User login
How to adapt cognitive-behavioral therapy for older adults
Some older patients with depression, anxiety, or insomnia may be reluctant to turn to pharmacotherapy and may prefer psychotherapeutic treatments.1 Evidence has established cognitive-behavioral therapy (CBT) as an effective intervention for several psychiatric disorders and CBT should be considered when treating geriatric patients (Table 1).2
Table 1
Indications for CBT
| Mild to moderate depression. In the case of severe depression, CBT can be combined with pharmacotherapy |
| Anxiety disorders, mixed anxiety states |
| Insomnia—both primary and comorbid with other medical and/or psychiatric conditions |
| CBT: cognitive-behavioral therapy |
Research evaluating the efficacy of CBT for depression in older adults was first published in the early 1980s. Since then, research and application of CBT with older adults has expanded to include other psychiatric disorders and researchers have suggested changes to increase the efficacy of CBT for these patients. This article provides:
- an overview of CBT’s efficacy for older adults with depression, anxiety, and insomnia
- modifications to employ when providing CBT to older patients.
The cognitive model of CBT
In the 1970s, Aaron T. Beck, MD, developed CBT while working with depressed patients. Beck’s patients reported thoughts characterized by inaccuracies and distortions in association with their depressed mood. He found these thoughts could be brought to the patient’s conscious attention and modified to improve the patient’s depression. This finding led to the development of CBT.
CBT is based on a cognitive model of the relationship among cognition, emotion, and behavior. Mood and behavior are viewed as determined by a person’s perception and interpretation of events, which manifest as a stream of automatically generated thoughts (Figure).3 These automatic thoughts have their origins in an underlying network of beliefs or schema. Patients with psychiatric disorders such as anxiety and depression typically have frequent automatic thoughts that characteristically lack validity because they arise from dysfunctional beliefs. The therapeutic process consists of helping the patient become aware of his or her internal stream of thoughts when distressed, and to identify and modify the dysfunctional thoughts. Behavioral techniques are used to bring about functional changes in behavior, regulate emotion, and help the cognitive restructuring process. Modifying the patient’s underlying dysfunctional beliefs leads to lasting improvements. In this structured therapy, the therapist and patient work collaboratively to use an approach that features reality testing and experimentation.4
Figure
The cognitive model of CBT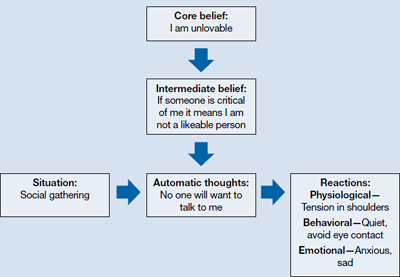
CBT: cognitive-behavioral therapy
Source: Adapted from reference 3
Indications for CBT in older adults
Depression. Among psychotherapies used in older adults, CBT has received the most research for late-life depression.5 Randomized controlled trials (RCTs) have found CBT is superior to treatment as usual in depressed adults age ≥60.6 It also has been found to be superior to wait-list control7 and talking as control.6,8 Meta-analyses have shown above-average effect sizes for CBT in treating late-life depression.9,10 A follow-up study found improvement was maintained up to 2 years after CBT, which suggests CBT’s impact is likely to be long lasting.11
Thompson et al12 compared 102 depressed patients age >60 who were treated with CBT alone, desipramine alone, or a combination of the 2. A combination of medication and CBT worked best for severely depressed patients; CBT alone or a combination of CBT and medication worked best for moderately depressed patients.
CBT is an option when treating depressed medically ill older adults. Research indicates that CBT could reduce depression in older patients with Parkinson’s disease13 and chronic obstructive pulmonary disease.14
As patients get older, cognitive impairment with comorbid depression can make treatment challenging. Limited research suggests CBT applied in a modified format that involves caregivers and uses problem solving and behavioral strategies can significantly reduce depression in patients with dementia.15
Anxiety. Researchers have examined the efficacy of variants of CBT in treating older adults with anxiety disorders—commonly, generalized anxiety disorder (GAD), panic disorder, agoraphobia, subjective anxiety, or a combination of these illnesses.16,17 Randomized trials have supported CBT’s efficacy for older patients with GAD and mixed anxiety states; gains made in CBT were maintained over a 1-year follow-up.18,19 In a meta-analysis of 15 studies using cognitive and behavioral methods of treating anxiety in older patients, Nordhus and Pallesen16 reported a significant effect size of 0.55. In a 2008 meta-analysis that included only RCTs, CBT was superior to wait-list conditions as well as active control conditions in treating anxious older patients.20
However, some research suggests that CBT for GAD may not be as effective for older adults as it is for younger adults. In a study of CBT for GAD in older adults, Stanley et al19 reported smaller effect sizes compared with CBT for younger adults. Researchers have found relatively few differences between CBT and comparison conditions—supportive psychotherapy or active control conditions—in treating GAD in older adults.21 Modified, more effective formats of CBT for GAD in older adults need to be established.22 Mohlman et al23 supplemented standard CBT for late-life GAD with memory and learning aids—weekly reading assignments, graphing exercises to chart mood ratings, reminder phone calls from therapists, and homework compliance requirement. This approach improved the response rate from 40% to 75%.23
Insomnia. Studies have found CBT to be an effective means of treating insomnia in geriatric patients. Although sleep problems occur more frequently among older patients, only 15% of chronic insomnia patients receive treatment; psychotherapy rarely is used.24 CBT for insomnia (CBT-I) should be considered for older adults because managing insomnia with medications may be problematic and these patients may prefer nonpharmacologic treatment.2 CBT-I typically incorporates cognitive strategies with established behavioral techniques, including sleep hygiene education, cognitive restructuring, relaxation training, stimulus control, and/or sleep restriction. The CBT-I multicomponent treatment package meets all criteria to be considered an evidence-based treatment for late-life insomnia.25
RCTs have reported significant improvements in late-life insomnia with CBT-I.26,27 Reviews and meta-analyses have also concluded that cognitive-behavioral treatments are effective for treating insomnia in older adults.25,28 Most insomnia cases in geriatric patients are reported to occur secondary to other medical or psychiatric conditions that are judged as causing the insomnia.25 In these cases, direct treatment of the insomnia usually is delayed or omitted.28 Studies evaluating the efficacy of CBT packages for treating insomnia occurring in conjunction with other medical or psychiatric illnesses have reported significant improvement of insomnia.28,29 Because insomnia frequently occurs in older patients with medical illnesses and psychiatric disorders, CBT-I could be beneficial for such patients.
Good candidates for CBT
Clinical experience indicates that older adults in relatively good health with no significant cognitive decline are good candidates for CBT. These patients tend to comply with their assignments, are interested in applying the learned strategies, and are motivated to read self-help books. CBT’s structured, goal-oriented approach makes it a short-term treatment, which makes it cost effective. Insomnia patients may improve after 6 to 8 CBT-I sessions and patients with anxiety or depression may need to undergo 15 to 20 CBT sessions. Patients age ≥65 have basic Medicare coverage that includes mental health care and psychotherapy.
There are no absolute contraindications for CBT, but the greater the cognitive impairment, the less the patient will benefit from CBT (Table 2). Similarly, severe depression and anxiety might make it difficult for patients to participate meaningfully, although CBT may be incorporated gradually as patients improve with medication. Severe medical illnesses and sensory losses such as visual and hearing loss would make it difficult to carry out CBT effectively.
Table 2
Contraindications for CBT
| High levels of cognitive impairment |
| Severe depression with psychotic features |
| Severe anxiety with high levels of agitation |
| Severe medical illness |
| Sensory losses |
| CBT: cognitive-behavioral therapy |
Adapting CBT for older patients
When using CBT with older patients, it is important to keep in mind characteristics that define the geriatric population. Laidlaw et al30 developed a model to help clinicians develop a more appropriate conceptualization of older patients that focuses on significant events and related cognitions associated with physical health, changes in role investments, and interactions with younger generations. It emphasizes the need to explore beliefs about aging viewed through each patient’s socio-cultural lens and examine cognitions in the context of the time period in which the individual has lived.
Losses and transitions. For many older patients, the latter years of life are characterized by losses and transitions.31 According to Thompson,31 these losses and transitions can trigger thoughts of missed opportunities or unresolved relationships and reflection on unachieved goals.31 CBT for older adults should focus on the meaning the patient gives to these losses and transitions. For example, depressed patients could view their retirement as a loss of self worth as they become less productive. CBT can help patients identify ways of thinking about the situation that will enable them to adapt to these losses and transitions.
Changes in cognition. Changes in cognitive functioning with aging are not universal and there’s considerable variability, but it’s important to make appropriate adaptations when needed. Patients may experience a decline in cognitive speed, working memory, selective attention, and fluid intelligence. This would require that information be presented slowly, with frequent repetitions and summaries. Also, it might be helpful to present information in alternate ways and to encourage patients to take notes during sessions. To accommodate for a decline in fluid intelligence, presenting new information in the context of previous experiences will help promote learning. Recordings of important information and conclusions from cognitive restructuring that patients can listen to between sessions could serve as helpful reminders that will help patients progress. Phone prompts or alarms can remind patients to carry out certain therapeutic measures, such as breathing exercises. Caretakers can attend sessions to become familiar with strategies performed during CBT and act as a co-therapist at home; however, their inclusion must be done with the consent of both parties and only if it’s viewed as necessary for the patient’s progress.
Additional strategies. For patients with substantial cognitive decline, cognitive restructuring might not be as effective as behavioral strategies—activity scheduling, graded task assignment, graded exposure, and rehearsals. Because older adults often have strengthened dysfunctional beliefs over a long time, modifying them takes longer, which is why the tapering process usually takes longer for older patients than for younger patients. The lengthier tapering ensures learning is well established and the process of modifying dysfunctional beliefs to functional beliefs continues. Collaborating with other professionals—physicians, social workers, and case managers—will help ensure a shared care process in which common goals are met.
The websites of the Academy of Cognitive Therapy, American Psychological Association, and Association for Behavioral and Cognitive Therapies can help clinicians who do not offer CBT to locate a qualified therapist for their patients (Related Resources).
- Academy of Cognitive Therapy. www.academyofct.org.
- American Psychological Association. www.apa.org.
- Association for Behavioral and Cognitive Therapies. www.abct.org.
- Laidlaw K, Thompson LW, Dick-Siskin L, et al. Cognitive behaviour therapy with older people. West Sussex, England: John Wiley & Sons, Ltd; 2003.
Drug Brand Name
- Desipramine • Norpramin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Landreville P, Landry J, Baillargeon L, et al. Older adults’ acceptance of psychological and pharmacological treatments for depression. J Gerontol B Psychol Sci Soc Sci. 2001;56(5):P285-P291.
2. Chambless DL, Ollendick TH. Empirically supported psychological interventions: controversies and evidence. Annu Rev Psychol. 2001;52:685-716.
3. Beck JS. Cognitive conceptualization. In: Cognitive therapy: basics and beyond. 2nd ed. New York NY: The Guilford Press; 2011:29–45.
4. Beck AT, Rush AJ, Shaw BF, et al. Cognitive therapy of depression. New York, NY: The Guilford Press; 1979.
5. Areán PA, Cook BL. Psychotherapy and combined psychotherapy/pharmacotherapy for late-life depression. Biol Psychiatry. 2002;52(3):293-303.
6. Laidlaw K, Davidson K, Toner H, et al. A randomised controlled trial of cognitive behaviour therapy vs treatment as usual in the treatment of mild to moderate late-life depression. Int J Geriatr Psychiatry. 2008;23(8):843-850.
7. Floyd M, Scogin F, McKendree-Smith NL, et al. Cognitive therapy for depression: a comparison of individual psychotherapy and bibliotherapy for depressed older adults. Behavior Modification. 2004;28(2):297-318.
8. Serfaty MA, Haworth D, Blanchard M, et al. Clinical effectiveness of individual cognitive behavioral therapy for depressed older people in primary care: a randomized controlled trial. Arch Gen Psychiatry. 2009;66(12):1332-1340.
9. Pinquart M, Sörensen S. How effective are psychotherapeutic and other psychosocial interventions with older adults? A meta-analysis. J Ment Health Aging. 2001;7(2):207-243.
10. Pinquart M, Duberstein PR, Lyness JM. Effects of psychotherapy and other behavioral interventions on clinically depressed older adults: a meta-analysis. Aging Ment Health. 2007;11(6):645-657.
11. Gallagher-Thompson D, Hanley-Peterson P, Thompson LW. Maintenance of gains versus relapse following brief psychotherapy for depression. J Consult Clin Psychol. 1990;58(3):371-374.
12. Thompson LW, Coon DW, Gallagher-Thompson D, et al. Comparison of desipramine and cognitive/behavioral therapy in the treatment of elderly outpatients with mild-to-moderate depression. Am J Geriatr Psychiatry. 2001;9(3):225-240.
13. Dobkin RD, Menza M, Allen LA, et al. Cognitive-behavioral therapy for depression in Parkinson’s disease: a randomized, controlled trial. Am J Psychiatry. 2011;168(10):1066-1074.
14. Kunik ME, Braun U, Stanley MA, et al. One session cognitive behavioural therapy for elderly patients with chronic obstructive pulmonary disease. Psychol Med. 2001;31(4):717-723.
15. Teri L, Logsdon RG, Uomoto J, et al. Behavioral treatment of depression in dementia patients: a controlled clinical trial. J Gerontol B Psychol Sci Soc Sci. 1997;52(4):P159-P166.
16. Nordhus IH, Pallesen S. Psychological treatment of late-life anxiety: an empirical review. J Consult Clin Psychol. 2003;71(4):643-651.
17. Gorenstein EE, Papp LA. Cognitive-behavioral therapy for anxiety in the elderly. Curr Psychiatry Rep. 2007;9(1):20-25.
18. Barrowclough C, King P, Colville J, et al. A randomized trial of the effectiveness of cognitive-behavioral therapy and supportive counseling for anxiety symptoms in older adults. J Consult Clin Psychol. 2001;69(5):756-762.
19. Stanley MA, Beck JG, Novy DM, et al. Cognitive-behavioral treatment of late-life generalized anxiety disorder. J Consult Clin Psychol. 2003;71(2):309-319.
20. Hendriks GJ, Oude Voshaar RC, Keijsers GP, et al. Cognitive-behavioural therapy for late-life anxiety disorders: a systematic review and meta-analysis. Acta Psychiatr Scand. 2008;117(6):403-411.
21. Wetherell JL, Gatz M, Craske MG. Treatment of generalized anxiety disorder in older adults. J Consult Clin Psychol. 2003;71(1):31-40.
22. Dugas MJ, Brillon P, Savard P, et al. A randomized clinical trial of cognitive-behavioral therapy and applied relaxation for adults with generalized anxiety disorder. Behav Ther. 2010;41(1):46-58.
23. Mohlman J, Gorenstein EE, Kleber M, et al. Standard and enhanced cognitive-behavior therapy for late-life generalized anxiety disorder: two pilot investigations. Am J Geriatr Psychiatry. 2003;11(1):24-32.
24. Flint AJ. Epidemiology and comorbidity of anxiety disorders in the elderly. Am J Psychiatry. 1994;151(5):640-649.
25. McCurry SM, Logsdon RG, Teri L, et al. Evidence-based psychological treatments for insomnia in older adults. Psychol Aging. 2007;22(1):18-27.
26. Sivertsen B, Omvik S, Pallesen S, et al. Cognitive behavioral therapy vs zopiclone for treatment of chronic primary insomnia in older adults: a randomized controlled trial. JAMA. 2006;295(24):2851-2858.
27. Morgan K, Dixon S, Mathers N, et al. Psychological treatment for insomnia in the regulation of long-term hypnotic drug use. Health Technol Assess. 2004;8(8):iii iv, 1-68.
28. Nau SD, McCrae CS, Cook KG, et al. Treatment of insomnia in older adults. Clin Psychol Rev. 2005;25(5):645-672.
29. Rybarczyk B, Stepanski E, Fogg L, et al. A placebo-controlled test of cognitive-behavioral therapy for comorbid insomnia in older adults. J Consult Clin Psychol. 2005;73(6):1164-1174.
30. Laidlaw K, Thompson LW, Gallagher-Thompson D. Comprehensive conceptualization of cognitive behaviour therapy for late life depression. Behav Cogn Psychother. 2004;32(4):389-399.
31. Thompson LW. Cognitive-behavioral therapy and treatment for late-life depression. J Clin Psychiatry. 1996;57(suppl 5):29-37.
Some older patients with depression, anxiety, or insomnia may be reluctant to turn to pharmacotherapy and may prefer psychotherapeutic treatments.1 Evidence has established cognitive-behavioral therapy (CBT) as an effective intervention for several psychiatric disorders and CBT should be considered when treating geriatric patients (Table 1).2
Table 1
Indications for CBT
| Mild to moderate depression. In the case of severe depression, CBT can be combined with pharmacotherapy |
| Anxiety disorders, mixed anxiety states |
| Insomnia—both primary and comorbid with other medical and/or psychiatric conditions |
| CBT: cognitive-behavioral therapy |
Research evaluating the efficacy of CBT for depression in older adults was first published in the early 1980s. Since then, research and application of CBT with older adults has expanded to include other psychiatric disorders and researchers have suggested changes to increase the efficacy of CBT for these patients. This article provides:
- an overview of CBT’s efficacy for older adults with depression, anxiety, and insomnia
- modifications to employ when providing CBT to older patients.
The cognitive model of CBT
In the 1970s, Aaron T. Beck, MD, developed CBT while working with depressed patients. Beck’s patients reported thoughts characterized by inaccuracies and distortions in association with their depressed mood. He found these thoughts could be brought to the patient’s conscious attention and modified to improve the patient’s depression. This finding led to the development of CBT.
CBT is based on a cognitive model of the relationship among cognition, emotion, and behavior. Mood and behavior are viewed as determined by a person’s perception and interpretation of events, which manifest as a stream of automatically generated thoughts (Figure).3 These automatic thoughts have their origins in an underlying network of beliefs or schema. Patients with psychiatric disorders such as anxiety and depression typically have frequent automatic thoughts that characteristically lack validity because they arise from dysfunctional beliefs. The therapeutic process consists of helping the patient become aware of his or her internal stream of thoughts when distressed, and to identify and modify the dysfunctional thoughts. Behavioral techniques are used to bring about functional changes in behavior, regulate emotion, and help the cognitive restructuring process. Modifying the patient’s underlying dysfunctional beliefs leads to lasting improvements. In this structured therapy, the therapist and patient work collaboratively to use an approach that features reality testing and experimentation.4
Figure
The cognitive model of CBT
CBT: cognitive-behavioral therapy
Source: Adapted from reference 3
Indications for CBT in older adults
Depression. Among psychotherapies used in older adults, CBT has received the most research for late-life depression.5 Randomized controlled trials (RCTs) have found CBT is superior to treatment as usual in depressed adults age ≥60.6 It also has been found to be superior to wait-list control7 and talking as control.6,8 Meta-analyses have shown above-average effect sizes for CBT in treating late-life depression.9,10 A follow-up study found improvement was maintained up to 2 years after CBT, which suggests CBT’s impact is likely to be long lasting.11
Thompson et al12 compared 102 depressed patients age >60 who were treated with CBT alone, desipramine alone, or a combination of the 2. A combination of medication and CBT worked best for severely depressed patients; CBT alone or a combination of CBT and medication worked best for moderately depressed patients.
CBT is an option when treating depressed medically ill older adults. Research indicates that CBT could reduce depression in older patients with Parkinson’s disease13 and chronic obstructive pulmonary disease.14
As patients get older, cognitive impairment with comorbid depression can make treatment challenging. Limited research suggests CBT applied in a modified format that involves caregivers and uses problem solving and behavioral strategies can significantly reduce depression in patients with dementia.15
Anxiety. Researchers have examined the efficacy of variants of CBT in treating older adults with anxiety disorders—commonly, generalized anxiety disorder (GAD), panic disorder, agoraphobia, subjective anxiety, or a combination of these illnesses.16,17 Randomized trials have supported CBT’s efficacy for older patients with GAD and mixed anxiety states; gains made in CBT were maintained over a 1-year follow-up.18,19 In a meta-analysis of 15 studies using cognitive and behavioral methods of treating anxiety in older patients, Nordhus and Pallesen16 reported a significant effect size of 0.55. In a 2008 meta-analysis that included only RCTs, CBT was superior to wait-list conditions as well as active control conditions in treating anxious older patients.20
However, some research suggests that CBT for GAD may not be as effective for older adults as it is for younger adults. In a study of CBT for GAD in older adults, Stanley et al19 reported smaller effect sizes compared with CBT for younger adults. Researchers have found relatively few differences between CBT and comparison conditions—supportive psychotherapy or active control conditions—in treating GAD in older adults.21 Modified, more effective formats of CBT for GAD in older adults need to be established.22 Mohlman et al23 supplemented standard CBT for late-life GAD with memory and learning aids—weekly reading assignments, graphing exercises to chart mood ratings, reminder phone calls from therapists, and homework compliance requirement. This approach improved the response rate from 40% to 75%.23
Insomnia. Studies have found CBT to be an effective means of treating insomnia in geriatric patients. Although sleep problems occur more frequently among older patients, only 15% of chronic insomnia patients receive treatment; psychotherapy rarely is used.24 CBT for insomnia (CBT-I) should be considered for older adults because managing insomnia with medications may be problematic and these patients may prefer nonpharmacologic treatment.2 CBT-I typically incorporates cognitive strategies with established behavioral techniques, including sleep hygiene education, cognitive restructuring, relaxation training, stimulus control, and/or sleep restriction. The CBT-I multicomponent treatment package meets all criteria to be considered an evidence-based treatment for late-life insomnia.25
RCTs have reported significant improvements in late-life insomnia with CBT-I.26,27 Reviews and meta-analyses have also concluded that cognitive-behavioral treatments are effective for treating insomnia in older adults.25,28 Most insomnia cases in geriatric patients are reported to occur secondary to other medical or psychiatric conditions that are judged as causing the insomnia.25 In these cases, direct treatment of the insomnia usually is delayed or omitted.28 Studies evaluating the efficacy of CBT packages for treating insomnia occurring in conjunction with other medical or psychiatric illnesses have reported significant improvement of insomnia.28,29 Because insomnia frequently occurs in older patients with medical illnesses and psychiatric disorders, CBT-I could be beneficial for such patients.
Good candidates for CBT
Clinical experience indicates that older adults in relatively good health with no significant cognitive decline are good candidates for CBT. These patients tend to comply with their assignments, are interested in applying the learned strategies, and are motivated to read self-help books. CBT’s structured, goal-oriented approach makes it a short-term treatment, which makes it cost effective. Insomnia patients may improve after 6 to 8 CBT-I sessions and patients with anxiety or depression may need to undergo 15 to 20 CBT sessions. Patients age ≥65 have basic Medicare coverage that includes mental health care and psychotherapy.
There are no absolute contraindications for CBT, but the greater the cognitive impairment, the less the patient will benefit from CBT (Table 2). Similarly, severe depression and anxiety might make it difficult for patients to participate meaningfully, although CBT may be incorporated gradually as patients improve with medication. Severe medical illnesses and sensory losses such as visual and hearing loss would make it difficult to carry out CBT effectively.
Table 2
Contraindications for CBT
| High levels of cognitive impairment |
| Severe depression with psychotic features |
| Severe anxiety with high levels of agitation |
| Severe medical illness |
| Sensory losses |
| CBT: cognitive-behavioral therapy |
Adapting CBT for older patients
When using CBT with older patients, it is important to keep in mind characteristics that define the geriatric population. Laidlaw et al30 developed a model to help clinicians develop a more appropriate conceptualization of older patients that focuses on significant events and related cognitions associated with physical health, changes in role investments, and interactions with younger generations. It emphasizes the need to explore beliefs about aging viewed through each patient’s socio-cultural lens and examine cognitions in the context of the time period in which the individual has lived.
Losses and transitions. For many older patients, the latter years of life are characterized by losses and transitions.31 According to Thompson,31 these losses and transitions can trigger thoughts of missed opportunities or unresolved relationships and reflection on unachieved goals.31 CBT for older adults should focus on the meaning the patient gives to these losses and transitions. For example, depressed patients could view their retirement as a loss of self worth as they become less productive. CBT can help patients identify ways of thinking about the situation that will enable them to adapt to these losses and transitions.
Changes in cognition. Changes in cognitive functioning with aging are not universal and there’s considerable variability, but it’s important to make appropriate adaptations when needed. Patients may experience a decline in cognitive speed, working memory, selective attention, and fluid intelligence. This would require that information be presented slowly, with frequent repetitions and summaries. Also, it might be helpful to present information in alternate ways and to encourage patients to take notes during sessions. To accommodate for a decline in fluid intelligence, presenting new information in the context of previous experiences will help promote learning. Recordings of important information and conclusions from cognitive restructuring that patients can listen to between sessions could serve as helpful reminders that will help patients progress. Phone prompts or alarms can remind patients to carry out certain therapeutic measures, such as breathing exercises. Caretakers can attend sessions to become familiar with strategies performed during CBT and act as a co-therapist at home; however, their inclusion must be done with the consent of both parties and only if it’s viewed as necessary for the patient’s progress.
Additional strategies. For patients with substantial cognitive decline, cognitive restructuring might not be as effective as behavioral strategies—activity scheduling, graded task assignment, graded exposure, and rehearsals. Because older adults often have strengthened dysfunctional beliefs over a long time, modifying them takes longer, which is why the tapering process usually takes longer for older patients than for younger patients. The lengthier tapering ensures learning is well established and the process of modifying dysfunctional beliefs to functional beliefs continues. Collaborating with other professionals—physicians, social workers, and case managers—will help ensure a shared care process in which common goals are met.
The websites of the Academy of Cognitive Therapy, American Psychological Association, and Association for Behavioral and Cognitive Therapies can help clinicians who do not offer CBT to locate a qualified therapist for their patients (Related Resources).
- Academy of Cognitive Therapy. www.academyofct.org.
- American Psychological Association. www.apa.org.
- Association for Behavioral and Cognitive Therapies. www.abct.org.
- Laidlaw K, Thompson LW, Dick-Siskin L, et al. Cognitive behaviour therapy with older people. West Sussex, England: John Wiley & Sons, Ltd; 2003.
Drug Brand Name
- Desipramine • Norpramin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Some older patients with depression, anxiety, or insomnia may be reluctant to turn to pharmacotherapy and may prefer psychotherapeutic treatments.1 Evidence has established cognitive-behavioral therapy (CBT) as an effective intervention for several psychiatric disorders and CBT should be considered when treating geriatric patients (Table 1).2
Table 1
Indications for CBT
| Mild to moderate depression. In the case of severe depression, CBT can be combined with pharmacotherapy |
| Anxiety disorders, mixed anxiety states |
| Insomnia—both primary and comorbid with other medical and/or psychiatric conditions |
| CBT: cognitive-behavioral therapy |
Research evaluating the efficacy of CBT for depression in older adults was first published in the early 1980s. Since then, research and application of CBT with older adults has expanded to include other psychiatric disorders and researchers have suggested changes to increase the efficacy of CBT for these patients. This article provides:
- an overview of CBT’s efficacy for older adults with depression, anxiety, and insomnia
- modifications to employ when providing CBT to older patients.
The cognitive model of CBT
In the 1970s, Aaron T. Beck, MD, developed CBT while working with depressed patients. Beck’s patients reported thoughts characterized by inaccuracies and distortions in association with their depressed mood. He found these thoughts could be brought to the patient’s conscious attention and modified to improve the patient’s depression. This finding led to the development of CBT.
CBT is based on a cognitive model of the relationship among cognition, emotion, and behavior. Mood and behavior are viewed as determined by a person’s perception and interpretation of events, which manifest as a stream of automatically generated thoughts (Figure).3 These automatic thoughts have their origins in an underlying network of beliefs or schema. Patients with psychiatric disorders such as anxiety and depression typically have frequent automatic thoughts that characteristically lack validity because they arise from dysfunctional beliefs. The therapeutic process consists of helping the patient become aware of his or her internal stream of thoughts when distressed, and to identify and modify the dysfunctional thoughts. Behavioral techniques are used to bring about functional changes in behavior, regulate emotion, and help the cognitive restructuring process. Modifying the patient’s underlying dysfunctional beliefs leads to lasting improvements. In this structured therapy, the therapist and patient work collaboratively to use an approach that features reality testing and experimentation.4
Figure
The cognitive model of CBT
CBT: cognitive-behavioral therapy
Source: Adapted from reference 3
Indications for CBT in older adults
Depression. Among psychotherapies used in older adults, CBT has received the most research for late-life depression.5 Randomized controlled trials (RCTs) have found CBT is superior to treatment as usual in depressed adults age ≥60.6 It also has been found to be superior to wait-list control7 and talking as control.6,8 Meta-analyses have shown above-average effect sizes for CBT in treating late-life depression.9,10 A follow-up study found improvement was maintained up to 2 years after CBT, which suggests CBT’s impact is likely to be long lasting.11
Thompson et al12 compared 102 depressed patients age >60 who were treated with CBT alone, desipramine alone, or a combination of the 2. A combination of medication and CBT worked best for severely depressed patients; CBT alone or a combination of CBT and medication worked best for moderately depressed patients.
CBT is an option when treating depressed medically ill older adults. Research indicates that CBT could reduce depression in older patients with Parkinson’s disease13 and chronic obstructive pulmonary disease.14
As patients get older, cognitive impairment with comorbid depression can make treatment challenging. Limited research suggests CBT applied in a modified format that involves caregivers and uses problem solving and behavioral strategies can significantly reduce depression in patients with dementia.15
Anxiety. Researchers have examined the efficacy of variants of CBT in treating older adults with anxiety disorders—commonly, generalized anxiety disorder (GAD), panic disorder, agoraphobia, subjective anxiety, or a combination of these illnesses.16,17 Randomized trials have supported CBT’s efficacy for older patients with GAD and mixed anxiety states; gains made in CBT were maintained over a 1-year follow-up.18,19 In a meta-analysis of 15 studies using cognitive and behavioral methods of treating anxiety in older patients, Nordhus and Pallesen16 reported a significant effect size of 0.55. In a 2008 meta-analysis that included only RCTs, CBT was superior to wait-list conditions as well as active control conditions in treating anxious older patients.20
However, some research suggests that CBT for GAD may not be as effective for older adults as it is for younger adults. In a study of CBT for GAD in older adults, Stanley et al19 reported smaller effect sizes compared with CBT for younger adults. Researchers have found relatively few differences between CBT and comparison conditions—supportive psychotherapy or active control conditions—in treating GAD in older adults.21 Modified, more effective formats of CBT for GAD in older adults need to be established.22 Mohlman et al23 supplemented standard CBT for late-life GAD with memory and learning aids—weekly reading assignments, graphing exercises to chart mood ratings, reminder phone calls from therapists, and homework compliance requirement. This approach improved the response rate from 40% to 75%.23
Insomnia. Studies have found CBT to be an effective means of treating insomnia in geriatric patients. Although sleep problems occur more frequently among older patients, only 15% of chronic insomnia patients receive treatment; psychotherapy rarely is used.24 CBT for insomnia (CBT-I) should be considered for older adults because managing insomnia with medications may be problematic and these patients may prefer nonpharmacologic treatment.2 CBT-I typically incorporates cognitive strategies with established behavioral techniques, including sleep hygiene education, cognitive restructuring, relaxation training, stimulus control, and/or sleep restriction. The CBT-I multicomponent treatment package meets all criteria to be considered an evidence-based treatment for late-life insomnia.25
RCTs have reported significant improvements in late-life insomnia with CBT-I.26,27 Reviews and meta-analyses have also concluded that cognitive-behavioral treatments are effective for treating insomnia in older adults.25,28 Most insomnia cases in geriatric patients are reported to occur secondary to other medical or psychiatric conditions that are judged as causing the insomnia.25 In these cases, direct treatment of the insomnia usually is delayed or omitted.28 Studies evaluating the efficacy of CBT packages for treating insomnia occurring in conjunction with other medical or psychiatric illnesses have reported significant improvement of insomnia.28,29 Because insomnia frequently occurs in older patients with medical illnesses and psychiatric disorders, CBT-I could be beneficial for such patients.
Good candidates for CBT
Clinical experience indicates that older adults in relatively good health with no significant cognitive decline are good candidates for CBT. These patients tend to comply with their assignments, are interested in applying the learned strategies, and are motivated to read self-help books. CBT’s structured, goal-oriented approach makes it a short-term treatment, which makes it cost effective. Insomnia patients may improve after 6 to 8 CBT-I sessions and patients with anxiety or depression may need to undergo 15 to 20 CBT sessions. Patients age ≥65 have basic Medicare coverage that includes mental health care and psychotherapy.
There are no absolute contraindications for CBT, but the greater the cognitive impairment, the less the patient will benefit from CBT (Table 2). Similarly, severe depression and anxiety might make it difficult for patients to participate meaningfully, although CBT may be incorporated gradually as patients improve with medication. Severe medical illnesses and sensory losses such as visual and hearing loss would make it difficult to carry out CBT effectively.
Table 2
Contraindications for CBT
| High levels of cognitive impairment |
| Severe depression with psychotic features |
| Severe anxiety with high levels of agitation |
| Severe medical illness |
| Sensory losses |
| CBT: cognitive-behavioral therapy |
Adapting CBT for older patients
When using CBT with older patients, it is important to keep in mind characteristics that define the geriatric population. Laidlaw et al30 developed a model to help clinicians develop a more appropriate conceptualization of older patients that focuses on significant events and related cognitions associated with physical health, changes in role investments, and interactions with younger generations. It emphasizes the need to explore beliefs about aging viewed through each patient’s socio-cultural lens and examine cognitions in the context of the time period in which the individual has lived.
Losses and transitions. For many older patients, the latter years of life are characterized by losses and transitions.31 According to Thompson,31 these losses and transitions can trigger thoughts of missed opportunities or unresolved relationships and reflection on unachieved goals.31 CBT for older adults should focus on the meaning the patient gives to these losses and transitions. For example, depressed patients could view their retirement as a loss of self worth as they become less productive. CBT can help patients identify ways of thinking about the situation that will enable them to adapt to these losses and transitions.
Changes in cognition. Changes in cognitive functioning with aging are not universal and there’s considerable variability, but it’s important to make appropriate adaptations when needed. Patients may experience a decline in cognitive speed, working memory, selective attention, and fluid intelligence. This would require that information be presented slowly, with frequent repetitions and summaries. Also, it might be helpful to present information in alternate ways and to encourage patients to take notes during sessions. To accommodate for a decline in fluid intelligence, presenting new information in the context of previous experiences will help promote learning. Recordings of important information and conclusions from cognitive restructuring that patients can listen to between sessions could serve as helpful reminders that will help patients progress. Phone prompts or alarms can remind patients to carry out certain therapeutic measures, such as breathing exercises. Caretakers can attend sessions to become familiar with strategies performed during CBT and act as a co-therapist at home; however, their inclusion must be done with the consent of both parties and only if it’s viewed as necessary for the patient’s progress.
Additional strategies. For patients with substantial cognitive decline, cognitive restructuring might not be as effective as behavioral strategies—activity scheduling, graded task assignment, graded exposure, and rehearsals. Because older adults often have strengthened dysfunctional beliefs over a long time, modifying them takes longer, which is why the tapering process usually takes longer for older patients than for younger patients. The lengthier tapering ensures learning is well established and the process of modifying dysfunctional beliefs to functional beliefs continues. Collaborating with other professionals—physicians, social workers, and case managers—will help ensure a shared care process in which common goals are met.
The websites of the Academy of Cognitive Therapy, American Psychological Association, and Association for Behavioral and Cognitive Therapies can help clinicians who do not offer CBT to locate a qualified therapist for their patients (Related Resources).
- Academy of Cognitive Therapy. www.academyofct.org.
- American Psychological Association. www.apa.org.
- Association for Behavioral and Cognitive Therapies. www.abct.org.
- Laidlaw K, Thompson LW, Dick-Siskin L, et al. Cognitive behaviour therapy with older people. West Sussex, England: John Wiley & Sons, Ltd; 2003.
Drug Brand Name
- Desipramine • Norpramin
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Landreville P, Landry J, Baillargeon L, et al. Older adults’ acceptance of psychological and pharmacological treatments for depression. J Gerontol B Psychol Sci Soc Sci. 2001;56(5):P285-P291.
2. Chambless DL, Ollendick TH. Empirically supported psychological interventions: controversies and evidence. Annu Rev Psychol. 2001;52:685-716.
3. Beck JS. Cognitive conceptualization. In: Cognitive therapy: basics and beyond. 2nd ed. New York NY: The Guilford Press; 2011:29–45.
4. Beck AT, Rush AJ, Shaw BF, et al. Cognitive therapy of depression. New York, NY: The Guilford Press; 1979.
5. Areán PA, Cook BL. Psychotherapy and combined psychotherapy/pharmacotherapy for late-life depression. Biol Psychiatry. 2002;52(3):293-303.
6. Laidlaw K, Davidson K, Toner H, et al. A randomised controlled trial of cognitive behaviour therapy vs treatment as usual in the treatment of mild to moderate late-life depression. Int J Geriatr Psychiatry. 2008;23(8):843-850.
7. Floyd M, Scogin F, McKendree-Smith NL, et al. Cognitive therapy for depression: a comparison of individual psychotherapy and bibliotherapy for depressed older adults. Behavior Modification. 2004;28(2):297-318.
8. Serfaty MA, Haworth D, Blanchard M, et al. Clinical effectiveness of individual cognitive behavioral therapy for depressed older people in primary care: a randomized controlled trial. Arch Gen Psychiatry. 2009;66(12):1332-1340.
9. Pinquart M, Sörensen S. How effective are psychotherapeutic and other psychosocial interventions with older adults? A meta-analysis. J Ment Health Aging. 2001;7(2):207-243.
10. Pinquart M, Duberstein PR, Lyness JM. Effects of psychotherapy and other behavioral interventions on clinically depressed older adults: a meta-analysis. Aging Ment Health. 2007;11(6):645-657.
11. Gallagher-Thompson D, Hanley-Peterson P, Thompson LW. Maintenance of gains versus relapse following brief psychotherapy for depression. J Consult Clin Psychol. 1990;58(3):371-374.
12. Thompson LW, Coon DW, Gallagher-Thompson D, et al. Comparison of desipramine and cognitive/behavioral therapy in the treatment of elderly outpatients with mild-to-moderate depression. Am J Geriatr Psychiatry. 2001;9(3):225-240.
13. Dobkin RD, Menza M, Allen LA, et al. Cognitive-behavioral therapy for depression in Parkinson’s disease: a randomized, controlled trial. Am J Psychiatry. 2011;168(10):1066-1074.
14. Kunik ME, Braun U, Stanley MA, et al. One session cognitive behavioural therapy for elderly patients with chronic obstructive pulmonary disease. Psychol Med. 2001;31(4):717-723.
15. Teri L, Logsdon RG, Uomoto J, et al. Behavioral treatment of depression in dementia patients: a controlled clinical trial. J Gerontol B Psychol Sci Soc Sci. 1997;52(4):P159-P166.
16. Nordhus IH, Pallesen S. Psychological treatment of late-life anxiety: an empirical review. J Consult Clin Psychol. 2003;71(4):643-651.
17. Gorenstein EE, Papp LA. Cognitive-behavioral therapy for anxiety in the elderly. Curr Psychiatry Rep. 2007;9(1):20-25.
18. Barrowclough C, King P, Colville J, et al. A randomized trial of the effectiveness of cognitive-behavioral therapy and supportive counseling for anxiety symptoms in older adults. J Consult Clin Psychol. 2001;69(5):756-762.
19. Stanley MA, Beck JG, Novy DM, et al. Cognitive-behavioral treatment of late-life generalized anxiety disorder. J Consult Clin Psychol. 2003;71(2):309-319.
20. Hendriks GJ, Oude Voshaar RC, Keijsers GP, et al. Cognitive-behavioural therapy for late-life anxiety disorders: a systematic review and meta-analysis. Acta Psychiatr Scand. 2008;117(6):403-411.
21. Wetherell JL, Gatz M, Craske MG. Treatment of generalized anxiety disorder in older adults. J Consult Clin Psychol. 2003;71(1):31-40.
22. Dugas MJ, Brillon P, Savard P, et al. A randomized clinical trial of cognitive-behavioral therapy and applied relaxation for adults with generalized anxiety disorder. Behav Ther. 2010;41(1):46-58.
23. Mohlman J, Gorenstein EE, Kleber M, et al. Standard and enhanced cognitive-behavior therapy for late-life generalized anxiety disorder: two pilot investigations. Am J Geriatr Psychiatry. 2003;11(1):24-32.
24. Flint AJ. Epidemiology and comorbidity of anxiety disorders in the elderly. Am J Psychiatry. 1994;151(5):640-649.
25. McCurry SM, Logsdon RG, Teri L, et al. Evidence-based psychological treatments for insomnia in older adults. Psychol Aging. 2007;22(1):18-27.
26. Sivertsen B, Omvik S, Pallesen S, et al. Cognitive behavioral therapy vs zopiclone for treatment of chronic primary insomnia in older adults: a randomized controlled trial. JAMA. 2006;295(24):2851-2858.
27. Morgan K, Dixon S, Mathers N, et al. Psychological treatment for insomnia in the regulation of long-term hypnotic drug use. Health Technol Assess. 2004;8(8):iii iv, 1-68.
28. Nau SD, McCrae CS, Cook KG, et al. Treatment of insomnia in older adults. Clin Psychol Rev. 2005;25(5):645-672.
29. Rybarczyk B, Stepanski E, Fogg L, et al. A placebo-controlled test of cognitive-behavioral therapy for comorbid insomnia in older adults. J Consult Clin Psychol. 2005;73(6):1164-1174.
30. Laidlaw K, Thompson LW, Gallagher-Thompson D. Comprehensive conceptualization of cognitive behaviour therapy for late life depression. Behav Cogn Psychother. 2004;32(4):389-399.
31. Thompson LW. Cognitive-behavioral therapy and treatment for late-life depression. J Clin Psychiatry. 1996;57(suppl 5):29-37.
1. Landreville P, Landry J, Baillargeon L, et al. Older adults’ acceptance of psychological and pharmacological treatments for depression. J Gerontol B Psychol Sci Soc Sci. 2001;56(5):P285-P291.
2. Chambless DL, Ollendick TH. Empirically supported psychological interventions: controversies and evidence. Annu Rev Psychol. 2001;52:685-716.
3. Beck JS. Cognitive conceptualization. In: Cognitive therapy: basics and beyond. 2nd ed. New York NY: The Guilford Press; 2011:29–45.
4. Beck AT, Rush AJ, Shaw BF, et al. Cognitive therapy of depression. New York, NY: The Guilford Press; 1979.
5. Areán PA, Cook BL. Psychotherapy and combined psychotherapy/pharmacotherapy for late-life depression. Biol Psychiatry. 2002;52(3):293-303.
6. Laidlaw K, Davidson K, Toner H, et al. A randomised controlled trial of cognitive behaviour therapy vs treatment as usual in the treatment of mild to moderate late-life depression. Int J Geriatr Psychiatry. 2008;23(8):843-850.
7. Floyd M, Scogin F, McKendree-Smith NL, et al. Cognitive therapy for depression: a comparison of individual psychotherapy and bibliotherapy for depressed older adults. Behavior Modification. 2004;28(2):297-318.
8. Serfaty MA, Haworth D, Blanchard M, et al. Clinical effectiveness of individual cognitive behavioral therapy for depressed older people in primary care: a randomized controlled trial. Arch Gen Psychiatry. 2009;66(12):1332-1340.
9. Pinquart M, Sörensen S. How effective are psychotherapeutic and other psychosocial interventions with older adults? A meta-analysis. J Ment Health Aging. 2001;7(2):207-243.
10. Pinquart M, Duberstein PR, Lyness JM. Effects of psychotherapy and other behavioral interventions on clinically depressed older adults: a meta-analysis. Aging Ment Health. 2007;11(6):645-657.
11. Gallagher-Thompson D, Hanley-Peterson P, Thompson LW. Maintenance of gains versus relapse following brief psychotherapy for depression. J Consult Clin Psychol. 1990;58(3):371-374.
12. Thompson LW, Coon DW, Gallagher-Thompson D, et al. Comparison of desipramine and cognitive/behavioral therapy in the treatment of elderly outpatients with mild-to-moderate depression. Am J Geriatr Psychiatry. 2001;9(3):225-240.
13. Dobkin RD, Menza M, Allen LA, et al. Cognitive-behavioral therapy for depression in Parkinson’s disease: a randomized, controlled trial. Am J Psychiatry. 2011;168(10):1066-1074.
14. Kunik ME, Braun U, Stanley MA, et al. One session cognitive behavioural therapy for elderly patients with chronic obstructive pulmonary disease. Psychol Med. 2001;31(4):717-723.
15. Teri L, Logsdon RG, Uomoto J, et al. Behavioral treatment of depression in dementia patients: a controlled clinical trial. J Gerontol B Psychol Sci Soc Sci. 1997;52(4):P159-P166.
16. Nordhus IH, Pallesen S. Psychological treatment of late-life anxiety: an empirical review. J Consult Clin Psychol. 2003;71(4):643-651.
17. Gorenstein EE, Papp LA. Cognitive-behavioral therapy for anxiety in the elderly. Curr Psychiatry Rep. 2007;9(1):20-25.
18. Barrowclough C, King P, Colville J, et al. A randomized trial of the effectiveness of cognitive-behavioral therapy and supportive counseling for anxiety symptoms in older adults. J Consult Clin Psychol. 2001;69(5):756-762.
19. Stanley MA, Beck JG, Novy DM, et al. Cognitive-behavioral treatment of late-life generalized anxiety disorder. J Consult Clin Psychol. 2003;71(2):309-319.
20. Hendriks GJ, Oude Voshaar RC, Keijsers GP, et al. Cognitive-behavioural therapy for late-life anxiety disorders: a systematic review and meta-analysis. Acta Psychiatr Scand. 2008;117(6):403-411.
21. Wetherell JL, Gatz M, Craske MG. Treatment of generalized anxiety disorder in older adults. J Consult Clin Psychol. 2003;71(1):31-40.
22. Dugas MJ, Brillon P, Savard P, et al. A randomized clinical trial of cognitive-behavioral therapy and applied relaxation for adults with generalized anxiety disorder. Behav Ther. 2010;41(1):46-58.
23. Mohlman J, Gorenstein EE, Kleber M, et al. Standard and enhanced cognitive-behavior therapy for late-life generalized anxiety disorder: two pilot investigations. Am J Geriatr Psychiatry. 2003;11(1):24-32.
24. Flint AJ. Epidemiology and comorbidity of anxiety disorders in the elderly. Am J Psychiatry. 1994;151(5):640-649.
25. McCurry SM, Logsdon RG, Teri L, et al. Evidence-based psychological treatments for insomnia in older adults. Psychol Aging. 2007;22(1):18-27.
26. Sivertsen B, Omvik S, Pallesen S, et al. Cognitive behavioral therapy vs zopiclone for treatment of chronic primary insomnia in older adults: a randomized controlled trial. JAMA. 2006;295(24):2851-2858.
27. Morgan K, Dixon S, Mathers N, et al. Psychological treatment for insomnia in the regulation of long-term hypnotic drug use. Health Technol Assess. 2004;8(8):iii iv, 1-68.
28. Nau SD, McCrae CS, Cook KG, et al. Treatment of insomnia in older adults. Clin Psychol Rev. 2005;25(5):645-672.
29. Rybarczyk B, Stepanski E, Fogg L, et al. A placebo-controlled test of cognitive-behavioral therapy for comorbid insomnia in older adults. J Consult Clin Psychol. 2005;73(6):1164-1174.
30. Laidlaw K, Thompson LW, Gallagher-Thompson D. Comprehensive conceptualization of cognitive behaviour therapy for late life depression. Behav Cogn Psychother. 2004;32(4):389-399.
31. Thompson LW. Cognitive-behavioral therapy and treatment for late-life depression. J Clin Psychiatry. 1996;57(suppl 5):29-37.
Antipsychotics for migraines, cluster headaches, and nausea
Most evidence supporting antipsychotics as a treatment for migraine headaches and cluster headaches is based on small studies and chart reviews. Some research suggests antipsychotics may effectively treat nausea but side effects such as akathisia may limit their use.
Migraine headaches
Antipsychotic treatment of migraines is supported by the theory that dopaminergic hyperactivity leads to migraine headaches (Table 1). Antipsychotics have been used off-label in migraine patients who do not tolerate triptans or have status migrainosus—intense, debilitating migraine lasting >72 hours.1 Primarily a result of D2 receptor blockade, the serotonergic effects of some second-generation antipsychotics (SGAs) may prevent migraine recurrence. The first-generation antipsychotics (FGAs) prochlorperazine, droperidol, haloperidol, and chlorpromazine have been used for migraine headaches (Table 2).1-27
Prochlorperazine may be an effective treatment of acute headaches9 and refractory chronic daily headache.10 Studies show that buccal prochlorperazine is more effective than oral ergotamine tartrate11 and IV prochlorperazine is more effective than IV ketorolac12 or valproate28 for treating acute headache.
Evidence suggests that chlorpromazine administered IM2 or IV3 is better than placebo for managing migraine pain. In a study comparing IV chlorpromazine, lidocaine, and dihydroergotamine, patients treated with chlorpromazine showed more persistent headache relief 12 to 24 hours post-dose.4 In another study, IV chlorpromazine, 25 mg, was as effective as IM ketorolac, 60 mg.5
Droperidol has been shown to be effective for managing headache, specifically status migrainosus.6 Patients with “benign headache”—headache not caused by an underlying medical disorder—who received droperidol reported greater reduction in visual analog pain scores within 1 hour of dosing compared with those taking prochlorperazine.7 In a randomized trial comparing IM droperidol and IM meperidine, patients with an acute migraine who received droperidol had improved scores on the visual pain analog scale and required less “rescue medication” for breakthrough pain.8 The FDA has issued a “black-box” warning of QTc prolongation with droperidol.
In a double blind, placebo-controlled trial, IV haloperidol, 5 mg, effectively treated migraine headache in 80% of patients compared with 15% of those who received placebo. However, 16% of patients considered the side effects—mainly sedation and akathisia—intolerable and 7% had symptom relapse.13 In an open-label trial of 6 patients with migraine headache, all patients achieved complete or substantial headache relief 25 to 65 minutes after receiving IV haloperidol, 5 mg.14
SGAs often antagonize 5-HT1D receptors and theoretically can render triptan therapy—which stimulates pre-synaptic 5-HT1D receptors—ineffective. This has not been seen clinically and instead, dose-related, non-specific headaches are a common adverse event with SGAs.29,30 A retrospective chart review found olanzapine provided relief for refractory headaches in patients who had failed ≥4 preventive medications. Olanzapine significantly decreased headache days, from 27.5±4.9 before treatment to 21.1±10.7 after treatment. Olanzapine also improved headache severity (measured on a 0 to 10 scale) from 8.7±1.6 before treatment to 2.2±2.1 after treatment.16 Researchers found that 2.5 or 5 mg of olanzapine relieved acute migraines for most patients, with repeat dosing as needed up to 20 mg/d. For prophylactic treatment, 5 or 10 mg of olanzapine was used. Olanzapine’s antinociceptive effect may be related to its action on α-2 adrenoreceptors and to a lesser extent on involvement of opioid and serotonergic receptors.17
In a case series, 3 migraine patients who met criteria for chronic daily headache and migraines but did not have a psychiatric disorder reported significant and sustained headache improvement when treated with risperidone.19 In a case series of 3 migraine patients with co-occurring psychiatric disorders, aripiprazole decreased migraine frequency and severity.15 Although limited data support quetiapine’s efficacy in treating acute migraines, in an open-label, pilot study, patients taking quetiapine, 25 to 75 mg/d, demonstrated a decrease in mean frequency of migraine days from 10.2 to 6.2 and decreased use of rescue medications from 2.3 to 1.2 days per week.18
Table 1
Possible rationale for antipsychotic use for headaches and nausea
| Condition | Possible rationale |
|---|---|
| Migraine | Patients are hypersensitive to dopamine agonists or dopamine transporter dysfunction. Some evidence that the dopamine D2 (DRD2) gene is involved |
| Cluster headache | Pain alleviation possibly related to dopamine receptor antagonism |
| Nausea | D2 and H1 receptor blockage |
Table 2
Antipsychotics for headache and nausea: Strength of the evidence
| Condition | Strength of evidencea |
|---|---|
| Migraine | Intermediate: Chlorpromazine,2-5 droperidol,6-8 prochlorperazine1,10-12 |
| Weak: Haloperidol13,14 | |
| Very weak: Aripiprazole,15 olanzapine,16,17 quetiapine,18 ziprasidone19 | |
| Cluster headache | Weak: Chlorpromazine20 |
| Very weak: Clozapine,21 olanzapine22 | |
| Nausea/vomiting | Intermediate: Droperidol,23 metoclopramide,24 prochlorperazine,25 promethazine25 |
| Weak: Olanzapine26,27 | |
| aStrong: Multiple, well-designed RCTs directly relevant to the recommendation, yielding consistent findings Intermediate: Some evidence from RCTs that support the recommendation, but the scientific support was not optimal Weak: Consensus recommendation in the absence of relevant randomized controlled trials and better evidence than case report or series Very weak: Case reports or case series or preliminary studies RCTs: randomized controlled trials | |
Cluster headaches
Subcutaneous sumatriptan and inhaled oxygen are first-line treatments for cluster headaches.31 A single, small study20 reported that chlorpromazine may prevent cluster headaches, which suggests that D2 receptor blockade may treat such headaches. However, limited supporting evidence relegates its use to a second- or third-line therapy.
In an open-label study (N = 5), olanzapine provided some relief of pain associated with cluster headache within 20 minutes of administration.22 In another study, patients with schizophrenia and comorbid cluster headaches improved with olanzapine.21
Because evidence is limited to small prospective studies, antipsychotic treatment of cluster headache is not well established.20-22 However, olanzapine may benefit patients with comorbid cluster headaches and schizophrenia.
Nausea
The signaling pathways that mediate emesis involve 5-HT3, D2, muscarinic, and histamine receptors.32 Before 5-HT3 antagonists were available, the FGAs metoclopramide, droperidol, prochlorperazine, and promethazine were used to manage acute emesis in emergency departments.23 A double-blind, placebo-controlled trial found IV droperidol, 1.25 mg, was more effective than metoclopramide, 10 mg, or prochlorperazine, 10 mg, for relieving moderate to severe nausea in adult patients.23 However, droperidol and prochlorperazine were associated with akathisia. In addition, this trial did not find a clinically significant difference between groups—including placebo—in anxiety, sedation, or need for rescue medications.23 Use of droperidol to treat nausea decreased after the drug received a “black-box” warning for QT prolongation and torsades de pointes.
Metoclopramide is effective for treating acute migraine and associated nausea24 and has been used to treat gastroparesis because of its effect on upper GI motility. Phenothiazines have been used to treat nausea and studies have shown prochlorperazine to be more effective than promethazine.25 Some studies of prochlorperazine have reported a 44% incidence of akathisia, which limits the drug’s use in patients who may be sensitive to such effects.33 Promethazine can cause sedation and risk of tissue necrosis at the injection site.34
Among SGAs, olanzapine effectively prevented acute and delayed chemotherapy-induced nausea and vomiting in a proof-of-concept study of patients receiving high and moderate emetogenic therapies.26,27 National Comprehensive Cancer Network guidelines cite olanzapine as a potential option for treating refractory and breakthrough emesis.35 In a small study (N = 50), olanzapine showed comparable anti-nausea effect to aprepitant—a neurokinin 1 receptor antagonist—and effectively prevented chemotherapy-induced nausea and vomiting in highly emetogenic chemotherapy.36
Related Resources
- Kelley NE, Tepper DE. Rescue therapy for acute migraine, part 2: neuroleptics, antihistamines, and others. Headache. 2012;52(2):292-306.
- Dusitanond P, Young WB. Neuroleptics and migraine. Cent Nerv Syst Agents Med Chem. 2009;9(1):63-70.
Drug Brand Names
- Aprepitant • Emend
- Aripiprazole • Abilify
- Chlorpromazine • Thorazine
- Dihydroergotamine • D.H.E 45
- Droperidol • Inapsine
- Ergotamine tartrate • Ergostat
- Haloperidol • Haldol
- Ketorolac • Toradol
- Lidocaine • Xylocaine, Lidoderm
- Meperidine • Demerol
- Metoclopramide • Reglan
- Olanzapine • Zyprexa
- Prochlorperazine • Compazine
- Promethazine • Phenergan
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sumatriptan • Imitrex
- Valproate • Depakote
Disclosures
Dr. Macaluso has received grant or research support from EnVivo Pharmaceuticals, Janssen L.P., and Pfizer, Inc.
Dr. Tripathi reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Dusitanond P, Young WB. Neuroleptics and migraine. Cent Nerv Syst Agents Med Chem. 2009;9(1):63-70.
2. McEwen JI, O’Connor HM, Dinsdale HB. Treatment of migraine with intramuscular chlorpromazine. Ann Emerg Med. 1987;16(7):758-763.
3. Bigal M, Bordini CA, Speciali JG. Intravenous chlorpromazine in the emergency department treatment of migraines: a randomized controlled trial. J Emerg Med. 2002;23(2):141-148.
4. Bell R, Montoya D, Shuaib A, et al. A comparative trial of three agents in the treatment of acute migraine headache. Ann Emerg Med. 1990;19(10):1079-1082.
5. Shrestha M, Singh R, Moreden J, et al. Ketorolac vs chlorpromazine in the treatment of acute migraine without aura. A prospective, randomized, double-blind trial. Arch Intern Med. 1996;156(15):1725-1728.
6. Wang SJ, Silberstein SD, Young WB. Droperidol treatment of status migrainosus and refractory migraine. Headache. 1997;37(6):377-382.
7. Miner JR, Fish SJ, Smith SW, et al. Droperidol vs. prochlorperazine for benign headaches in the emergency department. Acad Emerg Med. 2001;8(9):873-879.
8. Richman PB, Allegra J, Eskin B, et al. A randomized clinical trial to assess the efficacy of intramuscular droperidol for the treatment of acute migraine headache. Am J Emerg Med. 2002;20(1):39-42.
9. Jones J, Sklar D, Dougherty J, et al. Randomized double blind trial of intravenous prochlorperazine for the treatment of acute headache. JAMA. 1989;261(8):1174-1176.
10. Lu SR, Fuh JL, Juang KD, et al. Repetitive intravenous prochlorperazine treatment of patients with refractory chronic daily headache. Headache. 2000;40(9):724-729.
11. Sharma S, Prasad A, Nehru R, et al. Efficacy and tolerability of prochlorperazine buccal tablets in treatment of acute migraine. Headache. 2002;42(9):896-902.
12. Seim MB, March JA, Dunn KA. Intravenous ketorolac vs intravenous prochlorperazine for the treatment of migraine headaches. Acad Emerg Med. 1998;5(6):573-576.
13. Honkaniemi J, Liimatainen S, Rainesalo S, et al. Haloperidol in the acute treatment of migraine: a randomized, double-blind, placebo-controlled study. Headache. 2006;46(5):781-787.
14. Fisher H. A new approach to emergency department therapy of migraine headache with intravenous haloperidol: a case series. J Emerg Med. 1995;13(1):119-122.
15. LaPorta LD. Relief from migraine headache with aripiprazole treatment. Headache. 2007;47(6):922-926.
16. Silberstein SD, Peres MF, Hopkins MM, et al. Olanzapine in the treatment of refractory migraine and chronic daily headache. Headache. 2002;42(6):515-518.
17. Schreiber S, Getslev V, Backer MM, et al. The atypical neuroleptics clozapine and olanzapine differ regarding their antinociceptive mechanisms and potency. Pharmacol Biochem Behav. 1999;64(1):75-80.
18. Krymchantowski AV, Jevoux C. Quetiapine for the prevention of migraine refractory to the combination of atenolol + nortriptyline + flunarizine: an open pilot study. Arq Neuropsiquiatr. 2008;66(3B):615-618.
19. Cahill CM, Hardiman O, Murphy KC. Treatment of refractory chronic daily headache with the atypical antipsychotic ziprasidone-a case series. Cephalalgia. 2005;25(10):822-826.
20. Caviness VS, Jr, O’Brien P. Cluster headache: response to chlorpromazine. Headache. 1980;20(3):128-131.
21. Datta SS, Kumar S. Clozapine-responsive cluster headache. Neurol India. 2006;54(2):200-201.
22. Rozen TD. Olanzapine as an abortive agent for cluster headache. Headache. 2001;41(8):813-816.
23. Braude D, Soliz T, Crandall C, et al. Antiemetics in the ED: a randomized controlled trial comparing 3 common agents. Am J Emerg Med. 2006;24(2):177-182.
24. Colman I, Brown MD, Innes GD, et al. Parenteral metoclopramide for acute migraine: meta-analysis of randomised controlled trials. BMJ. 2004;329(7479):1369-1373.
25. Ernst AA, Weiss SJ, Park S, et al. Prochlorperazine versus promethazine for uncomplicated nausea and vomiting in the emergency department: a randomized, double-blind clinical trial. Ann Emerg Med. 2000;36(2):89-94.
26. Navari RM, Einhorn LH, Loehrer PJ Sr, et al. A phase II trial of olanzapine, dexamethasone, and palonosetron for the prevention of chemotherapy-induced nausea and vomiting: a Hoosier oncology group study. Support Care Cancer. 2007;15(11):1285-1291.
27. Passik SD, Navari RM, Jung SH, et al. A phase I trial of olanzapine (Zyprexa) for the prevention of delayed emesis in cancer patients: a Hoosier Oncology Group study. Cancer Invest. 2004;22(3):383-388.
28. Tanen DA, Miller S, French T, et al. Intravenous sodium valproate versus prochlorperazine for the emergency department treatment of acute migraine headaches: a prospective, randomized, double-blind trial. Ann Emerg Med. 2003;41(6):847-853.
29. Caley CF, Cooper CK. Ziprasidone: the fifth atypical antipsychotic. Ann Pharmacother. 2002;36(5):839-851.
30. Geodon [package insert]. New York NY. Pfizer Inc.; 2012.
31. Kudrow L. Response of cluster headache attacks to oxygen inhalation. Headache. 1981;21(1):1-4.
32. Scuderi PE. Pharmacology of antiemetics. Int Anesthesiol Clin. 2003;41(4):41-66.
33. Drotts DL, Vinson DR. Prochlorperazine induces akathisia in emergency patients. Ann Emerg Med. 1999;34(4):469-475.
34. Institute for Safe Medication Practices. Action needed to prevent serious tissue injury with IV promethazine. http://www.ismp.org/newsletters/acutecare/articles/20060810.asp?ptr_y. Published August 10 2006. Accessed November 28, 2012.
35. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. 2010. http://www.nccn.org/professionals/physician_gls/pdf/antiemesis.pdf. Accessed November 29 2012.
36. Navari R, Gray SE, Carr AC. Olanzapine versus aprepitant for the prevention of chemotherapy induced nausea and vomiting (CINV): a randomized phase III trial. J Clin Oncol. 2010;28(15 suppl):9020.-
Most evidence supporting antipsychotics as a treatment for migraine headaches and cluster headaches is based on small studies and chart reviews. Some research suggests antipsychotics may effectively treat nausea but side effects such as akathisia may limit their use.
Migraine headaches
Antipsychotic treatment of migraines is supported by the theory that dopaminergic hyperactivity leads to migraine headaches (Table 1). Antipsychotics have been used off-label in migraine patients who do not tolerate triptans or have status migrainosus—intense, debilitating migraine lasting >72 hours.1 Primarily a result of D2 receptor blockade, the serotonergic effects of some second-generation antipsychotics (SGAs) may prevent migraine recurrence. The first-generation antipsychotics (FGAs) prochlorperazine, droperidol, haloperidol, and chlorpromazine have been used for migraine headaches (Table 2).1-27
Prochlorperazine may be an effective treatment of acute headaches9 and refractory chronic daily headache.10 Studies show that buccal prochlorperazine is more effective than oral ergotamine tartrate11 and IV prochlorperazine is more effective than IV ketorolac12 or valproate28 for treating acute headache.
Evidence suggests that chlorpromazine administered IM2 or IV3 is better than placebo for managing migraine pain. In a study comparing IV chlorpromazine, lidocaine, and dihydroergotamine, patients treated with chlorpromazine showed more persistent headache relief 12 to 24 hours post-dose.4 In another study, IV chlorpromazine, 25 mg, was as effective as IM ketorolac, 60 mg.5
Droperidol has been shown to be effective for managing headache, specifically status migrainosus.6 Patients with “benign headache”—headache not caused by an underlying medical disorder—who received droperidol reported greater reduction in visual analog pain scores within 1 hour of dosing compared with those taking prochlorperazine.7 In a randomized trial comparing IM droperidol and IM meperidine, patients with an acute migraine who received droperidol had improved scores on the visual pain analog scale and required less “rescue medication” for breakthrough pain.8 The FDA has issued a “black-box” warning of QTc prolongation with droperidol.
In a double blind, placebo-controlled trial, IV haloperidol, 5 mg, effectively treated migraine headache in 80% of patients compared with 15% of those who received placebo. However, 16% of patients considered the side effects—mainly sedation and akathisia—intolerable and 7% had symptom relapse.13 In an open-label trial of 6 patients with migraine headache, all patients achieved complete or substantial headache relief 25 to 65 minutes after receiving IV haloperidol, 5 mg.14
SGAs often antagonize 5-HT1D receptors and theoretically can render triptan therapy—which stimulates pre-synaptic 5-HT1D receptors—ineffective. This has not been seen clinically and instead, dose-related, non-specific headaches are a common adverse event with SGAs.29,30 A retrospective chart review found olanzapine provided relief for refractory headaches in patients who had failed ≥4 preventive medications. Olanzapine significantly decreased headache days, from 27.5±4.9 before treatment to 21.1±10.7 after treatment. Olanzapine also improved headache severity (measured on a 0 to 10 scale) from 8.7±1.6 before treatment to 2.2±2.1 after treatment.16 Researchers found that 2.5 or 5 mg of olanzapine relieved acute migraines for most patients, with repeat dosing as needed up to 20 mg/d. For prophylactic treatment, 5 or 10 mg of olanzapine was used. Olanzapine’s antinociceptive effect may be related to its action on α-2 adrenoreceptors and to a lesser extent on involvement of opioid and serotonergic receptors.17
In a case series, 3 migraine patients who met criteria for chronic daily headache and migraines but did not have a psychiatric disorder reported significant and sustained headache improvement when treated with risperidone.19 In a case series of 3 migraine patients with co-occurring psychiatric disorders, aripiprazole decreased migraine frequency and severity.15 Although limited data support quetiapine’s efficacy in treating acute migraines, in an open-label, pilot study, patients taking quetiapine, 25 to 75 mg/d, demonstrated a decrease in mean frequency of migraine days from 10.2 to 6.2 and decreased use of rescue medications from 2.3 to 1.2 days per week.18
Table 1
Possible rationale for antipsychotic use for headaches and nausea
| Condition | Possible rationale |
|---|---|
| Migraine | Patients are hypersensitive to dopamine agonists or dopamine transporter dysfunction. Some evidence that the dopamine D2 (DRD2) gene is involved |
| Cluster headache | Pain alleviation possibly related to dopamine receptor antagonism |
| Nausea | D2 and H1 receptor blockage |
Table 2
Antipsychotics for headache and nausea: Strength of the evidence
| Condition | Strength of evidencea |
|---|---|
| Migraine | Intermediate: Chlorpromazine,2-5 droperidol,6-8 prochlorperazine1,10-12 |
| Weak: Haloperidol13,14 | |
| Very weak: Aripiprazole,15 olanzapine,16,17 quetiapine,18 ziprasidone19 | |
| Cluster headache | Weak: Chlorpromazine20 |
| Very weak: Clozapine,21 olanzapine22 | |
| Nausea/vomiting | Intermediate: Droperidol,23 metoclopramide,24 prochlorperazine,25 promethazine25 |
| Weak: Olanzapine26,27 | |
| aStrong: Multiple, well-designed RCTs directly relevant to the recommendation, yielding consistent findings Intermediate: Some evidence from RCTs that support the recommendation, but the scientific support was not optimal Weak: Consensus recommendation in the absence of relevant randomized controlled trials and better evidence than case report or series Very weak: Case reports or case series or preliminary studies RCTs: randomized controlled trials | |
Cluster headaches
Subcutaneous sumatriptan and inhaled oxygen are first-line treatments for cluster headaches.31 A single, small study20 reported that chlorpromazine may prevent cluster headaches, which suggests that D2 receptor blockade may treat such headaches. However, limited supporting evidence relegates its use to a second- or third-line therapy.
In an open-label study (N = 5), olanzapine provided some relief of pain associated with cluster headache within 20 minutes of administration.22 In another study, patients with schizophrenia and comorbid cluster headaches improved with olanzapine.21
Because evidence is limited to small prospective studies, antipsychotic treatment of cluster headache is not well established.20-22 However, olanzapine may benefit patients with comorbid cluster headaches and schizophrenia.
Nausea
The signaling pathways that mediate emesis involve 5-HT3, D2, muscarinic, and histamine receptors.32 Before 5-HT3 antagonists were available, the FGAs metoclopramide, droperidol, prochlorperazine, and promethazine were used to manage acute emesis in emergency departments.23 A double-blind, placebo-controlled trial found IV droperidol, 1.25 mg, was more effective than metoclopramide, 10 mg, or prochlorperazine, 10 mg, for relieving moderate to severe nausea in adult patients.23 However, droperidol and prochlorperazine were associated with akathisia. In addition, this trial did not find a clinically significant difference between groups—including placebo—in anxiety, sedation, or need for rescue medications.23 Use of droperidol to treat nausea decreased after the drug received a “black-box” warning for QT prolongation and torsades de pointes.
Metoclopramide is effective for treating acute migraine and associated nausea24 and has been used to treat gastroparesis because of its effect on upper GI motility. Phenothiazines have been used to treat nausea and studies have shown prochlorperazine to be more effective than promethazine.25 Some studies of prochlorperazine have reported a 44% incidence of akathisia, which limits the drug’s use in patients who may be sensitive to such effects.33 Promethazine can cause sedation and risk of tissue necrosis at the injection site.34
Among SGAs, olanzapine effectively prevented acute and delayed chemotherapy-induced nausea and vomiting in a proof-of-concept study of patients receiving high and moderate emetogenic therapies.26,27 National Comprehensive Cancer Network guidelines cite olanzapine as a potential option for treating refractory and breakthrough emesis.35 In a small study (N = 50), olanzapine showed comparable anti-nausea effect to aprepitant—a neurokinin 1 receptor antagonist—and effectively prevented chemotherapy-induced nausea and vomiting in highly emetogenic chemotherapy.36
Related Resources
- Kelley NE, Tepper DE. Rescue therapy for acute migraine, part 2: neuroleptics, antihistamines, and others. Headache. 2012;52(2):292-306.
- Dusitanond P, Young WB. Neuroleptics and migraine. Cent Nerv Syst Agents Med Chem. 2009;9(1):63-70.
Drug Brand Names
- Aprepitant • Emend
- Aripiprazole • Abilify
- Chlorpromazine • Thorazine
- Dihydroergotamine • D.H.E 45
- Droperidol • Inapsine
- Ergotamine tartrate • Ergostat
- Haloperidol • Haldol
- Ketorolac • Toradol
- Lidocaine • Xylocaine, Lidoderm
- Meperidine • Demerol
- Metoclopramide • Reglan
- Olanzapine • Zyprexa
- Prochlorperazine • Compazine
- Promethazine • Phenergan
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sumatriptan • Imitrex
- Valproate • Depakote
Disclosures
Dr. Macaluso has received grant or research support from EnVivo Pharmaceuticals, Janssen L.P., and Pfizer, Inc.
Dr. Tripathi reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Most evidence supporting antipsychotics as a treatment for migraine headaches and cluster headaches is based on small studies and chart reviews. Some research suggests antipsychotics may effectively treat nausea but side effects such as akathisia may limit their use.
Migraine headaches
Antipsychotic treatment of migraines is supported by the theory that dopaminergic hyperactivity leads to migraine headaches (Table 1). Antipsychotics have been used off-label in migraine patients who do not tolerate triptans or have status migrainosus—intense, debilitating migraine lasting >72 hours.1 Primarily a result of D2 receptor blockade, the serotonergic effects of some second-generation antipsychotics (SGAs) may prevent migraine recurrence. The first-generation antipsychotics (FGAs) prochlorperazine, droperidol, haloperidol, and chlorpromazine have been used for migraine headaches (Table 2).1-27
Prochlorperazine may be an effective treatment of acute headaches9 and refractory chronic daily headache.10 Studies show that buccal prochlorperazine is more effective than oral ergotamine tartrate11 and IV prochlorperazine is more effective than IV ketorolac12 or valproate28 for treating acute headache.
Evidence suggests that chlorpromazine administered IM2 or IV3 is better than placebo for managing migraine pain. In a study comparing IV chlorpromazine, lidocaine, and dihydroergotamine, patients treated with chlorpromazine showed more persistent headache relief 12 to 24 hours post-dose.4 In another study, IV chlorpromazine, 25 mg, was as effective as IM ketorolac, 60 mg.5
Droperidol has been shown to be effective for managing headache, specifically status migrainosus.6 Patients with “benign headache”—headache not caused by an underlying medical disorder—who received droperidol reported greater reduction in visual analog pain scores within 1 hour of dosing compared with those taking prochlorperazine.7 In a randomized trial comparing IM droperidol and IM meperidine, patients with an acute migraine who received droperidol had improved scores on the visual pain analog scale and required less “rescue medication” for breakthrough pain.8 The FDA has issued a “black-box” warning of QTc prolongation with droperidol.
In a double blind, placebo-controlled trial, IV haloperidol, 5 mg, effectively treated migraine headache in 80% of patients compared with 15% of those who received placebo. However, 16% of patients considered the side effects—mainly sedation and akathisia—intolerable and 7% had symptom relapse.13 In an open-label trial of 6 patients with migraine headache, all patients achieved complete or substantial headache relief 25 to 65 minutes after receiving IV haloperidol, 5 mg.14
SGAs often antagonize 5-HT1D receptors and theoretically can render triptan therapy—which stimulates pre-synaptic 5-HT1D receptors—ineffective. This has not been seen clinically and instead, dose-related, non-specific headaches are a common adverse event with SGAs.29,30 A retrospective chart review found olanzapine provided relief for refractory headaches in patients who had failed ≥4 preventive medications. Olanzapine significantly decreased headache days, from 27.5±4.9 before treatment to 21.1±10.7 after treatment. Olanzapine also improved headache severity (measured on a 0 to 10 scale) from 8.7±1.6 before treatment to 2.2±2.1 after treatment.16 Researchers found that 2.5 or 5 mg of olanzapine relieved acute migraines for most patients, with repeat dosing as needed up to 20 mg/d. For prophylactic treatment, 5 or 10 mg of olanzapine was used. Olanzapine’s antinociceptive effect may be related to its action on α-2 adrenoreceptors and to a lesser extent on involvement of opioid and serotonergic receptors.17
In a case series, 3 migraine patients who met criteria for chronic daily headache and migraines but did not have a psychiatric disorder reported significant and sustained headache improvement when treated with risperidone.19 In a case series of 3 migraine patients with co-occurring psychiatric disorders, aripiprazole decreased migraine frequency and severity.15 Although limited data support quetiapine’s efficacy in treating acute migraines, in an open-label, pilot study, patients taking quetiapine, 25 to 75 mg/d, demonstrated a decrease in mean frequency of migraine days from 10.2 to 6.2 and decreased use of rescue medications from 2.3 to 1.2 days per week.18
Table 1
Possible rationale for antipsychotic use for headaches and nausea
| Condition | Possible rationale |
|---|---|
| Migraine | Patients are hypersensitive to dopamine agonists or dopamine transporter dysfunction. Some evidence that the dopamine D2 (DRD2) gene is involved |
| Cluster headache | Pain alleviation possibly related to dopamine receptor antagonism |
| Nausea | D2 and H1 receptor blockage |
Table 2
Antipsychotics for headache and nausea: Strength of the evidence
| Condition | Strength of evidencea |
|---|---|
| Migraine | Intermediate: Chlorpromazine,2-5 droperidol,6-8 prochlorperazine1,10-12 |
| Weak: Haloperidol13,14 | |
| Very weak: Aripiprazole,15 olanzapine,16,17 quetiapine,18 ziprasidone19 | |
| Cluster headache | Weak: Chlorpromazine20 |
| Very weak: Clozapine,21 olanzapine22 | |
| Nausea/vomiting | Intermediate: Droperidol,23 metoclopramide,24 prochlorperazine,25 promethazine25 |
| Weak: Olanzapine26,27 | |
| aStrong: Multiple, well-designed RCTs directly relevant to the recommendation, yielding consistent findings Intermediate: Some evidence from RCTs that support the recommendation, but the scientific support was not optimal Weak: Consensus recommendation in the absence of relevant randomized controlled trials and better evidence than case report or series Very weak: Case reports or case series or preliminary studies RCTs: randomized controlled trials | |
Cluster headaches
Subcutaneous sumatriptan and inhaled oxygen are first-line treatments for cluster headaches.31 A single, small study20 reported that chlorpromazine may prevent cluster headaches, which suggests that D2 receptor blockade may treat such headaches. However, limited supporting evidence relegates its use to a second- or third-line therapy.
In an open-label study (N = 5), olanzapine provided some relief of pain associated with cluster headache within 20 minutes of administration.22 In another study, patients with schizophrenia and comorbid cluster headaches improved with olanzapine.21
Because evidence is limited to small prospective studies, antipsychotic treatment of cluster headache is not well established.20-22 However, olanzapine may benefit patients with comorbid cluster headaches and schizophrenia.
Nausea
The signaling pathways that mediate emesis involve 5-HT3, D2, muscarinic, and histamine receptors.32 Before 5-HT3 antagonists were available, the FGAs metoclopramide, droperidol, prochlorperazine, and promethazine were used to manage acute emesis in emergency departments.23 A double-blind, placebo-controlled trial found IV droperidol, 1.25 mg, was more effective than metoclopramide, 10 mg, or prochlorperazine, 10 mg, for relieving moderate to severe nausea in adult patients.23 However, droperidol and prochlorperazine were associated with akathisia. In addition, this trial did not find a clinically significant difference between groups—including placebo—in anxiety, sedation, or need for rescue medications.23 Use of droperidol to treat nausea decreased after the drug received a “black-box” warning for QT prolongation and torsades de pointes.
Metoclopramide is effective for treating acute migraine and associated nausea24 and has been used to treat gastroparesis because of its effect on upper GI motility. Phenothiazines have been used to treat nausea and studies have shown prochlorperazine to be more effective than promethazine.25 Some studies of prochlorperazine have reported a 44% incidence of akathisia, which limits the drug’s use in patients who may be sensitive to such effects.33 Promethazine can cause sedation and risk of tissue necrosis at the injection site.34
Among SGAs, olanzapine effectively prevented acute and delayed chemotherapy-induced nausea and vomiting in a proof-of-concept study of patients receiving high and moderate emetogenic therapies.26,27 National Comprehensive Cancer Network guidelines cite olanzapine as a potential option for treating refractory and breakthrough emesis.35 In a small study (N = 50), olanzapine showed comparable anti-nausea effect to aprepitant—a neurokinin 1 receptor antagonist—and effectively prevented chemotherapy-induced nausea and vomiting in highly emetogenic chemotherapy.36
Related Resources
- Kelley NE, Tepper DE. Rescue therapy for acute migraine, part 2: neuroleptics, antihistamines, and others. Headache. 2012;52(2):292-306.
- Dusitanond P, Young WB. Neuroleptics and migraine. Cent Nerv Syst Agents Med Chem. 2009;9(1):63-70.
Drug Brand Names
- Aprepitant • Emend
- Aripiprazole • Abilify
- Chlorpromazine • Thorazine
- Dihydroergotamine • D.H.E 45
- Droperidol • Inapsine
- Ergotamine tartrate • Ergostat
- Haloperidol • Haldol
- Ketorolac • Toradol
- Lidocaine • Xylocaine, Lidoderm
- Meperidine • Demerol
- Metoclopramide • Reglan
- Olanzapine • Zyprexa
- Prochlorperazine • Compazine
- Promethazine • Phenergan
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sumatriptan • Imitrex
- Valproate • Depakote
Disclosures
Dr. Macaluso has received grant or research support from EnVivo Pharmaceuticals, Janssen L.P., and Pfizer, Inc.
Dr. Tripathi reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Dusitanond P, Young WB. Neuroleptics and migraine. Cent Nerv Syst Agents Med Chem. 2009;9(1):63-70.
2. McEwen JI, O’Connor HM, Dinsdale HB. Treatment of migraine with intramuscular chlorpromazine. Ann Emerg Med. 1987;16(7):758-763.
3. Bigal M, Bordini CA, Speciali JG. Intravenous chlorpromazine in the emergency department treatment of migraines: a randomized controlled trial. J Emerg Med. 2002;23(2):141-148.
4. Bell R, Montoya D, Shuaib A, et al. A comparative trial of three agents in the treatment of acute migraine headache. Ann Emerg Med. 1990;19(10):1079-1082.
5. Shrestha M, Singh R, Moreden J, et al. Ketorolac vs chlorpromazine in the treatment of acute migraine without aura. A prospective, randomized, double-blind trial. Arch Intern Med. 1996;156(15):1725-1728.
6. Wang SJ, Silberstein SD, Young WB. Droperidol treatment of status migrainosus and refractory migraine. Headache. 1997;37(6):377-382.
7. Miner JR, Fish SJ, Smith SW, et al. Droperidol vs. prochlorperazine for benign headaches in the emergency department. Acad Emerg Med. 2001;8(9):873-879.
8. Richman PB, Allegra J, Eskin B, et al. A randomized clinical trial to assess the efficacy of intramuscular droperidol for the treatment of acute migraine headache. Am J Emerg Med. 2002;20(1):39-42.
9. Jones J, Sklar D, Dougherty J, et al. Randomized double blind trial of intravenous prochlorperazine for the treatment of acute headache. JAMA. 1989;261(8):1174-1176.
10. Lu SR, Fuh JL, Juang KD, et al. Repetitive intravenous prochlorperazine treatment of patients with refractory chronic daily headache. Headache. 2000;40(9):724-729.
11. Sharma S, Prasad A, Nehru R, et al. Efficacy and tolerability of prochlorperazine buccal tablets in treatment of acute migraine. Headache. 2002;42(9):896-902.
12. Seim MB, March JA, Dunn KA. Intravenous ketorolac vs intravenous prochlorperazine for the treatment of migraine headaches. Acad Emerg Med. 1998;5(6):573-576.
13. Honkaniemi J, Liimatainen S, Rainesalo S, et al. Haloperidol in the acute treatment of migraine: a randomized, double-blind, placebo-controlled study. Headache. 2006;46(5):781-787.
14. Fisher H. A new approach to emergency department therapy of migraine headache with intravenous haloperidol: a case series. J Emerg Med. 1995;13(1):119-122.
15. LaPorta LD. Relief from migraine headache with aripiprazole treatment. Headache. 2007;47(6):922-926.
16. Silberstein SD, Peres MF, Hopkins MM, et al. Olanzapine in the treatment of refractory migraine and chronic daily headache. Headache. 2002;42(6):515-518.
17. Schreiber S, Getslev V, Backer MM, et al. The atypical neuroleptics clozapine and olanzapine differ regarding their antinociceptive mechanisms and potency. Pharmacol Biochem Behav. 1999;64(1):75-80.
18. Krymchantowski AV, Jevoux C. Quetiapine for the prevention of migraine refractory to the combination of atenolol + nortriptyline + flunarizine: an open pilot study. Arq Neuropsiquiatr. 2008;66(3B):615-618.
19. Cahill CM, Hardiman O, Murphy KC. Treatment of refractory chronic daily headache with the atypical antipsychotic ziprasidone-a case series. Cephalalgia. 2005;25(10):822-826.
20. Caviness VS, Jr, O’Brien P. Cluster headache: response to chlorpromazine. Headache. 1980;20(3):128-131.
21. Datta SS, Kumar S. Clozapine-responsive cluster headache. Neurol India. 2006;54(2):200-201.
22. Rozen TD. Olanzapine as an abortive agent for cluster headache. Headache. 2001;41(8):813-816.
23. Braude D, Soliz T, Crandall C, et al. Antiemetics in the ED: a randomized controlled trial comparing 3 common agents. Am J Emerg Med. 2006;24(2):177-182.
24. Colman I, Brown MD, Innes GD, et al. Parenteral metoclopramide for acute migraine: meta-analysis of randomised controlled trials. BMJ. 2004;329(7479):1369-1373.
25. Ernst AA, Weiss SJ, Park S, et al. Prochlorperazine versus promethazine for uncomplicated nausea and vomiting in the emergency department: a randomized, double-blind clinical trial. Ann Emerg Med. 2000;36(2):89-94.
26. Navari RM, Einhorn LH, Loehrer PJ Sr, et al. A phase II trial of olanzapine, dexamethasone, and palonosetron for the prevention of chemotherapy-induced nausea and vomiting: a Hoosier oncology group study. Support Care Cancer. 2007;15(11):1285-1291.
27. Passik SD, Navari RM, Jung SH, et al. A phase I trial of olanzapine (Zyprexa) for the prevention of delayed emesis in cancer patients: a Hoosier Oncology Group study. Cancer Invest. 2004;22(3):383-388.
28. Tanen DA, Miller S, French T, et al. Intravenous sodium valproate versus prochlorperazine for the emergency department treatment of acute migraine headaches: a prospective, randomized, double-blind trial. Ann Emerg Med. 2003;41(6):847-853.
29. Caley CF, Cooper CK. Ziprasidone: the fifth atypical antipsychotic. Ann Pharmacother. 2002;36(5):839-851.
30. Geodon [package insert]. New York NY. Pfizer Inc.; 2012.
31. Kudrow L. Response of cluster headache attacks to oxygen inhalation. Headache. 1981;21(1):1-4.
32. Scuderi PE. Pharmacology of antiemetics. Int Anesthesiol Clin. 2003;41(4):41-66.
33. Drotts DL, Vinson DR. Prochlorperazine induces akathisia in emergency patients. Ann Emerg Med. 1999;34(4):469-475.
34. Institute for Safe Medication Practices. Action needed to prevent serious tissue injury with IV promethazine. http://www.ismp.org/newsletters/acutecare/articles/20060810.asp?ptr_y. Published August 10 2006. Accessed November 28, 2012.
35. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. 2010. http://www.nccn.org/professionals/physician_gls/pdf/antiemesis.pdf. Accessed November 29 2012.
36. Navari R, Gray SE, Carr AC. Olanzapine versus aprepitant for the prevention of chemotherapy induced nausea and vomiting (CINV): a randomized phase III trial. J Clin Oncol. 2010;28(15 suppl):9020.-
1. Dusitanond P, Young WB. Neuroleptics and migraine. Cent Nerv Syst Agents Med Chem. 2009;9(1):63-70.
2. McEwen JI, O’Connor HM, Dinsdale HB. Treatment of migraine with intramuscular chlorpromazine. Ann Emerg Med. 1987;16(7):758-763.
3. Bigal M, Bordini CA, Speciali JG. Intravenous chlorpromazine in the emergency department treatment of migraines: a randomized controlled trial. J Emerg Med. 2002;23(2):141-148.
4. Bell R, Montoya D, Shuaib A, et al. A comparative trial of three agents in the treatment of acute migraine headache. Ann Emerg Med. 1990;19(10):1079-1082.
5. Shrestha M, Singh R, Moreden J, et al. Ketorolac vs chlorpromazine in the treatment of acute migraine without aura. A prospective, randomized, double-blind trial. Arch Intern Med. 1996;156(15):1725-1728.
6. Wang SJ, Silberstein SD, Young WB. Droperidol treatment of status migrainosus and refractory migraine. Headache. 1997;37(6):377-382.
7. Miner JR, Fish SJ, Smith SW, et al. Droperidol vs. prochlorperazine for benign headaches in the emergency department. Acad Emerg Med. 2001;8(9):873-879.
8. Richman PB, Allegra J, Eskin B, et al. A randomized clinical trial to assess the efficacy of intramuscular droperidol for the treatment of acute migraine headache. Am J Emerg Med. 2002;20(1):39-42.
9. Jones J, Sklar D, Dougherty J, et al. Randomized double blind trial of intravenous prochlorperazine for the treatment of acute headache. JAMA. 1989;261(8):1174-1176.
10. Lu SR, Fuh JL, Juang KD, et al. Repetitive intravenous prochlorperazine treatment of patients with refractory chronic daily headache. Headache. 2000;40(9):724-729.
11. Sharma S, Prasad A, Nehru R, et al. Efficacy and tolerability of prochlorperazine buccal tablets in treatment of acute migraine. Headache. 2002;42(9):896-902.
12. Seim MB, March JA, Dunn KA. Intravenous ketorolac vs intravenous prochlorperazine for the treatment of migraine headaches. Acad Emerg Med. 1998;5(6):573-576.
13. Honkaniemi J, Liimatainen S, Rainesalo S, et al. Haloperidol in the acute treatment of migraine: a randomized, double-blind, placebo-controlled study. Headache. 2006;46(5):781-787.
14. Fisher H. A new approach to emergency department therapy of migraine headache with intravenous haloperidol: a case series. J Emerg Med. 1995;13(1):119-122.
15. LaPorta LD. Relief from migraine headache with aripiprazole treatment. Headache. 2007;47(6):922-926.
16. Silberstein SD, Peres MF, Hopkins MM, et al. Olanzapine in the treatment of refractory migraine and chronic daily headache. Headache. 2002;42(6):515-518.
17. Schreiber S, Getslev V, Backer MM, et al. The atypical neuroleptics clozapine and olanzapine differ regarding their antinociceptive mechanisms and potency. Pharmacol Biochem Behav. 1999;64(1):75-80.
18. Krymchantowski AV, Jevoux C. Quetiapine for the prevention of migraine refractory to the combination of atenolol + nortriptyline + flunarizine: an open pilot study. Arq Neuropsiquiatr. 2008;66(3B):615-618.
19. Cahill CM, Hardiman O, Murphy KC. Treatment of refractory chronic daily headache with the atypical antipsychotic ziprasidone-a case series. Cephalalgia. 2005;25(10):822-826.
20. Caviness VS, Jr, O’Brien P. Cluster headache: response to chlorpromazine. Headache. 1980;20(3):128-131.
21. Datta SS, Kumar S. Clozapine-responsive cluster headache. Neurol India. 2006;54(2):200-201.
22. Rozen TD. Olanzapine as an abortive agent for cluster headache. Headache. 2001;41(8):813-816.
23. Braude D, Soliz T, Crandall C, et al. Antiemetics in the ED: a randomized controlled trial comparing 3 common agents. Am J Emerg Med. 2006;24(2):177-182.
24. Colman I, Brown MD, Innes GD, et al. Parenteral metoclopramide for acute migraine: meta-analysis of randomised controlled trials. BMJ. 2004;329(7479):1369-1373.
25. Ernst AA, Weiss SJ, Park S, et al. Prochlorperazine versus promethazine for uncomplicated nausea and vomiting in the emergency department: a randomized, double-blind clinical trial. Ann Emerg Med. 2000;36(2):89-94.
26. Navari RM, Einhorn LH, Loehrer PJ Sr, et al. A phase II trial of olanzapine, dexamethasone, and palonosetron for the prevention of chemotherapy-induced nausea and vomiting: a Hoosier oncology group study. Support Care Cancer. 2007;15(11):1285-1291.
27. Passik SD, Navari RM, Jung SH, et al. A phase I trial of olanzapine (Zyprexa) for the prevention of delayed emesis in cancer patients: a Hoosier Oncology Group study. Cancer Invest. 2004;22(3):383-388.
28. Tanen DA, Miller S, French T, et al. Intravenous sodium valproate versus prochlorperazine for the emergency department treatment of acute migraine headaches: a prospective, randomized, double-blind trial. Ann Emerg Med. 2003;41(6):847-853.
29. Caley CF, Cooper CK. Ziprasidone: the fifth atypical antipsychotic. Ann Pharmacother. 2002;36(5):839-851.
30. Geodon [package insert]. New York NY. Pfizer Inc.; 2012.
31. Kudrow L. Response of cluster headache attacks to oxygen inhalation. Headache. 1981;21(1):1-4.
32. Scuderi PE. Pharmacology of antiemetics. Int Anesthesiol Clin. 2003;41(4):41-66.
33. Drotts DL, Vinson DR. Prochlorperazine induces akathisia in emergency patients. Ann Emerg Med. 1999;34(4):469-475.
34. Institute for Safe Medication Practices. Action needed to prevent serious tissue injury with IV promethazine. http://www.ismp.org/newsletters/acutecare/articles/20060810.asp?ptr_y. Published August 10 2006. Accessed November 28, 2012.
35. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. 2010. http://www.nccn.org/professionals/physician_gls/pdf/antiemesis.pdf. Accessed November 29 2012.
36. Navari R, Gray SE, Carr AC. Olanzapine versus aprepitant for the prevention of chemotherapy induced nausea and vomiting (CINV): a randomized phase III trial. J Clin Oncol. 2010;28(15 suppl):9020.-
Antipsychotics for nonpsychotic illness
Second-generation antipsychotics (SGAs) represent 5% of all U.S. drug expenditures.1 Their use for indications not approved by the FDA (“off-label” use) increased to a total of $6 billion in 2008, $5.4 billion of which was for uses with limited or uncertain evidence.1
Off-label use of antipsychotics usually is based on novel applications of known receptor binding affinities (Table 1).2-5 For example, antipsychotics with strong antihistamine effects may promote sedation and could be used to treat insomnia. Clinicians also might use antipsychotics to treat a specific symptom of an illness when other treatment options are limited6 or when patients do not respond to standard treatments.
Table 1
Possible rationales for antipsychotic use for nonpsychotic conditions
| Condition | Possible rationale |
|---|---|
| Insomnia2 | Effects on H1 α-1 adrenergic and muscarinic cholinergic receptors. 5-HT2 antagonism activity also has been implicated |
| Tics of Tourette’s disorder3 | By blocking dopamine receptors antipsychotics decrease the primarily dopaminergic input from the substantia nigra and ventral tegmentum to the basal ganglia |
| Delirium4 | Patients have reversible impairment of cerebral oxidative metabolism and multiple neurotransmitter abnormalities (dopamine acetylcholine CNS γ-aminobutyric acid and serotonin). Other hypotheses include inflammatory reactions damage to certain structural pathways and disruption of cortisol and β-endorphin circadian rhythms |
| Stuttering5 | Stutterers have a marked increase in dopaminergic afferent activity in the tail of the left caudate nucleus compared with healthy controls |
| H1: histamine | |
To safely use any medication off-label, clinicians should become familiar with literature on the proposed use. Clinicians should consider off-label use only after carefully weighing the potential therapeutic benefits against the risks. Patients should be aware that the prescribed use is not FDA-approved and informed consent should include a discussion of alternative treatments. The high cost of SGAs may be a limiting factor and should be discussed with patients.
This article reviews the evidence for using antipsychotics to treat insomnia, tics, delirium, and stuttering (Table 2). Click here for a review of the evidence supporting antipsychotics for treating migraine and cluster headaches and nausea
Table 2
Antipsychotics for nonpsychotic disorders: Strength of the evidence
| Condition | Strength of evidencea |
|---|---|
| Insomnia | Weak to intermediate: Haloperidol olanzapine quetiapine risperidone ziprasidone |
| Tics of Tourette’s disorder | Strong: Haloperidol pimozide |
| Intermediate: Chlorpromazine fluphenazine penfluridol perphenazine thioridazine trifluoperazine | |
| Weak: Risperidone | |
| Very weak: Aripiprazole olanzapine quetiapine ziprasidone | |
| Not effective: Clozapine | |
| Delirium | Intermediate: Haloperidol |
| Weak: Olanzapine quetiapine risperidone | |
| Very weak: Aripiprazole ziprasidone | |
| Stuttering | Very weak: Chlorpromazine haloperidol olanzapine risperidone |
| aStrong: Multiple well-designed RCTs directly relevant to the recommendation yielding consistent findings Intermediate: Some evidence from RCTs that support the recommendation but the scientific support was not optimal Weak: Consensus recommendation in the absence of relevant RCTs and better evidence than case report or series Very weak: Case reports case series or preliminary studies RCTs: randomized controlled trials INSOMNIA Arvanitis LA, Miller BG. Multiple fixed doses of “Seroquel” (quetiapine) in patients with acute exacerbation of schizophrenia: a comparison with haloperidol and placebo. The Seroquel Trial 13 Study Group. Biol Psychiatry. 1997;42(4):233-246. Beasley CM Jr, Tollefson G, Tran P, et al. Olanzapine versus placebo and haloperidol: acute phase results of the North American double-blind olanzapine trial. Neuropsychopharmacology. 1996;14(2):111-123. Cohrs S, Meier A, Neumann AC, et al. Improved sleep continuity and increased slow wave sleep and REM latency during ziprasidone treatment: a randomized, controlled, crossover trial of 12 healthy male subjects. J Clin Psychiatry. 2005;66(8):989-996. Cohrs S, Rodenbeck A, Guan Z, et al. Sleep-promoting properties of quetiapine in healthy subjects. Psychopharmacology. 2004;174(3):421-429. Juri C, Chaná P, Tapia J, et al. Quetiapine for insomnia in Parkinson’s disease: results from an open-label trial. Clin Neuropharmacol. 2005;28(4):185-187. Marder SR, Meibach RC. Risperidone in the treatment of schizophrenia. Am J Psychiatry. 1994;151(6):825-835. Pasquini M, Speca A, Biondi M. Quetiapine for tamoxifen-induced insomnia in women with breast cancer. Psychosomatics. 2009;50(2):159-161. Sharpley AL, Vassallo CM, Cowen PJ. Olanzapine increases slow-wave sleep: evidence for blockade of central 5-HT(2C) receptors in vivo. Biol Psychiatry. 2000;47(5):468-470. Terán A, Majadas S, Galan J. Quetiapine in the treatment of sleep disturbances associated with addictive conditions: a retrospective study. Subst Use Misuse. 2008;43(14):2169-2171. Wiegand MH, Landry F, Brückner T, et al. Quetiapine in primary insomnia: a pilot study. Psychopharmacology (Berl). 2008;196(2):337-338. TICS OF TOURETTE’S DISORDER Abuzzahab FS, Anderson FO. Gilles de la Tourette’s syndrome: international registry. Minn Med. 1973;56(6):492-496. Borison RL, Ang L, Chang S, et al. New pharmacological approaches in the treatment of Tourette’s syndrome. Adv Neurol. 1982;35:377-382. Bubl E, Perlov E, Tebartz Van Elst L. Aripiprazole in patients with Tourette syndrome. World Biol J Psychiatry. 2006;7(2):123-125. Caine ED, Polinsky RJ, Kartzinel R, et al. The trial use of clozapine for abnormal involuntary movement disorders. Am J Psychiatry. 1979;136(3):317-320. Dion Y, Annable L, Sabdor P, et al. Risperidone in the treatment of Tourette’s syndrome: a double-blind, placebo-controlled trial. J Clin Psychopharmacol. 2002;22(1):31-39. McCracken JT, Suddath R, Chang S, et al. Effectiveness and tolerability of open label olanzapine in children and adolescents with Tourette’s syndrome. J Child Adolesc Psychopharmacol. 2008;18(5):501-508. Mikkelsen EJ, Detlor J, Cohen DJ. School avoidance and social phobia triggered by haloperidol in patients with Tourette’s disorder. Am J Psychiatry. 1981;138(12):1572-1576. Murphy TK, Bengston MA, Soto O, et al. Case series on the use of aripiprazole for Tourette syndrome. Int J Neuropsychopharmacol. 2005;8(3):489-490. Párraga HC, Párraga M, Woodward R, et al. Quetiapine treatment of children with Tourette’s syndrome: report of two cases. J Child Adolesc Psychopharmacol. 2001;11(2):187-191. Regeur L, Pakkenberg B, Fog R, et al. Clinical features and long-term treatment with pimozide in 65 patients with Gilles de la Tourette’s syndrome. J Neurol Neurosurg Psychiatry. 1986;49(7):791-795. Sallee FR, Kurlan R, Goetz CG, et al. Ziprasidone treatment of children and adolescents with Tourette’s syndrome: a pilot study. J Am Acad Child Adolesc Psychiatry. 2000;39(3):292-299. Sallee FR, Nesbitt L, Jackson C, et al. Relative efficacy of haloperidol and pimozide in children and adolescents with Tourette’s disorder. Am J Psychiatry. 1997;154(8):1057-1062. Scahill L, Leckman JF, Schultz RT, et al. A placebo-controlled trial of risperidone in Tourette syndrome. Neurology. 2003; 60(7):1130-1135. Shapiro AK, Shapiro E, Eisenkraft GJ. Treatment of Tourette’s disorder with penfluridol. Compr Psychiatry. 1983;24(4): 327-331. Shapiro AK, Shapiro E, Wayne HL. Treatment of Tourette’s syndrome with haloperidol: review of 34 cases. Arch Gen Psychiatry. 1973;28(1):92-96. Shapiro AK, Shapiro E, Young JG, et al. Gilles de la Tourette’s syndrome. 2nd ed. New York, NY: Raven Press; 1998:387-390. Stephens RJ, Bassel C, Sandor P. Olanzapine in the treatment of aggression and tics in children with Tourette’s syndrome-a pilot study. J Child Adolesc Psychopharmacol. 2004;14(2):255-266. DELIRIUM Alao AO, Moskowitz L. Aripiprazole and delirium. Ann Clin Psychiatry. 2006;18(4):267-269. Al-Samarrai S, Dunn J, Newmark T, et al. Quetiapine for treatment-resistant delirium. Psychosomatics. 2003;44(4): 350-351. Bourgeois JA, Hilty DM. Prolonged delirium managed with risperidone. Psychosomatics. 2005;46(1):90-91. Breitbart W, Tremblay A, Gibson C. An open trial of olanzapine for the treatment of delirium in hospitalized cancer patients. Psychosomatics. 2002;43(3):175-182. Devlin JW, Roberts RJ, Fong JJ, et al. Efficacy and safety of quetiapine in critically ill patients with delirium: a prospective, multicenter, randomized, double-blind, placebo-controlled pilot study. Crit Care Med. 2010;38(2):419-427. Hans CS, Kim YK. A double-blind trial of risperidone and haloperidol for the treatment of delirium. Psychosomatics. 2004;45(4):297-301. Horikawa N, Yamazaki T, Miyamoto K, et al. Treatment for delirium with risperidone: results of a prospective open trial with 10 patients. Gen Hosp Psychiatry. 2003;25(4):289-292. Hu H, Deng W, Yang H. A prospective random control study comparison of olanzapine and haloperidol in senile delirium [in Chinese]. Chong’qing Medical Journal. 2004;8:1234-1237. Lacasse H, Perreault MM, Williamson DR. Systematic review of antipsychotics for the treatment of hospital-associated delirium in medically or surgically ill patients. Ann Pharmacother. 2006;40(11):1966-1973. Leso L, Schwartz TL. Ziprasidone treatment of delirium. Psychosomatics. 2002;43(1):61-62. Parellada E, Baeza I, de Pablo J, et al. Risperidone in the treatment of patients with delirium. J Clin Psychiatry. 2004;65(3):348-353. Sasaki Y, Matsuyama T, Inoue S, et al. A prospective, open-label, flexible-dose study of quetiapine in the treatment of delirium. J Clin Psychiatry. 2003;64(11):1316-1321. Sipahimalani A, Masand PS. Olanzapine in the treatment of delirium. Psychosomatics. 1998;39(5):422-430. Sipahimalani A, Masand PS. Use of risperidone in delirium: case reports. Ann Clin Psychiatry. 1997;9(2):105-107. Straker DA, Shapiro PA, Muskin PR. Aripiprazole in the treatment of delirium. Psychosomatics. 2006;47(5):385-391. Young CC, Lujan E. Intravenous ziprasidone for treatment of delirium in the intensive care unit. Anesthesiology. 2004;101(3): 794-795. STUTTERING Burr HG, Mullendore JM. Recent investigations on tranquilizers and stuttering. J Speech Hear Disord. 1960;25;33-37. Lavid N, Franklin DL, Maguire GA. Management of child and adolescent stuttering with olanzapine: three case reports. Ann Clin Psychiatry. 1999;11(4):233-236. Tapia F. Haldol in the treatment of children with tics and stutterers and an incidental finding. Behav Neuropsychiatry. 1969;1(3):28. van Wattum PJ. Stuttering improved with risperidone. J Am Acad Child Adolesc Psychiatry. 2006;45(2):133. | |
Current use of antipsychotics
Antipsychotics are divided into 2 major classes—first-generation antipsychotics (FGAs) and SGAs—and principally are FDA-approved for treating schizophrenia. Some antipsychotics have received FDA approval for maintenance treatment of schizophrenia and bipolar disorder (BD), and others have been approved to treat tic disorders (haloperidol and pimozide).
To varying degrees, all antipsychotics block D2 receptors, which is thought to be necessary for treating psychosis. However, some SGAs have significant affinity at other receptors—such as 5-HT2A and 5-HT1A—that confer additional properties that are not fully understood (Table 3). For example, it is believed that 5-HT2A blockade in the striatum reduces the potential for extrapyramidal symptoms (EPS).
Each antipsychotic blocks a unique set of receptors in the brain, leading to a specific set of intended and potentially untoward effects. For example, olanzapine’s effect on psychosis largely stems from its action at the D2 receptor, whereas its sedative and anticholinergic properties are a result of activity at histamine (H1) receptors and muscarinic receptors, respectively. Clinicians can make rational use of unintended effects by carefully selecting a medication based on receptor binding profile (eg, using an antipsychotic with sedating properties in a patient who has psychosis and insomnia). This approach can limit use of multiple medications and maximize a medication’s known effects while attempting to minimize side effects.
Table 3
Antipsychotics: Receptor pharmacology and common side effects
| Antipsychotic | Pharmacology | Common side effectsa |
|---|---|---|
| Prochlorperazinea,b | D2 receptor antagonist and α-1 adrenergic receptor antagonism | EPS, akathisia, prolactinemia, orthostatic hypotension, altered cardiac conduction, agranulocytosis, sexual dysfunction |
| Chlorpromazinea,b | D2 receptor antagonist. Also binds to H1 and cholinergic M1 | EPS, akathisia, prolactinemia, orthostatic hypotension, urinary retention, non-specific QT changes, agranulocytosis, sexual dysfunction |
| Droperidola,b | D2 receptor antagonist and antagonist at peripheral α-1 activity | EPS, akathisia, prolactinemia, orthostatic hypotension, urinary retention, QT changes (dose dependent) |
| Haloperidola,b | D2 receptor antagonist. Also binds to D1, 5-HT2, H1, and α-2 adrenergic receptors | EPS, akathisia, prolactinemia, QT changes (dose dependent) |
| Aripiprazolea,c,d | D2 and 5-HT1A partial agonism, 5-HT2A antagonism | Akathisia, EPS, sedation, restlessness, insomnia, tremor, anxiety, nausea, vomiting, possible weight gain (20% to 30%) |
| Clozapinea,c,e | 5-HT2, D1, D2, D3, D4, M1, H1, α-1, and α-2 antagonism | Sedation, dizziness, tachycardia, weight gain, nausea, vomiting, constipation |
| Olanzapinea,c | 5-HT2A, 5-HT2C, D1, D2, D3, D4, M1-5, H1, and α1- antagonism | Sedation, EPS, prolactinemia, weight gain, constipation |
| Quetiapinea,c,d | D1, D2, 5-HT2A, 5-HT1A, H1, α-1, and α-2 antagonism | Sedation, orthostatic hypotension, weight gain, triglyceride abnormalities, hypertension (frequently diastolic), constipation |
| Risperidonea,c | 5-HT2, D2, H1, α-1, and α-2 antagonism | Sedation, akathisia, EPS, prolactinemia, weight gain, tremor |
| Ziprasidonea,c | D2, D3, 5-HT2A, 5-HT2C, 5-HT1D, and α-1 antagonism; moderate inhibition of 5-HT and NE reuptake; 5-HT1A agonism | EPS, sedation, headache, dizziness, nausea |
| aSide effects and their prominence usually are based on receptor binding profile. All antipsychotics to varying degrees share the following symptoms: EPS, neuroleptic malignant syndrome, QTc prolongation, anticholinergic side effects (urinary retention, decreased gastrointestinal motility, xerostomia), sedation, orthostatic hypotension, blood dyscrasias, and problems with temperature regulation. The class as a whole also carries a “black-box” warning regarding increased mortality when treating geriatric patients with psychosis related to dementia bNo frequencies were available cOnly side effects with frequency >10% listed d”Black-box” warning for suicidal ideation and behavior in children, adolescents, and young adults (age 18 to 24) with major depressive disorder and other psychiatric disorders e”Black-box” warnings for agranulocytosis, myocarditis, orthostatic hypotension, seizure risk EPS: extrapyramidal symptoms; H1: histamine; M1: muscarinic; NE: norepinephrine | ||
Insomnia
Clinicians use FGAs and SGAs to treat insomnia because of their sedating effects, although evidence supporting this use is questionable. Among the FGAs, chlorpromazine produces moderate to severe sedation, whereas haloperidol is only mildly sedating. Clozapine is believed to be the most sedating SGA, whereas quetiapine and olanzapine produce moderate sedation.7
Most data on antipsychotics’ sedating effects comes from studies completed for schizophrenia or BD. Few studies have evaluated using antipsychotics to treat primary insomnia or other sleep disorders in otherwise healthy patients.2 However, data from phase I studies of antipsychotics has shown that schizophrenia patients tolerate a higher maximum dose compared with healthy volunteers, who often experience more sedation.
An antipsychotic’s potential for sedation is directly related to its affinity at H1 receptors and total drug concentration at the H1 receptor binding site. Because drugs with lower affinity for D2 receptors typically are prescribed at higher doses when treating psychiatric illness, the corresponding concentration at H1 receptors can lead to greater sedation compared with equivalent doses of higher-potency agents.
The same phenomenon is seen with high-potency agents. Haloperidol has a relatively weak binding affinity to the H1 receptor,8 but causes more sedation at higher doses. Haloperidol, 20 mg/d, produces sedation in more patients than a moderate dose of risperidone, 2 to 10 mg/d.8 These observations correlate with “the high milligram-low-potency” spectrum seen with FGAs.7
Among SGAs, a double-blind, placebo-controlled, crossover study of the effects of ziprasidone, 40 mg/d, on sleep in a group of healthy volunteers found a significant increase in total sleep time and sleep efficiency.9 A double-blind trial compared patients taking low, medium, or high daily doses of olanzapine with patients receiving haloperidol or placebo.10 Sedation was reported in 20% of patients taking low doses of olanzapine (5 ± 2.5 mg/d) compared with 29.7% on medium doses (10 ± 2.5 mg/d) and 39.1% on high doses (15 ± 2.5 mg/d).10
A double-blind, placebo-controlled, crossover study demonstrated that olanzapine produced significant increases in sleep continuity, slow wave sleep, and subjective ratings of sleep quality in healthy men.11 Similarly, a study comparing haloperidol, 12 mg/d, and quetiapine, 75 to 750 mg/d, for treating acute schizophrenia found an 8% to 11% incidence of somnolence in the quetiapine group compared with 6% and 8% in the haloperidol and placebo groups, respectively.12 Somnolence was reported as an adverse event in these studies, which were designed to examine the drug’s effect on acute schizophrenia and did not evaluate its effect on sleep.
A double-blind, placebo-controlled, crossover study examining quetiapine’s effects on sleep in 14 healthy patients demonstrated a significant difference in total sleep time, sleep period time, and sleep efficiency.13 Similarly, an open-label pilot study of quetiapine’s effect on primary insomnia showed significant improvement in total sleep time and sleep efficiency.14
Studies examining quetiapine’s effects on insomnia in patients with substance abuse15 and women with localized breast cancer16 showed improved sleep scores on multiple assessment tools, while an open-label study of quetiapine for Parkinson’s disease demonstrated decreased sleep latency.17 Adjunctive quetiapine administered over a 6-week, open-label trial in veterans with posttraumatic stress disorder revealed significant improvement from baseline in sleep quality and duration and diminished dreaming.18
Sedating antipsychotics such as thioridazine and chlorpromazine historically were used off-label for insomnia, but fell out of favor because of their associated cardiac risks. More recently, clinicians have been using SGAs in a similar manner19 even though SGAs are costly and have significant risks such as metabolic problems.
Studies supporting the use of SGAs for the short-term or long-term treatment of insomnia are limited by small sample sizes or open-label designs.20 In 2005 the National Institutes of Health State-of-the-Science Conference Panel did not recommend using SGAs for treating chronic insomnia.21
Tics in Tourette’s disorder
FGAs and SGAs have been used to treat tics associated with Tourette’s disorder (TD).22 Haloperidol is FDA-approved for treating tics in adult and pediatric patients with TD. Many studies have reported the efficacy of haloperidol in this population; however, cognitive blunting, weight gain, lethargy, and akathisia limit its use.23
Pimozide, the most widely used alternative to haloperidol for treating TD, can cause clinically significant QTc prolongation and sudden death. Penfluridol demonstrated significant symptomatic improvement compared with haloperidol in 1 study, but its carcinogenic potential limits its use.24
A double-blind, placebo-controlled study comparing fluphenazine and trifluoperazine with haloperidol for treating TD showed that both are significantly more effective than placebo, but none was more effective than the others.25 Studies show chlorpromazine, perphenazine, and thioridazine are less effective than haloperidol and their use is limited by photosensitivity, dermatitis, EPS, and blood and liver dyscrasias.26
Risperidone is superior to placebo for treating tics associated with TD.27 A placebo-controlled trial of ziprasidone showed the drug has efficacy similar to risperidone in reducing tics in children and adolescents with TD.28 However, ziprasidone is not FDA-approved for this use.
Evidence supporting the use of other SGAs for treating TD is more limited. Several small studies of olanzapine and aripiprazole had limited but favorable results. Quetiapine has not been studied for treating TD, but several case reports have indicated a positive response. In a double-blind, placebo-controlled trial, clozapine showed no therapeutic benefit for TD.29
Delirium
American Psychiatric Association practice guidelines suggest using psychotropic medications to treat neuropsychiatric symptoms of delirium.30 Antipsychotics are considered first-line agents that lower hospital mortality rates, decrease lengths of hospital stays, and improve delirium symptoms, in some cases before the underlying medical etiologies resolve.30,31 Available in liquid, oral, IM, and IV formulations, haloperidol is the mainstay of symptomatic treatment of delirium.31 Although not FDA-approved, it is recommended by the Society of Critical Care Medicine as a safe, cost-effective, and efficacious therapy for the psychiatric symptoms associated with delirium.
The most extensively studied SGA for treating delirium, risperidone often is used as an alternative to haloperidol. Case reports describe its potential efficacy.32 In a head-to-head study, risperidone was as effective as low-dose haloperidol for acute delirium treatment.33
Olanzapine was effective in managing delirium in several case studies.34 Also, in a 7-day, randomized, placebo-controlled study, olanzapine and haloperidol showed significantly greater and relatively equivalent improvement compared with placebo; patients treated with olanzapine experienced more rapid improvement in 1 study.35
Case reports and prospective studies also have described quetiapine as effective for treating delirium.36,37 In a prospective, double-blind, placebo-controlled study, patients taking quetiapine had a faster resolution of delirium with reduced overall duration and less agitation than those taking placebo.37 Mortality, intensive care unit length of stay, and incidence of QTc prolongation did not differ, but patients treated with quetiapine were more likely to have increased somnolence and were more frequently discharged to home or rehabilitation centers. One limitation of the study is that concomitant haloperidol use on an “as needed” basis was permitted.38
Evidence supporting the efficacy of ziprasidone for delirium is limited to case reports.39 In 1 case report, a patient with chronic HIV infection and acute cryptococcal meningitis experienced significant improvement of delirium symptoms but could not continue ziprasidone because of fluctuating QTc intervals.40
In 2 patients with delirium, aripiprazole, 15 and 30 mg/d, improved confusion, disorientation, and agitation within 7 days.41 In another study of delirium, 13 of 14 patients on flexibly dosed aripiprazole (5 to 15 mg/d) showed improvement in Clinical Global Impressions Scale scores, although 3 patients developed prolonged QTc intervals.42
Stuttering or stammering
Stuttering or stammering are age-inappropriate disturbances in normal fluency and time patterning of speech. The evidence for antipsychotics to treat stuttering or stammering speech mainly consists of case reports and does not include disfluency frequency data, which makes it difficult to accept claims of efficacy. Disfluency frequency data describe how often a patient has specific disfluencies (blocks, prolongations, interjection, and repetition of syllables, words, or phrases).
Two FGAs (chlorpromazine and haloperidol) and 2 SGAs (risperidone and olanzapine) have been evaluated for treating stuttering. Children were 2.5 times more likely to demonstrate significant improvement when taking chlorpromazine vs placebo.43 An open-label study of haloperidol lacked disfluency frequency data, therefore casting doubts on haloperidol’s reported efficacy in the study.44
In a case report, a 4-year-old boy with severe behavioral dyscontrol showed complete remission of stammering after 1 day of risperidone, 0.25 mg/d.45 The patient’s symptoms reappeared several days after the drug was stopped. In a case series of 2 patients with developmental stuttering, 1 patient reported significant improvement in fluency with olanzapine, 2.5 mg/d, and the other showed marked improvement in fluency with 5 mg/d.46
Related Resources
- Sipahimalani A, Masand PS. Use of risperidone in delirium: case reports. Ann Clin Psychiatry. 1997;9(2):105-107.
- Shapiro AK, Shapiro E, Wayne HL. Treatment of Tourette’s syndrome with haloperidol: review of 34 cases. Arch Gen Psychiatry. 1973;28(1):92-96.
- Sipahimalani A, Masand PS. Olanzapine in the treatment of delirium. Psychosomatics. 1998;39(5):422-430.
Drug Brand Names
- Aripiprazole • Abilify
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Fluphenazine • Permitil, Prolixin
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Pimozide • Orap
- Prochlorperazine • Compazine
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Trifluoperazine • Stelazine
- Ziprasidone • Geodon
Disclosure
Dr. Macaluso has received grant or research support from EnVivo Pharmaceuticals, Janssen, L.P., and Pfizer, Inc.
Dr. Tripathi reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Alexander GC, Gallagher SA, Mascola A, et al. Increasing off-label use of antipsychotic medications in the United States, 1995-2008. Pharmacoepidemiol Drug Saf. 2011;20(2):177-184.
2. DeMartinis N, Winokur A. Effects of psychiatric medications on sleep and sleep disorders. CNS Neurol Disord Drug Targets. 2007;6(1):17-29.
3. Leckman JF, Bloch MH, Smith ME, et al. Neurobiological substrates of Tourette’s disorder. J Child Adolesc Psychopharmacol. 2010;20(4):237-247.
4. Maldonado JR. Pathoetiological model of delirium: a comprehensive understanding of the neurobiology of delirium and an evidence-based approach to prevention and treatment. Crit Care Clin. 2008;24(4):789-856.
5. Wu JC, Maguire G, Riley G, et al. Increased dopamine activity associated with stuttering. Neuroreport. 1997;8(3):767-770.
6. Devulapalli K, Nasrallah HA. An analysis of the high psychotropic off-label use in psychiatric disorders: the majority of psychiatric diagnoses have no approved drug. Asian J Psychiatr. 2009;2(1):29-36.
7. Miller DD. Atypical antipsychotics: sleep sedation, and efficacy. Prim Care Companion J Clin Psychiatry. 2004;6(suppl 2):3-7.
8. Marder SR, Meibach RC. Risperidone in the treatment of schizophrenia. Am J Psychiatry. 1994;151(6):825-835.
9. Cohrs S, Meier A, Neumann AC, et al. Improved sleep continuity and increased slow wave sleep and REM latency during ziprasidone treatment: a randomized, controlled, crossover trial of 12 healthy male subjects. J Clin Psychiatry. 2005;66(8):989-996.
10. Beasley CM Jr, Tollefson G, Tran P, et al. Olanzapine versus placebo and haloperidol: acute phase results of the North American double-blind olanzapine trial. Neuropsychopharmacology. 1996;14(2):111-123.
11. Sharpley AL, Vassallo CM, Cowen PJ. Olanzapine increases slow-wave sleep: evidence for blockade of central 5-HT(2C) receptors in vivo. Biol Psychiatry. 2000;47(5):468-470.
12. Arvanitis LA, Miller BG. Multiple fixed doses of “Seroquel” (quetiapine) in patients with acute exacerbation of schizophrenia: a comparison with haloperidol and placebo. The Seroquel Trial 13 Study Group. Biol Psychiatry. 1997;42(4):233-246.
13. Cohrs S, Rodenbeck A, Guan Z, et al. Sleep-promoting properties of quetiapine in healthy subjects. Psychopharmacology. 2004;174(3):421-429.
14. Wiegand MH, Landry F, Brückner T, et al. Quetiapine in primary insomnia: a pilot study. Psychopharmacology (Berl). 2008;196(2):337-338.
15. Terán A, Majadas S, Galan J. Quetiapine in the treatment of sleep disturbances associated with addictive conditions: a retrospective study. Subst Use Misuse. 2008;43(14):2169-2171.
16. Pasquini M, Speca A, Biondi M. Quetiapine for tamoxifen-induced insomnia in women with breast cancer. Psychosomatics. 2009;50(2):159-161.
17. Juri C, Chaná P, Tapia J, et al. Quetiapine for insomnia in Parkinson’s disease: results from an open-label trial. Clin Neuropharmacol. 2005;28(4):185-187.
18. Robert S, Hamner MB, Kose S, et al. Quetiapine improves sleep disturbances in combat veterans with PTSD: sleep data from a prospective, open-label study. J Clin Psychopharmacol. 2005;25(4):387-388.
19. Wilson S, Nutt D. Management of insomnia: treatments and mechanisms. Br J Psychiatry. 2007;191:195-197.
20. Morin CM, Benca R. Chronic insomnia. Lancet. 2012;379(9821):1129-1141.
21. National Institutes of Health. National Institutes of Health State of the Science Conference statement on manifestations and management of chronic insomnia in adults June 13-15, 2005. Sleep. 2005;28(9):1049-1057.
22. Párraga HC, Harris KM, Párraga KL, et al. An overview of the treatment of Tourette’s disorder and tics. J Child Adolesc Psychopharmacol. 2010;20(4):249-262.
23. Mikkelsen EJ, Detlor J, Cohen DJ. School avoidance and social phobia triggered by haloperidol in patients with Tourette’s disorder. Am J Psychiatry. 1981;138(12):1572-1576.
24. Shapiro AK, Shapiro E, Eisenkraft GJ. Treatment of Tourette’s disorder with penfluridol. Compr Psychiatry. 1983;24(4):327-331.
25. Borison RL, Ang L, Chang S, et al. New pharmacological approaches in the treatment of Tourette’s syndrome. Adv Neurol. 1982;35:377-382.
26. Shapiro AK, Shapiro E, Young JG, et al. Gilles de la Tourette’s syndrome. 2nd ed. New York, NY: Raven Press; 1998:387–390.
27. Dion Y, Annable L, Sabdor P, et al. Risperidone in the treatment of Tourette’s syndrome: a double-blind, placebo-controlled trial. J Clin Psychopharmacol. 2002;22(1):31-39.
28. Sallee FR, Kurlan R, Goetz CG, et al. Ziprasidone treatment of children and adolescents with Tourette’s syndrome: a pilot study. J Am Acad Child Adolesc Psychiatry. 2000;39(3):292-299.
29. Caine ED, Polinsky RJ, Kartzinel R, et al. The trial use of clozapine for abnormal involuntary movement disorders. Am J Psychiatry. 1979;136(3):317-320.
30. American Psychiatric Association. Practice guideline for the treatment of patients with delirium. Am J Psychiatry. 1999;156(suppl 5):1-20.
31. Lacasse H, Perreault MM, Williamson DR. Systematic review of antipsychotics for the treatment of hospital-associated delirium in medically or surgically ill patients. Ann Pharmacother. 2006;40(11):1966-1973.
32. Parellada E, Baeza I, de Pablo J, et al. Risperidone in the treatment of patients with delirium. J Clin Psychiatry. 2004;65(3):348-353.
33. Hans CS, Kim YK. A double-blind trial of risperidone and haloperidol for the treatment of delirium. Psychosomatics. 2004;45(4):297-301.
34. Breitbart W, Tremblay A, Gibson C. An open trial of olanzapine for the treatment of delirium in hospitalized cancer patients. Psychosomatics. 2002;43(3):175-182.
35. Hu H, Deng W, Yang H. A prospective random control study comparison of olanzapine and haloperidol in senile delirium [in Chinese]. Chong’qing Medical Journal. 2004;8:1234-1237.
36. Al-Samarrai S, Dunn J, Newmark T, et al. Quetiapine for treatment-resistant delirium. Psychosomatics. 2003;44(4):350-351.
37. Sasaki Y, Matsuyama T, Inoue S, et al. A prospective, open-label, flexible-dose study of quetiapine in the treatment of delirium. J Clin Psychiatry. 2003;64(11):1316-1321.
38. Devlin JW, Roberts RJ, Fong JJ, et al. Efficacy and safety of quetiapine in critically ill patients with delirium: a prospective, multicenter, randomized, double-blind, placebo-controlled pilot study. Crit Care Med. 2010;38(2):419-427.
39. Young CC, Lujan E. Intravenous ziprasidone for treatment of delirium in the intensive care unit. Anesthesiology. 2004;101(3):794-795.
40. Leso L, Schwartz TL. Ziprasidone treatment of delirium. Psychosomatics. 2002;43(1):61-62.
41. Alao AO, Moskowitz L. Aripiprazole and delirium. Ann Clin Psychiatry. 2006;18(4):267-269.
42. Straker DA, Shapiro PA, Muskin PR. Aripiprazole in the treatment of delirium. Psychosomatics. 2006;47(5):385-391.
43. Burr HG, Mullendore JM. Recent investigations on tranquilizers and stuttering. J Speech Hear Disord. 1960;25:33-37.
44. Tapia F. Haldol in the treatment of children with tics and stutterers and an incidental finding. Behav Neuropsychiatry. 1969;1(3):28.-
45. van Wattum PJ. Stuttering improved with risperidone. J Am Acad Child Adolesc Psychiatry. 2006;45(2):133.-
46. Lavid N, Franklin DL, Maguire GA. Management of child and adolescent stuttering with olanzapine: three case reports. Ann Clin Psychiatry. 1999;11(4):233-236.
Second-generation antipsychotics (SGAs) represent 5% of all U.S. drug expenditures.1 Their use for indications not approved by the FDA (“off-label” use) increased to a total of $6 billion in 2008, $5.4 billion of which was for uses with limited or uncertain evidence.1
Off-label use of antipsychotics usually is based on novel applications of known receptor binding affinities (Table 1).2-5 For example, antipsychotics with strong antihistamine effects may promote sedation and could be used to treat insomnia. Clinicians also might use antipsychotics to treat a specific symptom of an illness when other treatment options are limited6 or when patients do not respond to standard treatments.
Table 1
Possible rationales for antipsychotic use for nonpsychotic conditions
| Condition | Possible rationale |
|---|---|
| Insomnia2 | Effects on H1 α-1 adrenergic and muscarinic cholinergic receptors. 5-HT2 antagonism activity also has been implicated |
| Tics of Tourette’s disorder3 | By blocking dopamine receptors antipsychotics decrease the primarily dopaminergic input from the substantia nigra and ventral tegmentum to the basal ganglia |
| Delirium4 | Patients have reversible impairment of cerebral oxidative metabolism and multiple neurotransmitter abnormalities (dopamine acetylcholine CNS γ-aminobutyric acid and serotonin). Other hypotheses include inflammatory reactions damage to certain structural pathways and disruption of cortisol and β-endorphin circadian rhythms |
| Stuttering5 | Stutterers have a marked increase in dopaminergic afferent activity in the tail of the left caudate nucleus compared with healthy controls |
| H1: histamine | |
To safely use any medication off-label, clinicians should become familiar with literature on the proposed use. Clinicians should consider off-label use only after carefully weighing the potential therapeutic benefits against the risks. Patients should be aware that the prescribed use is not FDA-approved and informed consent should include a discussion of alternative treatments. The high cost of SGAs may be a limiting factor and should be discussed with patients.
This article reviews the evidence for using antipsychotics to treat insomnia, tics, delirium, and stuttering (Table 2). Click here for a review of the evidence supporting antipsychotics for treating migraine and cluster headaches and nausea
Table 2
Antipsychotics for nonpsychotic disorders: Strength of the evidence
| Condition | Strength of evidencea |
|---|---|
| Insomnia | Weak to intermediate: Haloperidol olanzapine quetiapine risperidone ziprasidone |
| Tics of Tourette’s disorder | Strong: Haloperidol pimozide |
| Intermediate: Chlorpromazine fluphenazine penfluridol perphenazine thioridazine trifluoperazine | |
| Weak: Risperidone | |
| Very weak: Aripiprazole olanzapine quetiapine ziprasidone | |
| Not effective: Clozapine | |
| Delirium | Intermediate: Haloperidol |
| Weak: Olanzapine quetiapine risperidone | |
| Very weak: Aripiprazole ziprasidone | |
| Stuttering | Very weak: Chlorpromazine haloperidol olanzapine risperidone |
| aStrong: Multiple well-designed RCTs directly relevant to the recommendation yielding consistent findings Intermediate: Some evidence from RCTs that support the recommendation but the scientific support was not optimal Weak: Consensus recommendation in the absence of relevant RCTs and better evidence than case report or series Very weak: Case reports case series or preliminary studies RCTs: randomized controlled trials INSOMNIA Arvanitis LA, Miller BG. Multiple fixed doses of “Seroquel” (quetiapine) in patients with acute exacerbation of schizophrenia: a comparison with haloperidol and placebo. The Seroquel Trial 13 Study Group. Biol Psychiatry. 1997;42(4):233-246. Beasley CM Jr, Tollefson G, Tran P, et al. Olanzapine versus placebo and haloperidol: acute phase results of the North American double-blind olanzapine trial. Neuropsychopharmacology. 1996;14(2):111-123. Cohrs S, Meier A, Neumann AC, et al. Improved sleep continuity and increased slow wave sleep and REM latency during ziprasidone treatment: a randomized, controlled, crossover trial of 12 healthy male subjects. J Clin Psychiatry. 2005;66(8):989-996. Cohrs S, Rodenbeck A, Guan Z, et al. Sleep-promoting properties of quetiapine in healthy subjects. Psychopharmacology. 2004;174(3):421-429. Juri C, Chaná P, Tapia J, et al. Quetiapine for insomnia in Parkinson’s disease: results from an open-label trial. Clin Neuropharmacol. 2005;28(4):185-187. Marder SR, Meibach RC. Risperidone in the treatment of schizophrenia. Am J Psychiatry. 1994;151(6):825-835. Pasquini M, Speca A, Biondi M. Quetiapine for tamoxifen-induced insomnia in women with breast cancer. Psychosomatics. 2009;50(2):159-161. Sharpley AL, Vassallo CM, Cowen PJ. Olanzapine increases slow-wave sleep: evidence for blockade of central 5-HT(2C) receptors in vivo. Biol Psychiatry. 2000;47(5):468-470. Terán A, Majadas S, Galan J. Quetiapine in the treatment of sleep disturbances associated with addictive conditions: a retrospective study. Subst Use Misuse. 2008;43(14):2169-2171. Wiegand MH, Landry F, Brückner T, et al. Quetiapine in primary insomnia: a pilot study. Psychopharmacology (Berl). 2008;196(2):337-338. TICS OF TOURETTE’S DISORDER Abuzzahab FS, Anderson FO. Gilles de la Tourette’s syndrome: international registry. Minn Med. 1973;56(6):492-496. Borison RL, Ang L, Chang S, et al. New pharmacological approaches in the treatment of Tourette’s syndrome. Adv Neurol. 1982;35:377-382. Bubl E, Perlov E, Tebartz Van Elst L. Aripiprazole in patients with Tourette syndrome. World Biol J Psychiatry. 2006;7(2):123-125. Caine ED, Polinsky RJ, Kartzinel R, et al. The trial use of clozapine for abnormal involuntary movement disorders. Am J Psychiatry. 1979;136(3):317-320. Dion Y, Annable L, Sabdor P, et al. Risperidone in the treatment of Tourette’s syndrome: a double-blind, placebo-controlled trial. J Clin Psychopharmacol. 2002;22(1):31-39. McCracken JT, Suddath R, Chang S, et al. Effectiveness and tolerability of open label olanzapine in children and adolescents with Tourette’s syndrome. J Child Adolesc Psychopharmacol. 2008;18(5):501-508. Mikkelsen EJ, Detlor J, Cohen DJ. School avoidance and social phobia triggered by haloperidol in patients with Tourette’s disorder. Am J Psychiatry. 1981;138(12):1572-1576. Murphy TK, Bengston MA, Soto O, et al. Case series on the use of aripiprazole for Tourette syndrome. Int J Neuropsychopharmacol. 2005;8(3):489-490. Párraga HC, Párraga M, Woodward R, et al. Quetiapine treatment of children with Tourette’s syndrome: report of two cases. J Child Adolesc Psychopharmacol. 2001;11(2):187-191. Regeur L, Pakkenberg B, Fog R, et al. Clinical features and long-term treatment with pimozide in 65 patients with Gilles de la Tourette’s syndrome. J Neurol Neurosurg Psychiatry. 1986;49(7):791-795. Sallee FR, Kurlan R, Goetz CG, et al. Ziprasidone treatment of children and adolescents with Tourette’s syndrome: a pilot study. J Am Acad Child Adolesc Psychiatry. 2000;39(3):292-299. Sallee FR, Nesbitt L, Jackson C, et al. Relative efficacy of haloperidol and pimozide in children and adolescents with Tourette’s disorder. Am J Psychiatry. 1997;154(8):1057-1062. Scahill L, Leckman JF, Schultz RT, et al. A placebo-controlled trial of risperidone in Tourette syndrome. Neurology. 2003; 60(7):1130-1135. Shapiro AK, Shapiro E, Eisenkraft GJ. Treatment of Tourette’s disorder with penfluridol. Compr Psychiatry. 1983;24(4): 327-331. Shapiro AK, Shapiro E, Wayne HL. Treatment of Tourette’s syndrome with haloperidol: review of 34 cases. Arch Gen Psychiatry. 1973;28(1):92-96. Shapiro AK, Shapiro E, Young JG, et al. Gilles de la Tourette’s syndrome. 2nd ed. New York, NY: Raven Press; 1998:387-390. Stephens RJ, Bassel C, Sandor P. Olanzapine in the treatment of aggression and tics in children with Tourette’s syndrome-a pilot study. J Child Adolesc Psychopharmacol. 2004;14(2):255-266. DELIRIUM Alao AO, Moskowitz L. Aripiprazole and delirium. Ann Clin Psychiatry. 2006;18(4):267-269. Al-Samarrai S, Dunn J, Newmark T, et al. Quetiapine for treatment-resistant delirium. Psychosomatics. 2003;44(4): 350-351. Bourgeois JA, Hilty DM. Prolonged delirium managed with risperidone. Psychosomatics. 2005;46(1):90-91. Breitbart W, Tremblay A, Gibson C. An open trial of olanzapine for the treatment of delirium in hospitalized cancer patients. Psychosomatics. 2002;43(3):175-182. Devlin JW, Roberts RJ, Fong JJ, et al. Efficacy and safety of quetiapine in critically ill patients with delirium: a prospective, multicenter, randomized, double-blind, placebo-controlled pilot study. Crit Care Med. 2010;38(2):419-427. Hans CS, Kim YK. A double-blind trial of risperidone and haloperidol for the treatment of delirium. Psychosomatics. 2004;45(4):297-301. Horikawa N, Yamazaki T, Miyamoto K, et al. Treatment for delirium with risperidone: results of a prospective open trial with 10 patients. Gen Hosp Psychiatry. 2003;25(4):289-292. Hu H, Deng W, Yang H. A prospective random control study comparison of olanzapine and haloperidol in senile delirium [in Chinese]. Chong’qing Medical Journal. 2004;8:1234-1237. Lacasse H, Perreault MM, Williamson DR. Systematic review of antipsychotics for the treatment of hospital-associated delirium in medically or surgically ill patients. Ann Pharmacother. 2006;40(11):1966-1973. Leso L, Schwartz TL. Ziprasidone treatment of delirium. Psychosomatics. 2002;43(1):61-62. Parellada E, Baeza I, de Pablo J, et al. Risperidone in the treatment of patients with delirium. J Clin Psychiatry. 2004;65(3):348-353. Sasaki Y, Matsuyama T, Inoue S, et al. A prospective, open-label, flexible-dose study of quetiapine in the treatment of delirium. J Clin Psychiatry. 2003;64(11):1316-1321. Sipahimalani A, Masand PS. Olanzapine in the treatment of delirium. Psychosomatics. 1998;39(5):422-430. Sipahimalani A, Masand PS. Use of risperidone in delirium: case reports. Ann Clin Psychiatry. 1997;9(2):105-107. Straker DA, Shapiro PA, Muskin PR. Aripiprazole in the treatment of delirium. Psychosomatics. 2006;47(5):385-391. Young CC, Lujan E. Intravenous ziprasidone for treatment of delirium in the intensive care unit. Anesthesiology. 2004;101(3): 794-795. STUTTERING Burr HG, Mullendore JM. Recent investigations on tranquilizers and stuttering. J Speech Hear Disord. 1960;25;33-37. Lavid N, Franklin DL, Maguire GA. Management of child and adolescent stuttering with olanzapine: three case reports. Ann Clin Psychiatry. 1999;11(4):233-236. Tapia F. Haldol in the treatment of children with tics and stutterers and an incidental finding. Behav Neuropsychiatry. 1969;1(3):28. van Wattum PJ. Stuttering improved with risperidone. J Am Acad Child Adolesc Psychiatry. 2006;45(2):133. | |
Current use of antipsychotics
Antipsychotics are divided into 2 major classes—first-generation antipsychotics (FGAs) and SGAs—and principally are FDA-approved for treating schizophrenia. Some antipsychotics have received FDA approval for maintenance treatment of schizophrenia and bipolar disorder (BD), and others have been approved to treat tic disorders (haloperidol and pimozide).
To varying degrees, all antipsychotics block D2 receptors, which is thought to be necessary for treating psychosis. However, some SGAs have significant affinity at other receptors—such as 5-HT2A and 5-HT1A—that confer additional properties that are not fully understood (Table 3). For example, it is believed that 5-HT2A blockade in the striatum reduces the potential for extrapyramidal symptoms (EPS).
Each antipsychotic blocks a unique set of receptors in the brain, leading to a specific set of intended and potentially untoward effects. For example, olanzapine’s effect on psychosis largely stems from its action at the D2 receptor, whereas its sedative and anticholinergic properties are a result of activity at histamine (H1) receptors and muscarinic receptors, respectively. Clinicians can make rational use of unintended effects by carefully selecting a medication based on receptor binding profile (eg, using an antipsychotic with sedating properties in a patient who has psychosis and insomnia). This approach can limit use of multiple medications and maximize a medication’s known effects while attempting to minimize side effects.
Table 3
Antipsychotics: Receptor pharmacology and common side effects
| Antipsychotic | Pharmacology | Common side effectsa |
|---|---|---|
| Prochlorperazinea,b | D2 receptor antagonist and α-1 adrenergic receptor antagonism | EPS, akathisia, prolactinemia, orthostatic hypotension, altered cardiac conduction, agranulocytosis, sexual dysfunction |
| Chlorpromazinea,b | D2 receptor antagonist. Also binds to H1 and cholinergic M1 | EPS, akathisia, prolactinemia, orthostatic hypotension, urinary retention, non-specific QT changes, agranulocytosis, sexual dysfunction |
| Droperidola,b | D2 receptor antagonist and antagonist at peripheral α-1 activity | EPS, akathisia, prolactinemia, orthostatic hypotension, urinary retention, QT changes (dose dependent) |
| Haloperidola,b | D2 receptor antagonist. Also binds to D1, 5-HT2, H1, and α-2 adrenergic receptors | EPS, akathisia, prolactinemia, QT changes (dose dependent) |
| Aripiprazolea,c,d | D2 and 5-HT1A partial agonism, 5-HT2A antagonism | Akathisia, EPS, sedation, restlessness, insomnia, tremor, anxiety, nausea, vomiting, possible weight gain (20% to 30%) |
| Clozapinea,c,e | 5-HT2, D1, D2, D3, D4, M1, H1, α-1, and α-2 antagonism | Sedation, dizziness, tachycardia, weight gain, nausea, vomiting, constipation |
| Olanzapinea,c | 5-HT2A, 5-HT2C, D1, D2, D3, D4, M1-5, H1, and α1- antagonism | Sedation, EPS, prolactinemia, weight gain, constipation |
| Quetiapinea,c,d | D1, D2, 5-HT2A, 5-HT1A, H1, α-1, and α-2 antagonism | Sedation, orthostatic hypotension, weight gain, triglyceride abnormalities, hypertension (frequently diastolic), constipation |
| Risperidonea,c | 5-HT2, D2, H1, α-1, and α-2 antagonism | Sedation, akathisia, EPS, prolactinemia, weight gain, tremor |
| Ziprasidonea,c | D2, D3, 5-HT2A, 5-HT2C, 5-HT1D, and α-1 antagonism; moderate inhibition of 5-HT and NE reuptake; 5-HT1A agonism | EPS, sedation, headache, dizziness, nausea |
| aSide effects and their prominence usually are based on receptor binding profile. All antipsychotics to varying degrees share the following symptoms: EPS, neuroleptic malignant syndrome, QTc prolongation, anticholinergic side effects (urinary retention, decreased gastrointestinal motility, xerostomia), sedation, orthostatic hypotension, blood dyscrasias, and problems with temperature regulation. The class as a whole also carries a “black-box” warning regarding increased mortality when treating geriatric patients with psychosis related to dementia bNo frequencies were available cOnly side effects with frequency >10% listed d”Black-box” warning for suicidal ideation and behavior in children, adolescents, and young adults (age 18 to 24) with major depressive disorder and other psychiatric disorders e”Black-box” warnings for agranulocytosis, myocarditis, orthostatic hypotension, seizure risk EPS: extrapyramidal symptoms; H1: histamine; M1: muscarinic; NE: norepinephrine | ||
Insomnia
Clinicians use FGAs and SGAs to treat insomnia because of their sedating effects, although evidence supporting this use is questionable. Among the FGAs, chlorpromazine produces moderate to severe sedation, whereas haloperidol is only mildly sedating. Clozapine is believed to be the most sedating SGA, whereas quetiapine and olanzapine produce moderate sedation.7
Most data on antipsychotics’ sedating effects comes from studies completed for schizophrenia or BD. Few studies have evaluated using antipsychotics to treat primary insomnia or other sleep disorders in otherwise healthy patients.2 However, data from phase I studies of antipsychotics has shown that schizophrenia patients tolerate a higher maximum dose compared with healthy volunteers, who often experience more sedation.
An antipsychotic’s potential for sedation is directly related to its affinity at H1 receptors and total drug concentration at the H1 receptor binding site. Because drugs with lower affinity for D2 receptors typically are prescribed at higher doses when treating psychiatric illness, the corresponding concentration at H1 receptors can lead to greater sedation compared with equivalent doses of higher-potency agents.
The same phenomenon is seen with high-potency agents. Haloperidol has a relatively weak binding affinity to the H1 receptor,8 but causes more sedation at higher doses. Haloperidol, 20 mg/d, produces sedation in more patients than a moderate dose of risperidone, 2 to 10 mg/d.8 These observations correlate with “the high milligram-low-potency” spectrum seen with FGAs.7
Among SGAs, a double-blind, placebo-controlled, crossover study of the effects of ziprasidone, 40 mg/d, on sleep in a group of healthy volunteers found a significant increase in total sleep time and sleep efficiency.9 A double-blind trial compared patients taking low, medium, or high daily doses of olanzapine with patients receiving haloperidol or placebo.10 Sedation was reported in 20% of patients taking low doses of olanzapine (5 ± 2.5 mg/d) compared with 29.7% on medium doses (10 ± 2.5 mg/d) and 39.1% on high doses (15 ± 2.5 mg/d).10
A double-blind, placebo-controlled, crossover study demonstrated that olanzapine produced significant increases in sleep continuity, slow wave sleep, and subjective ratings of sleep quality in healthy men.11 Similarly, a study comparing haloperidol, 12 mg/d, and quetiapine, 75 to 750 mg/d, for treating acute schizophrenia found an 8% to 11% incidence of somnolence in the quetiapine group compared with 6% and 8% in the haloperidol and placebo groups, respectively.12 Somnolence was reported as an adverse event in these studies, which were designed to examine the drug’s effect on acute schizophrenia and did not evaluate its effect on sleep.
A double-blind, placebo-controlled, crossover study examining quetiapine’s effects on sleep in 14 healthy patients demonstrated a significant difference in total sleep time, sleep period time, and sleep efficiency.13 Similarly, an open-label pilot study of quetiapine’s effect on primary insomnia showed significant improvement in total sleep time and sleep efficiency.14
Studies examining quetiapine’s effects on insomnia in patients with substance abuse15 and women with localized breast cancer16 showed improved sleep scores on multiple assessment tools, while an open-label study of quetiapine for Parkinson’s disease demonstrated decreased sleep latency.17 Adjunctive quetiapine administered over a 6-week, open-label trial in veterans with posttraumatic stress disorder revealed significant improvement from baseline in sleep quality and duration and diminished dreaming.18
Sedating antipsychotics such as thioridazine and chlorpromazine historically were used off-label for insomnia, but fell out of favor because of their associated cardiac risks. More recently, clinicians have been using SGAs in a similar manner19 even though SGAs are costly and have significant risks such as metabolic problems.
Studies supporting the use of SGAs for the short-term or long-term treatment of insomnia are limited by small sample sizes or open-label designs.20 In 2005 the National Institutes of Health State-of-the-Science Conference Panel did not recommend using SGAs for treating chronic insomnia.21
Tics in Tourette’s disorder
FGAs and SGAs have been used to treat tics associated with Tourette’s disorder (TD).22 Haloperidol is FDA-approved for treating tics in adult and pediatric patients with TD. Many studies have reported the efficacy of haloperidol in this population; however, cognitive blunting, weight gain, lethargy, and akathisia limit its use.23
Pimozide, the most widely used alternative to haloperidol for treating TD, can cause clinically significant QTc prolongation and sudden death. Penfluridol demonstrated significant symptomatic improvement compared with haloperidol in 1 study, but its carcinogenic potential limits its use.24
A double-blind, placebo-controlled study comparing fluphenazine and trifluoperazine with haloperidol for treating TD showed that both are significantly more effective than placebo, but none was more effective than the others.25 Studies show chlorpromazine, perphenazine, and thioridazine are less effective than haloperidol and their use is limited by photosensitivity, dermatitis, EPS, and blood and liver dyscrasias.26
Risperidone is superior to placebo for treating tics associated with TD.27 A placebo-controlled trial of ziprasidone showed the drug has efficacy similar to risperidone in reducing tics in children and adolescents with TD.28 However, ziprasidone is not FDA-approved for this use.
Evidence supporting the use of other SGAs for treating TD is more limited. Several small studies of olanzapine and aripiprazole had limited but favorable results. Quetiapine has not been studied for treating TD, but several case reports have indicated a positive response. In a double-blind, placebo-controlled trial, clozapine showed no therapeutic benefit for TD.29
Delirium
American Psychiatric Association practice guidelines suggest using psychotropic medications to treat neuropsychiatric symptoms of delirium.30 Antipsychotics are considered first-line agents that lower hospital mortality rates, decrease lengths of hospital stays, and improve delirium symptoms, in some cases before the underlying medical etiologies resolve.30,31 Available in liquid, oral, IM, and IV formulations, haloperidol is the mainstay of symptomatic treatment of delirium.31 Although not FDA-approved, it is recommended by the Society of Critical Care Medicine as a safe, cost-effective, and efficacious therapy for the psychiatric symptoms associated with delirium.
The most extensively studied SGA for treating delirium, risperidone often is used as an alternative to haloperidol. Case reports describe its potential efficacy.32 In a head-to-head study, risperidone was as effective as low-dose haloperidol for acute delirium treatment.33
Olanzapine was effective in managing delirium in several case studies.34 Also, in a 7-day, randomized, placebo-controlled study, olanzapine and haloperidol showed significantly greater and relatively equivalent improvement compared with placebo; patients treated with olanzapine experienced more rapid improvement in 1 study.35
Case reports and prospective studies also have described quetiapine as effective for treating delirium.36,37 In a prospective, double-blind, placebo-controlled study, patients taking quetiapine had a faster resolution of delirium with reduced overall duration and less agitation than those taking placebo.37 Mortality, intensive care unit length of stay, and incidence of QTc prolongation did not differ, but patients treated with quetiapine were more likely to have increased somnolence and were more frequently discharged to home or rehabilitation centers. One limitation of the study is that concomitant haloperidol use on an “as needed” basis was permitted.38
Evidence supporting the efficacy of ziprasidone for delirium is limited to case reports.39 In 1 case report, a patient with chronic HIV infection and acute cryptococcal meningitis experienced significant improvement of delirium symptoms but could not continue ziprasidone because of fluctuating QTc intervals.40
In 2 patients with delirium, aripiprazole, 15 and 30 mg/d, improved confusion, disorientation, and agitation within 7 days.41 In another study of delirium, 13 of 14 patients on flexibly dosed aripiprazole (5 to 15 mg/d) showed improvement in Clinical Global Impressions Scale scores, although 3 patients developed prolonged QTc intervals.42
Stuttering or stammering
Stuttering or stammering are age-inappropriate disturbances in normal fluency and time patterning of speech. The evidence for antipsychotics to treat stuttering or stammering speech mainly consists of case reports and does not include disfluency frequency data, which makes it difficult to accept claims of efficacy. Disfluency frequency data describe how often a patient has specific disfluencies (blocks, prolongations, interjection, and repetition of syllables, words, or phrases).
Two FGAs (chlorpromazine and haloperidol) and 2 SGAs (risperidone and olanzapine) have been evaluated for treating stuttering. Children were 2.5 times more likely to demonstrate significant improvement when taking chlorpromazine vs placebo.43 An open-label study of haloperidol lacked disfluency frequency data, therefore casting doubts on haloperidol’s reported efficacy in the study.44
In a case report, a 4-year-old boy with severe behavioral dyscontrol showed complete remission of stammering after 1 day of risperidone, 0.25 mg/d.45 The patient’s symptoms reappeared several days after the drug was stopped. In a case series of 2 patients with developmental stuttering, 1 patient reported significant improvement in fluency with olanzapine, 2.5 mg/d, and the other showed marked improvement in fluency with 5 mg/d.46
Related Resources
- Sipahimalani A, Masand PS. Use of risperidone in delirium: case reports. Ann Clin Psychiatry. 1997;9(2):105-107.
- Shapiro AK, Shapiro E, Wayne HL. Treatment of Tourette’s syndrome with haloperidol: review of 34 cases. Arch Gen Psychiatry. 1973;28(1):92-96.
- Sipahimalani A, Masand PS. Olanzapine in the treatment of delirium. Psychosomatics. 1998;39(5):422-430.
Drug Brand Names
- Aripiprazole • Abilify
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Fluphenazine • Permitil, Prolixin
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Pimozide • Orap
- Prochlorperazine • Compazine
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Trifluoperazine • Stelazine
- Ziprasidone • Geodon
Disclosure
Dr. Macaluso has received grant or research support from EnVivo Pharmaceuticals, Janssen, L.P., and Pfizer, Inc.
Dr. Tripathi reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Second-generation antipsychotics (SGAs) represent 5% of all U.S. drug expenditures.1 Their use for indications not approved by the FDA (“off-label” use) increased to a total of $6 billion in 2008, $5.4 billion of which was for uses with limited or uncertain evidence.1
Off-label use of antipsychotics usually is based on novel applications of known receptor binding affinities (Table 1).2-5 For example, antipsychotics with strong antihistamine effects may promote sedation and could be used to treat insomnia. Clinicians also might use antipsychotics to treat a specific symptom of an illness when other treatment options are limited6 or when patients do not respond to standard treatments.
Table 1
Possible rationales for antipsychotic use for nonpsychotic conditions
| Condition | Possible rationale |
|---|---|
| Insomnia2 | Effects on H1 α-1 adrenergic and muscarinic cholinergic receptors. 5-HT2 antagonism activity also has been implicated |
| Tics of Tourette’s disorder3 | By blocking dopamine receptors antipsychotics decrease the primarily dopaminergic input from the substantia nigra and ventral tegmentum to the basal ganglia |
| Delirium4 | Patients have reversible impairment of cerebral oxidative metabolism and multiple neurotransmitter abnormalities (dopamine acetylcholine CNS γ-aminobutyric acid and serotonin). Other hypotheses include inflammatory reactions damage to certain structural pathways and disruption of cortisol and β-endorphin circadian rhythms |
| Stuttering5 | Stutterers have a marked increase in dopaminergic afferent activity in the tail of the left caudate nucleus compared with healthy controls |
| H1: histamine | |
To safely use any medication off-label, clinicians should become familiar with literature on the proposed use. Clinicians should consider off-label use only after carefully weighing the potential therapeutic benefits against the risks. Patients should be aware that the prescribed use is not FDA-approved and informed consent should include a discussion of alternative treatments. The high cost of SGAs may be a limiting factor and should be discussed with patients.
This article reviews the evidence for using antipsychotics to treat insomnia, tics, delirium, and stuttering (Table 2). Click here for a review of the evidence supporting antipsychotics for treating migraine and cluster headaches and nausea
Table 2
Antipsychotics for nonpsychotic disorders: Strength of the evidence
| Condition | Strength of evidencea |
|---|---|
| Insomnia | Weak to intermediate: Haloperidol olanzapine quetiapine risperidone ziprasidone |
| Tics of Tourette’s disorder | Strong: Haloperidol pimozide |
| Intermediate: Chlorpromazine fluphenazine penfluridol perphenazine thioridazine trifluoperazine | |
| Weak: Risperidone | |
| Very weak: Aripiprazole olanzapine quetiapine ziprasidone | |
| Not effective: Clozapine | |
| Delirium | Intermediate: Haloperidol |
| Weak: Olanzapine quetiapine risperidone | |
| Very weak: Aripiprazole ziprasidone | |
| Stuttering | Very weak: Chlorpromazine haloperidol olanzapine risperidone |
| aStrong: Multiple well-designed RCTs directly relevant to the recommendation yielding consistent findings Intermediate: Some evidence from RCTs that support the recommendation but the scientific support was not optimal Weak: Consensus recommendation in the absence of relevant RCTs and better evidence than case report or series Very weak: Case reports case series or preliminary studies RCTs: randomized controlled trials INSOMNIA Arvanitis LA, Miller BG. Multiple fixed doses of “Seroquel” (quetiapine) in patients with acute exacerbation of schizophrenia: a comparison with haloperidol and placebo. The Seroquel Trial 13 Study Group. Biol Psychiatry. 1997;42(4):233-246. Beasley CM Jr, Tollefson G, Tran P, et al. Olanzapine versus placebo and haloperidol: acute phase results of the North American double-blind olanzapine trial. Neuropsychopharmacology. 1996;14(2):111-123. Cohrs S, Meier A, Neumann AC, et al. Improved sleep continuity and increased slow wave sleep and REM latency during ziprasidone treatment: a randomized, controlled, crossover trial of 12 healthy male subjects. J Clin Psychiatry. 2005;66(8):989-996. Cohrs S, Rodenbeck A, Guan Z, et al. Sleep-promoting properties of quetiapine in healthy subjects. Psychopharmacology. 2004;174(3):421-429. Juri C, Chaná P, Tapia J, et al. Quetiapine for insomnia in Parkinson’s disease: results from an open-label trial. Clin Neuropharmacol. 2005;28(4):185-187. Marder SR, Meibach RC. Risperidone in the treatment of schizophrenia. Am J Psychiatry. 1994;151(6):825-835. Pasquini M, Speca A, Biondi M. Quetiapine for tamoxifen-induced insomnia in women with breast cancer. Psychosomatics. 2009;50(2):159-161. Sharpley AL, Vassallo CM, Cowen PJ. Olanzapine increases slow-wave sleep: evidence for blockade of central 5-HT(2C) receptors in vivo. Biol Psychiatry. 2000;47(5):468-470. Terán A, Majadas S, Galan J. Quetiapine in the treatment of sleep disturbances associated with addictive conditions: a retrospective study. Subst Use Misuse. 2008;43(14):2169-2171. Wiegand MH, Landry F, Brückner T, et al. Quetiapine in primary insomnia: a pilot study. Psychopharmacology (Berl). 2008;196(2):337-338. TICS OF TOURETTE’S DISORDER Abuzzahab FS, Anderson FO. Gilles de la Tourette’s syndrome: international registry. Minn Med. 1973;56(6):492-496. Borison RL, Ang L, Chang S, et al. New pharmacological approaches in the treatment of Tourette’s syndrome. Adv Neurol. 1982;35:377-382. Bubl E, Perlov E, Tebartz Van Elst L. Aripiprazole in patients with Tourette syndrome. World Biol J Psychiatry. 2006;7(2):123-125. Caine ED, Polinsky RJ, Kartzinel R, et al. The trial use of clozapine for abnormal involuntary movement disorders. Am J Psychiatry. 1979;136(3):317-320. Dion Y, Annable L, Sabdor P, et al. Risperidone in the treatment of Tourette’s syndrome: a double-blind, placebo-controlled trial. J Clin Psychopharmacol. 2002;22(1):31-39. McCracken JT, Suddath R, Chang S, et al. Effectiveness and tolerability of open label olanzapine in children and adolescents with Tourette’s syndrome. J Child Adolesc Psychopharmacol. 2008;18(5):501-508. Mikkelsen EJ, Detlor J, Cohen DJ. School avoidance and social phobia triggered by haloperidol in patients with Tourette’s disorder. Am J Psychiatry. 1981;138(12):1572-1576. Murphy TK, Bengston MA, Soto O, et al. Case series on the use of aripiprazole for Tourette syndrome. Int J Neuropsychopharmacol. 2005;8(3):489-490. Párraga HC, Párraga M, Woodward R, et al. Quetiapine treatment of children with Tourette’s syndrome: report of two cases. J Child Adolesc Psychopharmacol. 2001;11(2):187-191. Regeur L, Pakkenberg B, Fog R, et al. Clinical features and long-term treatment with pimozide in 65 patients with Gilles de la Tourette’s syndrome. J Neurol Neurosurg Psychiatry. 1986;49(7):791-795. Sallee FR, Kurlan R, Goetz CG, et al. Ziprasidone treatment of children and adolescents with Tourette’s syndrome: a pilot study. J Am Acad Child Adolesc Psychiatry. 2000;39(3):292-299. Sallee FR, Nesbitt L, Jackson C, et al. Relative efficacy of haloperidol and pimozide in children and adolescents with Tourette’s disorder. Am J Psychiatry. 1997;154(8):1057-1062. Scahill L, Leckman JF, Schultz RT, et al. A placebo-controlled trial of risperidone in Tourette syndrome. Neurology. 2003; 60(7):1130-1135. Shapiro AK, Shapiro E, Eisenkraft GJ. Treatment of Tourette’s disorder with penfluridol. Compr Psychiatry. 1983;24(4): 327-331. Shapiro AK, Shapiro E, Wayne HL. Treatment of Tourette’s syndrome with haloperidol: review of 34 cases. Arch Gen Psychiatry. 1973;28(1):92-96. Shapiro AK, Shapiro E, Young JG, et al. Gilles de la Tourette’s syndrome. 2nd ed. New York, NY: Raven Press; 1998:387-390. Stephens RJ, Bassel C, Sandor P. Olanzapine in the treatment of aggression and tics in children with Tourette’s syndrome-a pilot study. J Child Adolesc Psychopharmacol. 2004;14(2):255-266. DELIRIUM Alao AO, Moskowitz L. Aripiprazole and delirium. Ann Clin Psychiatry. 2006;18(4):267-269. Al-Samarrai S, Dunn J, Newmark T, et al. Quetiapine for treatment-resistant delirium. Psychosomatics. 2003;44(4): 350-351. Bourgeois JA, Hilty DM. Prolonged delirium managed with risperidone. Psychosomatics. 2005;46(1):90-91. Breitbart W, Tremblay A, Gibson C. An open trial of olanzapine for the treatment of delirium in hospitalized cancer patients. Psychosomatics. 2002;43(3):175-182. Devlin JW, Roberts RJ, Fong JJ, et al. Efficacy and safety of quetiapine in critically ill patients with delirium: a prospective, multicenter, randomized, double-blind, placebo-controlled pilot study. Crit Care Med. 2010;38(2):419-427. Hans CS, Kim YK. A double-blind trial of risperidone and haloperidol for the treatment of delirium. Psychosomatics. 2004;45(4):297-301. Horikawa N, Yamazaki T, Miyamoto K, et al. Treatment for delirium with risperidone: results of a prospective open trial with 10 patients. Gen Hosp Psychiatry. 2003;25(4):289-292. Hu H, Deng W, Yang H. A prospective random control study comparison of olanzapine and haloperidol in senile delirium [in Chinese]. Chong’qing Medical Journal. 2004;8:1234-1237. Lacasse H, Perreault MM, Williamson DR. Systematic review of antipsychotics for the treatment of hospital-associated delirium in medically or surgically ill patients. Ann Pharmacother. 2006;40(11):1966-1973. Leso L, Schwartz TL. Ziprasidone treatment of delirium. Psychosomatics. 2002;43(1):61-62. Parellada E, Baeza I, de Pablo J, et al. Risperidone in the treatment of patients with delirium. J Clin Psychiatry. 2004;65(3):348-353. Sasaki Y, Matsuyama T, Inoue S, et al. A prospective, open-label, flexible-dose study of quetiapine in the treatment of delirium. J Clin Psychiatry. 2003;64(11):1316-1321. Sipahimalani A, Masand PS. Olanzapine in the treatment of delirium. Psychosomatics. 1998;39(5):422-430. Sipahimalani A, Masand PS. Use of risperidone in delirium: case reports. Ann Clin Psychiatry. 1997;9(2):105-107. Straker DA, Shapiro PA, Muskin PR. Aripiprazole in the treatment of delirium. Psychosomatics. 2006;47(5):385-391. Young CC, Lujan E. Intravenous ziprasidone for treatment of delirium in the intensive care unit. Anesthesiology. 2004;101(3): 794-795. STUTTERING Burr HG, Mullendore JM. Recent investigations on tranquilizers and stuttering. J Speech Hear Disord. 1960;25;33-37. Lavid N, Franklin DL, Maguire GA. Management of child and adolescent stuttering with olanzapine: three case reports. Ann Clin Psychiatry. 1999;11(4):233-236. Tapia F. Haldol in the treatment of children with tics and stutterers and an incidental finding. Behav Neuropsychiatry. 1969;1(3):28. van Wattum PJ. Stuttering improved with risperidone. J Am Acad Child Adolesc Psychiatry. 2006;45(2):133. | |
Current use of antipsychotics
Antipsychotics are divided into 2 major classes—first-generation antipsychotics (FGAs) and SGAs—and principally are FDA-approved for treating schizophrenia. Some antipsychotics have received FDA approval for maintenance treatment of schizophrenia and bipolar disorder (BD), and others have been approved to treat tic disorders (haloperidol and pimozide).
To varying degrees, all antipsychotics block D2 receptors, which is thought to be necessary for treating psychosis. However, some SGAs have significant affinity at other receptors—such as 5-HT2A and 5-HT1A—that confer additional properties that are not fully understood (Table 3). For example, it is believed that 5-HT2A blockade in the striatum reduces the potential for extrapyramidal symptoms (EPS).
Each antipsychotic blocks a unique set of receptors in the brain, leading to a specific set of intended and potentially untoward effects. For example, olanzapine’s effect on psychosis largely stems from its action at the D2 receptor, whereas its sedative and anticholinergic properties are a result of activity at histamine (H1) receptors and muscarinic receptors, respectively. Clinicians can make rational use of unintended effects by carefully selecting a medication based on receptor binding profile (eg, using an antipsychotic with sedating properties in a patient who has psychosis and insomnia). This approach can limit use of multiple medications and maximize a medication’s known effects while attempting to minimize side effects.
Table 3
Antipsychotics: Receptor pharmacology and common side effects
| Antipsychotic | Pharmacology | Common side effectsa |
|---|---|---|
| Prochlorperazinea,b | D2 receptor antagonist and α-1 adrenergic receptor antagonism | EPS, akathisia, prolactinemia, orthostatic hypotension, altered cardiac conduction, agranulocytosis, sexual dysfunction |
| Chlorpromazinea,b | D2 receptor antagonist. Also binds to H1 and cholinergic M1 | EPS, akathisia, prolactinemia, orthostatic hypotension, urinary retention, non-specific QT changes, agranulocytosis, sexual dysfunction |
| Droperidola,b | D2 receptor antagonist and antagonist at peripheral α-1 activity | EPS, akathisia, prolactinemia, orthostatic hypotension, urinary retention, QT changes (dose dependent) |
| Haloperidola,b | D2 receptor antagonist. Also binds to D1, 5-HT2, H1, and α-2 adrenergic receptors | EPS, akathisia, prolactinemia, QT changes (dose dependent) |
| Aripiprazolea,c,d | D2 and 5-HT1A partial agonism, 5-HT2A antagonism | Akathisia, EPS, sedation, restlessness, insomnia, tremor, anxiety, nausea, vomiting, possible weight gain (20% to 30%) |
| Clozapinea,c,e | 5-HT2, D1, D2, D3, D4, M1, H1, α-1, and α-2 antagonism | Sedation, dizziness, tachycardia, weight gain, nausea, vomiting, constipation |
| Olanzapinea,c | 5-HT2A, 5-HT2C, D1, D2, D3, D4, M1-5, H1, and α1- antagonism | Sedation, EPS, prolactinemia, weight gain, constipation |
| Quetiapinea,c,d | D1, D2, 5-HT2A, 5-HT1A, H1, α-1, and α-2 antagonism | Sedation, orthostatic hypotension, weight gain, triglyceride abnormalities, hypertension (frequently diastolic), constipation |
| Risperidonea,c | 5-HT2, D2, H1, α-1, and α-2 antagonism | Sedation, akathisia, EPS, prolactinemia, weight gain, tremor |
| Ziprasidonea,c | D2, D3, 5-HT2A, 5-HT2C, 5-HT1D, and α-1 antagonism; moderate inhibition of 5-HT and NE reuptake; 5-HT1A agonism | EPS, sedation, headache, dizziness, nausea |
| aSide effects and their prominence usually are based on receptor binding profile. All antipsychotics to varying degrees share the following symptoms: EPS, neuroleptic malignant syndrome, QTc prolongation, anticholinergic side effects (urinary retention, decreased gastrointestinal motility, xerostomia), sedation, orthostatic hypotension, blood dyscrasias, and problems with temperature regulation. The class as a whole also carries a “black-box” warning regarding increased mortality when treating geriatric patients with psychosis related to dementia bNo frequencies were available cOnly side effects with frequency >10% listed d”Black-box” warning for suicidal ideation and behavior in children, adolescents, and young adults (age 18 to 24) with major depressive disorder and other psychiatric disorders e”Black-box” warnings for agranulocytosis, myocarditis, orthostatic hypotension, seizure risk EPS: extrapyramidal symptoms; H1: histamine; M1: muscarinic; NE: norepinephrine | ||
Insomnia
Clinicians use FGAs and SGAs to treat insomnia because of their sedating effects, although evidence supporting this use is questionable. Among the FGAs, chlorpromazine produces moderate to severe sedation, whereas haloperidol is only mildly sedating. Clozapine is believed to be the most sedating SGA, whereas quetiapine and olanzapine produce moderate sedation.7
Most data on antipsychotics’ sedating effects comes from studies completed for schizophrenia or BD. Few studies have evaluated using antipsychotics to treat primary insomnia or other sleep disorders in otherwise healthy patients.2 However, data from phase I studies of antipsychotics has shown that schizophrenia patients tolerate a higher maximum dose compared with healthy volunteers, who often experience more sedation.
An antipsychotic’s potential for sedation is directly related to its affinity at H1 receptors and total drug concentration at the H1 receptor binding site. Because drugs with lower affinity for D2 receptors typically are prescribed at higher doses when treating psychiatric illness, the corresponding concentration at H1 receptors can lead to greater sedation compared with equivalent doses of higher-potency agents.
The same phenomenon is seen with high-potency agents. Haloperidol has a relatively weak binding affinity to the H1 receptor,8 but causes more sedation at higher doses. Haloperidol, 20 mg/d, produces sedation in more patients than a moderate dose of risperidone, 2 to 10 mg/d.8 These observations correlate with “the high milligram-low-potency” spectrum seen with FGAs.7
Among SGAs, a double-blind, placebo-controlled, crossover study of the effects of ziprasidone, 40 mg/d, on sleep in a group of healthy volunteers found a significant increase in total sleep time and sleep efficiency.9 A double-blind trial compared patients taking low, medium, or high daily doses of olanzapine with patients receiving haloperidol or placebo.10 Sedation was reported in 20% of patients taking low doses of olanzapine (5 ± 2.5 mg/d) compared with 29.7% on medium doses (10 ± 2.5 mg/d) and 39.1% on high doses (15 ± 2.5 mg/d).10
A double-blind, placebo-controlled, crossover study demonstrated that olanzapine produced significant increases in sleep continuity, slow wave sleep, and subjective ratings of sleep quality in healthy men.11 Similarly, a study comparing haloperidol, 12 mg/d, and quetiapine, 75 to 750 mg/d, for treating acute schizophrenia found an 8% to 11% incidence of somnolence in the quetiapine group compared with 6% and 8% in the haloperidol and placebo groups, respectively.12 Somnolence was reported as an adverse event in these studies, which were designed to examine the drug’s effect on acute schizophrenia and did not evaluate its effect on sleep.
A double-blind, placebo-controlled, crossover study examining quetiapine’s effects on sleep in 14 healthy patients demonstrated a significant difference in total sleep time, sleep period time, and sleep efficiency.13 Similarly, an open-label pilot study of quetiapine’s effect on primary insomnia showed significant improvement in total sleep time and sleep efficiency.14
Studies examining quetiapine’s effects on insomnia in patients with substance abuse15 and women with localized breast cancer16 showed improved sleep scores on multiple assessment tools, while an open-label study of quetiapine for Parkinson’s disease demonstrated decreased sleep latency.17 Adjunctive quetiapine administered over a 6-week, open-label trial in veterans with posttraumatic stress disorder revealed significant improvement from baseline in sleep quality and duration and diminished dreaming.18
Sedating antipsychotics such as thioridazine and chlorpromazine historically were used off-label for insomnia, but fell out of favor because of their associated cardiac risks. More recently, clinicians have been using SGAs in a similar manner19 even though SGAs are costly and have significant risks such as metabolic problems.
Studies supporting the use of SGAs for the short-term or long-term treatment of insomnia are limited by small sample sizes or open-label designs.20 In 2005 the National Institutes of Health State-of-the-Science Conference Panel did not recommend using SGAs for treating chronic insomnia.21
Tics in Tourette’s disorder
FGAs and SGAs have been used to treat tics associated with Tourette’s disorder (TD).22 Haloperidol is FDA-approved for treating tics in adult and pediatric patients with TD. Many studies have reported the efficacy of haloperidol in this population; however, cognitive blunting, weight gain, lethargy, and akathisia limit its use.23
Pimozide, the most widely used alternative to haloperidol for treating TD, can cause clinically significant QTc prolongation and sudden death. Penfluridol demonstrated significant symptomatic improvement compared with haloperidol in 1 study, but its carcinogenic potential limits its use.24
A double-blind, placebo-controlled study comparing fluphenazine and trifluoperazine with haloperidol for treating TD showed that both are significantly more effective than placebo, but none was more effective than the others.25 Studies show chlorpromazine, perphenazine, and thioridazine are less effective than haloperidol and their use is limited by photosensitivity, dermatitis, EPS, and blood and liver dyscrasias.26
Risperidone is superior to placebo for treating tics associated with TD.27 A placebo-controlled trial of ziprasidone showed the drug has efficacy similar to risperidone in reducing tics in children and adolescents with TD.28 However, ziprasidone is not FDA-approved for this use.
Evidence supporting the use of other SGAs for treating TD is more limited. Several small studies of olanzapine and aripiprazole had limited but favorable results. Quetiapine has not been studied for treating TD, but several case reports have indicated a positive response. In a double-blind, placebo-controlled trial, clozapine showed no therapeutic benefit for TD.29
Delirium
American Psychiatric Association practice guidelines suggest using psychotropic medications to treat neuropsychiatric symptoms of delirium.30 Antipsychotics are considered first-line agents that lower hospital mortality rates, decrease lengths of hospital stays, and improve delirium symptoms, in some cases before the underlying medical etiologies resolve.30,31 Available in liquid, oral, IM, and IV formulations, haloperidol is the mainstay of symptomatic treatment of delirium.31 Although not FDA-approved, it is recommended by the Society of Critical Care Medicine as a safe, cost-effective, and efficacious therapy for the psychiatric symptoms associated with delirium.
The most extensively studied SGA for treating delirium, risperidone often is used as an alternative to haloperidol. Case reports describe its potential efficacy.32 In a head-to-head study, risperidone was as effective as low-dose haloperidol for acute delirium treatment.33
Olanzapine was effective in managing delirium in several case studies.34 Also, in a 7-day, randomized, placebo-controlled study, olanzapine and haloperidol showed significantly greater and relatively equivalent improvement compared with placebo; patients treated with olanzapine experienced more rapid improvement in 1 study.35
Case reports and prospective studies also have described quetiapine as effective for treating delirium.36,37 In a prospective, double-blind, placebo-controlled study, patients taking quetiapine had a faster resolution of delirium with reduced overall duration and less agitation than those taking placebo.37 Mortality, intensive care unit length of stay, and incidence of QTc prolongation did not differ, but patients treated with quetiapine were more likely to have increased somnolence and were more frequently discharged to home or rehabilitation centers. One limitation of the study is that concomitant haloperidol use on an “as needed” basis was permitted.38
Evidence supporting the efficacy of ziprasidone for delirium is limited to case reports.39 In 1 case report, a patient with chronic HIV infection and acute cryptococcal meningitis experienced significant improvement of delirium symptoms but could not continue ziprasidone because of fluctuating QTc intervals.40
In 2 patients with delirium, aripiprazole, 15 and 30 mg/d, improved confusion, disorientation, and agitation within 7 days.41 In another study of delirium, 13 of 14 patients on flexibly dosed aripiprazole (5 to 15 mg/d) showed improvement in Clinical Global Impressions Scale scores, although 3 patients developed prolonged QTc intervals.42
Stuttering or stammering
Stuttering or stammering are age-inappropriate disturbances in normal fluency and time patterning of speech. The evidence for antipsychotics to treat stuttering or stammering speech mainly consists of case reports and does not include disfluency frequency data, which makes it difficult to accept claims of efficacy. Disfluency frequency data describe how often a patient has specific disfluencies (blocks, prolongations, interjection, and repetition of syllables, words, or phrases).
Two FGAs (chlorpromazine and haloperidol) and 2 SGAs (risperidone and olanzapine) have been evaluated for treating stuttering. Children were 2.5 times more likely to demonstrate significant improvement when taking chlorpromazine vs placebo.43 An open-label study of haloperidol lacked disfluency frequency data, therefore casting doubts on haloperidol’s reported efficacy in the study.44
In a case report, a 4-year-old boy with severe behavioral dyscontrol showed complete remission of stammering after 1 day of risperidone, 0.25 mg/d.45 The patient’s symptoms reappeared several days after the drug was stopped. In a case series of 2 patients with developmental stuttering, 1 patient reported significant improvement in fluency with olanzapine, 2.5 mg/d, and the other showed marked improvement in fluency with 5 mg/d.46
Related Resources
- Sipahimalani A, Masand PS. Use of risperidone in delirium: case reports. Ann Clin Psychiatry. 1997;9(2):105-107.
- Shapiro AK, Shapiro E, Wayne HL. Treatment of Tourette’s syndrome with haloperidol: review of 34 cases. Arch Gen Psychiatry. 1973;28(1):92-96.
- Sipahimalani A, Masand PS. Olanzapine in the treatment of delirium. Psychosomatics. 1998;39(5):422-430.
Drug Brand Names
- Aripiprazole • Abilify
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Fluphenazine • Permitil, Prolixin
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Pimozide • Orap
- Prochlorperazine • Compazine
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Trifluoperazine • Stelazine
- Ziprasidone • Geodon
Disclosure
Dr. Macaluso has received grant or research support from EnVivo Pharmaceuticals, Janssen, L.P., and Pfizer, Inc.
Dr. Tripathi reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Alexander GC, Gallagher SA, Mascola A, et al. Increasing off-label use of antipsychotic medications in the United States, 1995-2008. Pharmacoepidemiol Drug Saf. 2011;20(2):177-184.
2. DeMartinis N, Winokur A. Effects of psychiatric medications on sleep and sleep disorders. CNS Neurol Disord Drug Targets. 2007;6(1):17-29.
3. Leckman JF, Bloch MH, Smith ME, et al. Neurobiological substrates of Tourette’s disorder. J Child Adolesc Psychopharmacol. 2010;20(4):237-247.
4. Maldonado JR. Pathoetiological model of delirium: a comprehensive understanding of the neurobiology of delirium and an evidence-based approach to prevention and treatment. Crit Care Clin. 2008;24(4):789-856.
5. Wu JC, Maguire G, Riley G, et al. Increased dopamine activity associated with stuttering. Neuroreport. 1997;8(3):767-770.
6. Devulapalli K, Nasrallah HA. An analysis of the high psychotropic off-label use in psychiatric disorders: the majority of psychiatric diagnoses have no approved drug. Asian J Psychiatr. 2009;2(1):29-36.
7. Miller DD. Atypical antipsychotics: sleep sedation, and efficacy. Prim Care Companion J Clin Psychiatry. 2004;6(suppl 2):3-7.
8. Marder SR, Meibach RC. Risperidone in the treatment of schizophrenia. Am J Psychiatry. 1994;151(6):825-835.
9. Cohrs S, Meier A, Neumann AC, et al. Improved sleep continuity and increased slow wave sleep and REM latency during ziprasidone treatment: a randomized, controlled, crossover trial of 12 healthy male subjects. J Clin Psychiatry. 2005;66(8):989-996.
10. Beasley CM Jr, Tollefson G, Tran P, et al. Olanzapine versus placebo and haloperidol: acute phase results of the North American double-blind olanzapine trial. Neuropsychopharmacology. 1996;14(2):111-123.
11. Sharpley AL, Vassallo CM, Cowen PJ. Olanzapine increases slow-wave sleep: evidence for blockade of central 5-HT(2C) receptors in vivo. Biol Psychiatry. 2000;47(5):468-470.
12. Arvanitis LA, Miller BG. Multiple fixed doses of “Seroquel” (quetiapine) in patients with acute exacerbation of schizophrenia: a comparison with haloperidol and placebo. The Seroquel Trial 13 Study Group. Biol Psychiatry. 1997;42(4):233-246.
13. Cohrs S, Rodenbeck A, Guan Z, et al. Sleep-promoting properties of quetiapine in healthy subjects. Psychopharmacology. 2004;174(3):421-429.
14. Wiegand MH, Landry F, Brückner T, et al. Quetiapine in primary insomnia: a pilot study. Psychopharmacology (Berl). 2008;196(2):337-338.
15. Terán A, Majadas S, Galan J. Quetiapine in the treatment of sleep disturbances associated with addictive conditions: a retrospective study. Subst Use Misuse. 2008;43(14):2169-2171.
16. Pasquini M, Speca A, Biondi M. Quetiapine for tamoxifen-induced insomnia in women with breast cancer. Psychosomatics. 2009;50(2):159-161.
17. Juri C, Chaná P, Tapia J, et al. Quetiapine for insomnia in Parkinson’s disease: results from an open-label trial. Clin Neuropharmacol. 2005;28(4):185-187.
18. Robert S, Hamner MB, Kose S, et al. Quetiapine improves sleep disturbances in combat veterans with PTSD: sleep data from a prospective, open-label study. J Clin Psychopharmacol. 2005;25(4):387-388.
19. Wilson S, Nutt D. Management of insomnia: treatments and mechanisms. Br J Psychiatry. 2007;191:195-197.
20. Morin CM, Benca R. Chronic insomnia. Lancet. 2012;379(9821):1129-1141.
21. National Institutes of Health. National Institutes of Health State of the Science Conference statement on manifestations and management of chronic insomnia in adults June 13-15, 2005. Sleep. 2005;28(9):1049-1057.
22. Párraga HC, Harris KM, Párraga KL, et al. An overview of the treatment of Tourette’s disorder and tics. J Child Adolesc Psychopharmacol. 2010;20(4):249-262.
23. Mikkelsen EJ, Detlor J, Cohen DJ. School avoidance and social phobia triggered by haloperidol in patients with Tourette’s disorder. Am J Psychiatry. 1981;138(12):1572-1576.
24. Shapiro AK, Shapiro E, Eisenkraft GJ. Treatment of Tourette’s disorder with penfluridol. Compr Psychiatry. 1983;24(4):327-331.
25. Borison RL, Ang L, Chang S, et al. New pharmacological approaches in the treatment of Tourette’s syndrome. Adv Neurol. 1982;35:377-382.
26. Shapiro AK, Shapiro E, Young JG, et al. Gilles de la Tourette’s syndrome. 2nd ed. New York, NY: Raven Press; 1998:387–390.
27. Dion Y, Annable L, Sabdor P, et al. Risperidone in the treatment of Tourette’s syndrome: a double-blind, placebo-controlled trial. J Clin Psychopharmacol. 2002;22(1):31-39.
28. Sallee FR, Kurlan R, Goetz CG, et al. Ziprasidone treatment of children and adolescents with Tourette’s syndrome: a pilot study. J Am Acad Child Adolesc Psychiatry. 2000;39(3):292-299.
29. Caine ED, Polinsky RJ, Kartzinel R, et al. The trial use of clozapine for abnormal involuntary movement disorders. Am J Psychiatry. 1979;136(3):317-320.
30. American Psychiatric Association. Practice guideline for the treatment of patients with delirium. Am J Psychiatry. 1999;156(suppl 5):1-20.
31. Lacasse H, Perreault MM, Williamson DR. Systematic review of antipsychotics for the treatment of hospital-associated delirium in medically or surgically ill patients. Ann Pharmacother. 2006;40(11):1966-1973.
32. Parellada E, Baeza I, de Pablo J, et al. Risperidone in the treatment of patients with delirium. J Clin Psychiatry. 2004;65(3):348-353.
33. Hans CS, Kim YK. A double-blind trial of risperidone and haloperidol for the treatment of delirium. Psychosomatics. 2004;45(4):297-301.
34. Breitbart W, Tremblay A, Gibson C. An open trial of olanzapine for the treatment of delirium in hospitalized cancer patients. Psychosomatics. 2002;43(3):175-182.
35. Hu H, Deng W, Yang H. A prospective random control study comparison of olanzapine and haloperidol in senile delirium [in Chinese]. Chong’qing Medical Journal. 2004;8:1234-1237.
36. Al-Samarrai S, Dunn J, Newmark T, et al. Quetiapine for treatment-resistant delirium. Psychosomatics. 2003;44(4):350-351.
37. Sasaki Y, Matsuyama T, Inoue S, et al. A prospective, open-label, flexible-dose study of quetiapine in the treatment of delirium. J Clin Psychiatry. 2003;64(11):1316-1321.
38. Devlin JW, Roberts RJ, Fong JJ, et al. Efficacy and safety of quetiapine in critically ill patients with delirium: a prospective, multicenter, randomized, double-blind, placebo-controlled pilot study. Crit Care Med. 2010;38(2):419-427.
39. Young CC, Lujan E. Intravenous ziprasidone for treatment of delirium in the intensive care unit. Anesthesiology. 2004;101(3):794-795.
40. Leso L, Schwartz TL. Ziprasidone treatment of delirium. Psychosomatics. 2002;43(1):61-62.
41. Alao AO, Moskowitz L. Aripiprazole and delirium. Ann Clin Psychiatry. 2006;18(4):267-269.
42. Straker DA, Shapiro PA, Muskin PR. Aripiprazole in the treatment of delirium. Psychosomatics. 2006;47(5):385-391.
43. Burr HG, Mullendore JM. Recent investigations on tranquilizers and stuttering. J Speech Hear Disord. 1960;25:33-37.
44. Tapia F. Haldol in the treatment of children with tics and stutterers and an incidental finding. Behav Neuropsychiatry. 1969;1(3):28.-
45. van Wattum PJ. Stuttering improved with risperidone. J Am Acad Child Adolesc Psychiatry. 2006;45(2):133.-
46. Lavid N, Franklin DL, Maguire GA. Management of child and adolescent stuttering with olanzapine: three case reports. Ann Clin Psychiatry. 1999;11(4):233-236.
1. Alexander GC, Gallagher SA, Mascola A, et al. Increasing off-label use of antipsychotic medications in the United States, 1995-2008. Pharmacoepidemiol Drug Saf. 2011;20(2):177-184.
2. DeMartinis N, Winokur A. Effects of psychiatric medications on sleep and sleep disorders. CNS Neurol Disord Drug Targets. 2007;6(1):17-29.
3. Leckman JF, Bloch MH, Smith ME, et al. Neurobiological substrates of Tourette’s disorder. J Child Adolesc Psychopharmacol. 2010;20(4):237-247.
4. Maldonado JR. Pathoetiological model of delirium: a comprehensive understanding of the neurobiology of delirium and an evidence-based approach to prevention and treatment. Crit Care Clin. 2008;24(4):789-856.
5. Wu JC, Maguire G, Riley G, et al. Increased dopamine activity associated with stuttering. Neuroreport. 1997;8(3):767-770.
6. Devulapalli K, Nasrallah HA. An analysis of the high psychotropic off-label use in psychiatric disorders: the majority of psychiatric diagnoses have no approved drug. Asian J Psychiatr. 2009;2(1):29-36.
7. Miller DD. Atypical antipsychotics: sleep sedation, and efficacy. Prim Care Companion J Clin Psychiatry. 2004;6(suppl 2):3-7.
8. Marder SR, Meibach RC. Risperidone in the treatment of schizophrenia. Am J Psychiatry. 1994;151(6):825-835.
9. Cohrs S, Meier A, Neumann AC, et al. Improved sleep continuity and increased slow wave sleep and REM latency during ziprasidone treatment: a randomized, controlled, crossover trial of 12 healthy male subjects. J Clin Psychiatry. 2005;66(8):989-996.
10. Beasley CM Jr, Tollefson G, Tran P, et al. Olanzapine versus placebo and haloperidol: acute phase results of the North American double-blind olanzapine trial. Neuropsychopharmacology. 1996;14(2):111-123.
11. Sharpley AL, Vassallo CM, Cowen PJ. Olanzapine increases slow-wave sleep: evidence for blockade of central 5-HT(2C) receptors in vivo. Biol Psychiatry. 2000;47(5):468-470.
12. Arvanitis LA, Miller BG. Multiple fixed doses of “Seroquel” (quetiapine) in patients with acute exacerbation of schizophrenia: a comparison with haloperidol and placebo. The Seroquel Trial 13 Study Group. Biol Psychiatry. 1997;42(4):233-246.
13. Cohrs S, Rodenbeck A, Guan Z, et al. Sleep-promoting properties of quetiapine in healthy subjects. Psychopharmacology. 2004;174(3):421-429.
14. Wiegand MH, Landry F, Brückner T, et al. Quetiapine in primary insomnia: a pilot study. Psychopharmacology (Berl). 2008;196(2):337-338.
15. Terán A, Majadas S, Galan J. Quetiapine in the treatment of sleep disturbances associated with addictive conditions: a retrospective study. Subst Use Misuse. 2008;43(14):2169-2171.
16. Pasquini M, Speca A, Biondi M. Quetiapine for tamoxifen-induced insomnia in women with breast cancer. Psychosomatics. 2009;50(2):159-161.
17. Juri C, Chaná P, Tapia J, et al. Quetiapine for insomnia in Parkinson’s disease: results from an open-label trial. Clin Neuropharmacol. 2005;28(4):185-187.
18. Robert S, Hamner MB, Kose S, et al. Quetiapine improves sleep disturbances in combat veterans with PTSD: sleep data from a prospective, open-label study. J Clin Psychopharmacol. 2005;25(4):387-388.
19. Wilson S, Nutt D. Management of insomnia: treatments and mechanisms. Br J Psychiatry. 2007;191:195-197.
20. Morin CM, Benca R. Chronic insomnia. Lancet. 2012;379(9821):1129-1141.
21. National Institutes of Health. National Institutes of Health State of the Science Conference statement on manifestations and management of chronic insomnia in adults June 13-15, 2005. Sleep. 2005;28(9):1049-1057.
22. Párraga HC, Harris KM, Párraga KL, et al. An overview of the treatment of Tourette’s disorder and tics. J Child Adolesc Psychopharmacol. 2010;20(4):249-262.
23. Mikkelsen EJ, Detlor J, Cohen DJ. School avoidance and social phobia triggered by haloperidol in patients with Tourette’s disorder. Am J Psychiatry. 1981;138(12):1572-1576.
24. Shapiro AK, Shapiro E, Eisenkraft GJ. Treatment of Tourette’s disorder with penfluridol. Compr Psychiatry. 1983;24(4):327-331.
25. Borison RL, Ang L, Chang S, et al. New pharmacological approaches in the treatment of Tourette’s syndrome. Adv Neurol. 1982;35:377-382.
26. Shapiro AK, Shapiro E, Young JG, et al. Gilles de la Tourette’s syndrome. 2nd ed. New York, NY: Raven Press; 1998:387–390.
27. Dion Y, Annable L, Sabdor P, et al. Risperidone in the treatment of Tourette’s syndrome: a double-blind, placebo-controlled trial. J Clin Psychopharmacol. 2002;22(1):31-39.
28. Sallee FR, Kurlan R, Goetz CG, et al. Ziprasidone treatment of children and adolescents with Tourette’s syndrome: a pilot study. J Am Acad Child Adolesc Psychiatry. 2000;39(3):292-299.
29. Caine ED, Polinsky RJ, Kartzinel R, et al. The trial use of clozapine for abnormal involuntary movement disorders. Am J Psychiatry. 1979;136(3):317-320.
30. American Psychiatric Association. Practice guideline for the treatment of patients with delirium. Am J Psychiatry. 1999;156(suppl 5):1-20.
31. Lacasse H, Perreault MM, Williamson DR. Systematic review of antipsychotics for the treatment of hospital-associated delirium in medically or surgically ill patients. Ann Pharmacother. 2006;40(11):1966-1973.
32. Parellada E, Baeza I, de Pablo J, et al. Risperidone in the treatment of patients with delirium. J Clin Psychiatry. 2004;65(3):348-353.
33. Hans CS, Kim YK. A double-blind trial of risperidone and haloperidol for the treatment of delirium. Psychosomatics. 2004;45(4):297-301.
34. Breitbart W, Tremblay A, Gibson C. An open trial of olanzapine for the treatment of delirium in hospitalized cancer patients. Psychosomatics. 2002;43(3):175-182.
35. Hu H, Deng W, Yang H. A prospective random control study comparison of olanzapine and haloperidol in senile delirium [in Chinese]. Chong’qing Medical Journal. 2004;8:1234-1237.
36. Al-Samarrai S, Dunn J, Newmark T, et al. Quetiapine for treatment-resistant delirium. Psychosomatics. 2003;44(4):350-351.
37. Sasaki Y, Matsuyama T, Inoue S, et al. A prospective, open-label, flexible-dose study of quetiapine in the treatment of delirium. J Clin Psychiatry. 2003;64(11):1316-1321.
38. Devlin JW, Roberts RJ, Fong JJ, et al. Efficacy and safety of quetiapine in critically ill patients with delirium: a prospective, multicenter, randomized, double-blind, placebo-controlled pilot study. Crit Care Med. 2010;38(2):419-427.
39. Young CC, Lujan E. Intravenous ziprasidone for treatment of delirium in the intensive care unit. Anesthesiology. 2004;101(3):794-795.
40. Leso L, Schwartz TL. Ziprasidone treatment of delirium. Psychosomatics. 2002;43(1):61-62.
41. Alao AO, Moskowitz L. Aripiprazole and delirium. Ann Clin Psychiatry. 2006;18(4):267-269.
42. Straker DA, Shapiro PA, Muskin PR. Aripiprazole in the treatment of delirium. Psychosomatics. 2006;47(5):385-391.
43. Burr HG, Mullendore JM. Recent investigations on tranquilizers and stuttering. J Speech Hear Disord. 1960;25:33-37.
44. Tapia F. Haldol in the treatment of children with tics and stutterers and an incidental finding. Behav Neuropsychiatry. 1969;1(3):28.-
45. van Wattum PJ. Stuttering improved with risperidone. J Am Acad Child Adolesc Psychiatry. 2006;45(2):133.-
46. Lavid N, Franklin DL, Maguire GA. Management of child and adolescent stuttering with olanzapine: three case reports. Ann Clin Psychiatry. 1999;11(4):233-236.
Maintenance of certification and licensing: What you need to know
Discuss this article at www.facebook.com/CurrentPsychiatry
In 2000, the American Board of Medical Specialties (ABMS) made a commitment to develop a maintenance of certification (MOC) system for their 24 specialty boards. MOC aims to keep physicians up to date because medical knowledge and practice are rapidly evolving and health care systems expect greater accountability linked with performance and outcomes. Previously, board certification for most specialties was limited to a 1-time board exam; upon passing, a clinician was considered board certified for life. The American Board of Psychiatry and Neurology (ABPN) first issued time-limited certificates for board certification in 1994; 2007 was the first year of initial MOC enrollment for ABPN. Diplomates whose certificates were issued before October 1, 1994 are not required to participate in the MOC program.
The ABPN time-limited certificates are on 10-year cycles and require diplomates to fulfill 4 MOC program components: Professional Standing, Self-Assessment and Continuing Medical Education (CME), Cognitive Expertise, and Performance in Practice (PIP) (Table).1 Requirement details are available at www.abpn.com.
The ABMS MOC initiative is closely aligned with other initiatives, such as maintenance of licensure (MOL), that will impact all physicians, including those who are not board certified and those who were certified before October 1, 1994 and therefore not required to participate in MOC. Licensure, reimbursement, and institutional credentials are developing required measures based on self-assessment and performance.
Table
Maintenance of certification: 4 components
| Component | Description |
|---|---|
| Professional Standing | Diplomates must hold an active and unrestricted license to practice medicine in ≥1 state commonwealth territory or possession of the United States or province of Canada |
| Self-Assessment and CME | Self-assessment: Diplomates must participate in ≥2 major broad-based self-assessment activities that must cover new knowledge and/or current best practices and provide feedback to the diplomate that can be used as the basis for focused CME lifelong learning and/or career development |
| CME activities: Diplomates are required to complete an average of 30 specialty or subspecialty Category 1 CME credits per year over the 10-year MOC cycle. At least an average of 8 of the CME credits per year (averaged over 2 to 5 years) should involve self-assessment | |
| Cognitive Expertise | Diplomates must pass a cognitive examination before the expiration date of their certificates |
| Performance in Practice (PIP) | Diplomates will be required to complete 3 PIP units over the 10-year MOC cycle each consisting of both a clinical module (chart review) and a feedback module (patient/peer second-party external review) |
| CME: continuing medical education; MOC: maintenance of certification Source: Adapted from reference 1 | |
MOC requirements
The ABMS developed its MOC program around 6 general competencies identified by the Accreditation Council for Graduate Medical Education:
- professionalism
- patient care and procedural skills
- medical knowledge
- practice-based learning and improvement
- interpersonal and communications skills
- systems-based practice.
Physicians with “lifetime” certificates are not required to participate in MOC; there are no consequences for physicians who are not required to participate in MOC and choose not to participate, because MOC is a voluntary system. Physicians with time-limited certificates can choose not to participate, but would forfeit their certification. Physicians with certifications in multiple specialties may consider the value of maintaining all of their certifications because it would require them to participate in multiple MOC programs.
Two of the 4 parts of MOC (Parts I and III) are extensions of existing board certification requirements. Part I stipulates a diplomate hold a valid and unrestricted license in ≥1 states or jurisdictions in the United States, its territories, or Canada. Part III (Cognitive Expertise) requires that he or she must pass a cognitive examination every 10 years. To qualify to take the cognitive exam, a diplomate must meet all current MOC requirements.
Parts II and IV integrate continuing education, self-assessment, and the ability to apply both to practice improvements. Part II requires an average of 8 CME credit hours that include a self-assessment component; this likely would eliminate most traditional CME activities. The ABPN stipulates that feedback from the self-assessment must include a comparison with peers and specific literature recommendations for each question in the self-assessment. A small but growing number of accredited CME providers have developed self-directed CME activities that meet these criteria. As of 2014, only ABPN-approved self-assessment activities can be used to meet Part II requirements.
Part IV, the PIP activity, has raised the most concern. The PIP component focuses on quality improvement in 2 parts: a clinical module and a feedback module. This targets active clinicians, and both modules focus on quality improvement activities. The clinical module consists of a baseline chart review by the physician MOC applicant in which results are compared with best practices or practice guidelines. The practitioner-applicant repeats a second chart review after a period of time to determine if intervening practice improvements had a positive impact.
The feedback module consists of reviews of clinical performance by patients, peers, or other second parties such as other practice staff or administrators. These are repeated after a period of time to determine whether practice improvements have been effective.
The PIP model (assessment, practice improvement, reassessment) parallels requirements for Performance Improvement CME (PICME) activities. The American Medical Association (AMA) developed PICME at approximately the same time ABMS was creating MOC. PICME is aimed at changing physician behavior within the context of their clinical practice and is divided into 3 stages:
- Stage A: learning from current practice performance assessment
- Stage B: learning from the application of performance improvement to patient care
- Stage C: learning from the evaluation of the PICME effort.
For example, a coalition of academic, nonprofit, and business organizations—the NOW Coalition for Bipolar Disorder— developed an online quality improvement activity (see Related Resources), which the ABPN certified for assessment and PIP points. It also is certified for 20 points toward the Self-Evaluation of Practice Performance MOC requirement through the American Board of Internal Medicine’s Approved Quality Improvement Pathway, 20 AMA PRA Category 1 Credits™, and 20 Prescribed Credits by the AAFP. Many physicians hold multiple board certificates, and this kind of activity can simultaneously meet requirements for licensure and several MOC programs.
Merging requirements
- reflective self-assessment
- assessment of knowledge and skills
- PIP.
Effects on reimbursement
In 2012, the Centers for Medicare and Medicaid Services’ Physician Quality Reporting System MOC Program Incentive provided a 0.5% incentive payment to physicians participating in a qualified MOC program.5 Other insurers are examining similar reimbursement incentives tied to practice assessment and improvement. Public reporting of quality metrics also is becoming more prevalent in practice and reimbursement incentives.
- Pinals DA. Ready or not, here it comes: maintenance of certification. J Am Acad Psychiatry Law. 2011;39(3):294-296.
- American Board of Psychiatry and Neurology, Inc. www.abpn.com.
- Maintenance of certification. American Board of Psychiatry and Neurology, Inc. www.abpn.com/moc_products.asp.
- NOW coalition performance improvement (PI) CME activity. NOW Coalition for Bipolar Disorder. www.nowbipolar.org/pi-cme.php.
Dr. Kues reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. American Board of Psychiatry and Neurology. Inc. Maintenance of Certification (10YR-MOC). http://www.abpn.com/moc_10yrmoc.html. Accessed December 18, 2012.
2. American Board of Psychiatry and Neurology. Inc. Maintenance of certification (CP-MOC). http://www.abpn.com/moc_cpmoc.html. Accessed December 18, 2012.
3. American Medical Association. The Physician’s Recognition Award and credit system. http://www.ama-assn.org/resources/doc/cme/pra-booklet.pdf. Published 2012. Accessed December 18 2012.
4. Federation of State Medical Boards. Maintenance of licensure (MOL) information center. http://www.fsmb.org/mol.html. Published 2012. Accessed December 18, 2012.
5. Centers for Medicare and Medicaid Services. Physician quality reporting system. http://www.cms.gov/Medicare/Quality-Initiatives-Patient-Assessment-Instruments/PQRS/index.html. Published September 27 2012. Accessed December 18, 2012.
Discuss this article at www.facebook.com/CurrentPsychiatry
In 2000, the American Board of Medical Specialties (ABMS) made a commitment to develop a maintenance of certification (MOC) system for their 24 specialty boards. MOC aims to keep physicians up to date because medical knowledge and practice are rapidly evolving and health care systems expect greater accountability linked with performance and outcomes. Previously, board certification for most specialties was limited to a 1-time board exam; upon passing, a clinician was considered board certified for life. The American Board of Psychiatry and Neurology (ABPN) first issued time-limited certificates for board certification in 1994; 2007 was the first year of initial MOC enrollment for ABPN. Diplomates whose certificates were issued before October 1, 1994 are not required to participate in the MOC program.
The ABPN time-limited certificates are on 10-year cycles and require diplomates to fulfill 4 MOC program components: Professional Standing, Self-Assessment and Continuing Medical Education (CME), Cognitive Expertise, and Performance in Practice (PIP) (Table).1 Requirement details are available at www.abpn.com.
The ABMS MOC initiative is closely aligned with other initiatives, such as maintenance of licensure (MOL), that will impact all physicians, including those who are not board certified and those who were certified before October 1, 1994 and therefore not required to participate in MOC. Licensure, reimbursement, and institutional credentials are developing required measures based on self-assessment and performance.
Table
Maintenance of certification: 4 components
| Component | Description |
|---|---|
| Professional Standing | Diplomates must hold an active and unrestricted license to practice medicine in ≥1 state commonwealth territory or possession of the United States or province of Canada |
| Self-Assessment and CME | Self-assessment: Diplomates must participate in ≥2 major broad-based self-assessment activities that must cover new knowledge and/or current best practices and provide feedback to the diplomate that can be used as the basis for focused CME lifelong learning and/or career development |
| CME activities: Diplomates are required to complete an average of 30 specialty or subspecialty Category 1 CME credits per year over the 10-year MOC cycle. At least an average of 8 of the CME credits per year (averaged over 2 to 5 years) should involve self-assessment | |
| Cognitive Expertise | Diplomates must pass a cognitive examination before the expiration date of their certificates |
| Performance in Practice (PIP) | Diplomates will be required to complete 3 PIP units over the 10-year MOC cycle each consisting of both a clinical module (chart review) and a feedback module (patient/peer second-party external review) |
| CME: continuing medical education; MOC: maintenance of certification Source: Adapted from reference 1 | |
MOC requirements
The ABMS developed its MOC program around 6 general competencies identified by the Accreditation Council for Graduate Medical Education:
- professionalism
- patient care and procedural skills
- medical knowledge
- practice-based learning and improvement
- interpersonal and communications skills
- systems-based practice.
Physicians with “lifetime” certificates are not required to participate in MOC; there are no consequences for physicians who are not required to participate in MOC and choose not to participate, because MOC is a voluntary system. Physicians with time-limited certificates can choose not to participate, but would forfeit their certification. Physicians with certifications in multiple specialties may consider the value of maintaining all of their certifications because it would require them to participate in multiple MOC programs.
Two of the 4 parts of MOC (Parts I and III) are extensions of existing board certification requirements. Part I stipulates a diplomate hold a valid and unrestricted license in ≥1 states or jurisdictions in the United States, its territories, or Canada. Part III (Cognitive Expertise) requires that he or she must pass a cognitive examination every 10 years. To qualify to take the cognitive exam, a diplomate must meet all current MOC requirements.
Parts II and IV integrate continuing education, self-assessment, and the ability to apply both to practice improvements. Part II requires an average of 8 CME credit hours that include a self-assessment component; this likely would eliminate most traditional CME activities. The ABPN stipulates that feedback from the self-assessment must include a comparison with peers and specific literature recommendations for each question in the self-assessment. A small but growing number of accredited CME providers have developed self-directed CME activities that meet these criteria. As of 2014, only ABPN-approved self-assessment activities can be used to meet Part II requirements.
Part IV, the PIP activity, has raised the most concern. The PIP component focuses on quality improvement in 2 parts: a clinical module and a feedback module. This targets active clinicians, and both modules focus on quality improvement activities. The clinical module consists of a baseline chart review by the physician MOC applicant in which results are compared with best practices or practice guidelines. The practitioner-applicant repeats a second chart review after a period of time to determine if intervening practice improvements had a positive impact.
The feedback module consists of reviews of clinical performance by patients, peers, or other second parties such as other practice staff or administrators. These are repeated after a period of time to determine whether practice improvements have been effective.
The PIP model (assessment, practice improvement, reassessment) parallels requirements for Performance Improvement CME (PICME) activities. The American Medical Association (AMA) developed PICME at approximately the same time ABMS was creating MOC. PICME is aimed at changing physician behavior within the context of their clinical practice and is divided into 3 stages:
- Stage A: learning from current practice performance assessment
- Stage B: learning from the application of performance improvement to patient care
- Stage C: learning from the evaluation of the PICME effort.
For example, a coalition of academic, nonprofit, and business organizations—the NOW Coalition for Bipolar Disorder— developed an online quality improvement activity (see Related Resources), which the ABPN certified for assessment and PIP points. It also is certified for 20 points toward the Self-Evaluation of Practice Performance MOC requirement through the American Board of Internal Medicine’s Approved Quality Improvement Pathway, 20 AMA PRA Category 1 Credits™, and 20 Prescribed Credits by the AAFP. Many physicians hold multiple board certificates, and this kind of activity can simultaneously meet requirements for licensure and several MOC programs.
Merging requirements
- reflective self-assessment
- assessment of knowledge and skills
- PIP.
Effects on reimbursement
In 2012, the Centers for Medicare and Medicaid Services’ Physician Quality Reporting System MOC Program Incentive provided a 0.5% incentive payment to physicians participating in a qualified MOC program.5 Other insurers are examining similar reimbursement incentives tied to practice assessment and improvement. Public reporting of quality metrics also is becoming more prevalent in practice and reimbursement incentives.
- Pinals DA. Ready or not, here it comes: maintenance of certification. J Am Acad Psychiatry Law. 2011;39(3):294-296.
- American Board of Psychiatry and Neurology, Inc. www.abpn.com.
- Maintenance of certification. American Board of Psychiatry and Neurology, Inc. www.abpn.com/moc_products.asp.
- NOW coalition performance improvement (PI) CME activity. NOW Coalition for Bipolar Disorder. www.nowbipolar.org/pi-cme.php.
Dr. Kues reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
In 2000, the American Board of Medical Specialties (ABMS) made a commitment to develop a maintenance of certification (MOC) system for their 24 specialty boards. MOC aims to keep physicians up to date because medical knowledge and practice are rapidly evolving and health care systems expect greater accountability linked with performance and outcomes. Previously, board certification for most specialties was limited to a 1-time board exam; upon passing, a clinician was considered board certified for life. The American Board of Psychiatry and Neurology (ABPN) first issued time-limited certificates for board certification in 1994; 2007 was the first year of initial MOC enrollment for ABPN. Diplomates whose certificates were issued before October 1, 1994 are not required to participate in the MOC program.
The ABPN time-limited certificates are on 10-year cycles and require diplomates to fulfill 4 MOC program components: Professional Standing, Self-Assessment and Continuing Medical Education (CME), Cognitive Expertise, and Performance in Practice (PIP) (Table).1 Requirement details are available at www.abpn.com.
The ABMS MOC initiative is closely aligned with other initiatives, such as maintenance of licensure (MOL), that will impact all physicians, including those who are not board certified and those who were certified before October 1, 1994 and therefore not required to participate in MOC. Licensure, reimbursement, and institutional credentials are developing required measures based on self-assessment and performance.
Table
Maintenance of certification: 4 components
| Component | Description |
|---|---|
| Professional Standing | Diplomates must hold an active and unrestricted license to practice medicine in ≥1 state commonwealth territory or possession of the United States or province of Canada |
| Self-Assessment and CME | Self-assessment: Diplomates must participate in ≥2 major broad-based self-assessment activities that must cover new knowledge and/or current best practices and provide feedback to the diplomate that can be used as the basis for focused CME lifelong learning and/or career development |
| CME activities: Diplomates are required to complete an average of 30 specialty or subspecialty Category 1 CME credits per year over the 10-year MOC cycle. At least an average of 8 of the CME credits per year (averaged over 2 to 5 years) should involve self-assessment | |
| Cognitive Expertise | Diplomates must pass a cognitive examination before the expiration date of their certificates |
| Performance in Practice (PIP) | Diplomates will be required to complete 3 PIP units over the 10-year MOC cycle each consisting of both a clinical module (chart review) and a feedback module (patient/peer second-party external review) |
| CME: continuing medical education; MOC: maintenance of certification Source: Adapted from reference 1 | |
MOC requirements
The ABMS developed its MOC program around 6 general competencies identified by the Accreditation Council for Graduate Medical Education:
- professionalism
- patient care and procedural skills
- medical knowledge
- practice-based learning and improvement
- interpersonal and communications skills
- systems-based practice.
Physicians with “lifetime” certificates are not required to participate in MOC; there are no consequences for physicians who are not required to participate in MOC and choose not to participate, because MOC is a voluntary system. Physicians with time-limited certificates can choose not to participate, but would forfeit their certification. Physicians with certifications in multiple specialties may consider the value of maintaining all of their certifications because it would require them to participate in multiple MOC programs.
Two of the 4 parts of MOC (Parts I and III) are extensions of existing board certification requirements. Part I stipulates a diplomate hold a valid and unrestricted license in ≥1 states or jurisdictions in the United States, its territories, or Canada. Part III (Cognitive Expertise) requires that he or she must pass a cognitive examination every 10 years. To qualify to take the cognitive exam, a diplomate must meet all current MOC requirements.
Parts II and IV integrate continuing education, self-assessment, and the ability to apply both to practice improvements. Part II requires an average of 8 CME credit hours that include a self-assessment component; this likely would eliminate most traditional CME activities. The ABPN stipulates that feedback from the self-assessment must include a comparison with peers and specific literature recommendations for each question in the self-assessment. A small but growing number of accredited CME providers have developed self-directed CME activities that meet these criteria. As of 2014, only ABPN-approved self-assessment activities can be used to meet Part II requirements.
Part IV, the PIP activity, has raised the most concern. The PIP component focuses on quality improvement in 2 parts: a clinical module and a feedback module. This targets active clinicians, and both modules focus on quality improvement activities. The clinical module consists of a baseline chart review by the physician MOC applicant in which results are compared with best practices or practice guidelines. The practitioner-applicant repeats a second chart review after a period of time to determine if intervening practice improvements had a positive impact.
The feedback module consists of reviews of clinical performance by patients, peers, or other second parties such as other practice staff or administrators. These are repeated after a period of time to determine whether practice improvements have been effective.
The PIP model (assessment, practice improvement, reassessment) parallels requirements for Performance Improvement CME (PICME) activities. The American Medical Association (AMA) developed PICME at approximately the same time ABMS was creating MOC. PICME is aimed at changing physician behavior within the context of their clinical practice and is divided into 3 stages:
- Stage A: learning from current practice performance assessment
- Stage B: learning from the application of performance improvement to patient care
- Stage C: learning from the evaluation of the PICME effort.
For example, a coalition of academic, nonprofit, and business organizations—the NOW Coalition for Bipolar Disorder— developed an online quality improvement activity (see Related Resources), which the ABPN certified for assessment and PIP points. It also is certified for 20 points toward the Self-Evaluation of Practice Performance MOC requirement through the American Board of Internal Medicine’s Approved Quality Improvement Pathway, 20 AMA PRA Category 1 Credits™, and 20 Prescribed Credits by the AAFP. Many physicians hold multiple board certificates, and this kind of activity can simultaneously meet requirements for licensure and several MOC programs.
Merging requirements
- reflective self-assessment
- assessment of knowledge and skills
- PIP.
Effects on reimbursement
In 2012, the Centers for Medicare and Medicaid Services’ Physician Quality Reporting System MOC Program Incentive provided a 0.5% incentive payment to physicians participating in a qualified MOC program.5 Other insurers are examining similar reimbursement incentives tied to practice assessment and improvement. Public reporting of quality metrics also is becoming more prevalent in practice and reimbursement incentives.
- Pinals DA. Ready or not, here it comes: maintenance of certification. J Am Acad Psychiatry Law. 2011;39(3):294-296.
- American Board of Psychiatry and Neurology, Inc. www.abpn.com.
- Maintenance of certification. American Board of Psychiatry and Neurology, Inc. www.abpn.com/moc_products.asp.
- NOW coalition performance improvement (PI) CME activity. NOW Coalition for Bipolar Disorder. www.nowbipolar.org/pi-cme.php.
Dr. Kues reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. American Board of Psychiatry and Neurology. Inc. Maintenance of Certification (10YR-MOC). http://www.abpn.com/moc_10yrmoc.html. Accessed December 18, 2012.
2. American Board of Psychiatry and Neurology. Inc. Maintenance of certification (CP-MOC). http://www.abpn.com/moc_cpmoc.html. Accessed December 18, 2012.
3. American Medical Association. The Physician’s Recognition Award and credit system. http://www.ama-assn.org/resources/doc/cme/pra-booklet.pdf. Published 2012. Accessed December 18 2012.
4. Federation of State Medical Boards. Maintenance of licensure (MOL) information center. http://www.fsmb.org/mol.html. Published 2012. Accessed December 18, 2012.
5. Centers for Medicare and Medicaid Services. Physician quality reporting system. http://www.cms.gov/Medicare/Quality-Initiatives-Patient-Assessment-Instruments/PQRS/index.html. Published September 27 2012. Accessed December 18, 2012.
1. American Board of Psychiatry and Neurology. Inc. Maintenance of Certification (10YR-MOC). http://www.abpn.com/moc_10yrmoc.html. Accessed December 18, 2012.
2. American Board of Psychiatry and Neurology. Inc. Maintenance of certification (CP-MOC). http://www.abpn.com/moc_cpmoc.html. Accessed December 18, 2012.
3. American Medical Association. The Physician’s Recognition Award and credit system. http://www.ama-assn.org/resources/doc/cme/pra-booklet.pdf. Published 2012. Accessed December 18 2012.
4. Federation of State Medical Boards. Maintenance of licensure (MOL) information center. http://www.fsmb.org/mol.html. Published 2012. Accessed December 18, 2012.
5. Centers for Medicare and Medicaid Services. Physician quality reporting system. http://www.cms.gov/Medicare/Quality-Initiatives-Patient-Assessment-Instruments/PQRS/index.html. Published September 27 2012. Accessed December 18, 2012.
Antidepressant use during pregnancy: How to avoid clinical and legal pitfalls
Discuss this article at www.facebook.com/CurrentPsychiatry
Recently there has been an increase in advertising soliciting participants for class-action lawsuits involving birth defects and antidepressants, particularly sertraline. Many psychiatrists are unsure why these ads are running in seemingly every medium because there has been no change in the FDA pregnancy classification for most selective serotonin reuptake inhibitors (SSRIs), except for paroxetine going from a C to a D rating in 2005.1 Some studies have found SSRIs increase the risk of adverse birth outcomes and others have not, which makes it difficult for clinicians to know what to discuss with patients regarding the risks and benefits of using antidepressants during pregnancy, as well as the risks of untreated major depressive disorder (MDD).
It can be hard to encourage some patients to take necessary medications in the best of circumstances, let alone suggest that a pregnant woman take a medication that has been labeled “dangerous.” This article seeks to alleviate physicians’ fears about being caught in a no-win situation by:
- explaining factors that may have led to this increase in class-action lawsuits
- clarifying the risks of using certain medications and not treating depression
- suggesting ways physicians can protect themselves and their patients.
The FDA’s position
In July 2006, the FDA issued a public health advisory regarding SSRI use during pregnancy and the possibility of persistent pulmonary hypertension (PPHN).2 This warning was based on a single study that found the risk of developing PPHN (baseline rate: 1 to 2 per 1,000 births) was 6 times greater for fetuses exposed to SSRIs in late pregnancy.3 Many legal websites highlight this 2006 warning as proof of SSRIs’ danger. However, because subsequent studies have had conflicting results, the FDA’s current position is that the risks of using SSRIs during pregnancy are “unknown” (Box).1-4
2006: In a warning about the risk of persistent pulmonary hypertension (PPHN) with antidepressant use during pregnancy, the FDA acknowledged “decisions about how to treat depression in pregnant women are increasingly complex.”2 The FDA issued the warning based on a study by Chambers et al,3 noting that the study was “too small” to look at individual medications.
This warning also cited a study by Cohen et al4 that found “women who stopped their [antidepressant] medicine were five times more likely to have a relapse of depression during their pregnancy than were the women who continued to take their antidepressant medicine while pregnant.”2 Although the warning identified a potential “rare” danger, the FDA guidance was that “women who are pregnant or thinking about becoming pregnant should not stop any antidepressant without first consulting their physician. The decision to continue medication or not should be made only after there has been careful consideration of the potential benefits and risks of the medication for each individual pregnant patient.”
2011: In this communication,1 the FDA stated “the initial Public Health Advisory in July 2006 on this potential risk was based on a single published study. Since then, there have been conflicting findings from new studies evaluating this potential risk, making it unclear whether use of [selective serotonin reuptake inhibitors (SSRIs)] during pregnancy can cause PPHN.” The FDA also said that the “potential risk with SSRI use during pregnancy remains unknown.”
Risks of depression
Although most physicians know the risks of untreated MDD, they tend to minimize or forget these risks when a woman becomes pregnant. Pregnant women with MDD face not only the expected risks of their psychiatric illness but additionally face risks of pre-eclampsia, suicide (20% of deaths in the postpartum period are due to suicide), and infanticide.5-10 Risks to the fetus include poor prenatal care, increased risk of intrauterine exposure to drugs or alcohol, increased exposure to maternal cortisol with resulting neurodevelopmental changes, preterm delivery, low birth weight, and failure to thrive.6-8 Later difficulties for the child of a mother with untreated depression may include poor stress adaptation, decreased cognitive performance, and behavioral difficulties because of poor mother-child bonding and other factors.6
See Table 1 for key statistics regarding pregnancy and depression.
Table 1
Statistics on pregnancy and depression
| There are approximately 6 million pregnancies each year in the United Statesa |
| There are approximately 4 million live births each year in the United Statesa |
| Two percent to 3% of healthy pregnancies result in a birth defect or miscarriageb-d |
| Sixty percent to 70% of birth complications occur due to an unknown caused |
| Rates of depression during pregnancy are 7% to 25%b,e,f |
| Approximately 13% of pregnant women take an antidepressant during pregnancye |
| Fifteen percent of women with untreated depression in pregnancy attempt suicideb |
| Twenty percent of deaths in the postpartum period are due to suicidee |
| Women who discontinue antidepressants are 5 times more likely than women who continue medications in pregnancy to have a relapse of depressiong,h |
| SSRIs are the antidepressant class most frequently prescribed to pregnant womeni |
| Sertraline is one of the most frequently prescribed antidepressants perinatally and has low concentration in breast milk and infant serumj,k |
| SSRIs: selective serotonin reuptake inhibitors
|
Limitations of research
Because of ethical difficulties in studying MDD treatment during pregnancy, most data are retrospective and prone to detection and confounding biases, such as11-15:
- the risks associated with depression
- comorbid conditions such as obesity
- maternal age
- poor prenatal care
- how the baby was delivered (eg, Caesarean sections have higher rates of PPHN)13
- illicit substance use
- effects of other medications (80% of pregnant women use medications, including nonsteroidal anti-inflammatory drugs [NSAIDs], which are associated with PPHN).11,16
There are several potential adverse outcomes to consider when prescribing psychotropics to a pregnant woman, including miscarriage, malformation, preterm delivery, perinatal toxicity, and behavioral teratogenesis (Table 2).6,7 SSRIs have been implicated in adverse outcomes, but there is no strong evidence that they increase the miscarriage rate, and several studies found no increase in birth defects.6,13,18-20 Regarding teratogenesis, the FDA switched paroxetine from class C to class D because of a potential 1.5% to 2% risk of fetal cardiac malformation, compared with a 1% baseline rate in the general population.21 Drug toxicity or withdrawal in a neonate also is a risk; however, this condition is self-limited and managed supportively by neonatology.22 Behavioral teratogenesis—neurobehavioral problems that develop later in a child’s life—remains a hypothetical concern; research has been conflicting, and studies often used flawed methodology.
- these databases were not designed to answer these types of exposure questions (eg, limitations in data collected, such as other potential causes not recorded)
- they have many confounding biases (undocumented illicit substance use, possible minimization of smoking history, publication basis for positive findings, etc.)
- individuals who provided the data did not follow a standardized method (eg, variability among individual clinicians).
Not to case aspersions on this group’s work, it should be noted that this study had limitations, including that the researchers:
- did not take into account SSRI dosage
- did not measure depression severity or remittance
- were not able to fully account for potential exposures (eg, over-the-counter NSAIDs)
- were unable to confirm that patients took their medications because the variable measured was prescriptions filled
- did not interview participants about their medication use or symptoms.
In a more recent study,24 33 of 11,014 infants exposed to SSRIs after gestational week 20 developed PPHN (absolute risk: 3 per 1,000 births, compared with an incidence of 1.2 per 1,000 births in the general population), with an adjusted OR of 2.1 (95% CI 1.5 to 3.0). Although the authors warned that the results suggest a “class effect,” the rate of PPHN also was higher for mothers with a history of a psychiatric hospitalization within the last 10 years who were not taking medication (OR=1.3, 95% CI 1.0 to 1.6) and the OR for escitalopram (1.5, CI 0.2 to 10.5) was not statistically significant. This study did include a control group, but the 10-year window may have been too wide to represent a group with similar comorbid risks. Similar to the previously discussed study, mothers prescribed SSRIs were older, 1.7 times more likely to be smokers, and twice as likely to be prescribed NSAIDs. The study did not analyze the risk factors of smoking and body mass index because of an initial subset analysis (which was not reported) finding that these known risk factors for PPHN “did not confound the results.”24
Table 2
Potential concerns when treating pregnant women with psychotropics
| Miscarriage (spontaneous abortion) |
| Malformation (teratogenesis) |
| Preterm delivery |
| Perinatal syndrome (toxicity or withdrawal in neonate; usually self-limited and related to serotonin overstimulation or withdrawal; symptoms may include disrupted sleep irritability jitteriness or abnormal breathing) |
| Behavioral teratogenesis (later behavioral problems in child eg lower IQ developmental delays or autism) |
| Lactation compatibility or plans to bottle-feed |
| Source: References 6,7 |
The basis of class-action lawsuits
Interest in class-action lawsuits involving birth defects and antidepressants, particularly sertraline, appears to be increasing. Many websites advertising these lawsuits quote unnamed articles from reputable medical journals to support the claim that the medications are dangerous and cause a wide range of birth defects. Although some of the birth defects mentioned are specific, others (eg, “breathing problems” or “gastrointestinal side effects”) are so broad that any problem or complication could conceivably be attributed to the antidepressant. The degree of causation—if any at all—for many of these conditions has not been determined. A national advertising campaign looking for any problem may be occurring because the exact risks are “unknown.”1
The 2009 U.S. Supreme Court ruling in Wyeth v Levine25 allows individuals to sue manufacturers of branded medications in state and federal court for lack of proper labeling. However, the 2011 U.S. Supreme Court case of PLIVA, Inc. v Mensing26 prohibits state lawsuits against manufacturers of generic medications over labeling because by federal (superseding) law, generic manufacturers must use the same warnings as the branded medication. This may in part explain why many medications targeted in commercials and websites for class-action lawsuits are branded products, even though generics are available.
Protect your patient and yourself
An estimated 13% of pregnant women take antidepressants; SSRIs are the most commonly used antidepressant during and after pregnancy.9 Although not every depressed pregnant woman requires medication, those with moderate to severe depression often do. Rational medication decisions, informed consent, and good documentation are important when treating these women. Discuss the risks of untreated illness as well as the risks of medications to ensure that the patient understands that avoiding medication does not guarantee a safe pregnancy. Suggest psychotherapy and electroconvulsive therapy as options when appropriate. When possible, include the patient’s partner and family in the discussion to help improve compliance and potentially reduce strife.29 The psychiatrist or patient should discuss the medication plan with the patient’s obstetrician or family physician.
6,22
Many women become pregnant while being treated for depression. Approximately one-half of all pregnancies are unplanned, so women using antidepressants may unknowingly expose their fetus to medication.30 For this reason, it is important to discuss potential pregnancy and birth control concerns with all women of childbearing age before initiating pharmacotherapy.31 If an unintended pregnancy occurs, tell your patient to contact you before stopping any medications. Lawsuits also can occur because of wrongful death by suicide or infanticide because of lack of treatment; risk of untreated illness should not be treated lightly.
Related Resources
- Motherisk. www.motherisk.org.
- Organization of Teratology Information Specialists. www.otispregnancy.org.
- Massachusetts General Hospital Center for Women’s Mental Health. www.womensmentalhealth.org.
- Escitalopram • Lexapro
- Paroxetine • Paxil
- Sertraline • Zoloft
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgments
The authors appreciate suggestions on prior versions of the manuscript from Miriam Rosenthal, Jaina Amin, Sarah Nagle-Yang, Sonal Moratschek, J.P. Shand, and Scott R. Miller.
1. U.S. Food and Drug Administration. FDA drug safety communication: selective serotonin reuptake inhibitor (SSRI) antidepressant use during pregnancy and reports of a rare heart and lung condition in newborn babies. http://www.fda.gov/Drugs/DrugSafety/ucm283375.htm. Published December 14, 2011. Accessed December 20, 2012.
2. U.S. Food and Drug Administration. Public health advisory: treatment challenges of depression in pregnancy and the possibility of persistent pulmonary hypertension in newborns. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsand
Providers/DrugSafetyInformationforHeathcareProfessionals/PublicHealthAdvisories/
ucm124348.htm. Published July 19, 2006. Accessed December 20, 2012.
3. Chambers CD, Hernandez-Diaz S, Van Marter LJ, et al. Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med. 2006;354(6):579-587.
4. Cohen LS, Altshuler LL, Harlow BL, et al. Relapse of major depression during pregnancy in women who maintain or discontinue antidepressant treatment. JAMA. 2006;295(5):499-507.
5. Muzik M, Hamilton S. Psychiatric illness during pregnancy. Current Psychiatry. 2012;11(2):23-32.
6. Hasser C, Brizendine L, Spielvogel A. SSRI use during pregnancy. Current Psychiatry. 2006;5(4):31-40.
7. Wisner KL, Sit DK, Hanusa BH, et al. Major depression and antidepressant treatment: impact on pregnancy and neonatal outcomes. Am J Psychiatry. 2009;166(5):557-566.
8. Friedman SH, Resnick PJ. Postpartum depression: an update. Women’s Health (Lond Engl). 2009;5(3):287-295.
9. Meltzer-Brody S. New insights into perinatal depression: pathogenesis and treatment during pregnancy and postpartum. Dialogues Clin Neurosci. 2011;13(1):89-100.
10. Friedman SH, Hall RCW. Treatment of mental illness in pregnancy and malpractice concerns. News Amer Acad Psychiatry Law. 2012;37(2):21-22.
11. Yonkers KA, Wisner KL, Stewart DE, et al. The management of depression during pregnancy: a report from the American Psychiatric Association and the American College of Obstetricians and Gynecologists. Gen Hosp Psychiatry. 2009;31(5):403-413.
12. Bar-Oz B, Einarson T, Einarson A, et al. Paroxetine and congenital malformations: meta-analysis and consideration of potential confounding factors. Clin Ther. 2007;29(5):918-926.
13. Wilson KL, Zelig CM, Harvey JP. Persistent pulmonary hypertension of the newborn is associated with mode of delivery and not with maternal use of selective serotonin reuptake inhibitors. Am J Perinatol. 2011;28(1):19-24.
14. Silvani P, Camporesi A. Drug-induced pulmonary hypertension in newborns: a review. Curr Vasc Pharmacol. 2007;5(2):129-133.
15. Occhiogrosso M, Omran SS, Altemus M. Persistent pulmonary hypertension of the newborn and selective serotonin reuptake inhibitors: lessons from clinical and translational studies. Am J Psychiatry. 2012;169(2):134-140.
16. Delaney C, Cornfield D. Risk factors for persistent pulmonary hypertension of the newborn. Pulm Circ. 2012;2(1):15-20.
17. Centers for Disease Control and Prevention. Key findings: updated national birth prevalence estimates for selected birth defects in the United States 2004-2006. http://www.cdc.gov/ncbddd/features/birthdefects-keyfindings.html. Published September 28, 2010. Accessed December 20, 2012.
18. Einarson A, Choi J, Einarson TR, et al. Incidence of major malformations in infants following antidepressant exposure in pregnancy: results of a large prospective cohort study. Can J Psychiatry. 2009;54(4):242-246.
19. Alwan S, Reefhuis J, Rasmussen SA, et al. National Birth Defects Prevention Study. Use of selective serotonin-reuptake inhibitors in pregnancy and the risk of birth defects. N Engl J Med. 2007;356(26):2684-2692.
20. Andrade SE, McPhillips H, Loren D. Antidepressant medication use and risk of persistent pulmonary hypertension of the newborn. Pharmacoepidemiol Drug Saf. 2009;18(3):246-252.
21. U.S. Food and Drug Administration. FDA advising of risk of birth defects with paxil. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2005/ucm108527.htm. Published December 8, 2005. Accessed December 20, 2012.
22. Koren G, Boucher N. Adverse effects in neonates exposed to SSRIs and SNRI in late gestation-Motherisk Update 2008. Can J Clin Pharmacol. 2009;16(1):e66-e67.
23. Pederson LH, Henriksen TB, Vestergaard M, et al. Selective serotonin reuptake inhibitors in pregnancy and congenital malformations: population based cohort study. BMJ. 2009;339:b3569.-doi:10.1136/bmj.b3569.
24. Kieler H, Artama M, Engeland A, et al. Selective serotonin reuptake inhibitors during pregnancy and risk of persistent pulmonary hypertension in the newborn: population based cohort study from the five Nordic countries. BMJ. 2012;344:d8012.-doi:10.1136/bmj.d801.
25. Wyeth v Levine, 555 US 555 (2009).
26. PLIVA, Inc. v Mensing, 588 F3d 603, 593 F3d 428 (2011).
27. Greenwood K. The mysteries of pregnancy: the role of law in solving the problem of unknown but knowable maternal–fetal medication risk. University of Cincinnati Law Review. 2011;79(1):267-322.
28. Lyam Kilker v SmithKline Beecham Corporation, Philadelphia Court of Common Pleas (2009).
29. Mulder E, Davis A, Gawley L, et al. Negative impact of non-evidence-based information received by women taking antidepressants during pregnancy from health care providers and others. J Obstet Gynaecol Can. 2012;34(1):66-71.
30. Henshaw SK. Unintended pregnancy in the United States. Fam Plann Perspect. 1998;30(1):24-29 46.
31. Altshuler L, Richards M, Yonkers K. Treating bipolar disorder during pregnancy. Current Psychiatry. 2003;2(7):14-26.
Discuss this article at www.facebook.com/CurrentPsychiatry
Recently there has been an increase in advertising soliciting participants for class-action lawsuits involving birth defects and antidepressants, particularly sertraline. Many psychiatrists are unsure why these ads are running in seemingly every medium because there has been no change in the FDA pregnancy classification for most selective serotonin reuptake inhibitors (SSRIs), except for paroxetine going from a C to a D rating in 2005.1 Some studies have found SSRIs increase the risk of adverse birth outcomes and others have not, which makes it difficult for clinicians to know what to discuss with patients regarding the risks and benefits of using antidepressants during pregnancy, as well as the risks of untreated major depressive disorder (MDD).
It can be hard to encourage some patients to take necessary medications in the best of circumstances, let alone suggest that a pregnant woman take a medication that has been labeled “dangerous.” This article seeks to alleviate physicians’ fears about being caught in a no-win situation by:
- explaining factors that may have led to this increase in class-action lawsuits
- clarifying the risks of using certain medications and not treating depression
- suggesting ways physicians can protect themselves and their patients.
The FDA’s position
In July 2006, the FDA issued a public health advisory regarding SSRI use during pregnancy and the possibility of persistent pulmonary hypertension (PPHN).2 This warning was based on a single study that found the risk of developing PPHN (baseline rate: 1 to 2 per 1,000 births) was 6 times greater for fetuses exposed to SSRIs in late pregnancy.3 Many legal websites highlight this 2006 warning as proof of SSRIs’ danger. However, because subsequent studies have had conflicting results, the FDA’s current position is that the risks of using SSRIs during pregnancy are “unknown” (Box).1-4
2006: In a warning about the risk of persistent pulmonary hypertension (PPHN) with antidepressant use during pregnancy, the FDA acknowledged “decisions about how to treat depression in pregnant women are increasingly complex.”2 The FDA issued the warning based on a study by Chambers et al,3 noting that the study was “too small” to look at individual medications.
This warning also cited a study by Cohen et al4 that found “women who stopped their [antidepressant] medicine were five times more likely to have a relapse of depression during their pregnancy than were the women who continued to take their antidepressant medicine while pregnant.”2 Although the warning identified a potential “rare” danger, the FDA guidance was that “women who are pregnant or thinking about becoming pregnant should not stop any antidepressant without first consulting their physician. The decision to continue medication or not should be made only after there has been careful consideration of the potential benefits and risks of the medication for each individual pregnant patient.”
2011: In this communication,1 the FDA stated “the initial Public Health Advisory in July 2006 on this potential risk was based on a single published study. Since then, there have been conflicting findings from new studies evaluating this potential risk, making it unclear whether use of [selective serotonin reuptake inhibitors (SSRIs)] during pregnancy can cause PPHN.” The FDA also said that the “potential risk with SSRI use during pregnancy remains unknown.”
Risks of depression
Although most physicians know the risks of untreated MDD, they tend to minimize or forget these risks when a woman becomes pregnant. Pregnant women with MDD face not only the expected risks of their psychiatric illness but additionally face risks of pre-eclampsia, suicide (20% of deaths in the postpartum period are due to suicide), and infanticide.5-10 Risks to the fetus include poor prenatal care, increased risk of intrauterine exposure to drugs or alcohol, increased exposure to maternal cortisol with resulting neurodevelopmental changes, preterm delivery, low birth weight, and failure to thrive.6-8 Later difficulties for the child of a mother with untreated depression may include poor stress adaptation, decreased cognitive performance, and behavioral difficulties because of poor mother-child bonding and other factors.6
See Table 1 for key statistics regarding pregnancy and depression.
Table 1
Statistics on pregnancy and depression
| There are approximately 6 million pregnancies each year in the United Statesa |
| There are approximately 4 million live births each year in the United Statesa |
| Two percent to 3% of healthy pregnancies result in a birth defect or miscarriageb-d |
| Sixty percent to 70% of birth complications occur due to an unknown caused |
| Rates of depression during pregnancy are 7% to 25%b,e,f |
| Approximately 13% of pregnant women take an antidepressant during pregnancye |
| Fifteen percent of women with untreated depression in pregnancy attempt suicideb |
| Twenty percent of deaths in the postpartum period are due to suicidee |
| Women who discontinue antidepressants are 5 times more likely than women who continue medications in pregnancy to have a relapse of depressiong,h |
| SSRIs are the antidepressant class most frequently prescribed to pregnant womeni |
| Sertraline is one of the most frequently prescribed antidepressants perinatally and has low concentration in breast milk and infant serumj,k |
| SSRIs: selective serotonin reuptake inhibitors
|
Limitations of research
Because of ethical difficulties in studying MDD treatment during pregnancy, most data are retrospective and prone to detection and confounding biases, such as11-15:
- the risks associated with depression
- comorbid conditions such as obesity
- maternal age
- poor prenatal care
- how the baby was delivered (eg, Caesarean sections have higher rates of PPHN)13
- illicit substance use
- effects of other medications (80% of pregnant women use medications, including nonsteroidal anti-inflammatory drugs [NSAIDs], which are associated with PPHN).11,16
There are several potential adverse outcomes to consider when prescribing psychotropics to a pregnant woman, including miscarriage, malformation, preterm delivery, perinatal toxicity, and behavioral teratogenesis (Table 2).6,7 SSRIs have been implicated in adverse outcomes, but there is no strong evidence that they increase the miscarriage rate, and several studies found no increase in birth defects.6,13,18-20 Regarding teratogenesis, the FDA switched paroxetine from class C to class D because of a potential 1.5% to 2% risk of fetal cardiac malformation, compared with a 1% baseline rate in the general population.21 Drug toxicity or withdrawal in a neonate also is a risk; however, this condition is self-limited and managed supportively by neonatology.22 Behavioral teratogenesis—neurobehavioral problems that develop later in a child’s life—remains a hypothetical concern; research has been conflicting, and studies often used flawed methodology.
- these databases were not designed to answer these types of exposure questions (eg, limitations in data collected, such as other potential causes not recorded)
- they have many confounding biases (undocumented illicit substance use, possible minimization of smoking history, publication basis for positive findings, etc.)
- individuals who provided the data did not follow a standardized method (eg, variability among individual clinicians).
Not to case aspersions on this group’s work, it should be noted that this study had limitations, including that the researchers:
- did not take into account SSRI dosage
- did not measure depression severity or remittance
- were not able to fully account for potential exposures (eg, over-the-counter NSAIDs)
- were unable to confirm that patients took their medications because the variable measured was prescriptions filled
- did not interview participants about their medication use or symptoms.
In a more recent study,24 33 of 11,014 infants exposed to SSRIs after gestational week 20 developed PPHN (absolute risk: 3 per 1,000 births, compared with an incidence of 1.2 per 1,000 births in the general population), with an adjusted OR of 2.1 (95% CI 1.5 to 3.0). Although the authors warned that the results suggest a “class effect,” the rate of PPHN also was higher for mothers with a history of a psychiatric hospitalization within the last 10 years who were not taking medication (OR=1.3, 95% CI 1.0 to 1.6) and the OR for escitalopram (1.5, CI 0.2 to 10.5) was not statistically significant. This study did include a control group, but the 10-year window may have been too wide to represent a group with similar comorbid risks. Similar to the previously discussed study, mothers prescribed SSRIs were older, 1.7 times more likely to be smokers, and twice as likely to be prescribed NSAIDs. The study did not analyze the risk factors of smoking and body mass index because of an initial subset analysis (which was not reported) finding that these known risk factors for PPHN “did not confound the results.”24
Table 2
Potential concerns when treating pregnant women with psychotropics
| Miscarriage (spontaneous abortion) |
| Malformation (teratogenesis) |
| Preterm delivery |
| Perinatal syndrome (toxicity or withdrawal in neonate; usually self-limited and related to serotonin overstimulation or withdrawal; symptoms may include disrupted sleep irritability jitteriness or abnormal breathing) |
| Behavioral teratogenesis (later behavioral problems in child eg lower IQ developmental delays or autism) |
| Lactation compatibility or plans to bottle-feed |
| Source: References 6,7 |
The basis of class-action lawsuits
Interest in class-action lawsuits involving birth defects and antidepressants, particularly sertraline, appears to be increasing. Many websites advertising these lawsuits quote unnamed articles from reputable medical journals to support the claim that the medications are dangerous and cause a wide range of birth defects. Although some of the birth defects mentioned are specific, others (eg, “breathing problems” or “gastrointestinal side effects”) are so broad that any problem or complication could conceivably be attributed to the antidepressant. The degree of causation—if any at all—for many of these conditions has not been determined. A national advertising campaign looking for any problem may be occurring because the exact risks are “unknown.”1
The 2009 U.S. Supreme Court ruling in Wyeth v Levine25 allows individuals to sue manufacturers of branded medications in state and federal court for lack of proper labeling. However, the 2011 U.S. Supreme Court case of PLIVA, Inc. v Mensing26 prohibits state lawsuits against manufacturers of generic medications over labeling because by federal (superseding) law, generic manufacturers must use the same warnings as the branded medication. This may in part explain why many medications targeted in commercials and websites for class-action lawsuits are branded products, even though generics are available.
Protect your patient and yourself
An estimated 13% of pregnant women take antidepressants; SSRIs are the most commonly used antidepressant during and after pregnancy.9 Although not every depressed pregnant woman requires medication, those with moderate to severe depression often do. Rational medication decisions, informed consent, and good documentation are important when treating these women. Discuss the risks of untreated illness as well as the risks of medications to ensure that the patient understands that avoiding medication does not guarantee a safe pregnancy. Suggest psychotherapy and electroconvulsive therapy as options when appropriate. When possible, include the patient’s partner and family in the discussion to help improve compliance and potentially reduce strife.29 The psychiatrist or patient should discuss the medication plan with the patient’s obstetrician or family physician.
6,22
Many women become pregnant while being treated for depression. Approximately one-half of all pregnancies are unplanned, so women using antidepressants may unknowingly expose their fetus to medication.30 For this reason, it is important to discuss potential pregnancy and birth control concerns with all women of childbearing age before initiating pharmacotherapy.31 If an unintended pregnancy occurs, tell your patient to contact you before stopping any medications. Lawsuits also can occur because of wrongful death by suicide or infanticide because of lack of treatment; risk of untreated illness should not be treated lightly.
Related Resources
- Motherisk. www.motherisk.org.
- Organization of Teratology Information Specialists. www.otispregnancy.org.
- Massachusetts General Hospital Center for Women’s Mental Health. www.womensmentalhealth.org.
- Escitalopram • Lexapro
- Paroxetine • Paxil
- Sertraline • Zoloft
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgments
The authors appreciate suggestions on prior versions of the manuscript from Miriam Rosenthal, Jaina Amin, Sarah Nagle-Yang, Sonal Moratschek, J.P. Shand, and Scott R. Miller.
Discuss this article at www.facebook.com/CurrentPsychiatry
Recently there has been an increase in advertising soliciting participants for class-action lawsuits involving birth defects and antidepressants, particularly sertraline. Many psychiatrists are unsure why these ads are running in seemingly every medium because there has been no change in the FDA pregnancy classification for most selective serotonin reuptake inhibitors (SSRIs), except for paroxetine going from a C to a D rating in 2005.1 Some studies have found SSRIs increase the risk of adverse birth outcomes and others have not, which makes it difficult for clinicians to know what to discuss with patients regarding the risks and benefits of using antidepressants during pregnancy, as well as the risks of untreated major depressive disorder (MDD).
It can be hard to encourage some patients to take necessary medications in the best of circumstances, let alone suggest that a pregnant woman take a medication that has been labeled “dangerous.” This article seeks to alleviate physicians’ fears about being caught in a no-win situation by:
- explaining factors that may have led to this increase in class-action lawsuits
- clarifying the risks of using certain medications and not treating depression
- suggesting ways physicians can protect themselves and their patients.
The FDA’s position
In July 2006, the FDA issued a public health advisory regarding SSRI use during pregnancy and the possibility of persistent pulmonary hypertension (PPHN).2 This warning was based on a single study that found the risk of developing PPHN (baseline rate: 1 to 2 per 1,000 births) was 6 times greater for fetuses exposed to SSRIs in late pregnancy.3 Many legal websites highlight this 2006 warning as proof of SSRIs’ danger. However, because subsequent studies have had conflicting results, the FDA’s current position is that the risks of using SSRIs during pregnancy are “unknown” (Box).1-4
2006: In a warning about the risk of persistent pulmonary hypertension (PPHN) with antidepressant use during pregnancy, the FDA acknowledged “decisions about how to treat depression in pregnant women are increasingly complex.”2 The FDA issued the warning based on a study by Chambers et al,3 noting that the study was “too small” to look at individual medications.
This warning also cited a study by Cohen et al4 that found “women who stopped their [antidepressant] medicine were five times more likely to have a relapse of depression during their pregnancy than were the women who continued to take their antidepressant medicine while pregnant.”2 Although the warning identified a potential “rare” danger, the FDA guidance was that “women who are pregnant or thinking about becoming pregnant should not stop any antidepressant without first consulting their physician. The decision to continue medication or not should be made only after there has been careful consideration of the potential benefits and risks of the medication for each individual pregnant patient.”
2011: In this communication,1 the FDA stated “the initial Public Health Advisory in July 2006 on this potential risk was based on a single published study. Since then, there have been conflicting findings from new studies evaluating this potential risk, making it unclear whether use of [selective serotonin reuptake inhibitors (SSRIs)] during pregnancy can cause PPHN.” The FDA also said that the “potential risk with SSRI use during pregnancy remains unknown.”
Risks of depression
Although most physicians know the risks of untreated MDD, they tend to minimize or forget these risks when a woman becomes pregnant. Pregnant women with MDD face not only the expected risks of their psychiatric illness but additionally face risks of pre-eclampsia, suicide (20% of deaths in the postpartum period are due to suicide), and infanticide.5-10 Risks to the fetus include poor prenatal care, increased risk of intrauterine exposure to drugs or alcohol, increased exposure to maternal cortisol with resulting neurodevelopmental changes, preterm delivery, low birth weight, and failure to thrive.6-8 Later difficulties for the child of a mother with untreated depression may include poor stress adaptation, decreased cognitive performance, and behavioral difficulties because of poor mother-child bonding and other factors.6
See Table 1 for key statistics regarding pregnancy and depression.
Table 1
Statistics on pregnancy and depression
| There are approximately 6 million pregnancies each year in the United Statesa |
| There are approximately 4 million live births each year in the United Statesa |
| Two percent to 3% of healthy pregnancies result in a birth defect or miscarriageb-d |
| Sixty percent to 70% of birth complications occur due to an unknown caused |
| Rates of depression during pregnancy are 7% to 25%b,e,f |
| Approximately 13% of pregnant women take an antidepressant during pregnancye |
| Fifteen percent of women with untreated depression in pregnancy attempt suicideb |
| Twenty percent of deaths in the postpartum period are due to suicidee |
| Women who discontinue antidepressants are 5 times more likely than women who continue medications in pregnancy to have a relapse of depressiong,h |
| SSRIs are the antidepressant class most frequently prescribed to pregnant womeni |
| Sertraline is one of the most frequently prescribed antidepressants perinatally and has low concentration in breast milk and infant serumj,k |
| SSRIs: selective serotonin reuptake inhibitors
|
Limitations of research
Because of ethical difficulties in studying MDD treatment during pregnancy, most data are retrospective and prone to detection and confounding biases, such as11-15:
- the risks associated with depression
- comorbid conditions such as obesity
- maternal age
- poor prenatal care
- how the baby was delivered (eg, Caesarean sections have higher rates of PPHN)13
- illicit substance use
- effects of other medications (80% of pregnant women use medications, including nonsteroidal anti-inflammatory drugs [NSAIDs], which are associated with PPHN).11,16
There are several potential adverse outcomes to consider when prescribing psychotropics to a pregnant woman, including miscarriage, malformation, preterm delivery, perinatal toxicity, and behavioral teratogenesis (Table 2).6,7 SSRIs have been implicated in adverse outcomes, but there is no strong evidence that they increase the miscarriage rate, and several studies found no increase in birth defects.6,13,18-20 Regarding teratogenesis, the FDA switched paroxetine from class C to class D because of a potential 1.5% to 2% risk of fetal cardiac malformation, compared with a 1% baseline rate in the general population.21 Drug toxicity or withdrawal in a neonate also is a risk; however, this condition is self-limited and managed supportively by neonatology.22 Behavioral teratogenesis—neurobehavioral problems that develop later in a child’s life—remains a hypothetical concern; research has been conflicting, and studies often used flawed methodology.
- these databases were not designed to answer these types of exposure questions (eg, limitations in data collected, such as other potential causes not recorded)
- they have many confounding biases (undocumented illicit substance use, possible minimization of smoking history, publication basis for positive findings, etc.)
- individuals who provided the data did not follow a standardized method (eg, variability among individual clinicians).
Not to case aspersions on this group’s work, it should be noted that this study had limitations, including that the researchers:
- did not take into account SSRI dosage
- did not measure depression severity or remittance
- were not able to fully account for potential exposures (eg, over-the-counter NSAIDs)
- were unable to confirm that patients took their medications because the variable measured was prescriptions filled
- did not interview participants about their medication use or symptoms.
In a more recent study,24 33 of 11,014 infants exposed to SSRIs after gestational week 20 developed PPHN (absolute risk: 3 per 1,000 births, compared with an incidence of 1.2 per 1,000 births in the general population), with an adjusted OR of 2.1 (95% CI 1.5 to 3.0). Although the authors warned that the results suggest a “class effect,” the rate of PPHN also was higher for mothers with a history of a psychiatric hospitalization within the last 10 years who were not taking medication (OR=1.3, 95% CI 1.0 to 1.6) and the OR for escitalopram (1.5, CI 0.2 to 10.5) was not statistically significant. This study did include a control group, but the 10-year window may have been too wide to represent a group with similar comorbid risks. Similar to the previously discussed study, mothers prescribed SSRIs were older, 1.7 times more likely to be smokers, and twice as likely to be prescribed NSAIDs. The study did not analyze the risk factors of smoking and body mass index because of an initial subset analysis (which was not reported) finding that these known risk factors for PPHN “did not confound the results.”24
Table 2
Potential concerns when treating pregnant women with psychotropics
| Miscarriage (spontaneous abortion) |
| Malformation (teratogenesis) |
| Preterm delivery |
| Perinatal syndrome (toxicity or withdrawal in neonate; usually self-limited and related to serotonin overstimulation or withdrawal; symptoms may include disrupted sleep irritability jitteriness or abnormal breathing) |
| Behavioral teratogenesis (later behavioral problems in child eg lower IQ developmental delays or autism) |
| Lactation compatibility or plans to bottle-feed |
| Source: References 6,7 |
The basis of class-action lawsuits
Interest in class-action lawsuits involving birth defects and antidepressants, particularly sertraline, appears to be increasing. Many websites advertising these lawsuits quote unnamed articles from reputable medical journals to support the claim that the medications are dangerous and cause a wide range of birth defects. Although some of the birth defects mentioned are specific, others (eg, “breathing problems” or “gastrointestinal side effects”) are so broad that any problem or complication could conceivably be attributed to the antidepressant. The degree of causation—if any at all—for many of these conditions has not been determined. A national advertising campaign looking for any problem may be occurring because the exact risks are “unknown.”1
The 2009 U.S. Supreme Court ruling in Wyeth v Levine25 allows individuals to sue manufacturers of branded medications in state and federal court for lack of proper labeling. However, the 2011 U.S. Supreme Court case of PLIVA, Inc. v Mensing26 prohibits state lawsuits against manufacturers of generic medications over labeling because by federal (superseding) law, generic manufacturers must use the same warnings as the branded medication. This may in part explain why many medications targeted in commercials and websites for class-action lawsuits are branded products, even though generics are available.
Protect your patient and yourself
An estimated 13% of pregnant women take antidepressants; SSRIs are the most commonly used antidepressant during and after pregnancy.9 Although not every depressed pregnant woman requires medication, those with moderate to severe depression often do. Rational medication decisions, informed consent, and good documentation are important when treating these women. Discuss the risks of untreated illness as well as the risks of medications to ensure that the patient understands that avoiding medication does not guarantee a safe pregnancy. Suggest psychotherapy and electroconvulsive therapy as options when appropriate. When possible, include the patient’s partner and family in the discussion to help improve compliance and potentially reduce strife.29 The psychiatrist or patient should discuss the medication plan with the patient’s obstetrician or family physician.
6,22
Many women become pregnant while being treated for depression. Approximately one-half of all pregnancies are unplanned, so women using antidepressants may unknowingly expose their fetus to medication.30 For this reason, it is important to discuss potential pregnancy and birth control concerns with all women of childbearing age before initiating pharmacotherapy.31 If an unintended pregnancy occurs, tell your patient to contact you before stopping any medications. Lawsuits also can occur because of wrongful death by suicide or infanticide because of lack of treatment; risk of untreated illness should not be treated lightly.
Related Resources
- Motherisk. www.motherisk.org.
- Organization of Teratology Information Specialists. www.otispregnancy.org.
- Massachusetts General Hospital Center for Women’s Mental Health. www.womensmentalhealth.org.
- Escitalopram • Lexapro
- Paroxetine • Paxil
- Sertraline • Zoloft
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgments
The authors appreciate suggestions on prior versions of the manuscript from Miriam Rosenthal, Jaina Amin, Sarah Nagle-Yang, Sonal Moratschek, J.P. Shand, and Scott R. Miller.
1. U.S. Food and Drug Administration. FDA drug safety communication: selective serotonin reuptake inhibitor (SSRI) antidepressant use during pregnancy and reports of a rare heart and lung condition in newborn babies. http://www.fda.gov/Drugs/DrugSafety/ucm283375.htm. Published December 14, 2011. Accessed December 20, 2012.
2. U.S. Food and Drug Administration. Public health advisory: treatment challenges of depression in pregnancy and the possibility of persistent pulmonary hypertension in newborns. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsand
Providers/DrugSafetyInformationforHeathcareProfessionals/PublicHealthAdvisories/
ucm124348.htm. Published July 19, 2006. Accessed December 20, 2012.
3. Chambers CD, Hernandez-Diaz S, Van Marter LJ, et al. Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med. 2006;354(6):579-587.
4. Cohen LS, Altshuler LL, Harlow BL, et al. Relapse of major depression during pregnancy in women who maintain or discontinue antidepressant treatment. JAMA. 2006;295(5):499-507.
5. Muzik M, Hamilton S. Psychiatric illness during pregnancy. Current Psychiatry. 2012;11(2):23-32.
6. Hasser C, Brizendine L, Spielvogel A. SSRI use during pregnancy. Current Psychiatry. 2006;5(4):31-40.
7. Wisner KL, Sit DK, Hanusa BH, et al. Major depression and antidepressant treatment: impact on pregnancy and neonatal outcomes. Am J Psychiatry. 2009;166(5):557-566.
8. Friedman SH, Resnick PJ. Postpartum depression: an update. Women’s Health (Lond Engl). 2009;5(3):287-295.
9. Meltzer-Brody S. New insights into perinatal depression: pathogenesis and treatment during pregnancy and postpartum. Dialogues Clin Neurosci. 2011;13(1):89-100.
10. Friedman SH, Hall RCW. Treatment of mental illness in pregnancy and malpractice concerns. News Amer Acad Psychiatry Law. 2012;37(2):21-22.
11. Yonkers KA, Wisner KL, Stewart DE, et al. The management of depression during pregnancy: a report from the American Psychiatric Association and the American College of Obstetricians and Gynecologists. Gen Hosp Psychiatry. 2009;31(5):403-413.
12. Bar-Oz B, Einarson T, Einarson A, et al. Paroxetine and congenital malformations: meta-analysis and consideration of potential confounding factors. Clin Ther. 2007;29(5):918-926.
13. Wilson KL, Zelig CM, Harvey JP. Persistent pulmonary hypertension of the newborn is associated with mode of delivery and not with maternal use of selective serotonin reuptake inhibitors. Am J Perinatol. 2011;28(1):19-24.
14. Silvani P, Camporesi A. Drug-induced pulmonary hypertension in newborns: a review. Curr Vasc Pharmacol. 2007;5(2):129-133.
15. Occhiogrosso M, Omran SS, Altemus M. Persistent pulmonary hypertension of the newborn and selective serotonin reuptake inhibitors: lessons from clinical and translational studies. Am J Psychiatry. 2012;169(2):134-140.
16. Delaney C, Cornfield D. Risk factors for persistent pulmonary hypertension of the newborn. Pulm Circ. 2012;2(1):15-20.
17. Centers for Disease Control and Prevention. Key findings: updated national birth prevalence estimates for selected birth defects in the United States 2004-2006. http://www.cdc.gov/ncbddd/features/birthdefects-keyfindings.html. Published September 28, 2010. Accessed December 20, 2012.
18. Einarson A, Choi J, Einarson TR, et al. Incidence of major malformations in infants following antidepressant exposure in pregnancy: results of a large prospective cohort study. Can J Psychiatry. 2009;54(4):242-246.
19. Alwan S, Reefhuis J, Rasmussen SA, et al. National Birth Defects Prevention Study. Use of selective serotonin-reuptake inhibitors in pregnancy and the risk of birth defects. N Engl J Med. 2007;356(26):2684-2692.
20. Andrade SE, McPhillips H, Loren D. Antidepressant medication use and risk of persistent pulmonary hypertension of the newborn. Pharmacoepidemiol Drug Saf. 2009;18(3):246-252.
21. U.S. Food and Drug Administration. FDA advising of risk of birth defects with paxil. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2005/ucm108527.htm. Published December 8, 2005. Accessed December 20, 2012.
22. Koren G, Boucher N. Adverse effects in neonates exposed to SSRIs and SNRI in late gestation-Motherisk Update 2008. Can J Clin Pharmacol. 2009;16(1):e66-e67.
23. Pederson LH, Henriksen TB, Vestergaard M, et al. Selective serotonin reuptake inhibitors in pregnancy and congenital malformations: population based cohort study. BMJ. 2009;339:b3569.-doi:10.1136/bmj.b3569.
24. Kieler H, Artama M, Engeland A, et al. Selective serotonin reuptake inhibitors during pregnancy and risk of persistent pulmonary hypertension in the newborn: population based cohort study from the five Nordic countries. BMJ. 2012;344:d8012.-doi:10.1136/bmj.d801.
25. Wyeth v Levine, 555 US 555 (2009).
26. PLIVA, Inc. v Mensing, 588 F3d 603, 593 F3d 428 (2011).
27. Greenwood K. The mysteries of pregnancy: the role of law in solving the problem of unknown but knowable maternal–fetal medication risk. University of Cincinnati Law Review. 2011;79(1):267-322.
28. Lyam Kilker v SmithKline Beecham Corporation, Philadelphia Court of Common Pleas (2009).
29. Mulder E, Davis A, Gawley L, et al. Negative impact of non-evidence-based information received by women taking antidepressants during pregnancy from health care providers and others. J Obstet Gynaecol Can. 2012;34(1):66-71.
30. Henshaw SK. Unintended pregnancy in the United States. Fam Plann Perspect. 1998;30(1):24-29 46.
31. Altshuler L, Richards M, Yonkers K. Treating bipolar disorder during pregnancy. Current Psychiatry. 2003;2(7):14-26.
1. U.S. Food and Drug Administration. FDA drug safety communication: selective serotonin reuptake inhibitor (SSRI) antidepressant use during pregnancy and reports of a rare heart and lung condition in newborn babies. http://www.fda.gov/Drugs/DrugSafety/ucm283375.htm. Published December 14, 2011. Accessed December 20, 2012.
2. U.S. Food and Drug Administration. Public health advisory: treatment challenges of depression in pregnancy and the possibility of persistent pulmonary hypertension in newborns. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsand
Providers/DrugSafetyInformationforHeathcareProfessionals/PublicHealthAdvisories/
ucm124348.htm. Published July 19, 2006. Accessed December 20, 2012.
3. Chambers CD, Hernandez-Diaz S, Van Marter LJ, et al. Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med. 2006;354(6):579-587.
4. Cohen LS, Altshuler LL, Harlow BL, et al. Relapse of major depression during pregnancy in women who maintain or discontinue antidepressant treatment. JAMA. 2006;295(5):499-507.
5. Muzik M, Hamilton S. Psychiatric illness during pregnancy. Current Psychiatry. 2012;11(2):23-32.
6. Hasser C, Brizendine L, Spielvogel A. SSRI use during pregnancy. Current Psychiatry. 2006;5(4):31-40.
7. Wisner KL, Sit DK, Hanusa BH, et al. Major depression and antidepressant treatment: impact on pregnancy and neonatal outcomes. Am J Psychiatry. 2009;166(5):557-566.
8. Friedman SH, Resnick PJ. Postpartum depression: an update. Women’s Health (Lond Engl). 2009;5(3):287-295.
9. Meltzer-Brody S. New insights into perinatal depression: pathogenesis and treatment during pregnancy and postpartum. Dialogues Clin Neurosci. 2011;13(1):89-100.
10. Friedman SH, Hall RCW. Treatment of mental illness in pregnancy and malpractice concerns. News Amer Acad Psychiatry Law. 2012;37(2):21-22.
11. Yonkers KA, Wisner KL, Stewart DE, et al. The management of depression during pregnancy: a report from the American Psychiatric Association and the American College of Obstetricians and Gynecologists. Gen Hosp Psychiatry. 2009;31(5):403-413.
12. Bar-Oz B, Einarson T, Einarson A, et al. Paroxetine and congenital malformations: meta-analysis and consideration of potential confounding factors. Clin Ther. 2007;29(5):918-926.
13. Wilson KL, Zelig CM, Harvey JP. Persistent pulmonary hypertension of the newborn is associated with mode of delivery and not with maternal use of selective serotonin reuptake inhibitors. Am J Perinatol. 2011;28(1):19-24.
14. Silvani P, Camporesi A. Drug-induced pulmonary hypertension in newborns: a review. Curr Vasc Pharmacol. 2007;5(2):129-133.
15. Occhiogrosso M, Omran SS, Altemus M. Persistent pulmonary hypertension of the newborn and selective serotonin reuptake inhibitors: lessons from clinical and translational studies. Am J Psychiatry. 2012;169(2):134-140.
16. Delaney C, Cornfield D. Risk factors for persistent pulmonary hypertension of the newborn. Pulm Circ. 2012;2(1):15-20.
17. Centers for Disease Control and Prevention. Key findings: updated national birth prevalence estimates for selected birth defects in the United States 2004-2006. http://www.cdc.gov/ncbddd/features/birthdefects-keyfindings.html. Published September 28, 2010. Accessed December 20, 2012.
18. Einarson A, Choi J, Einarson TR, et al. Incidence of major malformations in infants following antidepressant exposure in pregnancy: results of a large prospective cohort study. Can J Psychiatry. 2009;54(4):242-246.
19. Alwan S, Reefhuis J, Rasmussen SA, et al. National Birth Defects Prevention Study. Use of selective serotonin-reuptake inhibitors in pregnancy and the risk of birth defects. N Engl J Med. 2007;356(26):2684-2692.
20. Andrade SE, McPhillips H, Loren D. Antidepressant medication use and risk of persistent pulmonary hypertension of the newborn. Pharmacoepidemiol Drug Saf. 2009;18(3):246-252.
21. U.S. Food and Drug Administration. FDA advising of risk of birth defects with paxil. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2005/ucm108527.htm. Published December 8, 2005. Accessed December 20, 2012.
22. Koren G, Boucher N. Adverse effects in neonates exposed to SSRIs and SNRI in late gestation-Motherisk Update 2008. Can J Clin Pharmacol. 2009;16(1):e66-e67.
23. Pederson LH, Henriksen TB, Vestergaard M, et al. Selective serotonin reuptake inhibitors in pregnancy and congenital malformations: population based cohort study. BMJ. 2009;339:b3569.-doi:10.1136/bmj.b3569.
24. Kieler H, Artama M, Engeland A, et al. Selective serotonin reuptake inhibitors during pregnancy and risk of persistent pulmonary hypertension in the newborn: population based cohort study from the five Nordic countries. BMJ. 2012;344:d8012.-doi:10.1136/bmj.d801.
25. Wyeth v Levine, 555 US 555 (2009).
26. PLIVA, Inc. v Mensing, 588 F3d 603, 593 F3d 428 (2011).
27. Greenwood K. The mysteries of pregnancy: the role of law in solving the problem of unknown but knowable maternal–fetal medication risk. University of Cincinnati Law Review. 2011;79(1):267-322.
28. Lyam Kilker v SmithKline Beecham Corporation, Philadelphia Court of Common Pleas (2009).
29. Mulder E, Davis A, Gawley L, et al. Negative impact of non-evidence-based information received by women taking antidepressants during pregnancy from health care providers and others. J Obstet Gynaecol Can. 2012;34(1):66-71.
30. Henshaw SK. Unintended pregnancy in the United States. Fam Plann Perspect. 1998;30(1):24-29 46.
31. Altshuler L, Richards M, Yonkers K. Treating bipolar disorder during pregnancy. Current Psychiatry. 2003;2(7):14-26.
Pharmacotherapy for comorbid depression and alcohol dependence
Discuss this article at www.facebook.com/CurrentPsychiatry
With a lifetime prevalence of 30%, alcohol use disorders (AUDs)—which in DSM-IV-TR include alcohol abuse and alcohol dependence—are among the most common psychiatric disorders.1 Depressive disorders, including major depressive disorder (MDD) and dysthymia, frequently co-occur with AUDs.2-4 This pattern of comorbidity adversely affects the prognosis, course, and treatment of both MDD and AUDs.5 High severity in 1 of these disorders is associated with high severity in the other.2,6 Alcohol dependence appears to prolong the course of depression7,8 and increases the risk of suicidal symptoms and behaviors.9,10 Patients with depression and AUDs are at increased risk of relapse to heavy drinking.7,11
Whether the high comorbidity rate of depressive disorders and AUDs is a result of 1 disorder causing the other (ie, AUDs leading to depression or vice versa) or can be attributed to shared etiology is unknown. Clinicians need to consider this question when treating patients with this pattern of comorbidity because distinguishing primary depression from secondary depression influences treatment decisions.12
There is a great need for pharmacologic interventions that can concurrently treat both depression and AUDs. This article reviews the evidence for current treatments for dually diagnosed patients and highlights novel agents that are worthy of further study for this complex patient population.
Current treatment options
Pharmacotherapy for MDD and alcohol dependence is common when these conditions occur alone. FDA-approved medications for treating depression include monoamine oxidase inhibitors, tricyclic antidepressants (TCAs), tetracyclic antidepressants, selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors.
SSRIs are the most widely used class of antidepressants. They gained FDA approval based on studies conducted in non-comorbid patients because patients with comorbid conditions usually are excluded from research studies.13 Few trials have evaluated patients with depression and AUDs; TCAs and SSRIs are best studied in these patients.
Serotonergic antidepressants
SSRIs are first-line medications for MDD because of their low abuse potential, favorable side effect profile, and relative safety in overdose.
Table 1
Serotonergic antidepressants for patients with AUDs and depression
| Study | Sample | Results |
|---|---|---|
| Cornelius et al, 199714 | Outpatients with severe major depression and AD 1. Fluoxetine (20 to 40 mg/d; n = 25) 2. Placebo (n = 26) | Greater reductions in depressive symptoms and drinking in patients treated with fluoxetine compared with placebo |
| Roy, 199815 | Inpatients with current major depression and AD who were abstinent for ≥2 weeks 1. Sertraline (100 mg/d; n = 18) 2. Placebo (n = 18) | Greater reductions in depressive symptoms in patients treated with sertraline compared with placebo. Drinking outcomes were not emphasized because 35 of 36 patients reported continuous abstinence throughout the trial |
| Kranzler et al, 199516 | Outpatients with AD. Fourteen percent had current major depression. All received weekly individual or group CBT focused on relapse prevention and skills building 1. Fluoxetine (mean daily dose: 48 mg; n = 51) 2. Placebo (n = 50) | Significant decrease in alcohol consumption for both groups during the trial. No significant differences in alcohol consumption between groups. Among those with current depression, patients treated with fluoxetine experienced greater reduction in depressive symptoms vs placebo |
| Moak et al, 200317 | Currently depressed, actively drinking, alcohol-dependent outpatients. All received individual CBT 1. Sertraline (mean daily dose: 186 mg; n = 38) 2. Placebo (n = 44) | Sertraline had an advantage over placebo in reducing depressive symptoms in women but not in men. Sertraline reduced drinks per drinking day but not other drinking-related outcomes |
| Cornelius et al, 200918 | Adolescents (age 15 to 20) with AA or AD and MDD. All received intensive manual-based therapy (CBT for MDD and AUD, MET for AUD) almost weekly 1. Fluoxetine (20 mg/d; n = 24) 2. Placebo (n = 26) | All improved during the course of trial. No significant differences between fluoxetine and placebo groups in depression- or drinking-related outcomes |
| Roy-Byrne et al, 200019 | Actively drinking alcohol-dependent outpatients with history of ≥1 depressive episode. All received weekly group therapy for alcoholism 1. Nefazodone (mean daily dose: 460 mg; n = 32) 2. Placebo (n = 32) | Greater reduction in depressive symptoms but not in drinking-related outcomes in patients treated with nefazodone |
| Hernandez-Avila et al, 200420 | Outpatients with AD and current major depression. All received supportive psychotherapy for 10 weeks 1. Nefazodone (mean daily dose: 413 mg; n = 21) 2. Placebo (n = 20) | Depressive and anxiety symptoms declined significantly over time, but no statistically significant differences in depressive or anxiety symptoms between nefazodone and placebo. Patients treated with nefazodone had significantly greater reductions in heavy drinking days and in total drinks compared with placebo-treated patients |
| AA: alcohol abuse; AD: alcohol dependence; AUDs: alcohol use disorders; CBT: cognitive-behavioral therapy; MDD: major depressive disorder; MET: motivational enhancement therapy | ||
In a study of adolescents, Cornelius et al18 failed to find any differences between fluoxetine and placebo in any depression or drinking-related outcomes. This study compared the efficacy of fluoxetine, 20 mg/d, with placebo in 50 adolescents with MDD and AUDs who also received intensive, manual-based cognitive-behavioral therapy and motivational enhancement therapy. All patients improved during the trial, but there were no significant differences between fluoxetine- and placebo-treated adolescents.
Other serotonergic medications. Two studies have evaluated nefazodone, a serotonin (5-HT2) antagonist, in dually diagnosed patients. In a 12-week trial, Roy-Byrne et al19 evaluated the efficacy of nefazodone (mean daily dose: 460 mg) vs placebo in 64 actively drinking alcohol-dependent patients who had ≥1 prior episode of depression; all participated in a weekly psychoeducation group on alcoholism. Nefazodone was associated with significantly greater reduction in depressive symptoms but no reductions in drinking compared with placebo. However, a 10-week study of nefazodone20 (mean daily dose: 413 mg) vs placebo in 41 alcohol-dependent patients with current major depression found that those who received nefazodone significantly reduced heavy drinking days compared with the placebo group. There were no significant differences in depressive symptoms between groups.
Conflicting evidence on TCAs
Although several studies suggest TCAs may help reduce depressive symptoms in patients with AUDs, results on their ability to reduce drinking are conflicting (Table 2).24-26 In 1 study, 6 months of desipramine (mean daily dose: 200 mg) reduced drinking in 28 alcohol-dependent individuals with secondary depression24; in another, 12 weeks of imipramine plus weekly relapse prevention psychotherapy did not affect drinking-related outcomes in 69 actively drinking alcoholic outpatients with current depressive disorders.25
Table 2
Limited evidence supports TCAs for comorbid depression and AUDs
| Study | Sample | Results |
|---|---|---|
| Mason et al, 199624 | Outpatients with AD and secondary depression. Part of larger study including non-depressed patients with AD (N = 71) 1. Desipramine (mean daily dose 200 mg; n = 15) 2. Placebo (n = 13) | Greater reduction in depressive symptoms and drinking in desipramine-treated patients compared with placebo-treated patients |
| McGrath et al, 199625 | Outpatients with AD or AA and major depression, dysthymia, or depressive disorder NOS 1. Imipramine (mean daily dose 260 mg; n = 36) 2. Placebo (n = 33) | Greater reduction in depressive symptoms for imipramine-treated patients compared with placebo-treated patients. Drinking-related outcomes were not directly affected by medication except improvements in mood led to reduced alcohol use |
| Altintoprak et al, 200826 | Inpatients with AD and MDD 1. Mirtazapine (30 mg/d; n = 24) 2. Amitriptyline (100 mg/d; n = 20) | Drinking-related outcomes were not emphasized because all patients were required to abstain from drinking during the study. Both treatments reduced depressive symptoms; there were no significant differences between groups |
| AA: alcohol abuse; AD: alcohol dependence; AUDs: alcohol use disorders; MDD: major depressive disorder; NOS: not otherwise specified; TCAs: tricyclic antidepressants | ||
Altintoprak et al26 compared the efficacy of the antidepressant mirtazapine, 30 mg/d, with the TCA amitriptyline, 100 mg/d, in 44 patients with comorbid alcohol dependence and MDD. All patients were required to abstain from drinking alcohol during the study. Both medications resulted in steady reductions in depressive symptoms and alcohol cravings; however, researchers found no significant differences between the 2 treatment groups.
Analyses of combined studies
Pettinati27 conducted a qualitative review of antidepressants for patients with depression and alcohol dependence that included 8 controlled clinical trials (2 on TCAs and 6 on serotonergic medications) conducted between 1994 and 2004. In this review, both TCAs and serotonergic medications were similarly effective in reducing depressive symptoms but not consistently effective in reducing drinking.
27 this review suggested that antidepressants can reduce depressive symptoms but not drinking. The authors also found evidence that the more the antidepressant reduced depressive symptoms, the more it reduced alcohol use. Studies published after these reviews have not substantially altered these findings.
Alcohol abuse medications
Four medications are FDA-approved for treating alcohol dependence:
- disulfiram
- naltrexone (in 2 formulations: oral and long-acting injectable)
- acamprosate.
Table 3
Can medications that target alcohol use also improve depression?
| Study | Sample | Results |
|---|---|---|
| Petrakis et al, 200729 | Outpatients with AD and an axis I disorder, including depression (secondary analysis of Petrakis et al, 200531) 1. Naltrexone (50 mg/d; n = 34) 2. Disulfiram (250 mg/d; n = 43) 3. Naltrexone + disulfiram (n = 28) 4. Placebo (n = 34) | Generally, medication was more effective than no medication. No advantage of 1 medication over the other. There was no relationship between depression diagnosis and medication condition, which suggests that for patients with depression, there was no advantage to medication |
| Pettinati et al, 201030 | Outpatients with AD and MDD 1. Sertraline (200 mg/d; n = 40) 2. Naltrexone (100 mg/d; n = 49) 3. Sertraline + naltrexone (n = 42) 4. Placebo (n = 39) | Greater proportion of patients in combined medication group abstained from alcohol and refrained from heavy alcohol use during the trial compared with those in sertraline-only or naltrexone-only groups. No significant differences among groups on depression-related outcomes |
| AD: alcohol dependence; MDD: major depressive disorder | ||
Nevertheless, these studies suggest that medications for treating depression or AUDs have, at best, only a modest effect in patients with both disorders.
Novel agents
Several novel medications have been evaluated as possible treatments for comorbid depression and AUDs because they target the underlying neurobiology of both disorders:
- agents that target the neurotransmitter glutamate, including the N-methyl-d-aspartate glutamate receptor antagonists memantine and ketamine
- dopaminergic agents such as quetiapine
- corticotropin-releasing factor (CRF) receptor 1 (CRF1) antagonists.
In a case study, a 55-year-old man with treatment-resistant major depression and co-occurring alcohol and benzodiazepine dependence who received a single dose of IV ketamine, 0.5 mg/kg over 50 minutes, experienced “significant improvements” in depressive symptoms that lasted throughout the 7-day follow-up.33 This study did not report on ketamine’s effects on his alcohol use.
The atypical antipsychotic quetiapine acts as a serotonin (5-HT1A and 5-HT2) and dopamine (D1 and D2) antagonist, and reports suggest it reduces alcohol craving and affective symptoms in patients with AUDs.34,35 In a 16-week, open-label study, quetiapine decreased alcohol consumption, alcohol craving, and intensity of some psychiatric symptoms in 28 alcohol-dependent patients with bipolar disorder, schizoaffective disorder, or borderline personality disorder.36
See the Box for a description of the role CRF1 antagonists may play in treating patients with concurrent MDD and AUDs. See Table 4 for studies of memantine and quetiapine in treating depression with AUDs.
Corticotropin-releasing factor (CRF) has a well-established role in stress and has been implicated for treating anxiety and depressive disorders. Evidence also suggests that CRF receptor 1 (CRF1) may be a treatment target for alcohol use disorders (AUDs). Acute alcohol withdrawal and prolonged alcohol use are associated with elevated levels of extrahypothalamic CRF and correlated anxiety. CRF antagonists can reduce the anxiogenic effects of alcohol withdrawal and reduce some symptoms of alcohol dependence, including excessive alcohol self-administration and stress-induced relapse to alcohol use in rats with alcohol dependence, but not in those without dependence. Therefore, CRF1 receptor antagonists may be especially helpful for individuals who use alcohol to reduce negative emotional states such as anxiety or dysphoria, including those with concurrent major depressive disorder and AUDs.
Bibliography
Funk CK, Zorrilla EP, Lee MJ, et al. Corticotropin-releasing factor 1 antagonists selectively reduce ethanol self-administration in ethanol-dependent rats. Biol Psychiatry. 2007;61(1):78-86.
Gehlert DR, Cippitelli A, Thorsell A, et al. 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-8-(1-ethylpropyl)-2,6-dimethyl-imidazo[1,2-b]pyridazine: a novel brain-penetrant, orally available corticotropin-releasing factor receptor 1 antagonist with efficacy in animal models of alcoholism. J Neurosci. 2007;27(10):2718-2726.
Gilpin NW, Richardson HN, Koob GF. Effects of CRF1-receptor and opioid-receptor antagonists on dependence-induced increases in alcohol drinking by alcohol-preferring (P) rats. Alcohol Clin Exp Res. 2008;32(9):1535-1542.
Other agents may play a role in treating depression with AUDs
| Study | Sample | Results |
|---|---|---|
| Muhonen et al, 200832 | Outpatients with MDD and AD 1. Memantine (20 mg/d; n = 40) | Both treatments reduced depressive and anxiety symptoms. No significant differences between groups. Study did not examine alcohol-related outcomes |
| Martinotti et al, 200833 | Outpatients with comorbid AD and either bipolar disorder, schizoaffective disorder or borderline personality disorder. Open-label study 1. Quetiapine (300 to 800 mg/d; n = 28) | Quetiapine was associated with reduced alcohol consumption, alcohol craving, and intensity of psychiatric symptoms |
| AD: alcohol dependence; AUDs: alcohol use disorders; MDD: major depressive disorder | ||
Interpreting the evidence
These findings underscore the importance of thorough evaluations. SSRIs are a first-line treatment for depression and as such probably should be the first choice for patients with comorbid AUDs. Drinking should be monitored closely and abstinence encouraged. Using medications that target AUDs is safe and modestly effective in patients with comorbid depression. Evidence suggests that treating both disorders simultaneously is more effective than treating either alone. Medications should be prescribed as part of a comprehensive treatment plan that also includes psychotherapy.
Related Resources
- Pettinati H. Antidepressant treatment of co-occurring depression and alcohol dependence. Biol Psychiatry. 2004;56(10):785-792.
- Nunes EV, Levin FR. Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA. 2004;291(15):1887-1896.
- Acamprosate • Campral
- Amitriptyline • Elavil
- Desipramine • Norpramin
- Disulfiram • Antabuse
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Ketamine • Ketalar
- Memantine • Namenda
- Mirtazapine • Remeron
- Naltrexone • Revia, Vivitrol
- Nefazodone • Serzone
- Quetiapine • Seroquel
- Sertraline • Zoloft
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Petrakis receives research or grant support from the National Institute on Alcohol Abuse and Alcoholism, the National Institute on Drug Abuse, the U.S. Department of Defense, and the U.S. Department of Veterans Affairs.
This work has been supported by a grant from the Veterans Affairs New England Mental Illness Research, Education, and Clinical Center and by the National Institute of Mental Health (T32MH062994-07).
Acknowledgements
The authors thank Elizabeth Guidone for her helpful comments and Diana Limoncelli for her assistance in manuscript preparation.
1. Hasin DS, Stinson FS, Ogburn E, et al. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2007;64(7):830-842.
2. Grant BF, Harford TC. Comorbidity between DSM-IV alcohol use disorders and major depression: results of a national survey. Drug Alcohol Depend. 1995;39(3):197-206.
3. Kessler RC, Crum RM, Warner LA, et al. Lifetime co-occurrence of DSM-III-R alcohol abuse and dependence with other psychiatric disorders in the National Comorbidity Survey. Arch Gen Psychiatry. 1997;54(4):313-321.
4. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA. 1990;264(19):2511-2518.
5. Burns L, Teesson M, O’Neill K. The impact of comorbid anxiety and depression on alcohol treatment outcomes. Addiction. 2005;100(6):787-796.
6. Gilman SE, Abraham HD. A longitudinal study of the order of onset of alcohol dependence and major depression. Drug Alcohol Depend. 2001;63(3):277-286.
7. Hasin DS, Tsai WY, Endicott J, et al. Five-year course of major depression: effects of comorbid alcoholism. J Affect Disord. 1996;41(1):63-70.
8. Mueller TI, Lavori PW, Keller MB, et al. Prognostic effect of the variable course of alcoholism on the 10-year course of depression. Am J Psychiatry. 1994;151(5):701-706.
9. Cornelius JR, Salloum IM, Mezzich J, et al. Disproportionate suicidality in patients with comorbid major depression and alcoholism. Am J Psychiatry. 1995;152(3):358-364.
10. Sher L, Oquendo MA, Galfalvy HC, et al. The relationship of aggression to suicidal behavior in depressed patients with a history of alcoholism. Addict Behav. 2005;30(6):1144-1153.
11. Greenfield SF, Weiss RD, Muenz LR, et al. The effect of depression on return to drinking: a prospective study. Arch Gen Psychiatry. 1998;55(3):259-265.
12. Brady KT, Verduin ML, Tolliver BK. Treatment of patients comorbid for addiction and other psychiatric disorders. Curr Psychiatry Rep. 2007;9(5):374-380.
13. Oslin DW. Treatment of late-life depression complicated by alcohol dependence. Am J Geriatr Psychiatry. 2005;13(6):491-500.
14. Cornelius JR, Salloum IM, Ehler JG, et al. Fluoxetine in depressed alcoholics. A double-blind, placebo-controlled trial. Arch Gen Psychiatry. 1997;54(8):700-705.
15. Roy A. Placebo-controlled study of sertraline in depressed recently abstinent alcoholics. Biol Psychiatry. 1998;44(7):633-637.
16. Kranzler HR, Burleson JA, Korner P, et al. Placebo-controlled trial of fluoxetine as an adjunct to relapse prevention in alcoholics. Am J Psychiatry. 1995;152(3):391-397.
17. Moak DH, Anton RF, Latham PK, et al. Sertraline and cognitive behavioral therapy for depressed alcoholics: results of a placebo-controlled trial. J Clin Psychopharmacol. 2003;23(6):553-562.
18. Cornelius JR, Bukstein OG, Wood DS, et al. Double-blind placebo-controlled trial of fluoxetine in adolescents with comorbid major depression and an alcohol use disorder. Addict Behav. 2009;34(10):905-909.
19. Roy-Byrne PP, Pages KP, Russo JE, et al. Nefazodone treatment of major depression in alcohol-dependent patients: a double-blind, placebo-controlled trial. J Clin Psychopharmacol. 2000;20(2):129-136.
20. Hernandez-Avila CA, Modesto-Lowe V, Feinn R, et al. Nefazodone treatment of comorbid alcohol dependence and major depression. Alcohol Clin Exp Res. 2004;28(3):433-440.
21. Kranzler HR, Burleson JA, Brown J, et al. Fluoxetine treatment seems to reduce the beneficial effects of cognitive-behavioral therapy in type B alcoholics. Alcohol Clin Exp Res. 1996;20(9):1534-1541.
22. Naranjo CA, Bremner KE, Lanctôt KL. Effects of citalopram and a brief psycho-social intervention on alcohol intake dependence and problems. Addiction. 1995;90(1):87-99.
23. Pettinati HM, Volpicelli JR, Kranzler HR, et al. Sertraline treatment for alcohol dependence: interactive effects of medication and alcoholic subtype. Alcohol Clin Exp Res. 2000;24(7):1041-1049.
24. Mason BJ, Kocsis JH, Ritvo EC, et al. A double-blind, placebo-controlled trial of desipramine for primary alcohol dependence stratified on the presence or absence of major depression. JAMA. 1996;275(10):761-767.
25. McGrath PJ, Nunes EV, Stewart JW, et al. Imipramine treatment of alcoholics with primary depression: a placebo-controlled clinical trial. Arch Gen Psychiatry. 1996;53(3):232-240.
26. Altintoprak AE, Zorlu N, Coskunol H, et al. Effectiveness and tolerability of mirtazapine and amitriptyline in alcoholic patients with co-morbid depressive disorder: a randomized, double-blind study. Hum Psychopharmacol. 2008;23(4):313-319.
27. Pettinati HM. Antidepressant treatment of co-occurring depression and alcohol dependence. Biol Psychiatry. 2004;56(10):785-792.
28. Nunes EV, Levin FR. Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA. 2004;291(15):1887-1896.
29. Petrakis I, Ralevski E, Nich C, et al. Naltrexone and disulfiram in patients with alcohol dependence and current depression. J Clin Psychopharmacol. 2007;27(2):160-165.
30. Pettinati HM, Oslin DW, Kampman KM, et al. A double-blind, placebo-controlled trial combining sertraline and naltrexone for treating co-occurring depression and alcohol dependence. Am J Psychiatry. 2010;167(6):668-675.
31. Petrakis IL, Poling J, Levinson C, et al. Naltrexone and disulfiram in patients with alcohol dependence and comorbid psychiatric disorders. Biol Psychiatry. 2005;57(10):1128-1137.
32. Muhonen LH, Lönnqvist J, Juva K, et al. Double-blind, randomized comparison of memantine and escitalopram for the treatment of major depressive disorder comorbid with alcohol dependence. J Clin Psychiatry. 2008;69(3):392-399.
33. Liebrenz M, Borgeat A, Leisinger R, et al. Intravenous ketamine therapy in a patient with a treatment-resistant major depression. Swiss Med Wkly. 2007;137(15-16):234-236.
34. Kampman KM, Pettinati HM, Lynch KG, et al. A double-blind, placebo-controlled pilot trial of quetiapine for the treatment of Type A and Type B alcoholism. J Clin Psychopharmacol. 2007;27(4):344-351.
35. Croissant B, Klein O, Gehrlein L, et al. Quetiapine in relapse prevention in alcoholics suffering from craving and affective symptoms: a case series. Eur Psychiatry. 2006;21(8):570-573.
36. Martinotti G, Andreoli S, Di Nicola M, et al. Quetiapine decreases alcohol consumption, craving, and psychiatric symptoms in dually diagnosed alcoholics. Hum Psychopharmacol. 2008;23(5):417-424.
Discuss this article at www.facebook.com/CurrentPsychiatry
With a lifetime prevalence of 30%, alcohol use disorders (AUDs)—which in DSM-IV-TR include alcohol abuse and alcohol dependence—are among the most common psychiatric disorders.1 Depressive disorders, including major depressive disorder (MDD) and dysthymia, frequently co-occur with AUDs.2-4 This pattern of comorbidity adversely affects the prognosis, course, and treatment of both MDD and AUDs.5 High severity in 1 of these disorders is associated with high severity in the other.2,6 Alcohol dependence appears to prolong the course of depression7,8 and increases the risk of suicidal symptoms and behaviors.9,10 Patients with depression and AUDs are at increased risk of relapse to heavy drinking.7,11
Whether the high comorbidity rate of depressive disorders and AUDs is a result of 1 disorder causing the other (ie, AUDs leading to depression or vice versa) or can be attributed to shared etiology is unknown. Clinicians need to consider this question when treating patients with this pattern of comorbidity because distinguishing primary depression from secondary depression influences treatment decisions.12
There is a great need for pharmacologic interventions that can concurrently treat both depression and AUDs. This article reviews the evidence for current treatments for dually diagnosed patients and highlights novel agents that are worthy of further study for this complex patient population.
Current treatment options
Pharmacotherapy for MDD and alcohol dependence is common when these conditions occur alone. FDA-approved medications for treating depression include monoamine oxidase inhibitors, tricyclic antidepressants (TCAs), tetracyclic antidepressants, selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors.
SSRIs are the most widely used class of antidepressants. They gained FDA approval based on studies conducted in non-comorbid patients because patients with comorbid conditions usually are excluded from research studies.13 Few trials have evaluated patients with depression and AUDs; TCAs and SSRIs are best studied in these patients.
Serotonergic antidepressants
SSRIs are first-line medications for MDD because of their low abuse potential, favorable side effect profile, and relative safety in overdose.
Table 1
Serotonergic antidepressants for patients with AUDs and depression
| Study | Sample | Results |
|---|---|---|
| Cornelius et al, 199714 | Outpatients with severe major depression and AD 1. Fluoxetine (20 to 40 mg/d; n = 25) 2. Placebo (n = 26) | Greater reductions in depressive symptoms and drinking in patients treated with fluoxetine compared with placebo |
| Roy, 199815 | Inpatients with current major depression and AD who were abstinent for ≥2 weeks 1. Sertraline (100 mg/d; n = 18) 2. Placebo (n = 18) | Greater reductions in depressive symptoms in patients treated with sertraline compared with placebo. Drinking outcomes were not emphasized because 35 of 36 patients reported continuous abstinence throughout the trial |
| Kranzler et al, 199516 | Outpatients with AD. Fourteen percent had current major depression. All received weekly individual or group CBT focused on relapse prevention and skills building 1. Fluoxetine (mean daily dose: 48 mg; n = 51) 2. Placebo (n = 50) | Significant decrease in alcohol consumption for both groups during the trial. No significant differences in alcohol consumption between groups. Among those with current depression, patients treated with fluoxetine experienced greater reduction in depressive symptoms vs placebo |
| Moak et al, 200317 | Currently depressed, actively drinking, alcohol-dependent outpatients. All received individual CBT 1. Sertraline (mean daily dose: 186 mg; n = 38) 2. Placebo (n = 44) | Sertraline had an advantage over placebo in reducing depressive symptoms in women but not in men. Sertraline reduced drinks per drinking day but not other drinking-related outcomes |
| Cornelius et al, 200918 | Adolescents (age 15 to 20) with AA or AD and MDD. All received intensive manual-based therapy (CBT for MDD and AUD, MET for AUD) almost weekly 1. Fluoxetine (20 mg/d; n = 24) 2. Placebo (n = 26) | All improved during the course of trial. No significant differences between fluoxetine and placebo groups in depression- or drinking-related outcomes |
| Roy-Byrne et al, 200019 | Actively drinking alcohol-dependent outpatients with history of ≥1 depressive episode. All received weekly group therapy for alcoholism 1. Nefazodone (mean daily dose: 460 mg; n = 32) 2. Placebo (n = 32) | Greater reduction in depressive symptoms but not in drinking-related outcomes in patients treated with nefazodone |
| Hernandez-Avila et al, 200420 | Outpatients with AD and current major depression. All received supportive psychotherapy for 10 weeks 1. Nefazodone (mean daily dose: 413 mg; n = 21) 2. Placebo (n = 20) | Depressive and anxiety symptoms declined significantly over time, but no statistically significant differences in depressive or anxiety symptoms between nefazodone and placebo. Patients treated with nefazodone had significantly greater reductions in heavy drinking days and in total drinks compared with placebo-treated patients |
| AA: alcohol abuse; AD: alcohol dependence; AUDs: alcohol use disorders; CBT: cognitive-behavioral therapy; MDD: major depressive disorder; MET: motivational enhancement therapy | ||
In a study of adolescents, Cornelius et al18 failed to find any differences between fluoxetine and placebo in any depression or drinking-related outcomes. This study compared the efficacy of fluoxetine, 20 mg/d, with placebo in 50 adolescents with MDD and AUDs who also received intensive, manual-based cognitive-behavioral therapy and motivational enhancement therapy. All patients improved during the trial, but there were no significant differences between fluoxetine- and placebo-treated adolescents.
Other serotonergic medications. Two studies have evaluated nefazodone, a serotonin (5-HT2) antagonist, in dually diagnosed patients. In a 12-week trial, Roy-Byrne et al19 evaluated the efficacy of nefazodone (mean daily dose: 460 mg) vs placebo in 64 actively drinking alcohol-dependent patients who had ≥1 prior episode of depression; all participated in a weekly psychoeducation group on alcoholism. Nefazodone was associated with significantly greater reduction in depressive symptoms but no reductions in drinking compared with placebo. However, a 10-week study of nefazodone20 (mean daily dose: 413 mg) vs placebo in 41 alcohol-dependent patients with current major depression found that those who received nefazodone significantly reduced heavy drinking days compared with the placebo group. There were no significant differences in depressive symptoms between groups.
Conflicting evidence on TCAs
Although several studies suggest TCAs may help reduce depressive symptoms in patients with AUDs, results on their ability to reduce drinking are conflicting (Table 2).24-26 In 1 study, 6 months of desipramine (mean daily dose: 200 mg) reduced drinking in 28 alcohol-dependent individuals with secondary depression24; in another, 12 weeks of imipramine plus weekly relapse prevention psychotherapy did not affect drinking-related outcomes in 69 actively drinking alcoholic outpatients with current depressive disorders.25
Table 2
Limited evidence supports TCAs for comorbid depression and AUDs
| Study | Sample | Results |
|---|---|---|
| Mason et al, 199624 | Outpatients with AD and secondary depression. Part of larger study including non-depressed patients with AD (N = 71) 1. Desipramine (mean daily dose 200 mg; n = 15) 2. Placebo (n = 13) | Greater reduction in depressive symptoms and drinking in desipramine-treated patients compared with placebo-treated patients |
| McGrath et al, 199625 | Outpatients with AD or AA and major depression, dysthymia, or depressive disorder NOS 1. Imipramine (mean daily dose 260 mg; n = 36) 2. Placebo (n = 33) | Greater reduction in depressive symptoms for imipramine-treated patients compared with placebo-treated patients. Drinking-related outcomes were not directly affected by medication except improvements in mood led to reduced alcohol use |
| Altintoprak et al, 200826 | Inpatients with AD and MDD 1. Mirtazapine (30 mg/d; n = 24) 2. Amitriptyline (100 mg/d; n = 20) | Drinking-related outcomes were not emphasized because all patients were required to abstain from drinking during the study. Both treatments reduced depressive symptoms; there were no significant differences between groups |
| AA: alcohol abuse; AD: alcohol dependence; AUDs: alcohol use disorders; MDD: major depressive disorder; NOS: not otherwise specified; TCAs: tricyclic antidepressants | ||
Altintoprak et al26 compared the efficacy of the antidepressant mirtazapine, 30 mg/d, with the TCA amitriptyline, 100 mg/d, in 44 patients with comorbid alcohol dependence and MDD. All patients were required to abstain from drinking alcohol during the study. Both medications resulted in steady reductions in depressive symptoms and alcohol cravings; however, researchers found no significant differences between the 2 treatment groups.
Analyses of combined studies
Pettinati27 conducted a qualitative review of antidepressants for patients with depression and alcohol dependence that included 8 controlled clinical trials (2 on TCAs and 6 on serotonergic medications) conducted between 1994 and 2004. In this review, both TCAs and serotonergic medications were similarly effective in reducing depressive symptoms but not consistently effective in reducing drinking.
27 this review suggested that antidepressants can reduce depressive symptoms but not drinking. The authors also found evidence that the more the antidepressant reduced depressive symptoms, the more it reduced alcohol use. Studies published after these reviews have not substantially altered these findings.
Alcohol abuse medications
Four medications are FDA-approved for treating alcohol dependence:
- disulfiram
- naltrexone (in 2 formulations: oral and long-acting injectable)
- acamprosate.
Table 3
Can medications that target alcohol use also improve depression?
| Study | Sample | Results |
|---|---|---|
| Petrakis et al, 200729 | Outpatients with AD and an axis I disorder, including depression (secondary analysis of Petrakis et al, 200531) 1. Naltrexone (50 mg/d; n = 34) 2. Disulfiram (250 mg/d; n = 43) 3. Naltrexone + disulfiram (n = 28) 4. Placebo (n = 34) | Generally, medication was more effective than no medication. No advantage of 1 medication over the other. There was no relationship between depression diagnosis and medication condition, which suggests that for patients with depression, there was no advantage to medication |
| Pettinati et al, 201030 | Outpatients with AD and MDD 1. Sertraline (200 mg/d; n = 40) 2. Naltrexone (100 mg/d; n = 49) 3. Sertraline + naltrexone (n = 42) 4. Placebo (n = 39) | Greater proportion of patients in combined medication group abstained from alcohol and refrained from heavy alcohol use during the trial compared with those in sertraline-only or naltrexone-only groups. No significant differences among groups on depression-related outcomes |
| AD: alcohol dependence; MDD: major depressive disorder | ||
Nevertheless, these studies suggest that medications for treating depression or AUDs have, at best, only a modest effect in patients with both disorders.
Novel agents
Several novel medications have been evaluated as possible treatments for comorbid depression and AUDs because they target the underlying neurobiology of both disorders:
- agents that target the neurotransmitter glutamate, including the N-methyl-d-aspartate glutamate receptor antagonists memantine and ketamine
- dopaminergic agents such as quetiapine
- corticotropin-releasing factor (CRF) receptor 1 (CRF1) antagonists.
In a case study, a 55-year-old man with treatment-resistant major depression and co-occurring alcohol and benzodiazepine dependence who received a single dose of IV ketamine, 0.5 mg/kg over 50 minutes, experienced “significant improvements” in depressive symptoms that lasted throughout the 7-day follow-up.33 This study did not report on ketamine’s effects on his alcohol use.
The atypical antipsychotic quetiapine acts as a serotonin (5-HT1A and 5-HT2) and dopamine (D1 and D2) antagonist, and reports suggest it reduces alcohol craving and affective symptoms in patients with AUDs.34,35 In a 16-week, open-label study, quetiapine decreased alcohol consumption, alcohol craving, and intensity of some psychiatric symptoms in 28 alcohol-dependent patients with bipolar disorder, schizoaffective disorder, or borderline personality disorder.36
See the Box for a description of the role CRF1 antagonists may play in treating patients with concurrent MDD and AUDs. See Table 4 for studies of memantine and quetiapine in treating depression with AUDs.
Corticotropin-releasing factor (CRF) has a well-established role in stress and has been implicated for treating anxiety and depressive disorders. Evidence also suggests that CRF receptor 1 (CRF1) may be a treatment target for alcohol use disorders (AUDs). Acute alcohol withdrawal and prolonged alcohol use are associated with elevated levels of extrahypothalamic CRF and correlated anxiety. CRF antagonists can reduce the anxiogenic effects of alcohol withdrawal and reduce some symptoms of alcohol dependence, including excessive alcohol self-administration and stress-induced relapse to alcohol use in rats with alcohol dependence, but not in those without dependence. Therefore, CRF1 receptor antagonists may be especially helpful for individuals who use alcohol to reduce negative emotional states such as anxiety or dysphoria, including those with concurrent major depressive disorder and AUDs.
Bibliography
Funk CK, Zorrilla EP, Lee MJ, et al. Corticotropin-releasing factor 1 antagonists selectively reduce ethanol self-administration in ethanol-dependent rats. Biol Psychiatry. 2007;61(1):78-86.
Gehlert DR, Cippitelli A, Thorsell A, et al. 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-8-(1-ethylpropyl)-2,6-dimethyl-imidazo[1,2-b]pyridazine: a novel brain-penetrant, orally available corticotropin-releasing factor receptor 1 antagonist with efficacy in animal models of alcoholism. J Neurosci. 2007;27(10):2718-2726.
Gilpin NW, Richardson HN, Koob GF. Effects of CRF1-receptor and opioid-receptor antagonists on dependence-induced increases in alcohol drinking by alcohol-preferring (P) rats. Alcohol Clin Exp Res. 2008;32(9):1535-1542.
Other agents may play a role in treating depression with AUDs
| Study | Sample | Results |
|---|---|---|
| Muhonen et al, 200832 | Outpatients with MDD and AD 1. Memantine (20 mg/d; n = 40) | Both treatments reduced depressive and anxiety symptoms. No significant differences between groups. Study did not examine alcohol-related outcomes |
| Martinotti et al, 200833 | Outpatients with comorbid AD and either bipolar disorder, schizoaffective disorder or borderline personality disorder. Open-label study 1. Quetiapine (300 to 800 mg/d; n = 28) | Quetiapine was associated with reduced alcohol consumption, alcohol craving, and intensity of psychiatric symptoms |
| AD: alcohol dependence; AUDs: alcohol use disorders; MDD: major depressive disorder | ||
Interpreting the evidence
These findings underscore the importance of thorough evaluations. SSRIs are a first-line treatment for depression and as such probably should be the first choice for patients with comorbid AUDs. Drinking should be monitored closely and abstinence encouraged. Using medications that target AUDs is safe and modestly effective in patients with comorbid depression. Evidence suggests that treating both disorders simultaneously is more effective than treating either alone. Medications should be prescribed as part of a comprehensive treatment plan that also includes psychotherapy.
Related Resources
- Pettinati H. Antidepressant treatment of co-occurring depression and alcohol dependence. Biol Psychiatry. 2004;56(10):785-792.
- Nunes EV, Levin FR. Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA. 2004;291(15):1887-1896.
- Acamprosate • Campral
- Amitriptyline • Elavil
- Desipramine • Norpramin
- Disulfiram • Antabuse
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Ketamine • Ketalar
- Memantine • Namenda
- Mirtazapine • Remeron
- Naltrexone • Revia, Vivitrol
- Nefazodone • Serzone
- Quetiapine • Seroquel
- Sertraline • Zoloft
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Petrakis receives research or grant support from the National Institute on Alcohol Abuse and Alcoholism, the National Institute on Drug Abuse, the U.S. Department of Defense, and the U.S. Department of Veterans Affairs.
This work has been supported by a grant from the Veterans Affairs New England Mental Illness Research, Education, and Clinical Center and by the National Institute of Mental Health (T32MH062994-07).
Acknowledgements
The authors thank Elizabeth Guidone for her helpful comments and Diana Limoncelli for her assistance in manuscript preparation.
Discuss this article at www.facebook.com/CurrentPsychiatry
With a lifetime prevalence of 30%, alcohol use disorders (AUDs)—which in DSM-IV-TR include alcohol abuse and alcohol dependence—are among the most common psychiatric disorders.1 Depressive disorders, including major depressive disorder (MDD) and dysthymia, frequently co-occur with AUDs.2-4 This pattern of comorbidity adversely affects the prognosis, course, and treatment of both MDD and AUDs.5 High severity in 1 of these disorders is associated with high severity in the other.2,6 Alcohol dependence appears to prolong the course of depression7,8 and increases the risk of suicidal symptoms and behaviors.9,10 Patients with depression and AUDs are at increased risk of relapse to heavy drinking.7,11
Whether the high comorbidity rate of depressive disorders and AUDs is a result of 1 disorder causing the other (ie, AUDs leading to depression or vice versa) or can be attributed to shared etiology is unknown. Clinicians need to consider this question when treating patients with this pattern of comorbidity because distinguishing primary depression from secondary depression influences treatment decisions.12
There is a great need for pharmacologic interventions that can concurrently treat both depression and AUDs. This article reviews the evidence for current treatments for dually diagnosed patients and highlights novel agents that are worthy of further study for this complex patient population.
Current treatment options
Pharmacotherapy for MDD and alcohol dependence is common when these conditions occur alone. FDA-approved medications for treating depression include monoamine oxidase inhibitors, tricyclic antidepressants (TCAs), tetracyclic antidepressants, selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors.
SSRIs are the most widely used class of antidepressants. They gained FDA approval based on studies conducted in non-comorbid patients because patients with comorbid conditions usually are excluded from research studies.13 Few trials have evaluated patients with depression and AUDs; TCAs and SSRIs are best studied in these patients.
Serotonergic antidepressants
SSRIs are first-line medications for MDD because of their low abuse potential, favorable side effect profile, and relative safety in overdose.
Table 1
Serotonergic antidepressants for patients with AUDs and depression
| Study | Sample | Results |
|---|---|---|
| Cornelius et al, 199714 | Outpatients with severe major depression and AD 1. Fluoxetine (20 to 40 mg/d; n = 25) 2. Placebo (n = 26) | Greater reductions in depressive symptoms and drinking in patients treated with fluoxetine compared with placebo |
| Roy, 199815 | Inpatients with current major depression and AD who were abstinent for ≥2 weeks 1. Sertraline (100 mg/d; n = 18) 2. Placebo (n = 18) | Greater reductions in depressive symptoms in patients treated with sertraline compared with placebo. Drinking outcomes were not emphasized because 35 of 36 patients reported continuous abstinence throughout the trial |
| Kranzler et al, 199516 | Outpatients with AD. Fourteen percent had current major depression. All received weekly individual or group CBT focused on relapse prevention and skills building 1. Fluoxetine (mean daily dose: 48 mg; n = 51) 2. Placebo (n = 50) | Significant decrease in alcohol consumption for both groups during the trial. No significant differences in alcohol consumption between groups. Among those with current depression, patients treated with fluoxetine experienced greater reduction in depressive symptoms vs placebo |
| Moak et al, 200317 | Currently depressed, actively drinking, alcohol-dependent outpatients. All received individual CBT 1. Sertraline (mean daily dose: 186 mg; n = 38) 2. Placebo (n = 44) | Sertraline had an advantage over placebo in reducing depressive symptoms in women but not in men. Sertraline reduced drinks per drinking day but not other drinking-related outcomes |
| Cornelius et al, 200918 | Adolescents (age 15 to 20) with AA or AD and MDD. All received intensive manual-based therapy (CBT for MDD and AUD, MET for AUD) almost weekly 1. Fluoxetine (20 mg/d; n = 24) 2. Placebo (n = 26) | All improved during the course of trial. No significant differences between fluoxetine and placebo groups in depression- or drinking-related outcomes |
| Roy-Byrne et al, 200019 | Actively drinking alcohol-dependent outpatients with history of ≥1 depressive episode. All received weekly group therapy for alcoholism 1. Nefazodone (mean daily dose: 460 mg; n = 32) 2. Placebo (n = 32) | Greater reduction in depressive symptoms but not in drinking-related outcomes in patients treated with nefazodone |
| Hernandez-Avila et al, 200420 | Outpatients with AD and current major depression. All received supportive psychotherapy for 10 weeks 1. Nefazodone (mean daily dose: 413 mg; n = 21) 2. Placebo (n = 20) | Depressive and anxiety symptoms declined significantly over time, but no statistically significant differences in depressive or anxiety symptoms between nefazodone and placebo. Patients treated with nefazodone had significantly greater reductions in heavy drinking days and in total drinks compared with placebo-treated patients |
| AA: alcohol abuse; AD: alcohol dependence; AUDs: alcohol use disorders; CBT: cognitive-behavioral therapy; MDD: major depressive disorder; MET: motivational enhancement therapy | ||
In a study of adolescents, Cornelius et al18 failed to find any differences between fluoxetine and placebo in any depression or drinking-related outcomes. This study compared the efficacy of fluoxetine, 20 mg/d, with placebo in 50 adolescents with MDD and AUDs who also received intensive, manual-based cognitive-behavioral therapy and motivational enhancement therapy. All patients improved during the trial, but there were no significant differences between fluoxetine- and placebo-treated adolescents.
Other serotonergic medications. Two studies have evaluated nefazodone, a serotonin (5-HT2) antagonist, in dually diagnosed patients. In a 12-week trial, Roy-Byrne et al19 evaluated the efficacy of nefazodone (mean daily dose: 460 mg) vs placebo in 64 actively drinking alcohol-dependent patients who had ≥1 prior episode of depression; all participated in a weekly psychoeducation group on alcoholism. Nefazodone was associated with significantly greater reduction in depressive symptoms but no reductions in drinking compared with placebo. However, a 10-week study of nefazodone20 (mean daily dose: 413 mg) vs placebo in 41 alcohol-dependent patients with current major depression found that those who received nefazodone significantly reduced heavy drinking days compared with the placebo group. There were no significant differences in depressive symptoms between groups.
Conflicting evidence on TCAs
Although several studies suggest TCAs may help reduce depressive symptoms in patients with AUDs, results on their ability to reduce drinking are conflicting (Table 2).24-26 In 1 study, 6 months of desipramine (mean daily dose: 200 mg) reduced drinking in 28 alcohol-dependent individuals with secondary depression24; in another, 12 weeks of imipramine plus weekly relapse prevention psychotherapy did not affect drinking-related outcomes in 69 actively drinking alcoholic outpatients with current depressive disorders.25
Table 2
Limited evidence supports TCAs for comorbid depression and AUDs
| Study | Sample | Results |
|---|---|---|
| Mason et al, 199624 | Outpatients with AD and secondary depression. Part of larger study including non-depressed patients with AD (N = 71) 1. Desipramine (mean daily dose 200 mg; n = 15) 2. Placebo (n = 13) | Greater reduction in depressive symptoms and drinking in desipramine-treated patients compared with placebo-treated patients |
| McGrath et al, 199625 | Outpatients with AD or AA and major depression, dysthymia, or depressive disorder NOS 1. Imipramine (mean daily dose 260 mg; n = 36) 2. Placebo (n = 33) | Greater reduction in depressive symptoms for imipramine-treated patients compared with placebo-treated patients. Drinking-related outcomes were not directly affected by medication except improvements in mood led to reduced alcohol use |
| Altintoprak et al, 200826 | Inpatients with AD and MDD 1. Mirtazapine (30 mg/d; n = 24) 2. Amitriptyline (100 mg/d; n = 20) | Drinking-related outcomes were not emphasized because all patients were required to abstain from drinking during the study. Both treatments reduced depressive symptoms; there were no significant differences between groups |
| AA: alcohol abuse; AD: alcohol dependence; AUDs: alcohol use disorders; MDD: major depressive disorder; NOS: not otherwise specified; TCAs: tricyclic antidepressants | ||
Altintoprak et al26 compared the efficacy of the antidepressant mirtazapine, 30 mg/d, with the TCA amitriptyline, 100 mg/d, in 44 patients with comorbid alcohol dependence and MDD. All patients were required to abstain from drinking alcohol during the study. Both medications resulted in steady reductions in depressive symptoms and alcohol cravings; however, researchers found no significant differences between the 2 treatment groups.
Analyses of combined studies
Pettinati27 conducted a qualitative review of antidepressants for patients with depression and alcohol dependence that included 8 controlled clinical trials (2 on TCAs and 6 on serotonergic medications) conducted between 1994 and 2004. In this review, both TCAs and serotonergic medications were similarly effective in reducing depressive symptoms but not consistently effective in reducing drinking.
27 this review suggested that antidepressants can reduce depressive symptoms but not drinking. The authors also found evidence that the more the antidepressant reduced depressive symptoms, the more it reduced alcohol use. Studies published after these reviews have not substantially altered these findings.
Alcohol abuse medications
Four medications are FDA-approved for treating alcohol dependence:
- disulfiram
- naltrexone (in 2 formulations: oral and long-acting injectable)
- acamprosate.
Table 3
Can medications that target alcohol use also improve depression?
| Study | Sample | Results |
|---|---|---|
| Petrakis et al, 200729 | Outpatients with AD and an axis I disorder, including depression (secondary analysis of Petrakis et al, 200531) 1. Naltrexone (50 mg/d; n = 34) 2. Disulfiram (250 mg/d; n = 43) 3. Naltrexone + disulfiram (n = 28) 4. Placebo (n = 34) | Generally, medication was more effective than no medication. No advantage of 1 medication over the other. There was no relationship between depression diagnosis and medication condition, which suggests that for patients with depression, there was no advantage to medication |
| Pettinati et al, 201030 | Outpatients with AD and MDD 1. Sertraline (200 mg/d; n = 40) 2. Naltrexone (100 mg/d; n = 49) 3. Sertraline + naltrexone (n = 42) 4. Placebo (n = 39) | Greater proportion of patients in combined medication group abstained from alcohol and refrained from heavy alcohol use during the trial compared with those in sertraline-only or naltrexone-only groups. No significant differences among groups on depression-related outcomes |
| AD: alcohol dependence; MDD: major depressive disorder | ||
Nevertheless, these studies suggest that medications for treating depression or AUDs have, at best, only a modest effect in patients with both disorders.
Novel agents
Several novel medications have been evaluated as possible treatments for comorbid depression and AUDs because they target the underlying neurobiology of both disorders:
- agents that target the neurotransmitter glutamate, including the N-methyl-d-aspartate glutamate receptor antagonists memantine and ketamine
- dopaminergic agents such as quetiapine
- corticotropin-releasing factor (CRF) receptor 1 (CRF1) antagonists.
In a case study, a 55-year-old man with treatment-resistant major depression and co-occurring alcohol and benzodiazepine dependence who received a single dose of IV ketamine, 0.5 mg/kg over 50 minutes, experienced “significant improvements” in depressive symptoms that lasted throughout the 7-day follow-up.33 This study did not report on ketamine’s effects on his alcohol use.
The atypical antipsychotic quetiapine acts as a serotonin (5-HT1A and 5-HT2) and dopamine (D1 and D2) antagonist, and reports suggest it reduces alcohol craving and affective symptoms in patients with AUDs.34,35 In a 16-week, open-label study, quetiapine decreased alcohol consumption, alcohol craving, and intensity of some psychiatric symptoms in 28 alcohol-dependent patients with bipolar disorder, schizoaffective disorder, or borderline personality disorder.36
See the Box for a description of the role CRF1 antagonists may play in treating patients with concurrent MDD and AUDs. See Table 4 for studies of memantine and quetiapine in treating depression with AUDs.
Corticotropin-releasing factor (CRF) has a well-established role in stress and has been implicated for treating anxiety and depressive disorders. Evidence also suggests that CRF receptor 1 (CRF1) may be a treatment target for alcohol use disorders (AUDs). Acute alcohol withdrawal and prolonged alcohol use are associated with elevated levels of extrahypothalamic CRF and correlated anxiety. CRF antagonists can reduce the anxiogenic effects of alcohol withdrawal and reduce some symptoms of alcohol dependence, including excessive alcohol self-administration and stress-induced relapse to alcohol use in rats with alcohol dependence, but not in those without dependence. Therefore, CRF1 receptor antagonists may be especially helpful for individuals who use alcohol to reduce negative emotional states such as anxiety or dysphoria, including those with concurrent major depressive disorder and AUDs.
Bibliography
Funk CK, Zorrilla EP, Lee MJ, et al. Corticotropin-releasing factor 1 antagonists selectively reduce ethanol self-administration in ethanol-dependent rats. Biol Psychiatry. 2007;61(1):78-86.
Gehlert DR, Cippitelli A, Thorsell A, et al. 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-8-(1-ethylpropyl)-2,6-dimethyl-imidazo[1,2-b]pyridazine: a novel brain-penetrant, orally available corticotropin-releasing factor receptor 1 antagonist with efficacy in animal models of alcoholism. J Neurosci. 2007;27(10):2718-2726.
Gilpin NW, Richardson HN, Koob GF. Effects of CRF1-receptor and opioid-receptor antagonists on dependence-induced increases in alcohol drinking by alcohol-preferring (P) rats. Alcohol Clin Exp Res. 2008;32(9):1535-1542.
Other agents may play a role in treating depression with AUDs
| Study | Sample | Results |
|---|---|---|
| Muhonen et al, 200832 | Outpatients with MDD and AD 1. Memantine (20 mg/d; n = 40) | Both treatments reduced depressive and anxiety symptoms. No significant differences between groups. Study did not examine alcohol-related outcomes |
| Martinotti et al, 200833 | Outpatients with comorbid AD and either bipolar disorder, schizoaffective disorder or borderline personality disorder. Open-label study 1. Quetiapine (300 to 800 mg/d; n = 28) | Quetiapine was associated with reduced alcohol consumption, alcohol craving, and intensity of psychiatric symptoms |
| AD: alcohol dependence; AUDs: alcohol use disorders; MDD: major depressive disorder | ||
Interpreting the evidence
These findings underscore the importance of thorough evaluations. SSRIs are a first-line treatment for depression and as such probably should be the first choice for patients with comorbid AUDs. Drinking should be monitored closely and abstinence encouraged. Using medications that target AUDs is safe and modestly effective in patients with comorbid depression. Evidence suggests that treating both disorders simultaneously is more effective than treating either alone. Medications should be prescribed as part of a comprehensive treatment plan that also includes psychotherapy.
Related Resources
- Pettinati H. Antidepressant treatment of co-occurring depression and alcohol dependence. Biol Psychiatry. 2004;56(10):785-792.
- Nunes EV, Levin FR. Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA. 2004;291(15):1887-1896.
- Acamprosate • Campral
- Amitriptyline • Elavil
- Desipramine • Norpramin
- Disulfiram • Antabuse
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Ketamine • Ketalar
- Memantine • Namenda
- Mirtazapine • Remeron
- Naltrexone • Revia, Vivitrol
- Nefazodone • Serzone
- Quetiapine • Seroquel
- Sertraline • Zoloft
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Petrakis receives research or grant support from the National Institute on Alcohol Abuse and Alcoholism, the National Institute on Drug Abuse, the U.S. Department of Defense, and the U.S. Department of Veterans Affairs.
This work has been supported by a grant from the Veterans Affairs New England Mental Illness Research, Education, and Clinical Center and by the National Institute of Mental Health (T32MH062994-07).
Acknowledgements
The authors thank Elizabeth Guidone for her helpful comments and Diana Limoncelli for her assistance in manuscript preparation.
1. Hasin DS, Stinson FS, Ogburn E, et al. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2007;64(7):830-842.
2. Grant BF, Harford TC. Comorbidity between DSM-IV alcohol use disorders and major depression: results of a national survey. Drug Alcohol Depend. 1995;39(3):197-206.
3. Kessler RC, Crum RM, Warner LA, et al. Lifetime co-occurrence of DSM-III-R alcohol abuse and dependence with other psychiatric disorders in the National Comorbidity Survey. Arch Gen Psychiatry. 1997;54(4):313-321.
4. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA. 1990;264(19):2511-2518.
5. Burns L, Teesson M, O’Neill K. The impact of comorbid anxiety and depression on alcohol treatment outcomes. Addiction. 2005;100(6):787-796.
6. Gilman SE, Abraham HD. A longitudinal study of the order of onset of alcohol dependence and major depression. Drug Alcohol Depend. 2001;63(3):277-286.
7. Hasin DS, Tsai WY, Endicott J, et al. Five-year course of major depression: effects of comorbid alcoholism. J Affect Disord. 1996;41(1):63-70.
8. Mueller TI, Lavori PW, Keller MB, et al. Prognostic effect of the variable course of alcoholism on the 10-year course of depression. Am J Psychiatry. 1994;151(5):701-706.
9. Cornelius JR, Salloum IM, Mezzich J, et al. Disproportionate suicidality in patients with comorbid major depression and alcoholism. Am J Psychiatry. 1995;152(3):358-364.
10. Sher L, Oquendo MA, Galfalvy HC, et al. The relationship of aggression to suicidal behavior in depressed patients with a history of alcoholism. Addict Behav. 2005;30(6):1144-1153.
11. Greenfield SF, Weiss RD, Muenz LR, et al. The effect of depression on return to drinking: a prospective study. Arch Gen Psychiatry. 1998;55(3):259-265.
12. Brady KT, Verduin ML, Tolliver BK. Treatment of patients comorbid for addiction and other psychiatric disorders. Curr Psychiatry Rep. 2007;9(5):374-380.
13. Oslin DW. Treatment of late-life depression complicated by alcohol dependence. Am J Geriatr Psychiatry. 2005;13(6):491-500.
14. Cornelius JR, Salloum IM, Ehler JG, et al. Fluoxetine in depressed alcoholics. A double-blind, placebo-controlled trial. Arch Gen Psychiatry. 1997;54(8):700-705.
15. Roy A. Placebo-controlled study of sertraline in depressed recently abstinent alcoholics. Biol Psychiatry. 1998;44(7):633-637.
16. Kranzler HR, Burleson JA, Korner P, et al. Placebo-controlled trial of fluoxetine as an adjunct to relapse prevention in alcoholics. Am J Psychiatry. 1995;152(3):391-397.
17. Moak DH, Anton RF, Latham PK, et al. Sertraline and cognitive behavioral therapy for depressed alcoholics: results of a placebo-controlled trial. J Clin Psychopharmacol. 2003;23(6):553-562.
18. Cornelius JR, Bukstein OG, Wood DS, et al. Double-blind placebo-controlled trial of fluoxetine in adolescents with comorbid major depression and an alcohol use disorder. Addict Behav. 2009;34(10):905-909.
19. Roy-Byrne PP, Pages KP, Russo JE, et al. Nefazodone treatment of major depression in alcohol-dependent patients: a double-blind, placebo-controlled trial. J Clin Psychopharmacol. 2000;20(2):129-136.
20. Hernandez-Avila CA, Modesto-Lowe V, Feinn R, et al. Nefazodone treatment of comorbid alcohol dependence and major depression. Alcohol Clin Exp Res. 2004;28(3):433-440.
21. Kranzler HR, Burleson JA, Brown J, et al. Fluoxetine treatment seems to reduce the beneficial effects of cognitive-behavioral therapy in type B alcoholics. Alcohol Clin Exp Res. 1996;20(9):1534-1541.
22. Naranjo CA, Bremner KE, Lanctôt KL. Effects of citalopram and a brief psycho-social intervention on alcohol intake dependence and problems. Addiction. 1995;90(1):87-99.
23. Pettinati HM, Volpicelli JR, Kranzler HR, et al. Sertraline treatment for alcohol dependence: interactive effects of medication and alcoholic subtype. Alcohol Clin Exp Res. 2000;24(7):1041-1049.
24. Mason BJ, Kocsis JH, Ritvo EC, et al. A double-blind, placebo-controlled trial of desipramine for primary alcohol dependence stratified on the presence or absence of major depression. JAMA. 1996;275(10):761-767.
25. McGrath PJ, Nunes EV, Stewart JW, et al. Imipramine treatment of alcoholics with primary depression: a placebo-controlled clinical trial. Arch Gen Psychiatry. 1996;53(3):232-240.
26. Altintoprak AE, Zorlu N, Coskunol H, et al. Effectiveness and tolerability of mirtazapine and amitriptyline in alcoholic patients with co-morbid depressive disorder: a randomized, double-blind study. Hum Psychopharmacol. 2008;23(4):313-319.
27. Pettinati HM. Antidepressant treatment of co-occurring depression and alcohol dependence. Biol Psychiatry. 2004;56(10):785-792.
28. Nunes EV, Levin FR. Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA. 2004;291(15):1887-1896.
29. Petrakis I, Ralevski E, Nich C, et al. Naltrexone and disulfiram in patients with alcohol dependence and current depression. J Clin Psychopharmacol. 2007;27(2):160-165.
30. Pettinati HM, Oslin DW, Kampman KM, et al. A double-blind, placebo-controlled trial combining sertraline and naltrexone for treating co-occurring depression and alcohol dependence. Am J Psychiatry. 2010;167(6):668-675.
31. Petrakis IL, Poling J, Levinson C, et al. Naltrexone and disulfiram in patients with alcohol dependence and comorbid psychiatric disorders. Biol Psychiatry. 2005;57(10):1128-1137.
32. Muhonen LH, Lönnqvist J, Juva K, et al. Double-blind, randomized comparison of memantine and escitalopram for the treatment of major depressive disorder comorbid with alcohol dependence. J Clin Psychiatry. 2008;69(3):392-399.
33. Liebrenz M, Borgeat A, Leisinger R, et al. Intravenous ketamine therapy in a patient with a treatment-resistant major depression. Swiss Med Wkly. 2007;137(15-16):234-236.
34. Kampman KM, Pettinati HM, Lynch KG, et al. A double-blind, placebo-controlled pilot trial of quetiapine for the treatment of Type A and Type B alcoholism. J Clin Psychopharmacol. 2007;27(4):344-351.
35. Croissant B, Klein O, Gehrlein L, et al. Quetiapine in relapse prevention in alcoholics suffering from craving and affective symptoms: a case series. Eur Psychiatry. 2006;21(8):570-573.
36. Martinotti G, Andreoli S, Di Nicola M, et al. Quetiapine decreases alcohol consumption, craving, and psychiatric symptoms in dually diagnosed alcoholics. Hum Psychopharmacol. 2008;23(5):417-424.
1. Hasin DS, Stinson FS, Ogburn E, et al. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2007;64(7):830-842.
2. Grant BF, Harford TC. Comorbidity between DSM-IV alcohol use disorders and major depression: results of a national survey. Drug Alcohol Depend. 1995;39(3):197-206.
3. Kessler RC, Crum RM, Warner LA, et al. Lifetime co-occurrence of DSM-III-R alcohol abuse and dependence with other psychiatric disorders in the National Comorbidity Survey. Arch Gen Psychiatry. 1997;54(4):313-321.
4. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA. 1990;264(19):2511-2518.
5. Burns L, Teesson M, O’Neill K. The impact of comorbid anxiety and depression on alcohol treatment outcomes. Addiction. 2005;100(6):787-796.
6. Gilman SE, Abraham HD. A longitudinal study of the order of onset of alcohol dependence and major depression. Drug Alcohol Depend. 2001;63(3):277-286.
7. Hasin DS, Tsai WY, Endicott J, et al. Five-year course of major depression: effects of comorbid alcoholism. J Affect Disord. 1996;41(1):63-70.
8. Mueller TI, Lavori PW, Keller MB, et al. Prognostic effect of the variable course of alcoholism on the 10-year course of depression. Am J Psychiatry. 1994;151(5):701-706.
9. Cornelius JR, Salloum IM, Mezzich J, et al. Disproportionate suicidality in patients with comorbid major depression and alcoholism. Am J Psychiatry. 1995;152(3):358-364.
10. Sher L, Oquendo MA, Galfalvy HC, et al. The relationship of aggression to suicidal behavior in depressed patients with a history of alcoholism. Addict Behav. 2005;30(6):1144-1153.
11. Greenfield SF, Weiss RD, Muenz LR, et al. The effect of depression on return to drinking: a prospective study. Arch Gen Psychiatry. 1998;55(3):259-265.
12. Brady KT, Verduin ML, Tolliver BK. Treatment of patients comorbid for addiction and other psychiatric disorders. Curr Psychiatry Rep. 2007;9(5):374-380.
13. Oslin DW. Treatment of late-life depression complicated by alcohol dependence. Am J Geriatr Psychiatry. 2005;13(6):491-500.
14. Cornelius JR, Salloum IM, Ehler JG, et al. Fluoxetine in depressed alcoholics. A double-blind, placebo-controlled trial. Arch Gen Psychiatry. 1997;54(8):700-705.
15. Roy A. Placebo-controlled study of sertraline in depressed recently abstinent alcoholics. Biol Psychiatry. 1998;44(7):633-637.
16. Kranzler HR, Burleson JA, Korner P, et al. Placebo-controlled trial of fluoxetine as an adjunct to relapse prevention in alcoholics. Am J Psychiatry. 1995;152(3):391-397.
17. Moak DH, Anton RF, Latham PK, et al. Sertraline and cognitive behavioral therapy for depressed alcoholics: results of a placebo-controlled trial. J Clin Psychopharmacol. 2003;23(6):553-562.
18. Cornelius JR, Bukstein OG, Wood DS, et al. Double-blind placebo-controlled trial of fluoxetine in adolescents with comorbid major depression and an alcohol use disorder. Addict Behav. 2009;34(10):905-909.
19. Roy-Byrne PP, Pages KP, Russo JE, et al. Nefazodone treatment of major depression in alcohol-dependent patients: a double-blind, placebo-controlled trial. J Clin Psychopharmacol. 2000;20(2):129-136.
20. Hernandez-Avila CA, Modesto-Lowe V, Feinn R, et al. Nefazodone treatment of comorbid alcohol dependence and major depression. Alcohol Clin Exp Res. 2004;28(3):433-440.
21. Kranzler HR, Burleson JA, Brown J, et al. Fluoxetine treatment seems to reduce the beneficial effects of cognitive-behavioral therapy in type B alcoholics. Alcohol Clin Exp Res. 1996;20(9):1534-1541.
22. Naranjo CA, Bremner KE, Lanctôt KL. Effects of citalopram and a brief psycho-social intervention on alcohol intake dependence and problems. Addiction. 1995;90(1):87-99.
23. Pettinati HM, Volpicelli JR, Kranzler HR, et al. Sertraline treatment for alcohol dependence: interactive effects of medication and alcoholic subtype. Alcohol Clin Exp Res. 2000;24(7):1041-1049.
24. Mason BJ, Kocsis JH, Ritvo EC, et al. A double-blind, placebo-controlled trial of desipramine for primary alcohol dependence stratified on the presence or absence of major depression. JAMA. 1996;275(10):761-767.
25. McGrath PJ, Nunes EV, Stewart JW, et al. Imipramine treatment of alcoholics with primary depression: a placebo-controlled clinical trial. Arch Gen Psychiatry. 1996;53(3):232-240.
26. Altintoprak AE, Zorlu N, Coskunol H, et al. Effectiveness and tolerability of mirtazapine and amitriptyline in alcoholic patients with co-morbid depressive disorder: a randomized, double-blind study. Hum Psychopharmacol. 2008;23(4):313-319.
27. Pettinati HM. Antidepressant treatment of co-occurring depression and alcohol dependence. Biol Psychiatry. 2004;56(10):785-792.
28. Nunes EV, Levin FR. Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA. 2004;291(15):1887-1896.
29. Petrakis I, Ralevski E, Nich C, et al. Naltrexone and disulfiram in patients with alcohol dependence and current depression. J Clin Psychopharmacol. 2007;27(2):160-165.
30. Pettinati HM, Oslin DW, Kampman KM, et al. A double-blind, placebo-controlled trial combining sertraline and naltrexone for treating co-occurring depression and alcohol dependence. Am J Psychiatry. 2010;167(6):668-675.
31. Petrakis IL, Poling J, Levinson C, et al. Naltrexone and disulfiram in patients with alcohol dependence and comorbid psychiatric disorders. Biol Psychiatry. 2005;57(10):1128-1137.
32. Muhonen LH, Lönnqvist J, Juva K, et al. Double-blind, randomized comparison of memantine and escitalopram for the treatment of major depressive disorder comorbid with alcohol dependence. J Clin Psychiatry. 2008;69(3):392-399.
33. Liebrenz M, Borgeat A, Leisinger R, et al. Intravenous ketamine therapy in a patient with a treatment-resistant major depression. Swiss Med Wkly. 2007;137(15-16):234-236.
34. Kampman KM, Pettinati HM, Lynch KG, et al. A double-blind, placebo-controlled pilot trial of quetiapine for the treatment of Type A and Type B alcoholism. J Clin Psychopharmacol. 2007;27(4):344-351.
35. Croissant B, Klein O, Gehrlein L, et al. Quetiapine in relapse prevention in alcoholics suffering from craving and affective symptoms: a case series. Eur Psychiatry. 2006;21(8):570-573.
36. Martinotti G, Andreoli S, Di Nicola M, et al. Quetiapine decreases alcohol consumption, craving, and psychiatric symptoms in dually diagnosed alcoholics. Hum Psychopharmacol. 2008;23(5):417-424.
Vitamin deficiencies and mental health: How are they linked?
Discuss this article at www.facebook.com/CurrentPsychiatry
Patients today often are overfed but undernourished. A growing body of literature links dietary choices to brain health and the risk of psychiatric illness. Vitamin deficiencies can affect psychiatric patients in several ways:
- deficiencies may play a causative role in mental illness and exacerbate symptoms
- psychiatric symptoms can result in poor nutrition
- vitamin insufficiency—defined as subclinical deficiency—may compromise patient recovery.
Additionally, genetic differences may compromise vitamin and essential nutrient pathways.
Vitamins are dietary components other than carbohydrates, fats, minerals, and proteins that are necessary for life. B vitamins are required for proper functioning of the methylation cycle, monoamine production, DNA synthesis, and maintenance of phospholipids such as myelin (Figure). Fat-soluble vitamins A, D, and E play important roles in genetic transcription, antioxidant recycling, and inflammatory regulation in the brain.
Figure: The methylation cycle
Vitamins B2, B6, B9, and B12 directly impact the functioning of the methylation cycle. Deficiencies pertain to brain function, as neurotransmitters, myelin, and active glutathione are dependent on one-carbon metabolism
Illustration: Mala Nimalasuriya with permission from DrewRamseyMD.com
To help clinicians recognize and treat vitamin deficiencies among psychiatric patients, this article reviews the role of the 6 essential water-soluble vitamins (B1, B2, B6, B9, B12, and C; Table 1,1) and 3 fat-soluble vitamins (A, D, and E; Table 2,1) in brain metabolism and psychiatric pathology. Because numerous sources address using supplements to treat vitamin deficiencies, this article emphasizes food sources, which for many patients are adequate to sustain nutrient status.
Table 1
Water-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| B1 (thiamine): Glycolysis, tricarboxylic acid cycle | ||||
| Rare; 7% in heart failure patients | 5% total, 12% of older women | Wernicke-Korsakoff syndrome, memory impairment, confusion, lack of coordination, paralysis | Older adults, malabsorptive conditions, heavy alcohol use. Those with diabetes are at risk because of increased clearance | Pork, fish, beans, lentils, nuts, rice, and wheat germ. Raw fish, tea, and betel nuts impair absorption |
| B2 (riboflavin): FMN, FAD cofactors in glycolysis and oxidative pathways. B6, folate, and glutathione synthesis | ||||
| 10% to 27% of older adults | <3%; 95% of adolescent girls (measured by EGRAC) | Fatigue, cracked lips, sore throat, bloodshot eyes | Older adults, low intake of animal and dairy products, heavy alcohol use | Dairy, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes |
| B6 (pyridoxal): Methylation cycle | ||||
| 11% to 24% (<5 ng/mL); 38% of heart failure patients | 14% total, 26% of adults | Dermatitis, glossitis, convulsions, migraine, chronic pain, depression | Older adults, women who use oral contraceptives, alcoholism. 33% to 49% of women age >51 have inadequate intake | Bananas, beans, potatoes, navy beans, salmon, steak, and whole grains |
| B9 (folate): Methylation cycle | ||||
| 0.5% total; up to 50% of depressed patients | 16% of adults, 19% of adolescent girls | Loss of appetite, weight loss, weakness, heart palpitations, behavioral disorders | Depression, pregnancy and lactation, alcoholism, dialysis, liver disease. Deficiency during pregnancy is linked to neural tube defects | Leafy green vegetables, fruits, dried beans, and peas |
| B12 (cobalamin): Methylation cycle (cofactor methionine synthase) | ||||
| 10% to 15% of older adults | <3% to 9% | Depression, irritability, anemia, fatigue, shortness of breath, high blood pressure | Vegetarian or vegan diet, achlorhydria, older adults. Deficiency more often due to poor absorption than low consumption | Meat, seafood, eggs, and dairy |
| C (ascorbic acid): Antioxidant | ||||
| 7.1% | 31% | Scurvy, fatigue, anemia, joint pain, petechia. Symptoms develop after 1 to 3 months of no dietary intake | Smokers, infants fed boiled or evaporated milk, limited dietary variation, patients with malabsorption, chronic illnesses | Citrus fruits, tomatoes and tomato juice, and potatoes |
| EGRAC: erythrocyte glutathione reductase activation coefficient; FAD: flavin adenine dinucleotide; FMN: flavin mononucleotide Source: Reference 1 | ||||
Table 2
Fat-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| A (retinol): Transcription regulation, vision | ||||
| <5% of U.S. population | 44% | Blindness, decreased immunity, corneal and retinal damage | Pregnant women, individuals with strict dietary restrictions, heavy alcohol use, chronic diarrhea, fat malabsorptive conditions | Beef liver, dairy products. Convertible beta-carotene sources: sweet potatoes, carrots, spinach, butternut squash, greens, broccoli, cantaloupe |
| D (cholecalciferol): Hormone, transcriptional regulation | ||||
| ≥50%, 90% of adults age >50 | 69% | Rickets, osteoporosis, muscle twitching | Breast-fed infants, older adults, limited sun exposure, pigmented skin, fat malabsorption, obesity. Older adults have an impaired ability to make vitamin D from the sun. SPF 15 reduces production by 99% | Fatty fish and fish liver oils, sun-dried mushrooms |
| E (tocopherols and tocotrienols): Antioxidant, PUFA protectant, gene regulation | ||||
| Rare | 93% | Anemia, neuropathy, myopathy, abnormal eye movements, weakness, retinal damage | Malabsorptive conditions, HIV, depression | Sunflower, wheat germ, and safflower oils; meats; fish; dairy; green vegetables |
| HIV: human immunodeficiency virus; PUFA: polyunsaturated fatty acids; SPF: sun protection factor Source: Reference 1 | ||||
Water-soluble vitamins
Vitamin B1 (thiamine) is essential for glucose metabolism. Pregnancy, lactation, and fever increase the need for thiamine, and tea, coffee, and shellfish can impair its absorption. Although rare, severe B1 deficiency can lead to beriberi, Wernicke’s encephalopathy (confusion, ataxia, nystagmus), and Korsakoff’s psychosis (confabulation, lack of insight, retrograde and anterograde amnesia, and apathy). Confusion and disorientation stem from the brain’s inability to oxidize glucose for energy because B1 is a critical cofactor in glycolysis and the tricarboxylic acid cycle. Deficiency leads to an increase in reactive oxygen species, proinflammatory cytokines, and blood-brain barrier dysfunction.2 Wernicke’s encephalopathy is most frequently encountered in patients with chronic alcoholism, diabetes, or eating disorders, and after bariatric surgery.3 Iatrogenic Wernicke’s encephalopathy may occur when depleted patients receive IV saline with dextrose without receiving thiamine. Top dietary sources of B1 include pork, fish, beans, lentils, nuts, rice, and wheat germ.
Vitamin B2 (riboflavin) is essential for oxidative pathways, monoamine synthesis, and the methylation cycle. B2 is needed to create the essential flavoprotein coenzymes for synthesis of L-methylfolate—the active form of folate—and for proper utilization of B6. Deficiency can occur after 4 months of inadequate intake.
Although generally B2 deficiency is rare, surveys in the United States have found that 10% to 27% of older adults (age ≥65) are deficient.4 Low intake of dairy products and meat and chronic, excessive alcohol intake are associated with deficiency. Marginal B2 levels are more prevalent in depressed patients, possibly because of B2’s role in the function of glutathione, an endogenous antioxidant.5 Top dietary sources of B2 are dairy products, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes.
Vitamin B6 refers to 3 distinct compounds: pyridoxine, pyridoxal, and pyridoxamine. B6 is essential to glycolysis, the methylation cycle, and recharging glutathione, an innate antioxidant in the brain. Higher levels of vitamin B6 are associated with a lower prevalence of depression in adolescents,6 and low dietary and plasma B6 increases the risk and severity of depression in geriatric patients7 and predicts depression in prospective trials.8 Deficiency is common (24% to 56%) among patients receiving hemodialysis.9 Women who take oral contraceptives are at increased risk of vitamin B6 deficiency.10 Top dietary sources are fish, beef, poultry, potatoes, legumes, and spinach.
Vitamin B9 (folate) is needed for proper one-carbon metabolism and thus requisite in synthesis of serotonin, norepinephrine, dopamine, and DNA and in phospholipid production. Low maternal folate status increases the risk of neural tube defects in newborns. Folate deficiency and insufficiency are common among patients with mood disorders and correlate with illness severity.11 In a study of 2,682 Finnish men, those in the lowest one-third of folate consumption had a 67% increased relative risk of depression.12 A meta-analysis of 11 studies of 15,315 persons found those who had low folate levels had a significant risk of depression.13 Patients without deficiency but with folate levels near the low end of the normal range also report low mood.14 Compared with controls, patients experiencing a first episode of psychosis have lower levels of folate, B12, and docosahexaenoic acid.15
Dietary folate must be converted to L-methylfolate for use in the brain. Patients with a methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism produce a less active form of the enzyme. The TT genotype is associated with major depression and bipolar disorder.16 Clinical trials have shown that several forms of folate can enhance antidepressant treatment.17 Augmentation with L-methylfolate, which bypasses the MTHFR enzyme, can be an effective strategy for treating depression in these patients.18
Leafy greens and legumes such as lentils are top dietary sources of folate; supplemental folic acid has been linked to an increased risk of cancer and overall mortality.19,20
Vitamin B12 (cobalamin). An essential cofactor in one-carbon metabolism, B12 is needed to produce monoamine neurotransmitters and maintain myelin. Deficiency is found in up to one-third of depressed patients11 and compromises antidepressant response,21 whereas higher vitamin B12 levels are associated with better treatment outcomes.22 B12 deficiency can cause depression, irritability, agitation, psychosis, and obsessive symptoms.23,24 Low B12 levels and elevated homocysteine increase the risk of cognitive decline and Alzheimer’s disease and are linked to a 5-fold increase in the rate of brain atrophy.26
B12 deficiencies may be seen in patients with gastrointestinal illness, older adults with achlorhydria, and vegans and vegetarians, in whom B12 intake can be low. Proton pump inhibitors such as omeprazole interfere with B12 absorption from food.
Psychiatric symptoms of B12 deficiency may present before hematologic findings.23 Folic acid supplementation may mask a B12 deficiency by delaying anemia but will not delay psychiatric symptoms. Ten percent of patients with an insufficiency (low normal levels of 200 to 400 pg/mL) have elevated homocysteine, which increases the risk of psychiatric disorders as well as comorbid illnesses such as cardiovascular disease. Top dietary sources include fish, mollusks (oysters, mussels, and clams), meat, and dairy products.
Vitamin C is vital for the synthesis of monoamines such as serotonin and norepinephrine. Vitamin C’s primary role in the brain is as an antioxidant. As a necessary cofactor, it keeps the copper and iron in metalloenzymes reduced, and also recycles vitamin E. Proper function of the methylation cycle depends on vitamin C, as does collagen synthesis and metabolism of xenobiotics by the liver. It is concentrated in cerebrospinal fluid.
Humans cannot manufacture vitamin C. Although the need for vitamin C (90 mg/d) is thought to be met by diet, studies have found that up to 13.7% of healthy, middle class patients in the United States are depleted.27 Older adults and patients with a poor diet due to drug or alcohol abuse, eating disorders, or affective symptoms are at risk.
Scurvy is caused by vitamin C deficiency and leads to bleeding gums and petechiae. Patients with insufficiency report irritability, loss of appetite, weight loss, and hypochondriasis. Vitamin C intake is significantly lower in older adults (age ≥60) with depression.28 Some research indicates patients with schizophrenia have decreased vitamin C levels and dysfunction of antioxidant defenses.29 Citrus, potatoes, and tomatoes are top dietary sources of vitamin C.
Fat-soluble vitamins
Vitamin A. Although vitamin A activity in the brain is poorly understood, retinol—the active form of vitamin A—is crucial for formation of opsins, which are the basis for vision. Childhood vitamin A deficiency may lead to blindness. Vitamin A also plays an important role in maintaining bone growth, reproduction, cell division, and immune system integrity.30 Animal sources such as beef liver, dairy products, and eggs provide retinol, and plant sources such as carrots, sweet potatoes, and leafy greens provide provitamin A carotenoids that humans convert into retinol.
Deficiency rarely is observed in the United States but remains a common problem for developing nations. In the United States, vitamin A deficiency is most often seen with excessive alcohol use, rigorous dietary restrictions, and gastrointestinal diseases accompanied by poor fat absorption.
Excess vitamin A ingestion may result in bone abnormalities, liver damage, birth defects, and depression. Isotretinoin—a form of vitamin A used to treat severe acne—carries an FDA “black-box” warning for psychiatric adverse effects, including aggression, depression, psychosis, and suicide.
Vitamin D is produced from cholesterol in the epidermis through exposure to sunlight, namely ultraviolet B radiation. After dermal synthesis or ingestion, vitamin D is converted through a series of steps into the active form of vitamin D, calcitriol, which also is known as 25(OH)D3.
Although vitamin D is known for its role in bone growth and mineralization,31 increasing evidence reveals vitamin D’s role in brain function and development.32 Both glial and neuronal cells possess vitamin D receptors in the hippocampus, prefrontal cortex, hypothalamus, thalamus, and substantia nigra—all regions theorized to be linked to depression pathophysiology.33 A review of the association of vitamin D deficiency and psychiatric illnesses will be published in a future issue of Current Psychiatry.
Vitamin D exists in food as either D2 or D3, from plant and animal sources, respectively. Concentrated sources include oily fish, sun-dried or “UVB-irradiated” mushrooms, and milk.
Vitamin E. There are 8 isoforms of vitamin E—4 tocopherols and 4 tocotrienols—that function as fat-soluble antioxidants and also promote innate antioxidant enzymes. Because vitamin E protects neuronal membranes from oxidation, low levels may affect the brain via increased inflammation. Alpha-tocopherol is the most common form of vitamin E in humans, but emerging evidence suggests tocotrienols mediate disease by modifying transcription factors in the brain, such as glutathione reductase, superoxide dismutase, and nuclear factor-kappaB.34 Low plasma vitamin E levels are found in depressed patients, although some data suggest this may be caused by factors other than dietary intake.35 Low vitamin status has been found in up to 70% of older adults.36 Although deficiency is rare, most of the U.S. population (93%) has inadequate dietary intake of vitamin E.1 The reasons for this discrepancy are unclear. Foods rich in vitamin E include almonds, sunflower seeds, leafy greens, and wheat germ.
Recommendations
Patients with depression, alcohol abuse, eating disorders, obsessive-compulsive disorder, or schizophrenia may neglect to care for themselves or adopt particular eating patterns. Deficiencies are more common among geriatric patients and those who are medically ill. Because dietary patterns are linked to the risk of psychiatric disorders, nutritional inquiry often identifies multiple modifiable risk factors, such as folate, vitamin B12, and vitamin D intake.37,38 Nutritional counseling offers clinicians an intervention with minimal side effect risks and the opportunity to modify a behavior that patients engage in 3 times a day.
Psychiatrists should assess patients’ dietary patterns and vitamin status, particularly older adults and those with:
- lower socioeconomic status or food insecurity
- a history of treatment resistance
- restrictive dietary patterns such as veganism
- alcohol abuse.
On initial assessment, test or obtain from other health care providers your patient’s blood levels of folate and vitamins D and B12. In some patients, assessing B2 and B6 levels may provide etiological guidance regarding onset of psychiatric symptoms or failure to respond to pharmacologic treatment. Because treating vitamin deficiencies often includes using supplements, evaluate recent reviews of specific deficiencies and consider consulting with the patient’s primary care provider.
Conduct a simple assessment of dietary patterns by asking patients about a typical breakfast, lunch, and dinner, their favorite snacks and foods, and specific dietary habits or restrictions (eg, not consuming seafood, dairy, meat, etc.). Rudimentary nutritional recommendations can be effective in changing a patient’s eating habits, particularly when provided by a physician. Encourage patients to eat nutrient-dense foods such as leafy greens, beans and legumes, seafood, whole grains, and a variety of vegetables and fruits. For more complex patients, consult with a clinical nutritionist.
Related Resources
- Institute of Medicine. Dietary Reference Intakes: Recommended intakes for individuals. SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf" target="_blank">www.iom.edu/Activities/Nutrition/
SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf. - The Farmacy: Vitamins. http://drewramseymd.com/index.php/resources/farmacy/category/vitamins.
- Office of Dietary Supplements. National Institutes of Health. Dietary supplements fact sheets. http://ods.od.nih.gov/factsheets/list-all.
- Oregon State University. Linus Pauling Institute. Micronutrient information center. http://lpi.oregonstate.edu/infocenter/vitamins.html.
Drug Brand Names
- Isotretinoin • Accutane
- L-methylfolate • Deplin
- Omeprazole • Prilosec
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Moshfegh A, Goldman J, Cleveland L. United States Department of Agriculture, Agricultural Research Service. What we eat in America NHANES 2001-2002: Usual nutrient intakes from food compared to dietary reference intakes. http://www.ars.usda.gov/SP2UserFiles/Place/12355000/pdf/0102/usualintaketables2001-02.pdf. Published September 2005. Accessed November 27, 2012.
2. Page GL, Laight D, Cummings MH. Thiamine deficiency in diabetes mellitus and the impact of thiamine replacement on glucose metabolism and vascular disease. Int J Clin Pract. 2011;65(6):684-690.
3. McCormick LM, Buchanan JR, Onwuameze OE, et al. Beyond alcoholism: Wernicke-Korsakoff syndrome in patients with psychiatric disorders. Cogn Behav Neurol. 2011;24(4):209-216.
4. Powers HJ. Riboflavin (vitamin B-2) and health. Am J Clin Nutr. 2003;77(6):1352-1360.
5. Naghashpour M, Amani R, Nutr R, et al. Riboflavin status and its association with serum hs-CRP levels among clinical nurses with depression. J Am Coll Nutr. 2011;30(5):340-347.
6. Murakami K, Miyake Y, Sasaki S, et al. Dietary folate, riboflavin, vitamin B-6, and vitamin B-12 and depressive symptoms in early adolescence: the Ryukyus Child Health Study. Psychosom Med. 2010;72(8):763-768.
7. Merete C, Falcon LM, Tucker KL. Vitamin B6 is associated with depressive symptomatology in Massachusetts elders. J Am Coll Nutr. 2008;27(3):421-427.
8. Skarupski KA, Tangney C, Li H, et al. Longitudinal association of vitamin B-6, folate, and vitamin B-12 with depressive symptoms among older adults over time. Am J Clin Nutr. 2010;92(2):330-335.
9. Corken M, Porter J. Is vitamin B(6) deficiency an under-recognized risk in patients receiving haemodialysis? A systematic review: 2000-2010. Nephrology (Carlton). 2011;16(7):619-625.
10. Wilson SM, Bivins BN, Russell KA, et al. Oral contraceptive use: impact on folate, vitamin B6, and vitamin B12 status. Nutr Rev. 2011;69(10):572-583.
11. Coppen A, Bolander-Gouaille C. Treatment of depression: time to consider folic acid and vitamin B12. J Psychopharmacol. 2005;19(1):59-65.
12. Tolmunen T, Voutilainen S, Hintikka J, et al. Dietary folate and depressive symptoms are associated in middle-aged Finnish men. J Nutr. 2003;133(10):3233-3236.
13. Gilbody S, Lightfoot T, Sheldon T. Is low folate a risk factor for depression? A meta-analysis and exploration of heterogeneity. J Epidemiol Community Health. 2007;61(7):631-637.
14. Rösche J, Uhlmann C, Fröscher W. Low serum folate levels as a risk factor for depressive mood in patients with chronic epilepsy. J Neuropsychiatry Clin Neurosci. 2003;15(1):64-66.
15. Kale A, Naphade N, Sapkale S, et al. Reduced folic acid, vitamin B12 and docosahexaenoic acid and increased homocysteine and cortisol in never-medicated schizophrenia patients: implications for altered one-carbon metabolism. Psychiatry Res. 2010;175(1-2):47-53.
16. Gilbody S, Lewis S, Lightfoot T. Methylenetetrahydrofolate reductase (MTHFR) genetic polymorphisms and psychiatric disorders: a HuGE review. Am J Epidemiol. 2007;165(1):1-13.
17. Di Palma C, Urani R, Agricola R, et al. Is methylfolate effective in relieving major depression in chronic alcoholics? A hypothesis of treatment. Curr Ther Res Clin Exp. 1994;55(5):559-568.
18. Papakostas GI, Shelton RC, Zajecka JM, et al. l-Methylfolate as adjunctive therapy for ssri-resistant major depression: results of two randomized, double-blind, parallel-sequential trials. Am J Psychiatry. 2012;169(12):1267-1274.
19. Baggott JE, Oster RA, Tamura T. Meta-analysis of cancer risk in folic acid supplementation trials. Cancer Epidemiol. 2012;36(1):78-81.
20. Figueiredo JC, Grau MV, Haile RW, et al. Folic acid and risk of prostate cancer: results from a randomized clinical trial. J Natl Cancer Inst. 2009;101(6):432-435.
21. Kate N, Grover S, Agarwal M. Does B12 deficiency lead to lack of treatment response to conventional antidepressants? Psychiatry (Edgmont). 2010;7(11):42-44.
22. Hintikka J, Tolmunen T, Tanskanen A, et al. High vitamin B12 level and good treatment outcome may be associated in major depressive disorder. BMC Psychiatry. 2003;3:17.-
23. Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
24. Bar-Shai M, Gott D, Marmor S. Acute psychotic depression as a sole manifestation of vitamin B12 deficiency. Psychosomatics. 2011;52(4):384-386.
25. Sharma V, Biswas D. Cobalamin deficiency presenting as obsessive compulsive disorder: case report. Gen Hosp Psychiatry. 2012;34(5):578.e7-e8.
26. Vogiatzoglou A, Refsum H, Johnston C, et al. Vitamin B12 status and rate of brain volume loss in community-dwelling elderly. Neurology. 2008;71(11):826-832.
27. Smith A, Di Primio G, Humphrey-Murto S. Scurvy in the developed world. CMAJ. 2011;183(11):E752-E725.
28. Payne ME, Steck SE, George RR, et al. Fruit, vegetable, and antioxidant intakes are lower in older adults with depression. J Acad Nutr Diet. 2012;112(12):2022-2027.
29. Dadheech G, Mishra S, Gautam S, et al. Oxidative stress, α-tocopherol, ascorbic acid and reduced glutathione status in schizophrenics. Indian J Clin Biochem. 2006;21(2):34-38.
30. Hinds TS, West WL, Knight EM. Carotenoids and retinoids: a review of research clinical, and public health applications. J Clin Pharmacol. 1997;37(7):551-558.
31. Thacher TD, Clarke BL. Vitamin D insufficiency. Mayo Clin Proc. 2011;86(1):50-60.
32. Berk M, Sanders KM, Pasco JA, et al. Vitamin D deficiency may play a role in depression. Med Hypotheses. 2007;69(6):1316-1319.
33. Eyles DW, Smith S, Kinobe R, et al. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat. 2005;29(1):21-30.
34. Sen CK, Khanna S, Roy S. Tocotrienol: the natural vitamin E to defend the nervous system? Ann N Y Acad Sci. 2004;1031:127-142.
35. Owen AJ, Batterham MJ, Probst YC, et al. Low plasma vitamin E levels in major depression: diet or disease? Eur J Clin Nutr. 2005;59(2):304-306.
36. Panemangalore M, Lee CJ. Evaluation of the indices of retinol and alpha-tocopherol status in free-living elderly. J Gerontol. 1992;47(3):B98-B104.
37. Sánchez-Villegas A, Delgado-Rodríguez M, Alonso A, et al. Association of the Mediterranean dietary pattern with the incidence of depression: the Seguimiento Universidad de Navarra/University of Navarra follow-up (SUN) cohort. Arch Gen Psychiatry. 2009;66(10):1090-1098.
38. Jacka FN, Pasco JA, Mykletun A, et al. Association of Western and traditional diets with depression and anxiety in women. Am J Psychiatry. 2010;167(3):305-311.
Discuss this article at www.facebook.com/CurrentPsychiatry
Patients today often are overfed but undernourished. A growing body of literature links dietary choices to brain health and the risk of psychiatric illness. Vitamin deficiencies can affect psychiatric patients in several ways:
- deficiencies may play a causative role in mental illness and exacerbate symptoms
- psychiatric symptoms can result in poor nutrition
- vitamin insufficiency—defined as subclinical deficiency—may compromise patient recovery.
Additionally, genetic differences may compromise vitamin and essential nutrient pathways.
Vitamins are dietary components other than carbohydrates, fats, minerals, and proteins that are necessary for life. B vitamins are required for proper functioning of the methylation cycle, monoamine production, DNA synthesis, and maintenance of phospholipids such as myelin (Figure). Fat-soluble vitamins A, D, and E play important roles in genetic transcription, antioxidant recycling, and inflammatory regulation in the brain.
Figure: The methylation cycle
Vitamins B2, B6, B9, and B12 directly impact the functioning of the methylation cycle. Deficiencies pertain to brain function, as neurotransmitters, myelin, and active glutathione are dependent on one-carbon metabolism
Illustration: Mala Nimalasuriya with permission from DrewRamseyMD.com
To help clinicians recognize and treat vitamin deficiencies among psychiatric patients, this article reviews the role of the 6 essential water-soluble vitamins (B1, B2, B6, B9, B12, and C; Table 1,1) and 3 fat-soluble vitamins (A, D, and E; Table 2,1) in brain metabolism and psychiatric pathology. Because numerous sources address using supplements to treat vitamin deficiencies, this article emphasizes food sources, which for many patients are adequate to sustain nutrient status.
Table 1
Water-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| B1 (thiamine): Glycolysis, tricarboxylic acid cycle | ||||
| Rare; 7% in heart failure patients | 5% total, 12% of older women | Wernicke-Korsakoff syndrome, memory impairment, confusion, lack of coordination, paralysis | Older adults, malabsorptive conditions, heavy alcohol use. Those with diabetes are at risk because of increased clearance | Pork, fish, beans, lentils, nuts, rice, and wheat germ. Raw fish, tea, and betel nuts impair absorption |
| B2 (riboflavin): FMN, FAD cofactors in glycolysis and oxidative pathways. B6, folate, and glutathione synthesis | ||||
| 10% to 27% of older adults | <3%; 95% of adolescent girls (measured by EGRAC) | Fatigue, cracked lips, sore throat, bloodshot eyes | Older adults, low intake of animal and dairy products, heavy alcohol use | Dairy, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes |
| B6 (pyridoxal): Methylation cycle | ||||
| 11% to 24% (<5 ng/mL); 38% of heart failure patients | 14% total, 26% of adults | Dermatitis, glossitis, convulsions, migraine, chronic pain, depression | Older adults, women who use oral contraceptives, alcoholism. 33% to 49% of women age >51 have inadequate intake | Bananas, beans, potatoes, navy beans, salmon, steak, and whole grains |
| B9 (folate): Methylation cycle | ||||
| 0.5% total; up to 50% of depressed patients | 16% of adults, 19% of adolescent girls | Loss of appetite, weight loss, weakness, heart palpitations, behavioral disorders | Depression, pregnancy and lactation, alcoholism, dialysis, liver disease. Deficiency during pregnancy is linked to neural tube defects | Leafy green vegetables, fruits, dried beans, and peas |
| B12 (cobalamin): Methylation cycle (cofactor methionine synthase) | ||||
| 10% to 15% of older adults | <3% to 9% | Depression, irritability, anemia, fatigue, shortness of breath, high blood pressure | Vegetarian or vegan diet, achlorhydria, older adults. Deficiency more often due to poor absorption than low consumption | Meat, seafood, eggs, and dairy |
| C (ascorbic acid): Antioxidant | ||||
| 7.1% | 31% | Scurvy, fatigue, anemia, joint pain, petechia. Symptoms develop after 1 to 3 months of no dietary intake | Smokers, infants fed boiled or evaporated milk, limited dietary variation, patients with malabsorption, chronic illnesses | Citrus fruits, tomatoes and tomato juice, and potatoes |
| EGRAC: erythrocyte glutathione reductase activation coefficient; FAD: flavin adenine dinucleotide; FMN: flavin mononucleotide Source: Reference 1 | ||||
Table 2
Fat-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| A (retinol): Transcription regulation, vision | ||||
| <5% of U.S. population | 44% | Blindness, decreased immunity, corneal and retinal damage | Pregnant women, individuals with strict dietary restrictions, heavy alcohol use, chronic diarrhea, fat malabsorptive conditions | Beef liver, dairy products. Convertible beta-carotene sources: sweet potatoes, carrots, spinach, butternut squash, greens, broccoli, cantaloupe |
| D (cholecalciferol): Hormone, transcriptional regulation | ||||
| ≥50%, 90% of adults age >50 | 69% | Rickets, osteoporosis, muscle twitching | Breast-fed infants, older adults, limited sun exposure, pigmented skin, fat malabsorption, obesity. Older adults have an impaired ability to make vitamin D from the sun. SPF 15 reduces production by 99% | Fatty fish and fish liver oils, sun-dried mushrooms |
| E (tocopherols and tocotrienols): Antioxidant, PUFA protectant, gene regulation | ||||
| Rare | 93% | Anemia, neuropathy, myopathy, abnormal eye movements, weakness, retinal damage | Malabsorptive conditions, HIV, depression | Sunflower, wheat germ, and safflower oils; meats; fish; dairy; green vegetables |
| HIV: human immunodeficiency virus; PUFA: polyunsaturated fatty acids; SPF: sun protection factor Source: Reference 1 | ||||
Water-soluble vitamins
Vitamin B1 (thiamine) is essential for glucose metabolism. Pregnancy, lactation, and fever increase the need for thiamine, and tea, coffee, and shellfish can impair its absorption. Although rare, severe B1 deficiency can lead to beriberi, Wernicke’s encephalopathy (confusion, ataxia, nystagmus), and Korsakoff’s psychosis (confabulation, lack of insight, retrograde and anterograde amnesia, and apathy). Confusion and disorientation stem from the brain’s inability to oxidize glucose for energy because B1 is a critical cofactor in glycolysis and the tricarboxylic acid cycle. Deficiency leads to an increase in reactive oxygen species, proinflammatory cytokines, and blood-brain barrier dysfunction.2 Wernicke’s encephalopathy is most frequently encountered in patients with chronic alcoholism, diabetes, or eating disorders, and after bariatric surgery.3 Iatrogenic Wernicke’s encephalopathy may occur when depleted patients receive IV saline with dextrose without receiving thiamine. Top dietary sources of B1 include pork, fish, beans, lentils, nuts, rice, and wheat germ.
Vitamin B2 (riboflavin) is essential for oxidative pathways, monoamine synthesis, and the methylation cycle. B2 is needed to create the essential flavoprotein coenzymes for synthesis of L-methylfolate—the active form of folate—and for proper utilization of B6. Deficiency can occur after 4 months of inadequate intake.
Although generally B2 deficiency is rare, surveys in the United States have found that 10% to 27% of older adults (age ≥65) are deficient.4 Low intake of dairy products and meat and chronic, excessive alcohol intake are associated with deficiency. Marginal B2 levels are more prevalent in depressed patients, possibly because of B2’s role in the function of glutathione, an endogenous antioxidant.5 Top dietary sources of B2 are dairy products, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes.
Vitamin B6 refers to 3 distinct compounds: pyridoxine, pyridoxal, and pyridoxamine. B6 is essential to glycolysis, the methylation cycle, and recharging glutathione, an innate antioxidant in the brain. Higher levels of vitamin B6 are associated with a lower prevalence of depression in adolescents,6 and low dietary and plasma B6 increases the risk and severity of depression in geriatric patients7 and predicts depression in prospective trials.8 Deficiency is common (24% to 56%) among patients receiving hemodialysis.9 Women who take oral contraceptives are at increased risk of vitamin B6 deficiency.10 Top dietary sources are fish, beef, poultry, potatoes, legumes, and spinach.
Vitamin B9 (folate) is needed for proper one-carbon metabolism and thus requisite in synthesis of serotonin, norepinephrine, dopamine, and DNA and in phospholipid production. Low maternal folate status increases the risk of neural tube defects in newborns. Folate deficiency and insufficiency are common among patients with mood disorders and correlate with illness severity.11 In a study of 2,682 Finnish men, those in the lowest one-third of folate consumption had a 67% increased relative risk of depression.12 A meta-analysis of 11 studies of 15,315 persons found those who had low folate levels had a significant risk of depression.13 Patients without deficiency but with folate levels near the low end of the normal range also report low mood.14 Compared with controls, patients experiencing a first episode of psychosis have lower levels of folate, B12, and docosahexaenoic acid.15
Dietary folate must be converted to L-methylfolate for use in the brain. Patients with a methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism produce a less active form of the enzyme. The TT genotype is associated with major depression and bipolar disorder.16 Clinical trials have shown that several forms of folate can enhance antidepressant treatment.17 Augmentation with L-methylfolate, which bypasses the MTHFR enzyme, can be an effective strategy for treating depression in these patients.18
Leafy greens and legumes such as lentils are top dietary sources of folate; supplemental folic acid has been linked to an increased risk of cancer and overall mortality.19,20
Vitamin B12 (cobalamin). An essential cofactor in one-carbon metabolism, B12 is needed to produce monoamine neurotransmitters and maintain myelin. Deficiency is found in up to one-third of depressed patients11 and compromises antidepressant response,21 whereas higher vitamin B12 levels are associated with better treatment outcomes.22 B12 deficiency can cause depression, irritability, agitation, psychosis, and obsessive symptoms.23,24 Low B12 levels and elevated homocysteine increase the risk of cognitive decline and Alzheimer’s disease and are linked to a 5-fold increase in the rate of brain atrophy.26
B12 deficiencies may be seen in patients with gastrointestinal illness, older adults with achlorhydria, and vegans and vegetarians, in whom B12 intake can be low. Proton pump inhibitors such as omeprazole interfere with B12 absorption from food.
Psychiatric symptoms of B12 deficiency may present before hematologic findings.23 Folic acid supplementation may mask a B12 deficiency by delaying anemia but will not delay psychiatric symptoms. Ten percent of patients with an insufficiency (low normal levels of 200 to 400 pg/mL) have elevated homocysteine, which increases the risk of psychiatric disorders as well as comorbid illnesses such as cardiovascular disease. Top dietary sources include fish, mollusks (oysters, mussels, and clams), meat, and dairy products.
Vitamin C is vital for the synthesis of monoamines such as serotonin and norepinephrine. Vitamin C’s primary role in the brain is as an antioxidant. As a necessary cofactor, it keeps the copper and iron in metalloenzymes reduced, and also recycles vitamin E. Proper function of the methylation cycle depends on vitamin C, as does collagen synthesis and metabolism of xenobiotics by the liver. It is concentrated in cerebrospinal fluid.
Humans cannot manufacture vitamin C. Although the need for vitamin C (90 mg/d) is thought to be met by diet, studies have found that up to 13.7% of healthy, middle class patients in the United States are depleted.27 Older adults and patients with a poor diet due to drug or alcohol abuse, eating disorders, or affective symptoms are at risk.
Scurvy is caused by vitamin C deficiency and leads to bleeding gums and petechiae. Patients with insufficiency report irritability, loss of appetite, weight loss, and hypochondriasis. Vitamin C intake is significantly lower in older adults (age ≥60) with depression.28 Some research indicates patients with schizophrenia have decreased vitamin C levels and dysfunction of antioxidant defenses.29 Citrus, potatoes, and tomatoes are top dietary sources of vitamin C.
Fat-soluble vitamins
Vitamin A. Although vitamin A activity in the brain is poorly understood, retinol—the active form of vitamin A—is crucial for formation of opsins, which are the basis for vision. Childhood vitamin A deficiency may lead to blindness. Vitamin A also plays an important role in maintaining bone growth, reproduction, cell division, and immune system integrity.30 Animal sources such as beef liver, dairy products, and eggs provide retinol, and plant sources such as carrots, sweet potatoes, and leafy greens provide provitamin A carotenoids that humans convert into retinol.
Deficiency rarely is observed in the United States but remains a common problem for developing nations. In the United States, vitamin A deficiency is most often seen with excessive alcohol use, rigorous dietary restrictions, and gastrointestinal diseases accompanied by poor fat absorption.
Excess vitamin A ingestion may result in bone abnormalities, liver damage, birth defects, and depression. Isotretinoin—a form of vitamin A used to treat severe acne—carries an FDA “black-box” warning for psychiatric adverse effects, including aggression, depression, psychosis, and suicide.
Vitamin D is produced from cholesterol in the epidermis through exposure to sunlight, namely ultraviolet B radiation. After dermal synthesis or ingestion, vitamin D is converted through a series of steps into the active form of vitamin D, calcitriol, which also is known as 25(OH)D3.
Although vitamin D is known for its role in bone growth and mineralization,31 increasing evidence reveals vitamin D’s role in brain function and development.32 Both glial and neuronal cells possess vitamin D receptors in the hippocampus, prefrontal cortex, hypothalamus, thalamus, and substantia nigra—all regions theorized to be linked to depression pathophysiology.33 A review of the association of vitamin D deficiency and psychiatric illnesses will be published in a future issue of Current Psychiatry.
Vitamin D exists in food as either D2 or D3, from plant and animal sources, respectively. Concentrated sources include oily fish, sun-dried or “UVB-irradiated” mushrooms, and milk.
Vitamin E. There are 8 isoforms of vitamin E—4 tocopherols and 4 tocotrienols—that function as fat-soluble antioxidants and also promote innate antioxidant enzymes. Because vitamin E protects neuronal membranes from oxidation, low levels may affect the brain via increased inflammation. Alpha-tocopherol is the most common form of vitamin E in humans, but emerging evidence suggests tocotrienols mediate disease by modifying transcription factors in the brain, such as glutathione reductase, superoxide dismutase, and nuclear factor-kappaB.34 Low plasma vitamin E levels are found in depressed patients, although some data suggest this may be caused by factors other than dietary intake.35 Low vitamin status has been found in up to 70% of older adults.36 Although deficiency is rare, most of the U.S. population (93%) has inadequate dietary intake of vitamin E.1 The reasons for this discrepancy are unclear. Foods rich in vitamin E include almonds, sunflower seeds, leafy greens, and wheat germ.
Recommendations
Patients with depression, alcohol abuse, eating disorders, obsessive-compulsive disorder, or schizophrenia may neglect to care for themselves or adopt particular eating patterns. Deficiencies are more common among geriatric patients and those who are medically ill. Because dietary patterns are linked to the risk of psychiatric disorders, nutritional inquiry often identifies multiple modifiable risk factors, such as folate, vitamin B12, and vitamin D intake.37,38 Nutritional counseling offers clinicians an intervention with minimal side effect risks and the opportunity to modify a behavior that patients engage in 3 times a day.
Psychiatrists should assess patients’ dietary patterns and vitamin status, particularly older adults and those with:
- lower socioeconomic status or food insecurity
- a history of treatment resistance
- restrictive dietary patterns such as veganism
- alcohol abuse.
On initial assessment, test or obtain from other health care providers your patient’s blood levels of folate and vitamins D and B12. In some patients, assessing B2 and B6 levels may provide etiological guidance regarding onset of psychiatric symptoms or failure to respond to pharmacologic treatment. Because treating vitamin deficiencies often includes using supplements, evaluate recent reviews of specific deficiencies and consider consulting with the patient’s primary care provider.
Conduct a simple assessment of dietary patterns by asking patients about a typical breakfast, lunch, and dinner, their favorite snacks and foods, and specific dietary habits or restrictions (eg, not consuming seafood, dairy, meat, etc.). Rudimentary nutritional recommendations can be effective in changing a patient’s eating habits, particularly when provided by a physician. Encourage patients to eat nutrient-dense foods such as leafy greens, beans and legumes, seafood, whole grains, and a variety of vegetables and fruits. For more complex patients, consult with a clinical nutritionist.
Related Resources
- Institute of Medicine. Dietary Reference Intakes: Recommended intakes for individuals. SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf" target="_blank">www.iom.edu/Activities/Nutrition/
SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf. - The Farmacy: Vitamins. http://drewramseymd.com/index.php/resources/farmacy/category/vitamins.
- Office of Dietary Supplements. National Institutes of Health. Dietary supplements fact sheets. http://ods.od.nih.gov/factsheets/list-all.
- Oregon State University. Linus Pauling Institute. Micronutrient information center. http://lpi.oregonstate.edu/infocenter/vitamins.html.
Drug Brand Names
- Isotretinoin • Accutane
- L-methylfolate • Deplin
- Omeprazole • Prilosec
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Patients today often are overfed but undernourished. A growing body of literature links dietary choices to brain health and the risk of psychiatric illness. Vitamin deficiencies can affect psychiatric patients in several ways:
- deficiencies may play a causative role in mental illness and exacerbate symptoms
- psychiatric symptoms can result in poor nutrition
- vitamin insufficiency—defined as subclinical deficiency—may compromise patient recovery.
Additionally, genetic differences may compromise vitamin and essential nutrient pathways.
Vitamins are dietary components other than carbohydrates, fats, minerals, and proteins that are necessary for life. B vitamins are required for proper functioning of the methylation cycle, monoamine production, DNA synthesis, and maintenance of phospholipids such as myelin (Figure). Fat-soluble vitamins A, D, and E play important roles in genetic transcription, antioxidant recycling, and inflammatory regulation in the brain.
Figure: The methylation cycle
Vitamins B2, B6, B9, and B12 directly impact the functioning of the methylation cycle. Deficiencies pertain to brain function, as neurotransmitters, myelin, and active glutathione are dependent on one-carbon metabolism
Illustration: Mala Nimalasuriya with permission from DrewRamseyMD.com
To help clinicians recognize and treat vitamin deficiencies among psychiatric patients, this article reviews the role of the 6 essential water-soluble vitamins (B1, B2, B6, B9, B12, and C; Table 1,1) and 3 fat-soluble vitamins (A, D, and E; Table 2,1) in brain metabolism and psychiatric pathology. Because numerous sources address using supplements to treat vitamin deficiencies, this article emphasizes food sources, which for many patients are adequate to sustain nutrient status.
Table 1
Water-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| B1 (thiamine): Glycolysis, tricarboxylic acid cycle | ||||
| Rare; 7% in heart failure patients | 5% total, 12% of older women | Wernicke-Korsakoff syndrome, memory impairment, confusion, lack of coordination, paralysis | Older adults, malabsorptive conditions, heavy alcohol use. Those with diabetes are at risk because of increased clearance | Pork, fish, beans, lentils, nuts, rice, and wheat germ. Raw fish, tea, and betel nuts impair absorption |
| B2 (riboflavin): FMN, FAD cofactors in glycolysis and oxidative pathways. B6, folate, and glutathione synthesis | ||||
| 10% to 27% of older adults | <3%; 95% of adolescent girls (measured by EGRAC) | Fatigue, cracked lips, sore throat, bloodshot eyes | Older adults, low intake of animal and dairy products, heavy alcohol use | Dairy, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes |
| B6 (pyridoxal): Methylation cycle | ||||
| 11% to 24% (<5 ng/mL); 38% of heart failure patients | 14% total, 26% of adults | Dermatitis, glossitis, convulsions, migraine, chronic pain, depression | Older adults, women who use oral contraceptives, alcoholism. 33% to 49% of women age >51 have inadequate intake | Bananas, beans, potatoes, navy beans, salmon, steak, and whole grains |
| B9 (folate): Methylation cycle | ||||
| 0.5% total; up to 50% of depressed patients | 16% of adults, 19% of adolescent girls | Loss of appetite, weight loss, weakness, heart palpitations, behavioral disorders | Depression, pregnancy and lactation, alcoholism, dialysis, liver disease. Deficiency during pregnancy is linked to neural tube defects | Leafy green vegetables, fruits, dried beans, and peas |
| B12 (cobalamin): Methylation cycle (cofactor methionine synthase) | ||||
| 10% to 15% of older adults | <3% to 9% | Depression, irritability, anemia, fatigue, shortness of breath, high blood pressure | Vegetarian or vegan diet, achlorhydria, older adults. Deficiency more often due to poor absorption than low consumption | Meat, seafood, eggs, and dairy |
| C (ascorbic acid): Antioxidant | ||||
| 7.1% | 31% | Scurvy, fatigue, anemia, joint pain, petechia. Symptoms develop after 1 to 3 months of no dietary intake | Smokers, infants fed boiled or evaporated milk, limited dietary variation, patients with malabsorption, chronic illnesses | Citrus fruits, tomatoes and tomato juice, and potatoes |
| EGRAC: erythrocyte glutathione reductase activation coefficient; FAD: flavin adenine dinucleotide; FMN: flavin mononucleotide Source: Reference 1 | ||||
Table 2
Fat-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| A (retinol): Transcription regulation, vision | ||||
| <5% of U.S. population | 44% | Blindness, decreased immunity, corneal and retinal damage | Pregnant women, individuals with strict dietary restrictions, heavy alcohol use, chronic diarrhea, fat malabsorptive conditions | Beef liver, dairy products. Convertible beta-carotene sources: sweet potatoes, carrots, spinach, butternut squash, greens, broccoli, cantaloupe |
| D (cholecalciferol): Hormone, transcriptional regulation | ||||
| ≥50%, 90% of adults age >50 | 69% | Rickets, osteoporosis, muscle twitching | Breast-fed infants, older adults, limited sun exposure, pigmented skin, fat malabsorption, obesity. Older adults have an impaired ability to make vitamin D from the sun. SPF 15 reduces production by 99% | Fatty fish and fish liver oils, sun-dried mushrooms |
| E (tocopherols and tocotrienols): Antioxidant, PUFA protectant, gene regulation | ||||
| Rare | 93% | Anemia, neuropathy, myopathy, abnormal eye movements, weakness, retinal damage | Malabsorptive conditions, HIV, depression | Sunflower, wheat germ, and safflower oils; meats; fish; dairy; green vegetables |
| HIV: human immunodeficiency virus; PUFA: polyunsaturated fatty acids; SPF: sun protection factor Source: Reference 1 | ||||
Water-soluble vitamins
Vitamin B1 (thiamine) is essential for glucose metabolism. Pregnancy, lactation, and fever increase the need for thiamine, and tea, coffee, and shellfish can impair its absorption. Although rare, severe B1 deficiency can lead to beriberi, Wernicke’s encephalopathy (confusion, ataxia, nystagmus), and Korsakoff’s psychosis (confabulation, lack of insight, retrograde and anterograde amnesia, and apathy). Confusion and disorientation stem from the brain’s inability to oxidize glucose for energy because B1 is a critical cofactor in glycolysis and the tricarboxylic acid cycle. Deficiency leads to an increase in reactive oxygen species, proinflammatory cytokines, and blood-brain barrier dysfunction.2 Wernicke’s encephalopathy is most frequently encountered in patients with chronic alcoholism, diabetes, or eating disorders, and after bariatric surgery.3 Iatrogenic Wernicke’s encephalopathy may occur when depleted patients receive IV saline with dextrose without receiving thiamine. Top dietary sources of B1 include pork, fish, beans, lentils, nuts, rice, and wheat germ.
Vitamin B2 (riboflavin) is essential for oxidative pathways, monoamine synthesis, and the methylation cycle. B2 is needed to create the essential flavoprotein coenzymes for synthesis of L-methylfolate—the active form of folate—and for proper utilization of B6. Deficiency can occur after 4 months of inadequate intake.
Although generally B2 deficiency is rare, surveys in the United States have found that 10% to 27% of older adults (age ≥65) are deficient.4 Low intake of dairy products and meat and chronic, excessive alcohol intake are associated with deficiency. Marginal B2 levels are more prevalent in depressed patients, possibly because of B2’s role in the function of glutathione, an endogenous antioxidant.5 Top dietary sources of B2 are dairy products, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes.
Vitamin B6 refers to 3 distinct compounds: pyridoxine, pyridoxal, and pyridoxamine. B6 is essential to glycolysis, the methylation cycle, and recharging glutathione, an innate antioxidant in the brain. Higher levels of vitamin B6 are associated with a lower prevalence of depression in adolescents,6 and low dietary and plasma B6 increases the risk and severity of depression in geriatric patients7 and predicts depression in prospective trials.8 Deficiency is common (24% to 56%) among patients receiving hemodialysis.9 Women who take oral contraceptives are at increased risk of vitamin B6 deficiency.10 Top dietary sources are fish, beef, poultry, potatoes, legumes, and spinach.
Vitamin B9 (folate) is needed for proper one-carbon metabolism and thus requisite in synthesis of serotonin, norepinephrine, dopamine, and DNA and in phospholipid production. Low maternal folate status increases the risk of neural tube defects in newborns. Folate deficiency and insufficiency are common among patients with mood disorders and correlate with illness severity.11 In a study of 2,682 Finnish men, those in the lowest one-third of folate consumption had a 67% increased relative risk of depression.12 A meta-analysis of 11 studies of 15,315 persons found those who had low folate levels had a significant risk of depression.13 Patients without deficiency but with folate levels near the low end of the normal range also report low mood.14 Compared with controls, patients experiencing a first episode of psychosis have lower levels of folate, B12, and docosahexaenoic acid.15
Dietary folate must be converted to L-methylfolate for use in the brain. Patients with a methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism produce a less active form of the enzyme. The TT genotype is associated with major depression and bipolar disorder.16 Clinical trials have shown that several forms of folate can enhance antidepressant treatment.17 Augmentation with L-methylfolate, which bypasses the MTHFR enzyme, can be an effective strategy for treating depression in these patients.18
Leafy greens and legumes such as lentils are top dietary sources of folate; supplemental folic acid has been linked to an increased risk of cancer and overall mortality.19,20
Vitamin B12 (cobalamin). An essential cofactor in one-carbon metabolism, B12 is needed to produce monoamine neurotransmitters and maintain myelin. Deficiency is found in up to one-third of depressed patients11 and compromises antidepressant response,21 whereas higher vitamin B12 levels are associated with better treatment outcomes.22 B12 deficiency can cause depression, irritability, agitation, psychosis, and obsessive symptoms.23,24 Low B12 levels and elevated homocysteine increase the risk of cognitive decline and Alzheimer’s disease and are linked to a 5-fold increase in the rate of brain atrophy.26
B12 deficiencies may be seen in patients with gastrointestinal illness, older adults with achlorhydria, and vegans and vegetarians, in whom B12 intake can be low. Proton pump inhibitors such as omeprazole interfere with B12 absorption from food.
Psychiatric symptoms of B12 deficiency may present before hematologic findings.23 Folic acid supplementation may mask a B12 deficiency by delaying anemia but will not delay psychiatric symptoms. Ten percent of patients with an insufficiency (low normal levels of 200 to 400 pg/mL) have elevated homocysteine, which increases the risk of psychiatric disorders as well as comorbid illnesses such as cardiovascular disease. Top dietary sources include fish, mollusks (oysters, mussels, and clams), meat, and dairy products.
Vitamin C is vital for the synthesis of monoamines such as serotonin and norepinephrine. Vitamin C’s primary role in the brain is as an antioxidant. As a necessary cofactor, it keeps the copper and iron in metalloenzymes reduced, and also recycles vitamin E. Proper function of the methylation cycle depends on vitamin C, as does collagen synthesis and metabolism of xenobiotics by the liver. It is concentrated in cerebrospinal fluid.
Humans cannot manufacture vitamin C. Although the need for vitamin C (90 mg/d) is thought to be met by diet, studies have found that up to 13.7% of healthy, middle class patients in the United States are depleted.27 Older adults and patients with a poor diet due to drug or alcohol abuse, eating disorders, or affective symptoms are at risk.
Scurvy is caused by vitamin C deficiency and leads to bleeding gums and petechiae. Patients with insufficiency report irritability, loss of appetite, weight loss, and hypochondriasis. Vitamin C intake is significantly lower in older adults (age ≥60) with depression.28 Some research indicates patients with schizophrenia have decreased vitamin C levels and dysfunction of antioxidant defenses.29 Citrus, potatoes, and tomatoes are top dietary sources of vitamin C.
Fat-soluble vitamins
Vitamin A. Although vitamin A activity in the brain is poorly understood, retinol—the active form of vitamin A—is crucial for formation of opsins, which are the basis for vision. Childhood vitamin A deficiency may lead to blindness. Vitamin A also plays an important role in maintaining bone growth, reproduction, cell division, and immune system integrity.30 Animal sources such as beef liver, dairy products, and eggs provide retinol, and plant sources such as carrots, sweet potatoes, and leafy greens provide provitamin A carotenoids that humans convert into retinol.
Deficiency rarely is observed in the United States but remains a common problem for developing nations. In the United States, vitamin A deficiency is most often seen with excessive alcohol use, rigorous dietary restrictions, and gastrointestinal diseases accompanied by poor fat absorption.
Excess vitamin A ingestion may result in bone abnormalities, liver damage, birth defects, and depression. Isotretinoin—a form of vitamin A used to treat severe acne—carries an FDA “black-box” warning for psychiatric adverse effects, including aggression, depression, psychosis, and suicide.
Vitamin D is produced from cholesterol in the epidermis through exposure to sunlight, namely ultraviolet B radiation. After dermal synthesis or ingestion, vitamin D is converted through a series of steps into the active form of vitamin D, calcitriol, which also is known as 25(OH)D3.
Although vitamin D is known for its role in bone growth and mineralization,31 increasing evidence reveals vitamin D’s role in brain function and development.32 Both glial and neuronal cells possess vitamin D receptors in the hippocampus, prefrontal cortex, hypothalamus, thalamus, and substantia nigra—all regions theorized to be linked to depression pathophysiology.33 A review of the association of vitamin D deficiency and psychiatric illnesses will be published in a future issue of Current Psychiatry.
Vitamin D exists in food as either D2 or D3, from plant and animal sources, respectively. Concentrated sources include oily fish, sun-dried or “UVB-irradiated” mushrooms, and milk.
Vitamin E. There are 8 isoforms of vitamin E—4 tocopherols and 4 tocotrienols—that function as fat-soluble antioxidants and also promote innate antioxidant enzymes. Because vitamin E protects neuronal membranes from oxidation, low levels may affect the brain via increased inflammation. Alpha-tocopherol is the most common form of vitamin E in humans, but emerging evidence suggests tocotrienols mediate disease by modifying transcription factors in the brain, such as glutathione reductase, superoxide dismutase, and nuclear factor-kappaB.34 Low plasma vitamin E levels are found in depressed patients, although some data suggest this may be caused by factors other than dietary intake.35 Low vitamin status has been found in up to 70% of older adults.36 Although deficiency is rare, most of the U.S. population (93%) has inadequate dietary intake of vitamin E.1 The reasons for this discrepancy are unclear. Foods rich in vitamin E include almonds, sunflower seeds, leafy greens, and wheat germ.
Recommendations
Patients with depression, alcohol abuse, eating disorders, obsessive-compulsive disorder, or schizophrenia may neglect to care for themselves or adopt particular eating patterns. Deficiencies are more common among geriatric patients and those who are medically ill. Because dietary patterns are linked to the risk of psychiatric disorders, nutritional inquiry often identifies multiple modifiable risk factors, such as folate, vitamin B12, and vitamin D intake.37,38 Nutritional counseling offers clinicians an intervention with minimal side effect risks and the opportunity to modify a behavior that patients engage in 3 times a day.
Psychiatrists should assess patients’ dietary patterns and vitamin status, particularly older adults and those with:
- lower socioeconomic status or food insecurity
- a history of treatment resistance
- restrictive dietary patterns such as veganism
- alcohol abuse.
On initial assessment, test or obtain from other health care providers your patient’s blood levels of folate and vitamins D and B12. In some patients, assessing B2 and B6 levels may provide etiological guidance regarding onset of psychiatric symptoms or failure to respond to pharmacologic treatment. Because treating vitamin deficiencies often includes using supplements, evaluate recent reviews of specific deficiencies and consider consulting with the patient’s primary care provider.
Conduct a simple assessment of dietary patterns by asking patients about a typical breakfast, lunch, and dinner, their favorite snacks and foods, and specific dietary habits or restrictions (eg, not consuming seafood, dairy, meat, etc.). Rudimentary nutritional recommendations can be effective in changing a patient’s eating habits, particularly when provided by a physician. Encourage patients to eat nutrient-dense foods such as leafy greens, beans and legumes, seafood, whole grains, and a variety of vegetables and fruits. For more complex patients, consult with a clinical nutritionist.
Related Resources
- Institute of Medicine. Dietary Reference Intakes: Recommended intakes for individuals. SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf" target="_blank">www.iom.edu/Activities/Nutrition/
SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf. - The Farmacy: Vitamins. http://drewramseymd.com/index.php/resources/farmacy/category/vitamins.
- Office of Dietary Supplements. National Institutes of Health. Dietary supplements fact sheets. http://ods.od.nih.gov/factsheets/list-all.
- Oregon State University. Linus Pauling Institute. Micronutrient information center. http://lpi.oregonstate.edu/infocenter/vitamins.html.
Drug Brand Names
- Isotretinoin • Accutane
- L-methylfolate • Deplin
- Omeprazole • Prilosec
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Moshfegh A, Goldman J, Cleveland L. United States Department of Agriculture, Agricultural Research Service. What we eat in America NHANES 2001-2002: Usual nutrient intakes from food compared to dietary reference intakes. http://www.ars.usda.gov/SP2UserFiles/Place/12355000/pdf/0102/usualintaketables2001-02.pdf. Published September 2005. Accessed November 27, 2012.
2. Page GL, Laight D, Cummings MH. Thiamine deficiency in diabetes mellitus and the impact of thiamine replacement on glucose metabolism and vascular disease. Int J Clin Pract. 2011;65(6):684-690.
3. McCormick LM, Buchanan JR, Onwuameze OE, et al. Beyond alcoholism: Wernicke-Korsakoff syndrome in patients with psychiatric disorders. Cogn Behav Neurol. 2011;24(4):209-216.
4. Powers HJ. Riboflavin (vitamin B-2) and health. Am J Clin Nutr. 2003;77(6):1352-1360.
5. Naghashpour M, Amani R, Nutr R, et al. Riboflavin status and its association with serum hs-CRP levels among clinical nurses with depression. J Am Coll Nutr. 2011;30(5):340-347.
6. Murakami K, Miyake Y, Sasaki S, et al. Dietary folate, riboflavin, vitamin B-6, and vitamin B-12 and depressive symptoms in early adolescence: the Ryukyus Child Health Study. Psychosom Med. 2010;72(8):763-768.
7. Merete C, Falcon LM, Tucker KL. Vitamin B6 is associated with depressive symptomatology in Massachusetts elders. J Am Coll Nutr. 2008;27(3):421-427.
8. Skarupski KA, Tangney C, Li H, et al. Longitudinal association of vitamin B-6, folate, and vitamin B-12 with depressive symptoms among older adults over time. Am J Clin Nutr. 2010;92(2):330-335.
9. Corken M, Porter J. Is vitamin B(6) deficiency an under-recognized risk in patients receiving haemodialysis? A systematic review: 2000-2010. Nephrology (Carlton). 2011;16(7):619-625.
10. Wilson SM, Bivins BN, Russell KA, et al. Oral contraceptive use: impact on folate, vitamin B6, and vitamin B12 status. Nutr Rev. 2011;69(10):572-583.
11. Coppen A, Bolander-Gouaille C. Treatment of depression: time to consider folic acid and vitamin B12. J Psychopharmacol. 2005;19(1):59-65.
12. Tolmunen T, Voutilainen S, Hintikka J, et al. Dietary folate and depressive symptoms are associated in middle-aged Finnish men. J Nutr. 2003;133(10):3233-3236.
13. Gilbody S, Lightfoot T, Sheldon T. Is low folate a risk factor for depression? A meta-analysis and exploration of heterogeneity. J Epidemiol Community Health. 2007;61(7):631-637.
14. Rösche J, Uhlmann C, Fröscher W. Low serum folate levels as a risk factor for depressive mood in patients with chronic epilepsy. J Neuropsychiatry Clin Neurosci. 2003;15(1):64-66.
15. Kale A, Naphade N, Sapkale S, et al. Reduced folic acid, vitamin B12 and docosahexaenoic acid and increased homocysteine and cortisol in never-medicated schizophrenia patients: implications for altered one-carbon metabolism. Psychiatry Res. 2010;175(1-2):47-53.
16. Gilbody S, Lewis S, Lightfoot T. Methylenetetrahydrofolate reductase (MTHFR) genetic polymorphisms and psychiatric disorders: a HuGE review. Am J Epidemiol. 2007;165(1):1-13.
17. Di Palma C, Urani R, Agricola R, et al. Is methylfolate effective in relieving major depression in chronic alcoholics? A hypothesis of treatment. Curr Ther Res Clin Exp. 1994;55(5):559-568.
18. Papakostas GI, Shelton RC, Zajecka JM, et al. l-Methylfolate as adjunctive therapy for ssri-resistant major depression: results of two randomized, double-blind, parallel-sequential trials. Am J Psychiatry. 2012;169(12):1267-1274.
19. Baggott JE, Oster RA, Tamura T. Meta-analysis of cancer risk in folic acid supplementation trials. Cancer Epidemiol. 2012;36(1):78-81.
20. Figueiredo JC, Grau MV, Haile RW, et al. Folic acid and risk of prostate cancer: results from a randomized clinical trial. J Natl Cancer Inst. 2009;101(6):432-435.
21. Kate N, Grover S, Agarwal M. Does B12 deficiency lead to lack of treatment response to conventional antidepressants? Psychiatry (Edgmont). 2010;7(11):42-44.
22. Hintikka J, Tolmunen T, Tanskanen A, et al. High vitamin B12 level and good treatment outcome may be associated in major depressive disorder. BMC Psychiatry. 2003;3:17.-
23. Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
24. Bar-Shai M, Gott D, Marmor S. Acute psychotic depression as a sole manifestation of vitamin B12 deficiency. Psychosomatics. 2011;52(4):384-386.
25. Sharma V, Biswas D. Cobalamin deficiency presenting as obsessive compulsive disorder: case report. Gen Hosp Psychiatry. 2012;34(5):578.e7-e8.
26. Vogiatzoglou A, Refsum H, Johnston C, et al. Vitamin B12 status and rate of brain volume loss in community-dwelling elderly. Neurology. 2008;71(11):826-832.
27. Smith A, Di Primio G, Humphrey-Murto S. Scurvy in the developed world. CMAJ. 2011;183(11):E752-E725.
28. Payne ME, Steck SE, George RR, et al. Fruit, vegetable, and antioxidant intakes are lower in older adults with depression. J Acad Nutr Diet. 2012;112(12):2022-2027.
29. Dadheech G, Mishra S, Gautam S, et al. Oxidative stress, α-tocopherol, ascorbic acid and reduced glutathione status in schizophrenics. Indian J Clin Biochem. 2006;21(2):34-38.
30. Hinds TS, West WL, Knight EM. Carotenoids and retinoids: a review of research clinical, and public health applications. J Clin Pharmacol. 1997;37(7):551-558.
31. Thacher TD, Clarke BL. Vitamin D insufficiency. Mayo Clin Proc. 2011;86(1):50-60.
32. Berk M, Sanders KM, Pasco JA, et al. Vitamin D deficiency may play a role in depression. Med Hypotheses. 2007;69(6):1316-1319.
33. Eyles DW, Smith S, Kinobe R, et al. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat. 2005;29(1):21-30.
34. Sen CK, Khanna S, Roy S. Tocotrienol: the natural vitamin E to defend the nervous system? Ann N Y Acad Sci. 2004;1031:127-142.
35. Owen AJ, Batterham MJ, Probst YC, et al. Low plasma vitamin E levels in major depression: diet or disease? Eur J Clin Nutr. 2005;59(2):304-306.
36. Panemangalore M, Lee CJ. Evaluation of the indices of retinol and alpha-tocopherol status in free-living elderly. J Gerontol. 1992;47(3):B98-B104.
37. Sánchez-Villegas A, Delgado-Rodríguez M, Alonso A, et al. Association of the Mediterranean dietary pattern with the incidence of depression: the Seguimiento Universidad de Navarra/University of Navarra follow-up (SUN) cohort. Arch Gen Psychiatry. 2009;66(10):1090-1098.
38. Jacka FN, Pasco JA, Mykletun A, et al. Association of Western and traditional diets with depression and anxiety in women. Am J Psychiatry. 2010;167(3):305-311.
1. Moshfegh A, Goldman J, Cleveland L. United States Department of Agriculture, Agricultural Research Service. What we eat in America NHANES 2001-2002: Usual nutrient intakes from food compared to dietary reference intakes. http://www.ars.usda.gov/SP2UserFiles/Place/12355000/pdf/0102/usualintaketables2001-02.pdf. Published September 2005. Accessed November 27, 2012.
2. Page GL, Laight D, Cummings MH. Thiamine deficiency in diabetes mellitus and the impact of thiamine replacement on glucose metabolism and vascular disease. Int J Clin Pract. 2011;65(6):684-690.
3. McCormick LM, Buchanan JR, Onwuameze OE, et al. Beyond alcoholism: Wernicke-Korsakoff syndrome in patients with psychiatric disorders. Cogn Behav Neurol. 2011;24(4):209-216.
4. Powers HJ. Riboflavin (vitamin B-2) and health. Am J Clin Nutr. 2003;77(6):1352-1360.
5. Naghashpour M, Amani R, Nutr R, et al. Riboflavin status and its association with serum hs-CRP levels among clinical nurses with depression. J Am Coll Nutr. 2011;30(5):340-347.
6. Murakami K, Miyake Y, Sasaki S, et al. Dietary folate, riboflavin, vitamin B-6, and vitamin B-12 and depressive symptoms in early adolescence: the Ryukyus Child Health Study. Psychosom Med. 2010;72(8):763-768.
7. Merete C, Falcon LM, Tucker KL. Vitamin B6 is associated with depressive symptomatology in Massachusetts elders. J Am Coll Nutr. 2008;27(3):421-427.
8. Skarupski KA, Tangney C, Li H, et al. Longitudinal association of vitamin B-6, folate, and vitamin B-12 with depressive symptoms among older adults over time. Am J Clin Nutr. 2010;92(2):330-335.
9. Corken M, Porter J. Is vitamin B(6) deficiency an under-recognized risk in patients receiving haemodialysis? A systematic review: 2000-2010. Nephrology (Carlton). 2011;16(7):619-625.
10. Wilson SM, Bivins BN, Russell KA, et al. Oral contraceptive use: impact on folate, vitamin B6, and vitamin B12 status. Nutr Rev. 2011;69(10):572-583.
11. Coppen A, Bolander-Gouaille C. Treatment of depression: time to consider folic acid and vitamin B12. J Psychopharmacol. 2005;19(1):59-65.
12. Tolmunen T, Voutilainen S, Hintikka J, et al. Dietary folate and depressive symptoms are associated in middle-aged Finnish men. J Nutr. 2003;133(10):3233-3236.
13. Gilbody S, Lightfoot T, Sheldon T. Is low folate a risk factor for depression? A meta-analysis and exploration of heterogeneity. J Epidemiol Community Health. 2007;61(7):631-637.
14. Rösche J, Uhlmann C, Fröscher W. Low serum folate levels as a risk factor for depressive mood in patients with chronic epilepsy. J Neuropsychiatry Clin Neurosci. 2003;15(1):64-66.
15. Kale A, Naphade N, Sapkale S, et al. Reduced folic acid, vitamin B12 and docosahexaenoic acid and increased homocysteine and cortisol in never-medicated schizophrenia patients: implications for altered one-carbon metabolism. Psychiatry Res. 2010;175(1-2):47-53.
16. Gilbody S, Lewis S, Lightfoot T. Methylenetetrahydrofolate reductase (MTHFR) genetic polymorphisms and psychiatric disorders: a HuGE review. Am J Epidemiol. 2007;165(1):1-13.
17. Di Palma C, Urani R, Agricola R, et al. Is methylfolate effective in relieving major depression in chronic alcoholics? A hypothesis of treatment. Curr Ther Res Clin Exp. 1994;55(5):559-568.
18. Papakostas GI, Shelton RC, Zajecka JM, et al. l-Methylfolate as adjunctive therapy for ssri-resistant major depression: results of two randomized, double-blind, parallel-sequential trials. Am J Psychiatry. 2012;169(12):1267-1274.
19. Baggott JE, Oster RA, Tamura T. Meta-analysis of cancer risk in folic acid supplementation trials. Cancer Epidemiol. 2012;36(1):78-81.
20. Figueiredo JC, Grau MV, Haile RW, et al. Folic acid and risk of prostate cancer: results from a randomized clinical trial. J Natl Cancer Inst. 2009;101(6):432-435.
21. Kate N, Grover S, Agarwal M. Does B12 deficiency lead to lack of treatment response to conventional antidepressants? Psychiatry (Edgmont). 2010;7(11):42-44.
22. Hintikka J, Tolmunen T, Tanskanen A, et al. High vitamin B12 level and good treatment outcome may be associated in major depressive disorder. BMC Psychiatry. 2003;3:17.-
23. Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
24. Bar-Shai M, Gott D, Marmor S. Acute psychotic depression as a sole manifestation of vitamin B12 deficiency. Psychosomatics. 2011;52(4):384-386.
25. Sharma V, Biswas D. Cobalamin deficiency presenting as obsessive compulsive disorder: case report. Gen Hosp Psychiatry. 2012;34(5):578.e7-e8.
26. Vogiatzoglou A, Refsum H, Johnston C, et al. Vitamin B12 status and rate of brain volume loss in community-dwelling elderly. Neurology. 2008;71(11):826-832.
27. Smith A, Di Primio G, Humphrey-Murto S. Scurvy in the developed world. CMAJ. 2011;183(11):E752-E725.
28. Payne ME, Steck SE, George RR, et al. Fruit, vegetable, and antioxidant intakes are lower in older adults with depression. J Acad Nutr Diet. 2012;112(12):2022-2027.
29. Dadheech G, Mishra S, Gautam S, et al. Oxidative stress, α-tocopherol, ascorbic acid and reduced glutathione status in schizophrenics. Indian J Clin Biochem. 2006;21(2):34-38.
30. Hinds TS, West WL, Knight EM. Carotenoids and retinoids: a review of research clinical, and public health applications. J Clin Pharmacol. 1997;37(7):551-558.
31. Thacher TD, Clarke BL. Vitamin D insufficiency. Mayo Clin Proc. 2011;86(1):50-60.
32. Berk M, Sanders KM, Pasco JA, et al. Vitamin D deficiency may play a role in depression. Med Hypotheses. 2007;69(6):1316-1319.
33. Eyles DW, Smith S, Kinobe R, et al. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat. 2005;29(1):21-30.
34. Sen CK, Khanna S, Roy S. Tocotrienol: the natural vitamin E to defend the nervous system? Ann N Y Acad Sci. 2004;1031:127-142.
35. Owen AJ, Batterham MJ, Probst YC, et al. Low plasma vitamin E levels in major depression: diet or disease? Eur J Clin Nutr. 2005;59(2):304-306.
36. Panemangalore M, Lee CJ. Evaluation of the indices of retinol and alpha-tocopherol status in free-living elderly. J Gerontol. 1992;47(3):B98-B104.
37. Sánchez-Villegas A, Delgado-Rodríguez M, Alonso A, et al. Association of the Mediterranean dietary pattern with the incidence of depression: the Seguimiento Universidad de Navarra/University of Navarra follow-up (SUN) cohort. Arch Gen Psychiatry. 2009;66(10):1090-1098.
38. Jacka FN, Pasco JA, Mykletun A, et al. Association of Western and traditional diets with depression and anxiety in women. Am J Psychiatry. 2010;167(3):305-311.
Telepsychiatry: Overcoming barriers to implementation
Discuss this article at www.facebook.com/CurrentPsychiatry
Although many states have substantial health services in urban areas, these services—particularly mental health care—are relatively scarce in rural areas.1 Telepsychiatry, in which clinicians provide mental health care from a distance in real time by using interactive, 2-way, audio-video communication (videoconferencing), could mitigate workforce shortages that affect remote and underserved areas.2 Psychiatry is one of the biggest users of telemedicine, which refers to any combination of communication technology and medicine.3-5 This article discusses telepsychiatry’s effectiveness in providing psychiatric diagnosis and treatment, and the clinical implications of this technology, including improving access, cost, and quality of mental health services.
Outcomes comparable to face-to-face care
Telepsychiatry is used primarily in rural areas or correctional institutions or with underserved populations such as veterans with posttraumatic stress disorder or children. Although the literature generally is weak, there has been more research on psychiatry than other medical specialties because psychiatric clinicians rely on mental status examinations and verbal communications, not physical exams. Telepsychiatry can be considered a part of an evolving “connected health” system that offers many benefits to patients and clinicians (Table).
Table
Benefits of telepsychiatry as part of a ‘connected health’ system
| Available to everyone |
| Health care is provided at the point of convenience |
| Patients are informed and empowered |
| Facilitates patient compliance, continuing education, ease of access into the health care system, and healthy behaviors |
| Clinical data are integrated with longitudinal electronic health records |
| Data are available to patients via his or her personal electronic medical record and authorized clinical providers |
| Data and transactions are secure to greatest practical extent |
| Other telehealth applications with demonstrated efficacy—eg, telephone, internet, e-mail, and text messaging interventions—can be used as well |
Barriers to implementation
Although telepsychiatry offers tremendous promise, implementation has not been widespread or easy. Potential barriers to implementation, such as cost and resistance to change, are associated with acceptance of new technology or practice in health care. In addition, there are several legal, regulatory, and technical barriers.
Institutional barriers. Physicians and other providers may not have access to timely, evidence-based information and may face challenges, such as time constraints, access to technical support, and complexity of large health care institutions, when integrating this information into clinical practice.16 Two studies17 found that after controlling for other barriers—eg, reimbursement and regulatory issues—providers are the most significant initial gatekeepers that affect telemedicine use. When designing a telemedicine system, project managers should prioritize providers’ needs, such as ease of use and incentives.18
States do not cover services provided by other mental health providers, except for Utah’s coverage for social workers. The American Psychiatric Association has 2 suggestions regarding this issue3:
- reimbursement for telepsychiatry services should follow customary charges for delivering the appropriate current procedural terminology code(s)
- a structure for reimbursement of collateral charges, such as technician and line time, should be identified.
Licensure. A physician conducting a telemedicine session with a patient in another state must be licensed in both his or her state and the patient’s state. Nurses and other allied health professionals have similar state licensing constraints. Sanders22 suggests 3 potential solutions:
- establishing a national licensing system
- assigning the responsibility of care to the referring physician, with the consulting physician’s opinion serving as “recommendation only”
- determining that the patient is being “electronically transmitted” to the consultant’s state.
Although telemedicine has embraced many communication technologies, live, interactive, 2-way, audio-video communication—called videoconferencing—is broadly synonymous with telemedicine and, more specifically, telepsychiatry.
Telepsychiatry primarily uses interactive audiovisual conferencing systems over high-bandwidth networks. The central component of interactive telepsychiatry is the codec (coder/decoder), which provides compression, decompression, and synchronization of audio and video signals; both patients and clinicians need a codec. A codec can be a separate device, but personal computer-based codecs are being used more frequently. A typical setup also includes a video camera, microphone, speakers or headset, and 1 or 2 display monitors at both the clinician’s and patient’s end of the system. Often, separate displays or a picture-in-picture display are used to see both outgoing and incoming video. Another consideration is pan-zoom-tilt control of video cameras. This allows clinicians to remotely control his or her view of the patient’s site or control the view being transmitted to the patient.
Historically, interactive telepsychiatry applications have used point-to-point network connections, usually as full or fractional T-1 or integrated services digital network circuits. However, the rapid diffusion of internet and ethernet networks has led to the development of videoconferencing systems that can work over internet protocol (IP) networks. If using an IP network, ensure security by using encrypted codecs or by setting up a virtual private network and/or a virtual local area network (LAN). The principal advantage of IP networks is that by implementing proper security solutions, they can be shared by several applications—eg, internet, e-mail, LAN, etc. This means that the telecommunications network costs can be shared or considered a sunk cost (ie, not an additional cost of the telepsychiatry application).
The U.S. Federal Communications Commission’s (FCC) Universal Service Fund (USF) subsidies can reduce the cost of telepsychiatry network connections. The FCC implemented the USF to bring high bandwidth telecommunications to rural schools, libraries, and health care providers. Funding for the USF is generated from fees paid by telecommunications providers. However, the USF subsidies are not being widely used for several reasons, including a cumbersome application process, limitations on eligible facilities and locations, and questions regarding costs to the health care provider.19
Individual states also have developed funding streams to support telemedicine. The Centers for Medicare and Medicaid Services will pay a facility site fee to the host site (where the patient is located), but only if the site is in a rural area. Providers can charge patients a fee to support telepsychiatry infrastructure and maintenance, but typically this arrangement is not affordable and is not standard practice.
The future
Telepsychiatry’s ability to improve access to mental health care to underserved populations is becoming more evident. Technology is adequate for most uses and is constantly advancing. Numerous applications already have been defined, and more are ripe for exploration. Barriers to implementation are primarily of the human variety and will require a combination of consumer, provider, and governmental advocacy to overcome.
- American Telemedicine Association. www.americantelemed.org.
- International Society for Telemedicine & eHealth. www.isfteh.org.
- American Psychiatric Association. Telepsychiatry internet resources. www.psychiatry.org/practice/professional-interests/underserved-communities/telepsychiatry-internet-resources.
- Mossman D. Practicing psychiatry via Skype: Medicolegal considerations. Current Psychiatry. 2011;10(12):30-32,39.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. President’s New Freedom Commission on Mental Health. Subcommittee on rural issues: background paper. Rockville MD: Substance Abuse and Mental Health Administration; 2004.
2. Antonacci DJ, Bloch RM, Saeed SA, et al. Empirical evidence on the use and effectiveness of telepsychiatry via videoconferencing: implications for forensic and correctional psychiatry. Behav Sci Law. 2008;26(3):253-269.
3. American Psychiatric Association. Resource document on telepsychiatry via videoconferencing. http://www.psychiatry.med.uwo.ca/ecp/info/toronto/telepsych/Appendix%20II.htm. Accessed November 5 2012.
4. Grigsby J, Rigby M, Hiemstra A, et al. Telemedicine/telehealth: an international perspective. The diffusion of telemedicine. Telemed J E Health. 2002;8(1):79-94.
5. Krupinski E, Nypaver M, Poropatich R, et al. Telemedicine/telehealth: an international perspective. Clinical applications in telemedicine/telehealth. Telemed J E Health. 2002;8(1):13-34.
6. Diamond JM, Bloch RM. Telepsychiatry assessments of child or adolescent behavior disorders: a review of evidence and issues. Telemed J E Health. 2010;16(6):712-716.
7. Buono S, Città S. Tele-assistance in intellectual disability. J Telemed Telecare. 2007;13(5):241-245.
8. Manguno-Mire GM, Thompson JW, Jr, Shore JH, et al. The use of telemedicine to evaluate competency to stand trial: a preliminary randomized controlled study. J Am Acad Psychiatry Law. 2007;35(4):481-489.
9. Fortney JC, Maciejewski ML, Tripathi SP, et al. A budget impact analysis of telemedicine-based collaborative care for depression. Med Care. 2011;49(9):872-880.
10. Pyne JM, Fortney JC, Tripathi SP, et al. Cost-effectiveness analysis of a rural telemedicine collaborative care intervention for depression. Arch Gen Psychiatry. 2010;67(8):812-821.
11. Morland LA, Greene CJ, Rosen CS, et al. Telemedicine for anger management therapy in a rural population of combat veterans with posttraumatic stress disorder: a randomized noninferiority trial. J Clin Psychiatry. 2010;71(7):855-863.
12. Mitchell JE, Crosby RD, Wonderlich SA, et al. A randomized trial comparing the efficacy of cognitive-behavioral therapy for bulimia nervosa delivered via telemedicine versus face-to-face. Behav Res Ther. 2008;46(5):581-592.
13. Steel K, Cox D, Garry H. Therapeutic videoconferencing interventions for the treatment of long-term conditions. J Telemed Telecare. 2011;17(3):109-117.
14. Greene CJ, Morland LA, Macdonald A, et al. How does tele-mental health affect group therapy process? Secondary analysis of a noninferiority trial. J Consult Clin Psychol. 2010;78(5):746-750.
15. Morland LA, Greene CJ, Grubbs K, et al. Therapist adherence to manualized cognitive-behavioral therapy for anger management delivered to veterans with PTSD via videoconferencing. J Clin Psychol. 2011;67(6):629-638.
16. Saeed SA, Diamond J, Bloch RM. Use of telepsychiatry to improve care for people with mental illness in rural North Carolina. N C Med J. 2011;72(3):219-222.
17. Whitten PS, Mackert MS. Addressing telehealth’s foremost barrier: provider as initial gatekeeper. Int J Technol Assess Health Care. 2005;21(4):517-521.
18. Coleman JR. HMOs and the future of telemedicine and telehealth: part 2. Case Manager. 2002;13(4):38-43.
19. Puskin DS. Telemedicine: follow the money modalities. Online J Issues Nurs. 2001;6(3):2.-
20. American Telemedicine Association. Medicare payment of telemedicine and telehealth services. http://www.americantelemed.org/files/public/membergroups/businessfinance/reimbursement/
BF_MedicarePaymentofTelemedicine.pdf. Published May 15 2006. Accessed November 5, 2012.
21. Fishbein M. Developing effective behavior change interventions: some lessons learned from behavioral research. NIDA Res Monogr. 1995;155:246-261.
22. Sanders JH. Telemedicine: challenges to implementation. Paper presented at: Rural Telemedicine Workshop; November 4 1993; Washington, DC.
23. Kumekawa JK. Health information privacy protection: crisis or common sense? Online J Issues Nurs. 2001;6(3):3.-
Discuss this article at www.facebook.com/CurrentPsychiatry
Although many states have substantial health services in urban areas, these services—particularly mental health care—are relatively scarce in rural areas.1 Telepsychiatry, in which clinicians provide mental health care from a distance in real time by using interactive, 2-way, audio-video communication (videoconferencing), could mitigate workforce shortages that affect remote and underserved areas.2 Psychiatry is one of the biggest users of telemedicine, which refers to any combination of communication technology and medicine.3-5 This article discusses telepsychiatry’s effectiveness in providing psychiatric diagnosis and treatment, and the clinical implications of this technology, including improving access, cost, and quality of mental health services.
Outcomes comparable to face-to-face care
Telepsychiatry is used primarily in rural areas or correctional institutions or with underserved populations such as veterans with posttraumatic stress disorder or children. Although the literature generally is weak, there has been more research on psychiatry than other medical specialties because psychiatric clinicians rely on mental status examinations and verbal communications, not physical exams. Telepsychiatry can be considered a part of an evolving “connected health” system that offers many benefits to patients and clinicians (Table).
Table
Benefits of telepsychiatry as part of a ‘connected health’ system
| Available to everyone |
| Health care is provided at the point of convenience |
| Patients are informed and empowered |
| Facilitates patient compliance, continuing education, ease of access into the health care system, and healthy behaviors |
| Clinical data are integrated with longitudinal electronic health records |
| Data are available to patients via his or her personal electronic medical record and authorized clinical providers |
| Data and transactions are secure to greatest practical extent |
| Other telehealth applications with demonstrated efficacy—eg, telephone, internet, e-mail, and text messaging interventions—can be used as well |
Barriers to implementation
Although telepsychiatry offers tremendous promise, implementation has not been widespread or easy. Potential barriers to implementation, such as cost and resistance to change, are associated with acceptance of new technology or practice in health care. In addition, there are several legal, regulatory, and technical barriers.
Institutional barriers. Physicians and other providers may not have access to timely, evidence-based information and may face challenges, such as time constraints, access to technical support, and complexity of large health care institutions, when integrating this information into clinical practice.16 Two studies17 found that after controlling for other barriers—eg, reimbursement and regulatory issues—providers are the most significant initial gatekeepers that affect telemedicine use. When designing a telemedicine system, project managers should prioritize providers’ needs, such as ease of use and incentives.18
States do not cover services provided by other mental health providers, except for Utah’s coverage for social workers. The American Psychiatric Association has 2 suggestions regarding this issue3:
- reimbursement for telepsychiatry services should follow customary charges for delivering the appropriate current procedural terminology code(s)
- a structure for reimbursement of collateral charges, such as technician and line time, should be identified.
Licensure. A physician conducting a telemedicine session with a patient in another state must be licensed in both his or her state and the patient’s state. Nurses and other allied health professionals have similar state licensing constraints. Sanders22 suggests 3 potential solutions:
- establishing a national licensing system
- assigning the responsibility of care to the referring physician, with the consulting physician’s opinion serving as “recommendation only”
- determining that the patient is being “electronically transmitted” to the consultant’s state.
Although telemedicine has embraced many communication technologies, live, interactive, 2-way, audio-video communication—called videoconferencing—is broadly synonymous with telemedicine and, more specifically, telepsychiatry.
Telepsychiatry primarily uses interactive audiovisual conferencing systems over high-bandwidth networks. The central component of interactive telepsychiatry is the codec (coder/decoder), which provides compression, decompression, and synchronization of audio and video signals; both patients and clinicians need a codec. A codec can be a separate device, but personal computer-based codecs are being used more frequently. A typical setup also includes a video camera, microphone, speakers or headset, and 1 or 2 display monitors at both the clinician’s and patient’s end of the system. Often, separate displays or a picture-in-picture display are used to see both outgoing and incoming video. Another consideration is pan-zoom-tilt control of video cameras. This allows clinicians to remotely control his or her view of the patient’s site or control the view being transmitted to the patient.
Historically, interactive telepsychiatry applications have used point-to-point network connections, usually as full or fractional T-1 or integrated services digital network circuits. However, the rapid diffusion of internet and ethernet networks has led to the development of videoconferencing systems that can work over internet protocol (IP) networks. If using an IP network, ensure security by using encrypted codecs or by setting up a virtual private network and/or a virtual local area network (LAN). The principal advantage of IP networks is that by implementing proper security solutions, they can be shared by several applications—eg, internet, e-mail, LAN, etc. This means that the telecommunications network costs can be shared or considered a sunk cost (ie, not an additional cost of the telepsychiatry application).
The U.S. Federal Communications Commission’s (FCC) Universal Service Fund (USF) subsidies can reduce the cost of telepsychiatry network connections. The FCC implemented the USF to bring high bandwidth telecommunications to rural schools, libraries, and health care providers. Funding for the USF is generated from fees paid by telecommunications providers. However, the USF subsidies are not being widely used for several reasons, including a cumbersome application process, limitations on eligible facilities and locations, and questions regarding costs to the health care provider.19
Individual states also have developed funding streams to support telemedicine. The Centers for Medicare and Medicaid Services will pay a facility site fee to the host site (where the patient is located), but only if the site is in a rural area. Providers can charge patients a fee to support telepsychiatry infrastructure and maintenance, but typically this arrangement is not affordable and is not standard practice.
The future
Telepsychiatry’s ability to improve access to mental health care to underserved populations is becoming more evident. Technology is adequate for most uses and is constantly advancing. Numerous applications already have been defined, and more are ripe for exploration. Barriers to implementation are primarily of the human variety and will require a combination of consumer, provider, and governmental advocacy to overcome.
- American Telemedicine Association. www.americantelemed.org.
- International Society for Telemedicine & eHealth. www.isfteh.org.
- American Psychiatric Association. Telepsychiatry internet resources. www.psychiatry.org/practice/professional-interests/underserved-communities/telepsychiatry-internet-resources.
- Mossman D. Practicing psychiatry via Skype: Medicolegal considerations. Current Psychiatry. 2011;10(12):30-32,39.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Although many states have substantial health services in urban areas, these services—particularly mental health care—are relatively scarce in rural areas.1 Telepsychiatry, in which clinicians provide mental health care from a distance in real time by using interactive, 2-way, audio-video communication (videoconferencing), could mitigate workforce shortages that affect remote and underserved areas.2 Psychiatry is one of the biggest users of telemedicine, which refers to any combination of communication technology and medicine.3-5 This article discusses telepsychiatry’s effectiveness in providing psychiatric diagnosis and treatment, and the clinical implications of this technology, including improving access, cost, and quality of mental health services.
Outcomes comparable to face-to-face care
Telepsychiatry is used primarily in rural areas or correctional institutions or with underserved populations such as veterans with posttraumatic stress disorder or children. Although the literature generally is weak, there has been more research on psychiatry than other medical specialties because psychiatric clinicians rely on mental status examinations and verbal communications, not physical exams. Telepsychiatry can be considered a part of an evolving “connected health” system that offers many benefits to patients and clinicians (Table).
Table
Benefits of telepsychiatry as part of a ‘connected health’ system
| Available to everyone |
| Health care is provided at the point of convenience |
| Patients are informed and empowered |
| Facilitates patient compliance, continuing education, ease of access into the health care system, and healthy behaviors |
| Clinical data are integrated with longitudinal electronic health records |
| Data are available to patients via his or her personal electronic medical record and authorized clinical providers |
| Data and transactions are secure to greatest practical extent |
| Other telehealth applications with demonstrated efficacy—eg, telephone, internet, e-mail, and text messaging interventions—can be used as well |
Barriers to implementation
Although telepsychiatry offers tremendous promise, implementation has not been widespread or easy. Potential barriers to implementation, such as cost and resistance to change, are associated with acceptance of new technology or practice in health care. In addition, there are several legal, regulatory, and technical barriers.
Institutional barriers. Physicians and other providers may not have access to timely, evidence-based information and may face challenges, such as time constraints, access to technical support, and complexity of large health care institutions, when integrating this information into clinical practice.16 Two studies17 found that after controlling for other barriers—eg, reimbursement and regulatory issues—providers are the most significant initial gatekeepers that affect telemedicine use. When designing a telemedicine system, project managers should prioritize providers’ needs, such as ease of use and incentives.18
States do not cover services provided by other mental health providers, except for Utah’s coverage for social workers. The American Psychiatric Association has 2 suggestions regarding this issue3:
- reimbursement for telepsychiatry services should follow customary charges for delivering the appropriate current procedural terminology code(s)
- a structure for reimbursement of collateral charges, such as technician and line time, should be identified.
Licensure. A physician conducting a telemedicine session with a patient in another state must be licensed in both his or her state and the patient’s state. Nurses and other allied health professionals have similar state licensing constraints. Sanders22 suggests 3 potential solutions:
- establishing a national licensing system
- assigning the responsibility of care to the referring physician, with the consulting physician’s opinion serving as “recommendation only”
- determining that the patient is being “electronically transmitted” to the consultant’s state.
Although telemedicine has embraced many communication technologies, live, interactive, 2-way, audio-video communication—called videoconferencing—is broadly synonymous with telemedicine and, more specifically, telepsychiatry.
Telepsychiatry primarily uses interactive audiovisual conferencing systems over high-bandwidth networks. The central component of interactive telepsychiatry is the codec (coder/decoder), which provides compression, decompression, and synchronization of audio and video signals; both patients and clinicians need a codec. A codec can be a separate device, but personal computer-based codecs are being used more frequently. A typical setup also includes a video camera, microphone, speakers or headset, and 1 or 2 display monitors at both the clinician’s and patient’s end of the system. Often, separate displays or a picture-in-picture display are used to see both outgoing and incoming video. Another consideration is pan-zoom-tilt control of video cameras. This allows clinicians to remotely control his or her view of the patient’s site or control the view being transmitted to the patient.
Historically, interactive telepsychiatry applications have used point-to-point network connections, usually as full or fractional T-1 or integrated services digital network circuits. However, the rapid diffusion of internet and ethernet networks has led to the development of videoconferencing systems that can work over internet protocol (IP) networks. If using an IP network, ensure security by using encrypted codecs or by setting up a virtual private network and/or a virtual local area network (LAN). The principal advantage of IP networks is that by implementing proper security solutions, they can be shared by several applications—eg, internet, e-mail, LAN, etc. This means that the telecommunications network costs can be shared or considered a sunk cost (ie, not an additional cost of the telepsychiatry application).
The U.S. Federal Communications Commission’s (FCC) Universal Service Fund (USF) subsidies can reduce the cost of telepsychiatry network connections. The FCC implemented the USF to bring high bandwidth telecommunications to rural schools, libraries, and health care providers. Funding for the USF is generated from fees paid by telecommunications providers. However, the USF subsidies are not being widely used for several reasons, including a cumbersome application process, limitations on eligible facilities and locations, and questions regarding costs to the health care provider.19
Individual states also have developed funding streams to support telemedicine. The Centers for Medicare and Medicaid Services will pay a facility site fee to the host site (where the patient is located), but only if the site is in a rural area. Providers can charge patients a fee to support telepsychiatry infrastructure and maintenance, but typically this arrangement is not affordable and is not standard practice.
The future
Telepsychiatry’s ability to improve access to mental health care to underserved populations is becoming more evident. Technology is adequate for most uses and is constantly advancing. Numerous applications already have been defined, and more are ripe for exploration. Barriers to implementation are primarily of the human variety and will require a combination of consumer, provider, and governmental advocacy to overcome.
- American Telemedicine Association. www.americantelemed.org.
- International Society for Telemedicine & eHealth. www.isfteh.org.
- American Psychiatric Association. Telepsychiatry internet resources. www.psychiatry.org/practice/professional-interests/underserved-communities/telepsychiatry-internet-resources.
- Mossman D. Practicing psychiatry via Skype: Medicolegal considerations. Current Psychiatry. 2011;10(12):30-32,39.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. President’s New Freedom Commission on Mental Health. Subcommittee on rural issues: background paper. Rockville MD: Substance Abuse and Mental Health Administration; 2004.
2. Antonacci DJ, Bloch RM, Saeed SA, et al. Empirical evidence on the use and effectiveness of telepsychiatry via videoconferencing: implications for forensic and correctional psychiatry. Behav Sci Law. 2008;26(3):253-269.
3. American Psychiatric Association. Resource document on telepsychiatry via videoconferencing. http://www.psychiatry.med.uwo.ca/ecp/info/toronto/telepsych/Appendix%20II.htm. Accessed November 5 2012.
4. Grigsby J, Rigby M, Hiemstra A, et al. Telemedicine/telehealth: an international perspective. The diffusion of telemedicine. Telemed J E Health. 2002;8(1):79-94.
5. Krupinski E, Nypaver M, Poropatich R, et al. Telemedicine/telehealth: an international perspective. Clinical applications in telemedicine/telehealth. Telemed J E Health. 2002;8(1):13-34.
6. Diamond JM, Bloch RM. Telepsychiatry assessments of child or adolescent behavior disorders: a review of evidence and issues. Telemed J E Health. 2010;16(6):712-716.
7. Buono S, Città S. Tele-assistance in intellectual disability. J Telemed Telecare. 2007;13(5):241-245.
8. Manguno-Mire GM, Thompson JW, Jr, Shore JH, et al. The use of telemedicine to evaluate competency to stand trial: a preliminary randomized controlled study. J Am Acad Psychiatry Law. 2007;35(4):481-489.
9. Fortney JC, Maciejewski ML, Tripathi SP, et al. A budget impact analysis of telemedicine-based collaborative care for depression. Med Care. 2011;49(9):872-880.
10. Pyne JM, Fortney JC, Tripathi SP, et al. Cost-effectiveness analysis of a rural telemedicine collaborative care intervention for depression. Arch Gen Psychiatry. 2010;67(8):812-821.
11. Morland LA, Greene CJ, Rosen CS, et al. Telemedicine for anger management therapy in a rural population of combat veterans with posttraumatic stress disorder: a randomized noninferiority trial. J Clin Psychiatry. 2010;71(7):855-863.
12. Mitchell JE, Crosby RD, Wonderlich SA, et al. A randomized trial comparing the efficacy of cognitive-behavioral therapy for bulimia nervosa delivered via telemedicine versus face-to-face. Behav Res Ther. 2008;46(5):581-592.
13. Steel K, Cox D, Garry H. Therapeutic videoconferencing interventions for the treatment of long-term conditions. J Telemed Telecare. 2011;17(3):109-117.
14. Greene CJ, Morland LA, Macdonald A, et al. How does tele-mental health affect group therapy process? Secondary analysis of a noninferiority trial. J Consult Clin Psychol. 2010;78(5):746-750.
15. Morland LA, Greene CJ, Grubbs K, et al. Therapist adherence to manualized cognitive-behavioral therapy for anger management delivered to veterans with PTSD via videoconferencing. J Clin Psychol. 2011;67(6):629-638.
16. Saeed SA, Diamond J, Bloch RM. Use of telepsychiatry to improve care for people with mental illness in rural North Carolina. N C Med J. 2011;72(3):219-222.
17. Whitten PS, Mackert MS. Addressing telehealth’s foremost barrier: provider as initial gatekeeper. Int J Technol Assess Health Care. 2005;21(4):517-521.
18. Coleman JR. HMOs and the future of telemedicine and telehealth: part 2. Case Manager. 2002;13(4):38-43.
19. Puskin DS. Telemedicine: follow the money modalities. Online J Issues Nurs. 2001;6(3):2.-
20. American Telemedicine Association. Medicare payment of telemedicine and telehealth services. http://www.americantelemed.org/files/public/membergroups/businessfinance/reimbursement/
BF_MedicarePaymentofTelemedicine.pdf. Published May 15 2006. Accessed November 5, 2012.
21. Fishbein M. Developing effective behavior change interventions: some lessons learned from behavioral research. NIDA Res Monogr. 1995;155:246-261.
22. Sanders JH. Telemedicine: challenges to implementation. Paper presented at: Rural Telemedicine Workshop; November 4 1993; Washington, DC.
23. Kumekawa JK. Health information privacy protection: crisis or common sense? Online J Issues Nurs. 2001;6(3):3.-
1. President’s New Freedom Commission on Mental Health. Subcommittee on rural issues: background paper. Rockville MD: Substance Abuse and Mental Health Administration; 2004.
2. Antonacci DJ, Bloch RM, Saeed SA, et al. Empirical evidence on the use and effectiveness of telepsychiatry via videoconferencing: implications for forensic and correctional psychiatry. Behav Sci Law. 2008;26(3):253-269.
3. American Psychiatric Association. Resource document on telepsychiatry via videoconferencing. http://www.psychiatry.med.uwo.ca/ecp/info/toronto/telepsych/Appendix%20II.htm. Accessed November 5 2012.
4. Grigsby J, Rigby M, Hiemstra A, et al. Telemedicine/telehealth: an international perspective. The diffusion of telemedicine. Telemed J E Health. 2002;8(1):79-94.
5. Krupinski E, Nypaver M, Poropatich R, et al. Telemedicine/telehealth: an international perspective. Clinical applications in telemedicine/telehealth. Telemed J E Health. 2002;8(1):13-34.
6. Diamond JM, Bloch RM. Telepsychiatry assessments of child or adolescent behavior disorders: a review of evidence and issues. Telemed J E Health. 2010;16(6):712-716.
7. Buono S, Città S. Tele-assistance in intellectual disability. J Telemed Telecare. 2007;13(5):241-245.
8. Manguno-Mire GM, Thompson JW, Jr, Shore JH, et al. The use of telemedicine to evaluate competency to stand trial: a preliminary randomized controlled study. J Am Acad Psychiatry Law. 2007;35(4):481-489.
9. Fortney JC, Maciejewski ML, Tripathi SP, et al. A budget impact analysis of telemedicine-based collaborative care for depression. Med Care. 2011;49(9):872-880.
10. Pyne JM, Fortney JC, Tripathi SP, et al. Cost-effectiveness analysis of a rural telemedicine collaborative care intervention for depression. Arch Gen Psychiatry. 2010;67(8):812-821.
11. Morland LA, Greene CJ, Rosen CS, et al. Telemedicine for anger management therapy in a rural population of combat veterans with posttraumatic stress disorder: a randomized noninferiority trial. J Clin Psychiatry. 2010;71(7):855-863.
12. Mitchell JE, Crosby RD, Wonderlich SA, et al. A randomized trial comparing the efficacy of cognitive-behavioral therapy for bulimia nervosa delivered via telemedicine versus face-to-face. Behav Res Ther. 2008;46(5):581-592.
13. Steel K, Cox D, Garry H. Therapeutic videoconferencing interventions for the treatment of long-term conditions. J Telemed Telecare. 2011;17(3):109-117.
14. Greene CJ, Morland LA, Macdonald A, et al. How does tele-mental health affect group therapy process? Secondary analysis of a noninferiority trial. J Consult Clin Psychol. 2010;78(5):746-750.
15. Morland LA, Greene CJ, Grubbs K, et al. Therapist adherence to manualized cognitive-behavioral therapy for anger management delivered to veterans with PTSD via videoconferencing. J Clin Psychol. 2011;67(6):629-638.
16. Saeed SA, Diamond J, Bloch RM. Use of telepsychiatry to improve care for people with mental illness in rural North Carolina. N C Med J. 2011;72(3):219-222.
17. Whitten PS, Mackert MS. Addressing telehealth’s foremost barrier: provider as initial gatekeeper. Int J Technol Assess Health Care. 2005;21(4):517-521.
18. Coleman JR. HMOs and the future of telemedicine and telehealth: part 2. Case Manager. 2002;13(4):38-43.
19. Puskin DS. Telemedicine: follow the money modalities. Online J Issues Nurs. 2001;6(3):2.-
20. American Telemedicine Association. Medicare payment of telemedicine and telehealth services. http://www.americantelemed.org/files/public/membergroups/businessfinance/reimbursement/
BF_MedicarePaymentofTelemedicine.pdf. Published May 15 2006. Accessed November 5, 2012.
21. Fishbein M. Developing effective behavior change interventions: some lessons learned from behavioral research. NIDA Res Monogr. 1995;155:246-261.
22. Sanders JH. Telemedicine: challenges to implementation. Paper presented at: Rural Telemedicine Workshop; November 4 1993; Washington, DC.
23. Kumekawa JK. Health information privacy protection: crisis or common sense? Online J Issues Nurs. 2001;6(3):3.-
Monoamine oxidase inhibitors: Forgotten treatment for depression
Discuss this article at www.facebook.com/CurrentPsychiatry
For patients with major depressive disorder (MDD), monoamine oxidase inhibitors (MAOIs) have efficacy comparable to that of other antidepressants. However, concerns about side effects—particularly hypertensive crisis—and drug-drug interactions have led clinicians to prescribe MAOIs less often than newer antidepressants. A 1999 survey of 573 Michigan psychiatrists found that 30% had not prescribed an MAOI within the past 3 years, and 12% had never prescribed an MAOI.1 Although there are challenges to using these agents, we prefer prescribing MAOIs to depressed patients who have not responded to previous antidepressant trials over trying untested antidepressant combinations.
Currently, MAOIs are used primarily for patients who have not responded to other antidepressant trials and are considered treatment resistant. Treatment-resistant depression (TRD) typically is defined as nonresponse to ≥3 adequate antidepressant trials. TRD is a major cause of disability and loss of productivity. These patients tend to do poorly over the long term, with high rates of hospitalization and suicide attempts. Several controlled trials have shown that patients who fail other antidepressants may respond to MAOIs.2-4
Our knowledge regarding MAOIs has grown considerably. We have learned more about depression subtypes that MAOIs may help. As we learned more about dietary restrictions for patients taking MAOIs, the list of “forbidden foods” has decreased. Advances in treating a hypertensive crisis have decreased the need for hospitalization. By educating ourselves and our patients about MAOIs, we can provide another option for treating MDD.
An older antidepressant class
MAOIs were introduced approximately 60 years ago. Their potential for treating depression was discovered when a tuberculosis treatment—iproniazid—was found to reduce depressive symptoms. Researchers determined iproniazid’s antidepressant effects were the result of blocking removal of the amine group by monoamine oxidase (MAO) from dopamine, norepinephrine, and serotonin.5 A second MAOI, tranylcypromine, was discovered when it was found to be ineffective for treating decongestion.6
MAOI use in psychiatric practice has undergone significant changes since these medications were introduced. The discovery of hypertensive crises related to tyramine consumption led to decreased MAOI use, as did the rise of tricyclic antidepressants (TCAs) shortly thereafter. In the 1960s, research compared the relative efficacy of MAOIs to TCAs, and they became second-line antidepressants after the TCAs. In the late 1980s, the introduction of fluoxetine and other selective serotonin reuptake inhibitors (SSRIs) resulted in a significant drop-off in MAOI use.
Pharmacologic effects
MAO is a class of enzymes that initiate oxidation of extracellular neurotransmitters such as serotonin, norepinephrine, and dopamine. MAOIs can be classified based on their relative affinity to MAO as well as their enzyme selectivity. The first distinguishing characteristic is whether the drug binds to MAO in a reversible or irreversible manner. Currently, all MAOIs that are FDA-approved for treating depression bind irreversibly to MAO. As a result, the body must renew its MAO levels before a patient is no longer at risk for a hypertensive crisis, a process that may take up to 2 weeks. Clinicians must take care to ensure their patients avoid foods that contain tyramine and medications contraindicated with MAOIs during this period.
MAOIs also differ from each other in enzyme selectivity. There are 2 subtypes of MAO enzymes—MAOA and MAOB. Generally, the antidepressant activity of MAOIs appears to be directed toward MAOA inhibition. MAOA has been found to be more specific for binding to serotonin and norepinephrine and MAOB to be more specific for phenylethylamine. Dopamine is equally deaminated by both MAOA and MAOB.
Reversible MAOA inhibitors require fewer restrictions on diet or concurrent medications, but efficacy data of reversible MAOA inhibitors is mixed.
Clinical use of MAOIs
Four MAOIs are available in the United States: tranylcypromine, phenelzine, isocarboxazid, and selegiline. Selegiline is the only MAOI available as a transdermal patch. Transdermal administration results in fewer effects on MAO in the gastrointestinal tract, which means no dietary restrictions at the 6 mg/d starting dose, although the manufacturer recommends patients follow the MAO diet at 9 mg/d and 12 mg/d doses.7 Although selegiline is selective for MAOB at low doses, it becomes nonselective at therapeutic doses for depression. Recommended dosages for MAOIs can be found in Table 1.8
Table 1
Recommended dosages of monoamine oxidase inhibitors
| Medication | Starting dosages | Usual therapeutic dosage |
|---|---|---|
| Isocarboxazid | 10 mg twice a day | 30 to 60 mg/d |
| Phenelzine | 15 mg twice a day | 45 to 90 mg/d |
| Selegiline transdermal | 6 mg patch/d | 6 to 12 mg patch/d |
| Tranylcypromine | 10 mg, 2 or 3 times a day | 30 to 60 mg/d |
| Source: Adapted from reference 8 | ||
10,11
Several controlled trials have found a better response rate to MAOI therapy in outpatients with MDD who have not responded to other antidepressants.2,12 In a 6-week, double-blind trial, Vallejo et al10 reported that the TCA imipramine and high-dose phenelzine were equally efficacious in 32 patients with major depression with melancholia. In 32 dysthymic patients, high-dose phenelzine was superior to imipramine. Himmelhoch et al13 compared efficacy of tranylcypromine with that of imipramine in treating anergic bipolar depressive illness. Patients receiving tranylcypromine experienced significantly greater symptomatic improvement and higher global response without increased risk of treatment-emergent hypomania or mania.
Serum monitoring of MAOIs is not clinically indicated and there are no correlations between drug levels and effectiveness.14 In a study that examined the correlation of inhibiting platelet MAO and MAOIs’ antidepressant effects, researchers found that a higher dose of phenelzine (60 mg/d) was significantly better in treating depression and anxiety than a lower dose (30 mg/d), and only the higher dose achieved 80% of platelet MAO inhibition.15 Further studies with other MAOIs did not reproduce this effect and platelet MAO inhibition is not regularly used to assess adequate dosing.
A refined view of side effects
In a hypertensive crisis, patients experience significant hypertension, headaches, tachycardia, diaphoresis, and vomiting. Intravenous phentolamine—an α-adrenergic receptor blocker—can be used as an antidote; often a single dose is effective.16 Alternatively, calcium channel blockers such as nifedipine can be prescribed. A patient can take 10 mg/hour and be observed in the emergency room until symptoms are relieved (usually only 1 or 2 doses are needed) without being admitted to the hospital.
Table 2
Food restrictions with MAOIs
| Severe |
| Aged cheeses Aged meats (pepperoni, sausage, salami) Sauerkraut Soy sauce Fava or broad bean pods Banana peels All beers on tap |
| Use in moderation (≤2 servings/d) |
| Red wine (4 oz) White wine (4 oz) Bottled or canned beers (12 oz) |
| Mild to none |
| Avocados Banana pulp Bouillon Chocolate Fresh cheeses (cottage cheese, cream cheese, processed cheese slices) Fresh or processed meat |
| MAOIs: monoamine oxidase inhibitors Source: Adapted from references 4,17,18 |
Orthostatic hypotension is a common cardiovascular side effect of MAOIs that may lead to dizziness or syncope. Typically this is seen 2 to 3 weeks after initiating MAOI treatment. If hypotension remains a problem, mineralocorticoids can be prescribed with monitoring of serum potassium for hypokalemia. Increasing doses of tranylcypromine can increase supine—but not standing—diastolic blood pressure.19 Distinguish this type of blood pressure elevation from a hypertensive crisis by monitoring blood pressure with the patient sitting and standing and before and after he or she walks for 60 seconds.
Insomnia and day-night shifting—sleeping during the day and staying awake at night—are common and patients often cite these as reasons for discontinuing MAOIs. Many patients who respond to MAOIs report periods of substantial sleepiness in the mid to late afternoon. Table 320 provides a more complete list of reported side effects and their frequencies.
Table 3
MAOIs: Stay vigilant for side effects
| Medication | Common side effects |
|---|---|
| Isocarboxazid | Anxiety, blurred vision, constipation, dizziness, headache, insomnia, mania, somnolence, weight gain, xerostomia |
| Phenelzine | Constipation, disorder of ejaculation and/or orgasm, dizziness, edema, fatigue, headache, hyperreflexia, impotence, elevated values on liver function tests, orthostatic hypotension, sleep disorders, somnolence, tremor, weight gain, xerostomia |
| Selegiline transdermal | Application site reaction, decreased systolic blood pressure, diarrhea, headache, indigestion, insomnia, orthostatic hypotension, weight loss, xerostomia |
| Tranylcypromine | Agitation, anxiety, constipation, diarrhea, dizziness, headache, impotence, insomnia, loss of appetite, mania, nausea, orthostatic hypotension, somnolence, weight gain, xerostomia |
| MAOIs: monoamine oxidase inhibitors Source: Adapted from reference 20 | |
Practice guidelines
The American Psychiatric Association’s practice guidelines for treating major depression state that MAOIs are effective in treating subgroups of patients with MDD with atypical features who have failed TCA trials.21 These guidelines also state that MAOIs have been shown to be effective treatment for some patients who have failed other antidepressants. However, for TRD patients who have not responded to SSRIs or SNRIs, the effectiveness of MAOIs compared with other strategies is unclear.22
MAOIs have been used for >6 decades, and published studies continue to document their efficacy and safety when patients are monitored appropriately.11,12,14,15,25 However, based on our observations we suspect MAOIs are underutilized in clinical practice today. We are concerned that such practices can trickle down into residency training programs. Psychiatric residents typically do not receive adequate training in prescribing MAOIs, largely because many training faculty are not prescribing MAOIs themselves. Despite MAOIs’ limitations, concerns about an increased risk of suicide in patients with TRD26 and the high likelihood of a poor outcome associated with persistent nonresponse to prior treatments must be weighed against the relatively low risk of a hypertensive event with MAOIs.6
- McCabe-Sellers BJ, Staggs CG, Bogle ML. Tyramine in foods and monoamine oxidase inhibitor drugs: a crossroad where medicine, nutrition, pharmacy, and food industry converge. Journal of Food Composition and Analysis. 2006;19(suppl):S58-S65.
- Fiedorowicz JG, Swartz KL. The role of monoamine oxidase inhibitors in current psychiatric practice. J Psychiatr Pract. 2004;10(4):239-248.
- Clomipramine • Anafranil
- Epinephrine • Adrenalin, EpiPen
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Isocarboxazid • Marplan
- Lithium • Eskalith, Lithobid
- Nifedipine • Adalat, Afeditab
- Phenelzine • Nardil
- Phentolamine • OraVerse, Regitine
- Selegiline • EMSAM
- Tranylcypromine • Parnate
Dr. Kosinski reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Rothschild receives grant or research support from Cyberonics, the National Institute of Mental Health, St. Jude Medical, and Takeda, and is a consultant to Allergan, Eli Lilly and Company, GlaxoSmithKline, Noven Pharmaceuticals, Pfizer Inc., Shire Pharmaceuticals, and Sunovion.
1. Balon R, Mufti R, Arfken CL. A survey of prescribing practices for monoamine oxidase inhibitors. Psychiatr Serv. 1999;50(7):945-947.
2. Nolen WA, van de Putte JJ, Dijken WA, et al. Treatment strategy in depression. II. MAO inhibitors in depression resistant to cyclic antidepressants: two controlled crossover studies with tranylcypromine versus L-5-hydroxytryptophan and nomifensine. Acta Psychiatr Scand. 1988;78(6):676-683.
3. McGrath PJ, Stewart JW, Harrison W, et al. Treatment of tricyclic refractory depression with a monoamine oxidase inhibitor antidepressant. Psychopharmacol Bull. 1987;23(1):169-172.
4. Amsterdam JD. Monoamine oxidase inhibitor therapy in severe and resistant depression. Psychiatr Ann. 2006;36(9):607-613.
5. Schildkraut JJ. The catecholamine hypothesis of affective disorders: a review of supporting evidence. Am J Psychiatry. 1965;122(5):509-522.
6. Kennedy SH, Holt A, Baker GB. Monoamine oxidase inhibitors. In: Sadock BJ Sadock VA, eds. Kaplan and Sadock’s comprehensive textbook of psychiatry. 8th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005: 1076–1080.
7. EMSAM [package insert]. Napa CA: Dey Pharm LP; 2011.
8. Amsterdam JD, Chopra M. Monoamine oxidase inhibitors revisited. Psychiatric Ann. 2001;31(6):361-370.
9. Quitkin FM, Stewart JW, McGrath PJ, et al. Phenelzine versus imipramine in the treatment of probable atypical depression: defining syndrome boundaries of selective MAOI responders. Am J Psychiatry. 1988;145(3):306-311.
10. Vallejo J, Gasto C, Catalan R, et al. Double-blind study of imipramine versus phenelzine in melancholias and dysthymic disorders. Br J Psychiatry. 1987;151:639-642.
11. White K, Razani J, Cadow B, et al. Trancylpromine vs. nortriptyline vs. placebo in depressed outpatients: a controlled trial. Psychopharmacology (Berl). 1984;82(3):258-262.
12. Thase ME, Frank E, Mallinger AG, et al. Treatment of imipramine-resistant recurrent depression, III: efficacy of monoamine oxidase inhibitors. J Clin Psychiatry. 1992;53(1):5-11.
13. Himmelhoch JM, Thase ME, Mallinger AG, et al. Tranylcypromine versus imipramine in anergic bipolar depression. Am J Psychiatry. 1991;148(7):910-916.
14. Rothschild AJ. ed. The evidence-based guide to antidepressant medications. Arlington, VA: American Psychiatric Publishing, Inc.; 2012:15–20.
15. Ravaris CL, Nies A, Robinson DS, et al. A multiple-dose, controlled study of phenelzine in depression-anxiety states. Arch Gen Psychiatry. 1976;33(3):347-350.
16. Cockhill LA, Remick RA. Blood pressure effects of monoamine oxidase inhibitors—the highs and lows. Can J Psychiatry. 1987;32(9):803-808.
17. Shulman KI, Walker SE. A reevaluation of dietary restrictions for irreversible monoamine oxidase inhibitors. Psychiatr Ann. 2001;31(6):378-384.
18. Gardner DM, Shulman KI, Walker SE, et al. The making of a user friendly MAOI diet. J Clin Psychiatry. 1996;57(3):99-104.
19. Keck PE, Jr, Carter WP, Nierenberg AA, et al. Acute cardiovascular effects of tranylcypromine: correlation with plasma drug, metabolite, norepinephrine, and MHPG levels. J Clin Psychiatry. 1991;52(6):250-254.
20. Micromedex Healthcare Series [UMass Memorial Healthcare Intranet System]. Version 5.1. Greenwood Village CO: Thomson Reuters (Healthcare) Inc.
21. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder third edition. http://psychiatryonline.org/content.aspx?bookid=28§ionid=1667485. Published October 2010. Accessed October 26, 2012.
22. McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry. 2006;163(9):1531-1541; quiz 1666.
23. Nelson JC, Byck R. Rapid response to lithium in phenelzine non-responders. Br J Psychiatry. 1982;141:85-86.
24. Guze BH, Baxter LR, Jr, Rego J. Refractory depression treated with high doses of monoamine oxidase inhibitor. J Clin Psychiatry. 1987;48(1):31-32.
25. Robinson DS, Gilmor ML, Yang Y, et al. Treatment effects of selegiline transdermal system on symptoms of major depressive disorder: a meta analysis of short term, placebo controlled, efficacy trials. Psychopharmacol Bull. 2007;40(3):15-28.
26. Keller MB, Lavori PW, Rice J, et al. The persistent risk of chronicity in recurrent episodes of nonbipolar major depressive disorder: a prospective follow-up. Am J Psychiatry. 1986;143(1):24-28.
Discuss this article at www.facebook.com/CurrentPsychiatry
For patients with major depressive disorder (MDD), monoamine oxidase inhibitors (MAOIs) have efficacy comparable to that of other antidepressants. However, concerns about side effects—particularly hypertensive crisis—and drug-drug interactions have led clinicians to prescribe MAOIs less often than newer antidepressants. A 1999 survey of 573 Michigan psychiatrists found that 30% had not prescribed an MAOI within the past 3 years, and 12% had never prescribed an MAOI.1 Although there are challenges to using these agents, we prefer prescribing MAOIs to depressed patients who have not responded to previous antidepressant trials over trying untested antidepressant combinations.
Currently, MAOIs are used primarily for patients who have not responded to other antidepressant trials and are considered treatment resistant. Treatment-resistant depression (TRD) typically is defined as nonresponse to ≥3 adequate antidepressant trials. TRD is a major cause of disability and loss of productivity. These patients tend to do poorly over the long term, with high rates of hospitalization and suicide attempts. Several controlled trials have shown that patients who fail other antidepressants may respond to MAOIs.2-4
Our knowledge regarding MAOIs has grown considerably. We have learned more about depression subtypes that MAOIs may help. As we learned more about dietary restrictions for patients taking MAOIs, the list of “forbidden foods” has decreased. Advances in treating a hypertensive crisis have decreased the need for hospitalization. By educating ourselves and our patients about MAOIs, we can provide another option for treating MDD.
An older antidepressant class
MAOIs were introduced approximately 60 years ago. Their potential for treating depression was discovered when a tuberculosis treatment—iproniazid—was found to reduce depressive symptoms. Researchers determined iproniazid’s antidepressant effects were the result of blocking removal of the amine group by monoamine oxidase (MAO) from dopamine, norepinephrine, and serotonin.5 A second MAOI, tranylcypromine, was discovered when it was found to be ineffective for treating decongestion.6
MAOI use in psychiatric practice has undergone significant changes since these medications were introduced. The discovery of hypertensive crises related to tyramine consumption led to decreased MAOI use, as did the rise of tricyclic antidepressants (TCAs) shortly thereafter. In the 1960s, research compared the relative efficacy of MAOIs to TCAs, and they became second-line antidepressants after the TCAs. In the late 1980s, the introduction of fluoxetine and other selective serotonin reuptake inhibitors (SSRIs) resulted in a significant drop-off in MAOI use.
Pharmacologic effects
MAO is a class of enzymes that initiate oxidation of extracellular neurotransmitters such as serotonin, norepinephrine, and dopamine. MAOIs can be classified based on their relative affinity to MAO as well as their enzyme selectivity. The first distinguishing characteristic is whether the drug binds to MAO in a reversible or irreversible manner. Currently, all MAOIs that are FDA-approved for treating depression bind irreversibly to MAO. As a result, the body must renew its MAO levels before a patient is no longer at risk for a hypertensive crisis, a process that may take up to 2 weeks. Clinicians must take care to ensure their patients avoid foods that contain tyramine and medications contraindicated with MAOIs during this period.
MAOIs also differ from each other in enzyme selectivity. There are 2 subtypes of MAO enzymes—MAOA and MAOB. Generally, the antidepressant activity of MAOIs appears to be directed toward MAOA inhibition. MAOA has been found to be more specific for binding to serotonin and norepinephrine and MAOB to be more specific for phenylethylamine. Dopamine is equally deaminated by both MAOA and MAOB.
Reversible MAOA inhibitors require fewer restrictions on diet or concurrent medications, but efficacy data of reversible MAOA inhibitors is mixed.
Clinical use of MAOIs
Four MAOIs are available in the United States: tranylcypromine, phenelzine, isocarboxazid, and selegiline. Selegiline is the only MAOI available as a transdermal patch. Transdermal administration results in fewer effects on MAO in the gastrointestinal tract, which means no dietary restrictions at the 6 mg/d starting dose, although the manufacturer recommends patients follow the MAO diet at 9 mg/d and 12 mg/d doses.7 Although selegiline is selective for MAOB at low doses, it becomes nonselective at therapeutic doses for depression. Recommended dosages for MAOIs can be found in Table 1.8
Table 1
Recommended dosages of monoamine oxidase inhibitors
| Medication | Starting dosages | Usual therapeutic dosage |
|---|---|---|
| Isocarboxazid | 10 mg twice a day | 30 to 60 mg/d |
| Phenelzine | 15 mg twice a day | 45 to 90 mg/d |
| Selegiline transdermal | 6 mg patch/d | 6 to 12 mg patch/d |
| Tranylcypromine | 10 mg, 2 or 3 times a day | 30 to 60 mg/d |
| Source: Adapted from reference 8 | ||
10,11
Several controlled trials have found a better response rate to MAOI therapy in outpatients with MDD who have not responded to other antidepressants.2,12 In a 6-week, double-blind trial, Vallejo et al10 reported that the TCA imipramine and high-dose phenelzine were equally efficacious in 32 patients with major depression with melancholia. In 32 dysthymic patients, high-dose phenelzine was superior to imipramine. Himmelhoch et al13 compared efficacy of tranylcypromine with that of imipramine in treating anergic bipolar depressive illness. Patients receiving tranylcypromine experienced significantly greater symptomatic improvement and higher global response without increased risk of treatment-emergent hypomania or mania.
Serum monitoring of MAOIs is not clinically indicated and there are no correlations between drug levels and effectiveness.14 In a study that examined the correlation of inhibiting platelet MAO and MAOIs’ antidepressant effects, researchers found that a higher dose of phenelzine (60 mg/d) was significantly better in treating depression and anxiety than a lower dose (30 mg/d), and only the higher dose achieved 80% of platelet MAO inhibition.15 Further studies with other MAOIs did not reproduce this effect and platelet MAO inhibition is not regularly used to assess adequate dosing.
A refined view of side effects
In a hypertensive crisis, patients experience significant hypertension, headaches, tachycardia, diaphoresis, and vomiting. Intravenous phentolamine—an α-adrenergic receptor blocker—can be used as an antidote; often a single dose is effective.16 Alternatively, calcium channel blockers such as nifedipine can be prescribed. A patient can take 10 mg/hour and be observed in the emergency room until symptoms are relieved (usually only 1 or 2 doses are needed) without being admitted to the hospital.
Table 2
Food restrictions with MAOIs
| Severe |
| Aged cheeses Aged meats (pepperoni, sausage, salami) Sauerkraut Soy sauce Fava or broad bean pods Banana peels All beers on tap |
| Use in moderation (≤2 servings/d) |
| Red wine (4 oz) White wine (4 oz) Bottled or canned beers (12 oz) |
| Mild to none |
| Avocados Banana pulp Bouillon Chocolate Fresh cheeses (cottage cheese, cream cheese, processed cheese slices) Fresh or processed meat |
| MAOIs: monoamine oxidase inhibitors Source: Adapted from references 4,17,18 |
Orthostatic hypotension is a common cardiovascular side effect of MAOIs that may lead to dizziness or syncope. Typically this is seen 2 to 3 weeks after initiating MAOI treatment. If hypotension remains a problem, mineralocorticoids can be prescribed with monitoring of serum potassium for hypokalemia. Increasing doses of tranylcypromine can increase supine—but not standing—diastolic blood pressure.19 Distinguish this type of blood pressure elevation from a hypertensive crisis by monitoring blood pressure with the patient sitting and standing and before and after he or she walks for 60 seconds.
Insomnia and day-night shifting—sleeping during the day and staying awake at night—are common and patients often cite these as reasons for discontinuing MAOIs. Many patients who respond to MAOIs report periods of substantial sleepiness in the mid to late afternoon. Table 320 provides a more complete list of reported side effects and their frequencies.
Table 3
MAOIs: Stay vigilant for side effects
| Medication | Common side effects |
|---|---|
| Isocarboxazid | Anxiety, blurred vision, constipation, dizziness, headache, insomnia, mania, somnolence, weight gain, xerostomia |
| Phenelzine | Constipation, disorder of ejaculation and/or orgasm, dizziness, edema, fatigue, headache, hyperreflexia, impotence, elevated values on liver function tests, orthostatic hypotension, sleep disorders, somnolence, tremor, weight gain, xerostomia |
| Selegiline transdermal | Application site reaction, decreased systolic blood pressure, diarrhea, headache, indigestion, insomnia, orthostatic hypotension, weight loss, xerostomia |
| Tranylcypromine | Agitation, anxiety, constipation, diarrhea, dizziness, headache, impotence, insomnia, loss of appetite, mania, nausea, orthostatic hypotension, somnolence, weight gain, xerostomia |
| MAOIs: monoamine oxidase inhibitors Source: Adapted from reference 20 | |
Practice guidelines
The American Psychiatric Association’s practice guidelines for treating major depression state that MAOIs are effective in treating subgroups of patients with MDD with atypical features who have failed TCA trials.21 These guidelines also state that MAOIs have been shown to be effective treatment for some patients who have failed other antidepressants. However, for TRD patients who have not responded to SSRIs or SNRIs, the effectiveness of MAOIs compared with other strategies is unclear.22
MAOIs have been used for >6 decades, and published studies continue to document their efficacy and safety when patients are monitored appropriately.11,12,14,15,25 However, based on our observations we suspect MAOIs are underutilized in clinical practice today. We are concerned that such practices can trickle down into residency training programs. Psychiatric residents typically do not receive adequate training in prescribing MAOIs, largely because many training faculty are not prescribing MAOIs themselves. Despite MAOIs’ limitations, concerns about an increased risk of suicide in patients with TRD26 and the high likelihood of a poor outcome associated with persistent nonresponse to prior treatments must be weighed against the relatively low risk of a hypertensive event with MAOIs.6
- McCabe-Sellers BJ, Staggs CG, Bogle ML. Tyramine in foods and monoamine oxidase inhibitor drugs: a crossroad where medicine, nutrition, pharmacy, and food industry converge. Journal of Food Composition and Analysis. 2006;19(suppl):S58-S65.
- Fiedorowicz JG, Swartz KL. The role of monoamine oxidase inhibitors in current psychiatric practice. J Psychiatr Pract. 2004;10(4):239-248.
- Clomipramine • Anafranil
- Epinephrine • Adrenalin, EpiPen
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Isocarboxazid • Marplan
- Lithium • Eskalith, Lithobid
- Nifedipine • Adalat, Afeditab
- Phenelzine • Nardil
- Phentolamine • OraVerse, Regitine
- Selegiline • EMSAM
- Tranylcypromine • Parnate
Dr. Kosinski reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Rothschild receives grant or research support from Cyberonics, the National Institute of Mental Health, St. Jude Medical, and Takeda, and is a consultant to Allergan, Eli Lilly and Company, GlaxoSmithKline, Noven Pharmaceuticals, Pfizer Inc., Shire Pharmaceuticals, and Sunovion.
Discuss this article at www.facebook.com/CurrentPsychiatry
For patients with major depressive disorder (MDD), monoamine oxidase inhibitors (MAOIs) have efficacy comparable to that of other antidepressants. However, concerns about side effects—particularly hypertensive crisis—and drug-drug interactions have led clinicians to prescribe MAOIs less often than newer antidepressants. A 1999 survey of 573 Michigan psychiatrists found that 30% had not prescribed an MAOI within the past 3 years, and 12% had never prescribed an MAOI.1 Although there are challenges to using these agents, we prefer prescribing MAOIs to depressed patients who have not responded to previous antidepressant trials over trying untested antidepressant combinations.
Currently, MAOIs are used primarily for patients who have not responded to other antidepressant trials and are considered treatment resistant. Treatment-resistant depression (TRD) typically is defined as nonresponse to ≥3 adequate antidepressant trials. TRD is a major cause of disability and loss of productivity. These patients tend to do poorly over the long term, with high rates of hospitalization and suicide attempts. Several controlled trials have shown that patients who fail other antidepressants may respond to MAOIs.2-4
Our knowledge regarding MAOIs has grown considerably. We have learned more about depression subtypes that MAOIs may help. As we learned more about dietary restrictions for patients taking MAOIs, the list of “forbidden foods” has decreased. Advances in treating a hypertensive crisis have decreased the need for hospitalization. By educating ourselves and our patients about MAOIs, we can provide another option for treating MDD.
An older antidepressant class
MAOIs were introduced approximately 60 years ago. Their potential for treating depression was discovered when a tuberculosis treatment—iproniazid—was found to reduce depressive symptoms. Researchers determined iproniazid’s antidepressant effects were the result of blocking removal of the amine group by monoamine oxidase (MAO) from dopamine, norepinephrine, and serotonin.5 A second MAOI, tranylcypromine, was discovered when it was found to be ineffective for treating decongestion.6
MAOI use in psychiatric practice has undergone significant changes since these medications were introduced. The discovery of hypertensive crises related to tyramine consumption led to decreased MAOI use, as did the rise of tricyclic antidepressants (TCAs) shortly thereafter. In the 1960s, research compared the relative efficacy of MAOIs to TCAs, and they became second-line antidepressants after the TCAs. In the late 1980s, the introduction of fluoxetine and other selective serotonin reuptake inhibitors (SSRIs) resulted in a significant drop-off in MAOI use.
Pharmacologic effects
MAO is a class of enzymes that initiate oxidation of extracellular neurotransmitters such as serotonin, norepinephrine, and dopamine. MAOIs can be classified based on their relative affinity to MAO as well as their enzyme selectivity. The first distinguishing characteristic is whether the drug binds to MAO in a reversible or irreversible manner. Currently, all MAOIs that are FDA-approved for treating depression bind irreversibly to MAO. As a result, the body must renew its MAO levels before a patient is no longer at risk for a hypertensive crisis, a process that may take up to 2 weeks. Clinicians must take care to ensure their patients avoid foods that contain tyramine and medications contraindicated with MAOIs during this period.
MAOIs also differ from each other in enzyme selectivity. There are 2 subtypes of MAO enzymes—MAOA and MAOB. Generally, the antidepressant activity of MAOIs appears to be directed toward MAOA inhibition. MAOA has been found to be more specific for binding to serotonin and norepinephrine and MAOB to be more specific for phenylethylamine. Dopamine is equally deaminated by both MAOA and MAOB.
Reversible MAOA inhibitors require fewer restrictions on diet or concurrent medications, but efficacy data of reversible MAOA inhibitors is mixed.
Clinical use of MAOIs
Four MAOIs are available in the United States: tranylcypromine, phenelzine, isocarboxazid, and selegiline. Selegiline is the only MAOI available as a transdermal patch. Transdermal administration results in fewer effects on MAO in the gastrointestinal tract, which means no dietary restrictions at the 6 mg/d starting dose, although the manufacturer recommends patients follow the MAO diet at 9 mg/d and 12 mg/d doses.7 Although selegiline is selective for MAOB at low doses, it becomes nonselective at therapeutic doses for depression. Recommended dosages for MAOIs can be found in Table 1.8
Table 1
Recommended dosages of monoamine oxidase inhibitors
| Medication | Starting dosages | Usual therapeutic dosage |
|---|---|---|
| Isocarboxazid | 10 mg twice a day | 30 to 60 mg/d |
| Phenelzine | 15 mg twice a day | 45 to 90 mg/d |
| Selegiline transdermal | 6 mg patch/d | 6 to 12 mg patch/d |
| Tranylcypromine | 10 mg, 2 or 3 times a day | 30 to 60 mg/d |
| Source: Adapted from reference 8 | ||
10,11
Several controlled trials have found a better response rate to MAOI therapy in outpatients with MDD who have not responded to other antidepressants.2,12 In a 6-week, double-blind trial, Vallejo et al10 reported that the TCA imipramine and high-dose phenelzine were equally efficacious in 32 patients with major depression with melancholia. In 32 dysthymic patients, high-dose phenelzine was superior to imipramine. Himmelhoch et al13 compared efficacy of tranylcypromine with that of imipramine in treating anergic bipolar depressive illness. Patients receiving tranylcypromine experienced significantly greater symptomatic improvement and higher global response without increased risk of treatment-emergent hypomania or mania.
Serum monitoring of MAOIs is not clinically indicated and there are no correlations between drug levels and effectiveness.14 In a study that examined the correlation of inhibiting platelet MAO and MAOIs’ antidepressant effects, researchers found that a higher dose of phenelzine (60 mg/d) was significantly better in treating depression and anxiety than a lower dose (30 mg/d), and only the higher dose achieved 80% of platelet MAO inhibition.15 Further studies with other MAOIs did not reproduce this effect and platelet MAO inhibition is not regularly used to assess adequate dosing.
A refined view of side effects
In a hypertensive crisis, patients experience significant hypertension, headaches, tachycardia, diaphoresis, and vomiting. Intravenous phentolamine—an α-adrenergic receptor blocker—can be used as an antidote; often a single dose is effective.16 Alternatively, calcium channel blockers such as nifedipine can be prescribed. A patient can take 10 mg/hour and be observed in the emergency room until symptoms are relieved (usually only 1 or 2 doses are needed) without being admitted to the hospital.
Table 2
Food restrictions with MAOIs
| Severe |
| Aged cheeses Aged meats (pepperoni, sausage, salami) Sauerkraut Soy sauce Fava or broad bean pods Banana peels All beers on tap |
| Use in moderation (≤2 servings/d) |
| Red wine (4 oz) White wine (4 oz) Bottled or canned beers (12 oz) |
| Mild to none |
| Avocados Banana pulp Bouillon Chocolate Fresh cheeses (cottage cheese, cream cheese, processed cheese slices) Fresh or processed meat |
| MAOIs: monoamine oxidase inhibitors Source: Adapted from references 4,17,18 |
Orthostatic hypotension is a common cardiovascular side effect of MAOIs that may lead to dizziness or syncope. Typically this is seen 2 to 3 weeks after initiating MAOI treatment. If hypotension remains a problem, mineralocorticoids can be prescribed with monitoring of serum potassium for hypokalemia. Increasing doses of tranylcypromine can increase supine—but not standing—diastolic blood pressure.19 Distinguish this type of blood pressure elevation from a hypertensive crisis by monitoring blood pressure with the patient sitting and standing and before and after he or she walks for 60 seconds.
Insomnia and day-night shifting—sleeping during the day and staying awake at night—are common and patients often cite these as reasons for discontinuing MAOIs. Many patients who respond to MAOIs report periods of substantial sleepiness in the mid to late afternoon. Table 320 provides a more complete list of reported side effects and their frequencies.
Table 3
MAOIs: Stay vigilant for side effects
| Medication | Common side effects |
|---|---|
| Isocarboxazid | Anxiety, blurred vision, constipation, dizziness, headache, insomnia, mania, somnolence, weight gain, xerostomia |
| Phenelzine | Constipation, disorder of ejaculation and/or orgasm, dizziness, edema, fatigue, headache, hyperreflexia, impotence, elevated values on liver function tests, orthostatic hypotension, sleep disorders, somnolence, tremor, weight gain, xerostomia |
| Selegiline transdermal | Application site reaction, decreased systolic blood pressure, diarrhea, headache, indigestion, insomnia, orthostatic hypotension, weight loss, xerostomia |
| Tranylcypromine | Agitation, anxiety, constipation, diarrhea, dizziness, headache, impotence, insomnia, loss of appetite, mania, nausea, orthostatic hypotension, somnolence, weight gain, xerostomia |
| MAOIs: monoamine oxidase inhibitors Source: Adapted from reference 20 | |
Practice guidelines
The American Psychiatric Association’s practice guidelines for treating major depression state that MAOIs are effective in treating subgroups of patients with MDD with atypical features who have failed TCA trials.21 These guidelines also state that MAOIs have been shown to be effective treatment for some patients who have failed other antidepressants. However, for TRD patients who have not responded to SSRIs or SNRIs, the effectiveness of MAOIs compared with other strategies is unclear.22
MAOIs have been used for >6 decades, and published studies continue to document their efficacy and safety when patients are monitored appropriately.11,12,14,15,25 However, based on our observations we suspect MAOIs are underutilized in clinical practice today. We are concerned that such practices can trickle down into residency training programs. Psychiatric residents typically do not receive adequate training in prescribing MAOIs, largely because many training faculty are not prescribing MAOIs themselves. Despite MAOIs’ limitations, concerns about an increased risk of suicide in patients with TRD26 and the high likelihood of a poor outcome associated with persistent nonresponse to prior treatments must be weighed against the relatively low risk of a hypertensive event with MAOIs.6
- McCabe-Sellers BJ, Staggs CG, Bogle ML. Tyramine in foods and monoamine oxidase inhibitor drugs: a crossroad where medicine, nutrition, pharmacy, and food industry converge. Journal of Food Composition and Analysis. 2006;19(suppl):S58-S65.
- Fiedorowicz JG, Swartz KL. The role of monoamine oxidase inhibitors in current psychiatric practice. J Psychiatr Pract. 2004;10(4):239-248.
- Clomipramine • Anafranil
- Epinephrine • Adrenalin, EpiPen
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Isocarboxazid • Marplan
- Lithium • Eskalith, Lithobid
- Nifedipine • Adalat, Afeditab
- Phenelzine • Nardil
- Phentolamine • OraVerse, Regitine
- Selegiline • EMSAM
- Tranylcypromine • Parnate
Dr. Kosinski reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Rothschild receives grant or research support from Cyberonics, the National Institute of Mental Health, St. Jude Medical, and Takeda, and is a consultant to Allergan, Eli Lilly and Company, GlaxoSmithKline, Noven Pharmaceuticals, Pfizer Inc., Shire Pharmaceuticals, and Sunovion.
1. Balon R, Mufti R, Arfken CL. A survey of prescribing practices for monoamine oxidase inhibitors. Psychiatr Serv. 1999;50(7):945-947.
2. Nolen WA, van de Putte JJ, Dijken WA, et al. Treatment strategy in depression. II. MAO inhibitors in depression resistant to cyclic antidepressants: two controlled crossover studies with tranylcypromine versus L-5-hydroxytryptophan and nomifensine. Acta Psychiatr Scand. 1988;78(6):676-683.
3. McGrath PJ, Stewart JW, Harrison W, et al. Treatment of tricyclic refractory depression with a monoamine oxidase inhibitor antidepressant. Psychopharmacol Bull. 1987;23(1):169-172.
4. Amsterdam JD. Monoamine oxidase inhibitor therapy in severe and resistant depression. Psychiatr Ann. 2006;36(9):607-613.
5. Schildkraut JJ. The catecholamine hypothesis of affective disorders: a review of supporting evidence. Am J Psychiatry. 1965;122(5):509-522.
6. Kennedy SH, Holt A, Baker GB. Monoamine oxidase inhibitors. In: Sadock BJ Sadock VA, eds. Kaplan and Sadock’s comprehensive textbook of psychiatry. 8th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005: 1076–1080.
7. EMSAM [package insert]. Napa CA: Dey Pharm LP; 2011.
8. Amsterdam JD, Chopra M. Monoamine oxidase inhibitors revisited. Psychiatric Ann. 2001;31(6):361-370.
9. Quitkin FM, Stewart JW, McGrath PJ, et al. Phenelzine versus imipramine in the treatment of probable atypical depression: defining syndrome boundaries of selective MAOI responders. Am J Psychiatry. 1988;145(3):306-311.
10. Vallejo J, Gasto C, Catalan R, et al. Double-blind study of imipramine versus phenelzine in melancholias and dysthymic disorders. Br J Psychiatry. 1987;151:639-642.
11. White K, Razani J, Cadow B, et al. Trancylpromine vs. nortriptyline vs. placebo in depressed outpatients: a controlled trial. Psychopharmacology (Berl). 1984;82(3):258-262.
12. Thase ME, Frank E, Mallinger AG, et al. Treatment of imipramine-resistant recurrent depression, III: efficacy of monoamine oxidase inhibitors. J Clin Psychiatry. 1992;53(1):5-11.
13. Himmelhoch JM, Thase ME, Mallinger AG, et al. Tranylcypromine versus imipramine in anergic bipolar depression. Am J Psychiatry. 1991;148(7):910-916.
14. Rothschild AJ. ed. The evidence-based guide to antidepressant medications. Arlington, VA: American Psychiatric Publishing, Inc.; 2012:15–20.
15. Ravaris CL, Nies A, Robinson DS, et al. A multiple-dose, controlled study of phenelzine in depression-anxiety states. Arch Gen Psychiatry. 1976;33(3):347-350.
16. Cockhill LA, Remick RA. Blood pressure effects of monoamine oxidase inhibitors—the highs and lows. Can J Psychiatry. 1987;32(9):803-808.
17. Shulman KI, Walker SE. A reevaluation of dietary restrictions for irreversible monoamine oxidase inhibitors. Psychiatr Ann. 2001;31(6):378-384.
18. Gardner DM, Shulman KI, Walker SE, et al. The making of a user friendly MAOI diet. J Clin Psychiatry. 1996;57(3):99-104.
19. Keck PE, Jr, Carter WP, Nierenberg AA, et al. Acute cardiovascular effects of tranylcypromine: correlation with plasma drug, metabolite, norepinephrine, and MHPG levels. J Clin Psychiatry. 1991;52(6):250-254.
20. Micromedex Healthcare Series [UMass Memorial Healthcare Intranet System]. Version 5.1. Greenwood Village CO: Thomson Reuters (Healthcare) Inc.
21. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder third edition. http://psychiatryonline.org/content.aspx?bookid=28§ionid=1667485. Published October 2010. Accessed October 26, 2012.
22. McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry. 2006;163(9):1531-1541; quiz 1666.
23. Nelson JC, Byck R. Rapid response to lithium in phenelzine non-responders. Br J Psychiatry. 1982;141:85-86.
24. Guze BH, Baxter LR, Jr, Rego J. Refractory depression treated with high doses of monoamine oxidase inhibitor. J Clin Psychiatry. 1987;48(1):31-32.
25. Robinson DS, Gilmor ML, Yang Y, et al. Treatment effects of selegiline transdermal system on symptoms of major depressive disorder: a meta analysis of short term, placebo controlled, efficacy trials. Psychopharmacol Bull. 2007;40(3):15-28.
26. Keller MB, Lavori PW, Rice J, et al. The persistent risk of chronicity in recurrent episodes of nonbipolar major depressive disorder: a prospective follow-up. Am J Psychiatry. 1986;143(1):24-28.
1. Balon R, Mufti R, Arfken CL. A survey of prescribing practices for monoamine oxidase inhibitors. Psychiatr Serv. 1999;50(7):945-947.
2. Nolen WA, van de Putte JJ, Dijken WA, et al. Treatment strategy in depression. II. MAO inhibitors in depression resistant to cyclic antidepressants: two controlled crossover studies with tranylcypromine versus L-5-hydroxytryptophan and nomifensine. Acta Psychiatr Scand. 1988;78(6):676-683.
3. McGrath PJ, Stewart JW, Harrison W, et al. Treatment of tricyclic refractory depression with a monoamine oxidase inhibitor antidepressant. Psychopharmacol Bull. 1987;23(1):169-172.
4. Amsterdam JD. Monoamine oxidase inhibitor therapy in severe and resistant depression. Psychiatr Ann. 2006;36(9):607-613.
5. Schildkraut JJ. The catecholamine hypothesis of affective disorders: a review of supporting evidence. Am J Psychiatry. 1965;122(5):509-522.
6. Kennedy SH, Holt A, Baker GB. Monoamine oxidase inhibitors. In: Sadock BJ Sadock VA, eds. Kaplan and Sadock’s comprehensive textbook of psychiatry. 8th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005: 1076–1080.
7. EMSAM [package insert]. Napa CA: Dey Pharm LP; 2011.
8. Amsterdam JD, Chopra M. Monoamine oxidase inhibitors revisited. Psychiatric Ann. 2001;31(6):361-370.
9. Quitkin FM, Stewart JW, McGrath PJ, et al. Phenelzine versus imipramine in the treatment of probable atypical depression: defining syndrome boundaries of selective MAOI responders. Am J Psychiatry. 1988;145(3):306-311.
10. Vallejo J, Gasto C, Catalan R, et al. Double-blind study of imipramine versus phenelzine in melancholias and dysthymic disorders. Br J Psychiatry. 1987;151:639-642.
11. White K, Razani J, Cadow B, et al. Trancylpromine vs. nortriptyline vs. placebo in depressed outpatients: a controlled trial. Psychopharmacology (Berl). 1984;82(3):258-262.
12. Thase ME, Frank E, Mallinger AG, et al. Treatment of imipramine-resistant recurrent depression, III: efficacy of monoamine oxidase inhibitors. J Clin Psychiatry. 1992;53(1):5-11.
13. Himmelhoch JM, Thase ME, Mallinger AG, et al. Tranylcypromine versus imipramine in anergic bipolar depression. Am J Psychiatry. 1991;148(7):910-916.
14. Rothschild AJ. ed. The evidence-based guide to antidepressant medications. Arlington, VA: American Psychiatric Publishing, Inc.; 2012:15–20.
15. Ravaris CL, Nies A, Robinson DS, et al. A multiple-dose, controlled study of phenelzine in depression-anxiety states. Arch Gen Psychiatry. 1976;33(3):347-350.
16. Cockhill LA, Remick RA. Blood pressure effects of monoamine oxidase inhibitors—the highs and lows. Can J Psychiatry. 1987;32(9):803-808.
17. Shulman KI, Walker SE. A reevaluation of dietary restrictions for irreversible monoamine oxidase inhibitors. Psychiatr Ann. 2001;31(6):378-384.
18. Gardner DM, Shulman KI, Walker SE, et al. The making of a user friendly MAOI diet. J Clin Psychiatry. 1996;57(3):99-104.
19. Keck PE, Jr, Carter WP, Nierenberg AA, et al. Acute cardiovascular effects of tranylcypromine: correlation with plasma drug, metabolite, norepinephrine, and MHPG levels. J Clin Psychiatry. 1991;52(6):250-254.
20. Micromedex Healthcare Series [UMass Memorial Healthcare Intranet System]. Version 5.1. Greenwood Village CO: Thomson Reuters (Healthcare) Inc.
21. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder third edition. http://psychiatryonline.org/content.aspx?bookid=28§ionid=1667485. Published October 2010. Accessed October 26, 2012.
22. McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry. 2006;163(9):1531-1541; quiz 1666.
23. Nelson JC, Byck R. Rapid response to lithium in phenelzine non-responders. Br J Psychiatry. 1982;141:85-86.
24. Guze BH, Baxter LR, Jr, Rego J. Refractory depression treated with high doses of monoamine oxidase inhibitor. J Clin Psychiatry. 1987;48(1):31-32.
25. Robinson DS, Gilmor ML, Yang Y, et al. Treatment effects of selegiline transdermal system on symptoms of major depressive disorder: a meta analysis of short term, placebo controlled, efficacy trials. Psychopharmacol Bull. 2007;40(3):15-28.
26. Keller MB, Lavori PW, Rice J, et al. The persistent risk of chronicity in recurrent episodes of nonbipolar major depressive disorder: a prospective follow-up. Am J Psychiatry. 1986;143(1):24-28.
How to stabilize an acutely psychotic patient
Acute psychosis is a symptom that can be caused by many psychiatric and medical conditions. Psychotic patients might be unable to provide a history or participate in treatment if they are agitated, hostile, or violent. An appropriate workup may reveal the etiology of the psychosis; secondary causes, such as medical illness and substance use, are prevalent in the emergency room (ER) setting. If the patient has an underlying primary psychotic disorder, such as schizophrenia or mania, illness-specific intervention will help acutely and long-term. With agitated and uncooperative psychotic patients, clinicians often have to intervene quickly to ensure the safety of the patient and those nearby.
This article focuses on the initial evaluation and treatment of psychotic patients in the ER, either by a psychiatric emergency service or a psychiatric consultant. This process can be broken down into:
- triage or initial clinical assessment
- initial psychiatric stabilization, including pharmacologic interventions and agitation management
- diagnostic workup to evaluate medical and psychiatric conditions
- further psychiatric evaluation
- determining safe disposition.1
Triage determines the next step
Initial clinical assessment and triage are necessary to select the appropriate immediate intervention. When a patient arrives in the ER, determine if he or she requires urgent medical attention. Basic initial screening should include:
- vital signs
- finger stick blood glucose
- medical history
- signs or symptoms of intoxication or withdrawal
- signs of trauma (eg, neck ligature marks, gunshot wounds, lacerations)
- asking the patient to give a brief history leading up to the current presentation.
A review of medical records may reveal patients’ medical and psychiatric history and allergies. Collateral documentation—such as ambulance run sheets or police reports—may provide additional information. If no immediate medical intervention is warranted, determine if the patient can wait in an open, unlocked waiting area or if he or she needs to be in an unlocked area with a sitter, a locked open area, or a secluded room with access to restraints. In general, psychotic patients who pose a threat of harm to themselves or others or cannot care for themselves because of their psychosis need locked areas or observation.
Initial psychiatric stabilization
Agitation is diagnostically unspecific but can occur in patients with psychosis. Psychotic patients can become unpredictably and impulsively aggressive and assaultive. Rapid intervention is necessary to minimize risk of bodily harm to the patient and those around the patient. Physicians often must make quick interventions based on limited clinical information. It is important to recognize early signs and symptoms of agitation, including:
- restlessness (pacing, fidgeting, hand wringing, fist clenching, posturing)
- irritability
- decreased attention
- inappropriate or hostile behaviors.2
Pharmacologic interventions. The initial goals of pharmacologic treatment are to calm the patient without oversedation, thereby allowing the patient to take part in his or her care and begin treatment for the primary psychotic illness.3,4 Offering oral medications first and a choice of medications may help a patient feel more in control of the situation. If a patient has to be physically restrained, pharmacotherapy may limit the amount of time spent in restraints.
Medication choice depends on several factors, including onset of action, available formulation (eg, IM, liquid, rapidly dissolving), the patient’s previous medication response, side effect profile, allergies or adverse reactions to medications, and medical comorbidities.3 If a patient has a known psychotic illness, it may be helpful to administer the patient’s regular antipsychotic or anxiolytic medication. Some medications, such as lithium, are not effective in the acute setting and should be avoided. Additionally, benzodiazepines other than lorazepam or midazolam should not be administered IM because of erratic absorption.
Antipsychotics can be used for psychotic patients with or without agitation. Benzodiazepines may treat agitation, but are not specific for psychosis. Haloperidol can be used to treat acute psychosis and has proven efficacy for agitation. Benzodiazepines can decrease acute agitation and have efficacy similar to haloperidol, but with more sedation.5 A combination of lorazepam and haloperidol is thought to be superior to either medication alone.6 Lorazepam helps maintain sedation and decreases potential side effects caused by haloperidol. Consensus guidelines from 2001 and 2005 recommend combined haloperidol and lorazepam for first-line treatment of acute agitation.3,7 High-potency antipsychotics such as haloperidol have an increased risk for extrapyramidal symptoms (EPS), particularly acute dystonic reactions—involuntary, sustained muscle contractions—in susceptible patients (eg, antipsychotic-naïve patients); consider starting diphenhydramine, 25 to 50 mg, or benztropine, 0.5 to 2 mg, to prevent EPS from high-potency antipsychotics (Algorithm 1).
Algorithm 1: Treating acute psychosis: Choosing pharmacologic agents
EPS: extrapyramidal symptoms; PO: by mouth; SL: sublingual
Second-generation antipsychotics (SGAs) increasingly have been used for managing acute agitation in patients with an underlying psychotic disorder. Guidelines from a 2012 American Association for Emergency Psychiatry workgroup recommend using an SGA as monotherapy or in combination with another medication instead of haloperidol to treat agitated patients with a known psychotic disorder.8 Clinical policy guidelines from the American College of Emergency Physicians recommend antipsychotic monotherapy for agitation and initial treatment in patients with a known psychiatric illness for which antipsychotic treatment is indicated (eg, schizophrenia).9 For patients with known psychotic illness, expert opinion recommends oral risperidone or olanzapine.3,8 The combination of oral risperidone plus lorazepam may be as effective as the IM haloperidol and IM lorazepam combination.10 Patients who are too agitated to take oral doses may require parenteral medications. Ziprasidone, olanzapine, and aripiprazole are available in IM formulations. Ziprasidone, 20 mg IM, is well tolerated and has been shown to be effective in decreasing acute agitation symptoms in patients with psychotic disorders.11 Olanzapine is as effective as haloperidol in decreasing agitation in patients with schizophrenia, with lower rates of EPS.12 In a double-blind, placebo-controlled trial, psychotic symptoms in patients with schizophrenia or schizoaffective disorder decreased within 2 hours of IM olanzapine administration.13 Both IM ziprasidone and olanzapine have a relatively rapid onset of action (within 30 minutes), which makes them reasonable choices in the acute setting. Olanzapine has a long half-life (21 to 50 hours); therefore, patients’ comorbid medical conditions, such as cardiac abnormalities or hypotension, must be considered. If parenteral medication is required, IM olanzapine or IM ziprasidone is recommended.8 IM haloperidol with a benzodiazepine also can be considered.3
Coadministration of parenteral olanzapine and a benzodiazepine can lead to severe orthostatic hypotension and cardiac or respiratory depression and should be avoided in geriatric patients.14 Finally, it is important to rule out presentations that may worsen with antipsychotic treatment, including phencyclidine (PCP) toxicity (could worsen dystonic reactions), anticholinergic delirium, neuroleptic malignant syndrome (NMS), or catatonia.
If a patient does not respond to the initial dose of a medication, the dose may be repeated. However, doses should not be repeated until a patient is so sedated that he or she cannot take part in his or her care, or until he or she has developed significant EPS.
In addition to antipsychotics, consider loading with oral divalproex for patients who are acutely psychotic in the context of a manic episode (Table).15,16 Higher serum divalproex levels—target serum levels >94 μg/mL—are associated with greater efficacy as measured by change from baseline in Mania Rating Scale or Young Mania Rating Scale scores compared with placebo.15 For acutely psychotic schizophrenia patients, there is evidence of benefit with initial treatment with divalproex combined with an SGA. In a randomized, double-blind study, patients treated with divalproex plus olanzapine or risperidone showed quicker initial resolution of psychotic symptoms compared with olanzapine or risperidone monotherapy, but no better long-term benefit.16 Clinicians may consider this well-tolerated combination after an appropriate medical workup. This finding of early benefit was not replicated with divalproex extended-release.17
Table
Divalproex dosing for patients with acute psychosis and mania
| Initial dose | Titration | |
|---|---|---|
| Acute mania15 | Divalproex delayed-release: 750 mg/d Divalproex extended-release: 20 mg/kg/d | Increase to clinical effectiveness or maximum serum level of 125 μg/mL |
| Exacerbation of psychosis16 | Divalproex: 15 mg/kg/d (in 2 doses) | Increase to clinical effectiveness over 12 days or maximum dosage of 30 mg/kg/d |
Side effects and adverse reactions. Treatment with antipsychotics may cause QTc interval prolongation, which can lead to increased risk for torsades de pointes and sudden death due to ventricular fibrillation. However, there have been few cases of torsades de pointes after oral haloperidol and none with IM haloperidol compared with at least 30 cases of torsades de pointes after IV haloperidol treatment. Torsades de pointes after risperidone, olanzapine, or ziprasidone treatment has not been reported.18
Hypotension and bradycardia may occur in patients treated with olanzapine; however, these signs occur less frequently in agitated patients.18 Antipsychotic treatment increases risk for EPS, including acute dystonia, akathisia (subjective restlessness with desire to move), and parkinsonism (shuffling gait, resting tremor, rigidity and bradykinesia), as well as NMS.
Nonpharmacologic interventions. Verbal intervention to try to de-escalate an agitated, psychotic patient should be attempted first; however, this is not always possible. Other behavioral interventions include offering a meal, blanket, or pillow, or other comforting options to decrease the patient’s anxiety associated with psychosis.2 However, if agitated psychotic patients continue to display aggressive behaviors and pose a risk of harm to themselves or those around them, physical restraints should be considered because the clinician must balance protecting the patient’s rights with others’ safety. If physical restraints are used, medication also should be administered. Remove physical restraints as soon as safely possible; the Joint Commission has established standards for minimizing harm when using physical restraints.19
Diagnostic workup
Once a patient is medically stable in the ER, begin further workup of the etiology of the psychosis (Algorithm 2). All patients should have a physical exam, provided they are calm and in behavioral control. Monitor vital signs; patients at risk of withdrawal from substances should be monitored more frequently. Although there is no established standard for “medical clearance” of a psychiatric patient,20 all patients should undergo basic laboratory tests, including basic metabolic panel, complete blood count, and urine toxicology. The extent of the workup is determined by the clinical situation and suspected cause of psychosis.21
Algorithm 2: Diagnostic workup of an acutely psychotic patient
ER: emergency room; EEG: electroencephalography; LP: lumbar puncture; TSH: thyroid-stimulating hormone
If you suspect delirium, the underlying medical etiology must be identified and treated. Up to 40% of hospitalized patients with delirium may have psychosis.22 Psychosis in a delirious patient may be characterized by poorly formed delusions and visual hallucinations. Delirious patients often are inattentive, easily distracted, and disoriented, with a fluctuating clinical course. Patients with psychosis generally do not have impaired attention and are alert with intact memory. However, acutely psychotic patients may be quite disorganized and uncooperative, which makes it difficult to distinguish between these 2 diagnoses. Serial exams may help clarify the clinical picture. It is important to remember that patients with a history of a psychotic disorder may have a superimposed delirium.
In young patients (age 18 to 30) with new-onset psychosis, consider drug-induced psychosis; PCP, lysergic acid diethylamide, and methamphetamine intoxication and withdrawal can lead to psychotic presentations. Additionally, comorbid substance use is common among patients with primary psychotic disorders. One study found 37% of first-episode psychotic patients misused drugs or alcohol, similar to the lifetime rate of patients with chronic psychotic disorders.23,24 Check urine and serum toxicology screens and obtain relevant substance use history. Brain MRI may be considered for patients with first presentation of psychosis; however, there is little evidence to support head CT imaging unless there is known head trauma.25 Electroencephalography and lumbar puncture can be considered if clinically indicated.
Further psychiatric evaluation
Obtaining a psychiatric history is necessary to determine the etiology of the acute psychotic presentation. The timing and duration of psychotic symptoms are key. Acute symptom onset with fluctuating course and impaired attention suggests a delirious process. A gradual decline in functioning over several months to years in a young person suggests a first episode of a psychotic disorder (eg, schizophrenia). Drug abuse is common among young persons with a psychotic disorder and a positive drug screen for a psychogenic substance does not exclude a primary psychotic disorder.
If a patient has a history of schizophrenia, bipolar disorder, or psychotic depression, acutely worsening psychosis may be considered an acute or chronic presentation. Even in patients diagnosed with a psychotic illness, it is necessary to determine the cause of symptom exacerbation. Medication nonadherence (which can be partial), substance use, psychosocial stressors, or underlying medical illness should be considered. Collateral information from family or friends may be crucial to understanding a patient’s presentation.
Safe disposition
Patients who pose a risk of harm to themselves or others or who are so impaired by their psychosis that they cannot care for themselves generally should be admitted to an inpatient psychiatric facility. For some psychotic patients who are agreeable to treatment and not prone to violence, less restrictive settings—such as a crisis intervention unit or respite facility—may be appropriate. A patient with first-episode psychosis could be admitted for further diagnostic clarification and treatment initiation. Manic patients often have no insight into their illness and may need hospitalization for containment and assurance of medication adherence. Goals of inpatient care include initiating or resuming pharmacologic treatment to reduce psychotic symptoms and beginning the recovery process. Response rates—defined as ≥20% improvement in total score on a psychopathology scale such as the Positive and Negative Syndrome Scale—will vary, but can take ≥4 weeks in some patients with first-episode schizophrenia.26 However, most patients will be stabilized and ready for discharge before 4 weeks. Family education and alliance building with the patient and family are important during hospitalization.
Related Resources
- Schwartz S, Weathers, M. The psychotic patient. In: Riba MB, Ravindranath D, eds. Clinical manual of emergency psychiatry. Arlington, VA: American Psychiatric Publishing, Inc.; 2010:115-140.
- American Association for Emergency Psychiatry. http://emergencypsychiatry.org.
Drug Brand Names
- Aripiprazole • Abilify
- Benztropine • Cogentin
- Diphenhydramine • Benadryl
- Divalproex • Depakote
- Haloperidol • Haldol
- Lithium • Eskalith, Lithobid
- Lorazepam • Ativan
- Midazolam • Versed
- Olanzapine • Zyprexa
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr. Freudenreich receives grant or research support from Beacon Health Strategies, Global Medical Education, MGH Psychiatry Academy, Optimal Medicine, Pfizer Inc., and PsychoGenics.
Drs. Brown and Stoklosa report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Marco CA, Vaughan J. Emergency management of agitation in schizophrenia. Am J Emerg Med. 2005;23(6):767-776.
2. Freudenreich O. Emergency management of acute psychosis. In: Freudenreich O ed. Psychotic disorders: a practical guide. New York, NY: Wolter Kluwer/Lippincott Williams & Wilkins; 2008:72–78.
3. Allen MH, Currier GW, Carpenter D, et al. Expert Consensus Panel for Behavioral Emergencies 2005. The expert consensus guideline series. Treatment of behavioral emergencies 2005. J Psychiatr Pract. 2005;11(1 suppl):S5-S108.
4. National Institute for Health and Clinical Excellence. Schizophrenia: core interventions in the treatment and management of schizophrenia in primary and secondary care. London United Kingdom: National Institute for Clinical Excellence; 2002.
5. Allen MH. Managing the agitated psychotic patient: a reappraisal of the evidence. J Clin Psychiatry. 2000;61 (14 suppl):S11-S20.
6. Battaglia J, Moss S, Rush J, et al. Haloperidol, lorazepam, or both for psychotic agitation? A multicenter, prospective, double-blind, emergency department study. Am J Emerg Med. 1997;15(4):335-340.
7. Allen MH, Currier GW, Hughes DH, et al. Expert Consensus Panel for Behavioral Emergencies. The expert consensus guideline series. Treatment of behavioral emergencies. Postgrad Med. 2001;(Spec no):1-88.
8. Wilson MP, Pepper D, Currier GW, et al. The psychopharmacology of agitation: consensus statement of American Association for Emergency Psychiatry project BETA psychopharmacology workgroup. West J Emerg Med. 2012;13(1):26-34.
9. Lukens TW, Wolf SJ, Edlow JA, et al. American College of Emergency Physicians Clinical Policies Subcommittee (Writing Committee) on Critical Issues in the Diagnosis and Management of the Adult Psychiatric Patient in the Emergency Department. Clinical policy: critical issues in the diagnosis and management of the adult psychiatric patient in the emergency department. Ann Emerg Med. 2006;47(1):79-99.
10. Currier GW, Chou JC, Feifel D, et al. Acute treatment of psychotic agitation: a randomized comparison of oral treatment with risperidone and lorazepam versus intramuscular treatment with haloperidol and lorazepam. J Clin Psychiatry. 2004;65(3):386-394.
11. Daniel DG, Potkin SG, Reeves KR, et al. Intramuscular (IM) ziprasidone 20 mg is effective in reducing agitation associated with psychosis: a double-blind, randomized trial. Psychopharmacology (Berl). 2001;155(2):128-134.
12. Wright P, Birkett M, David SR, et al. Double-blind, placebo-controlled comparison of intramuscular olanzapine and intramuscular haloperidol in the treatment of acute agitation in schizophrenia. Am J Psychiatry. 2001;158(7):1149-1151.
13. Kapur S, Arenovic T, Agid O, et al. Evidence for onset of antipsychotic effects within the first 24 hours of treatment. Am J Psychiatry. 2005;162(5):939-946.
14. Marder SR, Sorasburu S, Dunayevic E, et al. Case reports of postmarketing adverse event experiences with olanzapine intramuscular treatment in patients with agitation. J Clin Psychiatry. 2010;71(4):433-441.
15. Allen MH, Hirschfeld RM, Wozniak PJ, et al. Linear relationship of valproate serum concentration to response and optimal serum levels for acute mania. Am J Psychiatry. 2006;163(2):272-275.
16. Casey DE, Daniel DG, Wassef AA, et al. Effect of divalproex combined with olanzapine or risperidone in patients with an acute exacerbation of schizophrenia. Neuropsychopharmacology. 2003;28(1):182-192.
17. Casey DE, Daniel DG, Tamminga C, et al. Divalproex ER combined with olanzapine or risperidone for treatment of acute exacerbations of schizophrenia. Neuropsychopharmacology. 2009;34(5):1330-1338.
18. Currier GW, Allen MH, Bunney EB, et al. Safety of medications used to treat acute agitation. J Emerg Med. 2004;27(4 suppl):S19-S24.
19. The Joint Commission. Sentinel event alert. Preventing restraint deaths. Published November 18 1998. http://www.jointcommission.org/assets/1/18/SEA_8.pdf. Accessed October 26, 2012
20. Janiak BD, Atteberry S. Medical clearance of the psychiatric patient in the emergency department. J Emerg Med. 2012;43(5):866-870.
21. Freudenreich O, Schulz SC, Goff DC. Initial medical work-up of first-episode psychosis: a conceptual review. Early Interv Psychiatry. 2009;3(1):10-18.
22. Webster R, Holroyd S. Prevalence of psychotic symptoms in delirium. Psychosomatics. 2000;41(6):519-522.
23. Cantwell R, Brewin J, Glazebrook C, et al. Prevalence of substance misuse in first-episode psychosis. Br J Psychiatry. 1999;174:150-153.
24. Green AI, Tohen MF, Hamer RM, et al. First episode schizophrenia-related psychosis and substance use disorders: acute response to olanzapine and haloperidol. Schizophr Res. 2004;66(2-3):125-135.
25. Goulet K, Deschamps B, Evoy F, et al. Use of brain imaging (computed tomography and magnetic resonance imaging) in first-episode psychosis: review and retrospective study. Can J Psychiatry. 2009;54(7):493-501.
26. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10):1785-1804.
Acute psychosis is a symptom that can be caused by many psychiatric and medical conditions. Psychotic patients might be unable to provide a history or participate in treatment if they are agitated, hostile, or violent. An appropriate workup may reveal the etiology of the psychosis; secondary causes, such as medical illness and substance use, are prevalent in the emergency room (ER) setting. If the patient has an underlying primary psychotic disorder, such as schizophrenia or mania, illness-specific intervention will help acutely and long-term. With agitated and uncooperative psychotic patients, clinicians often have to intervene quickly to ensure the safety of the patient and those nearby.
This article focuses on the initial evaluation and treatment of psychotic patients in the ER, either by a psychiatric emergency service or a psychiatric consultant. This process can be broken down into:
- triage or initial clinical assessment
- initial psychiatric stabilization, including pharmacologic interventions and agitation management
- diagnostic workup to evaluate medical and psychiatric conditions
- further psychiatric evaluation
- determining safe disposition.1
Triage determines the next step
Initial clinical assessment and triage are necessary to select the appropriate immediate intervention. When a patient arrives in the ER, determine if he or she requires urgent medical attention. Basic initial screening should include:
- vital signs
- finger stick blood glucose
- medical history
- signs or symptoms of intoxication or withdrawal
- signs of trauma (eg, neck ligature marks, gunshot wounds, lacerations)
- asking the patient to give a brief history leading up to the current presentation.
A review of medical records may reveal patients’ medical and psychiatric history and allergies. Collateral documentation—such as ambulance run sheets or police reports—may provide additional information. If no immediate medical intervention is warranted, determine if the patient can wait in an open, unlocked waiting area or if he or she needs to be in an unlocked area with a sitter, a locked open area, or a secluded room with access to restraints. In general, psychotic patients who pose a threat of harm to themselves or others or cannot care for themselves because of their psychosis need locked areas or observation.
Initial psychiatric stabilization
Agitation is diagnostically unspecific but can occur in patients with psychosis. Psychotic patients can become unpredictably and impulsively aggressive and assaultive. Rapid intervention is necessary to minimize risk of bodily harm to the patient and those around the patient. Physicians often must make quick interventions based on limited clinical information. It is important to recognize early signs and symptoms of agitation, including:
- restlessness (pacing, fidgeting, hand wringing, fist clenching, posturing)
- irritability
- decreased attention
- inappropriate or hostile behaviors.2
Pharmacologic interventions. The initial goals of pharmacologic treatment are to calm the patient without oversedation, thereby allowing the patient to take part in his or her care and begin treatment for the primary psychotic illness.3,4 Offering oral medications first and a choice of medications may help a patient feel more in control of the situation. If a patient has to be physically restrained, pharmacotherapy may limit the amount of time spent in restraints.
Medication choice depends on several factors, including onset of action, available formulation (eg, IM, liquid, rapidly dissolving), the patient’s previous medication response, side effect profile, allergies or adverse reactions to medications, and medical comorbidities.3 If a patient has a known psychotic illness, it may be helpful to administer the patient’s regular antipsychotic or anxiolytic medication. Some medications, such as lithium, are not effective in the acute setting and should be avoided. Additionally, benzodiazepines other than lorazepam or midazolam should not be administered IM because of erratic absorption.
Antipsychotics can be used for psychotic patients with or without agitation. Benzodiazepines may treat agitation, but are not specific for psychosis. Haloperidol can be used to treat acute psychosis and has proven efficacy for agitation. Benzodiazepines can decrease acute agitation and have efficacy similar to haloperidol, but with more sedation.5 A combination of lorazepam and haloperidol is thought to be superior to either medication alone.6 Lorazepam helps maintain sedation and decreases potential side effects caused by haloperidol. Consensus guidelines from 2001 and 2005 recommend combined haloperidol and lorazepam for first-line treatment of acute agitation.3,7 High-potency antipsychotics such as haloperidol have an increased risk for extrapyramidal symptoms (EPS), particularly acute dystonic reactions—involuntary, sustained muscle contractions—in susceptible patients (eg, antipsychotic-naïve patients); consider starting diphenhydramine, 25 to 50 mg, or benztropine, 0.5 to 2 mg, to prevent EPS from high-potency antipsychotics (Algorithm 1).
Algorithm 1: Treating acute psychosis: Choosing pharmacologic agents
EPS: extrapyramidal symptoms; PO: by mouth; SL: sublingual
Second-generation antipsychotics (SGAs) increasingly have been used for managing acute agitation in patients with an underlying psychotic disorder. Guidelines from a 2012 American Association for Emergency Psychiatry workgroup recommend using an SGA as monotherapy or in combination with another medication instead of haloperidol to treat agitated patients with a known psychotic disorder.8 Clinical policy guidelines from the American College of Emergency Physicians recommend antipsychotic monotherapy for agitation and initial treatment in patients with a known psychiatric illness for which antipsychotic treatment is indicated (eg, schizophrenia).9 For patients with known psychotic illness, expert opinion recommends oral risperidone or olanzapine.3,8 The combination of oral risperidone plus lorazepam may be as effective as the IM haloperidol and IM lorazepam combination.10 Patients who are too agitated to take oral doses may require parenteral medications. Ziprasidone, olanzapine, and aripiprazole are available in IM formulations. Ziprasidone, 20 mg IM, is well tolerated and has been shown to be effective in decreasing acute agitation symptoms in patients with psychotic disorders.11 Olanzapine is as effective as haloperidol in decreasing agitation in patients with schizophrenia, with lower rates of EPS.12 In a double-blind, placebo-controlled trial, psychotic symptoms in patients with schizophrenia or schizoaffective disorder decreased within 2 hours of IM olanzapine administration.13 Both IM ziprasidone and olanzapine have a relatively rapid onset of action (within 30 minutes), which makes them reasonable choices in the acute setting. Olanzapine has a long half-life (21 to 50 hours); therefore, patients’ comorbid medical conditions, such as cardiac abnormalities or hypotension, must be considered. If parenteral medication is required, IM olanzapine or IM ziprasidone is recommended.8 IM haloperidol with a benzodiazepine also can be considered.3
Coadministration of parenteral olanzapine and a benzodiazepine can lead to severe orthostatic hypotension and cardiac or respiratory depression and should be avoided in geriatric patients.14 Finally, it is important to rule out presentations that may worsen with antipsychotic treatment, including phencyclidine (PCP) toxicity (could worsen dystonic reactions), anticholinergic delirium, neuroleptic malignant syndrome (NMS), or catatonia.
If a patient does not respond to the initial dose of a medication, the dose may be repeated. However, doses should not be repeated until a patient is so sedated that he or she cannot take part in his or her care, or until he or she has developed significant EPS.
In addition to antipsychotics, consider loading with oral divalproex for patients who are acutely psychotic in the context of a manic episode (Table).15,16 Higher serum divalproex levels—target serum levels >94 μg/mL—are associated with greater efficacy as measured by change from baseline in Mania Rating Scale or Young Mania Rating Scale scores compared with placebo.15 For acutely psychotic schizophrenia patients, there is evidence of benefit with initial treatment with divalproex combined with an SGA. In a randomized, double-blind study, patients treated with divalproex plus olanzapine or risperidone showed quicker initial resolution of psychotic symptoms compared with olanzapine or risperidone monotherapy, but no better long-term benefit.16 Clinicians may consider this well-tolerated combination after an appropriate medical workup. This finding of early benefit was not replicated with divalproex extended-release.17
Table
Divalproex dosing for patients with acute psychosis and mania
| Initial dose | Titration | |
|---|---|---|
| Acute mania15 | Divalproex delayed-release: 750 mg/d Divalproex extended-release: 20 mg/kg/d | Increase to clinical effectiveness or maximum serum level of 125 μg/mL |
| Exacerbation of psychosis16 | Divalproex: 15 mg/kg/d (in 2 doses) | Increase to clinical effectiveness over 12 days or maximum dosage of 30 mg/kg/d |
Side effects and adverse reactions. Treatment with antipsychotics may cause QTc interval prolongation, which can lead to increased risk for torsades de pointes and sudden death due to ventricular fibrillation. However, there have been few cases of torsades de pointes after oral haloperidol and none with IM haloperidol compared with at least 30 cases of torsades de pointes after IV haloperidol treatment. Torsades de pointes after risperidone, olanzapine, or ziprasidone treatment has not been reported.18
Hypotension and bradycardia may occur in patients treated with olanzapine; however, these signs occur less frequently in agitated patients.18 Antipsychotic treatment increases risk for EPS, including acute dystonia, akathisia (subjective restlessness with desire to move), and parkinsonism (shuffling gait, resting tremor, rigidity and bradykinesia), as well as NMS.
Nonpharmacologic interventions. Verbal intervention to try to de-escalate an agitated, psychotic patient should be attempted first; however, this is not always possible. Other behavioral interventions include offering a meal, blanket, or pillow, or other comforting options to decrease the patient’s anxiety associated with psychosis.2 However, if agitated psychotic patients continue to display aggressive behaviors and pose a risk of harm to themselves or those around them, physical restraints should be considered because the clinician must balance protecting the patient’s rights with others’ safety. If physical restraints are used, medication also should be administered. Remove physical restraints as soon as safely possible; the Joint Commission has established standards for minimizing harm when using physical restraints.19
Diagnostic workup
Once a patient is medically stable in the ER, begin further workup of the etiology of the psychosis (Algorithm 2). All patients should have a physical exam, provided they are calm and in behavioral control. Monitor vital signs; patients at risk of withdrawal from substances should be monitored more frequently. Although there is no established standard for “medical clearance” of a psychiatric patient,20 all patients should undergo basic laboratory tests, including basic metabolic panel, complete blood count, and urine toxicology. The extent of the workup is determined by the clinical situation and suspected cause of psychosis.21
Algorithm 2: Diagnostic workup of an acutely psychotic patient
ER: emergency room; EEG: electroencephalography; LP: lumbar puncture; TSH: thyroid-stimulating hormone
If you suspect delirium, the underlying medical etiology must be identified and treated. Up to 40% of hospitalized patients with delirium may have psychosis.22 Psychosis in a delirious patient may be characterized by poorly formed delusions and visual hallucinations. Delirious patients often are inattentive, easily distracted, and disoriented, with a fluctuating clinical course. Patients with psychosis generally do not have impaired attention and are alert with intact memory. However, acutely psychotic patients may be quite disorganized and uncooperative, which makes it difficult to distinguish between these 2 diagnoses. Serial exams may help clarify the clinical picture. It is important to remember that patients with a history of a psychotic disorder may have a superimposed delirium.
In young patients (age 18 to 30) with new-onset psychosis, consider drug-induced psychosis; PCP, lysergic acid diethylamide, and methamphetamine intoxication and withdrawal can lead to psychotic presentations. Additionally, comorbid substance use is common among patients with primary psychotic disorders. One study found 37% of first-episode psychotic patients misused drugs or alcohol, similar to the lifetime rate of patients with chronic psychotic disorders.23,24 Check urine and serum toxicology screens and obtain relevant substance use history. Brain MRI may be considered for patients with first presentation of psychosis; however, there is little evidence to support head CT imaging unless there is known head trauma.25 Electroencephalography and lumbar puncture can be considered if clinically indicated.
Further psychiatric evaluation
Obtaining a psychiatric history is necessary to determine the etiology of the acute psychotic presentation. The timing and duration of psychotic symptoms are key. Acute symptom onset with fluctuating course and impaired attention suggests a delirious process. A gradual decline in functioning over several months to years in a young person suggests a first episode of a psychotic disorder (eg, schizophrenia). Drug abuse is common among young persons with a psychotic disorder and a positive drug screen for a psychogenic substance does not exclude a primary psychotic disorder.
If a patient has a history of schizophrenia, bipolar disorder, or psychotic depression, acutely worsening psychosis may be considered an acute or chronic presentation. Even in patients diagnosed with a psychotic illness, it is necessary to determine the cause of symptom exacerbation. Medication nonadherence (which can be partial), substance use, psychosocial stressors, or underlying medical illness should be considered. Collateral information from family or friends may be crucial to understanding a patient’s presentation.
Safe disposition
Patients who pose a risk of harm to themselves or others or who are so impaired by their psychosis that they cannot care for themselves generally should be admitted to an inpatient psychiatric facility. For some psychotic patients who are agreeable to treatment and not prone to violence, less restrictive settings—such as a crisis intervention unit or respite facility—may be appropriate. A patient with first-episode psychosis could be admitted for further diagnostic clarification and treatment initiation. Manic patients often have no insight into their illness and may need hospitalization for containment and assurance of medication adherence. Goals of inpatient care include initiating or resuming pharmacologic treatment to reduce psychotic symptoms and beginning the recovery process. Response rates—defined as ≥20% improvement in total score on a psychopathology scale such as the Positive and Negative Syndrome Scale—will vary, but can take ≥4 weeks in some patients with first-episode schizophrenia.26 However, most patients will be stabilized and ready for discharge before 4 weeks. Family education and alliance building with the patient and family are important during hospitalization.
Related Resources
- Schwartz S, Weathers, M. The psychotic patient. In: Riba MB, Ravindranath D, eds. Clinical manual of emergency psychiatry. Arlington, VA: American Psychiatric Publishing, Inc.; 2010:115-140.
- American Association for Emergency Psychiatry. http://emergencypsychiatry.org.
Drug Brand Names
- Aripiprazole • Abilify
- Benztropine • Cogentin
- Diphenhydramine • Benadryl
- Divalproex • Depakote
- Haloperidol • Haldol
- Lithium • Eskalith, Lithobid
- Lorazepam • Ativan
- Midazolam • Versed
- Olanzapine • Zyprexa
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr. Freudenreich receives grant or research support from Beacon Health Strategies, Global Medical Education, MGH Psychiatry Academy, Optimal Medicine, Pfizer Inc., and PsychoGenics.
Drs. Brown and Stoklosa report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acute psychosis is a symptom that can be caused by many psychiatric and medical conditions. Psychotic patients might be unable to provide a history or participate in treatment if they are agitated, hostile, or violent. An appropriate workup may reveal the etiology of the psychosis; secondary causes, such as medical illness and substance use, are prevalent in the emergency room (ER) setting. If the patient has an underlying primary psychotic disorder, such as schizophrenia or mania, illness-specific intervention will help acutely and long-term. With agitated and uncooperative psychotic patients, clinicians often have to intervene quickly to ensure the safety of the patient and those nearby.
This article focuses on the initial evaluation and treatment of psychotic patients in the ER, either by a psychiatric emergency service or a psychiatric consultant. This process can be broken down into:
- triage or initial clinical assessment
- initial psychiatric stabilization, including pharmacologic interventions and agitation management
- diagnostic workup to evaluate medical and psychiatric conditions
- further psychiatric evaluation
- determining safe disposition.1
Triage determines the next step
Initial clinical assessment and triage are necessary to select the appropriate immediate intervention. When a patient arrives in the ER, determine if he or she requires urgent medical attention. Basic initial screening should include:
- vital signs
- finger stick blood glucose
- medical history
- signs or symptoms of intoxication or withdrawal
- signs of trauma (eg, neck ligature marks, gunshot wounds, lacerations)
- asking the patient to give a brief history leading up to the current presentation.
A review of medical records may reveal patients’ medical and psychiatric history and allergies. Collateral documentation—such as ambulance run sheets or police reports—may provide additional information. If no immediate medical intervention is warranted, determine if the patient can wait in an open, unlocked waiting area or if he or she needs to be in an unlocked area with a sitter, a locked open area, or a secluded room with access to restraints. In general, psychotic patients who pose a threat of harm to themselves or others or cannot care for themselves because of their psychosis need locked areas or observation.
Initial psychiatric stabilization
Agitation is diagnostically unspecific but can occur in patients with psychosis. Psychotic patients can become unpredictably and impulsively aggressive and assaultive. Rapid intervention is necessary to minimize risk of bodily harm to the patient and those around the patient. Physicians often must make quick interventions based on limited clinical information. It is important to recognize early signs and symptoms of agitation, including:
- restlessness (pacing, fidgeting, hand wringing, fist clenching, posturing)
- irritability
- decreased attention
- inappropriate or hostile behaviors.2
Pharmacologic interventions. The initial goals of pharmacologic treatment are to calm the patient without oversedation, thereby allowing the patient to take part in his or her care and begin treatment for the primary psychotic illness.3,4 Offering oral medications first and a choice of medications may help a patient feel more in control of the situation. If a patient has to be physically restrained, pharmacotherapy may limit the amount of time spent in restraints.
Medication choice depends on several factors, including onset of action, available formulation (eg, IM, liquid, rapidly dissolving), the patient’s previous medication response, side effect profile, allergies or adverse reactions to medications, and medical comorbidities.3 If a patient has a known psychotic illness, it may be helpful to administer the patient’s regular antipsychotic or anxiolytic medication. Some medications, such as lithium, are not effective in the acute setting and should be avoided. Additionally, benzodiazepines other than lorazepam or midazolam should not be administered IM because of erratic absorption.
Antipsychotics can be used for psychotic patients with or without agitation. Benzodiazepines may treat agitation, but are not specific for psychosis. Haloperidol can be used to treat acute psychosis and has proven efficacy for agitation. Benzodiazepines can decrease acute agitation and have efficacy similar to haloperidol, but with more sedation.5 A combination of lorazepam and haloperidol is thought to be superior to either medication alone.6 Lorazepam helps maintain sedation and decreases potential side effects caused by haloperidol. Consensus guidelines from 2001 and 2005 recommend combined haloperidol and lorazepam for first-line treatment of acute agitation.3,7 High-potency antipsychotics such as haloperidol have an increased risk for extrapyramidal symptoms (EPS), particularly acute dystonic reactions—involuntary, sustained muscle contractions—in susceptible patients (eg, antipsychotic-naïve patients); consider starting diphenhydramine, 25 to 50 mg, or benztropine, 0.5 to 2 mg, to prevent EPS from high-potency antipsychotics (Algorithm 1).
Algorithm 1: Treating acute psychosis: Choosing pharmacologic agents
EPS: extrapyramidal symptoms; PO: by mouth; SL: sublingual
Second-generation antipsychotics (SGAs) increasingly have been used for managing acute agitation in patients with an underlying psychotic disorder. Guidelines from a 2012 American Association for Emergency Psychiatry workgroup recommend using an SGA as monotherapy or in combination with another medication instead of haloperidol to treat agitated patients with a known psychotic disorder.8 Clinical policy guidelines from the American College of Emergency Physicians recommend antipsychotic monotherapy for agitation and initial treatment in patients with a known psychiatric illness for which antipsychotic treatment is indicated (eg, schizophrenia).9 For patients with known psychotic illness, expert opinion recommends oral risperidone or olanzapine.3,8 The combination of oral risperidone plus lorazepam may be as effective as the IM haloperidol and IM lorazepam combination.10 Patients who are too agitated to take oral doses may require parenteral medications. Ziprasidone, olanzapine, and aripiprazole are available in IM formulations. Ziprasidone, 20 mg IM, is well tolerated and has been shown to be effective in decreasing acute agitation symptoms in patients with psychotic disorders.11 Olanzapine is as effective as haloperidol in decreasing agitation in patients with schizophrenia, with lower rates of EPS.12 In a double-blind, placebo-controlled trial, psychotic symptoms in patients with schizophrenia or schizoaffective disorder decreased within 2 hours of IM olanzapine administration.13 Both IM ziprasidone and olanzapine have a relatively rapid onset of action (within 30 minutes), which makes them reasonable choices in the acute setting. Olanzapine has a long half-life (21 to 50 hours); therefore, patients’ comorbid medical conditions, such as cardiac abnormalities or hypotension, must be considered. If parenteral medication is required, IM olanzapine or IM ziprasidone is recommended.8 IM haloperidol with a benzodiazepine also can be considered.3
Coadministration of parenteral olanzapine and a benzodiazepine can lead to severe orthostatic hypotension and cardiac or respiratory depression and should be avoided in geriatric patients.14 Finally, it is important to rule out presentations that may worsen with antipsychotic treatment, including phencyclidine (PCP) toxicity (could worsen dystonic reactions), anticholinergic delirium, neuroleptic malignant syndrome (NMS), or catatonia.
If a patient does not respond to the initial dose of a medication, the dose may be repeated. However, doses should not be repeated until a patient is so sedated that he or she cannot take part in his or her care, or until he or she has developed significant EPS.
In addition to antipsychotics, consider loading with oral divalproex for patients who are acutely psychotic in the context of a manic episode (Table).15,16 Higher serum divalproex levels—target serum levels >94 μg/mL—are associated with greater efficacy as measured by change from baseline in Mania Rating Scale or Young Mania Rating Scale scores compared with placebo.15 For acutely psychotic schizophrenia patients, there is evidence of benefit with initial treatment with divalproex combined with an SGA. In a randomized, double-blind study, patients treated with divalproex plus olanzapine or risperidone showed quicker initial resolution of psychotic symptoms compared with olanzapine or risperidone monotherapy, but no better long-term benefit.16 Clinicians may consider this well-tolerated combination after an appropriate medical workup. This finding of early benefit was not replicated with divalproex extended-release.17
Table
Divalproex dosing for patients with acute psychosis and mania
| Initial dose | Titration | |
|---|---|---|
| Acute mania15 | Divalproex delayed-release: 750 mg/d Divalproex extended-release: 20 mg/kg/d | Increase to clinical effectiveness or maximum serum level of 125 μg/mL |
| Exacerbation of psychosis16 | Divalproex: 15 mg/kg/d (in 2 doses) | Increase to clinical effectiveness over 12 days or maximum dosage of 30 mg/kg/d |
Side effects and adverse reactions. Treatment with antipsychotics may cause QTc interval prolongation, which can lead to increased risk for torsades de pointes and sudden death due to ventricular fibrillation. However, there have been few cases of torsades de pointes after oral haloperidol and none with IM haloperidol compared with at least 30 cases of torsades de pointes after IV haloperidol treatment. Torsades de pointes after risperidone, olanzapine, or ziprasidone treatment has not been reported.18
Hypotension and bradycardia may occur in patients treated with olanzapine; however, these signs occur less frequently in agitated patients.18 Antipsychotic treatment increases risk for EPS, including acute dystonia, akathisia (subjective restlessness with desire to move), and parkinsonism (shuffling gait, resting tremor, rigidity and bradykinesia), as well as NMS.
Nonpharmacologic interventions. Verbal intervention to try to de-escalate an agitated, psychotic patient should be attempted first; however, this is not always possible. Other behavioral interventions include offering a meal, blanket, or pillow, or other comforting options to decrease the patient’s anxiety associated with psychosis.2 However, if agitated psychotic patients continue to display aggressive behaviors and pose a risk of harm to themselves or those around them, physical restraints should be considered because the clinician must balance protecting the patient’s rights with others’ safety. If physical restraints are used, medication also should be administered. Remove physical restraints as soon as safely possible; the Joint Commission has established standards for minimizing harm when using physical restraints.19
Diagnostic workup
Once a patient is medically stable in the ER, begin further workup of the etiology of the psychosis (Algorithm 2). All patients should have a physical exam, provided they are calm and in behavioral control. Monitor vital signs; patients at risk of withdrawal from substances should be monitored more frequently. Although there is no established standard for “medical clearance” of a psychiatric patient,20 all patients should undergo basic laboratory tests, including basic metabolic panel, complete blood count, and urine toxicology. The extent of the workup is determined by the clinical situation and suspected cause of psychosis.21
Algorithm 2: Diagnostic workup of an acutely psychotic patient
ER: emergency room; EEG: electroencephalography; LP: lumbar puncture; TSH: thyroid-stimulating hormone
If you suspect delirium, the underlying medical etiology must be identified and treated. Up to 40% of hospitalized patients with delirium may have psychosis.22 Psychosis in a delirious patient may be characterized by poorly formed delusions and visual hallucinations. Delirious patients often are inattentive, easily distracted, and disoriented, with a fluctuating clinical course. Patients with psychosis generally do not have impaired attention and are alert with intact memory. However, acutely psychotic patients may be quite disorganized and uncooperative, which makes it difficult to distinguish between these 2 diagnoses. Serial exams may help clarify the clinical picture. It is important to remember that patients with a history of a psychotic disorder may have a superimposed delirium.
In young patients (age 18 to 30) with new-onset psychosis, consider drug-induced psychosis; PCP, lysergic acid diethylamide, and methamphetamine intoxication and withdrawal can lead to psychotic presentations. Additionally, comorbid substance use is common among patients with primary psychotic disorders. One study found 37% of first-episode psychotic patients misused drugs or alcohol, similar to the lifetime rate of patients with chronic psychotic disorders.23,24 Check urine and serum toxicology screens and obtain relevant substance use history. Brain MRI may be considered for patients with first presentation of psychosis; however, there is little evidence to support head CT imaging unless there is known head trauma.25 Electroencephalography and lumbar puncture can be considered if clinically indicated.
Further psychiatric evaluation
Obtaining a psychiatric history is necessary to determine the etiology of the acute psychotic presentation. The timing and duration of psychotic symptoms are key. Acute symptom onset with fluctuating course and impaired attention suggests a delirious process. A gradual decline in functioning over several months to years in a young person suggests a first episode of a psychotic disorder (eg, schizophrenia). Drug abuse is common among young persons with a psychotic disorder and a positive drug screen for a psychogenic substance does not exclude a primary psychotic disorder.
If a patient has a history of schizophrenia, bipolar disorder, or psychotic depression, acutely worsening psychosis may be considered an acute or chronic presentation. Even in patients diagnosed with a psychotic illness, it is necessary to determine the cause of symptom exacerbation. Medication nonadherence (which can be partial), substance use, psychosocial stressors, or underlying medical illness should be considered. Collateral information from family or friends may be crucial to understanding a patient’s presentation.
Safe disposition
Patients who pose a risk of harm to themselves or others or who are so impaired by their psychosis that they cannot care for themselves generally should be admitted to an inpatient psychiatric facility. For some psychotic patients who are agreeable to treatment and not prone to violence, less restrictive settings—such as a crisis intervention unit or respite facility—may be appropriate. A patient with first-episode psychosis could be admitted for further diagnostic clarification and treatment initiation. Manic patients often have no insight into their illness and may need hospitalization for containment and assurance of medication adherence. Goals of inpatient care include initiating or resuming pharmacologic treatment to reduce psychotic symptoms and beginning the recovery process. Response rates—defined as ≥20% improvement in total score on a psychopathology scale such as the Positive and Negative Syndrome Scale—will vary, but can take ≥4 weeks in some patients with first-episode schizophrenia.26 However, most patients will be stabilized and ready for discharge before 4 weeks. Family education and alliance building with the patient and family are important during hospitalization.
Related Resources
- Schwartz S, Weathers, M. The psychotic patient. In: Riba MB, Ravindranath D, eds. Clinical manual of emergency psychiatry. Arlington, VA: American Psychiatric Publishing, Inc.; 2010:115-140.
- American Association for Emergency Psychiatry. http://emergencypsychiatry.org.
Drug Brand Names
- Aripiprazole • Abilify
- Benztropine • Cogentin
- Diphenhydramine • Benadryl
- Divalproex • Depakote
- Haloperidol • Haldol
- Lithium • Eskalith, Lithobid
- Lorazepam • Ativan
- Midazolam • Versed
- Olanzapine • Zyprexa
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr. Freudenreich receives grant or research support from Beacon Health Strategies, Global Medical Education, MGH Psychiatry Academy, Optimal Medicine, Pfizer Inc., and PsychoGenics.
Drs. Brown and Stoklosa report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Marco CA, Vaughan J. Emergency management of agitation in schizophrenia. Am J Emerg Med. 2005;23(6):767-776.
2. Freudenreich O. Emergency management of acute psychosis. In: Freudenreich O ed. Psychotic disorders: a practical guide. New York, NY: Wolter Kluwer/Lippincott Williams & Wilkins; 2008:72–78.
3. Allen MH, Currier GW, Carpenter D, et al. Expert Consensus Panel for Behavioral Emergencies 2005. The expert consensus guideline series. Treatment of behavioral emergencies 2005. J Psychiatr Pract. 2005;11(1 suppl):S5-S108.
4. National Institute for Health and Clinical Excellence. Schizophrenia: core interventions in the treatment and management of schizophrenia in primary and secondary care. London United Kingdom: National Institute for Clinical Excellence; 2002.
5. Allen MH. Managing the agitated psychotic patient: a reappraisal of the evidence. J Clin Psychiatry. 2000;61 (14 suppl):S11-S20.
6. Battaglia J, Moss S, Rush J, et al. Haloperidol, lorazepam, or both for psychotic agitation? A multicenter, prospective, double-blind, emergency department study. Am J Emerg Med. 1997;15(4):335-340.
7. Allen MH, Currier GW, Hughes DH, et al. Expert Consensus Panel for Behavioral Emergencies. The expert consensus guideline series. Treatment of behavioral emergencies. Postgrad Med. 2001;(Spec no):1-88.
8. Wilson MP, Pepper D, Currier GW, et al. The psychopharmacology of agitation: consensus statement of American Association for Emergency Psychiatry project BETA psychopharmacology workgroup. West J Emerg Med. 2012;13(1):26-34.
9. Lukens TW, Wolf SJ, Edlow JA, et al. American College of Emergency Physicians Clinical Policies Subcommittee (Writing Committee) on Critical Issues in the Diagnosis and Management of the Adult Psychiatric Patient in the Emergency Department. Clinical policy: critical issues in the diagnosis and management of the adult psychiatric patient in the emergency department. Ann Emerg Med. 2006;47(1):79-99.
10. Currier GW, Chou JC, Feifel D, et al. Acute treatment of psychotic agitation: a randomized comparison of oral treatment with risperidone and lorazepam versus intramuscular treatment with haloperidol and lorazepam. J Clin Psychiatry. 2004;65(3):386-394.
11. Daniel DG, Potkin SG, Reeves KR, et al. Intramuscular (IM) ziprasidone 20 mg is effective in reducing agitation associated with psychosis: a double-blind, randomized trial. Psychopharmacology (Berl). 2001;155(2):128-134.
12. Wright P, Birkett M, David SR, et al. Double-blind, placebo-controlled comparison of intramuscular olanzapine and intramuscular haloperidol in the treatment of acute agitation in schizophrenia. Am J Psychiatry. 2001;158(7):1149-1151.
13. Kapur S, Arenovic T, Agid O, et al. Evidence for onset of antipsychotic effects within the first 24 hours of treatment. Am J Psychiatry. 2005;162(5):939-946.
14. Marder SR, Sorasburu S, Dunayevic E, et al. Case reports of postmarketing adverse event experiences with olanzapine intramuscular treatment in patients with agitation. J Clin Psychiatry. 2010;71(4):433-441.
15. Allen MH, Hirschfeld RM, Wozniak PJ, et al. Linear relationship of valproate serum concentration to response and optimal serum levels for acute mania. Am J Psychiatry. 2006;163(2):272-275.
16. Casey DE, Daniel DG, Wassef AA, et al. Effect of divalproex combined with olanzapine or risperidone in patients with an acute exacerbation of schizophrenia. Neuropsychopharmacology. 2003;28(1):182-192.
17. Casey DE, Daniel DG, Tamminga C, et al. Divalproex ER combined with olanzapine or risperidone for treatment of acute exacerbations of schizophrenia. Neuropsychopharmacology. 2009;34(5):1330-1338.
18. Currier GW, Allen MH, Bunney EB, et al. Safety of medications used to treat acute agitation. J Emerg Med. 2004;27(4 suppl):S19-S24.
19. The Joint Commission. Sentinel event alert. Preventing restraint deaths. Published November 18 1998. http://www.jointcommission.org/assets/1/18/SEA_8.pdf. Accessed October 26, 2012
20. Janiak BD, Atteberry S. Medical clearance of the psychiatric patient in the emergency department. J Emerg Med. 2012;43(5):866-870.
21. Freudenreich O, Schulz SC, Goff DC. Initial medical work-up of first-episode psychosis: a conceptual review. Early Interv Psychiatry. 2009;3(1):10-18.
22. Webster R, Holroyd S. Prevalence of psychotic symptoms in delirium. Psychosomatics. 2000;41(6):519-522.
23. Cantwell R, Brewin J, Glazebrook C, et al. Prevalence of substance misuse in first-episode psychosis. Br J Psychiatry. 1999;174:150-153.
24. Green AI, Tohen MF, Hamer RM, et al. First episode schizophrenia-related psychosis and substance use disorders: acute response to olanzapine and haloperidol. Schizophr Res. 2004;66(2-3):125-135.
25. Goulet K, Deschamps B, Evoy F, et al. Use of brain imaging (computed tomography and magnetic resonance imaging) in first-episode psychosis: review and retrospective study. Can J Psychiatry. 2009;54(7):493-501.
26. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10):1785-1804.
1. Marco CA, Vaughan J. Emergency management of agitation in schizophrenia. Am J Emerg Med. 2005;23(6):767-776.
2. Freudenreich O. Emergency management of acute psychosis. In: Freudenreich O ed. Psychotic disorders: a practical guide. New York, NY: Wolter Kluwer/Lippincott Williams & Wilkins; 2008:72–78.
3. Allen MH, Currier GW, Carpenter D, et al. Expert Consensus Panel for Behavioral Emergencies 2005. The expert consensus guideline series. Treatment of behavioral emergencies 2005. J Psychiatr Pract. 2005;11(1 suppl):S5-S108.
4. National Institute for Health and Clinical Excellence. Schizophrenia: core interventions in the treatment and management of schizophrenia in primary and secondary care. London United Kingdom: National Institute for Clinical Excellence; 2002.
5. Allen MH. Managing the agitated psychotic patient: a reappraisal of the evidence. J Clin Psychiatry. 2000;61 (14 suppl):S11-S20.
6. Battaglia J, Moss S, Rush J, et al. Haloperidol, lorazepam, or both for psychotic agitation? A multicenter, prospective, double-blind, emergency department study. Am J Emerg Med. 1997;15(4):335-340.
7. Allen MH, Currier GW, Hughes DH, et al. Expert Consensus Panel for Behavioral Emergencies. The expert consensus guideline series. Treatment of behavioral emergencies. Postgrad Med. 2001;(Spec no):1-88.
8. Wilson MP, Pepper D, Currier GW, et al. The psychopharmacology of agitation: consensus statement of American Association for Emergency Psychiatry project BETA psychopharmacology workgroup. West J Emerg Med. 2012;13(1):26-34.
9. Lukens TW, Wolf SJ, Edlow JA, et al. American College of Emergency Physicians Clinical Policies Subcommittee (Writing Committee) on Critical Issues in the Diagnosis and Management of the Adult Psychiatric Patient in the Emergency Department. Clinical policy: critical issues in the diagnosis and management of the adult psychiatric patient in the emergency department. Ann Emerg Med. 2006;47(1):79-99.
10. Currier GW, Chou JC, Feifel D, et al. Acute treatment of psychotic agitation: a randomized comparison of oral treatment with risperidone and lorazepam versus intramuscular treatment with haloperidol and lorazepam. J Clin Psychiatry. 2004;65(3):386-394.
11. Daniel DG, Potkin SG, Reeves KR, et al. Intramuscular (IM) ziprasidone 20 mg is effective in reducing agitation associated with psychosis: a double-blind, randomized trial. Psychopharmacology (Berl). 2001;155(2):128-134.
12. Wright P, Birkett M, David SR, et al. Double-blind, placebo-controlled comparison of intramuscular olanzapine and intramuscular haloperidol in the treatment of acute agitation in schizophrenia. Am J Psychiatry. 2001;158(7):1149-1151.
13. Kapur S, Arenovic T, Agid O, et al. Evidence for onset of antipsychotic effects within the first 24 hours of treatment. Am J Psychiatry. 2005;162(5):939-946.
14. Marder SR, Sorasburu S, Dunayevic E, et al. Case reports of postmarketing adverse event experiences with olanzapine intramuscular treatment in patients with agitation. J Clin Psychiatry. 2010;71(4):433-441.
15. Allen MH, Hirschfeld RM, Wozniak PJ, et al. Linear relationship of valproate serum concentration to response and optimal serum levels for acute mania. Am J Psychiatry. 2006;163(2):272-275.
16. Casey DE, Daniel DG, Wassef AA, et al. Effect of divalproex combined with olanzapine or risperidone in patients with an acute exacerbation of schizophrenia. Neuropsychopharmacology. 2003;28(1):182-192.
17. Casey DE, Daniel DG, Tamminga C, et al. Divalproex ER combined with olanzapine or risperidone for treatment of acute exacerbations of schizophrenia. Neuropsychopharmacology. 2009;34(5):1330-1338.
18. Currier GW, Allen MH, Bunney EB, et al. Safety of medications used to treat acute agitation. J Emerg Med. 2004;27(4 suppl):S19-S24.
19. The Joint Commission. Sentinel event alert. Preventing restraint deaths. Published November 18 1998. http://www.jointcommission.org/assets/1/18/SEA_8.pdf. Accessed October 26, 2012
20. Janiak BD, Atteberry S. Medical clearance of the psychiatric patient in the emergency department. J Emerg Med. 2012;43(5):866-870.
21. Freudenreich O, Schulz SC, Goff DC. Initial medical work-up of first-episode psychosis: a conceptual review. Early Interv Psychiatry. 2009;3(1):10-18.
22. Webster R, Holroyd S. Prevalence of psychotic symptoms in delirium. Psychosomatics. 2000;41(6):519-522.
23. Cantwell R, Brewin J, Glazebrook C, et al. Prevalence of substance misuse in first-episode psychosis. Br J Psychiatry. 1999;174:150-153.
24. Green AI, Tohen MF, Hamer RM, et al. First episode schizophrenia-related psychosis and substance use disorders: acute response to olanzapine and haloperidol. Schizophr Res. 2004;66(2-3):125-135.
25. Goulet K, Deschamps B, Evoy F, et al. Use of brain imaging (computed tomography and magnetic resonance imaging) in first-episode psychosis: review and retrospective study. Can J Psychiatry. 2009;54(7):493-501.
26. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10):1785-1804.