User login
Child of The New Gastroenterologist
Approach to dysphagia
Introduction
Dysphagia is the sensation of difficulty swallowing food or liquid in the acute or chronic setting. The prevalence of dysphagia ranges based on the type and etiology but may impact up to one in six adults.1,2 Dysphagia can cause a significant impact on a patient’s health and overall quality of life. A recent study found that only 50% of symptomatic adults seek medical care despite modifying their eating habits by either eating slowly or changing to softer foods or liquids.1 The most common, serious complications of dysphagia include aspiration pneumonia, malnutrition, and dehydration.3 According to the Agency for Healthcare Research and Quality, dysphagia may be responsible for up to 60,000 deaths annually.3

The diagnosis of esophageal dysphagia can be challenging. An initial, thorough history is essential to delineate between oropharyngeal and esophageal dysphagia and guide subsequent diagnostic testing. In recent years, there have been a number of advances in the approach to diagnosing dysphagia, including novel diagnostic modalities. The goal of this review article is to discuss the current approach to esophageal dysphagia and future direction to allow for timely diagnosis and management.
History
The diagnosis of dysphagia begins with a thorough history. Questions about the timing, onset, progression, localization of symptoms, and types of food that are difficult to swallow are essential in differentiating oropharyngeal and esophageal dysphagia.3,4 Further history taking must include medication and allergy review, smoking history, and review of prior radiation or surgical therapies to the head and neck.

Briefly, oropharyngeal dysphagia is difficulty initiating a swallow or passing food from the mouth or throat and can be caused by structural or functional etiologies.5 Clinical presentations include a sensation of food stuck in the back of the throat, coughing or choking while eating, or drooling. Structural causes include head and neck cancer, Zenker diverticulum, Killian Jamieson diverticula, prolonged intubation, or changes secondary to prior surgery or radiation.3 Functional causes may include neurologic, rheumatologic, or muscular disorders.6
Esophageal dysphagia refers to difficulty transporting food or liquid down the esophagus and can be caused by structural, inflammatory, or functional disorders.5 Patients typically localize symptoms of heartburn, regurgitation, nausea, vomiting, cough, or chest pain along the sternum or epigastric region. Alarm signs concerning for malignancy include unintentional weight loss, fevers, or night sweats.3,7 Aside from symptoms, medication review is essential, as dysphagia is a common side effect of antipsychotics, anticholinergics, antimuscarinics, narcotics, and immunosuppressant drugs.8 Larger pills such as NSAIDs, antibiotics, bisphosphonates, potassium supplements, and methylxanthines can cause drug-induced esophagitis, which can initially present as dysphagia.8 Inflammatory causes can be elucidated by obtaining a history about allergies, tobacco use, and recent infections such as thrush or pneumonia. Patients with a history of recurrent pneumonias may be silently aspirating, a complication of dysphagia.3 Once esophageal dysphagia is clinically suspected based on history, workup can begin.
Differentiating etiologies of esophageal dysphagia
The next step in diagnosing esophageal dysphagia is differentiating between structural, inflammatory, or dysmotility etiology (Figure 1).
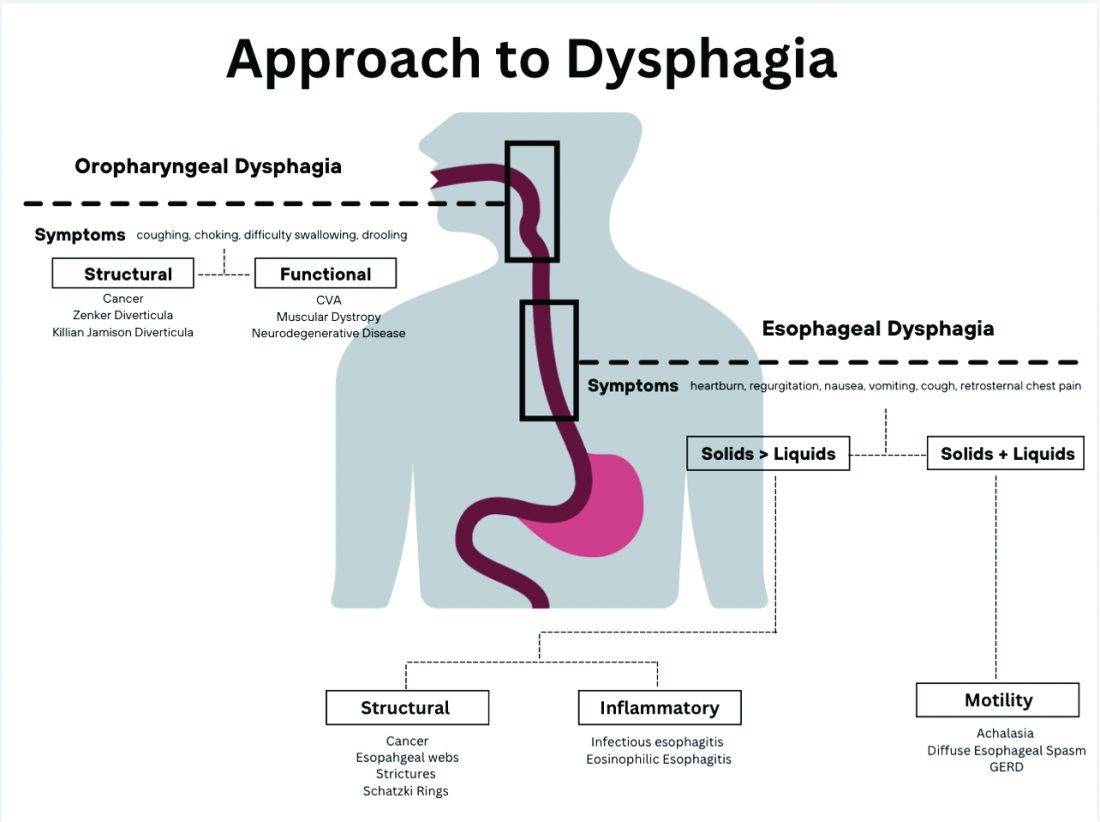
Patients with a structural cause typically have difficulty swallowing solids but are able to swallow liquids unless the disease progresses. Symptoms can rapidly worsen and lead to odynophagia, weight loss, and vomiting. In comparison, patients with motility disorders typically have difficulty swallowing both solids and liquids initially, and symptoms can be constant or intermittent.5
Prior to diagnostic studies, a 4-week trial of a proton pump inhibitor (PPI) is appropriate for patients with reflux symptoms who are younger than 50 with no alarm features concerning for malignancy.7,9 If symptoms persist after a PPI trial, then an upper endoscopy (EGD) is indicated. An EGD allows for visualization of structural etiologies, obtaining biopsies to rule out inflammatory etiologies, and the option to therapeutically treat reduced luminal diameter with dilatation.10 The most common structural and inflammatory etiologies noted on EGD include strictures, webs, carcinomas, Schatzki rings, and gastroesophageal reflux or eosinophilic esophagitis.4
If upper endoscopy is normal and clinical suspicion for an obstructive cause remains high, barium esophagram can be utilized as an adjunctive study. Previously, barium esophagram was the initial test to distinguish between structural and motility disorders. The benefits of endoscopy over barium esophagram as the first diagnostic study include higher diagnostic yield, higher sensitivity and specificity, and lower costs.7 However, barium studies may be more sensitive for lower esophageal rings or extrinsic esophageal compression.3
Evaluation of esophageal motility disorder
If a structural or inflammatory etiology of dysphagia is not identified, investigation for an esophageal motility disorder (EMD) is warranted. Examples of motility disorders include achalasia, ineffective esophageal motility, hypercontractility, spasticity, or esophagogastric junction outflow obstruction (EGJOO).10,11 High-resolution esophageal manometry (HRM) remains the gold standard in diagnosis of EMD.12 An HRM catheter utilizes 36 sensors placed two centimeters apart and is placed in the esophagus to evaluate pressure and peristalsis between the upper and lower esophageal sphincters.13 In 2009, the Chicago Classification System was developed to provide a diagnostic algorithm that categorizes EMD based on HRM testing, with the most recent version (4.0) being published in 2020.12,14 Motility diagnoses are divided into two general classifications of disorders of body peristalsis and disorders of EGJ outflow. The most recent updates also include changes in swallow protocols, patient positioning, targeted symptoms, addition of impedance sensors, and consideration of supplemental testing when HRM is inconclusive based on the clinical context.12 There are some limitations of HRM to highlight. One of the main diagnostic values used with HRM is the integrated relaxation pressure (IRP). Despite standardization, IRP measurements vary based on the recorder and patient position. A minority of patients with achalasia may have IRP that does not approach the accepted cutoff and, therefore, the EGJ is not accurately assessed on HRM.15,16 In addition, some swallow protocols have lower sensitivity and specificity for certain motility disorders, and the test can result as inconclusive.14 In these scenarios, supplemental testing with timed barium esophagram or functional luminal imaging probe (EndoFLIP) is indicated.10,11

Over the past decade, EndoFLIP has emerged as a novel diagnostic tool in evaluating EMD. EndoFLIP is usually completed during an upper endoscopy and utilizes impedance planimetry to measure cross-sectional area and esophageal distensibility and evaluate contractile patterns.16 During the procedure, a small catheter with an inflatable balloon is inserted into the esophagus with the distal end in the stomach, traversing the esophagogastric junction (EGJ). The pressure transducer has electrodes every centimeter to allow for a three-dimensional construction of the esophagus and EGJ.17 EndoFLIP has been shown to accurately measure pyloric diameter, pressure, and distensibility at certain balloon volumes.18 In addition, FLIP is being used to further identify aspects of esophageal dysmotility in patients with eosinophilic esophagitis, thought primarily to be an inflammatory disorder.19 However, limitations include minimal accessibility of EndoFLIP within clinical practice and a specific computer program needed to generate the topographic plots.20
When used in conjunction with HRM, EndoFLIP provides complementary data that can be used to better detect major motility disorders.15,20,21 Each study adds unique information about the different physiologic events comprising the esophageal response to distention. Overall, the benefits of EndoFLIP include expediting workup during index endoscopy, patient comfort with sedation, and real-time diagnostic data that supplement results obtained during HRM.10,16,20,2223
Of note, if the diagnostic evaluation for structural, inflammatory, and motility disorders are unrevealing, investigating for atypical reflux symptoms can be pursued for patients with persistent dysphagia. Studies investigating pH, or acidity in the esophagus, in relation to symptoms, can be conducted wirelessly via a capsule fixed to the mucosa or with a nasal catheter.3
Normal workup – hypervigilance
In a subset of patients, all diagnostic testing for structural, inflammatory, or motility disorders is normal. These patients are classified as having a functional esophageal disorder. Despite normal testing, patients still have significant symptoms including epigastric pain, chest pain, globus sensation, or difficulty swallowing. It is theorized that a degree of visceral hypersensitivity between the brain-gut axis contributes to ongoing symptoms.24 Studies for effective treatments are ongoing but typically include cognitive-behavioral therapy, brain-gut behavioral therapy, swallow therapy antidepressants, or short courses of proton pump inhibitors.9
Conclusion
In this review article, we discussed the diagnostic approach for esophageal dysphagia. Initial assessment requires a thorough history, differentiation between oropharyngeal and esophageal dysphagia, and determination of who warrants an upper endoscopy. Upper endoscopy may reveal structural or inflammatory causes of dysphagia, including strictures, masses, or esophagitis, to name a few. If a structural or inflammatory cause is ruled out, this warrants investigation for esophageal motility disorders. The current gold standard for diagnosing EMD is manometry, and supplemental studies, including EndoFLIP, barium esophagram, and pH studies, may provide complimentary data. If workup for dysphagia is normal, evaluation for esophageal hypervigilance causing increased sensitivity to normal or mild sensations may be warranted. In conclusion, the diagnosis of dysphagia is challenging and requires investigation with a systematic approach to ensure timely diagnosis and treatment
Dr. Ronnie and Dr. Bloomberg are in the department of internal medicine at Loyola University Chicago, Maywood, Ill. Dr. Venu is in the division of gastroenterology at Loyola. He is on the speakers bureau at Medtronic.
References
1. Adkins C et al. Clin Gastroenterol Hepatol. 2020;18(9):1970-9.e2.
2. Bhattacharyya N. Otolaryngol Head Neck Surg. 2014;151(5):765-9.
3. McCarty EB and Chao TN. Med Clin North Am. 2021;105(5):939-54.
4. Thiyagalingam S et al. Mayo Clin Proc. 2021;96(2):488-97.
5. Malagelada JR et al. J Clin Gastroenterol. 2015;49(5):370-8.
6. Rommel, N and Hamdy S. Nat Rev Gastroenterol Hepatol. 2016;13(1):49-59.
7. Liu LWC et al. J Can Assoc Gastroenterol. 2018;1(1):5-19.
8. Schwemmle C et al. HNO. 2015;63(7):504-10.
9. Moayyedi P et al. Am J Gastroenterol. 2017;112(7):988-1013.
10. Triggs J and Pandolfino J. F1000Res. 2019 Aug 29. doi: 10.12688/f1000research.18900.1.
11. Yadlapati R et al. Neurogastroenterol Motil. 2021;33(1):e14058.
12. Yadlapati R et al. Neurogastroenterol Motil. 2021;33(1):e14053.
13. Fox M et al. Neurogastroenterol Motil. 2004;16(5):533-42.
14. Sweis R and Fox M. Curr Gastroenterol Rep. 2020;22(10):49.
15. Carlson DA et al. Gastroenterology. 2015;149(7):1742-51.
16. Donnan EN and Pandolfino JE. Gastroenterol Clin North Am. 2020;49(3):427-35.
17. Carlson DA. Curr Opin Gastroenterol. 2016;32(4):310-8.
18. Zheng T et al. Neurogastroenterol Motil. 2022;34(10):e14386.
19. Carlson DA et al. Clin Gastroenterol Hepatol. 2022;20(8):1719-28.e3.
20. Carlson DA et al. Am J Gastroenterol. 2016;111(12):1726-35.
21. Carlson DA et al. Neurogastroenterol Motil. 2021;33(10):e14116.
22. Carlson DA et al. Gastrointest Endosc. 2019;90(6):915-923.e1.
23. Fox MR et al. Neurogastroenterol Motil. 2021;33(4):e14120.
24. Aziz Q et al. Gastroenterology. 2016 Feb 15. doi: 10.1053/j.gastro.2016.02.012.
Introduction
Dysphagia is the sensation of difficulty swallowing food or liquid in the acute or chronic setting. The prevalence of dysphagia ranges based on the type and etiology but may impact up to one in six adults.1,2 Dysphagia can cause a significant impact on a patient’s health and overall quality of life. A recent study found that only 50% of symptomatic adults seek medical care despite modifying their eating habits by either eating slowly or changing to softer foods or liquids.1 The most common, serious complications of dysphagia include aspiration pneumonia, malnutrition, and dehydration.3 According to the Agency for Healthcare Research and Quality, dysphagia may be responsible for up to 60,000 deaths annually.3
The diagnosis of esophageal dysphagia can be challenging. An initial, thorough history is essential to delineate between oropharyngeal and esophageal dysphagia and guide subsequent diagnostic testing. In recent years, there have been a number of advances in the approach to diagnosing dysphagia, including novel diagnostic modalities. The goal of this review article is to discuss the current approach to esophageal dysphagia and future direction to allow for timely diagnosis and management.
History
The diagnosis of dysphagia begins with a thorough history. Questions about the timing, onset, progression, localization of symptoms, and types of food that are difficult to swallow are essential in differentiating oropharyngeal and esophageal dysphagia.3,4 Further history taking must include medication and allergy review, smoking history, and review of prior radiation or surgical therapies to the head and neck.
Briefly, oropharyngeal dysphagia is difficulty initiating a swallow or passing food from the mouth or throat and can be caused by structural or functional etiologies.5 Clinical presentations include a sensation of food stuck in the back of the throat, coughing or choking while eating, or drooling. Structural causes include head and neck cancer, Zenker diverticulum, Killian Jamieson diverticula, prolonged intubation, or changes secondary to prior surgery or radiation.3 Functional causes may include neurologic, rheumatologic, or muscular disorders.6
Esophageal dysphagia refers to difficulty transporting food or liquid down the esophagus and can be caused by structural, inflammatory, or functional disorders.5 Patients typically localize symptoms of heartburn, regurgitation, nausea, vomiting, cough, or chest pain along the sternum or epigastric region. Alarm signs concerning for malignancy include unintentional weight loss, fevers, or night sweats.3,7 Aside from symptoms, medication review is essential, as dysphagia is a common side effect of antipsychotics, anticholinergics, antimuscarinics, narcotics, and immunosuppressant drugs.8 Larger pills such as NSAIDs, antibiotics, bisphosphonates, potassium supplements, and methylxanthines can cause drug-induced esophagitis, which can initially present as dysphagia.8 Inflammatory causes can be elucidated by obtaining a history about allergies, tobacco use, and recent infections such as thrush or pneumonia. Patients with a history of recurrent pneumonias may be silently aspirating, a complication of dysphagia.3 Once esophageal dysphagia is clinically suspected based on history, workup can begin.
Differentiating etiologies of esophageal dysphagia
The next step in diagnosing esophageal dysphagia is differentiating between structural, inflammatory, or dysmotility etiology (Figure 1).
Patients with a structural cause typically have difficulty swallowing solids but are able to swallow liquids unless the disease progresses. Symptoms can rapidly worsen and lead to odynophagia, weight loss, and vomiting. In comparison, patients with motility disorders typically have difficulty swallowing both solids and liquids initially, and symptoms can be constant or intermittent.5
Prior to diagnostic studies, a 4-week trial of a proton pump inhibitor (PPI) is appropriate for patients with reflux symptoms who are younger than 50 with no alarm features concerning for malignancy.7,9 If symptoms persist after a PPI trial, then an upper endoscopy (EGD) is indicated. An EGD allows for visualization of structural etiologies, obtaining biopsies to rule out inflammatory etiologies, and the option to therapeutically treat reduced luminal diameter with dilatation.10 The most common structural and inflammatory etiologies noted on EGD include strictures, webs, carcinomas, Schatzki rings, and gastroesophageal reflux or eosinophilic esophagitis.4
If upper endoscopy is normal and clinical suspicion for an obstructive cause remains high, barium esophagram can be utilized as an adjunctive study. Previously, barium esophagram was the initial test to distinguish between structural and motility disorders. The benefits of endoscopy over barium esophagram as the first diagnostic study include higher diagnostic yield, higher sensitivity and specificity, and lower costs.7 However, barium studies may be more sensitive for lower esophageal rings or extrinsic esophageal compression.3
Evaluation of esophageal motility disorder
If a structural or inflammatory etiology of dysphagia is not identified, investigation for an esophageal motility disorder (EMD) is warranted. Examples of motility disorders include achalasia, ineffective esophageal motility, hypercontractility, spasticity, or esophagogastric junction outflow obstruction (EGJOO).10,11 High-resolution esophageal manometry (HRM) remains the gold standard in diagnosis of EMD.12 An HRM catheter utilizes 36 sensors placed two centimeters apart and is placed in the esophagus to evaluate pressure and peristalsis between the upper and lower esophageal sphincters.13 In 2009, the Chicago Classification System was developed to provide a diagnostic algorithm that categorizes EMD based on HRM testing, with the most recent version (4.0) being published in 2020.12,14 Motility diagnoses are divided into two general classifications of disorders of body peristalsis and disorders of EGJ outflow. The most recent updates also include changes in swallow protocols, patient positioning, targeted symptoms, addition of impedance sensors, and consideration of supplemental testing when HRM is inconclusive based on the clinical context.12 There are some limitations of HRM to highlight. One of the main diagnostic values used with HRM is the integrated relaxation pressure (IRP). Despite standardization, IRP measurements vary based on the recorder and patient position. A minority of patients with achalasia may have IRP that does not approach the accepted cutoff and, therefore, the EGJ is not accurately assessed on HRM.15,16 In addition, some swallow protocols have lower sensitivity and specificity for certain motility disorders, and the test can result as inconclusive.14 In these scenarios, supplemental testing with timed barium esophagram or functional luminal imaging probe (EndoFLIP) is indicated.10,11
Over the past decade, EndoFLIP has emerged as a novel diagnostic tool in evaluating EMD. EndoFLIP is usually completed during an upper endoscopy and utilizes impedance planimetry to measure cross-sectional area and esophageal distensibility and evaluate contractile patterns.16 During the procedure, a small catheter with an inflatable balloon is inserted into the esophagus with the distal end in the stomach, traversing the esophagogastric junction (EGJ). The pressure transducer has electrodes every centimeter to allow for a three-dimensional construction of the esophagus and EGJ.17 EndoFLIP has been shown to accurately measure pyloric diameter, pressure, and distensibility at certain balloon volumes.18 In addition, FLIP is being used to further identify aspects of esophageal dysmotility in patients with eosinophilic esophagitis, thought primarily to be an inflammatory disorder.19 However, limitations include minimal accessibility of EndoFLIP within clinical practice and a specific computer program needed to generate the topographic plots.20
When used in conjunction with HRM, EndoFLIP provides complementary data that can be used to better detect major motility disorders.15,20,21 Each study adds unique information about the different physiologic events comprising the esophageal response to distention. Overall, the benefits of EndoFLIP include expediting workup during index endoscopy, patient comfort with sedation, and real-time diagnostic data that supplement results obtained during HRM.10,16,20,2223
Of note, if the diagnostic evaluation for structural, inflammatory, and motility disorders are unrevealing, investigating for atypical reflux symptoms can be pursued for patients with persistent dysphagia. Studies investigating pH, or acidity in the esophagus, in relation to symptoms, can be conducted wirelessly via a capsule fixed to the mucosa or with a nasal catheter.3
Normal workup – hypervigilance
In a subset of patients, all diagnostic testing for structural, inflammatory, or motility disorders is normal. These patients are classified as having a functional esophageal disorder. Despite normal testing, patients still have significant symptoms including epigastric pain, chest pain, globus sensation, or difficulty swallowing. It is theorized that a degree of visceral hypersensitivity between the brain-gut axis contributes to ongoing symptoms.24 Studies for effective treatments are ongoing but typically include cognitive-behavioral therapy, brain-gut behavioral therapy, swallow therapy antidepressants, or short courses of proton pump inhibitors.9
Conclusion
In this review article, we discussed the diagnostic approach for esophageal dysphagia. Initial assessment requires a thorough history, differentiation between oropharyngeal and esophageal dysphagia, and determination of who warrants an upper endoscopy. Upper endoscopy may reveal structural or inflammatory causes of dysphagia, including strictures, masses, or esophagitis, to name a few. If a structural or inflammatory cause is ruled out, this warrants investigation for esophageal motility disorders. The current gold standard for diagnosing EMD is manometry, and supplemental studies, including EndoFLIP, barium esophagram, and pH studies, may provide complimentary data. If workup for dysphagia is normal, evaluation for esophageal hypervigilance causing increased sensitivity to normal or mild sensations may be warranted. In conclusion, the diagnosis of dysphagia is challenging and requires investigation with a systematic approach to ensure timely diagnosis and treatment
Dr. Ronnie and Dr. Bloomberg are in the department of internal medicine at Loyola University Chicago, Maywood, Ill. Dr. Venu is in the division of gastroenterology at Loyola. He is on the speakers bureau at Medtronic.
References
1. Adkins C et al. Clin Gastroenterol Hepatol. 2020;18(9):1970-9.e2.
2. Bhattacharyya N. Otolaryngol Head Neck Surg. 2014;151(5):765-9.
3. McCarty EB and Chao TN. Med Clin North Am. 2021;105(5):939-54.
4. Thiyagalingam S et al. Mayo Clin Proc. 2021;96(2):488-97.
5. Malagelada JR et al. J Clin Gastroenterol. 2015;49(5):370-8.
6. Rommel, N and Hamdy S. Nat Rev Gastroenterol Hepatol. 2016;13(1):49-59.
7. Liu LWC et al. J Can Assoc Gastroenterol. 2018;1(1):5-19.
8. Schwemmle C et al. HNO. 2015;63(7):504-10.
9. Moayyedi P et al. Am J Gastroenterol. 2017;112(7):988-1013.
10. Triggs J and Pandolfino J. F1000Res. 2019 Aug 29. doi: 10.12688/f1000research.18900.1.
11. Yadlapati R et al. Neurogastroenterol Motil. 2021;33(1):e14058.
12. Yadlapati R et al. Neurogastroenterol Motil. 2021;33(1):e14053.
13. Fox M et al. Neurogastroenterol Motil. 2004;16(5):533-42.
14. Sweis R and Fox M. Curr Gastroenterol Rep. 2020;22(10):49.
15. Carlson DA et al. Gastroenterology. 2015;149(7):1742-51.
16. Donnan EN and Pandolfino JE. Gastroenterol Clin North Am. 2020;49(3):427-35.
17. Carlson DA. Curr Opin Gastroenterol. 2016;32(4):310-8.
18. Zheng T et al. Neurogastroenterol Motil. 2022;34(10):e14386.
19. Carlson DA et al. Clin Gastroenterol Hepatol. 2022;20(8):1719-28.e3.
20. Carlson DA et al. Am J Gastroenterol. 2016;111(12):1726-35.
21. Carlson DA et al. Neurogastroenterol Motil. 2021;33(10):e14116.
22. Carlson DA et al. Gastrointest Endosc. 2019;90(6):915-923.e1.
23. Fox MR et al. Neurogastroenterol Motil. 2021;33(4):e14120.
24. Aziz Q et al. Gastroenterology. 2016 Feb 15. doi: 10.1053/j.gastro.2016.02.012.
Introduction
Dysphagia is the sensation of difficulty swallowing food or liquid in the acute or chronic setting. The prevalence of dysphagia ranges based on the type and etiology but may impact up to one in six adults.1,2 Dysphagia can cause a significant impact on a patient’s health and overall quality of life. A recent study found that only 50% of symptomatic adults seek medical care despite modifying their eating habits by either eating slowly or changing to softer foods or liquids.1 The most common, serious complications of dysphagia include aspiration pneumonia, malnutrition, and dehydration.3 According to the Agency for Healthcare Research and Quality, dysphagia may be responsible for up to 60,000 deaths annually.3
The diagnosis of esophageal dysphagia can be challenging. An initial, thorough history is essential to delineate between oropharyngeal and esophageal dysphagia and guide subsequent diagnostic testing. In recent years, there have been a number of advances in the approach to diagnosing dysphagia, including novel diagnostic modalities. The goal of this review article is to discuss the current approach to esophageal dysphagia and future direction to allow for timely diagnosis and management.
History
The diagnosis of dysphagia begins with a thorough history. Questions about the timing, onset, progression, localization of symptoms, and types of food that are difficult to swallow are essential in differentiating oropharyngeal and esophageal dysphagia.3,4 Further history taking must include medication and allergy review, smoking history, and review of prior radiation or surgical therapies to the head and neck.
Briefly, oropharyngeal dysphagia is difficulty initiating a swallow or passing food from the mouth or throat and can be caused by structural or functional etiologies.5 Clinical presentations include a sensation of food stuck in the back of the throat, coughing or choking while eating, or drooling. Structural causes include head and neck cancer, Zenker diverticulum, Killian Jamieson diverticula, prolonged intubation, or changes secondary to prior surgery or radiation.3 Functional causes may include neurologic, rheumatologic, or muscular disorders.6
Esophageal dysphagia refers to difficulty transporting food or liquid down the esophagus and can be caused by structural, inflammatory, or functional disorders.5 Patients typically localize symptoms of heartburn, regurgitation, nausea, vomiting, cough, or chest pain along the sternum or epigastric region. Alarm signs concerning for malignancy include unintentional weight loss, fevers, or night sweats.3,7 Aside from symptoms, medication review is essential, as dysphagia is a common side effect of antipsychotics, anticholinergics, antimuscarinics, narcotics, and immunosuppressant drugs.8 Larger pills such as NSAIDs, antibiotics, bisphosphonates, potassium supplements, and methylxanthines can cause drug-induced esophagitis, which can initially present as dysphagia.8 Inflammatory causes can be elucidated by obtaining a history about allergies, tobacco use, and recent infections such as thrush or pneumonia. Patients with a history of recurrent pneumonias may be silently aspirating, a complication of dysphagia.3 Once esophageal dysphagia is clinically suspected based on history, workup can begin.
Differentiating etiologies of esophageal dysphagia
The next step in diagnosing esophageal dysphagia is differentiating between structural, inflammatory, or dysmotility etiology (Figure 1).
Patients with a structural cause typically have difficulty swallowing solids but are able to swallow liquids unless the disease progresses. Symptoms can rapidly worsen and lead to odynophagia, weight loss, and vomiting. In comparison, patients with motility disorders typically have difficulty swallowing both solids and liquids initially, and symptoms can be constant or intermittent.5
Prior to diagnostic studies, a 4-week trial of a proton pump inhibitor (PPI) is appropriate for patients with reflux symptoms who are younger than 50 with no alarm features concerning for malignancy.7,9 If symptoms persist after a PPI trial, then an upper endoscopy (EGD) is indicated. An EGD allows for visualization of structural etiologies, obtaining biopsies to rule out inflammatory etiologies, and the option to therapeutically treat reduced luminal diameter with dilatation.10 The most common structural and inflammatory etiologies noted on EGD include strictures, webs, carcinomas, Schatzki rings, and gastroesophageal reflux or eosinophilic esophagitis.4
If upper endoscopy is normal and clinical suspicion for an obstructive cause remains high, barium esophagram can be utilized as an adjunctive study. Previously, barium esophagram was the initial test to distinguish between structural and motility disorders. The benefits of endoscopy over barium esophagram as the first diagnostic study include higher diagnostic yield, higher sensitivity and specificity, and lower costs.7 However, barium studies may be more sensitive for lower esophageal rings or extrinsic esophageal compression.3
Evaluation of esophageal motility disorder
If a structural or inflammatory etiology of dysphagia is not identified, investigation for an esophageal motility disorder (EMD) is warranted. Examples of motility disorders include achalasia, ineffective esophageal motility, hypercontractility, spasticity, or esophagogastric junction outflow obstruction (EGJOO).10,11 High-resolution esophageal manometry (HRM) remains the gold standard in diagnosis of EMD.12 An HRM catheter utilizes 36 sensors placed two centimeters apart and is placed in the esophagus to evaluate pressure and peristalsis between the upper and lower esophageal sphincters.13 In 2009, the Chicago Classification System was developed to provide a diagnostic algorithm that categorizes EMD based on HRM testing, with the most recent version (4.0) being published in 2020.12,14 Motility diagnoses are divided into two general classifications of disorders of body peristalsis and disorders of EGJ outflow. The most recent updates also include changes in swallow protocols, patient positioning, targeted symptoms, addition of impedance sensors, and consideration of supplemental testing when HRM is inconclusive based on the clinical context.12 There are some limitations of HRM to highlight. One of the main diagnostic values used with HRM is the integrated relaxation pressure (IRP). Despite standardization, IRP measurements vary based on the recorder and patient position. A minority of patients with achalasia may have IRP that does not approach the accepted cutoff and, therefore, the EGJ is not accurately assessed on HRM.15,16 In addition, some swallow protocols have lower sensitivity and specificity for certain motility disorders, and the test can result as inconclusive.14 In these scenarios, supplemental testing with timed barium esophagram or functional luminal imaging probe (EndoFLIP) is indicated.10,11
Over the past decade, EndoFLIP has emerged as a novel diagnostic tool in evaluating EMD. EndoFLIP is usually completed during an upper endoscopy and utilizes impedance planimetry to measure cross-sectional area and esophageal distensibility and evaluate contractile patterns.16 During the procedure, a small catheter with an inflatable balloon is inserted into the esophagus with the distal end in the stomach, traversing the esophagogastric junction (EGJ). The pressure transducer has electrodes every centimeter to allow for a three-dimensional construction of the esophagus and EGJ.17 EndoFLIP has been shown to accurately measure pyloric diameter, pressure, and distensibility at certain balloon volumes.18 In addition, FLIP is being used to further identify aspects of esophageal dysmotility in patients with eosinophilic esophagitis, thought primarily to be an inflammatory disorder.19 However, limitations include minimal accessibility of EndoFLIP within clinical practice and a specific computer program needed to generate the topographic plots.20
When used in conjunction with HRM, EndoFLIP provides complementary data that can be used to better detect major motility disorders.15,20,21 Each study adds unique information about the different physiologic events comprising the esophageal response to distention. Overall, the benefits of EndoFLIP include expediting workup during index endoscopy, patient comfort with sedation, and real-time diagnostic data that supplement results obtained during HRM.10,16,20,2223
Of note, if the diagnostic evaluation for structural, inflammatory, and motility disorders are unrevealing, investigating for atypical reflux symptoms can be pursued for patients with persistent dysphagia. Studies investigating pH, or acidity in the esophagus, in relation to symptoms, can be conducted wirelessly via a capsule fixed to the mucosa or with a nasal catheter.3
Normal workup – hypervigilance
In a subset of patients, all diagnostic testing for structural, inflammatory, or motility disorders is normal. These patients are classified as having a functional esophageal disorder. Despite normal testing, patients still have significant symptoms including epigastric pain, chest pain, globus sensation, or difficulty swallowing. It is theorized that a degree of visceral hypersensitivity between the brain-gut axis contributes to ongoing symptoms.24 Studies for effective treatments are ongoing but typically include cognitive-behavioral therapy, brain-gut behavioral therapy, swallow therapy antidepressants, or short courses of proton pump inhibitors.9
Conclusion
In this review article, we discussed the diagnostic approach for esophageal dysphagia. Initial assessment requires a thorough history, differentiation between oropharyngeal and esophageal dysphagia, and determination of who warrants an upper endoscopy. Upper endoscopy may reveal structural or inflammatory causes of dysphagia, including strictures, masses, or esophagitis, to name a few. If a structural or inflammatory cause is ruled out, this warrants investigation for esophageal motility disorders. The current gold standard for diagnosing EMD is manometry, and supplemental studies, including EndoFLIP, barium esophagram, and pH studies, may provide complimentary data. If workup for dysphagia is normal, evaluation for esophageal hypervigilance causing increased sensitivity to normal or mild sensations may be warranted. In conclusion, the diagnosis of dysphagia is challenging and requires investigation with a systematic approach to ensure timely diagnosis and treatment
Dr. Ronnie and Dr. Bloomberg are in the department of internal medicine at Loyola University Chicago, Maywood, Ill. Dr. Venu is in the division of gastroenterology at Loyola. He is on the speakers bureau at Medtronic.
References
1. Adkins C et al. Clin Gastroenterol Hepatol. 2020;18(9):1970-9.e2.
2. Bhattacharyya N. Otolaryngol Head Neck Surg. 2014;151(5):765-9.
3. McCarty EB and Chao TN. Med Clin North Am. 2021;105(5):939-54.
4. Thiyagalingam S et al. Mayo Clin Proc. 2021;96(2):488-97.
5. Malagelada JR et al. J Clin Gastroenterol. 2015;49(5):370-8.
6. Rommel, N and Hamdy S. Nat Rev Gastroenterol Hepatol. 2016;13(1):49-59.
7. Liu LWC et al. J Can Assoc Gastroenterol. 2018;1(1):5-19.
8. Schwemmle C et al. HNO. 2015;63(7):504-10.
9. Moayyedi P et al. Am J Gastroenterol. 2017;112(7):988-1013.
10. Triggs J and Pandolfino J. F1000Res. 2019 Aug 29. doi: 10.12688/f1000research.18900.1.
11. Yadlapati R et al. Neurogastroenterol Motil. 2021;33(1):e14058.
12. Yadlapati R et al. Neurogastroenterol Motil. 2021;33(1):e14053.
13. Fox M et al. Neurogastroenterol Motil. 2004;16(5):533-42.
14. Sweis R and Fox M. Curr Gastroenterol Rep. 2020;22(10):49.
15. Carlson DA et al. Gastroenterology. 2015;149(7):1742-51.
16. Donnan EN and Pandolfino JE. Gastroenterol Clin North Am. 2020;49(3):427-35.
17. Carlson DA. Curr Opin Gastroenterol. 2016;32(4):310-8.
18. Zheng T et al. Neurogastroenterol Motil. 2022;34(10):e14386.
19. Carlson DA et al. Clin Gastroenterol Hepatol. 2022;20(8):1719-28.e3.
20. Carlson DA et al. Am J Gastroenterol. 2016;111(12):1726-35.
21. Carlson DA et al. Neurogastroenterol Motil. 2021;33(10):e14116.
22. Carlson DA et al. Gastrointest Endosc. 2019;90(6):915-923.e1.
23. Fox MR et al. Neurogastroenterol Motil. 2021;33(4):e14120.
24. Aziz Q et al. Gastroenterology. 2016 Feb 15. doi: 10.1053/j.gastro.2016.02.012.
Artificial intelligence applications in colonoscopy
Considerable advances in artificial intelligence (AI) and machine-learning (ML) methodologies have led to the emergence of promising tools in the field of gastrointestinal endoscopy. Computer vision is an application of AI/ML that has been successfully applied for the computer-aided detection (CADe) and computer-aided diagnosis (CADx) of colon polyps and numerous other conditions encountered during GI endoscopy. Outside of computer vision, a wide variety of other AI applications have been applied to gastroenterology, ranging from natural language processing (NLP) to optimize clinical documentation and endoscopy quality reporting to ML techniques that predict disease severity/treatment response and augment clinical decision-making.

In the United States, colonoscopy is the standard for colon cancer screening and prevention; however, precancerous polyps can be missed for various reasons, ranging from subtle surface appearance of the polyp or location behind a colonic fold to operator-dependent reasons such as inadequate mucosal inspection. Though clinical practice guidelines have set adenoma detection rate (ADR) thresholds at 20% for women and 30% for men, studies have shown a 4- to 10-fold variation in ADR among physicians in clinical practice settings,1 with an estimated adenoma miss rate (AMR) of 25% and a false-negative colonoscopy rate of 12%.2 Variability in adenoma detection affects the risk of interval colorectal cancer post colonoscopy.3,4
AI provides an opportunity for mitigating this risk. Advances in deep learning and computer vision have led to the development of CADe systems that automatically detect polyps in real time during colonoscopy, resulting in reduced adenoma miss rates (Table 1). In addition to polyp detection, deep-learning technologies are also being used in CADx systems for polyp diagnosis and characterization of malignancy risk. This could aid therapeutic decision-making: Unnecessary resection or histopathologic analysis could be obviated for benign hyperplastic polyps. On the other end of the polyp spectrum, an AI tool that could predict the presence or absence of submucosal invasion could be a powerful tool when evaluating early colon cancers for consideration of endoscopic submucosal dissection vs. surgery. Examples of CADe polyp detection and CADx polyp characterization are shown in Figure 1.

Other potential computer vision applications that may improve colonoscopy quality include tools that help measure adequacy of mucosal exposure, segmental inspection time, and a variety of other parameters associated with polyp detection performance. These are promising areas for future research. Beyond improving colonoscopy technique, natural language processing tools already are being used to optimize clinical documentation as well as extract information from colonoscopy and pathology reports that can facilitate reporting of colonoscopy quality metrics such as ADR, cecal intubation rate, withdrawal time, and bowel preparation adequacy. AI-powered analytics may help unlock large-scale reporting of colonoscopy quality metrics on a health-systems level5 or population-level,6 helping to ensure optimal performance and identifying avenues for colonoscopy quality improvement.
The majority of AI research in colonoscopy has focused on CADe for colon polyp detection and CADx for polyp diagnosis. Over the last few years, several randomized clinical trials – two in the United States – have shown that CADe significantly improves adenoma detection and reduces adenoma miss rates in comparison to standard colonoscopy. The existing data are summarized in Table 1, focusing on the two U.S. studies and an international meta-analysis.

In comparison, the data landscape for CADx is nascent and currently limited to several retrospective studies dating back to 2009 and a few prospective studies that have shown promising results.10,11 There is an expectation that integrated CADx also may support the adoption of “resect and discard” or “diagnose and leave” strategies for low-risk polyps. About two-thirds of polyps identified on average-risk screening colonoscopies are diminutive polyps (less than 5 mm in size), which rarely have advanced histologic features (about 0.5%) and are sometimes non-neoplastic (30%). Malignancy risk is even lower in the distal colon.12 As routine histopathologic assessment of such polyps is mostly of limited clinical utility and comes with added pathology costs, CADx technologies may offer a more cost-effective approach where polyps that are characterized in real-time as low-risk adenomas or non-neoplastic are “resected and discarded” or “left in” respectively. In 2011, prior to the development of current AI tools, the American Society for Gastrointestinal Endoscopy set performance thresholds for technologies supporting real-time endoscopic assessment of the histology of diminutive colorectal polyps. The ASGE recommended 90% histopathologic concordance for “resect and discard” tools and 90% negative predictive value for adenomatous histology for “diagnose and leave,” tools.13 Narrow-band imaging (NBI), for example, has been shown to meet these benchmarks14,15 with a modeling study suggesting that implementing “resect and discard” strategies with such tools could result in annual savings of $33 million without adversely affecting efficacy, although practical adoption has been limited.16 More recent work has directly explored the feasibility of leveraging CADx to support “leave-in-situ” and “resect-and-discard” strategies.17
Similarly, while CADe use in colonoscopy is associated with additional up-front costs, a modeling study suggests that its associated gains in ADR (as detailed in Table 1) make it a cost-saving strategy for colorectal cancer prevention in the long term.18 There is still uncertainty on whether the incremental CADe-associated gains in adenoma detection will necessarily translate to significant reductions in interval colorectal cancer risk, particularly for endoscopists who are already high-performing polyp detectors. A recent study suggests that, although higher ADRs were associated with lower rates of interval colorectal cancer, the gains in interval colorectal cancer risk reduction appeared to level off with ADRs above 35%-40% (this finding may be limited by statistical power).19 Further, most of the data from CADe trials suggest that gains in adenoma detection are not driven by increased detection of advanced lesions with high malignancy risk but by small polyps with long latency periods of about 5-10 years, which may not significantly alter interval cancer risk. It remains to be determined whether adoption of CADe will have an impact on hard outcomes, most importantly interval colorectal cancer risk, or merely result in increased resource utilization without moving the needle on colorectal cancer prevention. To answer this question, the OperA study – a large-scale randomized clinical trial of 200,000 patients across 18 centers from 13 countries – was launched in 2022. It will investigate the effect of colonoscopy with CADe on a number of critical measures, including long-term interval colon cancer risk.20
Despite commercial availability of regulatory-approved CADe systems and data supporting use for adenoma detection in colonoscopy, mainstream adoption in clinical practice has been sluggish. Physician survey studies have shown that, although there is considerable interest in integrating CADe into clinical practice, there are concerns about access, cost and reimbursement, integration into clinical work-flow, increased procedural times, over-reliance on AI, and algorithmic bias leading to errors.21,22 In addition, without mandatory requirements for ADR reporting or clinical practice guideline recommendations for CADe use, these systems may not be perceived as valuable or ready for prime time even though the evidence suggests otherwise.23,24 For CADe systems to see widespread adoption in clinical practice, it is important that future research studies rigorously investigate and characterize these potential barriers to better inform strategies to address AI hesitancy and implementation challenges. Such efforts can provide an integration framework for future AI applications in gastroenterology beyond colonoscopy, such as CADe of esophageal and gastric premalignant lesions in upper endoscopy, CADx for pancreatic cysts and liver lesions on imaging, NLP tools to optimizing efficient clinical documentation and reporting, and many others.
Dr. Uche-Anya is in the division of gastroenterology, Massachusetts General Hospital and Harvard Medical School, Boston. Dr. Berzin is with the Center for Advanced Endoscopy, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston. Dr. Berzin is a consultant for Wision AI, Medtronic, Magentiq Eye, RSIP Vision, and Docbot.
Corresponding Author: Eugenia Uche-Anya [email protected] Twitter: @UcheAnyaMD @tberzin
References
1. Corley DA et al. Can we improve adenoma detection rates? A systematic review of intervention studies. Gastrointest Endosc. Sep 2011;74(3):656-65. doi: 10.1016/j.gie.2011.04.017.
2. Zhao S et al. Magnitude, risk factors, and factors associated with adenoma miss rate of tandem colonoscopy: A systematic review and meta-analysis. Gastroenterology. 05 2019;156(6):1661-74.e11. doi: 10.1053/j.gastro.2019.01.260.
3. Kaminski MF et al. Quality indicators for colonoscopy and the risk of interval cancer. N Engl J Med. May 13 2010;362(19):1795-803. doi: 10.1056/NEJMoa0907667.
4. Corley DA et al. Adenoma detection rate and risk of colorectal cancer and death. N Engl J Med. Apr 03 2014;370(14):1298-306. doi: 10.1056/NEJMoa1309086.
5. Laique SN et al. Application of optical character recognition with natural language processing for large-scale quality metric data extraction in colonoscopy reports. Gastrointest Endosc. 03 2021;93(3):750-7. doi: 10.1016/j.gie.2020.08.038.
6. Tinmouth J et al. Validation of a natural language processing algorithm to identify adenomas and measure adenoma detection rates across a health system: a population-level study. Gastrointest Endosc. Jul 14 2022. doi: 10.1016/j.gie.2022.07.009.
7. Glissen Brown JR et al. Deep learning computer-aided polyp detection reduces adenoma miss rate: A United States multi-center randomized tandem colonoscopy study (CADeT-CS Trial). Clin Gastroenterol Hepatol. 07 2022;20(7):1499-1507.e4. doi: 10.1016/j.cgh.2021.09.009.
8. Wallace MB et al. Impact of artificial intelligence on miss rate of colorectal neoplasia. Gastroenterology. 07 2022;163(1):295-304.e5. doi: 10.1053/j.gastro.2022.03.007.
9. Hassan C et al. Performance of artificial intelligence in colonoscopy for adenoma and polyp detection: a systematic review and meta-analysis. Gastrointest Endosc. 01 2021;93(1):77-85.e6. doi: 10.1016/j.gie.2020.06.059.
10. Glissen Brown JR and Berzin TM. Adoption of new technologies: Artificial intelligence. Gastrointest Endosc Clin N Am. Oct 2021;31(4):743-58. doi: 10.1016/j.giec.2021.05.010.
11. Larsen SLV and Mori Y. Artificial intelligence in colonoscopy: A review on the current status. DEN open. Apr 2022;2(1):e109. doi: 10.1002/deo2.109.
12. Gupta N et al. Prevalence of advanced histological features in diminutive and small colon polyps. Gastrointest Endosc. May 2012;75(5):1022-30. doi: 10.1016/j.gie.2012.01.020.
13. Rex DK et al. The American Society for Gastrointestinal Endoscopy PIVI (Preservation and Incorporation of Valuable Endoscopic Innovations) on real-time endoscopic assessment of the histology of diminutive colorectal polyps. Gastrointest Endosc. Mar 2011;73(3):419-22. doi: 10.1016/j.gie.2011.01.023.
14. Abu Dayyeh BK et al. ASGE Technology Committee systematic review and meta-analysis assessing the ASGE PIVI thresholds for adopting real-time endoscopic assessment of the histology of diminutive colorectal polyps. Gastrointest Endosc. Mar 2015;81(3):502.e1-16. doi: 10.1016/j.gie.2014.12.022.
15. Mori Y et al. Real-time use of artificial intelligence in identification of diminutive polyps during colonoscopy: A prospective study. Ann Intern Med. Sep 18 2018;169(6):357-66. doi: 10.7326/M18-0249.
16. Hassan C et al.. A resect and discard strategy would improve cost-effectiveness of colorectal cancer screening. Clin Gastroenterol Hepatol. Oct 2010;8(10):865-9, 869.e1-3. doi: 10.1016/j.cgh.2010.05.018.
17. Hassan C et al. Artificial intelligence allows leaving-in-situ colorectal polyps. Clin Gastroenterol Hepatol. Nov 2022;20(11):2505-13.e4. doi: 10.1016/j.cgh.2022.04.045.
18. Areia M et al. Cost-effectiveness of artificial intelligence for screening colonoscopy: a modelling study. Lancet Digit Health. 06 2022;4(6):e436-44. doi: 10.1016/S2589-7500(22)00042-5.
19. Schottinger JE et al. Association of physician adenoma detection rates with postcolonoscopy colorectal cancer. JAMA. 2022 Jun 7;327(21):2114-22. doi: 10.1001/jama.2022.6644.
20. Oslo Uo. Optimising colorectal cancer prevention through personalised treatment with artificial intelligence. 2022.
21. Wadhwa V et al. Physician sentiment toward artificial intelligence (AI) in colonoscopic practice: a survey of US gastroenterologists. Endosc Int Open. Oct 2020;8(10):E1379-84. doi: 10.1055/a-1223-1926.
22. Kader R et al. Survey on the perceptions of UK gastroenterologists and endoscopists to artificial intelligence. Frontline Gastroenterol. 2022;13(5):423-9. doi: 10.1136/flgastro-2021-101994.
23. Rex DKet al. Artificial intelligence improves detection at colonoscopy: Why aren’t we all already using it? Gastroenterology. 07 2022;163(1):35-7. doi: 10.1053/j.gastro.2022.04.042.
24. Ahmad OF et al. Establishing key research questions for the implementation of artificial intelligence in colonoscopy: A modified Delphi method. Endoscopy. 09 2021;53(9):893-901. doi: 10.1055/a-1306-7590
Considerable advances in artificial intelligence (AI) and machine-learning (ML) methodologies have led to the emergence of promising tools in the field of gastrointestinal endoscopy. Computer vision is an application of AI/ML that has been successfully applied for the computer-aided detection (CADe) and computer-aided diagnosis (CADx) of colon polyps and numerous other conditions encountered during GI endoscopy. Outside of computer vision, a wide variety of other AI applications have been applied to gastroenterology, ranging from natural language processing (NLP) to optimize clinical documentation and endoscopy quality reporting to ML techniques that predict disease severity/treatment response and augment clinical decision-making.
In the United States, colonoscopy is the standard for colon cancer screening and prevention; however, precancerous polyps can be missed for various reasons, ranging from subtle surface appearance of the polyp or location behind a colonic fold to operator-dependent reasons such as inadequate mucosal inspection. Though clinical practice guidelines have set adenoma detection rate (ADR) thresholds at 20% for women and 30% for men, studies have shown a 4- to 10-fold variation in ADR among physicians in clinical practice settings,1 with an estimated adenoma miss rate (AMR) of 25% and a false-negative colonoscopy rate of 12%.2 Variability in adenoma detection affects the risk of interval colorectal cancer post colonoscopy.3,4
AI provides an opportunity for mitigating this risk. Advances in deep learning and computer vision have led to the development of CADe systems that automatically detect polyps in real time during colonoscopy, resulting in reduced adenoma miss rates (Table 1). In addition to polyp detection, deep-learning technologies are also being used in CADx systems for polyp diagnosis and characterization of malignancy risk. This could aid therapeutic decision-making: Unnecessary resection or histopathologic analysis could be obviated for benign hyperplastic polyps. On the other end of the polyp spectrum, an AI tool that could predict the presence or absence of submucosal invasion could be a powerful tool when evaluating early colon cancers for consideration of endoscopic submucosal dissection vs. surgery. Examples of CADe polyp detection and CADx polyp characterization are shown in Figure 1.
Other potential computer vision applications that may improve colonoscopy quality include tools that help measure adequacy of mucosal exposure, segmental inspection time, and a variety of other parameters associated with polyp detection performance. These are promising areas for future research. Beyond improving colonoscopy technique, natural language processing tools already are being used to optimize clinical documentation as well as extract information from colonoscopy and pathology reports that can facilitate reporting of colonoscopy quality metrics such as ADR, cecal intubation rate, withdrawal time, and bowel preparation adequacy. AI-powered analytics may help unlock large-scale reporting of colonoscopy quality metrics on a health-systems level5 or population-level,6 helping to ensure optimal performance and identifying avenues for colonoscopy quality improvement.
The majority of AI research in colonoscopy has focused on CADe for colon polyp detection and CADx for polyp diagnosis. Over the last few years, several randomized clinical trials – two in the United States – have shown that CADe significantly improves adenoma detection and reduces adenoma miss rates in comparison to standard colonoscopy. The existing data are summarized in Table 1, focusing on the two U.S. studies and an international meta-analysis.
In comparison, the data landscape for CADx is nascent and currently limited to several retrospective studies dating back to 2009 and a few prospective studies that have shown promising results.10,11 There is an expectation that integrated CADx also may support the adoption of “resect and discard” or “diagnose and leave” strategies for low-risk polyps. About two-thirds of polyps identified on average-risk screening colonoscopies are diminutive polyps (less than 5 mm in size), which rarely have advanced histologic features (about 0.5%) and are sometimes non-neoplastic (30%). Malignancy risk is even lower in the distal colon.12 As routine histopathologic assessment of such polyps is mostly of limited clinical utility and comes with added pathology costs, CADx technologies may offer a more cost-effective approach where polyps that are characterized in real-time as low-risk adenomas or non-neoplastic are “resected and discarded” or “left in” respectively. In 2011, prior to the development of current AI tools, the American Society for Gastrointestinal Endoscopy set performance thresholds for technologies supporting real-time endoscopic assessment of the histology of diminutive colorectal polyps. The ASGE recommended 90% histopathologic concordance for “resect and discard” tools and 90% negative predictive value for adenomatous histology for “diagnose and leave,” tools.13 Narrow-band imaging (NBI), for example, has been shown to meet these benchmarks14,15 with a modeling study suggesting that implementing “resect and discard” strategies with such tools could result in annual savings of $33 million without adversely affecting efficacy, although practical adoption has been limited.16 More recent work has directly explored the feasibility of leveraging CADx to support “leave-in-situ” and “resect-and-discard” strategies.17
Similarly, while CADe use in colonoscopy is associated with additional up-front costs, a modeling study suggests that its associated gains in ADR (as detailed in Table 1) make it a cost-saving strategy for colorectal cancer prevention in the long term.18 There is still uncertainty on whether the incremental CADe-associated gains in adenoma detection will necessarily translate to significant reductions in interval colorectal cancer risk, particularly for endoscopists who are already high-performing polyp detectors. A recent study suggests that, although higher ADRs were associated with lower rates of interval colorectal cancer, the gains in interval colorectal cancer risk reduction appeared to level off with ADRs above 35%-40% (this finding may be limited by statistical power).19 Further, most of the data from CADe trials suggest that gains in adenoma detection are not driven by increased detection of advanced lesions with high malignancy risk but by small polyps with long latency periods of about 5-10 years, which may not significantly alter interval cancer risk. It remains to be determined whether adoption of CADe will have an impact on hard outcomes, most importantly interval colorectal cancer risk, or merely result in increased resource utilization without moving the needle on colorectal cancer prevention. To answer this question, the OperA study – a large-scale randomized clinical trial of 200,000 patients across 18 centers from 13 countries – was launched in 2022. It will investigate the effect of colonoscopy with CADe on a number of critical measures, including long-term interval colon cancer risk.20
Despite commercial availability of regulatory-approved CADe systems and data supporting use for adenoma detection in colonoscopy, mainstream adoption in clinical practice has been sluggish. Physician survey studies have shown that, although there is considerable interest in integrating CADe into clinical practice, there are concerns about access, cost and reimbursement, integration into clinical work-flow, increased procedural times, over-reliance on AI, and algorithmic bias leading to errors.21,22 In addition, without mandatory requirements for ADR reporting or clinical practice guideline recommendations for CADe use, these systems may not be perceived as valuable or ready for prime time even though the evidence suggests otherwise.23,24 For CADe systems to see widespread adoption in clinical practice, it is important that future research studies rigorously investigate and characterize these potential barriers to better inform strategies to address AI hesitancy and implementation challenges. Such efforts can provide an integration framework for future AI applications in gastroenterology beyond colonoscopy, such as CADe of esophageal and gastric premalignant lesions in upper endoscopy, CADx for pancreatic cysts and liver lesions on imaging, NLP tools to optimizing efficient clinical documentation and reporting, and many others.
Dr. Uche-Anya is in the division of gastroenterology, Massachusetts General Hospital and Harvard Medical School, Boston. Dr. Berzin is with the Center for Advanced Endoscopy, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston. Dr. Berzin is a consultant for Wision AI, Medtronic, Magentiq Eye, RSIP Vision, and Docbot.
Corresponding Author: Eugenia Uche-Anya [email protected] Twitter: @UcheAnyaMD @tberzin
References
1. Corley DA et al. Can we improve adenoma detection rates? A systematic review of intervention studies. Gastrointest Endosc. Sep 2011;74(3):656-65. doi: 10.1016/j.gie.2011.04.017.
2. Zhao S et al. Magnitude, risk factors, and factors associated with adenoma miss rate of tandem colonoscopy: A systematic review and meta-analysis. Gastroenterology. 05 2019;156(6):1661-74.e11. doi: 10.1053/j.gastro.2019.01.260.
3. Kaminski MF et al. Quality indicators for colonoscopy and the risk of interval cancer. N Engl J Med. May 13 2010;362(19):1795-803. doi: 10.1056/NEJMoa0907667.
4. Corley DA et al. Adenoma detection rate and risk of colorectal cancer and death. N Engl J Med. Apr 03 2014;370(14):1298-306. doi: 10.1056/NEJMoa1309086.
5. Laique SN et al. Application of optical character recognition with natural language processing for large-scale quality metric data extraction in colonoscopy reports. Gastrointest Endosc. 03 2021;93(3):750-7. doi: 10.1016/j.gie.2020.08.038.
6. Tinmouth J et al. Validation of a natural language processing algorithm to identify adenomas and measure adenoma detection rates across a health system: a population-level study. Gastrointest Endosc. Jul 14 2022. doi: 10.1016/j.gie.2022.07.009.
7. Glissen Brown JR et al. Deep learning computer-aided polyp detection reduces adenoma miss rate: A United States multi-center randomized tandem colonoscopy study (CADeT-CS Trial). Clin Gastroenterol Hepatol. 07 2022;20(7):1499-1507.e4. doi: 10.1016/j.cgh.2021.09.009.
8. Wallace MB et al. Impact of artificial intelligence on miss rate of colorectal neoplasia. Gastroenterology. 07 2022;163(1):295-304.e5. doi: 10.1053/j.gastro.2022.03.007.
9. Hassan C et al. Performance of artificial intelligence in colonoscopy for adenoma and polyp detection: a systematic review and meta-analysis. Gastrointest Endosc. 01 2021;93(1):77-85.e6. doi: 10.1016/j.gie.2020.06.059.
10. Glissen Brown JR and Berzin TM. Adoption of new technologies: Artificial intelligence. Gastrointest Endosc Clin N Am. Oct 2021;31(4):743-58. doi: 10.1016/j.giec.2021.05.010.
11. Larsen SLV and Mori Y. Artificial intelligence in colonoscopy: A review on the current status. DEN open. Apr 2022;2(1):e109. doi: 10.1002/deo2.109.
12. Gupta N et al. Prevalence of advanced histological features in diminutive and small colon polyps. Gastrointest Endosc. May 2012;75(5):1022-30. doi: 10.1016/j.gie.2012.01.020.
13. Rex DK et al. The American Society for Gastrointestinal Endoscopy PIVI (Preservation and Incorporation of Valuable Endoscopic Innovations) on real-time endoscopic assessment of the histology of diminutive colorectal polyps. Gastrointest Endosc. Mar 2011;73(3):419-22. doi: 10.1016/j.gie.2011.01.023.
14. Abu Dayyeh BK et al. ASGE Technology Committee systematic review and meta-analysis assessing the ASGE PIVI thresholds for adopting real-time endoscopic assessment of the histology of diminutive colorectal polyps. Gastrointest Endosc. Mar 2015;81(3):502.e1-16. doi: 10.1016/j.gie.2014.12.022.
15. Mori Y et al. Real-time use of artificial intelligence in identification of diminutive polyps during colonoscopy: A prospective study. Ann Intern Med. Sep 18 2018;169(6):357-66. doi: 10.7326/M18-0249.
16. Hassan C et al.. A resect and discard strategy would improve cost-effectiveness of colorectal cancer screening. Clin Gastroenterol Hepatol. Oct 2010;8(10):865-9, 869.e1-3. doi: 10.1016/j.cgh.2010.05.018.
17. Hassan C et al. Artificial intelligence allows leaving-in-situ colorectal polyps. Clin Gastroenterol Hepatol. Nov 2022;20(11):2505-13.e4. doi: 10.1016/j.cgh.2022.04.045.
18. Areia M et al. Cost-effectiveness of artificial intelligence for screening colonoscopy: a modelling study. Lancet Digit Health. 06 2022;4(6):e436-44. doi: 10.1016/S2589-7500(22)00042-5.
19. Schottinger JE et al. Association of physician adenoma detection rates with postcolonoscopy colorectal cancer. JAMA. 2022 Jun 7;327(21):2114-22. doi: 10.1001/jama.2022.6644.
20. Oslo Uo. Optimising colorectal cancer prevention through personalised treatment with artificial intelligence. 2022.
21. Wadhwa V et al. Physician sentiment toward artificial intelligence (AI) in colonoscopic practice: a survey of US gastroenterologists. Endosc Int Open. Oct 2020;8(10):E1379-84. doi: 10.1055/a-1223-1926.
22. Kader R et al. Survey on the perceptions of UK gastroenterologists and endoscopists to artificial intelligence. Frontline Gastroenterol. 2022;13(5):423-9. doi: 10.1136/flgastro-2021-101994.
23. Rex DKet al. Artificial intelligence improves detection at colonoscopy: Why aren’t we all already using it? Gastroenterology. 07 2022;163(1):35-7. doi: 10.1053/j.gastro.2022.04.042.
24. Ahmad OF et al. Establishing key research questions for the implementation of artificial intelligence in colonoscopy: A modified Delphi method. Endoscopy. 09 2021;53(9):893-901. doi: 10.1055/a-1306-7590
Considerable advances in artificial intelligence (AI) and machine-learning (ML) methodologies have led to the emergence of promising tools in the field of gastrointestinal endoscopy. Computer vision is an application of AI/ML that has been successfully applied for the computer-aided detection (CADe) and computer-aided diagnosis (CADx) of colon polyps and numerous other conditions encountered during GI endoscopy. Outside of computer vision, a wide variety of other AI applications have been applied to gastroenterology, ranging from natural language processing (NLP) to optimize clinical documentation and endoscopy quality reporting to ML techniques that predict disease severity/treatment response and augment clinical decision-making.
In the United States, colonoscopy is the standard for colon cancer screening and prevention; however, precancerous polyps can be missed for various reasons, ranging from subtle surface appearance of the polyp or location behind a colonic fold to operator-dependent reasons such as inadequate mucosal inspection. Though clinical practice guidelines have set adenoma detection rate (ADR) thresholds at 20% for women and 30% for men, studies have shown a 4- to 10-fold variation in ADR among physicians in clinical practice settings,1 with an estimated adenoma miss rate (AMR) of 25% and a false-negative colonoscopy rate of 12%.2 Variability in adenoma detection affects the risk of interval colorectal cancer post colonoscopy.3,4
AI provides an opportunity for mitigating this risk. Advances in deep learning and computer vision have led to the development of CADe systems that automatically detect polyps in real time during colonoscopy, resulting in reduced adenoma miss rates (Table 1). In addition to polyp detection, deep-learning technologies are also being used in CADx systems for polyp diagnosis and characterization of malignancy risk. This could aid therapeutic decision-making: Unnecessary resection or histopathologic analysis could be obviated for benign hyperplastic polyps. On the other end of the polyp spectrum, an AI tool that could predict the presence or absence of submucosal invasion could be a powerful tool when evaluating early colon cancers for consideration of endoscopic submucosal dissection vs. surgery. Examples of CADe polyp detection and CADx polyp characterization are shown in Figure 1.
Other potential computer vision applications that may improve colonoscopy quality include tools that help measure adequacy of mucosal exposure, segmental inspection time, and a variety of other parameters associated with polyp detection performance. These are promising areas for future research. Beyond improving colonoscopy technique, natural language processing tools already are being used to optimize clinical documentation as well as extract information from colonoscopy and pathology reports that can facilitate reporting of colonoscopy quality metrics such as ADR, cecal intubation rate, withdrawal time, and bowel preparation adequacy. AI-powered analytics may help unlock large-scale reporting of colonoscopy quality metrics on a health-systems level5 or population-level,6 helping to ensure optimal performance and identifying avenues for colonoscopy quality improvement.
The majority of AI research in colonoscopy has focused on CADe for colon polyp detection and CADx for polyp diagnosis. Over the last few years, several randomized clinical trials – two in the United States – have shown that CADe significantly improves adenoma detection and reduces adenoma miss rates in comparison to standard colonoscopy. The existing data are summarized in Table 1, focusing on the two U.S. studies and an international meta-analysis.
In comparison, the data landscape for CADx is nascent and currently limited to several retrospective studies dating back to 2009 and a few prospective studies that have shown promising results.10,11 There is an expectation that integrated CADx also may support the adoption of “resect and discard” or “diagnose and leave” strategies for low-risk polyps. About two-thirds of polyps identified on average-risk screening colonoscopies are diminutive polyps (less than 5 mm in size), which rarely have advanced histologic features (about 0.5%) and are sometimes non-neoplastic (30%). Malignancy risk is even lower in the distal colon.12 As routine histopathologic assessment of such polyps is mostly of limited clinical utility and comes with added pathology costs, CADx technologies may offer a more cost-effective approach where polyps that are characterized in real-time as low-risk adenomas or non-neoplastic are “resected and discarded” or “left in” respectively. In 2011, prior to the development of current AI tools, the American Society for Gastrointestinal Endoscopy set performance thresholds for technologies supporting real-time endoscopic assessment of the histology of diminutive colorectal polyps. The ASGE recommended 90% histopathologic concordance for “resect and discard” tools and 90% negative predictive value for adenomatous histology for “diagnose and leave,” tools.13 Narrow-band imaging (NBI), for example, has been shown to meet these benchmarks14,15 with a modeling study suggesting that implementing “resect and discard” strategies with such tools could result in annual savings of $33 million without adversely affecting efficacy, although practical adoption has been limited.16 More recent work has directly explored the feasibility of leveraging CADx to support “leave-in-situ” and “resect-and-discard” strategies.17
Similarly, while CADe use in colonoscopy is associated with additional up-front costs, a modeling study suggests that its associated gains in ADR (as detailed in Table 1) make it a cost-saving strategy for colorectal cancer prevention in the long term.18 There is still uncertainty on whether the incremental CADe-associated gains in adenoma detection will necessarily translate to significant reductions in interval colorectal cancer risk, particularly for endoscopists who are already high-performing polyp detectors. A recent study suggests that, although higher ADRs were associated with lower rates of interval colorectal cancer, the gains in interval colorectal cancer risk reduction appeared to level off with ADRs above 35%-40% (this finding may be limited by statistical power).19 Further, most of the data from CADe trials suggest that gains in adenoma detection are not driven by increased detection of advanced lesions with high malignancy risk but by small polyps with long latency periods of about 5-10 years, which may not significantly alter interval cancer risk. It remains to be determined whether adoption of CADe will have an impact on hard outcomes, most importantly interval colorectal cancer risk, or merely result in increased resource utilization without moving the needle on colorectal cancer prevention. To answer this question, the OperA study – a large-scale randomized clinical trial of 200,000 patients across 18 centers from 13 countries – was launched in 2022. It will investigate the effect of colonoscopy with CADe on a number of critical measures, including long-term interval colon cancer risk.20
Despite commercial availability of regulatory-approved CADe systems and data supporting use for adenoma detection in colonoscopy, mainstream adoption in clinical practice has been sluggish. Physician survey studies have shown that, although there is considerable interest in integrating CADe into clinical practice, there are concerns about access, cost and reimbursement, integration into clinical work-flow, increased procedural times, over-reliance on AI, and algorithmic bias leading to errors.21,22 In addition, without mandatory requirements for ADR reporting or clinical practice guideline recommendations for CADe use, these systems may not be perceived as valuable or ready for prime time even though the evidence suggests otherwise.23,24 For CADe systems to see widespread adoption in clinical practice, it is important that future research studies rigorously investigate and characterize these potential barriers to better inform strategies to address AI hesitancy and implementation challenges. Such efforts can provide an integration framework for future AI applications in gastroenterology beyond colonoscopy, such as CADe of esophageal and gastric premalignant lesions in upper endoscopy, CADx for pancreatic cysts and liver lesions on imaging, NLP tools to optimizing efficient clinical documentation and reporting, and many others.
Dr. Uche-Anya is in the division of gastroenterology, Massachusetts General Hospital and Harvard Medical School, Boston. Dr. Berzin is with the Center for Advanced Endoscopy, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston. Dr. Berzin is a consultant for Wision AI, Medtronic, Magentiq Eye, RSIP Vision, and Docbot.
Corresponding Author: Eugenia Uche-Anya [email protected] Twitter: @UcheAnyaMD @tberzin
References
1. Corley DA et al. Can we improve adenoma detection rates? A systematic review of intervention studies. Gastrointest Endosc. Sep 2011;74(3):656-65. doi: 10.1016/j.gie.2011.04.017.
2. Zhao S et al. Magnitude, risk factors, and factors associated with adenoma miss rate of tandem colonoscopy: A systematic review and meta-analysis. Gastroenterology. 05 2019;156(6):1661-74.e11. doi: 10.1053/j.gastro.2019.01.260.
3. Kaminski MF et al. Quality indicators for colonoscopy and the risk of interval cancer. N Engl J Med. May 13 2010;362(19):1795-803. doi: 10.1056/NEJMoa0907667.
4. Corley DA et al. Adenoma detection rate and risk of colorectal cancer and death. N Engl J Med. Apr 03 2014;370(14):1298-306. doi: 10.1056/NEJMoa1309086.
5. Laique SN et al. Application of optical character recognition with natural language processing for large-scale quality metric data extraction in colonoscopy reports. Gastrointest Endosc. 03 2021;93(3):750-7. doi: 10.1016/j.gie.2020.08.038.
6. Tinmouth J et al. Validation of a natural language processing algorithm to identify adenomas and measure adenoma detection rates across a health system: a population-level study. Gastrointest Endosc. Jul 14 2022. doi: 10.1016/j.gie.2022.07.009.
7. Glissen Brown JR et al. Deep learning computer-aided polyp detection reduces adenoma miss rate: A United States multi-center randomized tandem colonoscopy study (CADeT-CS Trial). Clin Gastroenterol Hepatol. 07 2022;20(7):1499-1507.e4. doi: 10.1016/j.cgh.2021.09.009.
8. Wallace MB et al. Impact of artificial intelligence on miss rate of colorectal neoplasia. Gastroenterology. 07 2022;163(1):295-304.e5. doi: 10.1053/j.gastro.2022.03.007.
9. Hassan C et al. Performance of artificial intelligence in colonoscopy for adenoma and polyp detection: a systematic review and meta-analysis. Gastrointest Endosc. 01 2021;93(1):77-85.e6. doi: 10.1016/j.gie.2020.06.059.
10. Glissen Brown JR and Berzin TM. Adoption of new technologies: Artificial intelligence. Gastrointest Endosc Clin N Am. Oct 2021;31(4):743-58. doi: 10.1016/j.giec.2021.05.010.
11. Larsen SLV and Mori Y. Artificial intelligence in colonoscopy: A review on the current status. DEN open. Apr 2022;2(1):e109. doi: 10.1002/deo2.109.
12. Gupta N et al. Prevalence of advanced histological features in diminutive and small colon polyps. Gastrointest Endosc. May 2012;75(5):1022-30. doi: 10.1016/j.gie.2012.01.020.
13. Rex DK et al. The American Society for Gastrointestinal Endoscopy PIVI (Preservation and Incorporation of Valuable Endoscopic Innovations) on real-time endoscopic assessment of the histology of diminutive colorectal polyps. Gastrointest Endosc. Mar 2011;73(3):419-22. doi: 10.1016/j.gie.2011.01.023.
14. Abu Dayyeh BK et al. ASGE Technology Committee systematic review and meta-analysis assessing the ASGE PIVI thresholds for adopting real-time endoscopic assessment of the histology of diminutive colorectal polyps. Gastrointest Endosc. Mar 2015;81(3):502.e1-16. doi: 10.1016/j.gie.2014.12.022.
15. Mori Y et al. Real-time use of artificial intelligence in identification of diminutive polyps during colonoscopy: A prospective study. Ann Intern Med. Sep 18 2018;169(6):357-66. doi: 10.7326/M18-0249.
16. Hassan C et al.. A resect and discard strategy would improve cost-effectiveness of colorectal cancer screening. Clin Gastroenterol Hepatol. Oct 2010;8(10):865-9, 869.e1-3. doi: 10.1016/j.cgh.2010.05.018.
17. Hassan C et al. Artificial intelligence allows leaving-in-situ colorectal polyps. Clin Gastroenterol Hepatol. Nov 2022;20(11):2505-13.e4. doi: 10.1016/j.cgh.2022.04.045.
18. Areia M et al. Cost-effectiveness of artificial intelligence for screening colonoscopy: a modelling study. Lancet Digit Health. 06 2022;4(6):e436-44. doi: 10.1016/S2589-7500(22)00042-5.
19. Schottinger JE et al. Association of physician adenoma detection rates with postcolonoscopy colorectal cancer. JAMA. 2022 Jun 7;327(21):2114-22. doi: 10.1001/jama.2022.6644.
20. Oslo Uo. Optimising colorectal cancer prevention through personalised treatment with artificial intelligence. 2022.
21. Wadhwa V et al. Physician sentiment toward artificial intelligence (AI) in colonoscopic practice: a survey of US gastroenterologists. Endosc Int Open. Oct 2020;8(10):E1379-84. doi: 10.1055/a-1223-1926.
22. Kader R et al. Survey on the perceptions of UK gastroenterologists and endoscopists to artificial intelligence. Frontline Gastroenterol. 2022;13(5):423-9. doi: 10.1136/flgastro-2021-101994.
23. Rex DKet al. Artificial intelligence improves detection at colonoscopy: Why aren’t we all already using it? Gastroenterology. 07 2022;163(1):35-7. doi: 10.1053/j.gastro.2022.04.042.
24. Ahmad OF et al. Establishing key research questions for the implementation of artificial intelligence in colonoscopy: A modified Delphi method. Endoscopy. 09 2021;53(9):893-901. doi: 10.1055/a-1306-7590
Management of antithrombotic medications in elective endoscopy
Antithrombotic therapy is increasingly used to either reduce the risk of or treat thromboembolic episodes in patients with various medical conditions such as ischemic and valvular heart disease, atrial fibrillation (AF), cerebrovascular disease, peripheral arterial disease, venous thromboembolism (VTE) and hypercoagulable diseases. Antithrombotics include medications classified as anticoagulants or antiplatelets. Anticoagulants work by interfering with the native clotting cascade and consist of four main classes: vitamin K antagonists (VKA), heparin derivatives, direct factor Xa inhibitors, and direct thrombin inhibitors. Direct oral anticoagulants (DOACs) refer to dabigatran (a direct thrombin inhibitor) and the factor Xa inhibitors (apixaban, rivaroxaban, and edoxaban).
Antiplatelets, on the other hand, work by decreasing platelet aggregation and thus preventing thrombus formation; they include P2Y12 receptor inhibitors, protease-activated receptor-1 inhibitors, glycoprotein IIb/IIIa receptor inhibitors, acetylsalicylic acid (ASA), and nonsteroidal anti-inflammatory drugs. All of these agents may directly cause or increase the risk of gastrointestinal (GI) bleeding from luminal sources such as ulcers or diverticula, as well as after endoscopic interventions such as polypectomy. However, there is also a risk of thromboembolic consequences if some of these agents are withheld. Thus, the management of patients receiving antithrombotic agents and undergoing GI endoscopy represents an important clinical challenge and something that every GI physician has to deal with routinely.

The goal of this review is to discuss the optimal strategy for managing antithrombotics in patients undergoing elective endoscopy based on current available evidence and published clinical guidelines.1-4 Much of our discussion will review recommendations from the recently published joint American College of Gastroenterology (ACG) and Canadian Association of Gastroenterology (CAG) guidelines on management of anticoagulants and antiplatelets in the periendoscopic period by Abraham et al.4
Factors that guide decision-making
The two most vital factors to consider prior to performing endoscopic procedures in patients receiving antithrombotic therapy are to assess the risk of bleeding associated with the procedure and to assess the risk of thromboembolism associated with the underlying medical condition for which the antithrombotic agents are being used. In addition, it is also important to keep in mind the individual characteristics of the antithrombotic agent(s) used when making these decisions.
Estimating procedure-related bleeding risk
Various endoscopic procedures have different risks of associated bleeding. Although guidelines from GI societies may differ when classifying procedures into low or high risk, it is important to know that most of the original data on postprocedural bleeding risks are from studies conducted in patients who are not on complex antithrombotic regimens and thus may not accurately reflect the bleeding risk of patients using newer antithrombotic therapies.1,4-7

Traditionally, some of the common low-risk procedures have included diagnostic EGD and colonoscopy with or without biopsy, ERCP without sphincterotomy, biliary stent placement, and push or balloon-assisted enteroscopy. On the other hand, endoscopic procedures associated with interventions are known to have higher bleeding risk, and other procedural factors can influence this risk as well.8 For example, polypectomy, one of the most common interventions during endoscopy, is associated with bleeding risk ranging from 0.3% to 10% depending on multiple factors including polyp size, location, morphology (nonpolypoid, sessile, pedunculated), resection technique (cold or hot forceps, cold or hot snare), and type of cautery used.9 For some procedures, such as routine screening colonoscopy, however, the preprocedure estimate of bleeding risk can be uncertain because it is unclear if a high risk intervention (e.g., polypectomy of large polyp) will be necessary. For example, in the most recent ACG/CAG guidelines, colonoscopy with polypectomy < 1cm is considered a low/moderate risk bleeding procedure, whereas polypectomy > 1cm is considered high risk for bleeding.4 In these situations, the management of antithrombotic medications may depend on the individual patient’s risk of thrombosis and the specific antithrombotic agent. In the example of a patient undergoing colonoscopy while on antithrombotic medications, the bleeding risk associated with polypectomy can potentially be reduced by procedural techniques such as preferential use of cold snare polypectomy. Further high-quality data on the optimal procedural technique to reduce postpolypectomy bleeding in patients on antithrombotic medications is needed to help guide management.
Estimating thromboembolic risk
The risk of thromboembolic events in patients who are withholding their antithrombotic therapy for an endoscopic procedure depends on their underlying condition and individual characteristics. In patients who are on antithrombotic therapy for stroke prevention in non-valvular AF, the risk of cerebral thromboembolism in these patients is predictable using the CHA2DS2Vasc index.10 This scoring index includes heart failure, hypertension, age 75 years or older, diabetes mellitus, prior stroke or transient ischemic attack (TIA), vascular disease, age 65-74 years, and sex categories.
Patients with previous VTE on anticoagulation or those who have mechanical heart valves may have different risk factors for thromboembolic episodes. Among patients with VTE, time from initial VTE, history of recurrent VTE with antithrombotic interruption, and presence of underlying thrombophilia are most predictive of future thromboembolic risk. And for patients with mechanical heart valves, presence of a mitral valve prosthesis, and the presence or absence of associated heart failure and AF determine the annual risk of thromboembolic events. Bioprosthetic valves are generally considered low risk.
In patients with coronary artery disease (CAD), high thrombosis risk scenarios with holding antiplatelets include patients within 3 months of an acute coronary syndrome (ACS) event, within 6 months of a drug-eluting stent (DES) placement, or within 1 month of a bare metal coronary stent (BMS) placement. In addition, patients with ACS that occurred within the past 12 months of DES placement or within 2 months of BMS placement are also considered high risk.11,12 Even beyond these periods, certain patients may still be at high risk of stent occlusion. In particular, patients with a prior history of stent occlusion, ACS or ST elevation myocardial infection, prior multivessel percutaneous coronary intervention, diabetes, renal failure, or diffuse CAD are at higher risk of stent occlusion or ACS events with alteration of antithrombotic therapy.13 Thus, modification of antithrombotic regimens in these patients should be cautiously approached.
Management of antithrombotics prior to elective endoscopy
In patients who need elective endoscopic procedures, if the indication for antithrombotic therapy is short-term, the procedure is probably best delayed until after that period.13 For patients on long-term or lifelong antithrombotic treatment, the decision to temporarily hold the treatment for endoscopy should occur after a discussion with the patient and all of the involved providers. In some high-risk patients, these agents cannot be interrupted; therefore, clinicians must carefully weigh the risks and benefits of the procedure before proceeding with endoscopy. For patients who are known to be undergoing an elective endoscopic procedure, antithrombotic medications may or may not need to be held prior to the procedure depending on the type of therapy. For example, according to the recent ACG/CAG guidelines, warfarin should be continued, whereas DOACs should be temporarily stopped for patients who are undergoing elective/planned endoscopic GI procedures.
Unfractionated heparin (UFH) administered as a continuous intravenous infusion can generally be held 3-4 hours before the procedure, given its short half-life. Low molecular weight heparin (LMWH), including enoxaparin and dalteparin, should be stopped 24 hours prior to the procedure.2,14 Fondaparinux is a synthetic X-a inhibitor that requires discontinuation at least 36 hours preceding a high risk procedure. For patients on warfarin who are undergoing elective endoscopic procedures that are low risk for inducing bleeding, warfarin can be continued, as opposed to temporarily interrupted, although the dose should be omitted the morning of the procedure.4 For those who are undergoing high-risk endoscopic procedures (including colonoscopy with possible polypectomy > 1 cm), 5 days of temporary interruption without periprocedural bridging is appropriate in most patients. This is contrary to previous guidelines, which had recommended bridging for patients with a CHA2DS2Vasc score ≥ 2. Recent impactful randomized trials (BRIDGE and PERIOP-2) have called into question the benefit of periprocedural bridging with LMWH. Avoiding bridging anticoagulation was generally found to be similar to bridging in regard to prevention of thromboembolic complications, but importantly was associated with a decreased risk of major bleeding.15,16 Of note, periprocedural bridging may still be appropriate in a small subset of patients, including those with mechanical valves, AF with CHADS2 score > 5, and previous thromboembolism during temporary interruption of VKAs. The decision to bridge or not should ideally be made in a multidisciplinary fashion.15-20
Data are lacking on the ideal strategy for periendoscopic DOAC management. As mentioned above, for patients on DOACs who are undergoing elective endoscopic GI procedures, temporarily interrupting DOACs rather than continuing them is recommended. Currently, there are no randomized controlled trials addressing the management of DOACs in the periendoscopic period. However, based on five cohort studies, the ideal duration of DOAC interruption before endoscopic procedures may be between 1 and 2 days, excluding the day of the procedure.21-25 This strategy allows for a short preprocedural duration of DOAC interruption and likely provides a balance between bleeding and thromboembolism risk. Importantly, there are no reliable laboratory assays to assess the anticoagulant effect of DOACs, and an individual patient’s degree of renal dysfunction may impact how long the DOAC should be held. In general, the anti-Xa drugs should be held for 1-2 days if the creatinine clearance (CrCl) is ≥ 60 mL/min, for 3 days if the CrCl is between 30 mL/min and 59 mL/min, and for 4 days if the CrCl is less than 30 mL/min.26 For edoxaban, the recommendation is to hold at least 24 hours before high-risk procedures. The recommendation for stopping dabigatran is 2-3 days before a high-risk procedure in patients with CrCl more than 80 mL/min, 3-4 days prior if between 30 and 49 mL/min, and 4-6 days prior if less than 30 mL/min respectively.27
In regard to antiplatelet management, ASA and the P2Y12 receptor inhibitors (e.g. clopidogrel, prasugrel, and ticagrelor) are the most commonly utilized antiplatelets in patients undergoing endoscopic procedures. For patients who are on ASA monotherapy, whether 81 mg or 325 mg daily, for secondary cardiovascular prevention, no interruption of ASA therapy is necessary for elective procedures. The benefit of ASA for secondary cardiovascular prevention and the possible reduction in thrombotic events seen in RCTs of nonendoscopic surgical procedures is well known. However, there may be certain exceptions in which aspirin should be temporarily held. For example, short-term interruption of ASA could be considered in high risk procedures such as biliary or pancreatic sphincterotomy, ampullectomy, and peroral endoscopic myotomy. For patients on single antiplatelet therapy with a P2Y12 receptor inhibitor who are undergoing elective endoscopic GI procedures, the recent CAG/ACG guidelines did not provide a clear recommendation for or against temporary interruption of the P2Y12 receptor inhibitor. Although interruption of a P2Y12 receptor inhibitor should theoretically decrease a patient’s risk of bleeding, the available evidence reported a nonsignificant increased bleeding risk in patients who stop a P2Y12 receptor inhibitor for an elective endoscopic procedure compared with those who continue the medication.28,29 Therefore, until further data are available, for patients on P2Y12 receptor monotherapy, a reasonable strategy would be to temporarily hold therapy prior to high risk endoscopic procedures, assuming the patients are not at high cardiovascular risk. Clopidogrel and prasugrel have to be stopped 5-7 days prior to allow normal platelet aggregation to resume as opposed to ticagrelor, a reversible P2Y12 receptor inhibitor that can be stopped 3-5 days prior.30
Lastly, for patients who are on dual antiplatelet therapy (DAPT) for secondary prevention, continuation of ASA and temporary interruption of the P2Y12 receptor inhibitor is recommended while undergoing elective endoscopy. Studies have shown that those who discontinued both had a much higher incidence of stent thrombosis compared with those who remained on aspirin alone.4,28,31
Resumption of antithrombotic therapy after endoscopy
In general, antithrombotic therapy should be resumed upon completion of the procedure unless there remains a persistent risk of major bleeding.1,14 This consensus is based on studies available on warfarin and heparin products, with minimal literature available regarding the resumption of DOACs. The benefits of immediate re-initiation of antithrombotic therapy for the prevention of thromboembolic events should be weighed against the risk of hemorrhage associated with the specific agent, the time to onset of the medication, and procedure-specific circumstances. For the small subset of patients on warfarin with a high risk of thromboembolism (e.g., mechanical heart valve), bridging with LMWH should be started at the earliest possible time when there is no risk of major bleeding and continued until the international normalized ratio (INR) reaches a therapeutic level with warfarin. For patients at a lower risk of thromboembolism, warfarin should be restarted within 24 hours of the procedure. In addition, because of the shorter duration of DOACs, if treatment with these agents cannot resume within 24 hours of a high-risk procedure, bridge therapy should be considered with UFH or LMWH in patients with a high risk of thrombosis.18 In patients receiving DOACs for stroke prophylaxis in AF, the DOACS can be safely resumed 1 day after low-risk procedures and 2-3 days after high-risk procedures without the need for bridging.25 All antiplatelet agents should be resumed as soon as hemostasis is achieved.
Conclusion
Antithrombotic therapy is increasingly used given the aging population, widespread burden of cardiovascular comorbidities, and expanding indications for classes of medications such as direct oral anticoagulants. Given the association with antithrombotic medications and gastrointestinal bleeding, it is essential for gastroenterologists to understand the importance, necessity, and timing when holding these medications for endoscopic procedures. Even with the practice guidelines available today to help clinicians navigate certain situations, each patient’s antithrombotic management may be different, and communication with the prescribing physicians and including patients in the decision-making process is essential before planned procedures.
Dr. Wang is a gastroenterology fellow at the University of Chicago. Dr. Sengupta is an associate professor at the University of Chicago. They reported no funding or conflicts of interest.
References
1. ASGE Standards of Practice Committee, Acosta RD et al. The management of antithrombotic agents for patients undergoing GI endoscopy. Gastrointest Endosc. 2016;83(1):3-16.
2. Veitch AM et al. Endoscopy in patients on antiplatelet or anticoagulant therapy, including direct oral anticoagulants: British Society of Gastroenterology (BSG) and European Society of Gastrointestinal Endoscopy (ESGE) guidelines. Endoscopy. 2016;48(4):c1. doi: 10.1055/s-0042-122686.
3. Chan FKL et al. Management of patients on antithrombotic agents undergoing emergency and elective endoscopy: Joint Asian Pacific Association of Gastroenterology (APAGE) and Asian Pacific Society for Digestive Endoscopy (APSDE) practice guidelines. Gut. 2018;67(3):405-17.
4. Abraham NS et al. American College of Gastroenterology – Canadian Association of Gastroenterology clinical practice guideline: Management of anticoagulants and antiplatelets during acute gastrointestinal bleeding and the periendoscopic period. Am J Gastroenterol. 2022;117(4):542-58.
5. Boustière C et al. Endoscopy and antiplatelet agents. European Society of Gastrointestinal Endoscopy (ESGE) guideline. Endoscopy. 2011;43(5):445-61.
6. Fujimoto K et al. Guidelines for gastroenterological endoscopy in patients undergoing antithrombotic treatment. Dig Endosc. 2014;26(1):1-14.
7. Wilke T et al. Patient preferences for oral anticoagulation therapy in atrial fibrillation: A systematic literature review. Patient 2017;10(1):17-37.
8. Gerson LB et al. Adverse events associated with anticoagulation therapy in the periendoscopic period. Gastrointest Endosc. 2010 Jun;71(7):1211-17.e2.
9. Horiuchi A et al. Removal of small colorectal polyps in anticoagulated patients: A prospective randomized comparison of cold snare and conventional polypectomy. Gastrointest Endosc 2014;79(3):417-23.
10. Lip GYH et al. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: The euro heart survey on atrial fibrillation. Chest. 2010;137(2):263-72.
11. 2012 Writing Committee Members, Jneid H et al. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/non-ST-elevation myocardial infarction (Updating the 2007 guideline and replacing the 2011 focused update): A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2012;126(7):875-910.
12. Douketis JD et al. Perioperative management of antithrombotic therapy: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012 Feb;141(2 Suppl):e326S-e350S.
13. Becker RC et al. Management of platelet-directed pharmacotherapy in patients with atherosclerotic coronary artery disease undergoing elective endoscopic gastrointestinal procedures. J Am Coll Cardiol. 2009;54(24):2261-76.
14. Kwok A and Faigel DO. Management of anticoagulation before and after gastrointestinal endoscopy. Am J Gastroenterol. 2009;104(12):3085-97; quiz 3098.
15. Douketis JD et al. Perioperative bridging anticoagulation in patients with atrial fibrillation. N Engl J Med. 2015;373(9):823-33.
16. Kovacs MJ et al. Postoperative low molecular weight heparin bridging treatment for patients at high risk of arterial thromboembolism (PERIOP2): Double blind randomised controlled trial. BMJ 2021;373:n1205.
17. Tafur A and Douketis J. Perioperative management of anticoagulant and antiplatelet therapy. Heart 2018;104(17):1461-7.
18. Kato M et al. Guidelines for gastroenterological endoscopy in patients undergoing antithrombotic treatment: 2017 appendix on anticoagulants including direct oral anticoagulants. Dig Endosc. 2018;30(4):433-40.
19. Inoue T et al. Clinical features of postpolypectomy bleeding associated with heparin bridge therapy. Dig Endosc. 2014;26(2):243-9.
20. Takeuchi Y et al. Continuous anticoagulation and cold snare polypectomy versus heparin bridging and hot snare polypectomy in patients on anticoagulants with subcentimeter polyps: A randomized controlled trial. Ann Intern Med. 2019;171(4):229-37.
21. Ara N et al. Prospective analysis of risk for bleeding after endoscopic biopsy without cessation of antithrombotics in Japan. Dig Endosc. 2015;27(4):458-64.
22. Yanagisawa N et al. Postpolypectomy bleeding and thromboembolism risks associated with warfarin vs. direct oral anticoagulants. World J Gastroenterol. 2018;24(14):1540-9.
23. Arimoto J et al. Safety of cold snare polypectomy in patients receiving treatment with antithrombotic agents. Dig Dis Sci. 2019;64(11):3247-55.
24. Heublein V et al. Gastrointestinal endoscopy in patients receiving novel direct oral anticoagulants: Results from the prospective Dresden NOAC registry. J Gastroenterol. 2018;53(2):236-46.
25. Douketis JD et al. Perioperative management of patients with atrial fibrillation receiving a direct oral anticoagulant. JAMA Intern Med. 2019;179(11):1469-78.
26. Dubois V et al. Perioperative management of patients on direct oral anticoagulants. Thromb J. 2017;15:14.
27. Weitz JI et al. Periprocedural management and approach to bleeding in patients taking dabigatran. Circulation. 2012 Nov 13;126(20):2428-32.
28. Chan FKL et al. Risk of postpolypectomy bleeding with uninterrupted clopidogrel therapy in an industry-independent, double-blind, randomized trial. Gastroenterology. 2019;156(4):918-25.
29. Watanabe K et al. Effect of antiplatelet agent number, types, and pre-endoscopic management on postpolypectomy bleeding: Validation of endoscopy guidelines. Surg Endosc. 2021;35(1):317-25.
30. Gurbel PA et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: The ONSET/OFFSET study. Circulation. 2009;120(25):2577-85.
31. Eisenberg MJ et al. Safety of short-term discontinuation of antiplatelet therapy in patients with drug-eluting stents. Circulation. 2009;119(12):1634-42.
Antithrombotic therapy is increasingly used to either reduce the risk of or treat thromboembolic episodes in patients with various medical conditions such as ischemic and valvular heart disease, atrial fibrillation (AF), cerebrovascular disease, peripheral arterial disease, venous thromboembolism (VTE) and hypercoagulable diseases. Antithrombotics include medications classified as anticoagulants or antiplatelets. Anticoagulants work by interfering with the native clotting cascade and consist of four main classes: vitamin K antagonists (VKA), heparin derivatives, direct factor Xa inhibitors, and direct thrombin inhibitors. Direct oral anticoagulants (DOACs) refer to dabigatran (a direct thrombin inhibitor) and the factor Xa inhibitors (apixaban, rivaroxaban, and edoxaban).
Antiplatelets, on the other hand, work by decreasing platelet aggregation and thus preventing thrombus formation; they include P2Y12 receptor inhibitors, protease-activated receptor-1 inhibitors, glycoprotein IIb/IIIa receptor inhibitors, acetylsalicylic acid (ASA), and nonsteroidal anti-inflammatory drugs. All of these agents may directly cause or increase the risk of gastrointestinal (GI) bleeding from luminal sources such as ulcers or diverticula, as well as after endoscopic interventions such as polypectomy. However, there is also a risk of thromboembolic consequences if some of these agents are withheld. Thus, the management of patients receiving antithrombotic agents and undergoing GI endoscopy represents an important clinical challenge and something that every GI physician has to deal with routinely.
The goal of this review is to discuss the optimal strategy for managing antithrombotics in patients undergoing elective endoscopy based on current available evidence and published clinical guidelines.1-4 Much of our discussion will review recommendations from the recently published joint American College of Gastroenterology (ACG) and Canadian Association of Gastroenterology (CAG) guidelines on management of anticoagulants and antiplatelets in the periendoscopic period by Abraham et al.4
Factors that guide decision-making
The two most vital factors to consider prior to performing endoscopic procedures in patients receiving antithrombotic therapy are to assess the risk of bleeding associated with the procedure and to assess the risk of thromboembolism associated with the underlying medical condition for which the antithrombotic agents are being used. In addition, it is also important to keep in mind the individual characteristics of the antithrombotic agent(s) used when making these decisions.
Estimating procedure-related bleeding risk
Various endoscopic procedures have different risks of associated bleeding. Although guidelines from GI societies may differ when classifying procedures into low or high risk, it is important to know that most of the original data on postprocedural bleeding risks are from studies conducted in patients who are not on complex antithrombotic regimens and thus may not accurately reflect the bleeding risk of patients using newer antithrombotic therapies.1,4-7
Traditionally, some of the common low-risk procedures have included diagnostic EGD and colonoscopy with or without biopsy, ERCP without sphincterotomy, biliary stent placement, and push or balloon-assisted enteroscopy. On the other hand, endoscopic procedures associated with interventions are known to have higher bleeding risk, and other procedural factors can influence this risk as well.8 For example, polypectomy, one of the most common interventions during endoscopy, is associated with bleeding risk ranging from 0.3% to 10% depending on multiple factors including polyp size, location, morphology (nonpolypoid, sessile, pedunculated), resection technique (cold or hot forceps, cold or hot snare), and type of cautery used.9 For some procedures, such as routine screening colonoscopy, however, the preprocedure estimate of bleeding risk can be uncertain because it is unclear if a high risk intervention (e.g., polypectomy of large polyp) will be necessary. For example, in the most recent ACG/CAG guidelines, colonoscopy with polypectomy < 1cm is considered a low/moderate risk bleeding procedure, whereas polypectomy > 1cm is considered high risk for bleeding.4 In these situations, the management of antithrombotic medications may depend on the individual patient’s risk of thrombosis and the specific antithrombotic agent. In the example of a patient undergoing colonoscopy while on antithrombotic medications, the bleeding risk associated with polypectomy can potentially be reduced by procedural techniques such as preferential use of cold snare polypectomy. Further high-quality data on the optimal procedural technique to reduce postpolypectomy bleeding in patients on antithrombotic medications is needed to help guide management.
Estimating thromboembolic risk
The risk of thromboembolic events in patients who are withholding their antithrombotic therapy for an endoscopic procedure depends on their underlying condition and individual characteristics. In patients who are on antithrombotic therapy for stroke prevention in non-valvular AF, the risk of cerebral thromboembolism in these patients is predictable using the CHA2DS2Vasc index.10 This scoring index includes heart failure, hypertension, age 75 years or older, diabetes mellitus, prior stroke or transient ischemic attack (TIA), vascular disease, age 65-74 years, and sex categories.
Patients with previous VTE on anticoagulation or those who have mechanical heart valves may have different risk factors for thromboembolic episodes. Among patients with VTE, time from initial VTE, history of recurrent VTE with antithrombotic interruption, and presence of underlying thrombophilia are most predictive of future thromboembolic risk. And for patients with mechanical heart valves, presence of a mitral valve prosthesis, and the presence or absence of associated heart failure and AF determine the annual risk of thromboembolic events. Bioprosthetic valves are generally considered low risk.
In patients with coronary artery disease (CAD), high thrombosis risk scenarios with holding antiplatelets include patients within 3 months of an acute coronary syndrome (ACS) event, within 6 months of a drug-eluting stent (DES) placement, or within 1 month of a bare metal coronary stent (BMS) placement. In addition, patients with ACS that occurred within the past 12 months of DES placement or within 2 months of BMS placement are also considered high risk.11,12 Even beyond these periods, certain patients may still be at high risk of stent occlusion. In particular, patients with a prior history of stent occlusion, ACS or ST elevation myocardial infection, prior multivessel percutaneous coronary intervention, diabetes, renal failure, or diffuse CAD are at higher risk of stent occlusion or ACS events with alteration of antithrombotic therapy.13 Thus, modification of antithrombotic regimens in these patients should be cautiously approached.
Management of antithrombotics prior to elective endoscopy
In patients who need elective endoscopic procedures, if the indication for antithrombotic therapy is short-term, the procedure is probably best delayed until after that period.13 For patients on long-term or lifelong antithrombotic treatment, the decision to temporarily hold the treatment for endoscopy should occur after a discussion with the patient and all of the involved providers. In some high-risk patients, these agents cannot be interrupted; therefore, clinicians must carefully weigh the risks and benefits of the procedure before proceeding with endoscopy. For patients who are known to be undergoing an elective endoscopic procedure, antithrombotic medications may or may not need to be held prior to the procedure depending on the type of therapy. For example, according to the recent ACG/CAG guidelines, warfarin should be continued, whereas DOACs should be temporarily stopped for patients who are undergoing elective/planned endoscopic GI procedures.
Unfractionated heparin (UFH) administered as a continuous intravenous infusion can generally be held 3-4 hours before the procedure, given its short half-life. Low molecular weight heparin (LMWH), including enoxaparin and dalteparin, should be stopped 24 hours prior to the procedure.2,14 Fondaparinux is a synthetic X-a inhibitor that requires discontinuation at least 36 hours preceding a high risk procedure. For patients on warfarin who are undergoing elective endoscopic procedures that are low risk for inducing bleeding, warfarin can be continued, as opposed to temporarily interrupted, although the dose should be omitted the morning of the procedure.4 For those who are undergoing high-risk endoscopic procedures (including colonoscopy with possible polypectomy > 1 cm), 5 days of temporary interruption without periprocedural bridging is appropriate in most patients. This is contrary to previous guidelines, which had recommended bridging for patients with a CHA2DS2Vasc score ≥ 2. Recent impactful randomized trials (BRIDGE and PERIOP-2) have called into question the benefit of periprocedural bridging with LMWH. Avoiding bridging anticoagulation was generally found to be similar to bridging in regard to prevention of thromboembolic complications, but importantly was associated with a decreased risk of major bleeding.15,16 Of note, periprocedural bridging may still be appropriate in a small subset of patients, including those with mechanical valves, AF with CHADS2 score > 5, and previous thromboembolism during temporary interruption of VKAs. The decision to bridge or not should ideally be made in a multidisciplinary fashion.15-20
Data are lacking on the ideal strategy for periendoscopic DOAC management. As mentioned above, for patients on DOACs who are undergoing elective endoscopic GI procedures, temporarily interrupting DOACs rather than continuing them is recommended. Currently, there are no randomized controlled trials addressing the management of DOACs in the periendoscopic period. However, based on five cohort studies, the ideal duration of DOAC interruption before endoscopic procedures may be between 1 and 2 days, excluding the day of the procedure.21-25 This strategy allows for a short preprocedural duration of DOAC interruption and likely provides a balance between bleeding and thromboembolism risk. Importantly, there are no reliable laboratory assays to assess the anticoagulant effect of DOACs, and an individual patient’s degree of renal dysfunction may impact how long the DOAC should be held. In general, the anti-Xa drugs should be held for 1-2 days if the creatinine clearance (CrCl) is ≥ 60 mL/min, for 3 days if the CrCl is between 30 mL/min and 59 mL/min, and for 4 days if the CrCl is less than 30 mL/min.26 For edoxaban, the recommendation is to hold at least 24 hours before high-risk procedures. The recommendation for stopping dabigatran is 2-3 days before a high-risk procedure in patients with CrCl more than 80 mL/min, 3-4 days prior if between 30 and 49 mL/min, and 4-6 days prior if less than 30 mL/min respectively.27
In regard to antiplatelet management, ASA and the P2Y12 receptor inhibitors (e.g. clopidogrel, prasugrel, and ticagrelor) are the most commonly utilized antiplatelets in patients undergoing endoscopic procedures. For patients who are on ASA monotherapy, whether 81 mg or 325 mg daily, for secondary cardiovascular prevention, no interruption of ASA therapy is necessary for elective procedures. The benefit of ASA for secondary cardiovascular prevention and the possible reduction in thrombotic events seen in RCTs of nonendoscopic surgical procedures is well known. However, there may be certain exceptions in which aspirin should be temporarily held. For example, short-term interruption of ASA could be considered in high risk procedures such as biliary or pancreatic sphincterotomy, ampullectomy, and peroral endoscopic myotomy. For patients on single antiplatelet therapy with a P2Y12 receptor inhibitor who are undergoing elective endoscopic GI procedures, the recent CAG/ACG guidelines did not provide a clear recommendation for or against temporary interruption of the P2Y12 receptor inhibitor. Although interruption of a P2Y12 receptor inhibitor should theoretically decrease a patient’s risk of bleeding, the available evidence reported a nonsignificant increased bleeding risk in patients who stop a P2Y12 receptor inhibitor for an elective endoscopic procedure compared with those who continue the medication.28,29 Therefore, until further data are available, for patients on P2Y12 receptor monotherapy, a reasonable strategy would be to temporarily hold therapy prior to high risk endoscopic procedures, assuming the patients are not at high cardiovascular risk. Clopidogrel and prasugrel have to be stopped 5-7 days prior to allow normal platelet aggregation to resume as opposed to ticagrelor, a reversible P2Y12 receptor inhibitor that can be stopped 3-5 days prior.30
Lastly, for patients who are on dual antiplatelet therapy (DAPT) for secondary prevention, continuation of ASA and temporary interruption of the P2Y12 receptor inhibitor is recommended while undergoing elective endoscopy. Studies have shown that those who discontinued both had a much higher incidence of stent thrombosis compared with those who remained on aspirin alone.4,28,31
Resumption of antithrombotic therapy after endoscopy
In general, antithrombotic therapy should be resumed upon completion of the procedure unless there remains a persistent risk of major bleeding.1,14 This consensus is based on studies available on warfarin and heparin products, with minimal literature available regarding the resumption of DOACs. The benefits of immediate re-initiation of antithrombotic therapy for the prevention of thromboembolic events should be weighed against the risk of hemorrhage associated with the specific agent, the time to onset of the medication, and procedure-specific circumstances. For the small subset of patients on warfarin with a high risk of thromboembolism (e.g., mechanical heart valve), bridging with LMWH should be started at the earliest possible time when there is no risk of major bleeding and continued until the international normalized ratio (INR) reaches a therapeutic level with warfarin. For patients at a lower risk of thromboembolism, warfarin should be restarted within 24 hours of the procedure. In addition, because of the shorter duration of DOACs, if treatment with these agents cannot resume within 24 hours of a high-risk procedure, bridge therapy should be considered with UFH or LMWH in patients with a high risk of thrombosis.18 In patients receiving DOACs for stroke prophylaxis in AF, the DOACS can be safely resumed 1 day after low-risk procedures and 2-3 days after high-risk procedures without the need for bridging.25 All antiplatelet agents should be resumed as soon as hemostasis is achieved.
Conclusion
Antithrombotic therapy is increasingly used given the aging population, widespread burden of cardiovascular comorbidities, and expanding indications for classes of medications such as direct oral anticoagulants. Given the association with antithrombotic medications and gastrointestinal bleeding, it is essential for gastroenterologists to understand the importance, necessity, and timing when holding these medications for endoscopic procedures. Even with the practice guidelines available today to help clinicians navigate certain situations, each patient’s antithrombotic management may be different, and communication with the prescribing physicians and including patients in the decision-making process is essential before planned procedures.
Dr. Wang is a gastroenterology fellow at the University of Chicago. Dr. Sengupta is an associate professor at the University of Chicago. They reported no funding or conflicts of interest.
References
1. ASGE Standards of Practice Committee, Acosta RD et al. The management of antithrombotic agents for patients undergoing GI endoscopy. Gastrointest Endosc. 2016;83(1):3-16.
2. Veitch AM et al. Endoscopy in patients on antiplatelet or anticoagulant therapy, including direct oral anticoagulants: British Society of Gastroenterology (BSG) and European Society of Gastrointestinal Endoscopy (ESGE) guidelines. Endoscopy. 2016;48(4):c1. doi: 10.1055/s-0042-122686.
3. Chan FKL et al. Management of patients on antithrombotic agents undergoing emergency and elective endoscopy: Joint Asian Pacific Association of Gastroenterology (APAGE) and Asian Pacific Society for Digestive Endoscopy (APSDE) practice guidelines. Gut. 2018;67(3):405-17.
4. Abraham NS et al. American College of Gastroenterology – Canadian Association of Gastroenterology clinical practice guideline: Management of anticoagulants and antiplatelets during acute gastrointestinal bleeding and the periendoscopic period. Am J Gastroenterol. 2022;117(4):542-58.
5. Boustière C et al. Endoscopy and antiplatelet agents. European Society of Gastrointestinal Endoscopy (ESGE) guideline. Endoscopy. 2011;43(5):445-61.
6. Fujimoto K et al. Guidelines for gastroenterological endoscopy in patients undergoing antithrombotic treatment. Dig Endosc. 2014;26(1):1-14.
7. Wilke T et al. Patient preferences for oral anticoagulation therapy in atrial fibrillation: A systematic literature review. Patient 2017;10(1):17-37.
8. Gerson LB et al. Adverse events associated with anticoagulation therapy in the periendoscopic period. Gastrointest Endosc. 2010 Jun;71(7):1211-17.e2.
9. Horiuchi A et al. Removal of small colorectal polyps in anticoagulated patients: A prospective randomized comparison of cold snare and conventional polypectomy. Gastrointest Endosc 2014;79(3):417-23.
10. Lip GYH et al. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: The euro heart survey on atrial fibrillation. Chest. 2010;137(2):263-72.
11. 2012 Writing Committee Members, Jneid H et al. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/non-ST-elevation myocardial infarction (Updating the 2007 guideline and replacing the 2011 focused update): A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2012;126(7):875-910.
12. Douketis JD et al. Perioperative management of antithrombotic therapy: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012 Feb;141(2 Suppl):e326S-e350S.
13. Becker RC et al. Management of platelet-directed pharmacotherapy in patients with atherosclerotic coronary artery disease undergoing elective endoscopic gastrointestinal procedures. J Am Coll Cardiol. 2009;54(24):2261-76.
14. Kwok A and Faigel DO. Management of anticoagulation before and after gastrointestinal endoscopy. Am J Gastroenterol. 2009;104(12):3085-97; quiz 3098.
15. Douketis JD et al. Perioperative bridging anticoagulation in patients with atrial fibrillation. N Engl J Med. 2015;373(9):823-33.
16. Kovacs MJ et al. Postoperative low molecular weight heparin bridging treatment for patients at high risk of arterial thromboembolism (PERIOP2): Double blind randomised controlled trial. BMJ 2021;373:n1205.
17. Tafur A and Douketis J. Perioperative management of anticoagulant and antiplatelet therapy. Heart 2018;104(17):1461-7.
18. Kato M et al. Guidelines for gastroenterological endoscopy in patients undergoing antithrombotic treatment: 2017 appendix on anticoagulants including direct oral anticoagulants. Dig Endosc. 2018;30(4):433-40.
19. Inoue T et al. Clinical features of postpolypectomy bleeding associated with heparin bridge therapy. Dig Endosc. 2014;26(2):243-9.
20. Takeuchi Y et al. Continuous anticoagulation and cold snare polypectomy versus heparin bridging and hot snare polypectomy in patients on anticoagulants with subcentimeter polyps: A randomized controlled trial. Ann Intern Med. 2019;171(4):229-37.
21. Ara N et al. Prospective analysis of risk for bleeding after endoscopic biopsy without cessation of antithrombotics in Japan. Dig Endosc. 2015;27(4):458-64.
22. Yanagisawa N et al. Postpolypectomy bleeding and thromboembolism risks associated with warfarin vs. direct oral anticoagulants. World J Gastroenterol. 2018;24(14):1540-9.
23. Arimoto J et al. Safety of cold snare polypectomy in patients receiving treatment with antithrombotic agents. Dig Dis Sci. 2019;64(11):3247-55.
24. Heublein V et al. Gastrointestinal endoscopy in patients receiving novel direct oral anticoagulants: Results from the prospective Dresden NOAC registry. J Gastroenterol. 2018;53(2):236-46.
25. Douketis JD et al. Perioperative management of patients with atrial fibrillation receiving a direct oral anticoagulant. JAMA Intern Med. 2019;179(11):1469-78.
26. Dubois V et al. Perioperative management of patients on direct oral anticoagulants. Thromb J. 2017;15:14.
27. Weitz JI et al. Periprocedural management and approach to bleeding in patients taking dabigatran. Circulation. 2012 Nov 13;126(20):2428-32.
28. Chan FKL et al. Risk of postpolypectomy bleeding with uninterrupted clopidogrel therapy in an industry-independent, double-blind, randomized trial. Gastroenterology. 2019;156(4):918-25.
29. Watanabe K et al. Effect of antiplatelet agent number, types, and pre-endoscopic management on postpolypectomy bleeding: Validation of endoscopy guidelines. Surg Endosc. 2021;35(1):317-25.
30. Gurbel PA et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: The ONSET/OFFSET study. Circulation. 2009;120(25):2577-85.
31. Eisenberg MJ et al. Safety of short-term discontinuation of antiplatelet therapy in patients with drug-eluting stents. Circulation. 2009;119(12):1634-42.
Antithrombotic therapy is increasingly used to either reduce the risk of or treat thromboembolic episodes in patients with various medical conditions such as ischemic and valvular heart disease, atrial fibrillation (AF), cerebrovascular disease, peripheral arterial disease, venous thromboembolism (VTE) and hypercoagulable diseases. Antithrombotics include medications classified as anticoagulants or antiplatelets. Anticoagulants work by interfering with the native clotting cascade and consist of four main classes: vitamin K antagonists (VKA), heparin derivatives, direct factor Xa inhibitors, and direct thrombin inhibitors. Direct oral anticoagulants (DOACs) refer to dabigatran (a direct thrombin inhibitor) and the factor Xa inhibitors (apixaban, rivaroxaban, and edoxaban).
Antiplatelets, on the other hand, work by decreasing platelet aggregation and thus preventing thrombus formation; they include P2Y12 receptor inhibitors, protease-activated receptor-1 inhibitors, glycoprotein IIb/IIIa receptor inhibitors, acetylsalicylic acid (ASA), and nonsteroidal anti-inflammatory drugs. All of these agents may directly cause or increase the risk of gastrointestinal (GI) bleeding from luminal sources such as ulcers or diverticula, as well as after endoscopic interventions such as polypectomy. However, there is also a risk of thromboembolic consequences if some of these agents are withheld. Thus, the management of patients receiving antithrombotic agents and undergoing GI endoscopy represents an important clinical challenge and something that every GI physician has to deal with routinely.
The goal of this review is to discuss the optimal strategy for managing antithrombotics in patients undergoing elective endoscopy based on current available evidence and published clinical guidelines.1-4 Much of our discussion will review recommendations from the recently published joint American College of Gastroenterology (ACG) and Canadian Association of Gastroenterology (CAG) guidelines on management of anticoagulants and antiplatelets in the periendoscopic period by Abraham et al.4
Factors that guide decision-making
The two most vital factors to consider prior to performing endoscopic procedures in patients receiving antithrombotic therapy are to assess the risk of bleeding associated with the procedure and to assess the risk of thromboembolism associated with the underlying medical condition for which the antithrombotic agents are being used. In addition, it is also important to keep in mind the individual characteristics of the antithrombotic agent(s) used when making these decisions.
Estimating procedure-related bleeding risk
Various endoscopic procedures have different risks of associated bleeding. Although guidelines from GI societies may differ when classifying procedures into low or high risk, it is important to know that most of the original data on postprocedural bleeding risks are from studies conducted in patients who are not on complex antithrombotic regimens and thus may not accurately reflect the bleeding risk of patients using newer antithrombotic therapies.1,4-7
Traditionally, some of the common low-risk procedures have included diagnostic EGD and colonoscopy with or without biopsy, ERCP without sphincterotomy, biliary stent placement, and push or balloon-assisted enteroscopy. On the other hand, endoscopic procedures associated with interventions are known to have higher bleeding risk, and other procedural factors can influence this risk as well.8 For example, polypectomy, one of the most common interventions during endoscopy, is associated with bleeding risk ranging from 0.3% to 10% depending on multiple factors including polyp size, location, morphology (nonpolypoid, sessile, pedunculated), resection technique (cold or hot forceps, cold or hot snare), and type of cautery used.9 For some procedures, such as routine screening colonoscopy, however, the preprocedure estimate of bleeding risk can be uncertain because it is unclear if a high risk intervention (e.g., polypectomy of large polyp) will be necessary. For example, in the most recent ACG/CAG guidelines, colonoscopy with polypectomy < 1cm is considered a low/moderate risk bleeding procedure, whereas polypectomy > 1cm is considered high risk for bleeding.4 In these situations, the management of antithrombotic medications may depend on the individual patient’s risk of thrombosis and the specific antithrombotic agent. In the example of a patient undergoing colonoscopy while on antithrombotic medications, the bleeding risk associated with polypectomy can potentially be reduced by procedural techniques such as preferential use of cold snare polypectomy. Further high-quality data on the optimal procedural technique to reduce postpolypectomy bleeding in patients on antithrombotic medications is needed to help guide management.
Estimating thromboembolic risk
The risk of thromboembolic events in patients who are withholding their antithrombotic therapy for an endoscopic procedure depends on their underlying condition and individual characteristics. In patients who are on antithrombotic therapy for stroke prevention in non-valvular AF, the risk of cerebral thromboembolism in these patients is predictable using the CHA2DS2Vasc index.10 This scoring index includes heart failure, hypertension, age 75 years or older, diabetes mellitus, prior stroke or transient ischemic attack (TIA), vascular disease, age 65-74 years, and sex categories.
Patients with previous VTE on anticoagulation or those who have mechanical heart valves may have different risk factors for thromboembolic episodes. Among patients with VTE, time from initial VTE, history of recurrent VTE with antithrombotic interruption, and presence of underlying thrombophilia are most predictive of future thromboembolic risk. And for patients with mechanical heart valves, presence of a mitral valve prosthesis, and the presence or absence of associated heart failure and AF determine the annual risk of thromboembolic events. Bioprosthetic valves are generally considered low risk.
In patients with coronary artery disease (CAD), high thrombosis risk scenarios with holding antiplatelets include patients within 3 months of an acute coronary syndrome (ACS) event, within 6 months of a drug-eluting stent (DES) placement, or within 1 month of a bare metal coronary stent (BMS) placement. In addition, patients with ACS that occurred within the past 12 months of DES placement or within 2 months of BMS placement are also considered high risk.11,12 Even beyond these periods, certain patients may still be at high risk of stent occlusion. In particular, patients with a prior history of stent occlusion, ACS or ST elevation myocardial infection, prior multivessel percutaneous coronary intervention, diabetes, renal failure, or diffuse CAD are at higher risk of stent occlusion or ACS events with alteration of antithrombotic therapy.13 Thus, modification of antithrombotic regimens in these patients should be cautiously approached.
Management of antithrombotics prior to elective endoscopy
In patients who need elective endoscopic procedures, if the indication for antithrombotic therapy is short-term, the procedure is probably best delayed until after that period.13 For patients on long-term or lifelong antithrombotic treatment, the decision to temporarily hold the treatment for endoscopy should occur after a discussion with the patient and all of the involved providers. In some high-risk patients, these agents cannot be interrupted; therefore, clinicians must carefully weigh the risks and benefits of the procedure before proceeding with endoscopy. For patients who are known to be undergoing an elective endoscopic procedure, antithrombotic medications may or may not need to be held prior to the procedure depending on the type of therapy. For example, according to the recent ACG/CAG guidelines, warfarin should be continued, whereas DOACs should be temporarily stopped for patients who are undergoing elective/planned endoscopic GI procedures.
Unfractionated heparin (UFH) administered as a continuous intravenous infusion can generally be held 3-4 hours before the procedure, given its short half-life. Low molecular weight heparin (LMWH), including enoxaparin and dalteparin, should be stopped 24 hours prior to the procedure.2,14 Fondaparinux is a synthetic X-a inhibitor that requires discontinuation at least 36 hours preceding a high risk procedure. For patients on warfarin who are undergoing elective endoscopic procedures that are low risk for inducing bleeding, warfarin can be continued, as opposed to temporarily interrupted, although the dose should be omitted the morning of the procedure.4 For those who are undergoing high-risk endoscopic procedures (including colonoscopy with possible polypectomy > 1 cm), 5 days of temporary interruption without periprocedural bridging is appropriate in most patients. This is contrary to previous guidelines, which had recommended bridging for patients with a CHA2DS2Vasc score ≥ 2. Recent impactful randomized trials (BRIDGE and PERIOP-2) have called into question the benefit of periprocedural bridging with LMWH. Avoiding bridging anticoagulation was generally found to be similar to bridging in regard to prevention of thromboembolic complications, but importantly was associated with a decreased risk of major bleeding.15,16 Of note, periprocedural bridging may still be appropriate in a small subset of patients, including those with mechanical valves, AF with CHADS2 score > 5, and previous thromboembolism during temporary interruption of VKAs. The decision to bridge or not should ideally be made in a multidisciplinary fashion.15-20
Data are lacking on the ideal strategy for periendoscopic DOAC management. As mentioned above, for patients on DOACs who are undergoing elective endoscopic GI procedures, temporarily interrupting DOACs rather than continuing them is recommended. Currently, there are no randomized controlled trials addressing the management of DOACs in the periendoscopic period. However, based on five cohort studies, the ideal duration of DOAC interruption before endoscopic procedures may be between 1 and 2 days, excluding the day of the procedure.21-25 This strategy allows for a short preprocedural duration of DOAC interruption and likely provides a balance between bleeding and thromboembolism risk. Importantly, there are no reliable laboratory assays to assess the anticoagulant effect of DOACs, and an individual patient’s degree of renal dysfunction may impact how long the DOAC should be held. In general, the anti-Xa drugs should be held for 1-2 days if the creatinine clearance (CrCl) is ≥ 60 mL/min, for 3 days if the CrCl is between 30 mL/min and 59 mL/min, and for 4 days if the CrCl is less than 30 mL/min.26 For edoxaban, the recommendation is to hold at least 24 hours before high-risk procedures. The recommendation for stopping dabigatran is 2-3 days before a high-risk procedure in patients with CrCl more than 80 mL/min, 3-4 days prior if between 30 and 49 mL/min, and 4-6 days prior if less than 30 mL/min respectively.27
In regard to antiplatelet management, ASA and the P2Y12 receptor inhibitors (e.g. clopidogrel, prasugrel, and ticagrelor) are the most commonly utilized antiplatelets in patients undergoing endoscopic procedures. For patients who are on ASA monotherapy, whether 81 mg or 325 mg daily, for secondary cardiovascular prevention, no interruption of ASA therapy is necessary for elective procedures. The benefit of ASA for secondary cardiovascular prevention and the possible reduction in thrombotic events seen in RCTs of nonendoscopic surgical procedures is well known. However, there may be certain exceptions in which aspirin should be temporarily held. For example, short-term interruption of ASA could be considered in high risk procedures such as biliary or pancreatic sphincterotomy, ampullectomy, and peroral endoscopic myotomy. For patients on single antiplatelet therapy with a P2Y12 receptor inhibitor who are undergoing elective endoscopic GI procedures, the recent CAG/ACG guidelines did not provide a clear recommendation for or against temporary interruption of the P2Y12 receptor inhibitor. Although interruption of a P2Y12 receptor inhibitor should theoretically decrease a patient’s risk of bleeding, the available evidence reported a nonsignificant increased bleeding risk in patients who stop a P2Y12 receptor inhibitor for an elective endoscopic procedure compared with those who continue the medication.28,29 Therefore, until further data are available, for patients on P2Y12 receptor monotherapy, a reasonable strategy would be to temporarily hold therapy prior to high risk endoscopic procedures, assuming the patients are not at high cardiovascular risk. Clopidogrel and prasugrel have to be stopped 5-7 days prior to allow normal platelet aggregation to resume as opposed to ticagrelor, a reversible P2Y12 receptor inhibitor that can be stopped 3-5 days prior.30
Lastly, for patients who are on dual antiplatelet therapy (DAPT) for secondary prevention, continuation of ASA and temporary interruption of the P2Y12 receptor inhibitor is recommended while undergoing elective endoscopy. Studies have shown that those who discontinued both had a much higher incidence of stent thrombosis compared with those who remained on aspirin alone.4,28,31
Resumption of antithrombotic therapy after endoscopy
In general, antithrombotic therapy should be resumed upon completion of the procedure unless there remains a persistent risk of major bleeding.1,14 This consensus is based on studies available on warfarin and heparin products, with minimal literature available regarding the resumption of DOACs. The benefits of immediate re-initiation of antithrombotic therapy for the prevention of thromboembolic events should be weighed against the risk of hemorrhage associated with the specific agent, the time to onset of the medication, and procedure-specific circumstances. For the small subset of patients on warfarin with a high risk of thromboembolism (e.g., mechanical heart valve), bridging with LMWH should be started at the earliest possible time when there is no risk of major bleeding and continued until the international normalized ratio (INR) reaches a therapeutic level with warfarin. For patients at a lower risk of thromboembolism, warfarin should be restarted within 24 hours of the procedure. In addition, because of the shorter duration of DOACs, if treatment with these agents cannot resume within 24 hours of a high-risk procedure, bridge therapy should be considered with UFH or LMWH in patients with a high risk of thrombosis.18 In patients receiving DOACs for stroke prophylaxis in AF, the DOACS can be safely resumed 1 day after low-risk procedures and 2-3 days after high-risk procedures without the need for bridging.25 All antiplatelet agents should be resumed as soon as hemostasis is achieved.
Conclusion
Antithrombotic therapy is increasingly used given the aging population, widespread burden of cardiovascular comorbidities, and expanding indications for classes of medications such as direct oral anticoagulants. Given the association with antithrombotic medications and gastrointestinal bleeding, it is essential for gastroenterologists to understand the importance, necessity, and timing when holding these medications for endoscopic procedures. Even with the practice guidelines available today to help clinicians navigate certain situations, each patient’s antithrombotic management may be different, and communication with the prescribing physicians and including patients in the decision-making process is essential before planned procedures.
Dr. Wang is a gastroenterology fellow at the University of Chicago. Dr. Sengupta is an associate professor at the University of Chicago. They reported no funding or conflicts of interest.
References
1. ASGE Standards of Practice Committee, Acosta RD et al. The management of antithrombotic agents for patients undergoing GI endoscopy. Gastrointest Endosc. 2016;83(1):3-16.
2. Veitch AM et al. Endoscopy in patients on antiplatelet or anticoagulant therapy, including direct oral anticoagulants: British Society of Gastroenterology (BSG) and European Society of Gastrointestinal Endoscopy (ESGE) guidelines. Endoscopy. 2016;48(4):c1. doi: 10.1055/s-0042-122686.
3. Chan FKL et al. Management of patients on antithrombotic agents undergoing emergency and elective endoscopy: Joint Asian Pacific Association of Gastroenterology (APAGE) and Asian Pacific Society for Digestive Endoscopy (APSDE) practice guidelines. Gut. 2018;67(3):405-17.
4. Abraham NS et al. American College of Gastroenterology – Canadian Association of Gastroenterology clinical practice guideline: Management of anticoagulants and antiplatelets during acute gastrointestinal bleeding and the periendoscopic period. Am J Gastroenterol. 2022;117(4):542-58.
5. Boustière C et al. Endoscopy and antiplatelet agents. European Society of Gastrointestinal Endoscopy (ESGE) guideline. Endoscopy. 2011;43(5):445-61.
6. Fujimoto K et al. Guidelines for gastroenterological endoscopy in patients undergoing antithrombotic treatment. Dig Endosc. 2014;26(1):1-14.
7. Wilke T et al. Patient preferences for oral anticoagulation therapy in atrial fibrillation: A systematic literature review. Patient 2017;10(1):17-37.
8. Gerson LB et al. Adverse events associated with anticoagulation therapy in the periendoscopic period. Gastrointest Endosc. 2010 Jun;71(7):1211-17.e2.
9. Horiuchi A et al. Removal of small colorectal polyps in anticoagulated patients: A prospective randomized comparison of cold snare and conventional polypectomy. Gastrointest Endosc 2014;79(3):417-23.
10. Lip GYH et al. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: The euro heart survey on atrial fibrillation. Chest. 2010;137(2):263-72.
11. 2012 Writing Committee Members, Jneid H et al. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/non-ST-elevation myocardial infarction (Updating the 2007 guideline and replacing the 2011 focused update): A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2012;126(7):875-910.
12. Douketis JD et al. Perioperative management of antithrombotic therapy: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012 Feb;141(2 Suppl):e326S-e350S.
13. Becker RC et al. Management of platelet-directed pharmacotherapy in patients with atherosclerotic coronary artery disease undergoing elective endoscopic gastrointestinal procedures. J Am Coll Cardiol. 2009;54(24):2261-76.
14. Kwok A and Faigel DO. Management of anticoagulation before and after gastrointestinal endoscopy. Am J Gastroenterol. 2009;104(12):3085-97; quiz 3098.
15. Douketis JD et al. Perioperative bridging anticoagulation in patients with atrial fibrillation. N Engl J Med. 2015;373(9):823-33.
16. Kovacs MJ et al. Postoperative low molecular weight heparin bridging treatment for patients at high risk of arterial thromboembolism (PERIOP2): Double blind randomised controlled trial. BMJ 2021;373:n1205.
17. Tafur A and Douketis J. Perioperative management of anticoagulant and antiplatelet therapy. Heart 2018;104(17):1461-7.
18. Kato M et al. Guidelines for gastroenterological endoscopy in patients undergoing antithrombotic treatment: 2017 appendix on anticoagulants including direct oral anticoagulants. Dig Endosc. 2018;30(4):433-40.
19. Inoue T et al. Clinical features of postpolypectomy bleeding associated with heparin bridge therapy. Dig Endosc. 2014;26(2):243-9.
20. Takeuchi Y et al. Continuous anticoagulation and cold snare polypectomy versus heparin bridging and hot snare polypectomy in patients on anticoagulants with subcentimeter polyps: A randomized controlled trial. Ann Intern Med. 2019;171(4):229-37.
21. Ara N et al. Prospective analysis of risk for bleeding after endoscopic biopsy without cessation of antithrombotics in Japan. Dig Endosc. 2015;27(4):458-64.
22. Yanagisawa N et al. Postpolypectomy bleeding and thromboembolism risks associated with warfarin vs. direct oral anticoagulants. World J Gastroenterol. 2018;24(14):1540-9.
23. Arimoto J et al. Safety of cold snare polypectomy in patients receiving treatment with antithrombotic agents. Dig Dis Sci. 2019;64(11):3247-55.
24. Heublein V et al. Gastrointestinal endoscopy in patients receiving novel direct oral anticoagulants: Results from the prospective Dresden NOAC registry. J Gastroenterol. 2018;53(2):236-46.
25. Douketis JD et al. Perioperative management of patients with atrial fibrillation receiving a direct oral anticoagulant. JAMA Intern Med. 2019;179(11):1469-78.
26. Dubois V et al. Perioperative management of patients on direct oral anticoagulants. Thromb J. 2017;15:14.
27. Weitz JI et al. Periprocedural management and approach to bleeding in patients taking dabigatran. Circulation. 2012 Nov 13;126(20):2428-32.
28. Chan FKL et al. Risk of postpolypectomy bleeding with uninterrupted clopidogrel therapy in an industry-independent, double-blind, randomized trial. Gastroenterology. 2019;156(4):918-25.
29. Watanabe K et al. Effect of antiplatelet agent number, types, and pre-endoscopic management on postpolypectomy bleeding: Validation of endoscopy guidelines. Surg Endosc. 2021;35(1):317-25.
30. Gurbel PA et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: The ONSET/OFFSET study. Circulation. 2009;120(25):2577-85.
31. Eisenberg MJ et al. Safety of short-term discontinuation of antiplatelet therapy in patients with drug-eluting stents. Circulation. 2009;119(12):1634-42.
Therapeutic management of NAFLD
Nonalcoholic fatty liver disease (NAFLD) is defined by the presence of hepatic steatosis detected on either imaging or histology in the absence of secondary causes of fatty liver (e.g., excessive alcohol consumption) or other chronic liver diseases.1 For practical NAFLD diagnosis purposes, excessive alcohol intake can be defined as an active or recent history of more than 21 standard drinks per week in men and more than14 standard drinks per week in women. For the sake of terminology, NAFLD is characterized by fatty liver infiltration, affecting at least 5% of hepatocytes, with no evidence of hepatocyte injury, whereas nonalcoholic steatohepatitis (NASH) is defined as the presence of necroinflammation with or without fibrosis in a background of fatty liver.1

Natural history
NASH and the degree of fibrosis are the two most important determinants of the natural history of NAFLD. NASH can evolve into fibrosis and cirrhosis, whereas advanced fibrosis and cirrhosis (stages 3 or 4 of fibrosis) significantly increase the risk of liver-related decompensation and mortality. NAFLD, per se, has been associated with an increased risk of overall mortality, compared with that of the general population.2 The three most common causes of mortality for patients with NAFLD are cardiovascular diseases (CVD), extrahepatic malignancies, and liver-related deaths. Mortality and liver-related events, including hepatic decompensation and hepatocellular carcinoma (HCC), may significantly increase in a dose-dependent manner with increasing fibrosis stages, and stages 3 or 4 of fibrosis may display the highest rates of all-cause mortality and liver-related events.3,4 It is important to note, however, that almost 15% of HCCs occur in patients with NAFLD who do not have cirrhosis.5 The presence of commonly associated comorbidities such as obesity, insulin resistance or diabetes, dyslipidemia, hypothyroidism, polycystic ovary syndrome, and sleep apnea may contribute to an increased risk of NASH and advanced fibrosis and, therefore, an accelerated clinical course of NAFLD.
Nonpharmacological interventions
Lifestyle modification
Lifestyle modification to achieve weight loss remains a first-line intervention in patients with NAFLD. Weight loss achieved either by hypocaloric diet alone or in conjunction with increased physical activity can be beneficial for all patients with NAFLD. The benefits extend not only to those who are overweight and obese but also to those within normal body weight (lean NAFLD).1,6,7 Weight loss of approximately 3%-5% is necessary to improve hepatic steatosis, but a greater weight loss (7%-10%) is required to improve other histopathological features like necroinflammatory lesions and fibrosis.8-10 Individuals with higher BMI and/or type 2 diabetes (T2D) will require a larger weight reduction to achieve a similar benefit on NAFLD-related features.7,8 Weight loss via lifestyle changes can also decrease hepatic venous pressure gradient (HVPG), with greater declines reported among those with more than 10% weight loss.11

Weight loss can be achieved through a variety of modalities, but long-term maintenance of lost weight is much more challenging. A combination of a hypocaloric diet with a caloric deficit of 500-1,000 kcal/d, alongside moderate-intensity exercise and intensive on-site behavioral treatment, will likely increase the possibility of a sustained weight loss over time.1,12 A growing body of scientific evidence indicates that a healthy diet that includes a reduction of high-glycemic-index foods and refined carbohydrates; increased consumption of monounsaturated fatty acids, omega-3 fatty acids, and fibers; and high intakes of olive oil, nuts, vegetables, fruits, legumes, whole grains, and fish can have beneficial effects on NAFLD and its severity.13-16 Adherence to these healthy dietary patterns has been associated with a marked reduction in CVD morbidity and mortality and is, thus, a strategic lifestyle recommendation for patients with NAFLD in whom the leading cause of morbidity and death is CVD.1,3
Exercise alone in adults with NAFLD may reduce hepatic steatosis, but its ability to improve inflammation and fibrosis has not been proven in well-designed RCTs.17,18 Physical activity and exercise have been shown to curb both the development and the progression of NAFLD, and beneficial effects could be achieved independent of weight loss.17,19,20 Most importantly, moderate-to-vigorous physical activity is likely associated with lower all-cause and cardiovascular mortality in patients with NAFLD.21
Heavy alcohol intake should be avoided by patients with NAFLD or NASH, and those with cirrhotic NASH should avoid any alcohol consumption given the risk of HCC and hepatic decompensation.1,4,22 Limiting light-to-moderate alcohol intake among patients without cirrhosis is still under debate.1 People with NAFLD may be advised to drink an equivalent of two to three 8-oz cups of regular brewed coffee daily as it has shown certain antifibrotic effects in NAFLD patients.23
Bariatric surgery
Bariatric surgery is an attractive therapeutic option for eligible obese patients with NAFLD. Bariatric surgery has the potential for inducing great weight loss and, therefore, reverses not only the steatosis, inflammation, and fibrosis among NAFLD individuals but also important comorbid conditions like T2D. A recent systematic review and meta-analysis examining data on the effects of bariatric surgery on histologic features of NAFLD from 32 cohort studies (no RCTs included) showed that bariatric surgery was associated with significant improvements in steatosis (66%), lobular inflammation (50%), ballooning degeneration (76%), and fibrosis (40%), and the benefits were significantly higher in those who underwent Roux-en-Y gastric bypass (RYGB). Of note, worsening of liver histology, including fibrosis, could be seen in up to 12% of patients who underwent bariatric surgery.24 The postsurgical weight regained after RYGB could explain partly the lack of fibrosis improvement or even worsening of fibrosis, although further research is needed to clarify these controversial findings.
RYGB and sleeve gastrectomy (SG) are the most commonly performed bariatric surgeries worldwide. Patients who undergo RYGB achieve higher weight loss when compared with those treated with SG.25 Among all bariatric procedures, RYGB could result in a higher proportion of complete resolution of NAFLD than SG, although evidence is inconclusive on fibrosis improvement rates.24,26 Most recently, a single-center RCT has compared the effects of RYGB vs. SG on liver fat content and fibrosis in patients with severe obesity and T2D.27 Data showed that both surgical procedures were highly and equally effective in reducing fatty liver content (quantified by magnetic resonance imaging), with an almost complete resolution of the fatty liver at 1 year of both surgical interventions. The beneficial effects of both GB and SG on fibrosis (assessed by enhanced liver test [ELF]) were less evident with no substantial difference between the two groups. Importantly, 69% of participants had an increase in their ELF scores during the study, despite the majority of participants achieving significant reductions in their body weights and better glycemic control at the end of the study. These findings might be considered with caution as several factors, such as the duration of the study (only 1 year) and lack of a liver biopsy to confirm fibrosis changes over time, could be influencing the study results.
Among all NAFLD phenotypes, those with cirrhosis and, most importantly, hepatic decompensation appear to be at increased risk of perioperative mortality and inpatient hospital stays than those without cirrhosis.28-29 Bariatric surgery is an absolute contraindication in patients with decompensated cirrhosis (Child B and Child C). Among compensated -Child A- cirrhotics, those with portal hypertension are at increased risk of morbidity and perioperative mortality.30 A recent analysis of National Inpatient Sample data suggested that the rates of complications in those with cirrhosis have decreased with time, which could be due to a better selection process and the use of more restrictive bariatric surgery in those with cirrhosis. Low volume centers (defined as less than 50 procedures per year) and nonrestrictive bariatric surgery were associated with a higher mortality rate. These data may suggest that patients with cirrhosis should undergo bariatric surgery only in high-volume centers after a multidisciplinary evaluation.31 Bariatric endoscopy is emerging as a new treatment for obesity, but the long-term durability of its effects remains to be determined.
A recent retrospective cohort study, including 1,158 adult patients with biopsy-proven NASH, has investigated the benefits of bariatric surgery on the occurrence of major adverse liver and cardiovascular outcomes in 650 patients who underwent bariatric surgery, compared with 508 patients who received nonsurgical usual care. This study showed that bariatric surgery was associated with 88% lower risk of progression of fatty liver to cirrhosis, liver cancer, or liver-related death, and 70% lower risk of serious CVD events during a follow-up period of 10 years.32 Within 1 year after surgery, 0.6% of patients died from surgical complications. The potential benefits of bariatric surgery in patients with NAFLD must be balanced against surgical risk, especially in eligible obese individuals with established cirrhosis. Data from a retrospective cohort study have shown that bariatric surgery in obese cirrhotic patients does not seem to associate with excessive mortality, compared with noncirrhotic obese patients.33 More data on immediate complication rates and long-term outcomes in patients with NAFLD by type of bariatric surgery is also required.
NAFLD as a standalone is not an indication for bariatric surgery. However, it could be considered in NAFLD patients who have a BMI of 40 kg/m2 or more without coexisting comorbidities or with a BMI of 35 kg/m2 or more and one or more severe obesity-related comorbidities, including T2D, hypertension, hyperlipidemia, or obstructive sleep apnea. Bariatric surgery must always be offered in centers with an experienced bariatric surgery program.1
Management of comorbidities
Given the multiple comorbidities associated with NAFLD and the potential to influence its severity, a comprehensive and multidisciplinary approach is needed to ameliorate not only the progression of liver disease but also those complications related to metabolic syndrome, hyperlipidemia, hypertension, diabetes, and other related conditions. Of note, all patients with NAFLD should receive aggressive management of comorbidities regardless of the severity of NAFLD. Ideally, a multidisciplinary team – including a primary care provider, an endocrinologist for patients with T2D, and a gastroenterologist/hepatologist – is needed to successfully manage patients with NAFLD.
It is well recognized that individuals with biopsy-proven NAFLD are at a higher risk of coronary heart disease, stroke, congestive heart failure, and death resulting from CVD when compared with the non-NAFLD population, and excess in CVD morbidity and mortality is evident across all stages of NAFLD and increases with worsening disease severity.34 The strong association between CVD and NAFLD has important clinical implications that may influence the decision to initiate treatment for primary prevention, including lipid-lowering, antihypertensive, or antiplatelet therapies.35 Statins are widely used to reduce LDL cholesterol and have been proven to be safe in NAFLD, including for those with elevated liver enzymes and even in compensated cirrhosis, in several studies conducted during the last 15 years.36 Statins are characterized by anti-inflammatory, anti-oxidative, antifibrotic, and plaque-stabilizing effects, whereby they may improve vascular and hepatic function among patients with NAFLD and reduce cardiovascular risk.37 Statin use for the treatment of NAFLD is still controversial and off-label and is not specifically recommended to treat NASH, but positive results have been shown for reductions in liver enzymes.1 A recent meta-analysis of 13 studies showed that continued use of statin in cirrhosis was associated with a 46% and 44% risk reduction in hepatic decompensation and mortality, respectively.38
The Food and Drug Administration has approved omega-3 (n-3) fatty acid agents and fibrates for the treatment of very high triglycerides (500 mg/dL or higher); however, no specific indications exist to treat NAFLD.1 Fenofibrate is related to mild aminotransferase elevations and, in some cases, severe liver injury, so caution must be paid, especially within 2 days of taking the drug.39-40
NAFLD phenotypes that need liver pharmacotherapy
There are still no FDA-approved drugs or biological treatments for NASH. Pharmacological interventions aiming primarily at improving liver disease should generally be limited to those with biopsy-proven NASH and clinically significant fibrosis (fibrosis stages of 2 or greater).1,4 For FDA approval, medications used for treating NAFLD with fibrosis need to meet one of the following endpoint criteria: resolution of NASH without worsening of fibrosis, improvement in fibrosis without worsening of NASH, or both. In addition to those criteria, a new medication might improve the metabolic profile and have a tolerable safety profile. Table 1 displays those NAFLD phenotypes that will likely benefit from liver-directed therapy.
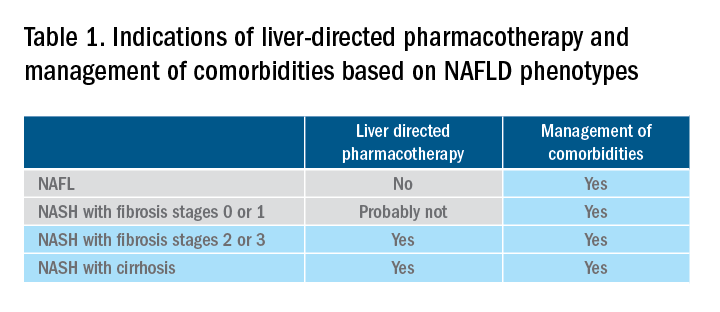
Obeticholic acid as an experimental therapy for NASH
A planned month-18 interim analysis of a multicentre, phase III RCT examined the efficacy and safety of obeticholic acid (OCA), a farnesoid X receptor agonist, in patients with NASH and stages 1-3 of fibrosis. The primary endpoint (fibrosis reduction 1 stage or more with no worsening of NASH) was met by 12% of patients in the placebo group, 18% of patients receiving OCA 10 mg (P = .045), and 23% of those receiving OCA 25 mg (P = .0002). An alternative primary endpoint of NASH resolution with no worsening of fibrosis was not met. OCA 25 mg led to the highest rates of pruritus and hyperlipidemia, compared with OCA 10 mg.42 These side effects seem to be related to the activation of the farnesoid X receptor.43
Currently available but off label medications
Vitamin E, an antioxidant, administered at a daily dose of 800 IU/day improves steatosis, inflammation, and ballooning, but not fibrosis in nondiabetic adults with biopsy-proven NASH.44 Vitamin E for 96 weeks was associated with a significantly higher rate of improvement in NASH (43% vs. 19%, P less than .01), compared with placebo.44 In the Treatment of Nonalcoholic Fatty Liver Disease in Children trial (TONIC), which examined vitamin E (800 IU/day) or metformin (500 mg twice daily) against placebo in children with biopsy-proven NAFLD, resolution of NASH was significantly greater in children treated with vitamin E than in children treated with placebo (58% vs. 28%, P less than .01). Metformin did not significantly improve the NASH resolution rates, compared with placebo (41% vs. 28%, P = .23). Vitamin E could be recommended for nondiabetic adults or children if lifestyle modifications do not produce the expected results as a result of noncompliance or ineffectiveness. Since continued use of vitamin E has been suggested to be associated with a very small increase in the risk for prostate cancer (an absolute increase of 1.6 per 1,000 person-years of vitamin E use) in men, risks and benefits should be discussed with each patient before starting therapy. A meta-analysis of nine placebo-controlled trials including roughly 119,000 patients reported that vitamin E supplementation increases the risk of hemorrhagic stroke by 20% while reducing ischemic stroke by 10%. It was estimated that vitamin E supplementation would prevent one ischemic stroke per 476 treated patients while inducing one hemorrhagic stroke for every 1,250 patients. It is noteworthy that the combination of vitamin E with anticoagulant and/or antiplatelet therapy was not examined in this trial, so we could not determine how combination therapy might affect the risk of ischemic or hemorrhagic stroke.45
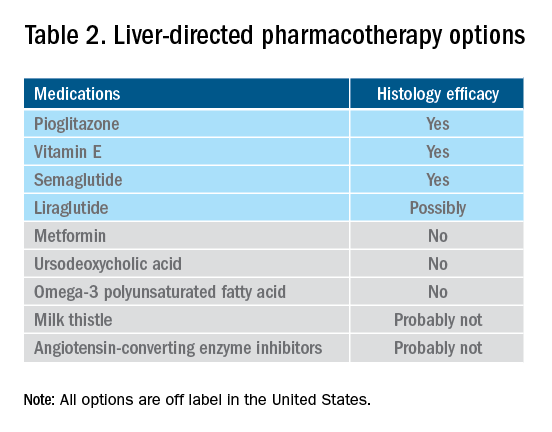
Thiazolidinediones drugs have been reported to be effective in improving NAFLD in many human studies. Evidence from RCTs suggests that pioglitazone could significantly improve glucose metabolism, alanine aminotransferase, and liver histology – such as hepatic steatosis, lobular inflammation, and ballooning degeneration – among patients with or without T2D. However, the beneficial effects on improving fibrosis remain to be verified.1,46 Because of safety concerns, the risk/benefit balance of using pioglitazone to treat NASH should be discussed with each patient.47-48 Pioglitazone has been associated with long-term risk of bladder cancer,49 congestive heart failure,50 and bone fractures.51 Data from the Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis (PIVENS) trial showed that pioglitazone was significantly associated with weight gain but with no other serious adverse events. However, this study was not powered to test any safety-related hypotheses.44
Glucagon-like peptide 1 analogs have been reported to induce weight loss and reduce insulin resistance, which may lead to improvements in NAFLD. Phase II RCTs of glucagon-like peptide 1 receptor agonists (liraglutide and semaglutide) for the treatment of biopsy-proven NASH showed significant improvements in serum liver enzymes, steatosis, and inflammation, as well as NASH resolution without worsening liver fibrosis, although no direct benefit was observed in reversing fibrosis.52-53 One of these studies explores the efficacy and safety of different doses of daily subcutaneous semaglutide vs. placebo on the rates of resolution of NASH with no worsening of fibrosis. The highest dose (0.4 mg) showed the greatest difference (59% vs. 17%, P less than .01), compared with the placebo arm. However, there was no difference in improvement in fibrosis stage between the two groups (43% in the 0.4-mg group vs. 33% in the placebo group, P = .48).53 Gastrointestinal adverse events were common in the semaglutide arm.
“Spontaneous” NASH resolution and fibrosis improvement are commonly seen in participants assigned to placebo arms in clinical trials. A recent meta-analysis of 43 RCTs including 2649 placebo-treated patients showed a pooled estimate of NASH resolution without worsening of fibrosis and 1 stage reduction or more in fibrosis of 12% and 19%, respectively. Relevant factors involved in “spontaneous” NASH improvement are unknown but could be related to changes in BMI resulting from lifestyle changes, race and ethnicity, age, and, likely, NAFLD-related genetic variations, although more data is needed to better understand the histologic response in placebo-treated patients.54
Semaglutide injections (2.4 mg once weekly) or (2.0 mg once weekly) have been recently approved by the FDA for chronic weight management in adults with obesity or overweight with at least one weight-related condition or glucose control of T2D, respectively. Of note, the semaglutide dose used in the NASH trial is not currently available for the treatment of patients who are overweight/obese or have T2D, but the beneficial effects on body weight reductions and glucose control are similar overall to the effects seen with currently available doses for management of obesity or diabetes. One may consider using semaglutide in patients who are overweight/obese or have T2D with NASH, but in the senior author’s experience, it has been quite challenging to receive the payer’s approval, as its use is not specifically approved to treat liver disease.1
How to follow patients with NAFLD in the clinic
Once a diagnosis of NAFLD is made, the use of noninvasive testing may aid to identify which patients are at high risk of fibrosis. Easy to use clinical tools, such as the NAFLD Fibrosis Score and the Fib-4 index, and liver stiffness measurements using vibration-controlled transient elastography (FibroScan) or magnetic resonance elastography (MRE) are clinically useful noninvasive tools for identifying patients with NAFLD who have a higher likelihood of progressing to advanced fibrosis.1,55 The use of either NAFLD Fibrosis Score (less than -1.455) or Fib-4 index (less than 1.30) low cutoffs may be particularly useful to rule out advanced fibrosis. People with a NAFLD Fibrosis Score (greater than –1.455) or Fib-4 index (greater than 1.30) should undergo liver stiffness measurement (LSM) via FibroScan. Those with an LSM of 8 kPa or higher should be referred to specialized care, where a decision to perform a liver biopsy and initiate monitoring and therapy will be taken. MRE is the most accurate noninvasive method for the estimation of liver fibrosis. When MRE is available, it can be a diagnostic alternative to accurately rule in and rule out patients with advanced fibrosis. This technique can be preferred in clinical trials, but it is rarely used in clinical practice because it is expensive and not easily available. Reassessment by noninvasive scores at 1-3 years’ follow-up will be considered for those with an LSM less than 8 kPa. Patients with NASH cirrhosis should be screened for both gastroesophageal varix and HCC according to the American Association for the Study of Liver Diseases guidelines.56-57
Dr. Vilar-Gomez is assistant professor in the division of gastroenterology and hepatology at Indiana University, Indianapolis. Dr. Chalasani is vice president for academic affairs at Indiana University Health, Indianapolis, and the David W. Crabb Professor of Gastroenterology and Hepatology and an adjunct professor of anatomy, cell biology, and physiology in the division of gastroenterology and hepatology at Indiana University. Dr. Vilar-Gomez reports no financial conflicts of interest. Dr. Chalasani serves as a paid consultant to AbbVie, Boehringer-Ingelheim, Altimmune, Madrigal, Lilly, Zydus, and Galectin. He receives research support from Galectin and DSM.
References
1. Chalasani N et al. Hepatology 2018;67:328-57.
2. Söderberg C et al. Hepatology 2010;51:595-602.
3. Sanyal AJ et al. N Engl J Med 2021;385:1559-69.
4. Vilar-Gomez E et al. Gastroenterology 2018;155:443-57.e17.
5. Younossi ZM et al. Hepatology 2016;64:73-84.
6. EASL-EASD-EASO. J Hepatol 2016;64:1388-402.
7. Wong VW et al. J Hepatol 2018; 69:1349-56.
8. Vilar-Gomez E et al. Gastroenterology 2015;149:367-78.e5; quiz e14-5.
9. Promrat K et al. Hepatology 2010;51:121-9.
10. Wong VW et al. J Hepatol 2013;59:536-42.
11. Berzigotti A et al. Hepatology 2017;65:1293-1305.
12. Sacks FM et al. N Engl J Med 2009;360:859-73.
13. Vilar-Gomez E et al. Hepatology 2022 Jun;75(6):1491-1506.
14. Zelber-Sagi S et al. Liver Int 2017;37:936-49.
15. Hassani Zadeh S et al. J Gastroenterol Hepatol 2021;36:1470-8.
16. Yaskolka Meir A et al. Gut 2021;70:2085-95.
17. Sung KC et al. J Hepatol 2016;65:791-7.
18. Orci LA et al. Clin Gastroenterol Hepatol 2016;14:1398-411.
19. Ryu S et al. J Hepatol 2015;63:1229-37.
20. Kim D et al. Hepatology 2020;72:1556-68.
21. Kim D et al. Clin Gastroenterol Hepatol 2021;19:1240-7.e5.
22. Ascha MS et al. Hepatology 2010;51:1972-8.
23. Bambha K et al. Liver Int 2014;34:1250-8.
24. Lee Y et al. Clin Gastroenterol Hepatol 2019;17:1040-60.e11.
25. Grönroos S et al. JAMA Surg 2021;156:137-46.
26. Fakhry TK et al. Surg Obes Relat Dis 2019;15:502-11.
27. Seeberg KA et al. Ann Intern Med 2022;175:74-83.
28. Bower G et al. Obes Surg 2015;25:2280-9.
29. Jan A et al. Obes Surg 2015;25:1518-26.
30. Hanipah ZN et al. Obes Surg 2018;28:3431-8.
31. Are VS et al. Am J Gastroenterol 2020;115:1849-56.
32. Aminian A et al. JAMA 2021;326:2031-42.
33. Vuppalanchi R et al. Ann Surg 2022;275:e174-80.
34. Simon TG et al. Gut 2021. doi: 10.1136/gutjnl-2021-325724.
35. Lonardo A et al. J Hepatol 2018;68:335-52.
36. Chalasani N et al. Gastroenterology 2004;126:1287-92.
37. Pastori D et al. Dig Liver Dis 2015;47:4-11.
38. Kim RG et al. Clin Gastroenterol Hepatol 2017;15:1521-30.e8.
39. Ahmad J et al. Dig Dis Sci 2017;62:3596-604.
40. Chalasani NP et al. Am J Gastroenterol 2021;116(5):878-98.
41. Rinella ME et al. Hepatology 2019;70:1424-36.
42. Younossi ZM et al. Lancet 2019;394:2184-96.
43. Ratziu V. Clin Liver Dis (Hoboken) 2021;17:398-400.
44. Sanyal AJ et al. N Engl J Med 2010;341:1675-85.
45. Schürks M et al. BMJ 2010;341:c5702.
46. Cusi K et al. Ann Intern Med 2016;165:305-15.
47. Lewis JD et al. JAMA 2015;314:265-77.
48. Billington EO et al. Diabetologia 2015;58:2238-46.
49. Lewis JD et al. Diabetes Care 2011;34:916-22.
50. Erdmann E et al. Diabetes Care 2007;30:2773-8.
51. Viscoli CM et al. J Clin Endocrinol Metab 2017;102:914-22.
52. Armstong MJ et al. Lancet 2016;387:679-90.
53. Newsome PN et al. N Engl J Med 2021;384:1113-24.
54. Ng CH et al. Hepatology 2022;75:1647-61.
55. Kanwal F et al. Gastroenterology 2021;161:1030-1042.e8.
56. Garcia-Tsao G et al. Hepatology 2017;65:310-35.
57. Heimbach JK et al. Hepatology 2018;67:358-80.
Nonalcoholic fatty liver disease (NAFLD) is defined by the presence of hepatic steatosis detected on either imaging or histology in the absence of secondary causes of fatty liver (e.g., excessive alcohol consumption) or other chronic liver diseases.1 For practical NAFLD diagnosis purposes, excessive alcohol intake can be defined as an active or recent history of more than 21 standard drinks per week in men and more than14 standard drinks per week in women. For the sake of terminology, NAFLD is characterized by fatty liver infiltration, affecting at least 5% of hepatocytes, with no evidence of hepatocyte injury, whereas nonalcoholic steatohepatitis (NASH) is defined as the presence of necroinflammation with or without fibrosis in a background of fatty liver.1
Natural history
NASH and the degree of fibrosis are the two most important determinants of the natural history of NAFLD. NASH can evolve into fibrosis and cirrhosis, whereas advanced fibrosis and cirrhosis (stages 3 or 4 of fibrosis) significantly increase the risk of liver-related decompensation and mortality. NAFLD, per se, has been associated with an increased risk of overall mortality, compared with that of the general population.2 The three most common causes of mortality for patients with NAFLD are cardiovascular diseases (CVD), extrahepatic malignancies, and liver-related deaths. Mortality and liver-related events, including hepatic decompensation and hepatocellular carcinoma (HCC), may significantly increase in a dose-dependent manner with increasing fibrosis stages, and stages 3 or 4 of fibrosis may display the highest rates of all-cause mortality and liver-related events.3,4 It is important to note, however, that almost 15% of HCCs occur in patients with NAFLD who do not have cirrhosis.5 The presence of commonly associated comorbidities such as obesity, insulin resistance or diabetes, dyslipidemia, hypothyroidism, polycystic ovary syndrome, and sleep apnea may contribute to an increased risk of NASH and advanced fibrosis and, therefore, an accelerated clinical course of NAFLD.
Nonpharmacological interventions
Lifestyle modification
Lifestyle modification to achieve weight loss remains a first-line intervention in patients with NAFLD. Weight loss achieved either by hypocaloric diet alone or in conjunction with increased physical activity can be beneficial for all patients with NAFLD. The benefits extend not only to those who are overweight and obese but also to those within normal body weight (lean NAFLD).1,6,7 Weight loss of approximately 3%-5% is necessary to improve hepatic steatosis, but a greater weight loss (7%-10%) is required to improve other histopathological features like necroinflammatory lesions and fibrosis.8-10 Individuals with higher BMI and/or type 2 diabetes (T2D) will require a larger weight reduction to achieve a similar benefit on NAFLD-related features.7,8 Weight loss via lifestyle changes can also decrease hepatic venous pressure gradient (HVPG), with greater declines reported among those with more than 10% weight loss.11
Weight loss can be achieved through a variety of modalities, but long-term maintenance of lost weight is much more challenging. A combination of a hypocaloric diet with a caloric deficit of 500-1,000 kcal/d, alongside moderate-intensity exercise and intensive on-site behavioral treatment, will likely increase the possibility of a sustained weight loss over time.1,12 A growing body of scientific evidence indicates that a healthy diet that includes a reduction of high-glycemic-index foods and refined carbohydrates; increased consumption of monounsaturated fatty acids, omega-3 fatty acids, and fibers; and high intakes of olive oil, nuts, vegetables, fruits, legumes, whole grains, and fish can have beneficial effects on NAFLD and its severity.13-16 Adherence to these healthy dietary patterns has been associated with a marked reduction in CVD morbidity and mortality and is, thus, a strategic lifestyle recommendation for patients with NAFLD in whom the leading cause of morbidity and death is CVD.1,3
Exercise alone in adults with NAFLD may reduce hepatic steatosis, but its ability to improve inflammation and fibrosis has not been proven in well-designed RCTs.17,18 Physical activity and exercise have been shown to curb both the development and the progression of NAFLD, and beneficial effects could be achieved independent of weight loss.17,19,20 Most importantly, moderate-to-vigorous physical activity is likely associated with lower all-cause and cardiovascular mortality in patients with NAFLD.21
Heavy alcohol intake should be avoided by patients with NAFLD or NASH, and those with cirrhotic NASH should avoid any alcohol consumption given the risk of HCC and hepatic decompensation.1,4,22 Limiting light-to-moderate alcohol intake among patients without cirrhosis is still under debate.1 People with NAFLD may be advised to drink an equivalent of two to three 8-oz cups of regular brewed coffee daily as it has shown certain antifibrotic effects in NAFLD patients.23
Bariatric surgery
Bariatric surgery is an attractive therapeutic option for eligible obese patients with NAFLD. Bariatric surgery has the potential for inducing great weight loss and, therefore, reverses not only the steatosis, inflammation, and fibrosis among NAFLD individuals but also important comorbid conditions like T2D. A recent systematic review and meta-analysis examining data on the effects of bariatric surgery on histologic features of NAFLD from 32 cohort studies (no RCTs included) showed that bariatric surgery was associated with significant improvements in steatosis (66%), lobular inflammation (50%), ballooning degeneration (76%), and fibrosis (40%), and the benefits were significantly higher in those who underwent Roux-en-Y gastric bypass (RYGB). Of note, worsening of liver histology, including fibrosis, could be seen in up to 12% of patients who underwent bariatric surgery.24 The postsurgical weight regained after RYGB could explain partly the lack of fibrosis improvement or even worsening of fibrosis, although further research is needed to clarify these controversial findings.
RYGB and sleeve gastrectomy (SG) are the most commonly performed bariatric surgeries worldwide. Patients who undergo RYGB achieve higher weight loss when compared with those treated with SG.25 Among all bariatric procedures, RYGB could result in a higher proportion of complete resolution of NAFLD than SG, although evidence is inconclusive on fibrosis improvement rates.24,26 Most recently, a single-center RCT has compared the effects of RYGB vs. SG on liver fat content and fibrosis in patients with severe obesity and T2D.27 Data showed that both surgical procedures were highly and equally effective in reducing fatty liver content (quantified by magnetic resonance imaging), with an almost complete resolution of the fatty liver at 1 year of both surgical interventions. The beneficial effects of both GB and SG on fibrosis (assessed by enhanced liver test [ELF]) were less evident with no substantial difference between the two groups. Importantly, 69% of participants had an increase in their ELF scores during the study, despite the majority of participants achieving significant reductions in their body weights and better glycemic control at the end of the study. These findings might be considered with caution as several factors, such as the duration of the study (only 1 year) and lack of a liver biopsy to confirm fibrosis changes over time, could be influencing the study results.
Among all NAFLD phenotypes, those with cirrhosis and, most importantly, hepatic decompensation appear to be at increased risk of perioperative mortality and inpatient hospital stays than those without cirrhosis.28-29 Bariatric surgery is an absolute contraindication in patients with decompensated cirrhosis (Child B and Child C). Among compensated -Child A- cirrhotics, those with portal hypertension are at increased risk of morbidity and perioperative mortality.30 A recent analysis of National Inpatient Sample data suggested that the rates of complications in those with cirrhosis have decreased with time, which could be due to a better selection process and the use of more restrictive bariatric surgery in those with cirrhosis. Low volume centers (defined as less than 50 procedures per year) and nonrestrictive bariatric surgery were associated with a higher mortality rate. These data may suggest that patients with cirrhosis should undergo bariatric surgery only in high-volume centers after a multidisciplinary evaluation.31 Bariatric endoscopy is emerging as a new treatment for obesity, but the long-term durability of its effects remains to be determined.
A recent retrospective cohort study, including 1,158 adult patients with biopsy-proven NASH, has investigated the benefits of bariatric surgery on the occurrence of major adverse liver and cardiovascular outcomes in 650 patients who underwent bariatric surgery, compared with 508 patients who received nonsurgical usual care. This study showed that bariatric surgery was associated with 88% lower risk of progression of fatty liver to cirrhosis, liver cancer, or liver-related death, and 70% lower risk of serious CVD events during a follow-up period of 10 years.32 Within 1 year after surgery, 0.6% of patients died from surgical complications. The potential benefits of bariatric surgery in patients with NAFLD must be balanced against surgical risk, especially in eligible obese individuals with established cirrhosis. Data from a retrospective cohort study have shown that bariatric surgery in obese cirrhotic patients does not seem to associate with excessive mortality, compared with noncirrhotic obese patients.33 More data on immediate complication rates and long-term outcomes in patients with NAFLD by type of bariatric surgery is also required.
NAFLD as a standalone is not an indication for bariatric surgery. However, it could be considered in NAFLD patients who have a BMI of 40 kg/m2 or more without coexisting comorbidities or with a BMI of 35 kg/m2 or more and one or more severe obesity-related comorbidities, including T2D, hypertension, hyperlipidemia, or obstructive sleep apnea. Bariatric surgery must always be offered in centers with an experienced bariatric surgery program.1
Management of comorbidities
Given the multiple comorbidities associated with NAFLD and the potential to influence its severity, a comprehensive and multidisciplinary approach is needed to ameliorate not only the progression of liver disease but also those complications related to metabolic syndrome, hyperlipidemia, hypertension, diabetes, and other related conditions. Of note, all patients with NAFLD should receive aggressive management of comorbidities regardless of the severity of NAFLD. Ideally, a multidisciplinary team – including a primary care provider, an endocrinologist for patients with T2D, and a gastroenterologist/hepatologist – is needed to successfully manage patients with NAFLD.
It is well recognized that individuals with biopsy-proven NAFLD are at a higher risk of coronary heart disease, stroke, congestive heart failure, and death resulting from CVD when compared with the non-NAFLD population, and excess in CVD morbidity and mortality is evident across all stages of NAFLD and increases with worsening disease severity.34 The strong association between CVD and NAFLD has important clinical implications that may influence the decision to initiate treatment for primary prevention, including lipid-lowering, antihypertensive, or antiplatelet therapies.35 Statins are widely used to reduce LDL cholesterol and have been proven to be safe in NAFLD, including for those with elevated liver enzymes and even in compensated cirrhosis, in several studies conducted during the last 15 years.36 Statins are characterized by anti-inflammatory, anti-oxidative, antifibrotic, and plaque-stabilizing effects, whereby they may improve vascular and hepatic function among patients with NAFLD and reduce cardiovascular risk.37 Statin use for the treatment of NAFLD is still controversial and off-label and is not specifically recommended to treat NASH, but positive results have been shown for reductions in liver enzymes.1 A recent meta-analysis of 13 studies showed that continued use of statin in cirrhosis was associated with a 46% and 44% risk reduction in hepatic decompensation and mortality, respectively.38
The Food and Drug Administration has approved omega-3 (n-3) fatty acid agents and fibrates for the treatment of very high triglycerides (500 mg/dL or higher); however, no specific indications exist to treat NAFLD.1 Fenofibrate is related to mild aminotransferase elevations and, in some cases, severe liver injury, so caution must be paid, especially within 2 days of taking the drug.39-40
NAFLD phenotypes that need liver pharmacotherapy
There are still no FDA-approved drugs or biological treatments for NASH. Pharmacological interventions aiming primarily at improving liver disease should generally be limited to those with biopsy-proven NASH and clinically significant fibrosis (fibrosis stages of 2 or greater).1,4 For FDA approval, medications used for treating NAFLD with fibrosis need to meet one of the following endpoint criteria: resolution of NASH without worsening of fibrosis, improvement in fibrosis without worsening of NASH, or both. In addition to those criteria, a new medication might improve the metabolic profile and have a tolerable safety profile. Table 1 displays those NAFLD phenotypes that will likely benefit from liver-directed therapy.
Obeticholic acid as an experimental therapy for NASH
A planned month-18 interim analysis of a multicentre, phase III RCT examined the efficacy and safety of obeticholic acid (OCA), a farnesoid X receptor agonist, in patients with NASH and stages 1-3 of fibrosis. The primary endpoint (fibrosis reduction 1 stage or more with no worsening of NASH) was met by 12% of patients in the placebo group, 18% of patients receiving OCA 10 mg (P = .045), and 23% of those receiving OCA 25 mg (P = .0002). An alternative primary endpoint of NASH resolution with no worsening of fibrosis was not met. OCA 25 mg led to the highest rates of pruritus and hyperlipidemia, compared with OCA 10 mg.42 These side effects seem to be related to the activation of the farnesoid X receptor.43
Currently available but off label medications
Vitamin E, an antioxidant, administered at a daily dose of 800 IU/day improves steatosis, inflammation, and ballooning, but not fibrosis in nondiabetic adults with biopsy-proven NASH.44 Vitamin E for 96 weeks was associated with a significantly higher rate of improvement in NASH (43% vs. 19%, P less than .01), compared with placebo.44 In the Treatment of Nonalcoholic Fatty Liver Disease in Children trial (TONIC), which examined vitamin E (800 IU/day) or metformin (500 mg twice daily) against placebo in children with biopsy-proven NAFLD, resolution of NASH was significantly greater in children treated with vitamin E than in children treated with placebo (58% vs. 28%, P less than .01). Metformin did not significantly improve the NASH resolution rates, compared with placebo (41% vs. 28%, P = .23). Vitamin E could be recommended for nondiabetic adults or children if lifestyle modifications do not produce the expected results as a result of noncompliance or ineffectiveness. Since continued use of vitamin E has been suggested to be associated with a very small increase in the risk for prostate cancer (an absolute increase of 1.6 per 1,000 person-years of vitamin E use) in men, risks and benefits should be discussed with each patient before starting therapy. A meta-analysis of nine placebo-controlled trials including roughly 119,000 patients reported that vitamin E supplementation increases the risk of hemorrhagic stroke by 20% while reducing ischemic stroke by 10%. It was estimated that vitamin E supplementation would prevent one ischemic stroke per 476 treated patients while inducing one hemorrhagic stroke for every 1,250 patients. It is noteworthy that the combination of vitamin E with anticoagulant and/or antiplatelet therapy was not examined in this trial, so we could not determine how combination therapy might affect the risk of ischemic or hemorrhagic stroke.45
Thiazolidinediones drugs have been reported to be effective in improving NAFLD in many human studies. Evidence from RCTs suggests that pioglitazone could significantly improve glucose metabolism, alanine aminotransferase, and liver histology – such as hepatic steatosis, lobular inflammation, and ballooning degeneration – among patients with or without T2D. However, the beneficial effects on improving fibrosis remain to be verified.1,46 Because of safety concerns, the risk/benefit balance of using pioglitazone to treat NASH should be discussed with each patient.47-48 Pioglitazone has been associated with long-term risk of bladder cancer,49 congestive heart failure,50 and bone fractures.51 Data from the Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis (PIVENS) trial showed that pioglitazone was significantly associated with weight gain but with no other serious adverse events. However, this study was not powered to test any safety-related hypotheses.44
Glucagon-like peptide 1 analogs have been reported to induce weight loss and reduce insulin resistance, which may lead to improvements in NAFLD. Phase II RCTs of glucagon-like peptide 1 receptor agonists (liraglutide and semaglutide) for the treatment of biopsy-proven NASH showed significant improvements in serum liver enzymes, steatosis, and inflammation, as well as NASH resolution without worsening liver fibrosis, although no direct benefit was observed in reversing fibrosis.52-53 One of these studies explores the efficacy and safety of different doses of daily subcutaneous semaglutide vs. placebo on the rates of resolution of NASH with no worsening of fibrosis. The highest dose (0.4 mg) showed the greatest difference (59% vs. 17%, P less than .01), compared with the placebo arm. However, there was no difference in improvement in fibrosis stage between the two groups (43% in the 0.4-mg group vs. 33% in the placebo group, P = .48).53 Gastrointestinal adverse events were common in the semaglutide arm.
“Spontaneous” NASH resolution and fibrosis improvement are commonly seen in participants assigned to placebo arms in clinical trials. A recent meta-analysis of 43 RCTs including 2649 placebo-treated patients showed a pooled estimate of NASH resolution without worsening of fibrosis and 1 stage reduction or more in fibrosis of 12% and 19%, respectively. Relevant factors involved in “spontaneous” NASH improvement are unknown but could be related to changes in BMI resulting from lifestyle changes, race and ethnicity, age, and, likely, NAFLD-related genetic variations, although more data is needed to better understand the histologic response in placebo-treated patients.54
Semaglutide injections (2.4 mg once weekly) or (2.0 mg once weekly) have been recently approved by the FDA for chronic weight management in adults with obesity or overweight with at least one weight-related condition or glucose control of T2D, respectively. Of note, the semaglutide dose used in the NASH trial is not currently available for the treatment of patients who are overweight/obese or have T2D, but the beneficial effects on body weight reductions and glucose control are similar overall to the effects seen with currently available doses for management of obesity or diabetes. One may consider using semaglutide in patients who are overweight/obese or have T2D with NASH, but in the senior author’s experience, it has been quite challenging to receive the payer’s approval, as its use is not specifically approved to treat liver disease.1
How to follow patients with NAFLD in the clinic
Once a diagnosis of NAFLD is made, the use of noninvasive testing may aid to identify which patients are at high risk of fibrosis. Easy to use clinical tools, such as the NAFLD Fibrosis Score and the Fib-4 index, and liver stiffness measurements using vibration-controlled transient elastography (FibroScan) or magnetic resonance elastography (MRE) are clinically useful noninvasive tools for identifying patients with NAFLD who have a higher likelihood of progressing to advanced fibrosis.1,55 The use of either NAFLD Fibrosis Score (less than -1.455) or Fib-4 index (less than 1.30) low cutoffs may be particularly useful to rule out advanced fibrosis. People with a NAFLD Fibrosis Score (greater than –1.455) or Fib-4 index (greater than 1.30) should undergo liver stiffness measurement (LSM) via FibroScan. Those with an LSM of 8 kPa or higher should be referred to specialized care, where a decision to perform a liver biopsy and initiate monitoring and therapy will be taken. MRE is the most accurate noninvasive method for the estimation of liver fibrosis. When MRE is available, it can be a diagnostic alternative to accurately rule in and rule out patients with advanced fibrosis. This technique can be preferred in clinical trials, but it is rarely used in clinical practice because it is expensive and not easily available. Reassessment by noninvasive scores at 1-3 years’ follow-up will be considered for those with an LSM less than 8 kPa. Patients with NASH cirrhosis should be screened for both gastroesophageal varix and HCC according to the American Association for the Study of Liver Diseases guidelines.56-57
Dr. Vilar-Gomez is assistant professor in the division of gastroenterology and hepatology at Indiana University, Indianapolis. Dr. Chalasani is vice president for academic affairs at Indiana University Health, Indianapolis, and the David W. Crabb Professor of Gastroenterology and Hepatology and an adjunct professor of anatomy, cell biology, and physiology in the division of gastroenterology and hepatology at Indiana University. Dr. Vilar-Gomez reports no financial conflicts of interest. Dr. Chalasani serves as a paid consultant to AbbVie, Boehringer-Ingelheim, Altimmune, Madrigal, Lilly, Zydus, and Galectin. He receives research support from Galectin and DSM.
References
1. Chalasani N et al. Hepatology 2018;67:328-57.
2. Söderberg C et al. Hepatology 2010;51:595-602.
3. Sanyal AJ et al. N Engl J Med 2021;385:1559-69.
4. Vilar-Gomez E et al. Gastroenterology 2018;155:443-57.e17.
5. Younossi ZM et al. Hepatology 2016;64:73-84.
6. EASL-EASD-EASO. J Hepatol 2016;64:1388-402.
7. Wong VW et al. J Hepatol 2018; 69:1349-56.
8. Vilar-Gomez E et al. Gastroenterology 2015;149:367-78.e5; quiz e14-5.
9. Promrat K et al. Hepatology 2010;51:121-9.
10. Wong VW et al. J Hepatol 2013;59:536-42.
11. Berzigotti A et al. Hepatology 2017;65:1293-1305.
12. Sacks FM et al. N Engl J Med 2009;360:859-73.
13. Vilar-Gomez E et al. Hepatology 2022 Jun;75(6):1491-1506.
14. Zelber-Sagi S et al. Liver Int 2017;37:936-49.
15. Hassani Zadeh S et al. J Gastroenterol Hepatol 2021;36:1470-8.
16. Yaskolka Meir A et al. Gut 2021;70:2085-95.
17. Sung KC et al. J Hepatol 2016;65:791-7.
18. Orci LA et al. Clin Gastroenterol Hepatol 2016;14:1398-411.
19. Ryu S et al. J Hepatol 2015;63:1229-37.
20. Kim D et al. Hepatology 2020;72:1556-68.
21. Kim D et al. Clin Gastroenterol Hepatol 2021;19:1240-7.e5.
22. Ascha MS et al. Hepatology 2010;51:1972-8.
23. Bambha K et al. Liver Int 2014;34:1250-8.
24. Lee Y et al. Clin Gastroenterol Hepatol 2019;17:1040-60.e11.
25. Grönroos S et al. JAMA Surg 2021;156:137-46.
26. Fakhry TK et al. Surg Obes Relat Dis 2019;15:502-11.
27. Seeberg KA et al. Ann Intern Med 2022;175:74-83.
28. Bower G et al. Obes Surg 2015;25:2280-9.
29. Jan A et al. Obes Surg 2015;25:1518-26.
30. Hanipah ZN et al. Obes Surg 2018;28:3431-8.
31. Are VS et al. Am J Gastroenterol 2020;115:1849-56.
32. Aminian A et al. JAMA 2021;326:2031-42.
33. Vuppalanchi R et al. Ann Surg 2022;275:e174-80.
34. Simon TG et al. Gut 2021. doi: 10.1136/gutjnl-2021-325724.
35. Lonardo A et al. J Hepatol 2018;68:335-52.
36. Chalasani N et al. Gastroenterology 2004;126:1287-92.
37. Pastori D et al. Dig Liver Dis 2015;47:4-11.
38. Kim RG et al. Clin Gastroenterol Hepatol 2017;15:1521-30.e8.
39. Ahmad J et al. Dig Dis Sci 2017;62:3596-604.
40. Chalasani NP et al. Am J Gastroenterol 2021;116(5):878-98.
41. Rinella ME et al. Hepatology 2019;70:1424-36.
42. Younossi ZM et al. Lancet 2019;394:2184-96.
43. Ratziu V. Clin Liver Dis (Hoboken) 2021;17:398-400.
44. Sanyal AJ et al. N Engl J Med 2010;341:1675-85.
45. Schürks M et al. BMJ 2010;341:c5702.
46. Cusi K et al. Ann Intern Med 2016;165:305-15.
47. Lewis JD et al. JAMA 2015;314:265-77.
48. Billington EO et al. Diabetologia 2015;58:2238-46.
49. Lewis JD et al. Diabetes Care 2011;34:916-22.
50. Erdmann E et al. Diabetes Care 2007;30:2773-8.
51. Viscoli CM et al. J Clin Endocrinol Metab 2017;102:914-22.
52. Armstong MJ et al. Lancet 2016;387:679-90.
53. Newsome PN et al. N Engl J Med 2021;384:1113-24.
54. Ng CH et al. Hepatology 2022;75:1647-61.
55. Kanwal F et al. Gastroenterology 2021;161:1030-1042.e8.
56. Garcia-Tsao G et al. Hepatology 2017;65:310-35.
57. Heimbach JK et al. Hepatology 2018;67:358-80.
Nonalcoholic fatty liver disease (NAFLD) is defined by the presence of hepatic steatosis detected on either imaging or histology in the absence of secondary causes of fatty liver (e.g., excessive alcohol consumption) or other chronic liver diseases.1 For practical NAFLD diagnosis purposes, excessive alcohol intake can be defined as an active or recent history of more than 21 standard drinks per week in men and more than14 standard drinks per week in women. For the sake of terminology, NAFLD is characterized by fatty liver infiltration, affecting at least 5% of hepatocytes, with no evidence of hepatocyte injury, whereas nonalcoholic steatohepatitis (NASH) is defined as the presence of necroinflammation with or without fibrosis in a background of fatty liver.1
Natural history
NASH and the degree of fibrosis are the two most important determinants of the natural history of NAFLD. NASH can evolve into fibrosis and cirrhosis, whereas advanced fibrosis and cirrhosis (stages 3 or 4 of fibrosis) significantly increase the risk of liver-related decompensation and mortality. NAFLD, per se, has been associated with an increased risk of overall mortality, compared with that of the general population.2 The three most common causes of mortality for patients with NAFLD are cardiovascular diseases (CVD), extrahepatic malignancies, and liver-related deaths. Mortality and liver-related events, including hepatic decompensation and hepatocellular carcinoma (HCC), may significantly increase in a dose-dependent manner with increasing fibrosis stages, and stages 3 or 4 of fibrosis may display the highest rates of all-cause mortality and liver-related events.3,4 It is important to note, however, that almost 15% of HCCs occur in patients with NAFLD who do not have cirrhosis.5 The presence of commonly associated comorbidities such as obesity, insulin resistance or diabetes, dyslipidemia, hypothyroidism, polycystic ovary syndrome, and sleep apnea may contribute to an increased risk of NASH and advanced fibrosis and, therefore, an accelerated clinical course of NAFLD.
Nonpharmacological interventions
Lifestyle modification
Lifestyle modification to achieve weight loss remains a first-line intervention in patients with NAFLD. Weight loss achieved either by hypocaloric diet alone or in conjunction with increased physical activity can be beneficial for all patients with NAFLD. The benefits extend not only to those who are overweight and obese but also to those within normal body weight (lean NAFLD).1,6,7 Weight loss of approximately 3%-5% is necessary to improve hepatic steatosis, but a greater weight loss (7%-10%) is required to improve other histopathological features like necroinflammatory lesions and fibrosis.8-10 Individuals with higher BMI and/or type 2 diabetes (T2D) will require a larger weight reduction to achieve a similar benefit on NAFLD-related features.7,8 Weight loss via lifestyle changes can also decrease hepatic venous pressure gradient (HVPG), with greater declines reported among those with more than 10% weight loss.11
Weight loss can be achieved through a variety of modalities, but long-term maintenance of lost weight is much more challenging. A combination of a hypocaloric diet with a caloric deficit of 500-1,000 kcal/d, alongside moderate-intensity exercise and intensive on-site behavioral treatment, will likely increase the possibility of a sustained weight loss over time.1,12 A growing body of scientific evidence indicates that a healthy diet that includes a reduction of high-glycemic-index foods and refined carbohydrates; increased consumption of monounsaturated fatty acids, omega-3 fatty acids, and fibers; and high intakes of olive oil, nuts, vegetables, fruits, legumes, whole grains, and fish can have beneficial effects on NAFLD and its severity.13-16 Adherence to these healthy dietary patterns has been associated with a marked reduction in CVD morbidity and mortality and is, thus, a strategic lifestyle recommendation for patients with NAFLD in whom the leading cause of morbidity and death is CVD.1,3
Exercise alone in adults with NAFLD may reduce hepatic steatosis, but its ability to improve inflammation and fibrosis has not been proven in well-designed RCTs.17,18 Physical activity and exercise have been shown to curb both the development and the progression of NAFLD, and beneficial effects could be achieved independent of weight loss.17,19,20 Most importantly, moderate-to-vigorous physical activity is likely associated with lower all-cause and cardiovascular mortality in patients with NAFLD.21
Heavy alcohol intake should be avoided by patients with NAFLD or NASH, and those with cirrhotic NASH should avoid any alcohol consumption given the risk of HCC and hepatic decompensation.1,4,22 Limiting light-to-moderate alcohol intake among patients without cirrhosis is still under debate.1 People with NAFLD may be advised to drink an equivalent of two to three 8-oz cups of regular brewed coffee daily as it has shown certain antifibrotic effects in NAFLD patients.23
Bariatric surgery
Bariatric surgery is an attractive therapeutic option for eligible obese patients with NAFLD. Bariatric surgery has the potential for inducing great weight loss and, therefore, reverses not only the steatosis, inflammation, and fibrosis among NAFLD individuals but also important comorbid conditions like T2D. A recent systematic review and meta-analysis examining data on the effects of bariatric surgery on histologic features of NAFLD from 32 cohort studies (no RCTs included) showed that bariatric surgery was associated with significant improvements in steatosis (66%), lobular inflammation (50%), ballooning degeneration (76%), and fibrosis (40%), and the benefits were significantly higher in those who underwent Roux-en-Y gastric bypass (RYGB). Of note, worsening of liver histology, including fibrosis, could be seen in up to 12% of patients who underwent bariatric surgery.24 The postsurgical weight regained after RYGB could explain partly the lack of fibrosis improvement or even worsening of fibrosis, although further research is needed to clarify these controversial findings.
RYGB and sleeve gastrectomy (SG) are the most commonly performed bariatric surgeries worldwide. Patients who undergo RYGB achieve higher weight loss when compared with those treated with SG.25 Among all bariatric procedures, RYGB could result in a higher proportion of complete resolution of NAFLD than SG, although evidence is inconclusive on fibrosis improvement rates.24,26 Most recently, a single-center RCT has compared the effects of RYGB vs. SG on liver fat content and fibrosis in patients with severe obesity and T2D.27 Data showed that both surgical procedures were highly and equally effective in reducing fatty liver content (quantified by magnetic resonance imaging), with an almost complete resolution of the fatty liver at 1 year of both surgical interventions. The beneficial effects of both GB and SG on fibrosis (assessed by enhanced liver test [ELF]) were less evident with no substantial difference between the two groups. Importantly, 69% of participants had an increase in their ELF scores during the study, despite the majority of participants achieving significant reductions in their body weights and better glycemic control at the end of the study. These findings might be considered with caution as several factors, such as the duration of the study (only 1 year) and lack of a liver biopsy to confirm fibrosis changes over time, could be influencing the study results.
Among all NAFLD phenotypes, those with cirrhosis and, most importantly, hepatic decompensation appear to be at increased risk of perioperative mortality and inpatient hospital stays than those without cirrhosis.28-29 Bariatric surgery is an absolute contraindication in patients with decompensated cirrhosis (Child B and Child C). Among compensated -Child A- cirrhotics, those with portal hypertension are at increased risk of morbidity and perioperative mortality.30 A recent analysis of National Inpatient Sample data suggested that the rates of complications in those with cirrhosis have decreased with time, which could be due to a better selection process and the use of more restrictive bariatric surgery in those with cirrhosis. Low volume centers (defined as less than 50 procedures per year) and nonrestrictive bariatric surgery were associated with a higher mortality rate. These data may suggest that patients with cirrhosis should undergo bariatric surgery only in high-volume centers after a multidisciplinary evaluation.31 Bariatric endoscopy is emerging as a new treatment for obesity, but the long-term durability of its effects remains to be determined.
A recent retrospective cohort study, including 1,158 adult patients with biopsy-proven NASH, has investigated the benefits of bariatric surgery on the occurrence of major adverse liver and cardiovascular outcomes in 650 patients who underwent bariatric surgery, compared with 508 patients who received nonsurgical usual care. This study showed that bariatric surgery was associated with 88% lower risk of progression of fatty liver to cirrhosis, liver cancer, or liver-related death, and 70% lower risk of serious CVD events during a follow-up period of 10 years.32 Within 1 year after surgery, 0.6% of patients died from surgical complications. The potential benefits of bariatric surgery in patients with NAFLD must be balanced against surgical risk, especially in eligible obese individuals with established cirrhosis. Data from a retrospective cohort study have shown that bariatric surgery in obese cirrhotic patients does not seem to associate with excessive mortality, compared with noncirrhotic obese patients.33 More data on immediate complication rates and long-term outcomes in patients with NAFLD by type of bariatric surgery is also required.
NAFLD as a standalone is not an indication for bariatric surgery. However, it could be considered in NAFLD patients who have a BMI of 40 kg/m2 or more without coexisting comorbidities or with a BMI of 35 kg/m2 or more and one or more severe obesity-related comorbidities, including T2D, hypertension, hyperlipidemia, or obstructive sleep apnea. Bariatric surgery must always be offered in centers with an experienced bariatric surgery program.1
Management of comorbidities
Given the multiple comorbidities associated with NAFLD and the potential to influence its severity, a comprehensive and multidisciplinary approach is needed to ameliorate not only the progression of liver disease but also those complications related to metabolic syndrome, hyperlipidemia, hypertension, diabetes, and other related conditions. Of note, all patients with NAFLD should receive aggressive management of comorbidities regardless of the severity of NAFLD. Ideally, a multidisciplinary team – including a primary care provider, an endocrinologist for patients with T2D, and a gastroenterologist/hepatologist – is needed to successfully manage patients with NAFLD.
It is well recognized that individuals with biopsy-proven NAFLD are at a higher risk of coronary heart disease, stroke, congestive heart failure, and death resulting from CVD when compared with the non-NAFLD population, and excess in CVD morbidity and mortality is evident across all stages of NAFLD and increases with worsening disease severity.34 The strong association between CVD and NAFLD has important clinical implications that may influence the decision to initiate treatment for primary prevention, including lipid-lowering, antihypertensive, or antiplatelet therapies.35 Statins are widely used to reduce LDL cholesterol and have been proven to be safe in NAFLD, including for those with elevated liver enzymes and even in compensated cirrhosis, in several studies conducted during the last 15 years.36 Statins are characterized by anti-inflammatory, anti-oxidative, antifibrotic, and plaque-stabilizing effects, whereby they may improve vascular and hepatic function among patients with NAFLD and reduce cardiovascular risk.37 Statin use for the treatment of NAFLD is still controversial and off-label and is not specifically recommended to treat NASH, but positive results have been shown for reductions in liver enzymes.1 A recent meta-analysis of 13 studies showed that continued use of statin in cirrhosis was associated with a 46% and 44% risk reduction in hepatic decompensation and mortality, respectively.38
The Food and Drug Administration has approved omega-3 (n-3) fatty acid agents and fibrates for the treatment of very high triglycerides (500 mg/dL or higher); however, no specific indications exist to treat NAFLD.1 Fenofibrate is related to mild aminotransferase elevations and, in some cases, severe liver injury, so caution must be paid, especially within 2 days of taking the drug.39-40
NAFLD phenotypes that need liver pharmacotherapy
There are still no FDA-approved drugs or biological treatments for NASH. Pharmacological interventions aiming primarily at improving liver disease should generally be limited to those with biopsy-proven NASH and clinically significant fibrosis (fibrosis stages of 2 or greater).1,4 For FDA approval, medications used for treating NAFLD with fibrosis need to meet one of the following endpoint criteria: resolution of NASH without worsening of fibrosis, improvement in fibrosis without worsening of NASH, or both. In addition to those criteria, a new medication might improve the metabolic profile and have a tolerable safety profile. Table 1 displays those NAFLD phenotypes that will likely benefit from liver-directed therapy.
Obeticholic acid as an experimental therapy for NASH
A planned month-18 interim analysis of a multicentre, phase III RCT examined the efficacy and safety of obeticholic acid (OCA), a farnesoid X receptor agonist, in patients with NASH and stages 1-3 of fibrosis. The primary endpoint (fibrosis reduction 1 stage or more with no worsening of NASH) was met by 12% of patients in the placebo group, 18% of patients receiving OCA 10 mg (P = .045), and 23% of those receiving OCA 25 mg (P = .0002). An alternative primary endpoint of NASH resolution with no worsening of fibrosis was not met. OCA 25 mg led to the highest rates of pruritus and hyperlipidemia, compared with OCA 10 mg.42 These side effects seem to be related to the activation of the farnesoid X receptor.43
Currently available but off label medications
Vitamin E, an antioxidant, administered at a daily dose of 800 IU/day improves steatosis, inflammation, and ballooning, but not fibrosis in nondiabetic adults with biopsy-proven NASH.44 Vitamin E for 96 weeks was associated with a significantly higher rate of improvement in NASH (43% vs. 19%, P less than .01), compared with placebo.44 In the Treatment of Nonalcoholic Fatty Liver Disease in Children trial (TONIC), which examined vitamin E (800 IU/day) or metformin (500 mg twice daily) against placebo in children with biopsy-proven NAFLD, resolution of NASH was significantly greater in children treated with vitamin E than in children treated with placebo (58% vs. 28%, P less than .01). Metformin did not significantly improve the NASH resolution rates, compared with placebo (41% vs. 28%, P = .23). Vitamin E could be recommended for nondiabetic adults or children if lifestyle modifications do not produce the expected results as a result of noncompliance or ineffectiveness. Since continued use of vitamin E has been suggested to be associated with a very small increase in the risk for prostate cancer (an absolute increase of 1.6 per 1,000 person-years of vitamin E use) in men, risks and benefits should be discussed with each patient before starting therapy. A meta-analysis of nine placebo-controlled trials including roughly 119,000 patients reported that vitamin E supplementation increases the risk of hemorrhagic stroke by 20% while reducing ischemic stroke by 10%. It was estimated that vitamin E supplementation would prevent one ischemic stroke per 476 treated patients while inducing one hemorrhagic stroke for every 1,250 patients. It is noteworthy that the combination of vitamin E with anticoagulant and/or antiplatelet therapy was not examined in this trial, so we could not determine how combination therapy might affect the risk of ischemic or hemorrhagic stroke.45
Thiazolidinediones drugs have been reported to be effective in improving NAFLD in many human studies. Evidence from RCTs suggests that pioglitazone could significantly improve glucose metabolism, alanine aminotransferase, and liver histology – such as hepatic steatosis, lobular inflammation, and ballooning degeneration – among patients with or without T2D. However, the beneficial effects on improving fibrosis remain to be verified.1,46 Because of safety concerns, the risk/benefit balance of using pioglitazone to treat NASH should be discussed with each patient.47-48 Pioglitazone has been associated with long-term risk of bladder cancer,49 congestive heart failure,50 and bone fractures.51 Data from the Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis (PIVENS) trial showed that pioglitazone was significantly associated with weight gain but with no other serious adverse events. However, this study was not powered to test any safety-related hypotheses.44
Glucagon-like peptide 1 analogs have been reported to induce weight loss and reduce insulin resistance, which may lead to improvements in NAFLD. Phase II RCTs of glucagon-like peptide 1 receptor agonists (liraglutide and semaglutide) for the treatment of biopsy-proven NASH showed significant improvements in serum liver enzymes, steatosis, and inflammation, as well as NASH resolution without worsening liver fibrosis, although no direct benefit was observed in reversing fibrosis.52-53 One of these studies explores the efficacy and safety of different doses of daily subcutaneous semaglutide vs. placebo on the rates of resolution of NASH with no worsening of fibrosis. The highest dose (0.4 mg) showed the greatest difference (59% vs. 17%, P less than .01), compared with the placebo arm. However, there was no difference in improvement in fibrosis stage between the two groups (43% in the 0.4-mg group vs. 33% in the placebo group, P = .48).53 Gastrointestinal adverse events were common in the semaglutide arm.
“Spontaneous” NASH resolution and fibrosis improvement are commonly seen in participants assigned to placebo arms in clinical trials. A recent meta-analysis of 43 RCTs including 2649 placebo-treated patients showed a pooled estimate of NASH resolution without worsening of fibrosis and 1 stage reduction or more in fibrosis of 12% and 19%, respectively. Relevant factors involved in “spontaneous” NASH improvement are unknown but could be related to changes in BMI resulting from lifestyle changes, race and ethnicity, age, and, likely, NAFLD-related genetic variations, although more data is needed to better understand the histologic response in placebo-treated patients.54
Semaglutide injections (2.4 mg once weekly) or (2.0 mg once weekly) have been recently approved by the FDA for chronic weight management in adults with obesity or overweight with at least one weight-related condition or glucose control of T2D, respectively. Of note, the semaglutide dose used in the NASH trial is not currently available for the treatment of patients who are overweight/obese or have T2D, but the beneficial effects on body weight reductions and glucose control are similar overall to the effects seen with currently available doses for management of obesity or diabetes. One may consider using semaglutide in patients who are overweight/obese or have T2D with NASH, but in the senior author’s experience, it has been quite challenging to receive the payer’s approval, as its use is not specifically approved to treat liver disease.1
How to follow patients with NAFLD in the clinic
Once a diagnosis of NAFLD is made, the use of noninvasive testing may aid to identify which patients are at high risk of fibrosis. Easy to use clinical tools, such as the NAFLD Fibrosis Score and the Fib-4 index, and liver stiffness measurements using vibration-controlled transient elastography (FibroScan) or magnetic resonance elastography (MRE) are clinically useful noninvasive tools for identifying patients with NAFLD who have a higher likelihood of progressing to advanced fibrosis.1,55 The use of either NAFLD Fibrosis Score (less than -1.455) or Fib-4 index (less than 1.30) low cutoffs may be particularly useful to rule out advanced fibrosis. People with a NAFLD Fibrosis Score (greater than –1.455) or Fib-4 index (greater than 1.30) should undergo liver stiffness measurement (LSM) via FibroScan. Those with an LSM of 8 kPa or higher should be referred to specialized care, where a decision to perform a liver biopsy and initiate monitoring and therapy will be taken. MRE is the most accurate noninvasive method for the estimation of liver fibrosis. When MRE is available, it can be a diagnostic alternative to accurately rule in and rule out patients with advanced fibrosis. This technique can be preferred in clinical trials, but it is rarely used in clinical practice because it is expensive and not easily available. Reassessment by noninvasive scores at 1-3 years’ follow-up will be considered for those with an LSM less than 8 kPa. Patients with NASH cirrhosis should be screened for both gastroesophageal varix and HCC according to the American Association for the Study of Liver Diseases guidelines.56-57
Dr. Vilar-Gomez is assistant professor in the division of gastroenterology and hepatology at Indiana University, Indianapolis. Dr. Chalasani is vice president for academic affairs at Indiana University Health, Indianapolis, and the David W. Crabb Professor of Gastroenterology and Hepatology and an adjunct professor of anatomy, cell biology, and physiology in the division of gastroenterology and hepatology at Indiana University. Dr. Vilar-Gomez reports no financial conflicts of interest. Dr. Chalasani serves as a paid consultant to AbbVie, Boehringer-Ingelheim, Altimmune, Madrigal, Lilly, Zydus, and Galectin. He receives research support from Galectin and DSM.
References
1. Chalasani N et al. Hepatology 2018;67:328-57.
2. Söderberg C et al. Hepatology 2010;51:595-602.
3. Sanyal AJ et al. N Engl J Med 2021;385:1559-69.
4. Vilar-Gomez E et al. Gastroenterology 2018;155:443-57.e17.
5. Younossi ZM et al. Hepatology 2016;64:73-84.
6. EASL-EASD-EASO. J Hepatol 2016;64:1388-402.
7. Wong VW et al. J Hepatol 2018; 69:1349-56.
8. Vilar-Gomez E et al. Gastroenterology 2015;149:367-78.e5; quiz e14-5.
9. Promrat K et al. Hepatology 2010;51:121-9.
10. Wong VW et al. J Hepatol 2013;59:536-42.
11. Berzigotti A et al. Hepatology 2017;65:1293-1305.
12. Sacks FM et al. N Engl J Med 2009;360:859-73.
13. Vilar-Gomez E et al. Hepatology 2022 Jun;75(6):1491-1506.
14. Zelber-Sagi S et al. Liver Int 2017;37:936-49.
15. Hassani Zadeh S et al. J Gastroenterol Hepatol 2021;36:1470-8.
16. Yaskolka Meir A et al. Gut 2021;70:2085-95.
17. Sung KC et al. J Hepatol 2016;65:791-7.
18. Orci LA et al. Clin Gastroenterol Hepatol 2016;14:1398-411.
19. Ryu S et al. J Hepatol 2015;63:1229-37.
20. Kim D et al. Hepatology 2020;72:1556-68.
21. Kim D et al. Clin Gastroenterol Hepatol 2021;19:1240-7.e5.
22. Ascha MS et al. Hepatology 2010;51:1972-8.
23. Bambha K et al. Liver Int 2014;34:1250-8.
24. Lee Y et al. Clin Gastroenterol Hepatol 2019;17:1040-60.e11.
25. Grönroos S et al. JAMA Surg 2021;156:137-46.
26. Fakhry TK et al. Surg Obes Relat Dis 2019;15:502-11.
27. Seeberg KA et al. Ann Intern Med 2022;175:74-83.
28. Bower G et al. Obes Surg 2015;25:2280-9.
29. Jan A et al. Obes Surg 2015;25:1518-26.
30. Hanipah ZN et al. Obes Surg 2018;28:3431-8.
31. Are VS et al. Am J Gastroenterol 2020;115:1849-56.
32. Aminian A et al. JAMA 2021;326:2031-42.
33. Vuppalanchi R et al. Ann Surg 2022;275:e174-80.
34. Simon TG et al. Gut 2021. doi: 10.1136/gutjnl-2021-325724.
35. Lonardo A et al. J Hepatol 2018;68:335-52.
36. Chalasani N et al. Gastroenterology 2004;126:1287-92.
37. Pastori D et al. Dig Liver Dis 2015;47:4-11.
38. Kim RG et al. Clin Gastroenterol Hepatol 2017;15:1521-30.e8.
39. Ahmad J et al. Dig Dis Sci 2017;62:3596-604.
40. Chalasani NP et al. Am J Gastroenterol 2021;116(5):878-98.
41. Rinella ME et al. Hepatology 2019;70:1424-36.
42. Younossi ZM et al. Lancet 2019;394:2184-96.
43. Ratziu V. Clin Liver Dis (Hoboken) 2021;17:398-400.
44. Sanyal AJ et al. N Engl J Med 2010;341:1675-85.
45. Schürks M et al. BMJ 2010;341:c5702.
46. Cusi K et al. Ann Intern Med 2016;165:305-15.
47. Lewis JD et al. JAMA 2015;314:265-77.
48. Billington EO et al. Diabetologia 2015;58:2238-46.
49. Lewis JD et al. Diabetes Care 2011;34:916-22.
50. Erdmann E et al. Diabetes Care 2007;30:2773-8.
51. Viscoli CM et al. J Clin Endocrinol Metab 2017;102:914-22.
52. Armstong MJ et al. Lancet 2016;387:679-90.
53. Newsome PN et al. N Engl J Med 2021;384:1113-24.
54. Ng CH et al. Hepatology 2022;75:1647-61.
55. Kanwal F et al. Gastroenterology 2021;161:1030-1042.e8.
56. Garcia-Tsao G et al. Hepatology 2017;65:310-35.
57. Heimbach JK et al. Hepatology 2018;67:358-80.
Management of gastroparesis in 2022
Introduction
Patients presenting with the symptoms of gastroparesis (Gp) are commonly seen in gastroenterology practice.
Presentation
Patients with foregut symptoms of Gp have characteristic presentations, with nausea, vomiting/retching, and abdominal pain often associated with bloating and distension, early satiety, anorexia, and heartburn. Mid- and hindgut gastrointestinal and/or urinary symptoms may be seen in patients with Gp as well.
The precise epidemiology of gastroparesis syndromes (GpS) is unknown. Classic gastroparesis, defined as delayed gastric emptying without known mechanical obstruction, has a prevalence of about 10 per 100,000 population in men and 30 per 100,000 in women with women being affected 3 to 4 times more than men.1,2 Some risk factors for GpS, such as diabetes mellitus (DM) in up to 5% of patients with Type 1 DM, are known.3 Caucasians have the highest prevalence of GpS, followed by African Americans.4,5

The classic definition of Gp has blurred with the realization that patients may have symptoms of Gp without delayed solid gastric emptying. Some patients have been described as having chronic unexplained nausea and vomiting or gastroparesis like syndrome.6 More recently the NIH Gastroparesis Consortium has proposed that disorders like functional dyspepsia may be a spectrum of the two disorders and classic Gp.7 Using this broadened definition, the number of patients with Gp symptoms is much greater, found in 10% or more of the U.S. population.8 For this discussion, GpS is used to encompass this spectrum of disorders.
The etiology of GpS is often unknown for a given patient, but clues to etiology exist in what is known about pathophysiology. Types of Gp are described as being idiopathic, diabetic, or postsurgical, each of which may have varying pathophysiology. Many patients with mild-to-moderate GpS symptoms are effectively treated with out-patient therapies; other patients may be refractory to available treatments. Refractory GpS patients have a high burden of illness affecting them, their families, providers, hospitals, and payers.
Pathophysiology
Specific types of gastroparesis syndromes have variable pathophysiology (Figure 1). In some cases, like GpS associated with DM, pathophysiology is partially related to diabetic autonomic dysfunction. GpS are multifactorial, however, and rather than focusing on subtypes, this discussion focuses on shared pathophysiology. Understanding pathophysiology is key to determining treatment options and potential future targets for therapy.
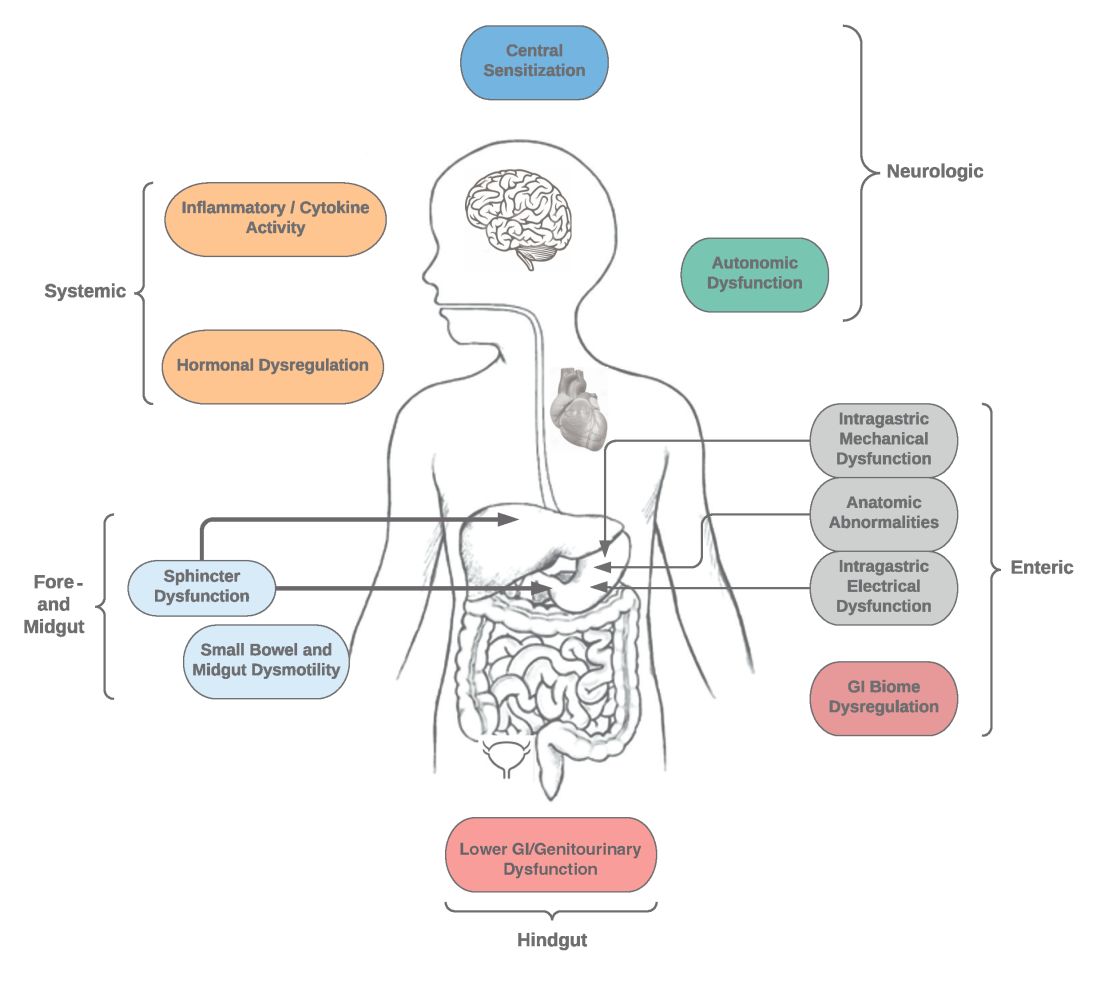
Intragastric mechanical dysfunction, both proximal (fundic relaxation and accommodation and/or lack of fundic contractility) and distal stomach (antral hypomotility) may be involved. Additionally, intragastric electrical disturbances in frequency, amplitude, and propagation of gastric electrical waves can be seen with low/high resolution gastric mapping.
Both gastroesophageal and gastropyloric sphincter dysfunction may be seen. Esophageal dysfunction is frequently seen but is not always categorized in GpS. Pyloric dysfunction is increasingly a focus of both diagnosis and therapy. GI anatomic abnormalities can be identified with gastric biopsies of full thickness muscle and mucosa. CD117/interstitial cells of Cajal, neural fibers, inflammatory and other cells can be evaluated by light microscopy, electron microscopy, and special staining techniques.
Small bowel, mid-, and hindgut dysmotility involvement has often been associated with pathologies of intragastric motility. Not only GI but genitourinary dysfunction may be associated with fore- and mid-gut dysfunction in GpS. Equally well described are abnormalities of the autonomic and sensory nervous system, which have recently been better quantified. Serologic measures, such as channelopathies and other antibody mediated abnormalities, have been recently noted.
Suspected for many years, immune dysregulation has now been documented in patients with GpS. Further investigation, including genetic dysregulation of immune measures, is ongoing. Other mechanisms include systemic and local inflammation, hormonal abnormalities, macro- and micronutrient deficiencies, dysregulation in GI microbiome, and physical frailty. The above factors may play a role in the pathophysiology of GpS, and it is likely that many of these are involved with a given patient presenting for care.9
Diagnosis of GpS
Diagnosis of GpS is often delayed and can be challenging; various tools have been developed, but not all are used. A diagnostic approach for patients with symptoms of Gp is listed below, and Figure 2 details a diagnostic approach and treatment options for symptomatic patients.
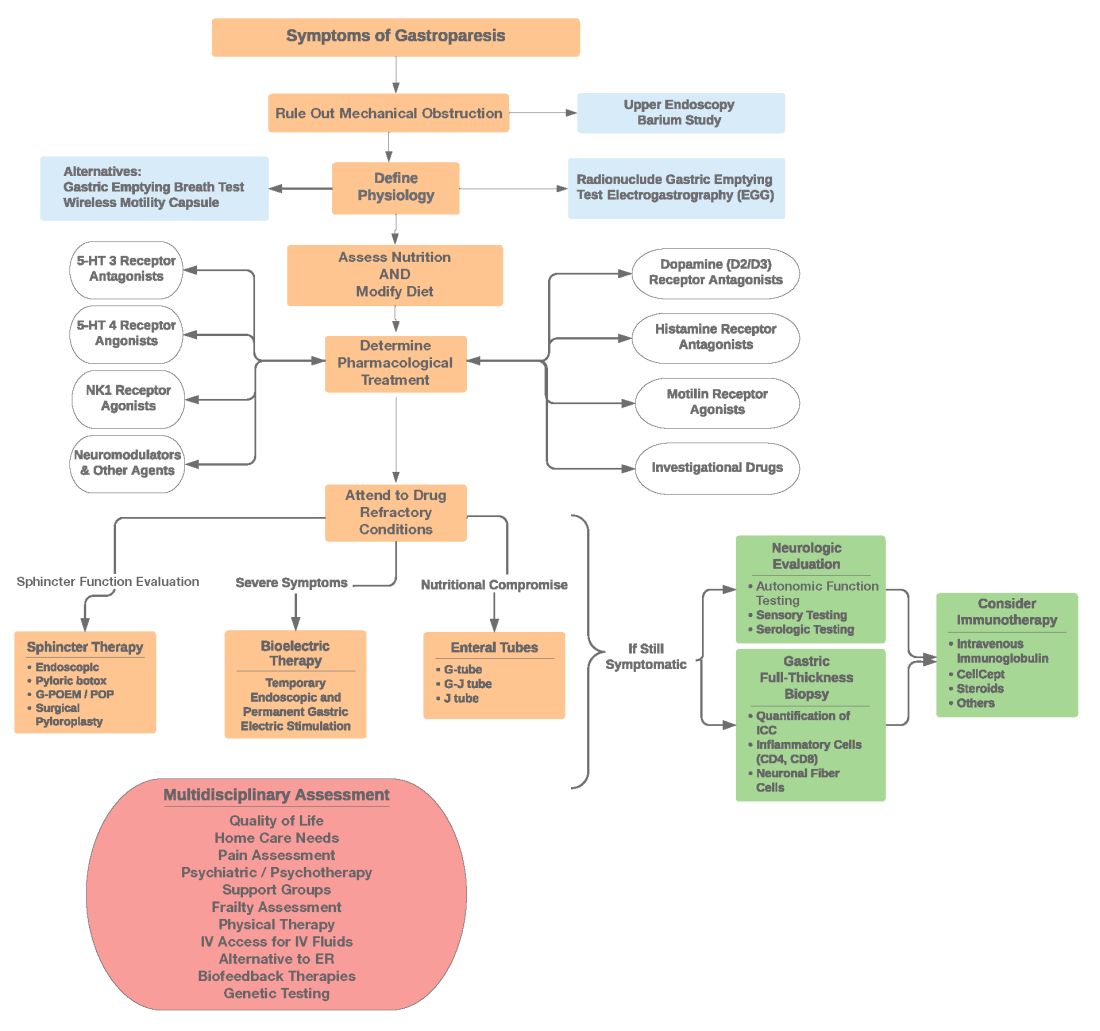
Symptom Assessment: Initially Gp symptoms can be assessed using Food and Drug Administration–approved patient-reported outcomes, including frequency and severity of nausea, vomiting, anorexia/early satiety, bloating/distention, and abdominal pain on a 0-4, 0-5 or 0-10 scale. The Gastrointestinal Cardinal Symptom Index or visual analog scales can also be used. It is also important to evaluate midgut and hindgut symptoms.9-11
Mechanical obstruction assessment: Mechanical obstruction can be ruled out using upper endoscopy or barium studies.
Physiologic testing: The most common is radionuclide gastric emptying testing (GET). Compliance with guidelines, standardization, and consistency of GETs is vital to help with an accurate diagnosis. Currently, two consensus recommendations for the standardized performance of GETs exist.12,13 Breath testing is FDA approved in the United States and can be used as an alternative. Wireless motility capsule testing can be complimentary.
Gastric dysrhythmias assessment: Assessment of gastric dysrhythmias can be performed in outpatient settings using cutaneous electrogastrogram, currently available in many referral centers. Most patients with GpS have an underlying gastric electrical abnormality.14,15
Sphincter dysfunction assessment: Both proximal and distal sphincter abnormalities have been described for many years and are of particular interest recently. Use of the functional luminal imaging probe (FLIP) shows patients with GpS may have decreased sphincter distensibility when examining the comparisons of the cross-sectional area relative to pressure Using this information, sphincter therapies can be offered.16-18
Other testing: Neurologic and autonomic testing, along with psychosocial, genetic and frailty assessments, are helpful to explore.19 Nutritional evaluation can be done using standardized scales, such as subjective global assessment and serologic testing for micronutrient deficiency or electrical impedance.20
Treatment of GpS
Therapies for GpS can be viewed as the five D’s: Diet, Drug, Disruption, Devices, and Details.
Diet and nutrition: The mainstay treatment of GpS remains dietary modification. The most common recommendation is to limit meal size, often with increased meal frequency, as well as nutrient composition, in areas that may retard gastric emptying. In addition, some patients with GpS report intolerances of specific foods, such as specific carbohydrates. Nutritional consultation can assist patients with meals tailored for their current nutritional needs. Nutritional supplementation is widely used for patients with GpS.20
Pharmacological treatment: The next tier of treatment for GpS is drugs. Review of a patient’s medications is important to minimize drugs that may retard gastric emptying such as opiates and GLP-1 agonists. A full discussion of medications is beyond the scope of this article, but classes of drugs available include: prokinetics, antiemetics, neuromodulators, and investigational agents.

There is only one approved prokinetic medication for gastroparesis – the dopamine blocker metoclopramide – and most providers are aware of metoclopramide’s limitations in terms of potential side effects, such as the risk of tardive dyskinesia and labeling on duration of therapy, with a maximum of 12 weeks recommended. Alternative prokinetics, such as domperidone, are not easily available in the United States; some mediations approved for other indications, such as the 5-HT drug prucalopride, are sometimes given for GpS off-label. Antiemetics such as promethazine and ondansetron are frequently used for symptomatic control in GpS. Despite lack of positive controlled trials in Gp, neuromodulator drugs, such as tricyclic or tetracyclic antidepressants like amitriptyline or mirtazapine are often used; their efficacy is more proven in the functional dyspepsia area. Other drugs such as the NK-1 drug aprepitant have been studied in Gp and are sometimes used off-label. Drugs such as scopolamine and related compounds can also provide symptomatic relief, as can the tetrahydrocannabinol-containing drug, dronabinol. New pharmacologic agents for GpS include investigational drugs such as ghrelin agonists and several novel compounds, none of which are currently FDA approved.21,22
Fortunately, the majority of patients with GpS respond to conservative therapies, such as dietary changes and/or medications. The last part of the section on treatment of GpS includes patients that are diet and drug refractory. Patients in this group are often referred to gastroenterologists and can be complex, time consuming, and frustrating to provide care for. Many of these patients are eventually seen in referral centers, and some travel great distances and have considerable medical expenses.
Pylorus-directed therapies: The recent renewed interest in pyloric dysfunction in patients with Gp symptoms has led to a great deal of clinical activity. Gastropyloric dysfunction in Gp has been documented for decades, originally in diabetic patients with autonomic and enteric neuropathy. The use of botulinum toxin in upper- and lower-gastric sphincters has led to continuing use of this therapy for patients with GpS. Despite initial negative controlled trials of botulinum toxin in the pyloric sphincter, newer studies indicate that physiologic measures, such as the FLIP, may help with patient selection. Other disruptive pyloric therapies, including pyloromyotomy, per oral pyloromyotomy, and gastric peroral endoscopic myotomy, are supported by open-label use, despite a lack of published positive controlled trials.17
Bioelectric therapy: Another approach for patients with symptomatic drug refractory GpS is bioelectric device therapies, which can be delivered several ways, including directly to the stomach or to the spinal cord or the vagus nerve in the neck or ear, as well as by electro-acupuncture. High-frequency, low-energy gastric electrical stimulation (GES) is the best studied. First done in 1992 as an experimental therapy, GES was investigational from 1995 to 2000, when it became FDA approved as a humanitarian-use device. GES has been used in over 10,000 patients worldwide; only a small number (greater than 700 study patients) have been in controlled trials. Nine controlled trials of GES have been primarily positive, and durability for over 10 years has been shown. Temporary GES can also be performed endoscopically, although that is an off-label procedure. It has been shown to predict long-term therapy outcome.23-26
Nutritional support: Nutritional abnormalities in some cases of GpS lead to consideration of enteral tubes, starting with a trial of feeding with an N-J tube placed endoscopically. An N-J trial is most often performed in patients who have macro-malnutrition and weight loss but can be considered for other highly symptomatic patients. Other endoscopic tubes can be PEG or PEG-J or direct PEJ tubes. Some patients may require surgical placement of enteral tubes, presenting an opportunity for a small bowel or gastric full-thickness biopsy. Enteral tubes are sometimes used for decompression in highly symptomatic patients.27
For patients presenting with neurological symptoms, findings and serologic abnormalities have led to interest in immunotherapies. One is intravenous immunoglobulin, given parenterally. Several open-label studies have been published, the most recent one with 47 patients showing better response if glutamic acid decarboxylase–65 antibodies were present and with longer therapeutic dosing.28 Drawbacks to immunotherapies like intravenous immunoglobulin are cost and requiring parenteral access.
Other evaluation/treatments for drug refractory patients can be detailed as follows: First, an overall quality of life assessment can be helpful, especially one that includes impact of GpS on the patients and family. Nutritional considerations, which may not have been fully assessed, can be examined in more detail. Frailty assessments may show the need for physical therapy. Assessment for home care needs may indicate, in severe patients, needs for IV fluids at home, either enteral or parenteral, if nutrition is not adequate. Psychosocial and/or psychiatric assessments may lead to the need for medications, psychotherapy, and/or support groups. Lastly, an assessment of overall health status may lead to approaches for minimizing visits to emergency rooms and hospitalizations.29,30
Conclusion
Patients with Gp symptoms are becoming increasingly recognized and referred to gastroenterologists. Better understandings of the pathophysiology of the spectrum of gastroparesis syndromes, assisted by innovations in diagnosis, have led to expansion of existing and new therapeutic approaches. Fortunately, most patients can benefit from a standardized diagnostic approach and directed noninvasive therapies. Patients with refractory gastroparesis symptoms, often with complex issues referred to gastroenterologists, remain a challenge, and novel approaches may improve their quality of life.
Dr. Mathur is a GI motility research fellow at the University of Louisville, Ky. He reports no conflicts of interest. Dr. Abell is the Arthur M. Schoen, MD, Chair in Gastroenterology at the University of Louisville. His main funding is NIH GpCRC and NIH Definitive Evaluation of Gastric Dysrhythmia. He is an investigator for Cindome, Vanda, Allergan, and Neurogastrx; a consultant for Censa, Nuvaira, and Takeda; a speaker for Takeda and Medtronic; and a reviewer for UpToDate. He is also the founder of ADEPT-GI, which holds IP related to mucosal stimulation and autonomic and enteric profiling.
References
1. Jung HK et al. Gastroenterology. 2009;136(4):1225-33.
2. Ye Y et al. Gut. 2021;70(4):644-53.
3. Oshima T et al. J Neurogastroenterol Motil. 2021;27(1):46-54.
4. Soykan I et al. Dig Dis Sci. 1998;43(11):2398-404.
5. Syed AR et al. J Clin Gastroenterol. 2020;54(1):50-4.
6.Pasricha PJ et al. Clin Gastroenterol Hepatol. 2011;9(7):567-76.e1-4.
7. Pasricha PJ et al. Gastroenterology. 2021;160(6):2006-17.
8. Almario CV et al. Am J Gastroenterol. 2018;113(11):1701-10.
9. Abell TL et al. Dig Dis Sci. 2021 Apr;66(4):1127-41.
10. Abell TL et al. Neurogastroenterol Motil. 2019;31(3):e13534.
11. Elmasry M et al. Neurogastroenterol Motil. 2021 Oct 26;e14274.
12. Maurer AH et al. J Nucl Med. 2020;61(3):11N-7N.
13. Abell TL et al. J Nucl Med Technol. 2008 Mar;36(1):44-54.
14. Shine A et al. Neuromodulation. 2022 Feb 16;S1094-7159(21)06986-5.
15. O’Grady G et al. Am J Physiol Gastrointest Liver Physiol. 2021;321(5):G527-g42.
16. Saadi M et al. Rev Gastroenterol Mex (Engl Ed). Oct-Dec 2018;83(4):375-84.
17. Kamal F et al. Aliment Pharmacol Ther. 2022;55(2):168-77.
18. Harberson J et al. Dig Dis Sci. 2010;55(2):359-70.
19. Winston J. Gastrointestinal Disorders. 2021;3(2):78-83.
20. Parkman HP et al. Gastroenterology. 2011;141(2):486-98, 98.e1-7.
21. Heckroth M et al. J Clin Gastroenterol. 2021;55(4):279-99.
22. Camilleri M. Clin Gastroenterol Hepatol. 2022;20(1):19-24.
23. Payne SC et al. Nat Rev Gastroenterol Hepatol. 2019;16(2):89-105.
24. Ducrotte P et al. Gastroenterology. 2020;158(3):506-14.e2.
25. Burlen J et al. Gastroenterology Res. 2018;11(5):349-54.
26. Hedjoudje A et al. Neurogastroenterol Motil. 2020;32(11):e13949.
27. Petrov RV et al. Gastroenterol Clin North Am. 2020;49(3):539-56.
28. Gala K et al. J Clin Gastroenterol. 2021 Dec 31. doi: 10.1097/MCG.0000000000001655.
29. Abell TL et al. Neurogastroenterol Motil. 2006;18(4):263-83.
30. Camilleri M et al. Am J Gastroenterol. 2013;108(1):18-37.
Introduction
Patients presenting with the symptoms of gastroparesis (Gp) are commonly seen in gastroenterology practice.
Presentation
Patients with foregut symptoms of Gp have characteristic presentations, with nausea, vomiting/retching, and abdominal pain often associated with bloating and distension, early satiety, anorexia, and heartburn. Mid- and hindgut gastrointestinal and/or urinary symptoms may be seen in patients with Gp as well.
The precise epidemiology of gastroparesis syndromes (GpS) is unknown. Classic gastroparesis, defined as delayed gastric emptying without known mechanical obstruction, has a prevalence of about 10 per 100,000 population in men and 30 per 100,000 in women with women being affected 3 to 4 times more than men.1,2 Some risk factors for GpS, such as diabetes mellitus (DM) in up to 5% of patients with Type 1 DM, are known.3 Caucasians have the highest prevalence of GpS, followed by African Americans.4,5
The classic definition of Gp has blurred with the realization that patients may have symptoms of Gp without delayed solid gastric emptying. Some patients have been described as having chronic unexplained nausea and vomiting or gastroparesis like syndrome.6 More recently the NIH Gastroparesis Consortium has proposed that disorders like functional dyspepsia may be a spectrum of the two disorders and classic Gp.7 Using this broadened definition, the number of patients with Gp symptoms is much greater, found in 10% or more of the U.S. population.8 For this discussion, GpS is used to encompass this spectrum of disorders.
The etiology of GpS is often unknown for a given patient, but clues to etiology exist in what is known about pathophysiology. Types of Gp are described as being idiopathic, diabetic, or postsurgical, each of which may have varying pathophysiology. Many patients with mild-to-moderate GpS symptoms are effectively treated with out-patient therapies; other patients may be refractory to available treatments. Refractory GpS patients have a high burden of illness affecting them, their families, providers, hospitals, and payers.
Pathophysiology
Specific types of gastroparesis syndromes have variable pathophysiology (Figure 1). In some cases, like GpS associated with DM, pathophysiology is partially related to diabetic autonomic dysfunction. GpS are multifactorial, however, and rather than focusing on subtypes, this discussion focuses on shared pathophysiology. Understanding pathophysiology is key to determining treatment options and potential future targets for therapy.
Intragastric mechanical dysfunction, both proximal (fundic relaxation and accommodation and/or lack of fundic contractility) and distal stomach (antral hypomotility) may be involved. Additionally, intragastric electrical disturbances in frequency, amplitude, and propagation of gastric electrical waves can be seen with low/high resolution gastric mapping.
Both gastroesophageal and gastropyloric sphincter dysfunction may be seen. Esophageal dysfunction is frequently seen but is not always categorized in GpS. Pyloric dysfunction is increasingly a focus of both diagnosis and therapy. GI anatomic abnormalities can be identified with gastric biopsies of full thickness muscle and mucosa. CD117/interstitial cells of Cajal, neural fibers, inflammatory and other cells can be evaluated by light microscopy, electron microscopy, and special staining techniques.
Small bowel, mid-, and hindgut dysmotility involvement has often been associated with pathologies of intragastric motility. Not only GI but genitourinary dysfunction may be associated with fore- and mid-gut dysfunction in GpS. Equally well described are abnormalities of the autonomic and sensory nervous system, which have recently been better quantified. Serologic measures, such as channelopathies and other antibody mediated abnormalities, have been recently noted.
Suspected for many years, immune dysregulation has now been documented in patients with GpS. Further investigation, including genetic dysregulation of immune measures, is ongoing. Other mechanisms include systemic and local inflammation, hormonal abnormalities, macro- and micronutrient deficiencies, dysregulation in GI microbiome, and physical frailty. The above factors may play a role in the pathophysiology of GpS, and it is likely that many of these are involved with a given patient presenting for care.9
Diagnosis of GpS
Diagnosis of GpS is often delayed and can be challenging; various tools have been developed, but not all are used. A diagnostic approach for patients with symptoms of Gp is listed below, and Figure 2 details a diagnostic approach and treatment options for symptomatic patients.
Symptom Assessment: Initially Gp symptoms can be assessed using Food and Drug Administration–approved patient-reported outcomes, including frequency and severity of nausea, vomiting, anorexia/early satiety, bloating/distention, and abdominal pain on a 0-4, 0-5 or 0-10 scale. The Gastrointestinal Cardinal Symptom Index or visual analog scales can also be used. It is also important to evaluate midgut and hindgut symptoms.9-11
Mechanical obstruction assessment: Mechanical obstruction can be ruled out using upper endoscopy or barium studies.
Physiologic testing: The most common is radionuclide gastric emptying testing (GET). Compliance with guidelines, standardization, and consistency of GETs is vital to help with an accurate diagnosis. Currently, two consensus recommendations for the standardized performance of GETs exist.12,13 Breath testing is FDA approved in the United States and can be used as an alternative. Wireless motility capsule testing can be complimentary.
Gastric dysrhythmias assessment: Assessment of gastric dysrhythmias can be performed in outpatient settings using cutaneous electrogastrogram, currently available in many referral centers. Most patients with GpS have an underlying gastric electrical abnormality.14,15
Sphincter dysfunction assessment: Both proximal and distal sphincter abnormalities have been described for many years and are of particular interest recently. Use of the functional luminal imaging probe (FLIP) shows patients with GpS may have decreased sphincter distensibility when examining the comparisons of the cross-sectional area relative to pressure Using this information, sphincter therapies can be offered.16-18
Other testing: Neurologic and autonomic testing, along with psychosocial, genetic and frailty assessments, are helpful to explore.19 Nutritional evaluation can be done using standardized scales, such as subjective global assessment and serologic testing for micronutrient deficiency or electrical impedance.20
Treatment of GpS
Therapies for GpS can be viewed as the five D’s: Diet, Drug, Disruption, Devices, and Details.
Diet and nutrition: The mainstay treatment of GpS remains dietary modification. The most common recommendation is to limit meal size, often with increased meal frequency, as well as nutrient composition, in areas that may retard gastric emptying. In addition, some patients with GpS report intolerances of specific foods, such as specific carbohydrates. Nutritional consultation can assist patients with meals tailored for their current nutritional needs. Nutritional supplementation is widely used for patients with GpS.20
Pharmacological treatment: The next tier of treatment for GpS is drugs. Review of a patient’s medications is important to minimize drugs that may retard gastric emptying such as opiates and GLP-1 agonists. A full discussion of medications is beyond the scope of this article, but classes of drugs available include: prokinetics, antiemetics, neuromodulators, and investigational agents.
There is only one approved prokinetic medication for gastroparesis – the dopamine blocker metoclopramide – and most providers are aware of metoclopramide’s limitations in terms of potential side effects, such as the risk of tardive dyskinesia and labeling on duration of therapy, with a maximum of 12 weeks recommended. Alternative prokinetics, such as domperidone, are not easily available in the United States; some mediations approved for other indications, such as the 5-HT drug prucalopride, are sometimes given for GpS off-label. Antiemetics such as promethazine and ondansetron are frequently used for symptomatic control in GpS. Despite lack of positive controlled trials in Gp, neuromodulator drugs, such as tricyclic or tetracyclic antidepressants like amitriptyline or mirtazapine are often used; their efficacy is more proven in the functional dyspepsia area. Other drugs such as the NK-1 drug aprepitant have been studied in Gp and are sometimes used off-label. Drugs such as scopolamine and related compounds can also provide symptomatic relief, as can the tetrahydrocannabinol-containing drug, dronabinol. New pharmacologic agents for GpS include investigational drugs such as ghrelin agonists and several novel compounds, none of which are currently FDA approved.21,22
Fortunately, the majority of patients with GpS respond to conservative therapies, such as dietary changes and/or medications. The last part of the section on treatment of GpS includes patients that are diet and drug refractory. Patients in this group are often referred to gastroenterologists and can be complex, time consuming, and frustrating to provide care for. Many of these patients are eventually seen in referral centers, and some travel great distances and have considerable medical expenses.
Pylorus-directed therapies: The recent renewed interest in pyloric dysfunction in patients with Gp symptoms has led to a great deal of clinical activity. Gastropyloric dysfunction in Gp has been documented for decades, originally in diabetic patients with autonomic and enteric neuropathy. The use of botulinum toxin in upper- and lower-gastric sphincters has led to continuing use of this therapy for patients with GpS. Despite initial negative controlled trials of botulinum toxin in the pyloric sphincter, newer studies indicate that physiologic measures, such as the FLIP, may help with patient selection. Other disruptive pyloric therapies, including pyloromyotomy, per oral pyloromyotomy, and gastric peroral endoscopic myotomy, are supported by open-label use, despite a lack of published positive controlled trials.17
Bioelectric therapy: Another approach for patients with symptomatic drug refractory GpS is bioelectric device therapies, which can be delivered several ways, including directly to the stomach or to the spinal cord or the vagus nerve in the neck or ear, as well as by electro-acupuncture. High-frequency, low-energy gastric electrical stimulation (GES) is the best studied. First done in 1992 as an experimental therapy, GES was investigational from 1995 to 2000, when it became FDA approved as a humanitarian-use device. GES has been used in over 10,000 patients worldwide; only a small number (greater than 700 study patients) have been in controlled trials. Nine controlled trials of GES have been primarily positive, and durability for over 10 years has been shown. Temporary GES can also be performed endoscopically, although that is an off-label procedure. It has been shown to predict long-term therapy outcome.23-26
Nutritional support: Nutritional abnormalities in some cases of GpS lead to consideration of enteral tubes, starting with a trial of feeding with an N-J tube placed endoscopically. An N-J trial is most often performed in patients who have macro-malnutrition and weight loss but can be considered for other highly symptomatic patients. Other endoscopic tubes can be PEG or PEG-J or direct PEJ tubes. Some patients may require surgical placement of enteral tubes, presenting an opportunity for a small bowel or gastric full-thickness biopsy. Enteral tubes are sometimes used for decompression in highly symptomatic patients.27
For patients presenting with neurological symptoms, findings and serologic abnormalities have led to interest in immunotherapies. One is intravenous immunoglobulin, given parenterally. Several open-label studies have been published, the most recent one with 47 patients showing better response if glutamic acid decarboxylase–65 antibodies were present and with longer therapeutic dosing.28 Drawbacks to immunotherapies like intravenous immunoglobulin are cost and requiring parenteral access.
Other evaluation/treatments for drug refractory patients can be detailed as follows: First, an overall quality of life assessment can be helpful, especially one that includes impact of GpS on the patients and family. Nutritional considerations, which may not have been fully assessed, can be examined in more detail. Frailty assessments may show the need for physical therapy. Assessment for home care needs may indicate, in severe patients, needs for IV fluids at home, either enteral or parenteral, if nutrition is not adequate. Psychosocial and/or psychiatric assessments may lead to the need for medications, psychotherapy, and/or support groups. Lastly, an assessment of overall health status may lead to approaches for minimizing visits to emergency rooms and hospitalizations.29,30
Conclusion
Patients with Gp symptoms are becoming increasingly recognized and referred to gastroenterologists. Better understandings of the pathophysiology of the spectrum of gastroparesis syndromes, assisted by innovations in diagnosis, have led to expansion of existing and new therapeutic approaches. Fortunately, most patients can benefit from a standardized diagnostic approach and directed noninvasive therapies. Patients with refractory gastroparesis symptoms, often with complex issues referred to gastroenterologists, remain a challenge, and novel approaches may improve their quality of life.
Dr. Mathur is a GI motility research fellow at the University of Louisville, Ky. He reports no conflicts of interest. Dr. Abell is the Arthur M. Schoen, MD, Chair in Gastroenterology at the University of Louisville. His main funding is NIH GpCRC and NIH Definitive Evaluation of Gastric Dysrhythmia. He is an investigator for Cindome, Vanda, Allergan, and Neurogastrx; a consultant for Censa, Nuvaira, and Takeda; a speaker for Takeda and Medtronic; and a reviewer for UpToDate. He is also the founder of ADEPT-GI, which holds IP related to mucosal stimulation and autonomic and enteric profiling.
References
1. Jung HK et al. Gastroenterology. 2009;136(4):1225-33.
2. Ye Y et al. Gut. 2021;70(4):644-53.
3. Oshima T et al. J Neurogastroenterol Motil. 2021;27(1):46-54.
4. Soykan I et al. Dig Dis Sci. 1998;43(11):2398-404.
5. Syed AR et al. J Clin Gastroenterol. 2020;54(1):50-4.
6.Pasricha PJ et al. Clin Gastroenterol Hepatol. 2011;9(7):567-76.e1-4.
7. Pasricha PJ et al. Gastroenterology. 2021;160(6):2006-17.
8. Almario CV et al. Am J Gastroenterol. 2018;113(11):1701-10.
9. Abell TL et al. Dig Dis Sci. 2021 Apr;66(4):1127-41.
10. Abell TL et al. Neurogastroenterol Motil. 2019;31(3):e13534.
11. Elmasry M et al. Neurogastroenterol Motil. 2021 Oct 26;e14274.
12. Maurer AH et al. J Nucl Med. 2020;61(3):11N-7N.
13. Abell TL et al. J Nucl Med Technol. 2008 Mar;36(1):44-54.
14. Shine A et al. Neuromodulation. 2022 Feb 16;S1094-7159(21)06986-5.
15. O’Grady G et al. Am J Physiol Gastrointest Liver Physiol. 2021;321(5):G527-g42.
16. Saadi M et al. Rev Gastroenterol Mex (Engl Ed). Oct-Dec 2018;83(4):375-84.
17. Kamal F et al. Aliment Pharmacol Ther. 2022;55(2):168-77.
18. Harberson J et al. Dig Dis Sci. 2010;55(2):359-70.
19. Winston J. Gastrointestinal Disorders. 2021;3(2):78-83.
20. Parkman HP et al. Gastroenterology. 2011;141(2):486-98, 98.e1-7.
21. Heckroth M et al. J Clin Gastroenterol. 2021;55(4):279-99.
22. Camilleri M. Clin Gastroenterol Hepatol. 2022;20(1):19-24.
23. Payne SC et al. Nat Rev Gastroenterol Hepatol. 2019;16(2):89-105.
24. Ducrotte P et al. Gastroenterology. 2020;158(3):506-14.e2.
25. Burlen J et al. Gastroenterology Res. 2018;11(5):349-54.
26. Hedjoudje A et al. Neurogastroenterol Motil. 2020;32(11):e13949.
27. Petrov RV et al. Gastroenterol Clin North Am. 2020;49(3):539-56.
28. Gala K et al. J Clin Gastroenterol. 2021 Dec 31. doi: 10.1097/MCG.0000000000001655.
29. Abell TL et al. Neurogastroenterol Motil. 2006;18(4):263-83.
30. Camilleri M et al. Am J Gastroenterol. 2013;108(1):18-37.
Introduction
Patients presenting with the symptoms of gastroparesis (Gp) are commonly seen in gastroenterology practice.
Presentation
Patients with foregut symptoms of Gp have characteristic presentations, with nausea, vomiting/retching, and abdominal pain often associated with bloating and distension, early satiety, anorexia, and heartburn. Mid- and hindgut gastrointestinal and/or urinary symptoms may be seen in patients with Gp as well.
The precise epidemiology of gastroparesis syndromes (GpS) is unknown. Classic gastroparesis, defined as delayed gastric emptying without known mechanical obstruction, has a prevalence of about 10 per 100,000 population in men and 30 per 100,000 in women with women being affected 3 to 4 times more than men.1,2 Some risk factors for GpS, such as diabetes mellitus (DM) in up to 5% of patients with Type 1 DM, are known.3 Caucasians have the highest prevalence of GpS, followed by African Americans.4,5
The classic definition of Gp has blurred with the realization that patients may have symptoms of Gp without delayed solid gastric emptying. Some patients have been described as having chronic unexplained nausea and vomiting or gastroparesis like syndrome.6 More recently the NIH Gastroparesis Consortium has proposed that disorders like functional dyspepsia may be a spectrum of the two disorders and classic Gp.7 Using this broadened definition, the number of patients with Gp symptoms is much greater, found in 10% or more of the U.S. population.8 For this discussion, GpS is used to encompass this spectrum of disorders.
The etiology of GpS is often unknown for a given patient, but clues to etiology exist in what is known about pathophysiology. Types of Gp are described as being idiopathic, diabetic, or postsurgical, each of which may have varying pathophysiology. Many patients with mild-to-moderate GpS symptoms are effectively treated with out-patient therapies; other patients may be refractory to available treatments. Refractory GpS patients have a high burden of illness affecting them, their families, providers, hospitals, and payers.
Pathophysiology
Specific types of gastroparesis syndromes have variable pathophysiology (Figure 1). In some cases, like GpS associated with DM, pathophysiology is partially related to diabetic autonomic dysfunction. GpS are multifactorial, however, and rather than focusing on subtypes, this discussion focuses on shared pathophysiology. Understanding pathophysiology is key to determining treatment options and potential future targets for therapy.
Intragastric mechanical dysfunction, both proximal (fundic relaxation and accommodation and/or lack of fundic contractility) and distal stomach (antral hypomotility) may be involved. Additionally, intragastric electrical disturbances in frequency, amplitude, and propagation of gastric electrical waves can be seen with low/high resolution gastric mapping.
Both gastroesophageal and gastropyloric sphincter dysfunction may be seen. Esophageal dysfunction is frequently seen but is not always categorized in GpS. Pyloric dysfunction is increasingly a focus of both diagnosis and therapy. GI anatomic abnormalities can be identified with gastric biopsies of full thickness muscle and mucosa. CD117/interstitial cells of Cajal, neural fibers, inflammatory and other cells can be evaluated by light microscopy, electron microscopy, and special staining techniques.
Small bowel, mid-, and hindgut dysmotility involvement has often been associated with pathologies of intragastric motility. Not only GI but genitourinary dysfunction may be associated with fore- and mid-gut dysfunction in GpS. Equally well described are abnormalities of the autonomic and sensory nervous system, which have recently been better quantified. Serologic measures, such as channelopathies and other antibody mediated abnormalities, have been recently noted.
Suspected for many years, immune dysregulation has now been documented in patients with GpS. Further investigation, including genetic dysregulation of immune measures, is ongoing. Other mechanisms include systemic and local inflammation, hormonal abnormalities, macro- and micronutrient deficiencies, dysregulation in GI microbiome, and physical frailty. The above factors may play a role in the pathophysiology of GpS, and it is likely that many of these are involved with a given patient presenting for care.9
Diagnosis of GpS
Diagnosis of GpS is often delayed and can be challenging; various tools have been developed, but not all are used. A diagnostic approach for patients with symptoms of Gp is listed below, and Figure 2 details a diagnostic approach and treatment options for symptomatic patients.
Symptom Assessment: Initially Gp symptoms can be assessed using Food and Drug Administration–approved patient-reported outcomes, including frequency and severity of nausea, vomiting, anorexia/early satiety, bloating/distention, and abdominal pain on a 0-4, 0-5 or 0-10 scale. The Gastrointestinal Cardinal Symptom Index or visual analog scales can also be used. It is also important to evaluate midgut and hindgut symptoms.9-11
Mechanical obstruction assessment: Mechanical obstruction can be ruled out using upper endoscopy or barium studies.
Physiologic testing: The most common is radionuclide gastric emptying testing (GET). Compliance with guidelines, standardization, and consistency of GETs is vital to help with an accurate diagnosis. Currently, two consensus recommendations for the standardized performance of GETs exist.12,13 Breath testing is FDA approved in the United States and can be used as an alternative. Wireless motility capsule testing can be complimentary.
Gastric dysrhythmias assessment: Assessment of gastric dysrhythmias can be performed in outpatient settings using cutaneous electrogastrogram, currently available in many referral centers. Most patients with GpS have an underlying gastric electrical abnormality.14,15
Sphincter dysfunction assessment: Both proximal and distal sphincter abnormalities have been described for many years and are of particular interest recently. Use of the functional luminal imaging probe (FLIP) shows patients with GpS may have decreased sphincter distensibility when examining the comparisons of the cross-sectional area relative to pressure Using this information, sphincter therapies can be offered.16-18
Other testing: Neurologic and autonomic testing, along with psychosocial, genetic and frailty assessments, are helpful to explore.19 Nutritional evaluation can be done using standardized scales, such as subjective global assessment and serologic testing for micronutrient deficiency or electrical impedance.20
Treatment of GpS
Therapies for GpS can be viewed as the five D’s: Diet, Drug, Disruption, Devices, and Details.
Diet and nutrition: The mainstay treatment of GpS remains dietary modification. The most common recommendation is to limit meal size, often with increased meal frequency, as well as nutrient composition, in areas that may retard gastric emptying. In addition, some patients with GpS report intolerances of specific foods, such as specific carbohydrates. Nutritional consultation can assist patients with meals tailored for their current nutritional needs. Nutritional supplementation is widely used for patients with GpS.20
Pharmacological treatment: The next tier of treatment for GpS is drugs. Review of a patient’s medications is important to minimize drugs that may retard gastric emptying such as opiates and GLP-1 agonists. A full discussion of medications is beyond the scope of this article, but classes of drugs available include: prokinetics, antiemetics, neuromodulators, and investigational agents.
There is only one approved prokinetic medication for gastroparesis – the dopamine blocker metoclopramide – and most providers are aware of metoclopramide’s limitations in terms of potential side effects, such as the risk of tardive dyskinesia and labeling on duration of therapy, with a maximum of 12 weeks recommended. Alternative prokinetics, such as domperidone, are not easily available in the United States; some mediations approved for other indications, such as the 5-HT drug prucalopride, are sometimes given for GpS off-label. Antiemetics such as promethazine and ondansetron are frequently used for symptomatic control in GpS. Despite lack of positive controlled trials in Gp, neuromodulator drugs, such as tricyclic or tetracyclic antidepressants like amitriptyline or mirtazapine are often used; their efficacy is more proven in the functional dyspepsia area. Other drugs such as the NK-1 drug aprepitant have been studied in Gp and are sometimes used off-label. Drugs such as scopolamine and related compounds can also provide symptomatic relief, as can the tetrahydrocannabinol-containing drug, dronabinol. New pharmacologic agents for GpS include investigational drugs such as ghrelin agonists and several novel compounds, none of which are currently FDA approved.21,22
Fortunately, the majority of patients with GpS respond to conservative therapies, such as dietary changes and/or medications. The last part of the section on treatment of GpS includes patients that are diet and drug refractory. Patients in this group are often referred to gastroenterologists and can be complex, time consuming, and frustrating to provide care for. Many of these patients are eventually seen in referral centers, and some travel great distances and have considerable medical expenses.
Pylorus-directed therapies: The recent renewed interest in pyloric dysfunction in patients with Gp symptoms has led to a great deal of clinical activity. Gastropyloric dysfunction in Gp has been documented for decades, originally in diabetic patients with autonomic and enteric neuropathy. The use of botulinum toxin in upper- and lower-gastric sphincters has led to continuing use of this therapy for patients with GpS. Despite initial negative controlled trials of botulinum toxin in the pyloric sphincter, newer studies indicate that physiologic measures, such as the FLIP, may help with patient selection. Other disruptive pyloric therapies, including pyloromyotomy, per oral pyloromyotomy, and gastric peroral endoscopic myotomy, are supported by open-label use, despite a lack of published positive controlled trials.17
Bioelectric therapy: Another approach for patients with symptomatic drug refractory GpS is bioelectric device therapies, which can be delivered several ways, including directly to the stomach or to the spinal cord or the vagus nerve in the neck or ear, as well as by electro-acupuncture. High-frequency, low-energy gastric electrical stimulation (GES) is the best studied. First done in 1992 as an experimental therapy, GES was investigational from 1995 to 2000, when it became FDA approved as a humanitarian-use device. GES has been used in over 10,000 patients worldwide; only a small number (greater than 700 study patients) have been in controlled trials. Nine controlled trials of GES have been primarily positive, and durability for over 10 years has been shown. Temporary GES can also be performed endoscopically, although that is an off-label procedure. It has been shown to predict long-term therapy outcome.23-26
Nutritional support: Nutritional abnormalities in some cases of GpS lead to consideration of enteral tubes, starting with a trial of feeding with an N-J tube placed endoscopically. An N-J trial is most often performed in patients who have macro-malnutrition and weight loss but can be considered for other highly symptomatic patients. Other endoscopic tubes can be PEG or PEG-J or direct PEJ tubes. Some patients may require surgical placement of enteral tubes, presenting an opportunity for a small bowel or gastric full-thickness biopsy. Enteral tubes are sometimes used for decompression in highly symptomatic patients.27
For patients presenting with neurological symptoms, findings and serologic abnormalities have led to interest in immunotherapies. One is intravenous immunoglobulin, given parenterally. Several open-label studies have been published, the most recent one with 47 patients showing better response if glutamic acid decarboxylase–65 antibodies were present and with longer therapeutic dosing.28 Drawbacks to immunotherapies like intravenous immunoglobulin are cost and requiring parenteral access.
Other evaluation/treatments for drug refractory patients can be detailed as follows: First, an overall quality of life assessment can be helpful, especially one that includes impact of GpS on the patients and family. Nutritional considerations, which may not have been fully assessed, can be examined in more detail. Frailty assessments may show the need for physical therapy. Assessment for home care needs may indicate, in severe patients, needs for IV fluids at home, either enteral or parenteral, if nutrition is not adequate. Psychosocial and/or psychiatric assessments may lead to the need for medications, psychotherapy, and/or support groups. Lastly, an assessment of overall health status may lead to approaches for minimizing visits to emergency rooms and hospitalizations.29,30
Conclusion
Patients with Gp symptoms are becoming increasingly recognized and referred to gastroenterologists. Better understandings of the pathophysiology of the spectrum of gastroparesis syndromes, assisted by innovations in diagnosis, have led to expansion of existing and new therapeutic approaches. Fortunately, most patients can benefit from a standardized diagnostic approach and directed noninvasive therapies. Patients with refractory gastroparesis symptoms, often with complex issues referred to gastroenterologists, remain a challenge, and novel approaches may improve their quality of life.
Dr. Mathur is a GI motility research fellow at the University of Louisville, Ky. He reports no conflicts of interest. Dr. Abell is the Arthur M. Schoen, MD, Chair in Gastroenterology at the University of Louisville. His main funding is NIH GpCRC and NIH Definitive Evaluation of Gastric Dysrhythmia. He is an investigator for Cindome, Vanda, Allergan, and Neurogastrx; a consultant for Censa, Nuvaira, and Takeda; a speaker for Takeda and Medtronic; and a reviewer for UpToDate. He is also the founder of ADEPT-GI, which holds IP related to mucosal stimulation and autonomic and enteric profiling.
References
1. Jung HK et al. Gastroenterology. 2009;136(4):1225-33.
2. Ye Y et al. Gut. 2021;70(4):644-53.
3. Oshima T et al. J Neurogastroenterol Motil. 2021;27(1):46-54.
4. Soykan I et al. Dig Dis Sci. 1998;43(11):2398-404.
5. Syed AR et al. J Clin Gastroenterol. 2020;54(1):50-4.
6.Pasricha PJ et al. Clin Gastroenterol Hepatol. 2011;9(7):567-76.e1-4.
7. Pasricha PJ et al. Gastroenterology. 2021;160(6):2006-17.
8. Almario CV et al. Am J Gastroenterol. 2018;113(11):1701-10.
9. Abell TL et al. Dig Dis Sci. 2021 Apr;66(4):1127-41.
10. Abell TL et al. Neurogastroenterol Motil. 2019;31(3):e13534.
11. Elmasry M et al. Neurogastroenterol Motil. 2021 Oct 26;e14274.
12. Maurer AH et al. J Nucl Med. 2020;61(3):11N-7N.
13. Abell TL et al. J Nucl Med Technol. 2008 Mar;36(1):44-54.
14. Shine A et al. Neuromodulation. 2022 Feb 16;S1094-7159(21)06986-5.
15. O’Grady G et al. Am J Physiol Gastrointest Liver Physiol. 2021;321(5):G527-g42.
16. Saadi M et al. Rev Gastroenterol Mex (Engl Ed). Oct-Dec 2018;83(4):375-84.
17. Kamal F et al. Aliment Pharmacol Ther. 2022;55(2):168-77.
18. Harberson J et al. Dig Dis Sci. 2010;55(2):359-70.
19. Winston J. Gastrointestinal Disorders. 2021;3(2):78-83.
20. Parkman HP et al. Gastroenterology. 2011;141(2):486-98, 98.e1-7.
21. Heckroth M et al. J Clin Gastroenterol. 2021;55(4):279-99.
22. Camilleri M. Clin Gastroenterol Hepatol. 2022;20(1):19-24.
23. Payne SC et al. Nat Rev Gastroenterol Hepatol. 2019;16(2):89-105.
24. Ducrotte P et al. Gastroenterology. 2020;158(3):506-14.e2.
25. Burlen J et al. Gastroenterology Res. 2018;11(5):349-54.
26. Hedjoudje A et al. Neurogastroenterol Motil. 2020;32(11):e13949.
27. Petrov RV et al. Gastroenterol Clin North Am. 2020;49(3):539-56.
28. Gala K et al. J Clin Gastroenterol. 2021 Dec 31. doi: 10.1097/MCG.0000000000001655.
29. Abell TL et al. Neurogastroenterol Motil. 2006;18(4):263-83.
30. Camilleri M et al. Am J Gastroenterol. 2013;108(1):18-37.
The management of inflammatory bowel disease in pregnancy
Inflammatory bowel disease (IBD) incidence is rising globally.1-3 In the United States, we have seen a 123% increase in prevalence of IBD among adults and a 133% increase among children from 2007 to 2016, with an annual percentage change of 9.9%.1 The rise of IBD in young people, and the overall higher prevalence in women compared with men, make pregnancy and IBD a topic of increasing importance for gastroenterologists.1 Here, we will discuss management and expectations in women with IBD before conception, during pregnancy, and post partum.
Preconception
Disease activity

Achieving both clinical and endoscopic remission of disease prior to conception is the key to ensuring the best maternal and fetal outcomes. Patients with IBD who conceive while in remission remain in remission 80% of the time.4,5 On the other hand, those who conceive while their disease is active may continue to have active or worsening disease in nearly 70% of cases.4 Active disease has been associated with an increased incidence of preterm birth, low birth weight, and small-for-gestational-age birth.6-8 Active disease can also exacerbate malnutrition and result in poor maternal weight gain, which is associated with intrauterine growth restriction.9,7 Pregnancy outcomes in patients with IBD and quiescent disease are similar to those in the general population.10,11
Health care maintenance
Optimizing maternal health prior to conception is critical. Alcohol, tobacco, recreational drugs, and marijuana should all be avoided. Opioids should be tapered off prior to conception, as continued use may result in neonatal opioid withdrawal syndrome and long-term neurodevelopmental consequences.12,13 In addition, aiming for a healthy body mass index between 18 and 25 months prior to conception allows for better overall pregnancy outcomes.13 Appropriate cancer screening includes colon cancer screening in those with more than 8 years of colitis, regular pap smear for cervical cancer, and annual total body skin cancer examinations for patients on thiopurines and biologic therapies.14

Nutrition
Folic acid supplementation with at least 400 micrograms (mcg) daily is necessary for all women planning pregnancy. Patients with small bowel involvement or history of small bowel resection should have a folate intake of a minimum of 2 grams per day. Adequate vitamin D levels (at least 20 ng/mL) are recommended in all women with IBD. Those with malabsorption should be screened for deficiencies in vitamin B12, folate, and iron.13 These nutritional markers should be evaluated prepregnancy, during the first trimester, and thereafter as needed.15-18
Preconception counseling
Steroid-free remission for at least 3 months prior to conception is recommended and is associated with reduced risk of flare during pregnancy.16,19 IBD medications needed to control disease activity are generally safe preconception and during pregnancy, with some exception (Table).
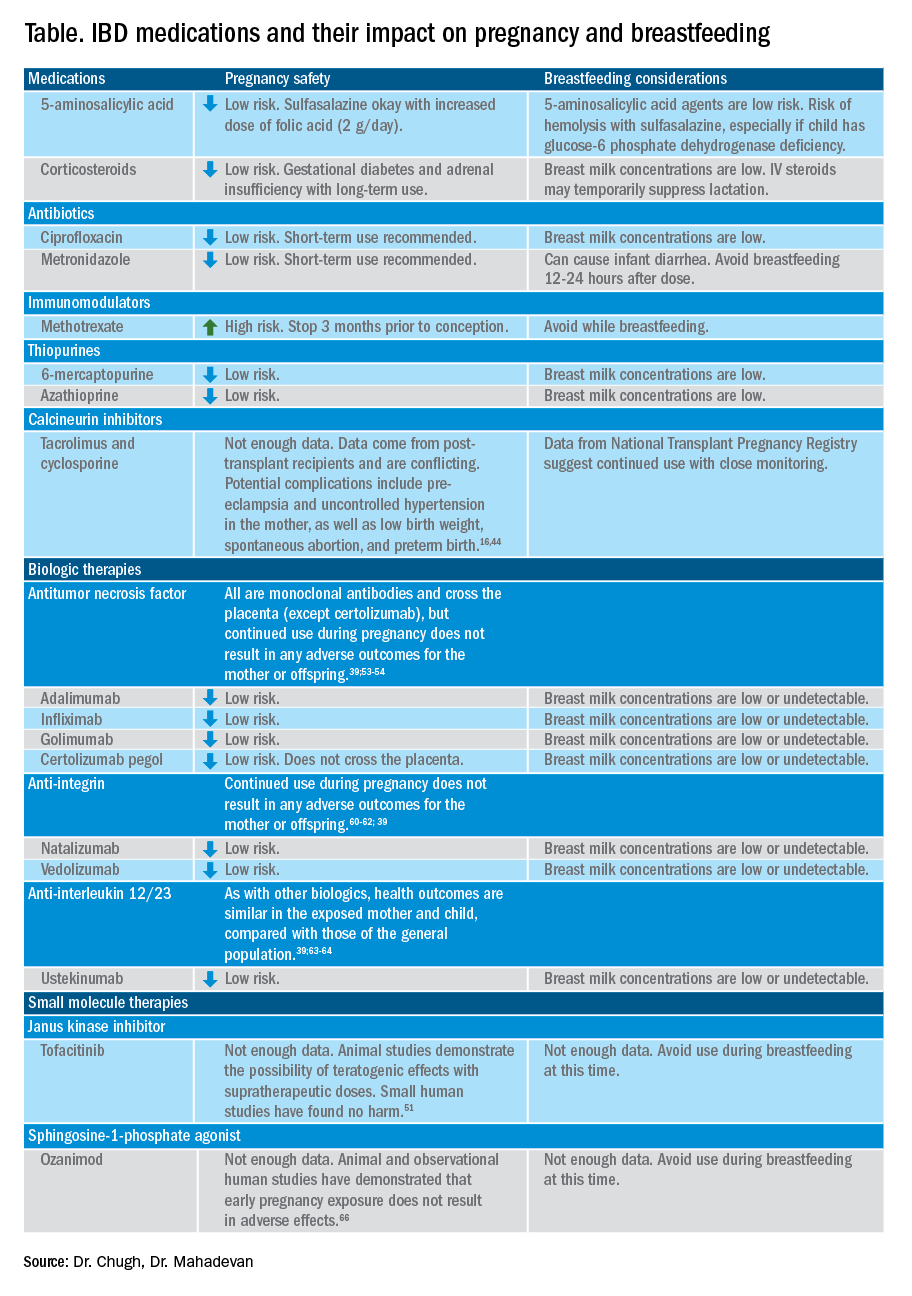
Misconceptions regarding heritability of IBD have sometimes discouraged men and women from having children. While genetics may increase susceptibility, environmental and other factors are involved as well. The concordance rates for monozygotic twins range from 33.3%-58.3% for Crohn’s disease and 13.4%-27.9% for ulcerative colitis (UC).20 The risk of a child developing IBD is higher in those who have multiple relatives with IBD and whose parents had IBD at the time of conception.21 While genetic testing for IBD loci is available, it is not commonly performed at this time as many genes are involved.22
Pregnancy
Coordinated care
A complete team of specialists with coordinated care among all providers is needed for optimal maternal and fetal outcomes.23,24 A gastroenterologist, ideally an IBD specialist, should follow the patient throughout pregnancy, seeing the patient at least once during the first or second trimester and as needed during pregnancy.16 A high-risk obstetrician or maternal-fetal medicine specialist should be involved early in pregnancy, as well. Open communication among all disciplines ensures that a common message is conveyed to the patient.16,24 A nutritionist, mental health provider, and lactation specialist knowledgeable about IBD drugs may be of assistance, as well.16
Disease activity
While women with IBD are at increased risk of spontaneous abortion, preterm birth, and labor complications, this risk is mitigated by controlling disease activity.25 The risk of preterm birth, small-for-gestational-age birth, and delivery via C-section is much higher in women with moderate-to-high disease activity, compared with those with low disease activity.26 The presence of active perianal disease mandates C-section over vaginal delivery. Fourth-degree lacerations following vaginal delivery are most common among those patients with perianal disease.26,27 Stillbirths were shown to be increased only in those with active IBD when compared with non-IBD comparators and inactive IBD.28-31;11
Noninvasive methods for disease monitoring are preferred in pregnancy, but serum markers such as erythrocyte sedimentation rate and C-reactive protein may not be reliable in the pregnant patient (Figure).32 Fecal calprotectin does rise in correlation with disease activity, but exact thresholds have not been validated in pregnancy.33,34

An unsedated, unprepped flexible sigmoidoscopy can be safely performed throughout pregnancy.35 When there is a strong indication, a complete colonoscopy can be performed in the pregnant patient as well.36 Current American Society for Gastrointestinal Endoscopy (ASGE) guidelines suggest placing the patient in the left lateral tilt position to avoid decreased maternal and placental perfusion via compression of the aorta or inferior vena cava and performing endoscopy during the second trimester, although trimester-specific timing is not always feasible by indication.37
Medication use and safety
IBD medications are a priority topic of concern among pregnant patients or those considering conception.38 Comprehensive data from the PIANO (Pregnancy in Inflammatory Bowel Disease and Neonatal Outcomes) registry has shown that most IBD drugs do not result in adverse pregnancy outcomes and should be continued.39 The use of biologics and thiopurines, either in combination or alone, is not related to an increased risk of congenital malformations, spontaneous abortion, preterm birth, low birth weight, or infections during the child’s first year of life.7,39 Developmental milestones also remain unaffected.39 Here, we will discuss safety considerations during pregnancy (see Table).
5-aminosalycylic acid. 5-aminosalicylic acid (5-ASA) agents are generally low risk during pregnancy and should be continued.40-41 Sulfasalazine does interfere with folate metabolism, but by increasing folic acid supplementation to 2 grams per day, sulfasalazine can be continued throughout pregnancy, as well.42
Corticosteroids. Intrapartum corticosteroid use is associated with an increased risk of gestational diabetes and adrenal insufficiency when used long term.43-45 Short-term use may, however, be necessary to control an acute flare. The lowest dose for the shortest duration possible is recommended. Because of its high first-pass metabolism, budesonide is considered low risk in pregnancy.
Methotrexate. Methotrexate needs to be stopped at least 3 months prior to conception and should be avoided throughout pregnancy. Use during pregnancy can result in spontaneous abortions, as well as embryotoxicity.46
Thiopurines (6-mercaptopurine and azathioprine). Patients who are taking thiopurines prior to conception to maintain remission can continue to do so. Data on thiopurines from the PIANO registry has shown no increase in spontaneous abortions, congenital malformations, low birth weight, preterm birth, rates of infection in the child, or developmental delays.47-51
Calcineurin inhibitors (cyclosporine and tacrolimus). Calcineurin inhibitors are reserved for the management of acute severe UC. Safety data on calcineurin inhibitors is conflicting, and there is not enough information at this time to identify risk during pregnancy. Cyclosporine can be used for salvage therapy if absolutely needed, and there are case reports of its successful using during pregnancy.16,52
Biologic therapies. With the exception of certolizumab, all of the currently used biologics are actively transported across the placenta.39,53,54 Intrapartum use of biologic therapies does not worsen pregnancy or neonatal outcomes, including the risk for intensive care unit admission, infections, and developmental milestones.39,47
While drug concentrations may vary slightly during pregnancy, these changes are not substantial enough to warrant more frequent monitoring or dose adjustments, and prepregnancy weight should be used for dosing.55,56
Antitumor necrosis factor agents used in IBD include infliximab, adalimumab, certolizumab, and golimumab.57 All are low risk for pregnant patients and their offspring. Dosage timings can be adjusted, but not stopped, to minimize exposure to the child; however, it cannot be adjusted for certolizumab pegol because of its lack of placental transfer.58-59
Natalizumab and vedolizumab are integrin receptor antagonists and are also low risk in pregnancy.57;60-62;39
Ustekinumab, an interleukin-12/23 antagonist, can be found in infant serum and cord blood, as well. Health outcomes are similar in the exposed mother and child, however, compared with those of the general population.39;63-64
Small molecule drugs. Unlike monoclonal antibodies, which do not cross the placenta in large amounts until early in the second trimester, small molecules can cross in the first trimester during the critical period of organogenesis.
The two small molecule agents currently approved for use in UC are tofacitinib, a janus kinase inhibitor, and ozanimod, a sphingosine-1-phosphate receptor agonist.65-66 Further data are still needed to make recommendations on the use of tofacitinib and ozanimod in pregnancy. At this time, we recommend weighing the risks (unknown risk to human pregnancy) vs. benefits (controlled disease activity with clear risk of harm to mother and baby from flare) in the individual patient before counseling on use in pregnancy.
Delivery
Mode of delivery
The mode of delivery should be determined by the obstetrician. C-section is recommended for patients with active perianal disease or, in some cases, a history of ileal pouch anal anastomosis (IPAA).67-68 Vaginal delivery in the setting of perianal disease has been shown to increase the risk of fourth-degree laceration and anal sphincter dysfunction in the future.26-27 Anorectal motility may be impacted by IPAA construction and vaginal delivery independently of each other. It is therefore suggested that vaginal delivery be avoided in patients with a history of IPAA to avoid compounding the risk. Some studies do not show clear harm from vaginal delivery in the setting of IPAA, however, and informed decision making among all stakeholders should be had.27;69-70
Anticoagulation
The incidence of venous thromboembolism (VTE) is elevated in patients with IBD during pregnancy, and up to 12 weeks postpartum, compared with pregnant patients without IBD.71-72 VTE for prophylaxis is indicated in the pregnant patient while hospitalized and potentially thereafter depending on the patient’s risk factors, which may include obesity, prior personal history of VTE, heart failure, and prolonged immobility. Unfractionated heparin, low molecular weight heparin, and warfarin are safe for breastfeeding women.16,73
Postpartum care of mother
There is a risk of postpartum flare, occurring in about one third of patients in the first 6 months postpartum.74-75 De-escalating therapy during delivery or immediately postpartum is a predictor of a postpartum flare.75 If no infection is present and the timing interval is appropriate, biologic therapies should be continued and can be resumed 24 hours after a vaginal delivery and 48 hours after a C-section.16,76
NSAIDs and opioids can be used for pain relief but should be avoided in the long-term to prevent flares (NSAIDs) and infant sedation (associated with opioids) when used while breastfeeding.77 The LactMed database is an excellent resource for clarification on risk of medication use while breastfeeding.78
In particular, contraception should be addressed postpartum. Exogenous estrogen use increases the risk of VTE, which is already increased in IBD; nonestrogen containing, long-acting reversible contraception is preferred.79-80 Progestin-only implants or intrauterine devices may be used first line. The efficacy of oral contraceptives is theoretically reduced in those with rapid bowel transit, active small bowel inflammation, and prior small bowel resection, so adding another form of contraception is recommended.16,81
Source: American Gastroenterological Association
Postdelivery care of baby
Breastfeeding
Guidelines regarding medication use during breastfeeding are similar to those in pregnancy (see Table). Breastfeeding on biologics and thiopurines can continue without interruption in the child. Thiopurine concentrations in breast milk are low or undetectable.82,78 TNF receptor antagonists, anti-integrin therapies, and ustekinumab are found in low to undetectable levels in breast milk, as well.78
On the other hand, the active metabolite of methotrexate is detectable in breast milk and most sources recommend not breastfeeding on methotrexate. At doses used in IBD (15-25 milligrams per week), some experts have suggested avoiding breastfeeding for 24 hours following a dose.57,78 It is the practice of this author to recommend not breastfeeding at all on methotrexate.
5-ASA therapies are low risk for breastfeeding, but alternatives to sulfasalazine are preferred. The sulfapyridine metabolite transfers to breast milk and may cause hemolysis in infants born with a glucose-6-phosphate dehydrogenase deficiency.78
With regards to calcineurin inhibitors, tacrolimus appears in breast milk in low quantities, while cyclosporine levels are variable. Data from the National Transplantation Pregnancy Registry suggest that these medications can be used at the time of breastfeeding with close monitoring.78
There is not enough data on small molecule therapies at this time to support breastfeeding safety, and it is our practice to not recommend breastfeeding in this scenario.
The transfer of steroids to the child via breast milk does occur but at subtherapeutic levels.16 Budesonide has high first pass metabolism and is low risk during breastfeeding.83-84 As far as is known, IBD maintenance medications do not suppress lactation. The use of intravenous corticosteroids can, however, temporarily decrease milk production.16,85
Vaccines
Vaccination of infants can proceed as indicated by the Center for Disease Control and Prevention guidelines, with one exception. If the child’s mother was exposed to any biologic agents (not including certolizumab) during the third trimester, any live vaccines should be withheld in the first 6 months of life. In the United States, this restriction currently only applies to the rotavirus vaccine, which is administered starting at the age of 2 months.16,86 Notably, inadvertent administration of the rotavirus vaccine in the biologic-exposed child does not appear to result in any adverse effects.87 Immunity is achieved even if the child is exposed to IBD therapies through breast milk.88
Developmental milestones
Infant exposure to biologics and thiopurines has not been shown to result in any developmental delays. The PIANO study measured developmental milestones at 48 months from birth and found no differences when compared with validated population norms.39 A separate study observing childhood development up to 7 years of age in patients born to mothers with IBD found similar cognitive scores and motor development when compared with those born to mothers without IBD.89
Conclusion
Women considering conception should be optimized prior to pregnancy and maintained on appropriate medications throughout pregnancy and lactation to achieve a healthy pregnancy for both mother and baby. To date, biologics and thiopurines are not associated with adverse pregnancy outcomes. More data are needed for small molecules.
Dr. Chugh is an advanced inflammatory bowel disease fellow in the division of gastroenterology at the University of California San Francisco. Dr. Mahadevan is professor of medicine and codirector at the Center for Colitis and Crohn’s Disease in the division of gastroenterology at the University of California San Francisco. Dr. Mahadevan has potential conflicts related to AbbVie, Janssen, BMS, Takeda, Pfizer, Lilly, Gilead, Arena, and Prometheus Biosciences.
References
1. Ye Y et al. Inflamm Bowel Dis. 2020;26:619-25.
2. Sykora J et al. World J Gastroenterol. 2018;24:2741-63.
3. Murakami Y et al. J Gastroenterol 2019;54:1070-7.
4. Hashash JG and Kane S. Gastroenterol Hepatol. (N Y) 2015;11:96-102.
5. Miller JP. J R Soc Med. 1986;79:221-5.
6. Cornish J et al. Gut. 2007;56:830-7.
7. Leung KK et al. Inflamm Bowel Dis. 2021;27:550-62.
8. O’Toole A et al. Dig Dis Sci. 2015;60:2750-61.
9. Nguyen GC et al. Inflamm Bowel Dis. 2008;14:1105-11.
10. Lee HH et al. Aliment Pharmacol Ther. 2020;51:861-9.
11. Kim MA et al. J Crohns Colitis. 2021;15:719-32.
12. Conradt E et al. Pediatrics. 2019;144.
13. ACOG Committee Opinion No. 762: Prepregnancy Counseling. Obstet Gynecol. 2019;133:e78-e89.
14. Farraye FA et al. Am J Gastroenterol. 2017;112:241-58.
15. Lee S et al. J Crohns Colitis. 2018;12:702-9.
16. Mahadevan U et al. Inflamm Bowel Dis. 2019;25:627-41.
17. Ward MG et al. Inflamm. Bowel Dis 2015;21:2839-47.
18. Battat R et al. Inflamm Bowel Dis. 2014;20:1120-8.
19. Pedersen N et al. Aliment Pharmacol Ther. 2013;38:501-12.
20. Annese V. Pharmacol Res. 2020;159:104892.
21. Bennett RA et al. Gastroenterology. 1991;100:1638-43.
22. Turpin W et al. Inflamm Bowel Dis. 2018;24:1133-48.
23. de Lima A et al. Clin Gastroenterol Hepatol. 2016;14:1285-92 e1.
24. Selinger C et al. Frontline Gastroenterol. 2021;12:182-7.
25. Mahadevan U et al. Gastroenterology. 2007;133:1106-12.
26. Hatch Q et al. Dis Colon Rectum. 2014;57:174-8.
27. Foulon A et al. Inflamm Bowel Dis. 2017;23:712-20.
28. Norgard B et al. Am J Gastroenterol. 2007;102:1947-54.
29. Broms G et al. Scand J Gastroenterol 2016;51:1462-9.
30. Meyer A et al. Aliment Pharmacol Ther. 2020;52:1480-90.
31. Kammerlander H et al. Inflamm Bowel Dis. 2017;23:1011-8.
32. Tandon P et al. J Clin Gastroenterol. 2019;53:574-81.
33. Kammerlander H et al. Inflamm Bowel Dis. 2018;24:839-48.
34. Julsgaard M et al. Inflamm Bowel Dis. 2017;23:1240-6.
35. Ko MS et al. Dig Dis Sci. 2020;65:2979-85.
36. Cappell MS et al. J Reprod Med. 2010;55:115-23.
37. Committee ASoP et al. Gastrointest Endosc. 2012;76:18-24.
38. Aboubakr A et al. Dig Dis Sci. 2021;66:1829-35.
39. Mahadevan U et al. Gastroenterology. 2021;160:1131-9.
40. Diav-Citrin O et al. Gastroenterology. 1998;114:23-8.
41. Rahimi R et al. Reprod Toxicol. 2008;25:271-5.
42. Norgard B et al. Aliment Pharmacol Ther. 2001;15:483-6.
43. Leung YP et al. J Crohns Colitis. 2015;9:223-30.
44. Schulze H et al. Aliment Pharmacol Ther. 2014;40:991-1008.
45. Szymanska E et al. J Gynecol Obstet Hum Reprod. 2021;50:101777.
46. Weber-Schoendorfer C et al. Arthritis Rheumatol. 2014;66:1101-10.
47. Nielsen OH et al. Clin Gastroenterol Hepatol. 2022 Jan;20(1):74-87.e3.
48. Coelho J et al. Gut. 2011;60:198-203.
49. Sheikh M et al. J Crohns Colitis. 2015;9:680-4.
50. Kanis SL et al. Clin Gastroenterol Hepatol. 2017;15:1232-41 e1.
51. Mahadevan U et al. Inflamm Bowel Dis. 2018;24:2494-500.
52. Rosen MH et al. Inflamm Bowel Dis. 2020;26:971-3.
53. Porter C et al. J Reprod Immunol. 2016;116:7-12.
54. Mahadevan U et al. Clin Gastroenterol Hepatol. 2013;11:286-92; quiz e24.
55. Picardo S and Seow CH. Best Pract Res Clin Gastroenterol. 2020;44-5:101670.
56. Flanagan E et al. Aliment Pharmacol Ther. 2020;52:1551-62.
57. Singh S et al. Gastroenterology. 2021;160:2512-56 e9.
58. de Lima A et al. Gut. 2016;65:1261-8.
59. Julsgaard M et al. Inflamm Bowel Dis. 2020;26:93-102.
60. Wils P et al. Aliment Pharmacol Ther. 2021;53:460-70.
61. Mahadevan U et al. Aliment Pharmacol Ther. 2017;45:941-50.
62. Bar-Gil Shitrit A et al. Am J Gastroenterol. 2019;114:1172-5.
63. Klenske E et al. J Crohns Colitis. 2019;13:267-9.
64. Matro R et al. Gastroenterology. 2018;155:696-704.
65. Feuerstein JD et al. Gastroenterology. 2020;158:1450-61.
66. Sandborn WJ et al. J Crohns Colitis. 2021 Jul 5;15(7):1120-1129.
67. Lamb CA et al. Gut. 2019;68:s1-s106.
68. Nguyen GC et al. Gastroenterology. 2016;150:734-57 e1.
69. Ravid A et al. Dis Colon Rectum. 2002;45:1283-8.
70. Seligman NS et al. J Matern Fetal Neonatal Med. 2011;24:525-30.
71. Kim YH et al. Medicine (Baltimore). 2019;98:e17309.
72. Hansen AT et al. J Thromb Haemost. 2017;15:702-8.
73. Bates SM et al. J Thromb Thrombolysis. 2016;41:92-128.
74. Bennett A et al. Inflamm Bowel Dis. 2021 May 17;izab104.
75. Yu A et al. Inflamm Bowel Dis. 2020;26:1926-32.
76. Mahadevan U et al. Gastroenterology. 2017;152:451-62 e2.
77. Long MD et al. J Clin Gastroenterol. 2016;50:152-6.
78. Drugs and Lactation Database (LactMed). 2006 ed. Bethesda, MD: National Library of Medicine (US), 2006-2021.
79. Khalili H et al. Gut. 2013;62:1153-9.
80. Long MD and Hutfless S. Gastroenterology. 2016;150:1518-20.
81. Centers for Disease Control and Prevention. U S. Medical Eligibility Criteria for Contraceptive Use, 2010. MMWR Recomm Rep. 2010;59:1-86.
82. Angelberger S et al. J Crohns Colitis. 2011;5:95-100.
83. Vestergaard T et al. Scand J Gastroenterol. 2018;53:1459-62.
84. Beaulieu DB et al. Inflamm Bowel Dis. 2009;15:25-8.
85. Anderson PO. Breastfeed Med. 2017;12:199-201.
86. Wodi AP et al. MMWR Morb Mortal Wkly Rep. 2021;70:189-92.
87. Chiarella-Redfern H et al. Inflamm Bowel Dis. 2022 Jan 5;28(1):79-86.
88. Beaulieu DB et al. Clin Gastroenterol Hepatol. 2018;16:99-105.
89. Friedman S et al. J Crohns Colitis. 2020 Dec 2;14(12):1709-1716.
Inflammatory bowel disease (IBD) incidence is rising globally.1-3 In the United States, we have seen a 123% increase in prevalence of IBD among adults and a 133% increase among children from 2007 to 2016, with an annual percentage change of 9.9%.1 The rise of IBD in young people, and the overall higher prevalence in women compared with men, make pregnancy and IBD a topic of increasing importance for gastroenterologists.1 Here, we will discuss management and expectations in women with IBD before conception, during pregnancy, and post partum.
Preconception
Disease activity
Achieving both clinical and endoscopic remission of disease prior to conception is the key to ensuring the best maternal and fetal outcomes. Patients with IBD who conceive while in remission remain in remission 80% of the time.4,5 On the other hand, those who conceive while their disease is active may continue to have active or worsening disease in nearly 70% of cases.4 Active disease has been associated with an increased incidence of preterm birth, low birth weight, and small-for-gestational-age birth.6-8 Active disease can also exacerbate malnutrition and result in poor maternal weight gain, which is associated with intrauterine growth restriction.9,7 Pregnancy outcomes in patients with IBD and quiescent disease are similar to those in the general population.10,11
Health care maintenance
Optimizing maternal health prior to conception is critical. Alcohol, tobacco, recreational drugs, and marijuana should all be avoided. Opioids should be tapered off prior to conception, as continued use may result in neonatal opioid withdrawal syndrome and long-term neurodevelopmental consequences.12,13 In addition, aiming for a healthy body mass index between 18 and 25 months prior to conception allows for better overall pregnancy outcomes.13 Appropriate cancer screening includes colon cancer screening in those with more than 8 years of colitis, regular pap smear for cervical cancer, and annual total body skin cancer examinations for patients on thiopurines and biologic therapies.14
Nutrition
Folic acid supplementation with at least 400 micrograms (mcg) daily is necessary for all women planning pregnancy. Patients with small bowel involvement or history of small bowel resection should have a folate intake of a minimum of 2 grams per day. Adequate vitamin D levels (at least 20 ng/mL) are recommended in all women with IBD. Those with malabsorption should be screened for deficiencies in vitamin B12, folate, and iron.13 These nutritional markers should be evaluated prepregnancy, during the first trimester, and thereafter as needed.15-18
Preconception counseling
Steroid-free remission for at least 3 months prior to conception is recommended and is associated with reduced risk of flare during pregnancy.16,19 IBD medications needed to control disease activity are generally safe preconception and during pregnancy, with some exception (Table).
Misconceptions regarding heritability of IBD have sometimes discouraged men and women from having children. While genetics may increase susceptibility, environmental and other factors are involved as well. The concordance rates for monozygotic twins range from 33.3%-58.3% for Crohn’s disease and 13.4%-27.9% for ulcerative colitis (UC).20 The risk of a child developing IBD is higher in those who have multiple relatives with IBD and whose parents had IBD at the time of conception.21 While genetic testing for IBD loci is available, it is not commonly performed at this time as many genes are involved.22
Pregnancy
Coordinated care
A complete team of specialists with coordinated care among all providers is needed for optimal maternal and fetal outcomes.23,24 A gastroenterologist, ideally an IBD specialist, should follow the patient throughout pregnancy, seeing the patient at least once during the first or second trimester and as needed during pregnancy.16 A high-risk obstetrician or maternal-fetal medicine specialist should be involved early in pregnancy, as well. Open communication among all disciplines ensures that a common message is conveyed to the patient.16,24 A nutritionist, mental health provider, and lactation specialist knowledgeable about IBD drugs may be of assistance, as well.16
Disease activity
While women with IBD are at increased risk of spontaneous abortion, preterm birth, and labor complications, this risk is mitigated by controlling disease activity.25 The risk of preterm birth, small-for-gestational-age birth, and delivery via C-section is much higher in women with moderate-to-high disease activity, compared with those with low disease activity.26 The presence of active perianal disease mandates C-section over vaginal delivery. Fourth-degree lacerations following vaginal delivery are most common among those patients with perianal disease.26,27 Stillbirths were shown to be increased only in those with active IBD when compared with non-IBD comparators and inactive IBD.28-31;11
Noninvasive methods for disease monitoring are preferred in pregnancy, but serum markers such as erythrocyte sedimentation rate and C-reactive protein may not be reliable in the pregnant patient (Figure).32 Fecal calprotectin does rise in correlation with disease activity, but exact thresholds have not been validated in pregnancy.33,34
An unsedated, unprepped flexible sigmoidoscopy can be safely performed throughout pregnancy.35 When there is a strong indication, a complete colonoscopy can be performed in the pregnant patient as well.36 Current American Society for Gastrointestinal Endoscopy (ASGE) guidelines suggest placing the patient in the left lateral tilt position to avoid decreased maternal and placental perfusion via compression of the aorta or inferior vena cava and performing endoscopy during the second trimester, although trimester-specific timing is not always feasible by indication.37
Medication use and safety
IBD medications are a priority topic of concern among pregnant patients or those considering conception.38 Comprehensive data from the PIANO (Pregnancy in Inflammatory Bowel Disease and Neonatal Outcomes) registry has shown that most IBD drugs do not result in adverse pregnancy outcomes and should be continued.39 The use of biologics and thiopurines, either in combination or alone, is not related to an increased risk of congenital malformations, spontaneous abortion, preterm birth, low birth weight, or infections during the child’s first year of life.7,39 Developmental milestones also remain unaffected.39 Here, we will discuss safety considerations during pregnancy (see Table).
5-aminosalycylic acid. 5-aminosalicylic acid (5-ASA) agents are generally low risk during pregnancy and should be continued.40-41 Sulfasalazine does interfere with folate metabolism, but by increasing folic acid supplementation to 2 grams per day, sulfasalazine can be continued throughout pregnancy, as well.42
Corticosteroids. Intrapartum corticosteroid use is associated with an increased risk of gestational diabetes and adrenal insufficiency when used long term.43-45 Short-term use may, however, be necessary to control an acute flare. The lowest dose for the shortest duration possible is recommended. Because of its high first-pass metabolism, budesonide is considered low risk in pregnancy.
Methotrexate. Methotrexate needs to be stopped at least 3 months prior to conception and should be avoided throughout pregnancy. Use during pregnancy can result in spontaneous abortions, as well as embryotoxicity.46
Thiopurines (6-mercaptopurine and azathioprine). Patients who are taking thiopurines prior to conception to maintain remission can continue to do so. Data on thiopurines from the PIANO registry has shown no increase in spontaneous abortions, congenital malformations, low birth weight, preterm birth, rates of infection in the child, or developmental delays.47-51
Calcineurin inhibitors (cyclosporine and tacrolimus). Calcineurin inhibitors are reserved for the management of acute severe UC. Safety data on calcineurin inhibitors is conflicting, and there is not enough information at this time to identify risk during pregnancy. Cyclosporine can be used for salvage therapy if absolutely needed, and there are case reports of its successful using during pregnancy.16,52
Biologic therapies. With the exception of certolizumab, all of the currently used biologics are actively transported across the placenta.39,53,54 Intrapartum use of biologic therapies does not worsen pregnancy or neonatal outcomes, including the risk for intensive care unit admission, infections, and developmental milestones.39,47
While drug concentrations may vary slightly during pregnancy, these changes are not substantial enough to warrant more frequent monitoring or dose adjustments, and prepregnancy weight should be used for dosing.55,56
Antitumor necrosis factor agents used in IBD include infliximab, adalimumab, certolizumab, and golimumab.57 All are low risk for pregnant patients and their offspring. Dosage timings can be adjusted, but not stopped, to minimize exposure to the child; however, it cannot be adjusted for certolizumab pegol because of its lack of placental transfer.58-59
Natalizumab and vedolizumab are integrin receptor antagonists and are also low risk in pregnancy.57;60-62;39
Ustekinumab, an interleukin-12/23 antagonist, can be found in infant serum and cord blood, as well. Health outcomes are similar in the exposed mother and child, however, compared with those of the general population.39;63-64
Small molecule drugs. Unlike monoclonal antibodies, which do not cross the placenta in large amounts until early in the second trimester, small molecules can cross in the first trimester during the critical period of organogenesis.
The two small molecule agents currently approved for use in UC are tofacitinib, a janus kinase inhibitor, and ozanimod, a sphingosine-1-phosphate receptor agonist.65-66 Further data are still needed to make recommendations on the use of tofacitinib and ozanimod in pregnancy. At this time, we recommend weighing the risks (unknown risk to human pregnancy) vs. benefits (controlled disease activity with clear risk of harm to mother and baby from flare) in the individual patient before counseling on use in pregnancy.
Delivery
Mode of delivery
The mode of delivery should be determined by the obstetrician. C-section is recommended for patients with active perianal disease or, in some cases, a history of ileal pouch anal anastomosis (IPAA).67-68 Vaginal delivery in the setting of perianal disease has been shown to increase the risk of fourth-degree laceration and anal sphincter dysfunction in the future.26-27 Anorectal motility may be impacted by IPAA construction and vaginal delivery independently of each other. It is therefore suggested that vaginal delivery be avoided in patients with a history of IPAA to avoid compounding the risk. Some studies do not show clear harm from vaginal delivery in the setting of IPAA, however, and informed decision making among all stakeholders should be had.27;69-70
Anticoagulation
The incidence of venous thromboembolism (VTE) is elevated in patients with IBD during pregnancy, and up to 12 weeks postpartum, compared with pregnant patients without IBD.71-72 VTE for prophylaxis is indicated in the pregnant patient while hospitalized and potentially thereafter depending on the patient’s risk factors, which may include obesity, prior personal history of VTE, heart failure, and prolonged immobility. Unfractionated heparin, low molecular weight heparin, and warfarin are safe for breastfeeding women.16,73
Postpartum care of mother
There is a risk of postpartum flare, occurring in about one third of patients in the first 6 months postpartum.74-75 De-escalating therapy during delivery or immediately postpartum is a predictor of a postpartum flare.75 If no infection is present and the timing interval is appropriate, biologic therapies should be continued and can be resumed 24 hours after a vaginal delivery and 48 hours after a C-section.16,76
NSAIDs and opioids can be used for pain relief but should be avoided in the long-term to prevent flares (NSAIDs) and infant sedation (associated with opioids) when used while breastfeeding.77 The LactMed database is an excellent resource for clarification on risk of medication use while breastfeeding.78
In particular, contraception should be addressed postpartum. Exogenous estrogen use increases the risk of VTE, which is already increased in IBD; nonestrogen containing, long-acting reversible contraception is preferred.79-80 Progestin-only implants or intrauterine devices may be used first line. The efficacy of oral contraceptives is theoretically reduced in those with rapid bowel transit, active small bowel inflammation, and prior small bowel resection, so adding another form of contraception is recommended.16,81
Source: American Gastroenterological Association
Postdelivery care of baby
Breastfeeding
Guidelines regarding medication use during breastfeeding are similar to those in pregnancy (see Table). Breastfeeding on biologics and thiopurines can continue without interruption in the child. Thiopurine concentrations in breast milk are low or undetectable.82,78 TNF receptor antagonists, anti-integrin therapies, and ustekinumab are found in low to undetectable levels in breast milk, as well.78
On the other hand, the active metabolite of methotrexate is detectable in breast milk and most sources recommend not breastfeeding on methotrexate. At doses used in IBD (15-25 milligrams per week), some experts have suggested avoiding breastfeeding for 24 hours following a dose.57,78 It is the practice of this author to recommend not breastfeeding at all on methotrexate.
5-ASA therapies are low risk for breastfeeding, but alternatives to sulfasalazine are preferred. The sulfapyridine metabolite transfers to breast milk and may cause hemolysis in infants born with a glucose-6-phosphate dehydrogenase deficiency.78
With regards to calcineurin inhibitors, tacrolimus appears in breast milk in low quantities, while cyclosporine levels are variable. Data from the National Transplantation Pregnancy Registry suggest that these medications can be used at the time of breastfeeding with close monitoring.78
There is not enough data on small molecule therapies at this time to support breastfeeding safety, and it is our practice to not recommend breastfeeding in this scenario.
The transfer of steroids to the child via breast milk does occur but at subtherapeutic levels.16 Budesonide has high first pass metabolism and is low risk during breastfeeding.83-84 As far as is known, IBD maintenance medications do not suppress lactation. The use of intravenous corticosteroids can, however, temporarily decrease milk production.16,85
Vaccines
Vaccination of infants can proceed as indicated by the Center for Disease Control and Prevention guidelines, with one exception. If the child’s mother was exposed to any biologic agents (not including certolizumab) during the third trimester, any live vaccines should be withheld in the first 6 months of life. In the United States, this restriction currently only applies to the rotavirus vaccine, which is administered starting at the age of 2 months.16,86 Notably, inadvertent administration of the rotavirus vaccine in the biologic-exposed child does not appear to result in any adverse effects.87 Immunity is achieved even if the child is exposed to IBD therapies through breast milk.88
Developmental milestones
Infant exposure to biologics and thiopurines has not been shown to result in any developmental delays. The PIANO study measured developmental milestones at 48 months from birth and found no differences when compared with validated population norms.39 A separate study observing childhood development up to 7 years of age in patients born to mothers with IBD found similar cognitive scores and motor development when compared with those born to mothers without IBD.89
Conclusion
Women considering conception should be optimized prior to pregnancy and maintained on appropriate medications throughout pregnancy and lactation to achieve a healthy pregnancy for both mother and baby. To date, biologics and thiopurines are not associated with adverse pregnancy outcomes. More data are needed for small molecules.
Dr. Chugh is an advanced inflammatory bowel disease fellow in the division of gastroenterology at the University of California San Francisco. Dr. Mahadevan is professor of medicine and codirector at the Center for Colitis and Crohn’s Disease in the division of gastroenterology at the University of California San Francisco. Dr. Mahadevan has potential conflicts related to AbbVie, Janssen, BMS, Takeda, Pfizer, Lilly, Gilead, Arena, and Prometheus Biosciences.
References
1. Ye Y et al. Inflamm Bowel Dis. 2020;26:619-25.
2. Sykora J et al. World J Gastroenterol. 2018;24:2741-63.
3. Murakami Y et al. J Gastroenterol 2019;54:1070-7.
4. Hashash JG and Kane S. Gastroenterol Hepatol. (N Y) 2015;11:96-102.
5. Miller JP. J R Soc Med. 1986;79:221-5.
6. Cornish J et al. Gut. 2007;56:830-7.
7. Leung KK et al. Inflamm Bowel Dis. 2021;27:550-62.
8. O’Toole A et al. Dig Dis Sci. 2015;60:2750-61.
9. Nguyen GC et al. Inflamm Bowel Dis. 2008;14:1105-11.
10. Lee HH et al. Aliment Pharmacol Ther. 2020;51:861-9.
11. Kim MA et al. J Crohns Colitis. 2021;15:719-32.
12. Conradt E et al. Pediatrics. 2019;144.
13. ACOG Committee Opinion No. 762: Prepregnancy Counseling. Obstet Gynecol. 2019;133:e78-e89.
14. Farraye FA et al. Am J Gastroenterol. 2017;112:241-58.
15. Lee S et al. J Crohns Colitis. 2018;12:702-9.
16. Mahadevan U et al. Inflamm Bowel Dis. 2019;25:627-41.
17. Ward MG et al. Inflamm. Bowel Dis 2015;21:2839-47.
18. Battat R et al. Inflamm Bowel Dis. 2014;20:1120-8.
19. Pedersen N et al. Aliment Pharmacol Ther. 2013;38:501-12.
20. Annese V. Pharmacol Res. 2020;159:104892.
21. Bennett RA et al. Gastroenterology. 1991;100:1638-43.
22. Turpin W et al. Inflamm Bowel Dis. 2018;24:1133-48.
23. de Lima A et al. Clin Gastroenterol Hepatol. 2016;14:1285-92 e1.
24. Selinger C et al. Frontline Gastroenterol. 2021;12:182-7.
25. Mahadevan U et al. Gastroenterology. 2007;133:1106-12.
26. Hatch Q et al. Dis Colon Rectum. 2014;57:174-8.
27. Foulon A et al. Inflamm Bowel Dis. 2017;23:712-20.
28. Norgard B et al. Am J Gastroenterol. 2007;102:1947-54.
29. Broms G et al. Scand J Gastroenterol 2016;51:1462-9.
30. Meyer A et al. Aliment Pharmacol Ther. 2020;52:1480-90.
31. Kammerlander H et al. Inflamm Bowel Dis. 2017;23:1011-8.
32. Tandon P et al. J Clin Gastroenterol. 2019;53:574-81.
33. Kammerlander H et al. Inflamm Bowel Dis. 2018;24:839-48.
34. Julsgaard M et al. Inflamm Bowel Dis. 2017;23:1240-6.
35. Ko MS et al. Dig Dis Sci. 2020;65:2979-85.
36. Cappell MS et al. J Reprod Med. 2010;55:115-23.
37. Committee ASoP et al. Gastrointest Endosc. 2012;76:18-24.
38. Aboubakr A et al. Dig Dis Sci. 2021;66:1829-35.
39. Mahadevan U et al. Gastroenterology. 2021;160:1131-9.
40. Diav-Citrin O et al. Gastroenterology. 1998;114:23-8.
41. Rahimi R et al. Reprod Toxicol. 2008;25:271-5.
42. Norgard B et al. Aliment Pharmacol Ther. 2001;15:483-6.
43. Leung YP et al. J Crohns Colitis. 2015;9:223-30.
44. Schulze H et al. Aliment Pharmacol Ther. 2014;40:991-1008.
45. Szymanska E et al. J Gynecol Obstet Hum Reprod. 2021;50:101777.
46. Weber-Schoendorfer C et al. Arthritis Rheumatol. 2014;66:1101-10.
47. Nielsen OH et al. Clin Gastroenterol Hepatol. 2022 Jan;20(1):74-87.e3.
48. Coelho J et al. Gut. 2011;60:198-203.
49. Sheikh M et al. J Crohns Colitis. 2015;9:680-4.
50. Kanis SL et al. Clin Gastroenterol Hepatol. 2017;15:1232-41 e1.
51. Mahadevan U et al. Inflamm Bowel Dis. 2018;24:2494-500.
52. Rosen MH et al. Inflamm Bowel Dis. 2020;26:971-3.
53. Porter C et al. J Reprod Immunol. 2016;116:7-12.
54. Mahadevan U et al. Clin Gastroenterol Hepatol. 2013;11:286-92; quiz e24.
55. Picardo S and Seow CH. Best Pract Res Clin Gastroenterol. 2020;44-5:101670.
56. Flanagan E et al. Aliment Pharmacol Ther. 2020;52:1551-62.
57. Singh S et al. Gastroenterology. 2021;160:2512-56 e9.
58. de Lima A et al. Gut. 2016;65:1261-8.
59. Julsgaard M et al. Inflamm Bowel Dis. 2020;26:93-102.
60. Wils P et al. Aliment Pharmacol Ther. 2021;53:460-70.
61. Mahadevan U et al. Aliment Pharmacol Ther. 2017;45:941-50.
62. Bar-Gil Shitrit A et al. Am J Gastroenterol. 2019;114:1172-5.
63. Klenske E et al. J Crohns Colitis. 2019;13:267-9.
64. Matro R et al. Gastroenterology. 2018;155:696-704.
65. Feuerstein JD et al. Gastroenterology. 2020;158:1450-61.
66. Sandborn WJ et al. J Crohns Colitis. 2021 Jul 5;15(7):1120-1129.
67. Lamb CA et al. Gut. 2019;68:s1-s106.
68. Nguyen GC et al. Gastroenterology. 2016;150:734-57 e1.
69. Ravid A et al. Dis Colon Rectum. 2002;45:1283-8.
70. Seligman NS et al. J Matern Fetal Neonatal Med. 2011;24:525-30.
71. Kim YH et al. Medicine (Baltimore). 2019;98:e17309.
72. Hansen AT et al. J Thromb Haemost. 2017;15:702-8.
73. Bates SM et al. J Thromb Thrombolysis. 2016;41:92-128.
74. Bennett A et al. Inflamm Bowel Dis. 2021 May 17;izab104.
75. Yu A et al. Inflamm Bowel Dis. 2020;26:1926-32.
76. Mahadevan U et al. Gastroenterology. 2017;152:451-62 e2.
77. Long MD et al. J Clin Gastroenterol. 2016;50:152-6.
78. Drugs and Lactation Database (LactMed). 2006 ed. Bethesda, MD: National Library of Medicine (US), 2006-2021.
79. Khalili H et al. Gut. 2013;62:1153-9.
80. Long MD and Hutfless S. Gastroenterology. 2016;150:1518-20.
81. Centers for Disease Control and Prevention. U S. Medical Eligibility Criteria for Contraceptive Use, 2010. MMWR Recomm Rep. 2010;59:1-86.
82. Angelberger S et al. J Crohns Colitis. 2011;5:95-100.
83. Vestergaard T et al. Scand J Gastroenterol. 2018;53:1459-62.
84. Beaulieu DB et al. Inflamm Bowel Dis. 2009;15:25-8.
85. Anderson PO. Breastfeed Med. 2017;12:199-201.
86. Wodi AP et al. MMWR Morb Mortal Wkly Rep. 2021;70:189-92.
87. Chiarella-Redfern H et al. Inflamm Bowel Dis. 2022 Jan 5;28(1):79-86.
88. Beaulieu DB et al. Clin Gastroenterol Hepatol. 2018;16:99-105.
89. Friedman S et al. J Crohns Colitis. 2020 Dec 2;14(12):1709-1716.
Inflammatory bowel disease (IBD) incidence is rising globally.1-3 In the United States, we have seen a 123% increase in prevalence of IBD among adults and a 133% increase among children from 2007 to 2016, with an annual percentage change of 9.9%.1 The rise of IBD in young people, and the overall higher prevalence in women compared with men, make pregnancy and IBD a topic of increasing importance for gastroenterologists.1 Here, we will discuss management and expectations in women with IBD before conception, during pregnancy, and post partum.
Preconception
Disease activity
Achieving both clinical and endoscopic remission of disease prior to conception is the key to ensuring the best maternal and fetal outcomes. Patients with IBD who conceive while in remission remain in remission 80% of the time.4,5 On the other hand, those who conceive while their disease is active may continue to have active or worsening disease in nearly 70% of cases.4 Active disease has been associated with an increased incidence of preterm birth, low birth weight, and small-for-gestational-age birth.6-8 Active disease can also exacerbate malnutrition and result in poor maternal weight gain, which is associated with intrauterine growth restriction.9,7 Pregnancy outcomes in patients with IBD and quiescent disease are similar to those in the general population.10,11
Health care maintenance
Optimizing maternal health prior to conception is critical. Alcohol, tobacco, recreational drugs, and marijuana should all be avoided. Opioids should be tapered off prior to conception, as continued use may result in neonatal opioid withdrawal syndrome and long-term neurodevelopmental consequences.12,13 In addition, aiming for a healthy body mass index between 18 and 25 months prior to conception allows for better overall pregnancy outcomes.13 Appropriate cancer screening includes colon cancer screening in those with more than 8 years of colitis, regular pap smear for cervical cancer, and annual total body skin cancer examinations for patients on thiopurines and biologic therapies.14
Nutrition
Folic acid supplementation with at least 400 micrograms (mcg) daily is necessary for all women planning pregnancy. Patients with small bowel involvement or history of small bowel resection should have a folate intake of a minimum of 2 grams per day. Adequate vitamin D levels (at least 20 ng/mL) are recommended in all women with IBD. Those with malabsorption should be screened for deficiencies in vitamin B12, folate, and iron.13 These nutritional markers should be evaluated prepregnancy, during the first trimester, and thereafter as needed.15-18
Preconception counseling
Steroid-free remission for at least 3 months prior to conception is recommended and is associated with reduced risk of flare during pregnancy.16,19 IBD medications needed to control disease activity are generally safe preconception and during pregnancy, with some exception (Table).
Misconceptions regarding heritability of IBD have sometimes discouraged men and women from having children. While genetics may increase susceptibility, environmental and other factors are involved as well. The concordance rates for monozygotic twins range from 33.3%-58.3% for Crohn’s disease and 13.4%-27.9% for ulcerative colitis (UC).20 The risk of a child developing IBD is higher in those who have multiple relatives with IBD and whose parents had IBD at the time of conception.21 While genetic testing for IBD loci is available, it is not commonly performed at this time as many genes are involved.22
Pregnancy
Coordinated care
A complete team of specialists with coordinated care among all providers is needed for optimal maternal and fetal outcomes.23,24 A gastroenterologist, ideally an IBD specialist, should follow the patient throughout pregnancy, seeing the patient at least once during the first or second trimester and as needed during pregnancy.16 A high-risk obstetrician or maternal-fetal medicine specialist should be involved early in pregnancy, as well. Open communication among all disciplines ensures that a common message is conveyed to the patient.16,24 A nutritionist, mental health provider, and lactation specialist knowledgeable about IBD drugs may be of assistance, as well.16
Disease activity
While women with IBD are at increased risk of spontaneous abortion, preterm birth, and labor complications, this risk is mitigated by controlling disease activity.25 The risk of preterm birth, small-for-gestational-age birth, and delivery via C-section is much higher in women with moderate-to-high disease activity, compared with those with low disease activity.26 The presence of active perianal disease mandates C-section over vaginal delivery. Fourth-degree lacerations following vaginal delivery are most common among those patients with perianal disease.26,27 Stillbirths were shown to be increased only in those with active IBD when compared with non-IBD comparators and inactive IBD.28-31;11
Noninvasive methods for disease monitoring are preferred in pregnancy, but serum markers such as erythrocyte sedimentation rate and C-reactive protein may not be reliable in the pregnant patient (Figure).32 Fecal calprotectin does rise in correlation with disease activity, but exact thresholds have not been validated in pregnancy.33,34
An unsedated, unprepped flexible sigmoidoscopy can be safely performed throughout pregnancy.35 When there is a strong indication, a complete colonoscopy can be performed in the pregnant patient as well.36 Current American Society for Gastrointestinal Endoscopy (ASGE) guidelines suggest placing the patient in the left lateral tilt position to avoid decreased maternal and placental perfusion via compression of the aorta or inferior vena cava and performing endoscopy during the second trimester, although trimester-specific timing is not always feasible by indication.37
Medication use and safety
IBD medications are a priority topic of concern among pregnant patients or those considering conception.38 Comprehensive data from the PIANO (Pregnancy in Inflammatory Bowel Disease and Neonatal Outcomes) registry has shown that most IBD drugs do not result in adverse pregnancy outcomes and should be continued.39 The use of biologics and thiopurines, either in combination or alone, is not related to an increased risk of congenital malformations, spontaneous abortion, preterm birth, low birth weight, or infections during the child’s first year of life.7,39 Developmental milestones also remain unaffected.39 Here, we will discuss safety considerations during pregnancy (see Table).
5-aminosalycylic acid. 5-aminosalicylic acid (5-ASA) agents are generally low risk during pregnancy and should be continued.40-41 Sulfasalazine does interfere with folate metabolism, but by increasing folic acid supplementation to 2 grams per day, sulfasalazine can be continued throughout pregnancy, as well.42
Corticosteroids. Intrapartum corticosteroid use is associated with an increased risk of gestational diabetes and adrenal insufficiency when used long term.43-45 Short-term use may, however, be necessary to control an acute flare. The lowest dose for the shortest duration possible is recommended. Because of its high first-pass metabolism, budesonide is considered low risk in pregnancy.
Methotrexate. Methotrexate needs to be stopped at least 3 months prior to conception and should be avoided throughout pregnancy. Use during pregnancy can result in spontaneous abortions, as well as embryotoxicity.46
Thiopurines (6-mercaptopurine and azathioprine). Patients who are taking thiopurines prior to conception to maintain remission can continue to do so. Data on thiopurines from the PIANO registry has shown no increase in spontaneous abortions, congenital malformations, low birth weight, preterm birth, rates of infection in the child, or developmental delays.47-51
Calcineurin inhibitors (cyclosporine and tacrolimus). Calcineurin inhibitors are reserved for the management of acute severe UC. Safety data on calcineurin inhibitors is conflicting, and there is not enough information at this time to identify risk during pregnancy. Cyclosporine can be used for salvage therapy if absolutely needed, and there are case reports of its successful using during pregnancy.16,52
Biologic therapies. With the exception of certolizumab, all of the currently used biologics are actively transported across the placenta.39,53,54 Intrapartum use of biologic therapies does not worsen pregnancy or neonatal outcomes, including the risk for intensive care unit admission, infections, and developmental milestones.39,47
While drug concentrations may vary slightly during pregnancy, these changes are not substantial enough to warrant more frequent monitoring or dose adjustments, and prepregnancy weight should be used for dosing.55,56
Antitumor necrosis factor agents used in IBD include infliximab, adalimumab, certolizumab, and golimumab.57 All are low risk for pregnant patients and their offspring. Dosage timings can be adjusted, but not stopped, to minimize exposure to the child; however, it cannot be adjusted for certolizumab pegol because of its lack of placental transfer.58-59
Natalizumab and vedolizumab are integrin receptor antagonists and are also low risk in pregnancy.57;60-62;39
Ustekinumab, an interleukin-12/23 antagonist, can be found in infant serum and cord blood, as well. Health outcomes are similar in the exposed mother and child, however, compared with those of the general population.39;63-64
Small molecule drugs. Unlike monoclonal antibodies, which do not cross the placenta in large amounts until early in the second trimester, small molecules can cross in the first trimester during the critical period of organogenesis.
The two small molecule agents currently approved for use in UC are tofacitinib, a janus kinase inhibitor, and ozanimod, a sphingosine-1-phosphate receptor agonist.65-66 Further data are still needed to make recommendations on the use of tofacitinib and ozanimod in pregnancy. At this time, we recommend weighing the risks (unknown risk to human pregnancy) vs. benefits (controlled disease activity with clear risk of harm to mother and baby from flare) in the individual patient before counseling on use in pregnancy.
Delivery
Mode of delivery
The mode of delivery should be determined by the obstetrician. C-section is recommended for patients with active perianal disease or, in some cases, a history of ileal pouch anal anastomosis (IPAA).67-68 Vaginal delivery in the setting of perianal disease has been shown to increase the risk of fourth-degree laceration and anal sphincter dysfunction in the future.26-27 Anorectal motility may be impacted by IPAA construction and vaginal delivery independently of each other. It is therefore suggested that vaginal delivery be avoided in patients with a history of IPAA to avoid compounding the risk. Some studies do not show clear harm from vaginal delivery in the setting of IPAA, however, and informed decision making among all stakeholders should be had.27;69-70
Anticoagulation
The incidence of venous thromboembolism (VTE) is elevated in patients with IBD during pregnancy, and up to 12 weeks postpartum, compared with pregnant patients without IBD.71-72 VTE for prophylaxis is indicated in the pregnant patient while hospitalized and potentially thereafter depending on the patient’s risk factors, which may include obesity, prior personal history of VTE, heart failure, and prolonged immobility. Unfractionated heparin, low molecular weight heparin, and warfarin are safe for breastfeeding women.16,73
Postpartum care of mother
There is a risk of postpartum flare, occurring in about one third of patients in the first 6 months postpartum.74-75 De-escalating therapy during delivery or immediately postpartum is a predictor of a postpartum flare.75 If no infection is present and the timing interval is appropriate, biologic therapies should be continued and can be resumed 24 hours after a vaginal delivery and 48 hours after a C-section.16,76
NSAIDs and opioids can be used for pain relief but should be avoided in the long-term to prevent flares (NSAIDs) and infant sedation (associated with opioids) when used while breastfeeding.77 The LactMed database is an excellent resource for clarification on risk of medication use while breastfeeding.78
In particular, contraception should be addressed postpartum. Exogenous estrogen use increases the risk of VTE, which is already increased in IBD; nonestrogen containing, long-acting reversible contraception is preferred.79-80 Progestin-only implants or intrauterine devices may be used first line. The efficacy of oral contraceptives is theoretically reduced in those with rapid bowel transit, active small bowel inflammation, and prior small bowel resection, so adding another form of contraception is recommended.16,81
Source: American Gastroenterological Association
Postdelivery care of baby
Breastfeeding
Guidelines regarding medication use during breastfeeding are similar to those in pregnancy (see Table). Breastfeeding on biologics and thiopurines can continue without interruption in the child. Thiopurine concentrations in breast milk are low or undetectable.82,78 TNF receptor antagonists, anti-integrin therapies, and ustekinumab are found in low to undetectable levels in breast milk, as well.78
On the other hand, the active metabolite of methotrexate is detectable in breast milk and most sources recommend not breastfeeding on methotrexate. At doses used in IBD (15-25 milligrams per week), some experts have suggested avoiding breastfeeding for 24 hours following a dose.57,78 It is the practice of this author to recommend not breastfeeding at all on methotrexate.
5-ASA therapies are low risk for breastfeeding, but alternatives to sulfasalazine are preferred. The sulfapyridine metabolite transfers to breast milk and may cause hemolysis in infants born with a glucose-6-phosphate dehydrogenase deficiency.78
With regards to calcineurin inhibitors, tacrolimus appears in breast milk in low quantities, while cyclosporine levels are variable. Data from the National Transplantation Pregnancy Registry suggest that these medications can be used at the time of breastfeeding with close monitoring.78
There is not enough data on small molecule therapies at this time to support breastfeeding safety, and it is our practice to not recommend breastfeeding in this scenario.
The transfer of steroids to the child via breast milk does occur but at subtherapeutic levels.16 Budesonide has high first pass metabolism and is low risk during breastfeeding.83-84 As far as is known, IBD maintenance medications do not suppress lactation. The use of intravenous corticosteroids can, however, temporarily decrease milk production.16,85
Vaccines
Vaccination of infants can proceed as indicated by the Center for Disease Control and Prevention guidelines, with one exception. If the child’s mother was exposed to any biologic agents (not including certolizumab) during the third trimester, any live vaccines should be withheld in the first 6 months of life. In the United States, this restriction currently only applies to the rotavirus vaccine, which is administered starting at the age of 2 months.16,86 Notably, inadvertent administration of the rotavirus vaccine in the biologic-exposed child does not appear to result in any adverse effects.87 Immunity is achieved even if the child is exposed to IBD therapies through breast milk.88
Developmental milestones
Infant exposure to biologics and thiopurines has not been shown to result in any developmental delays. The PIANO study measured developmental milestones at 48 months from birth and found no differences when compared with validated population norms.39 A separate study observing childhood development up to 7 years of age in patients born to mothers with IBD found similar cognitive scores and motor development when compared with those born to mothers without IBD.89
Conclusion
Women considering conception should be optimized prior to pregnancy and maintained on appropriate medications throughout pregnancy and lactation to achieve a healthy pregnancy for both mother and baby. To date, biologics and thiopurines are not associated with adverse pregnancy outcomes. More data are needed for small molecules.
Dr. Chugh is an advanced inflammatory bowel disease fellow in the division of gastroenterology at the University of California San Francisco. Dr. Mahadevan is professor of medicine and codirector at the Center for Colitis and Crohn’s Disease in the division of gastroenterology at the University of California San Francisco. Dr. Mahadevan has potential conflicts related to AbbVie, Janssen, BMS, Takeda, Pfizer, Lilly, Gilead, Arena, and Prometheus Biosciences.
References
1. Ye Y et al. Inflamm Bowel Dis. 2020;26:619-25.
2. Sykora J et al. World J Gastroenterol. 2018;24:2741-63.
3. Murakami Y et al. J Gastroenterol 2019;54:1070-7.
4. Hashash JG and Kane S. Gastroenterol Hepatol. (N Y) 2015;11:96-102.
5. Miller JP. J R Soc Med. 1986;79:221-5.
6. Cornish J et al. Gut. 2007;56:830-7.
7. Leung KK et al. Inflamm Bowel Dis. 2021;27:550-62.
8. O’Toole A et al. Dig Dis Sci. 2015;60:2750-61.
9. Nguyen GC et al. Inflamm Bowel Dis. 2008;14:1105-11.
10. Lee HH et al. Aliment Pharmacol Ther. 2020;51:861-9.
11. Kim MA et al. J Crohns Colitis. 2021;15:719-32.
12. Conradt E et al. Pediatrics. 2019;144.
13. ACOG Committee Opinion No. 762: Prepregnancy Counseling. Obstet Gynecol. 2019;133:e78-e89.
14. Farraye FA et al. Am J Gastroenterol. 2017;112:241-58.
15. Lee S et al. J Crohns Colitis. 2018;12:702-9.
16. Mahadevan U et al. Inflamm Bowel Dis. 2019;25:627-41.
17. Ward MG et al. Inflamm. Bowel Dis 2015;21:2839-47.
18. Battat R et al. Inflamm Bowel Dis. 2014;20:1120-8.
19. Pedersen N et al. Aliment Pharmacol Ther. 2013;38:501-12.
20. Annese V. Pharmacol Res. 2020;159:104892.
21. Bennett RA et al. Gastroenterology. 1991;100:1638-43.
22. Turpin W et al. Inflamm Bowel Dis. 2018;24:1133-48.
23. de Lima A et al. Clin Gastroenterol Hepatol. 2016;14:1285-92 e1.
24. Selinger C et al. Frontline Gastroenterol. 2021;12:182-7.
25. Mahadevan U et al. Gastroenterology. 2007;133:1106-12.
26. Hatch Q et al. Dis Colon Rectum. 2014;57:174-8.
27. Foulon A et al. Inflamm Bowel Dis. 2017;23:712-20.
28. Norgard B et al. Am J Gastroenterol. 2007;102:1947-54.
29. Broms G et al. Scand J Gastroenterol 2016;51:1462-9.
30. Meyer A et al. Aliment Pharmacol Ther. 2020;52:1480-90.
31. Kammerlander H et al. Inflamm Bowel Dis. 2017;23:1011-8.
32. Tandon P et al. J Clin Gastroenterol. 2019;53:574-81.
33. Kammerlander H et al. Inflamm Bowel Dis. 2018;24:839-48.
34. Julsgaard M et al. Inflamm Bowel Dis. 2017;23:1240-6.
35. Ko MS et al. Dig Dis Sci. 2020;65:2979-85.
36. Cappell MS et al. J Reprod Med. 2010;55:115-23.
37. Committee ASoP et al. Gastrointest Endosc. 2012;76:18-24.
38. Aboubakr A et al. Dig Dis Sci. 2021;66:1829-35.
39. Mahadevan U et al. Gastroenterology. 2021;160:1131-9.
40. Diav-Citrin O et al. Gastroenterology. 1998;114:23-8.
41. Rahimi R et al. Reprod Toxicol. 2008;25:271-5.
42. Norgard B et al. Aliment Pharmacol Ther. 2001;15:483-6.
43. Leung YP et al. J Crohns Colitis. 2015;9:223-30.
44. Schulze H et al. Aliment Pharmacol Ther. 2014;40:991-1008.
45. Szymanska E et al. J Gynecol Obstet Hum Reprod. 2021;50:101777.
46. Weber-Schoendorfer C et al. Arthritis Rheumatol. 2014;66:1101-10.
47. Nielsen OH et al. Clin Gastroenterol Hepatol. 2022 Jan;20(1):74-87.e3.
48. Coelho J et al. Gut. 2011;60:198-203.
49. Sheikh M et al. J Crohns Colitis. 2015;9:680-4.
50. Kanis SL et al. Clin Gastroenterol Hepatol. 2017;15:1232-41 e1.
51. Mahadevan U et al. Inflamm Bowel Dis. 2018;24:2494-500.
52. Rosen MH et al. Inflamm Bowel Dis. 2020;26:971-3.
53. Porter C et al. J Reprod Immunol. 2016;116:7-12.
54. Mahadevan U et al. Clin Gastroenterol Hepatol. 2013;11:286-92; quiz e24.
55. Picardo S and Seow CH. Best Pract Res Clin Gastroenterol. 2020;44-5:101670.
56. Flanagan E et al. Aliment Pharmacol Ther. 2020;52:1551-62.
57. Singh S et al. Gastroenterology. 2021;160:2512-56 e9.
58. de Lima A et al. Gut. 2016;65:1261-8.
59. Julsgaard M et al. Inflamm Bowel Dis. 2020;26:93-102.
60. Wils P et al. Aliment Pharmacol Ther. 2021;53:460-70.
61. Mahadevan U et al. Aliment Pharmacol Ther. 2017;45:941-50.
62. Bar-Gil Shitrit A et al. Am J Gastroenterol. 2019;114:1172-5.
63. Klenske E et al. J Crohns Colitis. 2019;13:267-9.
64. Matro R et al. Gastroenterology. 2018;155:696-704.
65. Feuerstein JD et al. Gastroenterology. 2020;158:1450-61.
66. Sandborn WJ et al. J Crohns Colitis. 2021 Jul 5;15(7):1120-1129.
67. Lamb CA et al. Gut. 2019;68:s1-s106.
68. Nguyen GC et al. Gastroenterology. 2016;150:734-57 e1.
69. Ravid A et al. Dis Colon Rectum. 2002;45:1283-8.
70. Seligman NS et al. J Matern Fetal Neonatal Med. 2011;24:525-30.
71. Kim YH et al. Medicine (Baltimore). 2019;98:e17309.
72. Hansen AT et al. J Thromb Haemost. 2017;15:702-8.
73. Bates SM et al. J Thromb Thrombolysis. 2016;41:92-128.
74. Bennett A et al. Inflamm Bowel Dis. 2021 May 17;izab104.
75. Yu A et al. Inflamm Bowel Dis. 2020;26:1926-32.
76. Mahadevan U et al. Gastroenterology. 2017;152:451-62 e2.
77. Long MD et al. J Clin Gastroenterol. 2016;50:152-6.
78. Drugs and Lactation Database (LactMed). 2006 ed. Bethesda, MD: National Library of Medicine (US), 2006-2021.
79. Khalili H et al. Gut. 2013;62:1153-9.
80. Long MD and Hutfless S. Gastroenterology. 2016;150:1518-20.
81. Centers for Disease Control and Prevention. U S. Medical Eligibility Criteria for Contraceptive Use, 2010. MMWR Recomm Rep. 2010;59:1-86.
82. Angelberger S et al. J Crohns Colitis. 2011;5:95-100.
83. Vestergaard T et al. Scand J Gastroenterol. 2018;53:1459-62.
84. Beaulieu DB et al. Inflamm Bowel Dis. 2009;15:25-8.
85. Anderson PO. Breastfeed Med. 2017;12:199-201.
86. Wodi AP et al. MMWR Morb Mortal Wkly Rep. 2021;70:189-92.
87. Chiarella-Redfern H et al. Inflamm Bowel Dis. 2022 Jan 5;28(1):79-86.
88. Beaulieu DB et al. Clin Gastroenterol Hepatol. 2018;16:99-105.
89. Friedman S et al. J Crohns Colitis. 2020 Dec 2;14(12):1709-1716.
Definitive diverticular hemorrhage: Diagnosis and management
Diverticular hemorrhage is the most common cause of colonic bleeding, accounting for 20%-65% of cases of severe lower intestinal bleeding in adults.1 Urgent colonoscopy after purging the colon of blood, clots, and stool is the most accurate method of diagnosing and guiding treatment of definitive diverticular hemorrhage.2-5 The diagnosis of definitive diverticular hemorrhage depends upon identification of some stigmata of recent hemorrhage (SRH) in a single diverticulum (TIC), which can include active arterial bleeding, oozing, non-bleeding visible vessel, adherent clot, or flat spot.2-4 Although other approaches, such as nuclear medicine scans and angiography of various types (CT, MRI, or standard angiography), for the early diagnosis of patients with severe hematochezia are utilized in many medical centers, only active bleeding can be detected by these techniques. However, as subsequently discussed, this SRH is documented in only 26% of definitive diverticular bleeds found on urgent colonoscopy, so diagnostic yields of these techniques will be low.2-5

The diagnosis of patients with severe hematochezia and diverticulosis, as well as triage of all of them to specific medical, endoscopic, radiologic, or surgical management, is facilitated by an urgent endoscopic approach.2-5 Patients who are diagnosed with definitive diverticular hemorrhage on colonoscopy represent about 30% of all true TIC bleeds when urgent colonoscopy is the management approach.2-5 That is because approximately 50% of all patients with colon diverticulosis and first presentation of severe hematochezia have incidental diverticulosis; they have colonic diverticulosis, but another site of bleeding is identified as the cause of hemorrhage in the gastrointestinal tract.2-4 Presumptive diverticular hemorrhage is diagnosed when colonic diverticulosis without TIC stigmata are found but no other GI bleeding source is found on colonoscopy, anoscopy, enteroscopy, or capsule endoscopy.2-5 In our experience with urgent colonoscopy, the presumptive diverticular bleed group accounts for about 70% of patients with documented diverticular hemorrhage (e.g., not including incidental diverticulosis bleeds but combining subgroups of patients with either definitive or presumptive TIC diagnoses as documented TIC hemorrhage).

Clinical presentation
Patients with diverticular hemorrhage present with severe, painless large volume hematochezia. Hematochezia may be self-limited and spontaneously resolve in 75%-80% of all patients but with high rebleeding rates up to 40%.5-7 Of all patients with diverticulosis, only about 3%-5% develop diverticular hemorrhage.8 Risk factors for diverticular hemorrhage include medications (e.g., nonsteroidal anti-inflammatory drugs – NSAIDs, antiplatelet drugs, and anticoagulants) and other clinical factors, such as older age, low-fiber diet, and chronic constipation.9,10 On urgent colonoscopy, more than 70% of diverticulosis in U.S. patients are located anatomically in the descending colon or more distally. In contrast, about 60% of definitive diverticular hemorrhage cases in our experience had diverticula with stigmata identified at or proximal to the splenic flexure.2,4,11

Pathophysiology
Colonic diverticula are herniations of mucosa and submucosa with colonic arteries that penetrate the muscular wall. Bleeding can occur when there is asymmetric rupture of the vasa recta at either the base of the diverticulum or the neck.4 Thinning of the mucosa on the luminal surface (such as that resulting from impacted fecaliths and stool) can cause injury to the site of the penetrating vessels, resulting in hemorrhage.12
Initial management
Patients with acute, severe hematochezia should be triaged to an inpatient setting with a monitored bed. Admission to an intensive care unit should be considered for patients with hemodynamic instability, persistent bleeding, and/or significant comorbidities. Patients with TIC hemorrhage often require resuscitation with crystalloids and packed red blood cell transfusions for hemoglobin less than 8 g/dl.4 Unlike upper GI hemorrhage, which has been extensively reported on, data regarding a more restrictive transfusion threshold, compared with a liberal transfusion threshold, in lower intestinal bleeding are very limited. Correction of underlying coagulopathies is recommended but should be individualized, particularly in those patients on antithrombotic agents or with underlying bleeding disorders.
Urgent diagnosis and hemostasis
Urgent colonoscopy within 24 hours is the most accurate way to make a diagnosis of definitive diverticular hemorrhage and to effectively and safely treat them.2-4,10,11 For patients with severe hematochezia, when the colonoscopy is either not available in a medical center or does not reveal the source of bleeding, nuclear scintigraphy or angiography (CT, MRI, or interventional radiology [IR]) are recommended. CT angiography may be particularly helpful to diagnose patients with hemodynamic instability who are suspected to have active TIC bleeding and are not able to complete a bowel preparation. However, these imaging techniques require active bleeding at the time of the study to be diagnostic. This SRH is also uncommon for definitive diverticular hemorrhage, so the diagnostic yield is usually quite low.2-5,10,11 An additional limitation of scintigraphy and CT or MRI angiography is that, if active bleeding is found, some other type of treatment, such as colonoscopy, IR angiography, or surgery, will be required for definitive hemostasis.
For urgent colonoscopy, adequate colon preparation with a large volume preparation (6-8 liters of polyethylene glycol-based solution) is recommended to clear stool, blood, and clots to allow endoscopic visualization and localization of the bleeding source. Use of a nasogastric tube should be considered if the patient is unable to drink enough prep.2-4,13 Additionally, administration of a prokinetic agent, such as Metoclopramide, may improve gastric emptying and tolerance of the prep. During colonoscopy, careful inspection of the colonic mucosa during insertion and withdrawal is important since lesions may bleed intermittently and SRH can be missed. An adult or pediatric colonoscope with a large working channel (at least 3.3 mm) is recommended to facilitate suctioning of blood clots and stool, as well as allow the passage of endoscopic hemostasis accessories. Targeted water-jet irrigation, an expert colonoscopist, a cap attachment, and adequate colon preparation are all predictors for improved diagnosis of definitive diverticular hemorrhage.4,14
SRH in definitive TIC bleeds all have a high risk of TIC rebleeding,2-4,10,11 including active bleeding, nonbleeding visible vessel, adherent clot, and a flat spot (See Figure).
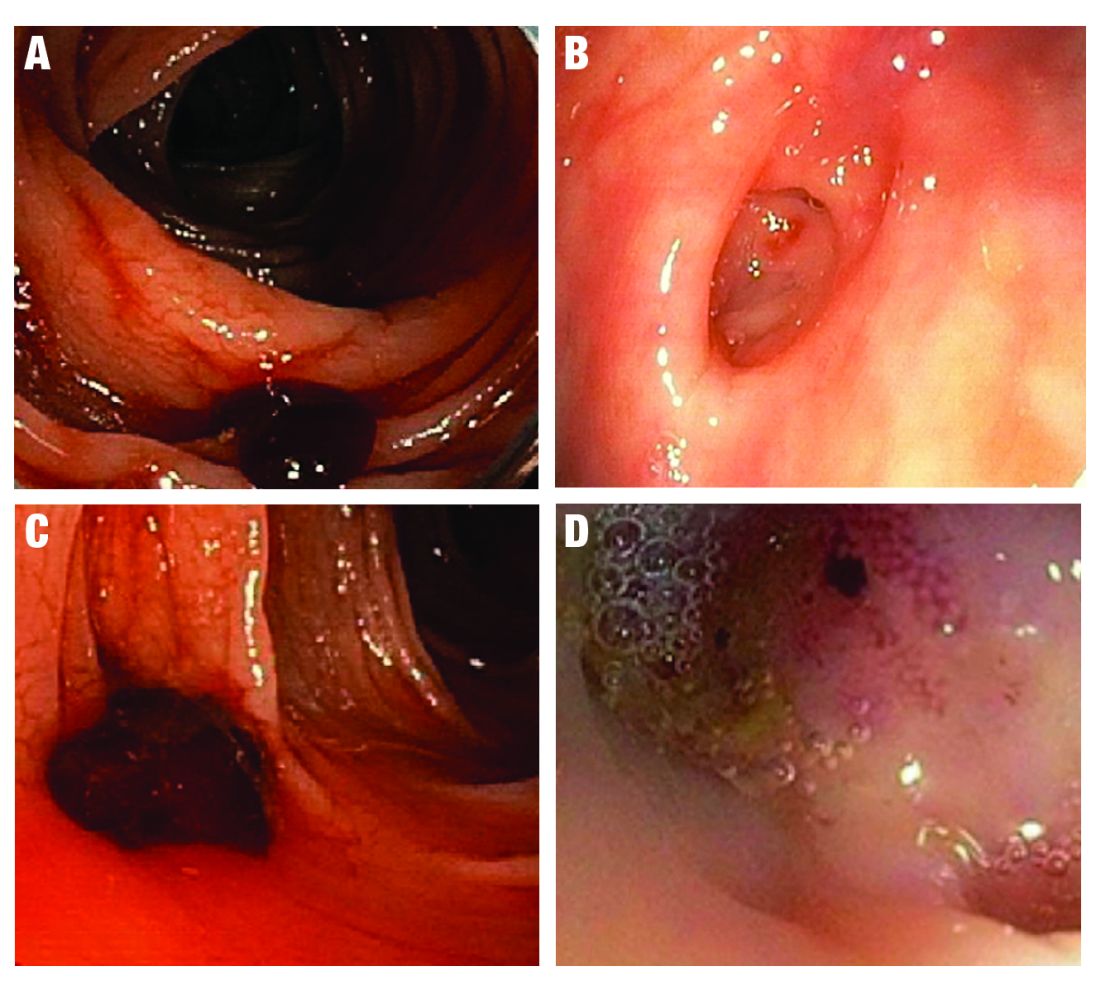
Based on CURE Hemostasis Group data of 118 definitive TIC bleeds, 26% had active bleeding, 24% had a nonbleeding visible vessel, 37% had an adherent clot, and 13% had a flat spot (with underlying arterial blood flow by Doppler probe monitoring).4 Approximately 50% of the SRH were found in the neck of the TIC and 50% at the base, with actively bleeding cases more often from the base. In CURE Doppler endoscopic probe studies, 90% of all stigmata had an underlying arterial blood flow detected with the Doppler probe.4,10 The Doppler probe is reported to be very useful for risk stratification and to confirm obliteration of the arterial blood flow underlying SRH for definitive hemostasis.4,10
Endoscopic treatment
Given high rates of rebleeding with medical management alone, definitive TIC hemorrhage can be effectively and safely treated with endoscopic therapies once SRH are localized.4,10 Endoscopic therapies that have been reported in the literature include electrocoagulation, hemoclip, band ligation, and over-the-scope clip. Four-quadrant injection of 1:20,000 epinephrine around the SRH can improve visualization of SRH and provide temporary control of bleeding, but it should be combined with other modalities because of risk of rebleeding with epinephrine alone.15 Results from studies reporting rates of both early rebleeding (occurring within 30 days) and late rebleeding (occurring after 30 days) are listed in the Table. 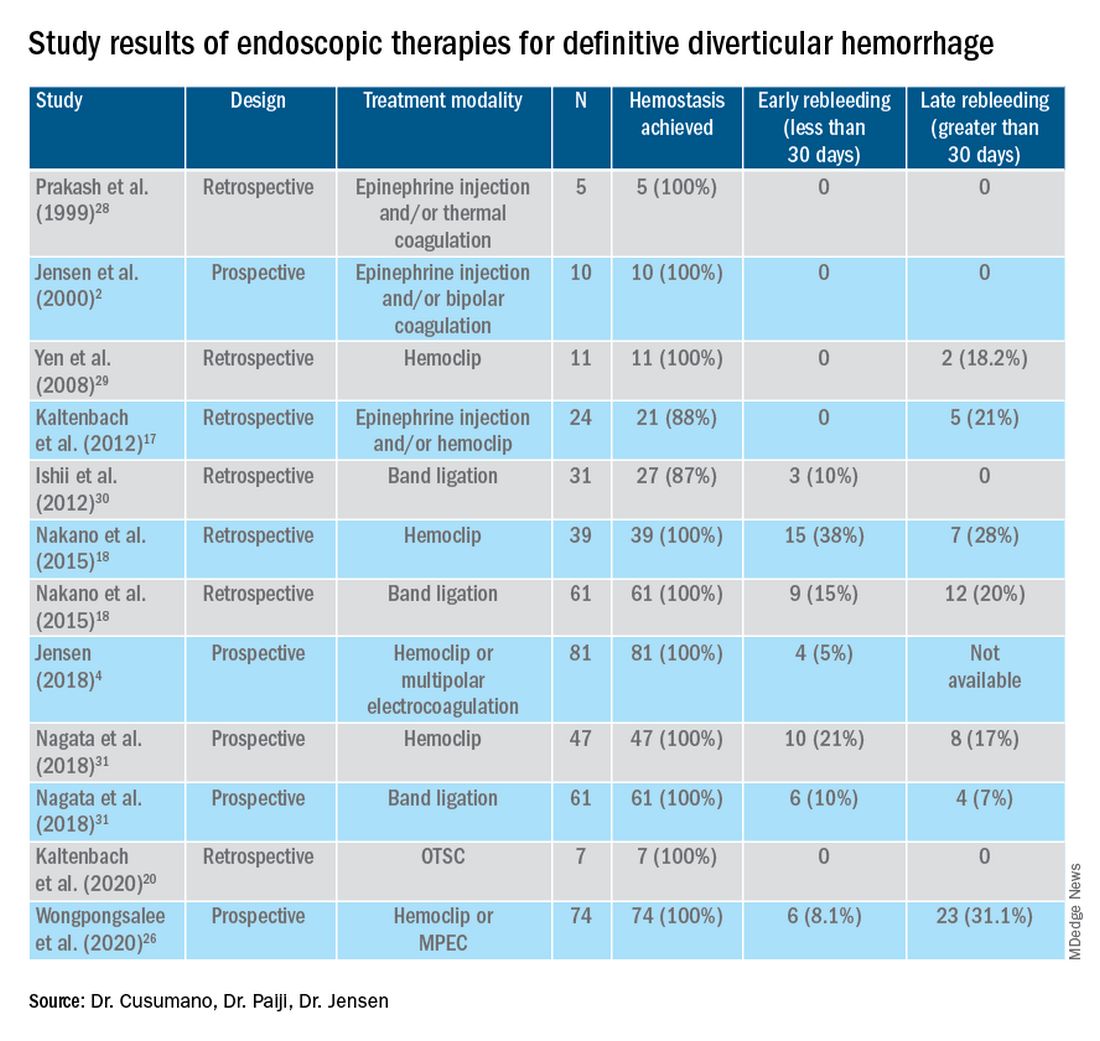
Multipolar electrocoagulation (MPEC), which utilizes a focal electric current to generate heat, can coaptively coagulate small TIC arteries.16 For SRH in the neck of TIC, MPEC is effective for coaptive coagulation at a power of 12-15 watts in 1-2 second pulses with moderate laterally applied tamponade pressure. MPEC should be avoided for treating SRH at the TIC base because of lack of muscularis propria and higher risk of perforation.
Hemoclip therapy has been reported to be safe and efficacious in treatment of definitive TIC hemorrhage, by causing mechanical hemostasis with occlusion of the bleeding artery.16 Hemoclips are recommended to treat stigmata in the base of TICs and should be targeted on either side of visible vessel in order to occlude the artery underneath it.4,10 With a cap on the tip of the colonoscope, suctioning can evert TICs, allowing more precise placement of hemoclip on SRH in the base of the TIC.17 Hemoclip retention rates vary with different models and can range from less than 7 days to more than 4 weeks. Hemoclips can also mark the site if early rebleeding occurs; then, reintervention (e.g., repeat endoscopy or angioembolization) is facilitated.
Another treatment is endoscopic band ligation, which provides mechanical hemostasis. Endoscopic band ligation has been reported to be efficacious for TIC hemorrhage.18 Suctioning the TIC with the SRH into the distal cap and deploying a band leads to obliteration of vessels and potentially necrosis and disappearance of banded TIC.16 This technique carries a risk of perforation because of the thin walls of TICs. This risk may be higher for right-sided colon lesions since an exvivo colon specimen study reported serosal entrapment and inclusion of muscularis propria postband ligation, both of which may result in ischemia of intestinal wall and delayed perforation.19
Over-the-scope clip (OTSC) has been reported in case series for treatment of definitive TIC hemorrhage. With a distal cap and large clip, suctioning can evert TICs and facilitate deployment over the SRH.20,21 OTSC can grasp an entire TIC with the SRH and obliterate the arterial blood flow with a single clip.20,21 No complications have been reported yet for treatment of TIC hemorrhage. However, the OTSC system is relatively expensive when compared with other modalities.
After endoscopic treatment is performed, four-quadrant spot tattooing is recommended adjacent to the TIC with the SRH. This step will facilitate localization and treatment in the case of TIC rebleeding.4,10
Outcomes following endoscopic treatment
Following endoscopic treatment, patients should be monitored for early and late rebleeding. In a pooled analysis of case series composed of 847 patients with TIC bleeding, among the 137 patients in which endoscopic hemostasis was initially achieved, early rebleeding occurred in 8% and late rebleeding occurred in 12% of patients.22 Risk factors for TIC rebleeding within 30 days were residual arterial blood flow following hemostasis and early reinitiation of antiplatelet agents.
Remote treatment of TIC hemorrhage distant from the SRH is a significant risk factor for early TIC rebleeding.4, 10 For example, using hemoclips to close the mouth of a TIC when active bleeding or an SRH is located in the TIC base often fails because arterial flow remains open in the base and the artery is larger there.4,10 This example highlights the importance of focal obliteration of arterial blood flow underlying SRH in order to achieve definitive hemostasis.4,10
Salvage treatments
For TIC hemorrhage that is not controlled by endoscopic therapy, transcatheter arterial embolization (TAE) is recommended. If bleeding rate is high enough (at least 0.5 milliliters per minute) to be detected by angiography, TAE can serve as an effective method of diagnosis and immediate hemostasis.23 However, the most common major complication of embolization is intestinal ischemia. The incidence of intestinal ischemia has been reported as high as 10%, with highest risk with embolization of at least three vasa recta.24
Surgery is also recommended if TIC hemorrhage cannot be controlled with endoscopic therapy or TAE. Segmental colectomy is recommended if the bleeding site can be localized before surgery with colonoscopy or angiography resulting from significantly lower perioperative morbidity than subtotal colectomy.25 However, subtotal colectomy may be necessary if preoperative localization of bleeding is unsuccessful.
There are very few reports of short- or long-term results that compare endoscopy, TAE, and surgery for management of TIC bleeding. However, a recent retrospective study reported better outcomes with endoscopic treatment of definitive TIC bleeding.26 Patients who underwent endoscopic treatment had fewer RBC transfusions, shorter hospitalizations, and lower rates of postprocedure complications.
Management after cessation of hemorrhage
Medical management is important following an episode of TIC hemorrhage. A mainstay is daily fiber supplementation every morning and stool softener in the evening. Furthermore, patients are advised to drink an extra liter of fluids (not containing alcohol or caffeine) daily. By reducing colon transit time and increasing stool weight, these measures can help control constipation and prevent future complications of TIC disease.27
Patients with recurrent TIC hemorrhage should undergo evaluation for elective surgery, provided they are appropriate surgical candidates. If preoperative localization of bleeding site is successful, segmental colectomy is preferred. Segmental resection is associated with significantly decreased rebleeding rate, with lower rates of morbidity compared with subtotal colectomy.32
Chronic NSAIDs, aspirin, and antiplatelet drugs are risk factors for recurrent TIC hemorrhage, and avoiding these medications is recommended if possible.33,34 Although anticoagulants have shown to be associated with increased risk of all-cause gastrointestinal bleeding, these agents have not been shown to increase risk of recurrent TIC hemorrhage in recent large retrospective studies. Since antiplatelet and anticoagulation agents serve to reduce risk of thromboembolic events, the clinician who recommended these medications should be consulted after a TIC bleed to re-evaluate whether these medications can be discontinued or reduced in dose.
Conclusion
The most effective way to diagnose and treat definitive TIC hemorrhage is to perform an urgent colonoscopy within 24 hours to identify and treat TIC SRH. This procedure requires thoroughly cleansing the colon first, as well as an experienced colonoscopist who can identify and treat TIC SRH to obliterate arterial blood flow underneath SRH and achieve definitive TIC hemostasis. Other approaches to early diagnosis include nuclear medicine scintigraphy or angiography (CT, MRI, or IR). However, these techniques can only detect active bleeding which is documented in only 26% of colonoscopically diagnosed definitive TIC hemorrhages. So, the expected diagnostic yield of these tests will be low. When urgent colonoscopy fails to make a diagnosis or TIC bleeding continues, TAE and/or surgery are recommended. After definitive hemostasis of TIC hemorrhage and for long term management, control of constipation and discontinuation of chronic NSAIDs and antiplatelet drugs (if possible) are recommended to prevent recurrent TIC hemorrhage.
Dr. Cusumano and Dr. Paiji are fellow physicians in the Vatche and Tamar Manoukian Division of Digestive Diseases at University of California Los Angeles. Dr. Jensen is a professor of medicine in Vatche and Tamar Manoukian Division of Digestive Diseases and is with the CURE Digestive Diseases Research Center at the VA Greater Los Angeles Healthcare System, Calif. All authors declare that they have no competing interests or disclosures.
References
1. Longstreth GF. Am J Gastroenterol. 1997;92(3):419-24.
2. Jensen DM et al. The New England Journal of Medicine. 2000;342(2):78-82.
3. Jensen DM et al. Techniques in Gastrointestinal Endoscopy. 2001;3(4):192-8.
4. Jensen DM. Am J Gastroenterol. 2018;113(11):1570-3.
5. Zuckerman GR et al. Gastrointestinal Endoscopy. 1999;49(2):228-38.
6. Stollman N et al. Lancet. 2004;363(9409):631-9.
7. McGuire HH et al. Ann Surg. 1994;220(5):653-6.
8. McGuire HH et al. Ann Surg. 1972;175(6):847-55.
9. Strate LL et al. Clinical gastroenterology and hepatol. 2008;6(9):1004-10.
10. Jensen DM et al. Gastrointestinal endoscopy. 2016;83(2):416-23.
11. Jensen DM et al. Gastrointest Endosc Clin N Am. 1997;7(3):477-98.
12. Maykel JA et al. Clin Colon Rectal Surg. 2004;17(3):195-204.
13. Green BT et al. Am J Gastroenterol. 2005;100(11):2395-402.
14. Niikura R et al. Journal of Clinical Gastroenterol. 2015;49(3):e24-30.
15. Bloomfeld RS et al. Am J Gastroenterol. 2001;96(8):2367-72.
16. Parsi MA,et al. VideoGIE. 2019;4(7):285-99.
17. Kaltenbach T et al. Clinical Gastroenterology and Hepatol. 2012;10(2):131-7.
18. Nakano K et al. Endosc Int Open. 2015;3(5):E529-33.
19. Barker KB et al. Gastrointestinal Endoscopy. 2005;62(2):224-7.
20. Kaltenbach T et al. Gastrointest Endosc Clin N Am. 2020;30(1):13-23.
21. Yamazaki K et al. VideoGIE. 2020;5(6):252-4.
22. Strate LL et al. Clinical Gastroenterology and Hepatol. 2010;8(4):333-43.
23. Evangelista et al. J Vasc Interv Radiol. 2000;11(5):601-6.
24. Kodani M et al. J Vasc Interv Radiol. 2016;27(6):824-30.
25. Mohammed et al. Clin Colon Rectal Surg. 2018;31(4):243-50.
26. Wongpongsalee T et al. Gastrointestinal Endoscopy. 2020;91(6):AB471-2.
27. Böhm SK. Viszeralmedizin. 2015;31(2):84-94.
28. Prakash C et al. Endoscopy. 1999;31(6):460-3.
29. Yen EF et al. Digestive Diseases and Sciences. 2008;53(9):2480-5.
30. Ishii N et al. Gastrointestinal Endoscopy. 2012;75(2):382-7.
31. Nagata N et al. Gastrointestinal Endoscopy. 2018;88(5):841-53.e4.
32. Parkes BM et al. Am Surg. 1993;59(10):676-8.
33. Vajravelu RK et al. Gastroenterology. 2018;155(5):1416-27.
34. Oakland K et al. Clin Gastroenterol Hepatol. 2019;17(7):1276-84.e3.
35. Yamada A et al. Dis Colon Rectum. 2008;51(1):116-20.
36. Coleman CI et al. Int J Clin Pract. 2012;66(1):53-63.
37. Holster IL et al. Gastroenterology. 2013;145(1):105-12.e15.
Diverticular hemorrhage is the most common cause of colonic bleeding, accounting for 20%-65% of cases of severe lower intestinal bleeding in adults.1 Urgent colonoscopy after purging the colon of blood, clots, and stool is the most accurate method of diagnosing and guiding treatment of definitive diverticular hemorrhage.2-5 The diagnosis of definitive diverticular hemorrhage depends upon identification of some stigmata of recent hemorrhage (SRH) in a single diverticulum (TIC), which can include active arterial bleeding, oozing, non-bleeding visible vessel, adherent clot, or flat spot.2-4 Although other approaches, such as nuclear medicine scans and angiography of various types (CT, MRI, or standard angiography), for the early diagnosis of patients with severe hematochezia are utilized in many medical centers, only active bleeding can be detected by these techniques. However, as subsequently discussed, this SRH is documented in only 26% of definitive diverticular bleeds found on urgent colonoscopy, so diagnostic yields of these techniques will be low.2-5
The diagnosis of patients with severe hematochezia and diverticulosis, as well as triage of all of them to specific medical, endoscopic, radiologic, or surgical management, is facilitated by an urgent endoscopic approach.2-5 Patients who are diagnosed with definitive diverticular hemorrhage on colonoscopy represent about 30% of all true TIC bleeds when urgent colonoscopy is the management approach.2-5 That is because approximately 50% of all patients with colon diverticulosis and first presentation of severe hematochezia have incidental diverticulosis; they have colonic diverticulosis, but another site of bleeding is identified as the cause of hemorrhage in the gastrointestinal tract.2-4 Presumptive diverticular hemorrhage is diagnosed when colonic diverticulosis without TIC stigmata are found but no other GI bleeding source is found on colonoscopy, anoscopy, enteroscopy, or capsule endoscopy.2-5 In our experience with urgent colonoscopy, the presumptive diverticular bleed group accounts for about 70% of patients with documented diverticular hemorrhage (e.g., not including incidental diverticulosis bleeds but combining subgroups of patients with either definitive or presumptive TIC diagnoses as documented TIC hemorrhage).
Clinical presentation
Patients with diverticular hemorrhage present with severe, painless large volume hematochezia. Hematochezia may be self-limited and spontaneously resolve in 75%-80% of all patients but with high rebleeding rates up to 40%.5-7 Of all patients with diverticulosis, only about 3%-5% develop diverticular hemorrhage.8 Risk factors for diverticular hemorrhage include medications (e.g., nonsteroidal anti-inflammatory drugs – NSAIDs, antiplatelet drugs, and anticoagulants) and other clinical factors, such as older age, low-fiber diet, and chronic constipation.9,10 On urgent colonoscopy, more than 70% of diverticulosis in U.S. patients are located anatomically in the descending colon or more distally. In contrast, about 60% of definitive diverticular hemorrhage cases in our experience had diverticula with stigmata identified at or proximal to the splenic flexure.2,4,11
Pathophysiology
Colonic diverticula are herniations of mucosa and submucosa with colonic arteries that penetrate the muscular wall. Bleeding can occur when there is asymmetric rupture of the vasa recta at either the base of the diverticulum or the neck.4 Thinning of the mucosa on the luminal surface (such as that resulting from impacted fecaliths and stool) can cause injury to the site of the penetrating vessels, resulting in hemorrhage.12
Initial management
Patients with acute, severe hematochezia should be triaged to an inpatient setting with a monitored bed. Admission to an intensive care unit should be considered for patients with hemodynamic instability, persistent bleeding, and/or significant comorbidities. Patients with TIC hemorrhage often require resuscitation with crystalloids and packed red blood cell transfusions for hemoglobin less than 8 g/dl.4 Unlike upper GI hemorrhage, which has been extensively reported on, data regarding a more restrictive transfusion threshold, compared with a liberal transfusion threshold, in lower intestinal bleeding are very limited. Correction of underlying coagulopathies is recommended but should be individualized, particularly in those patients on antithrombotic agents or with underlying bleeding disorders.
Urgent diagnosis and hemostasis
Urgent colonoscopy within 24 hours is the most accurate way to make a diagnosis of definitive diverticular hemorrhage and to effectively and safely treat them.2-4,10,11 For patients with severe hematochezia, when the colonoscopy is either not available in a medical center or does not reveal the source of bleeding, nuclear scintigraphy or angiography (CT, MRI, or interventional radiology [IR]) are recommended. CT angiography may be particularly helpful to diagnose patients with hemodynamic instability who are suspected to have active TIC bleeding and are not able to complete a bowel preparation. However, these imaging techniques require active bleeding at the time of the study to be diagnostic. This SRH is also uncommon for definitive diverticular hemorrhage, so the diagnostic yield is usually quite low.2-5,10,11 An additional limitation of scintigraphy and CT or MRI angiography is that, if active bleeding is found, some other type of treatment, such as colonoscopy, IR angiography, or surgery, will be required for definitive hemostasis.
For urgent colonoscopy, adequate colon preparation with a large volume preparation (6-8 liters of polyethylene glycol-based solution) is recommended to clear stool, blood, and clots to allow endoscopic visualization and localization of the bleeding source. Use of a nasogastric tube should be considered if the patient is unable to drink enough prep.2-4,13 Additionally, administration of a prokinetic agent, such as Metoclopramide, may improve gastric emptying and tolerance of the prep. During colonoscopy, careful inspection of the colonic mucosa during insertion and withdrawal is important since lesions may bleed intermittently and SRH can be missed. An adult or pediatric colonoscope with a large working channel (at least 3.3 mm) is recommended to facilitate suctioning of blood clots and stool, as well as allow the passage of endoscopic hemostasis accessories. Targeted water-jet irrigation, an expert colonoscopist, a cap attachment, and adequate colon preparation are all predictors for improved diagnosis of definitive diverticular hemorrhage.4,14
SRH in definitive TIC bleeds all have a high risk of TIC rebleeding,2-4,10,11 including active bleeding, nonbleeding visible vessel, adherent clot, and a flat spot (See Figure).
Based on CURE Hemostasis Group data of 118 definitive TIC bleeds, 26% had active bleeding, 24% had a nonbleeding visible vessel, 37% had an adherent clot, and 13% had a flat spot (with underlying arterial blood flow by Doppler probe monitoring).4 Approximately 50% of the SRH were found in the neck of the TIC and 50% at the base, with actively bleeding cases more often from the base. In CURE Doppler endoscopic probe studies, 90% of all stigmata had an underlying arterial blood flow detected with the Doppler probe.4,10 The Doppler probe is reported to be very useful for risk stratification and to confirm obliteration of the arterial blood flow underlying SRH for definitive hemostasis.4,10
Endoscopic treatment
Given high rates of rebleeding with medical management alone, definitive TIC hemorrhage can be effectively and safely treated with endoscopic therapies once SRH are localized.4,10 Endoscopic therapies that have been reported in the literature include electrocoagulation, hemoclip, band ligation, and over-the-scope clip. Four-quadrant injection of 1:20,000 epinephrine around the SRH can improve visualization of SRH and provide temporary control of bleeding, but it should be combined with other modalities because of risk of rebleeding with epinephrine alone.15 Results from studies reporting rates of both early rebleeding (occurring within 30 days) and late rebleeding (occurring after 30 days) are listed in the Table.
Multipolar electrocoagulation (MPEC), which utilizes a focal electric current to generate heat, can coaptively coagulate small TIC arteries.16 For SRH in the neck of TIC, MPEC is effective for coaptive coagulation at a power of 12-15 watts in 1-2 second pulses with moderate laterally applied tamponade pressure. MPEC should be avoided for treating SRH at the TIC base because of lack of muscularis propria and higher risk of perforation.
Hemoclip therapy has been reported to be safe and efficacious in treatment of definitive TIC hemorrhage, by causing mechanical hemostasis with occlusion of the bleeding artery.16 Hemoclips are recommended to treat stigmata in the base of TICs and should be targeted on either side of visible vessel in order to occlude the artery underneath it.4,10 With a cap on the tip of the colonoscope, suctioning can evert TICs, allowing more precise placement of hemoclip on SRH in the base of the TIC.17 Hemoclip retention rates vary with different models and can range from less than 7 days to more than 4 weeks. Hemoclips can also mark the site if early rebleeding occurs; then, reintervention (e.g., repeat endoscopy or angioembolization) is facilitated.
Another treatment is endoscopic band ligation, which provides mechanical hemostasis. Endoscopic band ligation has been reported to be efficacious for TIC hemorrhage.18 Suctioning the TIC with the SRH into the distal cap and deploying a band leads to obliteration of vessels and potentially necrosis and disappearance of banded TIC.16 This technique carries a risk of perforation because of the thin walls of TICs. This risk may be higher for right-sided colon lesions since an exvivo colon specimen study reported serosal entrapment and inclusion of muscularis propria postband ligation, both of which may result in ischemia of intestinal wall and delayed perforation.19
Over-the-scope clip (OTSC) has been reported in case series for treatment of definitive TIC hemorrhage. With a distal cap and large clip, suctioning can evert TICs and facilitate deployment over the SRH.20,21 OTSC can grasp an entire TIC with the SRH and obliterate the arterial blood flow with a single clip.20,21 No complications have been reported yet for treatment of TIC hemorrhage. However, the OTSC system is relatively expensive when compared with other modalities.
After endoscopic treatment is performed, four-quadrant spot tattooing is recommended adjacent to the TIC with the SRH. This step will facilitate localization and treatment in the case of TIC rebleeding.4,10
Outcomes following endoscopic treatment
Following endoscopic treatment, patients should be monitored for early and late rebleeding. In a pooled analysis of case series composed of 847 patients with TIC bleeding, among the 137 patients in which endoscopic hemostasis was initially achieved, early rebleeding occurred in 8% and late rebleeding occurred in 12% of patients.22 Risk factors for TIC rebleeding within 30 days were residual arterial blood flow following hemostasis and early reinitiation of antiplatelet agents.
Remote treatment of TIC hemorrhage distant from the SRH is a significant risk factor for early TIC rebleeding.4, 10 For example, using hemoclips to close the mouth of a TIC when active bleeding or an SRH is located in the TIC base often fails because arterial flow remains open in the base and the artery is larger there.4,10 This example highlights the importance of focal obliteration of arterial blood flow underlying SRH in order to achieve definitive hemostasis.4,10
Salvage treatments
For TIC hemorrhage that is not controlled by endoscopic therapy, transcatheter arterial embolization (TAE) is recommended. If bleeding rate is high enough (at least 0.5 milliliters per minute) to be detected by angiography, TAE can serve as an effective method of diagnosis and immediate hemostasis.23 However, the most common major complication of embolization is intestinal ischemia. The incidence of intestinal ischemia has been reported as high as 10%, with highest risk with embolization of at least three vasa recta.24
Surgery is also recommended if TIC hemorrhage cannot be controlled with endoscopic therapy or TAE. Segmental colectomy is recommended if the bleeding site can be localized before surgery with colonoscopy or angiography resulting from significantly lower perioperative morbidity than subtotal colectomy.25 However, subtotal colectomy may be necessary if preoperative localization of bleeding is unsuccessful.
There are very few reports of short- or long-term results that compare endoscopy, TAE, and surgery for management of TIC bleeding. However, a recent retrospective study reported better outcomes with endoscopic treatment of definitive TIC bleeding.26 Patients who underwent endoscopic treatment had fewer RBC transfusions, shorter hospitalizations, and lower rates of postprocedure complications.
Management after cessation of hemorrhage
Medical management is important following an episode of TIC hemorrhage. A mainstay is daily fiber supplementation every morning and stool softener in the evening. Furthermore, patients are advised to drink an extra liter of fluids (not containing alcohol or caffeine) daily. By reducing colon transit time and increasing stool weight, these measures can help control constipation and prevent future complications of TIC disease.27
Patients with recurrent TIC hemorrhage should undergo evaluation for elective surgery, provided they are appropriate surgical candidates. If preoperative localization of bleeding site is successful, segmental colectomy is preferred. Segmental resection is associated with significantly decreased rebleeding rate, with lower rates of morbidity compared with subtotal colectomy.32
Chronic NSAIDs, aspirin, and antiplatelet drugs are risk factors for recurrent TIC hemorrhage, and avoiding these medications is recommended if possible.33,34 Although anticoagulants have shown to be associated with increased risk of all-cause gastrointestinal bleeding, these agents have not been shown to increase risk of recurrent TIC hemorrhage in recent large retrospective studies. Since antiplatelet and anticoagulation agents serve to reduce risk of thromboembolic events, the clinician who recommended these medications should be consulted after a TIC bleed to re-evaluate whether these medications can be discontinued or reduced in dose.
Conclusion
The most effective way to diagnose and treat definitive TIC hemorrhage is to perform an urgent colonoscopy within 24 hours to identify and treat TIC SRH. This procedure requires thoroughly cleansing the colon first, as well as an experienced colonoscopist who can identify and treat TIC SRH to obliterate arterial blood flow underneath SRH and achieve definitive TIC hemostasis. Other approaches to early diagnosis include nuclear medicine scintigraphy or angiography (CT, MRI, or IR). However, these techniques can only detect active bleeding which is documented in only 26% of colonoscopically diagnosed definitive TIC hemorrhages. So, the expected diagnostic yield of these tests will be low. When urgent colonoscopy fails to make a diagnosis or TIC bleeding continues, TAE and/or surgery are recommended. After definitive hemostasis of TIC hemorrhage and for long term management, control of constipation and discontinuation of chronic NSAIDs and antiplatelet drugs (if possible) are recommended to prevent recurrent TIC hemorrhage.
Dr. Cusumano and Dr. Paiji are fellow physicians in the Vatche and Tamar Manoukian Division of Digestive Diseases at University of California Los Angeles. Dr. Jensen is a professor of medicine in Vatche and Tamar Manoukian Division of Digestive Diseases and is with the CURE Digestive Diseases Research Center at the VA Greater Los Angeles Healthcare System, Calif. All authors declare that they have no competing interests or disclosures.
References
1. Longstreth GF. Am J Gastroenterol. 1997;92(3):419-24.
2. Jensen DM et al. The New England Journal of Medicine. 2000;342(2):78-82.
3. Jensen DM et al. Techniques in Gastrointestinal Endoscopy. 2001;3(4):192-8.
4. Jensen DM. Am J Gastroenterol. 2018;113(11):1570-3.
5. Zuckerman GR et al. Gastrointestinal Endoscopy. 1999;49(2):228-38.
6. Stollman N et al. Lancet. 2004;363(9409):631-9.
7. McGuire HH et al. Ann Surg. 1994;220(5):653-6.
8. McGuire HH et al. Ann Surg. 1972;175(6):847-55.
9. Strate LL et al. Clinical gastroenterology and hepatol. 2008;6(9):1004-10.
10. Jensen DM et al. Gastrointestinal endoscopy. 2016;83(2):416-23.
11. Jensen DM et al. Gastrointest Endosc Clin N Am. 1997;7(3):477-98.
12. Maykel JA et al. Clin Colon Rectal Surg. 2004;17(3):195-204.
13. Green BT et al. Am J Gastroenterol. 2005;100(11):2395-402.
14. Niikura R et al. Journal of Clinical Gastroenterol. 2015;49(3):e24-30.
15. Bloomfeld RS et al. Am J Gastroenterol. 2001;96(8):2367-72.
16. Parsi MA,et al. VideoGIE. 2019;4(7):285-99.
17. Kaltenbach T et al. Clinical Gastroenterology and Hepatol. 2012;10(2):131-7.
18. Nakano K et al. Endosc Int Open. 2015;3(5):E529-33.
19. Barker KB et al. Gastrointestinal Endoscopy. 2005;62(2):224-7.
20. Kaltenbach T et al. Gastrointest Endosc Clin N Am. 2020;30(1):13-23.
21. Yamazaki K et al. VideoGIE. 2020;5(6):252-4.
22. Strate LL et al. Clinical Gastroenterology and Hepatol. 2010;8(4):333-43.
23. Evangelista et al. J Vasc Interv Radiol. 2000;11(5):601-6.
24. Kodani M et al. J Vasc Interv Radiol. 2016;27(6):824-30.
25. Mohammed et al. Clin Colon Rectal Surg. 2018;31(4):243-50.
26. Wongpongsalee T et al. Gastrointestinal Endoscopy. 2020;91(6):AB471-2.
27. Böhm SK. Viszeralmedizin. 2015;31(2):84-94.
28. Prakash C et al. Endoscopy. 1999;31(6):460-3.
29. Yen EF et al. Digestive Diseases and Sciences. 2008;53(9):2480-5.
30. Ishii N et al. Gastrointestinal Endoscopy. 2012;75(2):382-7.
31. Nagata N et al. Gastrointestinal Endoscopy. 2018;88(5):841-53.e4.
32. Parkes BM et al. Am Surg. 1993;59(10):676-8.
33. Vajravelu RK et al. Gastroenterology. 2018;155(5):1416-27.
34. Oakland K et al. Clin Gastroenterol Hepatol. 2019;17(7):1276-84.e3.
35. Yamada A et al. Dis Colon Rectum. 2008;51(1):116-20.
36. Coleman CI et al. Int J Clin Pract. 2012;66(1):53-63.
37. Holster IL et al. Gastroenterology. 2013;145(1):105-12.e15.
Diverticular hemorrhage is the most common cause of colonic bleeding, accounting for 20%-65% of cases of severe lower intestinal bleeding in adults.1 Urgent colonoscopy after purging the colon of blood, clots, and stool is the most accurate method of diagnosing and guiding treatment of definitive diverticular hemorrhage.2-5 The diagnosis of definitive diverticular hemorrhage depends upon identification of some stigmata of recent hemorrhage (SRH) in a single diverticulum (TIC), which can include active arterial bleeding, oozing, non-bleeding visible vessel, adherent clot, or flat spot.2-4 Although other approaches, such as nuclear medicine scans and angiography of various types (CT, MRI, or standard angiography), for the early diagnosis of patients with severe hematochezia are utilized in many medical centers, only active bleeding can be detected by these techniques. However, as subsequently discussed, this SRH is documented in only 26% of definitive diverticular bleeds found on urgent colonoscopy, so diagnostic yields of these techniques will be low.2-5
The diagnosis of patients with severe hematochezia and diverticulosis, as well as triage of all of them to specific medical, endoscopic, radiologic, or surgical management, is facilitated by an urgent endoscopic approach.2-5 Patients who are diagnosed with definitive diverticular hemorrhage on colonoscopy represent about 30% of all true TIC bleeds when urgent colonoscopy is the management approach.2-5 That is because approximately 50% of all patients with colon diverticulosis and first presentation of severe hematochezia have incidental diverticulosis; they have colonic diverticulosis, but another site of bleeding is identified as the cause of hemorrhage in the gastrointestinal tract.2-4 Presumptive diverticular hemorrhage is diagnosed when colonic diverticulosis without TIC stigmata are found but no other GI bleeding source is found on colonoscopy, anoscopy, enteroscopy, or capsule endoscopy.2-5 In our experience with urgent colonoscopy, the presumptive diverticular bleed group accounts for about 70% of patients with documented diverticular hemorrhage (e.g., not including incidental diverticulosis bleeds but combining subgroups of patients with either definitive or presumptive TIC diagnoses as documented TIC hemorrhage).
Clinical presentation
Patients with diverticular hemorrhage present with severe, painless large volume hematochezia. Hematochezia may be self-limited and spontaneously resolve in 75%-80% of all patients but with high rebleeding rates up to 40%.5-7 Of all patients with diverticulosis, only about 3%-5% develop diverticular hemorrhage.8 Risk factors for diverticular hemorrhage include medications (e.g., nonsteroidal anti-inflammatory drugs – NSAIDs, antiplatelet drugs, and anticoagulants) and other clinical factors, such as older age, low-fiber diet, and chronic constipation.9,10 On urgent colonoscopy, more than 70% of diverticulosis in U.S. patients are located anatomically in the descending colon or more distally. In contrast, about 60% of definitive diverticular hemorrhage cases in our experience had diverticula with stigmata identified at or proximal to the splenic flexure.2,4,11
Pathophysiology
Colonic diverticula are herniations of mucosa and submucosa with colonic arteries that penetrate the muscular wall. Bleeding can occur when there is asymmetric rupture of the vasa recta at either the base of the diverticulum or the neck.4 Thinning of the mucosa on the luminal surface (such as that resulting from impacted fecaliths and stool) can cause injury to the site of the penetrating vessels, resulting in hemorrhage.12
Initial management
Patients with acute, severe hematochezia should be triaged to an inpatient setting with a monitored bed. Admission to an intensive care unit should be considered for patients with hemodynamic instability, persistent bleeding, and/or significant comorbidities. Patients with TIC hemorrhage often require resuscitation with crystalloids and packed red blood cell transfusions for hemoglobin less than 8 g/dl.4 Unlike upper GI hemorrhage, which has been extensively reported on, data regarding a more restrictive transfusion threshold, compared with a liberal transfusion threshold, in lower intestinal bleeding are very limited. Correction of underlying coagulopathies is recommended but should be individualized, particularly in those patients on antithrombotic agents or with underlying bleeding disorders.
Urgent diagnosis and hemostasis
Urgent colonoscopy within 24 hours is the most accurate way to make a diagnosis of definitive diverticular hemorrhage and to effectively and safely treat them.2-4,10,11 For patients with severe hematochezia, when the colonoscopy is either not available in a medical center or does not reveal the source of bleeding, nuclear scintigraphy or angiography (CT, MRI, or interventional radiology [IR]) are recommended. CT angiography may be particularly helpful to diagnose patients with hemodynamic instability who are suspected to have active TIC bleeding and are not able to complete a bowel preparation. However, these imaging techniques require active bleeding at the time of the study to be diagnostic. This SRH is also uncommon for definitive diverticular hemorrhage, so the diagnostic yield is usually quite low.2-5,10,11 An additional limitation of scintigraphy and CT or MRI angiography is that, if active bleeding is found, some other type of treatment, such as colonoscopy, IR angiography, or surgery, will be required for definitive hemostasis.
For urgent colonoscopy, adequate colon preparation with a large volume preparation (6-8 liters of polyethylene glycol-based solution) is recommended to clear stool, blood, and clots to allow endoscopic visualization and localization of the bleeding source. Use of a nasogastric tube should be considered if the patient is unable to drink enough prep.2-4,13 Additionally, administration of a prokinetic agent, such as Metoclopramide, may improve gastric emptying and tolerance of the prep. During colonoscopy, careful inspection of the colonic mucosa during insertion and withdrawal is important since lesions may bleed intermittently and SRH can be missed. An adult or pediatric colonoscope with a large working channel (at least 3.3 mm) is recommended to facilitate suctioning of blood clots and stool, as well as allow the passage of endoscopic hemostasis accessories. Targeted water-jet irrigation, an expert colonoscopist, a cap attachment, and adequate colon preparation are all predictors for improved diagnosis of definitive diverticular hemorrhage.4,14
SRH in definitive TIC bleeds all have a high risk of TIC rebleeding,2-4,10,11 including active bleeding, nonbleeding visible vessel, adherent clot, and a flat spot (See Figure).
Based on CURE Hemostasis Group data of 118 definitive TIC bleeds, 26% had active bleeding, 24% had a nonbleeding visible vessel, 37% had an adherent clot, and 13% had a flat spot (with underlying arterial blood flow by Doppler probe monitoring).4 Approximately 50% of the SRH were found in the neck of the TIC and 50% at the base, with actively bleeding cases more often from the base. In CURE Doppler endoscopic probe studies, 90% of all stigmata had an underlying arterial blood flow detected with the Doppler probe.4,10 The Doppler probe is reported to be very useful for risk stratification and to confirm obliteration of the arterial blood flow underlying SRH for definitive hemostasis.4,10
Endoscopic treatment
Given high rates of rebleeding with medical management alone, definitive TIC hemorrhage can be effectively and safely treated with endoscopic therapies once SRH are localized.4,10 Endoscopic therapies that have been reported in the literature include electrocoagulation, hemoclip, band ligation, and over-the-scope clip. Four-quadrant injection of 1:20,000 epinephrine around the SRH can improve visualization of SRH and provide temporary control of bleeding, but it should be combined with other modalities because of risk of rebleeding with epinephrine alone.15 Results from studies reporting rates of both early rebleeding (occurring within 30 days) and late rebleeding (occurring after 30 days) are listed in the Table.
Multipolar electrocoagulation (MPEC), which utilizes a focal electric current to generate heat, can coaptively coagulate small TIC arteries.16 For SRH in the neck of TIC, MPEC is effective for coaptive coagulation at a power of 12-15 watts in 1-2 second pulses with moderate laterally applied tamponade pressure. MPEC should be avoided for treating SRH at the TIC base because of lack of muscularis propria and higher risk of perforation.
Hemoclip therapy has been reported to be safe and efficacious in treatment of definitive TIC hemorrhage, by causing mechanical hemostasis with occlusion of the bleeding artery.16 Hemoclips are recommended to treat stigmata in the base of TICs and should be targeted on either side of visible vessel in order to occlude the artery underneath it.4,10 With a cap on the tip of the colonoscope, suctioning can evert TICs, allowing more precise placement of hemoclip on SRH in the base of the TIC.17 Hemoclip retention rates vary with different models and can range from less than 7 days to more than 4 weeks. Hemoclips can also mark the site if early rebleeding occurs; then, reintervention (e.g., repeat endoscopy or angioembolization) is facilitated.
Another treatment is endoscopic band ligation, which provides mechanical hemostasis. Endoscopic band ligation has been reported to be efficacious for TIC hemorrhage.18 Suctioning the TIC with the SRH into the distal cap and deploying a band leads to obliteration of vessels and potentially necrosis and disappearance of banded TIC.16 This technique carries a risk of perforation because of the thin walls of TICs. This risk may be higher for right-sided colon lesions since an exvivo colon specimen study reported serosal entrapment and inclusion of muscularis propria postband ligation, both of which may result in ischemia of intestinal wall and delayed perforation.19
Over-the-scope clip (OTSC) has been reported in case series for treatment of definitive TIC hemorrhage. With a distal cap and large clip, suctioning can evert TICs and facilitate deployment over the SRH.20,21 OTSC can grasp an entire TIC with the SRH and obliterate the arterial blood flow with a single clip.20,21 No complications have been reported yet for treatment of TIC hemorrhage. However, the OTSC system is relatively expensive when compared with other modalities.
After endoscopic treatment is performed, four-quadrant spot tattooing is recommended adjacent to the TIC with the SRH. This step will facilitate localization and treatment in the case of TIC rebleeding.4,10
Outcomes following endoscopic treatment
Following endoscopic treatment, patients should be monitored for early and late rebleeding. In a pooled analysis of case series composed of 847 patients with TIC bleeding, among the 137 patients in which endoscopic hemostasis was initially achieved, early rebleeding occurred in 8% and late rebleeding occurred in 12% of patients.22 Risk factors for TIC rebleeding within 30 days were residual arterial blood flow following hemostasis and early reinitiation of antiplatelet agents.
Remote treatment of TIC hemorrhage distant from the SRH is a significant risk factor for early TIC rebleeding.4, 10 For example, using hemoclips to close the mouth of a TIC when active bleeding or an SRH is located in the TIC base often fails because arterial flow remains open in the base and the artery is larger there.4,10 This example highlights the importance of focal obliteration of arterial blood flow underlying SRH in order to achieve definitive hemostasis.4,10
Salvage treatments
For TIC hemorrhage that is not controlled by endoscopic therapy, transcatheter arterial embolization (TAE) is recommended. If bleeding rate is high enough (at least 0.5 milliliters per minute) to be detected by angiography, TAE can serve as an effective method of diagnosis and immediate hemostasis.23 However, the most common major complication of embolization is intestinal ischemia. The incidence of intestinal ischemia has been reported as high as 10%, with highest risk with embolization of at least three vasa recta.24
Surgery is also recommended if TIC hemorrhage cannot be controlled with endoscopic therapy or TAE. Segmental colectomy is recommended if the bleeding site can be localized before surgery with colonoscopy or angiography resulting from significantly lower perioperative morbidity than subtotal colectomy.25 However, subtotal colectomy may be necessary if preoperative localization of bleeding is unsuccessful.
There are very few reports of short- or long-term results that compare endoscopy, TAE, and surgery for management of TIC bleeding. However, a recent retrospective study reported better outcomes with endoscopic treatment of definitive TIC bleeding.26 Patients who underwent endoscopic treatment had fewer RBC transfusions, shorter hospitalizations, and lower rates of postprocedure complications.
Management after cessation of hemorrhage
Medical management is important following an episode of TIC hemorrhage. A mainstay is daily fiber supplementation every morning and stool softener in the evening. Furthermore, patients are advised to drink an extra liter of fluids (not containing alcohol or caffeine) daily. By reducing colon transit time and increasing stool weight, these measures can help control constipation and prevent future complications of TIC disease.27
Patients with recurrent TIC hemorrhage should undergo evaluation for elective surgery, provided they are appropriate surgical candidates. If preoperative localization of bleeding site is successful, segmental colectomy is preferred. Segmental resection is associated with significantly decreased rebleeding rate, with lower rates of morbidity compared with subtotal colectomy.32
Chronic NSAIDs, aspirin, and antiplatelet drugs are risk factors for recurrent TIC hemorrhage, and avoiding these medications is recommended if possible.33,34 Although anticoagulants have shown to be associated with increased risk of all-cause gastrointestinal bleeding, these agents have not been shown to increase risk of recurrent TIC hemorrhage in recent large retrospective studies. Since antiplatelet and anticoagulation agents serve to reduce risk of thromboembolic events, the clinician who recommended these medications should be consulted after a TIC bleed to re-evaluate whether these medications can be discontinued or reduced in dose.
Conclusion
The most effective way to diagnose and treat definitive TIC hemorrhage is to perform an urgent colonoscopy within 24 hours to identify and treat TIC SRH. This procedure requires thoroughly cleansing the colon first, as well as an experienced colonoscopist who can identify and treat TIC SRH to obliterate arterial blood flow underneath SRH and achieve definitive TIC hemostasis. Other approaches to early diagnosis include nuclear medicine scintigraphy or angiography (CT, MRI, or IR). However, these techniques can only detect active bleeding which is documented in only 26% of colonoscopically diagnosed definitive TIC hemorrhages. So, the expected diagnostic yield of these tests will be low. When urgent colonoscopy fails to make a diagnosis or TIC bleeding continues, TAE and/or surgery are recommended. After definitive hemostasis of TIC hemorrhage and for long term management, control of constipation and discontinuation of chronic NSAIDs and antiplatelet drugs (if possible) are recommended to prevent recurrent TIC hemorrhage.
Dr. Cusumano and Dr. Paiji are fellow physicians in the Vatche and Tamar Manoukian Division of Digestive Diseases at University of California Los Angeles. Dr. Jensen is a professor of medicine in Vatche and Tamar Manoukian Division of Digestive Diseases and is with the CURE Digestive Diseases Research Center at the VA Greater Los Angeles Healthcare System, Calif. All authors declare that they have no competing interests or disclosures.
References
1. Longstreth GF. Am J Gastroenterol. 1997;92(3):419-24.
2. Jensen DM et al. The New England Journal of Medicine. 2000;342(2):78-82.
3. Jensen DM et al. Techniques in Gastrointestinal Endoscopy. 2001;3(4):192-8.
4. Jensen DM. Am J Gastroenterol. 2018;113(11):1570-3.
5. Zuckerman GR et al. Gastrointestinal Endoscopy. 1999;49(2):228-38.
6. Stollman N et al. Lancet. 2004;363(9409):631-9.
7. McGuire HH et al. Ann Surg. 1994;220(5):653-6.
8. McGuire HH et al. Ann Surg. 1972;175(6):847-55.
9. Strate LL et al. Clinical gastroenterology and hepatol. 2008;6(9):1004-10.
10. Jensen DM et al. Gastrointestinal endoscopy. 2016;83(2):416-23.
11. Jensen DM et al. Gastrointest Endosc Clin N Am. 1997;7(3):477-98.
12. Maykel JA et al. Clin Colon Rectal Surg. 2004;17(3):195-204.
13. Green BT et al. Am J Gastroenterol. 2005;100(11):2395-402.
14. Niikura R et al. Journal of Clinical Gastroenterol. 2015;49(3):e24-30.
15. Bloomfeld RS et al. Am J Gastroenterol. 2001;96(8):2367-72.
16. Parsi MA,et al. VideoGIE. 2019;4(7):285-99.
17. Kaltenbach T et al. Clinical Gastroenterology and Hepatol. 2012;10(2):131-7.
18. Nakano K et al. Endosc Int Open. 2015;3(5):E529-33.
19. Barker KB et al. Gastrointestinal Endoscopy. 2005;62(2):224-7.
20. Kaltenbach T et al. Gastrointest Endosc Clin N Am. 2020;30(1):13-23.
21. Yamazaki K et al. VideoGIE. 2020;5(6):252-4.
22. Strate LL et al. Clinical Gastroenterology and Hepatol. 2010;8(4):333-43.
23. Evangelista et al. J Vasc Interv Radiol. 2000;11(5):601-6.
24. Kodani M et al. J Vasc Interv Radiol. 2016;27(6):824-30.
25. Mohammed et al. Clin Colon Rectal Surg. 2018;31(4):243-50.
26. Wongpongsalee T et al. Gastrointestinal Endoscopy. 2020;91(6):AB471-2.
27. Böhm SK. Viszeralmedizin. 2015;31(2):84-94.
28. Prakash C et al. Endoscopy. 1999;31(6):460-3.
29. Yen EF et al. Digestive Diseases and Sciences. 2008;53(9):2480-5.
30. Ishii N et al. Gastrointestinal Endoscopy. 2012;75(2):382-7.
31. Nagata N et al. Gastrointestinal Endoscopy. 2018;88(5):841-53.e4.
32. Parkes BM et al. Am Surg. 1993;59(10):676-8.
33. Vajravelu RK et al. Gastroenterology. 2018;155(5):1416-27.
34. Oakland K et al. Clin Gastroenterol Hepatol. 2019;17(7):1276-84.e3.
35. Yamada A et al. Dis Colon Rectum. 2008;51(1):116-20.
36. Coleman CI et al. Int J Clin Pract. 2012;66(1):53-63.
37. Holster IL et al. Gastroenterology. 2013;145(1):105-12.e15.
Microscopic colitis: A common, yet often overlooked, cause of chronic diarrhea
Microscopic colitis is an inflammatory disease of the colon and a frequent cause of chronic or recurrent watery diarrhea, particularly in older persons. MC consists of two subtypes, collagenous colitis (CC) and lymphocytic colitis (LC). While the primary symptom is diarrhea, other signs and symptoms such as abdominal pain, weight loss, and dehydration or electrolyte abnormalities may also be present depending on disease severity.1 In MC, the colonic mucosa usually appears normal on colonoscopy, and the diagnosis is made by histologic findings of intraepithelial lymphocytosis with (CC) or without (LC) a prominent subepithelial collagen band. The management approaches to CC and LC are similar and should be directed based on the severity of symptoms.2 We review the epidemiology, risk factors, pathophysiology, diagnosis, and clinical management for this condition, as well as novel therapeutic approaches.
Epidemiology

Although the incidence of MC increased in the late twentieth century, more recently, it has stabilized with an estimated incidence varying from 1 to 25 per 100,000 person-years.3-5 A recent meta-analysis revealed a pooled incidence of 4.85 per 100,000 persons for LC and 4.14 per 100,000 persons for CC.6 Proposed explanations for the rising incidence in the late twentieth century include improved clinical awareness of the disease, possible increased use of drugs associated with MC, and increased performance of diagnostic colonoscopies for chronic diarrhea. Since MC is now well-recognized, the recent plateau in incidence rates may reflect decreased detection bias.
The prevalence of MC ranges from 10%-20% in patients undergoing colonoscopy for chronic watery diarrhea.6,7 The prevalence of LC is approximately 63.1 cases per 100,000 person-years and, for CC, is 49.2 cases per 100,000 person-years.6-8 Recent studies have demonstrated increasing prevalence of MC likely resulting from an aging population.9,10
Risk stratification
Female gender, increasing age, concomitant autoimmune disease, and the use of certain drugs, including NSAIDs, proton pump inhibitors (PPIs), statins, and selective serotonin reuptake inhibitors (SSRIs), have been associated with an increased risk of MC.11,12 Autoimmune disorders, including celiac disease (CD), rheumatoid arthritis, hypothyroidism, and hyperthyroidism, are more common in patients with MC. The association with CD, in particular, is clinically important, as CD is associated with a 50-70 times greater risk of MC, and 2%-9% of patients with MC have CD.13,14
Several medications have been associated with MC. In a British multicenter prospective study, MC was associated with the use of NSAIDs, PPIs, and SSRIs;15 however, recent studies have questioned the association of MC with some of these medications, which might worsen diarrhea but not actually cause MC.16

An additional risk factor for MC is smoking. A recent meta-analysis demonstrated that current and former smokers had an increased risk of MC (odds ratio, 2.99; 95% confidence interval, 2.15-4.15 and OR, 1.63; 95% CI, 1.37-1.94, respectively), compared with nonsmokers.17 Smokers develop MC at a younger age, and smoking is associated with increased disease severity and decreased likelihood of attaining remission.18,19
Pathogenesis
The pathogenesis of MC remains largely unknown, although there are several hypotheses. The leading proposed mechanisms include reaction to luminal antigens, dysregulated collagen metabolism, genetic predisposition, autoimmunity, and bile acid malabsorption.
MC may be caused by abnormal epithelial barrier function, leading to increased permeability and reaction to luminal antigens, including dietary antigens, certain drugs, and bacterial products, 20,21 which themselves lead to the immune dysregulation and intestinal inflammation seen in MC. This mechanism may explain the association of several drugs with MC. Histological changes resembling LC are reported in patients with CD who consume gluten; however, large population-based studies have not found specific dietary associations with the development of MC.22
Another potential mechanism of MC is dysregulated collagen deposition. Collagen accumulation in the subepithelial layer in CC may result from increased levels of fibroblast growth factor, transforming growth factor–beta and vascular endothelial growth factor.23 Nonetheless, studies have not found an association between the severity of diarrhea in patients with CC and the thickness of the subepithelial collagen band.

Thirdly, autoimmunity and genetic predisposition have been postulated in the pathogenesis of MC. As previously discussed, MC is associated with several autoimmune diseases and predominantly occurs in women, a distinctive feature of autoimmune disorders. Several studies have demonstrated an association between MC and HLA-DQ2 and -DQ3 haplotypes,24 as well as potential polymorphisms in the serotonin transporter gene promoter.25 It is important to note, however, that only a few familial cases of MC have been reported to date.26
Lastly, bile acid malabsorption may play a role in the etiology of MC. Histologic findings of inflammation, along with villous atrophy and collagen deposition, have been reported in the ileum of patients with MC;27,28 however, because patients with MC without bile acid malabsorption may also respond to bile acid binders such as cholestyramine, these findings unlikely to be the sole mechanism explaining the development of the disease.
Despite the different proposed mechanisms for the pathogenesis of MC, no definite conclusions can be drawn because of the limited size of these studies and their often conflicting results.

Clinical features
Clinicians should suspect MC in patients with chronic or recurrent watery diarrhea, particularly in older persons. Other risk factors include female gender, use of certain culprit medications, smoking, and presence of other autoimmune diseases. The clinical manifestations of MC subtypes LC and CC are similar with no significant clinical differences.1,2 In addition to diarrhea, patients with MC may have abdominal pain, fatigue, and dehydration or electrolyte abnormalities depending on disease severity. Patients may also present with fecal urgency, incontinence, and nocturnal stools. Quality of life is often reduced in these patients, predominantly in those with severe or refractory symptoms.29,30 The natural course of MC is highly variable, with some patients achieving spontaneous resolution after one episode and others developing chronic symptoms.
Diagnosis
The differential diagnosis of chronic watery diarrhea is broad and includes malabsorption/maldigestion, inflammatory bowel disease (IBD), irritable bowel syndrome, and medication side effects. In addition, although gastrointestinal infections typically cause acute or subacute diarrhea, some can present with chronic diarrhea. Malabsorption/maldigestion may occur because of CD, lactose intolerance, and pancreatic insufficiency, among other conditions. A thorough history, regarding recent antibiotic and medication use, travel, and immunosuppression, should be obtained in patients with chronic diarrhea. Additionally, laboratory and endoscopic evaluation with random biopsies of the colon can further help differentiate these diseases from MC. A few studies suggest fecal calprotectin may be used to differentiate MC from other noninflammatory conditions such as irritable bowel syndrome, as well as to monitor disease activity. This test is not expected to distinguish MC from other inflammatory causes of diarrhea, such as IBD, and therefore, its role in clinical practice is uncertain.31
The diagnosis of MC is made by biopsy of the colonic mucosa demonstrating characteristic pathologic features.32 Unlike in diseases such as Crohn’s disease or ulcerative colitis, the colon usually appears normal in MC, although mild nonspecific changes, such as erythema or edema, may be visualized. There is no consensus on the ideal location to obtain biopsies for MC or whether biopsies from both the left and the right colon are required.2,33 The procedure of choice for the diagnosis of MC is colonoscopy with random biopsies taken throughout the colon. More limited evaluation by flexible sigmoidoscopy with biopsies may miss cases of MC as inflammation and collagen thickening are not necessarily uniform throughout the colon; however, in a patient that has undergone a recent colonoscopy for colon cancer screening without colon biopsies, a flexible sigmoidoscopy may be a reasonable next test for evaluation of MC, provided biopsies are obtained above the rectosigmoid colon.34
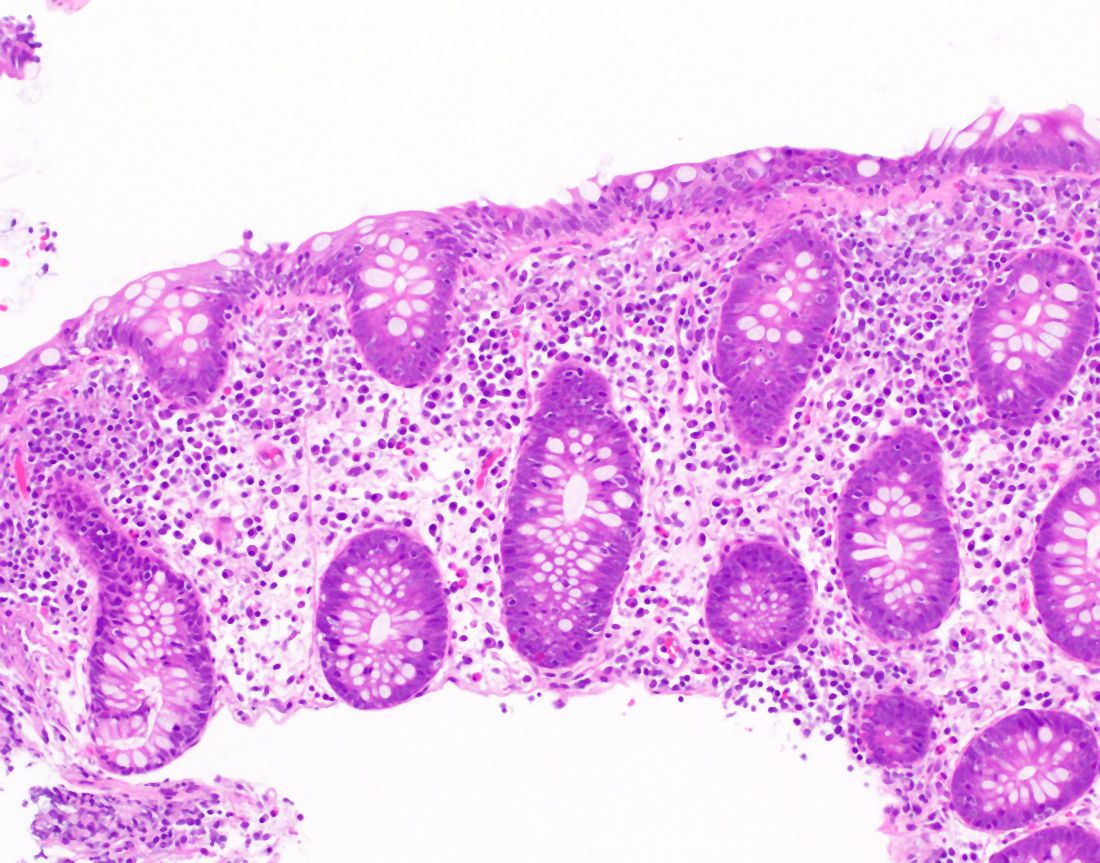
The MC subtypes are differentiated based on histology. The hallmark of LC is less than 20 intraepithelial lymphocytes per 100 surface epithelial cells (normal, less than 5) (Figure 1A). CC is characterized by a thickened subepithelial collagen band greater than 7-10 micrometers (normal, less than 5) (Figure 1B). For a subgroup of patients with milder abnormalities that do not meet these histological criteria, the terms “microscopic colitis, not otherwise specified” or “microscopic colitis, incomplete” may be used.35 These patients often respond to standard treatments for MC. There is an additional subset of patients with biopsy demonstrating features of both CC and LC simultaneously, as well as patients transitioning from one MC subtype to another over time.32,35
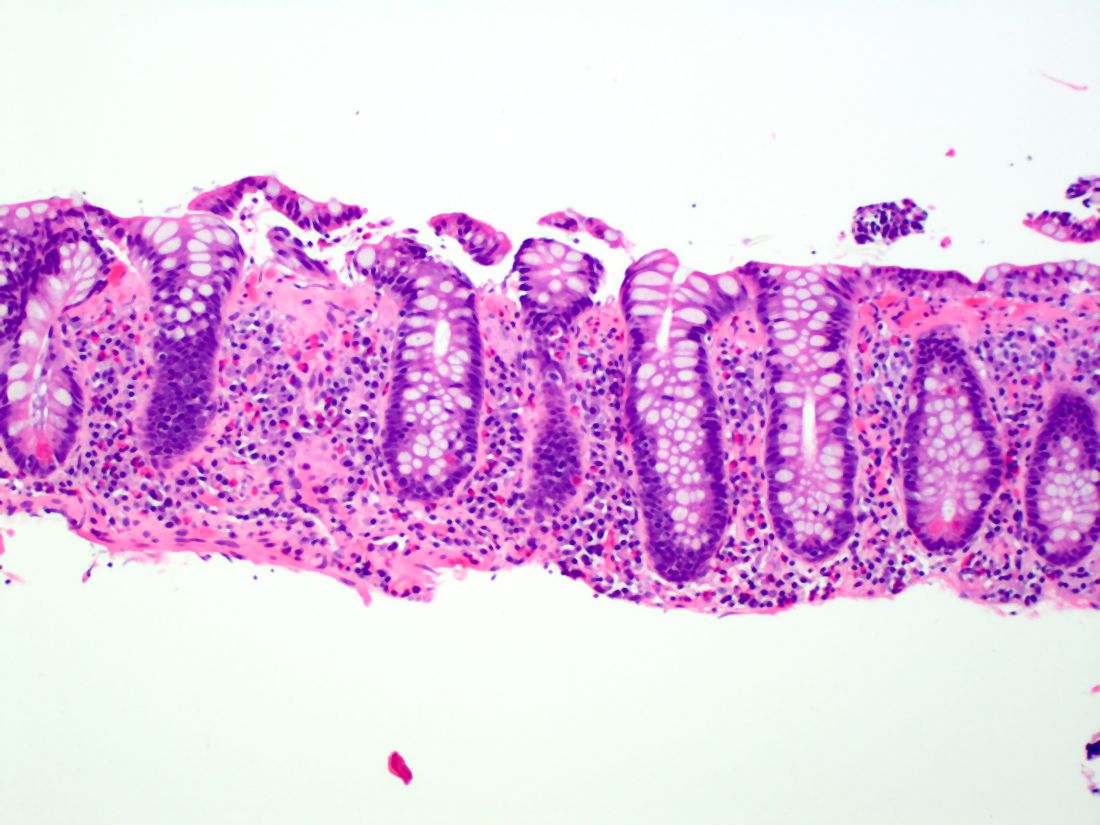
Management approach
The first step in management of patients with MC includes stopping culprit medications if there is a temporal relationship between the initiation of the medication and the onset of diarrhea, as well as encouraging smoking cessation. These steps alone, however, are unlikely to achieve clinical remission in most patients. A stepwise pharmacological approach is used in the management of MC based on disease severity (Figure 2). For patients with mild symptoms, antidiarrheal medications, such as loperamide, may be helpful.36 Long-term use of loperamide at therapeutic doses no greater than 16 mg daily appears to be safe if required to maintain symptom response. For those with persistent symptoms despite antidiarrheal medications, bismuth subsalicylate at three 262 mg tablets three times daily for 6-8 weeks can be considered. Long-term use of bismuth subsalicylate is not advised, especially at this dose, because of possible neurotoxicity.37
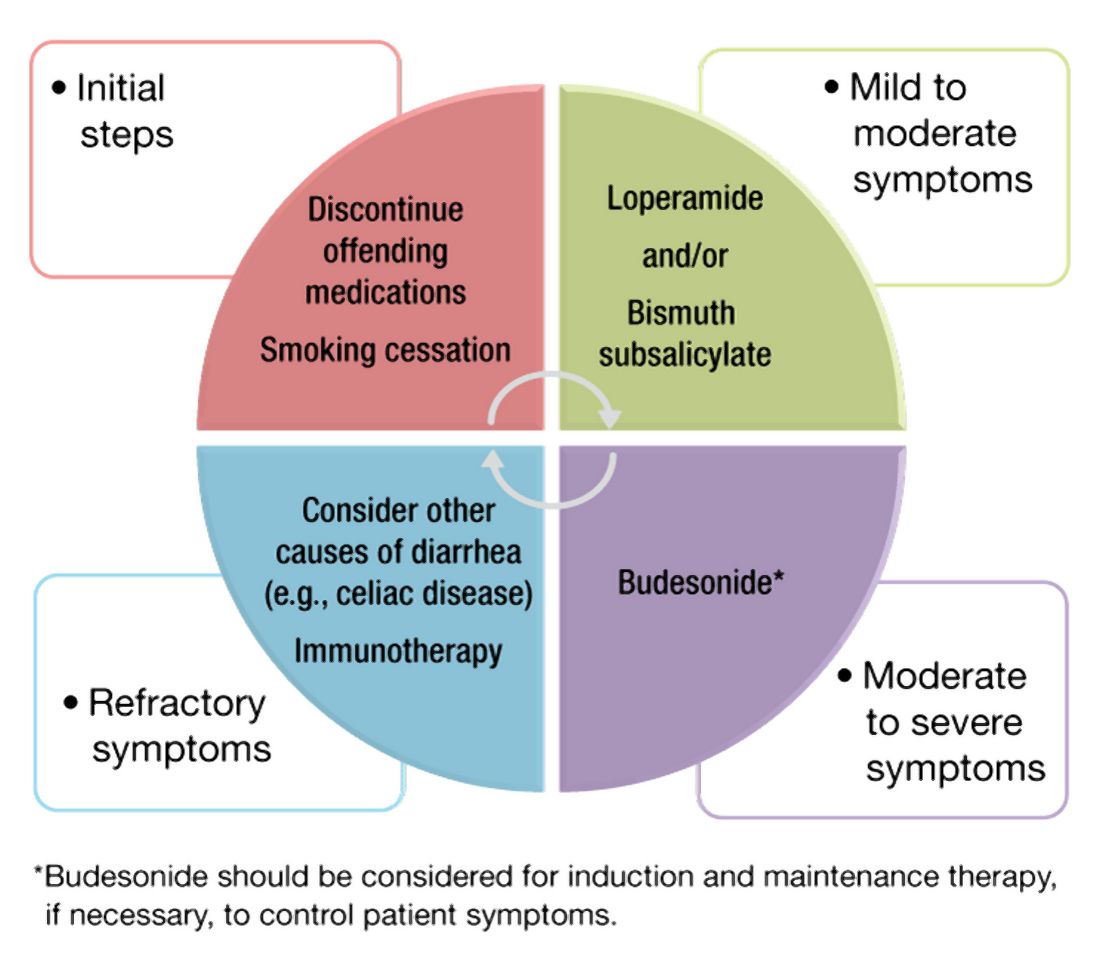
For patients refractory to the above treatments or those with moderate-to-severe symptoms, an 8-week course of budesonide at 9 mg daily is the first-line treatment.38 The dose was tapered before discontinuation in some studies but not in others. Both strategies appear effective. A recent meta-analysis of nine randomized trials demonstrated pooled ORs of 7.34 (95% CI, 4.08-13.19) and 8.35 (95% CI, 4.14-16.85) for response to budesonide induction and maintenance, respectively.39
Cholestyramine is another medication considered in the management of MC and warrants further investigation. To date, no randomized clinical trials have been conducted to evaluate bile acid sequestrants in MC, but they should be considered before placing patients on immunosuppressive medications. Some providers use mesalamine in this setting, although mesalamine is inferior to budesonide in the induction of clinical remission in MC.40
Despite high rates of response to budesonide, relapse after discontinuation is frequent (60%-80%), and time to relapse is variable41,42 The American Gastroenterological Association recommends budesonide for maintenance of remission in patients with recurrence following discontinuation of induction therapy. The lowest effective dose that maintains resolution of symptoms should be prescribed, ideally at 6 mg daily or lower.38 Although budesonide has a greater first-pass metabolism, compared with other glucocorticoids, patients should be monitored for possible side effects including hypertension, diabetes, and osteoporosis, as well as ophthalmologic disease, including cataracts and glaucoma.
For those who are intolerant to budesonide or have refractory symptoms, concomitant disorders such as CD that may be contributing to symptoms must be excluded. Immunosuppressive medications – such as thiopurines and biologic agents, including tumor necrosis factor–alpha inhibitors or vedolizumab – may be considered in refractory cases.43,44 Of note, there are limited studies evaluating the use of these medications for MC. Lastly, surgeries including ileostomy with or without colectomy have been performed in the most severe cases for resistant disease that has failed numerous pharmacological therapies.45
Patients should be counseled that, while symptoms from MC can be quite bothersome and disabling, there appears to be a normal life expectancy and no association between MC and colon cancer, unlike with other inflammatory conditions of the colon such as IBD.46,47
Conclusion and future outlook
As a common cause of chronic watery diarrhea, MC will be commonly encountered in primary care and gastroenterology practices. The diagnosis should be suspected in patients presenting with chronic or recurrent watery diarrhea, especially with female gender, autoimmune disease, and increasing age. The management of MC requires an algorithmic approach directed by symptom severity, with a subgroup of patients requiring maintenance therapy for relapsing symptoms. The care of patients with MC will continue to evolve in the future. Further work is needed to explore long-term safety outcomes with budesonide and the role of immunomodulators and newer biologic agents for patients with complex, refractory disease.
Dr. Tome is with the department of internal medicine at the Mayo Clinic, Rochester, Minn. Dr. Kamboj, and Dr. Pardi are with the division of gastroenterology and hepatology at the Mayo Clinic. Dr. Pardi has grant funding from Pfizer, Vedanta, Seres, Finch, Applied Molecular Transport, and Takeda and has consulted for Vedanta and Otsuka. The other authors have no conflicts of interest to report.
References
1. Nyhlin N et al. Aliment Pharmacol Ther. 2014;39:963-72.
2. Miehlke S et al. United European Gastroenterol J. 2020;20-8.
3. Pardi DS et al. Gut. 2007;56:504-8.
4. Fernández-Bañares F et al. J Crohn’s Colitis.2016;10(7):805-11.
5. Gentile NM et al. Clin Gastroenterol Hepatol. 2014;12(5):838-42.
6. Tong J et al. Am J Gastroenterol. 2015;110:265-76.
7. Olesen M et al. Gut. 2004;53(3):346-50.
8. Bergman D et al. Aliment Pharmacol Ther. 2019;49(11):1395-400.
9. Guagnozzi D et al. Dig Liver Dis. 2012;44(5):384-8.
10. Münch A et al. J Crohns Colitis. 2012;6(9):932-45.
11. Macaigne G et al. Am J Gastroenterol. 2014; 09(9):1461-70.
12. Verhaegh BP et al. Aliment Pharmacol Ther. 2016;43(9):1004-13.
13. Stewart M et al. Aliment Pharmacol Ther. 2011;33(12):1340-9.
14. Green PHR et al. Clin Gastroenterol Hepatol. 2009;7(11):1210-6.
15. Masclee GM et al. Am J Gastroenterol. 2015;110:749-59.
16. Zylberberg H et al. Ailment Pharmacol Ther. 2021 Jun;53(11)1209-15.
17. Jaruvongvanich V et al. Inflamm Bowel Dis. 2019;25(4):672-8.
18. Fernández-Bañares F et al. Inflamm Bowel Dis. 2013; 19(7):1470-6.
19. Yen EF et al. Inflamm Bowel Dis. 2012;18(10):1835-41.
20. Barmeyer C et al. J Gastroenterol. 2017;52(10):1090-100.
21. Morgan DM et al. Clin Gastroenterol Hepatol. 2020;18(4):984-6.
22. Larsson JK et al. Eur J Clin Nutr. 2016;70:1309-17.
23. Madisch A et al.. Inflamm Bowel Dis. 2011;17(11):2295-8.
24. Stahl E et al. Gastroenterology. 2020;159(2):549-61.
25. Sikander A et al. Dig Dis Sci. 2015; 60:887-94.
26. Abdo AA et al. Can J Gastroenterol. 2001;15(5):341-3.
27. Fernandez-Bañares F et al. Dig Dis Sci.2001;46(10):2231-8.
28. Lyutakov I et al. Eur J Gastroenterol Hepatol. 2021;1;33(3):380-7.
29. Hjortswang H et al. Dig Liver Dis. 2011 Feb;43(2):102-9.
30. Cotter TG= et al. Gut. 2018;67(3):441-6.
31. Von Arnim U et al. Clin Exp Gastroenterol. 2016;9:97-103.
32. Langner C et al. Histopathology. 2015;66:613-26.
33. ASGE Standards of Practice Committee and Sharaf RN et al. Gastrointest Endosc. 2013;78:216-24.
34. Macaigne G et al. Clin Res Hepatol Gastroenterol. 2017;41(3):333-40.
35. Bjørnbak C et al. Aliment Pharmacol Ther. 2011;34(10):1225-34.
36. Pardi DS et al. Gastroenterology. 2016;150(1):247-74.
37. Masannat Y and Nazer E. West Virginia Med J. 2013;109(3):32-4.
38. Nguyen GC et al. Gastroenterology. 2016; 150(1):242-6.
39. Sebastian S et al. Eur J Gastroenterol Hepatol. 2019 Aug;31(8):919-27.
40. Miehlke S et al. Gastroenterology. 2014;146(5):1222-30.
41. Gentile NM et al. Am J Gastroenterol. 2013;108:256-9.
42. Münch A et al. Gut. 2016; 65(1):47-56.
43. Cotter TG et al. Aliment Pharmacol Ther. 2017; 46(2):169-74.
44. Esteve M et al. J Crohns Colitis. 2011;5(6):612-8.
45. Cottreau J et al. Clin J Gastroenterol. 2016;9:140-4.
46. Kamboj AK et al. Program No. P1876. ACG 2018 Annual Scientific Meeting Abstracts. Philadelphia, Pennsylvania: American College of Gastroenterology.
47. Yen EF et al. Dig Dis Sci. 2012;57:161-9.
Microscopic colitis is an inflammatory disease of the colon and a frequent cause of chronic or recurrent watery diarrhea, particularly in older persons. MC consists of two subtypes, collagenous colitis (CC) and lymphocytic colitis (LC). While the primary symptom is diarrhea, other signs and symptoms such as abdominal pain, weight loss, and dehydration or electrolyte abnormalities may also be present depending on disease severity.1 In MC, the colonic mucosa usually appears normal on colonoscopy, and the diagnosis is made by histologic findings of intraepithelial lymphocytosis with (CC) or without (LC) a prominent subepithelial collagen band. The management approaches to CC and LC are similar and should be directed based on the severity of symptoms.2 We review the epidemiology, risk factors, pathophysiology, diagnosis, and clinical management for this condition, as well as novel therapeutic approaches.
Epidemiology
Although the incidence of MC increased in the late twentieth century, more recently, it has stabilized with an estimated incidence varying from 1 to 25 per 100,000 person-years.3-5 A recent meta-analysis revealed a pooled incidence of 4.85 per 100,000 persons for LC and 4.14 per 100,000 persons for CC.6 Proposed explanations for the rising incidence in the late twentieth century include improved clinical awareness of the disease, possible increased use of drugs associated with MC, and increased performance of diagnostic colonoscopies for chronic diarrhea. Since MC is now well-recognized, the recent plateau in incidence rates may reflect decreased detection bias.
The prevalence of MC ranges from 10%-20% in patients undergoing colonoscopy for chronic watery diarrhea.6,7 The prevalence of LC is approximately 63.1 cases per 100,000 person-years and, for CC, is 49.2 cases per 100,000 person-years.6-8 Recent studies have demonstrated increasing prevalence of MC likely resulting from an aging population.9,10
Risk stratification
Female gender, increasing age, concomitant autoimmune disease, and the use of certain drugs, including NSAIDs, proton pump inhibitors (PPIs), statins, and selective serotonin reuptake inhibitors (SSRIs), have been associated with an increased risk of MC.11,12 Autoimmune disorders, including celiac disease (CD), rheumatoid arthritis, hypothyroidism, and hyperthyroidism, are more common in patients with MC. The association with CD, in particular, is clinically important, as CD is associated with a 50-70 times greater risk of MC, and 2%-9% of patients with MC have CD.13,14
Several medications have been associated with MC. In a British multicenter prospective study, MC was associated with the use of NSAIDs, PPIs, and SSRIs;15 however, recent studies have questioned the association of MC with some of these medications, which might worsen diarrhea but not actually cause MC.16
An additional risk factor for MC is smoking. A recent meta-analysis demonstrated that current and former smokers had an increased risk of MC (odds ratio, 2.99; 95% confidence interval, 2.15-4.15 and OR, 1.63; 95% CI, 1.37-1.94, respectively), compared with nonsmokers.17 Smokers develop MC at a younger age, and smoking is associated with increased disease severity and decreased likelihood of attaining remission.18,19
Pathogenesis
The pathogenesis of MC remains largely unknown, although there are several hypotheses. The leading proposed mechanisms include reaction to luminal antigens, dysregulated collagen metabolism, genetic predisposition, autoimmunity, and bile acid malabsorption.
MC may be caused by abnormal epithelial barrier function, leading to increased permeability and reaction to luminal antigens, including dietary antigens, certain drugs, and bacterial products, 20,21 which themselves lead to the immune dysregulation and intestinal inflammation seen in MC. This mechanism may explain the association of several drugs with MC. Histological changes resembling LC are reported in patients with CD who consume gluten; however, large population-based studies have not found specific dietary associations with the development of MC.22
Another potential mechanism of MC is dysregulated collagen deposition. Collagen accumulation in the subepithelial layer in CC may result from increased levels of fibroblast growth factor, transforming growth factor–beta and vascular endothelial growth factor.23 Nonetheless, studies have not found an association between the severity of diarrhea in patients with CC and the thickness of the subepithelial collagen band.
Thirdly, autoimmunity and genetic predisposition have been postulated in the pathogenesis of MC. As previously discussed, MC is associated with several autoimmune diseases and predominantly occurs in women, a distinctive feature of autoimmune disorders. Several studies have demonstrated an association between MC and HLA-DQ2 and -DQ3 haplotypes,24 as well as potential polymorphisms in the serotonin transporter gene promoter.25 It is important to note, however, that only a few familial cases of MC have been reported to date.26
Lastly, bile acid malabsorption may play a role in the etiology of MC. Histologic findings of inflammation, along with villous atrophy and collagen deposition, have been reported in the ileum of patients with MC;27,28 however, because patients with MC without bile acid malabsorption may also respond to bile acid binders such as cholestyramine, these findings unlikely to be the sole mechanism explaining the development of the disease.
Despite the different proposed mechanisms for the pathogenesis of MC, no definite conclusions can be drawn because of the limited size of these studies and their often conflicting results.
Clinical features
Clinicians should suspect MC in patients with chronic or recurrent watery diarrhea, particularly in older persons. Other risk factors include female gender, use of certain culprit medications, smoking, and presence of other autoimmune diseases. The clinical manifestations of MC subtypes LC and CC are similar with no significant clinical differences.1,2 In addition to diarrhea, patients with MC may have abdominal pain, fatigue, and dehydration or electrolyte abnormalities depending on disease severity. Patients may also present with fecal urgency, incontinence, and nocturnal stools. Quality of life is often reduced in these patients, predominantly in those with severe or refractory symptoms.29,30 The natural course of MC is highly variable, with some patients achieving spontaneous resolution after one episode and others developing chronic symptoms.
Diagnosis
The differential diagnosis of chronic watery diarrhea is broad and includes malabsorption/maldigestion, inflammatory bowel disease (IBD), irritable bowel syndrome, and medication side effects. In addition, although gastrointestinal infections typically cause acute or subacute diarrhea, some can present with chronic diarrhea. Malabsorption/maldigestion may occur because of CD, lactose intolerance, and pancreatic insufficiency, among other conditions. A thorough history, regarding recent antibiotic and medication use, travel, and immunosuppression, should be obtained in patients with chronic diarrhea. Additionally, laboratory and endoscopic evaluation with random biopsies of the colon can further help differentiate these diseases from MC. A few studies suggest fecal calprotectin may be used to differentiate MC from other noninflammatory conditions such as irritable bowel syndrome, as well as to monitor disease activity. This test is not expected to distinguish MC from other inflammatory causes of diarrhea, such as IBD, and therefore, its role in clinical practice is uncertain.31
The diagnosis of MC is made by biopsy of the colonic mucosa demonstrating characteristic pathologic features.32 Unlike in diseases such as Crohn’s disease or ulcerative colitis, the colon usually appears normal in MC, although mild nonspecific changes, such as erythema or edema, may be visualized. There is no consensus on the ideal location to obtain biopsies for MC or whether biopsies from both the left and the right colon are required.2,33 The procedure of choice for the diagnosis of MC is colonoscopy with random biopsies taken throughout the colon. More limited evaluation by flexible sigmoidoscopy with biopsies may miss cases of MC as inflammation and collagen thickening are not necessarily uniform throughout the colon; however, in a patient that has undergone a recent colonoscopy for colon cancer screening without colon biopsies, a flexible sigmoidoscopy may be a reasonable next test for evaluation of MC, provided biopsies are obtained above the rectosigmoid colon.34
The MC subtypes are differentiated based on histology. The hallmark of LC is less than 20 intraepithelial lymphocytes per 100 surface epithelial cells (normal, less than 5) (Figure 1A). CC is characterized by a thickened subepithelial collagen band greater than 7-10 micrometers (normal, less than 5) (Figure 1B). For a subgroup of patients with milder abnormalities that do not meet these histological criteria, the terms “microscopic colitis, not otherwise specified” or “microscopic colitis, incomplete” may be used.35 These patients often respond to standard treatments for MC. There is an additional subset of patients with biopsy demonstrating features of both CC and LC simultaneously, as well as patients transitioning from one MC subtype to another over time.32,35
Management approach
The first step in management of patients with MC includes stopping culprit medications if there is a temporal relationship between the initiation of the medication and the onset of diarrhea, as well as encouraging smoking cessation. These steps alone, however, are unlikely to achieve clinical remission in most patients. A stepwise pharmacological approach is used in the management of MC based on disease severity (Figure 2). For patients with mild symptoms, antidiarrheal medications, such as loperamide, may be helpful.36 Long-term use of loperamide at therapeutic doses no greater than 16 mg daily appears to be safe if required to maintain symptom response. For those with persistent symptoms despite antidiarrheal medications, bismuth subsalicylate at three 262 mg tablets three times daily for 6-8 weeks can be considered. Long-term use of bismuth subsalicylate is not advised, especially at this dose, because of possible neurotoxicity.37
For patients refractory to the above treatments or those with moderate-to-severe symptoms, an 8-week course of budesonide at 9 mg daily is the first-line treatment.38 The dose was tapered before discontinuation in some studies but not in others. Both strategies appear effective. A recent meta-analysis of nine randomized trials demonstrated pooled ORs of 7.34 (95% CI, 4.08-13.19) and 8.35 (95% CI, 4.14-16.85) for response to budesonide induction and maintenance, respectively.39
Cholestyramine is another medication considered in the management of MC and warrants further investigation. To date, no randomized clinical trials have been conducted to evaluate bile acid sequestrants in MC, but they should be considered before placing patients on immunosuppressive medications. Some providers use mesalamine in this setting, although mesalamine is inferior to budesonide in the induction of clinical remission in MC.40
Despite high rates of response to budesonide, relapse after discontinuation is frequent (60%-80%), and time to relapse is variable41,42 The American Gastroenterological Association recommends budesonide for maintenance of remission in patients with recurrence following discontinuation of induction therapy. The lowest effective dose that maintains resolution of symptoms should be prescribed, ideally at 6 mg daily or lower.38 Although budesonide has a greater first-pass metabolism, compared with other glucocorticoids, patients should be monitored for possible side effects including hypertension, diabetes, and osteoporosis, as well as ophthalmologic disease, including cataracts and glaucoma.
For those who are intolerant to budesonide or have refractory symptoms, concomitant disorders such as CD that may be contributing to symptoms must be excluded. Immunosuppressive medications – such as thiopurines and biologic agents, including tumor necrosis factor–alpha inhibitors or vedolizumab – may be considered in refractory cases.43,44 Of note, there are limited studies evaluating the use of these medications for MC. Lastly, surgeries including ileostomy with or without colectomy have been performed in the most severe cases for resistant disease that has failed numerous pharmacological therapies.45
Patients should be counseled that, while symptoms from MC can be quite bothersome and disabling, there appears to be a normal life expectancy and no association between MC and colon cancer, unlike with other inflammatory conditions of the colon such as IBD.46,47
Conclusion and future outlook
As a common cause of chronic watery diarrhea, MC will be commonly encountered in primary care and gastroenterology practices. The diagnosis should be suspected in patients presenting with chronic or recurrent watery diarrhea, especially with female gender, autoimmune disease, and increasing age. The management of MC requires an algorithmic approach directed by symptom severity, with a subgroup of patients requiring maintenance therapy for relapsing symptoms. The care of patients with MC will continue to evolve in the future. Further work is needed to explore long-term safety outcomes with budesonide and the role of immunomodulators and newer biologic agents for patients with complex, refractory disease.
Dr. Tome is with the department of internal medicine at the Mayo Clinic, Rochester, Minn. Dr. Kamboj, and Dr. Pardi are with the division of gastroenterology and hepatology at the Mayo Clinic. Dr. Pardi has grant funding from Pfizer, Vedanta, Seres, Finch, Applied Molecular Transport, and Takeda and has consulted for Vedanta and Otsuka. The other authors have no conflicts of interest to report.
References
1. Nyhlin N et al. Aliment Pharmacol Ther. 2014;39:963-72.
2. Miehlke S et al. United European Gastroenterol J. 2020;20-8.
3. Pardi DS et al. Gut. 2007;56:504-8.
4. Fernández-Bañares F et al. J Crohn’s Colitis.2016;10(7):805-11.
5. Gentile NM et al. Clin Gastroenterol Hepatol. 2014;12(5):838-42.
6. Tong J et al. Am J Gastroenterol. 2015;110:265-76.
7. Olesen M et al. Gut. 2004;53(3):346-50.
8. Bergman D et al. Aliment Pharmacol Ther. 2019;49(11):1395-400.
9. Guagnozzi D et al. Dig Liver Dis. 2012;44(5):384-8.
10. Münch A et al. J Crohns Colitis. 2012;6(9):932-45.
11. Macaigne G et al. Am J Gastroenterol. 2014; 09(9):1461-70.
12. Verhaegh BP et al. Aliment Pharmacol Ther. 2016;43(9):1004-13.
13. Stewart M et al. Aliment Pharmacol Ther. 2011;33(12):1340-9.
14. Green PHR et al. Clin Gastroenterol Hepatol. 2009;7(11):1210-6.
15. Masclee GM et al. Am J Gastroenterol. 2015;110:749-59.
16. Zylberberg H et al. Ailment Pharmacol Ther. 2021 Jun;53(11)1209-15.
17. Jaruvongvanich V et al. Inflamm Bowel Dis. 2019;25(4):672-8.
18. Fernández-Bañares F et al. Inflamm Bowel Dis. 2013; 19(7):1470-6.
19. Yen EF et al. Inflamm Bowel Dis. 2012;18(10):1835-41.
20. Barmeyer C et al. J Gastroenterol. 2017;52(10):1090-100.
21. Morgan DM et al. Clin Gastroenterol Hepatol. 2020;18(4):984-6.
22. Larsson JK et al. Eur J Clin Nutr. 2016;70:1309-17.
23. Madisch A et al.. Inflamm Bowel Dis. 2011;17(11):2295-8.
24. Stahl E et al. Gastroenterology. 2020;159(2):549-61.
25. Sikander A et al. Dig Dis Sci. 2015; 60:887-94.
26. Abdo AA et al. Can J Gastroenterol. 2001;15(5):341-3.
27. Fernandez-Bañares F et al. Dig Dis Sci.2001;46(10):2231-8.
28. Lyutakov I et al. Eur J Gastroenterol Hepatol. 2021;1;33(3):380-7.
29. Hjortswang H et al. Dig Liver Dis. 2011 Feb;43(2):102-9.
30. Cotter TG= et al. Gut. 2018;67(3):441-6.
31. Von Arnim U et al. Clin Exp Gastroenterol. 2016;9:97-103.
32. Langner C et al. Histopathology. 2015;66:613-26.
33. ASGE Standards of Practice Committee and Sharaf RN et al. Gastrointest Endosc. 2013;78:216-24.
34. Macaigne G et al. Clin Res Hepatol Gastroenterol. 2017;41(3):333-40.
35. Bjørnbak C et al. Aliment Pharmacol Ther. 2011;34(10):1225-34.
36. Pardi DS et al. Gastroenterology. 2016;150(1):247-74.
37. Masannat Y and Nazer E. West Virginia Med J. 2013;109(3):32-4.
38. Nguyen GC et al. Gastroenterology. 2016; 150(1):242-6.
39. Sebastian S et al. Eur J Gastroenterol Hepatol. 2019 Aug;31(8):919-27.
40. Miehlke S et al. Gastroenterology. 2014;146(5):1222-30.
41. Gentile NM et al. Am J Gastroenterol. 2013;108:256-9.
42. Münch A et al. Gut. 2016; 65(1):47-56.
43. Cotter TG et al. Aliment Pharmacol Ther. 2017; 46(2):169-74.
44. Esteve M et al. J Crohns Colitis. 2011;5(6):612-8.
45. Cottreau J et al. Clin J Gastroenterol. 2016;9:140-4.
46. Kamboj AK et al. Program No. P1876. ACG 2018 Annual Scientific Meeting Abstracts. Philadelphia, Pennsylvania: American College of Gastroenterology.
47. Yen EF et al. Dig Dis Sci. 2012;57:161-9.
Microscopic colitis is an inflammatory disease of the colon and a frequent cause of chronic or recurrent watery diarrhea, particularly in older persons. MC consists of two subtypes, collagenous colitis (CC) and lymphocytic colitis (LC). While the primary symptom is diarrhea, other signs and symptoms such as abdominal pain, weight loss, and dehydration or electrolyte abnormalities may also be present depending on disease severity.1 In MC, the colonic mucosa usually appears normal on colonoscopy, and the diagnosis is made by histologic findings of intraepithelial lymphocytosis with (CC) or without (LC) a prominent subepithelial collagen band. The management approaches to CC and LC are similar and should be directed based on the severity of symptoms.2 We review the epidemiology, risk factors, pathophysiology, diagnosis, and clinical management for this condition, as well as novel therapeutic approaches.
Epidemiology
Although the incidence of MC increased in the late twentieth century, more recently, it has stabilized with an estimated incidence varying from 1 to 25 per 100,000 person-years.3-5 A recent meta-analysis revealed a pooled incidence of 4.85 per 100,000 persons for LC and 4.14 per 100,000 persons for CC.6 Proposed explanations for the rising incidence in the late twentieth century include improved clinical awareness of the disease, possible increased use of drugs associated with MC, and increased performance of diagnostic colonoscopies for chronic diarrhea. Since MC is now well-recognized, the recent plateau in incidence rates may reflect decreased detection bias.
The prevalence of MC ranges from 10%-20% in patients undergoing colonoscopy for chronic watery diarrhea.6,7 The prevalence of LC is approximately 63.1 cases per 100,000 person-years and, for CC, is 49.2 cases per 100,000 person-years.6-8 Recent studies have demonstrated increasing prevalence of MC likely resulting from an aging population.9,10
Risk stratification
Female gender, increasing age, concomitant autoimmune disease, and the use of certain drugs, including NSAIDs, proton pump inhibitors (PPIs), statins, and selective serotonin reuptake inhibitors (SSRIs), have been associated with an increased risk of MC.11,12 Autoimmune disorders, including celiac disease (CD), rheumatoid arthritis, hypothyroidism, and hyperthyroidism, are more common in patients with MC. The association with CD, in particular, is clinically important, as CD is associated with a 50-70 times greater risk of MC, and 2%-9% of patients with MC have CD.13,14
Several medications have been associated with MC. In a British multicenter prospective study, MC was associated with the use of NSAIDs, PPIs, and SSRIs;15 however, recent studies have questioned the association of MC with some of these medications, which might worsen diarrhea but not actually cause MC.16
An additional risk factor for MC is smoking. A recent meta-analysis demonstrated that current and former smokers had an increased risk of MC (odds ratio, 2.99; 95% confidence interval, 2.15-4.15 and OR, 1.63; 95% CI, 1.37-1.94, respectively), compared with nonsmokers.17 Smokers develop MC at a younger age, and smoking is associated with increased disease severity and decreased likelihood of attaining remission.18,19
Pathogenesis
The pathogenesis of MC remains largely unknown, although there are several hypotheses. The leading proposed mechanisms include reaction to luminal antigens, dysregulated collagen metabolism, genetic predisposition, autoimmunity, and bile acid malabsorption.
MC may be caused by abnormal epithelial barrier function, leading to increased permeability and reaction to luminal antigens, including dietary antigens, certain drugs, and bacterial products, 20,21 which themselves lead to the immune dysregulation and intestinal inflammation seen in MC. This mechanism may explain the association of several drugs with MC. Histological changes resembling LC are reported in patients with CD who consume gluten; however, large population-based studies have not found specific dietary associations with the development of MC.22
Another potential mechanism of MC is dysregulated collagen deposition. Collagen accumulation in the subepithelial layer in CC may result from increased levels of fibroblast growth factor, transforming growth factor–beta and vascular endothelial growth factor.23 Nonetheless, studies have not found an association between the severity of diarrhea in patients with CC and the thickness of the subepithelial collagen band.
Thirdly, autoimmunity and genetic predisposition have been postulated in the pathogenesis of MC. As previously discussed, MC is associated with several autoimmune diseases and predominantly occurs in women, a distinctive feature of autoimmune disorders. Several studies have demonstrated an association between MC and HLA-DQ2 and -DQ3 haplotypes,24 as well as potential polymorphisms in the serotonin transporter gene promoter.25 It is important to note, however, that only a few familial cases of MC have been reported to date.26
Lastly, bile acid malabsorption may play a role in the etiology of MC. Histologic findings of inflammation, along with villous atrophy and collagen deposition, have been reported in the ileum of patients with MC;27,28 however, because patients with MC without bile acid malabsorption may also respond to bile acid binders such as cholestyramine, these findings unlikely to be the sole mechanism explaining the development of the disease.
Despite the different proposed mechanisms for the pathogenesis of MC, no definite conclusions can be drawn because of the limited size of these studies and their often conflicting results.
Clinical features
Clinicians should suspect MC in patients with chronic or recurrent watery diarrhea, particularly in older persons. Other risk factors include female gender, use of certain culprit medications, smoking, and presence of other autoimmune diseases. The clinical manifestations of MC subtypes LC and CC are similar with no significant clinical differences.1,2 In addition to diarrhea, patients with MC may have abdominal pain, fatigue, and dehydration or electrolyte abnormalities depending on disease severity. Patients may also present with fecal urgency, incontinence, and nocturnal stools. Quality of life is often reduced in these patients, predominantly in those with severe or refractory symptoms.29,30 The natural course of MC is highly variable, with some patients achieving spontaneous resolution after one episode and others developing chronic symptoms.
Diagnosis
The differential diagnosis of chronic watery diarrhea is broad and includes malabsorption/maldigestion, inflammatory bowel disease (IBD), irritable bowel syndrome, and medication side effects. In addition, although gastrointestinal infections typically cause acute or subacute diarrhea, some can present with chronic diarrhea. Malabsorption/maldigestion may occur because of CD, lactose intolerance, and pancreatic insufficiency, among other conditions. A thorough history, regarding recent antibiotic and medication use, travel, and immunosuppression, should be obtained in patients with chronic diarrhea. Additionally, laboratory and endoscopic evaluation with random biopsies of the colon can further help differentiate these diseases from MC. A few studies suggest fecal calprotectin may be used to differentiate MC from other noninflammatory conditions such as irritable bowel syndrome, as well as to monitor disease activity. This test is not expected to distinguish MC from other inflammatory causes of diarrhea, such as IBD, and therefore, its role in clinical practice is uncertain.31
The diagnosis of MC is made by biopsy of the colonic mucosa demonstrating characteristic pathologic features.32 Unlike in diseases such as Crohn’s disease or ulcerative colitis, the colon usually appears normal in MC, although mild nonspecific changes, such as erythema or edema, may be visualized. There is no consensus on the ideal location to obtain biopsies for MC or whether biopsies from both the left and the right colon are required.2,33 The procedure of choice for the diagnosis of MC is colonoscopy with random biopsies taken throughout the colon. More limited evaluation by flexible sigmoidoscopy with biopsies may miss cases of MC as inflammation and collagen thickening are not necessarily uniform throughout the colon; however, in a patient that has undergone a recent colonoscopy for colon cancer screening without colon biopsies, a flexible sigmoidoscopy may be a reasonable next test for evaluation of MC, provided biopsies are obtained above the rectosigmoid colon.34
The MC subtypes are differentiated based on histology. The hallmark of LC is less than 20 intraepithelial lymphocytes per 100 surface epithelial cells (normal, less than 5) (Figure 1A). CC is characterized by a thickened subepithelial collagen band greater than 7-10 micrometers (normal, less than 5) (Figure 1B). For a subgroup of patients with milder abnormalities that do not meet these histological criteria, the terms “microscopic colitis, not otherwise specified” or “microscopic colitis, incomplete” may be used.35 These patients often respond to standard treatments for MC. There is an additional subset of patients with biopsy demonstrating features of both CC and LC simultaneously, as well as patients transitioning from one MC subtype to another over time.32,35
Management approach
The first step in management of patients with MC includes stopping culprit medications if there is a temporal relationship between the initiation of the medication and the onset of diarrhea, as well as encouraging smoking cessation. These steps alone, however, are unlikely to achieve clinical remission in most patients. A stepwise pharmacological approach is used in the management of MC based on disease severity (Figure 2). For patients with mild symptoms, antidiarrheal medications, such as loperamide, may be helpful.36 Long-term use of loperamide at therapeutic doses no greater than 16 mg daily appears to be safe if required to maintain symptom response. For those with persistent symptoms despite antidiarrheal medications, bismuth subsalicylate at three 262 mg tablets three times daily for 6-8 weeks can be considered. Long-term use of bismuth subsalicylate is not advised, especially at this dose, because of possible neurotoxicity.37
For patients refractory to the above treatments or those with moderate-to-severe symptoms, an 8-week course of budesonide at 9 mg daily is the first-line treatment.38 The dose was tapered before discontinuation in some studies but not in others. Both strategies appear effective. A recent meta-analysis of nine randomized trials demonstrated pooled ORs of 7.34 (95% CI, 4.08-13.19) and 8.35 (95% CI, 4.14-16.85) for response to budesonide induction and maintenance, respectively.39
Cholestyramine is another medication considered in the management of MC and warrants further investigation. To date, no randomized clinical trials have been conducted to evaluate bile acid sequestrants in MC, but they should be considered before placing patients on immunosuppressive medications. Some providers use mesalamine in this setting, although mesalamine is inferior to budesonide in the induction of clinical remission in MC.40
Despite high rates of response to budesonide, relapse after discontinuation is frequent (60%-80%), and time to relapse is variable41,42 The American Gastroenterological Association recommends budesonide for maintenance of remission in patients with recurrence following discontinuation of induction therapy. The lowest effective dose that maintains resolution of symptoms should be prescribed, ideally at 6 mg daily or lower.38 Although budesonide has a greater first-pass metabolism, compared with other glucocorticoids, patients should be monitored for possible side effects including hypertension, diabetes, and osteoporosis, as well as ophthalmologic disease, including cataracts and glaucoma.
For those who are intolerant to budesonide or have refractory symptoms, concomitant disorders such as CD that may be contributing to symptoms must be excluded. Immunosuppressive medications – such as thiopurines and biologic agents, including tumor necrosis factor–alpha inhibitors or vedolizumab – may be considered in refractory cases.43,44 Of note, there are limited studies evaluating the use of these medications for MC. Lastly, surgeries including ileostomy with or without colectomy have been performed in the most severe cases for resistant disease that has failed numerous pharmacological therapies.45
Patients should be counseled that, while symptoms from MC can be quite bothersome and disabling, there appears to be a normal life expectancy and no association between MC and colon cancer, unlike with other inflammatory conditions of the colon such as IBD.46,47
Conclusion and future outlook
As a common cause of chronic watery diarrhea, MC will be commonly encountered in primary care and gastroenterology practices. The diagnosis should be suspected in patients presenting with chronic or recurrent watery diarrhea, especially with female gender, autoimmune disease, and increasing age. The management of MC requires an algorithmic approach directed by symptom severity, with a subgroup of patients requiring maintenance therapy for relapsing symptoms. The care of patients with MC will continue to evolve in the future. Further work is needed to explore long-term safety outcomes with budesonide and the role of immunomodulators and newer biologic agents for patients with complex, refractory disease.
Dr. Tome is with the department of internal medicine at the Mayo Clinic, Rochester, Minn. Dr. Kamboj, and Dr. Pardi are with the division of gastroenterology and hepatology at the Mayo Clinic. Dr. Pardi has grant funding from Pfizer, Vedanta, Seres, Finch, Applied Molecular Transport, and Takeda and has consulted for Vedanta and Otsuka. The other authors have no conflicts of interest to report.
References
1. Nyhlin N et al. Aliment Pharmacol Ther. 2014;39:963-72.
2. Miehlke S et al. United European Gastroenterol J. 2020;20-8.
3. Pardi DS et al. Gut. 2007;56:504-8.
4. Fernández-Bañares F et al. J Crohn’s Colitis.2016;10(7):805-11.
5. Gentile NM et al. Clin Gastroenterol Hepatol. 2014;12(5):838-42.
6. Tong J et al. Am J Gastroenterol. 2015;110:265-76.
7. Olesen M et al. Gut. 2004;53(3):346-50.
8. Bergman D et al. Aliment Pharmacol Ther. 2019;49(11):1395-400.
9. Guagnozzi D et al. Dig Liver Dis. 2012;44(5):384-8.
10. Münch A et al. J Crohns Colitis. 2012;6(9):932-45.
11. Macaigne G et al. Am J Gastroenterol. 2014; 09(9):1461-70.
12. Verhaegh BP et al. Aliment Pharmacol Ther. 2016;43(9):1004-13.
13. Stewart M et al. Aliment Pharmacol Ther. 2011;33(12):1340-9.
14. Green PHR et al. Clin Gastroenterol Hepatol. 2009;7(11):1210-6.
15. Masclee GM et al. Am J Gastroenterol. 2015;110:749-59.
16. Zylberberg H et al. Ailment Pharmacol Ther. 2021 Jun;53(11)1209-15.
17. Jaruvongvanich V et al. Inflamm Bowel Dis. 2019;25(4):672-8.
18. Fernández-Bañares F et al. Inflamm Bowel Dis. 2013; 19(7):1470-6.
19. Yen EF et al. Inflamm Bowel Dis. 2012;18(10):1835-41.
20. Barmeyer C et al. J Gastroenterol. 2017;52(10):1090-100.
21. Morgan DM et al. Clin Gastroenterol Hepatol. 2020;18(4):984-6.
22. Larsson JK et al. Eur J Clin Nutr. 2016;70:1309-17.
23. Madisch A et al.. Inflamm Bowel Dis. 2011;17(11):2295-8.
24. Stahl E et al. Gastroenterology. 2020;159(2):549-61.
25. Sikander A et al. Dig Dis Sci. 2015; 60:887-94.
26. Abdo AA et al. Can J Gastroenterol. 2001;15(5):341-3.
27. Fernandez-Bañares F et al. Dig Dis Sci.2001;46(10):2231-8.
28. Lyutakov I et al. Eur J Gastroenterol Hepatol. 2021;1;33(3):380-7.
29. Hjortswang H et al. Dig Liver Dis. 2011 Feb;43(2):102-9.
30. Cotter TG= et al. Gut. 2018;67(3):441-6.
31. Von Arnim U et al. Clin Exp Gastroenterol. 2016;9:97-103.
32. Langner C et al. Histopathology. 2015;66:613-26.
33. ASGE Standards of Practice Committee and Sharaf RN et al. Gastrointest Endosc. 2013;78:216-24.
34. Macaigne G et al. Clin Res Hepatol Gastroenterol. 2017;41(3):333-40.
35. Bjørnbak C et al. Aliment Pharmacol Ther. 2011;34(10):1225-34.
36. Pardi DS et al. Gastroenterology. 2016;150(1):247-74.
37. Masannat Y and Nazer E. West Virginia Med J. 2013;109(3):32-4.
38. Nguyen GC et al. Gastroenterology. 2016; 150(1):242-6.
39. Sebastian S et al. Eur J Gastroenterol Hepatol. 2019 Aug;31(8):919-27.
40. Miehlke S et al. Gastroenterology. 2014;146(5):1222-30.
41. Gentile NM et al. Am J Gastroenterol. 2013;108:256-9.
42. Münch A et al. Gut. 2016; 65(1):47-56.
43. Cotter TG et al. Aliment Pharmacol Ther. 2017; 46(2):169-74.
44. Esteve M et al. J Crohns Colitis. 2011;5(6):612-8.
45. Cottreau J et al. Clin J Gastroenterol. 2016;9:140-4.
46. Kamboj AK et al. Program No. P1876. ACG 2018 Annual Scientific Meeting Abstracts. Philadelphia, Pennsylvania: American College of Gastroenterology.
47. Yen EF et al. Dig Dis Sci. 2012;57:161-9.
Dyssynergic defecation
Introduction
About 40% of the population experiences lower GI symptoms suggestive of gastrointestinal motility disorders.1,2 The global prevalence of chronic constipation is 18%, and the condition includes multiple overlapping subtypes.3 Evacuation disorders affect over half (59%) of patients and include dyssynergic defecation (DD).4 The inability to coordinate the abdominal, rectal, pelvic floor, and anal/puborectalis muscles to evacuate stools causes DD.5 The etiology of DD remains unclear and is often misdiagnosed. Clinically, the symptoms of DD overlap with other lower GI disorders, often leading to unnecessary and invasive procedures.2 We describe the clinical characteristics, diagnostic tools, treatment options, and evidence-based approach for the management of DD.

Clinical presentation
Over two-thirds of patients with DD acquire this disorder during adulthood, and one-third have symptoms from childhood.6 Though there is not usually an inciting event, 29% of patients report that symptoms began after events such as pregnancy or back injury,6 and opioid users have higher prevalence and severity of DD.7

Over 80% of patients report excessive straining, feelings of incomplete evacuation, and hard stools, and 50% report sensation of anal blockage or use of digital maneuvers.2 Other symptoms include infrequent bowel movements, abdominal pain, anal pain, and stool leakage.2 Evaluation of DD includes obtaining a detailed history utilizing the Bristol Stool Form Scale;8 however, patients’ recall of stool habit is often inaccurate, which results in suboptimal care.9,10 Prospective stool diaries can help to provide more objective assessment of patients’ symptoms, eliminate recall bias, and provide more reliable information. Several useful questionnaires are available for clinical and research purposes to characterize lower-GI symptoms, including the Constipation Scoring System,11 Patient Assessment of Constipation Symptoms (PAC-SYM),12 and Patient Assessment of Constipation Quality of Life (PAC-QOL).2,13 The Constipation Stool digital app enhances accuracy of data capture and offers a reliable and user-friendly method for recording bowel symptoms for patients, clinicians, and clinical investigators.14
Diagnosis
The diagnosis of DD requires careful physical and digital rectal examination together with anorectal manometry and a balloon expulsion test. Defecography and colonic transit studies provide additional assessment.

Physical examination
Abdominal examination should include palpation for stool in the colon and identification of abdominal mass or fecal impaction.2A high-quality digital rectal examination can help to identify patients who could benefit from physiological testing to confirm and treat DD.15 Rectal examination is performed by placing examiner’s lubricated gloved right index finger in a patient’s rectum, with the examiner’s left hand on patient’s abdomen, and asking the patient to push and bear down as if defecating.15 The contraction of the abdominal muscles is felt using the left hand, while the anal sphincter relaxation and degree of perineal descent are felt using the right-hand index finger.15 A diagnosis of dyssynergia is suspected if the digital rectal examination reveals two or more of the following abnormalities: inability to contract abdominal muscles (lack of push effort), inability to relax or paradoxical contraction of the anal sphincter and/or puborectalis, or absence of perineal descent.15 Digital rectal examination has good sensitivity (75%), specificity (87%), and positive predictive value (97%) for DD.16
High resolution anorectal manometry
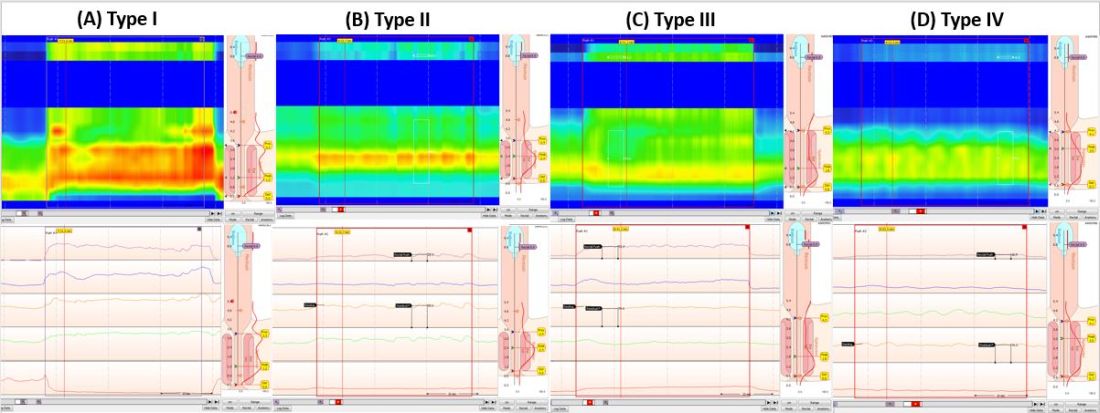
Anorectal manometry (ARM) is the preferred method for the evaluation of defecatory disorders.17,18 ARM is best performed using the high-resolution anorectal manometry (HRAM) systems19 that consist of a flexible probe – 0.5-cm diameter with multiple circumferential sensors along the anal canal – and another two sensors inside a rectal balloon.18 It provides a topographic and waveform display of manometric pressure data (Figure). The 3D high-definition ARM probe is a rigid 1-cm probe that provides 3D topographic profiles.18 ARM is typically performed in both the left lateral position and in a more physiological seated position.20,21 There is considerable variation amongst different institutions on how to perform HRAM, and a recent International Anorectal Physiology Working Group (IAPWG) has provided consensus recommendations for performing this test.22 The procedure for performing HRAM is reviewed elsewhere, but the key elements are summarized below.18
Push maneuver: On HRAM, after the assessment of resting and squeeze anal sphincter pressures, the patient is asked to push or bear down as if to defecate while lying in left lateral decubitus position. The best of two attempts that closely mimics a normal bearing down maneuver is used for categorizing patient’s defecatory pattern.18 In patients with DD, at least four distinct dyssynergia phenotypes have been recognized (Figure),23 though recent studies suggest eight patterns.24 Defecation index (maximum rectal pressure/minimum residual anal pressure when bearing down) greater than 1.2 is considered normal.18
Simulated defecation on commode: The subject is asked to attempt defecation while seated on a commode with intrarectal balloon filled with 60 cc of air, and both the defecation pattern(s) and defecation index are calculated. A lack of coordinated push effort is highly suggestive of DD.21
Rectoanal Inhibitory Reflex (RAIR): RAIR describes the reflex relaxation of the internal anal sphincter after rectal distension. RAIR is dependent on intact autonomic ganglia and myenteric plexus25and is mediated by the release of nitric oxide and vasoactive intestinal peptide.26 The absence of RAIR suggests Hirschsprung disease.22.27.28
Rectal sensory testing: Intermittent balloon distension of the rectum with incremental volumes of air induces a range of rectal sensations that include first sensation, desire to defecate, urgency to defecate, and maximum tolerable volume. Rectal hyposensitivity is diagnosed when two or more sensory thresholds are higher than those seen in normal subjects29.30 and likely results from disruption of afferent gut-brain pathways, cortical perception/rectal wall dysfunction, or both.29 Rectal hyposensitivity affects 40% of patients with constipation30and is associated with DD but not delayed colonic transit.31 Rectal hyposensitivity may also be seen in patients with diabetes or fecal incontinence.18 About two-thirds of patients with rectal hyposensitivity have rectal hypercompliance, and some have megarectum.32 Some patients with DD have coexisting irritable bowel syndrome (IBS) and may have rectal hypersensitivity.18,33 Rectal compliance is measured alongside rectal sensitivity analysis by plotting a graph between the change in intraballoon volume (mL) and change in intrarectal pressures (mm Hg) during incremental balloon distensions.18.34 Rectal hypercompliance may be seen in megarectum and dyssynergic defecation.34,35 Rectal hypocompliance may be seen in patients with inflammatory bowel disease, postpelvic radiation, chronic ischemia, and advanced age.18
Balloon expulsion test: This test is performed by placing a plastic probe with a balloon in the rectum and filling it with 50 cc of warm water. Patients are given 5 minutes to expel the balloon while sitting on a commode. Balloon expulsion time of more than 1 minute suggests a diagnosis of DD,21 although 2 minutes provides a higher level of agreement with manometric findings.36 Balloon type and body position can influence the results.37 Inability to expel the balloon with normal manometric findings is considered an inconclusive finding per the recent London Classification (i.e., it may be associated with generation of anorectal symptoms, but the clinical relevance of this finding is unclear as it may also be seen in healthy subjects).22
Defecography
Defecography is a dynamic fluoroscopic study performed in the sitting position after injecting 150 mL of barium paste into the patient’s rectum. Defecography provides useful information about structural changes (e.g., rectoceles, enteroceles, rectal prolapse, and intussusception), DD, and descending perineum syndrome.38 Methodological differences, radiation exposure, and poor interobserver agreement have limited its wider use; therefore, anorectal manometry and the balloon expulsion test are recommended for the initial evaluation of DD.39 Magnetic resonance defecography may be more useful.17,38
Colonic transit studies
Colonic transit study can be assessed using radiopaque markers, wireless motility capsule, or scintigraphy. Wireless motility capsule and scintigraphy have the advantage of determining gastric, small bowel, and whole gut transit times as well. About two-thirds of patients with DD have slow transit constipation (STC),6 which improves after treatment of DD.40 Hence, in patients with chronic constipation, evaluation and management of DD is recommended first. If symptoms persist, then consider colonic transit assessment.41 Given the overlapping nature of the conditions, documentation of STC at the outset could facilitate treatment of both.
Diagnostic criteria for DD
Patients should fulfill the following criteria for diagnosis of DD:42,43
- Fulfill symptom(s) diagnostic criteria for functional constipation and/or constipation-predominant IBS.
- Demonstrate dyssynergic pattern (Types I-IV; Figure) during attempted defecation on manometry recordings.
- Meet one or more of the following criteria:
- Inability to expel an artificial stool (50 mL water-filled balloon) within 1 minute.
- Inability to evacuate or retention of 50% or more of barium during defecography. (Some institutions use a prolonged colonic transit time: greater than 5 markers or 20% or higher marker retention on a plain abdominal x-Ray at 120 hours after ingestion of one radio-opaque marker capsule containing 24 radio-opaque markers.)
Treatment of DD
The treatment modalities for DD depend on several factors: patient’s age, comorbidities, underlying pathophysiology, and patient expectations. Treatment options include standard management of constipation, but biofeedback therapy is the mainstay.
Standard management
Medications that cause or worsen constipation should be avoided. The patient should consume adequate fluid and exercise regularly. Patients should receive instructions for timed toilet training (twice daily, 30 minutes after meals). Patients should push at about 50%-70% of their ability for no longer than 5 minutes and avoid postponing defecation or use of digital maneuvers to facilitate defecation.42 The patients should take 25 g of soluble fiber (e.g., psyllium) daily. Of note, the benefits of fiber can take days to weeks44 and may be limited in patients with STC and DD.45 Medications including laxatives and intestinal secretagogues (lubiprostone, linaclotide, plecanatide), and enterokinetic agents (prucalopride) can be used as adjunct therapy for management of DD.42 Their use is titrated during and after biofeedback therapy and may decrease after successful treatment.46
Biofeedback therapy
Biofeedback therapy involves operant conditioning techniques using either a solid state anorectal manometry system, electromyography, simulated balloon, or home biofeedback training devices.42,47 The goals of biofeedback therapy are to correct the abdominal pelvic muscle discoordination during defecation and improve rectal sensation to stool if impaired. Biofeedback therapy involves patient education and active training (typically six sessions, 1-2 weeks apart, with each about 30-60 minutes long), followed by a reinforcement stage (three sessions at 3, 6, and 12 months), though there are variations in training protocols.42
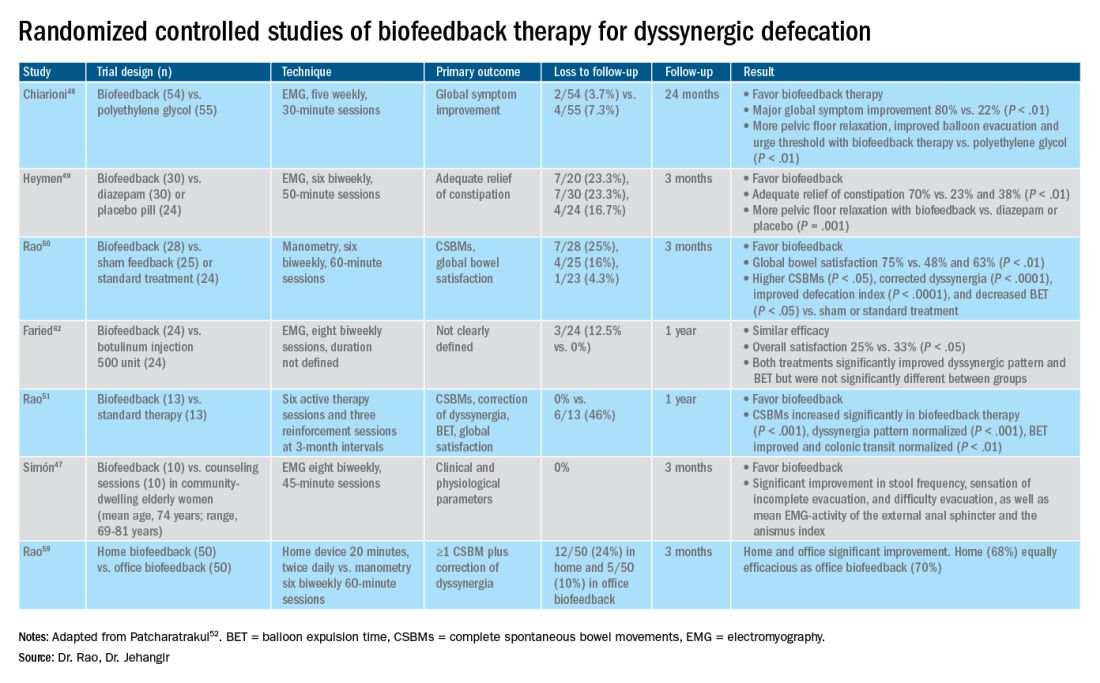
The success of biofeedback therapy depends on the patient’s motivation and the therapist’s skills.42 Compared with standard therapy (diet, exercise, pharmacotherapy), biofeedback therapy provides sustained improvement of bowel symptoms and anorectal function. Up to 70%-80% of DD patients show significant improvement of symptoms in randomized controlled trials (Table).48-52 Biofeedback therapy may also improve dyspeptic symptoms.53 Patients with harder stool consistency, greater willingness to participate, lower baseline bowel satisfaction, lower baseline anal sphincter relaxation, and prolonged balloon expulsion time, as well as patients who used digital maneuvers for defection, more commonly respond to biofeedback therapy.54,55 Longstanding laxative use has been associated with decreased response to biofeedback therapy.56 In patients with rectal hyposensitivity, barostat-assisted sensory training is more effective than a hand-held syringe technique.30 In patients with constipation predominant IBS and rectal hyposensitivity, sensory adaption training is more efficacious and better tolerated than escitalopram.30 Biofeedback therapy was afforded a grade A recommendation for treatment of DD by the American and European Societies of Neurogastroenterology and Motility.57
The access to office-based biofeedback therapy may be limited because of costs and low availability. The time required to attend multiple sessions may be burdensome for some patients, especially if they are taking time off from work. A recent study showed that patients with higher level of education may be less likely to adhere to biofeedback therapy.58 Recently, home biofeedback was shown to be noninferior to office biofeedback and was more cost-effective, which provides an alternative option for treating more patients.59
Endoscopic/surgical options
Other less effective treatment options for DD include botulinum toxin injection and myectomy.60-62 Botulinum toxin injection appears to have mixed effects with less than 50% of patients reporting symptomatic improvement, and it may cause fecal incontinence.60,63
Conclusion
DD is a common yet poorly recognized cause of constipation. Its clinical presentation overlaps with other lower-GI disorders. Its diagnosis requires detailed history, digital rectal examination, prospective stool diaries, anorectal manometry, and balloon expulsion tests. Biofeedback therapy offers excellent and sustained symptomatic improvement; however, access to office-based biofeedback is limited, and there is an urgent need for home-based biofeedback therapy programs.59
Dr. Rao is J. Harold Harrison Distinguished University Chair, professor of medicine, director of neurogastroenterology/motility, and director of digestive health at the Digestive Health Clinical Research Center Augusta (Georgia) University. He is supported by National Institutes of Health grants R01DK121003-02 and U01DK115572. Dr. Jehangir is a gastroenterology and Hepatology Fellow at the Digestive Health Clinical Research Center at Augusta University. They reported having no conflicts of interest.
References
1. Peery AF, et al. Gastroenterology. 2012;143(5):1179-1187.e3 .
2. Curtin B, et al. J Neurogastroenterol Motil. 2020 30;26(4):423-36.
3. Suares NC & Ford AC. Am J Gastroenterol. 2011 Sep;106(9):1582-91.
4. Mertz H, et al. Am J Gastroenterol. 1999;94(3):609-15.
5. Rao SS, et al. Am J Gastroenterol. 1998;93(7):1042-50.
6. Rao SSC, et al. J Clin Gastroenterol. 2004;38(8):680-5.
7. Nojkov B, et al. Am J Gastroenterol. 2019;114(11):1772-7.
8. Heaton KW, et al. Gut. 1992;33(6):818-24.
9. Prichard DO & Bharucha AE. 2018 Oct 15;7:F1000 Faculty Rev-1640.
10. Ashraf W, et al. Am J Gastroenterol. 1996;91(1):26-32.
11. Agachan F, et al.. Dis Colon Rectum. 1996;39(6):681-5.
12. Frank L, et al. Scand J Gastroenterol. 1999;34(9):870-7.
13. Marquis P, et al. Scand J Gastroenterol. 2005;40(5):540-51.
14. Yan Y, et al. Gastroenterology. 2020;158(6):S-400.
15. Rao SSC. Am J Gastroenterol. 2018;113(5):635-8.
16. Tantiphlachiva K, et al. Digital rectal examination is a useful tool for identifying patients with dyssynergia. Clin Gastroenterol Hepatol. 2010;8(11):955-60.
17. Carrington EV, et al. Nat Rev Gastroenterol Hepatol. 2018;15(5):309-23.
18. Tetangco EP, et al. Performing and analyzing high-resolution anorectal manometry. NeuroGastroLatam Rev. 2018;2:120-32.
19. Lee YY, et al. Curr Gastroenterol Rep. 2013;15(12):360.
20. Sharma M, et al. Neurogastroenterol Motil. 2020;32(10):e13910.
21. Rao SSC, et al.. Am J Gastroenterol. 2006;101(12):2790-6.
22. Carrington EV, et al. Neurogastroenterol Motil. 2020;32(1):e13679.
23. Rao SSC. Gastroenterol Clin North Am. 2008;37(3):569-86, viii.
24. Rao SSC, et al. Gastroenterology. 2016;150(4):S158-9.
25. Guinet A, et al. Int J Colorectal Dis. 2011;26(4):507-13.
26. Rattan S, et al. Gastroenterology. 1992;103(1):43-50.
27. Remes-Troche JM & Rao SSC. 2008;2(3):323-35.
28. Zaafouri H, et al..Int J Surgery. 2015. 2(1):9-17.
29. Remes-Troche JM, et al. Dis Colon Rectum. 2010;53(7):1047-54.
30. Rao SSC, et al. Gastroenterology. 2013;144(5):S-363.
31. Yu T, et al. Medicine (Baltimore). 2016;95(19):e3667.
32. Gladman MA, et al. Neurogastroenterol Motil. 2009;21(5):508-16, e4-5.
33. Lee KJ, et al. Digestion. 2006;73(2-3):133-41 .
34. Rao SSC, et al. Neurogastroenterol Motil. 2002;14(5):553-9.
35. Coss-Adame E, et al.. Clin Gastroenterol Hepatol. 2015;13(6):1143-1150.e1.
36. Chiarioni G, et al. Clin Gastroenterol Hepatol. 2014;12(12):2049-54.
37. Gu G, et al. Gastroenterology. 2018;154(6):S-545–S-546.
38. Savoye-Collet C, et al.. Gastroenterol Clin North Am. 2008;37(3):553-67, viii.
39. Videlock EJ, et al. Neurogastroenterol Motil. 2013;25(6):509-20.
40. Rao SSC, et al. Neurogastroenterol Motil. 2004;16(5):589-96.
41. Wald A, et al. Am J Gastroenterol. 2014;109(8):1141-57 ; (Quiz) 1058.
42. Rao SSC & Patcharatrakul T. J Neurogastroenterol Motil. 2016;22(3):423-35.
43. Rao SS, et al. Functional Anorectal Disorders. Gastroenterology. 2016. S0016-5085(16)00175-X.
44. Bharucha AE, et al.. Gastroenterology. 2013;144(1):218-38.
45. Voderholzer WA, et al. Am J Gastroenterol. 1997;92(1):95-8.
46. Lee HJ, et al. Neurogastroenterol Motil. 2015;27(6):787-95.
47. Simón MA & Bueno AM. J Clin Gastroenterol. 2017;51(10):e90-4.
48. Chiarioni G,et al.. Gastroenterology. 2006;130(3):657-64.
49. Heymen S, et al.. Dis Colon Rectum. 2007;50(4):428-41.
50. Rao SSC, et al. Clin Gastroenterol Hepatol. 2007;5(3):331-8.
51. Rao SSC, et al. Am J Gastroenterol. 2010;105(4):890-6.
52. Patcharatrakul T, et al. Biofeedback therapy. In Clinical and basic neurogastroenterology and motility. India: Stacy Masucci; 2020:517-32.
53. Huaman J-W, et al. Clin Gastroenterol Hepatol. 2020;18(11):2463-2470.e1.
54. Patcharatrakul T, et al. Clin Gastroenterol Hepatol. 2018;16(5):715-21.
55. Chaudhry A, et al. Gastroenterology. 2020;158(6):S-382–S-383.
56. Shim LSE, et al. Aliment Pharmacol Ther. 2011;33(11):1245-51.
57. Rao SSC, et al. Neurogastroenterol Motil. 2015;27(5):594-609.
58. Jangsirikul S, et al. Gastroenterology. 2020;158(6):S-383.
59. Rao SSC, et al. Am J Gastroenterol. 2019;114(6):938-44.
60. Ron Y, et al.. Dis Colon Rectum. 2001;44(12):1821-6.
61. Podzemny V, et al. World J Gastroenterol. 2015;21(4):1053-60.
62. Faried M, et al. J Gastrointest Surg. 2010;14(8):1235-43.
63. Hallan RI, et al. Lancet. 1988;2(8613):714-7.
Introduction
About 40% of the population experiences lower GI symptoms suggestive of gastrointestinal motility disorders.1,2 The global prevalence of chronic constipation is 18%, and the condition includes multiple overlapping subtypes.3 Evacuation disorders affect over half (59%) of patients and include dyssynergic defecation (DD).4 The inability to coordinate the abdominal, rectal, pelvic floor, and anal/puborectalis muscles to evacuate stools causes DD.5 The etiology of DD remains unclear and is often misdiagnosed. Clinically, the symptoms of DD overlap with other lower GI disorders, often leading to unnecessary and invasive procedures.2 We describe the clinical characteristics, diagnostic tools, treatment options, and evidence-based approach for the management of DD.
Clinical presentation
Over two-thirds of patients with DD acquire this disorder during adulthood, and one-third have symptoms from childhood.6 Though there is not usually an inciting event, 29% of patients report that symptoms began after events such as pregnancy or back injury,6 and opioid users have higher prevalence and severity of DD.7
Over 80% of patients report excessive straining, feelings of incomplete evacuation, and hard stools, and 50% report sensation of anal blockage or use of digital maneuvers.2 Other symptoms include infrequent bowel movements, abdominal pain, anal pain, and stool leakage.2 Evaluation of DD includes obtaining a detailed history utilizing the Bristol Stool Form Scale;8 however, patients’ recall of stool habit is often inaccurate, which results in suboptimal care.9,10 Prospective stool diaries can help to provide more objective assessment of patients’ symptoms, eliminate recall bias, and provide more reliable information. Several useful questionnaires are available for clinical and research purposes to characterize lower-GI symptoms, including the Constipation Scoring System,11 Patient Assessment of Constipation Symptoms (PAC-SYM),12 and Patient Assessment of Constipation Quality of Life (PAC-QOL).2,13 The Constipation Stool digital app enhances accuracy of data capture and offers a reliable and user-friendly method for recording bowel symptoms for patients, clinicians, and clinical investigators.14
Diagnosis
The diagnosis of DD requires careful physical and digital rectal examination together with anorectal manometry and a balloon expulsion test. Defecography and colonic transit studies provide additional assessment.
Physical examination
Abdominal examination should include palpation for stool in the colon and identification of abdominal mass or fecal impaction.2A high-quality digital rectal examination can help to identify patients who could benefit from physiological testing to confirm and treat DD.15 Rectal examination is performed by placing examiner’s lubricated gloved right index finger in a patient’s rectum, with the examiner’s left hand on patient’s abdomen, and asking the patient to push and bear down as if defecating.15 The contraction of the abdominal muscles is felt using the left hand, while the anal sphincter relaxation and degree of perineal descent are felt using the right-hand index finger.15 A diagnosis of dyssynergia is suspected if the digital rectal examination reveals two or more of the following abnormalities: inability to contract abdominal muscles (lack of push effort), inability to relax or paradoxical contraction of the anal sphincter and/or puborectalis, or absence of perineal descent.15 Digital rectal examination has good sensitivity (75%), specificity (87%), and positive predictive value (97%) for DD.16
High resolution anorectal manometry
Anorectal manometry (ARM) is the preferred method for the evaluation of defecatory disorders.17,18 ARM is best performed using the high-resolution anorectal manometry (HRAM) systems19 that consist of a flexible probe – 0.5-cm diameter with multiple circumferential sensors along the anal canal – and another two sensors inside a rectal balloon.18 It provides a topographic and waveform display of manometric pressure data (Figure). The 3D high-definition ARM probe is a rigid 1-cm probe that provides 3D topographic profiles.18 ARM is typically performed in both the left lateral position and in a more physiological seated position.20,21 There is considerable variation amongst different institutions on how to perform HRAM, and a recent International Anorectal Physiology Working Group (IAPWG) has provided consensus recommendations for performing this test.22 The procedure for performing HRAM is reviewed elsewhere, but the key elements are summarized below.18
Push maneuver: On HRAM, after the assessment of resting and squeeze anal sphincter pressures, the patient is asked to push or bear down as if to defecate while lying in left lateral decubitus position. The best of two attempts that closely mimics a normal bearing down maneuver is used for categorizing patient’s defecatory pattern.18 In patients with DD, at least four distinct dyssynergia phenotypes have been recognized (Figure),23 though recent studies suggest eight patterns.24 Defecation index (maximum rectal pressure/minimum residual anal pressure when bearing down) greater than 1.2 is considered normal.18
Simulated defecation on commode: The subject is asked to attempt defecation while seated on a commode with intrarectal balloon filled with 60 cc of air, and both the defecation pattern(s) and defecation index are calculated. A lack of coordinated push effort is highly suggestive of DD.21
Rectoanal Inhibitory Reflex (RAIR): RAIR describes the reflex relaxation of the internal anal sphincter after rectal distension. RAIR is dependent on intact autonomic ganglia and myenteric plexus25and is mediated by the release of nitric oxide and vasoactive intestinal peptide.26 The absence of RAIR suggests Hirschsprung disease.22.27.28
Rectal sensory testing: Intermittent balloon distension of the rectum with incremental volumes of air induces a range of rectal sensations that include first sensation, desire to defecate, urgency to defecate, and maximum tolerable volume. Rectal hyposensitivity is diagnosed when two or more sensory thresholds are higher than those seen in normal subjects29.30 and likely results from disruption of afferent gut-brain pathways, cortical perception/rectal wall dysfunction, or both.29 Rectal hyposensitivity affects 40% of patients with constipation30and is associated with DD but not delayed colonic transit.31 Rectal hyposensitivity may also be seen in patients with diabetes or fecal incontinence.18 About two-thirds of patients with rectal hyposensitivity have rectal hypercompliance, and some have megarectum.32 Some patients with DD have coexisting irritable bowel syndrome (IBS) and may have rectal hypersensitivity.18,33 Rectal compliance is measured alongside rectal sensitivity analysis by plotting a graph between the change in intraballoon volume (mL) and change in intrarectal pressures (mm Hg) during incremental balloon distensions.18.34 Rectal hypercompliance may be seen in megarectum and dyssynergic defecation.34,35 Rectal hypocompliance may be seen in patients with inflammatory bowel disease, postpelvic radiation, chronic ischemia, and advanced age.18
Balloon expulsion test: This test is performed by placing a plastic probe with a balloon in the rectum and filling it with 50 cc of warm water. Patients are given 5 minutes to expel the balloon while sitting on a commode. Balloon expulsion time of more than 1 minute suggests a diagnosis of DD,21 although 2 minutes provides a higher level of agreement with manometric findings.36 Balloon type and body position can influence the results.37 Inability to expel the balloon with normal manometric findings is considered an inconclusive finding per the recent London Classification (i.e., it may be associated with generation of anorectal symptoms, but the clinical relevance of this finding is unclear as it may also be seen in healthy subjects).22
Defecography
Defecography is a dynamic fluoroscopic study performed in the sitting position after injecting 150 mL of barium paste into the patient’s rectum. Defecography provides useful information about structural changes (e.g., rectoceles, enteroceles, rectal prolapse, and intussusception), DD, and descending perineum syndrome.38 Methodological differences, radiation exposure, and poor interobserver agreement have limited its wider use; therefore, anorectal manometry and the balloon expulsion test are recommended for the initial evaluation of DD.39 Magnetic resonance defecography may be more useful.17,38
Colonic transit studies
Colonic transit study can be assessed using radiopaque markers, wireless motility capsule, or scintigraphy. Wireless motility capsule and scintigraphy have the advantage of determining gastric, small bowel, and whole gut transit times as well. About two-thirds of patients with DD have slow transit constipation (STC),6 which improves after treatment of DD.40 Hence, in patients with chronic constipation, evaluation and management of DD is recommended first. If symptoms persist, then consider colonic transit assessment.41 Given the overlapping nature of the conditions, documentation of STC at the outset could facilitate treatment of both.
Diagnostic criteria for DD
Patients should fulfill the following criteria for diagnosis of DD:42,43
- Fulfill symptom(s) diagnostic criteria for functional constipation and/or constipation-predominant IBS.
- Demonstrate dyssynergic pattern (Types I-IV; Figure) during attempted defecation on manometry recordings.
- Meet one or more of the following criteria:
- Inability to expel an artificial stool (50 mL water-filled balloon) within 1 minute.
- Inability to evacuate or retention of 50% or more of barium during defecography. (Some institutions use a prolonged colonic transit time: greater than 5 markers or 20% or higher marker retention on a plain abdominal x-Ray at 120 hours after ingestion of one radio-opaque marker capsule containing 24 radio-opaque markers.)
Treatment of DD
The treatment modalities for DD depend on several factors: patient’s age, comorbidities, underlying pathophysiology, and patient expectations. Treatment options include standard management of constipation, but biofeedback therapy is the mainstay.
Standard management
Medications that cause or worsen constipation should be avoided. The patient should consume adequate fluid and exercise regularly. Patients should receive instructions for timed toilet training (twice daily, 30 minutes after meals). Patients should push at about 50%-70% of their ability for no longer than 5 minutes and avoid postponing defecation or use of digital maneuvers to facilitate defecation.42 The patients should take 25 g of soluble fiber (e.g., psyllium) daily. Of note, the benefits of fiber can take days to weeks44 and may be limited in patients with STC and DD.45 Medications including laxatives and intestinal secretagogues (lubiprostone, linaclotide, plecanatide), and enterokinetic agents (prucalopride) can be used as adjunct therapy for management of DD.42 Their use is titrated during and after biofeedback therapy and may decrease after successful treatment.46
Biofeedback therapy
Biofeedback therapy involves operant conditioning techniques using either a solid state anorectal manometry system, electromyography, simulated balloon, or home biofeedback training devices.42,47 The goals of biofeedback therapy are to correct the abdominal pelvic muscle discoordination during defecation and improve rectal sensation to stool if impaired. Biofeedback therapy involves patient education and active training (typically six sessions, 1-2 weeks apart, with each about 30-60 minutes long), followed by a reinforcement stage (three sessions at 3, 6, and 12 months), though there are variations in training protocols.42
The success of biofeedback therapy depends on the patient’s motivation and the therapist’s skills.42 Compared with standard therapy (diet, exercise, pharmacotherapy), biofeedback therapy provides sustained improvement of bowel symptoms and anorectal function. Up to 70%-80% of DD patients show significant improvement of symptoms in randomized controlled trials (Table).48-52 Biofeedback therapy may also improve dyspeptic symptoms.53 Patients with harder stool consistency, greater willingness to participate, lower baseline bowel satisfaction, lower baseline anal sphincter relaxation, and prolonged balloon expulsion time, as well as patients who used digital maneuvers for defection, more commonly respond to biofeedback therapy.54,55 Longstanding laxative use has been associated with decreased response to biofeedback therapy.56 In patients with rectal hyposensitivity, barostat-assisted sensory training is more effective than a hand-held syringe technique.30 In patients with constipation predominant IBS and rectal hyposensitivity, sensory adaption training is more efficacious and better tolerated than escitalopram.30 Biofeedback therapy was afforded a grade A recommendation for treatment of DD by the American and European Societies of Neurogastroenterology and Motility.57
The access to office-based biofeedback therapy may be limited because of costs and low availability. The time required to attend multiple sessions may be burdensome for some patients, especially if they are taking time off from work. A recent study showed that patients with higher level of education may be less likely to adhere to biofeedback therapy.58 Recently, home biofeedback was shown to be noninferior to office biofeedback and was more cost-effective, which provides an alternative option for treating more patients.59
Endoscopic/surgical options
Other less effective treatment options for DD include botulinum toxin injection and myectomy.60-62 Botulinum toxin injection appears to have mixed effects with less than 50% of patients reporting symptomatic improvement, and it may cause fecal incontinence.60,63
Conclusion
DD is a common yet poorly recognized cause of constipation. Its clinical presentation overlaps with other lower-GI disorders. Its diagnosis requires detailed history, digital rectal examination, prospective stool diaries, anorectal manometry, and balloon expulsion tests. Biofeedback therapy offers excellent and sustained symptomatic improvement; however, access to office-based biofeedback is limited, and there is an urgent need for home-based biofeedback therapy programs.59
Dr. Rao is J. Harold Harrison Distinguished University Chair, professor of medicine, director of neurogastroenterology/motility, and director of digestive health at the Digestive Health Clinical Research Center Augusta (Georgia) University. He is supported by National Institutes of Health grants R01DK121003-02 and U01DK115572. Dr. Jehangir is a gastroenterology and Hepatology Fellow at the Digestive Health Clinical Research Center at Augusta University. They reported having no conflicts of interest.
References
1. Peery AF, et al. Gastroenterology. 2012;143(5):1179-1187.e3 .
2. Curtin B, et al. J Neurogastroenterol Motil. 2020 30;26(4):423-36.
3. Suares NC & Ford AC. Am J Gastroenterol. 2011 Sep;106(9):1582-91.
4. Mertz H, et al. Am J Gastroenterol. 1999;94(3):609-15.
5. Rao SS, et al. Am J Gastroenterol. 1998;93(7):1042-50.
6. Rao SSC, et al. J Clin Gastroenterol. 2004;38(8):680-5.
7. Nojkov B, et al. Am J Gastroenterol. 2019;114(11):1772-7.
8. Heaton KW, et al. Gut. 1992;33(6):818-24.
9. Prichard DO & Bharucha AE. 2018 Oct 15;7:F1000 Faculty Rev-1640.
10. Ashraf W, et al. Am J Gastroenterol. 1996;91(1):26-32.
11. Agachan F, et al.. Dis Colon Rectum. 1996;39(6):681-5.
12. Frank L, et al. Scand J Gastroenterol. 1999;34(9):870-7.
13. Marquis P, et al. Scand J Gastroenterol. 2005;40(5):540-51.
14. Yan Y, et al. Gastroenterology. 2020;158(6):S-400.
15. Rao SSC. Am J Gastroenterol. 2018;113(5):635-8.
16. Tantiphlachiva K, et al. Digital rectal examination is a useful tool for identifying patients with dyssynergia. Clin Gastroenterol Hepatol. 2010;8(11):955-60.
17. Carrington EV, et al. Nat Rev Gastroenterol Hepatol. 2018;15(5):309-23.
18. Tetangco EP, et al. Performing and analyzing high-resolution anorectal manometry. NeuroGastroLatam Rev. 2018;2:120-32.
19. Lee YY, et al. Curr Gastroenterol Rep. 2013;15(12):360.
20. Sharma M, et al. Neurogastroenterol Motil. 2020;32(10):e13910.
21. Rao SSC, et al.. Am J Gastroenterol. 2006;101(12):2790-6.
22. Carrington EV, et al. Neurogastroenterol Motil. 2020;32(1):e13679.
23. Rao SSC. Gastroenterol Clin North Am. 2008;37(3):569-86, viii.
24. Rao SSC, et al. Gastroenterology. 2016;150(4):S158-9.
25. Guinet A, et al. Int J Colorectal Dis. 2011;26(4):507-13.
26. Rattan S, et al. Gastroenterology. 1992;103(1):43-50.
27. Remes-Troche JM & Rao SSC. 2008;2(3):323-35.
28. Zaafouri H, et al..Int J Surgery. 2015. 2(1):9-17.
29. Remes-Troche JM, et al. Dis Colon Rectum. 2010;53(7):1047-54.
30. Rao SSC, et al. Gastroenterology. 2013;144(5):S-363.
31. Yu T, et al. Medicine (Baltimore). 2016;95(19):e3667.
32. Gladman MA, et al. Neurogastroenterol Motil. 2009;21(5):508-16, e4-5.
33. Lee KJ, et al. Digestion. 2006;73(2-3):133-41 .
34. Rao SSC, et al. Neurogastroenterol Motil. 2002;14(5):553-9.
35. Coss-Adame E, et al.. Clin Gastroenterol Hepatol. 2015;13(6):1143-1150.e1.
36. Chiarioni G, et al. Clin Gastroenterol Hepatol. 2014;12(12):2049-54.
37. Gu G, et al. Gastroenterology. 2018;154(6):S-545–S-546.
38. Savoye-Collet C, et al.. Gastroenterol Clin North Am. 2008;37(3):553-67, viii.
39. Videlock EJ, et al. Neurogastroenterol Motil. 2013;25(6):509-20.
40. Rao SSC, et al. Neurogastroenterol Motil. 2004;16(5):589-96.
41. Wald A, et al. Am J Gastroenterol. 2014;109(8):1141-57 ; (Quiz) 1058.
42. Rao SSC & Patcharatrakul T. J Neurogastroenterol Motil. 2016;22(3):423-35.
43. Rao SS, et al. Functional Anorectal Disorders. Gastroenterology. 2016. S0016-5085(16)00175-X.
44. Bharucha AE, et al.. Gastroenterology. 2013;144(1):218-38.
45. Voderholzer WA, et al. Am J Gastroenterol. 1997;92(1):95-8.
46. Lee HJ, et al. Neurogastroenterol Motil. 2015;27(6):787-95.
47. Simón MA & Bueno AM. J Clin Gastroenterol. 2017;51(10):e90-4.
48. Chiarioni G,et al.. Gastroenterology. 2006;130(3):657-64.
49. Heymen S, et al.. Dis Colon Rectum. 2007;50(4):428-41.
50. Rao SSC, et al. Clin Gastroenterol Hepatol. 2007;5(3):331-8.
51. Rao SSC, et al. Am J Gastroenterol. 2010;105(4):890-6.
52. Patcharatrakul T, et al. Biofeedback therapy. In Clinical and basic neurogastroenterology and motility. India: Stacy Masucci; 2020:517-32.
53. Huaman J-W, et al. Clin Gastroenterol Hepatol. 2020;18(11):2463-2470.e1.
54. Patcharatrakul T, et al. Clin Gastroenterol Hepatol. 2018;16(5):715-21.
55. Chaudhry A, et al. Gastroenterology. 2020;158(6):S-382–S-383.
56. Shim LSE, et al. Aliment Pharmacol Ther. 2011;33(11):1245-51.
57. Rao SSC, et al. Neurogastroenterol Motil. 2015;27(5):594-609.
58. Jangsirikul S, et al. Gastroenterology. 2020;158(6):S-383.
59. Rao SSC, et al. Am J Gastroenterol. 2019;114(6):938-44.
60. Ron Y, et al.. Dis Colon Rectum. 2001;44(12):1821-6.
61. Podzemny V, et al. World J Gastroenterol. 2015;21(4):1053-60.
62. Faried M, et al. J Gastrointest Surg. 2010;14(8):1235-43.
63. Hallan RI, et al. Lancet. 1988;2(8613):714-7.
Introduction
About 40% of the population experiences lower GI symptoms suggestive of gastrointestinal motility disorders.1,2 The global prevalence of chronic constipation is 18%, and the condition includes multiple overlapping subtypes.3 Evacuation disorders affect over half (59%) of patients and include dyssynergic defecation (DD).4 The inability to coordinate the abdominal, rectal, pelvic floor, and anal/puborectalis muscles to evacuate stools causes DD.5 The etiology of DD remains unclear and is often misdiagnosed. Clinically, the symptoms of DD overlap with other lower GI disorders, often leading to unnecessary and invasive procedures.2 We describe the clinical characteristics, diagnostic tools, treatment options, and evidence-based approach for the management of DD.
Clinical presentation
Over two-thirds of patients with DD acquire this disorder during adulthood, and one-third have symptoms from childhood.6 Though there is not usually an inciting event, 29% of patients report that symptoms began after events such as pregnancy or back injury,6 and opioid users have higher prevalence and severity of DD.7
Over 80% of patients report excessive straining, feelings of incomplete evacuation, and hard stools, and 50% report sensation of anal blockage or use of digital maneuvers.2 Other symptoms include infrequent bowel movements, abdominal pain, anal pain, and stool leakage.2 Evaluation of DD includes obtaining a detailed history utilizing the Bristol Stool Form Scale;8 however, patients’ recall of stool habit is often inaccurate, which results in suboptimal care.9,10 Prospective stool diaries can help to provide more objective assessment of patients’ symptoms, eliminate recall bias, and provide more reliable information. Several useful questionnaires are available for clinical and research purposes to characterize lower-GI symptoms, including the Constipation Scoring System,11 Patient Assessment of Constipation Symptoms (PAC-SYM),12 and Patient Assessment of Constipation Quality of Life (PAC-QOL).2,13 The Constipation Stool digital app enhances accuracy of data capture and offers a reliable and user-friendly method for recording bowel symptoms for patients, clinicians, and clinical investigators.14
Diagnosis
The diagnosis of DD requires careful physical and digital rectal examination together with anorectal manometry and a balloon expulsion test. Defecography and colonic transit studies provide additional assessment.
Physical examination
Abdominal examination should include palpation for stool in the colon and identification of abdominal mass or fecal impaction.2A high-quality digital rectal examination can help to identify patients who could benefit from physiological testing to confirm and treat DD.15 Rectal examination is performed by placing examiner’s lubricated gloved right index finger in a patient’s rectum, with the examiner’s left hand on patient’s abdomen, and asking the patient to push and bear down as if defecating.15 The contraction of the abdominal muscles is felt using the left hand, while the anal sphincter relaxation and degree of perineal descent are felt using the right-hand index finger.15 A diagnosis of dyssynergia is suspected if the digital rectal examination reveals two or more of the following abnormalities: inability to contract abdominal muscles (lack of push effort), inability to relax or paradoxical contraction of the anal sphincter and/or puborectalis, or absence of perineal descent.15 Digital rectal examination has good sensitivity (75%), specificity (87%), and positive predictive value (97%) for DD.16
High resolution anorectal manometry
Anorectal manometry (ARM) is the preferred method for the evaluation of defecatory disorders.17,18 ARM is best performed using the high-resolution anorectal manometry (HRAM) systems19 that consist of a flexible probe – 0.5-cm diameter with multiple circumferential sensors along the anal canal – and another two sensors inside a rectal balloon.18 It provides a topographic and waveform display of manometric pressure data (Figure). The 3D high-definition ARM probe is a rigid 1-cm probe that provides 3D topographic profiles.18 ARM is typically performed in both the left lateral position and in a more physiological seated position.20,21 There is considerable variation amongst different institutions on how to perform HRAM, and a recent International Anorectal Physiology Working Group (IAPWG) has provided consensus recommendations for performing this test.22 The procedure for performing HRAM is reviewed elsewhere, but the key elements are summarized below.18
Push maneuver: On HRAM, after the assessment of resting and squeeze anal sphincter pressures, the patient is asked to push or bear down as if to defecate while lying in left lateral decubitus position. The best of two attempts that closely mimics a normal bearing down maneuver is used for categorizing patient’s defecatory pattern.18 In patients with DD, at least four distinct dyssynergia phenotypes have been recognized (Figure),23 though recent studies suggest eight patterns.24 Defecation index (maximum rectal pressure/minimum residual anal pressure when bearing down) greater than 1.2 is considered normal.18
Simulated defecation on commode: The subject is asked to attempt defecation while seated on a commode with intrarectal balloon filled with 60 cc of air, and both the defecation pattern(s) and defecation index are calculated. A lack of coordinated push effort is highly suggestive of DD.21
Rectoanal Inhibitory Reflex (RAIR): RAIR describes the reflex relaxation of the internal anal sphincter after rectal distension. RAIR is dependent on intact autonomic ganglia and myenteric plexus25and is mediated by the release of nitric oxide and vasoactive intestinal peptide.26 The absence of RAIR suggests Hirschsprung disease.22.27.28
Rectal sensory testing: Intermittent balloon distension of the rectum with incremental volumes of air induces a range of rectal sensations that include first sensation, desire to defecate, urgency to defecate, and maximum tolerable volume. Rectal hyposensitivity is diagnosed when two or more sensory thresholds are higher than those seen in normal subjects29.30 and likely results from disruption of afferent gut-brain pathways, cortical perception/rectal wall dysfunction, or both.29 Rectal hyposensitivity affects 40% of patients with constipation30and is associated with DD but not delayed colonic transit.31 Rectal hyposensitivity may also be seen in patients with diabetes or fecal incontinence.18 About two-thirds of patients with rectal hyposensitivity have rectal hypercompliance, and some have megarectum.32 Some patients with DD have coexisting irritable bowel syndrome (IBS) and may have rectal hypersensitivity.18,33 Rectal compliance is measured alongside rectal sensitivity analysis by plotting a graph between the change in intraballoon volume (mL) and change in intrarectal pressures (mm Hg) during incremental balloon distensions.18.34 Rectal hypercompliance may be seen in megarectum and dyssynergic defecation.34,35 Rectal hypocompliance may be seen in patients with inflammatory bowel disease, postpelvic radiation, chronic ischemia, and advanced age.18
Balloon expulsion test: This test is performed by placing a plastic probe with a balloon in the rectum and filling it with 50 cc of warm water. Patients are given 5 minutes to expel the balloon while sitting on a commode. Balloon expulsion time of more than 1 minute suggests a diagnosis of DD,21 although 2 minutes provides a higher level of agreement with manometric findings.36 Balloon type and body position can influence the results.37 Inability to expel the balloon with normal manometric findings is considered an inconclusive finding per the recent London Classification (i.e., it may be associated with generation of anorectal symptoms, but the clinical relevance of this finding is unclear as it may also be seen in healthy subjects).22
Defecography
Defecography is a dynamic fluoroscopic study performed in the sitting position after injecting 150 mL of barium paste into the patient’s rectum. Defecography provides useful information about structural changes (e.g., rectoceles, enteroceles, rectal prolapse, and intussusception), DD, and descending perineum syndrome.38 Methodological differences, radiation exposure, and poor interobserver agreement have limited its wider use; therefore, anorectal manometry and the balloon expulsion test are recommended for the initial evaluation of DD.39 Magnetic resonance defecography may be more useful.17,38
Colonic transit studies
Colonic transit study can be assessed using radiopaque markers, wireless motility capsule, or scintigraphy. Wireless motility capsule and scintigraphy have the advantage of determining gastric, small bowel, and whole gut transit times as well. About two-thirds of patients with DD have slow transit constipation (STC),6 which improves after treatment of DD.40 Hence, in patients with chronic constipation, evaluation and management of DD is recommended first. If symptoms persist, then consider colonic transit assessment.41 Given the overlapping nature of the conditions, documentation of STC at the outset could facilitate treatment of both.
Diagnostic criteria for DD
Patients should fulfill the following criteria for diagnosis of DD:42,43
- Fulfill symptom(s) diagnostic criteria for functional constipation and/or constipation-predominant IBS.
- Demonstrate dyssynergic pattern (Types I-IV; Figure) during attempted defecation on manometry recordings.
- Meet one or more of the following criteria:
- Inability to expel an artificial stool (50 mL water-filled balloon) within 1 minute.
- Inability to evacuate or retention of 50% or more of barium during defecography. (Some institutions use a prolonged colonic transit time: greater than 5 markers or 20% or higher marker retention on a plain abdominal x-Ray at 120 hours after ingestion of one radio-opaque marker capsule containing 24 radio-opaque markers.)
Treatment of DD
The treatment modalities for DD depend on several factors: patient’s age, comorbidities, underlying pathophysiology, and patient expectations. Treatment options include standard management of constipation, but biofeedback therapy is the mainstay.
Standard management
Medications that cause or worsen constipation should be avoided. The patient should consume adequate fluid and exercise regularly. Patients should receive instructions for timed toilet training (twice daily, 30 minutes after meals). Patients should push at about 50%-70% of their ability for no longer than 5 minutes and avoid postponing defecation or use of digital maneuvers to facilitate defecation.42 The patients should take 25 g of soluble fiber (e.g., psyllium) daily. Of note, the benefits of fiber can take days to weeks44 and may be limited in patients with STC and DD.45 Medications including laxatives and intestinal secretagogues (lubiprostone, linaclotide, plecanatide), and enterokinetic agents (prucalopride) can be used as adjunct therapy for management of DD.42 Their use is titrated during and after biofeedback therapy and may decrease after successful treatment.46
Biofeedback therapy
Biofeedback therapy involves operant conditioning techniques using either a solid state anorectal manometry system, electromyography, simulated balloon, or home biofeedback training devices.42,47 The goals of biofeedback therapy are to correct the abdominal pelvic muscle discoordination during defecation and improve rectal sensation to stool if impaired. Biofeedback therapy involves patient education and active training (typically six sessions, 1-2 weeks apart, with each about 30-60 minutes long), followed by a reinforcement stage (three sessions at 3, 6, and 12 months), though there are variations in training protocols.42
The success of biofeedback therapy depends on the patient’s motivation and the therapist’s skills.42 Compared with standard therapy (diet, exercise, pharmacotherapy), biofeedback therapy provides sustained improvement of bowel symptoms and anorectal function. Up to 70%-80% of DD patients show significant improvement of symptoms in randomized controlled trials (Table).48-52 Biofeedback therapy may also improve dyspeptic symptoms.53 Patients with harder stool consistency, greater willingness to participate, lower baseline bowel satisfaction, lower baseline anal sphincter relaxation, and prolonged balloon expulsion time, as well as patients who used digital maneuvers for defection, more commonly respond to biofeedback therapy.54,55 Longstanding laxative use has been associated with decreased response to biofeedback therapy.56 In patients with rectal hyposensitivity, barostat-assisted sensory training is more effective than a hand-held syringe technique.30 In patients with constipation predominant IBS and rectal hyposensitivity, sensory adaption training is more efficacious and better tolerated than escitalopram.30 Biofeedback therapy was afforded a grade A recommendation for treatment of DD by the American and European Societies of Neurogastroenterology and Motility.57
The access to office-based biofeedback therapy may be limited because of costs and low availability. The time required to attend multiple sessions may be burdensome for some patients, especially if they are taking time off from work. A recent study showed that patients with higher level of education may be less likely to adhere to biofeedback therapy.58 Recently, home biofeedback was shown to be noninferior to office biofeedback and was more cost-effective, which provides an alternative option for treating more patients.59
Endoscopic/surgical options
Other less effective treatment options for DD include botulinum toxin injection and myectomy.60-62 Botulinum toxin injection appears to have mixed effects with less than 50% of patients reporting symptomatic improvement, and it may cause fecal incontinence.60,63
Conclusion
DD is a common yet poorly recognized cause of constipation. Its clinical presentation overlaps with other lower-GI disorders. Its diagnosis requires detailed history, digital rectal examination, prospective stool diaries, anorectal manometry, and balloon expulsion tests. Biofeedback therapy offers excellent and sustained symptomatic improvement; however, access to office-based biofeedback is limited, and there is an urgent need for home-based biofeedback therapy programs.59
Dr. Rao is J. Harold Harrison Distinguished University Chair, professor of medicine, director of neurogastroenterology/motility, and director of digestive health at the Digestive Health Clinical Research Center Augusta (Georgia) University. He is supported by National Institutes of Health grants R01DK121003-02 and U01DK115572. Dr. Jehangir is a gastroenterology and Hepatology Fellow at the Digestive Health Clinical Research Center at Augusta University. They reported having no conflicts of interest.
References
1. Peery AF, et al. Gastroenterology. 2012;143(5):1179-1187.e3 .
2. Curtin B, et al. J Neurogastroenterol Motil. 2020 30;26(4):423-36.
3. Suares NC & Ford AC. Am J Gastroenterol. 2011 Sep;106(9):1582-91.
4. Mertz H, et al. Am J Gastroenterol. 1999;94(3):609-15.
5. Rao SS, et al. Am J Gastroenterol. 1998;93(7):1042-50.
6. Rao SSC, et al. J Clin Gastroenterol. 2004;38(8):680-5.
7. Nojkov B, et al. Am J Gastroenterol. 2019;114(11):1772-7.
8. Heaton KW, et al. Gut. 1992;33(6):818-24.
9. Prichard DO & Bharucha AE. 2018 Oct 15;7:F1000 Faculty Rev-1640.
10. Ashraf W, et al. Am J Gastroenterol. 1996;91(1):26-32.
11. Agachan F, et al.. Dis Colon Rectum. 1996;39(6):681-5.
12. Frank L, et al. Scand J Gastroenterol. 1999;34(9):870-7.
13. Marquis P, et al. Scand J Gastroenterol. 2005;40(5):540-51.
14. Yan Y, et al. Gastroenterology. 2020;158(6):S-400.
15. Rao SSC. Am J Gastroenterol. 2018;113(5):635-8.
16. Tantiphlachiva K, et al. Digital rectal examination is a useful tool for identifying patients with dyssynergia. Clin Gastroenterol Hepatol. 2010;8(11):955-60.
17. Carrington EV, et al. Nat Rev Gastroenterol Hepatol. 2018;15(5):309-23.
18. Tetangco EP, et al. Performing and analyzing high-resolution anorectal manometry. NeuroGastroLatam Rev. 2018;2:120-32.
19. Lee YY, et al. Curr Gastroenterol Rep. 2013;15(12):360.
20. Sharma M, et al. Neurogastroenterol Motil. 2020;32(10):e13910.
21. Rao SSC, et al.. Am J Gastroenterol. 2006;101(12):2790-6.
22. Carrington EV, et al. Neurogastroenterol Motil. 2020;32(1):e13679.
23. Rao SSC. Gastroenterol Clin North Am. 2008;37(3):569-86, viii.
24. Rao SSC, et al. Gastroenterology. 2016;150(4):S158-9.
25. Guinet A, et al. Int J Colorectal Dis. 2011;26(4):507-13.
26. Rattan S, et al. Gastroenterology. 1992;103(1):43-50.
27. Remes-Troche JM & Rao SSC. 2008;2(3):323-35.
28. Zaafouri H, et al..Int J Surgery. 2015. 2(1):9-17.
29. Remes-Troche JM, et al. Dis Colon Rectum. 2010;53(7):1047-54.
30. Rao SSC, et al. Gastroenterology. 2013;144(5):S-363.
31. Yu T, et al. Medicine (Baltimore). 2016;95(19):e3667.
32. Gladman MA, et al. Neurogastroenterol Motil. 2009;21(5):508-16, e4-5.
33. Lee KJ, et al. Digestion. 2006;73(2-3):133-41 .
34. Rao SSC, et al. Neurogastroenterol Motil. 2002;14(5):553-9.
35. Coss-Adame E, et al.. Clin Gastroenterol Hepatol. 2015;13(6):1143-1150.e1.
36. Chiarioni G, et al. Clin Gastroenterol Hepatol. 2014;12(12):2049-54.
37. Gu G, et al. Gastroenterology. 2018;154(6):S-545–S-546.
38. Savoye-Collet C, et al.. Gastroenterol Clin North Am. 2008;37(3):553-67, viii.
39. Videlock EJ, et al. Neurogastroenterol Motil. 2013;25(6):509-20.
40. Rao SSC, et al. Neurogastroenterol Motil. 2004;16(5):589-96.
41. Wald A, et al. Am J Gastroenterol. 2014;109(8):1141-57 ; (Quiz) 1058.
42. Rao SSC & Patcharatrakul T. J Neurogastroenterol Motil. 2016;22(3):423-35.
43. Rao SS, et al. Functional Anorectal Disorders. Gastroenterology. 2016. S0016-5085(16)00175-X.
44. Bharucha AE, et al.. Gastroenterology. 2013;144(1):218-38.
45. Voderholzer WA, et al. Am J Gastroenterol. 1997;92(1):95-8.
46. Lee HJ, et al. Neurogastroenterol Motil. 2015;27(6):787-95.
47. Simón MA & Bueno AM. J Clin Gastroenterol. 2017;51(10):e90-4.
48. Chiarioni G,et al.. Gastroenterology. 2006;130(3):657-64.
49. Heymen S, et al.. Dis Colon Rectum. 2007;50(4):428-41.
50. Rao SSC, et al. Clin Gastroenterol Hepatol. 2007;5(3):331-8.
51. Rao SSC, et al. Am J Gastroenterol. 2010;105(4):890-6.
52. Patcharatrakul T, et al. Biofeedback therapy. In Clinical and basic neurogastroenterology and motility. India: Stacy Masucci; 2020:517-32.
53. Huaman J-W, et al. Clin Gastroenterol Hepatol. 2020;18(11):2463-2470.e1.
54. Patcharatrakul T, et al. Clin Gastroenterol Hepatol. 2018;16(5):715-21.
55. Chaudhry A, et al. Gastroenterology. 2020;158(6):S-382–S-383.
56. Shim LSE, et al. Aliment Pharmacol Ther. 2011;33(11):1245-51.
57. Rao SSC, et al. Neurogastroenterol Motil. 2015;27(5):594-609.
58. Jangsirikul S, et al. Gastroenterology. 2020;158(6):S-383.
59. Rao SSC, et al. Am J Gastroenterol. 2019;114(6):938-44.
60. Ron Y, et al.. Dis Colon Rectum. 2001;44(12):1821-6.
61. Podzemny V, et al. World J Gastroenterol. 2015;21(4):1053-60.
62. Faried M, et al. J Gastrointest Surg. 2010;14(8):1235-43.
63. Hallan RI, et al. Lancet. 1988;2(8613):714-7.
Update on feeding tubes: Indications and troubleshooting complications
Introduction
Gastroenterologists are in a unique position to manage individuals with feeding tubes as their training underscores principles in digestion, absorption, nutrition support, and enteral tube placement. Adequate management of individuals with feeding tubes and, importantly, the complications that arise from feeding tube use and placement require a basic understanding of intestinal anatomy and physiology. Therefore, gastroenterologists are well suited to both place and manage individuals with feeding tubes in the long term.

Indications for tube feeding
When deciding on the appropriate route for artificial nutrition support, the first decision to be made is enteral access versus parenteral nutrition support. Enteral nutrition confers multiple benefits, including preservation of the mucosal lining, reductions in complicated infections, decreased costs, and improved patient compliance. All attempts at adequate enteral access should be made before deciding on the use of parenteral nutrition. Following the clinical decision to pursue artificial means of nutrition support and enteral access, the next common decision is the anticipated duration of nutrition support. Generally, the oral or nasal tubes are used for short durations (i.e., less than 4 weeks) with percutaneous placement into the stomach or small intestine for longer-term feeding (i.e., percutaneous endoscopic gastrostomy [PEG] or percutaneous endoscopic jejunostomy [PEJ]).

The most general indication for nutrition support is an inability to maintain adequate nutritional needs with oral intake alone. General categories of inadequate oral intake include neurologic disorders, malignancy, and gastrointestinal conditions affecting digestion and absorption (Table 1). Absolute and relative contraindications to PEG placement are listed in Table 2. If an endoscopic placement is not possible, alternative means of placement (i.e., surgery or interventional radiology) can be considered to avoid the consequences of prolonged malnutrition. In-hospital mortality following PEG placement has decreased 40% over the last 10 years, which can be attributed to improved patient selection, enhanced discharge practices, and exclusion of patients with the highest comorbidity and mortality rates, like those with advanced dementia or terminal cancer.1
PEG placement in patients with dementia is controversial, with previous studies not demonstrating improved outcomes and association with high mortality rates,2 so the practice is currently not recommended by the American Geriatrics Society in individuals with advanced dementia.3 However, a large Japanese study showed that careful selection of patients with mild dementia to undergo gastrostomy increased independence fourfold; therefore, multidisciplinary involvement is often necessary in the decision to pursue artificial means of nutrition support in this population.4
The recent coronavirus disease 2019 (COVID-19) pandemic has placed additional strains on endoscopic placement and has highlighted the effect of the severe acute respiratory syndrome coronavirus 2 (SARS-COV-2) on GI symptoms. A recent meta-analysis showed an overall incidence of GI symptoms of 17.6% in the following conditions in decreasing order of prevalence: anorexia, diarrhea, nausea, vomiting, and abdominal discomfort.5 In addition, the prolonged ventilatory requirements among a subset of individuals with the most severe COVID-19 results in extended periods of nutrition support via enteral tube placements. In individuals with ICU-acquired weakness and discharge to long-term care facilities, the placement of percutaneous endoscopic tubes may be required, although with the additional consideration of the need for an aerosolizing procedure. Delay of placement has been advocated, in addition to appropriate personal protective equipment, in order to ensure safe placement for the endoscopy staff.6
Types of feeding tubes
After deciding to feed a patient enterally and determining the anticipated duration of enteral support, the next decision is to determine the most appropriate location of feeding delivery: into the stomach or the small bowel. Gastric feeding is advantageous most commonly because of its increased capacity, allowing for larger volumes to be delivered over shorter durations. However, in the setting of postsurgical anatomy, gastroparesis, or obstructing tumors/pancreatic inflammation, distal delivery of tube feeds may be required into the jejunum. Additionally, percutaneous tubes placed into the stomach can have extenders into the small bowel (GJ tubes) to allow for feeding into the small bowel and decompression or delivery of medications into the stomach.

In general, gastric feeding is preferred over small bowel feeding as PEG tubes are more stable and have fewer complications than either PEG-J or direct PEJ tubes. Gastrostomy tubes are generally shorter and larger in diameter making them less likely to clog. PEG-J tubes have separate lumens for gastric and small intestinal access, but the smaller-bore jejunal extension tubes are more likely to clog or become dislodged. While direct PEJ is shown to have higher rates of tube patency and decreased rates of endoscopic re-intervention, compared with PEG-J,7 one limitation of a direct PEJ is difficulty in placement and site selection, which can be performed with a pediatric colonoscope or balloon enteroscopy system. Most commonly, this procedure is performed under general anesthesia.
In the case of a critically ill patient in the ICU, it is recommended to start enteral nutrition within 24-48 hours of arrival to avoid complications of prolonged calorie deficits. Nasally inserted feeding tubes (e.g., Cortrak, Avanos Medical Devices, Alpharetta, Ga.) are most commonly used at the bedside and can be placed blindly using electromagnetic image guidance, radiographically, or endoscopy. However, the small caliber of nasoenteric tubes comes with the common complication of clogging, which can be overcome with slightly larger bore gastric feeding tubes. If gastric feeding is not tolerated (e.g., in the case of vomiting, witnessed aspiration), small bowel feeding should be initiated and can be a more durable form of enteral feeding with fewer interruptions as feedings do not need to be held for procedures or symptomatic gastric intolerance. In clinical areas of question, or if there is a concern for intolerance of enteral feeding, a short trial with nasogastric or nasojejunal tube placement should be performed before a more definitive percutaneous placement.
With respect to percutaneous tubes, important characteristics to choose are the size (diameter in French units), type of internal retention device, and external appearance of the tube (standard or low profile). All percutaneous tubes contain an external retention device (i.e., bumper) that fits against the skin and an internal retention device that is either a balloon or plastic dome or funnel that prevents the tube from becoming dislodged. Balloon retention tubes require replacement every 3-6 months, while nonballoon tubes generally require replacement annually in order to prevent the plastic from cracking, which can make removal complicated. Low-profile tubes have an external cap, which, when opened, allows for extension tubing to be securely attached while in use and detached while not in use. Low-profile tubes are often preferred among younger, active patients and those with adequate dexterity to allow for attachment of the external extension tubing. These tubes are most often inserted as a replacement for an initially endoscopically placed tube, although one-step systems for initial placement are available. The size of the low-profile tube is chosen based on the size of the existing PEG tube and by measuring the length of the stoma tract using specialized measuring devices.8 Patients and caregivers can also be trained to replace balloon-type tubes on their own to limit complications of displaced or cracked tubes. Low-profile tubes are commercially available for both gastric placement and gastric placement with extension into the small bowel, which often requires fluoroscopy for secure placement.
All percutaneous enteral tubes are being transitioned to the ENfit connector system, which prevents connections from the enteral system to nonenteral systems (namely intravenous lines, chest tubes) and vice versa. Tubing misconnections have been rarely reported, and the EnFIT system is designed to prevent such misadventures that have resulted in serious complications and even mortality.9 Adapter devices are available that may be required for patients with feeding tubes who have not been transitioned yet. Most commonly with new tube placements and replacements, patients and providers will have to become familiar with the new syringes and feeding bags required with EnFIT connectors.
Gastrostomy placement can be considered a higher-risk endoscopic procedure. One complicating factor is the increased use of antiplatelet and anticoagulant therapies in individuals with a history of neurologic insults. The American Society for Gastrointestinal Endoscopy (ASGE) guidelines recommend that coumadin be held 5 days before the procedure and bridged with heparin if the patient is at high risk of thromboembolic complications. For patients on dual anti-platelet therapy, thienopyridines like clopidogrel are often stopped 5-7 days prior to procedure with continuation of aspirin,10 but there are more recent data that PEG insertion is safe with continued use of DAPT.11 Direct-acting anticoagulants (DOACs) are often stopped 24-48 hours prior to procedure and then restarted 48 hours after tube placement, but this is dependent on the half-life of the specific DOAC and the patient’s renal function. Patients with decreased creatinine clearance may need to hold the DOAC up to 3-4 days prior to the procedure. In this situation, referring to ASGE guidelines and consultation with a hematologist or managing anti-coagulation clinic is advised.10
Troubleshooting complications
Nasoenteric tubes: One of the most common and irritating complications with nasoenteric feeding tubes is clogging. To prevent clogging, the tube should be flushed frequently.12 At least 30 mL of free water should be used to flush the tube every 4-8 hours for continuous feedings or before and after bolus feeding. Additionally, 15-30 mL of water should be given with each separate medication administration, and if possible, medication administration via small-bore small bowel feeding tubes should be avoided.12 Water flushing is especially important with small-caliber tubes and pumps that deliver both feeding and water flushes. It is available for small bowel feeding in order to allow for programmed water delivery.
Warm water flushes can also help unclog the tube,12 and additional pharmacologic and mechanical devices have been promoted for clogged tubes. One common technique is mixing pancreatic enzymes (Viokase) with a crushed 325-mg tablet of nonenteric coated sodium bicarbonate and 5 mL of water to create a solution that has the alkaline properties allowing for both pancreatic enzyme activation and clog dissolution. Additionally, an endoscopic retrograde cholangiopancreatography (ERCP) catheter can be placed into longer feeding tubes to directly infuse the activated agent to the site of the clog.13 If water and enzymes are not successful in unclogging the tube, commercially available brushes can help remove clogs. The TubeClear® system (Actuated Medical, Bellefonte, Penna) has a single-use stem that is connected to AC power to create a jackhammerlike movement to remove clogs in longer nasoenteral and gastrojejunal tubes.
PEG tubes (short-term complications): Procedural and immediate postprocedural complications include bleeding, aspiration, pneumoperitoneum, and perforation. Pneumoperitoneum occurs in approximately 50% of cases and is generally clinically insignificant. The risk of pneumoperitoneum can be reduced by using CO2 insufflation.14 If the patient develops systemic signs of infection or peritoneal signs, CT scan with oral contrast is warranted for further evaluation and to assess for inadvertent perforation of overlying bowel or dislodged tube. Aspiration during or following endoscopy is another common complication of PEG placement and risk factors include over-sedation, supine positioning, advanced age, and neurologic dysfunction. This risk can be mitigated by avoiding over-sedation, immediately aspirating gastric contents when the stomach is reached, and avoiding excessive insufflation.15 In addition, elevating the head of the bed during the procedure and dedicating an assistant to perform oral suctioning during the entire procedure is recommended.
PEG tubes (long-term complications): More delayed complications of PEG insertion include wound infection, buried bumper syndrome, tumor seeding, peristomal leakage, and tube dislodgement. The prevalence of wound infection is 5%- 25%,16 and randomized controlled trials have demonstrated the efficacy of a single dose of an IV antibiotic (i.e., cephalosporin) in those not already receiving a broad spectrum antibiotic and administered prophylactically before tube placement.17 The significance of this reduction is such that antibiotic administration before tube placement should be considered a quality measure for the procedure. A small amount of redness around the tube site (less than 5 mm) is typical, but extension of erythema, warmth, tenderness, purulent drainage, or systemic symptoms is consistent with infection and warrants additional antibiotic administration. Minor infections can be treated with local antiseptics and oral antibiotics, and early intervention is important to prevent need for hospital admission, systemic antibiotics, and even surgical debridement.
Peristomal leakage is reported in approximately 1%-2% of patients.18 Photographs of the site can be very useful in evaluating and managing peristomal leakage and infections. Interventions include reducing gastric secretions with proton pump inhibitors and management of the skin with barrier creams, such as zinc oxide (Calmoseptine®) ointment. Placement of a larger-diameter tube only enlarges the stoma track and worsens the leakage. In such cases, thorough evaluations for delayed gastric emptying (gastroparesis), distal obstruction, or constipation should be performed and managed accordingly. Opiates are common contributors to constipation and delayed gastric emptying and often require reduction in use or directed antagonist therapy to reduce leaking. Continuous feeding over bolus feedings and delivering nutrition distally into the small bowel (PEG-J placement) can improve leaking from gastrostomy tubes. Additional means of management include stabilizing the tube by replacing a traditional tube with a low-profile tube or using right-angle external bumpers. If all measures fail, removing the tube and allowing for stomal closure can be attempted,16 although this option often requires parenteral nutrition support to prevent prolonged periods of inadequate nutrition.
Buried bumper syndrome (BBS) occurs in 1.5%-8.8% of PEG placements and is a common late complication of PEG placement, although early reports have been described.18 The development of BBS occurs when the internal bumper migrates from the gastric lumen through and into the stomach or abdominal wall. It occurs more frequently with solid nonballoon retention tubes and is caused by excessive compression of the external bumper against the skin and abdominal wall. Patients with BBS usually present with an immobile catheter, resistance with feeds (because of a closure of the stomach wall around the internal portion of the gastrostomy tube), abdominal pain, or peristomal leakage. Physicians should be aware of and assess tubes for BBS, in particular when replacing an immobile tube (cannot be pushed into the free stomach lumen) or when there is difficulty in flushing water into the tube. This complication can be easily prevented by allowing a minimum of 0.5-1.0 cm (1 finger breadth) between the external bumper and the abdominal wall. In particular, patients and caregivers should be warned that if the patient gains significant amounts of weight, the outer bumper will need to be loosened. Once BBS is diagnosed, the PEG tube requires removal and replacement as it can cause bleeding, infection, or fasciitis. The general steps to replacement include endoscopic removal of the existing tube and replacement of new PEG in the existing tract as long as the BBS is not severe. In most cases a replacement tube can be pulled into place using the pull-PEG technique at the same gastrostomy site as long as the stoma tract can be cannulated with a wire after the existing tube is removed.
Similar to nasoenteric tubes, PEG tubes can become clogged, although this complication is infrequent. The primary steps for prevention include adequately flushing with water before and after feeds and ensuring that all medications are liquid or well crushed and dissolved before instilling. Timely tube replacement also ensures that the internal portions of the gastrostomy tube remain free of debris. Management is similar to that of unclogging nasoenteral tubes, as discussed above, and specific commercial declogging devices for PEG tubes include the Bionix Declogger® (Bionix Development Corp., Toledo, Ohio) and the Bard® PEG cleaning brush (Bard Peripheral Vascular Inc., Tempe, Ariz.). The Bionix system has a plastic stem with a screw and thread design that will remove clogs in 14-24 French PEG tubes, while the Bard brush has a flexible nylon stem with soft bristles at the end to prevent mucosal injury and can be used for prophylaxis against clogs, as well as removing clogs themselves.12
Lastly, a rare but important complication of PEG placement is tumor seeding of the PEG site in patients with active head and neck or upper gastrointestinal cancer.19 The presumed mechanism is shearing of tumor cells as the PEG is pulled through the upper aerodigestive tract and through the wall of the stomach, as prior studies have demonstrated frequent seeding of tubes and incision sites as shown by brushing the tube for malignant cells after tube placement.20 It is important to recognize this complication and not misdiagnose it as granulation tissue, infection, or bleeding as the spread of the cancer generally portends a poor prognosis. Therefore, it is best to use a PEG insertion technique that does not involve pulling or pushing the PEG through the upper aerodigestive tract in patients with active cancer and instead place tubes via an external approach by colleagues in interventional radiology or via direct surgical placement.
Conclusion
Gastroenterologists occupy a unique role in evaluation, diagnosis, and management of patients requiring enteral feeding. In addition, they are best equipped to place, prevent, and manage complications of tube feeding. For this reason, it is imperative that gastroenterologists familiarize themselves with indications for enteral tubes and types of enteral tubes available, as well as the identification and management of common complications. Comprehensive understanding of these concepts will augment the practicing gastroenterologist’s ability to manage patients requiring enteral nutrition support with confidence.
References
1. Stein DJ et al. Dig Dis Sci. 2020 Jun 19. doi: 10.1007/s10620-020-06396-y.
2. American Geriatrics Society Ethics Committee and Clinical Practice and Models of Care Committee. J Am Geriatr Soc. 2014;62(8):1590-3.
3. Dietrich CG, Schoppmeyer K. World J Gastroenterol. 2020;26(20):2464-71.
4. Suzuki Y et al. T Gastroenterology Res.2012 Feb;5(1):10-20.
5. Cheung KS et al. Gastroenterology. 2020 Jul;159(1):81-95.
6. Micic D et al. Am J Gastroenterol. 2020 Sep;115(9):1367-70.
7. Fan AC et al. Gastrointest Endosc. 2002;56(6):890-4.
8. Tang SJ. Video J Encycl GI Endosc. 2014;2(2):70-3.
9. Guenter P, Lyman B. Nutr Clin Pract. 2016;31(6):769-72.
10. Acosta RD et al. Gastrointest Endosc. 2016;83(1):3-16.
11. Richter JA et al. Gastrointest Endosc. 2011;74(1):22-34.
12. Boullata JI et al. JPEN. 2017;41(1):15-103.
13. McClave SA. Tech Gastrointest Endosc. 2021;3(1):62-8.
14. Murphy CJ et al. Endosc Int Open. 2016;4(3):E292. doi: 10.1053/tgie.2001.19915.
15. Lynch CR et al. Pract Gastroenterology. 2004;28:66-77.
16. Hucl T et al. Best Pract Res Clin Gastroenterol. 2016;30(5):769-81. doi: 10.1016/j.bpg.2016.10.002.
17. Jafri NS et al. Aliment Pharmacol & Therapeut. 2007;25(6):647-56. doi: 10.1111/j.1365-2036.2007.03247.x.
18. Blumenstein I et al. World J Gastroenterol. 2014;20(26):8505-24. doi: 10.3748/wjg.v20.i26.8505.
19. Fung E et al. Surgical Endosc. 2017;31(9):3623-7. doi: 10.1007/s00464-016-5394-8.
20. Ellrichmann M et al. Endoscopy. 2013;45(07):526-31. doi: 10.1055/s-0033-1344023.
Dr. Toy is with the department of internal medicine at the University of Utah, Salt Lake City. Dr. Fang is with the division of gastroenterology and hepatology at the University of Utah.
Introduction
Gastroenterologists are in a unique position to manage individuals with feeding tubes as their training underscores principles in digestion, absorption, nutrition support, and enteral tube placement. Adequate management of individuals with feeding tubes and, importantly, the complications that arise from feeding tube use and placement require a basic understanding of intestinal anatomy and physiology. Therefore, gastroenterologists are well suited to both place and manage individuals with feeding tubes in the long term.
Indications for tube feeding
When deciding on the appropriate route for artificial nutrition support, the first decision to be made is enteral access versus parenteral nutrition support. Enteral nutrition confers multiple benefits, including preservation of the mucosal lining, reductions in complicated infections, decreased costs, and improved patient compliance. All attempts at adequate enteral access should be made before deciding on the use of parenteral nutrition. Following the clinical decision to pursue artificial means of nutrition support and enteral access, the next common decision is the anticipated duration of nutrition support. Generally, the oral or nasal tubes are used for short durations (i.e., less than 4 weeks) with percutaneous placement into the stomach or small intestine for longer-term feeding (i.e., percutaneous endoscopic gastrostomy [PEG] or percutaneous endoscopic jejunostomy [PEJ]).
The most general indication for nutrition support is an inability to maintain adequate nutritional needs with oral intake alone. General categories of inadequate oral intake include neurologic disorders, malignancy, and gastrointestinal conditions affecting digestion and absorption (Table 1). Absolute and relative contraindications to PEG placement are listed in Table 2. If an endoscopic placement is not possible, alternative means of placement (i.e., surgery or interventional radiology) can be considered to avoid the consequences of prolonged malnutrition. In-hospital mortality following PEG placement has decreased 40% over the last 10 years, which can be attributed to improved patient selection, enhanced discharge practices, and exclusion of patients with the highest comorbidity and mortality rates, like those with advanced dementia or terminal cancer.1
PEG placement in patients with dementia is controversial, with previous studies not demonstrating improved outcomes and association with high mortality rates,2 so the practice is currently not recommended by the American Geriatrics Society in individuals with advanced dementia.3 However, a large Japanese study showed that careful selection of patients with mild dementia to undergo gastrostomy increased independence fourfold; therefore, multidisciplinary involvement is often necessary in the decision to pursue artificial means of nutrition support in this population.4
The recent coronavirus disease 2019 (COVID-19) pandemic has placed additional strains on endoscopic placement and has highlighted the effect of the severe acute respiratory syndrome coronavirus 2 (SARS-COV-2) on GI symptoms. A recent meta-analysis showed an overall incidence of GI symptoms of 17.6% in the following conditions in decreasing order of prevalence: anorexia, diarrhea, nausea, vomiting, and abdominal discomfort.5 In addition, the prolonged ventilatory requirements among a subset of individuals with the most severe COVID-19 results in extended periods of nutrition support via enteral tube placements. In individuals with ICU-acquired weakness and discharge to long-term care facilities, the placement of percutaneous endoscopic tubes may be required, although with the additional consideration of the need for an aerosolizing procedure. Delay of placement has been advocated, in addition to appropriate personal protective equipment, in order to ensure safe placement for the endoscopy staff.6
Types of feeding tubes
After deciding to feed a patient enterally and determining the anticipated duration of enteral support, the next decision is to determine the most appropriate location of feeding delivery: into the stomach or the small bowel. Gastric feeding is advantageous most commonly because of its increased capacity, allowing for larger volumes to be delivered over shorter durations. However, in the setting of postsurgical anatomy, gastroparesis, or obstructing tumors/pancreatic inflammation, distal delivery of tube feeds may be required into the jejunum. Additionally, percutaneous tubes placed into the stomach can have extenders into the small bowel (GJ tubes) to allow for feeding into the small bowel and decompression or delivery of medications into the stomach.
In general, gastric feeding is preferred over small bowel feeding as PEG tubes are more stable and have fewer complications than either PEG-J or direct PEJ tubes. Gastrostomy tubes are generally shorter and larger in diameter making them less likely to clog. PEG-J tubes have separate lumens for gastric and small intestinal access, but the smaller-bore jejunal extension tubes are more likely to clog or become dislodged. While direct PEJ is shown to have higher rates of tube patency and decreased rates of endoscopic re-intervention, compared with PEG-J,7 one limitation of a direct PEJ is difficulty in placement and site selection, which can be performed with a pediatric colonoscope or balloon enteroscopy system. Most commonly, this procedure is performed under general anesthesia.
In the case of a critically ill patient in the ICU, it is recommended to start enteral nutrition within 24-48 hours of arrival to avoid complications of prolonged calorie deficits. Nasally inserted feeding tubes (e.g., Cortrak, Avanos Medical Devices, Alpharetta, Ga.) are most commonly used at the bedside and can be placed blindly using electromagnetic image guidance, radiographically, or endoscopy. However, the small caliber of nasoenteric tubes comes with the common complication of clogging, which can be overcome with slightly larger bore gastric feeding tubes. If gastric feeding is not tolerated (e.g., in the case of vomiting, witnessed aspiration), small bowel feeding should be initiated and can be a more durable form of enteral feeding with fewer interruptions as feedings do not need to be held for procedures or symptomatic gastric intolerance. In clinical areas of question, or if there is a concern for intolerance of enteral feeding, a short trial with nasogastric or nasojejunal tube placement should be performed before a more definitive percutaneous placement.
With respect to percutaneous tubes, important characteristics to choose are the size (diameter in French units), type of internal retention device, and external appearance of the tube (standard or low profile). All percutaneous tubes contain an external retention device (i.e., bumper) that fits against the skin and an internal retention device that is either a balloon or plastic dome or funnel that prevents the tube from becoming dislodged. Balloon retention tubes require replacement every 3-6 months, while nonballoon tubes generally require replacement annually in order to prevent the plastic from cracking, which can make removal complicated. Low-profile tubes have an external cap, which, when opened, allows for extension tubing to be securely attached while in use and detached while not in use. Low-profile tubes are often preferred among younger, active patients and those with adequate dexterity to allow for attachment of the external extension tubing. These tubes are most often inserted as a replacement for an initially endoscopically placed tube, although one-step systems for initial placement are available. The size of the low-profile tube is chosen based on the size of the existing PEG tube and by measuring the length of the stoma tract using specialized measuring devices.8 Patients and caregivers can also be trained to replace balloon-type tubes on their own to limit complications of displaced or cracked tubes. Low-profile tubes are commercially available for both gastric placement and gastric placement with extension into the small bowel, which often requires fluoroscopy for secure placement.
All percutaneous enteral tubes are being transitioned to the ENfit connector system, which prevents connections from the enteral system to nonenteral systems (namely intravenous lines, chest tubes) and vice versa. Tubing misconnections have been rarely reported, and the EnFIT system is designed to prevent such misadventures that have resulted in serious complications and even mortality.9 Adapter devices are available that may be required for patients with feeding tubes who have not been transitioned yet. Most commonly with new tube placements and replacements, patients and providers will have to become familiar with the new syringes and feeding bags required with EnFIT connectors.
Gastrostomy placement can be considered a higher-risk endoscopic procedure. One complicating factor is the increased use of antiplatelet and anticoagulant therapies in individuals with a history of neurologic insults. The American Society for Gastrointestinal Endoscopy (ASGE) guidelines recommend that coumadin be held 5 days before the procedure and bridged with heparin if the patient is at high risk of thromboembolic complications. For patients on dual anti-platelet therapy, thienopyridines like clopidogrel are often stopped 5-7 days prior to procedure with continuation of aspirin,10 but there are more recent data that PEG insertion is safe with continued use of DAPT.11 Direct-acting anticoagulants (DOACs) are often stopped 24-48 hours prior to procedure and then restarted 48 hours after tube placement, but this is dependent on the half-life of the specific DOAC and the patient’s renal function. Patients with decreased creatinine clearance may need to hold the DOAC up to 3-4 days prior to the procedure. In this situation, referring to ASGE guidelines and consultation with a hematologist or managing anti-coagulation clinic is advised.10
Troubleshooting complications
Nasoenteric tubes: One of the most common and irritating complications with nasoenteric feeding tubes is clogging. To prevent clogging, the tube should be flushed frequently.12 At least 30 mL of free water should be used to flush the tube every 4-8 hours for continuous feedings or before and after bolus feeding. Additionally, 15-30 mL of water should be given with each separate medication administration, and if possible, medication administration via small-bore small bowel feeding tubes should be avoided.12 Water flushing is especially important with small-caliber tubes and pumps that deliver both feeding and water flushes. It is available for small bowel feeding in order to allow for programmed water delivery.
Warm water flushes can also help unclog the tube,12 and additional pharmacologic and mechanical devices have been promoted for clogged tubes. One common technique is mixing pancreatic enzymes (Viokase) with a crushed 325-mg tablet of nonenteric coated sodium bicarbonate and 5 mL of water to create a solution that has the alkaline properties allowing for both pancreatic enzyme activation and clog dissolution. Additionally, an endoscopic retrograde cholangiopancreatography (ERCP) catheter can be placed into longer feeding tubes to directly infuse the activated agent to the site of the clog.13 If water and enzymes are not successful in unclogging the tube, commercially available brushes can help remove clogs. The TubeClear® system (Actuated Medical, Bellefonte, Penna) has a single-use stem that is connected to AC power to create a jackhammerlike movement to remove clogs in longer nasoenteral and gastrojejunal tubes.
PEG tubes (short-term complications): Procedural and immediate postprocedural complications include bleeding, aspiration, pneumoperitoneum, and perforation. Pneumoperitoneum occurs in approximately 50% of cases and is generally clinically insignificant. The risk of pneumoperitoneum can be reduced by using CO2 insufflation.14 If the patient develops systemic signs of infection or peritoneal signs, CT scan with oral contrast is warranted for further evaluation and to assess for inadvertent perforation of overlying bowel or dislodged tube. Aspiration during or following endoscopy is another common complication of PEG placement and risk factors include over-sedation, supine positioning, advanced age, and neurologic dysfunction. This risk can be mitigated by avoiding over-sedation, immediately aspirating gastric contents when the stomach is reached, and avoiding excessive insufflation.15 In addition, elevating the head of the bed during the procedure and dedicating an assistant to perform oral suctioning during the entire procedure is recommended.
PEG tubes (long-term complications): More delayed complications of PEG insertion include wound infection, buried bumper syndrome, tumor seeding, peristomal leakage, and tube dislodgement. The prevalence of wound infection is 5%- 25%,16 and randomized controlled trials have demonstrated the efficacy of a single dose of an IV antibiotic (i.e., cephalosporin) in those not already receiving a broad spectrum antibiotic and administered prophylactically before tube placement.17 The significance of this reduction is such that antibiotic administration before tube placement should be considered a quality measure for the procedure. A small amount of redness around the tube site (less than 5 mm) is typical, but extension of erythema, warmth, tenderness, purulent drainage, or systemic symptoms is consistent with infection and warrants additional antibiotic administration. Minor infections can be treated with local antiseptics and oral antibiotics, and early intervention is important to prevent need for hospital admission, systemic antibiotics, and even surgical debridement.
Peristomal leakage is reported in approximately 1%-2% of patients.18 Photographs of the site can be very useful in evaluating and managing peristomal leakage and infections. Interventions include reducing gastric secretions with proton pump inhibitors and management of the skin with barrier creams, such as zinc oxide (Calmoseptine®) ointment. Placement of a larger-diameter tube only enlarges the stoma track and worsens the leakage. In such cases, thorough evaluations for delayed gastric emptying (gastroparesis), distal obstruction, or constipation should be performed and managed accordingly. Opiates are common contributors to constipation and delayed gastric emptying and often require reduction in use or directed antagonist therapy to reduce leaking. Continuous feeding over bolus feedings and delivering nutrition distally into the small bowel (PEG-J placement) can improve leaking from gastrostomy tubes. Additional means of management include stabilizing the tube by replacing a traditional tube with a low-profile tube or using right-angle external bumpers. If all measures fail, removing the tube and allowing for stomal closure can be attempted,16 although this option often requires parenteral nutrition support to prevent prolonged periods of inadequate nutrition.
Buried bumper syndrome (BBS) occurs in 1.5%-8.8% of PEG placements and is a common late complication of PEG placement, although early reports have been described.18 The development of BBS occurs when the internal bumper migrates from the gastric lumen through and into the stomach or abdominal wall. It occurs more frequently with solid nonballoon retention tubes and is caused by excessive compression of the external bumper against the skin and abdominal wall. Patients with BBS usually present with an immobile catheter, resistance with feeds (because of a closure of the stomach wall around the internal portion of the gastrostomy tube), abdominal pain, or peristomal leakage. Physicians should be aware of and assess tubes for BBS, in particular when replacing an immobile tube (cannot be pushed into the free stomach lumen) or when there is difficulty in flushing water into the tube. This complication can be easily prevented by allowing a minimum of 0.5-1.0 cm (1 finger breadth) between the external bumper and the abdominal wall. In particular, patients and caregivers should be warned that if the patient gains significant amounts of weight, the outer bumper will need to be loosened. Once BBS is diagnosed, the PEG tube requires removal and replacement as it can cause bleeding, infection, or fasciitis. The general steps to replacement include endoscopic removal of the existing tube and replacement of new PEG in the existing tract as long as the BBS is not severe. In most cases a replacement tube can be pulled into place using the pull-PEG technique at the same gastrostomy site as long as the stoma tract can be cannulated with a wire after the existing tube is removed.
Similar to nasoenteric tubes, PEG tubes can become clogged, although this complication is infrequent. The primary steps for prevention include adequately flushing with water before and after feeds and ensuring that all medications are liquid or well crushed and dissolved before instilling. Timely tube replacement also ensures that the internal portions of the gastrostomy tube remain free of debris. Management is similar to that of unclogging nasoenteral tubes, as discussed above, and specific commercial declogging devices for PEG tubes include the Bionix Declogger® (Bionix Development Corp., Toledo, Ohio) and the Bard® PEG cleaning brush (Bard Peripheral Vascular Inc., Tempe, Ariz.). The Bionix system has a plastic stem with a screw and thread design that will remove clogs in 14-24 French PEG tubes, while the Bard brush has a flexible nylon stem with soft bristles at the end to prevent mucosal injury and can be used for prophylaxis against clogs, as well as removing clogs themselves.12
Lastly, a rare but important complication of PEG placement is tumor seeding of the PEG site in patients with active head and neck or upper gastrointestinal cancer.19 The presumed mechanism is shearing of tumor cells as the PEG is pulled through the upper aerodigestive tract and through the wall of the stomach, as prior studies have demonstrated frequent seeding of tubes and incision sites as shown by brushing the tube for malignant cells after tube placement.20 It is important to recognize this complication and not misdiagnose it as granulation tissue, infection, or bleeding as the spread of the cancer generally portends a poor prognosis. Therefore, it is best to use a PEG insertion technique that does not involve pulling or pushing the PEG through the upper aerodigestive tract in patients with active cancer and instead place tubes via an external approach by colleagues in interventional radiology or via direct surgical placement.
Conclusion
Gastroenterologists occupy a unique role in evaluation, diagnosis, and management of patients requiring enteral feeding. In addition, they are best equipped to place, prevent, and manage complications of tube feeding. For this reason, it is imperative that gastroenterologists familiarize themselves with indications for enteral tubes and types of enteral tubes available, as well as the identification and management of common complications. Comprehensive understanding of these concepts will augment the practicing gastroenterologist’s ability to manage patients requiring enteral nutrition support with confidence.
References
1. Stein DJ et al. Dig Dis Sci. 2020 Jun 19. doi: 10.1007/s10620-020-06396-y.
2. American Geriatrics Society Ethics Committee and Clinical Practice and Models of Care Committee. J Am Geriatr Soc. 2014;62(8):1590-3.
3. Dietrich CG, Schoppmeyer K. World J Gastroenterol. 2020;26(20):2464-71.
4. Suzuki Y et al. T Gastroenterology Res.2012 Feb;5(1):10-20.
5. Cheung KS et al. Gastroenterology. 2020 Jul;159(1):81-95.
6. Micic D et al. Am J Gastroenterol. 2020 Sep;115(9):1367-70.
7. Fan AC et al. Gastrointest Endosc. 2002;56(6):890-4.
8. Tang SJ. Video J Encycl GI Endosc. 2014;2(2):70-3.
9. Guenter P, Lyman B. Nutr Clin Pract. 2016;31(6):769-72.
10. Acosta RD et al. Gastrointest Endosc. 2016;83(1):3-16.
11. Richter JA et al. Gastrointest Endosc. 2011;74(1):22-34.
12. Boullata JI et al. JPEN. 2017;41(1):15-103.
13. McClave SA. Tech Gastrointest Endosc. 2021;3(1):62-8.
14. Murphy CJ et al. Endosc Int Open. 2016;4(3):E292. doi: 10.1053/tgie.2001.19915.
15. Lynch CR et al. Pract Gastroenterology. 2004;28:66-77.
16. Hucl T et al. Best Pract Res Clin Gastroenterol. 2016;30(5):769-81. doi: 10.1016/j.bpg.2016.10.002.
17. Jafri NS et al. Aliment Pharmacol & Therapeut. 2007;25(6):647-56. doi: 10.1111/j.1365-2036.2007.03247.x.
18. Blumenstein I et al. World J Gastroenterol. 2014;20(26):8505-24. doi: 10.3748/wjg.v20.i26.8505.
19. Fung E et al. Surgical Endosc. 2017;31(9):3623-7. doi: 10.1007/s00464-016-5394-8.
20. Ellrichmann M et al. Endoscopy. 2013;45(07):526-31. doi: 10.1055/s-0033-1344023.
Dr. Toy is with the department of internal medicine at the University of Utah, Salt Lake City. Dr. Fang is with the division of gastroenterology and hepatology at the University of Utah.
Introduction
Gastroenterologists are in a unique position to manage individuals with feeding tubes as their training underscores principles in digestion, absorption, nutrition support, and enteral tube placement. Adequate management of individuals with feeding tubes and, importantly, the complications that arise from feeding tube use and placement require a basic understanding of intestinal anatomy and physiology. Therefore, gastroenterologists are well suited to both place and manage individuals with feeding tubes in the long term.
Indications for tube feeding
When deciding on the appropriate route for artificial nutrition support, the first decision to be made is enteral access versus parenteral nutrition support. Enteral nutrition confers multiple benefits, including preservation of the mucosal lining, reductions in complicated infections, decreased costs, and improved patient compliance. All attempts at adequate enteral access should be made before deciding on the use of parenteral nutrition. Following the clinical decision to pursue artificial means of nutrition support and enteral access, the next common decision is the anticipated duration of nutrition support. Generally, the oral or nasal tubes are used for short durations (i.e., less than 4 weeks) with percutaneous placement into the stomach or small intestine for longer-term feeding (i.e., percutaneous endoscopic gastrostomy [PEG] or percutaneous endoscopic jejunostomy [PEJ]).
The most general indication for nutrition support is an inability to maintain adequate nutritional needs with oral intake alone. General categories of inadequate oral intake include neurologic disorders, malignancy, and gastrointestinal conditions affecting digestion and absorption (Table 1). Absolute and relative contraindications to PEG placement are listed in Table 2. If an endoscopic placement is not possible, alternative means of placement (i.e., surgery or interventional radiology) can be considered to avoid the consequences of prolonged malnutrition. In-hospital mortality following PEG placement has decreased 40% over the last 10 years, which can be attributed to improved patient selection, enhanced discharge practices, and exclusion of patients with the highest comorbidity and mortality rates, like those with advanced dementia or terminal cancer.1
PEG placement in patients with dementia is controversial, with previous studies not demonstrating improved outcomes and association with high mortality rates,2 so the practice is currently not recommended by the American Geriatrics Society in individuals with advanced dementia.3 However, a large Japanese study showed that careful selection of patients with mild dementia to undergo gastrostomy increased independence fourfold; therefore, multidisciplinary involvement is often necessary in the decision to pursue artificial means of nutrition support in this population.4
The recent coronavirus disease 2019 (COVID-19) pandemic has placed additional strains on endoscopic placement and has highlighted the effect of the severe acute respiratory syndrome coronavirus 2 (SARS-COV-2) on GI symptoms. A recent meta-analysis showed an overall incidence of GI symptoms of 17.6% in the following conditions in decreasing order of prevalence: anorexia, diarrhea, nausea, vomiting, and abdominal discomfort.5 In addition, the prolonged ventilatory requirements among a subset of individuals with the most severe COVID-19 results in extended periods of nutrition support via enteral tube placements. In individuals with ICU-acquired weakness and discharge to long-term care facilities, the placement of percutaneous endoscopic tubes may be required, although with the additional consideration of the need for an aerosolizing procedure. Delay of placement has been advocated, in addition to appropriate personal protective equipment, in order to ensure safe placement for the endoscopy staff.6
Types of feeding tubes
After deciding to feed a patient enterally and determining the anticipated duration of enteral support, the next decision is to determine the most appropriate location of feeding delivery: into the stomach or the small bowel. Gastric feeding is advantageous most commonly because of its increased capacity, allowing for larger volumes to be delivered over shorter durations. However, in the setting of postsurgical anatomy, gastroparesis, or obstructing tumors/pancreatic inflammation, distal delivery of tube feeds may be required into the jejunum. Additionally, percutaneous tubes placed into the stomach can have extenders into the small bowel (GJ tubes) to allow for feeding into the small bowel and decompression or delivery of medications into the stomach.
In general, gastric feeding is preferred over small bowel feeding as PEG tubes are more stable and have fewer complications than either PEG-J or direct PEJ tubes. Gastrostomy tubes are generally shorter and larger in diameter making them less likely to clog. PEG-J tubes have separate lumens for gastric and small intestinal access, but the smaller-bore jejunal extension tubes are more likely to clog or become dislodged. While direct PEJ is shown to have higher rates of tube patency and decreased rates of endoscopic re-intervention, compared with PEG-J,7 one limitation of a direct PEJ is difficulty in placement and site selection, which can be performed with a pediatric colonoscope or balloon enteroscopy system. Most commonly, this procedure is performed under general anesthesia.
In the case of a critically ill patient in the ICU, it is recommended to start enteral nutrition within 24-48 hours of arrival to avoid complications of prolonged calorie deficits. Nasally inserted feeding tubes (e.g., Cortrak, Avanos Medical Devices, Alpharetta, Ga.) are most commonly used at the bedside and can be placed blindly using electromagnetic image guidance, radiographically, or endoscopy. However, the small caliber of nasoenteric tubes comes with the common complication of clogging, which can be overcome with slightly larger bore gastric feeding tubes. If gastric feeding is not tolerated (e.g., in the case of vomiting, witnessed aspiration), small bowel feeding should be initiated and can be a more durable form of enteral feeding with fewer interruptions as feedings do not need to be held for procedures or symptomatic gastric intolerance. In clinical areas of question, or if there is a concern for intolerance of enteral feeding, a short trial with nasogastric or nasojejunal tube placement should be performed before a more definitive percutaneous placement.
With respect to percutaneous tubes, important characteristics to choose are the size (diameter in French units), type of internal retention device, and external appearance of the tube (standard or low profile). All percutaneous tubes contain an external retention device (i.e., bumper) that fits against the skin and an internal retention device that is either a balloon or plastic dome or funnel that prevents the tube from becoming dislodged. Balloon retention tubes require replacement every 3-6 months, while nonballoon tubes generally require replacement annually in order to prevent the plastic from cracking, which can make removal complicated. Low-profile tubes have an external cap, which, when opened, allows for extension tubing to be securely attached while in use and detached while not in use. Low-profile tubes are often preferred among younger, active patients and those with adequate dexterity to allow for attachment of the external extension tubing. These tubes are most often inserted as a replacement for an initially endoscopically placed tube, although one-step systems for initial placement are available. The size of the low-profile tube is chosen based on the size of the existing PEG tube and by measuring the length of the stoma tract using specialized measuring devices.8 Patients and caregivers can also be trained to replace balloon-type tubes on their own to limit complications of displaced or cracked tubes. Low-profile tubes are commercially available for both gastric placement and gastric placement with extension into the small bowel, which often requires fluoroscopy for secure placement.
All percutaneous enteral tubes are being transitioned to the ENfit connector system, which prevents connections from the enteral system to nonenteral systems (namely intravenous lines, chest tubes) and vice versa. Tubing misconnections have been rarely reported, and the EnFIT system is designed to prevent such misadventures that have resulted in serious complications and even mortality.9 Adapter devices are available that may be required for patients with feeding tubes who have not been transitioned yet. Most commonly with new tube placements and replacements, patients and providers will have to become familiar with the new syringes and feeding bags required with EnFIT connectors.
Gastrostomy placement can be considered a higher-risk endoscopic procedure. One complicating factor is the increased use of antiplatelet and anticoagulant therapies in individuals with a history of neurologic insults. The American Society for Gastrointestinal Endoscopy (ASGE) guidelines recommend that coumadin be held 5 days before the procedure and bridged with heparin if the patient is at high risk of thromboembolic complications. For patients on dual anti-platelet therapy, thienopyridines like clopidogrel are often stopped 5-7 days prior to procedure with continuation of aspirin,10 but there are more recent data that PEG insertion is safe with continued use of DAPT.11 Direct-acting anticoagulants (DOACs) are often stopped 24-48 hours prior to procedure and then restarted 48 hours after tube placement, but this is dependent on the half-life of the specific DOAC and the patient’s renal function. Patients with decreased creatinine clearance may need to hold the DOAC up to 3-4 days prior to the procedure. In this situation, referring to ASGE guidelines and consultation with a hematologist or managing anti-coagulation clinic is advised.10
Troubleshooting complications
Nasoenteric tubes: One of the most common and irritating complications with nasoenteric feeding tubes is clogging. To prevent clogging, the tube should be flushed frequently.12 At least 30 mL of free water should be used to flush the tube every 4-8 hours for continuous feedings or before and after bolus feeding. Additionally, 15-30 mL of water should be given with each separate medication administration, and if possible, medication administration via small-bore small bowel feeding tubes should be avoided.12 Water flushing is especially important with small-caliber tubes and pumps that deliver both feeding and water flushes. It is available for small bowel feeding in order to allow for programmed water delivery.
Warm water flushes can also help unclog the tube,12 and additional pharmacologic and mechanical devices have been promoted for clogged tubes. One common technique is mixing pancreatic enzymes (Viokase) with a crushed 325-mg tablet of nonenteric coated sodium bicarbonate and 5 mL of water to create a solution that has the alkaline properties allowing for both pancreatic enzyme activation and clog dissolution. Additionally, an endoscopic retrograde cholangiopancreatography (ERCP) catheter can be placed into longer feeding tubes to directly infuse the activated agent to the site of the clog.13 If water and enzymes are not successful in unclogging the tube, commercially available brushes can help remove clogs. The TubeClear® system (Actuated Medical, Bellefonte, Penna) has a single-use stem that is connected to AC power to create a jackhammerlike movement to remove clogs in longer nasoenteral and gastrojejunal tubes.
PEG tubes (short-term complications): Procedural and immediate postprocedural complications include bleeding, aspiration, pneumoperitoneum, and perforation. Pneumoperitoneum occurs in approximately 50% of cases and is generally clinically insignificant. The risk of pneumoperitoneum can be reduced by using CO2 insufflation.14 If the patient develops systemic signs of infection or peritoneal signs, CT scan with oral contrast is warranted for further evaluation and to assess for inadvertent perforation of overlying bowel or dislodged tube. Aspiration during or following endoscopy is another common complication of PEG placement and risk factors include over-sedation, supine positioning, advanced age, and neurologic dysfunction. This risk can be mitigated by avoiding over-sedation, immediately aspirating gastric contents when the stomach is reached, and avoiding excessive insufflation.15 In addition, elevating the head of the bed during the procedure and dedicating an assistant to perform oral suctioning during the entire procedure is recommended.
PEG tubes (long-term complications): More delayed complications of PEG insertion include wound infection, buried bumper syndrome, tumor seeding, peristomal leakage, and tube dislodgement. The prevalence of wound infection is 5%- 25%,16 and randomized controlled trials have demonstrated the efficacy of a single dose of an IV antibiotic (i.e., cephalosporin) in those not already receiving a broad spectrum antibiotic and administered prophylactically before tube placement.17 The significance of this reduction is such that antibiotic administration before tube placement should be considered a quality measure for the procedure. A small amount of redness around the tube site (less than 5 mm) is typical, but extension of erythema, warmth, tenderness, purulent drainage, or systemic symptoms is consistent with infection and warrants additional antibiotic administration. Minor infections can be treated with local antiseptics and oral antibiotics, and early intervention is important to prevent need for hospital admission, systemic antibiotics, and even surgical debridement.
Peristomal leakage is reported in approximately 1%-2% of patients.18 Photographs of the site can be very useful in evaluating and managing peristomal leakage and infections. Interventions include reducing gastric secretions with proton pump inhibitors and management of the skin with barrier creams, such as zinc oxide (Calmoseptine®) ointment. Placement of a larger-diameter tube only enlarges the stoma track and worsens the leakage. In such cases, thorough evaluations for delayed gastric emptying (gastroparesis), distal obstruction, or constipation should be performed and managed accordingly. Opiates are common contributors to constipation and delayed gastric emptying and often require reduction in use or directed antagonist therapy to reduce leaking. Continuous feeding over bolus feedings and delivering nutrition distally into the small bowel (PEG-J placement) can improve leaking from gastrostomy tubes. Additional means of management include stabilizing the tube by replacing a traditional tube with a low-profile tube or using right-angle external bumpers. If all measures fail, removing the tube and allowing for stomal closure can be attempted,16 although this option often requires parenteral nutrition support to prevent prolonged periods of inadequate nutrition.
Buried bumper syndrome (BBS) occurs in 1.5%-8.8% of PEG placements and is a common late complication of PEG placement, although early reports have been described.18 The development of BBS occurs when the internal bumper migrates from the gastric lumen through and into the stomach or abdominal wall. It occurs more frequently with solid nonballoon retention tubes and is caused by excessive compression of the external bumper against the skin and abdominal wall. Patients with BBS usually present with an immobile catheter, resistance with feeds (because of a closure of the stomach wall around the internal portion of the gastrostomy tube), abdominal pain, or peristomal leakage. Physicians should be aware of and assess tubes for BBS, in particular when replacing an immobile tube (cannot be pushed into the free stomach lumen) or when there is difficulty in flushing water into the tube. This complication can be easily prevented by allowing a minimum of 0.5-1.0 cm (1 finger breadth) between the external bumper and the abdominal wall. In particular, patients and caregivers should be warned that if the patient gains significant amounts of weight, the outer bumper will need to be loosened. Once BBS is diagnosed, the PEG tube requires removal and replacement as it can cause bleeding, infection, or fasciitis. The general steps to replacement include endoscopic removal of the existing tube and replacement of new PEG in the existing tract as long as the BBS is not severe. In most cases a replacement tube can be pulled into place using the pull-PEG technique at the same gastrostomy site as long as the stoma tract can be cannulated with a wire after the existing tube is removed.
Similar to nasoenteric tubes, PEG tubes can become clogged, although this complication is infrequent. The primary steps for prevention include adequately flushing with water before and after feeds and ensuring that all medications are liquid or well crushed and dissolved before instilling. Timely tube replacement also ensures that the internal portions of the gastrostomy tube remain free of debris. Management is similar to that of unclogging nasoenteral tubes, as discussed above, and specific commercial declogging devices for PEG tubes include the Bionix Declogger® (Bionix Development Corp., Toledo, Ohio) and the Bard® PEG cleaning brush (Bard Peripheral Vascular Inc., Tempe, Ariz.). The Bionix system has a plastic stem with a screw and thread design that will remove clogs in 14-24 French PEG tubes, while the Bard brush has a flexible nylon stem with soft bristles at the end to prevent mucosal injury and can be used for prophylaxis against clogs, as well as removing clogs themselves.12
Lastly, a rare but important complication of PEG placement is tumor seeding of the PEG site in patients with active head and neck or upper gastrointestinal cancer.19 The presumed mechanism is shearing of tumor cells as the PEG is pulled through the upper aerodigestive tract and through the wall of the stomach, as prior studies have demonstrated frequent seeding of tubes and incision sites as shown by brushing the tube for malignant cells after tube placement.20 It is important to recognize this complication and not misdiagnose it as granulation tissue, infection, or bleeding as the spread of the cancer generally portends a poor prognosis. Therefore, it is best to use a PEG insertion technique that does not involve pulling or pushing the PEG through the upper aerodigestive tract in patients with active cancer and instead place tubes via an external approach by colleagues in interventional radiology or via direct surgical placement.
Conclusion
Gastroenterologists occupy a unique role in evaluation, diagnosis, and management of patients requiring enteral feeding. In addition, they are best equipped to place, prevent, and manage complications of tube feeding. For this reason, it is imperative that gastroenterologists familiarize themselves with indications for enteral tubes and types of enteral tubes available, as well as the identification and management of common complications. Comprehensive understanding of these concepts will augment the practicing gastroenterologist’s ability to manage patients requiring enteral nutrition support with confidence.
References
1. Stein DJ et al. Dig Dis Sci. 2020 Jun 19. doi: 10.1007/s10620-020-06396-y.
2. American Geriatrics Society Ethics Committee and Clinical Practice and Models of Care Committee. J Am Geriatr Soc. 2014;62(8):1590-3.
3. Dietrich CG, Schoppmeyer K. World J Gastroenterol. 2020;26(20):2464-71.
4. Suzuki Y et al. T Gastroenterology Res.2012 Feb;5(1):10-20.
5. Cheung KS et al. Gastroenterology. 2020 Jul;159(1):81-95.
6. Micic D et al. Am J Gastroenterol. 2020 Sep;115(9):1367-70.
7. Fan AC et al. Gastrointest Endosc. 2002;56(6):890-4.
8. Tang SJ. Video J Encycl GI Endosc. 2014;2(2):70-3.
9. Guenter P, Lyman B. Nutr Clin Pract. 2016;31(6):769-72.
10. Acosta RD et al. Gastrointest Endosc. 2016;83(1):3-16.
11. Richter JA et al. Gastrointest Endosc. 2011;74(1):22-34.
12. Boullata JI et al. JPEN. 2017;41(1):15-103.
13. McClave SA. Tech Gastrointest Endosc. 2021;3(1):62-8.
14. Murphy CJ et al. Endosc Int Open. 2016;4(3):E292. doi: 10.1053/tgie.2001.19915.
15. Lynch CR et al. Pract Gastroenterology. 2004;28:66-77.
16. Hucl T et al. Best Pract Res Clin Gastroenterol. 2016;30(5):769-81. doi: 10.1016/j.bpg.2016.10.002.
17. Jafri NS et al. Aliment Pharmacol & Therapeut. 2007;25(6):647-56. doi: 10.1111/j.1365-2036.2007.03247.x.
18. Blumenstein I et al. World J Gastroenterol. 2014;20(26):8505-24. doi: 10.3748/wjg.v20.i26.8505.
19. Fung E et al. Surgical Endosc. 2017;31(9):3623-7. doi: 10.1007/s00464-016-5394-8.
20. Ellrichmann M et al. Endoscopy. 2013;45(07):526-31. doi: 10.1055/s-0033-1344023.
Dr. Toy is with the department of internal medicine at the University of Utah, Salt Lake City. Dr. Fang is with the division of gastroenterology and hepatology at the University of Utah.
