User login
Don’t touch that! You’ll get hurt! Fear in childhood
Fear is an intrinsic reaction evolved to protect us from harm. Unsurprisingly, anxiety disorders are common, affecting as many as 25% of children. On average, children have 2-14 fears typical for thinking at their age, from separation (1 year), animals (6 years), environment (dark or storms), medical intrusions or injury (9 years) to social disgrace (16 years). But about one-fifth of children with typical fear topics qualify as having a disorder; that is, they have impairment in functioning.
I wonder daily in my care of anxious children: Is this amount of fear really inevitable? Are there things we can do to avoid this burden on children?

For everyone, genetics predispose fear of things that are dangerous, such as snakes. (Tell me that they don’t make you startle!) Genetic influences account for about 50% of the variance in significant fearfulness as evidenced by parent-child patterns, and the fact that monozygotic twins are more highly concordant in fearfulness than dizygotic. Not much we can do about that!
So, if evolution armed humans with fear for protection, how is it that everyone is not impaired?
In combination with genetic vulnerabilities, fears are learned in three ways: experiential conditioning, modeling, and threat information transmission. These frequently co-occur because bad things happen, genetically anxious parents show a fear reaction, and the same parents warn their children frequently and expressively about potential dangers.
As for avoiding fear conditioning, all parents want to protect their children from scary experiences, but it is not always possible. Car crashes and other bad things happen. Even viewing events that threaten injury or death, such as 9/11, can be sufficient to induce post-traumatic stress disorder (18% of children in New York City). The closer and more severe a scary event is, the more it injures or has potential to injure the child or the child’s loved ones; the more expressive the family members are and the more it is repeated (abuse, for example), the greater the likelihood of it lasting and having impairing effects.
Conditioned fears from real experiences are not entirely random. Low-income children are more likely to experience frightening events from rat bites to house fires to domestic violence to gunshots. Asking about environmental factors or using screening tools such as Safe Environment for Every Kid to evaluate the home environment, and referring families for assistance are steps relevant to every child, but especially anxious ones.
You and I need to continue to advocate for safer communities for all children. In the meantime, it is important to know that encouraging a child to describe in detail to a caring adult – verbally and/or by drawing – traumas they experienced is significantly therapeutic. It might not seem intuitive to parents to promote “reliving the experience,” especially because they may have been traumatized themselves. So providing this opportunity ourselves or through a friend, teacher, or counselor who can calmly answer questions and put the event in perspective, is important advice.
But even simply viewing disasters, violence, or artificial frightening events on television or film can produce lasting fears. While inherently anxious children are more vulnerable to fears induced by media, 90% of undergraduates report at least one enduring fear that started this way, and 26% report persistence to the present. At least one-third of youth have fear reactions to media. Simply the number of hours watching television is associated with a child’s increased perception of personal vulnerability. While 8- to 10-year-olds had reduced fear when parents explained news events, more realistic and serious coverage (the Iraq War, for example) and older age predicted more severe fear reactions not similarly reassured. With this high prevalence of anxiety, I encourage parents to avoid media whose content is not known to them for all children, but especially for those already anxious or traumatized. It amazes me how many families of anxious children have the Weather Channel on constantly, showing devastation all over the world, oblivious that the child is internalizing the risk as though it was outside their window! When media trauma exposure can’t be avoided, parents need to show calm and provide explanation to the child to put it in perspective, as we saw the father do on TV after the Paris massacre.
Modeling of fearful reactions is the second powerful influence on the development of fears. How caregivers react when they encounter a situation such as an approaching dog is quickly modeled by the child. This vicarious learning by watching others’ reactions evolved as preferable to having to chance it yourself. Mothers’ voices and actions are especially salient to children, compared with fathers’ voices and actions. Unfortunately, females tend to be both more fearful and more expressive of fear than males. Some approaches you can suggest regarding modeling include coaching parents (sometimes even sitters) to dampen or mask their reactions, provide other adults without a similar fear to model for the child, or at least not tell the child why they are walking a different route to avoid a dog!
How information about threats is transmitted is the third and perhaps most modifiable influence on a child’s development of fears. Parents talk to children constantly, and a lot of it is warnings! This too may be genetic/cultural as evidenced by the 41% of nursery rhymes across cultures that include violence! Children who have been told potentially bad things about an animal, person, or event show a stronger fear response as measured by self-report, physiological reaction, and behavioral avoidance than when not primed. Conversely, children told positive things react with less fear immediately and are less likely to learn a fear response at later exposures. Once fear has been promoted by negative information, the child’s actual ways of thinking (cognitive biases) are shifted. Attention to forewarned stimuli is increased, the use of reasoning is limited to verifying that fear was warranted rather than alternatively looking for evidence against it, and over estimation of the likelihood of bad outcomes occurs. Children with an overly aroused brain behavioral inhibition system (inherent tendency to react to novelty with physiological arousal and fear) are more influenced by negative verbal information to have fear, cognitive distortions, and avoidance.1
Not surprisingly, anxious parents give more negative information, particularly about ambiguous situations, than other parents. Children living in homes with more negative interactions with fathers or more punitive or neglectful mothers also are more susceptible to increased fears from verbal threat information. Unfortunately, parents generally do not perceive their own role in transmitting threat information. In contrast, one-quarter to one-third of children with significant fears relate onset or intensification of their fears to things they heard. While possibly not relevant for innate fears such as of spiders, this is important information for prevention of fears in general. A child’s development of excessive fear can be somewhat dampened by adult verbal explanations, a focus on the positives, and reassurance, especially if this is done routinely.
The “30 Million Word Gap”2 in total word exposure before age 3 years of children in families on welfare vs. professionals found that higher-income parents provided far more words of praise and six encouragements for every discouragement vs. more total negative vocabulary and two discouragements for every encouragement. The same children more likely to be exposed to trauma also may have less positive preparation to reduce their development of significant fears with the associated stress effects. You and I see this during visits – take the opportunity to discuss and model an alternative.
References
1. Clin Child Fam Psychol Rev. 2010 Jun;13(2):129-50.
2. “The Early Catastrophe: The 30 Million Word Gap by Age 3” (Washington: American Educator, Spring 2003).
Dr. Howard is assistant professor of pediatrics at Johns Hopkins University School of Medicine, Baltimore, and creator of CHADIS. She has no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to Frontline Medical News. Email her at [email protected].
Fear is an intrinsic reaction evolved to protect us from harm. Unsurprisingly, anxiety disorders are common, affecting as many as 25% of children. On average, children have 2-14 fears typical for thinking at their age, from separation (1 year), animals (6 years), environment (dark or storms), medical intrusions or injury (9 years) to social disgrace (16 years). But about one-fifth of children with typical fear topics qualify as having a disorder; that is, they have impairment in functioning.
I wonder daily in my care of anxious children: Is this amount of fear really inevitable? Are there things we can do to avoid this burden on children?
For everyone, genetics predispose fear of things that are dangerous, such as snakes. (Tell me that they don’t make you startle!) Genetic influences account for about 50% of the variance in significant fearfulness as evidenced by parent-child patterns, and the fact that monozygotic twins are more highly concordant in fearfulness than dizygotic. Not much we can do about that!
So, if evolution armed humans with fear for protection, how is it that everyone is not impaired?
In combination with genetic vulnerabilities, fears are learned in three ways: experiential conditioning, modeling, and threat information transmission. These frequently co-occur because bad things happen, genetically anxious parents show a fear reaction, and the same parents warn their children frequently and expressively about potential dangers.
As for avoiding fear conditioning, all parents want to protect their children from scary experiences, but it is not always possible. Car crashes and other bad things happen. Even viewing events that threaten injury or death, such as 9/11, can be sufficient to induce post-traumatic stress disorder (18% of children in New York City). The closer and more severe a scary event is, the more it injures or has potential to injure the child or the child’s loved ones; the more expressive the family members are and the more it is repeated (abuse, for example), the greater the likelihood of it lasting and having impairing effects.
Conditioned fears from real experiences are not entirely random. Low-income children are more likely to experience frightening events from rat bites to house fires to domestic violence to gunshots. Asking about environmental factors or using screening tools such as Safe Environment for Every Kid to evaluate the home environment, and referring families for assistance are steps relevant to every child, but especially anxious ones.
You and I need to continue to advocate for safer communities for all children. In the meantime, it is important to know that encouraging a child to describe in detail to a caring adult – verbally and/or by drawing – traumas they experienced is significantly therapeutic. It might not seem intuitive to parents to promote “reliving the experience,” especially because they may have been traumatized themselves. So providing this opportunity ourselves or through a friend, teacher, or counselor who can calmly answer questions and put the event in perspective, is important advice.
But even simply viewing disasters, violence, or artificial frightening events on television or film can produce lasting fears. While inherently anxious children are more vulnerable to fears induced by media, 90% of undergraduates report at least one enduring fear that started this way, and 26% report persistence to the present. At least one-third of youth have fear reactions to media. Simply the number of hours watching television is associated with a child’s increased perception of personal vulnerability. While 8- to 10-year-olds had reduced fear when parents explained news events, more realistic and serious coverage (the Iraq War, for example) and older age predicted more severe fear reactions not similarly reassured. With this high prevalence of anxiety, I encourage parents to avoid media whose content is not known to them for all children, but especially for those already anxious or traumatized. It amazes me how many families of anxious children have the Weather Channel on constantly, showing devastation all over the world, oblivious that the child is internalizing the risk as though it was outside their window! When media trauma exposure can’t be avoided, parents need to show calm and provide explanation to the child to put it in perspective, as we saw the father do on TV after the Paris massacre.
Modeling of fearful reactions is the second powerful influence on the development of fears. How caregivers react when they encounter a situation such as an approaching dog is quickly modeled by the child. This vicarious learning by watching others’ reactions evolved as preferable to having to chance it yourself. Mothers’ voices and actions are especially salient to children, compared with fathers’ voices and actions. Unfortunately, females tend to be both more fearful and more expressive of fear than males. Some approaches you can suggest regarding modeling include coaching parents (sometimes even sitters) to dampen or mask their reactions, provide other adults without a similar fear to model for the child, or at least not tell the child why they are walking a different route to avoid a dog!
How information about threats is transmitted is the third and perhaps most modifiable influence on a child’s development of fears. Parents talk to children constantly, and a lot of it is warnings! This too may be genetic/cultural as evidenced by the 41% of nursery rhymes across cultures that include violence! Children who have been told potentially bad things about an animal, person, or event show a stronger fear response as measured by self-report, physiological reaction, and behavioral avoidance than when not primed. Conversely, children told positive things react with less fear immediately and are less likely to learn a fear response at later exposures. Once fear has been promoted by negative information, the child’s actual ways of thinking (cognitive biases) are shifted. Attention to forewarned stimuli is increased, the use of reasoning is limited to verifying that fear was warranted rather than alternatively looking for evidence against it, and over estimation of the likelihood of bad outcomes occurs. Children with an overly aroused brain behavioral inhibition system (inherent tendency to react to novelty with physiological arousal and fear) are more influenced by negative verbal information to have fear, cognitive distortions, and avoidance.1
Not surprisingly, anxious parents give more negative information, particularly about ambiguous situations, than other parents. Children living in homes with more negative interactions with fathers or more punitive or neglectful mothers also are more susceptible to increased fears from verbal threat information. Unfortunately, parents generally do not perceive their own role in transmitting threat information. In contrast, one-quarter to one-third of children with significant fears relate onset or intensification of their fears to things they heard. While possibly not relevant for innate fears such as of spiders, this is important information for prevention of fears in general. A child’s development of excessive fear can be somewhat dampened by adult verbal explanations, a focus on the positives, and reassurance, especially if this is done routinely.
The “30 Million Word Gap”2 in total word exposure before age 3 years of children in families on welfare vs. professionals found that higher-income parents provided far more words of praise and six encouragements for every discouragement vs. more total negative vocabulary and two discouragements for every encouragement. The same children more likely to be exposed to trauma also may have less positive preparation to reduce their development of significant fears with the associated stress effects. You and I see this during visits – take the opportunity to discuss and model an alternative.
References
1. Clin Child Fam Psychol Rev. 2010 Jun;13(2):129-50.
2. “The Early Catastrophe: The 30 Million Word Gap by Age 3” (Washington: American Educator, Spring 2003).
Dr. Howard is assistant professor of pediatrics at Johns Hopkins University School of Medicine, Baltimore, and creator of CHADIS. She has no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to Frontline Medical News. Email her at [email protected].
Fear is an intrinsic reaction evolved to protect us from harm. Unsurprisingly, anxiety disorders are common, affecting as many as 25% of children. On average, children have 2-14 fears typical for thinking at their age, from separation (1 year), animals (6 years), environment (dark or storms), medical intrusions or injury (9 years) to social disgrace (16 years). But about one-fifth of children with typical fear topics qualify as having a disorder; that is, they have impairment in functioning.
I wonder daily in my care of anxious children: Is this amount of fear really inevitable? Are there things we can do to avoid this burden on children?
For everyone, genetics predispose fear of things that are dangerous, such as snakes. (Tell me that they don’t make you startle!) Genetic influences account for about 50% of the variance in significant fearfulness as evidenced by parent-child patterns, and the fact that monozygotic twins are more highly concordant in fearfulness than dizygotic. Not much we can do about that!
So, if evolution armed humans with fear for protection, how is it that everyone is not impaired?
In combination with genetic vulnerabilities, fears are learned in three ways: experiential conditioning, modeling, and threat information transmission. These frequently co-occur because bad things happen, genetically anxious parents show a fear reaction, and the same parents warn their children frequently and expressively about potential dangers.
As for avoiding fear conditioning, all parents want to protect their children from scary experiences, but it is not always possible. Car crashes and other bad things happen. Even viewing events that threaten injury or death, such as 9/11, can be sufficient to induce post-traumatic stress disorder (18% of children in New York City). The closer and more severe a scary event is, the more it injures or has potential to injure the child or the child’s loved ones; the more expressive the family members are and the more it is repeated (abuse, for example), the greater the likelihood of it lasting and having impairing effects.
Conditioned fears from real experiences are not entirely random. Low-income children are more likely to experience frightening events from rat bites to house fires to domestic violence to gunshots. Asking about environmental factors or using screening tools such as Safe Environment for Every Kid to evaluate the home environment, and referring families for assistance are steps relevant to every child, but especially anxious ones.
You and I need to continue to advocate for safer communities for all children. In the meantime, it is important to know that encouraging a child to describe in detail to a caring adult – verbally and/or by drawing – traumas they experienced is significantly therapeutic. It might not seem intuitive to parents to promote “reliving the experience,” especially because they may have been traumatized themselves. So providing this opportunity ourselves or through a friend, teacher, or counselor who can calmly answer questions and put the event in perspective, is important advice.
But even simply viewing disasters, violence, or artificial frightening events on television or film can produce lasting fears. While inherently anxious children are more vulnerable to fears induced by media, 90% of undergraduates report at least one enduring fear that started this way, and 26% report persistence to the present. At least one-third of youth have fear reactions to media. Simply the number of hours watching television is associated with a child’s increased perception of personal vulnerability. While 8- to 10-year-olds had reduced fear when parents explained news events, more realistic and serious coverage (the Iraq War, for example) and older age predicted more severe fear reactions not similarly reassured. With this high prevalence of anxiety, I encourage parents to avoid media whose content is not known to them for all children, but especially for those already anxious or traumatized. It amazes me how many families of anxious children have the Weather Channel on constantly, showing devastation all over the world, oblivious that the child is internalizing the risk as though it was outside their window! When media trauma exposure can’t be avoided, parents need to show calm and provide explanation to the child to put it in perspective, as we saw the father do on TV after the Paris massacre.
Modeling of fearful reactions is the second powerful influence on the development of fears. How caregivers react when they encounter a situation such as an approaching dog is quickly modeled by the child. This vicarious learning by watching others’ reactions evolved as preferable to having to chance it yourself. Mothers’ voices and actions are especially salient to children, compared with fathers’ voices and actions. Unfortunately, females tend to be both more fearful and more expressive of fear than males. Some approaches you can suggest regarding modeling include coaching parents (sometimes even sitters) to dampen or mask their reactions, provide other adults without a similar fear to model for the child, or at least not tell the child why they are walking a different route to avoid a dog!
How information about threats is transmitted is the third and perhaps most modifiable influence on a child’s development of fears. Parents talk to children constantly, and a lot of it is warnings! This too may be genetic/cultural as evidenced by the 41% of nursery rhymes across cultures that include violence! Children who have been told potentially bad things about an animal, person, or event show a stronger fear response as measured by self-report, physiological reaction, and behavioral avoidance than when not primed. Conversely, children told positive things react with less fear immediately and are less likely to learn a fear response at later exposures. Once fear has been promoted by negative information, the child’s actual ways of thinking (cognitive biases) are shifted. Attention to forewarned stimuli is increased, the use of reasoning is limited to verifying that fear was warranted rather than alternatively looking for evidence against it, and over estimation of the likelihood of bad outcomes occurs. Children with an overly aroused brain behavioral inhibition system (inherent tendency to react to novelty with physiological arousal and fear) are more influenced by negative verbal information to have fear, cognitive distortions, and avoidance.1
Not surprisingly, anxious parents give more negative information, particularly about ambiguous situations, than other parents. Children living in homes with more negative interactions with fathers or more punitive or neglectful mothers also are more susceptible to increased fears from verbal threat information. Unfortunately, parents generally do not perceive their own role in transmitting threat information. In contrast, one-quarter to one-third of children with significant fears relate onset or intensification of their fears to things they heard. While possibly not relevant for innate fears such as of spiders, this is important information for prevention of fears in general. A child’s development of excessive fear can be somewhat dampened by adult verbal explanations, a focus on the positives, and reassurance, especially if this is done routinely.
The “30 Million Word Gap”2 in total word exposure before age 3 years of children in families on welfare vs. professionals found that higher-income parents provided far more words of praise and six encouragements for every discouragement vs. more total negative vocabulary and two discouragements for every encouragement. The same children more likely to be exposed to trauma also may have less positive preparation to reduce their development of significant fears with the associated stress effects. You and I see this during visits – take the opportunity to discuss and model an alternative.
References
1. Clin Child Fam Psychol Rev. 2010 Jun;13(2):129-50.
2. “The Early Catastrophe: The 30 Million Word Gap by Age 3” (Washington: American Educator, Spring 2003).
Dr. Howard is assistant professor of pediatrics at Johns Hopkins University School of Medicine, Baltimore, and creator of CHADIS. She has no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to Frontline Medical News. Email her at [email protected].

Early Signs May Precede Parkinson’s Disease Diagnosis by Six Years
VANCOUVER—Patients may have impairments in their daily activities six years before they receive a diagnosis of Parkinson’s disease, according to an analysis presented at the 68th Annual Meeting of the American Academy of Neurology. These problems typically involve instrumental activities, which require motor and nonmotor skills. Subtle motor and cognitive deficits also may be observable six years before diagnosis. As the time of diagnosis approaches, additional motor symptoms arise, and patients report symptoms of anxiety and depression more frequently than people who do not develop Parkinson’s disease, said Sirwan Darweesh, MD, of the Erasmus MC University Medical Center in Rotterdam, the Netherlands. In addition, patients’ problems in daily functioning begin to affect basic activities.

Among researchers, interest in defining earlier stages of Parkinson’s disease is increasing, under the assumption that it may enable effective neuroprotection. Dr. Darweesh and colleagues conducted a nested case–control study within the population-based Rotterdam Study. The latter study was initiated in 1990 among inhabitants of a district of Rotterdam. All persons age 55 or older were invited to participate. Dr. Darweesh and colleagues excluded people with Parkinson’s disease at the time of enrollment from their analyses.
Approximately 8,000 people (about 80% of the eligible population) participated in the study. Individuals who did not participate tended to be slightly older and to have more locomotor diseases and more cardiovascular diseases.
At baseline, participants underwent a battery of assessments for potential prediagnostic features of Parkinson’s disease. The tests represented the three broad categories of activities of daily living, motor features, and nonmotor features. The investigators also assessed symptoms of depression and anxiety with the Center for Epidemiologic Studies Depression Scale and the Hospital Anxiety and Depression Scale. These assessments were repeated every four years.
In addition, the researchers evaluated whether participants had developed Parkinson’s disease during follow-up. Research physicians conducted repeated in person examinations and had complete access to participants’ medical files, which included diagnostic codes, specialist letters, and free text entries by neurologists and other physicians.
To identify differences between prediagnostic patients with Parkinson’s disease and controls, Dr. Darweesh and colleagues matched every person with incident Parkinson’s disease with 15 controls, based on age and sex. They analyzed the data to determine when prediagnostic features differed significantly between cases and controls during the prediagnostic period. In all, 107 people (approximately 50% women) developed incident Parkinson’s disease at a mean age of 77.
Movement and Postural Problems Emerged
At six years before Parkinson’s disease diagnosis, prediagnostic patients reported problems with daily activities, specifically instrumental activities such as traveling, more frequently than controls did. At about three years before diagnosis, problems in daily functioning extended to basic tasks such as eating, said Dr. Darweesh.
At about five to six years before diagnosis, prediagnostic patients showed signs of hypokinesia, bradykinesia, or tremor more frequently than controls did. In the last few years before diagnosis, patients also showed signs of cogwheel rigidity, postural abnormality, and postural imbalance more frequently than controls did.
When the investigators examined nonmotor features, they found that at about five years before diagnosis, patients with Parkinson’s disease already had significantly lower Mini-Mental State Examination scores than controls did. In the years before diagnosis, patients were also more likely to report anxiety symptoms and depressive symptoms than controls were. During follow-up, the researchers found that patients with Parkinson’s disease used laxatives more frequently than controls did, although differences only became significant in the last few years before diagnosis. “We believe that these data add important information to our knowledge of prediagnostic Parkinson’s disease,” said Dr. Darweesh. “We hope that these data can contribute to a better understanding of how to define earlier stages of this disease.”
—Erik Greb
Suggested Readings
Arnulf I, Neutel D, Herlin B, et al. Sleepiness in idiopathic REM sleep behavior disorder and Parkinson disease. Sleep. 2015;38(10):1529-1535.
Beavan M, McNeill A, Proukakis C, et al. Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol. 2015;72(2):201-208.
Schrag A, Horsfall L, Walters K, et al. Prediagnostic presentations of Parkinson’s disease in primary care: a case-control study. Lancet Neurol. 2015;14(1):57-64.
VANCOUVER—Patients may have impairments in their daily activities six years before they receive a diagnosis of Parkinson’s disease, according to an analysis presented at the 68th Annual Meeting of the American Academy of Neurology. These problems typically involve instrumental activities, which require motor and nonmotor skills. Subtle motor and cognitive deficits also may be observable six years before diagnosis. As the time of diagnosis approaches, additional motor symptoms arise, and patients report symptoms of anxiety and depression more frequently than people who do not develop Parkinson’s disease, said Sirwan Darweesh, MD, of the Erasmus MC University Medical Center in Rotterdam, the Netherlands. In addition, patients’ problems in daily functioning begin to affect basic activities.
Among researchers, interest in defining earlier stages of Parkinson’s disease is increasing, under the assumption that it may enable effective neuroprotection. Dr. Darweesh and colleagues conducted a nested case–control study within the population-based Rotterdam Study. The latter study was initiated in 1990 among inhabitants of a district of Rotterdam. All persons age 55 or older were invited to participate. Dr. Darweesh and colleagues excluded people with Parkinson’s disease at the time of enrollment from their analyses.
Approximately 8,000 people (about 80% of the eligible population) participated in the study. Individuals who did not participate tended to be slightly older and to have more locomotor diseases and more cardiovascular diseases.
At baseline, participants underwent a battery of assessments for potential prediagnostic features of Parkinson’s disease. The tests represented the three broad categories of activities of daily living, motor features, and nonmotor features. The investigators also assessed symptoms of depression and anxiety with the Center for Epidemiologic Studies Depression Scale and the Hospital Anxiety and Depression Scale. These assessments were repeated every four years.
In addition, the researchers evaluated whether participants had developed Parkinson’s disease during follow-up. Research physicians conducted repeated in person examinations and had complete access to participants’ medical files, which included diagnostic codes, specialist letters, and free text entries by neurologists and other physicians.
To identify differences between prediagnostic patients with Parkinson’s disease and controls, Dr. Darweesh and colleagues matched every person with incident Parkinson’s disease with 15 controls, based on age and sex. They analyzed the data to determine when prediagnostic features differed significantly between cases and controls during the prediagnostic period. In all, 107 people (approximately 50% women) developed incident Parkinson’s disease at a mean age of 77.
Movement and Postural Problems Emerged
At six years before Parkinson’s disease diagnosis, prediagnostic patients reported problems with daily activities, specifically instrumental activities such as traveling, more frequently than controls did. At about three years before diagnosis, problems in daily functioning extended to basic tasks such as eating, said Dr. Darweesh.
At about five to six years before diagnosis, prediagnostic patients showed signs of hypokinesia, bradykinesia, or tremor more frequently than controls did. In the last few years before diagnosis, patients also showed signs of cogwheel rigidity, postural abnormality, and postural imbalance more frequently than controls did.
When the investigators examined nonmotor features, they found that at about five years before diagnosis, patients with Parkinson’s disease already had significantly lower Mini-Mental State Examination scores than controls did. In the years before diagnosis, patients were also more likely to report anxiety symptoms and depressive symptoms than controls were. During follow-up, the researchers found that patients with Parkinson’s disease used laxatives more frequently than controls did, although differences only became significant in the last few years before diagnosis. “We believe that these data add important information to our knowledge of prediagnostic Parkinson’s disease,” said Dr. Darweesh. “We hope that these data can contribute to a better understanding of how to define earlier stages of this disease.”
—Erik Greb
VANCOUVER—Patients may have impairments in their daily activities six years before they receive a diagnosis of Parkinson’s disease, according to an analysis presented at the 68th Annual Meeting of the American Academy of Neurology. These problems typically involve instrumental activities, which require motor and nonmotor skills. Subtle motor and cognitive deficits also may be observable six years before diagnosis. As the time of diagnosis approaches, additional motor symptoms arise, and patients report symptoms of anxiety and depression more frequently than people who do not develop Parkinson’s disease, said Sirwan Darweesh, MD, of the Erasmus MC University Medical Center in Rotterdam, the Netherlands. In addition, patients’ problems in daily functioning begin to affect basic activities.
Among researchers, interest in defining earlier stages of Parkinson’s disease is increasing, under the assumption that it may enable effective neuroprotection. Dr. Darweesh and colleagues conducted a nested case–control study within the population-based Rotterdam Study. The latter study was initiated in 1990 among inhabitants of a district of Rotterdam. All persons age 55 or older were invited to participate. Dr. Darweesh and colleagues excluded people with Parkinson’s disease at the time of enrollment from their analyses.
Approximately 8,000 people (about 80% of the eligible population) participated in the study. Individuals who did not participate tended to be slightly older and to have more locomotor diseases and more cardiovascular diseases.
At baseline, participants underwent a battery of assessments for potential prediagnostic features of Parkinson’s disease. The tests represented the three broad categories of activities of daily living, motor features, and nonmotor features. The investigators also assessed symptoms of depression and anxiety with the Center for Epidemiologic Studies Depression Scale and the Hospital Anxiety and Depression Scale. These assessments were repeated every four years.
In addition, the researchers evaluated whether participants had developed Parkinson’s disease during follow-up. Research physicians conducted repeated in person examinations and had complete access to participants’ medical files, which included diagnostic codes, specialist letters, and free text entries by neurologists and other physicians.
To identify differences between prediagnostic patients with Parkinson’s disease and controls, Dr. Darweesh and colleagues matched every person with incident Parkinson’s disease with 15 controls, based on age and sex. They analyzed the data to determine when prediagnostic features differed significantly between cases and controls during the prediagnostic period. In all, 107 people (approximately 50% women) developed incident Parkinson’s disease at a mean age of 77.
Movement and Postural Problems Emerged
At six years before Parkinson’s disease diagnosis, prediagnostic patients reported problems with daily activities, specifically instrumental activities such as traveling, more frequently than controls did. At about three years before diagnosis, problems in daily functioning extended to basic tasks such as eating, said Dr. Darweesh.
At about five to six years before diagnosis, prediagnostic patients showed signs of hypokinesia, bradykinesia, or tremor more frequently than controls did. In the last few years before diagnosis, patients also showed signs of cogwheel rigidity, postural abnormality, and postural imbalance more frequently than controls did.
When the investigators examined nonmotor features, they found that at about five years before diagnosis, patients with Parkinson’s disease already had significantly lower Mini-Mental State Examination scores than controls did. In the years before diagnosis, patients were also more likely to report anxiety symptoms and depressive symptoms than controls were. During follow-up, the researchers found that patients with Parkinson’s disease used laxatives more frequently than controls did, although differences only became significant in the last few years before diagnosis. “We believe that these data add important information to our knowledge of prediagnostic Parkinson’s disease,” said Dr. Darweesh. “We hope that these data can contribute to a better understanding of how to define earlier stages of this disease.”
—Erik Greb
Suggested Readings
Arnulf I, Neutel D, Herlin B, et al. Sleepiness in idiopathic REM sleep behavior disorder and Parkinson disease. Sleep. 2015;38(10):1529-1535.
Beavan M, McNeill A, Proukakis C, et al. Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol. 2015;72(2):201-208.
Schrag A, Horsfall L, Walters K, et al. Prediagnostic presentations of Parkinson’s disease in primary care: a case-control study. Lancet Neurol. 2015;14(1):57-64.
Suggested Readings
Arnulf I, Neutel D, Herlin B, et al. Sleepiness in idiopathic REM sleep behavior disorder and Parkinson disease. Sleep. 2015;38(10):1529-1535.
Beavan M, McNeill A, Proukakis C, et al. Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol. 2015;72(2):201-208.
Schrag A, Horsfall L, Walters K, et al. Prediagnostic presentations of Parkinson’s disease in primary care: a case-control study. Lancet Neurol. 2015;14(1):57-64.

ERAS protocol benefited colorectal surgery patients
LOS ANGELES – Implementation of a perioperative protocol designed to enhance recovery in patients undergoing elective laparoscopic colorectal surgery decreased hospital length of stay, the rate of complications, and overall direct costs, results from a single-center study showed.
“Until recently patients undergoing colorectal surgery were counseled to accept a 20%-25% risk of complications and a 7- to 10-day postoperative stay in the hospital,” lead study author Dr. Daniel S. Lavy said at the annual meeting of the American Society of Colon and Rectal Surgeons. “Studies from the 1990s have shown that length of stay rates improved when one single component of care was changed.”

Dr. Lavy of the department of surgery at Monmouth Medical Center, Long Branch, N.J., discussed results from a study of Enhanced Recovery After Surgery (ERAS), which he described as “a multimodal perioperative care pathway designed to achieve early recovery for patients undergoing major surgery. Many of its elements challenge existing surgical doctrine, including optimizing nutrition, standardized nonnarcotic and anesthetic regimens, early mobilization, and early initiation of enteral feeding.” The protocol also includes multimodal analgesia aimed at reducing the use of narcotics by intravenous Toradol (ketorolac), intravenous Tylenol (acetaminophen), and a transverse abdominis plane block; preoperative intravenous Solu-Medrol (methylprednisolone); prevention of fluid overload; preoperative and postoperative Entereg (alvimopan); preoperative enteral feedings and early postoperative diet initiation; and aggressive postoperative rehabilitation.
In an effort to evaluate the impact of the protocol in patients undergoing colorectal surgery, Dr. Lavy and his associates analyzed records from 283 elective laparoscopic colon procedures performed at Monmouth Medical Center from July 2013 to December 2015, a time period that included 11 months prior to implementation of ERAS and 18 months after implementation. The data were analyzed using control charts to assess for process changes, while open or emergent procedures were excluded from review. Key measures assessed included hospital length of stay, direct hospital costs, 30-day readmissions, and complications.
Dr. Lavy reported that following implementation of the ERAS protocol, the median length of stay decreased from 3.8 days to 2.8 days; the median direct hospital costs fell 8.5%, resulting in a savings of $876 per case; and the complication rate dropped from 20% to 16%. No changes were observed in the 30-day readmission rate, which held steady at 8%.
“This multifaceted approach has decreased hospital stay, decreased hospital cost and complication rate, did not change the 30-day readmission rate, and maintained patient safety while improving patient care,” Dr. Lavy said. “We suggest research be conducted to determine how this pathway can be altered to further improve quality of care given to patients while simultaneously reducing hospital costs. Also, these methods may be able to be applied to other surgical subspecialties, including ob.gyn., orthopedics, and urology.”
Dr. Lavy reported having no relevant financial disclosures.
LOS ANGELES – Implementation of a perioperative protocol designed to enhance recovery in patients undergoing elective laparoscopic colorectal surgery decreased hospital length of stay, the rate of complications, and overall direct costs, results from a single-center study showed.
“Until recently patients undergoing colorectal surgery were counseled to accept a 20%-25% risk of complications and a 7- to 10-day postoperative stay in the hospital,” lead study author Dr. Daniel S. Lavy said at the annual meeting of the American Society of Colon and Rectal Surgeons. “Studies from the 1990s have shown that length of stay rates improved when one single component of care was changed.”
Dr. Lavy of the department of surgery at Monmouth Medical Center, Long Branch, N.J., discussed results from a study of Enhanced Recovery After Surgery (ERAS), which he described as “a multimodal perioperative care pathway designed to achieve early recovery for patients undergoing major surgery. Many of its elements challenge existing surgical doctrine, including optimizing nutrition, standardized nonnarcotic and anesthetic regimens, early mobilization, and early initiation of enteral feeding.” The protocol also includes multimodal analgesia aimed at reducing the use of narcotics by intravenous Toradol (ketorolac), intravenous Tylenol (acetaminophen), and a transverse abdominis plane block; preoperative intravenous Solu-Medrol (methylprednisolone); prevention of fluid overload; preoperative and postoperative Entereg (alvimopan); preoperative enteral feedings and early postoperative diet initiation; and aggressive postoperative rehabilitation.
In an effort to evaluate the impact of the protocol in patients undergoing colorectal surgery, Dr. Lavy and his associates analyzed records from 283 elective laparoscopic colon procedures performed at Monmouth Medical Center from July 2013 to December 2015, a time period that included 11 months prior to implementation of ERAS and 18 months after implementation. The data were analyzed using control charts to assess for process changes, while open or emergent procedures were excluded from review. Key measures assessed included hospital length of stay, direct hospital costs, 30-day readmissions, and complications.
Dr. Lavy reported that following implementation of the ERAS protocol, the median length of stay decreased from 3.8 days to 2.8 days; the median direct hospital costs fell 8.5%, resulting in a savings of $876 per case; and the complication rate dropped from 20% to 16%. No changes were observed in the 30-day readmission rate, which held steady at 8%.
“This multifaceted approach has decreased hospital stay, decreased hospital cost and complication rate, did not change the 30-day readmission rate, and maintained patient safety while improving patient care,” Dr. Lavy said. “We suggest research be conducted to determine how this pathway can be altered to further improve quality of care given to patients while simultaneously reducing hospital costs. Also, these methods may be able to be applied to other surgical subspecialties, including ob.gyn., orthopedics, and urology.”
Dr. Lavy reported having no relevant financial disclosures.
LOS ANGELES – Implementation of a perioperative protocol designed to enhance recovery in patients undergoing elective laparoscopic colorectal surgery decreased hospital length of stay, the rate of complications, and overall direct costs, results from a single-center study showed.
“Until recently patients undergoing colorectal surgery were counseled to accept a 20%-25% risk of complications and a 7- to 10-day postoperative stay in the hospital,” lead study author Dr. Daniel S. Lavy said at the annual meeting of the American Society of Colon and Rectal Surgeons. “Studies from the 1990s have shown that length of stay rates improved when one single component of care was changed.”
Dr. Lavy of the department of surgery at Monmouth Medical Center, Long Branch, N.J., discussed results from a study of Enhanced Recovery After Surgery (ERAS), which he described as “a multimodal perioperative care pathway designed to achieve early recovery for patients undergoing major surgery. Many of its elements challenge existing surgical doctrine, including optimizing nutrition, standardized nonnarcotic and anesthetic regimens, early mobilization, and early initiation of enteral feeding.” The protocol also includes multimodal analgesia aimed at reducing the use of narcotics by intravenous Toradol (ketorolac), intravenous Tylenol (acetaminophen), and a transverse abdominis plane block; preoperative intravenous Solu-Medrol (methylprednisolone); prevention of fluid overload; preoperative and postoperative Entereg (alvimopan); preoperative enteral feedings and early postoperative diet initiation; and aggressive postoperative rehabilitation.
In an effort to evaluate the impact of the protocol in patients undergoing colorectal surgery, Dr. Lavy and his associates analyzed records from 283 elective laparoscopic colon procedures performed at Monmouth Medical Center from July 2013 to December 2015, a time period that included 11 months prior to implementation of ERAS and 18 months after implementation. The data were analyzed using control charts to assess for process changes, while open or emergent procedures were excluded from review. Key measures assessed included hospital length of stay, direct hospital costs, 30-day readmissions, and complications.
Dr. Lavy reported that following implementation of the ERAS protocol, the median length of stay decreased from 3.8 days to 2.8 days; the median direct hospital costs fell 8.5%, resulting in a savings of $876 per case; and the complication rate dropped from 20% to 16%. No changes were observed in the 30-day readmission rate, which held steady at 8%.
“This multifaceted approach has decreased hospital stay, decreased hospital cost and complication rate, did not change the 30-day readmission rate, and maintained patient safety while improving patient care,” Dr. Lavy said. “We suggest research be conducted to determine how this pathway can be altered to further improve quality of care given to patients while simultaneously reducing hospital costs. Also, these methods may be able to be applied to other surgical subspecialties, including ob.gyn., orthopedics, and urology.”
Dr. Lavy reported having no relevant financial disclosures.
AT THE ASCRS ANNUAL MEETING
Key clinical point: A multifaceted perioperative protocol benefited patients undergoing laparoscopic colorectal surgery.
Major finding: Following implementation of the protocol, the median length of stay decreased from 3.8 days to 2.8 days, the median direct hospital costs fell 8.5%, and the complication rate dropped from 20% to 16%.
Data source: A review of records from 283 elective laparoscopic colon procedures performed from July 2013 to December 2015.
Disclosures: Dr. Lavy reported having no relevant financial disclosures.

VIDEO: Integrated care effective in first-episode psychosis
ATLANTA – Increasingly, data support taking an integrated approach to care in the intervention of first-episode psychosis.
But what are the key components of such treatment?
Dr. Charles Schulz, who initiated an integrated care clinic for first-episode psychosis at the University of Minnesota, Minneapolis, outlined the steps in an integrated care approach.
In an interview at the annual meeting of the American Psychiatric Association, Dr. Schulz reviewed the importance of intervening in the prodromal phase whenever possible, as well as offering cognitive-behavioral and remediation therapies along with medication management.
He also addressed the need for family psychoeducation and group therapy. And Dr. Schulz explained what to do when there might be a differential that manifests with psychiatric presentations that are not psychosis.
Dr. Schulz said he has industry relationships with Forum Pharmaceuticals and Myriad.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @whitneymcknight
ATLANTA – Increasingly, data support taking an integrated approach to care in the intervention of first-episode psychosis.
But what are the key components of such treatment?
Dr. Charles Schulz, who initiated an integrated care clinic for first-episode psychosis at the University of Minnesota, Minneapolis, outlined the steps in an integrated care approach.
In an interview at the annual meeting of the American Psychiatric Association, Dr. Schulz reviewed the importance of intervening in the prodromal phase whenever possible, as well as offering cognitive-behavioral and remediation therapies along with medication management.
He also addressed the need for family psychoeducation and group therapy. And Dr. Schulz explained what to do when there might be a differential that manifests with psychiatric presentations that are not psychosis.
Dr. Schulz said he has industry relationships with Forum Pharmaceuticals and Myriad.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @whitneymcknight
ATLANTA – Increasingly, data support taking an integrated approach to care in the intervention of first-episode psychosis.
But what are the key components of such treatment?
Dr. Charles Schulz, who initiated an integrated care clinic for first-episode psychosis at the University of Minnesota, Minneapolis, outlined the steps in an integrated care approach.
In an interview at the annual meeting of the American Psychiatric Association, Dr. Schulz reviewed the importance of intervening in the prodromal phase whenever possible, as well as offering cognitive-behavioral and remediation therapies along with medication management.
He also addressed the need for family psychoeducation and group therapy. And Dr. Schulz explained what to do when there might be a differential that manifests with psychiatric presentations that are not psychosis.
Dr. Schulz said he has industry relationships with Forum Pharmaceuticals and Myriad.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @whitneymcknight
EXPERT ANALYSIS FROM THE APA ANNUAL MEETING

Itch, scratch, ad infinitum
1. A 62-year-old woman reports anogenital itching with pain on scratching and has developed introital dyspareunia. Physical exam reveals a well-demarcated white plaque of thickened, crinkled skin. A wet mount shows parabasal cells and no lactobacilli.
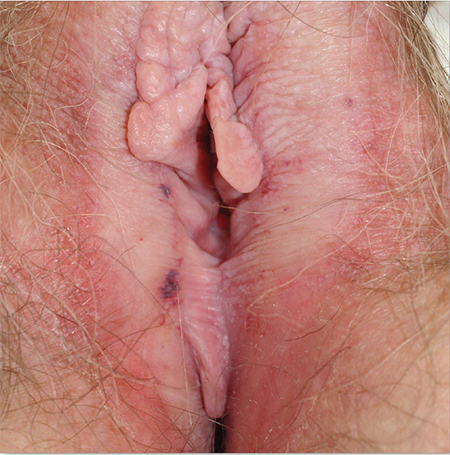
Diagnosis: Lichen sclerosus is a skin disease found most often on the vulva of postmenopausal women, although it also can affect prepubertal children and reproductive-age women. Lichen sclerosus is multifactorial in pathogenesis, including prominent autoimmune factors, local environmental factors, and genetic predisposition.1
Although there is no cure for lichen sclerosus, the symptoms and clinical abnormalities usually can be well managed with ultra-potent topical corticosteroids. However, scarring and architectural changes are not reversible. Moreover, poorly controlled lichen sclerosus exhibits malignant transformation on anogenital skin in about 3% of affected patients.
For more information on this case, see “Chronic vulvar symptoms and dermatologic disruptions: How to make the correct diagnosis.” OBG Manag. 2014;26(5):30-49.
For the next photograph, proceed to the next page >>
2. A 34-year-old patient reports excruciating itching, with disruption of daily activities and sleep. She has been treated for candidiasis on multiple occasions, but her wet mount and confirmatory culture are negative. Physical exam reveals a pink, lichenified plaque with excoriation.
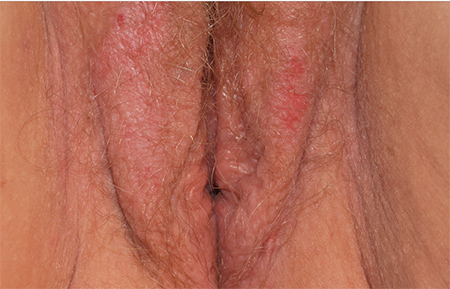
Diagnosis: Lichen simplex chronicus (formerly called squamous hyperplasia or hyperplastic dystrophy, and also known as eczema, neurodermatitis, or localized atopic dermatitis) occurs when irritation from any cause produces itching in a predisposed person. The subsequent scratching and rubbing both produce the rash and exacerbate the irritation that drives the itching, even after the original cause is gone. The rubbing and scratching perpetuate the irritation and itching, producing the “itch-scratch” cycle.
The appearance of lichen simplex chronicus is produced by rubbing (where the skin thickens and lichenifies) or scratching (where the skin becomes red with linear erosions, called excoriations, caused by fingernails).
For more information on this case, see “Chronic vulvar symptoms and dermatologic disruptions: How to make the correct diagnosis.” OBG Manag. 2014;26(5):30-49.
For the next photograph, proceed to the next page >>
3. A 36-year-old has a history of chronic yeast infection with introital burning, discharge, and dyspareunia. She is otherwise healthy, except for irritable bowel syndrome and fibromyalgia. A mild patchy redness appears on the vestibule and surrounding modified mucous membranes. Gentle probing triggers exquisite pain in the vestibule, with slight extension to the labia minora. Lactobacilli are abundant.
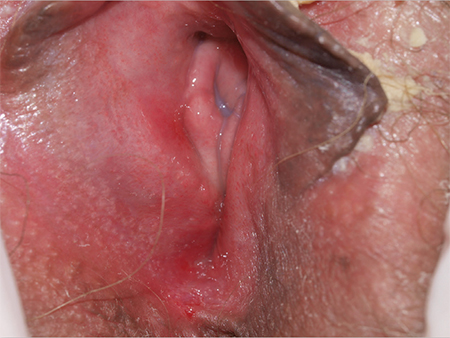
Diagnosis: Vulvodynia is a genital pain syndrome defined as sensations of chronic burning, irritation, rawness, and soreness in the absence of objective disease and infection that could explain the discomfort. Vulvodynia occurs in approximately 7% to 8% of women.
Vulvodynia generally is believed to be a multifactorial symptom, occurring as a result of pelvic floor dysfunction and neuropathic pain, with anxiety/depression issues exacerbating symptoms. Some recent studies have shown the presence of biochemical mediators of inflammation in the absence of clinical and histologic inflammation. Discomfort often is worsened by infections or the application of common irritants (creams, panty liners, soaps, some topical anesthetics). Estrogen deficiency is another common exacerbating factor.
For more information on this case, see “Chronic vulvar symptoms and dermatologic disruptions: How to make the correct diagnosis.” OBG Manag. 2014;26(5):30-49.
For the next photograph, proceed to the next page >>
4. A 73-year-old patient has longstanding introital itching and rawness, with dyspareunia. Topical estradiol cream intravaginally three times weekly with weekly fluconazole has not helped. The physical reveals deep red patches and erosions of the vestibule and anterior mucosal membranes, with loss of the labia minora. An oral exam shows deep redness of the gingivae and erosions of the buccal mucosae, with surrounding white, lacy papules.
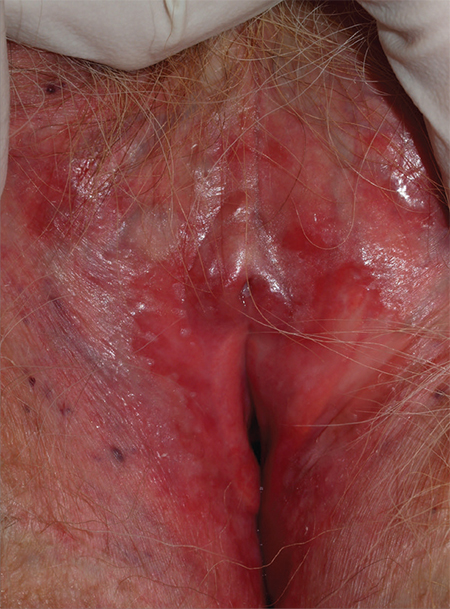
Diagnosis: After correlating the vulvar and oral findings, you make a diagnosis of lichen planus. Erosive multimucosal lichen planus is a disease of cell-mediated immunity that overwhelmingly affects menopausal women. The most common surfaces involved are the mouth, vagina, rectal mucosa, and vulva; usually, at least two surfaces are affected. The esophagus, extra-auditory canals, nasal mucosa, and eyes also can be involved. Dry, extragenital skin usually is not affected in the setting of erosive vulvovaginal lichen planus.
For more information on this case, see “Chronic vulvar symptoms and dermatologic disruptions: How to make the correct diagnosis.” OBG Manag. 2014;26(5):30-49.
1. A 62-year-old woman reports anogenital itching with pain on scratching and has developed introital dyspareunia. Physical exam reveals a well-demarcated white plaque of thickened, crinkled skin. A wet mount shows parabasal cells and no lactobacilli.
Diagnosis: Lichen sclerosus is a skin disease found most often on the vulva of postmenopausal women, although it also can affect prepubertal children and reproductive-age women. Lichen sclerosus is multifactorial in pathogenesis, including prominent autoimmune factors, local environmental factors, and genetic predisposition.1
Although there is no cure for lichen sclerosus, the symptoms and clinical abnormalities usually can be well managed with ultra-potent topical corticosteroids. However, scarring and architectural changes are not reversible. Moreover, poorly controlled lichen sclerosus exhibits malignant transformation on anogenital skin in about 3% of affected patients.
For more information on this case, see “Chronic vulvar symptoms and dermatologic disruptions: How to make the correct diagnosis.” OBG Manag. 2014;26(5):30-49.
For the next photograph, proceed to the next page >>
2. A 34-year-old patient reports excruciating itching, with disruption of daily activities and sleep. She has been treated for candidiasis on multiple occasions, but her wet mount and confirmatory culture are negative. Physical exam reveals a pink, lichenified plaque with excoriation.
Diagnosis: Lichen simplex chronicus (formerly called squamous hyperplasia or hyperplastic dystrophy, and also known as eczema, neurodermatitis, or localized atopic dermatitis) occurs when irritation from any cause produces itching in a predisposed person. The subsequent scratching and rubbing both produce the rash and exacerbate the irritation that drives the itching, even after the original cause is gone. The rubbing and scratching perpetuate the irritation and itching, producing the “itch-scratch” cycle.
The appearance of lichen simplex chronicus is produced by rubbing (where the skin thickens and lichenifies) or scratching (where the skin becomes red with linear erosions, called excoriations, caused by fingernails).
For more information on this case, see “Chronic vulvar symptoms and dermatologic disruptions: How to make the correct diagnosis.” OBG Manag. 2014;26(5):30-49.
For the next photograph, proceed to the next page >>
3. A 36-year-old has a history of chronic yeast infection with introital burning, discharge, and dyspareunia. She is otherwise healthy, except for irritable bowel syndrome and fibromyalgia. A mild patchy redness appears on the vestibule and surrounding modified mucous membranes. Gentle probing triggers exquisite pain in the vestibule, with slight extension to the labia minora. Lactobacilli are abundant.
Diagnosis: Vulvodynia is a genital pain syndrome defined as sensations of chronic burning, irritation, rawness, and soreness in the absence of objective disease and infection that could explain the discomfort. Vulvodynia occurs in approximately 7% to 8% of women.
Vulvodynia generally is believed to be a multifactorial symptom, occurring as a result of pelvic floor dysfunction and neuropathic pain, with anxiety/depression issues exacerbating symptoms. Some recent studies have shown the presence of biochemical mediators of inflammation in the absence of clinical and histologic inflammation. Discomfort often is worsened by infections or the application of common irritants (creams, panty liners, soaps, some topical anesthetics). Estrogen deficiency is another common exacerbating factor.
For more information on this case, see “Chronic vulvar symptoms and dermatologic disruptions: How to make the correct diagnosis.” OBG Manag. 2014;26(5):30-49.
For the next photograph, proceed to the next page >>
4. A 73-year-old patient has longstanding introital itching and rawness, with dyspareunia. Topical estradiol cream intravaginally three times weekly with weekly fluconazole has not helped. The physical reveals deep red patches and erosions of the vestibule and anterior mucosal membranes, with loss of the labia minora. An oral exam shows deep redness of the gingivae and erosions of the buccal mucosae, with surrounding white, lacy papules.
Diagnosis: After correlating the vulvar and oral findings, you make a diagnosis of lichen planus. Erosive multimucosal lichen planus is a disease of cell-mediated immunity that overwhelmingly affects menopausal women. The most common surfaces involved are the mouth, vagina, rectal mucosa, and vulva; usually, at least two surfaces are affected. The esophagus, extra-auditory canals, nasal mucosa, and eyes also can be involved. Dry, extragenital skin usually is not affected in the setting of erosive vulvovaginal lichen planus.
For more information on this case, see “Chronic vulvar symptoms and dermatologic disruptions: How to make the correct diagnosis.” OBG Manag. 2014;26(5):30-49.
1. A 62-year-old woman reports anogenital itching with pain on scratching and has developed introital dyspareunia. Physical exam reveals a well-demarcated white plaque of thickened, crinkled skin. A wet mount shows parabasal cells and no lactobacilli.
Diagnosis: Lichen sclerosus is a skin disease found most often on the vulva of postmenopausal women, although it also can affect prepubertal children and reproductive-age women. Lichen sclerosus is multifactorial in pathogenesis, including prominent autoimmune factors, local environmental factors, and genetic predisposition.1
Although there is no cure for lichen sclerosus, the symptoms and clinical abnormalities usually can be well managed with ultra-potent topical corticosteroids. However, scarring and architectural changes are not reversible. Moreover, poorly controlled lichen sclerosus exhibits malignant transformation on anogenital skin in about 3% of affected patients.
For more information on this case, see “Chronic vulvar symptoms and dermatologic disruptions: How to make the correct diagnosis.” OBG Manag. 2014;26(5):30-49.
For the next photograph, proceed to the next page >>
2. A 34-year-old patient reports excruciating itching, with disruption of daily activities and sleep. She has been treated for candidiasis on multiple occasions, but her wet mount and confirmatory culture are negative. Physical exam reveals a pink, lichenified plaque with excoriation.
Diagnosis: Lichen simplex chronicus (formerly called squamous hyperplasia or hyperplastic dystrophy, and also known as eczema, neurodermatitis, or localized atopic dermatitis) occurs when irritation from any cause produces itching in a predisposed person. The subsequent scratching and rubbing both produce the rash and exacerbate the irritation that drives the itching, even after the original cause is gone. The rubbing and scratching perpetuate the irritation and itching, producing the “itch-scratch” cycle.
The appearance of lichen simplex chronicus is produced by rubbing (where the skin thickens and lichenifies) or scratching (where the skin becomes red with linear erosions, called excoriations, caused by fingernails).
For more information on this case, see “Chronic vulvar symptoms and dermatologic disruptions: How to make the correct diagnosis.” OBG Manag. 2014;26(5):30-49.
For the next photograph, proceed to the next page >>
3. A 36-year-old has a history of chronic yeast infection with introital burning, discharge, and dyspareunia. She is otherwise healthy, except for irritable bowel syndrome and fibromyalgia. A mild patchy redness appears on the vestibule and surrounding modified mucous membranes. Gentle probing triggers exquisite pain in the vestibule, with slight extension to the labia minora. Lactobacilli are abundant.
Diagnosis: Vulvodynia is a genital pain syndrome defined as sensations of chronic burning, irritation, rawness, and soreness in the absence of objective disease and infection that could explain the discomfort. Vulvodynia occurs in approximately 7% to 8% of women.
Vulvodynia generally is believed to be a multifactorial symptom, occurring as a result of pelvic floor dysfunction and neuropathic pain, with anxiety/depression issues exacerbating symptoms. Some recent studies have shown the presence of biochemical mediators of inflammation in the absence of clinical and histologic inflammation. Discomfort often is worsened by infections or the application of common irritants (creams, panty liners, soaps, some topical anesthetics). Estrogen deficiency is another common exacerbating factor.
For more information on this case, see “Chronic vulvar symptoms and dermatologic disruptions: How to make the correct diagnosis.” OBG Manag. 2014;26(5):30-49.
For the next photograph, proceed to the next page >>
4. A 73-year-old patient has longstanding introital itching and rawness, with dyspareunia. Topical estradiol cream intravaginally three times weekly with weekly fluconazole has not helped. The physical reveals deep red patches and erosions of the vestibule and anterior mucosal membranes, with loss of the labia minora. An oral exam shows deep redness of the gingivae and erosions of the buccal mucosae, with surrounding white, lacy papules.
Diagnosis: After correlating the vulvar and oral findings, you make a diagnosis of lichen planus. Erosive multimucosal lichen planus is a disease of cell-mediated immunity that overwhelmingly affects menopausal women. The most common surfaces involved are the mouth, vagina, rectal mucosa, and vulva; usually, at least two surfaces are affected. The esophagus, extra-auditory canals, nasal mucosa, and eyes also can be involved. Dry, extragenital skin usually is not affected in the setting of erosive vulvovaginal lichen planus.
For more information on this case, see “Chronic vulvar symptoms and dermatologic disruptions: How to make the correct diagnosis.” OBG Manag. 2014;26(5):30-49.
Cangrelor offers advantages for antiplatelet management in PCI
PARIS – Parenteral cangrelor is a potent antiplatelet agent that provides similar anti-ischemic effects but a significantly lower risk of major bleeding than the popular combination of clopidogrel plus a glycoprotein IIb/IIIa inhibitor in patients undergoing elective or urgent percutaneous coronary intervention.
Moreover, cangrelor (Kengreal), a rapid-acting and reversible intravenous platelet P2Y12 inhibitor, cuts the blood transfusion requirement by more than half, Dr. Muthiah Vaduganathan reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.

These findings, based upon a propensity score–matched exploratory pooled analysis of individual patient-level data from the three phase III CHAMPION trials, suggest that cangrelor alone is an attractive alternative to clopidogrel plus a glycoprotein IIb/IIIa inhibitor (GPI) for antiplatelet management in percutaneous coronary intervention (PCI), according to Dr. Vaduganathan of Brigham and Women’s Hospital, Boston.
CHAMPION PLATFORM (N Engl J Med 2009 Dec 10;361[24]:2330-41), CHAMPION PCI (N Engl J Med 2009 Dec 10;361[24]:2318-29), and CHAMPION PHOENIX (N Engl J Med 2013 Apr 11;368[14]:1303-13) collectively included nearly 25,000 patients randomized to cangrelor versus standard therapy for platelet inhibition in elective or urgent PCI. The trials, which established that cangrelor led to fewer ischemic complications within 30 days post PCI than did clopidogrel plus a GPI, led to Food and Drug Administration approval of the drug in 2015. Cangrelor is also approved in Europe.
The cangrelor and clopidogrel groups differed in terms of baseline bleeding risk. For example, the clopidogrel-treated patients were younger, more likely to be male, had fewer comorbid conditions, and more often presented with an acute coronary syndrome. In order to accurately assess the bleeding risk associated with the two antiplatelet strategies, Dr. Vaduganathan and coinvestigators engaged in propensity score matching on the basis of 16 baseline clinical variables. They identified two closely matched cohorts consisting of 1,021 patient pairs.
The 1,021 patients in the cangrelor-alone group had a 2.6% rate of the primary composite efficacy endpoint consisting of all-cause mortality, MI, ischemia-driven revascularization, or stent thrombosis at 48 hours. Although this was 21% lower than the 3.3% rate in the clopidogrel plus GPI group, the difference didn’t achieve statistical significance because of the relatively small sample size.
In contrast, the rate of major bleeding within 48 hours by ACUITY (Acute Catheterization and Urgent Intervention Triage) criteria did differ significantly between the two groups: 3.6% with cangrelor and 5.8% with clopidogrel plus a GPI, for a 39% relative risk reduction in favor of the newer agent. Major bleeding rates were also lower in the cangrelor-treated patients on the GUSTO (Global Utilization of Streptokinase and TPA for Occluded Coronary Arteries) and TIMI (Thrombolysis in Myocardial Infarction) scales, although those trends didn’t achieve significance. The blood transfusion requirement was significantly lower in the cangrelor group: 1% versus 2.1%, for a 55% relative risk reduction.
Dr. Vaduganathan said the CHAMPION investigators’ future plans include examining the possibility of a favorable pharmacologic interaction between cangrelor and bivalirudin, a combination seeing increasing use in the United States.
“Finally, I think it’ll be important moving forward to look at comparative effectiveness data – either observational or randomized – as well as cost-effectiveness data between potent parenteral antiplatelet strategies,” he concluded.
Session chair Dr. Anthony Gershlick of Glenfield General Hospital in Leicester, England, commented that while he found this analysis of the CHAMPION data quite interesting, they are of little relevance in Europe, where cardiologists seldom use GPIs as part of their up-front PCI management strategy, instead resorting to those agents mainly as bailout therapy.
The U.S. situation is very different, according to Dr. Vaduganathan. American cardiologists use GPI therapy in about one-third of PCIs.
The CHAMPION trials were funded by the Medicines Co. Dr. Vaduganathan reported having no relevant financial conflicts.
PARIS – Parenteral cangrelor is a potent antiplatelet agent that provides similar anti-ischemic effects but a significantly lower risk of major bleeding than the popular combination of clopidogrel plus a glycoprotein IIb/IIIa inhibitor in patients undergoing elective or urgent percutaneous coronary intervention.
Moreover, cangrelor (Kengreal), a rapid-acting and reversible intravenous platelet P2Y12 inhibitor, cuts the blood transfusion requirement by more than half, Dr. Muthiah Vaduganathan reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
These findings, based upon a propensity score–matched exploratory pooled analysis of individual patient-level data from the three phase III CHAMPION trials, suggest that cangrelor alone is an attractive alternative to clopidogrel plus a glycoprotein IIb/IIIa inhibitor (GPI) for antiplatelet management in percutaneous coronary intervention (PCI), according to Dr. Vaduganathan of Brigham and Women’s Hospital, Boston.
CHAMPION PLATFORM (N Engl J Med 2009 Dec 10;361[24]:2330-41), CHAMPION PCI (N Engl J Med 2009 Dec 10;361[24]:2318-29), and CHAMPION PHOENIX (N Engl J Med 2013 Apr 11;368[14]:1303-13) collectively included nearly 25,000 patients randomized to cangrelor versus standard therapy for platelet inhibition in elective or urgent PCI. The trials, which established that cangrelor led to fewer ischemic complications within 30 days post PCI than did clopidogrel plus a GPI, led to Food and Drug Administration approval of the drug in 2015. Cangrelor is also approved in Europe.
The cangrelor and clopidogrel groups differed in terms of baseline bleeding risk. For example, the clopidogrel-treated patients were younger, more likely to be male, had fewer comorbid conditions, and more often presented with an acute coronary syndrome. In order to accurately assess the bleeding risk associated with the two antiplatelet strategies, Dr. Vaduganathan and coinvestigators engaged in propensity score matching on the basis of 16 baseline clinical variables. They identified two closely matched cohorts consisting of 1,021 patient pairs.
The 1,021 patients in the cangrelor-alone group had a 2.6% rate of the primary composite efficacy endpoint consisting of all-cause mortality, MI, ischemia-driven revascularization, or stent thrombosis at 48 hours. Although this was 21% lower than the 3.3% rate in the clopidogrel plus GPI group, the difference didn’t achieve statistical significance because of the relatively small sample size.
In contrast, the rate of major bleeding within 48 hours by ACUITY (Acute Catheterization and Urgent Intervention Triage) criteria did differ significantly between the two groups: 3.6% with cangrelor and 5.8% with clopidogrel plus a GPI, for a 39% relative risk reduction in favor of the newer agent. Major bleeding rates were also lower in the cangrelor-treated patients on the GUSTO (Global Utilization of Streptokinase and TPA for Occluded Coronary Arteries) and TIMI (Thrombolysis in Myocardial Infarction) scales, although those trends didn’t achieve significance. The blood transfusion requirement was significantly lower in the cangrelor group: 1% versus 2.1%, for a 55% relative risk reduction.
Dr. Vaduganathan said the CHAMPION investigators’ future plans include examining the possibility of a favorable pharmacologic interaction between cangrelor and bivalirudin, a combination seeing increasing use in the United States.
“Finally, I think it’ll be important moving forward to look at comparative effectiveness data – either observational or randomized – as well as cost-effectiveness data between potent parenteral antiplatelet strategies,” he concluded.
Session chair Dr. Anthony Gershlick of Glenfield General Hospital in Leicester, England, commented that while he found this analysis of the CHAMPION data quite interesting, they are of little relevance in Europe, where cardiologists seldom use GPIs as part of their up-front PCI management strategy, instead resorting to those agents mainly as bailout therapy.
The U.S. situation is very different, according to Dr. Vaduganathan. American cardiologists use GPI therapy in about one-third of PCIs.
The CHAMPION trials were funded by the Medicines Co. Dr. Vaduganathan reported having no relevant financial conflicts.
PARIS – Parenteral cangrelor is a potent antiplatelet agent that provides similar anti-ischemic effects but a significantly lower risk of major bleeding than the popular combination of clopidogrel plus a glycoprotein IIb/IIIa inhibitor in patients undergoing elective or urgent percutaneous coronary intervention.
Moreover, cangrelor (Kengreal), a rapid-acting and reversible intravenous platelet P2Y12 inhibitor, cuts the blood transfusion requirement by more than half, Dr. Muthiah Vaduganathan reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
These findings, based upon a propensity score–matched exploratory pooled analysis of individual patient-level data from the three phase III CHAMPION trials, suggest that cangrelor alone is an attractive alternative to clopidogrel plus a glycoprotein IIb/IIIa inhibitor (GPI) for antiplatelet management in percutaneous coronary intervention (PCI), according to Dr. Vaduganathan of Brigham and Women’s Hospital, Boston.
CHAMPION PLATFORM (N Engl J Med 2009 Dec 10;361[24]:2330-41), CHAMPION PCI (N Engl J Med 2009 Dec 10;361[24]:2318-29), and CHAMPION PHOENIX (N Engl J Med 2013 Apr 11;368[14]:1303-13) collectively included nearly 25,000 patients randomized to cangrelor versus standard therapy for platelet inhibition in elective or urgent PCI. The trials, which established that cangrelor led to fewer ischemic complications within 30 days post PCI than did clopidogrel plus a GPI, led to Food and Drug Administration approval of the drug in 2015. Cangrelor is also approved in Europe.
The cangrelor and clopidogrel groups differed in terms of baseline bleeding risk. For example, the clopidogrel-treated patients were younger, more likely to be male, had fewer comorbid conditions, and more often presented with an acute coronary syndrome. In order to accurately assess the bleeding risk associated with the two antiplatelet strategies, Dr. Vaduganathan and coinvestigators engaged in propensity score matching on the basis of 16 baseline clinical variables. They identified two closely matched cohorts consisting of 1,021 patient pairs.
The 1,021 patients in the cangrelor-alone group had a 2.6% rate of the primary composite efficacy endpoint consisting of all-cause mortality, MI, ischemia-driven revascularization, or stent thrombosis at 48 hours. Although this was 21% lower than the 3.3% rate in the clopidogrel plus GPI group, the difference didn’t achieve statistical significance because of the relatively small sample size.
In contrast, the rate of major bleeding within 48 hours by ACUITY (Acute Catheterization and Urgent Intervention Triage) criteria did differ significantly between the two groups: 3.6% with cangrelor and 5.8% with clopidogrel plus a GPI, for a 39% relative risk reduction in favor of the newer agent. Major bleeding rates were also lower in the cangrelor-treated patients on the GUSTO (Global Utilization of Streptokinase and TPA for Occluded Coronary Arteries) and TIMI (Thrombolysis in Myocardial Infarction) scales, although those trends didn’t achieve significance. The blood transfusion requirement was significantly lower in the cangrelor group: 1% versus 2.1%, for a 55% relative risk reduction.
Dr. Vaduganathan said the CHAMPION investigators’ future plans include examining the possibility of a favorable pharmacologic interaction between cangrelor and bivalirudin, a combination seeing increasing use in the United States.
“Finally, I think it’ll be important moving forward to look at comparative effectiveness data – either observational or randomized – as well as cost-effectiveness data between potent parenteral antiplatelet strategies,” he concluded.
Session chair Dr. Anthony Gershlick of Glenfield General Hospital in Leicester, England, commented that while he found this analysis of the CHAMPION data quite interesting, they are of little relevance in Europe, where cardiologists seldom use GPIs as part of their up-front PCI management strategy, instead resorting to those agents mainly as bailout therapy.
The U.S. situation is very different, according to Dr. Vaduganathan. American cardiologists use GPI therapy in about one-third of PCIs.
The CHAMPION trials were funded by the Medicines Co. Dr. Vaduganathan reported having no relevant financial conflicts.
AT EUROPCR 2016
Key clinical point: Cangrelor in patients undergoing PCI offers similar protection against ischemic events, compared with clopidogrel plus a glycoprotein IIb/IIIa inhibitor, but with significantly less major bleeding risk.
Major finding: The 48-hour rate of major bleeding as defined by the ACUITY scale was 3.6% with cangrelor alone, compared with 5.8% with clopidogrel plus a glycoprotein IIb/IIIa inhibitor.
Data source: This propensity score–matched analysis included 1,021 pairs of patients drawn from the three phase III CHAMPION trials.
Disclosures: The CHAMPION trials were funded by the Medicines Co. The presenter reported having no relevant financial conflicts.

Dr. Matt Kalaycio’s top 10 hematologic oncology abstracts for ASCO 2016
Hematology News’ Editor-in-Chief Matt Kalaycio selected the following as his “top 10” picks for hematologic oncology abstracts at ASCO 2016:
Abstract 7000: Final results of a phase III randomized trial of CPX-351 versus 7+3 in older patients with newly diagnosed high risk (secondary) AML
Comment: When any treatment appears to improve survival, compared with 7+3 for AML, all must take notice.
Abstract 7001: Treatment-free remission (TFR) in patients (pts) with chronic myeloid leukemia in chronic phase (CML-CP) treated with frontline nilotinib: Results from the ENESTFreedom study
Comment: About 50% of the CML patients treated with frontline nilotinib are eventually able to stop the drug and successfully stay off of it. That means more patients in treatment-free remission, compared with those initially treated with imatinib.
Link to abstract 7001
Abstract 7007: Phase Ib/2 study of venetoclax with low-dose cytarabine in treatment-naive patients age ≥ 65 with acute myelogenous leukemia
Abstract 7009: Results of a phase 1b study of venetoclax plus decitabine or azacitidine in untreated acute myeloid leukemia patients ≥ 65 years ineligible for standard induction therapy
Comment: The response rates in these older AML patients are remarkable and challenge results typically seen with 7+3 in a younger population.
Link to abstract 7007 and 7009
Abstract 7501: A prospective, multicenter, randomized study of anti-CCR4 monoclonal antibody mogamulizumab (moga) vs investigator’s choice (IC) in the treatment of patients (pts) with relapsed/refractory (R/R) adult T-cell leukemia-lymphoma (ATL)
Comment: The response rate to mogamulizumab was outstanding in the largest randomized clinical trial thus far conducted for this cancer. Although rare in the USA, ATL is more common in Asia.
Link to abstract 7501
Abstract 7507: Effect of bortezomib on complete remission (CR) rate when added to bendamustine-rituximab (BR) in previously untreated high-risk (HR) follicular lymphoma (FL): A randomized phase II trial of the ECOG-ACRIN Cancer Research Group (E2408)
Comment: This interesting observation of improved complete remission needs longer follow-up.
Link to abstract 7507
Abstract 7519: Venetoclax activity in CLL patients who have relapsed after or are refractory to ibrutinib or idelalisib
Comment: This study has implications for practice. Venetoclax elicits a 50%-60% response rate after patients with CLL progress during treatment with B-cell receptor pathway inhibitors.
Link to abstract 7519
Abstract 7521: Acalabrutinib, a second-generation bruton tyrosine kinase (Btk) inhibitor, in previously untreated chronic lymphocytic leukemia (CLL)
Comment: This next-generation variation on ibrutinib was associated with a 96% overall response rate with fewer adverse effects such as atrial fibrillation.
Link to abstract 7521
Abstract 8000: Upfront autologous stem cell transplantation (ASCT) versus novel agent-based therapy for multiple myeloma (MM): A randomized phase 3 study of the European Myeloma Network (EMN02/HO95 MM trial)
Comment: Other trials are underway to address the role of upfront ASCT for newly diagnosed multiple myeloma. While the last word on this issue has yet to be written, ASCT remains the standard of care for MM patients after induction.
Link to abstract 8000
LBA4: Phase III randomized controlled study of daratumumab, bortezomib, and dexamethasone (DVd) versus bortezomib and dexamethasone (Vd) in patients (pts) with relapsed or refractory multiple myeloma (RRMM): CASTOR study
Comment: As predicted by most, the addition of daratumumab to bortezomib-based therapy increases response rates, compared with bortezomib-based alone. Efficacy is becoming less of a concern with myeloma treatment than is economics..
Look for the full, final text of this abstract to be posted online at 7:30 AM (EDT) on Sunday, June 5.
Hematology News’ Editor-in-Chief Matt Kalaycio selected the following as his “top 10” picks for hematologic oncology abstracts at ASCO 2016:
Abstract 7000: Final results of a phase III randomized trial of CPX-351 versus 7+3 in older patients with newly diagnosed high risk (secondary) AML
Comment: When any treatment appears to improve survival, compared with 7+3 for AML, all must take notice.
Abstract 7001: Treatment-free remission (TFR) in patients (pts) with chronic myeloid leukemia in chronic phase (CML-CP) treated with frontline nilotinib: Results from the ENESTFreedom study
Comment: About 50% of the CML patients treated with frontline nilotinib are eventually able to stop the drug and successfully stay off of it. That means more patients in treatment-free remission, compared with those initially treated with imatinib.
Link to abstract 7001
Abstract 7007: Phase Ib/2 study of venetoclax with low-dose cytarabine in treatment-naive patients age ≥ 65 with acute myelogenous leukemia
Abstract 7009: Results of a phase 1b study of venetoclax plus decitabine or azacitidine in untreated acute myeloid leukemia patients ≥ 65 years ineligible for standard induction therapy
Comment: The response rates in these older AML patients are remarkable and challenge results typically seen with 7+3 in a younger population.
Link to abstract 7007 and 7009
Abstract 7501: A prospective, multicenter, randomized study of anti-CCR4 monoclonal antibody mogamulizumab (moga) vs investigator’s choice (IC) in the treatment of patients (pts) with relapsed/refractory (R/R) adult T-cell leukemia-lymphoma (ATL)
Comment: The response rate to mogamulizumab was outstanding in the largest randomized clinical trial thus far conducted for this cancer. Although rare in the USA, ATL is more common in Asia.
Link to abstract 7501
Abstract 7507: Effect of bortezomib on complete remission (CR) rate when added to bendamustine-rituximab (BR) in previously untreated high-risk (HR) follicular lymphoma (FL): A randomized phase II trial of the ECOG-ACRIN Cancer Research Group (E2408)
Comment: This interesting observation of improved complete remission needs longer follow-up.
Link to abstract 7507
Abstract 7519: Venetoclax activity in CLL patients who have relapsed after or are refractory to ibrutinib or idelalisib
Comment: This study has implications for practice. Venetoclax elicits a 50%-60% response rate after patients with CLL progress during treatment with B-cell receptor pathway inhibitors.
Link to abstract 7519
Abstract 7521: Acalabrutinib, a second-generation bruton tyrosine kinase (Btk) inhibitor, in previously untreated chronic lymphocytic leukemia (CLL)
Comment: This next-generation variation on ibrutinib was associated with a 96% overall response rate with fewer adverse effects such as atrial fibrillation.
Link to abstract 7521
Abstract 8000: Upfront autologous stem cell transplantation (ASCT) versus novel agent-based therapy for multiple myeloma (MM): A randomized phase 3 study of the European Myeloma Network (EMN02/HO95 MM trial)
Comment: Other trials are underway to address the role of upfront ASCT for newly diagnosed multiple myeloma. While the last word on this issue has yet to be written, ASCT remains the standard of care for MM patients after induction.
Link to abstract 8000
LBA4: Phase III randomized controlled study of daratumumab, bortezomib, and dexamethasone (DVd) versus bortezomib and dexamethasone (Vd) in patients (pts) with relapsed or refractory multiple myeloma (RRMM): CASTOR study
Comment: As predicted by most, the addition of daratumumab to bortezomib-based therapy increases response rates, compared with bortezomib-based alone. Efficacy is becoming less of a concern with myeloma treatment than is economics..
Look for the full, final text of this abstract to be posted online at 7:30 AM (EDT) on Sunday, June 5.
Hematology News’ Editor-in-Chief Matt Kalaycio selected the following as his “top 10” picks for hematologic oncology abstracts at ASCO 2016:
Abstract 7000: Final results of a phase III randomized trial of CPX-351 versus 7+3 in older patients with newly diagnosed high risk (secondary) AML
Comment: When any treatment appears to improve survival, compared with 7+3 for AML, all must take notice.
Abstract 7001: Treatment-free remission (TFR) in patients (pts) with chronic myeloid leukemia in chronic phase (CML-CP) treated with frontline nilotinib: Results from the ENESTFreedom study
Comment: About 50% of the CML patients treated with frontline nilotinib are eventually able to stop the drug and successfully stay off of it. That means more patients in treatment-free remission, compared with those initially treated with imatinib.
Link to abstract 7001
Abstract 7007: Phase Ib/2 study of venetoclax with low-dose cytarabine in treatment-naive patients age ≥ 65 with acute myelogenous leukemia
Abstract 7009: Results of a phase 1b study of venetoclax plus decitabine or azacitidine in untreated acute myeloid leukemia patients ≥ 65 years ineligible for standard induction therapy
Comment: The response rates in these older AML patients are remarkable and challenge results typically seen with 7+3 in a younger population.
Link to abstract 7007 and 7009
Abstract 7501: A prospective, multicenter, randomized study of anti-CCR4 monoclonal antibody mogamulizumab (moga) vs investigator’s choice (IC) in the treatment of patients (pts) with relapsed/refractory (R/R) adult T-cell leukemia-lymphoma (ATL)
Comment: The response rate to mogamulizumab was outstanding in the largest randomized clinical trial thus far conducted for this cancer. Although rare in the USA, ATL is more common in Asia.
Link to abstract 7501
Abstract 7507: Effect of bortezomib on complete remission (CR) rate when added to bendamustine-rituximab (BR) in previously untreated high-risk (HR) follicular lymphoma (FL): A randomized phase II trial of the ECOG-ACRIN Cancer Research Group (E2408)
Comment: This interesting observation of improved complete remission needs longer follow-up.
Link to abstract 7507
Abstract 7519: Venetoclax activity in CLL patients who have relapsed after or are refractory to ibrutinib or idelalisib
Comment: This study has implications for practice. Venetoclax elicits a 50%-60% response rate after patients with CLL progress during treatment with B-cell receptor pathway inhibitors.
Link to abstract 7519
Abstract 7521: Acalabrutinib, a second-generation bruton tyrosine kinase (Btk) inhibitor, in previously untreated chronic lymphocytic leukemia (CLL)
Comment: This next-generation variation on ibrutinib was associated with a 96% overall response rate with fewer adverse effects such as atrial fibrillation.
Link to abstract 7521
Abstract 8000: Upfront autologous stem cell transplantation (ASCT) versus novel agent-based therapy for multiple myeloma (MM): A randomized phase 3 study of the European Myeloma Network (EMN02/HO95 MM trial)
Comment: Other trials are underway to address the role of upfront ASCT for newly diagnosed multiple myeloma. While the last word on this issue has yet to be written, ASCT remains the standard of care for MM patients after induction.
Link to abstract 8000
LBA4: Phase III randomized controlled study of daratumumab, bortezomib, and dexamethasone (DVd) versus bortezomib and dexamethasone (Vd) in patients (pts) with relapsed or refractory multiple myeloma (RRMM): CASTOR study
Comment: As predicted by most, the addition of daratumumab to bortezomib-based therapy increases response rates, compared with bortezomib-based alone. Efficacy is becoming less of a concern with myeloma treatment than is economics..
Look for the full, final text of this abstract to be posted online at 7:30 AM (EDT) on Sunday, June 5.
Chronic Kidney Disease Risk with Proton Pump Inhibitors
Clinical question: What is the association between proton pump inhibitor (PPI) use and incident chronic kidney disease (CKD)?

Background: Medication use may play a potential role in the increasing prevalence of CKD. PPIs are commonly prescribed, and several observational studies have linked their use with multiple adverse outcomes, including acute interstitial nephritis. The risk for CKD with PPI use has never been evaluated.
Study design: Prospective cohort study.
Setting: U.S., multi-center.
Synopsis: Among 10,482 patients in the Atherosclerosis Risk in Communities study (ARIC) with an estimated glomerular filtration rate of at least 60 mL/min/1.73 m2, PPI use was associated with a 1.50 times risk of incident CKD (95% CI, 1.14–1.96; P=0.003) and a 1.64 times risk of incident acute kidney injury (95% CI, 1.22–2.21; P<0.001) when compared to nonusers. PPI use continued to have an association with incident CKD even when compared directly with H2 receptor antagonist users (adjusted HR, 1.39; 95% CI, 1.01–1.91). Findings were replicated in a cohort of 248,751 patients in the Geisinger Health System, and in all analyses, PPI use was associated with CKD.
One limitation is that this was an observational study and causality between PPI use and CKD cannot be established.
Bottom line: PPIs are associated with risk for CKD, and in patients on therapy, its use should be reevaluated.
Citation: Lazarus B, Chen Y, Wilson FP, et al. Proton pump inhibitor use and the risk of chronic kidney disease. JAMA Intern Med. 2016;176(2):238-246.
Clinical question: What is the association between proton pump inhibitor (PPI) use and incident chronic kidney disease (CKD)?
Background: Medication use may play a potential role in the increasing prevalence of CKD. PPIs are commonly prescribed, and several observational studies have linked their use with multiple adverse outcomes, including acute interstitial nephritis. The risk for CKD with PPI use has never been evaluated.
Study design: Prospective cohort study.
Setting: U.S., multi-center.
Synopsis: Among 10,482 patients in the Atherosclerosis Risk in Communities study (ARIC) with an estimated glomerular filtration rate of at least 60 mL/min/1.73 m2, PPI use was associated with a 1.50 times risk of incident CKD (95% CI, 1.14–1.96; P=0.003) and a 1.64 times risk of incident acute kidney injury (95% CI, 1.22–2.21; P<0.001) when compared to nonusers. PPI use continued to have an association with incident CKD even when compared directly with H2 receptor antagonist users (adjusted HR, 1.39; 95% CI, 1.01–1.91). Findings were replicated in a cohort of 248,751 patients in the Geisinger Health System, and in all analyses, PPI use was associated with CKD.
One limitation is that this was an observational study and causality between PPI use and CKD cannot be established.
Bottom line: PPIs are associated with risk for CKD, and in patients on therapy, its use should be reevaluated.
Citation: Lazarus B, Chen Y, Wilson FP, et al. Proton pump inhibitor use and the risk of chronic kidney disease. JAMA Intern Med. 2016;176(2):238-246.
Clinical question: What is the association between proton pump inhibitor (PPI) use and incident chronic kidney disease (CKD)?
Background: Medication use may play a potential role in the increasing prevalence of CKD. PPIs are commonly prescribed, and several observational studies have linked their use with multiple adverse outcomes, including acute interstitial nephritis. The risk for CKD with PPI use has never been evaluated.
Study design: Prospective cohort study.
Setting: U.S., multi-center.
Synopsis: Among 10,482 patients in the Atherosclerosis Risk in Communities study (ARIC) with an estimated glomerular filtration rate of at least 60 mL/min/1.73 m2, PPI use was associated with a 1.50 times risk of incident CKD (95% CI, 1.14–1.96; P=0.003) and a 1.64 times risk of incident acute kidney injury (95% CI, 1.22–2.21; P<0.001) when compared to nonusers. PPI use continued to have an association with incident CKD even when compared directly with H2 receptor antagonist users (adjusted HR, 1.39; 95% CI, 1.01–1.91). Findings were replicated in a cohort of 248,751 patients in the Geisinger Health System, and in all analyses, PPI use was associated with CKD.
One limitation is that this was an observational study and causality between PPI use and CKD cannot be established.
Bottom line: PPIs are associated with risk for CKD, and in patients on therapy, its use should be reevaluated.
Citation: Lazarus B, Chen Y, Wilson FP, et al. Proton pump inhibitor use and the risk of chronic kidney disease. JAMA Intern Med. 2016;176(2):238-246.

Risk Factors for Pseudomonas, MRSA in Healthcare-Associated Pneumonia
Clinical question: What risk factors could predict the likelihood of Pseudomonas and methicillin-resistant Staphylococcus aureus (MRSA) in patients hospitalized with healthcare-associated pneumonia (HCAP)?
Background: Patients identified with HCAP have an increased risk for multi-drug-resistant pathogens, such as gram-negative (GNR) organisms and MRSA. Meeting criteria for HCAP does not discriminate between the different infections, which require different antibiotic classes for treatment. Risk factors need to be identified to determine the most likely infectious organism to help guide initial empiric antibiotic therapy.
Study design: Retrospective cohort study.
Setting: Veterans Affairs hospitals.
Synopsis: Of 61,651 veterans with HCAP diagnosis, 1,156 (1.9%) had a discharge diagnosis of Pseudomonas pneumonia and were found to be younger and more likely to be immunocompromised; have hemiplegia; have a history of chronic obstructive pulmonary disease; have had corticosteroid exposure; and have been exposed to a fluoroquinolone, β-lactam, cephalosporin, or carbapenem antiobiotic within 90 days prior to admission. Pseudomonas pneumonia was negatively associated with age >84, drug abuse, diabetes, and higher socioeconomic status. A discharge diagnosis of MRSA pneumonia was found in 641 patients (1.0%), who also were positively associated with the male gender, age >74, recent nursing home stay, and recent exposure to fluoroquinolone antibiotics within 90 days prior to admission.
MRSA pneumonia was negatively associated with complicated diabetes. Neither diagnosis was present in 59,854 patients (97.1%).
This study was limited due to its predominantly male veteran population, low incidence of Pseudomonas and MRSA pneumonia being identified, and Pseudomonas as the only GNR organism analyzed.
Bottom line: Risk factors identified for Pseudomonas and MRSA pneumonia can help guide targeted antibiotics for HCAP patients.
Citation: Metersky ML, Frei CR, Mortenson EM. Predictors of Pseudomonas and methicillin-resistant Staphylococcus aureus in hospitalized patients with healthcare-associated pneumonia. Respirology. 2016;21(1):157-163.
Short Take
Hematuria as Marker of Urologic Cancer
Narrative literature review did not demonstrate beneficial role of screening urinalysis for cancer detection in asymptomatic patients, but it did suggest including gross hematuria as part of routine review of systems.
Citation: Nielsen M, Qaseem A, High Value Care Task Force of the American College of Physicians. Hematuria as a marker of occult urinary tract cancer: advice for high-value care from the American College of Physicians. Ann Intern Med. 2016;164(7):488-497. doi:10.7326/M15-1496.
Clinical question: What risk factors could predict the likelihood of Pseudomonas and methicillin-resistant Staphylococcus aureus (MRSA) in patients hospitalized with healthcare-associated pneumonia (HCAP)?
Background: Patients identified with HCAP have an increased risk for multi-drug-resistant pathogens, such as gram-negative (GNR) organisms and MRSA. Meeting criteria for HCAP does not discriminate between the different infections, which require different antibiotic classes for treatment. Risk factors need to be identified to determine the most likely infectious organism to help guide initial empiric antibiotic therapy.
Study design: Retrospective cohort study.
Setting: Veterans Affairs hospitals.
Synopsis: Of 61,651 veterans with HCAP diagnosis, 1,156 (1.9%) had a discharge diagnosis of Pseudomonas pneumonia and were found to be younger and more likely to be immunocompromised; have hemiplegia; have a history of chronic obstructive pulmonary disease; have had corticosteroid exposure; and have been exposed to a fluoroquinolone, β-lactam, cephalosporin, or carbapenem antiobiotic within 90 days prior to admission. Pseudomonas pneumonia was negatively associated with age >84, drug abuse, diabetes, and higher socioeconomic status. A discharge diagnosis of MRSA pneumonia was found in 641 patients (1.0%), who also were positively associated with the male gender, age >74, recent nursing home stay, and recent exposure to fluoroquinolone antibiotics within 90 days prior to admission.
MRSA pneumonia was negatively associated with complicated diabetes. Neither diagnosis was present in 59,854 patients (97.1%).
This study was limited due to its predominantly male veteran population, low incidence of Pseudomonas and MRSA pneumonia being identified, and Pseudomonas as the only GNR organism analyzed.
Bottom line: Risk factors identified for Pseudomonas and MRSA pneumonia can help guide targeted antibiotics for HCAP patients.
Citation: Metersky ML, Frei CR, Mortenson EM. Predictors of Pseudomonas and methicillin-resistant Staphylococcus aureus in hospitalized patients with healthcare-associated pneumonia. Respirology. 2016;21(1):157-163.
Short Take
Hematuria as Marker of Urologic Cancer
Narrative literature review did not demonstrate beneficial role of screening urinalysis for cancer detection in asymptomatic patients, but it did suggest including gross hematuria as part of routine review of systems.
Citation: Nielsen M, Qaseem A, High Value Care Task Force of the American College of Physicians. Hematuria as a marker of occult urinary tract cancer: advice for high-value care from the American College of Physicians. Ann Intern Med. 2016;164(7):488-497. doi:10.7326/M15-1496.
Clinical question: What risk factors could predict the likelihood of Pseudomonas and methicillin-resistant Staphylococcus aureus (MRSA) in patients hospitalized with healthcare-associated pneumonia (HCAP)?
Background: Patients identified with HCAP have an increased risk for multi-drug-resistant pathogens, such as gram-negative (GNR) organisms and MRSA. Meeting criteria for HCAP does not discriminate between the different infections, which require different antibiotic classes for treatment. Risk factors need to be identified to determine the most likely infectious organism to help guide initial empiric antibiotic therapy.
Study design: Retrospective cohort study.
Setting: Veterans Affairs hospitals.
Synopsis: Of 61,651 veterans with HCAP diagnosis, 1,156 (1.9%) had a discharge diagnosis of Pseudomonas pneumonia and were found to be younger and more likely to be immunocompromised; have hemiplegia; have a history of chronic obstructive pulmonary disease; have had corticosteroid exposure; and have been exposed to a fluoroquinolone, β-lactam, cephalosporin, or carbapenem antiobiotic within 90 days prior to admission. Pseudomonas pneumonia was negatively associated with age >84, drug abuse, diabetes, and higher socioeconomic status. A discharge diagnosis of MRSA pneumonia was found in 641 patients (1.0%), who also were positively associated with the male gender, age >74, recent nursing home stay, and recent exposure to fluoroquinolone antibiotics within 90 days prior to admission.
MRSA pneumonia was negatively associated with complicated diabetes. Neither diagnosis was present in 59,854 patients (97.1%).
This study was limited due to its predominantly male veteran population, low incidence of Pseudomonas and MRSA pneumonia being identified, and Pseudomonas as the only GNR organism analyzed.
Bottom line: Risk factors identified for Pseudomonas and MRSA pneumonia can help guide targeted antibiotics for HCAP patients.
Citation: Metersky ML, Frei CR, Mortenson EM. Predictors of Pseudomonas and methicillin-resistant Staphylococcus aureus in hospitalized patients with healthcare-associated pneumonia. Respirology. 2016;21(1):157-163.
Short Take
Hematuria as Marker of Urologic Cancer
Narrative literature review did not demonstrate beneficial role of screening urinalysis for cancer detection in asymptomatic patients, but it did suggest including gross hematuria as part of routine review of systems.
Citation: Nielsen M, Qaseem A, High Value Care Task Force of the American College of Physicians. Hematuria as a marker of occult urinary tract cancer: advice for high-value care from the American College of Physicians. Ann Intern Med. 2016;164(7):488-497. doi:10.7326/M15-1496.
Dose of transplanted HSCs affects their behavior

in the bone marrow
The dose of hematopoietic stem cells (HSCs) given in a transplant affects how those cells behave in the body, according to research published in Cell Reports.
To track the behavior of transplanted HSCs, researchers “barcoded” individual mouse HSCs with a genetic marker and observed their contributions to hematopoiesis.
The team found that only 20% to 30% of HSCs differentiated into all types of blood cells.
This relatively small group of HSCs produced a disproportionately large amount of blood.
The remaining 70% to 80% of HSCs were more strategic, and their behavior was dependent on the dose of HSCs transplanted.
At higher HSC doses, the “strategic majority” of HSCs opted to differentiate early, producing a balanced array of T cells and B cells. But at low HSC doses, the strategic HSCs prioritized T-cell production.
“The dose of transplanted bone marrow has strong and lasting effects on how HSCs specialize and coordinate their behavior,” said study author Rong Lu, PhD, of the University of Southern California in Los Angeles.
“This suggests that altering transplantation dose could be a tool for improving outcomes for patients—promoting bone marrow engraftment, reducing the risk of infection, and, ultimately, saving lives.” ![]()
in the bone marrow
The dose of hematopoietic stem cells (HSCs) given in a transplant affects how those cells behave in the body, according to research published in Cell Reports.
To track the behavior of transplanted HSCs, researchers “barcoded” individual mouse HSCs with a genetic marker and observed their contributions to hematopoiesis.
The team found that only 20% to 30% of HSCs differentiated into all types of blood cells.
This relatively small group of HSCs produced a disproportionately large amount of blood.
The remaining 70% to 80% of HSCs were more strategic, and their behavior was dependent on the dose of HSCs transplanted.
At higher HSC doses, the “strategic majority” of HSCs opted to differentiate early, producing a balanced array of T cells and B cells. But at low HSC doses, the strategic HSCs prioritized T-cell production.
“The dose of transplanted bone marrow has strong and lasting effects on how HSCs specialize and coordinate their behavior,” said study author Rong Lu, PhD, of the University of Southern California in Los Angeles.
“This suggests that altering transplantation dose could be a tool for improving outcomes for patients—promoting bone marrow engraftment, reducing the risk of infection, and, ultimately, saving lives.” ![]()
in the bone marrow
The dose of hematopoietic stem cells (HSCs) given in a transplant affects how those cells behave in the body, according to research published in Cell Reports.
To track the behavior of transplanted HSCs, researchers “barcoded” individual mouse HSCs with a genetic marker and observed their contributions to hematopoiesis.
The team found that only 20% to 30% of HSCs differentiated into all types of blood cells.
This relatively small group of HSCs produced a disproportionately large amount of blood.
The remaining 70% to 80% of HSCs were more strategic, and their behavior was dependent on the dose of HSCs transplanted.
At higher HSC doses, the “strategic majority” of HSCs opted to differentiate early, producing a balanced array of T cells and B cells. But at low HSC doses, the strategic HSCs prioritized T-cell production.
“The dose of transplanted bone marrow has strong and lasting effects on how HSCs specialize and coordinate their behavior,” said study author Rong Lu, PhD, of the University of Southern California in Los Angeles.
“This suggests that altering transplantation dose could be a tool for improving outcomes for patients—promoting bone marrow engraftment, reducing the risk of infection, and, ultimately, saving lives.” ![]()