User login
Long-term mogamulizumab appears safe, effective in CTCL
LA JOLLA, CALIF. — Prolonged exposure to mogamulizumab can improve responses without compromising safety in patients with cutaneous T-cell lymphoma (CTCL), according to a post hoc analysis of the MAVORIC trial.

Investigators found that exposure to mogamulizumab correlated with response. The highest response rate — 75.6% — was observed in patients exposed to the drug for at least 351 days, and the lowest — 1.9% — was observed in patients exposed to mogamulizumab for less than 72 days.
On the other hand, rates of adverse events (AEs) were similar regardless of how long patients were treated with mogamulizumab.
Youn H. Kim, MD, of Stanford Cancer Institute at Stanford (Calif.) University, and her colleagues presented these findings at the annual T-cell Lymphoma Forum.
The phase 3 MAVORIC trial (NCT01728805) included 372 adults with CTCL who had failed at least one systemic therapy. The patients were randomized to treatment with mogamulizumab or vorinostat.
Results from this comparison were previously reported at the 10th annual T-cell Lymphoma Forum.
At this year’s meeting, Dr. Kim and her colleagues reported results in 184 patients who were randomized to mogamulizumab — 105 of whom had mycosis fungoides (MF) and 79 of whom had Sézary syndrome (SS).
Patients were exposed to mogamulizumab for a mean of 275.2 days and a median of 170.0 days (range, 1-1,617 days).
The investigators divided patients into the following quartiles according to mogamulizumab exposure:
- Less than 72 days — 52 patients (28%)
- 72-170 days — 40 patients (22%)
- 171-351 days — 47 patients (26%)
- More than 351 days — 45 patients (24%).
Patients exposed to mogamulizumab for longer were more likely to have SS, stage III/IV disease, blood involvement, and a performance status of 0.
Dr. Kim said the SS patients “benefited a lot” from mogamulizumab and therefore remained on treatment longer.
Response
As expected, patients exposed to mogamulizumab for the longest period had the highest global response rates. Confirmed response rates according to drug exposure were as follows:
- Less than 72 days: 1.9% overall, 0% for SS, and 2.9% for MF
- 72-170 days: 10% overall, 18.8% for SS, and 4.2% for MF
- 171-351 days: 29.8% overall, 36.4% for SS, and 24% for MF
- More than 351 days: 75.6% overall, 83.3% for SS, and 66.7% for MF.
In addition, rates of complete response (CR) and partial response (PR) tended to increase with mogamulizumab exposure. Rates of CR, PR, and stable disease (SD) according to exposure time were as follows:
- Less than 72 days: 0% CR, 7.7% PR, and 38.5% SD
- 72-170 days: 2.5% CR, 20% PR, and 62.5% SD
- 171-351 days: 2.1% CR, 34% PR, and 57.4% SD
- More than 351 days: 6.7% CR, 71.1% PR, and 17.8% SD.
Safety
“The percentage of patients reporting adverse events was not different in the long-term treatment-exposure patients, compared to the short-term,” Dr. Kim said.
Percentages of treatment-emergent AEs (TEAEs) and serious AEs (SAEs) according to mogamulizumab exposure were as follows:
- Less than 72 days: 26.6% TEAEs and 6.5% SAEs
- 72-170 days: 18.5% TEAEs and 3.3% SAEs
- 171-351 days: 23.4% TEAEs and 6.0% SAEs
- More than 351 days: 21.7% TEAEs and 4.3% SAEs.
“The majority of the grade 3 events occurred in the first two quartiles, not later, which is important to show,” Dr. Kim said.
Most grade 3 AEs occurred within 170 days of treatment initiation, and the median time to a grade 3 or higher AE was 109 days.
The most common treatment-related AEs in the longest exposure cohort were drug eruption (20.0%), thrombocytopenia (11.1%), stomatitis (8.9%), and anemia (8.9%).
Of all patients in this analysis, 45 experienced drug eruption, which was defined as a skin rash possibly, probably, or definitely related to the study drug.
Nine drug eruption events were grade 3, and the rest were grade 1 or 2. The median time to drug eruption was 107 days.
While drug eruption “didn’t show up early,” there is no cumulative risk with longer exposure to mogamulizumab, Dr. Kim said. Likewise, she said, autoimmune AEs were not dose-cumulative events.
There were two patients with definite autoimmune disease — a 65-year-old man with Miller Fisher syndrome (occurring 199 days after mogamulizumab initiation) and a 40-year-old woman with myositis (151 days) and myocarditis (310 days).
The investigators also identified three patients with possible autoimmune disease, including:
- Pneumonitis (310 days) in a 74-year-old woman
- Polymyalgia rheumatica (209 days) and myopathy (not available) in an 84-year-old man
- Hepatitis (144 days), pneumonitis (about 174 days), and polymyositis (about 174 days) in a 73-year-old man.
Dr. Kim and her colleagues said these data suggest prolonged treatment with mogamulizumab is not associated with an increased safety risk in patients with MF or SS. And the manageable safety profile of mogamulizumab meant that patients who derived a clinical benefit could remain on the drug for an extended period of time.
The MAVORIC trial was sponsored by Kyowa Hakko Kirin Pharma. Dr. Kim reported relationships with Merck, Portola Pharmaceuticals, Soligenix, Takeda, TetraLogic Pharmaceuticals, Kyowa Kirin, Seattle Genetics, Medivir, Neumedicines, Eisai, Innate Pharma, Galderma, Miragen Therapeutics, Forty Seven, and Horizon Pharma. Her coinvestigators reported relationships with several companies.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. — Prolonged exposure to mogamulizumab can improve responses without compromising safety in patients with cutaneous T-cell lymphoma (CTCL), according to a post hoc analysis of the MAVORIC trial.
Investigators found that exposure to mogamulizumab correlated with response. The highest response rate — 75.6% — was observed in patients exposed to the drug for at least 351 days, and the lowest — 1.9% — was observed in patients exposed to mogamulizumab for less than 72 days.
On the other hand, rates of adverse events (AEs) were similar regardless of how long patients were treated with mogamulizumab.
Youn H. Kim, MD, of Stanford Cancer Institute at Stanford (Calif.) University, and her colleagues presented these findings at the annual T-cell Lymphoma Forum.
The phase 3 MAVORIC trial (NCT01728805) included 372 adults with CTCL who had failed at least one systemic therapy. The patients were randomized to treatment with mogamulizumab or vorinostat.
Results from this comparison were previously reported at the 10th annual T-cell Lymphoma Forum.
At this year’s meeting, Dr. Kim and her colleagues reported results in 184 patients who were randomized to mogamulizumab — 105 of whom had mycosis fungoides (MF) and 79 of whom had Sézary syndrome (SS).
Patients were exposed to mogamulizumab for a mean of 275.2 days and a median of 170.0 days (range, 1-1,617 days).
The investigators divided patients into the following quartiles according to mogamulizumab exposure:
- Less than 72 days — 52 patients (28%)
- 72-170 days — 40 patients (22%)
- 171-351 days — 47 patients (26%)
- More than 351 days — 45 patients (24%).
Patients exposed to mogamulizumab for longer were more likely to have SS, stage III/IV disease, blood involvement, and a performance status of 0.
Dr. Kim said the SS patients “benefited a lot” from mogamulizumab and therefore remained on treatment longer.
Response
As expected, patients exposed to mogamulizumab for the longest period had the highest global response rates. Confirmed response rates according to drug exposure were as follows:
- Less than 72 days: 1.9% overall, 0% for SS, and 2.9% for MF
- 72-170 days: 10% overall, 18.8% for SS, and 4.2% for MF
- 171-351 days: 29.8% overall, 36.4% for SS, and 24% for MF
- More than 351 days: 75.6% overall, 83.3% for SS, and 66.7% for MF.
In addition, rates of complete response (CR) and partial response (PR) tended to increase with mogamulizumab exposure. Rates of CR, PR, and stable disease (SD) according to exposure time were as follows:
- Less than 72 days: 0% CR, 7.7% PR, and 38.5% SD
- 72-170 days: 2.5% CR, 20% PR, and 62.5% SD
- 171-351 days: 2.1% CR, 34% PR, and 57.4% SD
- More than 351 days: 6.7% CR, 71.1% PR, and 17.8% SD.
Safety
“The percentage of patients reporting adverse events was not different in the long-term treatment-exposure patients, compared to the short-term,” Dr. Kim said.
Percentages of treatment-emergent AEs (TEAEs) and serious AEs (SAEs) according to mogamulizumab exposure were as follows:
- Less than 72 days: 26.6% TEAEs and 6.5% SAEs
- 72-170 days: 18.5% TEAEs and 3.3% SAEs
- 171-351 days: 23.4% TEAEs and 6.0% SAEs
- More than 351 days: 21.7% TEAEs and 4.3% SAEs.
“The majority of the grade 3 events occurred in the first two quartiles, not later, which is important to show,” Dr. Kim said.
Most grade 3 AEs occurred within 170 days of treatment initiation, and the median time to a grade 3 or higher AE was 109 days.
The most common treatment-related AEs in the longest exposure cohort were drug eruption (20.0%), thrombocytopenia (11.1%), stomatitis (8.9%), and anemia (8.9%).
Of all patients in this analysis, 45 experienced drug eruption, which was defined as a skin rash possibly, probably, or definitely related to the study drug.
Nine drug eruption events were grade 3, and the rest were grade 1 or 2. The median time to drug eruption was 107 days.
While drug eruption “didn’t show up early,” there is no cumulative risk with longer exposure to mogamulizumab, Dr. Kim said. Likewise, she said, autoimmune AEs were not dose-cumulative events.
There were two patients with definite autoimmune disease — a 65-year-old man with Miller Fisher syndrome (occurring 199 days after mogamulizumab initiation) and a 40-year-old woman with myositis (151 days) and myocarditis (310 days).
The investigators also identified three patients with possible autoimmune disease, including:
- Pneumonitis (310 days) in a 74-year-old woman
- Polymyalgia rheumatica (209 days) and myopathy (not available) in an 84-year-old man
- Hepatitis (144 days), pneumonitis (about 174 days), and polymyositis (about 174 days) in a 73-year-old man.
Dr. Kim and her colleagues said these data suggest prolonged treatment with mogamulizumab is not associated with an increased safety risk in patients with MF or SS. And the manageable safety profile of mogamulizumab meant that patients who derived a clinical benefit could remain on the drug for an extended period of time.
The MAVORIC trial was sponsored by Kyowa Hakko Kirin Pharma. Dr. Kim reported relationships with Merck, Portola Pharmaceuticals, Soligenix, Takeda, TetraLogic Pharmaceuticals, Kyowa Kirin, Seattle Genetics, Medivir, Neumedicines, Eisai, Innate Pharma, Galderma, Miragen Therapeutics, Forty Seven, and Horizon Pharma. Her coinvestigators reported relationships with several companies.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. — Prolonged exposure to mogamulizumab can improve responses without compromising safety in patients with cutaneous T-cell lymphoma (CTCL), according to a post hoc analysis of the MAVORIC trial.
Investigators found that exposure to mogamulizumab correlated with response. The highest response rate — 75.6% — was observed in patients exposed to the drug for at least 351 days, and the lowest — 1.9% — was observed in patients exposed to mogamulizumab for less than 72 days.
On the other hand, rates of adverse events (AEs) were similar regardless of how long patients were treated with mogamulizumab.
Youn H. Kim, MD, of Stanford Cancer Institute at Stanford (Calif.) University, and her colleagues presented these findings at the annual T-cell Lymphoma Forum.
The phase 3 MAVORIC trial (NCT01728805) included 372 adults with CTCL who had failed at least one systemic therapy. The patients were randomized to treatment with mogamulizumab or vorinostat.
Results from this comparison were previously reported at the 10th annual T-cell Lymphoma Forum.
At this year’s meeting, Dr. Kim and her colleagues reported results in 184 patients who were randomized to mogamulizumab — 105 of whom had mycosis fungoides (MF) and 79 of whom had Sézary syndrome (SS).
Patients were exposed to mogamulizumab for a mean of 275.2 days and a median of 170.0 days (range, 1-1,617 days).
The investigators divided patients into the following quartiles according to mogamulizumab exposure:
- Less than 72 days — 52 patients (28%)
- 72-170 days — 40 patients (22%)
- 171-351 days — 47 patients (26%)
- More than 351 days — 45 patients (24%).
Patients exposed to mogamulizumab for longer were more likely to have SS, stage III/IV disease, blood involvement, and a performance status of 0.
Dr. Kim said the SS patients “benefited a lot” from mogamulizumab and therefore remained on treatment longer.
Response
As expected, patients exposed to mogamulizumab for the longest period had the highest global response rates. Confirmed response rates according to drug exposure were as follows:
- Less than 72 days: 1.9% overall, 0% for SS, and 2.9% for MF
- 72-170 days: 10% overall, 18.8% for SS, and 4.2% for MF
- 171-351 days: 29.8% overall, 36.4% for SS, and 24% for MF
- More than 351 days: 75.6% overall, 83.3% for SS, and 66.7% for MF.
In addition, rates of complete response (CR) and partial response (PR) tended to increase with mogamulizumab exposure. Rates of CR, PR, and stable disease (SD) according to exposure time were as follows:
- Less than 72 days: 0% CR, 7.7% PR, and 38.5% SD
- 72-170 days: 2.5% CR, 20% PR, and 62.5% SD
- 171-351 days: 2.1% CR, 34% PR, and 57.4% SD
- More than 351 days: 6.7% CR, 71.1% PR, and 17.8% SD.
Safety
“The percentage of patients reporting adverse events was not different in the long-term treatment-exposure patients, compared to the short-term,” Dr. Kim said.
Percentages of treatment-emergent AEs (TEAEs) and serious AEs (SAEs) according to mogamulizumab exposure were as follows:
- Less than 72 days: 26.6% TEAEs and 6.5% SAEs
- 72-170 days: 18.5% TEAEs and 3.3% SAEs
- 171-351 days: 23.4% TEAEs and 6.0% SAEs
- More than 351 days: 21.7% TEAEs and 4.3% SAEs.
“The majority of the grade 3 events occurred in the first two quartiles, not later, which is important to show,” Dr. Kim said.
Most grade 3 AEs occurred within 170 days of treatment initiation, and the median time to a grade 3 or higher AE was 109 days.
The most common treatment-related AEs in the longest exposure cohort were drug eruption (20.0%), thrombocytopenia (11.1%), stomatitis (8.9%), and anemia (8.9%).
Of all patients in this analysis, 45 experienced drug eruption, which was defined as a skin rash possibly, probably, or definitely related to the study drug.
Nine drug eruption events were grade 3, and the rest were grade 1 or 2. The median time to drug eruption was 107 days.
While drug eruption “didn’t show up early,” there is no cumulative risk with longer exposure to mogamulizumab, Dr. Kim said. Likewise, she said, autoimmune AEs were not dose-cumulative events.
There were two patients with definite autoimmune disease — a 65-year-old man with Miller Fisher syndrome (occurring 199 days after mogamulizumab initiation) and a 40-year-old woman with myositis (151 days) and myocarditis (310 days).
The investigators also identified three patients with possible autoimmune disease, including:
- Pneumonitis (310 days) in a 74-year-old woman
- Polymyalgia rheumatica (209 days) and myopathy (not available) in an 84-year-old man
- Hepatitis (144 days), pneumonitis (about 174 days), and polymyositis (about 174 days) in a 73-year-old man.
Dr. Kim and her colleagues said these data suggest prolonged treatment with mogamulizumab is not associated with an increased safety risk in patients with MF or SS. And the manageable safety profile of mogamulizumab meant that patients who derived a clinical benefit could remain on the drug for an extended period of time.
The MAVORIC trial was sponsored by Kyowa Hakko Kirin Pharma. Dr. Kim reported relationships with Merck, Portola Pharmaceuticals, Soligenix, Takeda, TetraLogic Pharmaceuticals, Kyowa Kirin, Seattle Genetics, Medivir, Neumedicines, Eisai, Innate Pharma, Galderma, Miragen Therapeutics, Forty Seven, and Horizon Pharma. Her coinvestigators reported relationships with several companies.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
REPORTING FROM TCLF 2019
Key clinical point:
Major finding: The highest response rate – 75.6% – was observed in patients exposed to mogamulizumab for at least 351 days.
Study details: A post hoc analysis of the MAVORIC trial, including 184 patients treated with mogamulizumab.
Disclosures: The MAVORIC trial was sponsored by Kyowa Hakko Kirin Pharma. Investigators disclosed relationships with several companies.
Chidamide may be more effective in PTCL than previously thought
LA JOLLA, CALIF. – Real-world data suggest chidamide may be more effective against relapsed or refractory peripheral T-cell lymphoma (PTCL) than a pivotal study indicated.

Single-agent chidamide produced an overall response rate of 47.0% in a real-world study of more than 1,000 patients, compared with the 28.0% overall response rate that was observed in the phase 2 study of chidamide (Ann Oncol. 2015 Aug;26[8]:1766-71).
Yuqin Song, MD, PhD, of Peking University Cancer Hospital and Institute in Beijing, China, presented data from the real-world study at the annual T-cell Lymphoma Forum.
Dr. Song said this study is the largest cohort of real-world patients with relapsed or refractory PTCL. She and her colleagues analyzed data on 1,064 patients treated at 216 sites across China between February 2015 and December 2017.
The patients had a median age of 54 years, 63.9% were male, and 88.1% had stage III-IV disease.
Disease subtypes included PTCL not otherwise specified (NOS, 38.0%), angioimmunoblastic T-cell lymphoma (AITL, 29.1%), extranodal natural killer T-cell lymphoma (ENKTL, 13.4%), anaplastic large-cell lymphoma (ALCL, 9.1%), and others (10.3%), including cutaneous T-cell lymphoma (CTCL).
Fifty-two percent of patients (n = 553) received chidamide as a single agent, and 48% (n = 511) received the drug with other agents. The most common treatment regimens combined with chidamide were the following
- Cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP, 20.7%).
- Gemcitabine, dexamethasone, and cisplatin (GDP, 11.8%).
- Etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin (EPOCH, 9.8%).
- Patients with ENKTL received chidamide with L-asparaginase (35.4%) or without it (64.5%).
The median follow-up was 4.9 months (range, 0-36.2 months). Across disease subtypes, the overall response rate was 47.0% with single-agent chidamide and 65.4% when chidamide was given in combination with other agents (P less than .01).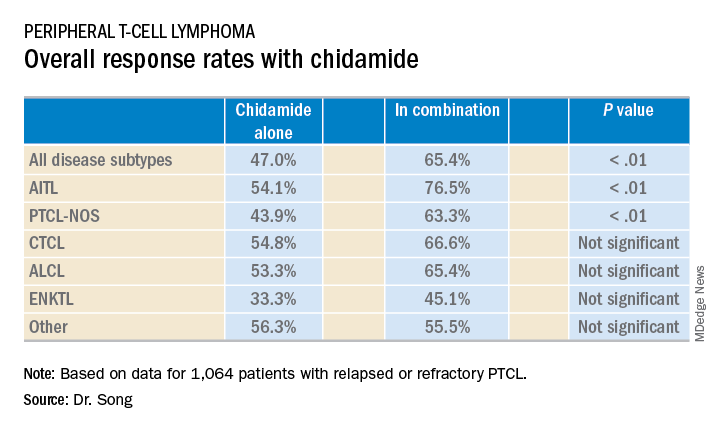
The median overall survival was 400 days for all patients, 342 days for patients treated with chidamide alone, and 457 days for patients who received combination therapy. The 1-year overall survival rates were 52%, 48%, and 56%, respectively.
Dr. Song said these data verify the efficacy of chidamide as a single agent and suggest chidamide might lead to improved survival in refractory or relapsed PTCLs.
Chidamide was generally well tolerated in this study, Dr. Song said. There were no unexpected adverse events (AEs) and most were grade 1 or 2.
The most common AEs (of any grade) observed with single-agent chidamide were neutropenia (42.9%), thrombocytopenia (40.5%), fatigue (38.3%), anemia (31.6%), and nausea/vomiting (21.0%).
The most common AEs observed with chidamide in combination were neutropenia (61.4%), thrombocytopenia (58.5%), fatigue (56.2%), anemia (54.2%), nausea/vomiting (30.7%), and fever (22.1%).
This study was supported by the Union for China Lymphoma Investigators and the Chinese Society of Clinical Oncology. Dr. Song did not disclose any conflicts of interest.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. – Real-world data suggest chidamide may be more effective against relapsed or refractory peripheral T-cell lymphoma (PTCL) than a pivotal study indicated.
Single-agent chidamide produced an overall response rate of 47.0% in a real-world study of more than 1,000 patients, compared with the 28.0% overall response rate that was observed in the phase 2 study of chidamide (Ann Oncol. 2015 Aug;26[8]:1766-71).
Yuqin Song, MD, PhD, of Peking University Cancer Hospital and Institute in Beijing, China, presented data from the real-world study at the annual T-cell Lymphoma Forum.
Dr. Song said this study is the largest cohort of real-world patients with relapsed or refractory PTCL. She and her colleagues analyzed data on 1,064 patients treated at 216 sites across China between February 2015 and December 2017.
The patients had a median age of 54 years, 63.9% were male, and 88.1% had stage III-IV disease.
Disease subtypes included PTCL not otherwise specified (NOS, 38.0%), angioimmunoblastic T-cell lymphoma (AITL, 29.1%), extranodal natural killer T-cell lymphoma (ENKTL, 13.4%), anaplastic large-cell lymphoma (ALCL, 9.1%), and others (10.3%), including cutaneous T-cell lymphoma (CTCL).
Fifty-two percent of patients (n = 553) received chidamide as a single agent, and 48% (n = 511) received the drug with other agents. The most common treatment regimens combined with chidamide were the following
- Cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP, 20.7%).
- Gemcitabine, dexamethasone, and cisplatin (GDP, 11.8%).
- Etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin (EPOCH, 9.8%).
- Patients with ENKTL received chidamide with L-asparaginase (35.4%) or without it (64.5%).
The median follow-up was 4.9 months (range, 0-36.2 months). Across disease subtypes, the overall response rate was 47.0% with single-agent chidamide and 65.4% when chidamide was given in combination with other agents (P less than .01).
The median overall survival was 400 days for all patients, 342 days for patients treated with chidamide alone, and 457 days for patients who received combination therapy. The 1-year overall survival rates were 52%, 48%, and 56%, respectively.
Dr. Song said these data verify the efficacy of chidamide as a single agent and suggest chidamide might lead to improved survival in refractory or relapsed PTCLs.
Chidamide was generally well tolerated in this study, Dr. Song said. There were no unexpected adverse events (AEs) and most were grade 1 or 2.
The most common AEs (of any grade) observed with single-agent chidamide were neutropenia (42.9%), thrombocytopenia (40.5%), fatigue (38.3%), anemia (31.6%), and nausea/vomiting (21.0%).
The most common AEs observed with chidamide in combination were neutropenia (61.4%), thrombocytopenia (58.5%), fatigue (56.2%), anemia (54.2%), nausea/vomiting (30.7%), and fever (22.1%).
This study was supported by the Union for China Lymphoma Investigators and the Chinese Society of Clinical Oncology. Dr. Song did not disclose any conflicts of interest.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. – Real-world data suggest chidamide may be more effective against relapsed or refractory peripheral T-cell lymphoma (PTCL) than a pivotal study indicated.
Single-agent chidamide produced an overall response rate of 47.0% in a real-world study of more than 1,000 patients, compared with the 28.0% overall response rate that was observed in the phase 2 study of chidamide (Ann Oncol. 2015 Aug;26[8]:1766-71).
Yuqin Song, MD, PhD, of Peking University Cancer Hospital and Institute in Beijing, China, presented data from the real-world study at the annual T-cell Lymphoma Forum.
Dr. Song said this study is the largest cohort of real-world patients with relapsed or refractory PTCL. She and her colleagues analyzed data on 1,064 patients treated at 216 sites across China between February 2015 and December 2017.
The patients had a median age of 54 years, 63.9% were male, and 88.1% had stage III-IV disease.
Disease subtypes included PTCL not otherwise specified (NOS, 38.0%), angioimmunoblastic T-cell lymphoma (AITL, 29.1%), extranodal natural killer T-cell lymphoma (ENKTL, 13.4%), anaplastic large-cell lymphoma (ALCL, 9.1%), and others (10.3%), including cutaneous T-cell lymphoma (CTCL).
Fifty-two percent of patients (n = 553) received chidamide as a single agent, and 48% (n = 511) received the drug with other agents. The most common treatment regimens combined with chidamide were the following
- Cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP, 20.7%).
- Gemcitabine, dexamethasone, and cisplatin (GDP, 11.8%).
- Etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin (EPOCH, 9.8%).
- Patients with ENKTL received chidamide with L-asparaginase (35.4%) or without it (64.5%).
The median follow-up was 4.9 months (range, 0-36.2 months). Across disease subtypes, the overall response rate was 47.0% with single-agent chidamide and 65.4% when chidamide was given in combination with other agents (P less than .01).
The median overall survival was 400 days for all patients, 342 days for patients treated with chidamide alone, and 457 days for patients who received combination therapy. The 1-year overall survival rates were 52%, 48%, and 56%, respectively.
Dr. Song said these data verify the efficacy of chidamide as a single agent and suggest chidamide might lead to improved survival in refractory or relapsed PTCLs.
Chidamide was generally well tolerated in this study, Dr. Song said. There were no unexpected adverse events (AEs) and most were grade 1 or 2.
The most common AEs (of any grade) observed with single-agent chidamide were neutropenia (42.9%), thrombocytopenia (40.5%), fatigue (38.3%), anemia (31.6%), and nausea/vomiting (21.0%).
The most common AEs observed with chidamide in combination were neutropenia (61.4%), thrombocytopenia (58.5%), fatigue (56.2%), anemia (54.2%), nausea/vomiting (30.7%), and fever (22.1%).
This study was supported by the Union for China Lymphoma Investigators and the Chinese Society of Clinical Oncology. Dr. Song did not disclose any conflicts of interest.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
REPORTING FROM TCLF 2019
Key clinical point:
Major finding: Single-agent chidamide had an overall response rate of 47.0% among relapsed/refractory PTCL patients, compared with 65.4% when used in combination with other agents (P less than .01).
Study details: A real-world cohort of 1,064 relapsed/refractory PTCL patients treated at 216 sites across China between February 2015 and December 2017.
Disclosures: The study was supported by the Union for China Lymphoma Investigators and the Chinese Society of Clinical Oncology. Dr. Song did not disclose any conflicts of interest.
Epigenetics is a hot topic at TCLF 2019
LA JOLLA, CALIF. –

In a video interview, meeting cochair Owen O’Connor, MD, PhD, of Columbia University Medical Center in New York, discussed a few presentations that addressed epigenetics in T-cell lymphomas.
Stephen Baylin, MD, of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins in Baltimore, gave the meeting’s keynote address, which focused on the idea that epigenetic therapy can enhance immune checkpoint therapy.
Susan Bates, MD, of Columbia University Medical Center, presented data that suggest romidepsin and other histone deacetylase inhibitors fight cutaneous T-cell lymphoma via epigenetic effects on gene expression, as well as DNA damage.
And Enrica Marchi, MD, PhD, of Columbia University Medical Center, discussed the use of epigenetic-based combination therapies to improve responses in T-cell lymphomas.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. –
In a video interview, meeting cochair Owen O’Connor, MD, PhD, of Columbia University Medical Center in New York, discussed a few presentations that addressed epigenetics in T-cell lymphomas.
Stephen Baylin, MD, of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins in Baltimore, gave the meeting’s keynote address, which focused on the idea that epigenetic therapy can enhance immune checkpoint therapy.
Susan Bates, MD, of Columbia University Medical Center, presented data that suggest romidepsin and other histone deacetylase inhibitors fight cutaneous T-cell lymphoma via epigenetic effects on gene expression, as well as DNA damage.
And Enrica Marchi, MD, PhD, of Columbia University Medical Center, discussed the use of epigenetic-based combination therapies to improve responses in T-cell lymphomas.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. –
In a video interview, meeting cochair Owen O’Connor, MD, PhD, of Columbia University Medical Center in New York, discussed a few presentations that addressed epigenetics in T-cell lymphomas.
Stephen Baylin, MD, of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins in Baltimore, gave the meeting’s keynote address, which focused on the idea that epigenetic therapy can enhance immune checkpoint therapy.
Susan Bates, MD, of Columbia University Medical Center, presented data that suggest romidepsin and other histone deacetylase inhibitors fight cutaneous T-cell lymphoma via epigenetic effects on gene expression, as well as DNA damage.
And Enrica Marchi, MD, PhD, of Columbia University Medical Center, discussed the use of epigenetic-based combination therapies to improve responses in T-cell lymphomas.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
REPORTING FROM TCLF 2019

Registry data favor CHOEP regimen for PTCL
LA JOLLA, CALIF. – Data from the Czech National Lymphoma Registry (NiHiL) suggest the CHOEP regimen (cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisone) prolongs survival in newly diagnosed patients with peripheral T-cell lymphoma (PTCL), but consolidation with autologous stem cell transplant (ASCT) does not.

“We failed to show that there is a real benefit for the patient to undergo autologous stem cell consolidation, so, nowadays, in the majority of the centers in our group, CHOEP is the chemotherapy of choice, and the patients are not consolidated,” said Marek Trneny, MD, of Charles University General Hospital in Prague.
Dr. Trneny presented results from NiHiL on behalf of the Czech Lymphoma Study Group at the annual T-cell Lymphoma Forum.
The NiHiL project is a prospective, observational study (NCT03199066) of patients newly diagnosed with non-Hodgkin lymphoma in the Czech Republic.
Dr. Trneny presented data on 838 PTCL patients who were diagnosed between 1999 and 2018, 462 of whom were included in a survival analysis (1999-2016).
The 462 patients had a median age of 61 years and more than half were men.
Patients had PTCL not otherwise specified (NOS, 43.9%), Anaplastic lymphoma kinase (ALK)–negative Anaplastic large-cell lymphoma (ALCL, 20.8%), ALK-positive ALCL (7.6%), unclassified ALCL (0.9%), angioimmunoblastic T-cell lymphoma (AITL, 10.6%), and other subtypes (16.2%).
Most patients (79.1%) had tumors measuring less than 7.5 cm, about half of patients (52.3%) had B symptoms, most (70.5%) had a performance status of 0-1, and most (68.5%) had stage III-IV disease. Half of patients were low or low/intermediate risk according to the International Prognostic Index.
For the entire cohort, the median progression-free survival (PFS) was 1.15 years, and the 5-year PFS rate was 34.2%. The median overall survival (OS) was 2.83 years, and the 5-year OS rate was 43.3%.
There was no significant difference in PFS or OS for patients diagnosed from 1999 to 2007 and those diagnosed from 2008 to 2016. The median PFS was 1.05 years and 1.23 years, respectively (P= .4487), and the median OS was 2.15 years and 3.39 years, respectively (P = .176).
Patients with ALK-positive ALCL had superior PFS and OS when compared to patients with the other disease subtypes (P = .0002 for PFS and P = .0009 for OS).
Treatment comparison
When Dr. Trneny and his colleagues compared the 223 patients who received CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) and the 65 patients who received CHOEP, the researchers found that patients who received CHOEP had superior PFS and OS.
In the CHOP group, the median PFS was 1.18 years and the 5-year PFS rate was 34.2%. In the CHOEP group, the median PFS was 3.67 years and the 5-year PFS rate was 49.0% (hazard ratio, 0.6781; P = .0373).
In the CHOP group, the median OS was 3.74 years and the 5-year OS was 45.5%. In the CHOEP group, the median OS was 7.46 years and the 5-year OS was 56.4% (HR, 0.6475; P = .0381).
Dr. Trneny noted that patients who received CHOP were significantly younger (P less than .0001), more likely to have AITL or ALK-positive ALCL (P = .0152), and more likely to have a performance status of 2-4 (P = .0385).
Dr. Trneny and his colleagues also compared survival in 56 ASCT recipients and 189 patients who received chemotherapy alone (either CHOP or CHOEP).
Patients who underwent ASCT were significantly younger (P less than .0001) and more likely to have stage III or IV disease (P = .0345).
However, the researchers found no significant survival differences between patients who underwent ASCT and those who did not.
ASCT recipients had a median PFS of 7.50 years and a 5-year PFS rate of 56.0%, while patients who received chemotherapy alone had a median PFS of 4.69 years and a 5-year PFS rate of 47.3% (P = .6537).
In ASCT recipients, the median OS was not reached, and the 5-year OS rate was 63.3%. Among patients who received chemotherapy alone, the median OS was 6.78 years, and the 5-year OS was 63.0% (P = .6201).
“For those patients who are eligible for a CHOP-like regimen, CHOEP gives a higher chance for longer progression-free survival and overall survival, at least in our ... analysis,” Dr. Trneny said. “Autologous stem cell transplant, on the other hand, doesn’t seem to prolong PFS or overall survival.”
This research is sponsored by the Czech Lymphoma Study Group. Dr. Trneny did not disclose any conflicts of interest.
The T-cell Lymphoma Forum is held by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. – Data from the Czech National Lymphoma Registry (NiHiL) suggest the CHOEP regimen (cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisone) prolongs survival in newly diagnosed patients with peripheral T-cell lymphoma (PTCL), but consolidation with autologous stem cell transplant (ASCT) does not.
“We failed to show that there is a real benefit for the patient to undergo autologous stem cell consolidation, so, nowadays, in the majority of the centers in our group, CHOEP is the chemotherapy of choice, and the patients are not consolidated,” said Marek Trneny, MD, of Charles University General Hospital in Prague.
Dr. Trneny presented results from NiHiL on behalf of the Czech Lymphoma Study Group at the annual T-cell Lymphoma Forum.
The NiHiL project is a prospective, observational study (NCT03199066) of patients newly diagnosed with non-Hodgkin lymphoma in the Czech Republic.
Dr. Trneny presented data on 838 PTCL patients who were diagnosed between 1999 and 2018, 462 of whom were included in a survival analysis (1999-2016).
The 462 patients had a median age of 61 years and more than half were men.
Patients had PTCL not otherwise specified (NOS, 43.9%), Anaplastic lymphoma kinase (ALK)–negative Anaplastic large-cell lymphoma (ALCL, 20.8%), ALK-positive ALCL (7.6%), unclassified ALCL (0.9%), angioimmunoblastic T-cell lymphoma (AITL, 10.6%), and other subtypes (16.2%).
Most patients (79.1%) had tumors measuring less than 7.5 cm, about half of patients (52.3%) had B symptoms, most (70.5%) had a performance status of 0-1, and most (68.5%) had stage III-IV disease. Half of patients were low or low/intermediate risk according to the International Prognostic Index.
For the entire cohort, the median progression-free survival (PFS) was 1.15 years, and the 5-year PFS rate was 34.2%. The median overall survival (OS) was 2.83 years, and the 5-year OS rate was 43.3%.
There was no significant difference in PFS or OS for patients diagnosed from 1999 to 2007 and those diagnosed from 2008 to 2016. The median PFS was 1.05 years and 1.23 years, respectively (P= .4487), and the median OS was 2.15 years and 3.39 years, respectively (P = .176).
Patients with ALK-positive ALCL had superior PFS and OS when compared to patients with the other disease subtypes (P = .0002 for PFS and P = .0009 for OS).
Treatment comparison
When Dr. Trneny and his colleagues compared the 223 patients who received CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) and the 65 patients who received CHOEP, the researchers found that patients who received CHOEP had superior PFS and OS.
In the CHOP group, the median PFS was 1.18 years and the 5-year PFS rate was 34.2%. In the CHOEP group, the median PFS was 3.67 years and the 5-year PFS rate was 49.0% (hazard ratio, 0.6781; P = .0373).
In the CHOP group, the median OS was 3.74 years and the 5-year OS was 45.5%. In the CHOEP group, the median OS was 7.46 years and the 5-year OS was 56.4% (HR, 0.6475; P = .0381).
Dr. Trneny noted that patients who received CHOP were significantly younger (P less than .0001), more likely to have AITL or ALK-positive ALCL (P = .0152), and more likely to have a performance status of 2-4 (P = .0385).
Dr. Trneny and his colleagues also compared survival in 56 ASCT recipients and 189 patients who received chemotherapy alone (either CHOP or CHOEP).
Patients who underwent ASCT were significantly younger (P less than .0001) and more likely to have stage III or IV disease (P = .0345).
However, the researchers found no significant survival differences between patients who underwent ASCT and those who did not.
ASCT recipients had a median PFS of 7.50 years and a 5-year PFS rate of 56.0%, while patients who received chemotherapy alone had a median PFS of 4.69 years and a 5-year PFS rate of 47.3% (P = .6537).
In ASCT recipients, the median OS was not reached, and the 5-year OS rate was 63.3%. Among patients who received chemotherapy alone, the median OS was 6.78 years, and the 5-year OS was 63.0% (P = .6201).
“For those patients who are eligible for a CHOP-like regimen, CHOEP gives a higher chance for longer progression-free survival and overall survival, at least in our ... analysis,” Dr. Trneny said. “Autologous stem cell transplant, on the other hand, doesn’t seem to prolong PFS or overall survival.”
This research is sponsored by the Czech Lymphoma Study Group. Dr. Trneny did not disclose any conflicts of interest.
The T-cell Lymphoma Forum is held by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. – Data from the Czech National Lymphoma Registry (NiHiL) suggest the CHOEP regimen (cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisone) prolongs survival in newly diagnosed patients with peripheral T-cell lymphoma (PTCL), but consolidation with autologous stem cell transplant (ASCT) does not.
“We failed to show that there is a real benefit for the patient to undergo autologous stem cell consolidation, so, nowadays, in the majority of the centers in our group, CHOEP is the chemotherapy of choice, and the patients are not consolidated,” said Marek Trneny, MD, of Charles University General Hospital in Prague.
Dr. Trneny presented results from NiHiL on behalf of the Czech Lymphoma Study Group at the annual T-cell Lymphoma Forum.
The NiHiL project is a prospective, observational study (NCT03199066) of patients newly diagnosed with non-Hodgkin lymphoma in the Czech Republic.
Dr. Trneny presented data on 838 PTCL patients who were diagnosed between 1999 and 2018, 462 of whom were included in a survival analysis (1999-2016).
The 462 patients had a median age of 61 years and more than half were men.
Patients had PTCL not otherwise specified (NOS, 43.9%), Anaplastic lymphoma kinase (ALK)–negative Anaplastic large-cell lymphoma (ALCL, 20.8%), ALK-positive ALCL (7.6%), unclassified ALCL (0.9%), angioimmunoblastic T-cell lymphoma (AITL, 10.6%), and other subtypes (16.2%).
Most patients (79.1%) had tumors measuring less than 7.5 cm, about half of patients (52.3%) had B symptoms, most (70.5%) had a performance status of 0-1, and most (68.5%) had stage III-IV disease. Half of patients were low or low/intermediate risk according to the International Prognostic Index.
For the entire cohort, the median progression-free survival (PFS) was 1.15 years, and the 5-year PFS rate was 34.2%. The median overall survival (OS) was 2.83 years, and the 5-year OS rate was 43.3%.
There was no significant difference in PFS or OS for patients diagnosed from 1999 to 2007 and those diagnosed from 2008 to 2016. The median PFS was 1.05 years and 1.23 years, respectively (P= .4487), and the median OS was 2.15 years and 3.39 years, respectively (P = .176).
Patients with ALK-positive ALCL had superior PFS and OS when compared to patients with the other disease subtypes (P = .0002 for PFS and P = .0009 for OS).
Treatment comparison
When Dr. Trneny and his colleagues compared the 223 patients who received CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) and the 65 patients who received CHOEP, the researchers found that patients who received CHOEP had superior PFS and OS.
In the CHOP group, the median PFS was 1.18 years and the 5-year PFS rate was 34.2%. In the CHOEP group, the median PFS was 3.67 years and the 5-year PFS rate was 49.0% (hazard ratio, 0.6781; P = .0373).
In the CHOP group, the median OS was 3.74 years and the 5-year OS was 45.5%. In the CHOEP group, the median OS was 7.46 years and the 5-year OS was 56.4% (HR, 0.6475; P = .0381).
Dr. Trneny noted that patients who received CHOP were significantly younger (P less than .0001), more likely to have AITL or ALK-positive ALCL (P = .0152), and more likely to have a performance status of 2-4 (P = .0385).
Dr. Trneny and his colleagues also compared survival in 56 ASCT recipients and 189 patients who received chemotherapy alone (either CHOP or CHOEP).
Patients who underwent ASCT were significantly younger (P less than .0001) and more likely to have stage III or IV disease (P = .0345).
However, the researchers found no significant survival differences between patients who underwent ASCT and those who did not.
ASCT recipients had a median PFS of 7.50 years and a 5-year PFS rate of 56.0%, while patients who received chemotherapy alone had a median PFS of 4.69 years and a 5-year PFS rate of 47.3% (P = .6537).
In ASCT recipients, the median OS was not reached, and the 5-year OS rate was 63.3%. Among patients who received chemotherapy alone, the median OS was 6.78 years, and the 5-year OS was 63.0% (P = .6201).
“For those patients who are eligible for a CHOP-like regimen, CHOEP gives a higher chance for longer progression-free survival and overall survival, at least in our ... analysis,” Dr. Trneny said. “Autologous stem cell transplant, on the other hand, doesn’t seem to prolong PFS or overall survival.”
This research is sponsored by the Czech Lymphoma Study Group. Dr. Trneny did not disclose any conflicts of interest.
The T-cell Lymphoma Forum is held by Jonathan Wood & Associates, which is owned by the same company as this news organization.
REPORTING FROM TCLF 2019
Key clinical point:
Major finding: The 5-year progression free survival rate was 49.0% in the group receiving CHOEP, compared with 34.2% in the group receiving CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone).
Study details: A survival analysis of 462 patients with PTCL who were part of the Czech National Lymphoma Registry.
Disclosures: This research is sponsored by the Czech Lymphoma Study Group. Dr. Trneny did not disclose any conflicts of interest.
CMT provides survival benefit in young HL patients
Combined modality therapy (CMT) can improve survival in young patients with early stage Hodgkin lymphoma (HL), according to research published in JAMA Oncology.
In a retrospective study, researchers compared chemotherapy followed by radiotherapy—CMT—to chemotherapy alone in more than 5,600 HL patients age 21 and younger.
There was a significant improvement in 5-year overall survival (OS) among patients who received CMT.
The treatment appeared particularly beneficial for adolescents and young adults as well as patients with low-risk disease.
However, the researchers observed a nearly 25% decrease in the use of CMT over the period studied.
“Nationwide, there has been a notable decrease in combined modality therapy, especially in clinical trials, many of which are designed to avoid this strategy,” said Rahul Parikh, MD, of Rutgers Cancer Institute of New Jersey in New Brunswick.
“This form of treatment has shown to be effective, with event-free survival rates greater than 80% and overall survival rates greater than 95%. The question then becomes, ‘does treatment benefit outweigh the risk of long-term side effects?”
With this in mind, Dr. Parikh and his colleagues compared CMT to chemotherapy alone using data from the National Cancer Database spanning the period from 2004 to 2015.
The researchers analyzed 5,657 patients with stage I/II classical HL who had a mean age of 17.1.
Roughly half of patients received CMT (50.3%, n=2845), and the other half received chemotherapy alone (49.7%, n=2812).
The median radiotherapy dose was 21.0 Gy, and the most common modality was photon therapy (59.0%).
Patients who received CMT were significantly more likely to be younger than 16 (P<0.001), be male (P<0.001), have stage II disease (P=0.02), and have private health insurance (P=0.002).
Results
The median follow-up was 5.1 years.
The 5-year OS was 94.5% for patients who received chemotherapy alone and 97.3% for patients treated with CMT.
CMT was significantly associated with improved OS in both univariate (hazard ratio [HR]=0.58, P<0.001) and multivariate analyses (HR=0.57, P<0.001).
In a sensitivity analysis, the researchers found the greatest benefits of CMT were in adolescents and young adults (age 14 and older, adjusted HR=0.47) and patients with low-risk disease (stage I-IIA, adjusted HR=0.59).
The researchers noted that this study was limited by their inability to control for unreported prognostic factors, such as the number of nodal sites and bulk of disease.
Another limitation was the duration of follow-up, which did not allow the researchers to fully assess secondary late effects of CMT and their potential impact on survival.
Still, Dr. Parikh said this study demonstrates a survival benefit for young HL patients treated with CMT.
“With that, physicians should be encouraged to discuss combined modality therapy as one of the many treatment options [for young HL patients],” he said.
“Investigators may also consider designing future clinical trials for this population to include combined modality therapy as a standard arm with the inclusion of interim treatment response assessment (PET scans, etc.). And as multiple disparities to the use of combined modality therapy have been identified through this work, future studies should address improving access to care for all pediatric patients.”
Dr. Parikh and his colleagues declared no conflicts of interest for the current study.
Combined modality therapy (CMT) can improve survival in young patients with early stage Hodgkin lymphoma (HL), according to research published in JAMA Oncology.
In a retrospective study, researchers compared chemotherapy followed by radiotherapy—CMT—to chemotherapy alone in more than 5,600 HL patients age 21 and younger.
There was a significant improvement in 5-year overall survival (OS) among patients who received CMT.
The treatment appeared particularly beneficial for adolescents and young adults as well as patients with low-risk disease.
However, the researchers observed a nearly 25% decrease in the use of CMT over the period studied.
“Nationwide, there has been a notable decrease in combined modality therapy, especially in clinical trials, many of which are designed to avoid this strategy,” said Rahul Parikh, MD, of Rutgers Cancer Institute of New Jersey in New Brunswick.
“This form of treatment has shown to be effective, with event-free survival rates greater than 80% and overall survival rates greater than 95%. The question then becomes, ‘does treatment benefit outweigh the risk of long-term side effects?”
With this in mind, Dr. Parikh and his colleagues compared CMT to chemotherapy alone using data from the National Cancer Database spanning the period from 2004 to 2015.
The researchers analyzed 5,657 patients with stage I/II classical HL who had a mean age of 17.1.
Roughly half of patients received CMT (50.3%, n=2845), and the other half received chemotherapy alone (49.7%, n=2812).
The median radiotherapy dose was 21.0 Gy, and the most common modality was photon therapy (59.0%).
Patients who received CMT were significantly more likely to be younger than 16 (P<0.001), be male (P<0.001), have stage II disease (P=0.02), and have private health insurance (P=0.002).
Results
The median follow-up was 5.1 years.
The 5-year OS was 94.5% for patients who received chemotherapy alone and 97.3% for patients treated with CMT.
CMT was significantly associated with improved OS in both univariate (hazard ratio [HR]=0.58, P<0.001) and multivariate analyses (HR=0.57, P<0.001).
In a sensitivity analysis, the researchers found the greatest benefits of CMT were in adolescents and young adults (age 14 and older, adjusted HR=0.47) and patients with low-risk disease (stage I-IIA, adjusted HR=0.59).
The researchers noted that this study was limited by their inability to control for unreported prognostic factors, such as the number of nodal sites and bulk of disease.
Another limitation was the duration of follow-up, which did not allow the researchers to fully assess secondary late effects of CMT and their potential impact on survival.
Still, Dr. Parikh said this study demonstrates a survival benefit for young HL patients treated with CMT.
“With that, physicians should be encouraged to discuss combined modality therapy as one of the many treatment options [for young HL patients],” he said.
“Investigators may also consider designing future clinical trials for this population to include combined modality therapy as a standard arm with the inclusion of interim treatment response assessment (PET scans, etc.). And as multiple disparities to the use of combined modality therapy have been identified through this work, future studies should address improving access to care for all pediatric patients.”
Dr. Parikh and his colleagues declared no conflicts of interest for the current study.
Combined modality therapy (CMT) can improve survival in young patients with early stage Hodgkin lymphoma (HL), according to research published in JAMA Oncology.
In a retrospective study, researchers compared chemotherapy followed by radiotherapy—CMT—to chemotherapy alone in more than 5,600 HL patients age 21 and younger.
There was a significant improvement in 5-year overall survival (OS) among patients who received CMT.
The treatment appeared particularly beneficial for adolescents and young adults as well as patients with low-risk disease.
However, the researchers observed a nearly 25% decrease in the use of CMT over the period studied.
“Nationwide, there has been a notable decrease in combined modality therapy, especially in clinical trials, many of which are designed to avoid this strategy,” said Rahul Parikh, MD, of Rutgers Cancer Institute of New Jersey in New Brunswick.
“This form of treatment has shown to be effective, with event-free survival rates greater than 80% and overall survival rates greater than 95%. The question then becomes, ‘does treatment benefit outweigh the risk of long-term side effects?”
With this in mind, Dr. Parikh and his colleagues compared CMT to chemotherapy alone using data from the National Cancer Database spanning the period from 2004 to 2015.
The researchers analyzed 5,657 patients with stage I/II classical HL who had a mean age of 17.1.
Roughly half of patients received CMT (50.3%, n=2845), and the other half received chemotherapy alone (49.7%, n=2812).
The median radiotherapy dose was 21.0 Gy, and the most common modality was photon therapy (59.0%).
Patients who received CMT were significantly more likely to be younger than 16 (P<0.001), be male (P<0.001), have stage II disease (P=0.02), and have private health insurance (P=0.002).
Results
The median follow-up was 5.1 years.
The 5-year OS was 94.5% for patients who received chemotherapy alone and 97.3% for patients treated with CMT.
CMT was significantly associated with improved OS in both univariate (hazard ratio [HR]=0.58, P<0.001) and multivariate analyses (HR=0.57, P<0.001).
In a sensitivity analysis, the researchers found the greatest benefits of CMT were in adolescents and young adults (age 14 and older, adjusted HR=0.47) and patients with low-risk disease (stage I-IIA, adjusted HR=0.59).
The researchers noted that this study was limited by their inability to control for unreported prognostic factors, such as the number of nodal sites and bulk of disease.
Another limitation was the duration of follow-up, which did not allow the researchers to fully assess secondary late effects of CMT and their potential impact on survival.
Still, Dr. Parikh said this study demonstrates a survival benefit for young HL patients treated with CMT.
“With that, physicians should be encouraged to discuss combined modality therapy as one of the many treatment options [for young HL patients],” he said.
“Investigators may also consider designing future clinical trials for this population to include combined modality therapy as a standard arm with the inclusion of interim treatment response assessment (PET scans, etc.). And as multiple disparities to the use of combined modality therapy have been identified through this work, future studies should address improving access to care for all pediatric patients.”
Dr. Parikh and his colleagues declared no conflicts of interest for the current study.
Chemo for solid tumors and risk of tMDS/AML

Chemotherapy for solid tumors is associated with an increased risk of therapy-related myelodysplastic syndromes or acute myeloid leukemia (tMDS/AML), according to a retrospective analysis.
Long-term, population-based cohort data showed the risk of tMDS/AML was significantly elevated after chemotherapy for 22 solid tumor types.
The relative risk of tMDS/AML was 1.5- to 39.0-fold greater among patients treated for these tumors than among the general population.
Lindsay M. Morton, PhD, of the National Institutes of Health in Rockville, Maryland, and her colleagues reported these findings in JAMA Oncology.
“We undertook an investigation to quantify tMDS/AML risks after chemotherapy for solid tumors in the modern treatment era, 2000-2014, using United States cancer registry data from the National Cancer Institute’s Surveillance, Epidemiology, and End Results Program,” the investigators wrote.
They retrospectively analyzed data from 1619 patients with tMDS/AML who were diagnosed with an initial primary solid tumor from 2000 to 2013.
Patients were given initial chemotherapy and lived for at least 1 year after treatment. Subsequently, Dr. Morton and her colleagues linked patient database records with Medicare insurance claim information to confirm the accuracy of chemotherapy data.
“Because registry data do not include treatment details, we used an alternative database to provide descriptive information on population-based patterns of chemotherapeutic drug use,” the investigators noted.
The team found the risk of developing tMDS/AML was significantly increased following chemotherapy administration for 22 of 23 solid tumor types, excluding colon cancer.
The standardized incidence ratio (SIR) for tMDS/AML ranged from 1.5 to 39.0, and the excess absolute risk (EAR) ranged from 1.4 to 23.6 cases per 10,000 person-years.
SIRs were greatest in patients who received chemotherapy for malignancy of the bone (SIR=39.0, EAR=23.6), testis (SIR, 12.3, EAR=4.4), soft tissue (SIR=10.4, EAR=12.6), fallopian tube (SIR=8.7, EAR=16.0), small cell lung (SIR=8.1, EAR=19.9), peritoneum (SIR=7.5, EAR=15.8), brain or central nervous system (SIR=7.2, EAR=6.0), and ovary (SIR=5.8, EAR=8.2).
The investigators also found that patients who were given chemotherapy at a young age had the highest risk of developing tMDS/AML.
“For patients treated with chemotherapy at the present time, approximately three-quarters of tMDS/AML cases expected to occur within the next 5 years will be attributable to chemotherapy,” the investigators said.
They acknowledged a key limitation of this study was the limited data on patient-specific chemotherapy and dosing information. Given these limitations, Dr. Morton and her colleagues said, “the exact magnitude of our risk estimates, including the proportions of excess cases, should therefore be interpreted cautiously.”
This study was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, and the California Department of Public Health. The authors reported no conflicts of interest.
Chemotherapy for solid tumors is associated with an increased risk of therapy-related myelodysplastic syndromes or acute myeloid leukemia (tMDS/AML), according to a retrospective analysis.
Long-term, population-based cohort data showed the risk of tMDS/AML was significantly elevated after chemotherapy for 22 solid tumor types.
The relative risk of tMDS/AML was 1.5- to 39.0-fold greater among patients treated for these tumors than among the general population.
Lindsay M. Morton, PhD, of the National Institutes of Health in Rockville, Maryland, and her colleagues reported these findings in JAMA Oncology.
“We undertook an investigation to quantify tMDS/AML risks after chemotherapy for solid tumors in the modern treatment era, 2000-2014, using United States cancer registry data from the National Cancer Institute’s Surveillance, Epidemiology, and End Results Program,” the investigators wrote.
They retrospectively analyzed data from 1619 patients with tMDS/AML who were diagnosed with an initial primary solid tumor from 2000 to 2013.
Patients were given initial chemotherapy and lived for at least 1 year after treatment. Subsequently, Dr. Morton and her colleagues linked patient database records with Medicare insurance claim information to confirm the accuracy of chemotherapy data.
“Because registry data do not include treatment details, we used an alternative database to provide descriptive information on population-based patterns of chemotherapeutic drug use,” the investigators noted.
The team found the risk of developing tMDS/AML was significantly increased following chemotherapy administration for 22 of 23 solid tumor types, excluding colon cancer.
The standardized incidence ratio (SIR) for tMDS/AML ranged from 1.5 to 39.0, and the excess absolute risk (EAR) ranged from 1.4 to 23.6 cases per 10,000 person-years.
SIRs were greatest in patients who received chemotherapy for malignancy of the bone (SIR=39.0, EAR=23.6), testis (SIR, 12.3, EAR=4.4), soft tissue (SIR=10.4, EAR=12.6), fallopian tube (SIR=8.7, EAR=16.0), small cell lung (SIR=8.1, EAR=19.9), peritoneum (SIR=7.5, EAR=15.8), brain or central nervous system (SIR=7.2, EAR=6.0), and ovary (SIR=5.8, EAR=8.2).
The investigators also found that patients who were given chemotherapy at a young age had the highest risk of developing tMDS/AML.
“For patients treated with chemotherapy at the present time, approximately three-quarters of tMDS/AML cases expected to occur within the next 5 years will be attributable to chemotherapy,” the investigators said.
They acknowledged a key limitation of this study was the limited data on patient-specific chemotherapy and dosing information. Given these limitations, Dr. Morton and her colleagues said, “the exact magnitude of our risk estimates, including the proportions of excess cases, should therefore be interpreted cautiously.”
This study was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, and the California Department of Public Health. The authors reported no conflicts of interest.
Chemotherapy for solid tumors is associated with an increased risk of therapy-related myelodysplastic syndromes or acute myeloid leukemia (tMDS/AML), according to a retrospective analysis.
Long-term, population-based cohort data showed the risk of tMDS/AML was significantly elevated after chemotherapy for 22 solid tumor types.
The relative risk of tMDS/AML was 1.5- to 39.0-fold greater among patients treated for these tumors than among the general population.
Lindsay M. Morton, PhD, of the National Institutes of Health in Rockville, Maryland, and her colleagues reported these findings in JAMA Oncology.
“We undertook an investigation to quantify tMDS/AML risks after chemotherapy for solid tumors in the modern treatment era, 2000-2014, using United States cancer registry data from the National Cancer Institute’s Surveillance, Epidemiology, and End Results Program,” the investigators wrote.
They retrospectively analyzed data from 1619 patients with tMDS/AML who were diagnosed with an initial primary solid tumor from 2000 to 2013.
Patients were given initial chemotherapy and lived for at least 1 year after treatment. Subsequently, Dr. Morton and her colleagues linked patient database records with Medicare insurance claim information to confirm the accuracy of chemotherapy data.
“Because registry data do not include treatment details, we used an alternative database to provide descriptive information on population-based patterns of chemotherapeutic drug use,” the investigators noted.
The team found the risk of developing tMDS/AML was significantly increased following chemotherapy administration for 22 of 23 solid tumor types, excluding colon cancer.
The standardized incidence ratio (SIR) for tMDS/AML ranged from 1.5 to 39.0, and the excess absolute risk (EAR) ranged from 1.4 to 23.6 cases per 10,000 person-years.
SIRs were greatest in patients who received chemotherapy for malignancy of the bone (SIR=39.0, EAR=23.6), testis (SIR, 12.3, EAR=4.4), soft tissue (SIR=10.4, EAR=12.6), fallopian tube (SIR=8.7, EAR=16.0), small cell lung (SIR=8.1, EAR=19.9), peritoneum (SIR=7.5, EAR=15.8), brain or central nervous system (SIR=7.2, EAR=6.0), and ovary (SIR=5.8, EAR=8.2).
The investigators also found that patients who were given chemotherapy at a young age had the highest risk of developing tMDS/AML.
“For patients treated with chemotherapy at the present time, approximately three-quarters of tMDS/AML cases expected to occur within the next 5 years will be attributable to chemotherapy,” the investigators said.
They acknowledged a key limitation of this study was the limited data on patient-specific chemotherapy and dosing information. Given these limitations, Dr. Morton and her colleagues said, “the exact magnitude of our risk estimates, including the proportions of excess cases, should therefore be interpreted cautiously.”
This study was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, and the California Department of Public Health. The authors reported no conflicts of interest.
FDA approves dasatinib for kids with Ph+ ALL

The U.S. Food and Drug Administration (FDA) has approved a second pediatric indication for dasatinib (Sprycel®).
The tyrosine kinase inhibitor is now approved for use in combination with chemotherapy to treat pediatric patients age 1 year and older who have newly diagnosed, Philadelphia-chromosome-positive (Ph+) acute lymphoblastic leukemia (ALL).
Dasatinib is also FDA-approved for use in children age 1 year and older who have chronic phase, Ph+ chronic myeloid leukemia (CML).
In adults, dasatinib is FDA-approved to treat:
- Newly diagnosed, Ph+, chronic phase CML
- Chronic, accelerated, or myeloid/lymphoid blast phase, Ph+ CML with resistance or intolerance to prior therapy including imatinib
- Ph+ ALL with resistance or intolerance to prior therapy.
Trial results
The FDA’s approval of dasatinib in children with Ph+ ALL is based on data from a phase 2 study (CA180-372, NCT01460160).
In this trial, researchers evaluated dasatinib in combination with the AIEOP-BFM ALL 2000 chemotherapy protocol in patients (ages 1 to 17) with newly diagnosed, B-cell precursor, Ph+ ALL.
There were 78 patients evaluated for efficacy in cohort 1. They had a median age of 10.4 years (range, 2.6 to 17.9 years). They received dasatinib at a daily dose of 60 mg/m2 for up to 24 months.
Patients with central nervous system 3 disease received cranial irradiation, and patients were assigned to stem cell transplant based on minimal residual disease if they were thought to have a high risk of relapse.
The 3-year event-free survival rate in the 78 patients was 64.1%.
There were 81 patients evaluable for safety who received dasatinib continuously in combination with chemotherapy. Their median duration of treatment was 24 months (range, 2 to 27 months).
The most common adverse events (AEs) in these patients were mucositis (93%), febrile neutropenia (86%), pyrexia (85%), diarrhea (84%), nausea (84%), vomiting (83%), musculoskeletal pain (83%), abdominal pain (78%), cough (78%), headache (77%), rash (68%), fatigue (59%), and constipation (57%).
Eight (10%) patients had AEs leading to treatment discontinuation. These included fungal sepsis, hepatotoxicity in the setting of graft-versus-host disease, thrombocytopenia, cytomegalovirus infection, pneumonia, nausea, enteritis, and drug hypersensitivity.
Three patients (4%) had fatal AEs, all infections.
This trial was sponsored by Bristol-Myers Squibb. Additional data are available in the prescribing information for dasatinib.
The U.S. Food and Drug Administration (FDA) has approved a second pediatric indication for dasatinib (Sprycel®).
The tyrosine kinase inhibitor is now approved for use in combination with chemotherapy to treat pediatric patients age 1 year and older who have newly diagnosed, Philadelphia-chromosome-positive (Ph+) acute lymphoblastic leukemia (ALL).
Dasatinib is also FDA-approved for use in children age 1 year and older who have chronic phase, Ph+ chronic myeloid leukemia (CML).
In adults, dasatinib is FDA-approved to treat:
- Newly diagnosed, Ph+, chronic phase CML
- Chronic, accelerated, or myeloid/lymphoid blast phase, Ph+ CML with resistance or intolerance to prior therapy including imatinib
- Ph+ ALL with resistance or intolerance to prior therapy.
Trial results
The FDA’s approval of dasatinib in children with Ph+ ALL is based on data from a phase 2 study (CA180-372, NCT01460160).
In this trial, researchers evaluated dasatinib in combination with the AIEOP-BFM ALL 2000 chemotherapy protocol in patients (ages 1 to 17) with newly diagnosed, B-cell precursor, Ph+ ALL.
There were 78 patients evaluated for efficacy in cohort 1. They had a median age of 10.4 years (range, 2.6 to 17.9 years). They received dasatinib at a daily dose of 60 mg/m2 for up to 24 months.
Patients with central nervous system 3 disease received cranial irradiation, and patients were assigned to stem cell transplant based on minimal residual disease if they were thought to have a high risk of relapse.
The 3-year event-free survival rate in the 78 patients was 64.1%.
There were 81 patients evaluable for safety who received dasatinib continuously in combination with chemotherapy. Their median duration of treatment was 24 months (range, 2 to 27 months).
The most common adverse events (AEs) in these patients were mucositis (93%), febrile neutropenia (86%), pyrexia (85%), diarrhea (84%), nausea (84%), vomiting (83%), musculoskeletal pain (83%), abdominal pain (78%), cough (78%), headache (77%), rash (68%), fatigue (59%), and constipation (57%).
Eight (10%) patients had AEs leading to treatment discontinuation. These included fungal sepsis, hepatotoxicity in the setting of graft-versus-host disease, thrombocytopenia, cytomegalovirus infection, pneumonia, nausea, enteritis, and drug hypersensitivity.
Three patients (4%) had fatal AEs, all infections.
This trial was sponsored by Bristol-Myers Squibb. Additional data are available in the prescribing information for dasatinib.
The U.S. Food and Drug Administration (FDA) has approved a second pediatric indication for dasatinib (Sprycel®).
The tyrosine kinase inhibitor is now approved for use in combination with chemotherapy to treat pediatric patients age 1 year and older who have newly diagnosed, Philadelphia-chromosome-positive (Ph+) acute lymphoblastic leukemia (ALL).
Dasatinib is also FDA-approved for use in children age 1 year and older who have chronic phase, Ph+ chronic myeloid leukemia (CML).
In adults, dasatinib is FDA-approved to treat:
- Newly diagnosed, Ph+, chronic phase CML
- Chronic, accelerated, or myeloid/lymphoid blast phase, Ph+ CML with resistance or intolerance to prior therapy including imatinib
- Ph+ ALL with resistance or intolerance to prior therapy.
Trial results
The FDA’s approval of dasatinib in children with Ph+ ALL is based on data from a phase 2 study (CA180-372, NCT01460160).
In this trial, researchers evaluated dasatinib in combination with the AIEOP-BFM ALL 2000 chemotherapy protocol in patients (ages 1 to 17) with newly diagnosed, B-cell precursor, Ph+ ALL.
There were 78 patients evaluated for efficacy in cohort 1. They had a median age of 10.4 years (range, 2.6 to 17.9 years). They received dasatinib at a daily dose of 60 mg/m2 for up to 24 months.
Patients with central nervous system 3 disease received cranial irradiation, and patients were assigned to stem cell transplant based on minimal residual disease if they were thought to have a high risk of relapse.
The 3-year event-free survival rate in the 78 patients was 64.1%.
There were 81 patients evaluable for safety who received dasatinib continuously in combination with chemotherapy. Their median duration of treatment was 24 months (range, 2 to 27 months).
The most common adverse events (AEs) in these patients were mucositis (93%), febrile neutropenia (86%), pyrexia (85%), diarrhea (84%), nausea (84%), vomiting (83%), musculoskeletal pain (83%), abdominal pain (78%), cough (78%), headache (77%), rash (68%), fatigue (59%), and constipation (57%).
Eight (10%) patients had AEs leading to treatment discontinuation. These included fungal sepsis, hepatotoxicity in the setting of graft-versus-host disease, thrombocytopenia, cytomegalovirus infection, pneumonia, nausea, enteritis, and drug hypersensitivity.
Three patients (4%) had fatal AEs, all infections.
This trial was sponsored by Bristol-Myers Squibb. Additional data are available in the prescribing information for dasatinib.
Group proposes new grading systems for CRS, neurotoxicity

A group of experts has proposed new consensus definitions and grading systems for cytokine release syndrome (CRS) and neurotoxicity related to immune effector cell therapies.
The group hopes their recommendations will be widely accepted and used in both trials and the clinical setting.
The recommendations were devised by 49 experts at a meeting supported by the American Society for Blood and Marrow Transplantation (ASBMT), compiled by a writing group, and reviewed by stakeholders.
Daniel W. Lee, MD, of the University of Virginia School of Medicine in Charlottesville, and his colleagues described the ASBMT consensus definitions and grading systems in Biology of Blood and Marrow Transplantation.
CRS
The ASBMT consensus definition for CRS is “a supraphysiologic response following any immune therapy that results in the activation or engagement of endogenous or infused T cells and/or other immune effector cells.”
To be diagnosed with CRS, a patient must have a fever and may have the following symptoms:
- Hypotension
- Capillary leak (hypoxia)
- End organ dysfunction.
The ASBMT consensus for grading CRS is as follows:
- Grade 1—Patient has a fever, defined as a temperature of 38.0°C or higher
- Grade 2—Patient has a fever, hypotension that doesn’t require vasopressors, and/or hypoxia that requires oxygen delivered by low-flow nasal cannula (≤6 L/min) or blow-by
- Grade 3—Patient has a fever, hypotension requiring one vasopressor (with or without vasopressin), and/or hypoxia (not attributable to any other cause) that requires high-flow nasal cannula (>6 L/min), facemask, non-rebreather mask, or venturi mask
- Grade 4—Patient has a fever, hypotension requiring multiple vasopressors (excluding vasopressin), and/or hypoxia (not attributable to any other cause) requiring positive-pressure ventilation
- Grade 5—Death due to CRS when there is no other “principle factor” leading to death.
Typically, severe CRS can be considered resolved if “fever, oxygen, and pressor requirements have resolved,” Dr. Lee and his coauthors said.
The authors also stressed that neurotoxicity that occurs with or after CRS “does not inform the grade of CRS but is instead captured separately in the neurotoxicity scale.”
Neurotoxicity
Dr. Lee and his coauthors said neurotoxicity in this setting is called “immune effector cell-associated neurotoxicity syndrome (ICANS).”
The ASBMT consensus definition for ICANs is “a disorder characterized by a pathologic process involving the central nervous system following any immune therapy that results in the activation or engagement of endogenous or infused T cells and/or other immune effector cells.”
Symptoms of ICANS may include:
- Aphasia
- Altered level of consciousness
- Impairment of cognitive skills
- Motor weakness
- Seizures
- Cerebral edema.
The ASBMT consensus for grading ICANS in adults and children age 12 and older is as follows:
- Grade 1—Patient has a score of 7-9 on the 10-point immune effector cell-associated encephalopathy (ICE) assessment and awakens spontaneously
- Grade 2—Patient has a score of 3-6 on the ICE assessment and will awaken to the sound of a voice
- Grade 3—Patient has a score of 0-2 on the ICE assessment, awakens only to tactile stimulus, has any clinical seizure that resolves rapidly or non-convulsive seizures that resolve with intervention, has focal/local edema on neuroimaging
- Grade 4—Patient is unable to perform the ICE assessment, is unarousable or requires “vigorous stimuli” to be aroused, has life-threatening seizure (lasting more than 5 minutes) or repetitive clinical or electrical seizures without return to baseline in between, has deep focal motor weakness, and/or has decerebrate or decorticate posturing, cranial nerve VI palsy, papilledema, Cushing’s triad, or signs of diffuse cerebral edema on neuroimaging
- Grade 5—Death due to ICANS when there is no other “principle factor” leading to death.
Dr. Lee and his coauthors noted that the ICE assessment is not suitable for children younger than 12. For these patients (and older patients with baseline developmental delays), ICANS can be assessed using the Cornell Assessment of Pediatric Delirium (CAPD).
The ASBMT consensus for grading ICANS in children younger than 12 (or older patients with developmental delays) is as follows:
- Grade 1—Patient has a CAPD score lower than 9 and awakens spontaneously
- Grade 2—Patient has a CAPD score lower than 9 and will awaken to the sound of a voice
- Grade 3—Patient has a CAPD score of 9 or higher, awakens only to tactile stimulus, has any clinical seizure that resolves rapidly or non-convulsive seizures that resolve with intervention, and/or has focal/local edema on neuroimaging
- Grade 4—Patient is unable to perform CAPD, is unarousable or requires “vigorous stimuli” to be aroused, has life-threatening seizure (lasting more than 5 minutes) or repetitive clinical or electrical seizures without return to baseline in between, has deep focal motor weakness, and/or has decerebrate or decorticate posturing, cranial nerve VI palsy, papilledema, Cushing’s triad, or signs of diffuse cerebral edema on neuroimaging
- Grade 5—Death due to ICANS when there is no other “principle factor” leading to death.
Dr. Lee and his coauthors reported relationships with a range of companies.
A group of experts has proposed new consensus definitions and grading systems for cytokine release syndrome (CRS) and neurotoxicity related to immune effector cell therapies.
The group hopes their recommendations will be widely accepted and used in both trials and the clinical setting.
The recommendations were devised by 49 experts at a meeting supported by the American Society for Blood and Marrow Transplantation (ASBMT), compiled by a writing group, and reviewed by stakeholders.
Daniel W. Lee, MD, of the University of Virginia School of Medicine in Charlottesville, and his colleagues described the ASBMT consensus definitions and grading systems in Biology of Blood and Marrow Transplantation.
CRS
The ASBMT consensus definition for CRS is “a supraphysiologic response following any immune therapy that results in the activation or engagement of endogenous or infused T cells and/or other immune effector cells.”
To be diagnosed with CRS, a patient must have a fever and may have the following symptoms:
- Hypotension
- Capillary leak (hypoxia)
- End organ dysfunction.
The ASBMT consensus for grading CRS is as follows:
- Grade 1—Patient has a fever, defined as a temperature of 38.0°C or higher
- Grade 2—Patient has a fever, hypotension that doesn’t require vasopressors, and/or hypoxia that requires oxygen delivered by low-flow nasal cannula (≤6 L/min) or blow-by
- Grade 3—Patient has a fever, hypotension requiring one vasopressor (with or without vasopressin), and/or hypoxia (not attributable to any other cause) that requires high-flow nasal cannula (>6 L/min), facemask, non-rebreather mask, or venturi mask
- Grade 4—Patient has a fever, hypotension requiring multiple vasopressors (excluding vasopressin), and/or hypoxia (not attributable to any other cause) requiring positive-pressure ventilation
- Grade 5—Death due to CRS when there is no other “principle factor” leading to death.
Typically, severe CRS can be considered resolved if “fever, oxygen, and pressor requirements have resolved,” Dr. Lee and his coauthors said.
The authors also stressed that neurotoxicity that occurs with or after CRS “does not inform the grade of CRS but is instead captured separately in the neurotoxicity scale.”
Neurotoxicity
Dr. Lee and his coauthors said neurotoxicity in this setting is called “immune effector cell-associated neurotoxicity syndrome (ICANS).”
The ASBMT consensus definition for ICANs is “a disorder characterized by a pathologic process involving the central nervous system following any immune therapy that results in the activation or engagement of endogenous or infused T cells and/or other immune effector cells.”
Symptoms of ICANS may include:
- Aphasia
- Altered level of consciousness
- Impairment of cognitive skills
- Motor weakness
- Seizures
- Cerebral edema.
The ASBMT consensus for grading ICANS in adults and children age 12 and older is as follows:
- Grade 1—Patient has a score of 7-9 on the 10-point immune effector cell-associated encephalopathy (ICE) assessment and awakens spontaneously
- Grade 2—Patient has a score of 3-6 on the ICE assessment and will awaken to the sound of a voice
- Grade 3—Patient has a score of 0-2 on the ICE assessment, awakens only to tactile stimulus, has any clinical seizure that resolves rapidly or non-convulsive seizures that resolve with intervention, has focal/local edema on neuroimaging
- Grade 4—Patient is unable to perform the ICE assessment, is unarousable or requires “vigorous stimuli” to be aroused, has life-threatening seizure (lasting more than 5 minutes) or repetitive clinical or electrical seizures without return to baseline in between, has deep focal motor weakness, and/or has decerebrate or decorticate posturing, cranial nerve VI palsy, papilledema, Cushing’s triad, or signs of diffuse cerebral edema on neuroimaging
- Grade 5—Death due to ICANS when there is no other “principle factor” leading to death.
Dr. Lee and his coauthors noted that the ICE assessment is not suitable for children younger than 12. For these patients (and older patients with baseline developmental delays), ICANS can be assessed using the Cornell Assessment of Pediatric Delirium (CAPD).
The ASBMT consensus for grading ICANS in children younger than 12 (or older patients with developmental delays) is as follows:
- Grade 1—Patient has a CAPD score lower than 9 and awakens spontaneously
- Grade 2—Patient has a CAPD score lower than 9 and will awaken to the sound of a voice
- Grade 3—Patient has a CAPD score of 9 or higher, awakens only to tactile stimulus, has any clinical seizure that resolves rapidly or non-convulsive seizures that resolve with intervention, and/or has focal/local edema on neuroimaging
- Grade 4—Patient is unable to perform CAPD, is unarousable or requires “vigorous stimuli” to be aroused, has life-threatening seizure (lasting more than 5 minutes) or repetitive clinical or electrical seizures without return to baseline in between, has deep focal motor weakness, and/or has decerebrate or decorticate posturing, cranial nerve VI palsy, papilledema, Cushing’s triad, or signs of diffuse cerebral edema on neuroimaging
- Grade 5—Death due to ICANS when there is no other “principle factor” leading to death.
Dr. Lee and his coauthors reported relationships with a range of companies.
A group of experts has proposed new consensus definitions and grading systems for cytokine release syndrome (CRS) and neurotoxicity related to immune effector cell therapies.
The group hopes their recommendations will be widely accepted and used in both trials and the clinical setting.
The recommendations were devised by 49 experts at a meeting supported by the American Society for Blood and Marrow Transplantation (ASBMT), compiled by a writing group, and reviewed by stakeholders.
Daniel W. Lee, MD, of the University of Virginia School of Medicine in Charlottesville, and his colleagues described the ASBMT consensus definitions and grading systems in Biology of Blood and Marrow Transplantation.
CRS
The ASBMT consensus definition for CRS is “a supraphysiologic response following any immune therapy that results in the activation or engagement of endogenous or infused T cells and/or other immune effector cells.”
To be diagnosed with CRS, a patient must have a fever and may have the following symptoms:
- Hypotension
- Capillary leak (hypoxia)
- End organ dysfunction.
The ASBMT consensus for grading CRS is as follows:
- Grade 1—Patient has a fever, defined as a temperature of 38.0°C or higher
- Grade 2—Patient has a fever, hypotension that doesn’t require vasopressors, and/or hypoxia that requires oxygen delivered by low-flow nasal cannula (≤6 L/min) or blow-by
- Grade 3—Patient has a fever, hypotension requiring one vasopressor (with or without vasopressin), and/or hypoxia (not attributable to any other cause) that requires high-flow nasal cannula (>6 L/min), facemask, non-rebreather mask, or venturi mask
- Grade 4—Patient has a fever, hypotension requiring multiple vasopressors (excluding vasopressin), and/or hypoxia (not attributable to any other cause) requiring positive-pressure ventilation
- Grade 5—Death due to CRS when there is no other “principle factor” leading to death.
Typically, severe CRS can be considered resolved if “fever, oxygen, and pressor requirements have resolved,” Dr. Lee and his coauthors said.
The authors also stressed that neurotoxicity that occurs with or after CRS “does not inform the grade of CRS but is instead captured separately in the neurotoxicity scale.”
Neurotoxicity
Dr. Lee and his coauthors said neurotoxicity in this setting is called “immune effector cell-associated neurotoxicity syndrome (ICANS).”
The ASBMT consensus definition for ICANs is “a disorder characterized by a pathologic process involving the central nervous system following any immune therapy that results in the activation or engagement of endogenous or infused T cells and/or other immune effector cells.”
Symptoms of ICANS may include:
- Aphasia
- Altered level of consciousness
- Impairment of cognitive skills
- Motor weakness
- Seizures
- Cerebral edema.
The ASBMT consensus for grading ICANS in adults and children age 12 and older is as follows:
- Grade 1—Patient has a score of 7-9 on the 10-point immune effector cell-associated encephalopathy (ICE) assessment and awakens spontaneously
- Grade 2—Patient has a score of 3-6 on the ICE assessment and will awaken to the sound of a voice
- Grade 3—Patient has a score of 0-2 on the ICE assessment, awakens only to tactile stimulus, has any clinical seizure that resolves rapidly or non-convulsive seizures that resolve with intervention, has focal/local edema on neuroimaging
- Grade 4—Patient is unable to perform the ICE assessment, is unarousable or requires “vigorous stimuli” to be aroused, has life-threatening seizure (lasting more than 5 minutes) or repetitive clinical or electrical seizures without return to baseline in between, has deep focal motor weakness, and/or has decerebrate or decorticate posturing, cranial nerve VI palsy, papilledema, Cushing’s triad, or signs of diffuse cerebral edema on neuroimaging
- Grade 5—Death due to ICANS when there is no other “principle factor” leading to death.
Dr. Lee and his coauthors noted that the ICE assessment is not suitable for children younger than 12. For these patients (and older patients with baseline developmental delays), ICANS can be assessed using the Cornell Assessment of Pediatric Delirium (CAPD).
The ASBMT consensus for grading ICANS in children younger than 12 (or older patients with developmental delays) is as follows:
- Grade 1—Patient has a CAPD score lower than 9 and awakens spontaneously
- Grade 2—Patient has a CAPD score lower than 9 and will awaken to the sound of a voice
- Grade 3—Patient has a CAPD score of 9 or higher, awakens only to tactile stimulus, has any clinical seizure that resolves rapidly or non-convulsive seizures that resolve with intervention, and/or has focal/local edema on neuroimaging
- Grade 4—Patient is unable to perform CAPD, is unarousable or requires “vigorous stimuli” to be aroused, has life-threatening seizure (lasting more than 5 minutes) or repetitive clinical or electrical seizures without return to baseline in between, has deep focal motor weakness, and/or has decerebrate or decorticate posturing, cranial nerve VI palsy, papilledema, Cushing’s triad, or signs of diffuse cerebral edema on neuroimaging
- Grade 5—Death due to ICANS when there is no other “principle factor” leading to death.
Dr. Lee and his coauthors reported relationships with a range of companies.
EC approves split dosing regimen for daratumumab

The European Commission (EC) has granted marketing authorization for a split dosing regimen for daratumumab (Darzalex®).
The approval provides healthcare professionals with the option to split the first infusion of daratumumab over 2 consecutive days.
“We are hopeful that the availability of this more flexible dosing option will make the first infusion of Darzalex more convenient for European multiple myeloma patients,” said Jan van de Winkel, PhD, chief executive officer of Genmab, which licensed daratumumab to Janssen Biotech, Inc.
Daratumumab is currently EC-approved for the following indications:
- For use in combination with bortezomib, melphalan, and prednisone to treat adults with newly diagnosed multiple myeloma (MM) who are ineligible for autologous stem cell transplant
- For use in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, for the treatment of adults with MM who have received at least one prior therapy
- As monotherapy for adults with relapsed and refractory MM whose prior therapy included a proteasome inhibitor and an immunomodulatory agent and who have demonstrated disease progression on their last therapy.
The EC’s approval of a split dosing regimen for daratumumab was based on data from the phase 1b EQUULEUS trial (MMY1001, NCT01998971), which was sponsored by Janssen.
This trial was designed to evaluate daratumumab in combination with bortezomib-dexamethasone, bortezomib-melphalan-prednisone, bortezomib-thalidomide-dexamethasone, pomalidomide-dexamethasone, carfilzomib-dexamethasone, and carfilzomib-lenalidomide-dexamethasone.
At the 2018 ASH Annual Meeting (abstract 1970), researchers presented data from this trial in MM patients who received their first 16 mg/kg daratumumab dose as a split dose of 8 mg/kg on day 1 of cycle 1 and 8 mg/kg on day 2 of cycle 1, compared to patients who received the full 16 mg/kg dose on day 1 of cycle 1.
The researchers said they observed “virtually identical” pharmacokinetics between the dosing groups.
Cmax on the first day of cycle 1 was lower in the split-dose group than in the full-dose group. However, after patients in the split-dose group received the second 8 mg/kg dose on day 2, concentrations were similar between the groups.
The researchers said they do not expect the initial difference they observed to have any impact on clinical outcomes.
The team also pointed out that there was no increase in infusion-related reactions among patients who received the split dose.
The researchers said split dosing of daratumumab is still being investigated in ongoing studies of MM patients, including CANDOR (NCT03158688) and LYRA (NCT02951819).
The European Commission (EC) has granted marketing authorization for a split dosing regimen for daratumumab (Darzalex®).
The approval provides healthcare professionals with the option to split the first infusion of daratumumab over 2 consecutive days.
“We are hopeful that the availability of this more flexible dosing option will make the first infusion of Darzalex more convenient for European multiple myeloma patients,” said Jan van de Winkel, PhD, chief executive officer of Genmab, which licensed daratumumab to Janssen Biotech, Inc.
Daratumumab is currently EC-approved for the following indications:
- For use in combination with bortezomib, melphalan, and prednisone to treat adults with newly diagnosed multiple myeloma (MM) who are ineligible for autologous stem cell transplant
- For use in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, for the treatment of adults with MM who have received at least one prior therapy
- As monotherapy for adults with relapsed and refractory MM whose prior therapy included a proteasome inhibitor and an immunomodulatory agent and who have demonstrated disease progression on their last therapy.
The EC’s approval of a split dosing regimen for daratumumab was based on data from the phase 1b EQUULEUS trial (MMY1001, NCT01998971), which was sponsored by Janssen.
This trial was designed to evaluate daratumumab in combination with bortezomib-dexamethasone, bortezomib-melphalan-prednisone, bortezomib-thalidomide-dexamethasone, pomalidomide-dexamethasone, carfilzomib-dexamethasone, and carfilzomib-lenalidomide-dexamethasone.
At the 2018 ASH Annual Meeting (abstract 1970), researchers presented data from this trial in MM patients who received their first 16 mg/kg daratumumab dose as a split dose of 8 mg/kg on day 1 of cycle 1 and 8 mg/kg on day 2 of cycle 1, compared to patients who received the full 16 mg/kg dose on day 1 of cycle 1.
The researchers said they observed “virtually identical” pharmacokinetics between the dosing groups.
Cmax on the first day of cycle 1 was lower in the split-dose group than in the full-dose group. However, after patients in the split-dose group received the second 8 mg/kg dose on day 2, concentrations were similar between the groups.
The researchers said they do not expect the initial difference they observed to have any impact on clinical outcomes.
The team also pointed out that there was no increase in infusion-related reactions among patients who received the split dose.
The researchers said split dosing of daratumumab is still being investigated in ongoing studies of MM patients, including CANDOR (NCT03158688) and LYRA (NCT02951819).
The European Commission (EC) has granted marketing authorization for a split dosing regimen for daratumumab (Darzalex®).
The approval provides healthcare professionals with the option to split the first infusion of daratumumab over 2 consecutive days.
“We are hopeful that the availability of this more flexible dosing option will make the first infusion of Darzalex more convenient for European multiple myeloma patients,” said Jan van de Winkel, PhD, chief executive officer of Genmab, which licensed daratumumab to Janssen Biotech, Inc.
Daratumumab is currently EC-approved for the following indications:
- For use in combination with bortezomib, melphalan, and prednisone to treat adults with newly diagnosed multiple myeloma (MM) who are ineligible for autologous stem cell transplant
- For use in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, for the treatment of adults with MM who have received at least one prior therapy
- As monotherapy for adults with relapsed and refractory MM whose prior therapy included a proteasome inhibitor and an immunomodulatory agent and who have demonstrated disease progression on their last therapy.
The EC’s approval of a split dosing regimen for daratumumab was based on data from the phase 1b EQUULEUS trial (MMY1001, NCT01998971), which was sponsored by Janssen.
This trial was designed to evaluate daratumumab in combination with bortezomib-dexamethasone, bortezomib-melphalan-prednisone, bortezomib-thalidomide-dexamethasone, pomalidomide-dexamethasone, carfilzomib-dexamethasone, and carfilzomib-lenalidomide-dexamethasone.
At the 2018 ASH Annual Meeting (abstract 1970), researchers presented data from this trial in MM patients who received their first 16 mg/kg daratumumab dose as a split dose of 8 mg/kg on day 1 of cycle 1 and 8 mg/kg on day 2 of cycle 1, compared to patients who received the full 16 mg/kg dose on day 1 of cycle 1.
The researchers said they observed “virtually identical” pharmacokinetics between the dosing groups.
Cmax on the first day of cycle 1 was lower in the split-dose group than in the full-dose group. However, after patients in the split-dose group received the second 8 mg/kg dose on day 2, concentrations were similar between the groups.
The researchers said they do not expect the initial difference they observed to have any impact on clinical outcomes.
The team also pointed out that there was no increase in infusion-related reactions among patients who received the split dose.
The researchers said split dosing of daratumumab is still being investigated in ongoing studies of MM patients, including CANDOR (NCT03158688) and LYRA (NCT02951819).
Potential treatment on the horizon for cold agglutinin disease

In a first-in-human trial, sutimlimab rapidly halted hemolysis, corrected anemia, precluded the need for transfusion, and caused no serious adverse effects in patients with cold agglutinin disease.
Sutimlimab also “induced clinically meaningful increases in hemoglobin levels, even in patients with multiple previous lines of therapy,” according to investigators.
The European Medicines Agency and the U.S. Food and Drug Administration (FDA) awarded sutimlimab orphan drug status based on these results. The FDA also granted sutimlimab breakthrough therapy designation.
Sutimlimab is a humanized anti-C1s IgG4 monoclonal antibody that blocks the classical complement pathway–specific protease C1s and prevents further hemolysis in patients with cold agglutinin disease.
Cold agglutinin disease is a rare, acquired chronic autoimmune hemolytic condition that destroys red blood cells. It leads to chronic anemia, severe fatigue, and potentially fatal thrombotic events. No drug has yet been approved to treat cold agglutinin disease.
The phase 1 trial of sutimlimab (formerly BIVV009 and TNT009) in cold agglutinin disease was conducted at the Medical University of Vienna in Austria and reported in Blood.
The study (NCT02502903) involved 10 patients, ages 56 to 76, who were previously treated with multiple lines of therapy, including two patients who failed treatment with eculizumab.
Of the 10 patients, eight were female, eight were Caucasian, one was Asian, and one was Hispanic. Patients had cold agglutinin disease for a median of 5 years (range, 1 – 20).
At baseline, the median hemoglobin level was 7.8 g/dL, the median number of reticulocytes was 133 x 109/L, the median bilirubin was 2.0 mg/dL, and the median haptoglobin was less than 12 mg/dL.
The patients received an initial dose of 10 mg/kg intravenous sutimlimab as a test dose to allow rapid wash-out of the drug if unforeseen adverse effects occurred with the first infusion.
One to 4 days later, they received the full dose of 60 mg/kg, followed by three additional weekly doses.
Investigators observed the patients for 49 to 53 days.
Results
Within the first week, patients’ median hemoglobin levels increased by 1.6 g/dL (P=0.007), and the median best response was an increase of 3.9 g/dL (P=0.005) after 6 weeks.
Seven patients had increased hemoglobin levels by more than 2 g/dL, and this included those who recently failed to respond or relapsed after rituximab, rituximab plus bendamustine, or eculizumab.
In five patients, hemoglobin increased by 4 g/dL or more. In four patients, it completely normalized to 12 g/dL.
In the first 24 hours after sutimlimab infusion, reticulocyte counts increased by a median of 41% and then gradually declined as hemoglobin levels rose.
In four patients, haptoglobin levels normalized within 1 to 2 weeks. In eight patients who had abnormal bilirubin levels at baseline, sutimlimab decreased the median bilirubin levels by 61%, normalizing levels in most patients within 24 hours of the first infusion (P=0.007).
When sutimlimab was washed out, bilirubin levels increased again, which demonstrated the recurrence of hemolysis.
Approximately 3 to 4 weeks after the last dose of sutimlimab, hemolysis and anemia recurred in all responders.
When patients were re-exposed to sutimlimab, rapid and complete inhibition of hemolysis occurred once again.
None of the patients required packed red blood cell transfusions during treatment.
Safety
The investigators reported that all infusions were well tolerated without premedication and without relevant drug-related adverse effects.
They reported few adverse events during the trial. All were mild or moderate in severity, and most were considered unrelated or unlikely related to sutimlimab.
Two adverse events—one mild purpural rash on both hands and one case of moderate hair loss (each occurring in one patient)—were possibly related to sutimlimab.
While the investigators considered the safety data encouraging, they recommended interpreting the data “cautiously in light of the limited duration of the trial.”
“Provided that safety results remain positive, sutimlimab could become the first approved treatment for cold agglutinin disease,” said corresponding author Bernd Jilma, MD, of the Medical University of Vienna in Austria.
“The drug clearly addresses an unmet medical need, as we have seen rapid, strong responses in patients for whom multiple prior therapies have failed.”
This study was funded by True North Therapeutics, Inc, now part of Bioverativ, a Sanofi company.
Some of the authors disclosed financial relationships, including employment, with True North Therapeutics and Bioverativ.
A phase 3 trial of sutimlimab is underway with top-line results due in 2019.
In a first-in-human trial, sutimlimab rapidly halted hemolysis, corrected anemia, precluded the need for transfusion, and caused no serious adverse effects in patients with cold agglutinin disease.
Sutimlimab also “induced clinically meaningful increases in hemoglobin levels, even in patients with multiple previous lines of therapy,” according to investigators.
The European Medicines Agency and the U.S. Food and Drug Administration (FDA) awarded sutimlimab orphan drug status based on these results. The FDA also granted sutimlimab breakthrough therapy designation.
Sutimlimab is a humanized anti-C1s IgG4 monoclonal antibody that blocks the classical complement pathway–specific protease C1s and prevents further hemolysis in patients with cold agglutinin disease.
Cold agglutinin disease is a rare, acquired chronic autoimmune hemolytic condition that destroys red blood cells. It leads to chronic anemia, severe fatigue, and potentially fatal thrombotic events. No drug has yet been approved to treat cold agglutinin disease.
The phase 1 trial of sutimlimab (formerly BIVV009 and TNT009) in cold agglutinin disease was conducted at the Medical University of Vienna in Austria and reported in Blood.
The study (NCT02502903) involved 10 patients, ages 56 to 76, who were previously treated with multiple lines of therapy, including two patients who failed treatment with eculizumab.
Of the 10 patients, eight were female, eight were Caucasian, one was Asian, and one was Hispanic. Patients had cold agglutinin disease for a median of 5 years (range, 1 – 20).
At baseline, the median hemoglobin level was 7.8 g/dL, the median number of reticulocytes was 133 x 109/L, the median bilirubin was 2.0 mg/dL, and the median haptoglobin was less than 12 mg/dL.
The patients received an initial dose of 10 mg/kg intravenous sutimlimab as a test dose to allow rapid wash-out of the drug if unforeseen adverse effects occurred with the first infusion.
One to 4 days later, they received the full dose of 60 mg/kg, followed by three additional weekly doses.
Investigators observed the patients for 49 to 53 days.
Results
Within the first week, patients’ median hemoglobin levels increased by 1.6 g/dL (P=0.007), and the median best response was an increase of 3.9 g/dL (P=0.005) after 6 weeks.
Seven patients had increased hemoglobin levels by more than 2 g/dL, and this included those who recently failed to respond or relapsed after rituximab, rituximab plus bendamustine, or eculizumab.
In five patients, hemoglobin increased by 4 g/dL or more. In four patients, it completely normalized to 12 g/dL.
In the first 24 hours after sutimlimab infusion, reticulocyte counts increased by a median of 41% and then gradually declined as hemoglobin levels rose.
In four patients, haptoglobin levels normalized within 1 to 2 weeks. In eight patients who had abnormal bilirubin levels at baseline, sutimlimab decreased the median bilirubin levels by 61%, normalizing levels in most patients within 24 hours of the first infusion (P=0.007).
When sutimlimab was washed out, bilirubin levels increased again, which demonstrated the recurrence of hemolysis.
Approximately 3 to 4 weeks after the last dose of sutimlimab, hemolysis and anemia recurred in all responders.
When patients were re-exposed to sutimlimab, rapid and complete inhibition of hemolysis occurred once again.
None of the patients required packed red blood cell transfusions during treatment.
Safety
The investigators reported that all infusions were well tolerated without premedication and without relevant drug-related adverse effects.
They reported few adverse events during the trial. All were mild or moderate in severity, and most were considered unrelated or unlikely related to sutimlimab.
Two adverse events—one mild purpural rash on both hands and one case of moderate hair loss (each occurring in one patient)—were possibly related to sutimlimab.
While the investigators considered the safety data encouraging, they recommended interpreting the data “cautiously in light of the limited duration of the trial.”
“Provided that safety results remain positive, sutimlimab could become the first approved treatment for cold agglutinin disease,” said corresponding author Bernd Jilma, MD, of the Medical University of Vienna in Austria.
“The drug clearly addresses an unmet medical need, as we have seen rapid, strong responses in patients for whom multiple prior therapies have failed.”
This study was funded by True North Therapeutics, Inc, now part of Bioverativ, a Sanofi company.
Some of the authors disclosed financial relationships, including employment, with True North Therapeutics and Bioverativ.
A phase 3 trial of sutimlimab is underway with top-line results due in 2019.
In a first-in-human trial, sutimlimab rapidly halted hemolysis, corrected anemia, precluded the need for transfusion, and caused no serious adverse effects in patients with cold agglutinin disease.
Sutimlimab also “induced clinically meaningful increases in hemoglobin levels, even in patients with multiple previous lines of therapy,” according to investigators.
The European Medicines Agency and the U.S. Food and Drug Administration (FDA) awarded sutimlimab orphan drug status based on these results. The FDA also granted sutimlimab breakthrough therapy designation.
Sutimlimab is a humanized anti-C1s IgG4 monoclonal antibody that blocks the classical complement pathway–specific protease C1s and prevents further hemolysis in patients with cold agglutinin disease.
Cold agglutinin disease is a rare, acquired chronic autoimmune hemolytic condition that destroys red blood cells. It leads to chronic anemia, severe fatigue, and potentially fatal thrombotic events. No drug has yet been approved to treat cold agglutinin disease.
The phase 1 trial of sutimlimab (formerly BIVV009 and TNT009) in cold agglutinin disease was conducted at the Medical University of Vienna in Austria and reported in Blood.
The study (NCT02502903) involved 10 patients, ages 56 to 76, who were previously treated with multiple lines of therapy, including two patients who failed treatment with eculizumab.
Of the 10 patients, eight were female, eight were Caucasian, one was Asian, and one was Hispanic. Patients had cold agglutinin disease for a median of 5 years (range, 1 – 20).
At baseline, the median hemoglobin level was 7.8 g/dL, the median number of reticulocytes was 133 x 109/L, the median bilirubin was 2.0 mg/dL, and the median haptoglobin was less than 12 mg/dL.
The patients received an initial dose of 10 mg/kg intravenous sutimlimab as a test dose to allow rapid wash-out of the drug if unforeseen adverse effects occurred with the first infusion.
One to 4 days later, they received the full dose of 60 mg/kg, followed by three additional weekly doses.
Investigators observed the patients for 49 to 53 days.
Results
Within the first week, patients’ median hemoglobin levels increased by 1.6 g/dL (P=0.007), and the median best response was an increase of 3.9 g/dL (P=0.005) after 6 weeks.
Seven patients had increased hemoglobin levels by more than 2 g/dL, and this included those who recently failed to respond or relapsed after rituximab, rituximab plus bendamustine, or eculizumab.
In five patients, hemoglobin increased by 4 g/dL or more. In four patients, it completely normalized to 12 g/dL.
In the first 24 hours after sutimlimab infusion, reticulocyte counts increased by a median of 41% and then gradually declined as hemoglobin levels rose.
In four patients, haptoglobin levels normalized within 1 to 2 weeks. In eight patients who had abnormal bilirubin levels at baseline, sutimlimab decreased the median bilirubin levels by 61%, normalizing levels in most patients within 24 hours of the first infusion (P=0.007).
When sutimlimab was washed out, bilirubin levels increased again, which demonstrated the recurrence of hemolysis.
Approximately 3 to 4 weeks after the last dose of sutimlimab, hemolysis and anemia recurred in all responders.
When patients were re-exposed to sutimlimab, rapid and complete inhibition of hemolysis occurred once again.
None of the patients required packed red blood cell transfusions during treatment.
Safety
The investigators reported that all infusions were well tolerated without premedication and without relevant drug-related adverse effects.
They reported few adverse events during the trial. All were mild or moderate in severity, and most were considered unrelated or unlikely related to sutimlimab.
Two adverse events—one mild purpural rash on both hands and one case of moderate hair loss (each occurring in one patient)—were possibly related to sutimlimab.
While the investigators considered the safety data encouraging, they recommended interpreting the data “cautiously in light of the limited duration of the trial.”
“Provided that safety results remain positive, sutimlimab could become the first approved treatment for cold agglutinin disease,” said corresponding author Bernd Jilma, MD, of the Medical University of Vienna in Austria.
“The drug clearly addresses an unmet medical need, as we have seen rapid, strong responses in patients for whom multiple prior therapies have failed.”
This study was funded by True North Therapeutics, Inc, now part of Bioverativ, a Sanofi company.
Some of the authors disclosed financial relationships, including employment, with True North Therapeutics and Bioverativ.
A phase 3 trial of sutimlimab is underway with top-line results due in 2019.