User login
Valbenazine for tardive dyskinesia
Despite improvements in the tolerability of antipsychotic medications, the development of tardive dyskinesia (TD) still is a significant area of concern; however, clinicians have had few treatment options. Valbenazine, a vesicular monoamine transport type 2 (VMAT2) inhibitor, is the only FDA-approved medication for TD (Table 1).1 By modulating dopamine transport into presynaptic vesicles, synaptic dopamine release is decreased, thereby reducing the postsynaptic stimulation of D2 receptors and the severity of dyskinetic movements.

In the pivotal 6-week clinical trial, valbenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) ratings.2 Study completion rates were high (87.6%), with only 2 dropouts because of adverse events in each of the placebo (n = 78) and 40-mg (n = 76) arms, and 3 in the 80-mg group (n = 80).
Before the development of valbenazine, tetrabenazine was the only effective option for treating TD. Despite tetrabenazine’s known efficacy for TD, it was not available in the United States until 2008 with the sole indication for movements related to Huntington’s disease. U.S. patients often were subjected to a litany of ineffective medications for TD, often at great expense. Moreover, tetrabenazine involved multiple daily dosing, required cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, had significant tolerability issues, and a monthly cost of $8,000 to $10,000. The availability of an agent that is effective for TD and does not have tetrabenazine’s kinetic limitations, adverse effect profile, or CYP2D6 monitoring requirements represents an enormous advance in the treatment of TD.
Clinical implications
Tardive dyskinesia remains a significant public health concern because of the increasing use of antipsychotics for disorders beyond the core indication for schizophrenia. Although exposure to dopamine D2 antagonism could result in postsynaptic receptor upregulation and supersensitivity, this process best explains what underlies withdrawal dyskinesia.3 The persistence of TD symptoms in 66% to 80% of patients after discontinuing offending agents has led to hypotheses that the underlying pathophysiology of TD might best be conceptualized as a problem with neuroplasticity. As with many disorders, environmental contributions (eg, oxidative stress) and genetic predisposition might play a role beyond that related to exposure to D2 antagonism.3
There have been trials of numerous agents, but no medication has been FDA-approved for treating TD, and limited data support the efficacy of a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]),4 albeit with small effect sizes. A medical food, consisting of branched-chain amino acids, received FDA approval for the dietary management of TD in males, but is no longer commercially available except from compounding pharmacies.5
Tetrabenazine, a molecule developed in the mid-1950s to improve on the tolerability of reserpine, was associated with significant adverse effects such as orthostasis.6 Like reserpine, tetrabenazine subsequently was found to be effective for TD7 but without the peripheral adverse effects of reserpine. However, the kinetics of tetrabenazine necessitated multiple daily doses, and required CYP2D6 genotyping for doses >50 mg/d.8
Receptor blocking. The mechanism that differentiated reserpine’s and tetrabenazine’s clinical properties became clearer in the 1980s when researchers discovered that transporters were necessary to package neurotransmitters into the synaptic vesicles of presynaptic neurons.9 The vesicular monoamine transporter (VMAT) exists in 2 isoforms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the central nervous system.10
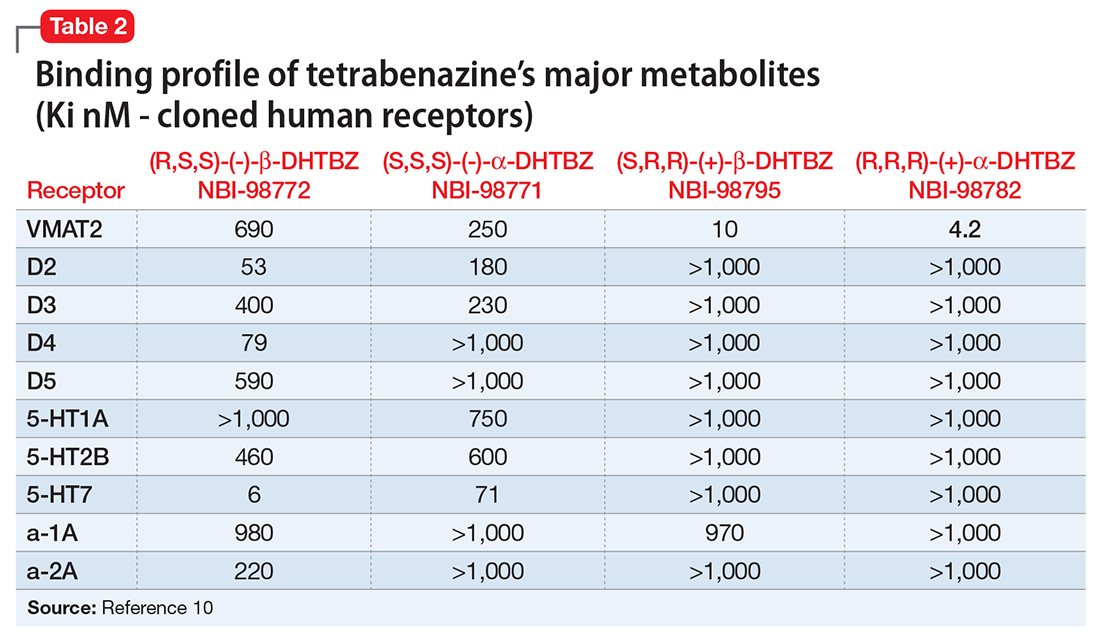
Tetrabenazine’s improved tolerability profile was related to the fact that it is a specific and reversible VMAT2 inhibitor, while reserpine is an irreversible and nonselective antagonist of both VMAT isoforms. Investigation of tetrabenazine’s metabolism revealed that it is rapidly and extensively converted into 2 isomers, α-dihydrotetrabenazine (DH-TBZ) and β-DH-TBZ. The isomeric forms of DH-TBZ have multiple chiral centers, and therefore numerous forms of which only 2 are significantly active at VMAT2.3 The α–DH-TBZ isomer is metabolized via CYP2D6 and 3A4 into inactive metabolites, while β-DH-TBZ is metabolized solely via 2D6.3 Because of the short half-life of DH-TBZ when generated from oral tetrabenazine, the existence of 2D6 polymorphisms, and the predominant activity deriving from only 2 isomers, a molecule was synthesized (valbenazine), that when metabolized would slowly be converted into the most active isomer of α–DH-TBZ designated as NBI-98782 (Table 2). This slower conversion to NBI-98782 from valbenazine (compared with its formation from oral tetrabenazine) yielded improved kinetics and permitted once-daily dosing; moreover, because the metabolism of NBI-98782 is not solely dependent on CYP2D6, the need for genotyping was removed. Neither of the 2 metabolites from valbenazine NBI-98782 and NB-136110 have significant affinity for targets other than VMAT2.11
Use in tardive dyskinesia. Recommended starting dosage is 40 mg once daily with or without food, increased to 80 mg after 1 week, based on the design and results from the phase-III clinical trial.12 The FDA granted breakthrough therapy designation for this compound, and only 1 phase-III trial was performed. Valbenazine produced significant improvement on the AIMS, with a mean 30% reduction in AIMS scores at the Week 6 endpoint from baseline of 10.4 ± 3.6.2 The effect size was large (Cohen’s d = 0.90) for the 80-mg dosage. Continuation of 40 mg/d may be considered for some patients based on tolerability, including those who are known CYP2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors. Patients taking strong 3A4 inhibitors should not exceed 40 mg/d. The maximum daily dose is 40 mg for those who have moderate or severe hepatic impairment (Child-Pugh score, 7 to 15). Dosage adjustment is not required for mild to moderate renal impairment (creatinine clearance, 30 to 90 mL/min).
Pharmacologic profile, adverse reactions
Valbenazine and its 2 metabolites lack affinity for receptors other than VMAT2, leading to an absence of orthostasis in clinical trials.1,2 In the phase-II trial, 76% of participants receiving valbenazine (n = 51) were titrated to the maximum dosage of 75 mg/d. Common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were headache (9.8% vs 4.1% placebo), fatigue (9.8% vs 4.1% placebo), and somnolence (5.9% vs 2% placebo).1 In the phase-III trial, participants were randomized 1:1:1 to valbenazine, 40 mg (n = 72), valbenazine, 80 mg (n = 79), or placebo (n = 76). In the clinical studies the most common diagnosis was schizophrenia or schizoaffective disorder, and 40% and 85% of participants in the phase-II and phase-III studies, respectively, remained on antipsychotics.1,2 There were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III trial.2
When data from all placebo-controlled studies were pooled, only 1 adverse effect occurred with an incidence ≥5% and twice that of placebo, somnolence with a rate of 10.9% for valbenazine vs 4.2% for placebo. The incidence of akathisia in the pooled analysis was 2.7% for valbenazine vs 0.5% for placebo. Importantly, in neither study was there a safety signal related to depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms.
The mean QT prolongation for valbenazine in healthy participants was 6.7 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 8.4 milliseconds. For those taking strong 2D6 or 3A4 inhibitors, or known 2D6 poor metabolizers, the mean QT prolongation was 11.7 milliseconds (14.7 milliseconds upper bound of double-sided 90% CI). In the controlled trials, there was a dose-related increase in prolactin, alkaline phosphatase, and bilirubin. Overall, 3% of valbenazine-treated patients and 2% of placebo-treated patients discontinued because of adverse reactions.
As noted above, there were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III valbenazine trial. Aggregate data across all placebo-controlled studies found that somnolence was the only adverse effect that occurred with an incidence ≥5% and twice that of placebo (10.9% for valbenazine vs 4.2% for placebo).2 As a comparsion, rates of sedation and akathisia for tetrabenazine were higher in the pivotal Huntington’s disease trial: sedation/somnolence 31% vs 3% for placebo, and akathisia 19% vs 0% for placebo.8
How it works
Tetrabenazine, a selective VMAT2 inhibitor, is the only agent that has demonstrated significant efficacy and tolerability for TD management; however, its complex metabolism generates numerous isomers of the metabolites α-DH-TBZ and β-DH-TBZ, of which only 2 are significantly active (Table 3). By choosing an active isomer (NBI-98782) as the metabolite of interest because of its selective and potent activity at VMAT2 and having a metabolism not solely dependent on CYP2D6, a compound was generated (valbenazine) that when metabolized slowly converts into NBI-98782.

Pharmacokinetics
Valbenazine demonstrates dose-proportional pharmacokinetics after single oral dosages from 40 to 300 mg with no impact of food or fasting status on levels of the active metabolite. Valbenazine has a Tmax of 0.5 to 1.0 hours, with 49% oral bioavailability. The plasma half-life for valbenazine and for NBI-98782 ranges from 15 to 22 hours. The Tmax for NBI-98782 when formed from valbenazine occurs between 4 and 8 hours, with a Cmax of approximately 30 ng/mL. It should be noted that when NBI-98782 is generated from oral tetrabenazine, the mean half-life and Tmax are considerably shorter (6 hours and 1.5 hours, respectively), while the Cmax is much higher (approximately 77 ng/mL) (Table 4).

Valbenazine is metabolized through endogenous esterases to NBI-98782 and NBI-136110. NBI-98782, the active metabolite, is further metabolized through multiple CYP pathways, predominantly 3A4 and 2D6. Neither valbenazine nor its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that valbenazine and its active metabolite are unlikely to inhibit most major drug transporters at clinically relevant concentrations. However, valbenazine increased digoxin levels because of inhibition of intestinal P-glycoprotein; therefore plasma digoxin level monitoring is recommended when these 2 are co-administered.
Efficacy
Efficacy was established in a 6-week, fixed-dosage, double-blind, placebo-controlled trial of adult patients with TD. Eligible participants had:
- DSM-IV diagnosis of antipsychotic-induced TD for ≥3 months before screening and moderate or severe TD, as indicated by AIMS item 8 (severity of abnormal movement), which was rated by a blinded, external reviewer using a video of the participant’s AIMS assessment at screening
- a DSM-IV diagnosis of schizophrenia or schizoaffective disorder or mood disorder (and stable per investigator)
- Brief Psychiatric Rating Scale score <50 at screening.
Exclusion criteria included clinically significant and unstable medical conditions within 1 month before screening; comorbid movement disorder (eg, parkinsonism, akathisia, truncal dystonia) that was more prominent than TD; and significant risk for active suicidal ideation, suicidal behavior, or violent behavior.2 Participants had a mean age of 56, 52% were male, and 65.7% of participants in the valbenazine 40-mg group had a schizophrenia spectrum disorder diagnosis, as did 65.8% in both the placebo and valbenazine 80-mg arms.
Antipsychotic treatments were permitted during the trial and >85% of participants continued taking these medications during the study. Participants (N = 234) were randomly allocated in a 1:1:1 manner to valbenazine 40 mg, 80 mg, or matched placebo. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. Baseline AIMS scores were 9.9 ± 4.3 in the placebo group, and 9.8 ± 4.1 and 10.4 ± 3.6 in the valbenazine 40-mg and 80-mg arms, respectively.2
Outcome. A fixed-sequence testing procedure to control for family-wise error rate and multiplicity was employed, and the primary endpoint was change from baseline to Week 6 in AIMS total score (items 1 to 7) for valbenazine 80 mg vs placebo. Valbenazine, 40 mg, was associated with a 1.9 point decrease in AIMS score, while valbenazine, 80 mg, was associated with a 3.2 point decrease in AIMS score, compared with 0.1 point decrease for placebo (P < .05 for valbenazine, 40 mg, P < .001 for valbenazine, 80 mg). This difference for the 40-mg dosage did not meet the prespecified analysis endpoints; however, for the 80-mg valbenazine dosage, the effect size for this difference (Cohen’s d) was large 0.90. There also were statistically significant differences between 40 mg and 80 mg at weeks 2, 4, and 6 in the intent-to-treat population. Of the 79 participants, 43 taking the 80-mg dosage completed a 48-week extension. Efficacy was sustained in this group; however, when valbenazine was discontinued at Week 48, AIMS scores returned to baseline after 4 weeks.
Tolerability
Of the 234 randomized patients, 205 (87.6%) completed the 6-week trial. Discontinuations due to adverse events were low across all treatment groups: 2.6% and 2.8% in the placebo and valbenazine 40-mg arms, respectively, and 3.8% in valbenazine 80-mg cohort. There was no safety signal based on changes in depression, suicidality, parkinsonism rating, or changes in schizophrenia symptoms. Because valbenazine can cause somnolence, patients should not perform activities requiring mental alertness (eg, operating a vehicle or hazardous machinery) until they know how they will be affected by valbenazine.
Valbenazine should be avoided in patients with congenital long QT syndrome or with arrhythmias associated with a prolonged QT interval. For patients at increased risk of a prolonged QT interval, assess the QT interval before increasing the dosage.
Clinical considerations
Unique properties. Valbenazine is metabolized slowly to a potent, selective VMAT2 antagonist (NBI-98782) in a manner that permits once daily dosing, removes the need for CYP2D6 genotyping, and provides significant efficacy.
Why Rx? The reasons to prescribe valbenazine for TD patients include:
- currently the only agent with FDA approval for TD
- fewer tolerability issues seen with the only other effective agent, tetrabenazine
- no signal for effects on mood parameters or rates of parkinsonism
- lack of multiple daily dosing and possible need for 2D6 genotyping involved with TBZ prescribing.
Dosing
The recommended dosage of valbenazine is 80 mg/d administered as a single dose with or without food, starting at 40 mg once daily for 1 week. There is no dosage adjustment required in those with mild to moderate renal impairment; however, valbenazine is not recommended in those with severe renal impairment. The maximum dose is 40 mg/d for those who with moderate or severe hepatic impairment (Child-Pugh score, 7 to 15) however, valbenazine is not recommended for patients with severe renal impairment (creatinine clearance <30 mL/min) because the exposure to the active metabolite is reduced by approximately 75%. The combined efficacy and tolerability of dosages >80 mg/d has not been evaluated. Adverse effects seen with tetrabenazine at higher dosages include akathisia, anxiety, insomnia, parkinsonism, fatigue, and depression.
A daily dose of 40 mg may be considered for some patients based on tolerability, including those who are known CYP 2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors.2 For those taking strong 3A4 inhibitors, the maximum daily dose is 40 mg. Concomitant use of valbenazine with strong 3A4 inducers is not recommended as the exposure to the active metabolite is reduced by approximately 75%.2 Lastly, because VMAT2 inhibition may alter synaptic levels of other monoamines, it is recommended that valbenazine not be administered with monoamine oxidase inhibitors, such as isocarboxazid, phenelzine, or selegiline.
Contraindications
There are no reported contraindications for valbenazine. As with most medications, there is limited available data on valbenazine use in pregnant women; however, administration of valbenazine to pregnant rats during organogenesis through lactation produced an increase in the number of stillborn pups and postnatal pup mortalities at doses under the maximum recommended human dose (MRHD) using body surface area based dosing (mg/m2). Pregnant women should be advised of the potential risk to a fetus. Valbenazine and its metabolites have been detected in rat milk at concentrations higher than in plasma after oral administration of valbenazine at doses 0.1 to 1.2 times the MRHD (based on mg/m2). Based on animal findings of increased perinatal mortality in exposed fetuses and pups, woman are advised not to breastfeed during valbenazine treatment and for 5 days after the final dose. No dosage adjustment is required for geriatric patients.
1. O’Brien CF, Jimenez R, Hauser RA, et al. NBI-98854, a selective monoamine transport inhibitor for the treatment of tardive dyskinesia: a randomized, double-blind, placebo-controlled study. Mov Disord. 2015;30(12):1681-1687.
2. Ingrezza [package insert]. San Diego, CA: Neurocrine Biosciences Inc.; 2017.
3. Marder S, Knesevich MA, Hauser RA, et al. KINECT 3: A randomized, double-blind, placebo-controlled phase 3 trial of valbenazine (NBI-98854) for tardive dyskinesia. Poster presented at the American Psychiatric Association Annual Meeting; May 14-18, 2016; Atlanta, GA.
4. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
5. Richardson MA, Bevans ML, Read LL, et al. Efficacy of the branched-chain amino acids in the treatment of tardive dyskinesia in men. Am J Psychiatry. 2003;160(6):1117-1124.
6. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
7. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
10. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
11. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
12. Grigoriadis DE, Smith E, Madan A, et al. Pharmacologic characteristics of valbenazine (NBI-98854) and its metabolites. Poster presented at the U.S. Psychiatric & Mental Health Congress, October 21-24, 2016; San Antonio, TX.
Despite improvements in the tolerability of antipsychotic medications, the development of tardive dyskinesia (TD) still is a significant area of concern; however, clinicians have had few treatment options. Valbenazine, a vesicular monoamine transport type 2 (VMAT2) inhibitor, is the only FDA-approved medication for TD (Table 1).1 By modulating dopamine transport into presynaptic vesicles, synaptic dopamine release is decreased, thereby reducing the postsynaptic stimulation of D2 receptors and the severity of dyskinetic movements.
In the pivotal 6-week clinical trial, valbenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) ratings.2 Study completion rates were high (87.6%), with only 2 dropouts because of adverse events in each of the placebo (n = 78) and 40-mg (n = 76) arms, and 3 in the 80-mg group (n = 80).
Before the development of valbenazine, tetrabenazine was the only effective option for treating TD. Despite tetrabenazine’s known efficacy for TD, it was not available in the United States until 2008 with the sole indication for movements related to Huntington’s disease. U.S. patients often were subjected to a litany of ineffective medications for TD, often at great expense. Moreover, tetrabenazine involved multiple daily dosing, required cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, had significant tolerability issues, and a monthly cost of $8,000 to $10,000. The availability of an agent that is effective for TD and does not have tetrabenazine’s kinetic limitations, adverse effect profile, or CYP2D6 monitoring requirements represents an enormous advance in the treatment of TD.
Clinical implications
Tardive dyskinesia remains a significant public health concern because of the increasing use of antipsychotics for disorders beyond the core indication for schizophrenia. Although exposure to dopamine D2 antagonism could result in postsynaptic receptor upregulation and supersensitivity, this process best explains what underlies withdrawal dyskinesia.3 The persistence of TD symptoms in 66% to 80% of patients after discontinuing offending agents has led to hypotheses that the underlying pathophysiology of TD might best be conceptualized as a problem with neuroplasticity. As with many disorders, environmental contributions (eg, oxidative stress) and genetic predisposition might play a role beyond that related to exposure to D2 antagonism.3
There have been trials of numerous agents, but no medication has been FDA-approved for treating TD, and limited data support the efficacy of a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]),4 albeit with small effect sizes. A medical food, consisting of branched-chain amino acids, received FDA approval for the dietary management of TD in males, but is no longer commercially available except from compounding pharmacies.5
Tetrabenazine, a molecule developed in the mid-1950s to improve on the tolerability of reserpine, was associated with significant adverse effects such as orthostasis.6 Like reserpine, tetrabenazine subsequently was found to be effective for TD7 but without the peripheral adverse effects of reserpine. However, the kinetics of tetrabenazine necessitated multiple daily doses, and required CYP2D6 genotyping for doses >50 mg/d.8
Receptor blocking. The mechanism that differentiated reserpine’s and tetrabenazine’s clinical properties became clearer in the 1980s when researchers discovered that transporters were necessary to package neurotransmitters into the synaptic vesicles of presynaptic neurons.9 The vesicular monoamine transporter (VMAT) exists in 2 isoforms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the central nervous system.10
Tetrabenazine’s improved tolerability profile was related to the fact that it is a specific and reversible VMAT2 inhibitor, while reserpine is an irreversible and nonselective antagonist of both VMAT isoforms. Investigation of tetrabenazine’s metabolism revealed that it is rapidly and extensively converted into 2 isomers, α-dihydrotetrabenazine (DH-TBZ) and β-DH-TBZ. The isomeric forms of DH-TBZ have multiple chiral centers, and therefore numerous forms of which only 2 are significantly active at VMAT2.3 The α–DH-TBZ isomer is metabolized via CYP2D6 and 3A4 into inactive metabolites, while β-DH-TBZ is metabolized solely via 2D6.3 Because of the short half-life of DH-TBZ when generated from oral tetrabenazine, the existence of 2D6 polymorphisms, and the predominant activity deriving from only 2 isomers, a molecule was synthesized (valbenazine), that when metabolized would slowly be converted into the most active isomer of α–DH-TBZ designated as NBI-98782 (Table 2). This slower conversion to NBI-98782 from valbenazine (compared with its formation from oral tetrabenazine) yielded improved kinetics and permitted once-daily dosing; moreover, because the metabolism of NBI-98782 is not solely dependent on CYP2D6, the need for genotyping was removed. Neither of the 2 metabolites from valbenazine NBI-98782 and NB-136110 have significant affinity for targets other than VMAT2.11
Use in tardive dyskinesia. Recommended starting dosage is 40 mg once daily with or without food, increased to 80 mg after 1 week, based on the design and results from the phase-III clinical trial.12 The FDA granted breakthrough therapy designation for this compound, and only 1 phase-III trial was performed. Valbenazine produced significant improvement on the AIMS, with a mean 30% reduction in AIMS scores at the Week 6 endpoint from baseline of 10.4 ± 3.6.2 The effect size was large (Cohen’s d = 0.90) for the 80-mg dosage. Continuation of 40 mg/d may be considered for some patients based on tolerability, including those who are known CYP2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors. Patients taking strong 3A4 inhibitors should not exceed 40 mg/d. The maximum daily dose is 40 mg for those who have moderate or severe hepatic impairment (Child-Pugh score, 7 to 15). Dosage adjustment is not required for mild to moderate renal impairment (creatinine clearance, 30 to 90 mL/min).
Pharmacologic profile, adverse reactions
Valbenazine and its 2 metabolites lack affinity for receptors other than VMAT2, leading to an absence of orthostasis in clinical trials.1,2 In the phase-II trial, 76% of participants receiving valbenazine (n = 51) were titrated to the maximum dosage of 75 mg/d. Common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were headache (9.8% vs 4.1% placebo), fatigue (9.8% vs 4.1% placebo), and somnolence (5.9% vs 2% placebo).1 In the phase-III trial, participants were randomized 1:1:1 to valbenazine, 40 mg (n = 72), valbenazine, 80 mg (n = 79), or placebo (n = 76). In the clinical studies the most common diagnosis was schizophrenia or schizoaffective disorder, and 40% and 85% of participants in the phase-II and phase-III studies, respectively, remained on antipsychotics.1,2 There were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III trial.2
When data from all placebo-controlled studies were pooled, only 1 adverse effect occurred with an incidence ≥5% and twice that of placebo, somnolence with a rate of 10.9% for valbenazine vs 4.2% for placebo. The incidence of akathisia in the pooled analysis was 2.7% for valbenazine vs 0.5% for placebo. Importantly, in neither study was there a safety signal related to depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms.
The mean QT prolongation for valbenazine in healthy participants was 6.7 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 8.4 milliseconds. For those taking strong 2D6 or 3A4 inhibitors, or known 2D6 poor metabolizers, the mean QT prolongation was 11.7 milliseconds (14.7 milliseconds upper bound of double-sided 90% CI). In the controlled trials, there was a dose-related increase in prolactin, alkaline phosphatase, and bilirubin. Overall, 3% of valbenazine-treated patients and 2% of placebo-treated patients discontinued because of adverse reactions.
As noted above, there were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III valbenazine trial. Aggregate data across all placebo-controlled studies found that somnolence was the only adverse effect that occurred with an incidence ≥5% and twice that of placebo (10.9% for valbenazine vs 4.2% for placebo).2 As a comparsion, rates of sedation and akathisia for tetrabenazine were higher in the pivotal Huntington’s disease trial: sedation/somnolence 31% vs 3% for placebo, and akathisia 19% vs 0% for placebo.8
How it works
Tetrabenazine, a selective VMAT2 inhibitor, is the only agent that has demonstrated significant efficacy and tolerability for TD management; however, its complex metabolism generates numerous isomers of the metabolites α-DH-TBZ and β-DH-TBZ, of which only 2 are significantly active (Table 3). By choosing an active isomer (NBI-98782) as the metabolite of interest because of its selective and potent activity at VMAT2 and having a metabolism not solely dependent on CYP2D6, a compound was generated (valbenazine) that when metabolized slowly converts into NBI-98782.
Pharmacokinetics
Valbenazine demonstrates dose-proportional pharmacokinetics after single oral dosages from 40 to 300 mg with no impact of food or fasting status on levels of the active metabolite. Valbenazine has a Tmax of 0.5 to 1.0 hours, with 49% oral bioavailability. The plasma half-life for valbenazine and for NBI-98782 ranges from 15 to 22 hours. The Tmax for NBI-98782 when formed from valbenazine occurs between 4 and 8 hours, with a Cmax of approximately 30 ng/mL. It should be noted that when NBI-98782 is generated from oral tetrabenazine, the mean half-life and Tmax are considerably shorter (6 hours and 1.5 hours, respectively), while the Cmax is much higher (approximately 77 ng/mL) (Table 4).
Valbenazine is metabolized through endogenous esterases to NBI-98782 and NBI-136110. NBI-98782, the active metabolite, is further metabolized through multiple CYP pathways, predominantly 3A4 and 2D6. Neither valbenazine nor its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that valbenazine and its active metabolite are unlikely to inhibit most major drug transporters at clinically relevant concentrations. However, valbenazine increased digoxin levels because of inhibition of intestinal P-glycoprotein; therefore plasma digoxin level monitoring is recommended when these 2 are co-administered.
Efficacy
Efficacy was established in a 6-week, fixed-dosage, double-blind, placebo-controlled trial of adult patients with TD. Eligible participants had:
- DSM-IV diagnosis of antipsychotic-induced TD for ≥3 months before screening and moderate or severe TD, as indicated by AIMS item 8 (severity of abnormal movement), which was rated by a blinded, external reviewer using a video of the participant’s AIMS assessment at screening
- a DSM-IV diagnosis of schizophrenia or schizoaffective disorder or mood disorder (and stable per investigator)
- Brief Psychiatric Rating Scale score <50 at screening.
Exclusion criteria included clinically significant and unstable medical conditions within 1 month before screening; comorbid movement disorder (eg, parkinsonism, akathisia, truncal dystonia) that was more prominent than TD; and significant risk for active suicidal ideation, suicidal behavior, or violent behavior.2 Participants had a mean age of 56, 52% were male, and 65.7% of participants in the valbenazine 40-mg group had a schizophrenia spectrum disorder diagnosis, as did 65.8% in both the placebo and valbenazine 80-mg arms.
Antipsychotic treatments were permitted during the trial and >85% of participants continued taking these medications during the study. Participants (N = 234) were randomly allocated in a 1:1:1 manner to valbenazine 40 mg, 80 mg, or matched placebo. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. Baseline AIMS scores were 9.9 ± 4.3 in the placebo group, and 9.8 ± 4.1 and 10.4 ± 3.6 in the valbenazine 40-mg and 80-mg arms, respectively.2
Outcome. A fixed-sequence testing procedure to control for family-wise error rate and multiplicity was employed, and the primary endpoint was change from baseline to Week 6 in AIMS total score (items 1 to 7) for valbenazine 80 mg vs placebo. Valbenazine, 40 mg, was associated with a 1.9 point decrease in AIMS score, while valbenazine, 80 mg, was associated with a 3.2 point decrease in AIMS score, compared with 0.1 point decrease for placebo (P < .05 for valbenazine, 40 mg, P < .001 for valbenazine, 80 mg). This difference for the 40-mg dosage did not meet the prespecified analysis endpoints; however, for the 80-mg valbenazine dosage, the effect size for this difference (Cohen’s d) was large 0.90. There also were statistically significant differences between 40 mg and 80 mg at weeks 2, 4, and 6 in the intent-to-treat population. Of the 79 participants, 43 taking the 80-mg dosage completed a 48-week extension. Efficacy was sustained in this group; however, when valbenazine was discontinued at Week 48, AIMS scores returned to baseline after 4 weeks.
Tolerability
Of the 234 randomized patients, 205 (87.6%) completed the 6-week trial. Discontinuations due to adverse events were low across all treatment groups: 2.6% and 2.8% in the placebo and valbenazine 40-mg arms, respectively, and 3.8% in valbenazine 80-mg cohort. There was no safety signal based on changes in depression, suicidality, parkinsonism rating, or changes in schizophrenia symptoms. Because valbenazine can cause somnolence, patients should not perform activities requiring mental alertness (eg, operating a vehicle or hazardous machinery) until they know how they will be affected by valbenazine.
Valbenazine should be avoided in patients with congenital long QT syndrome or with arrhythmias associated with a prolonged QT interval. For patients at increased risk of a prolonged QT interval, assess the QT interval before increasing the dosage.
Clinical considerations
Unique properties. Valbenazine is metabolized slowly to a potent, selective VMAT2 antagonist (NBI-98782) in a manner that permits once daily dosing, removes the need for CYP2D6 genotyping, and provides significant efficacy.
Why Rx? The reasons to prescribe valbenazine for TD patients include:
- currently the only agent with FDA approval for TD
- fewer tolerability issues seen with the only other effective agent, tetrabenazine
- no signal for effects on mood parameters or rates of parkinsonism
- lack of multiple daily dosing and possible need for 2D6 genotyping involved with TBZ prescribing.
Dosing
The recommended dosage of valbenazine is 80 mg/d administered as a single dose with or without food, starting at 40 mg once daily for 1 week. There is no dosage adjustment required in those with mild to moderate renal impairment; however, valbenazine is not recommended in those with severe renal impairment. The maximum dose is 40 mg/d for those who with moderate or severe hepatic impairment (Child-Pugh score, 7 to 15) however, valbenazine is not recommended for patients with severe renal impairment (creatinine clearance <30 mL/min) because the exposure to the active metabolite is reduced by approximately 75%. The combined efficacy and tolerability of dosages >80 mg/d has not been evaluated. Adverse effects seen with tetrabenazine at higher dosages include akathisia, anxiety, insomnia, parkinsonism, fatigue, and depression.
A daily dose of 40 mg may be considered for some patients based on tolerability, including those who are known CYP 2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors.2 For those taking strong 3A4 inhibitors, the maximum daily dose is 40 mg. Concomitant use of valbenazine with strong 3A4 inducers is not recommended as the exposure to the active metabolite is reduced by approximately 75%.2 Lastly, because VMAT2 inhibition may alter synaptic levels of other monoamines, it is recommended that valbenazine not be administered with monoamine oxidase inhibitors, such as isocarboxazid, phenelzine, or selegiline.
Contraindications
There are no reported contraindications for valbenazine. As with most medications, there is limited available data on valbenazine use in pregnant women; however, administration of valbenazine to pregnant rats during organogenesis through lactation produced an increase in the number of stillborn pups and postnatal pup mortalities at doses under the maximum recommended human dose (MRHD) using body surface area based dosing (mg/m2). Pregnant women should be advised of the potential risk to a fetus. Valbenazine and its metabolites have been detected in rat milk at concentrations higher than in plasma after oral administration of valbenazine at doses 0.1 to 1.2 times the MRHD (based on mg/m2). Based on animal findings of increased perinatal mortality in exposed fetuses and pups, woman are advised not to breastfeed during valbenazine treatment and for 5 days after the final dose. No dosage adjustment is required for geriatric patients.
Despite improvements in the tolerability of antipsychotic medications, the development of tardive dyskinesia (TD) still is a significant area of concern; however, clinicians have had few treatment options. Valbenazine, a vesicular monoamine transport type 2 (VMAT2) inhibitor, is the only FDA-approved medication for TD (Table 1).1 By modulating dopamine transport into presynaptic vesicles, synaptic dopamine release is decreased, thereby reducing the postsynaptic stimulation of D2 receptors and the severity of dyskinetic movements.
In the pivotal 6-week clinical trial, valbenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) ratings.2 Study completion rates were high (87.6%), with only 2 dropouts because of adverse events in each of the placebo (n = 78) and 40-mg (n = 76) arms, and 3 in the 80-mg group (n = 80).
Before the development of valbenazine, tetrabenazine was the only effective option for treating TD. Despite tetrabenazine’s known efficacy for TD, it was not available in the United States until 2008 with the sole indication for movements related to Huntington’s disease. U.S. patients often were subjected to a litany of ineffective medications for TD, often at great expense. Moreover, tetrabenazine involved multiple daily dosing, required cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, had significant tolerability issues, and a monthly cost of $8,000 to $10,000. The availability of an agent that is effective for TD and does not have tetrabenazine’s kinetic limitations, adverse effect profile, or CYP2D6 monitoring requirements represents an enormous advance in the treatment of TD.
Clinical implications
Tardive dyskinesia remains a significant public health concern because of the increasing use of antipsychotics for disorders beyond the core indication for schizophrenia. Although exposure to dopamine D2 antagonism could result in postsynaptic receptor upregulation and supersensitivity, this process best explains what underlies withdrawal dyskinesia.3 The persistence of TD symptoms in 66% to 80% of patients after discontinuing offending agents has led to hypotheses that the underlying pathophysiology of TD might best be conceptualized as a problem with neuroplasticity. As with many disorders, environmental contributions (eg, oxidative stress) and genetic predisposition might play a role beyond that related to exposure to D2 antagonism.3
There have been trials of numerous agents, but no medication has been FDA-approved for treating TD, and limited data support the efficacy of a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]),4 albeit with small effect sizes. A medical food, consisting of branched-chain amino acids, received FDA approval for the dietary management of TD in males, but is no longer commercially available except from compounding pharmacies.5
Tetrabenazine, a molecule developed in the mid-1950s to improve on the tolerability of reserpine, was associated with significant adverse effects such as orthostasis.6 Like reserpine, tetrabenazine subsequently was found to be effective for TD7 but without the peripheral adverse effects of reserpine. However, the kinetics of tetrabenazine necessitated multiple daily doses, and required CYP2D6 genotyping for doses >50 mg/d.8
Receptor blocking. The mechanism that differentiated reserpine’s and tetrabenazine’s clinical properties became clearer in the 1980s when researchers discovered that transporters were necessary to package neurotransmitters into the synaptic vesicles of presynaptic neurons.9 The vesicular monoamine transporter (VMAT) exists in 2 isoforms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the central nervous system.10
Tetrabenazine’s improved tolerability profile was related to the fact that it is a specific and reversible VMAT2 inhibitor, while reserpine is an irreversible and nonselective antagonist of both VMAT isoforms. Investigation of tetrabenazine’s metabolism revealed that it is rapidly and extensively converted into 2 isomers, α-dihydrotetrabenazine (DH-TBZ) and β-DH-TBZ. The isomeric forms of DH-TBZ have multiple chiral centers, and therefore numerous forms of which only 2 are significantly active at VMAT2.3 The α–DH-TBZ isomer is metabolized via CYP2D6 and 3A4 into inactive metabolites, while β-DH-TBZ is metabolized solely via 2D6.3 Because of the short half-life of DH-TBZ when generated from oral tetrabenazine, the existence of 2D6 polymorphisms, and the predominant activity deriving from only 2 isomers, a molecule was synthesized (valbenazine), that when metabolized would slowly be converted into the most active isomer of α–DH-TBZ designated as NBI-98782 (Table 2). This slower conversion to NBI-98782 from valbenazine (compared with its formation from oral tetrabenazine) yielded improved kinetics and permitted once-daily dosing; moreover, because the metabolism of NBI-98782 is not solely dependent on CYP2D6, the need for genotyping was removed. Neither of the 2 metabolites from valbenazine NBI-98782 and NB-136110 have significant affinity for targets other than VMAT2.11
Use in tardive dyskinesia. Recommended starting dosage is 40 mg once daily with or without food, increased to 80 mg after 1 week, based on the design and results from the phase-III clinical trial.12 The FDA granted breakthrough therapy designation for this compound, and only 1 phase-III trial was performed. Valbenazine produced significant improvement on the AIMS, with a mean 30% reduction in AIMS scores at the Week 6 endpoint from baseline of 10.4 ± 3.6.2 The effect size was large (Cohen’s d = 0.90) for the 80-mg dosage. Continuation of 40 mg/d may be considered for some patients based on tolerability, including those who are known CYP2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors. Patients taking strong 3A4 inhibitors should not exceed 40 mg/d. The maximum daily dose is 40 mg for those who have moderate or severe hepatic impairment (Child-Pugh score, 7 to 15). Dosage adjustment is not required for mild to moderate renal impairment (creatinine clearance, 30 to 90 mL/min).
Pharmacologic profile, adverse reactions
Valbenazine and its 2 metabolites lack affinity for receptors other than VMAT2, leading to an absence of orthostasis in clinical trials.1,2 In the phase-II trial, 76% of participants receiving valbenazine (n = 51) were titrated to the maximum dosage of 75 mg/d. Common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were headache (9.8% vs 4.1% placebo), fatigue (9.8% vs 4.1% placebo), and somnolence (5.9% vs 2% placebo).1 In the phase-III trial, participants were randomized 1:1:1 to valbenazine, 40 mg (n = 72), valbenazine, 80 mg (n = 79), or placebo (n = 76). In the clinical studies the most common diagnosis was schizophrenia or schizoaffective disorder, and 40% and 85% of participants in the phase-II and phase-III studies, respectively, remained on antipsychotics.1,2 There were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III trial.2
When data from all placebo-controlled studies were pooled, only 1 adverse effect occurred with an incidence ≥5% and twice that of placebo, somnolence with a rate of 10.9% for valbenazine vs 4.2% for placebo. The incidence of akathisia in the pooled analysis was 2.7% for valbenazine vs 0.5% for placebo. Importantly, in neither study was there a safety signal related to depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms.
The mean QT prolongation for valbenazine in healthy participants was 6.7 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 8.4 milliseconds. For those taking strong 2D6 or 3A4 inhibitors, or known 2D6 poor metabolizers, the mean QT prolongation was 11.7 milliseconds (14.7 milliseconds upper bound of double-sided 90% CI). In the controlled trials, there was a dose-related increase in prolactin, alkaline phosphatase, and bilirubin. Overall, 3% of valbenazine-treated patients and 2% of placebo-treated patients discontinued because of adverse reactions.
As noted above, there were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III valbenazine trial. Aggregate data across all placebo-controlled studies found that somnolence was the only adverse effect that occurred with an incidence ≥5% and twice that of placebo (10.9% for valbenazine vs 4.2% for placebo).2 As a comparsion, rates of sedation and akathisia for tetrabenazine were higher in the pivotal Huntington’s disease trial: sedation/somnolence 31% vs 3% for placebo, and akathisia 19% vs 0% for placebo.8
How it works
Tetrabenazine, a selective VMAT2 inhibitor, is the only agent that has demonstrated significant efficacy and tolerability for TD management; however, its complex metabolism generates numerous isomers of the metabolites α-DH-TBZ and β-DH-TBZ, of which only 2 are significantly active (Table 3). By choosing an active isomer (NBI-98782) as the metabolite of interest because of its selective and potent activity at VMAT2 and having a metabolism not solely dependent on CYP2D6, a compound was generated (valbenazine) that when metabolized slowly converts into NBI-98782.
Pharmacokinetics
Valbenazine demonstrates dose-proportional pharmacokinetics after single oral dosages from 40 to 300 mg with no impact of food or fasting status on levels of the active metabolite. Valbenazine has a Tmax of 0.5 to 1.0 hours, with 49% oral bioavailability. The plasma half-life for valbenazine and for NBI-98782 ranges from 15 to 22 hours. The Tmax for NBI-98782 when formed from valbenazine occurs between 4 and 8 hours, with a Cmax of approximately 30 ng/mL. It should be noted that when NBI-98782 is generated from oral tetrabenazine, the mean half-life and Tmax are considerably shorter (6 hours and 1.5 hours, respectively), while the Cmax is much higher (approximately 77 ng/mL) (Table 4).
Valbenazine is metabolized through endogenous esterases to NBI-98782 and NBI-136110. NBI-98782, the active metabolite, is further metabolized through multiple CYP pathways, predominantly 3A4 and 2D6. Neither valbenazine nor its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that valbenazine and its active metabolite are unlikely to inhibit most major drug transporters at clinically relevant concentrations. However, valbenazine increased digoxin levels because of inhibition of intestinal P-glycoprotein; therefore plasma digoxin level monitoring is recommended when these 2 are co-administered.
Efficacy
Efficacy was established in a 6-week, fixed-dosage, double-blind, placebo-controlled trial of adult patients with TD. Eligible participants had:
- DSM-IV diagnosis of antipsychotic-induced TD for ≥3 months before screening and moderate or severe TD, as indicated by AIMS item 8 (severity of abnormal movement), which was rated by a blinded, external reviewer using a video of the participant’s AIMS assessment at screening
- a DSM-IV diagnosis of schizophrenia or schizoaffective disorder or mood disorder (and stable per investigator)
- Brief Psychiatric Rating Scale score <50 at screening.
Exclusion criteria included clinically significant and unstable medical conditions within 1 month before screening; comorbid movement disorder (eg, parkinsonism, akathisia, truncal dystonia) that was more prominent than TD; and significant risk for active suicidal ideation, suicidal behavior, or violent behavior.2 Participants had a mean age of 56, 52% were male, and 65.7% of participants in the valbenazine 40-mg group had a schizophrenia spectrum disorder diagnosis, as did 65.8% in both the placebo and valbenazine 80-mg arms.
Antipsychotic treatments were permitted during the trial and >85% of participants continued taking these medications during the study. Participants (N = 234) were randomly allocated in a 1:1:1 manner to valbenazine 40 mg, 80 mg, or matched placebo. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. Baseline AIMS scores were 9.9 ± 4.3 in the placebo group, and 9.8 ± 4.1 and 10.4 ± 3.6 in the valbenazine 40-mg and 80-mg arms, respectively.2
Outcome. A fixed-sequence testing procedure to control for family-wise error rate and multiplicity was employed, and the primary endpoint was change from baseline to Week 6 in AIMS total score (items 1 to 7) for valbenazine 80 mg vs placebo. Valbenazine, 40 mg, was associated with a 1.9 point decrease in AIMS score, while valbenazine, 80 mg, was associated with a 3.2 point decrease in AIMS score, compared with 0.1 point decrease for placebo (P < .05 for valbenazine, 40 mg, P < .001 for valbenazine, 80 mg). This difference for the 40-mg dosage did not meet the prespecified analysis endpoints; however, for the 80-mg valbenazine dosage, the effect size for this difference (Cohen’s d) was large 0.90. There also were statistically significant differences between 40 mg and 80 mg at weeks 2, 4, and 6 in the intent-to-treat population. Of the 79 participants, 43 taking the 80-mg dosage completed a 48-week extension. Efficacy was sustained in this group; however, when valbenazine was discontinued at Week 48, AIMS scores returned to baseline after 4 weeks.
Tolerability
Of the 234 randomized patients, 205 (87.6%) completed the 6-week trial. Discontinuations due to adverse events were low across all treatment groups: 2.6% and 2.8% in the placebo and valbenazine 40-mg arms, respectively, and 3.8% in valbenazine 80-mg cohort. There was no safety signal based on changes in depression, suicidality, parkinsonism rating, or changes in schizophrenia symptoms. Because valbenazine can cause somnolence, patients should not perform activities requiring mental alertness (eg, operating a vehicle or hazardous machinery) until they know how they will be affected by valbenazine.
Valbenazine should be avoided in patients with congenital long QT syndrome or with arrhythmias associated with a prolonged QT interval. For patients at increased risk of a prolonged QT interval, assess the QT interval before increasing the dosage.
Clinical considerations
Unique properties. Valbenazine is metabolized slowly to a potent, selective VMAT2 antagonist (NBI-98782) in a manner that permits once daily dosing, removes the need for CYP2D6 genotyping, and provides significant efficacy.
Why Rx? The reasons to prescribe valbenazine for TD patients include:
- currently the only agent with FDA approval for TD
- fewer tolerability issues seen with the only other effective agent, tetrabenazine
- no signal for effects on mood parameters or rates of parkinsonism
- lack of multiple daily dosing and possible need for 2D6 genotyping involved with TBZ prescribing.
Dosing
The recommended dosage of valbenazine is 80 mg/d administered as a single dose with or without food, starting at 40 mg once daily for 1 week. There is no dosage adjustment required in those with mild to moderate renal impairment; however, valbenazine is not recommended in those with severe renal impairment. The maximum dose is 40 mg/d for those who with moderate or severe hepatic impairment (Child-Pugh score, 7 to 15) however, valbenazine is not recommended for patients with severe renal impairment (creatinine clearance <30 mL/min) because the exposure to the active metabolite is reduced by approximately 75%. The combined efficacy and tolerability of dosages >80 mg/d has not been evaluated. Adverse effects seen with tetrabenazine at higher dosages include akathisia, anxiety, insomnia, parkinsonism, fatigue, and depression.
A daily dose of 40 mg may be considered for some patients based on tolerability, including those who are known CYP 2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors.2 For those taking strong 3A4 inhibitors, the maximum daily dose is 40 mg. Concomitant use of valbenazine with strong 3A4 inducers is not recommended as the exposure to the active metabolite is reduced by approximately 75%.2 Lastly, because VMAT2 inhibition may alter synaptic levels of other monoamines, it is recommended that valbenazine not be administered with monoamine oxidase inhibitors, such as isocarboxazid, phenelzine, or selegiline.
Contraindications
There are no reported contraindications for valbenazine. As with most medications, there is limited available data on valbenazine use in pregnant women; however, administration of valbenazine to pregnant rats during organogenesis through lactation produced an increase in the number of stillborn pups and postnatal pup mortalities at doses under the maximum recommended human dose (MRHD) using body surface area based dosing (mg/m2). Pregnant women should be advised of the potential risk to a fetus. Valbenazine and its metabolites have been detected in rat milk at concentrations higher than in plasma after oral administration of valbenazine at doses 0.1 to 1.2 times the MRHD (based on mg/m2). Based on animal findings of increased perinatal mortality in exposed fetuses and pups, woman are advised not to breastfeed during valbenazine treatment and for 5 days after the final dose. No dosage adjustment is required for geriatric patients.
1. O’Brien CF, Jimenez R, Hauser RA, et al. NBI-98854, a selective monoamine transport inhibitor for the treatment of tardive dyskinesia: a randomized, double-blind, placebo-controlled study. Mov Disord. 2015;30(12):1681-1687.
2. Ingrezza [package insert]. San Diego, CA: Neurocrine Biosciences Inc.; 2017.
3. Marder S, Knesevich MA, Hauser RA, et al. KINECT 3: A randomized, double-blind, placebo-controlled phase 3 trial of valbenazine (NBI-98854) for tardive dyskinesia. Poster presented at the American Psychiatric Association Annual Meeting; May 14-18, 2016; Atlanta, GA.
4. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
5. Richardson MA, Bevans ML, Read LL, et al. Efficacy of the branched-chain amino acids in the treatment of tardive dyskinesia in men. Am J Psychiatry. 2003;160(6):1117-1124.
6. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
7. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
10. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
11. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
12. Grigoriadis DE, Smith E, Madan A, et al. Pharmacologic characteristics of valbenazine (NBI-98854) and its metabolites. Poster presented at the U.S. Psychiatric & Mental Health Congress, October 21-24, 2016; San Antonio, TX.
1. O’Brien CF, Jimenez R, Hauser RA, et al. NBI-98854, a selective monoamine transport inhibitor for the treatment of tardive dyskinesia: a randomized, double-blind, placebo-controlled study. Mov Disord. 2015;30(12):1681-1687.
2. Ingrezza [package insert]. San Diego, CA: Neurocrine Biosciences Inc.; 2017.
3. Marder S, Knesevich MA, Hauser RA, et al. KINECT 3: A randomized, double-blind, placebo-controlled phase 3 trial of valbenazine (NBI-98854) for tardive dyskinesia. Poster presented at the American Psychiatric Association Annual Meeting; May 14-18, 2016; Atlanta, GA.
4. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
5. Richardson MA, Bevans ML, Read LL, et al. Efficacy of the branched-chain amino acids in the treatment of tardive dyskinesia in men. Am J Psychiatry. 2003;160(6):1117-1124.
6. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
7. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
10. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
11. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
12. Grigoriadis DE, Smith E, Madan A, et al. Pharmacologic characteristics of valbenazine (NBI-98854) and its metabolites. Poster presented at the U.S. Psychiatric & Mental Health Congress, October 21-24, 2016; San Antonio, TX.
Pimavanserin for psychosis in patients with Parkinson’s disease
Pimavanserin is a potent 5-HT2A inverse agonist and 5-HT2C inverse agonist, with 5-fold greater affinity for the 5-HT2A receptor.1 Although antagonists block agonist actions at the receptor site, inverse agonists reduce the level of baseline constitutive activity seen in many G protein-coupled receptors. This medication is FDA approved for treating hallucinations and delusions associated with Parkinson’s disease (PD) psychosis (Table 1).1
In the pivotal 6-week clinical trial, pimavanserin significantly reduced positive symptoms seen in PD patients with psychosis (effect size = 0.50), with no evident impairment of motor function.2 Only 2 adverse effects occurred in ≥5% of pimavanserin-treated patients and at ≥2 times the rate of placebo: peripheral edema (7% vs 3% for placebo) and confusion (6% vs 3% for placebo). There was a mean increase in the QTc of 7.3 milliseconds compared with placebo in the pivotal phase III study.
Clinical implications
Despite numerous developments in the pharmacotherapeutics of psychotic disorders, patients with psychosis related to PD previously responded in a robust manner to only 1 antipsychotic, low-dosage clozapine (mean effect size, 0.80),2 with numerous failed trials for other atypical antipsychotics, including quetiapine.3,4 The pathophysiology of psychosis in PD patients is not related to dopamine agonist treatment, but is caused by the accumulation of cortical Lewy body burden, which results in loss of serotonergic signaling from dorsal raphe neurons. The net effect is up-regulation of postsynaptic 5-HT2A receptors.5 Psychosis is the most common cause of nursing home placement among PD patients without dementia.6
Receptor blocking. Based on the finding that clozapine in low dosages acts at 5-HT2A receptors,7 pimavanserin was designed to be a potent 5-HT2A inverse agonist, with more than 5-fold higher selectivity over 5-HT2C receptors, and no appreciable affinity for other serotonergic, adrenergic, dopaminergic, muscarinic, or histaminergic receptors8 (Table 2). The concept that 5-HT2A receptor stimulation can cause psychosis with prominent visual hallucinations is known from studies of LSD and other hallucinogenic compounds whose activity is blocked by 5-HT2A antagonists.

As an agent devoid of dopamine D2 antagonism, pimavanserin carries no risk of exacerbating motor symptoms, which was commonly seen with most atypical antipsychotics studied for psychosis in PD patients, except for clozapine and quetiapine.3 Although quetiapine did not cause motor effects, it proved ineffective in multiple studies (n = 153), likely because of the near absence of potent 5-HT2A binding.4
Pimavanserin also lacks:
- the hematologic monitoring requirement of clozapine
- clozapine’s risks of sedation, orthostasis, and anticholinergic and metabolic adverse effects.
Pimavanserin is significantly more potent than other non-antipsychotic psychotropics at the 5-HT2Areceptor, including doxepin (26 nM), trazodone (36 nM), and mirtazapine (60 nM).
Use in psychosis associated with PD. Recommended dosage is 34 mg once daily without titration (with or without food), based on results from a phase III clinical trial2 (because of the FDA breakthrough therapy designation for this compound, only 1 phase III trial was required). Pimavanserin produced significant improvement on the PD-adapted Scale for the Assessment of Positive Symptoms (SAPS-PD), a 9-item instrument extracted from the larger SAPS used in schizophrenia research. Specifically, pimavanserin was effective for both the hallucinations and delusions components of the SAPS-PD.
Pharmacologic profile, adverse effects. Pimavanserin lacks affinity for receptors other than 5-HT2A and 5-HT2C, leading to an absence of significant anticholinergic effects, orthostasis, or sedation in clinical trials.2 In all short-term clinical trials, the only common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were peripheral edema (7% vs 2% placebo) and confusional state (6% vs 3% placebo).2 More than 300 patients have been treated for >6 months, >270 have been treated for at least 12 months, and >150 have been treated for at least 24 months with no adverse effects other than those seen in the short-term trials.1
There is a measurable impact on cardiac conduction seen in phase III data and in the thorough QT study. In the thorough QT study, 252 healthy participants received multiple dosages in a randomized, double-blind manner with positive controls.1 The maximum mean change from baseline was 13.5 milliseconds at dosages twice the recommended dosage, and the upper limit of the 90% CI was only slightly greater at 16.6 milliseconds. Subsequent kinetic analyses suggested concentration-dependent QTc interval prolongation in the therapeutic range, with a recommendation to halve the daily dosage in patients taking potent cytochrome P450 (CYP) 3A4 inhibitors.
In the 6-week, placebo-controlled effectiveness studies, mean increases in QTc interval were in the range of 5 to 8 milliseconds. There were sporadic reports of QTcF values ≥500 milliseconds, or changes from baseline QTc values ≥60 milliseconds in pimavanserin-treated participants, although the incidence generally was the same for pimavanserin and placebo groups. There were no reports of torsades de pointes or any differences from placebo in the incidence of adverse reactions associated with delayed ventricular repolarization.
How it works
The theory behind development of pimavanserin rests in the finding that low-dosage clozapine (6.25 to 50 mg/d) was effective for PD patients with psychosis (effect size 0.80).8 Although clozapine has high affinity for multiple sites, including histamine H1 receptors (Ki = 1.13 nM), α-1A and a α-2C adrenergic receptors (Ki = 1.62 nM and 6 nM, respectively), 5-HT2A receptors (Ki = 5.35 nM), and muscarinic M1 receptors (Ki = 6 nM), the hypothesized primary mechanism of clozapine’s effectiveness for PD psychosis at low dosages focused on the 5-HT2Areceptor. This idea was based on the knowledge that hallucinogens such as mescaline, psilocybin, and LSD are 5-HT2A agonists.9 This hallucinogenic activity can be blocked with 5-HT2A antagonists. Because of pimavanserin’s binding profile, the compound was studied as a treatment for psychosis in PD patients.
Pharmacokinetics
Pimavanserin demonstrates dose-proportional pharmacokinetics after a single oral dose as much as 7.5 times the recommended dosage. The pharmacokinetics of pimavanserin were similar in study participants (mean age, 72.4) and healthy controls, and a high-fat meal had no impact on the maximum blood levels (Cmax) or total drug exposure (area under the curve [AUC]).
The mean plasma half-lives for pimavanserin and its metabolite N-desmethyl-pimavanserin (AC-279) are 57 hours and 200 hours, respectively. Although the metabolite appears active in in vitro assays, it does not cross the blood-brain barrier to any appreciable extent, therefore contributing little to the clinical effect. The median time to maximum concentration (Tmax) of pimavanserin is 6 hours with a range of 4 to 24 hours, while the median Tmax of the primary metabolite AC-279 is 6 hours. The bioavailability of pimavanserin in an oral tablet or solution essentially is identical.
Pimavanserin is primarily metabolized via CYP3A4 to AC-279, and strong CYP3A4 inhibitors (eg, ketoconazole, itraconazole, clarithromycin, indinavir) increase pimavanserin Cmax by 1.5-fold, and AUC by 3-fold. In patients taking strong CYP3A4 inhibitors, the dosage of pimavanserin should be reduced by 50% to 17 mg/d. Conversely, patients on CYP3A4 inducers (eg, rifampin, carbamazepine, phenytoin) should be monitored for lack of efficacy; consider a dosage increase as necessary. Neither pimavanserin nor its metabolite, AC-279, are inhibitors or inducers of major CYP enzymes or drug transporters.
Efficacy in PD psychosis
Study 1. This 6-week, fixed dosage, double-blind, placebo-controlled trial was performed in adult PD patients age ≥40 with PD psychosis.2 Participants had to have (1) a PD diagnosis for at least 1 year and (2) psychotic symptoms that developed after diagnosis. Psychotic symptoms had to be present for at least 1 month, occurring at least weekly in the month before screening, and severe enough to warrant antipsychotic treatment. Baseline Mini-Mental State Examination score had to be ≥21 out of 30, with no evidence of delirium. Patients with dementia preceding or concurrent with the PD diagnosis were excluded. Antipsychotic treatments were not permitted during the trial.
After a 2-week nonpharmacotherapeutic lead-in phase that included a brief, daily psychosocial intervention by a caregiver, 199 patients who still met severity criteria were randomly allocated in a 1:1 manner to pimavanserin (34 mg of active drug, reported in the paper as 40 mg of pimavanserin tartrate) or matched placebo. Based on kinetic modeling and earlier clinical data, lower dosages (ie, 17 mg) were not explored, because they achieved only 50% of the steady state plasma levels thought to be required for efficacy.
The primary outcome was assessed by central, independent raters using the PD-adapted SAPS-PD. The efficacy analysis included 95 pimavanserin-treated individuals and 90 taking placebo. Baseline SAPS-PD scores were 14.7 ± 5.55 in the placebo group, and 15.9 ± 6.12 in the pimavanserin arm. Participants had a mean age of 72.4 and 94% white ethnicity across both cohorts; 42% of the placebo group and 33% of the pimavanserin group were female. Antipsychotic exposure in the 21 days prior to study entry were reported in 17% (n = 15) and 19% (n = 18) of the placebo and pimavanserin groups, respectively, with the most common agent being quetiapine (13 of 15, placebo, 16 of 18, pimavanserin). Approximately one-third of all participants were taking a cholinesterase inhibitor throughout the study.
Efficacy outcome. Pimavanserin was associated with a 5.79-point decrease in SAPS-PD scores compared with 2.73-point decrease for placebo (difference −3.06, 95% CI −4.91 to −1.20; P = .001). The effect size for this difference (Cohen’s d) was 0.50. The significant effect of pimavanserin vs placebo also was seen in separate analyses of the SAPS-PD subscore for hallucinations and delusions (effect size 0.50), and individually for hallucinations (effect size 0.45) and delusions (effect size 0.33). Separation from placebo appeared after the second week of pimavanserin treatment, and continued through the end of the study. There is unpublished data showing efficacy through week 10, and longer term, uncontrolled data consistent with sustained response. An exploratory analysis of caregiver burden demonstrated an effect size of 0.50.
Tolerability
The discontinuation rate because of adverse events for pimavanserin and placebo-treated patients was 10 patients in the pimavanserin group (4 due to psychotic symptoms within 10 days of starting the study drug) compared with 2 in the placebo group. There was no evidence of motor worsening in either group, demonstrated by the score on part II of the Unified Parkinson’s Disease Rating Scale (UPDRS) that captures self-reported activities of daily living, or on UPDRS part III (motor examination). Pimavanserin has no contraindications.
Unique clinical issues
Binding properties. Pimavanserin possesses potent 5-HT2A inverse agonist properties required to manage psychosis in PD patients, but lacks clozapine’s affinities for α-1 adrenergic, muscarinic, or histaminergic receptors that contribute to clozapine’s poor tolerability. Moreover, pimavanserin has no appreciable affinity for dopaminergic receptors, and therefore does not induce motor adverse effects.
Clozapine aside, all available atypical antipsychotics have proved ineffective for psychosis in PD patients, and most caused significant motor worsening.3 Although quetiapine does not cause motor effects, it has been shown to be ineffective for psychosis in PD patients in multiple trials.4
The effect size for clozapine response is large (0.80) in PD patients with psychosis, but tolerability issues and administrative burdens regarding patient and prescriber registration and routine hematological monitoring pose significant clinical barriers. Clozapine also lacks an FDA indication for this purpose, which may pose a hurdle to its use in certain treatment settings.
Why Rx? The reasons to prescribe pimavanserin for PD patients with psychosis likely include:
- absence of tolerability issues seen with the only other effective agent, clozapine
- lack of motor effects
- lack of administrative and monitoring burden related to clozapine prescribing
- only agent with FDA approval for hallucinations and delusions in PD patients with psychosis.
Dosing
The recommended dosage of pimavanserin is 34 mg/d administered as a single dose with or without food. There is no need for titration, and none was performed in the pivotal clinical trial. Given the long half-life (57 hours), steady state is not achieved until day 12, therefore initiation with a lower dosage might prolong the time to efficacy. There is no dosage adjustment required in patients with mild or moderate renal impairment, but pimavanserin treatment is not recommended in patients with severe renal impairment. Pimavanserin has not been evaluated in patients with hepatic impairment (using Child-Pugh criteria), and is not recommended for these patients.
Other key aspects of dosing to keep in mind.
- Because pimavanserin is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a strong CYP3A4 inhibitor; the recommended dosage is 17 mg/d when administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of pimavanserin with CYP3A4 inducers, patients should be monitored for lack of efficacy during concomitant use with a CYP3A4 inducer, and consideration given to a dosage increase.
Use in pregnancy and lactation. There are no data on the use of pimavanserin in pregnant women, but no developmental effects were seen when the drug was administered orally at 10 or 12 times the maximum recommended human dosage to rats or rabbits during organogenesis. Pimavanserin was not teratogenic in pregnant rats and rabbits. There is no information regarding the presence of pimavanserin in human breast milk.
Geriatric patients. No dosage adjustment is required for older patients. The study population in the pivotal trial was mean age 72.4 years.
Summing up
Before development of pimavanserin, clozapine was the only effective treatment for psychosis in PD patients. Despite clozapine’s robust effects across several trials, patients often were given ineffective medications, such as quetiapine, because of the administrative and tolerability barriers posed by clozapine use. Because psychosis is the most common cause of nursing home placement in non-demented PD patients, an agent with demonstrated efficacy and without the adverse effect profile of clozapine or monitoring requirements represents an enormous advance in the treatment of psychosis in PD patients.
1. Nuplazid [package insert]. San Diego, CA: Acadia Pharmaceuticals Inc.; 2016.
2. Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. [Erratum in Lancet. 2014;384(9937):28]. Lancet. 2014;383(9916):533-540.
3. Borek LL, Friedman JH. Treating psychosis in movement disorder patients: a review. Expert Opin Pharmacother. 2014;15(11):1553-1564.
4. Desmarais P, Massoud F, Filion J, et al. Quetiapine for psychosis in Parkinson disease and neurodegenerative parkinsonian disorders: a systematic review. J Geriatr Psychiatry Neurol. 2016;29(4):227-236.
5. Ballanger B, Strafella AP, van Eimeren T, et al. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch Neurol. 2010;67(4):416-421.
6. Ravina B, Marder K, Fernandez HH, et al. Diagnostic criteria for psychosis in Parkinson’s disease: report of an NINDS, NIMH work group. Mov Disord. 2007;22(8):1061-1068.
7. Nordström AL, Farde L, Nyberg S, et al. D1, D2, and 5-HT2 receptor occupancy in relation to clozapine serum concentration: a PET study of schizophrenic patients. Am J Psychiatry. 1995;152(10):1444-1449.
8. Hacksell U, Burstein ES, McFarland K, et al. On the discovery and development of pimavanserin: a novel drug candidate for Parkinson’s psychosis. Neurochem Res. 2014;39(10):2008-2017.
9. Moreno JL, Holloway T, Albizu L, et al. Metabotropic glutamate mGlu2 receptor is necessary for the pharmacological and behavioral effects induced by hallucinogenic 5-HT2A receptor agonists. Neurosci Lett. 2011;493(3):76-79.
Pimavanserin is a potent 5-HT2A inverse agonist and 5-HT2C inverse agonist, with 5-fold greater affinity for the 5-HT2A receptor.1 Although antagonists block agonist actions at the receptor site, inverse agonists reduce the level of baseline constitutive activity seen in many G protein-coupled receptors. This medication is FDA approved for treating hallucinations and delusions associated with Parkinson’s disease (PD) psychosis (Table 1).1
In the pivotal 6-week clinical trial, pimavanserin significantly reduced positive symptoms seen in PD patients with psychosis (effect size = 0.50), with no evident impairment of motor function.2 Only 2 adverse effects occurred in ≥5% of pimavanserin-treated patients and at ≥2 times the rate of placebo: peripheral edema (7% vs 3% for placebo) and confusion (6% vs 3% for placebo). There was a mean increase in the QTc of 7.3 milliseconds compared with placebo in the pivotal phase III study.
Clinical implications
Despite numerous developments in the pharmacotherapeutics of psychotic disorders, patients with psychosis related to PD previously responded in a robust manner to only 1 antipsychotic, low-dosage clozapine (mean effect size, 0.80),2 with numerous failed trials for other atypical antipsychotics, including quetiapine.3,4 The pathophysiology of psychosis in PD patients is not related to dopamine agonist treatment, but is caused by the accumulation of cortical Lewy body burden, which results in loss of serotonergic signaling from dorsal raphe neurons. The net effect is up-regulation of postsynaptic 5-HT2A receptors.5 Psychosis is the most common cause of nursing home placement among PD patients without dementia.6
Receptor blocking. Based on the finding that clozapine in low dosages acts at 5-HT2A receptors,7 pimavanserin was designed to be a potent 5-HT2A inverse agonist, with more than 5-fold higher selectivity over 5-HT2C receptors, and no appreciable affinity for other serotonergic, adrenergic, dopaminergic, muscarinic, or histaminergic receptors8 (Table 2). The concept that 5-HT2A receptor stimulation can cause psychosis with prominent visual hallucinations is known from studies of LSD and other hallucinogenic compounds whose activity is blocked by 5-HT2A antagonists.
As an agent devoid of dopamine D2 antagonism, pimavanserin carries no risk of exacerbating motor symptoms, which was commonly seen with most atypical antipsychotics studied for psychosis in PD patients, except for clozapine and quetiapine.3 Although quetiapine did not cause motor effects, it proved ineffective in multiple studies (n = 153), likely because of the near absence of potent 5-HT2A binding.4
Pimavanserin also lacks:
- the hematologic monitoring requirement of clozapine
- clozapine’s risks of sedation, orthostasis, and anticholinergic and metabolic adverse effects.
Pimavanserin is significantly more potent than other non-antipsychotic psychotropics at the 5-HT2Areceptor, including doxepin (26 nM), trazodone (36 nM), and mirtazapine (60 nM).
Use in psychosis associated with PD. Recommended dosage is 34 mg once daily without titration (with or without food), based on results from a phase III clinical trial2 (because of the FDA breakthrough therapy designation for this compound, only 1 phase III trial was required). Pimavanserin produced significant improvement on the PD-adapted Scale for the Assessment of Positive Symptoms (SAPS-PD), a 9-item instrument extracted from the larger SAPS used in schizophrenia research. Specifically, pimavanserin was effective for both the hallucinations and delusions components of the SAPS-PD.
Pharmacologic profile, adverse effects. Pimavanserin lacks affinity for receptors other than 5-HT2A and 5-HT2C, leading to an absence of significant anticholinergic effects, orthostasis, or sedation in clinical trials.2 In all short-term clinical trials, the only common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were peripheral edema (7% vs 2% placebo) and confusional state (6% vs 3% placebo).2 More than 300 patients have been treated for >6 months, >270 have been treated for at least 12 months, and >150 have been treated for at least 24 months with no adverse effects other than those seen in the short-term trials.1
There is a measurable impact on cardiac conduction seen in phase III data and in the thorough QT study. In the thorough QT study, 252 healthy participants received multiple dosages in a randomized, double-blind manner with positive controls.1 The maximum mean change from baseline was 13.5 milliseconds at dosages twice the recommended dosage, and the upper limit of the 90% CI was only slightly greater at 16.6 milliseconds. Subsequent kinetic analyses suggested concentration-dependent QTc interval prolongation in the therapeutic range, with a recommendation to halve the daily dosage in patients taking potent cytochrome P450 (CYP) 3A4 inhibitors.
In the 6-week, placebo-controlled effectiveness studies, mean increases in QTc interval were in the range of 5 to 8 milliseconds. There were sporadic reports of QTcF values ≥500 milliseconds, or changes from baseline QTc values ≥60 milliseconds in pimavanserin-treated participants, although the incidence generally was the same for pimavanserin and placebo groups. There were no reports of torsades de pointes or any differences from placebo in the incidence of adverse reactions associated with delayed ventricular repolarization.
How it works
The theory behind development of pimavanserin rests in the finding that low-dosage clozapine (6.25 to 50 mg/d) was effective for PD patients with psychosis (effect size 0.80).8 Although clozapine has high affinity for multiple sites, including histamine H1 receptors (Ki = 1.13 nM), α-1A and a α-2C adrenergic receptors (Ki = 1.62 nM and 6 nM, respectively), 5-HT2A receptors (Ki = 5.35 nM), and muscarinic M1 receptors (Ki = 6 nM), the hypothesized primary mechanism of clozapine’s effectiveness for PD psychosis at low dosages focused on the 5-HT2Areceptor. This idea was based on the knowledge that hallucinogens such as mescaline, psilocybin, and LSD are 5-HT2A agonists.9 This hallucinogenic activity can be blocked with 5-HT2A antagonists. Because of pimavanserin’s binding profile, the compound was studied as a treatment for psychosis in PD patients.
Pharmacokinetics
Pimavanserin demonstrates dose-proportional pharmacokinetics after a single oral dose as much as 7.5 times the recommended dosage. The pharmacokinetics of pimavanserin were similar in study participants (mean age, 72.4) and healthy controls, and a high-fat meal had no impact on the maximum blood levels (Cmax) or total drug exposure (area under the curve [AUC]).
The mean plasma half-lives for pimavanserin and its metabolite N-desmethyl-pimavanserin (AC-279) are 57 hours and 200 hours, respectively. Although the metabolite appears active in in vitro assays, it does not cross the blood-brain barrier to any appreciable extent, therefore contributing little to the clinical effect. The median time to maximum concentration (Tmax) of pimavanserin is 6 hours with a range of 4 to 24 hours, while the median Tmax of the primary metabolite AC-279 is 6 hours. The bioavailability of pimavanserin in an oral tablet or solution essentially is identical.
Pimavanserin is primarily metabolized via CYP3A4 to AC-279, and strong CYP3A4 inhibitors (eg, ketoconazole, itraconazole, clarithromycin, indinavir) increase pimavanserin Cmax by 1.5-fold, and AUC by 3-fold. In patients taking strong CYP3A4 inhibitors, the dosage of pimavanserin should be reduced by 50% to 17 mg/d. Conversely, patients on CYP3A4 inducers (eg, rifampin, carbamazepine, phenytoin) should be monitored for lack of efficacy; consider a dosage increase as necessary. Neither pimavanserin nor its metabolite, AC-279, are inhibitors or inducers of major CYP enzymes or drug transporters.
Efficacy in PD psychosis
Study 1. This 6-week, fixed dosage, double-blind, placebo-controlled trial was performed in adult PD patients age ≥40 with PD psychosis.2 Participants had to have (1) a PD diagnosis for at least 1 year and (2) psychotic symptoms that developed after diagnosis. Psychotic symptoms had to be present for at least 1 month, occurring at least weekly in the month before screening, and severe enough to warrant antipsychotic treatment. Baseline Mini-Mental State Examination score had to be ≥21 out of 30, with no evidence of delirium. Patients with dementia preceding or concurrent with the PD diagnosis were excluded. Antipsychotic treatments were not permitted during the trial.
After a 2-week nonpharmacotherapeutic lead-in phase that included a brief, daily psychosocial intervention by a caregiver, 199 patients who still met severity criteria were randomly allocated in a 1:1 manner to pimavanserin (34 mg of active drug, reported in the paper as 40 mg of pimavanserin tartrate) or matched placebo. Based on kinetic modeling and earlier clinical data, lower dosages (ie, 17 mg) were not explored, because they achieved only 50% of the steady state plasma levels thought to be required for efficacy.
The primary outcome was assessed by central, independent raters using the PD-adapted SAPS-PD. The efficacy analysis included 95 pimavanserin-treated individuals and 90 taking placebo. Baseline SAPS-PD scores were 14.7 ± 5.55 in the placebo group, and 15.9 ± 6.12 in the pimavanserin arm. Participants had a mean age of 72.4 and 94% white ethnicity across both cohorts; 42% of the placebo group and 33% of the pimavanserin group were female. Antipsychotic exposure in the 21 days prior to study entry were reported in 17% (n = 15) and 19% (n = 18) of the placebo and pimavanserin groups, respectively, with the most common agent being quetiapine (13 of 15, placebo, 16 of 18, pimavanserin). Approximately one-third of all participants were taking a cholinesterase inhibitor throughout the study.
Efficacy outcome. Pimavanserin was associated with a 5.79-point decrease in SAPS-PD scores compared with 2.73-point decrease for placebo (difference −3.06, 95% CI −4.91 to −1.20; P = .001). The effect size for this difference (Cohen’s d) was 0.50. The significant effect of pimavanserin vs placebo also was seen in separate analyses of the SAPS-PD subscore for hallucinations and delusions (effect size 0.50), and individually for hallucinations (effect size 0.45) and delusions (effect size 0.33). Separation from placebo appeared after the second week of pimavanserin treatment, and continued through the end of the study. There is unpublished data showing efficacy through week 10, and longer term, uncontrolled data consistent with sustained response. An exploratory analysis of caregiver burden demonstrated an effect size of 0.50.
Tolerability
The discontinuation rate because of adverse events for pimavanserin and placebo-treated patients was 10 patients in the pimavanserin group (4 due to psychotic symptoms within 10 days of starting the study drug) compared with 2 in the placebo group. There was no evidence of motor worsening in either group, demonstrated by the score on part II of the Unified Parkinson’s Disease Rating Scale (UPDRS) that captures self-reported activities of daily living, or on UPDRS part III (motor examination). Pimavanserin has no contraindications.
Unique clinical issues
Binding properties. Pimavanserin possesses potent 5-HT2A inverse agonist properties required to manage psychosis in PD patients, but lacks clozapine’s affinities for α-1 adrenergic, muscarinic, or histaminergic receptors that contribute to clozapine’s poor tolerability. Moreover, pimavanserin has no appreciable affinity for dopaminergic receptors, and therefore does not induce motor adverse effects.
Clozapine aside, all available atypical antipsychotics have proved ineffective for psychosis in PD patients, and most caused significant motor worsening.3 Although quetiapine does not cause motor effects, it has been shown to be ineffective for psychosis in PD patients in multiple trials.4
The effect size for clozapine response is large (0.80) in PD patients with psychosis, but tolerability issues and administrative burdens regarding patient and prescriber registration and routine hematological monitoring pose significant clinical barriers. Clozapine also lacks an FDA indication for this purpose, which may pose a hurdle to its use in certain treatment settings.
Why Rx? The reasons to prescribe pimavanserin for PD patients with psychosis likely include:
- absence of tolerability issues seen with the only other effective agent, clozapine
- lack of motor effects
- lack of administrative and monitoring burden related to clozapine prescribing
- only agent with FDA approval for hallucinations and delusions in PD patients with psychosis.
Dosing
The recommended dosage of pimavanserin is 34 mg/d administered as a single dose with or without food. There is no need for titration, and none was performed in the pivotal clinical trial. Given the long half-life (57 hours), steady state is not achieved until day 12, therefore initiation with a lower dosage might prolong the time to efficacy. There is no dosage adjustment required in patients with mild or moderate renal impairment, but pimavanserin treatment is not recommended in patients with severe renal impairment. Pimavanserin has not been evaluated in patients with hepatic impairment (using Child-Pugh criteria), and is not recommended for these patients.
Other key aspects of dosing to keep in mind.
- Because pimavanserin is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a strong CYP3A4 inhibitor; the recommended dosage is 17 mg/d when administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of pimavanserin with CYP3A4 inducers, patients should be monitored for lack of efficacy during concomitant use with a CYP3A4 inducer, and consideration given to a dosage increase.
Use in pregnancy and lactation. There are no data on the use of pimavanserin in pregnant women, but no developmental effects were seen when the drug was administered orally at 10 or 12 times the maximum recommended human dosage to rats or rabbits during organogenesis. Pimavanserin was not teratogenic in pregnant rats and rabbits. There is no information regarding the presence of pimavanserin in human breast milk.
Geriatric patients. No dosage adjustment is required for older patients. The study population in the pivotal trial was mean age 72.4 years.
Summing up
Before development of pimavanserin, clozapine was the only effective treatment for psychosis in PD patients. Despite clozapine’s robust effects across several trials, patients often were given ineffective medications, such as quetiapine, because of the administrative and tolerability barriers posed by clozapine use. Because psychosis is the most common cause of nursing home placement in non-demented PD patients, an agent with demonstrated efficacy and without the adverse effect profile of clozapine or monitoring requirements represents an enormous advance in the treatment of psychosis in PD patients.
Pimavanserin is a potent 5-HT2A inverse agonist and 5-HT2C inverse agonist, with 5-fold greater affinity for the 5-HT2A receptor.1 Although antagonists block agonist actions at the receptor site, inverse agonists reduce the level of baseline constitutive activity seen in many G protein-coupled receptors. This medication is FDA approved for treating hallucinations and delusions associated with Parkinson’s disease (PD) psychosis (Table 1).1
In the pivotal 6-week clinical trial, pimavanserin significantly reduced positive symptoms seen in PD patients with psychosis (effect size = 0.50), with no evident impairment of motor function.2 Only 2 adverse effects occurred in ≥5% of pimavanserin-treated patients and at ≥2 times the rate of placebo: peripheral edema (7% vs 3% for placebo) and confusion (6% vs 3% for placebo). There was a mean increase in the QTc of 7.3 milliseconds compared with placebo in the pivotal phase III study.
Clinical implications
Despite numerous developments in the pharmacotherapeutics of psychotic disorders, patients with psychosis related to PD previously responded in a robust manner to only 1 antipsychotic, low-dosage clozapine (mean effect size, 0.80),2 with numerous failed trials for other atypical antipsychotics, including quetiapine.3,4 The pathophysiology of psychosis in PD patients is not related to dopamine agonist treatment, but is caused by the accumulation of cortical Lewy body burden, which results in loss of serotonergic signaling from dorsal raphe neurons. The net effect is up-regulation of postsynaptic 5-HT2A receptors.5 Psychosis is the most common cause of nursing home placement among PD patients without dementia.6
Receptor blocking. Based on the finding that clozapine in low dosages acts at 5-HT2A receptors,7 pimavanserin was designed to be a potent 5-HT2A inverse agonist, with more than 5-fold higher selectivity over 5-HT2C receptors, and no appreciable affinity for other serotonergic, adrenergic, dopaminergic, muscarinic, or histaminergic receptors8 (Table 2). The concept that 5-HT2A receptor stimulation can cause psychosis with prominent visual hallucinations is known from studies of LSD and other hallucinogenic compounds whose activity is blocked by 5-HT2A antagonists.
As an agent devoid of dopamine D2 antagonism, pimavanserin carries no risk of exacerbating motor symptoms, which was commonly seen with most atypical antipsychotics studied for psychosis in PD patients, except for clozapine and quetiapine.3 Although quetiapine did not cause motor effects, it proved ineffective in multiple studies (n = 153), likely because of the near absence of potent 5-HT2A binding.4
Pimavanserin also lacks:
- the hematologic monitoring requirement of clozapine
- clozapine’s risks of sedation, orthostasis, and anticholinergic and metabolic adverse effects.
Pimavanserin is significantly more potent than other non-antipsychotic psychotropics at the 5-HT2Areceptor, including doxepin (26 nM), trazodone (36 nM), and mirtazapine (60 nM).
Use in psychosis associated with PD. Recommended dosage is 34 mg once daily without titration (with or without food), based on results from a phase III clinical trial2 (because of the FDA breakthrough therapy designation for this compound, only 1 phase III trial was required). Pimavanserin produced significant improvement on the PD-adapted Scale for the Assessment of Positive Symptoms (SAPS-PD), a 9-item instrument extracted from the larger SAPS used in schizophrenia research. Specifically, pimavanserin was effective for both the hallucinations and delusions components of the SAPS-PD.
Pharmacologic profile, adverse effects. Pimavanserin lacks affinity for receptors other than 5-HT2A and 5-HT2C, leading to an absence of significant anticholinergic effects, orthostasis, or sedation in clinical trials.2 In all short-term clinical trials, the only common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were peripheral edema (7% vs 2% placebo) and confusional state (6% vs 3% placebo).2 More than 300 patients have been treated for >6 months, >270 have been treated for at least 12 months, and >150 have been treated for at least 24 months with no adverse effects other than those seen in the short-term trials.1
There is a measurable impact on cardiac conduction seen in phase III data and in the thorough QT study. In the thorough QT study, 252 healthy participants received multiple dosages in a randomized, double-blind manner with positive controls.1 The maximum mean change from baseline was 13.5 milliseconds at dosages twice the recommended dosage, and the upper limit of the 90% CI was only slightly greater at 16.6 milliseconds. Subsequent kinetic analyses suggested concentration-dependent QTc interval prolongation in the therapeutic range, with a recommendation to halve the daily dosage in patients taking potent cytochrome P450 (CYP) 3A4 inhibitors.
In the 6-week, placebo-controlled effectiveness studies, mean increases in QTc interval were in the range of 5 to 8 milliseconds. There were sporadic reports of QTcF values ≥500 milliseconds, or changes from baseline QTc values ≥60 milliseconds in pimavanserin-treated participants, although the incidence generally was the same for pimavanserin and placebo groups. There were no reports of torsades de pointes or any differences from placebo in the incidence of adverse reactions associated with delayed ventricular repolarization.
How it works
The theory behind development of pimavanserin rests in the finding that low-dosage clozapine (6.25 to 50 mg/d) was effective for PD patients with psychosis (effect size 0.80).8 Although clozapine has high affinity for multiple sites, including histamine H1 receptors (Ki = 1.13 nM), α-1A and a α-2C adrenergic receptors (Ki = 1.62 nM and 6 nM, respectively), 5-HT2A receptors (Ki = 5.35 nM), and muscarinic M1 receptors (Ki = 6 nM), the hypothesized primary mechanism of clozapine’s effectiveness for PD psychosis at low dosages focused on the 5-HT2Areceptor. This idea was based on the knowledge that hallucinogens such as mescaline, psilocybin, and LSD are 5-HT2A agonists.9 This hallucinogenic activity can be blocked with 5-HT2A antagonists. Because of pimavanserin’s binding profile, the compound was studied as a treatment for psychosis in PD patients.
Pharmacokinetics
Pimavanserin demonstrates dose-proportional pharmacokinetics after a single oral dose as much as 7.5 times the recommended dosage. The pharmacokinetics of pimavanserin were similar in study participants (mean age, 72.4) and healthy controls, and a high-fat meal had no impact on the maximum blood levels (Cmax) or total drug exposure (area under the curve [AUC]).
The mean plasma half-lives for pimavanserin and its metabolite N-desmethyl-pimavanserin (AC-279) are 57 hours and 200 hours, respectively. Although the metabolite appears active in in vitro assays, it does not cross the blood-brain barrier to any appreciable extent, therefore contributing little to the clinical effect. The median time to maximum concentration (Tmax) of pimavanserin is 6 hours with a range of 4 to 24 hours, while the median Tmax of the primary metabolite AC-279 is 6 hours. The bioavailability of pimavanserin in an oral tablet or solution essentially is identical.
Pimavanserin is primarily metabolized via CYP3A4 to AC-279, and strong CYP3A4 inhibitors (eg, ketoconazole, itraconazole, clarithromycin, indinavir) increase pimavanserin Cmax by 1.5-fold, and AUC by 3-fold. In patients taking strong CYP3A4 inhibitors, the dosage of pimavanserin should be reduced by 50% to 17 mg/d. Conversely, patients on CYP3A4 inducers (eg, rifampin, carbamazepine, phenytoin) should be monitored for lack of efficacy; consider a dosage increase as necessary. Neither pimavanserin nor its metabolite, AC-279, are inhibitors or inducers of major CYP enzymes or drug transporters.
Efficacy in PD psychosis
Study 1. This 6-week, fixed dosage, double-blind, placebo-controlled trial was performed in adult PD patients age ≥40 with PD psychosis.2 Participants had to have (1) a PD diagnosis for at least 1 year and (2) psychotic symptoms that developed after diagnosis. Psychotic symptoms had to be present for at least 1 month, occurring at least weekly in the month before screening, and severe enough to warrant antipsychotic treatment. Baseline Mini-Mental State Examination score had to be ≥21 out of 30, with no evidence of delirium. Patients with dementia preceding or concurrent with the PD diagnosis were excluded. Antipsychotic treatments were not permitted during the trial.
After a 2-week nonpharmacotherapeutic lead-in phase that included a brief, daily psychosocial intervention by a caregiver, 199 patients who still met severity criteria were randomly allocated in a 1:1 manner to pimavanserin (34 mg of active drug, reported in the paper as 40 mg of pimavanserin tartrate) or matched placebo. Based on kinetic modeling and earlier clinical data, lower dosages (ie, 17 mg) were not explored, because they achieved only 50% of the steady state plasma levels thought to be required for efficacy.
The primary outcome was assessed by central, independent raters using the PD-adapted SAPS-PD. The efficacy analysis included 95 pimavanserin-treated individuals and 90 taking placebo. Baseline SAPS-PD scores were 14.7 ± 5.55 in the placebo group, and 15.9 ± 6.12 in the pimavanserin arm. Participants had a mean age of 72.4 and 94% white ethnicity across both cohorts; 42% of the placebo group and 33% of the pimavanserin group were female. Antipsychotic exposure in the 21 days prior to study entry were reported in 17% (n = 15) and 19% (n = 18) of the placebo and pimavanserin groups, respectively, with the most common agent being quetiapine (13 of 15, placebo, 16 of 18, pimavanserin). Approximately one-third of all participants were taking a cholinesterase inhibitor throughout the study.
Efficacy outcome. Pimavanserin was associated with a 5.79-point decrease in SAPS-PD scores compared with 2.73-point decrease for placebo (difference −3.06, 95% CI −4.91 to −1.20; P = .001). The effect size for this difference (Cohen’s d) was 0.50. The significant effect of pimavanserin vs placebo also was seen in separate analyses of the SAPS-PD subscore for hallucinations and delusions (effect size 0.50), and individually for hallucinations (effect size 0.45) and delusions (effect size 0.33). Separation from placebo appeared after the second week of pimavanserin treatment, and continued through the end of the study. There is unpublished data showing efficacy through week 10, and longer term, uncontrolled data consistent with sustained response. An exploratory analysis of caregiver burden demonstrated an effect size of 0.50.
Tolerability
The discontinuation rate because of adverse events for pimavanserin and placebo-treated patients was 10 patients in the pimavanserin group (4 due to psychotic symptoms within 10 days of starting the study drug) compared with 2 in the placebo group. There was no evidence of motor worsening in either group, demonstrated by the score on part II of the Unified Parkinson’s Disease Rating Scale (UPDRS) that captures self-reported activities of daily living, or on UPDRS part III (motor examination). Pimavanserin has no contraindications.
Unique clinical issues
Binding properties. Pimavanserin possesses potent 5-HT2A inverse agonist properties required to manage psychosis in PD patients, but lacks clozapine’s affinities for α-1 adrenergic, muscarinic, or histaminergic receptors that contribute to clozapine’s poor tolerability. Moreover, pimavanserin has no appreciable affinity for dopaminergic receptors, and therefore does not induce motor adverse effects.
Clozapine aside, all available atypical antipsychotics have proved ineffective for psychosis in PD patients, and most caused significant motor worsening.3 Although quetiapine does not cause motor effects, it has been shown to be ineffective for psychosis in PD patients in multiple trials.4
The effect size for clozapine response is large (0.80) in PD patients with psychosis, but tolerability issues and administrative burdens regarding patient and prescriber registration and routine hematological monitoring pose significant clinical barriers. Clozapine also lacks an FDA indication for this purpose, which may pose a hurdle to its use in certain treatment settings.
Why Rx? The reasons to prescribe pimavanserin for PD patients with psychosis likely include:
- absence of tolerability issues seen with the only other effective agent, clozapine
- lack of motor effects
- lack of administrative and monitoring burden related to clozapine prescribing
- only agent with FDA approval for hallucinations and delusions in PD patients with psychosis.
Dosing
The recommended dosage of pimavanserin is 34 mg/d administered as a single dose with or without food. There is no need for titration, and none was performed in the pivotal clinical trial. Given the long half-life (57 hours), steady state is not achieved until day 12, therefore initiation with a lower dosage might prolong the time to efficacy. There is no dosage adjustment required in patients with mild or moderate renal impairment, but pimavanserin treatment is not recommended in patients with severe renal impairment. Pimavanserin has not been evaluated in patients with hepatic impairment (using Child-Pugh criteria), and is not recommended for these patients.
Other key aspects of dosing to keep in mind.
- Because pimavanserin is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a strong CYP3A4 inhibitor; the recommended dosage is 17 mg/d when administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of pimavanserin with CYP3A4 inducers, patients should be monitored for lack of efficacy during concomitant use with a CYP3A4 inducer, and consideration given to a dosage increase.
Use in pregnancy and lactation. There are no data on the use of pimavanserin in pregnant women, but no developmental effects were seen when the drug was administered orally at 10 or 12 times the maximum recommended human dosage to rats or rabbits during organogenesis. Pimavanserin was not teratogenic in pregnant rats and rabbits. There is no information regarding the presence of pimavanserin in human breast milk.
Geriatric patients. No dosage adjustment is required for older patients. The study population in the pivotal trial was mean age 72.4 years.
Summing up
Before development of pimavanserin, clozapine was the only effective treatment for psychosis in PD patients. Despite clozapine’s robust effects across several trials, patients often were given ineffective medications, such as quetiapine, because of the administrative and tolerability barriers posed by clozapine use. Because psychosis is the most common cause of nursing home placement in non-demented PD patients, an agent with demonstrated efficacy and without the adverse effect profile of clozapine or monitoring requirements represents an enormous advance in the treatment of psychosis in PD patients.
1. Nuplazid [package insert]. San Diego, CA: Acadia Pharmaceuticals Inc.; 2016.
2. Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. [Erratum in Lancet. 2014;384(9937):28]. Lancet. 2014;383(9916):533-540.
3. Borek LL, Friedman JH. Treating psychosis in movement disorder patients: a review. Expert Opin Pharmacother. 2014;15(11):1553-1564.
4. Desmarais P, Massoud F, Filion J, et al. Quetiapine for psychosis in Parkinson disease and neurodegenerative parkinsonian disorders: a systematic review. J Geriatr Psychiatry Neurol. 2016;29(4):227-236.
5. Ballanger B, Strafella AP, van Eimeren T, et al. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch Neurol. 2010;67(4):416-421.
6. Ravina B, Marder K, Fernandez HH, et al. Diagnostic criteria for psychosis in Parkinson’s disease: report of an NINDS, NIMH work group. Mov Disord. 2007;22(8):1061-1068.
7. Nordström AL, Farde L, Nyberg S, et al. D1, D2, and 5-HT2 receptor occupancy in relation to clozapine serum concentration: a PET study of schizophrenic patients. Am J Psychiatry. 1995;152(10):1444-1449.
8. Hacksell U, Burstein ES, McFarland K, et al. On the discovery and development of pimavanserin: a novel drug candidate for Parkinson’s psychosis. Neurochem Res. 2014;39(10):2008-2017.
9. Moreno JL, Holloway T, Albizu L, et al. Metabotropic glutamate mGlu2 receptor is necessary for the pharmacological and behavioral effects induced by hallucinogenic 5-HT2A receptor agonists. Neurosci Lett. 2011;493(3):76-79.
1. Nuplazid [package insert]. San Diego, CA: Acadia Pharmaceuticals Inc.; 2016.
2. Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. [Erratum in Lancet. 2014;384(9937):28]. Lancet. 2014;383(9916):533-540.
3. Borek LL, Friedman JH. Treating psychosis in movement disorder patients: a review. Expert Opin Pharmacother. 2014;15(11):1553-1564.
4. Desmarais P, Massoud F, Filion J, et al. Quetiapine for psychosis in Parkinson disease and neurodegenerative parkinsonian disorders: a systematic review. J Geriatr Psychiatry Neurol. 2016;29(4):227-236.
5. Ballanger B, Strafella AP, van Eimeren T, et al. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch Neurol. 2010;67(4):416-421.
6. Ravina B, Marder K, Fernandez HH, et al. Diagnostic criteria for psychosis in Parkinson’s disease: report of an NINDS, NIMH work group. Mov Disord. 2007;22(8):1061-1068.
7. Nordström AL, Farde L, Nyberg S, et al. D1, D2, and 5-HT2 receptor occupancy in relation to clozapine serum concentration: a PET study of schizophrenic patients. Am J Psychiatry. 1995;152(10):1444-1449.
8. Hacksell U, Burstein ES, McFarland K, et al. On the discovery and development of pimavanserin: a novel drug candidate for Parkinson’s psychosis. Neurochem Res. 2014;39(10):2008-2017.
9. Moreno JL, Holloway T, Albizu L, et al. Metabotropic glutamate mGlu2 receptor is necessary for the pharmacological and behavioral effects induced by hallucinogenic 5-HT2A receptor agonists. Neurosci Lett. 2011;493(3):76-79.
Long-acting injectable aripiprazole lauroxil for schizophrenia
Approximately 80% of patients with schizophrenia relapse within 5 years1 despite the availability and increased use of second-generation antipsychotics. Long-acting depot formulations are a proven, effective treatment option for patients with schizophrenia. In October 2015, another long-acting injectable antipsychotic, aripiprazole lauroxil, was FDA-approved for schizophrenia.2 Aripiprazole lauroxil is administered IM every 4 to 6 weeks in the deltoid or gluteal region and is available in multiple dosages (Table 1).
Mechanism of action
Aripiprazole lauroxil is a prodrug of aripiprazole. Prodrugs are chemical compounds that exert their pharmacological effects after they undergo a biologic transformation and transform into a more active metabolite.3 The development of prodrugs is an established method used to improve physio-chemical or pharmacokinetic properties of the pharmacologically active compound.
After IM injection, aripiprazole lauroxil is most likely converted by an enzyme-mediated hydrolysis to N-hydroxymethyl aripiprazole, which is then hydrolyzed to aripiprazole. Aripiprazole’s mechanism of action is mediated through a combination of partial agonist activity at D2 and 5-HT1A receptors and antagonistic activity at 5-HT2A receptors.2,4
Dosing and administration
If your patient has never taken aripiprazole, ensure that she (he) will tolerate the drug by initiating a trial of oral aripiprazole before beginning treatment with aripiprazole lauroxil; establishing tolerability might take as long as 2 weeks because of the half-life of aripiprazole.
Aripiprazole lauroxil can be started at 441 mg, 662 mg, or 882 mg administered monthly; these dosages correspond to 300 mg, 450 mg, and 600 mg of aripiprazole, or 10 mg/d, 15 mg/d, ≥20 mg/d of oral aripiprazole, respectively (Table 2).2 Aripiprazole lauroxil can be administered either in the deltoid muscle (441 mg only) or gluteal muscle (441 mg, 662 mg, or 882 mg).2,4,5 Treatment with the 441-mg, 662-mg, or 882-mg dosages can be given every 4 weeks but the 882-mg dosage can be given every 6 weeks and only in the gluteal muscle, which provides greater dosing flexibility compared with extended-release injectable aripiprazole.2,4,5

Supplementation with oral aripiprazole is required for 21 days before the first aripiprazole lauroxil injection.2,4 The next injection should not be given earlier than 14 days after the previous dose. When a dose is missed, follow the guidelines outlined in Table 3.2

After a single injection, aripiprazole starts to appear in the systemic circulation at Day 5 or Day 6 and continues to be released for another 36 days.2 Steady-state concentration will be reached after the fourth monthly injection. The termination half-life of aripiprazole lauroxil ranged from 29 to 35 days after each monthly injection.2
Packaging. Aripiprazole lauroxil is available as single-dose, pre-filled, color-coded syringes for IM injection at 441 mg (light blue), 662 mg (green), and 882 mg (burgundy); syringes do not require refrigeration (Table 2).2 The syringe needs to be tapped at least 10 times to dislodge any material that might have settled. Shake the syringe vigorously for at least 30 seconds to ensure a uniform suspension. Shake it again for 30 seconds if the syringe is not used within 15 minutes.2
Efficacy
The efficacy of aripiprazole lauroxil for treating patients with schizophrenia has been established, in part, on the basis of efficacy data from clinical trials of oral aripiprazole. In addition, efficacy has been established in a 12-week, multicenter, randomized, placebo-controlled, double-blind, fixed-dose study of 622 individuals age 18 to 70 with schizophrenia.4,5 All eligible patients were diagnosed with schizophrenia as defined by DSM-IV-TR criteria and confirmed by the Structured Clinical Interview for DSM-IV Disorders, Clinical Trial Version and were experiencing an acute exacerbation of their illness at the time of the study. To be eligible for the study, participants had to have a Positive and Negative Syndrome Scale (PANSS) total score of 70 to 120 and score of ≥4 for ≥2 of the selected positive items (delusions, conceptual disorganization, hallucinatory behavior, and suspiciousness/persecution). Individuals also were required to have a Clinical Global Impression-Severity scale score of ≥4. Efficacy was assessed using the PANSS and Clinical Global Impression–Improvement scale (CGI-I).
Patients were randomized in a 1:1:1 ratio to receive IM aripiprazole lauroxil, 441 mg, aripiprazole lauroxil, 882 mg, or placebo once monthly in the gluteal region for 12 weeks. The gluteal muscle was selected as the injection site to maintain blinding to the study drug.4,5 After establishing tolerability to oral aripiprazole, participants received oral aripiprazole or placebo daily for the first 3 weeks. The IM injections were administered on Days 1, 29, and 57.
Efficacy was measured primarily as change in total PANSS score from the baseline to day 854,5; secondary efficacy variable was the CGI-I score at day 85. Statistically significant separation in PANSS score was observed in each aripiprazole lauroxil dosage group (441 mg and 882 mg) compared with placebo. Significant improvement in both active treatment groups was observed as early as Day 8 and continued throughout the study (P ≤ .004). The number of patients who improved much or very much on the CGI-I was significantly greater in either aripiprazole lauroxil group, compared with placebo (P < .001).
Contraindications
Allergic reactions. Patients who are hypersensitive to oral aripiprazole should not receive aripiprazole lauroxil. Hypersensitivity reactions have ranged from pruritus and urticaria to anaphylaxis.2
Drug−drug interactions. Reduce aripiprazole lauroxil dosage to the next lower dosage when used in combination with strong cytochrome P450 (CYP) 3A4 inhibitors (eg, itraconazole, clarithromycin) or strong CYP2D6 inhibitors (eg, quinidine, fluoxetine, paroxetine) for more than 2 weeks or if the patient is known to be a poor metabolizer of CYP2D6, because concentration of aripiprazole lauroxil could increase. No dose adjustment is required if the patient is already taking 441 mg/month or if CYP450 modulators are added for less than 2 weeks.2 Similarly, a dosage increase is recommended when aripiprazole lauroxil is used in combination with strong CYP3A4 inducers (eg, carbamazepine, rifampin).2
Overdose
No data are available on aripiprazole lauroxil overdose. However, there is one known case of oral aripiprazole overdose in a patient who ingested 1,260 mg of oral aripiprazole (42 times the maximum recommended daily dosage) but recovered completely.2 Common side effects reported in at least 5% of all overdose cases include vomiting, somnolence, and tremor. If an overdose occurs, call a poison control center immediately.
‘Black-box’ warning for patients with dementia
Aripiprazole lauroxil, similar to all other atypical antipsychotics, has a “black-box” warning stating that (1) it is not approved for treating dementia-related psychosis, and (2) it is associated with an increased risk of death with off-label use to treat behavioral problems in older adults with dementia-related psychosis.2 Meta-analysis of 17 placebo-controlled trials in patients taking an atypical antipsychotic (olanzapine, aripiprazole, risperidone, or quetiapine) revealed a risk of death in drug-treated patients 1.6 to 1.7 times that of placebo-treated patients.6
Adverse reactions
The overall safety profile of aripiprazole lauroxil is similar to that of oral aripiprazole. Most commonly observed adverse reaction during clinical trials of aripiprazole lauroxil was akathisia (incidence ≥5% and at least twice rate seen with placebo).2 Other common adverse reactions are shown in Table 4.2 Recently, the FDA issued a warning that compulsive or uncontrollable urges to gamble, binge eat, shop, and have sex have been reported with all formulations of aripiprazole.7 According to reports, these urges stopped when the drug was discontinued or the dosage reduced. Although rare, these impulse-control problems could result in harm if they are not recognized. See the full prescribing information for a complete set of adverse reactions.
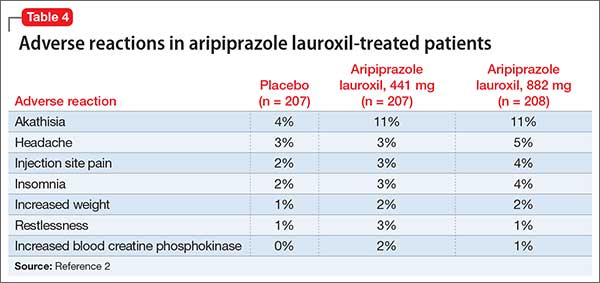
BOTTOM LINE
Aripiprazole lauroxil is a novel, long-acting second-generation antipsychotic that offers flexibility in terms of safe and effective dosing and can be administered in the deltoid (441 mg) or gluteal muscle (626 mg and 882 mg) and at dosing intervals of 4 to 6 weeks. Safety and tolerability profile of aripiprazole lauroxil are similar to that of oral aripiprazole. Aripiprazole lauroxil represents a new treatment option for patients with schizophrenia.
Related Resources
- Kennedy WK. When and how to use long-acting injectable antipsychotics. Current Psychiatry. 2012;11(8):40-43.
- Citrome L, Du Y, Risinger R, et al. Effect of aripiprazole lauroxil on agitation and hostility in patients with schizophrenia. Int Clin Psychopharmacol. 2016;31(2):69-75.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole extended-release • Abilify Maintena
Aripiprazole lauroxil • Aristada
Carbamazepine • Tegretol
Clarithromycin • Biaxin
Fluoxetine • Prozac
Itraconazole • Sporanox
Olanzapine • Zyprexa
Paroxetine • Paxil
Quetiapine • Seroquel
Quinidine • Quinidex
Rifampin • Rifadin
Risperidone • Risperdal
Acknowledgement
Maaz A. Khan, a student at the University of Oklahoma, Norman, Oklahoma, contributed to this article.
1. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry. 1999;56(3):241-247.
2. Aristada [package insert]. Waltham, MA; Alkermes; 2015.
3. Turncliff R, Hard M, Du Y, et al. Relative bioavailability and safety of aripiprazole lauroxil, a novel once-monthly, long-acting injectable atypical antipsychotic following deltoid and gluteal administration in adult subjects with schizophrenia. Schizophr Res. 2014;159(2-3):404-410.
4. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
5. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
6. U.S. Food and Drug Administration. Public health advisory: deaths with antipsychotics in elderly patients with behavioral disturbances. http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm053171. Published April 11, 2005. Accessed April 29, 2016.
7. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA warns about new impulse-control problems associated with mental health drug aripiprazole (Abilify, Abilify Maintena, Aristada). http://www.fda.gov/Drugs/DrugSafety/ucm498662.htm. Published May 3, 2016. Accessed June 20, 2016.
Approximately 80% of patients with schizophrenia relapse within 5 years1 despite the availability and increased use of second-generation antipsychotics. Long-acting depot formulations are a proven, effective treatment option for patients with schizophrenia. In October 2015, another long-acting injectable antipsychotic, aripiprazole lauroxil, was FDA-approved for schizophrenia.2 Aripiprazole lauroxil is administered IM every 4 to 6 weeks in the deltoid or gluteal region and is available in multiple dosages (Table 1).
Mechanism of action
Aripiprazole lauroxil is a prodrug of aripiprazole. Prodrugs are chemical compounds that exert their pharmacological effects after they undergo a biologic transformation and transform into a more active metabolite.3 The development of prodrugs is an established method used to improve physio-chemical or pharmacokinetic properties of the pharmacologically active compound.
After IM injection, aripiprazole lauroxil is most likely converted by an enzyme-mediated hydrolysis to N-hydroxymethyl aripiprazole, which is then hydrolyzed to aripiprazole. Aripiprazole’s mechanism of action is mediated through a combination of partial agonist activity at D2 and 5-HT1A receptors and antagonistic activity at 5-HT2A receptors.2,4
Dosing and administration
If your patient has never taken aripiprazole, ensure that she (he) will tolerate the drug by initiating a trial of oral aripiprazole before beginning treatment with aripiprazole lauroxil; establishing tolerability might take as long as 2 weeks because of the half-life of aripiprazole.
Aripiprazole lauroxil can be started at 441 mg, 662 mg, or 882 mg administered monthly; these dosages correspond to 300 mg, 450 mg, and 600 mg of aripiprazole, or 10 mg/d, 15 mg/d, ≥20 mg/d of oral aripiprazole, respectively (Table 2).2 Aripiprazole lauroxil can be administered either in the deltoid muscle (441 mg only) or gluteal muscle (441 mg, 662 mg, or 882 mg).2,4,5 Treatment with the 441-mg, 662-mg, or 882-mg dosages can be given every 4 weeks but the 882-mg dosage can be given every 6 weeks and only in the gluteal muscle, which provides greater dosing flexibility compared with extended-release injectable aripiprazole.2,4,5
Supplementation with oral aripiprazole is required for 21 days before the first aripiprazole lauroxil injection.2,4 The next injection should not be given earlier than 14 days after the previous dose. When a dose is missed, follow the guidelines outlined in Table 3.2
After a single injection, aripiprazole starts to appear in the systemic circulation at Day 5 or Day 6 and continues to be released for another 36 days.2 Steady-state concentration will be reached after the fourth monthly injection. The termination half-life of aripiprazole lauroxil ranged from 29 to 35 days after each monthly injection.2
Packaging. Aripiprazole lauroxil is available as single-dose, pre-filled, color-coded syringes for IM injection at 441 mg (light blue), 662 mg (green), and 882 mg (burgundy); syringes do not require refrigeration (Table 2).2 The syringe needs to be tapped at least 10 times to dislodge any material that might have settled. Shake the syringe vigorously for at least 30 seconds to ensure a uniform suspension. Shake it again for 30 seconds if the syringe is not used within 15 minutes.2
Efficacy
The efficacy of aripiprazole lauroxil for treating patients with schizophrenia has been established, in part, on the basis of efficacy data from clinical trials of oral aripiprazole. In addition, efficacy has been established in a 12-week, multicenter, randomized, placebo-controlled, double-blind, fixed-dose study of 622 individuals age 18 to 70 with schizophrenia.4,5 All eligible patients were diagnosed with schizophrenia as defined by DSM-IV-TR criteria and confirmed by the Structured Clinical Interview for DSM-IV Disorders, Clinical Trial Version and were experiencing an acute exacerbation of their illness at the time of the study. To be eligible for the study, participants had to have a Positive and Negative Syndrome Scale (PANSS) total score of 70 to 120 and score of ≥4 for ≥2 of the selected positive items (delusions, conceptual disorganization, hallucinatory behavior, and suspiciousness/persecution). Individuals also were required to have a Clinical Global Impression-Severity scale score of ≥4. Efficacy was assessed using the PANSS and Clinical Global Impression–Improvement scale (CGI-I).
Patients were randomized in a 1:1:1 ratio to receive IM aripiprazole lauroxil, 441 mg, aripiprazole lauroxil, 882 mg, or placebo once monthly in the gluteal region for 12 weeks. The gluteal muscle was selected as the injection site to maintain blinding to the study drug.4,5 After establishing tolerability to oral aripiprazole, participants received oral aripiprazole or placebo daily for the first 3 weeks. The IM injections were administered on Days 1, 29, and 57.
Efficacy was measured primarily as change in total PANSS score from the baseline to day 854,5; secondary efficacy variable was the CGI-I score at day 85. Statistically significant separation in PANSS score was observed in each aripiprazole lauroxil dosage group (441 mg and 882 mg) compared with placebo. Significant improvement in both active treatment groups was observed as early as Day 8 and continued throughout the study (P ≤ .004). The number of patients who improved much or very much on the CGI-I was significantly greater in either aripiprazole lauroxil group, compared with placebo (P < .001).
Contraindications
Allergic reactions. Patients who are hypersensitive to oral aripiprazole should not receive aripiprazole lauroxil. Hypersensitivity reactions have ranged from pruritus and urticaria to anaphylaxis.2
Drug−drug interactions. Reduce aripiprazole lauroxil dosage to the next lower dosage when used in combination with strong cytochrome P450 (CYP) 3A4 inhibitors (eg, itraconazole, clarithromycin) or strong CYP2D6 inhibitors (eg, quinidine, fluoxetine, paroxetine) for more than 2 weeks or if the patient is known to be a poor metabolizer of CYP2D6, because concentration of aripiprazole lauroxil could increase. No dose adjustment is required if the patient is already taking 441 mg/month or if CYP450 modulators are added for less than 2 weeks.2 Similarly, a dosage increase is recommended when aripiprazole lauroxil is used in combination with strong CYP3A4 inducers (eg, carbamazepine, rifampin).2
Overdose
No data are available on aripiprazole lauroxil overdose. However, there is one known case of oral aripiprazole overdose in a patient who ingested 1,260 mg of oral aripiprazole (42 times the maximum recommended daily dosage) but recovered completely.2 Common side effects reported in at least 5% of all overdose cases include vomiting, somnolence, and tremor. If an overdose occurs, call a poison control center immediately.
‘Black-box’ warning for patients with dementia
Aripiprazole lauroxil, similar to all other atypical antipsychotics, has a “black-box” warning stating that (1) it is not approved for treating dementia-related psychosis, and (2) it is associated with an increased risk of death with off-label use to treat behavioral problems in older adults with dementia-related psychosis.2 Meta-analysis of 17 placebo-controlled trials in patients taking an atypical antipsychotic (olanzapine, aripiprazole, risperidone, or quetiapine) revealed a risk of death in drug-treated patients 1.6 to 1.7 times that of placebo-treated patients.6
Adverse reactions
The overall safety profile of aripiprazole lauroxil is similar to that of oral aripiprazole. Most commonly observed adverse reaction during clinical trials of aripiprazole lauroxil was akathisia (incidence ≥5% and at least twice rate seen with placebo).2 Other common adverse reactions are shown in Table 4.2 Recently, the FDA issued a warning that compulsive or uncontrollable urges to gamble, binge eat, shop, and have sex have been reported with all formulations of aripiprazole.7 According to reports, these urges stopped when the drug was discontinued or the dosage reduced. Although rare, these impulse-control problems could result in harm if they are not recognized. See the full prescribing information for a complete set of adverse reactions.
BOTTOM LINE
Aripiprazole lauroxil is a novel, long-acting second-generation antipsychotic that offers flexibility in terms of safe and effective dosing and can be administered in the deltoid (441 mg) or gluteal muscle (626 mg and 882 mg) and at dosing intervals of 4 to 6 weeks. Safety and tolerability profile of aripiprazole lauroxil are similar to that of oral aripiprazole. Aripiprazole lauroxil represents a new treatment option for patients with schizophrenia.
Related Resources
- Kennedy WK. When and how to use long-acting injectable antipsychotics. Current Psychiatry. 2012;11(8):40-43.
- Citrome L, Du Y, Risinger R, et al. Effect of aripiprazole lauroxil on agitation and hostility in patients with schizophrenia. Int Clin Psychopharmacol. 2016;31(2):69-75.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole extended-release • Abilify Maintena
Aripiprazole lauroxil • Aristada
Carbamazepine • Tegretol
Clarithromycin • Biaxin
Fluoxetine • Prozac
Itraconazole • Sporanox
Olanzapine • Zyprexa
Paroxetine • Paxil
Quetiapine • Seroquel
Quinidine • Quinidex
Rifampin • Rifadin
Risperidone • Risperdal
Acknowledgement
Maaz A. Khan, a student at the University of Oklahoma, Norman, Oklahoma, contributed to this article.
Approximately 80% of patients with schizophrenia relapse within 5 years1 despite the availability and increased use of second-generation antipsychotics. Long-acting depot formulations are a proven, effective treatment option for patients with schizophrenia. In October 2015, another long-acting injectable antipsychotic, aripiprazole lauroxil, was FDA-approved for schizophrenia.2 Aripiprazole lauroxil is administered IM every 4 to 6 weeks in the deltoid or gluteal region and is available in multiple dosages (Table 1).
Mechanism of action
Aripiprazole lauroxil is a prodrug of aripiprazole. Prodrugs are chemical compounds that exert their pharmacological effects after they undergo a biologic transformation and transform into a more active metabolite.3 The development of prodrugs is an established method used to improve physio-chemical or pharmacokinetic properties of the pharmacologically active compound.
After IM injection, aripiprazole lauroxil is most likely converted by an enzyme-mediated hydrolysis to N-hydroxymethyl aripiprazole, which is then hydrolyzed to aripiprazole. Aripiprazole’s mechanism of action is mediated through a combination of partial agonist activity at D2 and 5-HT1A receptors and antagonistic activity at 5-HT2A receptors.2,4
Dosing and administration
If your patient has never taken aripiprazole, ensure that she (he) will tolerate the drug by initiating a trial of oral aripiprazole before beginning treatment with aripiprazole lauroxil; establishing tolerability might take as long as 2 weeks because of the half-life of aripiprazole.
Aripiprazole lauroxil can be started at 441 mg, 662 mg, or 882 mg administered monthly; these dosages correspond to 300 mg, 450 mg, and 600 mg of aripiprazole, or 10 mg/d, 15 mg/d, ≥20 mg/d of oral aripiprazole, respectively (Table 2).2 Aripiprazole lauroxil can be administered either in the deltoid muscle (441 mg only) or gluteal muscle (441 mg, 662 mg, or 882 mg).2,4,5 Treatment with the 441-mg, 662-mg, or 882-mg dosages can be given every 4 weeks but the 882-mg dosage can be given every 6 weeks and only in the gluteal muscle, which provides greater dosing flexibility compared with extended-release injectable aripiprazole.2,4,5
Supplementation with oral aripiprazole is required for 21 days before the first aripiprazole lauroxil injection.2,4 The next injection should not be given earlier than 14 days after the previous dose. When a dose is missed, follow the guidelines outlined in Table 3.2
After a single injection, aripiprazole starts to appear in the systemic circulation at Day 5 or Day 6 and continues to be released for another 36 days.2 Steady-state concentration will be reached after the fourth monthly injection. The termination half-life of aripiprazole lauroxil ranged from 29 to 35 days after each monthly injection.2
Packaging. Aripiprazole lauroxil is available as single-dose, pre-filled, color-coded syringes for IM injection at 441 mg (light blue), 662 mg (green), and 882 mg (burgundy); syringes do not require refrigeration (Table 2).2 The syringe needs to be tapped at least 10 times to dislodge any material that might have settled. Shake the syringe vigorously for at least 30 seconds to ensure a uniform suspension. Shake it again for 30 seconds if the syringe is not used within 15 minutes.2
Efficacy
The efficacy of aripiprazole lauroxil for treating patients with schizophrenia has been established, in part, on the basis of efficacy data from clinical trials of oral aripiprazole. In addition, efficacy has been established in a 12-week, multicenter, randomized, placebo-controlled, double-blind, fixed-dose study of 622 individuals age 18 to 70 with schizophrenia.4,5 All eligible patients were diagnosed with schizophrenia as defined by DSM-IV-TR criteria and confirmed by the Structured Clinical Interview for DSM-IV Disorders, Clinical Trial Version and were experiencing an acute exacerbation of their illness at the time of the study. To be eligible for the study, participants had to have a Positive and Negative Syndrome Scale (PANSS) total score of 70 to 120 and score of ≥4 for ≥2 of the selected positive items (delusions, conceptual disorganization, hallucinatory behavior, and suspiciousness/persecution). Individuals also were required to have a Clinical Global Impression-Severity scale score of ≥4. Efficacy was assessed using the PANSS and Clinical Global Impression–Improvement scale (CGI-I).
Patients were randomized in a 1:1:1 ratio to receive IM aripiprazole lauroxil, 441 mg, aripiprazole lauroxil, 882 mg, or placebo once monthly in the gluteal region for 12 weeks. The gluteal muscle was selected as the injection site to maintain blinding to the study drug.4,5 After establishing tolerability to oral aripiprazole, participants received oral aripiprazole or placebo daily for the first 3 weeks. The IM injections were administered on Days 1, 29, and 57.
Efficacy was measured primarily as change in total PANSS score from the baseline to day 854,5; secondary efficacy variable was the CGI-I score at day 85. Statistically significant separation in PANSS score was observed in each aripiprazole lauroxil dosage group (441 mg and 882 mg) compared with placebo. Significant improvement in both active treatment groups was observed as early as Day 8 and continued throughout the study (P ≤ .004). The number of patients who improved much or very much on the CGI-I was significantly greater in either aripiprazole lauroxil group, compared with placebo (P < .001).
Contraindications
Allergic reactions. Patients who are hypersensitive to oral aripiprazole should not receive aripiprazole lauroxil. Hypersensitivity reactions have ranged from pruritus and urticaria to anaphylaxis.2
Drug−drug interactions. Reduce aripiprazole lauroxil dosage to the next lower dosage when used in combination with strong cytochrome P450 (CYP) 3A4 inhibitors (eg, itraconazole, clarithromycin) or strong CYP2D6 inhibitors (eg, quinidine, fluoxetine, paroxetine) for more than 2 weeks or if the patient is known to be a poor metabolizer of CYP2D6, because concentration of aripiprazole lauroxil could increase. No dose adjustment is required if the patient is already taking 441 mg/month or if CYP450 modulators are added for less than 2 weeks.2 Similarly, a dosage increase is recommended when aripiprazole lauroxil is used in combination with strong CYP3A4 inducers (eg, carbamazepine, rifampin).2
Overdose
No data are available on aripiprazole lauroxil overdose. However, there is one known case of oral aripiprazole overdose in a patient who ingested 1,260 mg of oral aripiprazole (42 times the maximum recommended daily dosage) but recovered completely.2 Common side effects reported in at least 5% of all overdose cases include vomiting, somnolence, and tremor. If an overdose occurs, call a poison control center immediately.
‘Black-box’ warning for patients with dementia
Aripiprazole lauroxil, similar to all other atypical antipsychotics, has a “black-box” warning stating that (1) it is not approved for treating dementia-related psychosis, and (2) it is associated with an increased risk of death with off-label use to treat behavioral problems in older adults with dementia-related psychosis.2 Meta-analysis of 17 placebo-controlled trials in patients taking an atypical antipsychotic (olanzapine, aripiprazole, risperidone, or quetiapine) revealed a risk of death in drug-treated patients 1.6 to 1.7 times that of placebo-treated patients.6
Adverse reactions
The overall safety profile of aripiprazole lauroxil is similar to that of oral aripiprazole. Most commonly observed adverse reaction during clinical trials of aripiprazole lauroxil was akathisia (incidence ≥5% and at least twice rate seen with placebo).2 Other common adverse reactions are shown in Table 4.2 Recently, the FDA issued a warning that compulsive or uncontrollable urges to gamble, binge eat, shop, and have sex have been reported with all formulations of aripiprazole.7 According to reports, these urges stopped when the drug was discontinued or the dosage reduced. Although rare, these impulse-control problems could result in harm if they are not recognized. See the full prescribing information for a complete set of adverse reactions.
BOTTOM LINE
Aripiprazole lauroxil is a novel, long-acting second-generation antipsychotic that offers flexibility in terms of safe and effective dosing and can be administered in the deltoid (441 mg) or gluteal muscle (626 mg and 882 mg) and at dosing intervals of 4 to 6 weeks. Safety and tolerability profile of aripiprazole lauroxil are similar to that of oral aripiprazole. Aripiprazole lauroxil represents a new treatment option for patients with schizophrenia.
Related Resources
- Kennedy WK. When and how to use long-acting injectable antipsychotics. Current Psychiatry. 2012;11(8):40-43.
- Citrome L, Du Y, Risinger R, et al. Effect of aripiprazole lauroxil on agitation and hostility in patients with schizophrenia. Int Clin Psychopharmacol. 2016;31(2):69-75.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole extended-release • Abilify Maintena
Aripiprazole lauroxil • Aristada
Carbamazepine • Tegretol
Clarithromycin • Biaxin
Fluoxetine • Prozac
Itraconazole • Sporanox
Olanzapine • Zyprexa
Paroxetine • Paxil
Quetiapine • Seroquel
Quinidine • Quinidex
Rifampin • Rifadin
Risperidone • Risperdal
Acknowledgement
Maaz A. Khan, a student at the University of Oklahoma, Norman, Oklahoma, contributed to this article.
1. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry. 1999;56(3):241-247.
2. Aristada [package insert]. Waltham, MA; Alkermes; 2015.
3. Turncliff R, Hard M, Du Y, et al. Relative bioavailability and safety of aripiprazole lauroxil, a novel once-monthly, long-acting injectable atypical antipsychotic following deltoid and gluteal administration in adult subjects with schizophrenia. Schizophr Res. 2014;159(2-3):404-410.
4. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
5. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
6. U.S. Food and Drug Administration. Public health advisory: deaths with antipsychotics in elderly patients with behavioral disturbances. http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm053171. Published April 11, 2005. Accessed April 29, 2016.
7. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA warns about new impulse-control problems associated with mental health drug aripiprazole (Abilify, Abilify Maintena, Aristada). http://www.fda.gov/Drugs/DrugSafety/ucm498662.htm. Published May 3, 2016. Accessed June 20, 2016.
1. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry. 1999;56(3):241-247.
2. Aristada [package insert]. Waltham, MA; Alkermes; 2015.
3. Turncliff R, Hard M, Du Y, et al. Relative bioavailability and safety of aripiprazole lauroxil, a novel once-monthly, long-acting injectable atypical antipsychotic following deltoid and gluteal administration in adult subjects with schizophrenia. Schizophr Res. 2014;159(2-3):404-410.
4. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
5. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
6. U.S. Food and Drug Administration. Public health advisory: deaths with antipsychotics in elderly patients with behavioral disturbances. http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm053171. Published April 11, 2005. Accessed April 29, 2016.
7. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA warns about new impulse-control problems associated with mental health drug aripiprazole (Abilify, Abilify Maintena, Aristada). http://www.fda.gov/Drugs/DrugSafety/ucm498662.htm. Published May 3, 2016. Accessed June 20, 2016.
Extended-release, orally disintegrating mixed amphetamine salts for ADHD: New formulation
An amphetamine-based, extended-release, orally disintegrating tablet for patients age ≥6 diagnosed with attention-deficit/hyperactivity disorder (ADHD) won FDA approval on January 28, 2016 (Table).1

Adzenys XR-ODT is the first extended-release, orally disintegrating tablet for ADHD, Neos Therapeutics, Inc. the drug’s manufacturer, said in a statement.2 The newly approved agent is bioequivalent to Adderall XR (the capsule form of extended-release mixed amphetamine salts), and patients taking Adderall XR can be switched to the new drug. Equivalent dosages of the 2 drugs are outlined on the prescribing information.1
“The novel features of an extended-release orally disintegrating tablet ... make Adzenys XR-ODT attractive for use in both children (6 and older) and adults,” Alice R. Mao, MD, Medical Director, Memorial Park Psychiatry, Houston, Texas, said in the statement.2
As a condition of the approval, Neos must annually report the status of 3 post-marketing studies of children diagnosed with ADHD taking Adzenys XR-ODT, according to the approval letter.2 One is a single-dose, open-label study of children ages 4 and 5; the second is a randomized, double-blind, placebo-controlled titration study of children ages 4 and 5; and the third is a 1-year, open-label safety study of patients ages 4 and 5.
For patients age 6 to 17, the starting dosage is 6.3 mg once daily in the morning; for adults, it is 12.5 mg once daily in the morning, according to the label.1 The medication will be available in 4 other dose strengths: 3.1 mg, 9.4 mg, 15.7 mg, and 18.8 mg.
The most common adverse reactions to the drug among pediatric patients include loss of appetite, insomnia, and abdominal pain. Among adult patients, adverse reactions include dry mouth, loss of appetite, and insomnia.
1. Adzenys XR-ODT [prescription packet]. Grand Prairie, TX: Neos Therapeutics, LP; 2016.
2. Neos Therapeutics announces FDA approval of Adzenys XR-ODT (amphetamine extended-release orally disintegrating tablet) for the treatment of ADHD in patients 6 years and older [news release]. Dallas, TX: Neos Therapeutics, Inc; January 27, 2016. http://investors.neostx.com/phoenix.zhtml?c=254075&p=RssLanding&cat=news&id=2132931. Accessed February 3, 2016.
An amphetamine-based, extended-release, orally disintegrating tablet for patients age ≥6 diagnosed with attention-deficit/hyperactivity disorder (ADHD) won FDA approval on January 28, 2016 (Table).1
Adzenys XR-ODT is the first extended-release, orally disintegrating tablet for ADHD, Neos Therapeutics, Inc. the drug’s manufacturer, said in a statement.2 The newly approved agent is bioequivalent to Adderall XR (the capsule form of extended-release mixed amphetamine salts), and patients taking Adderall XR can be switched to the new drug. Equivalent dosages of the 2 drugs are outlined on the prescribing information.1
“The novel features of an extended-release orally disintegrating tablet ... make Adzenys XR-ODT attractive for use in both children (6 and older) and adults,” Alice R. Mao, MD, Medical Director, Memorial Park Psychiatry, Houston, Texas, said in the statement.2
As a condition of the approval, Neos must annually report the status of 3 post-marketing studies of children diagnosed with ADHD taking Adzenys XR-ODT, according to the approval letter.2 One is a single-dose, open-label study of children ages 4 and 5; the second is a randomized, double-blind, placebo-controlled titration study of children ages 4 and 5; and the third is a 1-year, open-label safety study of patients ages 4 and 5.
For patients age 6 to 17, the starting dosage is 6.3 mg once daily in the morning; for adults, it is 12.5 mg once daily in the morning, according to the label.1 The medication will be available in 4 other dose strengths: 3.1 mg, 9.4 mg, 15.7 mg, and 18.8 mg.
The most common adverse reactions to the drug among pediatric patients include loss of appetite, insomnia, and abdominal pain. Among adult patients, adverse reactions include dry mouth, loss of appetite, and insomnia.
An amphetamine-based, extended-release, orally disintegrating tablet for patients age ≥6 diagnosed with attention-deficit/hyperactivity disorder (ADHD) won FDA approval on January 28, 2016 (Table).1
Adzenys XR-ODT is the first extended-release, orally disintegrating tablet for ADHD, Neos Therapeutics, Inc. the drug’s manufacturer, said in a statement.2 The newly approved agent is bioequivalent to Adderall XR (the capsule form of extended-release mixed amphetamine salts), and patients taking Adderall XR can be switched to the new drug. Equivalent dosages of the 2 drugs are outlined on the prescribing information.1
“The novel features of an extended-release orally disintegrating tablet ... make Adzenys XR-ODT attractive for use in both children (6 and older) and adults,” Alice R. Mao, MD, Medical Director, Memorial Park Psychiatry, Houston, Texas, said in the statement.2
As a condition of the approval, Neos must annually report the status of 3 post-marketing studies of children diagnosed with ADHD taking Adzenys XR-ODT, according to the approval letter.2 One is a single-dose, open-label study of children ages 4 and 5; the second is a randomized, double-blind, placebo-controlled titration study of children ages 4 and 5; and the third is a 1-year, open-label safety study of patients ages 4 and 5.
For patients age 6 to 17, the starting dosage is 6.3 mg once daily in the morning; for adults, it is 12.5 mg once daily in the morning, according to the label.1 The medication will be available in 4 other dose strengths: 3.1 mg, 9.4 mg, 15.7 mg, and 18.8 mg.
The most common adverse reactions to the drug among pediatric patients include loss of appetite, insomnia, and abdominal pain. Among adult patients, adverse reactions include dry mouth, loss of appetite, and insomnia.
1. Adzenys XR-ODT [prescription packet]. Grand Prairie, TX: Neos Therapeutics, LP; 2016.
2. Neos Therapeutics announces FDA approval of Adzenys XR-ODT (amphetamine extended-release orally disintegrating tablet) for the treatment of ADHD in patients 6 years and older [news release]. Dallas, TX: Neos Therapeutics, Inc; January 27, 2016. http://investors.neostx.com/phoenix.zhtml?c=254075&p=RssLanding&cat=news&id=2132931. Accessed February 3, 2016.
1. Adzenys XR-ODT [prescription packet]. Grand Prairie, TX: Neos Therapeutics, LP; 2016.
2. Neos Therapeutics announces FDA approval of Adzenys XR-ODT (amphetamine extended-release orally disintegrating tablet) for the treatment of ADHD in patients 6 years and older [news release]. Dallas, TX: Neos Therapeutics, Inc; January 27, 2016. http://investors.neostx.com/phoenix.zhtml?c=254075&p=RssLanding&cat=news&id=2132931. Accessed February 3, 2016.
Flibanserin for hypoactive sexual desire disorder in premenopausal women
Flibanserin, FDA-approved in August 2015, is the first medication approved to treat acquired, generalized hypoactive sexual desire disorder (HSDD) in premenopausal women (Table 1). In clinical trials,1-4 the drug has shown modest efficacy in improving symptoms of low sexual desire (number of satisfying sexual events [SSEs], sexual desire, and overall sexual function). Flibanserin is not indicated to enhance sexual performance, for HSDD in postmenopausal women, or in men.

Clinical implications
Flibanserin could help premenopausal women who have distressing low sexual desire, which must be acquired and generalized:
- “Acquired low sexual desire” means that a patient had an adequate sexual desire that decreased or ceased for an unknown reason.
- “Generalized low sexual desire” means that lack of sexual desire occurs all the time and in all situations, not only with a certain partner or in some situations.
Women taking flibanserin could experience gradually increased sexual desire, increase in SSEs, and decrease of sexual distress. Flibanserin is indicated for long-term use; however, it should be discontinued after 8 weeks if the patient does not report any improvement in symptoms.
The number needed to treat with flibanserin likely would be rather large, but it is not available because of complex outcome measures in clinical trials. Flibanserin was not approved at 2 previous FDA committee hearings—mainly because of safety issues but also because of concerns about efficacy. For example, during the 2013 FDA hearing, the results presented showed statistically significant, but numerically small, treatment differences at 24 weeks compared with placebo. In an FDA responder analysis of the Phase-III trials, after accounting for the placebo effect, approximately 8% to 13% women were at least “much improved” on at least 1 of the primary outcomes.5
Flibanserin is not indicated for women whose sexual desire is due to (1) coexisting medical or psychiatric condition, (2) effects of medication or substance abuse, or (3) a relationship problem. It is unknown whether supplemental treatment would help these patients; however, it seems reasonable that combining flibanserin with psychosocial treatment, such as sex therapy or individual therapy, could be beneficial because it may be difficult to disentangle sexual dysfunction and relationship issues—2 problems that often are interwoven.
How it works
Flibanserin is a serotonin 1A receptor agonist and serotonin 2A receptor antagonist. In vitro, flibanserin demonstrated high affinity for the following 5-HT receptors:
- agonist activity at 5-HT1A
- antagonist activity at 5-HT2A, mostly in the prefrontal cortex.
Flibanserin also has moderate antagonist activities at the 5-HT2B, 5-HT2C, and dopamine D4 receptors. Flibanserin presumably acts centrally in the CNS; it has been suggested that flibanserin could rebalance neural circuitry involved in processing sexual desire by reducing serotonin activity and enhancing dopamine and epinephrine activity. The exact mechanism of how flibanserin improves sexual desire in women is unknown.
Pharmacokinetics
Flibanserin has a mean termination half-life of approximately 11 hours. It is administered once a day (50 to 100 mg) at bedtime. Steady state in healthy women was achieved after 3 days. Based on clinical observations, onset of action seems to be gradual and reaches maximum efficacy in approximately 8 weeks. Patients should discontinue the drug if no improvement is reported after 8 weeks. Flibanserin is readily absorbed from the gastrointestinal tract; however, food slows its absorption. The drug is 98% protein (mostly albumin)-bound.
Flibanserin is primarily metabolized in the liver by cytochrome P450 (CYP) 3A4 and to a lesser extent by CYP2C19. Co-administration of moderate (diltiazem, erythromycin, fluconazole, fosamprenavir, verapamil) or strong (eg, ketoconazole, clarithromycin, nefazodone, ritonavir) CYP3A4 inhibitors increases the concentration of flibanserin. This could lead to severe hypotension and syncope; therefore, co-administering flibanserin with a strong CYP3A4 inhibitor is contraindicated. Grapefruit juice is a moderate inhibitor of CYP3A4, and in a study of 26 healthy females, 240 mL of grapefruit juice increased flibanserin concentration 1.4-fold. Flibanserin is excreted though urine and feces. Flibanserin should be taken once a day at bedtime because of sedation, somnolence, and possible syncope.
Efficacy
The efficacy of flibanserin for treating HSDD was established in three 24-week, randomized, double-blind, placebo-controlled studies (Table 2). The target population in these studies was premenopausal women (mean age 36, range 19 to 55) with acquired HSDD lasting at least 6 months (mean duration, approximately 5 years). The 3 studies included 1,187 women who received flibanserin, 100 mg at bedtime, and 1,188 women who received placebo. Participants were mostly white (88.6%), and included black (9.6%) and Asian (1.5%) women. The completion rates were 69% for flibanserin and 78% for placebo. Some of the trials included arms with a lower dosage of flibanserin (25 mg and 50 mg), which are not included in this analysis.
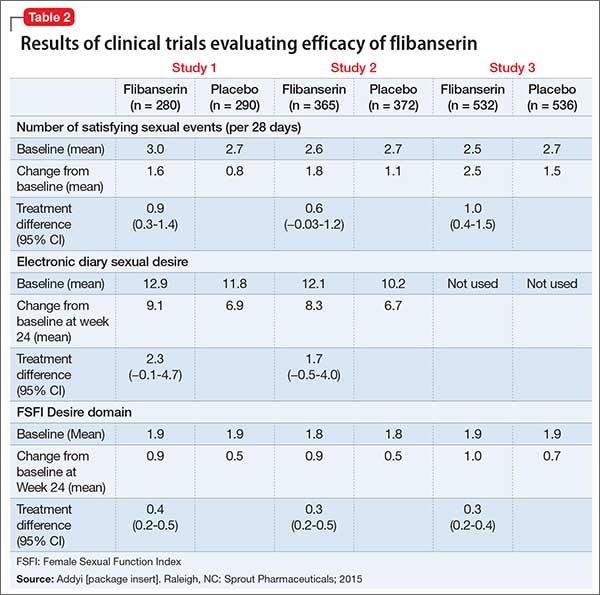
As noted in the package insert, these trials each had 2 co-primary efficacy endpoints, SSEs and sexual desire:
- change from baseline to Week 24 in the number of monthly SSEs (ie, sexual intercourse, oral sex, masturbation, or genital stimulation by the partner)
- change in sexual desire from baseline to 24-week endpoint.
In Study 1 and 2, change in sexual desire from baseline to Week 24 was measured daily by using an electronic diary. Every day, patients rated their sexual desire level by answering the question, “Indicate your most intense level of sexual desire” from 0 (no desire) to 3 (strong desire). These responses were totaled over a 28-day period to yield the monthly sexual desire score, which ranged from 0 to 84. These 2 studies also used the Female Sexual Function Index (FSFI) Desire domain as a secondary endpoint.
Study 3 used the FSFI Desire domain, comprising 2 questions, as the sexual desire co-primary endpoint:
- “Over the past 4 weeks, how often did you feel sexual desire or interest?” Responses ranged from 1 (almost never or never) to 5 (almost always or always).
- “Over the past 4 weeks, how would you rate your level (degree) of sexual desire or interest?” Responses ranged from 1 (very low or none at all) to 5 (very high).
In all 3 trials, flibanserin was associated with a small, yet statistically significant, improvement in change in monthly SSEs from baseline to Week 24 compared with placebo. In Study 1 and 2, there were no statistically significant differences between flibanserin and placebo for the electronic diary sexual desire endpoint. In the third study, there was statistically significant improvement in the change in sexual desire using the FSFI Desire domain with flibanserin compared with placebo. The FSFI Desire domain findings were consistent across all 3 trials. Flibanserin was associated with a decrease in sexual distress compared with placebo in all 3 studies.
Tolerability
Flibanserin was well tolerated in the 3 clinical trials. As the FDA noted, clinical trials are conducted under widely varying conditions and therefore adverse reaction rates observed in trials of flibanserin cannot be directly compared with those reported in clinical trials of another drug and might not reflect rates observed in clinical practice.
The discontinuation rate due to adverse reactions was 13% among patients treated with flibanserin, 100 mg at bedtime, and 6% among those taking placebo. The most common side effects were somnolence, dizziness, fatigue, nausea, insomnia, and dry mouth, which appear dose-dependent. Onset of most of these adverse events was within 14 days after the start of treatment.
Although hypotension and syncope rarely were seen with flibanserin alone in clinical trials, these adverse events occurred more frequently in the morning and when taken with alcohol and with some drugs (moderate or strong CYP3A4 inhibitors), and in patients with hepatic impairment. Therefore, women who drink alcohol or take a moderate or strong inhibitor of CYP3A4—both of which are contraindicated—and those with hepatic impairment should not take flibanserin.
Flibanserin should be taken at bedtime, because the risk of hypotension and syncope is higher when flibanserin is taken in the morning and because of associated sedation and somnolence.
Unique clinical issues
Flibanserin is the first FDA-approved medication for treating HSDD. It is important to note that the drug originally was developed as an antidepressant, but failed to show efficacy. Researchers noted that the drug was more effective than placebo when patients were asked, “How strong is your sexual desire?” The focus of development then shifted to a potential treatment of HSDD.
Flibanserin was not approved at 2 previous FDA hearings, mainly because of safety concerns. For the second hearing, the manufacturer, Boehringer Ingelheim, which sold the rights to the drug to Sprout Pharmaceuticals in 2011,6 did not present any new efficacy data, but provided additional safety data, such as research suggesting the absence of next-day driving impairment and data related to alcohol use (the study confirming hypotension associated with alcohol abuse used a small sample, and only 2 of 25 participants were women).
Contraindications
Flibanserin is contraindicated in patients using alcohol because of an increased risk of hypotension and syncope. A patient’s alcohol use should be evaluated before administering flibanserin, and patients should be counseled about the importance of abstaining from alcohol.
Similarly, concomitant use of flibanserin with a moderate or strong inhibitor of CYP3A4 increases the concentration of flibanserin and raises the risk of hypotension and syncope. Therefore, the use of a moderate or strong inhibitor of CYP3A4 in patients taking flibanserin is contraindicated. Similarly, patients with liver impairment should not take this drug.
Strong CYP2C19 inhibitors (proton-pump inhibitors, selective serotonin reuptake inhibitors, benzodiazepines, antifungals) could increase flibanserin exposure, which may increase risk of hypotension, syncope, and CNS depression. Discuss these risks with your patients; doing so is particularly important when treating women of Chinese heritage, and some other Asian women, because 20% of these populations are genotypic CYP2C19 poor metabolizers.
Because of the increased risk of hypotension and syncope with alcohol use, flibanserin is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the Addyi REMS Program. Flibanserin can be prescribed or dispensed only by physicians and pharmacists who watch this program’s online slide presentation and passed a comprehension test.a
Pregnant women should not take flibanserin because the effect on the fetus is unknown. Also, because the interaction with some oral contraceptives is unknown, patients should be cautioned about unwanted pregnancy. Women who are breastfeeding also should avoid using flibanserin because it is not known whether the drug is excreted in breast milk.
Women taking flibanserin also should avoid grapefruit juice, which increases flibanserin levels, and avoid using herbal products, resveratrol, and some over-the-counter drugs such as cimetidine. Women who have a depressive disorder also should avoid using flibanserin because their low sexual desire is more likely due to depression, which is not a therapeutic target for the drug.
Dosing
Flibanserin is provided in 100-mg film-coated tablets. It should be taken once a day at bedtime; titration is unnecessary. Length of treatment has not been determined, but it is recommended that patients stop flibanserin if they do not experience any benefit after 8 weeks. Although there is no guidance in the prescribing information, the medication probably could be stopped without tapering because withdrawal effects have not been observed.
Bottom Line
Flibanserin is FDA-approved for treating generalized, acquired hypoactive sexual desire disorder in premenopausal women. In clinical trials, the drug increased the number of satisfying sexual events and sexual desire, as measured by a diary and rating scales. Alcohol use and use of any moderate or strong inhibitor of cytochrome P450 3A4 are contraindicated in patients taking flibanserin because of an increased risk of hypotension and syncope.
1. Goldfisher ER, Breaux J, Katz M, et al. Continued efficacy and safety of flibanserin in premenopausal women with Hypoactive Sexual desire Disorder (HSDD): results from a randomized withdrawal trial. J Sex Med. 2011;8(11):3160- 3172.
2. Thorp J, Simon J, Dattani D, et al; DAISY trial investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the DAISY study. J Sex Med. 2012;9(3):793-804.
3. Derogatis LR, Komer L, Katz M, et al; VIOLET Trial Investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the VIOLET study. J Sex Med. 2012;9(4):1074-1085.
4. Katz M, DeRogatis LR, Ackerman R, et al; BEGONIA trial investigators. Efficacy of flibanserin in women with hypoactive sexual desire disorder: results from the BEGONIA trial. J Sex Med. 2013;10(7):1807-1815.
5. Gellad WF, Flynn KE, Alexander GC. Evaluation of flibanserin: science and advocacy at the FDA. JAMA. 2015;314(9):869-870
6. Joffe HV, Chang C, Sewell C, et al. FDA approval of flibanserin—treating hypoactive sexual desire disorder. N Engl J Med. 2016;374(2):101-104.
Flibanserin, FDA-approved in August 2015, is the first medication approved to treat acquired, generalized hypoactive sexual desire disorder (HSDD) in premenopausal women (Table 1). In clinical trials,1-4 the drug has shown modest efficacy in improving symptoms of low sexual desire (number of satisfying sexual events [SSEs], sexual desire, and overall sexual function). Flibanserin is not indicated to enhance sexual performance, for HSDD in postmenopausal women, or in men.
Clinical implications
Flibanserin could help premenopausal women who have distressing low sexual desire, which must be acquired and generalized:
- “Acquired low sexual desire” means that a patient had an adequate sexual desire that decreased or ceased for an unknown reason.
- “Generalized low sexual desire” means that lack of sexual desire occurs all the time and in all situations, not only with a certain partner or in some situations.
Women taking flibanserin could experience gradually increased sexual desire, increase in SSEs, and decrease of sexual distress. Flibanserin is indicated for long-term use; however, it should be discontinued after 8 weeks if the patient does not report any improvement in symptoms.
The number needed to treat with flibanserin likely would be rather large, but it is not available because of complex outcome measures in clinical trials. Flibanserin was not approved at 2 previous FDA committee hearings—mainly because of safety issues but also because of concerns about efficacy. For example, during the 2013 FDA hearing, the results presented showed statistically significant, but numerically small, treatment differences at 24 weeks compared with placebo. In an FDA responder analysis of the Phase-III trials, after accounting for the placebo effect, approximately 8% to 13% women were at least “much improved” on at least 1 of the primary outcomes.5
Flibanserin is not indicated for women whose sexual desire is due to (1) coexisting medical or psychiatric condition, (2) effects of medication or substance abuse, or (3) a relationship problem. It is unknown whether supplemental treatment would help these patients; however, it seems reasonable that combining flibanserin with psychosocial treatment, such as sex therapy or individual therapy, could be beneficial because it may be difficult to disentangle sexual dysfunction and relationship issues—2 problems that often are interwoven.
How it works
Flibanserin is a serotonin 1A receptor agonist and serotonin 2A receptor antagonist. In vitro, flibanserin demonstrated high affinity for the following 5-HT receptors:
- agonist activity at 5-HT1A
- antagonist activity at 5-HT2A, mostly in the prefrontal cortex.
Flibanserin also has moderate antagonist activities at the 5-HT2B, 5-HT2C, and dopamine D4 receptors. Flibanserin presumably acts centrally in the CNS; it has been suggested that flibanserin could rebalance neural circuitry involved in processing sexual desire by reducing serotonin activity and enhancing dopamine and epinephrine activity. The exact mechanism of how flibanserin improves sexual desire in women is unknown.
Pharmacokinetics
Flibanserin has a mean termination half-life of approximately 11 hours. It is administered once a day (50 to 100 mg) at bedtime. Steady state in healthy women was achieved after 3 days. Based on clinical observations, onset of action seems to be gradual and reaches maximum efficacy in approximately 8 weeks. Patients should discontinue the drug if no improvement is reported after 8 weeks. Flibanserin is readily absorbed from the gastrointestinal tract; however, food slows its absorption. The drug is 98% protein (mostly albumin)-bound.
Flibanserin is primarily metabolized in the liver by cytochrome P450 (CYP) 3A4 and to a lesser extent by CYP2C19. Co-administration of moderate (diltiazem, erythromycin, fluconazole, fosamprenavir, verapamil) or strong (eg, ketoconazole, clarithromycin, nefazodone, ritonavir) CYP3A4 inhibitors increases the concentration of flibanserin. This could lead to severe hypotension and syncope; therefore, co-administering flibanserin with a strong CYP3A4 inhibitor is contraindicated. Grapefruit juice is a moderate inhibitor of CYP3A4, and in a study of 26 healthy females, 240 mL of grapefruit juice increased flibanserin concentration 1.4-fold. Flibanserin is excreted though urine and feces. Flibanserin should be taken once a day at bedtime because of sedation, somnolence, and possible syncope.
Efficacy
The efficacy of flibanserin for treating HSDD was established in three 24-week, randomized, double-blind, placebo-controlled studies (Table 2). The target population in these studies was premenopausal women (mean age 36, range 19 to 55) with acquired HSDD lasting at least 6 months (mean duration, approximately 5 years). The 3 studies included 1,187 women who received flibanserin, 100 mg at bedtime, and 1,188 women who received placebo. Participants were mostly white (88.6%), and included black (9.6%) and Asian (1.5%) women. The completion rates were 69% for flibanserin and 78% for placebo. Some of the trials included arms with a lower dosage of flibanserin (25 mg and 50 mg), which are not included in this analysis.
As noted in the package insert, these trials each had 2 co-primary efficacy endpoints, SSEs and sexual desire:
- change from baseline to Week 24 in the number of monthly SSEs (ie, sexual intercourse, oral sex, masturbation, or genital stimulation by the partner)
- change in sexual desire from baseline to 24-week endpoint.
In Study 1 and 2, change in sexual desire from baseline to Week 24 was measured daily by using an electronic diary. Every day, patients rated their sexual desire level by answering the question, “Indicate your most intense level of sexual desire” from 0 (no desire) to 3 (strong desire). These responses were totaled over a 28-day period to yield the monthly sexual desire score, which ranged from 0 to 84. These 2 studies also used the Female Sexual Function Index (FSFI) Desire domain as a secondary endpoint.
Study 3 used the FSFI Desire domain, comprising 2 questions, as the sexual desire co-primary endpoint:
- “Over the past 4 weeks, how often did you feel sexual desire or interest?” Responses ranged from 1 (almost never or never) to 5 (almost always or always).
- “Over the past 4 weeks, how would you rate your level (degree) of sexual desire or interest?” Responses ranged from 1 (very low or none at all) to 5 (very high).
In all 3 trials, flibanserin was associated with a small, yet statistically significant, improvement in change in monthly SSEs from baseline to Week 24 compared with placebo. In Study 1 and 2, there were no statistically significant differences between flibanserin and placebo for the electronic diary sexual desire endpoint. In the third study, there was statistically significant improvement in the change in sexual desire using the FSFI Desire domain with flibanserin compared with placebo. The FSFI Desire domain findings were consistent across all 3 trials. Flibanserin was associated with a decrease in sexual distress compared with placebo in all 3 studies.
Tolerability
Flibanserin was well tolerated in the 3 clinical trials. As the FDA noted, clinical trials are conducted under widely varying conditions and therefore adverse reaction rates observed in trials of flibanserin cannot be directly compared with those reported in clinical trials of another drug and might not reflect rates observed in clinical practice.
The discontinuation rate due to adverse reactions was 13% among patients treated with flibanserin, 100 mg at bedtime, and 6% among those taking placebo. The most common side effects were somnolence, dizziness, fatigue, nausea, insomnia, and dry mouth, which appear dose-dependent. Onset of most of these adverse events was within 14 days after the start of treatment.
Although hypotension and syncope rarely were seen with flibanserin alone in clinical trials, these adverse events occurred more frequently in the morning and when taken with alcohol and with some drugs (moderate or strong CYP3A4 inhibitors), and in patients with hepatic impairment. Therefore, women who drink alcohol or take a moderate or strong inhibitor of CYP3A4—both of which are contraindicated—and those with hepatic impairment should not take flibanserin.
Flibanserin should be taken at bedtime, because the risk of hypotension and syncope is higher when flibanserin is taken in the morning and because of associated sedation and somnolence.
Unique clinical issues
Flibanserin is the first FDA-approved medication for treating HSDD. It is important to note that the drug originally was developed as an antidepressant, but failed to show efficacy. Researchers noted that the drug was more effective than placebo when patients were asked, “How strong is your sexual desire?” The focus of development then shifted to a potential treatment of HSDD.
Flibanserin was not approved at 2 previous FDA hearings, mainly because of safety concerns. For the second hearing, the manufacturer, Boehringer Ingelheim, which sold the rights to the drug to Sprout Pharmaceuticals in 2011,6 did not present any new efficacy data, but provided additional safety data, such as research suggesting the absence of next-day driving impairment and data related to alcohol use (the study confirming hypotension associated with alcohol abuse used a small sample, and only 2 of 25 participants were women).
Contraindications
Flibanserin is contraindicated in patients using alcohol because of an increased risk of hypotension and syncope. A patient’s alcohol use should be evaluated before administering flibanserin, and patients should be counseled about the importance of abstaining from alcohol.
Similarly, concomitant use of flibanserin with a moderate or strong inhibitor of CYP3A4 increases the concentration of flibanserin and raises the risk of hypotension and syncope. Therefore, the use of a moderate or strong inhibitor of CYP3A4 in patients taking flibanserin is contraindicated. Similarly, patients with liver impairment should not take this drug.
Strong CYP2C19 inhibitors (proton-pump inhibitors, selective serotonin reuptake inhibitors, benzodiazepines, antifungals) could increase flibanserin exposure, which may increase risk of hypotension, syncope, and CNS depression. Discuss these risks with your patients; doing so is particularly important when treating women of Chinese heritage, and some other Asian women, because 20% of these populations are genotypic CYP2C19 poor metabolizers.
Because of the increased risk of hypotension and syncope with alcohol use, flibanserin is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the Addyi REMS Program. Flibanserin can be prescribed or dispensed only by physicians and pharmacists who watch this program’s online slide presentation and passed a comprehension test.a
Pregnant women should not take flibanserin because the effect on the fetus is unknown. Also, because the interaction with some oral contraceptives is unknown, patients should be cautioned about unwanted pregnancy. Women who are breastfeeding also should avoid using flibanserin because it is not known whether the drug is excreted in breast milk.
Women taking flibanserin also should avoid grapefruit juice, which increases flibanserin levels, and avoid using herbal products, resveratrol, and some over-the-counter drugs such as cimetidine. Women who have a depressive disorder also should avoid using flibanserin because their low sexual desire is more likely due to depression, which is not a therapeutic target for the drug.
Dosing
Flibanserin is provided in 100-mg film-coated tablets. It should be taken once a day at bedtime; titration is unnecessary. Length of treatment has not been determined, but it is recommended that patients stop flibanserin if they do not experience any benefit after 8 weeks. Although there is no guidance in the prescribing information, the medication probably could be stopped without tapering because withdrawal effects have not been observed.
Bottom Line
Flibanserin is FDA-approved for treating generalized, acquired hypoactive sexual desire disorder in premenopausal women. In clinical trials, the drug increased the number of satisfying sexual events and sexual desire, as measured by a diary and rating scales. Alcohol use and use of any moderate or strong inhibitor of cytochrome P450 3A4 are contraindicated in patients taking flibanserin because of an increased risk of hypotension and syncope.
Flibanserin, FDA-approved in August 2015, is the first medication approved to treat acquired, generalized hypoactive sexual desire disorder (HSDD) in premenopausal women (Table 1). In clinical trials,1-4 the drug has shown modest efficacy in improving symptoms of low sexual desire (number of satisfying sexual events [SSEs], sexual desire, and overall sexual function). Flibanserin is not indicated to enhance sexual performance, for HSDD in postmenopausal women, or in men.
Clinical implications
Flibanserin could help premenopausal women who have distressing low sexual desire, which must be acquired and generalized:
- “Acquired low sexual desire” means that a patient had an adequate sexual desire that decreased or ceased for an unknown reason.
- “Generalized low sexual desire” means that lack of sexual desire occurs all the time and in all situations, not only with a certain partner or in some situations.
Women taking flibanserin could experience gradually increased sexual desire, increase in SSEs, and decrease of sexual distress. Flibanserin is indicated for long-term use; however, it should be discontinued after 8 weeks if the patient does not report any improvement in symptoms.
The number needed to treat with flibanserin likely would be rather large, but it is not available because of complex outcome measures in clinical trials. Flibanserin was not approved at 2 previous FDA committee hearings—mainly because of safety issues but also because of concerns about efficacy. For example, during the 2013 FDA hearing, the results presented showed statistically significant, but numerically small, treatment differences at 24 weeks compared with placebo. In an FDA responder analysis of the Phase-III trials, after accounting for the placebo effect, approximately 8% to 13% women were at least “much improved” on at least 1 of the primary outcomes.5
Flibanserin is not indicated for women whose sexual desire is due to (1) coexisting medical or psychiatric condition, (2) effects of medication or substance abuse, or (3) a relationship problem. It is unknown whether supplemental treatment would help these patients; however, it seems reasonable that combining flibanserin with psychosocial treatment, such as sex therapy or individual therapy, could be beneficial because it may be difficult to disentangle sexual dysfunction and relationship issues—2 problems that often are interwoven.
How it works
Flibanserin is a serotonin 1A receptor agonist and serotonin 2A receptor antagonist. In vitro, flibanserin demonstrated high affinity for the following 5-HT receptors:
- agonist activity at 5-HT1A
- antagonist activity at 5-HT2A, mostly in the prefrontal cortex.
Flibanserin also has moderate antagonist activities at the 5-HT2B, 5-HT2C, and dopamine D4 receptors. Flibanserin presumably acts centrally in the CNS; it has been suggested that flibanserin could rebalance neural circuitry involved in processing sexual desire by reducing serotonin activity and enhancing dopamine and epinephrine activity. The exact mechanism of how flibanserin improves sexual desire in women is unknown.
Pharmacokinetics
Flibanserin has a mean termination half-life of approximately 11 hours. It is administered once a day (50 to 100 mg) at bedtime. Steady state in healthy women was achieved after 3 days. Based on clinical observations, onset of action seems to be gradual and reaches maximum efficacy in approximately 8 weeks. Patients should discontinue the drug if no improvement is reported after 8 weeks. Flibanserin is readily absorbed from the gastrointestinal tract; however, food slows its absorption. The drug is 98% protein (mostly albumin)-bound.
Flibanserin is primarily metabolized in the liver by cytochrome P450 (CYP) 3A4 and to a lesser extent by CYP2C19. Co-administration of moderate (diltiazem, erythromycin, fluconazole, fosamprenavir, verapamil) or strong (eg, ketoconazole, clarithromycin, nefazodone, ritonavir) CYP3A4 inhibitors increases the concentration of flibanserin. This could lead to severe hypotension and syncope; therefore, co-administering flibanserin with a strong CYP3A4 inhibitor is contraindicated. Grapefruit juice is a moderate inhibitor of CYP3A4, and in a study of 26 healthy females, 240 mL of grapefruit juice increased flibanserin concentration 1.4-fold. Flibanserin is excreted though urine and feces. Flibanserin should be taken once a day at bedtime because of sedation, somnolence, and possible syncope.
Efficacy
The efficacy of flibanserin for treating HSDD was established in three 24-week, randomized, double-blind, placebo-controlled studies (Table 2). The target population in these studies was premenopausal women (mean age 36, range 19 to 55) with acquired HSDD lasting at least 6 months (mean duration, approximately 5 years). The 3 studies included 1,187 women who received flibanserin, 100 mg at bedtime, and 1,188 women who received placebo. Participants were mostly white (88.6%), and included black (9.6%) and Asian (1.5%) women. The completion rates were 69% for flibanserin and 78% for placebo. Some of the trials included arms with a lower dosage of flibanserin (25 mg and 50 mg), which are not included in this analysis.
As noted in the package insert, these trials each had 2 co-primary efficacy endpoints, SSEs and sexual desire:
- change from baseline to Week 24 in the number of monthly SSEs (ie, sexual intercourse, oral sex, masturbation, or genital stimulation by the partner)
- change in sexual desire from baseline to 24-week endpoint.
In Study 1 and 2, change in sexual desire from baseline to Week 24 was measured daily by using an electronic diary. Every day, patients rated their sexual desire level by answering the question, “Indicate your most intense level of sexual desire” from 0 (no desire) to 3 (strong desire). These responses were totaled over a 28-day period to yield the monthly sexual desire score, which ranged from 0 to 84. These 2 studies also used the Female Sexual Function Index (FSFI) Desire domain as a secondary endpoint.
Study 3 used the FSFI Desire domain, comprising 2 questions, as the sexual desire co-primary endpoint:
- “Over the past 4 weeks, how often did you feel sexual desire or interest?” Responses ranged from 1 (almost never or never) to 5 (almost always or always).
- “Over the past 4 weeks, how would you rate your level (degree) of sexual desire or interest?” Responses ranged from 1 (very low or none at all) to 5 (very high).
In all 3 trials, flibanserin was associated with a small, yet statistically significant, improvement in change in monthly SSEs from baseline to Week 24 compared with placebo. In Study 1 and 2, there were no statistically significant differences between flibanserin and placebo for the electronic diary sexual desire endpoint. In the third study, there was statistically significant improvement in the change in sexual desire using the FSFI Desire domain with flibanserin compared with placebo. The FSFI Desire domain findings were consistent across all 3 trials. Flibanserin was associated with a decrease in sexual distress compared with placebo in all 3 studies.
Tolerability
Flibanserin was well tolerated in the 3 clinical trials. As the FDA noted, clinical trials are conducted under widely varying conditions and therefore adverse reaction rates observed in trials of flibanserin cannot be directly compared with those reported in clinical trials of another drug and might not reflect rates observed in clinical practice.
The discontinuation rate due to adverse reactions was 13% among patients treated with flibanserin, 100 mg at bedtime, and 6% among those taking placebo. The most common side effects were somnolence, dizziness, fatigue, nausea, insomnia, and dry mouth, which appear dose-dependent. Onset of most of these adverse events was within 14 days after the start of treatment.
Although hypotension and syncope rarely were seen with flibanserin alone in clinical trials, these adverse events occurred more frequently in the morning and when taken with alcohol and with some drugs (moderate or strong CYP3A4 inhibitors), and in patients with hepatic impairment. Therefore, women who drink alcohol or take a moderate or strong inhibitor of CYP3A4—both of which are contraindicated—and those with hepatic impairment should not take flibanserin.
Flibanserin should be taken at bedtime, because the risk of hypotension and syncope is higher when flibanserin is taken in the morning and because of associated sedation and somnolence.
Unique clinical issues
Flibanserin is the first FDA-approved medication for treating HSDD. It is important to note that the drug originally was developed as an antidepressant, but failed to show efficacy. Researchers noted that the drug was more effective than placebo when patients were asked, “How strong is your sexual desire?” The focus of development then shifted to a potential treatment of HSDD.
Flibanserin was not approved at 2 previous FDA hearings, mainly because of safety concerns. For the second hearing, the manufacturer, Boehringer Ingelheim, which sold the rights to the drug to Sprout Pharmaceuticals in 2011,6 did not present any new efficacy data, but provided additional safety data, such as research suggesting the absence of next-day driving impairment and data related to alcohol use (the study confirming hypotension associated with alcohol abuse used a small sample, and only 2 of 25 participants were women).
Contraindications
Flibanserin is contraindicated in patients using alcohol because of an increased risk of hypotension and syncope. A patient’s alcohol use should be evaluated before administering flibanserin, and patients should be counseled about the importance of abstaining from alcohol.
Similarly, concomitant use of flibanserin with a moderate or strong inhibitor of CYP3A4 increases the concentration of flibanserin and raises the risk of hypotension and syncope. Therefore, the use of a moderate or strong inhibitor of CYP3A4 in patients taking flibanserin is contraindicated. Similarly, patients with liver impairment should not take this drug.
Strong CYP2C19 inhibitors (proton-pump inhibitors, selective serotonin reuptake inhibitors, benzodiazepines, antifungals) could increase flibanserin exposure, which may increase risk of hypotension, syncope, and CNS depression. Discuss these risks with your patients; doing so is particularly important when treating women of Chinese heritage, and some other Asian women, because 20% of these populations are genotypic CYP2C19 poor metabolizers.
Because of the increased risk of hypotension and syncope with alcohol use, flibanserin is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the Addyi REMS Program. Flibanserin can be prescribed or dispensed only by physicians and pharmacists who watch this program’s online slide presentation and passed a comprehension test.a
Pregnant women should not take flibanserin because the effect on the fetus is unknown. Also, because the interaction with some oral contraceptives is unknown, patients should be cautioned about unwanted pregnancy. Women who are breastfeeding also should avoid using flibanserin because it is not known whether the drug is excreted in breast milk.
Women taking flibanserin also should avoid grapefruit juice, which increases flibanserin levels, and avoid using herbal products, resveratrol, and some over-the-counter drugs such as cimetidine. Women who have a depressive disorder also should avoid using flibanserin because their low sexual desire is more likely due to depression, which is not a therapeutic target for the drug.
Dosing
Flibanserin is provided in 100-mg film-coated tablets. It should be taken once a day at bedtime; titration is unnecessary. Length of treatment has not been determined, but it is recommended that patients stop flibanserin if they do not experience any benefit after 8 weeks. Although there is no guidance in the prescribing information, the medication probably could be stopped without tapering because withdrawal effects have not been observed.
Bottom Line
Flibanserin is FDA-approved for treating generalized, acquired hypoactive sexual desire disorder in premenopausal women. In clinical trials, the drug increased the number of satisfying sexual events and sexual desire, as measured by a diary and rating scales. Alcohol use and use of any moderate or strong inhibitor of cytochrome P450 3A4 are contraindicated in patients taking flibanserin because of an increased risk of hypotension and syncope.
1. Goldfisher ER, Breaux J, Katz M, et al. Continued efficacy and safety of flibanserin in premenopausal women with Hypoactive Sexual desire Disorder (HSDD): results from a randomized withdrawal trial. J Sex Med. 2011;8(11):3160- 3172.
2. Thorp J, Simon J, Dattani D, et al; DAISY trial investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the DAISY study. J Sex Med. 2012;9(3):793-804.
3. Derogatis LR, Komer L, Katz M, et al; VIOLET Trial Investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the VIOLET study. J Sex Med. 2012;9(4):1074-1085.
4. Katz M, DeRogatis LR, Ackerman R, et al; BEGONIA trial investigators. Efficacy of flibanserin in women with hypoactive sexual desire disorder: results from the BEGONIA trial. J Sex Med. 2013;10(7):1807-1815.
5. Gellad WF, Flynn KE, Alexander GC. Evaluation of flibanserin: science and advocacy at the FDA. JAMA. 2015;314(9):869-870
6. Joffe HV, Chang C, Sewell C, et al. FDA approval of flibanserin—treating hypoactive sexual desire disorder. N Engl J Med. 2016;374(2):101-104.
1. Goldfisher ER, Breaux J, Katz M, et al. Continued efficacy and safety of flibanserin in premenopausal women with Hypoactive Sexual desire Disorder (HSDD): results from a randomized withdrawal trial. J Sex Med. 2011;8(11):3160- 3172.
2. Thorp J, Simon J, Dattani D, et al; DAISY trial investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the DAISY study. J Sex Med. 2012;9(3):793-804.
3. Derogatis LR, Komer L, Katz M, et al; VIOLET Trial Investigators. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the VIOLET study. J Sex Med. 2012;9(4):1074-1085.
4. Katz M, DeRogatis LR, Ackerman R, et al; BEGONIA trial investigators. Efficacy of flibanserin in women with hypoactive sexual desire disorder: results from the BEGONIA trial. J Sex Med. 2013;10(7):1807-1815.
5. Gellad WF, Flynn KE, Alexander GC. Evaluation of flibanserin: science and advocacy at the FDA. JAMA. 2015;314(9):869-870
6. Joffe HV, Chang C, Sewell C, et al. FDA approval of flibanserin—treating hypoactive sexual desire disorder. N Engl J Med. 2016;374(2):101-104.
Cariprazine for schizophrenia and bipolar I disorder
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
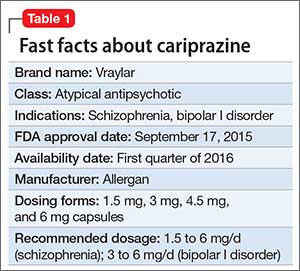
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
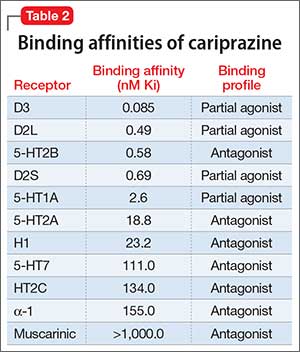
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
Cariprazine for schizophrenia and bipolar I disorder
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
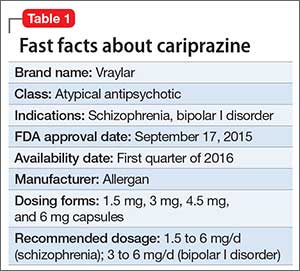
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
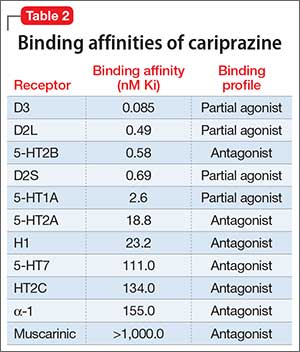
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
Brexpiprazole for schizophrenia and as adjunct for major depressive disorder
Brexpiprazole, FDA-approved in July 2015 to treat schizophrenia and as an adjunct for major depressive disorder (MDD) (Table 1), has shown efficacy in 2 phase-III acute trials for each indication.1-6 Although brexpiprazole is a dopamine D2 partial agonist, it differs from aripiprazole, the other available D2 partial agonist, because it is more potent at serotonin 5-HT1A and 5-HT2A receptors and displays less intrinsic activity at D2 receptors,7 which could mean better tolerability.
Clinical implications
Schizophrenia is heterogeneous, and individual response and tolerability to antipsychotics vary greatly8; therefore, new drug options are useful. For MDD, before the availability of brexpiprazole, only 3 antipsychotics were FDA-approved for adjunctive use with antidepressant therapy9; brexpiprazole represents another agent for patients whose depressive symptoms persist after standard antidepressant treatment.
Variables that limit the use of antipsychotics include extrapyramidal symptoms (EPS), akathisia, sedation/somnolence, weight gain, metabolic abnormalities, and hyperprolactinemia. If post-marketing studies and clinical experience confirm that brexpiprazole has an overall favorable side-effect profile regarding these tolerability obstacles, brexpiprazole would potentially have advantages over some other available agents, including aripiprazole.
How it works
In addition to a subnanomolar binding affinity (Ki < 1 nM) to dopamine D2 receptors as a partial agonist, brexpiprazole also exhibits similar binding affinities for serotonin 5-HT1A (partial agonist), 5-HT2A (antagonist), and adrenergic α1B (antagonist) and α2C (antagonist) receptors.7
Brexpiprazole also has high affinity (Ki < 5 nM) for dopamine D3 (partial ago nist), serotonin 5-HT2B (antagonist), and 5-HT7 (antagonist), and at adrenergic α1A (antagonist) and α1D (antagonist) receptors. Brexpiprazole has moderate affinity for histamine H1 receptors (Ki = 19 nM, antagonist), and low affinity for muscarinic M1 receptors (Ki > 1000 nM, antagonist).
Brexpiprazole’s pharmacodynamic profile differs from other available antipsychotics, including aripiprazole. Whether this translates to meaningful differences in efficacy and tolerability will depend on the outcomes of specifically designed clinical trials as well as “real-world” experience. Animal models have suggested amelioration of schizophrenia-like behavior, depression-like behavior, and anxiety-like behavior with brexipiprazole.6
Pharmacokinetics
At 91 hours, brexpiprazole’s half-life is relatively long; a steady-state concentration therefore is attained in approximately 2 weeks.1 In the phase-III clinical trials, brexpiprazole was titrated to target dosages, and therefore the product label recommends the same. Brexpiprazole can be administered with or without food.
In a study of brexpiprazole excretion, after a single oral dose of [14C]-labeled brexpiprazole, approximately 25% and 46% of the administered radioactivity was recovered in urine and feces, respectively. Less than 1% of unchanged brexpiprazole was excreted in the urine, and approximately 14% of the oral dose was recovered unchanged in the feces.
Exposure, as measured by maximum concentration and area under the concentration curve, is dose proportional.
Metabolism of brexpiprazole is mediated principally by cytochrome P450 (CYP) 3A4 and CYP2D6. Based on in vitro data, brexpiprazole shows little or no inhibition of CYP450 isozymes.
Efficacy
FDA approval for brexpiprazole for schizophrenia and for adjunctive use in MDD was based on 4 phase-III pivotal acute clinical trials conducted in adults, 2 studies each for each disorder.1-6 These studies are described in Table 2.2-5
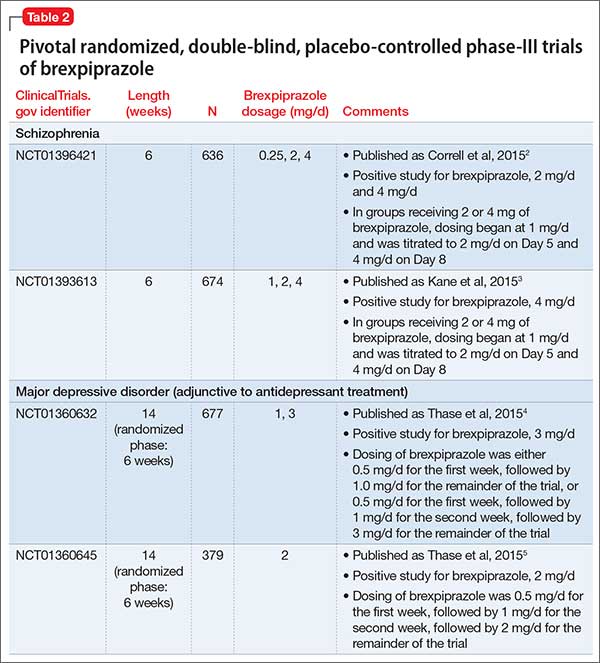
Schizophrenia. The primary outcome measure for the acute schizophrenia trials was change on the Positive and Negative Syndrome Scale (PANSS) total scores from baseline to 6-week endpoint. Statistically significant reductions in PANSS total score were observed for brexpiprazole dosages of 2 mg/d and 4 mg/d in one study,2 and 4 mg/d in another study.3 Responder rates also were measured, with response defined as a reduction of ≥30% from baseline in PANSS total score or a Clinical Global Impressions-Improvement score of 1 (very much improved) or 2 (much improved).2,3 Pooling together the available data for the recommended target dosage of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 phase-III studies, 45.5% of patients responded to the drug, compared with 31% for the pooled placebo groups, yielding a number needed to treat (NNT) of 7 (95% CI, 5-12).6
Although not described in product labeling, a phase-III 52-week maintenance study demonstrated brexpiprazole’s efficacy in preventing exacerbation of psychotic symptoms and impending relapse in patients with schizophrenia.10 Time from randomization to exacerbation of psychotic symptoms or impending relapse showed a beneficial effect with brexpiprazole compared with placebo (log-rank test: hazard ratio = 0.292, P < .0001). Significantly fewer patients in the brexpiprazole group relapsed compared with placebo (13.5% vs 38.5%, P < .0001), resulting in a NNT of 4 (95% CI, 3-8).
Major depressive disorder. The primary outcome measure for the acute MDD studies was change in Montgomery-Åsberg Depression Rating Scale (MADRS) scores from baseline to 6-week endpoint of the randomized treatment phase. All patients were required to have a history of inadequate response to 1 to 3 treatment trials of standard antidepressants for their current depressive episode. In addition, patients entered the randomized phase only if they had an inadequate response to antidepressant therapy during an 8-week prospective treatment trial of standard antidepressant treatment plus single-blind placebo.
Participants who responded adequately to the antidepressant in the prospective single-blind phase were not randomized, but instead continued on antidepressant treatment plus single-blind placebo for 6 weeks.
The phase-III studies showed positive results for brexpiprazole, 2 mg/d and 3 mg/d, with change in MADRS from baseline to endpoint superior to that observed with placebo.4,5
When examining treatment response, defined as a reduction of ≥50% in MADRS total score from baseline, NNT vs placebo for response were 12 at all dosages tested, however, NNT vs placebo for remission (defined as MADRS total score ≤10 and ≥50% improvement from baseline) ranged from 17 to 31 and were not statistically significant.6 When the results for brexpiprazole, 1 mg/d, 2 mg/d, and 3 mg/d, from the 2 phase-III trials are pooled together, 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI, 8-26); 14.4% of the brexpiprazole-treated patients met remission criteria, vs 9.6% for placebo, resulting in a NNT of 21 (95% CI, 12-138).6
Tolerability
Overall tolerability can be evaluated by examining the percentage of patients who discontinued the clinical trials because of an adverse event (AE). In the acute schizophrenia double-blind trials for the recommended dosage range of 2 to 4 mg, the discontinuation rates were lower overall for patients receiving brexpiprazole compared with placebo.2,3 In the acute MDD trials, 32.6% of brexpiprazole-treated patients and 10.7% of placebo-treated patients discontinued because of AEs,4,5 yielding a number needed to harm (NNH) of 53 (95% CI, 30-235).6
The most commonly encountered AEs for MDD (incidence ≥5% and at least twice the rate for placebo) were akathisia (8.6% vs 1.7% for brexpiprazole vs placebo, and dose-related) and weight gain (6.7% vs 1.9%),1 with NNH values of 15 (95% CI, 11-23), and 22 (95% CI, 15-42), respectively.6 The most commonly encountered AE for schizophrenia (incidence ≥4% and at least twice the rate for placebo) was weight gain (4% vs 2%),1 with a NNH of 50 (95% CI, 26-1773).6
Of note, rates of akathisia in the schizophrenia trials were 5.5% for brexpiprazole and 4.6% for placebo,1 yielding a non-statistically significant NNH of 112.6 In a 6-week exploratory study,11 the incidence of EPS-related AEs including akathisia was lower for brexpiprazole-treated patients (14.1%) compared with those receiving aripiprazole (30.3%), for a NNT advantage for brexpiprazole of 7 (not statistically significant).
Short-term weight gain appears modest; however, outliers with an increase of ≥7% of body weight were evident in open-label long-term safety studies.1,6 Effects on glucose and lipids were small. Minimal effects on prolactin were observed, and no clinically relevant effects on the QT interval were evident.
Contraindications
The only absolute contraindication for brexpiprazole is known hypersensitivity to brexpiprazole or any of its components. Reactions have included rash, facial swelling, urticaria, and anaphylaxis.1
As with all antipsychotics and antipsychotics with an indication for a depressive disorder:
• there is a bolded boxed warning in the product label regarding increased mortality in geriatric patients with dementia-related psychosis. Brexpiprazole is not approved for treating patients with dementia-related psychosis
• there is a bolded boxed warning in the product label about suicidal thoughts and behaviors in patients age ≤24. The safety and efficacy of brexpiprazole have not been established in pediatric patients.
Dosing
Schizophrenia. The recommended starting dosage for brexpiprazole for schizophrenia is 1 mg/d on Days 1 to 4. Brexpiprazole can be titrated to 2 mg/d on Day 5 through Day 7, then to 4 mg/d on Day 8 based on the patient’s response and ability to tolerate the medication. The recommended target dosage is 2 to 4 mg/d with a maximum recommended daily dosage of 4 mg.
Major depressive disorder. The recommended starting dosage for brexpiprazole as adjunctive treatment for MDD is 0.5 mg or 1 mg/d. Brexpiprazole can be titrated to 1 mg/d, then up to the target dosage of 2 mg/d, with dosage increases occurring at weekly intervals based on the patient’s clinical response and ability to tolerate the agent, with a maximum recommended dosage of 3 mg/d.
Other considerations. For patients with moderate to severe hepatic impairment, or moderate, severe, or end-stage renal impairment, the maximum recommended dosage is 3 mg/d for patients with schizophrenia, and 2 mg/d for patients with MDD.
In general, dosage adjustments are recommended in patients who are known CYP2D6 poor metabolizers and in those taking concomitant CYP3A4 inhibitors or CYP2D6 inhibitors or strong CYP3A4 inducers1:
• for strong CYP2D6 or CYP3A4 inhibitors, administer one-half the usual dosage
• for strong/moderate CYP2D6 with strong/moderate CYP3A4 inhibitors, administer a one-quarter of the usual dosage
• for known CYP2D6 poor metabolizers taking strong/moderate CYP3A4 inhibitors, also administer a one-quarter of the usual dosage
• for strong CYP3A4 inducers, double the usual dosage and further adjust based on clinical response.
In clinical trials for MDD, brexpiprazole dosage was not adjusted for strong CYP2D6 inhibitors (eg, paroxetine, fluoxetine). Therefore, CYP considerations are already factored into general dosing recommendations and brexpiprazole could be administered without dosage adjustment in patients with MDD; however, under these circumstances, it would be prudent to start brexpiprazole at 0.5 mg, which, although “on-label,” represents a low starting dosage. (Whenever 2 drugs are co-administered and 1 agent has the ability to disturb the metabolism of the other, using smaller increments to the target dosage and possibly waiting longer between dosage adjustments could help avoid potential drug–drug interactions.)
No dosage adjustment for brexpiprazole is required on the basis of sex, race or ethnicity, or smoking status. Although clinical studies did not include patients age ≥65, the product label recommends that in general, dose selection for a geriatric patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, and cardiac function, concomitant diseases, and other drug therapy.
Bottom Line
Brexpiprazole, an atypical antipsychotic, is FDA-approved for schizophrenia and as an adjunct to antidepressants in major depressive disorder. For both indications, brexpiprazole demonstrated positive results compared with placebo in phase-III trials. Brexpiprazole is more potent at serotonin 5-HT1A and 5-HT2A receptors and displays less intrinsic activity at D2 receptors than aripiprazole, which could mean that the drug may be better-tolerated.
Related Resources
• Citrome L. Brexpiprazole: a new dopamine D2 receptor partial agonist for the treatment of schizophrenia and major depressive disorder. Drugs Today (Barc). 2015;51(7):397-414.
• Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. In press.
Drug Brand Names
Aripiprazole • Abilify
Brexpiprazole • Rexulti
Fluoxetine • Prozac
Paroxetine • Paxil
Disclosure
Dr. Citrome is a consultant to Alexza Pharmaceuticals, Alkermes, Allergan, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly and Company, Forum Pharmaceuticals, Genentech, Janssen, Jazz Pharmaceuticals, Lundbeck, Merck, Medivation, Mylan, Novartis, Noven, Otsuka, Pfizer, Reckitt Benckiser, Reviva, Shire, Sunovion, Takeda, Teva, and Valeant Pharmaceuticals; and is a speaker for Allergan, AstraZeneca, Janssen, Jazz Pharmaceuticals, Lundbeck, Merck, Novartis, Otsuka, Pfizer, Shire, Sunovion, Takeda, and Teva.
1. Rexulti [package insert]. Rockville, MD: Otsuka; 2015.
2. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
3. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
4. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study [published online August 4, 2015]. J Clin Psychiatry. doi: 10.4088/ JCP.14m09689.
5. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants [published online August 4, 2015]. J Clin Psychiatry. doi: 10.4088/JCP.14m09688.
6. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
7. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
8. Volavka J, Citrome L. Oral antipsychotics for the treatment of schizophrenia: heterogeneity in efficacy and tolerability should drive decision-making. Expert Opin Pharmacother. 2009;10(12):1917-1928.
9. Citrome L. Adjunctive aripiprazole, olanzapine, or quetiapine for major depressive disorder: an analysis of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Postgrad Med. 2010;122(4):39-48.
10. Hobart M, Ouyang J, Forbes A, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Poster presented at: the American Society of Clinical Psychopharmacology Annual Meeting; June 22 to 25, 2015; Miami, FL.
11. Citrome L, Ota A, Nagamizu K, Perry P, et al. The effect of brexpiprazole (OPC‐34712) versus aripiprazole in adult patients with acute schizophrenia: an exploratory study. Poster presented at: the Society of Biological Psychiatry Annual Scientific Meeting and Convention; May 15, 2015; Toronto, Ontario, Canada.
Brexpiprazole, FDA-approved in July 2015 to treat schizophrenia and as an adjunct for major depressive disorder (MDD) (Table 1), has shown efficacy in 2 phase-III acute trials for each indication.1-6 Although brexpiprazole is a dopamine D2 partial agonist, it differs from aripiprazole, the other available D2 partial agonist, because it is more potent at serotonin 5-HT1A and 5-HT2A receptors and displays less intrinsic activity at D2 receptors,7 which could mean better tolerability.
Clinical implications
Schizophrenia is heterogeneous, and individual response and tolerability to antipsychotics vary greatly8; therefore, new drug options are useful. For MDD, before the availability of brexpiprazole, only 3 antipsychotics were FDA-approved for adjunctive use with antidepressant therapy9; brexpiprazole represents another agent for patients whose depressive symptoms persist after standard antidepressant treatment.
Variables that limit the use of antipsychotics include extrapyramidal symptoms (EPS), akathisia, sedation/somnolence, weight gain, metabolic abnormalities, and hyperprolactinemia. If post-marketing studies and clinical experience confirm that brexpiprazole has an overall favorable side-effect profile regarding these tolerability obstacles, brexpiprazole would potentially have advantages over some other available agents, including aripiprazole.
How it works
In addition to a subnanomolar binding affinity (Ki < 1 nM) to dopamine D2 receptors as a partial agonist, brexpiprazole also exhibits similar binding affinities for serotonin 5-HT1A (partial agonist), 5-HT2A (antagonist), and adrenergic α1B (antagonist) and α2C (antagonist) receptors.7
Brexpiprazole also has high affinity (Ki < 5 nM) for dopamine D3 (partial ago nist), serotonin 5-HT2B (antagonist), and 5-HT7 (antagonist), and at adrenergic α1A (antagonist) and α1D (antagonist) receptors. Brexpiprazole has moderate affinity for histamine H1 receptors (Ki = 19 nM, antagonist), and low affinity for muscarinic M1 receptors (Ki > 1000 nM, antagonist).
Brexpiprazole’s pharmacodynamic profile differs from other available antipsychotics, including aripiprazole. Whether this translates to meaningful differences in efficacy and tolerability will depend on the outcomes of specifically designed clinical trials as well as “real-world” experience. Animal models have suggested amelioration of schizophrenia-like behavior, depression-like behavior, and anxiety-like behavior with brexipiprazole.6
Pharmacokinetics
At 91 hours, brexpiprazole’s half-life is relatively long; a steady-state concentration therefore is attained in approximately 2 weeks.1 In the phase-III clinical trials, brexpiprazole was titrated to target dosages, and therefore the product label recommends the same. Brexpiprazole can be administered with or without food.
In a study of brexpiprazole excretion, after a single oral dose of [14C]-labeled brexpiprazole, approximately 25% and 46% of the administered radioactivity was recovered in urine and feces, respectively. Less than 1% of unchanged brexpiprazole was excreted in the urine, and approximately 14% of the oral dose was recovered unchanged in the feces.
Exposure, as measured by maximum concentration and area under the concentration curve, is dose proportional.
Metabolism of brexpiprazole is mediated principally by cytochrome P450 (CYP) 3A4 and CYP2D6. Based on in vitro data, brexpiprazole shows little or no inhibition of CYP450 isozymes.
Efficacy
FDA approval for brexpiprazole for schizophrenia and for adjunctive use in MDD was based on 4 phase-III pivotal acute clinical trials conducted in adults, 2 studies each for each disorder.1-6 These studies are described in Table 2.2-5
Schizophrenia. The primary outcome measure for the acute schizophrenia trials was change on the Positive and Negative Syndrome Scale (PANSS) total scores from baseline to 6-week endpoint. Statistically significant reductions in PANSS total score were observed for brexpiprazole dosages of 2 mg/d and 4 mg/d in one study,2 and 4 mg/d in another study.3 Responder rates also were measured, with response defined as a reduction of ≥30% from baseline in PANSS total score or a Clinical Global Impressions-Improvement score of 1 (very much improved) or 2 (much improved).2,3 Pooling together the available data for the recommended target dosage of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 phase-III studies, 45.5% of patients responded to the drug, compared with 31% for the pooled placebo groups, yielding a number needed to treat (NNT) of 7 (95% CI, 5-12).6
Although not described in product labeling, a phase-III 52-week maintenance study demonstrated brexpiprazole’s efficacy in preventing exacerbation of psychotic symptoms and impending relapse in patients with schizophrenia.10 Time from randomization to exacerbation of psychotic symptoms or impending relapse showed a beneficial effect with brexpiprazole compared with placebo (log-rank test: hazard ratio = 0.292, P < .0001). Significantly fewer patients in the brexpiprazole group relapsed compared with placebo (13.5% vs 38.5%, P < .0001), resulting in a NNT of 4 (95% CI, 3-8).
Major depressive disorder. The primary outcome measure for the acute MDD studies was change in Montgomery-Åsberg Depression Rating Scale (MADRS) scores from baseline to 6-week endpoint of the randomized treatment phase. All patients were required to have a history of inadequate response to 1 to 3 treatment trials of standard antidepressants for their current depressive episode. In addition, patients entered the randomized phase only if they had an inadequate response to antidepressant therapy during an 8-week prospective treatment trial of standard antidepressant treatment plus single-blind placebo.
Participants who responded adequately to the antidepressant in the prospective single-blind phase were not randomized, but instead continued on antidepressant treatment plus single-blind placebo for 6 weeks.
The phase-III studies showed positive results for brexpiprazole, 2 mg/d and 3 mg/d, with change in MADRS from baseline to endpoint superior to that observed with placebo.4,5
When examining treatment response, defined as a reduction of ≥50% in MADRS total score from baseline, NNT vs placebo for response were 12 at all dosages tested, however, NNT vs placebo for remission (defined as MADRS total score ≤10 and ≥50% improvement from baseline) ranged from 17 to 31 and were not statistically significant.6 When the results for brexpiprazole, 1 mg/d, 2 mg/d, and 3 mg/d, from the 2 phase-III trials are pooled together, 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI, 8-26); 14.4% of the brexpiprazole-treated patients met remission criteria, vs 9.6% for placebo, resulting in a NNT of 21 (95% CI, 12-138).6
Tolerability
Overall tolerability can be evaluated by examining the percentage of patients who discontinued the clinical trials because of an adverse event (AE). In the acute schizophrenia double-blind trials for the recommended dosage range of 2 to 4 mg, the discontinuation rates were lower overall for patients receiving brexpiprazole compared with placebo.2,3 In the acute MDD trials, 32.6% of brexpiprazole-treated patients and 10.7% of placebo-treated patients discontinued because of AEs,4,5 yielding a number needed to harm (NNH) of 53 (95% CI, 30-235).6
The most commonly encountered AEs for MDD (incidence ≥5% and at least twice the rate for placebo) were akathisia (8.6% vs 1.7% for brexpiprazole vs placebo, and dose-related) and weight gain (6.7% vs 1.9%),1 with NNH values of 15 (95% CI, 11-23), and 22 (95% CI, 15-42), respectively.6 The most commonly encountered AE for schizophrenia (incidence ≥4% and at least twice the rate for placebo) was weight gain (4% vs 2%),1 with a NNH of 50 (95% CI, 26-1773).6
Of note, rates of akathisia in the schizophrenia trials were 5.5% for brexpiprazole and 4.6% for placebo,1 yielding a non-statistically significant NNH of 112.6 In a 6-week exploratory study,11 the incidence of EPS-related AEs including akathisia was lower for brexpiprazole-treated patients (14.1%) compared with those receiving aripiprazole (30.3%), for a NNT advantage for brexpiprazole of 7 (not statistically significant).
Short-term weight gain appears modest; however, outliers with an increase of ≥7% of body weight were evident in open-label long-term safety studies.1,6 Effects on glucose and lipids were small. Minimal effects on prolactin were observed, and no clinically relevant effects on the QT interval were evident.
Contraindications
The only absolute contraindication for brexpiprazole is known hypersensitivity to brexpiprazole or any of its components. Reactions have included rash, facial swelling, urticaria, and anaphylaxis.1
As with all antipsychotics and antipsychotics with an indication for a depressive disorder:
• there is a bolded boxed warning in the product label regarding increased mortality in geriatric patients with dementia-related psychosis. Brexpiprazole is not approved for treating patients with dementia-related psychosis
• there is a bolded boxed warning in the product label about suicidal thoughts and behaviors in patients age ≤24. The safety and efficacy of brexpiprazole have not been established in pediatric patients.
Dosing
Schizophrenia. The recommended starting dosage for brexpiprazole for schizophrenia is 1 mg/d on Days 1 to 4. Brexpiprazole can be titrated to 2 mg/d on Day 5 through Day 7, then to 4 mg/d on Day 8 based on the patient’s response and ability to tolerate the medication. The recommended target dosage is 2 to 4 mg/d with a maximum recommended daily dosage of 4 mg.
Major depressive disorder. The recommended starting dosage for brexpiprazole as adjunctive treatment for MDD is 0.5 mg or 1 mg/d. Brexpiprazole can be titrated to 1 mg/d, then up to the target dosage of 2 mg/d, with dosage increases occurring at weekly intervals based on the patient’s clinical response and ability to tolerate the agent, with a maximum recommended dosage of 3 mg/d.
Other considerations. For patients with moderate to severe hepatic impairment, or moderate, severe, or end-stage renal impairment, the maximum recommended dosage is 3 mg/d for patients with schizophrenia, and 2 mg/d for patients with MDD.
In general, dosage adjustments are recommended in patients who are known CYP2D6 poor metabolizers and in those taking concomitant CYP3A4 inhibitors or CYP2D6 inhibitors or strong CYP3A4 inducers1:
• for strong CYP2D6 or CYP3A4 inhibitors, administer one-half the usual dosage
• for strong/moderate CYP2D6 with strong/moderate CYP3A4 inhibitors, administer a one-quarter of the usual dosage
• for known CYP2D6 poor metabolizers taking strong/moderate CYP3A4 inhibitors, also administer a one-quarter of the usual dosage
• for strong CYP3A4 inducers, double the usual dosage and further adjust based on clinical response.
In clinical trials for MDD, brexpiprazole dosage was not adjusted for strong CYP2D6 inhibitors (eg, paroxetine, fluoxetine). Therefore, CYP considerations are already factored into general dosing recommendations and brexpiprazole could be administered without dosage adjustment in patients with MDD; however, under these circumstances, it would be prudent to start brexpiprazole at 0.5 mg, which, although “on-label,” represents a low starting dosage. (Whenever 2 drugs are co-administered and 1 agent has the ability to disturb the metabolism of the other, using smaller increments to the target dosage and possibly waiting longer between dosage adjustments could help avoid potential drug–drug interactions.)
No dosage adjustment for brexpiprazole is required on the basis of sex, race or ethnicity, or smoking status. Although clinical studies did not include patients age ≥65, the product label recommends that in general, dose selection for a geriatric patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, and cardiac function, concomitant diseases, and other drug therapy.
Bottom Line
Brexpiprazole, an atypical antipsychotic, is FDA-approved for schizophrenia and as an adjunct to antidepressants in major depressive disorder. For both indications, brexpiprazole demonstrated positive results compared with placebo in phase-III trials. Brexpiprazole is more potent at serotonin 5-HT1A and 5-HT2A receptors and displays less intrinsic activity at D2 receptors than aripiprazole, which could mean that the drug may be better-tolerated.
Related Resources
• Citrome L. Brexpiprazole: a new dopamine D2 receptor partial agonist for the treatment of schizophrenia and major depressive disorder. Drugs Today (Barc). 2015;51(7):397-414.
• Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. In press.
Drug Brand Names
Aripiprazole • Abilify
Brexpiprazole • Rexulti
Fluoxetine • Prozac
Paroxetine • Paxil
Disclosure
Dr. Citrome is a consultant to Alexza Pharmaceuticals, Alkermes, Allergan, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly and Company, Forum Pharmaceuticals, Genentech, Janssen, Jazz Pharmaceuticals, Lundbeck, Merck, Medivation, Mylan, Novartis, Noven, Otsuka, Pfizer, Reckitt Benckiser, Reviva, Shire, Sunovion, Takeda, Teva, and Valeant Pharmaceuticals; and is a speaker for Allergan, AstraZeneca, Janssen, Jazz Pharmaceuticals, Lundbeck, Merck, Novartis, Otsuka, Pfizer, Shire, Sunovion, Takeda, and Teva.
Brexpiprazole, FDA-approved in July 2015 to treat schizophrenia and as an adjunct for major depressive disorder (MDD) (Table 1), has shown efficacy in 2 phase-III acute trials for each indication.1-6 Although brexpiprazole is a dopamine D2 partial agonist, it differs from aripiprazole, the other available D2 partial agonist, because it is more potent at serotonin 5-HT1A and 5-HT2A receptors and displays less intrinsic activity at D2 receptors,7 which could mean better tolerability.
Clinical implications
Schizophrenia is heterogeneous, and individual response and tolerability to antipsychotics vary greatly8; therefore, new drug options are useful. For MDD, before the availability of brexpiprazole, only 3 antipsychotics were FDA-approved for adjunctive use with antidepressant therapy9; brexpiprazole represents another agent for patients whose depressive symptoms persist after standard antidepressant treatment.
Variables that limit the use of antipsychotics include extrapyramidal symptoms (EPS), akathisia, sedation/somnolence, weight gain, metabolic abnormalities, and hyperprolactinemia. If post-marketing studies and clinical experience confirm that brexpiprazole has an overall favorable side-effect profile regarding these tolerability obstacles, brexpiprazole would potentially have advantages over some other available agents, including aripiprazole.
How it works
In addition to a subnanomolar binding affinity (Ki < 1 nM) to dopamine D2 receptors as a partial agonist, brexpiprazole also exhibits similar binding affinities for serotonin 5-HT1A (partial agonist), 5-HT2A (antagonist), and adrenergic α1B (antagonist) and α2C (antagonist) receptors.7
Brexpiprazole also has high affinity (Ki < 5 nM) for dopamine D3 (partial ago nist), serotonin 5-HT2B (antagonist), and 5-HT7 (antagonist), and at adrenergic α1A (antagonist) and α1D (antagonist) receptors. Brexpiprazole has moderate affinity for histamine H1 receptors (Ki = 19 nM, antagonist), and low affinity for muscarinic M1 receptors (Ki > 1000 nM, antagonist).
Brexpiprazole’s pharmacodynamic profile differs from other available antipsychotics, including aripiprazole. Whether this translates to meaningful differences in efficacy and tolerability will depend on the outcomes of specifically designed clinical trials as well as “real-world” experience. Animal models have suggested amelioration of schizophrenia-like behavior, depression-like behavior, and anxiety-like behavior with brexipiprazole.6
Pharmacokinetics
At 91 hours, brexpiprazole’s half-life is relatively long; a steady-state concentration therefore is attained in approximately 2 weeks.1 In the phase-III clinical trials, brexpiprazole was titrated to target dosages, and therefore the product label recommends the same. Brexpiprazole can be administered with or without food.
In a study of brexpiprazole excretion, after a single oral dose of [14C]-labeled brexpiprazole, approximately 25% and 46% of the administered radioactivity was recovered in urine and feces, respectively. Less than 1% of unchanged brexpiprazole was excreted in the urine, and approximately 14% of the oral dose was recovered unchanged in the feces.
Exposure, as measured by maximum concentration and area under the concentration curve, is dose proportional.
Metabolism of brexpiprazole is mediated principally by cytochrome P450 (CYP) 3A4 and CYP2D6. Based on in vitro data, brexpiprazole shows little or no inhibition of CYP450 isozymes.
Efficacy
FDA approval for brexpiprazole for schizophrenia and for adjunctive use in MDD was based on 4 phase-III pivotal acute clinical trials conducted in adults, 2 studies each for each disorder.1-6 These studies are described in Table 2.2-5
Schizophrenia. The primary outcome measure for the acute schizophrenia trials was change on the Positive and Negative Syndrome Scale (PANSS) total scores from baseline to 6-week endpoint. Statistically significant reductions in PANSS total score were observed for brexpiprazole dosages of 2 mg/d and 4 mg/d in one study,2 and 4 mg/d in another study.3 Responder rates also were measured, with response defined as a reduction of ≥30% from baseline in PANSS total score or a Clinical Global Impressions-Improvement score of 1 (very much improved) or 2 (much improved).2,3 Pooling together the available data for the recommended target dosage of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 phase-III studies, 45.5% of patients responded to the drug, compared with 31% for the pooled placebo groups, yielding a number needed to treat (NNT) of 7 (95% CI, 5-12).6
Although not described in product labeling, a phase-III 52-week maintenance study demonstrated brexpiprazole’s efficacy in preventing exacerbation of psychotic symptoms and impending relapse in patients with schizophrenia.10 Time from randomization to exacerbation of psychotic symptoms or impending relapse showed a beneficial effect with brexpiprazole compared with placebo (log-rank test: hazard ratio = 0.292, P < .0001). Significantly fewer patients in the brexpiprazole group relapsed compared with placebo (13.5% vs 38.5%, P < .0001), resulting in a NNT of 4 (95% CI, 3-8).
Major depressive disorder. The primary outcome measure for the acute MDD studies was change in Montgomery-Åsberg Depression Rating Scale (MADRS) scores from baseline to 6-week endpoint of the randomized treatment phase. All patients were required to have a history of inadequate response to 1 to 3 treatment trials of standard antidepressants for their current depressive episode. In addition, patients entered the randomized phase only if they had an inadequate response to antidepressant therapy during an 8-week prospective treatment trial of standard antidepressant treatment plus single-blind placebo.
Participants who responded adequately to the antidepressant in the prospective single-blind phase were not randomized, but instead continued on antidepressant treatment plus single-blind placebo for 6 weeks.
The phase-III studies showed positive results for brexpiprazole, 2 mg/d and 3 mg/d, with change in MADRS from baseline to endpoint superior to that observed with placebo.4,5
When examining treatment response, defined as a reduction of ≥50% in MADRS total score from baseline, NNT vs placebo for response were 12 at all dosages tested, however, NNT vs placebo for remission (defined as MADRS total score ≤10 and ≥50% improvement from baseline) ranged from 17 to 31 and were not statistically significant.6 When the results for brexpiprazole, 1 mg/d, 2 mg/d, and 3 mg/d, from the 2 phase-III trials are pooled together, 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI, 8-26); 14.4% of the brexpiprazole-treated patients met remission criteria, vs 9.6% for placebo, resulting in a NNT of 21 (95% CI, 12-138).6
Tolerability
Overall tolerability can be evaluated by examining the percentage of patients who discontinued the clinical trials because of an adverse event (AE). In the acute schizophrenia double-blind trials for the recommended dosage range of 2 to 4 mg, the discontinuation rates were lower overall for patients receiving brexpiprazole compared with placebo.2,3 In the acute MDD trials, 32.6% of brexpiprazole-treated patients and 10.7% of placebo-treated patients discontinued because of AEs,4,5 yielding a number needed to harm (NNH) of 53 (95% CI, 30-235).6
The most commonly encountered AEs for MDD (incidence ≥5% and at least twice the rate for placebo) were akathisia (8.6% vs 1.7% for brexpiprazole vs placebo, and dose-related) and weight gain (6.7% vs 1.9%),1 with NNH values of 15 (95% CI, 11-23), and 22 (95% CI, 15-42), respectively.6 The most commonly encountered AE for schizophrenia (incidence ≥4% and at least twice the rate for placebo) was weight gain (4% vs 2%),1 with a NNH of 50 (95% CI, 26-1773).6
Of note, rates of akathisia in the schizophrenia trials were 5.5% for brexpiprazole and 4.6% for placebo,1 yielding a non-statistically significant NNH of 112.6 In a 6-week exploratory study,11 the incidence of EPS-related AEs including akathisia was lower for brexpiprazole-treated patients (14.1%) compared with those receiving aripiprazole (30.3%), for a NNT advantage for brexpiprazole of 7 (not statistically significant).
Short-term weight gain appears modest; however, outliers with an increase of ≥7% of body weight were evident in open-label long-term safety studies.1,6 Effects on glucose and lipids were small. Minimal effects on prolactin were observed, and no clinically relevant effects on the QT interval were evident.
Contraindications
The only absolute contraindication for brexpiprazole is known hypersensitivity to brexpiprazole or any of its components. Reactions have included rash, facial swelling, urticaria, and anaphylaxis.1
As with all antipsychotics and antipsychotics with an indication for a depressive disorder:
• there is a bolded boxed warning in the product label regarding increased mortality in geriatric patients with dementia-related psychosis. Brexpiprazole is not approved for treating patients with dementia-related psychosis
• there is a bolded boxed warning in the product label about suicidal thoughts and behaviors in patients age ≤24. The safety and efficacy of brexpiprazole have not been established in pediatric patients.
Dosing
Schizophrenia. The recommended starting dosage for brexpiprazole for schizophrenia is 1 mg/d on Days 1 to 4. Brexpiprazole can be titrated to 2 mg/d on Day 5 through Day 7, then to 4 mg/d on Day 8 based on the patient’s response and ability to tolerate the medication. The recommended target dosage is 2 to 4 mg/d with a maximum recommended daily dosage of 4 mg.
Major depressive disorder. The recommended starting dosage for brexpiprazole as adjunctive treatment for MDD is 0.5 mg or 1 mg/d. Brexpiprazole can be titrated to 1 mg/d, then up to the target dosage of 2 mg/d, with dosage increases occurring at weekly intervals based on the patient’s clinical response and ability to tolerate the agent, with a maximum recommended dosage of 3 mg/d.
Other considerations. For patients with moderate to severe hepatic impairment, or moderate, severe, or end-stage renal impairment, the maximum recommended dosage is 3 mg/d for patients with schizophrenia, and 2 mg/d for patients with MDD.
In general, dosage adjustments are recommended in patients who are known CYP2D6 poor metabolizers and in those taking concomitant CYP3A4 inhibitors or CYP2D6 inhibitors or strong CYP3A4 inducers1:
• for strong CYP2D6 or CYP3A4 inhibitors, administer one-half the usual dosage
• for strong/moderate CYP2D6 with strong/moderate CYP3A4 inhibitors, administer a one-quarter of the usual dosage
• for known CYP2D6 poor metabolizers taking strong/moderate CYP3A4 inhibitors, also administer a one-quarter of the usual dosage
• for strong CYP3A4 inducers, double the usual dosage and further adjust based on clinical response.
In clinical trials for MDD, brexpiprazole dosage was not adjusted for strong CYP2D6 inhibitors (eg, paroxetine, fluoxetine). Therefore, CYP considerations are already factored into general dosing recommendations and brexpiprazole could be administered without dosage adjustment in patients with MDD; however, under these circumstances, it would be prudent to start brexpiprazole at 0.5 mg, which, although “on-label,” represents a low starting dosage. (Whenever 2 drugs are co-administered and 1 agent has the ability to disturb the metabolism of the other, using smaller increments to the target dosage and possibly waiting longer between dosage adjustments could help avoid potential drug–drug interactions.)
No dosage adjustment for brexpiprazole is required on the basis of sex, race or ethnicity, or smoking status. Although clinical studies did not include patients age ≥65, the product label recommends that in general, dose selection for a geriatric patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, and cardiac function, concomitant diseases, and other drug therapy.
Bottom Line
Brexpiprazole, an atypical antipsychotic, is FDA-approved for schizophrenia and as an adjunct to antidepressants in major depressive disorder. For both indications, brexpiprazole demonstrated positive results compared with placebo in phase-III trials. Brexpiprazole is more potent at serotonin 5-HT1A and 5-HT2A receptors and displays less intrinsic activity at D2 receptors than aripiprazole, which could mean that the drug may be better-tolerated.
Related Resources
• Citrome L. Brexpiprazole: a new dopamine D2 receptor partial agonist for the treatment of schizophrenia and major depressive disorder. Drugs Today (Barc). 2015;51(7):397-414.
• Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. In press.
Drug Brand Names
Aripiprazole • Abilify
Brexpiprazole • Rexulti
Fluoxetine • Prozac
Paroxetine • Paxil
Disclosure
Dr. Citrome is a consultant to Alexza Pharmaceuticals, Alkermes, Allergan, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly and Company, Forum Pharmaceuticals, Genentech, Janssen, Jazz Pharmaceuticals, Lundbeck, Merck, Medivation, Mylan, Novartis, Noven, Otsuka, Pfizer, Reckitt Benckiser, Reviva, Shire, Sunovion, Takeda, Teva, and Valeant Pharmaceuticals; and is a speaker for Allergan, AstraZeneca, Janssen, Jazz Pharmaceuticals, Lundbeck, Merck, Novartis, Otsuka, Pfizer, Shire, Sunovion, Takeda, and Teva.
1. Rexulti [package insert]. Rockville, MD: Otsuka; 2015.
2. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
3. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
4. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study [published online August 4, 2015]. J Clin Psychiatry. doi: 10.4088/ JCP.14m09689.
5. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants [published online August 4, 2015]. J Clin Psychiatry. doi: 10.4088/JCP.14m09688.
6. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
7. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
8. Volavka J, Citrome L. Oral antipsychotics for the treatment of schizophrenia: heterogeneity in efficacy and tolerability should drive decision-making. Expert Opin Pharmacother. 2009;10(12):1917-1928.
9. Citrome L. Adjunctive aripiprazole, olanzapine, or quetiapine for major depressive disorder: an analysis of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Postgrad Med. 2010;122(4):39-48.
10. Hobart M, Ouyang J, Forbes A, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Poster presented at: the American Society of Clinical Psychopharmacology Annual Meeting; June 22 to 25, 2015; Miami, FL.
11. Citrome L, Ota A, Nagamizu K, Perry P, et al. The effect of brexpiprazole (OPC‐34712) versus aripiprazole in adult patients with acute schizophrenia: an exploratory study. Poster presented at: the Society of Biological Psychiatry Annual Scientific Meeting and Convention; May 15, 2015; Toronto, Ontario, Canada.
1. Rexulti [package insert]. Rockville, MD: Otsuka; 2015.
2. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
3. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
4. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study [published online August 4, 2015]. J Clin Psychiatry. doi: 10.4088/ JCP.14m09689.
5. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants [published online August 4, 2015]. J Clin Psychiatry. doi: 10.4088/JCP.14m09688.
6. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
7. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
8. Volavka J, Citrome L. Oral antipsychotics for the treatment of schizophrenia: heterogeneity in efficacy and tolerability should drive decision-making. Expert Opin Pharmacother. 2009;10(12):1917-1928.
9. Citrome L. Adjunctive aripiprazole, olanzapine, or quetiapine for major depressive disorder: an analysis of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Postgrad Med. 2010;122(4):39-48.
10. Hobart M, Ouyang J, Forbes A, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Poster presented at: the American Society of Clinical Psychopharmacology Annual Meeting; June 22 to 25, 2015; Miami, FL.
11. Citrome L, Ota A, Nagamizu K, Perry P, et al. The effect of brexpiprazole (OPC‐34712) versus aripiprazole in adult patients with acute schizophrenia: an exploratory study. Poster presented at: the Society of Biological Psychiatry Annual Scientific Meeting and Convention; May 15, 2015; Toronto, Ontario, Canada.