia, I counteract my patients’ and my own frustration with this condition by structuring office visits, determining realistic treatment goals, and treating all symptoms as part of a common syndrome rather than individual illnesses.
Structure office visits. Before every visit, have patients rate each symptom domain and write their top 2 or 3 concerns for that day (Click here for a sample form). Focusing on the patient’s most troublesome symptoms can help both of you feel greater satisfaction with treatment.
Educate patients. Ask them to discuss their beliefs about fibromyalgia; many know others with this condition or have researched diagnosis and treatment. Before developing a treatment plan, explain that their symptoms are chronic and all part of the same syndrome. Describe their pain as a complex phenomenon with possible peripheral and CNS components. Guide them to reputable Web sites and resources (see Related Resources).
Set realistic expectations. Many patients expect to resume an energetic and pain-free life, which usually is not the case with fibromyalgia (Box). Most medications are considered successful if they reduce pain by 30% to 50%, and side effects can be problematic. Discuss side effects before treatment begins to reduce patients’ anxiety and improve compliance in the first weeks.
Cognitive-behavioral therapy (CBT) for fibromyalgia incorporates relaxation techniques, helping patients view symptoms as manageable, reinforcing adaptive coping skills, and teaching them how to monitor thoughts, feelings, and behavior to change the view that they are helpless victims. A modest course of 6 weekly group CBT sessions significantly improved physical functioning in 25% of fibromyalgia patients (n=76) compared with 12% in a standard-care group (n=69), even though patients’ pain severity did not improve.16
Lifestyle changes are as important as medications in controlling fibromyalgia symptoms. In addition to exercise, recommend that patients:
- follow a daily routine
- pace activity to avoid exacerbating symptoms
- reduce stress.
BELIEF: ‘A magic pill exists that will resolve all my symptoms and have no side effects’
Clinical evidence: Most medications that have been studied were effective in 30% to 50% of patients and reduced pain scores by 30% to 50%.
Patient education: Explain to the patient with a pain rating of 7 at the first visit that achieving a pain level of 3 to 4 may be possible with treatment. Even with successful treatment, symptoms may flare intermittently. As with any treatment, adverse effects may occur. Discuss these, so the patient is not surprised.
BELIEF: ‘I can’t exercise’
Clinical evidence: Most patients experience more fatigue and pain with physical activity, but exercise is important to maintain physical function.
Patient education: When discussing an exercise program, focus on what the patient can do. Most patients attempt too much, too soon; advise them to start at a tolerable level (such as 2 to 3 minutes of aerobic activity daily for the first week) and gradually increase as tolerated.
BELIEF: ‘You (the psychiatrist) can make me feel better’
Clinical evidence: Psychiatrists can help by prescribing appropriate medications, but much of the burden falls on the patient to maintain a healthy, active lifestyle and to manage stressors in an adaptive manner.
Patient education: A fibromyalgia patient may find relief with a medication, but symptoms may flare if they ‘overdo’ and take on too many physical or emotional stressors. A consistent, healthy routine is ideal.
BELIEF: ‘I will eventually become disabled by fibromyalgia’
Clinical evidence: Despite little long-term research on fibromyalgia patients, most evidence points to a chronic, fluctuating syndrome that does not worsen with age. Factors that may worsen symptoms include uncontrolled comorbid conditions, chronic opiate use, inactivity, and deconditioning.
Patient education: Discourage long-term physical disability; exercise and maintaining an active daily routine helps patients avoid focusing in a nonadaptive manner on their dysfunction and symptoms.
Source: Sharon B. (Shay) Stanford, MD
New direction with medications
Pregabalin is an anticonvulsant that binds to the alpha-2-delta subunits of neurons’ voltage-gated calcium channels. This activity reduces calcium influx at nerve terminals and inhibits release of excitatory neurotransmitters, such as substance P and glutamate.18 In June 2007, pregabalin was the first medication FDA-approved for fibromyalgia.
Two placebo-controlled trials19,20 showed that pregabalin at 150 mg bid, 225 mg bid, or 300 mg bid significantly reduced weekly mean pain scores in fibromyalgia patients. Click here for details of these trials. The most common side effects—dizziness, somnolence, peripheral edema, blurred vision, and weight gain—were regarded as mild to moderate in 87% of patients.21
Although a dosage of 300 mg bid also was studied, the FDA approved pregabalin at dosages of 150 mg bid and 225 mg bid for fibromyalgia.22
Duloxetine is a serotonin/norepinephrine reuptake inhibitor (SNRI) thought to inhibit dorsal horn neurons’ response to peripheral pain signals by increasing serotonin and norepinephrine in the brain and spinal cord. This SNRI was first FDA-approved for diabetic peripheral neuropathic pain and major depressive disorder. Approval for fibromyalgia at 60 mg/d in June 2008 was based on 2 placebo-controlled, double-blind, 12-week trials comprising 874 patients.23,24Click here for detailed findings of these studies and a 6-month fixed-dose trial.25
Milnacipran is an SNRI that was approved for treating fibromyalgia in January 2009 at dosages of 50 mg bid and 100 mg bid. Like other SNRIs, milnacipran is thought to work by inhibiting pain signals through increasing serotonin and norepinephrine in the brain and spinal cord. Milnacipran has a higher selectivity for norepinephrine reuptake compared with duloxetine, which may mean these medications will have different effects in different patients. Although milnacipran is approved as an antidepressant in other countries, the FDA has not approved it for treating depression in the United States.
Click here for details of a 15-week, double-blind, placebo-controlled trial of milnacipran in patients with fibromyalgia. Side effects in clinical trials were similar to those of duloxetine, with nausea, constipation, and increased sweating being most prominent.26
Other medications, such as the first-line agent amitriptyline, have shown beneficial effects in fibromyalgia but are not FDA-approved for this indication (Table 4).27-32
Choosing medications. When prescribing one of the FDA-approved medications to treat fibromyalgia, consider their benefits and side effects.
Pregabalin may be a beneficial first choice for patients who report pain and sleep as major issues. Although the medication’s label recommends starting with twice-daily dosing, patients might better tolerate an initial dose of 50 to 75 mg in the evening, with the morning dose added later. Pregabalin can be useful in patients taking multiple medications because of its renal clearance, resulting in low risk of interactions with drugs metabolized by liver enzymes. It also can be useful in patients who have not tolerated antidepressants in the past or in whom antidepressants are contraindicated.
If a patient has a history of depression or discontinuing medications because of sedating side effects, an antidepressant such as duloxetine or milnacipran may be more successful than starting with pregabalin. In general, if a patient does not respond to one of these SNRIs, moving on to the other might help. Milnacipran’s more selective effect on norepinephrine could be beneficial for some patients, especially those with excessive fatigue. Others, especially those with a high level of anxiety, might respond better to a more balanced SNRI such as duloxetine.
Table 4
Off-label medications shown to benefit patients with fibromyalgia
| Drug | Comment |
|---|---|
| Amitriptyline27,28 | Considered first-line because of studies supporting its use, low cost, and wide availability; may be associated with more side effects than newer medications |
| Gabapentin29 | Possible alternative to pregabalin but may not be as well tolerated |
| Tramadol30 | May help with breakthrough pain; use with extreme caution in patients taking antidepressants because of serotonin syndrome risk |
| Fluoxetine31 | Dosages of 40 to 60 mg/d may help patients who do not tolerate SNRIs |
| Venlafaxine32 | Dosages of 150 to 225 mg/d may be an alternative to other SNRIs |
| SNRIs: serotonin/norepinephrine reuptake inhibitors | |
CASE CONTINUED: Not as hopeless
Ms. D’s primary care physician confirms your presumptive diagnosis of fibromyalgia. He prescribes a trial of amitriptyline, which she does not tolerate well because of sedation and weight gain. At her next psychiatric visit, she tells you she remains very frustrated about her physical symptoms and reports that her doctor “has given up on me.”
You discuss what a fibromyalgia diagnosis means to her and educate her about the syndrome. You refer her to a colleague who does CBT with chronic pain patients and start her on a low dose of duloxetine (30 mg once daily) to minimize side effects. You discuss possible side effects and that she may need a higher dose to notice improvement in her pain. She seems receptive to starting a graded exercise program, and you encourage her to reduce physical and emotional stress in her life.
- Arthritis Foundation. Fibromyalgia. www.arthritis.org/disease-center.php?disease_id=10.
- National Fibromyalgia Association. www.fmaware.org.
- Self-management program for patients with fibromyalgia, cosponsored by the National Fibromyalgia Association and Eli Lilly and Company. www.knowfibro.com.
- Amitriptyline • Elavil, Endep
- Cyclobenzaprine • Flexeril
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Milnacipran • Savella
- Pregabalin • Lyrica
- Tramadol • Ultram, Ultracet
- Venlafaxine • Effexor, Effexor XR
Dr. Stanford receives grant/research support from Eli Lilly and Company, Pfizer, Cypress Bioscience, and Allergan.
1. Wolfe F, Smythe HA, Yunus MB, et al. The American College of Rheumatology 1990 criteria for the classification of fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum. 1990;33(2):160-172.
2. Aaron LA, Buchwald D. Chronic diffuse musculoskeletal pain, fibromyalgia and co-morbid unexplained clinical conditions. Best Prac Res Clin Rheumatol. 2003;17(4):563-574.
3. Arnold LM, Hudson JI, Keck PE, et al. Comorbidity of fibromyalgia and psychiatric disorders. J Clin Psychiatry. 2006;67(8):1219-1225.
4. Pope HG, Jr, Hudson JI. A supplemental interview for forms of “affective spectrum disorder.” Int J Psychiatry Med. 1991;21(3):205-232.
5. Wolfe F, Ross K, Anderson J, et al. The prevalence and characteristics of fibromyalgia in the general population. Arthritis Rheum. 1995;38(1):19-28.
6. Arnold LM, Hudson JI, Hess EV, et al. Family study of fibromyalgia. Arthritis Rheum. 2004;50(3):944-952.
7. Bondy B, Spaeth M, Offenbaecher M, et al. The T102C polymorphism of the 5-HT2A-receptor gene in fibromyalgia. Neurobiol Dis. 1999;6(5):433-439.
8. Offenbaecher M, Bondy B, de Jonge S, et al. Possible association of fibromyalgia with a polymorphism in the serotonin transporter gene regulatory region. Arthritis Rheum. 1999;42(11):2482-2488.
9. Gürsoy S, Erdal E, Herken H, et al. Significance of catechol-O-methyltransferase gene polymorphism in fibromyalgia syndrome. Rheumatol Int. 2003;23(3):104-107.
10. Russell IJ, Vaeroy H, Javors M, et al. Cerebrospinal fluid biogenic amine metabolites in fibromyalgia/fibrositis syndrome and rheumatoid arthritis. Arthritis Rheum. 1992;35(5):550-556.
11. Fields HL, Basbaum AI. In: Wall PD, Melzack R, eds. Textbook of pain. 4th ed. New York, NY: Churchill Livingstone; 1999: 309-329.
12. Clauw DJ, Crofford LJ. Chronic widespread pain and fibromyalgia: what we know, and what we need to know. Best Pract Res Clin Rheumatol. 2003;17(4):685-701.
13. Staud R, Cannon RC, Mauderli AP, et al. Temporal summation of pain from mechanical stimulation of muscle tissue in normal controls and subjects with fibromyalgia syndrome. Pain. 2003;102(1-2):87-95.
14. Russell IJ, Orr MD, Littman B, et al. Elevated cerebrospinal fluid levels of substance P in patients with the fibromyalgia syndrome. Arthritis Rheum. 1994;37(11):1593-1601.
15. Gracely RH, Petzke F, Wolf JM, et al. Functional magnetic resonance imaging evidence of augmented pain processing in fibromyalgia. Arthritis Rheum. 2002;46(5):1333-1343.
16. Williams DA, Cary MA, Groner KH. Improving physical functional status in patients with fibromyalgia: a brief cognitive behavioral intervention. J Rheumatol. 2002;29:1280-1286.
17. Busch AJ, Schachter CL, Overend TJ, et al. Exercise for fibromyalgia: a systematic review. J Rheumatol. 2008;35(6):1130-1144.
18. Field MJ, Cox PJ, Stott E. Identification of the alpha2-delta-1 subunit of voltage-dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. Proc Natl Acad Sci USA. 2006;103(46):17537-17542.
19. Arnold LM, Russel IJ, Diri EW, et al. A 14-week, randomized, double-blind, placebo-controlled, monotherapy trial of pregabalin in patients with fibromyalgia. J Pain. 2008;9(9):792-805.
20. Mease PJ, Russel IJ, Arnold LM, et al. A randomized, double-blind, placebo-controlled, phase III trial of pregabalin in the treatment of patients with fibromyalgia. J Rheumatol. 2008;35(3):502-514.
21. Crofford LJ, Mease J, Simpson SL, et al. Fibromyalgia relapse evaluation and efficacy for durability of meaningful relief (FREEDOM): a 6-month, double-blind, placebo-controlled trial with pregabalin. Pain. 2008;136(3):419-431.
22. Pregabalin [package insert]. New York, NY: Pfizer, Inc.; 2004.
23. Arnold LM, Pritchett YL, D’Souza DN, et al. Duloxetine for the treatment of fibromyalgia in women: pooled results from two randomized, placebo-controlled clinical trials. J Womens Health (Larchmt). 2007;16(8):1145-1156.
24. Arnold LM, Lu Y, Crofford LJ, et al. A double-blind, multicenter trial comparing duloxetine with placebo in the treatment of fibromyalgia patients with or without major depressive disorder. Arthritis Rheum. 2004;50(9):2974-2984.
25. Russell IJ, Mease PJ, Smith TR, et al. Efficacy and safety of duloxetine for treatment of fibromyalgia in patients with or without major depressive disorder: results from a 6-month, randomized, double-blind, placebo-controlled, fixed-dose trial. Pain. 2008;136(3):432-444.
26. Clauw DJ, Mease P, Palmer RH, et al. Milnacipran for the treatment of fibromyalgia in adults: a 15-week, multicenter, randomized, double-blind, placebo-controlled, multiple-dose clinical trial. Clin Ther. 2008;30(11):1988-2004.
27. Goldenberg DL, Felson DT, Dinerman H. A randomized, controlled trial of amitriptyline and naproxen in the treatment of patients with fibromyalgia. Arthritis Rheum. 1986;29(11):1371-1377.
28. Hannonen P, Malminiemi K, Yli-Kerttula U, et al. A randomized, double-blind, placebo-controlled study of moclobemide and amitriptyline in the treatment of fibromyalgia in females without psychiatric disorder. Br J Rheumatol. 1998;37:1279-1286.
29. Arnold LM, Goldenberg DL, Stanford SB, et al. Gabapentin in the treatment of fibromyalgia: a randomized, double-blind, placebo-controlled, multicenter trial. Arthritis Rheum. 2007;56:1336-1344.
30. Bennett RM, Schein J, Kosinski MR, et al. Impact of fibromyalgia pain on health-related quality of life before and after treatment with tramadol/ acetaminophen. Arthritis Rheum. 2005;53:519-527.
31. Arnold LM, Hess EV, Hudson JI, et al. Randomized, placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med. 2002;112:191-197.
32. Dwight MM, Arnold LM, O’Brien H, et al. An open clinical trial of venlafaxine treatment of fibromyalgia. Psychosomatics. 1998;39:14-17.
WEB AUDIO
Listen to Dr. Stanford discuss,
"Is fibromyalgia a somatoform disorder?
Patients with fibromyalgia are a heterogeneous group, yet many describe a common experience: seeing multiple physicians who seem unable or unwilling to provide a diagnosis or treat their symptoms. This situation may be changing with the recent FDA approval of an anticonvulsant and 2 antidepressants for managing fibromyalgia symptoms.
These medications—pregabalin, duloxetine, and milnacipran—reflect a revised understanding of fibromyalgia as a CNS condition, rather than an inflammatory process in the muscles or connective tissue. As a result, psychiatrists—because of our experience with CNS phenomena and managing antidepressant and anticonvulsant medications—are likely to play a larger role in treating fibromyalgia.
CASE REPORT: ‘Just too tired’
Ms. D, age 50, has a history of migraine headaches and is referred by her primary physician for evaluation of depression and anxiety. She reports deteriorating mood over 6 months, beginning when a minor car accident left her “very sore the next day.”
Ms. D reports she is depressed because she feels “just too tired” after work to keep up with social activities or housework. Her physician’s referral notes a normal physical exam except for tenderness over her upper back and hips. Laboratory testing is negative.
Making the diagnosis
American College of Rheumatology (ACR) criteria for fibromyalgia require widespread pain for at least 3 months. “Widespread” is defined as pain in the axial skeleton, left and right sides of the body, and above and below the waist. Pain must be found in at least 11 of 18 tender point sites on digital palpation using a force of approximately 4 kg/cm2.1 For many fibromyalgia patients, however, musculoskeletal pain is not their most problematic symptom (Table 1). They may suffer:
- migraine and tension headaches (10% to 80% of patients)
- irritable bowel syndrome (32% to 80%)2
- mood disorders (major depressive disorder [62%], bipolar disorder [11%])
- anxiety disorders (panic disorder [29%], posttraumatic stress disorder [21%], social phobia [19%]).3
Because patients with fibromyalgia often meet criteria for somatization or somatoform disorders, how to classify them—as medically or psychiatrically ill—is controversial. Some patients believe their mood or anxiety problem stems from the difficulty they experience dealing with their physical symptoms, and if they could feel better physically they would not be depressed or anxious. Others believe their psychiatric symptoms impede their ability to help themselves feel better.
Consider fibromyalgia in any patient with widespread pain of unknown cause. Before making the diagnosis, rule out other illnesses that present with similar symptoms (Table 3). Because many patients newly diagnosed with fibromyalgia worry that something “more serious” may be going on, confirm the diagnosis with appropriate testing and physical examination, usually by a rheumatologist or primary care physician.
Table 1
Medical and cognitive symptoms related to fibromyalgia
| Neurologic Tension/migraine headache |
| Psychiatric Memory and cognitive difficulties Mood disturbance Anxiety disorders |
| Ear, nose, throat Sicca symptoms Vasomotor rhinitis Vestibular complaints |
| Cardiovascular Neurally mediated hypotension Mitral valve prolapse Noncardiac chest pain |
| Gastrointestinal Esophageal dysmotility Irritable bowel syndrome |
| Urological Interstitial cystitis |
| Gynecological Vulvodynia Chronic pelvic pain |
| Oral/dental Temporomandibular joint syndrome |
| Other (general) Chronic fatigue syndrome Sleep disturbances Idiopathic low back pain Multiple chemical sensitivity |
Fibromyalgia: Structured interview for diagnosis
| A. Generalized pain affecting the axial, plus upper and lower segments, plus left and rights sides of the body |
| Either B or C: |
| B. At least 11 of 18 reproducible tender points |
C. At least 4 of the following symptoms:
|
| D. It cannot be established that disturbance was due to another systematic condition |
| Source: Reference 4 |
Differentiating fibromyalgia from illnesses with similar symptoms
| Illness | Tests to differentiate from primary fibromyalgia |
|---|---|
| Rheumatic diseases Osteoarthritis Spondyloarthropathies, rheumatoid arthritis Systemic lupus erythematosus, polymyalgia rheumatica Osteomalacia Myopathy | Radiographs Rheumatic markers (antinuclear antibody, rheumatoid factor, antibodies) Inflammatory markers (ESR, C-reactive protein) Vitamin D level CPK |
| Neurologic Multiple sclerosis, Chiari’s malformation, spinal stenosis, radiculopathy Neuropathy | MRI EMG |
| Endocrine Hypothyroidism Diabetes | TSH Basic chemistry panel with fasting glucose |
| Other Infectious Lyme disease Hepatitis Anemia Cancers | CBC Lyme titer Hepatitis antibody panel, liver function tests Hemoglobin/hematocrit Routine cancer screening tests, bone scan, blood chemistries specific for suspected primary cancer |
| ESR: erythrocyte sedimentation rate; CPK: creatine phosphokinase; EMG: electromyography; TSH: thyroid-stimulating hormone; CBC: complete blood count | |
CASE CONTINUED: Central pain sensitization
As you elicit more details about Ms. D’s mood, she continues to focus on her physical symptoms. She states that some days she wishes to die because her pain gets so bad, but she denies any plan or intent to harm herself. She worries that her symptoms will worsen and that she will become completely disabled.
Her primary physician attempted to relieve Ms. D’s pain with multiple trials of nonsteroidal anti-inflammatory drugs (NSAIDs) and cyclobenzaprine. She says she gained no benefit from the NSAIDs and discontinued the muscle relaxant because it made her too sleepy. Fibromyalgia affects 3.5% of women and 0.5% of men.5 It runs in families with histories of fibromyalgia and major mood and anxiety disorders, suggesting genetic links.6 Defects in genes controlling serotonin and norepinephrine have been implicated.7-9
Fibromyalgia patients show lower levels of serotonin, norepinephrine, and dopamine metabolites in cerebrospinal fluid (CSF), compared with controls.10 These neurotransmitters may inhibit descending pain pathways in the CNS, and low levels in the brain and spinal cord may inhibit CNS regulation of pain impulses from the periphery.11
Although many patients describe muscle pain, evidence suggests central pain augmentation rather than an abnormality of muscle or connective tissue.12 Some studies have found evidence of “windup,” in which second-order neurons in the spinal cord become sensitized by repeated signals from first-order neurons in the periphery, resulting in amplified and prolonged pain signals traveling to the brain.13
Levels of substance P—a primary transmitter of pain impulses—are significantly higher in CSF of fibromyalgia patients compared with controls.14 This finding, in addition to low levels of serotonin and norepinephrine, indicates that pain signals are ascending unchecked to be processed by the brain.
- Twice as much pressure was required before controls rated their pain at a level similar to that of fibromyalgia patients.
- When controls and fibromyalgia patients reported similar pain, a very high degree of overlap was seen in brain areas responsible for pain processing. This indicates that fibromyalgia patients and controls were experiencing the pain they reported in the same way.15
Treating the whole patient
As a clinician who specializes in fibromyalgia, I counteract my patients’ and my own frustration with this condition by structuring office visits, determining realistic treatment goals, and treating all symptoms as part of a common syndrome rather than individual illnesses.
Structure office visits. Before every visit, have patients rate each symptom domain and write their top 2 or 3 concerns for that day (Click here for a sample form). Focusing on the patient’s most troublesome symptoms can help both of you feel greater satisfaction with treatment.
Educate patients. Ask them to discuss their beliefs about fibromyalgia; many know others with this condition or have researched diagnosis and treatment. Before developing a treatment plan, explain that their symptoms are chronic and all part of the same syndrome. Describe their pain as a complex phenomenon with possible peripheral and CNS components. Guide them to reputable Web sites and resources (see Related Resources).
Set realistic expectations. Many patients expect to resume an energetic and pain-free life, which usually is not the case with fibromyalgia (Box). Most medications are considered successful if they reduce pain by 30% to 50%, and side effects can be problematic. Discuss side effects before treatment begins to reduce patients’ anxiety and improve compliance in the first weeks.
Cognitive-behavioral therapy (CBT) for fibromyalgia incorporates relaxation techniques, helping patients view symptoms as manageable, reinforcing adaptive coping skills, and teaching them how to monitor thoughts, feelings, and behavior to change the view that they are helpless victims. A modest course of 6 weekly group CBT sessions significantly improved physical functioning in 25% of fibromyalgia patients (n=76) compared with 12% in a standard-care group (n=69), even though patients’ pain severity did not improve.16
Lifestyle changes are as important as medications in controlling fibromyalgia symptoms. In addition to exercise, recommend that patients:
- follow a daily routine
- pace activity to avoid exacerbating symptoms
- reduce stress.
BELIEF: ‘A magic pill exists that will resolve all my symptoms and have no side effects’
Clinical evidence: Most medications that have been studied were effective in 30% to 50% of patients and reduced pain scores by 30% to 50%.
Patient education: Explain to the patient with a pain rating of 7 at the first visit that achieving a pain level of 3 to 4 may be possible with treatment. Even with successful treatment, symptoms may flare intermittently. As with any treatment, adverse effects may occur. Discuss these, so the patient is not surprised.
BELIEF: ‘I can’t exercise’
Clinical evidence: Most patients experience more fatigue and pain with physical activity, but exercise is important to maintain physical function.
Patient education: When discussing an exercise program, focus on what the patient can do. Most patients attempt too much, too soon; advise them to start at a tolerable level (such as 2 to 3 minutes of aerobic activity daily for the first week) and gradually increase as tolerated.
BELIEF: ‘You (the psychiatrist) can make me feel better’
Clinical evidence: Psychiatrists can help by prescribing appropriate medications, but much of the burden falls on the patient to maintain a healthy, active lifestyle and to manage stressors in an adaptive manner.
Patient education: A fibromyalgia patient may find relief with a medication, but symptoms may flare if they ‘overdo’ and take on too many physical or emotional stressors. A consistent, healthy routine is ideal.
BELIEF: ‘I will eventually become disabled by fibromyalgia’
Clinical evidence: Despite little long-term research on fibromyalgia patients, most evidence points to a chronic, fluctuating syndrome that does not worsen with age. Factors that may worsen symptoms include uncontrolled comorbid conditions, chronic opiate use, inactivity, and deconditioning.
Patient education: Discourage long-term physical disability; exercise and maintaining an active daily routine helps patients avoid focusing in a nonadaptive manner on their dysfunction and symptoms.
Source: Sharon B. (Shay) Stanford, MD
New direction with medications
Pregabalin is an anticonvulsant that binds to the alpha-2-delta subunits of neurons’ voltage-gated calcium channels. This activity reduces calcium influx at nerve terminals and inhibits release of excitatory neurotransmitters, such as substance P and glutamate.18 In June 2007, pregabalin was the first medication FDA-approved for fibromyalgia.
Two placebo-controlled trials19,20 showed that pregabalin at 150 mg bid, 225 mg bid, or 300 mg bid significantly reduced weekly mean pain scores in fibromyalgia patients. Click here for details of these trials. The most common side effects—dizziness, somnolence, peripheral edema, blurred vision, and weight gain—were regarded as mild to moderate in 87% of patients.21
Although a dosage of 300 mg bid also was studied, the FDA approved pregabalin at dosages of 150 mg bid and 225 mg bid for fibromyalgia.22
Duloxetine is a serotonin/norepinephrine reuptake inhibitor (SNRI) thought to inhibit dorsal horn neurons’ response to peripheral pain signals by increasing serotonin and norepinephrine in the brain and spinal cord. This SNRI was first FDA-approved for diabetic peripheral neuropathic pain and major depressive disorder. Approval for fibromyalgia at 60 mg/d in June 2008 was based on 2 placebo-controlled, double-blind, 12-week trials comprising 874 patients.23,24Click here for detailed findings of these studies and a 6-month fixed-dose trial.25
Milnacipran is an SNRI that was approved for treating fibromyalgia in January 2009 at dosages of 50 mg bid and 100 mg bid. Like other SNRIs, milnacipran is thought to work by inhibiting pain signals through increasing serotonin and norepinephrine in the brain and spinal cord. Milnacipran has a higher selectivity for norepinephrine reuptake compared with duloxetine, which may mean these medications will have different effects in different patients. Although milnacipran is approved as an antidepressant in other countries, the FDA has not approved it for treating depression in the United States.
Click here for details of a 15-week, double-blind, placebo-controlled trial of milnacipran in patients with fibromyalgia. Side effects in clinical trials were similar to those of duloxetine, with nausea, constipation, and increased sweating being most prominent.26
Other medications, such as the first-line agent amitriptyline, have shown beneficial effects in fibromyalgia but are not FDA-approved for this indication (Table 4).27-32
Choosing medications. When prescribing one of the FDA-approved medications to treat fibromyalgia, consider their benefits and side effects.
Pregabalin may be a beneficial first choice for patients who report pain and sleep as major issues. Although the medication’s label recommends starting with twice-daily dosing, patients might better tolerate an initial dose of 50 to 75 mg in the evening, with the morning dose added later. Pregabalin can be useful in patients taking multiple medications because of its renal clearance, resulting in low risk of interactions with drugs metabolized by liver enzymes. It also can be useful in patients who have not tolerated antidepressants in the past or in whom antidepressants are contraindicated.
If a patient has a history of depression or discontinuing medications because of sedating side effects, an antidepressant such as duloxetine or milnacipran may be more successful than starting with pregabalin. In general, if a patient does not respond to one of these SNRIs, moving on to the other might help. Milnacipran’s more selective effect on norepinephrine could be beneficial for some patients, especially those with excessive fatigue. Others, especially those with a high level of anxiety, might respond better to a more balanced SNRI such as duloxetine.
Table 4
Off-label medications shown to benefit patients with fibromyalgia
| Drug | Comment |
|---|---|
| Amitriptyline27,28 | Considered first-line because of studies supporting its use, low cost, and wide availability; may be associated with more side effects than newer medications |
| Gabapentin29 | Possible alternative to pregabalin but may not be as well tolerated |
| Tramadol30 | May help with breakthrough pain; use with extreme caution in patients taking antidepressants because of serotonin syndrome risk |
| Fluoxetine31 | Dosages of 40 to 60 mg/d may help patients who do not tolerate SNRIs |
| Venlafaxine32 | Dosages of 150 to 225 mg/d may be an alternative to other SNRIs |
| SNRIs: serotonin/norepinephrine reuptake inhibitors | |
CASE CONTINUED: Not as hopeless
Ms. D’s primary care physician confirms your presumptive diagnosis of fibromyalgia. He prescribes a trial of amitriptyline, which she does not tolerate well because of sedation and weight gain. At her next psychiatric visit, she tells you she remains very frustrated about her physical symptoms and reports that her doctor “has given up on me.”
You discuss what a fibromyalgia diagnosis means to her and educate her about the syndrome. You refer her to a colleague who does CBT with chronic pain patients and start her on a low dose of duloxetine (30 mg once daily) to minimize side effects. You discuss possible side effects and that she may need a higher dose to notice improvement in her pain. She seems receptive to starting a graded exercise program, and you encourage her to reduce physical and emotional stress in her life.
- Arthritis Foundation. Fibromyalgia. www.arthritis.org/disease-center.php?disease_id=10.
- National Fibromyalgia Association. www.fmaware.org.
- Self-management program for patients with fibromyalgia, cosponsored by the National Fibromyalgia Association and Eli Lilly and Company. www.knowfibro.com.
- Amitriptyline • Elavil, Endep
- Cyclobenzaprine • Flexeril
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Milnacipran • Savella
- Pregabalin • Lyrica
- Tramadol • Ultram, Ultracet
- Venlafaxine • Effexor, Effexor XR
Dr. Stanford receives grant/research support from Eli Lilly and Company, Pfizer, Cypress Bioscience, and Allergan.
WEB AUDIO
Listen to Dr. Stanford discuss,
"Is fibromyalgia a somatoform disorder?
Patients with fibromyalgia are a heterogeneous group, yet many describe a common experience: seeing multiple physicians who seem unable or unwilling to provide a diagnosis or treat their symptoms. This situation may be changing with the recent FDA approval of an anticonvulsant and 2 antidepressants for managing fibromyalgia symptoms.
These medications—pregabalin, duloxetine, and milnacipran—reflect a revised understanding of fibromyalgia as a CNS condition, rather than an inflammatory process in the muscles or connective tissue. As a result, psychiatrists—because of our experience with CNS phenomena and managing antidepressant and anticonvulsant medications—are likely to play a larger role in treating fibromyalgia.
CASE REPORT: ‘Just too tired’
Ms. D, age 50, has a history of migraine headaches and is referred by her primary physician for evaluation of depression and anxiety. She reports deteriorating mood over 6 months, beginning when a minor car accident left her “very sore the next day.”
Ms. D reports she is depressed because she feels “just too tired” after work to keep up with social activities or housework. Her physician’s referral notes a normal physical exam except for tenderness over her upper back and hips. Laboratory testing is negative.
Making the diagnosis
American College of Rheumatology (ACR) criteria for fibromyalgia require widespread pain for at least 3 months. “Widespread” is defined as pain in the axial skeleton, left and right sides of the body, and above and below the waist. Pain must be found in at least 11 of 18 tender point sites on digital palpation using a force of approximately 4 kg/cm2.1 For many fibromyalgia patients, however, musculoskeletal pain is not their most problematic symptom (Table 1). They may suffer:
- migraine and tension headaches (10% to 80% of patients)
- irritable bowel syndrome (32% to 80%)2
- mood disorders (major depressive disorder [62%], bipolar disorder [11%])
- anxiety disorders (panic disorder [29%], posttraumatic stress disorder [21%], social phobia [19%]).3
Because patients with fibromyalgia often meet criteria for somatization or somatoform disorders, how to classify them—as medically or psychiatrically ill—is controversial. Some patients believe their mood or anxiety problem stems from the difficulty they experience dealing with their physical symptoms, and if they could feel better physically they would not be depressed or anxious. Others believe their psychiatric symptoms impede their ability to help themselves feel better.
Consider fibromyalgia in any patient with widespread pain of unknown cause. Before making the diagnosis, rule out other illnesses that present with similar symptoms (Table 3). Because many patients newly diagnosed with fibromyalgia worry that something “more serious” may be going on, confirm the diagnosis with appropriate testing and physical examination, usually by a rheumatologist or primary care physician.
Table 1
Medical and cognitive symptoms related to fibromyalgia
| Neurologic Tension/migraine headache |
| Psychiatric Memory and cognitive difficulties Mood disturbance Anxiety disorders |
| Ear, nose, throat Sicca symptoms Vasomotor rhinitis Vestibular complaints |
| Cardiovascular Neurally mediated hypotension Mitral valve prolapse Noncardiac chest pain |
| Gastrointestinal Esophageal dysmotility Irritable bowel syndrome |
| Urological Interstitial cystitis |
| Gynecological Vulvodynia Chronic pelvic pain |
| Oral/dental Temporomandibular joint syndrome |
| Other (general) Chronic fatigue syndrome Sleep disturbances Idiopathic low back pain Multiple chemical sensitivity |
Fibromyalgia: Structured interview for diagnosis
| A. Generalized pain affecting the axial, plus upper and lower segments, plus left and rights sides of the body |
| Either B or C: |
| B. At least 11 of 18 reproducible tender points |
C. At least 4 of the following symptoms:
|
| D. It cannot be established that disturbance was due to another systematic condition |
| Source: Reference 4 |
Differentiating fibromyalgia from illnesses with similar symptoms
| Illness | Tests to differentiate from primary fibromyalgia |
|---|---|
| Rheumatic diseases Osteoarthritis Spondyloarthropathies, rheumatoid arthritis Systemic lupus erythematosus, polymyalgia rheumatica Osteomalacia Myopathy | Radiographs Rheumatic markers (antinuclear antibody, rheumatoid factor, antibodies) Inflammatory markers (ESR, C-reactive protein) Vitamin D level CPK |
| Neurologic Multiple sclerosis, Chiari’s malformation, spinal stenosis, radiculopathy Neuropathy | MRI EMG |
| Endocrine Hypothyroidism Diabetes | TSH Basic chemistry panel with fasting glucose |
| Other Infectious Lyme disease Hepatitis Anemia Cancers | CBC Lyme titer Hepatitis antibody panel, liver function tests Hemoglobin/hematocrit Routine cancer screening tests, bone scan, blood chemistries specific for suspected primary cancer |
| ESR: erythrocyte sedimentation rate; CPK: creatine phosphokinase; EMG: electromyography; TSH: thyroid-stimulating hormone; CBC: complete blood count | |
CASE CONTINUED: Central pain sensitization
As you elicit more details about Ms. D’s mood, she continues to focus on her physical symptoms. She states that some days she wishes to die because her pain gets so bad, but she denies any plan or intent to harm herself. She worries that her symptoms will worsen and that she will become completely disabled.
Her primary physician attempted to relieve Ms. D’s pain with multiple trials of nonsteroidal anti-inflammatory drugs (NSAIDs) and cyclobenzaprine. She says she gained no benefit from the NSAIDs and discontinued the muscle relaxant because it made her too sleepy. Fibromyalgia affects 3.5% of women and 0.5% of men.5 It runs in families with histories of fibromyalgia and major mood and anxiety disorders, suggesting genetic links.6 Defects in genes controlling serotonin and norepinephrine have been implicated.7-9
Fibromyalgia patients show lower levels of serotonin, norepinephrine, and dopamine metabolites in cerebrospinal fluid (CSF), compared with controls.10 These neurotransmitters may inhibit descending pain pathways in the CNS, and low levels in the brain and spinal cord may inhibit CNS regulation of pain impulses from the periphery.11
Although many patients describe muscle pain, evidence suggests central pain augmentation rather than an abnormality of muscle or connective tissue.12 Some studies have found evidence of “windup,” in which second-order neurons in the spinal cord become sensitized by repeated signals from first-order neurons in the periphery, resulting in amplified and prolonged pain signals traveling to the brain.13
Levels of substance P—a primary transmitter of pain impulses—are significantly higher in CSF of fibromyalgia patients compared with controls.14 This finding, in addition to low levels of serotonin and norepinephrine, indicates that pain signals are ascending unchecked to be processed by the brain.
- Twice as much pressure was required before controls rated their pain at a level similar to that of fibromyalgia patients.
- When controls and fibromyalgia patients reported similar pain, a very high degree of overlap was seen in brain areas responsible for pain processing. This indicates that fibromyalgia patients and controls were experiencing the pain they reported in the same way.15
Treating the whole patient
As a clinician who specializes in fibromyalgia, I counteract my patients’ and my own frustration with this condition by structuring office visits, determining realistic treatment goals, and treating all symptoms as part of a common syndrome rather than individual illnesses.
Structure office visits. Before every visit, have patients rate each symptom domain and write their top 2 or 3 concerns for that day (Click here for a sample form). Focusing on the patient’s most troublesome symptoms can help both of you feel greater satisfaction with treatment.
Educate patients. Ask them to discuss their beliefs about fibromyalgia; many know others with this condition or have researched diagnosis and treatment. Before developing a treatment plan, explain that their symptoms are chronic and all part of the same syndrome. Describe their pain as a complex phenomenon with possible peripheral and CNS components. Guide them to reputable Web sites and resources (see Related Resources).
Set realistic expectations. Many patients expect to resume an energetic and pain-free life, which usually is not the case with fibromyalgia (Box). Most medications are considered successful if they reduce pain by 30% to 50%, and side effects can be problematic. Discuss side effects before treatment begins to reduce patients’ anxiety and improve compliance in the first weeks.
Cognitive-behavioral therapy (CBT) for fibromyalgia incorporates relaxation techniques, helping patients view symptoms as manageable, reinforcing adaptive coping skills, and teaching them how to monitor thoughts, feelings, and behavior to change the view that they are helpless victims. A modest course of 6 weekly group CBT sessions significantly improved physical functioning in 25% of fibromyalgia patients (n=76) compared with 12% in a standard-care group (n=69), even though patients’ pain severity did not improve.16
Lifestyle changes are as important as medications in controlling fibromyalgia symptoms. In addition to exercise, recommend that patients:
- follow a daily routine
- pace activity to avoid exacerbating symptoms
- reduce stress.
BELIEF: ‘A magic pill exists that will resolve all my symptoms and have no side effects’
Clinical evidence: Most medications that have been studied were effective in 30% to 50% of patients and reduced pain scores by 30% to 50%.
Patient education: Explain to the patient with a pain rating of 7 at the first visit that achieving a pain level of 3 to 4 may be possible with treatment. Even with successful treatment, symptoms may flare intermittently. As with any treatment, adverse effects may occur. Discuss these, so the patient is not surprised.
BELIEF: ‘I can’t exercise’
Clinical evidence: Most patients experience more fatigue and pain with physical activity, but exercise is important to maintain physical function.
Patient education: When discussing an exercise program, focus on what the patient can do. Most patients attempt too much, too soon; advise them to start at a tolerable level (such as 2 to 3 minutes of aerobic activity daily for the first week) and gradually increase as tolerated.
BELIEF: ‘You (the psychiatrist) can make me feel better’
Clinical evidence: Psychiatrists can help by prescribing appropriate medications, but much of the burden falls on the patient to maintain a healthy, active lifestyle and to manage stressors in an adaptive manner.
Patient education: A fibromyalgia patient may find relief with a medication, but symptoms may flare if they ‘overdo’ and take on too many physical or emotional stressors. A consistent, healthy routine is ideal.
BELIEF: ‘I will eventually become disabled by fibromyalgia’
Clinical evidence: Despite little long-term research on fibromyalgia patients, most evidence points to a chronic, fluctuating syndrome that does not worsen with age. Factors that may worsen symptoms include uncontrolled comorbid conditions, chronic opiate use, inactivity, and deconditioning.
Patient education: Discourage long-term physical disability; exercise and maintaining an active daily routine helps patients avoid focusing in a nonadaptive manner on their dysfunction and symptoms.
Source: Sharon B. (Shay) Stanford, MD
New direction with medications
Pregabalin is an anticonvulsant that binds to the alpha-2-delta subunits of neurons’ voltage-gated calcium channels. This activity reduces calcium influx at nerve terminals and inhibits release of excitatory neurotransmitters, such as substance P and glutamate.18 In June 2007, pregabalin was the first medication FDA-approved for fibromyalgia.
Two placebo-controlled trials19,20 showed that pregabalin at 150 mg bid, 225 mg bid, or 300 mg bid significantly reduced weekly mean pain scores in fibromyalgia patients. Click here for details of these trials. The most common side effects—dizziness, somnolence, peripheral edema, blurred vision, and weight gain—were regarded as mild to moderate in 87% of patients.21
Although a dosage of 300 mg bid also was studied, the FDA approved pregabalin at dosages of 150 mg bid and 225 mg bid for fibromyalgia.22
Duloxetine is a serotonin/norepinephrine reuptake inhibitor (SNRI) thought to inhibit dorsal horn neurons’ response to peripheral pain signals by increasing serotonin and norepinephrine in the brain and spinal cord. This SNRI was first FDA-approved for diabetic peripheral neuropathic pain and major depressive disorder. Approval for fibromyalgia at 60 mg/d in June 2008 was based on 2 placebo-controlled, double-blind, 12-week trials comprising 874 patients.23,24Click here for detailed findings of these studies and a 6-month fixed-dose trial.25
Milnacipran is an SNRI that was approved for treating fibromyalgia in January 2009 at dosages of 50 mg bid and 100 mg bid. Like other SNRIs, milnacipran is thought to work by inhibiting pain signals through increasing serotonin and norepinephrine in the brain and spinal cord. Milnacipran has a higher selectivity for norepinephrine reuptake compared with duloxetine, which may mean these medications will have different effects in different patients. Although milnacipran is approved as an antidepressant in other countries, the FDA has not approved it for treating depression in the United States.
Click here for details of a 15-week, double-blind, placebo-controlled trial of milnacipran in patients with fibromyalgia. Side effects in clinical trials were similar to those of duloxetine, with nausea, constipation, and increased sweating being most prominent.26
Other medications, such as the first-line agent amitriptyline, have shown beneficial effects in fibromyalgia but are not FDA-approved for this indication (Table 4).27-32
Choosing medications. When prescribing one of the FDA-approved medications to treat fibromyalgia, consider their benefits and side effects.
Pregabalin may be a beneficial first choice for patients who report pain and sleep as major issues. Although the medication’s label recommends starting with twice-daily dosing, patients might better tolerate an initial dose of 50 to 75 mg in the evening, with the morning dose added later. Pregabalin can be useful in patients taking multiple medications because of its renal clearance, resulting in low risk of interactions with drugs metabolized by liver enzymes. It also can be useful in patients who have not tolerated antidepressants in the past or in whom antidepressants are contraindicated.
If a patient has a history of depression or discontinuing medications because of sedating side effects, an antidepressant such as duloxetine or milnacipran may be more successful than starting with pregabalin. In general, if a patient does not respond to one of these SNRIs, moving on to the other might help. Milnacipran’s more selective effect on norepinephrine could be beneficial for some patients, especially those with excessive fatigue. Others, especially those with a high level of anxiety, might respond better to a more balanced SNRI such as duloxetine.
Table 4
Off-label medications shown to benefit patients with fibromyalgia
| Drug | Comment |
|---|---|
| Amitriptyline27,28 | Considered first-line because of studies supporting its use, low cost, and wide availability; may be associated with more side effects than newer medications |
| Gabapentin29 | Possible alternative to pregabalin but may not be as well tolerated |
| Tramadol30 | May help with breakthrough pain; use with extreme caution in patients taking antidepressants because of serotonin syndrome risk |
| Fluoxetine31 | Dosages of 40 to 60 mg/d may help patients who do not tolerate SNRIs |
| Venlafaxine32 | Dosages of 150 to 225 mg/d may be an alternative to other SNRIs |
| SNRIs: serotonin/norepinephrine reuptake inhibitors | |
CASE CONTINUED: Not as hopeless
Ms. D’s primary care physician confirms your presumptive diagnosis of fibromyalgia. He prescribes a trial of amitriptyline, which she does not tolerate well because of sedation and weight gain. At her next psychiatric visit, she tells you she remains very frustrated about her physical symptoms and reports that her doctor “has given up on me.”
You discuss what a fibromyalgia diagnosis means to her and educate her about the syndrome. You refer her to a colleague who does CBT with chronic pain patients and start her on a low dose of duloxetine (30 mg once daily) to minimize side effects. You discuss possible side effects and that she may need a higher dose to notice improvement in her pain. She seems receptive to starting a graded exercise program, and you encourage her to reduce physical and emotional stress in her life.
- Arthritis Foundation. Fibromyalgia. www.arthritis.org/disease-center.php?disease_id=10.
- National Fibromyalgia Association. www.fmaware.org.
- Self-management program for patients with fibromyalgia, cosponsored by the National Fibromyalgia Association and Eli Lilly and Company. www.knowfibro.com.
- Amitriptyline • Elavil, Endep
- Cyclobenzaprine • Flexeril
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Milnacipran • Savella
- Pregabalin • Lyrica
- Tramadol • Ultram, Ultracet
- Venlafaxine • Effexor, Effexor XR
Dr. Stanford receives grant/research support from Eli Lilly and Company, Pfizer, Cypress Bioscience, and Allergan.
1. Wolfe F, Smythe HA, Yunus MB, et al. The American College of Rheumatology 1990 criteria for the classification of fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum. 1990;33(2):160-172.
2. Aaron LA, Buchwald D. Chronic diffuse musculoskeletal pain, fibromyalgia and co-morbid unexplained clinical conditions. Best Prac Res Clin Rheumatol. 2003;17(4):563-574.
3. Arnold LM, Hudson JI, Keck PE, et al. Comorbidity of fibromyalgia and psychiatric disorders. J Clin Psychiatry. 2006;67(8):1219-1225.
4. Pope HG, Jr, Hudson JI. A supplemental interview for forms of “affective spectrum disorder.” Int J Psychiatry Med. 1991;21(3):205-232.
5. Wolfe F, Ross K, Anderson J, et al. The prevalence and characteristics of fibromyalgia in the general population. Arthritis Rheum. 1995;38(1):19-28.
6. Arnold LM, Hudson JI, Hess EV, et al. Family study of fibromyalgia. Arthritis Rheum. 2004;50(3):944-952.
7. Bondy B, Spaeth M, Offenbaecher M, et al. The T102C polymorphism of the 5-HT2A-receptor gene in fibromyalgia. Neurobiol Dis. 1999;6(5):433-439.
8. Offenbaecher M, Bondy B, de Jonge S, et al. Possible association of fibromyalgia with a polymorphism in the serotonin transporter gene regulatory region. Arthritis Rheum. 1999;42(11):2482-2488.
9. Gürsoy S, Erdal E, Herken H, et al. Significance of catechol-O-methyltransferase gene polymorphism in fibromyalgia syndrome. Rheumatol Int. 2003;23(3):104-107.
10. Russell IJ, Vaeroy H, Javors M, et al. Cerebrospinal fluid biogenic amine metabolites in fibromyalgia/fibrositis syndrome and rheumatoid arthritis. Arthritis Rheum. 1992;35(5):550-556.
11. Fields HL, Basbaum AI. In: Wall PD, Melzack R, eds. Textbook of pain. 4th ed. New York, NY: Churchill Livingstone; 1999: 309-329.
12. Clauw DJ, Crofford LJ. Chronic widespread pain and fibromyalgia: what we know, and what we need to know. Best Pract Res Clin Rheumatol. 2003;17(4):685-701.
13. Staud R, Cannon RC, Mauderli AP, et al. Temporal summation of pain from mechanical stimulation of muscle tissue in normal controls and subjects with fibromyalgia syndrome. Pain. 2003;102(1-2):87-95.
14. Russell IJ, Orr MD, Littman B, et al. Elevated cerebrospinal fluid levels of substance P in patients with the fibromyalgia syndrome. Arthritis Rheum. 1994;37(11):1593-1601.
15. Gracely RH, Petzke F, Wolf JM, et al. Functional magnetic resonance imaging evidence of augmented pain processing in fibromyalgia. Arthritis Rheum. 2002;46(5):1333-1343.
16. Williams DA, Cary MA, Groner KH. Improving physical functional status in patients with fibromyalgia: a brief cognitive behavioral intervention. J Rheumatol. 2002;29:1280-1286.
17. Busch AJ, Schachter CL, Overend TJ, et al. Exercise for fibromyalgia: a systematic review. J Rheumatol. 2008;35(6):1130-1144.
18. Field MJ, Cox PJ, Stott E. Identification of the alpha2-delta-1 subunit of voltage-dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. Proc Natl Acad Sci USA. 2006;103(46):17537-17542.
19. Arnold LM, Russel IJ, Diri EW, et al. A 14-week, randomized, double-blind, placebo-controlled, monotherapy trial of pregabalin in patients with fibromyalgia. J Pain. 2008;9(9):792-805.
20. Mease PJ, Russel IJ, Arnold LM, et al. A randomized, double-blind, placebo-controlled, phase III trial of pregabalin in the treatment of patients with fibromyalgia. J Rheumatol. 2008;35(3):502-514.
21. Crofford LJ, Mease J, Simpson SL, et al. Fibromyalgia relapse evaluation and efficacy for durability of meaningful relief (FREEDOM): a 6-month, double-blind, placebo-controlled trial with pregabalin. Pain. 2008;136(3):419-431.
22. Pregabalin [package insert]. New York, NY: Pfizer, Inc.; 2004.
23. Arnold LM, Pritchett YL, D’Souza DN, et al. Duloxetine for the treatment of fibromyalgia in women: pooled results from two randomized, placebo-controlled clinical trials. J Womens Health (Larchmt). 2007;16(8):1145-1156.
24. Arnold LM, Lu Y, Crofford LJ, et al. A double-blind, multicenter trial comparing duloxetine with placebo in the treatment of fibromyalgia patients with or without major depressive disorder. Arthritis Rheum. 2004;50(9):2974-2984.
25. Russell IJ, Mease PJ, Smith TR, et al. Efficacy and safety of duloxetine for treatment of fibromyalgia in patients with or without major depressive disorder: results from a 6-month, randomized, double-blind, placebo-controlled, fixed-dose trial. Pain. 2008;136(3):432-444.
26. Clauw DJ, Mease P, Palmer RH, et al. Milnacipran for the treatment of fibromyalgia in adults: a 15-week, multicenter, randomized, double-blind, placebo-controlled, multiple-dose clinical trial. Clin Ther. 2008;30(11):1988-2004.
27. Goldenberg DL, Felson DT, Dinerman H. A randomized, controlled trial of amitriptyline and naproxen in the treatment of patients with fibromyalgia. Arthritis Rheum. 1986;29(11):1371-1377.
28. Hannonen P, Malminiemi K, Yli-Kerttula U, et al. A randomized, double-blind, placebo-controlled study of moclobemide and amitriptyline in the treatment of fibromyalgia in females without psychiatric disorder. Br J Rheumatol. 1998;37:1279-1286.
29. Arnold LM, Goldenberg DL, Stanford SB, et al. Gabapentin in the treatment of fibromyalgia: a randomized, double-blind, placebo-controlled, multicenter trial. Arthritis Rheum. 2007;56:1336-1344.
30. Bennett RM, Schein J, Kosinski MR, et al. Impact of fibromyalgia pain on health-related quality of life before and after treatment with tramadol/ acetaminophen. Arthritis Rheum. 2005;53:519-527.
31. Arnold LM, Hess EV, Hudson JI, et al. Randomized, placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med. 2002;112:191-197.
32. Dwight MM, Arnold LM, O’Brien H, et al. An open clinical trial of venlafaxine treatment of fibromyalgia. Psychosomatics. 1998;39:14-17.
1. Wolfe F, Smythe HA, Yunus MB, et al. The American College of Rheumatology 1990 criteria for the classification of fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum. 1990;33(2):160-172.
2. Aaron LA, Buchwald D. Chronic diffuse musculoskeletal pain, fibromyalgia and co-morbid unexplained clinical conditions. Best Prac Res Clin Rheumatol. 2003;17(4):563-574.
3. Arnold LM, Hudson JI, Keck PE, et al. Comorbidity of fibromyalgia and psychiatric disorders. J Clin Psychiatry. 2006;67(8):1219-1225.
4. Pope HG, Jr, Hudson JI. A supplemental interview for forms of “affective spectrum disorder.” Int J Psychiatry Med. 1991;21(3):205-232.
5. Wolfe F, Ross K, Anderson J, et al. The prevalence and characteristics of fibromyalgia in the general population. Arthritis Rheum. 1995;38(1):19-28.
6. Arnold LM, Hudson JI, Hess EV, et al. Family study of fibromyalgia. Arthritis Rheum. 2004;50(3):944-952.
7. Bondy B, Spaeth M, Offenbaecher M, et al. The T102C polymorphism of the 5-HT2A-receptor gene in fibromyalgia. Neurobiol Dis. 1999;6(5):433-439.
8. Offenbaecher M, Bondy B, de Jonge S, et al. Possible association of fibromyalgia with a polymorphism in the serotonin transporter gene regulatory region. Arthritis Rheum. 1999;42(11):2482-2488.
9. Gürsoy S, Erdal E, Herken H, et al. Significance of catechol-O-methyltransferase gene polymorphism in fibromyalgia syndrome. Rheumatol Int. 2003;23(3):104-107.
10. Russell IJ, Vaeroy H, Javors M, et al. Cerebrospinal fluid biogenic amine metabolites in fibromyalgia/fibrositis syndrome and rheumatoid arthritis. Arthritis Rheum. 1992;35(5):550-556.
11. Fields HL, Basbaum AI. In: Wall PD, Melzack R, eds. Textbook of pain. 4th ed. New York, NY: Churchill Livingstone; 1999: 309-329.
12. Clauw DJ, Crofford LJ. Chronic widespread pain and fibromyalgia: what we know, and what we need to know. Best Pract Res Clin Rheumatol. 2003;17(4):685-701.
13. Staud R, Cannon RC, Mauderli AP, et al. Temporal summation of pain from mechanical stimulation of muscle tissue in normal controls and subjects with fibromyalgia syndrome. Pain. 2003;102(1-2):87-95.
14. Russell IJ, Orr MD, Littman B, et al. Elevated cerebrospinal fluid levels of substance P in patients with the fibromyalgia syndrome. Arthritis Rheum. 1994;37(11):1593-1601.
15. Gracely RH, Petzke F, Wolf JM, et al. Functional magnetic resonance imaging evidence of augmented pain processing in fibromyalgia. Arthritis Rheum. 2002;46(5):1333-1343.
16. Williams DA, Cary MA, Groner KH. Improving physical functional status in patients with fibromyalgia: a brief cognitive behavioral intervention. J Rheumatol. 2002;29:1280-1286.
17. Busch AJ, Schachter CL, Overend TJ, et al. Exercise for fibromyalgia: a systematic review. J Rheumatol. 2008;35(6):1130-1144.
18. Field MJ, Cox PJ, Stott E. Identification of the alpha2-delta-1 subunit of voltage-dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. Proc Natl Acad Sci USA. 2006;103(46):17537-17542.
19. Arnold LM, Russel IJ, Diri EW, et al. A 14-week, randomized, double-blind, placebo-controlled, monotherapy trial of pregabalin in patients with fibromyalgia. J Pain. 2008;9(9):792-805.
20. Mease PJ, Russel IJ, Arnold LM, et al. A randomized, double-blind, placebo-controlled, phase III trial of pregabalin in the treatment of patients with fibromyalgia. J Rheumatol. 2008;35(3):502-514.
21. Crofford LJ, Mease J, Simpson SL, et al. Fibromyalgia relapse evaluation and efficacy for durability of meaningful relief (FREEDOM): a 6-month, double-blind, placebo-controlled trial with pregabalin. Pain. 2008;136(3):419-431.
22. Pregabalin [package insert]. New York, NY: Pfizer, Inc.; 2004.
23. Arnold LM, Pritchett YL, D’Souza DN, et al. Duloxetine for the treatment of fibromyalgia in women: pooled results from two randomized, placebo-controlled clinical trials. J Womens Health (Larchmt). 2007;16(8):1145-1156.
24. Arnold LM, Lu Y, Crofford LJ, et al. A double-blind, multicenter trial comparing duloxetine with placebo in the treatment of fibromyalgia patients with or without major depressive disorder. Arthritis Rheum. 2004;50(9):2974-2984.
25. Russell IJ, Mease PJ, Smith TR, et al. Efficacy and safety of duloxetine for treatment of fibromyalgia in patients with or without major depressive disorder: results from a 6-month, randomized, double-blind, placebo-controlled, fixed-dose trial. Pain. 2008;136(3):432-444.
26. Clauw DJ, Mease P, Palmer RH, et al. Milnacipran for the treatment of fibromyalgia in adults: a 15-week, multicenter, randomized, double-blind, placebo-controlled, multiple-dose clinical trial. Clin Ther. 2008;30(11):1988-2004.
27. Goldenberg DL, Felson DT, Dinerman H. A randomized, controlled trial of amitriptyline and naproxen in the treatment of patients with fibromyalgia. Arthritis Rheum. 1986;29(11):1371-1377.
28. Hannonen P, Malminiemi K, Yli-Kerttula U, et al. A randomized, double-blind, placebo-controlled study of moclobemide and amitriptyline in the treatment of fibromyalgia in females without psychiatric disorder. Br J Rheumatol. 1998;37:1279-1286.
29. Arnold LM, Goldenberg DL, Stanford SB, et al. Gabapentin in the treatment of fibromyalgia: a randomized, double-blind, placebo-controlled, multicenter trial. Arthritis Rheum. 2007;56:1336-1344.
30. Bennett RM, Schein J, Kosinski MR, et al. Impact of fibromyalgia pain on health-related quality of life before and after treatment with tramadol/ acetaminophen. Arthritis Rheum. 2005;53:519-527.
31. Arnold LM, Hess EV, Hudson JI, et al. Randomized, placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med. 2002;112:191-197.
32. Dwight MM, Arnold LM, O’Brien H, et al. An open clinical trial of venlafaxine treatment of fibromyalgia. Psychosomatics. 1998;39:14-17.
Compulsive bruxism: How to protect your patients’ teeth
Oral habits such as bruxism—compulsive grinding or clenching of the teeth—can be a manifestation of obsessive-compulsive disorder (OCD) and other anxiety disorders.1 Bruxism also may be a side effect of selective serotonin reuptake inhibitors (SSRIs)2,3 used to treat OCD4 and depression. Other oral conditions can complicate treatment of these disorders (Table 1).
Potentially serious sequelae of bruxism and similar behaviors include:
- wearing down of teeth (more common)
- necrosis of the pulpal tissues that results in non-vital teeth (less common).
The following case underlines the need for early referral to a dentist and close follow-up for patients who have tooth-related behaviors or are taking medications associated with a risk for such behaviors.
Table 1
Oral conditions associated with anxiety disorders and depression
| Bruxism |
| Canker sores |
| Dry mouth |
| Temporomandibular joint disorders |
| Lichen planus (redness or mouth ulcers) |
| Non-vital teeth |
| Tooth wear, fracture |
CASE REPORT: A compulsive oral habit
Mr. G, age 26, presents to our school of dental medicine with the chief complaint that he needs a crown. On clinical intraoral examination, we find he has multiple restorations, some areas of recurrent tooth decay, and a fractured cusp on a maxillary molar. His mandibular incisors show greater wear on their incisal surfaces than would be expected for a patient his age. This is especially true of his mandibular right lateral incisor (tooth #26) and canine (tooth #27) (Photo 1).
Clinical examination also reveals a restoration on tooth #26 that was consistent with an access cavity drilled for endodontic (root canal) therapy (Photo 2). This finding is consistent with his dental history. Soft tissue examination is within normal limits. He reports that he saw a dentist 7 months earlier but could not afford the fees.
During his medical history, Mr. G states he has mild Tourette’s syndrome that was diagnosed when he was 10. In the early 1990s he tried several medications, including haloperidol and pimozide, as a subject in research studies of Tourette’s. He could not recall the dosages of the medications or for how long he took them. Because these medications did not improve his symptoms, he stopped taking them after the studies ended.
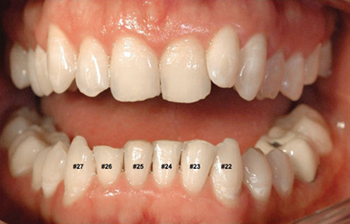
Photo 1 Unexpected tooth wear: A clue to an anxiety-related oral habit
Teeth of a 26-year-old man show greater than expected wear, particularly on the mandibular right lateral incisor (tooth #26) and canine (tooth #27).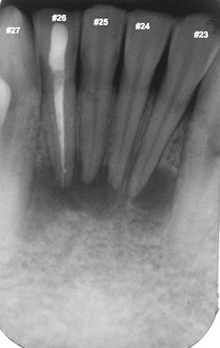
Photo 2 Radiographic evidence of tooth non-vitality
Radiolucencies (dark areas) in the bone at the apices of the tooth roots are a radiographic sign of non-vitality. Tooth #26 has undergone root canal.Mr. G reports that when he was in second grade, a psychiatrist diagnosed him with OCD, and has received treatment since then. We observe that while Mr. G is seated, he continually raises his right arm above his head and rubs his fingers together. He reports and demonstrates numerous other compulsive rituals, including head movements and rubbing his elbows against his side. His right elbow has a large scab.
He has been taking sertraline, 150 mg/d, for the past month. He says his psychiatrist prescribed this medication to help him break out of what he describes as episodes where he “gets into a mental loop.” Sertraline has improved Mr. G’s symptoms but they have not resolved.
He further reports that he has begun to “grind” his anterior teeth. Technically, he does not engage in grinding or bruxing; he has a habit of pushing his mandible forward so that his mandibular (lower) teeth are anterior to the maxillary (top) teeth, then forcefully pulling his mandible back so that the lingual (back) surfaces of the mandibular incisors push up against the buccal (outside) surfaces of the maxillary incisors (Photo 3). He states that he engaged in this habit frequently from approximately age 19 to 22. When Mr. G was 22, his dentist reduced the height of tooth #23, which Mr. G says he used “to set things in motion.” The dentist’s maneuver cut down but did not eliminate Mr. G’s habit.
Mr. G had not complained of nor had any clinician asked him about his bruxism-like behavior. He noted that the oral habit began prior to sertraline treatment, thus suggesting no relationship between the medication and the behavior. Interestingly, although some studies have reported bruxism as a side effect of SSRIs,2,3 at least 1 case report found that SSRIs reduced nocturnal bruxism.12
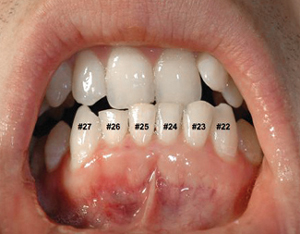
Photo 3 Obsessive-compulsive disorder manifested in Mr. G’s oral habit
Starting with his mandible pushed forward so that his mandibular (lower) teeth were anterior to the maxillary (upper) teeth, the patient would forcefully pull his mandible back so that the lingual (back) surfaces of the mandibular incisors pushed against the buccal (outside) surfaces of the maxillary incisors.As part of Mr. G’s dental examination, we take a full series of intraoral radiographs. These reveal radiolucencies at the apices of teeth #23 through #26 (Photo 2). The films also show root canal therapy on tooth #26.
Differential diagnosis for lesions in the periapical region of the mandibular incisors includes periapical cemental dysplasia (PCD), which typically is found in middle-aged African-American females,5 and lesions resulting from non-vitality of the teeth. Histopathologically, lesions resulting from the latter include an apical abscess, cyst, or granuloma.
As is customary when periapical lesions are noted, we test the vitality of the affected teeth. None of the affected teeth responded to cold or electric pulp testing, which indicated they were non-vital. Tooth vitality is not affected in PCD, which allowed us to exclude this condition.
Non-vital teeth indicate that the pulpal tissue is necrotic. Most commonly, non-vitality occurs when decay has penetrated the pulp chamber or as a complication of physical trauma. No decay was present on Mr. G’s mandibular anterior teeth and he denied a history of trauma such as a blow to the teeth. This left his oral habit as the likely cause of non-vitality.
Treatment for a non-vital tooth is a root canal, which had been done on tooth #26. We successfully performed root canal on Mr. G’s other non-vital teeth. We informed the patient of reason for his non-vital teeth, and made a protective occlusal guard to try to prevent additional trauma to the affected teeth.
Recognizing oral habits
Restoration of worn teeth, particularly those of the mandibular anterior, is technically difficult and—depending on the nature of the restoration—quite expensive. Endodontic therapy is more successful in teeth without periapical disease.6 Thus, preventing tooth-related problems in patients who grind their teeth or engage in other destructive dental behaviors is important.
As this case illustrates, teeth can become non-vital without clinical evidence of tooth wear; clinical evidence may be subtle or nonexistent (note teeth #23, #24, and #25 in Photo 2). Absence of tooth wear is not a reliable sign of tooth vitality. Mild to moderate tooth wear usually goes unnoticed by patients and clinicians.7
Patients with bruxism may complain of masticatory muscle soreness or increased wear of the teeth.7 In extreme cases, they may self-extract teeth as a result of bruxism.8
Screen patients who have anxiety disorders or depression for signs of bruxism or related behaviors (Table 2). If you detect signs of bruxism or related behavior, refer the patient to a dentist. Ask the dentist to look for signs of wear and perform vitality testing of teeth on a regular basis (twice a year is reasonable). Any signs of changes in pulp vitality should be followed up with intraoral periapical radiographs, which these patients might need more frequently than FDA guidelines recommend.9
Table 2
Screening for bruxism: 3 questions for patients
| 1. Do you have pain or discomfort in the jaw or facial muscles, headaches or earaches, or increased tooth sensitivity? |
| 2. Have you noticed changes in the way your teeth fit together or wearing down of your teeth? |
| 3. Has your sleeping partner noticed any noise at night that might be the result of teeth grinding? |
An occlusal guard may provide the most definitive tooth protection for patients who engage in bruxism or similar behaviors. Occlusal guards are made of material that is softer than enamel, so the patient will wear away the guard rather than tooth structure. When the guard is worn away, the patient needs a new one.
Pharmacologic strategies for bruxism or related oral habits involving the teeth are not well developed. One short-term, placebo-controlled trial for acute treatment in 10 drug-free patients with sleep bruxism consisted of a predrug night, a placebo night, and a clonazepam night. Clonazepam, 1 mg 30 minutes before bedtime, significantly improved bruxism and sleep quality as determined by objective and subjective measures.10
Kast11 reported 4 cases in which tiagabine suppressed nocturnal bruxism, trismus, and consequent morning pain in the teeth, masticatory musculature, jaw, and temporomandibular joint areas. This gammaaminobutyric acid reuptake inhibitor anticonvulsant approved for treating partial seizures was dosed at 4 to 8 mg at bedtime. These dosages are lower than those used to treat seizures.
Related resources
- American Dental Association. www.ada.org.
Drug brand names
- Clonazepam • Klonopin
- Haloperidol • Haldol
- Pimozide • Orap
- Sertraline • Zoloft
- Tiagabine • Gabitril
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Herren C, Lindroth J. Obsessive compulsive disorder: a case report. J Contemp Dent Pract. 2001;2:41-49.
2. Ellison JM, Stanziani P. SSRI-associated nocturnal bruxism in four patients. J Clin Psychiatry. 1993;54:432-434.
3. Gerber PE, Lynd LD. Selective serotonin-reuptake inhibitor-induced movement disorders. Ann Pharmacother. 1998;32:692-698.
4. Kent JM, Coplan JD, Gorman JM. Clinical utility of the selective serotonin reuptake inhibitors in the spectrum of anxiety. Biol Psychiatry. 1998;44:812-824.
5. White SC, Pharoah MJ. Oral radiology: principles and interpretation. St. Louis, MO: Mosby; 2004:492.
6. Salehrabi R, Rotstein I. Endodontic treatment outcome in a large patient population in the USA: an epidemiological study. J Endod. 2004;30:846-850.
7. Lobbezoo F, van Denderen RJ, Verheij JG, et al. Reports of SSRI-associated bruxism in the family physician’s office. J Orofac Pain. 2001;15:340-346.
8. Eisenhauer GL, Woody RC. Self-mutilation and Tourette’s disorder. J Child Neurol. 1987;2:265-267.
9. American Dental Association. Guidelines for prescribing dental radiographs. Available at: http://www.ada.org/prof/resources/topics/topics_radiography_chart.pdf. Accessed December 17, 2008.
10. Saletu A, Parapatics S, Saletu B, et al. On the pharmacotherapy of sleep bruxism: placebo-controlled polysomnographic and psychometric studies with clonazepam. Neuropsychobiology. 2005;51:214-225.
11. Kast RE. Tiagabine may reduce bruxism and associated temporomandibular joint pain. Anesth Prog. 2005;52:102-104.
12. Stein DJ, Van Greunen G, Niehaus D. Can bruxism respond to serotonin reuptake inhibitors? [letter]. J Clin Psychiatry. 1998;59:133.-
Oral habits such as bruxism—compulsive grinding or clenching of the teeth—can be a manifestation of obsessive-compulsive disorder (OCD) and other anxiety disorders.1 Bruxism also may be a side effect of selective serotonin reuptake inhibitors (SSRIs)2,3 used to treat OCD4 and depression. Other oral conditions can complicate treatment of these disorders (Table 1).
Potentially serious sequelae of bruxism and similar behaviors include:
- wearing down of teeth (more common)
- necrosis of the pulpal tissues that results in non-vital teeth (less common).
The following case underlines the need for early referral to a dentist and close follow-up for patients who have tooth-related behaviors or are taking medications associated with a risk for such behaviors.
Table 1
Oral conditions associated with anxiety disorders and depression
| Bruxism |
| Canker sores |
| Dry mouth |
| Temporomandibular joint disorders |
| Lichen planus (redness or mouth ulcers) |
| Non-vital teeth |
| Tooth wear, fracture |
CASE REPORT: A compulsive oral habit
Mr. G, age 26, presents to our school of dental medicine with the chief complaint that he needs a crown. On clinical intraoral examination, we find he has multiple restorations, some areas of recurrent tooth decay, and a fractured cusp on a maxillary molar. His mandibular incisors show greater wear on their incisal surfaces than would be expected for a patient his age. This is especially true of his mandibular right lateral incisor (tooth #26) and canine (tooth #27) (Photo 1).
Clinical examination also reveals a restoration on tooth #26 that was consistent with an access cavity drilled for endodontic (root canal) therapy (Photo 2). This finding is consistent with his dental history. Soft tissue examination is within normal limits. He reports that he saw a dentist 7 months earlier but could not afford the fees.
During his medical history, Mr. G states he has mild Tourette’s syndrome that was diagnosed when he was 10. In the early 1990s he tried several medications, including haloperidol and pimozide, as a subject in research studies of Tourette’s. He could not recall the dosages of the medications or for how long he took them. Because these medications did not improve his symptoms, he stopped taking them after the studies ended.
Photo 1 Unexpected tooth wear: A clue to an anxiety-related oral habit
Teeth of a 26-year-old man show greater than expected wear, particularly on the mandibular right lateral incisor (tooth #26) and canine (tooth #27).
Photo 2 Radiographic evidence of tooth non-vitality
Radiolucencies (dark areas) in the bone at the apices of the tooth roots are a radiographic sign of non-vitality. Tooth #26 has undergone root canal.Mr. G reports that when he was in second grade, a psychiatrist diagnosed him with OCD, and has received treatment since then. We observe that while Mr. G is seated, he continually raises his right arm above his head and rubs his fingers together. He reports and demonstrates numerous other compulsive rituals, including head movements and rubbing his elbows against his side. His right elbow has a large scab.
He has been taking sertraline, 150 mg/d, for the past month. He says his psychiatrist prescribed this medication to help him break out of what he describes as episodes where he “gets into a mental loop.” Sertraline has improved Mr. G’s symptoms but they have not resolved.
He further reports that he has begun to “grind” his anterior teeth. Technically, he does not engage in grinding or bruxing; he has a habit of pushing his mandible forward so that his mandibular (lower) teeth are anterior to the maxillary (top) teeth, then forcefully pulling his mandible back so that the lingual (back) surfaces of the mandibular incisors push up against the buccal (outside) surfaces of the maxillary incisors (Photo 3). He states that he engaged in this habit frequently from approximately age 19 to 22. When Mr. G was 22, his dentist reduced the height of tooth #23, which Mr. G says he used “to set things in motion.” The dentist’s maneuver cut down but did not eliminate Mr. G’s habit.
Mr. G had not complained of nor had any clinician asked him about his bruxism-like behavior. He noted that the oral habit began prior to sertraline treatment, thus suggesting no relationship between the medication and the behavior. Interestingly, although some studies have reported bruxism as a side effect of SSRIs,2,3 at least 1 case report found that SSRIs reduced nocturnal bruxism.12
Photo 3 Obsessive-compulsive disorder manifested in Mr. G’s oral habit
Starting with his mandible pushed forward so that his mandibular (lower) teeth were anterior to the maxillary (upper) teeth, the patient would forcefully pull his mandible back so that the lingual (back) surfaces of the mandibular incisors pushed against the buccal (outside) surfaces of the maxillary incisors.As part of Mr. G’s dental examination, we take a full series of intraoral radiographs. These reveal radiolucencies at the apices of teeth #23 through #26 (Photo 2). The films also show root canal therapy on tooth #26.
Differential diagnosis for lesions in the periapical region of the mandibular incisors includes periapical cemental dysplasia (PCD), which typically is found in middle-aged African-American females,5 and lesions resulting from non-vitality of the teeth. Histopathologically, lesions resulting from the latter include an apical abscess, cyst, or granuloma.
As is customary when periapical lesions are noted, we test the vitality of the affected teeth. None of the affected teeth responded to cold or electric pulp testing, which indicated they were non-vital. Tooth vitality is not affected in PCD, which allowed us to exclude this condition.
Non-vital teeth indicate that the pulpal tissue is necrotic. Most commonly, non-vitality occurs when decay has penetrated the pulp chamber or as a complication of physical trauma. No decay was present on Mr. G’s mandibular anterior teeth and he denied a history of trauma such as a blow to the teeth. This left his oral habit as the likely cause of non-vitality.
Treatment for a non-vital tooth is a root canal, which had been done on tooth #26. We successfully performed root canal on Mr. G’s other non-vital teeth. We informed the patient of reason for his non-vital teeth, and made a protective occlusal guard to try to prevent additional trauma to the affected teeth.
Recognizing oral habits
Restoration of worn teeth, particularly those of the mandibular anterior, is technically difficult and—depending on the nature of the restoration—quite expensive. Endodontic therapy is more successful in teeth without periapical disease.6 Thus, preventing tooth-related problems in patients who grind their teeth or engage in other destructive dental behaviors is important.
As this case illustrates, teeth can become non-vital without clinical evidence of tooth wear; clinical evidence may be subtle or nonexistent (note teeth #23, #24, and #25 in Photo 2). Absence of tooth wear is not a reliable sign of tooth vitality. Mild to moderate tooth wear usually goes unnoticed by patients and clinicians.7
Patients with bruxism may complain of masticatory muscle soreness or increased wear of the teeth.7 In extreme cases, they may self-extract teeth as a result of bruxism.8
Screen patients who have anxiety disorders or depression for signs of bruxism or related behaviors (Table 2). If you detect signs of bruxism or related behavior, refer the patient to a dentist. Ask the dentist to look for signs of wear and perform vitality testing of teeth on a regular basis (twice a year is reasonable). Any signs of changes in pulp vitality should be followed up with intraoral periapical radiographs, which these patients might need more frequently than FDA guidelines recommend.9
Table 2
Screening for bruxism: 3 questions for patients
| 1. Do you have pain or discomfort in the jaw or facial muscles, headaches or earaches, or increased tooth sensitivity? |
| 2. Have you noticed changes in the way your teeth fit together or wearing down of your teeth? |
| 3. Has your sleeping partner noticed any noise at night that might be the result of teeth grinding? |
An occlusal guard may provide the most definitive tooth protection for patients who engage in bruxism or similar behaviors. Occlusal guards are made of material that is softer than enamel, so the patient will wear away the guard rather than tooth structure. When the guard is worn away, the patient needs a new one.
Pharmacologic strategies for bruxism or related oral habits involving the teeth are not well developed. One short-term, placebo-controlled trial for acute treatment in 10 drug-free patients with sleep bruxism consisted of a predrug night, a placebo night, and a clonazepam night. Clonazepam, 1 mg 30 minutes before bedtime, significantly improved bruxism and sleep quality as determined by objective and subjective measures.10
Kast11 reported 4 cases in which tiagabine suppressed nocturnal bruxism, trismus, and consequent morning pain in the teeth, masticatory musculature, jaw, and temporomandibular joint areas. This gammaaminobutyric acid reuptake inhibitor anticonvulsant approved for treating partial seizures was dosed at 4 to 8 mg at bedtime. These dosages are lower than those used to treat seizures.
Related resources
- American Dental Association. www.ada.org.
Drug brand names
- Clonazepam • Klonopin
- Haloperidol • Haldol
- Pimozide • Orap
- Sertraline • Zoloft
- Tiagabine • Gabitril
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Oral habits such as bruxism—compulsive grinding or clenching of the teeth—can be a manifestation of obsessive-compulsive disorder (OCD) and other anxiety disorders.1 Bruxism also may be a side effect of selective serotonin reuptake inhibitors (SSRIs)2,3 used to treat OCD4 and depression. Other oral conditions can complicate treatment of these disorders (Table 1).
Potentially serious sequelae of bruxism and similar behaviors include:
- wearing down of teeth (more common)
- necrosis of the pulpal tissues that results in non-vital teeth (less common).
The following case underlines the need for early referral to a dentist and close follow-up for patients who have tooth-related behaviors or are taking medications associated with a risk for such behaviors.
Table 1
Oral conditions associated with anxiety disorders and depression
| Bruxism |
| Canker sores |
| Dry mouth |
| Temporomandibular joint disorders |
| Lichen planus (redness or mouth ulcers) |
| Non-vital teeth |
| Tooth wear, fracture |
CASE REPORT: A compulsive oral habit
Mr. G, age 26, presents to our school of dental medicine with the chief complaint that he needs a crown. On clinical intraoral examination, we find he has multiple restorations, some areas of recurrent tooth decay, and a fractured cusp on a maxillary molar. His mandibular incisors show greater wear on their incisal surfaces than would be expected for a patient his age. This is especially true of his mandibular right lateral incisor (tooth #26) and canine (tooth #27) (Photo 1).
Clinical examination also reveals a restoration on tooth #26 that was consistent with an access cavity drilled for endodontic (root canal) therapy (Photo 2). This finding is consistent with his dental history. Soft tissue examination is within normal limits. He reports that he saw a dentist 7 months earlier but could not afford the fees.
During his medical history, Mr. G states he has mild Tourette’s syndrome that was diagnosed when he was 10. In the early 1990s he tried several medications, including haloperidol and pimozide, as a subject in research studies of Tourette’s. He could not recall the dosages of the medications or for how long he took them. Because these medications did not improve his symptoms, he stopped taking them after the studies ended.
Photo 1 Unexpected tooth wear: A clue to an anxiety-related oral habit
Teeth of a 26-year-old man show greater than expected wear, particularly on the mandibular right lateral incisor (tooth #26) and canine (tooth #27).
Photo 2 Radiographic evidence of tooth non-vitality
Radiolucencies (dark areas) in the bone at the apices of the tooth roots are a radiographic sign of non-vitality. Tooth #26 has undergone root canal.Mr. G reports that when he was in second grade, a psychiatrist diagnosed him with OCD, and has received treatment since then. We observe that while Mr. G is seated, he continually raises his right arm above his head and rubs his fingers together. He reports and demonstrates numerous other compulsive rituals, including head movements and rubbing his elbows against his side. His right elbow has a large scab.
He has been taking sertraline, 150 mg/d, for the past month. He says his psychiatrist prescribed this medication to help him break out of what he describes as episodes where he “gets into a mental loop.” Sertraline has improved Mr. G’s symptoms but they have not resolved.
He further reports that he has begun to “grind” his anterior teeth. Technically, he does not engage in grinding or bruxing; he has a habit of pushing his mandible forward so that his mandibular (lower) teeth are anterior to the maxillary (top) teeth, then forcefully pulling his mandible back so that the lingual (back) surfaces of the mandibular incisors push up against the buccal (outside) surfaces of the maxillary incisors (Photo 3). He states that he engaged in this habit frequently from approximately age 19 to 22. When Mr. G was 22, his dentist reduced the height of tooth #23, which Mr. G says he used “to set things in motion.” The dentist’s maneuver cut down but did not eliminate Mr. G’s habit.
Mr. G had not complained of nor had any clinician asked him about his bruxism-like behavior. He noted that the oral habit began prior to sertraline treatment, thus suggesting no relationship between the medication and the behavior. Interestingly, although some studies have reported bruxism as a side effect of SSRIs,2,3 at least 1 case report found that SSRIs reduced nocturnal bruxism.12
Photo 3 Obsessive-compulsive disorder manifested in Mr. G’s oral habit
Starting with his mandible pushed forward so that his mandibular (lower) teeth were anterior to the maxillary (upper) teeth, the patient would forcefully pull his mandible back so that the lingual (back) surfaces of the mandibular incisors pushed against the buccal (outside) surfaces of the maxillary incisors.As part of Mr. G’s dental examination, we take a full series of intraoral radiographs. These reveal radiolucencies at the apices of teeth #23 through #26 (Photo 2). The films also show root canal therapy on tooth #26.
Differential diagnosis for lesions in the periapical region of the mandibular incisors includes periapical cemental dysplasia (PCD), which typically is found in middle-aged African-American females,5 and lesions resulting from non-vitality of the teeth. Histopathologically, lesions resulting from the latter include an apical abscess, cyst, or granuloma.
As is customary when periapical lesions are noted, we test the vitality of the affected teeth. None of the affected teeth responded to cold or electric pulp testing, which indicated they were non-vital. Tooth vitality is not affected in PCD, which allowed us to exclude this condition.
Non-vital teeth indicate that the pulpal tissue is necrotic. Most commonly, non-vitality occurs when decay has penetrated the pulp chamber or as a complication of physical trauma. No decay was present on Mr. G’s mandibular anterior teeth and he denied a history of trauma such as a blow to the teeth. This left his oral habit as the likely cause of non-vitality.
Treatment for a non-vital tooth is a root canal, which had been done on tooth #26. We successfully performed root canal on Mr. G’s other non-vital teeth. We informed the patient of reason for his non-vital teeth, and made a protective occlusal guard to try to prevent additional trauma to the affected teeth.
Recognizing oral habits
Restoration of worn teeth, particularly those of the mandibular anterior, is technically difficult and—depending on the nature of the restoration—quite expensive. Endodontic therapy is more successful in teeth without periapical disease.6 Thus, preventing tooth-related problems in patients who grind their teeth or engage in other destructive dental behaviors is important.
As this case illustrates, teeth can become non-vital without clinical evidence of tooth wear; clinical evidence may be subtle or nonexistent (note teeth #23, #24, and #25 in Photo 2). Absence of tooth wear is not a reliable sign of tooth vitality. Mild to moderate tooth wear usually goes unnoticed by patients and clinicians.7
Patients with bruxism may complain of masticatory muscle soreness or increased wear of the teeth.7 In extreme cases, they may self-extract teeth as a result of bruxism.8
Screen patients who have anxiety disorders or depression for signs of bruxism or related behaviors (Table 2). If you detect signs of bruxism or related behavior, refer the patient to a dentist. Ask the dentist to look for signs of wear and perform vitality testing of teeth on a regular basis (twice a year is reasonable). Any signs of changes in pulp vitality should be followed up with intraoral periapical radiographs, which these patients might need more frequently than FDA guidelines recommend.9
Table 2
Screening for bruxism: 3 questions for patients
| 1. Do you have pain or discomfort in the jaw or facial muscles, headaches or earaches, or increased tooth sensitivity? |
| 2. Have you noticed changes in the way your teeth fit together or wearing down of your teeth? |
| 3. Has your sleeping partner noticed any noise at night that might be the result of teeth grinding? |
An occlusal guard may provide the most definitive tooth protection for patients who engage in bruxism or similar behaviors. Occlusal guards are made of material that is softer than enamel, so the patient will wear away the guard rather than tooth structure. When the guard is worn away, the patient needs a new one.
Pharmacologic strategies for bruxism or related oral habits involving the teeth are not well developed. One short-term, placebo-controlled trial for acute treatment in 10 drug-free patients with sleep bruxism consisted of a predrug night, a placebo night, and a clonazepam night. Clonazepam, 1 mg 30 minutes before bedtime, significantly improved bruxism and sleep quality as determined by objective and subjective measures.10
Kast11 reported 4 cases in which tiagabine suppressed nocturnal bruxism, trismus, and consequent morning pain in the teeth, masticatory musculature, jaw, and temporomandibular joint areas. This gammaaminobutyric acid reuptake inhibitor anticonvulsant approved for treating partial seizures was dosed at 4 to 8 mg at bedtime. These dosages are lower than those used to treat seizures.
Related resources
- American Dental Association. www.ada.org.
Drug brand names
- Clonazepam • Klonopin
- Haloperidol • Haldol
- Pimozide • Orap
- Sertraline • Zoloft
- Tiagabine • Gabitril
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Herren C, Lindroth J. Obsessive compulsive disorder: a case report. J Contemp Dent Pract. 2001;2:41-49.
2. Ellison JM, Stanziani P. SSRI-associated nocturnal bruxism in four patients. J Clin Psychiatry. 1993;54:432-434.
3. Gerber PE, Lynd LD. Selective serotonin-reuptake inhibitor-induced movement disorders. Ann Pharmacother. 1998;32:692-698.
4. Kent JM, Coplan JD, Gorman JM. Clinical utility of the selective serotonin reuptake inhibitors in the spectrum of anxiety. Biol Psychiatry. 1998;44:812-824.
5. White SC, Pharoah MJ. Oral radiology: principles and interpretation. St. Louis, MO: Mosby; 2004:492.
6. Salehrabi R, Rotstein I. Endodontic treatment outcome in a large patient population in the USA: an epidemiological study. J Endod. 2004;30:846-850.
7. Lobbezoo F, van Denderen RJ, Verheij JG, et al. Reports of SSRI-associated bruxism in the family physician’s office. J Orofac Pain. 2001;15:340-346.
8. Eisenhauer GL, Woody RC. Self-mutilation and Tourette’s disorder. J Child Neurol. 1987;2:265-267.
9. American Dental Association. Guidelines for prescribing dental radiographs. Available at: http://www.ada.org/prof/resources/topics/topics_radiography_chart.pdf. Accessed December 17, 2008.
10. Saletu A, Parapatics S, Saletu B, et al. On the pharmacotherapy of sleep bruxism: placebo-controlled polysomnographic and psychometric studies with clonazepam. Neuropsychobiology. 2005;51:214-225.
11. Kast RE. Tiagabine may reduce bruxism and associated temporomandibular joint pain. Anesth Prog. 2005;52:102-104.
12. Stein DJ, Van Greunen G, Niehaus D. Can bruxism respond to serotonin reuptake inhibitors? [letter]. J Clin Psychiatry. 1998;59:133.-
1. Herren C, Lindroth J. Obsessive compulsive disorder: a case report. J Contemp Dent Pract. 2001;2:41-49.
2. Ellison JM, Stanziani P. SSRI-associated nocturnal bruxism in four patients. J Clin Psychiatry. 1993;54:432-434.
3. Gerber PE, Lynd LD. Selective serotonin-reuptake inhibitor-induced movement disorders. Ann Pharmacother. 1998;32:692-698.
4. Kent JM, Coplan JD, Gorman JM. Clinical utility of the selective serotonin reuptake inhibitors in the spectrum of anxiety. Biol Psychiatry. 1998;44:812-824.
5. White SC, Pharoah MJ. Oral radiology: principles and interpretation. St. Louis, MO: Mosby; 2004:492.
6. Salehrabi R, Rotstein I. Endodontic treatment outcome in a large patient population in the USA: an epidemiological study. J Endod. 2004;30:846-850.
7. Lobbezoo F, van Denderen RJ, Verheij JG, et al. Reports of SSRI-associated bruxism in the family physician’s office. J Orofac Pain. 2001;15:340-346.
8. Eisenhauer GL, Woody RC. Self-mutilation and Tourette’s disorder. J Child Neurol. 1987;2:265-267.
9. American Dental Association. Guidelines for prescribing dental radiographs. Available at: http://www.ada.org/prof/resources/topics/topics_radiography_chart.pdf. Accessed December 17, 2008.
10. Saletu A, Parapatics S, Saletu B, et al. On the pharmacotherapy of sleep bruxism: placebo-controlled polysomnographic and psychometric studies with clonazepam. Neuropsychobiology. 2005;51:214-225.
11. Kast RE. Tiagabine may reduce bruxism and associated temporomandibular joint pain. Anesth Prog. 2005;52:102-104.
12. Stein DJ, Van Greunen G, Niehaus D. Can bruxism respond to serotonin reuptake inhibitors? [letter]. J Clin Psychiatry. 1998;59:133.-
Unipolar depression or "soft" bipolar disorder? Tips to avoid misdiagnosis
Postpartum psychosis: Strategies to protect infant and mother from harm
In June 2001, Andrea Yates drowned her 5 children ages 6 months to 7 years in the bathtub of their home. She had delusions that her house was bugged and television cameras were monitoring her mothering skills. She came to believe that “the one and only Satan” was within her, and that her children would burn in hell if she did not save their souls while they were still innocent.
Her conviction of capital murder in her first trial was overturned on appeal. She was found not guilty by reason of insanity at her retrial in 2006 and committed to a Texas state mental hospital.1
Postpartum psychosis (PPP) presents dramatically days to weeks after delivery, with wide-ranging symptoms that can include dysphoric mania and delirium. Because untreated PPP has an estimated 4% risk of infanticide (murder of the infant in the first year of life),2 and a 5% risk of suicide,3 psychiatric hospitalization usually is required to protect the mother and her baby.
The diagnosis may be missed, however, because postpartum psychotic symptoms wax and wane and suspiciousness or poor insight cause some women—such as Andrea Yates—to hide their delusional thinking from their families. This article discusses the risk factors, prevention, and treatment of PPP, including a review of:
- infanticide and suicide risks in the postpartum period
- increased susceptibility to PPP in women with bipolar disorder and other psychiatric disorders
- hospitalization for support and safety of the mother and her infant.
Risks of infanticide and suicide
A number of motives exist for infanticide (Table 1).4 Psychiatric literature shows that mothers who kill their children often have experienced psychosis, suicidality, depression, and considerable life stress.5 Common factors include alcohol use, limited social support, and a personal history of abuse. Studies on infanticide found a significant increase in common psychiatric disorders and financial stress among the mothers. Neonaticide (murder of the infant in the first day of life) generally is not related to PPP because PPP usually does not begin until after the day of delivery.6
Among women who develop psychiatric illness, homicidal ideation is more frequent in those with a perinatal onset of psychopathology.7 Infanticidal ideas and behavior are associated with psychotic ideas about the infant.8 Suicide is the cause of up to 20% of postpartum deaths.9
Table 1
Motives for infanticide: Mental illness or something else?
| Motives | Examples |
|---|---|
| Likely related to postpartum psychosis or depression | |
| Altruistic | A depressed or psychotic mother may believe she is sending her baby to heaven to prevent suffering on earth |
| A suicidal mother may kill her infant along with herself rather than leave the child alone | |
| Acutely psychotic | A mother kills her baby for no comprehensible reason, such as in response to command hallucinations or the confusion of delirium |
| Rarely related to postpartum psychosis | |
| Fatal maltreatment | ‘Battered child’ syndrome is the most common cause of infanticide; death often occurs after chronic abuse or neglect |
| A minority of perpetrators are psychotic; a mother out of touch with reality may have difficulty providing for her infant’s needs | |
| Not likely related to postpartum psychosis | |
| Unwanted child | Parent does not want child because of inconvenience or out-of-wedlock birth |
| Spouse revenge | Murder of a child to cause emotional suffering for the other parent is the least frequent motive for infanticide |
| Source: Reference 4 | |
The bipolar connection
Many factors can elevate the risk of PPP, including sleep deprivation in susceptible women, the hormonal shifts after birth, and psychiatric comorbidity (Table 2). Nearly three-fourths (>72%) of mothers with PPP have bipolar disorder or schizoaffective disorder, whereas 12% have schizophrenia.10 Some authors consider PPP to be bipolar disorder until proven otherwise. Mothers with a history of bipolar disorder or PPP have a 100-fold increase in rates of psychiatric hospitalization in the postpartum period.11
Presentation. PPP is relatively rare, occurring at a rate of 1 to 3 cases per 1,000 births. Symptoms often have an abrupt onset, within days to weeks of delivery.10 In at least one-half of cases, symptoms begin by the third postpartum day,13 when many mothers have been discharged home and may be solely responsible for their infants.
Symptoms include confusion, bizarre behaviors, hallucinations (including rarer types such as tactile and olfactory), mood lability (ranging from euphoria to depression), decreased need for sleep or insomnia, restlessness, agitation, disorganized thinking, and bizarre delusions of relatively rapid onset.13 One mother might believe God wants her baby to be sacrificed as the second coming of the Messiah, a second may believe she has special powers, and a third that her baby is defective.
Table 2
Postpartum psychosis: Risk factors supported by evidence
| Sleep deprivation in susceptible women |
| Hormonal shifts after birth (primarily the rapid drop in estrogen) |
| Psychosocial stressors such as marital problems, older age, single motherhood, lower socioeconomic status |
| Bipolar disorder or schizoaffective disorder |
| Past history of postpartum psychosis |
| Family history of postpartum psychosis |
| Previous psychiatric hospitalization, especially during the prenatal period for a bipolar or psychotic condition |
| Menstruation or cessation of lactation |
Obstetric factors that can cause a small increase in relative risk:
|
| Source: For bibliographic citations |
Differential diagnosis
When evaluating a postpartum woman with psychotic symptoms, stay in contact with her obstetrician and the child’s pediatrician. Rule out delirium and organic causes of the mother’s symptoms (Box).11
Postpartum depression (PPD) occurs in approximately 10% to 15% of new mothers.14 Depressive symptoms occur within weeks to months after delivery and often coexist with anxious symptoms. Some women with severe depression may present with psychotic symptoms. A mother may experience insomnia, sometimes not being able to sleep when the baby is sleeping. She may lack interest in caring for her baby and experience difficulty bonding.
At times it can be difficult to distinguish PPD from PPP. When evaluating a mother who is referred for “postpartum depression,” consider PPP in the differential diagnosis. A woman with PPD or PPP may report depressed mood, but in PPP this symptom usually is related to rapid mood changes. Other clinical features that point toward PPP are abnormal hallucinations (such as olfactory or tactile), hypomanic or mixed mood symptoms, and confusion.
When evaluating a postpartum woman with psychotic symptoms, stay in contact with her obstetrician and the child’s pediatrician. Rule out delirium and organic causes of the mother’s symptoms, giving special consideration to metabolic, neurologic, cardiovascular, infectious, and substance- or medication-induced origins. The extensive differential diagnosis includes:
- thyroiditis
- tumor
- CNS infection
- head injury
- embolism
- eclampsia
- substance withdrawal
- medication-induced (such as corticosteroids)
- electrolyte anomalies
- anoxia
- vitamin B12 deficiency.11
Psychosis vs OCD. Psychotic thinking and behaviors also must be differentiated from obsessive thoughts and compulsions.10,16 Obsessive compulsive disorder (OCD) may be exacerbated or emerge for the first time during the perinatal period.17
In postpartum OCD, women may experience intrusive thoughts of accidental or purposeful harm to their baby. As opposed to women with PPP, mothers with OCD are not out of touch with reality and their thoughts are ego-dystonic.17 When these mothers have thoughts of their infants being harmed, they realize that these thoughts are not plans but fears and they try to avoid the thoughts.
Preventing PPP
Bipolar disorder is one of the most difficult disorders to treat during pregnancy because the serious risks of untreated illness must be balanced against the potential teratogenic risk of medications. Nevertheless, proactively managing bipolar disorder during pregnancy may reduce the risk of PPP.10
Closely monitor women with a history of bipolar disorder or PPP. During pregnancy, counsel them—and their partners—to:
- anticipate that depressive or psychotic symptoms could develop within days after delivery18
- seek treatment immediately if this occurs.
Postpartum medication. Whether or not a woman with bipolar disorder takes medication during pregnancy, consider treatment with mood stabilizers or atypical antipsychotics in the postpartum to prevent PPP (Table 3). Evidence is limited, but a search of PubMed found 1 study in which prophylactic lithium was given late in the third trimester or immediately after delivery to 21 women with a history of bipolar disorder or PPP. Only 2 patients had a psychotic recurrence while on prophylactic lithium; 1 unexplained stillbirth occurred.19
A retrospective study examined the course of women with bipolar disorder, some of whom were given prophylactic mood stabilizers immediately in the postpartum. One of 14 who received antimanic agents relapsed within the first 3 months postpartum, compared with 8 of 13 who were not so treated.18
Compared with antiepileptics, less information is available about the use of atypical antipsychotics in pregnancy and lactation. Antipsychotics’ potential advantage in women at risk for PPP is that these agents may help prevent or treat both manic and psychotic symptoms.
In a small, naturalistic, prospective study, 11 women at risk for PPP received olanzapine alone or with an antidepressant or mood stabilizer for at least 4 weeks after delivery. Two (18%) experienced a postpartum mood episode, compared with 8 (57%) of 14 other at-risk women who received antidepressants, mood stabilizers, or no medication.20
When you discuss breast-feeding, consider possible risks to the neonate as well as potential sleep interruption for the mother. If a mother has a supportive partner, the partner might be put in charge of night-time feedings in a routine combining breast-feeding and bottle-feeding. In some cases you may need to recommend cessation of lactation.21
Table 3
Treating postpartum psychosis: Consider 3 components
| Component | Recommendations |
|---|---|
| Hospitalization vs home care | Hospitalize in most cases because of emergent severe symptoms and fluctuating course; base decision on risk evaluation/safety issues for patient and infant After discharge, visiting nurses are useful to help monitor the mother’s condition at home |
| Psychoeducation | Educate patient, family, and social support network; address risks to mother and infant and risks in future pregnancies |
| Medication | When prescribing mood stabilizers and/or antipsychotics, consider:
|
Managing PPP
Early symptoms. Because of its severity and rapid evolution, PPP often presents as a psychiatric emergency. Monitor atrisk patients’ sleep patterns and mood for early signs of psychosis.22 Watch especially for hypomanic symptoms such as elevated or mixed mood and decreased judgment, which are common early in PPP.13
A mother with few signs of abnormal mood, good social support, and close follow-up may potentially be safely managed as an outpatient. Initial evaluation and management of PPP usually requires hospitalization, however, because of the risks of suicide, infanticide, and child maltreatment.23
Hospitalization. Mother-infant bonding is important, but safety is paramount if a mother is psychotic—especially if she is experiencing psychotic thoughts about her infant. If possible, the infant should remain with family members during the mother’s hospitalization. Supervised mother-infant visits are often arranged, as appropriate.
Mood-stabilizing medications, including antipsychotics, are mainstays of treatment.24 In some cases, conventional antipsychotics such as haloperidol may be useful because of a lower risk of weight gain or of sedation that could impair a mother’s ability to respond to her infant. Electroconvulsive therapy often yields rapid symptomatic improvement for mothers with postpartum mood or psychotic symptoms.25
Discharge planning. Assuming that the mother adheres to prescribed treatment, discharge may occur within 1 week. Plan discharge arrangements carefully (Table 4).28 A team approach can be very useful within the outpatient clinic. In the model of the Perinatal Psychiatry Clinic of Connections in suburban Cleveland, OH, the mother’s treatment team includes perinatal psychiatrists, nurses, counsellors, case managers (who do home visits), and peer counselors.
Outpatient civil commitment, in which patients are mandated to accept treatment, is an option in some jurisdictions and could help ensure that patients receive treatment consistently.
Table 4
Discharge planning for safety of mother and infant
| Notify child protective services (CPS) depending on the risk to the child. Case-by-case review is needed to assess whether the infant should be removed. CPS may put in place a plan for safety, short of removal. The plan may require that the woman continue psychiatric care |
| Meet with the patient and family to discuss her diagnosis, the risks, the importance of continued medication adherence, and the need for family or social supports to assist with child care |
| Consider engaging visiting nurses or doulas to provide help and support at home |
| Schedule frequent outpatient appointments for the mother after discharge |
| Consider family therapy after the mother has improved because of her risk for affective episodes outside the postpartum28 |
Related resources
- Altshuler L, Richards M, Yonkers K. Treating bipolar disorder during pregnancy. Current Psychiatry. 2003;2(7):14-26. www.CurrentPsychiatry.com.
- Gentile S. Infant safety with antipsychotic therapy in breastfeeding: a systematic review. J Clin Psychiatry. 2008;69(4):666-673.
- Miller LJ. Postpartum mood disorders. Washington, DC: American Psychiatric Publishing, Inc; 1999.
- Stowe ZN. The use of mood stabilizers during breastfeeding. J Clin Psychiatry. 2007;68(suppl 9):22-28.
- Toxicology Data Network (Toxnet). Literature on reproductive risks associated with psychotropics. National Library of Medicine. http://toxnet.nlm.nih.gov.
- Haloperidol • Haldol
- Lithium • various
- Olanzapine • Zyprexa
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
Dr. Resnick, a forensic psychiatrist and coauthor of this article, testified for the defense in both trials of Andrea Yates.
1. Resnick PJ. The Andrea Yates case: insanity on trial. Cleveland State Law Review. 2007;55(2):147-156.
2. Altshuler LL, Hendrick V, Cohen LS. Course of mood and anxiety disorders during pregnancy and the postpartum period. J Clin Psychiatry. 1998;59(suppl. 2):29-33.
3. Knops GG. Postpartum mood disorders. Postgrad Med. 1993;93:103-116.
4. Resnick PJ. Child murder by parents: a psychiatric review of filicide. Am J Psychiatry. 1969;126:73-82.
5. Friedman SH, Horwitz SM, Resnick PJ. Child murder by mothers: a critical analysis of the current state of knowledge and a research agenda. Am J Psychiatry. 2005;162:1578-1587.
6. Friedman SH, Resnick PJ. Neonaticide: phenomenology and considerations for prevention. Int J Law Psychiatry. In press.
7. Wisner K, Peindl K, Hanusa BH. Symptomatology of affective and psychotic illnesses related to childbearing. J Affect Disord. 1994;30:77-87.
8. Chandra PS, Venkatasubramanian G, Thomas T. Infanticidal ideas and infanticidal behaviour in Indian women with severe postpartum psychiatric disorders. J Nerv Ment Dis. 2002;190(7):457-461.
9. Lindahl V, Pearson JL, Colpe L. Prevalence of suicidality during pregnancy and the postpartum. Arch Womens Ment Health. 2005;8(2):77-87.
10. Sit D, Rothschild AJ, Wisner KL. A review of postpartum psychosis. J Women’s Health. 2006;15(4):352-368.
11. Attia E, Downey J, Oberman M. Postpartum psychoses. In: Miller LJ, ed. Postpartum mood disorders. Washington, DC: American Psychiatric Publishing Inc.; 1999:99-117.
12. Diagnostic and Statistical Manual of Mental Disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
13. Heron J, McGuinness M, Blackmore ER, et al. Early postpartum symptoms in puerperal psychosis. BJOG. 2008;115(3):348-353.
14. Meltzer-Brody S, Payne J, Rubinow D. Postpartum depression: what to tell patients who breast-feed. Current Psychiatry. 2008;7(5):87-95.
15. Jennings KD, Ross S, Popper S, et al. Thoughts of harming infants in depressed and nondepressed mothers. J Affect Disord. 1999;54:21-28.
16. Wisner KL, Gracious BL, Piontek CM, et al. Postpartum disorders: phenomenology, treatment approaches, and relationship to infanticide. In: Spinelli MG, ed. Infanticide: psychosocial and legal perspectives on mothers who kill. Washington, DC: American Psychiatric Publishing, Inc.; 2003.
17. Fairbrother N, Abramowitz JS. New parenthood as a risk factors for the development of obsessional problems. Behav Res Ther. 2007;45(9):2155-2163.
18. Cohen LS, Sichel DA, Robertson LM, et al. Postpartum prophylaxis for women with bipolar disorder. Am J Psychiatry. 1995;152(11):1641-1645.
19. Stewart DE, Klompenhouwer JL, Kendell RE, et al. Prophylactic lithium in puerperal psychosis. Br J Psychiatry. 1991;158:393-397.
20. Sharma V, Smith A, Mazmanian D. Olanzapine in the prevention of postpartum psychosis and mood episodes in bipolar disorder. Bipolar Disord. 2006;8(4):400-404.
21. Pfuhlmann B, Stoeber G, Beckmann H. Postpartum psychoses: prognosis, risk factors, and treatment. Curr Psychiatry Rep. 2002;4(3):185-190.
22. Sharma V, Mazmanian D. Sleep loss and postpartum psychosis. Bipolar Disord. 2003;5(2):98-105.
23. Lindahl V, Pearson JL, Colpe L. Prevalence of suicidality during pregnancy and the postpartum. Arch Womens Ment Health. 2005;8(2):77-87.
24. Connell M. The postpartum psychosis defense and feminism: more or less justice for women? Case Western Reserve Law Review. 2002;53:143.-
25. Forray A, Ostroff RB. The use of electroconvulsive therapy in postpartum affective disorders. J ECT. 2007;23(3):188-193.
26. Engqvist I, Nilsson A, Nilsson K, et al. Strategies in caring for women with postpartum psychosis—an interview study with psychiatric nurses. J Clin Nurs. 2007;16(7):1333-1342.
27. Robertson E, Lyons A. Living with puerperal psychosis: a qualitative analysis. Psychol Psychother. 2003;76(4):411-431.
28. Robertson E, Jones I, Haque S, et al. Risk of puerperal and non-puerperal recurrence of illness following bipolar affective puerperal (post-partum) psychosis. Br J Psychiatry. 2005;186:258-259.
In June 2001, Andrea Yates drowned her 5 children ages 6 months to 7 years in the bathtub of their home. She had delusions that her house was bugged and television cameras were monitoring her mothering skills. She came to believe that “the one and only Satan” was within her, and that her children would burn in hell if she did not save their souls while they were still innocent.
Her conviction of capital murder in her first trial was overturned on appeal. She was found not guilty by reason of insanity at her retrial in 2006 and committed to a Texas state mental hospital.1
Postpartum psychosis (PPP) presents dramatically days to weeks after delivery, with wide-ranging symptoms that can include dysphoric mania and delirium. Because untreated PPP has an estimated 4% risk of infanticide (murder of the infant in the first year of life),2 and a 5% risk of suicide,3 psychiatric hospitalization usually is required to protect the mother and her baby.
The diagnosis may be missed, however, because postpartum psychotic symptoms wax and wane and suspiciousness or poor insight cause some women—such as Andrea Yates—to hide their delusional thinking from their families. This article discusses the risk factors, prevention, and treatment of PPP, including a review of:
- infanticide and suicide risks in the postpartum period
- increased susceptibility to PPP in women with bipolar disorder and other psychiatric disorders
- hospitalization for support and safety of the mother and her infant.
Risks of infanticide and suicide
A number of motives exist for infanticide (Table 1).4 Psychiatric literature shows that mothers who kill their children often have experienced psychosis, suicidality, depression, and considerable life stress.5 Common factors include alcohol use, limited social support, and a personal history of abuse. Studies on infanticide found a significant increase in common psychiatric disorders and financial stress among the mothers. Neonaticide (murder of the infant in the first day of life) generally is not related to PPP because PPP usually does not begin until after the day of delivery.6
Among women who develop psychiatric illness, homicidal ideation is more frequent in those with a perinatal onset of psychopathology.7 Infanticidal ideas and behavior are associated with psychotic ideas about the infant.8 Suicide is the cause of up to 20% of postpartum deaths.9
Table 1
Motives for infanticide: Mental illness or something else?
| Motives | Examples |
|---|---|
| Likely related to postpartum psychosis or depression | |
| Altruistic | A depressed or psychotic mother may believe she is sending her baby to heaven to prevent suffering on earth |
| A suicidal mother may kill her infant along with herself rather than leave the child alone | |
| Acutely psychotic | A mother kills her baby for no comprehensible reason, such as in response to command hallucinations or the confusion of delirium |
| Rarely related to postpartum psychosis | |
| Fatal maltreatment | ‘Battered child’ syndrome is the most common cause of infanticide; death often occurs after chronic abuse or neglect |
| A minority of perpetrators are psychotic; a mother out of touch with reality may have difficulty providing for her infant’s needs | |
| Not likely related to postpartum psychosis | |
| Unwanted child | Parent does not want child because of inconvenience or out-of-wedlock birth |
| Spouse revenge | Murder of a child to cause emotional suffering for the other parent is the least frequent motive for infanticide |
| Source: Reference 4 | |
The bipolar connection
Many factors can elevate the risk of PPP, including sleep deprivation in susceptible women, the hormonal shifts after birth, and psychiatric comorbidity (Table 2). Nearly three-fourths (>72%) of mothers with PPP have bipolar disorder or schizoaffective disorder, whereas 12% have schizophrenia.10 Some authors consider PPP to be bipolar disorder until proven otherwise. Mothers with a history of bipolar disorder or PPP have a 100-fold increase in rates of psychiatric hospitalization in the postpartum period.11
Presentation. PPP is relatively rare, occurring at a rate of 1 to 3 cases per 1,000 births. Symptoms often have an abrupt onset, within days to weeks of delivery.10 In at least one-half of cases, symptoms begin by the third postpartum day,13 when many mothers have been discharged home and may be solely responsible for their infants.
Symptoms include confusion, bizarre behaviors, hallucinations (including rarer types such as tactile and olfactory), mood lability (ranging from euphoria to depression), decreased need for sleep or insomnia, restlessness, agitation, disorganized thinking, and bizarre delusions of relatively rapid onset.13 One mother might believe God wants her baby to be sacrificed as the second coming of the Messiah, a second may believe she has special powers, and a third that her baby is defective.
Table 2
Postpartum psychosis: Risk factors supported by evidence
| Sleep deprivation in susceptible women |
| Hormonal shifts after birth (primarily the rapid drop in estrogen) |
| Psychosocial stressors such as marital problems, older age, single motherhood, lower socioeconomic status |
| Bipolar disorder or schizoaffective disorder |
| Past history of postpartum psychosis |
| Family history of postpartum psychosis |
| Previous psychiatric hospitalization, especially during the prenatal period for a bipolar or psychotic condition |
| Menstruation or cessation of lactation |
Obstetric factors that can cause a small increase in relative risk:
|
| Source: For bibliographic citations |
Differential diagnosis
When evaluating a postpartum woman with psychotic symptoms, stay in contact with her obstetrician and the child’s pediatrician. Rule out delirium and organic causes of the mother’s symptoms (Box).11
Postpartum depression (PPD) occurs in approximately 10% to 15% of new mothers.14 Depressive symptoms occur within weeks to months after delivery and often coexist with anxious symptoms. Some women with severe depression may present with psychotic symptoms. A mother may experience insomnia, sometimes not being able to sleep when the baby is sleeping. She may lack interest in caring for her baby and experience difficulty bonding.
At times it can be difficult to distinguish PPD from PPP. When evaluating a mother who is referred for “postpartum depression,” consider PPP in the differential diagnosis. A woman with PPD or PPP may report depressed mood, but in PPP this symptom usually is related to rapid mood changes. Other clinical features that point toward PPP are abnormal hallucinations (such as olfactory or tactile), hypomanic or mixed mood symptoms, and confusion.
When evaluating a postpartum woman with psychotic symptoms, stay in contact with her obstetrician and the child’s pediatrician. Rule out delirium and organic causes of the mother’s symptoms, giving special consideration to metabolic, neurologic, cardiovascular, infectious, and substance- or medication-induced origins. The extensive differential diagnosis includes:
- thyroiditis
- tumor
- CNS infection
- head injury
- embolism
- eclampsia
- substance withdrawal
- medication-induced (such as corticosteroids)
- electrolyte anomalies
- anoxia
- vitamin B12 deficiency.11
Psychosis vs OCD. Psychotic thinking and behaviors also must be differentiated from obsessive thoughts and compulsions.10,16 Obsessive compulsive disorder (OCD) may be exacerbated or emerge for the first time during the perinatal period.17
In postpartum OCD, women may experience intrusive thoughts of accidental or purposeful harm to their baby. As opposed to women with PPP, mothers with OCD are not out of touch with reality and their thoughts are ego-dystonic.17 When these mothers have thoughts of their infants being harmed, they realize that these thoughts are not plans but fears and they try to avoid the thoughts.
Preventing PPP
Bipolar disorder is one of the most difficult disorders to treat during pregnancy because the serious risks of untreated illness must be balanced against the potential teratogenic risk of medications. Nevertheless, proactively managing bipolar disorder during pregnancy may reduce the risk of PPP.10
Closely monitor women with a history of bipolar disorder or PPP. During pregnancy, counsel them—and their partners—to:
- anticipate that depressive or psychotic symptoms could develop within days after delivery18
- seek treatment immediately if this occurs.
Postpartum medication. Whether or not a woman with bipolar disorder takes medication during pregnancy, consider treatment with mood stabilizers or atypical antipsychotics in the postpartum to prevent PPP (Table 3). Evidence is limited, but a search of PubMed found 1 study in which prophylactic lithium was given late in the third trimester or immediately after delivery to 21 women with a history of bipolar disorder or PPP. Only 2 patients had a psychotic recurrence while on prophylactic lithium; 1 unexplained stillbirth occurred.19
A retrospective study examined the course of women with bipolar disorder, some of whom were given prophylactic mood stabilizers immediately in the postpartum. One of 14 who received antimanic agents relapsed within the first 3 months postpartum, compared with 8 of 13 who were not so treated.18
Compared with antiepileptics, less information is available about the use of atypical antipsychotics in pregnancy and lactation. Antipsychotics’ potential advantage in women at risk for PPP is that these agents may help prevent or treat both manic and psychotic symptoms.
In a small, naturalistic, prospective study, 11 women at risk for PPP received olanzapine alone or with an antidepressant or mood stabilizer for at least 4 weeks after delivery. Two (18%) experienced a postpartum mood episode, compared with 8 (57%) of 14 other at-risk women who received antidepressants, mood stabilizers, or no medication.20
When you discuss breast-feeding, consider possible risks to the neonate as well as potential sleep interruption for the mother. If a mother has a supportive partner, the partner might be put in charge of night-time feedings in a routine combining breast-feeding and bottle-feeding. In some cases you may need to recommend cessation of lactation.21
Table 3
Treating postpartum psychosis: Consider 3 components
| Component | Recommendations |
|---|---|
| Hospitalization vs home care | Hospitalize in most cases because of emergent severe symptoms and fluctuating course; base decision on risk evaluation/safety issues for patient and infant After discharge, visiting nurses are useful to help monitor the mother’s condition at home |
| Psychoeducation | Educate patient, family, and social support network; address risks to mother and infant and risks in future pregnancies |
| Medication | When prescribing mood stabilizers and/or antipsychotics, consider:
|
Managing PPP
Early symptoms. Because of its severity and rapid evolution, PPP often presents as a psychiatric emergency. Monitor atrisk patients’ sleep patterns and mood for early signs of psychosis.22 Watch especially for hypomanic symptoms such as elevated or mixed mood and decreased judgment, which are common early in PPP.13
A mother with few signs of abnormal mood, good social support, and close follow-up may potentially be safely managed as an outpatient. Initial evaluation and management of PPP usually requires hospitalization, however, because of the risks of suicide, infanticide, and child maltreatment.23
Hospitalization. Mother-infant bonding is important, but safety is paramount if a mother is psychotic—especially if she is experiencing psychotic thoughts about her infant. If possible, the infant should remain with family members during the mother’s hospitalization. Supervised mother-infant visits are often arranged, as appropriate.
Mood-stabilizing medications, including antipsychotics, are mainstays of treatment.24 In some cases, conventional antipsychotics such as haloperidol may be useful because of a lower risk of weight gain or of sedation that could impair a mother’s ability to respond to her infant. Electroconvulsive therapy often yields rapid symptomatic improvement for mothers with postpartum mood or psychotic symptoms.25
Discharge planning. Assuming that the mother adheres to prescribed treatment, discharge may occur within 1 week. Plan discharge arrangements carefully (Table 4).28 A team approach can be very useful within the outpatient clinic. In the model of the Perinatal Psychiatry Clinic of Connections in suburban Cleveland, OH, the mother’s treatment team includes perinatal psychiatrists, nurses, counsellors, case managers (who do home visits), and peer counselors.
Outpatient civil commitment, in which patients are mandated to accept treatment, is an option in some jurisdictions and could help ensure that patients receive treatment consistently.
Table 4
Discharge planning for safety of mother and infant
| Notify child protective services (CPS) depending on the risk to the child. Case-by-case review is needed to assess whether the infant should be removed. CPS may put in place a plan for safety, short of removal. The plan may require that the woman continue psychiatric care |
| Meet with the patient and family to discuss her diagnosis, the risks, the importance of continued medication adherence, and the need for family or social supports to assist with child care |
| Consider engaging visiting nurses or doulas to provide help and support at home |
| Schedule frequent outpatient appointments for the mother after discharge |
| Consider family therapy after the mother has improved because of her risk for affective episodes outside the postpartum28 |
Related resources
- Altshuler L, Richards M, Yonkers K. Treating bipolar disorder during pregnancy. Current Psychiatry. 2003;2(7):14-26. www.CurrentPsychiatry.com.
- Gentile S. Infant safety with antipsychotic therapy in breastfeeding: a systematic review. J Clin Psychiatry. 2008;69(4):666-673.
- Miller LJ. Postpartum mood disorders. Washington, DC: American Psychiatric Publishing, Inc; 1999.
- Stowe ZN. The use of mood stabilizers during breastfeeding. J Clin Psychiatry. 2007;68(suppl 9):22-28.
- Toxicology Data Network (Toxnet). Literature on reproductive risks associated with psychotropics. National Library of Medicine. http://toxnet.nlm.nih.gov.
- Haloperidol • Haldol
- Lithium • various
- Olanzapine • Zyprexa
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
Dr. Resnick, a forensic psychiatrist and coauthor of this article, testified for the defense in both trials of Andrea Yates.
In June 2001, Andrea Yates drowned her 5 children ages 6 months to 7 years in the bathtub of their home. She had delusions that her house was bugged and television cameras were monitoring her mothering skills. She came to believe that “the one and only Satan” was within her, and that her children would burn in hell if she did not save their souls while they were still innocent.
Her conviction of capital murder in her first trial was overturned on appeal. She was found not guilty by reason of insanity at her retrial in 2006 and committed to a Texas state mental hospital.1
Postpartum psychosis (PPP) presents dramatically days to weeks after delivery, with wide-ranging symptoms that can include dysphoric mania and delirium. Because untreated PPP has an estimated 4% risk of infanticide (murder of the infant in the first year of life),2 and a 5% risk of suicide,3 psychiatric hospitalization usually is required to protect the mother and her baby.
The diagnosis may be missed, however, because postpartum psychotic symptoms wax and wane and suspiciousness or poor insight cause some women—such as Andrea Yates—to hide their delusional thinking from their families. This article discusses the risk factors, prevention, and treatment of PPP, including a review of:
- infanticide and suicide risks in the postpartum period
- increased susceptibility to PPP in women with bipolar disorder and other psychiatric disorders
- hospitalization for support and safety of the mother and her infant.
Risks of infanticide and suicide
A number of motives exist for infanticide (Table 1).4 Psychiatric literature shows that mothers who kill their children often have experienced psychosis, suicidality, depression, and considerable life stress.5 Common factors include alcohol use, limited social support, and a personal history of abuse. Studies on infanticide found a significant increase in common psychiatric disorders and financial stress among the mothers. Neonaticide (murder of the infant in the first day of life) generally is not related to PPP because PPP usually does not begin until after the day of delivery.6
Among women who develop psychiatric illness, homicidal ideation is more frequent in those with a perinatal onset of psychopathology.7 Infanticidal ideas and behavior are associated with psychotic ideas about the infant.8 Suicide is the cause of up to 20% of postpartum deaths.9
Table 1
Motives for infanticide: Mental illness or something else?
| Motives | Examples |
|---|---|
| Likely related to postpartum psychosis or depression | |
| Altruistic | A depressed or psychotic mother may believe she is sending her baby to heaven to prevent suffering on earth |
| A suicidal mother may kill her infant along with herself rather than leave the child alone | |
| Acutely psychotic | A mother kills her baby for no comprehensible reason, such as in response to command hallucinations or the confusion of delirium |
| Rarely related to postpartum psychosis | |
| Fatal maltreatment | ‘Battered child’ syndrome is the most common cause of infanticide; death often occurs after chronic abuse or neglect |
| A minority of perpetrators are psychotic; a mother out of touch with reality may have difficulty providing for her infant’s needs | |
| Not likely related to postpartum psychosis | |
| Unwanted child | Parent does not want child because of inconvenience or out-of-wedlock birth |
| Spouse revenge | Murder of a child to cause emotional suffering for the other parent is the least frequent motive for infanticide |
| Source: Reference 4 | |
The bipolar connection
Many factors can elevate the risk of PPP, including sleep deprivation in susceptible women, the hormonal shifts after birth, and psychiatric comorbidity (Table 2). Nearly three-fourths (>72%) of mothers with PPP have bipolar disorder or schizoaffective disorder, whereas 12% have schizophrenia.10 Some authors consider PPP to be bipolar disorder until proven otherwise. Mothers with a history of bipolar disorder or PPP have a 100-fold increase in rates of psychiatric hospitalization in the postpartum period.11
Presentation. PPP is relatively rare, occurring at a rate of 1 to 3 cases per 1,000 births. Symptoms often have an abrupt onset, within days to weeks of delivery.10 In at least one-half of cases, symptoms begin by the third postpartum day,13 when many mothers have been discharged home and may be solely responsible for their infants.
Symptoms include confusion, bizarre behaviors, hallucinations (including rarer types such as tactile and olfactory), mood lability (ranging from euphoria to depression), decreased need for sleep or insomnia, restlessness, agitation, disorganized thinking, and bizarre delusions of relatively rapid onset.13 One mother might believe God wants her baby to be sacrificed as the second coming of the Messiah, a second may believe she has special powers, and a third that her baby is defective.
Table 2
Postpartum psychosis: Risk factors supported by evidence
| Sleep deprivation in susceptible women |
| Hormonal shifts after birth (primarily the rapid drop in estrogen) |
| Psychosocial stressors such as marital problems, older age, single motherhood, lower socioeconomic status |
| Bipolar disorder or schizoaffective disorder |
| Past history of postpartum psychosis |
| Family history of postpartum psychosis |
| Previous psychiatric hospitalization, especially during the prenatal period for a bipolar or psychotic condition |
| Menstruation or cessation of lactation |
Obstetric factors that can cause a small increase in relative risk:
|
| Source: For bibliographic citations |
Differential diagnosis
When evaluating a postpartum woman with psychotic symptoms, stay in contact with her obstetrician and the child’s pediatrician. Rule out delirium and organic causes of the mother’s symptoms (Box).11
Postpartum depression (PPD) occurs in approximately 10% to 15% of new mothers.14 Depressive symptoms occur within weeks to months after delivery and often coexist with anxious symptoms. Some women with severe depression may present with psychotic symptoms. A mother may experience insomnia, sometimes not being able to sleep when the baby is sleeping. She may lack interest in caring for her baby and experience difficulty bonding.
At times it can be difficult to distinguish PPD from PPP. When evaluating a mother who is referred for “postpartum depression,” consider PPP in the differential diagnosis. A woman with PPD or PPP may report depressed mood, but in PPP this symptom usually is related to rapid mood changes. Other clinical features that point toward PPP are abnormal hallucinations (such as olfactory or tactile), hypomanic or mixed mood symptoms, and confusion.
When evaluating a postpartum woman with psychotic symptoms, stay in contact with her obstetrician and the child’s pediatrician. Rule out delirium and organic causes of the mother’s symptoms, giving special consideration to metabolic, neurologic, cardiovascular, infectious, and substance- or medication-induced origins. The extensive differential diagnosis includes:
- thyroiditis
- tumor
- CNS infection
- head injury
- embolism
- eclampsia
- substance withdrawal
- medication-induced (such as corticosteroids)
- electrolyte anomalies
- anoxia
- vitamin B12 deficiency.11
Psychosis vs OCD. Psychotic thinking and behaviors also must be differentiated from obsessive thoughts and compulsions.10,16 Obsessive compulsive disorder (OCD) may be exacerbated or emerge for the first time during the perinatal period.17
In postpartum OCD, women may experience intrusive thoughts of accidental or purposeful harm to their baby. As opposed to women with PPP, mothers with OCD are not out of touch with reality and their thoughts are ego-dystonic.17 When these mothers have thoughts of their infants being harmed, they realize that these thoughts are not plans but fears and they try to avoid the thoughts.
Preventing PPP
Bipolar disorder is one of the most difficult disorders to treat during pregnancy because the serious risks of untreated illness must be balanced against the potential teratogenic risk of medications. Nevertheless, proactively managing bipolar disorder during pregnancy may reduce the risk of PPP.10
Closely monitor women with a history of bipolar disorder or PPP. During pregnancy, counsel them—and their partners—to:
- anticipate that depressive or psychotic symptoms could develop within days after delivery18
- seek treatment immediately if this occurs.
Postpartum medication. Whether or not a woman with bipolar disorder takes medication during pregnancy, consider treatment with mood stabilizers or atypical antipsychotics in the postpartum to prevent PPP (Table 3). Evidence is limited, but a search of PubMed found 1 study in which prophylactic lithium was given late in the third trimester or immediately after delivery to 21 women with a history of bipolar disorder or PPP. Only 2 patients had a psychotic recurrence while on prophylactic lithium; 1 unexplained stillbirth occurred.19
A retrospective study examined the course of women with bipolar disorder, some of whom were given prophylactic mood stabilizers immediately in the postpartum. One of 14 who received antimanic agents relapsed within the first 3 months postpartum, compared with 8 of 13 who were not so treated.18
Compared with antiepileptics, less information is available about the use of atypical antipsychotics in pregnancy and lactation. Antipsychotics’ potential advantage in women at risk for PPP is that these agents may help prevent or treat both manic and psychotic symptoms.
In a small, naturalistic, prospective study, 11 women at risk for PPP received olanzapine alone or with an antidepressant or mood stabilizer for at least 4 weeks after delivery. Two (18%) experienced a postpartum mood episode, compared with 8 (57%) of 14 other at-risk women who received antidepressants, mood stabilizers, or no medication.20
When you discuss breast-feeding, consider possible risks to the neonate as well as potential sleep interruption for the mother. If a mother has a supportive partner, the partner might be put in charge of night-time feedings in a routine combining breast-feeding and bottle-feeding. In some cases you may need to recommend cessation of lactation.21
Table 3
Treating postpartum psychosis: Consider 3 components
| Component | Recommendations |
|---|---|
| Hospitalization vs home care | Hospitalize in most cases because of emergent severe symptoms and fluctuating course; base decision on risk evaluation/safety issues for patient and infant After discharge, visiting nurses are useful to help monitor the mother’s condition at home |
| Psychoeducation | Educate patient, family, and social support network; address risks to mother and infant and risks in future pregnancies |
| Medication | When prescribing mood stabilizers and/or antipsychotics, consider:
|
Managing PPP
Early symptoms. Because of its severity and rapid evolution, PPP often presents as a psychiatric emergency. Monitor atrisk patients’ sleep patterns and mood for early signs of psychosis.22 Watch especially for hypomanic symptoms such as elevated or mixed mood and decreased judgment, which are common early in PPP.13
A mother with few signs of abnormal mood, good social support, and close follow-up may potentially be safely managed as an outpatient. Initial evaluation and management of PPP usually requires hospitalization, however, because of the risks of suicide, infanticide, and child maltreatment.23
Hospitalization. Mother-infant bonding is important, but safety is paramount if a mother is psychotic—especially if she is experiencing psychotic thoughts about her infant. If possible, the infant should remain with family members during the mother’s hospitalization. Supervised mother-infant visits are often arranged, as appropriate.
Mood-stabilizing medications, including antipsychotics, are mainstays of treatment.24 In some cases, conventional antipsychotics such as haloperidol may be useful because of a lower risk of weight gain or of sedation that could impair a mother’s ability to respond to her infant. Electroconvulsive therapy often yields rapid symptomatic improvement for mothers with postpartum mood or psychotic symptoms.25
Discharge planning. Assuming that the mother adheres to prescribed treatment, discharge may occur within 1 week. Plan discharge arrangements carefully (Table 4).28 A team approach can be very useful within the outpatient clinic. In the model of the Perinatal Psychiatry Clinic of Connections in suburban Cleveland, OH, the mother’s treatment team includes perinatal psychiatrists, nurses, counsellors, case managers (who do home visits), and peer counselors.
Outpatient civil commitment, in which patients are mandated to accept treatment, is an option in some jurisdictions and could help ensure that patients receive treatment consistently.
Table 4
Discharge planning for safety of mother and infant
| Notify child protective services (CPS) depending on the risk to the child. Case-by-case review is needed to assess whether the infant should be removed. CPS may put in place a plan for safety, short of removal. The plan may require that the woman continue psychiatric care |
| Meet with the patient and family to discuss her diagnosis, the risks, the importance of continued medication adherence, and the need for family or social supports to assist with child care |
| Consider engaging visiting nurses or doulas to provide help and support at home |
| Schedule frequent outpatient appointments for the mother after discharge |
| Consider family therapy after the mother has improved because of her risk for affective episodes outside the postpartum28 |
Related resources
- Altshuler L, Richards M, Yonkers K. Treating bipolar disorder during pregnancy. Current Psychiatry. 2003;2(7):14-26. www.CurrentPsychiatry.com.
- Gentile S. Infant safety with antipsychotic therapy in breastfeeding: a systematic review. J Clin Psychiatry. 2008;69(4):666-673.
- Miller LJ. Postpartum mood disorders. Washington, DC: American Psychiatric Publishing, Inc; 1999.
- Stowe ZN. The use of mood stabilizers during breastfeeding. J Clin Psychiatry. 2007;68(suppl 9):22-28.
- Toxicology Data Network (Toxnet). Literature on reproductive risks associated with psychotropics. National Library of Medicine. http://toxnet.nlm.nih.gov.
- Haloperidol • Haldol
- Lithium • various
- Olanzapine • Zyprexa
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgement
Dr. Resnick, a forensic psychiatrist and coauthor of this article, testified for the defense in both trials of Andrea Yates.
1. Resnick PJ. The Andrea Yates case: insanity on trial. Cleveland State Law Review. 2007;55(2):147-156.
2. Altshuler LL, Hendrick V, Cohen LS. Course of mood and anxiety disorders during pregnancy and the postpartum period. J Clin Psychiatry. 1998;59(suppl. 2):29-33.
3. Knops GG. Postpartum mood disorders. Postgrad Med. 1993;93:103-116.
4. Resnick PJ. Child murder by parents: a psychiatric review of filicide. Am J Psychiatry. 1969;126:73-82.
5. Friedman SH, Horwitz SM, Resnick PJ. Child murder by mothers: a critical analysis of the current state of knowledge and a research agenda. Am J Psychiatry. 2005;162:1578-1587.
6. Friedman SH, Resnick PJ. Neonaticide: phenomenology and considerations for prevention. Int J Law Psychiatry. In press.
7. Wisner K, Peindl K, Hanusa BH. Symptomatology of affective and psychotic illnesses related to childbearing. J Affect Disord. 1994;30:77-87.
8. Chandra PS, Venkatasubramanian G, Thomas T. Infanticidal ideas and infanticidal behaviour in Indian women with severe postpartum psychiatric disorders. J Nerv Ment Dis. 2002;190(7):457-461.
9. Lindahl V, Pearson JL, Colpe L. Prevalence of suicidality during pregnancy and the postpartum. Arch Womens Ment Health. 2005;8(2):77-87.
10. Sit D, Rothschild AJ, Wisner KL. A review of postpartum psychosis. J Women’s Health. 2006;15(4):352-368.
11. Attia E, Downey J, Oberman M. Postpartum psychoses. In: Miller LJ, ed. Postpartum mood disorders. Washington, DC: American Psychiatric Publishing Inc.; 1999:99-117.
12. Diagnostic and Statistical Manual of Mental Disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
13. Heron J, McGuinness M, Blackmore ER, et al. Early postpartum symptoms in puerperal psychosis. BJOG. 2008;115(3):348-353.
14. Meltzer-Brody S, Payne J, Rubinow D. Postpartum depression: what to tell patients who breast-feed. Current Psychiatry. 2008;7(5):87-95.
15. Jennings KD, Ross S, Popper S, et al. Thoughts of harming infants in depressed and nondepressed mothers. J Affect Disord. 1999;54:21-28.
16. Wisner KL, Gracious BL, Piontek CM, et al. Postpartum disorders: phenomenology, treatment approaches, and relationship to infanticide. In: Spinelli MG, ed. Infanticide: psychosocial and legal perspectives on mothers who kill. Washington, DC: American Psychiatric Publishing, Inc.; 2003.
17. Fairbrother N, Abramowitz JS. New parenthood as a risk factors for the development of obsessional problems. Behav Res Ther. 2007;45(9):2155-2163.
18. Cohen LS, Sichel DA, Robertson LM, et al. Postpartum prophylaxis for women with bipolar disorder. Am J Psychiatry. 1995;152(11):1641-1645.
19. Stewart DE, Klompenhouwer JL, Kendell RE, et al. Prophylactic lithium in puerperal psychosis. Br J Psychiatry. 1991;158:393-397.
20. Sharma V, Smith A, Mazmanian D. Olanzapine in the prevention of postpartum psychosis and mood episodes in bipolar disorder. Bipolar Disord. 2006;8(4):400-404.
21. Pfuhlmann B, Stoeber G, Beckmann H. Postpartum psychoses: prognosis, risk factors, and treatment. Curr Psychiatry Rep. 2002;4(3):185-190.
22. Sharma V, Mazmanian D. Sleep loss and postpartum psychosis. Bipolar Disord. 2003;5(2):98-105.
23. Lindahl V, Pearson JL, Colpe L. Prevalence of suicidality during pregnancy and the postpartum. Arch Womens Ment Health. 2005;8(2):77-87.
24. Connell M. The postpartum psychosis defense and feminism: more or less justice for women? Case Western Reserve Law Review. 2002;53:143.-
25. Forray A, Ostroff RB. The use of electroconvulsive therapy in postpartum affective disorders. J ECT. 2007;23(3):188-193.
26. Engqvist I, Nilsson A, Nilsson K, et al. Strategies in caring for women with postpartum psychosis—an interview study with psychiatric nurses. J Clin Nurs. 2007;16(7):1333-1342.
27. Robertson E, Lyons A. Living with puerperal psychosis: a qualitative analysis. Psychol Psychother. 2003;76(4):411-431.
28. Robertson E, Jones I, Haque S, et al. Risk of puerperal and non-puerperal recurrence of illness following bipolar affective puerperal (post-partum) psychosis. Br J Psychiatry. 2005;186:258-259.
1. Resnick PJ. The Andrea Yates case: insanity on trial. Cleveland State Law Review. 2007;55(2):147-156.
2. Altshuler LL, Hendrick V, Cohen LS. Course of mood and anxiety disorders during pregnancy and the postpartum period. J Clin Psychiatry. 1998;59(suppl. 2):29-33.
3. Knops GG. Postpartum mood disorders. Postgrad Med. 1993;93:103-116.
4. Resnick PJ. Child murder by parents: a psychiatric review of filicide. Am J Psychiatry. 1969;126:73-82.
5. Friedman SH, Horwitz SM, Resnick PJ. Child murder by mothers: a critical analysis of the current state of knowledge and a research agenda. Am J Psychiatry. 2005;162:1578-1587.
6. Friedman SH, Resnick PJ. Neonaticide: phenomenology and considerations for prevention. Int J Law Psychiatry. In press.
7. Wisner K, Peindl K, Hanusa BH. Symptomatology of affective and psychotic illnesses related to childbearing. J Affect Disord. 1994;30:77-87.
8. Chandra PS, Venkatasubramanian G, Thomas T. Infanticidal ideas and infanticidal behaviour in Indian women with severe postpartum psychiatric disorders. J Nerv Ment Dis. 2002;190(7):457-461.
9. Lindahl V, Pearson JL, Colpe L. Prevalence of suicidality during pregnancy and the postpartum. Arch Womens Ment Health. 2005;8(2):77-87.
10. Sit D, Rothschild AJ, Wisner KL. A review of postpartum psychosis. J Women’s Health. 2006;15(4):352-368.
11. Attia E, Downey J, Oberman M. Postpartum psychoses. In: Miller LJ, ed. Postpartum mood disorders. Washington, DC: American Psychiatric Publishing Inc.; 1999:99-117.
12. Diagnostic and Statistical Manual of Mental Disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
13. Heron J, McGuinness M, Blackmore ER, et al. Early postpartum symptoms in puerperal psychosis. BJOG. 2008;115(3):348-353.
14. Meltzer-Brody S, Payne J, Rubinow D. Postpartum depression: what to tell patients who breast-feed. Current Psychiatry. 2008;7(5):87-95.
15. Jennings KD, Ross S, Popper S, et al. Thoughts of harming infants in depressed and nondepressed mothers. J Affect Disord. 1999;54:21-28.
16. Wisner KL, Gracious BL, Piontek CM, et al. Postpartum disorders: phenomenology, treatment approaches, and relationship to infanticide. In: Spinelli MG, ed. Infanticide: psychosocial and legal perspectives on mothers who kill. Washington, DC: American Psychiatric Publishing, Inc.; 2003.
17. Fairbrother N, Abramowitz JS. New parenthood as a risk factors for the development of obsessional problems. Behav Res Ther. 2007;45(9):2155-2163.
18. Cohen LS, Sichel DA, Robertson LM, et al. Postpartum prophylaxis for women with bipolar disorder. Am J Psychiatry. 1995;152(11):1641-1645.
19. Stewart DE, Klompenhouwer JL, Kendell RE, et al. Prophylactic lithium in puerperal psychosis. Br J Psychiatry. 1991;158:393-397.
20. Sharma V, Smith A, Mazmanian D. Olanzapine in the prevention of postpartum psychosis and mood episodes in bipolar disorder. Bipolar Disord. 2006;8(4):400-404.
21. Pfuhlmann B, Stoeber G, Beckmann H. Postpartum psychoses: prognosis, risk factors, and treatment. Curr Psychiatry Rep. 2002;4(3):185-190.
22. Sharma V, Mazmanian D. Sleep loss and postpartum psychosis. Bipolar Disord. 2003;5(2):98-105.
23. Lindahl V, Pearson JL, Colpe L. Prevalence of suicidality during pregnancy and the postpartum. Arch Womens Ment Health. 2005;8(2):77-87.
24. Connell M. The postpartum psychosis defense and feminism: more or less justice for women? Case Western Reserve Law Review. 2002;53:143.-
25. Forray A, Ostroff RB. The use of electroconvulsive therapy in postpartum affective disorders. J ECT. 2007;23(3):188-193.
26. Engqvist I, Nilsson A, Nilsson K, et al. Strategies in caring for women with postpartum psychosis—an interview study with psychiatric nurses. J Clin Nurs. 2007;16(7):1333-1342.
27. Robertson E, Lyons A. Living with puerperal psychosis: a qualitative analysis. Psychol Psychother. 2003;76(4):411-431.
28. Robertson E, Jones I, Haque S, et al. Risk of puerperal and non-puerperal recurrence of illness following bipolar affective puerperal (post-partum) psychosis. Br J Psychiatry. 2005;186:258-259.
Controversies in bipolar disorder: Trust evidence or experience?
Today’s buzzword in health care is evidence-based medicine. Most clinicians would agree that evidence from clinical research should guide decisions about treating bipolar disorder. In theory, randomized controlled trials should tell us how to manage bipolar patients and achieve therapeutic success. page 40.)
We rarely have encountered a patient with postpartum depression or psychosis who does not have a history of (often undiagnosed and untreated) recurrent mood episodes. For most of these patients, a mood stabilizer may be a better choice than an antidepressant.
The role of thyroid hormones
Adding a thyroid hormone—usually liothyronine—to an antidepressant has been demonstrated to accelerate, page 47.)
Atypical depression and the bipolar spectrum
Depressive episodes are considered either “typical” (a category that includes melancholic depression—in DSM-IV-TR, major depression with melancholic features) or “atypical” (in DSM-IV-TR, major depression with atypical features). Atypical features were originally associated with response to monoamine oxidase inhibitor antidepressants, whereas non atypical depression was thought more likely to respond to tricyclic antidepressants.34 The depression of bipolar disorder is usually atypical ( Box 4 ), especially in patients with softer variants of the illness.35
We believe that depressed patients with atypical symptoms aggregate into groups according to the presence, severity, and character of interdepressive manic or hypomanic episodes. Some patients experience recurrent depressive episodes with intervening euthymia (recurrent major depression), whereas others experience depressive episodes punctuated by brief subthreshold hypomanic episodes. Patients in these groups occasionally tolerate or even benefit from cautiously managed antidepressant monotherapy. Patients with atypical depressive episodes alternating with frank hypomanic, manic, mixed, or manic-psychotic episodes usually require a mood stabilizer and may benefit from cotreatment with an atypical antipsychotic.
Akiskol and Benazzi35 suggest that atypical depression may be a subtype of the bipolar spectrum. Our experience suggests that the bipolar spectrum is a continuum of degrees of risk for mood instability in persons with recurrent atypical depression.
DSM-IV-TR defines atypical depression as depression characterized by mood reactivity and at least 2 of these 4 features:
- hypersomnia
- increased appetite or weight gain
- leaden paralysis
- sensitivity to interpersonal rejection.
The term ‘hypersomnia’ is misleading. Many of these patients do not sleep excessively because work or school attendance prevents oversleeping. Instead, they experience an increased sleep requirement manifested by difficulty getting up in the morning and increased daytime sleepiness.
Increased appetite and weight gain (hyperphagia) often are present, but almost as often our patients report no change in appetite or weight or even anorexia and weight loss.
We rarely see a condition one would term ‘leaden paralysis.’ We also find that ‘sensitivity to interpersonal rejection’ is too narrow a construct. Our patients with atypical depression experience increased sensitivity to every stressor in their lives—work, school, family, and social stressors—not just interpersonal rejection.
Related resources
- Lieber AL. Bipolar spectrum disorder: an overview of the soft bipolar spectrum. www.psycom.net/depression.central.lieber.html.
- Phelps J. Why am I still depressed? Recognizing and managing the ups and downs of bipolar II and soft bipolar disorder. www.psycheducation.org.
- Maier T. Evidence-based psychiatry: understanding the limitations of a method. J Eval Clin Pract. 2006;12(3):325.
Drug brand names
- Liothyronine • Cytomel
- Sertraline • Zoloft
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Goldberg JF. What constitutes evidence-based pharmacotherapy for bipolar disorder? Part 1: First-line treatments. J Clin Psychiatry. 2007;68:1982-1983.
2. Goldberg JF. What constitutes evidence-based pharmacotherapy for bipolar disorder? Part 2: Complex presentations and clinical context. J Clin Psychiatry. 2008;69:495-496.
3. Levine R, Fink M. Why evidence-based medicine cannot be applied to psychiatry. Psychiatric Times. 2008;25(4):10.
4. Akiskol HS, Benazzi F. The DSM-IV and ICD-10 categories of recurrent [major] depressive and bipolar II disorders: evidence that they lie on a dimensional spectrum. J Affect Disord. 2006;92:45-54.
5. Goodwin FK, Jamison KR. Manic-depressive illness: bipolar disorders and recurrent depression. 2nd ed. New York, NY: Oxford University Press; 2007:3–27.
6. Hirschfeld RMA, Lewis L, Vornik L. Perceptions and impact of bipolar disorder: how far have we really come? Results of the National Depressive and Manic-Depressive Association 2000 survey of individuals with bipolar disorder. J Clin Psychiatry. 2003;64(2):161-167.
7. Blanco C, Laje G, Olfson M, et al. Trends in the treatment of bipolar disorder by outpatient psychiatrists. Am J Psychiatry. 2002;159:1005-1010.
8. Ghaemi SN, Lenox MS, Baldessarini RJ. Effectiveness and safety of long-term antidepressant treatment in bipolar disorder. J Clin Psychiatry. 2001;62:565-569.
9. Ghaemi SN, Boiman EE, Goodwin FK. Diagnosing bipolar disorder and the effect of antidepressants: a naturalistic study. J Clin Psychiatry. 2000;61:804-808.
10. Gijsman HF, Geddes JR, Rendell JM, et al. Antidepressants for bipolar depression: a systematic review of randomized, controlled trials. Am J Psychiatry. 2005;161:1537-1547.
11. Ghaemi SN, Sachs GS, Chiou AM, et al. Is bipolar disorder still underdiagnosed? Are antidepressants overutilized? J Affect Disord. 1999;52:134-144.
12. Sachs GS, Nierenberg AA, Calabrese JR, et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med. 2007;356:1711-1722.
13. Altshuler L, Suppes T, Black D, et al. Impact of antidepressant discontinuation after acute bipolar depression remission on rates of depressive relapse at 1-year follow-up. Am J Psychiatry. 2003;160:1252-1262.
14. Akiskol HS. Developmental pathways to bipolarity: are juvenile-onset depressions pre-bipolar? J Am Acad Child Adolesc Psychiatry. 1995;34(6):754-763.
15. Geller B, Zimmerman B, Williams M, et al. Bipolar disorder at prospective follow-up of adults who had prepubertal major depressive disorder. Am J Psychiatry. 2001;158:125-127.
16. Food and Drug Administration: Center for Drug Evaluation and Research. Revisions to product labeling. Available at: http://www.FDA.gov/cder/drug/antidepressants/default.htm. Accessed January 12, 2009.
17. McElroy S, Strakowski S, West S, et al. Phenomenology of adolescent and adult mania in hospitalized patients with bipolar disorder. Am J Psychiatry. 1997;154:44-49.
18. Olfson M, Marcus SC. A case-control study of antidepressants and attempted suicide during early phase treatment of major depressive episodes. J Clin Psychiatry. 2008;69:425-432.
19. Keck PE, Jr, McElroy SL, Havens JR, et al. Psychosis in bipolar disorder: phenomenology and impact on morbidity and course of illness. Compr Psychiatry. 2003;44:263-269.
20. Jones I, Craddock N. Familiarity of the puerperal trigger in bipolar disorder: results of a family study. Am J Psychiatry. 2001;158:913-917.
21. Chaudron LH, Pies RW. The relationship between postpartum psychosis and bipolar disorder: a review. J Clin Psychiatry. 2003;64:1284-1292.
22. Wisner KL, Peindl KS, Hanusa BH. Psychiatric episodes in women and young children. J Affect Disord. 1995;34:1-11.
23. Sharma V. A cautionary note on the use of antidepressants in postpartum depression. Bipolar Disord. 2006;8:411-414.
24. O’Malley S. “Are you there alone?” The unspeakable crime of Andrea Yates. New York, NY: Simon and Schuster; 2004.
25. Altshuler LL, Bauer M, Frye MA, et al. Does thyroid supplementation accelerate tricyclic antidepressant response? A review and meta-analysis of the literature. Am J Psychiatry. 2001;158:1617-1622.
26. Joffe RT. The use of thyroid supplements to augment antidepressant medication. J Clin Psychiatry. 2008;59:26-29.
27. Cooper-Kazaz R, Apter JT, Cohen R, et al. Combined treatment with sertraline and liothyronine in major depression: a randomized, double-blind, placebo-controlled trial. Arch Gen Psychiatry. 2007;64:679-688.
28. Gold MS, Pottash AL, Extein I. Hypothyroidism and depression: evidence from complete thyroid function evaluation. JAMA. 1981;245:28-31.
29. Kupka RW, Nolen WA, Post RM, et al. High rate of autoimmune thyroiditis in bipolar disorder: lack of association with lithium exposure. Biol Psychiatry. 2002;51:305-311.
30. Szuba MP, Amsterdam JD. Rapid antidepressant response after nocturnal TRH administration in patients with bipolar I and bipolar type II major depression. J Clin Psychopharmacol. 2005;25:325-330.
31. Extein I, Pottash AL, Gold MS. Does subclinical hypothyroidism predispose to tricyclic-induced rapid mood cycles? J Clin Psychiatry. 1982;43:32-33.
32. American Association of Clinical Endocrinologists. Medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. Endocr Prac. 2002;8:457-469.
33. El-Mallakh RS, Karippott A. Antidepressant-associated chronic irritable dysphoria (ACID) in bipolar disorder. J Affect Disord. 2005;84:267-272.
34. Henkl V, Mergl R, Antje-Kathrin A, et al. Treatment of depression with atypical features: a meta-analytic approach. Psychiatry Res. 2006;141(1):89-101.
35. Perugi G, Akiskal HS, Lattanzi D, et al. The high prevalence of “soft” bipolar (II) features in atypical depression. Compr Psychiatry. 1998;39(2):63-71.
Today’s buzzword in health care is evidence-based medicine. Most clinicians would agree that evidence from clinical research should guide decisions about treating bipolar disorder. In theory, randomized controlled trials should tell us how to manage bipolar patients and achieve therapeutic success. page 40.)
We rarely have encountered a patient with postpartum depression or psychosis who does not have a history of (often undiagnosed and untreated) recurrent mood episodes. For most of these patients, a mood stabilizer may be a better choice than an antidepressant.
The role of thyroid hormones
Adding a thyroid hormone—usually liothyronine—to an antidepressant has been demonstrated to accelerate, page 47.)
Atypical depression and the bipolar spectrum
Depressive episodes are considered either “typical” (a category that includes melancholic depression—in DSM-IV-TR, major depression with melancholic features) or “atypical” (in DSM-IV-TR, major depression with atypical features). Atypical features were originally associated with response to monoamine oxidase inhibitor antidepressants, whereas non atypical depression was thought more likely to respond to tricyclic antidepressants.34 The depression of bipolar disorder is usually atypical ( Box 4 ), especially in patients with softer variants of the illness.35
We believe that depressed patients with atypical symptoms aggregate into groups according to the presence, severity, and character of interdepressive manic or hypomanic episodes. Some patients experience recurrent depressive episodes with intervening euthymia (recurrent major depression), whereas others experience depressive episodes punctuated by brief subthreshold hypomanic episodes. Patients in these groups occasionally tolerate or even benefit from cautiously managed antidepressant monotherapy. Patients with atypical depressive episodes alternating with frank hypomanic, manic, mixed, or manic-psychotic episodes usually require a mood stabilizer and may benefit from cotreatment with an atypical antipsychotic.
Akiskol and Benazzi35 suggest that atypical depression may be a subtype of the bipolar spectrum. Our experience suggests that the bipolar spectrum is a continuum of degrees of risk for mood instability in persons with recurrent atypical depression.
DSM-IV-TR defines atypical depression as depression characterized by mood reactivity and at least 2 of these 4 features:
- hypersomnia
- increased appetite or weight gain
- leaden paralysis
- sensitivity to interpersonal rejection.
The term ‘hypersomnia’ is misleading. Many of these patients do not sleep excessively because work or school attendance prevents oversleeping. Instead, they experience an increased sleep requirement manifested by difficulty getting up in the morning and increased daytime sleepiness.
Increased appetite and weight gain (hyperphagia) often are present, but almost as often our patients report no change in appetite or weight or even anorexia and weight loss.
We rarely see a condition one would term ‘leaden paralysis.’ We also find that ‘sensitivity to interpersonal rejection’ is too narrow a construct. Our patients with atypical depression experience increased sensitivity to every stressor in their lives—work, school, family, and social stressors—not just interpersonal rejection.
Related resources
- Lieber AL. Bipolar spectrum disorder: an overview of the soft bipolar spectrum. www.psycom.net/depression.central.lieber.html.
- Phelps J. Why am I still depressed? Recognizing and managing the ups and downs of bipolar II and soft bipolar disorder. www.psycheducation.org.
- Maier T. Evidence-based psychiatry: understanding the limitations of a method. J Eval Clin Pract. 2006;12(3):325.
Drug brand names
- Liothyronine • Cytomel
- Sertraline • Zoloft
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Today’s buzzword in health care is evidence-based medicine. Most clinicians would agree that evidence from clinical research should guide decisions about treating bipolar disorder. In theory, randomized controlled trials should tell us how to manage bipolar patients and achieve therapeutic success. page 40.)
We rarely have encountered a patient with postpartum depression or psychosis who does not have a history of (often undiagnosed and untreated) recurrent mood episodes. For most of these patients, a mood stabilizer may be a better choice than an antidepressant.
The role of thyroid hormones
Adding a thyroid hormone—usually liothyronine—to an antidepressant has been demonstrated to accelerate, page 47.)
Atypical depression and the bipolar spectrum
Depressive episodes are considered either “typical” (a category that includes melancholic depression—in DSM-IV-TR, major depression with melancholic features) or “atypical” (in DSM-IV-TR, major depression with atypical features). Atypical features were originally associated with response to monoamine oxidase inhibitor antidepressants, whereas non atypical depression was thought more likely to respond to tricyclic antidepressants.34 The depression of bipolar disorder is usually atypical ( Box 4 ), especially in patients with softer variants of the illness.35
We believe that depressed patients with atypical symptoms aggregate into groups according to the presence, severity, and character of interdepressive manic or hypomanic episodes. Some patients experience recurrent depressive episodes with intervening euthymia (recurrent major depression), whereas others experience depressive episodes punctuated by brief subthreshold hypomanic episodes. Patients in these groups occasionally tolerate or even benefit from cautiously managed antidepressant monotherapy. Patients with atypical depressive episodes alternating with frank hypomanic, manic, mixed, or manic-psychotic episodes usually require a mood stabilizer and may benefit from cotreatment with an atypical antipsychotic.
Akiskol and Benazzi35 suggest that atypical depression may be a subtype of the bipolar spectrum. Our experience suggests that the bipolar spectrum is a continuum of degrees of risk for mood instability in persons with recurrent atypical depression.
DSM-IV-TR defines atypical depression as depression characterized by mood reactivity and at least 2 of these 4 features:
- hypersomnia
- increased appetite or weight gain
- leaden paralysis
- sensitivity to interpersonal rejection.
The term ‘hypersomnia’ is misleading. Many of these patients do not sleep excessively because work or school attendance prevents oversleeping. Instead, they experience an increased sleep requirement manifested by difficulty getting up in the morning and increased daytime sleepiness.
Increased appetite and weight gain (hyperphagia) often are present, but almost as often our patients report no change in appetite or weight or even anorexia and weight loss.
We rarely see a condition one would term ‘leaden paralysis.’ We also find that ‘sensitivity to interpersonal rejection’ is too narrow a construct. Our patients with atypical depression experience increased sensitivity to every stressor in their lives—work, school, family, and social stressors—not just interpersonal rejection.
Related resources
- Lieber AL. Bipolar spectrum disorder: an overview of the soft bipolar spectrum. www.psycom.net/depression.central.lieber.html.
- Phelps J. Why am I still depressed? Recognizing and managing the ups and downs of bipolar II and soft bipolar disorder. www.psycheducation.org.
- Maier T. Evidence-based psychiatry: understanding the limitations of a method. J Eval Clin Pract. 2006;12(3):325.
Drug brand names
- Liothyronine • Cytomel
- Sertraline • Zoloft
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Goldberg JF. What constitutes evidence-based pharmacotherapy for bipolar disorder? Part 1: First-line treatments. J Clin Psychiatry. 2007;68:1982-1983.
2. Goldberg JF. What constitutes evidence-based pharmacotherapy for bipolar disorder? Part 2: Complex presentations and clinical context. J Clin Psychiatry. 2008;69:495-496.
3. Levine R, Fink M. Why evidence-based medicine cannot be applied to psychiatry. Psychiatric Times. 2008;25(4):10.
4. Akiskol HS, Benazzi F. The DSM-IV and ICD-10 categories of recurrent [major] depressive and bipolar II disorders: evidence that they lie on a dimensional spectrum. J Affect Disord. 2006;92:45-54.
5. Goodwin FK, Jamison KR. Manic-depressive illness: bipolar disorders and recurrent depression. 2nd ed. New York, NY: Oxford University Press; 2007:3–27.
6. Hirschfeld RMA, Lewis L, Vornik L. Perceptions and impact of bipolar disorder: how far have we really come? Results of the National Depressive and Manic-Depressive Association 2000 survey of individuals with bipolar disorder. J Clin Psychiatry. 2003;64(2):161-167.
7. Blanco C, Laje G, Olfson M, et al. Trends in the treatment of bipolar disorder by outpatient psychiatrists. Am J Psychiatry. 2002;159:1005-1010.
8. Ghaemi SN, Lenox MS, Baldessarini RJ. Effectiveness and safety of long-term antidepressant treatment in bipolar disorder. J Clin Psychiatry. 2001;62:565-569.
9. Ghaemi SN, Boiman EE, Goodwin FK. Diagnosing bipolar disorder and the effect of antidepressants: a naturalistic study. J Clin Psychiatry. 2000;61:804-808.
10. Gijsman HF, Geddes JR, Rendell JM, et al. Antidepressants for bipolar depression: a systematic review of randomized, controlled trials. Am J Psychiatry. 2005;161:1537-1547.
11. Ghaemi SN, Sachs GS, Chiou AM, et al. Is bipolar disorder still underdiagnosed? Are antidepressants overutilized? J Affect Disord. 1999;52:134-144.
12. Sachs GS, Nierenberg AA, Calabrese JR, et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med. 2007;356:1711-1722.
13. Altshuler L, Suppes T, Black D, et al. Impact of antidepressant discontinuation after acute bipolar depression remission on rates of depressive relapse at 1-year follow-up. Am J Psychiatry. 2003;160:1252-1262.
14. Akiskol HS. Developmental pathways to bipolarity: are juvenile-onset depressions pre-bipolar? J Am Acad Child Adolesc Psychiatry. 1995;34(6):754-763.
15. Geller B, Zimmerman B, Williams M, et al. Bipolar disorder at prospective follow-up of adults who had prepubertal major depressive disorder. Am J Psychiatry. 2001;158:125-127.
16. Food and Drug Administration: Center for Drug Evaluation and Research. Revisions to product labeling. Available at: http://www.FDA.gov/cder/drug/antidepressants/default.htm. Accessed January 12, 2009.
17. McElroy S, Strakowski S, West S, et al. Phenomenology of adolescent and adult mania in hospitalized patients with bipolar disorder. Am J Psychiatry. 1997;154:44-49.
18. Olfson M, Marcus SC. A case-control study of antidepressants and attempted suicide during early phase treatment of major depressive episodes. J Clin Psychiatry. 2008;69:425-432.
19. Keck PE, Jr, McElroy SL, Havens JR, et al. Psychosis in bipolar disorder: phenomenology and impact on morbidity and course of illness. Compr Psychiatry. 2003;44:263-269.
20. Jones I, Craddock N. Familiarity of the puerperal trigger in bipolar disorder: results of a family study. Am J Psychiatry. 2001;158:913-917.
21. Chaudron LH, Pies RW. The relationship between postpartum psychosis and bipolar disorder: a review. J Clin Psychiatry. 2003;64:1284-1292.
22. Wisner KL, Peindl KS, Hanusa BH. Psychiatric episodes in women and young children. J Affect Disord. 1995;34:1-11.
23. Sharma V. A cautionary note on the use of antidepressants in postpartum depression. Bipolar Disord. 2006;8:411-414.
24. O’Malley S. “Are you there alone?” The unspeakable crime of Andrea Yates. New York, NY: Simon and Schuster; 2004.
25. Altshuler LL, Bauer M, Frye MA, et al. Does thyroid supplementation accelerate tricyclic antidepressant response? A review and meta-analysis of the literature. Am J Psychiatry. 2001;158:1617-1622.
26. Joffe RT. The use of thyroid supplements to augment antidepressant medication. J Clin Psychiatry. 2008;59:26-29.
27. Cooper-Kazaz R, Apter JT, Cohen R, et al. Combined treatment with sertraline and liothyronine in major depression: a randomized, double-blind, placebo-controlled trial. Arch Gen Psychiatry. 2007;64:679-688.
28. Gold MS, Pottash AL, Extein I. Hypothyroidism and depression: evidence from complete thyroid function evaluation. JAMA. 1981;245:28-31.
29. Kupka RW, Nolen WA, Post RM, et al. High rate of autoimmune thyroiditis in bipolar disorder: lack of association with lithium exposure. Biol Psychiatry. 2002;51:305-311.
30. Szuba MP, Amsterdam JD. Rapid antidepressant response after nocturnal TRH administration in patients with bipolar I and bipolar type II major depression. J Clin Psychopharmacol. 2005;25:325-330.
31. Extein I, Pottash AL, Gold MS. Does subclinical hypothyroidism predispose to tricyclic-induced rapid mood cycles? J Clin Psychiatry. 1982;43:32-33.
32. American Association of Clinical Endocrinologists. Medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. Endocr Prac. 2002;8:457-469.
33. El-Mallakh RS, Karippott A. Antidepressant-associated chronic irritable dysphoria (ACID) in bipolar disorder. J Affect Disord. 2005;84:267-272.
34. Henkl V, Mergl R, Antje-Kathrin A, et al. Treatment of depression with atypical features: a meta-analytic approach. Psychiatry Res. 2006;141(1):89-101.
35. Perugi G, Akiskal HS, Lattanzi D, et al. The high prevalence of “soft” bipolar (II) features in atypical depression. Compr Psychiatry. 1998;39(2):63-71.
1. Goldberg JF. What constitutes evidence-based pharmacotherapy for bipolar disorder? Part 1: First-line treatments. J Clin Psychiatry. 2007;68:1982-1983.
2. Goldberg JF. What constitutes evidence-based pharmacotherapy for bipolar disorder? Part 2: Complex presentations and clinical context. J Clin Psychiatry. 2008;69:495-496.
3. Levine R, Fink M. Why evidence-based medicine cannot be applied to psychiatry. Psychiatric Times. 2008;25(4):10.
4. Akiskol HS, Benazzi F. The DSM-IV and ICD-10 categories of recurrent [major] depressive and bipolar II disorders: evidence that they lie on a dimensional spectrum. J Affect Disord. 2006;92:45-54.
5. Goodwin FK, Jamison KR. Manic-depressive illness: bipolar disorders and recurrent depression. 2nd ed. New York, NY: Oxford University Press; 2007:3–27.
6. Hirschfeld RMA, Lewis L, Vornik L. Perceptions and impact of bipolar disorder: how far have we really come? Results of the National Depressive and Manic-Depressive Association 2000 survey of individuals with bipolar disorder. J Clin Psychiatry. 2003;64(2):161-167.
7. Blanco C, Laje G, Olfson M, et al. Trends in the treatment of bipolar disorder by outpatient psychiatrists. Am J Psychiatry. 2002;159:1005-1010.
8. Ghaemi SN, Lenox MS, Baldessarini RJ. Effectiveness and safety of long-term antidepressant treatment in bipolar disorder. J Clin Psychiatry. 2001;62:565-569.
9. Ghaemi SN, Boiman EE, Goodwin FK. Diagnosing bipolar disorder and the effect of antidepressants: a naturalistic study. J Clin Psychiatry. 2000;61:804-808.
10. Gijsman HF, Geddes JR, Rendell JM, et al. Antidepressants for bipolar depression: a systematic review of randomized, controlled trials. Am J Psychiatry. 2005;161:1537-1547.
11. Ghaemi SN, Sachs GS, Chiou AM, et al. Is bipolar disorder still underdiagnosed? Are antidepressants overutilized? J Affect Disord. 1999;52:134-144.
12. Sachs GS, Nierenberg AA, Calabrese JR, et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med. 2007;356:1711-1722.
13. Altshuler L, Suppes T, Black D, et al. Impact of antidepressant discontinuation after acute bipolar depression remission on rates of depressive relapse at 1-year follow-up. Am J Psychiatry. 2003;160:1252-1262.
14. Akiskol HS. Developmental pathways to bipolarity: are juvenile-onset depressions pre-bipolar? J Am Acad Child Adolesc Psychiatry. 1995;34(6):754-763.
15. Geller B, Zimmerman B, Williams M, et al. Bipolar disorder at prospective follow-up of adults who had prepubertal major depressive disorder. Am J Psychiatry. 2001;158:125-127.
16. Food and Drug Administration: Center for Drug Evaluation and Research. Revisions to product labeling. Available at: http://www.FDA.gov/cder/drug/antidepressants/default.htm. Accessed January 12, 2009.
17. McElroy S, Strakowski S, West S, et al. Phenomenology of adolescent and adult mania in hospitalized patients with bipolar disorder. Am J Psychiatry. 1997;154:44-49.
18. Olfson M, Marcus SC. A case-control study of antidepressants and attempted suicide during early phase treatment of major depressive episodes. J Clin Psychiatry. 2008;69:425-432.
19. Keck PE, Jr, McElroy SL, Havens JR, et al. Psychosis in bipolar disorder: phenomenology and impact on morbidity and course of illness. Compr Psychiatry. 2003;44:263-269.
20. Jones I, Craddock N. Familiarity of the puerperal trigger in bipolar disorder: results of a family study. Am J Psychiatry. 2001;158:913-917.
21. Chaudron LH, Pies RW. The relationship between postpartum psychosis and bipolar disorder: a review. J Clin Psychiatry. 2003;64:1284-1292.
22. Wisner KL, Peindl KS, Hanusa BH. Psychiatric episodes in women and young children. J Affect Disord. 1995;34:1-11.
23. Sharma V. A cautionary note on the use of antidepressants in postpartum depression. Bipolar Disord. 2006;8:411-414.
24. O’Malley S. “Are you there alone?” The unspeakable crime of Andrea Yates. New York, NY: Simon and Schuster; 2004.
25. Altshuler LL, Bauer M, Frye MA, et al. Does thyroid supplementation accelerate tricyclic antidepressant response? A review and meta-analysis of the literature. Am J Psychiatry. 2001;158:1617-1622.
26. Joffe RT. The use of thyroid supplements to augment antidepressant medication. J Clin Psychiatry. 2008;59:26-29.
27. Cooper-Kazaz R, Apter JT, Cohen R, et al. Combined treatment with sertraline and liothyronine in major depression: a randomized, double-blind, placebo-controlled trial. Arch Gen Psychiatry. 2007;64:679-688.
28. Gold MS, Pottash AL, Extein I. Hypothyroidism and depression: evidence from complete thyroid function evaluation. JAMA. 1981;245:28-31.
29. Kupka RW, Nolen WA, Post RM, et al. High rate of autoimmune thyroiditis in bipolar disorder: lack of association with lithium exposure. Biol Psychiatry. 2002;51:305-311.
30. Szuba MP, Amsterdam JD. Rapid antidepressant response after nocturnal TRH administration in patients with bipolar I and bipolar type II major depression. J Clin Psychopharmacol. 2005;25:325-330.
31. Extein I, Pottash AL, Gold MS. Does subclinical hypothyroidism predispose to tricyclic-induced rapid mood cycles? J Clin Psychiatry. 1982;43:32-33.
32. American Association of Clinical Endocrinologists. Medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. Endocr Prac. 2002;8:457-469.
33. El-Mallakh RS, Karippott A. Antidepressant-associated chronic irritable dysphoria (ACID) in bipolar disorder. J Affect Disord. 2005;84:267-272.
34. Henkl V, Mergl R, Antje-Kathrin A, et al. Treatment of depression with atypical features: a meta-analytic approach. Psychiatry Res. 2006;141(1):89-101.
35. Perugi G, Akiskal HS, Lattanzi D, et al. The high prevalence of “soft” bipolar (II) features in atypical depression. Compr Psychiatry. 1998;39(2):63-71.