User login
Generic imatinib launched with savings program

Photo by Rhoda Baer
Sun Pharma has announced the US launch of imatinib mesylate tablets, which are a generic version of Novartis’s Gleevec, for indications approved by the US Food and Drug Administration (FDA).
As part of this launch, Sun Pharma has rolled out a savings card program. The goal is to provide greater access to imatinib mesylate tablets for patients who have commercial insurance, but their out-of-pocket cost may exceed an affordable amount.
Sun Pharma’s Imatinib Mesylate Savings Card will reduce patient’s co-payment to $10. The card will also offer patients an additional savings benefit of up to $700 for a 30-day fill to offset any additional out-of-pocket cost should they be required to meet their deductible or co-insurance.
Participating pharmacies across the US can use the patient’s card as part of this program.
Eligible patients can participate in Sun Pharma’s Imatinib Mesylate Savings Card program by registering at www.imatinibrx.com or by requesting a savings card from their oncologist. Sun Pharma will be supplying its Imatinib Mesylate Savings Cards to more than 4500 oncologists.
Sun Pharma has established a Hub service so patients can call and speak with a trained healthcare professional about imatinib mesylate. The number is 1-844-502-5950.
In addition, qualifying patients can receive Sun Pharma’s imatinib mesylate at no cost. Based on qualifications for applying and including a doctor’s prescription, the Hub service will determine if a patient is qualified to receive imatinib mesylate for free. Upon acceptance, the prescription will be processed and delivered to the qualifying patient at no cost.
Sun Pharma’s imatinib mesylate was approved by the FDA in December 2015 and was granted 180 days of marketing exclusivity from the time of its launch. The drug is available in 100 mg and 400 mg tablets.
It is approved to treat:
- Newly diagnosed adult and pediatric patients with Philadelphia-chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase
- Patients with Ph+ CML in blast crisis, accelerated phase, or in chronic phase after failure of interferon-alpha therapy
- Adults with relapsed or refractory Ph+ acute lymphoblastic leukemia
- Adults with myelodysplastic/myeloproliferative diseases associated with PDGFR gene re-arrangements
- Adults with aggressive systemic mastocytosis without the D816V c-Kit mutation or with c-Kit mutational status unknown
- Adults with hypereosinophilic syndrome and/or chronic eosinophilic leukemia, including those who have the FIP1L1-PDGFRα fusion kinase
- Adult patients with unresectable, recurrent, and/or metastatic dermatofibrosarcoma protuberans.
Sun Pharma’s imatinib mesylate is not approved to treat patients with KIT (CD117)-positive unresectable and/or metastatic malignant gastrointestinal stromal tumors. ![]()
Photo by Rhoda Baer
Sun Pharma has announced the US launch of imatinib mesylate tablets, which are a generic version of Novartis’s Gleevec, for indications approved by the US Food and Drug Administration (FDA).
As part of this launch, Sun Pharma has rolled out a savings card program. The goal is to provide greater access to imatinib mesylate tablets for patients who have commercial insurance, but their out-of-pocket cost may exceed an affordable amount.
Sun Pharma’s Imatinib Mesylate Savings Card will reduce patient’s co-payment to $10. The card will also offer patients an additional savings benefit of up to $700 for a 30-day fill to offset any additional out-of-pocket cost should they be required to meet their deductible or co-insurance.
Participating pharmacies across the US can use the patient’s card as part of this program.
Eligible patients can participate in Sun Pharma’s Imatinib Mesylate Savings Card program by registering at www.imatinibrx.com or by requesting a savings card from their oncologist. Sun Pharma will be supplying its Imatinib Mesylate Savings Cards to more than 4500 oncologists.
Sun Pharma has established a Hub service so patients can call and speak with a trained healthcare professional about imatinib mesylate. The number is 1-844-502-5950.
In addition, qualifying patients can receive Sun Pharma’s imatinib mesylate at no cost. Based on qualifications for applying and including a doctor’s prescription, the Hub service will determine if a patient is qualified to receive imatinib mesylate for free. Upon acceptance, the prescription will be processed and delivered to the qualifying patient at no cost.
Sun Pharma’s imatinib mesylate was approved by the FDA in December 2015 and was granted 180 days of marketing exclusivity from the time of its launch. The drug is available in 100 mg and 400 mg tablets.
It is approved to treat:
- Newly diagnosed adult and pediatric patients with Philadelphia-chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase
- Patients with Ph+ CML in blast crisis, accelerated phase, or in chronic phase after failure of interferon-alpha therapy
- Adults with relapsed or refractory Ph+ acute lymphoblastic leukemia
- Adults with myelodysplastic/myeloproliferative diseases associated with PDGFR gene re-arrangements
- Adults with aggressive systemic mastocytosis without the D816V c-Kit mutation or with c-Kit mutational status unknown
- Adults with hypereosinophilic syndrome and/or chronic eosinophilic leukemia, including those who have the FIP1L1-PDGFRα fusion kinase
- Adult patients with unresectable, recurrent, and/or metastatic dermatofibrosarcoma protuberans.
Sun Pharma’s imatinib mesylate is not approved to treat patients with KIT (CD117)-positive unresectable and/or metastatic malignant gastrointestinal stromal tumors. ![]()
Photo by Rhoda Baer
Sun Pharma has announced the US launch of imatinib mesylate tablets, which are a generic version of Novartis’s Gleevec, for indications approved by the US Food and Drug Administration (FDA).
As part of this launch, Sun Pharma has rolled out a savings card program. The goal is to provide greater access to imatinib mesylate tablets for patients who have commercial insurance, but their out-of-pocket cost may exceed an affordable amount.
Sun Pharma’s Imatinib Mesylate Savings Card will reduce patient’s co-payment to $10. The card will also offer patients an additional savings benefit of up to $700 for a 30-day fill to offset any additional out-of-pocket cost should they be required to meet their deductible or co-insurance.
Participating pharmacies across the US can use the patient’s card as part of this program.
Eligible patients can participate in Sun Pharma’s Imatinib Mesylate Savings Card program by registering at www.imatinibrx.com or by requesting a savings card from their oncologist. Sun Pharma will be supplying its Imatinib Mesylate Savings Cards to more than 4500 oncologists.
Sun Pharma has established a Hub service so patients can call and speak with a trained healthcare professional about imatinib mesylate. The number is 1-844-502-5950.
In addition, qualifying patients can receive Sun Pharma’s imatinib mesylate at no cost. Based on qualifications for applying and including a doctor’s prescription, the Hub service will determine if a patient is qualified to receive imatinib mesylate for free. Upon acceptance, the prescription will be processed and delivered to the qualifying patient at no cost.
Sun Pharma’s imatinib mesylate was approved by the FDA in December 2015 and was granted 180 days of marketing exclusivity from the time of its launch. The drug is available in 100 mg and 400 mg tablets.
It is approved to treat:
- Newly diagnosed adult and pediatric patients with Philadelphia-chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase
- Patients with Ph+ CML in blast crisis, accelerated phase, or in chronic phase after failure of interferon-alpha therapy
- Adults with relapsed or refractory Ph+ acute lymphoblastic leukemia
- Adults with myelodysplastic/myeloproliferative diseases associated with PDGFR gene re-arrangements
- Adults with aggressive systemic mastocytosis without the D816V c-Kit mutation or with c-Kit mutational status unknown
- Adults with hypereosinophilic syndrome and/or chronic eosinophilic leukemia, including those who have the FIP1L1-PDGFRα fusion kinase
- Adult patients with unresectable, recurrent, and/or metastatic dermatofibrosarcoma protuberans.
Sun Pharma’s imatinib mesylate is not approved to treat patients with KIT (CD117)-positive unresectable and/or metastatic malignant gastrointestinal stromal tumors. ![]()
EHA creates ‘roadmap’ for hematology research

Photo by Daniel Sone
The European Hematology Association (EHA) has created a “roadmap” for hematology research in Europe.
This guidance document summarizes the current status of basic, translational, and clinical hematology research and identifies areas of unmet scientific and medical need in Europe.
It is intended to help European and national policy makers, funding agencies, charities, research institutes, and researchers make decisions on initiating, funding, or developing research.
The guidance, “The European Hematology Association Roadmap for European Hematology Research: A Consensus Document,” is published in this month’s issue of haematologica.
“For the first time, hematologists in Europe came together to develop a roadmap to guide hematology research in Europe” said Andreas Engert, MD, chair of the EHA Research Roadmap Task Force.
“Hematology in Europe has achieved a lot, but the discipline must focus and collaborate to be efficient and remain successful in improving patient outcomes. The roadmap does just that and will determine the research agenda in Europe in the coming years.”
Roughly 300 experts from more than 20 countries—including clinicians, basic researchers, and patients—contributed to the roadmap. Stakeholders such as national hematology societies, patient organizations, hematology trial groups, and other European organizations were consulted to comment on the final draft version.
The final roadmap has 9 sections: normal hematopoiesis, malignant lymphoid and myeloid diseases, anemias and related diseases, platelet disorders, blood coagulation and hemostatic disorders, transfusion medicine, infections in hematology, and hematopoietic stem cell transplantation.
The roadmap lists priorities and needs in these areas, including the need for targeted therapies based on genomic profiling and chemical biology, the need to eradicate minimal residual disease, and the need for treatments that are better tolerated by elderly patients.
“Now’s the time for Europe to pay attention,” said Ulrich Jäger, MD, chair of the EHA European Affairs Committee.
“With an aging population, the slow recovery from the financial and Euro crises, costly medical breakthroughs and innovations—quite a few of which involve hematology researchers—Europe faces increased health expenditures while budgets are limited.”
“Policy makers are rightfully cautious when spending the taxpayers’ money. So it is our responsibility to provide the policy makers with the information and evidence they need to decide where their support impacts knowledge and health most efficiently, to the benefit of patients and society. The Research Roadmap delivers on that. Now, it is up to the policy makers in the EU to deliver too.” ![]()
Photo by Daniel Sone
The European Hematology Association (EHA) has created a “roadmap” for hematology research in Europe.
This guidance document summarizes the current status of basic, translational, and clinical hematology research and identifies areas of unmet scientific and medical need in Europe.
It is intended to help European and national policy makers, funding agencies, charities, research institutes, and researchers make decisions on initiating, funding, or developing research.
The guidance, “The European Hematology Association Roadmap for European Hematology Research: A Consensus Document,” is published in this month’s issue of haematologica.
“For the first time, hematologists in Europe came together to develop a roadmap to guide hematology research in Europe” said Andreas Engert, MD, chair of the EHA Research Roadmap Task Force.
“Hematology in Europe has achieved a lot, but the discipline must focus and collaborate to be efficient and remain successful in improving patient outcomes. The roadmap does just that and will determine the research agenda in Europe in the coming years.”
Roughly 300 experts from more than 20 countries—including clinicians, basic researchers, and patients—contributed to the roadmap. Stakeholders such as national hematology societies, patient organizations, hematology trial groups, and other European organizations were consulted to comment on the final draft version.
The final roadmap has 9 sections: normal hematopoiesis, malignant lymphoid and myeloid diseases, anemias and related diseases, platelet disorders, blood coagulation and hemostatic disorders, transfusion medicine, infections in hematology, and hematopoietic stem cell transplantation.
The roadmap lists priorities and needs in these areas, including the need for targeted therapies based on genomic profiling and chemical biology, the need to eradicate minimal residual disease, and the need for treatments that are better tolerated by elderly patients.
“Now’s the time for Europe to pay attention,” said Ulrich Jäger, MD, chair of the EHA European Affairs Committee.
“With an aging population, the slow recovery from the financial and Euro crises, costly medical breakthroughs and innovations—quite a few of which involve hematology researchers—Europe faces increased health expenditures while budgets are limited.”
“Policy makers are rightfully cautious when spending the taxpayers’ money. So it is our responsibility to provide the policy makers with the information and evidence they need to decide where their support impacts knowledge and health most efficiently, to the benefit of patients and society. The Research Roadmap delivers on that. Now, it is up to the policy makers in the EU to deliver too.” ![]()
Photo by Daniel Sone
The European Hematology Association (EHA) has created a “roadmap” for hematology research in Europe.
This guidance document summarizes the current status of basic, translational, and clinical hematology research and identifies areas of unmet scientific and medical need in Europe.
It is intended to help European and national policy makers, funding agencies, charities, research institutes, and researchers make decisions on initiating, funding, or developing research.
The guidance, “The European Hematology Association Roadmap for European Hematology Research: A Consensus Document,” is published in this month’s issue of haematologica.
“For the first time, hematologists in Europe came together to develop a roadmap to guide hematology research in Europe” said Andreas Engert, MD, chair of the EHA Research Roadmap Task Force.
“Hematology in Europe has achieved a lot, but the discipline must focus and collaborate to be efficient and remain successful in improving patient outcomes. The roadmap does just that and will determine the research agenda in Europe in the coming years.”
Roughly 300 experts from more than 20 countries—including clinicians, basic researchers, and patients—contributed to the roadmap. Stakeholders such as national hematology societies, patient organizations, hematology trial groups, and other European organizations were consulted to comment on the final draft version.
The final roadmap has 9 sections: normal hematopoiesis, malignant lymphoid and myeloid diseases, anemias and related diseases, platelet disorders, blood coagulation and hemostatic disorders, transfusion medicine, infections in hematology, and hematopoietic stem cell transplantation.
The roadmap lists priorities and needs in these areas, including the need for targeted therapies based on genomic profiling and chemical biology, the need to eradicate minimal residual disease, and the need for treatments that are better tolerated by elderly patients.
“Now’s the time for Europe to pay attention,” said Ulrich Jäger, MD, chair of the EHA European Affairs Committee.
“With an aging population, the slow recovery from the financial and Euro crises, costly medical breakthroughs and innovations—quite a few of which involve hematology researchers—Europe faces increased health expenditures while budgets are limited.”
“Policy makers are rightfully cautious when spending the taxpayers’ money. So it is our responsibility to provide the policy makers with the information and evidence they need to decide where their support impacts knowledge and health most efficiently, to the benefit of patients and society. The Research Roadmap delivers on that. Now, it is up to the policy makers in the EU to deliver too.” ![]()
Protein’s role in thrombosis elucidated

Image by Andre E.X. Brown
Preclinical research helps explain how the protein lysyl oxidase (LOX) enhances platelet activation and thrombosis.
Previous studies showed that LOX is overexpressed in pathologies associated with thrombosis, including myeloproliferative neoplasms.
However, it wasn’t clear exactly how LOX leads to thrombosis.
So Shinobu Matsuuras, PhD, of Boston University School of Medicine in Massachusetts, and her colleagues attempted to find out.
The team reported their findings in Blood.
The researchers wanted to determine the role of LOX in thrombosis and platelet function without the compounding influences of other pathologies.
So they generated mice that expressed LOX in wild-type megakaryocytes and platelets—Pf4-Loxtg/tg mice.
These mice had the same number of platelets as control mice, but the platelets in Pf4-Loxtg/tg mice were more likely than normal platelets to form a thrombus.
The time to vessel occlusion after endothelial injury was significantly shorter in Pf4-Loxtg/tg mice than control mice. The average time to occlusion was about 35% shorter in Pf4-Loxtg/tg mice.
The reason for this, according to the researchers’ experiments, is that platelets from Pf4-Loxtg/tg mice adhere better to collagen and have a greater aggregation response to lower doses of collagen.
The researchers were also able to pinpoint the receptor on the platelets that was affected by the oxidation activity of LOX—integrin α2β1.
The team said this is the first study to show that LOX expression enhances platelet adhesion to collagen through integrin α2β1.
And the results suggest LOX could be a target for antithrombotic therapy in myeloproliferative neoplasms and other pathologies associated with LOX overexpression. ![]()
Image by Andre E.X. Brown
Preclinical research helps explain how the protein lysyl oxidase (LOX) enhances platelet activation and thrombosis.
Previous studies showed that LOX is overexpressed in pathologies associated with thrombosis, including myeloproliferative neoplasms.
However, it wasn’t clear exactly how LOX leads to thrombosis.
So Shinobu Matsuuras, PhD, of Boston University School of Medicine in Massachusetts, and her colleagues attempted to find out.
The team reported their findings in Blood.
The researchers wanted to determine the role of LOX in thrombosis and platelet function without the compounding influences of other pathologies.
So they generated mice that expressed LOX in wild-type megakaryocytes and platelets—Pf4-Loxtg/tg mice.
These mice had the same number of platelets as control mice, but the platelets in Pf4-Loxtg/tg mice were more likely than normal platelets to form a thrombus.
The time to vessel occlusion after endothelial injury was significantly shorter in Pf4-Loxtg/tg mice than control mice. The average time to occlusion was about 35% shorter in Pf4-Loxtg/tg mice.
The reason for this, according to the researchers’ experiments, is that platelets from Pf4-Loxtg/tg mice adhere better to collagen and have a greater aggregation response to lower doses of collagen.
The researchers were also able to pinpoint the receptor on the platelets that was affected by the oxidation activity of LOX—integrin α2β1.
The team said this is the first study to show that LOX expression enhances platelet adhesion to collagen through integrin α2β1.
And the results suggest LOX could be a target for antithrombotic therapy in myeloproliferative neoplasms and other pathologies associated with LOX overexpression. ![]()
Image by Andre E.X. Brown
Preclinical research helps explain how the protein lysyl oxidase (LOX) enhances platelet activation and thrombosis.
Previous studies showed that LOX is overexpressed in pathologies associated with thrombosis, including myeloproliferative neoplasms.
However, it wasn’t clear exactly how LOX leads to thrombosis.
So Shinobu Matsuuras, PhD, of Boston University School of Medicine in Massachusetts, and her colleagues attempted to find out.
The team reported their findings in Blood.
The researchers wanted to determine the role of LOX in thrombosis and platelet function without the compounding influences of other pathologies.
So they generated mice that expressed LOX in wild-type megakaryocytes and platelets—Pf4-Loxtg/tg mice.
These mice had the same number of platelets as control mice, but the platelets in Pf4-Loxtg/tg mice were more likely than normal platelets to form a thrombus.
The time to vessel occlusion after endothelial injury was significantly shorter in Pf4-Loxtg/tg mice than control mice. The average time to occlusion was about 35% shorter in Pf4-Loxtg/tg mice.
The reason for this, according to the researchers’ experiments, is that platelets from Pf4-Loxtg/tg mice adhere better to collagen and have a greater aggregation response to lower doses of collagen.
The researchers were also able to pinpoint the receptor on the platelets that was affected by the oxidation activity of LOX—integrin α2β1.
The team said this is the first study to show that LOX expression enhances platelet adhesion to collagen through integrin α2β1.
And the results suggest LOX could be a target for antithrombotic therapy in myeloproliferative neoplasms and other pathologies associated with LOX overexpression. ![]()
Team identifies potential target for XLP-1

Image courtesy of NIAID
A protein called diacylglycerol kinase alpha (DGKα) could be a therapeutic target for X-linked lymphoproliferative disease (XLP-1), according to research published in Science Translational Medicine.
Researchers have known for some time that XLP-1 is a heritable disorder caused by germline mutations in SH2D1A.
When this gene is affected, it leads to defects in an adaptor molecule known as SAP (signaling lymphocytic activation molecule-associated protein), which regulates T-cell receptor signaling and triggers cytotoxic T cells to self-destruct when they are no longer needed.
Without an effective SAP adaptor molecule, apoptosis is impaired, and DGKα is activated.
With the current study, researchers wanted to determine whether the over-activation of DGKα might contribute to the reduced apoptosis observed in T cells in patients with XLP-1 and the accumulation of T cells that occurs following infection with Epstein-Barr virus.
“Patients with X-linked lymphoproliferative disease are prone to severe Epstein-Barr virus infection due to a weakened immune system,” explained study author Kim Nichols, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Infection with Epstein-Barr virus can have potentially fatal consequences for these patients. This severe disease is a double-edged sword. On the one hand, the immune system is significantly weakened. However, detrimental side effects occur due to the expansion and hyper-activation of T cells.”
“[W]e wanted to establish the biochemical mechanism underlying these changes so that we could develop better treatments for X-linked lymphoproliferative disease patients experiencing hyper-inflammation.”
Studying T cells from XLP-1 patients, the researchers found that SAP and DGKα are both crucial for the regulation of T-cell death. Loss of SAP, which normally inhibits DGKα, led to unrestrained DGKα activity, resulting in impaired T-cell receptor signaling and resistance to apoptosis.
Pharmacologic inhibition of DGKα restored the sensitivity of XLP-1 T cells to cell death. Using small interfering RNA to knockout DGKα in cultured XLP-1 T cells had the same results.
And pharmacologic inhibition of DGKα curtailed the expansion of T cells in virus-infected mice that served as a model organism to study XLP-1.
Treating the mice with a DGKα inhibitor restored T cells’ sensitivity to cell death by boosting the expression of pro-apoptotic proteins, which prevented excessive T-cell buildup and reduced the severity of the disease.
“Our findings suggest that inhibition of DGKα could reverse some of the life-threatening effects linked to Epstein-Barr virus infection of patients with X-linked lymphoproliferative disease,” Dr Nichols concluded. ![]()
Image courtesy of NIAID
A protein called diacylglycerol kinase alpha (DGKα) could be a therapeutic target for X-linked lymphoproliferative disease (XLP-1), according to research published in Science Translational Medicine.
Researchers have known for some time that XLP-1 is a heritable disorder caused by germline mutations in SH2D1A.
When this gene is affected, it leads to defects in an adaptor molecule known as SAP (signaling lymphocytic activation molecule-associated protein), which regulates T-cell receptor signaling and triggers cytotoxic T cells to self-destruct when they are no longer needed.
Without an effective SAP adaptor molecule, apoptosis is impaired, and DGKα is activated.
With the current study, researchers wanted to determine whether the over-activation of DGKα might contribute to the reduced apoptosis observed in T cells in patients with XLP-1 and the accumulation of T cells that occurs following infection with Epstein-Barr virus.
“Patients with X-linked lymphoproliferative disease are prone to severe Epstein-Barr virus infection due to a weakened immune system,” explained study author Kim Nichols, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Infection with Epstein-Barr virus can have potentially fatal consequences for these patients. This severe disease is a double-edged sword. On the one hand, the immune system is significantly weakened. However, detrimental side effects occur due to the expansion and hyper-activation of T cells.”
“[W]e wanted to establish the biochemical mechanism underlying these changes so that we could develop better treatments for X-linked lymphoproliferative disease patients experiencing hyper-inflammation.”
Studying T cells from XLP-1 patients, the researchers found that SAP and DGKα are both crucial for the regulation of T-cell death. Loss of SAP, which normally inhibits DGKα, led to unrestrained DGKα activity, resulting in impaired T-cell receptor signaling and resistance to apoptosis.
Pharmacologic inhibition of DGKα restored the sensitivity of XLP-1 T cells to cell death. Using small interfering RNA to knockout DGKα in cultured XLP-1 T cells had the same results.
And pharmacologic inhibition of DGKα curtailed the expansion of T cells in virus-infected mice that served as a model organism to study XLP-1.
Treating the mice with a DGKα inhibitor restored T cells’ sensitivity to cell death by boosting the expression of pro-apoptotic proteins, which prevented excessive T-cell buildup and reduced the severity of the disease.
“Our findings suggest that inhibition of DGKα could reverse some of the life-threatening effects linked to Epstein-Barr virus infection of patients with X-linked lymphoproliferative disease,” Dr Nichols concluded. ![]()
Image courtesy of NIAID
A protein called diacylglycerol kinase alpha (DGKα) could be a therapeutic target for X-linked lymphoproliferative disease (XLP-1), according to research published in Science Translational Medicine.
Researchers have known for some time that XLP-1 is a heritable disorder caused by germline mutations in SH2D1A.
When this gene is affected, it leads to defects in an adaptor molecule known as SAP (signaling lymphocytic activation molecule-associated protein), which regulates T-cell receptor signaling and triggers cytotoxic T cells to self-destruct when they are no longer needed.
Without an effective SAP adaptor molecule, apoptosis is impaired, and DGKα is activated.
With the current study, researchers wanted to determine whether the over-activation of DGKα might contribute to the reduced apoptosis observed in T cells in patients with XLP-1 and the accumulation of T cells that occurs following infection with Epstein-Barr virus.
“Patients with X-linked lymphoproliferative disease are prone to severe Epstein-Barr virus infection due to a weakened immune system,” explained study author Kim Nichols, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Infection with Epstein-Barr virus can have potentially fatal consequences for these patients. This severe disease is a double-edged sword. On the one hand, the immune system is significantly weakened. However, detrimental side effects occur due to the expansion and hyper-activation of T cells.”
“[W]e wanted to establish the biochemical mechanism underlying these changes so that we could develop better treatments for X-linked lymphoproliferative disease patients experiencing hyper-inflammation.”
Studying T cells from XLP-1 patients, the researchers found that SAP and DGKα are both crucial for the regulation of T-cell death. Loss of SAP, which normally inhibits DGKα, led to unrestrained DGKα activity, resulting in impaired T-cell receptor signaling and resistance to apoptosis.
Pharmacologic inhibition of DGKα restored the sensitivity of XLP-1 T cells to cell death. Using small interfering RNA to knockout DGKα in cultured XLP-1 T cells had the same results.
And pharmacologic inhibition of DGKα curtailed the expansion of T cells in virus-infected mice that served as a model organism to study XLP-1.
Treating the mice with a DGKα inhibitor restored T cells’ sensitivity to cell death by boosting the expression of pro-apoptotic proteins, which prevented excessive T-cell buildup and reduced the severity of the disease.
“Our findings suggest that inhibition of DGKα could reverse some of the life-threatening effects linked to Epstein-Barr virus infection of patients with X-linked lymphoproliferative disease,” Dr Nichols concluded. ![]()
BM fibrosis grade may impact OS in PMF

ORLANDO, FL—Having a higher grade of bone marrow (BM) fibrosis may confer inferior overall survival (OS) in patients with primary myelofibrosis (PMF), according to a retrospective study.
Investigators found that having a fibrosis grade of 2 or higher at diagnosis was associated with “unique clinical and molecular variables” that suggested a more aggressive disease phenotype.
And the median OS was significantly shorter in patients with higher grades of fibrosis.
However, when the investigators divided patients according to their International Prognostic Scoring System (IPSS) risk group, having a fibrosis grade of 2 or higher was only significantly associated with reduced OS among patients in the low-risk or intermediate-1-risk categories.
Paola Guglielmelli, MD, PhD, of the University of Florence in Italy, presented these findings at the 2015 ASH Annual Meeting (abstract 351*).
Dr Guglielmelli noted that the prognostic significance of BM fibrosis grade in PMF has been debated. So she and her colleagues set out to analyze the prognostic impact of fibrosis in 540 PMF patients from 6 Italian centers belonging to AGIMM (AIRC-Gruppo Italiano Malattie Mieloproliferative).
BM biopsies were obtained at diagnosis and evaluated by local pathologists according to 2008 World Health Organization criteria. The European consensus scoring system was used to grade fibrosis on a scale of MF-0 to MF-3.
Fifty patients were classified as MF-0 (9.3%), 180 were MF-1 (33.3%), 196 were MF-2 (36.3%), and 114 were MF-3 (21.1%).
Patients in the MF-2 and MF-3 groups were significantly more likely to have constitutional symptoms (P<0.0001), splenomegaly ≥10 cm from left costal margin (P<0.0001), a peripheral blast count ≥1% (P<0.0001), a greater risk of anemia (P<0.0001) or thrombocytopenia (P=0.001), and belong to the intermediate-2 or high-risk IPSS categories (P<0.0001).
In addition, patients in the MF-2 and MF-3 groups were significantly more likely to qualify as high-molecular-risk (HMR), which was defined as having at least 1 mutation in ASXL1, EZH2, SRSF2, or IDH1/2 (P<0.0001). The frequency of HMR patients increased progressively according to fibrosis grade: MF-0 (16%), MF-1 (25.6%), MF-2 (33.7%), and MF-3 (44.7%).
Patients with 2 or more HMR mutated genes were preferentially MF-2 or MF-3. None of the MF-0 patients fell into this category, compared to 4.4% for MF-1, 10.2% for MF-2, and 10.5% for MF-3 (P<0.0001).
Survival
The median OS was significantly shorter in patients with higher BM fibrosis grades (P<0.0001). The median OS was 7.2 years in the MF-3 group (hazard ratio [HR]=8.7), 6.7 years in the MF-2 group (HR=7.3), 14.7 years in the MF-1 group (HR=3.9), and not reached in the MF-0 group (reference).
In multivariable analysis, having a BM fibrosis grade of 2 or greater was significantly associated with reduced OS (HR=3.8, P=0.01).
Other variables significantly associated with reduced OS were being in the intermediate-1 (HR=2.9, P<0.0001), intermedicate-2 (HR=10.0, P<0.0001), or high-risk IPSS categories (HR=9.7, P<0.0001); having CALR type 2 mutation (HR=3.4, P=0.010), JAK2/MPL mutation (HR=2.4, P=0.003), or being triple-negative (HR=4.5, P<0.0001); being classified as HMR (HR=2.4, P<0.0001); and having 2 or more HMR mutations (HR=4.3, P=0.009).
Dr Guglielmelli and her colleagues also assessed the impact of BM fibrosis grade according to IPSS risk score.
They found that, for patients in the low/intermediate-1-risk categories, the median OS was not reached in the MF-0 group, was 22.8 years in the MF-1 group (HR=3.9), and was 15.4 years in the MF-2 and -3 groups combined (HR=7.4, P=0.001).
In the intermediate-2/high-risk categories, the median OS was 11 years for the MF-0 group, 3.6 years for the MF-1 group (HR=2.2), and 3.6 years in the MF-2 and -3 groups (HR=2.7, P=0.28).
Dr Guglielmelli therefore concluded that BM fibrosis grade might help refine prognostic stratification for PMF patients in the lower-risk IPSS categories. However, she noted that this study had limitations, and the results should be confirmed with prospective research. ![]()
*Data in the abstract differ from the presentation.
ORLANDO, FL—Having a higher grade of bone marrow (BM) fibrosis may confer inferior overall survival (OS) in patients with primary myelofibrosis (PMF), according to a retrospective study.
Investigators found that having a fibrosis grade of 2 or higher at diagnosis was associated with “unique clinical and molecular variables” that suggested a more aggressive disease phenotype.
And the median OS was significantly shorter in patients with higher grades of fibrosis.
However, when the investigators divided patients according to their International Prognostic Scoring System (IPSS) risk group, having a fibrosis grade of 2 or higher was only significantly associated with reduced OS among patients in the low-risk or intermediate-1-risk categories.
Paola Guglielmelli, MD, PhD, of the University of Florence in Italy, presented these findings at the 2015 ASH Annual Meeting (abstract 351*).
Dr Guglielmelli noted that the prognostic significance of BM fibrosis grade in PMF has been debated. So she and her colleagues set out to analyze the prognostic impact of fibrosis in 540 PMF patients from 6 Italian centers belonging to AGIMM (AIRC-Gruppo Italiano Malattie Mieloproliferative).
BM biopsies were obtained at diagnosis and evaluated by local pathologists according to 2008 World Health Organization criteria. The European consensus scoring system was used to grade fibrosis on a scale of MF-0 to MF-3.
Fifty patients were classified as MF-0 (9.3%), 180 were MF-1 (33.3%), 196 were MF-2 (36.3%), and 114 were MF-3 (21.1%).
Patients in the MF-2 and MF-3 groups were significantly more likely to have constitutional symptoms (P<0.0001), splenomegaly ≥10 cm from left costal margin (P<0.0001), a peripheral blast count ≥1% (P<0.0001), a greater risk of anemia (P<0.0001) or thrombocytopenia (P=0.001), and belong to the intermediate-2 or high-risk IPSS categories (P<0.0001).
In addition, patients in the MF-2 and MF-3 groups were significantly more likely to qualify as high-molecular-risk (HMR), which was defined as having at least 1 mutation in ASXL1, EZH2, SRSF2, or IDH1/2 (P<0.0001). The frequency of HMR patients increased progressively according to fibrosis grade: MF-0 (16%), MF-1 (25.6%), MF-2 (33.7%), and MF-3 (44.7%).
Patients with 2 or more HMR mutated genes were preferentially MF-2 or MF-3. None of the MF-0 patients fell into this category, compared to 4.4% for MF-1, 10.2% for MF-2, and 10.5% for MF-3 (P<0.0001).
Survival
The median OS was significantly shorter in patients with higher BM fibrosis grades (P<0.0001). The median OS was 7.2 years in the MF-3 group (hazard ratio [HR]=8.7), 6.7 years in the MF-2 group (HR=7.3), 14.7 years in the MF-1 group (HR=3.9), and not reached in the MF-0 group (reference).
In multivariable analysis, having a BM fibrosis grade of 2 or greater was significantly associated with reduced OS (HR=3.8, P=0.01).
Other variables significantly associated with reduced OS were being in the intermediate-1 (HR=2.9, P<0.0001), intermedicate-2 (HR=10.0, P<0.0001), or high-risk IPSS categories (HR=9.7, P<0.0001); having CALR type 2 mutation (HR=3.4, P=0.010), JAK2/MPL mutation (HR=2.4, P=0.003), or being triple-negative (HR=4.5, P<0.0001); being classified as HMR (HR=2.4, P<0.0001); and having 2 or more HMR mutations (HR=4.3, P=0.009).
Dr Guglielmelli and her colleagues also assessed the impact of BM fibrosis grade according to IPSS risk score.
They found that, for patients in the low/intermediate-1-risk categories, the median OS was not reached in the MF-0 group, was 22.8 years in the MF-1 group (HR=3.9), and was 15.4 years in the MF-2 and -3 groups combined (HR=7.4, P=0.001).
In the intermediate-2/high-risk categories, the median OS was 11 years for the MF-0 group, 3.6 years for the MF-1 group (HR=2.2), and 3.6 years in the MF-2 and -3 groups (HR=2.7, P=0.28).
Dr Guglielmelli therefore concluded that BM fibrosis grade might help refine prognostic stratification for PMF patients in the lower-risk IPSS categories. However, she noted that this study had limitations, and the results should be confirmed with prospective research. ![]()
*Data in the abstract differ from the presentation.
ORLANDO, FL—Having a higher grade of bone marrow (BM) fibrosis may confer inferior overall survival (OS) in patients with primary myelofibrosis (PMF), according to a retrospective study.
Investigators found that having a fibrosis grade of 2 or higher at diagnosis was associated with “unique clinical and molecular variables” that suggested a more aggressive disease phenotype.
And the median OS was significantly shorter in patients with higher grades of fibrosis.
However, when the investigators divided patients according to their International Prognostic Scoring System (IPSS) risk group, having a fibrosis grade of 2 or higher was only significantly associated with reduced OS among patients in the low-risk or intermediate-1-risk categories.
Paola Guglielmelli, MD, PhD, of the University of Florence in Italy, presented these findings at the 2015 ASH Annual Meeting (abstract 351*).
Dr Guglielmelli noted that the prognostic significance of BM fibrosis grade in PMF has been debated. So she and her colleagues set out to analyze the prognostic impact of fibrosis in 540 PMF patients from 6 Italian centers belonging to AGIMM (AIRC-Gruppo Italiano Malattie Mieloproliferative).
BM biopsies were obtained at diagnosis and evaluated by local pathologists according to 2008 World Health Organization criteria. The European consensus scoring system was used to grade fibrosis on a scale of MF-0 to MF-3.
Fifty patients were classified as MF-0 (9.3%), 180 were MF-1 (33.3%), 196 were MF-2 (36.3%), and 114 were MF-3 (21.1%).
Patients in the MF-2 and MF-3 groups were significantly more likely to have constitutional symptoms (P<0.0001), splenomegaly ≥10 cm from left costal margin (P<0.0001), a peripheral blast count ≥1% (P<0.0001), a greater risk of anemia (P<0.0001) or thrombocytopenia (P=0.001), and belong to the intermediate-2 or high-risk IPSS categories (P<0.0001).
In addition, patients in the MF-2 and MF-3 groups were significantly more likely to qualify as high-molecular-risk (HMR), which was defined as having at least 1 mutation in ASXL1, EZH2, SRSF2, or IDH1/2 (P<0.0001). The frequency of HMR patients increased progressively according to fibrosis grade: MF-0 (16%), MF-1 (25.6%), MF-2 (33.7%), and MF-3 (44.7%).
Patients with 2 or more HMR mutated genes were preferentially MF-2 or MF-3. None of the MF-0 patients fell into this category, compared to 4.4% for MF-1, 10.2% for MF-2, and 10.5% for MF-3 (P<0.0001).
Survival
The median OS was significantly shorter in patients with higher BM fibrosis grades (P<0.0001). The median OS was 7.2 years in the MF-3 group (hazard ratio [HR]=8.7), 6.7 years in the MF-2 group (HR=7.3), 14.7 years in the MF-1 group (HR=3.9), and not reached in the MF-0 group (reference).
In multivariable analysis, having a BM fibrosis grade of 2 or greater was significantly associated with reduced OS (HR=3.8, P=0.01).
Other variables significantly associated with reduced OS were being in the intermediate-1 (HR=2.9, P<0.0001), intermedicate-2 (HR=10.0, P<0.0001), or high-risk IPSS categories (HR=9.7, P<0.0001); having CALR type 2 mutation (HR=3.4, P=0.010), JAK2/MPL mutation (HR=2.4, P=0.003), or being triple-negative (HR=4.5, P<0.0001); being classified as HMR (HR=2.4, P<0.0001); and having 2 or more HMR mutations (HR=4.3, P=0.009).
Dr Guglielmelli and her colleagues also assessed the impact of BM fibrosis grade according to IPSS risk score.
They found that, for patients in the low/intermediate-1-risk categories, the median OS was not reached in the MF-0 group, was 22.8 years in the MF-1 group (HR=3.9), and was 15.4 years in the MF-2 and -3 groups combined (HR=7.4, P=0.001).
In the intermediate-2/high-risk categories, the median OS was 11 years for the MF-0 group, 3.6 years for the MF-1 group (HR=2.2), and 3.6 years in the MF-2 and -3 groups (HR=2.7, P=0.28).
Dr Guglielmelli therefore concluded that BM fibrosis grade might help refine prognostic stratification for PMF patients in the lower-risk IPSS categories. However, she noted that this study had limitations, and the results should be confirmed with prospective research. ![]()
*Data in the abstract differ from the presentation.
Five-year data suggest ruxolitinib improves survival in MF

ASH Annual Meeting
Photo courtesy of ASH
ORLANDO, FL—Five-year results from the COMFORT-II trial appear to confirm that treatment with ruxolitinib can improve spleen size and survival in patients with myelofibrosis (MF).
“These results pave the way to use ruxolitinib earlier in the course of the disease,” said lead study author Claire Harrison, MD, a consultant hematologist at Guy’s and St. Thomas’ NHS Foundation Trust in London, UK.
Dr Harrison presented the results at the 2015 ASH Annual Meeting (abstract 59).
Ruxolitinib, a JAK1/JAK2 inhibitor, has demonstrated rapid, durable improvements in splenomegaly and MF symptoms, as well as improved survival in the phase 3 COMFORT-I and COMFORT-II studies.
In COMFORT-II, significantly more patients achieved the primary endpoint—a 35% or greater decrease in spleen volume from baseline at week 48—with ruxolitinib than with best available therapy (BAT).
The 3-year follow-up confirmed that spleen volume reductions were sustained, and ruxolitinib treatment remained tolerable with long-term use.
The randomized, open-label, multicenter study included 219 patients with primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
Two-thirds of patients received ruxolitinib twice daily, and one-third of patients received BAT, which was administered at doses and schedules determined by the investigator.
Almost two-thirds of the patients on the BAT arm crossed over to receive ruxolitinib upon protocol-defined progression following the primary analysis after week 48. All patients randomized to BAT have crossed over or discontinued, Dr Harrison said.
She presented the 5-year final study results, which showed that more than half of the patients (53.4%) experienced significant reductions in spleen size with ruxolitinib therapy and sustained this benefit over a median duration of 3.2 years.
“There was a 33% improvement in overall survival with ruxolitinib as compared to BAT,” she said.
Using a statistical model of survival if patients had not crossed-over to ruxolitinib, the survival benefit was 56% in favor of ruxolitinib.
“The plateau in spleen responses correlates well with the survival advantage,” Dr Harrison said.
She noted that the JAK allele burden was also reduced in the majority of patients who crossed over during the study. A recent bone marrow analysis shows a 20% improvement in fibrosis as well.
Nearly one-quarter of patients from both the ruxolitinib arm and those who crossed over from the BAT arm remained on treatment with ruxolitinib for 5 years.
All adverse events were consistent with previous analyses of treatment with ruxolitinib in MF, Dr Harrison said. The most common adverse events in ruxolitinib-treated patients were thrombocytopenia (52.4%), anemia (49.2%), diarrhea (35.6%), and peripheral edema (33%).
The most common grade 3/4 adverse events included anemia (22.5%), thrombocytopenia (15.2%), pneumonia (5.8%), general physical health deterioration (4.2%), and shortness of breath (4.2%).
“This long-term analysis after a vast number of patient-years shows the ongoing benefit, with no new safety signals and a strong survival message,” Dr Harrison said.
“Hematologists can be confident treating patients with ruxolitinib. It is safe, effective, and leads to significant long-term benefit. Myelofibrosis patients feel better, their spleens are smaller, and they may survive longer.”
COMFORT-II was sponsored by Novartis, which licensed ruxolitinib from Incyte Corporation for development and commercialization outside the US. COMFORT-I was sponsored by Incyte. ![]()
ASH Annual Meeting
Photo courtesy of ASH
ORLANDO, FL—Five-year results from the COMFORT-II trial appear to confirm that treatment with ruxolitinib can improve spleen size and survival in patients with myelofibrosis (MF).
“These results pave the way to use ruxolitinib earlier in the course of the disease,” said lead study author Claire Harrison, MD, a consultant hematologist at Guy’s and St. Thomas’ NHS Foundation Trust in London, UK.
Dr Harrison presented the results at the 2015 ASH Annual Meeting (abstract 59).
Ruxolitinib, a JAK1/JAK2 inhibitor, has demonstrated rapid, durable improvements in splenomegaly and MF symptoms, as well as improved survival in the phase 3 COMFORT-I and COMFORT-II studies.
In COMFORT-II, significantly more patients achieved the primary endpoint—a 35% or greater decrease in spleen volume from baseline at week 48—with ruxolitinib than with best available therapy (BAT).
The 3-year follow-up confirmed that spleen volume reductions were sustained, and ruxolitinib treatment remained tolerable with long-term use.
The randomized, open-label, multicenter study included 219 patients with primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
Two-thirds of patients received ruxolitinib twice daily, and one-third of patients received BAT, which was administered at doses and schedules determined by the investigator.
Almost two-thirds of the patients on the BAT arm crossed over to receive ruxolitinib upon protocol-defined progression following the primary analysis after week 48. All patients randomized to BAT have crossed over or discontinued, Dr Harrison said.
She presented the 5-year final study results, which showed that more than half of the patients (53.4%) experienced significant reductions in spleen size with ruxolitinib therapy and sustained this benefit over a median duration of 3.2 years.
“There was a 33% improvement in overall survival with ruxolitinib as compared to BAT,” she said.
Using a statistical model of survival if patients had not crossed-over to ruxolitinib, the survival benefit was 56% in favor of ruxolitinib.
“The plateau in spleen responses correlates well with the survival advantage,” Dr Harrison said.
She noted that the JAK allele burden was also reduced in the majority of patients who crossed over during the study. A recent bone marrow analysis shows a 20% improvement in fibrosis as well.
Nearly one-quarter of patients from both the ruxolitinib arm and those who crossed over from the BAT arm remained on treatment with ruxolitinib for 5 years.
All adverse events were consistent with previous analyses of treatment with ruxolitinib in MF, Dr Harrison said. The most common adverse events in ruxolitinib-treated patients were thrombocytopenia (52.4%), anemia (49.2%), diarrhea (35.6%), and peripheral edema (33%).
The most common grade 3/4 adverse events included anemia (22.5%), thrombocytopenia (15.2%), pneumonia (5.8%), general physical health deterioration (4.2%), and shortness of breath (4.2%).
“This long-term analysis after a vast number of patient-years shows the ongoing benefit, with no new safety signals and a strong survival message,” Dr Harrison said.
“Hematologists can be confident treating patients with ruxolitinib. It is safe, effective, and leads to significant long-term benefit. Myelofibrosis patients feel better, their spleens are smaller, and they may survive longer.”
COMFORT-II was sponsored by Novartis, which licensed ruxolitinib from Incyte Corporation for development and commercialization outside the US. COMFORT-I was sponsored by Incyte. ![]()
ASH Annual Meeting
Photo courtesy of ASH
ORLANDO, FL—Five-year results from the COMFORT-II trial appear to confirm that treatment with ruxolitinib can improve spleen size and survival in patients with myelofibrosis (MF).
“These results pave the way to use ruxolitinib earlier in the course of the disease,” said lead study author Claire Harrison, MD, a consultant hematologist at Guy’s and St. Thomas’ NHS Foundation Trust in London, UK.
Dr Harrison presented the results at the 2015 ASH Annual Meeting (abstract 59).
Ruxolitinib, a JAK1/JAK2 inhibitor, has demonstrated rapid, durable improvements in splenomegaly and MF symptoms, as well as improved survival in the phase 3 COMFORT-I and COMFORT-II studies.
In COMFORT-II, significantly more patients achieved the primary endpoint—a 35% or greater decrease in spleen volume from baseline at week 48—with ruxolitinib than with best available therapy (BAT).
The 3-year follow-up confirmed that spleen volume reductions were sustained, and ruxolitinib treatment remained tolerable with long-term use.
The randomized, open-label, multicenter study included 219 patients with primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
Two-thirds of patients received ruxolitinib twice daily, and one-third of patients received BAT, which was administered at doses and schedules determined by the investigator.
Almost two-thirds of the patients on the BAT arm crossed over to receive ruxolitinib upon protocol-defined progression following the primary analysis after week 48. All patients randomized to BAT have crossed over or discontinued, Dr Harrison said.
She presented the 5-year final study results, which showed that more than half of the patients (53.4%) experienced significant reductions in spleen size with ruxolitinib therapy and sustained this benefit over a median duration of 3.2 years.
“There was a 33% improvement in overall survival with ruxolitinib as compared to BAT,” she said.
Using a statistical model of survival if patients had not crossed-over to ruxolitinib, the survival benefit was 56% in favor of ruxolitinib.
“The plateau in spleen responses correlates well with the survival advantage,” Dr Harrison said.
She noted that the JAK allele burden was also reduced in the majority of patients who crossed over during the study. A recent bone marrow analysis shows a 20% improvement in fibrosis as well.
Nearly one-quarter of patients from both the ruxolitinib arm and those who crossed over from the BAT arm remained on treatment with ruxolitinib for 5 years.
All adverse events were consistent with previous analyses of treatment with ruxolitinib in MF, Dr Harrison said. The most common adverse events in ruxolitinib-treated patients were thrombocytopenia (52.4%), anemia (49.2%), diarrhea (35.6%), and peripheral edema (33%).
The most common grade 3/4 adverse events included anemia (22.5%), thrombocytopenia (15.2%), pneumonia (5.8%), general physical health deterioration (4.2%), and shortness of breath (4.2%).
“This long-term analysis after a vast number of patient-years shows the ongoing benefit, with no new safety signals and a strong survival message,” Dr Harrison said.
“Hematologists can be confident treating patients with ruxolitinib. It is safe, effective, and leads to significant long-term benefit. Myelofibrosis patients feel better, their spleens are smaller, and they may survive longer.”
COMFORT-II was sponsored by Novartis, which licensed ruxolitinib from Incyte Corporation for development and commercialization outside the US. COMFORT-I was sponsored by Incyte. ![]()
FDA approves generic imatinib

Photo by Steven Harbour
The US Food and Drug Administration (FDA) has approved the use of imatinib mesylate, a generic version of Novartis’s Gleevec being developed by a subsidiary of Sun Pharmaceuticals Limited.
Under the terms of a settlement agreement with Novartis, the Sun Pharma subsidiary is allowed to launch its generic imatinib in the US on February 1, 2016.
The drug will be available in 100 mg and 400 mg tablets.
The Sun Pharma subsidiary was the first company to file an abbreviated new drug application for generic imatinib with a para IV certification and is therefore eligible for 180 days of marketing exclusivity in the US.
Sun Pharma’s imatinib mesylate is approved for the following indications:
- Newly diagnosed adult and pediatric patients with Philadelphia chromosome positive chronic myeloid leukemia (Ph+ CML) in chronic phase
- Patients with (Ph+ CML) in blast crisis, accelerated phase, or in chronic phase after failure of interferon-alpha therapy
- Adult patients with relapsed or refractory Ph+ acute lymphoblastic leukemia
- Adult patients with myelodysplastic/myeloproliferative diseases associated with PDGFR gene re-arrangements
- Adult patients with aggressive systemic mastocytosis without the D816V c-Kit mutation or with c-Kit mutational status unknown
- Adult patients with hypereosinophilic syndrome (HES) and/or chronic eosinophilic leukemia (CEL) who have the FIP1L1-PDGFRα fusion kinase and for patients with HES and/or CEL who are FIP1L1- PDGFRα fusion kinase negative or unknown
- Adult patients with unresectable, recurrent and/or metastatic dermatofibrosarcoma protuberans.
The drug is not approved to treat patients with KIT (CD117)-positive unresectable and/or metastatic malignant gastrointestinal stromal tumors. ![]()
Photo by Steven Harbour
The US Food and Drug Administration (FDA) has approved the use of imatinib mesylate, a generic version of Novartis’s Gleevec being developed by a subsidiary of Sun Pharmaceuticals Limited.
Under the terms of a settlement agreement with Novartis, the Sun Pharma subsidiary is allowed to launch its generic imatinib in the US on February 1, 2016.
The drug will be available in 100 mg and 400 mg tablets.
The Sun Pharma subsidiary was the first company to file an abbreviated new drug application for generic imatinib with a para IV certification and is therefore eligible for 180 days of marketing exclusivity in the US.
Sun Pharma’s imatinib mesylate is approved for the following indications:
- Newly diagnosed adult and pediatric patients with Philadelphia chromosome positive chronic myeloid leukemia (Ph+ CML) in chronic phase
- Patients with (Ph+ CML) in blast crisis, accelerated phase, or in chronic phase after failure of interferon-alpha therapy
- Adult patients with relapsed or refractory Ph+ acute lymphoblastic leukemia
- Adult patients with myelodysplastic/myeloproliferative diseases associated with PDGFR gene re-arrangements
- Adult patients with aggressive systemic mastocytosis without the D816V c-Kit mutation or with c-Kit mutational status unknown
- Adult patients with hypereosinophilic syndrome (HES) and/or chronic eosinophilic leukemia (CEL) who have the FIP1L1-PDGFRα fusion kinase and for patients with HES and/or CEL who are FIP1L1- PDGFRα fusion kinase negative or unknown
- Adult patients with unresectable, recurrent and/or metastatic dermatofibrosarcoma protuberans.
The drug is not approved to treat patients with KIT (CD117)-positive unresectable and/or metastatic malignant gastrointestinal stromal tumors. ![]()
Photo by Steven Harbour
The US Food and Drug Administration (FDA) has approved the use of imatinib mesylate, a generic version of Novartis’s Gleevec being developed by a subsidiary of Sun Pharmaceuticals Limited.
Under the terms of a settlement agreement with Novartis, the Sun Pharma subsidiary is allowed to launch its generic imatinib in the US on February 1, 2016.
The drug will be available in 100 mg and 400 mg tablets.
The Sun Pharma subsidiary was the first company to file an abbreviated new drug application for generic imatinib with a para IV certification and is therefore eligible for 180 days of marketing exclusivity in the US.
Sun Pharma’s imatinib mesylate is approved for the following indications:
- Newly diagnosed adult and pediatric patients with Philadelphia chromosome positive chronic myeloid leukemia (Ph+ CML) in chronic phase
- Patients with (Ph+ CML) in blast crisis, accelerated phase, or in chronic phase after failure of interferon-alpha therapy
- Adult patients with relapsed or refractory Ph+ acute lymphoblastic leukemia
- Adult patients with myelodysplastic/myeloproliferative diseases associated with PDGFR gene re-arrangements
- Adult patients with aggressive systemic mastocytosis without the D816V c-Kit mutation or with c-Kit mutational status unknown
- Adult patients with hypereosinophilic syndrome (HES) and/or chronic eosinophilic leukemia (CEL) who have the FIP1L1-PDGFRα fusion kinase and for patients with HES and/or CEL who are FIP1L1- PDGFRα fusion kinase negative or unknown
- Adult patients with unresectable, recurrent and/or metastatic dermatofibrosarcoma protuberans.
The drug is not approved to treat patients with KIT (CD117)-positive unresectable and/or metastatic malignant gastrointestinal stromal tumors.
Team characterizes EMH niche
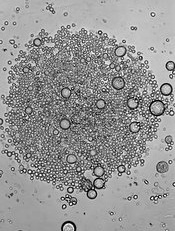
Image by John Perry
Previous studies have shown that hematopoietic stresses—such as myelofibrosis, anemia, and myeloablation—can induce extramedullary hematopoiesis (EMH), in which hematopoietic stem cells (HSCs) are mobilized to sites outside the bone marrow.
The splenic red pulp is known to be a prominent site of EMH in both mice and humans, but not much is known about the EMH niche.
Now, investigators say they have characterized this niche.
They detailed their findings in Nature.
The team used mouse models to examine the expression patterns of 2 known niche cell factors, SCF and CXCL12.
They discovered that the hematopoietic microenvironment in the spleen is found near sinusoidal blood vessels and is created by endothelial cells and perivascular stromal cells, just like the microenvironment in the bone marrow.
“Under emergency conditions, the endothelial cells and perivascular stromal cells that reside in the spleen are induced to proliferate so they can sustain all the new blood-forming stem cells that migrate into the spleen,” explained study author Sean Morrison, PhD, of the University of Texas Southwestern Medical Center, Dallas.
“We determined that this process in the spleen is physiologically important for responding to hematopoietic stress. Without it, the mice we studied could not maintain normal blood cell counts during pregnancy or quickly regenerate blood cell counts after bleeding or chemotherapy.”
The investigators believe these findings could aid the development of therapeutic interventions to enhance blood formation following chemotherapy or HSC transplant and thus accelerate the recovery of blood cell counts.
Image by John Perry
Previous studies have shown that hematopoietic stresses—such as myelofibrosis, anemia, and myeloablation—can induce extramedullary hematopoiesis (EMH), in which hematopoietic stem cells (HSCs) are mobilized to sites outside the bone marrow.
The splenic red pulp is known to be a prominent site of EMH in both mice and humans, but not much is known about the EMH niche.
Now, investigators say they have characterized this niche.
They detailed their findings in Nature.
The team used mouse models to examine the expression patterns of 2 known niche cell factors, SCF and CXCL12.
They discovered that the hematopoietic microenvironment in the spleen is found near sinusoidal blood vessels and is created by endothelial cells and perivascular stromal cells, just like the microenvironment in the bone marrow.
“Under emergency conditions, the endothelial cells and perivascular stromal cells that reside in the spleen are induced to proliferate so they can sustain all the new blood-forming stem cells that migrate into the spleen,” explained study author Sean Morrison, PhD, of the University of Texas Southwestern Medical Center, Dallas.
“We determined that this process in the spleen is physiologically important for responding to hematopoietic stress. Without it, the mice we studied could not maintain normal blood cell counts during pregnancy or quickly regenerate blood cell counts after bleeding or chemotherapy.”
The investigators believe these findings could aid the development of therapeutic interventions to enhance blood formation following chemotherapy or HSC transplant and thus accelerate the recovery of blood cell counts.
Image by John Perry
Previous studies have shown that hematopoietic stresses—such as myelofibrosis, anemia, and myeloablation—can induce extramedullary hematopoiesis (EMH), in which hematopoietic stem cells (HSCs) are mobilized to sites outside the bone marrow.
The splenic red pulp is known to be a prominent site of EMH in both mice and humans, but not much is known about the EMH niche.
Now, investigators say they have characterized this niche.
They detailed their findings in Nature.
The team used mouse models to examine the expression patterns of 2 known niche cell factors, SCF and CXCL12.
They discovered that the hematopoietic microenvironment in the spleen is found near sinusoidal blood vessels and is created by endothelial cells and perivascular stromal cells, just like the microenvironment in the bone marrow.
“Under emergency conditions, the endothelial cells and perivascular stromal cells that reside in the spleen are induced to proliferate so they can sustain all the new blood-forming stem cells that migrate into the spleen,” explained study author Sean Morrison, PhD, of the University of Texas Southwestern Medical Center, Dallas.
“We determined that this process in the spleen is physiologically important for responding to hematopoietic stress. Without it, the mice we studied could not maintain normal blood cell counts during pregnancy or quickly regenerate blood cell counts after bleeding or chemotherapy.”
The investigators believe these findings could aid the development of therapeutic interventions to enhance blood formation following chemotherapy or HSC transplant and thus accelerate the recovery of blood cell counts.
Haplo-HSCT appears comparable to fully matched HSCT
![]()
Photo by Chad McNeeley
A retrospective study suggests that, for patients with hematologic disorders, a haploidentical hematopoietic stem cell transplant (HSCT)
can be roughly as safe and effective as a fully matched HSCT.
The study showed that, when patients received an identical conditioning regimen, graft T-cell dose, and graft-vs-host disease (GVHD) prophylaxis, haploidentical and fully matched HSCTs produced comparable results.
Patients had similar rates of overall and progression-free survival, relapse, non-relapse mortality, and chronic GVHD.
However, patients who received haploidentical transplants had higher rates of grade 2-4 acute GVHD and cytomegalovirus reactivation.
Researchers reported these results in Biology of Blood and Marrow Transplantation.
“This is the first study to compare the gold standard to a half-match using an identical protocol,” said Neal Flomenberg, MD, of Thomas Jefferson University in Philadelphia, Pennsylvania.
“The field has debated whether the differences in outcomes between full and partial matches were caused by the quality of the match or by all the procedures the patient goes through before and after the donor cells are administered. We haven’t had a clear answer.”
With that in mind, Dr Flomenberg and his colleagues compared 3-year outcome data from patients who received haploidentical HSCTs (n=50) or fully matched HSCTs (n=27), when both groups of patients were treated with a 2-step protocol.
The patients had acute myeloid leukemia (n=38), acute lymphoblastic leukemia (n=20), myelodysplastic syndromes/myeloproliferative neoplasms (n=7), non-Hodgkin lymphoma (n=11), and aplastic anemia (n=1).
The 2-step protocol
All patients received a myeloablative conditioning regimen consisting of 12 Gy of total body irradiation administered in 8 fractions over 4 days. After the last fraction, they received a fixed T-cell dose (2 x 108 cells/kg), which was followed, 2 days later, by cyclophosphamide at 60 mg/kg/day for 2 days.
Twenty-four hours after they completed cyclophosphamide, patients received CD34-selected peripheral blood stem cells from a half-matched or fully matched donor.
On day -1, patients began taking tacrolimus and mycophenolate mofetil as GVHD prophylaxis. They also received growth factor support (granulocyte-macrophage colony-stimulating factor at 250 μg/m2) starting on day +1.
In the absence of GVHD, mycophenolate mofetil was discontinued on day 28 and tacrolimus was tapered, starting on day +60 after HSCT.
Results
The researchers said that early immune recovery was comparable between the patient groups in nearly all assessed T-cell subsets. The exception was the median CD3/CD8 cell count, which was significantly higher at day 28 in the fully matched group than the haploidentical group (P=0.029).
Survival rates were comparable between the groups. The estimated 3-year overall survival was 70% in the haploidentical group and 71% in the fully matched group (P=0.81). The 3-year progression-free survival was 68% and 70%, respectively (P=0.97).
The 3-year cumulative incidence of non-relapse mortality was 10% in the haploidentical group and 4% in the fully matched group (P=0.34). The 3-year cumulative incidence of relapse was 21% and 27%, respectively (P=0.93).
The 100-day cumulative incidence of grade 2-4 acute GVHD was significantly higher in the haploidentical group than the fully matched group—40% and 8%, respectively (P<0.001). But there was no significant difference in the incidence of grade 3-4 acute GVHD—6% and 4%, respectively (P=0.49).
The cumulative incidence of chronic GVHD at 2 years was not significantly different between the haploidentical and fully matched groups—19% and 12%, respectively (P=0.47). The same was true for severe chronic GVHD—4% and 8%, respectively (P=0.49).
The cumulative incidence of cytomegalovirus reactivation was significantly higher in the haploidentical group than the fully matched group—68% and 19%, respectively (P<0.001).
There were no deaths from infections or GVHD in either group.
“The results of the current study are certainly encouraging and suggest that outcomes from a half-matched, related donor are similar to fully matched donors,” said study author Sameh Gaballa, MD, also of Thomas Jefferson University.
“It might be time to reassess whether half-matched, related transplants can be considered the best alternative donor source for patients lacking a fully matched family member donor. For that, we’ll need more evidence from a randomly controlled, prospective trial, rather than studies that look at patient data retrospectively, to help solidify our findings here.”
![]()
Photo by Chad McNeeley
A retrospective study suggests that, for patients with hematologic disorders, a haploidentical hematopoietic stem cell transplant (HSCT)
can be roughly as safe and effective as a fully matched HSCT.
The study showed that, when patients received an identical conditioning regimen, graft T-cell dose, and graft-vs-host disease (GVHD) prophylaxis, haploidentical and fully matched HSCTs produced comparable results.
Patients had similar rates of overall and progression-free survival, relapse, non-relapse mortality, and chronic GVHD.
However, patients who received haploidentical transplants had higher rates of grade 2-4 acute GVHD and cytomegalovirus reactivation.
Researchers reported these results in Biology of Blood and Marrow Transplantation.
“This is the first study to compare the gold standard to a half-match using an identical protocol,” said Neal Flomenberg, MD, of Thomas Jefferson University in Philadelphia, Pennsylvania.
“The field has debated whether the differences in outcomes between full and partial matches were caused by the quality of the match or by all the procedures the patient goes through before and after the donor cells are administered. We haven’t had a clear answer.”
With that in mind, Dr Flomenberg and his colleagues compared 3-year outcome data from patients who received haploidentical HSCTs (n=50) or fully matched HSCTs (n=27), when both groups of patients were treated with a 2-step protocol.
The patients had acute myeloid leukemia (n=38), acute lymphoblastic leukemia (n=20), myelodysplastic syndromes/myeloproliferative neoplasms (n=7), non-Hodgkin lymphoma (n=11), and aplastic anemia (n=1).
The 2-step protocol
All patients received a myeloablative conditioning regimen consisting of 12 Gy of total body irradiation administered in 8 fractions over 4 days. After the last fraction, they received a fixed T-cell dose (2 x 108 cells/kg), which was followed, 2 days later, by cyclophosphamide at 60 mg/kg/day for 2 days.
Twenty-four hours after they completed cyclophosphamide, patients received CD34-selected peripheral blood stem cells from a half-matched or fully matched donor.
On day -1, patients began taking tacrolimus and mycophenolate mofetil as GVHD prophylaxis. They also received growth factor support (granulocyte-macrophage colony-stimulating factor at 250 μg/m2) starting on day +1.
In the absence of GVHD, mycophenolate mofetil was discontinued on day 28 and tacrolimus was tapered, starting on day +60 after HSCT.
Results
The researchers said that early immune recovery was comparable between the patient groups in nearly all assessed T-cell subsets. The exception was the median CD3/CD8 cell count, which was significantly higher at day 28 in the fully matched group than the haploidentical group (P=0.029).
Survival rates were comparable between the groups. The estimated 3-year overall survival was 70% in the haploidentical group and 71% in the fully matched group (P=0.81). The 3-year progression-free survival was 68% and 70%, respectively (P=0.97).
The 3-year cumulative incidence of non-relapse mortality was 10% in the haploidentical group and 4% in the fully matched group (P=0.34). The 3-year cumulative incidence of relapse was 21% and 27%, respectively (P=0.93).
The 100-day cumulative incidence of grade 2-4 acute GVHD was significantly higher in the haploidentical group than the fully matched group—40% and 8%, respectively (P<0.001). But there was no significant difference in the incidence of grade 3-4 acute GVHD—6% and 4%, respectively (P=0.49).
The cumulative incidence of chronic GVHD at 2 years was not significantly different between the haploidentical and fully matched groups—19% and 12%, respectively (P=0.47). The same was true for severe chronic GVHD—4% and 8%, respectively (P=0.49).
The cumulative incidence of cytomegalovirus reactivation was significantly higher in the haploidentical group than the fully matched group—68% and 19%, respectively (P<0.001).
There were no deaths from infections or GVHD in either group.
“The results of the current study are certainly encouraging and suggest that outcomes from a half-matched, related donor are similar to fully matched donors,” said study author Sameh Gaballa, MD, also of Thomas Jefferson University.
“It might be time to reassess whether half-matched, related transplants can be considered the best alternative donor source for patients lacking a fully matched family member donor. For that, we’ll need more evidence from a randomly controlled, prospective trial, rather than studies that look at patient data retrospectively, to help solidify our findings here.”
![]()
Photo by Chad McNeeley
A retrospective study suggests that, for patients with hematologic disorders, a haploidentical hematopoietic stem cell transplant (HSCT)
can be roughly as safe and effective as a fully matched HSCT.
The study showed that, when patients received an identical conditioning regimen, graft T-cell dose, and graft-vs-host disease (GVHD) prophylaxis, haploidentical and fully matched HSCTs produced comparable results.
Patients had similar rates of overall and progression-free survival, relapse, non-relapse mortality, and chronic GVHD.
However, patients who received haploidentical transplants had higher rates of grade 2-4 acute GVHD and cytomegalovirus reactivation.
Researchers reported these results in Biology of Blood and Marrow Transplantation.
“This is the first study to compare the gold standard to a half-match using an identical protocol,” said Neal Flomenberg, MD, of Thomas Jefferson University in Philadelphia, Pennsylvania.
“The field has debated whether the differences in outcomes between full and partial matches were caused by the quality of the match or by all the procedures the patient goes through before and after the donor cells are administered. We haven’t had a clear answer.”
With that in mind, Dr Flomenberg and his colleagues compared 3-year outcome data from patients who received haploidentical HSCTs (n=50) or fully matched HSCTs (n=27), when both groups of patients were treated with a 2-step protocol.
The patients had acute myeloid leukemia (n=38), acute lymphoblastic leukemia (n=20), myelodysplastic syndromes/myeloproliferative neoplasms (n=7), non-Hodgkin lymphoma (n=11), and aplastic anemia (n=1).
The 2-step protocol
All patients received a myeloablative conditioning regimen consisting of 12 Gy of total body irradiation administered in 8 fractions over 4 days. After the last fraction, they received a fixed T-cell dose (2 x 108 cells/kg), which was followed, 2 days later, by cyclophosphamide at 60 mg/kg/day for 2 days.
Twenty-four hours after they completed cyclophosphamide, patients received CD34-selected peripheral blood stem cells from a half-matched or fully matched donor.
On day -1, patients began taking tacrolimus and mycophenolate mofetil as GVHD prophylaxis. They also received growth factor support (granulocyte-macrophage colony-stimulating factor at 250 μg/m2) starting on day +1.
In the absence of GVHD, mycophenolate mofetil was discontinued on day 28 and tacrolimus was tapered, starting on day +60 after HSCT.
Results
The researchers said that early immune recovery was comparable between the patient groups in nearly all assessed T-cell subsets. The exception was the median CD3/CD8 cell count, which was significantly higher at day 28 in the fully matched group than the haploidentical group (P=0.029).
Survival rates were comparable between the groups. The estimated 3-year overall survival was 70% in the haploidentical group and 71% in the fully matched group (P=0.81). The 3-year progression-free survival was 68% and 70%, respectively (P=0.97).
The 3-year cumulative incidence of non-relapse mortality was 10% in the haploidentical group and 4% in the fully matched group (P=0.34). The 3-year cumulative incidence of relapse was 21% and 27%, respectively (P=0.93).
The 100-day cumulative incidence of grade 2-4 acute GVHD was significantly higher in the haploidentical group than the fully matched group—40% and 8%, respectively (P<0.001). But there was no significant difference in the incidence of grade 3-4 acute GVHD—6% and 4%, respectively (P=0.49).
The cumulative incidence of chronic GVHD at 2 years was not significantly different between the haploidentical and fully matched groups—19% and 12%, respectively (P=0.47). The same was true for severe chronic GVHD—4% and 8%, respectively (P=0.49).
The cumulative incidence of cytomegalovirus reactivation was significantly higher in the haploidentical group than the fully matched group—68% and 19%, respectively (P<0.001).
There were no deaths from infections or GVHD in either group.
“The results of the current study are certainly encouraging and suggest that outcomes from a half-matched, related donor are similar to fully matched donors,” said study author Sameh Gaballa, MD, also of Thomas Jefferson University.
“It might be time to reassess whether half-matched, related transplants can be considered the best alternative donor source for patients lacking a fully matched family member donor. For that, we’ll need more evidence from a randomly controlled, prospective trial, rather than studies that look at patient data retrospectively, to help solidify our findings here.”
Despite responses, imetelstat won’t move forward in ET
Although trial results suggest imetelstat is effective against essential thrombocythemia (ET), the telomerase inhibitor is only being developed to treat myelofibrosis (MF).
Imetelstat produced “rapid and durable” responses in a phase 2 trial of ET patients, but the drug also produced side effects that caused the US Food and Drug Administration (FDA) to place a full clinical hold on the drug.
The hold was lifted last November, but the company developing imetelstat decided not to pursue the drug as a treatment for ET or polycythemia vera.
Development continues for MF, however, and results of the pilot study of imetelstat in MF appear in NEJM.
Results from the trial of imetelstat in ET have been published in NEJM as well. Both studies were funded by Geron Corporation, the company developing imetelstat.
The ET trial included 18 patients with a median age of 59.5 (range, 21-83). All patients had received one or more prior treatments, including hydroxyurea (n=17, 94%), anagrelide (n=13, 72%), and interferon (n=4, 22%). Half of patients (n=9) were resistant to at least 1 prior therapy, and 78% (n=14) had experienced unacceptable side effects from a previous therapy.
The patients received imetelstat at an initial dose of 7.5 or 9.4 mg per kilogram of body weight intravenously once a week. Treatment continued until patients attained a platelet count of approximately 250,000 to 300,000 per cubic millimeter.
Responses
All 18 patients experienced hematologic responses, and 16 (89%) had a complete hematologic response. At a median follow-up of 17 months, 10 patients were still receiving treatment, and the median duration of response had not been reached (range, 5 to 30 months).
“[I]metelstat had a clinically significant effect on disease burden in ET patients,” said study investigator David Snyder, MD, of City of Hope in Duarte, California.
“This study was a first look at what happens when you treat ET patients with a drug that has a totally novel mechanism of action.”
Seven of the 8 patients (88%) who were positive for the JAK2 V617F mutation had a molecular response. All of the patients with CALR (n=5) or MPL (n=2) mutations saw a reduction in mutant allele burden, ranging from 15% to 66%.
“The molecular responses suggest that imetelstat may have broad activity across hematologic myeloid malignancies, which warrants further clinical study in other myeloproliferative neoplasms,” said investigator Gabriela M. Baerlocher, MD, of the University of Bern in Switzerland.
Toxicity, clinical hold, and discontinuation
The most common adverse events (≥50%) in this trial were fatigue (83%), diarrhea (78%), nausea (72%), dizziness (61%), increased alanine aminotransferase (ALT, 56%), increased aspartate aminotransferase (AST, 56%), constipation (50%), cough (50%), epistaxis (50%), and headache (50%).
Grade 3/4 adverse events included decreased neutrophil count (22%), neutropenia (22%), anemia (11%), syncope (11%), headache (11%), upper respiratory tract infection (6%), decreased white cell count (6%), myalgia (6%), hypokalemia (6%), fatigue (6%), cellulitis (6%), increased ALT (6%), increased AST (6%), and epistaxis (6%).
All 18 patients had at least one increase in grade, from baseline, in a liver-function value. Seventeen patients had an elevation in ALT, 17 in AST, 15 in alkaline phosphatase, and 8 in total bilirubin. Fourteen patients had persistent abnormalities (≥ 6 weeks), all of which were grade 1 or 2 in severity.
It was these liver-function abnormalities and the potential risk of chronic liver injury that prompted the FDA to place a clinical hold on imetelstat. The agency was concerned about whether the effects were reversible.
It turned out that, in many cases, abnormalities resolved after patients permanently discontinued treatment (as a result of the clinical hold). Sixteen of 17 patients with elevated ALT experienced a resolution, as did 12 of 17 patients with elevated AST, 9 of 15 with elevated alkaline phosphatase, and 7 of 8 with elevated bilirubin.
Of the 14 patients who had persistent abnormalities, 11 had their values reversed to normal or baseline values after stopping imetelstat. The median time to resolution after treatment discontinuation was 12 weeks.
Still, Geron Corporation decided not to develop imetelstat for patients with ET or polycythemia vera. And the FDA said the proposed clinical development plan for the drug, which is focused on MF and other high-risk myeloid disorders, was acceptable. So the clinical hold was lifted.
Although trial results suggest imetelstat is effective against essential thrombocythemia (ET), the telomerase inhibitor is only being developed to treat myelofibrosis (MF).
Imetelstat produced “rapid and durable” responses in a phase 2 trial of ET patients, but the drug also produced side effects that caused the US Food and Drug Administration (FDA) to place a full clinical hold on the drug.
The hold was lifted last November, but the company developing imetelstat decided not to pursue the drug as a treatment for ET or polycythemia vera.
Development continues for MF, however, and results of the pilot study of imetelstat in MF appear in NEJM.
Results from the trial of imetelstat in ET have been published in NEJM as well. Both studies were funded by Geron Corporation, the company developing imetelstat.
The ET trial included 18 patients with a median age of 59.5 (range, 21-83). All patients had received one or more prior treatments, including hydroxyurea (n=17, 94%), anagrelide (n=13, 72%), and interferon (n=4, 22%). Half of patients (n=9) were resistant to at least 1 prior therapy, and 78% (n=14) had experienced unacceptable side effects from a previous therapy.
The patients received imetelstat at an initial dose of 7.5 or 9.4 mg per kilogram of body weight intravenously once a week. Treatment continued until patients attained a platelet count of approximately 250,000 to 300,000 per cubic millimeter.
Responses
All 18 patients experienced hematologic responses, and 16 (89%) had a complete hematologic response. At a median follow-up of 17 months, 10 patients were still receiving treatment, and the median duration of response had not been reached (range, 5 to 30 months).
“[I]metelstat had a clinically significant effect on disease burden in ET patients,” said study investigator David Snyder, MD, of City of Hope in Duarte, California.
“This study was a first look at what happens when you treat ET patients with a drug that has a totally novel mechanism of action.”
Seven of the 8 patients (88%) who were positive for the JAK2 V617F mutation had a molecular response. All of the patients with CALR (n=5) or MPL (n=2) mutations saw a reduction in mutant allele burden, ranging from 15% to 66%.
“The molecular responses suggest that imetelstat may have broad activity across hematologic myeloid malignancies, which warrants further clinical study in other myeloproliferative neoplasms,” said investigator Gabriela M. Baerlocher, MD, of the University of Bern in Switzerland.
Toxicity, clinical hold, and discontinuation
The most common adverse events (≥50%) in this trial were fatigue (83%), diarrhea (78%), nausea (72%), dizziness (61%), increased alanine aminotransferase (ALT, 56%), increased aspartate aminotransferase (AST, 56%), constipation (50%), cough (50%), epistaxis (50%), and headache (50%).
Grade 3/4 adverse events included decreased neutrophil count (22%), neutropenia (22%), anemia (11%), syncope (11%), headache (11%), upper respiratory tract infection (6%), decreased white cell count (6%), myalgia (6%), hypokalemia (6%), fatigue (6%), cellulitis (6%), increased ALT (6%), increased AST (6%), and epistaxis (6%).
All 18 patients had at least one increase in grade, from baseline, in a liver-function value. Seventeen patients had an elevation in ALT, 17 in AST, 15 in alkaline phosphatase, and 8 in total bilirubin. Fourteen patients had persistent abnormalities (≥ 6 weeks), all of which were grade 1 or 2 in severity.
It was these liver-function abnormalities and the potential risk of chronic liver injury that prompted the FDA to place a clinical hold on imetelstat. The agency was concerned about whether the effects were reversible.
It turned out that, in many cases, abnormalities resolved after patients permanently discontinued treatment (as a result of the clinical hold). Sixteen of 17 patients with elevated ALT experienced a resolution, as did 12 of 17 patients with elevated AST, 9 of 15 with elevated alkaline phosphatase, and 7 of 8 with elevated bilirubin.
Of the 14 patients who had persistent abnormalities, 11 had their values reversed to normal or baseline values after stopping imetelstat. The median time to resolution after treatment discontinuation was 12 weeks.
Still, Geron Corporation decided not to develop imetelstat for patients with ET or polycythemia vera. And the FDA said the proposed clinical development plan for the drug, which is focused on MF and other high-risk myeloid disorders, was acceptable. So the clinical hold was lifted.
Although trial results suggest imetelstat is effective against essential thrombocythemia (ET), the telomerase inhibitor is only being developed to treat myelofibrosis (MF).
Imetelstat produced “rapid and durable” responses in a phase 2 trial of ET patients, but the drug also produced side effects that caused the US Food and Drug Administration (FDA) to place a full clinical hold on the drug.
The hold was lifted last November, but the company developing imetelstat decided not to pursue the drug as a treatment for ET or polycythemia vera.
Development continues for MF, however, and results of the pilot study of imetelstat in MF appear in NEJM.
Results from the trial of imetelstat in ET have been published in NEJM as well. Both studies were funded by Geron Corporation, the company developing imetelstat.
The ET trial included 18 patients with a median age of 59.5 (range, 21-83). All patients had received one or more prior treatments, including hydroxyurea (n=17, 94%), anagrelide (n=13, 72%), and interferon (n=4, 22%). Half of patients (n=9) were resistant to at least 1 prior therapy, and 78% (n=14) had experienced unacceptable side effects from a previous therapy.
The patients received imetelstat at an initial dose of 7.5 or 9.4 mg per kilogram of body weight intravenously once a week. Treatment continued until patients attained a platelet count of approximately 250,000 to 300,000 per cubic millimeter.
Responses
All 18 patients experienced hematologic responses, and 16 (89%) had a complete hematologic response. At a median follow-up of 17 months, 10 patients were still receiving treatment, and the median duration of response had not been reached (range, 5 to 30 months).
“[I]metelstat had a clinically significant effect on disease burden in ET patients,” said study investigator David Snyder, MD, of City of Hope in Duarte, California.
“This study was a first look at what happens when you treat ET patients with a drug that has a totally novel mechanism of action.”
Seven of the 8 patients (88%) who were positive for the JAK2 V617F mutation had a molecular response. All of the patients with CALR (n=5) or MPL (n=2) mutations saw a reduction in mutant allele burden, ranging from 15% to 66%.
“The molecular responses suggest that imetelstat may have broad activity across hematologic myeloid malignancies, which warrants further clinical study in other myeloproliferative neoplasms,” said investigator Gabriela M. Baerlocher, MD, of the University of Bern in Switzerland.
Toxicity, clinical hold, and discontinuation
The most common adverse events (≥50%) in this trial were fatigue (83%), diarrhea (78%), nausea (72%), dizziness (61%), increased alanine aminotransferase (ALT, 56%), increased aspartate aminotransferase (AST, 56%), constipation (50%), cough (50%), epistaxis (50%), and headache (50%).
Grade 3/4 adverse events included decreased neutrophil count (22%), neutropenia (22%), anemia (11%), syncope (11%), headache (11%), upper respiratory tract infection (6%), decreased white cell count (6%), myalgia (6%), hypokalemia (6%), fatigue (6%), cellulitis (6%), increased ALT (6%), increased AST (6%), and epistaxis (6%).
All 18 patients had at least one increase in grade, from baseline, in a liver-function value. Seventeen patients had an elevation in ALT, 17 in AST, 15 in alkaline phosphatase, and 8 in total bilirubin. Fourteen patients had persistent abnormalities (≥ 6 weeks), all of which were grade 1 or 2 in severity.
It was these liver-function abnormalities and the potential risk of chronic liver injury that prompted the FDA to place a clinical hold on imetelstat. The agency was concerned about whether the effects were reversible.
It turned out that, in many cases, abnormalities resolved after patients permanently discontinued treatment (as a result of the clinical hold). Sixteen of 17 patients with elevated ALT experienced a resolution, as did 12 of 17 patients with elevated AST, 9 of 15 with elevated alkaline phosphatase, and 7 of 8 with elevated bilirubin.
Of the 14 patients who had persistent abnormalities, 11 had their values reversed to normal or baseline values after stopping imetelstat. The median time to resolution after treatment discontinuation was 12 weeks.
Still, Geron Corporation decided not to develop imetelstat for patients with ET or polycythemia vera. And the FDA said the proposed clinical development plan for the drug, which is focused on MF and other high-risk myeloid disorders, was acceptable. So the clinical hold was lifted.