User login
Dasatinib outcomes similar to imatinib in pediatric Ph+ ALL

Dasatinib used during induction and consolidation in the Children’s Oncology Group (COG) AALL0622 trial provided early response rates for children with Ph-positive (Ph+) acute lymphoblastic leukemia (ALL), according to investigators.
But the early response rates did not improve event-free survival (EFS) compared to the use of consolidation imatinib in the AALL0031 study.
Incidence of cranial relapse was more than doubled in AALL0622 compared to AALL0031.
The investigators believe the incidence of cranial relapse may explain the results of AALL0622.
“We cannot yet conclude that the current dasatinib plus chemotherapy combination is better than imatinib plus chemotherapy,” the authors stated.
AALL0622 was designed to be an improvement on AALL0031, which demonstrated that adding the tyrosine kinase inhibitor (TKI) imatinib to intensive chemotherapy in the consolidation phase significantly improved survival for children with Ph+ ALL.
In AALL0622 dasatinib was given early in induction (day 15) and then in consolidation with the hope that patients could achieve early remission.
Another departure from AALL0031 was that cranial irradiation was not provided for control of central nervous system (CNS) metastasis. Because dasatinib accumulates in the CNS, which is a ‘sanctuary site’ for leukemia, it was presumed that patients could benefit from a TKI yet be spared from cranial irradiation.
As expected, adding dasatinib mid-induction provided a complete remission rate of 98% at the end of induction (day 29), which was better than the 89% seen in AALL0031.
In addition, more patients in AALL0622 showed minimal residual disease (MRD) <0.01% at the end of induction: 59% vs 25% in AALL0031 (P <0.001). At the end of consolidation, corresponding rates were 89% vs 71% for AALL0031.
For the primary outcome, 3-year EFS was 84.6% for patients in AALL0622 in standard-risk patients. Five-year OS and EFS rates were 86% and 60%, respectively.
In patients with overt brain metastasis (CNS3 status), 5-year CNS relapse was 15% for patients in the AALL0622 study vs 6.6% for patients in the AALL031 study.
However, 5-year OS rates were similar in the two groups of patients: 86% for AALL0622 vs 81% for AALL0031.
HSCT
AALL0622 allowed the use of hematopoietic stem cell transplantation (HSCT) in high-risk patients as well as in standard-risk patients with a sibling donor.
Five-year OS and EFS for standard-risk patients (19% underwent HSCT at first remission) and high-risk patients (91% underwent HSCT in first remission) were similar.
Children who did not undergo HSCT had a similar 5-year OS of 88%, which suggested that children with Ph+ ALL should not undergo transplantation at first remission.
Samples from a subset of patients was analyzed for IKZF1 mutations and correlated with outcomes.
Five-year OS was 80% in those harboring the mutation versus 100% who had the wild-type gene (P=0.04); 4-year EFS was also significantly lower—10% vs 82% (P=0.04).
Screening for IKZF1 may be used to identify high-risk patients suitable for HSCT and/or alternate treatment, the authors note.
The investigators reported their findings in The Journal of Clinical Oncology.
Dasatinib used during induction and consolidation in the Children’s Oncology Group (COG) AALL0622 trial provided early response rates for children with Ph-positive (Ph+) acute lymphoblastic leukemia (ALL), according to investigators.
But the early response rates did not improve event-free survival (EFS) compared to the use of consolidation imatinib in the AALL0031 study.
Incidence of cranial relapse was more than doubled in AALL0622 compared to AALL0031.
The investigators believe the incidence of cranial relapse may explain the results of AALL0622.
“We cannot yet conclude that the current dasatinib plus chemotherapy combination is better than imatinib plus chemotherapy,” the authors stated.
AALL0622 was designed to be an improvement on AALL0031, which demonstrated that adding the tyrosine kinase inhibitor (TKI) imatinib to intensive chemotherapy in the consolidation phase significantly improved survival for children with Ph+ ALL.
In AALL0622 dasatinib was given early in induction (day 15) and then in consolidation with the hope that patients could achieve early remission.
Another departure from AALL0031 was that cranial irradiation was not provided for control of central nervous system (CNS) metastasis. Because dasatinib accumulates in the CNS, which is a ‘sanctuary site’ for leukemia, it was presumed that patients could benefit from a TKI yet be spared from cranial irradiation.
As expected, adding dasatinib mid-induction provided a complete remission rate of 98% at the end of induction (day 29), which was better than the 89% seen in AALL0031.
In addition, more patients in AALL0622 showed minimal residual disease (MRD) <0.01% at the end of induction: 59% vs 25% in AALL0031 (P <0.001). At the end of consolidation, corresponding rates were 89% vs 71% for AALL0031.
For the primary outcome, 3-year EFS was 84.6% for patients in AALL0622 in standard-risk patients. Five-year OS and EFS rates were 86% and 60%, respectively.
In patients with overt brain metastasis (CNS3 status), 5-year CNS relapse was 15% for patients in the AALL0622 study vs 6.6% for patients in the AALL031 study.
However, 5-year OS rates were similar in the two groups of patients: 86% for AALL0622 vs 81% for AALL0031.
HSCT
AALL0622 allowed the use of hematopoietic stem cell transplantation (HSCT) in high-risk patients as well as in standard-risk patients with a sibling donor.
Five-year OS and EFS for standard-risk patients (19% underwent HSCT at first remission) and high-risk patients (91% underwent HSCT in first remission) were similar.
Children who did not undergo HSCT had a similar 5-year OS of 88%, which suggested that children with Ph+ ALL should not undergo transplantation at first remission.
Samples from a subset of patients was analyzed for IKZF1 mutations and correlated with outcomes.
Five-year OS was 80% in those harboring the mutation versus 100% who had the wild-type gene (P=0.04); 4-year EFS was also significantly lower—10% vs 82% (P=0.04).
Screening for IKZF1 may be used to identify high-risk patients suitable for HSCT and/or alternate treatment, the authors note.
The investigators reported their findings in The Journal of Clinical Oncology.
Dasatinib used during induction and consolidation in the Children’s Oncology Group (COG) AALL0622 trial provided early response rates for children with Ph-positive (Ph+) acute lymphoblastic leukemia (ALL), according to investigators.
But the early response rates did not improve event-free survival (EFS) compared to the use of consolidation imatinib in the AALL0031 study.
Incidence of cranial relapse was more than doubled in AALL0622 compared to AALL0031.
The investigators believe the incidence of cranial relapse may explain the results of AALL0622.
“We cannot yet conclude that the current dasatinib plus chemotherapy combination is better than imatinib plus chemotherapy,” the authors stated.
AALL0622 was designed to be an improvement on AALL0031, which demonstrated that adding the tyrosine kinase inhibitor (TKI) imatinib to intensive chemotherapy in the consolidation phase significantly improved survival for children with Ph+ ALL.
In AALL0622 dasatinib was given early in induction (day 15) and then in consolidation with the hope that patients could achieve early remission.
Another departure from AALL0031 was that cranial irradiation was not provided for control of central nervous system (CNS) metastasis. Because dasatinib accumulates in the CNS, which is a ‘sanctuary site’ for leukemia, it was presumed that patients could benefit from a TKI yet be spared from cranial irradiation.
As expected, adding dasatinib mid-induction provided a complete remission rate of 98% at the end of induction (day 29), which was better than the 89% seen in AALL0031.
In addition, more patients in AALL0622 showed minimal residual disease (MRD) <0.01% at the end of induction: 59% vs 25% in AALL0031 (P <0.001). At the end of consolidation, corresponding rates were 89% vs 71% for AALL0031.
For the primary outcome, 3-year EFS was 84.6% for patients in AALL0622 in standard-risk patients. Five-year OS and EFS rates were 86% and 60%, respectively.
In patients with overt brain metastasis (CNS3 status), 5-year CNS relapse was 15% for patients in the AALL0622 study vs 6.6% for patients in the AALL031 study.
However, 5-year OS rates were similar in the two groups of patients: 86% for AALL0622 vs 81% for AALL0031.
HSCT
AALL0622 allowed the use of hematopoietic stem cell transplantation (HSCT) in high-risk patients as well as in standard-risk patients with a sibling donor.
Five-year OS and EFS for standard-risk patients (19% underwent HSCT at first remission) and high-risk patients (91% underwent HSCT in first remission) were similar.
Children who did not undergo HSCT had a similar 5-year OS of 88%, which suggested that children with Ph+ ALL should not undergo transplantation at first remission.
Samples from a subset of patients was analyzed for IKZF1 mutations and correlated with outcomes.
Five-year OS was 80% in those harboring the mutation versus 100% who had the wild-type gene (P=0.04); 4-year EFS was also significantly lower—10% vs 82% (P=0.04).
Screening for IKZF1 may be used to identify high-risk patients suitable for HSCT and/or alternate treatment, the authors note.
The investigators reported their findings in The Journal of Clinical Oncology.
FDA grants regular approval to venetoclax for CLL/SLL
Venetoclax (Venclexta) has received regular approval from the Food and Drug Administration for the treatment of patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL), with or without 17p deletion, who have received at least one prior therapy.
The approval was based results from the MURANO trial of 389 patients, which was a randomized, multicenter, open-label trial of venetoclax plus rituximab versus bendamustine plus rituximab.
Neutropenia, diarrhea, upper respiratory tract infection, fatigue, cough, and nausea were the most common adverse events seen in the venetoclax arm. Grade 3 or 4 neutropenia developed in 64% of those patients, and grade 4 in 31%. The most common infection in venetoclax patients was pneumonia, but overall, 21% of patients in that arm experienced some kind of infection.
Because of the rapid reduction in tumor size, tumor lysis syndrome is possible with venetoclax treatment, the FDA noted.
In 2016, the FDA granted accelerated approval to venetoclax for treatment of patients with CLL with 17d deletion who had received at least one prior line of therapy.
Venetoclax (Venclexta) has received regular approval from the Food and Drug Administration for the treatment of patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL), with or without 17p deletion, who have received at least one prior therapy.
The approval was based results from the MURANO trial of 389 patients, which was a randomized, multicenter, open-label trial of venetoclax plus rituximab versus bendamustine plus rituximab.
Neutropenia, diarrhea, upper respiratory tract infection, fatigue, cough, and nausea were the most common adverse events seen in the venetoclax arm. Grade 3 or 4 neutropenia developed in 64% of those patients, and grade 4 in 31%. The most common infection in venetoclax patients was pneumonia, but overall, 21% of patients in that arm experienced some kind of infection.
Because of the rapid reduction in tumor size, tumor lysis syndrome is possible with venetoclax treatment, the FDA noted.
In 2016, the FDA granted accelerated approval to venetoclax for treatment of patients with CLL with 17d deletion who had received at least one prior line of therapy.
Venetoclax (Venclexta) has received regular approval from the Food and Drug Administration for the treatment of patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL), with or without 17p deletion, who have received at least one prior therapy.
The approval was based results from the MURANO trial of 389 patients, which was a randomized, multicenter, open-label trial of venetoclax plus rituximab versus bendamustine plus rituximab.
Neutropenia, diarrhea, upper respiratory tract infection, fatigue, cough, and nausea were the most common adverse events seen in the venetoclax arm. Grade 3 or 4 neutropenia developed in 64% of those patients, and grade 4 in 31%. The most common infection in venetoclax patients was pneumonia, but overall, 21% of patients in that arm experienced some kind of infection.
Because of the rapid reduction in tumor size, tumor lysis syndrome is possible with venetoclax treatment, the FDA noted.
In 2016, the FDA granted accelerated approval to venetoclax for treatment of patients with CLL with 17d deletion who had received at least one prior line of therapy.
Venetoclax plus ibrutinib yields encouraging MRD results in first-line CLL
CHICAGO – The combination of ibrutinib plus venetoclax yielded a high rate of undetectable minimal residual disease (MRD) when used as first-line treatment for chronic lymphocytic leukemia (CLL), according to preliminary results of the CAPTIVATE trial.
Of the first 30 patients in the trial, 23 (77%) had undetectable blood MRD after just six cycles of combined treatment, said investigator William G. Wierda, MD, PhD, of the University of Texas MD Anderson Cancer Center, Houston.

“These early results show a highly active and safe treatment with 12 cycles of combined treatment with ibrutinib and venetoclax,” Dr. Wierda said in a presentation of the CAPTIVATE results at the annual meeting of the American Society of Clinical Oncology.
Those MRD results are “at least as good as we can achieve with chemoimmunotherapy,” Bruce D. Cheson, MD, head of hematology at Georgetown University, Washington, said during a discussion of the CAPTIVATE study results.
Dr. Cheson referenced MRD results from a 2016 analysis of the CLL8 and CLL10 trials, which included patients treated with fludarabine, cyclophosphamide, and rituximab (FCR) and bendamustine plus rituximab (BR). In that analysis, 33.6% of patients achieved MRD-negative complete response and 29.1% achieved MRD-negative partial response.
In CAPTIVATE, by contrast, all of the complete remissions were MRD negative, as were a majority of the partial responders, Dr. Cheson noted.
Venetoclax and ibrutinib have “clinically complimentary activity” that provided a rationale for combining the two, Dr. Wierda said at ASCO. Ibrutinib is a BTK inhibitor that has a high rate of response and durable disease control, though continuous treatment is indicated, he said, because most patients achieve partial remissions as best response and continue to have residual disease in blood or bone marrow. Venetoclax, he added, is a BCL-2 inhibitor that produces durable partial remissions, though “residual disease is typically present in the form of persistently enlarged lymph nodes,” he said. “Venetoclax is highly effective at clearing disease from blood and bone marrow.”
The phase 2 CAPTIVATE trial includes a total of 164 patients younger than 70 years of age who receive a 3-cycle ibrutinib lead-in, followed by ibrutinib plus venetoclax for 12 cycles. At that point, patients are randomized according to MRD status. Patients with confirmed undetectable MRD are randomized to further treatment with ibrutinib or placebo, and those with undetectable MRD not confirmed are randomized to ibrutinib versus ibrutinib plus venetoclax.
In addition to early efficacy data, Dr. Wierda also reported some safety data. Compared with the single-agent ibrutinib lead-in period, combined ibrutinib plus venetoclax treatment had more gastrointestinal-associated events and neutropenia. Almost half of patients (45%) have had a treatment-related grade 3-4 adverse event, though just 18 (11%) have had treatment-related adverse events classified as serious, and there have been no adverse event-related deaths on study.
The high activity of ibrutinib plus venetoclax in CAPTIVATE supports further study of the combination, Dr. Wierda said. A randomized, open-label phase 3 trial of ibrutinib plus venetoclax versus chlorambucil plus obinutuzumab as first-line treatment for CLL is currently recruiting.
The study was sponsored by Pharmacyclics, an AbbVie company. Dr. Wierda reported consulting and research funding from Pharmacyclics, AbbVie, and several other companies.
SOURCE: Wierda WG et al. ASCO 2018, Abstract 7502.
CHICAGO – The combination of ibrutinib plus venetoclax yielded a high rate of undetectable minimal residual disease (MRD) when used as first-line treatment for chronic lymphocytic leukemia (CLL), according to preliminary results of the CAPTIVATE trial.
Of the first 30 patients in the trial, 23 (77%) had undetectable blood MRD after just six cycles of combined treatment, said investigator William G. Wierda, MD, PhD, of the University of Texas MD Anderson Cancer Center, Houston.
“These early results show a highly active and safe treatment with 12 cycles of combined treatment with ibrutinib and venetoclax,” Dr. Wierda said in a presentation of the CAPTIVATE results at the annual meeting of the American Society of Clinical Oncology.
Those MRD results are “at least as good as we can achieve with chemoimmunotherapy,” Bruce D. Cheson, MD, head of hematology at Georgetown University, Washington, said during a discussion of the CAPTIVATE study results.
Dr. Cheson referenced MRD results from a 2016 analysis of the CLL8 and CLL10 trials, which included patients treated with fludarabine, cyclophosphamide, and rituximab (FCR) and bendamustine plus rituximab (BR). In that analysis, 33.6% of patients achieved MRD-negative complete response and 29.1% achieved MRD-negative partial response.
In CAPTIVATE, by contrast, all of the complete remissions were MRD negative, as were a majority of the partial responders, Dr. Cheson noted.
Venetoclax and ibrutinib have “clinically complimentary activity” that provided a rationale for combining the two, Dr. Wierda said at ASCO. Ibrutinib is a BTK inhibitor that has a high rate of response and durable disease control, though continuous treatment is indicated, he said, because most patients achieve partial remissions as best response and continue to have residual disease in blood or bone marrow. Venetoclax, he added, is a BCL-2 inhibitor that produces durable partial remissions, though “residual disease is typically present in the form of persistently enlarged lymph nodes,” he said. “Venetoclax is highly effective at clearing disease from blood and bone marrow.”
The phase 2 CAPTIVATE trial includes a total of 164 patients younger than 70 years of age who receive a 3-cycle ibrutinib lead-in, followed by ibrutinib plus venetoclax for 12 cycles. At that point, patients are randomized according to MRD status. Patients with confirmed undetectable MRD are randomized to further treatment with ibrutinib or placebo, and those with undetectable MRD not confirmed are randomized to ibrutinib versus ibrutinib plus venetoclax.
In addition to early efficacy data, Dr. Wierda also reported some safety data. Compared with the single-agent ibrutinib lead-in period, combined ibrutinib plus venetoclax treatment had more gastrointestinal-associated events and neutropenia. Almost half of patients (45%) have had a treatment-related grade 3-4 adverse event, though just 18 (11%) have had treatment-related adverse events classified as serious, and there have been no adverse event-related deaths on study.
The high activity of ibrutinib plus venetoclax in CAPTIVATE supports further study of the combination, Dr. Wierda said. A randomized, open-label phase 3 trial of ibrutinib plus venetoclax versus chlorambucil plus obinutuzumab as first-line treatment for CLL is currently recruiting.
The study was sponsored by Pharmacyclics, an AbbVie company. Dr. Wierda reported consulting and research funding from Pharmacyclics, AbbVie, and several other companies.
SOURCE: Wierda WG et al. ASCO 2018, Abstract 7502.
CHICAGO – The combination of ibrutinib plus venetoclax yielded a high rate of undetectable minimal residual disease (MRD) when used as first-line treatment for chronic lymphocytic leukemia (CLL), according to preliminary results of the CAPTIVATE trial.
Of the first 30 patients in the trial, 23 (77%) had undetectable blood MRD after just six cycles of combined treatment, said investigator William G. Wierda, MD, PhD, of the University of Texas MD Anderson Cancer Center, Houston.
“These early results show a highly active and safe treatment with 12 cycles of combined treatment with ibrutinib and venetoclax,” Dr. Wierda said in a presentation of the CAPTIVATE results at the annual meeting of the American Society of Clinical Oncology.
Those MRD results are “at least as good as we can achieve with chemoimmunotherapy,” Bruce D. Cheson, MD, head of hematology at Georgetown University, Washington, said during a discussion of the CAPTIVATE study results.
Dr. Cheson referenced MRD results from a 2016 analysis of the CLL8 and CLL10 trials, which included patients treated with fludarabine, cyclophosphamide, and rituximab (FCR) and bendamustine plus rituximab (BR). In that analysis, 33.6% of patients achieved MRD-negative complete response and 29.1% achieved MRD-negative partial response.
In CAPTIVATE, by contrast, all of the complete remissions were MRD negative, as were a majority of the partial responders, Dr. Cheson noted.
Venetoclax and ibrutinib have “clinically complimentary activity” that provided a rationale for combining the two, Dr. Wierda said at ASCO. Ibrutinib is a BTK inhibitor that has a high rate of response and durable disease control, though continuous treatment is indicated, he said, because most patients achieve partial remissions as best response and continue to have residual disease in blood or bone marrow. Venetoclax, he added, is a BCL-2 inhibitor that produces durable partial remissions, though “residual disease is typically present in the form of persistently enlarged lymph nodes,” he said. “Venetoclax is highly effective at clearing disease from blood and bone marrow.”
The phase 2 CAPTIVATE trial includes a total of 164 patients younger than 70 years of age who receive a 3-cycle ibrutinib lead-in, followed by ibrutinib plus venetoclax for 12 cycles. At that point, patients are randomized according to MRD status. Patients with confirmed undetectable MRD are randomized to further treatment with ibrutinib or placebo, and those with undetectable MRD not confirmed are randomized to ibrutinib versus ibrutinib plus venetoclax.
In addition to early efficacy data, Dr. Wierda also reported some safety data. Compared with the single-agent ibrutinib lead-in period, combined ibrutinib plus venetoclax treatment had more gastrointestinal-associated events and neutropenia. Almost half of patients (45%) have had a treatment-related grade 3-4 adverse event, though just 18 (11%) have had treatment-related adverse events classified as serious, and there have been no adverse event-related deaths on study.
The high activity of ibrutinib plus venetoclax in CAPTIVATE supports further study of the combination, Dr. Wierda said. A randomized, open-label phase 3 trial of ibrutinib plus venetoclax versus chlorambucil plus obinutuzumab as first-line treatment for CLL is currently recruiting.
The study was sponsored by Pharmacyclics, an AbbVie company. Dr. Wierda reported consulting and research funding from Pharmacyclics, AbbVie, and several other companies.
SOURCE: Wierda WG et al. ASCO 2018, Abstract 7502.
REPORTING FROM ASCO 2018
Key clinical point:
Major finding: Of 14 patients, 12 (86%) who completed 12 cycles of treatment had undetectable bone marrow MRD.
Study details: Early results of the phase 2 CAPTIVATE trial including 164 patients younger than 70 years of age with previously untreated CLL.
Disclosures: The study was sponsored by Pharmacyclics, an Abbvie company. Dr. Wierda reported consulting and research funding from Pharmacyclics, AbbVie, and several other companies.
Source: Wierda WG et al. ASCO 2018, Abstract 7502.

Two agents could take AML therapy in new directions
CHICAGO—Two agents targeting novel pathways in myeloid malignancies—mivebresib and bencentinib—are showing promise in early studies, according to a speaker at the 2018 ASCO Annual Meeting.
“Both BET and AXL inhibition appear to be new and exciting targets in myeloid malignancies,” said Alice S. Mims, MD, and both have produced responses as single agents.
Dr Mims, of Ohio State University Wexner Medical Center in Columbus, made these observations in a poster discussion presentation that included commentary on the two agents.
Mivebresib (ABBV-075), an inhibitor of bromodomain and extra terminal (BET) proteins, yielded some responses in relapsed/refractory acute myeloid leukemia (AML) patients in a first-in-human study presented at the meeting (abstract 7019*).
Bemcentinib (BGB324), a first-in class selective inhibitor of the AXL tyrosine kinase, also showed activity in preliminary results of a study including patients with relapsed/refractory disease (abstract 7020*).
“It will be important to know individual patient characteristics to determine the potential response predictors,” Dr Mims said.
Mivebresib (NCT02391480)
Mivebresib is the subject of an ongoing phase 1 dose-escalation study in which 23 patients have been treated. That includes 12 who received the BET inhibitor as monotherapy, and 11 who got it in combination with the BCL-2 inhibitor venetoclax, which is indicated in CLL and has breakthrough therapy designation for AML.
Investigators observed responses in 3 of 17 evaluable patients (17.6%), including 1 complete remission with incomplete blood count recovery in a patient on mivebresib monotherapy, plus 1 partial response and 1 patient achieving a morphologic leukemia-free state with the combination.
The most common grade 3/4 treatment-emergent adverse events included anemia in 52%, thrombocytopenia in 44%, and febrile neutropenia in 26% of patients, with no dose-limiting toxicities noted as of this report.
Bemcentinib (NCT02488408)
Bemcentinib is being evaluated in a phase 1/2 trial including patients with relapsed/refractory AML and myelodysplastic syndromes (MDS).
For 32 patients treated so far, 3 patients achieved a complete remission, including 1 AML and 2 MDS patients.
In addition, 3 patients achieved partial response, including 1 MDS and 2 AML patients.
Treatment with bemcentinib was generally well-tolerated, and most adverse events were mild or moderate, investigators reported in their poster.
Pre-treatment levels of soluble AXL were lower in responders compared with non-responders, investigators also noted.
“Soluble AXL levels may be a predictive biomarker for AXL inhibition, but further assessment is necessary,” Dr Mims said.
*Data presented at the meeting differ from the abstracts.
CHICAGO—Two agents targeting novel pathways in myeloid malignancies—mivebresib and bencentinib—are showing promise in early studies, according to a speaker at the 2018 ASCO Annual Meeting.
“Both BET and AXL inhibition appear to be new and exciting targets in myeloid malignancies,” said Alice S. Mims, MD, and both have produced responses as single agents.
Dr Mims, of Ohio State University Wexner Medical Center in Columbus, made these observations in a poster discussion presentation that included commentary on the two agents.
Mivebresib (ABBV-075), an inhibitor of bromodomain and extra terminal (BET) proteins, yielded some responses in relapsed/refractory acute myeloid leukemia (AML) patients in a first-in-human study presented at the meeting (abstract 7019*).
Bemcentinib (BGB324), a first-in class selective inhibitor of the AXL tyrosine kinase, also showed activity in preliminary results of a study including patients with relapsed/refractory disease (abstract 7020*).
“It will be important to know individual patient characteristics to determine the potential response predictors,” Dr Mims said.
Mivebresib (NCT02391480)
Mivebresib is the subject of an ongoing phase 1 dose-escalation study in which 23 patients have been treated. That includes 12 who received the BET inhibitor as monotherapy, and 11 who got it in combination with the BCL-2 inhibitor venetoclax, which is indicated in CLL and has breakthrough therapy designation for AML.
Investigators observed responses in 3 of 17 evaluable patients (17.6%), including 1 complete remission with incomplete blood count recovery in a patient on mivebresib monotherapy, plus 1 partial response and 1 patient achieving a morphologic leukemia-free state with the combination.
The most common grade 3/4 treatment-emergent adverse events included anemia in 52%, thrombocytopenia in 44%, and febrile neutropenia in 26% of patients, with no dose-limiting toxicities noted as of this report.
Bemcentinib (NCT02488408)
Bemcentinib is being evaluated in a phase 1/2 trial including patients with relapsed/refractory AML and myelodysplastic syndromes (MDS).
For 32 patients treated so far, 3 patients achieved a complete remission, including 1 AML and 2 MDS patients.
In addition, 3 patients achieved partial response, including 1 MDS and 2 AML patients.
Treatment with bemcentinib was generally well-tolerated, and most adverse events were mild or moderate, investigators reported in their poster.
Pre-treatment levels of soluble AXL were lower in responders compared with non-responders, investigators also noted.
“Soluble AXL levels may be a predictive biomarker for AXL inhibition, but further assessment is necessary,” Dr Mims said.
*Data presented at the meeting differ from the abstracts.
CHICAGO—Two agents targeting novel pathways in myeloid malignancies—mivebresib and bencentinib—are showing promise in early studies, according to a speaker at the 2018 ASCO Annual Meeting.
“Both BET and AXL inhibition appear to be new and exciting targets in myeloid malignancies,” said Alice S. Mims, MD, and both have produced responses as single agents.
Dr Mims, of Ohio State University Wexner Medical Center in Columbus, made these observations in a poster discussion presentation that included commentary on the two agents.
Mivebresib (ABBV-075), an inhibitor of bromodomain and extra terminal (BET) proteins, yielded some responses in relapsed/refractory acute myeloid leukemia (AML) patients in a first-in-human study presented at the meeting (abstract 7019*).
Bemcentinib (BGB324), a first-in class selective inhibitor of the AXL tyrosine kinase, also showed activity in preliminary results of a study including patients with relapsed/refractory disease (abstract 7020*).
“It will be important to know individual patient characteristics to determine the potential response predictors,” Dr Mims said.
Mivebresib (NCT02391480)
Mivebresib is the subject of an ongoing phase 1 dose-escalation study in which 23 patients have been treated. That includes 12 who received the BET inhibitor as monotherapy, and 11 who got it in combination with the BCL-2 inhibitor venetoclax, which is indicated in CLL and has breakthrough therapy designation for AML.
Investigators observed responses in 3 of 17 evaluable patients (17.6%), including 1 complete remission with incomplete blood count recovery in a patient on mivebresib monotherapy, plus 1 partial response and 1 patient achieving a morphologic leukemia-free state with the combination.
The most common grade 3/4 treatment-emergent adverse events included anemia in 52%, thrombocytopenia in 44%, and febrile neutropenia in 26% of patients, with no dose-limiting toxicities noted as of this report.
Bemcentinib (NCT02488408)
Bemcentinib is being evaluated in a phase 1/2 trial including patients with relapsed/refractory AML and myelodysplastic syndromes (MDS).
For 32 patients treated so far, 3 patients achieved a complete remission, including 1 AML and 2 MDS patients.
In addition, 3 patients achieved partial response, including 1 MDS and 2 AML patients.
Treatment with bemcentinib was generally well-tolerated, and most adverse events were mild or moderate, investigators reported in their poster.
Pre-treatment levels of soluble AXL were lower in responders compared with non-responders, investigators also noted.
“Soluble AXL levels may be a predictive biomarker for AXL inhibition, but further assessment is necessary,” Dr Mims said.
*Data presented at the meeting differ from the abstracts.
MRD-negative status signals better outcomes in CAR T–treated ALL
CHICAGO – Minimal residual disease (MRD)–negative complete remission was strongly associated with improved survival outcomes in patients with B-cell acute lymphocytic leukemia (ALL) who received CD19 chimeric antigen receptor (CAR) T cells, results of a retrospective study showed.
Allogeneic hematopoietic stem cell transplant (HSCT) appeared to improve both disease-free and overall survival in those patients who had achieved MRD-negative complete remission, according to results of the study, which were presented at the annual meeting of the American Society of Clinical Oncology.
“Based upon our interaction testing, the potential benefit [of transplant] appears to exist in both good-risk and bad-risk patients as identified through multivariate modeling,” said study investigator Kevin Anthony Hay, MD, of Fred Hutchinson Cancer Research Center, Seattle.
In a comment on the results, Sarah Cooley, MD, noted that the benefits of allogeneic transplant were apparent regardless of whether the patients met criteria for the good-risk subgroup, which was defined by levels of lactate dehydrogenase (LDH) and platelets along with exposure to fludarabine as part of the conditioning regimen.
“I think this suggests that the goal at this point is to get patients to an MRD-negative state and to potentially curative transplant,” said Dr. Cooley, director of investigator-initiated research at Masonic Medical Center at the University of Minnesota, Minneapolis.
The retrospective analysis by Dr. Hay and his colleagues included 53 adults with relapsed or refractory ALL who had bone marrow or extramedullary disease at baseline and had received CD19 CAR T cells at or under the maximum tolerated dose at least 1 year prior to this analysis. Of that group, 45 (85%) achieved MRD-negative complete remission.
Those patients who did achieve MRD-negative complete remission had an improved median disease-free survival at 7.6 months versus 0.8 months (P less than .0001) and improved overall survival at 20.0 months versus 5.0 months (P = 0.014).
Most of the MRD-negative patients who relapsed did so within the first 6 months, an observation that led investigators to consider whether factors exist that could predict better outcomes.
In a multivariate analysis, they found three variables associated with disease free survival: higher LDH prior to lymphodepletion (hazard ratio, 1.39), along with higher platelet count prior to lymphodepletion and incorporation of fludarabine into the regimen, with hazard ratios of 0.65 and 0.34, respectively.
Using those three characteristics, investigators grouped patients as “good risk” if they had normal LDH, platelet count at or above 100 prior to lymphodepletion that included fludarabine. The 24-month disease-free survival for good-risk patients was 78%, and overall survival was 86%.
The role of allogeneic HSCT after ALL patients achieved MRD-negative complete remission with CAR T-cell therapy was one of the “major questions in the field,” Dr. Hay said.
In this analysis, Dr. Hay and colleagues found that patients who underwent transplant in MRD-negative complete remission had a 24-month disease free survival and overall survival of 61% and 72%, respectively, both of which were significantly higher than in patients with MRD-negative complete remission who had no transplant.
The disease-free survival benefit was not specific to the good-risk group, according to Dr. Hay, who said an interaction test demonstrated no significant interaction between risk group and allogeneic HSCT after CAR T-cell infusion (P = 0.53).
“This is a very important finding that should be further [studied] in an appropriately designed clinical trial,” Dr. Hay said during an oral presentation of the study results.
Dr. Hay and several coauthors reported financial disclosures related to Juno Therapeutics. Other disclosures reported by study coauthors included Cell Medica, Celgene, Eureka Therapeutics, Genentech/Roche, Gilead Sciences, Kite Pharma, Novartis, and others.
SOURCE: Hay KA. ASCO 2018, Abstract 7005.
CHICAGO – Minimal residual disease (MRD)–negative complete remission was strongly associated with improved survival outcomes in patients with B-cell acute lymphocytic leukemia (ALL) who received CD19 chimeric antigen receptor (CAR) T cells, results of a retrospective study showed.
Allogeneic hematopoietic stem cell transplant (HSCT) appeared to improve both disease-free and overall survival in those patients who had achieved MRD-negative complete remission, according to results of the study, which were presented at the annual meeting of the American Society of Clinical Oncology.
“Based upon our interaction testing, the potential benefit [of transplant] appears to exist in both good-risk and bad-risk patients as identified through multivariate modeling,” said study investigator Kevin Anthony Hay, MD, of Fred Hutchinson Cancer Research Center, Seattle.
In a comment on the results, Sarah Cooley, MD, noted that the benefits of allogeneic transplant were apparent regardless of whether the patients met criteria for the good-risk subgroup, which was defined by levels of lactate dehydrogenase (LDH) and platelets along with exposure to fludarabine as part of the conditioning regimen.
“I think this suggests that the goal at this point is to get patients to an MRD-negative state and to potentially curative transplant,” said Dr. Cooley, director of investigator-initiated research at Masonic Medical Center at the University of Minnesota, Minneapolis.
The retrospective analysis by Dr. Hay and his colleagues included 53 adults with relapsed or refractory ALL who had bone marrow or extramedullary disease at baseline and had received CD19 CAR T cells at or under the maximum tolerated dose at least 1 year prior to this analysis. Of that group, 45 (85%) achieved MRD-negative complete remission.
Those patients who did achieve MRD-negative complete remission had an improved median disease-free survival at 7.6 months versus 0.8 months (P less than .0001) and improved overall survival at 20.0 months versus 5.0 months (P = 0.014).
Most of the MRD-negative patients who relapsed did so within the first 6 months, an observation that led investigators to consider whether factors exist that could predict better outcomes.
In a multivariate analysis, they found three variables associated with disease free survival: higher LDH prior to lymphodepletion (hazard ratio, 1.39), along with higher platelet count prior to lymphodepletion and incorporation of fludarabine into the regimen, with hazard ratios of 0.65 and 0.34, respectively.
Using those three characteristics, investigators grouped patients as “good risk” if they had normal LDH, platelet count at or above 100 prior to lymphodepletion that included fludarabine. The 24-month disease-free survival for good-risk patients was 78%, and overall survival was 86%.
The role of allogeneic HSCT after ALL patients achieved MRD-negative complete remission with CAR T-cell therapy was one of the “major questions in the field,” Dr. Hay said.
In this analysis, Dr. Hay and colleagues found that patients who underwent transplant in MRD-negative complete remission had a 24-month disease free survival and overall survival of 61% and 72%, respectively, both of which were significantly higher than in patients with MRD-negative complete remission who had no transplant.
The disease-free survival benefit was not specific to the good-risk group, according to Dr. Hay, who said an interaction test demonstrated no significant interaction between risk group and allogeneic HSCT after CAR T-cell infusion (P = 0.53).
“This is a very important finding that should be further [studied] in an appropriately designed clinical trial,” Dr. Hay said during an oral presentation of the study results.
Dr. Hay and several coauthors reported financial disclosures related to Juno Therapeutics. Other disclosures reported by study coauthors included Cell Medica, Celgene, Eureka Therapeutics, Genentech/Roche, Gilead Sciences, Kite Pharma, Novartis, and others.
SOURCE: Hay KA. ASCO 2018, Abstract 7005.
CHICAGO – Minimal residual disease (MRD)–negative complete remission was strongly associated with improved survival outcomes in patients with B-cell acute lymphocytic leukemia (ALL) who received CD19 chimeric antigen receptor (CAR) T cells, results of a retrospective study showed.
Allogeneic hematopoietic stem cell transplant (HSCT) appeared to improve both disease-free and overall survival in those patients who had achieved MRD-negative complete remission, according to results of the study, which were presented at the annual meeting of the American Society of Clinical Oncology.
“Based upon our interaction testing, the potential benefit [of transplant] appears to exist in both good-risk and bad-risk patients as identified through multivariate modeling,” said study investigator Kevin Anthony Hay, MD, of Fred Hutchinson Cancer Research Center, Seattle.
In a comment on the results, Sarah Cooley, MD, noted that the benefits of allogeneic transplant were apparent regardless of whether the patients met criteria for the good-risk subgroup, which was defined by levels of lactate dehydrogenase (LDH) and platelets along with exposure to fludarabine as part of the conditioning regimen.
“I think this suggests that the goal at this point is to get patients to an MRD-negative state and to potentially curative transplant,” said Dr. Cooley, director of investigator-initiated research at Masonic Medical Center at the University of Minnesota, Minneapolis.
The retrospective analysis by Dr. Hay and his colleagues included 53 adults with relapsed or refractory ALL who had bone marrow or extramedullary disease at baseline and had received CD19 CAR T cells at or under the maximum tolerated dose at least 1 year prior to this analysis. Of that group, 45 (85%) achieved MRD-negative complete remission.
Those patients who did achieve MRD-negative complete remission had an improved median disease-free survival at 7.6 months versus 0.8 months (P less than .0001) and improved overall survival at 20.0 months versus 5.0 months (P = 0.014).
Most of the MRD-negative patients who relapsed did so within the first 6 months, an observation that led investigators to consider whether factors exist that could predict better outcomes.
In a multivariate analysis, they found three variables associated with disease free survival: higher LDH prior to lymphodepletion (hazard ratio, 1.39), along with higher platelet count prior to lymphodepletion and incorporation of fludarabine into the regimen, with hazard ratios of 0.65 and 0.34, respectively.
Using those three characteristics, investigators grouped patients as “good risk” if they had normal LDH, platelet count at or above 100 prior to lymphodepletion that included fludarabine. The 24-month disease-free survival for good-risk patients was 78%, and overall survival was 86%.
The role of allogeneic HSCT after ALL patients achieved MRD-negative complete remission with CAR T-cell therapy was one of the “major questions in the field,” Dr. Hay said.
In this analysis, Dr. Hay and colleagues found that patients who underwent transplant in MRD-negative complete remission had a 24-month disease free survival and overall survival of 61% and 72%, respectively, both of which were significantly higher than in patients with MRD-negative complete remission who had no transplant.
The disease-free survival benefit was not specific to the good-risk group, according to Dr. Hay, who said an interaction test demonstrated no significant interaction between risk group and allogeneic HSCT after CAR T-cell infusion (P = 0.53).
“This is a very important finding that should be further [studied] in an appropriately designed clinical trial,” Dr. Hay said during an oral presentation of the study results.
Dr. Hay and several coauthors reported financial disclosures related to Juno Therapeutics. Other disclosures reported by study coauthors included Cell Medica, Celgene, Eureka Therapeutics, Genentech/Roche, Gilead Sciences, Kite Pharma, Novartis, and others.
SOURCE: Hay KA. ASCO 2018, Abstract 7005.
REPORTING FROM ASCO 2018
Key clinical point:
Major finding: Patients who achieved MRD-negative complete remission had an improved median disease-free survival at 7.6 months versus 0.8 months (P less than .0001)
Study details: A retrospective analysis including 53 patients with ALL who had bone marrow or extramedullary disease at baseline and had received CD19 CAR T cells at or under the maximum tolerated dose at least 1 year prior to this analysis.
Disclosures: Researchers reported financial ties to Juno Therapeutics, Cell Medica, Celgene, Eureka Therapeutics, Genentech/Roche, Gilead Sciences, Kite Pharma, Novartis, and others.
Source: Hay KA. ASCO 2018, Abstract 7005.
FDA approves first biosimilar pegfilgrastim

The US Food and Drug Association (FDA) has approved pegfilgrastim-jmdb (Fulphila™) as the first biosimilar to Neulasta®.
The agents reduce the risk of infection or the duration of febrile neutropenia in patients treated with immunosuppressive chemotherapy for non-myeloid hematologic malignancies.
The FDA approved Fulphila based on evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamic data, clinical immunogenicity data, and other clinical safety and effectiveness data.
The evidence demonstrated that Fulphila is biosimilar to Amgen’s Neulasta. The FDA, in its announcement, noted that Fulphila has been approved as a biosimilar and not as an interchangeable product.
A biosimilar is a biological product approved based on data showing it is highly similar to a biological product already approved by the FDA, termed the reference product.
A biosimilar has no clinically meaningful differences from the reference product in terms of safety, purity, and effectiveness.
Common side effects of Fulphila include bone pain and pain in extremities.
The FDA cautions that patients with a history of serious allergic reaction to human granulocyte colony-stimulating factors, such as pegfilgrastim or filgrastim products, should not take Fulphila.
Serious side effects from Fulphila include:
- rupture of the spleen
- acute respiratory distress syndrome
- serious allergic reactions including anaphylaxis
- glomerulonephritis
- leukocytosis
- capillary leak syndrome
- potential for tumor growth
Fatal sickle cell crises have also occurred with Fulphila use.
Fulphila is not indicated for the mobilization of peripheral blood progenitor cells for hematopoietic stem cell transplantation.
The FDA is planning to release a comprehensive new plan to advance policy efforts that promote biosimilar product development, according to FDA Commissioner Scott Gotlieb, MD.
“We want to make sure that the pathway for developing biosimilar versions of approved biologics is efficient and effective, so that patients benefit from competition to existing biologics once lawful intellectual property has lapsed on these products,” he said in the announcement.
The FDA granted approval of Fulphila to Mylan GmbH. Mylan is co-developing Fulphila with Biocon.
Last fall, the agency had issued a complete response letter saying it could not approve the proposed biosimilar pending an update to the application.
The complete response letter did not raise any questions on the biosimilarity of Fulphila (investigational drug product MYL-1401H), pharmacokinetic/pharmacodynamic data, clinical data, or immunogenicity, however.
Mylan anticipates launching Fulphila in the coming weeks.
The US Food and Drug Association (FDA) has approved pegfilgrastim-jmdb (Fulphila™) as the first biosimilar to Neulasta®.
The agents reduce the risk of infection or the duration of febrile neutropenia in patients treated with immunosuppressive chemotherapy for non-myeloid hematologic malignancies.
The FDA approved Fulphila based on evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamic data, clinical immunogenicity data, and other clinical safety and effectiveness data.
The evidence demonstrated that Fulphila is biosimilar to Amgen’s Neulasta. The FDA, in its announcement, noted that Fulphila has been approved as a biosimilar and not as an interchangeable product.
A biosimilar is a biological product approved based on data showing it is highly similar to a biological product already approved by the FDA, termed the reference product.
A biosimilar has no clinically meaningful differences from the reference product in terms of safety, purity, and effectiveness.
Common side effects of Fulphila include bone pain and pain in extremities.
The FDA cautions that patients with a history of serious allergic reaction to human granulocyte colony-stimulating factors, such as pegfilgrastim or filgrastim products, should not take Fulphila.
Serious side effects from Fulphila include:
- rupture of the spleen
- acute respiratory distress syndrome
- serious allergic reactions including anaphylaxis
- glomerulonephritis
- leukocytosis
- capillary leak syndrome
- potential for tumor growth
Fatal sickle cell crises have also occurred with Fulphila use.
Fulphila is not indicated for the mobilization of peripheral blood progenitor cells for hematopoietic stem cell transplantation.
The FDA is planning to release a comprehensive new plan to advance policy efforts that promote biosimilar product development, according to FDA Commissioner Scott Gotlieb, MD.
“We want to make sure that the pathway for developing biosimilar versions of approved biologics is efficient and effective, so that patients benefit from competition to existing biologics once lawful intellectual property has lapsed on these products,” he said in the announcement.
The FDA granted approval of Fulphila to Mylan GmbH. Mylan is co-developing Fulphila with Biocon.
Last fall, the agency had issued a complete response letter saying it could not approve the proposed biosimilar pending an update to the application.
The complete response letter did not raise any questions on the biosimilarity of Fulphila (investigational drug product MYL-1401H), pharmacokinetic/pharmacodynamic data, clinical data, or immunogenicity, however.
Mylan anticipates launching Fulphila in the coming weeks.
The US Food and Drug Association (FDA) has approved pegfilgrastim-jmdb (Fulphila™) as the first biosimilar to Neulasta®.
The agents reduce the risk of infection or the duration of febrile neutropenia in patients treated with immunosuppressive chemotherapy for non-myeloid hematologic malignancies.
The FDA approved Fulphila based on evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamic data, clinical immunogenicity data, and other clinical safety and effectiveness data.
The evidence demonstrated that Fulphila is biosimilar to Amgen’s Neulasta. The FDA, in its announcement, noted that Fulphila has been approved as a biosimilar and not as an interchangeable product.
A biosimilar is a biological product approved based on data showing it is highly similar to a biological product already approved by the FDA, termed the reference product.
A biosimilar has no clinically meaningful differences from the reference product in terms of safety, purity, and effectiveness.
Common side effects of Fulphila include bone pain and pain in extremities.
The FDA cautions that patients with a history of serious allergic reaction to human granulocyte colony-stimulating factors, such as pegfilgrastim or filgrastim products, should not take Fulphila.
Serious side effects from Fulphila include:
- rupture of the spleen
- acute respiratory distress syndrome
- serious allergic reactions including anaphylaxis
- glomerulonephritis
- leukocytosis
- capillary leak syndrome
- potential for tumor growth
Fatal sickle cell crises have also occurred with Fulphila use.
Fulphila is not indicated for the mobilization of peripheral blood progenitor cells for hematopoietic stem cell transplantation.
The FDA is planning to release a comprehensive new plan to advance policy efforts that promote biosimilar product development, according to FDA Commissioner Scott Gotlieb, MD.
“We want to make sure that the pathway for developing biosimilar versions of approved biologics is efficient and effective, so that patients benefit from competition to existing biologics once lawful intellectual property has lapsed on these products,” he said in the announcement.
The FDA granted approval of Fulphila to Mylan GmbH. Mylan is co-developing Fulphila with Biocon.
Last fall, the agency had issued a complete response letter saying it could not approve the proposed biosimilar pending an update to the application.
The complete response letter did not raise any questions on the biosimilarity of Fulphila (investigational drug product MYL-1401H), pharmacokinetic/pharmacodynamic data, clinical data, or immunogenicity, however.
Mylan anticipates launching Fulphila in the coming weeks.
Over 1100 new meds, vaccines being developed to treat cancer

Currently, 1,120 new medicines and vaccines are being developed to treat cancer, according to a new report of the Pharmaceutical Research and Manufacturers of America (PhRMA).
And all of them, the organization states, are in clinical trials or awaiting review by the US Food and Drug Administration (FDA).
Leading the way are treatments for solid tumors, with 397 in development. Treatments for blood cancers are not far behind, with nearly 340 medicines in development: 137 for leukemias, 135 for lymphomas, and 62 for multiple myeloma.
Immuno-oncology and personalized medicine have a hand in this increase.
In the last year, according to PhRMA’s "Medicines in Development for Cancer 2018 Report," 47 new immune-oncology treatments have been added to the development pipeline, including CAR-T therapies and checkpoint inhibitors.
This brings the total to 295 immuno-oncology medicines and vaccines in the development pipeline this year.
The report also states that about 85% of these medicines in the oncology pipeline are first-in-class.
And PhRMA attributes the approximately 73% of survival gains in cancer to the new medicines.
Despite the bright picture, PhRMA acknowledges the financial burden and medical care challenges patients encounter.
It addresses them in a new chart pack, "Cancer Medicines: Value in Context," which puts cancer costs in perspective and offers solutions for improving the current system in the United States.
The association reports the top medical financial concerns of patients to be diagnostic tests or scans (53%), prescription medicines (43%), physician office visits (39%), outpatient treatments-including radiation (37%), and surgery (36%).
Spending on cancer medicines represents about 1% of overall healthcare spending, according to the organization, with cancer medications accounting for $49.8 billion of the $3.49 trillion healthcare spending in the United States.
Cancer medicines represent about 20% of spending on cancer, PhrMA notes, and some insurance plans place treatments for certain high-cost conditions on the highest drug formulary cost-sharing tier.
And patients with the highest copay were 5 times more likely to abandon treatment than the lowest copay group, PhRMA points out.
“No patient should struggle to afford their needed treatments,” PhRMA stated in a release, “and it is important that we address patient access challenges.”
Currently, 1,120 new medicines and vaccines are being developed to treat cancer, according to a new report of the Pharmaceutical Research and Manufacturers of America (PhRMA).
And all of them, the organization states, are in clinical trials or awaiting review by the US Food and Drug Administration (FDA).
Leading the way are treatments for solid tumors, with 397 in development. Treatments for blood cancers are not far behind, with nearly 340 medicines in development: 137 for leukemias, 135 for lymphomas, and 62 for multiple myeloma.
Immuno-oncology and personalized medicine have a hand in this increase.
In the last year, according to PhRMA’s "Medicines in Development for Cancer 2018 Report," 47 new immune-oncology treatments have been added to the development pipeline, including CAR-T therapies and checkpoint inhibitors.
This brings the total to 295 immuno-oncology medicines and vaccines in the development pipeline this year.
The report also states that about 85% of these medicines in the oncology pipeline are first-in-class.
And PhRMA attributes the approximately 73% of survival gains in cancer to the new medicines.
Despite the bright picture, PhRMA acknowledges the financial burden and medical care challenges patients encounter.
It addresses them in a new chart pack, "Cancer Medicines: Value in Context," which puts cancer costs in perspective and offers solutions for improving the current system in the United States.
The association reports the top medical financial concerns of patients to be diagnostic tests or scans (53%), prescription medicines (43%), physician office visits (39%), outpatient treatments-including radiation (37%), and surgery (36%).
Spending on cancer medicines represents about 1% of overall healthcare spending, according to the organization, with cancer medications accounting for $49.8 billion of the $3.49 trillion healthcare spending in the United States.
Cancer medicines represent about 20% of spending on cancer, PhrMA notes, and some insurance plans place treatments for certain high-cost conditions on the highest drug formulary cost-sharing tier.
And patients with the highest copay were 5 times more likely to abandon treatment than the lowest copay group, PhRMA points out.
“No patient should struggle to afford their needed treatments,” PhRMA stated in a release, “and it is important that we address patient access challenges.”
Currently, 1,120 new medicines and vaccines are being developed to treat cancer, according to a new report of the Pharmaceutical Research and Manufacturers of America (PhRMA).
And all of them, the organization states, are in clinical trials or awaiting review by the US Food and Drug Administration (FDA).
Leading the way are treatments for solid tumors, with 397 in development. Treatments for blood cancers are not far behind, with nearly 340 medicines in development: 137 for leukemias, 135 for lymphomas, and 62 for multiple myeloma.
Immuno-oncology and personalized medicine have a hand in this increase.
In the last year, according to PhRMA’s "Medicines in Development for Cancer 2018 Report," 47 new immune-oncology treatments have been added to the development pipeline, including CAR-T therapies and checkpoint inhibitors.
This brings the total to 295 immuno-oncology medicines and vaccines in the development pipeline this year.
The report also states that about 85% of these medicines in the oncology pipeline are first-in-class.
And PhRMA attributes the approximately 73% of survival gains in cancer to the new medicines.
Despite the bright picture, PhRMA acknowledges the financial burden and medical care challenges patients encounter.
It addresses them in a new chart pack, "Cancer Medicines: Value in Context," which puts cancer costs in perspective and offers solutions for improving the current system in the United States.
The association reports the top medical financial concerns of patients to be diagnostic tests or scans (53%), prescription medicines (43%), physician office visits (39%), outpatient treatments-including radiation (37%), and surgery (36%).
Spending on cancer medicines represents about 1% of overall healthcare spending, according to the organization, with cancer medications accounting for $49.8 billion of the $3.49 trillion healthcare spending in the United States.
Cancer medicines represent about 20% of spending on cancer, PhrMA notes, and some insurance plans place treatments for certain high-cost conditions on the highest drug formulary cost-sharing tier.
And patients with the highest copay were 5 times more likely to abandon treatment than the lowest copay group, PhRMA points out.
“No patient should struggle to afford their needed treatments,” PhRMA stated in a release, “and it is important that we address patient access challenges.”
Ivosidenib leads to complete responses in 32% of IDH1-mutated relapsed/refractory AML
CHICAGO – The investigational drug ivosidenib was associated with durable responses, transfusion independence, and a low rate of serious adverse events in patients with IDH1-mutated relapsed/refractory acute myeloid leukemia (AML), based on results of a phase 1 study presented at the annual meeting of the American Society of Clinical Oncology (ASCO).
Daily oral ivosidenib, which inhibits mutant IDH1 the gene that encodes for isocitrate dehydrogenase 1 and is present in 6%-10% of AML patients, was associated with a complete response rate of nearly 32%. In patients who achieved a complete response, median duration of response was 8.2 months and median overall survival was 18.8 months, Daniel A. Pollyea, MD, University of Colorado School of Medicine, Aurora, reported at the meeting.
A wide variety of study participants achieved transfusion independence, even patients who had lesser and no discernable responses, Dr. Pollyea said. “A significant minority of these patients (with lesser or no response) were able to achieve transfusion independence which is obviously a big achievement with respect to quality of life, and suggestive of the mechanism of action here with respect to differentiation.”
The conclusions of this study are “striking,” with response rates and tolerability “very similar” to results seen with the IDH2 inhibitor enasidenib, said Eunice S. Wang, MD, of Roswell Park Comprehensive Cancer Center, Buffalo, N.Y.
Ivosidenib is “likely to become the new standard of care treatment” for IDH1-mutant patients in the relapsed-refractory setting based on the data presented at ASCO, Dr. Wang said in a presentation commenting on the results.
“Given the development of ivosidenib and enasidenib, I think we should all carefully consider whether testing of IDH1 and IDH2 mutational status should also become standard of care in all relapsed and refractory patients, in order to offer them these novel and highly effective as well as well-tolerated targeted therapies,” she said.
Enasidenib was approved by the U.S. Food and Drug Administration (FDA) in 2017 for relapsed or refractory AML with an IDH2 mutation.
Here at ASCO, Dr. Pollyea reported results based on 179 patients with relapsed/refractory IDH1-mutant AML who received 500 mg of ivosidenib daily.
The overall response rate was 41.9% (75 patients), he said. The rate of complete remission (CR) or CR with partial hematologic recovery (CRh) was 31.8% (57 patients), with a median of 2.0 months to CR/CRh and a median duration of CR/CRh of 8.2 months, he said.
Grade 3 or greater leukocytosis was seen in 8% of patients, and grade 3 or greater QT prolongation was seen in 10% of patients, Dr. Pollyea reported.
IHD differentiation syndrome of any grade was seen in 19 patients (10.6%), and about half of those cases (5%) were grade 3 or greater. However, cases were managed with supportive care, and there were no dose reductions, permanent treatment discontinuations, or deaths due to the condition, Dr. Pollyea said.
Moreover, the majority of those patients (10 out of 19) went on to have a response. “Supportive care and continued treatment can allow for patients ultimately to respond who are experiencing differentiation syndrome,” Dr. Pollyea said.
Results of the phase 1 study were published simultaneously in the New England Journal of Medicine.The trial was sponsored by Agios. Dr. Pollyea reported disclosures related to Agios, as well as Abbvie, argenx, Celgene, Celyad, Curis, Pfizer, and Servier.
SOURCE: Pollyea DA, et al. J Clin Oncol36, 2018 (suppl; abstr 7000).
CHICAGO – The investigational drug ivosidenib was associated with durable responses, transfusion independence, and a low rate of serious adverse events in patients with IDH1-mutated relapsed/refractory acute myeloid leukemia (AML), based on results of a phase 1 study presented at the annual meeting of the American Society of Clinical Oncology (ASCO).
Daily oral ivosidenib, which inhibits mutant IDH1 the gene that encodes for isocitrate dehydrogenase 1 and is present in 6%-10% of AML patients, was associated with a complete response rate of nearly 32%. In patients who achieved a complete response, median duration of response was 8.2 months and median overall survival was 18.8 months, Daniel A. Pollyea, MD, University of Colorado School of Medicine, Aurora, reported at the meeting.
A wide variety of study participants achieved transfusion independence, even patients who had lesser and no discernable responses, Dr. Pollyea said. “A significant minority of these patients (with lesser or no response) were able to achieve transfusion independence which is obviously a big achievement with respect to quality of life, and suggestive of the mechanism of action here with respect to differentiation.”
The conclusions of this study are “striking,” with response rates and tolerability “very similar” to results seen with the IDH2 inhibitor enasidenib, said Eunice S. Wang, MD, of Roswell Park Comprehensive Cancer Center, Buffalo, N.Y.
Ivosidenib is “likely to become the new standard of care treatment” for IDH1-mutant patients in the relapsed-refractory setting based on the data presented at ASCO, Dr. Wang said in a presentation commenting on the results.
“Given the development of ivosidenib and enasidenib, I think we should all carefully consider whether testing of IDH1 and IDH2 mutational status should also become standard of care in all relapsed and refractory patients, in order to offer them these novel and highly effective as well as well-tolerated targeted therapies,” she said.
Enasidenib was approved by the U.S. Food and Drug Administration (FDA) in 2017 for relapsed or refractory AML with an IDH2 mutation.
Here at ASCO, Dr. Pollyea reported results based on 179 patients with relapsed/refractory IDH1-mutant AML who received 500 mg of ivosidenib daily.
The overall response rate was 41.9% (75 patients), he said. The rate of complete remission (CR) or CR with partial hematologic recovery (CRh) was 31.8% (57 patients), with a median of 2.0 months to CR/CRh and a median duration of CR/CRh of 8.2 months, he said.
Grade 3 or greater leukocytosis was seen in 8% of patients, and grade 3 or greater QT prolongation was seen in 10% of patients, Dr. Pollyea reported.
IHD differentiation syndrome of any grade was seen in 19 patients (10.6%), and about half of those cases (5%) were grade 3 or greater. However, cases were managed with supportive care, and there were no dose reductions, permanent treatment discontinuations, or deaths due to the condition, Dr. Pollyea said.
Moreover, the majority of those patients (10 out of 19) went on to have a response. “Supportive care and continued treatment can allow for patients ultimately to respond who are experiencing differentiation syndrome,” Dr. Pollyea said.
Results of the phase 1 study were published simultaneously in the New England Journal of Medicine.The trial was sponsored by Agios. Dr. Pollyea reported disclosures related to Agios, as well as Abbvie, argenx, Celgene, Celyad, Curis, Pfizer, and Servier.
SOURCE: Pollyea DA, et al. J Clin Oncol36, 2018 (suppl; abstr 7000).
CHICAGO – The investigational drug ivosidenib was associated with durable responses, transfusion independence, and a low rate of serious adverse events in patients with IDH1-mutated relapsed/refractory acute myeloid leukemia (AML), based on results of a phase 1 study presented at the annual meeting of the American Society of Clinical Oncology (ASCO).
Daily oral ivosidenib, which inhibits mutant IDH1 the gene that encodes for isocitrate dehydrogenase 1 and is present in 6%-10% of AML patients, was associated with a complete response rate of nearly 32%. In patients who achieved a complete response, median duration of response was 8.2 months and median overall survival was 18.8 months, Daniel A. Pollyea, MD, University of Colorado School of Medicine, Aurora, reported at the meeting.
A wide variety of study participants achieved transfusion independence, even patients who had lesser and no discernable responses, Dr. Pollyea said. “A significant minority of these patients (with lesser or no response) were able to achieve transfusion independence which is obviously a big achievement with respect to quality of life, and suggestive of the mechanism of action here with respect to differentiation.”
The conclusions of this study are “striking,” with response rates and tolerability “very similar” to results seen with the IDH2 inhibitor enasidenib, said Eunice S. Wang, MD, of Roswell Park Comprehensive Cancer Center, Buffalo, N.Y.
Ivosidenib is “likely to become the new standard of care treatment” for IDH1-mutant patients in the relapsed-refractory setting based on the data presented at ASCO, Dr. Wang said in a presentation commenting on the results.
“Given the development of ivosidenib and enasidenib, I think we should all carefully consider whether testing of IDH1 and IDH2 mutational status should also become standard of care in all relapsed and refractory patients, in order to offer them these novel and highly effective as well as well-tolerated targeted therapies,” she said.
Enasidenib was approved by the U.S. Food and Drug Administration (FDA) in 2017 for relapsed or refractory AML with an IDH2 mutation.
Here at ASCO, Dr. Pollyea reported results based on 179 patients with relapsed/refractory IDH1-mutant AML who received 500 mg of ivosidenib daily.
The overall response rate was 41.9% (75 patients), he said. The rate of complete remission (CR) or CR with partial hematologic recovery (CRh) was 31.8% (57 patients), with a median of 2.0 months to CR/CRh and a median duration of CR/CRh of 8.2 months, he said.
Grade 3 or greater leukocytosis was seen in 8% of patients, and grade 3 or greater QT prolongation was seen in 10% of patients, Dr. Pollyea reported.
IHD differentiation syndrome of any grade was seen in 19 patients (10.6%), and about half of those cases (5%) were grade 3 or greater. However, cases were managed with supportive care, and there were no dose reductions, permanent treatment discontinuations, or deaths due to the condition, Dr. Pollyea said.
Moreover, the majority of those patients (10 out of 19) went on to have a response. “Supportive care and continued treatment can allow for patients ultimately to respond who are experiencing differentiation syndrome,” Dr. Pollyea said.
Results of the phase 1 study were published simultaneously in the New England Journal of Medicine.The trial was sponsored by Agios. Dr. Pollyea reported disclosures related to Agios, as well as Abbvie, argenx, Celgene, Celyad, Curis, Pfizer, and Servier.
SOURCE: Pollyea DA, et al. J Clin Oncol36, 2018 (suppl; abstr 7000).
REPORTING FROM ASCO 2018
Key clinical point: Daily oral ivosidenib was associated with a complete response rate of nearly 32% and a low rate of serious adverse events in patients with IDH1-mutated relapsed/refractory acute myeloid leukemia (AML).
Major finding: Patients with complete responses had an 8.2-month duration of response, and a median overall survival of 18.8 months.
Study details: A phase 1 study including 179 patients who received 500 mg of ivosidenib daily.
Disclosures: Daniel A. Pollyea, MD, who presented the study, reported disclosures related to Abbvie, Agios, argenx, Celgene, Celyad, Curis, Pfizer, and Servier.
Source: Pollyea DA, et al. J Clin Oncol 36, 2018 (suppl; abstr 7000).
VF incidence increases significantly in kids after ALL treatment
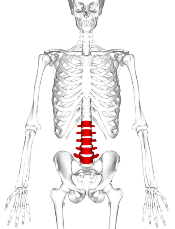
A 6-year prospective study from the Canadian STOPP investigators revealed that following treatment for acute lymphoblastic leukemia (ALL), approximately 1 out of 3 children experiences vertebral fractures (VF) and 1 out of 5 children shows non-VF.
Glucocorticoid use and vertebral fractures at diagnosis emerged as significant predictive factors.
Investigators reported a cumulative incidence of 32.5% for vertebral fractures (VF) following treatment and 23.0% for non-VF after 6 years.
Thirty-nine percent of children with VF were asymptomatic.
“The osteotoxicity of leukemia and its treatment are underscored by the observation that the non-VF incidence was more than 2.5 times higher than the general pediatric population,” the study authors wrote, “whereas the VF incidence was increased more than 2000 times.”
The STOPP investigators—the Steroid-Associated Osteoporosis in the Pediatric Population Consortium—published their findings in the Journal of Bone and Mineral Research.
To document the incidence and predictors of fractures and profile who is at greatest risk, the STOPP investigators enrolled 186 children who were diagnosed and treated at 10 children’s hospitals across Canada between 2005 and 2007.
Median age at diagnosis was 5.3 years, median duration of follow-up was 6 years, and 38.1% were boys.
Approximately 4 out of 5 children were treated with Children’s Oncology Group protocols and 1 out of 5 was treated with the Dana-Farber protocols.
In girls, glucocorticoid exposure was 7.0 g/m2 and in boys it was 8.9 g/m2.
The entire cohort received 54.3% of the total cumulative glucocorticoid dose in the first year and 83.1% in the second year.
For VF, incidence at baseline was 15.1%. Skeletal fractures at any site were reported in 36% of the children over the 6-year period, 71% occurring during the first 2 years. No VF was reported in the sixth year.
Of 38 children with VF, 32 sustained at least 1 VF in the first 2 years and 77% occurred while on therapy or within 6 months of the last glucocorticoid dose. None of the children had disease relapse.
Non-VF were reported in 1.6% of children at baseline; incidence peaked at years 2 (5.4%) and 5 (4.8%) and none occurred in the sixth year.
Thirty-one non-VFs were reported in 26 children; 18 occurred in the first 2 years and 23 occurred on therapy or within 6 months of the last glucocorticoid dose.
The STOPP investigators showed that glucocorticoid exposure, VF, and low spine bone mineral density Z-scores at baseline were significant predictors for risk of VF.
“[This] observation suggests the importance of baseline spine phenotype (both VF and low lumbar spine bone mineral density) in signaling the potential for future bone morbidity,” the STOPP investigators note.
“In revealing that vertebral fractures are frequent in children with ALL on chemotherapy, and that older children and those with more severe collapse are at risk for residual vertebral deformities, strategies to prevent vertebral fractures in those at greatest risk for permanent sequelae now merit further study,” said lead author Leanne Ward, MD, of the University of Ottawa, Canada, in a statement.
The study was primarily supported by an operating grant from the Canadian Institutes for Health Research (CIHR) with additional funding from a CIHR New Investigator Award, a Canadian Child Health Clinician Scientist Career Enhancement Award, a University of Ottawa Research Chair Award, a Children’s Hospital of Eastern Ontario (CHEO) Research Institute Capacity Building Award, and the CHEO Departments of Pediatrics and Surgery. This work was also supported by the University of Alberta Women and Children’s Health Research Institute.
A 6-year prospective study from the Canadian STOPP investigators revealed that following treatment for acute lymphoblastic leukemia (ALL), approximately 1 out of 3 children experiences vertebral fractures (VF) and 1 out of 5 children shows non-VF.
Glucocorticoid use and vertebral fractures at diagnosis emerged as significant predictive factors.
Investigators reported a cumulative incidence of 32.5% for vertebral fractures (VF) following treatment and 23.0% for non-VF after 6 years.
Thirty-nine percent of children with VF were asymptomatic.
“The osteotoxicity of leukemia and its treatment are underscored by the observation that the non-VF incidence was more than 2.5 times higher than the general pediatric population,” the study authors wrote, “whereas the VF incidence was increased more than 2000 times.”
The STOPP investigators—the Steroid-Associated Osteoporosis in the Pediatric Population Consortium—published their findings in the Journal of Bone and Mineral Research.
To document the incidence and predictors of fractures and profile who is at greatest risk, the STOPP investigators enrolled 186 children who were diagnosed and treated at 10 children’s hospitals across Canada between 2005 and 2007.
Median age at diagnosis was 5.3 years, median duration of follow-up was 6 years, and 38.1% were boys.
Approximately 4 out of 5 children were treated with Children’s Oncology Group protocols and 1 out of 5 was treated with the Dana-Farber protocols.
In girls, glucocorticoid exposure was 7.0 g/m2 and in boys it was 8.9 g/m2.
The entire cohort received 54.3% of the total cumulative glucocorticoid dose in the first year and 83.1% in the second year.
For VF, incidence at baseline was 15.1%. Skeletal fractures at any site were reported in 36% of the children over the 6-year period, 71% occurring during the first 2 years. No VF was reported in the sixth year.
Of 38 children with VF, 32 sustained at least 1 VF in the first 2 years and 77% occurred while on therapy or within 6 months of the last glucocorticoid dose. None of the children had disease relapse.
Non-VF were reported in 1.6% of children at baseline; incidence peaked at years 2 (5.4%) and 5 (4.8%) and none occurred in the sixth year.
Thirty-one non-VFs were reported in 26 children; 18 occurred in the first 2 years and 23 occurred on therapy or within 6 months of the last glucocorticoid dose.
The STOPP investigators showed that glucocorticoid exposure, VF, and low spine bone mineral density Z-scores at baseline were significant predictors for risk of VF.
“[This] observation suggests the importance of baseline spine phenotype (both VF and low lumbar spine bone mineral density) in signaling the potential for future bone morbidity,” the STOPP investigators note.
“In revealing that vertebral fractures are frequent in children with ALL on chemotherapy, and that older children and those with more severe collapse are at risk for residual vertebral deformities, strategies to prevent vertebral fractures in those at greatest risk for permanent sequelae now merit further study,” said lead author Leanne Ward, MD, of the University of Ottawa, Canada, in a statement.
The study was primarily supported by an operating grant from the Canadian Institutes for Health Research (CIHR) with additional funding from a CIHR New Investigator Award, a Canadian Child Health Clinician Scientist Career Enhancement Award, a University of Ottawa Research Chair Award, a Children’s Hospital of Eastern Ontario (CHEO) Research Institute Capacity Building Award, and the CHEO Departments of Pediatrics and Surgery. This work was also supported by the University of Alberta Women and Children’s Health Research Institute.
A 6-year prospective study from the Canadian STOPP investigators revealed that following treatment for acute lymphoblastic leukemia (ALL), approximately 1 out of 3 children experiences vertebral fractures (VF) and 1 out of 5 children shows non-VF.
Glucocorticoid use and vertebral fractures at diagnosis emerged as significant predictive factors.
Investigators reported a cumulative incidence of 32.5% for vertebral fractures (VF) following treatment and 23.0% for non-VF after 6 years.
Thirty-nine percent of children with VF were asymptomatic.
“The osteotoxicity of leukemia and its treatment are underscored by the observation that the non-VF incidence was more than 2.5 times higher than the general pediatric population,” the study authors wrote, “whereas the VF incidence was increased more than 2000 times.”
The STOPP investigators—the Steroid-Associated Osteoporosis in the Pediatric Population Consortium—published their findings in the Journal of Bone and Mineral Research.
To document the incidence and predictors of fractures and profile who is at greatest risk, the STOPP investigators enrolled 186 children who were diagnosed and treated at 10 children’s hospitals across Canada between 2005 and 2007.
Median age at diagnosis was 5.3 years, median duration of follow-up was 6 years, and 38.1% were boys.
Approximately 4 out of 5 children were treated with Children’s Oncology Group protocols and 1 out of 5 was treated with the Dana-Farber protocols.
In girls, glucocorticoid exposure was 7.0 g/m2 and in boys it was 8.9 g/m2.
The entire cohort received 54.3% of the total cumulative glucocorticoid dose in the first year and 83.1% in the second year.
For VF, incidence at baseline was 15.1%. Skeletal fractures at any site were reported in 36% of the children over the 6-year period, 71% occurring during the first 2 years. No VF was reported in the sixth year.
Of 38 children with VF, 32 sustained at least 1 VF in the first 2 years and 77% occurred while on therapy or within 6 months of the last glucocorticoid dose. None of the children had disease relapse.
Non-VF were reported in 1.6% of children at baseline; incidence peaked at years 2 (5.4%) and 5 (4.8%) and none occurred in the sixth year.
Thirty-one non-VFs were reported in 26 children; 18 occurred in the first 2 years and 23 occurred on therapy or within 6 months of the last glucocorticoid dose.
The STOPP investigators showed that glucocorticoid exposure, VF, and low spine bone mineral density Z-scores at baseline were significant predictors for risk of VF.
“[This] observation suggests the importance of baseline spine phenotype (both VF and low lumbar spine bone mineral density) in signaling the potential for future bone morbidity,” the STOPP investigators note.
“In revealing that vertebral fractures are frequent in children with ALL on chemotherapy, and that older children and those with more severe collapse are at risk for residual vertebral deformities, strategies to prevent vertebral fractures in those at greatest risk for permanent sequelae now merit further study,” said lead author Leanne Ward, MD, of the University of Ottawa, Canada, in a statement.
The study was primarily supported by an operating grant from the Canadian Institutes for Health Research (CIHR) with additional funding from a CIHR New Investigator Award, a Canadian Child Health Clinician Scientist Career Enhancement Award, a University of Ottawa Research Chair Award, a Children’s Hospital of Eastern Ontario (CHEO) Research Institute Capacity Building Award, and the CHEO Departments of Pediatrics and Surgery. This work was also supported by the University of Alberta Women and Children’s Health Research Institute.
FDA grants priority review to gilteritinib for R/R AML

The US Food and Drug Administration (FDA) has granted priority review to gilteritinib for the treatment of adult patients with relapsed or refractory (R/R) acute myeloid leukemia (AML) who have a FLT3 mutation.
At present, no FLT3-targeting agents are approved for the treatment of R/R FLT3-mutation-positive AML, according to Astellas, the developer of the drug.
The FDA previously granted gilteritinib orphan drug designation and fast track designation.
The European Commission (EC) also granted gilteritinib orphan designation and the Japan Ministry of Health, Labor and Welfare (MHLW) did likewise.
Gilteritinib has demonstrated inhibitory activity against FLT3 internal tandem duplication (ITD) and FLT tyrosine kinase domain (TKD) mutation. These 2 FLT3 mutations are present in approximately 1/3 of AML patients.
The priority review for the new drug application is based on the ongoing phase 3 ADMIRAL trial.
The FDA aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The agency grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The goal date for a decision by the FDA is the end of November.
This open-label multicenter randomized study compares gilteritinib with salvage chemotherapy in adult patients with FLT3 mutations who are refractory to or have relapsed after first-line AML therapy.
The study has enrolled 371 patients with FLT3 mutations present in bone marrow or whole blood, as determined by a central lab.
Patients were randomized in a 2:1 ratio to receive gilteritinib (120 mg) or salvage chemotherapy.
Salvage chemotherapy could consist of low-dose cytarabine, azacitidine, MEC (mitoxantrone, etoposide, cytarabine) induction chemotherapy, or FLAG-IDA (fludarabine, cytarabine, and granulocyte colony-stimulating factor [G-CSF] with idarubicin) induction chemotherapy.
The primary endpoints of the study are overall survival and complete remission or complete remission with partial hematologic recovery.
Secondary endpoints include event-free survival, complete remission rate, leukemia-free survival, duration of remission, transplantation rate, fatigue inventory, among other outcomes.
The study is estimated to be completed in October for its primary endpoint and February 2020 for the entire study.
The US Food and Drug Administration (FDA) has granted priority review to gilteritinib for the treatment of adult patients with relapsed or refractory (R/R) acute myeloid leukemia (AML) who have a FLT3 mutation.
At present, no FLT3-targeting agents are approved for the treatment of R/R FLT3-mutation-positive AML, according to Astellas, the developer of the drug.
The FDA previously granted gilteritinib orphan drug designation and fast track designation.
The European Commission (EC) also granted gilteritinib orphan designation and the Japan Ministry of Health, Labor and Welfare (MHLW) did likewise.
Gilteritinib has demonstrated inhibitory activity against FLT3 internal tandem duplication (ITD) and FLT tyrosine kinase domain (TKD) mutation. These 2 FLT3 mutations are present in approximately 1/3 of AML patients.
The priority review for the new drug application is based on the ongoing phase 3 ADMIRAL trial.
The FDA aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The agency grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The goal date for a decision by the FDA is the end of November.
This open-label multicenter randomized study compares gilteritinib with salvage chemotherapy in adult patients with FLT3 mutations who are refractory to or have relapsed after first-line AML therapy.
The study has enrolled 371 patients with FLT3 mutations present in bone marrow or whole blood, as determined by a central lab.
Patients were randomized in a 2:1 ratio to receive gilteritinib (120 mg) or salvage chemotherapy.
Salvage chemotherapy could consist of low-dose cytarabine, azacitidine, MEC (mitoxantrone, etoposide, cytarabine) induction chemotherapy, or FLAG-IDA (fludarabine, cytarabine, and granulocyte colony-stimulating factor [G-CSF] with idarubicin) induction chemotherapy.
The primary endpoints of the study are overall survival and complete remission or complete remission with partial hematologic recovery.
Secondary endpoints include event-free survival, complete remission rate, leukemia-free survival, duration of remission, transplantation rate, fatigue inventory, among other outcomes.
The study is estimated to be completed in October for its primary endpoint and February 2020 for the entire study.
The US Food and Drug Administration (FDA) has granted priority review to gilteritinib for the treatment of adult patients with relapsed or refractory (R/R) acute myeloid leukemia (AML) who have a FLT3 mutation.
At present, no FLT3-targeting agents are approved for the treatment of R/R FLT3-mutation-positive AML, according to Astellas, the developer of the drug.
The FDA previously granted gilteritinib orphan drug designation and fast track designation.
The European Commission (EC) also granted gilteritinib orphan designation and the Japan Ministry of Health, Labor and Welfare (MHLW) did likewise.
Gilteritinib has demonstrated inhibitory activity against FLT3 internal tandem duplication (ITD) and FLT tyrosine kinase domain (TKD) mutation. These 2 FLT3 mutations are present in approximately 1/3 of AML patients.
The priority review for the new drug application is based on the ongoing phase 3 ADMIRAL trial.
The FDA aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The agency grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The goal date for a decision by the FDA is the end of November.
This open-label multicenter randomized study compares gilteritinib with salvage chemotherapy in adult patients with FLT3 mutations who are refractory to or have relapsed after first-line AML therapy.
The study has enrolled 371 patients with FLT3 mutations present in bone marrow or whole blood, as determined by a central lab.
Patients were randomized in a 2:1 ratio to receive gilteritinib (120 mg) or salvage chemotherapy.
Salvage chemotherapy could consist of low-dose cytarabine, azacitidine, MEC (mitoxantrone, etoposide, cytarabine) induction chemotherapy, or FLAG-IDA (fludarabine, cytarabine, and granulocyte colony-stimulating factor [G-CSF] with idarubicin) induction chemotherapy.
The primary endpoints of the study are overall survival and complete remission or complete remission with partial hematologic recovery.
Secondary endpoints include event-free survival, complete remission rate, leukemia-free survival, duration of remission, transplantation rate, fatigue inventory, among other outcomes.
The study is estimated to be completed in October for its primary endpoint and February 2020 for the entire study.