User login
Study reveals potential target for AML treatment

New research has revealed a potential therapeutic target for acute myeloid leukemia (AML)—the methyl transferase enzyme METTL3.
Researchers found that inhibiting METTL3 destroys human and mouse AML cells without harming non-leukemic blood cells.
The team also discovered why METTL3 is required for AML cell survival by deciphering the mechanism it uses to regulate several other leukemia genes.
The researchers described this work in Nature.
“New treatments for AML are desperately needed, and we have been looking for genes that would be good drug targets,” said study author Tony Kouzarides, PhD, of University of Cambridge in the UK.
“We identified the methyl transferase enzyme METTL3 as a highly viable target against AML. Our study will inspire pharmaceutical efforts to find drugs that specifically inhibit METTL3 to treat AML.”
In their attempt to find therapeutic targets for AML, Dr Kouzarides and his colleagues used CRISPR-Cas9 to screen AML cells for vulnerable points.
The researchers created mouse leukemia cells with mutations in genes that may be targeted in human AML cells and systematically tested each gene, finding which were essential for AML survival.
The team ended up with 46 likely candidate genes, many of which produce proteins that could modify RNA. Among these, METTL3 was one of the genes with the strongest effect.
Experiments revealed that METTL3 was essential for the survival of AML cells, but it was not required for healthy blood cells.
Having found a potential target in METTL3, the researchers investigated how it worked.
They discovered that the protein produced by METTL3 bound to the beginning of 126 different genes, including several required for AML cell survival.
As RNAs were produced, the METTL3 protein added methyl groups to their middle section, something which had not been previously observed. These middle methyl groups increased the ability of the RNAs to be translated into proteins.
The researchers then found that when METTL3 was inhibited, no methyl groups were added to the RNA. This prevented the production of their essential proteins, so the AML cells started dying.
“This study uncovered an entirely new mechanism of gene regulation in AML that operates through modifications of RNA,” said study author Konstantinos Tzelepis, PhD, of Wellcome Trust Sanger Institute in Cambridge, UK.
“We discovered that inhibiting the methyl transferase activity of METTL3 would stop the translation of a whole set of proteins that the leukemia needs. This mechanism shows that a drug to inhibit methylation could be effective against AML without affecting normal cells.” ![]()
New research has revealed a potential therapeutic target for acute myeloid leukemia (AML)—the methyl transferase enzyme METTL3.
Researchers found that inhibiting METTL3 destroys human and mouse AML cells without harming non-leukemic blood cells.
The team also discovered why METTL3 is required for AML cell survival by deciphering the mechanism it uses to regulate several other leukemia genes.
The researchers described this work in Nature.
“New treatments for AML are desperately needed, and we have been looking for genes that would be good drug targets,” said study author Tony Kouzarides, PhD, of University of Cambridge in the UK.
“We identified the methyl transferase enzyme METTL3 as a highly viable target against AML. Our study will inspire pharmaceutical efforts to find drugs that specifically inhibit METTL3 to treat AML.”
In their attempt to find therapeutic targets for AML, Dr Kouzarides and his colleagues used CRISPR-Cas9 to screen AML cells for vulnerable points.
The researchers created mouse leukemia cells with mutations in genes that may be targeted in human AML cells and systematically tested each gene, finding which were essential for AML survival.
The team ended up with 46 likely candidate genes, many of which produce proteins that could modify RNA. Among these, METTL3 was one of the genes with the strongest effect.
Experiments revealed that METTL3 was essential for the survival of AML cells, but it was not required for healthy blood cells.
Having found a potential target in METTL3, the researchers investigated how it worked.
They discovered that the protein produced by METTL3 bound to the beginning of 126 different genes, including several required for AML cell survival.
As RNAs were produced, the METTL3 protein added methyl groups to their middle section, something which had not been previously observed. These middle methyl groups increased the ability of the RNAs to be translated into proteins.
The researchers then found that when METTL3 was inhibited, no methyl groups were added to the RNA. This prevented the production of their essential proteins, so the AML cells started dying.
“This study uncovered an entirely new mechanism of gene regulation in AML that operates through modifications of RNA,” said study author Konstantinos Tzelepis, PhD, of Wellcome Trust Sanger Institute in Cambridge, UK.
“We discovered that inhibiting the methyl transferase activity of METTL3 would stop the translation of a whole set of proteins that the leukemia needs. This mechanism shows that a drug to inhibit methylation could be effective against AML without affecting normal cells.” ![]()
New research has revealed a potential therapeutic target for acute myeloid leukemia (AML)—the methyl transferase enzyme METTL3.
Researchers found that inhibiting METTL3 destroys human and mouse AML cells without harming non-leukemic blood cells.
The team also discovered why METTL3 is required for AML cell survival by deciphering the mechanism it uses to regulate several other leukemia genes.
The researchers described this work in Nature.
“New treatments for AML are desperately needed, and we have been looking for genes that would be good drug targets,” said study author Tony Kouzarides, PhD, of University of Cambridge in the UK.
“We identified the methyl transferase enzyme METTL3 as a highly viable target against AML. Our study will inspire pharmaceutical efforts to find drugs that specifically inhibit METTL3 to treat AML.”
In their attempt to find therapeutic targets for AML, Dr Kouzarides and his colleagues used CRISPR-Cas9 to screen AML cells for vulnerable points.
The researchers created mouse leukemia cells with mutations in genes that may be targeted in human AML cells and systematically tested each gene, finding which were essential for AML survival.
The team ended up with 46 likely candidate genes, many of which produce proteins that could modify RNA. Among these, METTL3 was one of the genes with the strongest effect.
Experiments revealed that METTL3 was essential for the survival of AML cells, but it was not required for healthy blood cells.
Having found a potential target in METTL3, the researchers investigated how it worked.
They discovered that the protein produced by METTL3 bound to the beginning of 126 different genes, including several required for AML cell survival.
As RNAs were produced, the METTL3 protein added methyl groups to their middle section, something which had not been previously observed. These middle methyl groups increased the ability of the RNAs to be translated into proteins.
The researchers then found that when METTL3 was inhibited, no methyl groups were added to the RNA. This prevented the production of their essential proteins, so the AML cells started dying.
“This study uncovered an entirely new mechanism of gene regulation in AML that operates through modifications of RNA,” said study author Konstantinos Tzelepis, PhD, of Wellcome Trust Sanger Institute in Cambridge, UK.
“We discovered that inhibiting the methyl transferase activity of METTL3 would stop the translation of a whole set of proteins that the leukemia needs. This mechanism shows that a drug to inhibit methylation could be effective against AML without affecting normal cells.” ![]()
FDA grants drug orphan designation for AML, MDS

The US Food and Drug Administration (FDA) has granted orphan drug designation to AMV564, a CD33/CD3 bispecific antibody, for the treatment of acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS).
AMV564 is a T-cell engager, derived from human protein sequences, that binds both CD33 and CD3 to mediate T-cell directed lysis of CD33-positive cancer cells.
Amphivena Therapeutics Inc., is currently conducting a phase 1 trial of AMV564 in relapsed or refractory AML. The company plans to launch a phase 1 trial in patients with MDS in early 2018.
According to Amphivena, AMV564 has demonstrated “potent activity” in AML patient samples, and that activity was independent of CD33 expression level, disease stage, and cytogenetic risk.
AMV564 also eliminated nearly all blasts from the bone marrow and spleen in a stringent AML patient-derived xenograft murine model.
In addition, Amphivena established a therapeutic window for AMV564 in cynomolgus monkeys, with rapid and sustained elimination of CD33-expressing cells during AMV564 dosing and rapid hematopoietic recovery following dosing.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to AMV564, a CD33/CD3 bispecific antibody, for the treatment of acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS).
AMV564 is a T-cell engager, derived from human protein sequences, that binds both CD33 and CD3 to mediate T-cell directed lysis of CD33-positive cancer cells.
Amphivena Therapeutics Inc., is currently conducting a phase 1 trial of AMV564 in relapsed or refractory AML. The company plans to launch a phase 1 trial in patients with MDS in early 2018.
According to Amphivena, AMV564 has demonstrated “potent activity” in AML patient samples, and that activity was independent of CD33 expression level, disease stage, and cytogenetic risk.
AMV564 also eliminated nearly all blasts from the bone marrow and spleen in a stringent AML patient-derived xenograft murine model.
In addition, Amphivena established a therapeutic window for AMV564 in cynomolgus monkeys, with rapid and sustained elimination of CD33-expressing cells during AMV564 dosing and rapid hematopoietic recovery following dosing.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to AMV564, a CD33/CD3 bispecific antibody, for the treatment of acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS).
AMV564 is a T-cell engager, derived from human protein sequences, that binds both CD33 and CD3 to mediate T-cell directed lysis of CD33-positive cancer cells.
Amphivena Therapeutics Inc., is currently conducting a phase 1 trial of AMV564 in relapsed or refractory AML. The company plans to launch a phase 1 trial in patients with MDS in early 2018.
According to Amphivena, AMV564 has demonstrated “potent activity” in AML patient samples, and that activity was independent of CD33 expression level, disease stage, and cytogenetic risk.
AMV564 also eliminated nearly all blasts from the bone marrow and spleen in a stringent AML patient-derived xenograft murine model.
In addition, Amphivena established a therapeutic window for AMV564 in cynomolgus monkeys, with rapid and sustained elimination of CD33-expressing cells during AMV564 dosing and rapid hematopoietic recovery following dosing.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
Researchers identify potential gene target for AML drug development
Researchers in the United States and the United Kingdom believe they have found a new gene target that could aid in the development of more effective treatments for acute myeloid leukemia (AML).
Inhibition of the METTL3 gene allowed for the destruction of AML in human and mouse cells without damaging healthy blood cells, the researchers reported in a research letter published Nov. 27 in the journal Nature.
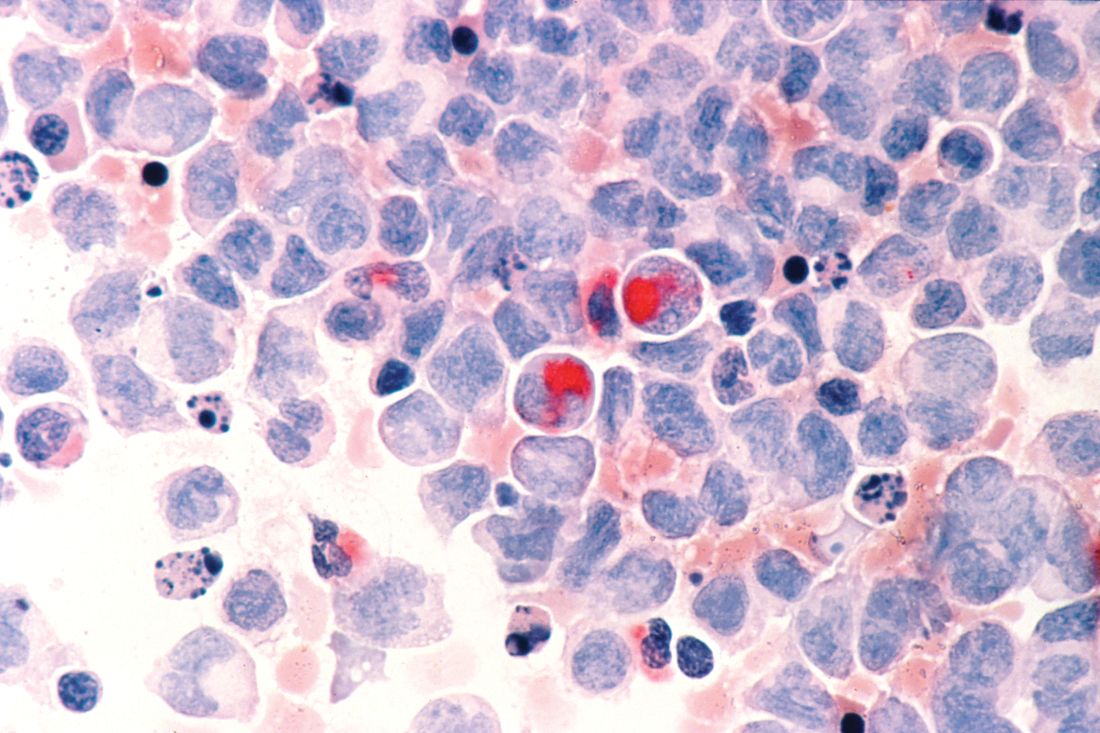
Using mouse cells, the researchers used CRISPR-Cas9 gene editing technology to identify RNA-modifying enzymes that are needed for the survival and proliferation of AML cells. They identified 46 potential candidate genes and further narrowed that to the METTL gene families. They next targeted METTL1, METTL3, METTL14, and METTL16 in 10 human AML cell lines and 10 cell lines from heterogeneous cancer types. METTL3 was shown to have the strongest effect.
Read the full research letter in Nature (doi: 10.1038/nature24678).
[email protected]
On Twitter @maryellenny
Researchers in the United States and the United Kingdom believe they have found a new gene target that could aid in the development of more effective treatments for acute myeloid leukemia (AML).
Inhibition of the METTL3 gene allowed for the destruction of AML in human and mouse cells without damaging healthy blood cells, the researchers reported in a research letter published Nov. 27 in the journal Nature.
Using mouse cells, the researchers used CRISPR-Cas9 gene editing technology to identify RNA-modifying enzymes that are needed for the survival and proliferation of AML cells. They identified 46 potential candidate genes and further narrowed that to the METTL gene families. They next targeted METTL1, METTL3, METTL14, and METTL16 in 10 human AML cell lines and 10 cell lines from heterogeneous cancer types. METTL3 was shown to have the strongest effect.
Read the full research letter in Nature (doi: 10.1038/nature24678).
[email protected]
On Twitter @maryellenny
Researchers in the United States and the United Kingdom believe they have found a new gene target that could aid in the development of more effective treatments for acute myeloid leukemia (AML).
Inhibition of the METTL3 gene allowed for the destruction of AML in human and mouse cells without damaging healthy blood cells, the researchers reported in a research letter published Nov. 27 in the journal Nature.
Using mouse cells, the researchers used CRISPR-Cas9 gene editing technology to identify RNA-modifying enzymes that are needed for the survival and proliferation of AML cells. They identified 46 potential candidate genes and further narrowed that to the METTL gene families. They next targeted METTL1, METTL3, METTL14, and METTL16 in 10 human AML cell lines and 10 cell lines from heterogeneous cancer types. METTL3 was shown to have the strongest effect.
Read the full research letter in Nature (doi: 10.1038/nature24678).
[email protected]
On Twitter @maryellenny
FROM NATURE
Study reveals predictor of early mortality in AML

New research has shown an increased risk of early death in patients with acute myeloid leukemia (AML) who have high levels of indoleamine 2,3 dioxygenase-1 (IDO-1), an enzyme known to suppress the immune system.
Researchers quantified IDO-1 expression in diagnostic samples from patients with AML and discovered that high levels of IDO-1 were significantly associated with induction failure and poor overall survival (OS).
Ravindra Kolhe, MD, PhD, of the Medical College of Georgia at Augusta University, and his colleagues recounted these findings in Scientific Reports.
The researchers reviewed data from 40 AML patients. They had a median age at diagnosis of 60 (range, 27–89), 60% were female, and 55% were self-reported as Caucasian.
Most patients (72.5%) received standard anthracycline and cytarabine as induction, 10% received hypomethylating agents, and 17.5% were untreated or had unknown treatment status.
Fifteen percent of patients underwent allogeneic hematopoietic stem cell transplant (allo-HSCT) at the time of first complete remission.
Half of all patients achieved remission, and half of those patients (n=10, 25%) had a subsequent relapse. The median OS was 283 days (range, 32–1941). Twenty percent of patients (n=8) were still alive at the time of data analysis.
IDO-1 analysis
“We wanted to look at what makes this leukemia so aggressive that initial induction chemotherapy is not working,” Dr Kolhe said. “Early relapse tends to predict early mortality in these patients, and one of the things we looked at was IDO.”
The researchers extracted IDO-1 mRNA from diagnostic bone marrow samples from 29 of the patients but assessed IDO-1 protein expression in all 40 patients via immunohistochemistry.
The team quantified IDO-1 expression using a “composite IDO-1 score.” They used a cut-off point of 0.45 and divided patients’ samples into 2 groups: high (≥0.45) and low (<0.45) IDO-1 score.
The researchers compared IDO-1 results across 4 survival groups, which included patients surviving:
- Less than 6 months
- More than 6 months to 1 year
- More than 1 year to less than 5 years
- Beyond 5 years.
The team found a direct correlation between poor OS and higher composite IDO-1 score (P=0.0005).
“The patients who died at 6 months had a high expression of IDO, while the blasts produced relatively little IDO in the patients who lived 5 years or more,” Dr Kolhe said.
Independent predictor
The researchers conducted a univariate analysis and identified several factors that were significantly associated with poor OS, including:
- Higher IDO-1 mRNA (P=0.005)
- Higher composite IDO-1 score (P<0.0001)
- Higher age (P=0.0018)
- Male gender (P=0.019)
- High-risk cytogenetics (P=0.002)
- Not undergoing allo-HSCT (P=0.0005).
In a multivariate analysis including the above variables, the researchers found that a higher composite IDO-1 score (P=0.007) and not undergoing allo-HSCT (P=0.007) were significantly associated with poor OS.
The team also found that patients who failed induction had a higher composite IDO-1 score (P=0.01).
“Most of the time, we don’t know why patients are not responding to chemotherapy,” Dr Kolhe noted. “But when the researchers adjusted for other risk factors for AML, like increased age and severe anemia, IDO levels were a standout. Right now, we know it’s high in patients who die at 6 months, and we show that it’s an independent indicator if you adjust for other known variables.”
Dr Kolhe said these results suggest IDO-1 expression should be measured when the diagnostic bone marrow biopsy is performed. This may help identify AML patients who could benefit from receiving an IDO inhibitor along with standard therapy.
Researchers are currently conducting a phase 1/2 trial of the IDO inhibitor indoximod in combination with idarubicin and cytarabine in patients with newly diagnosed AML (NCT02835729). ![]()
New research has shown an increased risk of early death in patients with acute myeloid leukemia (AML) who have high levels of indoleamine 2,3 dioxygenase-1 (IDO-1), an enzyme known to suppress the immune system.
Researchers quantified IDO-1 expression in diagnostic samples from patients with AML and discovered that high levels of IDO-1 were significantly associated with induction failure and poor overall survival (OS).
Ravindra Kolhe, MD, PhD, of the Medical College of Georgia at Augusta University, and his colleagues recounted these findings in Scientific Reports.
The researchers reviewed data from 40 AML patients. They had a median age at diagnosis of 60 (range, 27–89), 60% were female, and 55% were self-reported as Caucasian.
Most patients (72.5%) received standard anthracycline and cytarabine as induction, 10% received hypomethylating agents, and 17.5% were untreated or had unknown treatment status.
Fifteen percent of patients underwent allogeneic hematopoietic stem cell transplant (allo-HSCT) at the time of first complete remission.
Half of all patients achieved remission, and half of those patients (n=10, 25%) had a subsequent relapse. The median OS was 283 days (range, 32–1941). Twenty percent of patients (n=8) were still alive at the time of data analysis.
IDO-1 analysis
“We wanted to look at what makes this leukemia so aggressive that initial induction chemotherapy is not working,” Dr Kolhe said. “Early relapse tends to predict early mortality in these patients, and one of the things we looked at was IDO.”
The researchers extracted IDO-1 mRNA from diagnostic bone marrow samples from 29 of the patients but assessed IDO-1 protein expression in all 40 patients via immunohistochemistry.
The team quantified IDO-1 expression using a “composite IDO-1 score.” They used a cut-off point of 0.45 and divided patients’ samples into 2 groups: high (≥0.45) and low (<0.45) IDO-1 score.
The researchers compared IDO-1 results across 4 survival groups, which included patients surviving:
- Less than 6 months
- More than 6 months to 1 year
- More than 1 year to less than 5 years
- Beyond 5 years.
The team found a direct correlation between poor OS and higher composite IDO-1 score (P=0.0005).
“The patients who died at 6 months had a high expression of IDO, while the blasts produced relatively little IDO in the patients who lived 5 years or more,” Dr Kolhe said.
Independent predictor
The researchers conducted a univariate analysis and identified several factors that were significantly associated with poor OS, including:
- Higher IDO-1 mRNA (P=0.005)
- Higher composite IDO-1 score (P<0.0001)
- Higher age (P=0.0018)
- Male gender (P=0.019)
- High-risk cytogenetics (P=0.002)
- Not undergoing allo-HSCT (P=0.0005).
In a multivariate analysis including the above variables, the researchers found that a higher composite IDO-1 score (P=0.007) and not undergoing allo-HSCT (P=0.007) were significantly associated with poor OS.
The team also found that patients who failed induction had a higher composite IDO-1 score (P=0.01).
“Most of the time, we don’t know why patients are not responding to chemotherapy,” Dr Kolhe noted. “But when the researchers adjusted for other risk factors for AML, like increased age and severe anemia, IDO levels were a standout. Right now, we know it’s high in patients who die at 6 months, and we show that it’s an independent indicator if you adjust for other known variables.”
Dr Kolhe said these results suggest IDO-1 expression should be measured when the diagnostic bone marrow biopsy is performed. This may help identify AML patients who could benefit from receiving an IDO inhibitor along with standard therapy.
Researchers are currently conducting a phase 1/2 trial of the IDO inhibitor indoximod in combination with idarubicin and cytarabine in patients with newly diagnosed AML (NCT02835729). ![]()
New research has shown an increased risk of early death in patients with acute myeloid leukemia (AML) who have high levels of indoleamine 2,3 dioxygenase-1 (IDO-1), an enzyme known to suppress the immune system.
Researchers quantified IDO-1 expression in diagnostic samples from patients with AML and discovered that high levels of IDO-1 were significantly associated with induction failure and poor overall survival (OS).
Ravindra Kolhe, MD, PhD, of the Medical College of Georgia at Augusta University, and his colleagues recounted these findings in Scientific Reports.
The researchers reviewed data from 40 AML patients. They had a median age at diagnosis of 60 (range, 27–89), 60% were female, and 55% were self-reported as Caucasian.
Most patients (72.5%) received standard anthracycline and cytarabine as induction, 10% received hypomethylating agents, and 17.5% were untreated or had unknown treatment status.
Fifteen percent of patients underwent allogeneic hematopoietic stem cell transplant (allo-HSCT) at the time of first complete remission.
Half of all patients achieved remission, and half of those patients (n=10, 25%) had a subsequent relapse. The median OS was 283 days (range, 32–1941). Twenty percent of patients (n=8) were still alive at the time of data analysis.
IDO-1 analysis
“We wanted to look at what makes this leukemia so aggressive that initial induction chemotherapy is not working,” Dr Kolhe said. “Early relapse tends to predict early mortality in these patients, and one of the things we looked at was IDO.”
The researchers extracted IDO-1 mRNA from diagnostic bone marrow samples from 29 of the patients but assessed IDO-1 protein expression in all 40 patients via immunohistochemistry.
The team quantified IDO-1 expression using a “composite IDO-1 score.” They used a cut-off point of 0.45 and divided patients’ samples into 2 groups: high (≥0.45) and low (<0.45) IDO-1 score.
The researchers compared IDO-1 results across 4 survival groups, which included patients surviving:
- Less than 6 months
- More than 6 months to 1 year
- More than 1 year to less than 5 years
- Beyond 5 years.
The team found a direct correlation between poor OS and higher composite IDO-1 score (P=0.0005).
“The patients who died at 6 months had a high expression of IDO, while the blasts produced relatively little IDO in the patients who lived 5 years or more,” Dr Kolhe said.
Independent predictor
The researchers conducted a univariate analysis and identified several factors that were significantly associated with poor OS, including:
- Higher IDO-1 mRNA (P=0.005)
- Higher composite IDO-1 score (P<0.0001)
- Higher age (P=0.0018)
- Male gender (P=0.019)
- High-risk cytogenetics (P=0.002)
- Not undergoing allo-HSCT (P=0.0005).
In a multivariate analysis including the above variables, the researchers found that a higher composite IDO-1 score (P=0.007) and not undergoing allo-HSCT (P=0.007) were significantly associated with poor OS.
The team also found that patients who failed induction had a higher composite IDO-1 score (P=0.01).
“Most of the time, we don’t know why patients are not responding to chemotherapy,” Dr Kolhe noted. “But when the researchers adjusted for other risk factors for AML, like increased age and severe anemia, IDO levels were a standout. Right now, we know it’s high in patients who die at 6 months, and we show that it’s an independent indicator if you adjust for other known variables.”
Dr Kolhe said these results suggest IDO-1 expression should be measured when the diagnostic bone marrow biopsy is performed. This may help identify AML patients who could benefit from receiving an IDO inhibitor along with standard therapy.
Researchers are currently conducting a phase 1/2 trial of the IDO inhibitor indoximod in combination with idarubicin and cytarabine in patients with newly diagnosed AML (NCT02835729). ![]()
Method identifies effective treatments for leukemias, lymphomas

An ex vivo drug screening method can reveal optimal therapies for patients with hematologic malignancies, according to research published in The Lancet Haematology.
Researchers used a method called pharmacoscopy to measure single-cell responses to possible treatments in samples from patients with leukemias and lymphomas.
The team then used these results to guide treatment decisions and found that pharmacoscopy-guided treatment greatly improved response rates and progression-free survival (PFS).
“Having a robust, fast, and reliable predictive test at our disposal during the patient treatment process, especially at the time of relapse where a new intervention must be selected quickly, will change how medical doctors prioritize drugs to use for late-stage patients,” said study author Philipp Staber, MD, of Medical University of Vienna in Austria.
With pharmacoscopy, hundreds of drug options can be pre-tested ex vivo in small liquid biopsy samples collected from individual patients. The effects of each drug on the individual cells are quantified using high-throughput and high-content automated confocal microscopy.
In combination with specially developed analysis methods, machine learning, and other algorithms, pharmacoscopy allows quantification of never-before visualized phenotypes. The method was first described last April in Nature Chemical Biology.
Now, Dr Staber and his colleagues have reported, in The Lancet Haematology, an interim analysis of the first clinical trial testing pharmacoscopy-guided treatment.
There were 17 evaluable patients, all of whom had aggressive hematologic malignancies. This included diffuse large B-cell lymphoma (n=6), acute myeloid leukemia (n=3), B-cell acute lymphoblastic leukemia (n=2), precursor B-cell lymphoblastic lymphoma (n=1), peripheral T-cell lymphoma (n=1), primary mediastinal B-cell lymphoma (n=1), T-cell lymphoblastic lymphoma (n=1), follicular lymphoma (n=1), and T-cell prolymphocytic leukemia (n=1).
The researchers compared outcomes with pharmacoscopy-guided treatment to outcomes with the most recent regimen on which the patient had progressed.
The overall response rate was 88% with pharmacoscopy-guided treatment and 24% with the patients’ most recent previous treatment regimen (odds ratio=24.38; 95%, CI 3.99–125.4; P=0.0013).
None of the patients had progressive disease as their best overall response when they received pharmacoscopy-guided treatment. However, 7 patients had progressive disease in response to their most recent prior regimen.
At the time of analysis, 8 patients (47%) still had ongoing responses after pharmacoscopy-guided treatment.
In addition, pharmacoscopy-guided treatment significantly improved PFS. The median PFS was 22.6 weeks with pharmacoscopy-guided treatment and 5.7 weeks with the most recent prior regimen (hazard ratio=3.14; 95%, CI 1.37–7.22; P=0.0075).
“Evidence that the pharmacoscopy approach is helpful for clinical evaluation of therapy is wonderful,” said study author Giulio Superti-Furga, PhD, of CeMM Research Center for Molecular Medicine in Vienna, Austria.
“Single-cell functional analysis of primary material gives unprecedented resolution and precision that we are sure to further develop in the future to address yet more diseases.” ![]()
An ex vivo drug screening method can reveal optimal therapies for patients with hematologic malignancies, according to research published in The Lancet Haematology.
Researchers used a method called pharmacoscopy to measure single-cell responses to possible treatments in samples from patients with leukemias and lymphomas.
The team then used these results to guide treatment decisions and found that pharmacoscopy-guided treatment greatly improved response rates and progression-free survival (PFS).
“Having a robust, fast, and reliable predictive test at our disposal during the patient treatment process, especially at the time of relapse where a new intervention must be selected quickly, will change how medical doctors prioritize drugs to use for late-stage patients,” said study author Philipp Staber, MD, of Medical University of Vienna in Austria.
With pharmacoscopy, hundreds of drug options can be pre-tested ex vivo in small liquid biopsy samples collected from individual patients. The effects of each drug on the individual cells are quantified using high-throughput and high-content automated confocal microscopy.
In combination with specially developed analysis methods, machine learning, and other algorithms, pharmacoscopy allows quantification of never-before visualized phenotypes. The method was first described last April in Nature Chemical Biology.
Now, Dr Staber and his colleagues have reported, in The Lancet Haematology, an interim analysis of the first clinical trial testing pharmacoscopy-guided treatment.
There were 17 evaluable patients, all of whom had aggressive hematologic malignancies. This included diffuse large B-cell lymphoma (n=6), acute myeloid leukemia (n=3), B-cell acute lymphoblastic leukemia (n=2), precursor B-cell lymphoblastic lymphoma (n=1), peripheral T-cell lymphoma (n=1), primary mediastinal B-cell lymphoma (n=1), T-cell lymphoblastic lymphoma (n=1), follicular lymphoma (n=1), and T-cell prolymphocytic leukemia (n=1).
The researchers compared outcomes with pharmacoscopy-guided treatment to outcomes with the most recent regimen on which the patient had progressed.
The overall response rate was 88% with pharmacoscopy-guided treatment and 24% with the patients’ most recent previous treatment regimen (odds ratio=24.38; 95%, CI 3.99–125.4; P=0.0013).
None of the patients had progressive disease as their best overall response when they received pharmacoscopy-guided treatment. However, 7 patients had progressive disease in response to their most recent prior regimen.
At the time of analysis, 8 patients (47%) still had ongoing responses after pharmacoscopy-guided treatment.
In addition, pharmacoscopy-guided treatment significantly improved PFS. The median PFS was 22.6 weeks with pharmacoscopy-guided treatment and 5.7 weeks with the most recent prior regimen (hazard ratio=3.14; 95%, CI 1.37–7.22; P=0.0075).
“Evidence that the pharmacoscopy approach is helpful for clinical evaluation of therapy is wonderful,” said study author Giulio Superti-Furga, PhD, of CeMM Research Center for Molecular Medicine in Vienna, Austria.
“Single-cell functional analysis of primary material gives unprecedented resolution and precision that we are sure to further develop in the future to address yet more diseases.” ![]()
An ex vivo drug screening method can reveal optimal therapies for patients with hematologic malignancies, according to research published in The Lancet Haematology.
Researchers used a method called pharmacoscopy to measure single-cell responses to possible treatments in samples from patients with leukemias and lymphomas.
The team then used these results to guide treatment decisions and found that pharmacoscopy-guided treatment greatly improved response rates and progression-free survival (PFS).
“Having a robust, fast, and reliable predictive test at our disposal during the patient treatment process, especially at the time of relapse where a new intervention must be selected quickly, will change how medical doctors prioritize drugs to use for late-stage patients,” said study author Philipp Staber, MD, of Medical University of Vienna in Austria.
With pharmacoscopy, hundreds of drug options can be pre-tested ex vivo in small liquid biopsy samples collected from individual patients. The effects of each drug on the individual cells are quantified using high-throughput and high-content automated confocal microscopy.
In combination with specially developed analysis methods, machine learning, and other algorithms, pharmacoscopy allows quantification of never-before visualized phenotypes. The method was first described last April in Nature Chemical Biology.
Now, Dr Staber and his colleagues have reported, in The Lancet Haematology, an interim analysis of the first clinical trial testing pharmacoscopy-guided treatment.
There were 17 evaluable patients, all of whom had aggressive hematologic malignancies. This included diffuse large B-cell lymphoma (n=6), acute myeloid leukemia (n=3), B-cell acute lymphoblastic leukemia (n=2), precursor B-cell lymphoblastic lymphoma (n=1), peripheral T-cell lymphoma (n=1), primary mediastinal B-cell lymphoma (n=1), T-cell lymphoblastic lymphoma (n=1), follicular lymphoma (n=1), and T-cell prolymphocytic leukemia (n=1).
The researchers compared outcomes with pharmacoscopy-guided treatment to outcomes with the most recent regimen on which the patient had progressed.
The overall response rate was 88% with pharmacoscopy-guided treatment and 24% with the patients’ most recent previous treatment regimen (odds ratio=24.38; 95%, CI 3.99–125.4; P=0.0013).
None of the patients had progressive disease as their best overall response when they received pharmacoscopy-guided treatment. However, 7 patients had progressive disease in response to their most recent prior regimen.
At the time of analysis, 8 patients (47%) still had ongoing responses after pharmacoscopy-guided treatment.
In addition, pharmacoscopy-guided treatment significantly improved PFS. The median PFS was 22.6 weeks with pharmacoscopy-guided treatment and 5.7 weeks with the most recent prior regimen (hazard ratio=3.14; 95%, CI 1.37–7.22; P=0.0075).
“Evidence that the pharmacoscopy approach is helpful for clinical evaluation of therapy is wonderful,” said study author Giulio Superti-Furga, PhD, of CeMM Research Center for Molecular Medicine in Vienna, Austria.
“Single-cell functional analysis of primary material gives unprecedented resolution and precision that we are sure to further develop in the future to address yet more diseases.” ![]()
Novel agent to be studied in relapsed/refractory AML
Trovagene announced.
The aim of the phase 1 portion of the trial is to find out whether PCM-075 given orally daily for 5 consecutive days every 28 days is safe and tolerable in such patients or in those AML patients who are ineligible for intensive induction therapy. The researchers are also trying to determine the maximum tolerated dose of PCM-075 or recommended phase 2 dose of PCM-075 in combination with decitabine and/or PCM-075 in combination with low-dose cytarabine.
The primary outcomes of the phase 1 portion of the trial are the number of participants with dose-limiting toxicity and adverse events from baseline out to 30 days after the last dose of PCM-075, up to 27 months. The primary outcome of phase 2 , called PCM-075 in Combination With Either Low-Dose Cytarabine or Decitabine in Adult Patients With Acute Myeloid Leukemia, will be the rate of complete response plus complete response with incomplete blood count recovery out to 27 months.
The PLK1 enzyme is overexpressed in multiple hematologic and solid tumor cancers, and studies have shown that inhibition of polo-like kinases can lead to tumor cell death, Trovagene said in its statement.
Bill Welch, CEO of Trovagene, added that “PCM-075 is the first highly PLK1-selective competitive inhibitor administered orally to enter clinical trials with potential activity in both hematologic and solid tumor cancers.”
Trovagene announced.
The aim of the phase 1 portion of the trial is to find out whether PCM-075 given orally daily for 5 consecutive days every 28 days is safe and tolerable in such patients or in those AML patients who are ineligible for intensive induction therapy. The researchers are also trying to determine the maximum tolerated dose of PCM-075 or recommended phase 2 dose of PCM-075 in combination with decitabine and/or PCM-075 in combination with low-dose cytarabine.
The primary outcomes of the phase 1 portion of the trial are the number of participants with dose-limiting toxicity and adverse events from baseline out to 30 days after the last dose of PCM-075, up to 27 months. The primary outcome of phase 2 , called PCM-075 in Combination With Either Low-Dose Cytarabine or Decitabine in Adult Patients With Acute Myeloid Leukemia, will be the rate of complete response plus complete response with incomplete blood count recovery out to 27 months.
The PLK1 enzyme is overexpressed in multiple hematologic and solid tumor cancers, and studies have shown that inhibition of polo-like kinases can lead to tumor cell death, Trovagene said in its statement.
Bill Welch, CEO of Trovagene, added that “PCM-075 is the first highly PLK1-selective competitive inhibitor administered orally to enter clinical trials with potential activity in both hematologic and solid tumor cancers.”
Trovagene announced.
The aim of the phase 1 portion of the trial is to find out whether PCM-075 given orally daily for 5 consecutive days every 28 days is safe and tolerable in such patients or in those AML patients who are ineligible for intensive induction therapy. The researchers are also trying to determine the maximum tolerated dose of PCM-075 or recommended phase 2 dose of PCM-075 in combination with decitabine and/or PCM-075 in combination with low-dose cytarabine.
The primary outcomes of the phase 1 portion of the trial are the number of participants with dose-limiting toxicity and adverse events from baseline out to 30 days after the last dose of PCM-075, up to 27 months. The primary outcome of phase 2 , called PCM-075 in Combination With Either Low-Dose Cytarabine or Decitabine in Adult Patients With Acute Myeloid Leukemia, will be the rate of complete response plus complete response with incomplete blood count recovery out to 27 months.
The PLK1 enzyme is overexpressed in multiple hematologic and solid tumor cancers, and studies have shown that inhibition of polo-like kinases can lead to tumor cell death, Trovagene said in its statement.
Bill Welch, CEO of Trovagene, added that “PCM-075 is the first highly PLK1-selective competitive inhibitor administered orally to enter clinical trials with potential activity in both hematologic and solid tumor cancers.”
Start with fitness when deciding on treatment for elderly AML patients
When evaluating older patients with acute myeloid leukemia for treatment, start with their fitness levels.
ML is a disease of older adults, and with increasing age comes higher treatment-related mortality, lower complete remission rates, higher relapse risk, and shorter overall survival. So it may not be surprising that fewer than half of U.S. patients with newly diagnosed acute myeloid leukemia over age 65 receive any chemotherapy at all, wrote Li-Wen Huang, MD, and Rebecca L. Olin, MD, of the University of California, San Francisco.

Fitness is key: Older patients considered fit for intensive chemotherapy should receive standard induction therapy, and reduced-intensity allogeneic stem cell transplantation should then be considered. Patients considered unfit for intensive therapy, on the other hand, should receive hypomethylating agents.
Several new therapeutic agents have shown promising results either by improving intensive chemotherapy (CPX-351), by improving upon lower-intensity therapy (venetoclax, antibody drug conjugates), or by targeting somatic mutations (FLT3 inhibitors and others), the investigators concluded.
Dr. Huang reported no conflicts. Dr. Olin has received research funding from Daiichi Sankyo, Astellas, and Genentech.
When evaluating older patients with acute myeloid leukemia for treatment, start with their fitness levels.
ML is a disease of older adults, and with increasing age comes higher treatment-related mortality, lower complete remission rates, higher relapse risk, and shorter overall survival. So it may not be surprising that fewer than half of U.S. patients with newly diagnosed acute myeloid leukemia over age 65 receive any chemotherapy at all, wrote Li-Wen Huang, MD, and Rebecca L. Olin, MD, of the University of California, San Francisco.
Fitness is key: Older patients considered fit for intensive chemotherapy should receive standard induction therapy, and reduced-intensity allogeneic stem cell transplantation should then be considered. Patients considered unfit for intensive therapy, on the other hand, should receive hypomethylating agents.
Several new therapeutic agents have shown promising results either by improving intensive chemotherapy (CPX-351), by improving upon lower-intensity therapy (venetoclax, antibody drug conjugates), or by targeting somatic mutations (FLT3 inhibitors and others), the investigators concluded.
Dr. Huang reported no conflicts. Dr. Olin has received research funding from Daiichi Sankyo, Astellas, and Genentech.
When evaluating older patients with acute myeloid leukemia for treatment, start with their fitness levels.
ML is a disease of older adults, and with increasing age comes higher treatment-related mortality, lower complete remission rates, higher relapse risk, and shorter overall survival. So it may not be surprising that fewer than half of U.S. patients with newly diagnosed acute myeloid leukemia over age 65 receive any chemotherapy at all, wrote Li-Wen Huang, MD, and Rebecca L. Olin, MD, of the University of California, San Francisco.
Fitness is key: Older patients considered fit for intensive chemotherapy should receive standard induction therapy, and reduced-intensity allogeneic stem cell transplantation should then be considered. Patients considered unfit for intensive therapy, on the other hand, should receive hypomethylating agents.
Several new therapeutic agents have shown promising results either by improving intensive chemotherapy (CPX-351), by improving upon lower-intensity therapy (venetoclax, antibody drug conjugates), or by targeting somatic mutations (FLT3 inhibitors and others), the investigators concluded.
Dr. Huang reported no conflicts. Dr. Olin has received research funding from Daiichi Sankyo, Astellas, and Genentech.
FROM THE JOURNAL OF GERIATRIC ONCOLOGY
Team discovers mechanism of resistance in AML

Researchers say they have uncovered a target to overcome drug resistance in acute myeloid leukemia (AML).
The team discovered how a linkage between 2 proteins enables AML cells to resist chemotherapy and showed that disrupting the linkage could render the cells vulnerable to treatment.
The researchers believe their discovery could lead to drugs to enhance chemotherapy in patients with AML and other cancers.
John Schuetz, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues described this research in Nature Communications.
The team launched their experiments based on previous findings that a protein called ABCC4 was greatly elevated in aggressive cases of AML.
Dr Schuetz and his colleagues searched for other proteins that might interact with ABCC4 and enable its function. The team’s screening of candidate proteins yielded one, MPP1, which was also greatly increased in AML.
The researchers found the 2 proteins are connected, and the connection enables cells to assume the characteristics of highly proliferative leukemia cells.
These experiments involved genetically altering hematopoietic progenitor cells to have high MPP1 and ABCC4 levels. The cells were grown in culture and then replated to see if they would continue to grow, as such self-renewal is a hallmark of leukemia cells.
The researchers found that serial regrowth depended on the cells having high levels of both ABCC4 and MPP1.
“Typically, if you take normal progenitors and you replate, you could do that one time, maybe twice,” Dr Schuetz said. “But our big surprise was that overexpressing MPP1—analogous to what you would see in leukemia—allows those progenitors to self-renew, to be replated over and over, to form new colonies.”
The experiments also revealed that MPP1 and ABCC4 functioned at the cell membrane, where they could play a role in the machinery that would rid the leukemia cells of chemotherapy drugs.
“When we disrupted their interaction, ABCC4 moved off the membrane and the cells became more sensitive to drugs used in AML—drugs that are pumped out of the cell by ABCC4,” Dr Schuetz said.
By screening thousands of compounds, the researchers identified some that could disrupt the ABCC4-MPP1 connection. One, called Antimycin-A, reversed drug resistance in AML cell lines and in cells from AML patients.
Antimycin-A is too toxic to be used in chemotherapy, but the researchers believe identification of the compound should aid the search for other, less-toxic drugs to disrupt the ABCC4-MPP1 interaction.
The team’s findings could also enable clinicians to identify AML patients with high levels of ABCC4 and MPP1. In such patients, drugs that disrupt ABCC4-MPP1 might enhance the effectiveness of standard chemotherapy, Dr Schuetz said.
He also noted that other cancers, including breast and colon cancer and medulloblastoma, show high levels of both ABCC4 and MPP1. Chemotherapy for those cancers might also be enhanced by drugs that disrupt ABCC4-MPP1. ![]()
Researchers say they have uncovered a target to overcome drug resistance in acute myeloid leukemia (AML).
The team discovered how a linkage between 2 proteins enables AML cells to resist chemotherapy and showed that disrupting the linkage could render the cells vulnerable to treatment.
The researchers believe their discovery could lead to drugs to enhance chemotherapy in patients with AML and other cancers.
John Schuetz, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues described this research in Nature Communications.
The team launched their experiments based on previous findings that a protein called ABCC4 was greatly elevated in aggressive cases of AML.
Dr Schuetz and his colleagues searched for other proteins that might interact with ABCC4 and enable its function. The team’s screening of candidate proteins yielded one, MPP1, which was also greatly increased in AML.
The researchers found the 2 proteins are connected, and the connection enables cells to assume the characteristics of highly proliferative leukemia cells.
These experiments involved genetically altering hematopoietic progenitor cells to have high MPP1 and ABCC4 levels. The cells were grown in culture and then replated to see if they would continue to grow, as such self-renewal is a hallmark of leukemia cells.
The researchers found that serial regrowth depended on the cells having high levels of both ABCC4 and MPP1.
“Typically, if you take normal progenitors and you replate, you could do that one time, maybe twice,” Dr Schuetz said. “But our big surprise was that overexpressing MPP1—analogous to what you would see in leukemia—allows those progenitors to self-renew, to be replated over and over, to form new colonies.”
The experiments also revealed that MPP1 and ABCC4 functioned at the cell membrane, where they could play a role in the machinery that would rid the leukemia cells of chemotherapy drugs.
“When we disrupted their interaction, ABCC4 moved off the membrane and the cells became more sensitive to drugs used in AML—drugs that are pumped out of the cell by ABCC4,” Dr Schuetz said.
By screening thousands of compounds, the researchers identified some that could disrupt the ABCC4-MPP1 connection. One, called Antimycin-A, reversed drug resistance in AML cell lines and in cells from AML patients.
Antimycin-A is too toxic to be used in chemotherapy, but the researchers believe identification of the compound should aid the search for other, less-toxic drugs to disrupt the ABCC4-MPP1 interaction.
The team’s findings could also enable clinicians to identify AML patients with high levels of ABCC4 and MPP1. In such patients, drugs that disrupt ABCC4-MPP1 might enhance the effectiveness of standard chemotherapy, Dr Schuetz said.
He also noted that other cancers, including breast and colon cancer and medulloblastoma, show high levels of both ABCC4 and MPP1. Chemotherapy for those cancers might also be enhanced by drugs that disrupt ABCC4-MPP1. ![]()
Researchers say they have uncovered a target to overcome drug resistance in acute myeloid leukemia (AML).
The team discovered how a linkage between 2 proteins enables AML cells to resist chemotherapy and showed that disrupting the linkage could render the cells vulnerable to treatment.
The researchers believe their discovery could lead to drugs to enhance chemotherapy in patients with AML and other cancers.
John Schuetz, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues described this research in Nature Communications.
The team launched their experiments based on previous findings that a protein called ABCC4 was greatly elevated in aggressive cases of AML.
Dr Schuetz and his colleagues searched for other proteins that might interact with ABCC4 and enable its function. The team’s screening of candidate proteins yielded one, MPP1, which was also greatly increased in AML.
The researchers found the 2 proteins are connected, and the connection enables cells to assume the characteristics of highly proliferative leukemia cells.
These experiments involved genetically altering hematopoietic progenitor cells to have high MPP1 and ABCC4 levels. The cells were grown in culture and then replated to see if they would continue to grow, as such self-renewal is a hallmark of leukemia cells.
The researchers found that serial regrowth depended on the cells having high levels of both ABCC4 and MPP1.
“Typically, if you take normal progenitors and you replate, you could do that one time, maybe twice,” Dr Schuetz said. “But our big surprise was that overexpressing MPP1—analogous to what you would see in leukemia—allows those progenitors to self-renew, to be replated over and over, to form new colonies.”
The experiments also revealed that MPP1 and ABCC4 functioned at the cell membrane, where they could play a role in the machinery that would rid the leukemia cells of chemotherapy drugs.
“When we disrupted their interaction, ABCC4 moved off the membrane and the cells became more sensitive to drugs used in AML—drugs that are pumped out of the cell by ABCC4,” Dr Schuetz said.
By screening thousands of compounds, the researchers identified some that could disrupt the ABCC4-MPP1 connection. One, called Antimycin-A, reversed drug resistance in AML cell lines and in cells from AML patients.
Antimycin-A is too toxic to be used in chemotherapy, but the researchers believe identification of the compound should aid the search for other, less-toxic drugs to disrupt the ABCC4-MPP1 interaction.
The team’s findings could also enable clinicians to identify AML patients with high levels of ABCC4 and MPP1. In such patients, drugs that disrupt ABCC4-MPP1 might enhance the effectiveness of standard chemotherapy, Dr Schuetz said.
He also noted that other cancers, including breast and colon cancer and medulloblastoma, show high levels of both ABCC4 and MPP1. Chemotherapy for those cancers might also be enhanced by drugs that disrupt ABCC4-MPP1. ![]()
Rigosertib produces better OS in MDS than tAML
Rigosertib has demonstrated activity and tolerability in patients with myelodysplastic syndromes (MDS) and acute myeloid leukemia transformed from MDS (tAML), according to researchers.
In a phase 1/2 study, rigosertib produced responses in a quarter of MDS/tAML patients and enabled stable disease in another quarter.
Overall survival (OS) was about a year longer for responders than for non-responders.
MDS patients were more likely to respond to rigosertib and therefore enjoyed longer OS than tAML patients.
Overall, rigosertib was considered well-tolerated. There were no treatment-related deaths, though 18% of patients experienced treatment-related serious adverse events (AEs).
Lewis Silverman, MD, of Icahn School of Medicine at Mount Sinai in New York, New York, and his colleagues described these results in Leukemia Research.
The study was sponsored by Onconova Therapeutics, Inc., the company developing rigosertib.
Rigosertib is an inhibitor of Ras-effector pathways that interacts with the Ras binding domains common to several signaling proteins, including Raf and PI3 kinase.
Dr Silverman and his colleagues tested intravenous rigosertib in a dose-escalation, phase 1/2 study of 22 patients. Patients had tAML (n=13), high-risk MDS (n=6), intermediate-2-risk MDS (n=2), or chronic myelomonocytic leukemia (n=1).
All patients had relapsed or were refractory to standard therapy and had no approved options for second-line therapies. The patients’ median age was 78 (range, 59-84), and 90% were male.
Patients received 3- to 7-day continuous infusions of rigosertib at doses ranging from 650 mg/m2/day to 1700 mg/m2/day in 14-day cycles.
The mean number of treatment cycles was 5.6 ± 5.8 (range, 1-23). The maximum tolerated dose of rigosertib was 1700 mg/m2/day, and the recommended phase 2 dose was 1375 mg/m2/day.
Safety
All patients had at least 1 AE. The most common AEs of any grade were fatigue (n=16, 73%), diarrhea (n=12, 55%), pyrexia (n=12, 55%), dyspnea (n=11, 50%), insomnia (n=11, 50%), anemia (n=10, 46%), constipation (n=9, 41%), nausea (n=9, 41%), cough (n=9, 41%), and decreased appetite (n=9, 41%).
The most common grade 3 or higher AEs were anemia (n=9, 41%), thrombocytopenia (n=5, 23%), pneumonia (n=5, 23%), hypoglycemia (n=4, 18%), hyponatremia (n=4, 18%), and hypophosphatemia (n=4, 18%).
Four patients (18%) had treatment-related serious AEs. This included hematuria and pollakiuria (n=1), dysuria and pollakiuria (n=1), asthenia (n=1), and dyspnea (n=1). Thirteen patients (59%) stopped treatment due to AEs.
Ten patients, who remained on study from 1 to 19 months, died within 30 days of stopping rigosertib. There were no treatment-related deaths.
Efficacy
Nineteen patients were evaluable for efficacy.
Five patients responded to treatment. Four patients with MDS had a marrow complete response, and 1 with tAML had a marrow partial response. Two of the patients with marrow complete response also had hematologic improvements.
Five patients had stable disease, 3 with MDS and 2 with tAML.
The median OS was 15.7 months for responders and 2.0 months for non-responders (P=0.0070). The median OS was 12.0 months for MDS patients and 2.0 months for tAML patients (P<0.0001).
“The publication of results from this historical study provides support of the relationship between bone marrow blast response and improvement in overall survival in this group of patients with MDS and acute myeloid leukemia for whom no FDA-approved treatments are currently available,” said Ramesh Kumar, president and chief executive officer of Onconova Therapeutics, Inc.
He added that these data are “fundamental to the rationale” of ongoing studies of rigosertib in high-risk MDS patients. ![]()
Rigosertib has demonstrated activity and tolerability in patients with myelodysplastic syndromes (MDS) and acute myeloid leukemia transformed from MDS (tAML), according to researchers.
In a phase 1/2 study, rigosertib produced responses in a quarter of MDS/tAML patients and enabled stable disease in another quarter.
Overall survival (OS) was about a year longer for responders than for non-responders.
MDS patients were more likely to respond to rigosertib and therefore enjoyed longer OS than tAML patients.
Overall, rigosertib was considered well-tolerated. There were no treatment-related deaths, though 18% of patients experienced treatment-related serious adverse events (AEs).
Lewis Silverman, MD, of Icahn School of Medicine at Mount Sinai in New York, New York, and his colleagues described these results in Leukemia Research.
The study was sponsored by Onconova Therapeutics, Inc., the company developing rigosertib.
Rigosertib is an inhibitor of Ras-effector pathways that interacts with the Ras binding domains common to several signaling proteins, including Raf and PI3 kinase.
Dr Silverman and his colleagues tested intravenous rigosertib in a dose-escalation, phase 1/2 study of 22 patients. Patients had tAML (n=13), high-risk MDS (n=6), intermediate-2-risk MDS (n=2), or chronic myelomonocytic leukemia (n=1).
All patients had relapsed or were refractory to standard therapy and had no approved options for second-line therapies. The patients’ median age was 78 (range, 59-84), and 90% were male.
Patients received 3- to 7-day continuous infusions of rigosertib at doses ranging from 650 mg/m2/day to 1700 mg/m2/day in 14-day cycles.
The mean number of treatment cycles was 5.6 ± 5.8 (range, 1-23). The maximum tolerated dose of rigosertib was 1700 mg/m2/day, and the recommended phase 2 dose was 1375 mg/m2/day.
Safety
All patients had at least 1 AE. The most common AEs of any grade were fatigue (n=16, 73%), diarrhea (n=12, 55%), pyrexia (n=12, 55%), dyspnea (n=11, 50%), insomnia (n=11, 50%), anemia (n=10, 46%), constipation (n=9, 41%), nausea (n=9, 41%), cough (n=9, 41%), and decreased appetite (n=9, 41%).
The most common grade 3 or higher AEs were anemia (n=9, 41%), thrombocytopenia (n=5, 23%), pneumonia (n=5, 23%), hypoglycemia (n=4, 18%), hyponatremia (n=4, 18%), and hypophosphatemia (n=4, 18%).
Four patients (18%) had treatment-related serious AEs. This included hematuria and pollakiuria (n=1), dysuria and pollakiuria (n=1), asthenia (n=1), and dyspnea (n=1). Thirteen patients (59%) stopped treatment due to AEs.
Ten patients, who remained on study from 1 to 19 months, died within 30 days of stopping rigosertib. There were no treatment-related deaths.
Efficacy
Nineteen patients were evaluable for efficacy.
Five patients responded to treatment. Four patients with MDS had a marrow complete response, and 1 with tAML had a marrow partial response. Two of the patients with marrow complete response also had hematologic improvements.
Five patients had stable disease, 3 with MDS and 2 with tAML.
The median OS was 15.7 months for responders and 2.0 months for non-responders (P=0.0070). The median OS was 12.0 months for MDS patients and 2.0 months for tAML patients (P<0.0001).
“The publication of results from this historical study provides support of the relationship between bone marrow blast response and improvement in overall survival in this group of patients with MDS and acute myeloid leukemia for whom no FDA-approved treatments are currently available,” said Ramesh Kumar, president and chief executive officer of Onconova Therapeutics, Inc.
He added that these data are “fundamental to the rationale” of ongoing studies of rigosertib in high-risk MDS patients. ![]()
Rigosertib has demonstrated activity and tolerability in patients with myelodysplastic syndromes (MDS) and acute myeloid leukemia transformed from MDS (tAML), according to researchers.
In a phase 1/2 study, rigosertib produced responses in a quarter of MDS/tAML patients and enabled stable disease in another quarter.
Overall survival (OS) was about a year longer for responders than for non-responders.
MDS patients were more likely to respond to rigosertib and therefore enjoyed longer OS than tAML patients.
Overall, rigosertib was considered well-tolerated. There were no treatment-related deaths, though 18% of patients experienced treatment-related serious adverse events (AEs).
Lewis Silverman, MD, of Icahn School of Medicine at Mount Sinai in New York, New York, and his colleagues described these results in Leukemia Research.
The study was sponsored by Onconova Therapeutics, Inc., the company developing rigosertib.
Rigosertib is an inhibitor of Ras-effector pathways that interacts with the Ras binding domains common to several signaling proteins, including Raf and PI3 kinase.
Dr Silverman and his colleagues tested intravenous rigosertib in a dose-escalation, phase 1/2 study of 22 patients. Patients had tAML (n=13), high-risk MDS (n=6), intermediate-2-risk MDS (n=2), or chronic myelomonocytic leukemia (n=1).
All patients had relapsed or were refractory to standard therapy and had no approved options for second-line therapies. The patients’ median age was 78 (range, 59-84), and 90% were male.
Patients received 3- to 7-day continuous infusions of rigosertib at doses ranging from 650 mg/m2/day to 1700 mg/m2/day in 14-day cycles.
The mean number of treatment cycles was 5.6 ± 5.8 (range, 1-23). The maximum tolerated dose of rigosertib was 1700 mg/m2/day, and the recommended phase 2 dose was 1375 mg/m2/day.
Safety
All patients had at least 1 AE. The most common AEs of any grade were fatigue (n=16, 73%), diarrhea (n=12, 55%), pyrexia (n=12, 55%), dyspnea (n=11, 50%), insomnia (n=11, 50%), anemia (n=10, 46%), constipation (n=9, 41%), nausea (n=9, 41%), cough (n=9, 41%), and decreased appetite (n=9, 41%).
The most common grade 3 or higher AEs were anemia (n=9, 41%), thrombocytopenia (n=5, 23%), pneumonia (n=5, 23%), hypoglycemia (n=4, 18%), hyponatremia (n=4, 18%), and hypophosphatemia (n=4, 18%).
Four patients (18%) had treatment-related serious AEs. This included hematuria and pollakiuria (n=1), dysuria and pollakiuria (n=1), asthenia (n=1), and dyspnea (n=1). Thirteen patients (59%) stopped treatment due to AEs.
Ten patients, who remained on study from 1 to 19 months, died within 30 days of stopping rigosertib. There were no treatment-related deaths.
Efficacy
Nineteen patients were evaluable for efficacy.
Five patients responded to treatment. Four patients with MDS had a marrow complete response, and 1 with tAML had a marrow partial response. Two of the patients with marrow complete response also had hematologic improvements.
Five patients had stable disease, 3 with MDS and 2 with tAML.
The median OS was 15.7 months for responders and 2.0 months for non-responders (P=0.0070). The median OS was 12.0 months for MDS patients and 2.0 months for tAML patients (P<0.0001).
“The publication of results from this historical study provides support of the relationship between bone marrow blast response and improvement in overall survival in this group of patients with MDS and acute myeloid leukemia for whom no FDA-approved treatments are currently available,” said Ramesh Kumar, president and chief executive officer of Onconova Therapeutics, Inc.
He added that these data are “fundamental to the rationale” of ongoing studies of rigosertib in high-risk MDS patients. ![]()
Generic azacitidine approved in Canada

Health Canada has approved Dr. Reddy’s Laboratories Ltd.’s Azacitidine for Injection 100 mg/vial, a bioequivalent generic version of VIDAZA® (azacitidine for injection).
The generic drug is approved for the same indications as VIDAZA®.
This includes treating adults with intermediate-2 or high-risk myelodysplastic syndromes (according to the International Prognostic Scoring System) who are not eligible for hematopoietic stem cell transplant.
It also includes treating adults who have acute myeloid leukemia with 20% to 30% blasts and multi-lineage dysplasia (according to World Health Organization classification) who are not eligible for hematopoietic stem cell transplant.
“The approval and launch of Azacitidine for Injection is an important milestone for Dr. Reddy’s in Canada,” said Vinod Ramachandran, PhD, country manager, Dr. Reddy’s Canada.
“The launch of the first generic azacitidine for injection is another step in our long-term commitment to bring more cost-effective options to Canadian patients.” ![]()
Health Canada has approved Dr. Reddy’s Laboratories Ltd.’s Azacitidine for Injection 100 mg/vial, a bioequivalent generic version of VIDAZA® (azacitidine for injection).
The generic drug is approved for the same indications as VIDAZA®.
This includes treating adults with intermediate-2 or high-risk myelodysplastic syndromes (according to the International Prognostic Scoring System) who are not eligible for hematopoietic stem cell transplant.
It also includes treating adults who have acute myeloid leukemia with 20% to 30% blasts and multi-lineage dysplasia (according to World Health Organization classification) who are not eligible for hematopoietic stem cell transplant.
“The approval and launch of Azacitidine for Injection is an important milestone for Dr. Reddy’s in Canada,” said Vinod Ramachandran, PhD, country manager, Dr. Reddy’s Canada.
“The launch of the first generic azacitidine for injection is another step in our long-term commitment to bring more cost-effective options to Canadian patients.” ![]()
Health Canada has approved Dr. Reddy’s Laboratories Ltd.’s Azacitidine for Injection 100 mg/vial, a bioequivalent generic version of VIDAZA® (azacitidine for injection).
The generic drug is approved for the same indications as VIDAZA®.
This includes treating adults with intermediate-2 or high-risk myelodysplastic syndromes (according to the International Prognostic Scoring System) who are not eligible for hematopoietic stem cell transplant.
It also includes treating adults who have acute myeloid leukemia with 20% to 30% blasts and multi-lineage dysplasia (according to World Health Organization classification) who are not eligible for hematopoietic stem cell transplant.
“The approval and launch of Azacitidine for Injection is an important milestone for Dr. Reddy’s in Canada,” said Vinod Ramachandran, PhD, country manager, Dr. Reddy’s Canada.
“The launch of the first generic azacitidine for injection is another step in our long-term commitment to bring more cost-effective options to Canadian patients.”