User login
FDA issues CRL for proposed biosimilar rituximab

The US Food and Drug Administration (FDA) has issued a complete response letter (CRL) saying the agency cannot approve Sandoz’s proposed biosimilar rituximab.
Sandoz submitted the biologics licensing application for the product, known as GP2013, in September 2017.
The company was seeking approval for GP2013 to treat follicular lymphoma (FL), diffuse large B-cell lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis, and microscopic polyangiitis.
The drug already has approval for these indications in Europe. The European Commission approved GP2013 (Rixathon) in June 2017.
As for US approval, Sandoz said it is evaluating the content of the FDA’s CRL and “remains committed to further discussions with FDA in order to bring this important medicine to US patients as soon as possible.”
The company said it “stands behind the robust body of evidence included in the regulatory submission” for GP2013.
Part of this evidence is the ASSIST-FL trial, in which researchers compared GP2013 to the reference product, Roche’s MabThera. Results from this trial were published in The Lancet Haematology and presented at ESMO 2017 Congress.
This phase 3 trial included adults with previously untreated, advanced stage FL. Patients received 8 cycles of cyclophosphamide, vincristine, and prednisone with either GP2013 or reference rituximab. Responders then received GP2013 or rituximab monotherapy as maintenance for up to 2 years.
At a median follow-up of 11.6 months, the overall response rate was 87% (271/311) in the GP2013 arm and 88% in the rituximab arm (274/313). Complete response rates were 15% (n=46) and 13% (n=42), respectively.
Rates of adverse events (AEs) were 93% in the GP2013 arm and 91% in the rituximab arm. Rates of serious AEs were 23% and 20%, respectively. The rate of discontinuation due to AEs was 7% in both arms.
The most common AE was neutropenia, which occurred in 26% of patients in the GP2013 arm and 30% of those in the rituximab arm in the combination phase. Rates of neutropenia in the maintenance phase were 10% and 6%, respectively.
The US Food and Drug Administration (FDA) has issued a complete response letter (CRL) saying the agency cannot approve Sandoz’s proposed biosimilar rituximab.
Sandoz submitted the biologics licensing application for the product, known as GP2013, in September 2017.
The company was seeking approval for GP2013 to treat follicular lymphoma (FL), diffuse large B-cell lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis, and microscopic polyangiitis.
The drug already has approval for these indications in Europe. The European Commission approved GP2013 (Rixathon) in June 2017.
As for US approval, Sandoz said it is evaluating the content of the FDA’s CRL and “remains committed to further discussions with FDA in order to bring this important medicine to US patients as soon as possible.”
The company said it “stands behind the robust body of evidence included in the regulatory submission” for GP2013.
Part of this evidence is the ASSIST-FL trial, in which researchers compared GP2013 to the reference product, Roche’s MabThera. Results from this trial were published in The Lancet Haematology and presented at ESMO 2017 Congress.
This phase 3 trial included adults with previously untreated, advanced stage FL. Patients received 8 cycles of cyclophosphamide, vincristine, and prednisone with either GP2013 or reference rituximab. Responders then received GP2013 or rituximab monotherapy as maintenance for up to 2 years.
At a median follow-up of 11.6 months, the overall response rate was 87% (271/311) in the GP2013 arm and 88% in the rituximab arm (274/313). Complete response rates were 15% (n=46) and 13% (n=42), respectively.
Rates of adverse events (AEs) were 93% in the GP2013 arm and 91% in the rituximab arm. Rates of serious AEs were 23% and 20%, respectively. The rate of discontinuation due to AEs was 7% in both arms.
The most common AE was neutropenia, which occurred in 26% of patients in the GP2013 arm and 30% of those in the rituximab arm in the combination phase. Rates of neutropenia in the maintenance phase were 10% and 6%, respectively.
The US Food and Drug Administration (FDA) has issued a complete response letter (CRL) saying the agency cannot approve Sandoz’s proposed biosimilar rituximab.
Sandoz submitted the biologics licensing application for the product, known as GP2013, in September 2017.
The company was seeking approval for GP2013 to treat follicular lymphoma (FL), diffuse large B-cell lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis, and microscopic polyangiitis.
The drug already has approval for these indications in Europe. The European Commission approved GP2013 (Rixathon) in June 2017.
As for US approval, Sandoz said it is evaluating the content of the FDA’s CRL and “remains committed to further discussions with FDA in order to bring this important medicine to US patients as soon as possible.”
The company said it “stands behind the robust body of evidence included in the regulatory submission” for GP2013.
Part of this evidence is the ASSIST-FL trial, in which researchers compared GP2013 to the reference product, Roche’s MabThera. Results from this trial were published in The Lancet Haematology and presented at ESMO 2017 Congress.
This phase 3 trial included adults with previously untreated, advanced stage FL. Patients received 8 cycles of cyclophosphamide, vincristine, and prednisone with either GP2013 or reference rituximab. Responders then received GP2013 or rituximab monotherapy as maintenance for up to 2 years.
At a median follow-up of 11.6 months, the overall response rate was 87% (271/311) in the GP2013 arm and 88% in the rituximab arm (274/313). Complete response rates were 15% (n=46) and 13% (n=42), respectively.
Rates of adverse events (AEs) were 93% in the GP2013 arm and 91% in the rituximab arm. Rates of serious AEs were 23% and 20%, respectively. The rate of discontinuation due to AEs was 7% in both arms.
The most common AE was neutropenia, which occurred in 26% of patients in the GP2013 arm and 30% of those in the rituximab arm in the combination phase. Rates of neutropenia in the maintenance phase were 10% and 6%, respectively.
Five-year survival for non-Hodgkin lymphoma tops 71%
The overall 5-year survival rate for non-Hodgkin lymphoma (NHL) is 71.4%, according to the National Cancer Institute.
That number falls neatly into the middle of the range for survival by stage at diagnosis, with stage I (81.8%) and stage II (75.3%) disease on the high side and stage III (69.1%) and stage IV (61.7%) on the low side, the most recent data from the Surveillance, Epidemiology, and End Results (SEER) Program show. Five-year survival for NHL of unknown stage at diagnosis is 76.4%.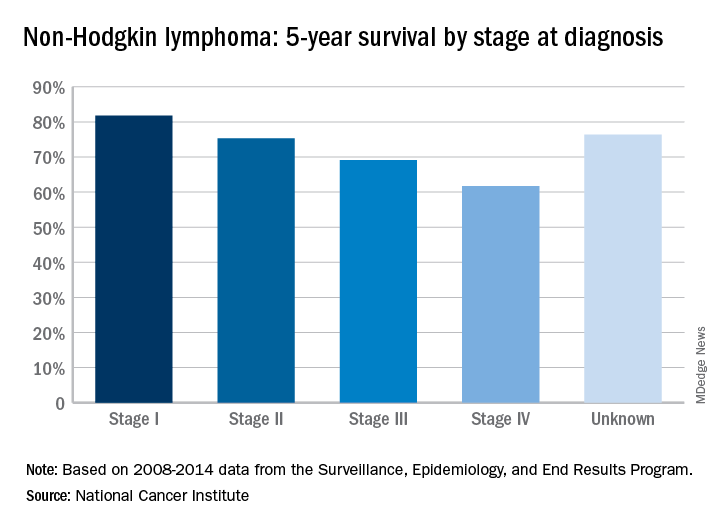
The overall 5-year survival rate for non-Hodgkin lymphoma (NHL) is 71.4%, according to the National Cancer Institute.
That number falls neatly into the middle of the range for survival by stage at diagnosis, with stage I (81.8%) and stage II (75.3%) disease on the high side and stage III (69.1%) and stage IV (61.7%) on the low side, the most recent data from the Surveillance, Epidemiology, and End Results (SEER) Program show. Five-year survival for NHL of unknown stage at diagnosis is 76.4%.
The overall 5-year survival rate for non-Hodgkin lymphoma (NHL) is 71.4%, according to the National Cancer Institute.
That number falls neatly into the middle of the range for survival by stage at diagnosis, with stage I (81.8%) and stage II (75.3%) disease on the high side and stage III (69.1%) and stage IV (61.7%) on the low side, the most recent data from the Surveillance, Epidemiology, and End Results (SEER) Program show. Five-year survival for NHL of unknown stage at diagnosis is 76.4%.
FDA approves CAR T-cell therapy for lymphoma

The US Food and Drug Administration (FDA) has approved tisagenlecleucel (Kymriah®) for its second indication.
The chimeric antigen receptor (CAR) T-cell therapy is now approved to treat adults with relapsed or refractory large B-cell lymphoma after 2 or more lines of systemic therapy.
This includes patients with diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.
The application for tisagenlecleucel in B-cell lymphoma was granted priority review. The FDA aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
Tisagenlecleucel is also FDA-approved to treat patients age 25 and younger who have B-cell precursor acute lymphoblastic leukemia that is refractory or in second or later relapse.
Access to tisagenlecleucel
The prescribing information for tisagenlecleucel includes a boxed warning detailing the risk of cytokine release syndrome (CRS) and neurological toxicities for patients receiving tisagenlecleucel.
Because of these risks, tisagenlecleucel is only available through a Risk Evaluation and Mitigation Strategy (REMS) program. The REMS program serves to inform and educate healthcare professionals about the risks associated with tisagenlecleucel treatment.
Novartis, the company marketing tisagenlecleucel, has established a network of certified treatment centers throughout the US. Staff at these centers are trained on the use of tisagenlecleucel and appropriate patient care.
Tisagenlecleucel is manufactured at a Novartis facility in Morris Plains, New Jersey. In the US, the target turnaround time for manufacturing tisagenlecleucel is 22 days.
Tisagenlecleucel costs $475,000 for a single course of treatment. However, Novartis said it is collaborating with the US Centers for Medicare and Medicaid Services on the creation of an appropriate value-based pricing approach.
The company also has a program called KYMRIAH CARES™, which offers financial assistance to eligible patients to help them gain access to tisagenlecleucel.
Phase 2 trial
The FDA approval of tisagenlecleucel for adults with relapsed/refractory B-cell lymphoma is based on results of the phase 2 JULIET trial.
The prescribing information for tisagenlecleucel includes data on 106 patients treated on this trial.
Only 68 of these patients were evaluable for efficacy. They had a median age of 56 (range, 22 to 74), and 71% were male.
Seventy-eight percent of patients had primary DLBCL not otherwise specified, and 22% had DLBCL following transformation from follicular lymphoma. Seventeen percent had high grade DLBCL.
Fifty-six percent of patients had refractory disease, and 44% had relapsed after their last therapy. The median number of prior therapies was 3 (range, 1 to 6), and 44% of patients had undergone autologous transplant.
Ninety percent of patients received lymphodepleting chemotherapy (66% fludarabine and 24% bendamustine) prior to tisagenlecleucel, and 10% did not. The median dose of tisagenlecleucel was 3.5 × 108 CAR+ T cells (range, 1.0 to 5.2 × 108).
The overall response rate was 50%, with 32% of patients achieving a complete response and 18% achieving a partial response. The median duration of response was not reached with a median follow-up of 9.4 months.
In all 106 patients infused with tisagenlecleucel, the most common grade 3/4 adverse events were infections (25%), CRS (23%), neurologic events (18%), febrile neutropenia (17%), encephalopathy (11%), lymphopenia (94%), neutropenia (81%), leukopenia (77%), anemia (58%), thrombocytopenia (54%), hypophosphatemia (24%), hypokalemia (12%), and hyponatremia (11%).
Three patients died within 30 days of tisagenlecleucel infusion. All of them had CRS and either stable or progressive disease. One of these patients developed bowel necrosis.
One patient died of infection. There were no deaths attributed to neurological events, and no fatal cases of cerebral edema.
The US Food and Drug Administration (FDA) has approved tisagenlecleucel (Kymriah®) for its second indication.
The chimeric antigen receptor (CAR) T-cell therapy is now approved to treat adults with relapsed or refractory large B-cell lymphoma after 2 or more lines of systemic therapy.
This includes patients with diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.
The application for tisagenlecleucel in B-cell lymphoma was granted priority review. The FDA aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
Tisagenlecleucel is also FDA-approved to treat patients age 25 and younger who have B-cell precursor acute lymphoblastic leukemia that is refractory or in second or later relapse.
Access to tisagenlecleucel
The prescribing information for tisagenlecleucel includes a boxed warning detailing the risk of cytokine release syndrome (CRS) and neurological toxicities for patients receiving tisagenlecleucel.
Because of these risks, tisagenlecleucel is only available through a Risk Evaluation and Mitigation Strategy (REMS) program. The REMS program serves to inform and educate healthcare professionals about the risks associated with tisagenlecleucel treatment.
Novartis, the company marketing tisagenlecleucel, has established a network of certified treatment centers throughout the US. Staff at these centers are trained on the use of tisagenlecleucel and appropriate patient care.
Tisagenlecleucel is manufactured at a Novartis facility in Morris Plains, New Jersey. In the US, the target turnaround time for manufacturing tisagenlecleucel is 22 days.
Tisagenlecleucel costs $475,000 for a single course of treatment. However, Novartis said it is collaborating with the US Centers for Medicare and Medicaid Services on the creation of an appropriate value-based pricing approach.
The company also has a program called KYMRIAH CARES™, which offers financial assistance to eligible patients to help them gain access to tisagenlecleucel.
Phase 2 trial
The FDA approval of tisagenlecleucel for adults with relapsed/refractory B-cell lymphoma is based on results of the phase 2 JULIET trial.
The prescribing information for tisagenlecleucel includes data on 106 patients treated on this trial.
Only 68 of these patients were evaluable for efficacy. They had a median age of 56 (range, 22 to 74), and 71% were male.
Seventy-eight percent of patients had primary DLBCL not otherwise specified, and 22% had DLBCL following transformation from follicular lymphoma. Seventeen percent had high grade DLBCL.
Fifty-six percent of patients had refractory disease, and 44% had relapsed after their last therapy. The median number of prior therapies was 3 (range, 1 to 6), and 44% of patients had undergone autologous transplant.
Ninety percent of patients received lymphodepleting chemotherapy (66% fludarabine and 24% bendamustine) prior to tisagenlecleucel, and 10% did not. The median dose of tisagenlecleucel was 3.5 × 108 CAR+ T cells (range, 1.0 to 5.2 × 108).
The overall response rate was 50%, with 32% of patients achieving a complete response and 18% achieving a partial response. The median duration of response was not reached with a median follow-up of 9.4 months.
In all 106 patients infused with tisagenlecleucel, the most common grade 3/4 adverse events were infections (25%), CRS (23%), neurologic events (18%), febrile neutropenia (17%), encephalopathy (11%), lymphopenia (94%), neutropenia (81%), leukopenia (77%), anemia (58%), thrombocytopenia (54%), hypophosphatemia (24%), hypokalemia (12%), and hyponatremia (11%).
Three patients died within 30 days of tisagenlecleucel infusion. All of them had CRS and either stable or progressive disease. One of these patients developed bowel necrosis.
One patient died of infection. There were no deaths attributed to neurological events, and no fatal cases of cerebral edema.
The US Food and Drug Administration (FDA) has approved tisagenlecleucel (Kymriah®) for its second indication.
The chimeric antigen receptor (CAR) T-cell therapy is now approved to treat adults with relapsed or refractory large B-cell lymphoma after 2 or more lines of systemic therapy.
This includes patients with diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.
The application for tisagenlecleucel in B-cell lymphoma was granted priority review. The FDA aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
Tisagenlecleucel is also FDA-approved to treat patients age 25 and younger who have B-cell precursor acute lymphoblastic leukemia that is refractory or in second or later relapse.
Access to tisagenlecleucel
The prescribing information for tisagenlecleucel includes a boxed warning detailing the risk of cytokine release syndrome (CRS) and neurological toxicities for patients receiving tisagenlecleucel.
Because of these risks, tisagenlecleucel is only available through a Risk Evaluation and Mitigation Strategy (REMS) program. The REMS program serves to inform and educate healthcare professionals about the risks associated with tisagenlecleucel treatment.
Novartis, the company marketing tisagenlecleucel, has established a network of certified treatment centers throughout the US. Staff at these centers are trained on the use of tisagenlecleucel and appropriate patient care.
Tisagenlecleucel is manufactured at a Novartis facility in Morris Plains, New Jersey. In the US, the target turnaround time for manufacturing tisagenlecleucel is 22 days.
Tisagenlecleucel costs $475,000 for a single course of treatment. However, Novartis said it is collaborating with the US Centers for Medicare and Medicaid Services on the creation of an appropriate value-based pricing approach.
The company also has a program called KYMRIAH CARES™, which offers financial assistance to eligible patients to help them gain access to tisagenlecleucel.
Phase 2 trial
The FDA approval of tisagenlecleucel for adults with relapsed/refractory B-cell lymphoma is based on results of the phase 2 JULIET trial.
The prescribing information for tisagenlecleucel includes data on 106 patients treated on this trial.
Only 68 of these patients were evaluable for efficacy. They had a median age of 56 (range, 22 to 74), and 71% were male.
Seventy-eight percent of patients had primary DLBCL not otherwise specified, and 22% had DLBCL following transformation from follicular lymphoma. Seventeen percent had high grade DLBCL.
Fifty-six percent of patients had refractory disease, and 44% had relapsed after their last therapy. The median number of prior therapies was 3 (range, 1 to 6), and 44% of patients had undergone autologous transplant.
Ninety percent of patients received lymphodepleting chemotherapy (66% fludarabine and 24% bendamustine) prior to tisagenlecleucel, and 10% did not. The median dose of tisagenlecleucel was 3.5 × 108 CAR+ T cells (range, 1.0 to 5.2 × 108).
The overall response rate was 50%, with 32% of patients achieving a complete response and 18% achieving a partial response. The median duration of response was not reached with a median follow-up of 9.4 months.
In all 106 patients infused with tisagenlecleucel, the most common grade 3/4 adverse events were infections (25%), CRS (23%), neurologic events (18%), febrile neutropenia (17%), encephalopathy (11%), lymphopenia (94%), neutropenia (81%), leukopenia (77%), anemia (58%), thrombocytopenia (54%), hypophosphatemia (24%), hypokalemia (12%), and hyponatremia (11%).
Three patients died within 30 days of tisagenlecleucel infusion. All of them had CRS and either stable or progressive disease. One of these patients developed bowel necrosis.
One patient died of infection. There were no deaths attributed to neurological events, and no fatal cases of cerebral edema.
Predicting response to CAR T-cell therapy in CLL

Researchers may have discovered why some patients with advanced chronic lymphocytic leukemia (CLL) don’t respond to chimeric antigen receptor (CAR) T-cell therapy.
The team found that CLL patients with elevated levels of “early memory” T cells prior to receiving CAR T-cell therapy had a partial or complete response to treatment, while patients with lower levels of these T cells did not respond.
The early memory T cells were marked by the expression of CD8 and CD27, as well as the absence of CD45RO.
The researchers validated the association between the early memory T cells and response in a small group of patients, predicting with 100% accuracy which patients would achieve a complete response.
Joseph A. Fraietta, PhD, of the University of Pennsylvania in Philadelphia, and his colleagues reported these findings in Nature Medicine. This research was supported, in part, by Novartis.
For this study, the researchers retrospectively analyzed 41 patients with advanced, heavily pretreated, high-risk CLL who received at least 1 dose of CD19-directed CAR T cells.
Consistent with the team’s previously reported findings, they were not able to identify patient or disease-specific factors that predict who responds best to the therapy.
Therefore, the researchers compared the gene expression profiles and phenotypes of T cells in patients who had a complete response, partial response, or no response to therapy.
The CAR T cells that persisted and expanded in complete responders were enriched in genes that regulate early memory and effector T cells and possess the IL-6/STAT3 signature.
Non-responders, on the other hand, expressed genes involved in late T-cell differentiation, glycolysis, exhaustion, and apoptosis. These characteristics make for a weaker set of T cells to persist, expand, and fight the CLL.
“Pre-existing T-cell qualities have previously been associated with poor clinical response to cancer therapy, as well differentiation in the T cells,” Dr Fraietta said. “What is special about what we have done here is finding that critical cell subset and signature.”
Elevated levels of the IL-6/STAT3 signaling pathway in these early T cells correlated with clinical responses to CAR T-cell therapy.
To validate these findings, the researchers screened for the early memory T cells in a group of 8 CLL patients, before and after CAR T-cell therapy. The team identified the complete responders with 100% specificity and sensitivity.
“With a very robust biomarker like this, we can take a blood sample, measure the frequency of this T-cell population, and decide with a degree of confidence whether we can apply this therapy and know the patient would have a response,” Dr Fraietta said.
“The ability to select patients most likely to respond would have tremendous clinical impact, as this therapy would be applied only to patients most likely to benefit, allowing patients unlikely to respond to pursue other options.”
These findings also suggest the possibility of improving CAR T-cell therapy by selecting for cell manufacturing the subpopulation of T cells responsible for driving responses. However, this approach would come with challenges.
“What we’ve seen in these non-responders is that the frequency of these T cells is low, so it would be very hard to infuse them as starting populations,” said study author J. Joseph Melenhorst, PhD, also of the University of Pennsylvania.
“But one way to potentially boost their efficacy is by adding checkpoint inhibitors with the therapy to block the negative regulation prior to CAR T-cell therapy, which a past, separate study has shown can help elicit responses in these patients.”
The researchers also noted that it’s unclear why some patients’ T cells are suboptimal prior to treatment. However, the team believes this could have to do with prior therapies.
Future studies with a larger group of CLL patients should be conducted to help answer these questions and validate the findings from this study, the researchers said.
Researchers may have discovered why some patients with advanced chronic lymphocytic leukemia (CLL) don’t respond to chimeric antigen receptor (CAR) T-cell therapy.
The team found that CLL patients with elevated levels of “early memory” T cells prior to receiving CAR T-cell therapy had a partial or complete response to treatment, while patients with lower levels of these T cells did not respond.
The early memory T cells were marked by the expression of CD8 and CD27, as well as the absence of CD45RO.
The researchers validated the association between the early memory T cells and response in a small group of patients, predicting with 100% accuracy which patients would achieve a complete response.
Joseph A. Fraietta, PhD, of the University of Pennsylvania in Philadelphia, and his colleagues reported these findings in Nature Medicine. This research was supported, in part, by Novartis.
For this study, the researchers retrospectively analyzed 41 patients with advanced, heavily pretreated, high-risk CLL who received at least 1 dose of CD19-directed CAR T cells.
Consistent with the team’s previously reported findings, they were not able to identify patient or disease-specific factors that predict who responds best to the therapy.
Therefore, the researchers compared the gene expression profiles and phenotypes of T cells in patients who had a complete response, partial response, or no response to therapy.
The CAR T cells that persisted and expanded in complete responders were enriched in genes that regulate early memory and effector T cells and possess the IL-6/STAT3 signature.
Non-responders, on the other hand, expressed genes involved in late T-cell differentiation, glycolysis, exhaustion, and apoptosis. These characteristics make for a weaker set of T cells to persist, expand, and fight the CLL.
“Pre-existing T-cell qualities have previously been associated with poor clinical response to cancer therapy, as well differentiation in the T cells,” Dr Fraietta said. “What is special about what we have done here is finding that critical cell subset and signature.”
Elevated levels of the IL-6/STAT3 signaling pathway in these early T cells correlated with clinical responses to CAR T-cell therapy.
To validate these findings, the researchers screened for the early memory T cells in a group of 8 CLL patients, before and after CAR T-cell therapy. The team identified the complete responders with 100% specificity and sensitivity.
“With a very robust biomarker like this, we can take a blood sample, measure the frequency of this T-cell population, and decide with a degree of confidence whether we can apply this therapy and know the patient would have a response,” Dr Fraietta said.
“The ability to select patients most likely to respond would have tremendous clinical impact, as this therapy would be applied only to patients most likely to benefit, allowing patients unlikely to respond to pursue other options.”
These findings also suggest the possibility of improving CAR T-cell therapy by selecting for cell manufacturing the subpopulation of T cells responsible for driving responses. However, this approach would come with challenges.
“What we’ve seen in these non-responders is that the frequency of these T cells is low, so it would be very hard to infuse them as starting populations,” said study author J. Joseph Melenhorst, PhD, also of the University of Pennsylvania.
“But one way to potentially boost their efficacy is by adding checkpoint inhibitors with the therapy to block the negative regulation prior to CAR T-cell therapy, which a past, separate study has shown can help elicit responses in these patients.”
The researchers also noted that it’s unclear why some patients’ T cells are suboptimal prior to treatment. However, the team believes this could have to do with prior therapies.
Future studies with a larger group of CLL patients should be conducted to help answer these questions and validate the findings from this study, the researchers said.
Researchers may have discovered why some patients with advanced chronic lymphocytic leukemia (CLL) don’t respond to chimeric antigen receptor (CAR) T-cell therapy.
The team found that CLL patients with elevated levels of “early memory” T cells prior to receiving CAR T-cell therapy had a partial or complete response to treatment, while patients with lower levels of these T cells did not respond.
The early memory T cells were marked by the expression of CD8 and CD27, as well as the absence of CD45RO.
The researchers validated the association between the early memory T cells and response in a small group of patients, predicting with 100% accuracy which patients would achieve a complete response.
Joseph A. Fraietta, PhD, of the University of Pennsylvania in Philadelphia, and his colleagues reported these findings in Nature Medicine. This research was supported, in part, by Novartis.
For this study, the researchers retrospectively analyzed 41 patients with advanced, heavily pretreated, high-risk CLL who received at least 1 dose of CD19-directed CAR T cells.
Consistent with the team’s previously reported findings, they were not able to identify patient or disease-specific factors that predict who responds best to the therapy.
Therefore, the researchers compared the gene expression profiles and phenotypes of T cells in patients who had a complete response, partial response, or no response to therapy.
The CAR T cells that persisted and expanded in complete responders were enriched in genes that regulate early memory and effector T cells and possess the IL-6/STAT3 signature.
Non-responders, on the other hand, expressed genes involved in late T-cell differentiation, glycolysis, exhaustion, and apoptosis. These characteristics make for a weaker set of T cells to persist, expand, and fight the CLL.
“Pre-existing T-cell qualities have previously been associated with poor clinical response to cancer therapy, as well differentiation in the T cells,” Dr Fraietta said. “What is special about what we have done here is finding that critical cell subset and signature.”
Elevated levels of the IL-6/STAT3 signaling pathway in these early T cells correlated with clinical responses to CAR T-cell therapy.
To validate these findings, the researchers screened for the early memory T cells in a group of 8 CLL patients, before and after CAR T-cell therapy. The team identified the complete responders with 100% specificity and sensitivity.
“With a very robust biomarker like this, we can take a blood sample, measure the frequency of this T-cell population, and decide with a degree of confidence whether we can apply this therapy and know the patient would have a response,” Dr Fraietta said.
“The ability to select patients most likely to respond would have tremendous clinical impact, as this therapy would be applied only to patients most likely to benefit, allowing patients unlikely to respond to pursue other options.”
These findings also suggest the possibility of improving CAR T-cell therapy by selecting for cell manufacturing the subpopulation of T cells responsible for driving responses. However, this approach would come with challenges.
“What we’ve seen in these non-responders is that the frequency of these T cells is low, so it would be very hard to infuse them as starting populations,” said study author J. Joseph Melenhorst, PhD, also of the University of Pennsylvania.
“But one way to potentially boost their efficacy is by adding checkpoint inhibitors with the therapy to block the negative regulation prior to CAR T-cell therapy, which a past, separate study has shown can help elicit responses in these patients.”
The researchers also noted that it’s unclear why some patients’ T cells are suboptimal prior to treatment. However, the team believes this could have to do with prior therapies.
Future studies with a larger group of CLL patients should be conducted to help answer these questions and validate the findings from this study, the researchers said.
Team identifies 5 subtypes of DLBCL

New research has revealed 5 genetic subtypes of diffuse large B-cell lymphoma (DLBCL).
Researchers identified a group of low-risk activated B-cell (ABC) DLBCLs, 2 subsets of germinal center B-cell (GCB) DLBCLs, a group of ABC/GCB-independent DLBCLs, and a group of ABC DLBCLs with genetic characteristics found in primary central nervous system lymphoma and testicular lymphoma.
The researchers believe these findings may have revealed new therapeutic targets for DLBCL, some of which could be inhibited by drugs that are already approved or under investigation in clinical trials.
Margaret Shipp, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and her colleagues conducted this research and reported the results in Nature Medicine.
The team performed genetic analyses on samples from 304 DLBCL patients and observed great genetic diversity. The median number of genetic driver alterations in individual tumors was 17.
The researchers integrated data on 3 types of genetic alterations—recurrent mutations, somatic copy number alterations, and structural variants—to define previously unappreciated DLBCL subtypes.
“Specific genes that were perturbed by mutations could also be altered by changes in gene copy numbers or by chromosomal rearrangements, underscoring the importance of evaluating all 3 types of genetic alterations,” Dr Shipp noted.
“Most importantly, we saw that there were 5 discrete types of DLBCL that were distinguished one from another on the basis of the specific types of genetic alterations that occurred in combination.”
The researchers classified these subtypes as clusters (C) 1 to 5.
C1 consisted of largely ABC-DLBCLs with genetic features of an extra-follicular, possibly marginal zone origin.
C2 included both ABC and GCB DLBCLs with biallelic inactivation of TP53, 9p21.3/CDKN2A, and associated genomic instability.
Most DLBCLs in C3 were of the GCB subtype and were characterized by BCL2 structural variants and alterations of PTEN and epigenetic enzymes.
C4 consisted largely of GCB DLBCLs with alterations in BCR/PI3K, JAK/STAT, and BRAF pathway components and multiple histones.
Most C5 DLBCLs were of the ABC subtype, and the researchers said the major components of the C5 signature—BCL2 gain, concordant MYD88L265P/CD79B mutations, and mutations of ETV6, PIM1, GRHPR, TBL1XR1, and BTG1—were similar to those observed in primary central nervous system and testicular lymphoma.
Dr Shipp and her colleagues also identified a sixth cluster of DLBCLs (dubbed C0) that “lacked defining genetic drivers.”
Finally, the team found that patients with C0, C1, and C4 DLBCLs had more favorable outcomes, while patients with C2, C3, and C5 DLBCLs had less favorable outcomes.
“We feel this research opens the door to a whole series of additional investigations to understand how the combinations of these genetic alterations work together, and then to use that information to benefit patients with targeted therapies,” Dr Shipp said.
She and her colleagues are now working on creating a clinical tool to identify these genetic signatures in patients. The team is also developing clinical trials that will match patients with given genetic signatures to targeted treatments.
Another group of researchers recently identified 4 genetic subtypes of DLBCL.
New research has revealed 5 genetic subtypes of diffuse large B-cell lymphoma (DLBCL).
Researchers identified a group of low-risk activated B-cell (ABC) DLBCLs, 2 subsets of germinal center B-cell (GCB) DLBCLs, a group of ABC/GCB-independent DLBCLs, and a group of ABC DLBCLs with genetic characteristics found in primary central nervous system lymphoma and testicular lymphoma.
The researchers believe these findings may have revealed new therapeutic targets for DLBCL, some of which could be inhibited by drugs that are already approved or under investigation in clinical trials.
Margaret Shipp, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and her colleagues conducted this research and reported the results in Nature Medicine.
The team performed genetic analyses on samples from 304 DLBCL patients and observed great genetic diversity. The median number of genetic driver alterations in individual tumors was 17.
The researchers integrated data on 3 types of genetic alterations—recurrent mutations, somatic copy number alterations, and structural variants—to define previously unappreciated DLBCL subtypes.
“Specific genes that were perturbed by mutations could also be altered by changes in gene copy numbers or by chromosomal rearrangements, underscoring the importance of evaluating all 3 types of genetic alterations,” Dr Shipp noted.
“Most importantly, we saw that there were 5 discrete types of DLBCL that were distinguished one from another on the basis of the specific types of genetic alterations that occurred in combination.”
The researchers classified these subtypes as clusters (C) 1 to 5.
C1 consisted of largely ABC-DLBCLs with genetic features of an extra-follicular, possibly marginal zone origin.
C2 included both ABC and GCB DLBCLs with biallelic inactivation of TP53, 9p21.3/CDKN2A, and associated genomic instability.
Most DLBCLs in C3 were of the GCB subtype and were characterized by BCL2 structural variants and alterations of PTEN and epigenetic enzymes.
C4 consisted largely of GCB DLBCLs with alterations in BCR/PI3K, JAK/STAT, and BRAF pathway components and multiple histones.
Most C5 DLBCLs were of the ABC subtype, and the researchers said the major components of the C5 signature—BCL2 gain, concordant MYD88L265P/CD79B mutations, and mutations of ETV6, PIM1, GRHPR, TBL1XR1, and BTG1—were similar to those observed in primary central nervous system and testicular lymphoma.
Dr Shipp and her colleagues also identified a sixth cluster of DLBCLs (dubbed C0) that “lacked defining genetic drivers.”
Finally, the team found that patients with C0, C1, and C4 DLBCLs had more favorable outcomes, while patients with C2, C3, and C5 DLBCLs had less favorable outcomes.
“We feel this research opens the door to a whole series of additional investigations to understand how the combinations of these genetic alterations work together, and then to use that information to benefit patients with targeted therapies,” Dr Shipp said.
She and her colleagues are now working on creating a clinical tool to identify these genetic signatures in patients. The team is also developing clinical trials that will match patients with given genetic signatures to targeted treatments.
Another group of researchers recently identified 4 genetic subtypes of DLBCL.
New research has revealed 5 genetic subtypes of diffuse large B-cell lymphoma (DLBCL).
Researchers identified a group of low-risk activated B-cell (ABC) DLBCLs, 2 subsets of germinal center B-cell (GCB) DLBCLs, a group of ABC/GCB-independent DLBCLs, and a group of ABC DLBCLs with genetic characteristics found in primary central nervous system lymphoma and testicular lymphoma.
The researchers believe these findings may have revealed new therapeutic targets for DLBCL, some of which could be inhibited by drugs that are already approved or under investigation in clinical trials.
Margaret Shipp, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and her colleagues conducted this research and reported the results in Nature Medicine.
The team performed genetic analyses on samples from 304 DLBCL patients and observed great genetic diversity. The median number of genetic driver alterations in individual tumors was 17.
The researchers integrated data on 3 types of genetic alterations—recurrent mutations, somatic copy number alterations, and structural variants—to define previously unappreciated DLBCL subtypes.
“Specific genes that were perturbed by mutations could also be altered by changes in gene copy numbers or by chromosomal rearrangements, underscoring the importance of evaluating all 3 types of genetic alterations,” Dr Shipp noted.
“Most importantly, we saw that there were 5 discrete types of DLBCL that were distinguished one from another on the basis of the specific types of genetic alterations that occurred in combination.”
The researchers classified these subtypes as clusters (C) 1 to 5.
C1 consisted of largely ABC-DLBCLs with genetic features of an extra-follicular, possibly marginal zone origin.
C2 included both ABC and GCB DLBCLs with biallelic inactivation of TP53, 9p21.3/CDKN2A, and associated genomic instability.
Most DLBCLs in C3 were of the GCB subtype and were characterized by BCL2 structural variants and alterations of PTEN and epigenetic enzymes.
C4 consisted largely of GCB DLBCLs with alterations in BCR/PI3K, JAK/STAT, and BRAF pathway components and multiple histones.
Most C5 DLBCLs were of the ABC subtype, and the researchers said the major components of the C5 signature—BCL2 gain, concordant MYD88L265P/CD79B mutations, and mutations of ETV6, PIM1, GRHPR, TBL1XR1, and BTG1—were similar to those observed in primary central nervous system and testicular lymphoma.
Dr Shipp and her colleagues also identified a sixth cluster of DLBCLs (dubbed C0) that “lacked defining genetic drivers.”
Finally, the team found that patients with C0, C1, and C4 DLBCLs had more favorable outcomes, while patients with C2, C3, and C5 DLBCLs had less favorable outcomes.
“We feel this research opens the door to a whole series of additional investigations to understand how the combinations of these genetic alterations work together, and then to use that information to benefit patients with targeted therapies,” Dr Shipp said.
She and her colleagues are now working on creating a clinical tool to identify these genetic signatures in patients. The team is also developing clinical trials that will match patients with given genetic signatures to targeted treatments.
Another group of researchers recently identified 4 genetic subtypes of DLBCL.
CHMP recommends approval for generic carmustine
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has adopted a positive opinion of Carmustine Obvius, a generic version of Carmubris.
The CHMP is recommending marketing authorization for Carmustine Obvius as second-line treatment for Hodgkin and non-Hodgkin lymphoma as well as to treat new or recurrent brain tumors, including glioblastoma, medulloblastoma, astrocytoma, and metastatic brain tumors.
The CHMP’s opinion on Carmustine Obvius will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP’s recommendation, the commission will grant a centralized marketing authorization that will be valid in the European Union.
The EC typically makes a decision on a product within 67 days of the CHMP’s recommendation.
If approved, Carmustine Obvius will be available as a 100 mg powder and solvent for solution for infusion.
The active substance of Carmustine Obvius is carmustine, an alkylating antineoplastic agent of the nitrosourea type, which prevents DNA replication and transcription by alkylating reactive sites on nucleoproteins.
Carmustine Obvius is a generic of Carmubris, which has been authorized in the European Union since July 31, 1996.
Since Carmustine Obvius is administered intravenously and is 100% bioavailable, a bioequivalence study of the drug versus Carmubris was not required.
Carmustine Obvius is a product of Obvius Investment B.V.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has adopted a positive opinion of Carmustine Obvius, a generic version of Carmubris.
The CHMP is recommending marketing authorization for Carmustine Obvius as second-line treatment for Hodgkin and non-Hodgkin lymphoma as well as to treat new or recurrent brain tumors, including glioblastoma, medulloblastoma, astrocytoma, and metastatic brain tumors.
The CHMP’s opinion on Carmustine Obvius will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP’s recommendation, the commission will grant a centralized marketing authorization that will be valid in the European Union.
The EC typically makes a decision on a product within 67 days of the CHMP’s recommendation.
If approved, Carmustine Obvius will be available as a 100 mg powder and solvent for solution for infusion.
The active substance of Carmustine Obvius is carmustine, an alkylating antineoplastic agent of the nitrosourea type, which prevents DNA replication and transcription by alkylating reactive sites on nucleoproteins.
Carmustine Obvius is a generic of Carmubris, which has been authorized in the European Union since July 31, 1996.
Since Carmustine Obvius is administered intravenously and is 100% bioavailable, a bioequivalence study of the drug versus Carmubris was not required.
Carmustine Obvius is a product of Obvius Investment B.V.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has adopted a positive opinion of Carmustine Obvius, a generic version of Carmubris.
The CHMP is recommending marketing authorization for Carmustine Obvius as second-line treatment for Hodgkin and non-Hodgkin lymphoma as well as to treat new or recurrent brain tumors, including glioblastoma, medulloblastoma, astrocytoma, and metastatic brain tumors.
The CHMP’s opinion on Carmustine Obvius will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP’s recommendation, the commission will grant a centralized marketing authorization that will be valid in the European Union.
The EC typically makes a decision on a product within 67 days of the CHMP’s recommendation.
If approved, Carmustine Obvius will be available as a 100 mg powder and solvent for solution for infusion.
The active substance of Carmustine Obvius is carmustine, an alkylating antineoplastic agent of the nitrosourea type, which prevents DNA replication and transcription by alkylating reactive sites on nucleoproteins.
Carmustine Obvius is a generic of Carmubris, which has been authorized in the European Union since July 31, 1996.
Since Carmustine Obvius is administered intravenously and is 100% bioavailable, a bioequivalence study of the drug versus Carmubris was not required.
Carmustine Obvius is a product of Obvius Investment B.V.
Radioimmunoconjugate shows activity in NHL

CHICAGO—The radioimmunoconjugate 177Lu-NNV003 has demonstrated activity against non-Hodgkin lymphomas (NHLs).
Experiments showed that 177Lu-NNV003 can inhibit proliferation in mantle cell lymphoma (MCL), diffuse large B-cell lymphoma (DLBCL), and chronic lymphocytic leukemia (CLL) cell lines.
177Lu-NNV003 also exhibited an antitumor effect and prolonged survival in mouse models of MCL, DLBCL, and CLL.
These results were presented at the AACR Annual Meeting 2018 (abstract 848).
This research was conducted by employees of Nordic Nanovector and other researchers. Nordic Nanovector is the company developing 177Lu-NNV003.
177Lu-NNV003 consists of a chimeric antibody targeting CD37 (NNV003) conjugated with p-SCN-Bn-DOTA, which chelates the β-emitting radionuclide lutetium-177.
The researchers found that 177Lu-NNV003 inhibited proliferation in all 3 NHL cell lines tested—MEC-2 (CLL), DOHH2 (DLBCL), and REC-1 (MCL). DOHH2 was the most radiosensitive cell line.
The unlabeled NNV003 antibody, on the other hand, did not exhibit an antiproliferative effect in these cell lines. However, NNV003 did induce antibody-dependent cellular cytotoxicity in MEC-2 and DOHH2 cells and antibody-dependent cellular phagocytosis in MEC-2 cells.
The researchers found that 177Lu-NNV003 targeted CD37-positive cells and demonstrated antitumor effects in mouse models of MCL, CLL, and DLBCL.
REC-1 model
177Lu-NNV003 prolonged survival in CB17 SCID mice injected with REC-1 cells and cured 50% to 60% of the mice.
Median survival times were 55 days in mice that received sodium chloride (NaCl), 85 days in mice that received NNV003 (0.167 mg/kg), and 61 days in mice that received 177Lu-IgG1 (100 MBq/kg).
On the other hand, mice treated with 177Lu-NNV003 had a median survival of 152 days (100 MBq/kg), or the median survival was not reached (50 MBq/kg).
The difference in survival was significant for 177Lu-NNV003 recipients compared to recipients of NaCl (P<0.001) or 177Lu-IgG1 (P<0.002).
MEC-2 model
177Lu-NNV003 also prolonged survival in NRG mice injected with MEC-2 cells.
The median survival was 21 days in mice that received NaCl, NNV003 (2 x 0.33 mg/kg), or 177Lu-IgG1 (200 MBq/kg).
However, mice treated with 177Lu-NNV003 had a median survival of 32 days (200 MBq/kg) or 29 days (2 x 200 MBq/kg).
The difference in survival was significant for 177Lu-NNV003 recipients compared to recipients of 177Lu-IgG1 (P<0.025) or 2 x NNV003 (P<0.02).
DOHH2 model
RAG-2 mice injected with DOHH2 cells had survival times surpassing 200 days after treatment with NNV003 or 177Lu-NNV003.
The median survival was not reached in mice that received NNV003 (at 2 or 30 mg/kg) or 177Lu-NNV003 (at 200, 300, or 400 MBq/kg). But the median survival was 46 days in mice that received NaCl and 47 days in mice that received 177Lu-IgG1 (300 MBq/kg).
The difference in survival was significant for recipients of NNV003 or 177Lu-NNV003 compared to recipients of 177Lu-IgG1 or NaCl (P<0.001 for all).
Toxicity and biodistribution
The researchers noted that the 177Lu-labeled antibodies caused transient hematologic toxicity in the mice.
In the MEC-2 and DOHH2 models, there was no redistribution of 177Lu-NNV003 in organs after initial uptake. (The researchers did not report on biodistribution in the REC-1 model.)
177Lu-NNV003 had low uptake in the liver, spleen, kidneys, and femur. The researchers said this suggests 177Lu-NNV003 is stable in vivo, as free lutetium-177 tends to accumulate in bones.
CHICAGO—The radioimmunoconjugate 177Lu-NNV003 has demonstrated activity against non-Hodgkin lymphomas (NHLs).
Experiments showed that 177Lu-NNV003 can inhibit proliferation in mantle cell lymphoma (MCL), diffuse large B-cell lymphoma (DLBCL), and chronic lymphocytic leukemia (CLL) cell lines.
177Lu-NNV003 also exhibited an antitumor effect and prolonged survival in mouse models of MCL, DLBCL, and CLL.
These results were presented at the AACR Annual Meeting 2018 (abstract 848).
This research was conducted by employees of Nordic Nanovector and other researchers. Nordic Nanovector is the company developing 177Lu-NNV003.
177Lu-NNV003 consists of a chimeric antibody targeting CD37 (NNV003) conjugated with p-SCN-Bn-DOTA, which chelates the β-emitting radionuclide lutetium-177.
The researchers found that 177Lu-NNV003 inhibited proliferation in all 3 NHL cell lines tested—MEC-2 (CLL), DOHH2 (DLBCL), and REC-1 (MCL). DOHH2 was the most radiosensitive cell line.
The unlabeled NNV003 antibody, on the other hand, did not exhibit an antiproliferative effect in these cell lines. However, NNV003 did induce antibody-dependent cellular cytotoxicity in MEC-2 and DOHH2 cells and antibody-dependent cellular phagocytosis in MEC-2 cells.
The researchers found that 177Lu-NNV003 targeted CD37-positive cells and demonstrated antitumor effects in mouse models of MCL, CLL, and DLBCL.
REC-1 model
177Lu-NNV003 prolonged survival in CB17 SCID mice injected with REC-1 cells and cured 50% to 60% of the mice.
Median survival times were 55 days in mice that received sodium chloride (NaCl), 85 days in mice that received NNV003 (0.167 mg/kg), and 61 days in mice that received 177Lu-IgG1 (100 MBq/kg).
On the other hand, mice treated with 177Lu-NNV003 had a median survival of 152 days (100 MBq/kg), or the median survival was not reached (50 MBq/kg).
The difference in survival was significant for 177Lu-NNV003 recipients compared to recipients of NaCl (P<0.001) or 177Lu-IgG1 (P<0.002).
MEC-2 model
177Lu-NNV003 also prolonged survival in NRG mice injected with MEC-2 cells.
The median survival was 21 days in mice that received NaCl, NNV003 (2 x 0.33 mg/kg), or 177Lu-IgG1 (200 MBq/kg).
However, mice treated with 177Lu-NNV003 had a median survival of 32 days (200 MBq/kg) or 29 days (2 x 200 MBq/kg).
The difference in survival was significant for 177Lu-NNV003 recipients compared to recipients of 177Lu-IgG1 (P<0.025) or 2 x NNV003 (P<0.02).
DOHH2 model
RAG-2 mice injected with DOHH2 cells had survival times surpassing 200 days after treatment with NNV003 or 177Lu-NNV003.
The median survival was not reached in mice that received NNV003 (at 2 or 30 mg/kg) or 177Lu-NNV003 (at 200, 300, or 400 MBq/kg). But the median survival was 46 days in mice that received NaCl and 47 days in mice that received 177Lu-IgG1 (300 MBq/kg).
The difference in survival was significant for recipients of NNV003 or 177Lu-NNV003 compared to recipients of 177Lu-IgG1 or NaCl (P<0.001 for all).
Toxicity and biodistribution
The researchers noted that the 177Lu-labeled antibodies caused transient hematologic toxicity in the mice.
In the MEC-2 and DOHH2 models, there was no redistribution of 177Lu-NNV003 in organs after initial uptake. (The researchers did not report on biodistribution in the REC-1 model.)
177Lu-NNV003 had low uptake in the liver, spleen, kidneys, and femur. The researchers said this suggests 177Lu-NNV003 is stable in vivo, as free lutetium-177 tends to accumulate in bones.
CHICAGO—The radioimmunoconjugate 177Lu-NNV003 has demonstrated activity against non-Hodgkin lymphomas (NHLs).
Experiments showed that 177Lu-NNV003 can inhibit proliferation in mantle cell lymphoma (MCL), diffuse large B-cell lymphoma (DLBCL), and chronic lymphocytic leukemia (CLL) cell lines.
177Lu-NNV003 also exhibited an antitumor effect and prolonged survival in mouse models of MCL, DLBCL, and CLL.
These results were presented at the AACR Annual Meeting 2018 (abstract 848).
This research was conducted by employees of Nordic Nanovector and other researchers. Nordic Nanovector is the company developing 177Lu-NNV003.
177Lu-NNV003 consists of a chimeric antibody targeting CD37 (NNV003) conjugated with p-SCN-Bn-DOTA, which chelates the β-emitting radionuclide lutetium-177.
The researchers found that 177Lu-NNV003 inhibited proliferation in all 3 NHL cell lines tested—MEC-2 (CLL), DOHH2 (DLBCL), and REC-1 (MCL). DOHH2 was the most radiosensitive cell line.
The unlabeled NNV003 antibody, on the other hand, did not exhibit an antiproliferative effect in these cell lines. However, NNV003 did induce antibody-dependent cellular cytotoxicity in MEC-2 and DOHH2 cells and antibody-dependent cellular phagocytosis in MEC-2 cells.
The researchers found that 177Lu-NNV003 targeted CD37-positive cells and demonstrated antitumor effects in mouse models of MCL, CLL, and DLBCL.
REC-1 model
177Lu-NNV003 prolonged survival in CB17 SCID mice injected with REC-1 cells and cured 50% to 60% of the mice.
Median survival times were 55 days in mice that received sodium chloride (NaCl), 85 days in mice that received NNV003 (0.167 mg/kg), and 61 days in mice that received 177Lu-IgG1 (100 MBq/kg).
On the other hand, mice treated with 177Lu-NNV003 had a median survival of 152 days (100 MBq/kg), or the median survival was not reached (50 MBq/kg).
The difference in survival was significant for 177Lu-NNV003 recipients compared to recipients of NaCl (P<0.001) or 177Lu-IgG1 (P<0.002).
MEC-2 model
177Lu-NNV003 also prolonged survival in NRG mice injected with MEC-2 cells.
The median survival was 21 days in mice that received NaCl, NNV003 (2 x 0.33 mg/kg), or 177Lu-IgG1 (200 MBq/kg).
However, mice treated with 177Lu-NNV003 had a median survival of 32 days (200 MBq/kg) or 29 days (2 x 200 MBq/kg).
The difference in survival was significant for 177Lu-NNV003 recipients compared to recipients of 177Lu-IgG1 (P<0.025) or 2 x NNV003 (P<0.02).
DOHH2 model
RAG-2 mice injected with DOHH2 cells had survival times surpassing 200 days after treatment with NNV003 or 177Lu-NNV003.
The median survival was not reached in mice that received NNV003 (at 2 or 30 mg/kg) or 177Lu-NNV003 (at 200, 300, or 400 MBq/kg). But the median survival was 46 days in mice that received NaCl and 47 days in mice that received 177Lu-IgG1 (300 MBq/kg).
The difference in survival was significant for recipients of NNV003 or 177Lu-NNV003 compared to recipients of 177Lu-IgG1 or NaCl (P<0.001 for all).
Toxicity and biodistribution
The researchers noted that the 177Lu-labeled antibodies caused transient hematologic toxicity in the mice.
In the MEC-2 and DOHH2 models, there was no redistribution of 177Lu-NNV003 in organs after initial uptake. (The researchers did not report on biodistribution in the REC-1 model.)
177Lu-NNV003 had low uptake in the liver, spleen, kidneys, and femur. The researchers said this suggests 177Lu-NNV003 is stable in vivo, as free lutetium-177 tends to accumulate in bones.
CDK inhibitor synergizes with venetoclax in CLL

CHICAGO—Researchers have reported “strong synergy” between the CDK2/9 inhibitor CYC065 and the Bcl-2 inhibitor venetoclax in chronic lymphocytic leukemia (CLL).
Experiments indicated that CYC065 and venetoclax target parallel mechanisms that promote survival in CLL cells, working together to induce apoptosis.
The drugs demonstrated synergy even in CLL samples that are inherently resistant to each drug alone.
William Plunkett, PhD, of The University of Texas MD Anderson Cancer Center in Houston, Texas, and his colleagues reported these findings at the AACR Annual Meeting 2018 (abstract 3905).
This research was supported by Cyclacel Pharmaceuticals, Inc., the company developing CYC065.
The researchers explained that CYC065 depletes Mcl-1 to induce apoptosis in CLL cells, while venetoclax induces apoptosis via inhibition of Bcl-2. However, upregulation of Mcl-1 is associated with resistance to venetoclax.
Therefore, the researchers theorized that combining CYC065 and venetoclax would serve to target 2 mechanisms that promote survival in CLL cells.
Experiments showed that CYC065 and venetoclax combined synergistically in CLL samples with or without 17p deletion. However, the researchers observed heterogeneity in response across samples.
The team said both drugs appeared to be less potent in some del(17p) samples. However, they also observed “great synergy” in del(17p) samples that were resistant to CYC065 or venetoclax alone.
The researchers noted differences in the kinetics of cell death in response to each drug and said this is consistent with the drugs’ different mechanisms of action.
Maximal cell death was reached at 6 to 8 hours with venetoclax but took at least 24 hours with CYC065.
The researchers also assessed the reversibility of CYC065 and venetoclax. They incubated CLL cells with each drug alone and in combination, then washed and incubated cells in drug-free media.
The team observed no additional cell death after the removal of CYC065, venetoclax, or the combination. They said this suggests an “adequate exposure time” is needed to maximize the induction of apoptosis with these drugs.
“[T]he combination of CYC065 and venetoclax is strongly synergistic in primary CLL cells from patients, including those with 17p deletions,” said Spiro Rombotis, president and chief executive officer of Cyclacel.
“In addition, the combination was active in 2 CLL samples which were resistant to either agent alone. These findings support the hypothesis that dual targeting of the Mcl-1- and Bcl-2-dependent mechanisms could induce synergistic cell death by apoptosis.”
Based on these results, Cyclacel is planning a trial of CYC065 and venetoclax in patients with relapsed/refractory CLL.
CHICAGO—Researchers have reported “strong synergy” between the CDK2/9 inhibitor CYC065 and the Bcl-2 inhibitor venetoclax in chronic lymphocytic leukemia (CLL).
Experiments indicated that CYC065 and venetoclax target parallel mechanisms that promote survival in CLL cells, working together to induce apoptosis.
The drugs demonstrated synergy even in CLL samples that are inherently resistant to each drug alone.
William Plunkett, PhD, of The University of Texas MD Anderson Cancer Center in Houston, Texas, and his colleagues reported these findings at the AACR Annual Meeting 2018 (abstract 3905).
This research was supported by Cyclacel Pharmaceuticals, Inc., the company developing CYC065.
The researchers explained that CYC065 depletes Mcl-1 to induce apoptosis in CLL cells, while venetoclax induces apoptosis via inhibition of Bcl-2. However, upregulation of Mcl-1 is associated with resistance to venetoclax.
Therefore, the researchers theorized that combining CYC065 and venetoclax would serve to target 2 mechanisms that promote survival in CLL cells.
Experiments showed that CYC065 and venetoclax combined synergistically in CLL samples with or without 17p deletion. However, the researchers observed heterogeneity in response across samples.
The team said both drugs appeared to be less potent in some del(17p) samples. However, they also observed “great synergy” in del(17p) samples that were resistant to CYC065 or venetoclax alone.
The researchers noted differences in the kinetics of cell death in response to each drug and said this is consistent with the drugs’ different mechanisms of action.
Maximal cell death was reached at 6 to 8 hours with venetoclax but took at least 24 hours with CYC065.
The researchers also assessed the reversibility of CYC065 and venetoclax. They incubated CLL cells with each drug alone and in combination, then washed and incubated cells in drug-free media.
The team observed no additional cell death after the removal of CYC065, venetoclax, or the combination. They said this suggests an “adequate exposure time” is needed to maximize the induction of apoptosis with these drugs.
“[T]he combination of CYC065 and venetoclax is strongly synergistic in primary CLL cells from patients, including those with 17p deletions,” said Spiro Rombotis, president and chief executive officer of Cyclacel.
“In addition, the combination was active in 2 CLL samples which were resistant to either agent alone. These findings support the hypothesis that dual targeting of the Mcl-1- and Bcl-2-dependent mechanisms could induce synergistic cell death by apoptosis.”
Based on these results, Cyclacel is planning a trial of CYC065 and venetoclax in patients with relapsed/refractory CLL.
CHICAGO—Researchers have reported “strong synergy” between the CDK2/9 inhibitor CYC065 and the Bcl-2 inhibitor venetoclax in chronic lymphocytic leukemia (CLL).
Experiments indicated that CYC065 and venetoclax target parallel mechanisms that promote survival in CLL cells, working together to induce apoptosis.
The drugs demonstrated synergy even in CLL samples that are inherently resistant to each drug alone.
William Plunkett, PhD, of The University of Texas MD Anderson Cancer Center in Houston, Texas, and his colleagues reported these findings at the AACR Annual Meeting 2018 (abstract 3905).
This research was supported by Cyclacel Pharmaceuticals, Inc., the company developing CYC065.
The researchers explained that CYC065 depletes Mcl-1 to induce apoptosis in CLL cells, while venetoclax induces apoptosis via inhibition of Bcl-2. However, upregulation of Mcl-1 is associated with resistance to venetoclax.
Therefore, the researchers theorized that combining CYC065 and venetoclax would serve to target 2 mechanisms that promote survival in CLL cells.
Experiments showed that CYC065 and venetoclax combined synergistically in CLL samples with or without 17p deletion. However, the researchers observed heterogeneity in response across samples.
The team said both drugs appeared to be less potent in some del(17p) samples. However, they also observed “great synergy” in del(17p) samples that were resistant to CYC065 or venetoclax alone.
The researchers noted differences in the kinetics of cell death in response to each drug and said this is consistent with the drugs’ different mechanisms of action.
Maximal cell death was reached at 6 to 8 hours with venetoclax but took at least 24 hours with CYC065.
The researchers also assessed the reversibility of CYC065 and venetoclax. They incubated CLL cells with each drug alone and in combination, then washed and incubated cells in drug-free media.
The team observed no additional cell death after the removal of CYC065, venetoclax, or the combination. They said this suggests an “adequate exposure time” is needed to maximize the induction of apoptosis with these drugs.
“[T]he combination of CYC065 and venetoclax is strongly synergistic in primary CLL cells from patients, including those with 17p deletions,” said Spiro Rombotis, president and chief executive officer of Cyclacel.
“In addition, the combination was active in 2 CLL samples which were resistant to either agent alone. These findings support the hypothesis that dual targeting of the Mcl-1- and Bcl-2-dependent mechanisms could induce synergistic cell death by apoptosis.”
Based on these results, Cyclacel is planning a trial of CYC065 and venetoclax in patients with relapsed/refractory CLL.
FDA places partial hold on trials after secondary lymphoma
The drugmaker after a pediatric patient developed a secondary T-cell lymphoma.
The Food and Drug Administration had issued a partial clinical hold in April on new enrollment of any patients with genetically defined solid tumors and hematologic malignancies. Patients already enrolled who have not had disease progression can continue to receive tazemetostat.
Tazemetostat is a first-in-class EZH2 inhibitor being studied as monotherapy in phase 1 and 2 trials for certain molecularly defined solid tumors, follicular lymphoma and diffuse large B-cell lymphoma, mesothelioma, and in combination studies of DLBCL and non–small cell lung cancer.
Epizyme is currently working to update informed consent, the investigator’s brochure, and study protocols, the company said in a statement.
The drugmaker after a pediatric patient developed a secondary T-cell lymphoma.
The Food and Drug Administration had issued a partial clinical hold in April on new enrollment of any patients with genetically defined solid tumors and hematologic malignancies. Patients already enrolled who have not had disease progression can continue to receive tazemetostat.
Tazemetostat is a first-in-class EZH2 inhibitor being studied as monotherapy in phase 1 and 2 trials for certain molecularly defined solid tumors, follicular lymphoma and diffuse large B-cell lymphoma, mesothelioma, and in combination studies of DLBCL and non–small cell lung cancer.
Epizyme is currently working to update informed consent, the investigator’s brochure, and study protocols, the company said in a statement.
The drugmaker after a pediatric patient developed a secondary T-cell lymphoma.
The Food and Drug Administration had issued a partial clinical hold in April on new enrollment of any patients with genetically defined solid tumors and hematologic malignancies. Patients already enrolled who have not had disease progression can continue to receive tazemetostat.
Tazemetostat is a first-in-class EZH2 inhibitor being studied as monotherapy in phase 1 and 2 trials for certain molecularly defined solid tumors, follicular lymphoma and diffuse large B-cell lymphoma, mesothelioma, and in combination studies of DLBCL and non–small cell lung cancer.
Epizyme is currently working to update informed consent, the investigator’s brochure, and study protocols, the company said in a statement.
FDA places tazemetostat trials on partial hold
The US Food and Drug Administration (FDA) has placed a partial hold on clinical trials of tazemetostat, an EZH2 inhibitor being developed to treat solid tumors and lymphomas.
The hold has halted enrollment in US-based trials of tazemetostat, but study subjects who have not experienced disease progression may continue to receive the drug.
The hold is due to an adverse event observed in a pediatric patient on a phase 1 study of tazemetostat.
The patient, who had advanced poorly differentiated chordoma, developed a secondary T-cell lymphoma while taking tazemetostat.
The patient had been on study for approximately 15 months and had achieved a confirmed partial response. Now, the patient has discontinued tazemetostat and is being treated for T-cell lymphoma.
More than 750 patients have been treated with tazemetostat to date, and this is the only case of secondary lymphoma that has been observed, according to Epizyme, Inc., the company developing tazemetostat.
The company also noted that doses of tazemetostat explored in its phase 1 pediatric study are higher than those used in the phase 2 adult studies.
Epizyme has begun taking steps to address the hold on tazemetostat trials—updating the informed consent, investigator’s brochure, and study protocols.
The company will need to confirm alignment with the FDA in order to resume US enrollment.
“We are working expeditiously with clinical trial investigators and regulatory authorities to initiate the appropriate steps to resume enrollment,” said Robert Bazemore, president and chief executive officer of Epizyme.
“Epizyme, along with our global investigator community, has been very encouraged by the clinical responses and tolerability of tazemetostat observed in pediatric and adult patients with hematological malignancies and solid tumors enrolled in our trials. We remain encouraged by the potential of tazemetostat to address the unmet needs of many patients living with cancer.”
The US Food and Drug Administration (FDA) has placed a partial hold on clinical trials of tazemetostat, an EZH2 inhibitor being developed to treat solid tumors and lymphomas.
The hold has halted enrollment in US-based trials of tazemetostat, but study subjects who have not experienced disease progression may continue to receive the drug.
The hold is due to an adverse event observed in a pediatric patient on a phase 1 study of tazemetostat.
The patient, who had advanced poorly differentiated chordoma, developed a secondary T-cell lymphoma while taking tazemetostat.
The patient had been on study for approximately 15 months and had achieved a confirmed partial response. Now, the patient has discontinued tazemetostat and is being treated for T-cell lymphoma.
More than 750 patients have been treated with tazemetostat to date, and this is the only case of secondary lymphoma that has been observed, according to Epizyme, Inc., the company developing tazemetostat.
The company also noted that doses of tazemetostat explored in its phase 1 pediatric study are higher than those used in the phase 2 adult studies.
Epizyme has begun taking steps to address the hold on tazemetostat trials—updating the informed consent, investigator’s brochure, and study protocols.
The company will need to confirm alignment with the FDA in order to resume US enrollment.
“We are working expeditiously with clinical trial investigators and regulatory authorities to initiate the appropriate steps to resume enrollment,” said Robert Bazemore, president and chief executive officer of Epizyme.
“Epizyme, along with our global investigator community, has been very encouraged by the clinical responses and tolerability of tazemetostat observed in pediatric and adult patients with hematological malignancies and solid tumors enrolled in our trials. We remain encouraged by the potential of tazemetostat to address the unmet needs of many patients living with cancer.”
The US Food and Drug Administration (FDA) has placed a partial hold on clinical trials of tazemetostat, an EZH2 inhibitor being developed to treat solid tumors and lymphomas.
The hold has halted enrollment in US-based trials of tazemetostat, but study subjects who have not experienced disease progression may continue to receive the drug.
The hold is due to an adverse event observed in a pediatric patient on a phase 1 study of tazemetostat.
The patient, who had advanced poorly differentiated chordoma, developed a secondary T-cell lymphoma while taking tazemetostat.
The patient had been on study for approximately 15 months and had achieved a confirmed partial response. Now, the patient has discontinued tazemetostat and is being treated for T-cell lymphoma.
More than 750 patients have been treated with tazemetostat to date, and this is the only case of secondary lymphoma that has been observed, according to Epizyme, Inc., the company developing tazemetostat.
The company also noted that doses of tazemetostat explored in its phase 1 pediatric study are higher than those used in the phase 2 adult studies.
Epizyme has begun taking steps to address the hold on tazemetostat trials—updating the informed consent, investigator’s brochure, and study protocols.
The company will need to confirm alignment with the FDA in order to resume US enrollment.
“We are working expeditiously with clinical trial investigators and regulatory authorities to initiate the appropriate steps to resume enrollment,” said Robert Bazemore, president and chief executive officer of Epizyme.
“Epizyme, along with our global investigator community, has been very encouraged by the clinical responses and tolerability of tazemetostat observed in pediatric and adult patients with hematological malignancies and solid tumors enrolled in our trials. We remain encouraged by the potential of tazemetostat to address the unmet needs of many patients living with cancer.”