User login
American Society of Hematology (ASH): ASH 2013
Real-world data backs rivaroxaban for postop VTE prevention
NEW ORLEANS – In routine practice, rivaroxaban was superior to low-molecular-weight heparin for venous thromboembolism prevention in older adults undergoing hip or knee arthroplasty, without an increase in bleeding risk.
Among 24,321 patients, aged 66 years or older, the 30-day VTE event rate was 0.47% for rivaroxaban (Xarelto) (61 events) and 0.81% for low-molecular-weight heparin (LMWH) (93 events).
These findings resulted in an unadjusted relative risk of 0.58, which was statistically significant (P = .001) and did not change after adjustment for significant covariates, Dr. Alejandro Lazo-Langner said during an antithrombotic therapy session at the annual meeting of the American Society of Hematology.
There were 23 major bleeding events in both the rivaroxaban (0.18%) and LMWH (0.20%) groups. The unadjusted relative risk was 0.89 (P = .700) and did not change after adjustment in the population-based, retrospective cohort analysis.

Rivaroxaban has been the subject of numerous randomized controlled trials, but "We don’t have much real-life data in these patients," said Dr. Lazo-Langner, a hematologist specializing in thromboembolic diseases at Western University, London, Ontario, Canada.
In a meta-analysis of eight randomized rivaroxaban trials, the factor Xa inhibitor was associated with a significant 52% reduction in thrombosis after total hip or knee replacement, compared with enoxaparin (Lovenox) in roughly 14,000 patients (BMJ 2012;344:e3675 [doi:10.1136/bmj.e3675]). This came at a cost, however, of a slightly increased risk of clinically significant bleeding (relative risk, 1.25), he noted.
For the current analysis,the investigators used linked health care databases in Ontario to identify 24,321 patients who received an outpatient prescription for rivaroxaban or subcutaneous LMWH including dalteparin (Fragmin), tinzaparin (Innohep), or enoxaparin on discharge after total hip or knee arthroplasty between 2002 and 2012 across 121 hospitals.
Their average age was 74 years, 59% were women, and 12,850 received rivaroxaban. The anticoagulants were prescribed for a median of 14 days. Patients were excluded if they had other indications for anticoagulation, prosthetic mechanical heart valves, required dialysis, or lived in a long-term-care facility.
At 90 days, the VTE event rate was significantly lower in the rivaroxaban group than in the LMWH group (0.71% vs. 1.20%; adjusted RR, 0.59; P = .001). Once again, major bleeding events were similar (0.24% vs. 0.27%; adjusted RR, 0.63; P = .138), Dr. Lazo-Langner reported.
No differences were observed between the two groups at 30 or 90 days for hospitalization with endoscopy or hospitalization with major bleeding or endoscopy.
All-cause mortality was not estimable at 30 days but was lower with rivaroxaban at 90 days (16 deaths vs. 25 deaths; adjusted RR, 0.52; P = .058).
Additional analyses were conducted to test the robustness of the findings and no differences in rates of thrombosis were found when the analysis was restricted to 2009 to 2012, by type of joint replacement, or different low-molecular-weight heparins, he said.
Finally, a cost analysis was performed that showed a modest, but significant increase in direct drug costs to patients prescribed LMWH rather than rivaroxaban (Canadian $242 vs. $228; P less than .001) and home-care costs, likely from increased nursing ($1,082 vs. $959; P less than .001), Dr. Lazo-Langner said.
During a discussion of the results, he said there was no difference in novel anticoagulant use across surgeons or hospital settings, which was academic for 21% of LMWH patients and 15% of rivaroxaban patients.
Session comoderator Dr. Elaine Hylek, professor of medicine at Boston University, called this reassuring, but also urged caution in extrapolating conclusions on treatment effect outside a randomized trial.
Dr. Lazo-Langner reported research funding from Alexion, serving as a speaker for Pfizer, and honoraria from Pfizer, Leo Pharma, and Boehringer Ingelheim.
NEW ORLEANS – In routine practice, rivaroxaban was superior to low-molecular-weight heparin for venous thromboembolism prevention in older adults undergoing hip or knee arthroplasty, without an increase in bleeding risk.
Among 24,321 patients, aged 66 years or older, the 30-day VTE event rate was 0.47% for rivaroxaban (Xarelto) (61 events) and 0.81% for low-molecular-weight heparin (LMWH) (93 events).
These findings resulted in an unadjusted relative risk of 0.58, which was statistically significant (P = .001) and did not change after adjustment for significant covariates, Dr. Alejandro Lazo-Langner said during an antithrombotic therapy session at the annual meeting of the American Society of Hematology.
There were 23 major bleeding events in both the rivaroxaban (0.18%) and LMWH (0.20%) groups. The unadjusted relative risk was 0.89 (P = .700) and did not change after adjustment in the population-based, retrospective cohort analysis.
Rivaroxaban has been the subject of numerous randomized controlled trials, but "We don’t have much real-life data in these patients," said Dr. Lazo-Langner, a hematologist specializing in thromboembolic diseases at Western University, London, Ontario, Canada.
In a meta-analysis of eight randomized rivaroxaban trials, the factor Xa inhibitor was associated with a significant 52% reduction in thrombosis after total hip or knee replacement, compared with enoxaparin (Lovenox) in roughly 14,000 patients (BMJ 2012;344:e3675 [doi:10.1136/bmj.e3675]). This came at a cost, however, of a slightly increased risk of clinically significant bleeding (relative risk, 1.25), he noted.
For the current analysis,the investigators used linked health care databases in Ontario to identify 24,321 patients who received an outpatient prescription for rivaroxaban or subcutaneous LMWH including dalteparin (Fragmin), tinzaparin (Innohep), or enoxaparin on discharge after total hip or knee arthroplasty between 2002 and 2012 across 121 hospitals.
Their average age was 74 years, 59% were women, and 12,850 received rivaroxaban. The anticoagulants were prescribed for a median of 14 days. Patients were excluded if they had other indications for anticoagulation, prosthetic mechanical heart valves, required dialysis, or lived in a long-term-care facility.
At 90 days, the VTE event rate was significantly lower in the rivaroxaban group than in the LMWH group (0.71% vs. 1.20%; adjusted RR, 0.59; P = .001). Once again, major bleeding events were similar (0.24% vs. 0.27%; adjusted RR, 0.63; P = .138), Dr. Lazo-Langner reported.
No differences were observed between the two groups at 30 or 90 days for hospitalization with endoscopy or hospitalization with major bleeding or endoscopy.
All-cause mortality was not estimable at 30 days but was lower with rivaroxaban at 90 days (16 deaths vs. 25 deaths; adjusted RR, 0.52; P = .058).
Additional analyses were conducted to test the robustness of the findings and no differences in rates of thrombosis were found when the analysis was restricted to 2009 to 2012, by type of joint replacement, or different low-molecular-weight heparins, he said.
Finally, a cost analysis was performed that showed a modest, but significant increase in direct drug costs to patients prescribed LMWH rather than rivaroxaban (Canadian $242 vs. $228; P less than .001) and home-care costs, likely from increased nursing ($1,082 vs. $959; P less than .001), Dr. Lazo-Langner said.
During a discussion of the results, he said there was no difference in novel anticoagulant use across surgeons or hospital settings, which was academic for 21% of LMWH patients and 15% of rivaroxaban patients.
Session comoderator Dr. Elaine Hylek, professor of medicine at Boston University, called this reassuring, but also urged caution in extrapolating conclusions on treatment effect outside a randomized trial.
Dr. Lazo-Langner reported research funding from Alexion, serving as a speaker for Pfizer, and honoraria from Pfizer, Leo Pharma, and Boehringer Ingelheim.
NEW ORLEANS – In routine practice, rivaroxaban was superior to low-molecular-weight heparin for venous thromboembolism prevention in older adults undergoing hip or knee arthroplasty, without an increase in bleeding risk.
Among 24,321 patients, aged 66 years or older, the 30-day VTE event rate was 0.47% for rivaroxaban (Xarelto) (61 events) and 0.81% for low-molecular-weight heparin (LMWH) (93 events).
These findings resulted in an unadjusted relative risk of 0.58, which was statistically significant (P = .001) and did not change after adjustment for significant covariates, Dr. Alejandro Lazo-Langner said during an antithrombotic therapy session at the annual meeting of the American Society of Hematology.
There were 23 major bleeding events in both the rivaroxaban (0.18%) and LMWH (0.20%) groups. The unadjusted relative risk was 0.89 (P = .700) and did not change after adjustment in the population-based, retrospective cohort analysis.
Rivaroxaban has been the subject of numerous randomized controlled trials, but "We don’t have much real-life data in these patients," said Dr. Lazo-Langner, a hematologist specializing in thromboembolic diseases at Western University, London, Ontario, Canada.
In a meta-analysis of eight randomized rivaroxaban trials, the factor Xa inhibitor was associated with a significant 52% reduction in thrombosis after total hip or knee replacement, compared with enoxaparin (Lovenox) in roughly 14,000 patients (BMJ 2012;344:e3675 [doi:10.1136/bmj.e3675]). This came at a cost, however, of a slightly increased risk of clinically significant bleeding (relative risk, 1.25), he noted.
For the current analysis,the investigators used linked health care databases in Ontario to identify 24,321 patients who received an outpatient prescription for rivaroxaban or subcutaneous LMWH including dalteparin (Fragmin), tinzaparin (Innohep), or enoxaparin on discharge after total hip or knee arthroplasty between 2002 and 2012 across 121 hospitals.
Their average age was 74 years, 59% were women, and 12,850 received rivaroxaban. The anticoagulants were prescribed for a median of 14 days. Patients were excluded if they had other indications for anticoagulation, prosthetic mechanical heart valves, required dialysis, or lived in a long-term-care facility.
At 90 days, the VTE event rate was significantly lower in the rivaroxaban group than in the LMWH group (0.71% vs. 1.20%; adjusted RR, 0.59; P = .001). Once again, major bleeding events were similar (0.24% vs. 0.27%; adjusted RR, 0.63; P = .138), Dr. Lazo-Langner reported.
No differences were observed between the two groups at 30 or 90 days for hospitalization with endoscopy or hospitalization with major bleeding or endoscopy.
All-cause mortality was not estimable at 30 days but was lower with rivaroxaban at 90 days (16 deaths vs. 25 deaths; adjusted RR, 0.52; P = .058).
Additional analyses were conducted to test the robustness of the findings and no differences in rates of thrombosis were found when the analysis was restricted to 2009 to 2012, by type of joint replacement, or different low-molecular-weight heparins, he said.
Finally, a cost analysis was performed that showed a modest, but significant increase in direct drug costs to patients prescribed LMWH rather than rivaroxaban (Canadian $242 vs. $228; P less than .001) and home-care costs, likely from increased nursing ($1,082 vs. $959; P less than .001), Dr. Lazo-Langner said.
During a discussion of the results, he said there was no difference in novel anticoagulant use across surgeons or hospital settings, which was academic for 21% of LMWH patients and 15% of rivaroxaban patients.
Session comoderator Dr. Elaine Hylek, professor of medicine at Boston University, called this reassuring, but also urged caution in extrapolating conclusions on treatment effect outside a randomized trial.
Dr. Lazo-Langner reported research funding from Alexion, serving as a speaker for Pfizer, and honoraria from Pfizer, Leo Pharma, and Boehringer Ingelheim.
AT ASH 2013
Major finding: The 30-day VTE event rate was 0.47% for rivaroxaban and 0.81% for low molecular weight heparin (unadjusted RR, 0.58; P = .001).
Data source: Population-based, retrospective cohort study of 24,321 patients undergoing hip or knee arthroplasty
Disclosures: Dr. Lazo-Langner reported research funding from Alexion, serving as a speaker for Pfizer, and honoraria from Pfizer, Leo Pharma, and Boehringer Ingelheim.

Low event rates with real-world use of rivaroxaban in NOAC
NEW ORLEANS – Real-world use of rivaroxaban produced comparatively low rates of cardiovascular and major bleeding events when used for stroke prevention in unselected patients with atrial fibrillation.
The centrally adjudicated annual stroke rate was 1.4% among 1,194 patients given once-daily rivaroxaban (Xarelto) by 230 physicians from private practices and community hospitals across Saxony, Germany, in the prospective Dresden Novel Oral Anticoagulant (NOAC) registry.
This result falls within the annual stroke rate of 1.7% seen in the ROCKET-AF trial, which served as the basis for rivaroxaban’s November 2011 U.S. approval in this setting, Dr. Jan Beyer-Westendorf reported at the annual meeting of the American Society of Hematology.
Nonmajor, clinically relevant bleeding occurred in 21.6% of patients per year in the NOAC registry, while major bleeding was 3.4% per year.

Again, this is within the range of the phase III ROCKET-AF results (3.6% per year) and lower than the rate of up to 8% seen in the daily care of patients on vitamin K antagonists, he said. Gastrointestinal bleeding, a known side effect of rivaroxaban, was the most common major bleed. Intracranial bleeds were rare at three events.
Adherence to the oral direct factor Xa inhibitor was high at 12 months (13.7%), compared with up to 25% of patients who discontinue vitamin K antagonist therapy in the first year.
"In our population, the discontinuation rate was 14%, so basically we don’t have any concern that we put these patients at more risk in a daily care situation by treating them with rivaroxaban," said Dr. Beyer-Westendorf of the University Hospital Carl Gustav Carus, Dresden, Germany.
According to the analysis, 2,345 patients have been prospectively enrolled in the NOAC registry since October 2011; 1,194 with atrial fibrillation have been treated with anticoagulation to prevent venous thromboembolism (VTE) or stroke. No patients have been lost to follow-up, which now stands at 2,313 patient-years.
NOAC patients were slightly older at baseline than were those in ROCKET-AF (74.8 years vs. 73 years), less likely to have had prior vitamin K antagonists (37% vs. 62%), and had lower CHADS2 (Cardiac failure, Hypertension, Age, Diabetes Stroke system) scores (mean, 2.4 vs. 3.48). Higher CHADS2 scores are associated with worse outcomes, including higher rates of major bleeding.
During follow-up through October 2013, 53 NOAC patients (4.4%) experienced a major vascular event including 22 strokes, transient ischemic attacks (TIA), systemic emboli; 15 acute coronary syndromes; and four VTEs, he said. In all, 56 patients died (4.7%).
Patients on rivaroxaban 20 mg/day vs. 15 mg/day had a significantly lower annual stroke rate (0.9% vs. 2.3%; P = .052), defined as a new stroke, TIA, or systemic embolism.
Compared with those on rivaroxaban 20 mg, patients on the lower dose had higher baseline CHADS2 scores (mean, 2.8 vs. 2.2), were older (79 years vs. 73 years), more likely to have had a prior stroke (17% vs. 12.5%) or prior vitamin K antagonist therapy (40% vs. 36%), and took more concomitant drugs (mean, 6.4 vs. 5.4).
"It’s no surprise that these patients get a dose reduction because they have more comorbidities; they are at high risk of bleeding, and so it’s not a surprise that they have a slightly higher event rate," said Dr. Beyer-Westendorf, who noted that the lower dose did not reduce the risk of bleeding.
The NOAC registry is supported by scientific grants from Bayer Healthcare, Boehringer Ingelheim, and Pfizer. Dr. Beyer-Westendorf reported research funding from and serving as a speaker for these firms.
Bleeding complications detailed
In a separate presentation at ASH, Dr. Beyer-Westendorf detailed the pattern and management of bleeding complications in NOAC patients treated for stroke prevention in atrial fibrillation or VTE with rivaroxaban, dabigatran (Pradaxa), or apixaban (Eliquis).
These complications are a major concern for practitioners because there isn’t an emergency lab test available or rescue medications.
Of the 1,241 bleeding events that have occurred so far in 879 patients, 742 were minor (60%), 425 were nonmajor clinically relevant (34.2%), and 74 were major (6%).
Major bleeds per year of therapy were reported with rivaroxaban in 3.4% of patients with atrial fibrillation and in 4.4% with VTE. The major bleeding event rate with dabigatran in patients with atrial fibrillation was 2.6/100 patient-years at the 110-mg dose and 2.0/100 patient-years at the 150-mg dose. Short-term follow-up and low numbers of dabigatran and apixaban patients did not allow for sound event-rate calculations, according to Dr. Beyer-Westendorf, who stressed that no direct comparisons should be made between event rates for the different agents since the patients were in different cohorts and not in a randomized trial.
Most bleeds (93.3%) were managed conservatively with watchful waiting, compression, tamponade, or red blood cell transfusion. None of the minor bleeds and 16% (83/499) of the nonmajor clinically relevant and major bleeds required surgical or interventional treatment, he said.
Fresh frozen plasma and prothrombin complex concentrate were rarely used (seven patients each). No patient received recombinant activated factor VII.
Mortality was 0.4% for all patients with bleeding complications and 6.8% in those with major bleeds.
Death from any cause at 30 days and 90 days post bleeding occurred in 1 of 19 (5.3%) and 3 of 17 (17.6%) patients on dabigatran, respectively; in 5 of 99 (5%) and 6 of 88 (6.8%) on rivaroxaban; and in 1 of 2 (50%) at each time point for the few patients on apixaban.
With vitamin K antagonists, the case-fatality rates of major bleeding reach 14% at 90 days after bleeding leading to hospitalization and 18% within a week of discharge in atrial fibrillation patients, Dr. Beyer-Westendorf observed.
NEW ORLEANS – Real-world use of rivaroxaban produced comparatively low rates of cardiovascular and major bleeding events when used for stroke prevention in unselected patients with atrial fibrillation.
The centrally adjudicated annual stroke rate was 1.4% among 1,194 patients given once-daily rivaroxaban (Xarelto) by 230 physicians from private practices and community hospitals across Saxony, Germany, in the prospective Dresden Novel Oral Anticoagulant (NOAC) registry.
This result falls within the annual stroke rate of 1.7% seen in the ROCKET-AF trial, which served as the basis for rivaroxaban’s November 2011 U.S. approval in this setting, Dr. Jan Beyer-Westendorf reported at the annual meeting of the American Society of Hematology.
Nonmajor, clinically relevant bleeding occurred in 21.6% of patients per year in the NOAC registry, while major bleeding was 3.4% per year.
Again, this is within the range of the phase III ROCKET-AF results (3.6% per year) and lower than the rate of up to 8% seen in the daily care of patients on vitamin K antagonists, he said. Gastrointestinal bleeding, a known side effect of rivaroxaban, was the most common major bleed. Intracranial bleeds were rare at three events.
Adherence to the oral direct factor Xa inhibitor was high at 12 months (13.7%), compared with up to 25% of patients who discontinue vitamin K antagonist therapy in the first year.
"In our population, the discontinuation rate was 14%, so basically we don’t have any concern that we put these patients at more risk in a daily care situation by treating them with rivaroxaban," said Dr. Beyer-Westendorf of the University Hospital Carl Gustav Carus, Dresden, Germany.
According to the analysis, 2,345 patients have been prospectively enrolled in the NOAC registry since October 2011; 1,194 with atrial fibrillation have been treated with anticoagulation to prevent venous thromboembolism (VTE) or stroke. No patients have been lost to follow-up, which now stands at 2,313 patient-years.
NOAC patients were slightly older at baseline than were those in ROCKET-AF (74.8 years vs. 73 years), less likely to have had prior vitamin K antagonists (37% vs. 62%), and had lower CHADS2 (Cardiac failure, Hypertension, Age, Diabetes Stroke system) scores (mean, 2.4 vs. 3.48). Higher CHADS2 scores are associated with worse outcomes, including higher rates of major bleeding.
During follow-up through October 2013, 53 NOAC patients (4.4%) experienced a major vascular event including 22 strokes, transient ischemic attacks (TIA), systemic emboli; 15 acute coronary syndromes; and four VTEs, he said. In all, 56 patients died (4.7%).
Patients on rivaroxaban 20 mg/day vs. 15 mg/day had a significantly lower annual stroke rate (0.9% vs. 2.3%; P = .052), defined as a new stroke, TIA, or systemic embolism.
Compared with those on rivaroxaban 20 mg, patients on the lower dose had higher baseline CHADS2 scores (mean, 2.8 vs. 2.2), were older (79 years vs. 73 years), more likely to have had a prior stroke (17% vs. 12.5%) or prior vitamin K antagonist therapy (40% vs. 36%), and took more concomitant drugs (mean, 6.4 vs. 5.4).
"It’s no surprise that these patients get a dose reduction because they have more comorbidities; they are at high risk of bleeding, and so it’s not a surprise that they have a slightly higher event rate," said Dr. Beyer-Westendorf, who noted that the lower dose did not reduce the risk of bleeding.
The NOAC registry is supported by scientific grants from Bayer Healthcare, Boehringer Ingelheim, and Pfizer. Dr. Beyer-Westendorf reported research funding from and serving as a speaker for these firms.
Bleeding complications detailed
In a separate presentation at ASH, Dr. Beyer-Westendorf detailed the pattern and management of bleeding complications in NOAC patients treated for stroke prevention in atrial fibrillation or VTE with rivaroxaban, dabigatran (Pradaxa), or apixaban (Eliquis).
These complications are a major concern for practitioners because there isn’t an emergency lab test available or rescue medications.
Of the 1,241 bleeding events that have occurred so far in 879 patients, 742 were minor (60%), 425 were nonmajor clinically relevant (34.2%), and 74 were major (6%).
Major bleeds per year of therapy were reported with rivaroxaban in 3.4% of patients with atrial fibrillation and in 4.4% with VTE. The major bleeding event rate with dabigatran in patients with atrial fibrillation was 2.6/100 patient-years at the 110-mg dose and 2.0/100 patient-years at the 150-mg dose. Short-term follow-up and low numbers of dabigatran and apixaban patients did not allow for sound event-rate calculations, according to Dr. Beyer-Westendorf, who stressed that no direct comparisons should be made between event rates for the different agents since the patients were in different cohorts and not in a randomized trial.
Most bleeds (93.3%) were managed conservatively with watchful waiting, compression, tamponade, or red blood cell transfusion. None of the minor bleeds and 16% (83/499) of the nonmajor clinically relevant and major bleeds required surgical or interventional treatment, he said.
Fresh frozen plasma and prothrombin complex concentrate were rarely used (seven patients each). No patient received recombinant activated factor VII.
Mortality was 0.4% for all patients with bleeding complications and 6.8% in those with major bleeds.
Death from any cause at 30 days and 90 days post bleeding occurred in 1 of 19 (5.3%) and 3 of 17 (17.6%) patients on dabigatran, respectively; in 5 of 99 (5%) and 6 of 88 (6.8%) on rivaroxaban; and in 1 of 2 (50%) at each time point for the few patients on apixaban.
With vitamin K antagonists, the case-fatality rates of major bleeding reach 14% at 90 days after bleeding leading to hospitalization and 18% within a week of discharge in atrial fibrillation patients, Dr. Beyer-Westendorf observed.
NEW ORLEANS – Real-world use of rivaroxaban produced comparatively low rates of cardiovascular and major bleeding events when used for stroke prevention in unselected patients with atrial fibrillation.
The centrally adjudicated annual stroke rate was 1.4% among 1,194 patients given once-daily rivaroxaban (Xarelto) by 230 physicians from private practices and community hospitals across Saxony, Germany, in the prospective Dresden Novel Oral Anticoagulant (NOAC) registry.
This result falls within the annual stroke rate of 1.7% seen in the ROCKET-AF trial, which served as the basis for rivaroxaban’s November 2011 U.S. approval in this setting, Dr. Jan Beyer-Westendorf reported at the annual meeting of the American Society of Hematology.
Nonmajor, clinically relevant bleeding occurred in 21.6% of patients per year in the NOAC registry, while major bleeding was 3.4% per year.
Again, this is within the range of the phase III ROCKET-AF results (3.6% per year) and lower than the rate of up to 8% seen in the daily care of patients on vitamin K antagonists, he said. Gastrointestinal bleeding, a known side effect of rivaroxaban, was the most common major bleed. Intracranial bleeds were rare at three events.
Adherence to the oral direct factor Xa inhibitor was high at 12 months (13.7%), compared with up to 25% of patients who discontinue vitamin K antagonist therapy in the first year.
"In our population, the discontinuation rate was 14%, so basically we don’t have any concern that we put these patients at more risk in a daily care situation by treating them with rivaroxaban," said Dr. Beyer-Westendorf of the University Hospital Carl Gustav Carus, Dresden, Germany.
According to the analysis, 2,345 patients have been prospectively enrolled in the NOAC registry since October 2011; 1,194 with atrial fibrillation have been treated with anticoagulation to prevent venous thromboembolism (VTE) or stroke. No patients have been lost to follow-up, which now stands at 2,313 patient-years.
NOAC patients were slightly older at baseline than were those in ROCKET-AF (74.8 years vs. 73 years), less likely to have had prior vitamin K antagonists (37% vs. 62%), and had lower CHADS2 (Cardiac failure, Hypertension, Age, Diabetes Stroke system) scores (mean, 2.4 vs. 3.48). Higher CHADS2 scores are associated with worse outcomes, including higher rates of major bleeding.
During follow-up through October 2013, 53 NOAC patients (4.4%) experienced a major vascular event including 22 strokes, transient ischemic attacks (TIA), systemic emboli; 15 acute coronary syndromes; and four VTEs, he said. In all, 56 patients died (4.7%).
Patients on rivaroxaban 20 mg/day vs. 15 mg/day had a significantly lower annual stroke rate (0.9% vs. 2.3%; P = .052), defined as a new stroke, TIA, or systemic embolism.
Compared with those on rivaroxaban 20 mg, patients on the lower dose had higher baseline CHADS2 scores (mean, 2.8 vs. 2.2), were older (79 years vs. 73 years), more likely to have had a prior stroke (17% vs. 12.5%) or prior vitamin K antagonist therapy (40% vs. 36%), and took more concomitant drugs (mean, 6.4 vs. 5.4).
"It’s no surprise that these patients get a dose reduction because they have more comorbidities; they are at high risk of bleeding, and so it’s not a surprise that they have a slightly higher event rate," said Dr. Beyer-Westendorf, who noted that the lower dose did not reduce the risk of bleeding.
The NOAC registry is supported by scientific grants from Bayer Healthcare, Boehringer Ingelheim, and Pfizer. Dr. Beyer-Westendorf reported research funding from and serving as a speaker for these firms.
Bleeding complications detailed
In a separate presentation at ASH, Dr. Beyer-Westendorf detailed the pattern and management of bleeding complications in NOAC patients treated for stroke prevention in atrial fibrillation or VTE with rivaroxaban, dabigatran (Pradaxa), or apixaban (Eliquis).
These complications are a major concern for practitioners because there isn’t an emergency lab test available or rescue medications.
Of the 1,241 bleeding events that have occurred so far in 879 patients, 742 were minor (60%), 425 were nonmajor clinically relevant (34.2%), and 74 were major (6%).
Major bleeds per year of therapy were reported with rivaroxaban in 3.4% of patients with atrial fibrillation and in 4.4% with VTE. The major bleeding event rate with dabigatran in patients with atrial fibrillation was 2.6/100 patient-years at the 110-mg dose and 2.0/100 patient-years at the 150-mg dose. Short-term follow-up and low numbers of dabigatran and apixaban patients did not allow for sound event-rate calculations, according to Dr. Beyer-Westendorf, who stressed that no direct comparisons should be made between event rates for the different agents since the patients were in different cohorts and not in a randomized trial.
Most bleeds (93.3%) were managed conservatively with watchful waiting, compression, tamponade, or red blood cell transfusion. None of the minor bleeds and 16% (83/499) of the nonmajor clinically relevant and major bleeds required surgical or interventional treatment, he said.
Fresh frozen plasma and prothrombin complex concentrate were rarely used (seven patients each). No patient received recombinant activated factor VII.
Mortality was 0.4% for all patients with bleeding complications and 6.8% in those with major bleeds.
Death from any cause at 30 days and 90 days post bleeding occurred in 1 of 19 (5.3%) and 3 of 17 (17.6%) patients on dabigatran, respectively; in 5 of 99 (5%) and 6 of 88 (6.8%) on rivaroxaban; and in 1 of 2 (50%) at each time point for the few patients on apixaban.
With vitamin K antagonists, the case-fatality rates of major bleeding reach 14% at 90 days after bleeding leading to hospitalization and 18% within a week of discharge in atrial fibrillation patients, Dr. Beyer-Westendorf observed.
AT ASH 2013
Major finding: The annual stroke rate with once-daily rivaroxaban was 1.4% and the major bleeding rate was 3.4% among 1,194 patients with atrial fibrillation.
Data source: A prospective database of 2,345 patients treated with anticoagulation.
Disclosures: The NOAC registry is supported by scientific grants from Bayer Healthcare, Boehringer Ingelheim, and Pfizer. Dr. Beyer-Westendorf reported research funding from and serving as a speaker for these firms.

ONO-4059 makes waves in heavily pretreated CLL
NEW ORLEANS – Early data suggest that the second-generation oral BTK inhibitor ONO-4059 may give ibrutinib a run for its money in chronic lymphocytic leukemia.
The response rate to ONO-4059 monotherapy was 89% overall and 71% in those with the deleterious 17p deletion among 18 heavily pretreated patients with relapsed/refractory or high-risk CLL in a phase I, dose-escalation study.
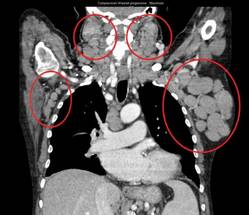
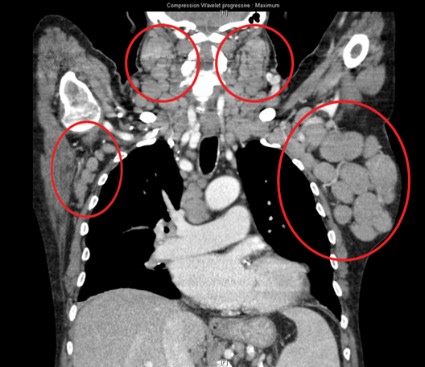
Patients had already received a median of three prior therapies, including rituximab (84%) and fludarabine (95%), and had no higher priority therapy available to them, said Dr. Gilles Salles of Hospices Civils de Lyon (France), Universite Claude Bernard Lyon.
All patients had improved hemoglobin and platelet counts after 3 months on treatment and rapid reductions in lymph node size within the first 28-day cycle. Tumor burden was reduced by 50% for most patients, and all but one patient experienced a response that was detectable on a CT scan.
"This was true whatever their FISH status or 17p or 11q deletion status," Dr. Salles said at the annual meeting of the American Society of Hematology.
ONO-4059 is a highly selective Bruton’s tyrosine kinase (BTK) inhibitor with antitumor activity in several preclinical models.
No patients had received prior treatment with a P13 kinase or a BTK inhibitor, including ibrutinib (Imbruvica), which recently gained accelerated approval for previously treated mantle cell lymphoma.
ONO-4059 was given at daily doses ranging from 20 mg to 320 mg for up to 6 months, with the option of additional dosing up to 2 years. Sustained BTK inhibition was established at doses of 40 mg and higher.
Overall, the best response was a partial response in 14 patients, as well as two partial responses with lymphocytosis and one stable disease, he said. No complete responses occurred.
One patient progressed roughly 1 month after showing an initial response and complete disappearance of all palpable disease on physical exam. Richter’s syndrome was suspected.
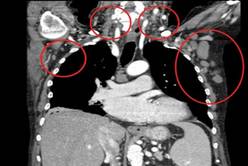
"It’s very promising efficacy in this highly pretreated population," Dr. Salles said.
Patients with relapsed/refractory mantle cell lymphoma and diffuse large B-cell lymphoma, especially the ABC subtype, also appear sensitive to ONO-4059. Overall response rates were 43% and 75%, respectively, including three complete responses reported from the phase I study in a separate poster presentation at the meeting.
ONO-4059 had a favorable safety profile with a single dose-limiting toxicity observed in a patient who had Waldenstrom’s macroglobulinemia, was on the 320-mg dose, and was intolerant to all prior therapies. The maximum tolerated dose has not yet been reached.
The majority of adverse events in the CLL patients were grades 1 and 2. There were no clinically significant bleeding events or bruising, and there was a low incidence of diarrhea and rash, Dr. Salles said.
ONO-4059–related grade 3-4 events were independent of dose and included one grade 3 neutropenia at 20 mg and two grade 4 events at 20 mg and 320 mg. Four serious adverse events (febrile neutropenia, pyrexia, rash, and neutropenia) occurred in three patients, all of whom are still in the study and showing good clinical response, Dr. Salles said. Of the 30 patients dosed to date, 22 remain in the study.
No other trials are firmly planned, and pharmacokinetics/pharmacodynamics data continue to be explored in order to assess a phase II dosage, he said in an interview.
Dr. Salles reported consulting for and receiving honoraria from Roche. Several coauthors have financial ties, including employment with the study sponsor, Ono Pharmaceutical, which is developing ONO-4059.
NEW ORLEANS – Early data suggest that the second-generation oral BTK inhibitor ONO-4059 may give ibrutinib a run for its money in chronic lymphocytic leukemia.
The response rate to ONO-4059 monotherapy was 89% overall and 71% in those with the deleterious 17p deletion among 18 heavily pretreated patients with relapsed/refractory or high-risk CLL in a phase I, dose-escalation study.
Patients had already received a median of three prior therapies, including rituximab (84%) and fludarabine (95%), and had no higher priority therapy available to them, said Dr. Gilles Salles of Hospices Civils de Lyon (France), Universite Claude Bernard Lyon.
All patients had improved hemoglobin and platelet counts after 3 months on treatment and rapid reductions in lymph node size within the first 28-day cycle. Tumor burden was reduced by 50% for most patients, and all but one patient experienced a response that was detectable on a CT scan.
"This was true whatever their FISH status or 17p or 11q deletion status," Dr. Salles said at the annual meeting of the American Society of Hematology.
ONO-4059 is a highly selective Bruton’s tyrosine kinase (BTK) inhibitor with antitumor activity in several preclinical models.
No patients had received prior treatment with a P13 kinase or a BTK inhibitor, including ibrutinib (Imbruvica), which recently gained accelerated approval for previously treated mantle cell lymphoma.
ONO-4059 was given at daily doses ranging from 20 mg to 320 mg for up to 6 months, with the option of additional dosing up to 2 years. Sustained BTK inhibition was established at doses of 40 mg and higher.
Overall, the best response was a partial response in 14 patients, as well as two partial responses with lymphocytosis and one stable disease, he said. No complete responses occurred.
One patient progressed roughly 1 month after showing an initial response and complete disappearance of all palpable disease on physical exam. Richter’s syndrome was suspected.
"It’s very promising efficacy in this highly pretreated population," Dr. Salles said.
Patients with relapsed/refractory mantle cell lymphoma and diffuse large B-cell lymphoma, especially the ABC subtype, also appear sensitive to ONO-4059. Overall response rates were 43% and 75%, respectively, including three complete responses reported from the phase I study in a separate poster presentation at the meeting.
ONO-4059 had a favorable safety profile with a single dose-limiting toxicity observed in a patient who had Waldenstrom’s macroglobulinemia, was on the 320-mg dose, and was intolerant to all prior therapies. The maximum tolerated dose has not yet been reached.
The majority of adverse events in the CLL patients were grades 1 and 2. There were no clinically significant bleeding events or bruising, and there was a low incidence of diarrhea and rash, Dr. Salles said.
ONO-4059–related grade 3-4 events were independent of dose and included one grade 3 neutropenia at 20 mg and two grade 4 events at 20 mg and 320 mg. Four serious adverse events (febrile neutropenia, pyrexia, rash, and neutropenia) occurred in three patients, all of whom are still in the study and showing good clinical response, Dr. Salles said. Of the 30 patients dosed to date, 22 remain in the study.
No other trials are firmly planned, and pharmacokinetics/pharmacodynamics data continue to be explored in order to assess a phase II dosage, he said in an interview.
Dr. Salles reported consulting for and receiving honoraria from Roche. Several coauthors have financial ties, including employment with the study sponsor, Ono Pharmaceutical, which is developing ONO-4059.
NEW ORLEANS – Early data suggest that the second-generation oral BTK inhibitor ONO-4059 may give ibrutinib a run for its money in chronic lymphocytic leukemia.
The response rate to ONO-4059 monotherapy was 89% overall and 71% in those with the deleterious 17p deletion among 18 heavily pretreated patients with relapsed/refractory or high-risk CLL in a phase I, dose-escalation study.
Patients had already received a median of three prior therapies, including rituximab (84%) and fludarabine (95%), and had no higher priority therapy available to them, said Dr. Gilles Salles of Hospices Civils de Lyon (France), Universite Claude Bernard Lyon.
All patients had improved hemoglobin and platelet counts after 3 months on treatment and rapid reductions in lymph node size within the first 28-day cycle. Tumor burden was reduced by 50% for most patients, and all but one patient experienced a response that was detectable on a CT scan.
"This was true whatever their FISH status or 17p or 11q deletion status," Dr. Salles said at the annual meeting of the American Society of Hematology.
ONO-4059 is a highly selective Bruton’s tyrosine kinase (BTK) inhibitor with antitumor activity in several preclinical models.
No patients had received prior treatment with a P13 kinase or a BTK inhibitor, including ibrutinib (Imbruvica), which recently gained accelerated approval for previously treated mantle cell lymphoma.
ONO-4059 was given at daily doses ranging from 20 mg to 320 mg for up to 6 months, with the option of additional dosing up to 2 years. Sustained BTK inhibition was established at doses of 40 mg and higher.
Overall, the best response was a partial response in 14 patients, as well as two partial responses with lymphocytosis and one stable disease, he said. No complete responses occurred.
One patient progressed roughly 1 month after showing an initial response and complete disappearance of all palpable disease on physical exam. Richter’s syndrome was suspected.
"It’s very promising efficacy in this highly pretreated population," Dr. Salles said.
Patients with relapsed/refractory mantle cell lymphoma and diffuse large B-cell lymphoma, especially the ABC subtype, also appear sensitive to ONO-4059. Overall response rates were 43% and 75%, respectively, including three complete responses reported from the phase I study in a separate poster presentation at the meeting.
ONO-4059 had a favorable safety profile with a single dose-limiting toxicity observed in a patient who had Waldenstrom’s macroglobulinemia, was on the 320-mg dose, and was intolerant to all prior therapies. The maximum tolerated dose has not yet been reached.
The majority of adverse events in the CLL patients were grades 1 and 2. There were no clinically significant bleeding events or bruising, and there was a low incidence of diarrhea and rash, Dr. Salles said.
ONO-4059–related grade 3-4 events were independent of dose and included one grade 3 neutropenia at 20 mg and two grade 4 events at 20 mg and 320 mg. Four serious adverse events (febrile neutropenia, pyrexia, rash, and neutropenia) occurred in three patients, all of whom are still in the study and showing good clinical response, Dr. Salles said. Of the 30 patients dosed to date, 22 remain in the study.
No other trials are firmly planned, and pharmacokinetics/pharmacodynamics data continue to be explored in order to assess a phase II dosage, he said in an interview.
Dr. Salles reported consulting for and receiving honoraria from Roche. Several coauthors have financial ties, including employment with the study sponsor, Ono Pharmaceutical, which is developing ONO-4059.
AT ASH 2013
Major finding: The response rate was 89% overall and 71% for patients with 17p deletion.
Data source: A prospective, phase I dose-escalation study in 18 patients with relapsed/refractory or high-risk CLL.
Disclosures: Dr. Salles reported honoraria from Janssen, Gilead, and Celgene. Several coauthors have financial ties, including employment with the study sponsor, Ono Pharmaceutical, which is developing ONO-4059.
Lower-dose quizartinib diminishes QT events
NEW ORLEANS – Lower doses of quizartinib reduced worrisome QT-interval prolongation events without a loss of efficacy in patients with FLT3-ITD–positive relapsed or refractory acute myeloid leukemia, a phase II study shows.
In a 76-patient study, grade 2 QT-interval prolongation (QTcF) of more than 480-500 msec occurred in two patients (5%) on oral quizartinib 30 mg/day and in five patients (14%) on 60 mg/day, with no differences between groups in QTcF events of more than 500 msec (5% vs. 3%).
In addition, an increase in QTcF from baseline of more than 60 msec was seen in 19% of patients on the 60-mg dose and in 3% of those on the 30-mg dose, Dr. Jorge Cortes reported at the annual meeting of the American Society of Hematology.
About a third of patients with acute myeloid leukemia (AML) will have FLT3 internal tandem duplications (FLT3-ITD), which are associated with early relapse and poor survival in AML. Quizartinib has shown the highest single-agent activity among FMS-like tyrosine kinase 3 (FLT3)-targeted agents in this population, according to Dr. Cortes.
At last year’s ASH meeting, investigators presented results from a phase II study in which the investigational agent elicited responses in both FLT3-positive and -negative relapsed/refractory AML. The unprecedented results at doses of 90, 135, and 200 mg were partially eclipsed, however, by respective 46%, 39%, and 92% increases in QTcF from baseline of more than 60 msec, noted Dr. Cortes, chair of the AML section, department of leukemia, University of Texas M.D. Anderson Cancer Center, Houston.
The current study randomized 76 patients to quizartinib 30 mg or 60 mg continuous daily dosing for primary AML or AML secondary to myelodysplastic syndrome that relapsed or was refractory to first-line salvage therapy or prior hematopoietic stem cell transplantation. The coprimary endpoints were rate of grade 2 QTc prolongation and the composite complete remission rate, which included complete remission (CR), CR with incomplete platelet recovery, and CR with incomplete hematologic recovery.
In all, 92% of patients had FLT3 internal tandem duplications, and 58 of 60 evaluable patients had intermediate or poor cytogenetic risk. Their mean age was 55 years. Two patients were randomized but not treated.
Treatment with the 30-mg and 60-mg doses resulted in a composite CR rate of 47%, Dr. Cortes said. The median duration of response was 4.1 weeks in the 30-mg group and 20 weeks in the 60-mg group.
Two patients (5%) on the 30-mg dose and 1 patient (3%) on the 60-mg dose achieved CR; 1 patient (3%) in the 60-mg group had a CR with incomplete platelet recovery; and 16 patients (42%) in each arm had a CR with incomplete hematologic recovery.
Partial responses were also seen in 5 patients (13%) in the 30-mg group and 9 (24%) in the 60-mg group.
These results compare favorably with composite CR rates of 47%, 45%, and 42% with the 90-, 135-, and 200-mg doses used in the earlier study, Dr. Cortes observed.
Median overall survival in the current study was 20.7 weeks in the lower-dose group and 25.4 weeks with the 60-mg dose.
Importantly, 34% of patients were successfully bridged to transplant, extending median survival to 31 weeks for those on 30 mg of quizartinib and to 28.1 weeks for those given 60 mg.
"This study demonstrates there is certainly sustained efficacy with these lower doses of quizartinib and a decreased QT signal at doses of 30 and 60 mg compared with the higher doses we’ve tested in the past," Dr. Cortes concluded.
Grade 3/4 adverse events were mainly anemia (39%) in the 30-mg group and febrile neutropenia (36%) in the 60-mg group. Three patients required dose reductions due to QTc prolongation.
A global phase III randomized study of quizartinib in FLT3-ITD–positive patients in first relapse is planned to start in early 2014, he said.
Development of quizartinib has been somewhat rocky, with Astellas Pharma announcing in March 2013 it was ending its collaboration with Ambit Biosciences to develop FLT3 inhibitors including quizartinib.
Dr. Cortes reported research funding from Astellas Pharma, Arog, Novartis, and Ambit Biosciences, which is developing quizartinib, and consulting for Astellas, Arog, and Ambit.
NEW ORLEANS – Lower doses of quizartinib reduced worrisome QT-interval prolongation events without a loss of efficacy in patients with FLT3-ITD–positive relapsed or refractory acute myeloid leukemia, a phase II study shows.
In a 76-patient study, grade 2 QT-interval prolongation (QTcF) of more than 480-500 msec occurred in two patients (5%) on oral quizartinib 30 mg/day and in five patients (14%) on 60 mg/day, with no differences between groups in QTcF events of more than 500 msec (5% vs. 3%).
In addition, an increase in QTcF from baseline of more than 60 msec was seen in 19% of patients on the 60-mg dose and in 3% of those on the 30-mg dose, Dr. Jorge Cortes reported at the annual meeting of the American Society of Hematology.
About a third of patients with acute myeloid leukemia (AML) will have FLT3 internal tandem duplications (FLT3-ITD), which are associated with early relapse and poor survival in AML. Quizartinib has shown the highest single-agent activity among FMS-like tyrosine kinase 3 (FLT3)-targeted agents in this population, according to Dr. Cortes.
At last year’s ASH meeting, investigators presented results from a phase II study in which the investigational agent elicited responses in both FLT3-positive and -negative relapsed/refractory AML. The unprecedented results at doses of 90, 135, and 200 mg were partially eclipsed, however, by respective 46%, 39%, and 92% increases in QTcF from baseline of more than 60 msec, noted Dr. Cortes, chair of the AML section, department of leukemia, University of Texas M.D. Anderson Cancer Center, Houston.
The current study randomized 76 patients to quizartinib 30 mg or 60 mg continuous daily dosing for primary AML or AML secondary to myelodysplastic syndrome that relapsed or was refractory to first-line salvage therapy or prior hematopoietic stem cell transplantation. The coprimary endpoints were rate of grade 2 QTc prolongation and the composite complete remission rate, which included complete remission (CR), CR with incomplete platelet recovery, and CR with incomplete hematologic recovery.
In all, 92% of patients had FLT3 internal tandem duplications, and 58 of 60 evaluable patients had intermediate or poor cytogenetic risk. Their mean age was 55 years. Two patients were randomized but not treated.
Treatment with the 30-mg and 60-mg doses resulted in a composite CR rate of 47%, Dr. Cortes said. The median duration of response was 4.1 weeks in the 30-mg group and 20 weeks in the 60-mg group.
Two patients (5%) on the 30-mg dose and 1 patient (3%) on the 60-mg dose achieved CR; 1 patient (3%) in the 60-mg group had a CR with incomplete platelet recovery; and 16 patients (42%) in each arm had a CR with incomplete hematologic recovery.
Partial responses were also seen in 5 patients (13%) in the 30-mg group and 9 (24%) in the 60-mg group.
These results compare favorably with composite CR rates of 47%, 45%, and 42% with the 90-, 135-, and 200-mg doses used in the earlier study, Dr. Cortes observed.
Median overall survival in the current study was 20.7 weeks in the lower-dose group and 25.4 weeks with the 60-mg dose.
Importantly, 34% of patients were successfully bridged to transplant, extending median survival to 31 weeks for those on 30 mg of quizartinib and to 28.1 weeks for those given 60 mg.
"This study demonstrates there is certainly sustained efficacy with these lower doses of quizartinib and a decreased QT signal at doses of 30 and 60 mg compared with the higher doses we’ve tested in the past," Dr. Cortes concluded.
Grade 3/4 adverse events were mainly anemia (39%) in the 30-mg group and febrile neutropenia (36%) in the 60-mg group. Three patients required dose reductions due to QTc prolongation.
A global phase III randomized study of quizartinib in FLT3-ITD–positive patients in first relapse is planned to start in early 2014, he said.
Development of quizartinib has been somewhat rocky, with Astellas Pharma announcing in March 2013 it was ending its collaboration with Ambit Biosciences to develop FLT3 inhibitors including quizartinib.
Dr. Cortes reported research funding from Astellas Pharma, Arog, Novartis, and Ambit Biosciences, which is developing quizartinib, and consulting for Astellas, Arog, and Ambit.
NEW ORLEANS – Lower doses of quizartinib reduced worrisome QT-interval prolongation events without a loss of efficacy in patients with FLT3-ITD–positive relapsed or refractory acute myeloid leukemia, a phase II study shows.
In a 76-patient study, grade 2 QT-interval prolongation (QTcF) of more than 480-500 msec occurred in two patients (5%) on oral quizartinib 30 mg/day and in five patients (14%) on 60 mg/day, with no differences between groups in QTcF events of more than 500 msec (5% vs. 3%).
In addition, an increase in QTcF from baseline of more than 60 msec was seen in 19% of patients on the 60-mg dose and in 3% of those on the 30-mg dose, Dr. Jorge Cortes reported at the annual meeting of the American Society of Hematology.
About a third of patients with acute myeloid leukemia (AML) will have FLT3 internal tandem duplications (FLT3-ITD), which are associated with early relapse and poor survival in AML. Quizartinib has shown the highest single-agent activity among FMS-like tyrosine kinase 3 (FLT3)-targeted agents in this population, according to Dr. Cortes.
At last year’s ASH meeting, investigators presented results from a phase II study in which the investigational agent elicited responses in both FLT3-positive and -negative relapsed/refractory AML. The unprecedented results at doses of 90, 135, and 200 mg were partially eclipsed, however, by respective 46%, 39%, and 92% increases in QTcF from baseline of more than 60 msec, noted Dr. Cortes, chair of the AML section, department of leukemia, University of Texas M.D. Anderson Cancer Center, Houston.
The current study randomized 76 patients to quizartinib 30 mg or 60 mg continuous daily dosing for primary AML or AML secondary to myelodysplastic syndrome that relapsed or was refractory to first-line salvage therapy or prior hematopoietic stem cell transplantation. The coprimary endpoints were rate of grade 2 QTc prolongation and the composite complete remission rate, which included complete remission (CR), CR with incomplete platelet recovery, and CR with incomplete hematologic recovery.
In all, 92% of patients had FLT3 internal tandem duplications, and 58 of 60 evaluable patients had intermediate or poor cytogenetic risk. Their mean age was 55 years. Two patients were randomized but not treated.
Treatment with the 30-mg and 60-mg doses resulted in a composite CR rate of 47%, Dr. Cortes said. The median duration of response was 4.1 weeks in the 30-mg group and 20 weeks in the 60-mg group.
Two patients (5%) on the 30-mg dose and 1 patient (3%) on the 60-mg dose achieved CR; 1 patient (3%) in the 60-mg group had a CR with incomplete platelet recovery; and 16 patients (42%) in each arm had a CR with incomplete hematologic recovery.
Partial responses were also seen in 5 patients (13%) in the 30-mg group and 9 (24%) in the 60-mg group.
These results compare favorably with composite CR rates of 47%, 45%, and 42% with the 90-, 135-, and 200-mg doses used in the earlier study, Dr. Cortes observed.
Median overall survival in the current study was 20.7 weeks in the lower-dose group and 25.4 weeks with the 60-mg dose.
Importantly, 34% of patients were successfully bridged to transplant, extending median survival to 31 weeks for those on 30 mg of quizartinib and to 28.1 weeks for those given 60 mg.
"This study demonstrates there is certainly sustained efficacy with these lower doses of quizartinib and a decreased QT signal at doses of 30 and 60 mg compared with the higher doses we’ve tested in the past," Dr. Cortes concluded.
Grade 3/4 adverse events were mainly anemia (39%) in the 30-mg group and febrile neutropenia (36%) in the 60-mg group. Three patients required dose reductions due to QTc prolongation.
A global phase III randomized study of quizartinib in FLT3-ITD–positive patients in first relapse is planned to start in early 2014, he said.
Development of quizartinib has been somewhat rocky, with Astellas Pharma announcing in March 2013 it was ending its collaboration with Ambit Biosciences to develop FLT3 inhibitors including quizartinib.
Dr. Cortes reported research funding from Astellas Pharma, Arog, Novartis, and Ambit Biosciences, which is developing quizartinib, and consulting for Astellas, Arog, and Ambit.
AT ASH 2013
Major finding: An increase in QTcF from baseline of more than 60 msec was seen in 19% of patients on quizartinib 60 mg and in 3% of those on 30 mg.
Data source: A prospective phase II study of 76 patients with relapsed/refractory AML.
Disclosures: Dr. Cortes reported research funding from Astellas Pharma, Arog, Novartis, and Ambit Biosciences, which is developing quizartinib, and consulting for Astellas, Arog, and Ambit.
Anticoagulation reaped survival benefit in leukemia patients with DVT
NEW ORLEANS – Continued anticoagulation proved surprisingly beneficial in acute leukemia patients with catheter-related deep vein thrombosis, based on a small retrospective study.
Significantly more patients on anticoagulation had DVT improvement than did those without anticoagulation (17/20 vs. 5/15; P = .03), but they also had significantly better survival (4/20 vs. 4/15).
Median survival has not been reached in the anticoagulation group and was 9 months in controls after a median follow-up of 6 months (P = .02), coauthors Dr. Briana Short and Dr. Nora Oliver reported at the annual meeting of the American Society of Hematology.
The study is one of few to tackle the risks and benefits of anticoagulation in leukemia patients who develop catheter-related DVTs.
There are no currently available guidelines, and the issue remains controversial because catheters increase the risk of DVT and pulmonary embolism, but anticoagulation raises the risk of bleeding in leukemia patients, particularly those with thrombocytopenia, the investigators noted. At many hospitals, the catheter is temporarily removed until the DVT resolves, but this can delay treatment and carries added risks with reinsertion of a central venous catheter.
Nonfatal bleeding events were more common with anticoagulation than without it, but the difference did not reach significance (5 patients vs. 1 patient; P = .21), according to Dr. Short and Dr. Oliver, chief residents at the University of Maryland, Baltimore.
The mechanism behind the increased survival is unknown, but it may be that leukemia patients who experience a DVT have microthrombi or inflammation after chemotherapy, said senior author and colleague Dr. Ashkan Emadi, who developed the novel strategy and also is at the university. A more favorable risk profile among anticoagulated patients was also a very real possibility, prompting the researchers to perform a slew of sensitivity analyses.
"We excluded people with APL [acute promyelocytic leukemia], and the data were still the same. We excluded people with bad cytogenetics, and the data still showed an overall survival benefit," he said in an interview. "We did a lot of rigorous, stingy sensitivity analyses, but wherever we looked, the survival advantage was still there."
The study, which attracted a crowd during the poster session, included 37 patients with acute leukemia who developed a DVT associated with a PICC (peripherally inserted central catheter) line. Half of these occurred within 18 days of catheter placement.
During hospitalization, 21 of the 22 patients in the anticoagulation group were started on unfractionated heparin or low-molecular-weight heparin (LMWH), with the remaining patient started on anticoagulation upon discharge. Two patients were anticoagulated with fondaparinux (Arixtra) and excluded from the analysis.
Two of the 15 controls were initially started on anticoagulation, but the therapies were discontinued during their hospital stay.
At discharge, 7 patients received enoxaparin (Lovenox) LMWH 0.5-0.75 mg/kg per day, and 13 received enoxaparin 1.0-1.5 mg/kg per day, both for 3 months. Patients were maintained at a platelet count of 50 x 103/mcL, and received platelet transfusions to decrease the risk of bleeding, if counts fell below this level. Controls were monitored post discharge by the treating physician without receiving any anticoagulation.
At baseline, the anticoagulated patients and controls were similar with respect to age (median, 56 vs. 51 years); presence of acute myeloid leukemia (12 each), acute promyelocytic leukemia (6 vs. 2 patients), or acute lymphoblastic leukemia (2 vs. 1 patient); poor-risk karyotype (5 vs. 4 patients); initial median white cell count (8.7 x 103/mcL vs. 7.6 x 103/mcL); and initial median platelet count (59 x 103/mcL vs. 45 x 103/mcL).
Though provocative, the findings need to be replicated in a prospective study, currently under discussion with researchers from Johns Hopkins Hospital, Baltimore, and Massachusetts General Hospital, Boston, Dr. Emadi said.
The authors reported having no financial disclosures.
NEW ORLEANS – Continued anticoagulation proved surprisingly beneficial in acute leukemia patients with catheter-related deep vein thrombosis, based on a small retrospective study.
Significantly more patients on anticoagulation had DVT improvement than did those without anticoagulation (17/20 vs. 5/15; P = .03), but they also had significantly better survival (4/20 vs. 4/15).
Median survival has not been reached in the anticoagulation group and was 9 months in controls after a median follow-up of 6 months (P = .02), coauthors Dr. Briana Short and Dr. Nora Oliver reported at the annual meeting of the American Society of Hematology.
The study is one of few to tackle the risks and benefits of anticoagulation in leukemia patients who develop catheter-related DVTs.
There are no currently available guidelines, and the issue remains controversial because catheters increase the risk of DVT and pulmonary embolism, but anticoagulation raises the risk of bleeding in leukemia patients, particularly those with thrombocytopenia, the investigators noted. At many hospitals, the catheter is temporarily removed until the DVT resolves, but this can delay treatment and carries added risks with reinsertion of a central venous catheter.
Nonfatal bleeding events were more common with anticoagulation than without it, but the difference did not reach significance (5 patients vs. 1 patient; P = .21), according to Dr. Short and Dr. Oliver, chief residents at the University of Maryland, Baltimore.
The mechanism behind the increased survival is unknown, but it may be that leukemia patients who experience a DVT have microthrombi or inflammation after chemotherapy, said senior author and colleague Dr. Ashkan Emadi, who developed the novel strategy and also is at the university. A more favorable risk profile among anticoagulated patients was also a very real possibility, prompting the researchers to perform a slew of sensitivity analyses.
"We excluded people with APL [acute promyelocytic leukemia], and the data were still the same. We excluded people with bad cytogenetics, and the data still showed an overall survival benefit," he said in an interview. "We did a lot of rigorous, stingy sensitivity analyses, but wherever we looked, the survival advantage was still there."
The study, which attracted a crowd during the poster session, included 37 patients with acute leukemia who developed a DVT associated with a PICC (peripherally inserted central catheter) line. Half of these occurred within 18 days of catheter placement.
During hospitalization, 21 of the 22 patients in the anticoagulation group were started on unfractionated heparin or low-molecular-weight heparin (LMWH), with the remaining patient started on anticoagulation upon discharge. Two patients were anticoagulated with fondaparinux (Arixtra) and excluded from the analysis.
Two of the 15 controls were initially started on anticoagulation, but the therapies were discontinued during their hospital stay.
At discharge, 7 patients received enoxaparin (Lovenox) LMWH 0.5-0.75 mg/kg per day, and 13 received enoxaparin 1.0-1.5 mg/kg per day, both for 3 months. Patients were maintained at a platelet count of 50 x 103/mcL, and received platelet transfusions to decrease the risk of bleeding, if counts fell below this level. Controls were monitored post discharge by the treating physician without receiving any anticoagulation.
At baseline, the anticoagulated patients and controls were similar with respect to age (median, 56 vs. 51 years); presence of acute myeloid leukemia (12 each), acute promyelocytic leukemia (6 vs. 2 patients), or acute lymphoblastic leukemia (2 vs. 1 patient); poor-risk karyotype (5 vs. 4 patients); initial median white cell count (8.7 x 103/mcL vs. 7.6 x 103/mcL); and initial median platelet count (59 x 103/mcL vs. 45 x 103/mcL).
Though provocative, the findings need to be replicated in a prospective study, currently under discussion with researchers from Johns Hopkins Hospital, Baltimore, and Massachusetts General Hospital, Boston, Dr. Emadi said.
The authors reported having no financial disclosures.
NEW ORLEANS – Continued anticoagulation proved surprisingly beneficial in acute leukemia patients with catheter-related deep vein thrombosis, based on a small retrospective study.
Significantly more patients on anticoagulation had DVT improvement than did those without anticoagulation (17/20 vs. 5/15; P = .03), but they also had significantly better survival (4/20 vs. 4/15).
Median survival has not been reached in the anticoagulation group and was 9 months in controls after a median follow-up of 6 months (P = .02), coauthors Dr. Briana Short and Dr. Nora Oliver reported at the annual meeting of the American Society of Hematology.
The study is one of few to tackle the risks and benefits of anticoagulation in leukemia patients who develop catheter-related DVTs.
There are no currently available guidelines, and the issue remains controversial because catheters increase the risk of DVT and pulmonary embolism, but anticoagulation raises the risk of bleeding in leukemia patients, particularly those with thrombocytopenia, the investigators noted. At many hospitals, the catheter is temporarily removed until the DVT resolves, but this can delay treatment and carries added risks with reinsertion of a central venous catheter.
Nonfatal bleeding events were more common with anticoagulation than without it, but the difference did not reach significance (5 patients vs. 1 patient; P = .21), according to Dr. Short and Dr. Oliver, chief residents at the University of Maryland, Baltimore.
The mechanism behind the increased survival is unknown, but it may be that leukemia patients who experience a DVT have microthrombi or inflammation after chemotherapy, said senior author and colleague Dr. Ashkan Emadi, who developed the novel strategy and also is at the university. A more favorable risk profile among anticoagulated patients was also a very real possibility, prompting the researchers to perform a slew of sensitivity analyses.
"We excluded people with APL [acute promyelocytic leukemia], and the data were still the same. We excluded people with bad cytogenetics, and the data still showed an overall survival benefit," he said in an interview. "We did a lot of rigorous, stingy sensitivity analyses, but wherever we looked, the survival advantage was still there."
The study, which attracted a crowd during the poster session, included 37 patients with acute leukemia who developed a DVT associated with a PICC (peripherally inserted central catheter) line. Half of these occurred within 18 days of catheter placement.
During hospitalization, 21 of the 22 patients in the anticoagulation group were started on unfractionated heparin or low-molecular-weight heparin (LMWH), with the remaining patient started on anticoagulation upon discharge. Two patients were anticoagulated with fondaparinux (Arixtra) and excluded from the analysis.
Two of the 15 controls were initially started on anticoagulation, but the therapies were discontinued during their hospital stay.
At discharge, 7 patients received enoxaparin (Lovenox) LMWH 0.5-0.75 mg/kg per day, and 13 received enoxaparin 1.0-1.5 mg/kg per day, both for 3 months. Patients were maintained at a platelet count of 50 x 103/mcL, and received platelet transfusions to decrease the risk of bleeding, if counts fell below this level. Controls were monitored post discharge by the treating physician without receiving any anticoagulation.
At baseline, the anticoagulated patients and controls were similar with respect to age (median, 56 vs. 51 years); presence of acute myeloid leukemia (12 each), acute promyelocytic leukemia (6 vs. 2 patients), or acute lymphoblastic leukemia (2 vs. 1 patient); poor-risk karyotype (5 vs. 4 patients); initial median white cell count (8.7 x 103/mcL vs. 7.6 x 103/mcL); and initial median platelet count (59 x 103/mcL vs. 45 x 103/mcL).
Though provocative, the findings need to be replicated in a prospective study, currently under discussion with researchers from Johns Hopkins Hospital, Baltimore, and Massachusetts General Hospital, Boston, Dr. Emadi said.
The authors reported having no financial disclosures.
AT ASH 2013
Major finding: Median survival has not been reached in the anticoagulation group and was 9 months in controls (P = .02).
Data source: A retrospective study of 37 patients with acute leukemia and catheter-related DVT.
Disclosures: The authors reported having no financial disclosures.
CAR-T cells drive ALL into remission
NEW ORLEANS – Modified T cells continue to show their mettle against treatment-refractory leukemias, based on study results presented at the annual meeting of the American Society of Hematology.
Among children and young adults with heavily pretreated relapsed or refractory acute lymphoblastic leukemia (ALL), chimeric antigen receptor (CAR) T cells targeted against the CD19 receptor produced complete responses in 10 of 16 patients, including 3 patients with primary, treatment-refractory ALL who had never previously been in remission, reported Dr. Daniel W. Lee III of the National Cancer Institute in Bethesda, Md.
"We were able to clear CNS [central nervous system] leukemia using CAR-T cells alone," Dr. Lee said.

In a second study, CD19-targeted T cells induced molecular remissions in adults with B-lineage ALL refractory to chemotherapy, said Dr. Marco L. Davila from the cellular therapeutics center at Memorial Sloan-Kettering Cancer Center (MSKCC) in New York City.
The immunotherapy produced a complete response (CR) in 12 of 16 patients, and a complete molecular response (CRm) in 12.
Dr. Davila commented that CAR-T cell therapy appears to be a good therapeutic choice for relapsed/refractory B-ALL.
"Especially in light of the data that we see in indolent lymphomas, where the response rates have not been nearly as great, I would speculate that there may be something about this disease that makes it particularly well suited to the second-generation CAR-T cell therapy," he said.
In both studies, therapy with CAR-T cells served as a bridge to stem cell transplantation for several patients.
Different CAR-T flavors
Each research team used its own variation on CAR-T cell therapy. The NCI investigators collected peripheral blood mononuclear cells (PBMCs) from patients via apheresis and used a gamma retrovirus to introduce into effector cells a genetic sequence targeting the CD19 receptor on malignant cells. The NCI version also uses CD28 costimulatory signaling to boost cell-killing effects.
The patients are conditioned with fludarabine and cyclophosphamide, and the treated T-cells are reinfused into the patients 11 days after harvesting.
In the phase I study, 15 patients with relapsed or refractory ALL and 1 with diffuse large B-cell lymphoma were treated. Eight of the patients had undergone at least one hematopoietic stem cell transplant, and all had received total body irradiation. Four had previously received another form of immunotherapy. The patients had to have been at least 100 days post transplant, with no graft-vs.-host disease.
T-cell expansion and transduction was feasible in this heavily pretreated population. All but two patients had an adequate or good expression of CAR-T cells. These patients were treated nonetheless, and one went on to have a minimal-residual disease (MRD) negative response, Dr. Lee noted.
In all, 10 of the patients had complete responses: All of these patients had ALL, and three had never previously achieved a remission. The patient with non-Hodgkin’s lymphoma did not have a significant treatment response.
Of eight patients who were negative for MRD after therapy, six went on to have stem cell transplants, with no unexpected toxicities.
As in other CAR-T cell studies, the chief toxicities seen included grade 4 neutropenia lasting longer than 2 weeks in nine patients, and the cytokine-release syndrome in four patients (grade 3 in two patients and grade 4 in two patients). One patient with the cytokine-release syndrome had cardiac arrest but was successfully resuscitated.
The cytokine-release syndrome was found to be associated with interleukin-6 (IL-6) and could be ameliorated with the IL-6 blocking agent tocilizumab (Actemra). The severity of cytokine-release syndrome did not correlate with tumor burden, the researchers noted.
MSKCC Study
Dr. Davila and his colleagues used a slightly different CAR-T cell construction, also with CD28 costimulation, to treat B-ALL in adults who either had refractory or relapsed disease (MRD-positive) or who were in their first complete remission. Patients who were positive for the Philadelphia chromosome (Ph+) and those who had extramedullary disease, CNS leukemia, or were in relapse after allogeneic stem cell transplant were all eligible.
He presented data on 16 patients with B-ALL with long-term follow-up: 14 patients had a complete response, with an average time to complete response of 24.5 days.
Seven patients in the MSKCC study have gone on to allogeneic stem cell transplants; three patients in complete remission were not eligible for transplant because of medical contraindications, and one additional patient was being evaluated for transplant. There have been no post-transplant relapses to date, with the longest follow-up out to 2 years, Dr. Davila said.
As in the NCI study, the cytokine-release syndrome was a common toxicity. The investigators initially tried to manage it with steroids but found that it came at the cost of lymphotoxicity that caused a marked decline in serum T-cells. They subsequently switched to tocilizumab, which was effective at treating the syndrome without lymphotoxicity.
Dr. Lee’s study was supported by the National Cancer Institute and St. Baldrick’s Foundation. He discussed off-label use of CAR-T cells. Dr. Lee reported having no conflicts of interest. Dr. Davila’s study was supported by MSKCC. He reported having no conflicts of interest.
NEW ORLEANS – Modified T cells continue to show their mettle against treatment-refractory leukemias, based on study results presented at the annual meeting of the American Society of Hematology.
Among children and young adults with heavily pretreated relapsed or refractory acute lymphoblastic leukemia (ALL), chimeric antigen receptor (CAR) T cells targeted against the CD19 receptor produced complete responses in 10 of 16 patients, including 3 patients with primary, treatment-refractory ALL who had never previously been in remission, reported Dr. Daniel W. Lee III of the National Cancer Institute in Bethesda, Md.
"We were able to clear CNS [central nervous system] leukemia using CAR-T cells alone," Dr. Lee said.
In a second study, CD19-targeted T cells induced molecular remissions in adults with B-lineage ALL refractory to chemotherapy, said Dr. Marco L. Davila from the cellular therapeutics center at Memorial Sloan-Kettering Cancer Center (MSKCC) in New York City.
The immunotherapy produced a complete response (CR) in 12 of 16 patients, and a complete molecular response (CRm) in 12.
Dr. Davila commented that CAR-T cell therapy appears to be a good therapeutic choice for relapsed/refractory B-ALL.
"Especially in light of the data that we see in indolent lymphomas, where the response rates have not been nearly as great, I would speculate that there may be something about this disease that makes it particularly well suited to the second-generation CAR-T cell therapy," he said.
In both studies, therapy with CAR-T cells served as a bridge to stem cell transplantation for several patients.
Different CAR-T flavors
Each research team used its own variation on CAR-T cell therapy. The NCI investigators collected peripheral blood mononuclear cells (PBMCs) from patients via apheresis and used a gamma retrovirus to introduce into effector cells a genetic sequence targeting the CD19 receptor on malignant cells. The NCI version also uses CD28 costimulatory signaling to boost cell-killing effects.
The patients are conditioned with fludarabine and cyclophosphamide, and the treated T-cells are reinfused into the patients 11 days after harvesting.
In the phase I study, 15 patients with relapsed or refractory ALL and 1 with diffuse large B-cell lymphoma were treated. Eight of the patients had undergone at least one hematopoietic stem cell transplant, and all had received total body irradiation. Four had previously received another form of immunotherapy. The patients had to have been at least 100 days post transplant, with no graft-vs.-host disease.
T-cell expansion and transduction was feasible in this heavily pretreated population. All but two patients had an adequate or good expression of CAR-T cells. These patients were treated nonetheless, and one went on to have a minimal-residual disease (MRD) negative response, Dr. Lee noted.
In all, 10 of the patients had complete responses: All of these patients had ALL, and three had never previously achieved a remission. The patient with non-Hodgkin’s lymphoma did not have a significant treatment response.
Of eight patients who were negative for MRD after therapy, six went on to have stem cell transplants, with no unexpected toxicities.
As in other CAR-T cell studies, the chief toxicities seen included grade 4 neutropenia lasting longer than 2 weeks in nine patients, and the cytokine-release syndrome in four patients (grade 3 in two patients and grade 4 in two patients). One patient with the cytokine-release syndrome had cardiac arrest but was successfully resuscitated.
The cytokine-release syndrome was found to be associated with interleukin-6 (IL-6) and could be ameliorated with the IL-6 blocking agent tocilizumab (Actemra). The severity of cytokine-release syndrome did not correlate with tumor burden, the researchers noted.
MSKCC Study
Dr. Davila and his colleagues used a slightly different CAR-T cell construction, also with CD28 costimulation, to treat B-ALL in adults who either had refractory or relapsed disease (MRD-positive) or who were in their first complete remission. Patients who were positive for the Philadelphia chromosome (Ph+) and those who had extramedullary disease, CNS leukemia, or were in relapse after allogeneic stem cell transplant were all eligible.
He presented data on 16 patients with B-ALL with long-term follow-up: 14 patients had a complete response, with an average time to complete response of 24.5 days.
Seven patients in the MSKCC study have gone on to allogeneic stem cell transplants; three patients in complete remission were not eligible for transplant because of medical contraindications, and one additional patient was being evaluated for transplant. There have been no post-transplant relapses to date, with the longest follow-up out to 2 years, Dr. Davila said.
As in the NCI study, the cytokine-release syndrome was a common toxicity. The investigators initially tried to manage it with steroids but found that it came at the cost of lymphotoxicity that caused a marked decline in serum T-cells. They subsequently switched to tocilizumab, which was effective at treating the syndrome without lymphotoxicity.
Dr. Lee’s study was supported by the National Cancer Institute and St. Baldrick’s Foundation. He discussed off-label use of CAR-T cells. Dr. Lee reported having no conflicts of interest. Dr. Davila’s study was supported by MSKCC. He reported having no conflicts of interest.
NEW ORLEANS – Modified T cells continue to show their mettle against treatment-refractory leukemias, based on study results presented at the annual meeting of the American Society of Hematology.
Among children and young adults with heavily pretreated relapsed or refractory acute lymphoblastic leukemia (ALL), chimeric antigen receptor (CAR) T cells targeted against the CD19 receptor produced complete responses in 10 of 16 patients, including 3 patients with primary, treatment-refractory ALL who had never previously been in remission, reported Dr. Daniel W. Lee III of the National Cancer Institute in Bethesda, Md.
"We were able to clear CNS [central nervous system] leukemia using CAR-T cells alone," Dr. Lee said.
In a second study, CD19-targeted T cells induced molecular remissions in adults with B-lineage ALL refractory to chemotherapy, said Dr. Marco L. Davila from the cellular therapeutics center at Memorial Sloan-Kettering Cancer Center (MSKCC) in New York City.
The immunotherapy produced a complete response (CR) in 12 of 16 patients, and a complete molecular response (CRm) in 12.
Dr. Davila commented that CAR-T cell therapy appears to be a good therapeutic choice for relapsed/refractory B-ALL.
"Especially in light of the data that we see in indolent lymphomas, where the response rates have not been nearly as great, I would speculate that there may be something about this disease that makes it particularly well suited to the second-generation CAR-T cell therapy," he said.
In both studies, therapy with CAR-T cells served as a bridge to stem cell transplantation for several patients.
Different CAR-T flavors
Each research team used its own variation on CAR-T cell therapy. The NCI investigators collected peripheral blood mononuclear cells (PBMCs) from patients via apheresis and used a gamma retrovirus to introduce into effector cells a genetic sequence targeting the CD19 receptor on malignant cells. The NCI version also uses CD28 costimulatory signaling to boost cell-killing effects.
The patients are conditioned with fludarabine and cyclophosphamide, and the treated T-cells are reinfused into the patients 11 days after harvesting.
In the phase I study, 15 patients with relapsed or refractory ALL and 1 with diffuse large B-cell lymphoma were treated. Eight of the patients had undergone at least one hematopoietic stem cell transplant, and all had received total body irradiation. Four had previously received another form of immunotherapy. The patients had to have been at least 100 days post transplant, with no graft-vs.-host disease.
T-cell expansion and transduction was feasible in this heavily pretreated population. All but two patients had an adequate or good expression of CAR-T cells. These patients were treated nonetheless, and one went on to have a minimal-residual disease (MRD) negative response, Dr. Lee noted.
In all, 10 of the patients had complete responses: All of these patients had ALL, and three had never previously achieved a remission. The patient with non-Hodgkin’s lymphoma did not have a significant treatment response.
Of eight patients who were negative for MRD after therapy, six went on to have stem cell transplants, with no unexpected toxicities.
As in other CAR-T cell studies, the chief toxicities seen included grade 4 neutropenia lasting longer than 2 weeks in nine patients, and the cytokine-release syndrome in four patients (grade 3 in two patients and grade 4 in two patients). One patient with the cytokine-release syndrome had cardiac arrest but was successfully resuscitated.
The cytokine-release syndrome was found to be associated with interleukin-6 (IL-6) and could be ameliorated with the IL-6 blocking agent tocilizumab (Actemra). The severity of cytokine-release syndrome did not correlate with tumor burden, the researchers noted.
MSKCC Study
Dr. Davila and his colleagues used a slightly different CAR-T cell construction, also with CD28 costimulation, to treat B-ALL in adults who either had refractory or relapsed disease (MRD-positive) or who were in their first complete remission. Patients who were positive for the Philadelphia chromosome (Ph+) and those who had extramedullary disease, CNS leukemia, or were in relapse after allogeneic stem cell transplant were all eligible.
He presented data on 16 patients with B-ALL with long-term follow-up: 14 patients had a complete response, with an average time to complete response of 24.5 days.
Seven patients in the MSKCC study have gone on to allogeneic stem cell transplants; three patients in complete remission were not eligible for transplant because of medical contraindications, and one additional patient was being evaluated for transplant. There have been no post-transplant relapses to date, with the longest follow-up out to 2 years, Dr. Davila said.
As in the NCI study, the cytokine-release syndrome was a common toxicity. The investigators initially tried to manage it with steroids but found that it came at the cost of lymphotoxicity that caused a marked decline in serum T-cells. They subsequently switched to tocilizumab, which was effective at treating the syndrome without lymphotoxicity.
Dr. Lee’s study was supported by the National Cancer Institute and St. Baldrick’s Foundation. He discussed off-label use of CAR-T cells. Dr. Lee reported having no conflicts of interest. Dr. Davila’s study was supported by MSKCC. He reported having no conflicts of interest.
AT ASH 2013
Major finding: Anti-CD19 chimeric antigen receptor T cells induced complete responses in 10 of 16 children and young adults with relapsed/refractory acute lymphoblastic leukemia. In a second study, CD19-targeted T cells induced complete molecular responses in 12 of 16 adults with B-lineage ALL refractory to chemotherapy.
Data source: Phase I studies of 2 novel CAR-T cell therapeutic strategies in a total of 32 patients.
Disclosures: Dr. Lee’s study was supported by the National Cancer Institute and St. Baldrick’s Foundation. He discussed off-label use of CAR-T cells. Dr. Lee reported having no conflicts of interest. Dr. Davila’s study was supported by Memorial Sloan-Kettering Cancer Center. He reported having no conflicts of interest.

So far, so good with dasatinib plus chemo in core binding factor AML
NEW ORLEANS – Adding dasatinib to standard first-line chemotherapy and continuing the KIT inhibitor as maintenance therapy are tolerable in core binding factor acute myeloid leukemia, even for older patients, a phase II study shows.
There were no unexpected toxicities, and clinical outcomes were at least comparable to those historically achieved in this population, Dr. Guido Marcucci said at the annual meeting of the American Society of Hematology.
Core binding factor (CBL) acute myeloid leukemia (AML) has a relatively favorable prognosis, though about 40% of patients will eventually relapse.

The Cancer and Leukemia Group B (CALGB) 10801 study investigators hypothesized that adding dasatinib (Sprycel) to chemotherapy would improve patient outcomes, because KIT mutations are present in about 25%-30% of CBL AML patients and are associated with a higher frequency of relapse, said Dr. Marcucci, professor of hematology at the Ohio State University, Columbus.
Molecular screening confirmed CBF AML in 69 patients, with 61 starting induction therapy with daunorubicin 60 mg/m2 per day on days 1-3, cytarabine 200 mg/m2 per day on days 1-7, and oral dasatinib 100 mg/day on days 8-21. Patients with residual disease on day 21 underwent reinduction chemotherapy, while those achieving complete remission received consolidation therapy with high-dose cytarabine and dasatinib for four cycles. Patients remaining in remission were maintained on dasatinib at 100 mg/day for 12 months.
The median age in the study was 51 years (range, 20-85 years), 65% were positive for the CBFB-MYH11 gene fusion, and 35% were positive for the RUNX1-RUNX1T1 gene fusion.
After a median follow-up of 14 months, 30-day survival was 97%. The complete remission rate was 89% overall – 91% for patients younger than 60 years, and 80% for those older than 60 years – Dr. Marcucci said.
To date, 20 patients remain on treatment and only 2 have relapsed. Four patients died during induction/consolidation therapy, and four died during follow-up (two after relapse).
Among 58 patients evaluable for toxicity, the most common grade 4 toxicities were sepsis (6 cases), increased alanine aminotransferase (3 cases), and 2 events each of pleural effusion, lung infection, elevated aspartate aminotransferase, hypocalcemia, and dyspnea. Grade 5 toxicities included sepsis in two patients and one respiratory failure. Nine patients discontinued treatment due to adverse events.
"We did not really see any adverse events that have not been seen before with chemotherapy or dasatinib," Dr. Marcucci said.
By 12 months, event-free survival was 85% and overall survival was 89%. So far, no survival difference has been seen between patients with chromosome 21 translocations and those with chromosome 16 inversions; however, younger patients seem to have better outcomes than older patients, he said.
The KIT inhibition strategy makes sense scientifically, but the follow-up is too short to enable investigators to draw conclusions, since the bulk of AML patients will relapse within 2 years, said Dr. Peter D. Emanuel, director of the Winthrop P. Rockefeller Cancer Institute at the University of Arkansas, Little Rock.
"At least so far, in early follow-up, it looks like they haven’t done any damage and time will tell if they’ve done good or not," he said in an interview.
Dr. Marcucci concluded that rapid screening for CBF AML is feasible within a cooperative group, noting that patients entered the trial at a median of just 4 days from diagnosis. This may not be feasible, just yet, in clinical practice.
"Most private practice hem/oncs send off the chromosomes from the bone marrow, and you’ll be way into induction chemo by the time you get those results back," Dr. Emanuel observed. "It’s got to be ramped up so it’s in a real-world setting beyond the university setting. I think we’ll get there because there are more and more labs capable of doing this."
The National Cancer Institute sponsored CALGB 10801. Dr. Marcucci reported research funding from Boehringer Ingelheim.
Core binding factor (CBL) acute myeloid leukemia (AML),
The Cancer and Leukemia Group B (CALGB) 10801 study, dasatinib, Sprycel, chemotherapy, improve patient outcomes, CBF AML, induction therapy with daunorubicin, cytarabine, oral dasatinib,
NEW ORLEANS – Adding dasatinib to standard first-line chemotherapy and continuing the KIT inhibitor as maintenance therapy are tolerable in core binding factor acute myeloid leukemia, even for older patients, a phase II study shows.
There were no unexpected toxicities, and clinical outcomes were at least comparable to those historically achieved in this population, Dr. Guido Marcucci said at the annual meeting of the American Society of Hematology.
Core binding factor (CBL) acute myeloid leukemia (AML) has a relatively favorable prognosis, though about 40% of patients will eventually relapse.
The Cancer and Leukemia Group B (CALGB) 10801 study investigators hypothesized that adding dasatinib (Sprycel) to chemotherapy would improve patient outcomes, because KIT mutations are present in about 25%-30% of CBL AML patients and are associated with a higher frequency of relapse, said Dr. Marcucci, professor of hematology at the Ohio State University, Columbus.
Molecular screening confirmed CBF AML in 69 patients, with 61 starting induction therapy with daunorubicin 60 mg/m2 per day on days 1-3, cytarabine 200 mg/m2 per day on days 1-7, and oral dasatinib 100 mg/day on days 8-21. Patients with residual disease on day 21 underwent reinduction chemotherapy, while those achieving complete remission received consolidation therapy with high-dose cytarabine and dasatinib for four cycles. Patients remaining in remission were maintained on dasatinib at 100 mg/day for 12 months.
The median age in the study was 51 years (range, 20-85 years), 65% were positive for the CBFB-MYH11 gene fusion, and 35% were positive for the RUNX1-RUNX1T1 gene fusion.
After a median follow-up of 14 months, 30-day survival was 97%. The complete remission rate was 89% overall – 91% for patients younger than 60 years, and 80% for those older than 60 years – Dr. Marcucci said.
To date, 20 patients remain on treatment and only 2 have relapsed. Four patients died during induction/consolidation therapy, and four died during follow-up (two after relapse).
Among 58 patients evaluable for toxicity, the most common grade 4 toxicities were sepsis (6 cases), increased alanine aminotransferase (3 cases), and 2 events each of pleural effusion, lung infection, elevated aspartate aminotransferase, hypocalcemia, and dyspnea. Grade 5 toxicities included sepsis in two patients and one respiratory failure. Nine patients discontinued treatment due to adverse events.
"We did not really see any adverse events that have not been seen before with chemotherapy or dasatinib," Dr. Marcucci said.
By 12 months, event-free survival was 85% and overall survival was 89%. So far, no survival difference has been seen between patients with chromosome 21 translocations and those with chromosome 16 inversions; however, younger patients seem to have better outcomes than older patients, he said.
The KIT inhibition strategy makes sense scientifically, but the follow-up is too short to enable investigators to draw conclusions, since the bulk of AML patients will relapse within 2 years, said Dr. Peter D. Emanuel, director of the Winthrop P. Rockefeller Cancer Institute at the University of Arkansas, Little Rock.
"At least so far, in early follow-up, it looks like they haven’t done any damage and time will tell if they’ve done good or not," he said in an interview.
Dr. Marcucci concluded that rapid screening for CBF AML is feasible within a cooperative group, noting that patients entered the trial at a median of just 4 days from diagnosis. This may not be feasible, just yet, in clinical practice.
"Most private practice hem/oncs send off the chromosomes from the bone marrow, and you’ll be way into induction chemo by the time you get those results back," Dr. Emanuel observed. "It’s got to be ramped up so it’s in a real-world setting beyond the university setting. I think we’ll get there because there are more and more labs capable of doing this."
The National Cancer Institute sponsored CALGB 10801. Dr. Marcucci reported research funding from Boehringer Ingelheim.
NEW ORLEANS – Adding dasatinib to standard first-line chemotherapy and continuing the KIT inhibitor as maintenance therapy are tolerable in core binding factor acute myeloid leukemia, even for older patients, a phase II study shows.
There were no unexpected toxicities, and clinical outcomes were at least comparable to those historically achieved in this population, Dr. Guido Marcucci said at the annual meeting of the American Society of Hematology.
Core binding factor (CBL) acute myeloid leukemia (AML) has a relatively favorable prognosis, though about 40% of patients will eventually relapse.
The Cancer and Leukemia Group B (CALGB) 10801 study investigators hypothesized that adding dasatinib (Sprycel) to chemotherapy would improve patient outcomes, because KIT mutations are present in about 25%-30% of CBL AML patients and are associated with a higher frequency of relapse, said Dr. Marcucci, professor of hematology at the Ohio State University, Columbus.
Molecular screening confirmed CBF AML in 69 patients, with 61 starting induction therapy with daunorubicin 60 mg/m2 per day on days 1-3, cytarabine 200 mg/m2 per day on days 1-7, and oral dasatinib 100 mg/day on days 8-21. Patients with residual disease on day 21 underwent reinduction chemotherapy, while those achieving complete remission received consolidation therapy with high-dose cytarabine and dasatinib for four cycles. Patients remaining in remission were maintained on dasatinib at 100 mg/day for 12 months.
The median age in the study was 51 years (range, 20-85 years), 65% were positive for the CBFB-MYH11 gene fusion, and 35% were positive for the RUNX1-RUNX1T1 gene fusion.
After a median follow-up of 14 months, 30-day survival was 97%. The complete remission rate was 89% overall – 91% for patients younger than 60 years, and 80% for those older than 60 years – Dr. Marcucci said.
To date, 20 patients remain on treatment and only 2 have relapsed. Four patients died during induction/consolidation therapy, and four died during follow-up (two after relapse).
Among 58 patients evaluable for toxicity, the most common grade 4 toxicities were sepsis (6 cases), increased alanine aminotransferase (3 cases), and 2 events each of pleural effusion, lung infection, elevated aspartate aminotransferase, hypocalcemia, and dyspnea. Grade 5 toxicities included sepsis in two patients and one respiratory failure. Nine patients discontinued treatment due to adverse events.
"We did not really see any adverse events that have not been seen before with chemotherapy or dasatinib," Dr. Marcucci said.
By 12 months, event-free survival was 85% and overall survival was 89%. So far, no survival difference has been seen between patients with chromosome 21 translocations and those with chromosome 16 inversions; however, younger patients seem to have better outcomes than older patients, he said.
The KIT inhibition strategy makes sense scientifically, but the follow-up is too short to enable investigators to draw conclusions, since the bulk of AML patients will relapse within 2 years, said Dr. Peter D. Emanuel, director of the Winthrop P. Rockefeller Cancer Institute at the University of Arkansas, Little Rock.
"At least so far, in early follow-up, it looks like they haven’t done any damage and time will tell if they’ve done good or not," he said in an interview.
Dr. Marcucci concluded that rapid screening for CBF AML is feasible within a cooperative group, noting that patients entered the trial at a median of just 4 days from diagnosis. This may not be feasible, just yet, in clinical practice.
"Most private practice hem/oncs send off the chromosomes from the bone marrow, and you’ll be way into induction chemo by the time you get those results back," Dr. Emanuel observed. "It’s got to be ramped up so it’s in a real-world setting beyond the university setting. I think we’ll get there because there are more and more labs capable of doing this."
The National Cancer Institute sponsored CALGB 10801. Dr. Marcucci reported research funding from Boehringer Ingelheim.
Core binding factor (CBL) acute myeloid leukemia (AML),
The Cancer and Leukemia Group B (CALGB) 10801 study, dasatinib, Sprycel, chemotherapy, improve patient outcomes, CBF AML, induction therapy with daunorubicin, cytarabine, oral dasatinib,
Core binding factor (CBL) acute myeloid leukemia (AML),
The Cancer and Leukemia Group B (CALGB) 10801 study, dasatinib, Sprycel, chemotherapy, improve patient outcomes, CBF AML, induction therapy with daunorubicin, cytarabine, oral dasatinib,
AT ASH 2013
Major finding: At 12 months, event-free survival was 85% and overall survival was 89%.
Data source: A prospective phase II study in 61 patients with core binding factor acute myeloid leukemia.
Disclosures: The National Cancer Institute sponsored CALGB 10801. Dr. Marcucci reported research funding from Boehringer Ingelheim.

Idelalisib plus rituximab boosts survival in relapsed chronic lymphocytic leukemia
NEW ORLEANS – Adding idelalisib to rituximab significantly extends survival in patients with heavily pretreated relapsed chronic lymphocytic leukemia unsuitable for chemotherapy, according to results from Study 116.
The primary endpoint of median progression-free survival (PFS) was 5.5 months with rituximab (Rituxan) plus placebo and had not been reached with rituximab plus idelalisib (hazard ratio, 0.15; P less than .0001).
The PFS advantage was seen across all subgroups including patients with the 17p deletion and TP53 mutations, Dr. Richard R. Furman reported during a late-breaking abstract session at the annual meeting of the American Society of Hematology.
Overall survival was not reached in either group, but was also significantly longer with idelalisib (HR, 0.28; P = .018).
New therapies are needed for relapsed chronic lymphocytic leukemia patients (CLL), especially those who are unfit for chemoimmunotherapy, observed Dr. Furman, head of the CLL and Waldenström’s macroglobulinemia program at Cornell University, New York

Relapsed CLL responds poorly to available therapies, and older patients and those with comorbidities or poor renal/bone marrow function often cannot tolerate standard chemoimmunotherapy.
A new drug application for refractory indolent non-Hodgkin lymphoma was submitted in September for idelalisib and the oral phosphoinositide 3-kinase–delta inhibitor was subsequently granted breakthrough therapy status for relapsed CLL based on the Study 116 findings.
The positive findings also prompted the phase III trial to be halted early in November.
Idelalisib had an acceptable safety profile, with adverse events leading 9 patients to stop idelalisib combination therapy vs. 11 controls, Dr. Furman said.
Infusion-related reactions were also lower, possibly because idelalisib had an impact on the tolerability of rituximab, he said. No patient on idelalisib had a grade 3 or higher infusion reaction vs. four on rituximab alone.
As seen in another idelalisib trial reported at the meeting, grade 3/4 transaminase elevations were more common with the addition of idelalisib to rituximab (six patients vs. one patient). Four of the six idelalisib patients with this event were successfully rechallenged, he said.
Concerns were raised by the audience, however, about whether single-agent rituximab was an appropriate comparator since 88% of controls had already been treated with rituximab.
In response to the criticism, Dr. Furman reminded the audience that all patients were unfit for chemotherapy and that physicians had to be comfortable with the protocol in order to enroll patients.
"You have to remember rituximab is actually, in practice, the most commonly used agent for these patients and by giving sort of a stepped-up dosing, they’d hopefully be getting sufficient therapy to better the patients’ outcome," he added.
Commenting on the results, Dr. Wendy Stock of the University of Chicago, who was a session co-moderator, said the choice of the control arm was her one concern, but that the study design would likely have not been improved if the previous standard, chlorambucil chemotherapy, had been used as the control.
"I think the data are very compelling in terms of the efficacy of this new agent, and we’ll have to see now how it gets incorporated with other novel agents and/or moved into the United States and Europe evenly randomized 220 patients to eight infusions of rituximab over 24 weeks plus either idelalisib 150 mg or placebo twice daily until disease progression. Rituximab was dosed at 375 mg/m2 in week 1, 500 mg/m2 every 2 weeks for four doses, followed by 500 mg/m2 every 4 weeks for three more doses.
Patients had received a median of three prior therapies, a median Cumulative Illness Rating Scale score of 8, and a median age of 71 years.
The addition of idelalisib to rituximab increased the overall response rate from 13% to 81% (P less than .0001) and the number of patients achieving at least a 50% reduction in lymph nodes from 4% to 93% (P less than .0001), Dr. Furman said.
Idelalisib is currently being evaluated in more than a dozen studies including a phase II study of combination therapy with the experimental spleen tyrosine kinase inhibitor GS-9973 in a variety of relapsed or refractory hematologic malignancies.
Dr. Furman reported research funding from and board of directors or advisory committee membership with Gilead Sciences, which is developing idelalisib.
NEW ORLEANS – Adding idelalisib to rituximab significantly extends survival in patients with heavily pretreated relapsed chronic lymphocytic leukemia unsuitable for chemotherapy, according to results from Study 116.
The primary endpoint of median progression-free survival (PFS) was 5.5 months with rituximab (Rituxan) plus placebo and had not been reached with rituximab plus idelalisib (hazard ratio, 0.15; P less than .0001).
The PFS advantage was seen across all subgroups including patients with the 17p deletion and TP53 mutations, Dr. Richard R. Furman reported during a late-breaking abstract session at the annual meeting of the American Society of Hematology.
Overall survival was not reached in either group, but was also significantly longer with idelalisib (HR, 0.28; P = .018).
New therapies are needed for relapsed chronic lymphocytic leukemia patients (CLL), especially those who are unfit for chemoimmunotherapy, observed Dr. Furman, head of the CLL and Waldenström’s macroglobulinemia program at Cornell University, New York
Relapsed CLL responds poorly to available therapies, and older patients and those with comorbidities or poor renal/bone marrow function often cannot tolerate standard chemoimmunotherapy.
A new drug application for refractory indolent non-Hodgkin lymphoma was submitted in September for idelalisib and the oral phosphoinositide 3-kinase–delta inhibitor was subsequently granted breakthrough therapy status for relapsed CLL based on the Study 116 findings.
The positive findings also prompted the phase III trial to be halted early in November.
Idelalisib had an acceptable safety profile, with adverse events leading 9 patients to stop idelalisib combination therapy vs. 11 controls, Dr. Furman said.
Infusion-related reactions were also lower, possibly because idelalisib had an impact on the tolerability of rituximab, he said. No patient on idelalisib had a grade 3 or higher infusion reaction vs. four on rituximab alone.
As seen in another idelalisib trial reported at the meeting, grade 3/4 transaminase elevations were more common with the addition of idelalisib to rituximab (six patients vs. one patient). Four of the six idelalisib patients with this event were successfully rechallenged, he said.
Concerns were raised by the audience, however, about whether single-agent rituximab was an appropriate comparator since 88% of controls had already been treated with rituximab.
In response to the criticism, Dr. Furman reminded the audience that all patients were unfit for chemotherapy and that physicians had to be comfortable with the protocol in order to enroll patients.
"You have to remember rituximab is actually, in practice, the most commonly used agent for these patients and by giving sort of a stepped-up dosing, they’d hopefully be getting sufficient therapy to better the patients’ outcome," he added.
Commenting on the results, Dr. Wendy Stock of the University of Chicago, who was a session co-moderator, said the choice of the control arm was her one concern, but that the study design would likely have not been improved if the previous standard, chlorambucil chemotherapy, had been used as the control.
"I think the data are very compelling in terms of the efficacy of this new agent, and we’ll have to see now how it gets incorporated with other novel agents and/or moved into the United States and Europe evenly randomized 220 patients to eight infusions of rituximab over 24 weeks plus either idelalisib 150 mg or placebo twice daily until disease progression. Rituximab was dosed at 375 mg/m2 in week 1, 500 mg/m2 every 2 weeks for four doses, followed by 500 mg/m2 every 4 weeks for three more doses.
Patients had received a median of three prior therapies, a median Cumulative Illness Rating Scale score of 8, and a median age of 71 years.
The addition of idelalisib to rituximab increased the overall response rate from 13% to 81% (P less than .0001) and the number of patients achieving at least a 50% reduction in lymph nodes from 4% to 93% (P less than .0001), Dr. Furman said.
Idelalisib is currently being evaluated in more than a dozen studies including a phase II study of combination therapy with the experimental spleen tyrosine kinase inhibitor GS-9973 in a variety of relapsed or refractory hematologic malignancies.
Dr. Furman reported research funding from and board of directors or advisory committee membership with Gilead Sciences, which is developing idelalisib.
NEW ORLEANS – Adding idelalisib to rituximab significantly extends survival in patients with heavily pretreated relapsed chronic lymphocytic leukemia unsuitable for chemotherapy, according to results from Study 116.
The primary endpoint of median progression-free survival (PFS) was 5.5 months with rituximab (Rituxan) plus placebo and had not been reached with rituximab plus idelalisib (hazard ratio, 0.15; P less than .0001).
The PFS advantage was seen across all subgroups including patients with the 17p deletion and TP53 mutations, Dr. Richard R. Furman reported during a late-breaking abstract session at the annual meeting of the American Society of Hematology.
Overall survival was not reached in either group, but was also significantly longer with idelalisib (HR, 0.28; P = .018).
New therapies are needed for relapsed chronic lymphocytic leukemia patients (CLL), especially those who are unfit for chemoimmunotherapy, observed Dr. Furman, head of the CLL and Waldenström’s macroglobulinemia program at Cornell University, New York
Relapsed CLL responds poorly to available therapies, and older patients and those with comorbidities or poor renal/bone marrow function often cannot tolerate standard chemoimmunotherapy.
A new drug application for refractory indolent non-Hodgkin lymphoma was submitted in September for idelalisib and the oral phosphoinositide 3-kinase–delta inhibitor was subsequently granted breakthrough therapy status for relapsed CLL based on the Study 116 findings.
The positive findings also prompted the phase III trial to be halted early in November.
Idelalisib had an acceptable safety profile, with adverse events leading 9 patients to stop idelalisib combination therapy vs. 11 controls, Dr. Furman said.
Infusion-related reactions were also lower, possibly because idelalisib had an impact on the tolerability of rituximab, he said. No patient on idelalisib had a grade 3 or higher infusion reaction vs. four on rituximab alone.
As seen in another idelalisib trial reported at the meeting, grade 3/4 transaminase elevations were more common with the addition of idelalisib to rituximab (six patients vs. one patient). Four of the six idelalisib patients with this event were successfully rechallenged, he said.
Concerns were raised by the audience, however, about whether single-agent rituximab was an appropriate comparator since 88% of controls had already been treated with rituximab.
In response to the criticism, Dr. Furman reminded the audience that all patients were unfit for chemotherapy and that physicians had to be comfortable with the protocol in order to enroll patients.
"You have to remember rituximab is actually, in practice, the most commonly used agent for these patients and by giving sort of a stepped-up dosing, they’d hopefully be getting sufficient therapy to better the patients’ outcome," he added.
Commenting on the results, Dr. Wendy Stock of the University of Chicago, who was a session co-moderator, said the choice of the control arm was her one concern, but that the study design would likely have not been improved if the previous standard, chlorambucil chemotherapy, had been used as the control.
"I think the data are very compelling in terms of the efficacy of this new agent, and we’ll have to see now how it gets incorporated with other novel agents and/or moved into the United States and Europe evenly randomized 220 patients to eight infusions of rituximab over 24 weeks plus either idelalisib 150 mg or placebo twice daily until disease progression. Rituximab was dosed at 375 mg/m2 in week 1, 500 mg/m2 every 2 weeks for four doses, followed by 500 mg/m2 every 4 weeks for three more doses.
Patients had received a median of three prior therapies, a median Cumulative Illness Rating Scale score of 8, and a median age of 71 years.
The addition of idelalisib to rituximab increased the overall response rate from 13% to 81% (P less than .0001) and the number of patients achieving at least a 50% reduction in lymph nodes from 4% to 93% (P less than .0001), Dr. Furman said.
Idelalisib is currently being evaluated in more than a dozen studies including a phase II study of combination therapy with the experimental spleen tyrosine kinase inhibitor GS-9973 in a variety of relapsed or refractory hematologic malignancies.
Dr. Furman reported research funding from and board of directors or advisory committee membership with Gilead Sciences, which is developing idelalisib.
AT ASH 2013
Major finding: Median progression-free survival was 5.5 months with rituximab and placebo and has not been reached with idelalisib plus rituximab. (P less than .0001).
Data source: A prospective phase III study of 220 patients with relapsed CLL unfit for chemotherapy.
Disclosures: Dr. Furman reported research funding from and board of directors or advisory committee membership with Gilead Sciences, which is developing idelalisib.

Sickle cell crises curtailed with experimental cellular adhesion inhibitor
NEW ORLEANS – An experimental cellular adhesion inhibitor was successful at reducing the severity and duration of vaso-occlusive crises in patients with sickle cell disease.
In a phase II trial of 76 patients with sickle cell disease, patients randomized to receive the pan-selectin inhibitor GMI 1070 early in their hospitalization for a vaso-occlusive crisis (VOC) had shorter lengths of stay and needed significantly lower cumulative doses of narcotics for pain control than did patients randomized to placebo, reported Dr. Marilyn J. Telen, chief of the hematology division at Duke University, Durham, N.C.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
"We see somewhere between 75,000 and 90,000 admissions [annually] for acute painful vaso-occlusive crisis among this patient population. Indeed, these crises are the most common and essentially the archetypal presentation of sickle cell disease. Nevertheless, up till this time, treatment for these crises or VOC in sickle cell disease, remain only supportive, focusing largely on using narcotics for symptom relief, and then other measures, some of which are used to counteract the ill effects of narcotics," said Dr. Telen at the annual meeting of the American Society of Hematology.
GMI 1070 (being developed by GlycoMimetics, in partnership with Pfizer) is a synthetic molecule designed to inhibit the glycoprotein cellular-adhesion molecules involved in inflammation. In previous studies, the drug has been shown to be safe, and in a mouse model of VOC, was successful at restoring blood flow, Dr. Telen said. The drug has received both orphan drug and fast-track status from the Food and Drug Administration, according to GlycoMimetics.
Dr. Telen and her colleagues enrolled 76 patients aged 12-51 years with sickle cell disease and randomized them to receive a loading dose of GMI 1070 delivered intravenously (43 patients), followed by up to 14 subsequent doses delivered every 12 hours, or placebo (33 patients), with other treatment left to the discretion of the participating institutions. After an interim pharmacokinetic analysis showed that the drug did not reach target nadir levels, the dose was doubled.
All 76 patients reached the primary endpoint of VOC resolution, defined as a composite of decreased pain, termination of the need for intravenous opioids, patient and physician agreement on the ability to discharge the patient, and actual hospital discharge.
A total of 58 patients continued on the assigned drug until they either reached the primary endpoint criteria or received the maximum number of doses allowed. The remaining 18 patients discontinued the drug either for adverse events, no improvement by day 5 on the assigned drug, or other reasons.
In an analysis pooling all patients assigned to GMI 1070, including those who started out on the lower dose, there was a consistent reduction over placebo in the mean time to resolution of VOC: 103 hours vs. 144 hours for patients treated with placebo. This difference was not statistically significant, however.
A Kaplan-Meier analysis showed a median time to resolution of 69.6 hours for GMI 1070, compared with 139 hours with placebo, a difference that was not significant.
There was an 83% reduction in the secondary endpoint of cumulative opioid analgesics administered during hospitalization, a difference that was statistically significant (P =.010). There was also a reduction by 84 hours in the median time to discharge, and by 55 hours in the mean time to discharge, among patients treated with the active drug, compared with those on placebo. These differences, while large, were not significant, Dr. Telen said.
She noted that although most of the endpoints in this study failed to reach statistical significance, the separation of the curves between the placebo- and GMI 1070–treated patients began early, usually within 2 days of the start of treatment.
Total adverse event rates, including serious events and those deemed to be treatment related, were comparable between the two study arms for all subgroups.
Dr. Telen noted that because the population of patients enrolled was more clinically diverse than the available literature would suggest, the study was underpowered to detect differences, given the size of the sample. She predicted that given the size of the effects seen, statistical significance would emerge in a larger study.
GlycoMimetics is currently working with Pfizer to develop a phase III trial of GM 1070 for this indication.
The study was supported by GlycoMimetics. Dr. Telen is a consultant to the company, and several coauthors are employees of the company.
NEW ORLEANS – An experimental cellular adhesion inhibitor was successful at reducing the severity and duration of vaso-occlusive crises in patients with sickle cell disease.
In a phase II trial of 76 patients with sickle cell disease, patients randomized to receive the pan-selectin inhibitor GMI 1070 early in their hospitalization for a vaso-occlusive crisis (VOC) had shorter lengths of stay and needed significantly lower cumulative doses of narcotics for pain control than did patients randomized to placebo, reported Dr. Marilyn J. Telen, chief of the hematology division at Duke University, Durham, N.C.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
"We see somewhere between 75,000 and 90,000 admissions [annually] for acute painful vaso-occlusive crisis among this patient population. Indeed, these crises are the most common and essentially the archetypal presentation of sickle cell disease. Nevertheless, up till this time, treatment for these crises or VOC in sickle cell disease, remain only supportive, focusing largely on using narcotics for symptom relief, and then other measures, some of which are used to counteract the ill effects of narcotics," said Dr. Telen at the annual meeting of the American Society of Hematology.
GMI 1070 (being developed by GlycoMimetics, in partnership with Pfizer) is a synthetic molecule designed to inhibit the glycoprotein cellular-adhesion molecules involved in inflammation. In previous studies, the drug has been shown to be safe, and in a mouse model of VOC, was successful at restoring blood flow, Dr. Telen said. The drug has received both orphan drug and fast-track status from the Food and Drug Administration, according to GlycoMimetics.
Dr. Telen and her colleagues enrolled 76 patients aged 12-51 years with sickle cell disease and randomized them to receive a loading dose of GMI 1070 delivered intravenously (43 patients), followed by up to 14 subsequent doses delivered every 12 hours, or placebo (33 patients), with other treatment left to the discretion of the participating institutions. After an interim pharmacokinetic analysis showed that the drug did not reach target nadir levels, the dose was doubled.
All 76 patients reached the primary endpoint of VOC resolution, defined as a composite of decreased pain, termination of the need for intravenous opioids, patient and physician agreement on the ability to discharge the patient, and actual hospital discharge.
A total of 58 patients continued on the assigned drug until they either reached the primary endpoint criteria or received the maximum number of doses allowed. The remaining 18 patients discontinued the drug either for adverse events, no improvement by day 5 on the assigned drug, or other reasons.
In an analysis pooling all patients assigned to GMI 1070, including those who started out on the lower dose, there was a consistent reduction over placebo in the mean time to resolution of VOC: 103 hours vs. 144 hours for patients treated with placebo. This difference was not statistically significant, however.
A Kaplan-Meier analysis showed a median time to resolution of 69.6 hours for GMI 1070, compared with 139 hours with placebo, a difference that was not significant.
There was an 83% reduction in the secondary endpoint of cumulative opioid analgesics administered during hospitalization, a difference that was statistically significant (P =.010). There was also a reduction by 84 hours in the median time to discharge, and by 55 hours in the mean time to discharge, among patients treated with the active drug, compared with those on placebo. These differences, while large, were not significant, Dr. Telen said.
She noted that although most of the endpoints in this study failed to reach statistical significance, the separation of the curves between the placebo- and GMI 1070–treated patients began early, usually within 2 days of the start of treatment.
Total adverse event rates, including serious events and those deemed to be treatment related, were comparable between the two study arms for all subgroups.
Dr. Telen noted that because the population of patients enrolled was more clinically diverse than the available literature would suggest, the study was underpowered to detect differences, given the size of the sample. She predicted that given the size of the effects seen, statistical significance would emerge in a larger study.
GlycoMimetics is currently working with Pfizer to develop a phase III trial of GM 1070 for this indication.
The study was supported by GlycoMimetics. Dr. Telen is a consultant to the company, and several coauthors are employees of the company.
NEW ORLEANS – An experimental cellular adhesion inhibitor was successful at reducing the severity and duration of vaso-occlusive crises in patients with sickle cell disease.
In a phase II trial of 76 patients with sickle cell disease, patients randomized to receive the pan-selectin inhibitor GMI 1070 early in their hospitalization for a vaso-occlusive crisis (VOC) had shorter lengths of stay and needed significantly lower cumulative doses of narcotics for pain control than did patients randomized to placebo, reported Dr. Marilyn J. Telen, chief of the hematology division at Duke University, Durham, N.C.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
"We see somewhere between 75,000 and 90,000 admissions [annually] for acute painful vaso-occlusive crisis among this patient population. Indeed, these crises are the most common and essentially the archetypal presentation of sickle cell disease. Nevertheless, up till this time, treatment for these crises or VOC in sickle cell disease, remain only supportive, focusing largely on using narcotics for symptom relief, and then other measures, some of which are used to counteract the ill effects of narcotics," said Dr. Telen at the annual meeting of the American Society of Hematology.
GMI 1070 (being developed by GlycoMimetics, in partnership with Pfizer) is a synthetic molecule designed to inhibit the glycoprotein cellular-adhesion molecules involved in inflammation. In previous studies, the drug has been shown to be safe, and in a mouse model of VOC, was successful at restoring blood flow, Dr. Telen said. The drug has received both orphan drug and fast-track status from the Food and Drug Administration, according to GlycoMimetics.
Dr. Telen and her colleagues enrolled 76 patients aged 12-51 years with sickle cell disease and randomized them to receive a loading dose of GMI 1070 delivered intravenously (43 patients), followed by up to 14 subsequent doses delivered every 12 hours, or placebo (33 patients), with other treatment left to the discretion of the participating institutions. After an interim pharmacokinetic analysis showed that the drug did not reach target nadir levels, the dose was doubled.
All 76 patients reached the primary endpoint of VOC resolution, defined as a composite of decreased pain, termination of the need for intravenous opioids, patient and physician agreement on the ability to discharge the patient, and actual hospital discharge.
A total of 58 patients continued on the assigned drug until they either reached the primary endpoint criteria or received the maximum number of doses allowed. The remaining 18 patients discontinued the drug either for adverse events, no improvement by day 5 on the assigned drug, or other reasons.
In an analysis pooling all patients assigned to GMI 1070, including those who started out on the lower dose, there was a consistent reduction over placebo in the mean time to resolution of VOC: 103 hours vs. 144 hours for patients treated with placebo. This difference was not statistically significant, however.
A Kaplan-Meier analysis showed a median time to resolution of 69.6 hours for GMI 1070, compared with 139 hours with placebo, a difference that was not significant.
There was an 83% reduction in the secondary endpoint of cumulative opioid analgesics administered during hospitalization, a difference that was statistically significant (P =.010). There was also a reduction by 84 hours in the median time to discharge, and by 55 hours in the mean time to discharge, among patients treated with the active drug, compared with those on placebo. These differences, while large, were not significant, Dr. Telen said.
She noted that although most of the endpoints in this study failed to reach statistical significance, the separation of the curves between the placebo- and GMI 1070–treated patients began early, usually within 2 days of the start of treatment.
Total adverse event rates, including serious events and those deemed to be treatment related, were comparable between the two study arms for all subgroups.
Dr. Telen noted that because the population of patients enrolled was more clinically diverse than the available literature would suggest, the study was underpowered to detect differences, given the size of the sample. She predicted that given the size of the effects seen, statistical significance would emerge in a larger study.
GlycoMimetics is currently working with Pfizer to develop a phase III trial of GM 1070 for this indication.
The study was supported by GlycoMimetics. Dr. Telen is a consultant to the company, and several coauthors are employees of the company.
AT ASH 2013
Major finding: The mean time to resolution of vaso-occlusive crisis in patients with sickle cell disease was 103 hours for patients treated with GMI 1070 vs. 144 for those treated with placebo.
Data source: A randomized, double blind multicenter study of 76 patients aged 12-51.
Disclosures: The study was supported by GlycoMimetics. Dr. Telen is a consultant to the company, and several coauthors are employees of the company.
Gene therapy for SCID-X1 may successfully reset immune system
NEW ORLEANS – Tweaking experimental gene therapy for X-linked severe combined immunodeficiency may help to restore patient immune function while reducing the risk for subsequent leukemias.
In a small, multinational phase I/II trial, seven of nine children with SCID-X1 showed evidence of T-cell recovery and function, as well as a lower risk for promoting growth of leukemic cells, when a self-inactivating gamma-retroviral vector (SCID-2) was used to promote reconstitution of the child’s immune system without insertional oncogenesis, reported Dr. Sung-Yun Pai at the annual meeting of the American Society of Hematology.
"Outcomes for boys who do not have well-matched donors are suboptimal, and it’s particularly for these boys that we are targeting gene therapy," said Dr. Pai, of Boston Children’s Hospital and the Dana-Farber Cancer Institute, Boston.
In previous gene therapy trials, investigators used the Moloney leukemia virus (MLV)-based gamma-retroviral vector (SCID-1) with strong promoters and enhancers to express an IL-2 receptor that reconstituted the immune system successfully in 18 of 20 boys.
However, 5 of the 20 boys developed T-cell acute lymphoblastic leukemia; 1 of the children died, and the remaining 4 were successfully treated.
The investigators found that in the patients with leukemia, the SCID-1 vector had inserted into a chromosomal region close to proto-oncogenes such as LMO2, and the enhancers were driving expression of the neighboring oncogene, promoting expression of aberrant T cells. The vector was subsequently modified with the goal of improved safety but similar efficacy to the original, said Dr. Pai. The strong viral enhancers were removed to prevent accident enhancement should the inserted genes manage to find their way into oncogenes.
The phase I/II study is being conducted in London, Paris, Boston, Cincinnati, and Los Angeles, and to date has enrolled 9 male children of a planned 20.
Of the 9, one child died from a preexisting adenoviral infection before immune recovery was complete, and one child did not have engraftment of the gene-marked cells and went on to transplant.
"The other patients have 9 months to 36 months of follow-up, they have evidence of T-cell recovery, of T-cell function, have cleared SCID-related infections, and are all out of hospital, healthy at home, [and] leading essentially normal lives."
When the investigators looked at the comparative efficacy of the SCID-1 and SCID-2 vectors, they saw that 6 months after gene therapy, there was no significant difference in the median number of T cells generated.
"It’s far too early to comment on whether this vector will truly be safer in terms of leukemia," Dr. Pai said, noting that in the SCID-1 trial the leukemias developed 3-5 years after gene therapy, and the longest follow-up in the SCID-2 study is only 3 years.
The investigators are, however, conducting molecular surrogate safety analyses looking at gene insertion sites from the blood of patients treated with SCID-1 and are comparing those sites with the vector-insertion sites in cells from patients in SCID-2.
Looking at a global genomewide map of integrations, they found no significant differences between SCID-1 and SCID-2. However, when they focused on 38 genes that are to be proto-oncogenes in lymphoid cancer, they found that significantly more integration of the modified genes occurred in proximity to the oncogenes in SCID-1 than in SCID-2 (P = .003).
"We hope that these data suggest that the modified SCID vector will show less capacity to drive aberrant cell growth and lead to less leukemogenesis," said Dr. Pai.
SCID-X1 is caused by inherited mutations in the gamma subunit of the interleukin (IL)-2 receptor. As a result, males are born without T lymphocytes or natural killer cells. Without a bone marrow or stem cell transplantation, children with the disease die early from opportunistic or community-acquired infections.
"These are really paradigm-changing results for mortally wounded children," said Dr. Laurence James Neil Cooper of the University of Texas M.D. Anderson Cancer Center in Houston, who moderated the briefing but was not involved in the study.
The trial is being sponsored by Children’s Hospital Boston, Cincinnati Children’s Hospital Medical Center, and the University of California, Los Angeles. Dr. Pai and Dr. Cooper reported having no relevant conflicts of interest.
NEW ORLEANS – Tweaking experimental gene therapy for X-linked severe combined immunodeficiency may help to restore patient immune function while reducing the risk for subsequent leukemias.
In a small, multinational phase I/II trial, seven of nine children with SCID-X1 showed evidence of T-cell recovery and function, as well as a lower risk for promoting growth of leukemic cells, when a self-inactivating gamma-retroviral vector (SCID-2) was used to promote reconstitution of the child’s immune system without insertional oncogenesis, reported Dr. Sung-Yun Pai at the annual meeting of the American Society of Hematology.
"Outcomes for boys who do not have well-matched donors are suboptimal, and it’s particularly for these boys that we are targeting gene therapy," said Dr. Pai, of Boston Children’s Hospital and the Dana-Farber Cancer Institute, Boston.
In previous gene therapy trials, investigators used the Moloney leukemia virus (MLV)-based gamma-retroviral vector (SCID-1) with strong promoters and enhancers to express an IL-2 receptor that reconstituted the immune system successfully in 18 of 20 boys.
However, 5 of the 20 boys developed T-cell acute lymphoblastic leukemia; 1 of the children died, and the remaining 4 were successfully treated.
The investigators found that in the patients with leukemia, the SCID-1 vector had inserted into a chromosomal region close to proto-oncogenes such as LMO2, and the enhancers were driving expression of the neighboring oncogene, promoting expression of aberrant T cells. The vector was subsequently modified with the goal of improved safety but similar efficacy to the original, said Dr. Pai. The strong viral enhancers were removed to prevent accident enhancement should the inserted genes manage to find their way into oncogenes.
The phase I/II study is being conducted in London, Paris, Boston, Cincinnati, and Los Angeles, and to date has enrolled 9 male children of a planned 20.
Of the 9, one child died from a preexisting adenoviral infection before immune recovery was complete, and one child did not have engraftment of the gene-marked cells and went on to transplant.
"The other patients have 9 months to 36 months of follow-up, they have evidence of T-cell recovery, of T-cell function, have cleared SCID-related infections, and are all out of hospital, healthy at home, [and] leading essentially normal lives."
When the investigators looked at the comparative efficacy of the SCID-1 and SCID-2 vectors, they saw that 6 months after gene therapy, there was no significant difference in the median number of T cells generated.
"It’s far too early to comment on whether this vector will truly be safer in terms of leukemia," Dr. Pai said, noting that in the SCID-1 trial the leukemias developed 3-5 years after gene therapy, and the longest follow-up in the SCID-2 study is only 3 years.
The investigators are, however, conducting molecular surrogate safety analyses looking at gene insertion sites from the blood of patients treated with SCID-1 and are comparing those sites with the vector-insertion sites in cells from patients in SCID-2.
Looking at a global genomewide map of integrations, they found no significant differences between SCID-1 and SCID-2. However, when they focused on 38 genes that are to be proto-oncogenes in lymphoid cancer, they found that significantly more integration of the modified genes occurred in proximity to the oncogenes in SCID-1 than in SCID-2 (P = .003).
"We hope that these data suggest that the modified SCID vector will show less capacity to drive aberrant cell growth and lead to less leukemogenesis," said Dr. Pai.
SCID-X1 is caused by inherited mutations in the gamma subunit of the interleukin (IL)-2 receptor. As a result, males are born without T lymphocytes or natural killer cells. Without a bone marrow or stem cell transplantation, children with the disease die early from opportunistic or community-acquired infections.
"These are really paradigm-changing results for mortally wounded children," said Dr. Laurence James Neil Cooper of the University of Texas M.D. Anderson Cancer Center in Houston, who moderated the briefing but was not involved in the study.
The trial is being sponsored by Children’s Hospital Boston, Cincinnati Children’s Hospital Medical Center, and the University of California, Los Angeles. Dr. Pai and Dr. Cooper reported having no relevant conflicts of interest.
NEW ORLEANS – Tweaking experimental gene therapy for X-linked severe combined immunodeficiency may help to restore patient immune function while reducing the risk for subsequent leukemias.
In a small, multinational phase I/II trial, seven of nine children with SCID-X1 showed evidence of T-cell recovery and function, as well as a lower risk for promoting growth of leukemic cells, when a self-inactivating gamma-retroviral vector (SCID-2) was used to promote reconstitution of the child’s immune system without insertional oncogenesis, reported Dr. Sung-Yun Pai at the annual meeting of the American Society of Hematology.
"Outcomes for boys who do not have well-matched donors are suboptimal, and it’s particularly for these boys that we are targeting gene therapy," said Dr. Pai, of Boston Children’s Hospital and the Dana-Farber Cancer Institute, Boston.
In previous gene therapy trials, investigators used the Moloney leukemia virus (MLV)-based gamma-retroviral vector (SCID-1) with strong promoters and enhancers to express an IL-2 receptor that reconstituted the immune system successfully in 18 of 20 boys.
However, 5 of the 20 boys developed T-cell acute lymphoblastic leukemia; 1 of the children died, and the remaining 4 were successfully treated.
The investigators found that in the patients with leukemia, the SCID-1 vector had inserted into a chromosomal region close to proto-oncogenes such as LMO2, and the enhancers were driving expression of the neighboring oncogene, promoting expression of aberrant T cells. The vector was subsequently modified with the goal of improved safety but similar efficacy to the original, said Dr. Pai. The strong viral enhancers were removed to prevent accident enhancement should the inserted genes manage to find their way into oncogenes.
The phase I/II study is being conducted in London, Paris, Boston, Cincinnati, and Los Angeles, and to date has enrolled 9 male children of a planned 20.
Of the 9, one child died from a preexisting adenoviral infection before immune recovery was complete, and one child did not have engraftment of the gene-marked cells and went on to transplant.
"The other patients have 9 months to 36 months of follow-up, they have evidence of T-cell recovery, of T-cell function, have cleared SCID-related infections, and are all out of hospital, healthy at home, [and] leading essentially normal lives."
When the investigators looked at the comparative efficacy of the SCID-1 and SCID-2 vectors, they saw that 6 months after gene therapy, there was no significant difference in the median number of T cells generated.
"It’s far too early to comment on whether this vector will truly be safer in terms of leukemia," Dr. Pai said, noting that in the SCID-1 trial the leukemias developed 3-5 years after gene therapy, and the longest follow-up in the SCID-2 study is only 3 years.
The investigators are, however, conducting molecular surrogate safety analyses looking at gene insertion sites from the blood of patients treated with SCID-1 and are comparing those sites with the vector-insertion sites in cells from patients in SCID-2.
Looking at a global genomewide map of integrations, they found no significant differences between SCID-1 and SCID-2. However, when they focused on 38 genes that are to be proto-oncogenes in lymphoid cancer, they found that significantly more integration of the modified genes occurred in proximity to the oncogenes in SCID-1 than in SCID-2 (P = .003).
"We hope that these data suggest that the modified SCID vector will show less capacity to drive aberrant cell growth and lead to less leukemogenesis," said Dr. Pai.
SCID-X1 is caused by inherited mutations in the gamma subunit of the interleukin (IL)-2 receptor. As a result, males are born without T lymphocytes or natural killer cells. Without a bone marrow or stem cell transplantation, children with the disease die early from opportunistic or community-acquired infections.
"These are really paradigm-changing results for mortally wounded children," said Dr. Laurence James Neil Cooper of the University of Texas M.D. Anderson Cancer Center in Houston, who moderated the briefing but was not involved in the study.
The trial is being sponsored by Children’s Hospital Boston, Cincinnati Children’s Hospital Medical Center, and the University of California, Los Angeles. Dr. Pai and Dr. Cooper reported having no relevant conflicts of interest.
AT ASH 2013
Major finding: Of nine boys with X-linked severe combined immunodeficiency who were treated with gene therapy, seven patients have evidence of T-cell function, have cleared SCID-related infections, and are out of hospital.
Data source: Preliminary results of a prospective phase I/II clinical trial of nine children.
Disclosures: The trial is being sponsored by Children’s Hospital Boston, Cincinnati Children’s Hospital Medical Center, and the University of California, Los Angeles. Dr. Pai and Dr. Cooper reported having no relevant conflicts of interest.