User login
The $64,000 Question/
A 40‐year‐old Sudanese man was admitted due to worsening abdominal pain with recurrent ascites. He had a history of hepatitis B (HBV) infection and diabetes. He previously drank 3 beers per day on the weekends, but he had not consumed alcohol in over a year. He was born in Sudan but lived in Egypt most of his adult life; he immigrated to the United States 6 years previously. He was hospitalized out of state 9 months ago for a swollen abdomen and underwent an exploratory laparotomy that reportedly was unremarkable except for ascites.
Portal hypertension due to liver disease is the most common cause of ascites. This patient has a known risk factor for liver disease (history of HBV infection). Although his reported alcohol consumption is low, there is a synergistic effect on liver injury in the setting of chronic hepatitis. Abdominal pain in the setting of ascites needs to be urgently evaluated to exclude spontaneous bacterial peritonitis (SBP). Also, because chronic HBV infection is the major risk factor for hepatocellular carcinoma in the world, malignant ascites is in the differential. Hepatic vascular thrombosis and tuberculous peritonitis (given the patient's country of origin and travel history) also should be considered. The most appropriate initial test would be a diagnostic paracentesis to support or exclude the presence of SBP and direct the evaluation toward liver disease or other less‐common causes of ascites.
The patient was seen as an outpatient 5 months prior to admission with transient fever and joint pains. Laboratory studies at that visit were notable for a serum albumin of 3.2 g/dL (normal 3.55), 2.4 g of predicted 24‐hour protein on urinalysis (normal 30 mg per 24 hours), creatinine of 0.5 mg/dL (normal 0.81.3), and a positive hepatitis B surface antibody. The working diagnosis was a nonspecific viral syndrome and his symptoms resolved without treatment. One month later, he developed ascites and mild lower extremity edema. Additional laboratory studies at that time showed a normocytic anemia with hemoglobin 11.7 g/dL (normal 13.517.5) and leukopenia with white blood cell count of 2.4 109/L (normal 3.510.5), neutrophil count of 1.45 109/L (normal 1.77.0), and lymphocyte count of 0.58 109/L (normal 0.902.90). Transaminases, serum bilirubin, prothrombin time, alpha fetoprotein, and peripheral blood smear were normal. Human immunodeficiency virus antibody screen and QuantiFERON‐TB assay were negative. Hemoglobin A1c was 6.2% (normal 4.06.0). Repeat urinalysis demonstrated 883 mg of predicted 24‐hour protein. Computed tomography (CT) of the abdomen showed a large amount of intra‐abdominal ascites; the liver and spleen were normal, and there were no varices or other evidence of portal hypertension. Echocardiogram was normal except for a small inferior vena cava (IVC) and a mildly increased right ventricular systolic pressure of 32 mm Hg (systolic blood pressure 98 mm Hg). Due to the indeterminate cause for the patient's ascites, referral was made for gastroenterology evaluation with consideration for a paracentesis.
Cirrhotic ascites seems less likely. Postsinusoidal causes of portal hypertension (eg, cardiomyopathy) are also less likely given the absence of suggestive findings on echocardiography. Malignant ascites also appears less probable in the absence of suggestive findings such as mass lesions, lymphadenopathy, or peritoneal carcinomatosis on CT imaging. The suspicion for tuberculous peritonitis is lower with the negative QuantiFERON‐TB test. Hypoalbuminemia, normocytic anemia, leukopenia, and proteinuria all suggest a systemic inflammatory condition (eg, systemic lupus erythematosus [SLE]) with inflammatory serositis causing ascites). Nephrotic syndrome can cause hypoalbuminemia, edema, and ascites, but his total urine protein losses of 3.5 grams per 24 hours are not in keeping with this diagnosis. Other uncommon causes of ascites such as chylous ascites have not yet been excluded. The most appropriate next step remains ascitic fluid analysis.
A paracentesis yielded 7.8 L of clear‐yellow fluid and improvement in his abdominal discomfort. Analysis showed 224 total nucleated cells/L with 2% neutrophils, 57% lymphocytes, and 37% monocytes. Ascites total protein was 3.8 g/dL and glucose was 55 mg/dL. Gram stain and culture were negative, and cytology was negative for malignancy but showed lymphocytes, plasma cells, monocytes, and reactive mesothelial cells interpreted as consistent with chronic inflammation. The serum‐ascites albumin gradient (SAAG) was not obtained.
With a low leukocyte count and a paucity of neutrophils, this is not SBP. The ascites fluid did not have a chylous appearance. The SAAG, which can distinguish between portal hypertensive and nonportal hypertensive causes for ascites using a cutoff of 1.1 g/dL, was not done. The total protein was high, arguing against cirrhosis. High protein ascites with a high SAAG would suggest a posthepatic source of portal hypertension (eg, Budd‐Chiari syndrome, constrictive pericarditis). High protein ascites with a low SAAG would suggest an inflammatory or malignant source of ascites. The relative lymphocytosis in the ascites fluid suggests an inflammatory process, but is a nonspecific finding. The negative cytology does not completely exclude a malignancy, but given the absence of findings on the CT, malignant ascites is less likely.
Three months before admission, the patient underwent a repeat large‐volume paracentesis and a liver biopsy. The biopsy showed ectopic portal vein branches consistent with hepatoportal sclerosis, but no actual sclerosis was identified. The pathologist concluded that the findings suggested noncirrhotic portal hypertension due to a vascular in‐flow abnormality. Abdominal ultrasound with Doppler was unremarkable other than slightly increased echogenicity of the liver. Magnetic resonance (MR) angiogram showed narrowing of the intra‐abdominal IVC at the level of the diaphragm. Because of concern that hepatic congestion from high pressures in the narrowed IVC was leading to poor vascular inflow as suggested by the biopsy findings, an inferior vena cavagram was performed. This study was normal, although no transhepatic pressure measurements were obtained. Three stool specimens and 2 urine specimens were negative for parasites. The patient required repeat large‐volume paracenteses monthly. SBP was again ruled out, but no other diagnostic labs were obtained. He had anorexia with poor oral intake each time his abdomen became distended.
The patient was started on furosemide 1 month prior to admission to the hospital but had only a slight improvement in the ascites. His other medications included insulin, tamsulosin, and hydrocodone‐acetaminophen. Five days prior to admission, he underwent a diagnostic laparoscopy, which showed only ascites and small adhesions to the anterior abdominal wall. There was no visual evidence of malignancy, and the surgeon commented that the liver was normal. No additional biopsies were obtained.
The liver biopsy findings could be seen in noncirrhotic portal hypertension, although this diagnosis would be unlikely without splenomegaly, varices, or other signs of portal hypertension. However, 2 possible etiologies for noncirrhotic portal hypertension in this patient would be hepatic congestion from the narrowed IVC (although the normal IVC study argues against this) and hepatic schistosomiasis. Schistosomiasis is an important cause of noncirrhotic portal hypertension in endemic areas like this patient's country of origin, but the negative stool and urine studies, combined with the lack of granulomas or fibrosis seen on biopsy, make this condition unlikely.
Systemic amyloidosis (primary or secondary) could also be a cause of ascites and could present with multiorgan involvement (diarrhea and nephrotic syndrome). Amyloid deposits would have probably been seen in the liver biopsy, if present, but may not have been apparent unless specific stains (Congo red) were performed.
Evaluation for systemic, inflammatory autoimmune processes is indicated. Serum autoantibodies (anti‐nuclear antibody [ANA] and extractable nuclear antigens), and a serum and 24‐hour urine protein electrophoresis would be appropriate diagnostic tests. Peritoneal biopsies would have been helpful to assess for serosal diseases.
The patient subsequently developed acute right‐sided abdominal pain requiring urgent evaluation and admission to the hospital. He was initially assessed by a general surgeon, who found no evidence of postoperative complications. His temperature was 36.7C, blood pressure 105/64, heart rate 82, respiratory rate 16, and oxygen saturation 97% on room air. He appeared chronically ill, but he was in no distress and he had a normal mental status. Cardiac exam was normal except for mild jugular venous distension. He had mild bibasilar lung crackles. His abdomen was distended with superficial abdominal tenderness and a fluid wave, but he had normal bowel sounds and no peritoneal signs. He had mild scrotal edema but no peripheral edema. Joint exam did not suggest synovitis and there were no rashes or oral ulcers. Lactate was 0.9 mmol/L (normal 0.62.3), albumin was 2.6 g/dL, and prealbumin was 9 mg/dL (normal 1938). Erythrocyte sedimentation rate and C‐reactive protein were 46 mm/hour (normal 22) and 33.1 mg/L (normal 8), respectively. He had a normocytic anemia and leukopenia. Liver tests and routine chemistries were normal. Serum protein electrophoresis indicated no monoclonal protein. Complete 24‐hour urine collection showed 1.2 g of protein (normal 102 mg). Paracentesis of 3.4 L demonstrated 227 total nucleated cells/L with 2% neutrophils. Following the fluid removal, he had improvement in his pain, which he felt was related to the ascites rather than the recent surgery. Ascites total protein was 3.9 g/dL and ascites albumin was 1.7 g/dL. Ascites culture was negative for infection. Serum Schistosoma immunoglobulin G (IgG) antibody was positive at 3.53 (normal 1.00).
Further history revealed prior episodes of polyarticular joint pain and swelling in his hands and knees 5 years before admission. At that time, he reported a diffuse, pruritic, papular body rash. In addition, he noticed that his fingertips and toes turned white with cold exposure.
Importantly, surgical and infectious complications have been excluded. High protein ascites with a low SAAG of 0.9 suggests an inflammatory source of ascites. The follow‐up clinical data (arthritis, normocytic anemia, leukopenia, rash, Raynaud's phenomenon) suggest a systemic inflammatory syndrome such as SLE, with accompanying serositis. Serologic testing for autoantibodies would be recommended. Peritoneal biopsies, if obtained, may have demonstrated chronic, inflammatory infiltrate (nonspecific) or leukocytoclastic vasculitis (strongly supportive).
ANA enzyme immunoassay was >12 U (normal 1.0 U). Extractable nuclear antigens revealed positive autoantibodies for anti‐SSA, anti‐SSB, and anti‐ribosomal P. Moreover, double‐stranded DNA IgG antibody was 120 IU/mL (normal 30 IU/mL) and C3, C4, and total complement levels were low.
The clinical data support a diagnosis of SLE with serositis. Treatment of the underlying connective tissue disease will typically result in resolution of the ascites; diuretic therapy is generally ineffective.
In consultation with rheumatology and gastroenterology specialists, the diagnosis of SLE was made based on criteria of serositis, persistent leukopenia, arthritis, renal disease (proteinuria), positive ANA, elevated ds‐DNA antibodies, and hypocomplementemia. MR imaging of the abdominal vasculature demonstrated no evidence of vasculitis. The patient was given intravenous methylprednisolone 1 g daily for 3 days followed by high‐dose oral corticosteroids with a gradual taper. He was also started on mycophenolate mofetil as a steroid‐sparing medication (which was later changed to leflunomide due to persistent leukopenia) and hydroxychloroquine. His isolated positive Schistosoma IgG antibody in the absence of other findings was consistent with past exposure or infection. The infectious disease specialist felt there was no evidence of active schistosomiasis, but recommended treatment with a single dose of praziquantel due to the potential benefit with low risk of side effects. The patient had ongoing improvement following dismissal. He had 1 additional paracentesis of 4.1 L, 10 days after his hospitalization, and his ascites and proteinuria resolved. At the 5‐year follow‐up visit, there had been no recurrence of abdominal ascites or abdominal pain. He remains on low‐dose prednisone at 5 mg daily, leflunomide, and hydroxychloroquine.
COMMENTARY
This patient had recurrent ascites with 29.6 L removed over the 4 months prior to admission and an additional 3.4 L during his hospitalization. His outpatient providers initially considered a portal hypertensive etiology of his ascites due to his history of HBV and prior alcohol use. They also appropriately investigated for a possible infectious process. They next directed their evaluation toward the liver biopsy findings, which raised concern for a vascular inflow abnormality. However, the evaluation could have been performed more rapidly and far more cost‐efficiently had a diagnostic paracentesis with calculation of the SAAG been performed early in the evaluation.
The SAAG, which was first described in 1983 by Par and colleagues, is a parameter reflecting the oncotic pressure gradient between the vascular bed and the interstitial splanchnic or ascitic fluid. [1] In the classic study by Runyon and colleagues, a SAAG difference of 1.1 g/dL correctly differentiated causes of ascites due to portal hypertension from those that were not due to portal hypertension 96.7% of the time. [2] Conditions such as nephrotic syndrome, peritoneal carcinomatosis, and serositis (lupus peritonitis) can cause ascites in patients without portal hypertension.
Serositis in the form of pleuritis and/or pericarditis is a common feature of SLE, and ascites has been described in 8% to 11% of SLE patients.[3] However, massive ascites due to lupus peritonitis as a presenting symptom is rare.[4] More common causes of ascites in the setting of SLE include nephrotic syndrome, heart failure, protein‐losing enteropathy, constrictive pericarditis, Budd‐Chiari syndrome, indolent infections such as tuberculosis, and chylous ascites.[5, 6, 7] Of note, lupus peritonitis may be chronic or acute. Chronic ascites develops insidiously with few manifestations of active lupus and may be painless, whereas ascites from acute lupus peritonitis typically develops rapidly and presents with acute abdominal pain and other signs of increased lupus activity.[3, 5, 6, 8, 9]
Ascites from lupus peritonitis may be due to marked serosal exudative accumulation with reduced absorptive capacity in the peritoneum.[3, 4, 10] Other possible causes include peritoneal inflammation from deposition of immune complexes or vasculitis of peritoneal vessels and visceral serous membranes.[4, 9, 11] Although subserosal and submucosal vasculitis have been found in acute ascites, chronic ascites may be related to scarring from vasculitis and serosal inflammation leading to poor venous and lymph drainage.[9] Ascitic fluid characteristics from lupus peritonitis include a SAAG 1.1, presence of white blood cells anywhere in a broad range from 10 to 1630/L, and a range of fluid protein from 3.4 to 4.7 mg/dL.[3] Although not tested in this patient, findings of low complement levels, positive ANA, and elevated anti‐DNA antibody in the ascitic fluid would be supportive of lupus peritonitis, but not specific.[5, 9, 12] Lupus erythematosus cells are occasionally found in the ascitic fluid, but do not rule out other causes of ascites.[9] On retrospective analysis, lupus erythematosus cells were not seen in this patient's pathology specimens.
Treatment of lupus peritonitis and ascites is with high‐dose glucocorticoid therapy, but many patients may need a second immunosuppressant, possibly because of impaired peritoneal circulation from chronic inflammation leading to decreased drug delivery.[13, 14] Chronic ascites may be recalcitrant to systemic glucocorticoids,[3] so a possible alternative therapy is intraperitoneal injection of triamcinolone, which successfully treated massive ascites in a patient who did not respond to oral glucocorticoid treatment.[13] Although ascites may be refractory in some patients, those with chronic lupus peritonitis can generally achieve remission, yet the overall prognosis depends on the presence and severity of multiorgan involvement from SLE. As with any SLE patient, there are also risks of infection from immunosuppression and increased cardiovascular risks.
This patient's evaluation and treatment could have been expedited if he had undergone a paracenteses with determination of the SAAG early in his workup. It is not known why the SAAG was not obtained despite multiple outpatient visits and paracenteses, his history of HBV, and prior alcohol use. This may have been simply an unfortunate oversight. Alternatively, it may have been that his outpatient providers focused on tantalizing clues such as his country of origin, which led to concern for schistosomiasis, and the biopsy findings suggestive of a vascular inflow abnormality that led to further extensive testing. In so doing, the clinicians committed several diagnostic errors, including multiple alternatives bias, anchoring, and confirmation bias.[15] As a result, the patient accrued excess charges of $64,000 from multiple tests, laparoscopic surgery, and 2 hospitalizations. This case highlights how cognitive errors introduce costly variability into patient care, especially when a simple and accurate test is at the beginning of the decision tree.
CLINICAL TEACHING POINTS
- Diagnostic paracentesis, with calculation of the serum‐ascites albumin gradient, should be the first test in the workup for ascites and can distinguish portal hypertensive causes from nonportal hypertensive causes.
- Ascites related to SLE can be acute or chronic and caused by bowel infarction, perforation, pancreatitis, mesenteric vasculitis, nephrotic syndrome, heart failure, protein‐losing enteropathy, constrictive pericarditis, lupus peritonitis, Budd‐Chiari syndrome, or serositis (lupus peritonitis).
- Ascites caused by lupus peritonitis is rare. Once treated, management should be directed toward keeping the SLE in remission.
ACKNOWLEDGMENTS
Disclosure: Nothing to report.
- , , . Serum‐ascites albumin concentration gradient: a physiologic approach to the differential diagnosis of ascites. Gastroenterology. 1983;85(2):240–244.
- , , , et al. The serum‐ascites albumin gradient is superior to the exudate‐transudate concept in the differential diagnosis of ascites. Ann Intern Med. 1992;117:215–220.
- , , . Insidious onset of massive painless ascites as initial manifestation of systemic lupus erythematosus. Lupus. 2011;20:754–757.
- , . Rapid onset of massive ascites as the initial presentation of systemic lupus erythematosus. Am J Gastroenterol. 2000;95:302–303.
- , . Gastrointestinal and hepatic manifestations of systemic lupus erythematosus. J Clin Gastroenterol. 2011;45:436–441.
- , , , . Massive ascites as a presenting feature of lupus. Int J Rheum Dis. 2012;15:e15–e16.
- , , , et al. Concurrent occurrence of chylothorax, chylous ascites, and protein‐losing enteropathy in systemic lupus erythematosus. J Rheumatol. 2002;29:1330–1333.
- , , , et al. Abdominal manifestations in childhood‐onset systemic lupus erythematosus. Ann Rheum Dis. 2007;66:174–178.
- , , . Chronic lupus peritonitis with ascites: review of the literature with a case report. Semin Arthritis Rheum. 1988;18:121–126.
- , . Nonhepatic Gastrointestinal Manifestations of Systemic Lupus Erythematosus. London, United Kingdom: Churchill Livingstone; 1987:747–760.
- , , , . Ascites due to lupus peritonitis: a rare form of onset of systemic lupus erythematosus. Rev Bras Reumatol. 2012;52(1):113–119.
- , , , . New‐onset lupus presenting as serositis in an 80‐year‐old woman: does a high‐titer ANA in pleural, pericardial, or peritoneal fluid help confirm the diagnosis? J Clin Rheum.2005:11(5):292–293.
- , , , . Successful treatment of massive ascites with intraperitoneal administration of a steroid in a case of systemic lupus erythematosus. Lupus. 2009;18:740–742.
- , , , et al. Chronic lupus peritonitis with massive ascites at elderly onset: case report and review of the literature. Intern Med. 2002;41:1056–1061.
- . The Importance of cognitive errors in diagnosis and strategies to minimize them. Acad Med. 2003;78:775–780.
A 40‐year‐old Sudanese man was admitted due to worsening abdominal pain with recurrent ascites. He had a history of hepatitis B (HBV) infection and diabetes. He previously drank 3 beers per day on the weekends, but he had not consumed alcohol in over a year. He was born in Sudan but lived in Egypt most of his adult life; he immigrated to the United States 6 years previously. He was hospitalized out of state 9 months ago for a swollen abdomen and underwent an exploratory laparotomy that reportedly was unremarkable except for ascites.
Portal hypertension due to liver disease is the most common cause of ascites. This patient has a known risk factor for liver disease (history of HBV infection). Although his reported alcohol consumption is low, there is a synergistic effect on liver injury in the setting of chronic hepatitis. Abdominal pain in the setting of ascites needs to be urgently evaluated to exclude spontaneous bacterial peritonitis (SBP). Also, because chronic HBV infection is the major risk factor for hepatocellular carcinoma in the world, malignant ascites is in the differential. Hepatic vascular thrombosis and tuberculous peritonitis (given the patient's country of origin and travel history) also should be considered. The most appropriate initial test would be a diagnostic paracentesis to support or exclude the presence of SBP and direct the evaluation toward liver disease or other less‐common causes of ascites.
The patient was seen as an outpatient 5 months prior to admission with transient fever and joint pains. Laboratory studies at that visit were notable for a serum albumin of 3.2 g/dL (normal 3.55), 2.4 g of predicted 24‐hour protein on urinalysis (normal 30 mg per 24 hours), creatinine of 0.5 mg/dL (normal 0.81.3), and a positive hepatitis B surface antibody. The working diagnosis was a nonspecific viral syndrome and his symptoms resolved without treatment. One month later, he developed ascites and mild lower extremity edema. Additional laboratory studies at that time showed a normocytic anemia with hemoglobin 11.7 g/dL (normal 13.517.5) and leukopenia with white blood cell count of 2.4 109/L (normal 3.510.5), neutrophil count of 1.45 109/L (normal 1.77.0), and lymphocyte count of 0.58 109/L (normal 0.902.90). Transaminases, serum bilirubin, prothrombin time, alpha fetoprotein, and peripheral blood smear were normal. Human immunodeficiency virus antibody screen and QuantiFERON‐TB assay were negative. Hemoglobin A1c was 6.2% (normal 4.06.0). Repeat urinalysis demonstrated 883 mg of predicted 24‐hour protein. Computed tomography (CT) of the abdomen showed a large amount of intra‐abdominal ascites; the liver and spleen were normal, and there were no varices or other evidence of portal hypertension. Echocardiogram was normal except for a small inferior vena cava (IVC) and a mildly increased right ventricular systolic pressure of 32 mm Hg (systolic blood pressure 98 mm Hg). Due to the indeterminate cause for the patient's ascites, referral was made for gastroenterology evaluation with consideration for a paracentesis.
Cirrhotic ascites seems less likely. Postsinusoidal causes of portal hypertension (eg, cardiomyopathy) are also less likely given the absence of suggestive findings on echocardiography. Malignant ascites also appears less probable in the absence of suggestive findings such as mass lesions, lymphadenopathy, or peritoneal carcinomatosis on CT imaging. The suspicion for tuberculous peritonitis is lower with the negative QuantiFERON‐TB test. Hypoalbuminemia, normocytic anemia, leukopenia, and proteinuria all suggest a systemic inflammatory condition (eg, systemic lupus erythematosus [SLE]) with inflammatory serositis causing ascites). Nephrotic syndrome can cause hypoalbuminemia, edema, and ascites, but his total urine protein losses of 3.5 grams per 24 hours are not in keeping with this diagnosis. Other uncommon causes of ascites such as chylous ascites have not yet been excluded. The most appropriate next step remains ascitic fluid analysis.
A paracentesis yielded 7.8 L of clear‐yellow fluid and improvement in his abdominal discomfort. Analysis showed 224 total nucleated cells/L with 2% neutrophils, 57% lymphocytes, and 37% monocytes. Ascites total protein was 3.8 g/dL and glucose was 55 mg/dL. Gram stain and culture were negative, and cytology was negative for malignancy but showed lymphocytes, plasma cells, monocytes, and reactive mesothelial cells interpreted as consistent with chronic inflammation. The serum‐ascites albumin gradient (SAAG) was not obtained.
With a low leukocyte count and a paucity of neutrophils, this is not SBP. The ascites fluid did not have a chylous appearance. The SAAG, which can distinguish between portal hypertensive and nonportal hypertensive causes for ascites using a cutoff of 1.1 g/dL, was not done. The total protein was high, arguing against cirrhosis. High protein ascites with a high SAAG would suggest a posthepatic source of portal hypertension (eg, Budd‐Chiari syndrome, constrictive pericarditis). High protein ascites with a low SAAG would suggest an inflammatory or malignant source of ascites. The relative lymphocytosis in the ascites fluid suggests an inflammatory process, but is a nonspecific finding. The negative cytology does not completely exclude a malignancy, but given the absence of findings on the CT, malignant ascites is less likely.
Three months before admission, the patient underwent a repeat large‐volume paracentesis and a liver biopsy. The biopsy showed ectopic portal vein branches consistent with hepatoportal sclerosis, but no actual sclerosis was identified. The pathologist concluded that the findings suggested noncirrhotic portal hypertension due to a vascular in‐flow abnormality. Abdominal ultrasound with Doppler was unremarkable other than slightly increased echogenicity of the liver. Magnetic resonance (MR) angiogram showed narrowing of the intra‐abdominal IVC at the level of the diaphragm. Because of concern that hepatic congestion from high pressures in the narrowed IVC was leading to poor vascular inflow as suggested by the biopsy findings, an inferior vena cavagram was performed. This study was normal, although no transhepatic pressure measurements were obtained. Three stool specimens and 2 urine specimens were negative for parasites. The patient required repeat large‐volume paracenteses monthly. SBP was again ruled out, but no other diagnostic labs were obtained. He had anorexia with poor oral intake each time his abdomen became distended.
The patient was started on furosemide 1 month prior to admission to the hospital but had only a slight improvement in the ascites. His other medications included insulin, tamsulosin, and hydrocodone‐acetaminophen. Five days prior to admission, he underwent a diagnostic laparoscopy, which showed only ascites and small adhesions to the anterior abdominal wall. There was no visual evidence of malignancy, and the surgeon commented that the liver was normal. No additional biopsies were obtained.
The liver biopsy findings could be seen in noncirrhotic portal hypertension, although this diagnosis would be unlikely without splenomegaly, varices, or other signs of portal hypertension. However, 2 possible etiologies for noncirrhotic portal hypertension in this patient would be hepatic congestion from the narrowed IVC (although the normal IVC study argues against this) and hepatic schistosomiasis. Schistosomiasis is an important cause of noncirrhotic portal hypertension in endemic areas like this patient's country of origin, but the negative stool and urine studies, combined with the lack of granulomas or fibrosis seen on biopsy, make this condition unlikely.
Systemic amyloidosis (primary or secondary) could also be a cause of ascites and could present with multiorgan involvement (diarrhea and nephrotic syndrome). Amyloid deposits would have probably been seen in the liver biopsy, if present, but may not have been apparent unless specific stains (Congo red) were performed.
Evaluation for systemic, inflammatory autoimmune processes is indicated. Serum autoantibodies (anti‐nuclear antibody [ANA] and extractable nuclear antigens), and a serum and 24‐hour urine protein electrophoresis would be appropriate diagnostic tests. Peritoneal biopsies would have been helpful to assess for serosal diseases.
The patient subsequently developed acute right‐sided abdominal pain requiring urgent evaluation and admission to the hospital. He was initially assessed by a general surgeon, who found no evidence of postoperative complications. His temperature was 36.7C, blood pressure 105/64, heart rate 82, respiratory rate 16, and oxygen saturation 97% on room air. He appeared chronically ill, but he was in no distress and he had a normal mental status. Cardiac exam was normal except for mild jugular venous distension. He had mild bibasilar lung crackles. His abdomen was distended with superficial abdominal tenderness and a fluid wave, but he had normal bowel sounds and no peritoneal signs. He had mild scrotal edema but no peripheral edema. Joint exam did not suggest synovitis and there were no rashes or oral ulcers. Lactate was 0.9 mmol/L (normal 0.62.3), albumin was 2.6 g/dL, and prealbumin was 9 mg/dL (normal 1938). Erythrocyte sedimentation rate and C‐reactive protein were 46 mm/hour (normal 22) and 33.1 mg/L (normal 8), respectively. He had a normocytic anemia and leukopenia. Liver tests and routine chemistries were normal. Serum protein electrophoresis indicated no monoclonal protein. Complete 24‐hour urine collection showed 1.2 g of protein (normal 102 mg). Paracentesis of 3.4 L demonstrated 227 total nucleated cells/L with 2% neutrophils. Following the fluid removal, he had improvement in his pain, which he felt was related to the ascites rather than the recent surgery. Ascites total protein was 3.9 g/dL and ascites albumin was 1.7 g/dL. Ascites culture was negative for infection. Serum Schistosoma immunoglobulin G (IgG) antibody was positive at 3.53 (normal 1.00).
Further history revealed prior episodes of polyarticular joint pain and swelling in his hands and knees 5 years before admission. At that time, he reported a diffuse, pruritic, papular body rash. In addition, he noticed that his fingertips and toes turned white with cold exposure.
Importantly, surgical and infectious complications have been excluded. High protein ascites with a low SAAG of 0.9 suggests an inflammatory source of ascites. The follow‐up clinical data (arthritis, normocytic anemia, leukopenia, rash, Raynaud's phenomenon) suggest a systemic inflammatory syndrome such as SLE, with accompanying serositis. Serologic testing for autoantibodies would be recommended. Peritoneal biopsies, if obtained, may have demonstrated chronic, inflammatory infiltrate (nonspecific) or leukocytoclastic vasculitis (strongly supportive).
ANA enzyme immunoassay was >12 U (normal 1.0 U). Extractable nuclear antigens revealed positive autoantibodies for anti‐SSA, anti‐SSB, and anti‐ribosomal P. Moreover, double‐stranded DNA IgG antibody was 120 IU/mL (normal 30 IU/mL) and C3, C4, and total complement levels were low.
The clinical data support a diagnosis of SLE with serositis. Treatment of the underlying connective tissue disease will typically result in resolution of the ascites; diuretic therapy is generally ineffective.
In consultation with rheumatology and gastroenterology specialists, the diagnosis of SLE was made based on criteria of serositis, persistent leukopenia, arthritis, renal disease (proteinuria), positive ANA, elevated ds‐DNA antibodies, and hypocomplementemia. MR imaging of the abdominal vasculature demonstrated no evidence of vasculitis. The patient was given intravenous methylprednisolone 1 g daily for 3 days followed by high‐dose oral corticosteroids with a gradual taper. He was also started on mycophenolate mofetil as a steroid‐sparing medication (which was later changed to leflunomide due to persistent leukopenia) and hydroxychloroquine. His isolated positive Schistosoma IgG antibody in the absence of other findings was consistent with past exposure or infection. The infectious disease specialist felt there was no evidence of active schistosomiasis, but recommended treatment with a single dose of praziquantel due to the potential benefit with low risk of side effects. The patient had ongoing improvement following dismissal. He had 1 additional paracentesis of 4.1 L, 10 days after his hospitalization, and his ascites and proteinuria resolved. At the 5‐year follow‐up visit, there had been no recurrence of abdominal ascites or abdominal pain. He remains on low‐dose prednisone at 5 mg daily, leflunomide, and hydroxychloroquine.
COMMENTARY
This patient had recurrent ascites with 29.6 L removed over the 4 months prior to admission and an additional 3.4 L during his hospitalization. His outpatient providers initially considered a portal hypertensive etiology of his ascites due to his history of HBV and prior alcohol use. They also appropriately investigated for a possible infectious process. They next directed their evaluation toward the liver biopsy findings, which raised concern for a vascular inflow abnormality. However, the evaluation could have been performed more rapidly and far more cost‐efficiently had a diagnostic paracentesis with calculation of the SAAG been performed early in the evaluation.
The SAAG, which was first described in 1983 by Par and colleagues, is a parameter reflecting the oncotic pressure gradient between the vascular bed and the interstitial splanchnic or ascitic fluid. [1] In the classic study by Runyon and colleagues, a SAAG difference of 1.1 g/dL correctly differentiated causes of ascites due to portal hypertension from those that were not due to portal hypertension 96.7% of the time. [2] Conditions such as nephrotic syndrome, peritoneal carcinomatosis, and serositis (lupus peritonitis) can cause ascites in patients without portal hypertension.
Serositis in the form of pleuritis and/or pericarditis is a common feature of SLE, and ascites has been described in 8% to 11% of SLE patients.[3] However, massive ascites due to lupus peritonitis as a presenting symptom is rare.[4] More common causes of ascites in the setting of SLE include nephrotic syndrome, heart failure, protein‐losing enteropathy, constrictive pericarditis, Budd‐Chiari syndrome, indolent infections such as tuberculosis, and chylous ascites.[5, 6, 7] Of note, lupus peritonitis may be chronic or acute. Chronic ascites develops insidiously with few manifestations of active lupus and may be painless, whereas ascites from acute lupus peritonitis typically develops rapidly and presents with acute abdominal pain and other signs of increased lupus activity.[3, 5, 6, 8, 9]
Ascites from lupus peritonitis may be due to marked serosal exudative accumulation with reduced absorptive capacity in the peritoneum.[3, 4, 10] Other possible causes include peritoneal inflammation from deposition of immune complexes or vasculitis of peritoneal vessels and visceral serous membranes.[4, 9, 11] Although subserosal and submucosal vasculitis have been found in acute ascites, chronic ascites may be related to scarring from vasculitis and serosal inflammation leading to poor venous and lymph drainage.[9] Ascitic fluid characteristics from lupus peritonitis include a SAAG 1.1, presence of white blood cells anywhere in a broad range from 10 to 1630/L, and a range of fluid protein from 3.4 to 4.7 mg/dL.[3] Although not tested in this patient, findings of low complement levels, positive ANA, and elevated anti‐DNA antibody in the ascitic fluid would be supportive of lupus peritonitis, but not specific.[5, 9, 12] Lupus erythematosus cells are occasionally found in the ascitic fluid, but do not rule out other causes of ascites.[9] On retrospective analysis, lupus erythematosus cells were not seen in this patient's pathology specimens.
Treatment of lupus peritonitis and ascites is with high‐dose glucocorticoid therapy, but many patients may need a second immunosuppressant, possibly because of impaired peritoneal circulation from chronic inflammation leading to decreased drug delivery.[13, 14] Chronic ascites may be recalcitrant to systemic glucocorticoids,[3] so a possible alternative therapy is intraperitoneal injection of triamcinolone, which successfully treated massive ascites in a patient who did not respond to oral glucocorticoid treatment.[13] Although ascites may be refractory in some patients, those with chronic lupus peritonitis can generally achieve remission, yet the overall prognosis depends on the presence and severity of multiorgan involvement from SLE. As with any SLE patient, there are also risks of infection from immunosuppression and increased cardiovascular risks.
This patient's evaluation and treatment could have been expedited if he had undergone a paracenteses with determination of the SAAG early in his workup. It is not known why the SAAG was not obtained despite multiple outpatient visits and paracenteses, his history of HBV, and prior alcohol use. This may have been simply an unfortunate oversight. Alternatively, it may have been that his outpatient providers focused on tantalizing clues such as his country of origin, which led to concern for schistosomiasis, and the biopsy findings suggestive of a vascular inflow abnormality that led to further extensive testing. In so doing, the clinicians committed several diagnostic errors, including multiple alternatives bias, anchoring, and confirmation bias.[15] As a result, the patient accrued excess charges of $64,000 from multiple tests, laparoscopic surgery, and 2 hospitalizations. This case highlights how cognitive errors introduce costly variability into patient care, especially when a simple and accurate test is at the beginning of the decision tree.
CLINICAL TEACHING POINTS
- Diagnostic paracentesis, with calculation of the serum‐ascites albumin gradient, should be the first test in the workup for ascites and can distinguish portal hypertensive causes from nonportal hypertensive causes.
- Ascites related to SLE can be acute or chronic and caused by bowel infarction, perforation, pancreatitis, mesenteric vasculitis, nephrotic syndrome, heart failure, protein‐losing enteropathy, constrictive pericarditis, lupus peritonitis, Budd‐Chiari syndrome, or serositis (lupus peritonitis).
- Ascites caused by lupus peritonitis is rare. Once treated, management should be directed toward keeping the SLE in remission.
ACKNOWLEDGMENTS
Disclosure: Nothing to report.
A 40‐year‐old Sudanese man was admitted due to worsening abdominal pain with recurrent ascites. He had a history of hepatitis B (HBV) infection and diabetes. He previously drank 3 beers per day on the weekends, but he had not consumed alcohol in over a year. He was born in Sudan but lived in Egypt most of his adult life; he immigrated to the United States 6 years previously. He was hospitalized out of state 9 months ago for a swollen abdomen and underwent an exploratory laparotomy that reportedly was unremarkable except for ascites.
Portal hypertension due to liver disease is the most common cause of ascites. This patient has a known risk factor for liver disease (history of HBV infection). Although his reported alcohol consumption is low, there is a synergistic effect on liver injury in the setting of chronic hepatitis. Abdominal pain in the setting of ascites needs to be urgently evaluated to exclude spontaneous bacterial peritonitis (SBP). Also, because chronic HBV infection is the major risk factor for hepatocellular carcinoma in the world, malignant ascites is in the differential. Hepatic vascular thrombosis and tuberculous peritonitis (given the patient's country of origin and travel history) also should be considered. The most appropriate initial test would be a diagnostic paracentesis to support or exclude the presence of SBP and direct the evaluation toward liver disease or other less‐common causes of ascites.
The patient was seen as an outpatient 5 months prior to admission with transient fever and joint pains. Laboratory studies at that visit were notable for a serum albumin of 3.2 g/dL (normal 3.55), 2.4 g of predicted 24‐hour protein on urinalysis (normal 30 mg per 24 hours), creatinine of 0.5 mg/dL (normal 0.81.3), and a positive hepatitis B surface antibody. The working diagnosis was a nonspecific viral syndrome and his symptoms resolved without treatment. One month later, he developed ascites and mild lower extremity edema. Additional laboratory studies at that time showed a normocytic anemia with hemoglobin 11.7 g/dL (normal 13.517.5) and leukopenia with white blood cell count of 2.4 109/L (normal 3.510.5), neutrophil count of 1.45 109/L (normal 1.77.0), and lymphocyte count of 0.58 109/L (normal 0.902.90). Transaminases, serum bilirubin, prothrombin time, alpha fetoprotein, and peripheral blood smear were normal. Human immunodeficiency virus antibody screen and QuantiFERON‐TB assay were negative. Hemoglobin A1c was 6.2% (normal 4.06.0). Repeat urinalysis demonstrated 883 mg of predicted 24‐hour protein. Computed tomography (CT) of the abdomen showed a large amount of intra‐abdominal ascites; the liver and spleen were normal, and there were no varices or other evidence of portal hypertension. Echocardiogram was normal except for a small inferior vena cava (IVC) and a mildly increased right ventricular systolic pressure of 32 mm Hg (systolic blood pressure 98 mm Hg). Due to the indeterminate cause for the patient's ascites, referral was made for gastroenterology evaluation with consideration for a paracentesis.
Cirrhotic ascites seems less likely. Postsinusoidal causes of portal hypertension (eg, cardiomyopathy) are also less likely given the absence of suggestive findings on echocardiography. Malignant ascites also appears less probable in the absence of suggestive findings such as mass lesions, lymphadenopathy, or peritoneal carcinomatosis on CT imaging. The suspicion for tuberculous peritonitis is lower with the negative QuantiFERON‐TB test. Hypoalbuminemia, normocytic anemia, leukopenia, and proteinuria all suggest a systemic inflammatory condition (eg, systemic lupus erythematosus [SLE]) with inflammatory serositis causing ascites). Nephrotic syndrome can cause hypoalbuminemia, edema, and ascites, but his total urine protein losses of 3.5 grams per 24 hours are not in keeping with this diagnosis. Other uncommon causes of ascites such as chylous ascites have not yet been excluded. The most appropriate next step remains ascitic fluid analysis.
A paracentesis yielded 7.8 L of clear‐yellow fluid and improvement in his abdominal discomfort. Analysis showed 224 total nucleated cells/L with 2% neutrophils, 57% lymphocytes, and 37% monocytes. Ascites total protein was 3.8 g/dL and glucose was 55 mg/dL. Gram stain and culture were negative, and cytology was negative for malignancy but showed lymphocytes, plasma cells, monocytes, and reactive mesothelial cells interpreted as consistent with chronic inflammation. The serum‐ascites albumin gradient (SAAG) was not obtained.
With a low leukocyte count and a paucity of neutrophils, this is not SBP. The ascites fluid did not have a chylous appearance. The SAAG, which can distinguish between portal hypertensive and nonportal hypertensive causes for ascites using a cutoff of 1.1 g/dL, was not done. The total protein was high, arguing against cirrhosis. High protein ascites with a high SAAG would suggest a posthepatic source of portal hypertension (eg, Budd‐Chiari syndrome, constrictive pericarditis). High protein ascites with a low SAAG would suggest an inflammatory or malignant source of ascites. The relative lymphocytosis in the ascites fluid suggests an inflammatory process, but is a nonspecific finding. The negative cytology does not completely exclude a malignancy, but given the absence of findings on the CT, malignant ascites is less likely.
Three months before admission, the patient underwent a repeat large‐volume paracentesis and a liver biopsy. The biopsy showed ectopic portal vein branches consistent with hepatoportal sclerosis, but no actual sclerosis was identified. The pathologist concluded that the findings suggested noncirrhotic portal hypertension due to a vascular in‐flow abnormality. Abdominal ultrasound with Doppler was unremarkable other than slightly increased echogenicity of the liver. Magnetic resonance (MR) angiogram showed narrowing of the intra‐abdominal IVC at the level of the diaphragm. Because of concern that hepatic congestion from high pressures in the narrowed IVC was leading to poor vascular inflow as suggested by the biopsy findings, an inferior vena cavagram was performed. This study was normal, although no transhepatic pressure measurements were obtained. Three stool specimens and 2 urine specimens were negative for parasites. The patient required repeat large‐volume paracenteses monthly. SBP was again ruled out, but no other diagnostic labs were obtained. He had anorexia with poor oral intake each time his abdomen became distended.
The patient was started on furosemide 1 month prior to admission to the hospital but had only a slight improvement in the ascites. His other medications included insulin, tamsulosin, and hydrocodone‐acetaminophen. Five days prior to admission, he underwent a diagnostic laparoscopy, which showed only ascites and small adhesions to the anterior abdominal wall. There was no visual evidence of malignancy, and the surgeon commented that the liver was normal. No additional biopsies were obtained.
The liver biopsy findings could be seen in noncirrhotic portal hypertension, although this diagnosis would be unlikely without splenomegaly, varices, or other signs of portal hypertension. However, 2 possible etiologies for noncirrhotic portal hypertension in this patient would be hepatic congestion from the narrowed IVC (although the normal IVC study argues against this) and hepatic schistosomiasis. Schistosomiasis is an important cause of noncirrhotic portal hypertension in endemic areas like this patient's country of origin, but the negative stool and urine studies, combined with the lack of granulomas or fibrosis seen on biopsy, make this condition unlikely.
Systemic amyloidosis (primary or secondary) could also be a cause of ascites and could present with multiorgan involvement (diarrhea and nephrotic syndrome). Amyloid deposits would have probably been seen in the liver biopsy, if present, but may not have been apparent unless specific stains (Congo red) were performed.
Evaluation for systemic, inflammatory autoimmune processes is indicated. Serum autoantibodies (anti‐nuclear antibody [ANA] and extractable nuclear antigens), and a serum and 24‐hour urine protein electrophoresis would be appropriate diagnostic tests. Peritoneal biopsies would have been helpful to assess for serosal diseases.
The patient subsequently developed acute right‐sided abdominal pain requiring urgent evaluation and admission to the hospital. He was initially assessed by a general surgeon, who found no evidence of postoperative complications. His temperature was 36.7C, blood pressure 105/64, heart rate 82, respiratory rate 16, and oxygen saturation 97% on room air. He appeared chronically ill, but he was in no distress and he had a normal mental status. Cardiac exam was normal except for mild jugular venous distension. He had mild bibasilar lung crackles. His abdomen was distended with superficial abdominal tenderness and a fluid wave, but he had normal bowel sounds and no peritoneal signs. He had mild scrotal edema but no peripheral edema. Joint exam did not suggest synovitis and there were no rashes or oral ulcers. Lactate was 0.9 mmol/L (normal 0.62.3), albumin was 2.6 g/dL, and prealbumin was 9 mg/dL (normal 1938). Erythrocyte sedimentation rate and C‐reactive protein were 46 mm/hour (normal 22) and 33.1 mg/L (normal 8), respectively. He had a normocytic anemia and leukopenia. Liver tests and routine chemistries were normal. Serum protein electrophoresis indicated no monoclonal protein. Complete 24‐hour urine collection showed 1.2 g of protein (normal 102 mg). Paracentesis of 3.4 L demonstrated 227 total nucleated cells/L with 2% neutrophils. Following the fluid removal, he had improvement in his pain, which he felt was related to the ascites rather than the recent surgery. Ascites total protein was 3.9 g/dL and ascites albumin was 1.7 g/dL. Ascites culture was negative for infection. Serum Schistosoma immunoglobulin G (IgG) antibody was positive at 3.53 (normal 1.00).
Further history revealed prior episodes of polyarticular joint pain and swelling in his hands and knees 5 years before admission. At that time, he reported a diffuse, pruritic, papular body rash. In addition, he noticed that his fingertips and toes turned white with cold exposure.
Importantly, surgical and infectious complications have been excluded. High protein ascites with a low SAAG of 0.9 suggests an inflammatory source of ascites. The follow‐up clinical data (arthritis, normocytic anemia, leukopenia, rash, Raynaud's phenomenon) suggest a systemic inflammatory syndrome such as SLE, with accompanying serositis. Serologic testing for autoantibodies would be recommended. Peritoneal biopsies, if obtained, may have demonstrated chronic, inflammatory infiltrate (nonspecific) or leukocytoclastic vasculitis (strongly supportive).
ANA enzyme immunoassay was >12 U (normal 1.0 U). Extractable nuclear antigens revealed positive autoantibodies for anti‐SSA, anti‐SSB, and anti‐ribosomal P. Moreover, double‐stranded DNA IgG antibody was 120 IU/mL (normal 30 IU/mL) and C3, C4, and total complement levels were low.
The clinical data support a diagnosis of SLE with serositis. Treatment of the underlying connective tissue disease will typically result in resolution of the ascites; diuretic therapy is generally ineffective.
In consultation with rheumatology and gastroenterology specialists, the diagnosis of SLE was made based on criteria of serositis, persistent leukopenia, arthritis, renal disease (proteinuria), positive ANA, elevated ds‐DNA antibodies, and hypocomplementemia. MR imaging of the abdominal vasculature demonstrated no evidence of vasculitis. The patient was given intravenous methylprednisolone 1 g daily for 3 days followed by high‐dose oral corticosteroids with a gradual taper. He was also started on mycophenolate mofetil as a steroid‐sparing medication (which was later changed to leflunomide due to persistent leukopenia) and hydroxychloroquine. His isolated positive Schistosoma IgG antibody in the absence of other findings was consistent with past exposure or infection. The infectious disease specialist felt there was no evidence of active schistosomiasis, but recommended treatment with a single dose of praziquantel due to the potential benefit with low risk of side effects. The patient had ongoing improvement following dismissal. He had 1 additional paracentesis of 4.1 L, 10 days after his hospitalization, and his ascites and proteinuria resolved. At the 5‐year follow‐up visit, there had been no recurrence of abdominal ascites or abdominal pain. He remains on low‐dose prednisone at 5 mg daily, leflunomide, and hydroxychloroquine.
COMMENTARY
This patient had recurrent ascites with 29.6 L removed over the 4 months prior to admission and an additional 3.4 L during his hospitalization. His outpatient providers initially considered a portal hypertensive etiology of his ascites due to his history of HBV and prior alcohol use. They also appropriately investigated for a possible infectious process. They next directed their evaluation toward the liver biopsy findings, which raised concern for a vascular inflow abnormality. However, the evaluation could have been performed more rapidly and far more cost‐efficiently had a diagnostic paracentesis with calculation of the SAAG been performed early in the evaluation.
The SAAG, which was first described in 1983 by Par and colleagues, is a parameter reflecting the oncotic pressure gradient between the vascular bed and the interstitial splanchnic or ascitic fluid. [1] In the classic study by Runyon and colleagues, a SAAG difference of 1.1 g/dL correctly differentiated causes of ascites due to portal hypertension from those that were not due to portal hypertension 96.7% of the time. [2] Conditions such as nephrotic syndrome, peritoneal carcinomatosis, and serositis (lupus peritonitis) can cause ascites in patients without portal hypertension.
Serositis in the form of pleuritis and/or pericarditis is a common feature of SLE, and ascites has been described in 8% to 11% of SLE patients.[3] However, massive ascites due to lupus peritonitis as a presenting symptom is rare.[4] More common causes of ascites in the setting of SLE include nephrotic syndrome, heart failure, protein‐losing enteropathy, constrictive pericarditis, Budd‐Chiari syndrome, indolent infections such as tuberculosis, and chylous ascites.[5, 6, 7] Of note, lupus peritonitis may be chronic or acute. Chronic ascites develops insidiously with few manifestations of active lupus and may be painless, whereas ascites from acute lupus peritonitis typically develops rapidly and presents with acute abdominal pain and other signs of increased lupus activity.[3, 5, 6, 8, 9]
Ascites from lupus peritonitis may be due to marked serosal exudative accumulation with reduced absorptive capacity in the peritoneum.[3, 4, 10] Other possible causes include peritoneal inflammation from deposition of immune complexes or vasculitis of peritoneal vessels and visceral serous membranes.[4, 9, 11] Although subserosal and submucosal vasculitis have been found in acute ascites, chronic ascites may be related to scarring from vasculitis and serosal inflammation leading to poor venous and lymph drainage.[9] Ascitic fluid characteristics from lupus peritonitis include a SAAG 1.1, presence of white blood cells anywhere in a broad range from 10 to 1630/L, and a range of fluid protein from 3.4 to 4.7 mg/dL.[3] Although not tested in this patient, findings of low complement levels, positive ANA, and elevated anti‐DNA antibody in the ascitic fluid would be supportive of lupus peritonitis, but not specific.[5, 9, 12] Lupus erythematosus cells are occasionally found in the ascitic fluid, but do not rule out other causes of ascites.[9] On retrospective analysis, lupus erythematosus cells were not seen in this patient's pathology specimens.
Treatment of lupus peritonitis and ascites is with high‐dose glucocorticoid therapy, but many patients may need a second immunosuppressant, possibly because of impaired peritoneal circulation from chronic inflammation leading to decreased drug delivery.[13, 14] Chronic ascites may be recalcitrant to systemic glucocorticoids,[3] so a possible alternative therapy is intraperitoneal injection of triamcinolone, which successfully treated massive ascites in a patient who did not respond to oral glucocorticoid treatment.[13] Although ascites may be refractory in some patients, those with chronic lupus peritonitis can generally achieve remission, yet the overall prognosis depends on the presence and severity of multiorgan involvement from SLE. As with any SLE patient, there are also risks of infection from immunosuppression and increased cardiovascular risks.
This patient's evaluation and treatment could have been expedited if he had undergone a paracenteses with determination of the SAAG early in his workup. It is not known why the SAAG was not obtained despite multiple outpatient visits and paracenteses, his history of HBV, and prior alcohol use. This may have been simply an unfortunate oversight. Alternatively, it may have been that his outpatient providers focused on tantalizing clues such as his country of origin, which led to concern for schistosomiasis, and the biopsy findings suggestive of a vascular inflow abnormality that led to further extensive testing. In so doing, the clinicians committed several diagnostic errors, including multiple alternatives bias, anchoring, and confirmation bias.[15] As a result, the patient accrued excess charges of $64,000 from multiple tests, laparoscopic surgery, and 2 hospitalizations. This case highlights how cognitive errors introduce costly variability into patient care, especially when a simple and accurate test is at the beginning of the decision tree.
CLINICAL TEACHING POINTS
- Diagnostic paracentesis, with calculation of the serum‐ascites albumin gradient, should be the first test in the workup for ascites and can distinguish portal hypertensive causes from nonportal hypertensive causes.
- Ascites related to SLE can be acute or chronic and caused by bowel infarction, perforation, pancreatitis, mesenteric vasculitis, nephrotic syndrome, heart failure, protein‐losing enteropathy, constrictive pericarditis, lupus peritonitis, Budd‐Chiari syndrome, or serositis (lupus peritonitis).
- Ascites caused by lupus peritonitis is rare. Once treated, management should be directed toward keeping the SLE in remission.
ACKNOWLEDGMENTS
Disclosure: Nothing to report.
- , , . Serum‐ascites albumin concentration gradient: a physiologic approach to the differential diagnosis of ascites. Gastroenterology. 1983;85(2):240–244.
- , , , et al. The serum‐ascites albumin gradient is superior to the exudate‐transudate concept in the differential diagnosis of ascites. Ann Intern Med. 1992;117:215–220.
- , , . Insidious onset of massive painless ascites as initial manifestation of systemic lupus erythematosus. Lupus. 2011;20:754–757.
- , . Rapid onset of massive ascites as the initial presentation of systemic lupus erythematosus. Am J Gastroenterol. 2000;95:302–303.
- , . Gastrointestinal and hepatic manifestations of systemic lupus erythematosus. J Clin Gastroenterol. 2011;45:436–441.
- , , , . Massive ascites as a presenting feature of lupus. Int J Rheum Dis. 2012;15:e15–e16.
- , , , et al. Concurrent occurrence of chylothorax, chylous ascites, and protein‐losing enteropathy in systemic lupus erythematosus. J Rheumatol. 2002;29:1330–1333.
- , , , et al. Abdominal manifestations in childhood‐onset systemic lupus erythematosus. Ann Rheum Dis. 2007;66:174–178.
- , , . Chronic lupus peritonitis with ascites: review of the literature with a case report. Semin Arthritis Rheum. 1988;18:121–126.
- , . Nonhepatic Gastrointestinal Manifestations of Systemic Lupus Erythematosus. London, United Kingdom: Churchill Livingstone; 1987:747–760.
- , , , . Ascites due to lupus peritonitis: a rare form of onset of systemic lupus erythematosus. Rev Bras Reumatol. 2012;52(1):113–119.
- , , , . New‐onset lupus presenting as serositis in an 80‐year‐old woman: does a high‐titer ANA in pleural, pericardial, or peritoneal fluid help confirm the diagnosis? J Clin Rheum.2005:11(5):292–293.
- , , , . Successful treatment of massive ascites with intraperitoneal administration of a steroid in a case of systemic lupus erythematosus. Lupus. 2009;18:740–742.
- , , , et al. Chronic lupus peritonitis with massive ascites at elderly onset: case report and review of the literature. Intern Med. 2002;41:1056–1061.
- . The Importance of cognitive errors in diagnosis and strategies to minimize them. Acad Med. 2003;78:775–780.
- , , . Serum‐ascites albumin concentration gradient: a physiologic approach to the differential diagnosis of ascites. Gastroenterology. 1983;85(2):240–244.
- , , , et al. The serum‐ascites albumin gradient is superior to the exudate‐transudate concept in the differential diagnosis of ascites. Ann Intern Med. 1992;117:215–220.
- , , . Insidious onset of massive painless ascites as initial manifestation of systemic lupus erythematosus. Lupus. 2011;20:754–757.
- , . Rapid onset of massive ascites as the initial presentation of systemic lupus erythematosus. Am J Gastroenterol. 2000;95:302–303.
- , . Gastrointestinal and hepatic manifestations of systemic lupus erythematosus. J Clin Gastroenterol. 2011;45:436–441.
- , , , . Massive ascites as a presenting feature of lupus. Int J Rheum Dis. 2012;15:e15–e16.
- , , , et al. Concurrent occurrence of chylothorax, chylous ascites, and protein‐losing enteropathy in systemic lupus erythematosus. J Rheumatol. 2002;29:1330–1333.
- , , , et al. Abdominal manifestations in childhood‐onset systemic lupus erythematosus. Ann Rheum Dis. 2007;66:174–178.
- , , . Chronic lupus peritonitis with ascites: review of the literature with a case report. Semin Arthritis Rheum. 1988;18:121–126.
- , . Nonhepatic Gastrointestinal Manifestations of Systemic Lupus Erythematosus. London, United Kingdom: Churchill Livingstone; 1987:747–760.
- , , , . Ascites due to lupus peritonitis: a rare form of onset of systemic lupus erythematosus. Rev Bras Reumatol. 2012;52(1):113–119.
- , , , . New‐onset lupus presenting as serositis in an 80‐year‐old woman: does a high‐titer ANA in pleural, pericardial, or peritoneal fluid help confirm the diagnosis? J Clin Rheum.2005:11(5):292–293.
- , , , . Successful treatment of massive ascites with intraperitoneal administration of a steroid in a case of systemic lupus erythematosus. Lupus. 2009;18:740–742.
- , , , et al. Chronic lupus peritonitis with massive ascites at elderly onset: case report and review of the literature. Intern Med. 2002;41:1056–1061.
- . The Importance of cognitive errors in diagnosis and strategies to minimize them. Acad Med. 2003;78:775–780.
Should Patients with an Unprovoked VTE Be Screened for Malignancy or a Hypercoagulable State?

Case
A 56-year-old woman with hypertension and diabetes presents to the hospital with acute onset of painful swelling in her right calf. She has had no recent surgeries, trauma, or travel, and takes lisinopril and metformin. An ultrasound of her right lower extremity demonstrates a venous thromboembolism (VTE). The patient’s last mammogram was three years ago, and she’s never undergone a screening colonoscopy. On lab workup, she is noted to have a microcytic anemia.
Should this patient be screened for an underlying hypercoagulable state or malignancy?
Background
An estimated 550,000 hospitalized adults are diagnosed with VTE each year.1 VTE can occur in the absence of known precipitants (unprovoked) or can be temporally associated with a known major risk factor (provoked). This practical division has implications for both treatment duration and risk of recurrence. A VTE is considered provoked if it occurs in the setting of surgery, leg trauma, fracture, pregnancy within the previous three months, estrogen therapy, immobility from an acute illness for more than one week, travel lasting more than six hours, or active malignancy.2 If none of these provoking factors is present, the VTE is considered unprovoked.2
Nearly 20% of first-time VTE events can be attributed to malignancy.3 Additionally, patients presenting with an unprovoked VTE possess a higher risk of being diagnosed with a cancer, raising the question of whether unprovoked VTEs should compel aggressive malignancy screening.4
Before the discovery of antithrombin deficiency in 1965, most unprovoked VTE events remained unexplained. Since then, numerous inherited coagulation abnormalities have been identified. It is now estimated that coagulation abnormalities can be found in up to half of patients with unprovoked thrombi.5
The increase in availability of molecular and genetic assays for hypercoagulability has been accompanied by a dramatic rise in the rate of testing for these disorders.6 Despite increased testing available for inherited thrombophilias, disagreement exists over the utility of this workup.6
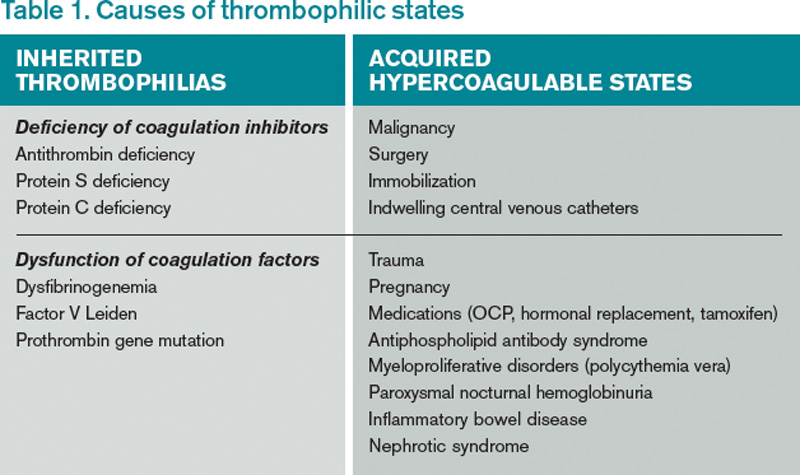
Review of the Data
Hypercoagulability leading to venous thrombosis can be broadly divided into two groups: acquired and hereditary (see Table 1). First, let’s examine acquired hypercoagulable states.
Malignancy: Armand Trousseau first suggested an association between thrombotic events and malignancy in 1865. Malignancy causes a hypercoagulable state; additionally, tumors can cause thromboemboli by other mechanisms, such as vascular invasion or external compression of vasculature.7
Multiple studies demonstrate that malignancy increases the chance of developing a VTE. A Danish cohort study of nearly 60,000 cancer patients compared with over 280,000 controls over nine years offered twice the incidence of VTE in patients with cancer.8 Other studies reveal that VTE rates peak in the first year after a cancer diagnosis; moreover, VTE events are associated with more advanced disease and worse prognosis.9 Approximately 11% of cancer patients will develop a clinically evident VTE during the course of their disease.10,11
The majority of cancers associated with VTE events are clinically evident; however, some patients with thrombi have an occult malignancy. During the two years following an unprovoked VTE, the rate of discovering a previously undiagnosed malignancy was three times higher when compared with provoked VTE.6
This potential to diagnose occult malignancy in patients with idiopathic thromboembolic events stimulates debate around the usefulness of extensive cancer screening for these patients. One large systematic review compared routine and extensive cancer screening strategies following an unprovoked VTE. An extensive screening strategy consisting of CT scans of the abdomen and pelvis significantly increased the proportion of previously undiagnosed cancers; however, the authors did not determine complication rates, cost effectiveness, or difference in morbidity and mortality associated with extensive screening strategies.7
Other studies have demonstrated that extensive screening with CT, endoscopy, and tumor markers finds more previously undetected cancers; however, up to half of these malignancies could have been identified without resorting to such expensive and invasive workups.12 Additionally, no prospective data demonstrate improved outcomes or increased survival from these diagnoses. Likewise, no cost-effectiveness data exist to support this expensive and aggressive screening approach.7

All patients with an idiopathic VTE should undergo a complete history and physical examination with attention to common areas of malignancy. Patients should have basic lab work and be recommended for age-appropriate cancer screening (see Table 2). Any abnormalities uncovered on this initial workup should be aggressively investigated.13 If overt cancer is detected, then low molecular weight heparin would be preferred over oral anticoagulation as treatment for the VTE.14 Extensive malignancy evaluation in all patients with unprovoked VTE is not warranted, however, given the lack of data regarding efficacy of extensive screening, the potential for increased harms, and the costs associated with this approach.
Antiphospholipid syndrome: Antiphospholipid syndrome is the most common acquired cause of thrombophilia.15 Characterized by the presence of antiphospholipid antibodies (e.g. lupus anticoagulant antibodies or anticardiolipin antibodies), this syndrome is usually secondary to cancer or an autoimmune disease.
Antiphospholipid antibody syndrome is a thrombophilic disorder in which both venous and arterial thrombosis may occur. Patients with this disorder are considered at high risk for thrombotic events. Data suggest that antiphospholipid antibody syndrome also increases the risk of VTE recurrence. In one retrospective study, cessation of warfarin therapy in patients with antiphospholipid antibodies after a VTE resulted in 69% of patients having recurrent thrombosis in the first year.16 Given this substantial risk, antiphospholipid antibody testing is recommended in those with a suggestive history, including patients with 1) recurrent fetal loss, 2) fetal loss after 10 weeks, or 3) known collagen vascular disease.16 Lifelong anticoagulation is recommended for these patients.
Inherited hypercoagulable states: The most frequent causes of an inherited hypercoagulable state are the factor V Leiden mutation and the prothrombin gene mutation, accounting for 50% to 60% of hereditary thrombophilias. Protein S, protein C, and antithrombin defects account for most of the remaining cases of inherited thrombophilias.15
Currently, there is no consensus regarding who should be tested for inherited thrombophilia. Testing for an inherited thrombophilia would be indicated if the results added prognostic information or changed management. Arguments against testing hinge on the fact that neither prognosis nor management is affected by the presence of an inherited thrombophilia.
The presence of a thrombophilia also does not change the method or intensity of anticoagulation.17 The risk of recurrence after discontinuing anticoagulation therapy is not affected.17,18 The strongest predictor of VTE recurrence is the unprovoked VTE itself, regardless of an underlying thrombophilia.15 Recurrent VTE is nearly twice as frequent in patients with idiopathic VTE compared to those with provoked VTE.15

The American College of Chest Physicians (ACCP) recommends treating a provoked VTE for three months.19 According to the same guidelines, an unprovoked VTE should be treated for a minimum of three months, and lifelong anticoagulation should be considered.19
Overall, the rate of recurrence after a first VTE is considerable after completion of anticoagulation, especially for an unprovoked thrombotic event. Studies show a 7%-15% recurrence rate during the two years following the index VTE (see Table 3).17,20,21 Currently, no data suggest that a hereditary thrombophilia substantially changes this baseline high recurrent risk. ACCP recommendations state that the presence of hereditary thrombophilia should not be used as a major factor to guide duration of anticoagulation.19
Back to the Case
Our patient presented with an unprovoked VTE. She should be started on anticoagulation therapy with low molecular weight heparin and transitioned to oral anticoagulation.
Her highest risk for VTE recurrence is the unprovoked VTE itself, regardless of an underlying thrombophilia. Since the presence of an inherited thrombophilia will not change duration or intensity of management, our patient should not be tested.
There are no prospective trials showing improved outcomes from aggressive workup for occult malignancy. Given this information, an extensive workup for occult malignancy should not be undertaken; however, this patient has an idiopathic VTE and should undergo a complete history, physical examination, and basic lab work, with attention to common areas of malignancy. Any abnormalities uncovered on this initial workup should be investigated more aggressively. Screening with mammography and Pap smear should be arranged in outpatient follow-up and communicated to the primary care physician, because she is not up to date with these age-appropriate screening tests.
Based on new evidence, a low-dose chest CT would be a consideration if she had a smoking history of at least 30 pack-years.22 Her microcytic anemia uncovered on routine lab work should be investigated further for a possible underlying gastrointestinal malignancy.
Bottom Line
An initial diagnosis of unprovoked VTE remains the strongest risk factor for recurrent thromboembolic events. The presence of an inherited thrombophilia does not significantly alter management. Aggressive workup for occult malignancy has not prospectively improved outcomes, but age-appropriate malignancy screening should be recommended.
Drs. Czernik and Anderson are hospitalists and instructors of medicine at the University of Colorado Denver (UCD). Dr. Wolfe is a hospitalist and assistant professor of medicine at UCD. Dr. Cumbler is a hospitalist and associate professor of medicine at UCD.
References
- Centers for Disease Control and Prevention. Venous thromboembolism in adult hospitalizations—United States, 2007–2009. MMWR Morb Mortal Wkly Rep. 2012;61(22);401-404.
- Baglin T, Gray E, Greaves M, et al. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol. 2010;149(2):209-220.
- Heit, JA, O’Fallon WM, Petterson TM, et al. Relative impact of risk factors for deep vein thrombosis and pulmonary embolism: a population-based study. Arch Intern Med. 2002;162(11):1245-1248.
- Iodice S, Gandini S, Löhr M, Lowenfels AB, Maisonneuve P. Venous thromboembolic events and organ-specific occult cancers: a review and meta-analysis. J Thromb Haemost. 2008;6(5):781-788.
- Coppens M, Reijnders JH, Middeldorp S, Doggen CJ, Rosendaal FR. Testing for inherited thrombophilia does not reduce the recurrence of venous thrombosis. J Thromb Haemost. 2008;6(9):1474-1477.
- Coppens M, van Mourik JA, Eckmann CM, Büller HR, Middeldorp S. Current practise of testing for inherited thrombophilia. J Thromb Haemost. 2007;5(9):1979-1981.
- Carrier M, Le Gal G, Wells PS, Fergusson D, Ramsay T, Rodger MA. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med. 2008;149(5):323-333.
- Cronin-Fenton DP, Søndergaard F, Pedersen LA, et al. Hospitalisation for venous thromboembolism in cancer patients and the general population: a population-based cohort study in Denmark, 1997-2006. Br J Cancer. 2010;103(7):947-953.
- Chew HK, Wun T, Harvey D, Zhou H, White RH. Incidence of venous thromboembolism and its effect on survival among patients with common cancers. Arch Intern Med. 2006;166(4):458-464.
- Lee JL, Lee JH, Kim MK, et al. A case of bone marrow necrosis with thrombotic thrombocytopenic purpura as a manifestation of occult colon cancer. Jpn J Clin Oncol. 2004;34(8):476-480.
- Sack GH Jr, Levin J, Bell WR. Trousseau’s syndrome and other manifestations of chronic disseminated coagulopathy in patients with neoplasms: clinical, pathophysiologic, and therapeutic features. Medicine (Baltimore). 1977;56(1):1-37.
- Prins MH, Hettiarachchi RJ, Lensing AW, Hirsh J. Newly diagnosed malignancy in patients with venous thromboembolism. Search or wait and see? Thromb Haemost. 1997;78(1):121-125.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med. 1996;125(10):785-793.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med. 2003;349(2):146-153.
- Dalen JE. Should patients with venous thromboembolism be screened for thrombophilia? Am J Med. 2008;121(6):458-463.
- Khamashta MA, Cuadrado MJ, Mujic F, Taub NA, Hunt BJ, Hughes GR. The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med. 1995;332:993-997.
- Ridker PM, Goldhaber SZ, Danielson E, et al. Long-term, low-intensity warfarin therapy for the prevention of recurrent venous thromboembolism. N Engl J Med. 2003;348(15):1425-1434.
- Hron G, Eichinger S, Weltermann A, et al. Family history for venous thromboembolism and the risk for recurrence. Am J Med. 2006;119(1):50-53.
- Kearon C, Akl EA, Comerota AJ, et al. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e419S-e494S.
- Douketis, James, Tosetto A, Marcucci M, et al. Risk of recurrence after venous thromboembolism in men and women: patient level meta-analysis. BMJ. 2011;342:d813.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA. 2005;293(19):2352-2361.
- American Cancer Society Guidelines for the Early Detection of Cancer. Available at: http://www.cancer.org/healthy/findcancerearly/cancerscreeningguidelines/american-cancer-society-guidelines-for-the-early-detection-of-cancer. Accessed November 15, 2014.
Case
A 56-year-old woman with hypertension and diabetes presents to the hospital with acute onset of painful swelling in her right calf. She has had no recent surgeries, trauma, or travel, and takes lisinopril and metformin. An ultrasound of her right lower extremity demonstrates a venous thromboembolism (VTE). The patient’s last mammogram was three years ago, and she’s never undergone a screening colonoscopy. On lab workup, she is noted to have a microcytic anemia.
Should this patient be screened for an underlying hypercoagulable state or malignancy?
Background
An estimated 550,000 hospitalized adults are diagnosed with VTE each year.1 VTE can occur in the absence of known precipitants (unprovoked) or can be temporally associated with a known major risk factor (provoked). This practical division has implications for both treatment duration and risk of recurrence. A VTE is considered provoked if it occurs in the setting of surgery, leg trauma, fracture, pregnancy within the previous three months, estrogen therapy, immobility from an acute illness for more than one week, travel lasting more than six hours, or active malignancy.2 If none of these provoking factors is present, the VTE is considered unprovoked.2
Nearly 20% of first-time VTE events can be attributed to malignancy.3 Additionally, patients presenting with an unprovoked VTE possess a higher risk of being diagnosed with a cancer, raising the question of whether unprovoked VTEs should compel aggressive malignancy screening.4
Before the discovery of antithrombin deficiency in 1965, most unprovoked VTE events remained unexplained. Since then, numerous inherited coagulation abnormalities have been identified. It is now estimated that coagulation abnormalities can be found in up to half of patients with unprovoked thrombi.5
The increase in availability of molecular and genetic assays for hypercoagulability has been accompanied by a dramatic rise in the rate of testing for these disorders.6 Despite increased testing available for inherited thrombophilias, disagreement exists over the utility of this workup.6
Review of the Data
Hypercoagulability leading to venous thrombosis can be broadly divided into two groups: acquired and hereditary (see Table 1). First, let’s examine acquired hypercoagulable states.
Malignancy: Armand Trousseau first suggested an association between thrombotic events and malignancy in 1865. Malignancy causes a hypercoagulable state; additionally, tumors can cause thromboemboli by other mechanisms, such as vascular invasion or external compression of vasculature.7
Multiple studies demonstrate that malignancy increases the chance of developing a VTE. A Danish cohort study of nearly 60,000 cancer patients compared with over 280,000 controls over nine years offered twice the incidence of VTE in patients with cancer.8 Other studies reveal that VTE rates peak in the first year after a cancer diagnosis; moreover, VTE events are associated with more advanced disease and worse prognosis.9 Approximately 11% of cancer patients will develop a clinically evident VTE during the course of their disease.10,11
The majority of cancers associated with VTE events are clinically evident; however, some patients with thrombi have an occult malignancy. During the two years following an unprovoked VTE, the rate of discovering a previously undiagnosed malignancy was three times higher when compared with provoked VTE.6
This potential to diagnose occult malignancy in patients with idiopathic thromboembolic events stimulates debate around the usefulness of extensive cancer screening for these patients. One large systematic review compared routine and extensive cancer screening strategies following an unprovoked VTE. An extensive screening strategy consisting of CT scans of the abdomen and pelvis significantly increased the proportion of previously undiagnosed cancers; however, the authors did not determine complication rates, cost effectiveness, or difference in morbidity and mortality associated with extensive screening strategies.7
Other studies have demonstrated that extensive screening with CT, endoscopy, and tumor markers finds more previously undetected cancers; however, up to half of these malignancies could have been identified without resorting to such expensive and invasive workups.12 Additionally, no prospective data demonstrate improved outcomes or increased survival from these diagnoses. Likewise, no cost-effectiveness data exist to support this expensive and aggressive screening approach.7
All patients with an idiopathic VTE should undergo a complete history and physical examination with attention to common areas of malignancy. Patients should have basic lab work and be recommended for age-appropriate cancer screening (see Table 2). Any abnormalities uncovered on this initial workup should be aggressively investigated.13 If overt cancer is detected, then low molecular weight heparin would be preferred over oral anticoagulation as treatment for the VTE.14 Extensive malignancy evaluation in all patients with unprovoked VTE is not warranted, however, given the lack of data regarding efficacy of extensive screening, the potential for increased harms, and the costs associated with this approach.
Antiphospholipid syndrome: Antiphospholipid syndrome is the most common acquired cause of thrombophilia.15 Characterized by the presence of antiphospholipid antibodies (e.g. lupus anticoagulant antibodies or anticardiolipin antibodies), this syndrome is usually secondary to cancer or an autoimmune disease.
Antiphospholipid antibody syndrome is a thrombophilic disorder in which both venous and arterial thrombosis may occur. Patients with this disorder are considered at high risk for thrombotic events. Data suggest that antiphospholipid antibody syndrome also increases the risk of VTE recurrence. In one retrospective study, cessation of warfarin therapy in patients with antiphospholipid antibodies after a VTE resulted in 69% of patients having recurrent thrombosis in the first year.16 Given this substantial risk, antiphospholipid antibody testing is recommended in those with a suggestive history, including patients with 1) recurrent fetal loss, 2) fetal loss after 10 weeks, or 3) known collagen vascular disease.16 Lifelong anticoagulation is recommended for these patients.
Inherited hypercoagulable states: The most frequent causes of an inherited hypercoagulable state are the factor V Leiden mutation and the prothrombin gene mutation, accounting for 50% to 60% of hereditary thrombophilias. Protein S, protein C, and antithrombin defects account for most of the remaining cases of inherited thrombophilias.15
Currently, there is no consensus regarding who should be tested for inherited thrombophilia. Testing for an inherited thrombophilia would be indicated if the results added prognostic information or changed management. Arguments against testing hinge on the fact that neither prognosis nor management is affected by the presence of an inherited thrombophilia.
The presence of a thrombophilia also does not change the method or intensity of anticoagulation.17 The risk of recurrence after discontinuing anticoagulation therapy is not affected.17,18 The strongest predictor of VTE recurrence is the unprovoked VTE itself, regardless of an underlying thrombophilia.15 Recurrent VTE is nearly twice as frequent in patients with idiopathic VTE compared to those with provoked VTE.15
The American College of Chest Physicians (ACCP) recommends treating a provoked VTE for three months.19 According to the same guidelines, an unprovoked VTE should be treated for a minimum of three months, and lifelong anticoagulation should be considered.19
Overall, the rate of recurrence after a first VTE is considerable after completion of anticoagulation, especially for an unprovoked thrombotic event. Studies show a 7%-15% recurrence rate during the two years following the index VTE (see Table 3).17,20,21 Currently, no data suggest that a hereditary thrombophilia substantially changes this baseline high recurrent risk. ACCP recommendations state that the presence of hereditary thrombophilia should not be used as a major factor to guide duration of anticoagulation.19
Back to the Case
Our patient presented with an unprovoked VTE. She should be started on anticoagulation therapy with low molecular weight heparin and transitioned to oral anticoagulation.
Her highest risk for VTE recurrence is the unprovoked VTE itself, regardless of an underlying thrombophilia. Since the presence of an inherited thrombophilia will not change duration or intensity of management, our patient should not be tested.
There are no prospective trials showing improved outcomes from aggressive workup for occult malignancy. Given this information, an extensive workup for occult malignancy should not be undertaken; however, this patient has an idiopathic VTE and should undergo a complete history, physical examination, and basic lab work, with attention to common areas of malignancy. Any abnormalities uncovered on this initial workup should be investigated more aggressively. Screening with mammography and Pap smear should be arranged in outpatient follow-up and communicated to the primary care physician, because she is not up to date with these age-appropriate screening tests.
Based on new evidence, a low-dose chest CT would be a consideration if she had a smoking history of at least 30 pack-years.22 Her microcytic anemia uncovered on routine lab work should be investigated further for a possible underlying gastrointestinal malignancy.
Bottom Line
An initial diagnosis of unprovoked VTE remains the strongest risk factor for recurrent thromboembolic events. The presence of an inherited thrombophilia does not significantly alter management. Aggressive workup for occult malignancy has not prospectively improved outcomes, but age-appropriate malignancy screening should be recommended.
Drs. Czernik and Anderson are hospitalists and instructors of medicine at the University of Colorado Denver (UCD). Dr. Wolfe is a hospitalist and assistant professor of medicine at UCD. Dr. Cumbler is a hospitalist and associate professor of medicine at UCD.
References
- Centers for Disease Control and Prevention. Venous thromboembolism in adult hospitalizations—United States, 2007–2009. MMWR Morb Mortal Wkly Rep. 2012;61(22);401-404.
- Baglin T, Gray E, Greaves M, et al. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol. 2010;149(2):209-220.
- Heit, JA, O’Fallon WM, Petterson TM, et al. Relative impact of risk factors for deep vein thrombosis and pulmonary embolism: a population-based study. Arch Intern Med. 2002;162(11):1245-1248.
- Iodice S, Gandini S, Löhr M, Lowenfels AB, Maisonneuve P. Venous thromboembolic events and organ-specific occult cancers: a review and meta-analysis. J Thromb Haemost. 2008;6(5):781-788.
- Coppens M, Reijnders JH, Middeldorp S, Doggen CJ, Rosendaal FR. Testing for inherited thrombophilia does not reduce the recurrence of venous thrombosis. J Thromb Haemost. 2008;6(9):1474-1477.
- Coppens M, van Mourik JA, Eckmann CM, Büller HR, Middeldorp S. Current practise of testing for inherited thrombophilia. J Thromb Haemost. 2007;5(9):1979-1981.
- Carrier M, Le Gal G, Wells PS, Fergusson D, Ramsay T, Rodger MA. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med. 2008;149(5):323-333.
- Cronin-Fenton DP, Søndergaard F, Pedersen LA, et al. Hospitalisation for venous thromboembolism in cancer patients and the general population: a population-based cohort study in Denmark, 1997-2006. Br J Cancer. 2010;103(7):947-953.
- Chew HK, Wun T, Harvey D, Zhou H, White RH. Incidence of venous thromboembolism and its effect on survival among patients with common cancers. Arch Intern Med. 2006;166(4):458-464.
- Lee JL, Lee JH, Kim MK, et al. A case of bone marrow necrosis with thrombotic thrombocytopenic purpura as a manifestation of occult colon cancer. Jpn J Clin Oncol. 2004;34(8):476-480.
- Sack GH Jr, Levin J, Bell WR. Trousseau’s syndrome and other manifestations of chronic disseminated coagulopathy in patients with neoplasms: clinical, pathophysiologic, and therapeutic features. Medicine (Baltimore). 1977;56(1):1-37.
- Prins MH, Hettiarachchi RJ, Lensing AW, Hirsh J. Newly diagnosed malignancy in patients with venous thromboembolism. Search or wait and see? Thromb Haemost. 1997;78(1):121-125.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med. 1996;125(10):785-793.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med. 2003;349(2):146-153.
- Dalen JE. Should patients with venous thromboembolism be screened for thrombophilia? Am J Med. 2008;121(6):458-463.
- Khamashta MA, Cuadrado MJ, Mujic F, Taub NA, Hunt BJ, Hughes GR. The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med. 1995;332:993-997.
- Ridker PM, Goldhaber SZ, Danielson E, et al. Long-term, low-intensity warfarin therapy for the prevention of recurrent venous thromboembolism. N Engl J Med. 2003;348(15):1425-1434.
- Hron G, Eichinger S, Weltermann A, et al. Family history for venous thromboembolism and the risk for recurrence. Am J Med. 2006;119(1):50-53.
- Kearon C, Akl EA, Comerota AJ, et al. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e419S-e494S.
- Douketis, James, Tosetto A, Marcucci M, et al. Risk of recurrence after venous thromboembolism in men and women: patient level meta-analysis. BMJ. 2011;342:d813.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA. 2005;293(19):2352-2361.
- American Cancer Society Guidelines for the Early Detection of Cancer. Available at: http://www.cancer.org/healthy/findcancerearly/cancerscreeningguidelines/american-cancer-society-guidelines-for-the-early-detection-of-cancer. Accessed November 15, 2014.
Case
A 56-year-old woman with hypertension and diabetes presents to the hospital with acute onset of painful swelling in her right calf. She has had no recent surgeries, trauma, or travel, and takes lisinopril and metformin. An ultrasound of her right lower extremity demonstrates a venous thromboembolism (VTE). The patient’s last mammogram was three years ago, and she’s never undergone a screening colonoscopy. On lab workup, she is noted to have a microcytic anemia.
Should this patient be screened for an underlying hypercoagulable state or malignancy?
Background
An estimated 550,000 hospitalized adults are diagnosed with VTE each year.1 VTE can occur in the absence of known precipitants (unprovoked) or can be temporally associated with a known major risk factor (provoked). This practical division has implications for both treatment duration and risk of recurrence. A VTE is considered provoked if it occurs in the setting of surgery, leg trauma, fracture, pregnancy within the previous three months, estrogen therapy, immobility from an acute illness for more than one week, travel lasting more than six hours, or active malignancy.2 If none of these provoking factors is present, the VTE is considered unprovoked.2
Nearly 20% of first-time VTE events can be attributed to malignancy.3 Additionally, patients presenting with an unprovoked VTE possess a higher risk of being diagnosed with a cancer, raising the question of whether unprovoked VTEs should compel aggressive malignancy screening.4
Before the discovery of antithrombin deficiency in 1965, most unprovoked VTE events remained unexplained. Since then, numerous inherited coagulation abnormalities have been identified. It is now estimated that coagulation abnormalities can be found in up to half of patients with unprovoked thrombi.5
The increase in availability of molecular and genetic assays for hypercoagulability has been accompanied by a dramatic rise in the rate of testing for these disorders.6 Despite increased testing available for inherited thrombophilias, disagreement exists over the utility of this workup.6
Review of the Data
Hypercoagulability leading to venous thrombosis can be broadly divided into two groups: acquired and hereditary (see Table 1). First, let’s examine acquired hypercoagulable states.
Malignancy: Armand Trousseau first suggested an association between thrombotic events and malignancy in 1865. Malignancy causes a hypercoagulable state; additionally, tumors can cause thromboemboli by other mechanisms, such as vascular invasion or external compression of vasculature.7
Multiple studies demonstrate that malignancy increases the chance of developing a VTE. A Danish cohort study of nearly 60,000 cancer patients compared with over 280,000 controls over nine years offered twice the incidence of VTE in patients with cancer.8 Other studies reveal that VTE rates peak in the first year after a cancer diagnosis; moreover, VTE events are associated with more advanced disease and worse prognosis.9 Approximately 11% of cancer patients will develop a clinically evident VTE during the course of their disease.10,11
The majority of cancers associated with VTE events are clinically evident; however, some patients with thrombi have an occult malignancy. During the two years following an unprovoked VTE, the rate of discovering a previously undiagnosed malignancy was three times higher when compared with provoked VTE.6
This potential to diagnose occult malignancy in patients with idiopathic thromboembolic events stimulates debate around the usefulness of extensive cancer screening for these patients. One large systematic review compared routine and extensive cancer screening strategies following an unprovoked VTE. An extensive screening strategy consisting of CT scans of the abdomen and pelvis significantly increased the proportion of previously undiagnosed cancers; however, the authors did not determine complication rates, cost effectiveness, or difference in morbidity and mortality associated with extensive screening strategies.7
Other studies have demonstrated that extensive screening with CT, endoscopy, and tumor markers finds more previously undetected cancers; however, up to half of these malignancies could have been identified without resorting to such expensive and invasive workups.12 Additionally, no prospective data demonstrate improved outcomes or increased survival from these diagnoses. Likewise, no cost-effectiveness data exist to support this expensive and aggressive screening approach.7
All patients with an idiopathic VTE should undergo a complete history and physical examination with attention to common areas of malignancy. Patients should have basic lab work and be recommended for age-appropriate cancer screening (see Table 2). Any abnormalities uncovered on this initial workup should be aggressively investigated.13 If overt cancer is detected, then low molecular weight heparin would be preferred over oral anticoagulation as treatment for the VTE.14 Extensive malignancy evaluation in all patients with unprovoked VTE is not warranted, however, given the lack of data regarding efficacy of extensive screening, the potential for increased harms, and the costs associated with this approach.
Antiphospholipid syndrome: Antiphospholipid syndrome is the most common acquired cause of thrombophilia.15 Characterized by the presence of antiphospholipid antibodies (e.g. lupus anticoagulant antibodies or anticardiolipin antibodies), this syndrome is usually secondary to cancer or an autoimmune disease.
Antiphospholipid antibody syndrome is a thrombophilic disorder in which both venous and arterial thrombosis may occur. Patients with this disorder are considered at high risk for thrombotic events. Data suggest that antiphospholipid antibody syndrome also increases the risk of VTE recurrence. In one retrospective study, cessation of warfarin therapy in patients with antiphospholipid antibodies after a VTE resulted in 69% of patients having recurrent thrombosis in the first year.16 Given this substantial risk, antiphospholipid antibody testing is recommended in those with a suggestive history, including patients with 1) recurrent fetal loss, 2) fetal loss after 10 weeks, or 3) known collagen vascular disease.16 Lifelong anticoagulation is recommended for these patients.
Inherited hypercoagulable states: The most frequent causes of an inherited hypercoagulable state are the factor V Leiden mutation and the prothrombin gene mutation, accounting for 50% to 60% of hereditary thrombophilias. Protein S, protein C, and antithrombin defects account for most of the remaining cases of inherited thrombophilias.15
Currently, there is no consensus regarding who should be tested for inherited thrombophilia. Testing for an inherited thrombophilia would be indicated if the results added prognostic information or changed management. Arguments against testing hinge on the fact that neither prognosis nor management is affected by the presence of an inherited thrombophilia.
The presence of a thrombophilia also does not change the method or intensity of anticoagulation.17 The risk of recurrence after discontinuing anticoagulation therapy is not affected.17,18 The strongest predictor of VTE recurrence is the unprovoked VTE itself, regardless of an underlying thrombophilia.15 Recurrent VTE is nearly twice as frequent in patients with idiopathic VTE compared to those with provoked VTE.15
The American College of Chest Physicians (ACCP) recommends treating a provoked VTE for three months.19 According to the same guidelines, an unprovoked VTE should be treated for a minimum of three months, and lifelong anticoagulation should be considered.19
Overall, the rate of recurrence after a first VTE is considerable after completion of anticoagulation, especially for an unprovoked thrombotic event. Studies show a 7%-15% recurrence rate during the two years following the index VTE (see Table 3).17,20,21 Currently, no data suggest that a hereditary thrombophilia substantially changes this baseline high recurrent risk. ACCP recommendations state that the presence of hereditary thrombophilia should not be used as a major factor to guide duration of anticoagulation.19
Back to the Case
Our patient presented with an unprovoked VTE. She should be started on anticoagulation therapy with low molecular weight heparin and transitioned to oral anticoagulation.
Her highest risk for VTE recurrence is the unprovoked VTE itself, regardless of an underlying thrombophilia. Since the presence of an inherited thrombophilia will not change duration or intensity of management, our patient should not be tested.
There are no prospective trials showing improved outcomes from aggressive workup for occult malignancy. Given this information, an extensive workup for occult malignancy should not be undertaken; however, this patient has an idiopathic VTE and should undergo a complete history, physical examination, and basic lab work, with attention to common areas of malignancy. Any abnormalities uncovered on this initial workup should be investigated more aggressively. Screening with mammography and Pap smear should be arranged in outpatient follow-up and communicated to the primary care physician, because she is not up to date with these age-appropriate screening tests.
Based on new evidence, a low-dose chest CT would be a consideration if she had a smoking history of at least 30 pack-years.22 Her microcytic anemia uncovered on routine lab work should be investigated further for a possible underlying gastrointestinal malignancy.
Bottom Line
An initial diagnosis of unprovoked VTE remains the strongest risk factor for recurrent thromboembolic events. The presence of an inherited thrombophilia does not significantly alter management. Aggressive workup for occult malignancy has not prospectively improved outcomes, but age-appropriate malignancy screening should be recommended.
Drs. Czernik and Anderson are hospitalists and instructors of medicine at the University of Colorado Denver (UCD). Dr. Wolfe is a hospitalist and assistant professor of medicine at UCD. Dr. Cumbler is a hospitalist and associate professor of medicine at UCD.
References
- Centers for Disease Control and Prevention. Venous thromboembolism in adult hospitalizations—United States, 2007–2009. MMWR Morb Mortal Wkly Rep. 2012;61(22);401-404.
- Baglin T, Gray E, Greaves M, et al. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol. 2010;149(2):209-220.
- Heit, JA, O’Fallon WM, Petterson TM, et al. Relative impact of risk factors for deep vein thrombosis and pulmonary embolism: a population-based study. Arch Intern Med. 2002;162(11):1245-1248.
- Iodice S, Gandini S, Löhr M, Lowenfels AB, Maisonneuve P. Venous thromboembolic events and organ-specific occult cancers: a review and meta-analysis. J Thromb Haemost. 2008;6(5):781-788.
- Coppens M, Reijnders JH, Middeldorp S, Doggen CJ, Rosendaal FR. Testing for inherited thrombophilia does not reduce the recurrence of venous thrombosis. J Thromb Haemost. 2008;6(9):1474-1477.
- Coppens M, van Mourik JA, Eckmann CM, Büller HR, Middeldorp S. Current practise of testing for inherited thrombophilia. J Thromb Haemost. 2007;5(9):1979-1981.
- Carrier M, Le Gal G, Wells PS, Fergusson D, Ramsay T, Rodger MA. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med. 2008;149(5):323-333.
- Cronin-Fenton DP, Søndergaard F, Pedersen LA, et al. Hospitalisation for venous thromboembolism in cancer patients and the general population: a population-based cohort study in Denmark, 1997-2006. Br J Cancer. 2010;103(7):947-953.
- Chew HK, Wun T, Harvey D, Zhou H, White RH. Incidence of venous thromboembolism and its effect on survival among patients with common cancers. Arch Intern Med. 2006;166(4):458-464.
- Lee JL, Lee JH, Kim MK, et al. A case of bone marrow necrosis with thrombotic thrombocytopenic purpura as a manifestation of occult colon cancer. Jpn J Clin Oncol. 2004;34(8):476-480.
- Sack GH Jr, Levin J, Bell WR. Trousseau’s syndrome and other manifestations of chronic disseminated coagulopathy in patients with neoplasms: clinical, pathophysiologic, and therapeutic features. Medicine (Baltimore). 1977;56(1):1-37.
- Prins MH, Hettiarachchi RJ, Lensing AW, Hirsh J. Newly diagnosed malignancy in patients with venous thromboembolism. Search or wait and see? Thromb Haemost. 1997;78(1):121-125.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med. 1996;125(10):785-793.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med. 2003;349(2):146-153.
- Dalen JE. Should patients with venous thromboembolism be screened for thrombophilia? Am J Med. 2008;121(6):458-463.
- Khamashta MA, Cuadrado MJ, Mujic F, Taub NA, Hunt BJ, Hughes GR. The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med. 1995;332:993-997.
- Ridker PM, Goldhaber SZ, Danielson E, et al. Long-term, low-intensity warfarin therapy for the prevention of recurrent venous thromboembolism. N Engl J Med. 2003;348(15):1425-1434.
- Hron G, Eichinger S, Weltermann A, et al. Family history for venous thromboembolism and the risk for recurrence. Am J Med. 2006;119(1):50-53.
- Kearon C, Akl EA, Comerota AJ, et al. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e419S-e494S.
- Douketis, James, Tosetto A, Marcucci M, et al. Risk of recurrence after venous thromboembolism in men and women: patient level meta-analysis. BMJ. 2011;342:d813.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA. 2005;293(19):2352-2361.
- American Cancer Society Guidelines for the Early Detection of Cancer. Available at: http://www.cancer.org/healthy/findcancerearly/cancerscreeningguidelines/american-cancer-society-guidelines-for-the-early-detection-of-cancer. Accessed November 15, 2014.
When Should You Decolonize Methicillin-Resistant Staphylococcus aureus (MRSA) in Hospitalized Patients?
Case
A 45-year-old previously healthy female was admitted to the ICU with sepsis caused by community-acquired pneumonia. Per hospital policy, all patients admitted to the ICU are screened for MRSA colonization. If the nasal screen is positive, contact isolation is initiated and the hospital’s MRSA decolonization protocol is implemented. Her nasal screen was positive for MRSA.
Overview
MRSA infections are associated with significant morbidity and mortality, and death occurs in almost 5% of patients who develop a MRSA infection. In 2005, invasive MRSA was responsible for approximately 278,000 hospitalizations and 19,000 deaths. MRSA is a common cause of healthcare-associated infections (HAIs) and is the most common pathogen in surgical site infections (SSIs) and ventilator-associated pneumonias. The cost of treating MRSA infections is substantial; in 2003, $14.5 billion was spent on MRSA-related hospitalizations.
It is well known that MRSA colonization is a risk factor for the subsequent development of a MRSA infection. This risk persists over time, and approximately 25% of individuals who are colonized with MRSA for more than one year will develop a late-onset MRSA infection.1 It is estimated that between 0.8% and 6% of people in the U.S. are asymptomatically colonized with MRSA.
One infection control strategy for reducing the transmission of MRSA among hospitalized patients involves screening for the presence of this organism and then placing colonized and/or infected patients in isolation; however, there is considerable controversy about which patients should be screened.
An additional element of many infection control strategies involves MRSA decolonization, but there is uncertainty about which patients benefit from it and significant variability in its reported success rates.2 Additionally, several studies have indicated that MRSA decolonization is only temporary and that patients become recolonized over time.
Treatment
It is estimated that 10% to 20% of MRSA carriers will develop an infection while they are hospitalized. Furthermore, even after they have been discharged from the hospital, their risk for developing a MRSA infection persists.
Most patients who develop a MRSA infection have been colonized prior to infection, and these patients usually develop an infection caused by the same strain as the colonization. In view of this fact, a primary goal of decolonization is reducing the likelihood of “auto-infection.” Another goal of decolonization is reducing the transmission of MRSA to other patients.
In order to determine whether MRSA colonization is present, patients undergo screening, and specimens are collected from the nares using nasal swabs. Specimens from extranasal sites, such as the groin, are sometimes also obtained for screening. These screening tests are usually done with either cultures or polymerase chain reaction testing.

There is significant variability in the details of screening and decolonization protocols among different healthcare facilities. Typically, the screening test costs more than the agents used for decolonization. Partly for this reason, some facilities forego screening altogether, instead treating all patients with a decolonization regimen; however, there is concern that administering decolonizing medications to all patients would lead to the unnecessary treatment of large numbers of patients. Such widespread use of the decolonizing agents might promote the development of resistance to these medications.
Medications. Decolonization typically involves the use of a topical antibiotic, most commonly mupirocin, which is applied to the nares. This may be used in conjunction with an oral antimicrobial agent. While the nares are the anatomical locations most commonly colonized by MRSA, extranasal colonization occurs in 50% of those who are nasally colonized.
Of the topical medications available for decolonization, mupirocin has the highest efficacy, with eradication of MRSA and methicillin-sensitive Staphylococcus aureus (MSSA) colonization ranging from 81% to 93%. To increase the likelihood of successful decolonization, an antiseptic agent, such as chlorhexidine gluconate, may also be applied to the skin. Chlorhexidine gluconate is also commonly used to prevent other HAIs.
Neomycin is sometimes used for decolonization, but its efficacy for this purpose is questionable. There are also concerns about resistance, but it may be an option in cases of documented mupirocin resistance. Preparations that contain tea tree oil appear to be more effective for decolonization of skin sites than for nasal decolonization. Table 1 lists the topical antibiotics and antiseptics that may be utilized for decolonization, while Table 2 lists the oral medications that can be used for this purpose. Table 3 lists investigational agents being evaluated for their ability to decolonize patients.

It has been suggested that the patients who might derive the most benefit from decolonization are those at increased risk for developing a MRSA infection during a specific time interval. This would include patients who are admitted to the ICU for an acute illness and cardiothoracic surgery patients. A benefit from decolonization has also been observed in hemodialysis patients, who have an incidence of invasive MRSA infections 100 times greater than the general population. Otherwise, there are no data to support the routine use of decolonization in nonsurgical patients.
It is not uncommon for hospitals to screen patients admitted to the ICU for MRSA nasal colonization; in fact, screening is mandatory in nine states. If the nasal screen is positive, contact precautions are instituted. The decision about whether or not to initiate a decolonization protocol varies among different ICUs, but most do not carry out universal decolonization.
Some studies show decolonization is beneficial for ICU patients. These studies include a large cluster-randomized trial called REDUCE MRSA,3 which took place in 43 hospitals and involved 74,256 patients in 74 ICUs. The study showed that universal (i.e., without screening) decolonization using mupirocin and chlorhexidine was effective in reducing rates of MRSA clinical isolates, as well as bloodstream infection from any pathogen. Other studies have demonstrated benefits from the decolonization of ICU patients.4,5
Surgical Site Infections. Meanwhile, SSIs are often associated with increased mortality rates and substantial healthcare costs, including increased hospital lengths of stay and readmission rates. Staphylococcus aureus is the pathogen most commonly isolated from SSIs. In surgical patients, colonization with MRSA is associated with an elevated rate of MRSA SSIs. The goal of decolonization in surgical patients is not to permanently eliminate MRSA but to prevent SSIs by suppressing the presence of this organism for a relatively brief duration.

There is evidence that decolonization reduces SSIs for cardiothoracic surgeries.6 For these patients, it is cost effective to screen for nasal carriage of MRSA and then treat carriers with a combination of pre-operative mupirocin and chlorhexidine. It may be reasonable to delay cardiothoracic surgery in colonized patients who will require implantation of prosthetic material until they complete MRSA decolonization.
In addition to reducing the risk of auto-infection, another goal of decolonization is limiting the possibility of transmission of MRSA from a colonized patient to a susceptible individual; however, there are only limited data available that measure the efficacy of decolonization for preventing transmission.
Concerns about the potential hazards of decolonization therapy have impacted its widespread implementation. The biggest concern is that patients may develop resistance to the antimicrobial agents used for decolonization, particularly if they are used at increased frequency. Mupirocin resistance monitoring is valuable, but, unfortunately, the susceptibility of Staphylococcus aureus to mupirocin is not routinely evaluated, so the prevalence of mupirocin resistance in local strains is often unknown. Another concern about decolonization is the cost of screening and decolonizing patients.
Back to the Case
The patient in this case required admission to an ICU and, based on the results of the REDUCE MRSA clinical trial, she would likely benefit from undergoing decolonization to reduce her risk of both MRSA-positive clinical cultures and bloodstream infections caused by any pathogen.
Bottom Line
Decolonization is beneficial for patients at increased risk of developing a MRSA infection during a specific period, such as patients admitted to the ICU and those undergoing cardiothoracic surgery.
Dr. Clarke is assistant professor in the division of hospital medicine at Emory University Hospital and a faculty member in the Emory University Department of Medicine, both in Atlanta.
References
- Dow G, Field D, Mancuso M, Allard J. Decolonization of methicillin-resistant Staphylococcus aureus during routine hospital care: Efficacy and long-term follow-up. Can J Infect Dis Med Microbiol. 2010;21(1):38-44.
- Simor AE. Staphylococcal decolonisation: An effective strategy for prevention of infection? Lancet Infect Dis. 2011;11(12):952-962.
- Huang SS, Septimus E, Kleinman K, et al. Targeted versus universal decolonization to prevent ICU infection. N Engl J Med. 2013;368(24):2255-2265.
- Fraser T, Fatica C, Scarpelli M, et al. Decrease in Staphylococcus aureus colonization and hospital-acquired infection in a medical intensive care unit after institution of an active surveillance and decolonization program. Infect Control Hosp Epidemiol. 2010;31(8):779-783.
- Robotham J, Graves N, Cookson B, et al. Screening, isolation, and decolonisation strategies in the control of methicillin-resistant Staphylococcus aureus in intensive care units: Cost effectiveness evaluation. BMJ. 2011;343:d5694.
- Schweizer M, Perencevich E, McDanel J, et al. Effectiveness of a bundled intervention of decolonization and prophylaxis to decrease Gram positive surgical site infections after cardiac or orthopedic surgery: Systematic review and meta-analysis. BMJ. 2013;346:f2743.
Case
A 45-year-old previously healthy female was admitted to the ICU with sepsis caused by community-acquired pneumonia. Per hospital policy, all patients admitted to the ICU are screened for MRSA colonization. If the nasal screen is positive, contact isolation is initiated and the hospital’s MRSA decolonization protocol is implemented. Her nasal screen was positive for MRSA.
Overview
MRSA infections are associated with significant morbidity and mortality, and death occurs in almost 5% of patients who develop a MRSA infection. In 2005, invasive MRSA was responsible for approximately 278,000 hospitalizations and 19,000 deaths. MRSA is a common cause of healthcare-associated infections (HAIs) and is the most common pathogen in surgical site infections (SSIs) and ventilator-associated pneumonias. The cost of treating MRSA infections is substantial; in 2003, $14.5 billion was spent on MRSA-related hospitalizations.
It is well known that MRSA colonization is a risk factor for the subsequent development of a MRSA infection. This risk persists over time, and approximately 25% of individuals who are colonized with MRSA for more than one year will develop a late-onset MRSA infection.1 It is estimated that between 0.8% and 6% of people in the U.S. are asymptomatically colonized with MRSA.
One infection control strategy for reducing the transmission of MRSA among hospitalized patients involves screening for the presence of this organism and then placing colonized and/or infected patients in isolation; however, there is considerable controversy about which patients should be screened.
An additional element of many infection control strategies involves MRSA decolonization, but there is uncertainty about which patients benefit from it and significant variability in its reported success rates.2 Additionally, several studies have indicated that MRSA decolonization is only temporary and that patients become recolonized over time.
Treatment
It is estimated that 10% to 20% of MRSA carriers will develop an infection while they are hospitalized. Furthermore, even after they have been discharged from the hospital, their risk for developing a MRSA infection persists.
Most patients who develop a MRSA infection have been colonized prior to infection, and these patients usually develop an infection caused by the same strain as the colonization. In view of this fact, a primary goal of decolonization is reducing the likelihood of “auto-infection.” Another goal of decolonization is reducing the transmission of MRSA to other patients.
In order to determine whether MRSA colonization is present, patients undergo screening, and specimens are collected from the nares using nasal swabs. Specimens from extranasal sites, such as the groin, are sometimes also obtained for screening. These screening tests are usually done with either cultures or polymerase chain reaction testing.
There is significant variability in the details of screening and decolonization protocols among different healthcare facilities. Typically, the screening test costs more than the agents used for decolonization. Partly for this reason, some facilities forego screening altogether, instead treating all patients with a decolonization regimen; however, there is concern that administering decolonizing medications to all patients would lead to the unnecessary treatment of large numbers of patients. Such widespread use of the decolonizing agents might promote the development of resistance to these medications.
Medications. Decolonization typically involves the use of a topical antibiotic, most commonly mupirocin, which is applied to the nares. This may be used in conjunction with an oral antimicrobial agent. While the nares are the anatomical locations most commonly colonized by MRSA, extranasal colonization occurs in 50% of those who are nasally colonized.
Of the topical medications available for decolonization, mupirocin has the highest efficacy, with eradication of MRSA and methicillin-sensitive Staphylococcus aureus (MSSA) colonization ranging from 81% to 93%. To increase the likelihood of successful decolonization, an antiseptic agent, such as chlorhexidine gluconate, may also be applied to the skin. Chlorhexidine gluconate is also commonly used to prevent other HAIs.
Neomycin is sometimes used for decolonization, but its efficacy for this purpose is questionable. There are also concerns about resistance, but it may be an option in cases of documented mupirocin resistance. Preparations that contain tea tree oil appear to be more effective for decolonization of skin sites than for nasal decolonization. Table 1 lists the topical antibiotics and antiseptics that may be utilized for decolonization, while Table 2 lists the oral medications that can be used for this purpose. Table 3 lists investigational agents being evaluated for their ability to decolonize patients.
It has been suggested that the patients who might derive the most benefit from decolonization are those at increased risk for developing a MRSA infection during a specific time interval. This would include patients who are admitted to the ICU for an acute illness and cardiothoracic surgery patients. A benefit from decolonization has also been observed in hemodialysis patients, who have an incidence of invasive MRSA infections 100 times greater than the general population. Otherwise, there are no data to support the routine use of decolonization in nonsurgical patients.
It is not uncommon for hospitals to screen patients admitted to the ICU for MRSA nasal colonization; in fact, screening is mandatory in nine states. If the nasal screen is positive, contact precautions are instituted. The decision about whether or not to initiate a decolonization protocol varies among different ICUs, but most do not carry out universal decolonization.
Some studies show decolonization is beneficial for ICU patients. These studies include a large cluster-randomized trial called REDUCE MRSA,3 which took place in 43 hospitals and involved 74,256 patients in 74 ICUs. The study showed that universal (i.e., without screening) decolonization using mupirocin and chlorhexidine was effective in reducing rates of MRSA clinical isolates, as well as bloodstream infection from any pathogen. Other studies have demonstrated benefits from the decolonization of ICU patients.4,5
Surgical Site Infections. Meanwhile, SSIs are often associated with increased mortality rates and substantial healthcare costs, including increased hospital lengths of stay and readmission rates. Staphylococcus aureus is the pathogen most commonly isolated from SSIs. In surgical patients, colonization with MRSA is associated with an elevated rate of MRSA SSIs. The goal of decolonization in surgical patients is not to permanently eliminate MRSA but to prevent SSIs by suppressing the presence of this organism for a relatively brief duration.
There is evidence that decolonization reduces SSIs for cardiothoracic surgeries.6 For these patients, it is cost effective to screen for nasal carriage of MRSA and then treat carriers with a combination of pre-operative mupirocin and chlorhexidine. It may be reasonable to delay cardiothoracic surgery in colonized patients who will require implantation of prosthetic material until they complete MRSA decolonization.
In addition to reducing the risk of auto-infection, another goal of decolonization is limiting the possibility of transmission of MRSA from a colonized patient to a susceptible individual; however, there are only limited data available that measure the efficacy of decolonization for preventing transmission.
Concerns about the potential hazards of decolonization therapy have impacted its widespread implementation. The biggest concern is that patients may develop resistance to the antimicrobial agents used for decolonization, particularly if they are used at increased frequency. Mupirocin resistance monitoring is valuable, but, unfortunately, the susceptibility of Staphylococcus aureus to mupirocin is not routinely evaluated, so the prevalence of mupirocin resistance in local strains is often unknown. Another concern about decolonization is the cost of screening and decolonizing patients.
Back to the Case
The patient in this case required admission to an ICU and, based on the results of the REDUCE MRSA clinical trial, she would likely benefit from undergoing decolonization to reduce her risk of both MRSA-positive clinical cultures and bloodstream infections caused by any pathogen.
Bottom Line
Decolonization is beneficial for patients at increased risk of developing a MRSA infection during a specific period, such as patients admitted to the ICU and those undergoing cardiothoracic surgery.
Dr. Clarke is assistant professor in the division of hospital medicine at Emory University Hospital and a faculty member in the Emory University Department of Medicine, both in Atlanta.
References
- Dow G, Field D, Mancuso M, Allard J. Decolonization of methicillin-resistant Staphylococcus aureus during routine hospital care: Efficacy and long-term follow-up. Can J Infect Dis Med Microbiol. 2010;21(1):38-44.
- Simor AE. Staphylococcal decolonisation: An effective strategy for prevention of infection? Lancet Infect Dis. 2011;11(12):952-962.
- Huang SS, Septimus E, Kleinman K, et al. Targeted versus universal decolonization to prevent ICU infection. N Engl J Med. 2013;368(24):2255-2265.
- Fraser T, Fatica C, Scarpelli M, et al. Decrease in Staphylococcus aureus colonization and hospital-acquired infection in a medical intensive care unit after institution of an active surveillance and decolonization program. Infect Control Hosp Epidemiol. 2010;31(8):779-783.
- Robotham J, Graves N, Cookson B, et al. Screening, isolation, and decolonisation strategies in the control of methicillin-resistant Staphylococcus aureus in intensive care units: Cost effectiveness evaluation. BMJ. 2011;343:d5694.
- Schweizer M, Perencevich E, McDanel J, et al. Effectiveness of a bundled intervention of decolonization and prophylaxis to decrease Gram positive surgical site infections after cardiac or orthopedic surgery: Systematic review and meta-analysis. BMJ. 2013;346:f2743.
Case
A 45-year-old previously healthy female was admitted to the ICU with sepsis caused by community-acquired pneumonia. Per hospital policy, all patients admitted to the ICU are screened for MRSA colonization. If the nasal screen is positive, contact isolation is initiated and the hospital’s MRSA decolonization protocol is implemented. Her nasal screen was positive for MRSA.
Overview
MRSA infections are associated with significant morbidity and mortality, and death occurs in almost 5% of patients who develop a MRSA infection. In 2005, invasive MRSA was responsible for approximately 278,000 hospitalizations and 19,000 deaths. MRSA is a common cause of healthcare-associated infections (HAIs) and is the most common pathogen in surgical site infections (SSIs) and ventilator-associated pneumonias. The cost of treating MRSA infections is substantial; in 2003, $14.5 billion was spent on MRSA-related hospitalizations.
It is well known that MRSA colonization is a risk factor for the subsequent development of a MRSA infection. This risk persists over time, and approximately 25% of individuals who are colonized with MRSA for more than one year will develop a late-onset MRSA infection.1 It is estimated that between 0.8% and 6% of people in the U.S. are asymptomatically colonized with MRSA.
One infection control strategy for reducing the transmission of MRSA among hospitalized patients involves screening for the presence of this organism and then placing colonized and/or infected patients in isolation; however, there is considerable controversy about which patients should be screened.
An additional element of many infection control strategies involves MRSA decolonization, but there is uncertainty about which patients benefit from it and significant variability in its reported success rates.2 Additionally, several studies have indicated that MRSA decolonization is only temporary and that patients become recolonized over time.
Treatment
It is estimated that 10% to 20% of MRSA carriers will develop an infection while they are hospitalized. Furthermore, even after they have been discharged from the hospital, their risk for developing a MRSA infection persists.
Most patients who develop a MRSA infection have been colonized prior to infection, and these patients usually develop an infection caused by the same strain as the colonization. In view of this fact, a primary goal of decolonization is reducing the likelihood of “auto-infection.” Another goal of decolonization is reducing the transmission of MRSA to other patients.
In order to determine whether MRSA colonization is present, patients undergo screening, and specimens are collected from the nares using nasal swabs. Specimens from extranasal sites, such as the groin, are sometimes also obtained for screening. These screening tests are usually done with either cultures or polymerase chain reaction testing.
There is significant variability in the details of screening and decolonization protocols among different healthcare facilities. Typically, the screening test costs more than the agents used for decolonization. Partly for this reason, some facilities forego screening altogether, instead treating all patients with a decolonization regimen; however, there is concern that administering decolonizing medications to all patients would lead to the unnecessary treatment of large numbers of patients. Such widespread use of the decolonizing agents might promote the development of resistance to these medications.
Medications. Decolonization typically involves the use of a topical antibiotic, most commonly mupirocin, which is applied to the nares. This may be used in conjunction with an oral antimicrobial agent. While the nares are the anatomical locations most commonly colonized by MRSA, extranasal colonization occurs in 50% of those who are nasally colonized.
Of the topical medications available for decolonization, mupirocin has the highest efficacy, with eradication of MRSA and methicillin-sensitive Staphylococcus aureus (MSSA) colonization ranging from 81% to 93%. To increase the likelihood of successful decolonization, an antiseptic agent, such as chlorhexidine gluconate, may also be applied to the skin. Chlorhexidine gluconate is also commonly used to prevent other HAIs.
Neomycin is sometimes used for decolonization, but its efficacy for this purpose is questionable. There are also concerns about resistance, but it may be an option in cases of documented mupirocin resistance. Preparations that contain tea tree oil appear to be more effective for decolonization of skin sites than for nasal decolonization. Table 1 lists the topical antibiotics and antiseptics that may be utilized for decolonization, while Table 2 lists the oral medications that can be used for this purpose. Table 3 lists investigational agents being evaluated for their ability to decolonize patients.
It has been suggested that the patients who might derive the most benefit from decolonization are those at increased risk for developing a MRSA infection during a specific time interval. This would include patients who are admitted to the ICU for an acute illness and cardiothoracic surgery patients. A benefit from decolonization has also been observed in hemodialysis patients, who have an incidence of invasive MRSA infections 100 times greater than the general population. Otherwise, there are no data to support the routine use of decolonization in nonsurgical patients.
It is not uncommon for hospitals to screen patients admitted to the ICU for MRSA nasal colonization; in fact, screening is mandatory in nine states. If the nasal screen is positive, contact precautions are instituted. The decision about whether or not to initiate a decolonization protocol varies among different ICUs, but most do not carry out universal decolonization.
Some studies show decolonization is beneficial for ICU patients. These studies include a large cluster-randomized trial called REDUCE MRSA,3 which took place in 43 hospitals and involved 74,256 patients in 74 ICUs. The study showed that universal (i.e., without screening) decolonization using mupirocin and chlorhexidine was effective in reducing rates of MRSA clinical isolates, as well as bloodstream infection from any pathogen. Other studies have demonstrated benefits from the decolonization of ICU patients.4,5
Surgical Site Infections. Meanwhile, SSIs are often associated with increased mortality rates and substantial healthcare costs, including increased hospital lengths of stay and readmission rates. Staphylococcus aureus is the pathogen most commonly isolated from SSIs. In surgical patients, colonization with MRSA is associated with an elevated rate of MRSA SSIs. The goal of decolonization in surgical patients is not to permanently eliminate MRSA but to prevent SSIs by suppressing the presence of this organism for a relatively brief duration.
There is evidence that decolonization reduces SSIs for cardiothoracic surgeries.6 For these patients, it is cost effective to screen for nasal carriage of MRSA and then treat carriers with a combination of pre-operative mupirocin and chlorhexidine. It may be reasonable to delay cardiothoracic surgery in colonized patients who will require implantation of prosthetic material until they complete MRSA decolonization.
In addition to reducing the risk of auto-infection, another goal of decolonization is limiting the possibility of transmission of MRSA from a colonized patient to a susceptible individual; however, there are only limited data available that measure the efficacy of decolonization for preventing transmission.
Concerns about the potential hazards of decolonization therapy have impacted its widespread implementation. The biggest concern is that patients may develop resistance to the antimicrobial agents used for decolonization, particularly if they are used at increased frequency. Mupirocin resistance monitoring is valuable, but, unfortunately, the susceptibility of Staphylococcus aureus to mupirocin is not routinely evaluated, so the prevalence of mupirocin resistance in local strains is often unknown. Another concern about decolonization is the cost of screening and decolonizing patients.
Back to the Case
The patient in this case required admission to an ICU and, based on the results of the REDUCE MRSA clinical trial, she would likely benefit from undergoing decolonization to reduce her risk of both MRSA-positive clinical cultures and bloodstream infections caused by any pathogen.
Bottom Line
Decolonization is beneficial for patients at increased risk of developing a MRSA infection during a specific period, such as patients admitted to the ICU and those undergoing cardiothoracic surgery.
Dr. Clarke is assistant professor in the division of hospital medicine at Emory University Hospital and a faculty member in the Emory University Department of Medicine, both in Atlanta.
References
- Dow G, Field D, Mancuso M, Allard J. Decolonization of methicillin-resistant Staphylococcus aureus during routine hospital care: Efficacy and long-term follow-up. Can J Infect Dis Med Microbiol. 2010;21(1):38-44.
- Simor AE. Staphylococcal decolonisation: An effective strategy for prevention of infection? Lancet Infect Dis. 2011;11(12):952-962.
- Huang SS, Septimus E, Kleinman K, et al. Targeted versus universal decolonization to prevent ICU infection. N Engl J Med. 2013;368(24):2255-2265.
- Fraser T, Fatica C, Scarpelli M, et al. Decrease in Staphylococcus aureus colonization and hospital-acquired infection in a medical intensive care unit after institution of an active surveillance and decolonization program. Infect Control Hosp Epidemiol. 2010;31(8):779-783.
- Robotham J, Graves N, Cookson B, et al. Screening, isolation, and decolonisation strategies in the control of methicillin-resistant Staphylococcus aureus in intensive care units: Cost effectiveness evaluation. BMJ. 2011;343:d5694.
- Schweizer M, Perencevich E, McDanel J, et al. Effectiveness of a bundled intervention of decolonization and prophylaxis to decrease Gram positive surgical site infections after cardiac or orthopedic surgery: Systematic review and meta-analysis. BMJ. 2013;346:f2743.
Antibiotic Prophylaxis Might Prevent Recurrent UTIs

Clinical question: Does antibiotic prophylaxis prevent future episodes of urinary tract infections?
Background: Recurrent urinary tract infections (UTI) in children might be associated with renal scarring and subsequent clinical consequences associated with long-term morbidity. Historically, antibiotic prophylaxis has been recommended for children who might have risk factors for recurrent infection, most commonly vesicoureteral reflux. However, scars may be present in the absence of known risk factors and upon first UTI. The efficacy of antibiotic prophylaxis in preventing recurrent UTIs is unclear.
Study design: Randomized, double-blind, placebo-controlled trial.
Setting: Four centers in Australia.
Synopsis: The study looked at 576 children under the age of 18 with a history of at least one symptomatic UTI. The patients were randomized to receive trimethoprim-sulfamethoxazole (TMP-SMX) or placebo for 12 months. Children with vesicoureteral reflux were included, but those with known neurologic, skeletal, or urologic predispositions were excluded.
Thirteen percent of patients in the antibiotic group developed a UTI compared with 19% of patients in the placebo group (P=0.02). The authors calculate that at 12 months, 14 patients would need to be treated to prevent one UTI.
This study was unable to enroll the planned number of children but remained adequately powered to show a reduction in the primary outcome (rate of symptomatic UTI). However, a significant number of patients (approximately 28%) in each arm stopped taking the medication, the majority for undisclosed reasons. Despite an intention-to-treat analysis, this degree of dropout raises questions about the true effect size. Additionally, this study does not answer the more important clinical question regarding the effect of prophylaxis on potential future renal damage, specifically in children with vesicoureteral reflux.
Bottom line: Antibiotic prophylaxis might be modestly effective in preventing recurrent UTIs.
Citation: Craig JC, Simpson JM, Williams GJ, et al. Antibiotic prophylaxis and recurrent urinary tract infection in children. N Engl J Med. 2009;361(18):1748-1759.
Clinical question: Does antibiotic prophylaxis prevent future episodes of urinary tract infections?
Background: Recurrent urinary tract infections (UTI) in children might be associated with renal scarring and subsequent clinical consequences associated with long-term morbidity. Historically, antibiotic prophylaxis has been recommended for children who might have risk factors for recurrent infection, most commonly vesicoureteral reflux. However, scars may be present in the absence of known risk factors and upon first UTI. The efficacy of antibiotic prophylaxis in preventing recurrent UTIs is unclear.
Study design: Randomized, double-blind, placebo-controlled trial.
Setting: Four centers in Australia.
Synopsis: The study looked at 576 children under the age of 18 with a history of at least one symptomatic UTI. The patients were randomized to receive trimethoprim-sulfamethoxazole (TMP-SMX) or placebo for 12 months. Children with vesicoureteral reflux were included, but those with known neurologic, skeletal, or urologic predispositions were excluded.
Thirteen percent of patients in the antibiotic group developed a UTI compared with 19% of patients in the placebo group (P=0.02). The authors calculate that at 12 months, 14 patients would need to be treated to prevent one UTI.
This study was unable to enroll the planned number of children but remained adequately powered to show a reduction in the primary outcome (rate of symptomatic UTI). However, a significant number of patients (approximately 28%) in each arm stopped taking the medication, the majority for undisclosed reasons. Despite an intention-to-treat analysis, this degree of dropout raises questions about the true effect size. Additionally, this study does not answer the more important clinical question regarding the effect of prophylaxis on potential future renal damage, specifically in children with vesicoureteral reflux.
Bottom line: Antibiotic prophylaxis might be modestly effective in preventing recurrent UTIs.
Citation: Craig JC, Simpson JM, Williams GJ, et al. Antibiotic prophylaxis and recurrent urinary tract infection in children. N Engl J Med. 2009;361(18):1748-1759.
Clinical question: Does antibiotic prophylaxis prevent future episodes of urinary tract infections?
Background: Recurrent urinary tract infections (UTI) in children might be associated with renal scarring and subsequent clinical consequences associated with long-term morbidity. Historically, antibiotic prophylaxis has been recommended for children who might have risk factors for recurrent infection, most commonly vesicoureteral reflux. However, scars may be present in the absence of known risk factors and upon first UTI. The efficacy of antibiotic prophylaxis in preventing recurrent UTIs is unclear.
Study design: Randomized, double-blind, placebo-controlled trial.
Setting: Four centers in Australia.
Synopsis: The study looked at 576 children under the age of 18 with a history of at least one symptomatic UTI. The patients were randomized to receive trimethoprim-sulfamethoxazole (TMP-SMX) or placebo for 12 months. Children with vesicoureteral reflux were included, but those with known neurologic, skeletal, or urologic predispositions were excluded.
Thirteen percent of patients in the antibiotic group developed a UTI compared with 19% of patients in the placebo group (P=0.02). The authors calculate that at 12 months, 14 patients would need to be treated to prevent one UTI.
This study was unable to enroll the planned number of children but remained adequately powered to show a reduction in the primary outcome (rate of symptomatic UTI). However, a significant number of patients (approximately 28%) in each arm stopped taking the medication, the majority for undisclosed reasons. Despite an intention-to-treat analysis, this degree of dropout raises questions about the true effect size. Additionally, this study does not answer the more important clinical question regarding the effect of prophylaxis on potential future renal damage, specifically in children with vesicoureteral reflux.
Bottom line: Antibiotic prophylaxis might be modestly effective in preventing recurrent UTIs.
Citation: Craig JC, Simpson JM, Williams GJ, et al. Antibiotic prophylaxis and recurrent urinary tract infection in children. N Engl J Med. 2009;361(18):1748-1759.
Advanced Dementia Is a Terminal Illness with High Morbidity and Mortality
Clinical question: Does understanding the expected clinical course of advanced dementia influence end-of-life decisions by proxy decision-makers?
Background: Advanced dementia is a leading cause of death in the United States, but the clinical course of advanced dementia has not been described in a rigorous, prospective manner. The lack of information might cause risk to be underestimated, and patients might receive suboptimal palliative care.
Study design: Multicenter prospective cohort study.
Setting: Twenty-two nursing homes in a single U.S. city.
Synopsis: The survey examined 323 nursing home residents with advanced dementia. The patients were clinically assessed at baseline and quarterly for 18 months through chart reviews, nursing interviews, and physical examinations. Additionally, their proxies were surveyed regarding their understanding of the subjects’ prognoses.
During the survey period, 41.1% of patients developed pneumonia, 52.6% of patients experienced a febrile episode, and 85.8% of patients developed an eating problem; cumulative all-cause mortality was 54.8%. Adjusted for age, sex, and disease duration, the six-month mortality rate for subjects who had pneumonia was 46.7%; a febrile episode, 44.5%; and an eating problem, 38.6%.
Distressing symptoms, including dyspnea (46.0%) and pain (39.1%), were common. In the last three months of life, 40.7% of subjects underwent at least one burdensome intervention (defined as hospitalization, ED visit, parenteral therapy, or tube feeding).
Subjects whose proxies reported an understanding of the poor prognosis and expected clinical complications of advanced dementia underwent significantly fewer burdensome interventions (adjusted odds ratio 0.12).
Bottom line: Advanced dementia is associated with frequent complications, including infections and eating problems, with high six-month mortality and significant associated morbidity. Patients whose healthcare proxies have a good understanding of the expected clinical course and prognosis receive less-aggressive end-of-life care.
Citation: Mitchell SL, Teno JM, Kiely DK, et al. The clinical course of advanced dementia. N Engl J Med. 2009;361(16):1529-1538. TH
Clinical question: Does understanding the expected clinical course of advanced dementia influence end-of-life decisions by proxy decision-makers?
Background: Advanced dementia is a leading cause of death in the United States, but the clinical course of advanced dementia has not been described in a rigorous, prospective manner. The lack of information might cause risk to be underestimated, and patients might receive suboptimal palliative care.
Study design: Multicenter prospective cohort study.
Setting: Twenty-two nursing homes in a single U.S. city.
Synopsis: The survey examined 323 nursing home residents with advanced dementia. The patients were clinically assessed at baseline and quarterly for 18 months through chart reviews, nursing interviews, and physical examinations. Additionally, their proxies were surveyed regarding their understanding of the subjects’ prognoses.
During the survey period, 41.1% of patients developed pneumonia, 52.6% of patients experienced a febrile episode, and 85.8% of patients developed an eating problem; cumulative all-cause mortality was 54.8%. Adjusted for age, sex, and disease duration, the six-month mortality rate for subjects who had pneumonia was 46.7%; a febrile episode, 44.5%; and an eating problem, 38.6%.
Distressing symptoms, including dyspnea (46.0%) and pain (39.1%), were common. In the last three months of life, 40.7% of subjects underwent at least one burdensome intervention (defined as hospitalization, ED visit, parenteral therapy, or tube feeding).
Subjects whose proxies reported an understanding of the poor prognosis and expected clinical complications of advanced dementia underwent significantly fewer burdensome interventions (adjusted odds ratio 0.12).
Bottom line: Advanced dementia is associated with frequent complications, including infections and eating problems, with high six-month mortality and significant associated morbidity. Patients whose healthcare proxies have a good understanding of the expected clinical course and prognosis receive less-aggressive end-of-life care.
Citation: Mitchell SL, Teno JM, Kiely DK, et al. The clinical course of advanced dementia. N Engl J Med. 2009;361(16):1529-1538. TH
Clinical question: Does understanding the expected clinical course of advanced dementia influence end-of-life decisions by proxy decision-makers?
Background: Advanced dementia is a leading cause of death in the United States, but the clinical course of advanced dementia has not been described in a rigorous, prospective manner. The lack of information might cause risk to be underestimated, and patients might receive suboptimal palliative care.
Study design: Multicenter prospective cohort study.
Setting: Twenty-two nursing homes in a single U.S. city.
Synopsis: The survey examined 323 nursing home residents with advanced dementia. The patients were clinically assessed at baseline and quarterly for 18 months through chart reviews, nursing interviews, and physical examinations. Additionally, their proxies were surveyed regarding their understanding of the subjects’ prognoses.
During the survey period, 41.1% of patients developed pneumonia, 52.6% of patients experienced a febrile episode, and 85.8% of patients developed an eating problem; cumulative all-cause mortality was 54.8%. Adjusted for age, sex, and disease duration, the six-month mortality rate for subjects who had pneumonia was 46.7%; a febrile episode, 44.5%; and an eating problem, 38.6%.
Distressing symptoms, including dyspnea (46.0%) and pain (39.1%), were common. In the last three months of life, 40.7% of subjects underwent at least one burdensome intervention (defined as hospitalization, ED visit, parenteral therapy, or tube feeding).
Subjects whose proxies reported an understanding of the poor prognosis and expected clinical complications of advanced dementia underwent significantly fewer burdensome interventions (adjusted odds ratio 0.12).
Bottom line: Advanced dementia is associated with frequent complications, including infections and eating problems, with high six-month mortality and significant associated morbidity. Patients whose healthcare proxies have a good understanding of the expected clinical course and prognosis receive less-aggressive end-of-life care.
Citation: Mitchell SL, Teno JM, Kiely DK, et al. The clinical course of advanced dementia. N Engl J Med. 2009;361(16):1529-1538. TH
Adding Basal Insulin to Oral Agents in Type 2 Diabetes Might Offer Best Glycemic Control
Clinical question: When added to oral diabetic agents, which insulin regimen (biphasic, prandial or basal) best achieves glycemic control in patients with Type 2 diabetes?
Background: Most patients with Type 2 diabetes mellitus (DM2) require insulin when oral agents provide suboptimal glycemic control. Little is known about which insulin regimen is most effective.
Study design: Three-year, open-label, multicenter trial.
Setting: Fifty-eight clinical centers in the United Kingdom and Ireland.
Synopsis: The authors randomized 708 insulin-naïve DM2 patients (median age 62 years) with HgbA1c 7% to 10% on maximum-dose metformin or sulfonylurea to one of three regimens: biphasic insulin twice daily; prandial insulin three times daily; or basal insulin once daily. Outcomes were HgbA1c, hypoglycemia rates, and weight gain. Sulfonylureas were replaced by another insulin if glycemic control was unacceptable.
The patients were mostly Caucasian and overweight. At three years of followup, median HgbA1c was similar in all groups (7.1% biphasic, 6.8% prandial, 6.9% basal); however, more patients who received prandial or basal insulin achieved HgbA1c less than 6.5% (45% and 43%, respectively) than in the biphasic group (32%).
Hypoglycemia was significantly less frequent in the basal insulin group (1.7 per patient per year versus 3.0 and 5.5 with biphasic and prandial, respectively). Patients gained weight in all groups; the greatest gain was with prandial insulin. At three years, there were no significant between-group differences in blood pressure, cholesterol, albuminuria, or quality of life.
Bottom line: Adding insulin to oral diabetic regimens improves glycemic control. Basal or prandial insulin regimens achieve glycemic targets more frequently than biphasic dosing.
Citation: Holman RR, Farmer AJ, Davies MJ, et al. Three-year efficacy of complex insulin regimens in type 2 diabetes. N Engl J Med. 2009;361(18):1736-1747.
Clinical question: When added to oral diabetic agents, which insulin regimen (biphasic, prandial or basal) best achieves glycemic control in patients with Type 2 diabetes?
Background: Most patients with Type 2 diabetes mellitus (DM2) require insulin when oral agents provide suboptimal glycemic control. Little is known about which insulin regimen is most effective.
Study design: Three-year, open-label, multicenter trial.
Setting: Fifty-eight clinical centers in the United Kingdom and Ireland.
Synopsis: The authors randomized 708 insulin-naïve DM2 patients (median age 62 years) with HgbA1c 7% to 10% on maximum-dose metformin or sulfonylurea to one of three regimens: biphasic insulin twice daily; prandial insulin three times daily; or basal insulin once daily. Outcomes were HgbA1c, hypoglycemia rates, and weight gain. Sulfonylureas were replaced by another insulin if glycemic control was unacceptable.
The patients were mostly Caucasian and overweight. At three years of followup, median HgbA1c was similar in all groups (7.1% biphasic, 6.8% prandial, 6.9% basal); however, more patients who received prandial or basal insulin achieved HgbA1c less than 6.5% (45% and 43%, respectively) than in the biphasic group (32%).
Hypoglycemia was significantly less frequent in the basal insulin group (1.7 per patient per year versus 3.0 and 5.5 with biphasic and prandial, respectively). Patients gained weight in all groups; the greatest gain was with prandial insulin. At three years, there were no significant between-group differences in blood pressure, cholesterol, albuminuria, or quality of life.
Bottom line: Adding insulin to oral diabetic regimens improves glycemic control. Basal or prandial insulin regimens achieve glycemic targets more frequently than biphasic dosing.
Citation: Holman RR, Farmer AJ, Davies MJ, et al. Three-year efficacy of complex insulin regimens in type 2 diabetes. N Engl J Med. 2009;361(18):1736-1747.
Clinical question: When added to oral diabetic agents, which insulin regimen (biphasic, prandial or basal) best achieves glycemic control in patients with Type 2 diabetes?
Background: Most patients with Type 2 diabetes mellitus (DM2) require insulin when oral agents provide suboptimal glycemic control. Little is known about which insulin regimen is most effective.
Study design: Three-year, open-label, multicenter trial.
Setting: Fifty-eight clinical centers in the United Kingdom and Ireland.
Synopsis: The authors randomized 708 insulin-naïve DM2 patients (median age 62 years) with HgbA1c 7% to 10% on maximum-dose metformin or sulfonylurea to one of three regimens: biphasic insulin twice daily; prandial insulin three times daily; or basal insulin once daily. Outcomes were HgbA1c, hypoglycemia rates, and weight gain. Sulfonylureas were replaced by another insulin if glycemic control was unacceptable.
The patients were mostly Caucasian and overweight. At three years of followup, median HgbA1c was similar in all groups (7.1% biphasic, 6.8% prandial, 6.9% basal); however, more patients who received prandial or basal insulin achieved HgbA1c less than 6.5% (45% and 43%, respectively) than in the biphasic group (32%).
Hypoglycemia was significantly less frequent in the basal insulin group (1.7 per patient per year versus 3.0 and 5.5 with biphasic and prandial, respectively). Patients gained weight in all groups; the greatest gain was with prandial insulin. At three years, there were no significant between-group differences in blood pressure, cholesterol, albuminuria, or quality of life.
Bottom line: Adding insulin to oral diabetic regimens improves glycemic control. Basal or prandial insulin regimens achieve glycemic targets more frequently than biphasic dosing.
Citation: Holman RR, Farmer AJ, Davies MJ, et al. Three-year efficacy of complex insulin regimens in type 2 diabetes. N Engl J Med. 2009;361(18):1736-1747.
Initiation of Dialysis Does Not Help Maintain Functional Status in Elderly
Clinical question: Is functional status in the elderly maintained over time after initiating long-term dialysis?
Background: Quality-of-life maintenance often is used as a goal when initiating long-term dialysis in elderly patients with end-stage renal disease. More elderly patients are being offered long-term dialysis treatment. Little is known about the functional status of elderly patients on long-term dialysis.
Study design: Retrospective cohort study.
Setting: U.S. nursing homes.
Synopsis: By cross-linking data from two population-based administrative datasets, this study identified 3,702 nursing home patients (mean 73.4 years) who had started long-term dialysis and whose functional status had been assessed. Activities of daily living assessments before and at three-month intervals after dialysis initiation were compared to see if functional status was maintained.
Within three months of starting dialysis, 61% of patients had a decline in functional status or had died. By one year, only 1 in 8 patients had maintained their pre-dialysis functional status.
Decline in functional status cannot be attributed solely to dialysis because study patients were not compared to patients with chronic kidney disease who were not dialyzed. In addition, these results might not apply to all elderly patients on dialysis, as the functional status of elderly nursing home patients might differ significantly from those living at home.
Bottom line: Functional status is not maintained in most elderly nursing home patients in the first 12 months after long-term dialysis is initiated. Elderly patients considering dialysis treatment should be aware that dialysis might not help maintain functional status and quality of life.
Citation: Kurella Tamura MK, Covinsky KE, Chertow GM, Yaffe C, Landefeld CS, McCulloch CE. Functional status of elderly adults before and after initiation of dialysis. N Engl J Med. 2009;361(16):1539-1547.
Clinical question: Is functional status in the elderly maintained over time after initiating long-term dialysis?
Background: Quality-of-life maintenance often is used as a goal when initiating long-term dialysis in elderly patients with end-stage renal disease. More elderly patients are being offered long-term dialysis treatment. Little is known about the functional status of elderly patients on long-term dialysis.
Study design: Retrospective cohort study.
Setting: U.S. nursing homes.
Synopsis: By cross-linking data from two population-based administrative datasets, this study identified 3,702 nursing home patients (mean 73.4 years) who had started long-term dialysis and whose functional status had been assessed. Activities of daily living assessments before and at three-month intervals after dialysis initiation were compared to see if functional status was maintained.
Within three months of starting dialysis, 61% of patients had a decline in functional status or had died. By one year, only 1 in 8 patients had maintained their pre-dialysis functional status.
Decline in functional status cannot be attributed solely to dialysis because study patients were not compared to patients with chronic kidney disease who were not dialyzed. In addition, these results might not apply to all elderly patients on dialysis, as the functional status of elderly nursing home patients might differ significantly from those living at home.
Bottom line: Functional status is not maintained in most elderly nursing home patients in the first 12 months after long-term dialysis is initiated. Elderly patients considering dialysis treatment should be aware that dialysis might not help maintain functional status and quality of life.
Citation: Kurella Tamura MK, Covinsky KE, Chertow GM, Yaffe C, Landefeld CS, McCulloch CE. Functional status of elderly adults before and after initiation of dialysis. N Engl J Med. 2009;361(16):1539-1547.
Clinical question: Is functional status in the elderly maintained over time after initiating long-term dialysis?
Background: Quality-of-life maintenance often is used as a goal when initiating long-term dialysis in elderly patients with end-stage renal disease. More elderly patients are being offered long-term dialysis treatment. Little is known about the functional status of elderly patients on long-term dialysis.
Study design: Retrospective cohort study.
Setting: U.S. nursing homes.
Synopsis: By cross-linking data from two population-based administrative datasets, this study identified 3,702 nursing home patients (mean 73.4 years) who had started long-term dialysis and whose functional status had been assessed. Activities of daily living assessments before and at three-month intervals after dialysis initiation were compared to see if functional status was maintained.
Within three months of starting dialysis, 61% of patients had a decline in functional status or had died. By one year, only 1 in 8 patients had maintained their pre-dialysis functional status.
Decline in functional status cannot be attributed solely to dialysis because study patients were not compared to patients with chronic kidney disease who were not dialyzed. In addition, these results might not apply to all elderly patients on dialysis, as the functional status of elderly nursing home patients might differ significantly from those living at home.
Bottom line: Functional status is not maintained in most elderly nursing home patients in the first 12 months after long-term dialysis is initiated. Elderly patients considering dialysis treatment should be aware that dialysis might not help maintain functional status and quality of life.
Citation: Kurella Tamura MK, Covinsky KE, Chertow GM, Yaffe C, Landefeld CS, McCulloch CE. Functional status of elderly adults before and after initiation of dialysis. N Engl J Med. 2009;361(16):1539-1547.
Inhaled Corticosteroids Decrease Inflammation in Moderate to Severe COPD
Clinical question: Does long-term inhaled corticosteroid therapy, with and without long-acting beta-agonists, decrease airway inflammation and improve lung function in patients with moderate to severe chronic obstructive pulmonary disease (COPD)?
Background: Guideline-recommended treatment of COPD with inhaled corticosteroids and long-acting beta-agonists improves symptoms and exacerbation rates; little is known about the impact of these therapies on inflammation and long-term lung function.
Study design: Randomized, double-blind, placebo-controlled trial.
Setting: Two university medical centers in the Netherlands.
Synopsis: One hundred one steroid-naïve patients, ages 45 to 75 who were current or former smokers with moderate to severe COPD, were randomized to one of four regimens: 1) fluticasone for six months, then placebo for 24 months; 2) fluticasone for 30 months; 3) fluticasone and salmeterol for 30 months; or 4) placebo for 30 months. The primary outcome was inflammatory cell counts in bronchial biopsies/induced sputum. Secondary outcomes included postbronchodilator spirometry, methacholine hyperresponsiveness, and self-reported symptoms and health status. Patients with asthma were excluded.
Short-term fluticasone therapy decreased inflammation and improved forced expiratory volume in one second (FEV1). Long-term therapy also decreased the rate of FEV1 decline, reduced dyspnea, and improved health status. Discontinuation of therapy at six months led to inflammation relapse with worsened symptoms and increased rate of FEV1 decline. The addition of long-acting beta-agonists did not provide additional anti-inflammatory benefits, but it did improve FEV1 and dyspnea at six months.
Additional studies are needed to further define clinical outcomes and assess the cost benefit of these therapies.
Bottom line: Inhaled corticosteroids decrease inflammation in steroid-naïve patients with moderate to severe COPD and might decrease the rate of lung function decline. Long-acting beta-agonists do not offer additional anti-inflammatory benefit.
Citation: Lapperre TS, Snoeck-Stroband JB, Gosman MM, et al. Effect of fluticasone with and without salmeterol on pulmonary outcomes in chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med. 2009;151(8):517-527.
Clinical question: Does long-term inhaled corticosteroid therapy, with and without long-acting beta-agonists, decrease airway inflammation and improve lung function in patients with moderate to severe chronic obstructive pulmonary disease (COPD)?
Background: Guideline-recommended treatment of COPD with inhaled corticosteroids and long-acting beta-agonists improves symptoms and exacerbation rates; little is known about the impact of these therapies on inflammation and long-term lung function.
Study design: Randomized, double-blind, placebo-controlled trial.
Setting: Two university medical centers in the Netherlands.
Synopsis: One hundred one steroid-naïve patients, ages 45 to 75 who were current or former smokers with moderate to severe COPD, were randomized to one of four regimens: 1) fluticasone for six months, then placebo for 24 months; 2) fluticasone for 30 months; 3) fluticasone and salmeterol for 30 months; or 4) placebo for 30 months. The primary outcome was inflammatory cell counts in bronchial biopsies/induced sputum. Secondary outcomes included postbronchodilator spirometry, methacholine hyperresponsiveness, and self-reported symptoms and health status. Patients with asthma were excluded.
Short-term fluticasone therapy decreased inflammation and improved forced expiratory volume in one second (FEV1). Long-term therapy also decreased the rate of FEV1 decline, reduced dyspnea, and improved health status. Discontinuation of therapy at six months led to inflammation relapse with worsened symptoms and increased rate of FEV1 decline. The addition of long-acting beta-agonists did not provide additional anti-inflammatory benefits, but it did improve FEV1 and dyspnea at six months.
Additional studies are needed to further define clinical outcomes and assess the cost benefit of these therapies.
Bottom line: Inhaled corticosteroids decrease inflammation in steroid-naïve patients with moderate to severe COPD and might decrease the rate of lung function decline. Long-acting beta-agonists do not offer additional anti-inflammatory benefit.
Citation: Lapperre TS, Snoeck-Stroband JB, Gosman MM, et al. Effect of fluticasone with and without salmeterol on pulmonary outcomes in chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med. 2009;151(8):517-527.
Clinical question: Does long-term inhaled corticosteroid therapy, with and without long-acting beta-agonists, decrease airway inflammation and improve lung function in patients with moderate to severe chronic obstructive pulmonary disease (COPD)?
Background: Guideline-recommended treatment of COPD with inhaled corticosteroids and long-acting beta-agonists improves symptoms and exacerbation rates; little is known about the impact of these therapies on inflammation and long-term lung function.
Study design: Randomized, double-blind, placebo-controlled trial.
Setting: Two university medical centers in the Netherlands.
Synopsis: One hundred one steroid-naïve patients, ages 45 to 75 who were current or former smokers with moderate to severe COPD, were randomized to one of four regimens: 1) fluticasone for six months, then placebo for 24 months; 2) fluticasone for 30 months; 3) fluticasone and salmeterol for 30 months; or 4) placebo for 30 months. The primary outcome was inflammatory cell counts in bronchial biopsies/induced sputum. Secondary outcomes included postbronchodilator spirometry, methacholine hyperresponsiveness, and self-reported symptoms and health status. Patients with asthma were excluded.
Short-term fluticasone therapy decreased inflammation and improved forced expiratory volume in one second (FEV1). Long-term therapy also decreased the rate of FEV1 decline, reduced dyspnea, and improved health status. Discontinuation of therapy at six months led to inflammation relapse with worsened symptoms and increased rate of FEV1 decline. The addition of long-acting beta-agonists did not provide additional anti-inflammatory benefits, but it did improve FEV1 and dyspnea at six months.
Additional studies are needed to further define clinical outcomes and assess the cost benefit of these therapies.
Bottom line: Inhaled corticosteroids decrease inflammation in steroid-naïve patients with moderate to severe COPD and might decrease the rate of lung function decline. Long-acting beta-agonists do not offer additional anti-inflammatory benefit.
Citation: Lapperre TS, Snoeck-Stroband JB, Gosman MM, et al. Effect of fluticasone with and without salmeterol on pulmonary outcomes in chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med. 2009;151(8):517-527.
Resident Fatigue and Distress Contribute to Perceived Medical Errors
Clinical question: Do resident fatigue and distress contribute to medical errors?
Background: In recent years, such measures as work-hour limitations have been implemented to decrease resident fatigue and, it is presumed, medical errors. However, few studies address the relationship between residents’ well-being and self-reported medical errors.
Study design: Prospective six-year longitudinal cohort study.
Setting: Single academic medical center.
Synopsis: The authors had 380 internal-medicine residents complete quarterly surveys to assess fatigue, quality of life, burnout, symptoms of depression, and frequency of perceived medical errors. In a univariate analysis, fatigue/sleepiness, burnout, depression, and overall quality of life measures correlated significantly with self-reported major medical errors. Fatigue/sleepiness and measures of distress additively increased the risk of self-reported errors. Increases in one or both domains were estimated to increase the risk of self-reported errors by as much as 15% to 28%.
The authors studied only self-reported medical errors. It is difficult to know whether these errors directly affected patient outcomes. Additionally, results of this single-site study might not be able to be generalized.
Bottom line: Fatigue and distress contribute to self-perceived medical errors among residents.
Citation: West CP, Tan AD, Habermann TM, Sloan JA, Shanafelt TD. Association of resident fatigue and distress with perceived medical errors. JAMA. 2009;302(12):1294-1300.
Clinical question: Do resident fatigue and distress contribute to medical errors?
Background: In recent years, such measures as work-hour limitations have been implemented to decrease resident fatigue and, it is presumed, medical errors. However, few studies address the relationship between residents’ well-being and self-reported medical errors.
Study design: Prospective six-year longitudinal cohort study.
Setting: Single academic medical center.
Synopsis: The authors had 380 internal-medicine residents complete quarterly surveys to assess fatigue, quality of life, burnout, symptoms of depression, and frequency of perceived medical errors. In a univariate analysis, fatigue/sleepiness, burnout, depression, and overall quality of life measures correlated significantly with self-reported major medical errors. Fatigue/sleepiness and measures of distress additively increased the risk of self-reported errors. Increases in one or both domains were estimated to increase the risk of self-reported errors by as much as 15% to 28%.
The authors studied only self-reported medical errors. It is difficult to know whether these errors directly affected patient outcomes. Additionally, results of this single-site study might not be able to be generalized.
Bottom line: Fatigue and distress contribute to self-perceived medical errors among residents.
Citation: West CP, Tan AD, Habermann TM, Sloan JA, Shanafelt TD. Association of resident fatigue and distress with perceived medical errors. JAMA. 2009;302(12):1294-1300.
Clinical question: Do resident fatigue and distress contribute to medical errors?
Background: In recent years, such measures as work-hour limitations have been implemented to decrease resident fatigue and, it is presumed, medical errors. However, few studies address the relationship between residents’ well-being and self-reported medical errors.
Study design: Prospective six-year longitudinal cohort study.
Setting: Single academic medical center.
Synopsis: The authors had 380 internal-medicine residents complete quarterly surveys to assess fatigue, quality of life, burnout, symptoms of depression, and frequency of perceived medical errors. In a univariate analysis, fatigue/sleepiness, burnout, depression, and overall quality of life measures correlated significantly with self-reported major medical errors. Fatigue/sleepiness and measures of distress additively increased the risk of self-reported errors. Increases in one or both domains were estimated to increase the risk of self-reported errors by as much as 15% to 28%.
The authors studied only self-reported medical errors. It is difficult to know whether these errors directly affected patient outcomes. Additionally, results of this single-site study might not be able to be generalized.
Bottom line: Fatigue and distress contribute to self-perceived medical errors among residents.
Citation: West CP, Tan AD, Habermann TM, Sloan JA, Shanafelt TD. Association of resident fatigue and distress with perceived medical errors. JAMA. 2009;302(12):1294-1300.
Dabigatran Is Not Inferior to Warfarin in Atrial Fibrillation
Clinical question: Is dabigatran, an oral thrombin inhibitor, an effective and safe alternative to warfarin in patients with atrial fibrillation?
Background: Warfarin reduces the risk of stroke among patients with atrial fibrillation (AF) but requires frequent laboratory monitoring. Dabigatran is an oral direct thrombin inhibitor given in fixed dosages without laboratory monitoring.
Study design: Randomized, multicenter, open-label, noninferiority trial.
Setting: 951 clinical centers in 44 countries.
Synopsis: More than 18,000 patients 65 and older with AF and at least one stroke risk factor were enrolled. The average CHADS2 score was 2.1. Patients were randomized to receive fixed doses of dabigatran (110 mg or 150 mg, twice daily) or warfarin adjusted to an INR of 2.0-3.0. The primary outcomes were a) stroke or systemic embolism and b) major hemorrhage. Median followup was two years.
The annual rates of stroke or systemic embolism for both doses of dabigatran were noninferior to warfarin (P<0.001); higher-dose dabigatran was statistically superior to warfarin (relative risk (RR)=0.66, P<0.001). The annual rate of major hemorrhage was lowest in the lower-dose dabigatran group (RR=0.80, P=0.003 compared with warfarin); the higher-dose dabigatran and warfarin groups had equivalent rates of major bleeding. No increased risk of liver function abnormalities was noted.
Bottom line: Dabigatran appears to be an effective and safe alternative to warfarin in AF patients. If the drug were to be FDA-approved, appropriate patient selection and cost will need to be established.
Citation: Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361(12):1139-1151.
Clinical question: Is dabigatran, an oral thrombin inhibitor, an effective and safe alternative to warfarin in patients with atrial fibrillation?
Background: Warfarin reduces the risk of stroke among patients with atrial fibrillation (AF) but requires frequent laboratory monitoring. Dabigatran is an oral direct thrombin inhibitor given in fixed dosages without laboratory monitoring.
Study design: Randomized, multicenter, open-label, noninferiority trial.
Setting: 951 clinical centers in 44 countries.
Synopsis: More than 18,000 patients 65 and older with AF and at least one stroke risk factor were enrolled. The average CHADS2 score was 2.1. Patients were randomized to receive fixed doses of dabigatran (110 mg or 150 mg, twice daily) or warfarin adjusted to an INR of 2.0-3.0. The primary outcomes were a) stroke or systemic embolism and b) major hemorrhage. Median followup was two years.
The annual rates of stroke or systemic embolism for both doses of dabigatran were noninferior to warfarin (P<0.001); higher-dose dabigatran was statistically superior to warfarin (relative risk (RR)=0.66, P<0.001). The annual rate of major hemorrhage was lowest in the lower-dose dabigatran group (RR=0.80, P=0.003 compared with warfarin); the higher-dose dabigatran and warfarin groups had equivalent rates of major bleeding. No increased risk of liver function abnormalities was noted.
Bottom line: Dabigatran appears to be an effective and safe alternative to warfarin in AF patients. If the drug were to be FDA-approved, appropriate patient selection and cost will need to be established.
Citation: Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361(12):1139-1151.
Clinical question: Is dabigatran, an oral thrombin inhibitor, an effective and safe alternative to warfarin in patients with atrial fibrillation?
Background: Warfarin reduces the risk of stroke among patients with atrial fibrillation (AF) but requires frequent laboratory monitoring. Dabigatran is an oral direct thrombin inhibitor given in fixed dosages without laboratory monitoring.
Study design: Randomized, multicenter, open-label, noninferiority trial.
Setting: 951 clinical centers in 44 countries.
Synopsis: More than 18,000 patients 65 and older with AF and at least one stroke risk factor were enrolled. The average CHADS2 score was 2.1. Patients were randomized to receive fixed doses of dabigatran (110 mg or 150 mg, twice daily) or warfarin adjusted to an INR of 2.0-3.0. The primary outcomes were a) stroke or systemic embolism and b) major hemorrhage. Median followup was two years.
The annual rates of stroke or systemic embolism for both doses of dabigatran were noninferior to warfarin (P<0.001); higher-dose dabigatran was statistically superior to warfarin (relative risk (RR)=0.66, P<0.001). The annual rate of major hemorrhage was lowest in the lower-dose dabigatran group (RR=0.80, P=0.003 compared with warfarin); the higher-dose dabigatran and warfarin groups had equivalent rates of major bleeding. No increased risk of liver function abnormalities was noted.
Bottom line: Dabigatran appears to be an effective and safe alternative to warfarin in AF patients. If the drug were to be FDA-approved, appropriate patient selection and cost will need to be established.
Citation: Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361(12):1139-1151.