User login
Health Canada expands approval of obinutuzumab

Health Canada has expanded the approved use of obinutuzumab (Gazyva®).
The anti-CD20 monoclonal antibody is now approved for use in combination with chemotherapy to treat patients with previously untreated follicular lymphoma (FL) that is advanced (stage II bulky, stage III, or stage IV).
In patients who respond to this treatment, obinutuzumab monotherapy can be given as maintenance.
Health Canada previously approved obinutuzumab for the following indications:
- In combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia
- First in combination with bendamustine, then as monotherapy, in FL patients who relapsed after or are refractory to a rituximab-containing regimen.
Phase 3 results
Health Canada’s latest approval of obinutuzumab is based on results from the phase 3 GALLIUM study, which were published in NEJM in October 2017. The following are updated data from the product monograph.
GALLIUM included 1385 patients with previously untreated non-Hodgkin lymphoma, and 1202 of these patients had previously untreated, advanced FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), and bendamustine.
At a median observation time of 41.1 months, the overall response rate was 91% in the obinutuzumab arm and 88% in the rituximab arm. The complete response rates were 28% and 27%, respectively.
The median progression-free survival was not reached in either arm. The hazard ratio, for obinutuzumab compared to rituximab, was 0.72 (95% CI, 0.56-0.93, P=0.0118).
The estimated 3-year progression-free survival was 78.9% in the rituximab arm and 83.4% in the obinutuzumab arm.
Safety was evaluated based on all 1385 patients in the study, 86% of whom had FL and 14% of whom had marginal zone lymphoma.
Serious adverse events (AEs) occurred in 50% of patients in the obinutuzumab arm and 43% in the rituximab arm. Fatal AEs occurred in 5% and 4%, respectively. Infections and second malignancies were the leading causes of these deaths.
During the monotherapy period, the most common AEs (≥ 5%) in patients treated with obinutuzumab were cough (21%), neutropenia (19%), upper respiratory tract infection (15%), viral upper respiratory tract infection (15%), diarrhea (13%), arthralgia (10%), fatigue (9%), sinusitis (9%), infusion reactions (8%), pneumonia (8%), herpes zoster (8%), lower respiratory tract infection (7%), pyrexia (7%), back pain (6%), headache (6%), urinary tract infection (6%), nausea (6%), bronchitis (5%), and vomiting (5%).
Grade 3-4 AEs (≥1%) in patients treated with obinutuzumab included neutropenia (17%), pneumonia (3%), and febrile neutropenia (2%). There were 2 deaths due to pneumonia in the obinutuzumab arm.
Health Canada has expanded the approved use of obinutuzumab (Gazyva®).
The anti-CD20 monoclonal antibody is now approved for use in combination with chemotherapy to treat patients with previously untreated follicular lymphoma (FL) that is advanced (stage II bulky, stage III, or stage IV).
In patients who respond to this treatment, obinutuzumab monotherapy can be given as maintenance.
Health Canada previously approved obinutuzumab for the following indications:
- In combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia
- First in combination with bendamustine, then as monotherapy, in FL patients who relapsed after or are refractory to a rituximab-containing regimen.
Phase 3 results
Health Canada’s latest approval of obinutuzumab is based on results from the phase 3 GALLIUM study, which were published in NEJM in October 2017. The following are updated data from the product monograph.
GALLIUM included 1385 patients with previously untreated non-Hodgkin lymphoma, and 1202 of these patients had previously untreated, advanced FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), and bendamustine.
At a median observation time of 41.1 months, the overall response rate was 91% in the obinutuzumab arm and 88% in the rituximab arm. The complete response rates were 28% and 27%, respectively.
The median progression-free survival was not reached in either arm. The hazard ratio, for obinutuzumab compared to rituximab, was 0.72 (95% CI, 0.56-0.93, P=0.0118).
The estimated 3-year progression-free survival was 78.9% in the rituximab arm and 83.4% in the obinutuzumab arm.
Safety was evaluated based on all 1385 patients in the study, 86% of whom had FL and 14% of whom had marginal zone lymphoma.
Serious adverse events (AEs) occurred in 50% of patients in the obinutuzumab arm and 43% in the rituximab arm. Fatal AEs occurred in 5% and 4%, respectively. Infections and second malignancies were the leading causes of these deaths.
During the monotherapy period, the most common AEs (≥ 5%) in patients treated with obinutuzumab were cough (21%), neutropenia (19%), upper respiratory tract infection (15%), viral upper respiratory tract infection (15%), diarrhea (13%), arthralgia (10%), fatigue (9%), sinusitis (9%), infusion reactions (8%), pneumonia (8%), herpes zoster (8%), lower respiratory tract infection (7%), pyrexia (7%), back pain (6%), headache (6%), urinary tract infection (6%), nausea (6%), bronchitis (5%), and vomiting (5%).
Grade 3-4 AEs (≥1%) in patients treated with obinutuzumab included neutropenia (17%), pneumonia (3%), and febrile neutropenia (2%). There were 2 deaths due to pneumonia in the obinutuzumab arm.
Health Canada has expanded the approved use of obinutuzumab (Gazyva®).
The anti-CD20 monoclonal antibody is now approved for use in combination with chemotherapy to treat patients with previously untreated follicular lymphoma (FL) that is advanced (stage II bulky, stage III, or stage IV).
In patients who respond to this treatment, obinutuzumab monotherapy can be given as maintenance.
Health Canada previously approved obinutuzumab for the following indications:
- In combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia
- First in combination with bendamustine, then as monotherapy, in FL patients who relapsed after or are refractory to a rituximab-containing regimen.
Phase 3 results
Health Canada’s latest approval of obinutuzumab is based on results from the phase 3 GALLIUM study, which were published in NEJM in October 2017. The following are updated data from the product monograph.
GALLIUM included 1385 patients with previously untreated non-Hodgkin lymphoma, and 1202 of these patients had previously untreated, advanced FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), and bendamustine.
At a median observation time of 41.1 months, the overall response rate was 91% in the obinutuzumab arm and 88% in the rituximab arm. The complete response rates were 28% and 27%, respectively.
The median progression-free survival was not reached in either arm. The hazard ratio, for obinutuzumab compared to rituximab, was 0.72 (95% CI, 0.56-0.93, P=0.0118).
The estimated 3-year progression-free survival was 78.9% in the rituximab arm and 83.4% in the obinutuzumab arm.
Safety was evaluated based on all 1385 patients in the study, 86% of whom had FL and 14% of whom had marginal zone lymphoma.
Serious adverse events (AEs) occurred in 50% of patients in the obinutuzumab arm and 43% in the rituximab arm. Fatal AEs occurred in 5% and 4%, respectively. Infections and second malignancies were the leading causes of these deaths.
During the monotherapy period, the most common AEs (≥ 5%) in patients treated with obinutuzumab were cough (21%), neutropenia (19%), upper respiratory tract infection (15%), viral upper respiratory tract infection (15%), diarrhea (13%), arthralgia (10%), fatigue (9%), sinusitis (9%), infusion reactions (8%), pneumonia (8%), herpes zoster (8%), lower respiratory tract infection (7%), pyrexia (7%), back pain (6%), headache (6%), urinary tract infection (6%), nausea (6%), bronchitis (5%), and vomiting (5%).
Grade 3-4 AEs (≥1%) in patients treated with obinutuzumab included neutropenia (17%), pneumonia (3%), and febrile neutropenia (2%). There were 2 deaths due to pneumonia in the obinutuzumab arm.
Transplant strategy not viable for aggressive B-NHL
Transplant with radioimmunotherapy (RIT)-based conditioning is a viable treatment option for patients with indolent—but not aggressive—B-cell non-Hodgkin lymphomas (NHLs), according to researchers.
Long-term follow-up data showed “excellent” outcomes in patients with indolent B-NHL who received conditioning with 90Y-ibritumomab tiuxetan plus fludarabine and low-dose total body irradiation (TBI) prior to HLA-matched hematopoietic stem cell transplant (HSCT).
However, long-term outcomes were inferior in patients with diffuse large B-cell lymphoma (DLBCL) and mantle cell lymphoma (MCL).
Camille E. Puronen, MD, of the University of Washington in Seattle, and her colleagues reported these results in Biology of Blood and Marrow Transplantation.
The study enrolled 40 patients with high-risk B-NHL. This included DLBCL (n=14), chronic lymphocytic leukemia (CLL; n=10), MCL (n=8), follicular lymphoma (FL; n=6); hairy cell leukemia (HCL; n=1), and marginal zone lymphoma (MZL; n=1).
Patients were treated with 0.4 mCi/kg 90Y-ibritumomab tiuxetan, given 2 weeks prior to HSCT, to a maximum dose of 32 mCi.
Patients also received fludarabine at 30 mg/m2 on day 5, 6, and 7 prior to HSCT and 2 Gy TBI given on the day of transplant.
In an earlier report, the objective response rate (ORR) was 60%, and 35% of patients had a complete response (CR) or unconfirmed CR.
The researchers said early responses were not associated with disease bulk or chemoresistance, as the ORR was 59% in patients with bulky or chemoresistant disease.
However, responses were associated with histology, as the ORR was 38% in patients with DLBCL, 50% in those with MCL, 83% in those with FL, and 90% in those with CLL.
Long-term survival
In the current report, 11 of 40 patients were still alive at a median follow up of 9 years (range, 5.3 to 10.2). Fourteen patients died of disease progression, and 14 died from complications of HSCT.
The 5-year overall survival (OS) was 40%, and the 5-year progression-free survival (PFS) was 28%.
The best survival rates were in patients with indolent histology. The 5-year PFS was 44% in these patients, and the 5-year OS was 67%.
The researchers said early CR was not associated with long-term survival. However, patients who had at least stable disease (SD) at earlier time points did have the opportunity to achieve long-term survival. All patients who progressed before day 84 were dead by the 1-year mark.
Of the 11 patients who were still alive at a median follow up of 9 years, 4 had a CR or unconfirmed CR at day 84 (FL: 1; CLL: 2; MCL: 1); 6 were in partial response (CLL: 3; FL: 1; MCL: 1; MZL: 1); and 1 patient with FL had SD.
Among the 18 patients with indolent NHL, long-term PFS was observed in 5 of the 7 patients who achieved early CR and 8 of the 11 patients who did not achieve early CR.
Two of the 4 MCL patients who achieved an early CR had long-term PFS, but none of the MCL patients without an early CR had long-term PFS.
Among DLBCL patients, 1 of the 4 who achieved early CR had long-term PFS, but none of the patients without an early CR had long-term PFS. Only 1 DLBCL patient survived beyond 5 years. None survived beyond 8 years.
The researchers said the favorable outcomes in patients with indolent B-NHL are consistent with the known efficacy of RIT and the graft-versus-leukemia effect in these patients.
The team also noted that, since this trial began, several novel agents have been approved for the treatment of indolent B-NHL, which means allogeneic HSCT is often moved to later in the disease course.
The researchers concluded that 90Y-ibritumomab tiuxetan-based conditioning could “continue to play an important role in these settings,” but “improved strategies are needed” for patients with MCL and DLBCL.
Transplant with radioimmunotherapy (RIT)-based conditioning is a viable treatment option for patients with indolent—but not aggressive—B-cell non-Hodgkin lymphomas (NHLs), according to researchers.
Long-term follow-up data showed “excellent” outcomes in patients with indolent B-NHL who received conditioning with 90Y-ibritumomab tiuxetan plus fludarabine and low-dose total body irradiation (TBI) prior to HLA-matched hematopoietic stem cell transplant (HSCT).
However, long-term outcomes were inferior in patients with diffuse large B-cell lymphoma (DLBCL) and mantle cell lymphoma (MCL).
Camille E. Puronen, MD, of the University of Washington in Seattle, and her colleagues reported these results in Biology of Blood and Marrow Transplantation.
The study enrolled 40 patients with high-risk B-NHL. This included DLBCL (n=14), chronic lymphocytic leukemia (CLL; n=10), MCL (n=8), follicular lymphoma (FL; n=6); hairy cell leukemia (HCL; n=1), and marginal zone lymphoma (MZL; n=1).
Patients were treated with 0.4 mCi/kg 90Y-ibritumomab tiuxetan, given 2 weeks prior to HSCT, to a maximum dose of 32 mCi.
Patients also received fludarabine at 30 mg/m2 on day 5, 6, and 7 prior to HSCT and 2 Gy TBI given on the day of transplant.
In an earlier report, the objective response rate (ORR) was 60%, and 35% of patients had a complete response (CR) or unconfirmed CR.
The researchers said early responses were not associated with disease bulk or chemoresistance, as the ORR was 59% in patients with bulky or chemoresistant disease.
However, responses were associated with histology, as the ORR was 38% in patients with DLBCL, 50% in those with MCL, 83% in those with FL, and 90% in those with CLL.
Long-term survival
In the current report, 11 of 40 patients were still alive at a median follow up of 9 years (range, 5.3 to 10.2). Fourteen patients died of disease progression, and 14 died from complications of HSCT.
The 5-year overall survival (OS) was 40%, and the 5-year progression-free survival (PFS) was 28%.
The best survival rates were in patients with indolent histology. The 5-year PFS was 44% in these patients, and the 5-year OS was 67%.
The researchers said early CR was not associated with long-term survival. However, patients who had at least stable disease (SD) at earlier time points did have the opportunity to achieve long-term survival. All patients who progressed before day 84 were dead by the 1-year mark.
Of the 11 patients who were still alive at a median follow up of 9 years, 4 had a CR or unconfirmed CR at day 84 (FL: 1; CLL: 2; MCL: 1); 6 were in partial response (CLL: 3; FL: 1; MCL: 1; MZL: 1); and 1 patient with FL had SD.
Among the 18 patients with indolent NHL, long-term PFS was observed in 5 of the 7 patients who achieved early CR and 8 of the 11 patients who did not achieve early CR.
Two of the 4 MCL patients who achieved an early CR had long-term PFS, but none of the MCL patients without an early CR had long-term PFS.
Among DLBCL patients, 1 of the 4 who achieved early CR had long-term PFS, but none of the patients without an early CR had long-term PFS. Only 1 DLBCL patient survived beyond 5 years. None survived beyond 8 years.
The researchers said the favorable outcomes in patients with indolent B-NHL are consistent with the known efficacy of RIT and the graft-versus-leukemia effect in these patients.
The team also noted that, since this trial began, several novel agents have been approved for the treatment of indolent B-NHL, which means allogeneic HSCT is often moved to later in the disease course.
The researchers concluded that 90Y-ibritumomab tiuxetan-based conditioning could “continue to play an important role in these settings,” but “improved strategies are needed” for patients with MCL and DLBCL.
Transplant with radioimmunotherapy (RIT)-based conditioning is a viable treatment option for patients with indolent—but not aggressive—B-cell non-Hodgkin lymphomas (NHLs), according to researchers.
Long-term follow-up data showed “excellent” outcomes in patients with indolent B-NHL who received conditioning with 90Y-ibritumomab tiuxetan plus fludarabine and low-dose total body irradiation (TBI) prior to HLA-matched hematopoietic stem cell transplant (HSCT).
However, long-term outcomes were inferior in patients with diffuse large B-cell lymphoma (DLBCL) and mantle cell lymphoma (MCL).
Camille E. Puronen, MD, of the University of Washington in Seattle, and her colleagues reported these results in Biology of Blood and Marrow Transplantation.
The study enrolled 40 patients with high-risk B-NHL. This included DLBCL (n=14), chronic lymphocytic leukemia (CLL; n=10), MCL (n=8), follicular lymphoma (FL; n=6); hairy cell leukemia (HCL; n=1), and marginal zone lymphoma (MZL; n=1).
Patients were treated with 0.4 mCi/kg 90Y-ibritumomab tiuxetan, given 2 weeks prior to HSCT, to a maximum dose of 32 mCi.
Patients also received fludarabine at 30 mg/m2 on day 5, 6, and 7 prior to HSCT and 2 Gy TBI given on the day of transplant.
In an earlier report, the objective response rate (ORR) was 60%, and 35% of patients had a complete response (CR) or unconfirmed CR.
The researchers said early responses were not associated with disease bulk or chemoresistance, as the ORR was 59% in patients with bulky or chemoresistant disease.
However, responses were associated with histology, as the ORR was 38% in patients with DLBCL, 50% in those with MCL, 83% in those with FL, and 90% in those with CLL.
Long-term survival
In the current report, 11 of 40 patients were still alive at a median follow up of 9 years (range, 5.3 to 10.2). Fourteen patients died of disease progression, and 14 died from complications of HSCT.
The 5-year overall survival (OS) was 40%, and the 5-year progression-free survival (PFS) was 28%.
The best survival rates were in patients with indolent histology. The 5-year PFS was 44% in these patients, and the 5-year OS was 67%.
The researchers said early CR was not associated with long-term survival. However, patients who had at least stable disease (SD) at earlier time points did have the opportunity to achieve long-term survival. All patients who progressed before day 84 were dead by the 1-year mark.
Of the 11 patients who were still alive at a median follow up of 9 years, 4 had a CR or unconfirmed CR at day 84 (FL: 1; CLL: 2; MCL: 1); 6 were in partial response (CLL: 3; FL: 1; MCL: 1; MZL: 1); and 1 patient with FL had SD.
Among the 18 patients with indolent NHL, long-term PFS was observed in 5 of the 7 patients who achieved early CR and 8 of the 11 patients who did not achieve early CR.
Two of the 4 MCL patients who achieved an early CR had long-term PFS, but none of the MCL patients without an early CR had long-term PFS.
Among DLBCL patients, 1 of the 4 who achieved early CR had long-term PFS, but none of the patients without an early CR had long-term PFS. Only 1 DLBCL patient survived beyond 5 years. None survived beyond 8 years.
The researchers said the favorable outcomes in patients with indolent B-NHL are consistent with the known efficacy of RIT and the graft-versus-leukemia effect in these patients.
The team also noted that, since this trial began, several novel agents have been approved for the treatment of indolent B-NHL, which means allogeneic HSCT is often moved to later in the disease course.
The researchers concluded that 90Y-ibritumomab tiuxetan-based conditioning could “continue to play an important role in these settings,” but “improved strategies are needed” for patients with MCL and DLBCL.
Rapid venetoclax dose escalation aids relapsed CLL
STOCKHOLM – Patients with chronic lymphocytic leukemia (CLL) who experience relapse after therapy with a B-cell receptor signaling inhibitor tend to have a swiftly progressive disease course that requires immediate intervention. For these patients, a rapid venetoclax dose-escalation protocol may be a safe way to quickly regain disease control, and possibly bridge to salvage therapies, investigators reported.
Of 15 patients with CLL who relapsed after treatment with a B-cell receptor inhibitor (BCRi), all were able to get to their target dose of venetoclax under close inpatient monitoring at a median of 12 days, compared with the 35 days usually required for venetoclax dose escalation, reported Farrukh T. Awan, MD, of Ohio State University Comprehensive Cancer Center in Columbus, and his colleagues.
Only two patients developed clinical tumor lysis syndrome (TLS), a common occurrence with venetoclax therapy, and this adverse event was manageable, Dr. Awan said at the annual congress of the European Hematology Association.
“The reason why we have been doing a slow ramp up on venetoclax is the original toxicity issues that we saw early on,” he said in an interview. “But unfortunately, a lot of patients are progressing on these new agents and have very rapid disease progression, and what we have seen is that if you stop the ibrutinib, the disease progresses very quickly, and by the time they can get up to the effective dose of venetoclax, they’re too sick to continue, or they might even die from disease progression.”

To combat this problem, Dr. Awan and his colleagues developed a rapid dose escalation protocol that would ramp up from 20 mg to 400 mg, with increases every 1 or 2 days depending on tolerability and incident TLS. Lab tests for TLS were evaluated every 4-8 hours.
All patients were closely monitored in the hospital, and all were started on allopurinol or other uric acid–lowering agents before starting on venetoclax.
The investigators reported safety and efficacy outcomes for the patients in a retrospective analysis.
The median age of the patients, 12 men and 3 women, was 65 years (range, 58-86 years). Seven patients had Eastern Cooperative Oncology Group Performance Status of 0, seven had an ECOG score of 1, and one had a score of 2-4.
Ten patients had most recently been treated with a BCRi, either a Bruton’s tyrosine kinase inhibitor (ibrutinib or acalabrutinib), idelalisib, or entospletinib. Three patients received ibrutinib plus chemotherapy, and two received rituximab and dexamethasone followed by rituximab maintenance.
The median time to full venetoclax dose was 12 days (range, 5-21 days) and all 15 patients reached the target dose. The mean length of stay during the ramp-up period was 9.5 days (range, 6-22 days).
The incidence of clinical TLS was 13.2%, occurring in two patients, one at the initial 20-mg dose, and one at the 200-mg dose level. Another five patients had asymptomatic TLS. Other treatment-related adverse events were anemia in seven patients, neutropenia in six patients, thrombocytopenia in five patients, and lung infection in one patient.
Twelve patients had a partial response, one had stable disease, and two had progressive disease. The mean time to best response was 71 days.
One-year progression-free survival was 49%, and 1-year overall survival was 68%.
The investigators found that for patients who still have some disease control with a BCRi, it may be possible to keep them on that drug while transitioning to venetoclax. The rapid dose escalation protocol should only be attempted in highly experience comprehensive cancer centers, Dr. Awan said.
“Under very close monitoring in an experienced inpatient setting, where the nurses are very used to doing this on a weekly basis in a very high volume center, I think that our data show that we could do this without affecting toxicity significantly or mortality,” he said.
Venetoclax therapy could buy enough time for patients to bridge to other options, such as chimeric antigen receptor (CAR) T-cell therapy or allogeneic stem cell transplant, he noted.
“But if we had waited 4 weeks, most of these patients would not have made it,” he said.
The study was internally funded. Dr. Awan reported research funding from Gilead, Pharmacyclics, AbbVie, and Janssen.
SOURCE: Koenig K et al. EHA Congress, Abstract PF357.
STOCKHOLM – Patients with chronic lymphocytic leukemia (CLL) who experience relapse after therapy with a B-cell receptor signaling inhibitor tend to have a swiftly progressive disease course that requires immediate intervention. For these patients, a rapid venetoclax dose-escalation protocol may be a safe way to quickly regain disease control, and possibly bridge to salvage therapies, investigators reported.
Of 15 patients with CLL who relapsed after treatment with a B-cell receptor inhibitor (BCRi), all were able to get to their target dose of venetoclax under close inpatient monitoring at a median of 12 days, compared with the 35 days usually required for venetoclax dose escalation, reported Farrukh T. Awan, MD, of Ohio State University Comprehensive Cancer Center in Columbus, and his colleagues.
Only two patients developed clinical tumor lysis syndrome (TLS), a common occurrence with venetoclax therapy, and this adverse event was manageable, Dr. Awan said at the annual congress of the European Hematology Association.
“The reason why we have been doing a slow ramp up on venetoclax is the original toxicity issues that we saw early on,” he said in an interview. “But unfortunately, a lot of patients are progressing on these new agents and have very rapid disease progression, and what we have seen is that if you stop the ibrutinib, the disease progresses very quickly, and by the time they can get up to the effective dose of venetoclax, they’re too sick to continue, or they might even die from disease progression.”
To combat this problem, Dr. Awan and his colleagues developed a rapid dose escalation protocol that would ramp up from 20 mg to 400 mg, with increases every 1 or 2 days depending on tolerability and incident TLS. Lab tests for TLS were evaluated every 4-8 hours.
All patients were closely monitored in the hospital, and all were started on allopurinol or other uric acid–lowering agents before starting on venetoclax.
The investigators reported safety and efficacy outcomes for the patients in a retrospective analysis.
The median age of the patients, 12 men and 3 women, was 65 years (range, 58-86 years). Seven patients had Eastern Cooperative Oncology Group Performance Status of 0, seven had an ECOG score of 1, and one had a score of 2-4.
Ten patients had most recently been treated with a BCRi, either a Bruton’s tyrosine kinase inhibitor (ibrutinib or acalabrutinib), idelalisib, or entospletinib. Three patients received ibrutinib plus chemotherapy, and two received rituximab and dexamethasone followed by rituximab maintenance.
The median time to full venetoclax dose was 12 days (range, 5-21 days) and all 15 patients reached the target dose. The mean length of stay during the ramp-up period was 9.5 days (range, 6-22 days).
The incidence of clinical TLS was 13.2%, occurring in two patients, one at the initial 20-mg dose, and one at the 200-mg dose level. Another five patients had asymptomatic TLS. Other treatment-related adverse events were anemia in seven patients, neutropenia in six patients, thrombocytopenia in five patients, and lung infection in one patient.
Twelve patients had a partial response, one had stable disease, and two had progressive disease. The mean time to best response was 71 days.
One-year progression-free survival was 49%, and 1-year overall survival was 68%.
The investigators found that for patients who still have some disease control with a BCRi, it may be possible to keep them on that drug while transitioning to venetoclax. The rapid dose escalation protocol should only be attempted in highly experience comprehensive cancer centers, Dr. Awan said.
“Under very close monitoring in an experienced inpatient setting, where the nurses are very used to doing this on a weekly basis in a very high volume center, I think that our data show that we could do this without affecting toxicity significantly or mortality,” he said.
Venetoclax therapy could buy enough time for patients to bridge to other options, such as chimeric antigen receptor (CAR) T-cell therapy or allogeneic stem cell transplant, he noted.
“But if we had waited 4 weeks, most of these patients would not have made it,” he said.
The study was internally funded. Dr. Awan reported research funding from Gilead, Pharmacyclics, AbbVie, and Janssen.
SOURCE: Koenig K et al. EHA Congress, Abstract PF357.
STOCKHOLM – Patients with chronic lymphocytic leukemia (CLL) who experience relapse after therapy with a B-cell receptor signaling inhibitor tend to have a swiftly progressive disease course that requires immediate intervention. For these patients, a rapid venetoclax dose-escalation protocol may be a safe way to quickly regain disease control, and possibly bridge to salvage therapies, investigators reported.
Of 15 patients with CLL who relapsed after treatment with a B-cell receptor inhibitor (BCRi), all were able to get to their target dose of venetoclax under close inpatient monitoring at a median of 12 days, compared with the 35 days usually required for venetoclax dose escalation, reported Farrukh T. Awan, MD, of Ohio State University Comprehensive Cancer Center in Columbus, and his colleagues.
Only two patients developed clinical tumor lysis syndrome (TLS), a common occurrence with venetoclax therapy, and this adverse event was manageable, Dr. Awan said at the annual congress of the European Hematology Association.
“The reason why we have been doing a slow ramp up on venetoclax is the original toxicity issues that we saw early on,” he said in an interview. “But unfortunately, a lot of patients are progressing on these new agents and have very rapid disease progression, and what we have seen is that if you stop the ibrutinib, the disease progresses very quickly, and by the time they can get up to the effective dose of venetoclax, they’re too sick to continue, or they might even die from disease progression.”
To combat this problem, Dr. Awan and his colleagues developed a rapid dose escalation protocol that would ramp up from 20 mg to 400 mg, with increases every 1 or 2 days depending on tolerability and incident TLS. Lab tests for TLS were evaluated every 4-8 hours.
All patients were closely monitored in the hospital, and all were started on allopurinol or other uric acid–lowering agents before starting on venetoclax.
The investigators reported safety and efficacy outcomes for the patients in a retrospective analysis.
The median age of the patients, 12 men and 3 women, was 65 years (range, 58-86 years). Seven patients had Eastern Cooperative Oncology Group Performance Status of 0, seven had an ECOG score of 1, and one had a score of 2-4.
Ten patients had most recently been treated with a BCRi, either a Bruton’s tyrosine kinase inhibitor (ibrutinib or acalabrutinib), idelalisib, or entospletinib. Three patients received ibrutinib plus chemotherapy, and two received rituximab and dexamethasone followed by rituximab maintenance.
The median time to full venetoclax dose was 12 days (range, 5-21 days) and all 15 patients reached the target dose. The mean length of stay during the ramp-up period was 9.5 days (range, 6-22 days).
The incidence of clinical TLS was 13.2%, occurring in two patients, one at the initial 20-mg dose, and one at the 200-mg dose level. Another five patients had asymptomatic TLS. Other treatment-related adverse events were anemia in seven patients, neutropenia in six patients, thrombocytopenia in five patients, and lung infection in one patient.
Twelve patients had a partial response, one had stable disease, and two had progressive disease. The mean time to best response was 71 days.
One-year progression-free survival was 49%, and 1-year overall survival was 68%.
The investigators found that for patients who still have some disease control with a BCRi, it may be possible to keep them on that drug while transitioning to venetoclax. The rapid dose escalation protocol should only be attempted in highly experience comprehensive cancer centers, Dr. Awan said.
“Under very close monitoring in an experienced inpatient setting, where the nurses are very used to doing this on a weekly basis in a very high volume center, I think that our data show that we could do this without affecting toxicity significantly or mortality,” he said.
Venetoclax therapy could buy enough time for patients to bridge to other options, such as chimeric antigen receptor (CAR) T-cell therapy or allogeneic stem cell transplant, he noted.
“But if we had waited 4 weeks, most of these patients would not have made it,” he said.
The study was internally funded. Dr. Awan reported research funding from Gilead, Pharmacyclics, AbbVie, and Janssen.
SOURCE: Koenig K et al. EHA Congress, Abstract PF357.
REPORTING FROM THE EHA CONGRESS
Key clinical point:
Major finding: All patients reached the target dose of venetoclax, with only two cases of manageable clinical tumor lysis syndrome.
Study details: Retrospective analysis of outcomes for 15 patients with CLL who relapsed after treatment with a B-cell receptor signaling inhibitor.
Disclosures: The study was internally funded. Dr. Awan reported research funding from Gilead, Pharmacyclics, AbbVie, and Janssen.
Source: Koenig K et al. EHA Congress, Abstract PF357.
JAK inhibition linked to B-cell lymphoma

New research indicates that JAK inhibitors may increase the risk of lymphoma in patients with myelofibrosis (MF).
The patients studied had a 15- to 25-fold higher risk of developing B-cell lymphoma if they received treatment with JAK inhibitors.
The researchers speculate that screening MF patients for a pre-existing B-cell clone before starting JAK inhibitor therapy may help prevent lymphoma development.
Heinz Gisslinger, MD, of the Medical University of Vienna in Austria, and his colleagues conducted this research and reported the findings in Blood.
“[W]e started noticing sporadic cases of lymphomas developing in patients being treated for myeloproliferative neoplasms and wanted to know if this phenomenon was connected to treatment,” Dr Gisslinger said.
Therefore, he and his colleagues assessed 626 patients receiving treatment for myeloproliferative neoplasms (MPNs) at the Medical University of Vienna.
The incidence of B-cell lymphoma was 5.8% (4/69) in patients treated with JAK inhibitors and 0.36% (2/557) in patients who did not receive JAK inhibitors. That amounts to a 16-fold increased risk of lymphoma in patients receiving JAK inhibitors.
When the researchers analyzed only patients with primary MF (n=216), the increased risk of B-cell lymphoma was even greater. The incidence of lymphoma was 9.68% (3/31) in patients treated with JAK inhibitors and 0.54% (1/185) in patients who did not receive JAK inhibitors.
That corresponds to a 19-fold increased risk of B-cell lymphoma in primary MF patients treated with JAK inhibitors. When the researchers adjusted for age, there was a 21-fold greater risk. When they adjusted for sex, the risk was 25 times higher.
In a second cohort of 929 MPN patients, the incidence of B-cell lymphoma was 3.51% (2/57) in patients who received JAK inhibitors and 0.23% (2/872) in patients who did not. This corresponds to a 15-fold increased risk of lymphoma in the JAK inhibitor recipients.
Lymphoma cases
In all, there were 6 patients who developed lymphoma after JAK inhibitor treatment. Five developed diffuse large B-cell lymphoma, and 1 had high-grade B-cell lymphoma not otherwise specified.
Four of the patients had primary MF, 1 had post-polycythemia vera MF, and 1 had post-essential thrombocythemia (ET) MF. Five patients had a JAK2V617F mutation, and 1 (the post-ET MF patient) had a CALR mutation.
All 6 patients had received treatment with ruxolitinib. One patient also received fedratinib.
B-cell clone
The researchers studied bone marrow samples from 54 of the 69 patients treated with JAK inhibitors in the first cohort. The team found a pre-existing B-cell clone in 3 of the 4 patients who developed lymphoma. Further investigation suggested this was the clone that later transformed into lymphoma.
The researchers also found an association between JAK inhibition and an increased frequency of aggressive B-cell lymphomas in mouse models.
“By replicating this link between this B-cell clone and aggressive lymphoma, we hope to speed the discovery of an alternative therapy for myelofibrosis,” said study author Veronica Sexl, MD, of the University of Veterinary Medicine in Vienna. “These findings are going to be valuable in clinical care.”
“We determined that patients with this pre-existing B-cell clone in their bone marrow are most at risk for developing aggressive lymphoma,” added study author Ulrich Jäger, MD, of the Medical University of Vienna.
“We also know that up to 16% of people with myelofibrosis have immunoglobulin gene rearrangements like this B-cell clone. Therefore, our findings suggest that all patients with myelofibrosis should be tested for such gene rearrangements before prescribing JAK inhibitors to treat their disease.”
New research indicates that JAK inhibitors may increase the risk of lymphoma in patients with myelofibrosis (MF).
The patients studied had a 15- to 25-fold higher risk of developing B-cell lymphoma if they received treatment with JAK inhibitors.
The researchers speculate that screening MF patients for a pre-existing B-cell clone before starting JAK inhibitor therapy may help prevent lymphoma development.
Heinz Gisslinger, MD, of the Medical University of Vienna in Austria, and his colleagues conducted this research and reported the findings in Blood.
“[W]e started noticing sporadic cases of lymphomas developing in patients being treated for myeloproliferative neoplasms and wanted to know if this phenomenon was connected to treatment,” Dr Gisslinger said.
Therefore, he and his colleagues assessed 626 patients receiving treatment for myeloproliferative neoplasms (MPNs) at the Medical University of Vienna.
The incidence of B-cell lymphoma was 5.8% (4/69) in patients treated with JAK inhibitors and 0.36% (2/557) in patients who did not receive JAK inhibitors. That amounts to a 16-fold increased risk of lymphoma in patients receiving JAK inhibitors.
When the researchers analyzed only patients with primary MF (n=216), the increased risk of B-cell lymphoma was even greater. The incidence of lymphoma was 9.68% (3/31) in patients treated with JAK inhibitors and 0.54% (1/185) in patients who did not receive JAK inhibitors.
That corresponds to a 19-fold increased risk of B-cell lymphoma in primary MF patients treated with JAK inhibitors. When the researchers adjusted for age, there was a 21-fold greater risk. When they adjusted for sex, the risk was 25 times higher.
In a second cohort of 929 MPN patients, the incidence of B-cell lymphoma was 3.51% (2/57) in patients who received JAK inhibitors and 0.23% (2/872) in patients who did not. This corresponds to a 15-fold increased risk of lymphoma in the JAK inhibitor recipients.
Lymphoma cases
In all, there were 6 patients who developed lymphoma after JAK inhibitor treatment. Five developed diffuse large B-cell lymphoma, and 1 had high-grade B-cell lymphoma not otherwise specified.
Four of the patients had primary MF, 1 had post-polycythemia vera MF, and 1 had post-essential thrombocythemia (ET) MF. Five patients had a JAK2V617F mutation, and 1 (the post-ET MF patient) had a CALR mutation.
All 6 patients had received treatment with ruxolitinib. One patient also received fedratinib.
B-cell clone
The researchers studied bone marrow samples from 54 of the 69 patients treated with JAK inhibitors in the first cohort. The team found a pre-existing B-cell clone in 3 of the 4 patients who developed lymphoma. Further investigation suggested this was the clone that later transformed into lymphoma.
The researchers also found an association between JAK inhibition and an increased frequency of aggressive B-cell lymphomas in mouse models.
“By replicating this link between this B-cell clone and aggressive lymphoma, we hope to speed the discovery of an alternative therapy for myelofibrosis,” said study author Veronica Sexl, MD, of the University of Veterinary Medicine in Vienna. “These findings are going to be valuable in clinical care.”
“We determined that patients with this pre-existing B-cell clone in their bone marrow are most at risk for developing aggressive lymphoma,” added study author Ulrich Jäger, MD, of the Medical University of Vienna.
“We also know that up to 16% of people with myelofibrosis have immunoglobulin gene rearrangements like this B-cell clone. Therefore, our findings suggest that all patients with myelofibrosis should be tested for such gene rearrangements before prescribing JAK inhibitors to treat their disease.”
New research indicates that JAK inhibitors may increase the risk of lymphoma in patients with myelofibrosis (MF).
The patients studied had a 15- to 25-fold higher risk of developing B-cell lymphoma if they received treatment with JAK inhibitors.
The researchers speculate that screening MF patients for a pre-existing B-cell clone before starting JAK inhibitor therapy may help prevent lymphoma development.
Heinz Gisslinger, MD, of the Medical University of Vienna in Austria, and his colleagues conducted this research and reported the findings in Blood.
“[W]e started noticing sporadic cases of lymphomas developing in patients being treated for myeloproliferative neoplasms and wanted to know if this phenomenon was connected to treatment,” Dr Gisslinger said.
Therefore, he and his colleagues assessed 626 patients receiving treatment for myeloproliferative neoplasms (MPNs) at the Medical University of Vienna.
The incidence of B-cell lymphoma was 5.8% (4/69) in patients treated with JAK inhibitors and 0.36% (2/557) in patients who did not receive JAK inhibitors. That amounts to a 16-fold increased risk of lymphoma in patients receiving JAK inhibitors.
When the researchers analyzed only patients with primary MF (n=216), the increased risk of B-cell lymphoma was even greater. The incidence of lymphoma was 9.68% (3/31) in patients treated with JAK inhibitors and 0.54% (1/185) in patients who did not receive JAK inhibitors.
That corresponds to a 19-fold increased risk of B-cell lymphoma in primary MF patients treated with JAK inhibitors. When the researchers adjusted for age, there was a 21-fold greater risk. When they adjusted for sex, the risk was 25 times higher.
In a second cohort of 929 MPN patients, the incidence of B-cell lymphoma was 3.51% (2/57) in patients who received JAK inhibitors and 0.23% (2/872) in patients who did not. This corresponds to a 15-fold increased risk of lymphoma in the JAK inhibitor recipients.
Lymphoma cases
In all, there were 6 patients who developed lymphoma after JAK inhibitor treatment. Five developed diffuse large B-cell lymphoma, and 1 had high-grade B-cell lymphoma not otherwise specified.
Four of the patients had primary MF, 1 had post-polycythemia vera MF, and 1 had post-essential thrombocythemia (ET) MF. Five patients had a JAK2V617F mutation, and 1 (the post-ET MF patient) had a CALR mutation.
All 6 patients had received treatment with ruxolitinib. One patient also received fedratinib.
B-cell clone
The researchers studied bone marrow samples from 54 of the 69 patients treated with JAK inhibitors in the first cohort. The team found a pre-existing B-cell clone in 3 of the 4 patients who developed lymphoma. Further investigation suggested this was the clone that later transformed into lymphoma.
The researchers also found an association between JAK inhibition and an increased frequency of aggressive B-cell lymphomas in mouse models.
“By replicating this link between this B-cell clone and aggressive lymphoma, we hope to speed the discovery of an alternative therapy for myelofibrosis,” said study author Veronica Sexl, MD, of the University of Veterinary Medicine in Vienna. “These findings are going to be valuable in clinical care.”
“We determined that patients with this pre-existing B-cell clone in their bone marrow are most at risk for developing aggressive lymphoma,” added study author Ulrich Jäger, MD, of the Medical University of Vienna.
“We also know that up to 16% of people with myelofibrosis have immunoglobulin gene rearrangements like this B-cell clone. Therefore, our findings suggest that all patients with myelofibrosis should be tested for such gene rearrangements before prescribing JAK inhibitors to treat their disease.”
CHMP recommends CAR T for ALL, DLBCL

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended the approval of tisagenlecleucel (Kymriah®, formerly CTL019) for 2 indications.
According to the CHMP, the chimeric antigen receptor (CAR) T-cell therapy should be approved to treat adults with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) who have received 2 or more lines of systemic therapy and patients up to 25 years of age who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post-transplant, or in second or later relapse.
The CHMP’s recommendation will be reviewed by the European Commission, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The European Commission usually makes a decision within 67 days of the CHMP’s recommendation.
The CHMP’s recommendation is based on results from a pair of phase 2 trials—ELIANA and JULIET.
JULIET trial
Updated results from JULIET were presented at the recent 23rd Annual Congress of the European Hematology Association (EHA) as abstract S799.
The trial enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Most of the patients who discontinued before dosing did so due to disease progression or clinical deterioration. The patients’ median age at baseline was 56 (range, 22-76).
Ninety-two percent of patients received bridging therapy, and 93% received lymphodepleting chemotherapy prior to tisagenlecleucel.
The median time from infusion to data cutoff was 13.9 months.
The overall response rate was 52%, and the complete response (CR) rate was 40%. Of the patients in CR at month 3, 83% remained in CR at month 12. The median duration of response was not reached.
At the time of data cutoff, none of the responders had proceeded to stem cell transplant.
For all infused patients (n=111), the 12-month overall survival (OS) rate was 49%, and the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome (CRS). Fifteen percent of patients received tocilizumab for CRS, including 3% of patients with grade 2 CRS and 50% of patients with grade 3 CRS.
Other adverse events (AEs) of interest included grade 3/4 neurologic events (12%), grade 3/4 cytopenias lasting more than 28 days (32%), grade 3/4 infections (20%), and grade 3/4 febrile neutropenia (15%).
ELIANA trial
Updated results from ELIANA were published in NEJM in February.
The trial included 75 children and young adults with relapsed/refractory ALL. The patients’ median age was 11 (range, 3 to 23).
All 75 patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The study’s primary endpoint was overall remission rate, which was defined as the rate of a best overall response of either CR or CR with incomplete hematologic recovery (CRi) within 3 months.
The overall remission rate was 81% (61/75), with 60% of patients (n=45) achieving a CR and 21% (n=16) achieving a CRi.
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to transplant while in remission. At last follow-up, 4 were still in remission, and 4 had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
All patients experienced at least 1 AE, and 95% had AEs thought to be related to tisagenlecleucel. The rate of grade 3/4 AEs was 88%, and the rate of related grade 3/4 AEs was 73%.
AEs of special interest included CRS (77%), neurologic events (40%), infections (43%), febrile neutropenia (35%), cytopenias not resolved by day 28 (37%), and tumor lysis syndrome (4%).
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended the approval of tisagenlecleucel (Kymriah®, formerly CTL019) for 2 indications.
According to the CHMP, the chimeric antigen receptor (CAR) T-cell therapy should be approved to treat adults with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) who have received 2 or more lines of systemic therapy and patients up to 25 years of age who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post-transplant, or in second or later relapse.
The CHMP’s recommendation will be reviewed by the European Commission, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The European Commission usually makes a decision within 67 days of the CHMP’s recommendation.
The CHMP’s recommendation is based on results from a pair of phase 2 trials—ELIANA and JULIET.
JULIET trial
Updated results from JULIET were presented at the recent 23rd Annual Congress of the European Hematology Association (EHA) as abstract S799.
The trial enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Most of the patients who discontinued before dosing did so due to disease progression or clinical deterioration. The patients’ median age at baseline was 56 (range, 22-76).
Ninety-two percent of patients received bridging therapy, and 93% received lymphodepleting chemotherapy prior to tisagenlecleucel.
The median time from infusion to data cutoff was 13.9 months.
The overall response rate was 52%, and the complete response (CR) rate was 40%. Of the patients in CR at month 3, 83% remained in CR at month 12. The median duration of response was not reached.
At the time of data cutoff, none of the responders had proceeded to stem cell transplant.
For all infused patients (n=111), the 12-month overall survival (OS) rate was 49%, and the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome (CRS). Fifteen percent of patients received tocilizumab for CRS, including 3% of patients with grade 2 CRS and 50% of patients with grade 3 CRS.
Other adverse events (AEs) of interest included grade 3/4 neurologic events (12%), grade 3/4 cytopenias lasting more than 28 days (32%), grade 3/4 infections (20%), and grade 3/4 febrile neutropenia (15%).
ELIANA trial
Updated results from ELIANA were published in NEJM in February.
The trial included 75 children and young adults with relapsed/refractory ALL. The patients’ median age was 11 (range, 3 to 23).
All 75 patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The study’s primary endpoint was overall remission rate, which was defined as the rate of a best overall response of either CR or CR with incomplete hematologic recovery (CRi) within 3 months.
The overall remission rate was 81% (61/75), with 60% of patients (n=45) achieving a CR and 21% (n=16) achieving a CRi.
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to transplant while in remission. At last follow-up, 4 were still in remission, and 4 had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
All patients experienced at least 1 AE, and 95% had AEs thought to be related to tisagenlecleucel. The rate of grade 3/4 AEs was 88%, and the rate of related grade 3/4 AEs was 73%.
AEs of special interest included CRS (77%), neurologic events (40%), infections (43%), febrile neutropenia (35%), cytopenias not resolved by day 28 (37%), and tumor lysis syndrome (4%).
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended the approval of tisagenlecleucel (Kymriah®, formerly CTL019) for 2 indications.
According to the CHMP, the chimeric antigen receptor (CAR) T-cell therapy should be approved to treat adults with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) who have received 2 or more lines of systemic therapy and patients up to 25 years of age who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post-transplant, or in second or later relapse.
The CHMP’s recommendation will be reviewed by the European Commission, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The European Commission usually makes a decision within 67 days of the CHMP’s recommendation.
The CHMP’s recommendation is based on results from a pair of phase 2 trials—ELIANA and JULIET.
JULIET trial
Updated results from JULIET were presented at the recent 23rd Annual Congress of the European Hematology Association (EHA) as abstract S799.
The trial enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Most of the patients who discontinued before dosing did so due to disease progression or clinical deterioration. The patients’ median age at baseline was 56 (range, 22-76).
Ninety-two percent of patients received bridging therapy, and 93% received lymphodepleting chemotherapy prior to tisagenlecleucel.
The median time from infusion to data cutoff was 13.9 months.
The overall response rate was 52%, and the complete response (CR) rate was 40%. Of the patients in CR at month 3, 83% remained in CR at month 12. The median duration of response was not reached.
At the time of data cutoff, none of the responders had proceeded to stem cell transplant.
For all infused patients (n=111), the 12-month overall survival (OS) rate was 49%, and the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome (CRS). Fifteen percent of patients received tocilizumab for CRS, including 3% of patients with grade 2 CRS and 50% of patients with grade 3 CRS.
Other adverse events (AEs) of interest included grade 3/4 neurologic events (12%), grade 3/4 cytopenias lasting more than 28 days (32%), grade 3/4 infections (20%), and grade 3/4 febrile neutropenia (15%).
ELIANA trial
Updated results from ELIANA were published in NEJM in February.
The trial included 75 children and young adults with relapsed/refractory ALL. The patients’ median age was 11 (range, 3 to 23).
All 75 patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The study’s primary endpoint was overall remission rate, which was defined as the rate of a best overall response of either CR or CR with incomplete hematologic recovery (CRi) within 3 months.
The overall remission rate was 81% (61/75), with 60% of patients (n=45) achieving a CR and 21% (n=16) achieving a CRi.
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to transplant while in remission. At last follow-up, 4 were still in remission, and 4 had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
All patients experienced at least 1 AE, and 95% had AEs thought to be related to tisagenlecleucel. The rate of grade 3/4 AEs was 88%, and the rate of related grade 3/4 AEs was 73%.
AEs of special interest included CRS (77%), neurologic events (40%), infections (43%), febrile neutropenia (35%), cytopenias not resolved by day 28 (37%), and tumor lysis syndrome (4%).
CHMP recommends CAR T for DLBCL, PMBCL

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for the chimeric antigen receptor (CAR) T-cell therapy axicabtagene ciloleucel (Yescarta®, formerly KTE-C19).
The recommendation pertains to axicabtagene ciloleucel as a treatment for adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) or primary mediastinal large B-cell lymphoma (PMBCL) who have received 2 or more lines of systemic therapy.
The CHMP’s recommendation will be reviewed by the European Commission, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The European Commission usually makes a decision within 67 days of the CHMP’s recommendation.
The marketing authorization application for axicabtagene ciloleucel is supported by data from the ZUMA-1 trial.
Results from this phase 2 trial were presented at the 2017 ASH Annual Meeting and published simultaneously in NEJM.
The trial enrolled 111 patients with relapsed/refractory B-cell lymphomas. There were 101 patients who received axicabtagene ciloleucel—77 with DLBCL, 8 with PMBCL, and 16 with transformed follicular lymphoma (TFL).
Patients received conditioning with low-dose cyclophosphamide and fludarabine, followed by axicabtagene ciloleucel.
The objective response rate (ORR) was 82% (n=83), and the complete response (CR) rate was 54% (n=55).
Among the DLBCL patients, the ORR was 82% (63/77), and the CR rate was 49% (38/77). In the patients with PMBCL or TFL, the ORR was 83% (20/24), and the CR rate was 71% (17/24).
With a median follow-up of 15.4 months, 42% of patients retained their response, and 40% retained a CR.
At 18 months, the overall survival was 52%. Most deaths were due to disease progression.
However, 2 patients died of adverse events related to axicabtagene ciloleucel, both cytokine release syndrome (CRS).
The most common grade 3 or higher adverse events were neutropenia (78%), anemia (43%), thrombocytopenia (38%), and febrile neutropenia (31%).
Grade 3 or higher CRS occurred in 13% of patients, and grade 3 or higher neurologic events occurred in 28%.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for the chimeric antigen receptor (CAR) T-cell therapy axicabtagene ciloleucel (Yescarta®, formerly KTE-C19).
The recommendation pertains to axicabtagene ciloleucel as a treatment for adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) or primary mediastinal large B-cell lymphoma (PMBCL) who have received 2 or more lines of systemic therapy.
The CHMP’s recommendation will be reviewed by the European Commission, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The European Commission usually makes a decision within 67 days of the CHMP’s recommendation.
The marketing authorization application for axicabtagene ciloleucel is supported by data from the ZUMA-1 trial.
Results from this phase 2 trial were presented at the 2017 ASH Annual Meeting and published simultaneously in NEJM.
The trial enrolled 111 patients with relapsed/refractory B-cell lymphomas. There were 101 patients who received axicabtagene ciloleucel—77 with DLBCL, 8 with PMBCL, and 16 with transformed follicular lymphoma (TFL).
Patients received conditioning with low-dose cyclophosphamide and fludarabine, followed by axicabtagene ciloleucel.
The objective response rate (ORR) was 82% (n=83), and the complete response (CR) rate was 54% (n=55).
Among the DLBCL patients, the ORR was 82% (63/77), and the CR rate was 49% (38/77). In the patients with PMBCL or TFL, the ORR was 83% (20/24), and the CR rate was 71% (17/24).
With a median follow-up of 15.4 months, 42% of patients retained their response, and 40% retained a CR.
At 18 months, the overall survival was 52%. Most deaths were due to disease progression.
However, 2 patients died of adverse events related to axicabtagene ciloleucel, both cytokine release syndrome (CRS).
The most common grade 3 or higher adverse events were neutropenia (78%), anemia (43%), thrombocytopenia (38%), and febrile neutropenia (31%).
Grade 3 or higher CRS occurred in 13% of patients, and grade 3 or higher neurologic events occurred in 28%.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for the chimeric antigen receptor (CAR) T-cell therapy axicabtagene ciloleucel (Yescarta®, formerly KTE-C19).
The recommendation pertains to axicabtagene ciloleucel as a treatment for adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) or primary mediastinal large B-cell lymphoma (PMBCL) who have received 2 or more lines of systemic therapy.
The CHMP’s recommendation will be reviewed by the European Commission, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein.
The European Commission usually makes a decision within 67 days of the CHMP’s recommendation.
The marketing authorization application for axicabtagene ciloleucel is supported by data from the ZUMA-1 trial.
Results from this phase 2 trial were presented at the 2017 ASH Annual Meeting and published simultaneously in NEJM.
The trial enrolled 111 patients with relapsed/refractory B-cell lymphomas. There were 101 patients who received axicabtagene ciloleucel—77 with DLBCL, 8 with PMBCL, and 16 with transformed follicular lymphoma (TFL).
Patients received conditioning with low-dose cyclophosphamide and fludarabine, followed by axicabtagene ciloleucel.
The objective response rate (ORR) was 82% (n=83), and the complete response (CR) rate was 54% (n=55).
Among the DLBCL patients, the ORR was 82% (63/77), and the CR rate was 49% (38/77). In the patients with PMBCL or TFL, the ORR was 83% (20/24), and the CR rate was 71% (17/24).
With a median follow-up of 15.4 months, 42% of patients retained their response, and 40% retained a CR.
At 18 months, the overall survival was 52%. Most deaths were due to disease progression.
However, 2 patients died of adverse events related to axicabtagene ciloleucel, both cytokine release syndrome (CRS).
The most common grade 3 or higher adverse events were neutropenia (78%), anemia (43%), thrombocytopenia (38%), and febrile neutropenia (31%).
Grade 3 or higher CRS occurred in 13% of patients, and grade 3 or higher neurologic events occurred in 28%.
European Medicines Agency recommends CAR T-cell approvals
The European Medicines Agency (EMA) has recommended a handful of hematology medications for approval, including two chimeric antigen receptor (CAR) T-cell therapies.
All of the drugs must next be approved by the European Commission in order to be marketed to patients throughout Europe.
At the end of June, the EMA’s Committee for Medicinal Products for Human Use tisagenlecleucel (Kymriah) and axicabtagene ciloleucel (Yescarta).
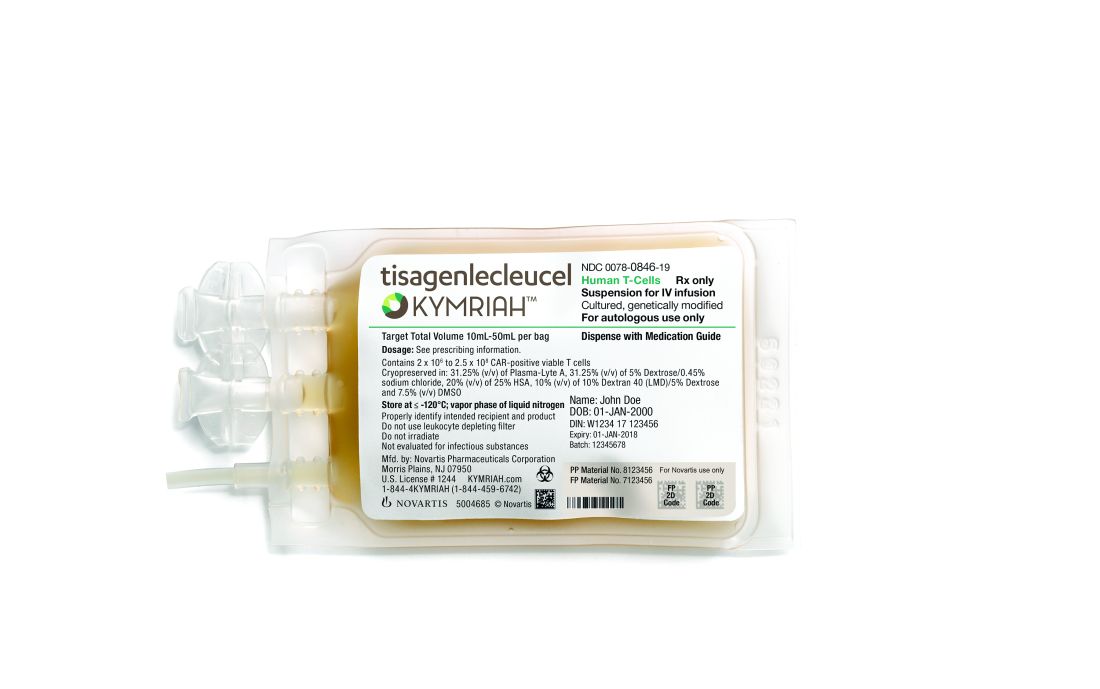
The EMA approval recommendations come with risk management measures to address the potential for cytokine release syndrome with both of these treatments. Drug makers must use a patient registry to monitor the long-term safety and efficacy of the therapies.
The EMA is also recommending approval of caplacizumab for acquired thrombotic thrombocytopenic purpura, vonicog alfa for the treatment of von Willebrand disease, and daunorubicin/cytarabine for the treatment of acute myeloid leukemia.
The European Medicines Agency (EMA) has recommended a handful of hematology medications for approval, including two chimeric antigen receptor (CAR) T-cell therapies.
All of the drugs must next be approved by the European Commission in order to be marketed to patients throughout Europe.
At the end of June, the EMA’s Committee for Medicinal Products for Human Use tisagenlecleucel (Kymriah) and axicabtagene ciloleucel (Yescarta).
The EMA approval recommendations come with risk management measures to address the potential for cytokine release syndrome with both of these treatments. Drug makers must use a patient registry to monitor the long-term safety and efficacy of the therapies.
The EMA is also recommending approval of caplacizumab for acquired thrombotic thrombocytopenic purpura, vonicog alfa for the treatment of von Willebrand disease, and daunorubicin/cytarabine for the treatment of acute myeloid leukemia.
The European Medicines Agency (EMA) has recommended a handful of hematology medications for approval, including two chimeric antigen receptor (CAR) T-cell therapies.
All of the drugs must next be approved by the European Commission in order to be marketed to patients throughout Europe.
At the end of June, the EMA’s Committee for Medicinal Products for Human Use tisagenlecleucel (Kymriah) and axicabtagene ciloleucel (Yescarta).
The EMA approval recommendations come with risk management measures to address the potential for cytokine release syndrome with both of these treatments. Drug makers must use a patient registry to monitor the long-term safety and efficacy of the therapies.
The EMA is also recommending approval of caplacizumab for acquired thrombotic thrombocytopenic purpura, vonicog alfa for the treatment of von Willebrand disease, and daunorubicin/cytarabine for the treatment of acute myeloid leukemia.
Bortezomib plus vorinostat shows modest response in MCL
but was less impressive among patients with diffuse large B-cell lymphoma (DLBCL).
Victor Yazbeck, MD, of the Massey Cancer Center at Virginia Commonwealth University in Richmond, and his colleagues reported the findings from the multicenter, nonrandomized, phase 2 trial with 65 treated patients. The trial included three cohorts: 22 patients with MCL and no prior treatment with bortezomib; 4 patients with MCL and prior treatment with bortezomib; and 39 patients with relapsed or refractory DLBCL and no prior bortezomib.
The best results were seen among MCL patients with no prior bortezomib treatment, with an overall response rate of 31.8% and a median progression-free survival (PFS) of 7.6 months. Responses were limited among the DLBCL cohort, which had an overall response rate of 7.7% and a median PFS of just 1.8 months. Among MCL patients who had received prior bortezomib treatment, there were no responses.
From a safety perspective, the combination treatment was well tolerated. The most common grade 3 and 4 hematologic toxicities were thrombocytopenia, lymphopenia, and neutropenia. There was one death among the DLBCL patients and it was unclear if it was related to treatment or progression of disease.
“Patients with MCL had a higher [overall response rate] compared to those with DLBCL, most likely due to the single-agent activity of bortezomib in MCL,” the researchers wrote. “Overall, the synergism previously demonstrated in preclinical models could not be confirmed.”
The study was supported by the Southeast Phase 2 Consortium and by a grant from the National Cancer Institute. Dr. Yazbeck reported having no financial disclosures. One of his coauthors is an employee of Amgen and owns Amgen stock. Another coauthor receives research support from Takeda, Celgene, Karyopharm Therapeutics, Bristol-Myers Squibb, Merck, and Signal Genetics.
SOURCE: Yazbeck V et al. Clin Lymphoma Myeloma Leuk. 2018 Jun 6. doi: 10.1016/j.clml.2018.05.023.
but was less impressive among patients with diffuse large B-cell lymphoma (DLBCL).
Victor Yazbeck, MD, of the Massey Cancer Center at Virginia Commonwealth University in Richmond, and his colleagues reported the findings from the multicenter, nonrandomized, phase 2 trial with 65 treated patients. The trial included three cohorts: 22 patients with MCL and no prior treatment with bortezomib; 4 patients with MCL and prior treatment with bortezomib; and 39 patients with relapsed or refractory DLBCL and no prior bortezomib.
The best results were seen among MCL patients with no prior bortezomib treatment, with an overall response rate of 31.8% and a median progression-free survival (PFS) of 7.6 months. Responses were limited among the DLBCL cohort, which had an overall response rate of 7.7% and a median PFS of just 1.8 months. Among MCL patients who had received prior bortezomib treatment, there were no responses.
From a safety perspective, the combination treatment was well tolerated. The most common grade 3 and 4 hematologic toxicities were thrombocytopenia, lymphopenia, and neutropenia. There was one death among the DLBCL patients and it was unclear if it was related to treatment or progression of disease.
“Patients with MCL had a higher [overall response rate] compared to those with DLBCL, most likely due to the single-agent activity of bortezomib in MCL,” the researchers wrote. “Overall, the synergism previously demonstrated in preclinical models could not be confirmed.”
The study was supported by the Southeast Phase 2 Consortium and by a grant from the National Cancer Institute. Dr. Yazbeck reported having no financial disclosures. One of his coauthors is an employee of Amgen and owns Amgen stock. Another coauthor receives research support from Takeda, Celgene, Karyopharm Therapeutics, Bristol-Myers Squibb, Merck, and Signal Genetics.
SOURCE: Yazbeck V et al. Clin Lymphoma Myeloma Leuk. 2018 Jun 6. doi: 10.1016/j.clml.2018.05.023.
but was less impressive among patients with diffuse large B-cell lymphoma (DLBCL).
Victor Yazbeck, MD, of the Massey Cancer Center at Virginia Commonwealth University in Richmond, and his colleagues reported the findings from the multicenter, nonrandomized, phase 2 trial with 65 treated patients. The trial included three cohorts: 22 patients with MCL and no prior treatment with bortezomib; 4 patients with MCL and prior treatment with bortezomib; and 39 patients with relapsed or refractory DLBCL and no prior bortezomib.
The best results were seen among MCL patients with no prior bortezomib treatment, with an overall response rate of 31.8% and a median progression-free survival (PFS) of 7.6 months. Responses were limited among the DLBCL cohort, which had an overall response rate of 7.7% and a median PFS of just 1.8 months. Among MCL patients who had received prior bortezomib treatment, there were no responses.
From a safety perspective, the combination treatment was well tolerated. The most common grade 3 and 4 hematologic toxicities were thrombocytopenia, lymphopenia, and neutropenia. There was one death among the DLBCL patients and it was unclear if it was related to treatment or progression of disease.
“Patients with MCL had a higher [overall response rate] compared to those with DLBCL, most likely due to the single-agent activity of bortezomib in MCL,” the researchers wrote. “Overall, the synergism previously demonstrated in preclinical models could not be confirmed.”
The study was supported by the Southeast Phase 2 Consortium and by a grant from the National Cancer Institute. Dr. Yazbeck reported having no financial disclosures. One of his coauthors is an employee of Amgen and owns Amgen stock. Another coauthor receives research support from Takeda, Celgene, Karyopharm Therapeutics, Bristol-Myers Squibb, Merck, and Signal Genetics.
SOURCE: Yazbeck V et al. Clin Lymphoma Myeloma Leuk. 2018 Jun 6. doi: 10.1016/j.clml.2018.05.023.
FROM CLINICAL LYMPHOMA, MYELOMA AND LEUKEMIA
Doc reports favorable results from trial on hold

STOCKHOLM—Interim trial results suggest the EZH2 inhibitor tazemetostat can produce durable responses in patients with relapsed or refractory follicular lymphoma (FL).
In patients with EZH2 mutations, the overall response rate (ORR) was 71%, and the median duration of response (DOR) was 32 weeks.
For patients with wild-type (WT) EZH2, the ORR was 33%, and the median DOR was 76 weeks.
Tazemetostat was considered generally well tolerated in this phase 2 trial, which is currently on partial clinical hold.
Gilles Salles, MD, PhD, of the University Hospital of Lyon France, presented results from the trial at the 23rd Congress of the European Hematology Association (EHA) as abstract S100.
The trial is sponsored by Epizyme, Inc.
In April, Epizyme announced that all US-based trials of tazemetostat had been placed on partial hold after a pediatric patient on a phase 1 trial developed secondary T-cell lymphoma.
Enrollment was stopped in all the trials, but patients could continue receiving tazemetostat if they had not progressed on the drug.
The phase 2 trial of tazemetostat in non-Hodgkin lymphoma has enrolled 89 adults with relapsed/refractory FL.
At EHA, Dr Salles presented results in 82 of these patients. There were 28 patients with EZH2-mutated FL and 54 with EZH2-WT FL.
The median age was 61 in both cohorts. Forty-three percent of EZH2-mutated and 63% of WT patients were male.
EZH2-mutated patients had a median of 3 prior therapies, and WT patients had a median of 4. Thirty-eight percent and 42%, respectively, were refractory to their last therapy. Eleven percent and 39%, respectively, had received prior transplant.
The median time from diagnosis was 5.1 years for EZH2-mutated patients and 6.4 years for WT patients. The median time from last prior therapy was 18.4 weeks and 28.1 weeks, respectively.
The patients received tazemetostat at 800 mg twice daily until disease progression or withdrawal.
Safety
In all 82 patients, the rate of treatment-emergent adverse events (AEs) was 95%, and the rate of treatment-related AEs was 78%. The rate of grade 3 or higher treatment-related AEs was 17%, and the rate of serious treatment-related AEs was 4%.
Six percent of patients discontinued treatment due to a related AE, 18% had a dose interruption, and 5% had a dose reduction due to a related AE.
Treatment-related AEs included nausea (20%), fatigue (13%), anemia (13%), diarrhea (11%), alopecia (11%), asthenia (10%), thrombocytopenia (10%), muscle spasms (6%), bronchitis (5%), vomiting (5%), headache (5%), abdominal pain (2%), pyrexia (1%), and cough (1%).
Grade 3 or higher treatment-related AEs included thrombocytopenia (4%), anemia (4%), fatigue (1%), and asthenia (1%).
Efficacy
In the EZH2-mutated cohort, the ORR was 71% (n=20). Eleven percent of patients (n=3) achieved a complete response, and 61% (n=17) had a partial response.
Twenty-nine percent (n=8) had stable disease as their best response. And 21% (n=6) of patients are still on study with stable disease.
All patients in this cohort experienced a reduction in tumor burden. None of the patients had progressive disease as their best response.
At the time of analysis (May 1, 2018), the median DOR was 32.3 weeks, and 55% of responders (n=11) had an ongoing response.
The median progression-free survival was 48.6 weeks.
In patients with WT EZH2 (n=54), the ORR was 33% (n=18). Six percent of patients (n=3) achieved a complete response, and 28% (n=15) had a partial response.
Thirty-one percent of patients (n=17) had stable disease as their best response, including 1 patient who is still receiving treatment.
Thirty-one percent of patients (n=17) progressed. For 4% (n=2), their response status was unknown.
At the time of analysis, the median DOR was 76 weeks, and 56% of responders (n=10) had an ongoing response.
The median progression-free survival was 29.9 weeks.
“I am impressed by the sustained clinical activity and the good tolerability of tazemetostat in this heavily pretreated patient population,” Dr Salles said. “This is important for patients with relapsed or refractory follicular lymphoma, as both the response rates and durations of response usually tend to decrease with each successive line of treatment.”
“I believe tazemetostat has the potential to fill a significant unmet need for these patients, and continued investigation of tazemetostat as a single agent or in combination with other agents is warranted.”
Epizyme’s president and chief executive officer, Robert Bazemore, said the company is still working to resolve the partial clinical hold on tazemetostat trials and is “making good progress.”
STOCKHOLM—Interim trial results suggest the EZH2 inhibitor tazemetostat can produce durable responses in patients with relapsed or refractory follicular lymphoma (FL).
In patients with EZH2 mutations, the overall response rate (ORR) was 71%, and the median duration of response (DOR) was 32 weeks.
For patients with wild-type (WT) EZH2, the ORR was 33%, and the median DOR was 76 weeks.
Tazemetostat was considered generally well tolerated in this phase 2 trial, which is currently on partial clinical hold.
Gilles Salles, MD, PhD, of the University Hospital of Lyon France, presented results from the trial at the 23rd Congress of the European Hematology Association (EHA) as abstract S100.
The trial is sponsored by Epizyme, Inc.
In April, Epizyme announced that all US-based trials of tazemetostat had been placed on partial hold after a pediatric patient on a phase 1 trial developed secondary T-cell lymphoma.
Enrollment was stopped in all the trials, but patients could continue receiving tazemetostat if they had not progressed on the drug.
The phase 2 trial of tazemetostat in non-Hodgkin lymphoma has enrolled 89 adults with relapsed/refractory FL.
At EHA, Dr Salles presented results in 82 of these patients. There were 28 patients with EZH2-mutated FL and 54 with EZH2-WT FL.
The median age was 61 in both cohorts. Forty-three percent of EZH2-mutated and 63% of WT patients were male.
EZH2-mutated patients had a median of 3 prior therapies, and WT patients had a median of 4. Thirty-eight percent and 42%, respectively, were refractory to their last therapy. Eleven percent and 39%, respectively, had received prior transplant.
The median time from diagnosis was 5.1 years for EZH2-mutated patients and 6.4 years for WT patients. The median time from last prior therapy was 18.4 weeks and 28.1 weeks, respectively.
The patients received tazemetostat at 800 mg twice daily until disease progression or withdrawal.
Safety
In all 82 patients, the rate of treatment-emergent adverse events (AEs) was 95%, and the rate of treatment-related AEs was 78%. The rate of grade 3 or higher treatment-related AEs was 17%, and the rate of serious treatment-related AEs was 4%.
Six percent of patients discontinued treatment due to a related AE, 18% had a dose interruption, and 5% had a dose reduction due to a related AE.
Treatment-related AEs included nausea (20%), fatigue (13%), anemia (13%), diarrhea (11%), alopecia (11%), asthenia (10%), thrombocytopenia (10%), muscle spasms (6%), bronchitis (5%), vomiting (5%), headache (5%), abdominal pain (2%), pyrexia (1%), and cough (1%).
Grade 3 or higher treatment-related AEs included thrombocytopenia (4%), anemia (4%), fatigue (1%), and asthenia (1%).
Efficacy
In the EZH2-mutated cohort, the ORR was 71% (n=20). Eleven percent of patients (n=3) achieved a complete response, and 61% (n=17) had a partial response.
Twenty-nine percent (n=8) had stable disease as their best response. And 21% (n=6) of patients are still on study with stable disease.
All patients in this cohort experienced a reduction in tumor burden. None of the patients had progressive disease as their best response.
At the time of analysis (May 1, 2018), the median DOR was 32.3 weeks, and 55% of responders (n=11) had an ongoing response.
The median progression-free survival was 48.6 weeks.
In patients with WT EZH2 (n=54), the ORR was 33% (n=18). Six percent of patients (n=3) achieved a complete response, and 28% (n=15) had a partial response.
Thirty-one percent of patients (n=17) had stable disease as their best response, including 1 patient who is still receiving treatment.
Thirty-one percent of patients (n=17) progressed. For 4% (n=2), their response status was unknown.
At the time of analysis, the median DOR was 76 weeks, and 56% of responders (n=10) had an ongoing response.
The median progression-free survival was 29.9 weeks.
“I am impressed by the sustained clinical activity and the good tolerability of tazemetostat in this heavily pretreated patient population,” Dr Salles said. “This is important for patients with relapsed or refractory follicular lymphoma, as both the response rates and durations of response usually tend to decrease with each successive line of treatment.”
“I believe tazemetostat has the potential to fill a significant unmet need for these patients, and continued investigation of tazemetostat as a single agent or in combination with other agents is warranted.”
Epizyme’s president and chief executive officer, Robert Bazemore, said the company is still working to resolve the partial clinical hold on tazemetostat trials and is “making good progress.”
STOCKHOLM—Interim trial results suggest the EZH2 inhibitor tazemetostat can produce durable responses in patients with relapsed or refractory follicular lymphoma (FL).
In patients with EZH2 mutations, the overall response rate (ORR) was 71%, and the median duration of response (DOR) was 32 weeks.
For patients with wild-type (WT) EZH2, the ORR was 33%, and the median DOR was 76 weeks.
Tazemetostat was considered generally well tolerated in this phase 2 trial, which is currently on partial clinical hold.
Gilles Salles, MD, PhD, of the University Hospital of Lyon France, presented results from the trial at the 23rd Congress of the European Hematology Association (EHA) as abstract S100.
The trial is sponsored by Epizyme, Inc.
In April, Epizyme announced that all US-based trials of tazemetostat had been placed on partial hold after a pediatric patient on a phase 1 trial developed secondary T-cell lymphoma.
Enrollment was stopped in all the trials, but patients could continue receiving tazemetostat if they had not progressed on the drug.
The phase 2 trial of tazemetostat in non-Hodgkin lymphoma has enrolled 89 adults with relapsed/refractory FL.
At EHA, Dr Salles presented results in 82 of these patients. There were 28 patients with EZH2-mutated FL and 54 with EZH2-WT FL.
The median age was 61 in both cohorts. Forty-three percent of EZH2-mutated and 63% of WT patients were male.
EZH2-mutated patients had a median of 3 prior therapies, and WT patients had a median of 4. Thirty-eight percent and 42%, respectively, were refractory to their last therapy. Eleven percent and 39%, respectively, had received prior transplant.
The median time from diagnosis was 5.1 years for EZH2-mutated patients and 6.4 years for WT patients. The median time from last prior therapy was 18.4 weeks and 28.1 weeks, respectively.
The patients received tazemetostat at 800 mg twice daily until disease progression or withdrawal.
Safety
In all 82 patients, the rate of treatment-emergent adverse events (AEs) was 95%, and the rate of treatment-related AEs was 78%. The rate of grade 3 or higher treatment-related AEs was 17%, and the rate of serious treatment-related AEs was 4%.
Six percent of patients discontinued treatment due to a related AE, 18% had a dose interruption, and 5% had a dose reduction due to a related AE.
Treatment-related AEs included nausea (20%), fatigue (13%), anemia (13%), diarrhea (11%), alopecia (11%), asthenia (10%), thrombocytopenia (10%), muscle spasms (6%), bronchitis (5%), vomiting (5%), headache (5%), abdominal pain (2%), pyrexia (1%), and cough (1%).
Grade 3 or higher treatment-related AEs included thrombocytopenia (4%), anemia (4%), fatigue (1%), and asthenia (1%).
Efficacy
In the EZH2-mutated cohort, the ORR was 71% (n=20). Eleven percent of patients (n=3) achieved a complete response, and 61% (n=17) had a partial response.
Twenty-nine percent (n=8) had stable disease as their best response. And 21% (n=6) of patients are still on study with stable disease.
All patients in this cohort experienced a reduction in tumor burden. None of the patients had progressive disease as their best response.
At the time of analysis (May 1, 2018), the median DOR was 32.3 weeks, and 55% of responders (n=11) had an ongoing response.
The median progression-free survival was 48.6 weeks.
In patients with WT EZH2 (n=54), the ORR was 33% (n=18). Six percent of patients (n=3) achieved a complete response, and 28% (n=15) had a partial response.
Thirty-one percent of patients (n=17) had stable disease as their best response, including 1 patient who is still receiving treatment.
Thirty-one percent of patients (n=17) progressed. For 4% (n=2), their response status was unknown.
At the time of analysis, the median DOR was 76 weeks, and 56% of responders (n=10) had an ongoing response.
The median progression-free survival was 29.9 weeks.
“I am impressed by the sustained clinical activity and the good tolerability of tazemetostat in this heavily pretreated patient population,” Dr Salles said. “This is important for patients with relapsed or refractory follicular lymphoma, as both the response rates and durations of response usually tend to decrease with each successive line of treatment.”
“I believe tazemetostat has the potential to fill a significant unmet need for these patients, and continued investigation of tazemetostat as a single agent or in combination with other agents is warranted.”
Epizyme’s president and chief executive officer, Robert Bazemore, said the company is still working to resolve the partial clinical hold on tazemetostat trials and is “making good progress.”
Group updates guidelines on CLL

Recent advances in chronic lymphocytic leukemia (CLL) have prompted an update to the 2008 International Workshop in Chronic Lymphocytic Leukemia (iwCLL) consensus guidelines.
The updated iwCLL guidelines include new information on genomic alterations, the use of clinical staging and prognostic markers/scores, response assessment, minimal residual disease (MRD), and viral diseases in CLL patients.
The update was recently published in Blood.
Diagnosis, prognosis, and staging
To verify CLL diagnosis, the iwCLL guidelines recommend obtaining complete blood counts and differential counts as well as immunophenotyping of peripheral blood lymphocytes. A panel of CD19, CD5, CD20, CD23, κ, and λ typically suffices to establish a diagnosis.
Other tests that may help in prognosis or assessing tumor burden include molecular cytogenetics for del(13q), del(11q), del(17p), and add(12) in peripheral blood lymphocytes and determining TP53 and IGHV mutational status.
These tests can help identify poor-prognosis patients who are not likely to benefit from standard chemotherapy but are likely to benefit from small-molecule inhibitors of BTK, PI3K, or BCL2.
In addition, serum markers such as β2-microglogulin provide insight into overall survival and progression-free survival.
With regard to clinical staging, the guidelines highlight the Binet and Rai systems, which are routinely used in clinical practice and clinical trials.
However, the guidelines also note that “there are a large number of biomarkers that can provide additional prognostic information,” and, recently, “several prognostic scores and stratification systems have been proposed based on multivariate analyses.”
For example, the CLL international prognostic index (CLL-IPI) provides a weighted score that uses clinical stage, age, IGHV mutational status, β2-microglogulin, and the presence of del(17p) and/or TP53 mutations.
Indications for treatment
The guidelines note that active disease must be documented to initiate therapy. At least 1 of the following criteria must be met:
- Evidence of progressive marrow failure
- Massive, progressive, or symptomatic splenomegaly
- Progressive/symptomatic lymphadenopathy or massive nodes
- Progressive lymphocytosis
- Autoimmune complications
- Extranodal involvement
- Disease-related symptoms (unintentional weight loss, significant fatigue, fevers, night sweats for over a month without evidence of infection).
Following relapse, subsequent lines of treatment should follow the same principles as those used for initial treatment decisions.
Response, MRD, and more
The guidelines say 2 groups of parameters must be assessed to determine response to therapy:
- Group A: lymphoid tumor load and constitutional symptoms, including liver and/or spleen size, lymph node evaluation, and circulating lymphocyte count
- Group B: the hematopoietic system (platelet count, hemoglobin, and marrow).
For therapies with a defined treatment duration, response should be assessed at least 2 months after treatment is completed. For continued therapies or maintenance, response should be assessed at a predefined time point or at least 2 months after patients achieve their maximum response.
The guidelines also say MRD should be assessed in clinical trials aimed at maximizing the depth of remission. Furthermore, it “may be important” to confirm MRD negativity in the blood and marrow, as there are therapies that preferentially clear the blood but not the marrow (such as monoclonal antibodies).
In addition to the aforementioned recommendations, the updated iwCLL guidelines also include information on patient eligibility for clinical trials, guidance regarding treatment-related toxicities, and recommendations for supportive care and managing complications.
Recent advances in chronic lymphocytic leukemia (CLL) have prompted an update to the 2008 International Workshop in Chronic Lymphocytic Leukemia (iwCLL) consensus guidelines.
The updated iwCLL guidelines include new information on genomic alterations, the use of clinical staging and prognostic markers/scores, response assessment, minimal residual disease (MRD), and viral diseases in CLL patients.
The update was recently published in Blood.
Diagnosis, prognosis, and staging
To verify CLL diagnosis, the iwCLL guidelines recommend obtaining complete blood counts and differential counts as well as immunophenotyping of peripheral blood lymphocytes. A panel of CD19, CD5, CD20, CD23, κ, and λ typically suffices to establish a diagnosis.
Other tests that may help in prognosis or assessing tumor burden include molecular cytogenetics for del(13q), del(11q), del(17p), and add(12) in peripheral blood lymphocytes and determining TP53 and IGHV mutational status.
These tests can help identify poor-prognosis patients who are not likely to benefit from standard chemotherapy but are likely to benefit from small-molecule inhibitors of BTK, PI3K, or BCL2.
In addition, serum markers such as β2-microglogulin provide insight into overall survival and progression-free survival.
With regard to clinical staging, the guidelines highlight the Binet and Rai systems, which are routinely used in clinical practice and clinical trials.
However, the guidelines also note that “there are a large number of biomarkers that can provide additional prognostic information,” and, recently, “several prognostic scores and stratification systems have been proposed based on multivariate analyses.”
For example, the CLL international prognostic index (CLL-IPI) provides a weighted score that uses clinical stage, age, IGHV mutational status, β2-microglogulin, and the presence of del(17p) and/or TP53 mutations.
Indications for treatment
The guidelines note that active disease must be documented to initiate therapy. At least 1 of the following criteria must be met:
- Evidence of progressive marrow failure
- Massive, progressive, or symptomatic splenomegaly
- Progressive/symptomatic lymphadenopathy or massive nodes
- Progressive lymphocytosis
- Autoimmune complications
- Extranodal involvement
- Disease-related symptoms (unintentional weight loss, significant fatigue, fevers, night sweats for over a month without evidence of infection).
Following relapse, subsequent lines of treatment should follow the same principles as those used for initial treatment decisions.
Response, MRD, and more
The guidelines say 2 groups of parameters must be assessed to determine response to therapy:
- Group A: lymphoid tumor load and constitutional symptoms, including liver and/or spleen size, lymph node evaluation, and circulating lymphocyte count
- Group B: the hematopoietic system (platelet count, hemoglobin, and marrow).
For therapies with a defined treatment duration, response should be assessed at least 2 months after treatment is completed. For continued therapies or maintenance, response should be assessed at a predefined time point or at least 2 months after patients achieve their maximum response.
The guidelines also say MRD should be assessed in clinical trials aimed at maximizing the depth of remission. Furthermore, it “may be important” to confirm MRD negativity in the blood and marrow, as there are therapies that preferentially clear the blood but not the marrow (such as monoclonal antibodies).
In addition to the aforementioned recommendations, the updated iwCLL guidelines also include information on patient eligibility for clinical trials, guidance regarding treatment-related toxicities, and recommendations for supportive care and managing complications.
Recent advances in chronic lymphocytic leukemia (CLL) have prompted an update to the 2008 International Workshop in Chronic Lymphocytic Leukemia (iwCLL) consensus guidelines.
The updated iwCLL guidelines include new information on genomic alterations, the use of clinical staging and prognostic markers/scores, response assessment, minimal residual disease (MRD), and viral diseases in CLL patients.
The update was recently published in Blood.
Diagnosis, prognosis, and staging
To verify CLL diagnosis, the iwCLL guidelines recommend obtaining complete blood counts and differential counts as well as immunophenotyping of peripheral blood lymphocytes. A panel of CD19, CD5, CD20, CD23, κ, and λ typically suffices to establish a diagnosis.
Other tests that may help in prognosis or assessing tumor burden include molecular cytogenetics for del(13q), del(11q), del(17p), and add(12) in peripheral blood lymphocytes and determining TP53 and IGHV mutational status.
These tests can help identify poor-prognosis patients who are not likely to benefit from standard chemotherapy but are likely to benefit from small-molecule inhibitors of BTK, PI3K, or BCL2.
In addition, serum markers such as β2-microglogulin provide insight into overall survival and progression-free survival.
With regard to clinical staging, the guidelines highlight the Binet and Rai systems, which are routinely used in clinical practice and clinical trials.
However, the guidelines also note that “there are a large number of biomarkers that can provide additional prognostic information,” and, recently, “several prognostic scores and stratification systems have been proposed based on multivariate analyses.”
For example, the CLL international prognostic index (CLL-IPI) provides a weighted score that uses clinical stage, age, IGHV mutational status, β2-microglogulin, and the presence of del(17p) and/or TP53 mutations.
Indications for treatment
The guidelines note that active disease must be documented to initiate therapy. At least 1 of the following criteria must be met:
- Evidence of progressive marrow failure
- Massive, progressive, or symptomatic splenomegaly
- Progressive/symptomatic lymphadenopathy or massive nodes
- Progressive lymphocytosis
- Autoimmune complications
- Extranodal involvement
- Disease-related symptoms (unintentional weight loss, significant fatigue, fevers, night sweats for over a month without evidence of infection).
Following relapse, subsequent lines of treatment should follow the same principles as those used for initial treatment decisions.
Response, MRD, and more
The guidelines say 2 groups of parameters must be assessed to determine response to therapy:
- Group A: lymphoid tumor load and constitutional symptoms, including liver and/or spleen size, lymph node evaluation, and circulating lymphocyte count
- Group B: the hematopoietic system (platelet count, hemoglobin, and marrow).
For therapies with a defined treatment duration, response should be assessed at least 2 months after treatment is completed. For continued therapies or maintenance, response should be assessed at a predefined time point or at least 2 months after patients achieve their maximum response.
The guidelines also say MRD should be assessed in clinical trials aimed at maximizing the depth of remission. Furthermore, it “may be important” to confirm MRD negativity in the blood and marrow, as there are therapies that preferentially clear the blood but not the marrow (such as monoclonal antibodies).
In addition to the aforementioned recommendations, the updated iwCLL guidelines also include information on patient eligibility for clinical trials, guidance regarding treatment-related toxicities, and recommendations for supportive care and managing complications.