User login
Nonmyeloablative conditioning gets a radiation boost for severe hemoglobinopathies
SALT LAKE CITY – A nonmyeloablative conditioning regimen with a boosted dose of total body irradiation yielded success for a cohort of patients with severe hemoglobinopathy and haploidentical donors.
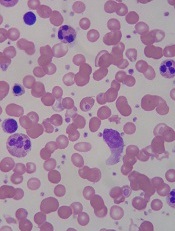
Of 17 patients with severe sickle cell disease or beta-thalassemia who received allogeneic bone marrow transplants, all but one had successful engraftment, and 13 have achieved full donor chimerism, said Javier Bolaños-Meade, MD.

“Cure of severe hemoglobinopathies is now possible for most patients,” said Dr. Bolaños-Meade. “It should no longer be considered as available to only a fraction of such patients,” such as those who come with a fully-matched donor and those able to tolerate myeloablative conditioning, he said.
Of the patients who received bone marrow transplants, five patients have stopped immunosuppressive therapy, and all patients are alive, having been followed for a median of 15 months (range, 3-34 months).
The rate of graft versus host disease (GVHD) was low: Two patients developed grade 2 acute GVHD, and one patient developed grade 3 acute GVHD; another three patients had mild to moderate chronic GVHD, but all GVHD has resolved, said Dr. Bolaños-Meade.
Historically, the difficulties with transplant in this population were numerous. “No. 1, it’s very difficult to find an HLA-matched donor,” said Dr. Bolaños-Meade. Also, since there’s no target for graft-versus-tumor effect post-transplant, any amount of chronic GVHD is also high on the list of concerns when considering a transplant for hemoglobinopathy.
“The other problem in this group of patients is their ability to tolerate myeloablation,” he said. The accumulated burden of disease, as well as sequelae of transfusion dependence for some, may make a myeloablative regime too risky.
Dr. Bolaños-Meade said that he and his collaborators at Johns Hopkins University, Baltimore, wanted to be able to address all of these concerns in one regimen. “We were trying to work out a system that may be able to solve all the problems – to use nonmyeloablation and to use whatever donor is available.”
His research group had previously shown that nonmyeloablative transplants were well tolerated in patients with sickle cell disease and that haploidentical donors could be used (Blood. 2012 Nov 22;120[22]:4285-91). “However, we had a very high incidence of graft failure,” Dr. Bolaños-Meade said.
A strategy to increase the engraftment rate while still limiting toxicity, he said, would be to increase the dose of total body irradiation used in the conditioning regimen, from 200 to 400 centigray (cGy); this higher dose was incorporated into the study protocol.
Patients were enrolled if they had severe sickle cell disease (SCD; n = 12) or beta-thalassemia (n = 5).
To enroll in the study, SCD patients had to have been hospitalized at least twice a year in the preceding 2 years. The patients with SCD were a median 26 years of age (range, 6-31 years); four were male, and eight were female. Three of the SCD patients were transfusion dependent, and several had such serious complications as osteonecrosis, brain changes seen in medical imaging, and acute coronary syndrome.
The beta-thalassemia patients were a median 7 years of age (range, 6-16 years); all but one were female, and all had been transfusion dependent since infancy.
Bone marrow donors were not all first degree relatives: There were five mothers, four fathers, four brothers, and three sisters, but also an aunt. Two pairs had major ABO incompatibility, and five had minor ABO incompatibility. Ten were ABO compatible.
The conditioning regimen for all patients involved rabbit antithymocyte globulin, fludarabine, and cyclophosphamide, and then total body irradiation given the day before transplant.
After transplant, in addition to standard supportive care, patients received cyclophosphamide on days 3 and 4. Beginning on day 5, patients received mycophenolate mofetil through day 35 and sirolimus for 1 full year after transplant.
The antithymocyte globulin induced sickle cell crises in all SCD patients, and one patient developed sirolimus-induced diabetes. One other patient had a worsening of Meniere disease, and another patient developed BK virus cystitis.
Breaking down outcomes by disease type, Dr. Bolaños-Meade said that the one engraftment failure occurred in an SCD patient. Of the remaining 11 engrafted patients, 9 have full donor chimerism, and all but 1 of the 11 are transfusion independent now. The patient who remains transfusion dependent has mixed chimerism and received bone marrow from a donor with major ABO mismatch. Although one of the five beta-thalassemia patients also has mixed chimerism, all are now transfusion independent.
The boost in hemoglobin post-transplant was relatively modest for the beta-thalassemia group, from a median 9.5 to 10.1 g/dL at the most recent visit. However, the pretransplant levels were boosted by transfusions for all patients in this group, Dr. Bolaños-Meade pointed out.
The SCD patients saw their hemoglobin go from a median 8.65 to 11.4 g/dL (P = .001). Median bilirubin for this group dropped from 2.4 to 0.2 mg/dL (P = .002) with the cessation of sickling-related hemolysis; significant improvements were also seen in absolute reticulocyte counts and lactate dehydrogenase levels.
Dr. Bolaños-Meade reported that he is on the data safety monitoring board of Incyte.
SOURCE: Bolaños-Meade J et al. BMT Tandem Meetings, Abstract LBA-3.
SALT LAKE CITY – A nonmyeloablative conditioning regimen with a boosted dose of total body irradiation yielded success for a cohort of patients with severe hemoglobinopathy and haploidentical donors.
Of 17 patients with severe sickle cell disease or beta-thalassemia who received allogeneic bone marrow transplants, all but one had successful engraftment, and 13 have achieved full donor chimerism, said Javier Bolaños-Meade, MD.
“Cure of severe hemoglobinopathies is now possible for most patients,” said Dr. Bolaños-Meade. “It should no longer be considered as available to only a fraction of such patients,” such as those who come with a fully-matched donor and those able to tolerate myeloablative conditioning, he said.
Of the patients who received bone marrow transplants, five patients have stopped immunosuppressive therapy, and all patients are alive, having been followed for a median of 15 months (range, 3-34 months).
The rate of graft versus host disease (GVHD) was low: Two patients developed grade 2 acute GVHD, and one patient developed grade 3 acute GVHD; another three patients had mild to moderate chronic GVHD, but all GVHD has resolved, said Dr. Bolaños-Meade.
Historically, the difficulties with transplant in this population were numerous. “No. 1, it’s very difficult to find an HLA-matched donor,” said Dr. Bolaños-Meade. Also, since there’s no target for graft-versus-tumor effect post-transplant, any amount of chronic GVHD is also high on the list of concerns when considering a transplant for hemoglobinopathy.
“The other problem in this group of patients is their ability to tolerate myeloablation,” he said. The accumulated burden of disease, as well as sequelae of transfusion dependence for some, may make a myeloablative regime too risky.
Dr. Bolaños-Meade said that he and his collaborators at Johns Hopkins University, Baltimore, wanted to be able to address all of these concerns in one regimen. “We were trying to work out a system that may be able to solve all the problems – to use nonmyeloablation and to use whatever donor is available.”
His research group had previously shown that nonmyeloablative transplants were well tolerated in patients with sickle cell disease and that haploidentical donors could be used (Blood. 2012 Nov 22;120[22]:4285-91). “However, we had a very high incidence of graft failure,” Dr. Bolaños-Meade said.
A strategy to increase the engraftment rate while still limiting toxicity, he said, would be to increase the dose of total body irradiation used in the conditioning regimen, from 200 to 400 centigray (cGy); this higher dose was incorporated into the study protocol.
Patients were enrolled if they had severe sickle cell disease (SCD; n = 12) or beta-thalassemia (n = 5).
To enroll in the study, SCD patients had to have been hospitalized at least twice a year in the preceding 2 years. The patients with SCD were a median 26 years of age (range, 6-31 years); four were male, and eight were female. Three of the SCD patients were transfusion dependent, and several had such serious complications as osteonecrosis, brain changes seen in medical imaging, and acute coronary syndrome.
The beta-thalassemia patients were a median 7 years of age (range, 6-16 years); all but one were female, and all had been transfusion dependent since infancy.
Bone marrow donors were not all first degree relatives: There were five mothers, four fathers, four brothers, and three sisters, but also an aunt. Two pairs had major ABO incompatibility, and five had minor ABO incompatibility. Ten were ABO compatible.
The conditioning regimen for all patients involved rabbit antithymocyte globulin, fludarabine, and cyclophosphamide, and then total body irradiation given the day before transplant.
After transplant, in addition to standard supportive care, patients received cyclophosphamide on days 3 and 4. Beginning on day 5, patients received mycophenolate mofetil through day 35 and sirolimus for 1 full year after transplant.
The antithymocyte globulin induced sickle cell crises in all SCD patients, and one patient developed sirolimus-induced diabetes. One other patient had a worsening of Meniere disease, and another patient developed BK virus cystitis.
Breaking down outcomes by disease type, Dr. Bolaños-Meade said that the one engraftment failure occurred in an SCD patient. Of the remaining 11 engrafted patients, 9 have full donor chimerism, and all but 1 of the 11 are transfusion independent now. The patient who remains transfusion dependent has mixed chimerism and received bone marrow from a donor with major ABO mismatch. Although one of the five beta-thalassemia patients also has mixed chimerism, all are now transfusion independent.
The boost in hemoglobin post-transplant was relatively modest for the beta-thalassemia group, from a median 9.5 to 10.1 g/dL at the most recent visit. However, the pretransplant levels were boosted by transfusions for all patients in this group, Dr. Bolaños-Meade pointed out.
The SCD patients saw their hemoglobin go from a median 8.65 to 11.4 g/dL (P = .001). Median bilirubin for this group dropped from 2.4 to 0.2 mg/dL (P = .002) with the cessation of sickling-related hemolysis; significant improvements were also seen in absolute reticulocyte counts and lactate dehydrogenase levels.
Dr. Bolaños-Meade reported that he is on the data safety monitoring board of Incyte.
SOURCE: Bolaños-Meade J et al. BMT Tandem Meetings, Abstract LBA-3.
SALT LAKE CITY – A nonmyeloablative conditioning regimen with a boosted dose of total body irradiation yielded success for a cohort of patients with severe hemoglobinopathy and haploidentical donors.
Of 17 patients with severe sickle cell disease or beta-thalassemia who received allogeneic bone marrow transplants, all but one had successful engraftment, and 13 have achieved full donor chimerism, said Javier Bolaños-Meade, MD.
“Cure of severe hemoglobinopathies is now possible for most patients,” said Dr. Bolaños-Meade. “It should no longer be considered as available to only a fraction of such patients,” such as those who come with a fully-matched donor and those able to tolerate myeloablative conditioning, he said.
Of the patients who received bone marrow transplants, five patients have stopped immunosuppressive therapy, and all patients are alive, having been followed for a median of 15 months (range, 3-34 months).
The rate of graft versus host disease (GVHD) was low: Two patients developed grade 2 acute GVHD, and one patient developed grade 3 acute GVHD; another three patients had mild to moderate chronic GVHD, but all GVHD has resolved, said Dr. Bolaños-Meade.
Historically, the difficulties with transplant in this population were numerous. “No. 1, it’s very difficult to find an HLA-matched donor,” said Dr. Bolaños-Meade. Also, since there’s no target for graft-versus-tumor effect post-transplant, any amount of chronic GVHD is also high on the list of concerns when considering a transplant for hemoglobinopathy.
“The other problem in this group of patients is their ability to tolerate myeloablation,” he said. The accumulated burden of disease, as well as sequelae of transfusion dependence for some, may make a myeloablative regime too risky.
Dr. Bolaños-Meade said that he and his collaborators at Johns Hopkins University, Baltimore, wanted to be able to address all of these concerns in one regimen. “We were trying to work out a system that may be able to solve all the problems – to use nonmyeloablation and to use whatever donor is available.”
His research group had previously shown that nonmyeloablative transplants were well tolerated in patients with sickle cell disease and that haploidentical donors could be used (Blood. 2012 Nov 22;120[22]:4285-91). “However, we had a very high incidence of graft failure,” Dr. Bolaños-Meade said.
A strategy to increase the engraftment rate while still limiting toxicity, he said, would be to increase the dose of total body irradiation used in the conditioning regimen, from 200 to 400 centigray (cGy); this higher dose was incorporated into the study protocol.
Patients were enrolled if they had severe sickle cell disease (SCD; n = 12) or beta-thalassemia (n = 5).
To enroll in the study, SCD patients had to have been hospitalized at least twice a year in the preceding 2 years. The patients with SCD were a median 26 years of age (range, 6-31 years); four were male, and eight were female. Three of the SCD patients were transfusion dependent, and several had such serious complications as osteonecrosis, brain changes seen in medical imaging, and acute coronary syndrome.
The beta-thalassemia patients were a median 7 years of age (range, 6-16 years); all but one were female, and all had been transfusion dependent since infancy.
Bone marrow donors were not all first degree relatives: There were five mothers, four fathers, four brothers, and three sisters, but also an aunt. Two pairs had major ABO incompatibility, and five had minor ABO incompatibility. Ten were ABO compatible.
The conditioning regimen for all patients involved rabbit antithymocyte globulin, fludarabine, and cyclophosphamide, and then total body irradiation given the day before transplant.
After transplant, in addition to standard supportive care, patients received cyclophosphamide on days 3 and 4. Beginning on day 5, patients received mycophenolate mofetil through day 35 and sirolimus for 1 full year after transplant.
The antithymocyte globulin induced sickle cell crises in all SCD patients, and one patient developed sirolimus-induced diabetes. One other patient had a worsening of Meniere disease, and another patient developed BK virus cystitis.
Breaking down outcomes by disease type, Dr. Bolaños-Meade said that the one engraftment failure occurred in an SCD patient. Of the remaining 11 engrafted patients, 9 have full donor chimerism, and all but 1 of the 11 are transfusion independent now. The patient who remains transfusion dependent has mixed chimerism and received bone marrow from a donor with major ABO mismatch. Although one of the five beta-thalassemia patients also has mixed chimerism, all are now transfusion independent.
The boost in hemoglobin post-transplant was relatively modest for the beta-thalassemia group, from a median 9.5 to 10.1 g/dL at the most recent visit. However, the pretransplant levels were boosted by transfusions for all patients in this group, Dr. Bolaños-Meade pointed out.
The SCD patients saw their hemoglobin go from a median 8.65 to 11.4 g/dL (P = .001). Median bilirubin for this group dropped from 2.4 to 0.2 mg/dL (P = .002) with the cessation of sickling-related hemolysis; significant improvements were also seen in absolute reticulocyte counts and lactate dehydrogenase levels.
Dr. Bolaños-Meade reported that he is on the data safety monitoring board of Incyte.
SOURCE: Bolaños-Meade J et al. BMT Tandem Meetings, Abstract LBA-3.
REPORTING FROM THE 2018 BMT TANDEM MEETINGS
Key clinical point:
Major finding: Of the 17 patients who received haploidentical bone marrow transplant, 13 have achieved full chimerism.
Study details: Report of 17 consecutive patients with severe sickle cell disease or beta-thalassemia who received nonmyeloablative conditioning and bone marrow transplant from haploidentical donors.
Disclosures: Dr. Bolaños-Meade reported no outside sources of funding for the study. He is on the data safety monitoring board of Incyte.
Source: Bolaños-Meade J et al. 2018 BMT Tandem Meetings, Abstract LBA-3.
Drug nets orphan designation for beta-thalassemia
The US Food and Drug Administration (FDA) has granted orphan drug designation to PTG-300, a subcutaneous injectable hepcidin mimetic, for the treatment of beta-thalassemia.
Protagonist Therapeutics, the company developing PTG-300, recently completed a phase 1 trial of the drug.
In this study, healthy volunteers treated with PTG-300 achieved dose-related and sustained reductions in serum iron levels.
In addition, PTG-300 was considered well tolerated, producing no serious adverse events or dose-limiting toxicities.
Protagonist Therapeutics intends to initiate a global clinical trial of PTG-300 in patients with beta-thalassemia following upcoming meetings with the FDA and European Medicines Agency.
“Beta-thalassemia is a rare genetic blood disorder that is characterized by impaired red blood cell production that can result in life-threatening chronic anemia, usually requiring regular and life-long blood transfusions for survival,” said David Y. Liu, PhD, chief scientific officer and head of research and development at Protagonist Therapeutics.
“Over time, these transfusions can lead to excessive iron levels in the body, which can be toxic and consequently lead to end-stage damage to vital organs such as the liver and the heart. As a hepcidin mimetic, PTG-300 is designed to help reduce these excessive iron levels, and, thereby, it may lead to improvements in anemia and decreased need for blood transfusions and chelation therapy.”
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The US Food and Drug Administration (FDA) has granted orphan drug designation to PTG-300, a subcutaneous injectable hepcidin mimetic, for the treatment of beta-thalassemia.
Protagonist Therapeutics, the company developing PTG-300, recently completed a phase 1 trial of the drug.
In this study, healthy volunteers treated with PTG-300 achieved dose-related and sustained reductions in serum iron levels.
In addition, PTG-300 was considered well tolerated, producing no serious adverse events or dose-limiting toxicities.
Protagonist Therapeutics intends to initiate a global clinical trial of PTG-300 in patients with beta-thalassemia following upcoming meetings with the FDA and European Medicines Agency.
“Beta-thalassemia is a rare genetic blood disorder that is characterized by impaired red blood cell production that can result in life-threatening chronic anemia, usually requiring regular and life-long blood transfusions for survival,” said David Y. Liu, PhD, chief scientific officer and head of research and development at Protagonist Therapeutics.
“Over time, these transfusions can lead to excessive iron levels in the body, which can be toxic and consequently lead to end-stage damage to vital organs such as the liver and the heart. As a hepcidin mimetic, PTG-300 is designed to help reduce these excessive iron levels, and, thereby, it may lead to improvements in anemia and decreased need for blood transfusions and chelation therapy.”
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The US Food and Drug Administration (FDA) has granted orphan drug designation to PTG-300, a subcutaneous injectable hepcidin mimetic, for the treatment of beta-thalassemia.
Protagonist Therapeutics, the company developing PTG-300, recently completed a phase 1 trial of the drug.
In this study, healthy volunteers treated with PTG-300 achieved dose-related and sustained reductions in serum iron levels.
In addition, PTG-300 was considered well tolerated, producing no serious adverse events or dose-limiting toxicities.
Protagonist Therapeutics intends to initiate a global clinical trial of PTG-300 in patients with beta-thalassemia following upcoming meetings with the FDA and European Medicines Agency.
“Beta-thalassemia is a rare genetic blood disorder that is characterized by impaired red blood cell production that can result in life-threatening chronic anemia, usually requiring regular and life-long blood transfusions for survival,” said David Y. Liu, PhD, chief scientific officer and head of research and development at Protagonist Therapeutics.
“Over time, these transfusions can lead to excessive iron levels in the body, which can be toxic and consequently lead to end-stage damage to vital organs such as the liver and the heart. As a hepcidin mimetic, PTG-300 is designed to help reduce these excessive iron levels, and, thereby, it may lead to improvements in anemia and decreased need for blood transfusions and chelation therapy.”
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
HCT-CI score may predict mortality for nonmalignant disease
SALT LAKE CITY – Scores of 3 or higher on the Hematopoietic Cell Transplantation Comorbidity Index (HCT-CI) are associated with an increased risk of posttransplant mortality in certain patients undergoing allogeneic HCT for nonmalignant disease, according to findings from a review of more than 4,000 patients.
The exception was in patients undergoing HCT for hemoglobinopathies, Larisa Broglie, MD, reported at the combined annual meetings of the Center for International Blood & Marrow Transplant Research and the American Society for Blood and Marrow Transplantation.
The findings of the study, which is the largest to date to validate the usefulness of the HCT-CI for risk assessment in HCT patients with nonmalignant disease, have important implications for patient counseling and decision making, said Dr. Broglie of the Medical College of Wisconsin, Milwaukee.
Of 4,083 children and adults who underwent a first allogeneic HCT for a nonmalignant disease between 2007 and 2014 and who had sufficient follow-up (median, 39 months), 61% had an HCT-CI score of 0, 20% had a score of 1-2, and 19% had a score of 3 or higher.
After adjustment for age, disease, donor, graft source, recipient cytomegalovirus status, and performance status, scores of 3 or greater were associated with an overall increased hazard ratio for poor survival (HRs of 1.33 for scores of 3-4 and 2.31 for scores of 5 or greater, vs. 1.0 and 1.127 for scores of 0 or 1-2, respectively), she said.
Patients with an HCT-CI score of 0 had estimated 2-year overall survival of 82.7%, compared with 80.3% for scores 1-2, 74.0% for scores 3-4, and 55.8% for scores of 5 or greater.
Patients included in this study were identified from the Center for International Blood & Marrow Transplant Research database. They ranged in age from under 1 year to 77 years but most were young; the median age was 9 years and 78% of patients were under age 20.
HCT was performed for acquired aplastic anemia in 33% of patients, immune deficiencies in 19%, hemoglobinopathies in 16%, bone marrow failure in 12%, histiocytic disorders in 11%, metabolic disease in 9%, or autoimmune disease in less than 1%, she said, noting that the most frequent comorbidities seen within the entire cohort were moderate pulmonary disease, hepatic disease, and infection requiring ongoing treatment.
The effect of HCT-CI score on survival was present regardless of conditioning intensity and graft-versus-host disease prophylaxis, but scores predicted mortality risk differently based on underlying disease, and different comorbidities predominated in each disease category, she said, explaining that this was apparent when patients were stratified by the seven represented nonmalignant disease categories to account for disease heterogeneity.
For example, HCT-CI score predicted mortality risk in patients with aplastic anemia (HRs of 1.00, 1.19, and 2.06 for scores of 0, 1-2, and 3 or greater, respectively), and in patients with immune deficiency (HRs of 1.00, 1.37, and 1.87 for scores of 0, 1-2, and 3 or greater, respectively), and the distribution of comorbidities in patients in these two disease categories was similar to that of the overall cohort.
However, HCT-CI score did not predict mortality risk in those undergoing HCT for hemoglobinopathies (HRs of 1.00, 0.46, and 0.59 for scores of 0, 1-2, and 3 or greater, respectively), Dr. Broglie said, noting that these patients had overall high survival rates regardless of HCT-CI scores, and they had comorbidities that differed from the overall cohort.
HCT is a curative treatment strategy for many patients with nonmalignant diseases but transplant-related mortality remains a concern, she said. While HCT-CI has been shown to be useful for discriminating the risks of nonrelapse and overall survival among patients with hematologic malignancies who undergo allogeneic HCT, its usefulness in patients undergoing HCT for nonmalignant diseases has been less clear.
The distinction is important, as patients with nonmalignant diseases have different pretransplant exposures and may have comorbidities specific to their underlying disease. Furthermore, transplantation approaches – including conditioning regimens and intensities – differ, she said.
“And the HCT-CI was developed to predict risk of nonrelapse mortality, but relapse in nonmalignant diseases can often be difficult to define,” she added.
The current findings demonstrate the value of the HCT-CI in nonmalignant diseases, and “offer the potential to intervene with transplantation prior to the onset of comorbidities, or with efforts to prevent comorbidities prior to transplantation,” she said, concluding that “future efforts could focus on further refining pretransplant risk assessment for patients with nonmalignant diseases, especially for patients with hemoglobinopathies and HCT-CI scores of less than 3.”
Dr. Broglie reported having no financial disclosures.
SOURCE: Broglie L et al. Abstract 16.
SALT LAKE CITY – Scores of 3 or higher on the Hematopoietic Cell Transplantation Comorbidity Index (HCT-CI) are associated with an increased risk of posttransplant mortality in certain patients undergoing allogeneic HCT for nonmalignant disease, according to findings from a review of more than 4,000 patients.
The exception was in patients undergoing HCT for hemoglobinopathies, Larisa Broglie, MD, reported at the combined annual meetings of the Center for International Blood & Marrow Transplant Research and the American Society for Blood and Marrow Transplantation.
The findings of the study, which is the largest to date to validate the usefulness of the HCT-CI for risk assessment in HCT patients with nonmalignant disease, have important implications for patient counseling and decision making, said Dr. Broglie of the Medical College of Wisconsin, Milwaukee.
Of 4,083 children and adults who underwent a first allogeneic HCT for a nonmalignant disease between 2007 and 2014 and who had sufficient follow-up (median, 39 months), 61% had an HCT-CI score of 0, 20% had a score of 1-2, and 19% had a score of 3 or higher.
After adjustment for age, disease, donor, graft source, recipient cytomegalovirus status, and performance status, scores of 3 or greater were associated with an overall increased hazard ratio for poor survival (HRs of 1.33 for scores of 3-4 and 2.31 for scores of 5 or greater, vs. 1.0 and 1.127 for scores of 0 or 1-2, respectively), she said.
Patients with an HCT-CI score of 0 had estimated 2-year overall survival of 82.7%, compared with 80.3% for scores 1-2, 74.0% for scores 3-4, and 55.8% for scores of 5 or greater.
Patients included in this study were identified from the Center for International Blood & Marrow Transplant Research database. They ranged in age from under 1 year to 77 years but most were young; the median age was 9 years and 78% of patients were under age 20.
HCT was performed for acquired aplastic anemia in 33% of patients, immune deficiencies in 19%, hemoglobinopathies in 16%, bone marrow failure in 12%, histiocytic disorders in 11%, metabolic disease in 9%, or autoimmune disease in less than 1%, she said, noting that the most frequent comorbidities seen within the entire cohort were moderate pulmonary disease, hepatic disease, and infection requiring ongoing treatment.
The effect of HCT-CI score on survival was present regardless of conditioning intensity and graft-versus-host disease prophylaxis, but scores predicted mortality risk differently based on underlying disease, and different comorbidities predominated in each disease category, she said, explaining that this was apparent when patients were stratified by the seven represented nonmalignant disease categories to account for disease heterogeneity.
For example, HCT-CI score predicted mortality risk in patients with aplastic anemia (HRs of 1.00, 1.19, and 2.06 for scores of 0, 1-2, and 3 or greater, respectively), and in patients with immune deficiency (HRs of 1.00, 1.37, and 1.87 for scores of 0, 1-2, and 3 or greater, respectively), and the distribution of comorbidities in patients in these two disease categories was similar to that of the overall cohort.
However, HCT-CI score did not predict mortality risk in those undergoing HCT for hemoglobinopathies (HRs of 1.00, 0.46, and 0.59 for scores of 0, 1-2, and 3 or greater, respectively), Dr. Broglie said, noting that these patients had overall high survival rates regardless of HCT-CI scores, and they had comorbidities that differed from the overall cohort.
HCT is a curative treatment strategy for many patients with nonmalignant diseases but transplant-related mortality remains a concern, she said. While HCT-CI has been shown to be useful for discriminating the risks of nonrelapse and overall survival among patients with hematologic malignancies who undergo allogeneic HCT, its usefulness in patients undergoing HCT for nonmalignant diseases has been less clear.
The distinction is important, as patients with nonmalignant diseases have different pretransplant exposures and may have comorbidities specific to their underlying disease. Furthermore, transplantation approaches – including conditioning regimens and intensities – differ, she said.
“And the HCT-CI was developed to predict risk of nonrelapse mortality, but relapse in nonmalignant diseases can often be difficult to define,” she added.
The current findings demonstrate the value of the HCT-CI in nonmalignant diseases, and “offer the potential to intervene with transplantation prior to the onset of comorbidities, or with efforts to prevent comorbidities prior to transplantation,” she said, concluding that “future efforts could focus on further refining pretransplant risk assessment for patients with nonmalignant diseases, especially for patients with hemoglobinopathies and HCT-CI scores of less than 3.”
Dr. Broglie reported having no financial disclosures.
SOURCE: Broglie L et al. Abstract 16.
SALT LAKE CITY – Scores of 3 or higher on the Hematopoietic Cell Transplantation Comorbidity Index (HCT-CI) are associated with an increased risk of posttransplant mortality in certain patients undergoing allogeneic HCT for nonmalignant disease, according to findings from a review of more than 4,000 patients.
The exception was in patients undergoing HCT for hemoglobinopathies, Larisa Broglie, MD, reported at the combined annual meetings of the Center for International Blood & Marrow Transplant Research and the American Society for Blood and Marrow Transplantation.
The findings of the study, which is the largest to date to validate the usefulness of the HCT-CI for risk assessment in HCT patients with nonmalignant disease, have important implications for patient counseling and decision making, said Dr. Broglie of the Medical College of Wisconsin, Milwaukee.
Of 4,083 children and adults who underwent a first allogeneic HCT for a nonmalignant disease between 2007 and 2014 and who had sufficient follow-up (median, 39 months), 61% had an HCT-CI score of 0, 20% had a score of 1-2, and 19% had a score of 3 or higher.
After adjustment for age, disease, donor, graft source, recipient cytomegalovirus status, and performance status, scores of 3 or greater were associated with an overall increased hazard ratio for poor survival (HRs of 1.33 for scores of 3-4 and 2.31 for scores of 5 or greater, vs. 1.0 and 1.127 for scores of 0 or 1-2, respectively), she said.
Patients with an HCT-CI score of 0 had estimated 2-year overall survival of 82.7%, compared with 80.3% for scores 1-2, 74.0% for scores 3-4, and 55.8% for scores of 5 or greater.
Patients included in this study were identified from the Center for International Blood & Marrow Transplant Research database. They ranged in age from under 1 year to 77 years but most were young; the median age was 9 years and 78% of patients were under age 20.
HCT was performed for acquired aplastic anemia in 33% of patients, immune deficiencies in 19%, hemoglobinopathies in 16%, bone marrow failure in 12%, histiocytic disorders in 11%, metabolic disease in 9%, or autoimmune disease in less than 1%, she said, noting that the most frequent comorbidities seen within the entire cohort were moderate pulmonary disease, hepatic disease, and infection requiring ongoing treatment.
The effect of HCT-CI score on survival was present regardless of conditioning intensity and graft-versus-host disease prophylaxis, but scores predicted mortality risk differently based on underlying disease, and different comorbidities predominated in each disease category, she said, explaining that this was apparent when patients were stratified by the seven represented nonmalignant disease categories to account for disease heterogeneity.
For example, HCT-CI score predicted mortality risk in patients with aplastic anemia (HRs of 1.00, 1.19, and 2.06 for scores of 0, 1-2, and 3 or greater, respectively), and in patients with immune deficiency (HRs of 1.00, 1.37, and 1.87 for scores of 0, 1-2, and 3 or greater, respectively), and the distribution of comorbidities in patients in these two disease categories was similar to that of the overall cohort.
However, HCT-CI score did not predict mortality risk in those undergoing HCT for hemoglobinopathies (HRs of 1.00, 0.46, and 0.59 for scores of 0, 1-2, and 3 or greater, respectively), Dr. Broglie said, noting that these patients had overall high survival rates regardless of HCT-CI scores, and they had comorbidities that differed from the overall cohort.
HCT is a curative treatment strategy for many patients with nonmalignant diseases but transplant-related mortality remains a concern, she said. While HCT-CI has been shown to be useful for discriminating the risks of nonrelapse and overall survival among patients with hematologic malignancies who undergo allogeneic HCT, its usefulness in patients undergoing HCT for nonmalignant diseases has been less clear.
The distinction is important, as patients with nonmalignant diseases have different pretransplant exposures and may have comorbidities specific to their underlying disease. Furthermore, transplantation approaches – including conditioning regimens and intensities – differ, she said.
“And the HCT-CI was developed to predict risk of nonrelapse mortality, but relapse in nonmalignant diseases can often be difficult to define,” she added.
The current findings demonstrate the value of the HCT-CI in nonmalignant diseases, and “offer the potential to intervene with transplantation prior to the onset of comorbidities, or with efforts to prevent comorbidities prior to transplantation,” she said, concluding that “future efforts could focus on further refining pretransplant risk assessment for patients with nonmalignant diseases, especially for patients with hemoglobinopathies and HCT-CI scores of less than 3.”
Dr. Broglie reported having no financial disclosures.
SOURCE: Broglie L et al. Abstract 16.
REPORTING FROM THE 2018 BMT TANDEM MEETINGS
Key clinical point:
Major finding: Hazard ratios for poor survival were 1.33 for scores of 3-4 and 2.31 for scores of 5 or greater, compared with 1.0 and 1.127 for scores of 0 or 1-2, respectively.
Study details: A review of 4,083 patients from the CIBMTR database.
Disclosures: Dr. Broglie reported having no financial disclosures.
Source: Broglie L et al. Abstract 16.
HSCT approach provides ‘excellent’ survival in FA

SALT LAKE CITY—A “risk-adjusted” approach leads to “excellent” survival in patients with Fanconi anemia (FA) undergoing alternative donor hematopoietic stem cell transplant (HSCT), according to a speaker at the 2018 BMT Tandem Meetings.
All FA patients who received personalized doses of busulfan in place of total body irradiation (TBI) were alive and disease-free after undergoing HSCT for bone marrow failure or myelodysplastic syndrome (MDS).
None of the patients developed graft-vs-host disease (GVHD), and the most common toxicity was viral infection.
Parinda A. Mehta, MD, of Cincinnati Children’s Hospital Medical Center in Ohio, presented these results at this year’s BMT Tandem Meetings as abstract 109.*
“We all know that inherent chemotherapy and radiation sensitivity makes transplant for patients with Fanconi anemia quite challenging,” Dr Mehta began. “In our recently published, prospective, multi-institutional study, we showed excellent outcomes of alternative donor transplant in patients with Fanconi anemia without using radiation.”
“In that study,** TBI was replaced by pharmacokinetically adjusted busulfan. It proved that, yes, we can do alternative donor transplant successfully without radiation by showing an overall survival of 80% for a total of 45 patients. We were quite ecstatic to see these numbers.”
The study also showed that younger patients fared better with this regimen, and younger patients did best with the lowest dose of busulfan tested (0.6 mg/kg vs 0.8 to 1.0 mg/kg). In addition, patients who underwent HSCT for bone marrow failure had better outcomes than patients who had MDS.
This led Dr Mehta and her colleagues to hypothesize that adjusting busulfan dosing based on a patient’s age and disease status at HSCT could minimize toxicity and improve outcomes.
Patients
The researchers tested their theory in 22 FA patients. They had a median age of 7 (range, 4-27), and most (n=13) were female.
Twelve patients had pancytopenia, 6 had severe single-lineage cytopenia, 3 had low-grade MDS, and 1 patient had acute myeloid leukemia (AML).
Eighteen patients had a history of transfusions, and 3 had a history of androgen use.
Treatment
The preparative regimen consisted of 4 doses of busulfan (every 12 hours on day -7 to -6), followed by cyclophosphamide at 10 mg/kg/day (on day -5 to -2), fludarabine at 35 mg/m2/day (on day -5 to -2), and rabbit antithymocyte globulin at 2.5 mg/kg/day (on day -5 to -2).
Busulfan doses were adjusted according to age and disease status.
Children (age 18 and younger) with bone marrow failure received busulfan at 0.6 to 0.8 mg/kg. Children with MDS/AML received busulfan at 0.8 to 1.0 mg/kg. Adults (19 and older) received the lowest dose of busulfan—0.4 mg/kg—regardless of disease status.
“At the first sight, this will look counterintuitive . . . ,” Dr Mehta said. “However, based on our previous experience, in general and also from results of our previous study, this was specifically designed to avoid upfront TRM [transplant-related mortality] for these adult patients.”
All 22 patients received CD34-selected, T-cell-depleted peripheral blood stem cells from unrelated donors. Eleven patients received a fully matched graft (10/10), 8 patients had a 9/10 match, and 3 had an 8/10 match.
The median number of CD34+ cells/kg was 23.9 x 106 (range, 4.9-76.6), and the median number of CD3 cells/kg was 1 x 104 (range, 0.003-3.1).
T-cell depletion was the only GVHD prophylaxis used.
Patients with MDS/AML could receive azacitidine at day 42 after HSCT, an option intended to prevent relapse in these patients.
Toxicity
There were no cases of acute or chronic GVHD.
Toxicities included infections (n=24), oral mucositis (n=14), hyperbilirubinemia (n=2), pulmonary hemorrhage (n=1), and sinusoidal obstruction syndrome (n=1).
There were 20 viral infections, 4 bacterial infections, and no fungal infections. Viral infections included BK virus (n=7), cytomegalovirus (n=6), Epstein-Barr virus (n=6), and adenovirus (n=1).
Dr Mehta noted that viral infections are “not unexpected in a T-cell-depleted graft setting.”
“Because we know this complication [can occur], and we worry about our patients, one of the things that, in recent years, we have done is, we manufacture viral-specific CTLs [cytotoxic T lymphocytes] for all of these patients ahead of time whenever possible,” she said.
“To give you an example, 19 out of these 20 patients’ viral infections—or rather, viremias—are completely under control with the use of either antivirals or donor-specific CTLs, including a third-party CTL in one of the patients.”
Response and survival
All 22 patients engrafted. The median time to neutrophil engraftment was 9 days (range, 8-10), and the median time to platelet engraftment was 16 days (range, 11-40).
Twenty-one of the 22 patients (95%) were alive and disease-free at last follow-up. The median follow-up was 21 months (range, 6-44).
The single AML patient achieved remission but died of post-transplant lymphoproliferative disorder (PTLD) on day 202 after HSCT. Dr Mehta said this was due to partial loss of follow-up and noncompliance with medical recommendations during PTLD treatment.
The AML patient also had “significant upfront toxicity” but “recovered very nicely,” according to Dr Mehta. He had severe mucositis, herpetic stomatitis, and sinusoidal obstruction syndrome that responded to defibrotide.
“Overall, we are quite excited to see 95% overall survival for this cohort and conclude that the current risk-adjusted approach leads to excellent overall survival and disease-free survival in patients undergoing alternative donor transplant either for marrow failure or MDS/AML,” Dr Mehta said.
“Enrollment is ongoing, and we hope to see continued success in patients with MDS/AML as well as in adult patients.”
*Data in the abstract differ from the presentation.
**Mehta PA et al. Radiation-free, alternative donor HCT for Fanconi anemia patients: results from a prospective multi-institutional study. Blood 2017; doi: https://doi.org/10.1182/blood-2016-09-743112.
SALT LAKE CITY—A “risk-adjusted” approach leads to “excellent” survival in patients with Fanconi anemia (FA) undergoing alternative donor hematopoietic stem cell transplant (HSCT), according to a speaker at the 2018 BMT Tandem Meetings.
All FA patients who received personalized doses of busulfan in place of total body irradiation (TBI) were alive and disease-free after undergoing HSCT for bone marrow failure or myelodysplastic syndrome (MDS).
None of the patients developed graft-vs-host disease (GVHD), and the most common toxicity was viral infection.
Parinda A. Mehta, MD, of Cincinnati Children’s Hospital Medical Center in Ohio, presented these results at this year’s BMT Tandem Meetings as abstract 109.*
“We all know that inherent chemotherapy and radiation sensitivity makes transplant for patients with Fanconi anemia quite challenging,” Dr Mehta began. “In our recently published, prospective, multi-institutional study, we showed excellent outcomes of alternative donor transplant in patients with Fanconi anemia without using radiation.”
“In that study,** TBI was replaced by pharmacokinetically adjusted busulfan. It proved that, yes, we can do alternative donor transplant successfully without radiation by showing an overall survival of 80% for a total of 45 patients. We were quite ecstatic to see these numbers.”
The study also showed that younger patients fared better with this regimen, and younger patients did best with the lowest dose of busulfan tested (0.6 mg/kg vs 0.8 to 1.0 mg/kg). In addition, patients who underwent HSCT for bone marrow failure had better outcomes than patients who had MDS.
This led Dr Mehta and her colleagues to hypothesize that adjusting busulfan dosing based on a patient’s age and disease status at HSCT could minimize toxicity and improve outcomes.
Patients
The researchers tested their theory in 22 FA patients. They had a median age of 7 (range, 4-27), and most (n=13) were female.
Twelve patients had pancytopenia, 6 had severe single-lineage cytopenia, 3 had low-grade MDS, and 1 patient had acute myeloid leukemia (AML).
Eighteen patients had a history of transfusions, and 3 had a history of androgen use.
Treatment
The preparative regimen consisted of 4 doses of busulfan (every 12 hours on day -7 to -6), followed by cyclophosphamide at 10 mg/kg/day (on day -5 to -2), fludarabine at 35 mg/m2/day (on day -5 to -2), and rabbit antithymocyte globulin at 2.5 mg/kg/day (on day -5 to -2).
Busulfan doses were adjusted according to age and disease status.
Children (age 18 and younger) with bone marrow failure received busulfan at 0.6 to 0.8 mg/kg. Children with MDS/AML received busulfan at 0.8 to 1.0 mg/kg. Adults (19 and older) received the lowest dose of busulfan—0.4 mg/kg—regardless of disease status.
“At the first sight, this will look counterintuitive . . . ,” Dr Mehta said. “However, based on our previous experience, in general and also from results of our previous study, this was specifically designed to avoid upfront TRM [transplant-related mortality] for these adult patients.”
All 22 patients received CD34-selected, T-cell-depleted peripheral blood stem cells from unrelated donors. Eleven patients received a fully matched graft (10/10), 8 patients had a 9/10 match, and 3 had an 8/10 match.
The median number of CD34+ cells/kg was 23.9 x 106 (range, 4.9-76.6), and the median number of CD3 cells/kg was 1 x 104 (range, 0.003-3.1).
T-cell depletion was the only GVHD prophylaxis used.
Patients with MDS/AML could receive azacitidine at day 42 after HSCT, an option intended to prevent relapse in these patients.
Toxicity
There were no cases of acute or chronic GVHD.
Toxicities included infections (n=24), oral mucositis (n=14), hyperbilirubinemia (n=2), pulmonary hemorrhage (n=1), and sinusoidal obstruction syndrome (n=1).
There were 20 viral infections, 4 bacterial infections, and no fungal infections. Viral infections included BK virus (n=7), cytomegalovirus (n=6), Epstein-Barr virus (n=6), and adenovirus (n=1).
Dr Mehta noted that viral infections are “not unexpected in a T-cell-depleted graft setting.”
“Because we know this complication [can occur], and we worry about our patients, one of the things that, in recent years, we have done is, we manufacture viral-specific CTLs [cytotoxic T lymphocytes] for all of these patients ahead of time whenever possible,” she said.
“To give you an example, 19 out of these 20 patients’ viral infections—or rather, viremias—are completely under control with the use of either antivirals or donor-specific CTLs, including a third-party CTL in one of the patients.”
Response and survival
All 22 patients engrafted. The median time to neutrophil engraftment was 9 days (range, 8-10), and the median time to platelet engraftment was 16 days (range, 11-40).
Twenty-one of the 22 patients (95%) were alive and disease-free at last follow-up. The median follow-up was 21 months (range, 6-44).
The single AML patient achieved remission but died of post-transplant lymphoproliferative disorder (PTLD) on day 202 after HSCT. Dr Mehta said this was due to partial loss of follow-up and noncompliance with medical recommendations during PTLD treatment.
The AML patient also had “significant upfront toxicity” but “recovered very nicely,” according to Dr Mehta. He had severe mucositis, herpetic stomatitis, and sinusoidal obstruction syndrome that responded to defibrotide.
“Overall, we are quite excited to see 95% overall survival for this cohort and conclude that the current risk-adjusted approach leads to excellent overall survival and disease-free survival in patients undergoing alternative donor transplant either for marrow failure or MDS/AML,” Dr Mehta said.
“Enrollment is ongoing, and we hope to see continued success in patients with MDS/AML as well as in adult patients.”
*Data in the abstract differ from the presentation.
**Mehta PA et al. Radiation-free, alternative donor HCT for Fanconi anemia patients: results from a prospective multi-institutional study. Blood 2017; doi: https://doi.org/10.1182/blood-2016-09-743112.
SALT LAKE CITY—A “risk-adjusted” approach leads to “excellent” survival in patients with Fanconi anemia (FA) undergoing alternative donor hematopoietic stem cell transplant (HSCT), according to a speaker at the 2018 BMT Tandem Meetings.
All FA patients who received personalized doses of busulfan in place of total body irradiation (TBI) were alive and disease-free after undergoing HSCT for bone marrow failure or myelodysplastic syndrome (MDS).
None of the patients developed graft-vs-host disease (GVHD), and the most common toxicity was viral infection.
Parinda A. Mehta, MD, of Cincinnati Children’s Hospital Medical Center in Ohio, presented these results at this year’s BMT Tandem Meetings as abstract 109.*
“We all know that inherent chemotherapy and radiation sensitivity makes transplant for patients with Fanconi anemia quite challenging,” Dr Mehta began. “In our recently published, prospective, multi-institutional study, we showed excellent outcomes of alternative donor transplant in patients with Fanconi anemia without using radiation.”
“In that study,** TBI was replaced by pharmacokinetically adjusted busulfan. It proved that, yes, we can do alternative donor transplant successfully without radiation by showing an overall survival of 80% for a total of 45 patients. We were quite ecstatic to see these numbers.”
The study also showed that younger patients fared better with this regimen, and younger patients did best with the lowest dose of busulfan tested (0.6 mg/kg vs 0.8 to 1.0 mg/kg). In addition, patients who underwent HSCT for bone marrow failure had better outcomes than patients who had MDS.
This led Dr Mehta and her colleagues to hypothesize that adjusting busulfan dosing based on a patient’s age and disease status at HSCT could minimize toxicity and improve outcomes.
Patients
The researchers tested their theory in 22 FA patients. They had a median age of 7 (range, 4-27), and most (n=13) were female.
Twelve patients had pancytopenia, 6 had severe single-lineage cytopenia, 3 had low-grade MDS, and 1 patient had acute myeloid leukemia (AML).
Eighteen patients had a history of transfusions, and 3 had a history of androgen use.
Treatment
The preparative regimen consisted of 4 doses of busulfan (every 12 hours on day -7 to -6), followed by cyclophosphamide at 10 mg/kg/day (on day -5 to -2), fludarabine at 35 mg/m2/day (on day -5 to -2), and rabbit antithymocyte globulin at 2.5 mg/kg/day (on day -5 to -2).
Busulfan doses were adjusted according to age and disease status.
Children (age 18 and younger) with bone marrow failure received busulfan at 0.6 to 0.8 mg/kg. Children with MDS/AML received busulfan at 0.8 to 1.0 mg/kg. Adults (19 and older) received the lowest dose of busulfan—0.4 mg/kg—regardless of disease status.
“At the first sight, this will look counterintuitive . . . ,” Dr Mehta said. “However, based on our previous experience, in general and also from results of our previous study, this was specifically designed to avoid upfront TRM [transplant-related mortality] for these adult patients.”
All 22 patients received CD34-selected, T-cell-depleted peripheral blood stem cells from unrelated donors. Eleven patients received a fully matched graft (10/10), 8 patients had a 9/10 match, and 3 had an 8/10 match.
The median number of CD34+ cells/kg was 23.9 x 106 (range, 4.9-76.6), and the median number of CD3 cells/kg was 1 x 104 (range, 0.003-3.1).
T-cell depletion was the only GVHD prophylaxis used.
Patients with MDS/AML could receive azacitidine at day 42 after HSCT, an option intended to prevent relapse in these patients.
Toxicity
There were no cases of acute or chronic GVHD.
Toxicities included infections (n=24), oral mucositis (n=14), hyperbilirubinemia (n=2), pulmonary hemorrhage (n=1), and sinusoidal obstruction syndrome (n=1).
There were 20 viral infections, 4 bacterial infections, and no fungal infections. Viral infections included BK virus (n=7), cytomegalovirus (n=6), Epstein-Barr virus (n=6), and adenovirus (n=1).
Dr Mehta noted that viral infections are “not unexpected in a T-cell-depleted graft setting.”
“Because we know this complication [can occur], and we worry about our patients, one of the things that, in recent years, we have done is, we manufacture viral-specific CTLs [cytotoxic T lymphocytes] for all of these patients ahead of time whenever possible,” she said.
“To give you an example, 19 out of these 20 patients’ viral infections—or rather, viremias—are completely under control with the use of either antivirals or donor-specific CTLs, including a third-party CTL in one of the patients.”
Response and survival
All 22 patients engrafted. The median time to neutrophil engraftment was 9 days (range, 8-10), and the median time to platelet engraftment was 16 days (range, 11-40).
Twenty-one of the 22 patients (95%) were alive and disease-free at last follow-up. The median follow-up was 21 months (range, 6-44).
The single AML patient achieved remission but died of post-transplant lymphoproliferative disorder (PTLD) on day 202 after HSCT. Dr Mehta said this was due to partial loss of follow-up and noncompliance with medical recommendations during PTLD treatment.
The AML patient also had “significant upfront toxicity” but “recovered very nicely,” according to Dr Mehta. He had severe mucositis, herpetic stomatitis, and sinusoidal obstruction syndrome that responded to defibrotide.
“Overall, we are quite excited to see 95% overall survival for this cohort and conclude that the current risk-adjusted approach leads to excellent overall survival and disease-free survival in patients undergoing alternative donor transplant either for marrow failure or MDS/AML,” Dr Mehta said.
“Enrollment is ongoing, and we hope to see continued success in patients with MDS/AML as well as in adult patients.”
*Data in the abstract differ from the presentation.
**Mehta PA et al. Radiation-free, alternative donor HCT for Fanconi anemia patients: results from a prospective multi-institutional study. Blood 2017; doi: https://doi.org/10.1182/blood-2016-09-743112.
CHMP endorses drug delivery system
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended a label variation for Neulasta® (pegfilgrastim) to include the Neulasta® Onpro® Kit.
The Neulasta Onpro Kit is an on-body injector (OBI) delivery system for Neulasta, which is approved in the European Union to reduce the duration of neutropenia and the incidence of febrile neutropenia in adults treated with cytotoxic chemotherapy for malignancy (with the exception of chronic myeloid leukemia and myelodysplastic syndromes).
The Neulasta Onpro Kit includes a specifically designed Neulasta pre-filled syringe along with a single-use OBI. The small, lightweight OBI is applied to a patient’s skin on the same day of chemotherapy.
The OBI is intended to facilitate timed delivery of the correct dose of Neulasta and eliminate the need for patients to return to a healthcare setting the day after they receive chemotherapy.
The CHMP’s opinion on the Neulasta Onpro Kit will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the centralized marketing authorization of Neulasta will be updated to include information on the Neulasta Onpro Kit in the drug’s label.
This change will be valid throughout the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions about Neulasta’s label on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended a label variation for Neulasta® (pegfilgrastim) to include the Neulasta® Onpro® Kit.
The Neulasta Onpro Kit is an on-body injector (OBI) delivery system for Neulasta, which is approved in the European Union to reduce the duration of neutropenia and the incidence of febrile neutropenia in adults treated with cytotoxic chemotherapy for malignancy (with the exception of chronic myeloid leukemia and myelodysplastic syndromes).
The Neulasta Onpro Kit includes a specifically designed Neulasta pre-filled syringe along with a single-use OBI. The small, lightweight OBI is applied to a patient’s skin on the same day of chemotherapy.
The OBI is intended to facilitate timed delivery of the correct dose of Neulasta and eliminate the need for patients to return to a healthcare setting the day after they receive chemotherapy.
The CHMP’s opinion on the Neulasta Onpro Kit will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the centralized marketing authorization of Neulasta will be updated to include information on the Neulasta Onpro Kit in the drug’s label.
This change will be valid throughout the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions about Neulasta’s label on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended a label variation for Neulasta® (pegfilgrastim) to include the Neulasta® Onpro® Kit.
The Neulasta Onpro Kit is an on-body injector (OBI) delivery system for Neulasta, which is approved in the European Union to reduce the duration of neutropenia and the incidence of febrile neutropenia in adults treated with cytotoxic chemotherapy for malignancy (with the exception of chronic myeloid leukemia and myelodysplastic syndromes).
The Neulasta Onpro Kit includes a specifically designed Neulasta pre-filled syringe along with a single-use OBI. The small, lightweight OBI is applied to a patient’s skin on the same day of chemotherapy.
The OBI is intended to facilitate timed delivery of the correct dose of Neulasta and eliminate the need for patients to return to a healthcare setting the day after they receive chemotherapy.
The CHMP’s opinion on the Neulasta Onpro Kit will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the centralized marketing authorization of Neulasta will be updated to include information on the Neulasta Onpro Kit in the drug’s label.
This change will be valid throughout the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions about Neulasta’s label on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
Expanded UCB product can stand alone

SALT LAKE CITY—The expanded umbilical cord blood (UCB) product NiCord can be used as a stand-alone graft, according to research presented at the 2018 BMT Tandem Meetings.
Researchers found that a single NiCord unit provided “robust” engraftment in a phase 1/2 study of patients with high-risk hematologic malignancies.
NiCord recipients had quicker neutrophil and platelet engraftment than matched control subjects who received standard myeloablative UCB transplant (single or double).
Mitchell Horwitz, MD, of the Duke University Medical Center in Durham, North Carolina, presented these results at the meeting as abstract 49.* The research was sponsored by Gamida Cell, the company developing NiCord.
“[NiCord] is an ex vivo expanded cell product that’s derived from an entire unit of umbilical cord blood,” Dr Horwitz explained. “It’s manufactured starting with a CD133-positive selection, which is the progenitor cell population that’s cultured, and a T-cell containing CD133-negative fraction that is provided also at the time of transplant.”
“The culture system contains nicotinamide—that’s the active ingredient in the culture. And that’s supplemented with cytokines—thrombopoietin, IL-6, FLT-3 ligand, and stem cell factor. The culture is 21 days.”
Previous research showed that double UCB transplant including a NiCord unit could provide benefits over standard double UCB transplant. This led Dr Horwitz and his colleagues to wonder if NiCord could be used as a stand-alone graft.
So the team evaluated the safety and efficacy of NiCord alone in 36 adolescents/adults with high-risk hematologic malignancies.
Patients had acute myelogenous leukemia (n=17), acute lymphoblastic leukemia (n=9), myelodysplastic syndrome (n=7), chronic myelogenous leukemia (n=2), and Hodgkin lymphoma (n=1).
Most patients had intermediate (n=15) or high-risk (n=13) disease. They had a median age of 44 (range, 13-63) and a median weight of 75 kg (range, 41-125).
Treatment
For conditioning, 19 patients received thiotepa, busulfan, and fludarabine. Fifteen patients received total body irradiation and fludarabine with or without cyclophosphamide or thiotepa. And 2 patients received clofarabine, fludarabine, and busulfan.
Most patients had a 4/6 human leukocyte antigen (HLA) match (n=26), 8 had a 5/6 HLA match, and 2 had a 6/6 HLA match.
The median total nucleated cell dose was 2.4 x 107/kg prior to expansion of the UCB unit and 3.7 x 107/kg after expansion. The median CD34+ cell dose was 0.2 x 106/kg and 6.3 x 106/kg, respectively.
“CD34 cells were expanded 33-fold in the 3-week culture system,” Dr Horwitz noted. “That translated to a median CD34 dose of 6.3 x 106/kg, a dose comparable to what would be obtained from an adult donor graft.”
Engraftment
There was 1 case of primary graft failure and 2 cases of secondary graft failure. One case of secondary graft failure was associated with an HHV-6 infection, and the other was due to a lethal adenovirus infection.
Of those patients who engrafted, 97% achieved full donor chimerism, and 3% had mixed chimerism.
Dr Horwitz and his colleagues compared engraftment results in the NiCord recipients to results in a cohort of patients from the CIBMTR registry who underwent UCB transplants from 2010 to 2013. They had similar characteristics as the NiCord patients—age, conditioning regimen, disease status, etc.
In total, there were 148 CIBMTR registry patients, 20% of whom received a single UCB unit.
The median time to neutrophil engraftment was 11.5 days (range, 6-26) with NiCord and 21 days in the CIBMTR matched control cohort (P<0.001). The cumulative incidence of neutrophil engraftment was 94.4% and 89.7%, respectively.
The median time to platelet engraftment was 34 days (range, 25-96) with NiCord and 46 days in the CIBMTR controls (P<0.001). The cumulative incidence of platelet engraftment was 80.6% and 67.1%, respectively.
“There’s a median 10-day reduction in neutrophil recovery [and] 12-day reduction in time to platelet recovery [with NiCord],” Dr Horwitz noted. “There is evidence of robust and durable engraftment with a NiCord unit, with one patient now over 7 years from his first transplant on the pilot trial.”
Relapse, survival, and GVHD
Dr Horwitz reported other outcomes in the NiCord recipients without making comparisons to the CIBMTR matched controls.
The estimated 2-year rate of non-relapse mortality in NiCord recipients was 23.8%, and the estimated 2-year incidence of relapse was 33.2%.
The estimated disease-free survival was 49.1% at 1 year and 43.0% at 2 years. The estimated overall survival was 51.2% at 1 year and 2 years.
At 100 days, the rate of grade 2-4 acute GVHD was 44.0%, and the rate of grade 3-4 acute GVHD was 11.1%.
The estimated 1-year rate of mild to severe chronic GVHD was 40.5%, and the estimated 2-year rate of moderate to severe chronic GVHD was 9.8%.
Dr Horwitz said these “promising results” have led to the launch of a phase 3 registration trial in which researchers are comparing NiCord to standard single or double UCB transplant. The trial is open for accrual.
*Information in the abstract differs from the presentation.
SALT LAKE CITY—The expanded umbilical cord blood (UCB) product NiCord can be used as a stand-alone graft, according to research presented at the 2018 BMT Tandem Meetings.
Researchers found that a single NiCord unit provided “robust” engraftment in a phase 1/2 study of patients with high-risk hematologic malignancies.
NiCord recipients had quicker neutrophil and platelet engraftment than matched control subjects who received standard myeloablative UCB transplant (single or double).
Mitchell Horwitz, MD, of the Duke University Medical Center in Durham, North Carolina, presented these results at the meeting as abstract 49.* The research was sponsored by Gamida Cell, the company developing NiCord.
“[NiCord] is an ex vivo expanded cell product that’s derived from an entire unit of umbilical cord blood,” Dr Horwitz explained. “It’s manufactured starting with a CD133-positive selection, which is the progenitor cell population that’s cultured, and a T-cell containing CD133-negative fraction that is provided also at the time of transplant.”
“The culture system contains nicotinamide—that’s the active ingredient in the culture. And that’s supplemented with cytokines—thrombopoietin, IL-6, FLT-3 ligand, and stem cell factor. The culture is 21 days.”
Previous research showed that double UCB transplant including a NiCord unit could provide benefits over standard double UCB transplant. This led Dr Horwitz and his colleagues to wonder if NiCord could be used as a stand-alone graft.
So the team evaluated the safety and efficacy of NiCord alone in 36 adolescents/adults with high-risk hematologic malignancies.
Patients had acute myelogenous leukemia (n=17), acute lymphoblastic leukemia (n=9), myelodysplastic syndrome (n=7), chronic myelogenous leukemia (n=2), and Hodgkin lymphoma (n=1).
Most patients had intermediate (n=15) or high-risk (n=13) disease. They had a median age of 44 (range, 13-63) and a median weight of 75 kg (range, 41-125).
Treatment
For conditioning, 19 patients received thiotepa, busulfan, and fludarabine. Fifteen patients received total body irradiation and fludarabine with or without cyclophosphamide or thiotepa. And 2 patients received clofarabine, fludarabine, and busulfan.
Most patients had a 4/6 human leukocyte antigen (HLA) match (n=26), 8 had a 5/6 HLA match, and 2 had a 6/6 HLA match.
The median total nucleated cell dose was 2.4 x 107/kg prior to expansion of the UCB unit and 3.7 x 107/kg after expansion. The median CD34+ cell dose was 0.2 x 106/kg and 6.3 x 106/kg, respectively.
“CD34 cells were expanded 33-fold in the 3-week culture system,” Dr Horwitz noted. “That translated to a median CD34 dose of 6.3 x 106/kg, a dose comparable to what would be obtained from an adult donor graft.”
Engraftment
There was 1 case of primary graft failure and 2 cases of secondary graft failure. One case of secondary graft failure was associated with an HHV-6 infection, and the other was due to a lethal adenovirus infection.
Of those patients who engrafted, 97% achieved full donor chimerism, and 3% had mixed chimerism.
Dr Horwitz and his colleagues compared engraftment results in the NiCord recipients to results in a cohort of patients from the CIBMTR registry who underwent UCB transplants from 2010 to 2013. They had similar characteristics as the NiCord patients—age, conditioning regimen, disease status, etc.
In total, there were 148 CIBMTR registry patients, 20% of whom received a single UCB unit.
The median time to neutrophil engraftment was 11.5 days (range, 6-26) with NiCord and 21 days in the CIBMTR matched control cohort (P<0.001). The cumulative incidence of neutrophil engraftment was 94.4% and 89.7%, respectively.
The median time to platelet engraftment was 34 days (range, 25-96) with NiCord and 46 days in the CIBMTR controls (P<0.001). The cumulative incidence of platelet engraftment was 80.6% and 67.1%, respectively.
“There’s a median 10-day reduction in neutrophil recovery [and] 12-day reduction in time to platelet recovery [with NiCord],” Dr Horwitz noted. “There is evidence of robust and durable engraftment with a NiCord unit, with one patient now over 7 years from his first transplant on the pilot trial.”
Relapse, survival, and GVHD
Dr Horwitz reported other outcomes in the NiCord recipients without making comparisons to the CIBMTR matched controls.
The estimated 2-year rate of non-relapse mortality in NiCord recipients was 23.8%, and the estimated 2-year incidence of relapse was 33.2%.
The estimated disease-free survival was 49.1% at 1 year and 43.0% at 2 years. The estimated overall survival was 51.2% at 1 year and 2 years.
At 100 days, the rate of grade 2-4 acute GVHD was 44.0%, and the rate of grade 3-4 acute GVHD was 11.1%.
The estimated 1-year rate of mild to severe chronic GVHD was 40.5%, and the estimated 2-year rate of moderate to severe chronic GVHD was 9.8%.
Dr Horwitz said these “promising results” have led to the launch of a phase 3 registration trial in which researchers are comparing NiCord to standard single or double UCB transplant. The trial is open for accrual.
*Information in the abstract differs from the presentation.
SALT LAKE CITY—The expanded umbilical cord blood (UCB) product NiCord can be used as a stand-alone graft, according to research presented at the 2018 BMT Tandem Meetings.
Researchers found that a single NiCord unit provided “robust” engraftment in a phase 1/2 study of patients with high-risk hematologic malignancies.
NiCord recipients had quicker neutrophil and platelet engraftment than matched control subjects who received standard myeloablative UCB transplant (single or double).
Mitchell Horwitz, MD, of the Duke University Medical Center in Durham, North Carolina, presented these results at the meeting as abstract 49.* The research was sponsored by Gamida Cell, the company developing NiCord.
“[NiCord] is an ex vivo expanded cell product that’s derived from an entire unit of umbilical cord blood,” Dr Horwitz explained. “It’s manufactured starting with a CD133-positive selection, which is the progenitor cell population that’s cultured, and a T-cell containing CD133-negative fraction that is provided also at the time of transplant.”
“The culture system contains nicotinamide—that’s the active ingredient in the culture. And that’s supplemented with cytokines—thrombopoietin, IL-6, FLT-3 ligand, and stem cell factor. The culture is 21 days.”
Previous research showed that double UCB transplant including a NiCord unit could provide benefits over standard double UCB transplant. This led Dr Horwitz and his colleagues to wonder if NiCord could be used as a stand-alone graft.
So the team evaluated the safety and efficacy of NiCord alone in 36 adolescents/adults with high-risk hematologic malignancies.
Patients had acute myelogenous leukemia (n=17), acute lymphoblastic leukemia (n=9), myelodysplastic syndrome (n=7), chronic myelogenous leukemia (n=2), and Hodgkin lymphoma (n=1).
Most patients had intermediate (n=15) or high-risk (n=13) disease. They had a median age of 44 (range, 13-63) and a median weight of 75 kg (range, 41-125).
Treatment
For conditioning, 19 patients received thiotepa, busulfan, and fludarabine. Fifteen patients received total body irradiation and fludarabine with or without cyclophosphamide or thiotepa. And 2 patients received clofarabine, fludarabine, and busulfan.
Most patients had a 4/6 human leukocyte antigen (HLA) match (n=26), 8 had a 5/6 HLA match, and 2 had a 6/6 HLA match.
The median total nucleated cell dose was 2.4 x 107/kg prior to expansion of the UCB unit and 3.7 x 107/kg after expansion. The median CD34+ cell dose was 0.2 x 106/kg and 6.3 x 106/kg, respectively.
“CD34 cells were expanded 33-fold in the 3-week culture system,” Dr Horwitz noted. “That translated to a median CD34 dose of 6.3 x 106/kg, a dose comparable to what would be obtained from an adult donor graft.”
Engraftment
There was 1 case of primary graft failure and 2 cases of secondary graft failure. One case of secondary graft failure was associated with an HHV-6 infection, and the other was due to a lethal adenovirus infection.
Of those patients who engrafted, 97% achieved full donor chimerism, and 3% had mixed chimerism.
Dr Horwitz and his colleagues compared engraftment results in the NiCord recipients to results in a cohort of patients from the CIBMTR registry who underwent UCB transplants from 2010 to 2013. They had similar characteristics as the NiCord patients—age, conditioning regimen, disease status, etc.
In total, there were 148 CIBMTR registry patients, 20% of whom received a single UCB unit.
The median time to neutrophil engraftment was 11.5 days (range, 6-26) with NiCord and 21 days in the CIBMTR matched control cohort (P<0.001). The cumulative incidence of neutrophil engraftment was 94.4% and 89.7%, respectively.
The median time to platelet engraftment was 34 days (range, 25-96) with NiCord and 46 days in the CIBMTR controls (P<0.001). The cumulative incidence of platelet engraftment was 80.6% and 67.1%, respectively.
“There’s a median 10-day reduction in neutrophil recovery [and] 12-day reduction in time to platelet recovery [with NiCord],” Dr Horwitz noted. “There is evidence of robust and durable engraftment with a NiCord unit, with one patient now over 7 years from his first transplant on the pilot trial.”
Relapse, survival, and GVHD
Dr Horwitz reported other outcomes in the NiCord recipients without making comparisons to the CIBMTR matched controls.
The estimated 2-year rate of non-relapse mortality in NiCord recipients was 23.8%, and the estimated 2-year incidence of relapse was 33.2%.
The estimated disease-free survival was 49.1% at 1 year and 43.0% at 2 years. The estimated overall survival was 51.2% at 1 year and 2 years.
At 100 days, the rate of grade 2-4 acute GVHD was 44.0%, and the rate of grade 3-4 acute GVHD was 11.1%.
The estimated 1-year rate of mild to severe chronic GVHD was 40.5%, and the estimated 2-year rate of moderate to severe chronic GVHD was 9.8%.
Dr Horwitz said these “promising results” have led to the launch of a phase 3 registration trial in which researchers are comparing NiCord to standard single or double UCB transplant. The trial is open for accrual.
*Information in the abstract differs from the presentation.
Minihaplo-BMT can cure severe hemoglobinopathies

SALT LAKE CITY—Cure of severe hemoglobinopathies is now possible for most patients, according to a speaker at the 2018 BMT Tandem Meetings.
Results of a small study suggest that nonmyeloablative haploidentical bone marrow transplant (minihaplo-BMT), when used with 400 cGy total body irradiation and post-transplant cyclophosphamide, can cure sickle cell disease (SCD) and beta-thalassemia while conferring limited toxicity.
“This is our second study showing that you can certainly get rid of these conditions using haploidentical donors and, also, that you can cure these conditions with a nonmyeloablative regimen,” said Javier Bolaños-Meade, MD, of Johns Hopkins University in Baltimore, Maryland.
“What we saw [in the current study] is that increasing the total body irradiation from 200 cGy to 400 cGy can overcome the very high rate of graft failure we observed in our prior study.”
However, the results also suggest that major ABO incompatible donors should be avoided.
Dr Bolaños-Meade presented these findings as a late-breaking abstract (LBA3) at this year’s BMT Tandem Meetings.
He began the presentation by pointing out that allogeneic BMT is the only potential cure for SCD and beta-thalassemia. Unfortunately, most of these patients have been excluded from transplant due to a lack of matched donors, concerns over graft-vs-host disease (GVHD), and/or compromised end organ function precluding them from myeloablative conditioning.
With a prior study, Dr Bolaños-Meade and his colleagues found that minihaplo-BMT with post-transplant cyclophosphamide expanded the donor pool and was well tolerated in patients with SCD. However, the rate of secondary graft failure was high, at 43%.
“We hypothesized that increasing the amount of radiation that was given to these patients, making the regimen absolutely more intensive, will help us to overcome this rate of graft failure and retain the tolerability of the regimen,” Dr Bolaños-Meade said.
He and his colleagues tested this theory in 12 patients with SCD and 5 with beta-thalassemia.
The SCD patients had a median age of 26 (range, 6-31), and most (n=8) were female. Ten were African-American, and 2 were of Arab descent.
All SCD patients had at least 2 hospital admissions in the last 2 years, 5 had osteonecrosis, 3 were transfusion-dependent, 2 had a history of acute chest syndrome, and 1 patient had changes in brain magnetic resonance imaging.
The thalassemia patients had a median age of 7 (range, 6-16). Four were female, and all 5 were of Arab descent. All patients were transfusion-dependent from infancy.
Treatment
All patients received antithymocyte globulin (rabbit) at 0.5 mg/kg on day -9 and 2 mg/kg on days -8 and -7, fludarabine at 30 mg/m2 on days -6 to -2, cyclophosphamide at 14.5 mg/kg on days -6 and -5, and total body irradiation at 400 cGy on day -1.
Patients received unmanipulated bone marrow that was collected and infused on day 0. Donors included 1 aunt, 4 brothers, 3 sisters, 5 mothers, and 4 fathers. There were 2 major ABO incompatible pairs, 5 minor ABO incompatible pairs, and 10 ABO compatible pairs.
For GVHD prophylaxis, patients received 50 mg/kg/day of cyclophosphamide on days 3 and 4, sirolimus (levels of 5-10 ng/dl) from days 5 to 365, and mycophenolate mofetil at 1 g every 8 hours from days 5 to 35.
Efficacy
All 17 patients are still alive at a median follow-up of 15 months (range, 3-34).
“Of all the patients transplanted, we only observed 1 graft failure,” Dr Bolaños-Meade said. “This is in sharp contrast to the 40% graft failure in our prior study.”
Thirteen patients achieved full donor chimerism, and 3 had mixed chimerism.
Four of the 5 beta-thalassemia patients achieved full donor chimerism, and 1 had mixed chimerism. All 5 patients became transfusion-independent.
Eleven of the 12 SCD patients engrafted, and 9 achieved full donor chimerism.
All but one of the engrafted SCD patients became transfusion-independent. The exception was a mixed chimeric patient who had a major ABO mismatched donor. The patient remains transfusion-dependent more than 30 months after BMT.
“This is the main problem we have seen in all our sickle cell and hemoglobinopathy experiences,” Dr Bolaños-Meade said. “If you have a major ABO incompatibility, and there’s mixed chimerism, you’re asking for trouble.”
Toxicity
All SCD patients had sickle cell crises after antithymocyte globulin. Other toxicities included sirolimus-induced diabetes in 1 patient, worsening of Meniere’s disease in 1 patient, and BK cystitis in 1 patient.
Two patients developed grade 2 acute GVHD, and 1 developed grade 3 acute GVHD. Three patients had chronic GVHD, 2 mild and 1 moderate.
However, all cases of GVHD resolved. And, of the 8 engrafted patients with more than 1 year of follow-up, 6 are off systemic immunosuppression.
SALT LAKE CITY—Cure of severe hemoglobinopathies is now possible for most patients, according to a speaker at the 2018 BMT Tandem Meetings.
Results of a small study suggest that nonmyeloablative haploidentical bone marrow transplant (minihaplo-BMT), when used with 400 cGy total body irradiation and post-transplant cyclophosphamide, can cure sickle cell disease (SCD) and beta-thalassemia while conferring limited toxicity.
“This is our second study showing that you can certainly get rid of these conditions using haploidentical donors and, also, that you can cure these conditions with a nonmyeloablative regimen,” said Javier Bolaños-Meade, MD, of Johns Hopkins University in Baltimore, Maryland.
“What we saw [in the current study] is that increasing the total body irradiation from 200 cGy to 400 cGy can overcome the very high rate of graft failure we observed in our prior study.”
However, the results also suggest that major ABO incompatible donors should be avoided.
Dr Bolaños-Meade presented these findings as a late-breaking abstract (LBA3) at this year’s BMT Tandem Meetings.
He began the presentation by pointing out that allogeneic BMT is the only potential cure for SCD and beta-thalassemia. Unfortunately, most of these patients have been excluded from transplant due to a lack of matched donors, concerns over graft-vs-host disease (GVHD), and/or compromised end organ function precluding them from myeloablative conditioning.
With a prior study, Dr Bolaños-Meade and his colleagues found that minihaplo-BMT with post-transplant cyclophosphamide expanded the donor pool and was well tolerated in patients with SCD. However, the rate of secondary graft failure was high, at 43%.
“We hypothesized that increasing the amount of radiation that was given to these patients, making the regimen absolutely more intensive, will help us to overcome this rate of graft failure and retain the tolerability of the regimen,” Dr Bolaños-Meade said.
He and his colleagues tested this theory in 12 patients with SCD and 5 with beta-thalassemia.
The SCD patients had a median age of 26 (range, 6-31), and most (n=8) were female. Ten were African-American, and 2 were of Arab descent.
All SCD patients had at least 2 hospital admissions in the last 2 years, 5 had osteonecrosis, 3 were transfusion-dependent, 2 had a history of acute chest syndrome, and 1 patient had changes in brain magnetic resonance imaging.
The thalassemia patients had a median age of 7 (range, 6-16). Four were female, and all 5 were of Arab descent. All patients were transfusion-dependent from infancy.
Treatment
All patients received antithymocyte globulin (rabbit) at 0.5 mg/kg on day -9 and 2 mg/kg on days -8 and -7, fludarabine at 30 mg/m2 on days -6 to -2, cyclophosphamide at 14.5 mg/kg on days -6 and -5, and total body irradiation at 400 cGy on day -1.
Patients received unmanipulated bone marrow that was collected and infused on day 0. Donors included 1 aunt, 4 brothers, 3 sisters, 5 mothers, and 4 fathers. There were 2 major ABO incompatible pairs, 5 minor ABO incompatible pairs, and 10 ABO compatible pairs.
For GVHD prophylaxis, patients received 50 mg/kg/day of cyclophosphamide on days 3 and 4, sirolimus (levels of 5-10 ng/dl) from days 5 to 365, and mycophenolate mofetil at 1 g every 8 hours from days 5 to 35.
Efficacy
All 17 patients are still alive at a median follow-up of 15 months (range, 3-34).
“Of all the patients transplanted, we only observed 1 graft failure,” Dr Bolaños-Meade said. “This is in sharp contrast to the 40% graft failure in our prior study.”
Thirteen patients achieved full donor chimerism, and 3 had mixed chimerism.
Four of the 5 beta-thalassemia patients achieved full donor chimerism, and 1 had mixed chimerism. All 5 patients became transfusion-independent.
Eleven of the 12 SCD patients engrafted, and 9 achieved full donor chimerism.
All but one of the engrafted SCD patients became transfusion-independent. The exception was a mixed chimeric patient who had a major ABO mismatched donor. The patient remains transfusion-dependent more than 30 months after BMT.
“This is the main problem we have seen in all our sickle cell and hemoglobinopathy experiences,” Dr Bolaños-Meade said. “If you have a major ABO incompatibility, and there’s mixed chimerism, you’re asking for trouble.”
Toxicity
All SCD patients had sickle cell crises after antithymocyte globulin. Other toxicities included sirolimus-induced diabetes in 1 patient, worsening of Meniere’s disease in 1 patient, and BK cystitis in 1 patient.
Two patients developed grade 2 acute GVHD, and 1 developed grade 3 acute GVHD. Three patients had chronic GVHD, 2 mild and 1 moderate.
However, all cases of GVHD resolved. And, of the 8 engrafted patients with more than 1 year of follow-up, 6 are off systemic immunosuppression.
SALT LAKE CITY—Cure of severe hemoglobinopathies is now possible for most patients, according to a speaker at the 2018 BMT Tandem Meetings.
Results of a small study suggest that nonmyeloablative haploidentical bone marrow transplant (minihaplo-BMT), when used with 400 cGy total body irradiation and post-transplant cyclophosphamide, can cure sickle cell disease (SCD) and beta-thalassemia while conferring limited toxicity.
“This is our second study showing that you can certainly get rid of these conditions using haploidentical donors and, also, that you can cure these conditions with a nonmyeloablative regimen,” said Javier Bolaños-Meade, MD, of Johns Hopkins University in Baltimore, Maryland.
“What we saw [in the current study] is that increasing the total body irradiation from 200 cGy to 400 cGy can overcome the very high rate of graft failure we observed in our prior study.”
However, the results also suggest that major ABO incompatible donors should be avoided.
Dr Bolaños-Meade presented these findings as a late-breaking abstract (LBA3) at this year’s BMT Tandem Meetings.
He began the presentation by pointing out that allogeneic BMT is the only potential cure for SCD and beta-thalassemia. Unfortunately, most of these patients have been excluded from transplant due to a lack of matched donors, concerns over graft-vs-host disease (GVHD), and/or compromised end organ function precluding them from myeloablative conditioning.
With a prior study, Dr Bolaños-Meade and his colleagues found that minihaplo-BMT with post-transplant cyclophosphamide expanded the donor pool and was well tolerated in patients with SCD. However, the rate of secondary graft failure was high, at 43%.
“We hypothesized that increasing the amount of radiation that was given to these patients, making the regimen absolutely more intensive, will help us to overcome this rate of graft failure and retain the tolerability of the regimen,” Dr Bolaños-Meade said.
He and his colleagues tested this theory in 12 patients with SCD and 5 with beta-thalassemia.
The SCD patients had a median age of 26 (range, 6-31), and most (n=8) were female. Ten were African-American, and 2 were of Arab descent.
All SCD patients had at least 2 hospital admissions in the last 2 years, 5 had osteonecrosis, 3 were transfusion-dependent, 2 had a history of acute chest syndrome, and 1 patient had changes in brain magnetic resonance imaging.
The thalassemia patients had a median age of 7 (range, 6-16). Four were female, and all 5 were of Arab descent. All patients were transfusion-dependent from infancy.
Treatment
All patients received antithymocyte globulin (rabbit) at 0.5 mg/kg on day -9 and 2 mg/kg on days -8 and -7, fludarabine at 30 mg/m2 on days -6 to -2, cyclophosphamide at 14.5 mg/kg on days -6 and -5, and total body irradiation at 400 cGy on day -1.
Patients received unmanipulated bone marrow that was collected and infused on day 0. Donors included 1 aunt, 4 brothers, 3 sisters, 5 mothers, and 4 fathers. There were 2 major ABO incompatible pairs, 5 minor ABO incompatible pairs, and 10 ABO compatible pairs.
For GVHD prophylaxis, patients received 50 mg/kg/day of cyclophosphamide on days 3 and 4, sirolimus (levels of 5-10 ng/dl) from days 5 to 365, and mycophenolate mofetil at 1 g every 8 hours from days 5 to 35.
Efficacy
All 17 patients are still alive at a median follow-up of 15 months (range, 3-34).
“Of all the patients transplanted, we only observed 1 graft failure,” Dr Bolaños-Meade said. “This is in sharp contrast to the 40% graft failure in our prior study.”
Thirteen patients achieved full donor chimerism, and 3 had mixed chimerism.
Four of the 5 beta-thalassemia patients achieved full donor chimerism, and 1 had mixed chimerism. All 5 patients became transfusion-independent.
Eleven of the 12 SCD patients engrafted, and 9 achieved full donor chimerism.
All but one of the engrafted SCD patients became transfusion-independent. The exception was a mixed chimeric patient who had a major ABO mismatched donor. The patient remains transfusion-dependent more than 30 months after BMT.
“This is the main problem we have seen in all our sickle cell and hemoglobinopathy experiences,” Dr Bolaños-Meade said. “If you have a major ABO incompatibility, and there’s mixed chimerism, you’re asking for trouble.”
Toxicity
All SCD patients had sickle cell crises after antithymocyte globulin. Other toxicities included sirolimus-induced diabetes in 1 patient, worsening of Meniere’s disease in 1 patient, and BK cystitis in 1 patient.
Two patients developed grade 2 acute GVHD, and 1 developed grade 3 acute GVHD. Three patients had chronic GVHD, 2 mild and 1 moderate.
However, all cases of GVHD resolved. And, of the 8 engrafted patients with more than 1 year of follow-up, 6 are off systemic immunosuppression.
NK-cell therapy in resistant MDS, AML

Results of a phase 1/2 trial suggest treatment with haploidentical natural killer (NK) cells can be effective against relapsed/refractory myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML).
NK-cell therapy elicited responses in 6 of the 16 patients studied and provided a bridge to transplant for 5 patients.
Three responders were still alive at more than 3 years of follow-up.
There were 4 grade 3 adverse events (AEs) and 2 grade 5 AEs considered possibly or probably related to NK-cell therapy.
Investigators reported these results in Clinical Cancer Research.
The trial enrolled 16 patients. Eight had MDS/AML, 3 had de novo AML, and 5 had high-risk MDS, including refractory anemia with excess blasts (RAEB) type 1 progressing toward type 2, RAEB-2, and chronic myelomonocytic leukemia type 2.
The patients’ median age was 64 (range, 40-70), and they had received a median of 3 prior therapies (range, 1-6). Six patients had received an allogeneic hematopoietic stem cell transplant (HSCT).
For this study, all patients received fludarabine, cyclophosphamide, and total lymphoid irradiation prior to receiving haploidentical NK cells.
The median follow-up was 8 months for all patients and 28 months for responders.
Efficacy
Six patients responded to treatment. One patient with de novo AML had a complete response (CR). Two high-risk MDS patients had a marrow CR (mCR), as did 2 MDS/AML patients. One MDS/AML patient had a partial response (PR).
Two patients had stable disease (SD)—1 with MDS and 1 with MDS/AML. One patient with de novo AML had a morphologic leukemia-free state after NK-cell therapy.
Five patients proceeded to HSCT—3 in mCR, 1 in PR, and 1 with SD.
Three patients were still alive at last follow-up—1 with MDS who achieved an mCR and went on to HSCT, 1 with MDS/AML who achieved an mCR and went on to HSCT, and 1 with MDS/AML who achieved an mCR and went on to receive chemotherapy and donor lymphocyte infusion.
One survivor has more than 5 years of follow-up (the MDS patient), and the other 2 have more than 3 years of follow-up.
“Our study shows that patients with MDS, AML, and MDS/AML can be treated with NK cell-based immunotherapy and that the therapy can be highly efficacious,” said study author Hans-Gustaf Ljunggren, MD, PhD, of Karolinska Institutet in Stockholm, Sweden.
Safety
The most common AEs of any grade considered possibly or probably related to NK-cell therapy were chills (n=13) and nausea (n=4).
Two patients had cytokine release syndrome (CRS) likely associated with hemophagocytic lymphohistiocytosis (HLH).
Each of the following potentially related AEs were reported once: headache, vomiting, encephalitis infection, sinus tachycardia, bone pain, pain in extremity, and maculopapular rash.
There were 4 grade 3 AEs—CRS/HLH (n=1), chills (n=1), and nausea (n=2)—but no grade 4 AEs.
There were 2 grade 5 AEs—CRS/HLH and encephalitis infection. These occurred in a single patient who died with HLH, human herpes virus-6 encephalitis, and AML relapse.
Two investigators involved in this study serve on the scientific advisory board of Fate Therapeutics. Dr Ljunggren serves on the scientific advisory board of CellProtect, Nordic Pharmaceuticals, and HOPE Bio-Sciences. He is also on the board of directors of Vycellix and is a collaborator with Fate Therapeutics.
Results of a phase 1/2 trial suggest treatment with haploidentical natural killer (NK) cells can be effective against relapsed/refractory myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML).
NK-cell therapy elicited responses in 6 of the 16 patients studied and provided a bridge to transplant for 5 patients.
Three responders were still alive at more than 3 years of follow-up.
There were 4 grade 3 adverse events (AEs) and 2 grade 5 AEs considered possibly or probably related to NK-cell therapy.
Investigators reported these results in Clinical Cancer Research.
The trial enrolled 16 patients. Eight had MDS/AML, 3 had de novo AML, and 5 had high-risk MDS, including refractory anemia with excess blasts (RAEB) type 1 progressing toward type 2, RAEB-2, and chronic myelomonocytic leukemia type 2.
The patients’ median age was 64 (range, 40-70), and they had received a median of 3 prior therapies (range, 1-6). Six patients had received an allogeneic hematopoietic stem cell transplant (HSCT).
For this study, all patients received fludarabine, cyclophosphamide, and total lymphoid irradiation prior to receiving haploidentical NK cells.
The median follow-up was 8 months for all patients and 28 months for responders.
Efficacy
Six patients responded to treatment. One patient with de novo AML had a complete response (CR). Two high-risk MDS patients had a marrow CR (mCR), as did 2 MDS/AML patients. One MDS/AML patient had a partial response (PR).
Two patients had stable disease (SD)—1 with MDS and 1 with MDS/AML. One patient with de novo AML had a morphologic leukemia-free state after NK-cell therapy.
Five patients proceeded to HSCT—3 in mCR, 1 in PR, and 1 with SD.
Three patients were still alive at last follow-up—1 with MDS who achieved an mCR and went on to HSCT, 1 with MDS/AML who achieved an mCR and went on to HSCT, and 1 with MDS/AML who achieved an mCR and went on to receive chemotherapy and donor lymphocyte infusion.
One survivor has more than 5 years of follow-up (the MDS patient), and the other 2 have more than 3 years of follow-up.
“Our study shows that patients with MDS, AML, and MDS/AML can be treated with NK cell-based immunotherapy and that the therapy can be highly efficacious,” said study author Hans-Gustaf Ljunggren, MD, PhD, of Karolinska Institutet in Stockholm, Sweden.
Safety
The most common AEs of any grade considered possibly or probably related to NK-cell therapy were chills (n=13) and nausea (n=4).
Two patients had cytokine release syndrome (CRS) likely associated with hemophagocytic lymphohistiocytosis (HLH).
Each of the following potentially related AEs were reported once: headache, vomiting, encephalitis infection, sinus tachycardia, bone pain, pain in extremity, and maculopapular rash.
There were 4 grade 3 AEs—CRS/HLH (n=1), chills (n=1), and nausea (n=2)—but no grade 4 AEs.
There were 2 grade 5 AEs—CRS/HLH and encephalitis infection. These occurred in a single patient who died with HLH, human herpes virus-6 encephalitis, and AML relapse.
Two investigators involved in this study serve on the scientific advisory board of Fate Therapeutics. Dr Ljunggren serves on the scientific advisory board of CellProtect, Nordic Pharmaceuticals, and HOPE Bio-Sciences. He is also on the board of directors of Vycellix and is a collaborator with Fate Therapeutics.
Results of a phase 1/2 trial suggest treatment with haploidentical natural killer (NK) cells can be effective against relapsed/refractory myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML).
NK-cell therapy elicited responses in 6 of the 16 patients studied and provided a bridge to transplant for 5 patients.
Three responders were still alive at more than 3 years of follow-up.
There were 4 grade 3 adverse events (AEs) and 2 grade 5 AEs considered possibly or probably related to NK-cell therapy.
Investigators reported these results in Clinical Cancer Research.
The trial enrolled 16 patients. Eight had MDS/AML, 3 had de novo AML, and 5 had high-risk MDS, including refractory anemia with excess blasts (RAEB) type 1 progressing toward type 2, RAEB-2, and chronic myelomonocytic leukemia type 2.
The patients’ median age was 64 (range, 40-70), and they had received a median of 3 prior therapies (range, 1-6). Six patients had received an allogeneic hematopoietic stem cell transplant (HSCT).
For this study, all patients received fludarabine, cyclophosphamide, and total lymphoid irradiation prior to receiving haploidentical NK cells.
The median follow-up was 8 months for all patients and 28 months for responders.
Efficacy
Six patients responded to treatment. One patient with de novo AML had a complete response (CR). Two high-risk MDS patients had a marrow CR (mCR), as did 2 MDS/AML patients. One MDS/AML patient had a partial response (PR).
Two patients had stable disease (SD)—1 with MDS and 1 with MDS/AML. One patient with de novo AML had a morphologic leukemia-free state after NK-cell therapy.
Five patients proceeded to HSCT—3 in mCR, 1 in PR, and 1 with SD.
Three patients were still alive at last follow-up—1 with MDS who achieved an mCR and went on to HSCT, 1 with MDS/AML who achieved an mCR and went on to HSCT, and 1 with MDS/AML who achieved an mCR and went on to receive chemotherapy and donor lymphocyte infusion.
One survivor has more than 5 years of follow-up (the MDS patient), and the other 2 have more than 3 years of follow-up.
“Our study shows that patients with MDS, AML, and MDS/AML can be treated with NK cell-based immunotherapy and that the therapy can be highly efficacious,” said study author Hans-Gustaf Ljunggren, MD, PhD, of Karolinska Institutet in Stockholm, Sweden.
Safety
The most common AEs of any grade considered possibly or probably related to NK-cell therapy were chills (n=13) and nausea (n=4).
Two patients had cytokine release syndrome (CRS) likely associated with hemophagocytic lymphohistiocytosis (HLH).
Each of the following potentially related AEs were reported once: headache, vomiting, encephalitis infection, sinus tachycardia, bone pain, pain in extremity, and maculopapular rash.
There were 4 grade 3 AEs—CRS/HLH (n=1), chills (n=1), and nausea (n=2)—but no grade 4 AEs.
There were 2 grade 5 AEs—CRS/HLH and encephalitis infection. These occurred in a single patient who died with HLH, human herpes virus-6 encephalitis, and AML relapse.
Two investigators involved in this study serve on the scientific advisory board of Fate Therapeutics. Dr Ljunggren serves on the scientific advisory board of CellProtect, Nordic Pharmaceuticals, and HOPE Bio-Sciences. He is also on the board of directors of Vycellix and is a collaborator with Fate Therapeutics.
Young kids with SCA not receiving recommended prophylaxis

Many young children with sickle cell anemia (SCA) may not be taking the recommended antibiotics to prevent invasive pneumococcal disease (IPD), according to research published in Pediatrics.
Results of a previous study indicated that daily treatment with penicillin could reduce the risk of IPD by 84% in young children with SCA.
In the current study, only 18% of young SCA patients received daily penicillin or an equivalent antibiotic as IPD prophylaxis.
“Most children with sickle cell anemia are not getting the antibiotics they should be to adequately protect against potentially deadly infections,” said study author Sarah Reeves, PhD, of the University of Michigan Medical School in Ann Arbor.
“Long-standing recommendations say children with sickle cell anemia should take antibiotics daily for their first 5 years of life. It can be life-saving.”
For this study, Dr Reeves and her colleagues analyzed data on 2821 SCA patients, ages 3 months to 5 years, living in Florida, Illinois, Louisiana, Michigan, South Carolina, and Texas.
The patients were continuously enrolled in the Medicaid program for at least 1 calendar year between 2005 and 2012. The researchers evaluated the receipt of antibiotics through the insurance claims for filled prescriptions.
The team found that, overall, 18% of patients received at least 300 days of antibiotics.
Sixteen percent of patients received at least 300 days of penicillin; 16% received at least 300 days of penicillin or erythromycin; 18% received at least 300 days of penicillin, erythromycin, or amoxicillin; and 22% received at least 300 days of any antibiotic to prevent Streptococcus pneumoniae.
On average, patients received 162 days of penicillin; 164 days of penicillin or erythromycin; 178 days of penicillin, erythromycin, or amoxicillin; and 193 days of any antibiotic to prevent S pneumoniae.
Multivariable analysis suggested that medical visits and a patient’s state of residence were associated with receiving at least 300 days of antibiotics.
The researchers said that each additional SCA-related outpatient visit and well-child visit was associated with incrementally increased odds of receiving at least 300 days of antibiotics. The odds ratio (OR) was 1.01 for SCA-related outpatient visits and 1.08 for well-child visits (P<0.05 for both).
Patients in Florida (OR=0.51, P<0.05), Louisiana (OR=0.57, P<0.05), Michigan (OR=0.60, P<0.05), and South Carolina (OR=0.62, P<0.05) had lower odds of receiving at least 300 days of antibiotics than patients in Illinois (OR=1.00) or Texas (OR=1.01).
The researchers did not investigate why children were not receiving recommended antibiotics, but Dr Reeves identified possible barriers to compliance. She noted that caregiver challenges include picking up prescriptions every 2 weeks from a pharmacy as well as remembering to administer an antibiotic to a young, healthy-appearing child twice a day.
“The types of challenges involved in making sure children get the recommended dose of antibiotics is exacerbated by the substantial burden of care already experienced by families to help control the symptoms of this disease,” Dr Reeves said.
She added that future studies should more deeply explore barriers preventing families from getting antibiotics and potential interventions to improve the rate of children receiving recommended prescriptions.
“Interventions to improve the receipt of antibiotics among children with sickle cell anemia should include enhanced collaboration between healthcare providers, pharmacists, and families,” Dr Reeves said.
“Doctors need to repeatedly discuss the importance of taking antibiotics with families of children with sickle cell anemia. Social factors that may impact receiving filled prescriptions should also be considered, such as the availability of transportation and time to travel to pharmacies to pick up the prescriptions.” ![]()
Many young children with sickle cell anemia (SCA) may not be taking the recommended antibiotics to prevent invasive pneumococcal disease (IPD), according to research published in Pediatrics.
Results of a previous study indicated that daily treatment with penicillin could reduce the risk of IPD by 84% in young children with SCA.
In the current study, only 18% of young SCA patients received daily penicillin or an equivalent antibiotic as IPD prophylaxis.
“Most children with sickle cell anemia are not getting the antibiotics they should be to adequately protect against potentially deadly infections,” said study author Sarah Reeves, PhD, of the University of Michigan Medical School in Ann Arbor.
“Long-standing recommendations say children with sickle cell anemia should take antibiotics daily for their first 5 years of life. It can be life-saving.”
For this study, Dr Reeves and her colleagues analyzed data on 2821 SCA patients, ages 3 months to 5 years, living in Florida, Illinois, Louisiana, Michigan, South Carolina, and Texas.
The patients were continuously enrolled in the Medicaid program for at least 1 calendar year between 2005 and 2012. The researchers evaluated the receipt of antibiotics through the insurance claims for filled prescriptions.
The team found that, overall, 18% of patients received at least 300 days of antibiotics.
Sixteen percent of patients received at least 300 days of penicillin; 16% received at least 300 days of penicillin or erythromycin; 18% received at least 300 days of penicillin, erythromycin, or amoxicillin; and 22% received at least 300 days of any antibiotic to prevent Streptococcus pneumoniae.
On average, patients received 162 days of penicillin; 164 days of penicillin or erythromycin; 178 days of penicillin, erythromycin, or amoxicillin; and 193 days of any antibiotic to prevent S pneumoniae.
Multivariable analysis suggested that medical visits and a patient’s state of residence were associated with receiving at least 300 days of antibiotics.
The researchers said that each additional SCA-related outpatient visit and well-child visit was associated with incrementally increased odds of receiving at least 300 days of antibiotics. The odds ratio (OR) was 1.01 for SCA-related outpatient visits and 1.08 for well-child visits (P<0.05 for both).
Patients in Florida (OR=0.51, P<0.05), Louisiana (OR=0.57, P<0.05), Michigan (OR=0.60, P<0.05), and South Carolina (OR=0.62, P<0.05) had lower odds of receiving at least 300 days of antibiotics than patients in Illinois (OR=1.00) or Texas (OR=1.01).
The researchers did not investigate why children were not receiving recommended antibiotics, but Dr Reeves identified possible barriers to compliance. She noted that caregiver challenges include picking up prescriptions every 2 weeks from a pharmacy as well as remembering to administer an antibiotic to a young, healthy-appearing child twice a day.
“The types of challenges involved in making sure children get the recommended dose of antibiotics is exacerbated by the substantial burden of care already experienced by families to help control the symptoms of this disease,” Dr Reeves said.
She added that future studies should more deeply explore barriers preventing families from getting antibiotics and potential interventions to improve the rate of children receiving recommended prescriptions.
“Interventions to improve the receipt of antibiotics among children with sickle cell anemia should include enhanced collaboration between healthcare providers, pharmacists, and families,” Dr Reeves said.
“Doctors need to repeatedly discuss the importance of taking antibiotics with families of children with sickle cell anemia. Social factors that may impact receiving filled prescriptions should also be considered, such as the availability of transportation and time to travel to pharmacies to pick up the prescriptions.” ![]()
Many young children with sickle cell anemia (SCA) may not be taking the recommended antibiotics to prevent invasive pneumococcal disease (IPD), according to research published in Pediatrics.
Results of a previous study indicated that daily treatment with penicillin could reduce the risk of IPD by 84% in young children with SCA.
In the current study, only 18% of young SCA patients received daily penicillin or an equivalent antibiotic as IPD prophylaxis.
“Most children with sickle cell anemia are not getting the antibiotics they should be to adequately protect against potentially deadly infections,” said study author Sarah Reeves, PhD, of the University of Michigan Medical School in Ann Arbor.
“Long-standing recommendations say children with sickle cell anemia should take antibiotics daily for their first 5 years of life. It can be life-saving.”
For this study, Dr Reeves and her colleagues analyzed data on 2821 SCA patients, ages 3 months to 5 years, living in Florida, Illinois, Louisiana, Michigan, South Carolina, and Texas.
The patients were continuously enrolled in the Medicaid program for at least 1 calendar year between 2005 and 2012. The researchers evaluated the receipt of antibiotics through the insurance claims for filled prescriptions.
The team found that, overall, 18% of patients received at least 300 days of antibiotics.
Sixteen percent of patients received at least 300 days of penicillin; 16% received at least 300 days of penicillin or erythromycin; 18% received at least 300 days of penicillin, erythromycin, or amoxicillin; and 22% received at least 300 days of any antibiotic to prevent Streptococcus pneumoniae.
On average, patients received 162 days of penicillin; 164 days of penicillin or erythromycin; 178 days of penicillin, erythromycin, or amoxicillin; and 193 days of any antibiotic to prevent S pneumoniae.
Multivariable analysis suggested that medical visits and a patient’s state of residence were associated with receiving at least 300 days of antibiotics.
The researchers said that each additional SCA-related outpatient visit and well-child visit was associated with incrementally increased odds of receiving at least 300 days of antibiotics. The odds ratio (OR) was 1.01 for SCA-related outpatient visits and 1.08 for well-child visits (P<0.05 for both).
Patients in Florida (OR=0.51, P<0.05), Louisiana (OR=0.57, P<0.05), Michigan (OR=0.60, P<0.05), and South Carolina (OR=0.62, P<0.05) had lower odds of receiving at least 300 days of antibiotics than patients in Illinois (OR=1.00) or Texas (OR=1.01).
The researchers did not investigate why children were not receiving recommended antibiotics, but Dr Reeves identified possible barriers to compliance. She noted that caregiver challenges include picking up prescriptions every 2 weeks from a pharmacy as well as remembering to administer an antibiotic to a young, healthy-appearing child twice a day.
“The types of challenges involved in making sure children get the recommended dose of antibiotics is exacerbated by the substantial burden of care already experienced by families to help control the symptoms of this disease,” Dr Reeves said.
She added that future studies should more deeply explore barriers preventing families from getting antibiotics and potential interventions to improve the rate of children receiving recommended prescriptions.
“Interventions to improve the receipt of antibiotics among children with sickle cell anemia should include enhanced collaboration between healthcare providers, pharmacists, and families,” Dr Reeves said.
“Doctors need to repeatedly discuss the importance of taking antibiotics with families of children with sickle cell anemia. Social factors that may impact receiving filled prescriptions should also be considered, such as the availability of transportation and time to travel to pharmacies to pick up the prescriptions.” ![]()
Azacitidine now available in China

Azacitidine for injection (Vidaza®) is now available in China.
The nucleoside metabolic inhibitor was approved in China to treat patients with intermediate-2/high-risk myelodysplastic syndromes (MDS), acute myeloid leukemia (AML) with 20% to 30% bone marrow blasts, and chronic myelomonocytic leukemia (CMML).
Azacitidine for injection is marketed in China by BeiGene Ltd. under an exclusive license from Celgene Corporation.
“Vidaza is the only approved hypomethylating agent shown to prolong survival for patients with MDS and the first new treatment for MDS patients approved in China since 2009,” said John V. Oyler, founder, chief executive officer, and chairman of BeiGene.
“We are excited to announce that the first prescription was made in January 2018. From now on, Chinese patients can benefit from Vidaza in hospitals around China.”
Azacitidine was evaluated in a global phase 3 trial of patients with intermediate-2- and high-risk MDS, CMML, or AML (AZA-001). Results from this trial were published in The Lancet Oncology in 2009.
Patients were randomized to receive azacitidine plus best supportive care (BSC, n=179) or conventional care regimens plus BSC (105 to BSC alone, 49 to low-dose cytarabine, and 25 to chemotherapy with cytarabine and anthracycline).
Azacitidine was given subcutaneously at a dose of 75 mg/m2 daily for 7 consecutive days every 28 days until disease progression, relapse after response, or unacceptable toxicity.
The median overall survival was 24.5 months with azacitidine, compared to 15 months for patients treated with conventional care regimens.
There was a higher hematologic response rate in the azacitidine arm than the conventional care arm—29% and 12%, respectively.
In the azacitidine group, 45% of patients who were dependent on red blood cell transfusions at baseline became transfusion independent, compared with 11% in the conventional care group.
Forty-six percent of patients in the azacitidine arm and 63% in the conventional care arm died.
Grade 3/4 hematologic toxicity (in the azacitidine and conventional care arms, respectively) included neutropenia (91% and 76%), thrombocytopenia (85% and 80%), and anemia (57% and 68%). ![]()
Azacitidine for injection (Vidaza®) is now available in China.
The nucleoside metabolic inhibitor was approved in China to treat patients with intermediate-2/high-risk myelodysplastic syndromes (MDS), acute myeloid leukemia (AML) with 20% to 30% bone marrow blasts, and chronic myelomonocytic leukemia (CMML).
Azacitidine for injection is marketed in China by BeiGene Ltd. under an exclusive license from Celgene Corporation.
“Vidaza is the only approved hypomethylating agent shown to prolong survival for patients with MDS and the first new treatment for MDS patients approved in China since 2009,” said John V. Oyler, founder, chief executive officer, and chairman of BeiGene.
“We are excited to announce that the first prescription was made in January 2018. From now on, Chinese patients can benefit from Vidaza in hospitals around China.”
Azacitidine was evaluated in a global phase 3 trial of patients with intermediate-2- and high-risk MDS, CMML, or AML (AZA-001). Results from this trial were published in The Lancet Oncology in 2009.
Patients were randomized to receive azacitidine plus best supportive care (BSC, n=179) or conventional care regimens plus BSC (105 to BSC alone, 49 to low-dose cytarabine, and 25 to chemotherapy with cytarabine and anthracycline).
Azacitidine was given subcutaneously at a dose of 75 mg/m2 daily for 7 consecutive days every 28 days until disease progression, relapse after response, or unacceptable toxicity.
The median overall survival was 24.5 months with azacitidine, compared to 15 months for patients treated with conventional care regimens.
There was a higher hematologic response rate in the azacitidine arm than the conventional care arm—29% and 12%, respectively.
In the azacitidine group, 45% of patients who were dependent on red blood cell transfusions at baseline became transfusion independent, compared with 11% in the conventional care group.
Forty-six percent of patients in the azacitidine arm and 63% in the conventional care arm died.
Grade 3/4 hematologic toxicity (in the azacitidine and conventional care arms, respectively) included neutropenia (91% and 76%), thrombocytopenia (85% and 80%), and anemia (57% and 68%). ![]()
Azacitidine for injection (Vidaza®) is now available in China.
The nucleoside metabolic inhibitor was approved in China to treat patients with intermediate-2/high-risk myelodysplastic syndromes (MDS), acute myeloid leukemia (AML) with 20% to 30% bone marrow blasts, and chronic myelomonocytic leukemia (CMML).
Azacitidine for injection is marketed in China by BeiGene Ltd. under an exclusive license from Celgene Corporation.
“Vidaza is the only approved hypomethylating agent shown to prolong survival for patients with MDS and the first new treatment for MDS patients approved in China since 2009,” said John V. Oyler, founder, chief executive officer, and chairman of BeiGene.
“We are excited to announce that the first prescription was made in January 2018. From now on, Chinese patients can benefit from Vidaza in hospitals around China.”
Azacitidine was evaluated in a global phase 3 trial of patients with intermediate-2- and high-risk MDS, CMML, or AML (AZA-001). Results from this trial were published in The Lancet Oncology in 2009.
Patients were randomized to receive azacitidine plus best supportive care (BSC, n=179) or conventional care regimens plus BSC (105 to BSC alone, 49 to low-dose cytarabine, and 25 to chemotherapy with cytarabine and anthracycline).
Azacitidine was given subcutaneously at a dose of 75 mg/m2 daily for 7 consecutive days every 28 days until disease progression, relapse after response, or unacceptable toxicity.
The median overall survival was 24.5 months with azacitidine, compared to 15 months for patients treated with conventional care regimens.
There was a higher hematologic response rate in the azacitidine arm than the conventional care arm—29% and 12%, respectively.
In the azacitidine group, 45% of patients who were dependent on red blood cell transfusions at baseline became transfusion independent, compared with 11% in the conventional care group.
Forty-six percent of patients in the azacitidine arm and 63% in the conventional care arm died.
Grade 3/4 hematologic toxicity (in the azacitidine and conventional care arms, respectively) included neutropenia (91% and 76%), thrombocytopenia (85% and 80%), and anemia (57% and 68%). ![]()