User login
Sandoz halts pursuit of U.S. approval for rituximab biosimilar
.
Sandoz, a division of Novartis, was seeking Food and Drug Administration approval of GP2013 for all the same indications as the reference product – B-cell non-Hodgkin lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis, microscopic polyangiitis, and pemphigus vulgaris.
GP2013 already is approved in the European Union and elsewhere.
The FDA had accepted the biologics license application (BLA) for GP2013 in September 2017. In May 2018, the agency issued a complete response letter saying it could not approve GP2013. The agency also requested additional information to complement the BLA submission.
At the time of the complete response letter, Sandoz said it was still committed to bringing GP2013 to the U.S. market.
“We appreciate the important conversations with the FDA, which have provided specific requirements for our potential U.S. biosimilar rituximab but believe the patient and marketplace needs in the U.S. will be satisfied before we can generate the data required,” Stefan Hendriks, global head of biopharmaceuticals at Sandoz, said in a statement.
“We are disappointed to have to make this decision and stand behind the safety, efficacy, and quality of our medicine, which met the stringent criteria for approval in the European Union, Switzerland, Japan, New Zealand, and Australia.”
The BLA for GP2013 was supported, in part, by results from the ASSIST-FL trial, in which researchers compared GP2013 with the reference product (Lancet Haematol. 2017 Aug;4[8]:e350-61).
The phase 3 trial included adults with previously untreated, advanced-stage follicular lymphoma. Patients received eight cycles of cyclophosphamide, vincristine, and prednisone with either GP2013 or reference rituximab. Responders then received GP2013 or rituximab monotherapy as maintenance for up to 2 years.
At a median follow-up of 11.6 months, the overall response rate was 87% in the GP2013 arm and 88% in the rituximab arm.
.
Sandoz, a division of Novartis, was seeking Food and Drug Administration approval of GP2013 for all the same indications as the reference product – B-cell non-Hodgkin lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis, microscopic polyangiitis, and pemphigus vulgaris.
GP2013 already is approved in the European Union and elsewhere.
The FDA had accepted the biologics license application (BLA) for GP2013 in September 2017. In May 2018, the agency issued a complete response letter saying it could not approve GP2013. The agency also requested additional information to complement the BLA submission.
At the time of the complete response letter, Sandoz said it was still committed to bringing GP2013 to the U.S. market.
“We appreciate the important conversations with the FDA, which have provided specific requirements for our potential U.S. biosimilar rituximab but believe the patient and marketplace needs in the U.S. will be satisfied before we can generate the data required,” Stefan Hendriks, global head of biopharmaceuticals at Sandoz, said in a statement.
“We are disappointed to have to make this decision and stand behind the safety, efficacy, and quality of our medicine, which met the stringent criteria for approval in the European Union, Switzerland, Japan, New Zealand, and Australia.”
The BLA for GP2013 was supported, in part, by results from the ASSIST-FL trial, in which researchers compared GP2013 with the reference product (Lancet Haematol. 2017 Aug;4[8]:e350-61).
The phase 3 trial included adults with previously untreated, advanced-stage follicular lymphoma. Patients received eight cycles of cyclophosphamide, vincristine, and prednisone with either GP2013 or reference rituximab. Responders then received GP2013 or rituximab monotherapy as maintenance for up to 2 years.
At a median follow-up of 11.6 months, the overall response rate was 87% in the GP2013 arm and 88% in the rituximab arm.
.
Sandoz, a division of Novartis, was seeking Food and Drug Administration approval of GP2013 for all the same indications as the reference product – B-cell non-Hodgkin lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis, microscopic polyangiitis, and pemphigus vulgaris.
GP2013 already is approved in the European Union and elsewhere.
The FDA had accepted the biologics license application (BLA) for GP2013 in September 2017. In May 2018, the agency issued a complete response letter saying it could not approve GP2013. The agency also requested additional information to complement the BLA submission.
At the time of the complete response letter, Sandoz said it was still committed to bringing GP2013 to the U.S. market.
“We appreciate the important conversations with the FDA, which have provided specific requirements for our potential U.S. biosimilar rituximab but believe the patient and marketplace needs in the U.S. will be satisfied before we can generate the data required,” Stefan Hendriks, global head of biopharmaceuticals at Sandoz, said in a statement.
“We are disappointed to have to make this decision and stand behind the safety, efficacy, and quality of our medicine, which met the stringent criteria for approval in the European Union, Switzerland, Japan, New Zealand, and Australia.”
The BLA for GP2013 was supported, in part, by results from the ASSIST-FL trial, in which researchers compared GP2013 with the reference product (Lancet Haematol. 2017 Aug;4[8]:e350-61).
The phase 3 trial included adults with previously untreated, advanced-stage follicular lymphoma. Patients received eight cycles of cyclophosphamide, vincristine, and prednisone with either GP2013 or reference rituximab. Responders then received GP2013 or rituximab monotherapy as maintenance for up to 2 years.
At a median follow-up of 11.6 months, the overall response rate was 87% in the GP2013 arm and 88% in the rituximab arm.
Checkpoint inhibitor plus rituximab is active in non-Hodgkin lymphoma
A macrophage-activating immune checkpoint inhibitor, combined with rituximab therapy, was safe and produced durable complete responses in patients with relapsed or refractory non-Hodgkin lymphoma, according to results of a phase 1b study.

Mainly low-grade toxic effects were seen on treatment with Hu5F9-G4 (5F9) and rituximab, which induced responses in more than half of patients, of which more than one-third were complete responses, the study investigators reported.
Most of the responses were ongoing at the time of data cutoff, suggesting durable responses with the combination of rituximab and 5F9 – a humanized monoclonal antibody that blocks CD47, an antiphagocytic or “do not eat me” signal overexpressed by most cancers, Ranjana Advani, MD, of Stanford (Calif.) University, and her coauthors wrote.
“The macrophage-mediated activity of 5F9 plus rituximab may serve as an effective new immunotherapy for stimulating the innate immune system,” Dr. Advani and her colleagues reported in the New England Journal of Medicine.
The study included 22 patients, including 15 with diffuse large B-cell lymphoma (DLBCL) and 7 with follicular lymphoma, who had received a median of four prior therapies. Almost all of the non-Hodgkin lymphomas (21, or 95%) were refractory to rituximab.
All patients received intravenous 5F9 starting with a priming dose of 1 mg/kg followed by weekly maintenance doses of 10-30 mg/kg in three dose-escalation cohorts, given until disease progression or lack of clinical benefit. Intravenous rituximab at 375 mg/m2 weekly was started on the second week of the first cycle, and then monthly for cycles 2 through 6.
“Substantial antitumor activity” was seen with this chemotherapy-free regimen in a group of heavily pretreated, largely rituximab-refractory patients, Dr. Advani and her coauthors wrote in their report.
The objective response rate was 50%, including a 36% complete response rate in the intent-to-treat analysis. For DLBCL, the rates of objective and complete responses were 40% and 33%, while for follicular lymphoma, they were 71% and 43%.
The median duration of response was not reached in either disease cohort with a median follow-up of 6.2 months for DLBCL and 8.1 months for follicular lymphoma. Of the 11 patients who responded, 10 (91%) were still in response at the time of data cutoff. “Longer follow-up is needed,” the investigators wrote.
Most adverse events were seen within the first few weeks of treatment and mainly included anemia and infusion-related reactions. The anemia was an expected, on-target effect of 5F9 because of selective clearance of older red cells, which was predictable, transient, and mitigated by the maintenance dosing strategy employed in this phase 1b trial.
“As red cells age, they lose CD47 expression and gain expression of prophagocytic signals, leading to homeostatic clearance,” they wrote.
The activity of 5F9 and rituximab is “synergistic” based on the results of previous, preclinical investigations in models of lymphoma, Dr. Advani and her coauthors added.
A phase 2 trial of 5F9 plus rituximab in relapsed or refractory B-cell non-Hodgkin lymphoma is ongoing, according to their report.
The study was supported by Forty Seven and the Leukemia & Lymphoma Society. Dr. Advani reported disclosures related to Forty Seven, Bristol-Myers Squibb, Pharmacyclics, Seattle Genetics, and Roche/Genentech, among others.
SOURCE: Advani R et al. N Engl J Med. 2018;379:1711-21.
A macrophage-activating immune checkpoint inhibitor, combined with rituximab therapy, was safe and produced durable complete responses in patients with relapsed or refractory non-Hodgkin lymphoma, according to results of a phase 1b study.
Mainly low-grade toxic effects were seen on treatment with Hu5F9-G4 (5F9) and rituximab, which induced responses in more than half of patients, of which more than one-third were complete responses, the study investigators reported.
Most of the responses were ongoing at the time of data cutoff, suggesting durable responses with the combination of rituximab and 5F9 – a humanized monoclonal antibody that blocks CD47, an antiphagocytic or “do not eat me” signal overexpressed by most cancers, Ranjana Advani, MD, of Stanford (Calif.) University, and her coauthors wrote.
“The macrophage-mediated activity of 5F9 plus rituximab may serve as an effective new immunotherapy for stimulating the innate immune system,” Dr. Advani and her colleagues reported in the New England Journal of Medicine.
The study included 22 patients, including 15 with diffuse large B-cell lymphoma (DLBCL) and 7 with follicular lymphoma, who had received a median of four prior therapies. Almost all of the non-Hodgkin lymphomas (21, or 95%) were refractory to rituximab.
All patients received intravenous 5F9 starting with a priming dose of 1 mg/kg followed by weekly maintenance doses of 10-30 mg/kg in three dose-escalation cohorts, given until disease progression or lack of clinical benefit. Intravenous rituximab at 375 mg/m2 weekly was started on the second week of the first cycle, and then monthly for cycles 2 through 6.
“Substantial antitumor activity” was seen with this chemotherapy-free regimen in a group of heavily pretreated, largely rituximab-refractory patients, Dr. Advani and her coauthors wrote in their report.
The objective response rate was 50%, including a 36% complete response rate in the intent-to-treat analysis. For DLBCL, the rates of objective and complete responses were 40% and 33%, while for follicular lymphoma, they were 71% and 43%.
The median duration of response was not reached in either disease cohort with a median follow-up of 6.2 months for DLBCL and 8.1 months for follicular lymphoma. Of the 11 patients who responded, 10 (91%) were still in response at the time of data cutoff. “Longer follow-up is needed,” the investigators wrote.
Most adverse events were seen within the first few weeks of treatment and mainly included anemia and infusion-related reactions. The anemia was an expected, on-target effect of 5F9 because of selective clearance of older red cells, which was predictable, transient, and mitigated by the maintenance dosing strategy employed in this phase 1b trial.
“As red cells age, they lose CD47 expression and gain expression of prophagocytic signals, leading to homeostatic clearance,” they wrote.
The activity of 5F9 and rituximab is “synergistic” based on the results of previous, preclinical investigations in models of lymphoma, Dr. Advani and her coauthors added.
A phase 2 trial of 5F9 plus rituximab in relapsed or refractory B-cell non-Hodgkin lymphoma is ongoing, according to their report.
The study was supported by Forty Seven and the Leukemia & Lymphoma Society. Dr. Advani reported disclosures related to Forty Seven, Bristol-Myers Squibb, Pharmacyclics, Seattle Genetics, and Roche/Genentech, among others.
SOURCE: Advani R et al. N Engl J Med. 2018;379:1711-21.
A macrophage-activating immune checkpoint inhibitor, combined with rituximab therapy, was safe and produced durable complete responses in patients with relapsed or refractory non-Hodgkin lymphoma, according to results of a phase 1b study.
Mainly low-grade toxic effects were seen on treatment with Hu5F9-G4 (5F9) and rituximab, which induced responses in more than half of patients, of which more than one-third were complete responses, the study investigators reported.
Most of the responses were ongoing at the time of data cutoff, suggesting durable responses with the combination of rituximab and 5F9 – a humanized monoclonal antibody that blocks CD47, an antiphagocytic or “do not eat me” signal overexpressed by most cancers, Ranjana Advani, MD, of Stanford (Calif.) University, and her coauthors wrote.
“The macrophage-mediated activity of 5F9 plus rituximab may serve as an effective new immunotherapy for stimulating the innate immune system,” Dr. Advani and her colleagues reported in the New England Journal of Medicine.
The study included 22 patients, including 15 with diffuse large B-cell lymphoma (DLBCL) and 7 with follicular lymphoma, who had received a median of four prior therapies. Almost all of the non-Hodgkin lymphomas (21, or 95%) were refractory to rituximab.
All patients received intravenous 5F9 starting with a priming dose of 1 mg/kg followed by weekly maintenance doses of 10-30 mg/kg in three dose-escalation cohorts, given until disease progression or lack of clinical benefit. Intravenous rituximab at 375 mg/m2 weekly was started on the second week of the first cycle, and then monthly for cycles 2 through 6.
“Substantial antitumor activity” was seen with this chemotherapy-free regimen in a group of heavily pretreated, largely rituximab-refractory patients, Dr. Advani and her coauthors wrote in their report.
The objective response rate was 50%, including a 36% complete response rate in the intent-to-treat analysis. For DLBCL, the rates of objective and complete responses were 40% and 33%, while for follicular lymphoma, they were 71% and 43%.
The median duration of response was not reached in either disease cohort with a median follow-up of 6.2 months for DLBCL and 8.1 months for follicular lymphoma. Of the 11 patients who responded, 10 (91%) were still in response at the time of data cutoff. “Longer follow-up is needed,” the investigators wrote.
Most adverse events were seen within the first few weeks of treatment and mainly included anemia and infusion-related reactions. The anemia was an expected, on-target effect of 5F9 because of selective clearance of older red cells, which was predictable, transient, and mitigated by the maintenance dosing strategy employed in this phase 1b trial.
“As red cells age, they lose CD47 expression and gain expression of prophagocytic signals, leading to homeostatic clearance,” they wrote.
The activity of 5F9 and rituximab is “synergistic” based on the results of previous, preclinical investigations in models of lymphoma, Dr. Advani and her coauthors added.
A phase 2 trial of 5F9 plus rituximab in relapsed or refractory B-cell non-Hodgkin lymphoma is ongoing, according to their report.
The study was supported by Forty Seven and the Leukemia & Lymphoma Society. Dr. Advani reported disclosures related to Forty Seven, Bristol-Myers Squibb, Pharmacyclics, Seattle Genetics, and Roche/Genentech, among others.
SOURCE: Advani R et al. N Engl J Med. 2018;379:1711-21.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: Rates of overall and complete responses were 50% and 36%, respectively, with most responses ongoing at the time of data cutoff.
Study details: A phase 1b study of 22 patients, including 15 with diffuse large B-cell lymphoma and 7 with follicular lymphoma.
Disclosures: The study was supported by Forty Seven and the Leukemia & Lymphoma Society. Study authors reported disclosures related to Forty Seven, Bristol-Myers Squibb, Pharmacyclics, Seattle Genetics, and Roche/Genentech, among others.
Source: Advani R et al. N Engl J Med. 2018;379:1711-21.
Older age predicts mortality after alloHCT in NHL, but not relapse
Elderly patients with non-Hodgkin lymphoma (NHL) are more likely to die, but not relapse, within 1 year of allogeneic hematopoietic cell transplantation (alloHCT), compared with younger or middle-age patients, according to investigators.

Comorbidities also increased risks of nonrelapse mortality (NRM) at 1 year, but to a lesser extent than that of elderly status, reported lead author Charalampia Kyriakou, MD, PhD, of the department of haematology at University College London Hospital and London North West University Healthcare NHS Trust, and her colleagues.
“Although alloHCT is feasible and effective in very old patients, the increased NRM risk must be taken into account when assessing the indication for alloHCT for NHL in this age group,” the investigators wrote in Biology of Blood and Marrow Transplantation.
This decision is becoming more common, they noted. “With the advent of reduced-intensity conditioning (RIC) strategies and other improvements in transplantation technology, alloHCT is being increasingly considered in elderly patients with [relapsed and refractory] NHL.”
The retrospective study analyzed 3,919 patients with NHL who underwent alloHCT between 2003 and 2013. Patients were sorted into three age groups: young (18-50 years), middle age (51-65 years), or elderly (66-77 years).
Disease types also were reported: 1,461 patients had follicular lymphoma (FL; 37%), 1,192 had diffuse large B cell lymphoma (DLBCL; 30%), 823 had mantle cell lymphoma (MCL; 21%), and 443 had peripheral T cell lymphoma (PTCL; 11%).
At the time of alloHCT, about 85% of patients were chemosensitive, with the remainder being chemorefractory. The age groups had similar patient characteristics, with exceptions noted for unrelated donors, MCL, and RIC, which became increasingly overrepresented with age.
The results showed that NRM at 1 year was 13% for young patients, 20% for middle-age patients, and 33% for elderly patients (P less than .001). Overall survival at 3 years followed an inverse trend, decreasing with age from 60% in young patients to 54% in middle-age patients, before dropping more dramatically to 38% in the elderly (P less than .001).
In contrast to these significant associations between age and survival, relapse risk at 3 years remained relatively consistent, with young patients at 30%, middle-age patients at 31%, and elderly patients at 28% (P = .355).
The investigators noted that the risk of NRM increased most dramatically between middle age and old age, with less significant differences between the middle-age and young groups. They suggested that “age per se should have a limited impact on the indication for alloHCT for NHL in patients up to age 65 years.”
The increased risk with elderly status could not be fully explained by comorbidities, although these were more common in elderly patients. After analyzing information from a subset of patients, the investigators concluded that “the presence of comorbidities is a significant risk factor for NRM and survival, but this does not fully explain the outcome disadvantages in our [elderly] group.” Therefore, age remains an independent risk factor.
“The information provided in this cohort of patients with NHL, the largest reported to date, is useful and relevant, especially in the era of evolving therapies,” the investigators wrote. They added that the information is “even more relevant now with the availability of treatment with ... chimeric antigen receptor (CAR) T cells ... after relapse post-alloHCT.”
The investigators reported having no financial disclosures.
SOURCE: Kyriakou C et al. Biol Blood Marrow Transplant. 2018 Sep 13. doi: 10.1016/j.bbmt.2018.08.025.
Elderly patients with non-Hodgkin lymphoma (NHL) are more likely to die, but not relapse, within 1 year of allogeneic hematopoietic cell transplantation (alloHCT), compared with younger or middle-age patients, according to investigators.
Comorbidities also increased risks of nonrelapse mortality (NRM) at 1 year, but to a lesser extent than that of elderly status, reported lead author Charalampia Kyriakou, MD, PhD, of the department of haematology at University College London Hospital and London North West University Healthcare NHS Trust, and her colleagues.
“Although alloHCT is feasible and effective in very old patients, the increased NRM risk must be taken into account when assessing the indication for alloHCT for NHL in this age group,” the investigators wrote in Biology of Blood and Marrow Transplantation.
This decision is becoming more common, they noted. “With the advent of reduced-intensity conditioning (RIC) strategies and other improvements in transplantation technology, alloHCT is being increasingly considered in elderly patients with [relapsed and refractory] NHL.”
The retrospective study analyzed 3,919 patients with NHL who underwent alloHCT between 2003 and 2013. Patients were sorted into three age groups: young (18-50 years), middle age (51-65 years), or elderly (66-77 years).
Disease types also were reported: 1,461 patients had follicular lymphoma (FL; 37%), 1,192 had diffuse large B cell lymphoma (DLBCL; 30%), 823 had mantle cell lymphoma (MCL; 21%), and 443 had peripheral T cell lymphoma (PTCL; 11%).
At the time of alloHCT, about 85% of patients were chemosensitive, with the remainder being chemorefractory. The age groups had similar patient characteristics, with exceptions noted for unrelated donors, MCL, and RIC, which became increasingly overrepresented with age.
The results showed that NRM at 1 year was 13% for young patients, 20% for middle-age patients, and 33% for elderly patients (P less than .001). Overall survival at 3 years followed an inverse trend, decreasing with age from 60% in young patients to 54% in middle-age patients, before dropping more dramatically to 38% in the elderly (P less than .001).
In contrast to these significant associations between age and survival, relapse risk at 3 years remained relatively consistent, with young patients at 30%, middle-age patients at 31%, and elderly patients at 28% (P = .355).
The investigators noted that the risk of NRM increased most dramatically between middle age and old age, with less significant differences between the middle-age and young groups. They suggested that “age per se should have a limited impact on the indication for alloHCT for NHL in patients up to age 65 years.”
The increased risk with elderly status could not be fully explained by comorbidities, although these were more common in elderly patients. After analyzing information from a subset of patients, the investigators concluded that “the presence of comorbidities is a significant risk factor for NRM and survival, but this does not fully explain the outcome disadvantages in our [elderly] group.” Therefore, age remains an independent risk factor.
“The information provided in this cohort of patients with NHL, the largest reported to date, is useful and relevant, especially in the era of evolving therapies,” the investigators wrote. They added that the information is “even more relevant now with the availability of treatment with ... chimeric antigen receptor (CAR) T cells ... after relapse post-alloHCT.”
The investigators reported having no financial disclosures.
SOURCE: Kyriakou C et al. Biol Blood Marrow Transplant. 2018 Sep 13. doi: 10.1016/j.bbmt.2018.08.025.
Elderly patients with non-Hodgkin lymphoma (NHL) are more likely to die, but not relapse, within 1 year of allogeneic hematopoietic cell transplantation (alloHCT), compared with younger or middle-age patients, according to investigators.
Comorbidities also increased risks of nonrelapse mortality (NRM) at 1 year, but to a lesser extent than that of elderly status, reported lead author Charalampia Kyriakou, MD, PhD, of the department of haematology at University College London Hospital and London North West University Healthcare NHS Trust, and her colleagues.
“Although alloHCT is feasible and effective in very old patients, the increased NRM risk must be taken into account when assessing the indication for alloHCT for NHL in this age group,” the investigators wrote in Biology of Blood and Marrow Transplantation.
This decision is becoming more common, they noted. “With the advent of reduced-intensity conditioning (RIC) strategies and other improvements in transplantation technology, alloHCT is being increasingly considered in elderly patients with [relapsed and refractory] NHL.”
The retrospective study analyzed 3,919 patients with NHL who underwent alloHCT between 2003 and 2013. Patients were sorted into three age groups: young (18-50 years), middle age (51-65 years), or elderly (66-77 years).
Disease types also were reported: 1,461 patients had follicular lymphoma (FL; 37%), 1,192 had diffuse large B cell lymphoma (DLBCL; 30%), 823 had mantle cell lymphoma (MCL; 21%), and 443 had peripheral T cell lymphoma (PTCL; 11%).
At the time of alloHCT, about 85% of patients were chemosensitive, with the remainder being chemorefractory. The age groups had similar patient characteristics, with exceptions noted for unrelated donors, MCL, and RIC, which became increasingly overrepresented with age.
The results showed that NRM at 1 year was 13% for young patients, 20% for middle-age patients, and 33% for elderly patients (P less than .001). Overall survival at 3 years followed an inverse trend, decreasing with age from 60% in young patients to 54% in middle-age patients, before dropping more dramatically to 38% in the elderly (P less than .001).
In contrast to these significant associations between age and survival, relapse risk at 3 years remained relatively consistent, with young patients at 30%, middle-age patients at 31%, and elderly patients at 28% (P = .355).
The investigators noted that the risk of NRM increased most dramatically between middle age and old age, with less significant differences between the middle-age and young groups. They suggested that “age per se should have a limited impact on the indication for alloHCT for NHL in patients up to age 65 years.”
The increased risk with elderly status could not be fully explained by comorbidities, although these were more common in elderly patients. After analyzing information from a subset of patients, the investigators concluded that “the presence of comorbidities is a significant risk factor for NRM and survival, but this does not fully explain the outcome disadvantages in our [elderly] group.” Therefore, age remains an independent risk factor.
“The information provided in this cohort of patients with NHL, the largest reported to date, is useful and relevant, especially in the era of evolving therapies,” the investigators wrote. They added that the information is “even more relevant now with the availability of treatment with ... chimeric antigen receptor (CAR) T cells ... after relapse post-alloHCT.”
The investigators reported having no financial disclosures.
SOURCE: Kyriakou C et al. Biol Blood Marrow Transplant. 2018 Sep 13. doi: 10.1016/j.bbmt.2018.08.025.
FROM BIOLOGY OF BLOOD AND MARROW TRANSPLANTATION
Key clinical point:
Major finding: One-year nonrelapse mortality (NRM) was 13% for young patients, 20% for middle-age patients, and 33% for elderly patients (P less than .001).
Study details: A retrospective analysis of 3,919 patients with NHL who underwent alloHCT between 2003 and 2013.
Disclosures: The researchers reported having no financial disclosures.
Source: Kyriakou C et al. Biol Blood Marrow Transplant. 2018 Sep 13. doi: 10.1016/j.bbmt.2018.08.025.
Entospletinib falls short in relapsed/refractory DLBCL
Entospletinib, a selective inhibitor of spleen tyrosine kinase (Syk), showed a dismal rate of progression-free survival and a high rate of adverse events in a cohort of previously treated patients with diffuse large B-cell lymphoma (DLBCL).
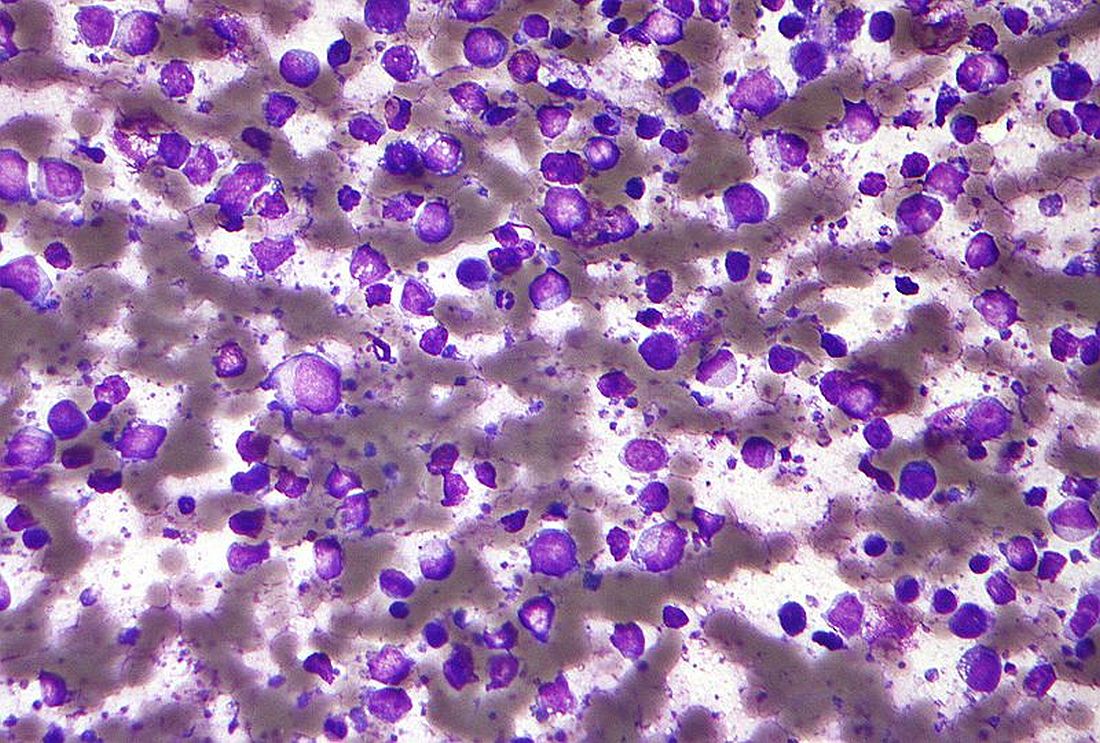
Entospletinib was evaluated in an open-label, single-agent, phase 2 trial (NCT01799889) with five relapsed/refractory patient cohorts: chronic lymphocytic leukemia (CLL), follicular lymphoma, other indolent non-Hodgkin lymphomas, mantle cell lymphoma, and DLBCL.
John M. Burke, MD, of Rocky Mountain Cancer Centers in Aurora, Colo., and his colleagues reported on the current analysis, which looked specifically at the 43 patients in the trial with previously treated DLBCL. Patients received at least one starting dose of 800 mg of entospletinib orally twice daily. The findings were published in Clinical Lymphoma, Myeloma & Leukemia.
In a previous report on the relapsed/refractory CLL cohort, the investigational agent demonstrated clinical activity with acceptable toxicity (Blood. 2015 Apr 9;125[15]:2336-43).
In the current report, the rate of progression-free survival (PFS) at 16 weeks was 3.6% and the median PFS was 1.5 months. None of the patients in the study achieved a complete or partial response to treatment, and just five patients had stable disease.
All patients in the study eventually discontinued treatment and the median treatment duration was 1 month.
“The lack of activity of Syk inhibition in patients with relapsed DLBCL is in contrast to what would have been expected from preclinical data,” the investigators wrote. “Although it is unclear why entospletinib monotherapy lacked activity in the present study, it is possible that resistance to Syk inhibition played a role. Potential mechanisms of resistance of DLBCL to Syk inhibition include transcriptional upregulation of Syk mediated by FOXO1 and PTEN depletion.”
The investigators said that Syk inhibition in combination with BCL2 inhibitors could potentially overcome this resistance. Another approach, they suggested, would be to offer entospletinib in combination with Janus kinase (JAK) 1/3 inhibition.
“Based on results of the preclinical data, the efficacy of entospletinib in combination will be evaluated in future clinical trials,” the investigators wrote.
The rate of adverse events was high in the DLBCL cohort. Forty-two patients (98%) experienced an adverse event and nearly three-quarters experienced a grade 3 event. Overall, 30% of the grade 3 adverse events were related to treatment. More than 40% of patients interrupted treatment because of adverse events, and 19% discontinued. Four patients experienced an adverse event that led to death.
While the lack of clinical activity may have surprised investigators, the safety profile was in line with other patient cohorts in the phase 2 study. In the CLL and indolent non-Hodgkin lymphoma cohorts, the rates of treatment interruption were 45% and 54%, respectively.
The study was supported by Gilead Sciences. Dr. Burke reported relationships with Gilead and other companies.
SOURCE: Burke JM et al. Clin Lymphoma Myeloma Leuk. 2018 Aug;18(8):e327-e331.
Entospletinib, a selective inhibitor of spleen tyrosine kinase (Syk), showed a dismal rate of progression-free survival and a high rate of adverse events in a cohort of previously treated patients with diffuse large B-cell lymphoma (DLBCL).
Entospletinib was evaluated in an open-label, single-agent, phase 2 trial (NCT01799889) with five relapsed/refractory patient cohorts: chronic lymphocytic leukemia (CLL), follicular lymphoma, other indolent non-Hodgkin lymphomas, mantle cell lymphoma, and DLBCL.
John M. Burke, MD, of Rocky Mountain Cancer Centers in Aurora, Colo., and his colleagues reported on the current analysis, which looked specifically at the 43 patients in the trial with previously treated DLBCL. Patients received at least one starting dose of 800 mg of entospletinib orally twice daily. The findings were published in Clinical Lymphoma, Myeloma & Leukemia.
In a previous report on the relapsed/refractory CLL cohort, the investigational agent demonstrated clinical activity with acceptable toxicity (Blood. 2015 Apr 9;125[15]:2336-43).
In the current report, the rate of progression-free survival (PFS) at 16 weeks was 3.6% and the median PFS was 1.5 months. None of the patients in the study achieved a complete or partial response to treatment, and just five patients had stable disease.
All patients in the study eventually discontinued treatment and the median treatment duration was 1 month.
“The lack of activity of Syk inhibition in patients with relapsed DLBCL is in contrast to what would have been expected from preclinical data,” the investigators wrote. “Although it is unclear why entospletinib monotherapy lacked activity in the present study, it is possible that resistance to Syk inhibition played a role. Potential mechanisms of resistance of DLBCL to Syk inhibition include transcriptional upregulation of Syk mediated by FOXO1 and PTEN depletion.”
The investigators said that Syk inhibition in combination with BCL2 inhibitors could potentially overcome this resistance. Another approach, they suggested, would be to offer entospletinib in combination with Janus kinase (JAK) 1/3 inhibition.
“Based on results of the preclinical data, the efficacy of entospletinib in combination will be evaluated in future clinical trials,” the investigators wrote.
The rate of adverse events was high in the DLBCL cohort. Forty-two patients (98%) experienced an adverse event and nearly three-quarters experienced a grade 3 event. Overall, 30% of the grade 3 adverse events were related to treatment. More than 40% of patients interrupted treatment because of adverse events, and 19% discontinued. Four patients experienced an adverse event that led to death.
While the lack of clinical activity may have surprised investigators, the safety profile was in line with other patient cohorts in the phase 2 study. In the CLL and indolent non-Hodgkin lymphoma cohorts, the rates of treatment interruption were 45% and 54%, respectively.
The study was supported by Gilead Sciences. Dr. Burke reported relationships with Gilead and other companies.
SOURCE: Burke JM et al. Clin Lymphoma Myeloma Leuk. 2018 Aug;18(8):e327-e331.
Entospletinib, a selective inhibitor of spleen tyrosine kinase (Syk), showed a dismal rate of progression-free survival and a high rate of adverse events in a cohort of previously treated patients with diffuse large B-cell lymphoma (DLBCL).
Entospletinib was evaluated in an open-label, single-agent, phase 2 trial (NCT01799889) with five relapsed/refractory patient cohorts: chronic lymphocytic leukemia (CLL), follicular lymphoma, other indolent non-Hodgkin lymphomas, mantle cell lymphoma, and DLBCL.
John M. Burke, MD, of Rocky Mountain Cancer Centers in Aurora, Colo., and his colleagues reported on the current analysis, which looked specifically at the 43 patients in the trial with previously treated DLBCL. Patients received at least one starting dose of 800 mg of entospletinib orally twice daily. The findings were published in Clinical Lymphoma, Myeloma & Leukemia.
In a previous report on the relapsed/refractory CLL cohort, the investigational agent demonstrated clinical activity with acceptable toxicity (Blood. 2015 Apr 9;125[15]:2336-43).
In the current report, the rate of progression-free survival (PFS) at 16 weeks was 3.6% and the median PFS was 1.5 months. None of the patients in the study achieved a complete or partial response to treatment, and just five patients had stable disease.
All patients in the study eventually discontinued treatment and the median treatment duration was 1 month.
“The lack of activity of Syk inhibition in patients with relapsed DLBCL is in contrast to what would have been expected from preclinical data,” the investigators wrote. “Although it is unclear why entospletinib monotherapy lacked activity in the present study, it is possible that resistance to Syk inhibition played a role. Potential mechanisms of resistance of DLBCL to Syk inhibition include transcriptional upregulation of Syk mediated by FOXO1 and PTEN depletion.”
The investigators said that Syk inhibition in combination with BCL2 inhibitors could potentially overcome this resistance. Another approach, they suggested, would be to offer entospletinib in combination with Janus kinase (JAK) 1/3 inhibition.
“Based on results of the preclinical data, the efficacy of entospletinib in combination will be evaluated in future clinical trials,” the investigators wrote.
The rate of adverse events was high in the DLBCL cohort. Forty-two patients (98%) experienced an adverse event and nearly three-quarters experienced a grade 3 event. Overall, 30% of the grade 3 adverse events were related to treatment. More than 40% of patients interrupted treatment because of adverse events, and 19% discontinued. Four patients experienced an adverse event that led to death.
While the lack of clinical activity may have surprised investigators, the safety profile was in line with other patient cohorts in the phase 2 study. In the CLL and indolent non-Hodgkin lymphoma cohorts, the rates of treatment interruption were 45% and 54%, respectively.
The study was supported by Gilead Sciences. Dr. Burke reported relationships with Gilead and other companies.
SOURCE: Burke JM et al. Clin Lymphoma Myeloma Leuk. 2018 Aug;18(8):e327-e331.
FROM CLINICAL LYMPHOMA, MYELOMA & LEUKEMIA
Key clinical point:
Major finding: The rate of progression-free survival at 16 weeks was 3.6% with a median PFS of 1.5 months.
Study details: An analysis of 43 relapsed/refractory DLBCL patients who received single-agent entospletinib.
Disclosures: The study was supported by Gilead Sciences. Dr. Burke reported relationships with Gilead and other companies.
Source: Burke JM et al. Clin Lymphoma Myeloma Leuk. 2018 Aug;18(8):e327-e331.
Inhibitor receives orphan designation for PTCL
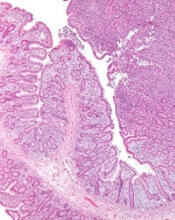
The U.S. Food and Drug Administration (FDA) has granted orphan drug designation to cerdulatinib for the treatment of peripheral T-cell lymphoma (PTCL).
Cerdulatinib is an oral Syk/JAK inhibitor being developed by Portola Pharmaceuticals, Inc.
Preclinical data have suggested an important role for Syk and JAK in PTCL tumor survival, and cerdulatinib is currently under evaluation in a phase 2a study of patients with PTCL and other non-Hodgkin lymphomas.
Results from this trial were presented at the 23rd Congress of the European Hematology Association (EHA) earlier this year.
At that time, the trial had enrolled 114 patients, 25 of them with PTCL. The patients received cerdulatinib at 25, 30, or 35 mg twice daily.
The objective response rate was 35% among the PTCL patients. All seven responders had a complete response, and 11 PTCL patients were still on cerdulatinib at the time of the presentation.
Grade 3 or higher adverse events observed in all evaluable patients included lipase increase (18%), neutropenia (17%), pneumonia/lung infection (11%), diarrhea (8%), fatigue (6%), amylase increase (5%), sepsis/septic shock (4%), hypertension (4%), anemia (4%), thrombocytopenia (4%), and hypophosphatemia (4%).
There were five deaths due to sepsis or septic shock (three of which were concomitant with pneumonia) that were considered related to cerdulatinib.
Three of the deaths occurred in patients with chronic lymphocytic leukemia, one in a patient with diffuse large B-cell lymphoma, and one in a patient with follicular lymphoma.
The deaths occurred early on in the trial, and researchers have since taken steps—dose reductions, monitoring, and antibiotic prophylaxis—to prevent additional deaths.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the United States.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The U.S. Food and Drug Administration (FDA) has granted orphan drug designation to cerdulatinib for the treatment of peripheral T-cell lymphoma (PTCL).
Cerdulatinib is an oral Syk/JAK inhibitor being developed by Portola Pharmaceuticals, Inc.
Preclinical data have suggested an important role for Syk and JAK in PTCL tumor survival, and cerdulatinib is currently under evaluation in a phase 2a study of patients with PTCL and other non-Hodgkin lymphomas.
Results from this trial were presented at the 23rd Congress of the European Hematology Association (EHA) earlier this year.
At that time, the trial had enrolled 114 patients, 25 of them with PTCL. The patients received cerdulatinib at 25, 30, or 35 mg twice daily.
The objective response rate was 35% among the PTCL patients. All seven responders had a complete response, and 11 PTCL patients were still on cerdulatinib at the time of the presentation.
Grade 3 or higher adverse events observed in all evaluable patients included lipase increase (18%), neutropenia (17%), pneumonia/lung infection (11%), diarrhea (8%), fatigue (6%), amylase increase (5%), sepsis/septic shock (4%), hypertension (4%), anemia (4%), thrombocytopenia (4%), and hypophosphatemia (4%).
There were five deaths due to sepsis or septic shock (three of which were concomitant with pneumonia) that were considered related to cerdulatinib.
Three of the deaths occurred in patients with chronic lymphocytic leukemia, one in a patient with diffuse large B-cell lymphoma, and one in a patient with follicular lymphoma.
The deaths occurred early on in the trial, and researchers have since taken steps—dose reductions, monitoring, and antibiotic prophylaxis—to prevent additional deaths.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the United States.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The U.S. Food and Drug Administration (FDA) has granted orphan drug designation to cerdulatinib for the treatment of peripheral T-cell lymphoma (PTCL).
Cerdulatinib is an oral Syk/JAK inhibitor being developed by Portola Pharmaceuticals, Inc.
Preclinical data have suggested an important role for Syk and JAK in PTCL tumor survival, and cerdulatinib is currently under evaluation in a phase 2a study of patients with PTCL and other non-Hodgkin lymphomas.
Results from this trial were presented at the 23rd Congress of the European Hematology Association (EHA) earlier this year.
At that time, the trial had enrolled 114 patients, 25 of them with PTCL. The patients received cerdulatinib at 25, 30, or 35 mg twice daily.
The objective response rate was 35% among the PTCL patients. All seven responders had a complete response, and 11 PTCL patients were still on cerdulatinib at the time of the presentation.
Grade 3 or higher adverse events observed in all evaluable patients included lipase increase (18%), neutropenia (17%), pneumonia/lung infection (11%), diarrhea (8%), fatigue (6%), amylase increase (5%), sepsis/septic shock (4%), hypertension (4%), anemia (4%), thrombocytopenia (4%), and hypophosphatemia (4%).
There were five deaths due to sepsis or septic shock (three of which were concomitant with pneumonia) that were considered related to cerdulatinib.
Three of the deaths occurred in patients with chronic lymphocytic leukemia, one in a patient with diffuse large B-cell lymphoma, and one in a patient with follicular lymphoma.
The deaths occurred early on in the trial, and researchers have since taken steps—dose reductions, monitoring, and antibiotic prophylaxis—to prevent additional deaths.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the United States.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
Frontline rituximab shows long-term success in indolent lymphoma
Advanced indolent lymphoma patients can be treated with a rituximab-containing regimen as first-line therapy and, in some cases, skip chemotherapy altogether, a study with 10 years of follow-up data suggests.
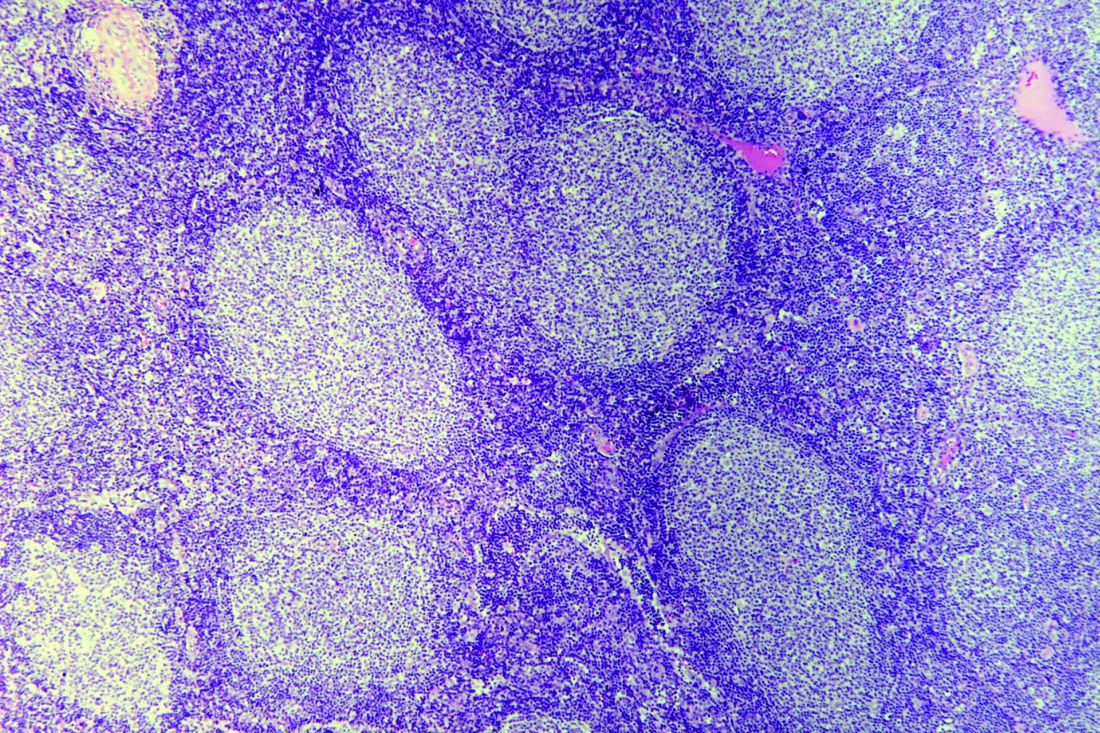
After a median of 10.6 years’ follow-up, almost three-quarters of patients (73%) in the study were alive, and 36% never required chemotherapy.
“This [overall survival] is at least as good as that observed in modern immunochemotherapy trials,” Sandra Lockmer, MD, of Karolinska University Hospital in Stockholm and her colleagues reported in the Journal of Clinical Oncology.
The study included 321 patients who were previously untreated and had been enrolled in two randomized clinical trials performed by the Nordic Lymphoma Group. The trials randomized patients to receive either rituximab monotherapy or rituximab combined with interferon alfa-2a. Neither trial used up-front chemotherapy.
Patients included in the follow-up analysis had follicular lymphoma, marginal zone lymphoma, small lymphocytic lymphoma, or indolent lymphoma not otherwise specified.
The overall survival rate at 10 years after trial assignment was 75% and 66% after 15 years. Similarly, the lymphoma-specific survival rate was 81% at 10 years after trial assignment and 77% at 15 years, the researchers reported.
Overall, 117 patients did not require treatment with chemotherapy, but 24 patients were further treated with antibodies and/or radiation. Of the 93 patients who received no additional therapies after frontline treatment, 9 patients died from causes unrelated to their lymphoma.
Among the 237 patients who failed initial treatment, the median time to treatment failure was 1.5 years.
In terms of transformation to aggressive lymphoma, the rate was 2.4%/person-year overall. The cumulative risk of transformation was 20% at 10 years after trial assignment and 24% at 15 years.
The study was funded in part by the Stockholm County Council and by the Nordic Lymphoma Group. The trials analyzed in the study were supported by Roche. Dr. Lockmer reported having no financial disclosures. Her coauthors reported relationships with Novartis, Gilead, Roche, and Takeda, among others.
[email protected]
SOURCE: Lockmer S et al. J Clin Oncol. 2018 Oct 4:JCO1800262. doi: 10.1200/JCO.18.00262.
Advanced indolent lymphoma patients can be treated with a rituximab-containing regimen as first-line therapy and, in some cases, skip chemotherapy altogether, a study with 10 years of follow-up data suggests.
After a median of 10.6 years’ follow-up, almost three-quarters of patients (73%) in the study were alive, and 36% never required chemotherapy.
“This [overall survival] is at least as good as that observed in modern immunochemotherapy trials,” Sandra Lockmer, MD, of Karolinska University Hospital in Stockholm and her colleagues reported in the Journal of Clinical Oncology.
The study included 321 patients who were previously untreated and had been enrolled in two randomized clinical trials performed by the Nordic Lymphoma Group. The trials randomized patients to receive either rituximab monotherapy or rituximab combined with interferon alfa-2a. Neither trial used up-front chemotherapy.
Patients included in the follow-up analysis had follicular lymphoma, marginal zone lymphoma, small lymphocytic lymphoma, or indolent lymphoma not otherwise specified.
The overall survival rate at 10 years after trial assignment was 75% and 66% after 15 years. Similarly, the lymphoma-specific survival rate was 81% at 10 years after trial assignment and 77% at 15 years, the researchers reported.
Overall, 117 patients did not require treatment with chemotherapy, but 24 patients were further treated with antibodies and/or radiation. Of the 93 patients who received no additional therapies after frontline treatment, 9 patients died from causes unrelated to their lymphoma.
Among the 237 patients who failed initial treatment, the median time to treatment failure was 1.5 years.
In terms of transformation to aggressive lymphoma, the rate was 2.4%/person-year overall. The cumulative risk of transformation was 20% at 10 years after trial assignment and 24% at 15 years.
The study was funded in part by the Stockholm County Council and by the Nordic Lymphoma Group. The trials analyzed in the study were supported by Roche. Dr. Lockmer reported having no financial disclosures. Her coauthors reported relationships with Novartis, Gilead, Roche, and Takeda, among others.
[email protected]
SOURCE: Lockmer S et al. J Clin Oncol. 2018 Oct 4:JCO1800262. doi: 10.1200/JCO.18.00262.
Advanced indolent lymphoma patients can be treated with a rituximab-containing regimen as first-line therapy and, in some cases, skip chemotherapy altogether, a study with 10 years of follow-up data suggests.
After a median of 10.6 years’ follow-up, almost three-quarters of patients (73%) in the study were alive, and 36% never required chemotherapy.
“This [overall survival] is at least as good as that observed in modern immunochemotherapy trials,” Sandra Lockmer, MD, of Karolinska University Hospital in Stockholm and her colleagues reported in the Journal of Clinical Oncology.
The study included 321 patients who were previously untreated and had been enrolled in two randomized clinical trials performed by the Nordic Lymphoma Group. The trials randomized patients to receive either rituximab monotherapy or rituximab combined with interferon alfa-2a. Neither trial used up-front chemotherapy.
Patients included in the follow-up analysis had follicular lymphoma, marginal zone lymphoma, small lymphocytic lymphoma, or indolent lymphoma not otherwise specified.
The overall survival rate at 10 years after trial assignment was 75% and 66% after 15 years. Similarly, the lymphoma-specific survival rate was 81% at 10 years after trial assignment and 77% at 15 years, the researchers reported.
Overall, 117 patients did not require treatment with chemotherapy, but 24 patients were further treated with antibodies and/or radiation. Of the 93 patients who received no additional therapies after frontline treatment, 9 patients died from causes unrelated to their lymphoma.
Among the 237 patients who failed initial treatment, the median time to treatment failure was 1.5 years.
In terms of transformation to aggressive lymphoma, the rate was 2.4%/person-year overall. The cumulative risk of transformation was 20% at 10 years after trial assignment and 24% at 15 years.
The study was funded in part by the Stockholm County Council and by the Nordic Lymphoma Group. The trials analyzed in the study were supported by Roche. Dr. Lockmer reported having no financial disclosures. Her coauthors reported relationships with Novartis, Gilead, Roche, and Takeda, among others.
[email protected]
SOURCE: Lockmer S et al. J Clin Oncol. 2018 Oct 4:JCO1800262. doi: 10.1200/JCO.18.00262.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point:
Major finding: After a median of 10.6 years’ follow up, 73% of patients were alive, and 36% did not require chemotherapy.
Study details: Ten-year follow-up data from two trials on 321 previously untreated patients who had follicular lymphoma, marginal zone lymphoma, small lymphocytic lymphoma, or indolent lymphoma not otherwise specified.
Disclosures: The study was funded in part by the Stockholm County Council and by the Nordic Lymphoma Group. The trials analyzed in the study were supported by Roche. Dr. Lockmer reported having no financial disclosures. Her coauthors reported relationships with Novartis, Gilead, Roche, and Takeda, among others.
Source: Lockmer S et al. J Clin Oncol. 2018 Oct 4:JCO1800262. doi: 10.1200/JCO.18.00262.
FDA lifts partial hold on tazemetostat trials
The U.S. Food and Drug Administration has lifted the partial clinical hold on trials of tazemetostat, an EZH2 inhibitor being developed to treat solid tumors and lymphomas, according to a press release from the drug’s developer Epizyme.

The patient had been on study for approximately 15 months and had achieved a confirmed partial response. The patient has since discontinued tazemetostat and responded to treatment for T-LBL.
“This remains the only case of T-LBL we’ve seen in more than 750 patients treated with tazemetostat,” Robert Bazemore, president and chief executive officer of Epizyme, said in a webcast on Sept. 24.
Epizyme assessed the risk of secondary malignancies, including T-LBL, as well as the overall risks and benefits of tazemetostat treatment, conducting a review of the published literature and an examination of efficacy and safety data across all of its tazemetostat trials. A panel of external scientific and medical experts who reviewed the findings concluded that T-LBL risks appear to be confined to pediatric patients who received higher doses of the drug. The phase 1 pediatric study in which the patient developed T-LBL included higher doses of tazemetostat than those used in the phase 2 adult studies.
“The team at Epizyme has worked diligently in collaboration with external experts and the FDA over the past several months,” Mr. Bazemore said.
The company is not making any substantial changes to trial designs or the patient populations involved in tazemetostat trials. However, Epizyme is modifying dosing in the pediatric studies, improving patient monitoring, and making changes to exclusion criteria to reduce the potential risk of T-LBL and other secondary malignancies. Mr. Bazemore said Epizyme hopes to submit a New Drug Application for tazemetostat in the treatment of epithelioid sarcoma.
Tazemetostat is under investigation as monotherapy in phase 2 trials of follicular lymphoma and solid-tumor malignancies. The drug is also being studied as part of combination therapy for non–small cell lung cancer and diffuse large B-cell lymphoma (DLBCL).
In August, Epizyme announced its decision to stop developing tazemetostat for use as monotherapy or in combination with prednisolone for patients with DLBCL. However, tazemetostat is still under investigation as a potential treatment for DLBCL as part of other combination regimens.
Epizyme is now working to resolve partial clinical holds placed on tazemetostat in France and Germany in order to resume trial enrollment in those countries.
The U.S. Food and Drug Administration has lifted the partial clinical hold on trials of tazemetostat, an EZH2 inhibitor being developed to treat solid tumors and lymphomas, according to a press release from the drug’s developer Epizyme.
The patient had been on study for approximately 15 months and had achieved a confirmed partial response. The patient has since discontinued tazemetostat and responded to treatment for T-LBL.
“This remains the only case of T-LBL we’ve seen in more than 750 patients treated with tazemetostat,” Robert Bazemore, president and chief executive officer of Epizyme, said in a webcast on Sept. 24.
Epizyme assessed the risk of secondary malignancies, including T-LBL, as well as the overall risks and benefits of tazemetostat treatment, conducting a review of the published literature and an examination of efficacy and safety data across all of its tazemetostat trials. A panel of external scientific and medical experts who reviewed the findings concluded that T-LBL risks appear to be confined to pediatric patients who received higher doses of the drug. The phase 1 pediatric study in which the patient developed T-LBL included higher doses of tazemetostat than those used in the phase 2 adult studies.
“The team at Epizyme has worked diligently in collaboration with external experts and the FDA over the past several months,” Mr. Bazemore said.
The company is not making any substantial changes to trial designs or the patient populations involved in tazemetostat trials. However, Epizyme is modifying dosing in the pediatric studies, improving patient monitoring, and making changes to exclusion criteria to reduce the potential risk of T-LBL and other secondary malignancies. Mr. Bazemore said Epizyme hopes to submit a New Drug Application for tazemetostat in the treatment of epithelioid sarcoma.
Tazemetostat is under investigation as monotherapy in phase 2 trials of follicular lymphoma and solid-tumor malignancies. The drug is also being studied as part of combination therapy for non–small cell lung cancer and diffuse large B-cell lymphoma (DLBCL).
In August, Epizyme announced its decision to stop developing tazemetostat for use as monotherapy or in combination with prednisolone for patients with DLBCL. However, tazemetostat is still under investigation as a potential treatment for DLBCL as part of other combination regimens.
Epizyme is now working to resolve partial clinical holds placed on tazemetostat in France and Germany in order to resume trial enrollment in those countries.
The U.S. Food and Drug Administration has lifted the partial clinical hold on trials of tazemetostat, an EZH2 inhibitor being developed to treat solid tumors and lymphomas, according to a press release from the drug’s developer Epizyme.
The patient had been on study for approximately 15 months and had achieved a confirmed partial response. The patient has since discontinued tazemetostat and responded to treatment for T-LBL.
“This remains the only case of T-LBL we’ve seen in more than 750 patients treated with tazemetostat,” Robert Bazemore, president and chief executive officer of Epizyme, said in a webcast on Sept. 24.
Epizyme assessed the risk of secondary malignancies, including T-LBL, as well as the overall risks and benefits of tazemetostat treatment, conducting a review of the published literature and an examination of efficacy and safety data across all of its tazemetostat trials. A panel of external scientific and medical experts who reviewed the findings concluded that T-LBL risks appear to be confined to pediatric patients who received higher doses of the drug. The phase 1 pediatric study in which the patient developed T-LBL included higher doses of tazemetostat than those used in the phase 2 adult studies.
“The team at Epizyme has worked diligently in collaboration with external experts and the FDA over the past several months,” Mr. Bazemore said.
The company is not making any substantial changes to trial designs or the patient populations involved in tazemetostat trials. However, Epizyme is modifying dosing in the pediatric studies, improving patient monitoring, and making changes to exclusion criteria to reduce the potential risk of T-LBL and other secondary malignancies. Mr. Bazemore said Epizyme hopes to submit a New Drug Application for tazemetostat in the treatment of epithelioid sarcoma.
Tazemetostat is under investigation as monotherapy in phase 2 trials of follicular lymphoma and solid-tumor malignancies. The drug is also being studied as part of combination therapy for non–small cell lung cancer and diffuse large B-cell lymphoma (DLBCL).
In August, Epizyme announced its decision to stop developing tazemetostat for use as monotherapy or in combination with prednisolone for patients with DLBCL. However, tazemetostat is still under investigation as a potential treatment for DLBCL as part of other combination regimens.
Epizyme is now working to resolve partial clinical holds placed on tazemetostat in France and Germany in order to resume trial enrollment in those countries.
FDA approves new drug for CLL/SLL and follicular lymphoma
The Food and Drug Administration has approved duvelisib (Copiktra), a dual PI3K delta/gamma inhibitor, for the treatment of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and follicular lymphoma.
who have received at least two prior therapies. Duvelisib also has accelerated approval to treat adults with relapsed or refractory follicular lymphoma (FL) who have received at least two prior systemic therapies.
Accelerated approval is based on a surrogate or intermediate endpoint – in this case, overall response rate – that is reasonably likely to predict clinical benefit. Continued approval of duvelisib in FL may be contingent upon results of confirmatory trials verifying that the drug provides a clinical benefit.
Duvelisib will be available in the U.S. immediately, according to Verastem, the company marketing the drug. The prescribing information for duvelisib includes a boxed warning detailing four fatal and/or serious toxicities associated with the drug – infections, diarrhea or colitis, cutaneous reactions, and pneumonitis. Verastem said it is implementing an informational risk evaluation and mitigation strategy to provide appropriate dosing and safety information for duvelisib.
The recommended dose of duvelisib is 25 mg orally twice daily, taken continuously in 28-day treatment cycles.
The FDA’s approval of duvelisib is supported by data from the phase 3 DUO trial and the phase 2 DYNAMO trial. The DUO trial included 319 patients with CLL (n=312) or SLL (n=7) who had received at least one prior therapy. They were randomized to receive either duvelisib (25 mg orally twice daily) or ofatumumab (initial infusion of 300 mg followed by 7 weekly infusions and 4 monthly infusions of 2,000 mg).
Efficacy results are based on patients who had received at least two prior therapies, including 95 patients in the duvelisib arm and 101 in the ofatumumab arm. The overall response rate was 78% in the duvelisib arm and 39% in the ofatumumab arm. All responses in both arms were partial responses.
The median progression-free survival was 16.4 months with duvelisib and 9.1 months with ofatumumab.
The safety results include all patients treated with duvelisib or ofatumumab in this trial. In the duvelisib arm, 12% of patients had fatal adverse events (AEs) within 30 days of the last dose. The same was true of 4% of patients treated with ofatumumab. Serious AEs occurred in 73% of patients treated with duvelisib. The most common were infection and diarrhea/colitis. The DYNAMO trial enrolled patients with indolent non-Hodgkin lymphoma whose disease was refractory to both rituximab and chemotherapy or radioimmunotherapy. There were 83 patients with FL.
Patients received duvelisib at 25 mg orally twice daily until disease progression or unacceptable toxicity.
The overall response rate was 42%. One patient achieved a complete response, and 34 had a partial response.
Forty-three percent of responders maintained their response at 6 months, and 17% maintained their response at 12 months.
Serious AEs occurred in 58% of FL patients. The most common were diarrhea/colitis, pneumonia, renal insufficiency, rash, and sepsis.
The Food and Drug Administration has approved duvelisib (Copiktra), a dual PI3K delta/gamma inhibitor, for the treatment of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and follicular lymphoma.
who have received at least two prior therapies. Duvelisib also has accelerated approval to treat adults with relapsed or refractory follicular lymphoma (FL) who have received at least two prior systemic therapies.
Accelerated approval is based on a surrogate or intermediate endpoint – in this case, overall response rate – that is reasonably likely to predict clinical benefit. Continued approval of duvelisib in FL may be contingent upon results of confirmatory trials verifying that the drug provides a clinical benefit.
Duvelisib will be available in the U.S. immediately, according to Verastem, the company marketing the drug. The prescribing information for duvelisib includes a boxed warning detailing four fatal and/or serious toxicities associated with the drug – infections, diarrhea or colitis, cutaneous reactions, and pneumonitis. Verastem said it is implementing an informational risk evaluation and mitigation strategy to provide appropriate dosing and safety information for duvelisib.
The recommended dose of duvelisib is 25 mg orally twice daily, taken continuously in 28-day treatment cycles.
The FDA’s approval of duvelisib is supported by data from the phase 3 DUO trial and the phase 2 DYNAMO trial. The DUO trial included 319 patients with CLL (n=312) or SLL (n=7) who had received at least one prior therapy. They were randomized to receive either duvelisib (25 mg orally twice daily) or ofatumumab (initial infusion of 300 mg followed by 7 weekly infusions and 4 monthly infusions of 2,000 mg).
Efficacy results are based on patients who had received at least two prior therapies, including 95 patients in the duvelisib arm and 101 in the ofatumumab arm. The overall response rate was 78% in the duvelisib arm and 39% in the ofatumumab arm. All responses in both arms were partial responses.
The median progression-free survival was 16.4 months with duvelisib and 9.1 months with ofatumumab.
The safety results include all patients treated with duvelisib or ofatumumab in this trial. In the duvelisib arm, 12% of patients had fatal adverse events (AEs) within 30 days of the last dose. The same was true of 4% of patients treated with ofatumumab. Serious AEs occurred in 73% of patients treated with duvelisib. The most common were infection and diarrhea/colitis. The DYNAMO trial enrolled patients with indolent non-Hodgkin lymphoma whose disease was refractory to both rituximab and chemotherapy or radioimmunotherapy. There were 83 patients with FL.
Patients received duvelisib at 25 mg orally twice daily until disease progression or unacceptable toxicity.
The overall response rate was 42%. One patient achieved a complete response, and 34 had a partial response.
Forty-three percent of responders maintained their response at 6 months, and 17% maintained their response at 12 months.
Serious AEs occurred in 58% of FL patients. The most common were diarrhea/colitis, pneumonia, renal insufficiency, rash, and sepsis.
The Food and Drug Administration has approved duvelisib (Copiktra), a dual PI3K delta/gamma inhibitor, for the treatment of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and follicular lymphoma.
who have received at least two prior therapies. Duvelisib also has accelerated approval to treat adults with relapsed or refractory follicular lymphoma (FL) who have received at least two prior systemic therapies.
Accelerated approval is based on a surrogate or intermediate endpoint – in this case, overall response rate – that is reasonably likely to predict clinical benefit. Continued approval of duvelisib in FL may be contingent upon results of confirmatory trials verifying that the drug provides a clinical benefit.
Duvelisib will be available in the U.S. immediately, according to Verastem, the company marketing the drug. The prescribing information for duvelisib includes a boxed warning detailing four fatal and/or serious toxicities associated with the drug – infections, diarrhea or colitis, cutaneous reactions, and pneumonitis. Verastem said it is implementing an informational risk evaluation and mitigation strategy to provide appropriate dosing and safety information for duvelisib.
The recommended dose of duvelisib is 25 mg orally twice daily, taken continuously in 28-day treatment cycles.
The FDA’s approval of duvelisib is supported by data from the phase 3 DUO trial and the phase 2 DYNAMO trial. The DUO trial included 319 patients with CLL (n=312) or SLL (n=7) who had received at least one prior therapy. They were randomized to receive either duvelisib (25 mg orally twice daily) or ofatumumab (initial infusion of 300 mg followed by 7 weekly infusions and 4 monthly infusions of 2,000 mg).
Efficacy results are based on patients who had received at least two prior therapies, including 95 patients in the duvelisib arm and 101 in the ofatumumab arm. The overall response rate was 78% in the duvelisib arm and 39% in the ofatumumab arm. All responses in both arms were partial responses.
The median progression-free survival was 16.4 months with duvelisib and 9.1 months with ofatumumab.
The safety results include all patients treated with duvelisib or ofatumumab in this trial. In the duvelisib arm, 12% of patients had fatal adverse events (AEs) within 30 days of the last dose. The same was true of 4% of patients treated with ofatumumab. Serious AEs occurred in 73% of patients treated with duvelisib. The most common were infection and diarrhea/colitis. The DYNAMO trial enrolled patients with indolent non-Hodgkin lymphoma whose disease was refractory to both rituximab and chemotherapy or radioimmunotherapy. There were 83 patients with FL.
Patients received duvelisib at 25 mg orally twice daily until disease progression or unacceptable toxicity.
The overall response rate was 42%. One patient achieved a complete response, and 34 had a partial response.
Forty-three percent of responders maintained their response at 6 months, and 17% maintained their response at 12 months.
Serious AEs occurred in 58% of FL patients. The most common were diarrhea/colitis, pneumonia, renal insufficiency, rash, and sepsis.
Risks of watchful waiting in follicular lymphoma

A subset of follicular lymphoma (FL) patients managed with watchful waiting are vulnerable to organ dysfunction and transformation, according to research published in Clinical Lymphoma, Myeloma & Leukemia.
In a retrospective study, about 24% of FL patients managed with watchful waiting developed significant organ dysfunction or transformation at first progression over 8.2 years of follow-up.
Organ dysfunction and transformation were associated with significantly worse overall survival (OS) that could not be predicted based on baseline characteristics.
Gwynivere A. Davies, MD, of the University of Calgary in Alberta, Canada, and her colleagues conducted this study using data from the Alberta Lymphoma Database. The team gathered data on patients with grade 1-3a FL who were diagnosed between 1994 and 2011.
The investigators identified 238 patients who were initially managed with watchful waiting. The patients had a median age of 54.1 years (range, 24.7-69.9) at diagnosis, and 83.2% were advanced stage.
The 10-year OS rate for these patients was 81.2%. At a median follow-up of 98.5 months, 71% (n=169) of patients had progressed and required therapy.
At the time of progression, 24.4% of patients (n=58) had organ dysfunction and/or transformation. The median time to organ dysfunction/transformation was 29.9 months.
These adverse outcomes were significantly associated with inferior OS. The 10-year OS rate was 65.4% for patients with transformation at progression and 83.2% for those without transformation (P=0.0017).
The 10-year OS rate was 71.5% for those with organ dysfunction at progression and 82.7% for those without organ dysfunction (P=0.028).
Comparison to treated patients
The investigators also looked at a comparison group of 236 FL patients managed with immediate rituximab-based chemotherapy (R-chemo), most of whom were scheduled to receive (72.9%) rituximab maintenance. Their median age was 52.1 (range, 27.3-65.4), and most (82.6%) had advanced stage disease.
At a median follow-up of 100.2 months, the median progression-free survival (PFS) was not reached. The 10-year OS rate was 84%.
The 10-year PFS rate after first R-chemo was 57.1% for patients who received immediate R-chemo (n=236) and 50.5% for patients who were initially managed with watchful waiting and proceeded to R-chemo (n=133; P=0.506). This was not affected by rituximab maintenance.
The investigators noted that OS measured from diagnosis was not affected by initial watchful waiting.
However, in a landmark analysis, OS was inferior when measured from R-chemo at first progression for watchful waiting recipients compared to patients who received immediate R-chemo. The 10-year OS rates were 74.4% and 84.0%, respectively (P=0.02).
The risk of transformation at first progression was significantly different between the groups. At 10 years, the rate of transformation was 25.5% in the watchful waiting group and 6.3% in the immediate R-chemo group (P<0.0001).
The investigators said these findings, taken together, suggest changes may be warranted for FL patients managed with watchful waiting.
“Consideration should be given to implementing standardized follow-up imaging, with early initiation of rituximab-based therapy if there is evidence of progression in an attempt to prevent these potentially clinically impactful events [i.e., organ dysfunction and transformation],” Dr. Davies and her coauthors wrote.
Dr. Davies reported no financial disclosures. Her coauthors reported disclosures related to Janssen, Gilead Sciences, Lundbeck, Roche, AbbVie, Amgen, Seattle Genetics, Bristol-Myers Squibb, Servier Laboratories, and Merck.
A subset of follicular lymphoma (FL) patients managed with watchful waiting are vulnerable to organ dysfunction and transformation, according to research published in Clinical Lymphoma, Myeloma & Leukemia.
In a retrospective study, about 24% of FL patients managed with watchful waiting developed significant organ dysfunction or transformation at first progression over 8.2 years of follow-up.
Organ dysfunction and transformation were associated with significantly worse overall survival (OS) that could not be predicted based on baseline characteristics.
Gwynivere A. Davies, MD, of the University of Calgary in Alberta, Canada, and her colleagues conducted this study using data from the Alberta Lymphoma Database. The team gathered data on patients with grade 1-3a FL who were diagnosed between 1994 and 2011.
The investigators identified 238 patients who were initially managed with watchful waiting. The patients had a median age of 54.1 years (range, 24.7-69.9) at diagnosis, and 83.2% were advanced stage.
The 10-year OS rate for these patients was 81.2%. At a median follow-up of 98.5 months, 71% (n=169) of patients had progressed and required therapy.
At the time of progression, 24.4% of patients (n=58) had organ dysfunction and/or transformation. The median time to organ dysfunction/transformation was 29.9 months.
These adverse outcomes were significantly associated with inferior OS. The 10-year OS rate was 65.4% for patients with transformation at progression and 83.2% for those without transformation (P=0.0017).
The 10-year OS rate was 71.5% for those with organ dysfunction at progression and 82.7% for those without organ dysfunction (P=0.028).
Comparison to treated patients
The investigators also looked at a comparison group of 236 FL patients managed with immediate rituximab-based chemotherapy (R-chemo), most of whom were scheduled to receive (72.9%) rituximab maintenance. Their median age was 52.1 (range, 27.3-65.4), and most (82.6%) had advanced stage disease.
At a median follow-up of 100.2 months, the median progression-free survival (PFS) was not reached. The 10-year OS rate was 84%.
The 10-year PFS rate after first R-chemo was 57.1% for patients who received immediate R-chemo (n=236) and 50.5% for patients who were initially managed with watchful waiting and proceeded to R-chemo (n=133; P=0.506). This was not affected by rituximab maintenance.
The investigators noted that OS measured from diagnosis was not affected by initial watchful waiting.
However, in a landmark analysis, OS was inferior when measured from R-chemo at first progression for watchful waiting recipients compared to patients who received immediate R-chemo. The 10-year OS rates were 74.4% and 84.0%, respectively (P=0.02).
The risk of transformation at first progression was significantly different between the groups. At 10 years, the rate of transformation was 25.5% in the watchful waiting group and 6.3% in the immediate R-chemo group (P<0.0001).
The investigators said these findings, taken together, suggest changes may be warranted for FL patients managed with watchful waiting.
“Consideration should be given to implementing standardized follow-up imaging, with early initiation of rituximab-based therapy if there is evidence of progression in an attempt to prevent these potentially clinically impactful events [i.e., organ dysfunction and transformation],” Dr. Davies and her coauthors wrote.
Dr. Davies reported no financial disclosures. Her coauthors reported disclosures related to Janssen, Gilead Sciences, Lundbeck, Roche, AbbVie, Amgen, Seattle Genetics, Bristol-Myers Squibb, Servier Laboratories, and Merck.
A subset of follicular lymphoma (FL) patients managed with watchful waiting are vulnerable to organ dysfunction and transformation, according to research published in Clinical Lymphoma, Myeloma & Leukemia.
In a retrospective study, about 24% of FL patients managed with watchful waiting developed significant organ dysfunction or transformation at first progression over 8.2 years of follow-up.
Organ dysfunction and transformation were associated with significantly worse overall survival (OS) that could not be predicted based on baseline characteristics.
Gwynivere A. Davies, MD, of the University of Calgary in Alberta, Canada, and her colleagues conducted this study using data from the Alberta Lymphoma Database. The team gathered data on patients with grade 1-3a FL who were diagnosed between 1994 and 2011.
The investigators identified 238 patients who were initially managed with watchful waiting. The patients had a median age of 54.1 years (range, 24.7-69.9) at diagnosis, and 83.2% were advanced stage.
The 10-year OS rate for these patients was 81.2%. At a median follow-up of 98.5 months, 71% (n=169) of patients had progressed and required therapy.
At the time of progression, 24.4% of patients (n=58) had organ dysfunction and/or transformation. The median time to organ dysfunction/transformation was 29.9 months.
These adverse outcomes were significantly associated with inferior OS. The 10-year OS rate was 65.4% for patients with transformation at progression and 83.2% for those without transformation (P=0.0017).
The 10-year OS rate was 71.5% for those with organ dysfunction at progression and 82.7% for those without organ dysfunction (P=0.028).
Comparison to treated patients
The investigators also looked at a comparison group of 236 FL patients managed with immediate rituximab-based chemotherapy (R-chemo), most of whom were scheduled to receive (72.9%) rituximab maintenance. Their median age was 52.1 (range, 27.3-65.4), and most (82.6%) had advanced stage disease.
At a median follow-up of 100.2 months, the median progression-free survival (PFS) was not reached. The 10-year OS rate was 84%.
The 10-year PFS rate after first R-chemo was 57.1% for patients who received immediate R-chemo (n=236) and 50.5% for patients who were initially managed with watchful waiting and proceeded to R-chemo (n=133; P=0.506). This was not affected by rituximab maintenance.
The investigators noted that OS measured from diagnosis was not affected by initial watchful waiting.
However, in a landmark analysis, OS was inferior when measured from R-chemo at first progression for watchful waiting recipients compared to patients who received immediate R-chemo. The 10-year OS rates were 74.4% and 84.0%, respectively (P=0.02).
The risk of transformation at first progression was significantly different between the groups. At 10 years, the rate of transformation was 25.5% in the watchful waiting group and 6.3% in the immediate R-chemo group (P<0.0001).
The investigators said these findings, taken together, suggest changes may be warranted for FL patients managed with watchful waiting.
“Consideration should be given to implementing standardized follow-up imaging, with early initiation of rituximab-based therapy if there is evidence of progression in an attempt to prevent these potentially clinically impactful events [i.e., organ dysfunction and transformation],” Dr. Davies and her coauthors wrote.
Dr. Davies reported no financial disclosures. Her coauthors reported disclosures related to Janssen, Gilead Sciences, Lundbeck, Roche, AbbVie, Amgen, Seattle Genetics, Bristol-Myers Squibb, Servier Laboratories, and Merck.
Be wary of watchful waiting in follicular lymphoma
A substantial proportion of patients with follicular lymphoma managed with watchful waiting develop organ dysfunction or transformation that may negatively impact survival outcomes, results of a retrospective study suggest.
About one-quarter of patients managed with watchful waiting developed significant organ dysfunction or transformation at first progression over 8.2 years of follow-up.
Organ dysfunction and transformation were associated with significantly worse overall survival that could not be predicted based on baseline characteristics, the study authors reported in Clinical Lymphoma, Myeloma & Leukemia.
The study confirmed certain benefits of watchful waiting, including a low risk of progression and an “excellent” rate of overall survival, the investigators said.
However, the substantial rate of organ dysfunction and transformation in a subset of patients is “clinically meaningful for informed decision making,” reported Gwynivere A. Davies, MD, of the University of Calgary (Alta.), and her coauthors.
“While consenting patients to initial [watchful waiting], patients need to be informed about the risk for these adverse events, as well as receiving education and the need for close monitoring regarding symptoms that may indicate serious progression events,” Dr. Davies and her coauthors wrote.
Alternatively, rituximab chemotherapy, with or without rituximab maintenance, might be warranted for watchful waiting patients with clear disease progression before organ dysfunction or transformation events, despite not meeting high-tumor burden therapy indications.
The retrospective study included data from the Alberta Lymphoma Database on patients with grade 1-3a follicular lymphoma aged 18-70 years who were diagnosed between 1994 and 2011. Investigators identified 238 patients initially managed with watchful waiting, with a median age of 54.1 years at diagnosis. More than 80% were advanced stage.
Only 71% of these patients progressed, with a median time to progression of about 30 months and a 10-year survival rate from diagnosis of 81.2%, investigators said. However, 58 patients (24.4%) had organ dysfunction or transformation at the time of progression.
Those adverse outcomes significantly affected overall survival. The 10-year overall survival was 65.4% for patients with transformation at progression versus 83.2% for those without (P = .0017). Likewise, 10-year overall survival was 71.5% and 82.7%, respectively, for those with organ dysfunction at progression and those without (P = .028).
Investigators also looked at a comparison group of 236 follicular lymphoma patients managed with immediate rituximab chemotherapy. They found survival outcomes in that group were similar to those in the subgroup of 56 watchful waiting patients who received primarily rituximab-containing regimens at the time of organ dysfunction or transformation.
Taken together, the findings suggest management changes may be warranted for follicular lymphoma patients managed according to a watchful waiting strategy, the investigators wrote. “Consideration should be given to implementing standardized follow-up imaging, with early initiation of rituximab-based therapy if there is evidence of progression in an attempt to prevent these potentially clinically impactful events.”
Dr. Davies reported having no financial disclosures. Study coauthors reported disclosures related to Janssen, Gilead Sciences, Lundbeck, Roche, AbbVie, Amgen, Seattle Genetics, Bristol-Myers Squibb, Servier Laboratories, and Merck.
SOURCE: Davies GA et al. Clin Lymphoma Myeloma Leuk. 2018 Aug 28. doi: 10.1016/j.clml.2018.08.015.
A substantial proportion of patients with follicular lymphoma managed with watchful waiting develop organ dysfunction or transformation that may negatively impact survival outcomes, results of a retrospective study suggest.
About one-quarter of patients managed with watchful waiting developed significant organ dysfunction or transformation at first progression over 8.2 years of follow-up.
Organ dysfunction and transformation were associated with significantly worse overall survival that could not be predicted based on baseline characteristics, the study authors reported in Clinical Lymphoma, Myeloma & Leukemia.
The study confirmed certain benefits of watchful waiting, including a low risk of progression and an “excellent” rate of overall survival, the investigators said.
However, the substantial rate of organ dysfunction and transformation in a subset of patients is “clinically meaningful for informed decision making,” reported Gwynivere A. Davies, MD, of the University of Calgary (Alta.), and her coauthors.
“While consenting patients to initial [watchful waiting], patients need to be informed about the risk for these adverse events, as well as receiving education and the need for close monitoring regarding symptoms that may indicate serious progression events,” Dr. Davies and her coauthors wrote.
Alternatively, rituximab chemotherapy, with or without rituximab maintenance, might be warranted for watchful waiting patients with clear disease progression before organ dysfunction or transformation events, despite not meeting high-tumor burden therapy indications.
The retrospective study included data from the Alberta Lymphoma Database on patients with grade 1-3a follicular lymphoma aged 18-70 years who were diagnosed between 1994 and 2011. Investigators identified 238 patients initially managed with watchful waiting, with a median age of 54.1 years at diagnosis. More than 80% were advanced stage.
Only 71% of these patients progressed, with a median time to progression of about 30 months and a 10-year survival rate from diagnosis of 81.2%, investigators said. However, 58 patients (24.4%) had organ dysfunction or transformation at the time of progression.
Those adverse outcomes significantly affected overall survival. The 10-year overall survival was 65.4% for patients with transformation at progression versus 83.2% for those without (P = .0017). Likewise, 10-year overall survival was 71.5% and 82.7%, respectively, for those with organ dysfunction at progression and those without (P = .028).
Investigators also looked at a comparison group of 236 follicular lymphoma patients managed with immediate rituximab chemotherapy. They found survival outcomes in that group were similar to those in the subgroup of 56 watchful waiting patients who received primarily rituximab-containing regimens at the time of organ dysfunction or transformation.
Taken together, the findings suggest management changes may be warranted for follicular lymphoma patients managed according to a watchful waiting strategy, the investigators wrote. “Consideration should be given to implementing standardized follow-up imaging, with early initiation of rituximab-based therapy if there is evidence of progression in an attempt to prevent these potentially clinically impactful events.”
Dr. Davies reported having no financial disclosures. Study coauthors reported disclosures related to Janssen, Gilead Sciences, Lundbeck, Roche, AbbVie, Amgen, Seattle Genetics, Bristol-Myers Squibb, Servier Laboratories, and Merck.
SOURCE: Davies GA et al. Clin Lymphoma Myeloma Leuk. 2018 Aug 28. doi: 10.1016/j.clml.2018.08.015.
A substantial proportion of patients with follicular lymphoma managed with watchful waiting develop organ dysfunction or transformation that may negatively impact survival outcomes, results of a retrospective study suggest.
About one-quarter of patients managed with watchful waiting developed significant organ dysfunction or transformation at first progression over 8.2 years of follow-up.
Organ dysfunction and transformation were associated with significantly worse overall survival that could not be predicted based on baseline characteristics, the study authors reported in Clinical Lymphoma, Myeloma & Leukemia.
The study confirmed certain benefits of watchful waiting, including a low risk of progression and an “excellent” rate of overall survival, the investigators said.
However, the substantial rate of organ dysfunction and transformation in a subset of patients is “clinically meaningful for informed decision making,” reported Gwynivere A. Davies, MD, of the University of Calgary (Alta.), and her coauthors.
“While consenting patients to initial [watchful waiting], patients need to be informed about the risk for these adverse events, as well as receiving education and the need for close monitoring regarding symptoms that may indicate serious progression events,” Dr. Davies and her coauthors wrote.
Alternatively, rituximab chemotherapy, with or without rituximab maintenance, might be warranted for watchful waiting patients with clear disease progression before organ dysfunction or transformation events, despite not meeting high-tumor burden therapy indications.
The retrospective study included data from the Alberta Lymphoma Database on patients with grade 1-3a follicular lymphoma aged 18-70 years who were diagnosed between 1994 and 2011. Investigators identified 238 patients initially managed with watchful waiting, with a median age of 54.1 years at diagnosis. More than 80% were advanced stage.
Only 71% of these patients progressed, with a median time to progression of about 30 months and a 10-year survival rate from diagnosis of 81.2%, investigators said. However, 58 patients (24.4%) had organ dysfunction or transformation at the time of progression.
Those adverse outcomes significantly affected overall survival. The 10-year overall survival was 65.4% for patients with transformation at progression versus 83.2% for those without (P = .0017). Likewise, 10-year overall survival was 71.5% and 82.7%, respectively, for those with organ dysfunction at progression and those without (P = .028).
Investigators also looked at a comparison group of 236 follicular lymphoma patients managed with immediate rituximab chemotherapy. They found survival outcomes in that group were similar to those in the subgroup of 56 watchful waiting patients who received primarily rituximab-containing regimens at the time of organ dysfunction or transformation.
Taken together, the findings suggest management changes may be warranted for follicular lymphoma patients managed according to a watchful waiting strategy, the investigators wrote. “Consideration should be given to implementing standardized follow-up imaging, with early initiation of rituximab-based therapy if there is evidence of progression in an attempt to prevent these potentially clinically impactful events.”
Dr. Davies reported having no financial disclosures. Study coauthors reported disclosures related to Janssen, Gilead Sciences, Lundbeck, Roche, AbbVie, Amgen, Seattle Genetics, Bristol-Myers Squibb, Servier Laboratories, and Merck.
SOURCE: Davies GA et al. Clin Lymphoma Myeloma Leuk. 2018 Aug 28. doi: 10.1016/j.clml.2018.08.015.
FROM CLINICAL LYMPHOMA, MYELOMA & LEUKEMIA
Key clinical point:
Major finding: A total of 58 patients (24.4%) had organ dysfunction or transformation at the time of progression and had worse survival outcomes, compared with patients who did not experience those events.
Study details: A retrospective study including data on 238 patients with grade 1-3a follicular lymphoma aged 18-70 years who were managed with watchful waiting.
Disclosures: Study authors reported disclosures related to Janssen, Gilead Sciences, Lundbeck, Roche, AbbVie, Amgen, Seattle Genetics, Bristol-Myers Squibb, Servier Laboratories, and Merck.
Source: Davies GA et al. Clin Lymphoma Myeloma Leuk. 2018 Aug 28. doi: 10.1016/j.clml.2018.08.015.