User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Are your electronic patient records secure?
Is your office computer system-and the confidential patient records it contains-safe from hackers?
Maintaining office computer security isn’t just good practice-it’s the law. The Health Insurance Portability and Accountability Act of 1996 (HIPAA) requires physicians to ensure that patient records are kept confidential.click here.
Hardware tokens are pocket-size devices that, when connected to the computer, allow access by entering the proper password. Hardware tokens are suitable for managing off-site remote access and add another layer of security for local access.
Medical records programs. Most software programs allow administrators to restrict access to medical records by setting up levels of ability to access and modifying electronic records for different users. Each user should have a unique password for authentication. The software also should have an audit trail capability, so that each user’s activity with electronic patient records can be reviewed.
Security breaches
An Internet connection-however brief-can invite security breaches that allow hackers to access patient information, delete programs, steal passwords, disrupt other Internet-connected computers, and erase the hard drive. Avoiding Internet connections altogether would increase security, but this is not feasible.
- Viruses reproduce using the host computer, most commonly by infiltrating the e-mail program and making it hard to detect corrupted files.
- Worms are similar to viruses but are self-contained, whereas a virus must attach to another file.
- Trojan horse programs, usually disguised within seemingly legitimate Internet programs, are less common than viruses or worms. They do not replicate but are equally dangerous. You unknowingly start the Trojan horse after downloading what looks like a useful program. The Trojan horse then self-installs silently, giving the hacker who created it access to your computer.
- Port attacks are malicious attempts to connect and eventually take over another computer. A ‘port’ is a software ‘location’ where a program on another computer can connect to a host computer.
- NEVER open e-mails from unfamiliar sources. Viruses are commonly sent as attachments, which you should never open unless you know they are safe.
- Turn off your computer or disconnect from the Internet when not in use.
- Back up your data regularly. Put patient files in one folder or directory, then copy them to a backup medium such as CD-ROM, zip drive, or portable hard drive. Of course, keep the disks in a secure place.
Antivirus programs can check for the latest viruses and their variants and remove them. To do this, automatically update the program with new virus signature files- files created by antivirus program vendors to help the software identify viruses. Most antivirus programs will automatically check the vendor’s Web site for updated files if the computer is connected to the Internet.
Virus signature files should be updated daily to provide maximum protection. Most companies provide a 1-year subscription to the updates, which must be renewed upon expiration for new virus definition files.
Manual updating is acceptable but may be too time-consuming for a busy office.
Well-known antivirus programs include Wireless Internet 101,” Psyber Psychiatry, December 2003)
If your computer is connected directly to the DSL or cable modem or a telephone line, you probably need a firewall. The most recent Microsoft Windows XP and Mac OS X versions each include a software firewall, which should be activated upon installation.
Windows-compatible firewall programs include ZoneAlarm, Sygate Personal Firewall, Symantec Norton Personal Firewall, and Tiny Software Personal. Mac OS-compatible firewalls include Intego NetBarrier, Sustainable Softworks IPNetSentryX, and Norton Personal Firewall.
Once your firewall is installed, check it to verify that all ports are protected. Gibson Research Corp. has two excellent (and free) security checks: ShieldsUP! and LeakTest. Run these tests, then follow the listed suggestions to secure your computer.
Disclosure
Dr. Luo reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Luo in this column are his own and do not necessarily reflect those of CURRENT PSYCHIATRY.
(all accessed July 13, 2004)
1. U. S. Department of Health and Human Services, Centers for Medicare and Medicaid Services. HIPAA administrative simplification - security. http://www.cms.hhs.gov/hipaa/hipaa2/regulations/security/default.asp
2. Microsoft: Creating stronger passwords. http://www.microsoft.com/security/articles/password.asp
3. SecureMac.com. Open firmware password protection. http://www.securemac.com/openfirmwarepasswordprotection.php
Is your office computer system-and the confidential patient records it contains-safe from hackers?
Maintaining office computer security isn’t just good practice-it’s the law. The Health Insurance Portability and Accountability Act of 1996 (HIPAA) requires physicians to ensure that patient records are kept confidential.click here.
Hardware tokens are pocket-size devices that, when connected to the computer, allow access by entering the proper password. Hardware tokens are suitable for managing off-site remote access and add another layer of security for local access.
Medical records programs. Most software programs allow administrators to restrict access to medical records by setting up levels of ability to access and modifying electronic records for different users. Each user should have a unique password for authentication. The software also should have an audit trail capability, so that each user’s activity with electronic patient records can be reviewed.
Security breaches
An Internet connection-however brief-can invite security breaches that allow hackers to access patient information, delete programs, steal passwords, disrupt other Internet-connected computers, and erase the hard drive. Avoiding Internet connections altogether would increase security, but this is not feasible.
- Viruses reproduce using the host computer, most commonly by infiltrating the e-mail program and making it hard to detect corrupted files.
- Worms are similar to viruses but are self-contained, whereas a virus must attach to another file.
- Trojan horse programs, usually disguised within seemingly legitimate Internet programs, are less common than viruses or worms. They do not replicate but are equally dangerous. You unknowingly start the Trojan horse after downloading what looks like a useful program. The Trojan horse then self-installs silently, giving the hacker who created it access to your computer.
- Port attacks are malicious attempts to connect and eventually take over another computer. A ‘port’ is a software ‘location’ where a program on another computer can connect to a host computer.
- NEVER open e-mails from unfamiliar sources. Viruses are commonly sent as attachments, which you should never open unless you know they are safe.
- Turn off your computer or disconnect from the Internet when not in use.
- Back up your data regularly. Put patient files in one folder or directory, then copy them to a backup medium such as CD-ROM, zip drive, or portable hard drive. Of course, keep the disks in a secure place.
Antivirus programs can check for the latest viruses and their variants and remove them. To do this, automatically update the program with new virus signature files- files created by antivirus program vendors to help the software identify viruses. Most antivirus programs will automatically check the vendor’s Web site for updated files if the computer is connected to the Internet.
Virus signature files should be updated daily to provide maximum protection. Most companies provide a 1-year subscription to the updates, which must be renewed upon expiration for new virus definition files.
Manual updating is acceptable but may be too time-consuming for a busy office.
Well-known antivirus programs include Wireless Internet 101,” Psyber Psychiatry, December 2003)
If your computer is connected directly to the DSL or cable modem or a telephone line, you probably need a firewall. The most recent Microsoft Windows XP and Mac OS X versions each include a software firewall, which should be activated upon installation.
Windows-compatible firewall programs include ZoneAlarm, Sygate Personal Firewall, Symantec Norton Personal Firewall, and Tiny Software Personal. Mac OS-compatible firewalls include Intego NetBarrier, Sustainable Softworks IPNetSentryX, and Norton Personal Firewall.
Once your firewall is installed, check it to verify that all ports are protected. Gibson Research Corp. has two excellent (and free) security checks: ShieldsUP! and LeakTest. Run these tests, then follow the listed suggestions to secure your computer.
Disclosure
Dr. Luo reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Luo in this column are his own and do not necessarily reflect those of CURRENT PSYCHIATRY.
Is your office computer system-and the confidential patient records it contains-safe from hackers?
Maintaining office computer security isn’t just good practice-it’s the law. The Health Insurance Portability and Accountability Act of 1996 (HIPAA) requires physicians to ensure that patient records are kept confidential.click here.
Hardware tokens are pocket-size devices that, when connected to the computer, allow access by entering the proper password. Hardware tokens are suitable for managing off-site remote access and add another layer of security for local access.
Medical records programs. Most software programs allow administrators to restrict access to medical records by setting up levels of ability to access and modifying electronic records for different users. Each user should have a unique password for authentication. The software also should have an audit trail capability, so that each user’s activity with electronic patient records can be reviewed.
Security breaches
An Internet connection-however brief-can invite security breaches that allow hackers to access patient information, delete programs, steal passwords, disrupt other Internet-connected computers, and erase the hard drive. Avoiding Internet connections altogether would increase security, but this is not feasible.
- Viruses reproduce using the host computer, most commonly by infiltrating the e-mail program and making it hard to detect corrupted files.
- Worms are similar to viruses but are self-contained, whereas a virus must attach to another file.
- Trojan horse programs, usually disguised within seemingly legitimate Internet programs, are less common than viruses or worms. They do not replicate but are equally dangerous. You unknowingly start the Trojan horse after downloading what looks like a useful program. The Trojan horse then self-installs silently, giving the hacker who created it access to your computer.
- Port attacks are malicious attempts to connect and eventually take over another computer. A ‘port’ is a software ‘location’ where a program on another computer can connect to a host computer.
- NEVER open e-mails from unfamiliar sources. Viruses are commonly sent as attachments, which you should never open unless you know they are safe.
- Turn off your computer or disconnect from the Internet when not in use.
- Back up your data regularly. Put patient files in one folder or directory, then copy them to a backup medium such as CD-ROM, zip drive, or portable hard drive. Of course, keep the disks in a secure place.
Antivirus programs can check for the latest viruses and their variants and remove them. To do this, automatically update the program with new virus signature files- files created by antivirus program vendors to help the software identify viruses. Most antivirus programs will automatically check the vendor’s Web site for updated files if the computer is connected to the Internet.
Virus signature files should be updated daily to provide maximum protection. Most companies provide a 1-year subscription to the updates, which must be renewed upon expiration for new virus definition files.
Manual updating is acceptable but may be too time-consuming for a busy office.
Well-known antivirus programs include Wireless Internet 101,” Psyber Psychiatry, December 2003)
If your computer is connected directly to the DSL or cable modem or a telephone line, you probably need a firewall. The most recent Microsoft Windows XP and Mac OS X versions each include a software firewall, which should be activated upon installation.
Windows-compatible firewall programs include ZoneAlarm, Sygate Personal Firewall, Symantec Norton Personal Firewall, and Tiny Software Personal. Mac OS-compatible firewalls include Intego NetBarrier, Sustainable Softworks IPNetSentryX, and Norton Personal Firewall.
Once your firewall is installed, check it to verify that all ports are protected. Gibson Research Corp. has two excellent (and free) security checks: ShieldsUP! and LeakTest. Run these tests, then follow the listed suggestions to secure your computer.
Disclosure
Dr. Luo reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Luo in this column are his own and do not necessarily reflect those of CURRENT PSYCHIATRY.
(all accessed July 13, 2004)
1. U. S. Department of Health and Human Services, Centers for Medicare and Medicaid Services. HIPAA administrative simplification - security. http://www.cms.hhs.gov/hipaa/hipaa2/regulations/security/default.asp
2. Microsoft: Creating stronger passwords. http://www.microsoft.com/security/articles/password.asp
3. SecureMac.com. Open firmware password protection. http://www.securemac.com/openfirmwarepasswordprotection.php
(all accessed July 13, 2004)
1. U. S. Department of Health and Human Services, Centers for Medicare and Medicaid Services. HIPAA administrative simplification - security. http://www.cms.hhs.gov/hipaa/hipaa2/regulations/security/default.asp
2. Microsoft: Creating stronger passwords. http://www.microsoft.com/security/articles/password.asp
3. SecureMac.com. Open firmware password protection. http://www.securemac.com/openfirmwarepasswordprotection.php
High-dose antipsychotics: Desperation or data-driven?
When nothing else works, desperate clinicians are resorting to progressively more-tenuous and unpredictable treatments, trying to improve the lives of patients with refractory schizophrenia. High-dose antipsychotics is a common strategy.
Does boosting antipsychotic doses beyond the recommended range—but short of the neuroleptic threshold—enhance efficacy? This article attempts to answer that question by presenting the evidence on higher-than-recommended doses of atypical antipsychotics.
Lessons from neuroleptics
Up to 30% of patients with schizophrenia do not respond to antipsychotics and are considered “treatment refractory.”1 Even among those who do respond, improving symptoms by 20%—as research defines “treatment response”—does not necessarily yield clinical or functional improvement. Clozapine is the only atypical antipsychotic with well-established efficacy in these chronically ill patients,2 but its daunting side effects greatly curtail its use.
Before atypical antipsychotics, patients who did not respond to usual dosages of the typical neuroleptics were treated with higher dosages or switched to another drug class. Although many clinicians embraced high-dose neuroleptics, subsequent research discredited “rapid neuroleptization” in any clinical circumstance and showed that exceeding an antipsychotic’s neuroleptic threshold—the dose at which extrapyramidal side effects (EPS) occur—reduces its efficacy (Figure 1).3-5 In some instances, reducing neuroleptic dosages improves treatment-resistant patients’ symptoms and reduces druginduced side effects.6
Figure 1 Typical antipsychotics’ dose-response curve
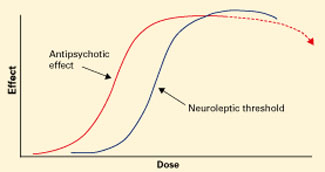
Narrow therapeutic window between antipsychotic effect and neuroleptic threshold. Dotted line indicates declining efficacy.
Figure 2 Atypical antipsychotics’ dose-response curve
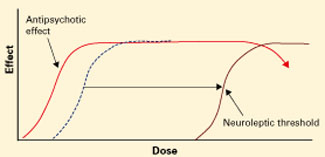
Wider therapeutic window with atypicals, compared with typical antipsychotics, as neuroleptic threshold (dotted line) moves right.Atypical antipsychotics are defined by their relative lack of EPS at recommended dosages (Figure 2). Because these agents can cause EPS if dosed too high, however, our historical habit of testing this dose limit risks losing “atypicality” and encountering other untoward events (Figure 3).
What is the safest, most effective dosage? Consider the evidence for each atypical antipsychotic.
Risperidone
Recommended dosage too high? When using atypicals at recommended doses, you are most likely to encounter the neuroleptic threshold with risperidone, with EPS risk increasing substantially at >6 mg/d.7 Post-approval studies set the most effective and safest dosage at approximately 4 mg/d, though this dosage was not studied in North American pre-approval trials. Dosages of 2 to 4 mg/d have been associated with more-favorable outcomes, suggesting that the initial recommendation to titrate to 6 mg/d within the first 3 days was ill-advised.8
In our study of patients with treatment-refractory schizophrenia,9 those treated with risperidone, 6 mg/d, improved significantly more after 4 weeks than did those receiving haloperidol, 15 mg/d, based on Brief Psychiatric Rating Scale (BPRS) scores. No additional benefit was seen after risperidone was increased to >6 mg/d at 8 weeks. Akathisia and tardive dyskinesia occurred significantly more often in the haloperidol group.
Conclusion. Some patients respond to higher-dose risperidone, but emerging EPS suggest the need to reduce the dosage rather than add an antiparkinsonian agent.
Figure 3 Unknown effects of high-dose atypical antipsychotic therapy
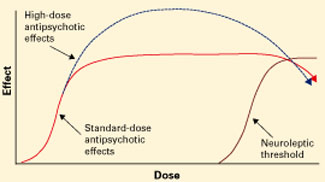
Dotted line indicates potential for greater antipsychotic effect with increasing dose.
Olanzapine
Mixed results. Case reports suggest that some patients who did not respond to previous antipsychotic trials or olanzapine, 20 mg/d, improved sig-nificantly—without substanial side effects—when olanzapine was increased up to 60 mg/d.10-14 Other case studies, however, report EPS, increased heart rate, increased transaminases, hyperprolactinemia, and prolonged QTc interval with high-dose olanzapine.14-16
In an open-label trial,17 43 patients with schizophrenia received olanzapine, up to 40 mg/d, after inadequate response to neuroleptics and risperidone or clozapine. Olanzapine was titrated to 20 mg/d by week 4 and increased 5 mg every 2 weeks if symptoms did not improve. After 14 weeks, improvement was modest and only 17% of patients met response criteria. However, >20 mg/d reduced symptoms more than did <20 mg/d, suggesting that high-dose olanzapine was more effective.
In a randomized trial,18 patients who did not respond to at least one atypical antipsychotic then received 8 weeks of fixed, standard-dose treatment with (mean dosages):
- haloperidol, 18.9 mg/d
- risperidone, 7.9 mg/d
- olanzapine, 19.6 mg/d
- clozapine, 401.6 mg/d.
Flexible dosing was then allowed for 6 weeks, and mean dosages were:
- haloperidol, 25.7 mg/d
- risperidone, 11.6 mg/d
- olanzapine, 30.4 mg/d
- clozapine, 526.6 mg/d.
Symptoms improved modestly at best for all medications, although patients taking olanzapine or clozapine improved significantly more than those treated with haloperidol as shown by mean changes in total Positive and Negative Syndrome Scale (PANSS) scores.
PANSS scores for olanzapine-treated patients showed additional improvement at week 14—when higher dosages were used—compared with week 8. This was not the case for the other medications, for which response plateaued. These findings suggest that high-dose risperidone and haloperidol are incrementally ineffective, but high-dose olanzapine could help some patients with refractory symptoms.
Results were different in a randomized, double-blind, 16-week, crossover study,19 when 13 patients with inadequate response to neuroleptics, risperidone, or conventional-dose olanzapine then received olanzapine, 50 mg/d, or clozapine, 450 mg/d. No olanzapine-treated patients and 20% of clozapine-treated patients met criteria for treatment response (20% improvement in BPRS score and final BPRS score <35 or 1-point improvement on Clinical Global Impressions-Severity of Illness scale).
Negative results don’t make headlines. Published clinical trials and case reports are subject to selective reporting of positive outcomes. Cases in which high-dose therapy proved ineffectivemay outnumber positive results but are less likely to be published.
Numbers don’t lie. Using high doses will almost always increase side effect risk and drug therapy costs, contributing to a poor risk-benefit ratio when efficacy remains unchanged. Resorting to an “if-it’s-not-working, double-it” strategy may seem reasonable, but two times zero is still zero.
Desperation warps perception. Clinicians tend to rely on observational experience. The desperation inherent in treating refractory patients, however, often creates a strong desire for improvement and therefore a potentially biased perception of outcome.
Likewise, patients may inaccurately portray themselves as improved to avoid disappointing their doctors. Controlled trials reduce these biases to better assess efficacy.
Antipsychotics work in 6 to 8 weeks. Improvements seen when pushing medications beyond recommended dosing may not be an effect of dose but of additional time on the medication. Antipsychotics usually take 6 to 8 weeks to produce maximal response, so high-dose therapy should not be started during this initial phase. This pace may not satisfy pressures for expedient stabilization and hospital discharge, but it is unrealistic to expect antipsychotics to work more quickly than they do.
Oversedation does not equal improvement. Patients who become excessively sedated from high-dose therapy or adjunctive medications may appear less psychotic but may not be so. The family or hospital staff may desire such sedation, but it can adversely affect the patient’s quality of life or medication adherence.
Polypharmacy clouds the issue. Many patients treated with high-dose antipsychotics are taking multiple agents, making it difficult to attribute improvement (or side effects) to any single one. A well-designed study of high-dose therapy would therefore:
- control for time
- examine concomitant medications’ effects
- determine whether “improvements” are related to sedation or reduced psychosis.
Medication may not need to change. When a patient decompensates, many forces pressure clinicians to change or add medications or increase dosages. Change may not be necessary, however, as nonadherence or substance abuse often trigger psychotic exacerbations. For example, Steingard et al27 added fluphenazine or placebo to antipsychotic regimens of newly hospitalized patients and found that increasing antipsychotic dosage did not improve outcome.
Subjects switching from clozapine to olanzapine tended to worsen, whereas those switching from olanzapine to clozapine tended to improve. Olanzapine-treated patients experienced more anticholinergic side effects and more weight gain than did clozapine-treated subjects.20
Conclusion. These mixed findings on high-dose olanzapine suggest questionable efficacy in patients with treatment-resistant schizophrenia and an uncertain risk of increased toxicity.
Quetiapine
Early placebo-controlled studies of quetiapine in schizophrenia concluded that statistically significant improvement begins at 150 mg/d and falls off after 600 mg/d.21 Although few high-dose quetiapine cases have been presented, clinical opinion holds that:
- most patients with chronic schizophrenia require 400 to 800 mg/d
- some treatment-refractory patients might benefit from >800 mg/d.
One patient responded to quetiapine, 1,600 mg/d, after not responding to olanzapine, 40 mg/d, and quetiapine, 800 mg/d. Constipation was the only reported side effect.22
Our group23 reported a series of 7 patients who responded (by clinician report) to quetiapine, 1,200 to 2,400 mg/d, after not responding to quetiapine, 800 mg/d, or to neuroleptics, risperidone, or olanzapine. Six responded to high-dose quetiapine and 1 to high-dose quetiapine plus risperidone, 2 mg/d; 4 received adjunctive dival-proex sodium, 1,500 to 3,000 mg/d. Psychopathology, violence, and behavioral disturbances were reduced throughout 5 to 14 months of monitoring. Side effects included sedation, orthostasis, and dysphagia.
When Nelson et al24 treated 13 subjects for 14 weeks with quetiapine, 1,000 to 1,400 mg/d, mean weight, glucose, total cholesterol, prolactin, and QTc interval duration did not change significantly. Heart rate increased significantly (though not to tachycardia), and headache, constipation, and lethargy were the most frequent side effects.
Summary. Although encouraging, these reports are preliminary, unpublished, and lack peer review. Controlled trials of high-dose quetiapine’s efficacy and safety are needed.
Ziprasidone and aripiprazole
No studies of high-dose ziprasidone or aripiprazole have been published. In premarketing trials:
- ziprasidone was studied at 200 mg/d and released with a maximum recommended dosage of 160 mg/d
- aripiprazole, 30 mg/d, was not more effective than 15 mg/d.25
Deutschman et al26 reviewed the charts of 31 patients who received ziprasidone, 240 to 320 mg/d, after an “incomplete” response to 160 mg/d. At the higher dosing:
- psychosis, affective symptoms, or anxiety improved in nearly one-half of patients
- 15% reported sedation, but most reported no side effects
- none developed QTc intervals >500 msec.
Caveats and precautions
These uncontrolled case reports and open-label studies do not “prove” efficacy or safety but reflect clinical practice. More than anything, they show that we need controlled trials to gauge high-dose antipsychotic therapy’s efficacy and safety and to curb our collective habit of relying on anecdotal experience and idiosyncratic beliefs.
Despite its side-effect profile, clozapine remains the treatment of choice for refractory schizophrenia. Given high-dose antipsychotic therapy’s uncertain efficacy and unknown risks, the evidence supports a clozapine trial before higher-than-recommended dosing is attempted.
Because educated guesswork plays a role in premarketing dosing studies, a medication’s optimal dose may be:
- overestimated (as with risperidone)
- underestimated (as perhaps with olanzapine and quetiapine).
Keep in mind some important caveats when you consider giving a patient high-dose antipsychotic therapy (Box).27 Of course, nonadherence is often the cause of apparent medication nonresponse. Increasing the dosage of a medication a patient is not taking rarely improves adherence. Interventions to enhance adherence—careful assessment, psychoeducation, and using longacting intramuscular medication—may be useful.
Related resources
- Marder SR, Essock SM, Miller AL, et al. The Mount Sinai Conference on the pharmacotherapy of schizophrenia. Schizophrenia Bull 2002;28:5-16.
- Practice guideline for the treatment of patients with schizophrenia (2nd ed). Am J Psychiatry 2004;161(suppl):1-56.
- Texas Medication Algorithm Project antipsychotic algorithm. http://www.mhmr.state.tx.us/centraloffice/medicaldirector/timascz1algo.pdf
Drug brand names
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Divalproex • Depakote
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr. Pierre receives research support from Cephalon Inc., and is a consultant to and/or speaker for Pfizer Inc., Bristol-Myers Squibb Co., AstraZeneca Pharmaceuticals, and Janssen Pharmaceutica.
Dr. Donna Wirshing receives research support from, is a consultant to, and/or is a speaker for Bristol-Myers Squibb Co., Pfizer Inc., Eli Lilly & Co., Janssen Pharmaceutica, AstraZeneca Pharmaceuticals, and Abbott Laboratories.
Dr. William Wirshing receives research support from, is a consultant to, and/or is a speaker for Bristol-Myers Squibb Co., Pfizer Inc., Eli Lilly & Co., Janssen Pharmaceutica, and AstraZeneca Pharmaceuticals.
1. Conley RR, Buchanan RW. Evaluation of treatment-resistant schizophrenia. Schizophr Bull 1997;23:663-74.
2. Chakos M, Lieberman J, Hoffman E, et al. Effectiveness of second-generation antipsychotics in patients with treatment-resistant schizophrenia: A review and meta-analysis of randomized trials. Am J Psychiatry 2001;158:518-26.
3. Baldessarini RJ, Cohen BM, Teicher MH. Significance of neuroleptic dose and plasma level in the pharmacological treatment of psychoses. Arch Gen Psych 1988;45:79-91.
4. McEvoy JP, Hogarty GE, Steingard S. Optimal dose of neuroleptic in acute schizophrenia: A controlled study of the neuroleptic threshold and higher haloperidol dose. Arch Gen Psychiatry 1991;48:739-45.
5. Van Putten T, Marder SR, Mintz J, Poland R. Haloperidol plasma levels and clinical response: A therapeutic window relationship. Am J Psychiatry 1992;149:500-5.
6. Van Putten T, Marshall BD, Liberman R, et al. Systematic dosage reduction in treatment-resistant schizophrenic patients. Psychopharmacol Bull 1993;29:315-20.
7. Marder SR, Meibach RC. Risperidone in the treatment of schizophrenia. Am J Psychiatry 1994;151:825-36.
8. Love RC, Conley RR, Kelly DL, Bartko JJ. A dose-outcome analysis of risperidone. J Clin Psychiatry 1999;60:771-5.
9. Wirshing DA, Marshall BD, Jr, Green MF, et al. Risperidone in treatment-refractory schizophrenia. Am J Psychiatry 1999;156:1374-9.
10. Fanous A, Lindenmayer JP. Schizophrenia and schizoaffective disorder treated with high doses of olanzapine. J Clin Psychopharmacol 1999;19:275-6.
11. Reich J. Use of high-dose olanzapine in refractory psychosis. Am J Psychiatry 1999;156:661.-
12. Dursun SM, Gardner DM, Bird DC, Flinn J. Olanzapine for patients with treatment-resistant schizophrenia: A naturalistic case-series outcome study. Can J Psychiatry 1999;44:701-4.
13. Lerner V. High-dose olanzapine for treatment-refractory schizophrenia. Clin Neuropharmacol 2003;26:58-61.
14. Sheitman BB, Lindgren JC, Early JE, Sved M. High-dose olanzapine for treatment-refractory schizophrenia. Am J Psychiatry 1997;154:1626.
15. Bronson BD, Lindenmayer JP. Adverse effects of high-dose olanzapine in treatment-refractory schizophrenia. J Clin Psychopharmacol 2000;20:383-4.
16. Dineen S, Withrow K, Voronovitch L, et al. QTc prolongation and high-dose olanzapine. Psychosomatics 2003;44:174-5.
17. Lindenmayer JP, Volavka J, Lieberman J, et al. Olanzapine for schizophrenia refractory to typical and atypical antipsychotics: An open-label, prospective trial. J Clin Psychopharmacol. 2001;21:448-53.
18. Volavka J, Czobor P, Sheitman B, et al. Clozapine, olanzapine, risperidone, and haloperidol in the treatment of patients with chronic schizophrenia and schizoaffective disorder. Am J Psychiatry 2002;159:255-62.
19. Conley RR, Kelly DL, Richardson CM, et al. The efficacy of high-dose olanzapine versus clozapine in treatment-resistant schizophrenia: A double-blind cross-over study. J Clin Psychopharmacol 2003;23:668-71.
20. Kelly DL, Conley RR, Richardson CM, et al. Adverse effects and laboratory parameters of high-dose olanzapine vs. clozapine in treatment-resistant schizophrenia. Ann Clin Psychiatry 2003;15:181-6.
21. Arvanitis LA, Miller BG. and the Seroquel Trial 13 Study Group. Multiple fixed doses of “Seroquel” (quetiapine) in patients with acute exacerbation of schizophrenia: A comparison with haloperidol and placebo. Biol Psychiatry 1997;42:233-46.
22. Bobes J, Garcia-Portilla MP, Saiz PA, et al. High degree of tolerability for monotherapy with high doses of quetiapine: A case report. J Clin Psychiatry 2002;63:1048-9.
23. Pierre JM, Wirshing DA, Cannell J, et al. High-dose quetiapine in treatment refractory schizophrenia (poster). Colorado Springs, CO: International Congress of Schizophrenia Research, 2003; abstracted in Schizophrenia Res 2003;60(supp):299.-
24. Nelson MW, Reynolds R, Kelly DL, et al. Safety and tolerability of high-dose quetiapine in treatment-refractory schizophrenia: Preliminary results from an open-label trial (poster). Colorado Springs, CO: International Congress of Schizophrenia Research, 2003; abstracted in Schizophrenia Res 2003;60(supp):363.-
25. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry 2003;60:681-90.
26. Deutschman DA, Deutschman DH. High-dose ziprasidone: effectiveness and tolerability in clinical practice (poster). Boston, MA: American Psychiatric Association Institute on Psychiatric Services annual meeting, 2003.
27. Steingard S, Allen M, Schooler MR. A study of pharmacologic treatment on medication-compliant schizophrenics who relapse. J Clin Psychiatry 1994;55:470-2.
When nothing else works, desperate clinicians are resorting to progressively more-tenuous and unpredictable treatments, trying to improve the lives of patients with refractory schizophrenia. High-dose antipsychotics is a common strategy.
Does boosting antipsychotic doses beyond the recommended range—but short of the neuroleptic threshold—enhance efficacy? This article attempts to answer that question by presenting the evidence on higher-than-recommended doses of atypical antipsychotics.
Lessons from neuroleptics
Up to 30% of patients with schizophrenia do not respond to antipsychotics and are considered “treatment refractory.”1 Even among those who do respond, improving symptoms by 20%—as research defines “treatment response”—does not necessarily yield clinical or functional improvement. Clozapine is the only atypical antipsychotic with well-established efficacy in these chronically ill patients,2 but its daunting side effects greatly curtail its use.
Before atypical antipsychotics, patients who did not respond to usual dosages of the typical neuroleptics were treated with higher dosages or switched to another drug class. Although many clinicians embraced high-dose neuroleptics, subsequent research discredited “rapid neuroleptization” in any clinical circumstance and showed that exceeding an antipsychotic’s neuroleptic threshold—the dose at which extrapyramidal side effects (EPS) occur—reduces its efficacy (Figure 1).3-5 In some instances, reducing neuroleptic dosages improves treatment-resistant patients’ symptoms and reduces druginduced side effects.6
Figure 1 Typical antipsychotics’ dose-response curve
Narrow therapeutic window between antipsychotic effect and neuroleptic threshold. Dotted line indicates declining efficacy.
Figure 2 Atypical antipsychotics’ dose-response curve
Wider therapeutic window with atypicals, compared with typical antipsychotics, as neuroleptic threshold (dotted line) moves right.Atypical antipsychotics are defined by their relative lack of EPS at recommended dosages (Figure 2). Because these agents can cause EPS if dosed too high, however, our historical habit of testing this dose limit risks losing “atypicality” and encountering other untoward events (Figure 3).
What is the safest, most effective dosage? Consider the evidence for each atypical antipsychotic.
Risperidone
Recommended dosage too high? When using atypicals at recommended doses, you are most likely to encounter the neuroleptic threshold with risperidone, with EPS risk increasing substantially at >6 mg/d.7 Post-approval studies set the most effective and safest dosage at approximately 4 mg/d, though this dosage was not studied in North American pre-approval trials. Dosages of 2 to 4 mg/d have been associated with more-favorable outcomes, suggesting that the initial recommendation to titrate to 6 mg/d within the first 3 days was ill-advised.8
In our study of patients with treatment-refractory schizophrenia,9 those treated with risperidone, 6 mg/d, improved significantly more after 4 weeks than did those receiving haloperidol, 15 mg/d, based on Brief Psychiatric Rating Scale (BPRS) scores. No additional benefit was seen after risperidone was increased to >6 mg/d at 8 weeks. Akathisia and tardive dyskinesia occurred significantly more often in the haloperidol group.
Conclusion. Some patients respond to higher-dose risperidone, but emerging EPS suggest the need to reduce the dosage rather than add an antiparkinsonian agent.
Figure 3 Unknown effects of high-dose atypical antipsychotic therapy
Dotted line indicates potential for greater antipsychotic effect with increasing dose.
Olanzapine
Mixed results. Case reports suggest that some patients who did not respond to previous antipsychotic trials or olanzapine, 20 mg/d, improved sig-nificantly—without substanial side effects—when olanzapine was increased up to 60 mg/d.10-14 Other case studies, however, report EPS, increased heart rate, increased transaminases, hyperprolactinemia, and prolonged QTc interval with high-dose olanzapine.14-16
In an open-label trial,17 43 patients with schizophrenia received olanzapine, up to 40 mg/d, after inadequate response to neuroleptics and risperidone or clozapine. Olanzapine was titrated to 20 mg/d by week 4 and increased 5 mg every 2 weeks if symptoms did not improve. After 14 weeks, improvement was modest and only 17% of patients met response criteria. However, >20 mg/d reduced symptoms more than did <20 mg/d, suggesting that high-dose olanzapine was more effective.
In a randomized trial,18 patients who did not respond to at least one atypical antipsychotic then received 8 weeks of fixed, standard-dose treatment with (mean dosages):
- haloperidol, 18.9 mg/d
- risperidone, 7.9 mg/d
- olanzapine, 19.6 mg/d
- clozapine, 401.6 mg/d.
Flexible dosing was then allowed for 6 weeks, and mean dosages were:
- haloperidol, 25.7 mg/d
- risperidone, 11.6 mg/d
- olanzapine, 30.4 mg/d
- clozapine, 526.6 mg/d.
Symptoms improved modestly at best for all medications, although patients taking olanzapine or clozapine improved significantly more than those treated with haloperidol as shown by mean changes in total Positive and Negative Syndrome Scale (PANSS) scores.
PANSS scores for olanzapine-treated patients showed additional improvement at week 14—when higher dosages were used—compared with week 8. This was not the case for the other medications, for which response plateaued. These findings suggest that high-dose risperidone and haloperidol are incrementally ineffective, but high-dose olanzapine could help some patients with refractory symptoms.
Results were different in a randomized, double-blind, 16-week, crossover study,19 when 13 patients with inadequate response to neuroleptics, risperidone, or conventional-dose olanzapine then received olanzapine, 50 mg/d, or clozapine, 450 mg/d. No olanzapine-treated patients and 20% of clozapine-treated patients met criteria for treatment response (20% improvement in BPRS score and final BPRS score <35 or 1-point improvement on Clinical Global Impressions-Severity of Illness scale).
Negative results don’t make headlines. Published clinical trials and case reports are subject to selective reporting of positive outcomes. Cases in which high-dose therapy proved ineffectivemay outnumber positive results but are less likely to be published.
Numbers don’t lie. Using high doses will almost always increase side effect risk and drug therapy costs, contributing to a poor risk-benefit ratio when efficacy remains unchanged. Resorting to an “if-it’s-not-working, double-it” strategy may seem reasonable, but two times zero is still zero.
Desperation warps perception. Clinicians tend to rely on observational experience. The desperation inherent in treating refractory patients, however, often creates a strong desire for improvement and therefore a potentially biased perception of outcome.
Likewise, patients may inaccurately portray themselves as improved to avoid disappointing their doctors. Controlled trials reduce these biases to better assess efficacy.
Antipsychotics work in 6 to 8 weeks. Improvements seen when pushing medications beyond recommended dosing may not be an effect of dose but of additional time on the medication. Antipsychotics usually take 6 to 8 weeks to produce maximal response, so high-dose therapy should not be started during this initial phase. This pace may not satisfy pressures for expedient stabilization and hospital discharge, but it is unrealistic to expect antipsychotics to work more quickly than they do.
Oversedation does not equal improvement. Patients who become excessively sedated from high-dose therapy or adjunctive medications may appear less psychotic but may not be so. The family or hospital staff may desire such sedation, but it can adversely affect the patient’s quality of life or medication adherence.
Polypharmacy clouds the issue. Many patients treated with high-dose antipsychotics are taking multiple agents, making it difficult to attribute improvement (or side effects) to any single one. A well-designed study of high-dose therapy would therefore:
- control for time
- examine concomitant medications’ effects
- determine whether “improvements” are related to sedation or reduced psychosis.
Medication may not need to change. When a patient decompensates, many forces pressure clinicians to change or add medications or increase dosages. Change may not be necessary, however, as nonadherence or substance abuse often trigger psychotic exacerbations. For example, Steingard et al27 added fluphenazine or placebo to antipsychotic regimens of newly hospitalized patients and found that increasing antipsychotic dosage did not improve outcome.
Subjects switching from clozapine to olanzapine tended to worsen, whereas those switching from olanzapine to clozapine tended to improve. Olanzapine-treated patients experienced more anticholinergic side effects and more weight gain than did clozapine-treated subjects.20
Conclusion. These mixed findings on high-dose olanzapine suggest questionable efficacy in patients with treatment-resistant schizophrenia and an uncertain risk of increased toxicity.
Quetiapine
Early placebo-controlled studies of quetiapine in schizophrenia concluded that statistically significant improvement begins at 150 mg/d and falls off after 600 mg/d.21 Although few high-dose quetiapine cases have been presented, clinical opinion holds that:
- most patients with chronic schizophrenia require 400 to 800 mg/d
- some treatment-refractory patients might benefit from >800 mg/d.
One patient responded to quetiapine, 1,600 mg/d, after not responding to olanzapine, 40 mg/d, and quetiapine, 800 mg/d. Constipation was the only reported side effect.22
Our group23 reported a series of 7 patients who responded (by clinician report) to quetiapine, 1,200 to 2,400 mg/d, after not responding to quetiapine, 800 mg/d, or to neuroleptics, risperidone, or olanzapine. Six responded to high-dose quetiapine and 1 to high-dose quetiapine plus risperidone, 2 mg/d; 4 received adjunctive dival-proex sodium, 1,500 to 3,000 mg/d. Psychopathology, violence, and behavioral disturbances were reduced throughout 5 to 14 months of monitoring. Side effects included sedation, orthostasis, and dysphagia.
When Nelson et al24 treated 13 subjects for 14 weeks with quetiapine, 1,000 to 1,400 mg/d, mean weight, glucose, total cholesterol, prolactin, and QTc interval duration did not change significantly. Heart rate increased significantly (though not to tachycardia), and headache, constipation, and lethargy were the most frequent side effects.
Summary. Although encouraging, these reports are preliminary, unpublished, and lack peer review. Controlled trials of high-dose quetiapine’s efficacy and safety are needed.
Ziprasidone and aripiprazole
No studies of high-dose ziprasidone or aripiprazole have been published. In premarketing trials:
- ziprasidone was studied at 200 mg/d and released with a maximum recommended dosage of 160 mg/d
- aripiprazole, 30 mg/d, was not more effective than 15 mg/d.25
Deutschman et al26 reviewed the charts of 31 patients who received ziprasidone, 240 to 320 mg/d, after an “incomplete” response to 160 mg/d. At the higher dosing:
- psychosis, affective symptoms, or anxiety improved in nearly one-half of patients
- 15% reported sedation, but most reported no side effects
- none developed QTc intervals >500 msec.
Caveats and precautions
These uncontrolled case reports and open-label studies do not “prove” efficacy or safety but reflect clinical practice. More than anything, they show that we need controlled trials to gauge high-dose antipsychotic therapy’s efficacy and safety and to curb our collective habit of relying on anecdotal experience and idiosyncratic beliefs.
Despite its side-effect profile, clozapine remains the treatment of choice for refractory schizophrenia. Given high-dose antipsychotic therapy’s uncertain efficacy and unknown risks, the evidence supports a clozapine trial before higher-than-recommended dosing is attempted.
Because educated guesswork plays a role in premarketing dosing studies, a medication’s optimal dose may be:
- overestimated (as with risperidone)
- underestimated (as perhaps with olanzapine and quetiapine).
Keep in mind some important caveats when you consider giving a patient high-dose antipsychotic therapy (Box).27 Of course, nonadherence is often the cause of apparent medication nonresponse. Increasing the dosage of a medication a patient is not taking rarely improves adherence. Interventions to enhance adherence—careful assessment, psychoeducation, and using longacting intramuscular medication—may be useful.
Related resources
- Marder SR, Essock SM, Miller AL, et al. The Mount Sinai Conference on the pharmacotherapy of schizophrenia. Schizophrenia Bull 2002;28:5-16.
- Practice guideline for the treatment of patients with schizophrenia (2nd ed). Am J Psychiatry 2004;161(suppl):1-56.
- Texas Medication Algorithm Project antipsychotic algorithm. http://www.mhmr.state.tx.us/centraloffice/medicaldirector/timascz1algo.pdf
Drug brand names
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Divalproex • Depakote
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr. Pierre receives research support from Cephalon Inc., and is a consultant to and/or speaker for Pfizer Inc., Bristol-Myers Squibb Co., AstraZeneca Pharmaceuticals, and Janssen Pharmaceutica.
Dr. Donna Wirshing receives research support from, is a consultant to, and/or is a speaker for Bristol-Myers Squibb Co., Pfizer Inc., Eli Lilly & Co., Janssen Pharmaceutica, AstraZeneca Pharmaceuticals, and Abbott Laboratories.
Dr. William Wirshing receives research support from, is a consultant to, and/or is a speaker for Bristol-Myers Squibb Co., Pfizer Inc., Eli Lilly & Co., Janssen Pharmaceutica, and AstraZeneca Pharmaceuticals.
When nothing else works, desperate clinicians are resorting to progressively more-tenuous and unpredictable treatments, trying to improve the lives of patients with refractory schizophrenia. High-dose antipsychotics is a common strategy.
Does boosting antipsychotic doses beyond the recommended range—but short of the neuroleptic threshold—enhance efficacy? This article attempts to answer that question by presenting the evidence on higher-than-recommended doses of atypical antipsychotics.
Lessons from neuroleptics
Up to 30% of patients with schizophrenia do not respond to antipsychotics and are considered “treatment refractory.”1 Even among those who do respond, improving symptoms by 20%—as research defines “treatment response”—does not necessarily yield clinical or functional improvement. Clozapine is the only atypical antipsychotic with well-established efficacy in these chronically ill patients,2 but its daunting side effects greatly curtail its use.
Before atypical antipsychotics, patients who did not respond to usual dosages of the typical neuroleptics were treated with higher dosages or switched to another drug class. Although many clinicians embraced high-dose neuroleptics, subsequent research discredited “rapid neuroleptization” in any clinical circumstance and showed that exceeding an antipsychotic’s neuroleptic threshold—the dose at which extrapyramidal side effects (EPS) occur—reduces its efficacy (Figure 1).3-5 In some instances, reducing neuroleptic dosages improves treatment-resistant patients’ symptoms and reduces druginduced side effects.6
Figure 1 Typical antipsychotics’ dose-response curve
Narrow therapeutic window between antipsychotic effect and neuroleptic threshold. Dotted line indicates declining efficacy.
Figure 2 Atypical antipsychotics’ dose-response curve
Wider therapeutic window with atypicals, compared with typical antipsychotics, as neuroleptic threshold (dotted line) moves right.Atypical antipsychotics are defined by their relative lack of EPS at recommended dosages (Figure 2). Because these agents can cause EPS if dosed too high, however, our historical habit of testing this dose limit risks losing “atypicality” and encountering other untoward events (Figure 3).
What is the safest, most effective dosage? Consider the evidence for each atypical antipsychotic.
Risperidone
Recommended dosage too high? When using atypicals at recommended doses, you are most likely to encounter the neuroleptic threshold with risperidone, with EPS risk increasing substantially at >6 mg/d.7 Post-approval studies set the most effective and safest dosage at approximately 4 mg/d, though this dosage was not studied in North American pre-approval trials. Dosages of 2 to 4 mg/d have been associated with more-favorable outcomes, suggesting that the initial recommendation to titrate to 6 mg/d within the first 3 days was ill-advised.8
In our study of patients with treatment-refractory schizophrenia,9 those treated with risperidone, 6 mg/d, improved significantly more after 4 weeks than did those receiving haloperidol, 15 mg/d, based on Brief Psychiatric Rating Scale (BPRS) scores. No additional benefit was seen after risperidone was increased to >6 mg/d at 8 weeks. Akathisia and tardive dyskinesia occurred significantly more often in the haloperidol group.
Conclusion. Some patients respond to higher-dose risperidone, but emerging EPS suggest the need to reduce the dosage rather than add an antiparkinsonian agent.
Figure 3 Unknown effects of high-dose atypical antipsychotic therapy
Dotted line indicates potential for greater antipsychotic effect with increasing dose.
Olanzapine
Mixed results. Case reports suggest that some patients who did not respond to previous antipsychotic trials or olanzapine, 20 mg/d, improved sig-nificantly—without substanial side effects—when olanzapine was increased up to 60 mg/d.10-14 Other case studies, however, report EPS, increased heart rate, increased transaminases, hyperprolactinemia, and prolonged QTc interval with high-dose olanzapine.14-16
In an open-label trial,17 43 patients with schizophrenia received olanzapine, up to 40 mg/d, after inadequate response to neuroleptics and risperidone or clozapine. Olanzapine was titrated to 20 mg/d by week 4 and increased 5 mg every 2 weeks if symptoms did not improve. After 14 weeks, improvement was modest and only 17% of patients met response criteria. However, >20 mg/d reduced symptoms more than did <20 mg/d, suggesting that high-dose olanzapine was more effective.
In a randomized trial,18 patients who did not respond to at least one atypical antipsychotic then received 8 weeks of fixed, standard-dose treatment with (mean dosages):
- haloperidol, 18.9 mg/d
- risperidone, 7.9 mg/d
- olanzapine, 19.6 mg/d
- clozapine, 401.6 mg/d.
Flexible dosing was then allowed for 6 weeks, and mean dosages were:
- haloperidol, 25.7 mg/d
- risperidone, 11.6 mg/d
- olanzapine, 30.4 mg/d
- clozapine, 526.6 mg/d.
Symptoms improved modestly at best for all medications, although patients taking olanzapine or clozapine improved significantly more than those treated with haloperidol as shown by mean changes in total Positive and Negative Syndrome Scale (PANSS) scores.
PANSS scores for olanzapine-treated patients showed additional improvement at week 14—when higher dosages were used—compared with week 8. This was not the case for the other medications, for which response plateaued. These findings suggest that high-dose risperidone and haloperidol are incrementally ineffective, but high-dose olanzapine could help some patients with refractory symptoms.
Results were different in a randomized, double-blind, 16-week, crossover study,19 when 13 patients with inadequate response to neuroleptics, risperidone, or conventional-dose olanzapine then received olanzapine, 50 mg/d, or clozapine, 450 mg/d. No olanzapine-treated patients and 20% of clozapine-treated patients met criteria for treatment response (20% improvement in BPRS score and final BPRS score <35 or 1-point improvement on Clinical Global Impressions-Severity of Illness scale).
Negative results don’t make headlines. Published clinical trials and case reports are subject to selective reporting of positive outcomes. Cases in which high-dose therapy proved ineffectivemay outnumber positive results but are less likely to be published.
Numbers don’t lie. Using high doses will almost always increase side effect risk and drug therapy costs, contributing to a poor risk-benefit ratio when efficacy remains unchanged. Resorting to an “if-it’s-not-working, double-it” strategy may seem reasonable, but two times zero is still zero.
Desperation warps perception. Clinicians tend to rely on observational experience. The desperation inherent in treating refractory patients, however, often creates a strong desire for improvement and therefore a potentially biased perception of outcome.
Likewise, patients may inaccurately portray themselves as improved to avoid disappointing their doctors. Controlled trials reduce these biases to better assess efficacy.
Antipsychotics work in 6 to 8 weeks. Improvements seen when pushing medications beyond recommended dosing may not be an effect of dose but of additional time on the medication. Antipsychotics usually take 6 to 8 weeks to produce maximal response, so high-dose therapy should not be started during this initial phase. This pace may not satisfy pressures for expedient stabilization and hospital discharge, but it is unrealistic to expect antipsychotics to work more quickly than they do.
Oversedation does not equal improvement. Patients who become excessively sedated from high-dose therapy or adjunctive medications may appear less psychotic but may not be so. The family or hospital staff may desire such sedation, but it can adversely affect the patient’s quality of life or medication adherence.
Polypharmacy clouds the issue. Many patients treated with high-dose antipsychotics are taking multiple agents, making it difficult to attribute improvement (or side effects) to any single one. A well-designed study of high-dose therapy would therefore:
- control for time
- examine concomitant medications’ effects
- determine whether “improvements” are related to sedation or reduced psychosis.
Medication may not need to change. When a patient decompensates, many forces pressure clinicians to change or add medications or increase dosages. Change may not be necessary, however, as nonadherence or substance abuse often trigger psychotic exacerbations. For example, Steingard et al27 added fluphenazine or placebo to antipsychotic regimens of newly hospitalized patients and found that increasing antipsychotic dosage did not improve outcome.
Subjects switching from clozapine to olanzapine tended to worsen, whereas those switching from olanzapine to clozapine tended to improve. Olanzapine-treated patients experienced more anticholinergic side effects and more weight gain than did clozapine-treated subjects.20
Conclusion. These mixed findings on high-dose olanzapine suggest questionable efficacy in patients with treatment-resistant schizophrenia and an uncertain risk of increased toxicity.
Quetiapine
Early placebo-controlled studies of quetiapine in schizophrenia concluded that statistically significant improvement begins at 150 mg/d and falls off after 600 mg/d.21 Although few high-dose quetiapine cases have been presented, clinical opinion holds that:
- most patients with chronic schizophrenia require 400 to 800 mg/d
- some treatment-refractory patients might benefit from >800 mg/d.
One patient responded to quetiapine, 1,600 mg/d, after not responding to olanzapine, 40 mg/d, and quetiapine, 800 mg/d. Constipation was the only reported side effect.22
Our group23 reported a series of 7 patients who responded (by clinician report) to quetiapine, 1,200 to 2,400 mg/d, after not responding to quetiapine, 800 mg/d, or to neuroleptics, risperidone, or olanzapine. Six responded to high-dose quetiapine and 1 to high-dose quetiapine plus risperidone, 2 mg/d; 4 received adjunctive dival-proex sodium, 1,500 to 3,000 mg/d. Psychopathology, violence, and behavioral disturbances were reduced throughout 5 to 14 months of monitoring. Side effects included sedation, orthostasis, and dysphagia.
When Nelson et al24 treated 13 subjects for 14 weeks with quetiapine, 1,000 to 1,400 mg/d, mean weight, glucose, total cholesterol, prolactin, and QTc interval duration did not change significantly. Heart rate increased significantly (though not to tachycardia), and headache, constipation, and lethargy were the most frequent side effects.
Summary. Although encouraging, these reports are preliminary, unpublished, and lack peer review. Controlled trials of high-dose quetiapine’s efficacy and safety are needed.
Ziprasidone and aripiprazole
No studies of high-dose ziprasidone or aripiprazole have been published. In premarketing trials:
- ziprasidone was studied at 200 mg/d and released with a maximum recommended dosage of 160 mg/d
- aripiprazole, 30 mg/d, was not more effective than 15 mg/d.25
Deutschman et al26 reviewed the charts of 31 patients who received ziprasidone, 240 to 320 mg/d, after an “incomplete” response to 160 mg/d. At the higher dosing:
- psychosis, affective symptoms, or anxiety improved in nearly one-half of patients
- 15% reported sedation, but most reported no side effects
- none developed QTc intervals >500 msec.
Caveats and precautions
These uncontrolled case reports and open-label studies do not “prove” efficacy or safety but reflect clinical practice. More than anything, they show that we need controlled trials to gauge high-dose antipsychotic therapy’s efficacy and safety and to curb our collective habit of relying on anecdotal experience and idiosyncratic beliefs.
Despite its side-effect profile, clozapine remains the treatment of choice for refractory schizophrenia. Given high-dose antipsychotic therapy’s uncertain efficacy and unknown risks, the evidence supports a clozapine trial before higher-than-recommended dosing is attempted.
Because educated guesswork plays a role in premarketing dosing studies, a medication’s optimal dose may be:
- overestimated (as with risperidone)
- underestimated (as perhaps with olanzapine and quetiapine).
Keep in mind some important caveats when you consider giving a patient high-dose antipsychotic therapy (Box).27 Of course, nonadherence is often the cause of apparent medication nonresponse. Increasing the dosage of a medication a patient is not taking rarely improves adherence. Interventions to enhance adherence—careful assessment, psychoeducation, and using longacting intramuscular medication—may be useful.
Related resources
- Marder SR, Essock SM, Miller AL, et al. The Mount Sinai Conference on the pharmacotherapy of schizophrenia. Schizophrenia Bull 2002;28:5-16.
- Practice guideline for the treatment of patients with schizophrenia (2nd ed). Am J Psychiatry 2004;161(suppl):1-56.
- Texas Medication Algorithm Project antipsychotic algorithm. http://www.mhmr.state.tx.us/centraloffice/medicaldirector/timascz1algo.pdf
Drug brand names
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Divalproex • Depakote
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr. Pierre receives research support from Cephalon Inc., and is a consultant to and/or speaker for Pfizer Inc., Bristol-Myers Squibb Co., AstraZeneca Pharmaceuticals, and Janssen Pharmaceutica.
Dr. Donna Wirshing receives research support from, is a consultant to, and/or is a speaker for Bristol-Myers Squibb Co., Pfizer Inc., Eli Lilly & Co., Janssen Pharmaceutica, AstraZeneca Pharmaceuticals, and Abbott Laboratories.
Dr. William Wirshing receives research support from, is a consultant to, and/or is a speaker for Bristol-Myers Squibb Co., Pfizer Inc., Eli Lilly & Co., Janssen Pharmaceutica, and AstraZeneca Pharmaceuticals.
1. Conley RR, Buchanan RW. Evaluation of treatment-resistant schizophrenia. Schizophr Bull 1997;23:663-74.
2. Chakos M, Lieberman J, Hoffman E, et al. Effectiveness of second-generation antipsychotics in patients with treatment-resistant schizophrenia: A review and meta-analysis of randomized trials. Am J Psychiatry 2001;158:518-26.
3. Baldessarini RJ, Cohen BM, Teicher MH. Significance of neuroleptic dose and plasma level in the pharmacological treatment of psychoses. Arch Gen Psych 1988;45:79-91.
4. McEvoy JP, Hogarty GE, Steingard S. Optimal dose of neuroleptic in acute schizophrenia: A controlled study of the neuroleptic threshold and higher haloperidol dose. Arch Gen Psychiatry 1991;48:739-45.
5. Van Putten T, Marder SR, Mintz J, Poland R. Haloperidol plasma levels and clinical response: A therapeutic window relationship. Am J Psychiatry 1992;149:500-5.
6. Van Putten T, Marshall BD, Liberman R, et al. Systematic dosage reduction in treatment-resistant schizophrenic patients. Psychopharmacol Bull 1993;29:315-20.
7. Marder SR, Meibach RC. Risperidone in the treatment of schizophrenia. Am J Psychiatry 1994;151:825-36.
8. Love RC, Conley RR, Kelly DL, Bartko JJ. A dose-outcome analysis of risperidone. J Clin Psychiatry 1999;60:771-5.
9. Wirshing DA, Marshall BD, Jr, Green MF, et al. Risperidone in treatment-refractory schizophrenia. Am J Psychiatry 1999;156:1374-9.
10. Fanous A, Lindenmayer JP. Schizophrenia and schizoaffective disorder treated with high doses of olanzapine. J Clin Psychopharmacol 1999;19:275-6.
11. Reich J. Use of high-dose olanzapine in refractory psychosis. Am J Psychiatry 1999;156:661.-
12. Dursun SM, Gardner DM, Bird DC, Flinn J. Olanzapine for patients with treatment-resistant schizophrenia: A naturalistic case-series outcome study. Can J Psychiatry 1999;44:701-4.
13. Lerner V. High-dose olanzapine for treatment-refractory schizophrenia. Clin Neuropharmacol 2003;26:58-61.
14. Sheitman BB, Lindgren JC, Early JE, Sved M. High-dose olanzapine for treatment-refractory schizophrenia. Am J Psychiatry 1997;154:1626.
15. Bronson BD, Lindenmayer JP. Adverse effects of high-dose olanzapine in treatment-refractory schizophrenia. J Clin Psychopharmacol 2000;20:383-4.
16. Dineen S, Withrow K, Voronovitch L, et al. QTc prolongation and high-dose olanzapine. Psychosomatics 2003;44:174-5.
17. Lindenmayer JP, Volavka J, Lieberman J, et al. Olanzapine for schizophrenia refractory to typical and atypical antipsychotics: An open-label, prospective trial. J Clin Psychopharmacol. 2001;21:448-53.
18. Volavka J, Czobor P, Sheitman B, et al. Clozapine, olanzapine, risperidone, and haloperidol in the treatment of patients with chronic schizophrenia and schizoaffective disorder. Am J Psychiatry 2002;159:255-62.
19. Conley RR, Kelly DL, Richardson CM, et al. The efficacy of high-dose olanzapine versus clozapine in treatment-resistant schizophrenia: A double-blind cross-over study. J Clin Psychopharmacol 2003;23:668-71.
20. Kelly DL, Conley RR, Richardson CM, et al. Adverse effects and laboratory parameters of high-dose olanzapine vs. clozapine in treatment-resistant schizophrenia. Ann Clin Psychiatry 2003;15:181-6.
21. Arvanitis LA, Miller BG. and the Seroquel Trial 13 Study Group. Multiple fixed doses of “Seroquel” (quetiapine) in patients with acute exacerbation of schizophrenia: A comparison with haloperidol and placebo. Biol Psychiatry 1997;42:233-46.
22. Bobes J, Garcia-Portilla MP, Saiz PA, et al. High degree of tolerability for monotherapy with high doses of quetiapine: A case report. J Clin Psychiatry 2002;63:1048-9.
23. Pierre JM, Wirshing DA, Cannell J, et al. High-dose quetiapine in treatment refractory schizophrenia (poster). Colorado Springs, CO: International Congress of Schizophrenia Research, 2003; abstracted in Schizophrenia Res 2003;60(supp):299.-
24. Nelson MW, Reynolds R, Kelly DL, et al. Safety and tolerability of high-dose quetiapine in treatment-refractory schizophrenia: Preliminary results from an open-label trial (poster). Colorado Springs, CO: International Congress of Schizophrenia Research, 2003; abstracted in Schizophrenia Res 2003;60(supp):363.-
25. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry 2003;60:681-90.
26. Deutschman DA, Deutschman DH. High-dose ziprasidone: effectiveness and tolerability in clinical practice (poster). Boston, MA: American Psychiatric Association Institute on Psychiatric Services annual meeting, 2003.
27. Steingard S, Allen M, Schooler MR. A study of pharmacologic treatment on medication-compliant schizophrenics who relapse. J Clin Psychiatry 1994;55:470-2.
1. Conley RR, Buchanan RW. Evaluation of treatment-resistant schizophrenia. Schizophr Bull 1997;23:663-74.
2. Chakos M, Lieberman J, Hoffman E, et al. Effectiveness of second-generation antipsychotics in patients with treatment-resistant schizophrenia: A review and meta-analysis of randomized trials. Am J Psychiatry 2001;158:518-26.
3. Baldessarini RJ, Cohen BM, Teicher MH. Significance of neuroleptic dose and plasma level in the pharmacological treatment of psychoses. Arch Gen Psych 1988;45:79-91.
4. McEvoy JP, Hogarty GE, Steingard S. Optimal dose of neuroleptic in acute schizophrenia: A controlled study of the neuroleptic threshold and higher haloperidol dose. Arch Gen Psychiatry 1991;48:739-45.
5. Van Putten T, Marder SR, Mintz J, Poland R. Haloperidol plasma levels and clinical response: A therapeutic window relationship. Am J Psychiatry 1992;149:500-5.
6. Van Putten T, Marshall BD, Liberman R, et al. Systematic dosage reduction in treatment-resistant schizophrenic patients. Psychopharmacol Bull 1993;29:315-20.
7. Marder SR, Meibach RC. Risperidone in the treatment of schizophrenia. Am J Psychiatry 1994;151:825-36.
8. Love RC, Conley RR, Kelly DL, Bartko JJ. A dose-outcome analysis of risperidone. J Clin Psychiatry 1999;60:771-5.
9. Wirshing DA, Marshall BD, Jr, Green MF, et al. Risperidone in treatment-refractory schizophrenia. Am J Psychiatry 1999;156:1374-9.
10. Fanous A, Lindenmayer JP. Schizophrenia and schizoaffective disorder treated with high doses of olanzapine. J Clin Psychopharmacol 1999;19:275-6.
11. Reich J. Use of high-dose olanzapine in refractory psychosis. Am J Psychiatry 1999;156:661.-
12. Dursun SM, Gardner DM, Bird DC, Flinn J. Olanzapine for patients with treatment-resistant schizophrenia: A naturalistic case-series outcome study. Can J Psychiatry 1999;44:701-4.
13. Lerner V. High-dose olanzapine for treatment-refractory schizophrenia. Clin Neuropharmacol 2003;26:58-61.
14. Sheitman BB, Lindgren JC, Early JE, Sved M. High-dose olanzapine for treatment-refractory schizophrenia. Am J Psychiatry 1997;154:1626.
15. Bronson BD, Lindenmayer JP. Adverse effects of high-dose olanzapine in treatment-refractory schizophrenia. J Clin Psychopharmacol 2000;20:383-4.
16. Dineen S, Withrow K, Voronovitch L, et al. QTc prolongation and high-dose olanzapine. Psychosomatics 2003;44:174-5.
17. Lindenmayer JP, Volavka J, Lieberman J, et al. Olanzapine for schizophrenia refractory to typical and atypical antipsychotics: An open-label, prospective trial. J Clin Psychopharmacol. 2001;21:448-53.
18. Volavka J, Czobor P, Sheitman B, et al. Clozapine, olanzapine, risperidone, and haloperidol in the treatment of patients with chronic schizophrenia and schizoaffective disorder. Am J Psychiatry 2002;159:255-62.
19. Conley RR, Kelly DL, Richardson CM, et al. The efficacy of high-dose olanzapine versus clozapine in treatment-resistant schizophrenia: A double-blind cross-over study. J Clin Psychopharmacol 2003;23:668-71.
20. Kelly DL, Conley RR, Richardson CM, et al. Adverse effects and laboratory parameters of high-dose olanzapine vs. clozapine in treatment-resistant schizophrenia. Ann Clin Psychiatry 2003;15:181-6.
21. Arvanitis LA, Miller BG. and the Seroquel Trial 13 Study Group. Multiple fixed doses of “Seroquel” (quetiapine) in patients with acute exacerbation of schizophrenia: A comparison with haloperidol and placebo. Biol Psychiatry 1997;42:233-46.
22. Bobes J, Garcia-Portilla MP, Saiz PA, et al. High degree of tolerability for monotherapy with high doses of quetiapine: A case report. J Clin Psychiatry 2002;63:1048-9.
23. Pierre JM, Wirshing DA, Cannell J, et al. High-dose quetiapine in treatment refractory schizophrenia (poster). Colorado Springs, CO: International Congress of Schizophrenia Research, 2003; abstracted in Schizophrenia Res 2003;60(supp):299.-
24. Nelson MW, Reynolds R, Kelly DL, et al. Safety and tolerability of high-dose quetiapine in treatment-refractory schizophrenia: Preliminary results from an open-label trial (poster). Colorado Springs, CO: International Congress of Schizophrenia Research, 2003; abstracted in Schizophrenia Res 2003;60(supp):363.-
25. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry 2003;60:681-90.
26. Deutschman DA, Deutschman DH. High-dose ziprasidone: effectiveness and tolerability in clinical practice (poster). Boston, MA: American Psychiatric Association Institute on Psychiatric Services annual meeting, 2003.
27. Steingard S, Allen M, Schooler MR. A study of pharmacologic treatment on medication-compliant schizophrenics who relapse. J Clin Psychiatry 1994;55:470-2.
Making ‘PEACE’ with hostile, unwilling patients
Often we encounter patients who are angry about having been “forced” to see a psychiatrist:
- In the emergency room or inpatient setting, patients who present with an apparent medical problem may become upset after learning that a psychiatric evaluation has been ordered without their knowledge or against their wishes.
- In outpatient clinics, patients who arrive under “coercion” from parents, spouses, or employers can also be hostile.
Defusing the hostility and engaging the patient are critical first steps toward a therapeutic alliance. When faced with a hostile patient, take a deep breath, control your emotions, and follow the PEACE principle: presence, empathy, acceptance, collaboration, and empowerment.
Presence. From the outset, make it clear that the patient has your undivided attention. Nonverbal cues such as sitting down, maintaining comfortable eye contact, and not writing notes during the interview’s initial stages give this impression.
Empathy. As you sit quietly and attentively, encourage the patient to vent his or her anger over being “forced” to see a psychiatrist. Most of us can empathize with a person who feels powerless, patronized, or coerced.
Acceptance. Acknowledging the patient’s distress can go far toward diminishing or defusing the anger. For example, tell the patient, “I understand that this is unsettling for you,” or “I, too, wish the circumstances were different because this is obviously difficult for you.”
Collaboration. Tell the patient you only want to help him, to be his partner in a therapeutic alliance.
Empower. Never force the evaluation. Rather, let the patient decide whether to proceed. Tell her, “I want to help you with what’s been going on in your life, but it’s totally up to you to continue. I cannot—and don’t want to—force you to do something you choose not to do.”
Then offer alternatives such as:
- a follow-up appointment
- a visit the next day if the patient is hospitalized
- or telling the emergency physician that the patient declined the psychiatric evaluation.
I find that when using this approach the patient usually agrees to a therapeutic assessment.
Dr. Christensen is associate professor of psychiatry, University of Florida College of Medicine, Jacksonville, and director of its community psychiatry program. Jacksonville
Often we encounter patients who are angry about having been “forced” to see a psychiatrist:
- In the emergency room or inpatient setting, patients who present with an apparent medical problem may become upset after learning that a psychiatric evaluation has been ordered without their knowledge or against their wishes.
- In outpatient clinics, patients who arrive under “coercion” from parents, spouses, or employers can also be hostile.
Defusing the hostility and engaging the patient are critical first steps toward a therapeutic alliance. When faced with a hostile patient, take a deep breath, control your emotions, and follow the PEACE principle: presence, empathy, acceptance, collaboration, and empowerment.
Presence. From the outset, make it clear that the patient has your undivided attention. Nonverbal cues such as sitting down, maintaining comfortable eye contact, and not writing notes during the interview’s initial stages give this impression.
Empathy. As you sit quietly and attentively, encourage the patient to vent his or her anger over being “forced” to see a psychiatrist. Most of us can empathize with a person who feels powerless, patronized, or coerced.
Acceptance. Acknowledging the patient’s distress can go far toward diminishing or defusing the anger. For example, tell the patient, “I understand that this is unsettling for you,” or “I, too, wish the circumstances were different because this is obviously difficult for you.”
Collaboration. Tell the patient you only want to help him, to be his partner in a therapeutic alliance.
Empower. Never force the evaluation. Rather, let the patient decide whether to proceed. Tell her, “I want to help you with what’s been going on in your life, but it’s totally up to you to continue. I cannot—and don’t want to—force you to do something you choose not to do.”
Then offer alternatives such as:
- a follow-up appointment
- a visit the next day if the patient is hospitalized
- or telling the emergency physician that the patient declined the psychiatric evaluation.
I find that when using this approach the patient usually agrees to a therapeutic assessment.
Often we encounter patients who are angry about having been “forced” to see a psychiatrist:
- In the emergency room or inpatient setting, patients who present with an apparent medical problem may become upset after learning that a psychiatric evaluation has been ordered without their knowledge or against their wishes.
- In outpatient clinics, patients who arrive under “coercion” from parents, spouses, or employers can also be hostile.
Defusing the hostility and engaging the patient are critical first steps toward a therapeutic alliance. When faced with a hostile patient, take a deep breath, control your emotions, and follow the PEACE principle: presence, empathy, acceptance, collaboration, and empowerment.
Presence. From the outset, make it clear that the patient has your undivided attention. Nonverbal cues such as sitting down, maintaining comfortable eye contact, and not writing notes during the interview’s initial stages give this impression.
Empathy. As you sit quietly and attentively, encourage the patient to vent his or her anger over being “forced” to see a psychiatrist. Most of us can empathize with a person who feels powerless, patronized, or coerced.
Acceptance. Acknowledging the patient’s distress can go far toward diminishing or defusing the anger. For example, tell the patient, “I understand that this is unsettling for you,” or “I, too, wish the circumstances were different because this is obviously difficult for you.”
Collaboration. Tell the patient you only want to help him, to be his partner in a therapeutic alliance.
Empower. Never force the evaluation. Rather, let the patient decide whether to proceed. Tell her, “I want to help you with what’s been going on in your life, but it’s totally up to you to continue. I cannot—and don’t want to—force you to do something you choose not to do.”
Then offer alternatives such as:
- a follow-up appointment
- a visit the next day if the patient is hospitalized
- or telling the emergency physician that the patient declined the psychiatric evaluation.
I find that when using this approach the patient usually agrees to a therapeutic assessment.
Dr. Christensen is associate professor of psychiatry, University of Florida College of Medicine, Jacksonville, and director of its community psychiatry program. Jacksonville
Dr. Christensen is associate professor of psychiatry, University of Florida College of Medicine, Jacksonville, and director of its community psychiatry program. Jacksonville
High-dose antipsychotics: A matter of opinion
Like you, I always worry about being sued for malpractice. I comfort myself by knowing that not every psychiatrist—or even the majority—has to agree with the way I practice. To meet the “standard of care” and fulfill my duty to patients, my practice just needs to be endorsed by a respectable minority of practitioners.
Two articles in this issue illustrate the legitimate diversity of opinion within psychiatry. Drs. Joseph Pierre, Donna Wirshing, and William Wirshing at UCLA provide an excellent review on higher-than-recommended antipsychotic dosages for patients with treatment-refractory schizophrenia. They conclude that:
- there is very little evidence that high dosages are more effective than usual dosages
- patients who do not respond to usual dosages should be switched to clozapine before high-dose therapy is tried.
We invited Sheldon Preskorn, MD—a Current Psychiatry associate editor—to review the article. Based on his research in clinical psychopharmacology, he wrote a commentary to explain the variables that determine patient response to drug therapy. He suggests that:
- some patients may need higher antipsychotic dosages, which might be tried before switching medications
- plasma levels should be considered before changing treatments.
We’ll let you decide which approach or synthesis of approaches works for you. Whichever you choose will be supported by at least a respectable minority of practitioners.

James Randolph Hillard, MD
Editor-in-Chief
James Randolph Hillard, MD
Editor-in-Chief
James Randolph Hillard, MD
Editor-in-Chief
Like you, I always worry about being sued for malpractice. I comfort myself by knowing that not every psychiatrist—or even the majority—has to agree with the way I practice. To meet the “standard of care” and fulfill my duty to patients, my practice just needs to be endorsed by a respectable minority of practitioners.
Two articles in this issue illustrate the legitimate diversity of opinion within psychiatry. Drs. Joseph Pierre, Donna Wirshing, and William Wirshing at UCLA provide an excellent review on higher-than-recommended antipsychotic dosages for patients with treatment-refractory schizophrenia. They conclude that:
- there is very little evidence that high dosages are more effective than usual dosages
- patients who do not respond to usual dosages should be switched to clozapine before high-dose therapy is tried.
We invited Sheldon Preskorn, MD—a Current Psychiatry associate editor—to review the article. Based on his research in clinical psychopharmacology, he wrote a commentary to explain the variables that determine patient response to drug therapy. He suggests that:
- some patients may need higher antipsychotic dosages, which might be tried before switching medications
- plasma levels should be considered before changing treatments.
We’ll let you decide which approach or synthesis of approaches works for you. Whichever you choose will be supported by at least a respectable minority of practitioners.
Like you, I always worry about being sued for malpractice. I comfort myself by knowing that not every psychiatrist—or even the majority—has to agree with the way I practice. To meet the “standard of care” and fulfill my duty to patients, my practice just needs to be endorsed by a respectable minority of practitioners.
Two articles in this issue illustrate the legitimate diversity of opinion within psychiatry. Drs. Joseph Pierre, Donna Wirshing, and William Wirshing at UCLA provide an excellent review on higher-than-recommended antipsychotic dosages for patients with treatment-refractory schizophrenia. They conclude that:
- there is very little evidence that high dosages are more effective than usual dosages
- patients who do not respond to usual dosages should be switched to clozapine before high-dose therapy is tried.
We invited Sheldon Preskorn, MD—a Current Psychiatry associate editor—to review the article. Based on his research in clinical psychopharmacology, he wrote a commentary to explain the variables that determine patient response to drug therapy. He suggests that:
- some patients may need higher antipsychotic dosages, which might be tried before switching medications
- plasma levels should be considered before changing treatments.
We’ll let you decide which approach or synthesis of approaches works for you. Whichever you choose will be supported by at least a respectable minority of practitioners.
Commentary: Why patients may not respond to usual recommended dosages
Psychiatrists may consider using higher than usually recommended dosages of antipsychotics when faced with nonresponse. In this issue , Pierre et al1 carefully and thoughtfully discuss the pros and cons of this practice in patients with schizophrenia. Having reviewed that article, I thought CURRENT PSYCHIATRY’s readers might benefit from a theoretical framework for analyzing drug response.
‘Usual’ vs ‘unusual’ patients
A clinical trial for drug registration is, in essence, a population pharmacokinetic study whose goal is to determine the usual dosage for the usual patient in the trial. Many patients seen in clinical practice, such as those with treatment-refractory psychotic disorders, are typically excluded from registration trials. Thus, the usual registration trial patient may be an unusual patient in a clinician’s practice, and the trial’s usual dosage may not produce an adequate response for the clinician’s usual patient. How, then, might a clinician approach inadequate response, except by:
- blindly exceeding the usually recommended dosage
- switching among available drugs
- adding drugs to create a complex cocktail?
This commentary dissects why a patient might not benefit from the usual recommended dosage and how that could lead to different courses of action.
Equation 1. Three variables (Table) determine response to any drug:
- affinity for and intrinsic action on a regulatory protein (such as a receptor)
- concentration (amount of drug reaching the site of action)
- biological variance, which can shift an individual’s dose-response curve relative to that of the “usual” patient, making that individual more or less sensitive to the drug’s effects.2
Table
3 variables that determine patient response to any drug
| Equation 1 | ||||||
| Effect | = | Affinity for and intrinsic activity at a site of action | X | Drug concentration (see Equation 2) Absorption Distribution Metabolism Elimination (ADME) | X | Biological variance Genetics Age Disease Environment (internal) (GADE) |
| Equation 2 | ||||||
| Drug concentration = dosing rate/clearance | ||||||
Equation 2. Drug concentration is dosing rate divided by clearance in a given patient. Dosing rate and clearance are equally important in determining drug concentration—which, in turn, determines the site of action engaged, to what degree, and the patient’s response to the drug.
Causes of inadequate response. Nonadherence is a common cause of inadequate response. When a patient repeatedly misses doses or stops taking the drug, the true dosing rate is lower than the prescribed dosing rate, resulting in reduced drug concentration and effect.
Pierre and colleagues focus on the “unusual” patient who does not respond optimally to antipsychotic dosages established in registration trials.1 As in Equation 1, sources of biological variance—genetics, age, disease, and environment (internal)—may distinguish the treatment-refractory patient from the responsive patient. The mnemonic GADE captures these variables:
Genetic variation refers to mutations in regulatory proteins that:
- determine the drug’s action (such mutations may change the drug’s binding affinity, so that a higher concentration is needed to adequately engage the site)
- determine what drug concentration reaches the site of action (such as drug-metabolizing enzymes that regulate clearance, or transporter proteins that prevent or facilitate the drug’s ability to reach the site of action).
Age refers to physiologic changes (pharmacodynamic or pharmacokinetic) that make the patient more or less sensitive to the drug’s effects.
Disease refers to differences in organ function related to pathophysiology. Patients with the same clinical presentation (in this case, psychosis) may respond differently to the same drug because they have different underlying pathophysiologies (such as schizophrenic syndrome due to differing genetic causes or to toxins or slow viruses).
Environment (internal) refers to exogenous substances in the body—such as drugs and dietary substances—that can interact with and influence response to other drugs.
Nonpsychiatric disease also can alter response to medication. For example, impaired hepatic, renal, or cardiac function can impair drug clearance, leading to greater-than-usual accumulation. Such a patient can be “sensitive” to the drug and experience a greater effect than is usually seen with the dosage given.
Dosing for clinical effect
Psychiatrists commonly titrate dosages based on clinical assessment of response.3 The clinician increases the dosage if a patient does not improve and has no obvious rate-limiting adverse effects.
Perhaps without realizing it, the clinician is assuming that the dosage is inadequate for a given patient because the concentration is inadequate due to rapid clearance. Other reasons are possible, however, such as:
- the drug is not reaching the site of action a mutation at the site of action is altering
- the drug’s binding affinity
- the concentration may be too high, but the resulting adverse effects resemble worsening of the disease being treated. For example, akathisia due to dopamine-2 receptor blockade can present as agitation, and the clinician may increase the dosage when it should be decreased.
In the first two instances, escalating the dosage may be beneficial or cause toxicity. High levels in a peripheral compartment can cause adverse effects that may be silent until they become deadly (such as torsades de pointes). In the third instance, dosage escalation is the wrong step because the level is already too high.
Recommended dosage range
Principal goals of phase I studies in drug development are to establish the optimal dosage range and a maximum tolerated dosage. This upper limit is rarely, if ever, exceeded in later trials. Because phase I trial results are rarely published, the prescriber often does not know the rationale for a recommended dosing range’s upper limit.
Clinicians who escalate a drug’s dosage above the recommended range are using an n=1 paradigm, in which the patient is his or her own control. Unfortunately, treating one patient at a time cannot detect infrequent (much less rare) adverse events.
Using higher-than-recommended dosages thus exposes patients to unknown risks, with less monitoring than in a typical phase I trial in which subjects are confined to a research unit before, during, and after drug exposure. During the study, participants undergo serial ECGs, laboratory tests, and plasma drug level monitoring.
Therapeutic drug monitoring (TDM) is based on the concept that a meaningful relationship exists between a drug’s plasma concentration and its concentration at the site of action. Clinicians can measure the drug’s plasma concentration relative to the presumed dosage a patient is taking.
When nonadherence is the reason for nonresponse to usual dosing, TDM measurements of drug concentration would be lower than expected—or nonexistent with complete nonadherence. Rapid clearance, however, can also cause lower-than-expected levels on a given dosage.
So, how can the clinician determine whether the problem is rapid clearance or noncompliance? One way is to repeat the plasma level after arranging for supervised dosing for at least five times the half-life of the drug being measured. A higher level on follow-up would indicate that noncompliance is the likely problem. If the repeat level remains low, then the problem is most likely rapid clearance. In the latter case, the patient would need a higher dosage to achieve the concentrations associated with response in clinical trials.
Although TDM’s results are often conceptualized as being relative to a therapeutic range, TDM is fundamentally a means of measuring a patient’s ability to clear the drug. If the dosing rate and plasma drug level are known, then the clinician can solve for clearance by rearranging Equation 2. Rather than formally solving for clearance, results can be considered as within, below, or above the expected range for the dosage given. The clinician can then adjust the dosage to compensate for clearance that is faster or slower than usual. Thus, TDM allows clinicians to individualize dosages, taking into account the biological variances (Equation 1) that affect a patient’s ability to clear a specific drug.
TDM has limitations. It cannot assess whether a genetic mutation may be altering a drug’s binding affinity at the receptor site or whether the drug is not reaching the target compartment because of an abnormality in distribution mechanisms. Those possibilities would need to be assessed by techniques not available to most clinicians today.
Many psychiatrists think the inability to show a correlation between plasma drug levels and response is a limitation of TDM. That is not a limitation of TDM as much as a reflection on clinical trials of psychiatric drugs. Many such trials fail because of poor “signal-to-noise ratio”—defined as the true specific response to the treatment versus either placebo response or nonresponse due to not having an illness that is responsive to the drug.
Consider instead that the usually effective dosage defines a usually expected plasma drug concentration range associated with response. Further discussion of this topic is beyond the scope of this commentary, but the interested reader is referred to articles at www.preskorn.com, including Clinical pharmacology of serotonin selective re-uptake inhibitors (chapter 5); the column, Understanding dose-response curves in psychiatry; and the discussion, Finding the signal through the noise.
Summary
Based on the review by Pierre et al, the evidence for high-dose atypical antipsychotics’ safety and tolerability is not encouraging. These authors found only a modest body of evidence, and most study designs were not rigorous enough to eliminate erroneous conclusions. My intent here is not to advocate the use of higher-than-recommended dosages but to explain reasons why the patient may not respond and to call for more research.
Investigators designing future studies of nonresponse could consider including procedures to first rule out noncompliance and then divide participants into two groups:
- patients who achieved usual plasma drug levels on the usual recommended dosages (normal clearance)
- those who achieved levels below the usual expected range, despite good compliance (rapid clearance).
These two groups could then be randomized to continued exposure to the usual dosing range or higher-than-usual dosing. Patients with rapid clearance would be predicted to have a greater response to higher-than-usual dosing, compared with those with usual clearance.
In the absence of such trials, the clinician should proceed cautiously—if at all—to use higher-than usual antipsychotic dosages in his or her patients. The prescriber must always consider whether the risks outweigh the potential benefits, taking into account:
- the drug’s therapeutic index
- evidence of safety and tolerability problems in the individual patient as the dosage is escalated.
Related resources
- Preskorn SH. The recommended dosage range: How is it established and why would it ever be exceeded? J Psychiatry Pract 2004;10(4):249-54.
1. Pierre JM, Wirshing DA, Wirshing WC. High-dose antipsychotic therapy: Desperation or data-driven. Current Psychiatry 2004;3(8):30-7.
2. Preskorn SH. Relating clinical trials to psychiatric practice: part I: the case of a 13-year-old on aripiprazole and fluoxetine. J Psychiatr Pract 2003;9(4):307-13. Also available at www.preskorn.com under Columns, Case studies.
3. Preskorn SH. Relating clinical trials to psychiatric practice: part II: the gap between the usual patient in registration trials and in practice. J Psychiatr Pract 2003;9(6):455-61. Also available at www.preskorn.com under Columns, Case studies.
Psychiatrists may consider using higher than usually recommended dosages of antipsychotics when faced with nonresponse. In this issue , Pierre et al1 carefully and thoughtfully discuss the pros and cons of this practice in patients with schizophrenia. Having reviewed that article, I thought CURRENT PSYCHIATRY’s readers might benefit from a theoretical framework for analyzing drug response.
‘Usual’ vs ‘unusual’ patients
A clinical trial for drug registration is, in essence, a population pharmacokinetic study whose goal is to determine the usual dosage for the usual patient in the trial. Many patients seen in clinical practice, such as those with treatment-refractory psychotic disorders, are typically excluded from registration trials. Thus, the usual registration trial patient may be an unusual patient in a clinician’s practice, and the trial’s usual dosage may not produce an adequate response for the clinician’s usual patient. How, then, might a clinician approach inadequate response, except by:
- blindly exceeding the usually recommended dosage
- switching among available drugs
- adding drugs to create a complex cocktail?
This commentary dissects why a patient might not benefit from the usual recommended dosage and how that could lead to different courses of action.
Equation 1. Three variables (Table) determine response to any drug:
- affinity for and intrinsic action on a regulatory protein (such as a receptor)
- concentration (amount of drug reaching the site of action)
- biological variance, which can shift an individual’s dose-response curve relative to that of the “usual” patient, making that individual more or less sensitive to the drug’s effects.2
Table
3 variables that determine patient response to any drug
| Equation 1 | ||||||
| Effect | = | Affinity for and intrinsic activity at a site of action | X | Drug concentration (see Equation 2) Absorption Distribution Metabolism Elimination (ADME) | X | Biological variance Genetics Age Disease Environment (internal) (GADE) |
| Equation 2 | ||||||
| Drug concentration = dosing rate/clearance | ||||||
Equation 2. Drug concentration is dosing rate divided by clearance in a given patient. Dosing rate and clearance are equally important in determining drug concentration—which, in turn, determines the site of action engaged, to what degree, and the patient’s response to the drug.
Causes of inadequate response. Nonadherence is a common cause of inadequate response. When a patient repeatedly misses doses or stops taking the drug, the true dosing rate is lower than the prescribed dosing rate, resulting in reduced drug concentration and effect.
Pierre and colleagues focus on the “unusual” patient who does not respond optimally to antipsychotic dosages established in registration trials.1 As in Equation 1, sources of biological variance—genetics, age, disease, and environment (internal)—may distinguish the treatment-refractory patient from the responsive patient. The mnemonic GADE captures these variables:
Genetic variation refers to mutations in regulatory proteins that:
- determine the drug’s action (such mutations may change the drug’s binding affinity, so that a higher concentration is needed to adequately engage the site)
- determine what drug concentration reaches the site of action (such as drug-metabolizing enzymes that regulate clearance, or transporter proteins that prevent or facilitate the drug’s ability to reach the site of action).
Age refers to physiologic changes (pharmacodynamic or pharmacokinetic) that make the patient more or less sensitive to the drug’s effects.
Disease refers to differences in organ function related to pathophysiology. Patients with the same clinical presentation (in this case, psychosis) may respond differently to the same drug because they have different underlying pathophysiologies (such as schizophrenic syndrome due to differing genetic causes or to toxins or slow viruses).
Environment (internal) refers to exogenous substances in the body—such as drugs and dietary substances—that can interact with and influence response to other drugs.
Nonpsychiatric disease also can alter response to medication. For example, impaired hepatic, renal, or cardiac function can impair drug clearance, leading to greater-than-usual accumulation. Such a patient can be “sensitive” to the drug and experience a greater effect than is usually seen with the dosage given.
Dosing for clinical effect
Psychiatrists commonly titrate dosages based on clinical assessment of response.3 The clinician increases the dosage if a patient does not improve and has no obvious rate-limiting adverse effects.
Perhaps without realizing it, the clinician is assuming that the dosage is inadequate for a given patient because the concentration is inadequate due to rapid clearance. Other reasons are possible, however, such as:
- the drug is not reaching the site of action a mutation at the site of action is altering
- the drug’s binding affinity
- the concentration may be too high, but the resulting adverse effects resemble worsening of the disease being treated. For example, akathisia due to dopamine-2 receptor blockade can present as agitation, and the clinician may increase the dosage when it should be decreased.
In the first two instances, escalating the dosage may be beneficial or cause toxicity. High levels in a peripheral compartment can cause adverse effects that may be silent until they become deadly (such as torsades de pointes). In the third instance, dosage escalation is the wrong step because the level is already too high.
Recommended dosage range
Principal goals of phase I studies in drug development are to establish the optimal dosage range and a maximum tolerated dosage. This upper limit is rarely, if ever, exceeded in later trials. Because phase I trial results are rarely published, the prescriber often does not know the rationale for a recommended dosing range’s upper limit.
Clinicians who escalate a drug’s dosage above the recommended range are using an n=1 paradigm, in which the patient is his or her own control. Unfortunately, treating one patient at a time cannot detect infrequent (much less rare) adverse events.
Using higher-than-recommended dosages thus exposes patients to unknown risks, with less monitoring than in a typical phase I trial in which subjects are confined to a research unit before, during, and after drug exposure. During the study, participants undergo serial ECGs, laboratory tests, and plasma drug level monitoring.
Therapeutic drug monitoring (TDM) is based on the concept that a meaningful relationship exists between a drug’s plasma concentration and its concentration at the site of action. Clinicians can measure the drug’s plasma concentration relative to the presumed dosage a patient is taking.
When nonadherence is the reason for nonresponse to usual dosing, TDM measurements of drug concentration would be lower than expected—or nonexistent with complete nonadherence. Rapid clearance, however, can also cause lower-than-expected levels on a given dosage.
So, how can the clinician determine whether the problem is rapid clearance or noncompliance? One way is to repeat the plasma level after arranging for supervised dosing for at least five times the half-life of the drug being measured. A higher level on follow-up would indicate that noncompliance is the likely problem. If the repeat level remains low, then the problem is most likely rapid clearance. In the latter case, the patient would need a higher dosage to achieve the concentrations associated with response in clinical trials.
Although TDM’s results are often conceptualized as being relative to a therapeutic range, TDM is fundamentally a means of measuring a patient’s ability to clear the drug. If the dosing rate and plasma drug level are known, then the clinician can solve for clearance by rearranging Equation 2. Rather than formally solving for clearance, results can be considered as within, below, or above the expected range for the dosage given. The clinician can then adjust the dosage to compensate for clearance that is faster or slower than usual. Thus, TDM allows clinicians to individualize dosages, taking into account the biological variances (Equation 1) that affect a patient’s ability to clear a specific drug.
TDM has limitations. It cannot assess whether a genetic mutation may be altering a drug’s binding affinity at the receptor site or whether the drug is not reaching the target compartment because of an abnormality in distribution mechanisms. Those possibilities would need to be assessed by techniques not available to most clinicians today.
Many psychiatrists think the inability to show a correlation between plasma drug levels and response is a limitation of TDM. That is not a limitation of TDM as much as a reflection on clinical trials of psychiatric drugs. Many such trials fail because of poor “signal-to-noise ratio”—defined as the true specific response to the treatment versus either placebo response or nonresponse due to not having an illness that is responsive to the drug.
Consider instead that the usually effective dosage defines a usually expected plasma drug concentration range associated with response. Further discussion of this topic is beyond the scope of this commentary, but the interested reader is referred to articles at www.preskorn.com, including Clinical pharmacology of serotonin selective re-uptake inhibitors (chapter 5); the column, Understanding dose-response curves in psychiatry; and the discussion, Finding the signal through the noise.
Summary
Based on the review by Pierre et al, the evidence for high-dose atypical antipsychotics’ safety and tolerability is not encouraging. These authors found only a modest body of evidence, and most study designs were not rigorous enough to eliminate erroneous conclusions. My intent here is not to advocate the use of higher-than-recommended dosages but to explain reasons why the patient may not respond and to call for more research.
Investigators designing future studies of nonresponse could consider including procedures to first rule out noncompliance and then divide participants into two groups:
- patients who achieved usual plasma drug levels on the usual recommended dosages (normal clearance)
- those who achieved levels below the usual expected range, despite good compliance (rapid clearance).
These two groups could then be randomized to continued exposure to the usual dosing range or higher-than-usual dosing. Patients with rapid clearance would be predicted to have a greater response to higher-than-usual dosing, compared with those with usual clearance.
In the absence of such trials, the clinician should proceed cautiously—if at all—to use higher-than usual antipsychotic dosages in his or her patients. The prescriber must always consider whether the risks outweigh the potential benefits, taking into account:
- the drug’s therapeutic index
- evidence of safety and tolerability problems in the individual patient as the dosage is escalated.
Related resources
- Preskorn SH. The recommended dosage range: How is it established and why would it ever be exceeded? J Psychiatry Pract 2004;10(4):249-54.
Psychiatrists may consider using higher than usually recommended dosages of antipsychotics when faced with nonresponse. In this issue , Pierre et al1 carefully and thoughtfully discuss the pros and cons of this practice in patients with schizophrenia. Having reviewed that article, I thought CURRENT PSYCHIATRY’s readers might benefit from a theoretical framework for analyzing drug response.
‘Usual’ vs ‘unusual’ patients
A clinical trial for drug registration is, in essence, a population pharmacokinetic study whose goal is to determine the usual dosage for the usual patient in the trial. Many patients seen in clinical practice, such as those with treatment-refractory psychotic disorders, are typically excluded from registration trials. Thus, the usual registration trial patient may be an unusual patient in a clinician’s practice, and the trial’s usual dosage may not produce an adequate response for the clinician’s usual patient. How, then, might a clinician approach inadequate response, except by:
- blindly exceeding the usually recommended dosage
- switching among available drugs
- adding drugs to create a complex cocktail?
This commentary dissects why a patient might not benefit from the usual recommended dosage and how that could lead to different courses of action.
Equation 1. Three variables (Table) determine response to any drug:
- affinity for and intrinsic action on a regulatory protein (such as a receptor)
- concentration (amount of drug reaching the site of action)
- biological variance, which can shift an individual’s dose-response curve relative to that of the “usual” patient, making that individual more or less sensitive to the drug’s effects.2
Table
3 variables that determine patient response to any drug
| Equation 1 | ||||||
| Effect | = | Affinity for and intrinsic activity at a site of action | X | Drug concentration (see Equation 2) Absorption Distribution Metabolism Elimination (ADME) | X | Biological variance Genetics Age Disease Environment (internal) (GADE) |
| Equation 2 | ||||||
| Drug concentration = dosing rate/clearance | ||||||
Equation 2. Drug concentration is dosing rate divided by clearance in a given patient. Dosing rate and clearance are equally important in determining drug concentration—which, in turn, determines the site of action engaged, to what degree, and the patient’s response to the drug.
Causes of inadequate response. Nonadherence is a common cause of inadequate response. When a patient repeatedly misses doses or stops taking the drug, the true dosing rate is lower than the prescribed dosing rate, resulting in reduced drug concentration and effect.
Pierre and colleagues focus on the “unusual” patient who does not respond optimally to antipsychotic dosages established in registration trials.1 As in Equation 1, sources of biological variance—genetics, age, disease, and environment (internal)—may distinguish the treatment-refractory patient from the responsive patient. The mnemonic GADE captures these variables:
Genetic variation refers to mutations in regulatory proteins that:
- determine the drug’s action (such mutations may change the drug’s binding affinity, so that a higher concentration is needed to adequately engage the site)
- determine what drug concentration reaches the site of action (such as drug-metabolizing enzymes that regulate clearance, or transporter proteins that prevent or facilitate the drug’s ability to reach the site of action).
Age refers to physiologic changes (pharmacodynamic or pharmacokinetic) that make the patient more or less sensitive to the drug’s effects.
Disease refers to differences in organ function related to pathophysiology. Patients with the same clinical presentation (in this case, psychosis) may respond differently to the same drug because they have different underlying pathophysiologies (such as schizophrenic syndrome due to differing genetic causes or to toxins or slow viruses).
Environment (internal) refers to exogenous substances in the body—such as drugs and dietary substances—that can interact with and influence response to other drugs.
Nonpsychiatric disease also can alter response to medication. For example, impaired hepatic, renal, or cardiac function can impair drug clearance, leading to greater-than-usual accumulation. Such a patient can be “sensitive” to the drug and experience a greater effect than is usually seen with the dosage given.
Dosing for clinical effect
Psychiatrists commonly titrate dosages based on clinical assessment of response.3 The clinician increases the dosage if a patient does not improve and has no obvious rate-limiting adverse effects.
Perhaps without realizing it, the clinician is assuming that the dosage is inadequate for a given patient because the concentration is inadequate due to rapid clearance. Other reasons are possible, however, such as:
- the drug is not reaching the site of action a mutation at the site of action is altering
- the drug’s binding affinity
- the concentration may be too high, but the resulting adverse effects resemble worsening of the disease being treated. For example, akathisia due to dopamine-2 receptor blockade can present as agitation, and the clinician may increase the dosage when it should be decreased.
In the first two instances, escalating the dosage may be beneficial or cause toxicity. High levels in a peripheral compartment can cause adverse effects that may be silent until they become deadly (such as torsades de pointes). In the third instance, dosage escalation is the wrong step because the level is already too high.
Recommended dosage range
Principal goals of phase I studies in drug development are to establish the optimal dosage range and a maximum tolerated dosage. This upper limit is rarely, if ever, exceeded in later trials. Because phase I trial results are rarely published, the prescriber often does not know the rationale for a recommended dosing range’s upper limit.
Clinicians who escalate a drug’s dosage above the recommended range are using an n=1 paradigm, in which the patient is his or her own control. Unfortunately, treating one patient at a time cannot detect infrequent (much less rare) adverse events.
Using higher-than-recommended dosages thus exposes patients to unknown risks, with less monitoring than in a typical phase I trial in which subjects are confined to a research unit before, during, and after drug exposure. During the study, participants undergo serial ECGs, laboratory tests, and plasma drug level monitoring.
Therapeutic drug monitoring (TDM) is based on the concept that a meaningful relationship exists between a drug’s plasma concentration and its concentration at the site of action. Clinicians can measure the drug’s plasma concentration relative to the presumed dosage a patient is taking.
When nonadherence is the reason for nonresponse to usual dosing, TDM measurements of drug concentration would be lower than expected—or nonexistent with complete nonadherence. Rapid clearance, however, can also cause lower-than-expected levels on a given dosage.
So, how can the clinician determine whether the problem is rapid clearance or noncompliance? One way is to repeat the plasma level after arranging for supervised dosing for at least five times the half-life of the drug being measured. A higher level on follow-up would indicate that noncompliance is the likely problem. If the repeat level remains low, then the problem is most likely rapid clearance. In the latter case, the patient would need a higher dosage to achieve the concentrations associated with response in clinical trials.
Although TDM’s results are often conceptualized as being relative to a therapeutic range, TDM is fundamentally a means of measuring a patient’s ability to clear the drug. If the dosing rate and plasma drug level are known, then the clinician can solve for clearance by rearranging Equation 2. Rather than formally solving for clearance, results can be considered as within, below, or above the expected range for the dosage given. The clinician can then adjust the dosage to compensate for clearance that is faster or slower than usual. Thus, TDM allows clinicians to individualize dosages, taking into account the biological variances (Equation 1) that affect a patient’s ability to clear a specific drug.
TDM has limitations. It cannot assess whether a genetic mutation may be altering a drug’s binding affinity at the receptor site or whether the drug is not reaching the target compartment because of an abnormality in distribution mechanisms. Those possibilities would need to be assessed by techniques not available to most clinicians today.
Many psychiatrists think the inability to show a correlation between plasma drug levels and response is a limitation of TDM. That is not a limitation of TDM as much as a reflection on clinical trials of psychiatric drugs. Many such trials fail because of poor “signal-to-noise ratio”—defined as the true specific response to the treatment versus either placebo response or nonresponse due to not having an illness that is responsive to the drug.
Consider instead that the usually effective dosage defines a usually expected plasma drug concentration range associated with response. Further discussion of this topic is beyond the scope of this commentary, but the interested reader is referred to articles at www.preskorn.com, including Clinical pharmacology of serotonin selective re-uptake inhibitors (chapter 5); the column, Understanding dose-response curves in psychiatry; and the discussion, Finding the signal through the noise.
Summary
Based on the review by Pierre et al, the evidence for high-dose atypical antipsychotics’ safety and tolerability is not encouraging. These authors found only a modest body of evidence, and most study designs were not rigorous enough to eliminate erroneous conclusions. My intent here is not to advocate the use of higher-than-recommended dosages but to explain reasons why the patient may not respond and to call for more research.
Investigators designing future studies of nonresponse could consider including procedures to first rule out noncompliance and then divide participants into two groups:
- patients who achieved usual plasma drug levels on the usual recommended dosages (normal clearance)
- those who achieved levels below the usual expected range, despite good compliance (rapid clearance).
These two groups could then be randomized to continued exposure to the usual dosing range or higher-than-usual dosing. Patients with rapid clearance would be predicted to have a greater response to higher-than-usual dosing, compared with those with usual clearance.
In the absence of such trials, the clinician should proceed cautiously—if at all—to use higher-than usual antipsychotic dosages in his or her patients. The prescriber must always consider whether the risks outweigh the potential benefits, taking into account:
- the drug’s therapeutic index
- evidence of safety and tolerability problems in the individual patient as the dosage is escalated.
Related resources
- Preskorn SH. The recommended dosage range: How is it established and why would it ever be exceeded? J Psychiatry Pract 2004;10(4):249-54.
1. Pierre JM, Wirshing DA, Wirshing WC. High-dose antipsychotic therapy: Desperation or data-driven. Current Psychiatry 2004;3(8):30-7.
2. Preskorn SH. Relating clinical trials to psychiatric practice: part I: the case of a 13-year-old on aripiprazole and fluoxetine. J Psychiatr Pract 2003;9(4):307-13. Also available at www.preskorn.com under Columns, Case studies.
3. Preskorn SH. Relating clinical trials to psychiatric practice: part II: the gap between the usual patient in registration trials and in practice. J Psychiatr Pract 2003;9(6):455-61. Also available at www.preskorn.com under Columns, Case studies.
1. Pierre JM, Wirshing DA, Wirshing WC. High-dose antipsychotic therapy: Desperation or data-driven. Current Psychiatry 2004;3(8):30-7.
2. Preskorn SH. Relating clinical trials to psychiatric practice: part I: the case of a 13-year-old on aripiprazole and fluoxetine. J Psychiatr Pract 2003;9(4):307-13. Also available at www.preskorn.com under Columns, Case studies.
3. Preskorn SH. Relating clinical trials to psychiatric practice: part II: the gap between the usual patient in registration trials and in practice. J Psychiatr Pract 2003;9(6):455-61. Also available at www.preskorn.com under Columns, Case studies.
Understanding the ‘joy’ of aggression
One of the most perplexing and shocking aspects of human behavior is the cruelty we can inflict on one another. Infamous events such as the Spanish Inquisition, slavery, lynching, conquest of the native Americans, and Nazi concentration camps are so sickening that many of us can barely tolerate hearing about them. How can people behave this way?
Recently, we’ve been shocked by photographs from the Abu Ghraib military prison in Iraq. American soldiers with unremarkable backgrounds—not Saddam’s henchmen—were shown humiliating and abusing Iraqi prisoners. Most remarkable was the joy on the Americans’ faces (Figure 1).
‘Good’ people, ‘bad’ circumstances
In experiments with college students, psychologists have shown that “regular” people can become sadistic under the right (or wrong) circumstances.1 One explanation is that it’s not just psychopaths who perpetrate crimes against humanity. Somehow, the psychopath within us all becomes unleashed.
Artificial situations such as the Stanford Prison Experiment (www.prisonexp.org) show that normal people can dissolve into cruelty but don’t explain why. A recent neuroscience experiment suggests a mechanism for this kind of aggression.
Figure 1 Deriving pleasure from abuse?

American soldiers appearing to enjoy themselves as they humiliate Iraqi prisoners in the Abu Ghraib military prison.
Source: Reprinted with permission of The New Yorker, which first published this photo.
Dopamine and aggression
The nucleus accumbens has been called the brain’s “pleasure center,” and dopamine is the neurotransmitter that activates it.2 Activities and substances that stimulate dopamine release include sex, gambling, and smoking as well as cocaine and alcohol. The good feeling a person gets from these activities/substances reinforces the behavior that produced the feeling. In some cases, problems develop when people cannot resist the urge for more.
Ferrari et al3 placed micropipettes in rats’ nucleus accumbens to measure extracellular dopamine before, during, and after an aggressive confrontation. When the rats were confronted with an intruder rat for 10 minutes, they attacked and bit the intruder an average of 5 times, despite being implanted, tethered, and sampled. During and after the fight, dopamine was increased in the rats’ nucleus accumbens (Figure 2). Clearly, fighting gave them a “squirt” of pleasure that lasted almost 2 hours.
If we can extrapolate from this study to humans, we may understand why people become aggressive. At some level, they enjoy it. The bully on the playground, the wife beater, the mean boss—they get pleasure from being aggressive. It’s not just serial killers.
It is important to acknowledge that other variables such as poor supervision and too much power affected the actions of American soldiers working as prison guards in Iraq. However, the neuroscientific studies show us that aggression can be pleasurable, and people often have a hard time resisting what feels good. This knowledge may help us treat war veterans struggling not only with traumatic memories of violence but also with socially and personally unacceptable feelings of pleasure.
Figure 2 A ‘squirt’ of dopamine during violence
A 10-minute fight increased extracellular dopamine levels in rats’ nucleus accumbens for approximately 2 hours, suggesting that aggressive behavior produced pleasure.
Source: Adapted and reprinted with permission from reference 3. Copyright 2003, Blackwell Publishing.
1. Shermer M. The science of good and evil: Why people cheat, gossip, care, share, and follow the Golden Rule. New York: Times Books, 2004.
2. Ikemoto S, Panksepp J. The role of nucleus accumbens dopamine in motivated behavior: a unifying interpretation with special reference to reward-seeking. Brain Res Brain Res Rev 1999;31(1):6-41.
3. Ferrari PF, van Erp AM, Tornatzky W, Miczek KA. Accumbal dopamine and serotonin in anticipation of the next aggressive episode in rats. Eur J Neurosci 2003;17:371-8.
One of the most perplexing and shocking aspects of human behavior is the cruelty we can inflict on one another. Infamous events such as the Spanish Inquisition, slavery, lynching, conquest of the native Americans, and Nazi concentration camps are so sickening that many of us can barely tolerate hearing about them. How can people behave this way?
Recently, we’ve been shocked by photographs from the Abu Ghraib military prison in Iraq. American soldiers with unremarkable backgrounds—not Saddam’s henchmen—were shown humiliating and abusing Iraqi prisoners. Most remarkable was the joy on the Americans’ faces (Figure 1).
‘Good’ people, ‘bad’ circumstances
In experiments with college students, psychologists have shown that “regular” people can become sadistic under the right (or wrong) circumstances.1 One explanation is that it’s not just psychopaths who perpetrate crimes against humanity. Somehow, the psychopath within us all becomes unleashed.
Artificial situations such as the Stanford Prison Experiment (www.prisonexp.org) show that normal people can dissolve into cruelty but don’t explain why. A recent neuroscience experiment suggests a mechanism for this kind of aggression.
Figure 1 Deriving pleasure from abuse?
American soldiers appearing to enjoy themselves as they humiliate Iraqi prisoners in the Abu Ghraib military prison.
Source: Reprinted with permission of The New Yorker, which first published this photo.
Dopamine and aggression
The nucleus accumbens has been called the brain’s “pleasure center,” and dopamine is the neurotransmitter that activates it.2 Activities and substances that stimulate dopamine release include sex, gambling, and smoking as well as cocaine and alcohol. The good feeling a person gets from these activities/substances reinforces the behavior that produced the feeling. In some cases, problems develop when people cannot resist the urge for more.
Ferrari et al3 placed micropipettes in rats’ nucleus accumbens to measure extracellular dopamine before, during, and after an aggressive confrontation. When the rats were confronted with an intruder rat for 10 minutes, they attacked and bit the intruder an average of 5 times, despite being implanted, tethered, and sampled. During and after the fight, dopamine was increased in the rats’ nucleus accumbens (Figure 2). Clearly, fighting gave them a “squirt” of pleasure that lasted almost 2 hours.
If we can extrapolate from this study to humans, we may understand why people become aggressive. At some level, they enjoy it. The bully on the playground, the wife beater, the mean boss—they get pleasure from being aggressive. It’s not just serial killers.
It is important to acknowledge that other variables such as poor supervision and too much power affected the actions of American soldiers working as prison guards in Iraq. However, the neuroscientific studies show us that aggression can be pleasurable, and people often have a hard time resisting what feels good. This knowledge may help us treat war veterans struggling not only with traumatic memories of violence but also with socially and personally unacceptable feelings of pleasure.
Figure 2 A ‘squirt’ of dopamine during violence
A 10-minute fight increased extracellular dopamine levels in rats’ nucleus accumbens for approximately 2 hours, suggesting that aggressive behavior produced pleasure.
Source: Adapted and reprinted with permission from reference 3. Copyright 2003, Blackwell Publishing.
One of the most perplexing and shocking aspects of human behavior is the cruelty we can inflict on one another. Infamous events such as the Spanish Inquisition, slavery, lynching, conquest of the native Americans, and Nazi concentration camps are so sickening that many of us can barely tolerate hearing about them. How can people behave this way?
Recently, we’ve been shocked by photographs from the Abu Ghraib military prison in Iraq. American soldiers with unremarkable backgrounds—not Saddam’s henchmen—were shown humiliating and abusing Iraqi prisoners. Most remarkable was the joy on the Americans’ faces (Figure 1).
‘Good’ people, ‘bad’ circumstances
In experiments with college students, psychologists have shown that “regular” people can become sadistic under the right (or wrong) circumstances.1 One explanation is that it’s not just psychopaths who perpetrate crimes against humanity. Somehow, the psychopath within us all becomes unleashed.
Artificial situations such as the Stanford Prison Experiment (www.prisonexp.org) show that normal people can dissolve into cruelty but don’t explain why. A recent neuroscience experiment suggests a mechanism for this kind of aggression.
Figure 1 Deriving pleasure from abuse?
American soldiers appearing to enjoy themselves as they humiliate Iraqi prisoners in the Abu Ghraib military prison.
Source: Reprinted with permission of The New Yorker, which first published this photo.
Dopamine and aggression
The nucleus accumbens has been called the brain’s “pleasure center,” and dopamine is the neurotransmitter that activates it.2 Activities and substances that stimulate dopamine release include sex, gambling, and smoking as well as cocaine and alcohol. The good feeling a person gets from these activities/substances reinforces the behavior that produced the feeling. In some cases, problems develop when people cannot resist the urge for more.
Ferrari et al3 placed micropipettes in rats’ nucleus accumbens to measure extracellular dopamine before, during, and after an aggressive confrontation. When the rats were confronted with an intruder rat for 10 minutes, they attacked and bit the intruder an average of 5 times, despite being implanted, tethered, and sampled. During and after the fight, dopamine was increased in the rats’ nucleus accumbens (Figure 2). Clearly, fighting gave them a “squirt” of pleasure that lasted almost 2 hours.
If we can extrapolate from this study to humans, we may understand why people become aggressive. At some level, they enjoy it. The bully on the playground, the wife beater, the mean boss—they get pleasure from being aggressive. It’s not just serial killers.
It is important to acknowledge that other variables such as poor supervision and too much power affected the actions of American soldiers working as prison guards in Iraq. However, the neuroscientific studies show us that aggression can be pleasurable, and people often have a hard time resisting what feels good. This knowledge may help us treat war veterans struggling not only with traumatic memories of violence but also with socially and personally unacceptable feelings of pleasure.
Figure 2 A ‘squirt’ of dopamine during violence
A 10-minute fight increased extracellular dopamine levels in rats’ nucleus accumbens for approximately 2 hours, suggesting that aggressive behavior produced pleasure.
Source: Adapted and reprinted with permission from reference 3. Copyright 2003, Blackwell Publishing.
1. Shermer M. The science of good and evil: Why people cheat, gossip, care, share, and follow the Golden Rule. New York: Times Books, 2004.
2. Ikemoto S, Panksepp J. The role of nucleus accumbens dopamine in motivated behavior: a unifying interpretation with special reference to reward-seeking. Brain Res Brain Res Rev 1999;31(1):6-41.
3. Ferrari PF, van Erp AM, Tornatzky W, Miczek KA. Accumbal dopamine and serotonin in anticipation of the next aggressive episode in rats. Eur J Neurosci 2003;17:371-8.
1. Shermer M. The science of good and evil: Why people cheat, gossip, care, share, and follow the Golden Rule. New York: Times Books, 2004.
2. Ikemoto S, Panksepp J. The role of nucleus accumbens dopamine in motivated behavior: a unifying interpretation with special reference to reward-seeking. Brain Res Brain Res Rev 1999;31(1):6-41.
3. Ferrari PF, van Erp AM, Tornatzky W, Miczek KA. Accumbal dopamine and serotonin in anticipation of the next aggressive episode in rats. Eur J Neurosci 2003;17:371-8.
Treating depression, chronic pain
We read with interest Dr. Nelson’s and Dr. Krahn’s article on treating chronic pain and comorbid major depression (Current Psychiatry, May 2004).
We treat many patients who present with depression and chronic pain—often as a partial cause of their depression. The article’s recommendations will be most useful.
We have found that two agents—lamotrigine and mirtazapine—have been particularly helpful. The authors, however, did not mention these agents or only briefly referred to them.
Lamotrigine, although not FDA-approved for these uses, has demonstrated efficacy in unipolar depression1 and chronic pain.2 Although the medication has not been studied for treating comorbid depression and chronic pain, we can attest to its usefulness for such patients.
Mirtazapine is FDA-approved for depression and has been compared favorably with selective serotonin reuptake inhibitors3-4 or venlafaxine.5 Fewer data support using mirtazapine for chronic pain, but its sedating effects make it an option for treating any syndrome associated with sleep disturbance—ie, major depression and chronic pain.
Vida Robertson, MD
Michael S. Wilson, II, MD
Department of psychiatry
Louisiana State University Health Sciences Center
New Orleans
- Barbosa L, Berk M, Vorster M. A double-blind, randomized, placebo-controlled trial of augmentation with lamotrigine or placebo in patients concomitantly treated with fluoxetine for resistant major depressive episodes. J Clin Psychiatry 2003;64(4):403–7.
- Eisenberg E, Lurie Y, Braker C, et al. Lamotrigine reduces painful diabetic neuropathy: a randomized, controlled study. Neurology 2001;57(3):505–9.
- Winokur A. DeMartinis NA 3rd, McNally DP, et al. Comparative effects of mirtazapine and fluoxetine on sleep physiology measures in patients with major depression and insomnia. J Clin Psychiatry 2003;64(10):1224–9.
- Wade A, Crawford GM, Angus M, et al. A randomized, double-blind, 24-week study comparing the efficacy and tolerability of mirtazapine and paroxetine in depressed patients in primary care. Int Clin Psychopharmacol 2003;18(3):133–41.
- Guelfi JD, Ansseau M, Timmerman L, Korsgaard S; Mirtazapine-Venlafaxine Study Group. Mirtazapine versus venlafaxine in hospitalized severely depressed patients with melancholic features. J Clin Psychopharmacol 2001;21(4):425–31.
We read with interest Dr. Nelson’s and Dr. Krahn’s article on treating chronic pain and comorbid major depression (Current Psychiatry, May 2004).
We treat many patients who present with depression and chronic pain—often as a partial cause of their depression. The article’s recommendations will be most useful.
We have found that two agents—lamotrigine and mirtazapine—have been particularly helpful. The authors, however, did not mention these agents or only briefly referred to them.
Lamotrigine, although not FDA-approved for these uses, has demonstrated efficacy in unipolar depression1 and chronic pain.2 Although the medication has not been studied for treating comorbid depression and chronic pain, we can attest to its usefulness for such patients.
Mirtazapine is FDA-approved for depression and has been compared favorably with selective serotonin reuptake inhibitors3-4 or venlafaxine.5 Fewer data support using mirtazapine for chronic pain, but its sedating effects make it an option for treating any syndrome associated with sleep disturbance—ie, major depression and chronic pain.
Vida Robertson, MD
Michael S. Wilson, II, MD
Department of psychiatry
Louisiana State University Health Sciences Center
New Orleans
- Barbosa L, Berk M, Vorster M. A double-blind, randomized, placebo-controlled trial of augmentation with lamotrigine or placebo in patients concomitantly treated with fluoxetine for resistant major depressive episodes. J Clin Psychiatry 2003;64(4):403–7.
- Eisenberg E, Lurie Y, Braker C, et al. Lamotrigine reduces painful diabetic neuropathy: a randomized, controlled study. Neurology 2001;57(3):505–9.
- Winokur A. DeMartinis NA 3rd, McNally DP, et al. Comparative effects of mirtazapine and fluoxetine on sleep physiology measures in patients with major depression and insomnia. J Clin Psychiatry 2003;64(10):1224–9.
- Wade A, Crawford GM, Angus M, et al. A randomized, double-blind, 24-week study comparing the efficacy and tolerability of mirtazapine and paroxetine in depressed patients in primary care. Int Clin Psychopharmacol 2003;18(3):133–41.
- Guelfi JD, Ansseau M, Timmerman L, Korsgaard S; Mirtazapine-Venlafaxine Study Group. Mirtazapine versus venlafaxine in hospitalized severely depressed patients with melancholic features. J Clin Psychopharmacol 2001;21(4):425–31.
We read with interest Dr. Nelson’s and Dr. Krahn’s article on treating chronic pain and comorbid major depression (Current Psychiatry, May 2004).
We treat many patients who present with depression and chronic pain—often as a partial cause of their depression. The article’s recommendations will be most useful.
We have found that two agents—lamotrigine and mirtazapine—have been particularly helpful. The authors, however, did not mention these agents or only briefly referred to them.
Lamotrigine, although not FDA-approved for these uses, has demonstrated efficacy in unipolar depression1 and chronic pain.2 Although the medication has not been studied for treating comorbid depression and chronic pain, we can attest to its usefulness for such patients.
Mirtazapine is FDA-approved for depression and has been compared favorably with selective serotonin reuptake inhibitors3-4 or venlafaxine.5 Fewer data support using mirtazapine for chronic pain, but its sedating effects make it an option for treating any syndrome associated with sleep disturbance—ie, major depression and chronic pain.
Vida Robertson, MD
Michael S. Wilson, II, MD
Department of psychiatry
Louisiana State University Health Sciences Center
New Orleans
- Barbosa L, Berk M, Vorster M. A double-blind, randomized, placebo-controlled trial of augmentation with lamotrigine or placebo in patients concomitantly treated with fluoxetine for resistant major depressive episodes. J Clin Psychiatry 2003;64(4):403–7.
- Eisenberg E, Lurie Y, Braker C, et al. Lamotrigine reduces painful diabetic neuropathy: a randomized, controlled study. Neurology 2001;57(3):505–9.
- Winokur A. DeMartinis NA 3rd, McNally DP, et al. Comparative effects of mirtazapine and fluoxetine on sleep physiology measures in patients with major depression and insomnia. J Clin Psychiatry 2003;64(10):1224–9.
- Wade A, Crawford GM, Angus M, et al. A randomized, double-blind, 24-week study comparing the efficacy and tolerability of mirtazapine and paroxetine in depressed patients in primary care. Int Clin Psychopharmacol 2003;18(3):133–41.
- Guelfi JD, Ansseau M, Timmerman L, Korsgaard S; Mirtazapine-Venlafaxine Study Group. Mirtazapine versus venlafaxine in hospitalized severely depressed patients with melancholic features. J Clin Psychopharmacol 2001;21(4):425–31.
Treating tardive dyskinesia
“Tardive Dyskinesia: How to prevent and treat a lingering nemesis” (Current Psychiatry, October 2003) was a very good, basic article. The algorithm on managing tardive dyskinesia was particularly helpful, and the information on possible reversible dyskinesias with mood stabilizers and with antihistamines such as Benadryl was a useful refresher.
For MDs such as I who practice in rural clinics, however, more-specific dosage information would be useful—even for experimental agents such as tetrabenazine—since we do not have ready access to higher-level movement disorder clinics. The nearest such clinic to my practice is 2 1/2 hours away, an impossible commute for many of my patients.
Sophia Bezirganian MD
Trumansburg, NY
“Tardive Dyskinesia: How to prevent and treat a lingering nemesis” (Current Psychiatry, October 2003) was a very good, basic article. The algorithm on managing tardive dyskinesia was particularly helpful, and the information on possible reversible dyskinesias with mood stabilizers and with antihistamines such as Benadryl was a useful refresher.
For MDs such as I who practice in rural clinics, however, more-specific dosage information would be useful—even for experimental agents such as tetrabenazine—since we do not have ready access to higher-level movement disorder clinics. The nearest such clinic to my practice is 2 1/2 hours away, an impossible commute for many of my patients.
Sophia Bezirganian MD
Trumansburg, NY
“Tardive Dyskinesia: How to prevent and treat a lingering nemesis” (Current Psychiatry, October 2003) was a very good, basic article. The algorithm on managing tardive dyskinesia was particularly helpful, and the information on possible reversible dyskinesias with mood stabilizers and with antihistamines such as Benadryl was a useful refresher.
For MDs such as I who practice in rural clinics, however, more-specific dosage information would be useful—even for experimental agents such as tetrabenazine—since we do not have ready access to higher-level movement disorder clinics. The nearest such clinic to my practice is 2 1/2 hours away, an impossible commute for many of my patients.
Sophia Bezirganian MD
Trumansburg, NY
New drugs: The whole story

Dr John Battaglia’s article about intramuscular (IM) olanzapine (Out of the Pipeline, Current Psychiatry, May 2004) appears biased because of his pharmaceutical company connection. He mentions four studies supporting its use in treating schizophrenia, bipolar type I mania, and dementia.
As a resident eager to learn what constitutes good clinical care, I feel the article does an injustice by mentioning no negative studies or those that recorded no significant change.
Getting all the facts is key to establishing how to best use a treatment. Debate or unbiased commentary should accompany articles on new medications/treatments.
Matthew Sager, MD
Chief resident
Brown University Psychiatry Program
Providence, RI
Dr. Battaglia responds
I appreciate the passion with which Dr. Sager is approaching his education; he might want to learn more about drug development.
For decades, the overwhelming majority of FDA approvals for psychiatric medications have resulted from industry-supported studies. Very few researchers are doing substantial psychopharmacology clinical trials without industry support. It is extremely difficult to publish “negative” studies or those that show “no significant change.” I am not aware of any such published studies with IM olanzapine.
The best “unbiased” commentary on IM olanzapine will occur when it is used widely in clinical practice. For now, we are limited to published studies.
John Battaglia, MD
Medical director, Meriter Hospital adult psychiatry program
Associate professor, department of psychiatry
University of Wisconsin Medical School
Madison
Dr John Battaglia’s article about intramuscular (IM) olanzapine (Out of the Pipeline, Current Psychiatry, May 2004) appears biased because of his pharmaceutical company connection. He mentions four studies supporting its use in treating schizophrenia, bipolar type I mania, and dementia.
As a resident eager to learn what constitutes good clinical care, I feel the article does an injustice by mentioning no negative studies or those that recorded no significant change.
Getting all the facts is key to establishing how to best use a treatment. Debate or unbiased commentary should accompany articles on new medications/treatments.
Matthew Sager, MD
Chief resident
Brown University Psychiatry Program
Providence, RI
Dr. Battaglia responds
I appreciate the passion with which Dr. Sager is approaching his education; he might want to learn more about drug development.
For decades, the overwhelming majority of FDA approvals for psychiatric medications have resulted from industry-supported studies. Very few researchers are doing substantial psychopharmacology clinical trials without industry support. It is extremely difficult to publish “negative” studies or those that show “no significant change.” I am not aware of any such published studies with IM olanzapine.
The best “unbiased” commentary on IM olanzapine will occur when it is used widely in clinical practice. For now, we are limited to published studies.
John Battaglia, MD
Medical director, Meriter Hospital adult psychiatry program
Associate professor, department of psychiatry
University of Wisconsin Medical School
Madison
Dr John Battaglia’s article about intramuscular (IM) olanzapine (Out of the Pipeline, Current Psychiatry, May 2004) appears biased because of his pharmaceutical company connection. He mentions four studies supporting its use in treating schizophrenia, bipolar type I mania, and dementia.
As a resident eager to learn what constitutes good clinical care, I feel the article does an injustice by mentioning no negative studies or those that recorded no significant change.
Getting all the facts is key to establishing how to best use a treatment. Debate or unbiased commentary should accompany articles on new medications/treatments.
Matthew Sager, MD
Chief resident
Brown University Psychiatry Program
Providence, RI
Dr. Battaglia responds
I appreciate the passion with which Dr. Sager is approaching his education; he might want to learn more about drug development.
For decades, the overwhelming majority of FDA approvals for psychiatric medications have resulted from industry-supported studies. Very few researchers are doing substantial psychopharmacology clinical trials without industry support. It is extremely difficult to publish “negative” studies or those that show “no significant change.” I am not aware of any such published studies with IM olanzapine.
The best “unbiased” commentary on IM olanzapine will occur when it is used widely in clinical practice. For now, we are limited to published studies.
John Battaglia, MD
Medical director, Meriter Hospital adult psychiatry program
Associate professor, department of psychiatry
University of Wisconsin Medical School
Madison
Are psychostimulants useful in pervasive developmental disorders?
Psychostimulants benefit many patients with attention-deficit/hyperactivity disorder (ADHD)1 and thus might seem a logical choice to manage hyperactivity and inattention in youths with a pervasive developmental disorder (PDD). Some PDD patients do respond to psychostimulant therapy, but others worsen—and side effects are common.
Youths with PDDs often exhibit maladaptive behaviors—aggression, self-injury, irritability, hyperactivity, inattention—with repetitive activity patterns and fundamentally impaired social interaction and communication.2 To help you treat youths with PDD, we draw on the evidence, clinical experience, and our research to suggest psychostimulants’ role in a multimodal approach.
Targeting hyperactivity and inattentions
Step 1. Our approach begins with behavioral therapy (Figure), which includes identifying situations that trigger maladaptive behavior and environments that yield optimum behavior. The therapist assesses the child’s baseline attention and works with him or her to gradually increase it, using reinforcement and visual token boards.
Algorithm Suggested approach to hyperactivity and/or inattention in patients with PDDs
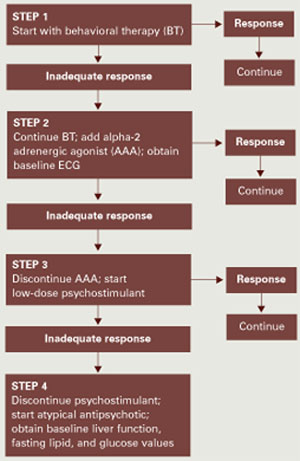
To set limits and expectations, the therapist introduces structure such as designating work and break areas and using visual schedules and timers to indicate activity duration. Minimizing distractions and understanding the child’s sensory needs may increase motivation and attention. Initially, the therapist allows numerous breaks and then may slowly decrease them as the child progresses. Tailoring work and play materials to the child’s interests can also help increase attention.
Step 2. Many patients will not respond to behavior therapy alone and will require added drug therapy. Based on evidence, we suggest starting with an alpha-2 adrenergic agonist. Guanfacine may be considered the drug of choice because of clonidine’s higher risk of adverse effects, such as hypotension and sedation. Obtain a baseline ECG with either agent, as clonidine has been associated with rare cardiovascular events.
Clonidine. Two small studies showed that clonidine may be of some benefit to patients with PDDs:
- Results were mixed in a 6-week, double-blind, placebo-controlled, crossover study of clonidine (4 to 10 μg/kg/d) in 8 autistic children ages 5 to 13.3 Teacher and parent rating instruments reflected significantly improved hyperactivity, irritability, and oppositional behavior. Clinician ratings, however, showed no significant difference between clonidine and placebo. Adverse effects with clonidine included hypotension, sedation, and decreased activity.
- In a 4-week, double-blind, placebo-con-trolled, crossover study of transdermal clonidine (0.16 to 0.48 mg/kg/d; mean: 3.6 μg/kg/d), clinician ratings showed significantly decreased hyperactivity, impulsivity, and anxiety in 9 autistic males ages 5 to 33. Sedation and fatigue were common adverse effects.
Guanfacine. In a recent retrospective review,5 we examined outcomes of 80 PDD patients ages 3 to 18 who received guanfacine (0.25 to 9 mg/d; mean: 2.6). Hyperactivity, inattention, and tics decreased in 19 patients (24%) treated for a mean 10 months.
Step 3. If clonidine or guanfacine fails to reduce hyperactivity and inattention, discontinue it and consider a psychostimulant trial.
Because psychostimulants’ efficacy in PDDs remains inconclusive, we suggest beginning with a low dosage and carefully monitoring the patient for worsening target symptoms and activation, such as emerging aggression or irritability.
Step 4. If hyperactivity and inattention remain prominent and treatment-refractory, we suggest that you discontinue the stimulant and consider an atypical antipsychotic trial. With the atypicals, monitor patients closely for adverse effects, including weight gain, extrapyramidal symptoms, and tardive dyskinesia. Fasting serum glucose and lipid profiles and liver function tests are recommended at least every 6 months and more often in individuals at risk for diabetes or hepatic disease.
Two studies provide evidence of atypicals’ efficacy in PDDs:
- In a 6-week open-label comparison,6 olanzapine significantly reduced hyperactivity and anger or uncooperativeness in 12 children with autistic disorder, but haloperidol did not. Average weight gain was 9 lbs in patients receiving olanzapine vs 3.2 lbs in those receiving haloperidol.
- An 8-week, double-blind study7 compared risperidone (0.5 to 3.5 mg/d; mean: 1.8) with placebo in 101 children and adolescents with autistic disorder. Response rates were 69% in the risperidone group and 12% in the control group. Risperidone reduced hyperactivity, aggression, agitation, and repetitive behavior. Adverse drug effects included weight gain (2.7 kg vs. 0.8 kg with placebo), increased appetite, and sedation.
Psychostimulant use in PDDs
Evidence is conflicting on psychostimulant use in patients with PDDs (Table). Early reviews suggested that stimulants were ineffective in PDDs and associated with adverse effects.8,9 Some preliminary studies supported that view, but recent reports have been mixed.
Dextroamphetamine. Campbell et al10 published a placebo-controlled study comparing triiodothyronine and dextroamphetamine (mean dosage, 4.8 mg/d; range 1.25 to 10 mg/d) in 16 children ages 3 to 6 (mean, 4.3 years) with diagnoses of autism, schizophrenia, and organic brain syndrome. All diagnostic groups worsened clinically with dextroamphetamine, and adverse effects—hyperactivity, worsened stereotypy, irritability, and decreased appetite—were common.
A subsequent case report11 found dex-troamphetamine effective when 2 patients ages 9 and 12 with PDD were treated with 10 and 5 mg/d, respectively. Hyperactivity, inattention, and impulsivity improved in both patients, and core PDD features did not worsen.
Levoamphetamine. In an 8-week, double-blind, crossover comparison with levodopa,12 levoamphetamine, 3.5 to 42 mg/d (mean, 13.4), worsened symptoms in 12 children ages 3 to 12 who had schizophrenia with autistic features. stereotypy emerged or increased in 9 of the 11 patients (82%) available for follow-up, and levoamphetamine was poorly tolerated.
Methylphenidate. In an early report, methylphenidate decreased hyperactivity and impulsivity in 9 of 15 children (60%) ages 2 to 13 with infantile autism.13 Dosages of 5 to 10 mg/d or 0.3 to 1 mg/kg/d were given for 2 to 60 weeks (mean, 26). Adverse effects included irritability, insomnia, and anorexia.
Table
Selected reports of stimulant use in pervasive developmental disorders
| Medication | Type of report | Dosage (mg/d); duration | Outcome | Adverse effects |
|---|---|---|---|---|
| Dextroamphetamine | Placebo-controlled10 (N=16) Case report11 (N=2) | Mean 4.8; N/A Mean 7.5; N/A | Clinical worsening Improved hyperactivity,inattention,impulsivity | Hyperactivity, irritability, decreased appetite, worsened stereotypy N/A |
| Levoamphetamine | Double-blind12 (N=12) | Mean 13.4 | Clinical worsening | Stereotypy emerged or worsened |
| Methylphenidate | Retrospective13 (N=15) Open-label14 (N=9) Case report15 (N=1) Double-blind, placebo-controlled, crossover16 (N=10) Double-blind, placebo-controlled, crossover17 (N=13) | 5 to 10; 26 weeks 10 to 50; 2 weeks 20; 4 weeks 20 mg/d for 2 weeks, 40 mg/d for 2 weeks 0.3 mg/kg and 0.6 mg/kg | Improved hyperactivity, impulsivity Improved hyperactivity Improved hyperactivity, concentration Modest benefit over placebo Improved hyperactivity, inattention | Irritability, insomnia, anorexia Initial mild insomnia Dysphoria, angry outbursts Statistically similar to placebo Social withdrawal, irritability |
| Methylphenidate, levoamphetamine, dextroamphetamine, or pemoline | Retrospective18 (N=195) | Various dosages, durations | Patients with, Asperger’s disorder were significantly more likely to respond | Agitation, dysphoria, irritability |
| N/A: not available | ||||
A subsequent open-label study and a case report also indicated that methylphenidate improved hyperactivity in patients with autistic disorder:
- In the 2-week, open-label study,14 9 patients ages 4 to 16 received methylphenidate, 10 to 50 mg/d. Two patients also received haloperidol, 4 and 5 mg/d. Hyperactivity improved significantly, as measured by the Conners Teacher Questionnaire.
- In the case report,15 one child, age 6, was. treated with methylphenidate, 10 mg bid, for 31 days. The drug significantly alleviated hyperactivity and improved concentration. Adverse effects included dysphoria and outbursts of anger.
Atomoxetine—a nonstimulant, selective norepinephrine reuptake inhibitor—has been approved to treat hyperactivity and inattention in ADHD, but no evidence has been published on its use in PDDs. A study of desipramine19 —also a norepinephrine reuptake inhibitor—may offer some insight into the possible efficacy and tolerability of atomoxetine in PDDs.
Desipramine (mean, 127 mg/d) was compared with the serotonin reuptake inhibitor clomipramine (mean, 153 mg/d) in a 10-week, double-blind, crossover study of 24 autistic patients ages 6 to 23. The agents were equally effective and superior to placebo in decreasing hyperactivity, although desipramine was associated with increased aggression and irritability.
Despite these results with desipramine, research is needed to understand atomoxetine’s potential role in treating hyperactivity and inattention in youths with PDDs.
Controlled trials. These early reports were followed by two double-blind, placebo-controlled, crossover studies of methylphenidate in children with autistic disorder.
- In the first trial,16 methylphenidate, 10 or 20 mg/d, improved irritability and hyperactivity in 10 children ages 7 to 11 but was only modestly more beneficial than placebo. Side-effect incidence—including decreased appetite, irritability, and insomnia—was similar during active and placebo treatments. Two patients required adjunctive haloperidol for prevailing behavioral problems.
- In the second trial,17 8 of 13 children (62%) ages 5 to 11 responded to methylphenidate, 0.3 and 0.6 mg/kg per dose. Hyperactivity and inattention improved significantly, as measured by a minimum 50% decrease in Conners Hyperactivity Index score. Ratings of stereotypy and inappropriate speech also decreased, but no changes were seen in the Child Autism Rating Scale. Adverse effects, which were more common with the 0.6 mg/kg dose, included social withdrawal and irritability.
Retrospective trial. Our group recently completed a retrospective study of 195 youth (mean age, 7.3 years; range, 2 to 19 years) with PDDs treated with a stimulant medication.18 As a whole, stimulants appeared ineffective.
Analysis of response by PDD subtype found that individuals with Asperger’s disorder—in contrast to those with autistic disorder or PDD not otherwise specified—were significantly more likely to respond to a stimulant medication. Gender, intelligence quotient (IQ), type of stimulant, and dosage did not significantly affect response. Adverse effects—including agitation, dysphoria, and irritability—occurred in 57.5% of the trials.
Atomoxetine. This nonstimulant medication has been approved for treating ADHD. However, research is needed to understand its use in patients with PDDs (Box)19
Summary. These mixed findings—combined with anecdotal reports from physicians describing the onset or exacerbation of hyperactivity, irritability, and aggression—indicate that much more evidence is needed regarding psychostimulant use in patients with PDDs.
To help meet this need, the National Institutes of Mental Health’s Research Units on Pediatric Psychopharmacology (RUPP) autism network recently completed a large, double-blind, placebo-controlled study to investigate methylphenidate’s efficacy and tolerability in PDDs. It is anticipated that the results will help us discern whether factors such as PDD subtype, patient age, dosage, or degree of mental retardation are associated with response.
Related resources
- Autism Society of America. www.autism-society.org
- McDougle CJ. Current and emerging therapeutics of autistic disorder and related pervasive developmental disorders. In: Davis KL, Charney D, Coyle JT, Nemeroff C (eds). Neuropsychopharmacology: The fifth generation of progress. Philadelphia: Lippincott Williams & Wilkins, 2002.
- McDougle CJ, Posey DJ. Autistic and other pervasive developmental disorders. In: Martin A, Scahill L, Charney DS, Leckman JF (eds). Pediatric psychopharmacology: Principles and practice.New York: Oxford University Press, 2002.
Drug brand names
- Atomoxetine • Strattera
- Clomipramine • Anafranil
- Clonidine • Catapres
- Desipramine • Norpramin
- Dextroamphetamine • Dexedrine, Dextrostat
- Guanfacine • Tenex
- Haloperidol • Haldol
- Levoamphetamine • Adderall
- Levodopa • Dopar, Laradopa
- Methylphenidate • Ritalin
- Olanzapine • Zyprexa
- Pemoline • Cylert
- Risperidone • Risperdal
Disclosure
Dr. Stigler reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Posey receives research support from Janssen Pharmaceutica and Eli Lilly and Co. and is a speaker for Janssen Pharmaceutica.
Dr. McDougle receives research support from Janssen Pharmaceutica, Pfizer Inc., Eli Lilly and Co., and Bristol-Myers Squibb Co. He is a consultant to or speaker for Janssen Pharmaceutica, Pfizer Inc., Eli Lilly and Co., RepliGen Corp., and Bristol-Myers Squibb Co.
Acknowledgments
This work was supported in part by a Daniel X. Freedman Psychiatric Research Fellowship Award (Dr. Posey), a National Alliance for Research in Schizophrenia and Depression (NARSAD) Young Investigator Award (Dr. Posey), a Research Units on Pediatric Psychopharmacology Grant (U10MH66766-02) from the National Institute of Mental Health (NIMH) to Indiana University (Dr. McDougle, Dr. Stigler, and Dr. Posey), a Research Career Development Award (K23-MH068627-01) from the NIMH (Dr. Posey), a National Institutes of Health Clinical Research Center grant to Indiana University (M01-RR00750), and a Department of Housing and Urban Development (HUD) grant (B-01-SP-IN-0200) (Dr. McDougle).
1. Greenhill LL, Pliszka S, Dulcan MK, et al. Practice parameter for the use of stimulant medications in the treatment of children, adolescents, and adults. J Am Acad Child Adolesc Psychiatry 2002;41(2 suppl):26S-49S.
2. Posey DJ, McDougle CJ. The pharmacotherapy of target symptoms associated with autistic disorder and other pervasive developmental disorders. Harv Rev Psychiatry 2000;8(2):45-63.
3. Jaselskis CA, Cook EH Jr, Fletcher KE, Leventhal BL. Clonidine treatment of hyperactive and impulsive children with autistic disorder. J Clin Psychopharmacol 1992;12(5):322-7.
4. Fankhauser MP, Karumanchi VC, German ML, et al. A double-blind, placebo-controlled study of the efficacy of transdermal clonidine in autism. J Clin Psychiatry 1992;53(3):77-82.
5. Posey DJ, Decker J, Sasher TM, et al. A retrospective analysis of guanfacine in the treatment of autism. J Child Adolesc.
6. Malone RP, Cater J, Sheikh RM, et al. Olanzapine versus haloperidol in children with autistic disorder: an open pilot study. J Am Acad Child Adolesc Psychiatry 2001;40(8):887-94.
7. McCracken JT, McGough J, Shah B, et al. Risperidone in children with autism and serious behavioral problems. N Engl J Med 2002;347(5):314-21.
8. Campbell M. Pharmacotherapy in early infantile autism. Biol Psychiatry 1975;10(4):399-423.
9. Aman MG. Stimulant drug effects in developmental disorders and hyperactivity—toward a resolution of disparate findings. J Autism Dev Disord 1982;12(4):385-98.
10. Campbell M, Fish B, David R, et al. Response to triiodothyronine and dextroamphetamine: a study of preschool schizophrenic children. J Autism Child Schizophr 1972;2(4):343-58.
11. Geller B, Guttmacher LB, Bleeg M. Coexistence of childhood onset pervasive developmental disorder and attention deficit disorder with hyperactivity. Am J Psychiatry 1981;138(3):388-9.
12. Campbell M, Small AM, Collins PJ, et al. Levodopa and levoamphetamine: a crossover study in young schizophrenic children. Curr Ther Res Clin Exp 1976;19(1):70-86.
13. Hoshino Y, Kumashiro H, Kaneko M, Takahashi Y. The effects of methylphenidate on early infantile autism and its relation to serum serotonin levels. Folia Psychiatr Neurol Jpn 1977;31(4):605-14.
14. Birmaher B, Quintana H, Greenhill LL. Methylphenidate treatment of hyperactive autistic children. J Am Acad Child Adolesc Psychiatry 1988;27(2):248-51.
15. Strayhorn JM Jr, Rapp N, Donina W, Strain PS. Randomized trial of methylphenidate for an autistic child. J Am Acad Child Adolesc Psychiatry 1988;27(2):244-7.
16. Quintana H, Birmaher B, Stedge D, et al. Use of methylphenidate in the treatment of children with autistic disorder. J Autism Dev Disord 1995;25(3):283-94.
17. Handen BL, Johnson CR, Lubetsky M. Efficacy of methylphenidate among children with autism and symptoms of attention-deficit hyperactivity disorder. J Autism Dev Disord 2000;30(3):245-55.
18. Stigler KA, Desmond LA, Posey DJ, et al. A naturalistic retrospective analysis of psychostimulants in pervasive developmental disorders. J Child Adolesc Psychopharmacol 2004;14(1):49-56.
19. Gordon CT, State RC, Nelson JE, et al. A double-blind comparison of clomipramine, desipramine, and placebo in the treatment of autistic disorder. Arch Gen Psychiatry 1993;50(6):441-7.
Psychostimulants benefit many patients with attention-deficit/hyperactivity disorder (ADHD)1 and thus might seem a logical choice to manage hyperactivity and inattention in youths with a pervasive developmental disorder (PDD). Some PDD patients do respond to psychostimulant therapy, but others worsen—and side effects are common.
Youths with PDDs often exhibit maladaptive behaviors—aggression, self-injury, irritability, hyperactivity, inattention—with repetitive activity patterns and fundamentally impaired social interaction and communication.2 To help you treat youths with PDD, we draw on the evidence, clinical experience, and our research to suggest psychostimulants’ role in a multimodal approach.
Targeting hyperactivity and inattentions
Step 1. Our approach begins with behavioral therapy (Figure), which includes identifying situations that trigger maladaptive behavior and environments that yield optimum behavior. The therapist assesses the child’s baseline attention and works with him or her to gradually increase it, using reinforcement and visual token boards.
Algorithm Suggested approach to hyperactivity and/or inattention in patients with PDDs
To set limits and expectations, the therapist introduces structure such as designating work and break areas and using visual schedules and timers to indicate activity duration. Minimizing distractions and understanding the child’s sensory needs may increase motivation and attention. Initially, the therapist allows numerous breaks and then may slowly decrease them as the child progresses. Tailoring work and play materials to the child’s interests can also help increase attention.
Step 2. Many patients will not respond to behavior therapy alone and will require added drug therapy. Based on evidence, we suggest starting with an alpha-2 adrenergic agonist. Guanfacine may be considered the drug of choice because of clonidine’s higher risk of adverse effects, such as hypotension and sedation. Obtain a baseline ECG with either agent, as clonidine has been associated with rare cardiovascular events.
Clonidine. Two small studies showed that clonidine may be of some benefit to patients with PDDs:
- Results were mixed in a 6-week, double-blind, placebo-controlled, crossover study of clonidine (4 to 10 μg/kg/d) in 8 autistic children ages 5 to 13.3 Teacher and parent rating instruments reflected significantly improved hyperactivity, irritability, and oppositional behavior. Clinician ratings, however, showed no significant difference between clonidine and placebo. Adverse effects with clonidine included hypotension, sedation, and decreased activity.
- In a 4-week, double-blind, placebo-con-trolled, crossover study of transdermal clonidine (0.16 to 0.48 mg/kg/d; mean: 3.6 μg/kg/d), clinician ratings showed significantly decreased hyperactivity, impulsivity, and anxiety in 9 autistic males ages 5 to 33. Sedation and fatigue were common adverse effects.
Guanfacine. In a recent retrospective review,5 we examined outcomes of 80 PDD patients ages 3 to 18 who received guanfacine (0.25 to 9 mg/d; mean: 2.6). Hyperactivity, inattention, and tics decreased in 19 patients (24%) treated for a mean 10 months.
Step 3. If clonidine or guanfacine fails to reduce hyperactivity and inattention, discontinue it and consider a psychostimulant trial.
Because psychostimulants’ efficacy in PDDs remains inconclusive, we suggest beginning with a low dosage and carefully monitoring the patient for worsening target symptoms and activation, such as emerging aggression or irritability.
Step 4. If hyperactivity and inattention remain prominent and treatment-refractory, we suggest that you discontinue the stimulant and consider an atypical antipsychotic trial. With the atypicals, monitor patients closely for adverse effects, including weight gain, extrapyramidal symptoms, and tardive dyskinesia. Fasting serum glucose and lipid profiles and liver function tests are recommended at least every 6 months and more often in individuals at risk for diabetes or hepatic disease.
Two studies provide evidence of atypicals’ efficacy in PDDs:
- In a 6-week open-label comparison,6 olanzapine significantly reduced hyperactivity and anger or uncooperativeness in 12 children with autistic disorder, but haloperidol did not. Average weight gain was 9 lbs in patients receiving olanzapine vs 3.2 lbs in those receiving haloperidol.
- An 8-week, double-blind study7 compared risperidone (0.5 to 3.5 mg/d; mean: 1.8) with placebo in 101 children and adolescents with autistic disorder. Response rates were 69% in the risperidone group and 12% in the control group. Risperidone reduced hyperactivity, aggression, agitation, and repetitive behavior. Adverse drug effects included weight gain (2.7 kg vs. 0.8 kg with placebo), increased appetite, and sedation.
Psychostimulant use in PDDs
Evidence is conflicting on psychostimulant use in patients with PDDs (Table). Early reviews suggested that stimulants were ineffective in PDDs and associated with adverse effects.8,9 Some preliminary studies supported that view, but recent reports have been mixed.
Dextroamphetamine. Campbell et al10 published a placebo-controlled study comparing triiodothyronine and dextroamphetamine (mean dosage, 4.8 mg/d; range 1.25 to 10 mg/d) in 16 children ages 3 to 6 (mean, 4.3 years) with diagnoses of autism, schizophrenia, and organic brain syndrome. All diagnostic groups worsened clinically with dextroamphetamine, and adverse effects—hyperactivity, worsened stereotypy, irritability, and decreased appetite—were common.
A subsequent case report11 found dex-troamphetamine effective when 2 patients ages 9 and 12 with PDD were treated with 10 and 5 mg/d, respectively. Hyperactivity, inattention, and impulsivity improved in both patients, and core PDD features did not worsen.
Levoamphetamine. In an 8-week, double-blind, crossover comparison with levodopa,12 levoamphetamine, 3.5 to 42 mg/d (mean, 13.4), worsened symptoms in 12 children ages 3 to 12 who had schizophrenia with autistic features. stereotypy emerged or increased in 9 of the 11 patients (82%) available for follow-up, and levoamphetamine was poorly tolerated.
Methylphenidate. In an early report, methylphenidate decreased hyperactivity and impulsivity in 9 of 15 children (60%) ages 2 to 13 with infantile autism.13 Dosages of 5 to 10 mg/d or 0.3 to 1 mg/kg/d were given for 2 to 60 weeks (mean, 26). Adverse effects included irritability, insomnia, and anorexia.
Table
Selected reports of stimulant use in pervasive developmental disorders
| Medication | Type of report | Dosage (mg/d); duration | Outcome | Adverse effects |
|---|---|---|---|---|
| Dextroamphetamine | Placebo-controlled10 (N=16) Case report11 (N=2) | Mean 4.8; N/A Mean 7.5; N/A | Clinical worsening Improved hyperactivity,inattention,impulsivity | Hyperactivity, irritability, decreased appetite, worsened stereotypy N/A |
| Levoamphetamine | Double-blind12 (N=12) | Mean 13.4 | Clinical worsening | Stereotypy emerged or worsened |
| Methylphenidate | Retrospective13 (N=15) Open-label14 (N=9) Case report15 (N=1) Double-blind, placebo-controlled, crossover16 (N=10) Double-blind, placebo-controlled, crossover17 (N=13) | 5 to 10; 26 weeks 10 to 50; 2 weeks 20; 4 weeks 20 mg/d for 2 weeks, 40 mg/d for 2 weeks 0.3 mg/kg and 0.6 mg/kg | Improved hyperactivity, impulsivity Improved hyperactivity Improved hyperactivity, concentration Modest benefit over placebo Improved hyperactivity, inattention | Irritability, insomnia, anorexia Initial mild insomnia Dysphoria, angry outbursts Statistically similar to placebo Social withdrawal, irritability |
| Methylphenidate, levoamphetamine, dextroamphetamine, or pemoline | Retrospective18 (N=195) | Various dosages, durations | Patients with, Asperger’s disorder were significantly more likely to respond | Agitation, dysphoria, irritability |
| N/A: not available | ||||
A subsequent open-label study and a case report also indicated that methylphenidate improved hyperactivity in patients with autistic disorder:
- In the 2-week, open-label study,14 9 patients ages 4 to 16 received methylphenidate, 10 to 50 mg/d. Two patients also received haloperidol, 4 and 5 mg/d. Hyperactivity improved significantly, as measured by the Conners Teacher Questionnaire.
- In the case report,15 one child, age 6, was. treated with methylphenidate, 10 mg bid, for 31 days. The drug significantly alleviated hyperactivity and improved concentration. Adverse effects included dysphoria and outbursts of anger.
Atomoxetine—a nonstimulant, selective norepinephrine reuptake inhibitor—has been approved to treat hyperactivity and inattention in ADHD, but no evidence has been published on its use in PDDs. A study of desipramine19 —also a norepinephrine reuptake inhibitor—may offer some insight into the possible efficacy and tolerability of atomoxetine in PDDs.
Desipramine (mean, 127 mg/d) was compared with the serotonin reuptake inhibitor clomipramine (mean, 153 mg/d) in a 10-week, double-blind, crossover study of 24 autistic patients ages 6 to 23. The agents were equally effective and superior to placebo in decreasing hyperactivity, although desipramine was associated with increased aggression and irritability.
Despite these results with desipramine, research is needed to understand atomoxetine’s potential role in treating hyperactivity and inattention in youths with PDDs.
Controlled trials. These early reports were followed by two double-blind, placebo-controlled, crossover studies of methylphenidate in children with autistic disorder.
- In the first trial,16 methylphenidate, 10 or 20 mg/d, improved irritability and hyperactivity in 10 children ages 7 to 11 but was only modestly more beneficial than placebo. Side-effect incidence—including decreased appetite, irritability, and insomnia—was similar during active and placebo treatments. Two patients required adjunctive haloperidol for prevailing behavioral problems.
- In the second trial,17 8 of 13 children (62%) ages 5 to 11 responded to methylphenidate, 0.3 and 0.6 mg/kg per dose. Hyperactivity and inattention improved significantly, as measured by a minimum 50% decrease in Conners Hyperactivity Index score. Ratings of stereotypy and inappropriate speech also decreased, but no changes were seen in the Child Autism Rating Scale. Adverse effects, which were more common with the 0.6 mg/kg dose, included social withdrawal and irritability.
Retrospective trial. Our group recently completed a retrospective study of 195 youth (mean age, 7.3 years; range, 2 to 19 years) with PDDs treated with a stimulant medication.18 As a whole, stimulants appeared ineffective.
Analysis of response by PDD subtype found that individuals with Asperger’s disorder—in contrast to those with autistic disorder or PDD not otherwise specified—were significantly more likely to respond to a stimulant medication. Gender, intelligence quotient (IQ), type of stimulant, and dosage did not significantly affect response. Adverse effects—including agitation, dysphoria, and irritability—occurred in 57.5% of the trials.
Atomoxetine. This nonstimulant medication has been approved for treating ADHD. However, research is needed to understand its use in patients with PDDs (Box)19
Summary. These mixed findings—combined with anecdotal reports from physicians describing the onset or exacerbation of hyperactivity, irritability, and aggression—indicate that much more evidence is needed regarding psychostimulant use in patients with PDDs.
To help meet this need, the National Institutes of Mental Health’s Research Units on Pediatric Psychopharmacology (RUPP) autism network recently completed a large, double-blind, placebo-controlled study to investigate methylphenidate’s efficacy and tolerability in PDDs. It is anticipated that the results will help us discern whether factors such as PDD subtype, patient age, dosage, or degree of mental retardation are associated with response.
Related resources
- Autism Society of America. www.autism-society.org
- McDougle CJ. Current and emerging therapeutics of autistic disorder and related pervasive developmental disorders. In: Davis KL, Charney D, Coyle JT, Nemeroff C (eds). Neuropsychopharmacology: The fifth generation of progress. Philadelphia: Lippincott Williams & Wilkins, 2002.
- McDougle CJ, Posey DJ. Autistic and other pervasive developmental disorders. In: Martin A, Scahill L, Charney DS, Leckman JF (eds). Pediatric psychopharmacology: Principles and practice.New York: Oxford University Press, 2002.
Drug brand names
- Atomoxetine • Strattera
- Clomipramine • Anafranil
- Clonidine • Catapres
- Desipramine • Norpramin
- Dextroamphetamine • Dexedrine, Dextrostat
- Guanfacine • Tenex
- Haloperidol • Haldol
- Levoamphetamine • Adderall
- Levodopa • Dopar, Laradopa
- Methylphenidate • Ritalin
- Olanzapine • Zyprexa
- Pemoline • Cylert
- Risperidone • Risperdal
Disclosure
Dr. Stigler reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Posey receives research support from Janssen Pharmaceutica and Eli Lilly and Co. and is a speaker for Janssen Pharmaceutica.
Dr. McDougle receives research support from Janssen Pharmaceutica, Pfizer Inc., Eli Lilly and Co., and Bristol-Myers Squibb Co. He is a consultant to or speaker for Janssen Pharmaceutica, Pfizer Inc., Eli Lilly and Co., RepliGen Corp., and Bristol-Myers Squibb Co.
Acknowledgments
This work was supported in part by a Daniel X. Freedman Psychiatric Research Fellowship Award (Dr. Posey), a National Alliance for Research in Schizophrenia and Depression (NARSAD) Young Investigator Award (Dr. Posey), a Research Units on Pediatric Psychopharmacology Grant (U10MH66766-02) from the National Institute of Mental Health (NIMH) to Indiana University (Dr. McDougle, Dr. Stigler, and Dr. Posey), a Research Career Development Award (K23-MH068627-01) from the NIMH (Dr. Posey), a National Institutes of Health Clinical Research Center grant to Indiana University (M01-RR00750), and a Department of Housing and Urban Development (HUD) grant (B-01-SP-IN-0200) (Dr. McDougle).
Psychostimulants benefit many patients with attention-deficit/hyperactivity disorder (ADHD)1 and thus might seem a logical choice to manage hyperactivity and inattention in youths with a pervasive developmental disorder (PDD). Some PDD patients do respond to psychostimulant therapy, but others worsen—and side effects are common.
Youths with PDDs often exhibit maladaptive behaviors—aggression, self-injury, irritability, hyperactivity, inattention—with repetitive activity patterns and fundamentally impaired social interaction and communication.2 To help you treat youths with PDD, we draw on the evidence, clinical experience, and our research to suggest psychostimulants’ role in a multimodal approach.
Targeting hyperactivity and inattentions
Step 1. Our approach begins with behavioral therapy (Figure), which includes identifying situations that trigger maladaptive behavior and environments that yield optimum behavior. The therapist assesses the child’s baseline attention and works with him or her to gradually increase it, using reinforcement and visual token boards.
Algorithm Suggested approach to hyperactivity and/or inattention in patients with PDDs
To set limits and expectations, the therapist introduces structure such as designating work and break areas and using visual schedules and timers to indicate activity duration. Minimizing distractions and understanding the child’s sensory needs may increase motivation and attention. Initially, the therapist allows numerous breaks and then may slowly decrease them as the child progresses. Tailoring work and play materials to the child’s interests can also help increase attention.
Step 2. Many patients will not respond to behavior therapy alone and will require added drug therapy. Based on evidence, we suggest starting with an alpha-2 adrenergic agonist. Guanfacine may be considered the drug of choice because of clonidine’s higher risk of adverse effects, such as hypotension and sedation. Obtain a baseline ECG with either agent, as clonidine has been associated with rare cardiovascular events.
Clonidine. Two small studies showed that clonidine may be of some benefit to patients with PDDs:
- Results were mixed in a 6-week, double-blind, placebo-controlled, crossover study of clonidine (4 to 10 μg/kg/d) in 8 autistic children ages 5 to 13.3 Teacher and parent rating instruments reflected significantly improved hyperactivity, irritability, and oppositional behavior. Clinician ratings, however, showed no significant difference between clonidine and placebo. Adverse effects with clonidine included hypotension, sedation, and decreased activity.
- In a 4-week, double-blind, placebo-con-trolled, crossover study of transdermal clonidine (0.16 to 0.48 mg/kg/d; mean: 3.6 μg/kg/d), clinician ratings showed significantly decreased hyperactivity, impulsivity, and anxiety in 9 autistic males ages 5 to 33. Sedation and fatigue were common adverse effects.
Guanfacine. In a recent retrospective review,5 we examined outcomes of 80 PDD patients ages 3 to 18 who received guanfacine (0.25 to 9 mg/d; mean: 2.6). Hyperactivity, inattention, and tics decreased in 19 patients (24%) treated for a mean 10 months.
Step 3. If clonidine or guanfacine fails to reduce hyperactivity and inattention, discontinue it and consider a psychostimulant trial.
Because psychostimulants’ efficacy in PDDs remains inconclusive, we suggest beginning with a low dosage and carefully monitoring the patient for worsening target symptoms and activation, such as emerging aggression or irritability.
Step 4. If hyperactivity and inattention remain prominent and treatment-refractory, we suggest that you discontinue the stimulant and consider an atypical antipsychotic trial. With the atypicals, monitor patients closely for adverse effects, including weight gain, extrapyramidal symptoms, and tardive dyskinesia. Fasting serum glucose and lipid profiles and liver function tests are recommended at least every 6 months and more often in individuals at risk for diabetes or hepatic disease.
Two studies provide evidence of atypicals’ efficacy in PDDs:
- In a 6-week open-label comparison,6 olanzapine significantly reduced hyperactivity and anger or uncooperativeness in 12 children with autistic disorder, but haloperidol did not. Average weight gain was 9 lbs in patients receiving olanzapine vs 3.2 lbs in those receiving haloperidol.
- An 8-week, double-blind study7 compared risperidone (0.5 to 3.5 mg/d; mean: 1.8) with placebo in 101 children and adolescents with autistic disorder. Response rates were 69% in the risperidone group and 12% in the control group. Risperidone reduced hyperactivity, aggression, agitation, and repetitive behavior. Adverse drug effects included weight gain (2.7 kg vs. 0.8 kg with placebo), increased appetite, and sedation.
Psychostimulant use in PDDs
Evidence is conflicting on psychostimulant use in patients with PDDs (Table). Early reviews suggested that stimulants were ineffective in PDDs and associated with adverse effects.8,9 Some preliminary studies supported that view, but recent reports have been mixed.
Dextroamphetamine. Campbell et al10 published a placebo-controlled study comparing triiodothyronine and dextroamphetamine (mean dosage, 4.8 mg/d; range 1.25 to 10 mg/d) in 16 children ages 3 to 6 (mean, 4.3 years) with diagnoses of autism, schizophrenia, and organic brain syndrome. All diagnostic groups worsened clinically with dextroamphetamine, and adverse effects—hyperactivity, worsened stereotypy, irritability, and decreased appetite—were common.
A subsequent case report11 found dex-troamphetamine effective when 2 patients ages 9 and 12 with PDD were treated with 10 and 5 mg/d, respectively. Hyperactivity, inattention, and impulsivity improved in both patients, and core PDD features did not worsen.
Levoamphetamine. In an 8-week, double-blind, crossover comparison with levodopa,12 levoamphetamine, 3.5 to 42 mg/d (mean, 13.4), worsened symptoms in 12 children ages 3 to 12 who had schizophrenia with autistic features. stereotypy emerged or increased in 9 of the 11 patients (82%) available for follow-up, and levoamphetamine was poorly tolerated.
Methylphenidate. In an early report, methylphenidate decreased hyperactivity and impulsivity in 9 of 15 children (60%) ages 2 to 13 with infantile autism.13 Dosages of 5 to 10 mg/d or 0.3 to 1 mg/kg/d were given for 2 to 60 weeks (mean, 26). Adverse effects included irritability, insomnia, and anorexia.
Table
Selected reports of stimulant use in pervasive developmental disorders
| Medication | Type of report | Dosage (mg/d); duration | Outcome | Adverse effects |
|---|---|---|---|---|
| Dextroamphetamine | Placebo-controlled10 (N=16) Case report11 (N=2) | Mean 4.8; N/A Mean 7.5; N/A | Clinical worsening Improved hyperactivity,inattention,impulsivity | Hyperactivity, irritability, decreased appetite, worsened stereotypy N/A |
| Levoamphetamine | Double-blind12 (N=12) | Mean 13.4 | Clinical worsening | Stereotypy emerged or worsened |
| Methylphenidate | Retrospective13 (N=15) Open-label14 (N=9) Case report15 (N=1) Double-blind, placebo-controlled, crossover16 (N=10) Double-blind, placebo-controlled, crossover17 (N=13) | 5 to 10; 26 weeks 10 to 50; 2 weeks 20; 4 weeks 20 mg/d for 2 weeks, 40 mg/d for 2 weeks 0.3 mg/kg and 0.6 mg/kg | Improved hyperactivity, impulsivity Improved hyperactivity Improved hyperactivity, concentration Modest benefit over placebo Improved hyperactivity, inattention | Irritability, insomnia, anorexia Initial mild insomnia Dysphoria, angry outbursts Statistically similar to placebo Social withdrawal, irritability |
| Methylphenidate, levoamphetamine, dextroamphetamine, or pemoline | Retrospective18 (N=195) | Various dosages, durations | Patients with, Asperger’s disorder were significantly more likely to respond | Agitation, dysphoria, irritability |
| N/A: not available | ||||
A subsequent open-label study and a case report also indicated that methylphenidate improved hyperactivity in patients with autistic disorder:
- In the 2-week, open-label study,14 9 patients ages 4 to 16 received methylphenidate, 10 to 50 mg/d. Two patients also received haloperidol, 4 and 5 mg/d. Hyperactivity improved significantly, as measured by the Conners Teacher Questionnaire.
- In the case report,15 one child, age 6, was. treated with methylphenidate, 10 mg bid, for 31 days. The drug significantly alleviated hyperactivity and improved concentration. Adverse effects included dysphoria and outbursts of anger.
Atomoxetine—a nonstimulant, selective norepinephrine reuptake inhibitor—has been approved to treat hyperactivity and inattention in ADHD, but no evidence has been published on its use in PDDs. A study of desipramine19 —also a norepinephrine reuptake inhibitor—may offer some insight into the possible efficacy and tolerability of atomoxetine in PDDs.
Desipramine (mean, 127 mg/d) was compared with the serotonin reuptake inhibitor clomipramine (mean, 153 mg/d) in a 10-week, double-blind, crossover study of 24 autistic patients ages 6 to 23. The agents were equally effective and superior to placebo in decreasing hyperactivity, although desipramine was associated with increased aggression and irritability.
Despite these results with desipramine, research is needed to understand atomoxetine’s potential role in treating hyperactivity and inattention in youths with PDDs.
Controlled trials. These early reports were followed by two double-blind, placebo-controlled, crossover studies of methylphenidate in children with autistic disorder.
- In the first trial,16 methylphenidate, 10 or 20 mg/d, improved irritability and hyperactivity in 10 children ages 7 to 11 but was only modestly more beneficial than placebo. Side-effect incidence—including decreased appetite, irritability, and insomnia—was similar during active and placebo treatments. Two patients required adjunctive haloperidol for prevailing behavioral problems.
- In the second trial,17 8 of 13 children (62%) ages 5 to 11 responded to methylphenidate, 0.3 and 0.6 mg/kg per dose. Hyperactivity and inattention improved significantly, as measured by a minimum 50% decrease in Conners Hyperactivity Index score. Ratings of stereotypy and inappropriate speech also decreased, but no changes were seen in the Child Autism Rating Scale. Adverse effects, which were more common with the 0.6 mg/kg dose, included social withdrawal and irritability.
Retrospective trial. Our group recently completed a retrospective study of 195 youth (mean age, 7.3 years; range, 2 to 19 years) with PDDs treated with a stimulant medication.18 As a whole, stimulants appeared ineffective.
Analysis of response by PDD subtype found that individuals with Asperger’s disorder—in contrast to those with autistic disorder or PDD not otherwise specified—were significantly more likely to respond to a stimulant medication. Gender, intelligence quotient (IQ), type of stimulant, and dosage did not significantly affect response. Adverse effects—including agitation, dysphoria, and irritability—occurred in 57.5% of the trials.
Atomoxetine. This nonstimulant medication has been approved for treating ADHD. However, research is needed to understand its use in patients with PDDs (Box)19
Summary. These mixed findings—combined with anecdotal reports from physicians describing the onset or exacerbation of hyperactivity, irritability, and aggression—indicate that much more evidence is needed regarding psychostimulant use in patients with PDDs.
To help meet this need, the National Institutes of Mental Health’s Research Units on Pediatric Psychopharmacology (RUPP) autism network recently completed a large, double-blind, placebo-controlled study to investigate methylphenidate’s efficacy and tolerability in PDDs. It is anticipated that the results will help us discern whether factors such as PDD subtype, patient age, dosage, or degree of mental retardation are associated with response.
Related resources
- Autism Society of America. www.autism-society.org
- McDougle CJ. Current and emerging therapeutics of autistic disorder and related pervasive developmental disorders. In: Davis KL, Charney D, Coyle JT, Nemeroff C (eds). Neuropsychopharmacology: The fifth generation of progress. Philadelphia: Lippincott Williams & Wilkins, 2002.
- McDougle CJ, Posey DJ. Autistic and other pervasive developmental disorders. In: Martin A, Scahill L, Charney DS, Leckman JF (eds). Pediatric psychopharmacology: Principles and practice.New York: Oxford University Press, 2002.
Drug brand names
- Atomoxetine • Strattera
- Clomipramine • Anafranil
- Clonidine • Catapres
- Desipramine • Norpramin
- Dextroamphetamine • Dexedrine, Dextrostat
- Guanfacine • Tenex
- Haloperidol • Haldol
- Levoamphetamine • Adderall
- Levodopa • Dopar, Laradopa
- Methylphenidate • Ritalin
- Olanzapine • Zyprexa
- Pemoline • Cylert
- Risperidone • Risperdal
Disclosure
Dr. Stigler reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Posey receives research support from Janssen Pharmaceutica and Eli Lilly and Co. and is a speaker for Janssen Pharmaceutica.
Dr. McDougle receives research support from Janssen Pharmaceutica, Pfizer Inc., Eli Lilly and Co., and Bristol-Myers Squibb Co. He is a consultant to or speaker for Janssen Pharmaceutica, Pfizer Inc., Eli Lilly and Co., RepliGen Corp., and Bristol-Myers Squibb Co.
Acknowledgments
This work was supported in part by a Daniel X. Freedman Psychiatric Research Fellowship Award (Dr. Posey), a National Alliance for Research in Schizophrenia and Depression (NARSAD) Young Investigator Award (Dr. Posey), a Research Units on Pediatric Psychopharmacology Grant (U10MH66766-02) from the National Institute of Mental Health (NIMH) to Indiana University (Dr. McDougle, Dr. Stigler, and Dr. Posey), a Research Career Development Award (K23-MH068627-01) from the NIMH (Dr. Posey), a National Institutes of Health Clinical Research Center grant to Indiana University (M01-RR00750), and a Department of Housing and Urban Development (HUD) grant (B-01-SP-IN-0200) (Dr. McDougle).
1. Greenhill LL, Pliszka S, Dulcan MK, et al. Practice parameter for the use of stimulant medications in the treatment of children, adolescents, and adults. J Am Acad Child Adolesc Psychiatry 2002;41(2 suppl):26S-49S.
2. Posey DJ, McDougle CJ. The pharmacotherapy of target symptoms associated with autistic disorder and other pervasive developmental disorders. Harv Rev Psychiatry 2000;8(2):45-63.
3. Jaselskis CA, Cook EH Jr, Fletcher KE, Leventhal BL. Clonidine treatment of hyperactive and impulsive children with autistic disorder. J Clin Psychopharmacol 1992;12(5):322-7.
4. Fankhauser MP, Karumanchi VC, German ML, et al. A double-blind, placebo-controlled study of the efficacy of transdermal clonidine in autism. J Clin Psychiatry 1992;53(3):77-82.
5. Posey DJ, Decker J, Sasher TM, et al. A retrospective analysis of guanfacine in the treatment of autism. J Child Adolesc.
6. Malone RP, Cater J, Sheikh RM, et al. Olanzapine versus haloperidol in children with autistic disorder: an open pilot study. J Am Acad Child Adolesc Psychiatry 2001;40(8):887-94.
7. McCracken JT, McGough J, Shah B, et al. Risperidone in children with autism and serious behavioral problems. N Engl J Med 2002;347(5):314-21.
8. Campbell M. Pharmacotherapy in early infantile autism. Biol Psychiatry 1975;10(4):399-423.
9. Aman MG. Stimulant drug effects in developmental disorders and hyperactivity—toward a resolution of disparate findings. J Autism Dev Disord 1982;12(4):385-98.
10. Campbell M, Fish B, David R, et al. Response to triiodothyronine and dextroamphetamine: a study of preschool schizophrenic children. J Autism Child Schizophr 1972;2(4):343-58.
11. Geller B, Guttmacher LB, Bleeg M. Coexistence of childhood onset pervasive developmental disorder and attention deficit disorder with hyperactivity. Am J Psychiatry 1981;138(3):388-9.
12. Campbell M, Small AM, Collins PJ, et al. Levodopa and levoamphetamine: a crossover study in young schizophrenic children. Curr Ther Res Clin Exp 1976;19(1):70-86.
13. Hoshino Y, Kumashiro H, Kaneko M, Takahashi Y. The effects of methylphenidate on early infantile autism and its relation to serum serotonin levels. Folia Psychiatr Neurol Jpn 1977;31(4):605-14.
14. Birmaher B, Quintana H, Greenhill LL. Methylphenidate treatment of hyperactive autistic children. J Am Acad Child Adolesc Psychiatry 1988;27(2):248-51.
15. Strayhorn JM Jr, Rapp N, Donina W, Strain PS. Randomized trial of methylphenidate for an autistic child. J Am Acad Child Adolesc Psychiatry 1988;27(2):244-7.
16. Quintana H, Birmaher B, Stedge D, et al. Use of methylphenidate in the treatment of children with autistic disorder. J Autism Dev Disord 1995;25(3):283-94.
17. Handen BL, Johnson CR, Lubetsky M. Efficacy of methylphenidate among children with autism and symptoms of attention-deficit hyperactivity disorder. J Autism Dev Disord 2000;30(3):245-55.
18. Stigler KA, Desmond LA, Posey DJ, et al. A naturalistic retrospective analysis of psychostimulants in pervasive developmental disorders. J Child Adolesc Psychopharmacol 2004;14(1):49-56.
19. Gordon CT, State RC, Nelson JE, et al. A double-blind comparison of clomipramine, desipramine, and placebo in the treatment of autistic disorder. Arch Gen Psychiatry 1993;50(6):441-7.
1. Greenhill LL, Pliszka S, Dulcan MK, et al. Practice parameter for the use of stimulant medications in the treatment of children, adolescents, and adults. J Am Acad Child Adolesc Psychiatry 2002;41(2 suppl):26S-49S.
2. Posey DJ, McDougle CJ. The pharmacotherapy of target symptoms associated with autistic disorder and other pervasive developmental disorders. Harv Rev Psychiatry 2000;8(2):45-63.
3. Jaselskis CA, Cook EH Jr, Fletcher KE, Leventhal BL. Clonidine treatment of hyperactive and impulsive children with autistic disorder. J Clin Psychopharmacol 1992;12(5):322-7.
4. Fankhauser MP, Karumanchi VC, German ML, et al. A double-blind, placebo-controlled study of the efficacy of transdermal clonidine in autism. J Clin Psychiatry 1992;53(3):77-82.
5. Posey DJ, Decker J, Sasher TM, et al. A retrospective analysis of guanfacine in the treatment of autism. J Child Adolesc.
6. Malone RP, Cater J, Sheikh RM, et al. Olanzapine versus haloperidol in children with autistic disorder: an open pilot study. J Am Acad Child Adolesc Psychiatry 2001;40(8):887-94.
7. McCracken JT, McGough J, Shah B, et al. Risperidone in children with autism and serious behavioral problems. N Engl J Med 2002;347(5):314-21.
8. Campbell M. Pharmacotherapy in early infantile autism. Biol Psychiatry 1975;10(4):399-423.
9. Aman MG. Stimulant drug effects in developmental disorders and hyperactivity—toward a resolution of disparate findings. J Autism Dev Disord 1982;12(4):385-98.
10. Campbell M, Fish B, David R, et al. Response to triiodothyronine and dextroamphetamine: a study of preschool schizophrenic children. J Autism Child Schizophr 1972;2(4):343-58.
11. Geller B, Guttmacher LB, Bleeg M. Coexistence of childhood onset pervasive developmental disorder and attention deficit disorder with hyperactivity. Am J Psychiatry 1981;138(3):388-9.
12. Campbell M, Small AM, Collins PJ, et al. Levodopa and levoamphetamine: a crossover study in young schizophrenic children. Curr Ther Res Clin Exp 1976;19(1):70-86.
13. Hoshino Y, Kumashiro H, Kaneko M, Takahashi Y. The effects of methylphenidate on early infantile autism and its relation to serum serotonin levels. Folia Psychiatr Neurol Jpn 1977;31(4):605-14.
14. Birmaher B, Quintana H, Greenhill LL. Methylphenidate treatment of hyperactive autistic children. J Am Acad Child Adolesc Psychiatry 1988;27(2):248-51.
15. Strayhorn JM Jr, Rapp N, Donina W, Strain PS. Randomized trial of methylphenidate for an autistic child. J Am Acad Child Adolesc Psychiatry 1988;27(2):244-7.
16. Quintana H, Birmaher B, Stedge D, et al. Use of methylphenidate in the treatment of children with autistic disorder. J Autism Dev Disord 1995;25(3):283-94.
17. Handen BL, Johnson CR, Lubetsky M. Efficacy of methylphenidate among children with autism and symptoms of attention-deficit hyperactivity disorder. J Autism Dev Disord 2000;30(3):245-55.
18. Stigler KA, Desmond LA, Posey DJ, et al. A naturalistic retrospective analysis of psychostimulants in pervasive developmental disorders. J Child Adolesc Psychopharmacol 2004;14(1):49-56.
19. Gordon CT, State RC, Nelson JE, et al. A double-blind comparison of clomipramine, desipramine, and placebo in the treatment of autistic disorder. Arch Gen Psychiatry 1993;50(6):441-7.