User login
ICD-10 Update
On April 17, the U.S. Department of Health and Human Services (HHS) published a proposed rule to delay the compliance date for the International Classification of Diseases, 10th Edition, diagnosis and procedure codes (ICD-10) from Oct. 1, 2013, to Oct. 1, 2014.2
Per HHS, the ICD-10 compliance date change is part of a proposed rule that would adopt a standard for a unique health plan identifier (HPID), adopt a data element that would serve as an “other entity” identifier (OEID), and add a National Provider Identifier (NPI) requirement. The proposed rule was developed by the Office of E-Health Standards and Services (OESS) as part of its ongoing role, delegated by HHS, to establish standards for electronic healthcare transactions under the Health Insurance Portability and Accountability Act of 1996 (HIPAA).
HHS proposes that covered entities must be in compliance with ICD-10 by Oct. 1, 2014.
On April 17, the U.S. Department of Health and Human Services (HHS) published a proposed rule to delay the compliance date for the International Classification of Diseases, 10th Edition, diagnosis and procedure codes (ICD-10) from Oct. 1, 2013, to Oct. 1, 2014.2
Per HHS, the ICD-10 compliance date change is part of a proposed rule that would adopt a standard for a unique health plan identifier (HPID), adopt a data element that would serve as an “other entity” identifier (OEID), and add a National Provider Identifier (NPI) requirement. The proposed rule was developed by the Office of E-Health Standards and Services (OESS) as part of its ongoing role, delegated by HHS, to establish standards for electronic healthcare transactions under the Health Insurance Portability and Accountability Act of 1996 (HIPAA).
HHS proposes that covered entities must be in compliance with ICD-10 by Oct. 1, 2014.
On April 17, the U.S. Department of Health and Human Services (HHS) published a proposed rule to delay the compliance date for the International Classification of Diseases, 10th Edition, diagnosis and procedure codes (ICD-10) from Oct. 1, 2013, to Oct. 1, 2014.2
Per HHS, the ICD-10 compliance date change is part of a proposed rule that would adopt a standard for a unique health plan identifier (HPID), adopt a data element that would serve as an “other entity” identifier (OEID), and add a National Provider Identifier (NPI) requirement. The proposed rule was developed by the Office of E-Health Standards and Services (OESS) as part of its ongoing role, delegated by HHS, to establish standards for electronic healthcare transactions under the Health Insurance Portability and Accountability Act of 1996 (HIPAA).
HHS proposes that covered entities must be in compliance with ICD-10 by Oct. 1, 2014.
Replenishing the Primary Care Physician Pipeline
A recent survey of nearly 1,000 students from three medical schools found that just 15% planned to become primary-care physicians, including 11.2% of first-year students.1
That startlingly low number might not be reflective of the whole country, and other national surveys have suggested significantly higher rates. But the responses underscore some important contributors beyond financial concerns that include a more negative overall view of PCPs’ work life compared to that of specialists. “Our data suggest that although medical school does not create these negative views of primary-care work life, it may reinforce them,” the authors write.
Conversely, the results suggest that time spent observing physicians could help break negative stereotypes about the ability to develop good relationships with patients, and that career plans might not be based on perceptions, but rather on values and goals. “The study reinforces the importance of admitting students with primary-care-oriented values and primary-care interest and reinforcing those values over the course of medical school,” the authors conclude.
“Maybe we’re not selecting medical students in the optimal way for what society needs,” says Elbert Huang, MD, associate professor of medicine at the University of Chicago. By emphasizing GPA and test scores, “maybe when you do that, you end with people who don’t want to actually take care of patients in primary care.”
Other studies suggest he’s on to something. Research conducted by the Washington, D.C.-based Robert Graham Center found that students in rural medical schools are significantly more likely to go into rural healthcare and primary care than students in urban medical schools.
“The problem there is that we’ve cut the number of people from rural areas going to medical school by half over the last 20 years,” center director Robert Phillips, MD, MSPH, says. “A lot of students just don’t have the background to make them competitive.” Many students in minority communities face similar challenges.
Ed Salsberg, director of the National Center for Health Workforce Analysis in the Health Resources and Services Administration, says many newer osteopathic schools are positioning themselves in rural communities, helping them attract students who might not have gone to medical school otherwise.
Reaching back even earlier into the pipeline to help mentor elementary and high school students might be another way to help build capacity. Medical organizations also seem to be getting the message. New MCAT recommendations by the Association of American Medical Colleges, for example, place less emphasis on scientific knowledge in favor of a more holistic assessment of critical analysis and reasoning skills. The association also is encouraging medical schools to pay more attention to such personal characteristics as integrity and service orientation.
“That’s more of a long-term strategy, but I think it has an impact on who gets recruited to medical school,” Salsberg says.
Reference
A recent survey of nearly 1,000 students from three medical schools found that just 15% planned to become primary-care physicians, including 11.2% of first-year students.1
That startlingly low number might not be reflective of the whole country, and other national surveys have suggested significantly higher rates. But the responses underscore some important contributors beyond financial concerns that include a more negative overall view of PCPs’ work life compared to that of specialists. “Our data suggest that although medical school does not create these negative views of primary-care work life, it may reinforce them,” the authors write.
Conversely, the results suggest that time spent observing physicians could help break negative stereotypes about the ability to develop good relationships with patients, and that career plans might not be based on perceptions, but rather on values and goals. “The study reinforces the importance of admitting students with primary-care-oriented values and primary-care interest and reinforcing those values over the course of medical school,” the authors conclude.
“Maybe we’re not selecting medical students in the optimal way for what society needs,” says Elbert Huang, MD, associate professor of medicine at the University of Chicago. By emphasizing GPA and test scores, “maybe when you do that, you end with people who don’t want to actually take care of patients in primary care.”
Other studies suggest he’s on to something. Research conducted by the Washington, D.C.-based Robert Graham Center found that students in rural medical schools are significantly more likely to go into rural healthcare and primary care than students in urban medical schools.
“The problem there is that we’ve cut the number of people from rural areas going to medical school by half over the last 20 years,” center director Robert Phillips, MD, MSPH, says. “A lot of students just don’t have the background to make them competitive.” Many students in minority communities face similar challenges.
Ed Salsberg, director of the National Center for Health Workforce Analysis in the Health Resources and Services Administration, says many newer osteopathic schools are positioning themselves in rural communities, helping them attract students who might not have gone to medical school otherwise.
Reaching back even earlier into the pipeline to help mentor elementary and high school students might be another way to help build capacity. Medical organizations also seem to be getting the message. New MCAT recommendations by the Association of American Medical Colleges, for example, place less emphasis on scientific knowledge in favor of a more holistic assessment of critical analysis and reasoning skills. The association also is encouraging medical schools to pay more attention to such personal characteristics as integrity and service orientation.
“That’s more of a long-term strategy, but I think it has an impact on who gets recruited to medical school,” Salsberg says.
Reference
A recent survey of nearly 1,000 students from three medical schools found that just 15% planned to become primary-care physicians, including 11.2% of first-year students.1
That startlingly low number might not be reflective of the whole country, and other national surveys have suggested significantly higher rates. But the responses underscore some important contributors beyond financial concerns that include a more negative overall view of PCPs’ work life compared to that of specialists. “Our data suggest that although medical school does not create these negative views of primary-care work life, it may reinforce them,” the authors write.
Conversely, the results suggest that time spent observing physicians could help break negative stereotypes about the ability to develop good relationships with patients, and that career plans might not be based on perceptions, but rather on values and goals. “The study reinforces the importance of admitting students with primary-care-oriented values and primary-care interest and reinforcing those values over the course of medical school,” the authors conclude.
“Maybe we’re not selecting medical students in the optimal way for what society needs,” says Elbert Huang, MD, associate professor of medicine at the University of Chicago. By emphasizing GPA and test scores, “maybe when you do that, you end with people who don’t want to actually take care of patients in primary care.”
Other studies suggest he’s on to something. Research conducted by the Washington, D.C.-based Robert Graham Center found that students in rural medical schools are significantly more likely to go into rural healthcare and primary care than students in urban medical schools.
“The problem there is that we’ve cut the number of people from rural areas going to medical school by half over the last 20 years,” center director Robert Phillips, MD, MSPH, says. “A lot of students just don’t have the background to make them competitive.” Many students in minority communities face similar challenges.
Ed Salsberg, director of the National Center for Health Workforce Analysis in the Health Resources and Services Administration, says many newer osteopathic schools are positioning themselves in rural communities, helping them attract students who might not have gone to medical school otherwise.
Reaching back even earlier into the pipeline to help mentor elementary and high school students might be another way to help build capacity. Medical organizations also seem to be getting the message. New MCAT recommendations by the Association of American Medical Colleges, for example, place less emphasis on scientific knowledge in favor of a more holistic assessment of critical analysis and reasoning skills. The association also is encouraging medical schools to pay more attention to such personal characteristics as integrity and service orientation.
“That’s more of a long-term strategy, but I think it has an impact on who gets recruited to medical school,” Salsberg says.
Reference
ONLINE EXCLUSIVE: Vineet Arora discusses primary-care workforce challenges
Click here to listen to Dr. Arora
Click here to listen to Dr. Arora
Click here to listen to Dr. Arora
ONLINE EXCLUSIVE: L.A. Care Health Plan's Z. Joseph Wanski discusses efforts to prevent 30-day readmissions
Click here to listen to Dr. Wanski
Click here to listen to Dr. Wanski
Click here to listen to Dr. Wanski
How to Diagnose Spells That Mimic Epilepsy
When parents say their child stopped breathing for a few seconds, suddenly fell to the floor, or sometimes stares off into space, you might immediately think "epilepsy." But several conditions can trigger these events and should be included in your differential diagnosis.
Breath-holding spells, staring off, and gastroesophageal reflux are probably the most common conditions that can mimic epilepsy, but there are many others. Awareness of all the possible etiologies is important because pediatricians are on the front line for diagnosis and initial management of these patients.

Begin by asking for a very detailed description of the spells, including circumstances, timing, and any triggers. Even neurologists who specialize in epilepsy may not spend enough time getting a very, very precise description from the patient and witnesses. Take a thorough medical history of the patient and family to narrow down your differential possibilities. Combined, this information will foster an accurate diagnosis or determine which tests or referrals are indicated.
Depending on your initial assessment, consider blood tests for glucose, calcium, electrolytes, and thyroid function. In some cases, a toxic screen also is appropriate. ECGs and EEGs can be diagnostic as well, although not every pediatrician’s office has these capabilities. It also may be appropriate to refer for polysomnography if you suspect a sleep disorder, including apnea.
In contrast, a CT scan is rarely helpful and in general should not be ordered for these patients. If epilepsy remains a consideration, a head MRI is the more appropriate test.
Because only rarely will you witness a spell in your office, you have to rely on patient and caregiver reports. A useful tip is to ask parents for a video of the event. This is very feasible given the widespread use of smart phones.
Most pediatricians feel comfortable managing the patient with breath-holding spells, tics (including those associated with Tourette syndrome), self-stimulatory behavior, head banging, and night terrors. In contrast, a specialist referral might be appropriate for the child with more severe gastroesophageal reflux, opsoclonus (rapid and irregular eye movements), or one of the psychiatric disorders with manifestations that can mimic epileptic seizures.
Do not hesitate to refer if you remain at all unsure after going through your differential diagnosis. Most pediatricians appropriately refer children to me for further evaluation; only infrequently do I assess a child who obviously does not have epilepsy.
Within the following broad categories are some specific conditions and potential concerns:
• Unusual movements. Newborns can experience seizures, jitteriness, and nonepileptic jerks. Benign sleep myoclonus is very common but parents may come in concerned about these jerky movements. Tics (including motor and vocal tics that characterize Tourette syndrome) can look like seizures. Tics also can point to epileptic myoclonus, in which case further evaluation with EEG is warranted.
Paroxysmal torticollis or head turning in infants is generally benign and well within the purview of pediatricians to diagnose and manage. Different eye movement disorders occur both with and without seizures as well. Be more concerned if you see opsoclonus or "dancing eyes," because you may need to rule out neuroblastoma.
"Paroxysmal kinesigenic dyskinesia" is a good example of a condition that can mimic epilepsy. The characteristic unusual writhing of extremities that is triggered by movement (such as rising from a chair) is a relatively rare condition.
Self-stimulatory behavior also is commonly mistaken for seizures. Typically children place their hands between their thighs, thrust their pelvis back and forth, and then after a few minutes fall asleep. Some refer to this behavior as "infantile masturbation.’"
• Loss of tone or consciousness. Syncope or fainting spells are common and can appear similar to seizures as well.
The typical loss of consciousness presentation is orthostatic. These patients reliably will say that they faint upon standing.
Occasionally, you may hear about patients whose knees buckle and they fall, right after a surge of negative emotion such anger. This is called cataplexy, and it’s one of the symptoms of narcolepsy; it is not epilepsy.
Hemiplegia and certain migraines can mimic seizures as well, so keep these in mind with your differential diagnosis.
You are more likely to hear reports about children who "stare off" for minutes at a time. If you hyperventilate a child in your office and this triggers a staring spell, the child might have absence seizures. In contrast, these spells are less concerning if parents report they can get the child’s attention during the staring. So, as part of your differential, ask parents if they can get the child’s attention during one of these spells. Another tip is to test recall: Instruct the parents to tell the child to remember a specific color and number during the spell; if the child can recall the information a few minutes later, this helps to rule out absence seizures.
• Respiratory disorders. Some parents may be alarmed about epilepsy, but it may help to describe a typical breath-holding spell for them. In general, it’s not epilepsy if a trigger (such as pain or frustration) causes the child to suddenly freeze, stop crying, and/or pass out. Such children may be so upset they just cannot move, but they are not having a seizure.
• Behavioral and sleep disorders. Night terrors are relatively common, and should be distinguished from seizures that occur at night and really frighten a child. Some children repeatedly bang their heads against the bed, but this behavior does not point to epilepsy.
Sleep walking, sleep apnea, and nightmares also can be mistaken for seizures. Ask parents about any excessive daytime sleepiness to raise your suspicion of sleep disorders, including apnea. Also consider confusional arousals as well as periodic limb movement disorder during sleep, both of which might require assessment by a sleep specialist.
• Psychiatric and mental disorders. Consider fugue state, panic attacks, and schizophrenia in your differential. Children can experience hallucinations as part of seizures or from psychiatric disorders.
Mannerisms and/or nonresponsiveness in your autistic patients can appear like seizures.
Münchausen syndrome by proxy is another condition to keep in mind. In rare cases, parents will provide a fabricated history and describe spells that did not happen. A parent who is dead set against supplying a video of a future event might raise your suspicion for this rare but important condition.
• Perceptual disturbances. Dizziness or vertigo can be described as part of a seizure, but these symptoms are general and can be associated with many other disorders.
• Episodic features of medical disorders. Hypoglycemia is sometimes confused for epilepsy if a child becomes sweaty, confused, or disoriented, and/or loses consciousness. Contractures associated with hypocalcemia also can mimic epilepsy.
In addition, paroxysmal changes can result from cardiac arrhythmias or long QT syndrome. Some congenital heart conditions (such as tetralogy of Fallot) cause events in which children pass out or turn blue. Another consideration is hydrocephaly, which can cause a sudden increase in intracranial pressure that causes fainting.
A very, very common condition – even for us – is gastroesophageal reflux or Sandifer’s syndrome. These infants may stiffen or arch in response to the reflux pain, which can look just like a tonic seizure. Pediatricians can do a great service in reassuring parents that their child has reflux, not epilepsy.
Dr. Bourgeois is the director of the division of epilepsy and clinical neurophysiology and the William G. Lennox Chair in pediatric epilepsy at Children’s Hospital Boston. He is also professor of neurology at Harvard Medical School, also in Boston. Dr. Bourgeois is a consultant for Upsher-Smith Laboratories and a principal investigator on a multicenter study sponsored by Ovation/Lundbeck Pharmaceuticals.
When parents say their child stopped breathing for a few seconds, suddenly fell to the floor, or sometimes stares off into space, you might immediately think "epilepsy." But several conditions can trigger these events and should be included in your differential diagnosis.
Breath-holding spells, staring off, and gastroesophageal reflux are probably the most common conditions that can mimic epilepsy, but there are many others. Awareness of all the possible etiologies is important because pediatricians are on the front line for diagnosis and initial management of these patients.
Begin by asking for a very detailed description of the spells, including circumstances, timing, and any triggers. Even neurologists who specialize in epilepsy may not spend enough time getting a very, very precise description from the patient and witnesses. Take a thorough medical history of the patient and family to narrow down your differential possibilities. Combined, this information will foster an accurate diagnosis or determine which tests or referrals are indicated.
Depending on your initial assessment, consider blood tests for glucose, calcium, electrolytes, and thyroid function. In some cases, a toxic screen also is appropriate. ECGs and EEGs can be diagnostic as well, although not every pediatrician’s office has these capabilities. It also may be appropriate to refer for polysomnography if you suspect a sleep disorder, including apnea.
In contrast, a CT scan is rarely helpful and in general should not be ordered for these patients. If epilepsy remains a consideration, a head MRI is the more appropriate test.
Because only rarely will you witness a spell in your office, you have to rely on patient and caregiver reports. A useful tip is to ask parents for a video of the event. This is very feasible given the widespread use of smart phones.
Most pediatricians feel comfortable managing the patient with breath-holding spells, tics (including those associated with Tourette syndrome), self-stimulatory behavior, head banging, and night terrors. In contrast, a specialist referral might be appropriate for the child with more severe gastroesophageal reflux, opsoclonus (rapid and irregular eye movements), or one of the psychiatric disorders with manifestations that can mimic epileptic seizures.
Do not hesitate to refer if you remain at all unsure after going through your differential diagnosis. Most pediatricians appropriately refer children to me for further evaluation; only infrequently do I assess a child who obviously does not have epilepsy.
Within the following broad categories are some specific conditions and potential concerns:
• Unusual movements. Newborns can experience seizures, jitteriness, and nonepileptic jerks. Benign sleep myoclonus is very common but parents may come in concerned about these jerky movements. Tics (including motor and vocal tics that characterize Tourette syndrome) can look like seizures. Tics also can point to epileptic myoclonus, in which case further evaluation with EEG is warranted.
Paroxysmal torticollis or head turning in infants is generally benign and well within the purview of pediatricians to diagnose and manage. Different eye movement disorders occur both with and without seizures as well. Be more concerned if you see opsoclonus or "dancing eyes," because you may need to rule out neuroblastoma.
"Paroxysmal kinesigenic dyskinesia" is a good example of a condition that can mimic epilepsy. The characteristic unusual writhing of extremities that is triggered by movement (such as rising from a chair) is a relatively rare condition.
Self-stimulatory behavior also is commonly mistaken for seizures. Typically children place their hands between their thighs, thrust their pelvis back and forth, and then after a few minutes fall asleep. Some refer to this behavior as "infantile masturbation.’"
• Loss of tone or consciousness. Syncope or fainting spells are common and can appear similar to seizures as well.
The typical loss of consciousness presentation is orthostatic. These patients reliably will say that they faint upon standing.
Occasionally, you may hear about patients whose knees buckle and they fall, right after a surge of negative emotion such anger. This is called cataplexy, and it’s one of the symptoms of narcolepsy; it is not epilepsy.
Hemiplegia and certain migraines can mimic seizures as well, so keep these in mind with your differential diagnosis.
You are more likely to hear reports about children who "stare off" for minutes at a time. If you hyperventilate a child in your office and this triggers a staring spell, the child might have absence seizures. In contrast, these spells are less concerning if parents report they can get the child’s attention during the staring. So, as part of your differential, ask parents if they can get the child’s attention during one of these spells. Another tip is to test recall: Instruct the parents to tell the child to remember a specific color and number during the spell; if the child can recall the information a few minutes later, this helps to rule out absence seizures.
• Respiratory disorders. Some parents may be alarmed about epilepsy, but it may help to describe a typical breath-holding spell for them. In general, it’s not epilepsy if a trigger (such as pain or frustration) causes the child to suddenly freeze, stop crying, and/or pass out. Such children may be so upset they just cannot move, but they are not having a seizure.
• Behavioral and sleep disorders. Night terrors are relatively common, and should be distinguished from seizures that occur at night and really frighten a child. Some children repeatedly bang their heads against the bed, but this behavior does not point to epilepsy.
Sleep walking, sleep apnea, and nightmares also can be mistaken for seizures. Ask parents about any excessive daytime sleepiness to raise your suspicion of sleep disorders, including apnea. Also consider confusional arousals as well as periodic limb movement disorder during sleep, both of which might require assessment by a sleep specialist.
• Psychiatric and mental disorders. Consider fugue state, panic attacks, and schizophrenia in your differential. Children can experience hallucinations as part of seizures or from psychiatric disorders.
Mannerisms and/or nonresponsiveness in your autistic patients can appear like seizures.
Münchausen syndrome by proxy is another condition to keep in mind. In rare cases, parents will provide a fabricated history and describe spells that did not happen. A parent who is dead set against supplying a video of a future event might raise your suspicion for this rare but important condition.
• Perceptual disturbances. Dizziness or vertigo can be described as part of a seizure, but these symptoms are general and can be associated with many other disorders.
• Episodic features of medical disorders. Hypoglycemia is sometimes confused for epilepsy if a child becomes sweaty, confused, or disoriented, and/or loses consciousness. Contractures associated with hypocalcemia also can mimic epilepsy.
In addition, paroxysmal changes can result from cardiac arrhythmias or long QT syndrome. Some congenital heart conditions (such as tetralogy of Fallot) cause events in which children pass out or turn blue. Another consideration is hydrocephaly, which can cause a sudden increase in intracranial pressure that causes fainting.
A very, very common condition – even for us – is gastroesophageal reflux or Sandifer’s syndrome. These infants may stiffen or arch in response to the reflux pain, which can look just like a tonic seizure. Pediatricians can do a great service in reassuring parents that their child has reflux, not epilepsy.
Dr. Bourgeois is the director of the division of epilepsy and clinical neurophysiology and the William G. Lennox Chair in pediatric epilepsy at Children’s Hospital Boston. He is also professor of neurology at Harvard Medical School, also in Boston. Dr. Bourgeois is a consultant for Upsher-Smith Laboratories and a principal investigator on a multicenter study sponsored by Ovation/Lundbeck Pharmaceuticals.
When parents say their child stopped breathing for a few seconds, suddenly fell to the floor, or sometimes stares off into space, you might immediately think "epilepsy." But several conditions can trigger these events and should be included in your differential diagnosis.
Breath-holding spells, staring off, and gastroesophageal reflux are probably the most common conditions that can mimic epilepsy, but there are many others. Awareness of all the possible etiologies is important because pediatricians are on the front line for diagnosis and initial management of these patients.
Begin by asking for a very detailed description of the spells, including circumstances, timing, and any triggers. Even neurologists who specialize in epilepsy may not spend enough time getting a very, very precise description from the patient and witnesses. Take a thorough medical history of the patient and family to narrow down your differential possibilities. Combined, this information will foster an accurate diagnosis or determine which tests or referrals are indicated.
Depending on your initial assessment, consider blood tests for glucose, calcium, electrolytes, and thyroid function. In some cases, a toxic screen also is appropriate. ECGs and EEGs can be diagnostic as well, although not every pediatrician’s office has these capabilities. It also may be appropriate to refer for polysomnography if you suspect a sleep disorder, including apnea.
In contrast, a CT scan is rarely helpful and in general should not be ordered for these patients. If epilepsy remains a consideration, a head MRI is the more appropriate test.
Because only rarely will you witness a spell in your office, you have to rely on patient and caregiver reports. A useful tip is to ask parents for a video of the event. This is very feasible given the widespread use of smart phones.
Most pediatricians feel comfortable managing the patient with breath-holding spells, tics (including those associated with Tourette syndrome), self-stimulatory behavior, head banging, and night terrors. In contrast, a specialist referral might be appropriate for the child with more severe gastroesophageal reflux, opsoclonus (rapid and irregular eye movements), or one of the psychiatric disorders with manifestations that can mimic epileptic seizures.
Do not hesitate to refer if you remain at all unsure after going through your differential diagnosis. Most pediatricians appropriately refer children to me for further evaluation; only infrequently do I assess a child who obviously does not have epilepsy.
Within the following broad categories are some specific conditions and potential concerns:
• Unusual movements. Newborns can experience seizures, jitteriness, and nonepileptic jerks. Benign sleep myoclonus is very common but parents may come in concerned about these jerky movements. Tics (including motor and vocal tics that characterize Tourette syndrome) can look like seizures. Tics also can point to epileptic myoclonus, in which case further evaluation with EEG is warranted.
Paroxysmal torticollis or head turning in infants is generally benign and well within the purview of pediatricians to diagnose and manage. Different eye movement disorders occur both with and without seizures as well. Be more concerned if you see opsoclonus or "dancing eyes," because you may need to rule out neuroblastoma.
"Paroxysmal kinesigenic dyskinesia" is a good example of a condition that can mimic epilepsy. The characteristic unusual writhing of extremities that is triggered by movement (such as rising from a chair) is a relatively rare condition.
Self-stimulatory behavior also is commonly mistaken for seizures. Typically children place their hands between their thighs, thrust their pelvis back and forth, and then after a few minutes fall asleep. Some refer to this behavior as "infantile masturbation.’"
• Loss of tone or consciousness. Syncope or fainting spells are common and can appear similar to seizures as well.
The typical loss of consciousness presentation is orthostatic. These patients reliably will say that they faint upon standing.
Occasionally, you may hear about patients whose knees buckle and they fall, right after a surge of negative emotion such anger. This is called cataplexy, and it’s one of the symptoms of narcolepsy; it is not epilepsy.
Hemiplegia and certain migraines can mimic seizures as well, so keep these in mind with your differential diagnosis.
You are more likely to hear reports about children who "stare off" for minutes at a time. If you hyperventilate a child in your office and this triggers a staring spell, the child might have absence seizures. In contrast, these spells are less concerning if parents report they can get the child’s attention during the staring. So, as part of your differential, ask parents if they can get the child’s attention during one of these spells. Another tip is to test recall: Instruct the parents to tell the child to remember a specific color and number during the spell; if the child can recall the information a few minutes later, this helps to rule out absence seizures.
• Respiratory disorders. Some parents may be alarmed about epilepsy, but it may help to describe a typical breath-holding spell for them. In general, it’s not epilepsy if a trigger (such as pain or frustration) causes the child to suddenly freeze, stop crying, and/or pass out. Such children may be so upset they just cannot move, but they are not having a seizure.
• Behavioral and sleep disorders. Night terrors are relatively common, and should be distinguished from seizures that occur at night and really frighten a child. Some children repeatedly bang their heads against the bed, but this behavior does not point to epilepsy.
Sleep walking, sleep apnea, and nightmares also can be mistaken for seizures. Ask parents about any excessive daytime sleepiness to raise your suspicion of sleep disorders, including apnea. Also consider confusional arousals as well as periodic limb movement disorder during sleep, both of which might require assessment by a sleep specialist.
• Psychiatric and mental disorders. Consider fugue state, panic attacks, and schizophrenia in your differential. Children can experience hallucinations as part of seizures or from psychiatric disorders.
Mannerisms and/or nonresponsiveness in your autistic patients can appear like seizures.
Münchausen syndrome by proxy is another condition to keep in mind. In rare cases, parents will provide a fabricated history and describe spells that did not happen. A parent who is dead set against supplying a video of a future event might raise your suspicion for this rare but important condition.
• Perceptual disturbances. Dizziness or vertigo can be described as part of a seizure, but these symptoms are general and can be associated with many other disorders.
• Episodic features of medical disorders. Hypoglycemia is sometimes confused for epilepsy if a child becomes sweaty, confused, or disoriented, and/or loses consciousness. Contractures associated with hypocalcemia also can mimic epilepsy.
In addition, paroxysmal changes can result from cardiac arrhythmias or long QT syndrome. Some congenital heart conditions (such as tetralogy of Fallot) cause events in which children pass out or turn blue. Another consideration is hydrocephaly, which can cause a sudden increase in intracranial pressure that causes fainting.
A very, very common condition – even for us – is gastroesophageal reflux or Sandifer’s syndrome. These infants may stiffen or arch in response to the reflux pain, which can look just like a tonic seizure. Pediatricians can do a great service in reassuring parents that their child has reflux, not epilepsy.
Dr. Bourgeois is the director of the division of epilepsy and clinical neurophysiology and the William G. Lennox Chair in pediatric epilepsy at Children’s Hospital Boston. He is also professor of neurology at Harvard Medical School, also in Boston. Dr. Bourgeois is a consultant for Upsher-Smith Laboratories and a principal investigator on a multicenter study sponsored by Ovation/Lundbeck Pharmaceuticals.

Treating Brain Tumors With Bacteria Gets Neurosurgeons Banned
Are a few animal studies and a handful of human case reports enough to let physicians skirt institutional review boards?
Two neurosurgeons in California did just that when they used Enterobacter aerogenes to infect the surgical wounds of three terminally ill glioblastoma patients. Two of the patients died from the infections.
Dr. J. Paul Muizelaar and Dr. Rudolph J. Schrot of the University of California, Davis, said their attempt to stimulate their patients’ immune response was not research but "a one-time procedure" – exempt from review, according to a report in the Sacramento Bee.
Now both are banned from human research projects and the institutional review board is the subject of its own investigation.
For an account of the scientific thinking behind the deployment of bacteria in these patients and of ongoing efforts to develop immunotherapies against cancer, see the journal Nature (2012 July 27 [doi:10.1038/nature.2012.11080]).
Are a few animal studies and a handful of human case reports enough to let physicians skirt institutional review boards?
Two neurosurgeons in California did just that when they used Enterobacter aerogenes to infect the surgical wounds of three terminally ill glioblastoma patients. Two of the patients died from the infections.
Dr. J. Paul Muizelaar and Dr. Rudolph J. Schrot of the University of California, Davis, said their attempt to stimulate their patients’ immune response was not research but "a one-time procedure" – exempt from review, according to a report in the Sacramento Bee.
Now both are banned from human research projects and the institutional review board is the subject of its own investigation.
For an account of the scientific thinking behind the deployment of bacteria in these patients and of ongoing efforts to develop immunotherapies against cancer, see the journal Nature (2012 July 27 [doi:10.1038/nature.2012.11080]).
Are a few animal studies and a handful of human case reports enough to let physicians skirt institutional review boards?
Two neurosurgeons in California did just that when they used Enterobacter aerogenes to infect the surgical wounds of three terminally ill glioblastoma patients. Two of the patients died from the infections.
Dr. J. Paul Muizelaar and Dr. Rudolph J. Schrot of the University of California, Davis, said their attempt to stimulate their patients’ immune response was not research but "a one-time procedure" – exempt from review, according to a report in the Sacramento Bee.
Now both are banned from human research projects and the institutional review board is the subject of its own investigation.
For an account of the scientific thinking behind the deployment of bacteria in these patients and of ongoing efforts to develop immunotherapies against cancer, see the journal Nature (2012 July 27 [doi:10.1038/nature.2012.11080]).
Rare Brainstem Glioma Doesn't Stop Former Marine
As the men and women who graciously serve our country return home, often times we can easily recognize the associated morbidity that resulted from their service. The physical injuries that can occur are obvious. But people have become more sensitive toward the injuries that are not so easily apparent, such as traumatic brain injury and posttraumatic stress disorder (PTSD). These injuries have been the targets of campaigns to increase awareness not only among practitioners, but also the lay population, particularly as they relate to concussive sports injuries.
Because these issues are on the forefront of our minds, it’s not hard to understand the misdiagnosis of PTSD in a young marine, Corporal Jordan Mills, who started having difficulty with left ptosis and episodic diplopia in 2008. It wasn’t until he became dysarthric and clumsy on the opposite side that he was transferred out of Afghanistan to a military facility in Germany, where an MRI of the brain revealed a mass raising concern for a brainstem glioma.

Brainstem glioma is a rare brain tumor that occurs mainly in the pediatric population and in young adults. Tissue diagnosis of brainstem glioma often is avoided in an attempt to first do no harm because of the tumor’s diffusely infiltrative nature and the distortion and expansion of the brainstem and its valuable inhabitants. Brainstem glioma is one of the rare instances in oncology when it has been accepted as appropriate to treat based on imaging alone without a tissue diagnosis. Conventional therapy includes radiation with or without the addition of chemotherapy. In spite of aggressive treatment strategies, this fulminating tumor is often fatal within months to years of diagnosis.
Colleagues at the Children’s National Medical Center have developed a protocol to collect serum, cerebrospinal fluid, urine, and tumor tissue of affected patients in an attempt to identify unique molecular abnormalities that would allow practitioners to target therapy more accurately to improve treatment efficacy. Brainstem glioma has been elusive given the lack of tissue to study up to this point, based on only tumor location and biopsy, as opposed to resection options.

In 2010, researchers at the Armed Forces Health Surveillance Center reviewed cancer data from 2000-2010 and found that service members have higher rates of melanoma, brain, non-Hodgkin’s lymphoma, and breast, prostate, and testicular cancers than civilians do. The strongest risk factor was associated with age. Interestingly, marines were found to have the lowest rate of cancer overall. Over the time period studied, 904 service members died of cancer, with 101 soldiers succumbing to brain or other nervous system types of cancer.
At the age of 22 years, it was accepted that this was Corp. Mills’s diagnosis and he was honorably discharged from his third and final tour with the marines. He was treated aggressively and went on to receive chemotherapy during his radiation phase and then received an additional 12 months of oral chemotherapy thereafter.

In addition to Jordan’s remarkable physical strength, his mental and emotional strength persevered. His attitude all the while was to continue to live life to the fullest and trust through his faith and his medical team that his tumor would be taken care of.
Jordan went on to marry, start a family, and enroll in the local community college where he graduated with an associate’s degree in accounting with honors. He was accepted into a prestigious school of business and is working to receive his bachelor’s degree in accounting.
Jordan’s tumor progressed in November 2011 and Jordan has reinitiated chemotherapy. His resolve is stronger than ever and he’s working to develop a foundation for marines with brain tumors. The goals of the foundation are to not only provide financial support to the marines and their immediate family, but also to support the education of military personnel on early detection of CNS disorders.
Dr. Porter is a neuro-oncologist in the department of neurology at the Mayo Clinic in Phoenix.
As the men and women who graciously serve our country return home, often times we can easily recognize the associated morbidity that resulted from their service. The physical injuries that can occur are obvious. But people have become more sensitive toward the injuries that are not so easily apparent, such as traumatic brain injury and posttraumatic stress disorder (PTSD). These injuries have been the targets of campaigns to increase awareness not only among practitioners, but also the lay population, particularly as they relate to concussive sports injuries.
Because these issues are on the forefront of our minds, it’s not hard to understand the misdiagnosis of PTSD in a young marine, Corporal Jordan Mills, who started having difficulty with left ptosis and episodic diplopia in 2008. It wasn’t until he became dysarthric and clumsy on the opposite side that he was transferred out of Afghanistan to a military facility in Germany, where an MRI of the brain revealed a mass raising concern for a brainstem glioma.
Brainstem glioma is a rare brain tumor that occurs mainly in the pediatric population and in young adults. Tissue diagnosis of brainstem glioma often is avoided in an attempt to first do no harm because of the tumor’s diffusely infiltrative nature and the distortion and expansion of the brainstem and its valuable inhabitants. Brainstem glioma is one of the rare instances in oncology when it has been accepted as appropriate to treat based on imaging alone without a tissue diagnosis. Conventional therapy includes radiation with or without the addition of chemotherapy. In spite of aggressive treatment strategies, this fulminating tumor is often fatal within months to years of diagnosis.
Colleagues at the Children’s National Medical Center have developed a protocol to collect serum, cerebrospinal fluid, urine, and tumor tissue of affected patients in an attempt to identify unique molecular abnormalities that would allow practitioners to target therapy more accurately to improve treatment efficacy. Brainstem glioma has been elusive given the lack of tissue to study up to this point, based on only tumor location and biopsy, as opposed to resection options.
In 2010, researchers at the Armed Forces Health Surveillance Center reviewed cancer data from 2000-2010 and found that service members have higher rates of melanoma, brain, non-Hodgkin’s lymphoma, and breast, prostate, and testicular cancers than civilians do. The strongest risk factor was associated with age. Interestingly, marines were found to have the lowest rate of cancer overall. Over the time period studied, 904 service members died of cancer, with 101 soldiers succumbing to brain or other nervous system types of cancer.
At the age of 22 years, it was accepted that this was Corp. Mills’s diagnosis and he was honorably discharged from his third and final tour with the marines. He was treated aggressively and went on to receive chemotherapy during his radiation phase and then received an additional 12 months of oral chemotherapy thereafter.
In addition to Jordan’s remarkable physical strength, his mental and emotional strength persevered. His attitude all the while was to continue to live life to the fullest and trust through his faith and his medical team that his tumor would be taken care of.
Jordan went on to marry, start a family, and enroll in the local community college where he graduated with an associate’s degree in accounting with honors. He was accepted into a prestigious school of business and is working to receive his bachelor’s degree in accounting.
Jordan’s tumor progressed in November 2011 and Jordan has reinitiated chemotherapy. His resolve is stronger than ever and he’s working to develop a foundation for marines with brain tumors. The goals of the foundation are to not only provide financial support to the marines and their immediate family, but also to support the education of military personnel on early detection of CNS disorders.
Dr. Porter is a neuro-oncologist in the department of neurology at the Mayo Clinic in Phoenix.
As the men and women who graciously serve our country return home, often times we can easily recognize the associated morbidity that resulted from their service. The physical injuries that can occur are obvious. But people have become more sensitive toward the injuries that are not so easily apparent, such as traumatic brain injury and posttraumatic stress disorder (PTSD). These injuries have been the targets of campaigns to increase awareness not only among practitioners, but also the lay population, particularly as they relate to concussive sports injuries.
Because these issues are on the forefront of our minds, it’s not hard to understand the misdiagnosis of PTSD in a young marine, Corporal Jordan Mills, who started having difficulty with left ptosis and episodic diplopia in 2008. It wasn’t until he became dysarthric and clumsy on the opposite side that he was transferred out of Afghanistan to a military facility in Germany, where an MRI of the brain revealed a mass raising concern for a brainstem glioma.
Brainstem glioma is a rare brain tumor that occurs mainly in the pediatric population and in young adults. Tissue diagnosis of brainstem glioma often is avoided in an attempt to first do no harm because of the tumor’s diffusely infiltrative nature and the distortion and expansion of the brainstem and its valuable inhabitants. Brainstem glioma is one of the rare instances in oncology when it has been accepted as appropriate to treat based on imaging alone without a tissue diagnosis. Conventional therapy includes radiation with or without the addition of chemotherapy. In spite of aggressive treatment strategies, this fulminating tumor is often fatal within months to years of diagnosis.
Colleagues at the Children’s National Medical Center have developed a protocol to collect serum, cerebrospinal fluid, urine, and tumor tissue of affected patients in an attempt to identify unique molecular abnormalities that would allow practitioners to target therapy more accurately to improve treatment efficacy. Brainstem glioma has been elusive given the lack of tissue to study up to this point, based on only tumor location and biopsy, as opposed to resection options.
In 2010, researchers at the Armed Forces Health Surveillance Center reviewed cancer data from 2000-2010 and found that service members have higher rates of melanoma, brain, non-Hodgkin’s lymphoma, and breast, prostate, and testicular cancers than civilians do. The strongest risk factor was associated with age. Interestingly, marines were found to have the lowest rate of cancer overall. Over the time period studied, 904 service members died of cancer, with 101 soldiers succumbing to brain or other nervous system types of cancer.
At the age of 22 years, it was accepted that this was Corp. Mills’s diagnosis and he was honorably discharged from his third and final tour with the marines. He was treated aggressively and went on to receive chemotherapy during his radiation phase and then received an additional 12 months of oral chemotherapy thereafter.
In addition to Jordan’s remarkable physical strength, his mental and emotional strength persevered. His attitude all the while was to continue to live life to the fullest and trust through his faith and his medical team that his tumor would be taken care of.
Jordan went on to marry, start a family, and enroll in the local community college where he graduated with an associate’s degree in accounting with honors. He was accepted into a prestigious school of business and is working to receive his bachelor’s degree in accounting.
Jordan’s tumor progressed in November 2011 and Jordan has reinitiated chemotherapy. His resolve is stronger than ever and he’s working to develop a foundation for marines with brain tumors. The goals of the foundation are to not only provide financial support to the marines and their immediate family, but also to support the education of military personnel on early detection of CNS disorders.
Dr. Porter is a neuro-oncologist in the department of neurology at the Mayo Clinic in Phoenix.

Skin Flaps Remedy Defects of the Ear
SAN DIEGO – In the clinical experience of Dr. Michael A. Keefe, 70%-80% of ear defects from auricular cancer treatment can be easily remedied with skin flaps.
The most common locations of auricular cancer are the helix, the posterior auricle skin, and the antihelix, Dr. Keefe said at a meeting on superficial anatomy and cutaneous surgery.

"More than 70% of lesions are smaller than 3 cm in size, and auricular lesions make up an estimated 8% of all skin cancers," said Dr. Keefe, a plastic surgeon with the division of head and neck surgery at Sharp Rees-Stealy Medical Group in San Diego. "The defects are unique, and the underlying cartilage structure makes it all the more interesting."
And challenging – defects may be located on the skin of the ear only, on the lateral side, or on the posterior side, or they may involve a combination of skin and cartilage. Healing by secondary intention is effective for concave defects, but the size of the defect drives the reconstruction options. "If there is no perichondrium, punch holes through cartilage with a 2-3 mm punch to allow granulation tissue to grow through, and then use a skin graft or allow it to heal with secondary intention," he said. "Keep the area moist with antibiotic ointment."
Options for reconstruction of defects in the middle one-third of the ear include primary closure, full-thickness skin grafts (FTSGs), the helical advancement flap, and the retroauricular composite advancement flap, while options for defects in the lower one-third of the ear include primary closure and the preauricular tubed flap. Options for reconstruction of defects in the upper one-third of the ear include primary closure, FTSGs, the helical advancement flap, the retroauricular and preauricular tubed flaps, and constructing an autogenous cartilage framework with FTSGs.
Dr. Keefe said that most small helical rim defects limited to the skin can be closed primarily. "There might be slight rim asymmetry [after closure]," he said at the meeting, which was sponsored by the University of California, San Diego, School of Medicine and the Scripps Clinic. "Some patients might not care [about this], but you have to advise them of that," he added.
A bilobed advancement flap is another option for helical rim defects limited to the skin. This flap "works well for cutaneous defects 2 cm or smaller in the helical rim or the posterior auricle," he said. "The other thing you can do with these bilobed flaps is advance them over the edge to correct helical rim defects."
The banner flap is another effective flap for helical rim defects, especially those located on the superior helix. It does not replace cartilage, but it conceals the incision well. For small composite helix and anterior defects, Dr. Keefe favors the chondrocutaneous advancement flap.
He said that he favors using FTSGs on the anterior surface of the helix for skin defects whenever possible. "You can use a composite skin graft as well, especially to replace cartilage or skin defects that are smaller than 1 cm in size," he said. "A FTSG is easy to harvest and has minimal contraction. Common donor sites include the preauricular, postauricular, supraclavicular, and clavicular regions. Make sure you trim off the fat." For posterior surface defects, the bilobe or advancement flaps work well.
Grafts must be placed on tissue with an adequate blood supply. Effective grafts establish imbibition in the first 24 hours, inosculation within 48-72 hours, and restoration of circulation within 4-7 days.
Dr. Keefe said that he had no relevant financial conflicts to disclose.
SAN DIEGO – In the clinical experience of Dr. Michael A. Keefe, 70%-80% of ear defects from auricular cancer treatment can be easily remedied with skin flaps.
The most common locations of auricular cancer are the helix, the posterior auricle skin, and the antihelix, Dr. Keefe said at a meeting on superficial anatomy and cutaneous surgery.
"More than 70% of lesions are smaller than 3 cm in size, and auricular lesions make up an estimated 8% of all skin cancers," said Dr. Keefe, a plastic surgeon with the division of head and neck surgery at Sharp Rees-Stealy Medical Group in San Diego. "The defects are unique, and the underlying cartilage structure makes it all the more interesting."
And challenging – defects may be located on the skin of the ear only, on the lateral side, or on the posterior side, or they may involve a combination of skin and cartilage. Healing by secondary intention is effective for concave defects, but the size of the defect drives the reconstruction options. "If there is no perichondrium, punch holes through cartilage with a 2-3 mm punch to allow granulation tissue to grow through, and then use a skin graft or allow it to heal with secondary intention," he said. "Keep the area moist with antibiotic ointment."
Options for reconstruction of defects in the middle one-third of the ear include primary closure, full-thickness skin grafts (FTSGs), the helical advancement flap, and the retroauricular composite advancement flap, while options for defects in the lower one-third of the ear include primary closure and the preauricular tubed flap. Options for reconstruction of defects in the upper one-third of the ear include primary closure, FTSGs, the helical advancement flap, the retroauricular and preauricular tubed flaps, and constructing an autogenous cartilage framework with FTSGs.
Dr. Keefe said that most small helical rim defects limited to the skin can be closed primarily. "There might be slight rim asymmetry [after closure]," he said at the meeting, which was sponsored by the University of California, San Diego, School of Medicine and the Scripps Clinic. "Some patients might not care [about this], but you have to advise them of that," he added.
A bilobed advancement flap is another option for helical rim defects limited to the skin. This flap "works well for cutaneous defects 2 cm or smaller in the helical rim or the posterior auricle," he said. "The other thing you can do with these bilobed flaps is advance them over the edge to correct helical rim defects."
The banner flap is another effective flap for helical rim defects, especially those located on the superior helix. It does not replace cartilage, but it conceals the incision well. For small composite helix and anterior defects, Dr. Keefe favors the chondrocutaneous advancement flap.
He said that he favors using FTSGs on the anterior surface of the helix for skin defects whenever possible. "You can use a composite skin graft as well, especially to replace cartilage or skin defects that are smaller than 1 cm in size," he said. "A FTSG is easy to harvest and has minimal contraction. Common donor sites include the preauricular, postauricular, supraclavicular, and clavicular regions. Make sure you trim off the fat." For posterior surface defects, the bilobe or advancement flaps work well.
Grafts must be placed on tissue with an adequate blood supply. Effective grafts establish imbibition in the first 24 hours, inosculation within 48-72 hours, and restoration of circulation within 4-7 days.
Dr. Keefe said that he had no relevant financial conflicts to disclose.
SAN DIEGO – In the clinical experience of Dr. Michael A. Keefe, 70%-80% of ear defects from auricular cancer treatment can be easily remedied with skin flaps.
The most common locations of auricular cancer are the helix, the posterior auricle skin, and the antihelix, Dr. Keefe said at a meeting on superficial anatomy and cutaneous surgery.
"More than 70% of lesions are smaller than 3 cm in size, and auricular lesions make up an estimated 8% of all skin cancers," said Dr. Keefe, a plastic surgeon with the division of head and neck surgery at Sharp Rees-Stealy Medical Group in San Diego. "The defects are unique, and the underlying cartilage structure makes it all the more interesting."
And challenging – defects may be located on the skin of the ear only, on the lateral side, or on the posterior side, or they may involve a combination of skin and cartilage. Healing by secondary intention is effective for concave defects, but the size of the defect drives the reconstruction options. "If there is no perichondrium, punch holes through cartilage with a 2-3 mm punch to allow granulation tissue to grow through, and then use a skin graft or allow it to heal with secondary intention," he said. "Keep the area moist with antibiotic ointment."
Options for reconstruction of defects in the middle one-third of the ear include primary closure, full-thickness skin grafts (FTSGs), the helical advancement flap, and the retroauricular composite advancement flap, while options for defects in the lower one-third of the ear include primary closure and the preauricular tubed flap. Options for reconstruction of defects in the upper one-third of the ear include primary closure, FTSGs, the helical advancement flap, the retroauricular and preauricular tubed flaps, and constructing an autogenous cartilage framework with FTSGs.
Dr. Keefe said that most small helical rim defects limited to the skin can be closed primarily. "There might be slight rim asymmetry [after closure]," he said at the meeting, which was sponsored by the University of California, San Diego, School of Medicine and the Scripps Clinic. "Some patients might not care [about this], but you have to advise them of that," he added.
A bilobed advancement flap is another option for helical rim defects limited to the skin. This flap "works well for cutaneous defects 2 cm or smaller in the helical rim or the posterior auricle," he said. "The other thing you can do with these bilobed flaps is advance them over the edge to correct helical rim defects."
The banner flap is another effective flap for helical rim defects, especially those located on the superior helix. It does not replace cartilage, but it conceals the incision well. For small composite helix and anterior defects, Dr. Keefe favors the chondrocutaneous advancement flap.
He said that he favors using FTSGs on the anterior surface of the helix for skin defects whenever possible. "You can use a composite skin graft as well, especially to replace cartilage or skin defects that are smaller than 1 cm in size," he said. "A FTSG is easy to harvest and has minimal contraction. Common donor sites include the preauricular, postauricular, supraclavicular, and clavicular regions. Make sure you trim off the fat." For posterior surface defects, the bilobe or advancement flaps work well.
Grafts must be placed on tissue with an adequate blood supply. Effective grafts establish imbibition in the first 24 hours, inosculation within 48-72 hours, and restoration of circulation within 4-7 days.
Dr. Keefe said that he had no relevant financial conflicts to disclose.
EXPERT ANALYSIS FROM A MEETING ON SUPERFICIAL ANATOMY AND CUTANEOUS SURGERY

Early Scar Treatment Is 'Critical'
SAN DIEGO – The future of treating hypertrophic and keloidal scars will involve earlier intervention with new and existing technologies – even at the genesis of scar formation, said Dr. E. Victor Ross.
"I think you’re going to see a lot more in the future about scars, not just in the laser area, but also in the biologic arena, because we’re learning more about the way scars behave," Dr. Ross said at a meeting on superficial anatomy and cutaneous surgery. "Some physicians are treating scars as early as the time of Mohs surgery, for example, by applying the PDL [pulsed-dye laser] at the time of suture placement. That’s perhaps a bit extreme, but I think you are going to see newer technologies and drugs used synergistically to give us a better fighting chance to prevent and treat scars."
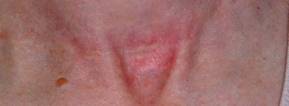
Dr. Ross of Scripps Clinic Laser and Cosmetic Dermatology Center in Carmel Valley, Calif., said that there is a lack of consensus regarding how the two main types of scars hypertrophic and keloidal – are defined. Historically, "we’ve said that hypertrophic scars don’t go beyond the boundary of where the scar tissue was, and keloidal scars go around the perimeter of where the scar boundaries were," he noted. "If the scar is red, even if it’s longstanding, I tend to call it a hypertrophic scar. If it tends to be more flesh colored, and aged like a fine wine, I tend to call it a keloidal scar. The critical thing with these scars is how long it takes the wound to heal. If an open wound takes more than 3-4 weeks to heal, often it will be hypertrophic."
Existing therapies that are commonly used to treat scars include intralesional steroids, intralesional 5-fluorouracil, oral antihistamines, cyclooxygenase-2 inhibitors, lasers, hydrogel sheeting, and compression. "The critical thing is to treat relatively early; you have to use all the weapons that are available to you," Dr. Ross said at the meeting, which was sponsored by the University of California San Diego School of Medicine and the Scripps Clinic.
He said that when treating scars, a modifiable approach should be taken. "You want to modify the scar. After it’s formed, you want to rehabilitate the scar and make it more like the skin around it."
When using intralesional steroids, Dr. Ross prefers to use very low volumes with a very high concentration of Kenalog, "typically 40 mg/mL in tiny amounts with a 3-gauge, half-inch needle," he said. "You want to keep the needle tip relatively superficial. If the steroid floats into the scar too easily you’re probably too deep or under the scar."
He favors using fractional lasers for scars whenever possible. These devices "create microscopic wounds in the skin," he said. "It turns out that if you fractionate a wound, the reservoirs of normal, undamaged skin act as ‘seeds’ to make the wounds heal quickly. I like to use purpuric settings with the pulsed-dye laser. They tend to give you better results than other settings."
For scars that form after thyroid surgery, Dr. Ross likes to use a PDL or IPL (intense pulsed light) to reduce the redness, followed by a nonablative fractional laser. With that tandem approach "you can almost make the scar go away, which is a complete rehabilitation of the scar," he said.
Innovative scar therapies include topical mitomycin C, which has worked well for postoperative keloids; oral and topical tamoxifen, which helps in the formation of fibroblasts; and oral methotrexate, which has demonstrated efficacy in the treatment and prevention of keloids. Imiquimod has also been used, "but I’m not a believer in it," Dr. Ross said. "We’ve tried it several times and we found that it irritated the skin most of the time. Retinoids are good and bad. They decrease fibroblast activity but also decrease collagenase."
Dr. Ross disclosed that he is a consultant for Cutera, Palomar Medical Technologies, and Lumenis. He has also received research support from Palomar, Sciton, and Syneron Medical.
SAN DIEGO – The future of treating hypertrophic and keloidal scars will involve earlier intervention with new and existing technologies – even at the genesis of scar formation, said Dr. E. Victor Ross.
"I think you’re going to see a lot more in the future about scars, not just in the laser area, but also in the biologic arena, because we’re learning more about the way scars behave," Dr. Ross said at a meeting on superficial anatomy and cutaneous surgery. "Some physicians are treating scars as early as the time of Mohs surgery, for example, by applying the PDL [pulsed-dye laser] at the time of suture placement. That’s perhaps a bit extreme, but I think you are going to see newer technologies and drugs used synergistically to give us a better fighting chance to prevent and treat scars."
Dr. Ross of Scripps Clinic Laser and Cosmetic Dermatology Center in Carmel Valley, Calif., said that there is a lack of consensus regarding how the two main types of scars hypertrophic and keloidal – are defined. Historically, "we’ve said that hypertrophic scars don’t go beyond the boundary of where the scar tissue was, and keloidal scars go around the perimeter of where the scar boundaries were," he noted. "If the scar is red, even if it’s longstanding, I tend to call it a hypertrophic scar. If it tends to be more flesh colored, and aged like a fine wine, I tend to call it a keloidal scar. The critical thing with these scars is how long it takes the wound to heal. If an open wound takes more than 3-4 weeks to heal, often it will be hypertrophic."
Existing therapies that are commonly used to treat scars include intralesional steroids, intralesional 5-fluorouracil, oral antihistamines, cyclooxygenase-2 inhibitors, lasers, hydrogel sheeting, and compression. "The critical thing is to treat relatively early; you have to use all the weapons that are available to you," Dr. Ross said at the meeting, which was sponsored by the University of California San Diego School of Medicine and the Scripps Clinic.
He said that when treating scars, a modifiable approach should be taken. "You want to modify the scar. After it’s formed, you want to rehabilitate the scar and make it more like the skin around it."
When using intralesional steroids, Dr. Ross prefers to use very low volumes with a very high concentration of Kenalog, "typically 40 mg/mL in tiny amounts with a 3-gauge, half-inch needle," he said. "You want to keep the needle tip relatively superficial. If the steroid floats into the scar too easily you’re probably too deep or under the scar."
He favors using fractional lasers for scars whenever possible. These devices "create microscopic wounds in the skin," he said. "It turns out that if you fractionate a wound, the reservoirs of normal, undamaged skin act as ‘seeds’ to make the wounds heal quickly. I like to use purpuric settings with the pulsed-dye laser. They tend to give you better results than other settings."
For scars that form after thyroid surgery, Dr. Ross likes to use a PDL or IPL (intense pulsed light) to reduce the redness, followed by a nonablative fractional laser. With that tandem approach "you can almost make the scar go away, which is a complete rehabilitation of the scar," he said.
Innovative scar therapies include topical mitomycin C, which has worked well for postoperative keloids; oral and topical tamoxifen, which helps in the formation of fibroblasts; and oral methotrexate, which has demonstrated efficacy in the treatment and prevention of keloids. Imiquimod has also been used, "but I’m not a believer in it," Dr. Ross said. "We’ve tried it several times and we found that it irritated the skin most of the time. Retinoids are good and bad. They decrease fibroblast activity but also decrease collagenase."
Dr. Ross disclosed that he is a consultant for Cutera, Palomar Medical Technologies, and Lumenis. He has also received research support from Palomar, Sciton, and Syneron Medical.
SAN DIEGO – The future of treating hypertrophic and keloidal scars will involve earlier intervention with new and existing technologies – even at the genesis of scar formation, said Dr. E. Victor Ross.
"I think you’re going to see a lot more in the future about scars, not just in the laser area, but also in the biologic arena, because we’re learning more about the way scars behave," Dr. Ross said at a meeting on superficial anatomy and cutaneous surgery. "Some physicians are treating scars as early as the time of Mohs surgery, for example, by applying the PDL [pulsed-dye laser] at the time of suture placement. That’s perhaps a bit extreme, but I think you are going to see newer technologies and drugs used synergistically to give us a better fighting chance to prevent and treat scars."
Dr. Ross of Scripps Clinic Laser and Cosmetic Dermatology Center in Carmel Valley, Calif., said that there is a lack of consensus regarding how the two main types of scars hypertrophic and keloidal – are defined. Historically, "we’ve said that hypertrophic scars don’t go beyond the boundary of where the scar tissue was, and keloidal scars go around the perimeter of where the scar boundaries were," he noted. "If the scar is red, even if it’s longstanding, I tend to call it a hypertrophic scar. If it tends to be more flesh colored, and aged like a fine wine, I tend to call it a keloidal scar. The critical thing with these scars is how long it takes the wound to heal. If an open wound takes more than 3-4 weeks to heal, often it will be hypertrophic."
Existing therapies that are commonly used to treat scars include intralesional steroids, intralesional 5-fluorouracil, oral antihistamines, cyclooxygenase-2 inhibitors, lasers, hydrogel sheeting, and compression. "The critical thing is to treat relatively early; you have to use all the weapons that are available to you," Dr. Ross said at the meeting, which was sponsored by the University of California San Diego School of Medicine and the Scripps Clinic.
He said that when treating scars, a modifiable approach should be taken. "You want to modify the scar. After it’s formed, you want to rehabilitate the scar and make it more like the skin around it."
When using intralesional steroids, Dr. Ross prefers to use very low volumes with a very high concentration of Kenalog, "typically 40 mg/mL in tiny amounts with a 3-gauge, half-inch needle," he said. "You want to keep the needle tip relatively superficial. If the steroid floats into the scar too easily you’re probably too deep or under the scar."
He favors using fractional lasers for scars whenever possible. These devices "create microscopic wounds in the skin," he said. "It turns out that if you fractionate a wound, the reservoirs of normal, undamaged skin act as ‘seeds’ to make the wounds heal quickly. I like to use purpuric settings with the pulsed-dye laser. They tend to give you better results than other settings."
For scars that form after thyroid surgery, Dr. Ross likes to use a PDL or IPL (intense pulsed light) to reduce the redness, followed by a nonablative fractional laser. With that tandem approach "you can almost make the scar go away, which is a complete rehabilitation of the scar," he said.
Innovative scar therapies include topical mitomycin C, which has worked well for postoperative keloids; oral and topical tamoxifen, which helps in the formation of fibroblasts; and oral methotrexate, which has demonstrated efficacy in the treatment and prevention of keloids. Imiquimod has also been used, "but I’m not a believer in it," Dr. Ross said. "We’ve tried it several times and we found that it irritated the skin most of the time. Retinoids are good and bad. They decrease fibroblast activity but also decrease collagenase."
Dr. Ross disclosed that he is a consultant for Cutera, Palomar Medical Technologies, and Lumenis. He has also received research support from Palomar, Sciton, and Syneron Medical.
EXPERT ANALYSIS FROM A MEETING ON SUPERFICIAL ANATOMY AND CUTANEOUS SURGERY
'Weekend Effect' Seen for Diverticulitis Procedures
Patients who were admitted for emergency surgery on a weekend to treat left-sided diverticulitis experience more short-term complications and are markedly more likely to undergo a Hartmann procedure than are those admitted on weekdays, according to results from a large population-based study.
Longer hospital stays, significantly higher treatment costs, and higher rates of reoperations were also associated with weekend admission. However, no differences in mortality were observed between the patient groups.
Previous studies have shown worse outcomes for patients with gastrointestinal hemorrhage, kidney injury, myocardial infarction, pulmonary embolism, and intracerebral hemorrhage when they were admitted on weekends. Although the current study, led by Dr. Mathias Worni of Duke University Medical Center in Durham, N.C., and Bern (Switzerland) University Hospital, was not designed to isolate the cause of the "weekend effect" for left-sided diverticulitis patients, the authors noted that hospital staffing tends to be reduced on weekends – especially among specialists such as colorectal surgeons.
Dr. Worni and his colleagues looked at records from the Nationwide Inpatient Sample between January 2002 and December 2008. Of the 31,832 patients who were treated surgically for left-sided diverticulitis, 7,066 (22.2%) were admitted on weekends and 24,766 (77.8%) on weekdays. Patients’ mean age was 60.8 years, and more than half were women.
Among patients who were admitted on a Saturday or Sunday, a Hartmann procedure was performed on 64.8% (n = 4,580), compared with only 53.9% (n = 13,351) for those admitted on a weekday (Arch. Surg. 2012;147:649-55). The Hartmann procedure – which involves formation of a colostomy – has long been the standard surgery for people presenting with left-sided diverticulitis, but is associated with long-term complications and a low rate of reversals.
Primary anastomosis, in which colostomy is avoided, is increasingly preferred, but only 35.2% of patients who were admitted on weekends underwent primary anastomosis, compared with 46.1% of patients admitted on weekdays.
The investigators found that patients admitted on weekends had significantly higher risk for any postoperative complication (odds ratio, 1.10; P = .005), compared with patients admitted on weekdays. Risk of reoperation was also higher among weekend admissions (OR, 1.50; P less than .001).
Furthermore, median total hospital charges were $3,734 higher among patients treated on weekends, and the median length of hospital stay was 0.5 days longer (P less than .001). The authors observed that these findings should motivate improvements in the quality of weekend care.
"Physicians working on weekends are thought to be less experienced than teams working during the week," they wrote. Experienced and specialized colorectal surgeons have been shown to perform more primary anastomoses, compared with trainees or general surgeons (Arch. Surg. 2010;145:79-86; Dis. Colon Rectum 2003;46:1461-8).
Limitations of the study include the fact that it did not capture long-term outcomes or severity of disease at presentation. The latter could be of potential importance: "Some patients, especially those with milder symptoms, may prefer weekend or weekday admission and may time their admission accordingly," the investigators noted.
In an invited critique that accompanied the article, Dr. Juerg Metzger, a surgeon at Lucerne (Switzerland) Cantonal Hospital, wrote that a disparity in experience among weekday and weekend surgical staff likely accounted for the higher rate of Hartmann procedures and complications following weekend admissions.
"Work-hour restrictions do not seem to have a negative influence on mortality and morbidity in surgical patients," Dr. Metzger wrote. "However, reduced experience owing to restricted working hours may negatively influence the practical skills of younger surgeons, resulting in more limited surgery [for example, a Hartmann procedure being performed instead of a primary anastomosis] and an increase in complications related to that surgery."
In the end, Dr. Metzger wrote, "quality is expensive, and our society has to decide if it is desirable and necessary to have the best surgical quality available all the time, especially when considering that health care costs will dramatically increase. It would be relevant to analyze additional large databases, asking similar questions about the outcomes of other common diseases [for example, appendicitis, cholecystitis, and strangulated hernias] and studying the effect of weekend admission on these illnesses."
Dr. Worni’s and colleagues’ was funded by a grant from the Swiss National Science Foundation. None of the investigators declared conflicts of interest. Dr. Metzger declared that he had no conflicts of interest related to his critique.
Patients who were admitted for emergency surgery on a weekend to treat left-sided diverticulitis experience more short-term complications and are markedly more likely to undergo a Hartmann procedure than are those admitted on weekdays, according to results from a large population-based study.
Longer hospital stays, significantly higher treatment costs, and higher rates of reoperations were also associated with weekend admission. However, no differences in mortality were observed between the patient groups.
Previous studies have shown worse outcomes for patients with gastrointestinal hemorrhage, kidney injury, myocardial infarction, pulmonary embolism, and intracerebral hemorrhage when they were admitted on weekends. Although the current study, led by Dr. Mathias Worni of Duke University Medical Center in Durham, N.C., and Bern (Switzerland) University Hospital, was not designed to isolate the cause of the "weekend effect" for left-sided diverticulitis patients, the authors noted that hospital staffing tends to be reduced on weekends – especially among specialists such as colorectal surgeons.
Dr. Worni and his colleagues looked at records from the Nationwide Inpatient Sample between January 2002 and December 2008. Of the 31,832 patients who were treated surgically for left-sided diverticulitis, 7,066 (22.2%) were admitted on weekends and 24,766 (77.8%) on weekdays. Patients’ mean age was 60.8 years, and more than half were women.
Among patients who were admitted on a Saturday or Sunday, a Hartmann procedure was performed on 64.8% (n = 4,580), compared with only 53.9% (n = 13,351) for those admitted on a weekday (Arch. Surg. 2012;147:649-55). The Hartmann procedure – which involves formation of a colostomy – has long been the standard surgery for people presenting with left-sided diverticulitis, but is associated with long-term complications and a low rate of reversals.
Primary anastomosis, in which colostomy is avoided, is increasingly preferred, but only 35.2% of patients who were admitted on weekends underwent primary anastomosis, compared with 46.1% of patients admitted on weekdays.
The investigators found that patients admitted on weekends had significantly higher risk for any postoperative complication (odds ratio, 1.10; P = .005), compared with patients admitted on weekdays. Risk of reoperation was also higher among weekend admissions (OR, 1.50; P less than .001).
Furthermore, median total hospital charges were $3,734 higher among patients treated on weekends, and the median length of hospital stay was 0.5 days longer (P less than .001). The authors observed that these findings should motivate improvements in the quality of weekend care.
"Physicians working on weekends are thought to be less experienced than teams working during the week," they wrote. Experienced and specialized colorectal surgeons have been shown to perform more primary anastomoses, compared with trainees or general surgeons (Arch. Surg. 2010;145:79-86; Dis. Colon Rectum 2003;46:1461-8).
Limitations of the study include the fact that it did not capture long-term outcomes or severity of disease at presentation. The latter could be of potential importance: "Some patients, especially those with milder symptoms, may prefer weekend or weekday admission and may time their admission accordingly," the investigators noted.
In an invited critique that accompanied the article, Dr. Juerg Metzger, a surgeon at Lucerne (Switzerland) Cantonal Hospital, wrote that a disparity in experience among weekday and weekend surgical staff likely accounted for the higher rate of Hartmann procedures and complications following weekend admissions.
"Work-hour restrictions do not seem to have a negative influence on mortality and morbidity in surgical patients," Dr. Metzger wrote. "However, reduced experience owing to restricted working hours may negatively influence the practical skills of younger surgeons, resulting in more limited surgery [for example, a Hartmann procedure being performed instead of a primary anastomosis] and an increase in complications related to that surgery."
In the end, Dr. Metzger wrote, "quality is expensive, and our society has to decide if it is desirable and necessary to have the best surgical quality available all the time, especially when considering that health care costs will dramatically increase. It would be relevant to analyze additional large databases, asking similar questions about the outcomes of other common diseases [for example, appendicitis, cholecystitis, and strangulated hernias] and studying the effect of weekend admission on these illnesses."
Dr. Worni’s and colleagues’ was funded by a grant from the Swiss National Science Foundation. None of the investigators declared conflicts of interest. Dr. Metzger declared that he had no conflicts of interest related to his critique.
Patients who were admitted for emergency surgery on a weekend to treat left-sided diverticulitis experience more short-term complications and are markedly more likely to undergo a Hartmann procedure than are those admitted on weekdays, according to results from a large population-based study.
Longer hospital stays, significantly higher treatment costs, and higher rates of reoperations were also associated with weekend admission. However, no differences in mortality were observed between the patient groups.
Previous studies have shown worse outcomes for patients with gastrointestinal hemorrhage, kidney injury, myocardial infarction, pulmonary embolism, and intracerebral hemorrhage when they were admitted on weekends. Although the current study, led by Dr. Mathias Worni of Duke University Medical Center in Durham, N.C., and Bern (Switzerland) University Hospital, was not designed to isolate the cause of the "weekend effect" for left-sided diverticulitis patients, the authors noted that hospital staffing tends to be reduced on weekends – especially among specialists such as colorectal surgeons.
Dr. Worni and his colleagues looked at records from the Nationwide Inpatient Sample between January 2002 and December 2008. Of the 31,832 patients who were treated surgically for left-sided diverticulitis, 7,066 (22.2%) were admitted on weekends and 24,766 (77.8%) on weekdays. Patients’ mean age was 60.8 years, and more than half were women.
Among patients who were admitted on a Saturday or Sunday, a Hartmann procedure was performed on 64.8% (n = 4,580), compared with only 53.9% (n = 13,351) for those admitted on a weekday (Arch. Surg. 2012;147:649-55). The Hartmann procedure – which involves formation of a colostomy – has long been the standard surgery for people presenting with left-sided diverticulitis, but is associated with long-term complications and a low rate of reversals.
Primary anastomosis, in which colostomy is avoided, is increasingly preferred, but only 35.2% of patients who were admitted on weekends underwent primary anastomosis, compared with 46.1% of patients admitted on weekdays.
The investigators found that patients admitted on weekends had significantly higher risk for any postoperative complication (odds ratio, 1.10; P = .005), compared with patients admitted on weekdays. Risk of reoperation was also higher among weekend admissions (OR, 1.50; P less than .001).
Furthermore, median total hospital charges were $3,734 higher among patients treated on weekends, and the median length of hospital stay was 0.5 days longer (P less than .001). The authors observed that these findings should motivate improvements in the quality of weekend care.
"Physicians working on weekends are thought to be less experienced than teams working during the week," they wrote. Experienced and specialized colorectal surgeons have been shown to perform more primary anastomoses, compared with trainees or general surgeons (Arch. Surg. 2010;145:79-86; Dis. Colon Rectum 2003;46:1461-8).
Limitations of the study include the fact that it did not capture long-term outcomes or severity of disease at presentation. The latter could be of potential importance: "Some patients, especially those with milder symptoms, may prefer weekend or weekday admission and may time their admission accordingly," the investigators noted.
In an invited critique that accompanied the article, Dr. Juerg Metzger, a surgeon at Lucerne (Switzerland) Cantonal Hospital, wrote that a disparity in experience among weekday and weekend surgical staff likely accounted for the higher rate of Hartmann procedures and complications following weekend admissions.
"Work-hour restrictions do not seem to have a negative influence on mortality and morbidity in surgical patients," Dr. Metzger wrote. "However, reduced experience owing to restricted working hours may negatively influence the practical skills of younger surgeons, resulting in more limited surgery [for example, a Hartmann procedure being performed instead of a primary anastomosis] and an increase in complications related to that surgery."
In the end, Dr. Metzger wrote, "quality is expensive, and our society has to decide if it is desirable and necessary to have the best surgical quality available all the time, especially when considering that health care costs will dramatically increase. It would be relevant to analyze additional large databases, asking similar questions about the outcomes of other common diseases [for example, appendicitis, cholecystitis, and strangulated hernias] and studying the effect of weekend admission on these illnesses."
Dr. Worni’s and colleagues’ was funded by a grant from the Swiss National Science Foundation. None of the investigators declared conflicts of interest. Dr. Metzger declared that he had no conflicts of interest related to his critique.
FROM ARCHIVES OF SURGERY
Major Finding: Weekend admission to the hospital for diverticulitis posed a significantly higher risk for any postoperative complication (OR, 1.10; P = .005) and risk of reoperation (OR, 1.50; P less than .001), compared with weekday admission.
Data Source: The findings are based on an analysis of NIS records for 31,832 patients who were treated surgically for left-sided diverticulitis.
Disclosures: Dr. Worni’s and colleagues’ study was funded by a grant from the Swiss National Science Foundation. None of the investigators declared conflicts of interest. Dr. Metzger declared that he had no conflicts of interest related to his critique.