User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Clarifying risk factors for violence
Dr. Battaglia’s article on patient violence is most useful. I’ve been waiting for an article that clarifies the risk factors we need to watch for in patients with a history of violence. To be able to copy and paste this article from currentpsychiatry.com into my psychiatric materials is great.
Maria S. Arrubla, MD
Veterans Administration Medical Center
Leeds, MA
Dr. Battaglia’s article on patient violence is most useful. I’ve been waiting for an article that clarifies the risk factors we need to watch for in patients with a history of violence. To be able to copy and paste this article from currentpsychiatry.com into my psychiatric materials is great.
Maria S. Arrubla, MD
Veterans Administration Medical Center
Leeds, MA
Dr. Battaglia’s article on patient violence is most useful. I’ve been waiting for an article that clarifies the risk factors we need to watch for in patients with a history of violence. To be able to copy and paste this article from currentpsychiatry.com into my psychiatric materials is great.
Maria S. Arrubla, MD
Veterans Administration Medical Center
Leeds, MA
Race and patient violence
John Battaglia, MD, describes many valid factors that could predict violence in an inpatient setting (Current Psychiatry, February 2004). However, we disagree that being “nonwhite” is among these factors.
Studies that cite race as a predictor of patient violence have not been adequately controlled for significant variables. By contrast, Silver1 showed that race does not predict violence among persons with mental disorders when neighborhood disadvantage is statistically well-controlled.
Using race to predict patient violence may explain why nonwhite patients inadvertently get excessive medication. In a retrospective study,2 African-American patients with schizophrenia were:
- 1.8 times more likely than their white counterparts to receive excessive doses of typical antipsychotics
- more likely than white patients to be treated with older, high-potency antipsychotics.2
Many researchers have demonstrated other differences in treatment of nonwhite vs. white inpatients and have proposed that nonwhites face barriers to diagnosis and drug management of psychiatric disorders. A review of 344 persons with schizophrenia3 found pronounced variations in treatment (such as use of atypical neuroleptics) based on race, even though the data were adjusted for demographic and clinical characteristics. After controlling for relevant variables, Allegra et al4 found that poor Latinos and African Americans not classified as poor are less likely to receive specialty psychiatric care than their white counterparts.
Using race as a variable in inpatient settings discourages objective clinical management, albeit not deliberately. In this way, a relatively inexperienced doctor subconsciously learns to consider race to explain a patient’s violent actions.
Babatunde A. Adetunji, MD
Maju Mathews, MD
Department of psychiatry
Drexel University College of Medicine
Philadelphia, PA
Kumar Budur, MD
Department of psychiatry
Cleveland Clinic Foundation, Cleveland, OH
- Silver E. Race, neighborhood disadvantage, and violence among persons with mental disorders: the importance of contextual measurement. Law Hum Behav 2000;24:449–56.
- Diaz FJ, De Leon J. Excessive antipsychotic dosing in 2 U.S. state hospitals [comment]. J Clin Psychiatry 2002;63:998.
- Kreyenbuhl J, Zito JM, Buchanan RW, et al. Racial disparity in the pharmacological management of schizophrenia. Schizophr Bull 2003;29:183–93.
- Allegra M, Canino G, Rios R, et al. Inequalities in use of specialty mental health services among Latinos, African Americans, and non-Latino whites. Psychiatr Serv 2002;53:1547–55.
Dr. Battaglia responds
Drs. Adetunji, Mathews, and Budur raise some interesting questions about race and statistics, and this of course is an area of intense scrutiny that requires further study.
Race and culture are inextricably linked, and studies designed to ferret out the differential aspects are often subject to the same criticisms they attempt to clarify. I agree that we must all keep an open mind for interpreting data in this intriguing area.
John Battaglia, MD
Medical director, Meriter Hospital adult psychiatry program
Associate professor, department of Psychiatry
University of Wisconsin Medical School
Madison
John Battaglia, MD, describes many valid factors that could predict violence in an inpatient setting (Current Psychiatry, February 2004). However, we disagree that being “nonwhite” is among these factors.
Studies that cite race as a predictor of patient violence have not been adequately controlled for significant variables. By contrast, Silver1 showed that race does not predict violence among persons with mental disorders when neighborhood disadvantage is statistically well-controlled.
Using race to predict patient violence may explain why nonwhite patients inadvertently get excessive medication. In a retrospective study,2 African-American patients with schizophrenia were:
- 1.8 times more likely than their white counterparts to receive excessive doses of typical antipsychotics
- more likely than white patients to be treated with older, high-potency antipsychotics.2
Many researchers have demonstrated other differences in treatment of nonwhite vs. white inpatients and have proposed that nonwhites face barriers to diagnosis and drug management of psychiatric disorders. A review of 344 persons with schizophrenia3 found pronounced variations in treatment (such as use of atypical neuroleptics) based on race, even though the data were adjusted for demographic and clinical characteristics. After controlling for relevant variables, Allegra et al4 found that poor Latinos and African Americans not classified as poor are less likely to receive specialty psychiatric care than their white counterparts.
Using race as a variable in inpatient settings discourages objective clinical management, albeit not deliberately. In this way, a relatively inexperienced doctor subconsciously learns to consider race to explain a patient’s violent actions.
Babatunde A. Adetunji, MD
Maju Mathews, MD
Department of psychiatry
Drexel University College of Medicine
Philadelphia, PA
Kumar Budur, MD
Department of psychiatry
Cleveland Clinic Foundation, Cleveland, OH
- Silver E. Race, neighborhood disadvantage, and violence among persons with mental disorders: the importance of contextual measurement. Law Hum Behav 2000;24:449–56.
- Diaz FJ, De Leon J. Excessive antipsychotic dosing in 2 U.S. state hospitals [comment]. J Clin Psychiatry 2002;63:998.
- Kreyenbuhl J, Zito JM, Buchanan RW, et al. Racial disparity in the pharmacological management of schizophrenia. Schizophr Bull 2003;29:183–93.
- Allegra M, Canino G, Rios R, et al. Inequalities in use of specialty mental health services among Latinos, African Americans, and non-Latino whites. Psychiatr Serv 2002;53:1547–55.
Dr. Battaglia responds
Drs. Adetunji, Mathews, and Budur raise some interesting questions about race and statistics, and this of course is an area of intense scrutiny that requires further study.
Race and culture are inextricably linked, and studies designed to ferret out the differential aspects are often subject to the same criticisms they attempt to clarify. I agree that we must all keep an open mind for interpreting data in this intriguing area.
John Battaglia, MD
Medical director, Meriter Hospital adult psychiatry program
Associate professor, department of Psychiatry
University of Wisconsin Medical School
Madison
John Battaglia, MD, describes many valid factors that could predict violence in an inpatient setting (Current Psychiatry, February 2004). However, we disagree that being “nonwhite” is among these factors.
Studies that cite race as a predictor of patient violence have not been adequately controlled for significant variables. By contrast, Silver1 showed that race does not predict violence among persons with mental disorders when neighborhood disadvantage is statistically well-controlled.
Using race to predict patient violence may explain why nonwhite patients inadvertently get excessive medication. In a retrospective study,2 African-American patients with schizophrenia were:
- 1.8 times more likely than their white counterparts to receive excessive doses of typical antipsychotics
- more likely than white patients to be treated with older, high-potency antipsychotics.2
Many researchers have demonstrated other differences in treatment of nonwhite vs. white inpatients and have proposed that nonwhites face barriers to diagnosis and drug management of psychiatric disorders. A review of 344 persons with schizophrenia3 found pronounced variations in treatment (such as use of atypical neuroleptics) based on race, even though the data were adjusted for demographic and clinical characteristics. After controlling for relevant variables, Allegra et al4 found that poor Latinos and African Americans not classified as poor are less likely to receive specialty psychiatric care than their white counterparts.
Using race as a variable in inpatient settings discourages objective clinical management, albeit not deliberately. In this way, a relatively inexperienced doctor subconsciously learns to consider race to explain a patient’s violent actions.
Babatunde A. Adetunji, MD
Maju Mathews, MD
Department of psychiatry
Drexel University College of Medicine
Philadelphia, PA
Kumar Budur, MD
Department of psychiatry
Cleveland Clinic Foundation, Cleveland, OH
- Silver E. Race, neighborhood disadvantage, and violence among persons with mental disorders: the importance of contextual measurement. Law Hum Behav 2000;24:449–56.
- Diaz FJ, De Leon J. Excessive antipsychotic dosing in 2 U.S. state hospitals [comment]. J Clin Psychiatry 2002;63:998.
- Kreyenbuhl J, Zito JM, Buchanan RW, et al. Racial disparity in the pharmacological management of schizophrenia. Schizophr Bull 2003;29:183–93.
- Allegra M, Canino G, Rios R, et al. Inequalities in use of specialty mental health services among Latinos, African Americans, and non-Latino whites. Psychiatr Serv 2002;53:1547–55.
Dr. Battaglia responds
Drs. Adetunji, Mathews, and Budur raise some interesting questions about race and statistics, and this of course is an area of intense scrutiny that requires further study.
Race and culture are inextricably linked, and studies designed to ferret out the differential aspects are often subject to the same criticisms they attempt to clarify. I agree that we must all keep an open mind for interpreting data in this intriguing area.
John Battaglia, MD
Medical director, Meriter Hospital adult psychiatry program
Associate professor, department of Psychiatry
University of Wisconsin Medical School
Madison
A ‘World’ of information in your pocket
With wireless Internet available in hospitals, coffee shops, airports, universities, and libraries, real-time Internet access away from the home or office is just a click away on your personal digital assistant (PDA). But what if you’re somewhere without wireless Internet-such as in flight or at the local department of motor vehicles?
Transferring and storing online content onto your PDA lets you access critical online information in places without a connection, making your down time more productive.
Portable online content
Much Web-based information can easily be captured or stored onto your PDA.
Electronic books, or e-books, have long been one of the pleasures of using PDAs and handheld devices such as the Beating the high cost of software,” Psyber Psychiatry, March 2004.)
Plucker also is free, but it creates documents primarily for Palm OS handhelds. A Pocket PC version of Plucker is in development. iSiloX documents can be viewed on Pocket PC or Palm OS devices, but the paid version of the viewer iSilo, available for $20, is required to use the navigational links. The free version of iSilo can read but cannot navigate with hyperlinks. Adobe Acrobat PDFs can be viewed on Pocket PC and Palm OS devices, but these PDFs must be distilled a second time for the handheld.
One disadvantage of all viewing systems is that Web pages with complicated formatting or specialized layers may not be accurately captured or well viewed on a PDA’s small screen.
Table
Sample systems for viewing Web-based content on PDAs
| Software | URL | Compatible PDA operating system(s) |
|---|---|---|
| Plucker | http://www.plkr.org; http://vade-mecum.sourceforge.net/ | Palm, Pocket PC |
| iSiloX | http://www.isilox.com http://www.isilo.com | Palm, Pocket PC |
| Adobe Acrobat | http://www.adobe.com | Palm, Pocket PC |
| PDF Creator | http://sector7g.wurzel6.de/pdfcreator/index_en.htm | Palm, Pocket PC |
Related Resources
- Microsoft Windows XP. Make web pages available for offline viewing.
- Kansas City Clinical Oncology Program. Mobile users help page.
- PDACorps discussion forum (topic: Plucker for Pocket PC).
If you have any questions about these products or comments about Current Psychiatry, click here to contact Dr. Luo or send an e-mail to [email protected].
Disclosure
Dr. Luo reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Luo in this column are his own and do not necessarily reflect those of Current Psychiatry.
With wireless Internet available in hospitals, coffee shops, airports, universities, and libraries, real-time Internet access away from the home or office is just a click away on your personal digital assistant (PDA). But what if you’re somewhere without wireless Internet-such as in flight or at the local department of motor vehicles?
Transferring and storing online content onto your PDA lets you access critical online information in places without a connection, making your down time more productive.
Portable online content
Much Web-based information can easily be captured or stored onto your PDA.
Electronic books, or e-books, have long been one of the pleasures of using PDAs and handheld devices such as the Beating the high cost of software,” Psyber Psychiatry, March 2004.)
Plucker also is free, but it creates documents primarily for Palm OS handhelds. A Pocket PC version of Plucker is in development. iSiloX documents can be viewed on Pocket PC or Palm OS devices, but the paid version of the viewer iSilo, available for $20, is required to use the navigational links. The free version of iSilo can read but cannot navigate with hyperlinks. Adobe Acrobat PDFs can be viewed on Pocket PC and Palm OS devices, but these PDFs must be distilled a second time for the handheld.
One disadvantage of all viewing systems is that Web pages with complicated formatting or specialized layers may not be accurately captured or well viewed on a PDA’s small screen.
Table
Sample systems for viewing Web-based content on PDAs
| Software | URL | Compatible PDA operating system(s) |
|---|---|---|
| Plucker | http://www.plkr.org; http://vade-mecum.sourceforge.net/ | Palm, Pocket PC |
| iSiloX | http://www.isilox.com http://www.isilo.com | Palm, Pocket PC |
| Adobe Acrobat | http://www.adobe.com | Palm, Pocket PC |
| PDF Creator | http://sector7g.wurzel6.de/pdfcreator/index_en.htm | Palm, Pocket PC |
Related Resources
- Microsoft Windows XP. Make web pages available for offline viewing.
- Kansas City Clinical Oncology Program. Mobile users help page.
- PDACorps discussion forum (topic: Plucker for Pocket PC).
If you have any questions about these products or comments about Current Psychiatry, click here to contact Dr. Luo or send an e-mail to [email protected].
Disclosure
Dr. Luo reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Luo in this column are his own and do not necessarily reflect those of Current Psychiatry.
With wireless Internet available in hospitals, coffee shops, airports, universities, and libraries, real-time Internet access away from the home or office is just a click away on your personal digital assistant (PDA). But what if you’re somewhere without wireless Internet-such as in flight or at the local department of motor vehicles?
Transferring and storing online content onto your PDA lets you access critical online information in places without a connection, making your down time more productive.
Portable online content
Much Web-based information can easily be captured or stored onto your PDA.
Electronic books, or e-books, have long been one of the pleasures of using PDAs and handheld devices such as the Beating the high cost of software,” Psyber Psychiatry, March 2004.)
Plucker also is free, but it creates documents primarily for Palm OS handhelds. A Pocket PC version of Plucker is in development. iSiloX documents can be viewed on Pocket PC or Palm OS devices, but the paid version of the viewer iSilo, available for $20, is required to use the navigational links. The free version of iSilo can read but cannot navigate with hyperlinks. Adobe Acrobat PDFs can be viewed on Pocket PC and Palm OS devices, but these PDFs must be distilled a second time for the handheld.
One disadvantage of all viewing systems is that Web pages with complicated formatting or specialized layers may not be accurately captured or well viewed on a PDA’s small screen.
Table
Sample systems for viewing Web-based content on PDAs
| Software | URL | Compatible PDA operating system(s) |
|---|---|---|
| Plucker | http://www.plkr.org; http://vade-mecum.sourceforge.net/ | Palm, Pocket PC |
| iSiloX | http://www.isilox.com http://www.isilo.com | Palm, Pocket PC |
| Adobe Acrobat | http://www.adobe.com | Palm, Pocket PC |
| PDF Creator | http://sector7g.wurzel6.de/pdfcreator/index_en.htm | Palm, Pocket PC |
Related Resources
- Microsoft Windows XP. Make web pages available for offline viewing.
- Kansas City Clinical Oncology Program. Mobile users help page.
- PDACorps discussion forum (topic: Plucker for Pocket PC).
If you have any questions about these products or comments about Current Psychiatry, click here to contact Dr. Luo or send an e-mail to [email protected].
Disclosure
Dr. Luo reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Luo in this column are his own and do not necessarily reflect those of Current Psychiatry.
Help night shift workers get enough sleep
Shift work sleep disorder is common among persons whose working hours fall between 6 PM and 7 AM. Some night or overnight shift workers cannot stay alert at work or sleep well when off duty, endangering others on the job or while driving.
When shift work sleep disorder is suspected, find out:
- Is the patient getting enough sleep? The average rotating shift worker sleeps 6 hours nightly1 while working the night shift.
- Is another sleep disorder present? Obstructive sleep apnea, restless legs syndrome, or other common comorbidities may also be disrupting sleep.
- Is an unrecognized comorbid psychiatric disorder present? Not surprisingly, major depression, chemical dependency, and other untreated psychiatric disorders impede adherence to a sleep schedule.
- Is caffeine being used appropriately? Shift workers can effectively use caffeine as an alerting agent but should only use it within 4 to 6 hours after arising. Advise patients against consuming beverages or foods containing caffeine within 8 to 10 hours of bedtime.
Promoting sleep
To help the patient get ample sleep, encourage him or her to:
- find time for uninterrupted sleep. Family time, social events, and errands must be scheduled so that they do not interfere.
- maintain a consistent sleep schedule when possible. Workers with long night shifts should try to stay awake all night and sleep during the day, even on days off.
- use bright lights during waking hours to promote alertness and prevent sleep disruption. Bright light has been shown to influence the human circadian clock.2
Some workplaces are installing artificial lights to increase light exposure during night work. Night shift workers traveling home in the morning should wear sunglasses to limit light exposure.
Also consider prescribing:
- a short-acting hypnotic. Although not specifically FDA-approved for shift work sleep disorder, medications such as zaleplon or zolpidem can reduce time to falling asleep and increase sleep without producing a hangover effect.
- a wakefulness-promoting agent. The FDA recently approved modafinil for reducing excessive daytime sleepiness in shift work sleep disorder. Patients take modafinil, 200 mg/d, shortly after arising to increase alertness at work. Be sure to advise patients that the medication is not a substitute for getting adequate sleep.
1. Colligan M, Tepas D. The stress of hours at work. Am Ind Hyg Assoc J 1986;47:686-95.
2. Horowitz TS, Tanigawa T. Circadian-based new technologies for night workers. Ind Health 2002;40(3):223-36.
Dr. Krahn is chair, department of psychiatry and psychology, Mayo Clinic, Scottsdale, AZ, and associate professor, Mayo Clinic College of Medicine. She is an Associate Editor of CURRENT PSYCHIATRY.
Shift work sleep disorder is common among persons whose working hours fall between 6 PM and 7 AM. Some night or overnight shift workers cannot stay alert at work or sleep well when off duty, endangering others on the job or while driving.
When shift work sleep disorder is suspected, find out:
- Is the patient getting enough sleep? The average rotating shift worker sleeps 6 hours nightly1 while working the night shift.
- Is another sleep disorder present? Obstructive sleep apnea, restless legs syndrome, or other common comorbidities may also be disrupting sleep.
- Is an unrecognized comorbid psychiatric disorder present? Not surprisingly, major depression, chemical dependency, and other untreated psychiatric disorders impede adherence to a sleep schedule.
- Is caffeine being used appropriately? Shift workers can effectively use caffeine as an alerting agent but should only use it within 4 to 6 hours after arising. Advise patients against consuming beverages or foods containing caffeine within 8 to 10 hours of bedtime.
Promoting sleep
To help the patient get ample sleep, encourage him or her to:
- find time for uninterrupted sleep. Family time, social events, and errands must be scheduled so that they do not interfere.
- maintain a consistent sleep schedule when possible. Workers with long night shifts should try to stay awake all night and sleep during the day, even on days off.
- use bright lights during waking hours to promote alertness and prevent sleep disruption. Bright light has been shown to influence the human circadian clock.2
Some workplaces are installing artificial lights to increase light exposure during night work. Night shift workers traveling home in the morning should wear sunglasses to limit light exposure.
Also consider prescribing:
- a short-acting hypnotic. Although not specifically FDA-approved for shift work sleep disorder, medications such as zaleplon or zolpidem can reduce time to falling asleep and increase sleep without producing a hangover effect.
- a wakefulness-promoting agent. The FDA recently approved modafinil for reducing excessive daytime sleepiness in shift work sleep disorder. Patients take modafinil, 200 mg/d, shortly after arising to increase alertness at work. Be sure to advise patients that the medication is not a substitute for getting adequate sleep.
Shift work sleep disorder is common among persons whose working hours fall between 6 PM and 7 AM. Some night or overnight shift workers cannot stay alert at work or sleep well when off duty, endangering others on the job or while driving.
When shift work sleep disorder is suspected, find out:
- Is the patient getting enough sleep? The average rotating shift worker sleeps 6 hours nightly1 while working the night shift.
- Is another sleep disorder present? Obstructive sleep apnea, restless legs syndrome, or other common comorbidities may also be disrupting sleep.
- Is an unrecognized comorbid psychiatric disorder present? Not surprisingly, major depression, chemical dependency, and other untreated psychiatric disorders impede adherence to a sleep schedule.
- Is caffeine being used appropriately? Shift workers can effectively use caffeine as an alerting agent but should only use it within 4 to 6 hours after arising. Advise patients against consuming beverages or foods containing caffeine within 8 to 10 hours of bedtime.
Promoting sleep
To help the patient get ample sleep, encourage him or her to:
- find time for uninterrupted sleep. Family time, social events, and errands must be scheduled so that they do not interfere.
- maintain a consistent sleep schedule when possible. Workers with long night shifts should try to stay awake all night and sleep during the day, even on days off.
- use bright lights during waking hours to promote alertness and prevent sleep disruption. Bright light has been shown to influence the human circadian clock.2
Some workplaces are installing artificial lights to increase light exposure during night work. Night shift workers traveling home in the morning should wear sunglasses to limit light exposure.
Also consider prescribing:
- a short-acting hypnotic. Although not specifically FDA-approved for shift work sleep disorder, medications such as zaleplon or zolpidem can reduce time to falling asleep and increase sleep without producing a hangover effect.
- a wakefulness-promoting agent. The FDA recently approved modafinil for reducing excessive daytime sleepiness in shift work sleep disorder. Patients take modafinil, 200 mg/d, shortly after arising to increase alertness at work. Be sure to advise patients that the medication is not a substitute for getting adequate sleep.
1. Colligan M, Tepas D. The stress of hours at work. Am Ind Hyg Assoc J 1986;47:686-95.
2. Horowitz TS, Tanigawa T. Circadian-based new technologies for night workers. Ind Health 2002;40(3):223-36.
Dr. Krahn is chair, department of psychiatry and psychology, Mayo Clinic, Scottsdale, AZ, and associate professor, Mayo Clinic College of Medicine. She is an Associate Editor of CURRENT PSYCHIATRY.
1. Colligan M, Tepas D. The stress of hours at work. Am Ind Hyg Assoc J 1986;47:686-95.
2. Horowitz TS, Tanigawa T. Circadian-based new technologies for night workers. Ind Health 2002;40(3):223-36.
Dr. Krahn is chair, department of psychiatry and psychology, Mayo Clinic, Scottsdale, AZ, and associate professor, Mayo Clinic College of Medicine. She is an Associate Editor of CURRENT PSYCHIATRY.
6 questions can reveal families’ cultural conflicts
Understanding how a patient’s cultural background intertwines with relationship concerns, communication issues, and family problems is key to diagnosis and to building a therapeutic alliance.
Asking patients from any culture these six questions can uncover subtleties of cultural interaction that may be contributing to an adjustment, anxiety, depressive, or other disorder.
- Are your parents content living in the United States? This is especially pertinent when treating children or second-generation adults for relationship problems or for an adjustment, anxiety, or depressive disorder. In clinical practice, we have seen that when parents are satisfied with their new home, children adapt more readily.
- What mainstream practices has your family adopted? Families tend to incorporate customs and values of the surrounding environment while maintaining much of their core culture. The family’s overriding desire to maintain cultural purity may turn some family members against their ethnicity. For example, children may marry outside their culture to escape a rigid or controlling family or ethnic environment. These divergences, however, are often fraught with guilt and consternation among family members.
- What value clashes persist in your family? Disagreements over dating or participation in athletics or cheerleading are common. Respect, which may be defined as acquiescing to elders’ opinions, can be an issue regarding personal relationships, occupational choices, or nursing home placement.
- Can everyone speak freely in your family? In many cultures, assertiveness is perceived as rude. Therefore, patients may need alternate methods of conflict resolution. For example, teaching the patient a more-direct communication pattern (such as politely asking the boss for a raise) may help him or her in the majority culture but can create problems within his or her native culture.
- Do you or a family member dread being alone? Individuals inured to a nuclear family may be uncomfortable with solitude. Also, some cultures define emotional closeness as the presence of multiple family members, rather than companionship between husband and wife.
- Is your family comfortable with people from the mainstream culture? Cultural integration requires multicultural contacts. Some families, however, try to maintain their culture at the expense of their stated values. For example, a dishonest, superficial friend from the native culture may be more highly valued than an honest person from the mainstream. These cultural distortions produce mixed messages for all involved.
Dr. Benjaminis a staff psychiatrist at the Oklahoma City Veterans Administration Medical Center and is clinical assistant professor, department of psychiatry and behavioral sciences, University of Oklahoma Health Sciences Center, Oklahoma City.
Dr. Mosalleaei-Benjamin is a third-year resident in internal medicine, University of Oklahoma Health Sciences Center.
Understanding how a patient’s cultural background intertwines with relationship concerns, communication issues, and family problems is key to diagnosis and to building a therapeutic alliance.
Asking patients from any culture these six questions can uncover subtleties of cultural interaction that may be contributing to an adjustment, anxiety, depressive, or other disorder.
- Are your parents content living in the United States? This is especially pertinent when treating children or second-generation adults for relationship problems or for an adjustment, anxiety, or depressive disorder. In clinical practice, we have seen that when parents are satisfied with their new home, children adapt more readily.
- What mainstream practices has your family adopted? Families tend to incorporate customs and values of the surrounding environment while maintaining much of their core culture. The family’s overriding desire to maintain cultural purity may turn some family members against their ethnicity. For example, children may marry outside their culture to escape a rigid or controlling family or ethnic environment. These divergences, however, are often fraught with guilt and consternation among family members.
- What value clashes persist in your family? Disagreements over dating or participation in athletics or cheerleading are common. Respect, which may be defined as acquiescing to elders’ opinions, can be an issue regarding personal relationships, occupational choices, or nursing home placement.
- Can everyone speak freely in your family? In many cultures, assertiveness is perceived as rude. Therefore, patients may need alternate methods of conflict resolution. For example, teaching the patient a more-direct communication pattern (such as politely asking the boss for a raise) may help him or her in the majority culture but can create problems within his or her native culture.
- Do you or a family member dread being alone? Individuals inured to a nuclear family may be uncomfortable with solitude. Also, some cultures define emotional closeness as the presence of multiple family members, rather than companionship between husband and wife.
- Is your family comfortable with people from the mainstream culture? Cultural integration requires multicultural contacts. Some families, however, try to maintain their culture at the expense of their stated values. For example, a dishonest, superficial friend from the native culture may be more highly valued than an honest person from the mainstream. These cultural distortions produce mixed messages for all involved.
Understanding how a patient’s cultural background intertwines with relationship concerns, communication issues, and family problems is key to diagnosis and to building a therapeutic alliance.
Asking patients from any culture these six questions can uncover subtleties of cultural interaction that may be contributing to an adjustment, anxiety, depressive, or other disorder.
- Are your parents content living in the United States? This is especially pertinent when treating children or second-generation adults for relationship problems or for an adjustment, anxiety, or depressive disorder. In clinical practice, we have seen that when parents are satisfied with their new home, children adapt more readily.
- What mainstream practices has your family adopted? Families tend to incorporate customs and values of the surrounding environment while maintaining much of their core culture. The family’s overriding desire to maintain cultural purity may turn some family members against their ethnicity. For example, children may marry outside their culture to escape a rigid or controlling family or ethnic environment. These divergences, however, are often fraught with guilt and consternation among family members.
- What value clashes persist in your family? Disagreements over dating or participation in athletics or cheerleading are common. Respect, which may be defined as acquiescing to elders’ opinions, can be an issue regarding personal relationships, occupational choices, or nursing home placement.
- Can everyone speak freely in your family? In many cultures, assertiveness is perceived as rude. Therefore, patients may need alternate methods of conflict resolution. For example, teaching the patient a more-direct communication pattern (such as politely asking the boss for a raise) may help him or her in the majority culture but can create problems within his or her native culture.
- Do you or a family member dread being alone? Individuals inured to a nuclear family may be uncomfortable with solitude. Also, some cultures define emotional closeness as the presence of multiple family members, rather than companionship between husband and wife.
- Is your family comfortable with people from the mainstream culture? Cultural integration requires multicultural contacts. Some families, however, try to maintain their culture at the expense of their stated values. For example, a dishonest, superficial friend from the native culture may be more highly valued than an honest person from the mainstream. These cultural distortions produce mixed messages for all involved.
Dr. Benjaminis a staff psychiatrist at the Oklahoma City Veterans Administration Medical Center and is clinical assistant professor, department of psychiatry and behavioral sciences, University of Oklahoma Health Sciences Center, Oklahoma City.
Dr. Mosalleaei-Benjamin is a third-year resident in internal medicine, University of Oklahoma Health Sciences Center.
Dr. Benjaminis a staff psychiatrist at the Oklahoma City Veterans Administration Medical Center and is clinical assistant professor, department of psychiatry and behavioral sciences, University of Oklahoma Health Sciences Center, Oklahoma City.
Dr. Mosalleaei-Benjamin is a third-year resident in internal medicine, University of Oklahoma Health Sciences Center.
No mystery about hypnosis
Hypnosis’ decline in psychiatric training programs is surprising, given today’s emphasis on short-term therapies. Hypnosis can be very useful—particularly for patients with anxiety disorders, phobias, and posttraumatic stress disorder—as David Spiegel, MD, of Stanford University writes in his thoughtful scientific review.
My first experience with hypnosis was as a University of North Carolina undergraduate, when a psychology professor did a group hypnotic induction. I scored 7 out of 12 on the Stanford Hypnotic Susceptibility Scale (SHSS), which indicated I was “moderately” hypnotizable. I felt good because the professor said hypnotizable people are curious, brave, and open to new experiences.
Later, as a Stanford University medical student, I was hired as a hypnotist at the Stanford Hypnotic Research Center. My job was to administer a new 5-point SHSS to undergraduates who had been screened with the 12-point version. The shorter version included a 30-minute hypnotic induction—instead of the regular 60 minutes—and was designed for clinical practice.
For 6 months I immediately hypnotized every student, and they all scored at the top of the scale. Naturally, I assumed I was God’s gift to hypnosis. My euphoria ended abruptly, however, when one student scored zero. Rather than going into a trance, he stared at me for a half-hour with pity and mild aggression in his eyes.
From then on, every subject behaved the same way, and none scored more than 1 point. I resigned because I couldn’t take it anymore. Later, I got over my narcissistic wound when I realized I had been involved in a blinded experiment to see how well the shorter scale evaluated students who scored very high or very low on the longer version.
Despite this setback, I have used hypnosis over the years to help treat a variety of psychiatric conditions, with varying degrees of success. I have never, however, regained a belief in my own singularity.

James Randolph Hillard, MD
Editor-in-Chief
James Randolph Hillard, MD
Editor-in-Chief
James Randolph Hillard, MD
Editor-in-Chief
Hypnosis’ decline in psychiatric training programs is surprising, given today’s emphasis on short-term therapies. Hypnosis can be very useful—particularly for patients with anxiety disorders, phobias, and posttraumatic stress disorder—as David Spiegel, MD, of Stanford University writes in his thoughtful scientific review.
My first experience with hypnosis was as a University of North Carolina undergraduate, when a psychology professor did a group hypnotic induction. I scored 7 out of 12 on the Stanford Hypnotic Susceptibility Scale (SHSS), which indicated I was “moderately” hypnotizable. I felt good because the professor said hypnotizable people are curious, brave, and open to new experiences.
Later, as a Stanford University medical student, I was hired as a hypnotist at the Stanford Hypnotic Research Center. My job was to administer a new 5-point SHSS to undergraduates who had been screened with the 12-point version. The shorter version included a 30-minute hypnotic induction—instead of the regular 60 minutes—and was designed for clinical practice.
For 6 months I immediately hypnotized every student, and they all scored at the top of the scale. Naturally, I assumed I was God’s gift to hypnosis. My euphoria ended abruptly, however, when one student scored zero. Rather than going into a trance, he stared at me for a half-hour with pity and mild aggression in his eyes.
From then on, every subject behaved the same way, and none scored more than 1 point. I resigned because I couldn’t take it anymore. Later, I got over my narcissistic wound when I realized I had been involved in a blinded experiment to see how well the shorter scale evaluated students who scored very high or very low on the longer version.
Despite this setback, I have used hypnosis over the years to help treat a variety of psychiatric conditions, with varying degrees of success. I have never, however, regained a belief in my own singularity.
Hypnosis’ decline in psychiatric training programs is surprising, given today’s emphasis on short-term therapies. Hypnosis can be very useful—particularly for patients with anxiety disorders, phobias, and posttraumatic stress disorder—as David Spiegel, MD, of Stanford University writes in his thoughtful scientific review.
My first experience with hypnosis was as a University of North Carolina undergraduate, when a psychology professor did a group hypnotic induction. I scored 7 out of 12 on the Stanford Hypnotic Susceptibility Scale (SHSS), which indicated I was “moderately” hypnotizable. I felt good because the professor said hypnotizable people are curious, brave, and open to new experiences.
Later, as a Stanford University medical student, I was hired as a hypnotist at the Stanford Hypnotic Research Center. My job was to administer a new 5-point SHSS to undergraduates who had been screened with the 12-point version. The shorter version included a 30-minute hypnotic induction—instead of the regular 60 minutes—and was designed for clinical practice.
For 6 months I immediately hypnotized every student, and they all scored at the top of the scale. Naturally, I assumed I was God’s gift to hypnosis. My euphoria ended abruptly, however, when one student scored zero. Rather than going into a trance, he stared at me for a half-hour with pity and mild aggression in his eyes.
From then on, every subject behaved the same way, and none scored more than 1 point. I resigned because I couldn’t take it anymore. Later, I got over my narcissistic wound when I realized I had been involved in a blinded experiment to see how well the shorter scale evaluated students who scored very high or very low on the longer version.
Despite this setback, I have used hypnosis over the years to help treat a variety of psychiatric conditions, with varying degrees of success. I have never, however, regained a belief in my own singularity.
Brain/body connection: Treating depression in patients with cardiovascular disease
Depression can exacerbate cardiovascular disease (CVD), and CVD can exacerbate depression (Figure). Thus, effectively treating depression enhances heart disease treatment, particularly if psychiatrists and medical physicians collaborate in providing patient care.
This article describes a patient with risk factors for heart disease, illustrates the physiologic pathways that link depression and CVD, and offers clinical tips to help you improve outcomes for patients with both disorders.
Case report: Trying to ‘get going’
Mr. D, age 51, presents with vegetative symptoms and a personal and family history of CVD, depression, and substance abuse disorders. He was born in a small town in Kentucky and raised in Louisville’s poorest neighborhood. After his mother died at age 42 of “hardening of the arteries,” his father started drinking more, working less, and “never really got going again.”
Figure Neuroendocrine pathways by which depression may cause or promote CVD
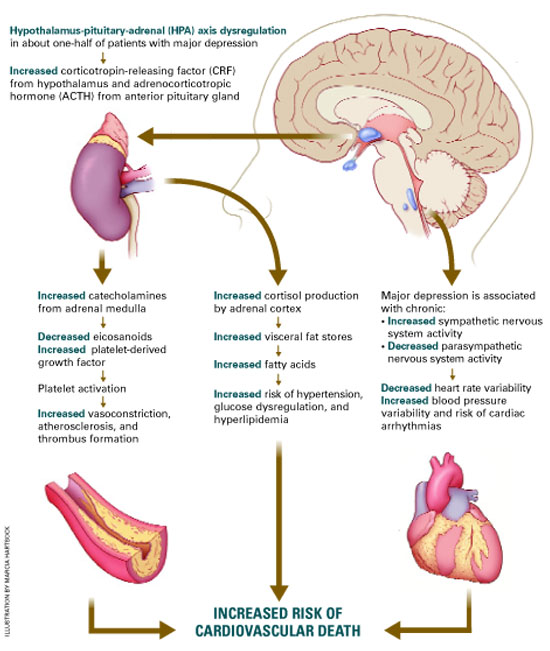
Among patients with a recent myocardial infarction (MI), as many as two-thirds report depressive symptoms.1 Major depression has been reported in:
- 16% to 22% of patients hospitalized post-MI,2,3 compared with 5% in the general population and 10% in the primary care population4
- 15% of patients with unstable angina5 and 20% of patients undergoing coronary artery bypass (CABG) surgery.6
Among the annual 1.5 million Americans who have an acute MI or unstable angina, 40% develop depression immediately thereafter. These 600,000 depressed patients are three times more likely to die within 6 months, compared with post-MI patients who are not depressed.7
Mr. D worked 20 years as a construction contractor, often running several work crews at once. At age 41, he slid into a depressive episode after his second divorce. He struggled with low energy, disturbed sleep, hopelessness, and increased smoking and drinking for 1 year, but he did not seek help.
Two years later, he suffered an inferior wall transmural myocardial infarction. His CVD risk factors included family history of early heart disease, smoking for 32 years, and elevated low-density lipoprotein (LDL) cholesterol. After subsequent episodes of unstable angina, stents were placed in two coronary arteries. Though his cardiologist cleared him to return to work, he felt able to work only part-time and erratically.
During a visit to their family doctor several years later, Mr. D’s wife suggested that her husband might be depressed. Reluctantly, Mr. D consulted a psychiatrist.
The psychiatrist diagnosed major depressive disorder and prescribed sertraline, 50 mg/d. Within 2 months, Mr. D’s symptoms had dropped by 50% on a symptom severity measure. He did not refill his prescription, however, because of concerns about sexual side effects. Two months later he was hospitalized for another episode of unstable angina. His depression had returned within 1 month of stopping sertraline.
The psychiatrist switched him to citalopram, 20 mg/d, and carefully monitored depressive symptoms, side effects, and medication adherence. Aside from talking with the psychiatrist for a half-hour in his family doctor’s office every few weeks, Mr. D refused to undergo psychotherapy. He eventually achieved depression remission with a combination of citalopram, 20 mg/d, and nefazodone, 200 mg/d.
Depression-CVD connection
As in Mr. D’s case, depression and CVD commonly occur together, often with serious consequences (Box). 1-7 The association between depression and CVD is not limited to depression’s effect on existing disease, however. Depression often precedes coronary disease by about 30 years—suggesting possible cause and effect. Two systematic reviews8,9 found that depression increased CVD risk by 64%.
Seven well-controlled studies5-7,10-13 compared the relative effect of depression on the cardiovascular system with that of established CVD predictors. All seven found depression’s independent effect to be significant and comparable to or greater than that of ejection fraction, previous MI history, or number of vessels with >50% narrowing.
Comorbid depression and CVD usually persists months or years,14 and most studies indicate a dose-response relationship; the more severe the depression, the greater the risk for CVD to develop or progress.8,15
The link between depression treatment and CVD risk has not been well-studied. The only randomized, controlled trial found that cognitive therapy for depression did not significantly reduce cardiac events among patients with known CVD.16
Possible mechanisms
Depression’s effect on CVD. How does depression affect CVD development and progression? Both behavioral and biological pathways may be involved.17 The behavioral pathway proposes that depression triggers behaviors—such as smoking, overeating, and sedentary lifestyles—that increase the risk of developing or worsening CVD. The biological pathway proposes that neuroendocrine changes during depression accelerate CVD development.
About one-half of persons with major depression exhibit hypothalamic-pituitary-adrenal (HPA) axis dysregulation, with excessive secretion of corticotropin releasing factor (CRF) and chronically elevated cortisol.18 This HPA dysregulation is related to defective negative feedback at the paraventricular nucleus of the hypothalamus. Chronic HPA axis dysregulation promotes vascular inflammation, and several studies have reported C-reactive protein elevation and cytokine changes in patients with major depression.19,20
Major depression is also associated with excessive sympathetic and diminished parasym-pathetic nervous system activity, potentially contributing to hypertension, increased resting heart rate, decreased heart rate variability, and altered endothelial function.2,21,22 Each of these factors facilitates arterial plaque formation.
Depression may also exacerbate chronic anxiety and other forms of distress. The combined effects of an overtaxed central nervous system, neuroendocrine dysregulation, and unhealthy behaviors may eventually overwhelm the cardiovascular system.
CVD’s effect on depression. How does CVD contribute to depression? The vascular depression hypothesis23 proposes that diffuse heart and brain atherosclerosis restricts perfusion of limbic and cortical structures that regulate mood. A first depressive episode after acute MI or CABG probably represents exacerbation of cerebrovascular insufficiency that preceded the coronary event.
Table
Four keys to effectively treat depression in patients with heart disease
|
In practical terms, this means that pathways linking depression and heart disease include not only biological factors but also:
- psychological factors such as depression, anxiety, and chronic stress
- behavioral factors such as smoking, physical inactivity, and high-fat diet.
How to improve outcomes
Patients with CVD commonly do not receive effective depression treatment:
- Internists and family physicians give preferential attention to physical illness.
- Patients may have insufficient access to mental health specialists.
- Physicians do not adequately monitor depression treatment.
- Patients are reluctant to accept the stigma of mental illness.
By collaborating with primary care physicians, you can improve the likelihood that depression treatment will achieve remission and prevent relapse (Table).
Risk factors for CVD. Depression contributes to heart disease by exacerbating four major CVD risk factors—smoking, diabetes, obesity, and physical inactivity. By effectively treating depression, you may help patients avoid common depressive symptoms—such as overeating and sedentary behaviors—that are related to low energy or fatigue.
Educate middle-aged patients with depression about CVD’s associated risk. Prochaska’s “stages of change” (see Related resources) can help them stop smoking, lose weight, and exercise.
Access to cardiac care. Depressed patients may be less motivated than nondepressed patients to pursue cardiac care.24 Therefore, you may need to:
- encourage your patients to take advantage of indicated state-of-the-art care, including stents, bypass surgery, and medications
- understand patients’ complex cardiac regimens and help them adhere when depression interferes with their motivation.
Effective depression treatment
Patient history. For depressed patients older than 40, take a careful inventory of CVD risk factors:
- family history of heart disease before age 60 for men and age 70 for women
- personal history of smoking, blood pressure >140/90 mm Hg, LDL cholesterol >100 mg/dL, type 2 diabetes, body mass index >30, or physical inactivity (<30 minutes of walking 3 days a week).
In general, the more risk factors, the greater the risk of CVD.
Antidepressant selection. Selective serotonin reuptake inhibitors (SSRIs) are safe and effective for treating major depression in CVD and congestive heart failure.25 Venlafaxine at doses >300 mg/d may increase blood pressure, so use this drug with caution in depressed patients with hypertension.
No controlled clinical trials have gauged the safety and efficacy of bupropion or mirtazapine in patients with CVD.
Tricyclic antidepressants are contraindicated for 6 months post-MI because they may contribute to arrhythmias. Avoid using them in depressed patients with CVD or conduction defects because of their quinidine-like effects on conduction.
Cardiac medications. Contrary to folk wisdom, beta blockers do not cause depression.26 Whether or not a patient is depressed, our primary care and cardiology colleagues can use beta blockers to help regulate the peripheral autonomic nervous system, reducing high blood pressure and the risk of arrhythmias.
SSRIs may increase blood levels of beta blockers, warfarin, and other cardiac medications via cytochrome P-450 isoenzyme inhibition. Make sure warfarin levels and other cardiac drug effects are well monitored when you adjust psychotropic dosages.
Divalproex and SSRIs also may reduce platelet aggregation. Patients who are receiving concomitant aspirin or warfarin may bruise or bleed easily and require dosage reductions or medication changes.
Psychotherapy. All patients with major or minor depression and CVD are considered high-risk and are candidates for a trial of brief psychotherapy. Therapeutic goals are to achieve full remission of depressive symptoms as rapidly as possible, prevent relapse, and maximize adherence to cardiac and depression drug regimens.
Collaborate closely with the cardiologist or primary care physician during the patient’s depressive episode and occasionally during maintenance treatment. Discuss or share notes on the patient’s depressive and cardiac disorders, medication management, symptom monitoring, and behavior changes needed to reduce cardiac risk.
With your added support, patients with depression and CVD are more likely to adhere to antidepressant medications and achieve symptom remission.
- National Institute of Mental Health. Depression and heart disease. www.nimh.nih.gov/publicat/depheart.cfm.
- Dewan NA, Suresh DP, Blomkalns A. Selecting safe psychotropics for post-MI patients. Current Psychiatry. 2003;2(3):15-21.
- Prochaska JO, Norcross JC, DiClemente CC. Changing for good. New York: Avon, 1994.
Drug brand names
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Paroxetine • Paxil
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
Dr. Wulsin is a consultant to Pfizer Inc. and Janssen Pharmaceutica.
Dr. Vieweg is a speaker for Janssen Pharmaceutica, Eli Lilly and Co., Pfizer Inc., Wyeth Pharmaceuticals, Forest Pharmaceuticals, and GlaxoSmithKline.
Dr. Fernandez reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Cassem N, Hackett T. Psychiatric condition in a coronary care unit. Ann Intern Med 1971;75:9-14.
2. Glassman A, Shapiro P. Depression and the course of coronary artery disease. Am J Psychiatry 1998;155:4-11.
3. Carney R, Freedland K, Sheline Y, Weiss E. Depression and coronary heart disease: a review for cardiologists. Clin Cardiol 1997;20:196-200.
4. Katon W, Schulbert H. Epidemiology of depression in primary care. Gen Hosp Psychiatry 1992;14:237-47.
5. Lesperance F, Frasure-Smith N, Theroux P. Depression and 1-year prognosis in unstable angina. Arch Intern Med 2000;160:1354-60.
6. Connerney I, Shapiro P, McLaughlin J, et al. Relation between depression after coronary artery bypass surgery and 12-month outcome: a prospective study. Lancet 2001;358:1766-71.
7. Frasure-Smith N, Lesperance F, Talajic M. Depression and 18-month prognosis after myocardial infarction. Circulation 1995;91:999-1005.
8. Rugulies R. Depression as a predictor for coronary heart disease. Am J Prev Med 2002;23:51-61.
9. Wulsin L, Singal B. Do depressive symptoms increase the risk for the onset of coronary disease? A systematic quantitative review. Psychosom Med 2003;65:201-10.
10. Carney R, Rich M, Freedland K, et al. Major depressive disorder predicts cardiac events in patients with coronary artery disease. Psychosom Med 1988;50:627-33.
11. Ladwig K, Roll G, Breithardt G, Borggrefe M. Extracardiac contributions to chest pain perception in patients 6 months after acute myocardial infarction. Am Heart J 1999;137:528-34.
12. Levine J, Covino N, Slack W, et al. Psychological predictors of subsequent medical care among patients hospitalized with cardiac disease. J Cardiopulm Rehabil 1996;16:109-16.
13. Lesperance F, Frasure-Smith N, Talajic M, Bourassa M. Five-year risk of cardiac mortality in relation to initial severity and one-year changes in depression symptoms after myocardial infarction. Circulation 2002;105:1049-53.
14. Dwight M, Stoudemire A. Effects of depressive disorders on coronary artery disease: a review. Harv Rev Psychiatry 1997;5:115-122.
15. Penninx B, Beekman A, Honig A, et al. Depression and cardiac mortality. Arch Gen Psychiatry 2001;58:221-7.
16. Writing committee of the ENRICHD investigators. Effects of treating depression and low perceived social support on clinical events after myocardial infarction. JAMA 2003;289:3106-16.
17. Carney RM, Freedland K, Miller G, Jaffe AS. Depression as a risk factor for cardiac mortality and morbidity: a review of potential mechanisms. J Psychosom Res 2002;53:897-902.
18. Musselman D, Evans D, Nemeroff C. The relationship of depression to cardiovascular disease. Arch Gen Psychiatry 1998;55:580-92.
19. Kop WJ. Chronic and acute psychological risk factors for clinical manifestations of coronary artery disease. Psychosom Med 1999;61:476-86.
20. Miller G, Cohen S, Herbert T. Pathways linking major depression and immunity in ambulatory female patients. Psychosom Med 1999;61:850-60.
21. Carney R, Freedland K, Stein P. Change in heart rate and heart rate variability during treatment for depression in patients with coronary heart disease. Psychosom Med 2000;62:639-47.
22. Carney R, Freedland K, Miller G, Jaffe A. Depression as a risk factor for cardiac mortality and morbidity: a review of potential mechanisms. J Psychosom Res 2002;53:897-902.
23. Alexopoulos G, Meyers B, Young R, et al. Vascular depression hypothesis. Psychosom Med 1997;58:113-121.
24. Ziegelstein R, Fauerbach J, Stevens S, et al. Patients with depression are less likely to follow recommendations to reduce cardiac risk during recovery from a myocardial infarction. Arch Intern Med 2000;160:1818-23.
25. Glassman AH, O’Connor C, Califf RM, et al. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002;288:701-9.
26. Ko D, Hebert P, Coffey C, et al. B-blocker therapy and symptoms of depression, fatigue, and sexual dysfunction. JAMA 2002;288:351-7.
Depression can exacerbate cardiovascular disease (CVD), and CVD can exacerbate depression (Figure). Thus, effectively treating depression enhances heart disease treatment, particularly if psychiatrists and medical physicians collaborate in providing patient care.
This article describes a patient with risk factors for heart disease, illustrates the physiologic pathways that link depression and CVD, and offers clinical tips to help you improve outcomes for patients with both disorders.
Case report: Trying to ‘get going’
Mr. D, age 51, presents with vegetative symptoms and a personal and family history of CVD, depression, and substance abuse disorders. He was born in a small town in Kentucky and raised in Louisville’s poorest neighborhood. After his mother died at age 42 of “hardening of the arteries,” his father started drinking more, working less, and “never really got going again.”
Figure Neuroendocrine pathways by which depression may cause or promote CVD
Among patients with a recent myocardial infarction (MI), as many as two-thirds report depressive symptoms.1 Major depression has been reported in:
- 16% to 22% of patients hospitalized post-MI,2,3 compared with 5% in the general population and 10% in the primary care population4
- 15% of patients with unstable angina5 and 20% of patients undergoing coronary artery bypass (CABG) surgery.6
Among the annual 1.5 million Americans who have an acute MI or unstable angina, 40% develop depression immediately thereafter. These 600,000 depressed patients are three times more likely to die within 6 months, compared with post-MI patients who are not depressed.7
Mr. D worked 20 years as a construction contractor, often running several work crews at once. At age 41, he slid into a depressive episode after his second divorce. He struggled with low energy, disturbed sleep, hopelessness, and increased smoking and drinking for 1 year, but he did not seek help.
Two years later, he suffered an inferior wall transmural myocardial infarction. His CVD risk factors included family history of early heart disease, smoking for 32 years, and elevated low-density lipoprotein (LDL) cholesterol. After subsequent episodes of unstable angina, stents were placed in two coronary arteries. Though his cardiologist cleared him to return to work, he felt able to work only part-time and erratically.
During a visit to their family doctor several years later, Mr. D’s wife suggested that her husband might be depressed. Reluctantly, Mr. D consulted a psychiatrist.
The psychiatrist diagnosed major depressive disorder and prescribed sertraline, 50 mg/d. Within 2 months, Mr. D’s symptoms had dropped by 50% on a symptom severity measure. He did not refill his prescription, however, because of concerns about sexual side effects. Two months later he was hospitalized for another episode of unstable angina. His depression had returned within 1 month of stopping sertraline.
The psychiatrist switched him to citalopram, 20 mg/d, and carefully monitored depressive symptoms, side effects, and medication adherence. Aside from talking with the psychiatrist for a half-hour in his family doctor’s office every few weeks, Mr. D refused to undergo psychotherapy. He eventually achieved depression remission with a combination of citalopram, 20 mg/d, and nefazodone, 200 mg/d.
Depression-CVD connection
As in Mr. D’s case, depression and CVD commonly occur together, often with serious consequences (Box). 1-7 The association between depression and CVD is not limited to depression’s effect on existing disease, however. Depression often precedes coronary disease by about 30 years—suggesting possible cause and effect. Two systematic reviews8,9 found that depression increased CVD risk by 64%.
Seven well-controlled studies5-7,10-13 compared the relative effect of depression on the cardiovascular system with that of established CVD predictors. All seven found depression’s independent effect to be significant and comparable to or greater than that of ejection fraction, previous MI history, or number of vessels with >50% narrowing.
Comorbid depression and CVD usually persists months or years,14 and most studies indicate a dose-response relationship; the more severe the depression, the greater the risk for CVD to develop or progress.8,15
The link between depression treatment and CVD risk has not been well-studied. The only randomized, controlled trial found that cognitive therapy for depression did not significantly reduce cardiac events among patients with known CVD.16
Possible mechanisms
Depression’s effect on CVD. How does depression affect CVD development and progression? Both behavioral and biological pathways may be involved.17 The behavioral pathway proposes that depression triggers behaviors—such as smoking, overeating, and sedentary lifestyles—that increase the risk of developing or worsening CVD. The biological pathway proposes that neuroendocrine changes during depression accelerate CVD development.
About one-half of persons with major depression exhibit hypothalamic-pituitary-adrenal (HPA) axis dysregulation, with excessive secretion of corticotropin releasing factor (CRF) and chronically elevated cortisol.18 This HPA dysregulation is related to defective negative feedback at the paraventricular nucleus of the hypothalamus. Chronic HPA axis dysregulation promotes vascular inflammation, and several studies have reported C-reactive protein elevation and cytokine changes in patients with major depression.19,20
Major depression is also associated with excessive sympathetic and diminished parasym-pathetic nervous system activity, potentially contributing to hypertension, increased resting heart rate, decreased heart rate variability, and altered endothelial function.2,21,22 Each of these factors facilitates arterial plaque formation.
Depression may also exacerbate chronic anxiety and other forms of distress. The combined effects of an overtaxed central nervous system, neuroendocrine dysregulation, and unhealthy behaviors may eventually overwhelm the cardiovascular system.
CVD’s effect on depression. How does CVD contribute to depression? The vascular depression hypothesis23 proposes that diffuse heart and brain atherosclerosis restricts perfusion of limbic and cortical structures that regulate mood. A first depressive episode after acute MI or CABG probably represents exacerbation of cerebrovascular insufficiency that preceded the coronary event.
Table
Four keys to effectively treat depression in patients with heart disease
|
In practical terms, this means that pathways linking depression and heart disease include not only biological factors but also:
- psychological factors such as depression, anxiety, and chronic stress
- behavioral factors such as smoking, physical inactivity, and high-fat diet.
How to improve outcomes
Patients with CVD commonly do not receive effective depression treatment:
- Internists and family physicians give preferential attention to physical illness.
- Patients may have insufficient access to mental health specialists.
- Physicians do not adequately monitor depression treatment.
- Patients are reluctant to accept the stigma of mental illness.
By collaborating with primary care physicians, you can improve the likelihood that depression treatment will achieve remission and prevent relapse (Table).
Risk factors for CVD. Depression contributes to heart disease by exacerbating four major CVD risk factors—smoking, diabetes, obesity, and physical inactivity. By effectively treating depression, you may help patients avoid common depressive symptoms—such as overeating and sedentary behaviors—that are related to low energy or fatigue.
Educate middle-aged patients with depression about CVD’s associated risk. Prochaska’s “stages of change” (see Related resources) can help them stop smoking, lose weight, and exercise.
Access to cardiac care. Depressed patients may be less motivated than nondepressed patients to pursue cardiac care.24 Therefore, you may need to:
- encourage your patients to take advantage of indicated state-of-the-art care, including stents, bypass surgery, and medications
- understand patients’ complex cardiac regimens and help them adhere when depression interferes with their motivation.
Effective depression treatment
Patient history. For depressed patients older than 40, take a careful inventory of CVD risk factors:
- family history of heart disease before age 60 for men and age 70 for women
- personal history of smoking, blood pressure >140/90 mm Hg, LDL cholesterol >100 mg/dL, type 2 diabetes, body mass index >30, or physical inactivity (<30 minutes of walking 3 days a week).
In general, the more risk factors, the greater the risk of CVD.
Antidepressant selection. Selective serotonin reuptake inhibitors (SSRIs) are safe and effective for treating major depression in CVD and congestive heart failure.25 Venlafaxine at doses >300 mg/d may increase blood pressure, so use this drug with caution in depressed patients with hypertension.
No controlled clinical trials have gauged the safety and efficacy of bupropion or mirtazapine in patients with CVD.
Tricyclic antidepressants are contraindicated for 6 months post-MI because they may contribute to arrhythmias. Avoid using them in depressed patients with CVD or conduction defects because of their quinidine-like effects on conduction.
Cardiac medications. Contrary to folk wisdom, beta blockers do not cause depression.26 Whether or not a patient is depressed, our primary care and cardiology colleagues can use beta blockers to help regulate the peripheral autonomic nervous system, reducing high blood pressure and the risk of arrhythmias.
SSRIs may increase blood levels of beta blockers, warfarin, and other cardiac medications via cytochrome P-450 isoenzyme inhibition. Make sure warfarin levels and other cardiac drug effects are well monitored when you adjust psychotropic dosages.
Divalproex and SSRIs also may reduce platelet aggregation. Patients who are receiving concomitant aspirin or warfarin may bruise or bleed easily and require dosage reductions or medication changes.
Psychotherapy. All patients with major or minor depression and CVD are considered high-risk and are candidates for a trial of brief psychotherapy. Therapeutic goals are to achieve full remission of depressive symptoms as rapidly as possible, prevent relapse, and maximize adherence to cardiac and depression drug regimens.
Collaborate closely with the cardiologist or primary care physician during the patient’s depressive episode and occasionally during maintenance treatment. Discuss or share notes on the patient’s depressive and cardiac disorders, medication management, symptom monitoring, and behavior changes needed to reduce cardiac risk.
With your added support, patients with depression and CVD are more likely to adhere to antidepressant medications and achieve symptom remission.
- National Institute of Mental Health. Depression and heart disease. www.nimh.nih.gov/publicat/depheart.cfm.
- Dewan NA, Suresh DP, Blomkalns A. Selecting safe psychotropics for post-MI patients. Current Psychiatry. 2003;2(3):15-21.
- Prochaska JO, Norcross JC, DiClemente CC. Changing for good. New York: Avon, 1994.
Drug brand names
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Paroxetine • Paxil
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
Dr. Wulsin is a consultant to Pfizer Inc. and Janssen Pharmaceutica.
Dr. Vieweg is a speaker for Janssen Pharmaceutica, Eli Lilly and Co., Pfizer Inc., Wyeth Pharmaceuticals, Forest Pharmaceuticals, and GlaxoSmithKline.
Dr. Fernandez reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Depression can exacerbate cardiovascular disease (CVD), and CVD can exacerbate depression (Figure). Thus, effectively treating depression enhances heart disease treatment, particularly if psychiatrists and medical physicians collaborate in providing patient care.
This article describes a patient with risk factors for heart disease, illustrates the physiologic pathways that link depression and CVD, and offers clinical tips to help you improve outcomes for patients with both disorders.
Case report: Trying to ‘get going’
Mr. D, age 51, presents with vegetative symptoms and a personal and family history of CVD, depression, and substance abuse disorders. He was born in a small town in Kentucky and raised in Louisville’s poorest neighborhood. After his mother died at age 42 of “hardening of the arteries,” his father started drinking more, working less, and “never really got going again.”
Figure Neuroendocrine pathways by which depression may cause or promote CVD
Among patients with a recent myocardial infarction (MI), as many as two-thirds report depressive symptoms.1 Major depression has been reported in:
- 16% to 22% of patients hospitalized post-MI,2,3 compared with 5% in the general population and 10% in the primary care population4
- 15% of patients with unstable angina5 and 20% of patients undergoing coronary artery bypass (CABG) surgery.6
Among the annual 1.5 million Americans who have an acute MI or unstable angina, 40% develop depression immediately thereafter. These 600,000 depressed patients are three times more likely to die within 6 months, compared with post-MI patients who are not depressed.7
Mr. D worked 20 years as a construction contractor, often running several work crews at once. At age 41, he slid into a depressive episode after his second divorce. He struggled with low energy, disturbed sleep, hopelessness, and increased smoking and drinking for 1 year, but he did not seek help.
Two years later, he suffered an inferior wall transmural myocardial infarction. His CVD risk factors included family history of early heart disease, smoking for 32 years, and elevated low-density lipoprotein (LDL) cholesterol. After subsequent episodes of unstable angina, stents were placed in two coronary arteries. Though his cardiologist cleared him to return to work, he felt able to work only part-time and erratically.
During a visit to their family doctor several years later, Mr. D’s wife suggested that her husband might be depressed. Reluctantly, Mr. D consulted a psychiatrist.
The psychiatrist diagnosed major depressive disorder and prescribed sertraline, 50 mg/d. Within 2 months, Mr. D’s symptoms had dropped by 50% on a symptom severity measure. He did not refill his prescription, however, because of concerns about sexual side effects. Two months later he was hospitalized for another episode of unstable angina. His depression had returned within 1 month of stopping sertraline.
The psychiatrist switched him to citalopram, 20 mg/d, and carefully monitored depressive symptoms, side effects, and medication adherence. Aside from talking with the psychiatrist for a half-hour in his family doctor’s office every few weeks, Mr. D refused to undergo psychotherapy. He eventually achieved depression remission with a combination of citalopram, 20 mg/d, and nefazodone, 200 mg/d.
Depression-CVD connection
As in Mr. D’s case, depression and CVD commonly occur together, often with serious consequences (Box). 1-7 The association between depression and CVD is not limited to depression’s effect on existing disease, however. Depression often precedes coronary disease by about 30 years—suggesting possible cause and effect. Two systematic reviews8,9 found that depression increased CVD risk by 64%.
Seven well-controlled studies5-7,10-13 compared the relative effect of depression on the cardiovascular system with that of established CVD predictors. All seven found depression’s independent effect to be significant and comparable to or greater than that of ejection fraction, previous MI history, or number of vessels with >50% narrowing.
Comorbid depression and CVD usually persists months or years,14 and most studies indicate a dose-response relationship; the more severe the depression, the greater the risk for CVD to develop or progress.8,15
The link between depression treatment and CVD risk has not been well-studied. The only randomized, controlled trial found that cognitive therapy for depression did not significantly reduce cardiac events among patients with known CVD.16
Possible mechanisms
Depression’s effect on CVD. How does depression affect CVD development and progression? Both behavioral and biological pathways may be involved.17 The behavioral pathway proposes that depression triggers behaviors—such as smoking, overeating, and sedentary lifestyles—that increase the risk of developing or worsening CVD. The biological pathway proposes that neuroendocrine changes during depression accelerate CVD development.
About one-half of persons with major depression exhibit hypothalamic-pituitary-adrenal (HPA) axis dysregulation, with excessive secretion of corticotropin releasing factor (CRF) and chronically elevated cortisol.18 This HPA dysregulation is related to defective negative feedback at the paraventricular nucleus of the hypothalamus. Chronic HPA axis dysregulation promotes vascular inflammation, and several studies have reported C-reactive protein elevation and cytokine changes in patients with major depression.19,20
Major depression is also associated with excessive sympathetic and diminished parasym-pathetic nervous system activity, potentially contributing to hypertension, increased resting heart rate, decreased heart rate variability, and altered endothelial function.2,21,22 Each of these factors facilitates arterial plaque formation.
Depression may also exacerbate chronic anxiety and other forms of distress. The combined effects of an overtaxed central nervous system, neuroendocrine dysregulation, and unhealthy behaviors may eventually overwhelm the cardiovascular system.
CVD’s effect on depression. How does CVD contribute to depression? The vascular depression hypothesis23 proposes that diffuse heart and brain atherosclerosis restricts perfusion of limbic and cortical structures that regulate mood. A first depressive episode after acute MI or CABG probably represents exacerbation of cerebrovascular insufficiency that preceded the coronary event.
Table
Four keys to effectively treat depression in patients with heart disease
|
In practical terms, this means that pathways linking depression and heart disease include not only biological factors but also:
- psychological factors such as depression, anxiety, and chronic stress
- behavioral factors such as smoking, physical inactivity, and high-fat diet.
How to improve outcomes
Patients with CVD commonly do not receive effective depression treatment:
- Internists and family physicians give preferential attention to physical illness.
- Patients may have insufficient access to mental health specialists.
- Physicians do not adequately monitor depression treatment.
- Patients are reluctant to accept the stigma of mental illness.
By collaborating with primary care physicians, you can improve the likelihood that depression treatment will achieve remission and prevent relapse (Table).
Risk factors for CVD. Depression contributes to heart disease by exacerbating four major CVD risk factors—smoking, diabetes, obesity, and physical inactivity. By effectively treating depression, you may help patients avoid common depressive symptoms—such as overeating and sedentary behaviors—that are related to low energy or fatigue.
Educate middle-aged patients with depression about CVD’s associated risk. Prochaska’s “stages of change” (see Related resources) can help them stop smoking, lose weight, and exercise.
Access to cardiac care. Depressed patients may be less motivated than nondepressed patients to pursue cardiac care.24 Therefore, you may need to:
- encourage your patients to take advantage of indicated state-of-the-art care, including stents, bypass surgery, and medications
- understand patients’ complex cardiac regimens and help them adhere when depression interferes with their motivation.
Effective depression treatment
Patient history. For depressed patients older than 40, take a careful inventory of CVD risk factors:
- family history of heart disease before age 60 for men and age 70 for women
- personal history of smoking, blood pressure >140/90 mm Hg, LDL cholesterol >100 mg/dL, type 2 diabetes, body mass index >30, or physical inactivity (<30 minutes of walking 3 days a week).
In general, the more risk factors, the greater the risk of CVD.
Antidepressant selection. Selective serotonin reuptake inhibitors (SSRIs) are safe and effective for treating major depression in CVD and congestive heart failure.25 Venlafaxine at doses >300 mg/d may increase blood pressure, so use this drug with caution in depressed patients with hypertension.
No controlled clinical trials have gauged the safety and efficacy of bupropion or mirtazapine in patients with CVD.
Tricyclic antidepressants are contraindicated for 6 months post-MI because they may contribute to arrhythmias. Avoid using them in depressed patients with CVD or conduction defects because of their quinidine-like effects on conduction.
Cardiac medications. Contrary to folk wisdom, beta blockers do not cause depression.26 Whether or not a patient is depressed, our primary care and cardiology colleagues can use beta blockers to help regulate the peripheral autonomic nervous system, reducing high blood pressure and the risk of arrhythmias.
SSRIs may increase blood levels of beta blockers, warfarin, and other cardiac medications via cytochrome P-450 isoenzyme inhibition. Make sure warfarin levels and other cardiac drug effects are well monitored when you adjust psychotropic dosages.
Divalproex and SSRIs also may reduce platelet aggregation. Patients who are receiving concomitant aspirin or warfarin may bruise or bleed easily and require dosage reductions or medication changes.
Psychotherapy. All patients with major or minor depression and CVD are considered high-risk and are candidates for a trial of brief psychotherapy. Therapeutic goals are to achieve full remission of depressive symptoms as rapidly as possible, prevent relapse, and maximize adherence to cardiac and depression drug regimens.
Collaborate closely with the cardiologist or primary care physician during the patient’s depressive episode and occasionally during maintenance treatment. Discuss or share notes on the patient’s depressive and cardiac disorders, medication management, symptom monitoring, and behavior changes needed to reduce cardiac risk.
With your added support, patients with depression and CVD are more likely to adhere to antidepressant medications and achieve symptom remission.
- National Institute of Mental Health. Depression and heart disease. www.nimh.nih.gov/publicat/depheart.cfm.
- Dewan NA, Suresh DP, Blomkalns A. Selecting safe psychotropics for post-MI patients. Current Psychiatry. 2003;2(3):15-21.
- Prochaska JO, Norcross JC, DiClemente CC. Changing for good. New York: Avon, 1994.
Drug brand names
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Paroxetine • Paxil
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
Dr. Wulsin is a consultant to Pfizer Inc. and Janssen Pharmaceutica.
Dr. Vieweg is a speaker for Janssen Pharmaceutica, Eli Lilly and Co., Pfizer Inc., Wyeth Pharmaceuticals, Forest Pharmaceuticals, and GlaxoSmithKline.
Dr. Fernandez reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Cassem N, Hackett T. Psychiatric condition in a coronary care unit. Ann Intern Med 1971;75:9-14.
2. Glassman A, Shapiro P. Depression and the course of coronary artery disease. Am J Psychiatry 1998;155:4-11.
3. Carney R, Freedland K, Sheline Y, Weiss E. Depression and coronary heart disease: a review for cardiologists. Clin Cardiol 1997;20:196-200.
4. Katon W, Schulbert H. Epidemiology of depression in primary care. Gen Hosp Psychiatry 1992;14:237-47.
5. Lesperance F, Frasure-Smith N, Theroux P. Depression and 1-year prognosis in unstable angina. Arch Intern Med 2000;160:1354-60.
6. Connerney I, Shapiro P, McLaughlin J, et al. Relation between depression after coronary artery bypass surgery and 12-month outcome: a prospective study. Lancet 2001;358:1766-71.
7. Frasure-Smith N, Lesperance F, Talajic M. Depression and 18-month prognosis after myocardial infarction. Circulation 1995;91:999-1005.
8. Rugulies R. Depression as a predictor for coronary heart disease. Am J Prev Med 2002;23:51-61.
9. Wulsin L, Singal B. Do depressive symptoms increase the risk for the onset of coronary disease? A systematic quantitative review. Psychosom Med 2003;65:201-10.
10. Carney R, Rich M, Freedland K, et al. Major depressive disorder predicts cardiac events in patients with coronary artery disease. Psychosom Med 1988;50:627-33.
11. Ladwig K, Roll G, Breithardt G, Borggrefe M. Extracardiac contributions to chest pain perception in patients 6 months after acute myocardial infarction. Am Heart J 1999;137:528-34.
12. Levine J, Covino N, Slack W, et al. Psychological predictors of subsequent medical care among patients hospitalized with cardiac disease. J Cardiopulm Rehabil 1996;16:109-16.
13. Lesperance F, Frasure-Smith N, Talajic M, Bourassa M. Five-year risk of cardiac mortality in relation to initial severity and one-year changes in depression symptoms after myocardial infarction. Circulation 2002;105:1049-53.
14. Dwight M, Stoudemire A. Effects of depressive disorders on coronary artery disease: a review. Harv Rev Psychiatry 1997;5:115-122.
15. Penninx B, Beekman A, Honig A, et al. Depression and cardiac mortality. Arch Gen Psychiatry 2001;58:221-7.
16. Writing committee of the ENRICHD investigators. Effects of treating depression and low perceived social support on clinical events after myocardial infarction. JAMA 2003;289:3106-16.
17. Carney RM, Freedland K, Miller G, Jaffe AS. Depression as a risk factor for cardiac mortality and morbidity: a review of potential mechanisms. J Psychosom Res 2002;53:897-902.
18. Musselman D, Evans D, Nemeroff C. The relationship of depression to cardiovascular disease. Arch Gen Psychiatry 1998;55:580-92.
19. Kop WJ. Chronic and acute psychological risk factors for clinical manifestations of coronary artery disease. Psychosom Med 1999;61:476-86.
20. Miller G, Cohen S, Herbert T. Pathways linking major depression and immunity in ambulatory female patients. Psychosom Med 1999;61:850-60.
21. Carney R, Freedland K, Stein P. Change in heart rate and heart rate variability during treatment for depression in patients with coronary heart disease. Psychosom Med 2000;62:639-47.
22. Carney R, Freedland K, Miller G, Jaffe A. Depression as a risk factor for cardiac mortality and morbidity: a review of potential mechanisms. J Psychosom Res 2002;53:897-902.
23. Alexopoulos G, Meyers B, Young R, et al. Vascular depression hypothesis. Psychosom Med 1997;58:113-121.
24. Ziegelstein R, Fauerbach J, Stevens S, et al. Patients with depression are less likely to follow recommendations to reduce cardiac risk during recovery from a myocardial infarction. Arch Intern Med 2000;160:1818-23.
25. Glassman AH, O’Connor C, Califf RM, et al. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002;288:701-9.
26. Ko D, Hebert P, Coffey C, et al. B-blocker therapy and symptoms of depression, fatigue, and sexual dysfunction. JAMA 2002;288:351-7.
1. Cassem N, Hackett T. Psychiatric condition in a coronary care unit. Ann Intern Med 1971;75:9-14.
2. Glassman A, Shapiro P. Depression and the course of coronary artery disease. Am J Psychiatry 1998;155:4-11.
3. Carney R, Freedland K, Sheline Y, Weiss E. Depression and coronary heart disease: a review for cardiologists. Clin Cardiol 1997;20:196-200.
4. Katon W, Schulbert H. Epidemiology of depression in primary care. Gen Hosp Psychiatry 1992;14:237-47.
5. Lesperance F, Frasure-Smith N, Theroux P. Depression and 1-year prognosis in unstable angina. Arch Intern Med 2000;160:1354-60.
6. Connerney I, Shapiro P, McLaughlin J, et al. Relation between depression after coronary artery bypass surgery and 12-month outcome: a prospective study. Lancet 2001;358:1766-71.
7. Frasure-Smith N, Lesperance F, Talajic M. Depression and 18-month prognosis after myocardial infarction. Circulation 1995;91:999-1005.
8. Rugulies R. Depression as a predictor for coronary heart disease. Am J Prev Med 2002;23:51-61.
9. Wulsin L, Singal B. Do depressive symptoms increase the risk for the onset of coronary disease? A systematic quantitative review. Psychosom Med 2003;65:201-10.
10. Carney R, Rich M, Freedland K, et al. Major depressive disorder predicts cardiac events in patients with coronary artery disease. Psychosom Med 1988;50:627-33.
11. Ladwig K, Roll G, Breithardt G, Borggrefe M. Extracardiac contributions to chest pain perception in patients 6 months after acute myocardial infarction. Am Heart J 1999;137:528-34.
12. Levine J, Covino N, Slack W, et al. Psychological predictors of subsequent medical care among patients hospitalized with cardiac disease. J Cardiopulm Rehabil 1996;16:109-16.
13. Lesperance F, Frasure-Smith N, Talajic M, Bourassa M. Five-year risk of cardiac mortality in relation to initial severity and one-year changes in depression symptoms after myocardial infarction. Circulation 2002;105:1049-53.
14. Dwight M, Stoudemire A. Effects of depressive disorders on coronary artery disease: a review. Harv Rev Psychiatry 1997;5:115-122.
15. Penninx B, Beekman A, Honig A, et al. Depression and cardiac mortality. Arch Gen Psychiatry 2001;58:221-7.
16. Writing committee of the ENRICHD investigators. Effects of treating depression and low perceived social support on clinical events after myocardial infarction. JAMA 2003;289:3106-16.
17. Carney RM, Freedland K, Miller G, Jaffe AS. Depression as a risk factor for cardiac mortality and morbidity: a review of potential mechanisms. J Psychosom Res 2002;53:897-902.
18. Musselman D, Evans D, Nemeroff C. The relationship of depression to cardiovascular disease. Arch Gen Psychiatry 1998;55:580-92.
19. Kop WJ. Chronic and acute psychological risk factors for clinical manifestations of coronary artery disease. Psychosom Med 1999;61:476-86.
20. Miller G, Cohen S, Herbert T. Pathways linking major depression and immunity in ambulatory female patients. Psychosom Med 1999;61:850-60.
21. Carney R, Freedland K, Stein P. Change in heart rate and heart rate variability during treatment for depression in patients with coronary heart disease. Psychosom Med 2000;62:639-47.
22. Carney R, Freedland K, Miller G, Jaffe A. Depression as a risk factor for cardiac mortality and morbidity: a review of potential mechanisms. J Psychosom Res 2002;53:897-902.
23. Alexopoulos G, Meyers B, Young R, et al. Vascular depression hypothesis. Psychosom Med 1997;58:113-121.
24. Ziegelstein R, Fauerbach J, Stevens S, et al. Patients with depression are less likely to follow recommendations to reduce cardiac risk during recovery from a myocardial infarction. Arch Intern Med 2000;160:1818-23.
25. Glassman AH, O’Connor C, Califf RM, et al. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002;288:701-9.
26. Ko D, Hebert P, Coffey C, et al. B-blocker therapy and symptoms of depression, fatigue, and sexual dysfunction. JAMA 2002;288:351-7.
SSRIs in children and adolescents: Where do we stand?
Caitlin McIntosh, 12, hanged herself with shoelaces weeks after starting treatment for depression with a selective serotonin reuptake inhibitor (SSRI). Matt Miller, 13, hanged himself in his bedroom closet after taking his seventh SSRI dose. Michael Shivak, 11, slashed his wrists in class—but survived—while taking an SSRI.
These adolescents’ parents testified at an FDA hearing Feb. 2 about possible increased risk of suicidality with SSRIs in depressed children and adolescents.
Other families related positive experiences. Sherri Walton said her daughter, Jordan, 14, has achieved “enormous benefit” from taking SSRIs for obsessive-compulsive disorder. Suzanne Vogel-Scibilia told the FDA panel she is convinced that her two children with psychiatric disorders lead full lives because of SSRIs.
Sensitive to the anguish of grieving families but not wanting to deprive seriously depressed children of effective treatment, the FDA is proceeding methodically with its inquiry—probably at least until summer.
In the meantime, this article offers resources to help you answer questions from parents concerned about their children starting or continuing SSRIs. We include pediatric antidepressant dosing recommendations and data on benefits and risks of SSRIs and other reuptake inhibitors in young patients.
Why the FDA inquiry?
SSRI-associated behavioral activation and suicidal ideation in children and adolescents were reported anecdotally, as case reports, in the early 1990s,1 and more recently.2 No convincing evidence has shown, however, that SSRIs increase the risk of suicide. In fact, widespread use of SSRIs has been associated with reduced suicide rates.3
In May 2003, unpublished data submitted to the FDA from placebo-controlled trials suggested an increased risk of “possibly suicide-related” events and “suicide attempts” in pediatric patients taking paroxetine for major depression. No suicides were reported.
The Medicines and Healthcare Products Regulatory Agency (MHRA)—the United Kingdom’s equivalent of the Food and Drug Administration—responded by warning British physicians against prescribing paroxetine for depressed patients younger than 18. It also ordered a labeling change for paroxetine, contraindicating its use in pediatric major depression.
The FDA advised U.S. doctors against using paroxetine for children under age 18 with major depressive disorder and began investigating data on pediatric use of antidepressants, including all approved SSRIs. Since then:
- In the United States, venlafaxine’s manufacturer changed the drug’s labeling to include increased reports of hostility and suicidality in pediatric trial data. A “Dear Health Care Professional” letter in August 2003 indicated that venlafaxine is not recommended in depressed pediatric patients.
- Britain’s MHRA added pediatric con-traindications to labeling of venlafaxine, sertraline, citalopram, and escitalopram in December 2003. The agency opined that fluoxetine is the only SSRI with a favorable risk-benefit profile for pediatric major depression.
- At press time, the FDA had not changed any SSRI labeling.
At the Feb. 2 public hearing, the FDA’s Psycho-pharmacological Drugs Advisory Committee and Pediatric Subcommittee of the Anti-Infective Drugs Advisory Committee heard public comments from patients, families, and physicians and reviewed the inquiry’s progress. To locate the Web site containing the FDA’s memorandum on this hearing, see Related resources.
For the next several months, an expert group at Columbia University is under contract with the FDA to develop a system to classify events that might represent suicidality. The group will then analyze data from 24 studies involving more than 4,000 depressed pediatric patients and nine antidepressants (paroxetine, fluoxetine, sertraline, fluvoxamine, citalopram, bupropion, venlafaxine, nefazodone, and mirtazapine).
Independent findings. In January, an American College of Neuropsychopharmacology (ACNP) report concluded that SSRIs do not increase suicidal thoughts or suicide attempts in youth (Table 1). An ACNP task force examined the use of SSRIs in more than 2,000 children and adolescents, including all published clinical trial data, unpublished data from several pharmaceutical companies, and data reported to Britain’s MHRA. An executive summary is available on the ACNP’s Web site (see Related resources).
Table 1
Suicide deaths, behavior, or ideation in clinical trials of youth with major depressive disorder
| Medication | Total youth* in trials | No. of suicide deaths | % of youth with suicidal behavior or ideation** | P value | Statistical significance | |
|---|---|---|---|---|---|---|
| Antidepressant | Placebo | |||||
| Citalopram | 418 | 0 | 8.9%(19) | 7.3% (15) | 0.5 | No |
| Fluoxetine | 458 | 0 | 3.6%(9) | 3.8% (8) | 0.9 | No |
| Paroxetine | 669 | 0 | 3.7%(14) | 2.5% (7) | 0.4 | No |
| Sertraline | 376 | 0 | 2.7%(5) | 1.1% (2) | 0.3 | No |
| Venlafaxine | 334 | 0 | 2%(NA) | 0% | 0.25 | No |
| * Total number of youth given antidepressants and placebo | ||||||
| ** Number inside parenthesis is actual number of youth | ||||||
| NA = Not available | ||||||
| Source: Data from published clinical trials, unpublished clinical trials provided by drug sponsor, and clinical data compiled by the Medicines and Healthcare Products Regulatory Agency of the United Kingdom. | ||||||
| Reprinted with permission of the American College of Neuropsychopharmacology from Preliminary report of the Task Force on SSRIs and Suicidal Behavior in Youth (executive summary), January 2004:18. | ||||||
Depression’s impact on children
The prevalence of depression in youth age 18 and younger is 8.3%,4 and the rate increases with patient age. Before puberty, common signs of depression include somatic symptoms such as abdominal pain, headaches, and irritability, whereas adolescents are more likely to express feelings of depression and exhibit suicidal behavior. Girls and boys are equally at risk for depression until puberty, when girls begin to outnumber boys and the presentation begins to resemble adult depression.
Untreated pediatric depression is associated with substantial morbidity, including reduced academic performance, substance abuse, interpersonal problems, social withdrawal, and a poor quality of life,4,5 It also increases the risk of suicide.
Early-onset depression is considered a more malignant illness than adult-onset depression because of its effect on development, potential for recurrence,6 and chronicity into adulthood.7 The quest to develop appropriate treatment for depressed children and adolescents has been spurred by:
- the substantial risks of morbidity and mortality from childhood depression
- advances in drug treatments for adult-onset depression
- recognition that early-onset depression is treatable.
Physiologically, children are not “mini-adults.” Thus, practicing evidence-based medicine requires separate empirical research for pediatric conditions.
Despite some efforts by the National Institute of Mental Health (NIMH) and the pharmaceutical industry, research in pediatric psychopharmacology and ancillary treatments lags decades behind evidence-based treatment in adults.
Table 2
Efficacy of SSRIs and other newer antidepressants in short-term, placebo-controlled pediatric studies in major depressive disorder
| Drug | Age range | Outcome* (drug vs. placebo) |
|---|---|---|
| Paroxetine | ||
| Study 1 Study 2 Study 3 | 12 to 18 13 to 18 7 to 17 | Negative† Negative Negative |
| Fluoxetine | ||
| Study 1 Study 2 | 8 to 17 8 to 17 | Positive Positive |
| Sertraline | ||
| Study 1 Study 2 | 6 to 17 6 to 17 | Trend Negative§ |
| Venlafaxine | ||
| Study 1 Study 2 | 7 to 17 7 to 17 | Negative Negative |
| Citalopram | ||
| Study 1 Study 2 | 7 to 17 13 to 18 | Positive Negative |
| Nefazodone | ||
| Study 1 Study 2 | 12 to 18 7 to 17 | Trend Negative |
| Mirtazapine | ||
| Study 1 Study 2 | 7 to 17 7 to 17 | Negative Negative |
| * Positive (P 0.05); Negative (P >0.10; Trend (0.05 | ||
| † Positive on most secondary endpoints | ||
| § Positive on pooling of 2 studies | ||
| Source: Adapted and reprinted from Laughren TP. Memorandum: Background comments for Feb. 2, 2004 meeting of Psychopharmacological Drugs Advisory Committee and Pediatric Subcommittee of the Anti-Infective Drugs Advisory Committee. Appendix I. Center for Drug Evaluation and Research. Food and Drug Administration, Public Health Service, Department of Health and Human Services. | ||
From tricyclics to SSRIS
Tricyclic antidepressants (TCAs) were the mainstay in treating childhood depression two decades ago, based on clinical and anecdotal evidence. However, double blind, placebo-controlled studies failed to show that TCAs were effective in treating depressed children and adolescents. The few controlled studies included small samples of children and adolescents, and the methodologies were often flawed.
In the late 1980s, TCA use in children dropped precipitously because of:
- episodes of sudden death in pediatric patients taking desipramine
- introduction of SSRIs as safer alternatives.
The causal link between sudden death and desipramine was tenuous; Biederman’s article comparing children exposed versus not exposed to TCAs did not demonstrate a statistically significant difference in sudden death.8 Even so, fear of cardiotoxicity in children curtailed TCAs’ use.
SSRIs are now are now usually considered firstline antidepressants for children and adolescents because they are presumed to be safer than TCAs. SSRIs are associated with minimal cardiotoxicity and anticholinergic effects, a wider margin of safety in overdose than TCAs, and demonstrated efficacy.
A 12-week, double-blind study compared paroxetine, 20 to 40 mg/d, imipramine, up to 300 mg/d, and placebo in 275 adolescents with major depression.9 Patients receiving paroxetine improved significantly more than patients receiving placebo, as measured by reductions in the Hamilton Rating Scale for Depression (HAM-D) total scores and other scales. Response to imipramine was not significantly different from placebo.
Discontinuation rates because of side effects were 9.7% for paroxetine and 31.5% (nearly one-third because of cardiovascular side effects) for imipramine. The average imipramine dosage of 200 mg/d was higher than usual, however.
Table 3
Recommended antidepressant dosages for children and adolescents
| Selective serotonin reuptake inhibitors | ||
| Drug | Dosage | Comments |
| Citalopram | Once-daily Children: 10 to 20 mg Adolescents: 10 to 40 mg | Antianxiety component Limited pediatric data Less effect on P-450 isoenzyme systems than other SSRIs, with fewer drug-drug interactions claimed |
| Escitalopram | Once-daily Children: 5 to 10 mg Adolescents: 10 mg | L-isomer of citalopram, purported to have lesser side effects than parent compound No data in children |
| Fluoxetine | Once-daily Children: 5 to 20 mg Adolescents: 10 to 60 mg | Antianxiety properties More effective than placebo in trials of adolescent depression11-12 Long half-life; watch for drug-drug interactions |
| Fluvoxamine | Divided Children: 50 to 100 mg/d Adolescents: 50 to 200 mg/d | Useful in depression with comorbid obsessive-compulsive symptoms No data for pediatric depression |
| Paroxetine | Once-daily Children: 10 to 20 mg Adolescents: 10 to 40 mg | Similar profile as fluoxetine but shorter half-life Not recommended in pediatric patients (FDA advisory) |
| Sertraline | Divided Children: 25 to100 mg/d Adolescents: 50 to 200 mg/d | Less activating and shorter half-life than fluoxetine |
| Other reuptake inhibitors | ||
| Drug | Dosage | Comment |
| Bupropion | Divided Children: 75 to 250 mg/d Adolescents: 75 to 400 mg/d | Useful in depression with comorbid attention- deficit/hyperactivity disorder No controlled data in children |
| Nefazodone | Divided Children: 100 to 300 mg/d Adolescents: 200 to 600 mg/d | 5HT2-receptor and serotonin-reuptake blocker |
| Venlafaxine | Divided or once-daily Children: 18.75 to 75 mg/d Adolescents: 37.5 to 150 mg/d | Noradrenergic and serotonergic effects Limited pediatric data; not recommended in depressed pediatric patients (manufacturer advisory) |
| Source: Adapted and reprinted with permission from Sood B, Sood R. Depression in children and adolescents. In: Levenson JL (ed). Depression: key diseases series. Philadelphia: American College of Physicians, 2000:244. | ||
Head-to-head studies of children with obsessive-compulsive disorder have shown fewer side effects with paroxetine compared with clomipramine. Paroxetine’s side effects included anxiety and headaches; clomipra-ALmine’s included headache, tremor, nausea, insomnia, dry mouth and anxiety.10
Efficacy data. Two double-blind, placebo-controlled studies have shown fluoxetine to be more effective than placebo in treating children and adolescents with depression.11-13 In general, however, not all SSRIs have shown consistent efficacy in placebo-controlled trials of pediatric major depression. Among 15 such trials submitted to the FDA, three (20%) showed positive results (Table 2). The success rate of drug therapy trials for adult major depression is about 50%.
The FDA’s Feb. 2 hearing memorandum notes several reasons why the agency does not view these findings as proof that SSRIs lack benefit for pediatric patients. For one, the FDA’s program allowing drug companies to apply for pediatric marketing exclusivity—for which the 15 studies were submitted—did not require positive efficacy results.
Clearly, more research is needed to demonstrate the benefits and risks of SSRIs in children and adolescents with major depression and other disorders.
Using SSRIs safely in youth
During its inquiry, the FDA recommended that prescribers observe standard antidepressant labeling language:
- Be cautious when using SSRIs or related antidepressants in major depressive disorder in children and adolescents.
- Supervise high-risk patients, especially during initial drug therapy.
Monitor suicidality. When treating depressed youth with SSRIs and other antidepressants, ask about suicide attempts, suicidal thinking, and plans for suicide. As part of informed consent, discuss with parents the potential for suicidal behavior in youth with untreated depression.
Discuss antidepressant side effects, which may include disinhibition and impulsivity. Individualize treatment plans; for example, aggressive and impulsive children require especially careful monitoring for risky or suicidal behavior.
Rule out bipolar depression and mixed episodes that are often characterized by marked irritability before prescribing antidepressants to depressed children and adolescents. Early-onset depression is a marker for bipolar disorder in pediatric populations, and bipolar illness may be a possible explanation for behavioral activation and dysphoria when antidepressants are prescribed.
Minimize side effects. Children can tolerate moderately high SSRI dosages but are usually started on lower dosages than are used in adolescents and adults (Table 3). SSRIs do not show a clear dose-response relationship, but their side effects are considered dose-dependent.14
Most-frequent SSRI side effects are nausea, diarrhea, decreased or increased appetite, headaches, restlessness, tremor, and insomnia or hypersomnia. Rare side effects include ecchymoses.15 Reduced growth, possibly related to growth hormone suppression, has been reported in four boys treated with SSRIs.16
Prevent drug interactions. SSRIs are rarely used as monotherapy in pediatric patients because of the high rates of comorbidity and severity of mental illness that presents in childhood. Using two or more medications is the rule, not the exception.17
SSRIs are highly protein-bound and are metabolized by the cytochrome P-450 isoenzyme system, which increases the likelihood of drug-drug interactions. Thus, be aware of the potential impact of combining SSRIs with other agents.
Some researchers suggest that paroxetine, sertraline, and citalopram’s relatively short half-lives (14 to 16 hours) in children may be a rationale for giving the medications twice daily.18
Avoid withdrawal. The withdrawal syndrome following abrupt cessation of paroxetine, venlafaxine, or fluvoxamine is well known, and its irritability and depression-like symptoms can be quite distressing for patients. Thus, make decisions thoughtfully when discontinuing SSRIs, and plan to taper for 1 to 2 weeks.
- Food and Drug Administration. Report prepared for public hearing Feb. 2, 2004. www.fda.gov/ohrms/dockets/ac/04/briefing/4006b1.htm (click on “Background Memorandum”)
- Preliminary report of the task force on SSRIs and suicidal behavior in youth, Jan. 21, 2004. American College of Neuropsychopharmacology. www.acnp.org/exec_summary.pdf
- Sood B, Sood R. Depression in children and adolescents. In: Levenson JL (ed). Depression: key diseases series. Philadelphia: American College of Physicians, 2000.
Drug brand names
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Nefazodone • Serzone
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
Dr. Sood is a speaker for AstraZeneca, Shire Pharmaceutical Group, and Eli Lilly and Co.
Dr. Elizabeth Weller has received research/grant support from Forest Pharmaceuticals, Organon, and Wyeth Pharmaceuticals and is a consultant to Johnson & Johnson, Novartis, AstraZeneca, and Otsuka Pharmaceutical.
Dr. Ronald Weller receives research/grant support from Wyeth Pharmaceuticals, Organon, and Forest Pharmaceuticals.
1. King RA, Riddle MA, Chappell PB, et al. Emergence of self-destructive phenomena in children and adolescents during fluoxetine treatment. J Am Acad Child Adolesc Psychiatry 1991;30:179-86.
2. Vorstman J, Lahuis B, Buitelaar JK. SSRIs associated with behavioral activation and suicidal ideation. J Am Acad Child Adolesc Psychiatry 2001;40:1364-5.
3. Hall WD, Mant A, Mitchell PB, et al. Association between antidepressant prescribing and suicide in Australia, 1991-2000. BMJ 2003;326(7397):1008.-
4. Shaffer D, Fisher P, Dulcan MK, et al. The NIMH Diagnostic Interview Schedule, Version 2.3 (DISC 2.3): description, acceptability, prevalence rates and performance of the MECA Study. Methods for the Epidemiology of Child and Adolescent Mental Disorders Study. J Am Acad Child Adolesc Psychiatry 1996a;35:865-77.
5. Harrington R, Bredenkamp D, Groothues C, et al. Adult outcomes of child and adolescent depression, III: Links with suicidal behaviors. J Child Psychol Psychiatry 1994;35:1309-19.
6. Kovacs M. Presentation of course and major depressive disorder during childhood and later years of the life span. J Am Acad Child Adolesc Psychiatry 1996;35:705-15.
7. Rao U, Hammen C, Daley SE. Continuity of depression during the transition through adulthood: a five-year longitudinal study of young women. J Am Acad Child Adolesc Psychiatry 1999;38:908-15.
8. Biederman J. Sudden death in children with a tricyclic antidepressant. J Am Acad Child Adolesc Psychiatry 1991;30(3):495-8.
9. Keller MB, Ryan ND, Strober M, et al. Efficacy of paroxetine in the treatment of adolescent major depression: a randomized, controlled study. J Am Acad Child Adolesc Psychiatry 2001;40:762-72.
10. Flament M, Cohen D. Childhood obsessive compulsive disorder. In: Maj M, Sartorius N (eds). Obsessive compulsive disorder: evidence and practice New York: World Psychiatric Association 2000;147-83.
11. Simeon J, Dinicola V, Ferguson H, Copping W. Adolescent depression: a placebo-controlled fluoxetine treatment study and follow up. Prog Neuropsychopharmacol Biol Psychiatry 1990;14:791-5.
12. Emslie G, Rush AJ, Weinberg AW, et al. A double-blind, randomized, placebo-controlled trial of fluoxetine in depressed children and adolescents. Arch Gen Psychiatry 1997;54:1031-7.
13. Emslie GJ, Heiligenstein JH, Wagner KD, et al. Fluoxetine for acute treatment of depression in children and adolescents: a placebo-controlled, randomized, clinical trial. J Am Acad Child Adolesc Psychiatry 2002;41(10):1205-15.
14. Preskorn SH. Outpatient management of depression: a guide for the practitioner (2nd ed). Caddo, OK: Professional Communications, 1999.
15. Lake MB, Birmaher B, Wassick S, et al. Bleeding and selective serotonin reuptake inhibitors in childhood and adolescence. J Child Adolesc Psychopharmacol 2000;10:35-8.
16. Weintrob N, Cohen D, Klipper-Aurbach Y, et al. Decreased growth during therapy with selective serotonin reuptake inhibitors. Arch Pediatr Adolesc Med 2002;156:696-701.
17. Rushton JL, Whitmire JT. Pediatric stimulant and selective serotonin reuptake inhibitor prescription trends. Arch Pediatr Adolesc Med 2001;155:560-5.
18. Axelson D, Perel J, Rudolph G, et al. Significant differences in pharmacokinetic/dynamics of citalopram between adolescents and adults: implications for clinical dosing (abstract). San Juan, PR: American College of Neuropsychopharmacology annual meeting, 2000.
Caitlin McIntosh, 12, hanged herself with shoelaces weeks after starting treatment for depression with a selective serotonin reuptake inhibitor (SSRI). Matt Miller, 13, hanged himself in his bedroom closet after taking his seventh SSRI dose. Michael Shivak, 11, slashed his wrists in class—but survived—while taking an SSRI.
These adolescents’ parents testified at an FDA hearing Feb. 2 about possible increased risk of suicidality with SSRIs in depressed children and adolescents.
Other families related positive experiences. Sherri Walton said her daughter, Jordan, 14, has achieved “enormous benefit” from taking SSRIs for obsessive-compulsive disorder. Suzanne Vogel-Scibilia told the FDA panel she is convinced that her two children with psychiatric disorders lead full lives because of SSRIs.
Sensitive to the anguish of grieving families but not wanting to deprive seriously depressed children of effective treatment, the FDA is proceeding methodically with its inquiry—probably at least until summer.
In the meantime, this article offers resources to help you answer questions from parents concerned about their children starting or continuing SSRIs. We include pediatric antidepressant dosing recommendations and data on benefits and risks of SSRIs and other reuptake inhibitors in young patients.
Why the FDA inquiry?
SSRI-associated behavioral activation and suicidal ideation in children and adolescents were reported anecdotally, as case reports, in the early 1990s,1 and more recently.2 No convincing evidence has shown, however, that SSRIs increase the risk of suicide. In fact, widespread use of SSRIs has been associated with reduced suicide rates.3
In May 2003, unpublished data submitted to the FDA from placebo-controlled trials suggested an increased risk of “possibly suicide-related” events and “suicide attempts” in pediatric patients taking paroxetine for major depression. No suicides were reported.
The Medicines and Healthcare Products Regulatory Agency (MHRA)—the United Kingdom’s equivalent of the Food and Drug Administration—responded by warning British physicians against prescribing paroxetine for depressed patients younger than 18. It also ordered a labeling change for paroxetine, contraindicating its use in pediatric major depression.
The FDA advised U.S. doctors against using paroxetine for children under age 18 with major depressive disorder and began investigating data on pediatric use of antidepressants, including all approved SSRIs. Since then:
- In the United States, venlafaxine’s manufacturer changed the drug’s labeling to include increased reports of hostility and suicidality in pediatric trial data. A “Dear Health Care Professional” letter in August 2003 indicated that venlafaxine is not recommended in depressed pediatric patients.
- Britain’s MHRA added pediatric con-traindications to labeling of venlafaxine, sertraline, citalopram, and escitalopram in December 2003. The agency opined that fluoxetine is the only SSRI with a favorable risk-benefit profile for pediatric major depression.
- At press time, the FDA had not changed any SSRI labeling.
At the Feb. 2 public hearing, the FDA’s Psycho-pharmacological Drugs Advisory Committee and Pediatric Subcommittee of the Anti-Infective Drugs Advisory Committee heard public comments from patients, families, and physicians and reviewed the inquiry’s progress. To locate the Web site containing the FDA’s memorandum on this hearing, see Related resources.
For the next several months, an expert group at Columbia University is under contract with the FDA to develop a system to classify events that might represent suicidality. The group will then analyze data from 24 studies involving more than 4,000 depressed pediatric patients and nine antidepressants (paroxetine, fluoxetine, sertraline, fluvoxamine, citalopram, bupropion, venlafaxine, nefazodone, and mirtazapine).
Independent findings. In January, an American College of Neuropsychopharmacology (ACNP) report concluded that SSRIs do not increase suicidal thoughts or suicide attempts in youth (Table 1). An ACNP task force examined the use of SSRIs in more than 2,000 children and adolescents, including all published clinical trial data, unpublished data from several pharmaceutical companies, and data reported to Britain’s MHRA. An executive summary is available on the ACNP’s Web site (see Related resources).
Table 1
Suicide deaths, behavior, or ideation in clinical trials of youth with major depressive disorder
| Medication | Total youth* in trials | No. of suicide deaths | % of youth with suicidal behavior or ideation** | P value | Statistical significance | |
|---|---|---|---|---|---|---|
| Antidepressant | Placebo | |||||
| Citalopram | 418 | 0 | 8.9%(19) | 7.3% (15) | 0.5 | No |
| Fluoxetine | 458 | 0 | 3.6%(9) | 3.8% (8) | 0.9 | No |
| Paroxetine | 669 | 0 | 3.7%(14) | 2.5% (7) | 0.4 | No |
| Sertraline | 376 | 0 | 2.7%(5) | 1.1% (2) | 0.3 | No |
| Venlafaxine | 334 | 0 | 2%(NA) | 0% | 0.25 | No |
| * Total number of youth given antidepressants and placebo | ||||||
| ** Number inside parenthesis is actual number of youth | ||||||
| NA = Not available | ||||||
| Source: Data from published clinical trials, unpublished clinical trials provided by drug sponsor, and clinical data compiled by the Medicines and Healthcare Products Regulatory Agency of the United Kingdom. | ||||||
| Reprinted with permission of the American College of Neuropsychopharmacology from Preliminary report of the Task Force on SSRIs and Suicidal Behavior in Youth (executive summary), January 2004:18. | ||||||
Depression’s impact on children
The prevalence of depression in youth age 18 and younger is 8.3%,4 and the rate increases with patient age. Before puberty, common signs of depression include somatic symptoms such as abdominal pain, headaches, and irritability, whereas adolescents are more likely to express feelings of depression and exhibit suicidal behavior. Girls and boys are equally at risk for depression until puberty, when girls begin to outnumber boys and the presentation begins to resemble adult depression.
Untreated pediatric depression is associated with substantial morbidity, including reduced academic performance, substance abuse, interpersonal problems, social withdrawal, and a poor quality of life,4,5 It also increases the risk of suicide.
Early-onset depression is considered a more malignant illness than adult-onset depression because of its effect on development, potential for recurrence,6 and chronicity into adulthood.7 The quest to develop appropriate treatment for depressed children and adolescents has been spurred by:
- the substantial risks of morbidity and mortality from childhood depression
- advances in drug treatments for adult-onset depression
- recognition that early-onset depression is treatable.
Physiologically, children are not “mini-adults.” Thus, practicing evidence-based medicine requires separate empirical research for pediatric conditions.
Despite some efforts by the National Institute of Mental Health (NIMH) and the pharmaceutical industry, research in pediatric psychopharmacology and ancillary treatments lags decades behind evidence-based treatment in adults.
Table 2
Efficacy of SSRIs and other newer antidepressants in short-term, placebo-controlled pediatric studies in major depressive disorder
| Drug | Age range | Outcome* (drug vs. placebo) |
|---|---|---|
| Paroxetine | ||
| Study 1 Study 2 Study 3 | 12 to 18 13 to 18 7 to 17 | Negative† Negative Negative |
| Fluoxetine | ||
| Study 1 Study 2 | 8 to 17 8 to 17 | Positive Positive |
| Sertraline | ||
| Study 1 Study 2 | 6 to 17 6 to 17 | Trend Negative§ |
| Venlafaxine | ||
| Study 1 Study 2 | 7 to 17 7 to 17 | Negative Negative |
| Citalopram | ||
| Study 1 Study 2 | 7 to 17 13 to 18 | Positive Negative |
| Nefazodone | ||
| Study 1 Study 2 | 12 to 18 7 to 17 | Trend Negative |
| Mirtazapine | ||
| Study 1 Study 2 | 7 to 17 7 to 17 | Negative Negative |
| * Positive (P 0.05); Negative (P >0.10; Trend (0.05 | ||
| † Positive on most secondary endpoints | ||
| § Positive on pooling of 2 studies | ||
| Source: Adapted and reprinted from Laughren TP. Memorandum: Background comments for Feb. 2, 2004 meeting of Psychopharmacological Drugs Advisory Committee and Pediatric Subcommittee of the Anti-Infective Drugs Advisory Committee. Appendix I. Center for Drug Evaluation and Research. Food and Drug Administration, Public Health Service, Department of Health and Human Services. | ||
From tricyclics to SSRIS
Tricyclic antidepressants (TCAs) were the mainstay in treating childhood depression two decades ago, based on clinical and anecdotal evidence. However, double blind, placebo-controlled studies failed to show that TCAs were effective in treating depressed children and adolescents. The few controlled studies included small samples of children and adolescents, and the methodologies were often flawed.
In the late 1980s, TCA use in children dropped precipitously because of:
- episodes of sudden death in pediatric patients taking desipramine
- introduction of SSRIs as safer alternatives.
The causal link between sudden death and desipramine was tenuous; Biederman’s article comparing children exposed versus not exposed to TCAs did not demonstrate a statistically significant difference in sudden death.8 Even so, fear of cardiotoxicity in children curtailed TCAs’ use.
SSRIs are now are now usually considered firstline antidepressants for children and adolescents because they are presumed to be safer than TCAs. SSRIs are associated with minimal cardiotoxicity and anticholinergic effects, a wider margin of safety in overdose than TCAs, and demonstrated efficacy.
A 12-week, double-blind study compared paroxetine, 20 to 40 mg/d, imipramine, up to 300 mg/d, and placebo in 275 adolescents with major depression.9 Patients receiving paroxetine improved significantly more than patients receiving placebo, as measured by reductions in the Hamilton Rating Scale for Depression (HAM-D) total scores and other scales. Response to imipramine was not significantly different from placebo.
Discontinuation rates because of side effects were 9.7% for paroxetine and 31.5% (nearly one-third because of cardiovascular side effects) for imipramine. The average imipramine dosage of 200 mg/d was higher than usual, however.
Table 3
Recommended antidepressant dosages for children and adolescents
| Selective serotonin reuptake inhibitors | ||
| Drug | Dosage | Comments |
| Citalopram | Once-daily Children: 10 to 20 mg Adolescents: 10 to 40 mg | Antianxiety component Limited pediatric data Less effect on P-450 isoenzyme systems than other SSRIs, with fewer drug-drug interactions claimed |
| Escitalopram | Once-daily Children: 5 to 10 mg Adolescents: 10 mg | L-isomer of citalopram, purported to have lesser side effects than parent compound No data in children |
| Fluoxetine | Once-daily Children: 5 to 20 mg Adolescents: 10 to 60 mg | Antianxiety properties More effective than placebo in trials of adolescent depression11-12 Long half-life; watch for drug-drug interactions |
| Fluvoxamine | Divided Children: 50 to 100 mg/d Adolescents: 50 to 200 mg/d | Useful in depression with comorbid obsessive-compulsive symptoms No data for pediatric depression |
| Paroxetine | Once-daily Children: 10 to 20 mg Adolescents: 10 to 40 mg | Similar profile as fluoxetine but shorter half-life Not recommended in pediatric patients (FDA advisory) |
| Sertraline | Divided Children: 25 to100 mg/d Adolescents: 50 to 200 mg/d | Less activating and shorter half-life than fluoxetine |
| Other reuptake inhibitors | ||
| Drug | Dosage | Comment |
| Bupropion | Divided Children: 75 to 250 mg/d Adolescents: 75 to 400 mg/d | Useful in depression with comorbid attention- deficit/hyperactivity disorder No controlled data in children |
| Nefazodone | Divided Children: 100 to 300 mg/d Adolescents: 200 to 600 mg/d | 5HT2-receptor and serotonin-reuptake blocker |
| Venlafaxine | Divided or once-daily Children: 18.75 to 75 mg/d Adolescents: 37.5 to 150 mg/d | Noradrenergic and serotonergic effects Limited pediatric data; not recommended in depressed pediatric patients (manufacturer advisory) |
| Source: Adapted and reprinted with permission from Sood B, Sood R. Depression in children and adolescents. In: Levenson JL (ed). Depression: key diseases series. Philadelphia: American College of Physicians, 2000:244. | ||
Head-to-head studies of children with obsessive-compulsive disorder have shown fewer side effects with paroxetine compared with clomipramine. Paroxetine’s side effects included anxiety and headaches; clomipra-ALmine’s included headache, tremor, nausea, insomnia, dry mouth and anxiety.10
Efficacy data. Two double-blind, placebo-controlled studies have shown fluoxetine to be more effective than placebo in treating children and adolescents with depression.11-13 In general, however, not all SSRIs have shown consistent efficacy in placebo-controlled trials of pediatric major depression. Among 15 such trials submitted to the FDA, three (20%) showed positive results (Table 2). The success rate of drug therapy trials for adult major depression is about 50%.
The FDA’s Feb. 2 hearing memorandum notes several reasons why the agency does not view these findings as proof that SSRIs lack benefit for pediatric patients. For one, the FDA’s program allowing drug companies to apply for pediatric marketing exclusivity—for which the 15 studies were submitted—did not require positive efficacy results.
Clearly, more research is needed to demonstrate the benefits and risks of SSRIs in children and adolescents with major depression and other disorders.
Using SSRIs safely in youth
During its inquiry, the FDA recommended that prescribers observe standard antidepressant labeling language:
- Be cautious when using SSRIs or related antidepressants in major depressive disorder in children and adolescents.
- Supervise high-risk patients, especially during initial drug therapy.
Monitor suicidality. When treating depressed youth with SSRIs and other antidepressants, ask about suicide attempts, suicidal thinking, and plans for suicide. As part of informed consent, discuss with parents the potential for suicidal behavior in youth with untreated depression.
Discuss antidepressant side effects, which may include disinhibition and impulsivity. Individualize treatment plans; for example, aggressive and impulsive children require especially careful monitoring for risky or suicidal behavior.
Rule out bipolar depression and mixed episodes that are often characterized by marked irritability before prescribing antidepressants to depressed children and adolescents. Early-onset depression is a marker for bipolar disorder in pediatric populations, and bipolar illness may be a possible explanation for behavioral activation and dysphoria when antidepressants are prescribed.
Minimize side effects. Children can tolerate moderately high SSRI dosages but are usually started on lower dosages than are used in adolescents and adults (Table 3). SSRIs do not show a clear dose-response relationship, but their side effects are considered dose-dependent.14
Most-frequent SSRI side effects are nausea, diarrhea, decreased or increased appetite, headaches, restlessness, tremor, and insomnia or hypersomnia. Rare side effects include ecchymoses.15 Reduced growth, possibly related to growth hormone suppression, has been reported in four boys treated with SSRIs.16
Prevent drug interactions. SSRIs are rarely used as monotherapy in pediatric patients because of the high rates of comorbidity and severity of mental illness that presents in childhood. Using two or more medications is the rule, not the exception.17
SSRIs are highly protein-bound and are metabolized by the cytochrome P-450 isoenzyme system, which increases the likelihood of drug-drug interactions. Thus, be aware of the potential impact of combining SSRIs with other agents.
Some researchers suggest that paroxetine, sertraline, and citalopram’s relatively short half-lives (14 to 16 hours) in children may be a rationale for giving the medications twice daily.18
Avoid withdrawal. The withdrawal syndrome following abrupt cessation of paroxetine, venlafaxine, or fluvoxamine is well known, and its irritability and depression-like symptoms can be quite distressing for patients. Thus, make decisions thoughtfully when discontinuing SSRIs, and plan to taper for 1 to 2 weeks.
- Food and Drug Administration. Report prepared for public hearing Feb. 2, 2004. www.fda.gov/ohrms/dockets/ac/04/briefing/4006b1.htm (click on “Background Memorandum”)
- Preliminary report of the task force on SSRIs and suicidal behavior in youth, Jan. 21, 2004. American College of Neuropsychopharmacology. www.acnp.org/exec_summary.pdf
- Sood B, Sood R. Depression in children and adolescents. In: Levenson JL (ed). Depression: key diseases series. Philadelphia: American College of Physicians, 2000.
Drug brand names
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Nefazodone • Serzone
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
Dr. Sood is a speaker for AstraZeneca, Shire Pharmaceutical Group, and Eli Lilly and Co.
Dr. Elizabeth Weller has received research/grant support from Forest Pharmaceuticals, Organon, and Wyeth Pharmaceuticals and is a consultant to Johnson & Johnson, Novartis, AstraZeneca, and Otsuka Pharmaceutical.
Dr. Ronald Weller receives research/grant support from Wyeth Pharmaceuticals, Organon, and Forest Pharmaceuticals.
Caitlin McIntosh, 12, hanged herself with shoelaces weeks after starting treatment for depression with a selective serotonin reuptake inhibitor (SSRI). Matt Miller, 13, hanged himself in his bedroom closet after taking his seventh SSRI dose. Michael Shivak, 11, slashed his wrists in class—but survived—while taking an SSRI.
These adolescents’ parents testified at an FDA hearing Feb. 2 about possible increased risk of suicidality with SSRIs in depressed children and adolescents.
Other families related positive experiences. Sherri Walton said her daughter, Jordan, 14, has achieved “enormous benefit” from taking SSRIs for obsessive-compulsive disorder. Suzanne Vogel-Scibilia told the FDA panel she is convinced that her two children with psychiatric disorders lead full lives because of SSRIs.
Sensitive to the anguish of grieving families but not wanting to deprive seriously depressed children of effective treatment, the FDA is proceeding methodically with its inquiry—probably at least until summer.
In the meantime, this article offers resources to help you answer questions from parents concerned about their children starting or continuing SSRIs. We include pediatric antidepressant dosing recommendations and data on benefits and risks of SSRIs and other reuptake inhibitors in young patients.
Why the FDA inquiry?
SSRI-associated behavioral activation and suicidal ideation in children and adolescents were reported anecdotally, as case reports, in the early 1990s,1 and more recently.2 No convincing evidence has shown, however, that SSRIs increase the risk of suicide. In fact, widespread use of SSRIs has been associated with reduced suicide rates.3
In May 2003, unpublished data submitted to the FDA from placebo-controlled trials suggested an increased risk of “possibly suicide-related” events and “suicide attempts” in pediatric patients taking paroxetine for major depression. No suicides were reported.
The Medicines and Healthcare Products Regulatory Agency (MHRA)—the United Kingdom’s equivalent of the Food and Drug Administration—responded by warning British physicians against prescribing paroxetine for depressed patients younger than 18. It also ordered a labeling change for paroxetine, contraindicating its use in pediatric major depression.
The FDA advised U.S. doctors against using paroxetine for children under age 18 with major depressive disorder and began investigating data on pediatric use of antidepressants, including all approved SSRIs. Since then:
- In the United States, venlafaxine’s manufacturer changed the drug’s labeling to include increased reports of hostility and suicidality in pediatric trial data. A “Dear Health Care Professional” letter in August 2003 indicated that venlafaxine is not recommended in depressed pediatric patients.
- Britain’s MHRA added pediatric con-traindications to labeling of venlafaxine, sertraline, citalopram, and escitalopram in December 2003. The agency opined that fluoxetine is the only SSRI with a favorable risk-benefit profile for pediatric major depression.
- At press time, the FDA had not changed any SSRI labeling.
At the Feb. 2 public hearing, the FDA’s Psycho-pharmacological Drugs Advisory Committee and Pediatric Subcommittee of the Anti-Infective Drugs Advisory Committee heard public comments from patients, families, and physicians and reviewed the inquiry’s progress. To locate the Web site containing the FDA’s memorandum on this hearing, see Related resources.
For the next several months, an expert group at Columbia University is under contract with the FDA to develop a system to classify events that might represent suicidality. The group will then analyze data from 24 studies involving more than 4,000 depressed pediatric patients and nine antidepressants (paroxetine, fluoxetine, sertraline, fluvoxamine, citalopram, bupropion, venlafaxine, nefazodone, and mirtazapine).
Independent findings. In January, an American College of Neuropsychopharmacology (ACNP) report concluded that SSRIs do not increase suicidal thoughts or suicide attempts in youth (Table 1). An ACNP task force examined the use of SSRIs in more than 2,000 children and adolescents, including all published clinical trial data, unpublished data from several pharmaceutical companies, and data reported to Britain’s MHRA. An executive summary is available on the ACNP’s Web site (see Related resources).
Table 1
Suicide deaths, behavior, or ideation in clinical trials of youth with major depressive disorder
| Medication | Total youth* in trials | No. of suicide deaths | % of youth with suicidal behavior or ideation** | P value | Statistical significance | |
|---|---|---|---|---|---|---|
| Antidepressant | Placebo | |||||
| Citalopram | 418 | 0 | 8.9%(19) | 7.3% (15) | 0.5 | No |
| Fluoxetine | 458 | 0 | 3.6%(9) | 3.8% (8) | 0.9 | No |
| Paroxetine | 669 | 0 | 3.7%(14) | 2.5% (7) | 0.4 | No |
| Sertraline | 376 | 0 | 2.7%(5) | 1.1% (2) | 0.3 | No |
| Venlafaxine | 334 | 0 | 2%(NA) | 0% | 0.25 | No |
| * Total number of youth given antidepressants and placebo | ||||||
| ** Number inside parenthesis is actual number of youth | ||||||
| NA = Not available | ||||||
| Source: Data from published clinical trials, unpublished clinical trials provided by drug sponsor, and clinical data compiled by the Medicines and Healthcare Products Regulatory Agency of the United Kingdom. | ||||||
| Reprinted with permission of the American College of Neuropsychopharmacology from Preliminary report of the Task Force on SSRIs and Suicidal Behavior in Youth (executive summary), January 2004:18. | ||||||
Depression’s impact on children
The prevalence of depression in youth age 18 and younger is 8.3%,4 and the rate increases with patient age. Before puberty, common signs of depression include somatic symptoms such as abdominal pain, headaches, and irritability, whereas adolescents are more likely to express feelings of depression and exhibit suicidal behavior. Girls and boys are equally at risk for depression until puberty, when girls begin to outnumber boys and the presentation begins to resemble adult depression.
Untreated pediatric depression is associated with substantial morbidity, including reduced academic performance, substance abuse, interpersonal problems, social withdrawal, and a poor quality of life,4,5 It also increases the risk of suicide.
Early-onset depression is considered a more malignant illness than adult-onset depression because of its effect on development, potential for recurrence,6 and chronicity into adulthood.7 The quest to develop appropriate treatment for depressed children and adolescents has been spurred by:
- the substantial risks of morbidity and mortality from childhood depression
- advances in drug treatments for adult-onset depression
- recognition that early-onset depression is treatable.
Physiologically, children are not “mini-adults.” Thus, practicing evidence-based medicine requires separate empirical research for pediatric conditions.
Despite some efforts by the National Institute of Mental Health (NIMH) and the pharmaceutical industry, research in pediatric psychopharmacology and ancillary treatments lags decades behind evidence-based treatment in adults.
Table 2
Efficacy of SSRIs and other newer antidepressants in short-term, placebo-controlled pediatric studies in major depressive disorder
| Drug | Age range | Outcome* (drug vs. placebo) |
|---|---|---|
| Paroxetine | ||
| Study 1 Study 2 Study 3 | 12 to 18 13 to 18 7 to 17 | Negative† Negative Negative |
| Fluoxetine | ||
| Study 1 Study 2 | 8 to 17 8 to 17 | Positive Positive |
| Sertraline | ||
| Study 1 Study 2 | 6 to 17 6 to 17 | Trend Negative§ |
| Venlafaxine | ||
| Study 1 Study 2 | 7 to 17 7 to 17 | Negative Negative |
| Citalopram | ||
| Study 1 Study 2 | 7 to 17 13 to 18 | Positive Negative |
| Nefazodone | ||
| Study 1 Study 2 | 12 to 18 7 to 17 | Trend Negative |
| Mirtazapine | ||
| Study 1 Study 2 | 7 to 17 7 to 17 | Negative Negative |
| * Positive (P 0.05); Negative (P >0.10; Trend (0.05 | ||
| † Positive on most secondary endpoints | ||
| § Positive on pooling of 2 studies | ||
| Source: Adapted and reprinted from Laughren TP. Memorandum: Background comments for Feb. 2, 2004 meeting of Psychopharmacological Drugs Advisory Committee and Pediatric Subcommittee of the Anti-Infective Drugs Advisory Committee. Appendix I. Center for Drug Evaluation and Research. Food and Drug Administration, Public Health Service, Department of Health and Human Services. | ||
From tricyclics to SSRIS
Tricyclic antidepressants (TCAs) were the mainstay in treating childhood depression two decades ago, based on clinical and anecdotal evidence. However, double blind, placebo-controlled studies failed to show that TCAs were effective in treating depressed children and adolescents. The few controlled studies included small samples of children and adolescents, and the methodologies were often flawed.
In the late 1980s, TCA use in children dropped precipitously because of:
- episodes of sudden death in pediatric patients taking desipramine
- introduction of SSRIs as safer alternatives.
The causal link between sudden death and desipramine was tenuous; Biederman’s article comparing children exposed versus not exposed to TCAs did not demonstrate a statistically significant difference in sudden death.8 Even so, fear of cardiotoxicity in children curtailed TCAs’ use.
SSRIs are now are now usually considered firstline antidepressants for children and adolescents because they are presumed to be safer than TCAs. SSRIs are associated with minimal cardiotoxicity and anticholinergic effects, a wider margin of safety in overdose than TCAs, and demonstrated efficacy.
A 12-week, double-blind study compared paroxetine, 20 to 40 mg/d, imipramine, up to 300 mg/d, and placebo in 275 adolescents with major depression.9 Patients receiving paroxetine improved significantly more than patients receiving placebo, as measured by reductions in the Hamilton Rating Scale for Depression (HAM-D) total scores and other scales. Response to imipramine was not significantly different from placebo.
Discontinuation rates because of side effects were 9.7% for paroxetine and 31.5% (nearly one-third because of cardiovascular side effects) for imipramine. The average imipramine dosage of 200 mg/d was higher than usual, however.
Table 3
Recommended antidepressant dosages for children and adolescents
| Selective serotonin reuptake inhibitors | ||
| Drug | Dosage | Comments |
| Citalopram | Once-daily Children: 10 to 20 mg Adolescents: 10 to 40 mg | Antianxiety component Limited pediatric data Less effect on P-450 isoenzyme systems than other SSRIs, with fewer drug-drug interactions claimed |
| Escitalopram | Once-daily Children: 5 to 10 mg Adolescents: 10 mg | L-isomer of citalopram, purported to have lesser side effects than parent compound No data in children |
| Fluoxetine | Once-daily Children: 5 to 20 mg Adolescents: 10 to 60 mg | Antianxiety properties More effective than placebo in trials of adolescent depression11-12 Long half-life; watch for drug-drug interactions |
| Fluvoxamine | Divided Children: 50 to 100 mg/d Adolescents: 50 to 200 mg/d | Useful in depression with comorbid obsessive-compulsive symptoms No data for pediatric depression |
| Paroxetine | Once-daily Children: 10 to 20 mg Adolescents: 10 to 40 mg | Similar profile as fluoxetine but shorter half-life Not recommended in pediatric patients (FDA advisory) |
| Sertraline | Divided Children: 25 to100 mg/d Adolescents: 50 to 200 mg/d | Less activating and shorter half-life than fluoxetine |
| Other reuptake inhibitors | ||
| Drug | Dosage | Comment |
| Bupropion | Divided Children: 75 to 250 mg/d Adolescents: 75 to 400 mg/d | Useful in depression with comorbid attention- deficit/hyperactivity disorder No controlled data in children |
| Nefazodone | Divided Children: 100 to 300 mg/d Adolescents: 200 to 600 mg/d | 5HT2-receptor and serotonin-reuptake blocker |
| Venlafaxine | Divided or once-daily Children: 18.75 to 75 mg/d Adolescents: 37.5 to 150 mg/d | Noradrenergic and serotonergic effects Limited pediatric data; not recommended in depressed pediatric patients (manufacturer advisory) |
| Source: Adapted and reprinted with permission from Sood B, Sood R. Depression in children and adolescents. In: Levenson JL (ed). Depression: key diseases series. Philadelphia: American College of Physicians, 2000:244. | ||
Head-to-head studies of children with obsessive-compulsive disorder have shown fewer side effects with paroxetine compared with clomipramine. Paroxetine’s side effects included anxiety and headaches; clomipra-ALmine’s included headache, tremor, nausea, insomnia, dry mouth and anxiety.10
Efficacy data. Two double-blind, placebo-controlled studies have shown fluoxetine to be more effective than placebo in treating children and adolescents with depression.11-13 In general, however, not all SSRIs have shown consistent efficacy in placebo-controlled trials of pediatric major depression. Among 15 such trials submitted to the FDA, three (20%) showed positive results (Table 2). The success rate of drug therapy trials for adult major depression is about 50%.
The FDA’s Feb. 2 hearing memorandum notes several reasons why the agency does not view these findings as proof that SSRIs lack benefit for pediatric patients. For one, the FDA’s program allowing drug companies to apply for pediatric marketing exclusivity—for which the 15 studies were submitted—did not require positive efficacy results.
Clearly, more research is needed to demonstrate the benefits and risks of SSRIs in children and adolescents with major depression and other disorders.
Using SSRIs safely in youth
During its inquiry, the FDA recommended that prescribers observe standard antidepressant labeling language:
- Be cautious when using SSRIs or related antidepressants in major depressive disorder in children and adolescents.
- Supervise high-risk patients, especially during initial drug therapy.
Monitor suicidality. When treating depressed youth with SSRIs and other antidepressants, ask about suicide attempts, suicidal thinking, and plans for suicide. As part of informed consent, discuss with parents the potential for suicidal behavior in youth with untreated depression.
Discuss antidepressant side effects, which may include disinhibition and impulsivity. Individualize treatment plans; for example, aggressive and impulsive children require especially careful monitoring for risky or suicidal behavior.
Rule out bipolar depression and mixed episodes that are often characterized by marked irritability before prescribing antidepressants to depressed children and adolescents. Early-onset depression is a marker for bipolar disorder in pediatric populations, and bipolar illness may be a possible explanation for behavioral activation and dysphoria when antidepressants are prescribed.
Minimize side effects. Children can tolerate moderately high SSRI dosages but are usually started on lower dosages than are used in adolescents and adults (Table 3). SSRIs do not show a clear dose-response relationship, but their side effects are considered dose-dependent.14
Most-frequent SSRI side effects are nausea, diarrhea, decreased or increased appetite, headaches, restlessness, tremor, and insomnia or hypersomnia. Rare side effects include ecchymoses.15 Reduced growth, possibly related to growth hormone suppression, has been reported in four boys treated with SSRIs.16
Prevent drug interactions. SSRIs are rarely used as monotherapy in pediatric patients because of the high rates of comorbidity and severity of mental illness that presents in childhood. Using two or more medications is the rule, not the exception.17
SSRIs are highly protein-bound and are metabolized by the cytochrome P-450 isoenzyme system, which increases the likelihood of drug-drug interactions. Thus, be aware of the potential impact of combining SSRIs with other agents.
Some researchers suggest that paroxetine, sertraline, and citalopram’s relatively short half-lives (14 to 16 hours) in children may be a rationale for giving the medications twice daily.18
Avoid withdrawal. The withdrawal syndrome following abrupt cessation of paroxetine, venlafaxine, or fluvoxamine is well known, and its irritability and depression-like symptoms can be quite distressing for patients. Thus, make decisions thoughtfully when discontinuing SSRIs, and plan to taper for 1 to 2 weeks.
- Food and Drug Administration. Report prepared for public hearing Feb. 2, 2004. www.fda.gov/ohrms/dockets/ac/04/briefing/4006b1.htm (click on “Background Memorandum”)
- Preliminary report of the task force on SSRIs and suicidal behavior in youth, Jan. 21, 2004. American College of Neuropsychopharmacology. www.acnp.org/exec_summary.pdf
- Sood B, Sood R. Depression in children and adolescents. In: Levenson JL (ed). Depression: key diseases series. Philadelphia: American College of Physicians, 2000.
Drug brand names
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Nefazodone • Serzone
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
Dr. Sood is a speaker for AstraZeneca, Shire Pharmaceutical Group, and Eli Lilly and Co.
Dr. Elizabeth Weller has received research/grant support from Forest Pharmaceuticals, Organon, and Wyeth Pharmaceuticals and is a consultant to Johnson & Johnson, Novartis, AstraZeneca, and Otsuka Pharmaceutical.
Dr. Ronald Weller receives research/grant support from Wyeth Pharmaceuticals, Organon, and Forest Pharmaceuticals.
1. King RA, Riddle MA, Chappell PB, et al. Emergence of self-destructive phenomena in children and adolescents during fluoxetine treatment. J Am Acad Child Adolesc Psychiatry 1991;30:179-86.
2. Vorstman J, Lahuis B, Buitelaar JK. SSRIs associated with behavioral activation and suicidal ideation. J Am Acad Child Adolesc Psychiatry 2001;40:1364-5.
3. Hall WD, Mant A, Mitchell PB, et al. Association between antidepressant prescribing and suicide in Australia, 1991-2000. BMJ 2003;326(7397):1008.-
4. Shaffer D, Fisher P, Dulcan MK, et al. The NIMH Diagnostic Interview Schedule, Version 2.3 (DISC 2.3): description, acceptability, prevalence rates and performance of the MECA Study. Methods for the Epidemiology of Child and Adolescent Mental Disorders Study. J Am Acad Child Adolesc Psychiatry 1996a;35:865-77.
5. Harrington R, Bredenkamp D, Groothues C, et al. Adult outcomes of child and adolescent depression, III: Links with suicidal behaviors. J Child Psychol Psychiatry 1994;35:1309-19.
6. Kovacs M. Presentation of course and major depressive disorder during childhood and later years of the life span. J Am Acad Child Adolesc Psychiatry 1996;35:705-15.
7. Rao U, Hammen C, Daley SE. Continuity of depression during the transition through adulthood: a five-year longitudinal study of young women. J Am Acad Child Adolesc Psychiatry 1999;38:908-15.
8. Biederman J. Sudden death in children with a tricyclic antidepressant. J Am Acad Child Adolesc Psychiatry 1991;30(3):495-8.
9. Keller MB, Ryan ND, Strober M, et al. Efficacy of paroxetine in the treatment of adolescent major depression: a randomized, controlled study. J Am Acad Child Adolesc Psychiatry 2001;40:762-72.
10. Flament M, Cohen D. Childhood obsessive compulsive disorder. In: Maj M, Sartorius N (eds). Obsessive compulsive disorder: evidence and practice New York: World Psychiatric Association 2000;147-83.
11. Simeon J, Dinicola V, Ferguson H, Copping W. Adolescent depression: a placebo-controlled fluoxetine treatment study and follow up. Prog Neuropsychopharmacol Biol Psychiatry 1990;14:791-5.
12. Emslie G, Rush AJ, Weinberg AW, et al. A double-blind, randomized, placebo-controlled trial of fluoxetine in depressed children and adolescents. Arch Gen Psychiatry 1997;54:1031-7.
13. Emslie GJ, Heiligenstein JH, Wagner KD, et al. Fluoxetine for acute treatment of depression in children and adolescents: a placebo-controlled, randomized, clinical trial. J Am Acad Child Adolesc Psychiatry 2002;41(10):1205-15.
14. Preskorn SH. Outpatient management of depression: a guide for the practitioner (2nd ed). Caddo, OK: Professional Communications, 1999.
15. Lake MB, Birmaher B, Wassick S, et al. Bleeding and selective serotonin reuptake inhibitors in childhood and adolescence. J Child Adolesc Psychopharmacol 2000;10:35-8.
16. Weintrob N, Cohen D, Klipper-Aurbach Y, et al. Decreased growth during therapy with selective serotonin reuptake inhibitors. Arch Pediatr Adolesc Med 2002;156:696-701.
17. Rushton JL, Whitmire JT. Pediatric stimulant and selective serotonin reuptake inhibitor prescription trends. Arch Pediatr Adolesc Med 2001;155:560-5.
18. Axelson D, Perel J, Rudolph G, et al. Significant differences in pharmacokinetic/dynamics of citalopram between adolescents and adults: implications for clinical dosing (abstract). San Juan, PR: American College of Neuropsychopharmacology annual meeting, 2000.
1. King RA, Riddle MA, Chappell PB, et al. Emergence of self-destructive phenomena in children and adolescents during fluoxetine treatment. J Am Acad Child Adolesc Psychiatry 1991;30:179-86.
2. Vorstman J, Lahuis B, Buitelaar JK. SSRIs associated with behavioral activation and suicidal ideation. J Am Acad Child Adolesc Psychiatry 2001;40:1364-5.
3. Hall WD, Mant A, Mitchell PB, et al. Association between antidepressant prescribing and suicide in Australia, 1991-2000. BMJ 2003;326(7397):1008.-
4. Shaffer D, Fisher P, Dulcan MK, et al. The NIMH Diagnostic Interview Schedule, Version 2.3 (DISC 2.3): description, acceptability, prevalence rates and performance of the MECA Study. Methods for the Epidemiology of Child and Adolescent Mental Disorders Study. J Am Acad Child Adolesc Psychiatry 1996a;35:865-77.
5. Harrington R, Bredenkamp D, Groothues C, et al. Adult outcomes of child and adolescent depression, III: Links with suicidal behaviors. J Child Psychol Psychiatry 1994;35:1309-19.
6. Kovacs M. Presentation of course and major depressive disorder during childhood and later years of the life span. J Am Acad Child Adolesc Psychiatry 1996;35:705-15.
7. Rao U, Hammen C, Daley SE. Continuity of depression during the transition through adulthood: a five-year longitudinal study of young women. J Am Acad Child Adolesc Psychiatry 1999;38:908-15.
8. Biederman J. Sudden death in children with a tricyclic antidepressant. J Am Acad Child Adolesc Psychiatry 1991;30(3):495-8.
9. Keller MB, Ryan ND, Strober M, et al. Efficacy of paroxetine in the treatment of adolescent major depression: a randomized, controlled study. J Am Acad Child Adolesc Psychiatry 2001;40:762-72.
10. Flament M, Cohen D. Childhood obsessive compulsive disorder. In: Maj M, Sartorius N (eds). Obsessive compulsive disorder: evidence and practice New York: World Psychiatric Association 2000;147-83.
11. Simeon J, Dinicola V, Ferguson H, Copping W. Adolescent depression: a placebo-controlled fluoxetine treatment study and follow up. Prog Neuropsychopharmacol Biol Psychiatry 1990;14:791-5.
12. Emslie G, Rush AJ, Weinberg AW, et al. A double-blind, randomized, placebo-controlled trial of fluoxetine in depressed children and adolescents. Arch Gen Psychiatry 1997;54:1031-7.
13. Emslie GJ, Heiligenstein JH, Wagner KD, et al. Fluoxetine for acute treatment of depression in children and adolescents: a placebo-controlled, randomized, clinical trial. J Am Acad Child Adolesc Psychiatry 2002;41(10):1205-15.
14. Preskorn SH. Outpatient management of depression: a guide for the practitioner (2nd ed). Caddo, OK: Professional Communications, 1999.
15. Lake MB, Birmaher B, Wassick S, et al. Bleeding and selective serotonin reuptake inhibitors in childhood and adolescence. J Child Adolesc Psychopharmacol 2000;10:35-8.
16. Weintrob N, Cohen D, Klipper-Aurbach Y, et al. Decreased growth during therapy with selective serotonin reuptake inhibitors. Arch Pediatr Adolesc Med 2002;156:696-701.
17. Rushton JL, Whitmire JT. Pediatric stimulant and selective serotonin reuptake inhibitor prescription trends. Arch Pediatr Adolesc Med 2001;155:560-5.
18. Axelson D, Perel J, Rudolph G, et al. Significant differences in pharmacokinetic/dynamics of citalopram between adolescents and adults: implications for clinical dosing (abstract). San Juan, PR: American College of Neuropsychopharmacology annual meeting, 2000.
Treatment-resistant depression: Switch or augment? Choices that improve response rates
When depression fails to respond to initial therapy—as it commonly does—we have many options but little evidence to guide our choices. We often wonder:
- Is this patient’s depression treatment-resistant?
- Would switching medications or augmenting the initial drug be more likely to achieve an adequate response?
- How effective is psychotherapy compared with medication for treatment-resistant depression?
This article offers insights into each question, based on available trial data, algorithmic approaches to major depressive disorder, and clinical experience. Included is a preview of an ongoing multicenter, treatment-resistant depression study that mimics clinical practice and a look at vagus nerve stimulation (VNS)—a novel somatic therapy being considered by the FDA.
Measuring treatment response
Sustained symptom remission—with normalization of function—is the aim of treating major depressive disorder. Outcomes are categorized as:
- remission (virtual absence of depressive symptoms)
- response with residual symptoms (>50% reduction in baseline symptom severity that does not qualify for remission)
- partial response (>25% but <50% decrease in baseline symptom severity)
- nonresponse (<25% reduction in baseline symptoms).
Major depressive disorder (MDD) is typically recurrent or chronic and characterized by marked disability and a life expectancy shortened by suicide and increased mortality from associated medical conditions. Lifetime prevalence is 16.2%.6
MDD is twice as likely to affect women as men and is common among adolescents, young adults, and persons with concurrent medical conditions.
Major depression’s course is characterized by:
- recurrent episodes (approximately every 5 years)
- or a persistent level of waxing and waning depressive symptoms (in 20% to 35% of cases).
Dysthymic disorder often heralds major depression. Within 1 year, 5% to 20% of persons with dysthymic disorder develop major depression.7
Disability associated with major depression
often exceeds that of other general medical conditions. Depression is the fourth most disabling condition worldwide and is projected to rank number two by 2020 because of its chronic and recurrent nature, high prevalence, and life-shortening effects.8
Consequences of unremitting depression include:
- poor day-to-day function (work, family)
- increased likelihood of recurrence
- psychiatric or medical complications, including substance abuse
- high use of mental health and general medical resources
- worsened prognosis of medical conditions
- high family burden.
In 8-week acute-phase trials, 7% to 15% of patients do not tolerate the initial medication, 25% show no response, 15% show partial response, 10% to 20% exhibit response with residual symptoms, and 30% to 40% achieve remission. Complicated depressions that may not respond as well include those concurrent with Axis I conditions—such as panic disorder or substance abuse—or Axis II or III conditions.1
Time-limited psychotherapies targeted at depressive symptoms (such as cognitive, interpersonal, and behavioral therapies) also typically achieve a 50% response rate in uncomplicated depression that is not treatment-resistant.
Recommendation. When treating depression, assess response at least every 4 weeks (preferably at each visit), using a self-report or clinician rating such as:
- Quick Inventory of Depressive Symptomatology2 (see Related resources)
- Beck Depression Inventory3,4
- Patient Health Questionnaire.5
Defining treatment resistance
A patient may not achieve remission for a variety of reasons, including poor adherence, inadequate medication trial or dosing, occult substance abuse, undiagnosed medical conditions (Box),6-8 concurrent Axis I or II disorders, or treatment resistance.
The general consensus is to consider depression “treatment-resistant” when at least two adequately delivered treatments do not achieve at least a response. A stricter definition—failure to achieve sustained remission with two or more treatments—has also been suggested.
Several schemes have proposed treatment resistance levels, such as the five stages identified in the Table. Recent studies9,10 suggest that increasing treatment resistance is associated with decreasing response or remission rates.
Therefore, when a patient’s treatment resistance is high, two appropriate strategies are to:
- persist with and use maximally tolerated dosages of the treatment you select
- aim for response because high resistance lowers the likelihood of remission.
Predicting response. A major clinical issue is determining whether remission will occur during an acute treatment trial. It is important to not declare treatment resistance unless there has been:
- adequate exposure (dosing and duration) to the treatment
- and adequate adherence.
Patients often have apparent but not actual resistance, meaning that the agent was not used long enough (at least 6 weeks) or at high enough doses. Remission typically follows response by several weeks or even 1 to 2 months for more-chronic depressions.11 Thus, treatment trials should continue at least 12 weeks to determine whether remission will occur.
On the other hand, not obtaining at least a signal of minimal benefit (at least a 20% reduction in baseline symptom severity) in 4 to 6 weeks often portends a low likelihood of response in the long run.12,13 Thus, continue a treatment at least 6 weeks before you decide that it will not achieve a response.
Recommendation. Measure symptoms at key decision points. If modest improvement (such as 20% reduction in baseline symptoms) is found at 4 to 6 weeks, continue treating another 4 to 6 weeks, increasing the dosage as tolerated.
Table
Simple system for staging antidepressant resistance
| Stage | Definition |
|---|---|
| I | Failure of at least one adequate trial of one major antidepressant class |
| II | Stage I resistance plus failure of an adequate trial of an antidepressant in a distinctly different class from that used in Stage I |
| III | Stage II resistance plus failure of an adequate trial of a tricyclic antidepressant |
| IV | Stage III resistance plus failure of an adequate trial of a monoamine oxidase inhibitor |
| V | Stage IV resistance plus failure of a course of bilateral electroconvulsive therapy |
| Source: Reprinted with permission from Thase ME, Rush AJ. When at first you don’t succeed: sequential strategies for antidepressant nonresponders. J Clin Psychiatry 1997;58(suppl 13):24. | |
Treatment options
When initial antidepressant treatment fails to achieve an adequate response—as it does in more than onehalf of major depression cases—the next step is to add a second agent or switch to another agent.
Available evidence14 relies almost exclusively on open, uncontrolled trials, which do not provide definitive answers. Even so, these trials indicate that nonresponse (or nonremission) with one agent does not predict nonresponse/nonremission with another.
Switching strategies. When a selective serotonin reuptake inhibitor (SSRI) is the first treatment, several open trials reveal an approximately 50% response rate to a second SSRI. However, opentrial evidence and retrospective chart review reports also indicate that switching out of class (such as from an SSRI to bupropion) is also approximately 50% effective.15
Some post hoc analyses of acute 8-week trials indicate that the dual-action agent venlafaxine at higher dosages (up to 225 mg/d of venlafaxine XR) is associated with higher remission rates than the more-selective SSRIs.16,17 On the other hand, unpublished data indicate that escitalopram, 10 mg/d, and venlafaxine XR, up to 150 mg/d, did not differ in efficacy among outpatients treated by primary care physicians.18
On the other hand, sertraline and imipramine (a dual-action agent) were equally effective in a 12-week acute-phase trial.19 Furthermore, response and remission rates were similar when nonresponders in each group switched to the other antidepressant.20 This suggests that the dual-action agent (imipramine) was not more effective than the more selective agent (sertraline) in this population.
Well-controlled trials show that monoamine oxidase inhibitors (MAOIs) can be effective when tricyclic antidepressants (TCAs) are not. Switches among the TCAs are associated with a 30% response rate, whereas switching from a TCA to an MAOI typically results in a 50% response rate.21
Controlled prospective comparisons of two or more alternate switch or augment treatments are needed to establish comparative efficacy and tolerability.
Augmentation strategies may include lithium, buspirone, thyroid hormone (T3), stimulants, or atypical antipsychotics. Although head-to-head comparisons are rare, a randomized, controlled trial found that combining olanzapine (mean 50 mg/d), with fluoxetine (mean 15 mg/d) was more effective than each agent used alone.22
Risperidone augmentation is supported by open trials, as is the use of modafinil, other stimulants, and bupropion. An important unanswered question with most augmentation strategies is how long to continue them if they are successful.
Psychotherapy may also play a key role in augmenting medication’s effects. Keller et al23 found in chronically depressed outpatients that 12 weeks of nefazodone, up to 600 mg/d, plus cognitive behavioral analytic system psychotherapy (CBASP) produced higher response and remission rates compared with either treatment alone. A subsequent report24 found that 50% of nefa-zodone and CBASP monotherapy nonresponders did respond when switched to the alternate treatment.
Thus, CBASP may be useful at least in chronic depression to augment medication or as a “switch” to monotherapy if medication alone fails. Interestingly, Nemeroff et al25 found CBASP more effective than nefazodone for patients with chronic major depression who had a childhood history of parental loss or physical, sexual, or emotional abuse.
Antidepressant tachyphylaxis—commonly referred to as “poop-out”—is reported with all antidepressants. That is, even while apparently taking their medications for 6 to 18 months, some patients lose the antidepressant effect, such that some symptoms return or a full relapse/recurrence ensues. Mechanisms of this phenomenon are unknown.
Clinically, some believe that “poop out” is more common with SSRIs than with other antidepressant classes, but no long-term comparative data support or challenge this view. Treatment options include a dosage increase, dosage reduction (especially for long half-life SSRIs such as fluoxetine), or augmentation with the options noted above (such as bupropion, buspirone, etc.).
Benefit of using algorithms
Algorithms (such as the Texas Medication Algorithm Project26 ) have suggested multiple treatment steps for major depression after initial treatment fails, with several options available at each step. Using medication algorithms has been found more effective than treatment-as-usual in outpatients with major depressive disorder.27 No studies have compared different algorithms.
STAR*D trial. The ongoing National Institute of Mental Health (NIMH) Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial may offer a new algorithmic approach to treating major depression.14,28 NIMH launched STAR*D in 1999, enrollment began in 2001, and results are expecte by May 2005 (see Related resources).
STAR*D—of which I am the study director—is a randomized, controlled, raterblinded, multisite trial of outpatients ages 18 to 75 with nonpsychotic major depression (17-item Hamilton Rating Scale for Depression score 14). The trial design includes four treatment levels and numerous antidepressant options (Figure).
The study’s aim is to enroll 4,000 patients into level 1, with 1,500 entering level 2. Patients who achieve an adequate response based on clinician judgment may continue the effective treatment for 12 months, during which their symptoms and other relevant information are monitored monthly by telephone. Patients who do not achieve an acceptable response in level 1 (or in subsequent levels) may proceed to the next level, which involves a randomized assignment.
STAR*D has an innovative design that mimics clinical practice and ensures high levels of patient participation. When patients agree to randomization, they may elect to exclude groups of treatments but may not pick a particular treatment (they must accept randomization to stay in the study).
Figure STAR*D treatment levels for major depressive disorder

For example, patient A entering level 2 may exclude switch treatments and elect to accept randomization to citalopram plus bupropion SR, citalopram plus buspirone, or citalopram and cognitive therapy. Conversely, patient B may exclude all augment options at level 2, and accept randomization to the four switch options.
Patients may exclude cognitive psychotherapy as an augment and/or switch option as long as they accept randomization to all available medication switches, or augments, or both. They may also choose cognitive therapy and exclude all medication switch and augment options. These patients must accept randomization to either cognitive therapy switch or cognitive therapy augmentation.
This so-called equipoise stratified randomized design29 allows us to compare all participants randomized to the treatments being compared. To date, only 1% of subjects have accepted randomization to all seven level-2 treatments. About one-half elect only the switch options, and about one-half elect only the augment options.
STAR*D’s goal is to determine whether there is a preferred next step for varying types and degrees of treatment-resistant repression.
Vagus nerve stimulation
Somatic therapies being investigated to expand our therapeutic options for major depressive disorder include magnetic seizure therapy, repetitive transcranial magnetic stimulation, and vagus nerve stimulation (VNS).
VNS—now indicated for treatment-resistant epilepsy—is being investigated as a potential augmentation for treatment-resistant depression. An application for this supplemental indication was submitted to the FDA in October 2003.
With VNS, a device implanted in the patient’s chest provides intermittent stimulation to the left vagus nerve (typically 30 seconds on and 5 minutes off, 24 hours a day). In an open trial10 and follow-up report,30 VNS was associated with a 30% to 45% response rate in 59 depressed patients with high levels of treatment resistance (inadequate response to an average of 16 treatment trials).
VNS is well tolerated, though it has not been prospectively studied in patients with diagnosed cardiovascular disease. Side effects that may occur when the stimulation is “on” include:
- voice alteration in about 60% of patients (the voice becomes more hoarse when the left recurrent laryngeal nerve is activated)
- paresthesias in the neck
- shortness of breath on heavy exertion.
These effects are usually absent in the 5-minute “off” phase.
- National Institute of Mental Health. Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial. Includes the Quick Inventory of Depressive Symptomatology. www.star-d.org
- HealthyPlace.Com Depression Community. Vagus nerve stimulation for treating depression. www.healthyplace.com/communities/depression/treatment/vns
Drug brand names
- Bupropion • Wellbutrin, Wellbutrin SR
- Buspirone • BuSpar
- Citalopram • Celexa
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Mirtazapine • Remeron
- Modafinil • Provigil
- Nefazodone • Serzone
- Nortriptyline • Pamelor, Aventyl
- Olanzapine • Zyprexa
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tranylcypromine • Parnate
- Venlafaxine • Effexor, Effexor XR
Disclosure
Dr. Rush receives grant/research support from the Robert Wood Johnson Foundation, National Institute of Mental Health, and The Stanley Foundation. He is a consultant to Bristol-Myers Squibb Co., Cyberonics, Eli Lilly & Co., Forest Laboratories, and GlaxoSmithKline, and a speaker for Bristol-Myers Squibb Co., Cyberonics, Eli Lilly & Co., Forest Laboratories, GlaxoSmithKline, and Wyeth Pharmaceuticals.
1. Depression Guideline Panel. Clinical practice guideline, number 5: depression in primary care: vol. 2. Treatment of major depression Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research. AHCPR publication no. 93-0551, 1993.
2. Rush AJ, Trivedi MH, Ibrahim HM, et al. The 16-Item Quick Inventory of Depressive Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biol Psychiatry 2003;54:573-83.
3. Beck AT, Steer RA, Brown GK. Beck Depression Inventory (2nd ed. manual). San Antonio, TX: The Psychological Corporation, 1996.
4. Beck AT, Ward CH, Mendelson M, et al. An inventory for measuring depression. Arch Gen Psychiatry 1961;4:561-71.
5. Kroenke K, Spitzer RL, Williams JBW. The PHQ-9. Validity of a brief depression severity measure. J Gen Intern Med 2001;16:606-13.
6. Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003;289(23):3095-105.
7. Depression Guideline Panel. Clinical practice guideline, number 5: Depression in primary care, vol. 1: detection and diagnosis Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research. AHCPR publication no. 93-0550, 1993.
8. Murray CJ, Lopez AD. (eds) The global burden of disease Boston: Harvard School of Public Health, 1996.
9. Barbee JG, Jamhour NJ. Lamotrigine as an augmentation agent in treatment-resistant depression. J Clin Psychiatry 2002;63(8):737-41.
10. Sackeim HA, Rush AJ, George MS, et al. Vagus nerve stimulation (VNS) for treatment-resistant depression: efficacy, side effects, and predictors of outcome. Neuropsychopharmacology 2001;25(5):713-28.
11. Koran LM, Gelenberg AJ, Kornstein SG, et al. Sertraline versus imipramine to prevent relapse in chronic depression. J Affect Disord 2001;65(1):27-36.
12. Nierenberg AA, Feighner JP, Rudolph R, et al. Venlafaxine for treatment-resistant unipolar depression. J Clin Psychopharmacol 1994;14(6):419-23.
13. Quitkin FM, Petkova E, McGrath PJ, et al. When should a trial of fluoxetine for major depression be declared failed? Am J Psychiatry 2003;160(4):734-40.
14. Fava M, Rush AJ, Trivedi MH, et al. Background and rationale for the sequenced treatment alternatives to relieve depression (STAR*D) study. Psychiatr Clin North Am 2003;26(2):457-94.
15. Fava M, Papakostas GI, Petersen T, et al. Switching to bupropion in fluoxetine-resistant major depressive disorder. Ann Clin Psychiatry 2003;15:17-22.
16. Entsuah AR, Huang H, Thase ME. Response and remission rates in different subpopulations with major depressive disorder administered venlafaxine, selective serotonin reuptake inhibitors, or placebo. J Clin Psychiatry 2001;62:869-77.
17. Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or selective serotonin reuptake inhibitors. Br J Psychiatry 2001;178:234-41.
18. Montgomery SA, Huusom A, Bothmer J. Flexible dose comparison of s-citalopram and venlafaxine XR. J Eur Neuropsychopharmacol 2002;12(suppl 3):S-254.
19. Keller MB, Gelenberg AJ, Hirschfeld RM, et al. The treatment of chronic depression, part 2: a double-blind, randomized trial of sertraline and imipramine. J Clin Psychiatry 1998;59(11):598-607.
20. Thase ME, Rush AJ, Howland RH, et al. Double-blind switch study of imipramine or sertraline treatment of antidepressant-resistant chronic depression. Arch Gen Psychiatry 2002;59(3):233-9.
21. Thase ME, Rush AJ. Treatment resistant depression. In: Bloom FE, Kupfer DJ (eds). Psychopharmacology: the fourth generation of progress New York: Raven Press, 1995;1081-97.
22. Shelton RC, Tollefson GD, Tohen M, et al. A novel augmentation strategy for treating resistant major depression. Am J Psychiatry 2001;158:131-4.
23. Keller MB, McCullough JP, Klein DN, et al. A comparison of nefazodone, the cognitive behavioral-analysis system of psychotherapy, and their combination for the treatment of chronic depression. N Engl J Med 2000;342:1462-70.
24. Schatzberg AF, Rush AJ, Arnow BA, et al. Medication or psychotherapy is effective when the other is not in chronic depression: empirical support. Arch Gen Psychiatry (submitted).
25. Nemeroff CB, Heim CM, Thase ME, et al. Differential responses to psychotherapy versus pharmacotherapy in patients with chronic forms of major depression and childhood trauma. Proc Natl Acad Sci USA 2003;100(24):14293-6.
26. Crismon ML, Trivedi M, Pigott TA, et al. The Texas Medication Algorithm Project: report of the Texas Consensus Conference Panel on Medication Treatment of Major Depressive Disorder. J Clin Psychiatry 1999;60(3):142-56.
27. Trivedi MH, Rush AJ, Crismon ML, et al. The Texas Medication Algorithm Project (TMAP): clinical results for patients with major depressive disorder. Arch Gen Psychiatry (in press).
28. Rush AJ, Fava M, Wisniewski SR, et al. for the STAR*D Investigators Group. Sequenced Treatment Alternatives to Relieve Depression (STAR*D): rationale and design. Control Clin Trials 2004;25(1):118-41.
29. Lavori PW, Rush AJ, Wisniewski SR, et al. Strengthening clinical effectiveness trials: equipoise-stratified randomization. Biol Psychiatry 2001;50:792-801.
30. Marangell LB, Rush AJ, George MS, et al. Vagus nerve stimulation (VNS) for major depressive episodes: one-year outcomes. Biol Psychiatry 2002;51(4):280-7.
When depression fails to respond to initial therapy—as it commonly does—we have many options but little evidence to guide our choices. We often wonder:
- Is this patient’s depression treatment-resistant?
- Would switching medications or augmenting the initial drug be more likely to achieve an adequate response?
- How effective is psychotherapy compared with medication for treatment-resistant depression?
This article offers insights into each question, based on available trial data, algorithmic approaches to major depressive disorder, and clinical experience. Included is a preview of an ongoing multicenter, treatment-resistant depression study that mimics clinical practice and a look at vagus nerve stimulation (VNS)—a novel somatic therapy being considered by the FDA.
Measuring treatment response
Sustained symptom remission—with normalization of function—is the aim of treating major depressive disorder. Outcomes are categorized as:
- remission (virtual absence of depressive symptoms)
- response with residual symptoms (>50% reduction in baseline symptom severity that does not qualify for remission)
- partial response (>25% but <50% decrease in baseline symptom severity)
- nonresponse (<25% reduction in baseline symptoms).
Major depressive disorder (MDD) is typically recurrent or chronic and characterized by marked disability and a life expectancy shortened by suicide and increased mortality from associated medical conditions. Lifetime prevalence is 16.2%.6
MDD is twice as likely to affect women as men and is common among adolescents, young adults, and persons with concurrent medical conditions.
Major depression’s course is characterized by:
- recurrent episodes (approximately every 5 years)
- or a persistent level of waxing and waning depressive symptoms (in 20% to 35% of cases).
Dysthymic disorder often heralds major depression. Within 1 year, 5% to 20% of persons with dysthymic disorder develop major depression.7
Disability associated with major depression
often exceeds that of other general medical conditions. Depression is the fourth most disabling condition worldwide and is projected to rank number two by 2020 because of its chronic and recurrent nature, high prevalence, and life-shortening effects.8
Consequences of unremitting depression include:
- poor day-to-day function (work, family)
- increased likelihood of recurrence
- psychiatric or medical complications, including substance abuse
- high use of mental health and general medical resources
- worsened prognosis of medical conditions
- high family burden.
In 8-week acute-phase trials, 7% to 15% of patients do not tolerate the initial medication, 25% show no response, 15% show partial response, 10% to 20% exhibit response with residual symptoms, and 30% to 40% achieve remission. Complicated depressions that may not respond as well include those concurrent with Axis I conditions—such as panic disorder or substance abuse—or Axis II or III conditions.1
Time-limited psychotherapies targeted at depressive symptoms (such as cognitive, interpersonal, and behavioral therapies) also typically achieve a 50% response rate in uncomplicated depression that is not treatment-resistant.
Recommendation. When treating depression, assess response at least every 4 weeks (preferably at each visit), using a self-report or clinician rating such as:
- Quick Inventory of Depressive Symptomatology2 (see Related resources)
- Beck Depression Inventory3,4
- Patient Health Questionnaire.5
Defining treatment resistance
A patient may not achieve remission for a variety of reasons, including poor adherence, inadequate medication trial or dosing, occult substance abuse, undiagnosed medical conditions (Box),6-8 concurrent Axis I or II disorders, or treatment resistance.
The general consensus is to consider depression “treatment-resistant” when at least two adequately delivered treatments do not achieve at least a response. A stricter definition—failure to achieve sustained remission with two or more treatments—has also been suggested.
Several schemes have proposed treatment resistance levels, such as the five stages identified in the Table. Recent studies9,10 suggest that increasing treatment resistance is associated with decreasing response or remission rates.
Therefore, when a patient’s treatment resistance is high, two appropriate strategies are to:
- persist with and use maximally tolerated dosages of the treatment you select
- aim for response because high resistance lowers the likelihood of remission.
Predicting response. A major clinical issue is determining whether remission will occur during an acute treatment trial. It is important to not declare treatment resistance unless there has been:
- adequate exposure (dosing and duration) to the treatment
- and adequate adherence.
Patients often have apparent but not actual resistance, meaning that the agent was not used long enough (at least 6 weeks) or at high enough doses. Remission typically follows response by several weeks or even 1 to 2 months for more-chronic depressions.11 Thus, treatment trials should continue at least 12 weeks to determine whether remission will occur.
On the other hand, not obtaining at least a signal of minimal benefit (at least a 20% reduction in baseline symptom severity) in 4 to 6 weeks often portends a low likelihood of response in the long run.12,13 Thus, continue a treatment at least 6 weeks before you decide that it will not achieve a response.
Recommendation. Measure symptoms at key decision points. If modest improvement (such as 20% reduction in baseline symptoms) is found at 4 to 6 weeks, continue treating another 4 to 6 weeks, increasing the dosage as tolerated.
Table
Simple system for staging antidepressant resistance
| Stage | Definition |
|---|---|
| I | Failure of at least one adequate trial of one major antidepressant class |
| II | Stage I resistance plus failure of an adequate trial of an antidepressant in a distinctly different class from that used in Stage I |
| III | Stage II resistance plus failure of an adequate trial of a tricyclic antidepressant |
| IV | Stage III resistance plus failure of an adequate trial of a monoamine oxidase inhibitor |
| V | Stage IV resistance plus failure of a course of bilateral electroconvulsive therapy |
| Source: Reprinted with permission from Thase ME, Rush AJ. When at first you don’t succeed: sequential strategies for antidepressant nonresponders. J Clin Psychiatry 1997;58(suppl 13):24. | |
Treatment options
When initial antidepressant treatment fails to achieve an adequate response—as it does in more than onehalf of major depression cases—the next step is to add a second agent or switch to another agent.
Available evidence14 relies almost exclusively on open, uncontrolled trials, which do not provide definitive answers. Even so, these trials indicate that nonresponse (or nonremission) with one agent does not predict nonresponse/nonremission with another.
Switching strategies. When a selective serotonin reuptake inhibitor (SSRI) is the first treatment, several open trials reveal an approximately 50% response rate to a second SSRI. However, opentrial evidence and retrospective chart review reports also indicate that switching out of class (such as from an SSRI to bupropion) is also approximately 50% effective.15
Some post hoc analyses of acute 8-week trials indicate that the dual-action agent venlafaxine at higher dosages (up to 225 mg/d of venlafaxine XR) is associated with higher remission rates than the more-selective SSRIs.16,17 On the other hand, unpublished data indicate that escitalopram, 10 mg/d, and venlafaxine XR, up to 150 mg/d, did not differ in efficacy among outpatients treated by primary care physicians.18
On the other hand, sertraline and imipramine (a dual-action agent) were equally effective in a 12-week acute-phase trial.19 Furthermore, response and remission rates were similar when nonresponders in each group switched to the other antidepressant.20 This suggests that the dual-action agent (imipramine) was not more effective than the more selective agent (sertraline) in this population.
Well-controlled trials show that monoamine oxidase inhibitors (MAOIs) can be effective when tricyclic antidepressants (TCAs) are not. Switches among the TCAs are associated with a 30% response rate, whereas switching from a TCA to an MAOI typically results in a 50% response rate.21
Controlled prospective comparisons of two or more alternate switch or augment treatments are needed to establish comparative efficacy and tolerability.
Augmentation strategies may include lithium, buspirone, thyroid hormone (T3), stimulants, or atypical antipsychotics. Although head-to-head comparisons are rare, a randomized, controlled trial found that combining olanzapine (mean 50 mg/d), with fluoxetine (mean 15 mg/d) was more effective than each agent used alone.22
Risperidone augmentation is supported by open trials, as is the use of modafinil, other stimulants, and bupropion. An important unanswered question with most augmentation strategies is how long to continue them if they are successful.
Psychotherapy may also play a key role in augmenting medication’s effects. Keller et al23 found in chronically depressed outpatients that 12 weeks of nefazodone, up to 600 mg/d, plus cognitive behavioral analytic system psychotherapy (CBASP) produced higher response and remission rates compared with either treatment alone. A subsequent report24 found that 50% of nefa-zodone and CBASP monotherapy nonresponders did respond when switched to the alternate treatment.
Thus, CBASP may be useful at least in chronic depression to augment medication or as a “switch” to monotherapy if medication alone fails. Interestingly, Nemeroff et al25 found CBASP more effective than nefazodone for patients with chronic major depression who had a childhood history of parental loss or physical, sexual, or emotional abuse.
Antidepressant tachyphylaxis—commonly referred to as “poop-out”—is reported with all antidepressants. That is, even while apparently taking their medications for 6 to 18 months, some patients lose the antidepressant effect, such that some symptoms return or a full relapse/recurrence ensues. Mechanisms of this phenomenon are unknown.
Clinically, some believe that “poop out” is more common with SSRIs than with other antidepressant classes, but no long-term comparative data support or challenge this view. Treatment options include a dosage increase, dosage reduction (especially for long half-life SSRIs such as fluoxetine), or augmentation with the options noted above (such as bupropion, buspirone, etc.).
Benefit of using algorithms
Algorithms (such as the Texas Medication Algorithm Project26 ) have suggested multiple treatment steps for major depression after initial treatment fails, with several options available at each step. Using medication algorithms has been found more effective than treatment-as-usual in outpatients with major depressive disorder.27 No studies have compared different algorithms.
STAR*D trial. The ongoing National Institute of Mental Health (NIMH) Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial may offer a new algorithmic approach to treating major depression.14,28 NIMH launched STAR*D in 1999, enrollment began in 2001, and results are expecte by May 2005 (see Related resources).
STAR*D—of which I am the study director—is a randomized, controlled, raterblinded, multisite trial of outpatients ages 18 to 75 with nonpsychotic major depression (17-item Hamilton Rating Scale for Depression score 14). The trial design includes four treatment levels and numerous antidepressant options (Figure).
The study’s aim is to enroll 4,000 patients into level 1, with 1,500 entering level 2. Patients who achieve an adequate response based on clinician judgment may continue the effective treatment for 12 months, during which their symptoms and other relevant information are monitored monthly by telephone. Patients who do not achieve an acceptable response in level 1 (or in subsequent levels) may proceed to the next level, which involves a randomized assignment.
STAR*D has an innovative design that mimics clinical practice and ensures high levels of patient participation. When patients agree to randomization, they may elect to exclude groups of treatments but may not pick a particular treatment (they must accept randomization to stay in the study).
Figure STAR*D treatment levels for major depressive disorder
For example, patient A entering level 2 may exclude switch treatments and elect to accept randomization to citalopram plus bupropion SR, citalopram plus buspirone, or citalopram and cognitive therapy. Conversely, patient B may exclude all augment options at level 2, and accept randomization to the four switch options.
Patients may exclude cognitive psychotherapy as an augment and/or switch option as long as they accept randomization to all available medication switches, or augments, or both. They may also choose cognitive therapy and exclude all medication switch and augment options. These patients must accept randomization to either cognitive therapy switch or cognitive therapy augmentation.
This so-called equipoise stratified randomized design29 allows us to compare all participants randomized to the treatments being compared. To date, only 1% of subjects have accepted randomization to all seven level-2 treatments. About one-half elect only the switch options, and about one-half elect only the augment options.
STAR*D’s goal is to determine whether there is a preferred next step for varying types and degrees of treatment-resistant repression.
Vagus nerve stimulation
Somatic therapies being investigated to expand our therapeutic options for major depressive disorder include magnetic seizure therapy, repetitive transcranial magnetic stimulation, and vagus nerve stimulation (VNS).
VNS—now indicated for treatment-resistant epilepsy—is being investigated as a potential augmentation for treatment-resistant depression. An application for this supplemental indication was submitted to the FDA in October 2003.
With VNS, a device implanted in the patient’s chest provides intermittent stimulation to the left vagus nerve (typically 30 seconds on and 5 minutes off, 24 hours a day). In an open trial10 and follow-up report,30 VNS was associated with a 30% to 45% response rate in 59 depressed patients with high levels of treatment resistance (inadequate response to an average of 16 treatment trials).
VNS is well tolerated, though it has not been prospectively studied in patients with diagnosed cardiovascular disease. Side effects that may occur when the stimulation is “on” include:
- voice alteration in about 60% of patients (the voice becomes more hoarse when the left recurrent laryngeal nerve is activated)
- paresthesias in the neck
- shortness of breath on heavy exertion.
These effects are usually absent in the 5-minute “off” phase.
- National Institute of Mental Health. Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial. Includes the Quick Inventory of Depressive Symptomatology. www.star-d.org
- HealthyPlace.Com Depression Community. Vagus nerve stimulation for treating depression. www.healthyplace.com/communities/depression/treatment/vns
Drug brand names
- Bupropion • Wellbutrin, Wellbutrin SR
- Buspirone • BuSpar
- Citalopram • Celexa
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Mirtazapine • Remeron
- Modafinil • Provigil
- Nefazodone • Serzone
- Nortriptyline • Pamelor, Aventyl
- Olanzapine • Zyprexa
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tranylcypromine • Parnate
- Venlafaxine • Effexor, Effexor XR
Disclosure
Dr. Rush receives grant/research support from the Robert Wood Johnson Foundation, National Institute of Mental Health, and The Stanley Foundation. He is a consultant to Bristol-Myers Squibb Co., Cyberonics, Eli Lilly & Co., Forest Laboratories, and GlaxoSmithKline, and a speaker for Bristol-Myers Squibb Co., Cyberonics, Eli Lilly & Co., Forest Laboratories, GlaxoSmithKline, and Wyeth Pharmaceuticals.
When depression fails to respond to initial therapy—as it commonly does—we have many options but little evidence to guide our choices. We often wonder:
- Is this patient’s depression treatment-resistant?
- Would switching medications or augmenting the initial drug be more likely to achieve an adequate response?
- How effective is psychotherapy compared with medication for treatment-resistant depression?
This article offers insights into each question, based on available trial data, algorithmic approaches to major depressive disorder, and clinical experience. Included is a preview of an ongoing multicenter, treatment-resistant depression study that mimics clinical practice and a look at vagus nerve stimulation (VNS)—a novel somatic therapy being considered by the FDA.
Measuring treatment response
Sustained symptom remission—with normalization of function—is the aim of treating major depressive disorder. Outcomes are categorized as:
- remission (virtual absence of depressive symptoms)
- response with residual symptoms (>50% reduction in baseline symptom severity that does not qualify for remission)
- partial response (>25% but <50% decrease in baseline symptom severity)
- nonresponse (<25% reduction in baseline symptoms).
Major depressive disorder (MDD) is typically recurrent or chronic and characterized by marked disability and a life expectancy shortened by suicide and increased mortality from associated medical conditions. Lifetime prevalence is 16.2%.6
MDD is twice as likely to affect women as men and is common among adolescents, young adults, and persons with concurrent medical conditions.
Major depression’s course is characterized by:
- recurrent episodes (approximately every 5 years)
- or a persistent level of waxing and waning depressive symptoms (in 20% to 35% of cases).
Dysthymic disorder often heralds major depression. Within 1 year, 5% to 20% of persons with dysthymic disorder develop major depression.7
Disability associated with major depression
often exceeds that of other general medical conditions. Depression is the fourth most disabling condition worldwide and is projected to rank number two by 2020 because of its chronic and recurrent nature, high prevalence, and life-shortening effects.8
Consequences of unremitting depression include:
- poor day-to-day function (work, family)
- increased likelihood of recurrence
- psychiatric or medical complications, including substance abuse
- high use of mental health and general medical resources
- worsened prognosis of medical conditions
- high family burden.
In 8-week acute-phase trials, 7% to 15% of patients do not tolerate the initial medication, 25% show no response, 15% show partial response, 10% to 20% exhibit response with residual symptoms, and 30% to 40% achieve remission. Complicated depressions that may not respond as well include those concurrent with Axis I conditions—such as panic disorder or substance abuse—or Axis II or III conditions.1
Time-limited psychotherapies targeted at depressive symptoms (such as cognitive, interpersonal, and behavioral therapies) also typically achieve a 50% response rate in uncomplicated depression that is not treatment-resistant.
Recommendation. When treating depression, assess response at least every 4 weeks (preferably at each visit), using a self-report or clinician rating such as:
- Quick Inventory of Depressive Symptomatology2 (see Related resources)
- Beck Depression Inventory3,4
- Patient Health Questionnaire.5
Defining treatment resistance
A patient may not achieve remission for a variety of reasons, including poor adherence, inadequate medication trial or dosing, occult substance abuse, undiagnosed medical conditions (Box),6-8 concurrent Axis I or II disorders, or treatment resistance.
The general consensus is to consider depression “treatment-resistant” when at least two adequately delivered treatments do not achieve at least a response. A stricter definition—failure to achieve sustained remission with two or more treatments—has also been suggested.
Several schemes have proposed treatment resistance levels, such as the five stages identified in the Table. Recent studies9,10 suggest that increasing treatment resistance is associated with decreasing response or remission rates.
Therefore, when a patient’s treatment resistance is high, two appropriate strategies are to:
- persist with and use maximally tolerated dosages of the treatment you select
- aim for response because high resistance lowers the likelihood of remission.
Predicting response. A major clinical issue is determining whether remission will occur during an acute treatment trial. It is important to not declare treatment resistance unless there has been:
- adequate exposure (dosing and duration) to the treatment
- and adequate adherence.
Patients often have apparent but not actual resistance, meaning that the agent was not used long enough (at least 6 weeks) or at high enough doses. Remission typically follows response by several weeks or even 1 to 2 months for more-chronic depressions.11 Thus, treatment trials should continue at least 12 weeks to determine whether remission will occur.
On the other hand, not obtaining at least a signal of minimal benefit (at least a 20% reduction in baseline symptom severity) in 4 to 6 weeks often portends a low likelihood of response in the long run.12,13 Thus, continue a treatment at least 6 weeks before you decide that it will not achieve a response.
Recommendation. Measure symptoms at key decision points. If modest improvement (such as 20% reduction in baseline symptoms) is found at 4 to 6 weeks, continue treating another 4 to 6 weeks, increasing the dosage as tolerated.
Table
Simple system for staging antidepressant resistance
| Stage | Definition |
|---|---|
| I | Failure of at least one adequate trial of one major antidepressant class |
| II | Stage I resistance plus failure of an adequate trial of an antidepressant in a distinctly different class from that used in Stage I |
| III | Stage II resistance plus failure of an adequate trial of a tricyclic antidepressant |
| IV | Stage III resistance plus failure of an adequate trial of a monoamine oxidase inhibitor |
| V | Stage IV resistance plus failure of a course of bilateral electroconvulsive therapy |
| Source: Reprinted with permission from Thase ME, Rush AJ. When at first you don’t succeed: sequential strategies for antidepressant nonresponders. J Clin Psychiatry 1997;58(suppl 13):24. | |
Treatment options
When initial antidepressant treatment fails to achieve an adequate response—as it does in more than onehalf of major depression cases—the next step is to add a second agent or switch to another agent.
Available evidence14 relies almost exclusively on open, uncontrolled trials, which do not provide definitive answers. Even so, these trials indicate that nonresponse (or nonremission) with one agent does not predict nonresponse/nonremission with another.
Switching strategies. When a selective serotonin reuptake inhibitor (SSRI) is the first treatment, several open trials reveal an approximately 50% response rate to a second SSRI. However, opentrial evidence and retrospective chart review reports also indicate that switching out of class (such as from an SSRI to bupropion) is also approximately 50% effective.15
Some post hoc analyses of acute 8-week trials indicate that the dual-action agent venlafaxine at higher dosages (up to 225 mg/d of venlafaxine XR) is associated with higher remission rates than the more-selective SSRIs.16,17 On the other hand, unpublished data indicate that escitalopram, 10 mg/d, and venlafaxine XR, up to 150 mg/d, did not differ in efficacy among outpatients treated by primary care physicians.18
On the other hand, sertraline and imipramine (a dual-action agent) were equally effective in a 12-week acute-phase trial.19 Furthermore, response and remission rates were similar when nonresponders in each group switched to the other antidepressant.20 This suggests that the dual-action agent (imipramine) was not more effective than the more selective agent (sertraline) in this population.
Well-controlled trials show that monoamine oxidase inhibitors (MAOIs) can be effective when tricyclic antidepressants (TCAs) are not. Switches among the TCAs are associated with a 30% response rate, whereas switching from a TCA to an MAOI typically results in a 50% response rate.21
Controlled prospective comparisons of two or more alternate switch or augment treatments are needed to establish comparative efficacy and tolerability.
Augmentation strategies may include lithium, buspirone, thyroid hormone (T3), stimulants, or atypical antipsychotics. Although head-to-head comparisons are rare, a randomized, controlled trial found that combining olanzapine (mean 50 mg/d), with fluoxetine (mean 15 mg/d) was more effective than each agent used alone.22
Risperidone augmentation is supported by open trials, as is the use of modafinil, other stimulants, and bupropion. An important unanswered question with most augmentation strategies is how long to continue them if they are successful.
Psychotherapy may also play a key role in augmenting medication’s effects. Keller et al23 found in chronically depressed outpatients that 12 weeks of nefazodone, up to 600 mg/d, plus cognitive behavioral analytic system psychotherapy (CBASP) produced higher response and remission rates compared with either treatment alone. A subsequent report24 found that 50% of nefa-zodone and CBASP monotherapy nonresponders did respond when switched to the alternate treatment.
Thus, CBASP may be useful at least in chronic depression to augment medication or as a “switch” to monotherapy if medication alone fails. Interestingly, Nemeroff et al25 found CBASP more effective than nefazodone for patients with chronic major depression who had a childhood history of parental loss or physical, sexual, or emotional abuse.
Antidepressant tachyphylaxis—commonly referred to as “poop-out”—is reported with all antidepressants. That is, even while apparently taking their medications for 6 to 18 months, some patients lose the antidepressant effect, such that some symptoms return or a full relapse/recurrence ensues. Mechanisms of this phenomenon are unknown.
Clinically, some believe that “poop out” is more common with SSRIs than with other antidepressant classes, but no long-term comparative data support or challenge this view. Treatment options include a dosage increase, dosage reduction (especially for long half-life SSRIs such as fluoxetine), or augmentation with the options noted above (such as bupropion, buspirone, etc.).
Benefit of using algorithms
Algorithms (such as the Texas Medication Algorithm Project26 ) have suggested multiple treatment steps for major depression after initial treatment fails, with several options available at each step. Using medication algorithms has been found more effective than treatment-as-usual in outpatients with major depressive disorder.27 No studies have compared different algorithms.
STAR*D trial. The ongoing National Institute of Mental Health (NIMH) Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial may offer a new algorithmic approach to treating major depression.14,28 NIMH launched STAR*D in 1999, enrollment began in 2001, and results are expecte by May 2005 (see Related resources).
STAR*D—of which I am the study director—is a randomized, controlled, raterblinded, multisite trial of outpatients ages 18 to 75 with nonpsychotic major depression (17-item Hamilton Rating Scale for Depression score 14). The trial design includes four treatment levels and numerous antidepressant options (Figure).
The study’s aim is to enroll 4,000 patients into level 1, with 1,500 entering level 2. Patients who achieve an adequate response based on clinician judgment may continue the effective treatment for 12 months, during which their symptoms and other relevant information are monitored monthly by telephone. Patients who do not achieve an acceptable response in level 1 (or in subsequent levels) may proceed to the next level, which involves a randomized assignment.
STAR*D has an innovative design that mimics clinical practice and ensures high levels of patient participation. When patients agree to randomization, they may elect to exclude groups of treatments but may not pick a particular treatment (they must accept randomization to stay in the study).
Figure STAR*D treatment levels for major depressive disorder
For example, patient A entering level 2 may exclude switch treatments and elect to accept randomization to citalopram plus bupropion SR, citalopram plus buspirone, or citalopram and cognitive therapy. Conversely, patient B may exclude all augment options at level 2, and accept randomization to the four switch options.
Patients may exclude cognitive psychotherapy as an augment and/or switch option as long as they accept randomization to all available medication switches, or augments, or both. They may also choose cognitive therapy and exclude all medication switch and augment options. These patients must accept randomization to either cognitive therapy switch or cognitive therapy augmentation.
This so-called equipoise stratified randomized design29 allows us to compare all participants randomized to the treatments being compared. To date, only 1% of subjects have accepted randomization to all seven level-2 treatments. About one-half elect only the switch options, and about one-half elect only the augment options.
STAR*D’s goal is to determine whether there is a preferred next step for varying types and degrees of treatment-resistant repression.
Vagus nerve stimulation
Somatic therapies being investigated to expand our therapeutic options for major depressive disorder include magnetic seizure therapy, repetitive transcranial magnetic stimulation, and vagus nerve stimulation (VNS).
VNS—now indicated for treatment-resistant epilepsy—is being investigated as a potential augmentation for treatment-resistant depression. An application for this supplemental indication was submitted to the FDA in October 2003.
With VNS, a device implanted in the patient’s chest provides intermittent stimulation to the left vagus nerve (typically 30 seconds on and 5 minutes off, 24 hours a day). In an open trial10 and follow-up report,30 VNS was associated with a 30% to 45% response rate in 59 depressed patients with high levels of treatment resistance (inadequate response to an average of 16 treatment trials).
VNS is well tolerated, though it has not been prospectively studied in patients with diagnosed cardiovascular disease. Side effects that may occur when the stimulation is “on” include:
- voice alteration in about 60% of patients (the voice becomes more hoarse when the left recurrent laryngeal nerve is activated)
- paresthesias in the neck
- shortness of breath on heavy exertion.
These effects are usually absent in the 5-minute “off” phase.
- National Institute of Mental Health. Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial. Includes the Quick Inventory of Depressive Symptomatology. www.star-d.org
- HealthyPlace.Com Depression Community. Vagus nerve stimulation for treating depression. www.healthyplace.com/communities/depression/treatment/vns
Drug brand names
- Bupropion • Wellbutrin, Wellbutrin SR
- Buspirone • BuSpar
- Citalopram • Celexa
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Mirtazapine • Remeron
- Modafinil • Provigil
- Nefazodone • Serzone
- Nortriptyline • Pamelor, Aventyl
- Olanzapine • Zyprexa
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tranylcypromine • Parnate
- Venlafaxine • Effexor, Effexor XR
Disclosure
Dr. Rush receives grant/research support from the Robert Wood Johnson Foundation, National Institute of Mental Health, and The Stanley Foundation. He is a consultant to Bristol-Myers Squibb Co., Cyberonics, Eli Lilly & Co., Forest Laboratories, and GlaxoSmithKline, and a speaker for Bristol-Myers Squibb Co., Cyberonics, Eli Lilly & Co., Forest Laboratories, GlaxoSmithKline, and Wyeth Pharmaceuticals.
1. Depression Guideline Panel. Clinical practice guideline, number 5: depression in primary care: vol. 2. Treatment of major depression Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research. AHCPR publication no. 93-0551, 1993.
2. Rush AJ, Trivedi MH, Ibrahim HM, et al. The 16-Item Quick Inventory of Depressive Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biol Psychiatry 2003;54:573-83.
3. Beck AT, Steer RA, Brown GK. Beck Depression Inventory (2nd ed. manual). San Antonio, TX: The Psychological Corporation, 1996.
4. Beck AT, Ward CH, Mendelson M, et al. An inventory for measuring depression. Arch Gen Psychiatry 1961;4:561-71.
5. Kroenke K, Spitzer RL, Williams JBW. The PHQ-9. Validity of a brief depression severity measure. J Gen Intern Med 2001;16:606-13.
6. Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003;289(23):3095-105.
7. Depression Guideline Panel. Clinical practice guideline, number 5: Depression in primary care, vol. 1: detection and diagnosis Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research. AHCPR publication no. 93-0550, 1993.
8. Murray CJ, Lopez AD. (eds) The global burden of disease Boston: Harvard School of Public Health, 1996.
9. Barbee JG, Jamhour NJ. Lamotrigine as an augmentation agent in treatment-resistant depression. J Clin Psychiatry 2002;63(8):737-41.
10. Sackeim HA, Rush AJ, George MS, et al. Vagus nerve stimulation (VNS) for treatment-resistant depression: efficacy, side effects, and predictors of outcome. Neuropsychopharmacology 2001;25(5):713-28.
11. Koran LM, Gelenberg AJ, Kornstein SG, et al. Sertraline versus imipramine to prevent relapse in chronic depression. J Affect Disord 2001;65(1):27-36.
12. Nierenberg AA, Feighner JP, Rudolph R, et al. Venlafaxine for treatment-resistant unipolar depression. J Clin Psychopharmacol 1994;14(6):419-23.
13. Quitkin FM, Petkova E, McGrath PJ, et al. When should a trial of fluoxetine for major depression be declared failed? Am J Psychiatry 2003;160(4):734-40.
14. Fava M, Rush AJ, Trivedi MH, et al. Background and rationale for the sequenced treatment alternatives to relieve depression (STAR*D) study. Psychiatr Clin North Am 2003;26(2):457-94.
15. Fava M, Papakostas GI, Petersen T, et al. Switching to bupropion in fluoxetine-resistant major depressive disorder. Ann Clin Psychiatry 2003;15:17-22.
16. Entsuah AR, Huang H, Thase ME. Response and remission rates in different subpopulations with major depressive disorder administered venlafaxine, selective serotonin reuptake inhibitors, or placebo. J Clin Psychiatry 2001;62:869-77.
17. Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or selective serotonin reuptake inhibitors. Br J Psychiatry 2001;178:234-41.
18. Montgomery SA, Huusom A, Bothmer J. Flexible dose comparison of s-citalopram and venlafaxine XR. J Eur Neuropsychopharmacol 2002;12(suppl 3):S-254.
19. Keller MB, Gelenberg AJ, Hirschfeld RM, et al. The treatment of chronic depression, part 2: a double-blind, randomized trial of sertraline and imipramine. J Clin Psychiatry 1998;59(11):598-607.
20. Thase ME, Rush AJ, Howland RH, et al. Double-blind switch study of imipramine or sertraline treatment of antidepressant-resistant chronic depression. Arch Gen Psychiatry 2002;59(3):233-9.
21. Thase ME, Rush AJ. Treatment resistant depression. In: Bloom FE, Kupfer DJ (eds). Psychopharmacology: the fourth generation of progress New York: Raven Press, 1995;1081-97.
22. Shelton RC, Tollefson GD, Tohen M, et al. A novel augmentation strategy for treating resistant major depression. Am J Psychiatry 2001;158:131-4.
23. Keller MB, McCullough JP, Klein DN, et al. A comparison of nefazodone, the cognitive behavioral-analysis system of psychotherapy, and their combination for the treatment of chronic depression. N Engl J Med 2000;342:1462-70.
24. Schatzberg AF, Rush AJ, Arnow BA, et al. Medication or psychotherapy is effective when the other is not in chronic depression: empirical support. Arch Gen Psychiatry (submitted).
25. Nemeroff CB, Heim CM, Thase ME, et al. Differential responses to psychotherapy versus pharmacotherapy in patients with chronic forms of major depression and childhood trauma. Proc Natl Acad Sci USA 2003;100(24):14293-6.
26. Crismon ML, Trivedi M, Pigott TA, et al. The Texas Medication Algorithm Project: report of the Texas Consensus Conference Panel on Medication Treatment of Major Depressive Disorder. J Clin Psychiatry 1999;60(3):142-56.
27. Trivedi MH, Rush AJ, Crismon ML, et al. The Texas Medication Algorithm Project (TMAP): clinical results for patients with major depressive disorder. Arch Gen Psychiatry (in press).
28. Rush AJ, Fava M, Wisniewski SR, et al. for the STAR*D Investigators Group. Sequenced Treatment Alternatives to Relieve Depression (STAR*D): rationale and design. Control Clin Trials 2004;25(1):118-41.
29. Lavori PW, Rush AJ, Wisniewski SR, et al. Strengthening clinical effectiveness trials: equipoise-stratified randomization. Biol Psychiatry 2001;50:792-801.
30. Marangell LB, Rush AJ, George MS, et al. Vagus nerve stimulation (VNS) for major depressive episodes: one-year outcomes. Biol Psychiatry 2002;51(4):280-7.
1. Depression Guideline Panel. Clinical practice guideline, number 5: depression in primary care: vol. 2. Treatment of major depression Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research. AHCPR publication no. 93-0551, 1993.
2. Rush AJ, Trivedi MH, Ibrahim HM, et al. The 16-Item Quick Inventory of Depressive Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biol Psychiatry 2003;54:573-83.
3. Beck AT, Steer RA, Brown GK. Beck Depression Inventory (2nd ed. manual). San Antonio, TX: The Psychological Corporation, 1996.
4. Beck AT, Ward CH, Mendelson M, et al. An inventory for measuring depression. Arch Gen Psychiatry 1961;4:561-71.
5. Kroenke K, Spitzer RL, Williams JBW. The PHQ-9. Validity of a brief depression severity measure. J Gen Intern Med 2001;16:606-13.
6. Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003;289(23):3095-105.
7. Depression Guideline Panel. Clinical practice guideline, number 5: Depression in primary care, vol. 1: detection and diagnosis Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research. AHCPR publication no. 93-0550, 1993.
8. Murray CJ, Lopez AD. (eds) The global burden of disease Boston: Harvard School of Public Health, 1996.
9. Barbee JG, Jamhour NJ. Lamotrigine as an augmentation agent in treatment-resistant depression. J Clin Psychiatry 2002;63(8):737-41.
10. Sackeim HA, Rush AJ, George MS, et al. Vagus nerve stimulation (VNS) for treatment-resistant depression: efficacy, side effects, and predictors of outcome. Neuropsychopharmacology 2001;25(5):713-28.
11. Koran LM, Gelenberg AJ, Kornstein SG, et al. Sertraline versus imipramine to prevent relapse in chronic depression. J Affect Disord 2001;65(1):27-36.
12. Nierenberg AA, Feighner JP, Rudolph R, et al. Venlafaxine for treatment-resistant unipolar depression. J Clin Psychopharmacol 1994;14(6):419-23.
13. Quitkin FM, Petkova E, McGrath PJ, et al. When should a trial of fluoxetine for major depression be declared failed? Am J Psychiatry 2003;160(4):734-40.
14. Fava M, Rush AJ, Trivedi MH, et al. Background and rationale for the sequenced treatment alternatives to relieve depression (STAR*D) study. Psychiatr Clin North Am 2003;26(2):457-94.
15. Fava M, Papakostas GI, Petersen T, et al. Switching to bupropion in fluoxetine-resistant major depressive disorder. Ann Clin Psychiatry 2003;15:17-22.
16. Entsuah AR, Huang H, Thase ME. Response and remission rates in different subpopulations with major depressive disorder administered venlafaxine, selective serotonin reuptake inhibitors, or placebo. J Clin Psychiatry 2001;62:869-77.
17. Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or selective serotonin reuptake inhibitors. Br J Psychiatry 2001;178:234-41.
18. Montgomery SA, Huusom A, Bothmer J. Flexible dose comparison of s-citalopram and venlafaxine XR. J Eur Neuropsychopharmacol 2002;12(suppl 3):S-254.
19. Keller MB, Gelenberg AJ, Hirschfeld RM, et al. The treatment of chronic depression, part 2: a double-blind, randomized trial of sertraline and imipramine. J Clin Psychiatry 1998;59(11):598-607.
20. Thase ME, Rush AJ, Howland RH, et al. Double-blind switch study of imipramine or sertraline treatment of antidepressant-resistant chronic depression. Arch Gen Psychiatry 2002;59(3):233-9.
21. Thase ME, Rush AJ. Treatment resistant depression. In: Bloom FE, Kupfer DJ (eds). Psychopharmacology: the fourth generation of progress New York: Raven Press, 1995;1081-97.
22. Shelton RC, Tollefson GD, Tohen M, et al. A novel augmentation strategy for treating resistant major depression. Am J Psychiatry 2001;158:131-4.
23. Keller MB, McCullough JP, Klein DN, et al. A comparison of nefazodone, the cognitive behavioral-analysis system of psychotherapy, and their combination for the treatment of chronic depression. N Engl J Med 2000;342:1462-70.
24. Schatzberg AF, Rush AJ, Arnow BA, et al. Medication or psychotherapy is effective when the other is not in chronic depression: empirical support. Arch Gen Psychiatry (submitted).
25. Nemeroff CB, Heim CM, Thase ME, et al. Differential responses to psychotherapy versus pharmacotherapy in patients with chronic forms of major depression and childhood trauma. Proc Natl Acad Sci USA 2003;100(24):14293-6.
26. Crismon ML, Trivedi M, Pigott TA, et al. The Texas Medication Algorithm Project: report of the Texas Consensus Conference Panel on Medication Treatment of Major Depressive Disorder. J Clin Psychiatry 1999;60(3):142-56.
27. Trivedi MH, Rush AJ, Crismon ML, et al. The Texas Medication Algorithm Project (TMAP): clinical results for patients with major depressive disorder. Arch Gen Psychiatry (in press).
28. Rush AJ, Fava M, Wisniewski SR, et al. for the STAR*D Investigators Group. Sequenced Treatment Alternatives to Relieve Depression (STAR*D): rationale and design. Control Clin Trials 2004;25(1):118-41.
29. Lavori PW, Rush AJ, Wisniewski SR, et al. Strengthening clinical effectiveness trials: equipoise-stratified randomization. Biol Psychiatry 2001;50:792-801.
30. Marangell LB, Rush AJ, George MS, et al. Vagus nerve stimulation (VNS) for major depressive episodes: one-year outcomes. Biol Psychiatry 2002;51(4):280-7.
Beating the high cost of software
Name-brand practice-management software can cost hundreds or even thousands of dollars-no small expense, especially for a new practice. Still, you and your staff need the word processing, spreadsheet, documentation, patient tracking, and appointment-scheduling capabilities these programs provide.
Open-source and general public licensing (GPL) software titles-available free or at minimal cost-may offer a budget-friendly alternative.
Open-source versus GPL
There are two types of “free” software:
- “free” as in no charge for its use. For example, Steve Gibson of Speeding up your Web search,” Psyber Psychiatry, May 2003). Use key words such as “freeware,” “free,” or “open source” in conjunction with the type of program (eg, “document” or “database”).
Many open-source or GPL projects are centrally stored at two sites, SourceForge and Freshmeat. Browse the categories for the type of program you need, such as databases, utilities, firewalls, etc. These sites offer source code and version information for programmers as well as precompiled software for the public.
Table Relevant open-source and GPL software titles
Related Resources (accessed Feb. 20, 2004) GNU Project. GNU General Public License (example of free-distribution license). http://www.gnu.org/copyleft/gpl.htmlType Software URL Office document OpenOffice * www.openoffice.org Instant messaging GAIM * http://gaim.sourceforge.net Web browser Firefox * www.mozilla.org/products/firefox/ Database Firebird * http://firebird.sourceforge.net Application server Zope * www.zope.org Practice management FreeMED www.freemed.org Clinical database SQL Clinic www.sqlclinic.net Electronic Medical Record Open EMR www.openemr.net * available in binary format and ready to use
GNU Project. Various licenses and comments about them. http://www.gnu.org/licenses/license-list.html.
GNU Project. Terms and conditions for copying, distribution, and modification http://www.gnu.org/copyleft/gpl.html#SEC3
Open Source Initiative. The open source definition. http://www.opensource.org/docs/definition.php
If you have questions about these products or comments about Psyber Psychiatry, click here to contact Dr. Luo or send an e-mail to [email protected].
Disclosure Dr. Luo reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Luo in this column are his own and do not necessarily reflect those of Current Psychiatry.
Name-brand practice-management software can cost hundreds or even thousands of dollars-no small expense, especially for a new practice. Still, you and your staff need the word processing, spreadsheet, documentation, patient tracking, and appointment-scheduling capabilities these programs provide.
Open-source and general public licensing (GPL) software titles-available free or at minimal cost-may offer a budget-friendly alternative.
Open-source versus GPL
There are two types of “free” software:
- “free” as in no charge for its use. For example, Steve Gibson of Speeding up your Web search,” Psyber Psychiatry, May 2003). Use key words such as “freeware,” “free,” or “open source” in conjunction with the type of program (eg, “document” or “database”).
Many open-source or GPL projects are centrally stored at two sites, SourceForge and Freshmeat. Browse the categories for the type of program you need, such as databases, utilities, firewalls, etc. These sites offer source code and version information for programmers as well as precompiled software for the public.
Table Relevant open-source and GPL software titles
Related Resources (accessed Feb. 20, 2004) GNU Project. GNU General Public License (example of free-distribution license). http://www.gnu.org/copyleft/gpl.htmlType Software URL Office document OpenOffice * www.openoffice.org Instant messaging GAIM * http://gaim.sourceforge.net Web browser Firefox * www.mozilla.org/products/firefox/ Database Firebird * http://firebird.sourceforge.net Application server Zope * www.zope.org Practice management FreeMED www.freemed.org Clinical database SQL Clinic www.sqlclinic.net Electronic Medical Record Open EMR www.openemr.net * available in binary format and ready to use
GNU Project. Various licenses and comments about them. http://www.gnu.org/licenses/license-list.html.
GNU Project. Terms and conditions for copying, distribution, and modification http://www.gnu.org/copyleft/gpl.html#SEC3
Open Source Initiative. The open source definition. http://www.opensource.org/docs/definition.php
If you have questions about these products or comments about Psyber Psychiatry, click here to contact Dr. Luo or send an e-mail to [email protected].
Disclosure Dr. Luo reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Luo in this column are his own and do not necessarily reflect those of Current Psychiatry.
Name-brand practice-management software can cost hundreds or even thousands of dollars-no small expense, especially for a new practice. Still, you and your staff need the word processing, spreadsheet, documentation, patient tracking, and appointment-scheduling capabilities these programs provide.
Open-source and general public licensing (GPL) software titles-available free or at minimal cost-may offer a budget-friendly alternative.
Open-source versus GPL
There are two types of “free” software:
- “free” as in no charge for its use. For example, Steve Gibson of Speeding up your Web search,” Psyber Psychiatry, May 2003). Use key words such as “freeware,” “free,” or “open source” in conjunction with the type of program (eg, “document” or “database”).
Many open-source or GPL projects are centrally stored at two sites, SourceForge and Freshmeat. Browse the categories for the type of program you need, such as databases, utilities, firewalls, etc. These sites offer source code and version information for programmers as well as precompiled software for the public.
Table Relevant open-source and GPL software titles
Related Resources (accessed Feb. 20, 2004) GNU Project. GNU General Public License (example of free-distribution license). http://www.gnu.org/copyleft/gpl.htmlType Software URL Office document OpenOffice * www.openoffice.org Instant messaging GAIM * http://gaim.sourceforge.net Web browser Firefox * www.mozilla.org/products/firefox/ Database Firebird * http://firebird.sourceforge.net Application server Zope * www.zope.org Practice management FreeMED www.freemed.org Clinical database SQL Clinic www.sqlclinic.net Electronic Medical Record Open EMR www.openemr.net * available in binary format and ready to use
GNU Project. Various licenses and comments about them. http://www.gnu.org/licenses/license-list.html.
GNU Project. Terms and conditions for copying, distribution, and modification http://www.gnu.org/copyleft/gpl.html#SEC3
Open Source Initiative. The open source definition. http://www.opensource.org/docs/definition.php
If you have questions about these products or comments about Psyber Psychiatry, click here to contact Dr. Luo or send an e-mail to [email protected].
Disclosure Dr. Luo reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Luo in this column are his own and do not necessarily reflect those of Current Psychiatry.