User login
Paradoxical Reaction to TNF-α Inhibitor Therapy in a Patient With Hidradenitis Suppurativa
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory condition of the pilosebaceous unit that occurs in concert with elevations of various cytokines, including tumor necrosis factor α (TNF-α), IL-1β, IL-10, and IL-17.1,2 Adalimumab is a TNF-α inhibitor approved by the US Food and Drug Administration for the treatment of HS. Although TNF-α inhibitors are effective for many immune-mediated inflammatory disorders, paradoxical drug reactions have been reported following treatment with these agents.3-6 True paradoxical drug reactions likely are immune mediated and directly lead to new onset of a pathologic condition that would otherwise respond to that drug. For example, there are reports of rheumatoid arthritis patients who were treated with a TNF-α inhibitor and developed psoriatic skin lesions.3,6 Paradoxical drug reactions also have been reported with acute-onset inflammatory bowel disease and HS or less commonly pyoderma gangrenosum (PG), uveitis, granulomatous reactions, and vasculitis.4,5 We present the case of a patient with HS who was treated with a TNF-α inhibitor and developed 2 distinct paradoxical drug reactions. We also provide an overview of paradoxical drug reactions associated with TNF-α inhibitors.
A 38-year-old woman developed a painful “boil” on the right leg that was previously treated in the emergency department with incision and drainage as well as oral clindamycin for 7 days, but the lesion spread and continued to worsen. She had a history of HS in the axillae and groin region that had been present since 12 years of age. The condition was poorly controlled despite multiple courses of oral antibiotics and surgical resections. An oral contraceptive also was attempted, but the patient discontinued treatment when liver enzyme levels became elevated. The patient had no other notable medical history, including skin disease. There was a family history of HS in her father and a sibling. Seeking more effective treatment, the patient was offered adalimumab approximately 4 months prior to clinical presentation and agreed to start a course of the drug. She received a loading dose of 160 mg on day 1 and 80 mg on day 15 followed by a maintenance dosage of 40 mg weekly. She experienced improvement in HS symptoms after 3 months on adalimumab; however, she developed scaly pruritic patches on the scalp, arms, and legs that were consistent with psoriasis. Because of the absence of a personal or family history of psoriasis, the patient was informed of the probability of paradoxical psoriasis resulting from adalimumab. She elected to continue adalimumab because of the improvement in HS symptoms, and the psoriatic lesions were mild and adequately controlled with a topical steroid.
At the current presentation 1 month later, physical examination revealed a large indurated and ulcerated area with jagged edges at the incision and drainage site (Figure 1). Pyoderma gangrenosum was clinically suspected; a biopsy was performed, and the patient was started on oral prednisone. At 2-week follow-up, the ulcer was found to be rapidly resolving with prednisone and healing with cribriform scarring (Figure 2). Histopathology revealed an undermining neutrophilic inflammatory process that was consistent with PG. A diagnosis of PG was made based on previously published criteria7 and the following major/minor criteria in the patient: pathology; absence of infection on histologic analysis; history of pathergy related to worsening ulceration at the site of incision and drainage of the initial boil; clinical findings of an ulcer with peripheral violaceous erythema; undermined borders and tenderness at the site; and rapid resolution of the ulcer with prednisone.

Cessation of adalimumab gradually led to clearance of both psoriasiform lesions and PG; however, HS lesions persisted.

Although the precise pathogenesis of HS is unclear, both genetic abnormalities of the pilosebaceous unit and a dysregulated immune reaction appear to lead to the clinical characteristics of chronic inflammation and scarring seen in HS. A key effector appears to be helper T-cell (TH17) lymphocyte activation, with increased secretion of TNF-α, IL-1β, and IL-17.1,2 In turn, IL-17 induces higher expression of TNF-α, leading to a persistent cycle of inflammation. Peripheral recruitment of IL-17–producing neutrophils also may contribute to chronic inflammation.8
Adalimumab is the only US Food and Drug Administration–approved biologic indicated for the treatment of HS. Our patient initially responded to adalimumab with improvement of HS; however, treatment had to be discontinued because of the unusual occurrence of 2 distinct paradoxical reactions in a short span of time. Psoriasis and PG are both considered true paradoxical reactions because primary occurrences of both diseases usually are responsive to treatment with adalimumab.
Tumor necrosis factor α inhibitor–induced psoriasis arises de novo and is estimated to occur in approximately 5% of patients with rheumatoid arthritis.3,6 Palmoplantar pustular psoriasiform reactions are the most common form of paradoxical psoriasis. Topical medications can be used to treat skin lesions, but systemic treatment is required in many cases. Switching to an alternate class of a biologic, such as an IL-17, IL-12/23, or IL-23 inhibitor, can improve the skin reaction; however, such treatment is inconsistently successful, and paradoxical drug reactions also have been seen with these other classes of biologics.4,9
Recent studies support distinct immune causes for classical and paradoxical psoriasis. In classical psoriasis, plasmacytoid dendritic cells (pDCs) produce IFN-α, which stimulates conventional dendritic cells to produce TNF-α. However, TNF-α matures both pDCs and conventional dendritic cells; upon maturation, both types of dendritic cells lose the ability to produce IFN-α, thus allowing TNF-α to become dominant.10 The blockade of TNF-α prevents pDC maturation, leading to uninhibited IFN-α, which appears to drive inflammation in paradoxical psoriasis. In classical psoriasis, oligoclonal dermal CD4+ T cells and epidermal CD8+ T cells remain, even in resolved skin lesions, and can cause disease recurrence through reactivation of skin-resident memory T cells.11 No relapse of paradoxical psoriasis occurs with discontinuation of anti-TNF-α therapy, which supports the notion of an absence of memory T cells.
The incidence of paradoxical psoriasis in patients receiving a TNF-α inhibitor for HS is unclear.12 There are case series in which patients who had concurrent psoriasis and HS were successfully treated with a TNF-α inhibitor.13 A recently recognized condition—PASH syndrome—encompasses the clinical triad of PG, acne, and HS.10
Our patient had no history of acne or PG, only a long-standing history of HS. New-onset PG occurred only after a TNF-α inhibitor was initiated. Notably, PASH syndrome has been successfully treated with TNF-α inhibitors, highlighting the shared inflammatory etiology of HS and PG.14 In patients with concurrent PG and HS, TNF-α inhibitors were more effective for treating PG than for HS.
Pyoderma gangrenosum is an inflammatory disorder that often occurs concomitantly with other conditions, such as inflammatory bowel disease. The exact underlying cause of PG is unclear, but there appears to be both neutrophil and T-cell dysfunction in PG, with excess inflammatory cytokine production (eg, IL-1β, TNF-α, IL-17).15
The mainstay of treatment of PG is systemic corticosteroids and immunosuppressives, such as cyclosporine. Tumor necrosis factor α inhibitors as well as other interleukin inhibitors are increasingly utilized as potential therapeutic alternatives for PG.16,17
Unlike paradoxical psoriasis, the underlying cause of paradoxical PG is unclear.18,19 A similar mechanism may be postulated whereby inhibition of TNF-α leads to excessive activation of alternative inflammatory pathways that result in paradoxical PG. In one study, the prevalence of PG among 68,232 patients with HS was 0.18% compared with 0.01% among those without HS; therefore, patients with HS appear to be more predisposed to PG.20
This case illustrates the complex, often conflicting effects of cytokine inhibition in the paradoxical elicitation of alternative inflammatory disorders as an unintended consequence of the initial cytokine blockade. It is likely that genetic predisposition allows for paradoxical reactions in some patients when there is predominant inhibition of one cytokine in the inflammatory pathway. In rare cases, multiple paradoxical reactions are possible.
1. Vossen ARJV, van der Zee HH, Prens EP. Hidradenitis suppurativa: a systematic review integrating inflammatory pathways into a cohesive pathogenic model. Front Immunol. 2018;9:2965. doi:10.3389/fimmu.2018.02965
2. Goldburg SR, Strober BE, Payette MJ. Hidradenitis suppurativa: epidemiology, clinical presentation and pathogenesis. J Am Acad Dermatol. 2020; 82:1045-1058. doi:10.1016/j.jaad.2019.08.090
3. Brown G, Wang E, Leon A, et al. Tumor necrosis factor-α inhibitor-induced psoriasis: systematic review of clinical features, histopathological findings, and management experience. J Am Acad Dermatol. 2017;76:334-341. doi:10.1016/j.jaad.2016.08.012
4. Puig L. Paradoxical reactions: anti-tumor necrosis factor alpha agents, ustekinumab, secukinumab, ixekizumab and others. Curr Prob Dermatol. 2018;53:49-63. doi:10.1159/000479475
5. Faivre C, Villani AP, Aubin F, et al; . Hidradenitis suppurativa (HS): an unrecognized paradoxical effect of biologic agents (BA) used in chronic inflammatory diseases. J Am Acad Dermatol. 2016;74:1153-1159. doi:10.1016/j.jaad.2016.01.018
6. Ko JM, Gottlieb AB, Kerbleski JF. Induction and exacerbation of psoriasis with TNF-blockade therapy: a review and analysis of 127 cases. J Dermatolog Treat. 2009;20:100-108. doi:10.1080/09546630802441234
7. Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a delphi consensus of international experts. JAMA Dermatol. 2018;154:461-466. doi:10.1001/jamadermatol.2017.5980
8. Lima AL, Karl I, Giner T, et al. Keratinocytes and neutrophils are important sources of proinflammatory molecules in hidradenitis suppurativa. Br J Dermatol. 2016;174:514-521. doi:10.1111/bjd.14214
9. Li SJ, Perez-Chada LM, Merola JF. TNF inhibitor-induced psoriasis: proposed algorithm for treatment and management. J Psoriasis Psoriatic Arthritis. 2019;4:70-80. doi:10.1177/2475530318810851
10. Conrad C, Di Domizio J, Mylonas A, et al. TNF blockade induces a dysregulated type I interferon response without autoimmunity in paradoxical psoriasis. Nat Commun. 2018;9:25. doi:10.1038/s41467-017-02466-4
11. Matos TR, O’Malley JT, Lowry EL, et al. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing αβ T cell clones. J Clin Invest. 2017;127:4031-4041. doi:10.1172/JCI93396
12. Faivre C, Villani AP, Aubin F, et al. Hidradenitis suppurativa (HS): an unrecognized paradoxical effect of biologic agents (BA) used in chronic inflammatory diseases. J Am Acad Dermatol. 2016;74:1153-1159. doi:10.1016/j.jaad.2016.01.018
13. Marzano AV, Damiani G, Ceccherini I, et al. Autoinflammation in pyoderma gangrenosum and its syndromic form (pyoderma gangrenosum, acne and suppurative hidradenitis). Br J Dermatol. 2017;176:1588-1598. doi:10.1111/bjd.15226
14. Cugno M, Borghi A, Marzano AV. PAPA, PASH, PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol. 2017;18:555-562. doi:10.1007/s40257-017-0265-1
15. Wang EA, Steel A, Luxardi G, et al. Classic ulcerative pyoderma gangrenosum is a T cell-mediated disease targeting follicular adnexal structures: a hypothesis based on molecular and clinicopathologic studies. Front Immunol. 2018;8:1980. doi:10.3389/fimmu.2017.01980
16. Patel F, Fitzmaurice S, Duong C, et al. Effective strategies for the management of pyoderma gangrenosum: a comprehensive review. Acta Derm Venereol. 2015;95:525-531. doi:10.2340/00015555-2008
17. Partridge ACR, Bai JW, Rosen CF, et al. Effectiveness of systemic treatments for pyoderma gangrenosum: a systematic review of observational studies and clinical trials. Br J Dermatol. 2018;179:290-295. doi:10.1111/bjd.16485
18. Benzaquen M, Monnier J, Beaussault Y, et al. Pyoderma gangrenosum arising during treatment of psoriasis with adalimumab: effectiveness of ustekinumab. Australas J Dermatol. 2017;58:e270-e271. doi:10.1111/ajd.12545
19. Fujimoto N, Yamasaki Y, Watanabe RJ. Paradoxical uveitis and pyoderma gangrenosum in a patient with psoriatic arthritis under infliximab treatment. J Dtsch Dermatol Ges. 2018;16:1139-1140. doi:10.1111/ddg.13632
20. Tannenbaum R, Strunk A, Garg A. Overall and subgroup prevalence of pyoderma gangrenosum among patients with hidradenitis suppurativa: a population-based analysis in the United States. J Am Acad Dermatol. 2019;80:1533-1537. doi:10.1016/j.jaad.2019.02.004
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory condition of the pilosebaceous unit that occurs in concert with elevations of various cytokines, including tumor necrosis factor α (TNF-α), IL-1β, IL-10, and IL-17.1,2 Adalimumab is a TNF-α inhibitor approved by the US Food and Drug Administration for the treatment of HS. Although TNF-α inhibitors are effective for many immune-mediated inflammatory disorders, paradoxical drug reactions have been reported following treatment with these agents.3-6 True paradoxical drug reactions likely are immune mediated and directly lead to new onset of a pathologic condition that would otherwise respond to that drug. For example, there are reports of rheumatoid arthritis patients who were treated with a TNF-α inhibitor and developed psoriatic skin lesions.3,6 Paradoxical drug reactions also have been reported with acute-onset inflammatory bowel disease and HS or less commonly pyoderma gangrenosum (PG), uveitis, granulomatous reactions, and vasculitis.4,5 We present the case of a patient with HS who was treated with a TNF-α inhibitor and developed 2 distinct paradoxical drug reactions. We also provide an overview of paradoxical drug reactions associated with TNF-α inhibitors.
A 38-year-old woman developed a painful “boil” on the right leg that was previously treated in the emergency department with incision and drainage as well as oral clindamycin for 7 days, but the lesion spread and continued to worsen. She had a history of HS in the axillae and groin region that had been present since 12 years of age. The condition was poorly controlled despite multiple courses of oral antibiotics and surgical resections. An oral contraceptive also was attempted, but the patient discontinued treatment when liver enzyme levels became elevated. The patient had no other notable medical history, including skin disease. There was a family history of HS in her father and a sibling. Seeking more effective treatment, the patient was offered adalimumab approximately 4 months prior to clinical presentation and agreed to start a course of the drug. She received a loading dose of 160 mg on day 1 and 80 mg on day 15 followed by a maintenance dosage of 40 mg weekly. She experienced improvement in HS symptoms after 3 months on adalimumab; however, she developed scaly pruritic patches on the scalp, arms, and legs that were consistent with psoriasis. Because of the absence of a personal or family history of psoriasis, the patient was informed of the probability of paradoxical psoriasis resulting from adalimumab. She elected to continue adalimumab because of the improvement in HS symptoms, and the psoriatic lesions were mild and adequately controlled with a topical steroid.
At the current presentation 1 month later, physical examination revealed a large indurated and ulcerated area with jagged edges at the incision and drainage site (Figure 1). Pyoderma gangrenosum was clinically suspected; a biopsy was performed, and the patient was started on oral prednisone. At 2-week follow-up, the ulcer was found to be rapidly resolving with prednisone and healing with cribriform scarring (Figure 2). Histopathology revealed an undermining neutrophilic inflammatory process that was consistent with PG. A diagnosis of PG was made based on previously published criteria7 and the following major/minor criteria in the patient: pathology; absence of infection on histologic analysis; history of pathergy related to worsening ulceration at the site of incision and drainage of the initial boil; clinical findings of an ulcer with peripheral violaceous erythema; undermined borders and tenderness at the site; and rapid resolution of the ulcer with prednisone.
Cessation of adalimumab gradually led to clearance of both psoriasiform lesions and PG; however, HS lesions persisted.
Although the precise pathogenesis of HS is unclear, both genetic abnormalities of the pilosebaceous unit and a dysregulated immune reaction appear to lead to the clinical characteristics of chronic inflammation and scarring seen in HS. A key effector appears to be helper T-cell (TH17) lymphocyte activation, with increased secretion of TNF-α, IL-1β, and IL-17.1,2 In turn, IL-17 induces higher expression of TNF-α, leading to a persistent cycle of inflammation. Peripheral recruitment of IL-17–producing neutrophils also may contribute to chronic inflammation.8
Adalimumab is the only US Food and Drug Administration–approved biologic indicated for the treatment of HS. Our patient initially responded to adalimumab with improvement of HS; however, treatment had to be discontinued because of the unusual occurrence of 2 distinct paradoxical reactions in a short span of time. Psoriasis and PG are both considered true paradoxical reactions because primary occurrences of both diseases usually are responsive to treatment with adalimumab.
Tumor necrosis factor α inhibitor–induced psoriasis arises de novo and is estimated to occur in approximately 5% of patients with rheumatoid arthritis.3,6 Palmoplantar pustular psoriasiform reactions are the most common form of paradoxical psoriasis. Topical medications can be used to treat skin lesions, but systemic treatment is required in many cases. Switching to an alternate class of a biologic, such as an IL-17, IL-12/23, or IL-23 inhibitor, can improve the skin reaction; however, such treatment is inconsistently successful, and paradoxical drug reactions also have been seen with these other classes of biologics.4,9
Recent studies support distinct immune causes for classical and paradoxical psoriasis. In classical psoriasis, plasmacytoid dendritic cells (pDCs) produce IFN-α, which stimulates conventional dendritic cells to produce TNF-α. However, TNF-α matures both pDCs and conventional dendritic cells; upon maturation, both types of dendritic cells lose the ability to produce IFN-α, thus allowing TNF-α to become dominant.10 The blockade of TNF-α prevents pDC maturation, leading to uninhibited IFN-α, which appears to drive inflammation in paradoxical psoriasis. In classical psoriasis, oligoclonal dermal CD4+ T cells and epidermal CD8+ T cells remain, even in resolved skin lesions, and can cause disease recurrence through reactivation of skin-resident memory T cells.11 No relapse of paradoxical psoriasis occurs with discontinuation of anti-TNF-α therapy, which supports the notion of an absence of memory T cells.
The incidence of paradoxical psoriasis in patients receiving a TNF-α inhibitor for HS is unclear.12 There are case series in which patients who had concurrent psoriasis and HS were successfully treated with a TNF-α inhibitor.13 A recently recognized condition—PASH syndrome—encompasses the clinical triad of PG, acne, and HS.10
Our patient had no history of acne or PG, only a long-standing history of HS. New-onset PG occurred only after a TNF-α inhibitor was initiated. Notably, PASH syndrome has been successfully treated with TNF-α inhibitors, highlighting the shared inflammatory etiology of HS and PG.14 In patients with concurrent PG and HS, TNF-α inhibitors were more effective for treating PG than for HS.
Pyoderma gangrenosum is an inflammatory disorder that often occurs concomitantly with other conditions, such as inflammatory bowel disease. The exact underlying cause of PG is unclear, but there appears to be both neutrophil and T-cell dysfunction in PG, with excess inflammatory cytokine production (eg, IL-1β, TNF-α, IL-17).15
The mainstay of treatment of PG is systemic corticosteroids and immunosuppressives, such as cyclosporine. Tumor necrosis factor α inhibitors as well as other interleukin inhibitors are increasingly utilized as potential therapeutic alternatives for PG.16,17
Unlike paradoxical psoriasis, the underlying cause of paradoxical PG is unclear.18,19 A similar mechanism may be postulated whereby inhibition of TNF-α leads to excessive activation of alternative inflammatory pathways that result in paradoxical PG. In one study, the prevalence of PG among 68,232 patients with HS was 0.18% compared with 0.01% among those without HS; therefore, patients with HS appear to be more predisposed to PG.20
This case illustrates the complex, often conflicting effects of cytokine inhibition in the paradoxical elicitation of alternative inflammatory disorders as an unintended consequence of the initial cytokine blockade. It is likely that genetic predisposition allows for paradoxical reactions in some patients when there is predominant inhibition of one cytokine in the inflammatory pathway. In rare cases, multiple paradoxical reactions are possible.
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory condition of the pilosebaceous unit that occurs in concert with elevations of various cytokines, including tumor necrosis factor α (TNF-α), IL-1β, IL-10, and IL-17.1,2 Adalimumab is a TNF-α inhibitor approved by the US Food and Drug Administration for the treatment of HS. Although TNF-α inhibitors are effective for many immune-mediated inflammatory disorders, paradoxical drug reactions have been reported following treatment with these agents.3-6 True paradoxical drug reactions likely are immune mediated and directly lead to new onset of a pathologic condition that would otherwise respond to that drug. For example, there are reports of rheumatoid arthritis patients who were treated with a TNF-α inhibitor and developed psoriatic skin lesions.3,6 Paradoxical drug reactions also have been reported with acute-onset inflammatory bowel disease and HS or less commonly pyoderma gangrenosum (PG), uveitis, granulomatous reactions, and vasculitis.4,5 We present the case of a patient with HS who was treated with a TNF-α inhibitor and developed 2 distinct paradoxical drug reactions. We also provide an overview of paradoxical drug reactions associated with TNF-α inhibitors.
A 38-year-old woman developed a painful “boil” on the right leg that was previously treated in the emergency department with incision and drainage as well as oral clindamycin for 7 days, but the lesion spread and continued to worsen. She had a history of HS in the axillae and groin region that had been present since 12 years of age. The condition was poorly controlled despite multiple courses of oral antibiotics and surgical resections. An oral contraceptive also was attempted, but the patient discontinued treatment when liver enzyme levels became elevated. The patient had no other notable medical history, including skin disease. There was a family history of HS in her father and a sibling. Seeking more effective treatment, the patient was offered adalimumab approximately 4 months prior to clinical presentation and agreed to start a course of the drug. She received a loading dose of 160 mg on day 1 and 80 mg on day 15 followed by a maintenance dosage of 40 mg weekly. She experienced improvement in HS symptoms after 3 months on adalimumab; however, she developed scaly pruritic patches on the scalp, arms, and legs that were consistent with psoriasis. Because of the absence of a personal or family history of psoriasis, the patient was informed of the probability of paradoxical psoriasis resulting from adalimumab. She elected to continue adalimumab because of the improvement in HS symptoms, and the psoriatic lesions were mild and adequately controlled with a topical steroid.
At the current presentation 1 month later, physical examination revealed a large indurated and ulcerated area with jagged edges at the incision and drainage site (Figure 1). Pyoderma gangrenosum was clinically suspected; a biopsy was performed, and the patient was started on oral prednisone. At 2-week follow-up, the ulcer was found to be rapidly resolving with prednisone and healing with cribriform scarring (Figure 2). Histopathology revealed an undermining neutrophilic inflammatory process that was consistent with PG. A diagnosis of PG was made based on previously published criteria7 and the following major/minor criteria in the patient: pathology; absence of infection on histologic analysis; history of pathergy related to worsening ulceration at the site of incision and drainage of the initial boil; clinical findings of an ulcer with peripheral violaceous erythema; undermined borders and tenderness at the site; and rapid resolution of the ulcer with prednisone.
Cessation of adalimumab gradually led to clearance of both psoriasiform lesions and PG; however, HS lesions persisted.
Although the precise pathogenesis of HS is unclear, both genetic abnormalities of the pilosebaceous unit and a dysregulated immune reaction appear to lead to the clinical characteristics of chronic inflammation and scarring seen in HS. A key effector appears to be helper T-cell (TH17) lymphocyte activation, with increased secretion of TNF-α, IL-1β, and IL-17.1,2 In turn, IL-17 induces higher expression of TNF-α, leading to a persistent cycle of inflammation. Peripheral recruitment of IL-17–producing neutrophils also may contribute to chronic inflammation.8
Adalimumab is the only US Food and Drug Administration–approved biologic indicated for the treatment of HS. Our patient initially responded to adalimumab with improvement of HS; however, treatment had to be discontinued because of the unusual occurrence of 2 distinct paradoxical reactions in a short span of time. Psoriasis and PG are both considered true paradoxical reactions because primary occurrences of both diseases usually are responsive to treatment with adalimumab.
Tumor necrosis factor α inhibitor–induced psoriasis arises de novo and is estimated to occur in approximately 5% of patients with rheumatoid arthritis.3,6 Palmoplantar pustular psoriasiform reactions are the most common form of paradoxical psoriasis. Topical medications can be used to treat skin lesions, but systemic treatment is required in many cases. Switching to an alternate class of a biologic, such as an IL-17, IL-12/23, or IL-23 inhibitor, can improve the skin reaction; however, such treatment is inconsistently successful, and paradoxical drug reactions also have been seen with these other classes of biologics.4,9
Recent studies support distinct immune causes for classical and paradoxical psoriasis. In classical psoriasis, plasmacytoid dendritic cells (pDCs) produce IFN-α, which stimulates conventional dendritic cells to produce TNF-α. However, TNF-α matures both pDCs and conventional dendritic cells; upon maturation, both types of dendritic cells lose the ability to produce IFN-α, thus allowing TNF-α to become dominant.10 The blockade of TNF-α prevents pDC maturation, leading to uninhibited IFN-α, which appears to drive inflammation in paradoxical psoriasis. In classical psoriasis, oligoclonal dermal CD4+ T cells and epidermal CD8+ T cells remain, even in resolved skin lesions, and can cause disease recurrence through reactivation of skin-resident memory T cells.11 No relapse of paradoxical psoriasis occurs with discontinuation of anti-TNF-α therapy, which supports the notion of an absence of memory T cells.
The incidence of paradoxical psoriasis in patients receiving a TNF-α inhibitor for HS is unclear.12 There are case series in which patients who had concurrent psoriasis and HS were successfully treated with a TNF-α inhibitor.13 A recently recognized condition—PASH syndrome—encompasses the clinical triad of PG, acne, and HS.10
Our patient had no history of acne or PG, only a long-standing history of HS. New-onset PG occurred only after a TNF-α inhibitor was initiated. Notably, PASH syndrome has been successfully treated with TNF-α inhibitors, highlighting the shared inflammatory etiology of HS and PG.14 In patients with concurrent PG and HS, TNF-α inhibitors were more effective for treating PG than for HS.
Pyoderma gangrenosum is an inflammatory disorder that often occurs concomitantly with other conditions, such as inflammatory bowel disease. The exact underlying cause of PG is unclear, but there appears to be both neutrophil and T-cell dysfunction in PG, with excess inflammatory cytokine production (eg, IL-1β, TNF-α, IL-17).15
The mainstay of treatment of PG is systemic corticosteroids and immunosuppressives, such as cyclosporine. Tumor necrosis factor α inhibitors as well as other interleukin inhibitors are increasingly utilized as potential therapeutic alternatives for PG.16,17
Unlike paradoxical psoriasis, the underlying cause of paradoxical PG is unclear.18,19 A similar mechanism may be postulated whereby inhibition of TNF-α leads to excessive activation of alternative inflammatory pathways that result in paradoxical PG. In one study, the prevalence of PG among 68,232 patients with HS was 0.18% compared with 0.01% among those without HS; therefore, patients with HS appear to be more predisposed to PG.20
This case illustrates the complex, often conflicting effects of cytokine inhibition in the paradoxical elicitation of alternative inflammatory disorders as an unintended consequence of the initial cytokine blockade. It is likely that genetic predisposition allows for paradoxical reactions in some patients when there is predominant inhibition of one cytokine in the inflammatory pathway. In rare cases, multiple paradoxical reactions are possible.
1. Vossen ARJV, van der Zee HH, Prens EP. Hidradenitis suppurativa: a systematic review integrating inflammatory pathways into a cohesive pathogenic model. Front Immunol. 2018;9:2965. doi:10.3389/fimmu.2018.02965
2. Goldburg SR, Strober BE, Payette MJ. Hidradenitis suppurativa: epidemiology, clinical presentation and pathogenesis. J Am Acad Dermatol. 2020; 82:1045-1058. doi:10.1016/j.jaad.2019.08.090
3. Brown G, Wang E, Leon A, et al. Tumor necrosis factor-α inhibitor-induced psoriasis: systematic review of clinical features, histopathological findings, and management experience. J Am Acad Dermatol. 2017;76:334-341. doi:10.1016/j.jaad.2016.08.012
4. Puig L. Paradoxical reactions: anti-tumor necrosis factor alpha agents, ustekinumab, secukinumab, ixekizumab and others. Curr Prob Dermatol. 2018;53:49-63. doi:10.1159/000479475
5. Faivre C, Villani AP, Aubin F, et al; . Hidradenitis suppurativa (HS): an unrecognized paradoxical effect of biologic agents (BA) used in chronic inflammatory diseases. J Am Acad Dermatol. 2016;74:1153-1159. doi:10.1016/j.jaad.2016.01.018
6. Ko JM, Gottlieb AB, Kerbleski JF. Induction and exacerbation of psoriasis with TNF-blockade therapy: a review and analysis of 127 cases. J Dermatolog Treat. 2009;20:100-108. doi:10.1080/09546630802441234
7. Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a delphi consensus of international experts. JAMA Dermatol. 2018;154:461-466. doi:10.1001/jamadermatol.2017.5980
8. Lima AL, Karl I, Giner T, et al. Keratinocytes and neutrophils are important sources of proinflammatory molecules in hidradenitis suppurativa. Br J Dermatol. 2016;174:514-521. doi:10.1111/bjd.14214
9. Li SJ, Perez-Chada LM, Merola JF. TNF inhibitor-induced psoriasis: proposed algorithm for treatment and management. J Psoriasis Psoriatic Arthritis. 2019;4:70-80. doi:10.1177/2475530318810851
10. Conrad C, Di Domizio J, Mylonas A, et al. TNF blockade induces a dysregulated type I interferon response without autoimmunity in paradoxical psoriasis. Nat Commun. 2018;9:25. doi:10.1038/s41467-017-02466-4
11. Matos TR, O’Malley JT, Lowry EL, et al. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing αβ T cell clones. J Clin Invest. 2017;127:4031-4041. doi:10.1172/JCI93396
12. Faivre C, Villani AP, Aubin F, et al. Hidradenitis suppurativa (HS): an unrecognized paradoxical effect of biologic agents (BA) used in chronic inflammatory diseases. J Am Acad Dermatol. 2016;74:1153-1159. doi:10.1016/j.jaad.2016.01.018
13. Marzano AV, Damiani G, Ceccherini I, et al. Autoinflammation in pyoderma gangrenosum and its syndromic form (pyoderma gangrenosum, acne and suppurative hidradenitis). Br J Dermatol. 2017;176:1588-1598. doi:10.1111/bjd.15226
14. Cugno M, Borghi A, Marzano AV. PAPA, PASH, PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol. 2017;18:555-562. doi:10.1007/s40257-017-0265-1
15. Wang EA, Steel A, Luxardi G, et al. Classic ulcerative pyoderma gangrenosum is a T cell-mediated disease targeting follicular adnexal structures: a hypothesis based on molecular and clinicopathologic studies. Front Immunol. 2018;8:1980. doi:10.3389/fimmu.2017.01980
16. Patel F, Fitzmaurice S, Duong C, et al. Effective strategies for the management of pyoderma gangrenosum: a comprehensive review. Acta Derm Venereol. 2015;95:525-531. doi:10.2340/00015555-2008
17. Partridge ACR, Bai JW, Rosen CF, et al. Effectiveness of systemic treatments for pyoderma gangrenosum: a systematic review of observational studies and clinical trials. Br J Dermatol. 2018;179:290-295. doi:10.1111/bjd.16485
18. Benzaquen M, Monnier J, Beaussault Y, et al. Pyoderma gangrenosum arising during treatment of psoriasis with adalimumab: effectiveness of ustekinumab. Australas J Dermatol. 2017;58:e270-e271. doi:10.1111/ajd.12545
19. Fujimoto N, Yamasaki Y, Watanabe RJ. Paradoxical uveitis and pyoderma gangrenosum in a patient with psoriatic arthritis under infliximab treatment. J Dtsch Dermatol Ges. 2018;16:1139-1140. doi:10.1111/ddg.13632
20. Tannenbaum R, Strunk A, Garg A. Overall and subgroup prevalence of pyoderma gangrenosum among patients with hidradenitis suppurativa: a population-based analysis in the United States. J Am Acad Dermatol. 2019;80:1533-1537. doi:10.1016/j.jaad.2019.02.004
1. Vossen ARJV, van der Zee HH, Prens EP. Hidradenitis suppurativa: a systematic review integrating inflammatory pathways into a cohesive pathogenic model. Front Immunol. 2018;9:2965. doi:10.3389/fimmu.2018.02965
2. Goldburg SR, Strober BE, Payette MJ. Hidradenitis suppurativa: epidemiology, clinical presentation and pathogenesis. J Am Acad Dermatol. 2020; 82:1045-1058. doi:10.1016/j.jaad.2019.08.090
3. Brown G, Wang E, Leon A, et al. Tumor necrosis factor-α inhibitor-induced psoriasis: systematic review of clinical features, histopathological findings, and management experience. J Am Acad Dermatol. 2017;76:334-341. doi:10.1016/j.jaad.2016.08.012
4. Puig L. Paradoxical reactions: anti-tumor necrosis factor alpha agents, ustekinumab, secukinumab, ixekizumab and others. Curr Prob Dermatol. 2018;53:49-63. doi:10.1159/000479475
5. Faivre C, Villani AP, Aubin F, et al; . Hidradenitis suppurativa (HS): an unrecognized paradoxical effect of biologic agents (BA) used in chronic inflammatory diseases. J Am Acad Dermatol. 2016;74:1153-1159. doi:10.1016/j.jaad.2016.01.018
6. Ko JM, Gottlieb AB, Kerbleski JF. Induction and exacerbation of psoriasis with TNF-blockade therapy: a review and analysis of 127 cases. J Dermatolog Treat. 2009;20:100-108. doi:10.1080/09546630802441234
7. Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a delphi consensus of international experts. JAMA Dermatol. 2018;154:461-466. doi:10.1001/jamadermatol.2017.5980
8. Lima AL, Karl I, Giner T, et al. Keratinocytes and neutrophils are important sources of proinflammatory molecules in hidradenitis suppurativa. Br J Dermatol. 2016;174:514-521. doi:10.1111/bjd.14214
9. Li SJ, Perez-Chada LM, Merola JF. TNF inhibitor-induced psoriasis: proposed algorithm for treatment and management. J Psoriasis Psoriatic Arthritis. 2019;4:70-80. doi:10.1177/2475530318810851
10. Conrad C, Di Domizio J, Mylonas A, et al. TNF blockade induces a dysregulated type I interferon response without autoimmunity in paradoxical psoriasis. Nat Commun. 2018;9:25. doi:10.1038/s41467-017-02466-4
11. Matos TR, O’Malley JT, Lowry EL, et al. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing αβ T cell clones. J Clin Invest. 2017;127:4031-4041. doi:10.1172/JCI93396
12. Faivre C, Villani AP, Aubin F, et al. Hidradenitis suppurativa (HS): an unrecognized paradoxical effect of biologic agents (BA) used in chronic inflammatory diseases. J Am Acad Dermatol. 2016;74:1153-1159. doi:10.1016/j.jaad.2016.01.018
13. Marzano AV, Damiani G, Ceccherini I, et al. Autoinflammation in pyoderma gangrenosum and its syndromic form (pyoderma gangrenosum, acne and suppurative hidradenitis). Br J Dermatol. 2017;176:1588-1598. doi:10.1111/bjd.15226
14. Cugno M, Borghi A, Marzano AV. PAPA, PASH, PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol. 2017;18:555-562. doi:10.1007/s40257-017-0265-1
15. Wang EA, Steel A, Luxardi G, et al. Classic ulcerative pyoderma gangrenosum is a T cell-mediated disease targeting follicular adnexal structures: a hypothesis based on molecular and clinicopathologic studies. Front Immunol. 2018;8:1980. doi:10.3389/fimmu.2017.01980
16. Patel F, Fitzmaurice S, Duong C, et al. Effective strategies for the management of pyoderma gangrenosum: a comprehensive review. Acta Derm Venereol. 2015;95:525-531. doi:10.2340/00015555-2008
17. Partridge ACR, Bai JW, Rosen CF, et al. Effectiveness of systemic treatments for pyoderma gangrenosum: a systematic review of observational studies and clinical trials. Br J Dermatol. 2018;179:290-295. doi:10.1111/bjd.16485
18. Benzaquen M, Monnier J, Beaussault Y, et al. Pyoderma gangrenosum arising during treatment of psoriasis with adalimumab: effectiveness of ustekinumab. Australas J Dermatol. 2017;58:e270-e271. doi:10.1111/ajd.12545
19. Fujimoto N, Yamasaki Y, Watanabe RJ. Paradoxical uveitis and pyoderma gangrenosum in a patient with psoriatic arthritis under infliximab treatment. J Dtsch Dermatol Ges. 2018;16:1139-1140. doi:10.1111/ddg.13632
20. Tannenbaum R, Strunk A, Garg A. Overall and subgroup prevalence of pyoderma gangrenosum among patients with hidradenitis suppurativa: a population-based analysis in the United States. J Am Acad Dermatol. 2019;80:1533-1537. doi:10.1016/j.jaad.2019.02.004
Practice Points
- Clinicians need to be aware of the potential risk for a paradoxical reaction in patients receiving a tumor necrosis factor α (TNF-α) inhibitor for hidradenitis suppurativa.
- Although uncommon, developing more than 1 type of paradoxical skin reaction is possible with a TNF-α inhibitor.
- Early recognition and appropriate management of these paradoxical reactions are critical.

Researchers link two genes to Raynaud’s disease
Researchers have identified two genes that may contribute to Raynaud’s phenomenon, a condition where blood vessels in the extremities constrict and limit blood flow.
Raynaud’s is a relatively common condition, affecting 2%-5% of the general population. Though Raynaud’s can be an annoyance for some, it can also cause severe pain and can require medication.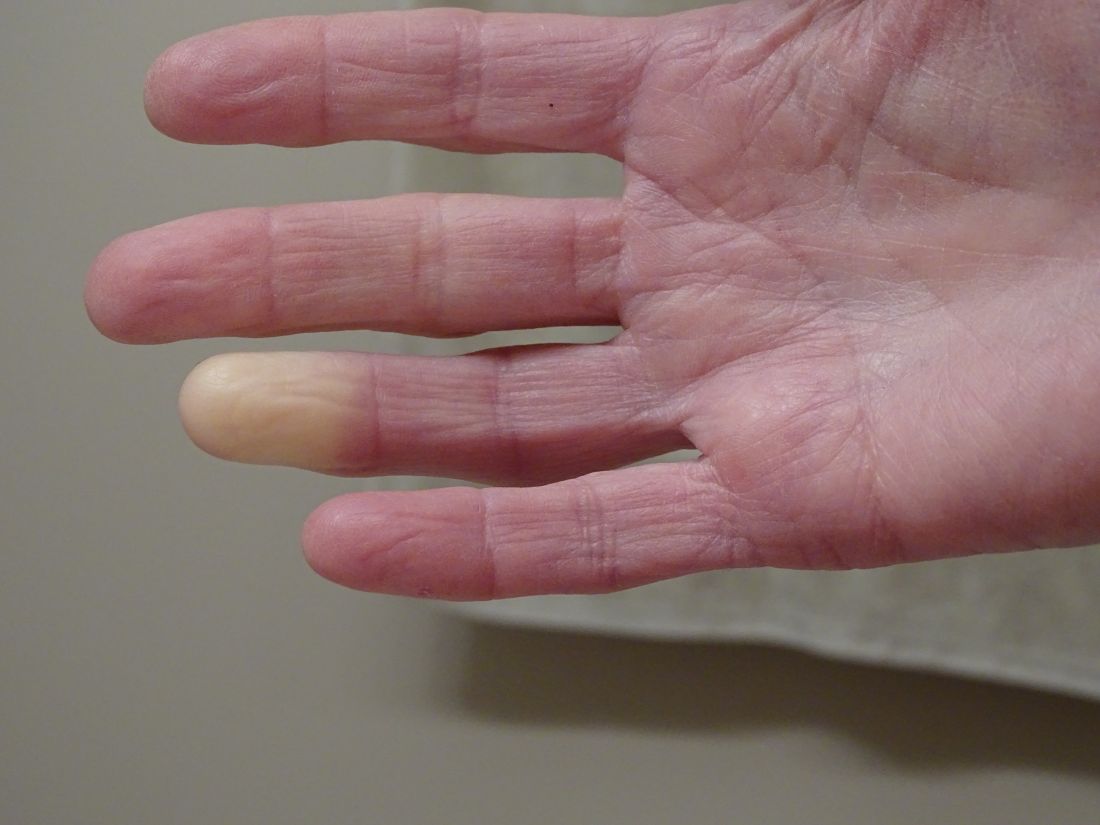
These newly identified genes will hopefully lead to new therapeutic options, said Maik Pietzner, PhD, chair in health data modeling at Queen Mary University of London’s Precision Healthcare University Research Institute (PHURI) and group leader in the Computational Medicine Group at the Berlin Institute of Health at Charité – Universitätsmedizin Berlin, Germany.

Dr. Pietzner led the research along with Claudia Langenberg, MD, PhD, director of PHURI.
The study was published in Nature Communications.
Largest genomic study of Raynaud’s to date
The researchers looked through electronic medical records from the UK Biobank, a large-scale database that contains genetic and health information on half a million participants. They identified more than 5,100 individuals with Raynaud’s, of which 68% had primary Raynaud’s. These participants were compared with more than 439,000 controls who did not have Raynaud’s.
In a secondary analysis, the team also used health records from the Queen Mary University of London Genes & Health Study, which contains health information on individuals of South Asian ancestry.
The researchers identified two genes that are likely involved with Raynaud’s. The first, ADRA2A, encodes for the alpha-2A adrenergic receptor that can cause vasoconstriction of small blood vessels in response to stress hormones. Researchers have long suspected that this type of receptor could be involved with Raynaud’s, but there was debate over which receptor subtype was responsible.
“Our finding of alpha-2A receptors is quite interesting because the focus has always been on alpha-2C receptors,” said Dr. Pietzner. “It’s only a letter, but it’s a massive difference in terms of biology and physiology,” he said, and could be why therapies targeting 2C receptors have been ineffective.
The second strongest association was for the transcription factor IRX1. Less is known about this gene, but the data we do have suggest that it is involved with regulating the dilation of blood vessels, Dr. Pietzner noted.
“There might be balance between the ADRA2A finding being responsible for constriction and the IRX1 finding indirectly linked to the dilation of those vessels following constrictions. Having both may explain why these prolonged episodes of vasoconstriction lead to a loss of oxygen to the tissues,” so they turn white and then blue, he said.
Because the Biobank cohort was European-centric, Dr. Pietzner and colleagues also identified 400 cases of Raynaud’s in British individuals of Bangladeshi and Pakistani ancestry and were able to replicate the association between IRX1 and Raynaud’s. Data on ADRA2A were unavailable.
The genes identified are associated with primary Raynaud’s. Secondary Raynaud’s is a rarer type of the condition that occurs along with autoimmune disorders, such as scleroderma, and is generally more severe.
It’s long been suspected that Raynaud’s had some genetic component, because half of patients with Raynaud’s have another family member with the same condition, said Laura Hummers, MD, who codirects the John Hopkins Scleroderma Center in Baltimore. She was not involved with the study.

This is “the largest study of this kind that’s been done,” she said, and the first to show a potential mechanism behind this genetic association.
The main gene finding, ADRA2A, “points to a receptor on the cells that regulate the tone of these blood vessels,” she continued. “It suggests maybe there’s too many of these receptors or they’re overly sensitive; something about them is different that makes patients more susceptible to these cold triggers. Knowing that is potentially really important, because it could give you a direct way to intervene, if true.”
New therapeutic avenues
The first-line treatment for primary Raynaud’s is behavioral interventions, such as maintaining body and extremity warmth and avoiding certain vasoconstricting drugs, said Kimberly Lakin, MD, a rheumatologist at the Hospital for Special Surgery in New York, who not involved in the research. These drugs could include over-the-counter decongestants and certain medications for attention-deficit/hyperactivity disorder.

If these behavioral interventions are not enough, clinicians most commonly prescribe calcium channel blockers. These medications are vasodilators but can be a concern for people with normal or already low blood pressure, Dr. Lakin said. They can also cause symptoms such as headache, leg swelling, constipation, and other gastrointestinal symptoms.
Other medications, such as fluoxetine, may also be considered as a later-line therapy, “but the effectiveness is fairly limited in Raynaud’s,” she said. “Certainly, other medication options that would be helpful and driven by the mechanisms of Raynaud’s would add to our ability to help patients.”
As it turns out, one of the genes identified in the study, ADRA2A, “is actually one of the most commonly targeted genes by drugs,” said Dr. Pietzner. Because the findings suggest that ADRA2A is overexpressed in Raynaud, a selective inhibitor like the antidepressant mirtazapine could be a promising candidate to repurpose for treating Raynaud’s, he said.
Limitations to electronic medical record analyses
Both Dr. Hummers and Dr. Lakin noted that research using diagnostic codes from medical records to identify cases has some limitations. The study may have included patients misdiagnosed with Raynaud’s when perhaps they had another condition. Patients with milder Raynaud’s who have not sought medical attention for the condition would not be represented in the study, Dr. Lakin said.
The UK Biobank includes individuals of mostly European descent, so an analysis confirming these findings in a more diverse population would be helpful, she said.
However, both Dr. Lakin and Dr. Hummers agreed that the study contributes to the understanding of the mechanisms behind Raynaud’s. Although the two identified genes were tied to primary Raynaud’s, the study’s findings could potentially apply to secondary Raynaud’s as well, Dr. Hummers said.
“Anything we learn about primary Raynaud’s may have implication for Raynaud’s more broadly,” she noted.
Dr. Hummers and Dr. Lakin disclosed no relevant financial relationships. Dr. Pietzner has received partnership funding for the MRC Clinical Pharmacology Training Scheme (cofunded by MRC and Roche, UCB, Eli Lilly, and Novartis) and a PhD studentship jointly funded by the UK Engineering and Physical Sciences Research Council and AstraZeneca. Dr. Pietzner also has unrestricted educational grant support for the UK Pharmacogenetics and Stratified Medicine Network from Bristol-Myers Squibb.
A version of this article appeared on Medscape.com.
Researchers have identified two genes that may contribute to Raynaud’s phenomenon, a condition where blood vessels in the extremities constrict and limit blood flow.
Raynaud’s is a relatively common condition, affecting 2%-5% of the general population. Though Raynaud’s can be an annoyance for some, it can also cause severe pain and can require medication.
These newly identified genes will hopefully lead to new therapeutic options, said Maik Pietzner, PhD, chair in health data modeling at Queen Mary University of London’s Precision Healthcare University Research Institute (PHURI) and group leader in the Computational Medicine Group at the Berlin Institute of Health at Charité – Universitätsmedizin Berlin, Germany.
Dr. Pietzner led the research along with Claudia Langenberg, MD, PhD, director of PHURI.
The study was published in Nature Communications.
Largest genomic study of Raynaud’s to date
The researchers looked through electronic medical records from the UK Biobank, a large-scale database that contains genetic and health information on half a million participants. They identified more than 5,100 individuals with Raynaud’s, of which 68% had primary Raynaud’s. These participants were compared with more than 439,000 controls who did not have Raynaud’s.
In a secondary analysis, the team also used health records from the Queen Mary University of London Genes & Health Study, which contains health information on individuals of South Asian ancestry.
The researchers identified two genes that are likely involved with Raynaud’s. The first, ADRA2A, encodes for the alpha-2A adrenergic receptor that can cause vasoconstriction of small blood vessels in response to stress hormones. Researchers have long suspected that this type of receptor could be involved with Raynaud’s, but there was debate over which receptor subtype was responsible.
“Our finding of alpha-2A receptors is quite interesting because the focus has always been on alpha-2C receptors,” said Dr. Pietzner. “It’s only a letter, but it’s a massive difference in terms of biology and physiology,” he said, and could be why therapies targeting 2C receptors have been ineffective.
The second strongest association was for the transcription factor IRX1. Less is known about this gene, but the data we do have suggest that it is involved with regulating the dilation of blood vessels, Dr. Pietzner noted.
“There might be balance between the ADRA2A finding being responsible for constriction and the IRX1 finding indirectly linked to the dilation of those vessels following constrictions. Having both may explain why these prolonged episodes of vasoconstriction lead to a loss of oxygen to the tissues,” so they turn white and then blue, he said.
Because the Biobank cohort was European-centric, Dr. Pietzner and colleagues also identified 400 cases of Raynaud’s in British individuals of Bangladeshi and Pakistani ancestry and were able to replicate the association between IRX1 and Raynaud’s. Data on ADRA2A were unavailable.
The genes identified are associated with primary Raynaud’s. Secondary Raynaud’s is a rarer type of the condition that occurs along with autoimmune disorders, such as scleroderma, and is generally more severe.
It’s long been suspected that Raynaud’s had some genetic component, because half of patients with Raynaud’s have another family member with the same condition, said Laura Hummers, MD, who codirects the John Hopkins Scleroderma Center in Baltimore. She was not involved with the study.
This is “the largest study of this kind that’s been done,” she said, and the first to show a potential mechanism behind this genetic association.
The main gene finding, ADRA2A, “points to a receptor on the cells that regulate the tone of these blood vessels,” she continued. “It suggests maybe there’s too many of these receptors or they’re overly sensitive; something about them is different that makes patients more susceptible to these cold triggers. Knowing that is potentially really important, because it could give you a direct way to intervene, if true.”
New therapeutic avenues
The first-line treatment for primary Raynaud’s is behavioral interventions, such as maintaining body and extremity warmth and avoiding certain vasoconstricting drugs, said Kimberly Lakin, MD, a rheumatologist at the Hospital for Special Surgery in New York, who not involved in the research. These drugs could include over-the-counter decongestants and certain medications for attention-deficit/hyperactivity disorder.
If these behavioral interventions are not enough, clinicians most commonly prescribe calcium channel blockers. These medications are vasodilators but can be a concern for people with normal or already low blood pressure, Dr. Lakin said. They can also cause symptoms such as headache, leg swelling, constipation, and other gastrointestinal symptoms.
Other medications, such as fluoxetine, may also be considered as a later-line therapy, “but the effectiveness is fairly limited in Raynaud’s,” she said. “Certainly, other medication options that would be helpful and driven by the mechanisms of Raynaud’s would add to our ability to help patients.”
As it turns out, one of the genes identified in the study, ADRA2A, “is actually one of the most commonly targeted genes by drugs,” said Dr. Pietzner. Because the findings suggest that ADRA2A is overexpressed in Raynaud, a selective inhibitor like the antidepressant mirtazapine could be a promising candidate to repurpose for treating Raynaud’s, he said.
Limitations to electronic medical record analyses
Both Dr. Hummers and Dr. Lakin noted that research using diagnostic codes from medical records to identify cases has some limitations. The study may have included patients misdiagnosed with Raynaud’s when perhaps they had another condition. Patients with milder Raynaud’s who have not sought medical attention for the condition would not be represented in the study, Dr. Lakin said.
The UK Biobank includes individuals of mostly European descent, so an analysis confirming these findings in a more diverse population would be helpful, she said.
However, both Dr. Lakin and Dr. Hummers agreed that the study contributes to the understanding of the mechanisms behind Raynaud’s. Although the two identified genes were tied to primary Raynaud’s, the study’s findings could potentially apply to secondary Raynaud’s as well, Dr. Hummers said.
“Anything we learn about primary Raynaud’s may have implication for Raynaud’s more broadly,” she noted.
Dr. Hummers and Dr. Lakin disclosed no relevant financial relationships. Dr. Pietzner has received partnership funding for the MRC Clinical Pharmacology Training Scheme (cofunded by MRC and Roche, UCB, Eli Lilly, and Novartis) and a PhD studentship jointly funded by the UK Engineering and Physical Sciences Research Council and AstraZeneca. Dr. Pietzner also has unrestricted educational grant support for the UK Pharmacogenetics and Stratified Medicine Network from Bristol-Myers Squibb.
A version of this article appeared on Medscape.com.
Researchers have identified two genes that may contribute to Raynaud’s phenomenon, a condition where blood vessels in the extremities constrict and limit blood flow.
Raynaud’s is a relatively common condition, affecting 2%-5% of the general population. Though Raynaud’s can be an annoyance for some, it can also cause severe pain and can require medication.
These newly identified genes will hopefully lead to new therapeutic options, said Maik Pietzner, PhD, chair in health data modeling at Queen Mary University of London’s Precision Healthcare University Research Institute (PHURI) and group leader in the Computational Medicine Group at the Berlin Institute of Health at Charité – Universitätsmedizin Berlin, Germany.
Dr. Pietzner led the research along with Claudia Langenberg, MD, PhD, director of PHURI.
The study was published in Nature Communications.
Largest genomic study of Raynaud’s to date
The researchers looked through electronic medical records from the UK Biobank, a large-scale database that contains genetic and health information on half a million participants. They identified more than 5,100 individuals with Raynaud’s, of which 68% had primary Raynaud’s. These participants were compared with more than 439,000 controls who did not have Raynaud’s.
In a secondary analysis, the team also used health records from the Queen Mary University of London Genes & Health Study, which contains health information on individuals of South Asian ancestry.
The researchers identified two genes that are likely involved with Raynaud’s. The first, ADRA2A, encodes for the alpha-2A adrenergic receptor that can cause vasoconstriction of small blood vessels in response to stress hormones. Researchers have long suspected that this type of receptor could be involved with Raynaud’s, but there was debate over which receptor subtype was responsible.
“Our finding of alpha-2A receptors is quite interesting because the focus has always been on alpha-2C receptors,” said Dr. Pietzner. “It’s only a letter, but it’s a massive difference in terms of biology and physiology,” he said, and could be why therapies targeting 2C receptors have been ineffective.
The second strongest association was for the transcription factor IRX1. Less is known about this gene, but the data we do have suggest that it is involved with regulating the dilation of blood vessels, Dr. Pietzner noted.
“There might be balance between the ADRA2A finding being responsible for constriction and the IRX1 finding indirectly linked to the dilation of those vessels following constrictions. Having both may explain why these prolonged episodes of vasoconstriction lead to a loss of oxygen to the tissues,” so they turn white and then blue, he said.
Because the Biobank cohort was European-centric, Dr. Pietzner and colleagues also identified 400 cases of Raynaud’s in British individuals of Bangladeshi and Pakistani ancestry and were able to replicate the association between IRX1 and Raynaud’s. Data on ADRA2A were unavailable.
The genes identified are associated with primary Raynaud’s. Secondary Raynaud’s is a rarer type of the condition that occurs along with autoimmune disorders, such as scleroderma, and is generally more severe.
It’s long been suspected that Raynaud’s had some genetic component, because half of patients with Raynaud’s have another family member with the same condition, said Laura Hummers, MD, who codirects the John Hopkins Scleroderma Center in Baltimore. She was not involved with the study.
This is “the largest study of this kind that’s been done,” she said, and the first to show a potential mechanism behind this genetic association.
The main gene finding, ADRA2A, “points to a receptor on the cells that regulate the tone of these blood vessels,” she continued. “It suggests maybe there’s too many of these receptors or they’re overly sensitive; something about them is different that makes patients more susceptible to these cold triggers. Knowing that is potentially really important, because it could give you a direct way to intervene, if true.”
New therapeutic avenues
The first-line treatment for primary Raynaud’s is behavioral interventions, such as maintaining body and extremity warmth and avoiding certain vasoconstricting drugs, said Kimberly Lakin, MD, a rheumatologist at the Hospital for Special Surgery in New York, who not involved in the research. These drugs could include over-the-counter decongestants and certain medications for attention-deficit/hyperactivity disorder.
If these behavioral interventions are not enough, clinicians most commonly prescribe calcium channel blockers. These medications are vasodilators but can be a concern for people with normal or already low blood pressure, Dr. Lakin said. They can also cause symptoms such as headache, leg swelling, constipation, and other gastrointestinal symptoms.
Other medications, such as fluoxetine, may also be considered as a later-line therapy, “but the effectiveness is fairly limited in Raynaud’s,” she said. “Certainly, other medication options that would be helpful and driven by the mechanisms of Raynaud’s would add to our ability to help patients.”
As it turns out, one of the genes identified in the study, ADRA2A, “is actually one of the most commonly targeted genes by drugs,” said Dr. Pietzner. Because the findings suggest that ADRA2A is overexpressed in Raynaud, a selective inhibitor like the antidepressant mirtazapine could be a promising candidate to repurpose for treating Raynaud’s, he said.
Limitations to electronic medical record analyses
Both Dr. Hummers and Dr. Lakin noted that research using diagnostic codes from medical records to identify cases has some limitations. The study may have included patients misdiagnosed with Raynaud’s when perhaps they had another condition. Patients with milder Raynaud’s who have not sought medical attention for the condition would not be represented in the study, Dr. Lakin said.
The UK Biobank includes individuals of mostly European descent, so an analysis confirming these findings in a more diverse population would be helpful, she said.
However, both Dr. Lakin and Dr. Hummers agreed that the study contributes to the understanding of the mechanisms behind Raynaud’s. Although the two identified genes were tied to primary Raynaud’s, the study’s findings could potentially apply to secondary Raynaud’s as well, Dr. Hummers said.
“Anything we learn about primary Raynaud’s may have implication for Raynaud’s more broadly,” she noted.
Dr. Hummers and Dr. Lakin disclosed no relevant financial relationships. Dr. Pietzner has received partnership funding for the MRC Clinical Pharmacology Training Scheme (cofunded by MRC and Roche, UCB, Eli Lilly, and Novartis) and a PhD studentship jointly funded by the UK Engineering and Physical Sciences Research Council and AstraZeneca. Dr. Pietzner also has unrestricted educational grant support for the UK Pharmacogenetics and Stratified Medicine Network from Bristol-Myers Squibb.
A version of this article appeared on Medscape.com.
FROM NATURE COMMUNICATIONS
3-D stereophotogrammetry helps detect progression of craniofacial morphea
TOPLINE:
over time.
METHODOLOGY:
- Existing tools that detect disease progression in patients with CM are limited.
- In a prospective cohort study, researchers evaluated the use of 3-D stereophotogrammetry, a noninvasive, radiation-free imaging modality, to detect disease progression in 27 consecutive patients with CM seen at Boston Children’s Hospital and Brigham and Women’s Hospital from April 1, 2019, to March 1, 2023.
- After clinical and 3-D stereophotogrammetry assessments were performed at 2- to 12-month intervals, the 3-D images were rated by an expert (a board-certified plastic craniofacial surgeon) and a nonexpert (a board-certified dermatologist) as demonstrating progression or no progression.
- Kappa coefficients were used to calculate inter-rater reliability.
TAKEAWAY:
- Most of the study participants (73%) were female, their median age was 14 years (range, 5-40 years), and each underwent 3-D stereophotogrammetry imaging at least two times spaced a median of 3 months apart.
- On the basis of clinical assessments during the 48-month study period, 10 patients (37%) experienced progression of their disease.
- 3-D stereophotogrammetry not only corroborated clinical impressions of disease progression with strong inter-rater reliability (kappa = 0.80; 95% confidence interval, 0.61-0.99), but it also detected occult progression of asymmetry not noted on clinical examination in three additional patients.
- In subgroup analyses, assessment of 3-D images demonstrated substantial to near-perfect inter-rater reliability in patients with Fitzpatrick skin types IV-VI.
IN PRACTICE:
“Further work is necessary to validate this measure in a larger cohort and to guide its incorporation into medical decision-making for patients with CM,” the researchers wrote.
SOURCE:
Katharina S. Shaw, MD, of the department of dermatology at the University of Pennsylvania, Philadelphia, led the research. The study was published online in JAMA Dermatology.
LIMITATIONS:
The sample was small, and a criterion standard for assessing CM was lacking.
DISCLOSURES:
The researchers reported having no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
TOPLINE:
over time.
METHODOLOGY:
- Existing tools that detect disease progression in patients with CM are limited.
- In a prospective cohort study, researchers evaluated the use of 3-D stereophotogrammetry, a noninvasive, radiation-free imaging modality, to detect disease progression in 27 consecutive patients with CM seen at Boston Children’s Hospital and Brigham and Women’s Hospital from April 1, 2019, to March 1, 2023.
- After clinical and 3-D stereophotogrammetry assessments were performed at 2- to 12-month intervals, the 3-D images were rated by an expert (a board-certified plastic craniofacial surgeon) and a nonexpert (a board-certified dermatologist) as demonstrating progression or no progression.
- Kappa coefficients were used to calculate inter-rater reliability.
TAKEAWAY:
- Most of the study participants (73%) were female, their median age was 14 years (range, 5-40 years), and each underwent 3-D stereophotogrammetry imaging at least two times spaced a median of 3 months apart.
- On the basis of clinical assessments during the 48-month study period, 10 patients (37%) experienced progression of their disease.
- 3-D stereophotogrammetry not only corroborated clinical impressions of disease progression with strong inter-rater reliability (kappa = 0.80; 95% confidence interval, 0.61-0.99), but it also detected occult progression of asymmetry not noted on clinical examination in three additional patients.
- In subgroup analyses, assessment of 3-D images demonstrated substantial to near-perfect inter-rater reliability in patients with Fitzpatrick skin types IV-VI.
IN PRACTICE:
“Further work is necessary to validate this measure in a larger cohort and to guide its incorporation into medical decision-making for patients with CM,” the researchers wrote.
SOURCE:
Katharina S. Shaw, MD, of the department of dermatology at the University of Pennsylvania, Philadelphia, led the research. The study was published online in JAMA Dermatology.
LIMITATIONS:
The sample was small, and a criterion standard for assessing CM was lacking.
DISCLOSURES:
The researchers reported having no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
TOPLINE:
over time.
METHODOLOGY:
- Existing tools that detect disease progression in patients with CM are limited.
- In a prospective cohort study, researchers evaluated the use of 3-D stereophotogrammetry, a noninvasive, radiation-free imaging modality, to detect disease progression in 27 consecutive patients with CM seen at Boston Children’s Hospital and Brigham and Women’s Hospital from April 1, 2019, to March 1, 2023.
- After clinical and 3-D stereophotogrammetry assessments were performed at 2- to 12-month intervals, the 3-D images were rated by an expert (a board-certified plastic craniofacial surgeon) and a nonexpert (a board-certified dermatologist) as demonstrating progression or no progression.
- Kappa coefficients were used to calculate inter-rater reliability.
TAKEAWAY:
- Most of the study participants (73%) were female, their median age was 14 years (range, 5-40 years), and each underwent 3-D stereophotogrammetry imaging at least two times spaced a median of 3 months apart.
- On the basis of clinical assessments during the 48-month study period, 10 patients (37%) experienced progression of their disease.
- 3-D stereophotogrammetry not only corroborated clinical impressions of disease progression with strong inter-rater reliability (kappa = 0.80; 95% confidence interval, 0.61-0.99), but it also detected occult progression of asymmetry not noted on clinical examination in three additional patients.
- In subgroup analyses, assessment of 3-D images demonstrated substantial to near-perfect inter-rater reliability in patients with Fitzpatrick skin types IV-VI.
IN PRACTICE:
“Further work is necessary to validate this measure in a larger cohort and to guide its incorporation into medical decision-making for patients with CM,” the researchers wrote.
SOURCE:
Katharina S. Shaw, MD, of the department of dermatology at the University of Pennsylvania, Philadelphia, led the research. The study was published online in JAMA Dermatology.
LIMITATIONS:
The sample was small, and a criterion standard for assessing CM was lacking.
DISCLOSURES:
The researchers reported having no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
Anti-acid meds lower strength of systemic sclerosis drug
TOPLINE:
Anti-acid drugs used by patients with systemic sclerosis reduce the bioavailability of mycophenolate mofetil (MMF).
METHODOLOGY:
- Researchers conducted an open-label, pragmatic crossover study of 20 patients (all female) with systemic sclerosis at a single center who were on a stable MMF dose (1.5-2 g/day) for the last 3 months or more.
- Participants sequentially took MMF alone for 1 month, then with the H2 receptor blocker (HRB) ranitidine 300 mg/day in the second month, then with the proton pump inhibitor (PPI) esomeprazole 40 mg/day in the third month.
- Researchers measured the bioavailability of MMF in the patients during treatment with ranitidine or esomeprazole and the impact of the drugs on the total GI score of the UCLA Scleroderma Clinical Trial Consortium Gastrointestinal Tract 2.0 instrument.
- Patients were excluded if they were receiving co-prescription of cholestyramine, magnesium- or aluminum-containing antacids, and rifampicin; taking prednisolone-equivalent dose > 5 mg/day; taking MMF plus a PPI or an HRB at baseline; living with chronic kidney disease with a glomerular filtration rate < 30 mL/min; positive for HIV, HCV, or HBV; or living with end-stage lung disease or gastroduodenal ulcers.
TAKEAWAY:
- Mean estimated 12-hour area under curve levels of mycophenolic acid dropped by 32.7% (mean difference = 22.28 mcg h mL–1) when patients added esomeprazole, and they dipped by 21.97% (mean difference = 14.93 mcg h mL–1) when they added ranitidine vs. MMF alone.
- The pharmacokinetic parameter T-max did not differ significantly between MMF alone vs. MMF plus ranitidine but was significantly different with esomeprazole. C-max significantly declined with administration of ranitidine or esomeprazole vs. MMF alone.
- Total GI scores dipped when patients added esomeprazole or ranitidine.
IN PRACTICE:
In patients with significant gastroesophageal reflux disease symptoms who need to take MMF, management options may include monitoring MMF drug levels, switching to enteric-coated mycophenolate sodium, and spacing doses with anti-acid drugs.
SOURCE:
Glaxon Alex, MD, and colleagues from the Center for Arthritis and Rheumatism Excellence in Kochi, India, conducted the study, which was published online in Seminars in Arthritis & Rheumatism.
LIMITATIONS:
The sample size is small, and the optimum dose of MMF is unknown.
DISCLOSURES:
The study had no outside funding. The authors report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
TOPLINE:
Anti-acid drugs used by patients with systemic sclerosis reduce the bioavailability of mycophenolate mofetil (MMF).
METHODOLOGY:
- Researchers conducted an open-label, pragmatic crossover study of 20 patients (all female) with systemic sclerosis at a single center who were on a stable MMF dose (1.5-2 g/day) for the last 3 months or more.
- Participants sequentially took MMF alone for 1 month, then with the H2 receptor blocker (HRB) ranitidine 300 mg/day in the second month, then with the proton pump inhibitor (PPI) esomeprazole 40 mg/day in the third month.
- Researchers measured the bioavailability of MMF in the patients during treatment with ranitidine or esomeprazole and the impact of the drugs on the total GI score of the UCLA Scleroderma Clinical Trial Consortium Gastrointestinal Tract 2.0 instrument.
- Patients were excluded if they were receiving co-prescription of cholestyramine, magnesium- or aluminum-containing antacids, and rifampicin; taking prednisolone-equivalent dose > 5 mg/day; taking MMF plus a PPI or an HRB at baseline; living with chronic kidney disease with a glomerular filtration rate < 30 mL/min; positive for HIV, HCV, or HBV; or living with end-stage lung disease or gastroduodenal ulcers.
TAKEAWAY:
- Mean estimated 12-hour area under curve levels of mycophenolic acid dropped by 32.7% (mean difference = 22.28 mcg h mL–1) when patients added esomeprazole, and they dipped by 21.97% (mean difference = 14.93 mcg h mL–1) when they added ranitidine vs. MMF alone.
- The pharmacokinetic parameter T-max did not differ significantly between MMF alone vs. MMF plus ranitidine but was significantly different with esomeprazole. C-max significantly declined with administration of ranitidine or esomeprazole vs. MMF alone.
- Total GI scores dipped when patients added esomeprazole or ranitidine.
IN PRACTICE:
In patients with significant gastroesophageal reflux disease symptoms who need to take MMF, management options may include monitoring MMF drug levels, switching to enteric-coated mycophenolate sodium, and spacing doses with anti-acid drugs.
SOURCE:
Glaxon Alex, MD, and colleagues from the Center for Arthritis and Rheumatism Excellence in Kochi, India, conducted the study, which was published online in Seminars in Arthritis & Rheumatism.
LIMITATIONS:
The sample size is small, and the optimum dose of MMF is unknown.
DISCLOSURES:
The study had no outside funding. The authors report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
TOPLINE:
Anti-acid drugs used by patients with systemic sclerosis reduce the bioavailability of mycophenolate mofetil (MMF).
METHODOLOGY:
- Researchers conducted an open-label, pragmatic crossover study of 20 patients (all female) with systemic sclerosis at a single center who were on a stable MMF dose (1.5-2 g/day) for the last 3 months or more.
- Participants sequentially took MMF alone for 1 month, then with the H2 receptor blocker (HRB) ranitidine 300 mg/day in the second month, then with the proton pump inhibitor (PPI) esomeprazole 40 mg/day in the third month.
- Researchers measured the bioavailability of MMF in the patients during treatment with ranitidine or esomeprazole and the impact of the drugs on the total GI score of the UCLA Scleroderma Clinical Trial Consortium Gastrointestinal Tract 2.0 instrument.
- Patients were excluded if they were receiving co-prescription of cholestyramine, magnesium- or aluminum-containing antacids, and rifampicin; taking prednisolone-equivalent dose > 5 mg/day; taking MMF plus a PPI or an HRB at baseline; living with chronic kidney disease with a glomerular filtration rate < 30 mL/min; positive for HIV, HCV, or HBV; or living with end-stage lung disease or gastroduodenal ulcers.
TAKEAWAY:
- Mean estimated 12-hour area under curve levels of mycophenolic acid dropped by 32.7% (mean difference = 22.28 mcg h mL–1) when patients added esomeprazole, and they dipped by 21.97% (mean difference = 14.93 mcg h mL–1) when they added ranitidine vs. MMF alone.
- The pharmacokinetic parameter T-max did not differ significantly between MMF alone vs. MMF plus ranitidine but was significantly different with esomeprazole. C-max significantly declined with administration of ranitidine or esomeprazole vs. MMF alone.
- Total GI scores dipped when patients added esomeprazole or ranitidine.
IN PRACTICE:
In patients with significant gastroesophageal reflux disease symptoms who need to take MMF, management options may include monitoring MMF drug levels, switching to enteric-coated mycophenolate sodium, and spacing doses with anti-acid drugs.
SOURCE:
Glaxon Alex, MD, and colleagues from the Center for Arthritis and Rheumatism Excellence in Kochi, India, conducted the study, which was published online in Seminars in Arthritis & Rheumatism.
LIMITATIONS:
The sample size is small, and the optimum dose of MMF is unknown.
DISCLOSURES:
The study had no outside funding. The authors report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Tapering lupus drugs in stable patients: Large study outlines risks, benefits
The question looms large for patients with stable systemic lupus erythematosus (SLE): to taper or not to taper corticosteroids or immunosuppressive therapy? For patients and the physicians treating them, the evidence points in both directions. Flares are exacerbated by tapering, but simultaneously organ damage is tempered. Where is the balance? What competing factors together inform decision-making?

A recent multinational, observational cohort study conducted by Jiacai Cho, MBBS, of National University Hospital, Singapore, and colleagues, and published in The Lancet Rheumatology concluded that, given the odds of excess flares associated with tapering of corticosteroids and immunosuppressive therapy in patients with stable SLE, drug tapering warrants careful consideration of risks and benefits and is best reserved for those in complete clinical and serological remission with stable disease for at least 6 months. However, in an accompanying editorial, Yann Nguyen, MD, MPH, and Nathalie Costedoat-Chalumeau, MD, PhD, of the National Referral Center for Rare Autoimmune and Systemic Diseases at Cochin Hospital, Paris, and the Center for Research in Epidemiology and Statistics at Paris City University, argued for tipping the scale back from some of those expressed cautions.

In interviews, experts in the field expressed both strong appreciation for the cohort study and, like the editorialists, cognizance of its limitations.
Dr. Cho and colleagues recruited 3,002 adult patients with SLE (92.2% female, median age 39.5 years), from 25 sites across 13 Asia-Pacific countries. They were receiving routine clinical care and had achieved stable disease in at least one of two or more visits. Stable disease was defined by meeting criteria for Lupus Low Disease Activity State (LLDAS; SLE Disease Activity Index 2000 [SLEDAI-2K] score ≤ 4, Physician Global Assessment [PGA] ≤ 1, and prednisolone ≤ 7.5 mg/day), the 2021 DORIS definition of remission (clinical SLEDAI-2K score 0, PGA score < 0.5, and prednisolone dose ≤ 5 mg/day), or DORIS complete remission on therapy (SLEDAI-2K score 0, PGA score < 0.5, and prednisolone dose ≤ 5 mg/day). Any decrease in dose of corticosteroids or immunosuppressive therapy (mycophenolate mofetil, calcineurin inhibitors, azathioprine, leflunomide, or methotrexate) defined tapering. The investigators compared the odds of disease flares (SELENA-SLEDAI Flare Index) at the visit following tapering among those with tapering versus those who had continued the same drug doses.
Higher odds of flare with tapering
Tapering, compared with continuing with the same dose, was clearly associated with higher odds of flare at the next visit (11.4% with continuing vs. 17.0% with tapering; odds ratio, 1.24; 95% confidence interval, 1.10-1.39; P = .0005). Flares among patients who tapered were also slightly more often severe than with continuing the same dose (21.5% of flares vs. 19.7%). The level of remission at the time of tapering also mattered. Of 2,095 continuous tapering attempts, 860 (41.1%) were initiated in LLDAS, 596 (28.4%) in remission, and 639 (30.5%) in complete remission. Tapering when in LLDAS or remission, compared with complete remission, was associated with a higher likelihood of flare by 1 year (LLDAS: OR, 1.37; 95% CI, 1.03-1.81; P = .029; and remission: OR, 1.45; 95% CI, 1.08-1.94; P = .013). Time to first flare followed the same pattern. Also, sustained LLDAS, remission, or complete remission for at least 6 months just before the time of taper was associated with lower odds of flare at next visit and flares in 1 year, and longer time to flare.
Take baseline disease status, hydroxychloroquine’s effect into account
Dr. Nguyen and Dr. Costedoat-Chalumeau underscored several factors that may soften the risk for flares seen with tapering. They pointed to higher baseline doses of prednisone and immunosuppressants (and thus likely more severe disease that is more likely to flare) in the patients with tapering. Also, the SELENA-SLEDAI Flare Index used in the study classifies some clinically insignificant flares as mild to moderate and ignores the benefit of tapering. (It classifies patients as having a severe flare even when starting a new immunosuppressant prescription, such as azathioprine, methotrexate, or both, in an effort to reduce corticosteroid use.) They wrote that the study did not assess the rate of clinically meaningful flares (“essentially renal flares”), nor did it highlight that the “tiny” increase in absolute risk of severe flares (from 2.2% to 3.7%) could be further contextualized by the offset of the smaller, unmeasured rate of clinically significant flares and the “extremely relevant” risk of concomitant damage from prolonged treatment.
Dr. Nguyen and Dr. Costedoat-Chalumeau urged hydroxychloroquine use for all patients unless clearly contraindicated. In their own research, they have detailed hydroxychloroquine benefits in reducing not only flare risk, but also comorbidities, damage, and mortality. In the current study, the prevalence of hydroxychloroquine use in all the patient visits was only 63.3%. “We can assume that if more patients had been treated with hydroxychloroquine, both the number of flares and the difference between the two strategies would have been lower,” they wrote. They cited findings from a study of patients in remission for 2 years or longer in the Toronto Lupus Cohort in which a gradual taper of corticosteroids over 1 year was safe and feasible and resulted in less damage accrual at 24 months than not tapering. Optimizing tapering can minimize flare risk, they concluded.

Tapering SLE medications always involves some chance of flare and has to be considered a calculated risk, Sasha Bernatsky, MD, the James McGill professor of medicine in the division of rheumatology at McGill University, Montreal, said in an interview. “Long-term prednisone is not good for patients. I have heard it called ‘the miracle drug from hell’ – meaning that, yes, it controls disease, but at a cost of long-term complications. So we must be conscientious about tapering prednisone.” She observed that in the short-term, there may not be a huge risk to keeping a patient on an antimalarial and counseling patients to stay on it because their risk of flare is higher if they taper. Rheumatologists usually agree, however, that after 10 years or more, there is a real chance of retinal toxicity. “In our Montreal cohort, the risk of retinal toxicity was 5% after an average of 12.8 years of antimalarial use. My concern is that if a patient develops SLE in their 20s, how do we decide if we should keep them on an antimalarial for the next 60 or 70 years? If we keep them on the drug from age 25 to 45, and they then get retinal toxicity, they would essentially never be able to be on the drug again. So I do try to keep patients on the lowest dose of an antimalarial that is possible.”
Dr. Bernatsky pointed out further, “We think about tapering other immunosuppressants (such as methotrexate or mycophenolate or azathioprine) quite differently than prednisone tapering. We take our time a bit more, since many patients will tolerate being on standard doses of these drugs fairly well. If or when we do consider tapering these drugs, both our intuition and the literature suggests that someone with worse baseline disease activity or severity, who has needed a lot of steroids and multiple combinations of drugs to control disease, has a higher chance of flaring than someone with milder disease. As the editorial points out, lupus physicians (and their patients) need to think carefully about the patient’s risk profile, and be sure to tailor follow-up based on flare risk.”
Frank discussions with patients about the risks of tapering are needed, she said. “On one hand, there is consensus about how some aspects of lupus should be managed (for example, aggressive treatment of severe nephritis), but on the other hand, when it comes to long-term management and especially discussing tapering, we must have good discussions with patients. When a patient asks if they can taper a drug – many just lower or stop their drugs without asking – I am as honest as I can be, but ultimately have to admit any taper could be associated with a flare. It’s helpful to have actual figures to discuss with patients.”
No surprises
“This is an interesting study, which did not produce any surprises,” Dafna D. Gladman, MD, professor of medicine at University of Toronto and senior scientist at the university’s Schroeder Arthritis Institute, said when asked to comment. “We already knew from previous studies that abrupt withdrawal is not a good idea, and that if you taper when a patient is under conditions of remission, the rate of flare is actually lower than the usual rate of flare that occurs in people who continue on these medications. But the major limitation is that they did not specifically look at those who we would taper in clinical practice. In addition, they do not specify that the patients had to be on low-dose glucocorticoids before tapering, and they combined both immunosuppressive and steroids. It is not clear from the study what the excess flare rate was, or whether the flares were mild or severe. Most flares in patients with SLE are mild, consisting of skin and joint manifestations, while only a few patients have flares in kidney or neurologic manifestations.”
Dr. Gladman described her approach to tapering: “We aim for our patients to be taking no more than 5 mg of prednisone and to be in at least clinical remission with a SLEDAI-2K of 0 for at least 2 years before we would taper to glucocorticoids withdrawal. We always withdraw glucocorticoids first and immunosuppressives later, and keep patients on antimalarials the longest, unless there are specific side effects to the immunosuppressive or antimalarials which require their cessation earlier.”
Uncertainty persists
Other SLE experts weighing in confirmed the view that future research should aim to achieve clarity about the relative risks and benefits of tapering SLE drug regimens to maintain disease remission while minimizing potential for organ damage.

“Steroids are our friend and our enemy,” Joan T. Merrill, MD, professor of medicine at the University of Oklahoma Health Sciences Center, Oklahoma City, said in an interview. “If a person with lupus is in a lot of trouble, corticosteroids are almost universally a good option to get them out. But for too many decades, for too many patients, despite all the improvements we have made in better understanding the disease and developing some promising new treatments, we have yet to shed the inexorable toxicity in multiple organs of steroid dependence.” She continued, “Corticosteroids, even at low dose, may have broad-spectrum effects. But, in fact, so do many of the more ‘targeted’ agents. If all patients were lined up at the beginning of a study while being given azathioprine or a calcineurin inhibitor or belimumab at a stable, tolerable dose, you might see the same data if you tapered that agent down. What we really need is improved individualized guidance about when and how fast to remove immune modulators from stable patients with lupus without disturbing the balance that had been achieved in such a quiescent patient.”

That enduring uncertainty was echoed by Daniel J. Wallace, MD, professor of medicine at Cedars-Sinai Medical Center, Los Angeles: “The take-home message from this interesting paper,” he commented, “is that current lupus biomarkers are not adequate. They do not guide the practitioner well enough, so that all too often medication regimens are tapered even though the risks are not really well known. Also, there is evidence in the literature that fibrosis and ‘damage’ progress even if acute phase reactants such as sedimentation rate, [C-reactive protein], complement 3 and 4, and anti-dsDNA are normal. We don’t have a good metric to detect them.”
Dr. Cho and colleagues’ study was funded by AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Janssen, Merck Serono, GlaxoSmithKline, and UCB. Dr. Gladman disclosed consulting and/or research support from AbbVie, Amgen, Bristol-Myers Squibb, Eli Lilly, Janssen, Novartis, Pfizer, and UCB.
The question looms large for patients with stable systemic lupus erythematosus (SLE): to taper or not to taper corticosteroids or immunosuppressive therapy? For patients and the physicians treating them, the evidence points in both directions. Flares are exacerbated by tapering, but simultaneously organ damage is tempered. Where is the balance? What competing factors together inform decision-making?
A recent multinational, observational cohort study conducted by Jiacai Cho, MBBS, of National University Hospital, Singapore, and colleagues, and published in The Lancet Rheumatology concluded that, given the odds of excess flares associated with tapering of corticosteroids and immunosuppressive therapy in patients with stable SLE, drug tapering warrants careful consideration of risks and benefits and is best reserved for those in complete clinical and serological remission with stable disease for at least 6 months. However, in an accompanying editorial, Yann Nguyen, MD, MPH, and Nathalie Costedoat-Chalumeau, MD, PhD, of the National Referral Center for Rare Autoimmune and Systemic Diseases at Cochin Hospital, Paris, and the Center for Research in Epidemiology and Statistics at Paris City University, argued for tipping the scale back from some of those expressed cautions.
In interviews, experts in the field expressed both strong appreciation for the cohort study and, like the editorialists, cognizance of its limitations.
Dr. Cho and colleagues recruited 3,002 adult patients with SLE (92.2% female, median age 39.5 years), from 25 sites across 13 Asia-Pacific countries. They were receiving routine clinical care and had achieved stable disease in at least one of two or more visits. Stable disease was defined by meeting criteria for Lupus Low Disease Activity State (LLDAS; SLE Disease Activity Index 2000 [SLEDAI-2K] score ≤ 4, Physician Global Assessment [PGA] ≤ 1, and prednisolone ≤ 7.5 mg/day), the 2021 DORIS definition of remission (clinical SLEDAI-2K score 0, PGA score < 0.5, and prednisolone dose ≤ 5 mg/day), or DORIS complete remission on therapy (SLEDAI-2K score 0, PGA score < 0.5, and prednisolone dose ≤ 5 mg/day). Any decrease in dose of corticosteroids or immunosuppressive therapy (mycophenolate mofetil, calcineurin inhibitors, azathioprine, leflunomide, or methotrexate) defined tapering. The investigators compared the odds of disease flares (SELENA-SLEDAI Flare Index) at the visit following tapering among those with tapering versus those who had continued the same drug doses.
Higher odds of flare with tapering
Tapering, compared with continuing with the same dose, was clearly associated with higher odds of flare at the next visit (11.4% with continuing vs. 17.0% with tapering; odds ratio, 1.24; 95% confidence interval, 1.10-1.39; P = .0005). Flares among patients who tapered were also slightly more often severe than with continuing the same dose (21.5% of flares vs. 19.7%). The level of remission at the time of tapering also mattered. Of 2,095 continuous tapering attempts, 860 (41.1%) were initiated in LLDAS, 596 (28.4%) in remission, and 639 (30.5%) in complete remission. Tapering when in LLDAS or remission, compared with complete remission, was associated with a higher likelihood of flare by 1 year (LLDAS: OR, 1.37; 95% CI, 1.03-1.81; P = .029; and remission: OR, 1.45; 95% CI, 1.08-1.94; P = .013). Time to first flare followed the same pattern. Also, sustained LLDAS, remission, or complete remission for at least 6 months just before the time of taper was associated with lower odds of flare at next visit and flares in 1 year, and longer time to flare.
Take baseline disease status, hydroxychloroquine’s effect into account
Dr. Nguyen and Dr. Costedoat-Chalumeau underscored several factors that may soften the risk for flares seen with tapering. They pointed to higher baseline doses of prednisone and immunosuppressants (and thus likely more severe disease that is more likely to flare) in the patients with tapering. Also, the SELENA-SLEDAI Flare Index used in the study classifies some clinically insignificant flares as mild to moderate and ignores the benefit of tapering. (It classifies patients as having a severe flare even when starting a new immunosuppressant prescription, such as azathioprine, methotrexate, or both, in an effort to reduce corticosteroid use.) They wrote that the study did not assess the rate of clinically meaningful flares (“essentially renal flares”), nor did it highlight that the “tiny” increase in absolute risk of severe flares (from 2.2% to 3.7%) could be further contextualized by the offset of the smaller, unmeasured rate of clinically significant flares and the “extremely relevant” risk of concomitant damage from prolonged treatment.
Dr. Nguyen and Dr. Costedoat-Chalumeau urged hydroxychloroquine use for all patients unless clearly contraindicated. In their own research, they have detailed hydroxychloroquine benefits in reducing not only flare risk, but also comorbidities, damage, and mortality. In the current study, the prevalence of hydroxychloroquine use in all the patient visits was only 63.3%. “We can assume that if more patients had been treated with hydroxychloroquine, both the number of flares and the difference between the two strategies would have been lower,” they wrote. They cited findings from a study of patients in remission for 2 years or longer in the Toronto Lupus Cohort in which a gradual taper of corticosteroids over 1 year was safe and feasible and resulted in less damage accrual at 24 months than not tapering. Optimizing tapering can minimize flare risk, they concluded.
Tapering SLE medications always involves some chance of flare and has to be considered a calculated risk, Sasha Bernatsky, MD, the James McGill professor of medicine in the division of rheumatology at McGill University, Montreal, said in an interview. “Long-term prednisone is not good for patients. I have heard it called ‘the miracle drug from hell’ – meaning that, yes, it controls disease, but at a cost of long-term complications. So we must be conscientious about tapering prednisone.” She observed that in the short-term, there may not be a huge risk to keeping a patient on an antimalarial and counseling patients to stay on it because their risk of flare is higher if they taper. Rheumatologists usually agree, however, that after 10 years or more, there is a real chance of retinal toxicity. “In our Montreal cohort, the risk of retinal toxicity was 5% after an average of 12.8 years of antimalarial use. My concern is that if a patient develops SLE in their 20s, how do we decide if we should keep them on an antimalarial for the next 60 or 70 years? If we keep them on the drug from age 25 to 45, and they then get retinal toxicity, they would essentially never be able to be on the drug again. So I do try to keep patients on the lowest dose of an antimalarial that is possible.”
Dr. Bernatsky pointed out further, “We think about tapering other immunosuppressants (such as methotrexate or mycophenolate or azathioprine) quite differently than prednisone tapering. We take our time a bit more, since many patients will tolerate being on standard doses of these drugs fairly well. If or when we do consider tapering these drugs, both our intuition and the literature suggests that someone with worse baseline disease activity or severity, who has needed a lot of steroids and multiple combinations of drugs to control disease, has a higher chance of flaring than someone with milder disease. As the editorial points out, lupus physicians (and their patients) need to think carefully about the patient’s risk profile, and be sure to tailor follow-up based on flare risk.”
Frank discussions with patients about the risks of tapering are needed, she said. “On one hand, there is consensus about how some aspects of lupus should be managed (for example, aggressive treatment of severe nephritis), but on the other hand, when it comes to long-term management and especially discussing tapering, we must have good discussions with patients. When a patient asks if they can taper a drug – many just lower or stop their drugs without asking – I am as honest as I can be, but ultimately have to admit any taper could be associated with a flare. It’s helpful to have actual figures to discuss with patients.”
No surprises
“This is an interesting study, which did not produce any surprises,” Dafna D. Gladman, MD, professor of medicine at University of Toronto and senior scientist at the university’s Schroeder Arthritis Institute, said when asked to comment. “We already knew from previous studies that abrupt withdrawal is not a good idea, and that if you taper when a patient is under conditions of remission, the rate of flare is actually lower than the usual rate of flare that occurs in people who continue on these medications. But the major limitation is that they did not specifically look at those who we would taper in clinical practice. In addition, they do not specify that the patients had to be on low-dose glucocorticoids before tapering, and they combined both immunosuppressive and steroids. It is not clear from the study what the excess flare rate was, or whether the flares were mild or severe. Most flares in patients with SLE are mild, consisting of skin and joint manifestations, while only a few patients have flares in kidney or neurologic manifestations.”
Dr. Gladman described her approach to tapering: “We aim for our patients to be taking no more than 5 mg of prednisone and to be in at least clinical remission with a SLEDAI-2K of 0 for at least 2 years before we would taper to glucocorticoids withdrawal. We always withdraw glucocorticoids first and immunosuppressives later, and keep patients on antimalarials the longest, unless there are specific side effects to the immunosuppressive or antimalarials which require their cessation earlier.”
Uncertainty persists
Other SLE experts weighing in confirmed the view that future research should aim to achieve clarity about the relative risks and benefits of tapering SLE drug regimens to maintain disease remission while minimizing potential for organ damage.
“Steroids are our friend and our enemy,” Joan T. Merrill, MD, professor of medicine at the University of Oklahoma Health Sciences Center, Oklahoma City, said in an interview. “If a person with lupus is in a lot of trouble, corticosteroids are almost universally a good option to get them out. But for too many decades, for too many patients, despite all the improvements we have made in better understanding the disease and developing some promising new treatments, we have yet to shed the inexorable toxicity in multiple organs of steroid dependence.” She continued, “Corticosteroids, even at low dose, may have broad-spectrum effects. But, in fact, so do many of the more ‘targeted’ agents. If all patients were lined up at the beginning of a study while being given azathioprine or a calcineurin inhibitor or belimumab at a stable, tolerable dose, you might see the same data if you tapered that agent down. What we really need is improved individualized guidance about when and how fast to remove immune modulators from stable patients with lupus without disturbing the balance that had been achieved in such a quiescent patient.”
That enduring uncertainty was echoed by Daniel J. Wallace, MD, professor of medicine at Cedars-Sinai Medical Center, Los Angeles: “The take-home message from this interesting paper,” he commented, “is that current lupus biomarkers are not adequate. They do not guide the practitioner well enough, so that all too often medication regimens are tapered even though the risks are not really well known. Also, there is evidence in the literature that fibrosis and ‘damage’ progress even if acute phase reactants such as sedimentation rate, [C-reactive protein], complement 3 and 4, and anti-dsDNA are normal. We don’t have a good metric to detect them.”
Dr. Cho and colleagues’ study was funded by AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Janssen, Merck Serono, GlaxoSmithKline, and UCB. Dr. Gladman disclosed consulting and/or research support from AbbVie, Amgen, Bristol-Myers Squibb, Eli Lilly, Janssen, Novartis, Pfizer, and UCB.
The question looms large for patients with stable systemic lupus erythematosus (SLE): to taper or not to taper corticosteroids or immunosuppressive therapy? For patients and the physicians treating them, the evidence points in both directions. Flares are exacerbated by tapering, but simultaneously organ damage is tempered. Where is the balance? What competing factors together inform decision-making?
A recent multinational, observational cohort study conducted by Jiacai Cho, MBBS, of National University Hospital, Singapore, and colleagues, and published in The Lancet Rheumatology concluded that, given the odds of excess flares associated with tapering of corticosteroids and immunosuppressive therapy in patients with stable SLE, drug tapering warrants careful consideration of risks and benefits and is best reserved for those in complete clinical and serological remission with stable disease for at least 6 months. However, in an accompanying editorial, Yann Nguyen, MD, MPH, and Nathalie Costedoat-Chalumeau, MD, PhD, of the National Referral Center for Rare Autoimmune and Systemic Diseases at Cochin Hospital, Paris, and the Center for Research in Epidemiology and Statistics at Paris City University, argued for tipping the scale back from some of those expressed cautions.
In interviews, experts in the field expressed both strong appreciation for the cohort study and, like the editorialists, cognizance of its limitations.
Dr. Cho and colleagues recruited 3,002 adult patients with SLE (92.2% female, median age 39.5 years), from 25 sites across 13 Asia-Pacific countries. They were receiving routine clinical care and had achieved stable disease in at least one of two or more visits. Stable disease was defined by meeting criteria for Lupus Low Disease Activity State (LLDAS; SLE Disease Activity Index 2000 [SLEDAI-2K] score ≤ 4, Physician Global Assessment [PGA] ≤ 1, and prednisolone ≤ 7.5 mg/day), the 2021 DORIS definition of remission (clinical SLEDAI-2K score 0, PGA score < 0.5, and prednisolone dose ≤ 5 mg/day), or DORIS complete remission on therapy (SLEDAI-2K score 0, PGA score < 0.5, and prednisolone dose ≤ 5 mg/day). Any decrease in dose of corticosteroids or immunosuppressive therapy (mycophenolate mofetil, calcineurin inhibitors, azathioprine, leflunomide, or methotrexate) defined tapering. The investigators compared the odds of disease flares (SELENA-SLEDAI Flare Index) at the visit following tapering among those with tapering versus those who had continued the same drug doses.
Higher odds of flare with tapering
Tapering, compared with continuing with the same dose, was clearly associated with higher odds of flare at the next visit (11.4% with continuing vs. 17.0% with tapering; odds ratio, 1.24; 95% confidence interval, 1.10-1.39; P = .0005). Flares among patients who tapered were also slightly more often severe than with continuing the same dose (21.5% of flares vs. 19.7%). The level of remission at the time of tapering also mattered. Of 2,095 continuous tapering attempts, 860 (41.1%) were initiated in LLDAS, 596 (28.4%) in remission, and 639 (30.5%) in complete remission. Tapering when in LLDAS or remission, compared with complete remission, was associated with a higher likelihood of flare by 1 year (LLDAS: OR, 1.37; 95% CI, 1.03-1.81; P = .029; and remission: OR, 1.45; 95% CI, 1.08-1.94; P = .013). Time to first flare followed the same pattern. Also, sustained LLDAS, remission, or complete remission for at least 6 months just before the time of taper was associated with lower odds of flare at next visit and flares in 1 year, and longer time to flare.
Take baseline disease status, hydroxychloroquine’s effect into account
Dr. Nguyen and Dr. Costedoat-Chalumeau underscored several factors that may soften the risk for flares seen with tapering. They pointed to higher baseline doses of prednisone and immunosuppressants (and thus likely more severe disease that is more likely to flare) in the patients with tapering. Also, the SELENA-SLEDAI Flare Index used in the study classifies some clinically insignificant flares as mild to moderate and ignores the benefit of tapering. (It classifies patients as having a severe flare even when starting a new immunosuppressant prescription, such as azathioprine, methotrexate, or both, in an effort to reduce corticosteroid use.) They wrote that the study did not assess the rate of clinically meaningful flares (“essentially renal flares”), nor did it highlight that the “tiny” increase in absolute risk of severe flares (from 2.2% to 3.7%) could be further contextualized by the offset of the smaller, unmeasured rate of clinically significant flares and the “extremely relevant” risk of concomitant damage from prolonged treatment.
Dr. Nguyen and Dr. Costedoat-Chalumeau urged hydroxychloroquine use for all patients unless clearly contraindicated. In their own research, they have detailed hydroxychloroquine benefits in reducing not only flare risk, but also comorbidities, damage, and mortality. In the current study, the prevalence of hydroxychloroquine use in all the patient visits was only 63.3%. “We can assume that if more patients had been treated with hydroxychloroquine, both the number of flares and the difference between the two strategies would have been lower,” they wrote. They cited findings from a study of patients in remission for 2 years or longer in the Toronto Lupus Cohort in which a gradual taper of corticosteroids over 1 year was safe and feasible and resulted in less damage accrual at 24 months than not tapering. Optimizing tapering can minimize flare risk, they concluded.
Tapering SLE medications always involves some chance of flare and has to be considered a calculated risk, Sasha Bernatsky, MD, the James McGill professor of medicine in the division of rheumatology at McGill University, Montreal, said in an interview. “Long-term prednisone is not good for patients. I have heard it called ‘the miracle drug from hell’ – meaning that, yes, it controls disease, but at a cost of long-term complications. So we must be conscientious about tapering prednisone.” She observed that in the short-term, there may not be a huge risk to keeping a patient on an antimalarial and counseling patients to stay on it because their risk of flare is higher if they taper. Rheumatologists usually agree, however, that after 10 years or more, there is a real chance of retinal toxicity. “In our Montreal cohort, the risk of retinal toxicity was 5% after an average of 12.8 years of antimalarial use. My concern is that if a patient develops SLE in their 20s, how do we decide if we should keep them on an antimalarial for the next 60 or 70 years? If we keep them on the drug from age 25 to 45, and they then get retinal toxicity, they would essentially never be able to be on the drug again. So I do try to keep patients on the lowest dose of an antimalarial that is possible.”
Dr. Bernatsky pointed out further, “We think about tapering other immunosuppressants (such as methotrexate or mycophenolate or azathioprine) quite differently than prednisone tapering. We take our time a bit more, since many patients will tolerate being on standard doses of these drugs fairly well. If or when we do consider tapering these drugs, both our intuition and the literature suggests that someone with worse baseline disease activity or severity, who has needed a lot of steroids and multiple combinations of drugs to control disease, has a higher chance of flaring than someone with milder disease. As the editorial points out, lupus physicians (and their patients) need to think carefully about the patient’s risk profile, and be sure to tailor follow-up based on flare risk.”
Frank discussions with patients about the risks of tapering are needed, she said. “On one hand, there is consensus about how some aspects of lupus should be managed (for example, aggressive treatment of severe nephritis), but on the other hand, when it comes to long-term management and especially discussing tapering, we must have good discussions with patients. When a patient asks if they can taper a drug – many just lower or stop their drugs without asking – I am as honest as I can be, but ultimately have to admit any taper could be associated with a flare. It’s helpful to have actual figures to discuss with patients.”
No surprises
“This is an interesting study, which did not produce any surprises,” Dafna D. Gladman, MD, professor of medicine at University of Toronto and senior scientist at the university’s Schroeder Arthritis Institute, said when asked to comment. “We already knew from previous studies that abrupt withdrawal is not a good idea, and that if you taper when a patient is under conditions of remission, the rate of flare is actually lower than the usual rate of flare that occurs in people who continue on these medications. But the major limitation is that they did not specifically look at those who we would taper in clinical practice. In addition, they do not specify that the patients had to be on low-dose glucocorticoids before tapering, and they combined both immunosuppressive and steroids. It is not clear from the study what the excess flare rate was, or whether the flares were mild or severe. Most flares in patients with SLE are mild, consisting of skin and joint manifestations, while only a few patients have flares in kidney or neurologic manifestations.”
Dr. Gladman described her approach to tapering: “We aim for our patients to be taking no more than 5 mg of prednisone and to be in at least clinical remission with a SLEDAI-2K of 0 for at least 2 years before we would taper to glucocorticoids withdrawal. We always withdraw glucocorticoids first and immunosuppressives later, and keep patients on antimalarials the longest, unless there are specific side effects to the immunosuppressive or antimalarials which require their cessation earlier.”
Uncertainty persists
Other SLE experts weighing in confirmed the view that future research should aim to achieve clarity about the relative risks and benefits of tapering SLE drug regimens to maintain disease remission while minimizing potential for organ damage.
“Steroids are our friend and our enemy,” Joan T. Merrill, MD, professor of medicine at the University of Oklahoma Health Sciences Center, Oklahoma City, said in an interview. “If a person with lupus is in a lot of trouble, corticosteroids are almost universally a good option to get them out. But for too many decades, for too many patients, despite all the improvements we have made in better understanding the disease and developing some promising new treatments, we have yet to shed the inexorable toxicity in multiple organs of steroid dependence.” She continued, “Corticosteroids, even at low dose, may have broad-spectrum effects. But, in fact, so do many of the more ‘targeted’ agents. If all patients were lined up at the beginning of a study while being given azathioprine or a calcineurin inhibitor or belimumab at a stable, tolerable dose, you might see the same data if you tapered that agent down. What we really need is improved individualized guidance about when and how fast to remove immune modulators from stable patients with lupus without disturbing the balance that had been achieved in such a quiescent patient.”
That enduring uncertainty was echoed by Daniel J. Wallace, MD, professor of medicine at Cedars-Sinai Medical Center, Los Angeles: “The take-home message from this interesting paper,” he commented, “is that current lupus biomarkers are not adequate. They do not guide the practitioner well enough, so that all too often medication regimens are tapered even though the risks are not really well known. Also, there is evidence in the literature that fibrosis and ‘damage’ progress even if acute phase reactants such as sedimentation rate, [C-reactive protein], complement 3 and 4, and anti-dsDNA are normal. We don’t have a good metric to detect them.”
Dr. Cho and colleagues’ study was funded by AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Janssen, Merck Serono, GlaxoSmithKline, and UCB. Dr. Gladman disclosed consulting and/or research support from AbbVie, Amgen, Bristol-Myers Squibb, Eli Lilly, Janssen, Novartis, Pfizer, and UCB.
FROM THE LANCET RHEUMATOLOGY
Lupus may overlap in many patients with systemic sclerosis
TOPLINE:
Patients with both systemic sclerosis (SSc) and systemic lupus erythematosus (SLE) are more likely to be female, Black, and diagnosed with limited cutaneous SSc.
METHODOLOGY:
- Researchers used the 2019 SLE classification criteria from the European Alliance of Associations for Rheumatology and American College of Rheumatology to identify patients with SSc who also met criteria for SLE at a single academic center.
- The study population included 402 adults with SSc.
- The researchers compared demographics, laboratory data, clinical features, and mortality between patients with SSc-SLE and patients with SSc only.
TAKEAWAY:
- Among the 402 patients with SSc who were analyzed, 40 (10%) met the 2019 EULAR/ACR Classification Criteria for SLE.
- Patients with both SSc and SLE were significantly more likely to be female and Black, which is consistent with previous studies; patients with both conditions also were more likely than those with SSc alone to have limited cutaneous SSc (75% vs. 52.2%; P = .006).
- The prevalence of anti-U1-RNP antibody positivity, a classic marker for mixed connective tissue disease, was 30% in SSc-SLE patients and 6.6% in those with SSc only (P < .001).
- Mortality was similar between the two groups, and similar rates were also seen between the two for severe SSc-related end-organ damage, including pulmonary fibrosis, pulmonary hypertension, and scleroderma renal crisis.
IN PRACTICE:
The results highlight the need for clinicians to recognize the SSc-SLE overlap syndrome and to watch for scleroderma organ involvement in patients with features of SLE, Raynaud syndrome, anti-U1-RNP antibody positivity, or an isolated nucleolar pattern of antinuclear antibodies.
SOURCE:
First author Ronald D. Bass, MD, MBA, of Georgetown University, Washington, and colleagues published their report online in Arthritis Care & Research.
LIMITATIONS:
The primary cohort was designed to compare Black to non-Black patients with SSc, and the process of matching these patients may have introduced unmeasured selection bias. Also, since the study was based on classification criteria and not diagnostic criteria, the overlapping patients may not reflect patients with true overlapping of both conditions.
DISCLOSURES:
No outside funding source was listed by the authors. The researchers report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
TOPLINE:
Patients with both systemic sclerosis (SSc) and systemic lupus erythematosus (SLE) are more likely to be female, Black, and diagnosed with limited cutaneous SSc.
METHODOLOGY:
- Researchers used the 2019 SLE classification criteria from the European Alliance of Associations for Rheumatology and American College of Rheumatology to identify patients with SSc who also met criteria for SLE at a single academic center.
- The study population included 402 adults with SSc.
- The researchers compared demographics, laboratory data, clinical features, and mortality between patients with SSc-SLE and patients with SSc only.
TAKEAWAY:
- Among the 402 patients with SSc who were analyzed, 40 (10%) met the 2019 EULAR/ACR Classification Criteria for SLE.
- Patients with both SSc and SLE were significantly more likely to be female and Black, which is consistent with previous studies; patients with both conditions also were more likely than those with SSc alone to have limited cutaneous SSc (75% vs. 52.2%; P = .006).
- The prevalence of anti-U1-RNP antibody positivity, a classic marker for mixed connective tissue disease, was 30% in SSc-SLE patients and 6.6% in those with SSc only (P < .001).
- Mortality was similar between the two groups, and similar rates were also seen between the two for severe SSc-related end-organ damage, including pulmonary fibrosis, pulmonary hypertension, and scleroderma renal crisis.
IN PRACTICE:
The results highlight the need for clinicians to recognize the SSc-SLE overlap syndrome and to watch for scleroderma organ involvement in patients with features of SLE, Raynaud syndrome, anti-U1-RNP antibody positivity, or an isolated nucleolar pattern of antinuclear antibodies.
SOURCE:
First author Ronald D. Bass, MD, MBA, of Georgetown University, Washington, and colleagues published their report online in Arthritis Care & Research.
LIMITATIONS:
The primary cohort was designed to compare Black to non-Black patients with SSc, and the process of matching these patients may have introduced unmeasured selection bias. Also, since the study was based on classification criteria and not diagnostic criteria, the overlapping patients may not reflect patients with true overlapping of both conditions.
DISCLOSURES:
No outside funding source was listed by the authors. The researchers report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
TOPLINE:
Patients with both systemic sclerosis (SSc) and systemic lupus erythematosus (SLE) are more likely to be female, Black, and diagnosed with limited cutaneous SSc.
METHODOLOGY:
- Researchers used the 2019 SLE classification criteria from the European Alliance of Associations for Rheumatology and American College of Rheumatology to identify patients with SSc who also met criteria for SLE at a single academic center.
- The study population included 402 adults with SSc.
- The researchers compared demographics, laboratory data, clinical features, and mortality between patients with SSc-SLE and patients with SSc only.
TAKEAWAY:
- Among the 402 patients with SSc who were analyzed, 40 (10%) met the 2019 EULAR/ACR Classification Criteria for SLE.
- Patients with both SSc and SLE were significantly more likely to be female and Black, which is consistent with previous studies; patients with both conditions also were more likely than those with SSc alone to have limited cutaneous SSc (75% vs. 52.2%; P = .006).
- The prevalence of anti-U1-RNP antibody positivity, a classic marker for mixed connective tissue disease, was 30% in SSc-SLE patients and 6.6% in those with SSc only (P < .001).
- Mortality was similar between the two groups, and similar rates were also seen between the two for severe SSc-related end-organ damage, including pulmonary fibrosis, pulmonary hypertension, and scleroderma renal crisis.
IN PRACTICE:
The results highlight the need for clinicians to recognize the SSc-SLE overlap syndrome and to watch for scleroderma organ involvement in patients with features of SLE, Raynaud syndrome, anti-U1-RNP antibody positivity, or an isolated nucleolar pattern of antinuclear antibodies.
SOURCE:
First author Ronald D. Bass, MD, MBA, of Georgetown University, Washington, and colleagues published their report online in Arthritis Care & Research.
LIMITATIONS:
The primary cohort was designed to compare Black to non-Black patients with SSc, and the process of matching these patients may have introduced unmeasured selection bias. Also, since the study was based on classification criteria and not diagnostic criteria, the overlapping patients may not reflect patients with true overlapping of both conditions.
DISCLOSURES:
No outside funding source was listed by the authors. The researchers report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Hydroxychloroquine blood level ‘sweet spot’ may maximize efficacy in lupus
A blood-level reference range of 750-1,200 ng/mL of hydroxychloroquine (HCQ) has been linked with 71% lower odds of active lupus, new research suggests.
Researchers, led by Shivani Garg, MD, assistant professor of rheumatology at the University of Wisconsin–Madison, also found that maintaining levels within that range lowered the odds for flares by 26% over 9 months of follow-up.
The findings, published in Arthritis Care & Research, could help clinicians personalize HCQ doses to maximize efficacy for each patient.

HCQ levels in whole blood and the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) were measured during a baseline visit and again during a routine follow-up visit.
Among 158 baseline patient visits, 19% of the patients had active lupus. Researchers longitudinally followed 42 patients using convenience sampling, and among those patients, 7 (17%) had flares at the follow-up visit.
Michelle Petri, MD, MPH, director of the Johns Hopkins Lupus Center in Baltimore, called the findings that suggest upper and lower efficacy and safety boundaries “very important.”
The findings highlight that guidelines for dosing don’t match efficacy needs, said Dr. Petri, who was not involved with the study.
“HCQ dosing has been under threat by guidelines insisting that the dose should be < 5 mg/kg even though this does not correlate with efficacy,” she said. “Basically, if we dose too low, the patient loses efficacy. If we dose too high, the risk of retinopathy increases, so this paper hones down the sweet spot.”
A 2014 study identified a higher eye toxicity risk with HCQ doses > 5 mg/kg per day, and the American Academy of Ophthalmology followed with guidelines for HCQ retinopathy screening that recommended reducing HCQ to ≤ 5 mg/kg per day.
Dr. Petri said that the range Dr. Garg and colleagues identified corroborates findings in one of her team’s studies.
That paper showed that thrombotic events dropped by 69% in patients with average HCQ blood levels ≥ 1,068 ng/mL vs. those with levels < 648 ng/mL (relative risk, 0.31; 95% confidence interval, 0.11-0.86; P = .024).
Dr. Garg and colleagues write that current lupus treatment guidelines do not universally recommend blood level monitoring for HCQ “as different cut-points have been used to define therapeutic HCQ blood levels and an effective range of HCQ levels with upper and lower bounds for efficacy has not been extensively examined.”
When to start checking levels
Blood levels of HCQ can be checked for any patient, although 1-3 months after starting the medication may be best to get steady levels, Dr. Garg told this news organization.
Dr. Petri said that she recommends HCQ whole blood levels be checked routinely for maximum dosing efficacy “but also to identify patients who are missing so many doses that they are subtherapeutic.”
She noted that nonadherence is a major issue among patients with systemic lupus erythematosus, especially among those who are younger and newly diagnosed.
Dr. Garg and Dr. Petri both said that insurance does not automatically cover the costs of checking HCQ levels in the blood, which has been a consistent frustration in the field.
“Having more data validates the reason to do it,” Dr. Garg said.
She added that “HCQ blood levels are still not done routinely in all patients, and at times the test needs to be sent to outside laboratories.”
Importance for patients with CKD
Many patient factors can affect how the body absorbs HCQ, Dr. Garg said, so finding the right level that is safe and maximizes benefit individually is important.
The findings are particularly important for patients with chronic kidney disease (CKD) of stage 3 or higher, Dr. Garg said.
The authors write that because kidneys clear more than half of all HCQ, impaired kidney function could boost HCQ blood levels, risking toxicity.
“Our study found a sixfold higher odds of having supratherapeutic HCQ blood levels in patients with CKD stage ≥ 3,” they write.
Dr. Garg added that if blood levels cannot be analyzed in all patients, they could be prioritized in patients with CKD stage 3 or above because these patients are at “higher risk of being underdosed with arbitrary reductions in HCQ doses and carry higher risk of toxicity if HCQ doses are not adjusted.”
More research will uncover other high-risk groups who would benefit most from close monitoring of HCQ blood levels, she said.
The study was supported by an award from the University of Wisconsin–Madison, and by an award to the institution from the National Institutes of Health National Center for Advancing Translational Sciences. Dr. Garg and coauthors as well as Dr. Petri report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A blood-level reference range of 750-1,200 ng/mL of hydroxychloroquine (HCQ) has been linked with 71% lower odds of active lupus, new research suggests.
Researchers, led by Shivani Garg, MD, assistant professor of rheumatology at the University of Wisconsin–Madison, also found that maintaining levels within that range lowered the odds for flares by 26% over 9 months of follow-up.
The findings, published in Arthritis Care & Research, could help clinicians personalize HCQ doses to maximize efficacy for each patient.
HCQ levels in whole blood and the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) were measured during a baseline visit and again during a routine follow-up visit.
Among 158 baseline patient visits, 19% of the patients had active lupus. Researchers longitudinally followed 42 patients using convenience sampling, and among those patients, 7 (17%) had flares at the follow-up visit.
Michelle Petri, MD, MPH, director of the Johns Hopkins Lupus Center in Baltimore, called the findings that suggest upper and lower efficacy and safety boundaries “very important.”
The findings highlight that guidelines for dosing don’t match efficacy needs, said Dr. Petri, who was not involved with the study.
“HCQ dosing has been under threat by guidelines insisting that the dose should be < 5 mg/kg even though this does not correlate with efficacy,” she said. “Basically, if we dose too low, the patient loses efficacy. If we dose too high, the risk of retinopathy increases, so this paper hones down the sweet spot.”
A 2014 study identified a higher eye toxicity risk with HCQ doses > 5 mg/kg per day, and the American Academy of Ophthalmology followed with guidelines for HCQ retinopathy screening that recommended reducing HCQ to ≤ 5 mg/kg per day.
Dr. Petri said that the range Dr. Garg and colleagues identified corroborates findings in one of her team’s studies.
That paper showed that thrombotic events dropped by 69% in patients with average HCQ blood levels ≥ 1,068 ng/mL vs. those with levels < 648 ng/mL (relative risk, 0.31; 95% confidence interval, 0.11-0.86; P = .024).
Dr. Garg and colleagues write that current lupus treatment guidelines do not universally recommend blood level monitoring for HCQ “as different cut-points have been used to define therapeutic HCQ blood levels and an effective range of HCQ levels with upper and lower bounds for efficacy has not been extensively examined.”
When to start checking levels
Blood levels of HCQ can be checked for any patient, although 1-3 months after starting the medication may be best to get steady levels, Dr. Garg told this news organization.
Dr. Petri said that she recommends HCQ whole blood levels be checked routinely for maximum dosing efficacy “but also to identify patients who are missing so many doses that they are subtherapeutic.”
She noted that nonadherence is a major issue among patients with systemic lupus erythematosus, especially among those who are younger and newly diagnosed.
Dr. Garg and Dr. Petri both said that insurance does not automatically cover the costs of checking HCQ levels in the blood, which has been a consistent frustration in the field.
“Having more data validates the reason to do it,” Dr. Garg said.
She added that “HCQ blood levels are still not done routinely in all patients, and at times the test needs to be sent to outside laboratories.”
Importance for patients with CKD
Many patient factors can affect how the body absorbs HCQ, Dr. Garg said, so finding the right level that is safe and maximizes benefit individually is important.
The findings are particularly important for patients with chronic kidney disease (CKD) of stage 3 or higher, Dr. Garg said.
The authors write that because kidneys clear more than half of all HCQ, impaired kidney function could boost HCQ blood levels, risking toxicity.
“Our study found a sixfold higher odds of having supratherapeutic HCQ blood levels in patients with CKD stage ≥ 3,” they write.
Dr. Garg added that if blood levels cannot be analyzed in all patients, they could be prioritized in patients with CKD stage 3 or above because these patients are at “higher risk of being underdosed with arbitrary reductions in HCQ doses and carry higher risk of toxicity if HCQ doses are not adjusted.”
More research will uncover other high-risk groups who would benefit most from close monitoring of HCQ blood levels, she said.
The study was supported by an award from the University of Wisconsin–Madison, and by an award to the institution from the National Institutes of Health National Center for Advancing Translational Sciences. Dr. Garg and coauthors as well as Dr. Petri report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A blood-level reference range of 750-1,200 ng/mL of hydroxychloroquine (HCQ) has been linked with 71% lower odds of active lupus, new research suggests.
Researchers, led by Shivani Garg, MD, assistant professor of rheumatology at the University of Wisconsin–Madison, also found that maintaining levels within that range lowered the odds for flares by 26% over 9 months of follow-up.
The findings, published in Arthritis Care & Research, could help clinicians personalize HCQ doses to maximize efficacy for each patient.
HCQ levels in whole blood and the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) were measured during a baseline visit and again during a routine follow-up visit.
Among 158 baseline patient visits, 19% of the patients had active lupus. Researchers longitudinally followed 42 patients using convenience sampling, and among those patients, 7 (17%) had flares at the follow-up visit.
Michelle Petri, MD, MPH, director of the Johns Hopkins Lupus Center in Baltimore, called the findings that suggest upper and lower efficacy and safety boundaries “very important.”
The findings highlight that guidelines for dosing don’t match efficacy needs, said Dr. Petri, who was not involved with the study.
“HCQ dosing has been under threat by guidelines insisting that the dose should be < 5 mg/kg even though this does not correlate with efficacy,” she said. “Basically, if we dose too low, the patient loses efficacy. If we dose too high, the risk of retinopathy increases, so this paper hones down the sweet spot.”
A 2014 study identified a higher eye toxicity risk with HCQ doses > 5 mg/kg per day, and the American Academy of Ophthalmology followed with guidelines for HCQ retinopathy screening that recommended reducing HCQ to ≤ 5 mg/kg per day.
Dr. Petri said that the range Dr. Garg and colleagues identified corroborates findings in one of her team’s studies.
That paper showed that thrombotic events dropped by 69% in patients with average HCQ blood levels ≥ 1,068 ng/mL vs. those with levels < 648 ng/mL (relative risk, 0.31; 95% confidence interval, 0.11-0.86; P = .024).
Dr. Garg and colleagues write that current lupus treatment guidelines do not universally recommend blood level monitoring for HCQ “as different cut-points have been used to define therapeutic HCQ blood levels and an effective range of HCQ levels with upper and lower bounds for efficacy has not been extensively examined.”
When to start checking levels
Blood levels of HCQ can be checked for any patient, although 1-3 months after starting the medication may be best to get steady levels, Dr. Garg told this news organization.
Dr. Petri said that she recommends HCQ whole blood levels be checked routinely for maximum dosing efficacy “but also to identify patients who are missing so many doses that they are subtherapeutic.”
She noted that nonadherence is a major issue among patients with systemic lupus erythematosus, especially among those who are younger and newly diagnosed.
Dr. Garg and Dr. Petri both said that insurance does not automatically cover the costs of checking HCQ levels in the blood, which has been a consistent frustration in the field.
“Having more data validates the reason to do it,” Dr. Garg said.
She added that “HCQ blood levels are still not done routinely in all patients, and at times the test needs to be sent to outside laboratories.”
Importance for patients with CKD
Many patient factors can affect how the body absorbs HCQ, Dr. Garg said, so finding the right level that is safe and maximizes benefit individually is important.
The findings are particularly important for patients with chronic kidney disease (CKD) of stage 3 or higher, Dr. Garg said.
The authors write that because kidneys clear more than half of all HCQ, impaired kidney function could boost HCQ blood levels, risking toxicity.
“Our study found a sixfold higher odds of having supratherapeutic HCQ blood levels in patients with CKD stage ≥ 3,” they write.
Dr. Garg added that if blood levels cannot be analyzed in all patients, they could be prioritized in patients with CKD stage 3 or above because these patients are at “higher risk of being underdosed with arbitrary reductions in HCQ doses and carry higher risk of toxicity if HCQ doses are not adjusted.”
More research will uncover other high-risk groups who would benefit most from close monitoring of HCQ blood levels, she said.
The study was supported by an award from the University of Wisconsin–Madison, and by an award to the institution from the National Institutes of Health National Center for Advancing Translational Sciences. Dr. Garg and coauthors as well as Dr. Petri report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM ARTHRITIS CARE & RESEARCH
Sarcoidosis in Post–9/11 Military Veterans
Sarcoidosis is a chronic inflammatory disease characterized by noncaseating granulomas that can affect many organ systems, most commonly the lungs and skin, with cutaneous involvement in 25% to 30% of patients in the United States.1 The etiology of sarcoidosis largely is unknown and likely is multifactorial; however, specific environmental, infectious, and pharmaceutical triggers may contribute to its pathogenesis. Sarcoidosis secondary to occupational exposures in US Military veterans historically has been discussed and investigated. Still, it was not considered a service-connected disability until the passing of the Promise to Address Comprehensive Toxics (PACT) Act2 in 2022. In this article, we review the risk factors and incidence of sarcoidosis in post–9/11 veterans as well as provide recommendations for managing presumptive service-connected sarcoidosis covered under the recently enacted PACT Act.
The PACT Act and Post–9/11 Military Veterans
Veterans of Operation Iraqi Freedom (OIF) and Operation Enduring Freedom (OEF) have a history of occupational exposures to open-air burn pits, gun smoke, and recurrent high-intensity sandstorms that may cause chronic disease.3 Burn pits, which were used to dispose of solid waste on forward operating bases, released antigenic particulate matter that was detectable on air sampling.4,5 Increased respiratory disease rates in veterans that were deployed post–9/11 are well documented, but a causal relationship has not been established.6 Although burn pits cannot be directly associated with any disease at this time,5 veterans with assumed exposures can now receive a Veterans Affairs (VA) Disability Rating for presumptive conditions under the PACT Act.2 The major points of this legislation include expanding and extending eligibility for veterans with toxic exposures, providing access to toxic exposure screening for all veterans receiving VA health care, and increasing research related to toxic exposures in US servicemembers. The PACT Act expands health care benefits, making it easier for veterans exposed post–9/11 to receive coverage for 24 new presumptive diagnoses.2 Of these diagnoses, several are relevant to the practicing dermatologist. Patients with metastasis of primary cancers to the skin as well as melanoma or sarcoidosis may be eligible for coverage depending on the location and time of service. The Table lists service locations where the VA has determined servicemembers may have been exposed to burn pits or other toxins. Servicemembers with a presumptive diagnosis who served in these locations may be eligible for care under the PACT Act. Sarcoidosis is of particular concern due to its increased incidence and prevalence in military veterans compared to civilian populations. An analysis of more than 13 million veterans who received health care benefits through the Veterans Health Administration in 2019 found an annual incidence of sarcoidosis of 52 cases per 100,000 person-years and an annual prevalence of 141 cases per 100,000 individuals.7 In contrast, the United States has a reported annual incidence of sarcoidosis of 4.9 cases per 100,000 person-years and an annual prevalence of 60 cases per 100,000 individuals.8 Although the increased rates of sarcoidosis in veterans have been noted for decades, only recently have investigations provided insights into the etiology of sarcoidosis in this population.

Sarcoidosis and Environmental Factors
Sarcoidosis is a multisystem granulomatous inflammatory disease that can present in any organ system9; however, it most commonly affects the lungs, skin, and eyes—all of which are subjected to direct contact with environmental toxins. The cause of sarcoidosis is unknown, but environmental exposures are theorized to play a role.9,10 It has been hypothesized that exposure to various immunologically active triggers may invoke the granulomatous inflammatory response that characterizes the disease.11 The World Trade Center disaster on 9/11 has provided insight into the potential environmental component of sarcoidosis. Firefighters who spent extensive amounts of time at the World Trade Center site experienced intense exposure to inorganic particulate matter; it was later found that there was a marked increase in the incidence of sarcoidosis or sarcoidosislike granulomatous pulmonary disease in exposed firefighters. It has been speculated that the elevated exposure to potentially antigenic particulates may have induced granulomatous inflammation, resulting in the manifestation of the disease.12 Other known occupational exposures associated with an increased risk for sarcoidosis or sarcoidosislike illness include mold, silicates, metal dust, and microbial contaminants.11 Servicemembers commonly are exposed to several of these aerosolized toxins, which theoretically could increase their risk for developing sarcoidosis.
Sarcoidosis in the Military
Servicemembers historically have faced unique environmental hazards that may increase their risk for developing sarcoidosis. Studies of naval veterans have shown relationships between occupational location and increased rates of sarcoidosis. Sailors assigned to aircraft carriers with nonskid coatings containing particulate matter such as aluminum, titanium, and silicates had a higher prevalence of sarcoidosis than those stationed on “clean” ships.13,14 Although no one trigger was identified, the increased rates of sarcoidosis in populations with extensive exposure to toxins raise concern for the possibility of occupationally induced sarcoidosis in post–9/11 veterans.
Environmental exposures during OIF and OEF may be associated with sarcoidosis. A retrospective review of lung biopsy data collected from Department of Defense military treatment facilities was conducted to identify associations between lung disease and deployment to the Middle East.15 The study included 391 military patients divided into deployed and nondeployed groups undergoing lung biopsies for various reasons from 2005 to 2012. An analysis of the reported lung histology showed an increased frequency of nonnecrotizing granulomas in those with a history of deployment to the Middle East compared to those who had never been deployed. Development of disease was not associated with confounding factors such as age, ethnicity, sex, or tobacco use, raising suspicion about similar shared toxic exposures among deployed servicemembers.15 A 2020 study of sarcoidosis in active-duty military personnel reported that the incidence of observed cases was 2-times those seen in civilian Department of Defense employees from 2005 to 2010; however, data collected in this study did not indicate an increased risk for developing sarcoidosis based on deployment to the Middle East. Still, the higher prevalence of sarcoidosis in active-duty military personnel suggests similar shared exposures in this group.16
Identification of exposures that may potentially trigger sarcoidosis is difficult due to many confounding variables; however, the Airborne Hazards and Open Burn Pit Registry questionnaire has been used to extrapolate prospective hazards of concern. Results from the questionnaire identified that only veterans exposed to convoy activity had a statistically significant (odds ratio, 1.16; 95% CI, 1.00-1.35; P=.046) increased risk for developing sarcoidosis.17 Interestingly, enlisted personnel had a higher rate of sarcoidosis than officers, comprising upwards of 78% of cases in the Military Health System from 2004 to 2013.9 This finding requires further study, but increased exposure to toxins due to occupational specialty may be the cause.
Veterans with sarcoidosis may have a unique pathophysiology, which may point to occupational exposure. Studies show that affected veterans have unique plasma metabolites and metal ions compared to civilians, with lower anti-inflammatory amino acid concentrations and downregulated GABA synthesis. The environmental exposures in OIF and OEF may have primed deployed servicemembers to develop a distinct subtype of sarcoidosis.3 Overall, there is a dearth of literature on post–9/11 veterans with sarcoidosis; therefore, further investigation is necessary to determine the actual risk for developing the disease following exposures related to military service.
Clinical Presentation and Diagnosis
Cutaneous sarcoidosis protean morphology is considered an imitator of many other skin diseases. The most common sarcoidosis-specific skin lesions include papules and papulonodules (Figure, A), lupus pernio (Figure, B), plaques (Figure, C), and subcutaneous nodules. Lesions typically present on the face, neck, trunk, and extremities and are associated with a favorable prognosis. Lupus pernio presents as centrofacial, bluish-red or violaceous nodules and can be disfiguring (Figure, B). Subcutaneous nodules occur in the subcutaneous tissue or deep dermis with minimal surface changes. Sarcoidal lesions also can occur at sites of scar tissue or trauma, within tattoos, and around foreign bodies. Other uncommon sarcoidosis-specific skin lesions include ichthyosiform, hypopigmented, atrophic, ulcerative and mucosal lesions; erythroderma; alopecia; and nail sarcoidosis.18

When cutaneous sarcoidosis is suspected, the skin serves as an easily accessible organ for biopsy to confirm the diagnosis.1 Sarcoidosis-specific skin lesions are histologically characterized as sarcoidal granulomas with a classic noncaseating naked appearance comprised of epithelioid histocytes with giant cells amidst a mild lymphocytic inflammatory infiltrate. Nonspecific sarcoidosis skin lesions do not contain characteristic noncaseating granulomas. Erythema nodosum is the most common nonspecific lesion and is associated with a favorable prognosis. Other nonspecific sarcoidosis skin findings include calcinosis cutis, clubbing, and vasculitis.18
Workup
Due to the systemic nature of sarcoidosis, dermatologists should initiate a comprehensive workup upon diagnosis of cutaneous sarcoidosis, which should include the following: a complete in-depth history, including occupational/environmental exposures; a complete review of systems; a military history, including time of service and location of deployments; physical examination; pulmonary function test; high-resolution chest computed tomography19; pulmonology referral for additional pulmonary function tests, including diffusion capacity for carbon monoxide and 6-minute walk test; ophthalmology referral for full ophthalmologic examination; initial cardiac screening with electrocardiogram; and a review of symptoms including assessment of heart palpitations. Any abnormalities should prompt cardiology referral for evaluation of cardiac involvement with a workup that may include transthoracic echocardiogram, Holter monitor, cardiac magnetic resonance imaging with gadolinium contrast, or cardiac positron emission tomography/computed tomography; a complete blood cell count; comprehensive metabolic panel; urinalysis, with a 24-hour urine calcium if there is a history of a kidney stone; tuberculin skin test or IFN-γ release assay to rule out tuberculosis on a case-by-case basis; thyroid testing; and 25-hydroxy vitamin D and 1,25-dihydroxy vitamin D screening.1
Treatment
Cutaneous sarcoidosis is treated with topical or intralesional anti-inflammatory medications, immunomodulators, systemic immunosuppressants, and biologic agents. Management of cutaneous sarcoidosis should be done in an escalating approach guided by treatment response, location on the body, and patient preference. Response to therapy can take upwards of 3 months, and appropriate patient counseling is necessary to manage expectations.20 Most cutaneous sarcoidosis treatments are not approved by the US Food and Drug Administration for this purpose, and off-label use is based on available evidence and expert consensus (eTable).

An important consideration for treating sarcoidosis in active-duty servicemembers is the use of immunosuppressants or biologics requiring refrigeration or continuous monitoring. According to Department of Defense retention standards, an active-duty servicemember may be disqualified from future service if their condition persists despite appropriate treatment and impairs their ability to perform required military duties. A medical evaluation board typically is initiated on any servicemember who starts a medication while on active duty that requires frequent monitoring by a medical provider, including immunomodulating and immunosuppressant medications.21
Final Thoughts
Military servicemembers put themselves at risk for acute bodily harm during deployment and also expose themselves to occupational hazards that may result in chronic health conditions. The VA’s coverage of new presumptive diagnoses means that veterans will receive extended care for conditions presumptively acquired during military service, including sarcoidosis. Although there are no conclusive data on whether exposure while on deployment overseas causes sarcoidosis, environmental exposures should be considered a potential cause. Patients with confirmed cutaneous sarcoidosis should undergo a complete workup for systemic sarcoidosis and be asked about their history of military service to evaluate for coverage under the PACT Act.
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;36:685-702. doi:10.1016/j.ccm.2015.08.010
- US Department of Veterans Affairs. The Pact Act and your VA benefits. Updated August 15, 2023. Accessed August 18, 2023. https://www.va.gov/resources/the-pact-act-and-your-va-benefits/
- Banoei MM, Iupe I, Bazaz RD, et al. Metabolomic and metallomic profile differences between veterans and civilians with pulmonary sarcoidosis. Sci Rep. 2019;9:19584. doi:10.1038/s41598-019-56174-8
- Bith-Melander P, Ratliff J, Poisson C, et al. Slow burns: a qualitative study of burn pit and toxic exposures among military veterans serving in Afghanistan, Iraq and throughout the Middle East. Ann Psychiatry Clin Neurosci. 2021;4:1042.
- Military burn pits and cancer risk. American Cancer Society website. Revised August 25, 2022. Accessed August 18, 2023. https://www.cancer.org/healthy/cancer-causes/chemicals/burn-pits.html
- McLean J, Anderson D, Capra G, et al. The potential effects of burn pit exposure on the respiratory tract: a systematic review. Mil Med. 2021;186:672-681. doi: 10.1093/milmed/usab070
- Seedahmed MI, Baugh AD, Albirair MT, et al. Epidemiology of sarcoidosis in U.S. veterans from 2003 to 2019 [published online February 1, 2023]. Ann Am Thorac Soc. 2023. doi:10.1513/AnnalsATS.202206-515OC
- Arkema EV, Cozier YC. Sarcoidosis epidemiology: recent estimates of incidence, prevalence and risk factors. Curr Opin Pulm Med. 2020;26:527-534. doi:10.1097/MCP.0000000000000715
- Parrish SC, Lin TK, Sicignano NM, et al. Sarcoidosis in the United States Military Health System. Sarcoidosis Vasc Diffuse Lung Dis. 2018;35:261-267. doi:10.36141/svdld.v35i3.6949
- Jain R, Yadav D, Puranik N, et al. Sarcoidosis: causes, diagnosis, clinical features, and treatments. J Clin Med. 2020;9:1081. doi:10.3390/jcm9041081
- Newman KL, Newman LS. Occupational causes of sarcoidosis. Curr Opin Allergy Clin Immunol. 2012;12:145-150. doi:10.1097/ACI.0b013e3283515173
- Izbicki G, Chavko R, Banauch GI, et al. World Trade Center “sarcoid-like” granulomatous pulmonary disease in New York City Fire Department rescue workers. Chest. 2007;131:1414-1423. doi:10.1378/chest.06-2114
- Jajosky P. Sarcoidosis diagnoses among U.S. military personnel: trends and ship assignment associations. Am J Prev Med. 1998;14:176-183. doi:10.1016/s0749-3797(97)00063-9
- Gorham ED, Garland CF, Garland FC, et al. Trends and occupational associations in incidence of hospitalized pulmonary sarcoidosis and other lung diseases in Navy personnel: a 27-year historical prospective study, 1975-2001. Chest. 2004;126:1431-1438. doi:10.1378/chest.126.5.1431
- Madar CS, Lewin-Smith MR, Franks TJ, et al. Histological diagnoses of military personnel undergoing lung biopsy after deployment to southwest Asia. Lung. 2017;195:507-515. doi:10.1007/s00408-017-0009-2
- Forbes DA, Anderson JT, Hamilton JA, et al. Relationship to deployment on sarcoidosis staging and severity in military personnel. Mil Med. 2020;185:E804-E810. doi:10.1093/milmed/usz407
- Jani N, Christie IC, Wu TD, et al. Factors associated with a diagnosis of sarcoidosis among US veterans of Iraq and Afghanistan. Sci Rep. 2022;12:22045. doi:10.1038/s41598-022-24853-8
- Sève P, Pacheco Y, Durupt F, et al. Sarcoidosis: a clinical overview from symptoms to diagnosis. Cells. 2021;10:766. doi:10.3390/cells10040766
- Motamedi M, Ferrara G, Yacyshyn E, et al. Skin disorders and interstitial lung disease: part I—screening, diagnosis, and therapeutic principles. J Am Acad Dermatol. 2023;88:751-764. doi:10.1016/j.jaad.2022.10.001
- Wu JH, Imadojemu S, Caplan AS. The evolving landscape of cutaneous sarcoidosis: pathogenic insight, clinical challenges, and new frontiers in therapy. Am J Clin Dermatol. 2022;23:499-514. doi:10.1007/s40257-022-00693-0
- US Department of Defense. DoD Instruction 6130.03, Volume 2. Medical Standards for Military Service: Retention. Published September 4, 2020. Accessed August 18, 2023. https://www.med.navy.mil/Portals/62/Documents/NMFSC/NMOTC/NAMI/ARWG/Miscellaneous/613003v2p_MEDICAL_STANDARDS_RETENTION.PDF?ver=7gMDUq1G1dOupje6wf_-DQ%3D%3D
Sarcoidosis is a chronic inflammatory disease characterized by noncaseating granulomas that can affect many organ systems, most commonly the lungs and skin, with cutaneous involvement in 25% to 30% of patients in the United States.1 The etiology of sarcoidosis largely is unknown and likely is multifactorial; however, specific environmental, infectious, and pharmaceutical triggers may contribute to its pathogenesis. Sarcoidosis secondary to occupational exposures in US Military veterans historically has been discussed and investigated. Still, it was not considered a service-connected disability until the passing of the Promise to Address Comprehensive Toxics (PACT) Act2 in 2022. In this article, we review the risk factors and incidence of sarcoidosis in post–9/11 veterans as well as provide recommendations for managing presumptive service-connected sarcoidosis covered under the recently enacted PACT Act.
The PACT Act and Post–9/11 Military Veterans
Veterans of Operation Iraqi Freedom (OIF) and Operation Enduring Freedom (OEF) have a history of occupational exposures to open-air burn pits, gun smoke, and recurrent high-intensity sandstorms that may cause chronic disease.3 Burn pits, which were used to dispose of solid waste on forward operating bases, released antigenic particulate matter that was detectable on air sampling.4,5 Increased respiratory disease rates in veterans that were deployed post–9/11 are well documented, but a causal relationship has not been established.6 Although burn pits cannot be directly associated with any disease at this time,5 veterans with assumed exposures can now receive a Veterans Affairs (VA) Disability Rating for presumptive conditions under the PACT Act.2 The major points of this legislation include expanding and extending eligibility for veterans with toxic exposures, providing access to toxic exposure screening for all veterans receiving VA health care, and increasing research related to toxic exposures in US servicemembers. The PACT Act expands health care benefits, making it easier for veterans exposed post–9/11 to receive coverage for 24 new presumptive diagnoses.2 Of these diagnoses, several are relevant to the practicing dermatologist. Patients with metastasis of primary cancers to the skin as well as melanoma or sarcoidosis may be eligible for coverage depending on the location and time of service. The Table lists service locations where the VA has determined servicemembers may have been exposed to burn pits or other toxins. Servicemembers with a presumptive diagnosis who served in these locations may be eligible for care under the PACT Act. Sarcoidosis is of particular concern due to its increased incidence and prevalence in military veterans compared to civilian populations. An analysis of more than 13 million veterans who received health care benefits through the Veterans Health Administration in 2019 found an annual incidence of sarcoidosis of 52 cases per 100,000 person-years and an annual prevalence of 141 cases per 100,000 individuals.7 In contrast, the United States has a reported annual incidence of sarcoidosis of 4.9 cases per 100,000 person-years and an annual prevalence of 60 cases per 100,000 individuals.8 Although the increased rates of sarcoidosis in veterans have been noted for decades, only recently have investigations provided insights into the etiology of sarcoidosis in this population.
Sarcoidosis and Environmental Factors
Sarcoidosis is a multisystem granulomatous inflammatory disease that can present in any organ system9; however, it most commonly affects the lungs, skin, and eyes—all of which are subjected to direct contact with environmental toxins. The cause of sarcoidosis is unknown, but environmental exposures are theorized to play a role.9,10 It has been hypothesized that exposure to various immunologically active triggers may invoke the granulomatous inflammatory response that characterizes the disease.11 The World Trade Center disaster on 9/11 has provided insight into the potential environmental component of sarcoidosis. Firefighters who spent extensive amounts of time at the World Trade Center site experienced intense exposure to inorganic particulate matter; it was later found that there was a marked increase in the incidence of sarcoidosis or sarcoidosislike granulomatous pulmonary disease in exposed firefighters. It has been speculated that the elevated exposure to potentially antigenic particulates may have induced granulomatous inflammation, resulting in the manifestation of the disease.12 Other known occupational exposures associated with an increased risk for sarcoidosis or sarcoidosislike illness include mold, silicates, metal dust, and microbial contaminants.11 Servicemembers commonly are exposed to several of these aerosolized toxins, which theoretically could increase their risk for developing sarcoidosis.
Sarcoidosis in the Military
Servicemembers historically have faced unique environmental hazards that may increase their risk for developing sarcoidosis. Studies of naval veterans have shown relationships between occupational location and increased rates of sarcoidosis. Sailors assigned to aircraft carriers with nonskid coatings containing particulate matter such as aluminum, titanium, and silicates had a higher prevalence of sarcoidosis than those stationed on “clean” ships.13,14 Although no one trigger was identified, the increased rates of sarcoidosis in populations with extensive exposure to toxins raise concern for the possibility of occupationally induced sarcoidosis in post–9/11 veterans.
Environmental exposures during OIF and OEF may be associated with sarcoidosis. A retrospective review of lung biopsy data collected from Department of Defense military treatment facilities was conducted to identify associations between lung disease and deployment to the Middle East.15 The study included 391 military patients divided into deployed and nondeployed groups undergoing lung biopsies for various reasons from 2005 to 2012. An analysis of the reported lung histology showed an increased frequency of nonnecrotizing granulomas in those with a history of deployment to the Middle East compared to those who had never been deployed. Development of disease was not associated with confounding factors such as age, ethnicity, sex, or tobacco use, raising suspicion about similar shared toxic exposures among deployed servicemembers.15 A 2020 study of sarcoidosis in active-duty military personnel reported that the incidence of observed cases was 2-times those seen in civilian Department of Defense employees from 2005 to 2010; however, data collected in this study did not indicate an increased risk for developing sarcoidosis based on deployment to the Middle East. Still, the higher prevalence of sarcoidosis in active-duty military personnel suggests similar shared exposures in this group.16
Identification of exposures that may potentially trigger sarcoidosis is difficult due to many confounding variables; however, the Airborne Hazards and Open Burn Pit Registry questionnaire has been used to extrapolate prospective hazards of concern. Results from the questionnaire identified that only veterans exposed to convoy activity had a statistically significant (odds ratio, 1.16; 95% CI, 1.00-1.35; P=.046) increased risk for developing sarcoidosis.17 Interestingly, enlisted personnel had a higher rate of sarcoidosis than officers, comprising upwards of 78% of cases in the Military Health System from 2004 to 2013.9 This finding requires further study, but increased exposure to toxins due to occupational specialty may be the cause.
Veterans with sarcoidosis may have a unique pathophysiology, which may point to occupational exposure. Studies show that affected veterans have unique plasma metabolites and metal ions compared to civilians, with lower anti-inflammatory amino acid concentrations and downregulated GABA synthesis. The environmental exposures in OIF and OEF may have primed deployed servicemembers to develop a distinct subtype of sarcoidosis.3 Overall, there is a dearth of literature on post–9/11 veterans with sarcoidosis; therefore, further investigation is necessary to determine the actual risk for developing the disease following exposures related to military service.
Clinical Presentation and Diagnosis
Cutaneous sarcoidosis protean morphology is considered an imitator of many other skin diseases. The most common sarcoidosis-specific skin lesions include papules and papulonodules (Figure, A), lupus pernio (Figure, B), plaques (Figure, C), and subcutaneous nodules. Lesions typically present on the face, neck, trunk, and extremities and are associated with a favorable prognosis. Lupus pernio presents as centrofacial, bluish-red or violaceous nodules and can be disfiguring (Figure, B). Subcutaneous nodules occur in the subcutaneous tissue or deep dermis with minimal surface changes. Sarcoidal lesions also can occur at sites of scar tissue or trauma, within tattoos, and around foreign bodies. Other uncommon sarcoidosis-specific skin lesions include ichthyosiform, hypopigmented, atrophic, ulcerative and mucosal lesions; erythroderma; alopecia; and nail sarcoidosis.18
When cutaneous sarcoidosis is suspected, the skin serves as an easily accessible organ for biopsy to confirm the diagnosis.1 Sarcoidosis-specific skin lesions are histologically characterized as sarcoidal granulomas with a classic noncaseating naked appearance comprised of epithelioid histocytes with giant cells amidst a mild lymphocytic inflammatory infiltrate. Nonspecific sarcoidosis skin lesions do not contain characteristic noncaseating granulomas. Erythema nodosum is the most common nonspecific lesion and is associated with a favorable prognosis. Other nonspecific sarcoidosis skin findings include calcinosis cutis, clubbing, and vasculitis.18
Workup
Due to the systemic nature of sarcoidosis, dermatologists should initiate a comprehensive workup upon diagnosis of cutaneous sarcoidosis, which should include the following: a complete in-depth history, including occupational/environmental exposures; a complete review of systems; a military history, including time of service and location of deployments; physical examination; pulmonary function test; high-resolution chest computed tomography19; pulmonology referral for additional pulmonary function tests, including diffusion capacity for carbon monoxide and 6-minute walk test; ophthalmology referral for full ophthalmologic examination; initial cardiac screening with electrocardiogram; and a review of symptoms including assessment of heart palpitations. Any abnormalities should prompt cardiology referral for evaluation of cardiac involvement with a workup that may include transthoracic echocardiogram, Holter monitor, cardiac magnetic resonance imaging with gadolinium contrast, or cardiac positron emission tomography/computed tomography; a complete blood cell count; comprehensive metabolic panel; urinalysis, with a 24-hour urine calcium if there is a history of a kidney stone; tuberculin skin test or IFN-γ release assay to rule out tuberculosis on a case-by-case basis; thyroid testing; and 25-hydroxy vitamin D and 1,25-dihydroxy vitamin D screening.1
Treatment
Cutaneous sarcoidosis is treated with topical or intralesional anti-inflammatory medications, immunomodulators, systemic immunosuppressants, and biologic agents. Management of cutaneous sarcoidosis should be done in an escalating approach guided by treatment response, location on the body, and patient preference. Response to therapy can take upwards of 3 months, and appropriate patient counseling is necessary to manage expectations.20 Most cutaneous sarcoidosis treatments are not approved by the US Food and Drug Administration for this purpose, and off-label use is based on available evidence and expert consensus (eTable).
An important consideration for treating sarcoidosis in active-duty servicemembers is the use of immunosuppressants or biologics requiring refrigeration or continuous monitoring. According to Department of Defense retention standards, an active-duty servicemember may be disqualified from future service if their condition persists despite appropriate treatment and impairs their ability to perform required military duties. A medical evaluation board typically is initiated on any servicemember who starts a medication while on active duty that requires frequent monitoring by a medical provider, including immunomodulating and immunosuppressant medications.21
Final Thoughts
Military servicemembers put themselves at risk for acute bodily harm during deployment and also expose themselves to occupational hazards that may result in chronic health conditions. The VA’s coverage of new presumptive diagnoses means that veterans will receive extended care for conditions presumptively acquired during military service, including sarcoidosis. Although there are no conclusive data on whether exposure while on deployment overseas causes sarcoidosis, environmental exposures should be considered a potential cause. Patients with confirmed cutaneous sarcoidosis should undergo a complete workup for systemic sarcoidosis and be asked about their history of military service to evaluate for coverage under the PACT Act.
Sarcoidosis is a chronic inflammatory disease characterized by noncaseating granulomas that can affect many organ systems, most commonly the lungs and skin, with cutaneous involvement in 25% to 30% of patients in the United States.1 The etiology of sarcoidosis largely is unknown and likely is multifactorial; however, specific environmental, infectious, and pharmaceutical triggers may contribute to its pathogenesis. Sarcoidosis secondary to occupational exposures in US Military veterans historically has been discussed and investigated. Still, it was not considered a service-connected disability until the passing of the Promise to Address Comprehensive Toxics (PACT) Act2 in 2022. In this article, we review the risk factors and incidence of sarcoidosis in post–9/11 veterans as well as provide recommendations for managing presumptive service-connected sarcoidosis covered under the recently enacted PACT Act.
The PACT Act and Post–9/11 Military Veterans
Veterans of Operation Iraqi Freedom (OIF) and Operation Enduring Freedom (OEF) have a history of occupational exposures to open-air burn pits, gun smoke, and recurrent high-intensity sandstorms that may cause chronic disease.3 Burn pits, which were used to dispose of solid waste on forward operating bases, released antigenic particulate matter that was detectable on air sampling.4,5 Increased respiratory disease rates in veterans that were deployed post–9/11 are well documented, but a causal relationship has not been established.6 Although burn pits cannot be directly associated with any disease at this time,5 veterans with assumed exposures can now receive a Veterans Affairs (VA) Disability Rating for presumptive conditions under the PACT Act.2 The major points of this legislation include expanding and extending eligibility for veterans with toxic exposures, providing access to toxic exposure screening for all veterans receiving VA health care, and increasing research related to toxic exposures in US servicemembers. The PACT Act expands health care benefits, making it easier for veterans exposed post–9/11 to receive coverage for 24 new presumptive diagnoses.2 Of these diagnoses, several are relevant to the practicing dermatologist. Patients with metastasis of primary cancers to the skin as well as melanoma or sarcoidosis may be eligible for coverage depending on the location and time of service. The Table lists service locations where the VA has determined servicemembers may have been exposed to burn pits or other toxins. Servicemembers with a presumptive diagnosis who served in these locations may be eligible for care under the PACT Act. Sarcoidosis is of particular concern due to its increased incidence and prevalence in military veterans compared to civilian populations. An analysis of more than 13 million veterans who received health care benefits through the Veterans Health Administration in 2019 found an annual incidence of sarcoidosis of 52 cases per 100,000 person-years and an annual prevalence of 141 cases per 100,000 individuals.7 In contrast, the United States has a reported annual incidence of sarcoidosis of 4.9 cases per 100,000 person-years and an annual prevalence of 60 cases per 100,000 individuals.8 Although the increased rates of sarcoidosis in veterans have been noted for decades, only recently have investigations provided insights into the etiology of sarcoidosis in this population.
Sarcoidosis and Environmental Factors
Sarcoidosis is a multisystem granulomatous inflammatory disease that can present in any organ system9; however, it most commonly affects the lungs, skin, and eyes—all of which are subjected to direct contact with environmental toxins. The cause of sarcoidosis is unknown, but environmental exposures are theorized to play a role.9,10 It has been hypothesized that exposure to various immunologically active triggers may invoke the granulomatous inflammatory response that characterizes the disease.11 The World Trade Center disaster on 9/11 has provided insight into the potential environmental component of sarcoidosis. Firefighters who spent extensive amounts of time at the World Trade Center site experienced intense exposure to inorganic particulate matter; it was later found that there was a marked increase in the incidence of sarcoidosis or sarcoidosislike granulomatous pulmonary disease in exposed firefighters. It has been speculated that the elevated exposure to potentially antigenic particulates may have induced granulomatous inflammation, resulting in the manifestation of the disease.12 Other known occupational exposures associated with an increased risk for sarcoidosis or sarcoidosislike illness include mold, silicates, metal dust, and microbial contaminants.11 Servicemembers commonly are exposed to several of these aerosolized toxins, which theoretically could increase their risk for developing sarcoidosis.
Sarcoidosis in the Military
Servicemembers historically have faced unique environmental hazards that may increase their risk for developing sarcoidosis. Studies of naval veterans have shown relationships between occupational location and increased rates of sarcoidosis. Sailors assigned to aircraft carriers with nonskid coatings containing particulate matter such as aluminum, titanium, and silicates had a higher prevalence of sarcoidosis than those stationed on “clean” ships.13,14 Although no one trigger was identified, the increased rates of sarcoidosis in populations with extensive exposure to toxins raise concern for the possibility of occupationally induced sarcoidosis in post–9/11 veterans.
Environmental exposures during OIF and OEF may be associated with sarcoidosis. A retrospective review of lung biopsy data collected from Department of Defense military treatment facilities was conducted to identify associations between lung disease and deployment to the Middle East.15 The study included 391 military patients divided into deployed and nondeployed groups undergoing lung biopsies for various reasons from 2005 to 2012. An analysis of the reported lung histology showed an increased frequency of nonnecrotizing granulomas in those with a history of deployment to the Middle East compared to those who had never been deployed. Development of disease was not associated with confounding factors such as age, ethnicity, sex, or tobacco use, raising suspicion about similar shared toxic exposures among deployed servicemembers.15 A 2020 study of sarcoidosis in active-duty military personnel reported that the incidence of observed cases was 2-times those seen in civilian Department of Defense employees from 2005 to 2010; however, data collected in this study did not indicate an increased risk for developing sarcoidosis based on deployment to the Middle East. Still, the higher prevalence of sarcoidosis in active-duty military personnel suggests similar shared exposures in this group.16
Identification of exposures that may potentially trigger sarcoidosis is difficult due to many confounding variables; however, the Airborne Hazards and Open Burn Pit Registry questionnaire has been used to extrapolate prospective hazards of concern. Results from the questionnaire identified that only veterans exposed to convoy activity had a statistically significant (odds ratio, 1.16; 95% CI, 1.00-1.35; P=.046) increased risk for developing sarcoidosis.17 Interestingly, enlisted personnel had a higher rate of sarcoidosis than officers, comprising upwards of 78% of cases in the Military Health System from 2004 to 2013.9 This finding requires further study, but increased exposure to toxins due to occupational specialty may be the cause.
Veterans with sarcoidosis may have a unique pathophysiology, which may point to occupational exposure. Studies show that affected veterans have unique plasma metabolites and metal ions compared to civilians, with lower anti-inflammatory amino acid concentrations and downregulated GABA synthesis. The environmental exposures in OIF and OEF may have primed deployed servicemembers to develop a distinct subtype of sarcoidosis.3 Overall, there is a dearth of literature on post–9/11 veterans with sarcoidosis; therefore, further investigation is necessary to determine the actual risk for developing the disease following exposures related to military service.
Clinical Presentation and Diagnosis
Cutaneous sarcoidosis protean morphology is considered an imitator of many other skin diseases. The most common sarcoidosis-specific skin lesions include papules and papulonodules (Figure, A), lupus pernio (Figure, B), plaques (Figure, C), and subcutaneous nodules. Lesions typically present on the face, neck, trunk, and extremities and are associated with a favorable prognosis. Lupus pernio presents as centrofacial, bluish-red or violaceous nodules and can be disfiguring (Figure, B). Subcutaneous nodules occur in the subcutaneous tissue or deep dermis with minimal surface changes. Sarcoidal lesions also can occur at sites of scar tissue or trauma, within tattoos, and around foreign bodies. Other uncommon sarcoidosis-specific skin lesions include ichthyosiform, hypopigmented, atrophic, ulcerative and mucosal lesions; erythroderma; alopecia; and nail sarcoidosis.18
When cutaneous sarcoidosis is suspected, the skin serves as an easily accessible organ for biopsy to confirm the diagnosis.1 Sarcoidosis-specific skin lesions are histologically characterized as sarcoidal granulomas with a classic noncaseating naked appearance comprised of epithelioid histocytes with giant cells amidst a mild lymphocytic inflammatory infiltrate. Nonspecific sarcoidosis skin lesions do not contain characteristic noncaseating granulomas. Erythema nodosum is the most common nonspecific lesion and is associated with a favorable prognosis. Other nonspecific sarcoidosis skin findings include calcinosis cutis, clubbing, and vasculitis.18
Workup
Due to the systemic nature of sarcoidosis, dermatologists should initiate a comprehensive workup upon diagnosis of cutaneous sarcoidosis, which should include the following: a complete in-depth history, including occupational/environmental exposures; a complete review of systems; a military history, including time of service and location of deployments; physical examination; pulmonary function test; high-resolution chest computed tomography19; pulmonology referral for additional pulmonary function tests, including diffusion capacity for carbon monoxide and 6-minute walk test; ophthalmology referral for full ophthalmologic examination; initial cardiac screening with electrocardiogram; and a review of symptoms including assessment of heart palpitations. Any abnormalities should prompt cardiology referral for evaluation of cardiac involvement with a workup that may include transthoracic echocardiogram, Holter monitor, cardiac magnetic resonance imaging with gadolinium contrast, or cardiac positron emission tomography/computed tomography; a complete blood cell count; comprehensive metabolic panel; urinalysis, with a 24-hour urine calcium if there is a history of a kidney stone; tuberculin skin test or IFN-γ release assay to rule out tuberculosis on a case-by-case basis; thyroid testing; and 25-hydroxy vitamin D and 1,25-dihydroxy vitamin D screening.1
Treatment
Cutaneous sarcoidosis is treated with topical or intralesional anti-inflammatory medications, immunomodulators, systemic immunosuppressants, and biologic agents. Management of cutaneous sarcoidosis should be done in an escalating approach guided by treatment response, location on the body, and patient preference. Response to therapy can take upwards of 3 months, and appropriate patient counseling is necessary to manage expectations.20 Most cutaneous sarcoidosis treatments are not approved by the US Food and Drug Administration for this purpose, and off-label use is based on available evidence and expert consensus (eTable).
An important consideration for treating sarcoidosis in active-duty servicemembers is the use of immunosuppressants or biologics requiring refrigeration or continuous monitoring. According to Department of Defense retention standards, an active-duty servicemember may be disqualified from future service if their condition persists despite appropriate treatment and impairs their ability to perform required military duties. A medical evaluation board typically is initiated on any servicemember who starts a medication while on active duty that requires frequent monitoring by a medical provider, including immunomodulating and immunosuppressant medications.21
Final Thoughts
Military servicemembers put themselves at risk for acute bodily harm during deployment and also expose themselves to occupational hazards that may result in chronic health conditions. The VA’s coverage of new presumptive diagnoses means that veterans will receive extended care for conditions presumptively acquired during military service, including sarcoidosis. Although there are no conclusive data on whether exposure while on deployment overseas causes sarcoidosis, environmental exposures should be considered a potential cause. Patients with confirmed cutaneous sarcoidosis should undergo a complete workup for systemic sarcoidosis and be asked about their history of military service to evaluate for coverage under the PACT Act.
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;36:685-702. doi:10.1016/j.ccm.2015.08.010
- US Department of Veterans Affairs. The Pact Act and your VA benefits. Updated August 15, 2023. Accessed August 18, 2023. https://www.va.gov/resources/the-pact-act-and-your-va-benefits/
- Banoei MM, Iupe I, Bazaz RD, et al. Metabolomic and metallomic profile differences between veterans and civilians with pulmonary sarcoidosis. Sci Rep. 2019;9:19584. doi:10.1038/s41598-019-56174-8
- Bith-Melander P, Ratliff J, Poisson C, et al. Slow burns: a qualitative study of burn pit and toxic exposures among military veterans serving in Afghanistan, Iraq and throughout the Middle East. Ann Psychiatry Clin Neurosci. 2021;4:1042.
- Military burn pits and cancer risk. American Cancer Society website. Revised August 25, 2022. Accessed August 18, 2023. https://www.cancer.org/healthy/cancer-causes/chemicals/burn-pits.html
- McLean J, Anderson D, Capra G, et al. The potential effects of burn pit exposure on the respiratory tract: a systematic review. Mil Med. 2021;186:672-681. doi: 10.1093/milmed/usab070
- Seedahmed MI, Baugh AD, Albirair MT, et al. Epidemiology of sarcoidosis in U.S. veterans from 2003 to 2019 [published online February 1, 2023]. Ann Am Thorac Soc. 2023. doi:10.1513/AnnalsATS.202206-515OC
- Arkema EV, Cozier YC. Sarcoidosis epidemiology: recent estimates of incidence, prevalence and risk factors. Curr Opin Pulm Med. 2020;26:527-534. doi:10.1097/MCP.0000000000000715
- Parrish SC, Lin TK, Sicignano NM, et al. Sarcoidosis in the United States Military Health System. Sarcoidosis Vasc Diffuse Lung Dis. 2018;35:261-267. doi:10.36141/svdld.v35i3.6949
- Jain R, Yadav D, Puranik N, et al. Sarcoidosis: causes, diagnosis, clinical features, and treatments. J Clin Med. 2020;9:1081. doi:10.3390/jcm9041081
- Newman KL, Newman LS. Occupational causes of sarcoidosis. Curr Opin Allergy Clin Immunol. 2012;12:145-150. doi:10.1097/ACI.0b013e3283515173
- Izbicki G, Chavko R, Banauch GI, et al. World Trade Center “sarcoid-like” granulomatous pulmonary disease in New York City Fire Department rescue workers. Chest. 2007;131:1414-1423. doi:10.1378/chest.06-2114
- Jajosky P. Sarcoidosis diagnoses among U.S. military personnel: trends and ship assignment associations. Am J Prev Med. 1998;14:176-183. doi:10.1016/s0749-3797(97)00063-9
- Gorham ED, Garland CF, Garland FC, et al. Trends and occupational associations in incidence of hospitalized pulmonary sarcoidosis and other lung diseases in Navy personnel: a 27-year historical prospective study, 1975-2001. Chest. 2004;126:1431-1438. doi:10.1378/chest.126.5.1431
- Madar CS, Lewin-Smith MR, Franks TJ, et al. Histological diagnoses of military personnel undergoing lung biopsy after deployment to southwest Asia. Lung. 2017;195:507-515. doi:10.1007/s00408-017-0009-2
- Forbes DA, Anderson JT, Hamilton JA, et al. Relationship to deployment on sarcoidosis staging and severity in military personnel. Mil Med. 2020;185:E804-E810. doi:10.1093/milmed/usz407
- Jani N, Christie IC, Wu TD, et al. Factors associated with a diagnosis of sarcoidosis among US veterans of Iraq and Afghanistan. Sci Rep. 2022;12:22045. doi:10.1038/s41598-022-24853-8
- Sève P, Pacheco Y, Durupt F, et al. Sarcoidosis: a clinical overview from symptoms to diagnosis. Cells. 2021;10:766. doi:10.3390/cells10040766
- Motamedi M, Ferrara G, Yacyshyn E, et al. Skin disorders and interstitial lung disease: part I—screening, diagnosis, and therapeutic principles. J Am Acad Dermatol. 2023;88:751-764. doi:10.1016/j.jaad.2022.10.001
- Wu JH, Imadojemu S, Caplan AS. The evolving landscape of cutaneous sarcoidosis: pathogenic insight, clinical challenges, and new frontiers in therapy. Am J Clin Dermatol. 2022;23:499-514. doi:10.1007/s40257-022-00693-0
- US Department of Defense. DoD Instruction 6130.03, Volume 2. Medical Standards for Military Service: Retention. Published September 4, 2020. Accessed August 18, 2023. https://www.med.navy.mil/Portals/62/Documents/NMFSC/NMOTC/NAMI/ARWG/Miscellaneous/613003v2p_MEDICAL_STANDARDS_RETENTION.PDF?ver=7gMDUq1G1dOupje6wf_-DQ%3D%3D
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;36:685-702. doi:10.1016/j.ccm.2015.08.010
- US Department of Veterans Affairs. The Pact Act and your VA benefits. Updated August 15, 2023. Accessed August 18, 2023. https://www.va.gov/resources/the-pact-act-and-your-va-benefits/
- Banoei MM, Iupe I, Bazaz RD, et al. Metabolomic and metallomic profile differences between veterans and civilians with pulmonary sarcoidosis. Sci Rep. 2019;9:19584. doi:10.1038/s41598-019-56174-8
- Bith-Melander P, Ratliff J, Poisson C, et al. Slow burns: a qualitative study of burn pit and toxic exposures among military veterans serving in Afghanistan, Iraq and throughout the Middle East. Ann Psychiatry Clin Neurosci. 2021;4:1042.
- Military burn pits and cancer risk. American Cancer Society website. Revised August 25, 2022. Accessed August 18, 2023. https://www.cancer.org/healthy/cancer-causes/chemicals/burn-pits.html
- McLean J, Anderson D, Capra G, et al. The potential effects of burn pit exposure on the respiratory tract: a systematic review. Mil Med. 2021;186:672-681. doi: 10.1093/milmed/usab070
- Seedahmed MI, Baugh AD, Albirair MT, et al. Epidemiology of sarcoidosis in U.S. veterans from 2003 to 2019 [published online February 1, 2023]. Ann Am Thorac Soc. 2023. doi:10.1513/AnnalsATS.202206-515OC
- Arkema EV, Cozier YC. Sarcoidosis epidemiology: recent estimates of incidence, prevalence and risk factors. Curr Opin Pulm Med. 2020;26:527-534. doi:10.1097/MCP.0000000000000715
- Parrish SC, Lin TK, Sicignano NM, et al. Sarcoidosis in the United States Military Health System. Sarcoidosis Vasc Diffuse Lung Dis. 2018;35:261-267. doi:10.36141/svdld.v35i3.6949
- Jain R, Yadav D, Puranik N, et al. Sarcoidosis: causes, diagnosis, clinical features, and treatments. J Clin Med. 2020;9:1081. doi:10.3390/jcm9041081
- Newman KL, Newman LS. Occupational causes of sarcoidosis. Curr Opin Allergy Clin Immunol. 2012;12:145-150. doi:10.1097/ACI.0b013e3283515173
- Izbicki G, Chavko R, Banauch GI, et al. World Trade Center “sarcoid-like” granulomatous pulmonary disease in New York City Fire Department rescue workers. Chest. 2007;131:1414-1423. doi:10.1378/chest.06-2114
- Jajosky P. Sarcoidosis diagnoses among U.S. military personnel: trends and ship assignment associations. Am J Prev Med. 1998;14:176-183. doi:10.1016/s0749-3797(97)00063-9
- Gorham ED, Garland CF, Garland FC, et al. Trends and occupational associations in incidence of hospitalized pulmonary sarcoidosis and other lung diseases in Navy personnel: a 27-year historical prospective study, 1975-2001. Chest. 2004;126:1431-1438. doi:10.1378/chest.126.5.1431
- Madar CS, Lewin-Smith MR, Franks TJ, et al. Histological diagnoses of military personnel undergoing lung biopsy after deployment to southwest Asia. Lung. 2017;195:507-515. doi:10.1007/s00408-017-0009-2
- Forbes DA, Anderson JT, Hamilton JA, et al. Relationship to deployment on sarcoidosis staging and severity in military personnel. Mil Med. 2020;185:E804-E810. doi:10.1093/milmed/usz407
- Jani N, Christie IC, Wu TD, et al. Factors associated with a diagnosis of sarcoidosis among US veterans of Iraq and Afghanistan. Sci Rep. 2022;12:22045. doi:10.1038/s41598-022-24853-8
- Sève P, Pacheco Y, Durupt F, et al. Sarcoidosis: a clinical overview from symptoms to diagnosis. Cells. 2021;10:766. doi:10.3390/cells10040766
- Motamedi M, Ferrara G, Yacyshyn E, et al. Skin disorders and interstitial lung disease: part I—screening, diagnosis, and therapeutic principles. J Am Acad Dermatol. 2023;88:751-764. doi:10.1016/j.jaad.2022.10.001
- Wu JH, Imadojemu S, Caplan AS. The evolving landscape of cutaneous sarcoidosis: pathogenic insight, clinical challenges, and new frontiers in therapy. Am J Clin Dermatol. 2022;23:499-514. doi:10.1007/s40257-022-00693-0
- US Department of Defense. DoD Instruction 6130.03, Volume 2. Medical Standards for Military Service: Retention. Published September 4, 2020. Accessed August 18, 2023. https://www.med.navy.mil/Portals/62/Documents/NMFSC/NMOTC/NAMI/ARWG/Miscellaneous/613003v2p_MEDICAL_STANDARDS_RETENTION.PDF?ver=7gMDUq1G1dOupje6wf_-DQ%3D%3D
Practice Points
- Cutaneous sarcoidosis is the most common extrapulmonary manifestation of the disease.
- Cutaneous sarcoidosis can precede systemic manifestations of the disease and should prompt further workup.
- Sarcoidosis is a presumptive diagnosis under the PACT Act and may be a service-connected condition. Veterans with presumptive exposures should be referred to the US Department of Veterans Affairs.

ACR releases guideline for managing ILD in patients with rheumatic disease
The American College of Rheumatology has released a summary of upcoming guidelines on screening, monitoring, and treatment for interstitial lung disease (ILD) in patients with systemic autoimmune rheumatic disease.
The recommendations apply to adults with rheumatic diseases at greater risk for ILD: rheumatoid arthritis, systemic sclerosis (SSc), mixed connective tissue disease (MCTD), Sjögren’s disease (SjD), and idiopathic inflammatory myopathies (IIM).
“Interstitial lung disease is a major cause of morbidity and mortality across several systemic autoimmune rheumatic diseases,” Sindhu R. Johnson, MD, PhD, lead author of the new guidelines and director of the clinical epidemiology and health care research program at the University of Toronto, said in an ACR press release. “Guidance was needed for which tests to use for screening and monitoring this particular disease.”
The two documents are summaries of part of a larger manuscript currently awaiting peer review, according to the ACR, and the final guidelines are anticipated to be published by early 2024.
The recommendations were developed using “the best available evidence and consensus across a range of expert opinions and incorporated patient values and preferences,” according to the press release.
Highlights of recommendations for screening and monitoring ILD are:
- Providers can screen patients at higher risk for ILD with pulmonary function tests (PFTs) and high-resolution CT of the chest.
- PFTs, chest high-resolution CT, and ambulatory desaturation testing are conditionally recommended for monitoring ILD progression.
- It is conditionally recommended that providers do not use 6-minute walk test distance, chest radiography, or bronchoscopy for screening or monitoring disease.
- It is suggested that patients with IIM-ILD and SSc-ILD receive PFTs for monitoring every 3-6 months during the first year, then less frequently once stable.
- It is suggested that patients with RA-ILD, SjD-ILD, and MCTD-ILD receive PFTs every 3-12 months for the first year, then less frequently once stable.

Suggestions on how often to screen for ILD were not present in the summary documents, but will be made available in the larger manuscript, said Elana Bernstein, MD, director of the Columbia University Medical Center/New York–Presbyterian Hospital scleroderma program, New York. She is co–first author of the guidelines.
Nearly all recommendations are conditional, primarily because the certainty of evidence behind many of these recommendations is low or very low, she said in an interview. More clinical data on ILD in patients with rheumatic disease would help strengthen evidence, she said, particularly for best practices in frequency of testing. “We need more research on how often patients should be screened for ILD and how often they should be monitored for ILD progression,” she said. “That would enable us to provide recommendations, rather than just suggestions.”
Highlights of recommendations for ILD treatment are:
- The guidelines strongly recommend against using glucocorticoids for first-line ILD treatment in patients with SSc-ILD.
- Short-term glucocorticoids are conditionally recommended as a first-line ILD treatment for patients with systemic autoimmune rheumatic disease–related ILD (SARD-ILD), excluding SSc-ILD.
- Mycophenolate, azathioprine, rituximab, and cyclophosphamide are all potential first-line ILD treatment options for patients with SARD-ILD.
- It is conditionally recommended that patients with SARD-ILD do not receive leflunomide, methotrexate, tumor necrosis factor inhibitors, or abatacept as first-line ILD treatment.
- If SARD-ILD progresses despite first-line therapy, mycophenolate, rituximab, cyclophosphamide, and nintedanib are potential secondary treatment options.
- If RA-ILD progresses following initial therapy, pirfenidone is a treatment option.
- The guidelines conditionally recommend against pirfenidone as a secondary treatment option for SARD-ILD other than RA-ILD.

These summary guidelines appear “comprehensive,” but there has yet to be information published on the basis of these recommendations, Elizabeth Volkmann, MD, said in an interview.
“It’s important to understand that we don’t know whether most of these recommendations were just driven by expert opinion versus actual evidence from randomized, controlled clinical trials,” said Dr. Volkmann, who codirects the connective tissue disease–related interstitial lung disease program at the University of California, Los Angeles. She was not involved with creating the guidelines.
She expects that many of the recommendations for first- and second-line ILD treatment options were based on expert opinion, as there have been no randomized clinical trials looking at that specific topic, she said. For example, nintedanib is conditionally recommended as a first-line treatment option for SSc-ILD, but as a second-line treatment for SjD-ILD, IIM-ILD, and MCTD-ILD. “There’s no literature to support one or the other – whether nintedanib is first-line or second-line [treatment].”
The decision to publish the summary recommendations online prior to peer review is unusual, she said, as these recommendations could be altered during that process; however, Dr. Bernstein noted that was not likely.
By releasing the summary guideline now, the ACR can “get the needed information to clinicians earlier as the manuscript goes through its remaining stages and is finalized,” an ACR representative explained.
Prior to the expected publication of these guidelines in early 2024, Dr. Volkmann noted that the American Thoracic Society will be publishing guidelines on the treatment of SSc-ILD in the American Journal of Respiratory and Critical Care Medicine in September.
Dr. Bernstein reported grants/contracts with the Department of Defense, the Scleroderma Research Foundation, the National Institutes of Health, Eicos, Boehringer Ingelheim, Kadmon, and Pfizer. Dr. Volkmann has received consulting and speaking fees from Boehringer Ingelheim and GlaxoSmithKline and institutional support for performing studies on systemic sclerosis for Kadmon, Boehringer Ingelheim, Horizon, and Prometheus.
A version of this article first appeared on Medscape.com.
The American College of Rheumatology has released a summary of upcoming guidelines on screening, monitoring, and treatment for interstitial lung disease (ILD) in patients with systemic autoimmune rheumatic disease.
The recommendations apply to adults with rheumatic diseases at greater risk for ILD: rheumatoid arthritis, systemic sclerosis (SSc), mixed connective tissue disease (MCTD), Sjögren’s disease (SjD), and idiopathic inflammatory myopathies (IIM).
“Interstitial lung disease is a major cause of morbidity and mortality across several systemic autoimmune rheumatic diseases,” Sindhu R. Johnson, MD, PhD, lead author of the new guidelines and director of the clinical epidemiology and health care research program at the University of Toronto, said in an ACR press release. “Guidance was needed for which tests to use for screening and monitoring this particular disease.”
The two documents are summaries of part of a larger manuscript currently awaiting peer review, according to the ACR, and the final guidelines are anticipated to be published by early 2024.
The recommendations were developed using “the best available evidence and consensus across a range of expert opinions and incorporated patient values and preferences,” according to the press release.
Highlights of recommendations for screening and monitoring ILD are:
- Providers can screen patients at higher risk for ILD with pulmonary function tests (PFTs) and high-resolution CT of the chest.
- PFTs, chest high-resolution CT, and ambulatory desaturation testing are conditionally recommended for monitoring ILD progression.
- It is conditionally recommended that providers do not use 6-minute walk test distance, chest radiography, or bronchoscopy for screening or monitoring disease.
- It is suggested that patients with IIM-ILD and SSc-ILD receive PFTs for monitoring every 3-6 months during the first year, then less frequently once stable.
- It is suggested that patients with RA-ILD, SjD-ILD, and MCTD-ILD receive PFTs every 3-12 months for the first year, then less frequently once stable.
Suggestions on how often to screen for ILD were not present in the summary documents, but will be made available in the larger manuscript, said Elana Bernstein, MD, director of the Columbia University Medical Center/New York–Presbyterian Hospital scleroderma program, New York. She is co–first author of the guidelines.
Nearly all recommendations are conditional, primarily because the certainty of evidence behind many of these recommendations is low or very low, she said in an interview. More clinical data on ILD in patients with rheumatic disease would help strengthen evidence, she said, particularly for best practices in frequency of testing. “We need more research on how often patients should be screened for ILD and how often they should be monitored for ILD progression,” she said. “That would enable us to provide recommendations, rather than just suggestions.”
Highlights of recommendations for ILD treatment are:
- The guidelines strongly recommend against using glucocorticoids for first-line ILD treatment in patients with SSc-ILD.
- Short-term glucocorticoids are conditionally recommended as a first-line ILD treatment for patients with systemic autoimmune rheumatic disease–related ILD (SARD-ILD), excluding SSc-ILD.
- Mycophenolate, azathioprine, rituximab, and cyclophosphamide are all potential first-line ILD treatment options for patients with SARD-ILD.
- It is conditionally recommended that patients with SARD-ILD do not receive leflunomide, methotrexate, tumor necrosis factor inhibitors, or abatacept as first-line ILD treatment.
- If SARD-ILD progresses despite first-line therapy, mycophenolate, rituximab, cyclophosphamide, and nintedanib are potential secondary treatment options.
- If RA-ILD progresses following initial therapy, pirfenidone is a treatment option.
- The guidelines conditionally recommend against pirfenidone as a secondary treatment option for SARD-ILD other than RA-ILD.
These summary guidelines appear “comprehensive,” but there has yet to be information published on the basis of these recommendations, Elizabeth Volkmann, MD, said in an interview.
“It’s important to understand that we don’t know whether most of these recommendations were just driven by expert opinion versus actual evidence from randomized, controlled clinical trials,” said Dr. Volkmann, who codirects the connective tissue disease–related interstitial lung disease program at the University of California, Los Angeles. She was not involved with creating the guidelines.
She expects that many of the recommendations for first- and second-line ILD treatment options were based on expert opinion, as there have been no randomized clinical trials looking at that specific topic, she said. For example, nintedanib is conditionally recommended as a first-line treatment option for SSc-ILD, but as a second-line treatment for SjD-ILD, IIM-ILD, and MCTD-ILD. “There’s no literature to support one or the other – whether nintedanib is first-line or second-line [treatment].”
The decision to publish the summary recommendations online prior to peer review is unusual, she said, as these recommendations could be altered during that process; however, Dr. Bernstein noted that was not likely.
By releasing the summary guideline now, the ACR can “get the needed information to clinicians earlier as the manuscript goes through its remaining stages and is finalized,” an ACR representative explained.
Prior to the expected publication of these guidelines in early 2024, Dr. Volkmann noted that the American Thoracic Society will be publishing guidelines on the treatment of SSc-ILD in the American Journal of Respiratory and Critical Care Medicine in September.
Dr. Bernstein reported grants/contracts with the Department of Defense, the Scleroderma Research Foundation, the National Institutes of Health, Eicos, Boehringer Ingelheim, Kadmon, and Pfizer. Dr. Volkmann has received consulting and speaking fees from Boehringer Ingelheim and GlaxoSmithKline and institutional support for performing studies on systemic sclerosis for Kadmon, Boehringer Ingelheim, Horizon, and Prometheus.
A version of this article first appeared on Medscape.com.
The American College of Rheumatology has released a summary of upcoming guidelines on screening, monitoring, and treatment for interstitial lung disease (ILD) in patients with systemic autoimmune rheumatic disease.
The recommendations apply to adults with rheumatic diseases at greater risk for ILD: rheumatoid arthritis, systemic sclerosis (SSc), mixed connective tissue disease (MCTD), Sjögren’s disease (SjD), and idiopathic inflammatory myopathies (IIM).
“Interstitial lung disease is a major cause of morbidity and mortality across several systemic autoimmune rheumatic diseases,” Sindhu R. Johnson, MD, PhD, lead author of the new guidelines and director of the clinical epidemiology and health care research program at the University of Toronto, said in an ACR press release. “Guidance was needed for which tests to use for screening and monitoring this particular disease.”
The two documents are summaries of part of a larger manuscript currently awaiting peer review, according to the ACR, and the final guidelines are anticipated to be published by early 2024.
The recommendations were developed using “the best available evidence and consensus across a range of expert opinions and incorporated patient values and preferences,” according to the press release.
Highlights of recommendations for screening and monitoring ILD are:
- Providers can screen patients at higher risk for ILD with pulmonary function tests (PFTs) and high-resolution CT of the chest.
- PFTs, chest high-resolution CT, and ambulatory desaturation testing are conditionally recommended for monitoring ILD progression.
- It is conditionally recommended that providers do not use 6-minute walk test distance, chest radiography, or bronchoscopy for screening or monitoring disease.
- It is suggested that patients with IIM-ILD and SSc-ILD receive PFTs for monitoring every 3-6 months during the first year, then less frequently once stable.
- It is suggested that patients with RA-ILD, SjD-ILD, and MCTD-ILD receive PFTs every 3-12 months for the first year, then less frequently once stable.
Suggestions on how often to screen for ILD were not present in the summary documents, but will be made available in the larger manuscript, said Elana Bernstein, MD, director of the Columbia University Medical Center/New York–Presbyterian Hospital scleroderma program, New York. She is co–first author of the guidelines.
Nearly all recommendations are conditional, primarily because the certainty of evidence behind many of these recommendations is low or very low, she said in an interview. More clinical data on ILD in patients with rheumatic disease would help strengthen evidence, she said, particularly for best practices in frequency of testing. “We need more research on how often patients should be screened for ILD and how often they should be monitored for ILD progression,” she said. “That would enable us to provide recommendations, rather than just suggestions.”
Highlights of recommendations for ILD treatment are:
- The guidelines strongly recommend against using glucocorticoids for first-line ILD treatment in patients with SSc-ILD.
- Short-term glucocorticoids are conditionally recommended as a first-line ILD treatment for patients with systemic autoimmune rheumatic disease–related ILD (SARD-ILD), excluding SSc-ILD.
- Mycophenolate, azathioprine, rituximab, and cyclophosphamide are all potential first-line ILD treatment options for patients with SARD-ILD.
- It is conditionally recommended that patients with SARD-ILD do not receive leflunomide, methotrexate, tumor necrosis factor inhibitors, or abatacept as first-line ILD treatment.
- If SARD-ILD progresses despite first-line therapy, mycophenolate, rituximab, cyclophosphamide, and nintedanib are potential secondary treatment options.
- If RA-ILD progresses following initial therapy, pirfenidone is a treatment option.
- The guidelines conditionally recommend against pirfenidone as a secondary treatment option for SARD-ILD other than RA-ILD.
These summary guidelines appear “comprehensive,” but there has yet to be information published on the basis of these recommendations, Elizabeth Volkmann, MD, said in an interview.
“It’s important to understand that we don’t know whether most of these recommendations were just driven by expert opinion versus actual evidence from randomized, controlled clinical trials,” said Dr. Volkmann, who codirects the connective tissue disease–related interstitial lung disease program at the University of California, Los Angeles. She was not involved with creating the guidelines.
She expects that many of the recommendations for first- and second-line ILD treatment options were based on expert opinion, as there have been no randomized clinical trials looking at that specific topic, she said. For example, nintedanib is conditionally recommended as a first-line treatment option for SSc-ILD, but as a second-line treatment for SjD-ILD, IIM-ILD, and MCTD-ILD. “There’s no literature to support one or the other – whether nintedanib is first-line or second-line [treatment].”
The decision to publish the summary recommendations online prior to peer review is unusual, she said, as these recommendations could be altered during that process; however, Dr. Bernstein noted that was not likely.
By releasing the summary guideline now, the ACR can “get the needed information to clinicians earlier as the manuscript goes through its remaining stages and is finalized,” an ACR representative explained.
Prior to the expected publication of these guidelines in early 2024, Dr. Volkmann noted that the American Thoracic Society will be publishing guidelines on the treatment of SSc-ILD in the American Journal of Respiratory and Critical Care Medicine in September.
Dr. Bernstein reported grants/contracts with the Department of Defense, the Scleroderma Research Foundation, the National Institutes of Health, Eicos, Boehringer Ingelheim, Kadmon, and Pfizer. Dr. Volkmann has received consulting and speaking fees from Boehringer Ingelheim and GlaxoSmithKline and institutional support for performing studies on systemic sclerosis for Kadmon, Boehringer Ingelheim, Horizon, and Prometheus.
A version of this article first appeared on Medscape.com.
Diffuse Annular Plaques in an Infant
The Diagnosis: Neonatal Lupus Erythematosus
A review of the medical records of the patient’s mother from her first pregnancy revealed positive anti-Ro/SSA (Sjögren syndrome A) (>8.0 U [reference range <1.0 U]) and anti-La/SSB (Sjögren syndrome B) antibodies (>8.0 U [reference range <1.0 U]), which were reconfirmed during her pregnancy with our patient (the second child). The patient’s older brother was diagnosed with neonatal lupus erythematosus (NLE) 2 years prior at 1 month of age; therefore, the mother took hydroxychloroquine during the pregnancy with the second child to help prevent heart block if the child was diagnosed with NLE. Given the family history, positive antibodies in the mother, and clinical presentation, our patient was diagnosed with NLE. He was referred to a pediatric cardiologist and pediatrician to continue the workup of systemic manifestations of NLE and to rule out the presence of congenital heart block. The rash resolved 6 months after the initial presentation, and he did not develop any systemic manifestations of NLE.
Neonatal lupus erythematosus is a rare acquired autoimmune disorder caused by the placental transfer of anti-Ro/SSA and anti-La/SSB antibodies and less commonly anti-U1 ribonucleoprotein antinuclear autoantibodies.1,2 Approximately 1% to 2% of mothers with these positive antibodies will have infants affected with NLE.2 The annual prevalence of NLE in the United States is approximately 1 in 20,000 live births. Mothers of children with NLE most commonly have clinical Sjögren syndrome; however, anti-Ro/SSA and anti-LA/SSB antibodies may be present in 0.1% to 1.5% of healthy women, and 25% to 60% of women with autoimmune disease may be asymptomatic.1 As demonstrated in our case, when there is a family history of NLE in an infant from an earlier pregnancy, the risk for NLE increases to 17% to 20% in subsequent pregnancies1,3 and up to 25% in subsequent pregnancies if the initial child was diagnosed with a congenital heart block in the setting of NLE.1
Neonatal lupus erythematosus classically presents as annular erythematous macules and plaques with central scaling, telangictasia, atrophy, and pigmentary changes. It may start on the scalp and face and spread caudally.1,2 Patients may develop these lesions after UV exposure, and 80% of infants may not have dermatologic findings at birth. Importantly, 40% to 60% of mothers may be asymptomatic at the time of presentation of their child’s NLE.1 The diagnosis can be confirmed via antibody testing in the mother and/or infant. If performed, a punch biopsy shows interface dermatitis, vacuolar degeneration, and possible periadnexal lymphocytic infiltrates on histopathology.1,2
Management of cutaneous NLE includes sun protection (eg, application of sunscreen) and topical corticosteroids. Most dermatologic manifestations of NLE are transient, resolving after clearance of maternal IgG antibodies in 6 to 9 months; however, some telangiectasia, dyspigmentation, and atrophic scarring may persist.1-3
Neonatal lupus erythematosus also may have hepatobiliary, cardiac, hematologic, and less commonly neurologic manifestations. Hepatobiliary manifestations usually present as hepatomegaly or asymptomatic elevated transaminases or γ-glutamyl transferase.1,3 Approximately 10% to 20% of infants with NLE may present with transient anemia and thrombocytopenia.1 Cardiac manifestations are permanent and may require pacemaker implantation.1,3 The incidence of a congenital heart block in infants with NLE is 15% to 30%.3 Cardiac NLE most commonly injures the conductive tissue, leading to a congenital atrioventricular block. The development of a congenital heart block develops in the 18th to 24th week of gestation. Manifestations of a more advanced condition can include dilation of the ascending aorta and dilated cardiomyopathy.1 As such, patients need to be followed by a pediatric cardiologist for monitoring and treatment of any cardiac manifestations.
The overall prognosis of infants affected with NLE varies. Cardiac involvement is associated with a poor prognosis, while isolated cutaneous involvement requires little treatment and portends a favorable prognosis. It is critical for dermatologists to recognize NLE to refer patients to appropriate specialists to investigate and further monitor possible extracutaneous manifestations. With an understanding of the increased risk for a congenital heart block and NLE in subsequent pregnancies, mothers with positive anti-Ro/La antibodies should receive timely counseling and screening. In expectant mothers with suspected autoimmune disease, testing for antinuclear antibodies and SSA and SSB antibodies can be considered, as administration of hydroxychloroquine or prenatal systemic corticosteroids has proven to be effective in preventing a congenital heart block.1 Our patient was followed by pediatric cardiology and was not found to have a congenital heart block.
The differential diagnosis includes other causes of annular erythema in infants, as NLE can mimic several conditions. Tinea corporis may present as scaly annular plaques with central clearing; however, it rarely is encountered fulminantly in neonates.4 Erythema multiforme is a mucocutaneous hypersensitivy reaction distinguished by targetoid morphology.5 It is an exceedingly rare diagnosis in neonates; the average pediatric age of onset is 5.6 years.6 Erythema multiforme often is associated with an infection, most commonly herpes simplex virus,5 and mucosal involvement is common.6 Urticaria multiforme (also known as acute annular urticaria) is a benign disease that appears between 2 months to 3 years of age with blanchable urticarial plaques that likely are triggered by viral or bacterial infections, antibiotics, or vaccines.6 Specific lesions usually will resolve within 24 hours. Annular erythema of infancy is a benign and asymptomatic gyrate erythema that presents as annular plaques with palpable borders that spread centrifugally in patients younger than 1 year. Notably, lesions should periodically fade and may reappear cyclically for months to years. Evaluation for underlying disease usually is negative.6
- Derdulska JM, Rudnicka L, Szykut-Badaczewska A, et al. Neonatal lupus erythematosus—practical guidelines. J Perinat Med. 2021;49:529-538. doi:10.1515/jpm-2020-0543
- Wu J, Berk-Krauss J, Glick SA. Neonatal lupus erythematosus. JAMA Dermatol. 2021;157:590. doi:10.1001/jamadermatol.2021.0041
- Hon KL, Leung AK. Neonatal lupus erythematosus. Autoimmune Dis. 2012;2012:301274. doi:10.1155/2012/301274
- Khare AK, Gupta LK, Mittal A, et al. Neonatal tinea corporis. Indian J Dermatol. 2010;55:201. doi:10.4103/0019-5154.6274
- Ang-Tiu CU, Nicolas ME. Erythema multiforme in a 25-day old neonate. Pediatr Dermatol. 2013;30:E118-E120. doi:10.1111 /j.1525-1470.2012.01873.x
- Agnihotri G, Tsoukas MM. Annular skin lesions in infancy [published online February 3, 2022]. Clin Dermatol. 2022;40:505-512. doi:10.1016/j.clindermatol.2021.12.011
The Diagnosis: Neonatal Lupus Erythematosus
A review of the medical records of the patient’s mother from her first pregnancy revealed positive anti-Ro/SSA (Sjögren syndrome A) (>8.0 U [reference range <1.0 U]) and anti-La/SSB (Sjögren syndrome B) antibodies (>8.0 U [reference range <1.0 U]), which were reconfirmed during her pregnancy with our patient (the second child). The patient’s older brother was diagnosed with neonatal lupus erythematosus (NLE) 2 years prior at 1 month of age; therefore, the mother took hydroxychloroquine during the pregnancy with the second child to help prevent heart block if the child was diagnosed with NLE. Given the family history, positive antibodies in the mother, and clinical presentation, our patient was diagnosed with NLE. He was referred to a pediatric cardiologist and pediatrician to continue the workup of systemic manifestations of NLE and to rule out the presence of congenital heart block. The rash resolved 6 months after the initial presentation, and he did not develop any systemic manifestations of NLE.
Neonatal lupus erythematosus is a rare acquired autoimmune disorder caused by the placental transfer of anti-Ro/SSA and anti-La/SSB antibodies and less commonly anti-U1 ribonucleoprotein antinuclear autoantibodies.1,2 Approximately 1% to 2% of mothers with these positive antibodies will have infants affected with NLE.2 The annual prevalence of NLE in the United States is approximately 1 in 20,000 live births. Mothers of children with NLE most commonly have clinical Sjögren syndrome; however, anti-Ro/SSA and anti-LA/SSB antibodies may be present in 0.1% to 1.5% of healthy women, and 25% to 60% of women with autoimmune disease may be asymptomatic.1 As demonstrated in our case, when there is a family history of NLE in an infant from an earlier pregnancy, the risk for NLE increases to 17% to 20% in subsequent pregnancies1,3 and up to 25% in subsequent pregnancies if the initial child was diagnosed with a congenital heart block in the setting of NLE.1
Neonatal lupus erythematosus classically presents as annular erythematous macules and plaques with central scaling, telangictasia, atrophy, and pigmentary changes. It may start on the scalp and face and spread caudally.1,2 Patients may develop these lesions after UV exposure, and 80% of infants may not have dermatologic findings at birth. Importantly, 40% to 60% of mothers may be asymptomatic at the time of presentation of their child’s NLE.1 The diagnosis can be confirmed via antibody testing in the mother and/or infant. If performed, a punch biopsy shows interface dermatitis, vacuolar degeneration, and possible periadnexal lymphocytic infiltrates on histopathology.1,2
Management of cutaneous NLE includes sun protection (eg, application of sunscreen) and topical corticosteroids. Most dermatologic manifestations of NLE are transient, resolving after clearance of maternal IgG antibodies in 6 to 9 months; however, some telangiectasia, dyspigmentation, and atrophic scarring may persist.1-3
Neonatal lupus erythematosus also may have hepatobiliary, cardiac, hematologic, and less commonly neurologic manifestations. Hepatobiliary manifestations usually present as hepatomegaly or asymptomatic elevated transaminases or γ-glutamyl transferase.1,3 Approximately 10% to 20% of infants with NLE may present with transient anemia and thrombocytopenia.1 Cardiac manifestations are permanent and may require pacemaker implantation.1,3 The incidence of a congenital heart block in infants with NLE is 15% to 30%.3 Cardiac NLE most commonly injures the conductive tissue, leading to a congenital atrioventricular block. The development of a congenital heart block develops in the 18th to 24th week of gestation. Manifestations of a more advanced condition can include dilation of the ascending aorta and dilated cardiomyopathy.1 As such, patients need to be followed by a pediatric cardiologist for monitoring and treatment of any cardiac manifestations.
The overall prognosis of infants affected with NLE varies. Cardiac involvement is associated with a poor prognosis, while isolated cutaneous involvement requires little treatment and portends a favorable prognosis. It is critical for dermatologists to recognize NLE to refer patients to appropriate specialists to investigate and further monitor possible extracutaneous manifestations. With an understanding of the increased risk for a congenital heart block and NLE in subsequent pregnancies, mothers with positive anti-Ro/La antibodies should receive timely counseling and screening. In expectant mothers with suspected autoimmune disease, testing for antinuclear antibodies and SSA and SSB antibodies can be considered, as administration of hydroxychloroquine or prenatal systemic corticosteroids has proven to be effective in preventing a congenital heart block.1 Our patient was followed by pediatric cardiology and was not found to have a congenital heart block.
The differential diagnosis includes other causes of annular erythema in infants, as NLE can mimic several conditions. Tinea corporis may present as scaly annular plaques with central clearing; however, it rarely is encountered fulminantly in neonates.4 Erythema multiforme is a mucocutaneous hypersensitivy reaction distinguished by targetoid morphology.5 It is an exceedingly rare diagnosis in neonates; the average pediatric age of onset is 5.6 years.6 Erythema multiforme often is associated with an infection, most commonly herpes simplex virus,5 and mucosal involvement is common.6 Urticaria multiforme (also known as acute annular urticaria) is a benign disease that appears between 2 months to 3 years of age with blanchable urticarial plaques that likely are triggered by viral or bacterial infections, antibiotics, or vaccines.6 Specific lesions usually will resolve within 24 hours. Annular erythema of infancy is a benign and asymptomatic gyrate erythema that presents as annular plaques with palpable borders that spread centrifugally in patients younger than 1 year. Notably, lesions should periodically fade and may reappear cyclically for months to years. Evaluation for underlying disease usually is negative.6
The Diagnosis: Neonatal Lupus Erythematosus
A review of the medical records of the patient’s mother from her first pregnancy revealed positive anti-Ro/SSA (Sjögren syndrome A) (>8.0 U [reference range <1.0 U]) and anti-La/SSB (Sjögren syndrome B) antibodies (>8.0 U [reference range <1.0 U]), which were reconfirmed during her pregnancy with our patient (the second child). The patient’s older brother was diagnosed with neonatal lupus erythematosus (NLE) 2 years prior at 1 month of age; therefore, the mother took hydroxychloroquine during the pregnancy with the second child to help prevent heart block if the child was diagnosed with NLE. Given the family history, positive antibodies in the mother, and clinical presentation, our patient was diagnosed with NLE. He was referred to a pediatric cardiologist and pediatrician to continue the workup of systemic manifestations of NLE and to rule out the presence of congenital heart block. The rash resolved 6 months after the initial presentation, and he did not develop any systemic manifestations of NLE.
Neonatal lupus erythematosus is a rare acquired autoimmune disorder caused by the placental transfer of anti-Ro/SSA and anti-La/SSB antibodies and less commonly anti-U1 ribonucleoprotein antinuclear autoantibodies.1,2 Approximately 1% to 2% of mothers with these positive antibodies will have infants affected with NLE.2 The annual prevalence of NLE in the United States is approximately 1 in 20,000 live births. Mothers of children with NLE most commonly have clinical Sjögren syndrome; however, anti-Ro/SSA and anti-LA/SSB antibodies may be present in 0.1% to 1.5% of healthy women, and 25% to 60% of women with autoimmune disease may be asymptomatic.1 As demonstrated in our case, when there is a family history of NLE in an infant from an earlier pregnancy, the risk for NLE increases to 17% to 20% in subsequent pregnancies1,3 and up to 25% in subsequent pregnancies if the initial child was diagnosed with a congenital heart block in the setting of NLE.1
Neonatal lupus erythematosus classically presents as annular erythematous macules and plaques with central scaling, telangictasia, atrophy, and pigmentary changes. It may start on the scalp and face and spread caudally.1,2 Patients may develop these lesions after UV exposure, and 80% of infants may not have dermatologic findings at birth. Importantly, 40% to 60% of mothers may be asymptomatic at the time of presentation of their child’s NLE.1 The diagnosis can be confirmed via antibody testing in the mother and/or infant. If performed, a punch biopsy shows interface dermatitis, vacuolar degeneration, and possible periadnexal lymphocytic infiltrates on histopathology.1,2
Management of cutaneous NLE includes sun protection (eg, application of sunscreen) and topical corticosteroids. Most dermatologic manifestations of NLE are transient, resolving after clearance of maternal IgG antibodies in 6 to 9 months; however, some telangiectasia, dyspigmentation, and atrophic scarring may persist.1-3
Neonatal lupus erythematosus also may have hepatobiliary, cardiac, hematologic, and less commonly neurologic manifestations. Hepatobiliary manifestations usually present as hepatomegaly or asymptomatic elevated transaminases or γ-glutamyl transferase.1,3 Approximately 10% to 20% of infants with NLE may present with transient anemia and thrombocytopenia.1 Cardiac manifestations are permanent and may require pacemaker implantation.1,3 The incidence of a congenital heart block in infants with NLE is 15% to 30%.3 Cardiac NLE most commonly injures the conductive tissue, leading to a congenital atrioventricular block. The development of a congenital heart block develops in the 18th to 24th week of gestation. Manifestations of a more advanced condition can include dilation of the ascending aorta and dilated cardiomyopathy.1 As such, patients need to be followed by a pediatric cardiologist for monitoring and treatment of any cardiac manifestations.
The overall prognosis of infants affected with NLE varies. Cardiac involvement is associated with a poor prognosis, while isolated cutaneous involvement requires little treatment and portends a favorable prognosis. It is critical for dermatologists to recognize NLE to refer patients to appropriate specialists to investigate and further monitor possible extracutaneous manifestations. With an understanding of the increased risk for a congenital heart block and NLE in subsequent pregnancies, mothers with positive anti-Ro/La antibodies should receive timely counseling and screening. In expectant mothers with suspected autoimmune disease, testing for antinuclear antibodies and SSA and SSB antibodies can be considered, as administration of hydroxychloroquine or prenatal systemic corticosteroids has proven to be effective in preventing a congenital heart block.1 Our patient was followed by pediatric cardiology and was not found to have a congenital heart block.
The differential diagnosis includes other causes of annular erythema in infants, as NLE can mimic several conditions. Tinea corporis may present as scaly annular plaques with central clearing; however, it rarely is encountered fulminantly in neonates.4 Erythema multiforme is a mucocutaneous hypersensitivy reaction distinguished by targetoid morphology.5 It is an exceedingly rare diagnosis in neonates; the average pediatric age of onset is 5.6 years.6 Erythema multiforme often is associated with an infection, most commonly herpes simplex virus,5 and mucosal involvement is common.6 Urticaria multiforme (also known as acute annular urticaria) is a benign disease that appears between 2 months to 3 years of age with blanchable urticarial plaques that likely are triggered by viral or bacterial infections, antibiotics, or vaccines.6 Specific lesions usually will resolve within 24 hours. Annular erythema of infancy is a benign and asymptomatic gyrate erythema that presents as annular plaques with palpable borders that spread centrifugally in patients younger than 1 year. Notably, lesions should periodically fade and may reappear cyclically for months to years. Evaluation for underlying disease usually is negative.6
- Derdulska JM, Rudnicka L, Szykut-Badaczewska A, et al. Neonatal lupus erythematosus—practical guidelines. J Perinat Med. 2021;49:529-538. doi:10.1515/jpm-2020-0543
- Wu J, Berk-Krauss J, Glick SA. Neonatal lupus erythematosus. JAMA Dermatol. 2021;157:590. doi:10.1001/jamadermatol.2021.0041
- Hon KL, Leung AK. Neonatal lupus erythematosus. Autoimmune Dis. 2012;2012:301274. doi:10.1155/2012/301274
- Khare AK, Gupta LK, Mittal A, et al. Neonatal tinea corporis. Indian J Dermatol. 2010;55:201. doi:10.4103/0019-5154.6274
- Ang-Tiu CU, Nicolas ME. Erythema multiforme in a 25-day old neonate. Pediatr Dermatol. 2013;30:E118-E120. doi:10.1111 /j.1525-1470.2012.01873.x
- Agnihotri G, Tsoukas MM. Annular skin lesions in infancy [published online February 3, 2022]. Clin Dermatol. 2022;40:505-512. doi:10.1016/j.clindermatol.2021.12.011
- Derdulska JM, Rudnicka L, Szykut-Badaczewska A, et al. Neonatal lupus erythematosus—practical guidelines. J Perinat Med. 2021;49:529-538. doi:10.1515/jpm-2020-0543
- Wu J, Berk-Krauss J, Glick SA. Neonatal lupus erythematosus. JAMA Dermatol. 2021;157:590. doi:10.1001/jamadermatol.2021.0041
- Hon KL, Leung AK. Neonatal lupus erythematosus. Autoimmune Dis. 2012;2012:301274. doi:10.1155/2012/301274
- Khare AK, Gupta LK, Mittal A, et al. Neonatal tinea corporis. Indian J Dermatol. 2010;55:201. doi:10.4103/0019-5154.6274
- Ang-Tiu CU, Nicolas ME. Erythema multiforme in a 25-day old neonate. Pediatr Dermatol. 2013;30:E118-E120. doi:10.1111 /j.1525-1470.2012.01873.x
- Agnihotri G, Tsoukas MM. Annular skin lesions in infancy [published online February 3, 2022]. Clin Dermatol. 2022;40:505-512. doi:10.1016/j.clindermatol.2021.12.011
A 5-week-old infant boy presented with a rash at birth (left). The pregnancy was full term without complications, and he was otherwise healthy. A family history revealed that his older brother developed a similar rash 2 weeks after birth (right). Physical examination revealed polycyclic annular patches with an erythematous border and central clearing diffusely located on the trunk, extremities, scalp, and face with periorbital edema.

