User login
Brexanolone injection for postpartum depression
Postpartum depression (PPD) is one of the most prevalent complications associated with pregnancy and childbirth in the United States, affecting more than 400,000 women annually.1 Postpartum depression is most commonly treated with psychotherapy and antidepressants approved for the treatment of major depressive disorder. Until recently, there was no pharmacologic therapy approved by the FDA specifically for the treatment of PPD. Considering the adverse outcomes associated with untreated or inadequately treated PPD, and the limitations of existing therapies, there is a significant unmet need for pharmacologic treatment options for PPD.2 To help address this need, the FDA recently approved brexanolone injection (brand name: ZULRESSO™) (Table 13) as a first-in-class therapy for the treatment of adults with PPD.3

Clinical implications
Postpartum depression can result in adverse outcomes for the patient, baby, and family when under- or untreated, and the need for rapid resolution of symptoms cannot be overstated.2 Suicide is strongly associated with depression and is a leading cause of pregnancy-related deaths.4 Additionally, PPD can impact the health, safety, and well-being of the child, with both short- and long-term consequences, including greater rates of psychological or behavioral difficulties among children of patients with PPD.5 Postpartum depression can also have negative effects on the patient’s partner, with 24% to 50% of partners experiencing depression.6 Current PPD management strategies include the use of psychotherapy and pharmacologic interventions for major depressive disorder that may take up to 4 to 6 weeks for some patients, and may not achieve remission for all patients.7-9
Brexanolone injection is a first-in-class medication with a novel mechanism of action. In clinical studies, it achieved rapid (by Hour 60) and sustained (through Day 30) reductions in depressive symptoms and could provide a meaningful new treatment option for adult women with PPD.10,11
How it works
Animal and human studies have established the re

Brexanolone is a neuroactive steroid that is chemically identical to endogenous allopregnanolone produced in the CNS. Brexanolone potentiates GABA-mediated currents from recombinant human GABAARs in mammalian cells expressing α1β2γ2 receptor subunits, α4β3δ receptor subunits, and α6β3δ receptor subunits.3 Positive allosteric modulation of both synaptic and extrasynaptic GABAARs differentiates brexanolone from other GABAAR modulators, such as benzodiazepines.10,11
Brexanolone’s mechanism of action in the treatment of PPD is not fully understood, but it is thought to be related to GABAAR PAM activity.3
Supporting evidence
The FDA approval of brexanolone injection was based on the efficacy demonstrated in 2 Phase III multicenter, randomized, double-blind, placebo-controlled studies in adult women (age 18 to 45) with PPD (defined by DSM-IV criteria for a major depressive episode, with onset of symptoms in the third trimester or within 4 weeks of delivery). Exclusion criteria included the presence of bipolar disorder or psychosis. In these studies, 60-hour continuous IV infusions of brexanolone or placebo were given, followed by 4 weeks of observation. Study 1 (202B) enrolled patients with severe PPD (Hamilton Rating Scale for Depression [HAM-D] total score ≥26), and Study 2 (202C) enrolled patients with moderate PPD (HAM-D score 20 to 25). A titration to the recommended target dosage of 90 μg/kg/hour was evaluated in both studies. BRX90 patients received 30 μg/kg/hour for 4 hours, 60 μg/kg/hour for 20 hours, 90 μg/kg/hour for 28 hours, followed by a taper to 60 μg/kg/hour for 4 hours and then 30 μg/kg/hour for 4 hours. The primary endpoint in both studies was the mean change from baseline in depressive symptoms as measured by HAM-D total score at the end of the 60-hour infusion. A pre-specified secondary efficacy endpoint was the mean change from baseline in HAM-D total score at Day 30.
Continue to: Efficacy
Efficacy. In both placebo-controlled studies, titration to a target dose of brexanolone 90 μg/kg/hour was superior to placebo in improvement of depressive symptoms (Table 33).
")
Pharmacological profile
Brexanolone exposure-response relationships and the time course of pharmacodynamic response are unknown.3
Adverse reactions. Safety was evaluated from all patients receiving brexanolone injection, regardless of dosing regimen (N = 140, including patients from a Phase IIb study, 202A).3,11
The most common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were sedation/somnolence, dry mouth, loss of consciousness, and flushing/hot flush.3 The incidence of patients discontinuing due to any adverse reaction was 2% for brexanolone vs 1% for placebo.3
Sedation, somnolence, and loss of consciousness. In clinical studies, brexanolone caused sedation and somnolence that required dose interruption or reduction in some patients during the infusion (5% of brexanolone-treated patients compared with 0% of placebo-treated patients).3 Some patients were also reported to have loss of consciousness or altered state of consciousness during the brexanolone infusion (4% of patients treated with brexanolone compared with 0% of patients treated with placebo).3 All patients with loss of or altered state of consciousness recovered fully 15 to 60 minutes after dose interruption.3 There was no clear association between loss or alteration of consciousness and pattern or timing of dose, and not all patients who experienced a loss or alteration of consciousness reported sedation or somnolence before the episode.
Continue to: Suicidality
Suicidality. The risk of developing suicidal thoughts and behaviors with brexanolone is unknown, due to the relatively low number of exposures to brexanolone injection during clinical development and a mechanism of action distinct from that of existing antidepressant medications.3
Pharmacokinetics
In clinical trials, brexanolone exhibited dose-proportional pharmacokinetics, and the terminal half-life is approximately 9 hours (Table 43). Brexanolone is metabolized by non-cytochrome P450 (CYP)-based pathways, including keto-reduction, glucuronidation, and sulfation.3 No clinically significant differences in the pharmacokinetics of brexanolone were observed based on renal or hepatic impairment, and no studies were conducted to evaluate the effects of other drugs on brexanolone.3

Lactation. A population pharmacokinetics model constructed from studies in the clinical development program calculated the maximum relative infant dose for brexanolone during infusion as 1.3%.3 Given the low oral bioavailability of brexanolone (<5%) in adults, the potential for breastfed infant exposure is considered low.3
Clinical considerations
Risk Evaluation and Mitigation Strategies (REMS) requirements. Brexanolone injection is a Schedule IV controlled substance. It has a “black-box” warning regarding excessive sedation and sudden loss of consciousness, which has been taken into account within the REMS drug safety program. Health care facilities and pharmacies must enroll in the REMS program and ensure that brexanolone is administered only to patients who are enrolled in the REMS program. Staff must be trained on the processes and procedures to administer brexanolone, and the facility must have a fall precautions protocol in place and be equipped with a programmable peristaltic IV infusion pump and continuous pulse oximetry with alarms.3
Monitoring. A REMS-trained clinician must be available continuously on-site to oversee each patient for the duration of the continuous IV infusion, which lasts 60 hours (2.5 days) and should be initiated early enough in the day to allow for recognition of excessive sedation. Patients must be monitored for hypoxia using continuous pulse oximetry equipped with an alarm and should also be assessed for excessive sedation every 2 hours during planned, non-sleep periods. If excessive sedation occurs, the infusion should be stopped until symptoms resolve, after which the infusion may be resumed at the same or a lower dose as clinically appropriate. In case of overdosage, the infusion should be stopped immediately and supportive measures initiated as necessary. Patients must not be the primary caregiver of dependents, and must be accompanied during interactions with their child(ren).
Continue to: Contraindications
Contraindications. There are no contraindications for the use of brexanolone in adults with PPD.
End-stage renal disease (ESRD). Avoid using brexanolone in patients with ESRD because of the potential accumulation of the solubilizing agent, betadex sulfobutyl ether sodium.
Pregnancy. Brexanolone has not been studied in pregnant patients. Pregnant women and women of reproductive age should be informed of the potential risk to a fetus based on data from other drugs that enhance GABAergic inhibition.
Breastfeeding. There are no data on the effects of brexanolone on a breastfed infant. Breastfeeding should be a discussion of risk and benefit between the patient and her doctor. The developmental and health benefits of breastfeeding should be considered, along with the mother’s clinical need for brexanolone and any potential adverse effects on the breastfed child from brexanolone or from the underlying maternal condition. However, based on the low relative infant dose (<2%) and the low oral bioavailability in adults, the risk to breastfed infants is thought to be low.16
Potential for abuse. Brexanolone injection is a Schedule IV controlled substance. Although it was not possible to assess physical dependency in the registrational trials due to dose tapering at the end of treatment, clinicians should advise patients about the theoretical possibility for brexanolone to be abused or lead to dependence based on other medications with similar primary pharmacology.
Continue to: Concomitant medications
Concomitant medications. Caution patients that taking opioids or other CNS depressants, such as benzodiazepines, in combination with brexanolone may increase the severity of sedative effects.
Suicidal thoughts and behaviors. Advise patients and caregivers to look for the emergence of suicidal thoughts and behavior and instruct them to report such symptoms to their clinician. Consider changing the therapeutic regimen, including discontinuing brexanolone, in patients whose depression becomes worse or who experience emergent suicidal thoughts and behaviors.
Why Rx?
Postpartum depression is a common and often devastating medical complication of childbirth that can result in adverse outcomes for the patient, baby, and family when left undertreated or untreated. There is a great need to identify and treat women who develop PPD. Rapid and sustained resolution of symptoms in women who experience PPD should be the goal of treatment, and consequently, brexanolone injection presents an important new tool in available treatment options for PPD.
Bottom Line
Brexanolone injection is a neuroactive steroid gamma-aminobutyric acid (GABA) A receptor positive allosteric modulator that’s been FDA-approved for the treatment of postpartum depression (PPD). It is administered as a continuous IV infusion over 60 hours. The rapid and sustained improvement of PPD observed in clinical trials with brexanolone injection may support a new treatment paradigm for women with PPD.
1. Ko JY, Rockhill KM, Tong VT, et al. Trends in postpartum depressive symptoms - 27 states, 2004, 2008, and 2012. MMWR Morb Mortal Wkly Rep. 2017;66(6):153-158.
2. Frieder A, Fersh M, Hainline R, et al. Pharmacotherapy of postpartum depression: current approaches and novel drug development. CNS Drugs. 2019;33(3):265-282.
3. Brexanolone injection [package insert]. Cambridge, MA: Sage Therapeutics, Inc.; 2019.
4. Bodnar-Deren S, Klipstein K, Fersh M, et al. Suicidal ideation during the postpartum period. J Womens Health (Larchmt). 2016;25(12):1219-1224.
5. Netsi E, Pearson RM, Murray L, et al. Association of persistent and severe postnatal depression with child outcomes. JAMA Psychiatry. 2018;75(3):247-253.
6. Goodman JH. Paternal postpartum depression, its relationship to maternal postpartum depression, and implications for family health. J Adv Nurs. 2004;45(1):26-35.
7. Gelenberg AJ, Freeman MP, Markowitz JC, et al; American Psychiatric Association Work Group on Major Depressive Disorder. Practice guidelines for the treatment of patients with major depressive disorder. 3rd ed. Washington, DC: American Psychiatric Association; 2010.
8. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905-1917.
9. Molyneaux E, Telesia LA, Henshaw C, et al. Antidepressants for preventing postnatal depression. Cochrane Database Syst Rev. 2018;4:CD004363.
10. Kanes S, Colquhoun H, Gunduz-Bruce H, et al. Brexanolone (SAGE-547 injection) in post-partum depression: a randomised controlled trial. Lancet. 2017;390(10093):480-489.
11. Meltzer-Brody S, Colquhoun H, Riesenberg R, et al. Brexanolone injection in post-partum depression: two multicentre, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet. 2018;392(10152):1058-1070.
12. Melon LC, Hooper A, Yang X, et al. Inability to suppress the stress-induced activation of the HPA axis during the peripartum period engenders deficits in postpartum behaviors in mice. Psychoneuroendocrinology. 2018;90:182-193.
13. Deligiannidis KM, Fales CL, Kroll-Desrosiers AR, et al. Resting-state functional connectivity, cortical GABA, and neuroactive steroids in peripartum and peripartum depressed women: a functional magnetic resonance imaging and spectroscopy study. Neuropsychopharmacology. 2019;44(3):546-554.
14. Licheri V, Talani G, Gorule AA, et al. Plasticity of GABAA receptors during pregnancy and postpartum period: from gene to function. Neural Plast. 2015;2015:170435. doi: 10.1155/2015/170435.
15. Luisi S, Petraglia F, Benedetto C, et al. Serum allopregnanolone levels in pregnant women: changes during pregnancy, at delivery, and in hypertensive patients. J Clin Endocrinol Metab. 2000;85(7):2429-2433.
16. Hoffmann E, Wald J, Dray D, et al. Brexanolone injection administration to lactating women: breast milk allopregnanolone levels [30J]. Obstetrics & Gynecology. 2019;133:115S.
Postpartum depression (PPD) is one of the most prevalent complications associated with pregnancy and childbirth in the United States, affecting more than 400,000 women annually.1 Postpartum depression is most commonly treated with psychotherapy and antidepressants approved for the treatment of major depressive disorder. Until recently, there was no pharmacologic therapy approved by the FDA specifically for the treatment of PPD. Considering the adverse outcomes associated with untreated or inadequately treated PPD, and the limitations of existing therapies, there is a significant unmet need for pharmacologic treatment options for PPD.2 To help address this need, the FDA recently approved brexanolone injection (brand name: ZULRESSO™) (Table 13) as a first-in-class therapy for the treatment of adults with PPD.3
Clinical implications
Postpartum depression can result in adverse outcomes for the patient, baby, and family when under- or untreated, and the need for rapid resolution of symptoms cannot be overstated.2 Suicide is strongly associated with depression and is a leading cause of pregnancy-related deaths.4 Additionally, PPD can impact the health, safety, and well-being of the child, with both short- and long-term consequences, including greater rates of psychological or behavioral difficulties among children of patients with PPD.5 Postpartum depression can also have negative effects on the patient’s partner, with 24% to 50% of partners experiencing depression.6 Current PPD management strategies include the use of psychotherapy and pharmacologic interventions for major depressive disorder that may take up to 4 to 6 weeks for some patients, and may not achieve remission for all patients.7-9
Brexanolone injection is a first-in-class medication with a novel mechanism of action. In clinical studies, it achieved rapid (by Hour 60) and sustained (through Day 30) reductions in depressive symptoms and could provide a meaningful new treatment option for adult women with PPD.10,11
How it works
Animal and human studies have established the re
Brexanolone is a neuroactive steroid that is chemically identical to endogenous allopregnanolone produced in the CNS. Brexanolone potentiates GABA-mediated currents from recombinant human GABAARs in mammalian cells expressing α1β2γ2 receptor subunits, α4β3δ receptor subunits, and α6β3δ receptor subunits.3 Positive allosteric modulation of both synaptic and extrasynaptic GABAARs differentiates brexanolone from other GABAAR modulators, such as benzodiazepines.10,11
Brexanolone’s mechanism of action in the treatment of PPD is not fully understood, but it is thought to be related to GABAAR PAM activity.3
Supporting evidence
The FDA approval of brexanolone injection was based on the efficacy demonstrated in 2 Phase III multicenter, randomized, double-blind, placebo-controlled studies in adult women (age 18 to 45) with PPD (defined by DSM-IV criteria for a major depressive episode, with onset of symptoms in the third trimester or within 4 weeks of delivery). Exclusion criteria included the presence of bipolar disorder or psychosis. In these studies, 60-hour continuous IV infusions of brexanolone or placebo were given, followed by 4 weeks of observation. Study 1 (202B) enrolled patients with severe PPD (Hamilton Rating Scale for Depression [HAM-D] total score ≥26), and Study 2 (202C) enrolled patients with moderate PPD (HAM-D score 20 to 25). A titration to the recommended target dosage of 90 μg/kg/hour was evaluated in both studies. BRX90 patients received 30 μg/kg/hour for 4 hours, 60 μg/kg/hour for 20 hours, 90 μg/kg/hour for 28 hours, followed by a taper to 60 μg/kg/hour for 4 hours and then 30 μg/kg/hour for 4 hours. The primary endpoint in both studies was the mean change from baseline in depressive symptoms as measured by HAM-D total score at the end of the 60-hour infusion. A pre-specified secondary efficacy endpoint was the mean change from baseline in HAM-D total score at Day 30.
Continue to: Efficacy
Efficacy. In both placebo-controlled studies, titration to a target dose of brexanolone 90 μg/kg/hour was superior to placebo in improvement of depressive symptoms (Table 33).
Pharmacological profile
Brexanolone exposure-response relationships and the time course of pharmacodynamic response are unknown.3
Adverse reactions. Safety was evaluated from all patients receiving brexanolone injection, regardless of dosing regimen (N = 140, including patients from a Phase IIb study, 202A).3,11
The most common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were sedation/somnolence, dry mouth, loss of consciousness, and flushing/hot flush.3 The incidence of patients discontinuing due to any adverse reaction was 2% for brexanolone vs 1% for placebo.3
Sedation, somnolence, and loss of consciousness. In clinical studies, brexanolone caused sedation and somnolence that required dose interruption or reduction in some patients during the infusion (5% of brexanolone-treated patients compared with 0% of placebo-treated patients).3 Some patients were also reported to have loss of consciousness or altered state of consciousness during the brexanolone infusion (4% of patients treated with brexanolone compared with 0% of patients treated with placebo).3 All patients with loss of or altered state of consciousness recovered fully 15 to 60 minutes after dose interruption.3 There was no clear association between loss or alteration of consciousness and pattern or timing of dose, and not all patients who experienced a loss or alteration of consciousness reported sedation or somnolence before the episode.
Continue to: Suicidality
Suicidality. The risk of developing suicidal thoughts and behaviors with brexanolone is unknown, due to the relatively low number of exposures to brexanolone injection during clinical development and a mechanism of action distinct from that of existing antidepressant medications.3
Pharmacokinetics
In clinical trials, brexanolone exhibited dose-proportional pharmacokinetics, and the terminal half-life is approximately 9 hours (Table 43). Brexanolone is metabolized by non-cytochrome P450 (CYP)-based pathways, including keto-reduction, glucuronidation, and sulfation.3 No clinically significant differences in the pharmacokinetics of brexanolone were observed based on renal or hepatic impairment, and no studies were conducted to evaluate the effects of other drugs on brexanolone.3
Lactation. A population pharmacokinetics model constructed from studies in the clinical development program calculated the maximum relative infant dose for brexanolone during infusion as 1.3%.3 Given the low oral bioavailability of brexanolone (<5%) in adults, the potential for breastfed infant exposure is considered low.3
Clinical considerations
Risk Evaluation and Mitigation Strategies (REMS) requirements. Brexanolone injection is a Schedule IV controlled substance. It has a “black-box” warning regarding excessive sedation and sudden loss of consciousness, which has been taken into account within the REMS drug safety program. Health care facilities and pharmacies must enroll in the REMS program and ensure that brexanolone is administered only to patients who are enrolled in the REMS program. Staff must be trained on the processes and procedures to administer brexanolone, and the facility must have a fall precautions protocol in place and be equipped with a programmable peristaltic IV infusion pump and continuous pulse oximetry with alarms.3
Monitoring. A REMS-trained clinician must be available continuously on-site to oversee each patient for the duration of the continuous IV infusion, which lasts 60 hours (2.5 days) and should be initiated early enough in the day to allow for recognition of excessive sedation. Patients must be monitored for hypoxia using continuous pulse oximetry equipped with an alarm and should also be assessed for excessive sedation every 2 hours during planned, non-sleep periods. If excessive sedation occurs, the infusion should be stopped until symptoms resolve, after which the infusion may be resumed at the same or a lower dose as clinically appropriate. In case of overdosage, the infusion should be stopped immediately and supportive measures initiated as necessary. Patients must not be the primary caregiver of dependents, and must be accompanied during interactions with their child(ren).
Continue to: Contraindications
Contraindications. There are no contraindications for the use of brexanolone in adults with PPD.
End-stage renal disease (ESRD). Avoid using brexanolone in patients with ESRD because of the potential accumulation of the solubilizing agent, betadex sulfobutyl ether sodium.
Pregnancy. Brexanolone has not been studied in pregnant patients. Pregnant women and women of reproductive age should be informed of the potential risk to a fetus based on data from other drugs that enhance GABAergic inhibition.
Breastfeeding. There are no data on the effects of brexanolone on a breastfed infant. Breastfeeding should be a discussion of risk and benefit between the patient and her doctor. The developmental and health benefits of breastfeeding should be considered, along with the mother’s clinical need for brexanolone and any potential adverse effects on the breastfed child from brexanolone or from the underlying maternal condition. However, based on the low relative infant dose (<2%) and the low oral bioavailability in adults, the risk to breastfed infants is thought to be low.16
Potential for abuse. Brexanolone injection is a Schedule IV controlled substance. Although it was not possible to assess physical dependency in the registrational trials due to dose tapering at the end of treatment, clinicians should advise patients about the theoretical possibility for brexanolone to be abused or lead to dependence based on other medications with similar primary pharmacology.
Continue to: Concomitant medications
Concomitant medications. Caution patients that taking opioids or other CNS depressants, such as benzodiazepines, in combination with brexanolone may increase the severity of sedative effects.
Suicidal thoughts and behaviors. Advise patients and caregivers to look for the emergence of suicidal thoughts and behavior and instruct them to report such symptoms to their clinician. Consider changing the therapeutic regimen, including discontinuing brexanolone, in patients whose depression becomes worse or who experience emergent suicidal thoughts and behaviors.
Why Rx?
Postpartum depression is a common and often devastating medical complication of childbirth that can result in adverse outcomes for the patient, baby, and family when left undertreated or untreated. There is a great need to identify and treat women who develop PPD. Rapid and sustained resolution of symptoms in women who experience PPD should be the goal of treatment, and consequently, brexanolone injection presents an important new tool in available treatment options for PPD.
Bottom Line
Brexanolone injection is a neuroactive steroid gamma-aminobutyric acid (GABA) A receptor positive allosteric modulator that’s been FDA-approved for the treatment of postpartum depression (PPD). It is administered as a continuous IV infusion over 60 hours. The rapid and sustained improvement of PPD observed in clinical trials with brexanolone injection may support a new treatment paradigm for women with PPD.
Postpartum depression (PPD) is one of the most prevalent complications associated with pregnancy and childbirth in the United States, affecting more than 400,000 women annually.1 Postpartum depression is most commonly treated with psychotherapy and antidepressants approved for the treatment of major depressive disorder. Until recently, there was no pharmacologic therapy approved by the FDA specifically for the treatment of PPD. Considering the adverse outcomes associated with untreated or inadequately treated PPD, and the limitations of existing therapies, there is a significant unmet need for pharmacologic treatment options for PPD.2 To help address this need, the FDA recently approved brexanolone injection (brand name: ZULRESSO™) (Table 13) as a first-in-class therapy for the treatment of adults with PPD.3
Clinical implications
Postpartum depression can result in adverse outcomes for the patient, baby, and family when under- or untreated, and the need for rapid resolution of symptoms cannot be overstated.2 Suicide is strongly associated with depression and is a leading cause of pregnancy-related deaths.4 Additionally, PPD can impact the health, safety, and well-being of the child, with both short- and long-term consequences, including greater rates of psychological or behavioral difficulties among children of patients with PPD.5 Postpartum depression can also have negative effects on the patient’s partner, with 24% to 50% of partners experiencing depression.6 Current PPD management strategies include the use of psychotherapy and pharmacologic interventions for major depressive disorder that may take up to 4 to 6 weeks for some patients, and may not achieve remission for all patients.7-9
Brexanolone injection is a first-in-class medication with a novel mechanism of action. In clinical studies, it achieved rapid (by Hour 60) and sustained (through Day 30) reductions in depressive symptoms and could provide a meaningful new treatment option for adult women with PPD.10,11
How it works
Animal and human studies have established the re
Brexanolone is a neuroactive steroid that is chemically identical to endogenous allopregnanolone produced in the CNS. Brexanolone potentiates GABA-mediated currents from recombinant human GABAARs in mammalian cells expressing α1β2γ2 receptor subunits, α4β3δ receptor subunits, and α6β3δ receptor subunits.3 Positive allosteric modulation of both synaptic and extrasynaptic GABAARs differentiates brexanolone from other GABAAR modulators, such as benzodiazepines.10,11
Brexanolone’s mechanism of action in the treatment of PPD is not fully understood, but it is thought to be related to GABAAR PAM activity.3
Supporting evidence
The FDA approval of brexanolone injection was based on the efficacy demonstrated in 2 Phase III multicenter, randomized, double-blind, placebo-controlled studies in adult women (age 18 to 45) with PPD (defined by DSM-IV criteria for a major depressive episode, with onset of symptoms in the third trimester or within 4 weeks of delivery). Exclusion criteria included the presence of bipolar disorder or psychosis. In these studies, 60-hour continuous IV infusions of brexanolone or placebo were given, followed by 4 weeks of observation. Study 1 (202B) enrolled patients with severe PPD (Hamilton Rating Scale for Depression [HAM-D] total score ≥26), and Study 2 (202C) enrolled patients with moderate PPD (HAM-D score 20 to 25). A titration to the recommended target dosage of 90 μg/kg/hour was evaluated in both studies. BRX90 patients received 30 μg/kg/hour for 4 hours, 60 μg/kg/hour for 20 hours, 90 μg/kg/hour for 28 hours, followed by a taper to 60 μg/kg/hour for 4 hours and then 30 μg/kg/hour for 4 hours. The primary endpoint in both studies was the mean change from baseline in depressive symptoms as measured by HAM-D total score at the end of the 60-hour infusion. A pre-specified secondary efficacy endpoint was the mean change from baseline in HAM-D total score at Day 30.
Continue to: Efficacy
Efficacy. In both placebo-controlled studies, titration to a target dose of brexanolone 90 μg/kg/hour was superior to placebo in improvement of depressive symptoms (Table 33).
Pharmacological profile
Brexanolone exposure-response relationships and the time course of pharmacodynamic response are unknown.3
Adverse reactions. Safety was evaluated from all patients receiving brexanolone injection, regardless of dosing regimen (N = 140, including patients from a Phase IIb study, 202A).3,11
The most common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were sedation/somnolence, dry mouth, loss of consciousness, and flushing/hot flush.3 The incidence of patients discontinuing due to any adverse reaction was 2% for brexanolone vs 1% for placebo.3
Sedation, somnolence, and loss of consciousness. In clinical studies, brexanolone caused sedation and somnolence that required dose interruption or reduction in some patients during the infusion (5% of brexanolone-treated patients compared with 0% of placebo-treated patients).3 Some patients were also reported to have loss of consciousness or altered state of consciousness during the brexanolone infusion (4% of patients treated with brexanolone compared with 0% of patients treated with placebo).3 All patients with loss of or altered state of consciousness recovered fully 15 to 60 minutes after dose interruption.3 There was no clear association between loss or alteration of consciousness and pattern or timing of dose, and not all patients who experienced a loss or alteration of consciousness reported sedation or somnolence before the episode.
Continue to: Suicidality
Suicidality. The risk of developing suicidal thoughts and behaviors with brexanolone is unknown, due to the relatively low number of exposures to brexanolone injection during clinical development and a mechanism of action distinct from that of existing antidepressant medications.3
Pharmacokinetics
In clinical trials, brexanolone exhibited dose-proportional pharmacokinetics, and the terminal half-life is approximately 9 hours (Table 43). Brexanolone is metabolized by non-cytochrome P450 (CYP)-based pathways, including keto-reduction, glucuronidation, and sulfation.3 No clinically significant differences in the pharmacokinetics of brexanolone were observed based on renal or hepatic impairment, and no studies were conducted to evaluate the effects of other drugs on brexanolone.3
Lactation. A population pharmacokinetics model constructed from studies in the clinical development program calculated the maximum relative infant dose for brexanolone during infusion as 1.3%.3 Given the low oral bioavailability of brexanolone (<5%) in adults, the potential for breastfed infant exposure is considered low.3
Clinical considerations
Risk Evaluation and Mitigation Strategies (REMS) requirements. Brexanolone injection is a Schedule IV controlled substance. It has a “black-box” warning regarding excessive sedation and sudden loss of consciousness, which has been taken into account within the REMS drug safety program. Health care facilities and pharmacies must enroll in the REMS program and ensure that brexanolone is administered only to patients who are enrolled in the REMS program. Staff must be trained on the processes and procedures to administer brexanolone, and the facility must have a fall precautions protocol in place and be equipped with a programmable peristaltic IV infusion pump and continuous pulse oximetry with alarms.3
Monitoring. A REMS-trained clinician must be available continuously on-site to oversee each patient for the duration of the continuous IV infusion, which lasts 60 hours (2.5 days) and should be initiated early enough in the day to allow for recognition of excessive sedation. Patients must be monitored for hypoxia using continuous pulse oximetry equipped with an alarm and should also be assessed for excessive sedation every 2 hours during planned, non-sleep periods. If excessive sedation occurs, the infusion should be stopped until symptoms resolve, after which the infusion may be resumed at the same or a lower dose as clinically appropriate. In case of overdosage, the infusion should be stopped immediately and supportive measures initiated as necessary. Patients must not be the primary caregiver of dependents, and must be accompanied during interactions with their child(ren).
Continue to: Contraindications
Contraindications. There are no contraindications for the use of brexanolone in adults with PPD.
End-stage renal disease (ESRD). Avoid using brexanolone in patients with ESRD because of the potential accumulation of the solubilizing agent, betadex sulfobutyl ether sodium.
Pregnancy. Brexanolone has not been studied in pregnant patients. Pregnant women and women of reproductive age should be informed of the potential risk to a fetus based on data from other drugs that enhance GABAergic inhibition.
Breastfeeding. There are no data on the effects of brexanolone on a breastfed infant. Breastfeeding should be a discussion of risk and benefit between the patient and her doctor. The developmental and health benefits of breastfeeding should be considered, along with the mother’s clinical need for brexanolone and any potential adverse effects on the breastfed child from brexanolone or from the underlying maternal condition. However, based on the low relative infant dose (<2%) and the low oral bioavailability in adults, the risk to breastfed infants is thought to be low.16
Potential for abuse. Brexanolone injection is a Schedule IV controlled substance. Although it was not possible to assess physical dependency in the registrational trials due to dose tapering at the end of treatment, clinicians should advise patients about the theoretical possibility for brexanolone to be abused or lead to dependence based on other medications with similar primary pharmacology.
Continue to: Concomitant medications
Concomitant medications. Caution patients that taking opioids or other CNS depressants, such as benzodiazepines, in combination with brexanolone may increase the severity of sedative effects.
Suicidal thoughts and behaviors. Advise patients and caregivers to look for the emergence of suicidal thoughts and behavior and instruct them to report such symptoms to their clinician. Consider changing the therapeutic regimen, including discontinuing brexanolone, in patients whose depression becomes worse or who experience emergent suicidal thoughts and behaviors.
Why Rx?
Postpartum depression is a common and often devastating medical complication of childbirth that can result in adverse outcomes for the patient, baby, and family when left undertreated or untreated. There is a great need to identify and treat women who develop PPD. Rapid and sustained resolution of symptoms in women who experience PPD should be the goal of treatment, and consequently, brexanolone injection presents an important new tool in available treatment options for PPD.
Bottom Line
Brexanolone injection is a neuroactive steroid gamma-aminobutyric acid (GABA) A receptor positive allosteric modulator that’s been FDA-approved for the treatment of postpartum depression (PPD). It is administered as a continuous IV infusion over 60 hours. The rapid and sustained improvement of PPD observed in clinical trials with brexanolone injection may support a new treatment paradigm for women with PPD.
1. Ko JY, Rockhill KM, Tong VT, et al. Trends in postpartum depressive symptoms - 27 states, 2004, 2008, and 2012. MMWR Morb Mortal Wkly Rep. 2017;66(6):153-158.
2. Frieder A, Fersh M, Hainline R, et al. Pharmacotherapy of postpartum depression: current approaches and novel drug development. CNS Drugs. 2019;33(3):265-282.
3. Brexanolone injection [package insert]. Cambridge, MA: Sage Therapeutics, Inc.; 2019.
4. Bodnar-Deren S, Klipstein K, Fersh M, et al. Suicidal ideation during the postpartum period. J Womens Health (Larchmt). 2016;25(12):1219-1224.
5. Netsi E, Pearson RM, Murray L, et al. Association of persistent and severe postnatal depression with child outcomes. JAMA Psychiatry. 2018;75(3):247-253.
6. Goodman JH. Paternal postpartum depression, its relationship to maternal postpartum depression, and implications for family health. J Adv Nurs. 2004;45(1):26-35.
7. Gelenberg AJ, Freeman MP, Markowitz JC, et al; American Psychiatric Association Work Group on Major Depressive Disorder. Practice guidelines for the treatment of patients with major depressive disorder. 3rd ed. Washington, DC: American Psychiatric Association; 2010.
8. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905-1917.
9. Molyneaux E, Telesia LA, Henshaw C, et al. Antidepressants for preventing postnatal depression. Cochrane Database Syst Rev. 2018;4:CD004363.
10. Kanes S, Colquhoun H, Gunduz-Bruce H, et al. Brexanolone (SAGE-547 injection) in post-partum depression: a randomised controlled trial. Lancet. 2017;390(10093):480-489.
11. Meltzer-Brody S, Colquhoun H, Riesenberg R, et al. Brexanolone injection in post-partum depression: two multicentre, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet. 2018;392(10152):1058-1070.
12. Melon LC, Hooper A, Yang X, et al. Inability to suppress the stress-induced activation of the HPA axis during the peripartum period engenders deficits in postpartum behaviors in mice. Psychoneuroendocrinology. 2018;90:182-193.
13. Deligiannidis KM, Fales CL, Kroll-Desrosiers AR, et al. Resting-state functional connectivity, cortical GABA, and neuroactive steroids in peripartum and peripartum depressed women: a functional magnetic resonance imaging and spectroscopy study. Neuropsychopharmacology. 2019;44(3):546-554.
14. Licheri V, Talani G, Gorule AA, et al. Plasticity of GABAA receptors during pregnancy and postpartum period: from gene to function. Neural Plast. 2015;2015:170435. doi: 10.1155/2015/170435.
15. Luisi S, Petraglia F, Benedetto C, et al. Serum allopregnanolone levels in pregnant women: changes during pregnancy, at delivery, and in hypertensive patients. J Clin Endocrinol Metab. 2000;85(7):2429-2433.
16. Hoffmann E, Wald J, Dray D, et al. Brexanolone injection administration to lactating women: breast milk allopregnanolone levels [30J]. Obstetrics & Gynecology. 2019;133:115S.
1. Ko JY, Rockhill KM, Tong VT, et al. Trends in postpartum depressive symptoms - 27 states, 2004, 2008, and 2012. MMWR Morb Mortal Wkly Rep. 2017;66(6):153-158.
2. Frieder A, Fersh M, Hainline R, et al. Pharmacotherapy of postpartum depression: current approaches and novel drug development. CNS Drugs. 2019;33(3):265-282.
3. Brexanolone injection [package insert]. Cambridge, MA: Sage Therapeutics, Inc.; 2019.
4. Bodnar-Deren S, Klipstein K, Fersh M, et al. Suicidal ideation during the postpartum period. J Womens Health (Larchmt). 2016;25(12):1219-1224.
5. Netsi E, Pearson RM, Murray L, et al. Association of persistent and severe postnatal depression with child outcomes. JAMA Psychiatry. 2018;75(3):247-253.
6. Goodman JH. Paternal postpartum depression, its relationship to maternal postpartum depression, and implications for family health. J Adv Nurs. 2004;45(1):26-35.
7. Gelenberg AJ, Freeman MP, Markowitz JC, et al; American Psychiatric Association Work Group on Major Depressive Disorder. Practice guidelines for the treatment of patients with major depressive disorder. 3rd ed. Washington, DC: American Psychiatric Association; 2010.
8. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905-1917.
9. Molyneaux E, Telesia LA, Henshaw C, et al. Antidepressants for preventing postnatal depression. Cochrane Database Syst Rev. 2018;4:CD004363.
10. Kanes S, Colquhoun H, Gunduz-Bruce H, et al. Brexanolone (SAGE-547 injection) in post-partum depression: a randomised controlled trial. Lancet. 2017;390(10093):480-489.
11. Meltzer-Brody S, Colquhoun H, Riesenberg R, et al. Brexanolone injection in post-partum depression: two multicentre, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet. 2018;392(10152):1058-1070.
12. Melon LC, Hooper A, Yang X, et al. Inability to suppress the stress-induced activation of the HPA axis during the peripartum period engenders deficits in postpartum behaviors in mice. Psychoneuroendocrinology. 2018;90:182-193.
13. Deligiannidis KM, Fales CL, Kroll-Desrosiers AR, et al. Resting-state functional connectivity, cortical GABA, and neuroactive steroids in peripartum and peripartum depressed women: a functional magnetic resonance imaging and spectroscopy study. Neuropsychopharmacology. 2019;44(3):546-554.
14. Licheri V, Talani G, Gorule AA, et al. Plasticity of GABAA receptors during pregnancy and postpartum period: from gene to function. Neural Plast. 2015;2015:170435. doi: 10.1155/2015/170435.
15. Luisi S, Petraglia F, Benedetto C, et al. Serum allopregnanolone levels in pregnant women: changes during pregnancy, at delivery, and in hypertensive patients. J Clin Endocrinol Metab. 2000;85(7):2429-2433.
16. Hoffmann E, Wald J, Dray D, et al. Brexanolone injection administration to lactating women: breast milk allopregnanolone levels [30J]. Obstetrics & Gynecology. 2019;133:115S.

Intranasal esketamine
Treatment-resistant depression (TRD) is a common clinical struggle that practicing clinicians address on a daily basis. Major depressive disorder affects nearly 1 in 5 Americans at some point in their life and, by definition, impairs social and occupational functioning. Historic treatments have focused on the monoamine theories of depression—modulating the monoamines serotonin, norepinephrine, and/or dopamine. Limitations of currently available antidepressants include delayed onset of effect and low remission rates. To further complicate the matter, numerous studies have shown that with each subsequent antidepressant trial, patients have a decreasing likelihood of responding to subsequent antidepressant treatment options. For example, in the classic STAR*D trial, by the time a patient had not responded to the first 2 antidepressant options, the chance that they would respond to a third or fourth antidepressant had decreased to approximately 15% per antidepressant treatment course.1
To address the need for new treatments for patients with TRD, on March 5, 2019 the FDA-approved intranasal

How it works
Modern research has looked beyond the monoamine system to explore the neuro-modulatory effects of glutamate and gamma-aminobutyric acid (GABA).3 The yin and yang of glutamate and GABA revolves around neural excitation vs neural inhibition at a local synaptic level. The primary effects of the glutamate and GABA systems (Table 2) can be broken down into several key areas of understanding.

Glutamate modulates ionotropic N-methyl-
Esketamine, the S-enantiomer of ketamine, has a higher affinity for the NMDA receptor than the R-enantiomer and has been developed as an intranasal adjunctive treatment for TRD. Esketamine blocks NMDA receptors on GABA interneurons. This allows for increased pulsatile release of glutamate into the synapse. Intrasynaptic glutamate then stimulates postsynaptic AMPA receptors. Glutamate stimulation of postsynaptic AMPA receptors results in an intracellular cascade that activates the enzymes tropomyosin receptor kinase B (TrkB) and mammalian target of rapamycin (mTOR). TrkB stimulation results in increased production and release of BDNF. mTor stimulation increases neuronal membrane protein formation with subsequent increased neural plasticity. Taken together, preclinical models show that esketamine’s inhibition of the NMDA receptor on the GABA interneuron results in a cascade of increased BDNF release and synaptogenesis with increased neuroplasticity (Table 3).

Clinical implications
Treatment-resistant depression affects nearly one-third of patients currently receiving standard antidepressant treatment. Major depressive disorder is currently the second leading cause of disability for working adults within the United States and one of the largest causes of disability worldwide. The esketamine nasal spray could be beneficial for patients who have experienced TRD with standard monoamine antidepressants.
Supporting evidence
Clinical trials examining intranasal esketamine include both short- and long-term studies of patients with TRD.
Continue to: Esketamine was evaluated...
Esketamine was evaluated in a randomized, placebo-controlled, double-blind, multicenter, short-term (4-week) phase III study in adult patients age 18 to 65 with TRD (they had not responded to at least 2 different antidepressants of adequate dose and duration).4 After discontinuing prior antidepressant treatments, all patients were started on a newly initiated antidepressant and were also randomized to concomitant intranasal esketamine or intranasal placebo as follows:
- 114 patients were randomized to the intranasal esketamine plus newly initiated oral antidepressant arm
- 109 patients were randomized to the placebo nasal spray plus newly initiated oral antidepressant arm
- The mean baseline Montgomery-Åsberg Depression Rating Scale (MADRS) score for each group was 37 (ie, moderately to severely depressed).
Newly started antidepressants included esc

A long-term, double-blind multicenter maintenance-of-effect trial examined adults age 18 to 65 with TRD.5-6 Patients in this study were responders in 1 of 2 short-term studies or in an open-label direct enrollment study. Stable remission was defined as a MADRS total score <12 for at least 3 of the last 4 weeks of the study, and stable response was defined as a MADRS reduction of >50% but not in remission. After 16 weeks of intranasal esketamine plus an oral antidepressant, stable remitters and stable responders were then randomized separately to continue intranasal esketamine or switch to placebo nasal spray, with both groups continuing on their concomitant oral antidepressant. The primary study endpoint was time to relapse. Relapse was defined as a MADRS total score >22 for more than 2 consecutive weeks, hospitalization for worsening of depression, or any other clinically relevant event. The median age was 48, 66% were female, 90% were White and 4% were black. Patients in stable response or stable remission experienced a significantly longer time to relapse compared with patients who continued their oral antidepressant but were switched to placebo intranasal spray. In this remission response study, patients could receive intranasal treatment weekly or bi-weekly based on symptom severity (Figure 22).

Impact on driving. Two studies examined the impact of esketamine on driving performance. One examined adults with major depressive disorder and the other examined healthy participants. The effects of a single 84-mg dose of esketamine nasal spray on a patient’s ability to drive was assessed in 23 healthy adults. In this study, mirt
A second study evaluated the effects of repeated esketamine administration on driving performance in 25 adults with major depressive disorder. In this study, an ethanol-containing beverage was used as an active control. After administration of a single 84-mg dose of intranasal esketamine, driving performance was the same as a placebo at 18 hours. In the multiple dose phase, standard driving performance was similar for esketamine nasal spray and placebo at 6 hours postdose on Days 11, 18, and 25.
Continue to: Pharmacologic profile
Pharmacologic profile
Adverse events. The most common adverse events in patients treated with esketamine nasal spray were dissociation (41%), dizziness (29%), nausea (28%), sedation (23%), and vertigo (23%).2 The majority of these effects were short-term and resolved during the 2-hour observation period.
In addition to spontaneously reported events, sedation and dissociation were further monitored with specific scales. Sedation was measured with the Modified Observer’s Alertness and Sedation Scale. Using this scale, 50% of patients receiving 56 mg and 61% of patients receiving 84 mg of esketamine met criteria for sedation.
Similarly, dissociation/perceptional changes were measured with spontaneously reported events and also with the Clinician Administered Dissociative State Scale. On this scale, 61% of patients receiving the 56-mg dose, and 69% of patients receiving the 84-mg dose met criteria for dissociation/perceptional changes after dose administration.
Increases in blod pressure. Esketamine intranasal spray was associated with a 7 to 9 mm Hg increase in systolic blood pressure and a 4 to 6 mm Hg increase in diastolic blood pressure, both of which peaked 40 minutes post-dose.
Nausea and vomiting. Intranasal esketamine was associated with a 27% rate of nausea at 56 mg, and 32% at 84 mg, with a 6% rate of vomiting at 56 mg and 12% at 84 mg.
Continue to: Pharmacokinetics
Pharmacokinetics
Esketamine exposure increases from 28 to 84 mg in a fairly dose-proportional range. No accumulation of esketamine was observed in the plasma following twice-weekly administration. Bioavailability is approximately 48% following nasal administration. The Tmax for esketamine plasma concentration is 20 to 40 minutes after the last nasal spray. Protein binding of esketamine is approximately 43% to 45%. The brain-to-plasma ratio of noresketamine is 4 to 6 times lower than that of esketamine. The half-life of esketamine ranged from 7 to 12 hours. The mean half-life of nore
Potential drug interactions
Central nervous system depressants. Concomitant use of esketamine and other CNS depressants (ie, benzodiazepines, opioids, alcohol) may increase sedation. Patients receiving esketamine with concomitant use of other CNS depressants should be closely monitored for sedation.
Psychostimulants. Concomitant use of esketamine and psychostimulants (ie, amphetamines, methylphenidates, moda
Monoamine oxidase inhibitors. Concomitant use of esketamine with monoamine oxidase inhibitors may increase blood pressure. Closely monitor blood pressure with concomitant use of esketamine and monoamine oxidase inhibitors.
Use in special populations. Because of concerns of increased sedation, intranasal esketamine should be administered cautiously in patients receiving other CNS depressants, such as benzodiazepines. In patients with psychosis or a prior history of psychosis, esketamine should be used with increased caution and the risk/benefit ratio should be carefully considered.
Continue to: Because of potential teratogenicity...
Because of potential teratogenicity, esketamine is not recommended in women who are pregnant, may become pregnant, or who are currently nursing.
Intranasal esketamine was examined in a phase III trial of 194 patients age ≥65. At the end of 4 weeks, there was no statistically significant difference in groups on the MADRS, the primary efficacy endpoint. There were no overall differences in the safety profile in patients >65 years compared with younger patients; however, the mean esketamine Cmax and area under the curve were higher in older patients compared with younger adults. The mean esketamine half-life was longer in patients with moderate hepatic impairment.
Abuse liability
Esketamine is a CIII controlled substance and concerns about abuse, misuse, and diversion have been taken into account within the REMS drug safety program.2 Patients with a prior history of substance abuse or misuse should be considered with regard to the risk/benefit ratio.
The REMS drug safety program
Due to the nature of its usually transient adverse effects, including sedation, dissociation, hypertension, and nausea, intranasal esketamine will be administered through a REMS drug safety program at certified REMS treatment centers. Certified REMS treatment centers will receive training on how to safely and effectively counsel and monitor patients. Prior to treatment, patients will receive blood pressure monitoring and anticipated adverse effects will be discussed. Patients will be instructed to not eat solid food for 2 hours pre-dose and to not drink anything for 30 minutes prior.
A treatment session consists of nasal administration and a minimum 2-hour post-administration observation period. Blood pressure must be assessed prior to administration and if elevated, (ie, systolic blood pressure >140 mm Hg, diastolic >90 mm Hg), clinicians should consider the risk of short-term increases in blood pressure that may occur. Do not administer if increases in blood pressure or intracranial pressure pose a serious risk.
Continue to: After each intranasal...
After each intranasal administration the patient will be observed for 5 minutes before the second nasal inhaler is utilized and for another 5 minutes when the patient is receiving 84 mg (ie, each inhaler equals 28 mg). After administering, blood pressure should be reassessed at approximately 40 minutes, which corresponds to the Cmax of intranasal esketamine, and periodically thereafter as warranted.
The patient will then be monitored in a quiet environment for a minimum of 2 hours to make sure that dissociative phenomenon, sedation, and hypertensive reactions have normalized prior to discharge from a certified REMS treatment center.
Dosing and administration
Each intranasal device is primed for 2 infusions (1 in each nostril) for a total dose of 28 mg of esketamine. Combinations of devices can be used to adjust the dose as appropriate for individual patients. The recommended starting dose is 56 mg (ie, 2 devices, with a 5-minute gap between devices). The dose can be increased to 84 mg (ie, 3 intranasal devices spaced at 5-minute intervals) by the second dose based on clinical judgment.
The patient will be instructed to recline the head to a 45° angle, clear his or her nostrils prior to the first treatment, and then self-administer a dose to each nostril while holding the reciprocal nostril closed and inhaling. This process is then repeated every 5 minutes for each subsequent device, with a maximum total dose of 3 devices, or 84 mg (Figure 32). The patient will then be monitored for blood pressure, heart rate, and signs of psychologic or physiologic changes for the next 2 hours. Patients may not drive a car or operate any type of motor equipment until the following day after receiving a normal night’s sleep. Patients will be released from the REMS treatment center after 2 hours if both psychological and physical adverse effects have normalized.

Missed treatment sessions. If a patient misses a treatment session and there is worsening of depressive symptoms, consider returning the patient to the previous dosing schedule (ie, every 2 weeks to once weekly, or weekly to twice weekly).
Continue to: Contraindications for...
Contraindications for intranasal esketamine include:
- aneurysmal vascular disease, including thoracic and abdominal aortic, intracranial, and peripheral arterial vessels, or arterial venous malformations
- history of intracerebral hemorrhage
- hypersensitivity to esketamine, ketamine, or any of the excipients.
Clinical considerations
Intranasal esketamine represents a unique delivery system for the first glutamatergic treatment approved for patients with TRD.
Why Rx? Treatment-resistant depression is found in nearly 1 out of 3 patients with currently available monoaminergic antidepressant treatment options. Patients with TRD are at increased risk of physical and psychological impairment, subsequent worsening of their condition, and social and occupational disability.
Bottom Line
Intranasal esketamine is the first glutamatergic treatment option FDA-approved for patients with treatment-resistant depression who have not responded to standard antidepressant treatment options. In short-term trials, intranasal esketamine significantly improved depressive symptoms as quickly as 24 hours after treatment, with significant improvement maintained through 4 weeks of ongoing administration. In addition, intranasal esketamine was shown to significantly decrease time to relapse for patients who had achieved stable remission or stable response.
Related Resource
- Sullivan MG. FDA approves intranasal esketamine for refractory major depressive disorder. Clinical Psychiatry News. https://www.mdedge.com/psychiatry/article/195712/depression/fda-approves-intranasal-esketamine-refractory-major-depressive. Published March 5, 2019.
Drug Brand Names
Armodafinil • Nuvigil
Duloxetine • Cymbalta
Escitalopram • Lexapro
Esketamine • Spravato
Mirtazapine • Remeron
Modafinil • Provigil
Sertraline • Zoloft
Venlafaxine • Effexor
1. Rush AG, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR D Report. Am J Psychiatry. 2006;163(11):1905-1917.
2. Spravato [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2019.
3. Duman RS, Aghajanian GK, Sanacora G, et al. Synaptic plasticity and depression: new insights from stress and rapid-acting anti-depression. Nat Med. 2016;22(3):238-249.
4. Daly EJ, Singh JB, Fedgchin M, et al. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2018;75(2):139-148.
5. Daly EJ, Trivedi M, Janik A, et al. A randomized withdrawal, double-blind, multicenter study of esketamine nasal spray plus an oral antidepressant for relapse prevention in treatment-resistant depression. Poster presented at the 2018 American Society of Clinical Psychopharmacology Annual Meeting; May 2018; Miami, Florida.
6. Wajs E, Aluisio L, Morrison R, et al. Long-term safety of esketamine nasal spray plus oral antidepressants in patients with treatment-resistant depression: phase III open-label safety and efficacy study. Poster presented at the 2018 American Society of Clinical Psychopharmacology Annual Meeting; May 2018; Miami, Florida.
Treatment-resistant depression (TRD) is a common clinical struggle that practicing clinicians address on a daily basis. Major depressive disorder affects nearly 1 in 5 Americans at some point in their life and, by definition, impairs social and occupational functioning. Historic treatments have focused on the monoamine theories of depression—modulating the monoamines serotonin, norepinephrine, and/or dopamine. Limitations of currently available antidepressants include delayed onset of effect and low remission rates. To further complicate the matter, numerous studies have shown that with each subsequent antidepressant trial, patients have a decreasing likelihood of responding to subsequent antidepressant treatment options. For example, in the classic STAR*D trial, by the time a patient had not responded to the first 2 antidepressant options, the chance that they would respond to a third or fourth antidepressant had decreased to approximately 15% per antidepressant treatment course.1
To address the need for new treatments for patients with TRD, on March 5, 2019 the FDA-approved intranasal
How it works
Modern research has looked beyond the monoamine system to explore the neuro-modulatory effects of glutamate and gamma-aminobutyric acid (GABA).3 The yin and yang of glutamate and GABA revolves around neural excitation vs neural inhibition at a local synaptic level. The primary effects of the glutamate and GABA systems (Table 2) can be broken down into several key areas of understanding.
Glutamate modulates ionotropic N-methyl-
Esketamine, the S-enantiomer of ketamine, has a higher affinity for the NMDA receptor than the R-enantiomer and has been developed as an intranasal adjunctive treatment for TRD. Esketamine blocks NMDA receptors on GABA interneurons. This allows for increased pulsatile release of glutamate into the synapse. Intrasynaptic glutamate then stimulates postsynaptic AMPA receptors. Glutamate stimulation of postsynaptic AMPA receptors results in an intracellular cascade that activates the enzymes tropomyosin receptor kinase B (TrkB) and mammalian target of rapamycin (mTOR). TrkB stimulation results in increased production and release of BDNF. mTor stimulation increases neuronal membrane protein formation with subsequent increased neural plasticity. Taken together, preclinical models show that esketamine’s inhibition of the NMDA receptor on the GABA interneuron results in a cascade of increased BDNF release and synaptogenesis with increased neuroplasticity (Table 3).
Clinical implications
Treatment-resistant depression affects nearly one-third of patients currently receiving standard antidepressant treatment. Major depressive disorder is currently the second leading cause of disability for working adults within the United States and one of the largest causes of disability worldwide. The esketamine nasal spray could be beneficial for patients who have experienced TRD with standard monoamine antidepressants.
Supporting evidence
Clinical trials examining intranasal esketamine include both short- and long-term studies of patients with TRD.
Continue to: Esketamine was evaluated...
Esketamine was evaluated in a randomized, placebo-controlled, double-blind, multicenter, short-term (4-week) phase III study in adult patients age 18 to 65 with TRD (they had not responded to at least 2 different antidepressants of adequate dose and duration).4 After discontinuing prior antidepressant treatments, all patients were started on a newly initiated antidepressant and were also randomized to concomitant intranasal esketamine or intranasal placebo as follows:
- 114 patients were randomized to the intranasal esketamine plus newly initiated oral antidepressant arm
- 109 patients were randomized to the placebo nasal spray plus newly initiated oral antidepressant arm
- The mean baseline Montgomery-Åsberg Depression Rating Scale (MADRS) score for each group was 37 (ie, moderately to severely depressed).
Newly started antidepressants included esc
A long-term, double-blind multicenter maintenance-of-effect trial examined adults age 18 to 65 with TRD.5-6 Patients in this study were responders in 1 of 2 short-term studies or in an open-label direct enrollment study. Stable remission was defined as a MADRS total score <12 for at least 3 of the last 4 weeks of the study, and stable response was defined as a MADRS reduction of >50% but not in remission. After 16 weeks of intranasal esketamine plus an oral antidepressant, stable remitters and stable responders were then randomized separately to continue intranasal esketamine or switch to placebo nasal spray, with both groups continuing on their concomitant oral antidepressant. The primary study endpoint was time to relapse. Relapse was defined as a MADRS total score >22 for more than 2 consecutive weeks, hospitalization for worsening of depression, or any other clinically relevant event. The median age was 48, 66% were female, 90% were White and 4% were black. Patients in stable response or stable remission experienced a significantly longer time to relapse compared with patients who continued their oral antidepressant but were switched to placebo intranasal spray. In this remission response study, patients could receive intranasal treatment weekly or bi-weekly based on symptom severity (Figure 22).
Impact on driving. Two studies examined the impact of esketamine on driving performance. One examined adults with major depressive disorder and the other examined healthy participants. The effects of a single 84-mg dose of esketamine nasal spray on a patient’s ability to drive was assessed in 23 healthy adults. In this study, mirt
A second study evaluated the effects of repeated esketamine administration on driving performance in 25 adults with major depressive disorder. In this study, an ethanol-containing beverage was used as an active control. After administration of a single 84-mg dose of intranasal esketamine, driving performance was the same as a placebo at 18 hours. In the multiple dose phase, standard driving performance was similar for esketamine nasal spray and placebo at 6 hours postdose on Days 11, 18, and 25.
Continue to: Pharmacologic profile
Pharmacologic profile
Adverse events. The most common adverse events in patients treated with esketamine nasal spray were dissociation (41%), dizziness (29%), nausea (28%), sedation (23%), and vertigo (23%).2 The majority of these effects were short-term and resolved during the 2-hour observation period.
In addition to spontaneously reported events, sedation and dissociation were further monitored with specific scales. Sedation was measured with the Modified Observer’s Alertness and Sedation Scale. Using this scale, 50% of patients receiving 56 mg and 61% of patients receiving 84 mg of esketamine met criteria for sedation.
Similarly, dissociation/perceptional changes were measured with spontaneously reported events and also with the Clinician Administered Dissociative State Scale. On this scale, 61% of patients receiving the 56-mg dose, and 69% of patients receiving the 84-mg dose met criteria for dissociation/perceptional changes after dose administration.
Increases in blod pressure. Esketamine intranasal spray was associated with a 7 to 9 mm Hg increase in systolic blood pressure and a 4 to 6 mm Hg increase in diastolic blood pressure, both of which peaked 40 minutes post-dose.
Nausea and vomiting. Intranasal esketamine was associated with a 27% rate of nausea at 56 mg, and 32% at 84 mg, with a 6% rate of vomiting at 56 mg and 12% at 84 mg.
Continue to: Pharmacokinetics
Pharmacokinetics
Esketamine exposure increases from 28 to 84 mg in a fairly dose-proportional range. No accumulation of esketamine was observed in the plasma following twice-weekly administration. Bioavailability is approximately 48% following nasal administration. The Tmax for esketamine plasma concentration is 20 to 40 minutes after the last nasal spray. Protein binding of esketamine is approximately 43% to 45%. The brain-to-plasma ratio of noresketamine is 4 to 6 times lower than that of esketamine. The half-life of esketamine ranged from 7 to 12 hours. The mean half-life of nore
Potential drug interactions
Central nervous system depressants. Concomitant use of esketamine and other CNS depressants (ie, benzodiazepines, opioids, alcohol) may increase sedation. Patients receiving esketamine with concomitant use of other CNS depressants should be closely monitored for sedation.
Psychostimulants. Concomitant use of esketamine and psychostimulants (ie, amphetamines, methylphenidates, moda
Monoamine oxidase inhibitors. Concomitant use of esketamine with monoamine oxidase inhibitors may increase blood pressure. Closely monitor blood pressure with concomitant use of esketamine and monoamine oxidase inhibitors.
Use in special populations. Because of concerns of increased sedation, intranasal esketamine should be administered cautiously in patients receiving other CNS depressants, such as benzodiazepines. In patients with psychosis or a prior history of psychosis, esketamine should be used with increased caution and the risk/benefit ratio should be carefully considered.
Continue to: Because of potential teratogenicity...
Because of potential teratogenicity, esketamine is not recommended in women who are pregnant, may become pregnant, or who are currently nursing.
Intranasal esketamine was examined in a phase III trial of 194 patients age ≥65. At the end of 4 weeks, there was no statistically significant difference in groups on the MADRS, the primary efficacy endpoint. There were no overall differences in the safety profile in patients >65 years compared with younger patients; however, the mean esketamine Cmax and area under the curve were higher in older patients compared with younger adults. The mean esketamine half-life was longer in patients with moderate hepatic impairment.
Abuse liability
Esketamine is a CIII controlled substance and concerns about abuse, misuse, and diversion have been taken into account within the REMS drug safety program.2 Patients with a prior history of substance abuse or misuse should be considered with regard to the risk/benefit ratio.
The REMS drug safety program
Due to the nature of its usually transient adverse effects, including sedation, dissociation, hypertension, and nausea, intranasal esketamine will be administered through a REMS drug safety program at certified REMS treatment centers. Certified REMS treatment centers will receive training on how to safely and effectively counsel and monitor patients. Prior to treatment, patients will receive blood pressure monitoring and anticipated adverse effects will be discussed. Patients will be instructed to not eat solid food for 2 hours pre-dose and to not drink anything for 30 minutes prior.
A treatment session consists of nasal administration and a minimum 2-hour post-administration observation period. Blood pressure must be assessed prior to administration and if elevated, (ie, systolic blood pressure >140 mm Hg, diastolic >90 mm Hg), clinicians should consider the risk of short-term increases in blood pressure that may occur. Do not administer if increases in blood pressure or intracranial pressure pose a serious risk.
Continue to: After each intranasal...
After each intranasal administration the patient will be observed for 5 minutes before the second nasal inhaler is utilized and for another 5 minutes when the patient is receiving 84 mg (ie, each inhaler equals 28 mg). After administering, blood pressure should be reassessed at approximately 40 minutes, which corresponds to the Cmax of intranasal esketamine, and periodically thereafter as warranted.
The patient will then be monitored in a quiet environment for a minimum of 2 hours to make sure that dissociative phenomenon, sedation, and hypertensive reactions have normalized prior to discharge from a certified REMS treatment center.
Dosing and administration
Each intranasal device is primed for 2 infusions (1 in each nostril) for a total dose of 28 mg of esketamine. Combinations of devices can be used to adjust the dose as appropriate for individual patients. The recommended starting dose is 56 mg (ie, 2 devices, with a 5-minute gap between devices). The dose can be increased to 84 mg (ie, 3 intranasal devices spaced at 5-minute intervals) by the second dose based on clinical judgment.
The patient will be instructed to recline the head to a 45° angle, clear his or her nostrils prior to the first treatment, and then self-administer a dose to each nostril while holding the reciprocal nostril closed and inhaling. This process is then repeated every 5 minutes for each subsequent device, with a maximum total dose of 3 devices, or 84 mg (Figure 32). The patient will then be monitored for blood pressure, heart rate, and signs of psychologic or physiologic changes for the next 2 hours. Patients may not drive a car or operate any type of motor equipment until the following day after receiving a normal night’s sleep. Patients will be released from the REMS treatment center after 2 hours if both psychological and physical adverse effects have normalized.
Missed treatment sessions. If a patient misses a treatment session and there is worsening of depressive symptoms, consider returning the patient to the previous dosing schedule (ie, every 2 weeks to once weekly, or weekly to twice weekly).
Continue to: Contraindications for...
Contraindications for intranasal esketamine include:
- aneurysmal vascular disease, including thoracic and abdominal aortic, intracranial, and peripheral arterial vessels, or arterial venous malformations
- history of intracerebral hemorrhage
- hypersensitivity to esketamine, ketamine, or any of the excipients.
Clinical considerations
Intranasal esketamine represents a unique delivery system for the first glutamatergic treatment approved for patients with TRD.
Why Rx? Treatment-resistant depression is found in nearly 1 out of 3 patients with currently available monoaminergic antidepressant treatment options. Patients with TRD are at increased risk of physical and psychological impairment, subsequent worsening of their condition, and social and occupational disability.
Bottom Line
Intranasal esketamine is the first glutamatergic treatment option FDA-approved for patients with treatment-resistant depression who have not responded to standard antidepressant treatment options. In short-term trials, intranasal esketamine significantly improved depressive symptoms as quickly as 24 hours after treatment, with significant improvement maintained through 4 weeks of ongoing administration. In addition, intranasal esketamine was shown to significantly decrease time to relapse for patients who had achieved stable remission or stable response.
Related Resource
- Sullivan MG. FDA approves intranasal esketamine for refractory major depressive disorder. Clinical Psychiatry News. https://www.mdedge.com/psychiatry/article/195712/depression/fda-approves-intranasal-esketamine-refractory-major-depressive. Published March 5, 2019.
Drug Brand Names
Armodafinil • Nuvigil
Duloxetine • Cymbalta
Escitalopram • Lexapro
Esketamine • Spravato
Mirtazapine • Remeron
Modafinil • Provigil
Sertraline • Zoloft
Venlafaxine • Effexor
Treatment-resistant depression (TRD) is a common clinical struggle that practicing clinicians address on a daily basis. Major depressive disorder affects nearly 1 in 5 Americans at some point in their life and, by definition, impairs social and occupational functioning. Historic treatments have focused on the monoamine theories of depression—modulating the monoamines serotonin, norepinephrine, and/or dopamine. Limitations of currently available antidepressants include delayed onset of effect and low remission rates. To further complicate the matter, numerous studies have shown that with each subsequent antidepressant trial, patients have a decreasing likelihood of responding to subsequent antidepressant treatment options. For example, in the classic STAR*D trial, by the time a patient had not responded to the first 2 antidepressant options, the chance that they would respond to a third or fourth antidepressant had decreased to approximately 15% per antidepressant treatment course.1
To address the need for new treatments for patients with TRD, on March 5, 2019 the FDA-approved intranasal
How it works
Modern research has looked beyond the monoamine system to explore the neuro-modulatory effects of glutamate and gamma-aminobutyric acid (GABA).3 The yin and yang of glutamate and GABA revolves around neural excitation vs neural inhibition at a local synaptic level. The primary effects of the glutamate and GABA systems (Table 2) can be broken down into several key areas of understanding.
Glutamate modulates ionotropic N-methyl-
Esketamine, the S-enantiomer of ketamine, has a higher affinity for the NMDA receptor than the R-enantiomer and has been developed as an intranasal adjunctive treatment for TRD. Esketamine blocks NMDA receptors on GABA interneurons. This allows for increased pulsatile release of glutamate into the synapse. Intrasynaptic glutamate then stimulates postsynaptic AMPA receptors. Glutamate stimulation of postsynaptic AMPA receptors results in an intracellular cascade that activates the enzymes tropomyosin receptor kinase B (TrkB) and mammalian target of rapamycin (mTOR). TrkB stimulation results in increased production and release of BDNF. mTor stimulation increases neuronal membrane protein formation with subsequent increased neural plasticity. Taken together, preclinical models show that esketamine’s inhibition of the NMDA receptor on the GABA interneuron results in a cascade of increased BDNF release and synaptogenesis with increased neuroplasticity (Table 3).
Clinical implications
Treatment-resistant depression affects nearly one-third of patients currently receiving standard antidepressant treatment. Major depressive disorder is currently the second leading cause of disability for working adults within the United States and one of the largest causes of disability worldwide. The esketamine nasal spray could be beneficial for patients who have experienced TRD with standard monoamine antidepressants.
Supporting evidence
Clinical trials examining intranasal esketamine include both short- and long-term studies of patients with TRD.
Continue to: Esketamine was evaluated...
Esketamine was evaluated in a randomized, placebo-controlled, double-blind, multicenter, short-term (4-week) phase III study in adult patients age 18 to 65 with TRD (they had not responded to at least 2 different antidepressants of adequate dose and duration).4 After discontinuing prior antidepressant treatments, all patients were started on a newly initiated antidepressant and were also randomized to concomitant intranasal esketamine or intranasal placebo as follows:
- 114 patients were randomized to the intranasal esketamine plus newly initiated oral antidepressant arm
- 109 patients were randomized to the placebo nasal spray plus newly initiated oral antidepressant arm
- The mean baseline Montgomery-Åsberg Depression Rating Scale (MADRS) score for each group was 37 (ie, moderately to severely depressed).
Newly started antidepressants included esc
A long-term, double-blind multicenter maintenance-of-effect trial examined adults age 18 to 65 with TRD.5-6 Patients in this study were responders in 1 of 2 short-term studies or in an open-label direct enrollment study. Stable remission was defined as a MADRS total score <12 for at least 3 of the last 4 weeks of the study, and stable response was defined as a MADRS reduction of >50% but not in remission. After 16 weeks of intranasal esketamine plus an oral antidepressant, stable remitters and stable responders were then randomized separately to continue intranasal esketamine or switch to placebo nasal spray, with both groups continuing on their concomitant oral antidepressant. The primary study endpoint was time to relapse. Relapse was defined as a MADRS total score >22 for more than 2 consecutive weeks, hospitalization for worsening of depression, or any other clinically relevant event. The median age was 48, 66% were female, 90% were White and 4% were black. Patients in stable response or stable remission experienced a significantly longer time to relapse compared with patients who continued their oral antidepressant but were switched to placebo intranasal spray. In this remission response study, patients could receive intranasal treatment weekly or bi-weekly based on symptom severity (Figure 22).
Impact on driving. Two studies examined the impact of esketamine on driving performance. One examined adults with major depressive disorder and the other examined healthy participants. The effects of a single 84-mg dose of esketamine nasal spray on a patient’s ability to drive was assessed in 23 healthy adults. In this study, mirt
A second study evaluated the effects of repeated esketamine administration on driving performance in 25 adults with major depressive disorder. In this study, an ethanol-containing beverage was used as an active control. After administration of a single 84-mg dose of intranasal esketamine, driving performance was the same as a placebo at 18 hours. In the multiple dose phase, standard driving performance was similar for esketamine nasal spray and placebo at 6 hours postdose on Days 11, 18, and 25.
Continue to: Pharmacologic profile
Pharmacologic profile
Adverse events. The most common adverse events in patients treated with esketamine nasal spray were dissociation (41%), dizziness (29%), nausea (28%), sedation (23%), and vertigo (23%).2 The majority of these effects were short-term and resolved during the 2-hour observation period.
In addition to spontaneously reported events, sedation and dissociation were further monitored with specific scales. Sedation was measured with the Modified Observer’s Alertness and Sedation Scale. Using this scale, 50% of patients receiving 56 mg and 61% of patients receiving 84 mg of esketamine met criteria for sedation.
Similarly, dissociation/perceptional changes were measured with spontaneously reported events and also with the Clinician Administered Dissociative State Scale. On this scale, 61% of patients receiving the 56-mg dose, and 69% of patients receiving the 84-mg dose met criteria for dissociation/perceptional changes after dose administration.
Increases in blod pressure. Esketamine intranasal spray was associated with a 7 to 9 mm Hg increase in systolic blood pressure and a 4 to 6 mm Hg increase in diastolic blood pressure, both of which peaked 40 minutes post-dose.
Nausea and vomiting. Intranasal esketamine was associated with a 27% rate of nausea at 56 mg, and 32% at 84 mg, with a 6% rate of vomiting at 56 mg and 12% at 84 mg.
Continue to: Pharmacokinetics
Pharmacokinetics
Esketamine exposure increases from 28 to 84 mg in a fairly dose-proportional range. No accumulation of esketamine was observed in the plasma following twice-weekly administration. Bioavailability is approximately 48% following nasal administration. The Tmax for esketamine plasma concentration is 20 to 40 minutes after the last nasal spray. Protein binding of esketamine is approximately 43% to 45%. The brain-to-plasma ratio of noresketamine is 4 to 6 times lower than that of esketamine. The half-life of esketamine ranged from 7 to 12 hours. The mean half-life of nore
Potential drug interactions
Central nervous system depressants. Concomitant use of esketamine and other CNS depressants (ie, benzodiazepines, opioids, alcohol) may increase sedation. Patients receiving esketamine with concomitant use of other CNS depressants should be closely monitored for sedation.
Psychostimulants. Concomitant use of esketamine and psychostimulants (ie, amphetamines, methylphenidates, moda
Monoamine oxidase inhibitors. Concomitant use of esketamine with monoamine oxidase inhibitors may increase blood pressure. Closely monitor blood pressure with concomitant use of esketamine and monoamine oxidase inhibitors.
Use in special populations. Because of concerns of increased sedation, intranasal esketamine should be administered cautiously in patients receiving other CNS depressants, such as benzodiazepines. In patients with psychosis or a prior history of psychosis, esketamine should be used with increased caution and the risk/benefit ratio should be carefully considered.
Continue to: Because of potential teratogenicity...
Because of potential teratogenicity, esketamine is not recommended in women who are pregnant, may become pregnant, or who are currently nursing.
Intranasal esketamine was examined in a phase III trial of 194 patients age ≥65. At the end of 4 weeks, there was no statistically significant difference in groups on the MADRS, the primary efficacy endpoint. There were no overall differences in the safety profile in patients >65 years compared with younger patients; however, the mean esketamine Cmax and area under the curve were higher in older patients compared with younger adults. The mean esketamine half-life was longer in patients with moderate hepatic impairment.
Abuse liability
Esketamine is a CIII controlled substance and concerns about abuse, misuse, and diversion have been taken into account within the REMS drug safety program.2 Patients with a prior history of substance abuse or misuse should be considered with regard to the risk/benefit ratio.
The REMS drug safety program
Due to the nature of its usually transient adverse effects, including sedation, dissociation, hypertension, and nausea, intranasal esketamine will be administered through a REMS drug safety program at certified REMS treatment centers. Certified REMS treatment centers will receive training on how to safely and effectively counsel and monitor patients. Prior to treatment, patients will receive blood pressure monitoring and anticipated adverse effects will be discussed. Patients will be instructed to not eat solid food for 2 hours pre-dose and to not drink anything for 30 minutes prior.
A treatment session consists of nasal administration and a minimum 2-hour post-administration observation period. Blood pressure must be assessed prior to administration and if elevated, (ie, systolic blood pressure >140 mm Hg, diastolic >90 mm Hg), clinicians should consider the risk of short-term increases in blood pressure that may occur. Do not administer if increases in blood pressure or intracranial pressure pose a serious risk.
Continue to: After each intranasal...
After each intranasal administration the patient will be observed for 5 minutes before the second nasal inhaler is utilized and for another 5 minutes when the patient is receiving 84 mg (ie, each inhaler equals 28 mg). After administering, blood pressure should be reassessed at approximately 40 minutes, which corresponds to the Cmax of intranasal esketamine, and periodically thereafter as warranted.
The patient will then be monitored in a quiet environment for a minimum of 2 hours to make sure that dissociative phenomenon, sedation, and hypertensive reactions have normalized prior to discharge from a certified REMS treatment center.
Dosing and administration
Each intranasal device is primed for 2 infusions (1 in each nostril) for a total dose of 28 mg of esketamine. Combinations of devices can be used to adjust the dose as appropriate for individual patients. The recommended starting dose is 56 mg (ie, 2 devices, with a 5-minute gap between devices). The dose can be increased to 84 mg (ie, 3 intranasal devices spaced at 5-minute intervals) by the second dose based on clinical judgment.
The patient will be instructed to recline the head to a 45° angle, clear his or her nostrils prior to the first treatment, and then self-administer a dose to each nostril while holding the reciprocal nostril closed and inhaling. This process is then repeated every 5 minutes for each subsequent device, with a maximum total dose of 3 devices, or 84 mg (Figure 32). The patient will then be monitored for blood pressure, heart rate, and signs of psychologic or physiologic changes for the next 2 hours. Patients may not drive a car or operate any type of motor equipment until the following day after receiving a normal night’s sleep. Patients will be released from the REMS treatment center after 2 hours if both psychological and physical adverse effects have normalized.
Missed treatment sessions. If a patient misses a treatment session and there is worsening of depressive symptoms, consider returning the patient to the previous dosing schedule (ie, every 2 weeks to once weekly, or weekly to twice weekly).
Continue to: Contraindications for...
Contraindications for intranasal esketamine include:
- aneurysmal vascular disease, including thoracic and abdominal aortic, intracranial, and peripheral arterial vessels, or arterial venous malformations
- history of intracerebral hemorrhage
- hypersensitivity to esketamine, ketamine, or any of the excipients.
Clinical considerations
Intranasal esketamine represents a unique delivery system for the first glutamatergic treatment approved for patients with TRD.
Why Rx? Treatment-resistant depression is found in nearly 1 out of 3 patients with currently available monoaminergic antidepressant treatment options. Patients with TRD are at increased risk of physical and psychological impairment, subsequent worsening of their condition, and social and occupational disability.
Bottom Line
Intranasal esketamine is the first glutamatergic treatment option FDA-approved for patients with treatment-resistant depression who have not responded to standard antidepressant treatment options. In short-term trials, intranasal esketamine significantly improved depressive symptoms as quickly as 24 hours after treatment, with significant improvement maintained through 4 weeks of ongoing administration. In addition, intranasal esketamine was shown to significantly decrease time to relapse for patients who had achieved stable remission or stable response.
Related Resource
- Sullivan MG. FDA approves intranasal esketamine for refractory major depressive disorder. Clinical Psychiatry News. https://www.mdedge.com/psychiatry/article/195712/depression/fda-approves-intranasal-esketamine-refractory-major-depressive. Published March 5, 2019.
Drug Brand Names
Armodafinil • Nuvigil
Duloxetine • Cymbalta
Escitalopram • Lexapro
Esketamine • Spravato
Mirtazapine • Remeron
Modafinil • Provigil
Sertraline • Zoloft
Venlafaxine • Effexor
1. Rush AG, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR D Report. Am J Psychiatry. 2006;163(11):1905-1917.
2. Spravato [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2019.
3. Duman RS, Aghajanian GK, Sanacora G, et al. Synaptic plasticity and depression: new insights from stress and rapid-acting anti-depression. Nat Med. 2016;22(3):238-249.
4. Daly EJ, Singh JB, Fedgchin M, et al. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2018;75(2):139-148.
5. Daly EJ, Trivedi M, Janik A, et al. A randomized withdrawal, double-blind, multicenter study of esketamine nasal spray plus an oral antidepressant for relapse prevention in treatment-resistant depression. Poster presented at the 2018 American Society of Clinical Psychopharmacology Annual Meeting; May 2018; Miami, Florida.
6. Wajs E, Aluisio L, Morrison R, et al. Long-term safety of esketamine nasal spray plus oral antidepressants in patients with treatment-resistant depression: phase III open-label safety and efficacy study. Poster presented at the 2018 American Society of Clinical Psychopharmacology Annual Meeting; May 2018; Miami, Florida.
1. Rush AG, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR D Report. Am J Psychiatry. 2006;163(11):1905-1917.
2. Spravato [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2019.
3. Duman RS, Aghajanian GK, Sanacora G, et al. Synaptic plasticity and depression: new insights from stress and rapid-acting anti-depression. Nat Med. 2016;22(3):238-249.
4. Daly EJ, Singh JB, Fedgchin M, et al. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2018;75(2):139-148.
5. Daly EJ, Trivedi M, Janik A, et al. A randomized withdrawal, double-blind, multicenter study of esketamine nasal spray plus an oral antidepressant for relapse prevention in treatment-resistant depression. Poster presented at the 2018 American Society of Clinical Psychopharmacology Annual Meeting; May 2018; Miami, Florida.
6. Wajs E, Aluisio L, Morrison R, et al. Long-term safety of esketamine nasal spray plus oral antidepressants in patients with treatment-resistant depression: phase III open-label safety and efficacy study. Poster presented at the 2018 American Society of Clinical Psychopharmacology Annual Meeting; May 2018; Miami, Florida.

Risperidone extended-release injectable suspension
Oral antipsychotic nonadherence is a significant contributor to relapse in patients with schizophrenia spectrum disorders. Long-acting injectable (LAI) antipsychotics have been developed to provide sustained antipsychotic exposure, with evidence that use of LAIs significantly reduces hospitalization rates.1 One limiting factor in transitioning patients to certain LAIs is the need for prolonged oral coverage at the onset of treatment for agents that cannot be loaded. Nonadherence with this bridging oral therapy places the patient at risk for symptom exacerbation until effective antipsychotic plasma levels are achieved from the LAI.2 Although risperidone is one of the more widely used antipsychotics for treating schizophrenia, until recently the only available LAI preparation, risperidone microspheres (Risperdal Consta), required 3 weeks of oral coverage upon initiation.3

Clinical implications
Oral medication nonadherence remains a significant public health issue for patients with schizophrenia, with an estimated 50% of patients failing to achieve 80% adherence even when enrolled in clinical trials specifically designed to track adherence.5 Although LAI atypical antipsychotics have been available since the approval of Risperdal Consta, the LAI form of risperidone, and both LAI forms of aripiprazole, were not designed to be loaded. A 1-day initiation regimen for aripiprazole lauroxil has been developed to avoid the need for 3 weeks of oral medication coverage,6,7 but aripiprazole monohydrate and risperidone microspheres mandate oral bridging of 2 and 3 weeks, respectively.2 Because one of the primary indications for LAI antipsychotic therapy is oral medication nonadherence, this prolonged period of oral coverage creates a risk for symptom exacerbation when the bridging period occurs outside of a controlled setting, as is common when patients are discharged from inpatient hospitalization.
One solution to this problem has its antecedents in the development of the Atrigel biodegradable injectable polymer, which was designed to deliver prolonged medication exposure after subcutaneous injection.8 This biodegradable polymer drug delivery system suspends and dissolves the medication of interest (in this case, risperidone) in a poly DL-lactide-coglycolide gel and its biocompatible carrier.9 The viscous liquid undergoes a phase transition upon contact with tissue fluids after subcutaneous injection, resulting in an implant that releases risperidone in a controlled manner as it is resorbed. Importantly, the kinetic parameters of RBP-7000 are such that effective drug levels are seen within the first week without the need for oral coverage.10
Use in adults with schizophrenia. After establishing tolerability with oral risperidone, the recommended doses are 90 mg or 120 mg monthly, which correspond to oral daily risperidone doses of 3 mg or 4 mg. RBP-7000 must be administered as a subcutaneous abdominal injection by a health care professional. It is recommended that the patient be in the supine position for the injection and that the injection sites be rotated monthly among 4 quadrants in the abdominal region. The injection volumes for the 90 mg and 120 mg doses are 0.6 mL and 0.8 mL, respectively.10 As the gel implant becomes firmer, the patient will notice a lump for several weeks that will decrease in size over time. Patients should be advised not to rub or massage the injection site, and to be aware of the placement of any belts or clothing with waistbands.10
Pharmacologic profile, adverse reactions
Risperidone is an atypical antipsychotic that has been commercially available in the U.S. since December 29, 1993, and its adverse effect profile is well characterized. The most common adverse effects associated with risperidone include those related to dopamine D2 antagonism, metabolic adverse effects, and an increase in serum prolactin. In the 12-month long-term safety study of RBP-7000, 1-minute post-dose injection site pain scores (on a 100-point scale) were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following the last injection).10
Continue to: How the Atrigel system works
How the Atrigel system works. The Atrigel system was developed in the late 1980s and consists of a solution of a resorbable polymer in a biocompatible carrier.11 After in vivo administration (typically via subcutaneous injection), the polymer undergoes a phase change from a liquid to a formed implant (Figure 1). Being in liquid form, this system provides the advantage of placement by simple means, such as injection by syringes. The absorption rates of various polymers and the release rates for various drugs are tailored to the desired indication. Approved uses for Atrigel include the subgingival delivery of the antibiotic doxycycline for chronic adult periodontitis (approved September 1998), and the monthly subcutaneous injectable form of the anti-androgen leuprolide, which was approved in January 2002.8,12 Release periods up to 4 months have been achieved with Atrigel; 1 month is the most often desired release period. The biodegradable polymer used for RBP-7000 is designed to provide effective plasma drug levels during the first week of treatment, and sustained levels with a 1-month dosing interval. The small subcutaneous implant that is formed is gradually resorbed over the course of 1 month.
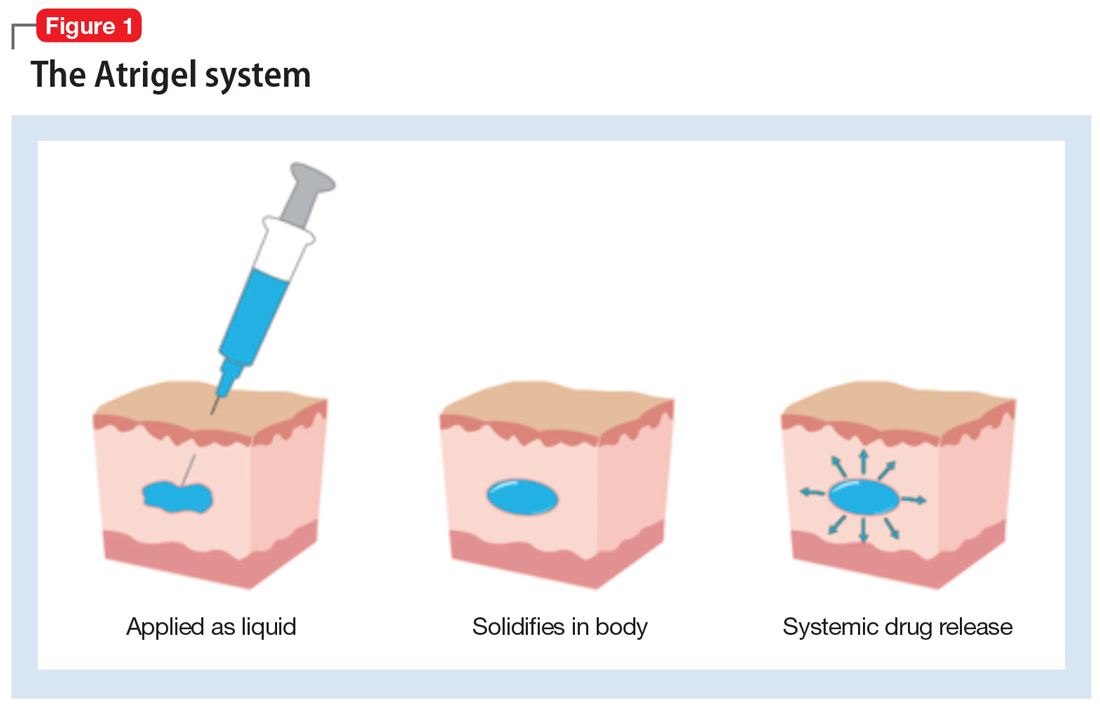
Pharmacokinetics. As with all LAI medications, the half-life with repeated dosing vastly exceeds that achieved with oral administration. Following oral administration, mean peak plasma levels of risperidone occur at 1 hour, and those for the active metabolite 9-OH risperidone occur at 3 hours.13 Oral risperidone has a mean half-life of 3 hours, while the active metabolite 9-OH risperidone has a mean half-life of 21 hours.14 Due to its longer half-life, the metabolite comprises 83% of the active drug levels at steady state.14 Although risperidone is susceptible to interactions via cytochrome P450 (CYP) inhibitors and inducers, particularly CYP2D6 (Table 210), the pharmacokinetics of the combined total of risperidone and 9-OH risperidone levels (deemed the active moiety) are similar in CYP2D6 extensive and poor metabolizers, with an overall mean elimination half-life of approximately 20 hours.13

The kinetics for RBP-7000 are markedly different than those for oral risperidone (Figure 215). After a single subcutaneous injection, RBP-7000 shows 2 absorption peaks for risperidone. The first lower peak occurs with a Tmax of 4 to 6 hours due to initial release of risperidone during the implant formation process; a second risperidone peak occurs after 10 to 14 days and is associated with slow release from the subcutaneous depot.9,16,17 For both 9-OH risperidone levels and the total active moiety (risperidone plus 9-OH risperidone levels), the median Tmax of the first peak ranges from 4 to 48 hours and the second peak ranges from 7 to 11 days. Following a single subcutaneous injection of RBP-7000, the apparent terminal half-life of risperidone ranges from 9 to 11 days, on average. The mean apparent terminal half-life of the active moiety ranges from 8 to 9 days.9,16,17 Based on population pharmacokinetic modeling, the 90 mg and 120 mg doses of RBP-7000 are estimated to provide drug exposure equivalent to 3 mg/d and 4 mg/d of oral risperidone, respectively.9,16,17
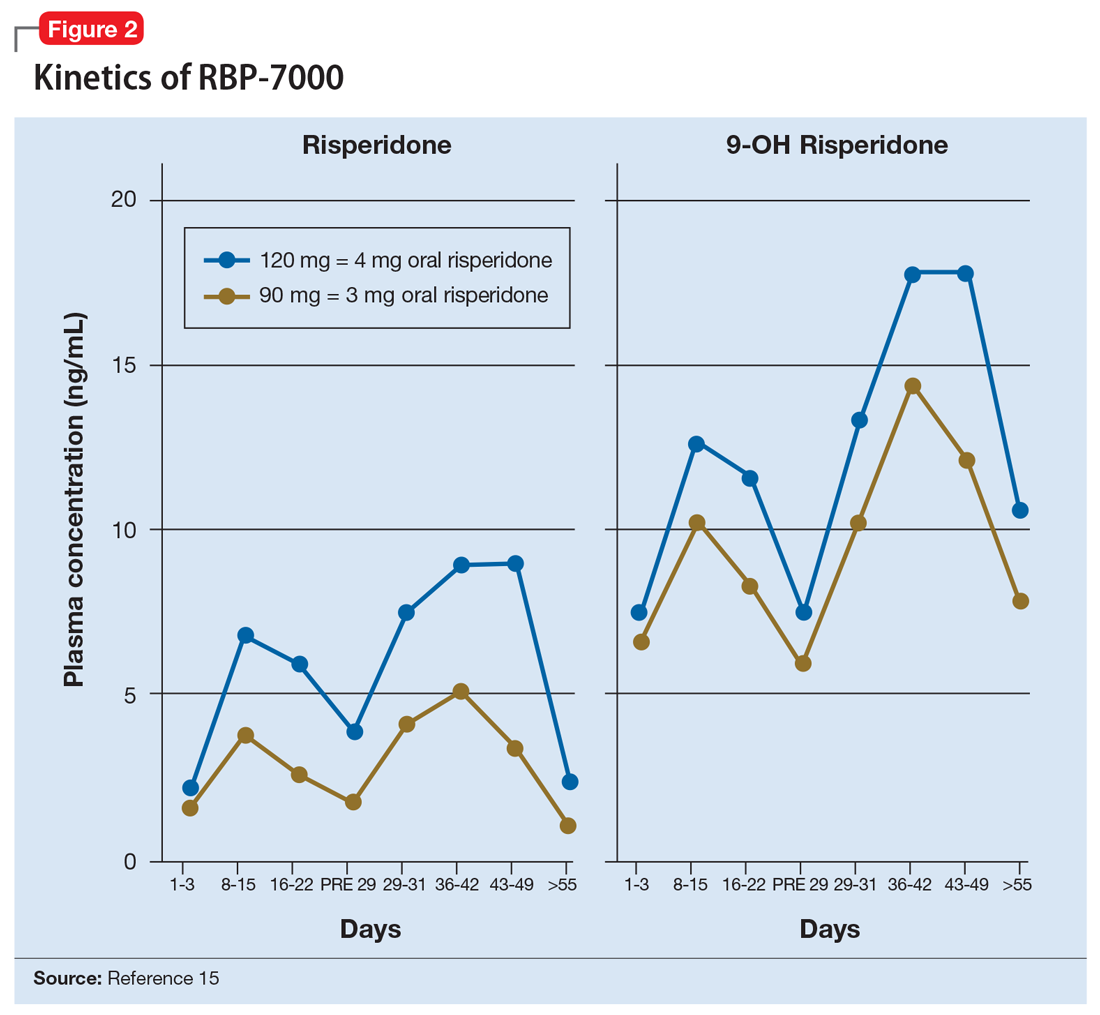
Continue to: Efficacy of RBP-7000
Efficacy of RBP-7000 was established in an 8-week, double-blind, placebo-controlled trial of adult patients experiencing an acute exacerbation of schizophrenia (age 18 to 55).4 Eligible participants had:
- An acute exacerbation of schizophrenia that occurred ≤8 weeks before the screening visit and would have benefited from psychiatric hospitalization or continued hospitalization
- Positive and Negative Syndrome Scale (PANSS) total score between 80 and 120 at visit 1 and a score of >4 on at least 2 of the following 4 items: hallucinatory behavior, delusions, conceptual disorganization, or suspiciousness/persecution
- The diagnosis of acute exacerbation of schizophrenia and PANSS total score were confirmed through an independent video-conference interview conducted by an experienced rater.
Participants were excluded if they:
- Experienced a ≥20% improvement in PANSS total score between the initial screening visit and the first injection
- had been treated at any time with clozapine for treatment-resistant schizophrenia
- had met DSM-IV-TR criteria for substance dependence (with the exception of nicotine or caffeine) before screening.
During the initial screening visit, participants received a 0.25-mg tablet of oral risperidone on 2 consecutive days to assess the tolerability of risperidone.
Outcome. Participants were randomized in a 1:1:1 manner to placebo (n = 112) or 1 of 2 monthly doses of RBP-7000: 90 mg (n = 111) or 120 mg (n = 114). Using the least squares means of repeated-measures changes from baseline in PANSS total scores, there was a significant improvement in the difference in PANSS total scores from baseline to the end of the study compared with placebo: 90-mg RBP-7000, -6.148 points (95% confidence interval [CI], -9.982 to -2.314, P = .0004); 120-mg RBP-7000, -7.237 points (95% CI, -11.045 to -3.429, P < .0001). The absolute change from baseline in PANSS total score was -15.367 points for the 90-mg dose and -16.456 points for the 120-mg dose.4 Completion rates across all 3 arms were comparable: placebo 70.6%, RBP-7000 90 mg 77.6%, and RBP-7000 120 mg 71.4%.
Tolerability. In the 8-week phase III efficacy trial of RBP-7000, adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo were weight gain (placebo 3.4%, 90 mg 13.0%, 120 mg 12.8%) and sedation (placebo 0%, 90 mg 7.0%, 120 mg 7.7%).10 Compared with baseline, participants had a mean weight gain at the end of the study of 2.83 kg in the placebo group, 5.15 kg in the 90-mg RBP-7000 group, and 4.69 kg in the 120-mg RBP-7000 group. There were no clinically significant differences at study endpoint in glucose and lipid parameters. Consistent with the known effects of risperidone, there were increases in mean prolactin levels during the 8-week study, the effects of which were greater for women. For men, mean prolactin levels from baseline to study end were: placebo: 9.8 ± 7.9 vs 9.9 ± 8.0 ng/mL; 90 mg: 8.9 ± 6.9 vs 22.4 ± 11.2 ng/mL; and 120 mg: 8.2 ± 5.2 vs 31.3 ± 14.8 ng/mL. For women, mean prolactin levels from baseline to study end were: placebo: 12.8 ± 11.7 vs 10.4 ± 8.0 ng/mL; 90 mg: 7.7 ± 5.3 vs 60.3 ± 46.9 ng/mL; and 120 mg: 10.9 ± 8.6 vs 85.5 ± 55.1 ng/mL. In the pivotal study, discontinuations due to adverse events were low across all treatment groups: 2.5% for placebo vs 0% for 90 mg and 1.7% for 120 mg.4 There was no single adverse reaction leading to discontinuation that occurred at a rate of ≥2% and greater than placebo in patients treated with RBP-7000.10 There were no clinically relevant differences in mean changes from baseline in corrected QT, QRS, and PR intervals, and in heart rate. Similarly, in the 12-month, long-term safety study, there were no clinically relevant changes in mean electrocardiography interval values from baseline to post-dose assessments.10
Using a 100-point visual analog scale (VAS), injection site pain scores 1 minute after the first dose decreased from a mean of 27 to the range of 3 to 7 for scores obtained 30 to 60 minutes post-dose. In the 12-month long-term safety study, 1-minute post-dose injection site pain VAS scores were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following last injection).10
Clinical considerations
Unique properties. RBP-7000 uses the established Atrigel system to provide effective antipsychotic levels in the first week of treatment, without the need for bridging oral coverage or a second loading injection. The abdominal subcutaneous injection volume is relatively small (0.6 mL or 0.8 mL).
Why Rx? The reasons to prescribe RBP-7000 for adult patients with schizophrenia include:
- no oral coverage required at the initiation of treatment
- effective plasma active moiety levels are seen within the first week without the need for a second loading injection
- monthly injection schedule.
Dosing. The recommended dosage of RBP-7000 is 90 mg or 120 mg once monthly, equivalent to 3 mg/d or 4 mg/d of oral risperidone, respectively. Oral risperidone tolerability should be established before the first injection. No oral risperidone coverage is required. RBP-7000 has not been studied in patients with renal or hepatic impairment and should be used with caution in these patients. Prior to initiating treatment in these patients, it is advised to carefully titrate up to at least 3 mg/d of oral risperidone. If a patient can tolerate 3 mg/d of oral risperidone and is psychiatrically stable, then the 90-mg dose of RBP-7000 can be considered.10
Contraindications. The only contraindications for RBP-7000 are known hypersensitivity to risperidone, paliperidone (9-OH risperidone), or other components of the injection.
Bottom Line
RBP-7000 (Perseris) is the second long-acting injectable (LAI) form of risperidone approved in the U.S. Unlike risperidone microspheres (Consta), RBP-7000 does not require any oral risperidone coverage at the beginning of therapy, provides effective drug levels within the first week of treatment with a single injection, and uses a monthly dosing interval. RBP-7000 does not require loading upon initiation. The monthly injection is <1 mL, is administered in abdominal subcutaneous tissue, and uses the Atrigel system.
Related Resource
- Carpenter J, Wong KK. Long-acting injectable antipsychotics: What to do about missed doses. Current Psychiatry. 2018;17(7):10-12,14-19,56.
Drug Brand Names
Aripiprazole • Abilify
Carbamazepine • Carbatrol, Tegretol
Doxycycline • Atridox
Leuprolide acetate injectable suspension • Eligard
Paliperidone palmitate • Invega Sustenna
Risperidone • Risperdal
Risperidone extended-release injectable suspension • Perseris
Risperidone long-acting injection • Risperdal Consta
1. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
2. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
3. Risperdal Consta [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
4. Nasser AF, Henderson DC, Fava M, et al. Efficacy, safety, and tolerability of RBP-7000 once-monthly risperidone for the treatment of acute schizophrenia: an 8-week, randomized, double-blind, placebo-controlled, multicenter phase 3 study. J Clin Psychopharmacol. 2016;36(2):130-140.
5. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
6. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
7. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
8. Southard GL, Dunn RL, Garrett S. The drug delivery and biomaterial attributes of the ATRIGEL technology in the treatment of periodontal disease. Expert Opin Investig Drugs. 1998;7(9):1483-1491.
9. Gomeni R, Heidbreder C, Fudala PJ, Nasser AF. A model-based approach to characterize the population pharmacokinetics and the relationship between the pharmacokinetic and safety profiles of RBP-7000, a new, long-acting, sustained-released formulation of risperidone. J Clin Pharmacol. 2013;53(10):1010-1019.
10. Perseris [package insert]. North Chesterfield, VA: Indivior Inc; 2018.
11. Malik K, Singh I, Nagpal M, et al. Atrigel: a potential parenteral controlled drug delivery system. Der Pharmacia Sinica. 2010;1(1):74-81.
12. Sartor O. Eligard: leuprolide acetate in a novel sustained-release delivery system. Urology. 2003;61(2 Suppl 1):25-31.
13. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
14. de Leon J, Wynn G, Sandson NB. The pharmacokinetics of paliperidone versus risperidone. Psychosomatics. 2010;51(1):80-88.
15. Ivaturi V, Gopalakrishnan M, Gobburu JVS, et al. Exposure-response analysis after subcutaneous administration of RBP-7000, a once-a-month long-acting Atrigel formulation of risperidone. Br J Clin Pharmacol. 2017;83(7):1476-1498.
16. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetics and prediction of dopamine D2 receptor occupancy after multiple doses of RBP-7000, a new sustained-release formulation of risperidone, in schizophrenia patients on stable oral risperidone treatment. Clin Pharmacokinet. 2014;53(6):533-543.
17. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetic modeling and simulation to guide dose selection for RBP-7000, a new sustained-release formulation of risperidone. J Clin Pharmacol. 2015;55(1):93-103.
Oral antipsychotic nonadherence is a significant contributor to relapse in patients with schizophrenia spectrum disorders. Long-acting injectable (LAI) antipsychotics have been developed to provide sustained antipsychotic exposure, with evidence that use of LAIs significantly reduces hospitalization rates.1 One limiting factor in transitioning patients to certain LAIs is the need for prolonged oral coverage at the onset of treatment for agents that cannot be loaded. Nonadherence with this bridging oral therapy places the patient at risk for symptom exacerbation until effective antipsychotic plasma levels are achieved from the LAI.2 Although risperidone is one of the more widely used antipsychotics for treating schizophrenia, until recently the only available LAI preparation, risperidone microspheres (Risperdal Consta), required 3 weeks of oral coverage upon initiation.3
Clinical implications
Oral medication nonadherence remains a significant public health issue for patients with schizophrenia, with an estimated 50% of patients failing to achieve 80% adherence even when enrolled in clinical trials specifically designed to track adherence.5 Although LAI atypical antipsychotics have been available since the approval of Risperdal Consta, the LAI form of risperidone, and both LAI forms of aripiprazole, were not designed to be loaded. A 1-day initiation regimen for aripiprazole lauroxil has been developed to avoid the need for 3 weeks of oral medication coverage,6,7 but aripiprazole monohydrate and risperidone microspheres mandate oral bridging of 2 and 3 weeks, respectively.2 Because one of the primary indications for LAI antipsychotic therapy is oral medication nonadherence, this prolonged period of oral coverage creates a risk for symptom exacerbation when the bridging period occurs outside of a controlled setting, as is common when patients are discharged from inpatient hospitalization.
One solution to this problem has its antecedents in the development of the Atrigel biodegradable injectable polymer, which was designed to deliver prolonged medication exposure after subcutaneous injection.8 This biodegradable polymer drug delivery system suspends and dissolves the medication of interest (in this case, risperidone) in a poly DL-lactide-coglycolide gel and its biocompatible carrier.9 The viscous liquid undergoes a phase transition upon contact with tissue fluids after subcutaneous injection, resulting in an implant that releases risperidone in a controlled manner as it is resorbed. Importantly, the kinetic parameters of RBP-7000 are such that effective drug levels are seen within the first week without the need for oral coverage.10
Use in adults with schizophrenia. After establishing tolerability with oral risperidone, the recommended doses are 90 mg or 120 mg monthly, which correspond to oral daily risperidone doses of 3 mg or 4 mg. RBP-7000 must be administered as a subcutaneous abdominal injection by a health care professional. It is recommended that the patient be in the supine position for the injection and that the injection sites be rotated monthly among 4 quadrants in the abdominal region. The injection volumes for the 90 mg and 120 mg doses are 0.6 mL and 0.8 mL, respectively.10 As the gel implant becomes firmer, the patient will notice a lump for several weeks that will decrease in size over time. Patients should be advised not to rub or massage the injection site, and to be aware of the placement of any belts or clothing with waistbands.10
Pharmacologic profile, adverse reactions
Risperidone is an atypical antipsychotic that has been commercially available in the U.S. since December 29, 1993, and its adverse effect profile is well characterized. The most common adverse effects associated with risperidone include those related to dopamine D2 antagonism, metabolic adverse effects, and an increase in serum prolactin. In the 12-month long-term safety study of RBP-7000, 1-minute post-dose injection site pain scores (on a 100-point scale) were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following the last injection).10
Continue to: How the Atrigel system works
How the Atrigel system works. The Atrigel system was developed in the late 1980s and consists of a solution of a resorbable polymer in a biocompatible carrier.11 After in vivo administration (typically via subcutaneous injection), the polymer undergoes a phase change from a liquid to a formed implant (Figure 1). Being in liquid form, this system provides the advantage of placement by simple means, such as injection by syringes. The absorption rates of various polymers and the release rates for various drugs are tailored to the desired indication. Approved uses for Atrigel include the subgingival delivery of the antibiotic doxycycline for chronic adult periodontitis (approved September 1998), and the monthly subcutaneous injectable form of the anti-androgen leuprolide, which was approved in January 2002.8,12 Release periods up to 4 months have been achieved with Atrigel; 1 month is the most often desired release period. The biodegradable polymer used for RBP-7000 is designed to provide effective plasma drug levels during the first week of treatment, and sustained levels with a 1-month dosing interval. The small subcutaneous implant that is formed is gradually resorbed over the course of 1 month.
Pharmacokinetics. As with all LAI medications, the half-life with repeated dosing vastly exceeds that achieved with oral administration. Following oral administration, mean peak plasma levels of risperidone occur at 1 hour, and those for the active metabolite 9-OH risperidone occur at 3 hours.13 Oral risperidone has a mean half-life of 3 hours, while the active metabolite 9-OH risperidone has a mean half-life of 21 hours.14 Due to its longer half-life, the metabolite comprises 83% of the active drug levels at steady state.14 Although risperidone is susceptible to interactions via cytochrome P450 (CYP) inhibitors and inducers, particularly CYP2D6 (Table 210), the pharmacokinetics of the combined total of risperidone and 9-OH risperidone levels (deemed the active moiety) are similar in CYP2D6 extensive and poor metabolizers, with an overall mean elimination half-life of approximately 20 hours.13
The kinetics for RBP-7000 are markedly different than those for oral risperidone (Figure 215). After a single subcutaneous injection, RBP-7000 shows 2 absorption peaks for risperidone. The first lower peak occurs with a Tmax of 4 to 6 hours due to initial release of risperidone during the implant formation process; a second risperidone peak occurs after 10 to 14 days and is associated with slow release from the subcutaneous depot.9,16,17 For both 9-OH risperidone levels and the total active moiety (risperidone plus 9-OH risperidone levels), the median Tmax of the first peak ranges from 4 to 48 hours and the second peak ranges from 7 to 11 days. Following a single subcutaneous injection of RBP-7000, the apparent terminal half-life of risperidone ranges from 9 to 11 days, on average. The mean apparent terminal half-life of the active moiety ranges from 8 to 9 days.9,16,17 Based on population pharmacokinetic modeling, the 90 mg and 120 mg doses of RBP-7000 are estimated to provide drug exposure equivalent to 3 mg/d and 4 mg/d of oral risperidone, respectively.9,16,17
Continue to: Efficacy of RBP-7000
Efficacy of RBP-7000 was established in an 8-week, double-blind, placebo-controlled trial of adult patients experiencing an acute exacerbation of schizophrenia (age 18 to 55).4 Eligible participants had:
- An acute exacerbation of schizophrenia that occurred ≤8 weeks before the screening visit and would have benefited from psychiatric hospitalization or continued hospitalization
- Positive and Negative Syndrome Scale (PANSS) total score between 80 and 120 at visit 1 and a score of >4 on at least 2 of the following 4 items: hallucinatory behavior, delusions, conceptual disorganization, or suspiciousness/persecution
- The diagnosis of acute exacerbation of schizophrenia and PANSS total score were confirmed through an independent video-conference interview conducted by an experienced rater.
Participants were excluded if they:
- Experienced a ≥20% improvement in PANSS total score between the initial screening visit and the first injection
- had been treated at any time with clozapine for treatment-resistant schizophrenia
- had met DSM-IV-TR criteria for substance dependence (with the exception of nicotine or caffeine) before screening.
During the initial screening visit, participants received a 0.25-mg tablet of oral risperidone on 2 consecutive days to assess the tolerability of risperidone.
Outcome. Participants were randomized in a 1:1:1 manner to placebo (n = 112) or 1 of 2 monthly doses of RBP-7000: 90 mg (n = 111) or 120 mg (n = 114). Using the least squares means of repeated-measures changes from baseline in PANSS total scores, there was a significant improvement in the difference in PANSS total scores from baseline to the end of the study compared with placebo: 90-mg RBP-7000, -6.148 points (95% confidence interval [CI], -9.982 to -2.314, P = .0004); 120-mg RBP-7000, -7.237 points (95% CI, -11.045 to -3.429, P < .0001). The absolute change from baseline in PANSS total score was -15.367 points for the 90-mg dose and -16.456 points for the 120-mg dose.4 Completion rates across all 3 arms were comparable: placebo 70.6%, RBP-7000 90 mg 77.6%, and RBP-7000 120 mg 71.4%.
Tolerability. In the 8-week phase III efficacy trial of RBP-7000, adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo were weight gain (placebo 3.4%, 90 mg 13.0%, 120 mg 12.8%) and sedation (placebo 0%, 90 mg 7.0%, 120 mg 7.7%).10 Compared with baseline, participants had a mean weight gain at the end of the study of 2.83 kg in the placebo group, 5.15 kg in the 90-mg RBP-7000 group, and 4.69 kg in the 120-mg RBP-7000 group. There were no clinically significant differences at study endpoint in glucose and lipid parameters. Consistent with the known effects of risperidone, there were increases in mean prolactin levels during the 8-week study, the effects of which were greater for women. For men, mean prolactin levels from baseline to study end were: placebo: 9.8 ± 7.9 vs 9.9 ± 8.0 ng/mL; 90 mg: 8.9 ± 6.9 vs 22.4 ± 11.2 ng/mL; and 120 mg: 8.2 ± 5.2 vs 31.3 ± 14.8 ng/mL. For women, mean prolactin levels from baseline to study end were: placebo: 12.8 ± 11.7 vs 10.4 ± 8.0 ng/mL; 90 mg: 7.7 ± 5.3 vs 60.3 ± 46.9 ng/mL; and 120 mg: 10.9 ± 8.6 vs 85.5 ± 55.1 ng/mL. In the pivotal study, discontinuations due to adverse events were low across all treatment groups: 2.5% for placebo vs 0% for 90 mg and 1.7% for 120 mg.4 There was no single adverse reaction leading to discontinuation that occurred at a rate of ≥2% and greater than placebo in patients treated with RBP-7000.10 There were no clinically relevant differences in mean changes from baseline in corrected QT, QRS, and PR intervals, and in heart rate. Similarly, in the 12-month, long-term safety study, there were no clinically relevant changes in mean electrocardiography interval values from baseline to post-dose assessments.10
Using a 100-point visual analog scale (VAS), injection site pain scores 1 minute after the first dose decreased from a mean of 27 to the range of 3 to 7 for scores obtained 30 to 60 minutes post-dose. In the 12-month long-term safety study, 1-minute post-dose injection site pain VAS scores were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following last injection).10
Clinical considerations
Unique properties. RBP-7000 uses the established Atrigel system to provide effective antipsychotic levels in the first week of treatment, without the need for bridging oral coverage or a second loading injection. The abdominal subcutaneous injection volume is relatively small (0.6 mL or 0.8 mL).
Why Rx? The reasons to prescribe RBP-7000 for adult patients with schizophrenia include:
- no oral coverage required at the initiation of treatment
- effective plasma active moiety levels are seen within the first week without the need for a second loading injection
- monthly injection schedule.
Dosing. The recommended dosage of RBP-7000 is 90 mg or 120 mg once monthly, equivalent to 3 mg/d or 4 mg/d of oral risperidone, respectively. Oral risperidone tolerability should be established before the first injection. No oral risperidone coverage is required. RBP-7000 has not been studied in patients with renal or hepatic impairment and should be used with caution in these patients. Prior to initiating treatment in these patients, it is advised to carefully titrate up to at least 3 mg/d of oral risperidone. If a patient can tolerate 3 mg/d of oral risperidone and is psychiatrically stable, then the 90-mg dose of RBP-7000 can be considered.10
Contraindications. The only contraindications for RBP-7000 are known hypersensitivity to risperidone, paliperidone (9-OH risperidone), or other components of the injection.
Bottom Line
RBP-7000 (Perseris) is the second long-acting injectable (LAI) form of risperidone approved in the U.S. Unlike risperidone microspheres (Consta), RBP-7000 does not require any oral risperidone coverage at the beginning of therapy, provides effective drug levels within the first week of treatment with a single injection, and uses a monthly dosing interval. RBP-7000 does not require loading upon initiation. The monthly injection is <1 mL, is administered in abdominal subcutaneous tissue, and uses the Atrigel system.
Related Resource
- Carpenter J, Wong KK. Long-acting injectable antipsychotics: What to do about missed doses. Current Psychiatry. 2018;17(7):10-12,14-19,56.
Drug Brand Names
Aripiprazole • Abilify
Carbamazepine • Carbatrol, Tegretol
Doxycycline • Atridox
Leuprolide acetate injectable suspension • Eligard
Paliperidone palmitate • Invega Sustenna
Risperidone • Risperdal
Risperidone extended-release injectable suspension • Perseris
Risperidone long-acting injection • Risperdal Consta
Oral antipsychotic nonadherence is a significant contributor to relapse in patients with schizophrenia spectrum disorders. Long-acting injectable (LAI) antipsychotics have been developed to provide sustained antipsychotic exposure, with evidence that use of LAIs significantly reduces hospitalization rates.1 One limiting factor in transitioning patients to certain LAIs is the need for prolonged oral coverage at the onset of treatment for agents that cannot be loaded. Nonadherence with this bridging oral therapy places the patient at risk for symptom exacerbation until effective antipsychotic plasma levels are achieved from the LAI.2 Although risperidone is one of the more widely used antipsychotics for treating schizophrenia, until recently the only available LAI preparation, risperidone microspheres (Risperdal Consta), required 3 weeks of oral coverage upon initiation.3
Clinical implications
Oral medication nonadherence remains a significant public health issue for patients with schizophrenia, with an estimated 50% of patients failing to achieve 80% adherence even when enrolled in clinical trials specifically designed to track adherence.5 Although LAI atypical antipsychotics have been available since the approval of Risperdal Consta, the LAI form of risperidone, and both LAI forms of aripiprazole, were not designed to be loaded. A 1-day initiation regimen for aripiprazole lauroxil has been developed to avoid the need for 3 weeks of oral medication coverage,6,7 but aripiprazole monohydrate and risperidone microspheres mandate oral bridging of 2 and 3 weeks, respectively.2 Because one of the primary indications for LAI antipsychotic therapy is oral medication nonadherence, this prolonged period of oral coverage creates a risk for symptom exacerbation when the bridging period occurs outside of a controlled setting, as is common when patients are discharged from inpatient hospitalization.
One solution to this problem has its antecedents in the development of the Atrigel biodegradable injectable polymer, which was designed to deliver prolonged medication exposure after subcutaneous injection.8 This biodegradable polymer drug delivery system suspends and dissolves the medication of interest (in this case, risperidone) in a poly DL-lactide-coglycolide gel and its biocompatible carrier.9 The viscous liquid undergoes a phase transition upon contact with tissue fluids after subcutaneous injection, resulting in an implant that releases risperidone in a controlled manner as it is resorbed. Importantly, the kinetic parameters of RBP-7000 are such that effective drug levels are seen within the first week without the need for oral coverage.10
Use in adults with schizophrenia. After establishing tolerability with oral risperidone, the recommended doses are 90 mg or 120 mg monthly, which correspond to oral daily risperidone doses of 3 mg or 4 mg. RBP-7000 must be administered as a subcutaneous abdominal injection by a health care professional. It is recommended that the patient be in the supine position for the injection and that the injection sites be rotated monthly among 4 quadrants in the abdominal region. The injection volumes for the 90 mg and 120 mg doses are 0.6 mL and 0.8 mL, respectively.10 As the gel implant becomes firmer, the patient will notice a lump for several weeks that will decrease in size over time. Patients should be advised not to rub or massage the injection site, and to be aware of the placement of any belts or clothing with waistbands.10
Pharmacologic profile, adverse reactions
Risperidone is an atypical antipsychotic that has been commercially available in the U.S. since December 29, 1993, and its adverse effect profile is well characterized. The most common adverse effects associated with risperidone include those related to dopamine D2 antagonism, metabolic adverse effects, and an increase in serum prolactin. In the 12-month long-term safety study of RBP-7000, 1-minute post-dose injection site pain scores (on a 100-point scale) were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following the last injection).10
Continue to: How the Atrigel system works
How the Atrigel system works. The Atrigel system was developed in the late 1980s and consists of a solution of a resorbable polymer in a biocompatible carrier.11 After in vivo administration (typically via subcutaneous injection), the polymer undergoes a phase change from a liquid to a formed implant (Figure 1). Being in liquid form, this system provides the advantage of placement by simple means, such as injection by syringes. The absorption rates of various polymers and the release rates for various drugs are tailored to the desired indication. Approved uses for Atrigel include the subgingival delivery of the antibiotic doxycycline for chronic adult periodontitis (approved September 1998), and the monthly subcutaneous injectable form of the anti-androgen leuprolide, which was approved in January 2002.8,12 Release periods up to 4 months have been achieved with Atrigel; 1 month is the most often desired release period. The biodegradable polymer used for RBP-7000 is designed to provide effective plasma drug levels during the first week of treatment, and sustained levels with a 1-month dosing interval. The small subcutaneous implant that is formed is gradually resorbed over the course of 1 month.
Pharmacokinetics. As with all LAI medications, the half-life with repeated dosing vastly exceeds that achieved with oral administration. Following oral administration, mean peak plasma levels of risperidone occur at 1 hour, and those for the active metabolite 9-OH risperidone occur at 3 hours.13 Oral risperidone has a mean half-life of 3 hours, while the active metabolite 9-OH risperidone has a mean half-life of 21 hours.14 Due to its longer half-life, the metabolite comprises 83% of the active drug levels at steady state.14 Although risperidone is susceptible to interactions via cytochrome P450 (CYP) inhibitors and inducers, particularly CYP2D6 (Table 210), the pharmacokinetics of the combined total of risperidone and 9-OH risperidone levels (deemed the active moiety) are similar in CYP2D6 extensive and poor metabolizers, with an overall mean elimination half-life of approximately 20 hours.13
The kinetics for RBP-7000 are markedly different than those for oral risperidone (Figure 215). After a single subcutaneous injection, RBP-7000 shows 2 absorption peaks for risperidone. The first lower peak occurs with a Tmax of 4 to 6 hours due to initial release of risperidone during the implant formation process; a second risperidone peak occurs after 10 to 14 days and is associated with slow release from the subcutaneous depot.9,16,17 For both 9-OH risperidone levels and the total active moiety (risperidone plus 9-OH risperidone levels), the median Tmax of the first peak ranges from 4 to 48 hours and the second peak ranges from 7 to 11 days. Following a single subcutaneous injection of RBP-7000, the apparent terminal half-life of risperidone ranges from 9 to 11 days, on average. The mean apparent terminal half-life of the active moiety ranges from 8 to 9 days.9,16,17 Based on population pharmacokinetic modeling, the 90 mg and 120 mg doses of RBP-7000 are estimated to provide drug exposure equivalent to 3 mg/d and 4 mg/d of oral risperidone, respectively.9,16,17
Continue to: Efficacy of RBP-7000
Efficacy of RBP-7000 was established in an 8-week, double-blind, placebo-controlled trial of adult patients experiencing an acute exacerbation of schizophrenia (age 18 to 55).4 Eligible participants had:
- An acute exacerbation of schizophrenia that occurred ≤8 weeks before the screening visit and would have benefited from psychiatric hospitalization or continued hospitalization
- Positive and Negative Syndrome Scale (PANSS) total score between 80 and 120 at visit 1 and a score of >4 on at least 2 of the following 4 items: hallucinatory behavior, delusions, conceptual disorganization, or suspiciousness/persecution
- The diagnosis of acute exacerbation of schizophrenia and PANSS total score were confirmed through an independent video-conference interview conducted by an experienced rater.
Participants were excluded if they:
- Experienced a ≥20% improvement in PANSS total score between the initial screening visit and the first injection
- had been treated at any time with clozapine for treatment-resistant schizophrenia
- had met DSM-IV-TR criteria for substance dependence (with the exception of nicotine or caffeine) before screening.
During the initial screening visit, participants received a 0.25-mg tablet of oral risperidone on 2 consecutive days to assess the tolerability of risperidone.
Outcome. Participants were randomized in a 1:1:1 manner to placebo (n = 112) or 1 of 2 monthly doses of RBP-7000: 90 mg (n = 111) or 120 mg (n = 114). Using the least squares means of repeated-measures changes from baseline in PANSS total scores, there was a significant improvement in the difference in PANSS total scores from baseline to the end of the study compared with placebo: 90-mg RBP-7000, -6.148 points (95% confidence interval [CI], -9.982 to -2.314, P = .0004); 120-mg RBP-7000, -7.237 points (95% CI, -11.045 to -3.429, P < .0001). The absolute change from baseline in PANSS total score was -15.367 points for the 90-mg dose and -16.456 points for the 120-mg dose.4 Completion rates across all 3 arms were comparable: placebo 70.6%, RBP-7000 90 mg 77.6%, and RBP-7000 120 mg 71.4%.
Tolerability. In the 8-week phase III efficacy trial of RBP-7000, adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo were weight gain (placebo 3.4%, 90 mg 13.0%, 120 mg 12.8%) and sedation (placebo 0%, 90 mg 7.0%, 120 mg 7.7%).10 Compared with baseline, participants had a mean weight gain at the end of the study of 2.83 kg in the placebo group, 5.15 kg in the 90-mg RBP-7000 group, and 4.69 kg in the 120-mg RBP-7000 group. There were no clinically significant differences at study endpoint in glucose and lipid parameters. Consistent with the known effects of risperidone, there were increases in mean prolactin levels during the 8-week study, the effects of which were greater for women. For men, mean prolactin levels from baseline to study end were: placebo: 9.8 ± 7.9 vs 9.9 ± 8.0 ng/mL; 90 mg: 8.9 ± 6.9 vs 22.4 ± 11.2 ng/mL; and 120 mg: 8.2 ± 5.2 vs 31.3 ± 14.8 ng/mL. For women, mean prolactin levels from baseline to study end were: placebo: 12.8 ± 11.7 vs 10.4 ± 8.0 ng/mL; 90 mg: 7.7 ± 5.3 vs 60.3 ± 46.9 ng/mL; and 120 mg: 10.9 ± 8.6 vs 85.5 ± 55.1 ng/mL. In the pivotal study, discontinuations due to adverse events were low across all treatment groups: 2.5% for placebo vs 0% for 90 mg and 1.7% for 120 mg.4 There was no single adverse reaction leading to discontinuation that occurred at a rate of ≥2% and greater than placebo in patients treated with RBP-7000.10 There were no clinically relevant differences in mean changes from baseline in corrected QT, QRS, and PR intervals, and in heart rate. Similarly, in the 12-month, long-term safety study, there were no clinically relevant changes in mean electrocardiography interval values from baseline to post-dose assessments.10
Using a 100-point visual analog scale (VAS), injection site pain scores 1 minute after the first dose decreased from a mean of 27 to the range of 3 to 7 for scores obtained 30 to 60 minutes post-dose. In the 12-month long-term safety study, 1-minute post-dose injection site pain VAS scores were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following last injection).10
Clinical considerations
Unique properties. RBP-7000 uses the established Atrigel system to provide effective antipsychotic levels in the first week of treatment, without the need for bridging oral coverage or a second loading injection. The abdominal subcutaneous injection volume is relatively small (0.6 mL or 0.8 mL).
Why Rx? The reasons to prescribe RBP-7000 for adult patients with schizophrenia include:
- no oral coverage required at the initiation of treatment
- effective plasma active moiety levels are seen within the first week without the need for a second loading injection
- monthly injection schedule.
Dosing. The recommended dosage of RBP-7000 is 90 mg or 120 mg once monthly, equivalent to 3 mg/d or 4 mg/d of oral risperidone, respectively. Oral risperidone tolerability should be established before the first injection. No oral risperidone coverage is required. RBP-7000 has not been studied in patients with renal or hepatic impairment and should be used with caution in these patients. Prior to initiating treatment in these patients, it is advised to carefully titrate up to at least 3 mg/d of oral risperidone. If a patient can tolerate 3 mg/d of oral risperidone and is psychiatrically stable, then the 90-mg dose of RBP-7000 can be considered.10
Contraindications. The only contraindications for RBP-7000 are known hypersensitivity to risperidone, paliperidone (9-OH risperidone), or other components of the injection.
Bottom Line
RBP-7000 (Perseris) is the second long-acting injectable (LAI) form of risperidone approved in the U.S. Unlike risperidone microspheres (Consta), RBP-7000 does not require any oral risperidone coverage at the beginning of therapy, provides effective drug levels within the first week of treatment with a single injection, and uses a monthly dosing interval. RBP-7000 does not require loading upon initiation. The monthly injection is <1 mL, is administered in abdominal subcutaneous tissue, and uses the Atrigel system.
Related Resource
- Carpenter J, Wong KK. Long-acting injectable antipsychotics: What to do about missed doses. Current Psychiatry. 2018;17(7):10-12,14-19,56.
Drug Brand Names
Aripiprazole • Abilify
Carbamazepine • Carbatrol, Tegretol
Doxycycline • Atridox
Leuprolide acetate injectable suspension • Eligard
Paliperidone palmitate • Invega Sustenna
Risperidone • Risperdal
Risperidone extended-release injectable suspension • Perseris
Risperidone long-acting injection • Risperdal Consta
1. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
2. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
3. Risperdal Consta [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
4. Nasser AF, Henderson DC, Fava M, et al. Efficacy, safety, and tolerability of RBP-7000 once-monthly risperidone for the treatment of acute schizophrenia: an 8-week, randomized, double-blind, placebo-controlled, multicenter phase 3 study. J Clin Psychopharmacol. 2016;36(2):130-140.
5. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
6. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
7. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
8. Southard GL, Dunn RL, Garrett S. The drug delivery and biomaterial attributes of the ATRIGEL technology in the treatment of periodontal disease. Expert Opin Investig Drugs. 1998;7(9):1483-1491.
9. Gomeni R, Heidbreder C, Fudala PJ, Nasser AF. A model-based approach to characterize the population pharmacokinetics and the relationship between the pharmacokinetic and safety profiles of RBP-7000, a new, long-acting, sustained-released formulation of risperidone. J Clin Pharmacol. 2013;53(10):1010-1019.
10. Perseris [package insert]. North Chesterfield, VA: Indivior Inc; 2018.
11. Malik K, Singh I, Nagpal M, et al. Atrigel: a potential parenteral controlled drug delivery system. Der Pharmacia Sinica. 2010;1(1):74-81.
12. Sartor O. Eligard: leuprolide acetate in a novel sustained-release delivery system. Urology. 2003;61(2 Suppl 1):25-31.
13. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
14. de Leon J, Wynn G, Sandson NB. The pharmacokinetics of paliperidone versus risperidone. Psychosomatics. 2010;51(1):80-88.
15. Ivaturi V, Gopalakrishnan M, Gobburu JVS, et al. Exposure-response analysis after subcutaneous administration of RBP-7000, a once-a-month long-acting Atrigel formulation of risperidone. Br J Clin Pharmacol. 2017;83(7):1476-1498.
16. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetics and prediction of dopamine D2 receptor occupancy after multiple doses of RBP-7000, a new sustained-release formulation of risperidone, in schizophrenia patients on stable oral risperidone treatment. Clin Pharmacokinet. 2014;53(6):533-543.
17. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetic modeling and simulation to guide dose selection for RBP-7000, a new sustained-release formulation of risperidone. J Clin Pharmacol. 2015;55(1):93-103.
1. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
2. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
3. Risperdal Consta [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
4. Nasser AF, Henderson DC, Fava M, et al. Efficacy, safety, and tolerability of RBP-7000 once-monthly risperidone for the treatment of acute schizophrenia: an 8-week, randomized, double-blind, placebo-controlled, multicenter phase 3 study. J Clin Psychopharmacol. 2016;36(2):130-140.
5. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
6. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
7. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
8. Southard GL, Dunn RL, Garrett S. The drug delivery and biomaterial attributes of the ATRIGEL technology in the treatment of periodontal disease. Expert Opin Investig Drugs. 1998;7(9):1483-1491.
9. Gomeni R, Heidbreder C, Fudala PJ, Nasser AF. A model-based approach to characterize the population pharmacokinetics and the relationship between the pharmacokinetic and safety profiles of RBP-7000, a new, long-acting, sustained-released formulation of risperidone. J Clin Pharmacol. 2013;53(10):1010-1019.
10. Perseris [package insert]. North Chesterfield, VA: Indivior Inc; 2018.
11. Malik K, Singh I, Nagpal M, et al. Atrigel: a potential parenteral controlled drug delivery system. Der Pharmacia Sinica. 2010;1(1):74-81.
12. Sartor O. Eligard: leuprolide acetate in a novel sustained-release delivery system. Urology. 2003;61(2 Suppl 1):25-31.
13. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2018.
14. de Leon J, Wynn G, Sandson NB. The pharmacokinetics of paliperidone versus risperidone. Psychosomatics. 2010;51(1):80-88.
15. Ivaturi V, Gopalakrishnan M, Gobburu JVS, et al. Exposure-response analysis after subcutaneous administration of RBP-7000, a once-a-month long-acting Atrigel formulation of risperidone. Br J Clin Pharmacol. 2017;83(7):1476-1498.
16. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetics and prediction of dopamine D2 receptor occupancy after multiple doses of RBP-7000, a new sustained-release formulation of risperidone, in schizophrenia patients on stable oral risperidone treatment. Clin Pharmacokinet. 2014;53(6):533-543.
17. Laffont CM, Gomeni R, Zheng B, et al. Population pharmacokinetic modeling and simulation to guide dose selection for RBP-7000, a new sustained-release formulation of risperidone. J Clin Pharmacol. 2015;55(1):93-103.
Aripiprazole lauroxil nanocrystal suspension

Clinical implications
Nonadherence with oral antipsychotics is a common problem for patients with schizophrenia, one that is often underappreciated by clinicians.5 Whether one uses 70% or 80% as the measure of oral medication adherence, at least 50% of schizophrenia patients are nonadherent, with resultant increased risks for symptom exacerbation and hospitalization.5,6 Although 2 LAI forms of aripiprazole have been introduced over the past few years, neither was designed to be loaded, resulting in the need for 2 or 3 weeks of oral antipsychotic coverage following the first injectable dose.1 The primary reason for LAI antipsychotic therapy is oral medication nonadherence, and thus the need for 14 to 21 days of oral coverage at the outset of treatment creates a risk for symptom exacerbation if the patient is nonadherent with this oral bridging therapy which is needed to achieve the necessary serum concentrations until the long-acting formulation takes over.
One approach was to create a new form of AL using smaller nanomolecular particles rather than the micron-sized particles used for maintenance AL injections.3,4 This nanocrystal suspension is called Aristada Initio (AL

Use in adults with schizophrenia. After establishing tolerability with oral aripiprazole, AL
Continue to: Pharmacologic profile, adverse reactions
Pharmacologic profile, adverse reactions
Aripiprazole is a dopamine partial agonist atypical antipsychotic that has been commercially available in the United States since November 15, 2002, and its adverse effect profile is well characterized. The LAI formulation AL was approved on October 5, 2015. In the pivotal, 12-week, fixed-dose, placebo-controlled clinical trial of AL 441 mg or 882 mg monthly for adults with an acute exacerbation of schizophrenia, the only adverse effect that occurred in ≥5% of AL-treated patients and a rate at least twice that of placebo was akathisia (441 mg: 11%; 882 mg: 11%; placebo: 4%).10 Only 2 of 415 AL-treated patients discontinued the study due to akathisia. Injection-site reactions were reported by 4%, 5%, and 2% of patients treated with AL 441 mg, AL 882 mg, and placebo, respectively. Most of these were injection-site pain associated with the first injection, and decreased with each subsequent injection. Other injection-site reactions (induration, swelling, and redness) occurred at rates <1%.11
Having established that the range of plasma aripiprazole levels consistent with effective treatment is bounded by levels seen with AL 441 mg or 882 mg monthly, the FDA did not require additional efficacy studies for new AL doses provided that pharmacokinetic (PK) studies could demonstrate levels within the effective range. This is consistent with how new doses of other LAI antipsychotic preparations have been addressed in the past. For example, the 37.5 mg dose of risperidone microspheres was approved based on PK data, although the pivotal efficacy trials included doses of 25 mg, 50 mg, and 75 mg.12 Based on PK studies, AL doses of 662 mg monthly, 882 mg every 6 weeks, and 1,064 mg every 8 weeks were previously approved.13 The approval process for AL
Pharmacokinetic outcomes. A comparative phase 1 PK study was performed to evaluate initiation regimens: either 21 days of oral aripiprazole (15 mg/d) and one AL dose (n = 81) or one injection of AL
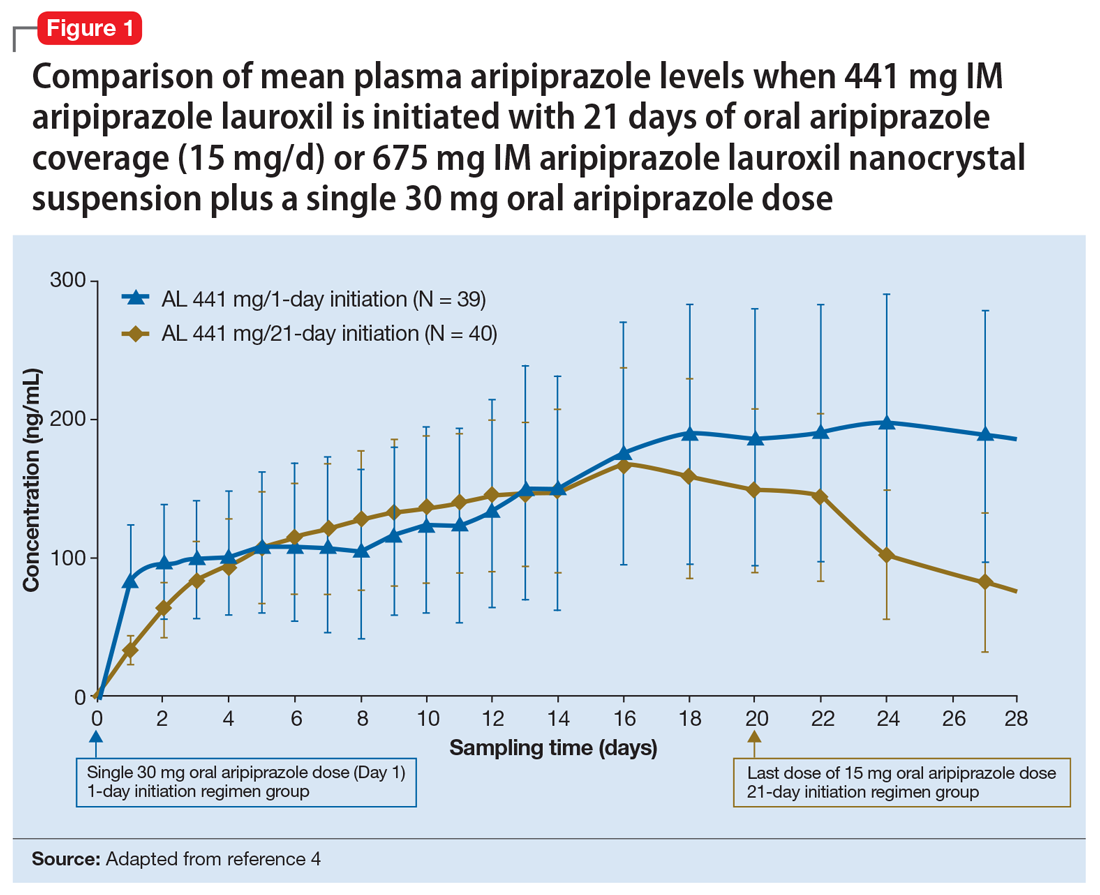
Tolerability. In PK studies, the safety profile and incidences of injection site reactions of AL
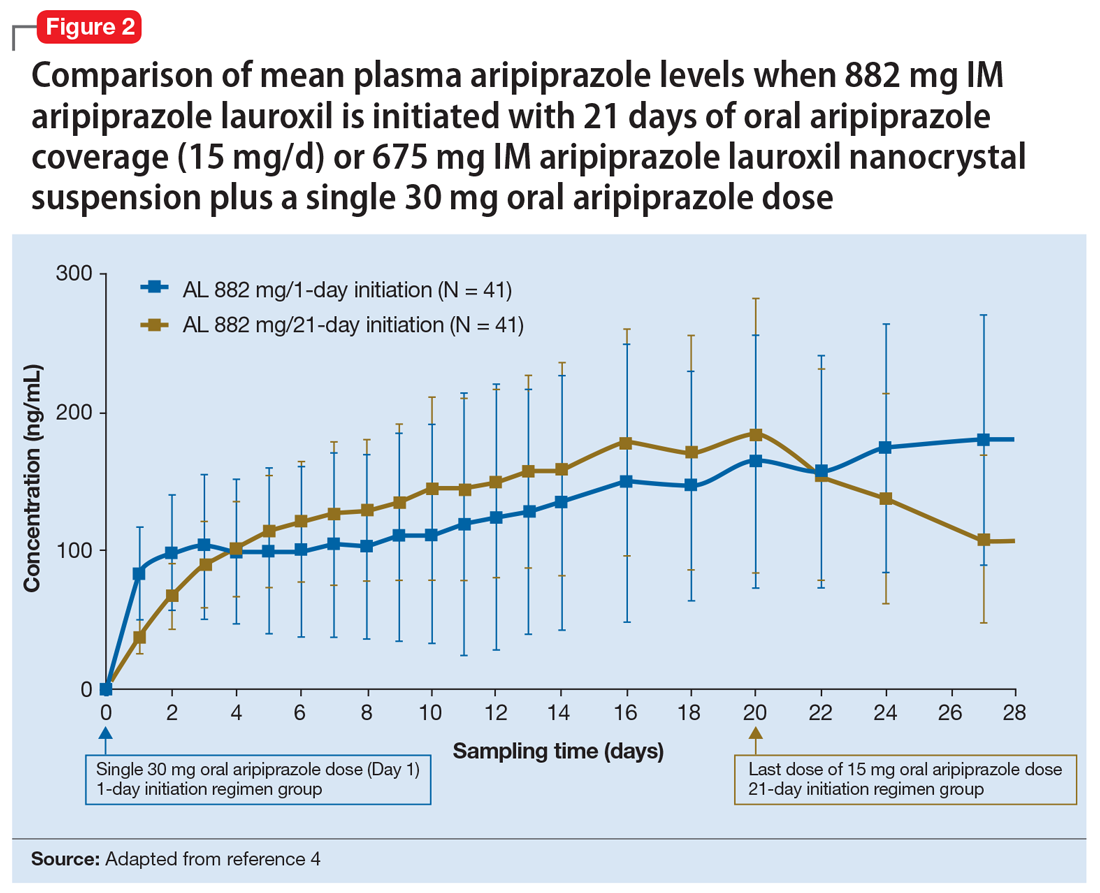
Continue to: Clinical considerations
Clinical considerations
AL
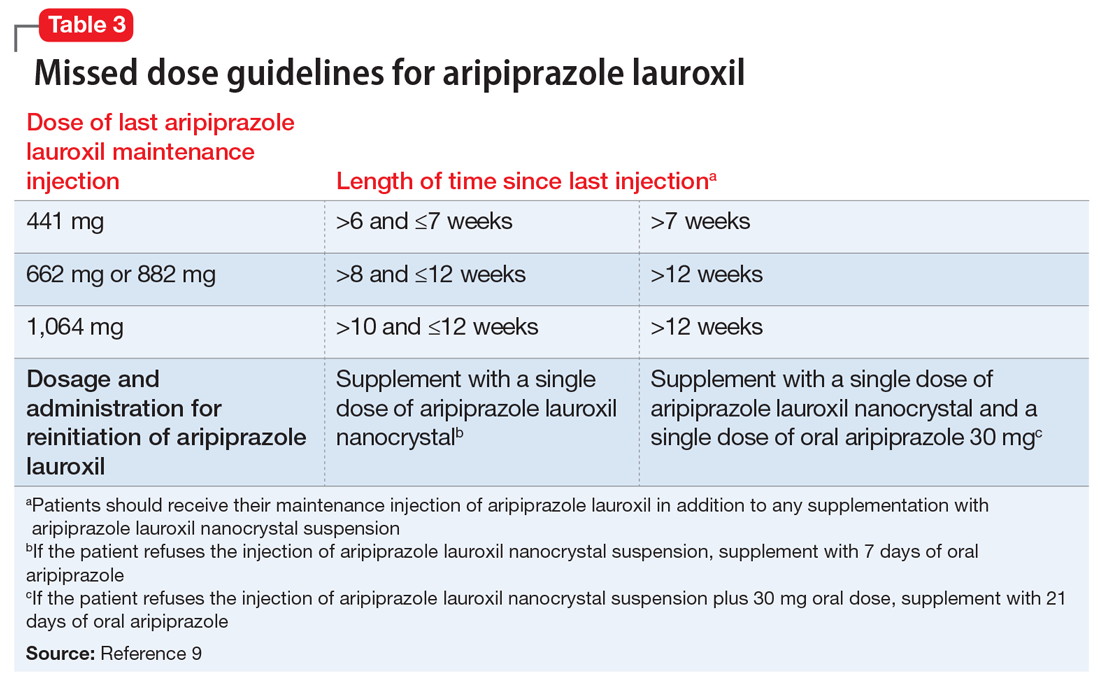
Unique properties. When combined with a single 30 mg oral dose, AL
Why Rx? The reasons to prescribe AL
- it obviates the need for 21 days of oral coverage previously required at the initiation of AL treatment
- clinically relevant plasma levels are seen within the first week when AL
ncd is combined with a single 30 mg oral aripiprazole dose - per the revised missed dose guidelines for AL, it can be used in those situations that previously demanded 7 days of oral coverage, and, when combined with a single 30 mg oral dose, can be used for resumption of therapy after prolonged absences that required 21 days of oral coverage. In all instances, the patient will also receive their usual maintenance dose of AL.
Dosing. There is only one dose available for AL
Contraindications. The only contraindication is a known hypersensitivity to aripiprazole.
Bottom Line
Aripiprazole lauroxil nanocrystal suspension (Aristada Initio) was specifically developed to obviate the need for 21 days of oral aripiprazole coverage when commencing treatment with aripiprazole lauroxil (Aristada). The plasma levels achieved when an injection of aripiprazole lauroxil nanocrystal suspension is combined with a single 30 mg oral dose are comparable to those achieved with 21 days of oral coverage. This initiation regimen, including a aripiprazole lauroxil nanocrystal injection and a 30 mg oral dose, should be administered on the same day as the maintenance aripiprazole lauroxil injection, although the latter can be administered on any of the next 10 days.
Related Resource
- Khan AY, Ovais DM. Long-acting injectable aripiprazole lauroxil for schizophrenia. Current Psychiatry. 2016;15(7):50-52,58.
Drug Brand Names
Aripiprazole lauroxil • Aristada
Aripiprazole lauroxil nanocrystal • Aristada Initio
Risperidone microspheres • Risperdal Consta
1. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
2. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
3. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
4. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
5. Byerly MJ, Thompson A, Carmody T, et al. Validity of electronically monitored medication adherence and conventional adherence measures in schizophrenia. Psychiatric Services. 2007;58(6):844-847.
6. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
7. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
8. Hard ML, Mills RJ, Sadler BM, et al. Aripiprazole lauroxil: pharmacokinetic profile of this long-acting injectable antipsychotic in persons with schizophrenia. J Clin Psychopharmacol. 2017;37(3):289-295.
9. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
10. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
11. Aristada [package insert]. Waltham, MA: Alkermes Inc; 2018.
12. Fleischhacker WW, Eerdekens M, Karcher K, et al. Treatment of schizophrenia with long-acting injectable risperidone: a 12-month open-label trial of the first long-acting second-generation antipsychotic. J Clin Psychiatry. 2003;64(10):1250-1257.
13. Hard ML, Mills RJ, Sadler BM, et al. Pharmacokinetic profile of a 2-month dose regimen of aripiprazole lauroxil: a phase I study and a population pharmacokinetic model. CNS Drugs. 2017;31(7):617-624.
Clinical implications
Nonadherence with oral antipsychotics is a common problem for patients with schizophrenia, one that is often underappreciated by clinicians.5 Whether one uses 70% or 80% as the measure of oral medication adherence, at least 50% of schizophrenia patients are nonadherent, with resultant increased risks for symptom exacerbation and hospitalization.5,6 Although 2 LAI forms of aripiprazole have been introduced over the past few years, neither was designed to be loaded, resulting in the need for 2 or 3 weeks of oral antipsychotic coverage following the first injectable dose.1 The primary reason for LAI antipsychotic therapy is oral medication nonadherence, and thus the need for 14 to 21 days of oral coverage at the outset of treatment creates a risk for symptom exacerbation if the patient is nonadherent with this oral bridging therapy which is needed to achieve the necessary serum concentrations until the long-acting formulation takes over.
One approach was to create a new form of AL using smaller nanomolecular particles rather than the micron-sized particles used for maintenance AL injections.3,4 This nanocrystal suspension is called Aristada Initio (AL
Use in adults with schizophrenia. After establishing tolerability with oral aripiprazole, AL
Continue to: Pharmacologic profile, adverse reactions
Pharmacologic profile, adverse reactions
Aripiprazole is a dopamine partial agonist atypical antipsychotic that has been commercially available in the United States since November 15, 2002, and its adverse effect profile is well characterized. The LAI formulation AL was approved on October 5, 2015. In the pivotal, 12-week, fixed-dose, placebo-controlled clinical trial of AL 441 mg or 882 mg monthly for adults with an acute exacerbation of schizophrenia, the only adverse effect that occurred in ≥5% of AL-treated patients and a rate at least twice that of placebo was akathisia (441 mg: 11%; 882 mg: 11%; placebo: 4%).10 Only 2 of 415 AL-treated patients discontinued the study due to akathisia. Injection-site reactions were reported by 4%, 5%, and 2% of patients treated with AL 441 mg, AL 882 mg, and placebo, respectively. Most of these were injection-site pain associated with the first injection, and decreased with each subsequent injection. Other injection-site reactions (induration, swelling, and redness) occurred at rates <1%.11
Having established that the range of plasma aripiprazole levels consistent with effective treatment is bounded by levels seen with AL 441 mg or 882 mg monthly, the FDA did not require additional efficacy studies for new AL doses provided that pharmacokinetic (PK) studies could demonstrate levels within the effective range. This is consistent with how new doses of other LAI antipsychotic preparations have been addressed in the past. For example, the 37.5 mg dose of risperidone microspheres was approved based on PK data, although the pivotal efficacy trials included doses of 25 mg, 50 mg, and 75 mg.12 Based on PK studies, AL doses of 662 mg monthly, 882 mg every 6 weeks, and 1,064 mg every 8 weeks were previously approved.13 The approval process for AL
Pharmacokinetic outcomes. A comparative phase 1 PK study was performed to evaluate initiation regimens: either 21 days of oral aripiprazole (15 mg/d) and one AL dose (n = 81) or one injection of AL
Tolerability. In PK studies, the safety profile and incidences of injection site reactions of AL
Continue to: Clinical considerations
Clinical considerations
AL
Unique properties. When combined with a single 30 mg oral dose, AL
Why Rx? The reasons to prescribe AL
- it obviates the need for 21 days of oral coverage previously required at the initiation of AL treatment
- clinically relevant plasma levels are seen within the first week when AL
ncd is combined with a single 30 mg oral aripiprazole dose - per the revised missed dose guidelines for AL, it can be used in those situations that previously demanded 7 days of oral coverage, and, when combined with a single 30 mg oral dose, can be used for resumption of therapy after prolonged absences that required 21 days of oral coverage. In all instances, the patient will also receive their usual maintenance dose of AL.
Dosing. There is only one dose available for AL
Contraindications. The only contraindication is a known hypersensitivity to aripiprazole.
Bottom Line
Aripiprazole lauroxil nanocrystal suspension (Aristada Initio) was specifically developed to obviate the need for 21 days of oral aripiprazole coverage when commencing treatment with aripiprazole lauroxil (Aristada). The plasma levels achieved when an injection of aripiprazole lauroxil nanocrystal suspension is combined with a single 30 mg oral dose are comparable to those achieved with 21 days of oral coverage. This initiation regimen, including a aripiprazole lauroxil nanocrystal injection and a 30 mg oral dose, should be administered on the same day as the maintenance aripiprazole lauroxil injection, although the latter can be administered on any of the next 10 days.
Related Resource
- Khan AY, Ovais DM. Long-acting injectable aripiprazole lauroxil for schizophrenia. Current Psychiatry. 2016;15(7):50-52,58.
Drug Brand Names
Aripiprazole lauroxil • Aristada
Aripiprazole lauroxil nanocrystal • Aristada Initio
Risperidone microspheres • Risperdal Consta
Clinical implications
Nonadherence with oral antipsychotics is a common problem for patients with schizophrenia, one that is often underappreciated by clinicians.5 Whether one uses 70% or 80% as the measure of oral medication adherence, at least 50% of schizophrenia patients are nonadherent, with resultant increased risks for symptom exacerbation and hospitalization.5,6 Although 2 LAI forms of aripiprazole have been introduced over the past few years, neither was designed to be loaded, resulting in the need for 2 or 3 weeks of oral antipsychotic coverage following the first injectable dose.1 The primary reason for LAI antipsychotic therapy is oral medication nonadherence, and thus the need for 14 to 21 days of oral coverage at the outset of treatment creates a risk for symptom exacerbation if the patient is nonadherent with this oral bridging therapy which is needed to achieve the necessary serum concentrations until the long-acting formulation takes over.
One approach was to create a new form of AL using smaller nanomolecular particles rather than the micron-sized particles used for maintenance AL injections.3,4 This nanocrystal suspension is called Aristada Initio (AL
Use in adults with schizophrenia. After establishing tolerability with oral aripiprazole, AL
Continue to: Pharmacologic profile, adverse reactions
Pharmacologic profile, adverse reactions
Aripiprazole is a dopamine partial agonist atypical antipsychotic that has been commercially available in the United States since November 15, 2002, and its adverse effect profile is well characterized. The LAI formulation AL was approved on October 5, 2015. In the pivotal, 12-week, fixed-dose, placebo-controlled clinical trial of AL 441 mg or 882 mg monthly for adults with an acute exacerbation of schizophrenia, the only adverse effect that occurred in ≥5% of AL-treated patients and a rate at least twice that of placebo was akathisia (441 mg: 11%; 882 mg: 11%; placebo: 4%).10 Only 2 of 415 AL-treated patients discontinued the study due to akathisia. Injection-site reactions were reported by 4%, 5%, and 2% of patients treated with AL 441 mg, AL 882 mg, and placebo, respectively. Most of these were injection-site pain associated with the first injection, and decreased with each subsequent injection. Other injection-site reactions (induration, swelling, and redness) occurred at rates <1%.11
Having established that the range of plasma aripiprazole levels consistent with effective treatment is bounded by levels seen with AL 441 mg or 882 mg monthly, the FDA did not require additional efficacy studies for new AL doses provided that pharmacokinetic (PK) studies could demonstrate levels within the effective range. This is consistent with how new doses of other LAI antipsychotic preparations have been addressed in the past. For example, the 37.5 mg dose of risperidone microspheres was approved based on PK data, although the pivotal efficacy trials included doses of 25 mg, 50 mg, and 75 mg.12 Based on PK studies, AL doses of 662 mg monthly, 882 mg every 6 weeks, and 1,064 mg every 8 weeks were previously approved.13 The approval process for AL
Pharmacokinetic outcomes. A comparative phase 1 PK study was performed to evaluate initiation regimens: either 21 days of oral aripiprazole (15 mg/d) and one AL dose (n = 81) or one injection of AL
Tolerability. In PK studies, the safety profile and incidences of injection site reactions of AL
Continue to: Clinical considerations
Clinical considerations
AL
Unique properties. When combined with a single 30 mg oral dose, AL
Why Rx? The reasons to prescribe AL
- it obviates the need for 21 days of oral coverage previously required at the initiation of AL treatment
- clinically relevant plasma levels are seen within the first week when AL
ncd is combined with a single 30 mg oral aripiprazole dose - per the revised missed dose guidelines for AL, it can be used in those situations that previously demanded 7 days of oral coverage, and, when combined with a single 30 mg oral dose, can be used for resumption of therapy after prolonged absences that required 21 days of oral coverage. In all instances, the patient will also receive their usual maintenance dose of AL.
Dosing. There is only one dose available for AL
Contraindications. The only contraindication is a known hypersensitivity to aripiprazole.
Bottom Line
Aripiprazole lauroxil nanocrystal suspension (Aristada Initio) was specifically developed to obviate the need for 21 days of oral aripiprazole coverage when commencing treatment with aripiprazole lauroxil (Aristada). The plasma levels achieved when an injection of aripiprazole lauroxil nanocrystal suspension is combined with a single 30 mg oral dose are comparable to those achieved with 21 days of oral coverage. This initiation regimen, including a aripiprazole lauroxil nanocrystal injection and a 30 mg oral dose, should be administered on the same day as the maintenance aripiprazole lauroxil injection, although the latter can be administered on any of the next 10 days.
Related Resource
- Khan AY, Ovais DM. Long-acting injectable aripiprazole lauroxil for schizophrenia. Current Psychiatry. 2016;15(7):50-52,58.
Drug Brand Names
Aripiprazole lauroxil • Aristada
Aripiprazole lauroxil nanocrystal • Aristada Initio
Risperidone microspheres • Risperdal Consta
1. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
2. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
3. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
4. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
5. Byerly MJ, Thompson A, Carmody T, et al. Validity of electronically monitored medication adherence and conventional adherence measures in schizophrenia. Psychiatric Services. 2007;58(6):844-847.
6. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
7. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
8. Hard ML, Mills RJ, Sadler BM, et al. Aripiprazole lauroxil: pharmacokinetic profile of this long-acting injectable antipsychotic in persons with schizophrenia. J Clin Psychopharmacol. 2017;37(3):289-295.
9. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
10. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
11. Aristada [package insert]. Waltham, MA: Alkermes Inc; 2018.
12. Fleischhacker WW, Eerdekens M, Karcher K, et al. Treatment of schizophrenia with long-acting injectable risperidone: a 12-month open-label trial of the first long-acting second-generation antipsychotic. J Clin Psychiatry. 2003;64(10):1250-1257.
13. Hard ML, Mills RJ, Sadler BM, et al. Pharmacokinetic profile of a 2-month dose regimen of aripiprazole lauroxil: a phase I study and a population pharmacokinetic model. CNS Drugs. 2017;31(7):617-624.
1. Meyer JM. Converting oral to long acting injectable antipsychotics: a guide for the perplexed. CNS Spectrums. 2017;22(S1):14-28.
2. Kishimoto T, Hagi K, Nitta M, et al. Effectiveness of long-acting injectable vs oral antipsychotics in patients with schizophrenia: a meta-analysis of prospective and retrospective cohort studies. Schizophr Bull. 2018;44(3):603-619.
3. Hard ML, Wehr AY, Sadler BM, et al. Population pharmacokinetic analysis and model-based simulations of aripiprazole for a 1-day initiation regimen for the long-acting antipsychotic aripiprazole lauroxil. Eur J Drug Metab Pharmacokinet. 2018;43(4):461-469.
4. Hard ML, Wehr AY, Du Y, et al. Pharmacokinetic evaluation of a 1-day treatment initiation option for starting long-acting aripiprazole lauroxil for schizophrenia. J Clin Psychopharmacol. 2018;38(5):435-441.
5. Byerly MJ, Thompson A, Carmody T, et al. Validity of electronically monitored medication adherence and conventional adherence measures in schizophrenia. Psychiatric Services. 2007;58(6):844-847.
6. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
7. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
8. Hard ML, Mills RJ, Sadler BM, et al. Aripiprazole lauroxil: pharmacokinetic profile of this long-acting injectable antipsychotic in persons with schizophrenia. J Clin Psychopharmacol. 2017;37(3):289-295.
9. Aristada Initio [package insert]. Waltham, MA: Alkermes Inc; 2018.
10. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
11. Aristada [package insert]. Waltham, MA: Alkermes Inc; 2018.
12. Fleischhacker WW, Eerdekens M, Karcher K, et al. Treatment of schizophrenia with long-acting injectable risperidone: a 12-month open-label trial of the first long-acting second-generation antipsychotic. J Clin Psychiatry. 2003;64(10):1250-1257.
13. Hard ML, Mills RJ, Sadler BM, et al. Pharmacokinetic profile of a 2-month dose regimen of aripiprazole lauroxil: a phase I study and a population pharmacokinetic model. CNS Drugs. 2017;31(7):617-624.
Deutetrabenazine for tardive dyskinesia
Compared with first-generation antipsychotics, second-generation antipsychotics (SGAs) have a lower risk for extrapyramidal symptoms. Yet tardive dyskinesia (TD) remains a concern because of the widespread use of SGAs for multiple indications.1 Prior to April 2017, clinicians had no FDA-approved TD treatment options. The most widely used agent worldwide, tetrabenazine, had positive efficacy data in TD trials over the past 45 years but was not available in the United States until 2008, and its sole indication was for chorea associated with Huntington’s disease.2 Moreover, the use of tetrabenazine involved slow titration, multiple daily dosing, cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, and tolerability issues.
Tetrabenazine is an inhibitor of vesicular monoamine transport type 2 (VMAT2), a transport protein located almost exclusively in the CNS whose role is to place monoamine neurotransmitters (dopamine, serotonin, norepinephrine) into presynaptic vesicles. By decreasing dopamine transport into these presynaptic vesicles, synaptic dopamine release is lessened, thus reducing postsynaptic dopamine D2 receptor activity and the severity of dyskinetic movements.1
In 2 pivotal 12-week clinical trials, deutetrabenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) scores (see Efficacy).6,7
Clinical implications
TD remains a substantial public health concern due to the increasing use of antipsychotics for mood and other disorders beyond the initial indications for schizophrenia.1 Although exposure to dopamine D2antagonism results in postsynaptic receptor upregulation and supersensitivity that underlies the development of dyskinesia, this process is often rapidly reversible in animal models.1 The persistence of TD symptoms in up to 80% of patients after dopamine receptor blocking agents (DRBAs) are stopped has led to hypotheses that the underlying pathophysiology of TD is also a problem with neuroplasticity. Aside from DRBA exposure, environmental factors (eg, oxidative stress) and genetic predisposition might contribute to TD risk.1
Before 2017, only 1 medication (branched-chain amino acids) had been FDA-approved for treating TD in the United States, and only a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]) had positive results from controlled trials, most with small effect sizes.8 Moreover, there was only 1 controlled trial each for clonazepam and EGb-761.1 A branched-chain amino acid preparation received FDA approval for managing TD in male patients, but is no longer commercially available, except from compounding pharmacies.9
Tetrabenazine was developed in the mid-1950s to avoid orthostasis and sedation associated with reserpine.10 Both reserpine and tetrabenazine proved effective for TD,11 but tetrabenazine lacked reserpine’s peripheral adverse effects. However, the kinetics of tetrabenazine necessitated multiple daily doses, and CYP2D6 genotyping was required for doses >50 mg/d.2
Receptor blocking. The mechanism that distinguishes the clinical profiles of reserpine and tetrabenazine relates to their differential properties at VMAT.12 VMAT exists in 2 forms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the CNS.13 Tetrabenazine is a specific and reversible VMAT2 inhibitor, whereas reserpine is an irreversible and nonselective antagonist of VMAT1 and VMAT2. It is reserpine’s VMAT1 inhibition that results in peripheral adverse effects such as orthostasis. Tetrabenazine is rapidly and extensively converted into 2 isomers, alpha-dihydrotetrabenazine (α-DHTBZ) and beta-dihydrotetrabenazine (β-DHTBZ), both of which are metabolized by CYP2D6, with a role for CYP3A4 in α-DHTBZ metabolism.1 These DHTBZ metabolites have a short half-life when generated from oral tetrabenazine, a feature that necessitates multiple daily dosing; moreover, the existence of 2D6 polymorphisms led to FDA-mandated CYP2D6 genotyping for tetrabenazine doses >50 mg/d when it was approved for Huntington’s chorea. The concern is that 2D6 poor metabolizers will have excessive exposure to the VMAT2 effects of DHTBZ, resulting in sedation, akathisia, parkinsonism, and mood symptoms.2
How deuterium impacts medication kinetics. Deuterium is a naturally occurring, stable, nontoxic isotope of hydrogen. Humans have 5 g of deuterium in their body at any time, mostly in the form of heavy water (D2O).14 When deuterium is used to replace selected hydrogen atoms, the resulting molecule will have similar configuration and receptor-binding properties but markedly different kinetics. Because the carbon–deuterium covalent bond requires 8 times more energy to break than a carbon–hydrogen bond, the half-life is prolonged.15 Utilizing this knowledge, a deuterated form of tetrabenazine, deutetrabenazine, was synthesized with such a purpose in mind. While the active metabolites of deutetrabenazine retain the VMAT2 affinity of non-deuterated tetrabenazine, the substitution of deuterium for hydrogen at specific positions slows the breakdown of metabolites, resulting in sustained duration of action, greater active drug exposure, and less impact of 2D6 genotype on drug exposure, thus eliminating the need for genotyping, unless one wants to exceed 36 mg/d.
Deutetrabenazine was first studied in Huntington’s chorea in a 13-week, double-blind, placebo-controlled, parallel-group study (N = 90).4 The maximum daily deutetrabenazine dose was 48 mg, but reduced to 36 mg in those taking strong CYP2D6 inhibitors (bupropion, fluoxetine, or paroxetine). Blinded 2D6 genotyping was performed, but there was no dose modification required based on 2D6 genotype. There was a 36.4% reduction in total maximal chorea score for deutetrabenazine compared with 14.4% for placebo (P < .001).4 Importantly, adverse effects were comparable between both groups, with 1 drop-out in the deutetrabenazine arm vs 2 in the placebo arm. The only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo was somnolence: 11.1% for deutetrabenazine vs 4.4% for placebo.4 The mean deutetrabenazine daily dose at the end of the treatment period was 39.7 ± 9.3 mg, and for those with impaired CYP2D6 function (poor metabolizers or those taking strong CYP2D6 inhibiting medications), the mean daily dose was 34.8 mg ± 3.8 mg.4
Use in tardive dyskinesia. The recommended starting dosage for TD treatment is 6 mg, twice daily with food. The dose may be increased at weekly intervals in increments of 6 mg/d to a maximum recommended daily dosage of 48 mg.5 The maximum daily dose is 36 mg (18 mg, twice daily) in patients receiving strong CYP2D6 inhibitors or who are 2D6 poor metabolizers.5
Deutetrabenazine has not been studied in those with moderate or severe hepatic impairment, and its use is contraindicated in these patients.5 No clinical studies have been conducted to assess the effect of renal impairment on the pharmacokinetics of deutetrabenazine.5
Pharmacologic profile, adverse reactions
When the data from the two 12-week, phase 3 placebo-controlled studies were pooled, the most common adverse reactions occurring in >3% of deutetrabenazine patients and greater than placebo were nasopharyngeal symptoms (4% vs 2% placebo) and insomnia (4% vs 1% placebo).5 Importantly, in neither TD study were there clinically significant changes in rating scales for depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms. The mean QT prolongation for a single 24 mg dose of deutetrabenazine in healthy volunteers was 4.5 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 6.5 milliseconds.5 For tetrabenazine, single 50 mg doses administered to volunteers resulted in mean QT prolongation of 8 milliseconds.5 In patients requiring deutetrabenazine doses >24 mg/d who are taking other medications known to prolong QTc, assess the QTc interval before and after increasing the dose of deutetrabenazine or other medications that are known to prolong QTc.5
How it works
Tetrabenazine is the only agent that has demonstrated significant efficacy for TD management, but its use involves slow titration, multiple daily dosing, CYP2D6 genotyping for doses >50 mg/d, and tolerability issues. For example, the most common adverse effects in the pivotal tetrabenazine Huntington’s disease trial were sedation/somnolence (tetrabenazine 31% vs 3% for placebo), insomnia (tetrabenazine 22% vs 0% for placebo), depression (tetrabenazine 19% vs 0% for placebo), fatigue (tetrabenazine 22% vs 13% for placebo), and akathisia (tetrabenazine 19% vs 0% for placebo).2 For comparison, the only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo in the pivotal Huntington’s disease trial was somnolence (11.1% for deutetrabenazine vs 4.4% for placebo).4
Pharmacokinetics
Deutetrabenazine has 80% oral bioavailability, and is rapidly converted to its active metabolites after oral dosing (Table 2).5 Linear dose dependence of Cmax and area under the curve (AUC) was observed for the active metabolites following single or multiple doses of deutetrabenazine (6 to 24 mg and 7.5 to 22.5 mg, twice daily).15 Cmax of deuterated α-DHTBZ and β-DHTBZ is reached within 3 to 4 hours after dosing, with a steady state ratio of 3:1 for the α-DHTBZ vs the β-DHTBZ form. Food had no effect on AUC, but did increase Cmax by 50%.5
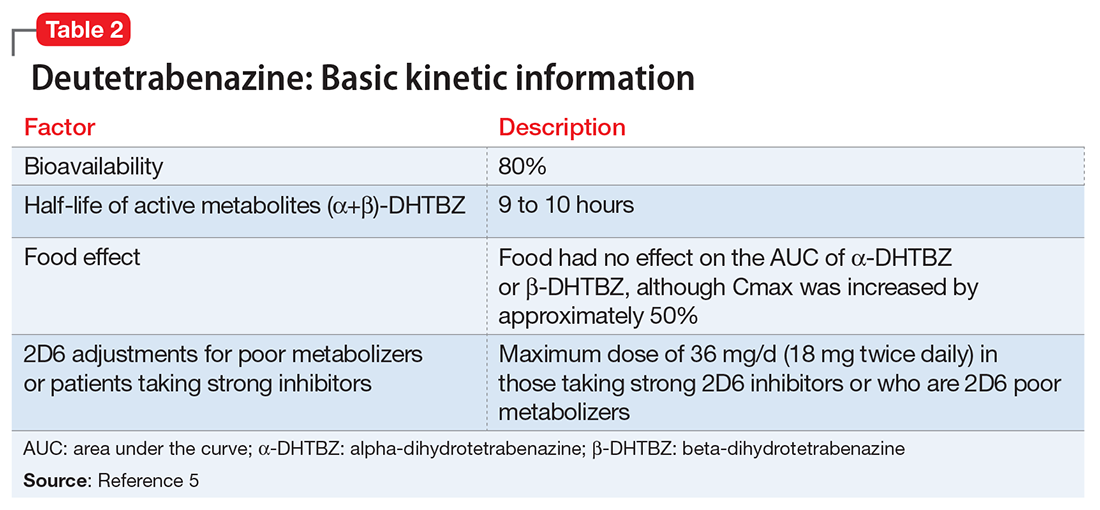
Deutetrabenazine is metabolized through carbonyl reductase enzymes to its active metabolites, and these are further metabolized through multiple CYP pathways, predominantly 2D6 and to a lesser extent 3A4. The effect of CYP2D6 inhibition on the pharmacokinetics of deutetrabenazine and its α-DHTBZ and β-DHTBZ metabolites was studied in 24 healthy participants following a single 22.5 mg dose of deutetrabenazine given after 8 days of administration of the strong CYP2D6 inhibitor paroxetine, 20 mg/d. In the presence of paroxetine, systemic exposure (AUC) of α-DHTBZ was 1.9-fold higher and β-DHTBZ was 6.5-fold higher, resulting in an approximately 3-fold increase in AUC for total (α+β)-DHTBZ, with corresponding increases in mean half-life of approximately 1.5-fold and 2.7-fold, respectively.5 Neither deutetrabenazine or its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that deutetrabenazine and its active metabolites are unlikely to inhibit most major drug transporters at clinically relevant concentrations.
Efficacy
Efficacy was established in two 12-week, double-blind, placebo-controlled trials of adult patients with TD (ages 18 to 80).6,7 Eligible participants had:
- TD diagnosis for ≥3 months before screening and a history of DRBA treatment for ≥3 months (≥1 month if age ≥60)
- Total AIMS motor score ≥6 (items 1 to 7) at both screening and baseline, verified by a blinded central rater at screening via central video rating
- Patients with an underlying psychiatric illness had to be stable. Psychoactive medication use, including antipsychotics, was allowed if stable for ≥30 days before screening (antidepressants, ≥45 days).
Exclusion criteria included treatment with tetrabenazine, reserpine, α-methyl-p-tyrosine, strong anticholinergic medications, dopamine antagonizing antiemetics (eg, metoclopramide, prochlorperazine, promethazine), dopamine agonists, levodopa, stimulants, or a monoamine oxidase inhibitor (MAOI) within 30 days of screening or baseline, or treatment with botulinum toxin within 3 months of screening; and presence of a neurologic condition that could confound TD assessments, serious untreated or undertreated psychiatric illness, or unstable medical illness. Patients with a history of or active suicidal ideation or behavior within 6 months of screening or score ≥11 on the depression subscale of the Hospital Anxiety and Depression Scale were excluded. Those participants with Fridericia-corrected QT interval values >450 milliseconds in men, >460 milliseconds in women, or >480 milliseconds in patients with a right bundle branch block on electrocardiography at screening also were excluded.
The flexible-dose TD study was performed in 117 participants randomized in a 1:1 manner to deutetrabenazine or placebo, both administered twice daily, titrated to optimal dosage (12 to 48 mg/d) over 6 weeks, and then administered at that dose for another 6 weeks.7 The population demographics were: mean age, 54.6 ± 10.3 years, 52.1% female, 69.2% white, and 80.3% receiving ongoing dopamine antagonists, with a mean TD duration of 74.7 ± 81.5 months. Sixty-eight percent had schizophrenia spectrum disorders, and 30% had mood disorders. The primary outcome was change in total AIMS score (items 1 to 7) assessed by central, independent raters. The mean baseline AIMS score for items 1 to 7 was 9.6 ± 3.9, with 82.9% of participants with baseline AIMS scores ≥6. Study treatment retention was high: placebo 88.1%, deutetrabenazine 89.7%.7 There was a mean 3 point decrease in AIMS score for deutetrabenazine compared with 1.4 for placebo (P = .019). Among those with baseline AIMS scores ≥6, there was a 3.4 point decrease in AIMS scores for deutetrabenazine compared with a 1.9 point decrease for placebo (P = .027). The only adverse effects that occurred in ≥5% of deutetrabenazine participants and at a rate ≥2 times the rate in placebo were insomnia (deutetrabenazine 6.9% vs placebo 1.7%) and akathisia (deutetrabenazine 5.2% vs placebo 0%).
The fixed-dose TD study was performed in 293 participants randomized in 1:1:1:1 manner to 1 of 3 fixed doses of deutetrabenazine (12 mg/d, 24 mg/d, or 36 mg/d) or placebo, both administered twice daily.6 The starting dose of deutetrabenazine was 6 mg twice daily. During the dose escalation period (through Week 4), the dose of study drug was increased weekly in increments of 6 mg/d until the randomized dose was achieved. Patients continued to receive the dose they were assigned to over a maintenance period of 8 weeks.6 The population demographics were: mean age, 56.4 ± 11.3 years, 55% female, 79% white, 76% receiving ongoing dopamine antagonists, with a mean TD duration of 67.2 ± 66 months. Sixty percent had schizophrenia spectrum disorders, and 36% had mood disorders. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. The mean AIMS score at baseline was 9.5 ± 2.7 in the placebo group, and for deutetrabenazine: 9.6 ± 2.4 in the 12 mg/d group, 9.4 ± 2.9 in the 24 mg/d group, and 10.1 ± 3.2 in the 36 mg/d group. The 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). Study treatment retention rates were high: placebo 90.5%, deutetrabenazine 88%. Across all doses, only 1 adverse effect occurred in ≥5% of deutetrabenazine participants: headache (5% deutetrabenazine vs 6% placebo). At the highest dose, 36 mg/d, the only adverse effects that occurred in ≥5% of participants were diarrhea (7% deutetrabenazine vs 3% placebo) and headache (7% deutetrabenazine vs 6% placebo).
Outcome. In the flexible-dose study (mean dose 38.8 ± 7.92 mg/d), the deutetrabenazine arm experienced a mean 30% reduction in AIMS scores from baseline at the Week 12 endpoint. Compared with placebo, the mean reduction in AIMS scores (standard error) was: −3.0 (0.45) deutetrabenazine vs −1.6 (0.46) placebo (P = .019).7 For the fixed-dose study, the 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). In addition to these mean changes from baseline, 35% of the 24 mg/d group and 33% of the 36 mg/d group demonstrated ≥50% reduction in AIMS scores.6
Tolerability
In the 2 phase 3 trials, there were no adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo.5 Discontinuations because of adverse events were low in both pivotal studies across all treatment groups: 3.4% for placebo vs 1.7% for deutetrabenazine in the flexible-dose trial,7 and 3% for placebo vs 4% for deutetrabenazine in the fixed-dose study.6 In neither trial were there clinically significant changes in ratings of depression, suicidality, parkinsonism, or schizophrenia symptoms. The mean QT prolongation in healthy volunteers is described above.
Clinical considerations
Unique properties. Deutetrabenazine utilizes the greater bond strength of the carbon–deuterium bond to slow CYP metabolism, resulting in prolonged duration of action that is well tolerated, and provides significant efficacy.
Why Rx? The reasons to prescribe deutetrabenazine for TD patients include:
- only 1 of 2 agents with FDA approval for TD
- fewer tolerability issues than with tetrabenazine
- lower sedation rates in TD trials than with valbenazine
- no signal for effects on mood parameters or rates of parkinsonism when used for TD.
Dosing
The recommended starting dosage of deutetrabenazine is 6 mg twice daily taken with food, increasing by 6 mg/d weekly as needed, with a maximum dose of 48 mg/d or 36 mg/d in those taking strong CYP2D6 inhibitors or who are 2D6 poor metabolizers. Deutetrabenazine is contraindicated in patients with hepatic impairment (as determined by Child-Pugh criteria16). There are no data in patients with renal impairment. The combined efficacy and tolerability of dosages >48 mg/d has not been evaluated. Overdoses of tetrabenazine ranging from 100 to 1,000 mg have been reported in the literature and were associated with acute dystonia, oculogyric crisis, nausea and vomiting, sweating, sedation, hypotension, confusion, diarrhea, hallucinations, rubor, and tremor.5
Contraindications
When used for TD, deutetrabenazine is contraindicated for patients taking reserpine, tetrabenazine, valbenazine, or MAOIs, and for patients with hepatic impairment. As with most medications, there are no data on deutetrabenazine use in pregnant women; however, oral administration of deutetrabenazine (5, 10, or 30 mg/kg/d) or tetrabenazine (30 mg/kg/d) to pregnant rats during organogenesis had no clear effect on embryofetal development. The highest dose tested was 6 times the maximum recommended human dose of 48 mg/d on a body surface area (mg/m2) basis. There are no data on the presence of deutetrabenazine or its metabolites in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.
1. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
2. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
3. Meyer JM. Valbenazine for tardive dyskinesia. Current Psychiatry. 2017;16(5):40-46.
4. Huntington Study Group; Frank S, Testa CM, Stamler D, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA. 2016;316(1):40-50.
5. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals USA, Inc.; 2017.
6. Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595-604.
7. Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003-2010.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Richardson MA, Small AM, Read LL, et al. Branched chain amino acid treatment of tardive dyskinesia in children and adolescents. J Clin Psychiatry. 2004;65(1):92-96.
10. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
11. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
12. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
13. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
14. Kushner DJ, Baker A, Dunstall TG. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can J Physiol Pharmacol. 1999;77(2):79-88.
15. United States Securities and Exchange Commission. Form S-1 Registration Statement of Auspex Pharmaceuticals, Inc. https://www.sec.gov/Archives/edgar/data/1454189/000119312513481239/d627086ds1.htm. Published December 20, 2013. Accessed July 1, 2016.
16. Cholongitas E, Papatheodoridis GV, Vangeli M, et al. Systematic review: the model for end-stage liver disease—should it replace Child-Pugh’s classification for assessing prognosis in cirrhosis? Aliment Pharmacol Ther. 2005;22(11-12):1079-1089.
Compared with first-generation antipsychotics, second-generation antipsychotics (SGAs) have a lower risk for extrapyramidal symptoms. Yet tardive dyskinesia (TD) remains a concern because of the widespread use of SGAs for multiple indications.1 Prior to April 2017, clinicians had no FDA-approved TD treatment options. The most widely used agent worldwide, tetrabenazine, had positive efficacy data in TD trials over the past 45 years but was not available in the United States until 2008, and its sole indication was for chorea associated with Huntington’s disease.2 Moreover, the use of tetrabenazine involved slow titration, multiple daily dosing, cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, and tolerability issues.
Tetrabenazine is an inhibitor of vesicular monoamine transport type 2 (VMAT2), a transport protein located almost exclusively in the CNS whose role is to place monoamine neurotransmitters (dopamine, serotonin, norepinephrine) into presynaptic vesicles. By decreasing dopamine transport into these presynaptic vesicles, synaptic dopamine release is lessened, thus reducing postsynaptic dopamine D2 receptor activity and the severity of dyskinetic movements.1
In 2 pivotal 12-week clinical trials, deutetrabenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) scores (see Efficacy).6,7
Clinical implications
TD remains a substantial public health concern due to the increasing use of antipsychotics for mood and other disorders beyond the initial indications for schizophrenia.1 Although exposure to dopamine D2antagonism results in postsynaptic receptor upregulation and supersensitivity that underlies the development of dyskinesia, this process is often rapidly reversible in animal models.1 The persistence of TD symptoms in up to 80% of patients after dopamine receptor blocking agents (DRBAs) are stopped has led to hypotheses that the underlying pathophysiology of TD is also a problem with neuroplasticity. Aside from DRBA exposure, environmental factors (eg, oxidative stress) and genetic predisposition might contribute to TD risk.1
Before 2017, only 1 medication (branched-chain amino acids) had been FDA-approved for treating TD in the United States, and only a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]) had positive results from controlled trials, most with small effect sizes.8 Moreover, there was only 1 controlled trial each for clonazepam and EGb-761.1 A branched-chain amino acid preparation received FDA approval for managing TD in male patients, but is no longer commercially available, except from compounding pharmacies.9
Tetrabenazine was developed in the mid-1950s to avoid orthostasis and sedation associated with reserpine.10 Both reserpine and tetrabenazine proved effective for TD,11 but tetrabenazine lacked reserpine’s peripheral adverse effects. However, the kinetics of tetrabenazine necessitated multiple daily doses, and CYP2D6 genotyping was required for doses >50 mg/d.2
Receptor blocking. The mechanism that distinguishes the clinical profiles of reserpine and tetrabenazine relates to their differential properties at VMAT.12 VMAT exists in 2 forms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the CNS.13 Tetrabenazine is a specific and reversible VMAT2 inhibitor, whereas reserpine is an irreversible and nonselective antagonist of VMAT1 and VMAT2. It is reserpine’s VMAT1 inhibition that results in peripheral adverse effects such as orthostasis. Tetrabenazine is rapidly and extensively converted into 2 isomers, alpha-dihydrotetrabenazine (α-DHTBZ) and beta-dihydrotetrabenazine (β-DHTBZ), both of which are metabolized by CYP2D6, with a role for CYP3A4 in α-DHTBZ metabolism.1 These DHTBZ metabolites have a short half-life when generated from oral tetrabenazine, a feature that necessitates multiple daily dosing; moreover, the existence of 2D6 polymorphisms led to FDA-mandated CYP2D6 genotyping for tetrabenazine doses >50 mg/d when it was approved for Huntington’s chorea. The concern is that 2D6 poor metabolizers will have excessive exposure to the VMAT2 effects of DHTBZ, resulting in sedation, akathisia, parkinsonism, and mood symptoms.2
How deuterium impacts medication kinetics. Deuterium is a naturally occurring, stable, nontoxic isotope of hydrogen. Humans have 5 g of deuterium in their body at any time, mostly in the form of heavy water (D2O).14 When deuterium is used to replace selected hydrogen atoms, the resulting molecule will have similar configuration and receptor-binding properties but markedly different kinetics. Because the carbon–deuterium covalent bond requires 8 times more energy to break than a carbon–hydrogen bond, the half-life is prolonged.15 Utilizing this knowledge, a deuterated form of tetrabenazine, deutetrabenazine, was synthesized with such a purpose in mind. While the active metabolites of deutetrabenazine retain the VMAT2 affinity of non-deuterated tetrabenazine, the substitution of deuterium for hydrogen at specific positions slows the breakdown of metabolites, resulting in sustained duration of action, greater active drug exposure, and less impact of 2D6 genotype on drug exposure, thus eliminating the need for genotyping, unless one wants to exceed 36 mg/d.
Deutetrabenazine was first studied in Huntington’s chorea in a 13-week, double-blind, placebo-controlled, parallel-group study (N = 90).4 The maximum daily deutetrabenazine dose was 48 mg, but reduced to 36 mg in those taking strong CYP2D6 inhibitors (bupropion, fluoxetine, or paroxetine). Blinded 2D6 genotyping was performed, but there was no dose modification required based on 2D6 genotype. There was a 36.4% reduction in total maximal chorea score for deutetrabenazine compared with 14.4% for placebo (P < .001).4 Importantly, adverse effects were comparable between both groups, with 1 drop-out in the deutetrabenazine arm vs 2 in the placebo arm. The only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo was somnolence: 11.1% for deutetrabenazine vs 4.4% for placebo.4 The mean deutetrabenazine daily dose at the end of the treatment period was 39.7 ± 9.3 mg, and for those with impaired CYP2D6 function (poor metabolizers or those taking strong CYP2D6 inhibiting medications), the mean daily dose was 34.8 mg ± 3.8 mg.4
Use in tardive dyskinesia. The recommended starting dosage for TD treatment is 6 mg, twice daily with food. The dose may be increased at weekly intervals in increments of 6 mg/d to a maximum recommended daily dosage of 48 mg.5 The maximum daily dose is 36 mg (18 mg, twice daily) in patients receiving strong CYP2D6 inhibitors or who are 2D6 poor metabolizers.5
Deutetrabenazine has not been studied in those with moderate or severe hepatic impairment, and its use is contraindicated in these patients.5 No clinical studies have been conducted to assess the effect of renal impairment on the pharmacokinetics of deutetrabenazine.5
Pharmacologic profile, adverse reactions
When the data from the two 12-week, phase 3 placebo-controlled studies were pooled, the most common adverse reactions occurring in >3% of deutetrabenazine patients and greater than placebo were nasopharyngeal symptoms (4% vs 2% placebo) and insomnia (4% vs 1% placebo).5 Importantly, in neither TD study were there clinically significant changes in rating scales for depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms. The mean QT prolongation for a single 24 mg dose of deutetrabenazine in healthy volunteers was 4.5 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 6.5 milliseconds.5 For tetrabenazine, single 50 mg doses administered to volunteers resulted in mean QT prolongation of 8 milliseconds.5 In patients requiring deutetrabenazine doses >24 mg/d who are taking other medications known to prolong QTc, assess the QTc interval before and after increasing the dose of deutetrabenazine or other medications that are known to prolong QTc.5
How it works
Tetrabenazine is the only agent that has demonstrated significant efficacy for TD management, but its use involves slow titration, multiple daily dosing, CYP2D6 genotyping for doses >50 mg/d, and tolerability issues. For example, the most common adverse effects in the pivotal tetrabenazine Huntington’s disease trial were sedation/somnolence (tetrabenazine 31% vs 3% for placebo), insomnia (tetrabenazine 22% vs 0% for placebo), depression (tetrabenazine 19% vs 0% for placebo), fatigue (tetrabenazine 22% vs 13% for placebo), and akathisia (tetrabenazine 19% vs 0% for placebo).2 For comparison, the only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo in the pivotal Huntington’s disease trial was somnolence (11.1% for deutetrabenazine vs 4.4% for placebo).4
Pharmacokinetics
Deutetrabenazine has 80% oral bioavailability, and is rapidly converted to its active metabolites after oral dosing (Table 2).5 Linear dose dependence of Cmax and area under the curve (AUC) was observed for the active metabolites following single or multiple doses of deutetrabenazine (6 to 24 mg and 7.5 to 22.5 mg, twice daily).15 Cmax of deuterated α-DHTBZ and β-DHTBZ is reached within 3 to 4 hours after dosing, with a steady state ratio of 3:1 for the α-DHTBZ vs the β-DHTBZ form. Food had no effect on AUC, but did increase Cmax by 50%.5
Deutetrabenazine is metabolized through carbonyl reductase enzymes to its active metabolites, and these are further metabolized through multiple CYP pathways, predominantly 2D6 and to a lesser extent 3A4. The effect of CYP2D6 inhibition on the pharmacokinetics of deutetrabenazine and its α-DHTBZ and β-DHTBZ metabolites was studied in 24 healthy participants following a single 22.5 mg dose of deutetrabenazine given after 8 days of administration of the strong CYP2D6 inhibitor paroxetine, 20 mg/d. In the presence of paroxetine, systemic exposure (AUC) of α-DHTBZ was 1.9-fold higher and β-DHTBZ was 6.5-fold higher, resulting in an approximately 3-fold increase in AUC for total (α+β)-DHTBZ, with corresponding increases in mean half-life of approximately 1.5-fold and 2.7-fold, respectively.5 Neither deutetrabenazine or its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that deutetrabenazine and its active metabolites are unlikely to inhibit most major drug transporters at clinically relevant concentrations.
Efficacy
Efficacy was established in two 12-week, double-blind, placebo-controlled trials of adult patients with TD (ages 18 to 80).6,7 Eligible participants had:
- TD diagnosis for ≥3 months before screening and a history of DRBA treatment for ≥3 months (≥1 month if age ≥60)
- Total AIMS motor score ≥6 (items 1 to 7) at both screening and baseline, verified by a blinded central rater at screening via central video rating
- Patients with an underlying psychiatric illness had to be stable. Psychoactive medication use, including antipsychotics, was allowed if stable for ≥30 days before screening (antidepressants, ≥45 days).
Exclusion criteria included treatment with tetrabenazine, reserpine, α-methyl-p-tyrosine, strong anticholinergic medications, dopamine antagonizing antiemetics (eg, metoclopramide, prochlorperazine, promethazine), dopamine agonists, levodopa, stimulants, or a monoamine oxidase inhibitor (MAOI) within 30 days of screening or baseline, or treatment with botulinum toxin within 3 months of screening; and presence of a neurologic condition that could confound TD assessments, serious untreated or undertreated psychiatric illness, or unstable medical illness. Patients with a history of or active suicidal ideation or behavior within 6 months of screening or score ≥11 on the depression subscale of the Hospital Anxiety and Depression Scale were excluded. Those participants with Fridericia-corrected QT interval values >450 milliseconds in men, >460 milliseconds in women, or >480 milliseconds in patients with a right bundle branch block on electrocardiography at screening also were excluded.
The flexible-dose TD study was performed in 117 participants randomized in a 1:1 manner to deutetrabenazine or placebo, both administered twice daily, titrated to optimal dosage (12 to 48 mg/d) over 6 weeks, and then administered at that dose for another 6 weeks.7 The population demographics were: mean age, 54.6 ± 10.3 years, 52.1% female, 69.2% white, and 80.3% receiving ongoing dopamine antagonists, with a mean TD duration of 74.7 ± 81.5 months. Sixty-eight percent had schizophrenia spectrum disorders, and 30% had mood disorders. The primary outcome was change in total AIMS score (items 1 to 7) assessed by central, independent raters. The mean baseline AIMS score for items 1 to 7 was 9.6 ± 3.9, with 82.9% of participants with baseline AIMS scores ≥6. Study treatment retention was high: placebo 88.1%, deutetrabenazine 89.7%.7 There was a mean 3 point decrease in AIMS score for deutetrabenazine compared with 1.4 for placebo (P = .019). Among those with baseline AIMS scores ≥6, there was a 3.4 point decrease in AIMS scores for deutetrabenazine compared with a 1.9 point decrease for placebo (P = .027). The only adverse effects that occurred in ≥5% of deutetrabenazine participants and at a rate ≥2 times the rate in placebo were insomnia (deutetrabenazine 6.9% vs placebo 1.7%) and akathisia (deutetrabenazine 5.2% vs placebo 0%).
The fixed-dose TD study was performed in 293 participants randomized in 1:1:1:1 manner to 1 of 3 fixed doses of deutetrabenazine (12 mg/d, 24 mg/d, or 36 mg/d) or placebo, both administered twice daily.6 The starting dose of deutetrabenazine was 6 mg twice daily. During the dose escalation period (through Week 4), the dose of study drug was increased weekly in increments of 6 mg/d until the randomized dose was achieved. Patients continued to receive the dose they were assigned to over a maintenance period of 8 weeks.6 The population demographics were: mean age, 56.4 ± 11.3 years, 55% female, 79% white, 76% receiving ongoing dopamine antagonists, with a mean TD duration of 67.2 ± 66 months. Sixty percent had schizophrenia spectrum disorders, and 36% had mood disorders. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. The mean AIMS score at baseline was 9.5 ± 2.7 in the placebo group, and for deutetrabenazine: 9.6 ± 2.4 in the 12 mg/d group, 9.4 ± 2.9 in the 24 mg/d group, and 10.1 ± 3.2 in the 36 mg/d group. The 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). Study treatment retention rates were high: placebo 90.5%, deutetrabenazine 88%. Across all doses, only 1 adverse effect occurred in ≥5% of deutetrabenazine participants: headache (5% deutetrabenazine vs 6% placebo). At the highest dose, 36 mg/d, the only adverse effects that occurred in ≥5% of participants were diarrhea (7% deutetrabenazine vs 3% placebo) and headache (7% deutetrabenazine vs 6% placebo).
Outcome. In the flexible-dose study (mean dose 38.8 ± 7.92 mg/d), the deutetrabenazine arm experienced a mean 30% reduction in AIMS scores from baseline at the Week 12 endpoint. Compared with placebo, the mean reduction in AIMS scores (standard error) was: −3.0 (0.45) deutetrabenazine vs −1.6 (0.46) placebo (P = .019).7 For the fixed-dose study, the 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). In addition to these mean changes from baseline, 35% of the 24 mg/d group and 33% of the 36 mg/d group demonstrated ≥50% reduction in AIMS scores.6
Tolerability
In the 2 phase 3 trials, there were no adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo.5 Discontinuations because of adverse events were low in both pivotal studies across all treatment groups: 3.4% for placebo vs 1.7% for deutetrabenazine in the flexible-dose trial,7 and 3% for placebo vs 4% for deutetrabenazine in the fixed-dose study.6 In neither trial were there clinically significant changes in ratings of depression, suicidality, parkinsonism, or schizophrenia symptoms. The mean QT prolongation in healthy volunteers is described above.
Clinical considerations
Unique properties. Deutetrabenazine utilizes the greater bond strength of the carbon–deuterium bond to slow CYP metabolism, resulting in prolonged duration of action that is well tolerated, and provides significant efficacy.
Why Rx? The reasons to prescribe deutetrabenazine for TD patients include:
- only 1 of 2 agents with FDA approval for TD
- fewer tolerability issues than with tetrabenazine
- lower sedation rates in TD trials than with valbenazine
- no signal for effects on mood parameters or rates of parkinsonism when used for TD.
Dosing
The recommended starting dosage of deutetrabenazine is 6 mg twice daily taken with food, increasing by 6 mg/d weekly as needed, with a maximum dose of 48 mg/d or 36 mg/d in those taking strong CYP2D6 inhibitors or who are 2D6 poor metabolizers. Deutetrabenazine is contraindicated in patients with hepatic impairment (as determined by Child-Pugh criteria16). There are no data in patients with renal impairment. The combined efficacy and tolerability of dosages >48 mg/d has not been evaluated. Overdoses of tetrabenazine ranging from 100 to 1,000 mg have been reported in the literature and were associated with acute dystonia, oculogyric crisis, nausea and vomiting, sweating, sedation, hypotension, confusion, diarrhea, hallucinations, rubor, and tremor.5
Contraindications
When used for TD, deutetrabenazine is contraindicated for patients taking reserpine, tetrabenazine, valbenazine, or MAOIs, and for patients with hepatic impairment. As with most medications, there are no data on deutetrabenazine use in pregnant women; however, oral administration of deutetrabenazine (5, 10, or 30 mg/kg/d) or tetrabenazine (30 mg/kg/d) to pregnant rats during organogenesis had no clear effect on embryofetal development. The highest dose tested was 6 times the maximum recommended human dose of 48 mg/d on a body surface area (mg/m2) basis. There are no data on the presence of deutetrabenazine or its metabolites in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.
Compared with first-generation antipsychotics, second-generation antipsychotics (SGAs) have a lower risk for extrapyramidal symptoms. Yet tardive dyskinesia (TD) remains a concern because of the widespread use of SGAs for multiple indications.1 Prior to April 2017, clinicians had no FDA-approved TD treatment options. The most widely used agent worldwide, tetrabenazine, had positive efficacy data in TD trials over the past 45 years but was not available in the United States until 2008, and its sole indication was for chorea associated with Huntington’s disease.2 Moreover, the use of tetrabenazine involved slow titration, multiple daily dosing, cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, and tolerability issues.
Tetrabenazine is an inhibitor of vesicular monoamine transport type 2 (VMAT2), a transport protein located almost exclusively in the CNS whose role is to place monoamine neurotransmitters (dopamine, serotonin, norepinephrine) into presynaptic vesicles. By decreasing dopamine transport into these presynaptic vesicles, synaptic dopamine release is lessened, thus reducing postsynaptic dopamine D2 receptor activity and the severity of dyskinetic movements.1
In 2 pivotal 12-week clinical trials, deutetrabenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) scores (see Efficacy).6,7
Clinical implications
TD remains a substantial public health concern due to the increasing use of antipsychotics for mood and other disorders beyond the initial indications for schizophrenia.1 Although exposure to dopamine D2antagonism results in postsynaptic receptor upregulation and supersensitivity that underlies the development of dyskinesia, this process is often rapidly reversible in animal models.1 The persistence of TD symptoms in up to 80% of patients after dopamine receptor blocking agents (DRBAs) are stopped has led to hypotheses that the underlying pathophysiology of TD is also a problem with neuroplasticity. Aside from DRBA exposure, environmental factors (eg, oxidative stress) and genetic predisposition might contribute to TD risk.1
Before 2017, only 1 medication (branched-chain amino acids) had been FDA-approved for treating TD in the United States, and only a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]) had positive results from controlled trials, most with small effect sizes.8 Moreover, there was only 1 controlled trial each for clonazepam and EGb-761.1 A branched-chain amino acid preparation received FDA approval for managing TD in male patients, but is no longer commercially available, except from compounding pharmacies.9
Tetrabenazine was developed in the mid-1950s to avoid orthostasis and sedation associated with reserpine.10 Both reserpine and tetrabenazine proved effective for TD,11 but tetrabenazine lacked reserpine’s peripheral adverse effects. However, the kinetics of tetrabenazine necessitated multiple daily doses, and CYP2D6 genotyping was required for doses >50 mg/d.2
Receptor blocking. The mechanism that distinguishes the clinical profiles of reserpine and tetrabenazine relates to their differential properties at VMAT.12 VMAT exists in 2 forms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the CNS.13 Tetrabenazine is a specific and reversible VMAT2 inhibitor, whereas reserpine is an irreversible and nonselective antagonist of VMAT1 and VMAT2. It is reserpine’s VMAT1 inhibition that results in peripheral adverse effects such as orthostasis. Tetrabenazine is rapidly and extensively converted into 2 isomers, alpha-dihydrotetrabenazine (α-DHTBZ) and beta-dihydrotetrabenazine (β-DHTBZ), both of which are metabolized by CYP2D6, with a role for CYP3A4 in α-DHTBZ metabolism.1 These DHTBZ metabolites have a short half-life when generated from oral tetrabenazine, a feature that necessitates multiple daily dosing; moreover, the existence of 2D6 polymorphisms led to FDA-mandated CYP2D6 genotyping for tetrabenazine doses >50 mg/d when it was approved for Huntington’s chorea. The concern is that 2D6 poor metabolizers will have excessive exposure to the VMAT2 effects of DHTBZ, resulting in sedation, akathisia, parkinsonism, and mood symptoms.2
How deuterium impacts medication kinetics. Deuterium is a naturally occurring, stable, nontoxic isotope of hydrogen. Humans have 5 g of deuterium in their body at any time, mostly in the form of heavy water (D2O).14 When deuterium is used to replace selected hydrogen atoms, the resulting molecule will have similar configuration and receptor-binding properties but markedly different kinetics. Because the carbon–deuterium covalent bond requires 8 times more energy to break than a carbon–hydrogen bond, the half-life is prolonged.15 Utilizing this knowledge, a deuterated form of tetrabenazine, deutetrabenazine, was synthesized with such a purpose in mind. While the active metabolites of deutetrabenazine retain the VMAT2 affinity of non-deuterated tetrabenazine, the substitution of deuterium for hydrogen at specific positions slows the breakdown of metabolites, resulting in sustained duration of action, greater active drug exposure, and less impact of 2D6 genotype on drug exposure, thus eliminating the need for genotyping, unless one wants to exceed 36 mg/d.
Deutetrabenazine was first studied in Huntington’s chorea in a 13-week, double-blind, placebo-controlled, parallel-group study (N = 90).4 The maximum daily deutetrabenazine dose was 48 mg, but reduced to 36 mg in those taking strong CYP2D6 inhibitors (bupropion, fluoxetine, or paroxetine). Blinded 2D6 genotyping was performed, but there was no dose modification required based on 2D6 genotype. There was a 36.4% reduction in total maximal chorea score for deutetrabenazine compared with 14.4% for placebo (P < .001).4 Importantly, adverse effects were comparable between both groups, with 1 drop-out in the deutetrabenazine arm vs 2 in the placebo arm. The only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo was somnolence: 11.1% for deutetrabenazine vs 4.4% for placebo.4 The mean deutetrabenazine daily dose at the end of the treatment period was 39.7 ± 9.3 mg, and for those with impaired CYP2D6 function (poor metabolizers or those taking strong CYP2D6 inhibiting medications), the mean daily dose was 34.8 mg ± 3.8 mg.4
Use in tardive dyskinesia. The recommended starting dosage for TD treatment is 6 mg, twice daily with food. The dose may be increased at weekly intervals in increments of 6 mg/d to a maximum recommended daily dosage of 48 mg.5 The maximum daily dose is 36 mg (18 mg, twice daily) in patients receiving strong CYP2D6 inhibitors or who are 2D6 poor metabolizers.5
Deutetrabenazine has not been studied in those with moderate or severe hepatic impairment, and its use is contraindicated in these patients.5 No clinical studies have been conducted to assess the effect of renal impairment on the pharmacokinetics of deutetrabenazine.5
Pharmacologic profile, adverse reactions
When the data from the two 12-week, phase 3 placebo-controlled studies were pooled, the most common adverse reactions occurring in >3% of deutetrabenazine patients and greater than placebo were nasopharyngeal symptoms (4% vs 2% placebo) and insomnia (4% vs 1% placebo).5 Importantly, in neither TD study were there clinically significant changes in rating scales for depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms. The mean QT prolongation for a single 24 mg dose of deutetrabenazine in healthy volunteers was 4.5 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 6.5 milliseconds.5 For tetrabenazine, single 50 mg doses administered to volunteers resulted in mean QT prolongation of 8 milliseconds.5 In patients requiring deutetrabenazine doses >24 mg/d who are taking other medications known to prolong QTc, assess the QTc interval before and after increasing the dose of deutetrabenazine or other medications that are known to prolong QTc.5
How it works
Tetrabenazine is the only agent that has demonstrated significant efficacy for TD management, but its use involves slow titration, multiple daily dosing, CYP2D6 genotyping for doses >50 mg/d, and tolerability issues. For example, the most common adverse effects in the pivotal tetrabenazine Huntington’s disease trial were sedation/somnolence (tetrabenazine 31% vs 3% for placebo), insomnia (tetrabenazine 22% vs 0% for placebo), depression (tetrabenazine 19% vs 0% for placebo), fatigue (tetrabenazine 22% vs 13% for placebo), and akathisia (tetrabenazine 19% vs 0% for placebo).2 For comparison, the only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo in the pivotal Huntington’s disease trial was somnolence (11.1% for deutetrabenazine vs 4.4% for placebo).4
Pharmacokinetics
Deutetrabenazine has 80% oral bioavailability, and is rapidly converted to its active metabolites after oral dosing (Table 2).5 Linear dose dependence of Cmax and area under the curve (AUC) was observed for the active metabolites following single or multiple doses of deutetrabenazine (6 to 24 mg and 7.5 to 22.5 mg, twice daily).15 Cmax of deuterated α-DHTBZ and β-DHTBZ is reached within 3 to 4 hours after dosing, with a steady state ratio of 3:1 for the α-DHTBZ vs the β-DHTBZ form. Food had no effect on AUC, but did increase Cmax by 50%.5
Deutetrabenazine is metabolized through carbonyl reductase enzymes to its active metabolites, and these are further metabolized through multiple CYP pathways, predominantly 2D6 and to a lesser extent 3A4. The effect of CYP2D6 inhibition on the pharmacokinetics of deutetrabenazine and its α-DHTBZ and β-DHTBZ metabolites was studied in 24 healthy participants following a single 22.5 mg dose of deutetrabenazine given after 8 days of administration of the strong CYP2D6 inhibitor paroxetine, 20 mg/d. In the presence of paroxetine, systemic exposure (AUC) of α-DHTBZ was 1.9-fold higher and β-DHTBZ was 6.5-fold higher, resulting in an approximately 3-fold increase in AUC for total (α+β)-DHTBZ, with corresponding increases in mean half-life of approximately 1.5-fold and 2.7-fold, respectively.5 Neither deutetrabenazine or its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that deutetrabenazine and its active metabolites are unlikely to inhibit most major drug transporters at clinically relevant concentrations.
Efficacy
Efficacy was established in two 12-week, double-blind, placebo-controlled trials of adult patients with TD (ages 18 to 80).6,7 Eligible participants had:
- TD diagnosis for ≥3 months before screening and a history of DRBA treatment for ≥3 months (≥1 month if age ≥60)
- Total AIMS motor score ≥6 (items 1 to 7) at both screening and baseline, verified by a blinded central rater at screening via central video rating
- Patients with an underlying psychiatric illness had to be stable. Psychoactive medication use, including antipsychotics, was allowed if stable for ≥30 days before screening (antidepressants, ≥45 days).
Exclusion criteria included treatment with tetrabenazine, reserpine, α-methyl-p-tyrosine, strong anticholinergic medications, dopamine antagonizing antiemetics (eg, metoclopramide, prochlorperazine, promethazine), dopamine agonists, levodopa, stimulants, or a monoamine oxidase inhibitor (MAOI) within 30 days of screening or baseline, or treatment with botulinum toxin within 3 months of screening; and presence of a neurologic condition that could confound TD assessments, serious untreated or undertreated psychiatric illness, or unstable medical illness. Patients with a history of or active suicidal ideation or behavior within 6 months of screening or score ≥11 on the depression subscale of the Hospital Anxiety and Depression Scale were excluded. Those participants with Fridericia-corrected QT interval values >450 milliseconds in men, >460 milliseconds in women, or >480 milliseconds in patients with a right bundle branch block on electrocardiography at screening also were excluded.
The flexible-dose TD study was performed in 117 participants randomized in a 1:1 manner to deutetrabenazine or placebo, both administered twice daily, titrated to optimal dosage (12 to 48 mg/d) over 6 weeks, and then administered at that dose for another 6 weeks.7 The population demographics were: mean age, 54.6 ± 10.3 years, 52.1% female, 69.2% white, and 80.3% receiving ongoing dopamine antagonists, with a mean TD duration of 74.7 ± 81.5 months. Sixty-eight percent had schizophrenia spectrum disorders, and 30% had mood disorders. The primary outcome was change in total AIMS score (items 1 to 7) assessed by central, independent raters. The mean baseline AIMS score for items 1 to 7 was 9.6 ± 3.9, with 82.9% of participants with baseline AIMS scores ≥6. Study treatment retention was high: placebo 88.1%, deutetrabenazine 89.7%.7 There was a mean 3 point decrease in AIMS score for deutetrabenazine compared with 1.4 for placebo (P = .019). Among those with baseline AIMS scores ≥6, there was a 3.4 point decrease in AIMS scores for deutetrabenazine compared with a 1.9 point decrease for placebo (P = .027). The only adverse effects that occurred in ≥5% of deutetrabenazine participants and at a rate ≥2 times the rate in placebo were insomnia (deutetrabenazine 6.9% vs placebo 1.7%) and akathisia (deutetrabenazine 5.2% vs placebo 0%).
The fixed-dose TD study was performed in 293 participants randomized in 1:1:1:1 manner to 1 of 3 fixed doses of deutetrabenazine (12 mg/d, 24 mg/d, or 36 mg/d) or placebo, both administered twice daily.6 The starting dose of deutetrabenazine was 6 mg twice daily. During the dose escalation period (through Week 4), the dose of study drug was increased weekly in increments of 6 mg/d until the randomized dose was achieved. Patients continued to receive the dose they were assigned to over a maintenance period of 8 weeks.6 The population demographics were: mean age, 56.4 ± 11.3 years, 55% female, 79% white, 76% receiving ongoing dopamine antagonists, with a mean TD duration of 67.2 ± 66 months. Sixty percent had schizophrenia spectrum disorders, and 36% had mood disorders. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. The mean AIMS score at baseline was 9.5 ± 2.7 in the placebo group, and for deutetrabenazine: 9.6 ± 2.4 in the 12 mg/d group, 9.4 ± 2.9 in the 24 mg/d group, and 10.1 ± 3.2 in the 36 mg/d group. The 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). Study treatment retention rates were high: placebo 90.5%, deutetrabenazine 88%. Across all doses, only 1 adverse effect occurred in ≥5% of deutetrabenazine participants: headache (5% deutetrabenazine vs 6% placebo). At the highest dose, 36 mg/d, the only adverse effects that occurred in ≥5% of participants were diarrhea (7% deutetrabenazine vs 3% placebo) and headache (7% deutetrabenazine vs 6% placebo).
Outcome. In the flexible-dose study (mean dose 38.8 ± 7.92 mg/d), the deutetrabenazine arm experienced a mean 30% reduction in AIMS scores from baseline at the Week 12 endpoint. Compared with placebo, the mean reduction in AIMS scores (standard error) was: −3.0 (0.45) deutetrabenazine vs −1.6 (0.46) placebo (P = .019).7 For the fixed-dose study, the 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). In addition to these mean changes from baseline, 35% of the 24 mg/d group and 33% of the 36 mg/d group demonstrated ≥50% reduction in AIMS scores.6
Tolerability
In the 2 phase 3 trials, there were no adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo.5 Discontinuations because of adverse events were low in both pivotal studies across all treatment groups: 3.4% for placebo vs 1.7% for deutetrabenazine in the flexible-dose trial,7 and 3% for placebo vs 4% for deutetrabenazine in the fixed-dose study.6 In neither trial were there clinically significant changes in ratings of depression, suicidality, parkinsonism, or schizophrenia symptoms. The mean QT prolongation in healthy volunteers is described above.
Clinical considerations
Unique properties. Deutetrabenazine utilizes the greater bond strength of the carbon–deuterium bond to slow CYP metabolism, resulting in prolonged duration of action that is well tolerated, and provides significant efficacy.
Why Rx? The reasons to prescribe deutetrabenazine for TD patients include:
- only 1 of 2 agents with FDA approval for TD
- fewer tolerability issues than with tetrabenazine
- lower sedation rates in TD trials than with valbenazine
- no signal for effects on mood parameters or rates of parkinsonism when used for TD.
Dosing
The recommended starting dosage of deutetrabenazine is 6 mg twice daily taken with food, increasing by 6 mg/d weekly as needed, with a maximum dose of 48 mg/d or 36 mg/d in those taking strong CYP2D6 inhibitors or who are 2D6 poor metabolizers. Deutetrabenazine is contraindicated in patients with hepatic impairment (as determined by Child-Pugh criteria16). There are no data in patients with renal impairment. The combined efficacy and tolerability of dosages >48 mg/d has not been evaluated. Overdoses of tetrabenazine ranging from 100 to 1,000 mg have been reported in the literature and were associated with acute dystonia, oculogyric crisis, nausea and vomiting, sweating, sedation, hypotension, confusion, diarrhea, hallucinations, rubor, and tremor.5
Contraindications
When used for TD, deutetrabenazine is contraindicated for patients taking reserpine, tetrabenazine, valbenazine, or MAOIs, and for patients with hepatic impairment. As with most medications, there are no data on deutetrabenazine use in pregnant women; however, oral administration of deutetrabenazine (5, 10, or 30 mg/kg/d) or tetrabenazine (30 mg/kg/d) to pregnant rats during organogenesis had no clear effect on embryofetal development. The highest dose tested was 6 times the maximum recommended human dose of 48 mg/d on a body surface area (mg/m2) basis. There are no data on the presence of deutetrabenazine or its metabolites in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.
1. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
2. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
3. Meyer JM. Valbenazine for tardive dyskinesia. Current Psychiatry. 2017;16(5):40-46.
4. Huntington Study Group; Frank S, Testa CM, Stamler D, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA. 2016;316(1):40-50.
5. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals USA, Inc.; 2017.
6. Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595-604.
7. Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003-2010.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Richardson MA, Small AM, Read LL, et al. Branched chain amino acid treatment of tardive dyskinesia in children and adolescents. J Clin Psychiatry. 2004;65(1):92-96.
10. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
11. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
12. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
13. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
14. Kushner DJ, Baker A, Dunstall TG. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can J Physiol Pharmacol. 1999;77(2):79-88.
15. United States Securities and Exchange Commission. Form S-1 Registration Statement of Auspex Pharmaceuticals, Inc. https://www.sec.gov/Archives/edgar/data/1454189/000119312513481239/d627086ds1.htm. Published December 20, 2013. Accessed July 1, 2016.
16. Cholongitas E, Papatheodoridis GV, Vangeli M, et al. Systematic review: the model for end-stage liver disease—should it replace Child-Pugh’s classification for assessing prognosis in cirrhosis? Aliment Pharmacol Ther. 2005;22(11-12):1079-1089.
1. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
2. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
3. Meyer JM. Valbenazine for tardive dyskinesia. Current Psychiatry. 2017;16(5):40-46.
4. Huntington Study Group; Frank S, Testa CM, Stamler D, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA. 2016;316(1):40-50.
5. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals USA, Inc.; 2017.
6. Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595-604.
7. Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003-2010.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Richardson MA, Small AM, Read LL, et al. Branched chain amino acid treatment of tardive dyskinesia in children and adolescents. J Clin Psychiatry. 2004;65(1):92-96.
10. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
11. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
12. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
13. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
14. Kushner DJ, Baker A, Dunstall TG. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can J Physiol Pharmacol. 1999;77(2):79-88.
15. United States Securities and Exchange Commission. Form S-1 Registration Statement of Auspex Pharmaceuticals, Inc. https://www.sec.gov/Archives/edgar/data/1454189/000119312513481239/d627086ds1.htm. Published December 20, 2013. Accessed July 1, 2016.
16. Cholongitas E, Papatheodoridis GV, Vangeli M, et al. Systematic review: the model for end-stage liver disease—should it replace Child-Pugh’s classification for assessing prognosis in cirrhosis? Aliment Pharmacol Ther. 2005;22(11-12):1079-1089.
Triple-bead mixed amphetamine salt for ADHD
Stimulants are first-line psychopharmacologic interventions for attention-deficit/hyperactivity disorder (ADHD), and their efficacy is supported by clinical trials and meta-analyses in children and adolescents1 as well as adults.2 Despite decades of tolerability and efficacy data supporting their use, a major drawback of stimulants is that their salutary therapeutic effects wane once the medication is cleared or metabolized. Both mixed amphetamine- and methylphenidate-based preparations have short half-lives, necessitating multiple doses per day (eg, 3 or 4 times a day) when short-acting preparations are used. Over the past 15 years, nearly a dozen formulations were developed that extend the duration of action through delayed release, delayed absorption, or utilizing prodrugs.

The encapsulated preparation contains 3 MAS beads: an immediate-release amphetamine salt bead, a pulsed-delayed release bead, and an extended-release bead (Figure 1), which give rise to a unique pharmacokinetic profile (Figure 2).3
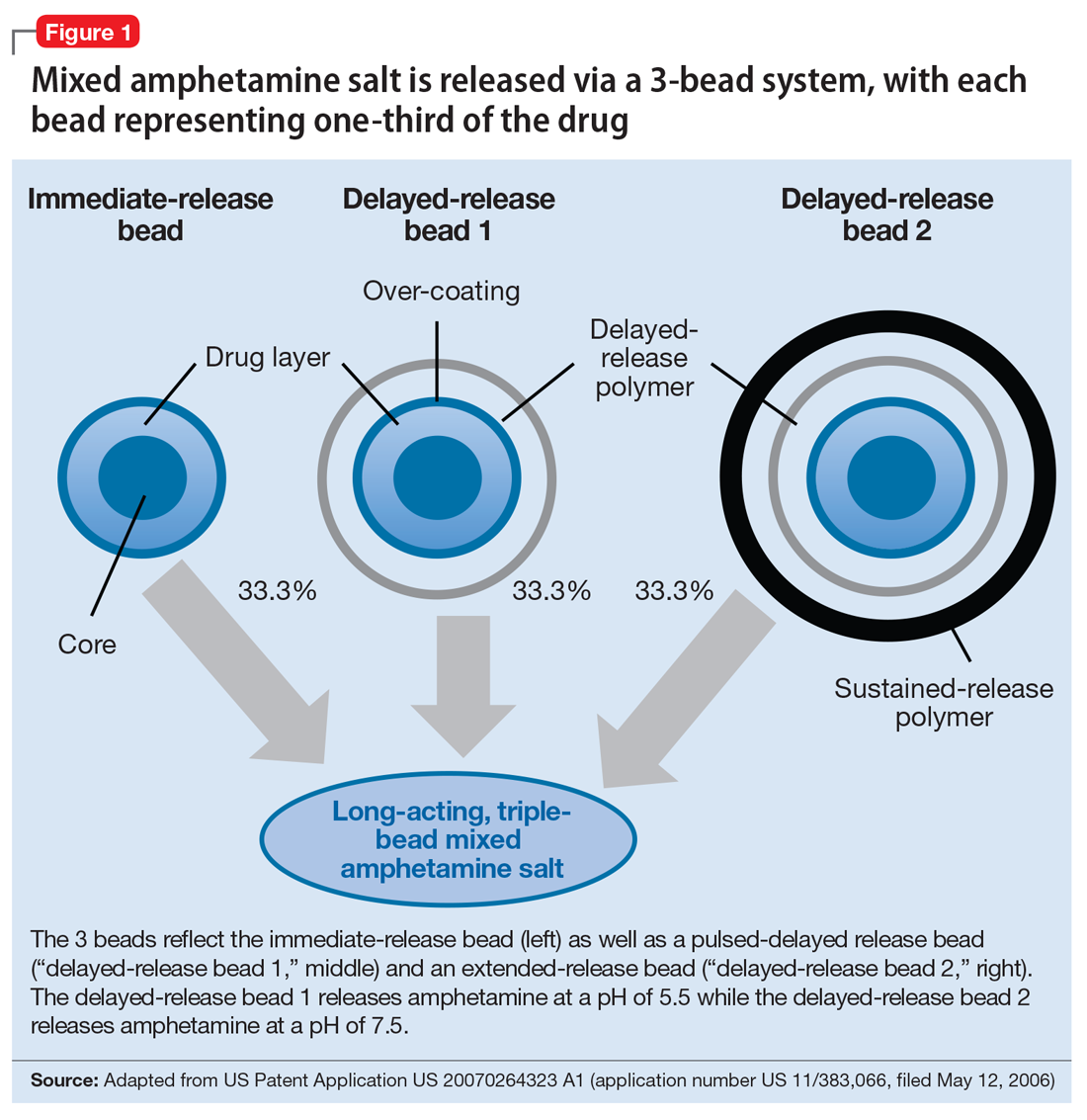
Mechanism of action
Like all MAS, this formulation blocks the reuptake of norepinephrine and dopamine, increasing synaptic concentrations of these monoamine neurotransmitters. Additionally, amphetamine salts may inhibit the activity of monoamine oxidase (MAO), further increasing synaptic levels of monoamines.4 Enhancing noradrenergic, dopaminergic neurotransmission, particularly within the prefrontal cortex, increases attention, working memory, and processing speed in patients with ADHD.4
Pharmacokinetics
Cmax occurs approximately 7 to 10 hours and 8 hours following administration in adolescent and adult patients, respectively (Figure 2).3 In adolescents who were administered a single dose of long-acting, triple-bead MAS, Cmax and area under the curve (AUC) for d- and l-amphetamine were both 21% to 31% higher compared with adults3 and did not appear to be affected by sex or race.3
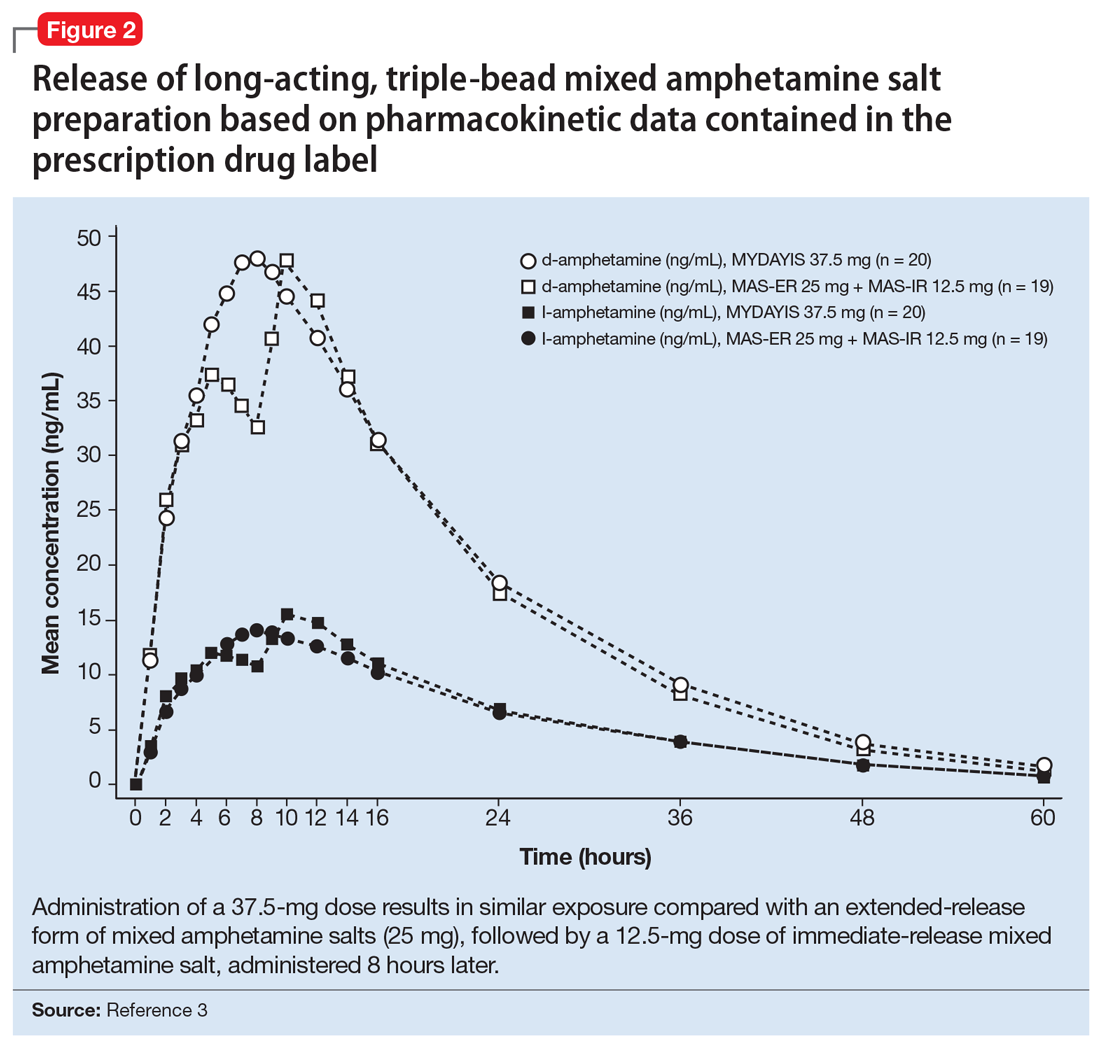
Half-life is 10 to 11 hours for d-amphetamine and 10 to 13 hours for l-amphetamine and does not statistically differ between pediatric and adult studies.3
Metabolism and elimination. Amphetamines are partially metabolized through cytochrome 450 (CYP) 2D6-dependent mechanisms, and thus in CYP2D6 poor metabolizers medication exposure may be increased, while decreased exposure may occur in ultra-rapid metabolizers; however, there are no guidelines from the Clinical Pharmacogenetics Implementation Consortium regarding alternate dosing strategies for patients based on CYP2D6 genotype or activity phenotype.5 Because amphetamines are renally excreted, dosages should be adjusted in patients with renal impairment.
Drug interactions. Medications that affect gastrointestinal and urinary pH may affect serum concentrations of amphetamine. Specifically, agents that increase gastric pH (eg, proton pump inhibitors) as well as urinary alkalinizing agents (eg, acetazolamide, some thiazide diuretics) increase serum amphetamine concentrations.3 Because amphetamine is a weak MAOI, there is a theoretical risk of serotonin syndrome when amphetamine-based preparations are used concurrently with SSRIs, TCAs, and MAOIs. However, the concurrent use of MAS and SSRIs generally is considered safe and common practice in patients with ADHD and co-occurring anxiety6,7 or depressive disorders.
Dosing
Long-acting, triple-bead MAS is available in 12.5-, 25-, 37.5-, and 50-mg capsules. The capsule may be opened and sprinkled in food for patients who cannot swallow capsules. Opening of the capsule results in similar absorption relative to oral administration of the intact capsule.3
In adults with ADHD, long-acting, triple-bead MAS should be initiated at 12.5 mg in the morning (Table 2). However, in some individuals, long-acting, triple-bead MAS may be initiated at 25 mg each morning. Titration should occur in 12.5-mg weekly increments to a maximum dosage of 50 mg/d.3

In adults with severe renal impairment (glomerular filtrate rate, 15 to 30 mL/min/1.73 m2), the recommended starting dose is 12.5 mg/d, with a maximum dosage of 25 mg/d.3
The efficacy of long-acting, triple-bead MAS in adults with ADHD was demonstrated in 3 studies involving adults ages 18 to 55, and the effectiveness of the medication, with regard to duration of action, was assessed using the Time-Sensitive ADHD Symptom Scale—a self-report scale that consists of items indexed by the ADHD Rating Scale-IV (ADHD-RS-IV) which assesses ADHD symptom severity. Doses up to 75 mg/d were studied; however, there were no significant effects. It should be noted that this maximum daily dose was not determined by any safety parameter.
Study 1 (dose-optimization, triple-bead MAS, n = 137; placebo, n = 135, dosing: 12.5 to 75 mg) and Study 2 (forced dose-titration study, triple-bead MAS, n = 308; placebo, n = 104, dosing: 25 mg, 50 mg, 75 mg) demonstrated efficacy of triple-bead MAS for treating ADHD in adults. Despite differences in study designs, statistically significant and similar clinically relevant improvement was observed with triple-bead MAS (vs placebo) on ADHD-RS-IV total scores in both Study 1 and Study 2.8 An additional study in adults ages 18 to 55 (N = 275) with ADHD (DSM-5 criteria) involved randomization to either 12.5 mg (fixed dose) or forced titration (12.5 to 37.5 mg) or placebo and, as with the first 2 studies, improvement in ADHD symptoms was observed in triple-bead MAS-treated patients relative to those who had received placebo. (See Reference 3 for a summary of the clinical trials of triple-bead MAS in adults with ADHD.)
The tolerability of this medication was evaluated in a 12-month open-label study of adults with ADHD (DSM-IV-TR criteria) in which discontinuation was higher at doses >25 mg/d.7 Treatment-related increases in blood pressure and heart rate were consistent with the known hemodynamic adverse effect profile of stimulants.9
In adolescents with ADHD ages 13 to 17, long-acting, triple-bead MAS should be initiated at 12.5 mg/d and may be increased to 25 mg/d (Table 2). Importantly, in younger patients, including those younger than age 12, triple-bead MAS was associated with an increased risk of adverse events including insomnia and anorexia, and this was thought to be related to increased drug exposure (ie, AUC).
The efficacy of long-acting, triple-bead MAS was evaluated in 2 studies of adolescents ages 13 to 17, including 1 fixed-dose trial (25 mg/d) and 1 flexibly-dosed trial (12.5 to 25 mg/d). These unpublished studies utilized the ADHD-RS-IV score and the Average Permanent Product Measure of Performance, an age-adjusted math test and measure of sustained attention, and revealed statistically significant differences between medication and placebo in the primary outcomes.3
Adverse effects
Long-acting, triple-bead MAS was developed to treat ADHD symptoms throughout the day, and serum concentrations of the medication may be higher with this formulation compared with other long-acting preparations. Therefore, adverse effects that are directly related to plasma exposure (eg, insomnia and appetite suppression) may occur at higher rates with this preparation compared with alternatives. For example, in some of the registration trials, insomnia occurred in more than one-third of patients receiving the active medication (38%).9 Although insomnia was the most frequently reported adverse event in adults with ADHD, most reports of insomnia occurred early in the course of treatment. Of these insomnia-related adverse events, 94% were mild to moderate and resulted in discontinuation of the medication in approximately 2% of patients. Further, 73.9% of treatment-emergent, insomnia–related adverse events resolved during the course of the study. It is also important to note that the Pittsburgh Sleep Quality Index did not differ from placebo in studies of triple-bead MAS in adults with ADHD.10 Similarly, rates of stimulant-induced appetite suppression may be higher with this preparation compared with other long-acting preparations.9
Adverse effects observed in adults with ADHD that occurred in ≥2% of patients receiving triple-bead MAS and at least twice the incidence in patients randomized to placebo included:
- anxiety (7% vs 3%)
- feeling jittery (2% vs 1%)
- agitation (2% vs 0%)
- insomnia (31% vs 8%)
- depression (3% vs 0%)
- decreased appetite (30% vs 4%)
- weight loss (9% vs 0%)
- xerostomia (23% vs 4%)
- diarrhea (3% vs 0%)
- increased heart rate (9% vs 0%)
- palpitations (4% vs 2%)
- dysmenorrhea (4% vs 2%)
- erectile dysfunction (2% vs 1%).
In adolescents receiving triple-bea
1. Punja S, Shamseer L, Hartling L, et al. Amphetamines for attention deficit hyperactivity disorder (ADHD) in children and adolescents. Cochrane Database Syst Rev. 2016;2016(2):CD009996.
2. Castells X, Ramos-Quiroga J, Bosch R, et al. Amphetamines for attention deficit hyperactivity disorder (ADHD) in adults. Cochrane Database Syst Rev. 2011;(6):CD007813.
3. Mydayis [package insert]. Lexington, MA: Shire; 2017.
4. Heal DJ, Smith SL, Gosden J, et al. Amphetamine, past and present—a pharmacological and clinical perspective. J Psychopharmacol. 2013;27(6):479-496.
5. Hoffman JM, Dunnenberger HM, Kevin Hicks J, et al. Developing knowledge resources to support precision medicine: principles from the Clinical Pharmacogenetics Implementation Consortium (CPIC). J Am Med Inform Assoc. 2016;23(4):766-801.
6. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
7. Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
8. Goodman DW, Spencer TJ, Adler LA, et al. Clinical evaluation of triple-bead mixed amphetamine salts in adult ADHD. Presented at: 54th Annual Meeting of the American Academy of Child and Adolescent Psychiatry; October 25, 2007; Boston, MA.
9. Adler LA, Frick G, Yan B. A Long-term, open-label, safety study of triple-bead mixed amphetamine salts (SHP465) in adults with ADHD [published online April 1, 2017]. J Atten Disord. doi: 10.1177/1087054717696770.
10. Backhaus J, Junghanns K, Broocks A, et al. test-retest reliability and validity of the Pittsburgh Sleep Quality Index in primary insomnia. J Psychosom Res. 2002;53(3):737-740.
Stimulants are first-line psychopharmacologic interventions for attention-deficit/hyperactivity disorder (ADHD), and their efficacy is supported by clinical trials and meta-analyses in children and adolescents1 as well as adults.2 Despite decades of tolerability and efficacy data supporting their use, a major drawback of stimulants is that their salutary therapeutic effects wane once the medication is cleared or metabolized. Both mixed amphetamine- and methylphenidate-based preparations have short half-lives, necessitating multiple doses per day (eg, 3 or 4 times a day) when short-acting preparations are used. Over the past 15 years, nearly a dozen formulations were developed that extend the duration of action through delayed release, delayed absorption, or utilizing prodrugs.
The encapsulated preparation contains 3 MAS beads: an immediate-release amphetamine salt bead, a pulsed-delayed release bead, and an extended-release bead (Figure 1), which give rise to a unique pharmacokinetic profile (Figure 2).3
Mechanism of action
Like all MAS, this formulation blocks the reuptake of norepinephrine and dopamine, increasing synaptic concentrations of these monoamine neurotransmitters. Additionally, amphetamine salts may inhibit the activity of monoamine oxidase (MAO), further increasing synaptic levels of monoamines.4 Enhancing noradrenergic, dopaminergic neurotransmission, particularly within the prefrontal cortex, increases attention, working memory, and processing speed in patients with ADHD.4
Pharmacokinetics
Cmax occurs approximately 7 to 10 hours and 8 hours following administration in adolescent and adult patients, respectively (Figure 2).3 In adolescents who were administered a single dose of long-acting, triple-bead MAS, Cmax and area under the curve (AUC) for d- and l-amphetamine were both 21% to 31% higher compared with adults3 and did not appear to be affected by sex or race.3
Half-life is 10 to 11 hours for d-amphetamine and 10 to 13 hours for l-amphetamine and does not statistically differ between pediatric and adult studies.3
Metabolism and elimination. Amphetamines are partially metabolized through cytochrome 450 (CYP) 2D6-dependent mechanisms, and thus in CYP2D6 poor metabolizers medication exposure may be increased, while decreased exposure may occur in ultra-rapid metabolizers; however, there are no guidelines from the Clinical Pharmacogenetics Implementation Consortium regarding alternate dosing strategies for patients based on CYP2D6 genotype or activity phenotype.5 Because amphetamines are renally excreted, dosages should be adjusted in patients with renal impairment.
Drug interactions. Medications that affect gastrointestinal and urinary pH may affect serum concentrations of amphetamine. Specifically, agents that increase gastric pH (eg, proton pump inhibitors) as well as urinary alkalinizing agents (eg, acetazolamide, some thiazide diuretics) increase serum amphetamine concentrations.3 Because amphetamine is a weak MAOI, there is a theoretical risk of serotonin syndrome when amphetamine-based preparations are used concurrently with SSRIs, TCAs, and MAOIs. However, the concurrent use of MAS and SSRIs generally is considered safe and common practice in patients with ADHD and co-occurring anxiety6,7 or depressive disorders.
Dosing
Long-acting, triple-bead MAS is available in 12.5-, 25-, 37.5-, and 50-mg capsules. The capsule may be opened and sprinkled in food for patients who cannot swallow capsules. Opening of the capsule results in similar absorption relative to oral administration of the intact capsule.3
In adults with ADHD, long-acting, triple-bead MAS should be initiated at 12.5 mg in the morning (Table 2). However, in some individuals, long-acting, triple-bead MAS may be initiated at 25 mg each morning. Titration should occur in 12.5-mg weekly increments to a maximum dosage of 50 mg/d.3
In adults with severe renal impairment (glomerular filtrate rate, 15 to 30 mL/min/1.73 m2), the recommended starting dose is 12.5 mg/d, with a maximum dosage of 25 mg/d.3
The efficacy of long-acting, triple-bead MAS in adults with ADHD was demonstrated in 3 studies involving adults ages 18 to 55, and the effectiveness of the medication, with regard to duration of action, was assessed using the Time-Sensitive ADHD Symptom Scale—a self-report scale that consists of items indexed by the ADHD Rating Scale-IV (ADHD-RS-IV) which assesses ADHD symptom severity. Doses up to 75 mg/d were studied; however, there were no significant effects. It should be noted that this maximum daily dose was not determined by any safety parameter.
Study 1 (dose-optimization, triple-bead MAS, n = 137; placebo, n = 135, dosing: 12.5 to 75 mg) and Study 2 (forced dose-titration study, triple-bead MAS, n = 308; placebo, n = 104, dosing: 25 mg, 50 mg, 75 mg) demonstrated efficacy of triple-bead MAS for treating ADHD in adults. Despite differences in study designs, statistically significant and similar clinically relevant improvement was observed with triple-bead MAS (vs placebo) on ADHD-RS-IV total scores in both Study 1 and Study 2.8 An additional study in adults ages 18 to 55 (N = 275) with ADHD (DSM-5 criteria) involved randomization to either 12.5 mg (fixed dose) or forced titration (12.5 to 37.5 mg) or placebo and, as with the first 2 studies, improvement in ADHD symptoms was observed in triple-bead MAS-treated patients relative to those who had received placebo. (See Reference 3 for a summary of the clinical trials of triple-bead MAS in adults with ADHD.)
The tolerability of this medication was evaluated in a 12-month open-label study of adults with ADHD (DSM-IV-TR criteria) in which discontinuation was higher at doses >25 mg/d.7 Treatment-related increases in blood pressure and heart rate were consistent with the known hemodynamic adverse effect profile of stimulants.9
In adolescents with ADHD ages 13 to 17, long-acting, triple-bead MAS should be initiated at 12.5 mg/d and may be increased to 25 mg/d (Table 2). Importantly, in younger patients, including those younger than age 12, triple-bead MAS was associated with an increased risk of adverse events including insomnia and anorexia, and this was thought to be related to increased drug exposure (ie, AUC).
The efficacy of long-acting, triple-bead MAS was evaluated in 2 studies of adolescents ages 13 to 17, including 1 fixed-dose trial (25 mg/d) and 1 flexibly-dosed trial (12.5 to 25 mg/d). These unpublished studies utilized the ADHD-RS-IV score and the Average Permanent Product Measure of Performance, an age-adjusted math test and measure of sustained attention, and revealed statistically significant differences between medication and placebo in the primary outcomes.3
Adverse effects
Long-acting, triple-bead MAS was developed to treat ADHD symptoms throughout the day, and serum concentrations of the medication may be higher with this formulation compared with other long-acting preparations. Therefore, adverse effects that are directly related to plasma exposure (eg, insomnia and appetite suppression) may occur at higher rates with this preparation compared with alternatives. For example, in some of the registration trials, insomnia occurred in more than one-third of patients receiving the active medication (38%).9 Although insomnia was the most frequently reported adverse event in adults with ADHD, most reports of insomnia occurred early in the course of treatment. Of these insomnia-related adverse events, 94% were mild to moderate and resulted in discontinuation of the medication in approximately 2% of patients. Further, 73.9% of treatment-emergent, insomnia–related adverse events resolved during the course of the study. It is also important to note that the Pittsburgh Sleep Quality Index did not differ from placebo in studies of triple-bead MAS in adults with ADHD.10 Similarly, rates of stimulant-induced appetite suppression may be higher with this preparation compared with other long-acting preparations.9
Adverse effects observed in adults with ADHD that occurred in ≥2% of patients receiving triple-bead MAS and at least twice the incidence in patients randomized to placebo included:
- anxiety (7% vs 3%)
- feeling jittery (2% vs 1%)
- agitation (2% vs 0%)
- insomnia (31% vs 8%)
- depression (3% vs 0%)
- decreased appetite (30% vs 4%)
- weight loss (9% vs 0%)
- xerostomia (23% vs 4%)
- diarrhea (3% vs 0%)
- increased heart rate (9% vs 0%)
- palpitations (4% vs 2%)
- dysmenorrhea (4% vs 2%)
- erectile dysfunction (2% vs 1%).
In adolescents receiving triple-bea
Stimulants are first-line psychopharmacologic interventions for attention-deficit/hyperactivity disorder (ADHD), and their efficacy is supported by clinical trials and meta-analyses in children and adolescents1 as well as adults.2 Despite decades of tolerability and efficacy data supporting their use, a major drawback of stimulants is that their salutary therapeutic effects wane once the medication is cleared or metabolized. Both mixed amphetamine- and methylphenidate-based preparations have short half-lives, necessitating multiple doses per day (eg, 3 or 4 times a day) when short-acting preparations are used. Over the past 15 years, nearly a dozen formulations were developed that extend the duration of action through delayed release, delayed absorption, or utilizing prodrugs.
The encapsulated preparation contains 3 MAS beads: an immediate-release amphetamine salt bead, a pulsed-delayed release bead, and an extended-release bead (Figure 1), which give rise to a unique pharmacokinetic profile (Figure 2).3
Mechanism of action
Like all MAS, this formulation blocks the reuptake of norepinephrine and dopamine, increasing synaptic concentrations of these monoamine neurotransmitters. Additionally, amphetamine salts may inhibit the activity of monoamine oxidase (MAO), further increasing synaptic levels of monoamines.4 Enhancing noradrenergic, dopaminergic neurotransmission, particularly within the prefrontal cortex, increases attention, working memory, and processing speed in patients with ADHD.4
Pharmacokinetics
Cmax occurs approximately 7 to 10 hours and 8 hours following administration in adolescent and adult patients, respectively (Figure 2).3 In adolescents who were administered a single dose of long-acting, triple-bead MAS, Cmax and area under the curve (AUC) for d- and l-amphetamine were both 21% to 31% higher compared with adults3 and did not appear to be affected by sex or race.3
Half-life is 10 to 11 hours for d-amphetamine and 10 to 13 hours for l-amphetamine and does not statistically differ between pediatric and adult studies.3
Metabolism and elimination. Amphetamines are partially metabolized through cytochrome 450 (CYP) 2D6-dependent mechanisms, and thus in CYP2D6 poor metabolizers medication exposure may be increased, while decreased exposure may occur in ultra-rapid metabolizers; however, there are no guidelines from the Clinical Pharmacogenetics Implementation Consortium regarding alternate dosing strategies for patients based on CYP2D6 genotype or activity phenotype.5 Because amphetamines are renally excreted, dosages should be adjusted in patients with renal impairment.
Drug interactions. Medications that affect gastrointestinal and urinary pH may affect serum concentrations of amphetamine. Specifically, agents that increase gastric pH (eg, proton pump inhibitors) as well as urinary alkalinizing agents (eg, acetazolamide, some thiazide diuretics) increase serum amphetamine concentrations.3 Because amphetamine is a weak MAOI, there is a theoretical risk of serotonin syndrome when amphetamine-based preparations are used concurrently with SSRIs, TCAs, and MAOIs. However, the concurrent use of MAS and SSRIs generally is considered safe and common practice in patients with ADHD and co-occurring anxiety6,7 or depressive disorders.
Dosing
Long-acting, triple-bead MAS is available in 12.5-, 25-, 37.5-, and 50-mg capsules. The capsule may be opened and sprinkled in food for patients who cannot swallow capsules. Opening of the capsule results in similar absorption relative to oral administration of the intact capsule.3
In adults with ADHD, long-acting, triple-bead MAS should be initiated at 12.5 mg in the morning (Table 2). However, in some individuals, long-acting, triple-bead MAS may be initiated at 25 mg each morning. Titration should occur in 12.5-mg weekly increments to a maximum dosage of 50 mg/d.3
In adults with severe renal impairment (glomerular filtrate rate, 15 to 30 mL/min/1.73 m2), the recommended starting dose is 12.5 mg/d, with a maximum dosage of 25 mg/d.3
The efficacy of long-acting, triple-bead MAS in adults with ADHD was demonstrated in 3 studies involving adults ages 18 to 55, and the effectiveness of the medication, with regard to duration of action, was assessed using the Time-Sensitive ADHD Symptom Scale—a self-report scale that consists of items indexed by the ADHD Rating Scale-IV (ADHD-RS-IV) which assesses ADHD symptom severity. Doses up to 75 mg/d were studied; however, there were no significant effects. It should be noted that this maximum daily dose was not determined by any safety parameter.
Study 1 (dose-optimization, triple-bead MAS, n = 137; placebo, n = 135, dosing: 12.5 to 75 mg) and Study 2 (forced dose-titration study, triple-bead MAS, n = 308; placebo, n = 104, dosing: 25 mg, 50 mg, 75 mg) demonstrated efficacy of triple-bead MAS for treating ADHD in adults. Despite differences in study designs, statistically significant and similar clinically relevant improvement was observed with triple-bead MAS (vs placebo) on ADHD-RS-IV total scores in both Study 1 and Study 2.8 An additional study in adults ages 18 to 55 (N = 275) with ADHD (DSM-5 criteria) involved randomization to either 12.5 mg (fixed dose) or forced titration (12.5 to 37.5 mg) or placebo and, as with the first 2 studies, improvement in ADHD symptoms was observed in triple-bead MAS-treated patients relative to those who had received placebo. (See Reference 3 for a summary of the clinical trials of triple-bead MAS in adults with ADHD.)
The tolerability of this medication was evaluated in a 12-month open-label study of adults with ADHD (DSM-IV-TR criteria) in which discontinuation was higher at doses >25 mg/d.7 Treatment-related increases in blood pressure and heart rate were consistent with the known hemodynamic adverse effect profile of stimulants.9
In adolescents with ADHD ages 13 to 17, long-acting, triple-bead MAS should be initiated at 12.5 mg/d and may be increased to 25 mg/d (Table 2). Importantly, in younger patients, including those younger than age 12, triple-bead MAS was associated with an increased risk of adverse events including insomnia and anorexia, and this was thought to be related to increased drug exposure (ie, AUC).
The efficacy of long-acting, triple-bead MAS was evaluated in 2 studies of adolescents ages 13 to 17, including 1 fixed-dose trial (25 mg/d) and 1 flexibly-dosed trial (12.5 to 25 mg/d). These unpublished studies utilized the ADHD-RS-IV score and the Average Permanent Product Measure of Performance, an age-adjusted math test and measure of sustained attention, and revealed statistically significant differences between medication and placebo in the primary outcomes.3
Adverse effects
Long-acting, triple-bead MAS was developed to treat ADHD symptoms throughout the day, and serum concentrations of the medication may be higher with this formulation compared with other long-acting preparations. Therefore, adverse effects that are directly related to plasma exposure (eg, insomnia and appetite suppression) may occur at higher rates with this preparation compared with alternatives. For example, in some of the registration trials, insomnia occurred in more than one-third of patients receiving the active medication (38%).9 Although insomnia was the most frequently reported adverse event in adults with ADHD, most reports of insomnia occurred early in the course of treatment. Of these insomnia-related adverse events, 94% were mild to moderate and resulted in discontinuation of the medication in approximately 2% of patients. Further, 73.9% of treatment-emergent, insomnia–related adverse events resolved during the course of the study. It is also important to note that the Pittsburgh Sleep Quality Index did not differ from placebo in studies of triple-bead MAS in adults with ADHD.10 Similarly, rates of stimulant-induced appetite suppression may be higher with this preparation compared with other long-acting preparations.9
Adverse effects observed in adults with ADHD that occurred in ≥2% of patients receiving triple-bead MAS and at least twice the incidence in patients randomized to placebo included:
- anxiety (7% vs 3%)
- feeling jittery (2% vs 1%)
- agitation (2% vs 0%)
- insomnia (31% vs 8%)
- depression (3% vs 0%)
- decreased appetite (30% vs 4%)
- weight loss (9% vs 0%)
- xerostomia (23% vs 4%)
- diarrhea (3% vs 0%)
- increased heart rate (9% vs 0%)
- palpitations (4% vs 2%)
- dysmenorrhea (4% vs 2%)
- erectile dysfunction (2% vs 1%).
In adolescents receiving triple-bea
1. Punja S, Shamseer L, Hartling L, et al. Amphetamines for attention deficit hyperactivity disorder (ADHD) in children and adolescents. Cochrane Database Syst Rev. 2016;2016(2):CD009996.
2. Castells X, Ramos-Quiroga J, Bosch R, et al. Amphetamines for attention deficit hyperactivity disorder (ADHD) in adults. Cochrane Database Syst Rev. 2011;(6):CD007813.
3. Mydayis [package insert]. Lexington, MA: Shire; 2017.
4. Heal DJ, Smith SL, Gosden J, et al. Amphetamine, past and present—a pharmacological and clinical perspective. J Psychopharmacol. 2013;27(6):479-496.
5. Hoffman JM, Dunnenberger HM, Kevin Hicks J, et al. Developing knowledge resources to support precision medicine: principles from the Clinical Pharmacogenetics Implementation Consortium (CPIC). J Am Med Inform Assoc. 2016;23(4):766-801.
6. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
7. Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
8. Goodman DW, Spencer TJ, Adler LA, et al. Clinical evaluation of triple-bead mixed amphetamine salts in adult ADHD. Presented at: 54th Annual Meeting of the American Academy of Child and Adolescent Psychiatry; October 25, 2007; Boston, MA.
9. Adler LA, Frick G, Yan B. A Long-term, open-label, safety study of triple-bead mixed amphetamine salts (SHP465) in adults with ADHD [published online April 1, 2017]. J Atten Disord. doi: 10.1177/1087054717696770.
10. Backhaus J, Junghanns K, Broocks A, et al. test-retest reliability and validity of the Pittsburgh Sleep Quality Index in primary insomnia. J Psychosom Res. 2002;53(3):737-740.
1. Punja S, Shamseer L, Hartling L, et al. Amphetamines for attention deficit hyperactivity disorder (ADHD) in children and adolescents. Cochrane Database Syst Rev. 2016;2016(2):CD009996.
2. Castells X, Ramos-Quiroga J, Bosch R, et al. Amphetamines for attention deficit hyperactivity disorder (ADHD) in adults. Cochrane Database Syst Rev. 2011;(6):CD007813.
3. Mydayis [package insert]. Lexington, MA: Shire; 2017.
4. Heal DJ, Smith SL, Gosden J, et al. Amphetamine, past and present—a pharmacological and clinical perspective. J Psychopharmacol. 2013;27(6):479-496.
5. Hoffman JM, Dunnenberger HM, Kevin Hicks J, et al. Developing knowledge resources to support precision medicine: principles from the Clinical Pharmacogenetics Implementation Consortium (CPIC). J Am Med Inform Assoc. 2016;23(4):766-801.
6. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
7. Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
8. Goodman DW, Spencer TJ, Adler LA, et al. Clinical evaluation of triple-bead mixed amphetamine salts in adult ADHD. Presented at: 54th Annual Meeting of the American Academy of Child and Adolescent Psychiatry; October 25, 2007; Boston, MA.
9. Adler LA, Frick G, Yan B. A Long-term, open-label, safety study of triple-bead mixed amphetamine salts (SHP465) in adults with ADHD [published online April 1, 2017]. J Atten Disord. doi: 10.1177/1087054717696770.
10. Backhaus J, Junghanns K, Broocks A, et al. test-retest reliability and validity of the Pittsburgh Sleep Quality Index in primary insomnia. J Psychosom Res. 2002;53(3):737-740.