User login
Depression and inflammation: Examining the link
Sneezing, coughing, and a sore throat are hallmark symptoms of a common cold, but what keeps you in bed are the accompanying fatigue, inattentiveness, loss of appetite, change in sleep pattern, heightened perception of pain, and apathetic withdrawal. This “sickness behavior” is induced by inflammatory markers released in response to illness.1,2 These symptoms are similar to the constellation of symptoms that define depression. Within the inflammatory response to illness, we see the shadow of depression, but the precise relationship remains murky.
Is depression part of a normal somatic inflammatory response run amok? Some researchers have argued that “sickness behavior” is adaptive, forcing the body into a constricted pattern in order to funnel energy into healing.1,3 If depression and inflammation are related, depression pushes past these adaptive roots and is less a forced pause than a debilitating withdrawal. Perhaps depression, or a subtype, is a sign of inflammation along with heat, pain, redness, and swelling. In some instances, depression may be a sign of an underlying inflammatory process.4
In our progression toward understanding depression’s pathophysiology, we see factors that point to a relationship between depression and inflammation:
• depression frequently is comorbid with many inflammatory illnesses
• increased inflammatory biomarkers are associated with major depressive disorder (MDD)
• exposure to immunomodulating agents may increase the risk of developing depression
• stress can activate proinflammatory pathways
• antidepressants can decrease inflammatory response
• inhibition of inflammatory pathways can improve mood.
Exploring these factors and a possible pathway linking inflammation and neurobiologic changes found in depression allows us to look closer at the possible integration of the inflammatory process and depressive symptoms.
Illness and depression rates
Individuals with inflammatory illnesses—autoimmune diseases, cardiovascular disease, diabetes, and cancer—often struggle with depression. Nearly 1 in 5 persons with cardiovascular disease experiences MDD.5 A diabetes diagnosis doubles the odds of having depression.6 Up to 70% of patients with autoimmune diseases, such as rheumatoid arthritis or systemic lupus erythematosus, experience depression.7,8 In a large-scale longitudinal study, having a prior autoimmune disease increased the risk of depression by 45% and history of hospitalization with infection increased a patient’s risk by 62%; the risk more than doubled in individuals with both.9 Several studies show that 15% to 25% of cancer patients experience depression,10 compared with 9% in the general population.11
Role of inflammatory markers
During an inflammatory episode the body releases cytokines, which are small, cell-signaling protein molecules. These inflammatory markers launch signaling cascades that incite the immune system into action. Type 1 cytokines (interferon-ã, tumor necrosis factor-á [TNF-á], interleukin [IL]-1) enhance cellular immune responses, and type 2 cytokines (IL-6, IL-10, IL-13) engage antibody responses. These cytokines also induce acute phase proteins, such as C-reactive protein (CRP), which can activate the immune system. Significantly higher levels of inflammatory markers are associated with a range of depressive symptoms, which grants insight into disease severity and treatment response.3,12,13
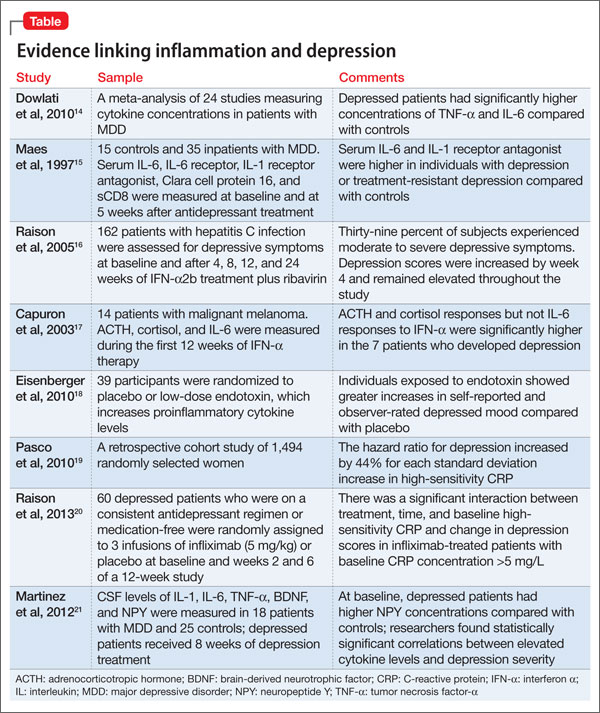
Multiple studies have explored the link between depression and inflammatory markers (Table).14-21 Peripheral inflammatory markers such as IL-6, IL-1â, CRP, and TNF-á are elevated in inflammatory diseases and in otherwise healthy individuals with MDD.12 In a meta-analysis of 24 studies measuring cytokines in depressed patients, Dowlati et al14 found individuals with MDD had significantly higher concentrations of TNF-á and IL-6 compared with controls. Increased peripheral inflammatory markers were found among antidepressant nonresponders more often than those who responded to treatment.15,22
Cytokines and depression risk
Administering immunomodulating agents has been shown to increase the risk of developing depression. Injecting animals with IL-1â or TNF-á causes sickness behavior in a dose- and time-related manner.1 As these inflammatory signaling proteins increase, sickness behaviors become more pronounced.
In humans, a natural model arises in the use of the cytokine interferon-á (INF-á) for treating hepatitis C, multiple sclerosis, malignant melanoma, and some blood cancers. Patients receiving INF-á have higher rates of depression than those not administered interferon.16 Patients receiving chronic immunotherapy treatment show long-term changes in monoamine neurotransmitters and along the HPA axis; these changes mimic those seen in depressed individuals.17,23 Acutely administered immunotherapeutic agents, such as the typhoid vaccine, have led to depressive symptoms with brain changes similar to those seen in MDD.18 Low levels of IL-6 and CRP independently predicted development of depression over several years.19
Immunotherapy-induced depression looks similar to any other major depressive episode through our current diagnostic framework and at the molecular and anatomical level.
Stress and inflammation
Depression can develop in the absence of inflammatory illness. Knowing that depressive symptoms may be associated with increased peripheral inflammatory markers, what induces the inflammatory process in some persons who are depressed but medically healthy? One theory is that psychological stress can activate inflammation.
Acute and chronic stress is associated with increased availability of proinflammatory cytokines and decreases in anti-inflammatory cytokines.3,24 One theory looks to glucocorticoid response to stress as an explanation. Miller et al25 found glucocorticoid sensitivity decreased among depressed women after exposure to a mock job interview stressor and increased among nondepressed controls. Because glucocorticoids normally stop the inflammatory cascade, this finding suggests depressed individuals may not be able to control inflammation during stress.26 At the level of genetic expression, there is increased transcription of proinflammatory genes in response to stress as a result of increased activation of nuclear factor kappa B.3,27
Shared pathways
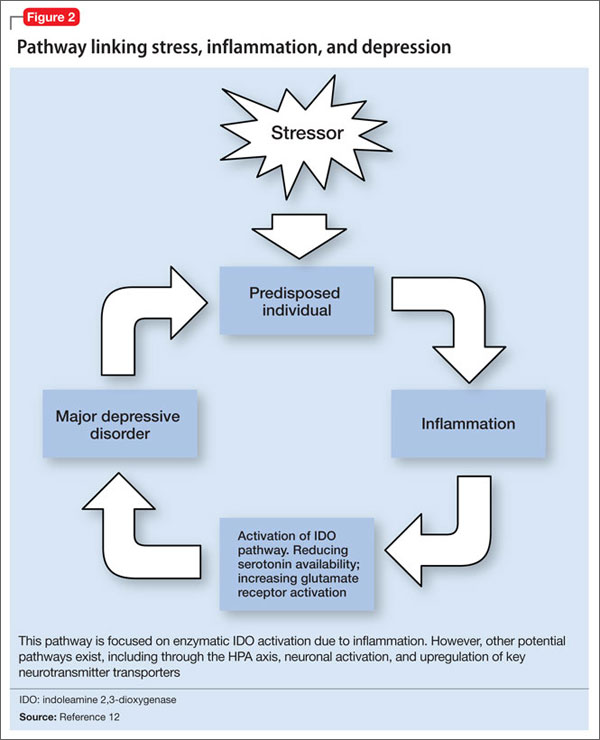
If there is a relationship between inflammation and depression, what is the possible shared pathway?
There are 4 pathways by which cytokines effect changes in the CNS:12
• cytokines can activate primary afferent neurons (eg, vagal nerve)
• cytokines, released by macrophage-like cells in response to pathogens, diffuse through the brain’s circumventricular organs
• cytokine transporters saturate the blood-brain barrier
• cytokine IL-1 activates receptors on perivascular macrophages and endothelial cells of brain venules, causing local release of prostaglandin E2.
Through these pathways, cytokines initiate a cascade of reactions that lower serotonin levels and boost glutamatergic actions, possibly contributing to development of depressive symptoms. Depression correlates with a deficiency in serotonergic neurotransmission and increased glutamate receptor N-methyl-d-aspartate (NMDA) activation.28
Proinflammatory cytokines activate theextrahepatic enzyme indoleamine 2,3-dioxygenase (IDO), which degrades tryptophan, a precursor to serotonin (Figure 1). Tryptophan is channeled increasingly toward production of kynurenine via IDO degradation, competing with the serotonin pathway. Within the microglia, which are preferentially activated over astrocytes during inflammatory states, kynurenine is metabolized into quinolinic acid, which is an agonist of glutamatergic NMDA receptors.28 Therefore, there is a serotonergic deficiency and glutamatergic overdrive in proinflammatory states that paves the way toward a likely depressive syndrome (Figure 2).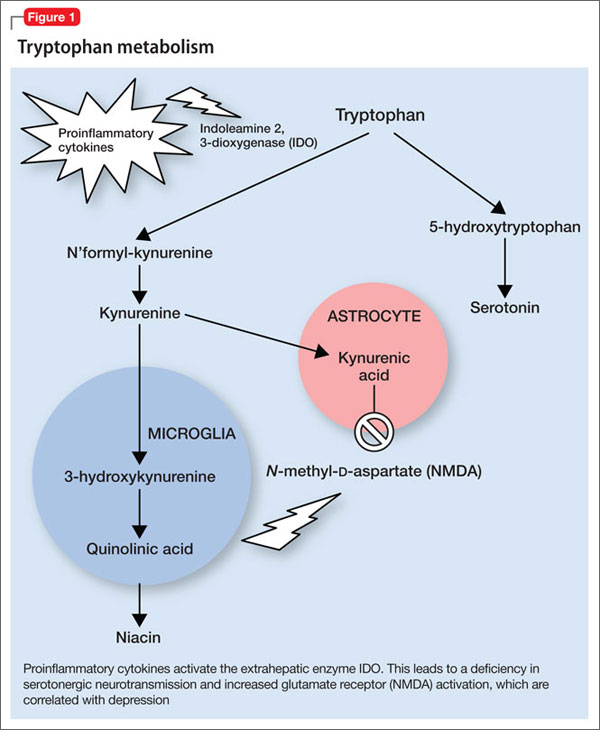
Antidepressants’ effects
The symptoms of cytokine-induced depression are no different from MDD with unknown etiology29 and both are effectively treated with antidepressants. Even sickness behavior can be improved with antidepressant treatment.30
Antidepressants not only decrease immunotherapy-induced depressive symptoms but have been shown to decrease inflammatory response and lower proinflammatory factors (IL-2, IL-6, TNF-á, and INF-ã).31-33 Electroconvulsive therapy has been shown to normalize elevated TNF-á levels.34
Enhancing depression treatment
Researchers are investigating whether treatment with anti-inflammatory agents can ease depressive symptoms. In animal studies, normal behavioral reactions to a stressor—similar to sickness behavior and overlapping with several features of depression—were reduced with administration of cytokine antagonists or anti-inflammatory cytokines directly into the brain.35 However, there have been few successful trials in humans. Both anti-inflammatory agents such as cyclooxygenase-2 (COX-2) inhibitors, acetylsalicylic acid (aspirin), and TNF receptor antagonists can enhance depression treatments. Persoons et al36 found that Crohn’s disease patients who had higher pretreatment CRP levels and MDD had greater remission of depressive symptoms after treatment with the TNF-á antagonist infliximab. In studies, depression within the context of other autoimmune disorders or any condition with increased inflammation has responded to treatment with TNF-á antagonists.37,38 COX-2 inhibitors added to a standard antidepressant regimen improved depressive symptoms in medically healthy individuals during an acute depressive episode.39 Aspirin has shown some benefits as an adjuvant agent in persons who have failed selective serotonin reuptake inhibitor monotherapy.40,41
These anti-inflammatory agents have shown benefits in treating depression in some persons, but not in all. The key difference between those subsets of patients is elusive, mired in the complex interactions of the many systems that contribute to the symptoms we label as depression.
Future clinical applications
The association between depression and inflammation raises the possibility of a tantalizing line of future theories and treatment options. However, when considered individually, these pieces are limited in defining the precise relationship - a task nearly impossible for such a diffuse symptom as inflammation and such a complex disease as depression.
It is evident that inflammation and depression form a strong relationship to each other in individuals, which suggests the possibility of an inflammatory subtype of depression. At least within that limited group, there is the possibility of successful intervention and treatment of depression by directly treating inflammation with anti-inflammatory agents.
Perhaps once the relationship between depression and inflammation is further defined and a high-risk population identified—maybe even by genotype—depressive symptoms might be used to flag a provider’s attention to a possible disease process and serve as a new tool for identifying dangerous inflammatory activity at an early stage. Managing stress and depression may become the next tool to prevent inflammatory diseases.
Given our current knowledge, clinicians treating patients with inflammatory conditions should be aware of the increased risk of depression and ensure that depression screening is routinely completed and treatment is initiated or referrals made as needed. Ensuring appropriate depression treatment may help improve patients’ quality of life and ease the inflammatory response itself.
For psychiatrists seeing patients with an inflammatory condition, brief explanations of the known links between depression and inflammation can provide patients—particularly those ambivalent about seeking mental health care—support for engaging in treatment and adhering to medication. Describing the links between inflammation and depression also can help encourage regular exercise and healthy diets rich in fruits, vegetables, and omega-3 fatty acids. In cases of treatment-resistant depression, particularly in those with known high inflammatory factors, it may be worthwhile to consider anti-inflammatory agents, such as infliximab, as an adjuvant treatment.
The relationship between inflammation and depression is rapidly unfolding, but the full intricacies have not yet described. However, this beginning awareness of the interplay among stress, inflammation, and depression can broaden our approach to care and treatment.
Bottom Line
Depression and inflammation are linked in many ways, although neither appears to be wholly necessary or sufficient for the other. Most likely there exists a particular subset of patients for whom inflammation will precipitate and perpetuate depression.
Related Resources
- The Emory University Mind-Body Program. www.
psychiatry.emory.edu/PROGRAMS/mindbody/index.html. - Gabriel B. The evolutionary advantage of depression. The Atlantic. October 2, 2012. www.theatlantic.com/health/archive/2012/10/the-evolutionary-advantage-of-depression/263124.
Drug Brand Names
Infliximab • Remicade Ribavirin • Rebetol, Virazole
Interferon-α • Intron
Disclosure
Dr. Almond reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
References
1. Dantzer R, O’Connor JC, Freund GG, et al. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46-56.
2. Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12(2):123-137.
3. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27(1):24-31.
4. Lamers F, Vogelzangs N, Merikangas KR, et al. Evidence for a differential role of HPA-axis function, inflammation and metabolic syndrome in melancholic versus atypical depression [published online October 23, 2012]. Mol Psychiatry. doi: 10.1038/mp.2012.144.
5. Hoen P, Kupper N, de Jonge P. Depression and cardiovascular disease progression: epidemiology, mechanisms and treatment. In: Hjemdahl P, Rosengren A, Steptoe A, eds. Stress and cardiovascular disease. London, United Kingdom: Springer; 2012:211-233.
6. Anderson RJ, Freedland KE, Clouse RE, et al. The prevalence of comorbid depression in adults with diabetes: a meta-analysis. Diabetes Care. 2001;24(6):1069-1078.
7. Bachen EA, Chesney MA, Criswell LA. Prevalence of mood and anxiety disorders in women with systemic lupus erythematosus. Arthritis Rheum. 2009;61(6):822-829.
8. Dickens C, McGowan L, Clark-Carter D, et al. Depression in rheumatoid arthritis: a systematic review of the literature with meta-analysis. Psychosom Med. 2002;64(1):52-60.
9. Benros ME, Waltoft BL, Nordentoft M, et al. Autoimmunity and infections as risk factors for depression and other severe mental illnesses. Neurology, Psychiatry and Brain Research. 2012;18(2):40-41.
10. National Cancer Institute. Depression (PDQ). http://www.cancer.gov/cancertopics/pdq/supportivecare/depression/HealthProfessional/page1. Updated January 9, 2013. Accessed April 23, 2013.
11. Centers for Disease Control and Prevention. Current depression among adults—United States, 2006 and 2008. Morb Mortal Wkly Rep. 2010;59(38):1229-1235.
12. Krishnadas R, Cavanagh J. Depression: an inflammatory illness? J Neurol Neurosurg Psychiatry. 2012;83(5):495-502.
13. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732-741.
14. Dowlati Y, Herrmann N, Swardfager W, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67(5):446-457.
15. Maes M, Bosmans E, De Jongh R, et al. Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine. 1997;9(11):853-858.
16. Raison CL, Borisov AS, Broadwell SD, et al. Depression during pegylated interferon-alpha plus ribavirin therapy: prevalence and prediction. J Clin Psychiatry. 2005;66(1):41-48.
17. Capuron L, Raison CL, Musselman DL, et al. Association of exaggerated HPA axis response to the initial injection of interferon-alpha with development of depression during interferon-alpha therapy. Am J Psychiatry. 2003;160(7):1342-1345.
18. Eisenberger NI, Berkman ET, Inagaki TK, et al. Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry. 2010;68(8):748-754.
19. Pasco JA, Nicholson GC, Williams LJ, et al. Association of high-sensitivity C-reactive protein with de novo major depression. Br J Psychiatry. 2010;197(5):372-377.
20. Raison CL, Rutherford RE, Woolwine BJ, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry. 2013;70(1):31-41.
21. Martinez JM, Garakani A, Yehuda R, et al. Proinflammatory and “resiliency” proteins in the CSF of patients with major depression. Depress Anxiety. 2012;29(1):32-38.
22. Lanquillon S, Krieg JC, Bening-Abu-Shach U, et al. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology. 2000;22(4):370-379.
23. Raison CL, Miller AH. Is depression an inflammatory disorder? Curr Psychiatry Rep. 2011;13(6):467-775.
24. Maes M, Song C, Lin A, et al. The effects of psychological stress on humans: increased production of pro-inflammatory cytokines and Th1-like response in stress-induced anxiety. Cytokine. 1998;10(4):313-318.
25. Miller GE, Rohleder N, Stetler C, et al. Clinical depression and regulation of the inflammatory response during acute stress. Psychosom Med. 2005;67(5):679-687.
26. Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. 2003;160(9):1554-1565.
27. Tak PP, Firestein GS. NF-êB: a key role in inflammatory diseases. J Clin Invest. 2001;107(1):7-12.
28. Müller N, Schwarz MJ. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry. 2007;12(11):988-1000.
29. Capuron L, Fornwalt FB, Knight BT, et al. Does cytokine-induced depression differ from idiopathic major depression in medically healthy individuals? J Affect Disord. 2009;119(1-3):181-185.
30. Yirmiya R, Pollak Y, Morag M, et al. Illness, cytokines, and depression. Ann N Y Acad Sci. 2000;917(1):478-487.
31. Maes M. The immunoregulatory effects of antidepressants. Hum Psychopharmacol. 2001;16(1):95-103.
32. Szuster-Ciesielska A, Tustanowska-Stachura A, Słotwin`ska M, et al. In vitro immunoregulatory effects of antidepressants in healthy volunteers. Pol J Pharmacol. 2003;55(3):353-362.
33. Maes M, Berk M, Goehler L, et al. Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med. 2012;10(1):66.
34. Hestad KA, Tønseth S, Støen CD, et al. Raised plasma levels of tumor necrosis factor [alpha] in patients with depression: normalization during electroconvulsive therapy. J ECT. 2003;19(4):183-188.
35. Maier SF, Watkins LR. Intracerebroventricular interleukin-1 receptor antagonist blocks the enhancement of fear conditioning and interference with escape produced by inescapable shock. Brain Res. 1995;695(2):279-282.
36. Persoons P, Vermeire S, Demyttenaere K, et al. The impact of major depressive disorder on the short- and long-term outcome of Crohn’s disease treatment with infliximab. Aliment Pharmacol Ther. 2005;22(2):101-110.
37. Mathias SD, Colwell HH, Miller DP, et al. Health-related quality of life and functional status of patients with rheumatoid arthritis randomly assigned to receive etanercept or placebo. Clin Ther. 2000;22(1):128-139.
38. Raison C, Rutherford RE, Woolwine B, et al. The tumor necrosis factor-alpha antagonist infliximab reduces depressive symptoms in patients with treatment resistant depression and high inflammation. Brain, Behavior, and Immunity. 2012;26(suppl 1):S49.
39. Müller N, Schwarz MJ, Dehning S, et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry. 2006;11(7):680-684.
40. Mendlewicz J, Kriwin P, Oswald P, et al. Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: a pilot open-label study. Int Clin Psychopharmacol. 2006;21(4):227-231.
41. Brunello N, Alboni S, Capone G, et al. Acetylsalicylic acid accelerates the antidepressant effect of fluoxetine in the chronic escape deficit model of depression. Int Clin Psychopharmacol. 2006;21(4):219-225.
Sneezing, coughing, and a sore throat are hallmark symptoms of a common cold, but what keeps you in bed are the accompanying fatigue, inattentiveness, loss of appetite, change in sleep pattern, heightened perception of pain, and apathetic withdrawal. This “sickness behavior” is induced by inflammatory markers released in response to illness.1,2 These symptoms are similar to the constellation of symptoms that define depression. Within the inflammatory response to illness, we see the shadow of depression, but the precise relationship remains murky.
Is depression part of a normal somatic inflammatory response run amok? Some researchers have argued that “sickness behavior” is adaptive, forcing the body into a constricted pattern in order to funnel energy into healing.1,3 If depression and inflammation are related, depression pushes past these adaptive roots and is less a forced pause than a debilitating withdrawal. Perhaps depression, or a subtype, is a sign of inflammation along with heat, pain, redness, and swelling. In some instances, depression may be a sign of an underlying inflammatory process.4
In our progression toward understanding depression’s pathophysiology, we see factors that point to a relationship between depression and inflammation:
• depression frequently is comorbid with many inflammatory illnesses
• increased inflammatory biomarkers are associated with major depressive disorder (MDD)
• exposure to immunomodulating agents may increase the risk of developing depression
• stress can activate proinflammatory pathways
• antidepressants can decrease inflammatory response
• inhibition of inflammatory pathways can improve mood.
Exploring these factors and a possible pathway linking inflammation and neurobiologic changes found in depression allows us to look closer at the possible integration of the inflammatory process and depressive symptoms.
Illness and depression rates
Individuals with inflammatory illnesses—autoimmune diseases, cardiovascular disease, diabetes, and cancer—often struggle with depression. Nearly 1 in 5 persons with cardiovascular disease experiences MDD.5 A diabetes diagnosis doubles the odds of having depression.6 Up to 70% of patients with autoimmune diseases, such as rheumatoid arthritis or systemic lupus erythematosus, experience depression.7,8 In a large-scale longitudinal study, having a prior autoimmune disease increased the risk of depression by 45% and history of hospitalization with infection increased a patient’s risk by 62%; the risk more than doubled in individuals with both.9 Several studies show that 15% to 25% of cancer patients experience depression,10 compared with 9% in the general population.11
Role of inflammatory markers
During an inflammatory episode the body releases cytokines, which are small, cell-signaling protein molecules. These inflammatory markers launch signaling cascades that incite the immune system into action. Type 1 cytokines (interferon-ã, tumor necrosis factor-á [TNF-á], interleukin [IL]-1) enhance cellular immune responses, and type 2 cytokines (IL-6, IL-10, IL-13) engage antibody responses. These cytokines also induce acute phase proteins, such as C-reactive protein (CRP), which can activate the immune system. Significantly higher levels of inflammatory markers are associated with a range of depressive symptoms, which grants insight into disease severity and treatment response.3,12,13
Multiple studies have explored the link between depression and inflammatory markers (Table).14-21 Peripheral inflammatory markers such as IL-6, IL-1â, CRP, and TNF-á are elevated in inflammatory diseases and in otherwise healthy individuals with MDD.12 In a meta-analysis of 24 studies measuring cytokines in depressed patients, Dowlati et al14 found individuals with MDD had significantly higher concentrations of TNF-á and IL-6 compared with controls. Increased peripheral inflammatory markers were found among antidepressant nonresponders more often than those who responded to treatment.15,22
Cytokines and depression risk
Administering immunomodulating agents has been shown to increase the risk of developing depression. Injecting animals with IL-1â or TNF-á causes sickness behavior in a dose- and time-related manner.1 As these inflammatory signaling proteins increase, sickness behaviors become more pronounced.
In humans, a natural model arises in the use of the cytokine interferon-á (INF-á) for treating hepatitis C, multiple sclerosis, malignant melanoma, and some blood cancers. Patients receiving INF-á have higher rates of depression than those not administered interferon.16 Patients receiving chronic immunotherapy treatment show long-term changes in monoamine neurotransmitters and along the HPA axis; these changes mimic those seen in depressed individuals.17,23 Acutely administered immunotherapeutic agents, such as the typhoid vaccine, have led to depressive symptoms with brain changes similar to those seen in MDD.18 Low levels of IL-6 and CRP independently predicted development of depression over several years.19
Immunotherapy-induced depression looks similar to any other major depressive episode through our current diagnostic framework and at the molecular and anatomical level.
Stress and inflammation
Depression can develop in the absence of inflammatory illness. Knowing that depressive symptoms may be associated with increased peripheral inflammatory markers, what induces the inflammatory process in some persons who are depressed but medically healthy? One theory is that psychological stress can activate inflammation.
Acute and chronic stress is associated with increased availability of proinflammatory cytokines and decreases in anti-inflammatory cytokines.3,24 One theory looks to glucocorticoid response to stress as an explanation. Miller et al25 found glucocorticoid sensitivity decreased among depressed women after exposure to a mock job interview stressor and increased among nondepressed controls. Because glucocorticoids normally stop the inflammatory cascade, this finding suggests depressed individuals may not be able to control inflammation during stress.26 At the level of genetic expression, there is increased transcription of proinflammatory genes in response to stress as a result of increased activation of nuclear factor kappa B.3,27
Shared pathways
If there is a relationship between inflammation and depression, what is the possible shared pathway?
There are 4 pathways by which cytokines effect changes in the CNS:12
• cytokines can activate primary afferent neurons (eg, vagal nerve)
• cytokines, released by macrophage-like cells in response to pathogens, diffuse through the brain’s circumventricular organs
• cytokine transporters saturate the blood-brain barrier
• cytokine IL-1 activates receptors on perivascular macrophages and endothelial cells of brain venules, causing local release of prostaglandin E2.
Through these pathways, cytokines initiate a cascade of reactions that lower serotonin levels and boost glutamatergic actions, possibly contributing to development of depressive symptoms. Depression correlates with a deficiency in serotonergic neurotransmission and increased glutamate receptor N-methyl-d-aspartate (NMDA) activation.28
Proinflammatory cytokines activate theextrahepatic enzyme indoleamine 2,3-dioxygenase (IDO), which degrades tryptophan, a precursor to serotonin (Figure 1). Tryptophan is channeled increasingly toward production of kynurenine via IDO degradation, competing with the serotonin pathway. Within the microglia, which are preferentially activated over astrocytes during inflammatory states, kynurenine is metabolized into quinolinic acid, which is an agonist of glutamatergic NMDA receptors.28 Therefore, there is a serotonergic deficiency and glutamatergic overdrive in proinflammatory states that paves the way toward a likely depressive syndrome (Figure 2).
Antidepressants’ effects
The symptoms of cytokine-induced depression are no different from MDD with unknown etiology29 and both are effectively treated with antidepressants. Even sickness behavior can be improved with antidepressant treatment.30
Antidepressants not only decrease immunotherapy-induced depressive symptoms but have been shown to decrease inflammatory response and lower proinflammatory factors (IL-2, IL-6, TNF-á, and INF-ã).31-33 Electroconvulsive therapy has been shown to normalize elevated TNF-á levels.34
Enhancing depression treatment
Researchers are investigating whether treatment with anti-inflammatory agents can ease depressive symptoms. In animal studies, normal behavioral reactions to a stressor—similar to sickness behavior and overlapping with several features of depression—were reduced with administration of cytokine antagonists or anti-inflammatory cytokines directly into the brain.35 However, there have been few successful trials in humans. Both anti-inflammatory agents such as cyclooxygenase-2 (COX-2) inhibitors, acetylsalicylic acid (aspirin), and TNF receptor antagonists can enhance depression treatments. Persoons et al36 found that Crohn’s disease patients who had higher pretreatment CRP levels and MDD had greater remission of depressive symptoms after treatment with the TNF-á antagonist infliximab. In studies, depression within the context of other autoimmune disorders or any condition with increased inflammation has responded to treatment with TNF-á antagonists.37,38 COX-2 inhibitors added to a standard antidepressant regimen improved depressive symptoms in medically healthy individuals during an acute depressive episode.39 Aspirin has shown some benefits as an adjuvant agent in persons who have failed selective serotonin reuptake inhibitor monotherapy.40,41
These anti-inflammatory agents have shown benefits in treating depression in some persons, but not in all. The key difference between those subsets of patients is elusive, mired in the complex interactions of the many systems that contribute to the symptoms we label as depression.
Future clinical applications
The association between depression and inflammation raises the possibility of a tantalizing line of future theories and treatment options. However, when considered individually, these pieces are limited in defining the precise relationship - a task nearly impossible for such a diffuse symptom as inflammation and such a complex disease as depression.
It is evident that inflammation and depression form a strong relationship to each other in individuals, which suggests the possibility of an inflammatory subtype of depression. At least within that limited group, there is the possibility of successful intervention and treatment of depression by directly treating inflammation with anti-inflammatory agents.
Perhaps once the relationship between depression and inflammation is further defined and a high-risk population identified—maybe even by genotype—depressive symptoms might be used to flag a provider’s attention to a possible disease process and serve as a new tool for identifying dangerous inflammatory activity at an early stage. Managing stress and depression may become the next tool to prevent inflammatory diseases.
Given our current knowledge, clinicians treating patients with inflammatory conditions should be aware of the increased risk of depression and ensure that depression screening is routinely completed and treatment is initiated or referrals made as needed. Ensuring appropriate depression treatment may help improve patients’ quality of life and ease the inflammatory response itself.
For psychiatrists seeing patients with an inflammatory condition, brief explanations of the known links between depression and inflammation can provide patients—particularly those ambivalent about seeking mental health care—support for engaging in treatment and adhering to medication. Describing the links between inflammation and depression also can help encourage regular exercise and healthy diets rich in fruits, vegetables, and omega-3 fatty acids. In cases of treatment-resistant depression, particularly in those with known high inflammatory factors, it may be worthwhile to consider anti-inflammatory agents, such as infliximab, as an adjuvant treatment.
The relationship between inflammation and depression is rapidly unfolding, but the full intricacies have not yet described. However, this beginning awareness of the interplay among stress, inflammation, and depression can broaden our approach to care and treatment.
Bottom Line
Depression and inflammation are linked in many ways, although neither appears to be wholly necessary or sufficient for the other. Most likely there exists a particular subset of patients for whom inflammation will precipitate and perpetuate depression.
Related Resources
- The Emory University Mind-Body Program. www.
psychiatry.emory.edu/PROGRAMS/mindbody/index.html. - Gabriel B. The evolutionary advantage of depression. The Atlantic. October 2, 2012. www.theatlantic.com/health/archive/2012/10/the-evolutionary-advantage-of-depression/263124.
Drug Brand Names
Infliximab • Remicade Ribavirin • Rebetol, Virazole
Interferon-α • Intron
Disclosure
Dr. Almond reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
References
1. Dantzer R, O’Connor JC, Freund GG, et al. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46-56.
2. Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12(2):123-137.
3. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27(1):24-31.
4. Lamers F, Vogelzangs N, Merikangas KR, et al. Evidence for a differential role of HPA-axis function, inflammation and metabolic syndrome in melancholic versus atypical depression [published online October 23, 2012]. Mol Psychiatry. doi: 10.1038/mp.2012.144.
5. Hoen P, Kupper N, de Jonge P. Depression and cardiovascular disease progression: epidemiology, mechanisms and treatment. In: Hjemdahl P, Rosengren A, Steptoe A, eds. Stress and cardiovascular disease. London, United Kingdom: Springer; 2012:211-233.
6. Anderson RJ, Freedland KE, Clouse RE, et al. The prevalence of comorbid depression in adults with diabetes: a meta-analysis. Diabetes Care. 2001;24(6):1069-1078.
7. Bachen EA, Chesney MA, Criswell LA. Prevalence of mood and anxiety disorders in women with systemic lupus erythematosus. Arthritis Rheum. 2009;61(6):822-829.
8. Dickens C, McGowan L, Clark-Carter D, et al. Depression in rheumatoid arthritis: a systematic review of the literature with meta-analysis. Psychosom Med. 2002;64(1):52-60.
9. Benros ME, Waltoft BL, Nordentoft M, et al. Autoimmunity and infections as risk factors for depression and other severe mental illnesses. Neurology, Psychiatry and Brain Research. 2012;18(2):40-41.
10. National Cancer Institute. Depression (PDQ). http://www.cancer.gov/cancertopics/pdq/supportivecare/depression/HealthProfessional/page1. Updated January 9, 2013. Accessed April 23, 2013.
11. Centers for Disease Control and Prevention. Current depression among adults—United States, 2006 and 2008. Morb Mortal Wkly Rep. 2010;59(38):1229-1235.
12. Krishnadas R, Cavanagh J. Depression: an inflammatory illness? J Neurol Neurosurg Psychiatry. 2012;83(5):495-502.
13. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732-741.
14. Dowlati Y, Herrmann N, Swardfager W, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67(5):446-457.
15. Maes M, Bosmans E, De Jongh R, et al. Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine. 1997;9(11):853-858.
16. Raison CL, Borisov AS, Broadwell SD, et al. Depression during pegylated interferon-alpha plus ribavirin therapy: prevalence and prediction. J Clin Psychiatry. 2005;66(1):41-48.
17. Capuron L, Raison CL, Musselman DL, et al. Association of exaggerated HPA axis response to the initial injection of interferon-alpha with development of depression during interferon-alpha therapy. Am J Psychiatry. 2003;160(7):1342-1345.
18. Eisenberger NI, Berkman ET, Inagaki TK, et al. Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry. 2010;68(8):748-754.
19. Pasco JA, Nicholson GC, Williams LJ, et al. Association of high-sensitivity C-reactive protein with de novo major depression. Br J Psychiatry. 2010;197(5):372-377.
20. Raison CL, Rutherford RE, Woolwine BJ, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry. 2013;70(1):31-41.
21. Martinez JM, Garakani A, Yehuda R, et al. Proinflammatory and “resiliency” proteins in the CSF of patients with major depression. Depress Anxiety. 2012;29(1):32-38.
22. Lanquillon S, Krieg JC, Bening-Abu-Shach U, et al. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology. 2000;22(4):370-379.
23. Raison CL, Miller AH. Is depression an inflammatory disorder? Curr Psychiatry Rep. 2011;13(6):467-775.
24. Maes M, Song C, Lin A, et al. The effects of psychological stress on humans: increased production of pro-inflammatory cytokines and Th1-like response in stress-induced anxiety. Cytokine. 1998;10(4):313-318.
25. Miller GE, Rohleder N, Stetler C, et al. Clinical depression and regulation of the inflammatory response during acute stress. Psychosom Med. 2005;67(5):679-687.
26. Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. 2003;160(9):1554-1565.
27. Tak PP, Firestein GS. NF-êB: a key role in inflammatory diseases. J Clin Invest. 2001;107(1):7-12.
28. Müller N, Schwarz MJ. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry. 2007;12(11):988-1000.
29. Capuron L, Fornwalt FB, Knight BT, et al. Does cytokine-induced depression differ from idiopathic major depression in medically healthy individuals? J Affect Disord. 2009;119(1-3):181-185.
30. Yirmiya R, Pollak Y, Morag M, et al. Illness, cytokines, and depression. Ann N Y Acad Sci. 2000;917(1):478-487.
31. Maes M. The immunoregulatory effects of antidepressants. Hum Psychopharmacol. 2001;16(1):95-103.
32. Szuster-Ciesielska A, Tustanowska-Stachura A, Słotwin`ska M, et al. In vitro immunoregulatory effects of antidepressants in healthy volunteers. Pol J Pharmacol. 2003;55(3):353-362.
33. Maes M, Berk M, Goehler L, et al. Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med. 2012;10(1):66.
34. Hestad KA, Tønseth S, Støen CD, et al. Raised plasma levels of tumor necrosis factor [alpha] in patients with depression: normalization during electroconvulsive therapy. J ECT. 2003;19(4):183-188.
35. Maier SF, Watkins LR. Intracerebroventricular interleukin-1 receptor antagonist blocks the enhancement of fear conditioning and interference with escape produced by inescapable shock. Brain Res. 1995;695(2):279-282.
36. Persoons P, Vermeire S, Demyttenaere K, et al. The impact of major depressive disorder on the short- and long-term outcome of Crohn’s disease treatment with infliximab. Aliment Pharmacol Ther. 2005;22(2):101-110.
37. Mathias SD, Colwell HH, Miller DP, et al. Health-related quality of life and functional status of patients with rheumatoid arthritis randomly assigned to receive etanercept or placebo. Clin Ther. 2000;22(1):128-139.
38. Raison C, Rutherford RE, Woolwine B, et al. The tumor necrosis factor-alpha antagonist infliximab reduces depressive symptoms in patients with treatment resistant depression and high inflammation. Brain, Behavior, and Immunity. 2012;26(suppl 1):S49.
39. Müller N, Schwarz MJ, Dehning S, et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry. 2006;11(7):680-684.
40. Mendlewicz J, Kriwin P, Oswald P, et al. Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: a pilot open-label study. Int Clin Psychopharmacol. 2006;21(4):227-231.
41. Brunello N, Alboni S, Capone G, et al. Acetylsalicylic acid accelerates the antidepressant effect of fluoxetine in the chronic escape deficit model of depression. Int Clin Psychopharmacol. 2006;21(4):219-225.
Sneezing, coughing, and a sore throat are hallmark symptoms of a common cold, but what keeps you in bed are the accompanying fatigue, inattentiveness, loss of appetite, change in sleep pattern, heightened perception of pain, and apathetic withdrawal. This “sickness behavior” is induced by inflammatory markers released in response to illness.1,2 These symptoms are similar to the constellation of symptoms that define depression. Within the inflammatory response to illness, we see the shadow of depression, but the precise relationship remains murky.
Is depression part of a normal somatic inflammatory response run amok? Some researchers have argued that “sickness behavior” is adaptive, forcing the body into a constricted pattern in order to funnel energy into healing.1,3 If depression and inflammation are related, depression pushes past these adaptive roots and is less a forced pause than a debilitating withdrawal. Perhaps depression, or a subtype, is a sign of inflammation along with heat, pain, redness, and swelling. In some instances, depression may be a sign of an underlying inflammatory process.4
In our progression toward understanding depression’s pathophysiology, we see factors that point to a relationship between depression and inflammation:
• depression frequently is comorbid with many inflammatory illnesses
• increased inflammatory biomarkers are associated with major depressive disorder (MDD)
• exposure to immunomodulating agents may increase the risk of developing depression
• stress can activate proinflammatory pathways
• antidepressants can decrease inflammatory response
• inhibition of inflammatory pathways can improve mood.
Exploring these factors and a possible pathway linking inflammation and neurobiologic changes found in depression allows us to look closer at the possible integration of the inflammatory process and depressive symptoms.
Illness and depression rates
Individuals with inflammatory illnesses—autoimmune diseases, cardiovascular disease, diabetes, and cancer—often struggle with depression. Nearly 1 in 5 persons with cardiovascular disease experiences MDD.5 A diabetes diagnosis doubles the odds of having depression.6 Up to 70% of patients with autoimmune diseases, such as rheumatoid arthritis or systemic lupus erythematosus, experience depression.7,8 In a large-scale longitudinal study, having a prior autoimmune disease increased the risk of depression by 45% and history of hospitalization with infection increased a patient’s risk by 62%; the risk more than doubled in individuals with both.9 Several studies show that 15% to 25% of cancer patients experience depression,10 compared with 9% in the general population.11
Role of inflammatory markers
During an inflammatory episode the body releases cytokines, which are small, cell-signaling protein molecules. These inflammatory markers launch signaling cascades that incite the immune system into action. Type 1 cytokines (interferon-ã, tumor necrosis factor-á [TNF-á], interleukin [IL]-1) enhance cellular immune responses, and type 2 cytokines (IL-6, IL-10, IL-13) engage antibody responses. These cytokines also induce acute phase proteins, such as C-reactive protein (CRP), which can activate the immune system. Significantly higher levels of inflammatory markers are associated with a range of depressive symptoms, which grants insight into disease severity and treatment response.3,12,13
Multiple studies have explored the link between depression and inflammatory markers (Table).14-21 Peripheral inflammatory markers such as IL-6, IL-1â, CRP, and TNF-á are elevated in inflammatory diseases and in otherwise healthy individuals with MDD.12 In a meta-analysis of 24 studies measuring cytokines in depressed patients, Dowlati et al14 found individuals with MDD had significantly higher concentrations of TNF-á and IL-6 compared with controls. Increased peripheral inflammatory markers were found among antidepressant nonresponders more often than those who responded to treatment.15,22
Cytokines and depression risk
Administering immunomodulating agents has been shown to increase the risk of developing depression. Injecting animals with IL-1â or TNF-á causes sickness behavior in a dose- and time-related manner.1 As these inflammatory signaling proteins increase, sickness behaviors become more pronounced.
In humans, a natural model arises in the use of the cytokine interferon-á (INF-á) for treating hepatitis C, multiple sclerosis, malignant melanoma, and some blood cancers. Patients receiving INF-á have higher rates of depression than those not administered interferon.16 Patients receiving chronic immunotherapy treatment show long-term changes in monoamine neurotransmitters and along the HPA axis; these changes mimic those seen in depressed individuals.17,23 Acutely administered immunotherapeutic agents, such as the typhoid vaccine, have led to depressive symptoms with brain changes similar to those seen in MDD.18 Low levels of IL-6 and CRP independently predicted development of depression over several years.19
Immunotherapy-induced depression looks similar to any other major depressive episode through our current diagnostic framework and at the molecular and anatomical level.
Stress and inflammation
Depression can develop in the absence of inflammatory illness. Knowing that depressive symptoms may be associated with increased peripheral inflammatory markers, what induces the inflammatory process in some persons who are depressed but medically healthy? One theory is that psychological stress can activate inflammation.
Acute and chronic stress is associated with increased availability of proinflammatory cytokines and decreases in anti-inflammatory cytokines.3,24 One theory looks to glucocorticoid response to stress as an explanation. Miller et al25 found glucocorticoid sensitivity decreased among depressed women after exposure to a mock job interview stressor and increased among nondepressed controls. Because glucocorticoids normally stop the inflammatory cascade, this finding suggests depressed individuals may not be able to control inflammation during stress.26 At the level of genetic expression, there is increased transcription of proinflammatory genes in response to stress as a result of increased activation of nuclear factor kappa B.3,27
Shared pathways
If there is a relationship between inflammation and depression, what is the possible shared pathway?
There are 4 pathways by which cytokines effect changes in the CNS:12
• cytokines can activate primary afferent neurons (eg, vagal nerve)
• cytokines, released by macrophage-like cells in response to pathogens, diffuse through the brain’s circumventricular organs
• cytokine transporters saturate the blood-brain barrier
• cytokine IL-1 activates receptors on perivascular macrophages and endothelial cells of brain venules, causing local release of prostaglandin E2.
Through these pathways, cytokines initiate a cascade of reactions that lower serotonin levels and boost glutamatergic actions, possibly contributing to development of depressive symptoms. Depression correlates with a deficiency in serotonergic neurotransmission and increased glutamate receptor N-methyl-d-aspartate (NMDA) activation.28
Proinflammatory cytokines activate theextrahepatic enzyme indoleamine 2,3-dioxygenase (IDO), which degrades tryptophan, a precursor to serotonin (Figure 1). Tryptophan is channeled increasingly toward production of kynurenine via IDO degradation, competing with the serotonin pathway. Within the microglia, which are preferentially activated over astrocytes during inflammatory states, kynurenine is metabolized into quinolinic acid, which is an agonist of glutamatergic NMDA receptors.28 Therefore, there is a serotonergic deficiency and glutamatergic overdrive in proinflammatory states that paves the way toward a likely depressive syndrome (Figure 2).
Antidepressants’ effects
The symptoms of cytokine-induced depression are no different from MDD with unknown etiology29 and both are effectively treated with antidepressants. Even sickness behavior can be improved with antidepressant treatment.30
Antidepressants not only decrease immunotherapy-induced depressive symptoms but have been shown to decrease inflammatory response and lower proinflammatory factors (IL-2, IL-6, TNF-á, and INF-ã).31-33 Electroconvulsive therapy has been shown to normalize elevated TNF-á levels.34
Enhancing depression treatment
Researchers are investigating whether treatment with anti-inflammatory agents can ease depressive symptoms. In animal studies, normal behavioral reactions to a stressor—similar to sickness behavior and overlapping with several features of depression—were reduced with administration of cytokine antagonists or anti-inflammatory cytokines directly into the brain.35 However, there have been few successful trials in humans. Both anti-inflammatory agents such as cyclooxygenase-2 (COX-2) inhibitors, acetylsalicylic acid (aspirin), and TNF receptor antagonists can enhance depression treatments. Persoons et al36 found that Crohn’s disease patients who had higher pretreatment CRP levels and MDD had greater remission of depressive symptoms after treatment with the TNF-á antagonist infliximab. In studies, depression within the context of other autoimmune disorders or any condition with increased inflammation has responded to treatment with TNF-á antagonists.37,38 COX-2 inhibitors added to a standard antidepressant regimen improved depressive symptoms in medically healthy individuals during an acute depressive episode.39 Aspirin has shown some benefits as an adjuvant agent in persons who have failed selective serotonin reuptake inhibitor monotherapy.40,41
These anti-inflammatory agents have shown benefits in treating depression in some persons, but not in all. The key difference between those subsets of patients is elusive, mired in the complex interactions of the many systems that contribute to the symptoms we label as depression.
Future clinical applications
The association between depression and inflammation raises the possibility of a tantalizing line of future theories and treatment options. However, when considered individually, these pieces are limited in defining the precise relationship - a task nearly impossible for such a diffuse symptom as inflammation and such a complex disease as depression.
It is evident that inflammation and depression form a strong relationship to each other in individuals, which suggests the possibility of an inflammatory subtype of depression. At least within that limited group, there is the possibility of successful intervention and treatment of depression by directly treating inflammation with anti-inflammatory agents.
Perhaps once the relationship between depression and inflammation is further defined and a high-risk population identified—maybe even by genotype—depressive symptoms might be used to flag a provider’s attention to a possible disease process and serve as a new tool for identifying dangerous inflammatory activity at an early stage. Managing stress and depression may become the next tool to prevent inflammatory diseases.
Given our current knowledge, clinicians treating patients with inflammatory conditions should be aware of the increased risk of depression and ensure that depression screening is routinely completed and treatment is initiated or referrals made as needed. Ensuring appropriate depression treatment may help improve patients’ quality of life and ease the inflammatory response itself.
For psychiatrists seeing patients with an inflammatory condition, brief explanations of the known links between depression and inflammation can provide patients—particularly those ambivalent about seeking mental health care—support for engaging in treatment and adhering to medication. Describing the links between inflammation and depression also can help encourage regular exercise and healthy diets rich in fruits, vegetables, and omega-3 fatty acids. In cases of treatment-resistant depression, particularly in those with known high inflammatory factors, it may be worthwhile to consider anti-inflammatory agents, such as infliximab, as an adjuvant treatment.
The relationship between inflammation and depression is rapidly unfolding, but the full intricacies have not yet described. However, this beginning awareness of the interplay among stress, inflammation, and depression can broaden our approach to care and treatment.
Bottom Line
Depression and inflammation are linked in many ways, although neither appears to be wholly necessary or sufficient for the other. Most likely there exists a particular subset of patients for whom inflammation will precipitate and perpetuate depression.
Related Resources
- The Emory University Mind-Body Program. www.
psychiatry.emory.edu/PROGRAMS/mindbody/index.html. - Gabriel B. The evolutionary advantage of depression. The Atlantic. October 2, 2012. www.theatlantic.com/health/archive/2012/10/the-evolutionary-advantage-of-depression/263124.
Drug Brand Names
Infliximab • Remicade Ribavirin • Rebetol, Virazole
Interferon-α • Intron
Disclosure
Dr. Almond reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
References
1. Dantzer R, O’Connor JC, Freund GG, et al. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46-56.
2. Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12(2):123-137.
3. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27(1):24-31.
4. Lamers F, Vogelzangs N, Merikangas KR, et al. Evidence for a differential role of HPA-axis function, inflammation and metabolic syndrome in melancholic versus atypical depression [published online October 23, 2012]. Mol Psychiatry. doi: 10.1038/mp.2012.144.
5. Hoen P, Kupper N, de Jonge P. Depression and cardiovascular disease progression: epidemiology, mechanisms and treatment. In: Hjemdahl P, Rosengren A, Steptoe A, eds. Stress and cardiovascular disease. London, United Kingdom: Springer; 2012:211-233.
6. Anderson RJ, Freedland KE, Clouse RE, et al. The prevalence of comorbid depression in adults with diabetes: a meta-analysis. Diabetes Care. 2001;24(6):1069-1078.
7. Bachen EA, Chesney MA, Criswell LA. Prevalence of mood and anxiety disorders in women with systemic lupus erythematosus. Arthritis Rheum. 2009;61(6):822-829.
8. Dickens C, McGowan L, Clark-Carter D, et al. Depression in rheumatoid arthritis: a systematic review of the literature with meta-analysis. Psychosom Med. 2002;64(1):52-60.
9. Benros ME, Waltoft BL, Nordentoft M, et al. Autoimmunity and infections as risk factors for depression and other severe mental illnesses. Neurology, Psychiatry and Brain Research. 2012;18(2):40-41.
10. National Cancer Institute. Depression (PDQ). http://www.cancer.gov/cancertopics/pdq/supportivecare/depression/HealthProfessional/page1. Updated January 9, 2013. Accessed April 23, 2013.
11. Centers for Disease Control and Prevention. Current depression among adults—United States, 2006 and 2008. Morb Mortal Wkly Rep. 2010;59(38):1229-1235.
12. Krishnadas R, Cavanagh J. Depression: an inflammatory illness? J Neurol Neurosurg Psychiatry. 2012;83(5):495-502.
13. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732-741.
14. Dowlati Y, Herrmann N, Swardfager W, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67(5):446-457.
15. Maes M, Bosmans E, De Jongh R, et al. Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine. 1997;9(11):853-858.
16. Raison CL, Borisov AS, Broadwell SD, et al. Depression during pegylated interferon-alpha plus ribavirin therapy: prevalence and prediction. J Clin Psychiatry. 2005;66(1):41-48.
17. Capuron L, Raison CL, Musselman DL, et al. Association of exaggerated HPA axis response to the initial injection of interferon-alpha with development of depression during interferon-alpha therapy. Am J Psychiatry. 2003;160(7):1342-1345.
18. Eisenberger NI, Berkman ET, Inagaki TK, et al. Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry. 2010;68(8):748-754.
19. Pasco JA, Nicholson GC, Williams LJ, et al. Association of high-sensitivity C-reactive protein with de novo major depression. Br J Psychiatry. 2010;197(5):372-377.
20. Raison CL, Rutherford RE, Woolwine BJ, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry. 2013;70(1):31-41.
21. Martinez JM, Garakani A, Yehuda R, et al. Proinflammatory and “resiliency” proteins in the CSF of patients with major depression. Depress Anxiety. 2012;29(1):32-38.
22. Lanquillon S, Krieg JC, Bening-Abu-Shach U, et al. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology. 2000;22(4):370-379.
23. Raison CL, Miller AH. Is depression an inflammatory disorder? Curr Psychiatry Rep. 2011;13(6):467-775.
24. Maes M, Song C, Lin A, et al. The effects of psychological stress on humans: increased production of pro-inflammatory cytokines and Th1-like response in stress-induced anxiety. Cytokine. 1998;10(4):313-318.
25. Miller GE, Rohleder N, Stetler C, et al. Clinical depression and regulation of the inflammatory response during acute stress. Psychosom Med. 2005;67(5):679-687.
26. Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. 2003;160(9):1554-1565.
27. Tak PP, Firestein GS. NF-êB: a key role in inflammatory diseases. J Clin Invest. 2001;107(1):7-12.
28. Müller N, Schwarz MJ. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry. 2007;12(11):988-1000.
29. Capuron L, Fornwalt FB, Knight BT, et al. Does cytokine-induced depression differ from idiopathic major depression in medically healthy individuals? J Affect Disord. 2009;119(1-3):181-185.
30. Yirmiya R, Pollak Y, Morag M, et al. Illness, cytokines, and depression. Ann N Y Acad Sci. 2000;917(1):478-487.
31. Maes M. The immunoregulatory effects of antidepressants. Hum Psychopharmacol. 2001;16(1):95-103.
32. Szuster-Ciesielska A, Tustanowska-Stachura A, Słotwin`ska M, et al. In vitro immunoregulatory effects of antidepressants in healthy volunteers. Pol J Pharmacol. 2003;55(3):353-362.
33. Maes M, Berk M, Goehler L, et al. Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med. 2012;10(1):66.
34. Hestad KA, Tønseth S, Støen CD, et al. Raised plasma levels of tumor necrosis factor [alpha] in patients with depression: normalization during electroconvulsive therapy. J ECT. 2003;19(4):183-188.
35. Maier SF, Watkins LR. Intracerebroventricular interleukin-1 receptor antagonist blocks the enhancement of fear conditioning and interference with escape produced by inescapable shock. Brain Res. 1995;695(2):279-282.
36. Persoons P, Vermeire S, Demyttenaere K, et al. The impact of major depressive disorder on the short- and long-term outcome of Crohn’s disease treatment with infliximab. Aliment Pharmacol Ther. 2005;22(2):101-110.
37. Mathias SD, Colwell HH, Miller DP, et al. Health-related quality of life and functional status of patients with rheumatoid arthritis randomly assigned to receive etanercept or placebo. Clin Ther. 2000;22(1):128-139.
38. Raison C, Rutherford RE, Woolwine B, et al. The tumor necrosis factor-alpha antagonist infliximab reduces depressive symptoms in patients with treatment resistant depression and high inflammation. Brain, Behavior, and Immunity. 2012;26(suppl 1):S49.
39. Müller N, Schwarz MJ, Dehning S, et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry. 2006;11(7):680-684.
40. Mendlewicz J, Kriwin P, Oswald P, et al. Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: a pilot open-label study. Int Clin Psychopharmacol. 2006;21(4):227-231.
41. Brunello N, Alboni S, Capone G, et al. Acetylsalicylic acid accelerates the antidepressant effect of fluoxetine in the chronic escape deficit model of depression. Int Clin Psychopharmacol. 2006;21(4):219-225.

HIV: How to provide compassionate care
The prevalence of HIV in persons with untreated psychiatric illness may be 10 to 20 times that of the general population.1 The U.S. Preventive Services Task Force has recommended HIV screening of all persons age 15 to 65 because 20% to 25% of individuals with HIV infection are unaware that they are HIV-positive.2 Because >20% of new HIV infections in the United States are undiagnosed,3 it is crucial to educate patients with mental illness about HIV prevention, make condoms available, and offer HIV testing.
As psychiatrists, we have a unique role in caring for patients at risk for or infected with HIV because in addition to comprehensive medical and psychiatric histories, we routinely take histories of substance use, sexual activities, relationships, and trauma, including childhood neglect and emotional, physical, and sexual abuse. We develop long-term, trusting relationships and work with individuals to change behaviors and maximize life potential.
Increasing awareness of stigma, discrimination, and psychiatric factors involved with the HIV pandemic can lead to decreased transmission of HIV infection and early diagnosis and treatment. Compassionate medical and psychiatric care can mitigate suffering in persons at risk for, infected with, or affected by HIV.
Preventing HIV transmission
AIDS differs from other complex, severe illnesses in 2 ways that are relevant to psychiatrists:
• it is almost entirely preventable
• HIV and AIDS are associated with sex, drugs, and AIDS-associated stigma and discrimination (“AIDSism”).4-6
Unsafe exposure of mucosal surfaces to the virus—primarily from exchanging body fluids in unprotected sexual encounters—accounts for 80% of new HIV infections.7 HIV transmission via sexual encounters is preventable with condoms. Percutaneous or intravenous infection with HIV—primarily from sharing needles in injection drug use—accounts for 20% of new infections.7 Use of alcohol or other substances can lead to sexual coercion, unprotected sex, and exchange of sex for drugs or money. Hence, treating substance use disorders can prevent HIV transmission.
Early diagnosis of HIV can lead to appropriate medical care, quicker onset of antiretroviral (ARV) treatment, and better outcomes. Recent research has shown that pre-exposure prophylaxis with ARV treatment can prevent transmission of HIV8; therefore, becoming aware of risk behaviors and prevention can be lifesaving for serodiscordant couples.
One of the most important ways to prevent HIV’s impact on the brain and CNS is to diagnose HIV shortly after transmission at onset of acute infection. If HIV is diagnosed very early—preferably as soon as possible after inoculation with HIV or at onset of the first flu-like symptoms—and treated with ARVs, the brain has less of an opportunity to act as an independent reservoir for HIV-infected cells and therefore to develop HIV-associated neurocognitive disorders.9,10Table 1 outlines steps psychiatrists can take to help prevent HIV transmission.

Psychiatric disorders and HIV
Psychiatric disorders and distress play a significant role in transmission of, exposure to, and infection with HIV (Table 2).4-6,11 They are relevant for prevention, clinical care, and adherence throughout every aspect of illness.

Comprehensive, compassionate, nonjudgmental care of persons at risk for or infected with HIV begins with a thorough psychiatric evaluation designed to provide an ego-supportive, sensitive, and comprehensive assessment that can guide other clinicians in providing care.12 Setting the tone and demonstrating compassion and respect includes shaking hands, which takes on special relevance in the context of AIDSism and stigma. Assessing the impact of HIV seropositivity or AIDS is best done by asking about the individual’s understanding of his or her diagnosis or illness and its impact. For some persons with HIV, verbalizing this understanding can be relieving as well as revealing. It is a chance for the patient to reveal painful experiences encountered in the home, school, camp, workplace, or community and the anguish of AIDSism and stigma.
Pay attention to sensitive and sometimes painful issues related to sexual history and sexuality. Questions related to sexual history and sexuality in heterosexual men and women as well as gay, lesbian, bisexual, and transgender individuals—such as “What is your sexual function like since you have been ill?” “Do feelings about your sexual identity play a role in your current level of distress?” and “What kind of barrier contraception are you using?”—are included in the comprehensive assessment described by Cohen et al.12
Comprehensive psychiatric evaluations can provide diagnoses, inform treatment, and mitigate anguish, distress, depression, anxiety, and substance use in persons with HIV and AIDS.12 A thorough and comprehensive assessment is crucial because HIV has an affinity for brain and neural tissue and can cause CNS complications such as HIV-associated neurocognitive disorders (HAND), even in otherwise healthy HIV-seropositive individuals. See this article at CurrentPsychiatry.com for a discussion of HAND and delirium in patients with HIV.
Some persons with HIV and AIDS do not have a psychiatric disorder, while others have multiple complex psychiatric disorders that are responses to illness or treatments or are associated with HIV/AIDS (such as HAND) or other medical illnesses and treatments (such as hepatitis C, cirrhosis, end-stage liver disease, HIV nephropathy, end-stage renal disease, anemia, coronary artery disease, and cancer). See this article at CurrentPsychiatry.com for case studies of HIV patients with delirium, depression, posttraumatic stress disorder (PTSD), and substance dependence.
Mood disorders. Depression is common among persons with HIV. Demoralization and bereavement may masquerade as depression and can complicate diagnosis and treatment. Depression and other mood disorders may be related to stigma and AIDSism as well as to biologic, psychological, social, and genetic factors. Because suicide is prevalent among persons with HIV and AIDS,13 every patient with HIV should be evaluated for depression and suicidal ideation.
PTSD is prevalent among persons with HIV. It is a risky diagnosis because it is associated with a sense of a foreshortened future, which leads to a lack of adequate self-care, poor adherence to medical care, risky behaviors, and comorbid substance dependence to help numb the pain of trauma.14,15 Persons with PTSD may have difficulty trusting clinicians and other authority figures if their trauma was a high-betrayal trauma, such as incest or military trauma.14,15
In patients with HIV, PTSD often is overlooked because it may be overshadowed by other psychiatric diagnoses. Intimate partner violence, history of childhood trauma, and childhood sexual abuse are risk factors for HIV infection and PTSD. Increased severity of HIV-related PTSD symptoms is associated with having a greater number of HIV-related physical symptoms, history of pre-HIV trauma, decreased social support, increased perception of stigma, and negative life events.
PTSD also is associated with nonadherence to risk reduction strategies and medical care.14,15 Diagnosis is further complicated by repression or retrograde amnesia of traumatic events and difficulties forming trusting relationships and disclosing HIV status to sexual partners or potential sexual partners because of fear of rejection.
Substance use disorders. Dependence on alcohol and other drugs complicates and perpetuates the HIV pandemic. Sharing needles and other drug paraphernalia is instrumental in HIV transmission. The indirect effects of alcohol and substance abuse include:
• the impact of intimate partner violence, child abuse, neglect, and/or abandonment
• development of PTSD in adults, with early childhood trauma leading to repeating their own history
• lack of self-care
• unhealthy partner choices
• use of drugs and alcohol to numb the pain associated with trauma.
Persons who are using alcohol or other drugs may have difficulty attending to their health, and substance dependence may prevent persons at risk from seeking HIV testing.
Intoxication from alcohol and drug use frequently leads to inappropriate partner choice, violent and coercive sexual behaviors, and lack of condom use. Substance dependence also may lead individuals to exchange sex for drugs and to fail to adhere to safer sexual practices or use sterile drug paraphernalia.
Treating persons with HIV/AIDS
Several organizations publish evidence-based clinical guidelines for treating depression, anxiety, substance abuse, and other psychiatric disorders in patients with HIV/AIDS. One such set of guidelines is available from the New York State Department of Health AIDS Institute at www.hivguidelines.org. As is the case with patients who do not have HIV, psychotherapy and pharmacotherapy are common first-line treatments.
Psychotherapy. Patients with HIV/AIDS with psychiatric comorbidities generally respond well to psychotherapeutic treatments.16,17 The choice of therapy needs to be tailored to the needs of individuals, couples, and families coping with AIDS. Options include:
• individual, couple, family, and group psychotherapy
• crisis intervention
• 12-step programs (Alcohol Anonymous, Narcotics Anonymous, etc.)
• adult survivors of child abuse programs (www.ascasupport.org), groups, and workbooks
• palliative psychiatry
• bereavement therapy
• spiritual support
• relaxation response
• wellness interventions such as exercise, yoga, keeping a journal, writing a life narrative, reading, artwork, movement therapy, listening to music or books on tape, and working on crossword puzzles and jigsaw puzzles.
Psychopharmacotherapy. Accurate diagnosis and awareness of drug-drug and drug-illness interactions are important when treating patients with HIV/AIDS; consult resources in the literature18 and online resources that are updated regularly (see Related Resources). Because persons with AIDS are particularly vulnerable to extrapyramidal and anticholinergic side effects of psychotropics, the principle start very low and go very slow is critical. For patients who are opioid-dependent, be cautious when prescribing medications that are cytochrome P450 3A4 inducers—such as carbamazepine, efavirenz, nevirapine, and ritonavir—because these medications can lower methadone levels in persons receiving agonist treatment and might lead to opioid withdrawal symptoms, discontinuation of ARVs, or relapse to opioids.18 When a person with AIDS is experiencing pain and is on a maintenance dose of methadone for heroin withdrawal, pain should be treated as a separate problem with additional opioids. Methadone for relapse prevention will target opioid tolerance needs and prevent withdrawal but will not provide analgesia for pain.
HIV through the life cycle
From prevention of prenatal transmission to the care of children with HIV to reproductive issues in serodiscordant couples, HIV complicates patients’ development. Table 3 outlines concerns regarding HIV transmission and treatment at different stages of a patient’s life.

Bottom Line
HIV transmission and effective treatment are complicated by a high prevalence of psychiatric comorbidities, including depression and other mood disorders, posttraumatic stress disorder, substance use disorders, and cognitive disorders. With an increased understanding of the issues faced by patients at risk for or infected with HIV, psychiatrists can help prevent HIV transmission, improve adherence to medical care, and diminish suffering, morbidity, and mortality.
Related Resources
- Academy of Psychosomatic Medicine HIV/AIDS Psychiatry Special Interest Group. www.apm.org/sigs/oap.
- New York State Department of Health AIDS Institute. HIV Clinical Resource. www.hivguidelines.org.
- University of Liverpool. HIV drug interactions list. www.hiv-druginteractions.org.
- Toronto General Hospital Immunodeficiency Clinic. Drug interactions tables. www.hivclinic.ca/main/drugs_interact.html.
Drug Brand Names
Bupropion • Wellbutrin, Zyban
Nevirapine • Viramune
Carbamazepine • Carbatrol, Tegretol, others
Olanzapine • Zyprexa
Quetiapine • Seroquel
Clonazepam • Klonopin
Ritonavir • Norvir
Efavirenz • Sustiva
Venlafaxine • Effexor
Escitalopram • Lexapro
Disclosure
Dr. Cohen reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
References
1. Blank MB, Mandell DS, Aiken L, et al. Co-occurrence of HIV and serious mental illness among Medicaid recipients. Psychiatr Serv. 2002;53(7):868-873.
2.Moyer VA, on behalf of the U.S. Preventive Services Task Force. Screening for HIV: U.S. Preventive Services Task Force recommendation statement [published online April 30, 2013]. Ann Intern Med. doi:10.7326/0003-4819-159-1-201307020-00645.
3. Hall HI, Song R, Rhodes P, et al. Estimation of HIV incidence in the United States. JAMA. 2008;300(5):520-529.
4. Cohen MA. AIDSism, a new form of discrimination. Am Med News. 1989;32:43.
5. Cohen MA, Gorman JM. Comprehensive textbook of AIDS psychiatry. New York, NY: Oxford University Press; 2008.
6. Cohen MA, Goforth HW, Lux JZ, et al, eds. Handbook of AIDS psychiatry. New York, NY: Oxford University Press; 2010.
7. World Health Organization, United Nations Children’s Fund, Joint United Nations Programme on HIV/AIDS. Global HIV/AIDS response. Epidemic update and health sector progress towards universal access. Progress report 2011. http://www.unaids.org/en/media/unaids/
contentassets/documents/unaidspublication/2011/
20111130_UA_Report_en.pdf. Accessed April 25, 2013.
8. Centers for Disease Control and Prevention (CDC). Interim guidance for clinicians considering the use of preexposure prophylaxis for the prevention of HIV infection in heterosexually active adults. MMWR Morb Mortal Wkly Rep. 2012;61(31):586-589.
9. Cysique LA, Murray JM, Dunbar M, et al. A screening algorithm for HIV-associated neurocognitive disorders. HIV Med. 2010;11(10):642-649.
10. Simioni S, Cavassini M, Annoni JM, et al. Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS. 2010;24(9):1243-1250.
11. Cohen M, Hoffman RG, Cromwell C, et al. The prevalence of distress in persons with human immunodeficiency virus infection. Psychosomatics. 2002;43(1):10-15.
12. Cohen MA, Batista SM, Lux JZ. A biopsychosocial approach to psychiatric consultation in persons with HIV and AIDS. In: Cohen MA, Goforth HW, Lux JZ, et al, eds. Handbook of AIDS psychiatry. New York, NY: Oxford University Press; 2010:33-60.
13. Carrico AW. Elevated suicide rate among HIV-positive persons despite benefits of antiretroviral therapy: implications for a stress and coping model of suicide. Am J Psychiatry. 2010;167(2):117-119.
14. Cohen MA, Alfonso CA, Hoffman RG, et al. The impact of PTSD on treatment adherence in persons with HIV infection. Gen Hosp Psychiatry. 2001;23(5):294-296.
15. Boarts JM, Sledjeski EM, Bogart LM, et al. The differential impact of PTSD and depression on HIV disease markers and adherence to HAART in people living with HIV. AIDS Behav. 2006;10(3):253-261.
16. Sikkema KJ, Hansen NB, Ghebremichael M, et al. A randomized controlled trial of a coping group intervention for adults with HIV who are AIDS bereaved: longitudinal effects on grief. Health Psychol. 2006;25(5):563-570.
17. Cohen MA. Psychodynamic psychotherapy in an AIDS nursing home. J Am Acad Psychoanal. 1999;27(1):121-133.
18. Cozza KL, Goforth HW, Batista SM. Psychopharmacologic treatment issues in AIDS psychiatry. In: Cohen MA, Goforth HW, Lux JZ, et al, eds. Handbook of AIDS psychiatry. New York, NY: Oxford University Press; 2010:147-199.
The prevalence of HIV in persons with untreated psychiatric illness may be 10 to 20 times that of the general population.1 The U.S. Preventive Services Task Force has recommended HIV screening of all persons age 15 to 65 because 20% to 25% of individuals with HIV infection are unaware that they are HIV-positive.2 Because >20% of new HIV infections in the United States are undiagnosed,3 it is crucial to educate patients with mental illness about HIV prevention, make condoms available, and offer HIV testing.
As psychiatrists, we have a unique role in caring for patients at risk for or infected with HIV because in addition to comprehensive medical and psychiatric histories, we routinely take histories of substance use, sexual activities, relationships, and trauma, including childhood neglect and emotional, physical, and sexual abuse. We develop long-term, trusting relationships and work with individuals to change behaviors and maximize life potential.
Increasing awareness of stigma, discrimination, and psychiatric factors involved with the HIV pandemic can lead to decreased transmission of HIV infection and early diagnosis and treatment. Compassionate medical and psychiatric care can mitigate suffering in persons at risk for, infected with, or affected by HIV.
Preventing HIV transmission
AIDS differs from other complex, severe illnesses in 2 ways that are relevant to psychiatrists:
• it is almost entirely preventable
• HIV and AIDS are associated with sex, drugs, and AIDS-associated stigma and discrimination (“AIDSism”).4-6
Unsafe exposure of mucosal surfaces to the virus—primarily from exchanging body fluids in unprotected sexual encounters—accounts for 80% of new HIV infections.7 HIV transmission via sexual encounters is preventable with condoms. Percutaneous or intravenous infection with HIV—primarily from sharing needles in injection drug use—accounts for 20% of new infections.7 Use of alcohol or other substances can lead to sexual coercion, unprotected sex, and exchange of sex for drugs or money. Hence, treating substance use disorders can prevent HIV transmission.
Early diagnosis of HIV can lead to appropriate medical care, quicker onset of antiretroviral (ARV) treatment, and better outcomes. Recent research has shown that pre-exposure prophylaxis with ARV treatment can prevent transmission of HIV8; therefore, becoming aware of risk behaviors and prevention can be lifesaving for serodiscordant couples.
One of the most important ways to prevent HIV’s impact on the brain and CNS is to diagnose HIV shortly after transmission at onset of acute infection. If HIV is diagnosed very early—preferably as soon as possible after inoculation with HIV or at onset of the first flu-like symptoms—and treated with ARVs, the brain has less of an opportunity to act as an independent reservoir for HIV-infected cells and therefore to develop HIV-associated neurocognitive disorders.9,10Table 1 outlines steps psychiatrists can take to help prevent HIV transmission.
Psychiatric disorders and HIV
Psychiatric disorders and distress play a significant role in transmission of, exposure to, and infection with HIV (Table 2).4-6,11 They are relevant for prevention, clinical care, and adherence throughout every aspect of illness.
Comprehensive, compassionate, nonjudgmental care of persons at risk for or infected with HIV begins with a thorough psychiatric evaluation designed to provide an ego-supportive, sensitive, and comprehensive assessment that can guide other clinicians in providing care.12 Setting the tone and demonstrating compassion and respect includes shaking hands, which takes on special relevance in the context of AIDSism and stigma. Assessing the impact of HIV seropositivity or AIDS is best done by asking about the individual’s understanding of his or her diagnosis or illness and its impact. For some persons with HIV, verbalizing this understanding can be relieving as well as revealing. It is a chance for the patient to reveal painful experiences encountered in the home, school, camp, workplace, or community and the anguish of AIDSism and stigma.
Pay attention to sensitive and sometimes painful issues related to sexual history and sexuality. Questions related to sexual history and sexuality in heterosexual men and women as well as gay, lesbian, bisexual, and transgender individuals—such as “What is your sexual function like since you have been ill?” “Do feelings about your sexual identity play a role in your current level of distress?” and “What kind of barrier contraception are you using?”—are included in the comprehensive assessment described by Cohen et al.12
Comprehensive psychiatric evaluations can provide diagnoses, inform treatment, and mitigate anguish, distress, depression, anxiety, and substance use in persons with HIV and AIDS.12 A thorough and comprehensive assessment is crucial because HIV has an affinity for brain and neural tissue and can cause CNS complications such as HIV-associated neurocognitive disorders (HAND), even in otherwise healthy HIV-seropositive individuals. See this article at CurrentPsychiatry.com for a discussion of HAND and delirium in patients with HIV.
Some persons with HIV and AIDS do not have a psychiatric disorder, while others have multiple complex psychiatric disorders that are responses to illness or treatments or are associated with HIV/AIDS (such as HAND) or other medical illnesses and treatments (such as hepatitis C, cirrhosis, end-stage liver disease, HIV nephropathy, end-stage renal disease, anemia, coronary artery disease, and cancer). See this article at CurrentPsychiatry.com for case studies of HIV patients with delirium, depression, posttraumatic stress disorder (PTSD), and substance dependence.
Mood disorders. Depression is common among persons with HIV. Demoralization and bereavement may masquerade as depression and can complicate diagnosis and treatment. Depression and other mood disorders may be related to stigma and AIDSism as well as to biologic, psychological, social, and genetic factors. Because suicide is prevalent among persons with HIV and AIDS,13 every patient with HIV should be evaluated for depression and suicidal ideation.
PTSD is prevalent among persons with HIV. It is a risky diagnosis because it is associated with a sense of a foreshortened future, which leads to a lack of adequate self-care, poor adherence to medical care, risky behaviors, and comorbid substance dependence to help numb the pain of trauma.14,15 Persons with PTSD may have difficulty trusting clinicians and other authority figures if their trauma was a high-betrayal trauma, such as incest or military trauma.14,15
In patients with HIV, PTSD often is overlooked because it may be overshadowed by other psychiatric diagnoses. Intimate partner violence, history of childhood trauma, and childhood sexual abuse are risk factors for HIV infection and PTSD. Increased severity of HIV-related PTSD symptoms is associated with having a greater number of HIV-related physical symptoms, history of pre-HIV trauma, decreased social support, increased perception of stigma, and negative life events.
PTSD also is associated with nonadherence to risk reduction strategies and medical care.14,15 Diagnosis is further complicated by repression or retrograde amnesia of traumatic events and difficulties forming trusting relationships and disclosing HIV status to sexual partners or potential sexual partners because of fear of rejection.
Substance use disorders. Dependence on alcohol and other drugs complicates and perpetuates the HIV pandemic. Sharing needles and other drug paraphernalia is instrumental in HIV transmission. The indirect effects of alcohol and substance abuse include:
• the impact of intimate partner violence, child abuse, neglect, and/or abandonment
• development of PTSD in adults, with early childhood trauma leading to repeating their own history
• lack of self-care
• unhealthy partner choices
• use of drugs and alcohol to numb the pain associated with trauma.
Persons who are using alcohol or other drugs may have difficulty attending to their health, and substance dependence may prevent persons at risk from seeking HIV testing.
Intoxication from alcohol and drug use frequently leads to inappropriate partner choice, violent and coercive sexual behaviors, and lack of condom use. Substance dependence also may lead individuals to exchange sex for drugs and to fail to adhere to safer sexual practices or use sterile drug paraphernalia.
Treating persons with HIV/AIDS
Several organizations publish evidence-based clinical guidelines for treating depression, anxiety, substance abuse, and other psychiatric disorders in patients with HIV/AIDS. One such set of guidelines is available from the New York State Department of Health AIDS Institute at www.hivguidelines.org. As is the case with patients who do not have HIV, psychotherapy and pharmacotherapy are common first-line treatments.
Psychotherapy. Patients with HIV/AIDS with psychiatric comorbidities generally respond well to psychotherapeutic treatments.16,17 The choice of therapy needs to be tailored to the needs of individuals, couples, and families coping with AIDS. Options include:
• individual, couple, family, and group psychotherapy
• crisis intervention
• 12-step programs (Alcohol Anonymous, Narcotics Anonymous, etc.)
• adult survivors of child abuse programs (www.ascasupport.org), groups, and workbooks
• palliative psychiatry
• bereavement therapy
• spiritual support
• relaxation response
• wellness interventions such as exercise, yoga, keeping a journal, writing a life narrative, reading, artwork, movement therapy, listening to music or books on tape, and working on crossword puzzles and jigsaw puzzles.
Psychopharmacotherapy. Accurate diagnosis and awareness of drug-drug and drug-illness interactions are important when treating patients with HIV/AIDS; consult resources in the literature18 and online resources that are updated regularly (see Related Resources). Because persons with AIDS are particularly vulnerable to extrapyramidal and anticholinergic side effects of psychotropics, the principle start very low and go very slow is critical. For patients who are opioid-dependent, be cautious when prescribing medications that are cytochrome P450 3A4 inducers—such as carbamazepine, efavirenz, nevirapine, and ritonavir—because these medications can lower methadone levels in persons receiving agonist treatment and might lead to opioid withdrawal symptoms, discontinuation of ARVs, or relapse to opioids.18 When a person with AIDS is experiencing pain and is on a maintenance dose of methadone for heroin withdrawal, pain should be treated as a separate problem with additional opioids. Methadone for relapse prevention will target opioid tolerance needs and prevent withdrawal but will not provide analgesia for pain.
HIV through the life cycle
From prevention of prenatal transmission to the care of children with HIV to reproductive issues in serodiscordant couples, HIV complicates patients’ development. Table 3 outlines concerns regarding HIV transmission and treatment at different stages of a patient’s life.
Bottom Line
HIV transmission and effective treatment are complicated by a high prevalence of psychiatric comorbidities, including depression and other mood disorders, posttraumatic stress disorder, substance use disorders, and cognitive disorders. With an increased understanding of the issues faced by patients at risk for or infected with HIV, psychiatrists can help prevent HIV transmission, improve adherence to medical care, and diminish suffering, morbidity, and mortality.
Related Resources
- Academy of Psychosomatic Medicine HIV/AIDS Psychiatry Special Interest Group. www.apm.org/sigs/oap.
- New York State Department of Health AIDS Institute. HIV Clinical Resource. www.hivguidelines.org.
- University of Liverpool. HIV drug interactions list. www.hiv-druginteractions.org.
- Toronto General Hospital Immunodeficiency Clinic. Drug interactions tables. www.hivclinic.ca/main/drugs_interact.html.
Drug Brand Names
Bupropion • Wellbutrin, Zyban
Nevirapine • Viramune
Carbamazepine • Carbatrol, Tegretol, others
Olanzapine • Zyprexa
Quetiapine • Seroquel
Clonazepam • Klonopin
Ritonavir • Norvir
Efavirenz • Sustiva
Venlafaxine • Effexor
Escitalopram • Lexapro
Disclosure
Dr. Cohen reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
References
1. Blank MB, Mandell DS, Aiken L, et al. Co-occurrence of HIV and serious mental illness among Medicaid recipients. Psychiatr Serv. 2002;53(7):868-873.
2.Moyer VA, on behalf of the U.S. Preventive Services Task Force. Screening for HIV: U.S. Preventive Services Task Force recommendation statement [published online April 30, 2013]. Ann Intern Med. doi:10.7326/0003-4819-159-1-201307020-00645.
3. Hall HI, Song R, Rhodes P, et al. Estimation of HIV incidence in the United States. JAMA. 2008;300(5):520-529.
4. Cohen MA. AIDSism, a new form of discrimination. Am Med News. 1989;32:43.
5. Cohen MA, Gorman JM. Comprehensive textbook of AIDS psychiatry. New York, NY: Oxford University Press; 2008.
6. Cohen MA, Goforth HW, Lux JZ, et al, eds. Handbook of AIDS psychiatry. New York, NY: Oxford University Press; 2010.
7. World Health Organization, United Nations Children’s Fund, Joint United Nations Programme on HIV/AIDS. Global HIV/AIDS response. Epidemic update and health sector progress towards universal access. Progress report 2011. http://www.unaids.org/en/media/unaids/
contentassets/documents/unaidspublication/2011/
20111130_UA_Report_en.pdf. Accessed April 25, 2013.
8. Centers for Disease Control and Prevention (CDC). Interim guidance for clinicians considering the use of preexposure prophylaxis for the prevention of HIV infection in heterosexually active adults. MMWR Morb Mortal Wkly Rep. 2012;61(31):586-589.
9. Cysique LA, Murray JM, Dunbar M, et al. A screening algorithm for HIV-associated neurocognitive disorders. HIV Med. 2010;11(10):642-649.
10. Simioni S, Cavassini M, Annoni JM, et al. Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS. 2010;24(9):1243-1250.
11. Cohen M, Hoffman RG, Cromwell C, et al. The prevalence of distress in persons with human immunodeficiency virus infection. Psychosomatics. 2002;43(1):10-15.
12. Cohen MA, Batista SM, Lux JZ. A biopsychosocial approach to psychiatric consultation in persons with HIV and AIDS. In: Cohen MA, Goforth HW, Lux JZ, et al, eds. Handbook of AIDS psychiatry. New York, NY: Oxford University Press; 2010:33-60.
13. Carrico AW. Elevated suicide rate among HIV-positive persons despite benefits of antiretroviral therapy: implications for a stress and coping model of suicide. Am J Psychiatry. 2010;167(2):117-119.
14. Cohen MA, Alfonso CA, Hoffman RG, et al. The impact of PTSD on treatment adherence in persons with HIV infection. Gen Hosp Psychiatry. 2001;23(5):294-296.
15. Boarts JM, Sledjeski EM, Bogart LM, et al. The differential impact of PTSD and depression on HIV disease markers and adherence to HAART in people living with HIV. AIDS Behav. 2006;10(3):253-261.
16. Sikkema KJ, Hansen NB, Ghebremichael M, et al. A randomized controlled trial of a coping group intervention for adults with HIV who are AIDS bereaved: longitudinal effects on grief. Health Psychol. 2006;25(5):563-570.
17. Cohen MA. Psychodynamic psychotherapy in an AIDS nursing home. J Am Acad Psychoanal. 1999;27(1):121-133.
18. Cozza KL, Goforth HW, Batista SM. Psychopharmacologic treatment issues in AIDS psychiatry. In: Cohen MA, Goforth HW, Lux JZ, et al, eds. Handbook of AIDS psychiatry. New York, NY: Oxford University Press; 2010:147-199.
The prevalence of HIV in persons with untreated psychiatric illness may be 10 to 20 times that of the general population.1 The U.S. Preventive Services Task Force has recommended HIV screening of all persons age 15 to 65 because 20% to 25% of individuals with HIV infection are unaware that they are HIV-positive.2 Because >20% of new HIV infections in the United States are undiagnosed,3 it is crucial to educate patients with mental illness about HIV prevention, make condoms available, and offer HIV testing.
As psychiatrists, we have a unique role in caring for patients at risk for or infected with HIV because in addition to comprehensive medical and psychiatric histories, we routinely take histories of substance use, sexual activities, relationships, and trauma, including childhood neglect and emotional, physical, and sexual abuse. We develop long-term, trusting relationships and work with individuals to change behaviors and maximize life potential.
Increasing awareness of stigma, discrimination, and psychiatric factors involved with the HIV pandemic can lead to decreased transmission of HIV infection and early diagnosis and treatment. Compassionate medical and psychiatric care can mitigate suffering in persons at risk for, infected with, or affected by HIV.
Preventing HIV transmission
AIDS differs from other complex, severe illnesses in 2 ways that are relevant to psychiatrists:
• it is almost entirely preventable
• HIV and AIDS are associated with sex, drugs, and AIDS-associated stigma and discrimination (“AIDSism”).4-6
Unsafe exposure of mucosal surfaces to the virus—primarily from exchanging body fluids in unprotected sexual encounters—accounts for 80% of new HIV infections.7 HIV transmission via sexual encounters is preventable with condoms. Percutaneous or intravenous infection with HIV—primarily from sharing needles in injection drug use—accounts for 20% of new infections.7 Use of alcohol or other substances can lead to sexual coercion, unprotected sex, and exchange of sex for drugs or money. Hence, treating substance use disorders can prevent HIV transmission.
Early diagnosis of HIV can lead to appropriate medical care, quicker onset of antiretroviral (ARV) treatment, and better outcomes. Recent research has shown that pre-exposure prophylaxis with ARV treatment can prevent transmission of HIV8; therefore, becoming aware of risk behaviors and prevention can be lifesaving for serodiscordant couples.
One of the most important ways to prevent HIV’s impact on the brain and CNS is to diagnose HIV shortly after transmission at onset of acute infection. If HIV is diagnosed very early—preferably as soon as possible after inoculation with HIV or at onset of the first flu-like symptoms—and treated with ARVs, the brain has less of an opportunity to act as an independent reservoir for HIV-infected cells and therefore to develop HIV-associated neurocognitive disorders.9,10Table 1 outlines steps psychiatrists can take to help prevent HIV transmission.
Psychiatric disorders and HIV
Psychiatric disorders and distress play a significant role in transmission of, exposure to, and infection with HIV (Table 2).4-6,11 They are relevant for prevention, clinical care, and adherence throughout every aspect of illness.
Comprehensive, compassionate, nonjudgmental care of persons at risk for or infected with HIV begins with a thorough psychiatric evaluation designed to provide an ego-supportive, sensitive, and comprehensive assessment that can guide other clinicians in providing care.12 Setting the tone and demonstrating compassion and respect includes shaking hands, which takes on special relevance in the context of AIDSism and stigma. Assessing the impact of HIV seropositivity or AIDS is best done by asking about the individual’s understanding of his or her diagnosis or illness and its impact. For some persons with HIV, verbalizing this understanding can be relieving as well as revealing. It is a chance for the patient to reveal painful experiences encountered in the home, school, camp, workplace, or community and the anguish of AIDSism and stigma.
Pay attention to sensitive and sometimes painful issues related to sexual history and sexuality. Questions related to sexual history and sexuality in heterosexual men and women as well as gay, lesbian, bisexual, and transgender individuals—such as “What is your sexual function like since you have been ill?” “Do feelings about your sexual identity play a role in your current level of distress?” and “What kind of barrier contraception are you using?”—are included in the comprehensive assessment described by Cohen et al.12
Comprehensive psychiatric evaluations can provide diagnoses, inform treatment, and mitigate anguish, distress, depression, anxiety, and substance use in persons with HIV and AIDS.12 A thorough and comprehensive assessment is crucial because HIV has an affinity for brain and neural tissue and can cause CNS complications such as HIV-associated neurocognitive disorders (HAND), even in otherwise healthy HIV-seropositive individuals. See this article at CurrentPsychiatry.com for a discussion of HAND and delirium in patients with HIV.
Some persons with HIV and AIDS do not have a psychiatric disorder, while others have multiple complex psychiatric disorders that are responses to illness or treatments or are associated with HIV/AIDS (such as HAND) or other medical illnesses and treatments (such as hepatitis C, cirrhosis, end-stage liver disease, HIV nephropathy, end-stage renal disease, anemia, coronary artery disease, and cancer). See this article at CurrentPsychiatry.com for case studies of HIV patients with delirium, depression, posttraumatic stress disorder (PTSD), and substance dependence.
Mood disorders. Depression is common among persons with HIV. Demoralization and bereavement may masquerade as depression and can complicate diagnosis and treatment. Depression and other mood disorders may be related to stigma and AIDSism as well as to biologic, psychological, social, and genetic factors. Because suicide is prevalent among persons with HIV and AIDS,13 every patient with HIV should be evaluated for depression and suicidal ideation.
PTSD is prevalent among persons with HIV. It is a risky diagnosis because it is associated with a sense of a foreshortened future, which leads to a lack of adequate self-care, poor adherence to medical care, risky behaviors, and comorbid substance dependence to help numb the pain of trauma.14,15 Persons with PTSD may have difficulty trusting clinicians and other authority figures if their trauma was a high-betrayal trauma, such as incest or military trauma.14,15
In patients with HIV, PTSD often is overlooked because it may be overshadowed by other psychiatric diagnoses. Intimate partner violence, history of childhood trauma, and childhood sexual abuse are risk factors for HIV infection and PTSD. Increased severity of HIV-related PTSD symptoms is associated with having a greater number of HIV-related physical symptoms, history of pre-HIV trauma, decreased social support, increased perception of stigma, and negative life events.
PTSD also is associated with nonadherence to risk reduction strategies and medical care.14,15 Diagnosis is further complicated by repression or retrograde amnesia of traumatic events and difficulties forming trusting relationships and disclosing HIV status to sexual partners or potential sexual partners because of fear of rejection.
Substance use disorders. Dependence on alcohol and other drugs complicates and perpetuates the HIV pandemic. Sharing needles and other drug paraphernalia is instrumental in HIV transmission. The indirect effects of alcohol and substance abuse include:
• the impact of intimate partner violence, child abuse, neglect, and/or abandonment
• development of PTSD in adults, with early childhood trauma leading to repeating their own history
• lack of self-care
• unhealthy partner choices
• use of drugs and alcohol to numb the pain associated with trauma.
Persons who are using alcohol or other drugs may have difficulty attending to their health, and substance dependence may prevent persons at risk from seeking HIV testing.
Intoxication from alcohol and drug use frequently leads to inappropriate partner choice, violent and coercive sexual behaviors, and lack of condom use. Substance dependence also may lead individuals to exchange sex for drugs and to fail to adhere to safer sexual practices or use sterile drug paraphernalia.
Treating persons with HIV/AIDS
Several organizations publish evidence-based clinical guidelines for treating depression, anxiety, substance abuse, and other psychiatric disorders in patients with HIV/AIDS. One such set of guidelines is available from the New York State Department of Health AIDS Institute at www.hivguidelines.org. As is the case with patients who do not have HIV, psychotherapy and pharmacotherapy are common first-line treatments.
Psychotherapy. Patients with HIV/AIDS with psychiatric comorbidities generally respond well to psychotherapeutic treatments.16,17 The choice of therapy needs to be tailored to the needs of individuals, couples, and families coping with AIDS. Options include:
• individual, couple, family, and group psychotherapy
• crisis intervention
• 12-step programs (Alcohol Anonymous, Narcotics Anonymous, etc.)
• adult survivors of child abuse programs (www.ascasupport.org), groups, and workbooks
• palliative psychiatry
• bereavement therapy
• spiritual support
• relaxation response
• wellness interventions such as exercise, yoga, keeping a journal, writing a life narrative, reading, artwork, movement therapy, listening to music or books on tape, and working on crossword puzzles and jigsaw puzzles.
Psychopharmacotherapy. Accurate diagnosis and awareness of drug-drug and drug-illness interactions are important when treating patients with HIV/AIDS; consult resources in the literature18 and online resources that are updated regularly (see Related Resources). Because persons with AIDS are particularly vulnerable to extrapyramidal and anticholinergic side effects of psychotropics, the principle start very low and go very slow is critical. For patients who are opioid-dependent, be cautious when prescribing medications that are cytochrome P450 3A4 inducers—such as carbamazepine, efavirenz, nevirapine, and ritonavir—because these medications can lower methadone levels in persons receiving agonist treatment and might lead to opioid withdrawal symptoms, discontinuation of ARVs, or relapse to opioids.18 When a person with AIDS is experiencing pain and is on a maintenance dose of methadone for heroin withdrawal, pain should be treated as a separate problem with additional opioids. Methadone for relapse prevention will target opioid tolerance needs and prevent withdrawal but will not provide analgesia for pain.
HIV through the life cycle
From prevention of prenatal transmission to the care of children with HIV to reproductive issues in serodiscordant couples, HIV complicates patients’ development. Table 3 outlines concerns regarding HIV transmission and treatment at different stages of a patient’s life.
Bottom Line
HIV transmission and effective treatment are complicated by a high prevalence of psychiatric comorbidities, including depression and other mood disorders, posttraumatic stress disorder, substance use disorders, and cognitive disorders. With an increased understanding of the issues faced by patients at risk for or infected with HIV, psychiatrists can help prevent HIV transmission, improve adherence to medical care, and diminish suffering, morbidity, and mortality.
Related Resources
- Academy of Psychosomatic Medicine HIV/AIDS Psychiatry Special Interest Group. www.apm.org/sigs/oap.
- New York State Department of Health AIDS Institute. HIV Clinical Resource. www.hivguidelines.org.
- University of Liverpool. HIV drug interactions list. www.hiv-druginteractions.org.
- Toronto General Hospital Immunodeficiency Clinic. Drug interactions tables. www.hivclinic.ca/main/drugs_interact.html.
Drug Brand Names
Bupropion • Wellbutrin, Zyban
Nevirapine • Viramune
Carbamazepine • Carbatrol, Tegretol, others
Olanzapine • Zyprexa
Quetiapine • Seroquel
Clonazepam • Klonopin
Ritonavir • Norvir
Efavirenz • Sustiva
Venlafaxine • Effexor
Escitalopram • Lexapro
Disclosure
Dr. Cohen reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
References
1. Blank MB, Mandell DS, Aiken L, et al. Co-occurrence of HIV and serious mental illness among Medicaid recipients. Psychiatr Serv. 2002;53(7):868-873.
2.Moyer VA, on behalf of the U.S. Preventive Services Task Force. Screening for HIV: U.S. Preventive Services Task Force recommendation statement [published online April 30, 2013]. Ann Intern Med. doi:10.7326/0003-4819-159-1-201307020-00645.
3. Hall HI, Song R, Rhodes P, et al. Estimation of HIV incidence in the United States. JAMA. 2008;300(5):520-529.
4. Cohen MA. AIDSism, a new form of discrimination. Am Med News. 1989;32:43.
5. Cohen MA, Gorman JM. Comprehensive textbook of AIDS psychiatry. New York, NY: Oxford University Press; 2008.
6. Cohen MA, Goforth HW, Lux JZ, et al, eds. Handbook of AIDS psychiatry. New York, NY: Oxford University Press; 2010.
7. World Health Organization, United Nations Children’s Fund, Joint United Nations Programme on HIV/AIDS. Global HIV/AIDS response. Epidemic update and health sector progress towards universal access. Progress report 2011. http://www.unaids.org/en/media/unaids/
contentassets/documents/unaidspublication/2011/
20111130_UA_Report_en.pdf. Accessed April 25, 2013.
8. Centers for Disease Control and Prevention (CDC). Interim guidance for clinicians considering the use of preexposure prophylaxis for the prevention of HIV infection in heterosexually active adults. MMWR Morb Mortal Wkly Rep. 2012;61(31):586-589.
9. Cysique LA, Murray JM, Dunbar M, et al. A screening algorithm for HIV-associated neurocognitive disorders. HIV Med. 2010;11(10):642-649.
10. Simioni S, Cavassini M, Annoni JM, et al. Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS. 2010;24(9):1243-1250.
11. Cohen M, Hoffman RG, Cromwell C, et al. The prevalence of distress in persons with human immunodeficiency virus infection. Psychosomatics. 2002;43(1):10-15.
12. Cohen MA, Batista SM, Lux JZ. A biopsychosocial approach to psychiatric consultation in persons with HIV and AIDS. In: Cohen MA, Goforth HW, Lux JZ, et al, eds. Handbook of AIDS psychiatry. New York, NY: Oxford University Press; 2010:33-60.
13. Carrico AW. Elevated suicide rate among HIV-positive persons despite benefits of antiretroviral therapy: implications for a stress and coping model of suicide. Am J Psychiatry. 2010;167(2):117-119.
14. Cohen MA, Alfonso CA, Hoffman RG, et al. The impact of PTSD on treatment adherence in persons with HIV infection. Gen Hosp Psychiatry. 2001;23(5):294-296.
15. Boarts JM, Sledjeski EM, Bogart LM, et al. The differential impact of PTSD and depression on HIV disease markers and adherence to HAART in people living with HIV. AIDS Behav. 2006;10(3):253-261.
16. Sikkema KJ, Hansen NB, Ghebremichael M, et al. A randomized controlled trial of a coping group intervention for adults with HIV who are AIDS bereaved: longitudinal effects on grief. Health Psychol. 2006;25(5):563-570.
17. Cohen MA. Psychodynamic psychotherapy in an AIDS nursing home. J Am Acad Psychoanal. 1999;27(1):121-133.
18. Cozza KL, Goforth HW, Batista SM. Psychopharmacologic treatment issues in AIDS psychiatry. In: Cohen MA, Goforth HW, Lux JZ, et al, eds. Handbook of AIDS psychiatry. New York, NY: Oxford University Press; 2010:147-199.

Evaluating psychotic patients' risk of violence: A practical guide
When evaluating a patient’s risk of violence, the presence of psychosis is a crucial concern. Douglas et al1 found that psychosis was the most important predictor of violent behavior in an analysis of 204 studies examining the relationship between psychopathology and aggression. Clinicians need to be familiar with aspects of persecutory delusions and command auditory hallucinations that are associated with an increased risk of aggression because accurately assessing patients who are experiencing these 2 symptoms is an important part of a comprehensive violence risk assessment.
This article highlights the importance of investigating persecutory delusions and command auditory hallucinations when evaluating a psychotic patient’s risk for violence. We provide specific questions to ask to help gauge risk associated with these 2 symptoms.
Evaluating persecutory delusions
Do persecutory delusions increase the risk that a person will behave violently? Research examining delusions’ contribution to violent behavior does not provide a clear answer. Earlier studies suggested that persecutory delusions were associated with an increased risk of aggression.2 Delusions noted to increase the risk of violence were characterized by threat/control-override (TCO) symptoms. TCO symptoms are beliefs that one is being threatened (eg, being followed or poisoned) or is losing control to an external source (eg, one’s mind is dominated by forces beyond his or her control).3 Similarly, using data from the Epidemiologic Catchment Area surveys, Swanson et al4 found that patients who reported TCO symptoms were approximately twice as likely to engage in assaultive behavior compared with patients with other psychotic symptoms.
In contrast, the MacArthur Study of Mental Disorder and Violence5,6 showed that the presence of delusions did not predict higher rates of violence among recently discharged psychiatric patients. In particular, researchers did not find a relationship between the presence of TCO delusions and violent behavior. In a study comparing male criminal offenders with schizophrenia found not guilty by reason of insanity with matched non-offending schizophrenia patients, Stompe et al7 found no significant association between TCO symptoms and severity of violent behavior; prevalence of TCO symptoms did not differ between the 2 groups. However, nondelusional suspiciousness—such as misperceiving others’ behavior as indicating hostile intent—was associated with subsequent violence.6
Nederlof et al8 conducted a cross-sectional multicenter study to further examine whether TCO symptoms are related to aggressive behavior. Their study included 124 patients (88% men) who had paranoid schizophrenia (70%), “other forms” of schizophrenia (16%), schizoaffective disorder (3%), delusional disorder (1%), and psychosis not otherwise specified (10%). To measure TCO symptoms in a more detailed manner than in previous research, these researchers developed the Threat/Control-Override Questionnaire (TCOQ), a 14-item, self-report scale. The 7 threat items specific to the TCOQ are:8
- I am under the control of an external force that determines my actions.
- Other people have tried to poison me or to do me harm.
- Someone has deliberately tried to make me ill.
- Other people have been secretly plotting to ruin me.
- Someone has had evil intentions against me.
- I have the thought that I was being followed for a special reason.
- People have tried to drive me insane.
The 7 control-override items on the TCOQ are:8
- Other people control my way of movements.
- Other people can insert thoughts into my head.
- My thoughts are dominated by an external force.
- I have the feeling that other people can determine my thoughts.
- Other people can insert thoughts into my mind.
- I have the feeling that other people have control over me.
- My life is being determined by something or someone except for myself.
Nederlof et al8 determined that TCO symptoms were a significant correlate of aggression in their study sample. When the 2 domains of TCO symptoms were evaluated separately, only threat symptoms made a significant contribution to aggressive behavior. These researchers suggested that varying methods of measuring TCO symptoms may underlie previous studies’ seemingly contradictory findings.8 These recent findings indicate that the debate regarding the contribution of TCO symptoms, particularly threat symptoms, to future violence remains active.
Appelbaum et al9 used the MacArthur-Maudsley Delusions Assessment Schedule to examine the contribution of non-content-related delusional material to violence in interviews with 328 delusional hospitalized psychiatric patients. The 7 dimensions of the MacArthur-Maudsley Delusions Assessment Schedule are:
- Conviction—the degree of certainty about the delusional belief
- Negative affect—whether the delusional belief makes the patient unhappy, frightened, anxious, or angry
- Action—the extent to which the patient’s actions are motivated by the delusional belief
- Inaction—whether the patient has refrained from any action as a result of the delusional belief
- Preoccupation—the extent to which the patient indicates his or her thoughts focus exclusively on the delusion
- Pervasiveness—the degree to which the delusional belief penetrates all aspects of the patient’s experiences
- Fluidity—the degree to which the delusional belief changed frequently during the interview.
Patients with persecutory delusions had significantly higher scores on “action” and “negative affect” dimensions, indicating that those with persecutory delusions may be more likely to react in response to the dysphoric aspects of their symptoms.9 Subsequent research has demonstrated that patients who suffer from persecutory delusions and negative affect are more likely to act on their delusions2,10 and to act violently11 than patients without these symptoms.
When evaluating a patient who experiences persecutory delusions, inquire if he or she has employed “safety actions.” These are specific behaviors—such as avoiding a perceived persecutor or escaping a fearful situation—the individual has employed with the intention of minimizing a misperceived threat. In a study of 100 patients with persecutory delusions, 96% reported using safety behaviors in the past month.12 In this study, individuals with a history of violence reported a greater use of safety behaviors.
Table 1 lists 10 questions to ask patients to explore persecutory delusions and associated risk factors for aggression.
Table 1
Evaluating persecutory delusions: 10 questions
| 1. | Who or what do you believe wants to harm you? |
| 2. | How is this person attempting to harm you? (Ask about specific threat/control-override beliefs) |
| 3. | How certain are you that this is happening? |
| 4. | Is there anything that could convince you that this isn’t true? |
| 5. | How does your belief make you feel (eg, unhappy, frightened, anxious, or angry)? |
| 6. | Have you thought about any actions to take as a result of these beliefs? If so, what? |
| 7. | Have you taken any action as a result of your beliefs? If so, what specific actions? |
| 8. | Has your concern about being harmed stopped you from doing any action that you would normally do? Have you changed your routine in any way? |
| 9. | How much time do you spend thinking about this each day? |
| 10. | In what ways have these beliefs impacted your life? |
Assessing auditory hallucinations
A careful inquiry about hallucinations can help determine whether their presence increases a patient’s risk of committing a violent act. Command hallucinations provide some type of directive to the patient. Approximately 50% of hallucinating psychiatric patients experience command hallucinations.13 Most command hallucinations are nonviolent, and patients are more likely to obey nonviolent instructions than violent commands.14
Research on factors associated with a patient acting on harmful command hallucinations has been mixed. In a review of 7 controlled studies, no study demonstrated a positive relationship between command hallucinations and violence, and 1 found an inverse relationship.15 In contrast, in a study of 103 psychiatric inpatients, McNiel et al16 found 30% reported having command hallucinations to harm others during the past year and 22% reported they complied with such commands. These researchers concluded that compared with those without command hallucinations, patients in their study who experienced command hallucinations to harm others were more than twice as likely to be violent.
Much of the literature examining the relationship between a patient’s actions and command hallucinations has examined the patient’s response to all command hallucinations, without delineating factors specific to violent commands. Seven factors are associated with acting on command hallucinations:13
- the presence of coexisting delusions17
- having delusions that relate to the hallucination18
- knowing the voice’s identity18
- believing the voices to be real19
- believing that the voices are benevolent20
- having few coping strategies to deal with the voices17
- not feeling in control over the voices.20
These factors also have been found to indicate increased compliance with acting on violent command hallucinations.18,20 Studies that have examined compliance specific to harmful command hallucinations provide additional guidance when evaluating the patient’s risk of harm. Aspects relevant to increased compliance to violent command hallucinations include a belief that the voice is powerful,13,21 a patient’s sense of personal superiority,21 a belief that command hallucinations benefit the patient,13 delusions that were congruent with the action described,13 and hallucinations that generate negative emotions such as anger, anxiety, and sadness.11
Table 2 lists 10 questions to ask to further investigate general command auditory hallucinations and violent command auditory hallucinations.
Table 2
Evaluating command auditory hallucinations: 10 questions
| 1. | What are the voices telling you to do? |
| 2. | Do you have any thoughts or beliefs that are associated with what you are hearing? If so, what are they? |
| 3. | Do you know the voice’s identity? If so, who is it? |
| 4. | How convinced are you that these voices are real? |
| 5. | Are these voices wishing you well or do you think that they wish you harm? |
| 6. | Have you done anything to help make the voices go away? If so, what? |
| 7. | Do you feel you have control of the voices or do you feel they control you? |
| 8. | Do you believe the voice is powerful? |
| 9. | How do the voices make you feel? |
| 10. | Have you ever done what the voice has told you to do? If so, describe what you did. |
Related Resources
- MacArthur Research Network on Mental Health and the Law. The MacArthur Violence Risk Assessment Study.http://macarthur.virginia.edu/risk.html.
- Witt K, van Dorn R, Fazel S. Risk factors for violence in psychosis: systematic review and meta-regression analysis of 110 studies [published online February 13, 2013]. PLoS One. 2013;8(2):e55942. doi: 10.1371/journal.pone.0055942.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Douglas KS, Guy LS, Hart SD. Psychosis as a risk factor for violence to others: a meta-analysis. Psychol Bull. 2009;135(5):679-706.
2. Wessely S, Buchanan A, Reed A, et al. Acting on delusions. I: Prevalence. Br J Psychiatry. 1993;163:69-76.
3. Link BG, Stueve A. Evidence bearing on mental illness as a possible cause of violent behavior. Epidemiol Rev. 1995;17(1):172-181.
4. Swanson JW, Borum R, Swartz MS, et al. Psychotic symptoms and disorders and the risk of violent behaviour in the community. Crim Behav Ment Health. 1996;6(4):309-329.
5. MacArthur Research Network on Mental Health and the Law. The MacArthur Violence Risk Assessment Study. http://macarthur.virginia.edu/risk.html. Published April 2001. Accessed March 21 2013.
6. Monahan J, Steadman HJ, Silver E, et al. Rethinking risk assessment: the MacArthur study of mental disorder and violence. New York, NY: Oxford University Press, Inc.; 2001.
7. Stompe T, Ortwein-Swoboda G, Schanda H. Schizophrenia delusional symptoms, and violence: the threat/control override concept reexamined. Schizophr Bull. 2004;30(1):31-44.
8. Nederlof AF, Muris P, Hovens JE. Threat/control-override symptoms and emotional reactions to positive symptoms as correlates of aggressive behavior in psychotic patients. J Nerv Ment Dis. 2011;199(5):342-347.
9. Appelbaum PS, Robbins PC, Roth LH. Dimensional approach to delusions: comparison across types and diagnoses. Am J Psychiatry. 1999;156(12):1938-1943.
10. Buchanan A, Reed A, Wessely S, et al. Acting on delusions. II: The phenomenological correlates of acting on delusions. Br J Psychiatry. 1993;163:77-81.
11. Cheung P, Schweitzer I, Crowley K, et al. Violence in schizophrenia: role of hallucinations and delusions. Schizophr Res. 1997;26(2-3):181-190.
12. Freeman D, Garety PA, Kuipers E, et al. Acting on persecutory delusions: the importance of safety seeking. Behav Res Ther. 2007;45(1):89-99.
13. Shawyer F, MacKinnon A, Farhall J, et al. Command hallucinations and violence: implications for detention and treatment. Psychiatr Psychol Law. 2003;10(1):97-107.
14. Chadwick P, Birchwood M. The omnipotence of voices. A cognitive approach to auditory hallucinations. Br J Psychiatry. 1994;164(2):190-201.
15. Rudnick A. Relation between command hallucinations and dangerous behavior. J Am Acad Psychiatry Law. 1999;27(2):253-257.
16. McNiel DE, Eisner JP, Binder RL. The relationship between command hallucinations and violence. Psychiatr Serv. 2000;51(10):1288-1292.
17. Mackinnon A, Copolov DL, Trauer T. Factors associated with compliance and resistance to command hallucinations. J Nerv Ment Dis. 2004;192(5):357-362.
18. Junginger J. Predicting compliance with command hallucinations. Am J Psychiatry. 1990;147(2):245-247.
19. Erkwoh R, Willmes K, Eming-Erdmann A, et al. Command hallucinations: who obeys and who resists when? Psychopathology. 2002;35(5):272-279.
20. Beck-Sander A, Birchwood M, Chadwick P. Acting on command hallucinations: a cognitive approach. Br J Clin Psychol. 1997;36(pt 1):139-148.
21. Fox JRE, Gray NS, Lewis H. Factors determining compliance with command hallucinations with violent content: the role of social rank perceived power of the voice and voice malevolence. J Forens Psychiatry Psychol. 2004;15(3):511-531.
When evaluating a patient’s risk of violence, the presence of psychosis is a crucial concern. Douglas et al1 found that psychosis was the most important predictor of violent behavior in an analysis of 204 studies examining the relationship between psychopathology and aggression. Clinicians need to be familiar with aspects of persecutory delusions and command auditory hallucinations that are associated with an increased risk of aggression because accurately assessing patients who are experiencing these 2 symptoms is an important part of a comprehensive violence risk assessment.
This article highlights the importance of investigating persecutory delusions and command auditory hallucinations when evaluating a psychotic patient’s risk for violence. We provide specific questions to ask to help gauge risk associated with these 2 symptoms.
Evaluating persecutory delusions
Do persecutory delusions increase the risk that a person will behave violently? Research examining delusions’ contribution to violent behavior does not provide a clear answer. Earlier studies suggested that persecutory delusions were associated with an increased risk of aggression.2 Delusions noted to increase the risk of violence were characterized by threat/control-override (TCO) symptoms. TCO symptoms are beliefs that one is being threatened (eg, being followed or poisoned) or is losing control to an external source (eg, one’s mind is dominated by forces beyond his or her control).3 Similarly, using data from the Epidemiologic Catchment Area surveys, Swanson et al4 found that patients who reported TCO symptoms were approximately twice as likely to engage in assaultive behavior compared with patients with other psychotic symptoms.
In contrast, the MacArthur Study of Mental Disorder and Violence5,6 showed that the presence of delusions did not predict higher rates of violence among recently discharged psychiatric patients. In particular, researchers did not find a relationship between the presence of TCO delusions and violent behavior. In a study comparing male criminal offenders with schizophrenia found not guilty by reason of insanity with matched non-offending schizophrenia patients, Stompe et al7 found no significant association between TCO symptoms and severity of violent behavior; prevalence of TCO symptoms did not differ between the 2 groups. However, nondelusional suspiciousness—such as misperceiving others’ behavior as indicating hostile intent—was associated with subsequent violence.6
Nederlof et al8 conducted a cross-sectional multicenter study to further examine whether TCO symptoms are related to aggressive behavior. Their study included 124 patients (88% men) who had paranoid schizophrenia (70%), “other forms” of schizophrenia (16%), schizoaffective disorder (3%), delusional disorder (1%), and psychosis not otherwise specified (10%). To measure TCO symptoms in a more detailed manner than in previous research, these researchers developed the Threat/Control-Override Questionnaire (TCOQ), a 14-item, self-report scale. The 7 threat items specific to the TCOQ are:8
- I am under the control of an external force that determines my actions.
- Other people have tried to poison me or to do me harm.
- Someone has deliberately tried to make me ill.
- Other people have been secretly plotting to ruin me.
- Someone has had evil intentions against me.
- I have the thought that I was being followed for a special reason.
- People have tried to drive me insane.
The 7 control-override items on the TCOQ are:8
- Other people control my way of movements.
- Other people can insert thoughts into my head.
- My thoughts are dominated by an external force.
- I have the feeling that other people can determine my thoughts.
- Other people can insert thoughts into my mind.
- I have the feeling that other people have control over me.
- My life is being determined by something or someone except for myself.
Nederlof et al8 determined that TCO symptoms were a significant correlate of aggression in their study sample. When the 2 domains of TCO symptoms were evaluated separately, only threat symptoms made a significant contribution to aggressive behavior. These researchers suggested that varying methods of measuring TCO symptoms may underlie previous studies’ seemingly contradictory findings.8 These recent findings indicate that the debate regarding the contribution of TCO symptoms, particularly threat symptoms, to future violence remains active.
Appelbaum et al9 used the MacArthur-Maudsley Delusions Assessment Schedule to examine the contribution of non-content-related delusional material to violence in interviews with 328 delusional hospitalized psychiatric patients. The 7 dimensions of the MacArthur-Maudsley Delusions Assessment Schedule are:
- Conviction—the degree of certainty about the delusional belief
- Negative affect—whether the delusional belief makes the patient unhappy, frightened, anxious, or angry
- Action—the extent to which the patient’s actions are motivated by the delusional belief
- Inaction—whether the patient has refrained from any action as a result of the delusional belief
- Preoccupation—the extent to which the patient indicates his or her thoughts focus exclusively on the delusion
- Pervasiveness—the degree to which the delusional belief penetrates all aspects of the patient’s experiences
- Fluidity—the degree to which the delusional belief changed frequently during the interview.
Patients with persecutory delusions had significantly higher scores on “action” and “negative affect” dimensions, indicating that those with persecutory delusions may be more likely to react in response to the dysphoric aspects of their symptoms.9 Subsequent research has demonstrated that patients who suffer from persecutory delusions and negative affect are more likely to act on their delusions2,10 and to act violently11 than patients without these symptoms.
When evaluating a patient who experiences persecutory delusions, inquire if he or she has employed “safety actions.” These are specific behaviors—such as avoiding a perceived persecutor or escaping a fearful situation—the individual has employed with the intention of minimizing a misperceived threat. In a study of 100 patients with persecutory delusions, 96% reported using safety behaviors in the past month.12 In this study, individuals with a history of violence reported a greater use of safety behaviors.
Table 1 lists 10 questions to ask patients to explore persecutory delusions and associated risk factors for aggression.
Table 1
Evaluating persecutory delusions: 10 questions
| 1. | Who or what do you believe wants to harm you? |
| 2. | How is this person attempting to harm you? (Ask about specific threat/control-override beliefs) |
| 3. | How certain are you that this is happening? |
| 4. | Is there anything that could convince you that this isn’t true? |
| 5. | How does your belief make you feel (eg, unhappy, frightened, anxious, or angry)? |
| 6. | Have you thought about any actions to take as a result of these beliefs? If so, what? |
| 7. | Have you taken any action as a result of your beliefs? If so, what specific actions? |
| 8. | Has your concern about being harmed stopped you from doing any action that you would normally do? Have you changed your routine in any way? |
| 9. | How much time do you spend thinking about this each day? |
| 10. | In what ways have these beliefs impacted your life? |
Assessing auditory hallucinations
A careful inquiry about hallucinations can help determine whether their presence increases a patient’s risk of committing a violent act. Command hallucinations provide some type of directive to the patient. Approximately 50% of hallucinating psychiatric patients experience command hallucinations.13 Most command hallucinations are nonviolent, and patients are more likely to obey nonviolent instructions than violent commands.14
Research on factors associated with a patient acting on harmful command hallucinations has been mixed. In a review of 7 controlled studies, no study demonstrated a positive relationship between command hallucinations and violence, and 1 found an inverse relationship.15 In contrast, in a study of 103 psychiatric inpatients, McNiel et al16 found 30% reported having command hallucinations to harm others during the past year and 22% reported they complied with such commands. These researchers concluded that compared with those without command hallucinations, patients in their study who experienced command hallucinations to harm others were more than twice as likely to be violent.
Much of the literature examining the relationship between a patient’s actions and command hallucinations has examined the patient’s response to all command hallucinations, without delineating factors specific to violent commands. Seven factors are associated with acting on command hallucinations:13
- the presence of coexisting delusions17
- having delusions that relate to the hallucination18
- knowing the voice’s identity18
- believing the voices to be real19
- believing that the voices are benevolent20
- having few coping strategies to deal with the voices17
- not feeling in control over the voices.20
These factors also have been found to indicate increased compliance with acting on violent command hallucinations.18,20 Studies that have examined compliance specific to harmful command hallucinations provide additional guidance when evaluating the patient’s risk of harm. Aspects relevant to increased compliance to violent command hallucinations include a belief that the voice is powerful,13,21 a patient’s sense of personal superiority,21 a belief that command hallucinations benefit the patient,13 delusions that were congruent with the action described,13 and hallucinations that generate negative emotions such as anger, anxiety, and sadness.11
Table 2 lists 10 questions to ask to further investigate general command auditory hallucinations and violent command auditory hallucinations.
Table 2
Evaluating command auditory hallucinations: 10 questions
| 1. | What are the voices telling you to do? |
| 2. | Do you have any thoughts or beliefs that are associated with what you are hearing? If so, what are they? |
| 3. | Do you know the voice’s identity? If so, who is it? |
| 4. | How convinced are you that these voices are real? |
| 5. | Are these voices wishing you well or do you think that they wish you harm? |
| 6. | Have you done anything to help make the voices go away? If so, what? |
| 7. | Do you feel you have control of the voices or do you feel they control you? |
| 8. | Do you believe the voice is powerful? |
| 9. | How do the voices make you feel? |
| 10. | Have you ever done what the voice has told you to do? If so, describe what you did. |
Related Resources
- MacArthur Research Network on Mental Health and the Law. The MacArthur Violence Risk Assessment Study.http://macarthur.virginia.edu/risk.html.
- Witt K, van Dorn R, Fazel S. Risk factors for violence in psychosis: systematic review and meta-regression analysis of 110 studies [published online February 13, 2013]. PLoS One. 2013;8(2):e55942. doi: 10.1371/journal.pone.0055942.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
When evaluating a patient’s risk of violence, the presence of psychosis is a crucial concern. Douglas et al1 found that psychosis was the most important predictor of violent behavior in an analysis of 204 studies examining the relationship between psychopathology and aggression. Clinicians need to be familiar with aspects of persecutory delusions and command auditory hallucinations that are associated with an increased risk of aggression because accurately assessing patients who are experiencing these 2 symptoms is an important part of a comprehensive violence risk assessment.
This article highlights the importance of investigating persecutory delusions and command auditory hallucinations when evaluating a psychotic patient’s risk for violence. We provide specific questions to ask to help gauge risk associated with these 2 symptoms.
Evaluating persecutory delusions
Do persecutory delusions increase the risk that a person will behave violently? Research examining delusions’ contribution to violent behavior does not provide a clear answer. Earlier studies suggested that persecutory delusions were associated with an increased risk of aggression.2 Delusions noted to increase the risk of violence were characterized by threat/control-override (TCO) symptoms. TCO symptoms are beliefs that one is being threatened (eg, being followed or poisoned) or is losing control to an external source (eg, one’s mind is dominated by forces beyond his or her control).3 Similarly, using data from the Epidemiologic Catchment Area surveys, Swanson et al4 found that patients who reported TCO symptoms were approximately twice as likely to engage in assaultive behavior compared with patients with other psychotic symptoms.
In contrast, the MacArthur Study of Mental Disorder and Violence5,6 showed that the presence of delusions did not predict higher rates of violence among recently discharged psychiatric patients. In particular, researchers did not find a relationship between the presence of TCO delusions and violent behavior. In a study comparing male criminal offenders with schizophrenia found not guilty by reason of insanity with matched non-offending schizophrenia patients, Stompe et al7 found no significant association between TCO symptoms and severity of violent behavior; prevalence of TCO symptoms did not differ between the 2 groups. However, nondelusional suspiciousness—such as misperceiving others’ behavior as indicating hostile intent—was associated with subsequent violence.6
Nederlof et al8 conducted a cross-sectional multicenter study to further examine whether TCO symptoms are related to aggressive behavior. Their study included 124 patients (88% men) who had paranoid schizophrenia (70%), “other forms” of schizophrenia (16%), schizoaffective disorder (3%), delusional disorder (1%), and psychosis not otherwise specified (10%). To measure TCO symptoms in a more detailed manner than in previous research, these researchers developed the Threat/Control-Override Questionnaire (TCOQ), a 14-item, self-report scale. The 7 threat items specific to the TCOQ are:8
- I am under the control of an external force that determines my actions.
- Other people have tried to poison me or to do me harm.
- Someone has deliberately tried to make me ill.
- Other people have been secretly plotting to ruin me.
- Someone has had evil intentions against me.
- I have the thought that I was being followed for a special reason.
- People have tried to drive me insane.
The 7 control-override items on the TCOQ are:8
- Other people control my way of movements.
- Other people can insert thoughts into my head.
- My thoughts are dominated by an external force.
- I have the feeling that other people can determine my thoughts.
- Other people can insert thoughts into my mind.
- I have the feeling that other people have control over me.
- My life is being determined by something or someone except for myself.
Nederlof et al8 determined that TCO symptoms were a significant correlate of aggression in their study sample. When the 2 domains of TCO symptoms were evaluated separately, only threat symptoms made a significant contribution to aggressive behavior. These researchers suggested that varying methods of measuring TCO symptoms may underlie previous studies’ seemingly contradictory findings.8 These recent findings indicate that the debate regarding the contribution of TCO symptoms, particularly threat symptoms, to future violence remains active.
Appelbaum et al9 used the MacArthur-Maudsley Delusions Assessment Schedule to examine the contribution of non-content-related delusional material to violence in interviews with 328 delusional hospitalized psychiatric patients. The 7 dimensions of the MacArthur-Maudsley Delusions Assessment Schedule are:
- Conviction—the degree of certainty about the delusional belief
- Negative affect—whether the delusional belief makes the patient unhappy, frightened, anxious, or angry
- Action—the extent to which the patient’s actions are motivated by the delusional belief
- Inaction—whether the patient has refrained from any action as a result of the delusional belief
- Preoccupation—the extent to which the patient indicates his or her thoughts focus exclusively on the delusion
- Pervasiveness—the degree to which the delusional belief penetrates all aspects of the patient’s experiences
- Fluidity—the degree to which the delusional belief changed frequently during the interview.
Patients with persecutory delusions had significantly higher scores on “action” and “negative affect” dimensions, indicating that those with persecutory delusions may be more likely to react in response to the dysphoric aspects of their symptoms.9 Subsequent research has demonstrated that patients who suffer from persecutory delusions and negative affect are more likely to act on their delusions2,10 and to act violently11 than patients without these symptoms.
When evaluating a patient who experiences persecutory delusions, inquire if he or she has employed “safety actions.” These are specific behaviors—such as avoiding a perceived persecutor or escaping a fearful situation—the individual has employed with the intention of minimizing a misperceived threat. In a study of 100 patients with persecutory delusions, 96% reported using safety behaviors in the past month.12 In this study, individuals with a history of violence reported a greater use of safety behaviors.
Table 1 lists 10 questions to ask patients to explore persecutory delusions and associated risk factors for aggression.
Table 1
Evaluating persecutory delusions: 10 questions
| 1. | Who or what do you believe wants to harm you? |
| 2. | How is this person attempting to harm you? (Ask about specific threat/control-override beliefs) |
| 3. | How certain are you that this is happening? |
| 4. | Is there anything that could convince you that this isn’t true? |
| 5. | How does your belief make you feel (eg, unhappy, frightened, anxious, or angry)? |
| 6. | Have you thought about any actions to take as a result of these beliefs? If so, what? |
| 7. | Have you taken any action as a result of your beliefs? If so, what specific actions? |
| 8. | Has your concern about being harmed stopped you from doing any action that you would normally do? Have you changed your routine in any way? |
| 9. | How much time do you spend thinking about this each day? |
| 10. | In what ways have these beliefs impacted your life? |
Assessing auditory hallucinations
A careful inquiry about hallucinations can help determine whether their presence increases a patient’s risk of committing a violent act. Command hallucinations provide some type of directive to the patient. Approximately 50% of hallucinating psychiatric patients experience command hallucinations.13 Most command hallucinations are nonviolent, and patients are more likely to obey nonviolent instructions than violent commands.14
Research on factors associated with a patient acting on harmful command hallucinations has been mixed. In a review of 7 controlled studies, no study demonstrated a positive relationship between command hallucinations and violence, and 1 found an inverse relationship.15 In contrast, in a study of 103 psychiatric inpatients, McNiel et al16 found 30% reported having command hallucinations to harm others during the past year and 22% reported they complied with such commands. These researchers concluded that compared with those without command hallucinations, patients in their study who experienced command hallucinations to harm others were more than twice as likely to be violent.
Much of the literature examining the relationship between a patient’s actions and command hallucinations has examined the patient’s response to all command hallucinations, without delineating factors specific to violent commands. Seven factors are associated with acting on command hallucinations:13
- the presence of coexisting delusions17
- having delusions that relate to the hallucination18
- knowing the voice’s identity18
- believing the voices to be real19
- believing that the voices are benevolent20
- having few coping strategies to deal with the voices17
- not feeling in control over the voices.20
These factors also have been found to indicate increased compliance with acting on violent command hallucinations.18,20 Studies that have examined compliance specific to harmful command hallucinations provide additional guidance when evaluating the patient’s risk of harm. Aspects relevant to increased compliance to violent command hallucinations include a belief that the voice is powerful,13,21 a patient’s sense of personal superiority,21 a belief that command hallucinations benefit the patient,13 delusions that were congruent with the action described,13 and hallucinations that generate negative emotions such as anger, anxiety, and sadness.11
Table 2 lists 10 questions to ask to further investigate general command auditory hallucinations and violent command auditory hallucinations.
Table 2
Evaluating command auditory hallucinations: 10 questions
| 1. | What are the voices telling you to do? |
| 2. | Do you have any thoughts or beliefs that are associated with what you are hearing? If so, what are they? |
| 3. | Do you know the voice’s identity? If so, who is it? |
| 4. | How convinced are you that these voices are real? |
| 5. | Are these voices wishing you well or do you think that they wish you harm? |
| 6. | Have you done anything to help make the voices go away? If so, what? |
| 7. | Do you feel you have control of the voices or do you feel they control you? |
| 8. | Do you believe the voice is powerful? |
| 9. | How do the voices make you feel? |
| 10. | Have you ever done what the voice has told you to do? If so, describe what you did. |
Related Resources
- MacArthur Research Network on Mental Health and the Law. The MacArthur Violence Risk Assessment Study.http://macarthur.virginia.edu/risk.html.
- Witt K, van Dorn R, Fazel S. Risk factors for violence in psychosis: systematic review and meta-regression analysis of 110 studies [published online February 13, 2013]. PLoS One. 2013;8(2):e55942. doi: 10.1371/journal.pone.0055942.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Douglas KS, Guy LS, Hart SD. Psychosis as a risk factor for violence to others: a meta-analysis. Psychol Bull. 2009;135(5):679-706.
2. Wessely S, Buchanan A, Reed A, et al. Acting on delusions. I: Prevalence. Br J Psychiatry. 1993;163:69-76.
3. Link BG, Stueve A. Evidence bearing on mental illness as a possible cause of violent behavior. Epidemiol Rev. 1995;17(1):172-181.
4. Swanson JW, Borum R, Swartz MS, et al. Psychotic symptoms and disorders and the risk of violent behaviour in the community. Crim Behav Ment Health. 1996;6(4):309-329.
5. MacArthur Research Network on Mental Health and the Law. The MacArthur Violence Risk Assessment Study. http://macarthur.virginia.edu/risk.html. Published April 2001. Accessed March 21 2013.
6. Monahan J, Steadman HJ, Silver E, et al. Rethinking risk assessment: the MacArthur study of mental disorder and violence. New York, NY: Oxford University Press, Inc.; 2001.
7. Stompe T, Ortwein-Swoboda G, Schanda H. Schizophrenia delusional symptoms, and violence: the threat/control override concept reexamined. Schizophr Bull. 2004;30(1):31-44.
8. Nederlof AF, Muris P, Hovens JE. Threat/control-override symptoms and emotional reactions to positive symptoms as correlates of aggressive behavior in psychotic patients. J Nerv Ment Dis. 2011;199(5):342-347.
9. Appelbaum PS, Robbins PC, Roth LH. Dimensional approach to delusions: comparison across types and diagnoses. Am J Psychiatry. 1999;156(12):1938-1943.
10. Buchanan A, Reed A, Wessely S, et al. Acting on delusions. II: The phenomenological correlates of acting on delusions. Br J Psychiatry. 1993;163:77-81.
11. Cheung P, Schweitzer I, Crowley K, et al. Violence in schizophrenia: role of hallucinations and delusions. Schizophr Res. 1997;26(2-3):181-190.
12. Freeman D, Garety PA, Kuipers E, et al. Acting on persecutory delusions: the importance of safety seeking. Behav Res Ther. 2007;45(1):89-99.
13. Shawyer F, MacKinnon A, Farhall J, et al. Command hallucinations and violence: implications for detention and treatment. Psychiatr Psychol Law. 2003;10(1):97-107.
14. Chadwick P, Birchwood M. The omnipotence of voices. A cognitive approach to auditory hallucinations. Br J Psychiatry. 1994;164(2):190-201.
15. Rudnick A. Relation between command hallucinations and dangerous behavior. J Am Acad Psychiatry Law. 1999;27(2):253-257.
16. McNiel DE, Eisner JP, Binder RL. The relationship between command hallucinations and violence. Psychiatr Serv. 2000;51(10):1288-1292.
17. Mackinnon A, Copolov DL, Trauer T. Factors associated with compliance and resistance to command hallucinations. J Nerv Ment Dis. 2004;192(5):357-362.
18. Junginger J. Predicting compliance with command hallucinations. Am J Psychiatry. 1990;147(2):245-247.
19. Erkwoh R, Willmes K, Eming-Erdmann A, et al. Command hallucinations: who obeys and who resists when? Psychopathology. 2002;35(5):272-279.
20. Beck-Sander A, Birchwood M, Chadwick P. Acting on command hallucinations: a cognitive approach. Br J Clin Psychol. 1997;36(pt 1):139-148.
21. Fox JRE, Gray NS, Lewis H. Factors determining compliance with command hallucinations with violent content: the role of social rank perceived power of the voice and voice malevolence. J Forens Psychiatry Psychol. 2004;15(3):511-531.
1. Douglas KS, Guy LS, Hart SD. Psychosis as a risk factor for violence to others: a meta-analysis. Psychol Bull. 2009;135(5):679-706.
2. Wessely S, Buchanan A, Reed A, et al. Acting on delusions. I: Prevalence. Br J Psychiatry. 1993;163:69-76.
3. Link BG, Stueve A. Evidence bearing on mental illness as a possible cause of violent behavior. Epidemiol Rev. 1995;17(1):172-181.
4. Swanson JW, Borum R, Swartz MS, et al. Psychotic symptoms and disorders and the risk of violent behaviour in the community. Crim Behav Ment Health. 1996;6(4):309-329.
5. MacArthur Research Network on Mental Health and the Law. The MacArthur Violence Risk Assessment Study. http://macarthur.virginia.edu/risk.html. Published April 2001. Accessed March 21 2013.
6. Monahan J, Steadman HJ, Silver E, et al. Rethinking risk assessment: the MacArthur study of mental disorder and violence. New York, NY: Oxford University Press, Inc.; 2001.
7. Stompe T, Ortwein-Swoboda G, Schanda H. Schizophrenia delusional symptoms, and violence: the threat/control override concept reexamined. Schizophr Bull. 2004;30(1):31-44.
8. Nederlof AF, Muris P, Hovens JE. Threat/control-override symptoms and emotional reactions to positive symptoms as correlates of aggressive behavior in psychotic patients. J Nerv Ment Dis. 2011;199(5):342-347.
9. Appelbaum PS, Robbins PC, Roth LH. Dimensional approach to delusions: comparison across types and diagnoses. Am J Psychiatry. 1999;156(12):1938-1943.
10. Buchanan A, Reed A, Wessely S, et al. Acting on delusions. II: The phenomenological correlates of acting on delusions. Br J Psychiatry. 1993;163:77-81.
11. Cheung P, Schweitzer I, Crowley K, et al. Violence in schizophrenia: role of hallucinations and delusions. Schizophr Res. 1997;26(2-3):181-190.
12. Freeman D, Garety PA, Kuipers E, et al. Acting on persecutory delusions: the importance of safety seeking. Behav Res Ther. 2007;45(1):89-99.
13. Shawyer F, MacKinnon A, Farhall J, et al. Command hallucinations and violence: implications for detention and treatment. Psychiatr Psychol Law. 2003;10(1):97-107.
14. Chadwick P, Birchwood M. The omnipotence of voices. A cognitive approach to auditory hallucinations. Br J Psychiatry. 1994;164(2):190-201.
15. Rudnick A. Relation between command hallucinations and dangerous behavior. J Am Acad Psychiatry Law. 1999;27(2):253-257.
16. McNiel DE, Eisner JP, Binder RL. The relationship between command hallucinations and violence. Psychiatr Serv. 2000;51(10):1288-1292.
17. Mackinnon A, Copolov DL, Trauer T. Factors associated with compliance and resistance to command hallucinations. J Nerv Ment Dis. 2004;192(5):357-362.
18. Junginger J. Predicting compliance with command hallucinations. Am J Psychiatry. 1990;147(2):245-247.
19. Erkwoh R, Willmes K, Eming-Erdmann A, et al. Command hallucinations: who obeys and who resists when? Psychopathology. 2002;35(5):272-279.
20. Beck-Sander A, Birchwood M, Chadwick P. Acting on command hallucinations: a cognitive approach. Br J Clin Psychol. 1997;36(pt 1):139-148.
21. Fox JRE, Gray NS, Lewis H. Factors determining compliance with command hallucinations with violent content: the role of social rank perceived power of the voice and voice malevolence. J Forens Psychiatry Psychol. 2004;15(3):511-531.

Suicide, depression, and CYP2D6: How are they linked?
Genetic variations in drug-metabolizing enzymes dramatically affect drug pharmacokinetics and can result in clinically relevant differences in drug efficacy or toxicity. Cytochrome P450 (CYP) enzymes such as CYP2D6 are involved in metabolism of antidepressants, including selective serotonin reuptake inhibitors (SSRIs), which often are a first-line choice for patients with major depressive disorder (MDD).1,2 CYP2D6 is a highly polymorphic gene with 75 allelic variants (CYP2D6*1 to *75) and >30 additional subvariants.3 These variants are associated with phenotypes where CYP2D6 activity is increased, reduced, or lost, which can increase the risk of adverse drug reactions, decrease efficacy, and possibly influence a patient’s suicide risk.
In this article, we review the pharmacogenetics of CYP2D6 and discuss a possible relationship between CYP2D6 genotype and suicidal events during antidepressant treatment for MDD.
CYP2D6: Many variants
CYP450 enzymes are a group of 57 proteins, each coded by a different gene. Five subfamilies in the CYP450 family metabolize most drugs: CYP1A2, CYP3A4, CYP2C19, CYP2E1, and CYP2D6.4
Researchers discovered CYP2D6 in studies of nonpsychotropics (Box).5-9 CYP2D6 is widely expressed in many tissues, with dominant expression in the liver. Although CYP2D6 accounts for 2% of the total CYP450 liver enzyme content, it mediates metabolism in 25% to 30% of drugs in common clinical use and has a major influence on the biotransformation of SSRIs (Table).10
I the late 1970s, 2 groups of researchers noted unexpected serious adverse reactions in studies of debrisoquine,5 a sympatholytic antihypertensive drug, and sparteine,6 an antiarrhythmic and oxytocic alkaloid drug. They observed that 5% to 10% of patients were unable to efficiently metabolize debrisoquine and sparteine and went on to define a genetic polymorphism responsible for these metabolic differences. They also observed that metabolism of antidepressants, antipsychotics, and beta blockers also was defective in these patients.
Further investigations established that the enzyme responsible for debrisoquine metabolism was a cytochrome P450 (CYP) enzyme that is now termed CYP2D6.7 In addition to biochemical evidence, the colocalization of sparteine oxidation deficiency and of the CYP2D6 locus at chromosome 22q13.1 confirmed CYP2D6 as the target gene of the debrisoquine/sparteine polymorphism.8,9
Table
CYP450 enzymes involved in biotransformation of SSRIs
| SSRI | Enzymes involved in biotransformation |
|---|---|
| Citalopram | CYP2C19, CYP2D6, CYP3A4 |
| Escitalopram | CYP2C19, CYP2D6, CYP3A4 |
| Fluoxetine | CYP2D6, CYP2C9, CYP2C19, CYP3A4 |
| Fluvoxamine | CYP1A2, CYP2D6 |
| Paroxetine | CYP2D6, CYP3A4 |
| Sertraline | CYP2C9, CYP2C19, CYP2D6, CYP3A4 |
| CYP: cytochrome P450; SSRI: selective serotonin reuptake inhibitors Source: Reference 10 | |
Approximately 100 polymorphic CYP2D6 alleles (variants) have been identified.3 These alleles are active, resulting in normal CYP2D6 enzyme activity, or inactive, leading to decreased enzyme activity. Genotyping for most common CYP2D6 alleles in ethnically defined populations can predict poor metabolizers (PMs), intermediate metabolizers (IMs), extensive metabolizers (EMs), and ultra-rapid metabolizers (UMs) with high accuracy.11 PMs are compound heterozygous for inactivating alleles or homozygous for an inactivating variant. IMs carry one functional allele and one nonfunctional allele but may demonstrate a range of enzyme activity levels. EMs have 2 functional gene copies and UMs have >2 functional genes from gene duplication, resulting in ultra-rapid metabolism.
Suicide and CYP2D6 status
The widespread use of antidepressants appears to have led to significant decline in suicide rates in many countries.12 Based on an investigation of suicide mortality in 27 countries from 1980 to 2000, Ludwig and Marcotte12 found that faster growth in SSRI sales per capita was associated with larger declines in suicide rates. This finding was not confounded by other suicide risk factors such as unemployment, sex, age, or divorce rate.12 Countries such as Germany, Austria, Estonia, Switzerland, Sweden, Denmark, Hungary, and Slovenia—which had the highest suicide rate in the world 20 years ago (20 to 46 per 100,000 per year)—have had impressive declines in suicide rates (24% to 57% in the last 2 decades) with a marked (6- to 8-fold) increase in SSRI prescriptions during the same period.13-15 On the other hand, a few countries, such as Portugal and Spain, have experienced dramatic increases (58% and 86%, respectively) in the suicide rate with a similar increase in SSRI prescribing during the same 20-year period.16
A review of the distribution of CYP2D6 genotype among countries indicates a south/north gradient of CYP2D6 gene duplications, which indicate UM status.16 The proportion of UMs increases by almost 2-fold in southern European countries (8.4% and 7% to 10% for Portugal and Spain, respectively) compared with northern European countries (1% to 2% and 3.6% for Sweden and Germany, respectively); this south/north trend extends to Africa.17 The prevalence of CYP2D6 UMs is lower in northern countries, where increased anti-depressant use appears to have reduced suicide rates, and higher in southern countries, where suicide rates increased despite higher antidepressant use.
Case reports and observational studies18-21 suggest that compared with other CYP2D6 phenotypes, UMs may need to take higher doses of antidepressants to achieve therapeutic response. In a case report, Bertilsson et al18 described 2 patients who were UMs and required high doses of nortriptyline and clomipramine to obtain appropriate plasma drug concentrations. Baumann et al19 described a depressed patient with CYP2D6 gene duplication who required higher-than-usual doses of clomipramine. Rau et al20 found a 3-fold increase in the frequency of UMs in a group of 16 depressed German patients who did not respond to SSRIs or serotonin–norepinephrine reuptake inhibitors, both of which are metabolized by CYP2D6. Kawanishi et al21 found a significantly greater prevalence of UMs among 81 Nordic patients who did not respond to SSRIs compared with the general population.
Because suicidality may be caused by inadequately treated depressive illness, MDD patients who are UMs may be more likely to commit suicide because of suboptimal antidepressant levels. In a 2010 Swedish study, Zackrisson et al22 found that compared with those who died of other causes, significantly more individuals who committed suicide had >2 active CYP2D6 genes. Stingl et al23 found that among 285 depressed German patients, UMs had an elevated risk of having a high suicidality score compared with individuals with other genotypes, after adjusting for sex, baseline score on the Hamilton Depression Rating Scale (after excluding item 3 for suicidality), and number of previous depressive episodes. Other researchers found that patients with eating disorders who are UMs have a greater risk of suicidal behavior.24 Although none of these 3 studies specified if these patients were treated with antidepressants, the association between CYP2D6 gene duplication and suicide risk suggests CYP2D6’s role in suicide risk might not be related solely to antidepressant metabolism.
Effects on serotonin, dopamine
CYP2D6 is expressed in the brain and localized primarily in large principle cells of the hippocampus and Purkinje cells of the cerebellum, with no expression in other brain regions such as glial cells.25 This heterogeneous expression among brain regions and cell types indicates that in addition to its role in metabolizing drugs, CYP2D6 might influence neurotransmitter levels. In vitro and in vivo animal studies suggest that CYP2D6 plays a role in biotransformation of serotonin and dopamine.26,27
Serotonin is likely to play a causal role in the pathophysiology of depression, and depressed patients have abnormalities in serotonin activity.28 Serotonin is generated primarily from the transformation of tryptophan by tryptophan decarboxylase and tryptamine 5-hydroxylase.29 Yu et al27 found that CYP2D6 may be an additional pathway to regenerate serotonin through O-demethylation from 5-methoxytryptamine, but it is unclear what proportion of the physiologic pool of serotonin in synaptic nerve terminals is generated through the CYP2D6 pathway. However, this discovery provides a mechanistic basis of CYP2D6 involvement in the endogenous serotonin balance and by extension, in serotonergic physiology and neuropsychiatric disorders such as depression.30 Because SSRIs target the serotonergic pathway, baseline levels of serotonin and all related components of this pathway—including CYP2D6—are likely to help determine a patient’s response to SSRIs.
Dopamine also is generated from tyramine through CYP2D6,31 and distribution of CYP2D6 in the brain follows that of dopamine nerve terminals.32 The serotonergic system has strong anatomical and functional interaction with the dopaminergic system,33 and imbalance between serotonin and dopamine activity is thought to give rise to behavioral changes,2 which play an important role in the development of anxiety and impulsivity.
CYP2D6 in clinical practice
Although research into a possible link between CYP2D6 status and suicide risk in depressed patients treated with antidepressants is ongoing, at present this connection is speculative. More studies are warranted to reveal the exact role of CYP2D6 in response to SSRI treatment and suicide risk.
Knowledge of this potential association can help clinicians keep CYP450 genotyping in mind when prescribing antidepressants to depressed patients. The FDA has approved a pharmacogenetic test to analyze polymorphisms of CYP2D6 and CYP2C19.34 The results of such testing might guide pharmacotherapy for depressed patients, including medication selection and dosing. For example, a patient who is a PM might be started at a lower antidepressant dosage to avoid potential adverse drug effects, whereas it might be appropriate to prescribe a higher starting dose for a UM patient to achieve an effective drug concentration.
Related Resources
- Peñas-Lledó EM, Blasco-Fontecilla H, Dorado P, et al. CYP2D6 and the severity of suicide attempts. Pharmacogenomics. 2012;13(2):179-184.
- Blasco-Fontecilla H, Peñas-Lledó E, Vaquero-Lorenzo C, et al. CYP2D6 polymorphism and mental and personality disorders in suicide attempters [published online February 11, 2013]. J Pers Disord. doi: 10.1521/pedi_2013_27_080.
Drug Brand Names
- Citalopram • Celexa
- Clomipramine • Anafranil
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Nortriptyline • Aventyl, Pamelor
- Paroxetine • Paxil
- Sertraline • Zoloft
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
The authors thank Marwah Shahid and Ijlal Yazdani for their assistance with this article.
1. Meyer UA, Amrein R, Balant LP, et al. Antidepressants and drug-metabolizing enzymes—expert group report. Acta Psychiatr Scand. 1996;93(2):71-79.
2. Kroemer HK, Eichelbaum M. “It’s the genes stupid”. Molecular bases and clinical consequences of genetic cytochrome P450 2D6 polymorphism. Life Sci. 1995;56(26):2285-2298.
3. The Human Cytochrome P450 (CYP) Allele Nomenclature Database. CYP2D6 allele nomenclature. http://www.cypalleles.ki.se/cyp2d6.htm. Accessed February 25, 2013.
4. Hemeryck A, Belpaire FM. Selective serotonin reuptake inhibitors and cytochrome P-450 mediated drug-drug interactions: an update. Curr Drug Metab. 2002;3(1):13-37.
5. Mahgoub A, Idle JR, Dring LG, et al. Polymorphic hydroxylation of debrisoquine in man. Lancet. 1977;2(8038):584-586.
6. Eichelbaum M, Spannbrucker N, Steincke B, et al. Defective N-oxidation of sparteine in man: a new pharmacogenetic defect. Eur J Clin Pharmacol. 1979;16(3):183-187.
7. Distlerath LM, Reilly PE, Martin MV, et al. Purification and characterization of the human liver cytochromes P-450 involved in debrisoquine 4-hydroxylation and phenacetin O-deethylation, two prototypes for genetic polymorphism in oxidative drug metabolism. J Biol Chem. 1985;260(15):9057-9067.
8. Eichelbaum M, Baur MP, Dengler HJ, et al. Chromosomal assignment of human cytochrome P-450 (debrisoquine/sparteine type) to chromosome 22. Br J Clin Pharmacol. 1987;23(4):455-458.
9. Gonzalez FJ, Vilbois F, Hardwick JP, et al. Human debrisoquine 4-hydroxylase (P450IID1): cDNA and deduced amino acid sequence and assignment of the CYP2D locus to chromosome 22. Geonomics. 1988;2(2):174-179.
10. Spina E, Santoro V, D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008;30(7):1206-1227.
11. Roses AD. Pharmacogenetics and the practice of medicine. Nature. 2000;405(6788):857-865.
12. Ludwig J, Marcotte DE. Anti-depressants suicide, and drug regulation. J Policy Anal Manage. 2005;24(2):249-272.
13. Isacsson G. Suicide prevention—a medical breakthrough? Acta Psychiatr Scand. 2000;102(2):113-117.
14. Rihmer Z. Can better recognition and treatment of depression reduce suicide rates? A brief review. Eur Psychiatry. 2001;16(7):406-409.
15. Rihmer Z. Decreasing national suicide rates—fact or fiction? World J Biol Psychiatry. 2004;5(1):55-56.
16. Rihmer Z, Akiskal H. Do antidepressants t(h)reat(en) depressives? Toward a clinically judicious formulation of the antidepressant-suicidality FDA advisory in light of declining national suicide statistics from many countries. J Affect Disord. 2006;94(1-3):3-13.
17. Correia C, Santos P, Coutinho AM, et al. Characterization of pharmacogenetically relevant CYP2D6 and ABCB1 gene polymorphisms in a Portuguese population sample. Cell Biochem Funct. 2009;27(4):251-255.
18. Bertilsson L, Dahl ML, Sjöqvist F, et al. Molecular basis for rational megaprescribing in ultrarapid hydroxylators of debrisoquine. Lancet. 1993;341(8836):63.-
19. Baumann P, Broly F, Kosel M, et al. Ultrarapid metabolism of clomipramine in a therapy-resistant depressive patient, as confirmed by CYP2 D6 genotyping. Pharmacopsychiatry. 1998;31(2):72.-
20. Rau T, Wohlleben G, Wuttke H, et al. CYP2D6 genotype: impact on adverse effects and nonresponse during treatment with antidepressants-a pilot study. Clin Pharmacol Ther. 2004;75(5):386-393.
21. Kawanishi C, Lundgren S, Agren H, et al. Increased incidence of CYP2D6 gene duplication in patients with persistent mood disorders: ultrarapid metabolism of antidepressants as a cause of nonresponse. A pilot study. Eur J Clin Pharmacol. 2004;59(11):803-807.
22. Zackrisson AL, Lindblom B, Ahlner J. High frequency of occurrence of CYP2D6 gene duplication/multiduplication indicating ultrarapid metabolism among suicide cases. Clin Pharmacol Ther. 2010;88(3):354-359.
23. Stingl JC, Viviani R. CYP2D6 in the brain: impact on suicidality. Clin Pharmacol Ther. 2011;89(3):352-353.
24. Peñas-Lledó EM, Dorado P, Agüera Z, et al. High risk of lifetime history of suicide attempts among CYP2D6 ultrarapid metabolizers with eating disorders. Mol Psychiatry. 2011;16(7):691-692.
25. Siegle I, Fritz P, Eckhardt K, et al. Cellular localization and regional distribution of CYP2D6 mRNA and protein expression in human brain. Pharmacogenetics. 2001;11(3):237-245.
26. Eichelbaum M. In search of endogenous CYP2D6 substrates. Pharmacogenetics. 2003;13(6):305-306.
27. Yu AM, Idle JR, Gonzalez FJ. Polymorphic cytochrome P450 2D6: humanized mouse model and endogenous substrates. Drug Metab Rev. 2004;36(2):243-277.
28. Cowen PJ. Serotonin and depression: pathophysiological mechanism or marketing myth? Trends Pharmacol Sci. 2008;29(9):433-436.
29. Kang S, Kang K, Lee K, et al. Characterization of tryptamine 5-hydroxylase and serotonin synthesis in rice plants. Plant Cell Rep. 2007;26(11):2009-2015.
30. Yu AM, Idle JR, Herraiz T, et al. Screening for endogenous substrates reveals that CYP2D6 is a 5-methoxyindolethylamine O-demethylase. Pharmacogenetics. 2003;13(6):307-319.
31. Hiroi T, Imaoka S, Funae Y. Dopamine formation from tyramine by CYP2D6. Biochem Biophys Res Commun. 1998;249(3):838-843.
32. Niznik HB, Tyndale RF, Sallee FR, et al. The dopamine transporter and cytochrome P45OIID1 (debrisoquine 4-hydroxylase) in brain: resolution and identification of two distinct [3H]GBR-12935 binding proteins. Arch Biochem Biophys. 1990;276(2):424-432.
33. Kapur S, Remington G. Serotonin-dopamine interaction and its relevance to schizophrenia. Am J Psychiatry. 1996;153(4):466-476.
34. Jain KK. Applications of AmpliChip CYP450. Mol Diagn. 2005;9(3):119-127.
Genetic variations in drug-metabolizing enzymes dramatically affect drug pharmacokinetics and can result in clinically relevant differences in drug efficacy or toxicity. Cytochrome P450 (CYP) enzymes such as CYP2D6 are involved in metabolism of antidepressants, including selective serotonin reuptake inhibitors (SSRIs), which often are a first-line choice for patients with major depressive disorder (MDD).1,2 CYP2D6 is a highly polymorphic gene with 75 allelic variants (CYP2D6*1 to *75) and >30 additional subvariants.3 These variants are associated with phenotypes where CYP2D6 activity is increased, reduced, or lost, which can increase the risk of adverse drug reactions, decrease efficacy, and possibly influence a patient’s suicide risk.
In this article, we review the pharmacogenetics of CYP2D6 and discuss a possible relationship between CYP2D6 genotype and suicidal events during antidepressant treatment for MDD.
CYP2D6: Many variants
CYP450 enzymes are a group of 57 proteins, each coded by a different gene. Five subfamilies in the CYP450 family metabolize most drugs: CYP1A2, CYP3A4, CYP2C19, CYP2E1, and CYP2D6.4
Researchers discovered CYP2D6 in studies of nonpsychotropics (Box).5-9 CYP2D6 is widely expressed in many tissues, with dominant expression in the liver. Although CYP2D6 accounts for 2% of the total CYP450 liver enzyme content, it mediates metabolism in 25% to 30% of drugs in common clinical use and has a major influence on the biotransformation of SSRIs (Table).10
I the late 1970s, 2 groups of researchers noted unexpected serious adverse reactions in studies of debrisoquine,5 a sympatholytic antihypertensive drug, and sparteine,6 an antiarrhythmic and oxytocic alkaloid drug. They observed that 5% to 10% of patients were unable to efficiently metabolize debrisoquine and sparteine and went on to define a genetic polymorphism responsible for these metabolic differences. They also observed that metabolism of antidepressants, antipsychotics, and beta blockers also was defective in these patients.
Further investigations established that the enzyme responsible for debrisoquine metabolism was a cytochrome P450 (CYP) enzyme that is now termed CYP2D6.7 In addition to biochemical evidence, the colocalization of sparteine oxidation deficiency and of the CYP2D6 locus at chromosome 22q13.1 confirmed CYP2D6 as the target gene of the debrisoquine/sparteine polymorphism.8,9
Table
CYP450 enzymes involved in biotransformation of SSRIs
| SSRI | Enzymes involved in biotransformation |
|---|---|
| Citalopram | CYP2C19, CYP2D6, CYP3A4 |
| Escitalopram | CYP2C19, CYP2D6, CYP3A4 |
| Fluoxetine | CYP2D6, CYP2C9, CYP2C19, CYP3A4 |
| Fluvoxamine | CYP1A2, CYP2D6 |
| Paroxetine | CYP2D6, CYP3A4 |
| Sertraline | CYP2C9, CYP2C19, CYP2D6, CYP3A4 |
| CYP: cytochrome P450; SSRI: selective serotonin reuptake inhibitors Source: Reference 10 | |
Approximately 100 polymorphic CYP2D6 alleles (variants) have been identified.3 These alleles are active, resulting in normal CYP2D6 enzyme activity, or inactive, leading to decreased enzyme activity. Genotyping for most common CYP2D6 alleles in ethnically defined populations can predict poor metabolizers (PMs), intermediate metabolizers (IMs), extensive metabolizers (EMs), and ultra-rapid metabolizers (UMs) with high accuracy.11 PMs are compound heterozygous for inactivating alleles or homozygous for an inactivating variant. IMs carry one functional allele and one nonfunctional allele but may demonstrate a range of enzyme activity levels. EMs have 2 functional gene copies and UMs have >2 functional genes from gene duplication, resulting in ultra-rapid metabolism.
Suicide and CYP2D6 status
The widespread use of antidepressants appears to have led to significant decline in suicide rates in many countries.12 Based on an investigation of suicide mortality in 27 countries from 1980 to 2000, Ludwig and Marcotte12 found that faster growth in SSRI sales per capita was associated with larger declines in suicide rates. This finding was not confounded by other suicide risk factors such as unemployment, sex, age, or divorce rate.12 Countries such as Germany, Austria, Estonia, Switzerland, Sweden, Denmark, Hungary, and Slovenia—which had the highest suicide rate in the world 20 years ago (20 to 46 per 100,000 per year)—have had impressive declines in suicide rates (24% to 57% in the last 2 decades) with a marked (6- to 8-fold) increase in SSRI prescriptions during the same period.13-15 On the other hand, a few countries, such as Portugal and Spain, have experienced dramatic increases (58% and 86%, respectively) in the suicide rate with a similar increase in SSRI prescribing during the same 20-year period.16
A review of the distribution of CYP2D6 genotype among countries indicates a south/north gradient of CYP2D6 gene duplications, which indicate UM status.16 The proportion of UMs increases by almost 2-fold in southern European countries (8.4% and 7% to 10% for Portugal and Spain, respectively) compared with northern European countries (1% to 2% and 3.6% for Sweden and Germany, respectively); this south/north trend extends to Africa.17 The prevalence of CYP2D6 UMs is lower in northern countries, where increased anti-depressant use appears to have reduced suicide rates, and higher in southern countries, where suicide rates increased despite higher antidepressant use.
Case reports and observational studies18-21 suggest that compared with other CYP2D6 phenotypes, UMs may need to take higher doses of antidepressants to achieve therapeutic response. In a case report, Bertilsson et al18 described 2 patients who were UMs and required high doses of nortriptyline and clomipramine to obtain appropriate plasma drug concentrations. Baumann et al19 described a depressed patient with CYP2D6 gene duplication who required higher-than-usual doses of clomipramine. Rau et al20 found a 3-fold increase in the frequency of UMs in a group of 16 depressed German patients who did not respond to SSRIs or serotonin–norepinephrine reuptake inhibitors, both of which are metabolized by CYP2D6. Kawanishi et al21 found a significantly greater prevalence of UMs among 81 Nordic patients who did not respond to SSRIs compared with the general population.
Because suicidality may be caused by inadequately treated depressive illness, MDD patients who are UMs may be more likely to commit suicide because of suboptimal antidepressant levels. In a 2010 Swedish study, Zackrisson et al22 found that compared with those who died of other causes, significantly more individuals who committed suicide had >2 active CYP2D6 genes. Stingl et al23 found that among 285 depressed German patients, UMs had an elevated risk of having a high suicidality score compared with individuals with other genotypes, after adjusting for sex, baseline score on the Hamilton Depression Rating Scale (after excluding item 3 for suicidality), and number of previous depressive episodes. Other researchers found that patients with eating disorders who are UMs have a greater risk of suicidal behavior.24 Although none of these 3 studies specified if these patients were treated with antidepressants, the association between CYP2D6 gene duplication and suicide risk suggests CYP2D6’s role in suicide risk might not be related solely to antidepressant metabolism.
Effects on serotonin, dopamine
CYP2D6 is expressed in the brain and localized primarily in large principle cells of the hippocampus and Purkinje cells of the cerebellum, with no expression in other brain regions such as glial cells.25 This heterogeneous expression among brain regions and cell types indicates that in addition to its role in metabolizing drugs, CYP2D6 might influence neurotransmitter levels. In vitro and in vivo animal studies suggest that CYP2D6 plays a role in biotransformation of serotonin and dopamine.26,27
Serotonin is likely to play a causal role in the pathophysiology of depression, and depressed patients have abnormalities in serotonin activity.28 Serotonin is generated primarily from the transformation of tryptophan by tryptophan decarboxylase and tryptamine 5-hydroxylase.29 Yu et al27 found that CYP2D6 may be an additional pathway to regenerate serotonin through O-demethylation from 5-methoxytryptamine, but it is unclear what proportion of the physiologic pool of serotonin in synaptic nerve terminals is generated through the CYP2D6 pathway. However, this discovery provides a mechanistic basis of CYP2D6 involvement in the endogenous serotonin balance and by extension, in serotonergic physiology and neuropsychiatric disorders such as depression.30 Because SSRIs target the serotonergic pathway, baseline levels of serotonin and all related components of this pathway—including CYP2D6—are likely to help determine a patient’s response to SSRIs.
Dopamine also is generated from tyramine through CYP2D6,31 and distribution of CYP2D6 in the brain follows that of dopamine nerve terminals.32 The serotonergic system has strong anatomical and functional interaction with the dopaminergic system,33 and imbalance between serotonin and dopamine activity is thought to give rise to behavioral changes,2 which play an important role in the development of anxiety and impulsivity.
CYP2D6 in clinical practice
Although research into a possible link between CYP2D6 status and suicide risk in depressed patients treated with antidepressants is ongoing, at present this connection is speculative. More studies are warranted to reveal the exact role of CYP2D6 in response to SSRI treatment and suicide risk.
Knowledge of this potential association can help clinicians keep CYP450 genotyping in mind when prescribing antidepressants to depressed patients. The FDA has approved a pharmacogenetic test to analyze polymorphisms of CYP2D6 and CYP2C19.34 The results of such testing might guide pharmacotherapy for depressed patients, including medication selection and dosing. For example, a patient who is a PM might be started at a lower antidepressant dosage to avoid potential adverse drug effects, whereas it might be appropriate to prescribe a higher starting dose for a UM patient to achieve an effective drug concentration.
Related Resources
- Peñas-Lledó EM, Blasco-Fontecilla H, Dorado P, et al. CYP2D6 and the severity of suicide attempts. Pharmacogenomics. 2012;13(2):179-184.
- Blasco-Fontecilla H, Peñas-Lledó E, Vaquero-Lorenzo C, et al. CYP2D6 polymorphism and mental and personality disorders in suicide attempters [published online February 11, 2013]. J Pers Disord. doi: 10.1521/pedi_2013_27_080.
Drug Brand Names
- Citalopram • Celexa
- Clomipramine • Anafranil
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Nortriptyline • Aventyl, Pamelor
- Paroxetine • Paxil
- Sertraline • Zoloft
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
The authors thank Marwah Shahid and Ijlal Yazdani for their assistance with this article.
Genetic variations in drug-metabolizing enzymes dramatically affect drug pharmacokinetics and can result in clinically relevant differences in drug efficacy or toxicity. Cytochrome P450 (CYP) enzymes such as CYP2D6 are involved in metabolism of antidepressants, including selective serotonin reuptake inhibitors (SSRIs), which often are a first-line choice for patients with major depressive disorder (MDD).1,2 CYP2D6 is a highly polymorphic gene with 75 allelic variants (CYP2D6*1 to *75) and >30 additional subvariants.3 These variants are associated with phenotypes where CYP2D6 activity is increased, reduced, or lost, which can increase the risk of adverse drug reactions, decrease efficacy, and possibly influence a patient’s suicide risk.
In this article, we review the pharmacogenetics of CYP2D6 and discuss a possible relationship between CYP2D6 genotype and suicidal events during antidepressant treatment for MDD.
CYP2D6: Many variants
CYP450 enzymes are a group of 57 proteins, each coded by a different gene. Five subfamilies in the CYP450 family metabolize most drugs: CYP1A2, CYP3A4, CYP2C19, CYP2E1, and CYP2D6.4
Researchers discovered CYP2D6 in studies of nonpsychotropics (Box).5-9 CYP2D6 is widely expressed in many tissues, with dominant expression in the liver. Although CYP2D6 accounts for 2% of the total CYP450 liver enzyme content, it mediates metabolism in 25% to 30% of drugs in common clinical use and has a major influence on the biotransformation of SSRIs (Table).10
I the late 1970s, 2 groups of researchers noted unexpected serious adverse reactions in studies of debrisoquine,5 a sympatholytic antihypertensive drug, and sparteine,6 an antiarrhythmic and oxytocic alkaloid drug. They observed that 5% to 10% of patients were unable to efficiently metabolize debrisoquine and sparteine and went on to define a genetic polymorphism responsible for these metabolic differences. They also observed that metabolism of antidepressants, antipsychotics, and beta blockers also was defective in these patients.
Further investigations established that the enzyme responsible for debrisoquine metabolism was a cytochrome P450 (CYP) enzyme that is now termed CYP2D6.7 In addition to biochemical evidence, the colocalization of sparteine oxidation deficiency and of the CYP2D6 locus at chromosome 22q13.1 confirmed CYP2D6 as the target gene of the debrisoquine/sparteine polymorphism.8,9
Table
CYP450 enzymes involved in biotransformation of SSRIs
| SSRI | Enzymes involved in biotransformation |
|---|---|
| Citalopram | CYP2C19, CYP2D6, CYP3A4 |
| Escitalopram | CYP2C19, CYP2D6, CYP3A4 |
| Fluoxetine | CYP2D6, CYP2C9, CYP2C19, CYP3A4 |
| Fluvoxamine | CYP1A2, CYP2D6 |
| Paroxetine | CYP2D6, CYP3A4 |
| Sertraline | CYP2C9, CYP2C19, CYP2D6, CYP3A4 |
| CYP: cytochrome P450; SSRI: selective serotonin reuptake inhibitors Source: Reference 10 | |
Approximately 100 polymorphic CYP2D6 alleles (variants) have been identified.3 These alleles are active, resulting in normal CYP2D6 enzyme activity, or inactive, leading to decreased enzyme activity. Genotyping for most common CYP2D6 alleles in ethnically defined populations can predict poor metabolizers (PMs), intermediate metabolizers (IMs), extensive metabolizers (EMs), and ultra-rapid metabolizers (UMs) with high accuracy.11 PMs are compound heterozygous for inactivating alleles or homozygous for an inactivating variant. IMs carry one functional allele and one nonfunctional allele but may demonstrate a range of enzyme activity levels. EMs have 2 functional gene copies and UMs have >2 functional genes from gene duplication, resulting in ultra-rapid metabolism.
Suicide and CYP2D6 status
The widespread use of antidepressants appears to have led to significant decline in suicide rates in many countries.12 Based on an investigation of suicide mortality in 27 countries from 1980 to 2000, Ludwig and Marcotte12 found that faster growth in SSRI sales per capita was associated with larger declines in suicide rates. This finding was not confounded by other suicide risk factors such as unemployment, sex, age, or divorce rate.12 Countries such as Germany, Austria, Estonia, Switzerland, Sweden, Denmark, Hungary, and Slovenia—which had the highest suicide rate in the world 20 years ago (20 to 46 per 100,000 per year)—have had impressive declines in suicide rates (24% to 57% in the last 2 decades) with a marked (6- to 8-fold) increase in SSRI prescriptions during the same period.13-15 On the other hand, a few countries, such as Portugal and Spain, have experienced dramatic increases (58% and 86%, respectively) in the suicide rate with a similar increase in SSRI prescribing during the same 20-year period.16
A review of the distribution of CYP2D6 genotype among countries indicates a south/north gradient of CYP2D6 gene duplications, which indicate UM status.16 The proportion of UMs increases by almost 2-fold in southern European countries (8.4% and 7% to 10% for Portugal and Spain, respectively) compared with northern European countries (1% to 2% and 3.6% for Sweden and Germany, respectively); this south/north trend extends to Africa.17 The prevalence of CYP2D6 UMs is lower in northern countries, where increased anti-depressant use appears to have reduced suicide rates, and higher in southern countries, where suicide rates increased despite higher antidepressant use.
Case reports and observational studies18-21 suggest that compared with other CYP2D6 phenotypes, UMs may need to take higher doses of antidepressants to achieve therapeutic response. In a case report, Bertilsson et al18 described 2 patients who were UMs and required high doses of nortriptyline and clomipramine to obtain appropriate plasma drug concentrations. Baumann et al19 described a depressed patient with CYP2D6 gene duplication who required higher-than-usual doses of clomipramine. Rau et al20 found a 3-fold increase in the frequency of UMs in a group of 16 depressed German patients who did not respond to SSRIs or serotonin–norepinephrine reuptake inhibitors, both of which are metabolized by CYP2D6. Kawanishi et al21 found a significantly greater prevalence of UMs among 81 Nordic patients who did not respond to SSRIs compared with the general population.
Because suicidality may be caused by inadequately treated depressive illness, MDD patients who are UMs may be more likely to commit suicide because of suboptimal antidepressant levels. In a 2010 Swedish study, Zackrisson et al22 found that compared with those who died of other causes, significantly more individuals who committed suicide had >2 active CYP2D6 genes. Stingl et al23 found that among 285 depressed German patients, UMs had an elevated risk of having a high suicidality score compared with individuals with other genotypes, after adjusting for sex, baseline score on the Hamilton Depression Rating Scale (after excluding item 3 for suicidality), and number of previous depressive episodes. Other researchers found that patients with eating disorders who are UMs have a greater risk of suicidal behavior.24 Although none of these 3 studies specified if these patients were treated with antidepressants, the association between CYP2D6 gene duplication and suicide risk suggests CYP2D6’s role in suicide risk might not be related solely to antidepressant metabolism.
Effects on serotonin, dopamine
CYP2D6 is expressed in the brain and localized primarily in large principle cells of the hippocampus and Purkinje cells of the cerebellum, with no expression in other brain regions such as glial cells.25 This heterogeneous expression among brain regions and cell types indicates that in addition to its role in metabolizing drugs, CYP2D6 might influence neurotransmitter levels. In vitro and in vivo animal studies suggest that CYP2D6 plays a role in biotransformation of serotonin and dopamine.26,27
Serotonin is likely to play a causal role in the pathophysiology of depression, and depressed patients have abnormalities in serotonin activity.28 Serotonin is generated primarily from the transformation of tryptophan by tryptophan decarboxylase and tryptamine 5-hydroxylase.29 Yu et al27 found that CYP2D6 may be an additional pathway to regenerate serotonin through O-demethylation from 5-methoxytryptamine, but it is unclear what proportion of the physiologic pool of serotonin in synaptic nerve terminals is generated through the CYP2D6 pathway. However, this discovery provides a mechanistic basis of CYP2D6 involvement in the endogenous serotonin balance and by extension, in serotonergic physiology and neuropsychiatric disorders such as depression.30 Because SSRIs target the serotonergic pathway, baseline levels of serotonin and all related components of this pathway—including CYP2D6—are likely to help determine a patient’s response to SSRIs.
Dopamine also is generated from tyramine through CYP2D6,31 and distribution of CYP2D6 in the brain follows that of dopamine nerve terminals.32 The serotonergic system has strong anatomical and functional interaction with the dopaminergic system,33 and imbalance between serotonin and dopamine activity is thought to give rise to behavioral changes,2 which play an important role in the development of anxiety and impulsivity.
CYP2D6 in clinical practice
Although research into a possible link between CYP2D6 status and suicide risk in depressed patients treated with antidepressants is ongoing, at present this connection is speculative. More studies are warranted to reveal the exact role of CYP2D6 in response to SSRI treatment and suicide risk.
Knowledge of this potential association can help clinicians keep CYP450 genotyping in mind when prescribing antidepressants to depressed patients. The FDA has approved a pharmacogenetic test to analyze polymorphisms of CYP2D6 and CYP2C19.34 The results of such testing might guide pharmacotherapy for depressed patients, including medication selection and dosing. For example, a patient who is a PM might be started at a lower antidepressant dosage to avoid potential adverse drug effects, whereas it might be appropriate to prescribe a higher starting dose for a UM patient to achieve an effective drug concentration.
Related Resources
- Peñas-Lledó EM, Blasco-Fontecilla H, Dorado P, et al. CYP2D6 and the severity of suicide attempts. Pharmacogenomics. 2012;13(2):179-184.
- Blasco-Fontecilla H, Peñas-Lledó E, Vaquero-Lorenzo C, et al. CYP2D6 polymorphism and mental and personality disorders in suicide attempters [published online February 11, 2013]. J Pers Disord. doi: 10.1521/pedi_2013_27_080.
Drug Brand Names
- Citalopram • Celexa
- Clomipramine • Anafranil
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Nortriptyline • Aventyl, Pamelor
- Paroxetine • Paxil
- Sertraline • Zoloft
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
The authors thank Marwah Shahid and Ijlal Yazdani for their assistance with this article.
1. Meyer UA, Amrein R, Balant LP, et al. Antidepressants and drug-metabolizing enzymes—expert group report. Acta Psychiatr Scand. 1996;93(2):71-79.
2. Kroemer HK, Eichelbaum M. “It’s the genes stupid”. Molecular bases and clinical consequences of genetic cytochrome P450 2D6 polymorphism. Life Sci. 1995;56(26):2285-2298.
3. The Human Cytochrome P450 (CYP) Allele Nomenclature Database. CYP2D6 allele nomenclature. http://www.cypalleles.ki.se/cyp2d6.htm. Accessed February 25, 2013.
4. Hemeryck A, Belpaire FM. Selective serotonin reuptake inhibitors and cytochrome P-450 mediated drug-drug interactions: an update. Curr Drug Metab. 2002;3(1):13-37.
5. Mahgoub A, Idle JR, Dring LG, et al. Polymorphic hydroxylation of debrisoquine in man. Lancet. 1977;2(8038):584-586.
6. Eichelbaum M, Spannbrucker N, Steincke B, et al. Defective N-oxidation of sparteine in man: a new pharmacogenetic defect. Eur J Clin Pharmacol. 1979;16(3):183-187.
7. Distlerath LM, Reilly PE, Martin MV, et al. Purification and characterization of the human liver cytochromes P-450 involved in debrisoquine 4-hydroxylation and phenacetin O-deethylation, two prototypes for genetic polymorphism in oxidative drug metabolism. J Biol Chem. 1985;260(15):9057-9067.
8. Eichelbaum M, Baur MP, Dengler HJ, et al. Chromosomal assignment of human cytochrome P-450 (debrisoquine/sparteine type) to chromosome 22. Br J Clin Pharmacol. 1987;23(4):455-458.
9. Gonzalez FJ, Vilbois F, Hardwick JP, et al. Human debrisoquine 4-hydroxylase (P450IID1): cDNA and deduced amino acid sequence and assignment of the CYP2D locus to chromosome 22. Geonomics. 1988;2(2):174-179.
10. Spina E, Santoro V, D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008;30(7):1206-1227.
11. Roses AD. Pharmacogenetics and the practice of medicine. Nature. 2000;405(6788):857-865.
12. Ludwig J, Marcotte DE. Anti-depressants suicide, and drug regulation. J Policy Anal Manage. 2005;24(2):249-272.
13. Isacsson G. Suicide prevention—a medical breakthrough? Acta Psychiatr Scand. 2000;102(2):113-117.
14. Rihmer Z. Can better recognition and treatment of depression reduce suicide rates? A brief review. Eur Psychiatry. 2001;16(7):406-409.
15. Rihmer Z. Decreasing national suicide rates—fact or fiction? World J Biol Psychiatry. 2004;5(1):55-56.
16. Rihmer Z, Akiskal H. Do antidepressants t(h)reat(en) depressives? Toward a clinically judicious formulation of the antidepressant-suicidality FDA advisory in light of declining national suicide statistics from many countries. J Affect Disord. 2006;94(1-3):3-13.
17. Correia C, Santos P, Coutinho AM, et al. Characterization of pharmacogenetically relevant CYP2D6 and ABCB1 gene polymorphisms in a Portuguese population sample. Cell Biochem Funct. 2009;27(4):251-255.
18. Bertilsson L, Dahl ML, Sjöqvist F, et al. Molecular basis for rational megaprescribing in ultrarapid hydroxylators of debrisoquine. Lancet. 1993;341(8836):63.-
19. Baumann P, Broly F, Kosel M, et al. Ultrarapid metabolism of clomipramine in a therapy-resistant depressive patient, as confirmed by CYP2 D6 genotyping. Pharmacopsychiatry. 1998;31(2):72.-
20. Rau T, Wohlleben G, Wuttke H, et al. CYP2D6 genotype: impact on adverse effects and nonresponse during treatment with antidepressants-a pilot study. Clin Pharmacol Ther. 2004;75(5):386-393.
21. Kawanishi C, Lundgren S, Agren H, et al. Increased incidence of CYP2D6 gene duplication in patients with persistent mood disorders: ultrarapid metabolism of antidepressants as a cause of nonresponse. A pilot study. Eur J Clin Pharmacol. 2004;59(11):803-807.
22. Zackrisson AL, Lindblom B, Ahlner J. High frequency of occurrence of CYP2D6 gene duplication/multiduplication indicating ultrarapid metabolism among suicide cases. Clin Pharmacol Ther. 2010;88(3):354-359.
23. Stingl JC, Viviani R. CYP2D6 in the brain: impact on suicidality. Clin Pharmacol Ther. 2011;89(3):352-353.
24. Peñas-Lledó EM, Dorado P, Agüera Z, et al. High risk of lifetime history of suicide attempts among CYP2D6 ultrarapid metabolizers with eating disorders. Mol Psychiatry. 2011;16(7):691-692.
25. Siegle I, Fritz P, Eckhardt K, et al. Cellular localization and regional distribution of CYP2D6 mRNA and protein expression in human brain. Pharmacogenetics. 2001;11(3):237-245.
26. Eichelbaum M. In search of endogenous CYP2D6 substrates. Pharmacogenetics. 2003;13(6):305-306.
27. Yu AM, Idle JR, Gonzalez FJ. Polymorphic cytochrome P450 2D6: humanized mouse model and endogenous substrates. Drug Metab Rev. 2004;36(2):243-277.
28. Cowen PJ. Serotonin and depression: pathophysiological mechanism or marketing myth? Trends Pharmacol Sci. 2008;29(9):433-436.
29. Kang S, Kang K, Lee K, et al. Characterization of tryptamine 5-hydroxylase and serotonin synthesis in rice plants. Plant Cell Rep. 2007;26(11):2009-2015.
30. Yu AM, Idle JR, Herraiz T, et al. Screening for endogenous substrates reveals that CYP2D6 is a 5-methoxyindolethylamine O-demethylase. Pharmacogenetics. 2003;13(6):307-319.
31. Hiroi T, Imaoka S, Funae Y. Dopamine formation from tyramine by CYP2D6. Biochem Biophys Res Commun. 1998;249(3):838-843.
32. Niznik HB, Tyndale RF, Sallee FR, et al. The dopamine transporter and cytochrome P45OIID1 (debrisoquine 4-hydroxylase) in brain: resolution and identification of two distinct [3H]GBR-12935 binding proteins. Arch Biochem Biophys. 1990;276(2):424-432.
33. Kapur S, Remington G. Serotonin-dopamine interaction and its relevance to schizophrenia. Am J Psychiatry. 1996;153(4):466-476.
34. Jain KK. Applications of AmpliChip CYP450. Mol Diagn. 2005;9(3):119-127.
1. Meyer UA, Amrein R, Balant LP, et al. Antidepressants and drug-metabolizing enzymes—expert group report. Acta Psychiatr Scand. 1996;93(2):71-79.
2. Kroemer HK, Eichelbaum M. “It’s the genes stupid”. Molecular bases and clinical consequences of genetic cytochrome P450 2D6 polymorphism. Life Sci. 1995;56(26):2285-2298.
3. The Human Cytochrome P450 (CYP) Allele Nomenclature Database. CYP2D6 allele nomenclature. http://www.cypalleles.ki.se/cyp2d6.htm. Accessed February 25, 2013.
4. Hemeryck A, Belpaire FM. Selective serotonin reuptake inhibitors and cytochrome P-450 mediated drug-drug interactions: an update. Curr Drug Metab. 2002;3(1):13-37.
5. Mahgoub A, Idle JR, Dring LG, et al. Polymorphic hydroxylation of debrisoquine in man. Lancet. 1977;2(8038):584-586.
6. Eichelbaum M, Spannbrucker N, Steincke B, et al. Defective N-oxidation of sparteine in man: a new pharmacogenetic defect. Eur J Clin Pharmacol. 1979;16(3):183-187.
7. Distlerath LM, Reilly PE, Martin MV, et al. Purification and characterization of the human liver cytochromes P-450 involved in debrisoquine 4-hydroxylation and phenacetin O-deethylation, two prototypes for genetic polymorphism in oxidative drug metabolism. J Biol Chem. 1985;260(15):9057-9067.
8. Eichelbaum M, Baur MP, Dengler HJ, et al. Chromosomal assignment of human cytochrome P-450 (debrisoquine/sparteine type) to chromosome 22. Br J Clin Pharmacol. 1987;23(4):455-458.
9. Gonzalez FJ, Vilbois F, Hardwick JP, et al. Human debrisoquine 4-hydroxylase (P450IID1): cDNA and deduced amino acid sequence and assignment of the CYP2D locus to chromosome 22. Geonomics. 1988;2(2):174-179.
10. Spina E, Santoro V, D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008;30(7):1206-1227.
11. Roses AD. Pharmacogenetics and the practice of medicine. Nature. 2000;405(6788):857-865.
12. Ludwig J, Marcotte DE. Anti-depressants suicide, and drug regulation. J Policy Anal Manage. 2005;24(2):249-272.
13. Isacsson G. Suicide prevention—a medical breakthrough? Acta Psychiatr Scand. 2000;102(2):113-117.
14. Rihmer Z. Can better recognition and treatment of depression reduce suicide rates? A brief review. Eur Psychiatry. 2001;16(7):406-409.
15. Rihmer Z. Decreasing national suicide rates—fact or fiction? World J Biol Psychiatry. 2004;5(1):55-56.
16. Rihmer Z, Akiskal H. Do antidepressants t(h)reat(en) depressives? Toward a clinically judicious formulation of the antidepressant-suicidality FDA advisory in light of declining national suicide statistics from many countries. J Affect Disord. 2006;94(1-3):3-13.
17. Correia C, Santos P, Coutinho AM, et al. Characterization of pharmacogenetically relevant CYP2D6 and ABCB1 gene polymorphisms in a Portuguese population sample. Cell Biochem Funct. 2009;27(4):251-255.
18. Bertilsson L, Dahl ML, Sjöqvist F, et al. Molecular basis for rational megaprescribing in ultrarapid hydroxylators of debrisoquine. Lancet. 1993;341(8836):63.-
19. Baumann P, Broly F, Kosel M, et al. Ultrarapid metabolism of clomipramine in a therapy-resistant depressive patient, as confirmed by CYP2 D6 genotyping. Pharmacopsychiatry. 1998;31(2):72.-
20. Rau T, Wohlleben G, Wuttke H, et al. CYP2D6 genotype: impact on adverse effects and nonresponse during treatment with antidepressants-a pilot study. Clin Pharmacol Ther. 2004;75(5):386-393.
21. Kawanishi C, Lundgren S, Agren H, et al. Increased incidence of CYP2D6 gene duplication in patients with persistent mood disorders: ultrarapid metabolism of antidepressants as a cause of nonresponse. A pilot study. Eur J Clin Pharmacol. 2004;59(11):803-807.
22. Zackrisson AL, Lindblom B, Ahlner J. High frequency of occurrence of CYP2D6 gene duplication/multiduplication indicating ultrarapid metabolism among suicide cases. Clin Pharmacol Ther. 2010;88(3):354-359.
23. Stingl JC, Viviani R. CYP2D6 in the brain: impact on suicidality. Clin Pharmacol Ther. 2011;89(3):352-353.
24. Peñas-Lledó EM, Dorado P, Agüera Z, et al. High risk of lifetime history of suicide attempts among CYP2D6 ultrarapid metabolizers with eating disorders. Mol Psychiatry. 2011;16(7):691-692.
25. Siegle I, Fritz P, Eckhardt K, et al. Cellular localization and regional distribution of CYP2D6 mRNA and protein expression in human brain. Pharmacogenetics. 2001;11(3):237-245.
26. Eichelbaum M. In search of endogenous CYP2D6 substrates. Pharmacogenetics. 2003;13(6):305-306.
27. Yu AM, Idle JR, Gonzalez FJ. Polymorphic cytochrome P450 2D6: humanized mouse model and endogenous substrates. Drug Metab Rev. 2004;36(2):243-277.
28. Cowen PJ. Serotonin and depression: pathophysiological mechanism or marketing myth? Trends Pharmacol Sci. 2008;29(9):433-436.
29. Kang S, Kang K, Lee K, et al. Characterization of tryptamine 5-hydroxylase and serotonin synthesis in rice plants. Plant Cell Rep. 2007;26(11):2009-2015.
30. Yu AM, Idle JR, Herraiz T, et al. Screening for endogenous substrates reveals that CYP2D6 is a 5-methoxyindolethylamine O-demethylase. Pharmacogenetics. 2003;13(6):307-319.
31. Hiroi T, Imaoka S, Funae Y. Dopamine formation from tyramine by CYP2D6. Biochem Biophys Res Commun. 1998;249(3):838-843.
32. Niznik HB, Tyndale RF, Sallee FR, et al. The dopamine transporter and cytochrome P45OIID1 (debrisoquine 4-hydroxylase) in brain: resolution and identification of two distinct [3H]GBR-12935 binding proteins. Arch Biochem Biophys. 1990;276(2):424-432.
33. Kapur S, Remington G. Serotonin-dopamine interaction and its relevance to schizophrenia. Am J Psychiatry. 1996;153(4):466-476.
34. Jain KK. Applications of AmpliChip CYP450. Mol Diagn. 2005;9(3):119-127.

Vitamin D deficiency and psychiatric illness
In the United States, >50% of psychiatric inpatients have vitamin D deficiency—<30 nmol/L (<12 ng/mL).1 A growing body of literature has found associations between vitamin D deficiency and psychiatric illnesses, particularly depression. Several randomized controlled trials (RCTs) have demonstrated that vitamin D supplementation can benefit depression symptoms. In this article, we discuss the current literature on vitamin D and psychiatric illness, and provide practical information for clinicians on the use of vitamin D supplementation.
Biosynthesis of vitamin D
Biosynthesis of vitamin D begins with the sterol provitamin D3 molecule 7-dehydrocholesterol (Figure).2 When skin is exposed to sunlight, 7-dehydrocholesterol absorbs UV radiation and forms provitamin D3, which undergoes rapid transformation to vitamin D3.2 Vitamin D3 is released from the plasma membrane and enters systemic circulation in a protein-bound form that has a serum half-life of 36 to 78 hours.3 Vitamin D3 can be taken up by adipocytes and stored in fat deposits, where it has a half-life of approximately 2 months.4
Figure: Biosynthesis of vitamin D
Provitamin D3 (7-dehydrocholesterol) in the skin absorbs UV radiation and undergoes isomerization to form vitamin D3. Endogenously produced vitamin D3 along with dietary vitamin D2 and vitamin D3 absorbed in the gastrointestinal tract are metabolized in the liver to 25-hydroxyvitamin D (25[OH]D), which re-enters the circulation and is metabolized in the kidney and other tissues to the active metabolite 1,25-dihydroxyvitamin D (1,25[OH]2D). Catabolism of 25(OH)D and 1,25(OH)2D into biologically-inactive molecules is primarily mediated by the cytochrome P450 (CYP) enzymes CYP24 and CYP3A4.
Source: Reference 2Circulating vitamin D3 is metabolized in the liver by the enzyme vitamin D-25-hydroxylase to 25-hydroxyvitamin D (25[OH]D3), which has a serum half-life of approximately 15 days.4 Circulating 25(OH)D3 is not biologically active at the physiological level, and requires activation by conversion to 1,25-dihydroxyvitamin D (1,25[OH]2D3) in the kidneys by the enzyme 25(OH)D-1α-hydroxylase. Production of 1,25(OH)2D3 is regulated by serum phosphorus and parathyroid hormone levels and other factors.5 Catabolism of 1,25(OH)2D3 is rapid, with a serum half-life of 3.5 to 21 hours.6 Vitamin D2 is structurally similar to vitamin D3, but occurs primarily in fungi, yeasts, and some invertebrates.
Risk factors for deficiency
A patient’s vitamin D status is determined by measuring 25(OH)D (Box 1). Risk factors for vitamin D deficiency include conditions that affect cutaneous production (insufficient sunlight exposure), obesity, gastrointestinal disorders, aging, renal disorders, and medications (Table 1). 2,5,7,8 The link between sunscreen use, either alone or in cosmetics, and vitamin D deficiency continues to be debated. While controlled studies have found that application of sunscreen with high sun protection factor can significantly reduce vitamin D production, 9 studies in clinical populations have failed to confirm these findings. 10,11 See Box 2 for a discussion of these risk factors and Box 3 for a discussion of acute and long-term medical manifestations of deficiency.
Although 1,25-dihydroxyvitamin D (1,25[OH]2D3) is the biologically active form of vitamin D, its circulating half-life is only 4 to 6 hours.a,b Therefore, 25-hydroxyvitamin D (25[OH]D) is the principal vitamin D metabolite measured to determine vitamin D status. Vitamin D levels commonly are expressed as ng/mL or nmol/L; the conversion factor from ng/mL to nmol/L is 2.496. The Institute of Medicine has defined vitamin D deficiency as a serum 25(OH)D level of <30 nmol/L (<12 ng/mL).c However, many experts define vitamin D insufficiency as a 25(OH)D level of 21 to 29 ng/ml, and deficiency as <20 ng/mL.a,d The upper limit is more difficult to define, but symptoms of vitamin D intoxication appear with blood levels >150 to 200 ng/mL.a
References
- Holick MF. High prevalence of vitamin D inadequacy and implications for health. Mayo Clin Proc. 2006;81(3):353-373.
- Holick MF. Vitamin D status: measurement, interpretation, and clinical application. Ann Epidemiol. 2009;19(2):73-78.
- Aloia JF. Clinical review: the 2011 report on dietary reference intake for vitamin D: where do we go from here? J Clin Endocrinol Metab. 2011;96(10):2987-2996.
- Bischoff-Ferrari HA, Giovannucci E, Willett WC, et al. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84(1):18-28.
Table 1
Risk factors associated with vitamin D deficiency
| Age (>65) |
| Insufficient sunlight |
| Breastfeeding |
| Dark skin |
| Malabsorption diseases |
| Obesity (BMI >30 kg/m2) |
| Use of medications that alter vitamin D metabolism (eg, anticonvulsants, glucocorticoids) |
| Hepatobiliary disease |
| Renal disease |
| BMI: body mass index Source: References 2,5,7,8 |
Any factor that diminishes UV radiation penetration into the skin will affect cutaneous synthesis of vitamin D.a,b For example, sunscreen with a sun protection factor of 15 can decrease vitamin D synthesis by 98%.c Geography and its impact on yearly sunlight exposure is a well-known factor in vitamin D deficiency. Individuals who live below a latitude of approximately 35° North—approximately the southern border of Tennessee and through Albuquerque, NM—receive sufficient UV radiation exposure to ensure adequate vitamin D production throughout the year, but at higher latitudes, adequate vitamin D is not produced during winter months.d Melanin affects UV radiation absorption in a manner that prevents vitamin D production, and increased skin pigmentation markedly reduces vitamin D synthesis.e African Americans with very dark skin have significantly diminished cutaneous production of vitamin D.e,f
Renal 1α-hydroxylase activity decreases with aging in parallel with age-related decreases in glomerular filtration.g In addition, aging is associated with increased clearance of 1,25-dihydroxyvitamin D (1,25[OH]2D3).h However, vitamin D absorption generally is adequate even at older ages.i Studies have shown that obese individuals tend to have lower serum concentrations of vitamin D and 25-hydroxyvitamin D (25[OH]D) than those at a normal weight.j,k Obese patients have been shown to have lower cutaneous production of vitamin D3 and display lower bioavailability of orally administered vitamin D2.j
For patients with chronic renal insufficiency, creatinine clearance is positively correlated with serum 1,25(OH)2D levels.l Any process that results in malabsorption of intestinal fat may impair vitamin D absorption. In patients with celiac disease, biliary obstruction, or chronic pancreatitis, absorption consistently is reduced.m Individuals taking bile acid-binding medications, such as cholestyramine for hypercholesterolemia, also may have impaired vitamin D absorption.n In addition, hepatobiliary disease is associated with low levels of 25(OH)D.o Some drugs that alter hepatic metabolism are associated with vitamin D deficiency, including anticonvulsants or glucocorticoids, which can increase catabolism or vitamin D.p
References
- Holick MF. The vitamin D deficiency pandemic and consequences for nonskeletal health: mechanisms of action. Mol Aspects Med. 2008;29(6):361-368.
- Holick MF, Chen TC. Vitamin D deficiency: a worldwide problem with health consequences. Am J Clin Nutr. 2008;87(4):1080S-1086S.
- Matsuoka LY, Ide L, Wortsman J, et al. Sunscreens suppress cutaneous vitamin D3 synthesis. J Clin Endocrinol Metab. 1987;64(6):1165-1168.
- Holick MF. Vitamin D: importance in the prevention of cancers, type 1 diabetes, heart disease, and osteoporosis. Am J Clin Nutr. 2004;79(3):362-371.
- Clemens TL, Adams JS, Henderson SL, et al. Increased skin pigment reduces the capacity of skin to synthesise vitamin D3. Lancet. 1982;1(8263):74-76.
- Chen TC, Chimeh F, Lu Z, et al. Factors that influence the cutaneous synthesis and dietary sources of vitamin D. Arch Biochem Biophys. 2007;460(2):213-217.
- Slovik DM, Adams JS, Neer RM, et al. Deficient production of 1,25-dihydroxyvitamin D in elderly osteoporotic patients. N Engl J Med. 1981;305(7):372-374.
- Armbrecht HJ, Zenser TV, Davis BB. Effect of age on the conversion of 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3 by kidney of rat. J Clin Invest. 1980;66(5):1118-1123.
- Lips P, Wiersinga A, van Ginkel FC, et al. The effect of vitamin D supplementation on vitamin D status and parathyroid function in elderly subjects. J Clin Endocrinol Metab. 1988;67(4):644-650.
- Wortsman J, Matsuoka LY, Chen TC, et al. Decreased bioavailability of vitamin D in obesity. Am J Clin Nutr. 2000;72(3):690-693.
- Bell NH, Epstein S, Greene A, et al. Evidence for alteration of the vitamin D-endocrine system in obese subjects. J Clin Invest. 1985;76(1):370-373.
- Pitts TO, Piraino BH, Mitro R, et al. Hyperparathyroidism and 1,25-dihydroxyvitamin D deficiency in mild, moderate, and severe renal failure. J Clin Endocrinol Metab. 1988;67(5):876-881.
- Thompson GR, Lewis B, Booth CC. Absorption of vitamin D3-3H in control subjects and patients with intestinal malabsorption. J Clin Invest. 1966;45(1):94-102.
- Lo CW, Paris PW, Clemens TL, et al. Vitamin D absorption in healthy subjects and in patients with intestinal malabsorption syndromes. Am J Clin Nutr. 1985;42(4):644-649.
- Pappa HM, Bern E, Kamin D, et al. Vitamin D status in gastrointestinal and liver disease. Curr Opin Gastroenterol. 2008;24(2):176-183.
- Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266-281.
Acute effects. Vitamin D deficiency produces a range of clinical effects.a-c One well-documented consequence of vitamin D deficiency is osteomalacia—bone demineralization—which produces characteristic bone deformity and growth retardation in children.d,e In adults, osteomalacia may manifest as diffuse pain bone discomfort and muscle aches that may resemble fibromyalgia or arthritis.f Because vitamin D receptors are present in skeletal muscle, deficiency also may lead to proximal muscle weakness; an increased risk of falls; global bone discomfort, often elicited with pressure over the sternum or tibia; and low back pain in older women.c,f
Long-term effects. A large epidemiologic study found that adults with 25-hydroxyvitamin D (25[OH]D) levels <21 ng/mL had an increased risk of hypertension, diabetes, obesity, and dyslipidemia.g Cardiovascular mortality was higher in individuals with 25(OH)D levels <10 ng/mL compared with those with >40 ng/mL.h Adolescents in the National Health and Nutrition Examination Survey-III with serum 25(OH)D levels <15 ng/mL were more likely to have elevated blood glucose levels than those with >26 ng/mL.i Other epidemiologic data have demonstrated associations of vitamin D deficiency with multiple sclerosis, seasonal allergies, asthma, and various infectious diseases.j,k
Because vitamin D is known to promote cellular differentiation and inhibit cellular proliferation, its role in cancer has been studied extensively. A recent meta-analysis of case-control studies found that the odds of colon cancer were reduced by >40% for each 20 ng/mL increase in serum 25(OH)D levels.l Another meta-analysis reported a lower risk of breast cancer among women in the highest quartile of 25(OH)D values compared with the lowest quartile.m
References
- Holick MF. High prevalence of vitamin D inadequacy and implications for health. Mayo Clin Proc. 2006;81(3):353-373.
- Holick MF. Vitamin D status: measurement, interpretation, and clinical application. Ann Epidemiol. 2009;19(2):73-78.
- Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266-281.
- Holick MF, Chen TC. Vitamin D deficiency: a worldwide problem with health consequences. Am J Clin Nutr. 2008;87(4):1080S-1086S.
- Bordelon P, Ghetu MV, Langan RC. Recognition and management of vitamin D deficiency. Am Fam Physician. 2009;80(8):841-846.
- Hicks GE, Shardell M, Miller RR, et al. Associations between vitamin D status and pain in older adults: the Invecchiare in Chianti study. J Am Geriatr Soc. 2008;56(5):785-791.
- Martins D, Wolf M, Pan D, et al. Prevalence of cardiovascular risk factors and the serum levels of 25-hydroxyvitamin D in the United States: data from the Third National Health and Nutrition Examination Survey. Arch Intern Med. 2007;167(11):1159-1165.
- Ginde AA, Scragg R, Schwartz RS, et al. Prospective study of serum 25-hydroxyvitamin D level, cardiovascular disease mortality, and all-cause mortality in older U.S. adults. J Am Geriatr Soc. 2009;57(9):1595-1603.
- Reis JP, von Mühlen D, Miller ER 3rd, et al. Vitamin D status and cardiometabolic risk factors in the United States adolescent population. Pediatrics. 2009;124(3):e371-379.
- Thacher TD, Clarke BL. Vitamin D insufficiency. Mayo Clin Proc. 2011;86(1):50-60.
- Munger KL, Levin LI, Hollis BW, et al. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA. 2006;296(23):2832-2838.
- Yin L, Grandi N, Raum E, et al. Meta-analysis: longitudinal studies of serum vitamin D and colorectal cancer risk. Aliment Pharmacol Ther. 2009;30(2):113-125.
- Chen P, Hu P, Xie D, et al. Meta-analysis of vitamin D, calcium and the prevention of breast cancer. Breast Cancer Res Treat. 2010;121(2):469-477.
Vitamin D’s role in the brain
Vitamin D’s role in psychiatric illnesses is suggested by region-specific expression of vitamin D receptors (VDR) in the cingulate cortex, thalamus, cerebellum, amygdala, and hippocampus.12 Most of these regions also express 1α-hydroxylase enzymes capable of metabolizing 25(OH)D to 1,25(OH)2D3, which suggests that vitamin D may have an autocrine or paracrine function in brain.13
Vitamin D regulates expression of tyrosine hydroxylase, the rate-limiting enzyme in the biosynthesis of dopamine, norepinephrine, and epinephrine.14 Vitamin D also promotes survival of monoaminergic neurons through upregulation of glial cell line-derived neurotrophic factor, which supports survival of midbrain dopaminergic neurons and confers resistance to neurotoxins that deplete dopaminergic neurons in Parkinson’s disease.15 Vitamin D also promotes neuronal survival by inhibiting oxidative pathways in the brain through inhibition of inducible nitric oxide synthase (reducing free radical formation)16 and upregulation of γ-glutamyl transpeptidase (increasing antioxidant production).17 Vitamin D may play a neuroprotective role through regulation of calcium channels. In vitro studies have shown that vitamin D downregulates expression of L-type calcium channels, conferring protection against excitatory neurotoxins in cultured neurons.18 Proteomic analysis of brain tissue in a rat model of developmental vitamin D revealed dysregulation of 36 brain proteins involved in many biologic pathways involved in calcium homeostasis, synaptic plasticity, and neurotransmission.19 Taken together, these findings suggest vitamin D has a neurosteroid-like role in the CNS.
Psychotic disorders
Several epidemiologic studies have linked low vitamin D levels to schizophrenia and other psychotic disorders. Researchers in Norway who used a structured clinical interview to identify psychosis consistently found low levels of 25(OH)D among immigrants and native Norwegians with psychotic symptoms.20 A study of 8,411 Swedish women found low vitamin D levels were associated with psychotic symptoms.21 The Finnish birth cohort study found that use of vitamin D supplementation during the first year of life reduced the incidence of schizophrenia.22 In another pilot study, researchers measured third-trimester serum 25(OH)D levels and found that low levels of maternal vitamin D may be associated with an increased risk of schizophrenia.23 These studies suggest that low prenatal vitamin D levels may adversely impact the developing brain, increasing the risk for adult-onset schizophrenia.
Cognitive dysfunction
Low vitamin D concentrations have been associated with impairments in cognitive functions such as memory and orientation,24 executive function impairments,25 and Alzheimer’s disease (AD).26 A large study conducted from 1998 to 2006 in Italy concluded that persons with severe vitamin D deficiency (<25 nmol/L) had a higher risk of substantial decline on Mini-Mental State Examination than those with sufficient levels (≥75 nmol/L).27 Other studies have linked low vitamin D levels to poor cognitive performance in depressed older adults.28 Low vitamin D levels in older women have been associated with risk of AD, but not with other dementias.29 Polymorphisms of VDR have been associated with depression and poor cognitive performance.30
Depression
Epidemiologic studies evaluating vitamin D deficiency have had conflicting results. The Third National Health and Nutrition Examination Survey, which used a sample of 7,970 non-institutionalized U.S. residents age 15 to 39, demonstrated that individuals with serum vitamin D ≤50 nmol/L are at a significantly higher risk of developing depression than those with vitamin D ≥75 nmol/L.31 A study of 1,282 adults age 65 to 95 in the Netherlands found that 25(OH)D levels were 14% lower in depressed patients compared with controls.32 However, a large epidemiologic study in China did not detect a relationship between vitamin D and depression in 3,262 men and women age 50 to 70.33 After researchers adjusted for geography, body mass index, physical activity, and smoking, 25(OH)D levels did not correlate significantly with the presence or severity of depression. In a case series,34 after 48 vitamin D-deficient depressed adolescents were given vitamin D3 over 3 months, there was a significant improvement in well-being, depressive symptoms, irritability, and fatigue.34 Other small, cross-sectional studies have examined associations between vitamin D status and depression with divergent results, which may reflect differences in population and methodology.
Prospective interventional studies. Although direct causal relationships are difficult to establish, several prospective studies have tested the hypothesis that treating vitamin D deficiency can improve depressive symptoms.
In a double-blind, controlled trial, Jorde et al35 randomized 441 individuals age 21 to 70 to vitamin D, 20,000 IU per week; vitamin D, 40,000 IU per week; or placebo for 1 year. Individuals with serum 25(OH)D levels <40 nmol/L scored significantly higher on depression rating scales than those with serum 25(OH)D levels ≥40 nmol/L at the end of the study. There was no significant improvement in depression ratings in the placebo group (Table 2).35 These results must be interpreted with care because depressive symptoms were secondary endpoints in this study.
Table 2
Effect of vitamin D supplementation on depressive symptoms in a controlled trial
| Vitamin D Supplementation | Serum 25(OH)D levels at baseline | BDI total score, Median and range at end of study | After 1 year of vitamin D supplementation |
|---|---|---|---|
| 20,000 IU/week | <40 nmol/L | Significantly higher (more depressive traits), 6.0 (0 to 23) | Significantly improved BDI score |
| 40,000 IU/week | ≥40 nmol/L | 4.5 (0 to 28) | Significantly improved BDI score |
| Placebo | - | - | No improvement in BDI score |
| 25(OH)D: 25-hydroxyvitamin D; BDI: Beck Depression Inventory Source: Reference 35 | |||
Kjærgaard et al36 systematically examined vitamin D levels in a case-control study followed by a randomized controlled trial (RCT) of vitamin D supplementation. In the case-control phase, participants with low 25(OH)D levels at baseline were significantly more depressed than participants with high 25(OH)D levels. Participants with low 25(OH)D levels were randomized to placebo or 40,000 IU vitamin D3 per week for 6 months. Low levels of vitamin D were strongly associated with depressive symptoms, but vitamin D supplementation did not have a significant effect on depressive symptom scores.
Seasonal affective disorder (SAD). Seasonal variation in vitamin D levels suggests that supplementation may help patients who have seasonal mood disturbances. In a randomized, double-blind study, 44 healthy individuals received vitamin D3, 400 IU/d, 800 IU/d, or no vitamin D3 for 5 days during late winter. Based on self-reports, vitamin D3 significantly enhanced positive affect and there was some evidence it reduced negative affect.37 In a pilot study of 9 women with serum vitamin D levels <40 ng/ml, vitamin D supplementation during winter was associated with an average 10-point decline in Beck Depression Inventory-II scores.38 In a prospective RCT of 15 individuals with SAD, all patients who received vitamin D improved in all outcome measures.39 Vieth40 randomized 82 adults with vitamin D deficiency to 600 IU/d or 4,000 IU/d of vitamin D3 for 3 months over 2 consecutive winters. Patients taking the higher dose showed some evidence of improved well-being compared with those taking the lower dose, although results were not significant for all comparisons. Two other trials did not observe any improvement in SAD symptoms with vitamin D treatment.41,42
Treating vitamin D deficiency
The Endocrine Society recently developed consensus guidelines for diagnosing and managing vitamin D deficiency.43 In addition, the Institute of Medicine of the National Academies recommends daily vitamin D supplementation to prevent deficiency:
- age <70: 400 IU/d
- age >70: 800 IU/d
- pregnant or lactating women: 600 IU/d
- upper limit: 4,000 IU/d.7
Higher doses may be used for patients deprived of sun exposure.8 A typical replacement regimen consists of oral ergocalciferol, 50,000 IU per week for 8 weeks.44 The optimal time for rechecking serum levels after repletion has not been clearly defined, but serum 25(OH)D levels should be measured again after therapy is completed. If values have not reached or exceeded 20 ng/mL, consider a second 8-week course of ergocalciferol (see the Box 1 for a discussion of measuring vitamin D levels). If serum 25(OH)D levels have not increased, the most likely cause is nonadherence or malabsorption.
Contraindications and toxicity. Contraindications to vitamin D supplementation include granulomatous diseases, sarcoidosis, metastatic bone disease, and Williams syndrome.45Table 345 lists signs of vitamin D toxicity. There is little risk of toxicity at dosages of up to 2,000 IU/d.46
Table 3
Signs of vitamin D toxicity
| Headache |
| Metallic taste |
| Nephrocalcinosis or vascular calcinosis |
| Pancreatitis |
| Nausea |
| Vomiting |
| Source: Reference 45 |
Related Resources
- LaFerney MC. Vitamin D deficiency in older adults. Current Psychiatry. 2012;11(11):63.
- National Institutes of Health Office of Dietary Supplements. Dietary supplement fact sheet: vitamin D. http://ods.od.nih.gov/factsheets/VitaminD-HealthProfessional.
Drug Brand Names
- Cholestyramine • Questran
- Ergocalciferol • Calciferol, Drisdol
Disclosures
Dr. Harris is an employee of Rho, Chapel Hill, NC.
Dr. Jaiswal reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Holmes receives research support from Bristol-Myers Squibb, Elan, Merck, Otsuka, Shire, Takeda, and Theravance, and is on the speaker’s bureau for Forest Pharmaceuticals and PamLab.
Dr. Patkar is a consultant for Dey Pharmaceuticals, Forest, Gilead, and TTK Pharma and is on the speaker’s bureau and received honoraria from Alkermes, Bristol-Myers Squibb, Dey Pharmaceuticals, Pfizer, and Sunovion; and has received grant support from the Duke Endowment, Dey Pharmaceuticals, Envivo, Forest, Janssen, Lundbeck, The National Institutes of Health, the National Institute on Drug Abuse, National Institute on Alcohol Abuse and Alcoholism, Pfizer Inc., Shire, Sunovion, and Titan.
Dr. Weisler has been a consultant to, on the speaker’s bureaus of, and/or received research support from Abbott, Agency for Toxic Substances and Disease Registry, AstraZeneca, Biovail, Bristol-Myers Squibb, Burroughs Wellcome, Cenerx, Centers for Disease Control and Prevention, Cephalon, Ciba Geigy, CoMentis, Corcept, Cortex, Dainippon Sumitomo Pharma America, Eisai, Elan, Eli Lilly and Company, Forest Pharmaceuticals, GlaxoSmithKline, Janssen, Johnson & Johnson, Lundbeck, McNeil Pharmaceuticals, Medicinova, Medscape Advisory Board, Merck, National Institute of Mental Health, Neurochem, New River Pharmaceuticals, Novartis, Organon, Otsuka America Pharma, Pfizer Inc., Pharmacia, Repligen, Saegis, Sandoz, Sanofi, Sanofi-Synthelabo, Schwabe/Ingenix, Sepracor, Shire, Solvay, Sunovion, Synaptic, Takeda, TAP, Theravance, Transcept Pharma, TransTech, UCB Pharma, Validus, Vela, and Wyeth.
1. McCue RE, Charles RA, Orendain GC, et al. Vitamin D deficiency among psychiatric inpatients [published online April 19, 2012]. Prim Care Companion CNS Disord. doi: 10.4088/PCC.11m01230.
2. Tsiaras WG, Weinstock MA. Factors influencing vitamin D status. Acta Derm Venereol. 2011;91(2):115-124.
3. Adams JS, Clemens TL, Parrish JA, et al. Vitamin-D synthesis and metabolism after ultraviolet irradiation of normal and vitamin-D-deficient subjects. N Engl J Med. 1982;306(12):722-725.
4. Jones G. Pharmacokinetics of vitamin D toxicity. Am J Clin Nutr. 2008;88(2):582S-586S.
5. Holick MF. Vitamin D: importance in the prevention of cancers type 1 diabetes, heart disease, and osteoporosis. Am J Clin Nutr. 2004;79(3):362-371.
6. Fakih MG, Trump DL, Muindi JR, et al. A phase I pharmacokinetic and pharmacodynamic study of intravenous calcitriol in combination with oral gefitinib in patients with advanced solid tumors. Clin Cancer Res. 2007;13(4):1216-1223.
7. Holick MF, Chen TC. Vitamin D deficiency: a worldwide problem with health consequences. Am J Clin Nutr. 2008;87(4):1080S-1086S.
8. Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266-281.
9. Matsuoka LY, Ide L, Wortsman J, et al. Sunscreens suppress cutaneous vitamin D3 synthesis. J Clin Endocrinol Metab. 1987;64(6):1165-1168.
10. Norval M, Wulf HC. Does chronic sunscreen use reduce vitamin D production to insufficient levels? Br J Dermatol. 2009;161(4):732-736.
11. Linos E, Keiser E, Kanzler M, et al. Sun protective behaviors and vitamin D levels in the US population: NHANES 2003-2006. Cancer Causes Control. 2012;23(1):133-140.
12. Prüfer K, Veenstra TD, Jirikowski GF, et al. Distribution of 1,25-dihydroxyvitamin D3 receptor immunoreactivity in the rat brain and spinal cord. J Chem Neuroanat. 1999;16(2):135-145.
13. Eyles DW, Smith S, Kinobe R, et al. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat. 2005;29(1):21-30.
14. Garcion E, Wion-Barbot N, Montero-Menei CN, et al. New clues about vitamin D functions in the nervous system. Trends Endocrinol Metab. 2002;13(3):100-105.
15. Smith MP, Fletcher-Turner A, Yurek DM, et al. Calcitriol protection against dopamine loss induced by intracerebroventricular administration of 6-hydroxydopamine. Neurochem Res. 2006;31(4):533-539.
16. Garcion E, Nataf S, Berod A, et al. 1,25-dihydroxyvitamin D3 inhibits the expression of inducible nitric oxide synthase in rat central nervous system during experimental allergic encephalomyelitis. Brain Res Mol Brain Res. 1997;45(2):255-267.
17. Baas D, Prüfer K, Ittel ME, et al. Rat oligodendrocytes express the vitamin D(3) receptor and respond to 1,25-dihydroxyvitamin D(3). Glia. 2000;31(1):59-68.
18. Brewer LD, Thibault V, Chen KC, et al. Vitamin D hormone confers neuroprotection in parallel with downregulation of L-type calcium channel expression in hippocampal neurons. J Neurosci. 2001;21(1):98-108.
19. Almeras L, Eyles D, Benech P, et al. Developmental vitamin D deficiency alters brain protein expression in the adult rat: implications for neuropsychiatric disorders. Proteomics. 2007;7(5):769-780.
20. Berg AO, Melle I, Torjesen PA, et al. A cross-sectional study of vitamin D deficiency among immigrants and Norwegians with psychosis compared to the general population. J Clin Psychiatry. 2010;71(12):1598-1604.
21. Hedelin M, Löf M, Olsson M, et al. Dietary intake of fish, omega-3, omega-6 polyunsaturated fatty acids and vitamin D and the prevalence of psychotic-like symptoms in a cohort of 33,000 women from the general population. BMC Psychiatry. 2010;10:38.-
22. McGrath J, Saari K, Hakko H, et al. Vitamin D supplementation during the first year of life and risk of schizophrenia: a Finnish birth cohort study. Schizophr Res. 2004;67(2-3):237-245.
23. McGrath J, Eyles D, Mowry B, et al. Low maternal vitamin D as a risk factor for schizophrenia: a pilot study using banked sera. Schizophr Res. 2003;63(1-2):73-78.
24. Przybelski RJ, Binkley NC. Is vitamin D important for preserving cognition? A positive correlation of serum 25-hydroxyvitamin D concentration with cognitive function. Arch Biochem Biophys. 2007;460(2):202-205.
25. Lee DM, Tajar A, Ulubaev A, et al. Association between 25-hydroxyvitamin D levels and cognitive performance in middle-aged and older European men. J Neurol Neurosurg Psychiatry. 2009;80(7):722-729.
26. Buell JS, Dawson-Hughes B, Scott TM, et al. 25-hydroxyvitamin D, dementia, and cerebrovascular pathology in elders receiving home services. Neurology. 2010;74(1):18-26.
27. Llewellyn DJ, Lang IA, Langa KM, et al. Vitamin D and risk of cognitive decline in elderly persons. Arch Intern Med. 2010;170(13):1135-1141.
28. Wilkins CH, Sheline YI, Roe CM, et al. Vitamin D deficiency is associated with low mood and worse cognitive performance in older adults. Am J Geriatr Psychiatry. 2006;14(12):1032-1040.
29. Annweiler C, Rolland Y, Schott AM, et al. Higher vitamin D dietary intake is associated with lower risk of Alzheimer’s disease: a 7-year follow-up. J Gerontol A Biol Sci Med Sci. 2012;67(11):1205-1211.
30. Kuningas M, Mooijaart SP, Jolles J, et al. VDR gene variants associate with cognitive function and depressive symptoms in old age. Neurobiol Aging. 2009;30(3):466-473.
31. Ganji V, Milone C, Cody MM, et al. Serum vitamin D concentrations are related to depression in young adult US population: the Third National Health and Nutrition Examination Survey. Int Arch Med. 2010;3:29.-
32. Hoogendijk WJ, Lips P, Dik MG, et al. Depression is associated with decreased 25-hydroxyvitamin D and increased parathyroid hormone levels in older adults. Arch Gen Psychiatry. 2008;65(5):508-512.
33. Pan A, Lu L, Franco OH, et al. Association between depressive symptoms and 25-hydroxyvitamin D in middle-aged and elderly Chinese. J Affect Disord. 2009;118(1-3):240-243.
34. Högberg G, Gustafsson SA, Hällström T, et al. Depressed adolescents in a case-series were low in vitamin D and depression was ameliorated by vitamin D supplementation. Acta Paediatr. 2012;101(7):779-783.
35. Jorde R, Sneve M, Figenschau Y, et al. Effects of vitamin D supplementation on symptoms of depression in overweight and obese subjects: randomized double blind trial. J Intern Med. 2008;264(6):599-609.
36. Kjærgaard M, Waterloo K, Wang CE, et al. Effect of vitamin D supplement on depression scores in people with low levels of serum 25-hydroxyvitamin D: nested case-control study and randomised clinical trial. Br J Psychiatry. 2012;201(5):360-368.
37. Lansdowne AT, Provost SC. Vitamin D3 enhances mood in healthy subjects during winter. Psychopharmacology (Berl). 1998;135(4):319-223.
38. Shipowick CD, Moore CB, Corbett C, et al. Vitamin D and depressive symptoms in women during the winter: a pilot study. Appl Nurs Res. 2009;22(3):221-225.
39. Gloth FM 3rd, Alam W, Hollis B. Vitamin D vs broad spectrum phototherapy in the treatment of seasonal affective disorder. J Nutr Health Aging. 1999;3(1):5-7.
40. Vieth R, Kimball S, Hu A, et al. Randomized comparison of the effects of the vitamin D3 adequate intake versus 100 mcg (4000 IU) per day on biochemical responses and the wellbeing of patients. Nutr J. 2004;3:8.-
41. Harris S, Dawson-Hughes B. Seasonal mood changes in 250 normal women. Psychiatry Res. 1993;49(1):77-87.
42. Dumville JC, Miles JN, Porthouse J, et al. Can vitamin D supplementation prevent winter-time blues? A randomised trial among older women. J Nutr Health Aging. 2006;10(2):151-153.
43. Holick MF, Binkley NC, Bischoff-Ferrari HA, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96(7):1911-1930.
44. Thacher TD, Clarke BL. Vitamin D insufficiency. Mayo Clin Proc. 2011;86(1):50-60.
45. Schwalfenberg G. Not enough vitamin D: health consequences for Canadians. Can Fam Physician. 2007;53(5):841-854.
46. Norman AW, Bouillon R, Whiting SJ, et al. 13th Workshop consensus for vitamin D nutritional guidelines. J Steroid Biochem Mol Biol. 2007;103(3-5):204-205.
In the United States, >50% of psychiatric inpatients have vitamin D deficiency—<30 nmol/L (<12 ng/mL).1 A growing body of literature has found associations between vitamin D deficiency and psychiatric illnesses, particularly depression. Several randomized controlled trials (RCTs) have demonstrated that vitamin D supplementation can benefit depression symptoms. In this article, we discuss the current literature on vitamin D and psychiatric illness, and provide practical information for clinicians on the use of vitamin D supplementation.
Biosynthesis of vitamin D
Biosynthesis of vitamin D begins with the sterol provitamin D3 molecule 7-dehydrocholesterol (Figure).2 When skin is exposed to sunlight, 7-dehydrocholesterol absorbs UV radiation and forms provitamin D3, which undergoes rapid transformation to vitamin D3.2 Vitamin D3 is released from the plasma membrane and enters systemic circulation in a protein-bound form that has a serum half-life of 36 to 78 hours.3 Vitamin D3 can be taken up by adipocytes and stored in fat deposits, where it has a half-life of approximately 2 months.4
Figure: Biosynthesis of vitamin D
Provitamin D3 (7-dehydrocholesterol) in the skin absorbs UV radiation and undergoes isomerization to form vitamin D3. Endogenously produced vitamin D3 along with dietary vitamin D2 and vitamin D3 absorbed in the gastrointestinal tract are metabolized in the liver to 25-hydroxyvitamin D (25[OH]D), which re-enters the circulation and is metabolized in the kidney and other tissues to the active metabolite 1,25-dihydroxyvitamin D (1,25[OH]2D). Catabolism of 25(OH)D and 1,25(OH)2D into biologically-inactive molecules is primarily mediated by the cytochrome P450 (CYP) enzymes CYP24 and CYP3A4.
Source: Reference 2Circulating vitamin D3 is metabolized in the liver by the enzyme vitamin D-25-hydroxylase to 25-hydroxyvitamin D (25[OH]D3), which has a serum half-life of approximately 15 days.4 Circulating 25(OH)D3 is not biologically active at the physiological level, and requires activation by conversion to 1,25-dihydroxyvitamin D (1,25[OH]2D3) in the kidneys by the enzyme 25(OH)D-1α-hydroxylase. Production of 1,25(OH)2D3 is regulated by serum phosphorus and parathyroid hormone levels and other factors.5 Catabolism of 1,25(OH)2D3 is rapid, with a serum half-life of 3.5 to 21 hours.6 Vitamin D2 is structurally similar to vitamin D3, but occurs primarily in fungi, yeasts, and some invertebrates.
Risk factors for deficiency
A patient’s vitamin D status is determined by measuring 25(OH)D (Box 1). Risk factors for vitamin D deficiency include conditions that affect cutaneous production (insufficient sunlight exposure), obesity, gastrointestinal disorders, aging, renal disorders, and medications (Table 1). 2,5,7,8 The link between sunscreen use, either alone or in cosmetics, and vitamin D deficiency continues to be debated. While controlled studies have found that application of sunscreen with high sun protection factor can significantly reduce vitamin D production, 9 studies in clinical populations have failed to confirm these findings. 10,11 See Box 2 for a discussion of these risk factors and Box 3 for a discussion of acute and long-term medical manifestations of deficiency.
Although 1,25-dihydroxyvitamin D (1,25[OH]2D3) is the biologically active form of vitamin D, its circulating half-life is only 4 to 6 hours.a,b Therefore, 25-hydroxyvitamin D (25[OH]D) is the principal vitamin D metabolite measured to determine vitamin D status. Vitamin D levels commonly are expressed as ng/mL or nmol/L; the conversion factor from ng/mL to nmol/L is 2.496. The Institute of Medicine has defined vitamin D deficiency as a serum 25(OH)D level of <30 nmol/L (<12 ng/mL).c However, many experts define vitamin D insufficiency as a 25(OH)D level of 21 to 29 ng/ml, and deficiency as <20 ng/mL.a,d The upper limit is more difficult to define, but symptoms of vitamin D intoxication appear with blood levels >150 to 200 ng/mL.a
References
- Holick MF. High prevalence of vitamin D inadequacy and implications for health. Mayo Clin Proc. 2006;81(3):353-373.
- Holick MF. Vitamin D status: measurement, interpretation, and clinical application. Ann Epidemiol. 2009;19(2):73-78.
- Aloia JF. Clinical review: the 2011 report on dietary reference intake for vitamin D: where do we go from here? J Clin Endocrinol Metab. 2011;96(10):2987-2996.
- Bischoff-Ferrari HA, Giovannucci E, Willett WC, et al. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84(1):18-28.
Table 1
Risk factors associated with vitamin D deficiency
| Age (>65) |
| Insufficient sunlight |
| Breastfeeding |
| Dark skin |
| Malabsorption diseases |
| Obesity (BMI >30 kg/m2) |
| Use of medications that alter vitamin D metabolism (eg, anticonvulsants, glucocorticoids) |
| Hepatobiliary disease |
| Renal disease |
| BMI: body mass index Source: References 2,5,7,8 |
Any factor that diminishes UV radiation penetration into the skin will affect cutaneous synthesis of vitamin D.a,b For example, sunscreen with a sun protection factor of 15 can decrease vitamin D synthesis by 98%.c Geography and its impact on yearly sunlight exposure is a well-known factor in vitamin D deficiency. Individuals who live below a latitude of approximately 35° North—approximately the southern border of Tennessee and through Albuquerque, NM—receive sufficient UV radiation exposure to ensure adequate vitamin D production throughout the year, but at higher latitudes, adequate vitamin D is not produced during winter months.d Melanin affects UV radiation absorption in a manner that prevents vitamin D production, and increased skin pigmentation markedly reduces vitamin D synthesis.e African Americans with very dark skin have significantly diminished cutaneous production of vitamin D.e,f
Renal 1α-hydroxylase activity decreases with aging in parallel with age-related decreases in glomerular filtration.g In addition, aging is associated with increased clearance of 1,25-dihydroxyvitamin D (1,25[OH]2D3).h However, vitamin D absorption generally is adequate even at older ages.i Studies have shown that obese individuals tend to have lower serum concentrations of vitamin D and 25-hydroxyvitamin D (25[OH]D) than those at a normal weight.j,k Obese patients have been shown to have lower cutaneous production of vitamin D3 and display lower bioavailability of orally administered vitamin D2.j
For patients with chronic renal insufficiency, creatinine clearance is positively correlated with serum 1,25(OH)2D levels.l Any process that results in malabsorption of intestinal fat may impair vitamin D absorption. In patients with celiac disease, biliary obstruction, or chronic pancreatitis, absorption consistently is reduced.m Individuals taking bile acid-binding medications, such as cholestyramine for hypercholesterolemia, also may have impaired vitamin D absorption.n In addition, hepatobiliary disease is associated with low levels of 25(OH)D.o Some drugs that alter hepatic metabolism are associated with vitamin D deficiency, including anticonvulsants or glucocorticoids, which can increase catabolism or vitamin D.p
References
- Holick MF. The vitamin D deficiency pandemic and consequences for nonskeletal health: mechanisms of action. Mol Aspects Med. 2008;29(6):361-368.
- Holick MF, Chen TC. Vitamin D deficiency: a worldwide problem with health consequences. Am J Clin Nutr. 2008;87(4):1080S-1086S.
- Matsuoka LY, Ide L, Wortsman J, et al. Sunscreens suppress cutaneous vitamin D3 synthesis. J Clin Endocrinol Metab. 1987;64(6):1165-1168.
- Holick MF. Vitamin D: importance in the prevention of cancers, type 1 diabetes, heart disease, and osteoporosis. Am J Clin Nutr. 2004;79(3):362-371.
- Clemens TL, Adams JS, Henderson SL, et al. Increased skin pigment reduces the capacity of skin to synthesise vitamin D3. Lancet. 1982;1(8263):74-76.
- Chen TC, Chimeh F, Lu Z, et al. Factors that influence the cutaneous synthesis and dietary sources of vitamin D. Arch Biochem Biophys. 2007;460(2):213-217.
- Slovik DM, Adams JS, Neer RM, et al. Deficient production of 1,25-dihydroxyvitamin D in elderly osteoporotic patients. N Engl J Med. 1981;305(7):372-374.
- Armbrecht HJ, Zenser TV, Davis BB. Effect of age on the conversion of 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3 by kidney of rat. J Clin Invest. 1980;66(5):1118-1123.
- Lips P, Wiersinga A, van Ginkel FC, et al. The effect of vitamin D supplementation on vitamin D status and parathyroid function in elderly subjects. J Clin Endocrinol Metab. 1988;67(4):644-650.
- Wortsman J, Matsuoka LY, Chen TC, et al. Decreased bioavailability of vitamin D in obesity. Am J Clin Nutr. 2000;72(3):690-693.
- Bell NH, Epstein S, Greene A, et al. Evidence for alteration of the vitamin D-endocrine system in obese subjects. J Clin Invest. 1985;76(1):370-373.
- Pitts TO, Piraino BH, Mitro R, et al. Hyperparathyroidism and 1,25-dihydroxyvitamin D deficiency in mild, moderate, and severe renal failure. J Clin Endocrinol Metab. 1988;67(5):876-881.
- Thompson GR, Lewis B, Booth CC. Absorption of vitamin D3-3H in control subjects and patients with intestinal malabsorption. J Clin Invest. 1966;45(1):94-102.
- Lo CW, Paris PW, Clemens TL, et al. Vitamin D absorption in healthy subjects and in patients with intestinal malabsorption syndromes. Am J Clin Nutr. 1985;42(4):644-649.
- Pappa HM, Bern E, Kamin D, et al. Vitamin D status in gastrointestinal and liver disease. Curr Opin Gastroenterol. 2008;24(2):176-183.
- Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266-281.
Acute effects. Vitamin D deficiency produces a range of clinical effects.a-c One well-documented consequence of vitamin D deficiency is osteomalacia—bone demineralization—which produces characteristic bone deformity and growth retardation in children.d,e In adults, osteomalacia may manifest as diffuse pain bone discomfort and muscle aches that may resemble fibromyalgia or arthritis.f Because vitamin D receptors are present in skeletal muscle, deficiency also may lead to proximal muscle weakness; an increased risk of falls; global bone discomfort, often elicited with pressure over the sternum or tibia; and low back pain in older women.c,f
Long-term effects. A large epidemiologic study found that adults with 25-hydroxyvitamin D (25[OH]D) levels <21 ng/mL had an increased risk of hypertension, diabetes, obesity, and dyslipidemia.g Cardiovascular mortality was higher in individuals with 25(OH)D levels <10 ng/mL compared with those with >40 ng/mL.h Adolescents in the National Health and Nutrition Examination Survey-III with serum 25(OH)D levels <15 ng/mL were more likely to have elevated blood glucose levels than those with >26 ng/mL.i Other epidemiologic data have demonstrated associations of vitamin D deficiency with multiple sclerosis, seasonal allergies, asthma, and various infectious diseases.j,k
Because vitamin D is known to promote cellular differentiation and inhibit cellular proliferation, its role in cancer has been studied extensively. A recent meta-analysis of case-control studies found that the odds of colon cancer were reduced by >40% for each 20 ng/mL increase in serum 25(OH)D levels.l Another meta-analysis reported a lower risk of breast cancer among women in the highest quartile of 25(OH)D values compared with the lowest quartile.m
References
- Holick MF. High prevalence of vitamin D inadequacy and implications for health. Mayo Clin Proc. 2006;81(3):353-373.
- Holick MF. Vitamin D status: measurement, interpretation, and clinical application. Ann Epidemiol. 2009;19(2):73-78.
- Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266-281.
- Holick MF, Chen TC. Vitamin D deficiency: a worldwide problem with health consequences. Am J Clin Nutr. 2008;87(4):1080S-1086S.
- Bordelon P, Ghetu MV, Langan RC. Recognition and management of vitamin D deficiency. Am Fam Physician. 2009;80(8):841-846.
- Hicks GE, Shardell M, Miller RR, et al. Associations between vitamin D status and pain in older adults: the Invecchiare in Chianti study. J Am Geriatr Soc. 2008;56(5):785-791.
- Martins D, Wolf M, Pan D, et al. Prevalence of cardiovascular risk factors and the serum levels of 25-hydroxyvitamin D in the United States: data from the Third National Health and Nutrition Examination Survey. Arch Intern Med. 2007;167(11):1159-1165.
- Ginde AA, Scragg R, Schwartz RS, et al. Prospective study of serum 25-hydroxyvitamin D level, cardiovascular disease mortality, and all-cause mortality in older U.S. adults. J Am Geriatr Soc. 2009;57(9):1595-1603.
- Reis JP, von Mühlen D, Miller ER 3rd, et al. Vitamin D status and cardiometabolic risk factors in the United States adolescent population. Pediatrics. 2009;124(3):e371-379.
- Thacher TD, Clarke BL. Vitamin D insufficiency. Mayo Clin Proc. 2011;86(1):50-60.
- Munger KL, Levin LI, Hollis BW, et al. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA. 2006;296(23):2832-2838.
- Yin L, Grandi N, Raum E, et al. Meta-analysis: longitudinal studies of serum vitamin D and colorectal cancer risk. Aliment Pharmacol Ther. 2009;30(2):113-125.
- Chen P, Hu P, Xie D, et al. Meta-analysis of vitamin D, calcium and the prevention of breast cancer. Breast Cancer Res Treat. 2010;121(2):469-477.
Vitamin D’s role in the brain
Vitamin D’s role in psychiatric illnesses is suggested by region-specific expression of vitamin D receptors (VDR) in the cingulate cortex, thalamus, cerebellum, amygdala, and hippocampus.12 Most of these regions also express 1α-hydroxylase enzymes capable of metabolizing 25(OH)D to 1,25(OH)2D3, which suggests that vitamin D may have an autocrine or paracrine function in brain.13
Vitamin D regulates expression of tyrosine hydroxylase, the rate-limiting enzyme in the biosynthesis of dopamine, norepinephrine, and epinephrine.14 Vitamin D also promotes survival of monoaminergic neurons through upregulation of glial cell line-derived neurotrophic factor, which supports survival of midbrain dopaminergic neurons and confers resistance to neurotoxins that deplete dopaminergic neurons in Parkinson’s disease.15 Vitamin D also promotes neuronal survival by inhibiting oxidative pathways in the brain through inhibition of inducible nitric oxide synthase (reducing free radical formation)16 and upregulation of γ-glutamyl transpeptidase (increasing antioxidant production).17 Vitamin D may play a neuroprotective role through regulation of calcium channels. In vitro studies have shown that vitamin D downregulates expression of L-type calcium channels, conferring protection against excitatory neurotoxins in cultured neurons.18 Proteomic analysis of brain tissue in a rat model of developmental vitamin D revealed dysregulation of 36 brain proteins involved in many biologic pathways involved in calcium homeostasis, synaptic plasticity, and neurotransmission.19 Taken together, these findings suggest vitamin D has a neurosteroid-like role in the CNS.
Psychotic disorders
Several epidemiologic studies have linked low vitamin D levels to schizophrenia and other psychotic disorders. Researchers in Norway who used a structured clinical interview to identify psychosis consistently found low levels of 25(OH)D among immigrants and native Norwegians with psychotic symptoms.20 A study of 8,411 Swedish women found low vitamin D levels were associated with psychotic symptoms.21 The Finnish birth cohort study found that use of vitamin D supplementation during the first year of life reduced the incidence of schizophrenia.22 In another pilot study, researchers measured third-trimester serum 25(OH)D levels and found that low levels of maternal vitamin D may be associated with an increased risk of schizophrenia.23 These studies suggest that low prenatal vitamin D levels may adversely impact the developing brain, increasing the risk for adult-onset schizophrenia.
Cognitive dysfunction
Low vitamin D concentrations have been associated with impairments in cognitive functions such as memory and orientation,24 executive function impairments,25 and Alzheimer’s disease (AD).26 A large study conducted from 1998 to 2006 in Italy concluded that persons with severe vitamin D deficiency (<25 nmol/L) had a higher risk of substantial decline on Mini-Mental State Examination than those with sufficient levels (≥75 nmol/L).27 Other studies have linked low vitamin D levels to poor cognitive performance in depressed older adults.28 Low vitamin D levels in older women have been associated with risk of AD, but not with other dementias.29 Polymorphisms of VDR have been associated with depression and poor cognitive performance.30
Depression
Epidemiologic studies evaluating vitamin D deficiency have had conflicting results. The Third National Health and Nutrition Examination Survey, which used a sample of 7,970 non-institutionalized U.S. residents age 15 to 39, demonstrated that individuals with serum vitamin D ≤50 nmol/L are at a significantly higher risk of developing depression than those with vitamin D ≥75 nmol/L.31 A study of 1,282 adults age 65 to 95 in the Netherlands found that 25(OH)D levels were 14% lower in depressed patients compared with controls.32 However, a large epidemiologic study in China did not detect a relationship between vitamin D and depression in 3,262 men and women age 50 to 70.33 After researchers adjusted for geography, body mass index, physical activity, and smoking, 25(OH)D levels did not correlate significantly with the presence or severity of depression. In a case series,34 after 48 vitamin D-deficient depressed adolescents were given vitamin D3 over 3 months, there was a significant improvement in well-being, depressive symptoms, irritability, and fatigue.34 Other small, cross-sectional studies have examined associations between vitamin D status and depression with divergent results, which may reflect differences in population and methodology.
Prospective interventional studies. Although direct causal relationships are difficult to establish, several prospective studies have tested the hypothesis that treating vitamin D deficiency can improve depressive symptoms.
In a double-blind, controlled trial, Jorde et al35 randomized 441 individuals age 21 to 70 to vitamin D, 20,000 IU per week; vitamin D, 40,000 IU per week; or placebo for 1 year. Individuals with serum 25(OH)D levels <40 nmol/L scored significantly higher on depression rating scales than those with serum 25(OH)D levels ≥40 nmol/L at the end of the study. There was no significant improvement in depression ratings in the placebo group (Table 2).35 These results must be interpreted with care because depressive symptoms were secondary endpoints in this study.
Table 2
Effect of vitamin D supplementation on depressive symptoms in a controlled trial
| Vitamin D Supplementation | Serum 25(OH)D levels at baseline | BDI total score, Median and range at end of study | After 1 year of vitamin D supplementation |
|---|---|---|---|
| 20,000 IU/week | <40 nmol/L | Significantly higher (more depressive traits), 6.0 (0 to 23) | Significantly improved BDI score |
| 40,000 IU/week | ≥40 nmol/L | 4.5 (0 to 28) | Significantly improved BDI score |
| Placebo | - | - | No improvement in BDI score |
| 25(OH)D: 25-hydroxyvitamin D; BDI: Beck Depression Inventory Source: Reference 35 | |||
Kjærgaard et al36 systematically examined vitamin D levels in a case-control study followed by a randomized controlled trial (RCT) of vitamin D supplementation. In the case-control phase, participants with low 25(OH)D levels at baseline were significantly more depressed than participants with high 25(OH)D levels. Participants with low 25(OH)D levels were randomized to placebo or 40,000 IU vitamin D3 per week for 6 months. Low levels of vitamin D were strongly associated with depressive symptoms, but vitamin D supplementation did not have a significant effect on depressive symptom scores.
Seasonal affective disorder (SAD). Seasonal variation in vitamin D levels suggests that supplementation may help patients who have seasonal mood disturbances. In a randomized, double-blind study, 44 healthy individuals received vitamin D3, 400 IU/d, 800 IU/d, or no vitamin D3 for 5 days during late winter. Based on self-reports, vitamin D3 significantly enhanced positive affect and there was some evidence it reduced negative affect.37 In a pilot study of 9 women with serum vitamin D levels <40 ng/ml, vitamin D supplementation during winter was associated with an average 10-point decline in Beck Depression Inventory-II scores.38 In a prospective RCT of 15 individuals with SAD, all patients who received vitamin D improved in all outcome measures.39 Vieth40 randomized 82 adults with vitamin D deficiency to 600 IU/d or 4,000 IU/d of vitamin D3 for 3 months over 2 consecutive winters. Patients taking the higher dose showed some evidence of improved well-being compared with those taking the lower dose, although results were not significant for all comparisons. Two other trials did not observe any improvement in SAD symptoms with vitamin D treatment.41,42
Treating vitamin D deficiency
The Endocrine Society recently developed consensus guidelines for diagnosing and managing vitamin D deficiency.43 In addition, the Institute of Medicine of the National Academies recommends daily vitamin D supplementation to prevent deficiency:
- age <70: 400 IU/d
- age >70: 800 IU/d
- pregnant or lactating women: 600 IU/d
- upper limit: 4,000 IU/d.7
Higher doses may be used for patients deprived of sun exposure.8 A typical replacement regimen consists of oral ergocalciferol, 50,000 IU per week for 8 weeks.44 The optimal time for rechecking serum levels after repletion has not been clearly defined, but serum 25(OH)D levels should be measured again after therapy is completed. If values have not reached or exceeded 20 ng/mL, consider a second 8-week course of ergocalciferol (see the Box 1 for a discussion of measuring vitamin D levels). If serum 25(OH)D levels have not increased, the most likely cause is nonadherence or malabsorption.
Contraindications and toxicity. Contraindications to vitamin D supplementation include granulomatous diseases, sarcoidosis, metastatic bone disease, and Williams syndrome.45Table 345 lists signs of vitamin D toxicity. There is little risk of toxicity at dosages of up to 2,000 IU/d.46
Table 3
Signs of vitamin D toxicity
| Headache |
| Metallic taste |
| Nephrocalcinosis or vascular calcinosis |
| Pancreatitis |
| Nausea |
| Vomiting |
| Source: Reference 45 |
Related Resources
- LaFerney MC. Vitamin D deficiency in older adults. Current Psychiatry. 2012;11(11):63.
- National Institutes of Health Office of Dietary Supplements. Dietary supplement fact sheet: vitamin D. http://ods.od.nih.gov/factsheets/VitaminD-HealthProfessional.
Drug Brand Names
- Cholestyramine • Questran
- Ergocalciferol • Calciferol, Drisdol
Disclosures
Dr. Harris is an employee of Rho, Chapel Hill, NC.
Dr. Jaiswal reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Holmes receives research support from Bristol-Myers Squibb, Elan, Merck, Otsuka, Shire, Takeda, and Theravance, and is on the speaker’s bureau for Forest Pharmaceuticals and PamLab.
Dr. Patkar is a consultant for Dey Pharmaceuticals, Forest, Gilead, and TTK Pharma and is on the speaker’s bureau and received honoraria from Alkermes, Bristol-Myers Squibb, Dey Pharmaceuticals, Pfizer, and Sunovion; and has received grant support from the Duke Endowment, Dey Pharmaceuticals, Envivo, Forest, Janssen, Lundbeck, The National Institutes of Health, the National Institute on Drug Abuse, National Institute on Alcohol Abuse and Alcoholism, Pfizer Inc., Shire, Sunovion, and Titan.
Dr. Weisler has been a consultant to, on the speaker’s bureaus of, and/or received research support from Abbott, Agency for Toxic Substances and Disease Registry, AstraZeneca, Biovail, Bristol-Myers Squibb, Burroughs Wellcome, Cenerx, Centers for Disease Control and Prevention, Cephalon, Ciba Geigy, CoMentis, Corcept, Cortex, Dainippon Sumitomo Pharma America, Eisai, Elan, Eli Lilly and Company, Forest Pharmaceuticals, GlaxoSmithKline, Janssen, Johnson & Johnson, Lundbeck, McNeil Pharmaceuticals, Medicinova, Medscape Advisory Board, Merck, National Institute of Mental Health, Neurochem, New River Pharmaceuticals, Novartis, Organon, Otsuka America Pharma, Pfizer Inc., Pharmacia, Repligen, Saegis, Sandoz, Sanofi, Sanofi-Synthelabo, Schwabe/Ingenix, Sepracor, Shire, Solvay, Sunovion, Synaptic, Takeda, TAP, Theravance, Transcept Pharma, TransTech, UCB Pharma, Validus, Vela, and Wyeth.
In the United States, >50% of psychiatric inpatients have vitamin D deficiency—<30 nmol/L (<12 ng/mL).1 A growing body of literature has found associations between vitamin D deficiency and psychiatric illnesses, particularly depression. Several randomized controlled trials (RCTs) have demonstrated that vitamin D supplementation can benefit depression symptoms. In this article, we discuss the current literature on vitamin D and psychiatric illness, and provide practical information for clinicians on the use of vitamin D supplementation.
Biosynthesis of vitamin D
Biosynthesis of vitamin D begins with the sterol provitamin D3 molecule 7-dehydrocholesterol (Figure).2 When skin is exposed to sunlight, 7-dehydrocholesterol absorbs UV radiation and forms provitamin D3, which undergoes rapid transformation to vitamin D3.2 Vitamin D3 is released from the plasma membrane and enters systemic circulation in a protein-bound form that has a serum half-life of 36 to 78 hours.3 Vitamin D3 can be taken up by adipocytes and stored in fat deposits, where it has a half-life of approximately 2 months.4
Figure: Biosynthesis of vitamin D
Provitamin D3 (7-dehydrocholesterol) in the skin absorbs UV radiation and undergoes isomerization to form vitamin D3. Endogenously produced vitamin D3 along with dietary vitamin D2 and vitamin D3 absorbed in the gastrointestinal tract are metabolized in the liver to 25-hydroxyvitamin D (25[OH]D), which re-enters the circulation and is metabolized in the kidney and other tissues to the active metabolite 1,25-dihydroxyvitamin D (1,25[OH]2D). Catabolism of 25(OH)D and 1,25(OH)2D into biologically-inactive molecules is primarily mediated by the cytochrome P450 (CYP) enzymes CYP24 and CYP3A4.
Source: Reference 2Circulating vitamin D3 is metabolized in the liver by the enzyme vitamin D-25-hydroxylase to 25-hydroxyvitamin D (25[OH]D3), which has a serum half-life of approximately 15 days.4 Circulating 25(OH)D3 is not biologically active at the physiological level, and requires activation by conversion to 1,25-dihydroxyvitamin D (1,25[OH]2D3) in the kidneys by the enzyme 25(OH)D-1α-hydroxylase. Production of 1,25(OH)2D3 is regulated by serum phosphorus and parathyroid hormone levels and other factors.5 Catabolism of 1,25(OH)2D3 is rapid, with a serum half-life of 3.5 to 21 hours.6 Vitamin D2 is structurally similar to vitamin D3, but occurs primarily in fungi, yeasts, and some invertebrates.
Risk factors for deficiency
A patient’s vitamin D status is determined by measuring 25(OH)D (Box 1). Risk factors for vitamin D deficiency include conditions that affect cutaneous production (insufficient sunlight exposure), obesity, gastrointestinal disorders, aging, renal disorders, and medications (Table 1). 2,5,7,8 The link between sunscreen use, either alone or in cosmetics, and vitamin D deficiency continues to be debated. While controlled studies have found that application of sunscreen with high sun protection factor can significantly reduce vitamin D production, 9 studies in clinical populations have failed to confirm these findings. 10,11 See Box 2 for a discussion of these risk factors and Box 3 for a discussion of acute and long-term medical manifestations of deficiency.
Although 1,25-dihydroxyvitamin D (1,25[OH]2D3) is the biologically active form of vitamin D, its circulating half-life is only 4 to 6 hours.a,b Therefore, 25-hydroxyvitamin D (25[OH]D) is the principal vitamin D metabolite measured to determine vitamin D status. Vitamin D levels commonly are expressed as ng/mL or nmol/L; the conversion factor from ng/mL to nmol/L is 2.496. The Institute of Medicine has defined vitamin D deficiency as a serum 25(OH)D level of <30 nmol/L (<12 ng/mL).c However, many experts define vitamin D insufficiency as a 25(OH)D level of 21 to 29 ng/ml, and deficiency as <20 ng/mL.a,d The upper limit is more difficult to define, but symptoms of vitamin D intoxication appear with blood levels >150 to 200 ng/mL.a
References
- Holick MF. High prevalence of vitamin D inadequacy and implications for health. Mayo Clin Proc. 2006;81(3):353-373.
- Holick MF. Vitamin D status: measurement, interpretation, and clinical application. Ann Epidemiol. 2009;19(2):73-78.
- Aloia JF. Clinical review: the 2011 report on dietary reference intake for vitamin D: where do we go from here? J Clin Endocrinol Metab. 2011;96(10):2987-2996.
- Bischoff-Ferrari HA, Giovannucci E, Willett WC, et al. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84(1):18-28.
Table 1
Risk factors associated with vitamin D deficiency
| Age (>65) |
| Insufficient sunlight |
| Breastfeeding |
| Dark skin |
| Malabsorption diseases |
| Obesity (BMI >30 kg/m2) |
| Use of medications that alter vitamin D metabolism (eg, anticonvulsants, glucocorticoids) |
| Hepatobiliary disease |
| Renal disease |
| BMI: body mass index Source: References 2,5,7,8 |
Any factor that diminishes UV radiation penetration into the skin will affect cutaneous synthesis of vitamin D.a,b For example, sunscreen with a sun protection factor of 15 can decrease vitamin D synthesis by 98%.c Geography and its impact on yearly sunlight exposure is a well-known factor in vitamin D deficiency. Individuals who live below a latitude of approximately 35° North—approximately the southern border of Tennessee and through Albuquerque, NM—receive sufficient UV radiation exposure to ensure adequate vitamin D production throughout the year, but at higher latitudes, adequate vitamin D is not produced during winter months.d Melanin affects UV radiation absorption in a manner that prevents vitamin D production, and increased skin pigmentation markedly reduces vitamin D synthesis.e African Americans with very dark skin have significantly diminished cutaneous production of vitamin D.e,f
Renal 1α-hydroxylase activity decreases with aging in parallel with age-related decreases in glomerular filtration.g In addition, aging is associated with increased clearance of 1,25-dihydroxyvitamin D (1,25[OH]2D3).h However, vitamin D absorption generally is adequate even at older ages.i Studies have shown that obese individuals tend to have lower serum concentrations of vitamin D and 25-hydroxyvitamin D (25[OH]D) than those at a normal weight.j,k Obese patients have been shown to have lower cutaneous production of vitamin D3 and display lower bioavailability of orally administered vitamin D2.j
For patients with chronic renal insufficiency, creatinine clearance is positively correlated with serum 1,25(OH)2D levels.l Any process that results in malabsorption of intestinal fat may impair vitamin D absorption. In patients with celiac disease, biliary obstruction, or chronic pancreatitis, absorption consistently is reduced.m Individuals taking bile acid-binding medications, such as cholestyramine for hypercholesterolemia, also may have impaired vitamin D absorption.n In addition, hepatobiliary disease is associated with low levels of 25(OH)D.o Some drugs that alter hepatic metabolism are associated with vitamin D deficiency, including anticonvulsants or glucocorticoids, which can increase catabolism or vitamin D.p
References
- Holick MF. The vitamin D deficiency pandemic and consequences for nonskeletal health: mechanisms of action. Mol Aspects Med. 2008;29(6):361-368.
- Holick MF, Chen TC. Vitamin D deficiency: a worldwide problem with health consequences. Am J Clin Nutr. 2008;87(4):1080S-1086S.
- Matsuoka LY, Ide L, Wortsman J, et al. Sunscreens suppress cutaneous vitamin D3 synthesis. J Clin Endocrinol Metab. 1987;64(6):1165-1168.
- Holick MF. Vitamin D: importance in the prevention of cancers, type 1 diabetes, heart disease, and osteoporosis. Am J Clin Nutr. 2004;79(3):362-371.
- Clemens TL, Adams JS, Henderson SL, et al. Increased skin pigment reduces the capacity of skin to synthesise vitamin D3. Lancet. 1982;1(8263):74-76.
- Chen TC, Chimeh F, Lu Z, et al. Factors that influence the cutaneous synthesis and dietary sources of vitamin D. Arch Biochem Biophys. 2007;460(2):213-217.
- Slovik DM, Adams JS, Neer RM, et al. Deficient production of 1,25-dihydroxyvitamin D in elderly osteoporotic patients. N Engl J Med. 1981;305(7):372-374.
- Armbrecht HJ, Zenser TV, Davis BB. Effect of age on the conversion of 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3 by kidney of rat. J Clin Invest. 1980;66(5):1118-1123.
- Lips P, Wiersinga A, van Ginkel FC, et al. The effect of vitamin D supplementation on vitamin D status and parathyroid function in elderly subjects. J Clin Endocrinol Metab. 1988;67(4):644-650.
- Wortsman J, Matsuoka LY, Chen TC, et al. Decreased bioavailability of vitamin D in obesity. Am J Clin Nutr. 2000;72(3):690-693.
- Bell NH, Epstein S, Greene A, et al. Evidence for alteration of the vitamin D-endocrine system in obese subjects. J Clin Invest. 1985;76(1):370-373.
- Pitts TO, Piraino BH, Mitro R, et al. Hyperparathyroidism and 1,25-dihydroxyvitamin D deficiency in mild, moderate, and severe renal failure. J Clin Endocrinol Metab. 1988;67(5):876-881.
- Thompson GR, Lewis B, Booth CC. Absorption of vitamin D3-3H in control subjects and patients with intestinal malabsorption. J Clin Invest. 1966;45(1):94-102.
- Lo CW, Paris PW, Clemens TL, et al. Vitamin D absorption in healthy subjects and in patients with intestinal malabsorption syndromes. Am J Clin Nutr. 1985;42(4):644-649.
- Pappa HM, Bern E, Kamin D, et al. Vitamin D status in gastrointestinal and liver disease. Curr Opin Gastroenterol. 2008;24(2):176-183.
- Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266-281.
Acute effects. Vitamin D deficiency produces a range of clinical effects.a-c One well-documented consequence of vitamin D deficiency is osteomalacia—bone demineralization—which produces characteristic bone deformity and growth retardation in children.d,e In adults, osteomalacia may manifest as diffuse pain bone discomfort and muscle aches that may resemble fibromyalgia or arthritis.f Because vitamin D receptors are present in skeletal muscle, deficiency also may lead to proximal muscle weakness; an increased risk of falls; global bone discomfort, often elicited with pressure over the sternum or tibia; and low back pain in older women.c,f
Long-term effects. A large epidemiologic study found that adults with 25-hydroxyvitamin D (25[OH]D) levels <21 ng/mL had an increased risk of hypertension, diabetes, obesity, and dyslipidemia.g Cardiovascular mortality was higher in individuals with 25(OH)D levels <10 ng/mL compared with those with >40 ng/mL.h Adolescents in the National Health and Nutrition Examination Survey-III with serum 25(OH)D levels <15 ng/mL were more likely to have elevated blood glucose levels than those with >26 ng/mL.i Other epidemiologic data have demonstrated associations of vitamin D deficiency with multiple sclerosis, seasonal allergies, asthma, and various infectious diseases.j,k
Because vitamin D is known to promote cellular differentiation and inhibit cellular proliferation, its role in cancer has been studied extensively. A recent meta-analysis of case-control studies found that the odds of colon cancer were reduced by >40% for each 20 ng/mL increase in serum 25(OH)D levels.l Another meta-analysis reported a lower risk of breast cancer among women in the highest quartile of 25(OH)D values compared with the lowest quartile.m
References
- Holick MF. High prevalence of vitamin D inadequacy and implications for health. Mayo Clin Proc. 2006;81(3):353-373.
- Holick MF. Vitamin D status: measurement, interpretation, and clinical application. Ann Epidemiol. 2009;19(2):73-78.
- Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266-281.
- Holick MF, Chen TC. Vitamin D deficiency: a worldwide problem with health consequences. Am J Clin Nutr. 2008;87(4):1080S-1086S.
- Bordelon P, Ghetu MV, Langan RC. Recognition and management of vitamin D deficiency. Am Fam Physician. 2009;80(8):841-846.
- Hicks GE, Shardell M, Miller RR, et al. Associations between vitamin D status and pain in older adults: the Invecchiare in Chianti study. J Am Geriatr Soc. 2008;56(5):785-791.
- Martins D, Wolf M, Pan D, et al. Prevalence of cardiovascular risk factors and the serum levels of 25-hydroxyvitamin D in the United States: data from the Third National Health and Nutrition Examination Survey. Arch Intern Med. 2007;167(11):1159-1165.
- Ginde AA, Scragg R, Schwartz RS, et al. Prospective study of serum 25-hydroxyvitamin D level, cardiovascular disease mortality, and all-cause mortality in older U.S. adults. J Am Geriatr Soc. 2009;57(9):1595-1603.
- Reis JP, von Mühlen D, Miller ER 3rd, et al. Vitamin D status and cardiometabolic risk factors in the United States adolescent population. Pediatrics. 2009;124(3):e371-379.
- Thacher TD, Clarke BL. Vitamin D insufficiency. Mayo Clin Proc. 2011;86(1):50-60.
- Munger KL, Levin LI, Hollis BW, et al. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA. 2006;296(23):2832-2838.
- Yin L, Grandi N, Raum E, et al. Meta-analysis: longitudinal studies of serum vitamin D and colorectal cancer risk. Aliment Pharmacol Ther. 2009;30(2):113-125.
- Chen P, Hu P, Xie D, et al. Meta-analysis of vitamin D, calcium and the prevention of breast cancer. Breast Cancer Res Treat. 2010;121(2):469-477.
Vitamin D’s role in the brain
Vitamin D’s role in psychiatric illnesses is suggested by region-specific expression of vitamin D receptors (VDR) in the cingulate cortex, thalamus, cerebellum, amygdala, and hippocampus.12 Most of these regions also express 1α-hydroxylase enzymes capable of metabolizing 25(OH)D to 1,25(OH)2D3, which suggests that vitamin D may have an autocrine or paracrine function in brain.13
Vitamin D regulates expression of tyrosine hydroxylase, the rate-limiting enzyme in the biosynthesis of dopamine, norepinephrine, and epinephrine.14 Vitamin D also promotes survival of monoaminergic neurons through upregulation of glial cell line-derived neurotrophic factor, which supports survival of midbrain dopaminergic neurons and confers resistance to neurotoxins that deplete dopaminergic neurons in Parkinson’s disease.15 Vitamin D also promotes neuronal survival by inhibiting oxidative pathways in the brain through inhibition of inducible nitric oxide synthase (reducing free radical formation)16 and upregulation of γ-glutamyl transpeptidase (increasing antioxidant production).17 Vitamin D may play a neuroprotective role through regulation of calcium channels. In vitro studies have shown that vitamin D downregulates expression of L-type calcium channels, conferring protection against excitatory neurotoxins in cultured neurons.18 Proteomic analysis of brain tissue in a rat model of developmental vitamin D revealed dysregulation of 36 brain proteins involved in many biologic pathways involved in calcium homeostasis, synaptic plasticity, and neurotransmission.19 Taken together, these findings suggest vitamin D has a neurosteroid-like role in the CNS.
Psychotic disorders
Several epidemiologic studies have linked low vitamin D levels to schizophrenia and other psychotic disorders. Researchers in Norway who used a structured clinical interview to identify psychosis consistently found low levels of 25(OH)D among immigrants and native Norwegians with psychotic symptoms.20 A study of 8,411 Swedish women found low vitamin D levels were associated with psychotic symptoms.21 The Finnish birth cohort study found that use of vitamin D supplementation during the first year of life reduced the incidence of schizophrenia.22 In another pilot study, researchers measured third-trimester serum 25(OH)D levels and found that low levels of maternal vitamin D may be associated with an increased risk of schizophrenia.23 These studies suggest that low prenatal vitamin D levels may adversely impact the developing brain, increasing the risk for adult-onset schizophrenia.
Cognitive dysfunction
Low vitamin D concentrations have been associated with impairments in cognitive functions such as memory and orientation,24 executive function impairments,25 and Alzheimer’s disease (AD).26 A large study conducted from 1998 to 2006 in Italy concluded that persons with severe vitamin D deficiency (<25 nmol/L) had a higher risk of substantial decline on Mini-Mental State Examination than those with sufficient levels (≥75 nmol/L).27 Other studies have linked low vitamin D levels to poor cognitive performance in depressed older adults.28 Low vitamin D levels in older women have been associated with risk of AD, but not with other dementias.29 Polymorphisms of VDR have been associated with depression and poor cognitive performance.30
Depression
Epidemiologic studies evaluating vitamin D deficiency have had conflicting results. The Third National Health and Nutrition Examination Survey, which used a sample of 7,970 non-institutionalized U.S. residents age 15 to 39, demonstrated that individuals with serum vitamin D ≤50 nmol/L are at a significantly higher risk of developing depression than those with vitamin D ≥75 nmol/L.31 A study of 1,282 adults age 65 to 95 in the Netherlands found that 25(OH)D levels were 14% lower in depressed patients compared with controls.32 However, a large epidemiologic study in China did not detect a relationship between vitamin D and depression in 3,262 men and women age 50 to 70.33 After researchers adjusted for geography, body mass index, physical activity, and smoking, 25(OH)D levels did not correlate significantly with the presence or severity of depression. In a case series,34 after 48 vitamin D-deficient depressed adolescents were given vitamin D3 over 3 months, there was a significant improvement in well-being, depressive symptoms, irritability, and fatigue.34 Other small, cross-sectional studies have examined associations between vitamin D status and depression with divergent results, which may reflect differences in population and methodology.
Prospective interventional studies. Although direct causal relationships are difficult to establish, several prospective studies have tested the hypothesis that treating vitamin D deficiency can improve depressive symptoms.
In a double-blind, controlled trial, Jorde et al35 randomized 441 individuals age 21 to 70 to vitamin D, 20,000 IU per week; vitamin D, 40,000 IU per week; or placebo for 1 year. Individuals with serum 25(OH)D levels <40 nmol/L scored significantly higher on depression rating scales than those with serum 25(OH)D levels ≥40 nmol/L at the end of the study. There was no significant improvement in depression ratings in the placebo group (Table 2).35 These results must be interpreted with care because depressive symptoms were secondary endpoints in this study.
Table 2
Effect of vitamin D supplementation on depressive symptoms in a controlled trial
| Vitamin D Supplementation | Serum 25(OH)D levels at baseline | BDI total score, Median and range at end of study | After 1 year of vitamin D supplementation |
|---|---|---|---|
| 20,000 IU/week | <40 nmol/L | Significantly higher (more depressive traits), 6.0 (0 to 23) | Significantly improved BDI score |
| 40,000 IU/week | ≥40 nmol/L | 4.5 (0 to 28) | Significantly improved BDI score |
| Placebo | - | - | No improvement in BDI score |
| 25(OH)D: 25-hydroxyvitamin D; BDI: Beck Depression Inventory Source: Reference 35 | |||
Kjærgaard et al36 systematically examined vitamin D levels in a case-control study followed by a randomized controlled trial (RCT) of vitamin D supplementation. In the case-control phase, participants with low 25(OH)D levels at baseline were significantly more depressed than participants with high 25(OH)D levels. Participants with low 25(OH)D levels were randomized to placebo or 40,000 IU vitamin D3 per week for 6 months. Low levels of vitamin D were strongly associated with depressive symptoms, but vitamin D supplementation did not have a significant effect on depressive symptom scores.
Seasonal affective disorder (SAD). Seasonal variation in vitamin D levels suggests that supplementation may help patients who have seasonal mood disturbances. In a randomized, double-blind study, 44 healthy individuals received vitamin D3, 400 IU/d, 800 IU/d, or no vitamin D3 for 5 days during late winter. Based on self-reports, vitamin D3 significantly enhanced positive affect and there was some evidence it reduced negative affect.37 In a pilot study of 9 women with serum vitamin D levels <40 ng/ml, vitamin D supplementation during winter was associated with an average 10-point decline in Beck Depression Inventory-II scores.38 In a prospective RCT of 15 individuals with SAD, all patients who received vitamin D improved in all outcome measures.39 Vieth40 randomized 82 adults with vitamin D deficiency to 600 IU/d or 4,000 IU/d of vitamin D3 for 3 months over 2 consecutive winters. Patients taking the higher dose showed some evidence of improved well-being compared with those taking the lower dose, although results were not significant for all comparisons. Two other trials did not observe any improvement in SAD symptoms with vitamin D treatment.41,42
Treating vitamin D deficiency
The Endocrine Society recently developed consensus guidelines for diagnosing and managing vitamin D deficiency.43 In addition, the Institute of Medicine of the National Academies recommends daily vitamin D supplementation to prevent deficiency:
- age <70: 400 IU/d
- age >70: 800 IU/d
- pregnant or lactating women: 600 IU/d
- upper limit: 4,000 IU/d.7
Higher doses may be used for patients deprived of sun exposure.8 A typical replacement regimen consists of oral ergocalciferol, 50,000 IU per week for 8 weeks.44 The optimal time for rechecking serum levels after repletion has not been clearly defined, but serum 25(OH)D levels should be measured again after therapy is completed. If values have not reached or exceeded 20 ng/mL, consider a second 8-week course of ergocalciferol (see the Box 1 for a discussion of measuring vitamin D levels). If serum 25(OH)D levels have not increased, the most likely cause is nonadherence or malabsorption.
Contraindications and toxicity. Contraindications to vitamin D supplementation include granulomatous diseases, sarcoidosis, metastatic bone disease, and Williams syndrome.45Table 345 lists signs of vitamin D toxicity. There is little risk of toxicity at dosages of up to 2,000 IU/d.46
Table 3
Signs of vitamin D toxicity
| Headache |
| Metallic taste |
| Nephrocalcinosis or vascular calcinosis |
| Pancreatitis |
| Nausea |
| Vomiting |
| Source: Reference 45 |
Related Resources
- LaFerney MC. Vitamin D deficiency in older adults. Current Psychiatry. 2012;11(11):63.
- National Institutes of Health Office of Dietary Supplements. Dietary supplement fact sheet: vitamin D. http://ods.od.nih.gov/factsheets/VitaminD-HealthProfessional.
Drug Brand Names
- Cholestyramine • Questran
- Ergocalciferol • Calciferol, Drisdol
Disclosures
Dr. Harris is an employee of Rho, Chapel Hill, NC.
Dr. Jaiswal reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Holmes receives research support from Bristol-Myers Squibb, Elan, Merck, Otsuka, Shire, Takeda, and Theravance, and is on the speaker’s bureau for Forest Pharmaceuticals and PamLab.
Dr. Patkar is a consultant for Dey Pharmaceuticals, Forest, Gilead, and TTK Pharma and is on the speaker’s bureau and received honoraria from Alkermes, Bristol-Myers Squibb, Dey Pharmaceuticals, Pfizer, and Sunovion; and has received grant support from the Duke Endowment, Dey Pharmaceuticals, Envivo, Forest, Janssen, Lundbeck, The National Institutes of Health, the National Institute on Drug Abuse, National Institute on Alcohol Abuse and Alcoholism, Pfizer Inc., Shire, Sunovion, and Titan.
Dr. Weisler has been a consultant to, on the speaker’s bureaus of, and/or received research support from Abbott, Agency for Toxic Substances and Disease Registry, AstraZeneca, Biovail, Bristol-Myers Squibb, Burroughs Wellcome, Cenerx, Centers for Disease Control and Prevention, Cephalon, Ciba Geigy, CoMentis, Corcept, Cortex, Dainippon Sumitomo Pharma America, Eisai, Elan, Eli Lilly and Company, Forest Pharmaceuticals, GlaxoSmithKline, Janssen, Johnson & Johnson, Lundbeck, McNeil Pharmaceuticals, Medicinova, Medscape Advisory Board, Merck, National Institute of Mental Health, Neurochem, New River Pharmaceuticals, Novartis, Organon, Otsuka America Pharma, Pfizer Inc., Pharmacia, Repligen, Saegis, Sandoz, Sanofi, Sanofi-Synthelabo, Schwabe/Ingenix, Sepracor, Shire, Solvay, Sunovion, Synaptic, Takeda, TAP, Theravance, Transcept Pharma, TransTech, UCB Pharma, Validus, Vela, and Wyeth.
1. McCue RE, Charles RA, Orendain GC, et al. Vitamin D deficiency among psychiatric inpatients [published online April 19, 2012]. Prim Care Companion CNS Disord. doi: 10.4088/PCC.11m01230.
2. Tsiaras WG, Weinstock MA. Factors influencing vitamin D status. Acta Derm Venereol. 2011;91(2):115-124.
3. Adams JS, Clemens TL, Parrish JA, et al. Vitamin-D synthesis and metabolism after ultraviolet irradiation of normal and vitamin-D-deficient subjects. N Engl J Med. 1982;306(12):722-725.
4. Jones G. Pharmacokinetics of vitamin D toxicity. Am J Clin Nutr. 2008;88(2):582S-586S.
5. Holick MF. Vitamin D: importance in the prevention of cancers type 1 diabetes, heart disease, and osteoporosis. Am J Clin Nutr. 2004;79(3):362-371.
6. Fakih MG, Trump DL, Muindi JR, et al. A phase I pharmacokinetic and pharmacodynamic study of intravenous calcitriol in combination with oral gefitinib in patients with advanced solid tumors. Clin Cancer Res. 2007;13(4):1216-1223.
7. Holick MF, Chen TC. Vitamin D deficiency: a worldwide problem with health consequences. Am J Clin Nutr. 2008;87(4):1080S-1086S.
8. Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266-281.
9. Matsuoka LY, Ide L, Wortsman J, et al. Sunscreens suppress cutaneous vitamin D3 synthesis. J Clin Endocrinol Metab. 1987;64(6):1165-1168.
10. Norval M, Wulf HC. Does chronic sunscreen use reduce vitamin D production to insufficient levels? Br J Dermatol. 2009;161(4):732-736.
11. Linos E, Keiser E, Kanzler M, et al. Sun protective behaviors and vitamin D levels in the US population: NHANES 2003-2006. Cancer Causes Control. 2012;23(1):133-140.
12. Prüfer K, Veenstra TD, Jirikowski GF, et al. Distribution of 1,25-dihydroxyvitamin D3 receptor immunoreactivity in the rat brain and spinal cord. J Chem Neuroanat. 1999;16(2):135-145.
13. Eyles DW, Smith S, Kinobe R, et al. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat. 2005;29(1):21-30.
14. Garcion E, Wion-Barbot N, Montero-Menei CN, et al. New clues about vitamin D functions in the nervous system. Trends Endocrinol Metab. 2002;13(3):100-105.
15. Smith MP, Fletcher-Turner A, Yurek DM, et al. Calcitriol protection against dopamine loss induced by intracerebroventricular administration of 6-hydroxydopamine. Neurochem Res. 2006;31(4):533-539.
16. Garcion E, Nataf S, Berod A, et al. 1,25-dihydroxyvitamin D3 inhibits the expression of inducible nitric oxide synthase in rat central nervous system during experimental allergic encephalomyelitis. Brain Res Mol Brain Res. 1997;45(2):255-267.
17. Baas D, Prüfer K, Ittel ME, et al. Rat oligodendrocytes express the vitamin D(3) receptor and respond to 1,25-dihydroxyvitamin D(3). Glia. 2000;31(1):59-68.
18. Brewer LD, Thibault V, Chen KC, et al. Vitamin D hormone confers neuroprotection in parallel with downregulation of L-type calcium channel expression in hippocampal neurons. J Neurosci. 2001;21(1):98-108.
19. Almeras L, Eyles D, Benech P, et al. Developmental vitamin D deficiency alters brain protein expression in the adult rat: implications for neuropsychiatric disorders. Proteomics. 2007;7(5):769-780.
20. Berg AO, Melle I, Torjesen PA, et al. A cross-sectional study of vitamin D deficiency among immigrants and Norwegians with psychosis compared to the general population. J Clin Psychiatry. 2010;71(12):1598-1604.
21. Hedelin M, Löf M, Olsson M, et al. Dietary intake of fish, omega-3, omega-6 polyunsaturated fatty acids and vitamin D and the prevalence of psychotic-like symptoms in a cohort of 33,000 women from the general population. BMC Psychiatry. 2010;10:38.-
22. McGrath J, Saari K, Hakko H, et al. Vitamin D supplementation during the first year of life and risk of schizophrenia: a Finnish birth cohort study. Schizophr Res. 2004;67(2-3):237-245.
23. McGrath J, Eyles D, Mowry B, et al. Low maternal vitamin D as a risk factor for schizophrenia: a pilot study using banked sera. Schizophr Res. 2003;63(1-2):73-78.
24. Przybelski RJ, Binkley NC. Is vitamin D important for preserving cognition? A positive correlation of serum 25-hydroxyvitamin D concentration with cognitive function. Arch Biochem Biophys. 2007;460(2):202-205.
25. Lee DM, Tajar A, Ulubaev A, et al. Association between 25-hydroxyvitamin D levels and cognitive performance in middle-aged and older European men. J Neurol Neurosurg Psychiatry. 2009;80(7):722-729.
26. Buell JS, Dawson-Hughes B, Scott TM, et al. 25-hydroxyvitamin D, dementia, and cerebrovascular pathology in elders receiving home services. Neurology. 2010;74(1):18-26.
27. Llewellyn DJ, Lang IA, Langa KM, et al. Vitamin D and risk of cognitive decline in elderly persons. Arch Intern Med. 2010;170(13):1135-1141.
28. Wilkins CH, Sheline YI, Roe CM, et al. Vitamin D deficiency is associated with low mood and worse cognitive performance in older adults. Am J Geriatr Psychiatry. 2006;14(12):1032-1040.
29. Annweiler C, Rolland Y, Schott AM, et al. Higher vitamin D dietary intake is associated with lower risk of Alzheimer’s disease: a 7-year follow-up. J Gerontol A Biol Sci Med Sci. 2012;67(11):1205-1211.
30. Kuningas M, Mooijaart SP, Jolles J, et al. VDR gene variants associate with cognitive function and depressive symptoms in old age. Neurobiol Aging. 2009;30(3):466-473.
31. Ganji V, Milone C, Cody MM, et al. Serum vitamin D concentrations are related to depression in young adult US population: the Third National Health and Nutrition Examination Survey. Int Arch Med. 2010;3:29.-
32. Hoogendijk WJ, Lips P, Dik MG, et al. Depression is associated with decreased 25-hydroxyvitamin D and increased parathyroid hormone levels in older adults. Arch Gen Psychiatry. 2008;65(5):508-512.
33. Pan A, Lu L, Franco OH, et al. Association between depressive symptoms and 25-hydroxyvitamin D in middle-aged and elderly Chinese. J Affect Disord. 2009;118(1-3):240-243.
34. Högberg G, Gustafsson SA, Hällström T, et al. Depressed adolescents in a case-series were low in vitamin D and depression was ameliorated by vitamin D supplementation. Acta Paediatr. 2012;101(7):779-783.
35. Jorde R, Sneve M, Figenschau Y, et al. Effects of vitamin D supplementation on symptoms of depression in overweight and obese subjects: randomized double blind trial. J Intern Med. 2008;264(6):599-609.
36. Kjærgaard M, Waterloo K, Wang CE, et al. Effect of vitamin D supplement on depression scores in people with low levels of serum 25-hydroxyvitamin D: nested case-control study and randomised clinical trial. Br J Psychiatry. 2012;201(5):360-368.
37. Lansdowne AT, Provost SC. Vitamin D3 enhances mood in healthy subjects during winter. Psychopharmacology (Berl). 1998;135(4):319-223.
38. Shipowick CD, Moore CB, Corbett C, et al. Vitamin D and depressive symptoms in women during the winter: a pilot study. Appl Nurs Res. 2009;22(3):221-225.
39. Gloth FM 3rd, Alam W, Hollis B. Vitamin D vs broad spectrum phototherapy in the treatment of seasonal affective disorder. J Nutr Health Aging. 1999;3(1):5-7.
40. Vieth R, Kimball S, Hu A, et al. Randomized comparison of the effects of the vitamin D3 adequate intake versus 100 mcg (4000 IU) per day on biochemical responses and the wellbeing of patients. Nutr J. 2004;3:8.-
41. Harris S, Dawson-Hughes B. Seasonal mood changes in 250 normal women. Psychiatry Res. 1993;49(1):77-87.
42. Dumville JC, Miles JN, Porthouse J, et al. Can vitamin D supplementation prevent winter-time blues? A randomised trial among older women. J Nutr Health Aging. 2006;10(2):151-153.
43. Holick MF, Binkley NC, Bischoff-Ferrari HA, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96(7):1911-1930.
44. Thacher TD, Clarke BL. Vitamin D insufficiency. Mayo Clin Proc. 2011;86(1):50-60.
45. Schwalfenberg G. Not enough vitamin D: health consequences for Canadians. Can Fam Physician. 2007;53(5):841-854.
46. Norman AW, Bouillon R, Whiting SJ, et al. 13th Workshop consensus for vitamin D nutritional guidelines. J Steroid Biochem Mol Biol. 2007;103(3-5):204-205.
1. McCue RE, Charles RA, Orendain GC, et al. Vitamin D deficiency among psychiatric inpatients [published online April 19, 2012]. Prim Care Companion CNS Disord. doi: 10.4088/PCC.11m01230.
2. Tsiaras WG, Weinstock MA. Factors influencing vitamin D status. Acta Derm Venereol. 2011;91(2):115-124.
3. Adams JS, Clemens TL, Parrish JA, et al. Vitamin-D synthesis and metabolism after ultraviolet irradiation of normal and vitamin-D-deficient subjects. N Engl J Med. 1982;306(12):722-725.
4. Jones G. Pharmacokinetics of vitamin D toxicity. Am J Clin Nutr. 2008;88(2):582S-586S.
5. Holick MF. Vitamin D: importance in the prevention of cancers type 1 diabetes, heart disease, and osteoporosis. Am J Clin Nutr. 2004;79(3):362-371.
6. Fakih MG, Trump DL, Muindi JR, et al. A phase I pharmacokinetic and pharmacodynamic study of intravenous calcitriol in combination with oral gefitinib in patients with advanced solid tumors. Clin Cancer Res. 2007;13(4):1216-1223.
7. Holick MF, Chen TC. Vitamin D deficiency: a worldwide problem with health consequences. Am J Clin Nutr. 2008;87(4):1080S-1086S.
8. Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266-281.
9. Matsuoka LY, Ide L, Wortsman J, et al. Sunscreens suppress cutaneous vitamin D3 synthesis. J Clin Endocrinol Metab. 1987;64(6):1165-1168.
10. Norval M, Wulf HC. Does chronic sunscreen use reduce vitamin D production to insufficient levels? Br J Dermatol. 2009;161(4):732-736.
11. Linos E, Keiser E, Kanzler M, et al. Sun protective behaviors and vitamin D levels in the US population: NHANES 2003-2006. Cancer Causes Control. 2012;23(1):133-140.
12. Prüfer K, Veenstra TD, Jirikowski GF, et al. Distribution of 1,25-dihydroxyvitamin D3 receptor immunoreactivity in the rat brain and spinal cord. J Chem Neuroanat. 1999;16(2):135-145.
13. Eyles DW, Smith S, Kinobe R, et al. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat. 2005;29(1):21-30.
14. Garcion E, Wion-Barbot N, Montero-Menei CN, et al. New clues about vitamin D functions in the nervous system. Trends Endocrinol Metab. 2002;13(3):100-105.
15. Smith MP, Fletcher-Turner A, Yurek DM, et al. Calcitriol protection against dopamine loss induced by intracerebroventricular administration of 6-hydroxydopamine. Neurochem Res. 2006;31(4):533-539.
16. Garcion E, Nataf S, Berod A, et al. 1,25-dihydroxyvitamin D3 inhibits the expression of inducible nitric oxide synthase in rat central nervous system during experimental allergic encephalomyelitis. Brain Res Mol Brain Res. 1997;45(2):255-267.
17. Baas D, Prüfer K, Ittel ME, et al. Rat oligodendrocytes express the vitamin D(3) receptor and respond to 1,25-dihydroxyvitamin D(3). Glia. 2000;31(1):59-68.
18. Brewer LD, Thibault V, Chen KC, et al. Vitamin D hormone confers neuroprotection in parallel with downregulation of L-type calcium channel expression in hippocampal neurons. J Neurosci. 2001;21(1):98-108.
19. Almeras L, Eyles D, Benech P, et al. Developmental vitamin D deficiency alters brain protein expression in the adult rat: implications for neuropsychiatric disorders. Proteomics. 2007;7(5):769-780.
20. Berg AO, Melle I, Torjesen PA, et al. A cross-sectional study of vitamin D deficiency among immigrants and Norwegians with psychosis compared to the general population. J Clin Psychiatry. 2010;71(12):1598-1604.
21. Hedelin M, Löf M, Olsson M, et al. Dietary intake of fish, omega-3, omega-6 polyunsaturated fatty acids and vitamin D and the prevalence of psychotic-like symptoms in a cohort of 33,000 women from the general population. BMC Psychiatry. 2010;10:38.-
22. McGrath J, Saari K, Hakko H, et al. Vitamin D supplementation during the first year of life and risk of schizophrenia: a Finnish birth cohort study. Schizophr Res. 2004;67(2-3):237-245.
23. McGrath J, Eyles D, Mowry B, et al. Low maternal vitamin D as a risk factor for schizophrenia: a pilot study using banked sera. Schizophr Res. 2003;63(1-2):73-78.
24. Przybelski RJ, Binkley NC. Is vitamin D important for preserving cognition? A positive correlation of serum 25-hydroxyvitamin D concentration with cognitive function. Arch Biochem Biophys. 2007;460(2):202-205.
25. Lee DM, Tajar A, Ulubaev A, et al. Association between 25-hydroxyvitamin D levels and cognitive performance in middle-aged and older European men. J Neurol Neurosurg Psychiatry. 2009;80(7):722-729.
26. Buell JS, Dawson-Hughes B, Scott TM, et al. 25-hydroxyvitamin D, dementia, and cerebrovascular pathology in elders receiving home services. Neurology. 2010;74(1):18-26.
27. Llewellyn DJ, Lang IA, Langa KM, et al. Vitamin D and risk of cognitive decline in elderly persons. Arch Intern Med. 2010;170(13):1135-1141.
28. Wilkins CH, Sheline YI, Roe CM, et al. Vitamin D deficiency is associated with low mood and worse cognitive performance in older adults. Am J Geriatr Psychiatry. 2006;14(12):1032-1040.
29. Annweiler C, Rolland Y, Schott AM, et al. Higher vitamin D dietary intake is associated with lower risk of Alzheimer’s disease: a 7-year follow-up. J Gerontol A Biol Sci Med Sci. 2012;67(11):1205-1211.
30. Kuningas M, Mooijaart SP, Jolles J, et al. VDR gene variants associate with cognitive function and depressive symptoms in old age. Neurobiol Aging. 2009;30(3):466-473.
31. Ganji V, Milone C, Cody MM, et al. Serum vitamin D concentrations are related to depression in young adult US population: the Third National Health and Nutrition Examination Survey. Int Arch Med. 2010;3:29.-
32. Hoogendijk WJ, Lips P, Dik MG, et al. Depression is associated with decreased 25-hydroxyvitamin D and increased parathyroid hormone levels in older adults. Arch Gen Psychiatry. 2008;65(5):508-512.
33. Pan A, Lu L, Franco OH, et al. Association between depressive symptoms and 25-hydroxyvitamin D in middle-aged and elderly Chinese. J Affect Disord. 2009;118(1-3):240-243.
34. Högberg G, Gustafsson SA, Hällström T, et al. Depressed adolescents in a case-series were low in vitamin D and depression was ameliorated by vitamin D supplementation. Acta Paediatr. 2012;101(7):779-783.
35. Jorde R, Sneve M, Figenschau Y, et al. Effects of vitamin D supplementation on symptoms of depression in overweight and obese subjects: randomized double blind trial. J Intern Med. 2008;264(6):599-609.
36. Kjærgaard M, Waterloo K, Wang CE, et al. Effect of vitamin D supplement on depression scores in people with low levels of serum 25-hydroxyvitamin D: nested case-control study and randomised clinical trial. Br J Psychiatry. 2012;201(5):360-368.
37. Lansdowne AT, Provost SC. Vitamin D3 enhances mood in healthy subjects during winter. Psychopharmacology (Berl). 1998;135(4):319-223.
38. Shipowick CD, Moore CB, Corbett C, et al. Vitamin D and depressive symptoms in women during the winter: a pilot study. Appl Nurs Res. 2009;22(3):221-225.
39. Gloth FM 3rd, Alam W, Hollis B. Vitamin D vs broad spectrum phototherapy in the treatment of seasonal affective disorder. J Nutr Health Aging. 1999;3(1):5-7.
40. Vieth R, Kimball S, Hu A, et al. Randomized comparison of the effects of the vitamin D3 adequate intake versus 100 mcg (4000 IU) per day on biochemical responses and the wellbeing of patients. Nutr J. 2004;3:8.-
41. Harris S, Dawson-Hughes B. Seasonal mood changes in 250 normal women. Psychiatry Res. 1993;49(1):77-87.
42. Dumville JC, Miles JN, Porthouse J, et al. Can vitamin D supplementation prevent winter-time blues? A randomised trial among older women. J Nutr Health Aging. 2006;10(2):151-153.
43. Holick MF, Binkley NC, Bischoff-Ferrari HA, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96(7):1911-1930.
44. Thacher TD, Clarke BL. Vitamin D insufficiency. Mayo Clin Proc. 2011;86(1):50-60.
45. Schwalfenberg G. Not enough vitamin D: health consequences for Canadians. Can Fam Physician. 2007;53(5):841-854.
46. Norman AW, Bouillon R, Whiting SJ, et al. 13th Workshop consensus for vitamin D nutritional guidelines. J Steroid Biochem Mol Biol. 2007;103(3-5):204-205.
Strategies for treating depression in patients with hepatitis C

Mr. P, age 31, has been using heroin intravenously for 9 years. He smokes 1 pack of cigarettes daily, but denies using other substances, including alcohol. After an unintentional heroin overdose, Mr. P enrolls in a methadone maintenance treatment program (MMTP) that includes primary medical care and addiction medicine and psychiatric specialists, where he undergoes medical evaluation and screening for hepatitis C virus (HCV) and human immunodeficiency virus (HIV). Laboratory data reveal that although Mr. P is HIV negative, he has been exposed to HCV and treatment is indicated.
Among the approximately 3 million people in the United States with chronic HCV—an enveloped, single-stranded RNA virus—there’s a high prevalence of premorbid psychopathology and substance abuse, as well as neuropsychiatric effects caused by HCV treatment.1-3 Because underdiagnosing and undertreating psychiatric disorders contributes to morbidity and mortality in HCV patients, early identification and prompt treatment is critical.
IV drug use is the most common route for HCV infection, accounting for 65% to 70% of infections.1 The prevalence of HCV among IV drug users is 28% to 90%.1 Once exposed to HCV, 75% to 85% of patients do not clear the initial infection and become chronically infected.
This article reviews the pathophysiology, identification, and management of psychiatric manifestations found among HCV patients and provides an understanding of how psychiatric symptoms manifest in HCV patients. This article also discusses HCV treatment and its neuropsychiatric side effects.
Testing for HCV
Chronically infected HCV patients may have few, if any, specific physical complaints, and often are diagnosed during screenings or other routine laboratory evaluations. The presence of risk factors, such as a history of injection drug use or receiving a blood transfusion before 1992,1 guides the decision to screen for HCV. Normal liver function test results should not preclude testing because many HCV-positive patients have transaminases within the normal range.4 Initial screening is via an antibody-mediated immunoassay that is highly specific and sensitive for past exposure to HCV (Table 1).4 However, a positive screen does not indicate the presence of active infection. Evidence of the virus via a viral assay will identify active HCV, but does not indicate need for treatment. Liver biopsy confirms the presence of liver injury and quantifies its extent. The severity of liver damage will determine whether treatment is needed. HCV genotyping determines the appropriate duration and dosage of pharmacotherapy.
Table 1
Tests to diagnose and evaluate HCV
| Test | Results |
|---|---|
| HCV antibody | Determines prior exposure to HCV |
| HCV viral assay | Evaluates for current HCV infection |
| Liver biopsy | Assesses level of liver damage |
| HCV genotyping | Provides data to determine duration and intensity of treatment and likelihood of treatment success |
| HCV: hepatitis C virus Source: Reference 4 | |
CASE CONTINUED: Mood improves, but fatigue persists
As part of pre-HCV treatment evaluation, Mr. P undergoes a psychiatric evaluation. He describes periods of low mood while actively engaged in drug use but has never received psychiatric treatment, experienced suicidal ideation, or attempted suicide. Since starting opioid agonist therapy, he reports improved mood but endorses continued mild fatigue and difficulty falling sleep. The psychiatrist determines Mr. P does not meet criteria for an axis I diagnosis other than a substance use disorder.
Although most HCV patients have few, if any, nonspecific physical symptoms, many have psychiatric symptoms or disorders before the HCV diagnosis is made or treatment is initiated; substance use disorders are most common. Batki et al1 found that 56% of HCV patients in an MMTP met criteria for a nonsubstance axis I disorder and 82% met criteria for such a disorder during their lifetime. Additionally, 66% of patients were taking psychiatric medications. Table 21,5,6 lists the rates of other psychiatric disorders found in patients with untreated HCV.
Table 2
Rates of psychiatric disorders in patients with untreated hepatitis C virus
| Disorder(s) | Current rate | Lifetime rate |
|---|---|---|
| Mood disorders | 34% to 35% | 67% |
| Major depressive disorder | 22% to 28% | 42% |
| Anxiety disorders | 26% to 44% | 63% |
| Antisocial personality disorder | No rates; lifetime diagnosis | 16% to 40% |
| Psychotic disorders | 9% to 17% | 11% |
| Substance use disorder | 56% | 56% to 86% |
| Source: References 1,5,6 | ||
Many patients with chronic HCV complain of chronic fatigue and deficiencies in attention, concentration, higher executive functions, learning ability, and memory that result in significant reduction in quality of life (Box 1).7-9 These findings have been found to be independent of the degree of liver disease and are seen in HCV patients with normal liver function.7,8
The pathophysiology of fatigue and neurocognitive dysfunction in hepatitis C virus (HCV) infection is unclear. However, the improvement of chronic fatigue in patients with HCV who receive ondansetron, a 5-hydroxytryptophan-3 receptor antagonist, has implicated abnormal monoaminergic function. Single-photon emission CT studies have found decreased midbrain serotonergic and striatal dopaminergic transmission in some HCV patients with cognitive deficits.7
Recently, data have been mounting on a direct neuropathic effect of HCV, with viral elements found in autopsy brain tissue and cerebrospinal fluid.8 Researchers have suggested that HCV may enter the CNS via a Trojan horse-like mechanism inside infected mononuclear cells.8 More recently, human brain microvascular endothelium, the major component of the blood-brain barrier, has been found to express all major viral receptors that would allow HCV infection of the CNS.9
CASE CONTINUED: Motivated and compliant
Since joining the MMTP 6 months ago, Mr. P has been motivated and compliant with all appointments and treatments. Routine urine toxicology screening supports his claim of abstinence. Mr. P begins HCV treatment while continuing follow-up with addiction medicine and psychiatric clinicians and maintains open communication with all treatment providers.
For many years the standard HCV treatment was pegylated interferon-α (IFN-α) and ribavirin. IFN-α is a proinflammatory cytokine with antiproliferative, antiviral, and immunoregulatory properties. The half-life of IFN-α significantly increases with pegylation, which allows for weekly injections.10,11 IFN-α usually is combined with ribavirin, which increases its efficacy as measured by the sustained virological response (SVR) compared with IFN-α alone. Depending on the virus genotype, treatment lasts 24 to 48 weeks; SVR rates range from 40% to 82%.11-13 In 2011, the FDA approved 2 agents—telaprevir and boceprevir—for adjunctive treatment of HCV genotype 1 infection. These 2 agents are protease inhibitors that when added to IFN-α and ribavirin increase the SVR rate in genotype 1 infection from 40% to 50% to approximately 75%.14,15
Although the neuropsychiatric side effects of telaprevir and boceprevir have not been determined, treating chronic HCV with IFN-α and ribavirin has been associated with multiple psychiatric symptoms, including depression, mania, suicidality, anxiety, and psychosis.11-14 Psychiatric symptoms are a common reason for discontinuing or reducing HCV treatment. Because of the high frequency of neuropsychiatric complications, some clinicians believe HCV patients with preexisting affective, psychotic, or substance use disorders should be excluded from HCV treatment. This has led to many HCV patients being untreated despite a lack of prospective, controlled data to support this opinion.12 To improve outcomes and decrease morbidity, providing appropriate psychiatric services appears to be more important than attempting to select lower-risk patients for antiviral therapy.1,12,16 The goals of psychiatric treatment should be to alleviate symptoms and allow patients to complete IFN-α therapy without interruption.16,17
Studies of high-risk patients who attend multidisciplinary treatment programs that can monitor adherence and efficacy and control side effects before and during HCV treatment have found psychiatric patients have similar adherence, compliance, and SVR rates and were not at increased risk of worsening depressive or psychotic symptoms compared with patients without a psychiatric history.12,18 Additionally, HCV patients with a psychiatric history are not at an increased risk of suicide.13,16 Similar findings have been observed in patients with active IV drug use or those receiving opioid agonist therapy. When HCV and substance use are treated simultaneously, patients can successfully complete HCV treatment with SVR rates comparable to those of patients not receiving opioid agonist therapy.19-21
CASE CONTINUED: Worsening symptoms
During a psychiatric follow-up 12 weeks after starting HCV treatment, Mr. P reports worsening depressive symptoms with low mood, decreased enjoyment of activities, poor sleep, low appetite, and fatigue. He shows no evidence of psychosis and denies suicidal ideation. We continue his HCV treatment, schedule more frequent psychiatric visits, and initiate citalopram, titrated to 40 mg/d.
Depressive symptoms, the most common neuropsychiatric manifestation of HCV, typically begin early in treatment, usually within the first 12 weeks. Two distinct symptom clusters are noted. A neurovegetative cluster characterized by reduced energy, anorexia, and psychomotor retardation typically begins within the first few months of treatment. Months later, a depression-specific syndrome appears that includes depressed mood, anxiety, and cognitive impairment.22
Depressive symptoms may occur in up to 60% of patients treated with IFN-α.11 When more rigorous depression measures are used, rates decrease to approximately 20% to 30%.11,13 Accurate diagnosis and treatment of emerging depressive symptoms is essential because untreated depression can lead to postponing or excluding patients from antiviral treatment.2 Screening instruments such as the Beck Depression Inventory-Second Edition (BDI-II) can be used to measure depressive symptoms in HCV patients with high sensitivity. However, because specificity has been low and somatic symptoms of chronic illness and depression often overlap, the BDI-II and other inventories may overestimate depression. Some researchers have suggested that focusing on questions targeting cognitive and affective symptoms rather than somatic ones may be a more valid measure of depression in patients undergoing immunotherapy for HCV.2
The immune system is implicated in IFN-α-induced depression because depressive symptoms share many features with a constellation of somatic and behavioral symptoms termed “sickness behavior.”11 These behaviors can occur when patients are exposed to cytokines that lead to a depressed level of functioning, which may allow the body to devote more energy to fighting illness. IFN-α, a cytokine, stimulates the immune system, which can lead to increases of interleukin (IL)-2, IL-6, and IL-10. Increased circulating levels of these ILs have been correlated with higher depression scores. Additionally, studies have found that patients who develop depression during IFN-α treatment have higher SVR rates, suggesting a more robust immune response.11,22 For a discussion of how serotonin metabolism and genetic polymorphisms also may help explain the prevalence of depression in patients with HCV, see Box 2.
Altered serotonin metabolism has been linked to depression in hepatitis C virus (HCV) patients treated with interferon-α (IFN-α). Tryptophan can be metabolized towards serotonin via tryptophan hydroxylase and niacin via indoleamine-2,3-dioxygenase (IDO) with kynurenine (KYN) and quinolinic acid (QUIN) as intermediaries. Introduction of IFN-α activates IDO, causing preferential conversion of tryptophan towards the niacin arm away from serotonin and leads to elevated levels of KYN and QUIN. KYN and QUIN are available centrally, are neurotoxic, and have been correlated with increased depressive symptoms in IFN-α-treated patients.a,b A tryptophan-deficient state is created, with less tryptophan being converted to serotonin and subsequently to its metabolite, 5-hydroxyindoleacetic acid (5-HIAA). Decreased levels of 5-HIAA in cerebrospinal fluid have been associated with higher depressive symptoms and higher rates of suicide.a,b
Several genetic polymorphisms may help identify patients at risk for developing IFN-α-induced depression. Genes for the 5’ promoter of the serotonin transporter (5-HTTLPR) have been investigated for roles in depression development in patients undergoing immunotherapy. Studies have found that persons with the short allele in the 5-HTTLPR gene are more likely to develop depression than those with the long allele. However, this has not been consistent across racial or ethnic groups.a,b Research also has associated the serotonin (5-HT) transporter, interferon receptor-A1, apolipoprotein ε4 allele, cyclooxygenase 2, and phospholipase A2 with development of a specific subgroup of symptoms.a
References
a. Smith KJ, Norris S, O’Farrelly C, et al. Risk factors for the development of depression in patients with hepatitis C taking interferon-α. Neuropsychiatr Dis Treat. 2011;7:275-292.
b. Sockalingam S, Links PS, Abbey SE. Suicide risk in hepatitis C and during interferon-alpha therapy: a review and clinical update. J Viral Hepat. 2011;18(3):153-160.
Treating depressed HCV patients
Antidepressants are the treatment of choice for IFN-α-induced depression. Most currently used antidepressants are effective22 and selective serotonin reuptake inhibitors are considered first choice.16 Antidepressant choice should be guided by principles similar to those used for patients without HCV: using side effects profiles to target specific symptoms and being mindful of pharmacokinetic properties.
Two treatment approaches have been investigated: prophylactic and symptomatic. A 2012 study23 of 181 HCV patients with no history of mental illness determined escitalopram, 10 mg/d, effectively reduced the incidence and severity of interferon-associated depression. Other studies examining prophylactic treatment of all patients who were to undergo interferon treatment found this approach did not prevent depressive episodes.24,25 However, antidepressants have been beneficial for patients with subsyndromal depressive symptoms at baseline26 and after clinically significant depressive symptoms emerge.27 Electroconvulsive therapy also has been reported to effectively treat depression in HCV patients undergoing antiviral therapy.28
CASE CONTINUED: Lingering symptoms
Mr. P responds to citalopram with an improvement in mood, anhedonia, and appetite, but he continues to complain of low energy and poor concentration. In an effort to target these symptoms, methylphenidate, titrated to 30 mg/d in divided doses, is added to his regimen, which rapidly improves his symptoms. Insomnia is treated successfully with trazodone, 50 mg/d. Mr. P frequently visits his psychiatrist, who monitors his depressive symptoms using the BDI-II. Mr. P completes HCV treatment without recurrence of depressive symptoms or relapse to heroin use.
Although antidepressants are effective for treating affective and cognitive symptoms, they are not as effective for fatigue and other neurovegetative symptoms.16,29 The psychostimulants methylphenidate and dextroamphetamine and the nonstimulant modafinil have been studied for treating depressive symptoms in medically ill patients and can be used to treat IFN-α-induced fatigue.16,22,29
IFN-α’s effect on serotonin metabolism leads to a tryptophan-deficient state because of increased catabolism as a result of activation of indoleamine-2,3-dioxygenase (IDO). This has led to use of tryptophan supplementation, either as augmentation or monotherapy, for managing depressive symptoms in patients treated with IFN-α. Schaefer et al30 reported 3 cases where tryptophan supplementation significantly decreased depressive symptoms. Other researchers have argued that supplementing tryptophan in the context of IDO activation can lead to greater production of kynurenine and quinolinic acid, which have been linked to increased depressive symptoms in patients receiving IFN-α.31 They argue that supplementation of 5-HTP, which is available as a dietary supplement without a prescription, can lead to increased serotonin levels and improvement in depressive symptoms.31
IFN-α treatment also is associated with mania and psychosis. The incidence, pathophysiology, and management of these treatment-emergent symptoms are not as well studied as IFN-α-induced depression. Mania and hypomania have been reported with interferon treatment, discontinuation of interferon, and use of antidepressants for interferon-induced depression.29,32 Psychosis, in association with mood symptoms or alone, has been reported to occur in <1% of treated patients.33 Treatment for mania and psychosis consists of decreasing or discontinuing immunotherapy and adding mood stabilizers and antipsychotics. Once immunotherapy is discontinued, mania and psychosis usually resolve, but prolonged duration of symptoms has been reported.29,32,33
Related Resources
- Centers for Disease Control and Prevention. Hepatitis C information for health professionals. www.cdc.gov/hepatitis/hcv/index.htm.
- Hep C Connection. www.hepc-connection.org.
- United States Department of Veterans Affairs. Hepatitis C. www.hepatitis.va.gov.
Drug Brand Names
- Boceprevir • Victrelis
- Citalopram • Celexa
- Dextroamphetamine • Dexedrine
- Escitalopram • Lexapro
- Interferon-α • Intron
- Methadone • Dolophine, Methadose
- Methylphenidate • Ritalin, Methylin, others
- Modafinil • Provigil
- Ondansetron • Zofran
- Ribavirin • Copegus, Rebetol, others
- Telaprevir • Incivek
- Trazodone • Desyrel, Oleptro
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Batki SL, Canfield KM, Ploutz-Snyder R. Psychiatric and substance use disorders among methadone maintenance patients with chronic hepatitis C infection: effects on eligibility for hepatitis C treatment. Am J Addict. 2011;20(4):312-318.
2. Patterson AL, Morasco BJ, Fuller BE, et al. Screening for depression in patients with hepatitis C using the Beck Depression Inventory-II: do somatic symptoms compromise validity? Gen Hosp Psychiatry. 2011;33(4):354-362.
3. Maddur H, Kwo PY. Boceprevir. Hepatology. 2011;54(6):2254-2257.
4. Sylvestre D. Hepatitis C for addiction professionals. Addict Sci Clin Pract. 2007;4(1):34-41.
5. Dwight MM, Kowdley KV, Russo JE, et al. Depression, fatigue, and functional disability in patients with chronic hepatitis C. J Psychosom Res. 2000;49(5):311-317.
6. Yovtcheva SP, Rifai MA, Moles JK, et al. Psychiatric comorbidity among hepatitis C-positive patients. Psychosomatics. 2001;42(5):411-415.
7. Weissenborn K, Ennen JC, Bokemeyer M, et al. Monoaminergic neurotransmission is altered in hepatitis C virus infected patients with chronic fatigue and cognitive impairment. Gut. 2006;55(11):1624-1630.
8. Weissenborn K, Tryc AB, Heeren M, et al. Hepatitis C virus infection and the brain. Metab Brain Dis. 2009;24(1):197-210.
9. Fletcher NF, Wilson GK, Murray J, et al. Hepatitis C virus infects the endothelial cells of the blood-brain barrier. Gastroenterology. 2012;142(3):634-643.e6.
10. Pawlotsky JM. Therapy of hepatitis C: from empiricism to eradication. Hepatology. 2006;43(2 suppl 1):S207-S220.
11. Smith KJ, Norris S, O’Farrelly C, et al. Risk factors for the development of depression in patients with hepatitis C taking interferon-α. Neuropsychiatr Dis Treat. 2011;7:275-292.
12. Schaefer M, Hinzpeter A, Mohmand A, et al. Hepatitis C treatment in “difficult-to-treat” psychiatric patients with pegylated interferon-alpha and ribavirin: response and psychiatric side effects. Hepatology. 2007;46(4):991-998.
13. Sockalingam S, Links PS, Abbey SE. Suicide risk in hepatitis C and during interferon-alpha therapy: a review and clinical update. J Viral Hepat. 2011;18(3):153-160.
14. Telaprevir (Incivek) and boceprevir (Victrelis) for chronic hepatitis C. Med Lett Drugs Ther. 2011;53(1369):57-59.
15. Nelson DR. The role of triple therapy with protease inhibitors in hepatitis C virus genotype 1 naïve patients. Liver Int. 2011;31(suppl 1):53-57.
16. Spennati A, Pariante CM. Withdrawing interferon-α from psychiatric patients: clinical care or unjustifiable stigma? [published online September 14 2012] Psychol Med. doi: 10. 1017/S0033291712001808.
17. Baraldi S, Hepgul N, Mondelli V, et al. Symptomatic treatment of interferon-α-induced depression in hepatitis C: a systematic review. J Clin Psychopharmacol. 2012;32(4):531-543.
18. Schaefer M, Schmidt F, Folwaczny C, et al. Adherence and mental side effects during hepatitis C treatment with interferon alfa and ribavirin in psychiatric risk groups. Hepatology. 2003;37(2):443-451.
19. Harris KA, Jr, Arnsten JH, Litwin AH. Successful integration of hepatitis C evaluation and treatment services with methadone maintenance. J Addict Med. 2010;4(1):20-26.
20. Litwin AH, Harris KA, Jr, Nahvi S, et al. Successful treatment of chronic hepatitis C with pegylated interferon in combination with ribavirin in a methadone maintenance treatment program. J Subst Abuse Treat. 2009;37(1):32-40.
21. Sasadeusz JJ, Dore G, Kronborg I, et al. Clinical experience with the treatment of hepatitis C infection in patients on opioid pharmacotherapy. Addiction. 2011;106(5):977-984.
22. Sockalingam S, Abbey SE. Managing depression during hepatitis C treatment. Can J Psychiatry. 2009;54(9):614-625.
23. Schaefer M, Sarkar R, Knop V, et al. Escitalopram for the prevention of peginterferon-α2a-associated depression in hepatitis C virus-infected patients without previous psychiatric disease: a randomized trial. Ann Intern Med. 2012;157(2):94-103.
24. Galvão-de Almeida A, Guindalini C, Batista-Neves S, et al. Can antidepressants prevent interferon-alpha-induced depression? A review of the literature. Gen Hosp Psychiatry. 2010;32(4):401-405.
25. Morasco BJ, Loftis JM, Indest DW, et al. Prophylactic antidepressant treatment in patients with hepatitis C on antiviral therapy: a double-blind, placebo-controlled trial. Psychosomatics. 2010;51(5):401-408.
26. Raison CL, Woolwine BJ, Demetrashvili MF, et al. Paroxetine for prevention of depressive symptoms induced by interferon-alpha and ribavirin for hepatitis C. Aliment Pharmacol Ther. 2007;25(10):1163-1174.
27. Kraus MR, Schäfer A, Schöttker K, et al. Therapy of interferon-induced depression in chronic hepatitis C with citalopram: a randomised, double-blind, placebo-controlled study. Gut. 2008;57(4):531-536.
28. Zincke MT, Kurani A, Istafanous R, et al. The successful use of electroconvulsive therapy in a patient with interferon-induced psychotic depression. J ECT. 2007;23(4):291-292.
29. Crone CC, Gabriel GM, Wise TN. Managing the neuropsychiatric side effects of interferon-based therapy for hepatitis C. Cleve Clin J Med. 2004;71(suppl 3):S27-S32.
30. Schaefer M, Winterer J, Sarkar R, et al. Three cases of successful tryptophan add-on or monotherapy of hepatitis C and IFNa-associated mood disorders. Psychosomatics. 2008;49(5):442-446.
31. Turner EH, Blackwell AD. 5-Hydroxytryptophan plus SSRIs for interferon-induced depression: synergistic mechanisms for normalizing synaptic serotonin. Med Hypotheses. 2005;65(1):138-144.
32. Onyike CU, Bonner JO, Lyketsos CG, et al. Mania during treatment of chronic hepatitis C with pegylated interferon and ribavirin. Am J Psychiatry. 2004;161(3):429-435.
33. Cheng YC, Chen CC, Ho AS, et al. Prolonged psychosis associated with interferon therapy in a patient with hepatitis C: case study and literature review. Psychosomatics. 2009;50(5):538-542.
Mr. P, age 31, has been using heroin intravenously for 9 years. He smokes 1 pack of cigarettes daily, but denies using other substances, including alcohol. After an unintentional heroin overdose, Mr. P enrolls in a methadone maintenance treatment program (MMTP) that includes primary medical care and addiction medicine and psychiatric specialists, where he undergoes medical evaluation and screening for hepatitis C virus (HCV) and human immunodeficiency virus (HIV). Laboratory data reveal that although Mr. P is HIV negative, he has been exposed to HCV and treatment is indicated.
Among the approximately 3 million people in the United States with chronic HCV—an enveloped, single-stranded RNA virus—there’s a high prevalence of premorbid psychopathology and substance abuse, as well as neuropsychiatric effects caused by HCV treatment.1-3 Because underdiagnosing and undertreating psychiatric disorders contributes to morbidity and mortality in HCV patients, early identification and prompt treatment is critical.
IV drug use is the most common route for HCV infection, accounting for 65% to 70% of infections.1 The prevalence of HCV among IV drug users is 28% to 90%.1 Once exposed to HCV, 75% to 85% of patients do not clear the initial infection and become chronically infected.
This article reviews the pathophysiology, identification, and management of psychiatric manifestations found among HCV patients and provides an understanding of how psychiatric symptoms manifest in HCV patients. This article also discusses HCV treatment and its neuropsychiatric side effects.
Testing for HCV
Chronically infected HCV patients may have few, if any, specific physical complaints, and often are diagnosed during screenings or other routine laboratory evaluations. The presence of risk factors, such as a history of injection drug use or receiving a blood transfusion before 1992,1 guides the decision to screen for HCV. Normal liver function test results should not preclude testing because many HCV-positive patients have transaminases within the normal range.4 Initial screening is via an antibody-mediated immunoassay that is highly specific and sensitive for past exposure to HCV (Table 1).4 However, a positive screen does not indicate the presence of active infection. Evidence of the virus via a viral assay will identify active HCV, but does not indicate need for treatment. Liver biopsy confirms the presence of liver injury and quantifies its extent. The severity of liver damage will determine whether treatment is needed. HCV genotyping determines the appropriate duration and dosage of pharmacotherapy.
Table 1
Tests to diagnose and evaluate HCV
| Test | Results |
|---|---|
| HCV antibody | Determines prior exposure to HCV |
| HCV viral assay | Evaluates for current HCV infection |
| Liver biopsy | Assesses level of liver damage |
| HCV genotyping | Provides data to determine duration and intensity of treatment and likelihood of treatment success |
| HCV: hepatitis C virus Source: Reference 4 | |
CASE CONTINUED: Mood improves, but fatigue persists
As part of pre-HCV treatment evaluation, Mr. P undergoes a psychiatric evaluation. He describes periods of low mood while actively engaged in drug use but has never received psychiatric treatment, experienced suicidal ideation, or attempted suicide. Since starting opioid agonist therapy, he reports improved mood but endorses continued mild fatigue and difficulty falling sleep. The psychiatrist determines Mr. P does not meet criteria for an axis I diagnosis other than a substance use disorder.
Although most HCV patients have few, if any, nonspecific physical symptoms, many have psychiatric symptoms or disorders before the HCV diagnosis is made or treatment is initiated; substance use disorders are most common. Batki et al1 found that 56% of HCV patients in an MMTP met criteria for a nonsubstance axis I disorder and 82% met criteria for such a disorder during their lifetime. Additionally, 66% of patients were taking psychiatric medications. Table 21,5,6 lists the rates of other psychiatric disorders found in patients with untreated HCV.
Table 2
Rates of psychiatric disorders in patients with untreated hepatitis C virus
| Disorder(s) | Current rate | Lifetime rate |
|---|---|---|
| Mood disorders | 34% to 35% | 67% |
| Major depressive disorder | 22% to 28% | 42% |
| Anxiety disorders | 26% to 44% | 63% |
| Antisocial personality disorder | No rates; lifetime diagnosis | 16% to 40% |
| Psychotic disorders | 9% to 17% | 11% |
| Substance use disorder | 56% | 56% to 86% |
| Source: References 1,5,6 | ||
Many patients with chronic HCV complain of chronic fatigue and deficiencies in attention, concentration, higher executive functions, learning ability, and memory that result in significant reduction in quality of life (Box 1).7-9 These findings have been found to be independent of the degree of liver disease and are seen in HCV patients with normal liver function.7,8
The pathophysiology of fatigue and neurocognitive dysfunction in hepatitis C virus (HCV) infection is unclear. However, the improvement of chronic fatigue in patients with HCV who receive ondansetron, a 5-hydroxytryptophan-3 receptor antagonist, has implicated abnormal monoaminergic function. Single-photon emission CT studies have found decreased midbrain serotonergic and striatal dopaminergic transmission in some HCV patients with cognitive deficits.7
Recently, data have been mounting on a direct neuropathic effect of HCV, with viral elements found in autopsy brain tissue and cerebrospinal fluid.8 Researchers have suggested that HCV may enter the CNS via a Trojan horse-like mechanism inside infected mononuclear cells.8 More recently, human brain microvascular endothelium, the major component of the blood-brain barrier, has been found to express all major viral receptors that would allow HCV infection of the CNS.9
CASE CONTINUED: Motivated and compliant
Since joining the MMTP 6 months ago, Mr. P has been motivated and compliant with all appointments and treatments. Routine urine toxicology screening supports his claim of abstinence. Mr. P begins HCV treatment while continuing follow-up with addiction medicine and psychiatric clinicians and maintains open communication with all treatment providers.
For many years the standard HCV treatment was pegylated interferon-α (IFN-α) and ribavirin. IFN-α is a proinflammatory cytokine with antiproliferative, antiviral, and immunoregulatory properties. The half-life of IFN-α significantly increases with pegylation, which allows for weekly injections.10,11 IFN-α usually is combined with ribavirin, which increases its efficacy as measured by the sustained virological response (SVR) compared with IFN-α alone. Depending on the virus genotype, treatment lasts 24 to 48 weeks; SVR rates range from 40% to 82%.11-13 In 2011, the FDA approved 2 agents—telaprevir and boceprevir—for adjunctive treatment of HCV genotype 1 infection. These 2 agents are protease inhibitors that when added to IFN-α and ribavirin increase the SVR rate in genotype 1 infection from 40% to 50% to approximately 75%.14,15
Although the neuropsychiatric side effects of telaprevir and boceprevir have not been determined, treating chronic HCV with IFN-α and ribavirin has been associated with multiple psychiatric symptoms, including depression, mania, suicidality, anxiety, and psychosis.11-14 Psychiatric symptoms are a common reason for discontinuing or reducing HCV treatment. Because of the high frequency of neuropsychiatric complications, some clinicians believe HCV patients with preexisting affective, psychotic, or substance use disorders should be excluded from HCV treatment. This has led to many HCV patients being untreated despite a lack of prospective, controlled data to support this opinion.12 To improve outcomes and decrease morbidity, providing appropriate psychiatric services appears to be more important than attempting to select lower-risk patients for antiviral therapy.1,12,16 The goals of psychiatric treatment should be to alleviate symptoms and allow patients to complete IFN-α therapy without interruption.16,17
Studies of high-risk patients who attend multidisciplinary treatment programs that can monitor adherence and efficacy and control side effects before and during HCV treatment have found psychiatric patients have similar adherence, compliance, and SVR rates and were not at increased risk of worsening depressive or psychotic symptoms compared with patients without a psychiatric history.12,18 Additionally, HCV patients with a psychiatric history are not at an increased risk of suicide.13,16 Similar findings have been observed in patients with active IV drug use or those receiving opioid agonist therapy. When HCV and substance use are treated simultaneously, patients can successfully complete HCV treatment with SVR rates comparable to those of patients not receiving opioid agonist therapy.19-21
CASE CONTINUED: Worsening symptoms
During a psychiatric follow-up 12 weeks after starting HCV treatment, Mr. P reports worsening depressive symptoms with low mood, decreased enjoyment of activities, poor sleep, low appetite, and fatigue. He shows no evidence of psychosis and denies suicidal ideation. We continue his HCV treatment, schedule more frequent psychiatric visits, and initiate citalopram, titrated to 40 mg/d.
Depressive symptoms, the most common neuropsychiatric manifestation of HCV, typically begin early in treatment, usually within the first 12 weeks. Two distinct symptom clusters are noted. A neurovegetative cluster characterized by reduced energy, anorexia, and psychomotor retardation typically begins within the first few months of treatment. Months later, a depression-specific syndrome appears that includes depressed mood, anxiety, and cognitive impairment.22
Depressive symptoms may occur in up to 60% of patients treated with IFN-α.11 When more rigorous depression measures are used, rates decrease to approximately 20% to 30%.11,13 Accurate diagnosis and treatment of emerging depressive symptoms is essential because untreated depression can lead to postponing or excluding patients from antiviral treatment.2 Screening instruments such as the Beck Depression Inventory-Second Edition (BDI-II) can be used to measure depressive symptoms in HCV patients with high sensitivity. However, because specificity has been low and somatic symptoms of chronic illness and depression often overlap, the BDI-II and other inventories may overestimate depression. Some researchers have suggested that focusing on questions targeting cognitive and affective symptoms rather than somatic ones may be a more valid measure of depression in patients undergoing immunotherapy for HCV.2
The immune system is implicated in IFN-α-induced depression because depressive symptoms share many features with a constellation of somatic and behavioral symptoms termed “sickness behavior.”11 These behaviors can occur when patients are exposed to cytokines that lead to a depressed level of functioning, which may allow the body to devote more energy to fighting illness. IFN-α, a cytokine, stimulates the immune system, which can lead to increases of interleukin (IL)-2, IL-6, and IL-10. Increased circulating levels of these ILs have been correlated with higher depression scores. Additionally, studies have found that patients who develop depression during IFN-α treatment have higher SVR rates, suggesting a more robust immune response.11,22 For a discussion of how serotonin metabolism and genetic polymorphisms also may help explain the prevalence of depression in patients with HCV, see Box 2.
Altered serotonin metabolism has been linked to depression in hepatitis C virus (HCV) patients treated with interferon-α (IFN-α). Tryptophan can be metabolized towards serotonin via tryptophan hydroxylase and niacin via indoleamine-2,3-dioxygenase (IDO) with kynurenine (KYN) and quinolinic acid (QUIN) as intermediaries. Introduction of IFN-α activates IDO, causing preferential conversion of tryptophan towards the niacin arm away from serotonin and leads to elevated levels of KYN and QUIN. KYN and QUIN are available centrally, are neurotoxic, and have been correlated with increased depressive symptoms in IFN-α-treated patients.a,b A tryptophan-deficient state is created, with less tryptophan being converted to serotonin and subsequently to its metabolite, 5-hydroxyindoleacetic acid (5-HIAA). Decreased levels of 5-HIAA in cerebrospinal fluid have been associated with higher depressive symptoms and higher rates of suicide.a,b
Several genetic polymorphisms may help identify patients at risk for developing IFN-α-induced depression. Genes for the 5’ promoter of the serotonin transporter (5-HTTLPR) have been investigated for roles in depression development in patients undergoing immunotherapy. Studies have found that persons with the short allele in the 5-HTTLPR gene are more likely to develop depression than those with the long allele. However, this has not been consistent across racial or ethnic groups.a,b Research also has associated the serotonin (5-HT) transporter, interferon receptor-A1, apolipoprotein ε4 allele, cyclooxygenase 2, and phospholipase A2 with development of a specific subgroup of symptoms.a
References
a. Smith KJ, Norris S, O’Farrelly C, et al. Risk factors for the development of depression in patients with hepatitis C taking interferon-α. Neuropsychiatr Dis Treat. 2011;7:275-292.
b. Sockalingam S, Links PS, Abbey SE. Suicide risk in hepatitis C and during interferon-alpha therapy: a review and clinical update. J Viral Hepat. 2011;18(3):153-160.
Treating depressed HCV patients
Antidepressants are the treatment of choice for IFN-α-induced depression. Most currently used antidepressants are effective22 and selective serotonin reuptake inhibitors are considered first choice.16 Antidepressant choice should be guided by principles similar to those used for patients without HCV: using side effects profiles to target specific symptoms and being mindful of pharmacokinetic properties.
Two treatment approaches have been investigated: prophylactic and symptomatic. A 2012 study23 of 181 HCV patients with no history of mental illness determined escitalopram, 10 mg/d, effectively reduced the incidence and severity of interferon-associated depression. Other studies examining prophylactic treatment of all patients who were to undergo interferon treatment found this approach did not prevent depressive episodes.24,25 However, antidepressants have been beneficial for patients with subsyndromal depressive symptoms at baseline26 and after clinically significant depressive symptoms emerge.27 Electroconvulsive therapy also has been reported to effectively treat depression in HCV patients undergoing antiviral therapy.28
CASE CONTINUED: Lingering symptoms
Mr. P responds to citalopram with an improvement in mood, anhedonia, and appetite, but he continues to complain of low energy and poor concentration. In an effort to target these symptoms, methylphenidate, titrated to 30 mg/d in divided doses, is added to his regimen, which rapidly improves his symptoms. Insomnia is treated successfully with trazodone, 50 mg/d. Mr. P frequently visits his psychiatrist, who monitors his depressive symptoms using the BDI-II. Mr. P completes HCV treatment without recurrence of depressive symptoms or relapse to heroin use.
Although antidepressants are effective for treating affective and cognitive symptoms, they are not as effective for fatigue and other neurovegetative symptoms.16,29 The psychostimulants methylphenidate and dextroamphetamine and the nonstimulant modafinil have been studied for treating depressive symptoms in medically ill patients and can be used to treat IFN-α-induced fatigue.16,22,29
IFN-α’s effect on serotonin metabolism leads to a tryptophan-deficient state because of increased catabolism as a result of activation of indoleamine-2,3-dioxygenase (IDO). This has led to use of tryptophan supplementation, either as augmentation or monotherapy, for managing depressive symptoms in patients treated with IFN-α. Schaefer et al30 reported 3 cases where tryptophan supplementation significantly decreased depressive symptoms. Other researchers have argued that supplementing tryptophan in the context of IDO activation can lead to greater production of kynurenine and quinolinic acid, which have been linked to increased depressive symptoms in patients receiving IFN-α.31 They argue that supplementation of 5-HTP, which is available as a dietary supplement without a prescription, can lead to increased serotonin levels and improvement in depressive symptoms.31
IFN-α treatment also is associated with mania and psychosis. The incidence, pathophysiology, and management of these treatment-emergent symptoms are not as well studied as IFN-α-induced depression. Mania and hypomania have been reported with interferon treatment, discontinuation of interferon, and use of antidepressants for interferon-induced depression.29,32 Psychosis, in association with mood symptoms or alone, has been reported to occur in <1% of treated patients.33 Treatment for mania and psychosis consists of decreasing or discontinuing immunotherapy and adding mood stabilizers and antipsychotics. Once immunotherapy is discontinued, mania and psychosis usually resolve, but prolonged duration of symptoms has been reported.29,32,33
Related Resources
- Centers for Disease Control and Prevention. Hepatitis C information for health professionals. www.cdc.gov/hepatitis/hcv/index.htm.
- Hep C Connection. www.hepc-connection.org.
- United States Department of Veterans Affairs. Hepatitis C. www.hepatitis.va.gov.
Drug Brand Names
- Boceprevir • Victrelis
- Citalopram • Celexa
- Dextroamphetamine • Dexedrine
- Escitalopram • Lexapro
- Interferon-α • Intron
- Methadone • Dolophine, Methadose
- Methylphenidate • Ritalin, Methylin, others
- Modafinil • Provigil
- Ondansetron • Zofran
- Ribavirin • Copegus, Rebetol, others
- Telaprevir • Incivek
- Trazodone • Desyrel, Oleptro
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Mr. P, age 31, has been using heroin intravenously for 9 years. He smokes 1 pack of cigarettes daily, but denies using other substances, including alcohol. After an unintentional heroin overdose, Mr. P enrolls in a methadone maintenance treatment program (MMTP) that includes primary medical care and addiction medicine and psychiatric specialists, where he undergoes medical evaluation and screening for hepatitis C virus (HCV) and human immunodeficiency virus (HIV). Laboratory data reveal that although Mr. P is HIV negative, he has been exposed to HCV and treatment is indicated.
Among the approximately 3 million people in the United States with chronic HCV—an enveloped, single-stranded RNA virus—there’s a high prevalence of premorbid psychopathology and substance abuse, as well as neuropsychiatric effects caused by HCV treatment.1-3 Because underdiagnosing and undertreating psychiatric disorders contributes to morbidity and mortality in HCV patients, early identification and prompt treatment is critical.
IV drug use is the most common route for HCV infection, accounting for 65% to 70% of infections.1 The prevalence of HCV among IV drug users is 28% to 90%.1 Once exposed to HCV, 75% to 85% of patients do not clear the initial infection and become chronically infected.
This article reviews the pathophysiology, identification, and management of psychiatric manifestations found among HCV patients and provides an understanding of how psychiatric symptoms manifest in HCV patients. This article also discusses HCV treatment and its neuropsychiatric side effects.
Testing for HCV
Chronically infected HCV patients may have few, if any, specific physical complaints, and often are diagnosed during screenings or other routine laboratory evaluations. The presence of risk factors, such as a history of injection drug use or receiving a blood transfusion before 1992,1 guides the decision to screen for HCV. Normal liver function test results should not preclude testing because many HCV-positive patients have transaminases within the normal range.4 Initial screening is via an antibody-mediated immunoassay that is highly specific and sensitive for past exposure to HCV (Table 1).4 However, a positive screen does not indicate the presence of active infection. Evidence of the virus via a viral assay will identify active HCV, but does not indicate need for treatment. Liver biopsy confirms the presence of liver injury and quantifies its extent. The severity of liver damage will determine whether treatment is needed. HCV genotyping determines the appropriate duration and dosage of pharmacotherapy.
Table 1
Tests to diagnose and evaluate HCV
| Test | Results |
|---|---|
| HCV antibody | Determines prior exposure to HCV |
| HCV viral assay | Evaluates for current HCV infection |
| Liver biopsy | Assesses level of liver damage |
| HCV genotyping | Provides data to determine duration and intensity of treatment and likelihood of treatment success |
| HCV: hepatitis C virus Source: Reference 4 | |
CASE CONTINUED: Mood improves, but fatigue persists
As part of pre-HCV treatment evaluation, Mr. P undergoes a psychiatric evaluation. He describes periods of low mood while actively engaged in drug use but has never received psychiatric treatment, experienced suicidal ideation, or attempted suicide. Since starting opioid agonist therapy, he reports improved mood but endorses continued mild fatigue and difficulty falling sleep. The psychiatrist determines Mr. P does not meet criteria for an axis I diagnosis other than a substance use disorder.
Although most HCV patients have few, if any, nonspecific physical symptoms, many have psychiatric symptoms or disorders before the HCV diagnosis is made or treatment is initiated; substance use disorders are most common. Batki et al1 found that 56% of HCV patients in an MMTP met criteria for a nonsubstance axis I disorder and 82% met criteria for such a disorder during their lifetime. Additionally, 66% of patients were taking psychiatric medications. Table 21,5,6 lists the rates of other psychiatric disorders found in patients with untreated HCV.
Table 2
Rates of psychiatric disorders in patients with untreated hepatitis C virus
| Disorder(s) | Current rate | Lifetime rate |
|---|---|---|
| Mood disorders | 34% to 35% | 67% |
| Major depressive disorder | 22% to 28% | 42% |
| Anxiety disorders | 26% to 44% | 63% |
| Antisocial personality disorder | No rates; lifetime diagnosis | 16% to 40% |
| Psychotic disorders | 9% to 17% | 11% |
| Substance use disorder | 56% | 56% to 86% |
| Source: References 1,5,6 | ||
Many patients with chronic HCV complain of chronic fatigue and deficiencies in attention, concentration, higher executive functions, learning ability, and memory that result in significant reduction in quality of life (Box 1).7-9 These findings have been found to be independent of the degree of liver disease and are seen in HCV patients with normal liver function.7,8
The pathophysiology of fatigue and neurocognitive dysfunction in hepatitis C virus (HCV) infection is unclear. However, the improvement of chronic fatigue in patients with HCV who receive ondansetron, a 5-hydroxytryptophan-3 receptor antagonist, has implicated abnormal monoaminergic function. Single-photon emission CT studies have found decreased midbrain serotonergic and striatal dopaminergic transmission in some HCV patients with cognitive deficits.7
Recently, data have been mounting on a direct neuropathic effect of HCV, with viral elements found in autopsy brain tissue and cerebrospinal fluid.8 Researchers have suggested that HCV may enter the CNS via a Trojan horse-like mechanism inside infected mononuclear cells.8 More recently, human brain microvascular endothelium, the major component of the blood-brain barrier, has been found to express all major viral receptors that would allow HCV infection of the CNS.9
CASE CONTINUED: Motivated and compliant
Since joining the MMTP 6 months ago, Mr. P has been motivated and compliant with all appointments and treatments. Routine urine toxicology screening supports his claim of abstinence. Mr. P begins HCV treatment while continuing follow-up with addiction medicine and psychiatric clinicians and maintains open communication with all treatment providers.
For many years the standard HCV treatment was pegylated interferon-α (IFN-α) and ribavirin. IFN-α is a proinflammatory cytokine with antiproliferative, antiviral, and immunoregulatory properties. The half-life of IFN-α significantly increases with pegylation, which allows for weekly injections.10,11 IFN-α usually is combined with ribavirin, which increases its efficacy as measured by the sustained virological response (SVR) compared with IFN-α alone. Depending on the virus genotype, treatment lasts 24 to 48 weeks; SVR rates range from 40% to 82%.11-13 In 2011, the FDA approved 2 agents—telaprevir and boceprevir—for adjunctive treatment of HCV genotype 1 infection. These 2 agents are protease inhibitors that when added to IFN-α and ribavirin increase the SVR rate in genotype 1 infection from 40% to 50% to approximately 75%.14,15
Although the neuropsychiatric side effects of telaprevir and boceprevir have not been determined, treating chronic HCV with IFN-α and ribavirin has been associated with multiple psychiatric symptoms, including depression, mania, suicidality, anxiety, and psychosis.11-14 Psychiatric symptoms are a common reason for discontinuing or reducing HCV treatment. Because of the high frequency of neuropsychiatric complications, some clinicians believe HCV patients with preexisting affective, psychotic, or substance use disorders should be excluded from HCV treatment. This has led to many HCV patients being untreated despite a lack of prospective, controlled data to support this opinion.12 To improve outcomes and decrease morbidity, providing appropriate psychiatric services appears to be more important than attempting to select lower-risk patients for antiviral therapy.1,12,16 The goals of psychiatric treatment should be to alleviate symptoms and allow patients to complete IFN-α therapy without interruption.16,17
Studies of high-risk patients who attend multidisciplinary treatment programs that can monitor adherence and efficacy and control side effects before and during HCV treatment have found psychiatric patients have similar adherence, compliance, and SVR rates and were not at increased risk of worsening depressive or psychotic symptoms compared with patients without a psychiatric history.12,18 Additionally, HCV patients with a psychiatric history are not at an increased risk of suicide.13,16 Similar findings have been observed in patients with active IV drug use or those receiving opioid agonist therapy. When HCV and substance use are treated simultaneously, patients can successfully complete HCV treatment with SVR rates comparable to those of patients not receiving opioid agonist therapy.19-21
CASE CONTINUED: Worsening symptoms
During a psychiatric follow-up 12 weeks after starting HCV treatment, Mr. P reports worsening depressive symptoms with low mood, decreased enjoyment of activities, poor sleep, low appetite, and fatigue. He shows no evidence of psychosis and denies suicidal ideation. We continue his HCV treatment, schedule more frequent psychiatric visits, and initiate citalopram, titrated to 40 mg/d.
Depressive symptoms, the most common neuropsychiatric manifestation of HCV, typically begin early in treatment, usually within the first 12 weeks. Two distinct symptom clusters are noted. A neurovegetative cluster characterized by reduced energy, anorexia, and psychomotor retardation typically begins within the first few months of treatment. Months later, a depression-specific syndrome appears that includes depressed mood, anxiety, and cognitive impairment.22
Depressive symptoms may occur in up to 60% of patients treated with IFN-α.11 When more rigorous depression measures are used, rates decrease to approximately 20% to 30%.11,13 Accurate diagnosis and treatment of emerging depressive symptoms is essential because untreated depression can lead to postponing or excluding patients from antiviral treatment.2 Screening instruments such as the Beck Depression Inventory-Second Edition (BDI-II) can be used to measure depressive symptoms in HCV patients with high sensitivity. However, because specificity has been low and somatic symptoms of chronic illness and depression often overlap, the BDI-II and other inventories may overestimate depression. Some researchers have suggested that focusing on questions targeting cognitive and affective symptoms rather than somatic ones may be a more valid measure of depression in patients undergoing immunotherapy for HCV.2
The immune system is implicated in IFN-α-induced depression because depressive symptoms share many features with a constellation of somatic and behavioral symptoms termed “sickness behavior.”11 These behaviors can occur when patients are exposed to cytokines that lead to a depressed level of functioning, which may allow the body to devote more energy to fighting illness. IFN-α, a cytokine, stimulates the immune system, which can lead to increases of interleukin (IL)-2, IL-6, and IL-10. Increased circulating levels of these ILs have been correlated with higher depression scores. Additionally, studies have found that patients who develop depression during IFN-α treatment have higher SVR rates, suggesting a more robust immune response.11,22 For a discussion of how serotonin metabolism and genetic polymorphisms also may help explain the prevalence of depression in patients with HCV, see Box 2.
Altered serotonin metabolism has been linked to depression in hepatitis C virus (HCV) patients treated with interferon-α (IFN-α). Tryptophan can be metabolized towards serotonin via tryptophan hydroxylase and niacin via indoleamine-2,3-dioxygenase (IDO) with kynurenine (KYN) and quinolinic acid (QUIN) as intermediaries. Introduction of IFN-α activates IDO, causing preferential conversion of tryptophan towards the niacin arm away from serotonin and leads to elevated levels of KYN and QUIN. KYN and QUIN are available centrally, are neurotoxic, and have been correlated with increased depressive symptoms in IFN-α-treated patients.a,b A tryptophan-deficient state is created, with less tryptophan being converted to serotonin and subsequently to its metabolite, 5-hydroxyindoleacetic acid (5-HIAA). Decreased levels of 5-HIAA in cerebrospinal fluid have been associated with higher depressive symptoms and higher rates of suicide.a,b
Several genetic polymorphisms may help identify patients at risk for developing IFN-α-induced depression. Genes for the 5’ promoter of the serotonin transporter (5-HTTLPR) have been investigated for roles in depression development in patients undergoing immunotherapy. Studies have found that persons with the short allele in the 5-HTTLPR gene are more likely to develop depression than those with the long allele. However, this has not been consistent across racial or ethnic groups.a,b Research also has associated the serotonin (5-HT) transporter, interferon receptor-A1, apolipoprotein ε4 allele, cyclooxygenase 2, and phospholipase A2 with development of a specific subgroup of symptoms.a
References
a. Smith KJ, Norris S, O’Farrelly C, et al. Risk factors for the development of depression in patients with hepatitis C taking interferon-α. Neuropsychiatr Dis Treat. 2011;7:275-292.
b. Sockalingam S, Links PS, Abbey SE. Suicide risk in hepatitis C and during interferon-alpha therapy: a review and clinical update. J Viral Hepat. 2011;18(3):153-160.
Treating depressed HCV patients
Antidepressants are the treatment of choice for IFN-α-induced depression. Most currently used antidepressants are effective22 and selective serotonin reuptake inhibitors are considered first choice.16 Antidepressant choice should be guided by principles similar to those used for patients without HCV: using side effects profiles to target specific symptoms and being mindful of pharmacokinetic properties.
Two treatment approaches have been investigated: prophylactic and symptomatic. A 2012 study23 of 181 HCV patients with no history of mental illness determined escitalopram, 10 mg/d, effectively reduced the incidence and severity of interferon-associated depression. Other studies examining prophylactic treatment of all patients who were to undergo interferon treatment found this approach did not prevent depressive episodes.24,25 However, antidepressants have been beneficial for patients with subsyndromal depressive symptoms at baseline26 and after clinically significant depressive symptoms emerge.27 Electroconvulsive therapy also has been reported to effectively treat depression in HCV patients undergoing antiviral therapy.28
CASE CONTINUED: Lingering symptoms
Mr. P responds to citalopram with an improvement in mood, anhedonia, and appetite, but he continues to complain of low energy and poor concentration. In an effort to target these symptoms, methylphenidate, titrated to 30 mg/d in divided doses, is added to his regimen, which rapidly improves his symptoms. Insomnia is treated successfully with trazodone, 50 mg/d. Mr. P frequently visits his psychiatrist, who monitors his depressive symptoms using the BDI-II. Mr. P completes HCV treatment without recurrence of depressive symptoms or relapse to heroin use.
Although antidepressants are effective for treating affective and cognitive symptoms, they are not as effective for fatigue and other neurovegetative symptoms.16,29 The psychostimulants methylphenidate and dextroamphetamine and the nonstimulant modafinil have been studied for treating depressive symptoms in medically ill patients and can be used to treat IFN-α-induced fatigue.16,22,29
IFN-α’s effect on serotonin metabolism leads to a tryptophan-deficient state because of increased catabolism as a result of activation of indoleamine-2,3-dioxygenase (IDO). This has led to use of tryptophan supplementation, either as augmentation or monotherapy, for managing depressive symptoms in patients treated with IFN-α. Schaefer et al30 reported 3 cases where tryptophan supplementation significantly decreased depressive symptoms. Other researchers have argued that supplementing tryptophan in the context of IDO activation can lead to greater production of kynurenine and quinolinic acid, which have been linked to increased depressive symptoms in patients receiving IFN-α.31 They argue that supplementation of 5-HTP, which is available as a dietary supplement without a prescription, can lead to increased serotonin levels and improvement in depressive symptoms.31
IFN-α treatment also is associated with mania and psychosis. The incidence, pathophysiology, and management of these treatment-emergent symptoms are not as well studied as IFN-α-induced depression. Mania and hypomania have been reported with interferon treatment, discontinuation of interferon, and use of antidepressants for interferon-induced depression.29,32 Psychosis, in association with mood symptoms or alone, has been reported to occur in <1% of treated patients.33 Treatment for mania and psychosis consists of decreasing or discontinuing immunotherapy and adding mood stabilizers and antipsychotics. Once immunotherapy is discontinued, mania and psychosis usually resolve, but prolonged duration of symptoms has been reported.29,32,33
Related Resources
- Centers for Disease Control and Prevention. Hepatitis C information for health professionals. www.cdc.gov/hepatitis/hcv/index.htm.
- Hep C Connection. www.hepc-connection.org.
- United States Department of Veterans Affairs. Hepatitis C. www.hepatitis.va.gov.
Drug Brand Names
- Boceprevir • Victrelis
- Citalopram • Celexa
- Dextroamphetamine • Dexedrine
- Escitalopram • Lexapro
- Interferon-α • Intron
- Methadone • Dolophine, Methadose
- Methylphenidate • Ritalin, Methylin, others
- Modafinil • Provigil
- Ondansetron • Zofran
- Ribavirin • Copegus, Rebetol, others
- Telaprevir • Incivek
- Trazodone • Desyrel, Oleptro
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Batki SL, Canfield KM, Ploutz-Snyder R. Psychiatric and substance use disorders among methadone maintenance patients with chronic hepatitis C infection: effects on eligibility for hepatitis C treatment. Am J Addict. 2011;20(4):312-318.
2. Patterson AL, Morasco BJ, Fuller BE, et al. Screening for depression in patients with hepatitis C using the Beck Depression Inventory-II: do somatic symptoms compromise validity? Gen Hosp Psychiatry. 2011;33(4):354-362.
3. Maddur H, Kwo PY. Boceprevir. Hepatology. 2011;54(6):2254-2257.
4. Sylvestre D. Hepatitis C for addiction professionals. Addict Sci Clin Pract. 2007;4(1):34-41.
5. Dwight MM, Kowdley KV, Russo JE, et al. Depression, fatigue, and functional disability in patients with chronic hepatitis C. J Psychosom Res. 2000;49(5):311-317.
6. Yovtcheva SP, Rifai MA, Moles JK, et al. Psychiatric comorbidity among hepatitis C-positive patients. Psychosomatics. 2001;42(5):411-415.
7. Weissenborn K, Ennen JC, Bokemeyer M, et al. Monoaminergic neurotransmission is altered in hepatitis C virus infected patients with chronic fatigue and cognitive impairment. Gut. 2006;55(11):1624-1630.
8. Weissenborn K, Tryc AB, Heeren M, et al. Hepatitis C virus infection and the brain. Metab Brain Dis. 2009;24(1):197-210.
9. Fletcher NF, Wilson GK, Murray J, et al. Hepatitis C virus infects the endothelial cells of the blood-brain barrier. Gastroenterology. 2012;142(3):634-643.e6.
10. Pawlotsky JM. Therapy of hepatitis C: from empiricism to eradication. Hepatology. 2006;43(2 suppl 1):S207-S220.
11. Smith KJ, Norris S, O’Farrelly C, et al. Risk factors for the development of depression in patients with hepatitis C taking interferon-α. Neuropsychiatr Dis Treat. 2011;7:275-292.
12. Schaefer M, Hinzpeter A, Mohmand A, et al. Hepatitis C treatment in “difficult-to-treat” psychiatric patients with pegylated interferon-alpha and ribavirin: response and psychiatric side effects. Hepatology. 2007;46(4):991-998.
13. Sockalingam S, Links PS, Abbey SE. Suicide risk in hepatitis C and during interferon-alpha therapy: a review and clinical update. J Viral Hepat. 2011;18(3):153-160.
14. Telaprevir (Incivek) and boceprevir (Victrelis) for chronic hepatitis C. Med Lett Drugs Ther. 2011;53(1369):57-59.
15. Nelson DR. The role of triple therapy with protease inhibitors in hepatitis C virus genotype 1 naïve patients. Liver Int. 2011;31(suppl 1):53-57.
16. Spennati A, Pariante CM. Withdrawing interferon-α from psychiatric patients: clinical care or unjustifiable stigma? [published online September 14 2012] Psychol Med. doi: 10. 1017/S0033291712001808.
17. Baraldi S, Hepgul N, Mondelli V, et al. Symptomatic treatment of interferon-α-induced depression in hepatitis C: a systematic review. J Clin Psychopharmacol. 2012;32(4):531-543.
18. Schaefer M, Schmidt F, Folwaczny C, et al. Adherence and mental side effects during hepatitis C treatment with interferon alfa and ribavirin in psychiatric risk groups. Hepatology. 2003;37(2):443-451.
19. Harris KA, Jr, Arnsten JH, Litwin AH. Successful integration of hepatitis C evaluation and treatment services with methadone maintenance. J Addict Med. 2010;4(1):20-26.
20. Litwin AH, Harris KA, Jr, Nahvi S, et al. Successful treatment of chronic hepatitis C with pegylated interferon in combination with ribavirin in a methadone maintenance treatment program. J Subst Abuse Treat. 2009;37(1):32-40.
21. Sasadeusz JJ, Dore G, Kronborg I, et al. Clinical experience with the treatment of hepatitis C infection in patients on opioid pharmacotherapy. Addiction. 2011;106(5):977-984.
22. Sockalingam S, Abbey SE. Managing depression during hepatitis C treatment. Can J Psychiatry. 2009;54(9):614-625.
23. Schaefer M, Sarkar R, Knop V, et al. Escitalopram for the prevention of peginterferon-α2a-associated depression in hepatitis C virus-infected patients without previous psychiatric disease: a randomized trial. Ann Intern Med. 2012;157(2):94-103.
24. Galvão-de Almeida A, Guindalini C, Batista-Neves S, et al. Can antidepressants prevent interferon-alpha-induced depression? A review of the literature. Gen Hosp Psychiatry. 2010;32(4):401-405.
25. Morasco BJ, Loftis JM, Indest DW, et al. Prophylactic antidepressant treatment in patients with hepatitis C on antiviral therapy: a double-blind, placebo-controlled trial. Psychosomatics. 2010;51(5):401-408.
26. Raison CL, Woolwine BJ, Demetrashvili MF, et al. Paroxetine for prevention of depressive symptoms induced by interferon-alpha and ribavirin for hepatitis C. Aliment Pharmacol Ther. 2007;25(10):1163-1174.
27. Kraus MR, Schäfer A, Schöttker K, et al. Therapy of interferon-induced depression in chronic hepatitis C with citalopram: a randomised, double-blind, placebo-controlled study. Gut. 2008;57(4):531-536.
28. Zincke MT, Kurani A, Istafanous R, et al. The successful use of electroconvulsive therapy in a patient with interferon-induced psychotic depression. J ECT. 2007;23(4):291-292.
29. Crone CC, Gabriel GM, Wise TN. Managing the neuropsychiatric side effects of interferon-based therapy for hepatitis C. Cleve Clin J Med. 2004;71(suppl 3):S27-S32.
30. Schaefer M, Winterer J, Sarkar R, et al. Three cases of successful tryptophan add-on or monotherapy of hepatitis C and IFNa-associated mood disorders. Psychosomatics. 2008;49(5):442-446.
31. Turner EH, Blackwell AD. 5-Hydroxytryptophan plus SSRIs for interferon-induced depression: synergistic mechanisms for normalizing synaptic serotonin. Med Hypotheses. 2005;65(1):138-144.
32. Onyike CU, Bonner JO, Lyketsos CG, et al. Mania during treatment of chronic hepatitis C with pegylated interferon and ribavirin. Am J Psychiatry. 2004;161(3):429-435.
33. Cheng YC, Chen CC, Ho AS, et al. Prolonged psychosis associated with interferon therapy in a patient with hepatitis C: case study and literature review. Psychosomatics. 2009;50(5):538-542.
1. Batki SL, Canfield KM, Ploutz-Snyder R. Psychiatric and substance use disorders among methadone maintenance patients with chronic hepatitis C infection: effects on eligibility for hepatitis C treatment. Am J Addict. 2011;20(4):312-318.
2. Patterson AL, Morasco BJ, Fuller BE, et al. Screening for depression in patients with hepatitis C using the Beck Depression Inventory-II: do somatic symptoms compromise validity? Gen Hosp Psychiatry. 2011;33(4):354-362.
3. Maddur H, Kwo PY. Boceprevir. Hepatology. 2011;54(6):2254-2257.
4. Sylvestre D. Hepatitis C for addiction professionals. Addict Sci Clin Pract. 2007;4(1):34-41.
5. Dwight MM, Kowdley KV, Russo JE, et al. Depression, fatigue, and functional disability in patients with chronic hepatitis C. J Psychosom Res. 2000;49(5):311-317.
6. Yovtcheva SP, Rifai MA, Moles JK, et al. Psychiatric comorbidity among hepatitis C-positive patients. Psychosomatics. 2001;42(5):411-415.
7. Weissenborn K, Ennen JC, Bokemeyer M, et al. Monoaminergic neurotransmission is altered in hepatitis C virus infected patients with chronic fatigue and cognitive impairment. Gut. 2006;55(11):1624-1630.
8. Weissenborn K, Tryc AB, Heeren M, et al. Hepatitis C virus infection and the brain. Metab Brain Dis. 2009;24(1):197-210.
9. Fletcher NF, Wilson GK, Murray J, et al. Hepatitis C virus infects the endothelial cells of the blood-brain barrier. Gastroenterology. 2012;142(3):634-643.e6.
10. Pawlotsky JM. Therapy of hepatitis C: from empiricism to eradication. Hepatology. 2006;43(2 suppl 1):S207-S220.
11. Smith KJ, Norris S, O’Farrelly C, et al. Risk factors for the development of depression in patients with hepatitis C taking interferon-α. Neuropsychiatr Dis Treat. 2011;7:275-292.
12. Schaefer M, Hinzpeter A, Mohmand A, et al. Hepatitis C treatment in “difficult-to-treat” psychiatric patients with pegylated interferon-alpha and ribavirin: response and psychiatric side effects. Hepatology. 2007;46(4):991-998.
13. Sockalingam S, Links PS, Abbey SE. Suicide risk in hepatitis C and during interferon-alpha therapy: a review and clinical update. J Viral Hepat. 2011;18(3):153-160.
14. Telaprevir (Incivek) and boceprevir (Victrelis) for chronic hepatitis C. Med Lett Drugs Ther. 2011;53(1369):57-59.
15. Nelson DR. The role of triple therapy with protease inhibitors in hepatitis C virus genotype 1 naïve patients. Liver Int. 2011;31(suppl 1):53-57.
16. Spennati A, Pariante CM. Withdrawing interferon-α from psychiatric patients: clinical care or unjustifiable stigma? [published online September 14 2012] Psychol Med. doi: 10. 1017/S0033291712001808.
17. Baraldi S, Hepgul N, Mondelli V, et al. Symptomatic treatment of interferon-α-induced depression in hepatitis C: a systematic review. J Clin Psychopharmacol. 2012;32(4):531-543.
18. Schaefer M, Schmidt F, Folwaczny C, et al. Adherence and mental side effects during hepatitis C treatment with interferon alfa and ribavirin in psychiatric risk groups. Hepatology. 2003;37(2):443-451.
19. Harris KA, Jr, Arnsten JH, Litwin AH. Successful integration of hepatitis C evaluation and treatment services with methadone maintenance. J Addict Med. 2010;4(1):20-26.
20. Litwin AH, Harris KA, Jr, Nahvi S, et al. Successful treatment of chronic hepatitis C with pegylated interferon in combination with ribavirin in a methadone maintenance treatment program. J Subst Abuse Treat. 2009;37(1):32-40.
21. Sasadeusz JJ, Dore G, Kronborg I, et al. Clinical experience with the treatment of hepatitis C infection in patients on opioid pharmacotherapy. Addiction. 2011;106(5):977-984.
22. Sockalingam S, Abbey SE. Managing depression during hepatitis C treatment. Can J Psychiatry. 2009;54(9):614-625.
23. Schaefer M, Sarkar R, Knop V, et al. Escitalopram for the prevention of peginterferon-α2a-associated depression in hepatitis C virus-infected patients without previous psychiatric disease: a randomized trial. Ann Intern Med. 2012;157(2):94-103.
24. Galvão-de Almeida A, Guindalini C, Batista-Neves S, et al. Can antidepressants prevent interferon-alpha-induced depression? A review of the literature. Gen Hosp Psychiatry. 2010;32(4):401-405.
25. Morasco BJ, Loftis JM, Indest DW, et al. Prophylactic antidepressant treatment in patients with hepatitis C on antiviral therapy: a double-blind, placebo-controlled trial. Psychosomatics. 2010;51(5):401-408.
26. Raison CL, Woolwine BJ, Demetrashvili MF, et al. Paroxetine for prevention of depressive symptoms induced by interferon-alpha and ribavirin for hepatitis C. Aliment Pharmacol Ther. 2007;25(10):1163-1174.
27. Kraus MR, Schäfer A, Schöttker K, et al. Therapy of interferon-induced depression in chronic hepatitis C with citalopram: a randomised, double-blind, placebo-controlled study. Gut. 2008;57(4):531-536.
28. Zincke MT, Kurani A, Istafanous R, et al. The successful use of electroconvulsive therapy in a patient with interferon-induced psychotic depression. J ECT. 2007;23(4):291-292.
29. Crone CC, Gabriel GM, Wise TN. Managing the neuropsychiatric side effects of interferon-based therapy for hepatitis C. Cleve Clin J Med. 2004;71(suppl 3):S27-S32.
30. Schaefer M, Winterer J, Sarkar R, et al. Three cases of successful tryptophan add-on or monotherapy of hepatitis C and IFNa-associated mood disorders. Psychosomatics. 2008;49(5):442-446.
31. Turner EH, Blackwell AD. 5-Hydroxytryptophan plus SSRIs for interferon-induced depression: synergistic mechanisms for normalizing synaptic serotonin. Med Hypotheses. 2005;65(1):138-144.
32. Onyike CU, Bonner JO, Lyketsos CG, et al. Mania during treatment of chronic hepatitis C with pegylated interferon and ribavirin. Am J Psychiatry. 2004;161(3):429-435.
33. Cheng YC, Chen CC, Ho AS, et al. Prolonged psychosis associated with interferon therapy in a patient with hepatitis C: case study and literature review. Psychosomatics. 2009;50(5):538-542.
Genetics of schizophrenia: What do we know?
Discuss this article at www.facebook.com/CurrentPsychiatry
Genetic factors play a major role in the etiology and development of schizophrenia. Genetic linkage studies and twin studies have estimated the heritability of schizophrenia to be 70% to 90%.1 Research on the genetic underpinnings of schizophrenia has accelerated since the Human Genome Project was completed in 2001, which opened the door to expanding our understanding of molecular mechanisms of human diseases. Experts have hailed the dawn of personalized medicine,2 hoping that we will be able to use knowledge of the human genome to tailor individual treatment.
In this article we review some significant recent findings in genetics of schizophrenia. Gene names are italicized and proteins coded by genes are not. The names, functions, and locations of all genes included in this article appear in Table 1. For a glossary of genetic terms, see Table 2.
Table 1
Select genes and their functions
| Gene | Name | Location | Function(s) |
|---|---|---|---|
| CACNA1C | Calcium channel, voltage-dependent, L type, alpha 1C subunit | 12p13.3 | Calcium channels mediate the influx of calcium ions into the cell upon membrane polarization |
| COMT | Catechol-O-methyltransferase | 22q11.21 | Key enzyme in degradation of dopamine and norepinephrine |
| CSMD1 | CUB and Sushi multiple domains 1 | 8p23.2 | One of the proteins that modulate the classical complement pathway, part of the immune system |
| CYP2D6 | Cytochrome P450 2D6 | 22q13.1 | Key enzyme in drug metabolism |
| C10orf26 | Chromosome 10 open reading frame 26 | 10q24.32 | Unknown |
| DISC1 | Disrupted in schizophrenia 1 | 1q42 | Neurite outgrowth, cortical development, synaptic function |
| DRD1 | Dopamine receptor D1 | 5q35.1 | D1 receptors regulate neuronal growth and development, mediate behavioral responses, and modulate D2 receptor-mediated events |
| DRD2 | Dopamine receptor D2 | 11q23 | D2 receptors regulate motor activities and information processing in the brain |
| DTNBP1 | Dystrobrevin binding protein 1 | 6p22 | Neurodevelopment and synaptic transmission |
| HLA-DQB1 | Major histocompatibility complex, class II, DQ beta 1 | 6p21.3 | Plays a central role in the immune system by presenting peptides derived from extracellular proteins |
| HTR2C | Serotonin receptor 2C | Xq24 | Modulate mood, food intake behavior, and feeling of satiety |
| MC4R | Melanocortin 4 receptor | 18q22 | Modulate food intake behavior and feeling of satiety |
| MHC region | Major histocompatibility complex | 6p21-22 | Immune function; neurodevelopment, synaptic plasticity |
| MIR137 | MicroRNA 137 | 1p23.3 | Post-transcriptional regulation of messenger RNAs; neuron maturation, adult neurogenesis |
| MTHFR | Methylenetetrahydrofolate reductase | 1p36.3 | Key enzyme in folate metabolism |
| TCF4 | Transcription factor 4 | 18q21.2 | Neuronal transcriptional factor, neurogenesis |
| TPH1 | Tryptophan hydroxylase 1 | 11p15.3 | Key enzyme in biosynthesis of serotonin |
| ZNF804A | Zinc finger protein 804A | 2q32.1 | Transcription factor, neuronal connectivity in the dorsolateral prefrontal cortex |
Table 2
Glossary of genetic terms
| Allele: One of several variants of a gene, usually referring to a specific site within the gene |
| Association study: Genetic association refers to the association between a particular genotype and a phenotypic trait in the population. Genetic association studies aim to test whether single-locus alleles genotype frequencies or multi-locus haplotype frequencies differ between 2 groups (such as cases and controls) |
| Candidate gene study: A study that evaluates association of specific genetic variants with outcomes or traits of interest, selecting variants to be tested according to explicit considerations (known or postulated biology or function, previous studies, etc.) |
| Case-control design: An association study design in which the primary comparison is between a group of individuals (cases) ascertained for the phenotype of interest (eg, patients with schizophrenia) and a second group (control) ascertained for not having the phenotype (eg, healthy controls) |
| Copy number variation: A class of DNA sequence variant (including deletions and duplications) in which the result is a departure from the expected 2-copy representation of DNA sequence (ie, each person has 2 copies of the same chromosome) |
| Endophenotype: Phenotypes that are genetically determined, directly measurable traits as part of a complex illness. This term is used to connect the pathway from genes to a disease (eg, impairment in working memory is an endophenotype of schizophrenia) |
| Genetic association: A relationship that is defined by the nonrandom occurrence of a genetic marker with a trait, which suggests an association between the genetic marker (or a marker close to it) and disease pathogenesis |
| Genetic marker: A specific genetic variant known to be associated with a recognizable trait or disease |
| Genome: The entire collection of genetic information (or genes) that an organism possesses |
| Genome-wide association study: A study that evaluates association of genetic variation with outcomes or traits of interest by using 300,000 to 1,000,000 markers across the whole genome. No hypothesis about any particular gene is required for GWAS |
| Genotype: The genetic constitution of an individual, either overall or at a specific gene |
| Heritability (h2): A measure of the strength of genetic effects on a trait. It is defined as the proportion of the phenotypic variation in a trait that is attributable to genetic effects |
| Linkage disequilibrium (LD): Two polymorphic loci are in LD when they are co-located, and alleles at those loci are distributed non-randomly with respect to each other on chromosomes in the population |
| Linkage study: A technique used in genetic epidemiology that focuses on linking a chromosome region to transmission of a particular trait across multiple familial generations |
| Phenotype: The observable characteristics of a cell or organism, usually being the results of the product coded by a gene (genotype) |
| Polymorphism: The existence of ≥2 variants of a gene, occurring in a population, with at least 1% frequency of the less common variant |
| Recombination hotspot: Recombination is breaking and rejoining of DNA strands to form new DNA molecules encoding a novel set of genetic information. Recombination hotspots are individual regions within the genome that have frequent recombination events (eg, the human leukocyte antigen region is a recombination hotspot) |
| Single nucleotide polymorphism: A single base pair change in the DNA sequence at a particular point, compared with the “common” or “wild type” sequence |
| Translocation: A type of chromosomal abnormality resulted by rearrangement of parts between nonhomologous chromosomes, often leading to cancer or developmental abnormalities |
Focusing on single nucleotide polymorphisms
Genetic research of diseases previously relied on linkage studies, which focus on linking a chromosome region to transmission of a particular trait across multiple familial generations. This approach has identified several genomic regions that may be associated with schizophrenia, but most of these regions contain multiple genes and are not specific to schizophrenia.
Today, many genetic studies examine variations of a single nucleotide in the DNA sequence, ie, a change of 1 letter in a particular location on the DNA chain. Single nucleotide polymorphisms (SNPs)—relatively common DNA variations found in >5% of the population—have been a major focus of psychiatric genetics in the past decade. Technology now allows researchers to simultaneously genotype millions of SNPs across the genome, producing tremendous power to investigate the entire genome in relation to a phenotype (a disease or a trait) in genome-wide association studies (GWAS).3 GWAS do not require an a priori hypothesis regarding which regions or genes may be important, and have yielded many novel genetic variants implicated in schizophrenia.
Susceptibility genes
Genetic researchers initially hoped to find that one or a few genes are responsible for schizophrenia. However, recent research revealed that many genes may be involved in susceptibility to schizophrenia, and that a particular gene may contribute to the risk of not only schizophrenia but also other psychiatric disorders such as bipolar disorder (BD).
Discovery of the DISC1 gene is an example of how our understanding of the complex genetic architecture in psychiatric disorders has evolved. In 2000, a linkage study in a Scottish family cohort found a translocation on chromosome 1, t(1:11), highly correlated with schizophrenia.4 Later studies found that this translocation directly disrupts a gene, which researchers named “disrupted in schizophrenia 1.” The protein encoded by DISC1 appears to provide a scaffold to other proteins involved in multiple cellular functions, particularly regulation of brain development and maturation. It is involved in neuronal proliferation, differentiation, and migration via various signaling pathways by interacting with many other proteins.5 Disruption of DISC1 results in dysfunction in multiple neurodevelopmental processes, significantly increasing susceptibility not only for schizophrenia but also for BD and depression.
Many common variants of DISC1 slightly alter expression levels of the gene, which may exert subtle but pervasive effects on neural circuitry development. DISC1 knockout mouse models showed close interactions between DISC1 and N-methyl-d-aspartate receptors and dopamine D2 receptors, linking to the glutamate hypothesis of schizophrenia and the common site of action of antipsychotics. Despite advances in understanding the biology of DISC1, large case-control studies have not found a consistent association between DISC1 and schizophrenia.6,7 It is possible that DISC1 pathology represents one subtype of schizophrenia that is not prevalent among the general population; therefore, large-scale epidemiologic studies could not find evidence to support DISC1’s role in schizophrenia.
DTNBP1 is another schizophrenia susceptibility gene discovered in linkage studies. Originally found in a large Irish cohort, several SNPs of DTNBP1 were significantly associated with schizophrenia.8 A meta-analysis of candidate genes identified DTNBP1 as one of 4 genes with the strongest evidence for association with schizophrenia (the other 3 are DRD1, MTHFR, and TPH1).9 DTNBP1 is widely expressed in the brain and is present in presynaptic, postsynaptic, and microtubule locations implicated in a number of brain functions, including synaptic transmission and neurite outgrowth in a developing organism. Furthermore, DTNBP1 is associated with cognitive functions in schizophrenia patients10 as well as in control subjects.11 Cognitive impairment is considered an endophenotype for schizophrenia. Similar to DISC1 and other candidate genes, DTNBP1 has not emerged as a significant hit in later, large-scale GWAS studies.
Since the first schizophrenia GWAS in 2007,12 >15 GWAS have been published, with increasingly larger samples sizes. GWAS are based on the “common disease/common variant hypothesis” that common disorders such as diabetes, macular degeneration, and schizophrenia are caused by multiple common variants in the genome. Because GWAS can analyze hundreds of thousands of SNPs simultaneously, a stringent criterion (usually P < 5×10-8) is used to gauge statistical significance to correct for multiple testing. Because most effect sizes associated with genetic markers in psychiatry are fairly small (odds ratios [ORs] are approximately 1.1 to 1.2), large samples are required to detect significant effects. Several international consortia have accumulated large samples. The Psychiatric GWAS Consortium has >17,000 patients with schizophrenia, >11,000 with BD, >16,000 with major depression, and >50,000 healthy controls. This wave of GWAS has implicated several novel genomic regions in schizophrenia pathophysiology, including ZNF804A, the major histocompatibility complex (MHC) region, and MIR137.
ZNF804A was the first gene that reached genome-wide significance in a large GWAS,13 and this finding has been replicated. The function of this novel gene largely is unknown. ZNF804A is widely expressed in the brain, especially in the developing hippocampus and the cortex as well as in the adult cerebellum. Recent studies found that ZNF804A is a putative transcription factor, upregulating expression of catechol-O-methyltransferase while downregulating dopamine D2 receptors in animal studies.14 The minor allele of SNP rs1344706 was associated with impaired brain functional connectivity in a human study.15 More work is needed to understand how this gene increases schizophrenia susceptibility.
The MHC region on chromosome 6p22.1,1 also was significant in schizophrenia GWAS,16,17 and this may be the most replicated schizophrenia GWAS finding. This region is a recombination hotspot and harbors many genetic variants. Many immune-related genes previously were associated with autoimmune and infectious disorders, which may suggest that the immunologic system plays a role in schizophrenia pathogenesis. These genes also may involve neurodevelopment, synaptic plasticity, and other neuronal processes.18 However, the complex gene composition in the region makes it difficult to pinpoint the exact signal to schizophrenia pathophysiology.
The most recent finding from the largest GWAS is MIR137,19 coding for microRNA 137, which was associated with schizophrenia at P=1.6×10-11 in 17,836 patients and 33,859 controls. MicroRNAs are small, noncoding RNA fragments that are involved in post-transcriptional regulation of messenger RNAs. MIR137 plays important roles in neuron maturation and adult neurogenesis by acting at the level of dendritic morphogenesis and spine development.20 More interestingly, the other 4 loci achieving genome-wide significance in the same GWAS (TCF4, CACNA1C, CSMD1, and C10orf26) contain predicted target sites of MIR137. This suggests MIR137-mediated dysregulation may be an etiologic mechanism in schizophrenia.
Limitations of these findings. The effect sizes of these genetic variants are small, explaining only 1% to 2% of genetic risks of schizophrenia. However, this is not unique to schizophrenia or psychiatry. “Missing heritability” is puzzling in other branches of medicine.21 Future research will focus on gene-environment interactions as well as gene-gene interactions in relation to schizophrenia’s neurodevelopmental processes.
In addition, many top hits in GWAS are SNPs that are not functional or located in intergenic regions with unknown functions. They may be proxies of causal variants that truly play causal roles in pathogenesis of diseases but were not genotyped in those studies. Recently, researchers have grown increasingly interested in copy number variations (CNVs) in the etiology of complex diseases. Compared with SNPs, CNVs usually are much larger changes in the DNA sequence, including deletions and duplications of a large chunk of DNA segments. Disease-causing CNVs are rare but have large effect sizes. Recent studies have examined the role of CNVs in schizophrenia.22,23
Although genes such as DISC1 and CACNA1C are linked to schizophrenia, they are neither necessary nor sufficient for developing the disorder, and also are linked equally, if not more strongly, to other neuropsychiatric disorders, including BD and autism. Therefore, they are not “schizophrenia genes.” Variations in multiple genes likely cause slight deviations in neurodevelopment that interact with environmental variables and lead to development of schizophrenia.
Nevertheless, these schizophrenia GWAS findings provide insight into this complex disorder. Much work is needed to move from these association signals to understanding the function and regulation of these genes to turn basic biologic knowledge into targets for new drugs or other interventions.
Antipsychotic pharmacogenetics
Genetic research of schizophrenia also contributes to our knowledge of how to best use existing drugs. Medications for treating schizophrenia often need to be changed because patients experience lack of efficacy or intolerable side effects, which may lead them to discontinue treatment. Clinical predictors of which medication would work for an individual patient are lacking. Pharmacogenetics may be able to fulfill the promise of personalized medicine in psychiatry by using genetic information to guide drug selection to maximize therapeutic efficacy and minimize drug-induced side effects.
Researchers first attempted to find genetic predictors of antipsychotic efficacy in the early 1990s. One replicated finding is that DRD2, the gene coding for dopamine receptor D2, is associated with antipsychotic efficacy. This may not be surprising because D2 receptor antagonism is a common and necessary drug action mechanism for all antipsychotics. One SNP, -141C Ins/Del (rs1799732), represents a deletion (vs insertion) of cytosine at position -141, located in the 5’ promoter region of DRD2. Pre-clinical studies showed that this SNP might modulate DRD2 gene expression and influence D2 receptor density in the brain. Del allele carriers had poor response to clozapine among a treatment-refractory sample24 and took longer to respond to olanzapine and risperidone among first-episode schizophrenia patients.25 A 2010 meta-analysis of approximately 700 patients26 showed that the -141C Ins/Del polymorphism is significantly associated with antipsychotic response. Patients who carry 1 or 2 Del alleles tend to have a less favorable antipsychotic response than patients with the Ins/Ins genotype. Patients with the Ins/Ins genotype are 54% more likely to respond to antipsychotics than those with ≥1 copy of the Del allele.
Researchers have studied other genes in relation to antipsychotic efficacy, but have yielded few consistent findings.27 Some have looked at combining multiple SNPs across several genes to predict antipsychotic efficacy, but these findings have not been replicated. For example, a combination of variants in the HTR2A, HTR2C, and 5-HTTLPR genes and genes coding for H2 receptors was found to correctly predict clozapine response in 76% of patients.28 However, this finding was not replicated in an independent sample.29 A recent GWAS30 found that a combination of 6 genetic markers—NPAS3, XKR4, TNR, GRIA4, GFRA2, and NUDT9P1—predicted treatment response to iloperidone. Although promising, this finding needs to be validated in independent samples.
Predicting adverse drug events
In other branches of medicine, researchers have used pharmacogenetics to successfully identify predictors of drug-induced adverse events. A GWAS found that a specific human leukocyte antigen (HLA) allele markedly increases the risk of liver toxicity from flucloxacillin (OR=80.6).31 This HLA marker also is related to hypersensitivity reaction to abacavir, a common medication for treating AIDS, and lamotrigine-induced Stevens-Johnson syndrome.
Clozapine-induced granulocytosis also may be related to genetic variation in the HLA region. Despite superior efficacy, clozapine remains underutilized in part because it carries the risk of potentially fatal agranulocytosis. Identifying a genetic marker for agranulocytosis would lift the burden of weekly blood monitoring. A recent pharmacogenetic study detected a replicated association of an allele at the HLA-DQB1 locus with risk of agranulocytosis in 2 small groups of clozapine-treated schizophrenia patients.32 Effect sizes were extremely high (OR=16.86); nearly 90% of allele carriers developed agranulocytosis. Unfortunately, the overall sensitivity of the marker was 21%, indicating that most individuals who develop agranulocytosis are not carriers of the allele and presumably have other genetic risk factors. A more comprehensive risk profile would be necessary to obviate the need for weekly blood monitoring.
Weight gain and metabolic syndrome are common side effects of antipsychotics, and no clear clinical predictors have been identified. Researchers have examined potential genetic markers in association with antipsychotic-induced weight gain. One consistent finding has been that a single SNP in the promoter region of the HTR2C gene (serotonin receptor 2C), C-759T (rs3813929), affects antipsychotic-induced weight gain. The 5-HT2C receptor is involved in regulating food intake in rodents and is related to late-onset diabetes and obesity in humans. HTR2C knockout mice display chronic hyperphagia that leads to obesity and hyperinsulinemia. Since the original finding in 2002,33 at least 17 studies have reported on the association between the C-759T SNP in HTR2C and antipsychotic-induced weight gain. A meta-analysis found that the T allele was significantly protective against antipsychotic-induced weight gain.34 The C allele was associated with >2-fold increase of risk for clinically significant weight gain (gaining >7% of baseline body weight).
In a GWAS of antipsychotic-induced weight gain in pediatric patients who were prescribed antipsychotics for the first time, researchers discovered a single top signal at a marginally genome-wide significant level (P=1.6×10-7).35 This was replicated in 3 other independent samples. The peak signal is located on chromosome 18q21, overlapping a peak identified as a predictor of obesity. This locus is approximately 150 kb downstream from MC4R, the melanocortin 4 receptor gene, which has long been suspected as a candidate for weight-related phenotypes, including antipsychotic-induced weight gain.36 Mutations in this gene are linked with extreme obesity in humans, and MC4R knockout mice develop obesity. MC4R-expressing neurons in the ventromedial hypothalamus are regulated by circulating levels of leptin via pathways in the arcuate nucleus. In turn, MC4R regulates 5-HT2C receptors, which are implicated in weight gain. In the discovery sample, risk allele homozygotes gained twice as much weight as other patients after 12 weeks of treatment, and the genetic effect was not drug-specific. The consistency of HTR2C-MC4R findings poses a possibility that a drug may be developed at these targets to treat or prevent antipsychotic-induced weight gain.
Drug metabolism. Pharmacogenetic studies of antipsychotic drug response also have focused on genes that code for enzymes in drug metabolism, particularly cytochrome (CYP) 450 enzymes, which are responsible for the metabolism of many drugs. CYP2D6 is the main metabolic pathway for several antipsychotics, including risperidone, aripiprazole, haloperidol, and perphenazine. The CYP2D6 gene contains >100 variants, many of which yield nonfunctional or reduced-function enzymes. There are 4 phenotypes of CYP2D6 produced by combinations of various alleles with different degrees of enzymatic activities: poor (PM), intermediate (IM), extensive (EM), and ultrarapid metabolizers (UM). Compared with EMs with normal CYP2D6 enzyme activity, PMs and IMs have minimal or reduced activity, respectively. UMs have duplicate or multiple copies of the gene that result in increased enzyme activity. Approximately 7% to 10% of whites and 1% to 2% of Asians are PMs, who tend to accumulate higher serum drug levels and, theoretically, require lower doses to achieve therapeutic effects. UMs, in contrast, consist of 1% of the population and may require higher doses because of faster drug elimination.37 Therefore, CYP2D6 metabolic status could play an important role in determining patients’ antipsychotic response. So far, no empirical data support the association between CYP2D6 and antipsychotic efficacy, although studies have found significant relationships between PMs and higher rates of drug-induced side effects such as tardive dyskinesia (TD), extrapyramidal symptoms, and weight gain. A meta-analysis38 of 8 studies showed that PMs had a 43% higher risk of developing TD compared with EMs. An FDA-approved pharmacogenetic test, AmpliChip® CYP450 Test, is available to assess CYP2D6 and CYP2C19 genotypes,39 but its use is limited, perhaps because of clinician concerns about how to interpret test results, paucity of prospective data suggesting that using the test can improve clinical outcomes, and lack of reimbursement.
Implications for clinical practice
Although schizophrenia genetic research has made tremendous progress in the past decade, most findings are at basic science level and clinical applications are limited. It is premature to attempt to use genetic markers to help diagnose schizophrenia or other psychiatric disorders.40 Researchers hope that new gene discovery will translate to better understanding of the pathophysiological mechanisms underlying schizophrenia, which in turn lead to finding novel molecular targets for new drug development. Furthermore, pharmacogenetics helps clinicians use existing drugs more efficiently by maximizing efficacy and minimizing side effects. Several institutions have experimented with genotyping CYP450 in routine clinical practice,41 but prospective pharmacogenetic clinical trials are needed to validate the utility and cost-effectiveness of genetic testing-guided treatment algorithms.42
Bottom Line
Variations in multiple genes likely cause slight deviations in neurodevelopment that interact with environmental variables and lead to development of schizophrenia. Genome-wide association studies are allowing researchers to gain insight into which patients may have increased susceptibility to the disorder, identify potential molecular targets for new drugs, and expand their knowledge of how to best use medications.
Related Resource
- National Institute of Mental Health Center for Collaborative Genomic Studies on Mental Disorders. Schizophrenia. www.nimhgenetics.org/available_data/schizophrenia.
Drug Brand Names
- Abacavir • Ziagen
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Haloperidol • Haldol
- Iloperidone • Fanapt
- Lamotrigine • Lamictal
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Risperidone • Risperdal
Disclosures
Dr. Zhang reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Malhotra is a consultant to Genomind, Inc.
This work was partly supported by a Young Investigator Award from the Brain and Behavior Research Foundation (Dr. Zhang), and by the National Institute of Mental Health (P50MH080173 to Dr. Malhotra and 1K23MH097108 to Dr. Zhang).
1. Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60(12):1187-1192.
2. de Leon J. AmpliChip CYP450 test: personalized medicine has arrived in psychiatry. Expert Rev Mol Diagn. 2006;6(3):277-286.
3. Psychiatric GWAS Consortium Coordinating Committee; Cichon S, Craddock N, Daly M, et al. Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am J Psychiatry. 2009;166(5):540-556.
4. Millar JK, Wilson-Annan JC, Anderson S, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9(9):1415-1423.
5. Porteous DJ, Millar JK, Brandon NJ, et al. DISC1 at 10: connecting psychiatric genetics and neuroscience. Trends Mol Med. 2011;17(12):699-706.
6. Schumacher J, Laje G, Abou Jamra R, et al. The DISC locus and schizophrenia: evidence from an association study in a central European sample and from a meta-analysis across different European populations. Hum Mol Genet. 2009;18(14):2719-2727.
7. Mathieson I, Munafò MR, Flint J, et al. Meta-analysis indicates that common variants at the DISC1 locus are not associated with schizophrenia. Mol Psychiatry. 2012;17(6):634-641.
8. Straub RE, Jiang Y, MacLean CJ, et al. Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet. 2002;71(2):337-348.
9. Allen NC, Bagade S, McQueen MB, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40(7):827-834.
10. Burdick KE, Lencz T, Funke B, et al. Genetic variation in DTNBP1 influences general cognitive ability. Hum Mol Genet. 2006;15(10):1563-1568.
11. Zhang JP, Burdick KE, Lencz T, et al. Meta-analysis of genetic variation in DTNBP1 and general cognitive ability. Biol Psychiatry. 2010;68(12):1126-1133.
12. Lencz T, Morgan TV, Athanasiou M, et al. Converging evidence for a pseudoautosomal cytokine receptor gene locus in schizophrenia. Mol Psychiatry. 2007;12(6):572-580.
13. O’Donovan MC, Craddock N, Norton N, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008;40(9):1053-1055.
14. Girgenti MJ, LoTurco JJ, Maher BJ. ZNF804a regulates expression of the schizophrenia-associated genes PRSS16 COMT, PDE4B, and DRD2. PLoS One. 2012;7(2):e32404.-
15. Lencz T, Szeszko PR, DeRosse P, et al. A schizophrenia risk gene, ZNF804A, influences neuroanatomical and neurocognitive phenotypes. Neuropsychopharmacology. 2010;35(11):2284-2291.
16. International Schizophrenia Consortium; Purcell SM, Wray NR, Stone JL, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748-752.
17. Stefansson H, Ophoff RA, Steinberg S, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460(7256):744-747.
18. Handel AE, Ramagopalan SV. The potential role of major histocompatibility complex class I in schizophrenia. Biol Psychiatry. 2010;68(7):e29-e30.
19. Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43(10):969-976.
20. Gallego JA, Gordon ML, Claycomb K, et al. In vivo microRNA detection and quantitation in cerebrospinal fluid. J Mol Neurosci. 2012;47(2):243-248.
21. Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747-753.
22. Walsh T, McClellan JM, McCarthy SE, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320(5875):539-543.
23. Rees E, Kirov G, O’Donovan MC, et al. De novo mutation in schizophrenia. Schizophr Bull. 2012;38(3):377-381.
24. Malhotra AK, Buchanan RW, Kim S. Allelic variation in the promotor region of the dopamine D2 receptor gene and clozapine response. Schizophr Res. 1999;36:92-93.
25. Lencz T, Robinson DG, Xu K, et al. DRD2 promoter region variation as a predictor of sustained response to antipsychotic medication in first-episode schizophrenia patients. Am J Psychiatry. 2006;163(3):529-531.
26. Zhang JP, Lencz T, Malhotra AK. D2 receptor genetic variation and clinical response to antipsychotic drug treatment: a meta-analysis. Am J Psychiatry. 2010;167(7):763-772.
27. Zhang JP, Malhotra AK. Pharmacogenetics and antipsychotics: therapeutic efficacy and side effects prediction. Expert Opin Drug Metab Toxicol. 2011;7(1):9-37.
28. Arranz MJ, Munro J, Birkett J, et al. Pharmacogenetic prediction of clozapine response. Lancet. 2000;355(9215):1615-1616.
29. Schumacher J, Schulze TG, Wienker TF, et al. Pharmacogenetics of the clozapine response. Lancet. 2000;356(9228):506-507.
30. Lavedan C, Licamele L, Volpi S, et al. Association of the NPAS3 gene and five other loci with response to the antipsychotic iloperidone identified in a whole genome association study. Mol Psychiatry. 2009;14(8):804-819.
31. Daly AK, Donaldson PT, Bhatnagar P, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41(7):816-819.
32. Athanasiou MC, Dettling M, Cascorbi I, et al. Candidate gene analysis identifies a polymorphism in HLA-DQB1 associated with clozapine-induced agranulocytosis. J Clin Psychiatry. 2011;72(4):458-463.
33. Reynolds GP, Zhang ZJ, Zhang XB. Association of antipsychotic drug-induced weight gain with a 5-HT2C receptor gene polymorphism. Lancet. 2002;359(9323):2086-2087.
34. Sicard MN, Zai CC, Tiwari AK, et al. Polymorphisms of the HTR2C gene and antipsychotic-induced weight gain: an update and meta-analysis. Pharmacogenomics. 2010;11(11):1561-1571.
35. Malhotra AK, Correll CU, Chowdhury NI, et al. Association between common variants near the melanocortin 4 receptor gene and severe antipsychotic drug-induced weight gain. Arch Gen Psychiatry. 2012;69(9):904-912.
36. Correll CU, Malhotra AK. Pharmacogenetics of antipsychotic-induced weight gain. Psychopharmacology (Berl). 2004;174(4):477-489.
37. Zhang JP, Malhotra AK. Pharmacogenetics and antipsychotics: therapeutic efficacy and side effects prediction. Expert Opin Drug Metab Toxicol. 2011;7(1):9-37.
38. Patsopoulos NA, Ntzani EE, Zintzaras E, et al. CYP2D6 polymorphisms and the risk of tardive dyskinesia in schizophrenia: a meta-analysis. Pharmacogenet Genomics. 2005;15(3):151-158.
39. de Leon J. AmpliChip CYP450 test: personalized medicine has arrived in psychiatry. Expert Rev Mol Diagn. 2006;6(3):277-286.
40. Mitchell PB, Meiser B, Wilde A, et al. Predictive and diagnostic genetic testing in psychiatry. Psychiatr Clin North Am. 2010;33(1):225-243.
41. Rundell JR, Staab JP, Shinozaki G, et al. Pharmacogenomic testing in a tertiary care outpatient psychosomatic medicine practice. Psychosomatics. 2011;52(2):141-146.
42. Malhotra AK, Zhang JP, Lencz T. Pharmacogenetics in psychiatry: translating research into clinical practice. Mol Psychiatry. 2012;17(8):760-769.
Discuss this article at www.facebook.com/CurrentPsychiatry
Genetic factors play a major role in the etiology and development of schizophrenia. Genetic linkage studies and twin studies have estimated the heritability of schizophrenia to be 70% to 90%.1 Research on the genetic underpinnings of schizophrenia has accelerated since the Human Genome Project was completed in 2001, which opened the door to expanding our understanding of molecular mechanisms of human diseases. Experts have hailed the dawn of personalized medicine,2 hoping that we will be able to use knowledge of the human genome to tailor individual treatment.
In this article we review some significant recent findings in genetics of schizophrenia. Gene names are italicized and proteins coded by genes are not. The names, functions, and locations of all genes included in this article appear in Table 1. For a glossary of genetic terms, see Table 2.
Table 1
Select genes and their functions
| Gene | Name | Location | Function(s) |
|---|---|---|---|
| CACNA1C | Calcium channel, voltage-dependent, L type, alpha 1C subunit | 12p13.3 | Calcium channels mediate the influx of calcium ions into the cell upon membrane polarization |
| COMT | Catechol-O-methyltransferase | 22q11.21 | Key enzyme in degradation of dopamine and norepinephrine |
| CSMD1 | CUB and Sushi multiple domains 1 | 8p23.2 | One of the proteins that modulate the classical complement pathway, part of the immune system |
| CYP2D6 | Cytochrome P450 2D6 | 22q13.1 | Key enzyme in drug metabolism |
| C10orf26 | Chromosome 10 open reading frame 26 | 10q24.32 | Unknown |
| DISC1 | Disrupted in schizophrenia 1 | 1q42 | Neurite outgrowth, cortical development, synaptic function |
| DRD1 | Dopamine receptor D1 | 5q35.1 | D1 receptors regulate neuronal growth and development, mediate behavioral responses, and modulate D2 receptor-mediated events |
| DRD2 | Dopamine receptor D2 | 11q23 | D2 receptors regulate motor activities and information processing in the brain |
| DTNBP1 | Dystrobrevin binding protein 1 | 6p22 | Neurodevelopment and synaptic transmission |
| HLA-DQB1 | Major histocompatibility complex, class II, DQ beta 1 | 6p21.3 | Plays a central role in the immune system by presenting peptides derived from extracellular proteins |
| HTR2C | Serotonin receptor 2C | Xq24 | Modulate mood, food intake behavior, and feeling of satiety |
| MC4R | Melanocortin 4 receptor | 18q22 | Modulate food intake behavior and feeling of satiety |
| MHC region | Major histocompatibility complex | 6p21-22 | Immune function; neurodevelopment, synaptic plasticity |
| MIR137 | MicroRNA 137 | 1p23.3 | Post-transcriptional regulation of messenger RNAs; neuron maturation, adult neurogenesis |
| MTHFR | Methylenetetrahydrofolate reductase | 1p36.3 | Key enzyme in folate metabolism |
| TCF4 | Transcription factor 4 | 18q21.2 | Neuronal transcriptional factor, neurogenesis |
| TPH1 | Tryptophan hydroxylase 1 | 11p15.3 | Key enzyme in biosynthesis of serotonin |
| ZNF804A | Zinc finger protein 804A | 2q32.1 | Transcription factor, neuronal connectivity in the dorsolateral prefrontal cortex |
Table 2
Glossary of genetic terms
| Allele: One of several variants of a gene, usually referring to a specific site within the gene |
| Association study: Genetic association refers to the association between a particular genotype and a phenotypic trait in the population. Genetic association studies aim to test whether single-locus alleles genotype frequencies or multi-locus haplotype frequencies differ between 2 groups (such as cases and controls) |
| Candidate gene study: A study that evaluates association of specific genetic variants with outcomes or traits of interest, selecting variants to be tested according to explicit considerations (known or postulated biology or function, previous studies, etc.) |
| Case-control design: An association study design in which the primary comparison is between a group of individuals (cases) ascertained for the phenotype of interest (eg, patients with schizophrenia) and a second group (control) ascertained for not having the phenotype (eg, healthy controls) |
| Copy number variation: A class of DNA sequence variant (including deletions and duplications) in which the result is a departure from the expected 2-copy representation of DNA sequence (ie, each person has 2 copies of the same chromosome) |
| Endophenotype: Phenotypes that are genetically determined, directly measurable traits as part of a complex illness. This term is used to connect the pathway from genes to a disease (eg, impairment in working memory is an endophenotype of schizophrenia) |
| Genetic association: A relationship that is defined by the nonrandom occurrence of a genetic marker with a trait, which suggests an association between the genetic marker (or a marker close to it) and disease pathogenesis |
| Genetic marker: A specific genetic variant known to be associated with a recognizable trait or disease |
| Genome: The entire collection of genetic information (or genes) that an organism possesses |
| Genome-wide association study: A study that evaluates association of genetic variation with outcomes or traits of interest by using 300,000 to 1,000,000 markers across the whole genome. No hypothesis about any particular gene is required for GWAS |
| Genotype: The genetic constitution of an individual, either overall or at a specific gene |
| Heritability (h2): A measure of the strength of genetic effects on a trait. It is defined as the proportion of the phenotypic variation in a trait that is attributable to genetic effects |
| Linkage disequilibrium (LD): Two polymorphic loci are in LD when they are co-located, and alleles at those loci are distributed non-randomly with respect to each other on chromosomes in the population |
| Linkage study: A technique used in genetic epidemiology that focuses on linking a chromosome region to transmission of a particular trait across multiple familial generations |
| Phenotype: The observable characteristics of a cell or organism, usually being the results of the product coded by a gene (genotype) |
| Polymorphism: The existence of ≥2 variants of a gene, occurring in a population, with at least 1% frequency of the less common variant |
| Recombination hotspot: Recombination is breaking and rejoining of DNA strands to form new DNA molecules encoding a novel set of genetic information. Recombination hotspots are individual regions within the genome that have frequent recombination events (eg, the human leukocyte antigen region is a recombination hotspot) |
| Single nucleotide polymorphism: A single base pair change in the DNA sequence at a particular point, compared with the “common” or “wild type” sequence |
| Translocation: A type of chromosomal abnormality resulted by rearrangement of parts between nonhomologous chromosomes, often leading to cancer or developmental abnormalities |
Focusing on single nucleotide polymorphisms
Genetic research of diseases previously relied on linkage studies, which focus on linking a chromosome region to transmission of a particular trait across multiple familial generations. This approach has identified several genomic regions that may be associated with schizophrenia, but most of these regions contain multiple genes and are not specific to schizophrenia.
Today, many genetic studies examine variations of a single nucleotide in the DNA sequence, ie, a change of 1 letter in a particular location on the DNA chain. Single nucleotide polymorphisms (SNPs)—relatively common DNA variations found in >5% of the population—have been a major focus of psychiatric genetics in the past decade. Technology now allows researchers to simultaneously genotype millions of SNPs across the genome, producing tremendous power to investigate the entire genome in relation to a phenotype (a disease or a trait) in genome-wide association studies (GWAS).3 GWAS do not require an a priori hypothesis regarding which regions or genes may be important, and have yielded many novel genetic variants implicated in schizophrenia.
Susceptibility genes
Genetic researchers initially hoped to find that one or a few genes are responsible for schizophrenia. However, recent research revealed that many genes may be involved in susceptibility to schizophrenia, and that a particular gene may contribute to the risk of not only schizophrenia but also other psychiatric disorders such as bipolar disorder (BD).
Discovery of the DISC1 gene is an example of how our understanding of the complex genetic architecture in psychiatric disorders has evolved. In 2000, a linkage study in a Scottish family cohort found a translocation on chromosome 1, t(1:11), highly correlated with schizophrenia.4 Later studies found that this translocation directly disrupts a gene, which researchers named “disrupted in schizophrenia 1.” The protein encoded by DISC1 appears to provide a scaffold to other proteins involved in multiple cellular functions, particularly regulation of brain development and maturation. It is involved in neuronal proliferation, differentiation, and migration via various signaling pathways by interacting with many other proteins.5 Disruption of DISC1 results in dysfunction in multiple neurodevelopmental processes, significantly increasing susceptibility not only for schizophrenia but also for BD and depression.
Many common variants of DISC1 slightly alter expression levels of the gene, which may exert subtle but pervasive effects on neural circuitry development. DISC1 knockout mouse models showed close interactions between DISC1 and N-methyl-d-aspartate receptors and dopamine D2 receptors, linking to the glutamate hypothesis of schizophrenia and the common site of action of antipsychotics. Despite advances in understanding the biology of DISC1, large case-control studies have not found a consistent association between DISC1 and schizophrenia.6,7 It is possible that DISC1 pathology represents one subtype of schizophrenia that is not prevalent among the general population; therefore, large-scale epidemiologic studies could not find evidence to support DISC1’s role in schizophrenia.
DTNBP1 is another schizophrenia susceptibility gene discovered in linkage studies. Originally found in a large Irish cohort, several SNPs of DTNBP1 were significantly associated with schizophrenia.8 A meta-analysis of candidate genes identified DTNBP1 as one of 4 genes with the strongest evidence for association with schizophrenia (the other 3 are DRD1, MTHFR, and TPH1).9 DTNBP1 is widely expressed in the brain and is present in presynaptic, postsynaptic, and microtubule locations implicated in a number of brain functions, including synaptic transmission and neurite outgrowth in a developing organism. Furthermore, DTNBP1 is associated with cognitive functions in schizophrenia patients10 as well as in control subjects.11 Cognitive impairment is considered an endophenotype for schizophrenia. Similar to DISC1 and other candidate genes, DTNBP1 has not emerged as a significant hit in later, large-scale GWAS studies.
Since the first schizophrenia GWAS in 2007,12 >15 GWAS have been published, with increasingly larger samples sizes. GWAS are based on the “common disease/common variant hypothesis” that common disorders such as diabetes, macular degeneration, and schizophrenia are caused by multiple common variants in the genome. Because GWAS can analyze hundreds of thousands of SNPs simultaneously, a stringent criterion (usually P < 5×10-8) is used to gauge statistical significance to correct for multiple testing. Because most effect sizes associated with genetic markers in psychiatry are fairly small (odds ratios [ORs] are approximately 1.1 to 1.2), large samples are required to detect significant effects. Several international consortia have accumulated large samples. The Psychiatric GWAS Consortium has >17,000 patients with schizophrenia, >11,000 with BD, >16,000 with major depression, and >50,000 healthy controls. This wave of GWAS has implicated several novel genomic regions in schizophrenia pathophysiology, including ZNF804A, the major histocompatibility complex (MHC) region, and MIR137.
ZNF804A was the first gene that reached genome-wide significance in a large GWAS,13 and this finding has been replicated. The function of this novel gene largely is unknown. ZNF804A is widely expressed in the brain, especially in the developing hippocampus and the cortex as well as in the adult cerebellum. Recent studies found that ZNF804A is a putative transcription factor, upregulating expression of catechol-O-methyltransferase while downregulating dopamine D2 receptors in animal studies.14 The minor allele of SNP rs1344706 was associated with impaired brain functional connectivity in a human study.15 More work is needed to understand how this gene increases schizophrenia susceptibility.
The MHC region on chromosome 6p22.1,1 also was significant in schizophrenia GWAS,16,17 and this may be the most replicated schizophrenia GWAS finding. This region is a recombination hotspot and harbors many genetic variants. Many immune-related genes previously were associated with autoimmune and infectious disorders, which may suggest that the immunologic system plays a role in schizophrenia pathogenesis. These genes also may involve neurodevelopment, synaptic plasticity, and other neuronal processes.18 However, the complex gene composition in the region makes it difficult to pinpoint the exact signal to schizophrenia pathophysiology.
The most recent finding from the largest GWAS is MIR137,19 coding for microRNA 137, which was associated with schizophrenia at P=1.6×10-11 in 17,836 patients and 33,859 controls. MicroRNAs are small, noncoding RNA fragments that are involved in post-transcriptional regulation of messenger RNAs. MIR137 plays important roles in neuron maturation and adult neurogenesis by acting at the level of dendritic morphogenesis and spine development.20 More interestingly, the other 4 loci achieving genome-wide significance in the same GWAS (TCF4, CACNA1C, CSMD1, and C10orf26) contain predicted target sites of MIR137. This suggests MIR137-mediated dysregulation may be an etiologic mechanism in schizophrenia.
Limitations of these findings. The effect sizes of these genetic variants are small, explaining only 1% to 2% of genetic risks of schizophrenia. However, this is not unique to schizophrenia or psychiatry. “Missing heritability” is puzzling in other branches of medicine.21 Future research will focus on gene-environment interactions as well as gene-gene interactions in relation to schizophrenia’s neurodevelopmental processes.
In addition, many top hits in GWAS are SNPs that are not functional or located in intergenic regions with unknown functions. They may be proxies of causal variants that truly play causal roles in pathogenesis of diseases but were not genotyped in those studies. Recently, researchers have grown increasingly interested in copy number variations (CNVs) in the etiology of complex diseases. Compared with SNPs, CNVs usually are much larger changes in the DNA sequence, including deletions and duplications of a large chunk of DNA segments. Disease-causing CNVs are rare but have large effect sizes. Recent studies have examined the role of CNVs in schizophrenia.22,23
Although genes such as DISC1 and CACNA1C are linked to schizophrenia, they are neither necessary nor sufficient for developing the disorder, and also are linked equally, if not more strongly, to other neuropsychiatric disorders, including BD and autism. Therefore, they are not “schizophrenia genes.” Variations in multiple genes likely cause slight deviations in neurodevelopment that interact with environmental variables and lead to development of schizophrenia.
Nevertheless, these schizophrenia GWAS findings provide insight into this complex disorder. Much work is needed to move from these association signals to understanding the function and regulation of these genes to turn basic biologic knowledge into targets for new drugs or other interventions.
Antipsychotic pharmacogenetics
Genetic research of schizophrenia also contributes to our knowledge of how to best use existing drugs. Medications for treating schizophrenia often need to be changed because patients experience lack of efficacy or intolerable side effects, which may lead them to discontinue treatment. Clinical predictors of which medication would work for an individual patient are lacking. Pharmacogenetics may be able to fulfill the promise of personalized medicine in psychiatry by using genetic information to guide drug selection to maximize therapeutic efficacy and minimize drug-induced side effects.
Researchers first attempted to find genetic predictors of antipsychotic efficacy in the early 1990s. One replicated finding is that DRD2, the gene coding for dopamine receptor D2, is associated with antipsychotic efficacy. This may not be surprising because D2 receptor antagonism is a common and necessary drug action mechanism for all antipsychotics. One SNP, -141C Ins/Del (rs1799732), represents a deletion (vs insertion) of cytosine at position -141, located in the 5’ promoter region of DRD2. Pre-clinical studies showed that this SNP might modulate DRD2 gene expression and influence D2 receptor density in the brain. Del allele carriers had poor response to clozapine among a treatment-refractory sample24 and took longer to respond to olanzapine and risperidone among first-episode schizophrenia patients.25 A 2010 meta-analysis of approximately 700 patients26 showed that the -141C Ins/Del polymorphism is significantly associated with antipsychotic response. Patients who carry 1 or 2 Del alleles tend to have a less favorable antipsychotic response than patients with the Ins/Ins genotype. Patients with the Ins/Ins genotype are 54% more likely to respond to antipsychotics than those with ≥1 copy of the Del allele.
Researchers have studied other genes in relation to antipsychotic efficacy, but have yielded few consistent findings.27 Some have looked at combining multiple SNPs across several genes to predict antipsychotic efficacy, but these findings have not been replicated. For example, a combination of variants in the HTR2A, HTR2C, and 5-HTTLPR genes and genes coding for H2 receptors was found to correctly predict clozapine response in 76% of patients.28 However, this finding was not replicated in an independent sample.29 A recent GWAS30 found that a combination of 6 genetic markers—NPAS3, XKR4, TNR, GRIA4, GFRA2, and NUDT9P1—predicted treatment response to iloperidone. Although promising, this finding needs to be validated in independent samples.
Predicting adverse drug events
In other branches of medicine, researchers have used pharmacogenetics to successfully identify predictors of drug-induced adverse events. A GWAS found that a specific human leukocyte antigen (HLA) allele markedly increases the risk of liver toxicity from flucloxacillin (OR=80.6).31 This HLA marker also is related to hypersensitivity reaction to abacavir, a common medication for treating AIDS, and lamotrigine-induced Stevens-Johnson syndrome.
Clozapine-induced granulocytosis also may be related to genetic variation in the HLA region. Despite superior efficacy, clozapine remains underutilized in part because it carries the risk of potentially fatal agranulocytosis. Identifying a genetic marker for agranulocytosis would lift the burden of weekly blood monitoring. A recent pharmacogenetic study detected a replicated association of an allele at the HLA-DQB1 locus with risk of agranulocytosis in 2 small groups of clozapine-treated schizophrenia patients.32 Effect sizes were extremely high (OR=16.86); nearly 90% of allele carriers developed agranulocytosis. Unfortunately, the overall sensitivity of the marker was 21%, indicating that most individuals who develop agranulocytosis are not carriers of the allele and presumably have other genetic risk factors. A more comprehensive risk profile would be necessary to obviate the need for weekly blood monitoring.
Weight gain and metabolic syndrome are common side effects of antipsychotics, and no clear clinical predictors have been identified. Researchers have examined potential genetic markers in association with antipsychotic-induced weight gain. One consistent finding has been that a single SNP in the promoter region of the HTR2C gene (serotonin receptor 2C), C-759T (rs3813929), affects antipsychotic-induced weight gain. The 5-HT2C receptor is involved in regulating food intake in rodents and is related to late-onset diabetes and obesity in humans. HTR2C knockout mice display chronic hyperphagia that leads to obesity and hyperinsulinemia. Since the original finding in 2002,33 at least 17 studies have reported on the association between the C-759T SNP in HTR2C and antipsychotic-induced weight gain. A meta-analysis found that the T allele was significantly protective against antipsychotic-induced weight gain.34 The C allele was associated with >2-fold increase of risk for clinically significant weight gain (gaining >7% of baseline body weight).
In a GWAS of antipsychotic-induced weight gain in pediatric patients who were prescribed antipsychotics for the first time, researchers discovered a single top signal at a marginally genome-wide significant level (P=1.6×10-7).35 This was replicated in 3 other independent samples. The peak signal is located on chromosome 18q21, overlapping a peak identified as a predictor of obesity. This locus is approximately 150 kb downstream from MC4R, the melanocortin 4 receptor gene, which has long been suspected as a candidate for weight-related phenotypes, including antipsychotic-induced weight gain.36 Mutations in this gene are linked with extreme obesity in humans, and MC4R knockout mice develop obesity. MC4R-expressing neurons in the ventromedial hypothalamus are regulated by circulating levels of leptin via pathways in the arcuate nucleus. In turn, MC4R regulates 5-HT2C receptors, which are implicated in weight gain. In the discovery sample, risk allele homozygotes gained twice as much weight as other patients after 12 weeks of treatment, and the genetic effect was not drug-specific. The consistency of HTR2C-MC4R findings poses a possibility that a drug may be developed at these targets to treat or prevent antipsychotic-induced weight gain.
Drug metabolism. Pharmacogenetic studies of antipsychotic drug response also have focused on genes that code for enzymes in drug metabolism, particularly cytochrome (CYP) 450 enzymes, which are responsible for the metabolism of many drugs. CYP2D6 is the main metabolic pathway for several antipsychotics, including risperidone, aripiprazole, haloperidol, and perphenazine. The CYP2D6 gene contains >100 variants, many of which yield nonfunctional or reduced-function enzymes. There are 4 phenotypes of CYP2D6 produced by combinations of various alleles with different degrees of enzymatic activities: poor (PM), intermediate (IM), extensive (EM), and ultrarapid metabolizers (UM). Compared with EMs with normal CYP2D6 enzyme activity, PMs and IMs have minimal or reduced activity, respectively. UMs have duplicate or multiple copies of the gene that result in increased enzyme activity. Approximately 7% to 10% of whites and 1% to 2% of Asians are PMs, who tend to accumulate higher serum drug levels and, theoretically, require lower doses to achieve therapeutic effects. UMs, in contrast, consist of 1% of the population and may require higher doses because of faster drug elimination.37 Therefore, CYP2D6 metabolic status could play an important role in determining patients’ antipsychotic response. So far, no empirical data support the association between CYP2D6 and antipsychotic efficacy, although studies have found significant relationships between PMs and higher rates of drug-induced side effects such as tardive dyskinesia (TD), extrapyramidal symptoms, and weight gain. A meta-analysis38 of 8 studies showed that PMs had a 43% higher risk of developing TD compared with EMs. An FDA-approved pharmacogenetic test, AmpliChip® CYP450 Test, is available to assess CYP2D6 and CYP2C19 genotypes,39 but its use is limited, perhaps because of clinician concerns about how to interpret test results, paucity of prospective data suggesting that using the test can improve clinical outcomes, and lack of reimbursement.
Implications for clinical practice
Although schizophrenia genetic research has made tremendous progress in the past decade, most findings are at basic science level and clinical applications are limited. It is premature to attempt to use genetic markers to help diagnose schizophrenia or other psychiatric disorders.40 Researchers hope that new gene discovery will translate to better understanding of the pathophysiological mechanisms underlying schizophrenia, which in turn lead to finding novel molecular targets for new drug development. Furthermore, pharmacogenetics helps clinicians use existing drugs more efficiently by maximizing efficacy and minimizing side effects. Several institutions have experimented with genotyping CYP450 in routine clinical practice,41 but prospective pharmacogenetic clinical trials are needed to validate the utility and cost-effectiveness of genetic testing-guided treatment algorithms.42
Bottom Line
Variations in multiple genes likely cause slight deviations in neurodevelopment that interact with environmental variables and lead to development of schizophrenia. Genome-wide association studies are allowing researchers to gain insight into which patients may have increased susceptibility to the disorder, identify potential molecular targets for new drugs, and expand their knowledge of how to best use medications.
Related Resource
- National Institute of Mental Health Center for Collaborative Genomic Studies on Mental Disorders. Schizophrenia. www.nimhgenetics.org/available_data/schizophrenia.
Drug Brand Names
- Abacavir • Ziagen
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Haloperidol • Haldol
- Iloperidone • Fanapt
- Lamotrigine • Lamictal
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Risperidone • Risperdal
Disclosures
Dr. Zhang reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Malhotra is a consultant to Genomind, Inc.
This work was partly supported by a Young Investigator Award from the Brain and Behavior Research Foundation (Dr. Zhang), and by the National Institute of Mental Health (P50MH080173 to Dr. Malhotra and 1K23MH097108 to Dr. Zhang).
Discuss this article at www.facebook.com/CurrentPsychiatry
Genetic factors play a major role in the etiology and development of schizophrenia. Genetic linkage studies and twin studies have estimated the heritability of schizophrenia to be 70% to 90%.1 Research on the genetic underpinnings of schizophrenia has accelerated since the Human Genome Project was completed in 2001, which opened the door to expanding our understanding of molecular mechanisms of human diseases. Experts have hailed the dawn of personalized medicine,2 hoping that we will be able to use knowledge of the human genome to tailor individual treatment.
In this article we review some significant recent findings in genetics of schizophrenia. Gene names are italicized and proteins coded by genes are not. The names, functions, and locations of all genes included in this article appear in Table 1. For a glossary of genetic terms, see Table 2.
Table 1
Select genes and their functions
| Gene | Name | Location | Function(s) |
|---|---|---|---|
| CACNA1C | Calcium channel, voltage-dependent, L type, alpha 1C subunit | 12p13.3 | Calcium channels mediate the influx of calcium ions into the cell upon membrane polarization |
| COMT | Catechol-O-methyltransferase | 22q11.21 | Key enzyme in degradation of dopamine and norepinephrine |
| CSMD1 | CUB and Sushi multiple domains 1 | 8p23.2 | One of the proteins that modulate the classical complement pathway, part of the immune system |
| CYP2D6 | Cytochrome P450 2D6 | 22q13.1 | Key enzyme in drug metabolism |
| C10orf26 | Chromosome 10 open reading frame 26 | 10q24.32 | Unknown |
| DISC1 | Disrupted in schizophrenia 1 | 1q42 | Neurite outgrowth, cortical development, synaptic function |
| DRD1 | Dopamine receptor D1 | 5q35.1 | D1 receptors regulate neuronal growth and development, mediate behavioral responses, and modulate D2 receptor-mediated events |
| DRD2 | Dopamine receptor D2 | 11q23 | D2 receptors regulate motor activities and information processing in the brain |
| DTNBP1 | Dystrobrevin binding protein 1 | 6p22 | Neurodevelopment and synaptic transmission |
| HLA-DQB1 | Major histocompatibility complex, class II, DQ beta 1 | 6p21.3 | Plays a central role in the immune system by presenting peptides derived from extracellular proteins |
| HTR2C | Serotonin receptor 2C | Xq24 | Modulate mood, food intake behavior, and feeling of satiety |
| MC4R | Melanocortin 4 receptor | 18q22 | Modulate food intake behavior and feeling of satiety |
| MHC region | Major histocompatibility complex | 6p21-22 | Immune function; neurodevelopment, synaptic plasticity |
| MIR137 | MicroRNA 137 | 1p23.3 | Post-transcriptional regulation of messenger RNAs; neuron maturation, adult neurogenesis |
| MTHFR | Methylenetetrahydrofolate reductase | 1p36.3 | Key enzyme in folate metabolism |
| TCF4 | Transcription factor 4 | 18q21.2 | Neuronal transcriptional factor, neurogenesis |
| TPH1 | Tryptophan hydroxylase 1 | 11p15.3 | Key enzyme in biosynthesis of serotonin |
| ZNF804A | Zinc finger protein 804A | 2q32.1 | Transcription factor, neuronal connectivity in the dorsolateral prefrontal cortex |
Table 2
Glossary of genetic terms
| Allele: One of several variants of a gene, usually referring to a specific site within the gene |
| Association study: Genetic association refers to the association between a particular genotype and a phenotypic trait in the population. Genetic association studies aim to test whether single-locus alleles genotype frequencies or multi-locus haplotype frequencies differ between 2 groups (such as cases and controls) |
| Candidate gene study: A study that evaluates association of specific genetic variants with outcomes or traits of interest, selecting variants to be tested according to explicit considerations (known or postulated biology or function, previous studies, etc.) |
| Case-control design: An association study design in which the primary comparison is between a group of individuals (cases) ascertained for the phenotype of interest (eg, patients with schizophrenia) and a second group (control) ascertained for not having the phenotype (eg, healthy controls) |
| Copy number variation: A class of DNA sequence variant (including deletions and duplications) in which the result is a departure from the expected 2-copy representation of DNA sequence (ie, each person has 2 copies of the same chromosome) |
| Endophenotype: Phenotypes that are genetically determined, directly measurable traits as part of a complex illness. This term is used to connect the pathway from genes to a disease (eg, impairment in working memory is an endophenotype of schizophrenia) |
| Genetic association: A relationship that is defined by the nonrandom occurrence of a genetic marker with a trait, which suggests an association between the genetic marker (or a marker close to it) and disease pathogenesis |
| Genetic marker: A specific genetic variant known to be associated with a recognizable trait or disease |
| Genome: The entire collection of genetic information (or genes) that an organism possesses |
| Genome-wide association study: A study that evaluates association of genetic variation with outcomes or traits of interest by using 300,000 to 1,000,000 markers across the whole genome. No hypothesis about any particular gene is required for GWAS |
| Genotype: The genetic constitution of an individual, either overall or at a specific gene |
| Heritability (h2): A measure of the strength of genetic effects on a trait. It is defined as the proportion of the phenotypic variation in a trait that is attributable to genetic effects |
| Linkage disequilibrium (LD): Two polymorphic loci are in LD when they are co-located, and alleles at those loci are distributed non-randomly with respect to each other on chromosomes in the population |
| Linkage study: A technique used in genetic epidemiology that focuses on linking a chromosome region to transmission of a particular trait across multiple familial generations |
| Phenotype: The observable characteristics of a cell or organism, usually being the results of the product coded by a gene (genotype) |
| Polymorphism: The existence of ≥2 variants of a gene, occurring in a population, with at least 1% frequency of the less common variant |
| Recombination hotspot: Recombination is breaking and rejoining of DNA strands to form new DNA molecules encoding a novel set of genetic information. Recombination hotspots are individual regions within the genome that have frequent recombination events (eg, the human leukocyte antigen region is a recombination hotspot) |
| Single nucleotide polymorphism: A single base pair change in the DNA sequence at a particular point, compared with the “common” or “wild type” sequence |
| Translocation: A type of chromosomal abnormality resulted by rearrangement of parts between nonhomologous chromosomes, often leading to cancer or developmental abnormalities |
Focusing on single nucleotide polymorphisms
Genetic research of diseases previously relied on linkage studies, which focus on linking a chromosome region to transmission of a particular trait across multiple familial generations. This approach has identified several genomic regions that may be associated with schizophrenia, but most of these regions contain multiple genes and are not specific to schizophrenia.
Today, many genetic studies examine variations of a single nucleotide in the DNA sequence, ie, a change of 1 letter in a particular location on the DNA chain. Single nucleotide polymorphisms (SNPs)—relatively common DNA variations found in >5% of the population—have been a major focus of psychiatric genetics in the past decade. Technology now allows researchers to simultaneously genotype millions of SNPs across the genome, producing tremendous power to investigate the entire genome in relation to a phenotype (a disease or a trait) in genome-wide association studies (GWAS).3 GWAS do not require an a priori hypothesis regarding which regions or genes may be important, and have yielded many novel genetic variants implicated in schizophrenia.
Susceptibility genes
Genetic researchers initially hoped to find that one or a few genes are responsible for schizophrenia. However, recent research revealed that many genes may be involved in susceptibility to schizophrenia, and that a particular gene may contribute to the risk of not only schizophrenia but also other psychiatric disorders such as bipolar disorder (BD).
Discovery of the DISC1 gene is an example of how our understanding of the complex genetic architecture in psychiatric disorders has evolved. In 2000, a linkage study in a Scottish family cohort found a translocation on chromosome 1, t(1:11), highly correlated with schizophrenia.4 Later studies found that this translocation directly disrupts a gene, which researchers named “disrupted in schizophrenia 1.” The protein encoded by DISC1 appears to provide a scaffold to other proteins involved in multiple cellular functions, particularly regulation of brain development and maturation. It is involved in neuronal proliferation, differentiation, and migration via various signaling pathways by interacting with many other proteins.5 Disruption of DISC1 results in dysfunction in multiple neurodevelopmental processes, significantly increasing susceptibility not only for schizophrenia but also for BD and depression.
Many common variants of DISC1 slightly alter expression levels of the gene, which may exert subtle but pervasive effects on neural circuitry development. DISC1 knockout mouse models showed close interactions between DISC1 and N-methyl-d-aspartate receptors and dopamine D2 receptors, linking to the glutamate hypothesis of schizophrenia and the common site of action of antipsychotics. Despite advances in understanding the biology of DISC1, large case-control studies have not found a consistent association between DISC1 and schizophrenia.6,7 It is possible that DISC1 pathology represents one subtype of schizophrenia that is not prevalent among the general population; therefore, large-scale epidemiologic studies could not find evidence to support DISC1’s role in schizophrenia.
DTNBP1 is another schizophrenia susceptibility gene discovered in linkage studies. Originally found in a large Irish cohort, several SNPs of DTNBP1 were significantly associated with schizophrenia.8 A meta-analysis of candidate genes identified DTNBP1 as one of 4 genes with the strongest evidence for association with schizophrenia (the other 3 are DRD1, MTHFR, and TPH1).9 DTNBP1 is widely expressed in the brain and is present in presynaptic, postsynaptic, and microtubule locations implicated in a number of brain functions, including synaptic transmission and neurite outgrowth in a developing organism. Furthermore, DTNBP1 is associated with cognitive functions in schizophrenia patients10 as well as in control subjects.11 Cognitive impairment is considered an endophenotype for schizophrenia. Similar to DISC1 and other candidate genes, DTNBP1 has not emerged as a significant hit in later, large-scale GWAS studies.
Since the first schizophrenia GWAS in 2007,12 >15 GWAS have been published, with increasingly larger samples sizes. GWAS are based on the “common disease/common variant hypothesis” that common disorders such as diabetes, macular degeneration, and schizophrenia are caused by multiple common variants in the genome. Because GWAS can analyze hundreds of thousands of SNPs simultaneously, a stringent criterion (usually P < 5×10-8) is used to gauge statistical significance to correct for multiple testing. Because most effect sizes associated with genetic markers in psychiatry are fairly small (odds ratios [ORs] are approximately 1.1 to 1.2), large samples are required to detect significant effects. Several international consortia have accumulated large samples. The Psychiatric GWAS Consortium has >17,000 patients with schizophrenia, >11,000 with BD, >16,000 with major depression, and >50,000 healthy controls. This wave of GWAS has implicated several novel genomic regions in schizophrenia pathophysiology, including ZNF804A, the major histocompatibility complex (MHC) region, and MIR137.
ZNF804A was the first gene that reached genome-wide significance in a large GWAS,13 and this finding has been replicated. The function of this novel gene largely is unknown. ZNF804A is widely expressed in the brain, especially in the developing hippocampus and the cortex as well as in the adult cerebellum. Recent studies found that ZNF804A is a putative transcription factor, upregulating expression of catechol-O-methyltransferase while downregulating dopamine D2 receptors in animal studies.14 The minor allele of SNP rs1344706 was associated with impaired brain functional connectivity in a human study.15 More work is needed to understand how this gene increases schizophrenia susceptibility.
The MHC region on chromosome 6p22.1,1 also was significant in schizophrenia GWAS,16,17 and this may be the most replicated schizophrenia GWAS finding. This region is a recombination hotspot and harbors many genetic variants. Many immune-related genes previously were associated with autoimmune and infectious disorders, which may suggest that the immunologic system plays a role in schizophrenia pathogenesis. These genes also may involve neurodevelopment, synaptic plasticity, and other neuronal processes.18 However, the complex gene composition in the region makes it difficult to pinpoint the exact signal to schizophrenia pathophysiology.
The most recent finding from the largest GWAS is MIR137,19 coding for microRNA 137, which was associated with schizophrenia at P=1.6×10-11 in 17,836 patients and 33,859 controls. MicroRNAs are small, noncoding RNA fragments that are involved in post-transcriptional regulation of messenger RNAs. MIR137 plays important roles in neuron maturation and adult neurogenesis by acting at the level of dendritic morphogenesis and spine development.20 More interestingly, the other 4 loci achieving genome-wide significance in the same GWAS (TCF4, CACNA1C, CSMD1, and C10orf26) contain predicted target sites of MIR137. This suggests MIR137-mediated dysregulation may be an etiologic mechanism in schizophrenia.
Limitations of these findings. The effect sizes of these genetic variants are small, explaining only 1% to 2% of genetic risks of schizophrenia. However, this is not unique to schizophrenia or psychiatry. “Missing heritability” is puzzling in other branches of medicine.21 Future research will focus on gene-environment interactions as well as gene-gene interactions in relation to schizophrenia’s neurodevelopmental processes.
In addition, many top hits in GWAS are SNPs that are not functional or located in intergenic regions with unknown functions. They may be proxies of causal variants that truly play causal roles in pathogenesis of diseases but were not genotyped in those studies. Recently, researchers have grown increasingly interested in copy number variations (CNVs) in the etiology of complex diseases. Compared with SNPs, CNVs usually are much larger changes in the DNA sequence, including deletions and duplications of a large chunk of DNA segments. Disease-causing CNVs are rare but have large effect sizes. Recent studies have examined the role of CNVs in schizophrenia.22,23
Although genes such as DISC1 and CACNA1C are linked to schizophrenia, they are neither necessary nor sufficient for developing the disorder, and also are linked equally, if not more strongly, to other neuropsychiatric disorders, including BD and autism. Therefore, they are not “schizophrenia genes.” Variations in multiple genes likely cause slight deviations in neurodevelopment that interact with environmental variables and lead to development of schizophrenia.
Nevertheless, these schizophrenia GWAS findings provide insight into this complex disorder. Much work is needed to move from these association signals to understanding the function and regulation of these genes to turn basic biologic knowledge into targets for new drugs or other interventions.
Antipsychotic pharmacogenetics
Genetic research of schizophrenia also contributes to our knowledge of how to best use existing drugs. Medications for treating schizophrenia often need to be changed because patients experience lack of efficacy or intolerable side effects, which may lead them to discontinue treatment. Clinical predictors of which medication would work for an individual patient are lacking. Pharmacogenetics may be able to fulfill the promise of personalized medicine in psychiatry by using genetic information to guide drug selection to maximize therapeutic efficacy and minimize drug-induced side effects.
Researchers first attempted to find genetic predictors of antipsychotic efficacy in the early 1990s. One replicated finding is that DRD2, the gene coding for dopamine receptor D2, is associated with antipsychotic efficacy. This may not be surprising because D2 receptor antagonism is a common and necessary drug action mechanism for all antipsychotics. One SNP, -141C Ins/Del (rs1799732), represents a deletion (vs insertion) of cytosine at position -141, located in the 5’ promoter region of DRD2. Pre-clinical studies showed that this SNP might modulate DRD2 gene expression and influence D2 receptor density in the brain. Del allele carriers had poor response to clozapine among a treatment-refractory sample24 and took longer to respond to olanzapine and risperidone among first-episode schizophrenia patients.25 A 2010 meta-analysis of approximately 700 patients26 showed that the -141C Ins/Del polymorphism is significantly associated with antipsychotic response. Patients who carry 1 or 2 Del alleles tend to have a less favorable antipsychotic response than patients with the Ins/Ins genotype. Patients with the Ins/Ins genotype are 54% more likely to respond to antipsychotics than those with ≥1 copy of the Del allele.
Researchers have studied other genes in relation to antipsychotic efficacy, but have yielded few consistent findings.27 Some have looked at combining multiple SNPs across several genes to predict antipsychotic efficacy, but these findings have not been replicated. For example, a combination of variants in the HTR2A, HTR2C, and 5-HTTLPR genes and genes coding for H2 receptors was found to correctly predict clozapine response in 76% of patients.28 However, this finding was not replicated in an independent sample.29 A recent GWAS30 found that a combination of 6 genetic markers—NPAS3, XKR4, TNR, GRIA4, GFRA2, and NUDT9P1—predicted treatment response to iloperidone. Although promising, this finding needs to be validated in independent samples.
Predicting adverse drug events
In other branches of medicine, researchers have used pharmacogenetics to successfully identify predictors of drug-induced adverse events. A GWAS found that a specific human leukocyte antigen (HLA) allele markedly increases the risk of liver toxicity from flucloxacillin (OR=80.6).31 This HLA marker also is related to hypersensitivity reaction to abacavir, a common medication for treating AIDS, and lamotrigine-induced Stevens-Johnson syndrome.
Clozapine-induced granulocytosis also may be related to genetic variation in the HLA region. Despite superior efficacy, clozapine remains underutilized in part because it carries the risk of potentially fatal agranulocytosis. Identifying a genetic marker for agranulocytosis would lift the burden of weekly blood monitoring. A recent pharmacogenetic study detected a replicated association of an allele at the HLA-DQB1 locus with risk of agranulocytosis in 2 small groups of clozapine-treated schizophrenia patients.32 Effect sizes were extremely high (OR=16.86); nearly 90% of allele carriers developed agranulocytosis. Unfortunately, the overall sensitivity of the marker was 21%, indicating that most individuals who develop agranulocytosis are not carriers of the allele and presumably have other genetic risk factors. A more comprehensive risk profile would be necessary to obviate the need for weekly blood monitoring.
Weight gain and metabolic syndrome are common side effects of antipsychotics, and no clear clinical predictors have been identified. Researchers have examined potential genetic markers in association with antipsychotic-induced weight gain. One consistent finding has been that a single SNP in the promoter region of the HTR2C gene (serotonin receptor 2C), C-759T (rs3813929), affects antipsychotic-induced weight gain. The 5-HT2C receptor is involved in regulating food intake in rodents and is related to late-onset diabetes and obesity in humans. HTR2C knockout mice display chronic hyperphagia that leads to obesity and hyperinsulinemia. Since the original finding in 2002,33 at least 17 studies have reported on the association between the C-759T SNP in HTR2C and antipsychotic-induced weight gain. A meta-analysis found that the T allele was significantly protective against antipsychotic-induced weight gain.34 The C allele was associated with >2-fold increase of risk for clinically significant weight gain (gaining >7% of baseline body weight).
In a GWAS of antipsychotic-induced weight gain in pediatric patients who were prescribed antipsychotics for the first time, researchers discovered a single top signal at a marginally genome-wide significant level (P=1.6×10-7).35 This was replicated in 3 other independent samples. The peak signal is located on chromosome 18q21, overlapping a peak identified as a predictor of obesity. This locus is approximately 150 kb downstream from MC4R, the melanocortin 4 receptor gene, which has long been suspected as a candidate for weight-related phenotypes, including antipsychotic-induced weight gain.36 Mutations in this gene are linked with extreme obesity in humans, and MC4R knockout mice develop obesity. MC4R-expressing neurons in the ventromedial hypothalamus are regulated by circulating levels of leptin via pathways in the arcuate nucleus. In turn, MC4R regulates 5-HT2C receptors, which are implicated in weight gain. In the discovery sample, risk allele homozygotes gained twice as much weight as other patients after 12 weeks of treatment, and the genetic effect was not drug-specific. The consistency of HTR2C-MC4R findings poses a possibility that a drug may be developed at these targets to treat or prevent antipsychotic-induced weight gain.
Drug metabolism. Pharmacogenetic studies of antipsychotic drug response also have focused on genes that code for enzymes in drug metabolism, particularly cytochrome (CYP) 450 enzymes, which are responsible for the metabolism of many drugs. CYP2D6 is the main metabolic pathway for several antipsychotics, including risperidone, aripiprazole, haloperidol, and perphenazine. The CYP2D6 gene contains >100 variants, many of which yield nonfunctional or reduced-function enzymes. There are 4 phenotypes of CYP2D6 produced by combinations of various alleles with different degrees of enzymatic activities: poor (PM), intermediate (IM), extensive (EM), and ultrarapid metabolizers (UM). Compared with EMs with normal CYP2D6 enzyme activity, PMs and IMs have minimal or reduced activity, respectively. UMs have duplicate or multiple copies of the gene that result in increased enzyme activity. Approximately 7% to 10% of whites and 1% to 2% of Asians are PMs, who tend to accumulate higher serum drug levels and, theoretically, require lower doses to achieve therapeutic effects. UMs, in contrast, consist of 1% of the population and may require higher doses because of faster drug elimination.37 Therefore, CYP2D6 metabolic status could play an important role in determining patients’ antipsychotic response. So far, no empirical data support the association between CYP2D6 and antipsychotic efficacy, although studies have found significant relationships between PMs and higher rates of drug-induced side effects such as tardive dyskinesia (TD), extrapyramidal symptoms, and weight gain. A meta-analysis38 of 8 studies showed that PMs had a 43% higher risk of developing TD compared with EMs. An FDA-approved pharmacogenetic test, AmpliChip® CYP450 Test, is available to assess CYP2D6 and CYP2C19 genotypes,39 but its use is limited, perhaps because of clinician concerns about how to interpret test results, paucity of prospective data suggesting that using the test can improve clinical outcomes, and lack of reimbursement.
Implications for clinical practice
Although schizophrenia genetic research has made tremendous progress in the past decade, most findings are at basic science level and clinical applications are limited. It is premature to attempt to use genetic markers to help diagnose schizophrenia or other psychiatric disorders.40 Researchers hope that new gene discovery will translate to better understanding of the pathophysiological mechanisms underlying schizophrenia, which in turn lead to finding novel molecular targets for new drug development. Furthermore, pharmacogenetics helps clinicians use existing drugs more efficiently by maximizing efficacy and minimizing side effects. Several institutions have experimented with genotyping CYP450 in routine clinical practice,41 but prospective pharmacogenetic clinical trials are needed to validate the utility and cost-effectiveness of genetic testing-guided treatment algorithms.42
Bottom Line
Variations in multiple genes likely cause slight deviations in neurodevelopment that interact with environmental variables and lead to development of schizophrenia. Genome-wide association studies are allowing researchers to gain insight into which patients may have increased susceptibility to the disorder, identify potential molecular targets for new drugs, and expand their knowledge of how to best use medications.
Related Resource
- National Institute of Mental Health Center for Collaborative Genomic Studies on Mental Disorders. Schizophrenia. www.nimhgenetics.org/available_data/schizophrenia.
Drug Brand Names
- Abacavir • Ziagen
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Haloperidol • Haldol
- Iloperidone • Fanapt
- Lamotrigine • Lamictal
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Risperidone • Risperdal
Disclosures
Dr. Zhang reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Malhotra is a consultant to Genomind, Inc.
This work was partly supported by a Young Investigator Award from the Brain and Behavior Research Foundation (Dr. Zhang), and by the National Institute of Mental Health (P50MH080173 to Dr. Malhotra and 1K23MH097108 to Dr. Zhang).
1. Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60(12):1187-1192.
2. de Leon J. AmpliChip CYP450 test: personalized medicine has arrived in psychiatry. Expert Rev Mol Diagn. 2006;6(3):277-286.
3. Psychiatric GWAS Consortium Coordinating Committee; Cichon S, Craddock N, Daly M, et al. Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am J Psychiatry. 2009;166(5):540-556.
4. Millar JK, Wilson-Annan JC, Anderson S, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9(9):1415-1423.
5. Porteous DJ, Millar JK, Brandon NJ, et al. DISC1 at 10: connecting psychiatric genetics and neuroscience. Trends Mol Med. 2011;17(12):699-706.
6. Schumacher J, Laje G, Abou Jamra R, et al. The DISC locus and schizophrenia: evidence from an association study in a central European sample and from a meta-analysis across different European populations. Hum Mol Genet. 2009;18(14):2719-2727.
7. Mathieson I, Munafò MR, Flint J, et al. Meta-analysis indicates that common variants at the DISC1 locus are not associated with schizophrenia. Mol Psychiatry. 2012;17(6):634-641.
8. Straub RE, Jiang Y, MacLean CJ, et al. Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet. 2002;71(2):337-348.
9. Allen NC, Bagade S, McQueen MB, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40(7):827-834.
10. Burdick KE, Lencz T, Funke B, et al. Genetic variation in DTNBP1 influences general cognitive ability. Hum Mol Genet. 2006;15(10):1563-1568.
11. Zhang JP, Burdick KE, Lencz T, et al. Meta-analysis of genetic variation in DTNBP1 and general cognitive ability. Biol Psychiatry. 2010;68(12):1126-1133.
12. Lencz T, Morgan TV, Athanasiou M, et al. Converging evidence for a pseudoautosomal cytokine receptor gene locus in schizophrenia. Mol Psychiatry. 2007;12(6):572-580.
13. O’Donovan MC, Craddock N, Norton N, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008;40(9):1053-1055.
14. Girgenti MJ, LoTurco JJ, Maher BJ. ZNF804a regulates expression of the schizophrenia-associated genes PRSS16 COMT, PDE4B, and DRD2. PLoS One. 2012;7(2):e32404.-
15. Lencz T, Szeszko PR, DeRosse P, et al. A schizophrenia risk gene, ZNF804A, influences neuroanatomical and neurocognitive phenotypes. Neuropsychopharmacology. 2010;35(11):2284-2291.
16. International Schizophrenia Consortium; Purcell SM, Wray NR, Stone JL, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748-752.
17. Stefansson H, Ophoff RA, Steinberg S, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460(7256):744-747.
18. Handel AE, Ramagopalan SV. The potential role of major histocompatibility complex class I in schizophrenia. Biol Psychiatry. 2010;68(7):e29-e30.
19. Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43(10):969-976.
20. Gallego JA, Gordon ML, Claycomb K, et al. In vivo microRNA detection and quantitation in cerebrospinal fluid. J Mol Neurosci. 2012;47(2):243-248.
21. Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747-753.
22. Walsh T, McClellan JM, McCarthy SE, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320(5875):539-543.
23. Rees E, Kirov G, O’Donovan MC, et al. De novo mutation in schizophrenia. Schizophr Bull. 2012;38(3):377-381.
24. Malhotra AK, Buchanan RW, Kim S. Allelic variation in the promotor region of the dopamine D2 receptor gene and clozapine response. Schizophr Res. 1999;36:92-93.
25. Lencz T, Robinson DG, Xu K, et al. DRD2 promoter region variation as a predictor of sustained response to antipsychotic medication in first-episode schizophrenia patients. Am J Psychiatry. 2006;163(3):529-531.
26. Zhang JP, Lencz T, Malhotra AK. D2 receptor genetic variation and clinical response to antipsychotic drug treatment: a meta-analysis. Am J Psychiatry. 2010;167(7):763-772.
27. Zhang JP, Malhotra AK. Pharmacogenetics and antipsychotics: therapeutic efficacy and side effects prediction. Expert Opin Drug Metab Toxicol. 2011;7(1):9-37.
28. Arranz MJ, Munro J, Birkett J, et al. Pharmacogenetic prediction of clozapine response. Lancet. 2000;355(9215):1615-1616.
29. Schumacher J, Schulze TG, Wienker TF, et al. Pharmacogenetics of the clozapine response. Lancet. 2000;356(9228):506-507.
30. Lavedan C, Licamele L, Volpi S, et al. Association of the NPAS3 gene and five other loci with response to the antipsychotic iloperidone identified in a whole genome association study. Mol Psychiatry. 2009;14(8):804-819.
31. Daly AK, Donaldson PT, Bhatnagar P, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41(7):816-819.
32. Athanasiou MC, Dettling M, Cascorbi I, et al. Candidate gene analysis identifies a polymorphism in HLA-DQB1 associated with clozapine-induced agranulocytosis. J Clin Psychiatry. 2011;72(4):458-463.
33. Reynolds GP, Zhang ZJ, Zhang XB. Association of antipsychotic drug-induced weight gain with a 5-HT2C receptor gene polymorphism. Lancet. 2002;359(9323):2086-2087.
34. Sicard MN, Zai CC, Tiwari AK, et al. Polymorphisms of the HTR2C gene and antipsychotic-induced weight gain: an update and meta-analysis. Pharmacogenomics. 2010;11(11):1561-1571.
35. Malhotra AK, Correll CU, Chowdhury NI, et al. Association between common variants near the melanocortin 4 receptor gene and severe antipsychotic drug-induced weight gain. Arch Gen Psychiatry. 2012;69(9):904-912.
36. Correll CU, Malhotra AK. Pharmacogenetics of antipsychotic-induced weight gain. Psychopharmacology (Berl). 2004;174(4):477-489.
37. Zhang JP, Malhotra AK. Pharmacogenetics and antipsychotics: therapeutic efficacy and side effects prediction. Expert Opin Drug Metab Toxicol. 2011;7(1):9-37.
38. Patsopoulos NA, Ntzani EE, Zintzaras E, et al. CYP2D6 polymorphisms and the risk of tardive dyskinesia in schizophrenia: a meta-analysis. Pharmacogenet Genomics. 2005;15(3):151-158.
39. de Leon J. AmpliChip CYP450 test: personalized medicine has arrived in psychiatry. Expert Rev Mol Diagn. 2006;6(3):277-286.
40. Mitchell PB, Meiser B, Wilde A, et al. Predictive and diagnostic genetic testing in psychiatry. Psychiatr Clin North Am. 2010;33(1):225-243.
41. Rundell JR, Staab JP, Shinozaki G, et al. Pharmacogenomic testing in a tertiary care outpatient psychosomatic medicine practice. Psychosomatics. 2011;52(2):141-146.
42. Malhotra AK, Zhang JP, Lencz T. Pharmacogenetics in psychiatry: translating research into clinical practice. Mol Psychiatry. 2012;17(8):760-769.
1. Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60(12):1187-1192.
2. de Leon J. AmpliChip CYP450 test: personalized medicine has arrived in psychiatry. Expert Rev Mol Diagn. 2006;6(3):277-286.
3. Psychiatric GWAS Consortium Coordinating Committee; Cichon S, Craddock N, Daly M, et al. Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am J Psychiatry. 2009;166(5):540-556.
4. Millar JK, Wilson-Annan JC, Anderson S, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9(9):1415-1423.
5. Porteous DJ, Millar JK, Brandon NJ, et al. DISC1 at 10: connecting psychiatric genetics and neuroscience. Trends Mol Med. 2011;17(12):699-706.
6. Schumacher J, Laje G, Abou Jamra R, et al. The DISC locus and schizophrenia: evidence from an association study in a central European sample and from a meta-analysis across different European populations. Hum Mol Genet. 2009;18(14):2719-2727.
7. Mathieson I, Munafò MR, Flint J, et al. Meta-analysis indicates that common variants at the DISC1 locus are not associated with schizophrenia. Mol Psychiatry. 2012;17(6):634-641.
8. Straub RE, Jiang Y, MacLean CJ, et al. Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet. 2002;71(2):337-348.
9. Allen NC, Bagade S, McQueen MB, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40(7):827-834.
10. Burdick KE, Lencz T, Funke B, et al. Genetic variation in DTNBP1 influences general cognitive ability. Hum Mol Genet. 2006;15(10):1563-1568.
11. Zhang JP, Burdick KE, Lencz T, et al. Meta-analysis of genetic variation in DTNBP1 and general cognitive ability. Biol Psychiatry. 2010;68(12):1126-1133.
12. Lencz T, Morgan TV, Athanasiou M, et al. Converging evidence for a pseudoautosomal cytokine receptor gene locus in schizophrenia. Mol Psychiatry. 2007;12(6):572-580.
13. O’Donovan MC, Craddock N, Norton N, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008;40(9):1053-1055.
14. Girgenti MJ, LoTurco JJ, Maher BJ. ZNF804a regulates expression of the schizophrenia-associated genes PRSS16 COMT, PDE4B, and DRD2. PLoS One. 2012;7(2):e32404.-
15. Lencz T, Szeszko PR, DeRosse P, et al. A schizophrenia risk gene, ZNF804A, influences neuroanatomical and neurocognitive phenotypes. Neuropsychopharmacology. 2010;35(11):2284-2291.
16. International Schizophrenia Consortium; Purcell SM, Wray NR, Stone JL, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748-752.
17. Stefansson H, Ophoff RA, Steinberg S, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460(7256):744-747.
18. Handel AE, Ramagopalan SV. The potential role of major histocompatibility complex class I in schizophrenia. Biol Psychiatry. 2010;68(7):e29-e30.
19. Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43(10):969-976.
20. Gallego JA, Gordon ML, Claycomb K, et al. In vivo microRNA detection and quantitation in cerebrospinal fluid. J Mol Neurosci. 2012;47(2):243-248.
21. Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747-753.
22. Walsh T, McClellan JM, McCarthy SE, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320(5875):539-543.
23. Rees E, Kirov G, O’Donovan MC, et al. De novo mutation in schizophrenia. Schizophr Bull. 2012;38(3):377-381.
24. Malhotra AK, Buchanan RW, Kim S. Allelic variation in the promotor region of the dopamine D2 receptor gene and clozapine response. Schizophr Res. 1999;36:92-93.
25. Lencz T, Robinson DG, Xu K, et al. DRD2 promoter region variation as a predictor of sustained response to antipsychotic medication in first-episode schizophrenia patients. Am J Psychiatry. 2006;163(3):529-531.
26. Zhang JP, Lencz T, Malhotra AK. D2 receptor genetic variation and clinical response to antipsychotic drug treatment: a meta-analysis. Am J Psychiatry. 2010;167(7):763-772.
27. Zhang JP, Malhotra AK. Pharmacogenetics and antipsychotics: therapeutic efficacy and side effects prediction. Expert Opin Drug Metab Toxicol. 2011;7(1):9-37.
28. Arranz MJ, Munro J, Birkett J, et al. Pharmacogenetic prediction of clozapine response. Lancet. 2000;355(9215):1615-1616.
29. Schumacher J, Schulze TG, Wienker TF, et al. Pharmacogenetics of the clozapine response. Lancet. 2000;356(9228):506-507.
30. Lavedan C, Licamele L, Volpi S, et al. Association of the NPAS3 gene and five other loci with response to the antipsychotic iloperidone identified in a whole genome association study. Mol Psychiatry. 2009;14(8):804-819.
31. Daly AK, Donaldson PT, Bhatnagar P, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41(7):816-819.
32. Athanasiou MC, Dettling M, Cascorbi I, et al. Candidate gene analysis identifies a polymorphism in HLA-DQB1 associated with clozapine-induced agranulocytosis. J Clin Psychiatry. 2011;72(4):458-463.
33. Reynolds GP, Zhang ZJ, Zhang XB. Association of antipsychotic drug-induced weight gain with a 5-HT2C receptor gene polymorphism. Lancet. 2002;359(9323):2086-2087.
34. Sicard MN, Zai CC, Tiwari AK, et al. Polymorphisms of the HTR2C gene and antipsychotic-induced weight gain: an update and meta-analysis. Pharmacogenomics. 2010;11(11):1561-1571.
35. Malhotra AK, Correll CU, Chowdhury NI, et al. Association between common variants near the melanocortin 4 receptor gene and severe antipsychotic drug-induced weight gain. Arch Gen Psychiatry. 2012;69(9):904-912.
36. Correll CU, Malhotra AK. Pharmacogenetics of antipsychotic-induced weight gain. Psychopharmacology (Berl). 2004;174(4):477-489.
37. Zhang JP, Malhotra AK. Pharmacogenetics and antipsychotics: therapeutic efficacy and side effects prediction. Expert Opin Drug Metab Toxicol. 2011;7(1):9-37.
38. Patsopoulos NA, Ntzani EE, Zintzaras E, et al. CYP2D6 polymorphisms and the risk of tardive dyskinesia in schizophrenia: a meta-analysis. Pharmacogenet Genomics. 2005;15(3):151-158.
39. de Leon J. AmpliChip CYP450 test: personalized medicine has arrived in psychiatry. Expert Rev Mol Diagn. 2006;6(3):277-286.
40. Mitchell PB, Meiser B, Wilde A, et al. Predictive and diagnostic genetic testing in psychiatry. Psychiatr Clin North Am. 2010;33(1):225-243.
41. Rundell JR, Staab JP, Shinozaki G, et al. Pharmacogenomic testing in a tertiary care outpatient psychosomatic medicine practice. Psychosomatics. 2011;52(2):141-146.
42. Malhotra AK, Zhang JP, Lencz T. Pharmacogenetics in psychiatry: translating research into clinical practice. Mol Psychiatry. 2012;17(8):760-769.
Psychiatric ‘holds’ for nonpsychiatric patients
Dear Dr. Mossman,
At the general hospital where I work, doctors and nurses sometimes ask me to fill out psychiatric “hold” documents to keep seriously ill medical or surgical patients from leaving the hospital. Last week, they asked me to stop Mr. J, a man with diabetes and a gangrenous lower leg, from leaving against medical advice (AMA). If he left, he would die. But if I filled out the psychiatric “hold,” I’d be saying the man needed civil commitment for a mental illness, which wasn’t true. If this happens again, what should I do?
Submitted by “Dr. Q”
“It is tempting, if the only tool you have is a hammer, to treat everything as if it were a nail,” wrote Abraham Maslow.1 The situation Dr. Q describes is one that psychiatrists frequently encounter because in some situations, a psychiatric “hold” can seem like the only way to stop a physically ill patient from leaving the hospital AMA. But pounding on this problem with a civil commitment hammer is the wrong response.
What’s wrong with using psychiatric holds in these situations? Do doctors have any other equipment in their medical toolbox for stopping an improvident AMA departure? To find out, we’ll look at:
- what a psychiatric hold does
- why holds don’t apply to medical-surgical treatment
- alternative responses to patients who lack capacity to refuse care.
Psychiatric holds
All states have laws that permit involuntary psychiatric hospitalization. The wording and procedural details in these laws vary across jurisdictions, but all states allow civil (ie, noncriminal) commitment of mentally ill persons who have gross impairments of judgment, behavior, reality-testing, or everyday functioning if their recent behavior show that they pose a danger because of their mental illness.2 Table 13 lists examples of the types of dangers that are potential reasons for civil commitment.
Table 1
Types of risks covered in civil commitment statutes
| All states |
|
| In some jurisdictions |
|
| Source: Adapted from reference 3 |
State laws also allow certain individuals (eg, police) to apprehend and transport mentally ill persons to facilities for psychiatric evaluation. Doctors may hold these persons temporarily until a court decides whether a longer involuntary hospitalization is justified. The documents used to initiate psychiatric holds have various informal names—”5150” (California), “pink slip” (Ohio), “pink paper” (Massachusetts), “Baker Act Form” (Florida)—but their function is the same: permitting lawful restraint of patients whose dangerousness results from their mental illness.
Urgent medical and surgical care
What about medical or surgical patients who refuse care despite being told they’ll die without it? Might involuntary psychiatric hospitalization procedures be a convenient way to keep them from coming to harm?
The answer: probably not, for 4 reasons:
- Once a psychiatric hold has been executed, the person who is subject to detention must be transferred to an appropriate facility within a specified period (usually 24 hours) for further evaluation and care.4,5 In this context, “appropriate facility” means a state-approved psychiatric treatment setting. A hospital’s medical or surgical unit usually would not qualify.
- The lawful use of a psychiatric hold is to declare that someone needs involuntary psychiatric examination for dangerousness arising “as a result of mental illness”—not for danger from a nonpsychiatric medical problem.6 Some civil commitment statutes specify that persons who have serious nonpsychiatric illness but no mental health problems that satisfy civil commitment criteria are to be offered voluntary treatment only.7
- A psychiatric hold only authorizes short-term detention. It does not allow forcing what patients such as Mr. J need: medical or surgical treatment. A psychiatric hold would not solve the problem that Mr. J’s doctors are facing.
- Doctors who execute psychiatric holds in good faith—sincerely believing a patient meets the legal criteria—enjoy statutory immunity from later accusations of malpractice or false imprisonment.8 Using civil commitment mechanisms when one does not actually believe those mechanisms apply might void this immunity.
Nonconsent: 2 varieties
For present purposes, let’s think of nonconsenting medical-surgical patients as coming in 2 varieties:
Variety 1: patients with compromised mental status. Often, medical-surgical patients cannot express objections to treatment because they are unconscious, delirious, or incoherent. Nurses and doctors assume such patients would want proper care and proceed with what they believe is in the patients’ best interest, often with input from family members.
Variety 2: lucid patients who refuse treatment. Patients who do not have obvious psychiatric problems may refuse necessary medical or surgical treatment for various reasons: obstinacy, distrust of doctors, fear, ignorance, incorrect but firmly held ideas about body functioning, cultural differences, or religious beliefs. None of these reasons is necessarily psychopathological, and none provides justification for a psychiatric hold.
Key determinant: Competence
Refusing treatment may be a bad choice and sometimes is evidence of a mental disorder, but it is not, by itself, a mental disorder. When a Variety 2 adult patient refuses care, the key question is, “Is this a competent refusal?” Assessment of a patient’s capacity to make medical decisions is not a skill unique to psychiatrists. Other specialists make judgments about capacity routinely—if only implicitly—when they elicit their patients’ informed consent for care. But when, as in Mr. J’s case, a seriously ill medical-surgical patient refuses lifesaving treatment, our medical colleagues often get psychiatrists involved. Consulting a psychiatrist in such circumstances makes sense, for at least 4 reasons:
- Although assessment of decision-making isn’t the special province of psychiatry, psychiatrists often have more experience assessing the capacity of persons whose thinking seems impaired.
- Psychiatrists also have more experience in detecting subtle indications of mental disorders (eg, mild dementia, depression, psychosis) that can compromise decision-making capacity.
- A nonpsychiatrist may believe that a patient is making a competent refusal but still wants a psychiatrist’s perspective to better understand the patient’s reasoning or to confirm the initial belief.
- Getting an independent opinion is a prudent way to make sure one’s emotions are not adversely influencing a critical judgment about a patient’s treatment.
Determining whether a patient has the requisite capacity to refuse care involves a situation-specific assessment of 4 aspects of mental functioning: expressing a choice coherently, understanding relevant information, appreciating this information, and using the information rationally. Table 29 describes these functional areas in more detail.
Table 2
Evaluating the quality of a patient’s decision: 4 dimensions
| 1. Can the patient communicate a choice and express a consistent preference? |
2. Can the patient grasp relevant information about:
|
| 3. Does the patient appreciate the illness and its consequences? Does he recognize he is ill and acknowledge how the information applies to his situation? |
| 4. Does the patient use the information rationally? Can he explain his decision-making and reasoning? Does he apply information to his situation in light of rational beliefs and desires? |
| Source: Adapted from reference 9 |
If capacity is lacking, what next?
As Judge Benjamin Cardozo ruled nearly a century ago, “Every human being of adult years and sound mind has a right to determine what shall be done with his own body.”10 In a case such as Mr. J’s, where a patient wants to leave the hospital or refuses medical treatment despite grave risk to himself, staff members should not let him leave until his treating doctors have tried to clarify his reasons for leaving and determined whether he has the capacity to give informed consent and refuse treatment. Psychiatrists may be consulted in this process, although the final judgment about capacity rests with the responsible physician. If an assessment shows that the patient has the capacity to make medical decisions, his treatment refusal is binding, even when it creates a clear risk of death.
What should happen if an assessment shows that a gravely ill patient lacks capacity to refuse treatment? Clinicians should consult with the hospital attorney about their facility’s policies and how to implement them properly.
Thinking about the possible legal implications of their actions, treating clinicians might worry that if they detain an unwilling patient without authorization from a court or guardian, they would risk being sued later for false imprisonment. But attorneys are likely to advise clinicians that they have more to fear liability-wise from letting incompetent patients leave the hospital than from detaining them for their own safety. As an Ohio court commented about a police officer who stopped a patient from leaving the hospital:
- What in the name of all that is reasonable should the officer have done? The court finds that the officer acted properly under the circumstances known to him at the time—and the reasonableness of an officer’s actions must be judged at the exigent split second on the street…11
Rather than allowing an incompetent patient to come to harm, attorneys may advise physicians to write an order to keep the patient in the hospital. Then, physicians can obtain consent for treatment from family members, making them aware of any physical or chemical restraint that might be needed to continue the patient’s treatment. Depending on the situation and the reasons for the lack of capacity, hospital staff members may later need to help a family member obtain a court’s authorization for emergency guardianship to allow non-urgent care to continue.
Treating physicians also should document the thinking and findings that support their actions. Table 3 provides an outline for this documentation.
Table 3
Detaining a patient for medical-surgical care: 7 components of documentation
| 1. Description of the patient’s refusal or efforts to leave the hospital |
| 2. Patient’s stated reasons for refusing or wanting to leave |
| 3. Reasonable alternatives to discharge that were offered |
| 4. Description of how refusing medical treatment would create a clear risk of physical harm or death |
| 5. Evidence that the patient lacks capacity to give informed consent or to refuse treatment |
| 6. Actions taken by the treating physician (eg, obtaining psychiatric consultation, enlisting other patient services, instituting physical restraint) |
| 7. Person who provided consent to continue treatment and that person’s relationship to patient |
Related Resources
- Appelbaum PS. Clinical practice. Assessment of patients’ competence to consent to treatment. N Engl J Med. 2007; 357(18):1834-1840.
- Disability Rights California. Involuntary psychiatric treatment: California’s 72-hour hold and 14-day certification. www.disabilityrightsca.org/pubs/502401.pdf.
- Treatment Advocacy Center. Know the laws in your state. www.treatmentadvocacycenter.org/get-help/know-the-laws-in-your-state.
Disclosure
Dr. Mossman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Dr. Mossman thanks David Schwallie, Esq, for his helpful insights about the topics discussed in this article.
1. Maslow AH. The psychology of science: a reconnaissance. New York NY: Harper & Row; 1966.
2. Mossman D, Schwartz AH, Elam ER. Risky business versus overt acts: what relevance do “actuarial” probabilistic risk assessments have for judicial decisions on involuntary psychiatric hospitalization? Houston Journal of Health Law & Policy. 2011;11:365-453.
3. Pinals DA, Mossman D. Evaluation for civil commitment. New York NY: Oxford University Press; 2012.
4. Ohio Revised Code § 5122.10.
5. Oregon Revised Statutes § 426.060.
6. California Welfare and Institutions Code § 5150.
7. Florida statutes § 394.463.
8. Cruze v National Psychiatric Services, Inc., 105 Cal. App. 4th 48 (2003).
9. Appelbaum PS, Grisso T. Assessing patients’ capacities to consent to treatment. N Engl J Med. 1988;319(25):1635-1638.
10. Schloendorff v Society of New York Hospital, 211 N.Y. 125, 105 N.E. 92 (1914).
11. State v Clay, 43 Ohio Misc. 2d 5, 539 N.E.2d 1168 (1988).

Douglas Mossman, MD
Series Editor
Professor and Program Director, University of Cincinnati Forensic Psychiatry Fellowship, Cincinnati, OH
Douglas Mossman, MD
Series Editor
Professor and Program Director, University of Cincinnati Forensic Psychiatry Fellowship, Cincinnati, OH
Douglas Mossman, MD
Series Editor
Professor and Program Director, University of Cincinnati Forensic Psychiatry Fellowship, Cincinnati, OH
Dear Dr. Mossman,
At the general hospital where I work, doctors and nurses sometimes ask me to fill out psychiatric “hold” documents to keep seriously ill medical or surgical patients from leaving the hospital. Last week, they asked me to stop Mr. J, a man with diabetes and a gangrenous lower leg, from leaving against medical advice (AMA). If he left, he would die. But if I filled out the psychiatric “hold,” I’d be saying the man needed civil commitment for a mental illness, which wasn’t true. If this happens again, what should I do?
Submitted by “Dr. Q”
“It is tempting, if the only tool you have is a hammer, to treat everything as if it were a nail,” wrote Abraham Maslow.1 The situation Dr. Q describes is one that psychiatrists frequently encounter because in some situations, a psychiatric “hold” can seem like the only way to stop a physically ill patient from leaving the hospital AMA. But pounding on this problem with a civil commitment hammer is the wrong response.
What’s wrong with using psychiatric holds in these situations? Do doctors have any other equipment in their medical toolbox for stopping an improvident AMA departure? To find out, we’ll look at:
- what a psychiatric hold does
- why holds don’t apply to medical-surgical treatment
- alternative responses to patients who lack capacity to refuse care.
Psychiatric holds
All states have laws that permit involuntary psychiatric hospitalization. The wording and procedural details in these laws vary across jurisdictions, but all states allow civil (ie, noncriminal) commitment of mentally ill persons who have gross impairments of judgment, behavior, reality-testing, or everyday functioning if their recent behavior show that they pose a danger because of their mental illness.2 Table 13 lists examples of the types of dangers that are potential reasons for civil commitment.
Table 1
Types of risks covered in civil commitment statutes
| All states |
|
| In some jurisdictions |
|
| Source: Adapted from reference 3 |
State laws also allow certain individuals (eg, police) to apprehend and transport mentally ill persons to facilities for psychiatric evaluation. Doctors may hold these persons temporarily until a court decides whether a longer involuntary hospitalization is justified. The documents used to initiate psychiatric holds have various informal names—”5150” (California), “pink slip” (Ohio), “pink paper” (Massachusetts), “Baker Act Form” (Florida)—but their function is the same: permitting lawful restraint of patients whose dangerousness results from their mental illness.
Urgent medical and surgical care
What about medical or surgical patients who refuse care despite being told they’ll die without it? Might involuntary psychiatric hospitalization procedures be a convenient way to keep them from coming to harm?
The answer: probably not, for 4 reasons:
- Once a psychiatric hold has been executed, the person who is subject to detention must be transferred to an appropriate facility within a specified period (usually 24 hours) for further evaluation and care.4,5 In this context, “appropriate facility” means a state-approved psychiatric treatment setting. A hospital’s medical or surgical unit usually would not qualify.
- The lawful use of a psychiatric hold is to declare that someone needs involuntary psychiatric examination for dangerousness arising “as a result of mental illness”—not for danger from a nonpsychiatric medical problem.6 Some civil commitment statutes specify that persons who have serious nonpsychiatric illness but no mental health problems that satisfy civil commitment criteria are to be offered voluntary treatment only.7
- A psychiatric hold only authorizes short-term detention. It does not allow forcing what patients such as Mr. J need: medical or surgical treatment. A psychiatric hold would not solve the problem that Mr. J’s doctors are facing.
- Doctors who execute psychiatric holds in good faith—sincerely believing a patient meets the legal criteria—enjoy statutory immunity from later accusations of malpractice or false imprisonment.8 Using civil commitment mechanisms when one does not actually believe those mechanisms apply might void this immunity.
Nonconsent: 2 varieties
For present purposes, let’s think of nonconsenting medical-surgical patients as coming in 2 varieties:
Variety 1: patients with compromised mental status. Often, medical-surgical patients cannot express objections to treatment because they are unconscious, delirious, or incoherent. Nurses and doctors assume such patients would want proper care and proceed with what they believe is in the patients’ best interest, often with input from family members.
Variety 2: lucid patients who refuse treatment. Patients who do not have obvious psychiatric problems may refuse necessary medical or surgical treatment for various reasons: obstinacy, distrust of doctors, fear, ignorance, incorrect but firmly held ideas about body functioning, cultural differences, or religious beliefs. None of these reasons is necessarily psychopathological, and none provides justification for a psychiatric hold.
Key determinant: Competence
Refusing treatment may be a bad choice and sometimes is evidence of a mental disorder, but it is not, by itself, a mental disorder. When a Variety 2 adult patient refuses care, the key question is, “Is this a competent refusal?” Assessment of a patient’s capacity to make medical decisions is not a skill unique to psychiatrists. Other specialists make judgments about capacity routinely—if only implicitly—when they elicit their patients’ informed consent for care. But when, as in Mr. J’s case, a seriously ill medical-surgical patient refuses lifesaving treatment, our medical colleagues often get psychiatrists involved. Consulting a psychiatrist in such circumstances makes sense, for at least 4 reasons:
- Although assessment of decision-making isn’t the special province of psychiatry, psychiatrists often have more experience assessing the capacity of persons whose thinking seems impaired.
- Psychiatrists also have more experience in detecting subtle indications of mental disorders (eg, mild dementia, depression, psychosis) that can compromise decision-making capacity.
- A nonpsychiatrist may believe that a patient is making a competent refusal but still wants a psychiatrist’s perspective to better understand the patient’s reasoning or to confirm the initial belief.
- Getting an independent opinion is a prudent way to make sure one’s emotions are not adversely influencing a critical judgment about a patient’s treatment.
Determining whether a patient has the requisite capacity to refuse care involves a situation-specific assessment of 4 aspects of mental functioning: expressing a choice coherently, understanding relevant information, appreciating this information, and using the information rationally. Table 29 describes these functional areas in more detail.
Table 2
Evaluating the quality of a patient’s decision: 4 dimensions
| 1. Can the patient communicate a choice and express a consistent preference? |
2. Can the patient grasp relevant information about:
|
| 3. Does the patient appreciate the illness and its consequences? Does he recognize he is ill and acknowledge how the information applies to his situation? |
| 4. Does the patient use the information rationally? Can he explain his decision-making and reasoning? Does he apply information to his situation in light of rational beliefs and desires? |
| Source: Adapted from reference 9 |
If capacity is lacking, what next?
As Judge Benjamin Cardozo ruled nearly a century ago, “Every human being of adult years and sound mind has a right to determine what shall be done with his own body.”10 In a case such as Mr. J’s, where a patient wants to leave the hospital or refuses medical treatment despite grave risk to himself, staff members should not let him leave until his treating doctors have tried to clarify his reasons for leaving and determined whether he has the capacity to give informed consent and refuse treatment. Psychiatrists may be consulted in this process, although the final judgment about capacity rests with the responsible physician. If an assessment shows that the patient has the capacity to make medical decisions, his treatment refusal is binding, even when it creates a clear risk of death.
What should happen if an assessment shows that a gravely ill patient lacks capacity to refuse treatment? Clinicians should consult with the hospital attorney about their facility’s policies and how to implement them properly.
Thinking about the possible legal implications of their actions, treating clinicians might worry that if they detain an unwilling patient without authorization from a court or guardian, they would risk being sued later for false imprisonment. But attorneys are likely to advise clinicians that they have more to fear liability-wise from letting incompetent patients leave the hospital than from detaining them for their own safety. As an Ohio court commented about a police officer who stopped a patient from leaving the hospital:
- What in the name of all that is reasonable should the officer have done? The court finds that the officer acted properly under the circumstances known to him at the time—and the reasonableness of an officer’s actions must be judged at the exigent split second on the street…11
Rather than allowing an incompetent patient to come to harm, attorneys may advise physicians to write an order to keep the patient in the hospital. Then, physicians can obtain consent for treatment from family members, making them aware of any physical or chemical restraint that might be needed to continue the patient’s treatment. Depending on the situation and the reasons for the lack of capacity, hospital staff members may later need to help a family member obtain a court’s authorization for emergency guardianship to allow non-urgent care to continue.
Treating physicians also should document the thinking and findings that support their actions. Table 3 provides an outline for this documentation.
Table 3
Detaining a patient for medical-surgical care: 7 components of documentation
| 1. Description of the patient’s refusal or efforts to leave the hospital |
| 2. Patient’s stated reasons for refusing or wanting to leave |
| 3. Reasonable alternatives to discharge that were offered |
| 4. Description of how refusing medical treatment would create a clear risk of physical harm or death |
| 5. Evidence that the patient lacks capacity to give informed consent or to refuse treatment |
| 6. Actions taken by the treating physician (eg, obtaining psychiatric consultation, enlisting other patient services, instituting physical restraint) |
| 7. Person who provided consent to continue treatment and that person’s relationship to patient |
Related Resources
- Appelbaum PS. Clinical practice. Assessment of patients’ competence to consent to treatment. N Engl J Med. 2007; 357(18):1834-1840.
- Disability Rights California. Involuntary psychiatric treatment: California’s 72-hour hold and 14-day certification. www.disabilityrightsca.org/pubs/502401.pdf.
- Treatment Advocacy Center. Know the laws in your state. www.treatmentadvocacycenter.org/get-help/know-the-laws-in-your-state.
Disclosure
Dr. Mossman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Dr. Mossman thanks David Schwallie, Esq, for his helpful insights about the topics discussed in this article.
Dear Dr. Mossman,
At the general hospital where I work, doctors and nurses sometimes ask me to fill out psychiatric “hold” documents to keep seriously ill medical or surgical patients from leaving the hospital. Last week, they asked me to stop Mr. J, a man with diabetes and a gangrenous lower leg, from leaving against medical advice (AMA). If he left, he would die. But if I filled out the psychiatric “hold,” I’d be saying the man needed civil commitment for a mental illness, which wasn’t true. If this happens again, what should I do?
Submitted by “Dr. Q”
“It is tempting, if the only tool you have is a hammer, to treat everything as if it were a nail,” wrote Abraham Maslow.1 The situation Dr. Q describes is one that psychiatrists frequently encounter because in some situations, a psychiatric “hold” can seem like the only way to stop a physically ill patient from leaving the hospital AMA. But pounding on this problem with a civil commitment hammer is the wrong response.
What’s wrong with using psychiatric holds in these situations? Do doctors have any other equipment in their medical toolbox for stopping an improvident AMA departure? To find out, we’ll look at:
- what a psychiatric hold does
- why holds don’t apply to medical-surgical treatment
- alternative responses to patients who lack capacity to refuse care.
Psychiatric holds
All states have laws that permit involuntary psychiatric hospitalization. The wording and procedural details in these laws vary across jurisdictions, but all states allow civil (ie, noncriminal) commitment of mentally ill persons who have gross impairments of judgment, behavior, reality-testing, or everyday functioning if their recent behavior show that they pose a danger because of their mental illness.2 Table 13 lists examples of the types of dangers that are potential reasons for civil commitment.
Table 1
Types of risks covered in civil commitment statutes
| All states |
|
| In some jurisdictions |
|
| Source: Adapted from reference 3 |
State laws also allow certain individuals (eg, police) to apprehend and transport mentally ill persons to facilities for psychiatric evaluation. Doctors may hold these persons temporarily until a court decides whether a longer involuntary hospitalization is justified. The documents used to initiate psychiatric holds have various informal names—”5150” (California), “pink slip” (Ohio), “pink paper” (Massachusetts), “Baker Act Form” (Florida)—but their function is the same: permitting lawful restraint of patients whose dangerousness results from their mental illness.
Urgent medical and surgical care
What about medical or surgical patients who refuse care despite being told they’ll die without it? Might involuntary psychiatric hospitalization procedures be a convenient way to keep them from coming to harm?
The answer: probably not, for 4 reasons:
- Once a psychiatric hold has been executed, the person who is subject to detention must be transferred to an appropriate facility within a specified period (usually 24 hours) for further evaluation and care.4,5 In this context, “appropriate facility” means a state-approved psychiatric treatment setting. A hospital’s medical or surgical unit usually would not qualify.
- The lawful use of a psychiatric hold is to declare that someone needs involuntary psychiatric examination for dangerousness arising “as a result of mental illness”—not for danger from a nonpsychiatric medical problem.6 Some civil commitment statutes specify that persons who have serious nonpsychiatric illness but no mental health problems that satisfy civil commitment criteria are to be offered voluntary treatment only.7
- A psychiatric hold only authorizes short-term detention. It does not allow forcing what patients such as Mr. J need: medical or surgical treatment. A psychiatric hold would not solve the problem that Mr. J’s doctors are facing.
- Doctors who execute psychiatric holds in good faith—sincerely believing a patient meets the legal criteria—enjoy statutory immunity from later accusations of malpractice or false imprisonment.8 Using civil commitment mechanisms when one does not actually believe those mechanisms apply might void this immunity.
Nonconsent: 2 varieties
For present purposes, let’s think of nonconsenting medical-surgical patients as coming in 2 varieties:
Variety 1: patients with compromised mental status. Often, medical-surgical patients cannot express objections to treatment because they are unconscious, delirious, or incoherent. Nurses and doctors assume such patients would want proper care and proceed with what they believe is in the patients’ best interest, often with input from family members.
Variety 2: lucid patients who refuse treatment. Patients who do not have obvious psychiatric problems may refuse necessary medical or surgical treatment for various reasons: obstinacy, distrust of doctors, fear, ignorance, incorrect but firmly held ideas about body functioning, cultural differences, or religious beliefs. None of these reasons is necessarily psychopathological, and none provides justification for a psychiatric hold.
Key determinant: Competence
Refusing treatment may be a bad choice and sometimes is evidence of a mental disorder, but it is not, by itself, a mental disorder. When a Variety 2 adult patient refuses care, the key question is, “Is this a competent refusal?” Assessment of a patient’s capacity to make medical decisions is not a skill unique to psychiatrists. Other specialists make judgments about capacity routinely—if only implicitly—when they elicit their patients’ informed consent for care. But when, as in Mr. J’s case, a seriously ill medical-surgical patient refuses lifesaving treatment, our medical colleagues often get psychiatrists involved. Consulting a psychiatrist in such circumstances makes sense, for at least 4 reasons:
- Although assessment of decision-making isn’t the special province of psychiatry, psychiatrists often have more experience assessing the capacity of persons whose thinking seems impaired.
- Psychiatrists also have more experience in detecting subtle indications of mental disorders (eg, mild dementia, depression, psychosis) that can compromise decision-making capacity.
- A nonpsychiatrist may believe that a patient is making a competent refusal but still wants a psychiatrist’s perspective to better understand the patient’s reasoning or to confirm the initial belief.
- Getting an independent opinion is a prudent way to make sure one’s emotions are not adversely influencing a critical judgment about a patient’s treatment.
Determining whether a patient has the requisite capacity to refuse care involves a situation-specific assessment of 4 aspects of mental functioning: expressing a choice coherently, understanding relevant information, appreciating this information, and using the information rationally. Table 29 describes these functional areas in more detail.
Table 2
Evaluating the quality of a patient’s decision: 4 dimensions
| 1. Can the patient communicate a choice and express a consistent preference? |
2. Can the patient grasp relevant information about:
|
| 3. Does the patient appreciate the illness and its consequences? Does he recognize he is ill and acknowledge how the information applies to his situation? |
| 4. Does the patient use the information rationally? Can he explain his decision-making and reasoning? Does he apply information to his situation in light of rational beliefs and desires? |
| Source: Adapted from reference 9 |
If capacity is lacking, what next?
As Judge Benjamin Cardozo ruled nearly a century ago, “Every human being of adult years and sound mind has a right to determine what shall be done with his own body.”10 In a case such as Mr. J’s, where a patient wants to leave the hospital or refuses medical treatment despite grave risk to himself, staff members should not let him leave until his treating doctors have tried to clarify his reasons for leaving and determined whether he has the capacity to give informed consent and refuse treatment. Psychiatrists may be consulted in this process, although the final judgment about capacity rests with the responsible physician. If an assessment shows that the patient has the capacity to make medical decisions, his treatment refusal is binding, even when it creates a clear risk of death.
What should happen if an assessment shows that a gravely ill patient lacks capacity to refuse treatment? Clinicians should consult with the hospital attorney about their facility’s policies and how to implement them properly.
Thinking about the possible legal implications of their actions, treating clinicians might worry that if they detain an unwilling patient without authorization from a court or guardian, they would risk being sued later for false imprisonment. But attorneys are likely to advise clinicians that they have more to fear liability-wise from letting incompetent patients leave the hospital than from detaining them for their own safety. As an Ohio court commented about a police officer who stopped a patient from leaving the hospital:
- What in the name of all that is reasonable should the officer have done? The court finds that the officer acted properly under the circumstances known to him at the time—and the reasonableness of an officer’s actions must be judged at the exigent split second on the street…11
Rather than allowing an incompetent patient to come to harm, attorneys may advise physicians to write an order to keep the patient in the hospital. Then, physicians can obtain consent for treatment from family members, making them aware of any physical or chemical restraint that might be needed to continue the patient’s treatment. Depending on the situation and the reasons for the lack of capacity, hospital staff members may later need to help a family member obtain a court’s authorization for emergency guardianship to allow non-urgent care to continue.
Treating physicians also should document the thinking and findings that support their actions. Table 3 provides an outline for this documentation.
Table 3
Detaining a patient for medical-surgical care: 7 components of documentation
| 1. Description of the patient’s refusal or efforts to leave the hospital |
| 2. Patient’s stated reasons for refusing or wanting to leave |
| 3. Reasonable alternatives to discharge that were offered |
| 4. Description of how refusing medical treatment would create a clear risk of physical harm or death |
| 5. Evidence that the patient lacks capacity to give informed consent or to refuse treatment |
| 6. Actions taken by the treating physician (eg, obtaining psychiatric consultation, enlisting other patient services, instituting physical restraint) |
| 7. Person who provided consent to continue treatment and that person’s relationship to patient |
Related Resources
- Appelbaum PS. Clinical practice. Assessment of patients’ competence to consent to treatment. N Engl J Med. 2007; 357(18):1834-1840.
- Disability Rights California. Involuntary psychiatric treatment: California’s 72-hour hold and 14-day certification. www.disabilityrightsca.org/pubs/502401.pdf.
- Treatment Advocacy Center. Know the laws in your state. www.treatmentadvocacycenter.org/get-help/know-the-laws-in-your-state.
Disclosure
Dr. Mossman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Dr. Mossman thanks David Schwallie, Esq, for his helpful insights about the topics discussed in this article.
1. Maslow AH. The psychology of science: a reconnaissance. New York NY: Harper & Row; 1966.
2. Mossman D, Schwartz AH, Elam ER. Risky business versus overt acts: what relevance do “actuarial” probabilistic risk assessments have for judicial decisions on involuntary psychiatric hospitalization? Houston Journal of Health Law & Policy. 2011;11:365-453.
3. Pinals DA, Mossman D. Evaluation for civil commitment. New York NY: Oxford University Press; 2012.
4. Ohio Revised Code § 5122.10.
5. Oregon Revised Statutes § 426.060.
6. California Welfare and Institutions Code § 5150.
7. Florida statutes § 394.463.
8. Cruze v National Psychiatric Services, Inc., 105 Cal. App. 4th 48 (2003).
9. Appelbaum PS, Grisso T. Assessing patients’ capacities to consent to treatment. N Engl J Med. 1988;319(25):1635-1638.
10. Schloendorff v Society of New York Hospital, 211 N.Y. 125, 105 N.E. 92 (1914).
11. State v Clay, 43 Ohio Misc. 2d 5, 539 N.E.2d 1168 (1988).
1. Maslow AH. The psychology of science: a reconnaissance. New York NY: Harper & Row; 1966.
2. Mossman D, Schwartz AH, Elam ER. Risky business versus overt acts: what relevance do “actuarial” probabilistic risk assessments have for judicial decisions on involuntary psychiatric hospitalization? Houston Journal of Health Law & Policy. 2011;11:365-453.
3. Pinals DA, Mossman D. Evaluation for civil commitment. New York NY: Oxford University Press; 2012.
4. Ohio Revised Code § 5122.10.
5. Oregon Revised Statutes § 426.060.
6. California Welfare and Institutions Code § 5150.
7. Florida statutes § 394.463.
8. Cruze v National Psychiatric Services, Inc., 105 Cal. App. 4th 48 (2003).
9. Appelbaum PS, Grisso T. Assessing patients’ capacities to consent to treatment. N Engl J Med. 1988;319(25):1635-1638.
10. Schloendorff v Society of New York Hospital, 211 N.Y. 125, 105 N.E. 92 (1914).
11. State v Clay, 43 Ohio Misc. 2d 5, 539 N.E.2d 1168 (1988).
Metabolic disturbance and dementia: A modifiable link
Discuss this article at www.facebook.com/CurrentPsychiatry
In addition to increasing patients’ risk for cardiovascular disease, stroke, and cancer, obesity and metabolic disturbance contribute to age-related cognitive decline and dementia. In particular, insulin resistance and hyperinsulinemia promote neurocognitive dysfunction and neurodegenerative changes during the extended, preclinical phase of Alzheimer’s disease (AD). However, with dietary modification it may be possible to resensitize insulin receptors, correct hyperinsulinemia, and improve memory function.
Metabolic disturbance and neurodegeneration
In the United States, 5.4 million people have AD, and there will be an estimated 16 million cases by 2050.1 Simultaneously we are experiencing an epidemic of metabolic disturbance and obesity. Approximately, 64% of adults in the United States are overweight (body mass index [BMI]: 25.0 to 29.9 kg/m2) and 34% are obese (BMI: ≥30 kg/m2).2 By 2030, 86% of adults will be overweight and 51% will be obese.3 This confluence of epidemics is not coincidental but instead reflects the fact that metabolic disturbance is a fundamental factor contributing to cognitive decline and neurodegeneration.4
Ninety-six percent of AD cases are classified as late onset, sporadic AD, occurring after age 64.1 Mild cognitive impairment (MCI) is a clinical construct that entails greater than expected memory impairment for the patient’s age and identifies older adults who are at increased risk for dementia. MCI represents the first clinical manifestation of neurodegeneration for a subset of patients who will progress to AD.5,6 MCI is distinguished from age-associated memory impairment (AAMI), which originally was conceptualized as normal or benign memory decline with aging.7,8 Recent data indicate that Alzheimer’s-type neuropathologic changes are the basis for subjective memory complaints and objectively assessed age-related cognitive decline,9 and early neurodegeneration is present in many patients with AAMI or MCI.10 This is consistent with the idea that an extended preclinical phase precedes AD onset. The preclinical phase can persist for a decade or more and precedes MCI and overt functional decline. However, neuropathologic changes accumulate during the preclinical phase of AD11 and during the preclinical phase of type 2 diabetes mellitus (T2DM).
Hyperinsulinemia and dementia
Insulin resistance and hyperinsulinemia occur in >40% of individuals age ≥60 and prevalence increases with age.4,12 Hyperinsulinemia develops to compensate for insulin resistance to overcome receptor insensitivity and maintain glucose homeostasis. Insulin receptors are densely expressed in brain regions vulnerable to neurodegeneration, including the medial temporal lobe and prefrontal cortex, which mediate long-term memory and working memory. However, insulin must be transported into the CNS from the periphery because little is synthesized in the brain. Paradoxically, peripheral compensatory hyperinsulinemia resulting from insulin resistance is associated with central (brain) hypoinsulinemia because of insensitivity and saturation of the receptor-mediated blood-brain barrier transport mechanism.13-15
Hyperinsulinemia is the precursor to T2DM. However, hyperinsulinemia is not well recognized in clinical contexts and generally is not a treatment target. Nonetheless, it contributes to several health problems, and insulin resistance in middle age is associated with age-related diseases such as hypertension, coronary artery disease, stroke, and cancer, while insulin sensitivity protects against such disorders.16
Chronic insulin resistance may contribute more to dementia development than T2DM because of the extended period of hyperinsulinemia that precedes T2DM onset. In population studies,17 insulin resistance syndrome increases risk for developing AD independent of apolipoprotein E (APOE e4) allele status, and in a longitudinal study,18 the risk for AD solely attributable to peripheral hyperinsulinemia was up to 39%. Being overweight in midlife increases risk for dementia in late life, and APOE e4 allele status does not contribute additional risk after accounting for BMI.19 Middle-aged individuals with hyperinsulinemia show memory decline, and obesity in middle age was associated with greater cognitive impairment after 6-year follow-up.20 Even in older adults who seem cognitively unimpaired, BMI and fasting insulin are positively correlated with atrophy in frontal, temporal, and subcortical brain regions, and obesity is an independent risk for atrophy in several brain regions, including the hippocampus.21
Compared with healthy older adults, individuals with AD have lower ratios of cerebrospinal fluid to plasma insulin.22 This lower ratio reflects the peripheral-to-central gradient of insulin levels in AD and suggests an etiological role for such metabolic disturbance. Insulin resistance has downstream effects that potentiate neurodegenerative factors, and central hypoinsulinemia can accelerate neurodegenerative processes and cognitive decline.4,23 Brain insulin plays a direct role in regulating proinflammatory cytokines and neurotrophic and neuroplastic factors essential for memory function. Insulin degrading enzyme, which varies with insulin levels,24 regulates the generation and clearance of amyloid β (Aβ) from the brain.25
Hyperinsulinemia typically is evident in increasing waist circumference and body weight.26 Waist circumference of ≥100 cm (39 inches) is a sensitive, specific, and independent predictor of hyperinsulinemia for men and women and a stronger predictor than BMI, waist-to-hip ratio, and other measures of body fat.27 Unpublished data derived from our clinical research with MCI subjects supports the association of metabolic disturbance with age-related cognitive decline. Our subjects are recruited from the community on the basis of mild memory decline and—other than excluding those with diabetes—weight and metabolic status are not considered in evaluating individuals for enrollment. The Table contains data on waist circumference and metabolic function in 122 older adults (age ≥68) with MCI. On average, these individuals exhibited fasting insulin values in the hyperinsulinemia range and elevated fasting glucose levels that indicated borderline diabetes. Waist circumference also was high, indicating excessive visceral fat deposition. We also observed a relationship between waist circumference and insulin, a consistent observation in older adults with memory decline. These data would not be surprising in any sample of older adults because of the population base rates for these conditions. However, we also found that waist circumference was a significant predictor of memory performance in patients with MCI. Abdominal adiposity is highly correlated with intrahepatic fat.28 Given this and recent indications that Alzheimer’s-type neuropathologic factors are generated in the liver,29,30 the predictive value of waist circumference to memory performance may reflect the fact that it is a proxy for downstream actions of liver fat.
Table
Waist circumference and metabolic factors in 122 older adults with MCIa
| Metabolic indicator | Value |
|---|---|
| Mean (SD) fasting glucose, mg/dL | 99.5 (11.2) |
| Mean (SD) fasting insulin, μIU/mL | 15.2 (8.1) |
| Mean (SD) waist, cm | 96.4 (13.3) |
| Waist-insulin correlation | r=0.51, P < .001 |
| aOlder adult patients (age ≥68) with subjective memory complaints were recruited from the community and screened with instruments assessing everyday functioning and objective memory performance to establish the presence of MCI MCI: mild cognitive impairment; SD: standard deviation | |
Dietary interventions
There is no cure for dementia, and it is not clear when effective therapy might be developed. Prevention and risk mitigation represent the best means of reducing the impact of this public health problem. Researchers have proposed that interventions initiated when individuals have predementia conditions such as AAMI and MCI might stall progression of cognitive decline, and MCI may be the last point when interventions might be effective because of the self-reinforcing neuropathologic cascades of AD.31 Because central hypoinsulinemia may promote central inflammation, Aβ generation, and reduced neuroplasticity, approaches aimed at improving metabolic function (and in particular correcting hyperinsulinemia) could influence fundamental neurodegenerative processes. Dietary approaches to preventing dementia are effective, low-risk, yet underutilized interventions. Reducing insulin by restricting calories32 or maintaining a ketogenic diet33 has been associated with improved memory function in middle-aged and older adults.
Carbohydrate consumption is the principal determinant of insulin secretion. Eliminating high-glycemic foods, including processed carbohydrates and sweets, would sensitize insulin receptors and correct hyperinsulinemia. In addition, replacing high glycemic foods with fruits and vegetables would increase polyphenol intake. Epidemiologic evidence supports the idea that greater consumption of polyphenol-containing vegetables and fruits mitigates risk for neurocognitive decline and dementia.34,35 Preclinical evidence suggests that such protection may be related to neuronal signaling effects and anti- inflammatory and antioxidant actions.36 In addition, certain polyphenol compounds, such as those found in berries, enhance metabolic function.37,38 In a 12-week pilot trial, older adults with early memory changes (N=9, mean age 76) who drank supplemental blueberry juice showed enhanced memory and improved metabolic parameters.39
Dietary changes that preserve insulin receptor sensitivity can help ensure general health with aging and substantially mitigate risk for neurodegeneration. The Western diet is particularly insulinogenic and dietary habits are difficult to change. However, the substantial benefits, absence of adverse effects, and low cost make dietary intervention the optimal means of protecting against neurodegeneration and other age-related diseases. Embarking on such a program early in life would be best, although late-life intervention can be effective.
Related Resources
- Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3(3):169-178.
- Luchsinger JA, Tang MX, Shea S, et al. Hyperinsulinemia and risk of Alzheimer’s disease. Neurology. 2004; 63(7):1187-1192.
- Krikorian R, Shidler MD, Dangelo K, et al. Dietary ketosis enhances memory in mild cognitive impairment. Neurbiol Aging. 2012;33(2):425.e19-e27.
Disclosure
Dr. Krikorian receives grant support from the National Institutes of Health, 1R01AG034617-01.
1. Alzheimer’s Association; Thies W, Bleiler L. 2011 Alzheimer’s disease facts and figures. Alzheimers Dement. 2011;7(2):208-244.
2. Flegal KM, Carroll MD, Ogden CL, et al. Prevalence and trends in obesity among US adults, 1999-2008. JAMA. 2010;303(3):235-241.
3. Wang Y, Beydoun MA, Liang L, et al. Will all Americans become overweight or obese? Estimating the progression and cost of the US obesity epidemic. Obesity (Silver Spring). 2008;16(10):2323-2330.
4. Craft S. Insulin resistance syndrome and Alzheimer’s disease: age- and obesity-related effect on memory amyloid, and inflammation. Neurobiol Aging. 2005;26(suppl 1):S65-S69.
5. Mitchell AJ, Shiri-Feshki M. Rate of progression of mild cognitive impairment to dementia – meta-analysis of 41 robust inception cohort studies. Acta Psychiat Scand. 2009;119(4):252-265.
6. Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256(3):183-194.
7. Crook TH, Bartus RT, Ferris SH, et al. Age-associated memory impairment: proposed diagnostic criteria and measures of clinical change—report of a National Institute of Mental Health work group. Dev Neuropsychol. 1986;2(4):261-276.
8. Neilsen H, Lolk A, Kragh-Sørensen P. Age-associated memory impairment–pathological memory decline or normal aging? Scand J Psychol. 1998;39(1):33-37.
9. Wilson RS, Leurgans SE, Boyle PA, et al. Neurodegenerative basis of age related cognitive decline. Neurology. 2010;75(12):1070-1078.
10. Saykin AJ, Wishart HA, Rabin LA, et al. Older adults with cognitive complaints show brain atrophy similar to that of amnestic MCI. Neurology. 2006;67(5):834-842.
11. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280-292.
12. Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287(3):356-359.
13. Baura GD, Foster DM, Kaiyala K, et al. Insulin transport from plasma into the central nervous system is inhibited by dexamethasone in dogs. Diabetes. 1996;45(1):86-90.
14. Wallum BJ, Taborsky GJ, Jr, Porte D Jr, et al. Cerebrospinal fluid insulin levels increase during intravenous insulin infusions in man. J Clin Endocr Metab. 1987;64(1):190-194.
15. Woods SC, Seeley RJ, Baskin DG, et al. Insulin and the blood-brain barrier. Curr Pharm Des. 2003;9(10):795-800.
16. Facchini FS, Hua N, Abbasi F, et al. Insulin resistance as a predictor of age-related diseases. J Clin Endocrinol Metab. 2001;86(8):3574-3578.
17. Kuusisto J, Koivisto K, Mykkänen L, et al. Association between features of the insulin resistance syndrome and Alzheimer’s disease independently of apolipoprotein E4 phenotype. BMJ. 1997;315(7115):1045-1049.
18. Luchsinger JA, Tang MX, Shea S, et al. Hyperinsulinemia and risk of Alzheimer’s disease. Neurology. 2004;63(7):1187-1192.
19. Hassing LB, Dahl AK, Thorvaldsson V, et al. Overweight in midlife and risk of dementia: a 40-year follow up study. Int J Obesity (Lond). 2009;33(8):893-898.
20. Young SE, Mainous AG 3rd, Carnemolla M. Hyperinsulinemia and cognitive decline in a middle-aged cohort. Diabetes Care. 2006;29(12):2688-2693.
21. Raji CA, Ho AJ, Parikshak NN, et al. Brain structure and obesity. Hum Brain Mapp. 2009;31(3):353-364.
22. Craft S, Peskind E, Schwartz MW, et al. Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease. Neurology. 1998;50(1):164-168.
23. Craft S, Asthana S, Cook DG, et al. Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: interactions with apolipoprotein E genotype. Psychoneuroendocrinology. 2003;28(6):809-822.
24. Zhao L, Teter B, Morihara T, et al. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer’s disease intervention. J Neurosci. 2004;24(49):11120-11126.
25. Farris W, Mansourian S, Chang Y, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100(7):4162-4167.
26. Tabata S, Yoshimitsu S, Hamachi T, et al. Waist circumference and insulin resistance: a cross-sectional study of Japanese men. BMC Endocr Disord. 2009;9:1.-doi: 10.1186/1472-6823-9-1.
27. Wahrenberg H, Hertel K, Leijonhufvud B, et al. Use of waist circumference to predict insulin resistance: retrospective study. BMJ. 2005;330(7504):1363-1364.
28. Jang S, Lee CH, Choi KM, et al. Correlation of fatty liver and abdominal fat distribution using a simple fat computed tomography protocol. World J Gastroenterol. 2011;17(28):3335-3341.
29. Sutcliffe JG, Hedlund PB, Thomas EA, et al. Peripheral reduction of ß-amyloid is sufficient to reduce brain ß-amyloid: implications for Alzheimer’s disease. J Neurosci Res. 2011;89(6):808-814.
30. Marques MA, Kulstad JJ, Savard CE, et al. Peripheral amyloid-β levels regulate amyloid-β clearance from the central nervous system. J Alzheimers Dis. 2009;16(2):325-329.
31. Cotman CW. Homeostatic processes in brain aging: the role of apoptosis inflammation, and oxidative stress in regulating healthy neural circuitry in the aging brain. In: Stern P, Carstensen L, eds. The aging mind: opportunities in cognitive research. Washington, DC: National Academy Press; 2000:114–143.
32. Witte AV, Fobker M, Gellner R, et al. Caloric restriction improves memory in elderly humans. Proc Natl Acad Sci U S A. 2009;106(4):1255-1260.
33. Krikorian R, Shidler MD, Dangelo K, et al. Dietary ketosis enhances memory in mild cognitive impairment. Neurbiol Aging. 2012;33(2):425.e19-e27.
34. Letenneur L, Proust-Lima C, Le Gouge A, et al. Flavonoid intake and cognitive decline over a 10-year period. Am J Epidemiol. 2007;165(2):1364-1371.
35. Solfrizzi V, Panza F, Capurso A. The role of diet in cognitive decline. J Neural Transm. 2003;110(3):95-110.
36. Williams CM, El Mohsen MA, Vauzour D, et al. Blueberry-induced changes in spatial working memory correlate with changes in hippocampal CREB phosphorylation and brain-derived neurotrophic factor (BDNF) levels. Free Radical Bio Med. 2008;45(3):295-305.
37. Martineau LC, Couture A, Spoor D, et al. Anti-diabetic properties of the Canadian lowbush blueberry Vaccinium angustifolium Ait. Phytomedicine. 2006;13(9-10):612-623.
38. Tsuda T. Regulation of adipocyte function by anthocyanins; possibility of preventing the metabolic syndrome. J Agr Food Chem. 2008;56(3):642-646.
39. Krikorian R, Shidler MD, Nash TA, et al. Blueberry supplementation improves memory in older adults. J Agric Food Chem. 2010;58(7):3996-4000.

Robert Krikorian, PhD
Professor, Department of Psychiatry and Behavioral Neuroscience, University of Cincinnati Academic Health Center, Cincinnati, OH
Robert Krikorian, PhD
Professor, Department of Psychiatry and Behavioral Neuroscience, University of Cincinnati Academic Health Center, Cincinnati, OH
Robert Krikorian, PhD
Professor, Department of Psychiatry and Behavioral Neuroscience, University of Cincinnati Academic Health Center, Cincinnati, OH
Discuss this article at www.facebook.com/CurrentPsychiatry
In addition to increasing patients’ risk for cardiovascular disease, stroke, and cancer, obesity and metabolic disturbance contribute to age-related cognitive decline and dementia. In particular, insulin resistance and hyperinsulinemia promote neurocognitive dysfunction and neurodegenerative changes during the extended, preclinical phase of Alzheimer’s disease (AD). However, with dietary modification it may be possible to resensitize insulin receptors, correct hyperinsulinemia, and improve memory function.
Metabolic disturbance and neurodegeneration
In the United States, 5.4 million people have AD, and there will be an estimated 16 million cases by 2050.1 Simultaneously we are experiencing an epidemic of metabolic disturbance and obesity. Approximately, 64% of adults in the United States are overweight (body mass index [BMI]: 25.0 to 29.9 kg/m2) and 34% are obese (BMI: ≥30 kg/m2).2 By 2030, 86% of adults will be overweight and 51% will be obese.3 This confluence of epidemics is not coincidental but instead reflects the fact that metabolic disturbance is a fundamental factor contributing to cognitive decline and neurodegeneration.4
Ninety-six percent of AD cases are classified as late onset, sporadic AD, occurring after age 64.1 Mild cognitive impairment (MCI) is a clinical construct that entails greater than expected memory impairment for the patient’s age and identifies older adults who are at increased risk for dementia. MCI represents the first clinical manifestation of neurodegeneration for a subset of patients who will progress to AD.5,6 MCI is distinguished from age-associated memory impairment (AAMI), which originally was conceptualized as normal or benign memory decline with aging.7,8 Recent data indicate that Alzheimer’s-type neuropathologic changes are the basis for subjective memory complaints and objectively assessed age-related cognitive decline,9 and early neurodegeneration is present in many patients with AAMI or MCI.10 This is consistent with the idea that an extended preclinical phase precedes AD onset. The preclinical phase can persist for a decade or more and precedes MCI and overt functional decline. However, neuropathologic changes accumulate during the preclinical phase of AD11 and during the preclinical phase of type 2 diabetes mellitus (T2DM).
Hyperinsulinemia and dementia
Insulin resistance and hyperinsulinemia occur in >40% of individuals age ≥60 and prevalence increases with age.4,12 Hyperinsulinemia develops to compensate for insulin resistance to overcome receptor insensitivity and maintain glucose homeostasis. Insulin receptors are densely expressed in brain regions vulnerable to neurodegeneration, including the medial temporal lobe and prefrontal cortex, which mediate long-term memory and working memory. However, insulin must be transported into the CNS from the periphery because little is synthesized in the brain. Paradoxically, peripheral compensatory hyperinsulinemia resulting from insulin resistance is associated with central (brain) hypoinsulinemia because of insensitivity and saturation of the receptor-mediated blood-brain barrier transport mechanism.13-15
Hyperinsulinemia is the precursor to T2DM. However, hyperinsulinemia is not well recognized in clinical contexts and generally is not a treatment target. Nonetheless, it contributes to several health problems, and insulin resistance in middle age is associated with age-related diseases such as hypertension, coronary artery disease, stroke, and cancer, while insulin sensitivity protects against such disorders.16
Chronic insulin resistance may contribute more to dementia development than T2DM because of the extended period of hyperinsulinemia that precedes T2DM onset. In population studies,17 insulin resistance syndrome increases risk for developing AD independent of apolipoprotein E (APOE e4) allele status, and in a longitudinal study,18 the risk for AD solely attributable to peripheral hyperinsulinemia was up to 39%. Being overweight in midlife increases risk for dementia in late life, and APOE e4 allele status does not contribute additional risk after accounting for BMI.19 Middle-aged individuals with hyperinsulinemia show memory decline, and obesity in middle age was associated with greater cognitive impairment after 6-year follow-up.20 Even in older adults who seem cognitively unimpaired, BMI and fasting insulin are positively correlated with atrophy in frontal, temporal, and subcortical brain regions, and obesity is an independent risk for atrophy in several brain regions, including the hippocampus.21
Compared with healthy older adults, individuals with AD have lower ratios of cerebrospinal fluid to plasma insulin.22 This lower ratio reflects the peripheral-to-central gradient of insulin levels in AD and suggests an etiological role for such metabolic disturbance. Insulin resistance has downstream effects that potentiate neurodegenerative factors, and central hypoinsulinemia can accelerate neurodegenerative processes and cognitive decline.4,23 Brain insulin plays a direct role in regulating proinflammatory cytokines and neurotrophic and neuroplastic factors essential for memory function. Insulin degrading enzyme, which varies with insulin levels,24 regulates the generation and clearance of amyloid β (Aβ) from the brain.25
Hyperinsulinemia typically is evident in increasing waist circumference and body weight.26 Waist circumference of ≥100 cm (39 inches) is a sensitive, specific, and independent predictor of hyperinsulinemia for men and women and a stronger predictor than BMI, waist-to-hip ratio, and other measures of body fat.27 Unpublished data derived from our clinical research with MCI subjects supports the association of metabolic disturbance with age-related cognitive decline. Our subjects are recruited from the community on the basis of mild memory decline and—other than excluding those with diabetes—weight and metabolic status are not considered in evaluating individuals for enrollment. The Table contains data on waist circumference and metabolic function in 122 older adults (age ≥68) with MCI. On average, these individuals exhibited fasting insulin values in the hyperinsulinemia range and elevated fasting glucose levels that indicated borderline diabetes. Waist circumference also was high, indicating excessive visceral fat deposition. We also observed a relationship between waist circumference and insulin, a consistent observation in older adults with memory decline. These data would not be surprising in any sample of older adults because of the population base rates for these conditions. However, we also found that waist circumference was a significant predictor of memory performance in patients with MCI. Abdominal adiposity is highly correlated with intrahepatic fat.28 Given this and recent indications that Alzheimer’s-type neuropathologic factors are generated in the liver,29,30 the predictive value of waist circumference to memory performance may reflect the fact that it is a proxy for downstream actions of liver fat.
Table
Waist circumference and metabolic factors in 122 older adults with MCIa
| Metabolic indicator | Value |
|---|---|
| Mean (SD) fasting glucose, mg/dL | 99.5 (11.2) |
| Mean (SD) fasting insulin, μIU/mL | 15.2 (8.1) |
| Mean (SD) waist, cm | 96.4 (13.3) |
| Waist-insulin correlation | r=0.51, P < .001 |
| aOlder adult patients (age ≥68) with subjective memory complaints were recruited from the community and screened with instruments assessing everyday functioning and objective memory performance to establish the presence of MCI MCI: mild cognitive impairment; SD: standard deviation | |
Dietary interventions
There is no cure for dementia, and it is not clear when effective therapy might be developed. Prevention and risk mitigation represent the best means of reducing the impact of this public health problem. Researchers have proposed that interventions initiated when individuals have predementia conditions such as AAMI and MCI might stall progression of cognitive decline, and MCI may be the last point when interventions might be effective because of the self-reinforcing neuropathologic cascades of AD.31 Because central hypoinsulinemia may promote central inflammation, Aβ generation, and reduced neuroplasticity, approaches aimed at improving metabolic function (and in particular correcting hyperinsulinemia) could influence fundamental neurodegenerative processes. Dietary approaches to preventing dementia are effective, low-risk, yet underutilized interventions. Reducing insulin by restricting calories32 or maintaining a ketogenic diet33 has been associated with improved memory function in middle-aged and older adults.
Carbohydrate consumption is the principal determinant of insulin secretion. Eliminating high-glycemic foods, including processed carbohydrates and sweets, would sensitize insulin receptors and correct hyperinsulinemia. In addition, replacing high glycemic foods with fruits and vegetables would increase polyphenol intake. Epidemiologic evidence supports the idea that greater consumption of polyphenol-containing vegetables and fruits mitigates risk for neurocognitive decline and dementia.34,35 Preclinical evidence suggests that such protection may be related to neuronal signaling effects and anti- inflammatory and antioxidant actions.36 In addition, certain polyphenol compounds, such as those found in berries, enhance metabolic function.37,38 In a 12-week pilot trial, older adults with early memory changes (N=9, mean age 76) who drank supplemental blueberry juice showed enhanced memory and improved metabolic parameters.39
Dietary changes that preserve insulin receptor sensitivity can help ensure general health with aging and substantially mitigate risk for neurodegeneration. The Western diet is particularly insulinogenic and dietary habits are difficult to change. However, the substantial benefits, absence of adverse effects, and low cost make dietary intervention the optimal means of protecting against neurodegeneration and other age-related diseases. Embarking on such a program early in life would be best, although late-life intervention can be effective.
Related Resources
- Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3(3):169-178.
- Luchsinger JA, Tang MX, Shea S, et al. Hyperinsulinemia and risk of Alzheimer’s disease. Neurology. 2004; 63(7):1187-1192.
- Krikorian R, Shidler MD, Dangelo K, et al. Dietary ketosis enhances memory in mild cognitive impairment. Neurbiol Aging. 2012;33(2):425.e19-e27.
Disclosure
Dr. Krikorian receives grant support from the National Institutes of Health, 1R01AG034617-01.
Discuss this article at www.facebook.com/CurrentPsychiatry
In addition to increasing patients’ risk for cardiovascular disease, stroke, and cancer, obesity and metabolic disturbance contribute to age-related cognitive decline and dementia. In particular, insulin resistance and hyperinsulinemia promote neurocognitive dysfunction and neurodegenerative changes during the extended, preclinical phase of Alzheimer’s disease (AD). However, with dietary modification it may be possible to resensitize insulin receptors, correct hyperinsulinemia, and improve memory function.
Metabolic disturbance and neurodegeneration
In the United States, 5.4 million people have AD, and there will be an estimated 16 million cases by 2050.1 Simultaneously we are experiencing an epidemic of metabolic disturbance and obesity. Approximately, 64% of adults in the United States are overweight (body mass index [BMI]: 25.0 to 29.9 kg/m2) and 34% are obese (BMI: ≥30 kg/m2).2 By 2030, 86% of adults will be overweight and 51% will be obese.3 This confluence of epidemics is not coincidental but instead reflects the fact that metabolic disturbance is a fundamental factor contributing to cognitive decline and neurodegeneration.4
Ninety-six percent of AD cases are classified as late onset, sporadic AD, occurring after age 64.1 Mild cognitive impairment (MCI) is a clinical construct that entails greater than expected memory impairment for the patient’s age and identifies older adults who are at increased risk for dementia. MCI represents the first clinical manifestation of neurodegeneration for a subset of patients who will progress to AD.5,6 MCI is distinguished from age-associated memory impairment (AAMI), which originally was conceptualized as normal or benign memory decline with aging.7,8 Recent data indicate that Alzheimer’s-type neuropathologic changes are the basis for subjective memory complaints and objectively assessed age-related cognitive decline,9 and early neurodegeneration is present in many patients with AAMI or MCI.10 This is consistent with the idea that an extended preclinical phase precedes AD onset. The preclinical phase can persist for a decade or more and precedes MCI and overt functional decline. However, neuropathologic changes accumulate during the preclinical phase of AD11 and during the preclinical phase of type 2 diabetes mellitus (T2DM).
Hyperinsulinemia and dementia
Insulin resistance and hyperinsulinemia occur in >40% of individuals age ≥60 and prevalence increases with age.4,12 Hyperinsulinemia develops to compensate for insulin resistance to overcome receptor insensitivity and maintain glucose homeostasis. Insulin receptors are densely expressed in brain regions vulnerable to neurodegeneration, including the medial temporal lobe and prefrontal cortex, which mediate long-term memory and working memory. However, insulin must be transported into the CNS from the periphery because little is synthesized in the brain. Paradoxically, peripheral compensatory hyperinsulinemia resulting from insulin resistance is associated with central (brain) hypoinsulinemia because of insensitivity and saturation of the receptor-mediated blood-brain barrier transport mechanism.13-15
Hyperinsulinemia is the precursor to T2DM. However, hyperinsulinemia is not well recognized in clinical contexts and generally is not a treatment target. Nonetheless, it contributes to several health problems, and insulin resistance in middle age is associated with age-related diseases such as hypertension, coronary artery disease, stroke, and cancer, while insulin sensitivity protects against such disorders.16
Chronic insulin resistance may contribute more to dementia development than T2DM because of the extended period of hyperinsulinemia that precedes T2DM onset. In population studies,17 insulin resistance syndrome increases risk for developing AD independent of apolipoprotein E (APOE e4) allele status, and in a longitudinal study,18 the risk for AD solely attributable to peripheral hyperinsulinemia was up to 39%. Being overweight in midlife increases risk for dementia in late life, and APOE e4 allele status does not contribute additional risk after accounting for BMI.19 Middle-aged individuals with hyperinsulinemia show memory decline, and obesity in middle age was associated with greater cognitive impairment after 6-year follow-up.20 Even in older adults who seem cognitively unimpaired, BMI and fasting insulin are positively correlated with atrophy in frontal, temporal, and subcortical brain regions, and obesity is an independent risk for atrophy in several brain regions, including the hippocampus.21
Compared with healthy older adults, individuals with AD have lower ratios of cerebrospinal fluid to plasma insulin.22 This lower ratio reflects the peripheral-to-central gradient of insulin levels in AD and suggests an etiological role for such metabolic disturbance. Insulin resistance has downstream effects that potentiate neurodegenerative factors, and central hypoinsulinemia can accelerate neurodegenerative processes and cognitive decline.4,23 Brain insulin plays a direct role in regulating proinflammatory cytokines and neurotrophic and neuroplastic factors essential for memory function. Insulin degrading enzyme, which varies with insulin levels,24 regulates the generation and clearance of amyloid β (Aβ) from the brain.25
Hyperinsulinemia typically is evident in increasing waist circumference and body weight.26 Waist circumference of ≥100 cm (39 inches) is a sensitive, specific, and independent predictor of hyperinsulinemia for men and women and a stronger predictor than BMI, waist-to-hip ratio, and other measures of body fat.27 Unpublished data derived from our clinical research with MCI subjects supports the association of metabolic disturbance with age-related cognitive decline. Our subjects are recruited from the community on the basis of mild memory decline and—other than excluding those with diabetes—weight and metabolic status are not considered in evaluating individuals for enrollment. The Table contains data on waist circumference and metabolic function in 122 older adults (age ≥68) with MCI. On average, these individuals exhibited fasting insulin values in the hyperinsulinemia range and elevated fasting glucose levels that indicated borderline diabetes. Waist circumference also was high, indicating excessive visceral fat deposition. We also observed a relationship between waist circumference and insulin, a consistent observation in older adults with memory decline. These data would not be surprising in any sample of older adults because of the population base rates for these conditions. However, we also found that waist circumference was a significant predictor of memory performance in patients with MCI. Abdominal adiposity is highly correlated with intrahepatic fat.28 Given this and recent indications that Alzheimer’s-type neuropathologic factors are generated in the liver,29,30 the predictive value of waist circumference to memory performance may reflect the fact that it is a proxy for downstream actions of liver fat.
Table
Waist circumference and metabolic factors in 122 older adults with MCIa
| Metabolic indicator | Value |
|---|---|
| Mean (SD) fasting glucose, mg/dL | 99.5 (11.2) |
| Mean (SD) fasting insulin, μIU/mL | 15.2 (8.1) |
| Mean (SD) waist, cm | 96.4 (13.3) |
| Waist-insulin correlation | r=0.51, P < .001 |
| aOlder adult patients (age ≥68) with subjective memory complaints were recruited from the community and screened with instruments assessing everyday functioning and objective memory performance to establish the presence of MCI MCI: mild cognitive impairment; SD: standard deviation | |
Dietary interventions
There is no cure for dementia, and it is not clear when effective therapy might be developed. Prevention and risk mitigation represent the best means of reducing the impact of this public health problem. Researchers have proposed that interventions initiated when individuals have predementia conditions such as AAMI and MCI might stall progression of cognitive decline, and MCI may be the last point when interventions might be effective because of the self-reinforcing neuropathologic cascades of AD.31 Because central hypoinsulinemia may promote central inflammation, Aβ generation, and reduced neuroplasticity, approaches aimed at improving metabolic function (and in particular correcting hyperinsulinemia) could influence fundamental neurodegenerative processes. Dietary approaches to preventing dementia are effective, low-risk, yet underutilized interventions. Reducing insulin by restricting calories32 or maintaining a ketogenic diet33 has been associated with improved memory function in middle-aged and older adults.
Carbohydrate consumption is the principal determinant of insulin secretion. Eliminating high-glycemic foods, including processed carbohydrates and sweets, would sensitize insulin receptors and correct hyperinsulinemia. In addition, replacing high glycemic foods with fruits and vegetables would increase polyphenol intake. Epidemiologic evidence supports the idea that greater consumption of polyphenol-containing vegetables and fruits mitigates risk for neurocognitive decline and dementia.34,35 Preclinical evidence suggests that such protection may be related to neuronal signaling effects and anti- inflammatory and antioxidant actions.36 In addition, certain polyphenol compounds, such as those found in berries, enhance metabolic function.37,38 In a 12-week pilot trial, older adults with early memory changes (N=9, mean age 76) who drank supplemental blueberry juice showed enhanced memory and improved metabolic parameters.39
Dietary changes that preserve insulin receptor sensitivity can help ensure general health with aging and substantially mitigate risk for neurodegeneration. The Western diet is particularly insulinogenic and dietary habits are difficult to change. However, the substantial benefits, absence of adverse effects, and low cost make dietary intervention the optimal means of protecting against neurodegeneration and other age-related diseases. Embarking on such a program early in life would be best, although late-life intervention can be effective.
Related Resources
- Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3(3):169-178.
- Luchsinger JA, Tang MX, Shea S, et al. Hyperinsulinemia and risk of Alzheimer’s disease. Neurology. 2004; 63(7):1187-1192.
- Krikorian R, Shidler MD, Dangelo K, et al. Dietary ketosis enhances memory in mild cognitive impairment. Neurbiol Aging. 2012;33(2):425.e19-e27.
Disclosure
Dr. Krikorian receives grant support from the National Institutes of Health, 1R01AG034617-01.
1. Alzheimer’s Association; Thies W, Bleiler L. 2011 Alzheimer’s disease facts and figures. Alzheimers Dement. 2011;7(2):208-244.
2. Flegal KM, Carroll MD, Ogden CL, et al. Prevalence and trends in obesity among US adults, 1999-2008. JAMA. 2010;303(3):235-241.
3. Wang Y, Beydoun MA, Liang L, et al. Will all Americans become overweight or obese? Estimating the progression and cost of the US obesity epidemic. Obesity (Silver Spring). 2008;16(10):2323-2330.
4. Craft S. Insulin resistance syndrome and Alzheimer’s disease: age- and obesity-related effect on memory amyloid, and inflammation. Neurobiol Aging. 2005;26(suppl 1):S65-S69.
5. Mitchell AJ, Shiri-Feshki M. Rate of progression of mild cognitive impairment to dementia – meta-analysis of 41 robust inception cohort studies. Acta Psychiat Scand. 2009;119(4):252-265.
6. Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256(3):183-194.
7. Crook TH, Bartus RT, Ferris SH, et al. Age-associated memory impairment: proposed diagnostic criteria and measures of clinical change—report of a National Institute of Mental Health work group. Dev Neuropsychol. 1986;2(4):261-276.
8. Neilsen H, Lolk A, Kragh-Sørensen P. Age-associated memory impairment–pathological memory decline or normal aging? Scand J Psychol. 1998;39(1):33-37.
9. Wilson RS, Leurgans SE, Boyle PA, et al. Neurodegenerative basis of age related cognitive decline. Neurology. 2010;75(12):1070-1078.
10. Saykin AJ, Wishart HA, Rabin LA, et al. Older adults with cognitive complaints show brain atrophy similar to that of amnestic MCI. Neurology. 2006;67(5):834-842.
11. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280-292.
12. Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287(3):356-359.
13. Baura GD, Foster DM, Kaiyala K, et al. Insulin transport from plasma into the central nervous system is inhibited by dexamethasone in dogs. Diabetes. 1996;45(1):86-90.
14. Wallum BJ, Taborsky GJ, Jr, Porte D Jr, et al. Cerebrospinal fluid insulin levels increase during intravenous insulin infusions in man. J Clin Endocr Metab. 1987;64(1):190-194.
15. Woods SC, Seeley RJ, Baskin DG, et al. Insulin and the blood-brain barrier. Curr Pharm Des. 2003;9(10):795-800.
16. Facchini FS, Hua N, Abbasi F, et al. Insulin resistance as a predictor of age-related diseases. J Clin Endocrinol Metab. 2001;86(8):3574-3578.
17. Kuusisto J, Koivisto K, Mykkänen L, et al. Association between features of the insulin resistance syndrome and Alzheimer’s disease independently of apolipoprotein E4 phenotype. BMJ. 1997;315(7115):1045-1049.
18. Luchsinger JA, Tang MX, Shea S, et al. Hyperinsulinemia and risk of Alzheimer’s disease. Neurology. 2004;63(7):1187-1192.
19. Hassing LB, Dahl AK, Thorvaldsson V, et al. Overweight in midlife and risk of dementia: a 40-year follow up study. Int J Obesity (Lond). 2009;33(8):893-898.
20. Young SE, Mainous AG 3rd, Carnemolla M. Hyperinsulinemia and cognitive decline in a middle-aged cohort. Diabetes Care. 2006;29(12):2688-2693.
21. Raji CA, Ho AJ, Parikshak NN, et al. Brain structure and obesity. Hum Brain Mapp. 2009;31(3):353-364.
22. Craft S, Peskind E, Schwartz MW, et al. Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease. Neurology. 1998;50(1):164-168.
23. Craft S, Asthana S, Cook DG, et al. Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: interactions with apolipoprotein E genotype. Psychoneuroendocrinology. 2003;28(6):809-822.
24. Zhao L, Teter B, Morihara T, et al. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer’s disease intervention. J Neurosci. 2004;24(49):11120-11126.
25. Farris W, Mansourian S, Chang Y, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100(7):4162-4167.
26. Tabata S, Yoshimitsu S, Hamachi T, et al. Waist circumference and insulin resistance: a cross-sectional study of Japanese men. BMC Endocr Disord. 2009;9:1.-doi: 10.1186/1472-6823-9-1.
27. Wahrenberg H, Hertel K, Leijonhufvud B, et al. Use of waist circumference to predict insulin resistance: retrospective study. BMJ. 2005;330(7504):1363-1364.
28. Jang S, Lee CH, Choi KM, et al. Correlation of fatty liver and abdominal fat distribution using a simple fat computed tomography protocol. World J Gastroenterol. 2011;17(28):3335-3341.
29. Sutcliffe JG, Hedlund PB, Thomas EA, et al. Peripheral reduction of ß-amyloid is sufficient to reduce brain ß-amyloid: implications for Alzheimer’s disease. J Neurosci Res. 2011;89(6):808-814.
30. Marques MA, Kulstad JJ, Savard CE, et al. Peripheral amyloid-β levels regulate amyloid-β clearance from the central nervous system. J Alzheimers Dis. 2009;16(2):325-329.
31. Cotman CW. Homeostatic processes in brain aging: the role of apoptosis inflammation, and oxidative stress in regulating healthy neural circuitry in the aging brain. In: Stern P, Carstensen L, eds. The aging mind: opportunities in cognitive research. Washington, DC: National Academy Press; 2000:114–143.
32. Witte AV, Fobker M, Gellner R, et al. Caloric restriction improves memory in elderly humans. Proc Natl Acad Sci U S A. 2009;106(4):1255-1260.
33. Krikorian R, Shidler MD, Dangelo K, et al. Dietary ketosis enhances memory in mild cognitive impairment. Neurbiol Aging. 2012;33(2):425.e19-e27.
34. Letenneur L, Proust-Lima C, Le Gouge A, et al. Flavonoid intake and cognitive decline over a 10-year period. Am J Epidemiol. 2007;165(2):1364-1371.
35. Solfrizzi V, Panza F, Capurso A. The role of diet in cognitive decline. J Neural Transm. 2003;110(3):95-110.
36. Williams CM, El Mohsen MA, Vauzour D, et al. Blueberry-induced changes in spatial working memory correlate with changes in hippocampal CREB phosphorylation and brain-derived neurotrophic factor (BDNF) levels. Free Radical Bio Med. 2008;45(3):295-305.
37. Martineau LC, Couture A, Spoor D, et al. Anti-diabetic properties of the Canadian lowbush blueberry Vaccinium angustifolium Ait. Phytomedicine. 2006;13(9-10):612-623.
38. Tsuda T. Regulation of adipocyte function by anthocyanins; possibility of preventing the metabolic syndrome. J Agr Food Chem. 2008;56(3):642-646.
39. Krikorian R, Shidler MD, Nash TA, et al. Blueberry supplementation improves memory in older adults. J Agric Food Chem. 2010;58(7):3996-4000.
1. Alzheimer’s Association; Thies W, Bleiler L. 2011 Alzheimer’s disease facts and figures. Alzheimers Dement. 2011;7(2):208-244.
2. Flegal KM, Carroll MD, Ogden CL, et al. Prevalence and trends in obesity among US adults, 1999-2008. JAMA. 2010;303(3):235-241.
3. Wang Y, Beydoun MA, Liang L, et al. Will all Americans become overweight or obese? Estimating the progression and cost of the US obesity epidemic. Obesity (Silver Spring). 2008;16(10):2323-2330.
4. Craft S. Insulin resistance syndrome and Alzheimer’s disease: age- and obesity-related effect on memory amyloid, and inflammation. Neurobiol Aging. 2005;26(suppl 1):S65-S69.
5. Mitchell AJ, Shiri-Feshki M. Rate of progression of mild cognitive impairment to dementia – meta-analysis of 41 robust inception cohort studies. Acta Psychiat Scand. 2009;119(4):252-265.
6. Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256(3):183-194.
7. Crook TH, Bartus RT, Ferris SH, et al. Age-associated memory impairment: proposed diagnostic criteria and measures of clinical change—report of a National Institute of Mental Health work group. Dev Neuropsychol. 1986;2(4):261-276.
8. Neilsen H, Lolk A, Kragh-Sørensen P. Age-associated memory impairment–pathological memory decline or normal aging? Scand J Psychol. 1998;39(1):33-37.
9. Wilson RS, Leurgans SE, Boyle PA, et al. Neurodegenerative basis of age related cognitive decline. Neurology. 2010;75(12):1070-1078.
10. Saykin AJ, Wishart HA, Rabin LA, et al. Older adults with cognitive complaints show brain atrophy similar to that of amnestic MCI. Neurology. 2006;67(5):834-842.
11. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280-292.
12. Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287(3):356-359.
13. Baura GD, Foster DM, Kaiyala K, et al. Insulin transport from plasma into the central nervous system is inhibited by dexamethasone in dogs. Diabetes. 1996;45(1):86-90.
14. Wallum BJ, Taborsky GJ, Jr, Porte D Jr, et al. Cerebrospinal fluid insulin levels increase during intravenous insulin infusions in man. J Clin Endocr Metab. 1987;64(1):190-194.
15. Woods SC, Seeley RJ, Baskin DG, et al. Insulin and the blood-brain barrier. Curr Pharm Des. 2003;9(10):795-800.
16. Facchini FS, Hua N, Abbasi F, et al. Insulin resistance as a predictor of age-related diseases. J Clin Endocrinol Metab. 2001;86(8):3574-3578.
17. Kuusisto J, Koivisto K, Mykkänen L, et al. Association between features of the insulin resistance syndrome and Alzheimer’s disease independently of apolipoprotein E4 phenotype. BMJ. 1997;315(7115):1045-1049.
18. Luchsinger JA, Tang MX, Shea S, et al. Hyperinsulinemia and risk of Alzheimer’s disease. Neurology. 2004;63(7):1187-1192.
19. Hassing LB, Dahl AK, Thorvaldsson V, et al. Overweight in midlife and risk of dementia: a 40-year follow up study. Int J Obesity (Lond). 2009;33(8):893-898.
20. Young SE, Mainous AG 3rd, Carnemolla M. Hyperinsulinemia and cognitive decline in a middle-aged cohort. Diabetes Care. 2006;29(12):2688-2693.
21. Raji CA, Ho AJ, Parikshak NN, et al. Brain structure and obesity. Hum Brain Mapp. 2009;31(3):353-364.
22. Craft S, Peskind E, Schwartz MW, et al. Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease. Neurology. 1998;50(1):164-168.
23. Craft S, Asthana S, Cook DG, et al. Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: interactions with apolipoprotein E genotype. Psychoneuroendocrinology. 2003;28(6):809-822.
24. Zhao L, Teter B, Morihara T, et al. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer’s disease intervention. J Neurosci. 2004;24(49):11120-11126.
25. Farris W, Mansourian S, Chang Y, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100(7):4162-4167.
26. Tabata S, Yoshimitsu S, Hamachi T, et al. Waist circumference and insulin resistance: a cross-sectional study of Japanese men. BMC Endocr Disord. 2009;9:1.-doi: 10.1186/1472-6823-9-1.
27. Wahrenberg H, Hertel K, Leijonhufvud B, et al. Use of waist circumference to predict insulin resistance: retrospective study. BMJ. 2005;330(7504):1363-1364.
28. Jang S, Lee CH, Choi KM, et al. Correlation of fatty liver and abdominal fat distribution using a simple fat computed tomography protocol. World J Gastroenterol. 2011;17(28):3335-3341.
29. Sutcliffe JG, Hedlund PB, Thomas EA, et al. Peripheral reduction of ß-amyloid is sufficient to reduce brain ß-amyloid: implications for Alzheimer’s disease. J Neurosci Res. 2011;89(6):808-814.
30. Marques MA, Kulstad JJ, Savard CE, et al. Peripheral amyloid-β levels regulate amyloid-β clearance from the central nervous system. J Alzheimers Dis. 2009;16(2):325-329.
31. Cotman CW. Homeostatic processes in brain aging: the role of apoptosis inflammation, and oxidative stress in regulating healthy neural circuitry in the aging brain. In: Stern P, Carstensen L, eds. The aging mind: opportunities in cognitive research. Washington, DC: National Academy Press; 2000:114–143.
32. Witte AV, Fobker M, Gellner R, et al. Caloric restriction improves memory in elderly humans. Proc Natl Acad Sci U S A. 2009;106(4):1255-1260.
33. Krikorian R, Shidler MD, Dangelo K, et al. Dietary ketosis enhances memory in mild cognitive impairment. Neurbiol Aging. 2012;33(2):425.e19-e27.
34. Letenneur L, Proust-Lima C, Le Gouge A, et al. Flavonoid intake and cognitive decline over a 10-year period. Am J Epidemiol. 2007;165(2):1364-1371.
35. Solfrizzi V, Panza F, Capurso A. The role of diet in cognitive decline. J Neural Transm. 2003;110(3):95-110.
36. Williams CM, El Mohsen MA, Vauzour D, et al. Blueberry-induced changes in spatial working memory correlate with changes in hippocampal CREB phosphorylation and brain-derived neurotrophic factor (BDNF) levels. Free Radical Bio Med. 2008;45(3):295-305.
37. Martineau LC, Couture A, Spoor D, et al. Anti-diabetic properties of the Canadian lowbush blueberry Vaccinium angustifolium Ait. Phytomedicine. 2006;13(9-10):612-623.
38. Tsuda T. Regulation of adipocyte function by anthocyanins; possibility of preventing the metabolic syndrome. J Agr Food Chem. 2008;56(3):642-646.
39. Krikorian R, Shidler MD, Nash TA, et al. Blueberry supplementation improves memory in older adults. J Agric Food Chem. 2010;58(7):3996-4000.
Can topiramate reduce nightmares in posttraumatic stress disorder?
Re-experiencing a previous life-threatening stress through nightmares or recurrent memories is a hallmark of posttraumatic stress disorder (PTSD). In the United States, the lifetime risk of PTSD is 10.1% and the 12-month prevalence is 3.7%.1 The selective serotonin reuptake inhibitors (SSRIs) sertraline and paroxetine are FDA-approved for treating PTSD; clinicians commonly use any SSRI for this disorder. Although SSRIs can alleviate many PTSD symptoms, at times patients experience only a partial response, which necessitates other interventions.
Rationale for using topiramate
The anticonvulsant topiramate blocks voltage-sensitive sodium channels, augments γ-aminobutyric acid type A, antagonizes the glutamate receptor, and inhibits carbonic anhydrase. Researchers have hypothesized that limbic nuclei become sensitized and “kindled” after exposure to a traumatic event. Anticonvulsants such as topiramate may help mitigate stress-activated kindling in PTSD.2,3
What does the evidence say?
Although less compelling than double-blind, placebo-controlled trials, small open-label studies and some case reports indicate a potential role for topiramate in PTSD for specific populations.4,5 In an 8-week open- label study, Alderman et al6 found adjunctive topiramate led to a statistically significant reduction in Clinician-Administered PTSD Scale (CAPS) scores and nightmares in 43 male veterans with combat-related PTSD. There was a nonsignificant decrease in high-risk alcohol use.
In a 2002 retrospective case series, Berlant et al7 found topiramate as monotherapy or adjunctive therapy reduced nightmares in 35 patients with chronic, non-combat PTSD. Nightmares decreased in 79% of patients and flashbacks decreased in 86%, with symptom improvement in a median of 4 days. Limitations of this study included lack of placebo control, a low number of participants, and a high dropout rate (9/35).
Two years later, Berlant8 used the PTSD Checklist-Civilian version (PCL-C) to assess response to topiramate in an open-label study of 33 patients with chronic, non-hallucinatory PTSD. Twenty-eight patients used topiramate as add-on therapy. PCL-C scores decreased by ≥30% in 77% of patients in 4 weeks, with a median dose of 50 mg/d and a median response time of 9 days.
In a double-blind, placebo-controlled trial, Tucker et al9 assessed 38 civilian patients who took topiramate monotherapy for PTSD. Using the CAPS, researchers concluded that topiramate reduced re-experiencing symptoms, but the effect was not statistically significant.9
Lindley et al10 conducted a randomized, double-blind, placebo-controlled trial to study the effect of add-on topiramate in 40 patients with chronic, combat-related PTSD. Because many patients in this study had a history of depression and substance use disorders, topiramate was added to antidepressants; no anticonvulsants, antipsychotics, or benzodiazepines were used. Similar to previous studies, researchers found no statistically significant effect on PTSD symptom severity or global symptom improvement. However, the small number of participants and a high dropout rate limited this study.10
In a 12-week, double-blind, placebo-controlled study of 35 men and women age 18 to 62 with PTSD, Yeh et al11 found that topiramate (mean dose: 102.94 mg/d) lead to a statistically significant overall CAPS score reduction, with significant improvements in re-experiencing symptoms, such as nightmares.
Our opinion
FDA-approved treatments such as SSRIs should be the first pharmacologic intervention for PTSD. If a patient’s response is partial or inadequate, consider additional treatment options. For patients with persistent re-experiencing symptoms, evidence and experience with prazosin and trazodone are more robust than that for topiramate.12
Using topiramate to reduce re-experiencing symptoms such as nightmares in PTSD is not supported by statistically significant evidence from double-blind, placebo- controlled trials. However, numerous open-label studies and case reports suggest that there may be a role for topiramate in PTSD patients who do not respond to other treatments. Data indicate that topiramate may be helpful for PTSD patients who have high-risk alcohol use6 or migraine headaches.13 Because some patients who take topiramate lose weight, the medication may be useful for PTSD patients who are overweight.13
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Related Resource
- U.S. Department of Veterans Affairs. Nightmares and PTSD. www.ptsd.va.gov/public/pages/nightmares.asp.
Drug Brand Names
- Paroxetine • Paxil
- Sertraline • Zoloft
- Prazosin • Minipress
- Topiramate • Topamax
- Trazodone • Desyrel, Oleptro
1. Kessler RC, Petukhova M, Sampson NA, et al. Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int J Methods Psychiatr Res. 2012;21(3):169-184.
2. Berlin HA. Antiepileptic drugs for the treatment of post-traumatic stress disorder. Curr Psychiatry Rep. 2007;9(4):291-300.
3. Khan S, Liberzon I. Topiramate attenuates exaggerated acoustic startle in an animal model of PTSD. Psychopharmacology (Berl). 2004;172(2):225-229.
4. Berlant JL. Topiramate in posttraumatic stress disorder: preliminary clinical observations. J Clin Psychiatry. 2001;62(suppl 17):60-63.
5. Tucker P, Masters B, Nawar O. Topiramate in the treatment of comorbid night eating syndrome and PTSD: a case study. Eat Disord. 2004;12(1):75-78.
6. Alderman CP, McCarthy LC, Condon JT, et al. Topiramate in combat-related posttraumatic stress disorder. Ann Pharmacother. 2009;43(4):635-641.
7. Berlant J, van Kammen DP. Open-label topiramate as primary or adjunctive therapy in chronic civilian posttraumatic stress disorder: a preliminary report. J Clin Psychiatry. 2002;63(1):15-20.
8. Berlant JL. Prospective open-label study of add-on and monotherapy topiramate in civilians with chronic nonhallucinatory posttraumatic stress disorder. BMC Psychiatry. 2004;4:24.-
9. Tucker P, Trautman RP, Wyatt DB, et al. Efficacy and safety of topiramate monotherapy in civilian posttraumatic stress disorder: a randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(2):201-206.
10. Lindley SE, Carlson EB, Hill K. A randomized double-blind, placebo-controlled trial of augmentation topiramate for chronic combat-related posttraumatic stress disorder. J Clin Psychopharmacol. 2007;27(6):677-681.
11. Yeh MS, Mari JJ, Costa MC, et al. A double-blind randomized controlled trial to study the efficacy of topiramate in a civilian sample of PTSD. CNW Neurosci Ther. 2011;17(5):305-310.
12. Bajor LA, Ticlea AN, Osser DN. The Psychopharmacology Algorithm Project at the Harvard South Shore Program: an update on posttraumatic stress disorder. Harv Rev Psychiatry. 2011;19(5):240-258.
13. Topax [package insert]. Titusville NJ: Janssen Pharmaceuticals; 2009.
Re-experiencing a previous life-threatening stress through nightmares or recurrent memories is a hallmark of posttraumatic stress disorder (PTSD). In the United States, the lifetime risk of PTSD is 10.1% and the 12-month prevalence is 3.7%.1 The selective serotonin reuptake inhibitors (SSRIs) sertraline and paroxetine are FDA-approved for treating PTSD; clinicians commonly use any SSRI for this disorder. Although SSRIs can alleviate many PTSD symptoms, at times patients experience only a partial response, which necessitates other interventions.
Rationale for using topiramate
The anticonvulsant topiramate blocks voltage-sensitive sodium channels, augments γ-aminobutyric acid type A, antagonizes the glutamate receptor, and inhibits carbonic anhydrase. Researchers have hypothesized that limbic nuclei become sensitized and “kindled” after exposure to a traumatic event. Anticonvulsants such as topiramate may help mitigate stress-activated kindling in PTSD.2,3
What does the evidence say?
Although less compelling than double-blind, placebo-controlled trials, small open-label studies and some case reports indicate a potential role for topiramate in PTSD for specific populations.4,5 In an 8-week open- label study, Alderman et al6 found adjunctive topiramate led to a statistically significant reduction in Clinician-Administered PTSD Scale (CAPS) scores and nightmares in 43 male veterans with combat-related PTSD. There was a nonsignificant decrease in high-risk alcohol use.
In a 2002 retrospective case series, Berlant et al7 found topiramate as monotherapy or adjunctive therapy reduced nightmares in 35 patients with chronic, non-combat PTSD. Nightmares decreased in 79% of patients and flashbacks decreased in 86%, with symptom improvement in a median of 4 days. Limitations of this study included lack of placebo control, a low number of participants, and a high dropout rate (9/35).
Two years later, Berlant8 used the PTSD Checklist-Civilian version (PCL-C) to assess response to topiramate in an open-label study of 33 patients with chronic, non-hallucinatory PTSD. Twenty-eight patients used topiramate as add-on therapy. PCL-C scores decreased by ≥30% in 77% of patients in 4 weeks, with a median dose of 50 mg/d and a median response time of 9 days.
In a double-blind, placebo-controlled trial, Tucker et al9 assessed 38 civilian patients who took topiramate monotherapy for PTSD. Using the CAPS, researchers concluded that topiramate reduced re-experiencing symptoms, but the effect was not statistically significant.9
Lindley et al10 conducted a randomized, double-blind, placebo-controlled trial to study the effect of add-on topiramate in 40 patients with chronic, combat-related PTSD. Because many patients in this study had a history of depression and substance use disorders, topiramate was added to antidepressants; no anticonvulsants, antipsychotics, or benzodiazepines were used. Similar to previous studies, researchers found no statistically significant effect on PTSD symptom severity or global symptom improvement. However, the small number of participants and a high dropout rate limited this study.10
In a 12-week, double-blind, placebo-controlled study of 35 men and women age 18 to 62 with PTSD, Yeh et al11 found that topiramate (mean dose: 102.94 mg/d) lead to a statistically significant overall CAPS score reduction, with significant improvements in re-experiencing symptoms, such as nightmares.
Our opinion
FDA-approved treatments such as SSRIs should be the first pharmacologic intervention for PTSD. If a patient’s response is partial or inadequate, consider additional treatment options. For patients with persistent re-experiencing symptoms, evidence and experience with prazosin and trazodone are more robust than that for topiramate.12
Using topiramate to reduce re-experiencing symptoms such as nightmares in PTSD is not supported by statistically significant evidence from double-blind, placebo- controlled trials. However, numerous open-label studies and case reports suggest that there may be a role for topiramate in PTSD patients who do not respond to other treatments. Data indicate that topiramate may be helpful for PTSD patients who have high-risk alcohol use6 or migraine headaches.13 Because some patients who take topiramate lose weight, the medication may be useful for PTSD patients who are overweight.13
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Related Resource
- U.S. Department of Veterans Affairs. Nightmares and PTSD. www.ptsd.va.gov/public/pages/nightmares.asp.
Drug Brand Names
- Paroxetine • Paxil
- Sertraline • Zoloft
- Prazosin • Minipress
- Topiramate • Topamax
- Trazodone • Desyrel, Oleptro
Re-experiencing a previous life-threatening stress through nightmares or recurrent memories is a hallmark of posttraumatic stress disorder (PTSD). In the United States, the lifetime risk of PTSD is 10.1% and the 12-month prevalence is 3.7%.1 The selective serotonin reuptake inhibitors (SSRIs) sertraline and paroxetine are FDA-approved for treating PTSD; clinicians commonly use any SSRI for this disorder. Although SSRIs can alleviate many PTSD symptoms, at times patients experience only a partial response, which necessitates other interventions.
Rationale for using topiramate
The anticonvulsant topiramate blocks voltage-sensitive sodium channels, augments γ-aminobutyric acid type A, antagonizes the glutamate receptor, and inhibits carbonic anhydrase. Researchers have hypothesized that limbic nuclei become sensitized and “kindled” after exposure to a traumatic event. Anticonvulsants such as topiramate may help mitigate stress-activated kindling in PTSD.2,3
What does the evidence say?
Although less compelling than double-blind, placebo-controlled trials, small open-label studies and some case reports indicate a potential role for topiramate in PTSD for specific populations.4,5 In an 8-week open- label study, Alderman et al6 found adjunctive topiramate led to a statistically significant reduction in Clinician-Administered PTSD Scale (CAPS) scores and nightmares in 43 male veterans with combat-related PTSD. There was a nonsignificant decrease in high-risk alcohol use.
In a 2002 retrospective case series, Berlant et al7 found topiramate as monotherapy or adjunctive therapy reduced nightmares in 35 patients with chronic, non-combat PTSD. Nightmares decreased in 79% of patients and flashbacks decreased in 86%, with symptom improvement in a median of 4 days. Limitations of this study included lack of placebo control, a low number of participants, and a high dropout rate (9/35).
Two years later, Berlant8 used the PTSD Checklist-Civilian version (PCL-C) to assess response to topiramate in an open-label study of 33 patients with chronic, non-hallucinatory PTSD. Twenty-eight patients used topiramate as add-on therapy. PCL-C scores decreased by ≥30% in 77% of patients in 4 weeks, with a median dose of 50 mg/d and a median response time of 9 days.
In a double-blind, placebo-controlled trial, Tucker et al9 assessed 38 civilian patients who took topiramate monotherapy for PTSD. Using the CAPS, researchers concluded that topiramate reduced re-experiencing symptoms, but the effect was not statistically significant.9
Lindley et al10 conducted a randomized, double-blind, placebo-controlled trial to study the effect of add-on topiramate in 40 patients with chronic, combat-related PTSD. Because many patients in this study had a history of depression and substance use disorders, topiramate was added to antidepressants; no anticonvulsants, antipsychotics, or benzodiazepines were used. Similar to previous studies, researchers found no statistically significant effect on PTSD symptom severity or global symptom improvement. However, the small number of participants and a high dropout rate limited this study.10
In a 12-week, double-blind, placebo-controlled study of 35 men and women age 18 to 62 with PTSD, Yeh et al11 found that topiramate (mean dose: 102.94 mg/d) lead to a statistically significant overall CAPS score reduction, with significant improvements in re-experiencing symptoms, such as nightmares.
Our opinion
FDA-approved treatments such as SSRIs should be the first pharmacologic intervention for PTSD. If a patient’s response is partial or inadequate, consider additional treatment options. For patients with persistent re-experiencing symptoms, evidence and experience with prazosin and trazodone are more robust than that for topiramate.12
Using topiramate to reduce re-experiencing symptoms such as nightmares in PTSD is not supported by statistically significant evidence from double-blind, placebo- controlled trials. However, numerous open-label studies and case reports suggest that there may be a role for topiramate in PTSD patients who do not respond to other treatments. Data indicate that topiramate may be helpful for PTSD patients who have high-risk alcohol use6 or migraine headaches.13 Because some patients who take topiramate lose weight, the medication may be useful for PTSD patients who are overweight.13
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Related Resource
- U.S. Department of Veterans Affairs. Nightmares and PTSD. www.ptsd.va.gov/public/pages/nightmares.asp.
Drug Brand Names
- Paroxetine • Paxil
- Sertraline • Zoloft
- Prazosin • Minipress
- Topiramate • Topamax
- Trazodone • Desyrel, Oleptro
1. Kessler RC, Petukhova M, Sampson NA, et al. Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int J Methods Psychiatr Res. 2012;21(3):169-184.
2. Berlin HA. Antiepileptic drugs for the treatment of post-traumatic stress disorder. Curr Psychiatry Rep. 2007;9(4):291-300.
3. Khan S, Liberzon I. Topiramate attenuates exaggerated acoustic startle in an animal model of PTSD. Psychopharmacology (Berl). 2004;172(2):225-229.
4. Berlant JL. Topiramate in posttraumatic stress disorder: preliminary clinical observations. J Clin Psychiatry. 2001;62(suppl 17):60-63.
5. Tucker P, Masters B, Nawar O. Topiramate in the treatment of comorbid night eating syndrome and PTSD: a case study. Eat Disord. 2004;12(1):75-78.
6. Alderman CP, McCarthy LC, Condon JT, et al. Topiramate in combat-related posttraumatic stress disorder. Ann Pharmacother. 2009;43(4):635-641.
7. Berlant J, van Kammen DP. Open-label topiramate as primary or adjunctive therapy in chronic civilian posttraumatic stress disorder: a preliminary report. J Clin Psychiatry. 2002;63(1):15-20.
8. Berlant JL. Prospective open-label study of add-on and monotherapy topiramate in civilians with chronic nonhallucinatory posttraumatic stress disorder. BMC Psychiatry. 2004;4:24.-
9. Tucker P, Trautman RP, Wyatt DB, et al. Efficacy and safety of topiramate monotherapy in civilian posttraumatic stress disorder: a randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(2):201-206.
10. Lindley SE, Carlson EB, Hill K. A randomized double-blind, placebo-controlled trial of augmentation topiramate for chronic combat-related posttraumatic stress disorder. J Clin Psychopharmacol. 2007;27(6):677-681.
11. Yeh MS, Mari JJ, Costa MC, et al. A double-blind randomized controlled trial to study the efficacy of topiramate in a civilian sample of PTSD. CNW Neurosci Ther. 2011;17(5):305-310.
12. Bajor LA, Ticlea AN, Osser DN. The Psychopharmacology Algorithm Project at the Harvard South Shore Program: an update on posttraumatic stress disorder. Harv Rev Psychiatry. 2011;19(5):240-258.
13. Topax [package insert]. Titusville NJ: Janssen Pharmaceuticals; 2009.
1. Kessler RC, Petukhova M, Sampson NA, et al. Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int J Methods Psychiatr Res. 2012;21(3):169-184.
2. Berlin HA. Antiepileptic drugs for the treatment of post-traumatic stress disorder. Curr Psychiatry Rep. 2007;9(4):291-300.
3. Khan S, Liberzon I. Topiramate attenuates exaggerated acoustic startle in an animal model of PTSD. Psychopharmacology (Berl). 2004;172(2):225-229.
4. Berlant JL. Topiramate in posttraumatic stress disorder: preliminary clinical observations. J Clin Psychiatry. 2001;62(suppl 17):60-63.
5. Tucker P, Masters B, Nawar O. Topiramate in the treatment of comorbid night eating syndrome and PTSD: a case study. Eat Disord. 2004;12(1):75-78.
6. Alderman CP, McCarthy LC, Condon JT, et al. Topiramate in combat-related posttraumatic stress disorder. Ann Pharmacother. 2009;43(4):635-641.
7. Berlant J, van Kammen DP. Open-label topiramate as primary or adjunctive therapy in chronic civilian posttraumatic stress disorder: a preliminary report. J Clin Psychiatry. 2002;63(1):15-20.
8. Berlant JL. Prospective open-label study of add-on and monotherapy topiramate in civilians with chronic nonhallucinatory posttraumatic stress disorder. BMC Psychiatry. 2004;4:24.-
9. Tucker P, Trautman RP, Wyatt DB, et al. Efficacy and safety of topiramate monotherapy in civilian posttraumatic stress disorder: a randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(2):201-206.
10. Lindley SE, Carlson EB, Hill K. A randomized double-blind, placebo-controlled trial of augmentation topiramate for chronic combat-related posttraumatic stress disorder. J Clin Psychopharmacol. 2007;27(6):677-681.
11. Yeh MS, Mari JJ, Costa MC, et al. A double-blind randomized controlled trial to study the efficacy of topiramate in a civilian sample of PTSD. CNW Neurosci Ther. 2011;17(5):305-310.
12. Bajor LA, Ticlea AN, Osser DN. The Psychopharmacology Algorithm Project at the Harvard South Shore Program: an update on posttraumatic stress disorder. Harv Rev Psychiatry. 2011;19(5):240-258.
13. Topax [package insert]. Titusville NJ: Janssen Pharmaceuticals; 2009.