User login
Resistant hypertension hits SLE patients hard
at a tertiary care center.

A patient with resistant hypertension either has blood pressure remaining above 140/90 mm Hg while taking three antihypertensive medications or requires the use of four or more antihypertensives to attain blood pressure control. Resistant hypertension, which was more likely to occur among blacks and patients with lower renal function, hypercholesterolemia, and increased inflammatory markers, increased the risk of death nearly threefold (hazard ratio, 2.91; P = .0005) when compared with those who didn’t have this condition.
The results of this analysis were published March 15 in Arthritis Care & Research (doi: 10.1002/acr.23880). We covered this study at the 2018 annual meeting of the American College of Rheumatology in Chicago before it was published in the journal. Read our previous story at the link above.
at a tertiary care center.
A patient with resistant hypertension either has blood pressure remaining above 140/90 mm Hg while taking three antihypertensive medications or requires the use of four or more antihypertensives to attain blood pressure control. Resistant hypertension, which was more likely to occur among blacks and patients with lower renal function, hypercholesterolemia, and increased inflammatory markers, increased the risk of death nearly threefold (hazard ratio, 2.91; P = .0005) when compared with those who didn’t have this condition.
The results of this analysis were published March 15 in Arthritis Care & Research (doi: 10.1002/acr.23880). We covered this study at the 2018 annual meeting of the American College of Rheumatology in Chicago before it was published in the journal. Read our previous story at the link above.
at a tertiary care center.
A patient with resistant hypertension either has blood pressure remaining above 140/90 mm Hg while taking three antihypertensive medications or requires the use of four or more antihypertensives to attain blood pressure control. Resistant hypertension, which was more likely to occur among blacks and patients with lower renal function, hypercholesterolemia, and increased inflammatory markers, increased the risk of death nearly threefold (hazard ratio, 2.91; P = .0005) when compared with those who didn’t have this condition.
The results of this analysis were published March 15 in Arthritis Care & Research (doi: 10.1002/acr.23880). We covered this study at the 2018 annual meeting of the American College of Rheumatology in Chicago before it was published in the journal. Read our previous story at the link above.
FROM ARTHRITIS CARE & RESEARCH
BTK inhibitor calms pemphigus vulgaris with low-dose steroids
WASHINGTON – An investigational molecule that blocks the downstream proinflammatory effects of B cells controlled disease activity and induced clinical remission in patients with pemphigus by 12 weeks.
At the end of a 24-week, open-label trial, a key driver of the sometimes-fatal blistering disease, Deedee Murrell, MD, said at the annual meeting of the American Academy of Dermatology.
The clinical efficacy plus a favorable safety profile supports the further development of the molecule, designed and manufactured by Principia Biopharma in San Francisco. The company is currently recruiting for a pivotal phase 3 trial of PRN1008 in 120 patients with moderate to severe pemphigus vulgaris.
Despite the recent approval of rituximab (Rituxan) for moderate to severe pemphigus, there remains an unmet need for a quick-acting, steroid-sparing, anti-inflammatory treatment, said Dr. Murrell, professor and head of the department of dermatology at the University of New South Wales, Sydney.
“We need something to use instead of high-dose steroids while we are waiting for rituximab to kick in, which can take 3 months,” and rituximab, which depletes B cells, puts patients at risk for infection, she said. “We need something that has rapid onset, is steroid sparing, safe for chronic administration, avoids B-cell depletion, and is convenient.”
Blocking the BTK receptor on B cells puts the brakes on the B-cell mediated inflammatory pathway, preventing activation of monocytes, macrophages, mast cells, basophils, and neutrophils. At the same time, however, it does not deplete the B-cell population, said Dr. Murrell, the lead investigator.
The BELIEVE study comprised 27 patients with mild to severe pemphigus of an average 6 years’ duration. Most (18) had relapsing disease; the remainder had newly diagnosed pemphigus. A majority (16) had severe disease, as measured by a score of 15 or more on the Pemphigus Disease Activity Index (PDAI). Almost all (23) were positive for antidesmoglein antibodies. Only one patient was negative for antibodies.
The mean corticosteroid dose at baseline was 14 mg/day, although that ranged from no steroids to 30 mg/day.
The study consisted of a 12-week treatment phase and a 12-week follow-up phase. During treatment, patients could take no more than 0.5 mg/kg of prednisone daily, although with 400 mg PRN1008 twice a day. They were allowed to undertake rescue immunosuppression if they experienced a disease flare.
The primary endpoint was disease control by day 29 as evidenced by no new lesions. Secondary endpoints were complete remission, minimization of prednisone, quality of life, antibody levels, and clinician measures including the PDAI and the Autoimmune Bullous Skin Disorder Intensity Score.
By the end of week 4, 54% of patients had achieved the primary endpoint. The benefit continued to expand, with 73% reaching that response by the end of week 12. During this period, the mean prednisone dose was 12 mg/day.
Among the 24 patients who completed the study, complete remission occurred in 17% by week 12. However, patients continued to respond through the follow-up period, even after the study medication was stopped. By week 24, 25% of these patients experienced a complete remission. At the point of remission, the mean steroid dose was 8 mg/day. The median duration of remission was 2 months after stopping PRN1008.
The PDAI fell by a median of 70% by week 12 and was maintained at that level by the end of week 24. The median level of antidesmoglein autoantibodies fell by up to 65%. Again, the improvement continued throughout the off-drug follow-up period. In subgroup analyses, PRN1008 was more effective in patients with moderate to severe disease than those with mild disease (80% response vs. 64%). It was equally effective in those with newly diagnosed disease (75% vs. 72%) and regardless of antibody level at baseline.
The adverse event profile was relatively benign. Most side effects were mild and transient, and included upper abdominal pain, headache, and nausea. There were two mild infections and one serious infection, which presented in a patient with a long-standing localized cellulitis that activated and was associated a high fever. It was culture negative and PRN1008 was restarted without issue.
There was also one serious adverse event and one death, both unrelated to the study drug. One patient developed a pancreatic cyst that was discovered on day 29. The patient dropped out of the study to have elective surgery. The death occurred in a patient who developed acute respiratory failure on day 8 of treatment, caused by an undiagnosed congenital pulmonary sequestration. The patient died of a brain embolism shortly after lung surgery.
Dr. Murrell designed the study and was an investigator. She reported a financial relationship with Principia, as well as with numerous other pharmaceutical companies.
SOURCE: Murrell D et al. AAD 2019, Session S034.
WASHINGTON – An investigational molecule that blocks the downstream proinflammatory effects of B cells controlled disease activity and induced clinical remission in patients with pemphigus by 12 weeks.
At the end of a 24-week, open-label trial, a key driver of the sometimes-fatal blistering disease, Deedee Murrell, MD, said at the annual meeting of the American Academy of Dermatology.
The clinical efficacy plus a favorable safety profile supports the further development of the molecule, designed and manufactured by Principia Biopharma in San Francisco. The company is currently recruiting for a pivotal phase 3 trial of PRN1008 in 120 patients with moderate to severe pemphigus vulgaris.
Despite the recent approval of rituximab (Rituxan) for moderate to severe pemphigus, there remains an unmet need for a quick-acting, steroid-sparing, anti-inflammatory treatment, said Dr. Murrell, professor and head of the department of dermatology at the University of New South Wales, Sydney.
“We need something to use instead of high-dose steroids while we are waiting for rituximab to kick in, which can take 3 months,” and rituximab, which depletes B cells, puts patients at risk for infection, she said. “We need something that has rapid onset, is steroid sparing, safe for chronic administration, avoids B-cell depletion, and is convenient.”
Blocking the BTK receptor on B cells puts the brakes on the B-cell mediated inflammatory pathway, preventing activation of monocytes, macrophages, mast cells, basophils, and neutrophils. At the same time, however, it does not deplete the B-cell population, said Dr. Murrell, the lead investigator.
The BELIEVE study comprised 27 patients with mild to severe pemphigus of an average 6 years’ duration. Most (18) had relapsing disease; the remainder had newly diagnosed pemphigus. A majority (16) had severe disease, as measured by a score of 15 or more on the Pemphigus Disease Activity Index (PDAI). Almost all (23) were positive for antidesmoglein antibodies. Only one patient was negative for antibodies.
The mean corticosteroid dose at baseline was 14 mg/day, although that ranged from no steroids to 30 mg/day.
The study consisted of a 12-week treatment phase and a 12-week follow-up phase. During treatment, patients could take no more than 0.5 mg/kg of prednisone daily, although with 400 mg PRN1008 twice a day. They were allowed to undertake rescue immunosuppression if they experienced a disease flare.
The primary endpoint was disease control by day 29 as evidenced by no new lesions. Secondary endpoints were complete remission, minimization of prednisone, quality of life, antibody levels, and clinician measures including the PDAI and the Autoimmune Bullous Skin Disorder Intensity Score.
By the end of week 4, 54% of patients had achieved the primary endpoint. The benefit continued to expand, with 73% reaching that response by the end of week 12. During this period, the mean prednisone dose was 12 mg/day.
Among the 24 patients who completed the study, complete remission occurred in 17% by week 12. However, patients continued to respond through the follow-up period, even after the study medication was stopped. By week 24, 25% of these patients experienced a complete remission. At the point of remission, the mean steroid dose was 8 mg/day. The median duration of remission was 2 months after stopping PRN1008.
The PDAI fell by a median of 70% by week 12 and was maintained at that level by the end of week 24. The median level of antidesmoglein autoantibodies fell by up to 65%. Again, the improvement continued throughout the off-drug follow-up period. In subgroup analyses, PRN1008 was more effective in patients with moderate to severe disease than those with mild disease (80% response vs. 64%). It was equally effective in those with newly diagnosed disease (75% vs. 72%) and regardless of antibody level at baseline.
The adverse event profile was relatively benign. Most side effects were mild and transient, and included upper abdominal pain, headache, and nausea. There were two mild infections and one serious infection, which presented in a patient with a long-standing localized cellulitis that activated and was associated a high fever. It was culture negative and PRN1008 was restarted without issue.
There was also one serious adverse event and one death, both unrelated to the study drug. One patient developed a pancreatic cyst that was discovered on day 29. The patient dropped out of the study to have elective surgery. The death occurred in a patient who developed acute respiratory failure on day 8 of treatment, caused by an undiagnosed congenital pulmonary sequestration. The patient died of a brain embolism shortly after lung surgery.
Dr. Murrell designed the study and was an investigator. She reported a financial relationship with Principia, as well as with numerous other pharmaceutical companies.
SOURCE: Murrell D et al. AAD 2019, Session S034.
WASHINGTON – An investigational molecule that blocks the downstream proinflammatory effects of B cells controlled disease activity and induced clinical remission in patients with pemphigus by 12 weeks.
At the end of a 24-week, open-label trial, a key driver of the sometimes-fatal blistering disease, Deedee Murrell, MD, said at the annual meeting of the American Academy of Dermatology.
The clinical efficacy plus a favorable safety profile supports the further development of the molecule, designed and manufactured by Principia Biopharma in San Francisco. The company is currently recruiting for a pivotal phase 3 trial of PRN1008 in 120 patients with moderate to severe pemphigus vulgaris.
Despite the recent approval of rituximab (Rituxan) for moderate to severe pemphigus, there remains an unmet need for a quick-acting, steroid-sparing, anti-inflammatory treatment, said Dr. Murrell, professor and head of the department of dermatology at the University of New South Wales, Sydney.
“We need something to use instead of high-dose steroids while we are waiting for rituximab to kick in, which can take 3 months,” and rituximab, which depletes B cells, puts patients at risk for infection, she said. “We need something that has rapid onset, is steroid sparing, safe for chronic administration, avoids B-cell depletion, and is convenient.”
Blocking the BTK receptor on B cells puts the brakes on the B-cell mediated inflammatory pathway, preventing activation of monocytes, macrophages, mast cells, basophils, and neutrophils. At the same time, however, it does not deplete the B-cell population, said Dr. Murrell, the lead investigator.
The BELIEVE study comprised 27 patients with mild to severe pemphigus of an average 6 years’ duration. Most (18) had relapsing disease; the remainder had newly diagnosed pemphigus. A majority (16) had severe disease, as measured by a score of 15 or more on the Pemphigus Disease Activity Index (PDAI). Almost all (23) were positive for antidesmoglein antibodies. Only one patient was negative for antibodies.
The mean corticosteroid dose at baseline was 14 mg/day, although that ranged from no steroids to 30 mg/day.
The study consisted of a 12-week treatment phase and a 12-week follow-up phase. During treatment, patients could take no more than 0.5 mg/kg of prednisone daily, although with 400 mg PRN1008 twice a day. They were allowed to undertake rescue immunosuppression if they experienced a disease flare.
The primary endpoint was disease control by day 29 as evidenced by no new lesions. Secondary endpoints were complete remission, minimization of prednisone, quality of life, antibody levels, and clinician measures including the PDAI and the Autoimmune Bullous Skin Disorder Intensity Score.
By the end of week 4, 54% of patients had achieved the primary endpoint. The benefit continued to expand, with 73% reaching that response by the end of week 12. During this period, the mean prednisone dose was 12 mg/day.
Among the 24 patients who completed the study, complete remission occurred in 17% by week 12. However, patients continued to respond through the follow-up period, even after the study medication was stopped. By week 24, 25% of these patients experienced a complete remission. At the point of remission, the mean steroid dose was 8 mg/day. The median duration of remission was 2 months after stopping PRN1008.
The PDAI fell by a median of 70% by week 12 and was maintained at that level by the end of week 24. The median level of antidesmoglein autoantibodies fell by up to 65%. Again, the improvement continued throughout the off-drug follow-up period. In subgroup analyses, PRN1008 was more effective in patients with moderate to severe disease than those with mild disease (80% response vs. 64%). It was equally effective in those with newly diagnosed disease (75% vs. 72%) and regardless of antibody level at baseline.
The adverse event profile was relatively benign. Most side effects were mild and transient, and included upper abdominal pain, headache, and nausea. There were two mild infections and one serious infection, which presented in a patient with a long-standing localized cellulitis that activated and was associated a high fever. It was culture negative and PRN1008 was restarted without issue.
There was also one serious adverse event and one death, both unrelated to the study drug. One patient developed a pancreatic cyst that was discovered on day 29. The patient dropped out of the study to have elective surgery. The death occurred in a patient who developed acute respiratory failure on day 8 of treatment, caused by an undiagnosed congenital pulmonary sequestration. The patient died of a brain embolism shortly after lung surgery.
Dr. Murrell designed the study and was an investigator. She reported a financial relationship with Principia, as well as with numerous other pharmaceutical companies.
SOURCE: Murrell D et al. AAD 2019, Session S034.
REPORTING FROM AAD 2019
Vagus nerve stimulation for rheumatology? Maybe
The work is being led by SetPoint Medical, a small company in Valencia, Calif., just north of Los Angeles. Its vagus nerve stimulation (VNS) device, dubbed the microregulator, has been implanted in 14 patients with refractory rheumatoid arthritis (RA) in the company’s initial safety study.

The microregulator is a small lithium ion battery encased in an inert silastic pod; it’s surgically implanted to sit atop the vagus nerve in the left side of the neck, and delivers an electrical pulse at set intervals. Data from the 12-week, sham-controlled safety study is set to be unblinded in coming weeks. A pivotal trial also is in the works, perhaps to start in late 2019, according to rheumatologist and SetPoint’s Chief Medical Officer David Chernoff, MD.
Although SetPoint is ahead of the pack, it’s not alone. ElectroCore, a biotech company in Basking Ridge, N.J., has expressed interest in pursuing rheumatoid arthritis and Sjögren’s syndrome indications for its gammaCore device, a vagus nerve stimulator patients apply to the neck. It’s already on the market for migraines and cluster headaches.
Researchers recently reported a small decrease in 28-joint Disease Activity Score using C-reactive protein (DAS28-CRP) results after 16 RA patients with flares used the device for 4 days (Ann Rheum Dis. 2018;77:1401. Abstract AB0481). In another recent open-label study, 15 women with Sjögren’s reported less fatigue while using the device for a month (Arthritis Rheumatol. 2017;69[suppl 10]: Abstract 563).
Meanwhile, The Feinstein Institute for Medical Research, based in Manhasset, N.Y., on Long Island, recently reported positive outcomes in 18 patients with systemic lupus erythematosus, using its own novel device, which stimulates the vagus nerve through the ear lobe. VNS was delivered for 5 minutes per day for 4 days (Arthritis Rheumatol. 2018;70[suppl 10]: Abstract 2652).
On day 5, patients who received VNS, versus sham patients in whom the device was not turned on, had a significant decrease in pain, fatigue, and joint scores. The investigators concluded that “additional studies evaluating this promising intervention and its potential mechanisms are warranted.”
“We are clearly ahead of everybody because we’ve already implanted people, but I think it’s good for the field if more people are chasing this. The more resources that are put into it, the more we can show that this approach actually works,” said SetPoint’s Dr. Chernoff.
The hope
In general, interest in VNS for rheumatology is being driven by the possibility that it may reduce proinflammatory cytokines, which opens the door for VNS as an alternative to biologics. The hope is that instead of going after tumor necrosis factor and other cytokines one at a time, VNS could be used to target a range of cytokines all at once, without the cost and side effects of biologics.
“It seems so dramatically different” from what rheumatologists have done in the past, “that our first instinct is to say ‘oh, that’s ridiculous,’ but the science behind it is actually not bad. There may indeed be something to this,” said rheumatologist Joel Kremer, MD, Pfaff Family Professor of Medicine at Albany (N.Y.) Medical College.
Dr. Kremer reviewed SetPoint’s early scientific data after being asked by the company to participate in the safety study; he declined for logistical reasons.
He noted that “there are some strange interactions between the CNS and inflammatory disease.” When RA patients have a stroke, for instance, RA goes into remission on the side of their body affected by the stroke. “That’s been known for decades, but we really don’t understand what’s going on there,” Dr. Kremer said.
The evidence
Perhaps the strongest evidence to date for VNS as a cytokine blocker in rheumatology comes from an open-label, 12-week study, also conducted by SetPoint, in 17 patients with active RA despite methotrexate treatment; some had failed biologics (Proc Natl Acad Sci U S A. 2016 Jul 19;113[29]:8284-9. doi: 10.1073/pnas.160563511).
The microregulator wasn’t ready yet, so investigators implanted a VNS system commercially available for epilepsy and reprogrammed it to deliver a 60-second pulse once a day to the left cervical vagus nerve, which was increased after a month to four 60-second stimulations a day in nonresponders.
The investigators “observed that TNF production in cultured peripheral blood obtained ... on day 42 was significantly reduced from” 21 days before the study was started (TNF 2,900 pg/mL on day –21, versus 1,776 pg/mL on day 42; P less than .05).
When VNS was shut off, TNF production increased; when it was turned back on, it dropped. Interleukin 6 also fell significantly among responders. Overall, DAS28-CRP scores fell about 1.5 points on the 10-point scale from baseline to week 12.
Two-year outcomes were recently reported (Ann Rheum Dis. 2018;77:981-2. Abstract SAT0240). All 17 patients elected to continue treatment after the initial 12 weeks. Biologics were added in nine subjects (53%), because of no or limited response to VNS. Investigators were free to change the VNS dosing regimen, which varied during the study extension up to eight 60-second bursts a day. The roughly 1.5-point improvement in DAS28-CRP was maintained at 2 years.
“These long-term data suggest that bioelectronic therapy may be used as an alternative to, or in combination with, biological[s],” concluded Dr. Chernoff and other study team members.
Awaiting more data
When asked for comment, Daniel E. Furst, MD, professor of medicine (emeritus) at the University of California, Los Angeles, said “there certainly are neurotropic factors” at play in rheumatology, “so there’s sort of a potential reason why” VNS might work, “but we need to understand far more about its mechanism, and [remember] that open-label studies are not to be believed until” large, randomized, blinded, placebo-controlled studies are done.

Dr. Furst also is an adjunct professor at the University of Washington, Seattle, and a research professor at the University of Florence (Italy). He is in part-time practice in Los Angeles and Seattle.
If everything works out, however, “the vagus nerve may give us a much wider opportunity to block a host of cytokines; it may change the whole paradigm of how we manage rheumatoid arthritis. I think this is possibly a groundbreaking new therapeutic area, much in the way the biologics were” 20 years ago, said rheumatologist Norman B. Gaylis, MD.
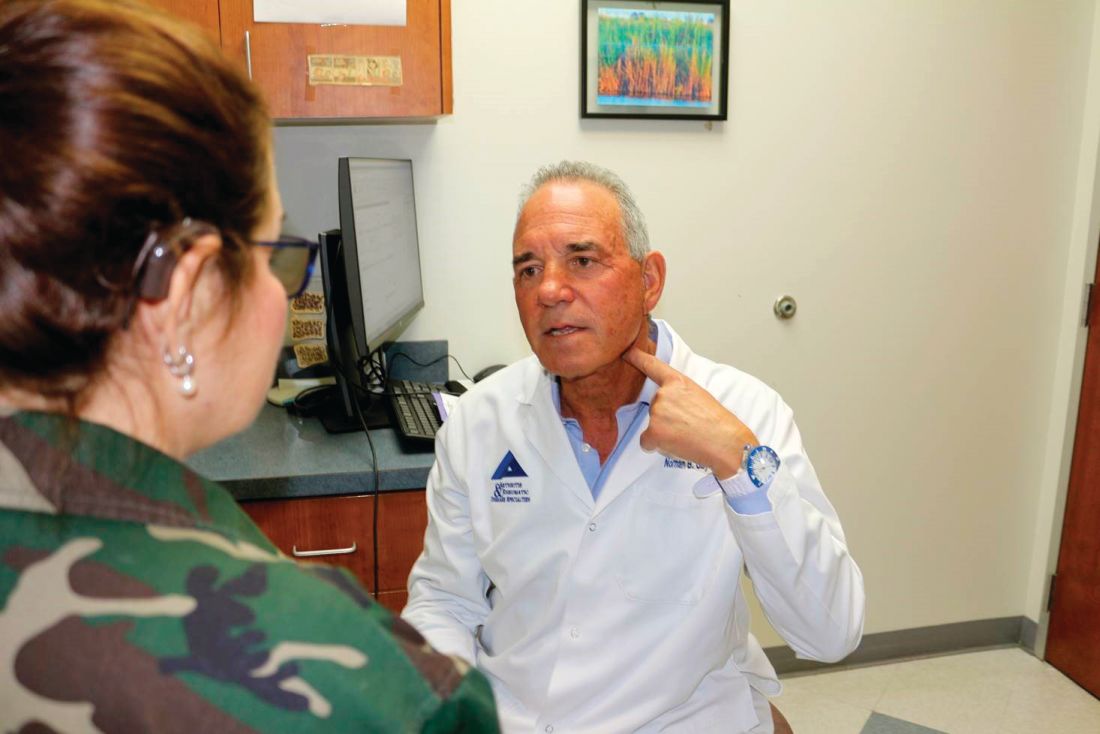
Several of the 14 patients in SetPoint’s safety study were enrolled at Dr. Gaylis’s practice in Aventura, Fla., just north of Miami; he said he is eagerly awaiting for the results to be unblinded. If clinical response in that study and others correlates with a cytokine response, “that’s going to be big, and very significant” in the rheumatology community, he said.
SetPoint’s microregulator is charged wirelessly through a collar patients wear for a few minutes once a week. Dosing can also be adjusted through the collar with the help of a computer application.
The device wasn’t turned on in 4 of the 14 patients in the safety study, as a sham control, but shamming was problematic because patients can potentially feel VNS as a buzz or a change in their voice. To get around that potential confounder, both sham and treated patients were told they might or might not feel something during the study.
Implantation takes about an hour, and is much less complex than implanting currently available epilepsy VNS systems, which require implantation of both a power source on the chest wall and wire coils on the vagus nerve.
Cardiac concerns are the main safety issue with VNS, beyond the surgery itself. Cardiac monitoring was done in the safety study to “ensure that we did not cause things like bradycardia, heart block, syncope, etc.” Dr. Chernoff said. So far, they haven’t turned out to be a problem.
Dr. Furst and Dr. Kremer had no relevant disclosures. Dr. Gaylis was compensated by SetPoint for participating in the safety study; he is a consultant and investigator for Electrocore. Dr. Furst and Dr. Gaylis are members of the editorial advisory board for MDedge Rheumatology/Rheumatology News.
The work is being led by SetPoint Medical, a small company in Valencia, Calif., just north of Los Angeles. Its vagus nerve stimulation (VNS) device, dubbed the microregulator, has been implanted in 14 patients with refractory rheumatoid arthritis (RA) in the company’s initial safety study.
The microregulator is a small lithium ion battery encased in an inert silastic pod; it’s surgically implanted to sit atop the vagus nerve in the left side of the neck, and delivers an electrical pulse at set intervals. Data from the 12-week, sham-controlled safety study is set to be unblinded in coming weeks. A pivotal trial also is in the works, perhaps to start in late 2019, according to rheumatologist and SetPoint’s Chief Medical Officer David Chernoff, MD.
Although SetPoint is ahead of the pack, it’s not alone. ElectroCore, a biotech company in Basking Ridge, N.J., has expressed interest in pursuing rheumatoid arthritis and Sjögren’s syndrome indications for its gammaCore device, a vagus nerve stimulator patients apply to the neck. It’s already on the market for migraines and cluster headaches.
Researchers recently reported a small decrease in 28-joint Disease Activity Score using C-reactive protein (DAS28-CRP) results after 16 RA patients with flares used the device for 4 days (Ann Rheum Dis. 2018;77:1401. Abstract AB0481). In another recent open-label study, 15 women with Sjögren’s reported less fatigue while using the device for a month (Arthritis Rheumatol. 2017;69[suppl 10]: Abstract 563).
Meanwhile, The Feinstein Institute for Medical Research, based in Manhasset, N.Y., on Long Island, recently reported positive outcomes in 18 patients with systemic lupus erythematosus, using its own novel device, which stimulates the vagus nerve through the ear lobe. VNS was delivered for 5 minutes per day for 4 days (Arthritis Rheumatol. 2018;70[suppl 10]: Abstract 2652).
On day 5, patients who received VNS, versus sham patients in whom the device was not turned on, had a significant decrease in pain, fatigue, and joint scores. The investigators concluded that “additional studies evaluating this promising intervention and its potential mechanisms are warranted.”
“We are clearly ahead of everybody because we’ve already implanted people, but I think it’s good for the field if more people are chasing this. The more resources that are put into it, the more we can show that this approach actually works,” said SetPoint’s Dr. Chernoff.
The hope
In general, interest in VNS for rheumatology is being driven by the possibility that it may reduce proinflammatory cytokines, which opens the door for VNS as an alternative to biologics. The hope is that instead of going after tumor necrosis factor and other cytokines one at a time, VNS could be used to target a range of cytokines all at once, without the cost and side effects of biologics.
“It seems so dramatically different” from what rheumatologists have done in the past, “that our first instinct is to say ‘oh, that’s ridiculous,’ but the science behind it is actually not bad. There may indeed be something to this,” said rheumatologist Joel Kremer, MD, Pfaff Family Professor of Medicine at Albany (N.Y.) Medical College.
Dr. Kremer reviewed SetPoint’s early scientific data after being asked by the company to participate in the safety study; he declined for logistical reasons.
He noted that “there are some strange interactions between the CNS and inflammatory disease.” When RA patients have a stroke, for instance, RA goes into remission on the side of their body affected by the stroke. “That’s been known for decades, but we really don’t understand what’s going on there,” Dr. Kremer said.
The evidence
Perhaps the strongest evidence to date for VNS as a cytokine blocker in rheumatology comes from an open-label, 12-week study, also conducted by SetPoint, in 17 patients with active RA despite methotrexate treatment; some had failed biologics (Proc Natl Acad Sci U S A. 2016 Jul 19;113[29]:8284-9. doi: 10.1073/pnas.160563511).
The microregulator wasn’t ready yet, so investigators implanted a VNS system commercially available for epilepsy and reprogrammed it to deliver a 60-second pulse once a day to the left cervical vagus nerve, which was increased after a month to four 60-second stimulations a day in nonresponders.
The investigators “observed that TNF production in cultured peripheral blood obtained ... on day 42 was significantly reduced from” 21 days before the study was started (TNF 2,900 pg/mL on day –21, versus 1,776 pg/mL on day 42; P less than .05).
When VNS was shut off, TNF production increased; when it was turned back on, it dropped. Interleukin 6 also fell significantly among responders. Overall, DAS28-CRP scores fell about 1.5 points on the 10-point scale from baseline to week 12.
Two-year outcomes were recently reported (Ann Rheum Dis. 2018;77:981-2. Abstract SAT0240). All 17 patients elected to continue treatment after the initial 12 weeks. Biologics were added in nine subjects (53%), because of no or limited response to VNS. Investigators were free to change the VNS dosing regimen, which varied during the study extension up to eight 60-second bursts a day. The roughly 1.5-point improvement in DAS28-CRP was maintained at 2 years.
“These long-term data suggest that bioelectronic therapy may be used as an alternative to, or in combination with, biological[s],” concluded Dr. Chernoff and other study team members.
Awaiting more data
When asked for comment, Daniel E. Furst, MD, professor of medicine (emeritus) at the University of California, Los Angeles, said “there certainly are neurotropic factors” at play in rheumatology, “so there’s sort of a potential reason why” VNS might work, “but we need to understand far more about its mechanism, and [remember] that open-label studies are not to be believed until” large, randomized, blinded, placebo-controlled studies are done.
Dr. Furst also is an adjunct professor at the University of Washington, Seattle, and a research professor at the University of Florence (Italy). He is in part-time practice in Los Angeles and Seattle.
If everything works out, however, “the vagus nerve may give us a much wider opportunity to block a host of cytokines; it may change the whole paradigm of how we manage rheumatoid arthritis. I think this is possibly a groundbreaking new therapeutic area, much in the way the biologics were” 20 years ago, said rheumatologist Norman B. Gaylis, MD.
Several of the 14 patients in SetPoint’s safety study were enrolled at Dr. Gaylis’s practice in Aventura, Fla., just north of Miami; he said he is eagerly awaiting for the results to be unblinded. If clinical response in that study and others correlates with a cytokine response, “that’s going to be big, and very significant” in the rheumatology community, he said.
SetPoint’s microregulator is charged wirelessly through a collar patients wear for a few minutes once a week. Dosing can also be adjusted through the collar with the help of a computer application.
The device wasn’t turned on in 4 of the 14 patients in the safety study, as a sham control, but shamming was problematic because patients can potentially feel VNS as a buzz or a change in their voice. To get around that potential confounder, both sham and treated patients were told they might or might not feel something during the study.
Implantation takes about an hour, and is much less complex than implanting currently available epilepsy VNS systems, which require implantation of both a power source on the chest wall and wire coils on the vagus nerve.
Cardiac concerns are the main safety issue with VNS, beyond the surgery itself. Cardiac monitoring was done in the safety study to “ensure that we did not cause things like bradycardia, heart block, syncope, etc.” Dr. Chernoff said. So far, they haven’t turned out to be a problem.
Dr. Furst and Dr. Kremer had no relevant disclosures. Dr. Gaylis was compensated by SetPoint for participating in the safety study; he is a consultant and investigator for Electrocore. Dr. Furst and Dr. Gaylis are members of the editorial advisory board for MDedge Rheumatology/Rheumatology News.
The work is being led by SetPoint Medical, a small company in Valencia, Calif., just north of Los Angeles. Its vagus nerve stimulation (VNS) device, dubbed the microregulator, has been implanted in 14 patients with refractory rheumatoid arthritis (RA) in the company’s initial safety study.
The microregulator is a small lithium ion battery encased in an inert silastic pod; it’s surgically implanted to sit atop the vagus nerve in the left side of the neck, and delivers an electrical pulse at set intervals. Data from the 12-week, sham-controlled safety study is set to be unblinded in coming weeks. A pivotal trial also is in the works, perhaps to start in late 2019, according to rheumatologist and SetPoint’s Chief Medical Officer David Chernoff, MD.
Although SetPoint is ahead of the pack, it’s not alone. ElectroCore, a biotech company in Basking Ridge, N.J., has expressed interest in pursuing rheumatoid arthritis and Sjögren’s syndrome indications for its gammaCore device, a vagus nerve stimulator patients apply to the neck. It’s already on the market for migraines and cluster headaches.
Researchers recently reported a small decrease in 28-joint Disease Activity Score using C-reactive protein (DAS28-CRP) results after 16 RA patients with flares used the device for 4 days (Ann Rheum Dis. 2018;77:1401. Abstract AB0481). In another recent open-label study, 15 women with Sjögren’s reported less fatigue while using the device for a month (Arthritis Rheumatol. 2017;69[suppl 10]: Abstract 563).
Meanwhile, The Feinstein Institute for Medical Research, based in Manhasset, N.Y., on Long Island, recently reported positive outcomes in 18 patients with systemic lupus erythematosus, using its own novel device, which stimulates the vagus nerve through the ear lobe. VNS was delivered for 5 minutes per day for 4 days (Arthritis Rheumatol. 2018;70[suppl 10]: Abstract 2652).
On day 5, patients who received VNS, versus sham patients in whom the device was not turned on, had a significant decrease in pain, fatigue, and joint scores. The investigators concluded that “additional studies evaluating this promising intervention and its potential mechanisms are warranted.”
“We are clearly ahead of everybody because we’ve already implanted people, but I think it’s good for the field if more people are chasing this. The more resources that are put into it, the more we can show that this approach actually works,” said SetPoint’s Dr. Chernoff.
The hope
In general, interest in VNS for rheumatology is being driven by the possibility that it may reduce proinflammatory cytokines, which opens the door for VNS as an alternative to biologics. The hope is that instead of going after tumor necrosis factor and other cytokines one at a time, VNS could be used to target a range of cytokines all at once, without the cost and side effects of biologics.
“It seems so dramatically different” from what rheumatologists have done in the past, “that our first instinct is to say ‘oh, that’s ridiculous,’ but the science behind it is actually not bad. There may indeed be something to this,” said rheumatologist Joel Kremer, MD, Pfaff Family Professor of Medicine at Albany (N.Y.) Medical College.
Dr. Kremer reviewed SetPoint’s early scientific data after being asked by the company to participate in the safety study; he declined for logistical reasons.
He noted that “there are some strange interactions between the CNS and inflammatory disease.” When RA patients have a stroke, for instance, RA goes into remission on the side of their body affected by the stroke. “That’s been known for decades, but we really don’t understand what’s going on there,” Dr. Kremer said.
The evidence
Perhaps the strongest evidence to date for VNS as a cytokine blocker in rheumatology comes from an open-label, 12-week study, also conducted by SetPoint, in 17 patients with active RA despite methotrexate treatment; some had failed biologics (Proc Natl Acad Sci U S A. 2016 Jul 19;113[29]:8284-9. doi: 10.1073/pnas.160563511).
The microregulator wasn’t ready yet, so investigators implanted a VNS system commercially available for epilepsy and reprogrammed it to deliver a 60-second pulse once a day to the left cervical vagus nerve, which was increased after a month to four 60-second stimulations a day in nonresponders.
The investigators “observed that TNF production in cultured peripheral blood obtained ... on day 42 was significantly reduced from” 21 days before the study was started (TNF 2,900 pg/mL on day –21, versus 1,776 pg/mL on day 42; P less than .05).
When VNS was shut off, TNF production increased; when it was turned back on, it dropped. Interleukin 6 also fell significantly among responders. Overall, DAS28-CRP scores fell about 1.5 points on the 10-point scale from baseline to week 12.
Two-year outcomes were recently reported (Ann Rheum Dis. 2018;77:981-2. Abstract SAT0240). All 17 patients elected to continue treatment after the initial 12 weeks. Biologics were added in nine subjects (53%), because of no or limited response to VNS. Investigators were free to change the VNS dosing regimen, which varied during the study extension up to eight 60-second bursts a day. The roughly 1.5-point improvement in DAS28-CRP was maintained at 2 years.
“These long-term data suggest that bioelectronic therapy may be used as an alternative to, or in combination with, biological[s],” concluded Dr. Chernoff and other study team members.
Awaiting more data
When asked for comment, Daniel E. Furst, MD, professor of medicine (emeritus) at the University of California, Los Angeles, said “there certainly are neurotropic factors” at play in rheumatology, “so there’s sort of a potential reason why” VNS might work, “but we need to understand far more about its mechanism, and [remember] that open-label studies are not to be believed until” large, randomized, blinded, placebo-controlled studies are done.
Dr. Furst also is an adjunct professor at the University of Washington, Seattle, and a research professor at the University of Florence (Italy). He is in part-time practice in Los Angeles and Seattle.
If everything works out, however, “the vagus nerve may give us a much wider opportunity to block a host of cytokines; it may change the whole paradigm of how we manage rheumatoid arthritis. I think this is possibly a groundbreaking new therapeutic area, much in the way the biologics were” 20 years ago, said rheumatologist Norman B. Gaylis, MD.
Several of the 14 patients in SetPoint’s safety study were enrolled at Dr. Gaylis’s practice in Aventura, Fla., just north of Miami; he said he is eagerly awaiting for the results to be unblinded. If clinical response in that study and others correlates with a cytokine response, “that’s going to be big, and very significant” in the rheumatology community, he said.
SetPoint’s microregulator is charged wirelessly through a collar patients wear for a few minutes once a week. Dosing can also be adjusted through the collar with the help of a computer application.
The device wasn’t turned on in 4 of the 14 patients in the safety study, as a sham control, but shamming was problematic because patients can potentially feel VNS as a buzz or a change in their voice. To get around that potential confounder, both sham and treated patients were told they might or might not feel something during the study.
Implantation takes about an hour, and is much less complex than implanting currently available epilepsy VNS systems, which require implantation of both a power source on the chest wall and wire coils on the vagus nerve.
Cardiac concerns are the main safety issue with VNS, beyond the surgery itself. Cardiac monitoring was done in the safety study to “ensure that we did not cause things like bradycardia, heart block, syncope, etc.” Dr. Chernoff said. So far, they haven’t turned out to be a problem.
Dr. Furst and Dr. Kremer had no relevant disclosures. Dr. Gaylis was compensated by SetPoint for participating in the safety study; he is a consultant and investigator for Electrocore. Dr. Furst and Dr. Gaylis are members of the editorial advisory board for MDedge Rheumatology/Rheumatology News.
Belimumab data out to 13 years show continued safety, efficacy
New data from the longest continuous belimumab treatment in a clinical trial of patients with systemic lupus erythematosus (SLE) indicate similar or lower adverse events each year and maintenance of efficacy for up to 13 years in those who initially respond to and stay on treatment.
First author Daniel J. Wallace, MD, and his colleagues reported in Arthritis & Rheumatology on 298 patients who continued from a phase 2 trial of 476 patients and its extension phase to a continuation study with belimumab (Benlysta) plus standard of care. These patients entered the continuation study having gone from placebo to 10 mg/kg belimumab or continued on 1, 4, or 10 mg/kg belimumab or escalated treatment up to 10 mg/kg. They needed to have an improvement in Physician’s Global Assessment (PGA) score, compared with baseline, and had no severe flare in the last 30 days of the extension study.
At year 5, 70% of patients were still in the study, and this declined to 44% at year 10 and 32% (96 patients) at the end of the study. There were stable or declining rates of the most common adverse events from year 1 to year 11 or later, and serious infections and infestations occurred at a stable rate, from 3.7 per 100 patient-years in year 1 to 6.7 per 100 patients-years through year 11, despite a reduction in immunoglobulin G levels during the study. A total of 15% of patients overall withdrew because of adverse events.
The overall SLE Responder Index response rate rose as the number of participants declined, going from 33% at 1 year and 16 weeks to 76% at 12 years and 32 weeks.
In addition to consistently low flare rates starting at year 5, “those patients remaining had reduced requirements for corticosteroids, and the percentage achieving low disease activity increased. Furthermore, patients continued to have serological improvements. ” Dr. Wallace and his coauthors wrote.
“Patients who remained in the study were likely to be those who responded better or tolerated belimumab better than patients who withdrew; hence, the findings may not be representative of all patients with SLE,” they said.
GlaxoSmithKline and Human Genome Sciences funded the study. Most of the investigators received grants, research support, or consulting fees; held shares in; or were employees of GlaxoSmithKline.
New data from the longest continuous belimumab treatment in a clinical trial of patients with systemic lupus erythematosus (SLE) indicate similar or lower adverse events each year and maintenance of efficacy for up to 13 years in those who initially respond to and stay on treatment.
First author Daniel J. Wallace, MD, and his colleagues reported in Arthritis & Rheumatology on 298 patients who continued from a phase 2 trial of 476 patients and its extension phase to a continuation study with belimumab (Benlysta) plus standard of care. These patients entered the continuation study having gone from placebo to 10 mg/kg belimumab or continued on 1, 4, or 10 mg/kg belimumab or escalated treatment up to 10 mg/kg. They needed to have an improvement in Physician’s Global Assessment (PGA) score, compared with baseline, and had no severe flare in the last 30 days of the extension study.
At year 5, 70% of patients were still in the study, and this declined to 44% at year 10 and 32% (96 patients) at the end of the study. There were stable or declining rates of the most common adverse events from year 1 to year 11 or later, and serious infections and infestations occurred at a stable rate, from 3.7 per 100 patient-years in year 1 to 6.7 per 100 patients-years through year 11, despite a reduction in immunoglobulin G levels during the study. A total of 15% of patients overall withdrew because of adverse events.
The overall SLE Responder Index response rate rose as the number of participants declined, going from 33% at 1 year and 16 weeks to 76% at 12 years and 32 weeks.
In addition to consistently low flare rates starting at year 5, “those patients remaining had reduced requirements for corticosteroids, and the percentage achieving low disease activity increased. Furthermore, patients continued to have serological improvements. ” Dr. Wallace and his coauthors wrote.
“Patients who remained in the study were likely to be those who responded better or tolerated belimumab better than patients who withdrew; hence, the findings may not be representative of all patients with SLE,” they said.
GlaxoSmithKline and Human Genome Sciences funded the study. Most of the investigators received grants, research support, or consulting fees; held shares in; or were employees of GlaxoSmithKline.
New data from the longest continuous belimumab treatment in a clinical trial of patients with systemic lupus erythematosus (SLE) indicate similar or lower adverse events each year and maintenance of efficacy for up to 13 years in those who initially respond to and stay on treatment.
First author Daniel J. Wallace, MD, and his colleagues reported in Arthritis & Rheumatology on 298 patients who continued from a phase 2 trial of 476 patients and its extension phase to a continuation study with belimumab (Benlysta) plus standard of care. These patients entered the continuation study having gone from placebo to 10 mg/kg belimumab or continued on 1, 4, or 10 mg/kg belimumab or escalated treatment up to 10 mg/kg. They needed to have an improvement in Physician’s Global Assessment (PGA) score, compared with baseline, and had no severe flare in the last 30 days of the extension study.
At year 5, 70% of patients were still in the study, and this declined to 44% at year 10 and 32% (96 patients) at the end of the study. There were stable or declining rates of the most common adverse events from year 1 to year 11 or later, and serious infections and infestations occurred at a stable rate, from 3.7 per 100 patient-years in year 1 to 6.7 per 100 patients-years through year 11, despite a reduction in immunoglobulin G levels during the study. A total of 15% of patients overall withdrew because of adverse events.
The overall SLE Responder Index response rate rose as the number of participants declined, going from 33% at 1 year and 16 weeks to 76% at 12 years and 32 weeks.
In addition to consistently low flare rates starting at year 5, “those patients remaining had reduced requirements for corticosteroids, and the percentage achieving low disease activity increased. Furthermore, patients continued to have serological improvements. ” Dr. Wallace and his coauthors wrote.
“Patients who remained in the study were likely to be those who responded better or tolerated belimumab better than patients who withdrew; hence, the findings may not be representative of all patients with SLE,” they said.
GlaxoSmithKline and Human Genome Sciences funded the study. Most of the investigators received grants, research support, or consulting fees; held shares in; or were employees of GlaxoSmithKline.
FROM ARTHRITIS & RHEUMATOLOGY
Rheumatologist involvement often reclassifies interstitial lung disease
MAUI, HAWAII – The latest practice guidelines on the diagnosis of interstitial lung disease issued by the American Thoracic Society and allied organizations recommend as the standard of care a review of all cases by a multidisciplinary team consisting of a pulmonologist, radiologist, and pathologist to ensure accurate diagnosis and classification.

That’s not good enough. A rheumatologist needs to routinely be involved in those multidisciplinary discussions as well, Aryeh Fischer, MD, asserted at the 2019 Rheumatology Winter Clinical Symposium.
Why? Because a rheumatologist’s input often leads to a change in diagnosis. And that change can have important prognostic and therapeutic implications.
“We want to distinguish the IPF [idiopathic pulmonary fibrosis] patients from everybody else. The most important thing with regards to therapy is to identify the IPF patient. The IPF patients are the only ones who are able to be treated with antifibrotic agents: pirfenidone or nintedanib. And we know that immunosuppression can make patients with IPF worse; their risks of hospitalization and mortality are higher on immunosuppression. But everybody else, including anyone with any of the autoimmune diseases along with ILD [interstitial lung disease] or any other causes of ILD, gets treated with immunosuppression,” explained Dr. Fischer, a rheumatologist with a special interest in autoimmune lung disease at the University of Colorado, Denver.
He cited a recent prospective blinded study in which 60 newly diagnosed ILD patients were evaluated separately by a multidisciplinary team comprising a pulmonologist, radiologist, and pathologist and once again with the involvement of a rheumatologist. The rheumatologic assessment reclassified 21% of patients from IPF – that is, lung disease unrelated to connective tissue disease (CTD) or exposure to asbestos, bird droppings, or other triggers – to ILD with connective tissue disease (CTD-ILD). And the number of patients classified as having ILD with autoimmune features without meeting full diagnostic criteria for a major CTD, a category that includes antisynthetase syndrome and IgG4-related ILD, jumped by 77%. Also, the investigators determined that adding a rheumatologist to the multidisciplinary team would have resulted in seven fewer bronchoscopies and one less surgical biopsy among this 60-patient cohort (J Rheumatol. 2018 Nov;45[11]:1509-14).
It’s not at all uncommon to identify a new occult CTD in patients presenting with ILD. Dr. Fischer noted that in a series of 114 consecutive patients evaluated at the interdisciplinary ILD program at Johns Hopkins University, Baltimore, 30% of them had a well-defined CTD, half of whom, or 15% of the total, were newly diagnosed with CTD by virtue of their ILD evaluation (Respir Med. 2009 Aug;103[8]:1152-8).
A CTD-ILD diagnosis has prognostic implications: Survival is significantly better than with IPF, with the exception of ILD with rheumatoid arthritis (RA-ILD), where the prognosis seems to be worse than for other forms of CTD-ILD and is more akin to that of IPF. Indeed, the risk of death is threefold higher in patients with RA-ILD than in those with RA without ILD. While the predominant pattern of lung injury in RA-ILD is usual interstitial pneumonia, marked by fibrosis and honeycombing, the predominant pattern in all other forms of CTD-ILD is nonspecific interstitial pneumonia.
A French study of 778 consecutive patients with ILD highlighted how common autoimmune disease is in the ILD population. Nearly one-third of the ILD patients had autoimmune disease: 22% had CTD-ILD and another 7% had interstitial pneumonia with autoimmune features, or IPAF (Respir Med. 2017 Feb;123:56-62).
Dr. Fischer was lead author of a report by a European Respiratory Society/American Thoracic Society task force that first put forth the term IPAF to describe ILD patients with subtle serologic, clinical, and/or morphologic findings that don’t rise to the level required for formal diagnosis of a defined CTD (Eur Respir J. 2015 Oct;46[4]:976-87).
IPAF is a research construct. Patients who fall into this category are the focus of ongoing prospective studies aimed at better understanding their prognosis and appropriate treatment.
The big three autoimmune diseases comorbid with ILD as reported by Dr. Fischer and coinvestigators in a study of 237 patients with an autoimmune phenotype in a Denver ILD clinic were scleroderma, present in 37%; rheumatoid arthritis, present in 18%; and myositis, with a prevalence of 11%. Another 24% had IPAF.
“There was surprisingly little SLE [3%], and not much Sjögren’s [6%],” he observed.
When pulmonologists come knocking on Dr. Fischer’s door asking if their patients with ILD have occult CTD, he finds it helpful to look for quantifiable specific extrathoracic features suggestive of rheumatologic disease, such as Raynaud’s, sclerodactyly, mechanic hands, and Gottron’s papules.
The ILD practice guidelines recently issued jointly by the American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society recommend that serologic evaluation for a long list of autoantibodies “should be performed even in the absence of signs or symptoms of connective tissue disease” (Am J Respir Crit Care Med. 2018 Sep 1;198[5]:e44-e68). Dr. Fischer indicated he has a problem with that recommendation since nonrheumatologists aren’t typically adept in interpreting the significance of autoantibodies.
“I will just tell you, I see a lot of patients for ILD evaluation, and autoimmune serologies often lead to more questions than answers. They always need to be considered within the clinical context,” the rheumatologist said.
High-resolution CT (HRCT) is the imaging method of choice for detecting ILD and classifying its severity. HRCT also holds clues for detection of CTD-ILD.
“There are no upper lung–predominant ILDs that really make you think CTD. Our diseases like the lower lung zones. We like to see multicompartment involvement. When you hear ‘airways and parenchymal,’ ‘pleural,’ ‘pericardial thickening,’ that sounds a lot like autoimmune disease,” according to Dr. Fischer.
Also, the presence of a dilated esophagus on HRCT is strongly suggestive of scleroderma, he added.
In the event a pathology report is available, it’s important to read the fine print. Secondary histopathologic features of CTD-ILD include extensive pleuritis, lymphoid aggregates with germinal center formation, dense perivascular collagen, and/or prominent plasmacytic infiltration.
While ILD can be the first manifestation of an underlying occult CTD, it’s also common for a patient with an established CTD to subsequently develop ILD.
Rheumatologists are no strangers to ambiguity, so it should come as no surprise that while audible bibasilar crackles on physical examination are strongly predictive of ILD, their absence doesn’t indicate absence of ILD. Similarly, dyspnea in a patient with established CTD can be tough to interpret, and absence of dyspnea doesn’t imply absence of ILD. If a patient with CTD is on immunosuppressive therapy, bronchoalveolar lavage is of value in ruling out infection.
When to order a surgical lung biopsy
“If you’re pretty sure your patient had a CTD and then you get a characteristic HRCT and there are no exposures to account for the imaging findings – if we have a scenario where it all fits – we often don’t biopsy. Biopsy usually means a 2-night hospital stay, and it’s reportedly associated with about a 1% mortality risk. And the clinical reality is the biopsy may not impact treatment. They’re going to give you azathioprine, mycophenolate, cyclophosphamide, or prednisone for the ILD and the extrathoracic disease irrespective of the ILD pattern,” the rheumatologist said.
He reserves surgical biopsy for patients with an atypical HRCT, those with known CTD and a possible alternative etiology for the ILD, such as exposure to asbestos or owning pet birds, and patients where he’s just not sure a CTD is actually present.
Treatment of CTD-ILD
“The available controlled efficacy data are limited to scleroderma-ILD, where cyclophosphamide and mycophenolate work a little bit. And that’s it. We don’t have good data to guide us on which agent for which CTD or ILD pattern, for how long, or what dose,” Dr. Fischer said. “We have good drugs for the joints, nothing for the lungs. Treatment is not evidence based. We initiate with high-dose steroids, switch to a steroid-sparing agent, we evaluate the response to treatment with surveillance every 3-6 months by 6-minute walking, lung function tests, and sometimes imaging, and we treat for a long time. Oftentimes stability equals success.”
That being said, it must be emphasized that not all CTD-ILD warrants treatment of the pulmonary disease. If the patient’s ILD is mild and indolent, clinical surveillance is often appropriate, he continued.
More important than the pharmacotherapy available at present are adjunctive nonpharmacologic approaches: supplemental oxygen, pulmonary rehabilitation, treatment of comorbid GERD, immunizations, and addressing mental health issues related to these devastating diseases.
“We really don’t do these things well,” Dr. Fischer said.
On the horizon
An investigator-driven phase 2 clinical trial of the antifibrotic drug pirfenidone (Esbriet), now approved for IPF, is underway in patients with RA-ILD. Results of a trial of antifibrotic therapy in patients with scleroderma-ILD are due to be presented this year at EULAR. And a small study of antifibrotic therapy in patients with myositis-ILD is ongoing.
As for the biologics, there is a signal in the literature that tumor necrosis factor inhibitors may be associated with increased risk of rapidly progressive lung disease in patients with RA-ILD. An influential report from the British Society for Rheumatology Biologics Register on 299 patients with preexisting RA-ILD treated with anti-TNF therapy and 68 who received traditional disease-modifying antirrheumatic drugs showed that while mortality during follow-up was similar in the two groups, the proportion of deaths attributed to RA-ILD was 21% in the group on anti-TNF therapy, compared with only 7% in those on traditional DMARDs (Ann Rheum Dis. 2010 Jun;69[6]:1086-91).
Paul Emery, MD, rose from the audience at Dr. Fischer’s request to share his extensive experience with anti-TNF therapy and rituximab (Rituxan) in the setting of RA-ILD. When he was involved in several of the pivotal clinical trials of anti-TNF agents for RA he encountered a couple of cases of rapidly progressive ILD in patients on treatment.
“We had never before seen rapidly progressive ILD that didn’t respond to cyclophosphamide. Both patients died,” recalled Dr. Emery, professor of rheumatology and director of the University of Leeds (England) Musculoskeletal Biomedical Research Center.
“Our experience is if you’ve got mild ILD – and if you look hard enough you can find it in many rheumatoids – TNF inhibitor therapy doesn’t affect it. But if there’s any hint of deterioration we move away from anti-TNF therapy. Our preference has been for rituximab,” he said.
Dr. Emery was senior author of the largest study to date of rituximab in patients with RA-ILD (Rheumatology [Oxford]. 2017 Aug 1;56[8]:1348-57). While he and his coinvestigators concluded that rituximab “appears to be an acceptable therapeutic choice for patients with RA-ILD,” Dr. Fischer didn’t find persuasive evidence from this relatively small retrospective observational study in which only 44 patients had lung data available.
“My own conclusion is evidence is lacking to support a role of rituximab for treating ILD in RA,” Dr. Fischer said.
He reported receiving research grants from Boehringer-Ingelheim and Corbus and serving as a consultant to a handful of other pharmaceutical companies.
MAUI, HAWAII – The latest practice guidelines on the diagnosis of interstitial lung disease issued by the American Thoracic Society and allied organizations recommend as the standard of care a review of all cases by a multidisciplinary team consisting of a pulmonologist, radiologist, and pathologist to ensure accurate diagnosis and classification.
That’s not good enough. A rheumatologist needs to routinely be involved in those multidisciplinary discussions as well, Aryeh Fischer, MD, asserted at the 2019 Rheumatology Winter Clinical Symposium.
Why? Because a rheumatologist’s input often leads to a change in diagnosis. And that change can have important prognostic and therapeutic implications.
“We want to distinguish the IPF [idiopathic pulmonary fibrosis] patients from everybody else. The most important thing with regards to therapy is to identify the IPF patient. The IPF patients are the only ones who are able to be treated with antifibrotic agents: pirfenidone or nintedanib. And we know that immunosuppression can make patients with IPF worse; their risks of hospitalization and mortality are higher on immunosuppression. But everybody else, including anyone with any of the autoimmune diseases along with ILD [interstitial lung disease] or any other causes of ILD, gets treated with immunosuppression,” explained Dr. Fischer, a rheumatologist with a special interest in autoimmune lung disease at the University of Colorado, Denver.
He cited a recent prospective blinded study in which 60 newly diagnosed ILD patients were evaluated separately by a multidisciplinary team comprising a pulmonologist, radiologist, and pathologist and once again with the involvement of a rheumatologist. The rheumatologic assessment reclassified 21% of patients from IPF – that is, lung disease unrelated to connective tissue disease (CTD) or exposure to asbestos, bird droppings, or other triggers – to ILD with connective tissue disease (CTD-ILD). And the number of patients classified as having ILD with autoimmune features without meeting full diagnostic criteria for a major CTD, a category that includes antisynthetase syndrome and IgG4-related ILD, jumped by 77%. Also, the investigators determined that adding a rheumatologist to the multidisciplinary team would have resulted in seven fewer bronchoscopies and one less surgical biopsy among this 60-patient cohort (J Rheumatol. 2018 Nov;45[11]:1509-14).
It’s not at all uncommon to identify a new occult CTD in patients presenting with ILD. Dr. Fischer noted that in a series of 114 consecutive patients evaluated at the interdisciplinary ILD program at Johns Hopkins University, Baltimore, 30% of them had a well-defined CTD, half of whom, or 15% of the total, were newly diagnosed with CTD by virtue of their ILD evaluation (Respir Med. 2009 Aug;103[8]:1152-8).
A CTD-ILD diagnosis has prognostic implications: Survival is significantly better than with IPF, with the exception of ILD with rheumatoid arthritis (RA-ILD), where the prognosis seems to be worse than for other forms of CTD-ILD and is more akin to that of IPF. Indeed, the risk of death is threefold higher in patients with RA-ILD than in those with RA without ILD. While the predominant pattern of lung injury in RA-ILD is usual interstitial pneumonia, marked by fibrosis and honeycombing, the predominant pattern in all other forms of CTD-ILD is nonspecific interstitial pneumonia.
A French study of 778 consecutive patients with ILD highlighted how common autoimmune disease is in the ILD population. Nearly one-third of the ILD patients had autoimmune disease: 22% had CTD-ILD and another 7% had interstitial pneumonia with autoimmune features, or IPAF (Respir Med. 2017 Feb;123:56-62).
Dr. Fischer was lead author of a report by a European Respiratory Society/American Thoracic Society task force that first put forth the term IPAF to describe ILD patients with subtle serologic, clinical, and/or morphologic findings that don’t rise to the level required for formal diagnosis of a defined CTD (Eur Respir J. 2015 Oct;46[4]:976-87).
IPAF is a research construct. Patients who fall into this category are the focus of ongoing prospective studies aimed at better understanding their prognosis and appropriate treatment.
The big three autoimmune diseases comorbid with ILD as reported by Dr. Fischer and coinvestigators in a study of 237 patients with an autoimmune phenotype in a Denver ILD clinic were scleroderma, present in 37%; rheumatoid arthritis, present in 18%; and myositis, with a prevalence of 11%. Another 24% had IPAF.
“There was surprisingly little SLE [3%], and not much Sjögren’s [6%],” he observed.
When pulmonologists come knocking on Dr. Fischer’s door asking if their patients with ILD have occult CTD, he finds it helpful to look for quantifiable specific extrathoracic features suggestive of rheumatologic disease, such as Raynaud’s, sclerodactyly, mechanic hands, and Gottron’s papules.
The ILD practice guidelines recently issued jointly by the American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society recommend that serologic evaluation for a long list of autoantibodies “should be performed even in the absence of signs or symptoms of connective tissue disease” (Am J Respir Crit Care Med. 2018 Sep 1;198[5]:e44-e68). Dr. Fischer indicated he has a problem with that recommendation since nonrheumatologists aren’t typically adept in interpreting the significance of autoantibodies.
“I will just tell you, I see a lot of patients for ILD evaluation, and autoimmune serologies often lead to more questions than answers. They always need to be considered within the clinical context,” the rheumatologist said.
High-resolution CT (HRCT) is the imaging method of choice for detecting ILD and classifying its severity. HRCT also holds clues for detection of CTD-ILD.
“There are no upper lung–predominant ILDs that really make you think CTD. Our diseases like the lower lung zones. We like to see multicompartment involvement. When you hear ‘airways and parenchymal,’ ‘pleural,’ ‘pericardial thickening,’ that sounds a lot like autoimmune disease,” according to Dr. Fischer.
Also, the presence of a dilated esophagus on HRCT is strongly suggestive of scleroderma, he added.
In the event a pathology report is available, it’s important to read the fine print. Secondary histopathologic features of CTD-ILD include extensive pleuritis, lymphoid aggregates with germinal center formation, dense perivascular collagen, and/or prominent plasmacytic infiltration.
While ILD can be the first manifestation of an underlying occult CTD, it’s also common for a patient with an established CTD to subsequently develop ILD.
Rheumatologists are no strangers to ambiguity, so it should come as no surprise that while audible bibasilar crackles on physical examination are strongly predictive of ILD, their absence doesn’t indicate absence of ILD. Similarly, dyspnea in a patient with established CTD can be tough to interpret, and absence of dyspnea doesn’t imply absence of ILD. If a patient with CTD is on immunosuppressive therapy, bronchoalveolar lavage is of value in ruling out infection.
When to order a surgical lung biopsy
“If you’re pretty sure your patient had a CTD and then you get a characteristic HRCT and there are no exposures to account for the imaging findings – if we have a scenario where it all fits – we often don’t biopsy. Biopsy usually means a 2-night hospital stay, and it’s reportedly associated with about a 1% mortality risk. And the clinical reality is the biopsy may not impact treatment. They’re going to give you azathioprine, mycophenolate, cyclophosphamide, or prednisone for the ILD and the extrathoracic disease irrespective of the ILD pattern,” the rheumatologist said.
He reserves surgical biopsy for patients with an atypical HRCT, those with known CTD and a possible alternative etiology for the ILD, such as exposure to asbestos or owning pet birds, and patients where he’s just not sure a CTD is actually present.
Treatment of CTD-ILD
“The available controlled efficacy data are limited to scleroderma-ILD, where cyclophosphamide and mycophenolate work a little bit. And that’s it. We don’t have good data to guide us on which agent for which CTD or ILD pattern, for how long, or what dose,” Dr. Fischer said. “We have good drugs for the joints, nothing for the lungs. Treatment is not evidence based. We initiate with high-dose steroids, switch to a steroid-sparing agent, we evaluate the response to treatment with surveillance every 3-6 months by 6-minute walking, lung function tests, and sometimes imaging, and we treat for a long time. Oftentimes stability equals success.”
That being said, it must be emphasized that not all CTD-ILD warrants treatment of the pulmonary disease. If the patient’s ILD is mild and indolent, clinical surveillance is often appropriate, he continued.
More important than the pharmacotherapy available at present are adjunctive nonpharmacologic approaches: supplemental oxygen, pulmonary rehabilitation, treatment of comorbid GERD, immunizations, and addressing mental health issues related to these devastating diseases.
“We really don’t do these things well,” Dr. Fischer said.
On the horizon
An investigator-driven phase 2 clinical trial of the antifibrotic drug pirfenidone (Esbriet), now approved for IPF, is underway in patients with RA-ILD. Results of a trial of antifibrotic therapy in patients with scleroderma-ILD are due to be presented this year at EULAR. And a small study of antifibrotic therapy in patients with myositis-ILD is ongoing.
As for the biologics, there is a signal in the literature that tumor necrosis factor inhibitors may be associated with increased risk of rapidly progressive lung disease in patients with RA-ILD. An influential report from the British Society for Rheumatology Biologics Register on 299 patients with preexisting RA-ILD treated with anti-TNF therapy and 68 who received traditional disease-modifying antirrheumatic drugs showed that while mortality during follow-up was similar in the two groups, the proportion of deaths attributed to RA-ILD was 21% in the group on anti-TNF therapy, compared with only 7% in those on traditional DMARDs (Ann Rheum Dis. 2010 Jun;69[6]:1086-91).
Paul Emery, MD, rose from the audience at Dr. Fischer’s request to share his extensive experience with anti-TNF therapy and rituximab (Rituxan) in the setting of RA-ILD. When he was involved in several of the pivotal clinical trials of anti-TNF agents for RA he encountered a couple of cases of rapidly progressive ILD in patients on treatment.
“We had never before seen rapidly progressive ILD that didn’t respond to cyclophosphamide. Both patients died,” recalled Dr. Emery, professor of rheumatology and director of the University of Leeds (England) Musculoskeletal Biomedical Research Center.
“Our experience is if you’ve got mild ILD – and if you look hard enough you can find it in many rheumatoids – TNF inhibitor therapy doesn’t affect it. But if there’s any hint of deterioration we move away from anti-TNF therapy. Our preference has been for rituximab,” he said.
Dr. Emery was senior author of the largest study to date of rituximab in patients with RA-ILD (Rheumatology [Oxford]. 2017 Aug 1;56[8]:1348-57). While he and his coinvestigators concluded that rituximab “appears to be an acceptable therapeutic choice for patients with RA-ILD,” Dr. Fischer didn’t find persuasive evidence from this relatively small retrospective observational study in which only 44 patients had lung data available.
“My own conclusion is evidence is lacking to support a role of rituximab for treating ILD in RA,” Dr. Fischer said.
He reported receiving research grants from Boehringer-Ingelheim and Corbus and serving as a consultant to a handful of other pharmaceutical companies.
MAUI, HAWAII – The latest practice guidelines on the diagnosis of interstitial lung disease issued by the American Thoracic Society and allied organizations recommend as the standard of care a review of all cases by a multidisciplinary team consisting of a pulmonologist, radiologist, and pathologist to ensure accurate diagnosis and classification.
That’s not good enough. A rheumatologist needs to routinely be involved in those multidisciplinary discussions as well, Aryeh Fischer, MD, asserted at the 2019 Rheumatology Winter Clinical Symposium.
Why? Because a rheumatologist’s input often leads to a change in diagnosis. And that change can have important prognostic and therapeutic implications.
“We want to distinguish the IPF [idiopathic pulmonary fibrosis] patients from everybody else. The most important thing with regards to therapy is to identify the IPF patient. The IPF patients are the only ones who are able to be treated with antifibrotic agents: pirfenidone or nintedanib. And we know that immunosuppression can make patients with IPF worse; their risks of hospitalization and mortality are higher on immunosuppression. But everybody else, including anyone with any of the autoimmune diseases along with ILD [interstitial lung disease] or any other causes of ILD, gets treated with immunosuppression,” explained Dr. Fischer, a rheumatologist with a special interest in autoimmune lung disease at the University of Colorado, Denver.
He cited a recent prospective blinded study in which 60 newly diagnosed ILD patients were evaluated separately by a multidisciplinary team comprising a pulmonologist, radiologist, and pathologist and once again with the involvement of a rheumatologist. The rheumatologic assessment reclassified 21% of patients from IPF – that is, lung disease unrelated to connective tissue disease (CTD) or exposure to asbestos, bird droppings, or other triggers – to ILD with connective tissue disease (CTD-ILD). And the number of patients classified as having ILD with autoimmune features without meeting full diagnostic criteria for a major CTD, a category that includes antisynthetase syndrome and IgG4-related ILD, jumped by 77%. Also, the investigators determined that adding a rheumatologist to the multidisciplinary team would have resulted in seven fewer bronchoscopies and one less surgical biopsy among this 60-patient cohort (J Rheumatol. 2018 Nov;45[11]:1509-14).
It’s not at all uncommon to identify a new occult CTD in patients presenting with ILD. Dr. Fischer noted that in a series of 114 consecutive patients evaluated at the interdisciplinary ILD program at Johns Hopkins University, Baltimore, 30% of them had a well-defined CTD, half of whom, or 15% of the total, were newly diagnosed with CTD by virtue of their ILD evaluation (Respir Med. 2009 Aug;103[8]:1152-8).
A CTD-ILD diagnosis has prognostic implications: Survival is significantly better than with IPF, with the exception of ILD with rheumatoid arthritis (RA-ILD), where the prognosis seems to be worse than for other forms of CTD-ILD and is more akin to that of IPF. Indeed, the risk of death is threefold higher in patients with RA-ILD than in those with RA without ILD. While the predominant pattern of lung injury in RA-ILD is usual interstitial pneumonia, marked by fibrosis and honeycombing, the predominant pattern in all other forms of CTD-ILD is nonspecific interstitial pneumonia.
A French study of 778 consecutive patients with ILD highlighted how common autoimmune disease is in the ILD population. Nearly one-third of the ILD patients had autoimmune disease: 22% had CTD-ILD and another 7% had interstitial pneumonia with autoimmune features, or IPAF (Respir Med. 2017 Feb;123:56-62).
Dr. Fischer was lead author of a report by a European Respiratory Society/American Thoracic Society task force that first put forth the term IPAF to describe ILD patients with subtle serologic, clinical, and/or morphologic findings that don’t rise to the level required for formal diagnosis of a defined CTD (Eur Respir J. 2015 Oct;46[4]:976-87).
IPAF is a research construct. Patients who fall into this category are the focus of ongoing prospective studies aimed at better understanding their prognosis and appropriate treatment.
The big three autoimmune diseases comorbid with ILD as reported by Dr. Fischer and coinvestigators in a study of 237 patients with an autoimmune phenotype in a Denver ILD clinic were scleroderma, present in 37%; rheumatoid arthritis, present in 18%; and myositis, with a prevalence of 11%. Another 24% had IPAF.
“There was surprisingly little SLE [3%], and not much Sjögren’s [6%],” he observed.
When pulmonologists come knocking on Dr. Fischer’s door asking if their patients with ILD have occult CTD, he finds it helpful to look for quantifiable specific extrathoracic features suggestive of rheumatologic disease, such as Raynaud’s, sclerodactyly, mechanic hands, and Gottron’s papules.
The ILD practice guidelines recently issued jointly by the American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society recommend that serologic evaluation for a long list of autoantibodies “should be performed even in the absence of signs or symptoms of connective tissue disease” (Am J Respir Crit Care Med. 2018 Sep 1;198[5]:e44-e68). Dr. Fischer indicated he has a problem with that recommendation since nonrheumatologists aren’t typically adept in interpreting the significance of autoantibodies.
“I will just tell you, I see a lot of patients for ILD evaluation, and autoimmune serologies often lead to more questions than answers. They always need to be considered within the clinical context,” the rheumatologist said.
High-resolution CT (HRCT) is the imaging method of choice for detecting ILD and classifying its severity. HRCT also holds clues for detection of CTD-ILD.
“There are no upper lung–predominant ILDs that really make you think CTD. Our diseases like the lower lung zones. We like to see multicompartment involvement. When you hear ‘airways and parenchymal,’ ‘pleural,’ ‘pericardial thickening,’ that sounds a lot like autoimmune disease,” according to Dr. Fischer.
Also, the presence of a dilated esophagus on HRCT is strongly suggestive of scleroderma, he added.
In the event a pathology report is available, it’s important to read the fine print. Secondary histopathologic features of CTD-ILD include extensive pleuritis, lymphoid aggregates with germinal center formation, dense perivascular collagen, and/or prominent plasmacytic infiltration.
While ILD can be the first manifestation of an underlying occult CTD, it’s also common for a patient with an established CTD to subsequently develop ILD.
Rheumatologists are no strangers to ambiguity, so it should come as no surprise that while audible bibasilar crackles on physical examination are strongly predictive of ILD, their absence doesn’t indicate absence of ILD. Similarly, dyspnea in a patient with established CTD can be tough to interpret, and absence of dyspnea doesn’t imply absence of ILD. If a patient with CTD is on immunosuppressive therapy, bronchoalveolar lavage is of value in ruling out infection.
When to order a surgical lung biopsy
“If you’re pretty sure your patient had a CTD and then you get a characteristic HRCT and there are no exposures to account for the imaging findings – if we have a scenario where it all fits – we often don’t biopsy. Biopsy usually means a 2-night hospital stay, and it’s reportedly associated with about a 1% mortality risk. And the clinical reality is the biopsy may not impact treatment. They’re going to give you azathioprine, mycophenolate, cyclophosphamide, or prednisone for the ILD and the extrathoracic disease irrespective of the ILD pattern,” the rheumatologist said.
He reserves surgical biopsy for patients with an atypical HRCT, those with known CTD and a possible alternative etiology for the ILD, such as exposure to asbestos or owning pet birds, and patients where he’s just not sure a CTD is actually present.
Treatment of CTD-ILD
“The available controlled efficacy data are limited to scleroderma-ILD, where cyclophosphamide and mycophenolate work a little bit. And that’s it. We don’t have good data to guide us on which agent for which CTD or ILD pattern, for how long, or what dose,” Dr. Fischer said. “We have good drugs for the joints, nothing for the lungs. Treatment is not evidence based. We initiate with high-dose steroids, switch to a steroid-sparing agent, we evaluate the response to treatment with surveillance every 3-6 months by 6-minute walking, lung function tests, and sometimes imaging, and we treat for a long time. Oftentimes stability equals success.”
That being said, it must be emphasized that not all CTD-ILD warrants treatment of the pulmonary disease. If the patient’s ILD is mild and indolent, clinical surveillance is often appropriate, he continued.
More important than the pharmacotherapy available at present are adjunctive nonpharmacologic approaches: supplemental oxygen, pulmonary rehabilitation, treatment of comorbid GERD, immunizations, and addressing mental health issues related to these devastating diseases.
“We really don’t do these things well,” Dr. Fischer said.
On the horizon
An investigator-driven phase 2 clinical trial of the antifibrotic drug pirfenidone (Esbriet), now approved for IPF, is underway in patients with RA-ILD. Results of a trial of antifibrotic therapy in patients with scleroderma-ILD are due to be presented this year at EULAR. And a small study of antifibrotic therapy in patients with myositis-ILD is ongoing.
As for the biologics, there is a signal in the literature that tumor necrosis factor inhibitors may be associated with increased risk of rapidly progressive lung disease in patients with RA-ILD. An influential report from the British Society for Rheumatology Biologics Register on 299 patients with preexisting RA-ILD treated with anti-TNF therapy and 68 who received traditional disease-modifying antirrheumatic drugs showed that while mortality during follow-up was similar in the two groups, the proportion of deaths attributed to RA-ILD was 21% in the group on anti-TNF therapy, compared with only 7% in those on traditional DMARDs (Ann Rheum Dis. 2010 Jun;69[6]:1086-91).
Paul Emery, MD, rose from the audience at Dr. Fischer’s request to share his extensive experience with anti-TNF therapy and rituximab (Rituxan) in the setting of RA-ILD. When he was involved in several of the pivotal clinical trials of anti-TNF agents for RA he encountered a couple of cases of rapidly progressive ILD in patients on treatment.
“We had never before seen rapidly progressive ILD that didn’t respond to cyclophosphamide. Both patients died,” recalled Dr. Emery, professor of rheumatology and director of the University of Leeds (England) Musculoskeletal Biomedical Research Center.
“Our experience is if you’ve got mild ILD – and if you look hard enough you can find it in many rheumatoids – TNF inhibitor therapy doesn’t affect it. But if there’s any hint of deterioration we move away from anti-TNF therapy. Our preference has been for rituximab,” he said.
Dr. Emery was senior author of the largest study to date of rituximab in patients with RA-ILD (Rheumatology [Oxford]. 2017 Aug 1;56[8]:1348-57). While he and his coinvestigators concluded that rituximab “appears to be an acceptable therapeutic choice for patients with RA-ILD,” Dr. Fischer didn’t find persuasive evidence from this relatively small retrospective observational study in which only 44 patients had lung data available.
“My own conclusion is evidence is lacking to support a role of rituximab for treating ILD in RA,” Dr. Fischer said.
He reported receiving research grants from Boehringer-Ingelheim and Corbus and serving as a consultant to a handful of other pharmaceutical companies.
REPORTING FROM RWCS 2019
Myositis mimics: Clues for making the right diagnosis
A number of conditions can mimic myositis, but clues that can point to the correct diagnosis are often present in cases involving the mimics, according to Lisa Christopher-Stine, MD.

For example, elevated levels of certain muscle enzymes are an important source of diagnostic information, Dr. Christopher-Stine, director of the Johns Hopkins Myositis Center, Baltimore, said at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
Isolated elevations in aldolase can be seen in connective tissue–associated interstitial lung disease or in patients with fascial edema, and aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase (LDH), and creatine kinase (CK) levels can also be helpful, she explained.
The latter can also be elevated in the absence of muscle disease, for example, in healthy individuals following exercise. CK peaks at 24 hours after exercise before returning to baseline by 72 hours. In an experimental setting, a threefold increase in CK levels has been seen at 8-24 hours after exercise, Dr. Christopher-Stine said.
“HyperCKemia”
Trauma from causes such as intramuscular injection, electromyography (EMG), major surgery, or biopsy can also lead to increased CK levels. Motor neuron disease can also cause such increases. In one study, 75% of patients with amyotrophic lateral sclerosis had a mean twofold increase in CK levels, she said.
Asymptomatic CK elevations may also represent presymptomatic myopathies, type 1 or 2 macro-CK, manual labor occupations, or they may be idiopathic.
Race can play a role in CK levels as well. Black people tend to have higher CK levels than white people, she said, noting that one study of more than 10,000 adults showed that black race was strongly associated with CK, and that body composition largely explained differences in CK by age, but not by race/ethnicity (Medicine. Aug 2016;95[33]:e4344).
“So elevated CK may not herald any discernible illness,” she said.
Dr. Christopher-Stine described a case involving an otherwise healthy 30-year-old man with a CK level of 695 IU/L that was found incidentally. He had a desk job, no recent travel, and denied weakness, myalgias, joint pain, dysphagia, shortness of breath, and fevers. In this case, the elevated CK was felt to be secondary to his African American race given that other causes were ruled out.
Another case involved a 72-year-old man with left-arm pain. A cardiac event was ruled out, and CK was found to be about 4,500 IU/L. He reported “flare-ups” of diffuse swelling of the hands and feet. X-rays showed concerning signs of erosions. His transaminases and electromyogram were normal; he reported no weakness or myalgia; and an MRI showed no muscle edema. He was diagnosed with macro-CK, which refers to CK with an increased molecular weight. A clue to this diagnosis is a normal liver function test. In some cases, muscle/brain CK levels (CK-MB) are elevated and higher than total CK, she noted.
She presented an algorithm for the diagnostic work-up of patients presenting with elevated CK of unclear significance. Her recommended approach involves repeat CK assessment and a closer look at family history, medication, drug/toxin history, examination for weakness and neurologic abnormalities, and additional lab assessments in those whose levels remain elevated. In those in whom a diagnosis is not identified, the algorithm calls for observation every 3 months – including physical examination and labs – in asymptomatic patients with levels at less than five times the upper limit of normal, and further evaluation, including EMG with nerve-conduction velocity testing, muscle biopsy, and MRI in those with (or who later develop) marked elevation greater than five times the upper limit of normal and/or symptoms.
Patient assessment
The physical examination should involve localization and quantification of weakness, and assessment for fever, rash, atrophy/wasting/scooping of forearms, fasciculations, cranial nerve involvement, Raynaud’s phenomenon, nailfold capillary changes, arthritis, calcinosis, “mechanic’s hands,” signs of other autoimmune diseases, and lung crackles. Initial laboratory testing should include HIV and hepatitis B and C testing; measurement of CK, AST, ALT, aldolase, thyroid-stimulating hormone, and magnesium levels; a comprehensive metabolic panel and complete blood count; and measurement of erythrocyte sedimentation rate and C-reactive protein.
“Weakness may be secondary to a neuropathy, myopathy, or a problem at the neuropathic junction. Many causes of weakness can be readily identified by careful history taking, focused physical examination, and directed laboratory evaluation,” she said.
Features pointing toward a diagnosis of myositis include characteristic rashes, gradual symptom onset, proximal limb and truncal weakness, other connective tissue disease features such as Raynaud’s and arthritis, and the presence of lung disease, including interstitial lung disease or unexplained infiltrates, she said.
Features pointing away from a diagnosis of myositis include a family history of a similar illness, weakness that is associated with eating or fasting, neurologic signs, cranial nerve involvement, fasciculations, severe muscle cramping, early atrophy, and creatine phosphokinase levels that are either less than 2 times or more than 100 times the upper limit of normal.
Among the conditions to consider in the presence of the features that point away from a myositis diagnosis are muscular dystrophies, metabolic myopathies, and toxic (drug-induced) myopathies, to name a few, Dr. Christopher-Stine said.
She described a number of other cases to illustrate the need for – and to help develop – a differential diagnosis in patients presenting with apparent myositis.
Muscular dystrophies
A 38-year-old woman with limited scleroderma and anti-PM/Scl autoantibodies developed proximal weakness over 9 months and was eventually unable to walk up a flight of stairs. She had heliotrope rash and Gottron’s sign, her serum CK was 723 IU/L, and EMG showed an irritable myopathy.
Muscle biopsy showed inflammation, and she was treated with prednisone, but this led to worsening weakness. She complained of prominent fatigue and double vision at the end of the day, and these symptoms did not improve with steroids.
Anti-AChR and anti-MuSK antibodies were negative, but she had a decrement on repetitive nerve stimulation testing.
She was treated with pyridostigmine and experienced near-complete resolution of her proximal weakness and double vision. A chest CT scan showed thymic hyperplasia; thymectomy was recommended.
In another case, a 19-year-old woman who complained of leg pain after exercise was found to have intact strength but asymmetric calf hypertrophy. Her CK level was 5,000 IU/L, and she was referred to rule out acute myositis.
A quadriceps biopsy was performed and showed abnormal dystrophin immunostaining but no inflammation. A molecular genetic analysis showed deletions in Xp21 and she was diagnosed as a manifesting carrier of Duchenne muscular dystrophy. It was recommended that she be evaluated for cardiomyopathy and receive genetic counseling.
A number of other cases presented by Dr. Christopher-Stine highlighted other muscular dystrophies that can mimic myositis, such as:
- Myotonic dystrophies. These are more often type 2 than type 1. Myotonia may be subtle, cataracts are seen early in all patients, and cardiac arrhythmias are common.
- Limb girdle muscular dystrophy type 2 B (dysferlinopathy). In the legs, this often affects the gastrocnemius muscle, and this will be visible on MRI. In the arms, it most often affects the biceps, sparing the deltoids. CKs are typically very high.
- Facioscapulohumeral muscular dystrophy (FSHD). This involves facial weakness, especially obicularis oris, in 95% of cases, as well as scapular weakness and winging, inflammation on muscle biopsy in 75% of cases, and typically is endomysial or perivascular.
Metabolic myopathies
Among metabolic myopathies that can mimic myositis are disorders of carbohydrate metabolism such as McArdle’s disease, 6-phosphofructokinase deficiency, and Pompe’s disease (adult acid maltase deficiency); disorders of lipid metabolism such as carnitine deficiency and carnitine palmitoyltransferase 2 (CPT2) deficiency; and disorders of purine metabolism, such as myoadenylate deaminase deficiency.
A 27-year-old patient who complained of weakness with activity was referred for possible myositis and was found to have a CK of 3,650 IU/L that never normalized. Physical examination showed intact strength and no muscle atrophy or fasciculations, and an enzyme stain for myophosphorylase showed a normal staining pattern and complete absence of the enzyme on quadricep biopsy. A 22-year-old man with similar symptoms plus recent onset of brown/black urine after physical activity had CK of 110,000 IU/L when symptomatic, and also underwent biopsy after being referred for possible myopathy. Both patients were ultimately diagnosed with CPT2 deficiency, which is associated with risk of rhabdomyolysis triggered by prolonged exercise, diets low in carbohydrates and high in fat, or by fasting.
Myalgias are common, and CK levels are normal or only mildly elevated between episodes in CPT2 deficiency, Dr. Christopher-Stine noted.
Toxic myopathies
Drug-induced myopathies are among the most common etiologies of myopathy and can range from mild myalgia to massive rhabdomyolysis. They can cause mild to severe weakness and may be chronic. The mechanism of toxic injury is direct via myotoxins such as ethyl alcohol, glucocorticoids, lipid-lowering drugs, cocaine, antimalarial drugs, antipsychotic drugs, colchicine, and Ipecac syrup.
One case described by Dr. Christopher-Stine involved “statin myopathy.”
A 55-year-old man on atorvastatin complained of myalgias and brown urine, but had no definitive weakness. He had intact strength and diffuse myalgias that weren’t reproducible. His CK was 45,000 IU/L.
Statin myopathy, as seen in this patient, is usually self-limited and is not associated with autoimmunity or with anti-HMGCR autoantibody positivity.
The mechanism is unknown, but statin myopathy has an incidence of 1.2 per 10,000 patient-years. Myalgias, myositis, rhabdomyolysis, and asymptomatic hyperCKemia are commonly seen. This is in contrast to the immune-mediated necrotizing myelitis that can be secondary to statins and is responsive to immunosuppression, she noted.
Other myositis mimics
In addition to these common myositis mimics, certain other neurologic diseases (such as ALS and cervical myelopathy), endocrinopathies (such as hypothyroidism), and infections (like toxoplasmosis) can also be mistaken for myositis, Dr. Christopher-Stine said, noting that cases illustrating these mimics underscore the need for careful consideration of possible alternate diagnoses.
“While most noninflammatory myopathies are self-limited or have no therapies available, knowing the diagnosis can be helpful for genetic counseling of the patient and family, for mitigating risk factors, and for precluding the use of unwarranted immunosuppressive agents,” she said.
Dr. Christopher-Stine reported having intellectual property interest in a novel Inova Diagnostics autoantibody assay detection for anti-HMGCR. She was also the safety officer for the JBT-101 Trial sponsored by Corbus and funded by the National Institutes of Health.
A number of conditions can mimic myositis, but clues that can point to the correct diagnosis are often present in cases involving the mimics, according to Lisa Christopher-Stine, MD.
For example, elevated levels of certain muscle enzymes are an important source of diagnostic information, Dr. Christopher-Stine, director of the Johns Hopkins Myositis Center, Baltimore, said at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
Isolated elevations in aldolase can be seen in connective tissue–associated interstitial lung disease or in patients with fascial edema, and aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase (LDH), and creatine kinase (CK) levels can also be helpful, she explained.
The latter can also be elevated in the absence of muscle disease, for example, in healthy individuals following exercise. CK peaks at 24 hours after exercise before returning to baseline by 72 hours. In an experimental setting, a threefold increase in CK levels has been seen at 8-24 hours after exercise, Dr. Christopher-Stine said.
“HyperCKemia”
Trauma from causes such as intramuscular injection, electromyography (EMG), major surgery, or biopsy can also lead to increased CK levels. Motor neuron disease can also cause such increases. In one study, 75% of patients with amyotrophic lateral sclerosis had a mean twofold increase in CK levels, she said.
Asymptomatic CK elevations may also represent presymptomatic myopathies, type 1 or 2 macro-CK, manual labor occupations, or they may be idiopathic.
Race can play a role in CK levels as well. Black people tend to have higher CK levels than white people, she said, noting that one study of more than 10,000 adults showed that black race was strongly associated with CK, and that body composition largely explained differences in CK by age, but not by race/ethnicity (Medicine. Aug 2016;95[33]:e4344).
“So elevated CK may not herald any discernible illness,” she said.
Dr. Christopher-Stine described a case involving an otherwise healthy 30-year-old man with a CK level of 695 IU/L that was found incidentally. He had a desk job, no recent travel, and denied weakness, myalgias, joint pain, dysphagia, shortness of breath, and fevers. In this case, the elevated CK was felt to be secondary to his African American race given that other causes were ruled out.
Another case involved a 72-year-old man with left-arm pain. A cardiac event was ruled out, and CK was found to be about 4,500 IU/L. He reported “flare-ups” of diffuse swelling of the hands and feet. X-rays showed concerning signs of erosions. His transaminases and electromyogram were normal; he reported no weakness or myalgia; and an MRI showed no muscle edema. He was diagnosed with macro-CK, which refers to CK with an increased molecular weight. A clue to this diagnosis is a normal liver function test. In some cases, muscle/brain CK levels (CK-MB) are elevated and higher than total CK, she noted.
She presented an algorithm for the diagnostic work-up of patients presenting with elevated CK of unclear significance. Her recommended approach involves repeat CK assessment and a closer look at family history, medication, drug/toxin history, examination for weakness and neurologic abnormalities, and additional lab assessments in those whose levels remain elevated. In those in whom a diagnosis is not identified, the algorithm calls for observation every 3 months – including physical examination and labs – in asymptomatic patients with levels at less than five times the upper limit of normal, and further evaluation, including EMG with nerve-conduction velocity testing, muscle biopsy, and MRI in those with (or who later develop) marked elevation greater than five times the upper limit of normal and/or symptoms.
Patient assessment
The physical examination should involve localization and quantification of weakness, and assessment for fever, rash, atrophy/wasting/scooping of forearms, fasciculations, cranial nerve involvement, Raynaud’s phenomenon, nailfold capillary changes, arthritis, calcinosis, “mechanic’s hands,” signs of other autoimmune diseases, and lung crackles. Initial laboratory testing should include HIV and hepatitis B and C testing; measurement of CK, AST, ALT, aldolase, thyroid-stimulating hormone, and magnesium levels; a comprehensive metabolic panel and complete blood count; and measurement of erythrocyte sedimentation rate and C-reactive protein.
“Weakness may be secondary to a neuropathy, myopathy, or a problem at the neuropathic junction. Many causes of weakness can be readily identified by careful history taking, focused physical examination, and directed laboratory evaluation,” she said.
Features pointing toward a diagnosis of myositis include characteristic rashes, gradual symptom onset, proximal limb and truncal weakness, other connective tissue disease features such as Raynaud’s and arthritis, and the presence of lung disease, including interstitial lung disease or unexplained infiltrates, she said.
Features pointing away from a diagnosis of myositis include a family history of a similar illness, weakness that is associated with eating or fasting, neurologic signs, cranial nerve involvement, fasciculations, severe muscle cramping, early atrophy, and creatine phosphokinase levels that are either less than 2 times or more than 100 times the upper limit of normal.
Among the conditions to consider in the presence of the features that point away from a myositis diagnosis are muscular dystrophies, metabolic myopathies, and toxic (drug-induced) myopathies, to name a few, Dr. Christopher-Stine said.
She described a number of other cases to illustrate the need for – and to help develop – a differential diagnosis in patients presenting with apparent myositis.
Muscular dystrophies
A 38-year-old woman with limited scleroderma and anti-PM/Scl autoantibodies developed proximal weakness over 9 months and was eventually unable to walk up a flight of stairs. She had heliotrope rash and Gottron’s sign, her serum CK was 723 IU/L, and EMG showed an irritable myopathy.
Muscle biopsy showed inflammation, and she was treated with prednisone, but this led to worsening weakness. She complained of prominent fatigue and double vision at the end of the day, and these symptoms did not improve with steroids.
Anti-AChR and anti-MuSK antibodies were negative, but she had a decrement on repetitive nerve stimulation testing.
She was treated with pyridostigmine and experienced near-complete resolution of her proximal weakness and double vision. A chest CT scan showed thymic hyperplasia; thymectomy was recommended.
In another case, a 19-year-old woman who complained of leg pain after exercise was found to have intact strength but asymmetric calf hypertrophy. Her CK level was 5,000 IU/L, and she was referred to rule out acute myositis.
A quadriceps biopsy was performed and showed abnormal dystrophin immunostaining but no inflammation. A molecular genetic analysis showed deletions in Xp21 and she was diagnosed as a manifesting carrier of Duchenne muscular dystrophy. It was recommended that she be evaluated for cardiomyopathy and receive genetic counseling.
A number of other cases presented by Dr. Christopher-Stine highlighted other muscular dystrophies that can mimic myositis, such as:
- Myotonic dystrophies. These are more often type 2 than type 1. Myotonia may be subtle, cataracts are seen early in all patients, and cardiac arrhythmias are common.
- Limb girdle muscular dystrophy type 2 B (dysferlinopathy). In the legs, this often affects the gastrocnemius muscle, and this will be visible on MRI. In the arms, it most often affects the biceps, sparing the deltoids. CKs are typically very high.
- Facioscapulohumeral muscular dystrophy (FSHD). This involves facial weakness, especially obicularis oris, in 95% of cases, as well as scapular weakness and winging, inflammation on muscle biopsy in 75% of cases, and typically is endomysial or perivascular.
Metabolic myopathies
Among metabolic myopathies that can mimic myositis are disorders of carbohydrate metabolism such as McArdle’s disease, 6-phosphofructokinase deficiency, and Pompe’s disease (adult acid maltase deficiency); disorders of lipid metabolism such as carnitine deficiency and carnitine palmitoyltransferase 2 (CPT2) deficiency; and disorders of purine metabolism, such as myoadenylate deaminase deficiency.
A 27-year-old patient who complained of weakness with activity was referred for possible myositis and was found to have a CK of 3,650 IU/L that never normalized. Physical examination showed intact strength and no muscle atrophy or fasciculations, and an enzyme stain for myophosphorylase showed a normal staining pattern and complete absence of the enzyme on quadricep biopsy. A 22-year-old man with similar symptoms plus recent onset of brown/black urine after physical activity had CK of 110,000 IU/L when symptomatic, and also underwent biopsy after being referred for possible myopathy. Both patients were ultimately diagnosed with CPT2 deficiency, which is associated with risk of rhabdomyolysis triggered by prolonged exercise, diets low in carbohydrates and high in fat, or by fasting.
Myalgias are common, and CK levels are normal or only mildly elevated between episodes in CPT2 deficiency, Dr. Christopher-Stine noted.
Toxic myopathies
Drug-induced myopathies are among the most common etiologies of myopathy and can range from mild myalgia to massive rhabdomyolysis. They can cause mild to severe weakness and may be chronic. The mechanism of toxic injury is direct via myotoxins such as ethyl alcohol, glucocorticoids, lipid-lowering drugs, cocaine, antimalarial drugs, antipsychotic drugs, colchicine, and Ipecac syrup.
One case described by Dr. Christopher-Stine involved “statin myopathy.”
A 55-year-old man on atorvastatin complained of myalgias and brown urine, but had no definitive weakness. He had intact strength and diffuse myalgias that weren’t reproducible. His CK was 45,000 IU/L.
Statin myopathy, as seen in this patient, is usually self-limited and is not associated with autoimmunity or with anti-HMGCR autoantibody positivity.
The mechanism is unknown, but statin myopathy has an incidence of 1.2 per 10,000 patient-years. Myalgias, myositis, rhabdomyolysis, and asymptomatic hyperCKemia are commonly seen. This is in contrast to the immune-mediated necrotizing myelitis that can be secondary to statins and is responsive to immunosuppression, she noted.
Other myositis mimics
In addition to these common myositis mimics, certain other neurologic diseases (such as ALS and cervical myelopathy), endocrinopathies (such as hypothyroidism), and infections (like toxoplasmosis) can also be mistaken for myositis, Dr. Christopher-Stine said, noting that cases illustrating these mimics underscore the need for careful consideration of possible alternate diagnoses.
“While most noninflammatory myopathies are self-limited or have no therapies available, knowing the diagnosis can be helpful for genetic counseling of the patient and family, for mitigating risk factors, and for precluding the use of unwarranted immunosuppressive agents,” she said.
Dr. Christopher-Stine reported having intellectual property interest in a novel Inova Diagnostics autoantibody assay detection for anti-HMGCR. She was also the safety officer for the JBT-101 Trial sponsored by Corbus and funded by the National Institutes of Health.
A number of conditions can mimic myositis, but clues that can point to the correct diagnosis are often present in cases involving the mimics, according to Lisa Christopher-Stine, MD.
For example, elevated levels of certain muscle enzymes are an important source of diagnostic information, Dr. Christopher-Stine, director of the Johns Hopkins Myositis Center, Baltimore, said at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
Isolated elevations in aldolase can be seen in connective tissue–associated interstitial lung disease or in patients with fascial edema, and aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase (LDH), and creatine kinase (CK) levels can also be helpful, she explained.
The latter can also be elevated in the absence of muscle disease, for example, in healthy individuals following exercise. CK peaks at 24 hours after exercise before returning to baseline by 72 hours. In an experimental setting, a threefold increase in CK levels has been seen at 8-24 hours after exercise, Dr. Christopher-Stine said.
“HyperCKemia”
Trauma from causes such as intramuscular injection, electromyography (EMG), major surgery, or biopsy can also lead to increased CK levels. Motor neuron disease can also cause such increases. In one study, 75% of patients with amyotrophic lateral sclerosis had a mean twofold increase in CK levels, she said.
Asymptomatic CK elevations may also represent presymptomatic myopathies, type 1 or 2 macro-CK, manual labor occupations, or they may be idiopathic.
Race can play a role in CK levels as well. Black people tend to have higher CK levels than white people, she said, noting that one study of more than 10,000 adults showed that black race was strongly associated with CK, and that body composition largely explained differences in CK by age, but not by race/ethnicity (Medicine. Aug 2016;95[33]:e4344).
“So elevated CK may not herald any discernible illness,” she said.
Dr. Christopher-Stine described a case involving an otherwise healthy 30-year-old man with a CK level of 695 IU/L that was found incidentally. He had a desk job, no recent travel, and denied weakness, myalgias, joint pain, dysphagia, shortness of breath, and fevers. In this case, the elevated CK was felt to be secondary to his African American race given that other causes were ruled out.
Another case involved a 72-year-old man with left-arm pain. A cardiac event was ruled out, and CK was found to be about 4,500 IU/L. He reported “flare-ups” of diffuse swelling of the hands and feet. X-rays showed concerning signs of erosions. His transaminases and electromyogram were normal; he reported no weakness or myalgia; and an MRI showed no muscle edema. He was diagnosed with macro-CK, which refers to CK with an increased molecular weight. A clue to this diagnosis is a normal liver function test. In some cases, muscle/brain CK levels (CK-MB) are elevated and higher than total CK, she noted.
She presented an algorithm for the diagnostic work-up of patients presenting with elevated CK of unclear significance. Her recommended approach involves repeat CK assessment and a closer look at family history, medication, drug/toxin history, examination for weakness and neurologic abnormalities, and additional lab assessments in those whose levels remain elevated. In those in whom a diagnosis is not identified, the algorithm calls for observation every 3 months – including physical examination and labs – in asymptomatic patients with levels at less than five times the upper limit of normal, and further evaluation, including EMG with nerve-conduction velocity testing, muscle biopsy, and MRI in those with (or who later develop) marked elevation greater than five times the upper limit of normal and/or symptoms.
Patient assessment
The physical examination should involve localization and quantification of weakness, and assessment for fever, rash, atrophy/wasting/scooping of forearms, fasciculations, cranial nerve involvement, Raynaud’s phenomenon, nailfold capillary changes, arthritis, calcinosis, “mechanic’s hands,” signs of other autoimmune diseases, and lung crackles. Initial laboratory testing should include HIV and hepatitis B and C testing; measurement of CK, AST, ALT, aldolase, thyroid-stimulating hormone, and magnesium levels; a comprehensive metabolic panel and complete blood count; and measurement of erythrocyte sedimentation rate and C-reactive protein.
“Weakness may be secondary to a neuropathy, myopathy, or a problem at the neuropathic junction. Many causes of weakness can be readily identified by careful history taking, focused physical examination, and directed laboratory evaluation,” she said.
Features pointing toward a diagnosis of myositis include characteristic rashes, gradual symptom onset, proximal limb and truncal weakness, other connective tissue disease features such as Raynaud’s and arthritis, and the presence of lung disease, including interstitial lung disease or unexplained infiltrates, she said.
Features pointing away from a diagnosis of myositis include a family history of a similar illness, weakness that is associated with eating or fasting, neurologic signs, cranial nerve involvement, fasciculations, severe muscle cramping, early atrophy, and creatine phosphokinase levels that are either less than 2 times or more than 100 times the upper limit of normal.
Among the conditions to consider in the presence of the features that point away from a myositis diagnosis are muscular dystrophies, metabolic myopathies, and toxic (drug-induced) myopathies, to name a few, Dr. Christopher-Stine said.
She described a number of other cases to illustrate the need for – and to help develop – a differential diagnosis in patients presenting with apparent myositis.
Muscular dystrophies
A 38-year-old woman with limited scleroderma and anti-PM/Scl autoantibodies developed proximal weakness over 9 months and was eventually unable to walk up a flight of stairs. She had heliotrope rash and Gottron’s sign, her serum CK was 723 IU/L, and EMG showed an irritable myopathy.
Muscle biopsy showed inflammation, and she was treated with prednisone, but this led to worsening weakness. She complained of prominent fatigue and double vision at the end of the day, and these symptoms did not improve with steroids.
Anti-AChR and anti-MuSK antibodies were negative, but she had a decrement on repetitive nerve stimulation testing.
She was treated with pyridostigmine and experienced near-complete resolution of her proximal weakness and double vision. A chest CT scan showed thymic hyperplasia; thymectomy was recommended.
In another case, a 19-year-old woman who complained of leg pain after exercise was found to have intact strength but asymmetric calf hypertrophy. Her CK level was 5,000 IU/L, and she was referred to rule out acute myositis.
A quadriceps biopsy was performed and showed abnormal dystrophin immunostaining but no inflammation. A molecular genetic analysis showed deletions in Xp21 and she was diagnosed as a manifesting carrier of Duchenne muscular dystrophy. It was recommended that she be evaluated for cardiomyopathy and receive genetic counseling.
A number of other cases presented by Dr. Christopher-Stine highlighted other muscular dystrophies that can mimic myositis, such as:
- Myotonic dystrophies. These are more often type 2 than type 1. Myotonia may be subtle, cataracts are seen early in all patients, and cardiac arrhythmias are common.
- Limb girdle muscular dystrophy type 2 B (dysferlinopathy). In the legs, this often affects the gastrocnemius muscle, and this will be visible on MRI. In the arms, it most often affects the biceps, sparing the deltoids. CKs are typically very high.
- Facioscapulohumeral muscular dystrophy (FSHD). This involves facial weakness, especially obicularis oris, in 95% of cases, as well as scapular weakness and winging, inflammation on muscle biopsy in 75% of cases, and typically is endomysial or perivascular.
Metabolic myopathies
Among metabolic myopathies that can mimic myositis are disorders of carbohydrate metabolism such as McArdle’s disease, 6-phosphofructokinase deficiency, and Pompe’s disease (adult acid maltase deficiency); disorders of lipid metabolism such as carnitine deficiency and carnitine palmitoyltransferase 2 (CPT2) deficiency; and disorders of purine metabolism, such as myoadenylate deaminase deficiency.
A 27-year-old patient who complained of weakness with activity was referred for possible myositis and was found to have a CK of 3,650 IU/L that never normalized. Physical examination showed intact strength and no muscle atrophy or fasciculations, and an enzyme stain for myophosphorylase showed a normal staining pattern and complete absence of the enzyme on quadricep biopsy. A 22-year-old man with similar symptoms plus recent onset of brown/black urine after physical activity had CK of 110,000 IU/L when symptomatic, and also underwent biopsy after being referred for possible myopathy. Both patients were ultimately diagnosed with CPT2 deficiency, which is associated with risk of rhabdomyolysis triggered by prolonged exercise, diets low in carbohydrates and high in fat, or by fasting.
Myalgias are common, and CK levels are normal or only mildly elevated between episodes in CPT2 deficiency, Dr. Christopher-Stine noted.
Toxic myopathies
Drug-induced myopathies are among the most common etiologies of myopathy and can range from mild myalgia to massive rhabdomyolysis. They can cause mild to severe weakness and may be chronic. The mechanism of toxic injury is direct via myotoxins such as ethyl alcohol, glucocorticoids, lipid-lowering drugs, cocaine, antimalarial drugs, antipsychotic drugs, colchicine, and Ipecac syrup.
One case described by Dr. Christopher-Stine involved “statin myopathy.”
A 55-year-old man on atorvastatin complained of myalgias and brown urine, but had no definitive weakness. He had intact strength and diffuse myalgias that weren’t reproducible. His CK was 45,000 IU/L.
Statin myopathy, as seen in this patient, is usually self-limited and is not associated with autoimmunity or with anti-HMGCR autoantibody positivity.
The mechanism is unknown, but statin myopathy has an incidence of 1.2 per 10,000 patient-years. Myalgias, myositis, rhabdomyolysis, and asymptomatic hyperCKemia are commonly seen. This is in contrast to the immune-mediated necrotizing myelitis that can be secondary to statins and is responsive to immunosuppression, she noted.
Other myositis mimics
In addition to these common myositis mimics, certain other neurologic diseases (such as ALS and cervical myelopathy), endocrinopathies (such as hypothyroidism), and infections (like toxoplasmosis) can also be mistaken for myositis, Dr. Christopher-Stine said, noting that cases illustrating these mimics underscore the need for careful consideration of possible alternate diagnoses.
“While most noninflammatory myopathies are self-limited or have no therapies available, knowing the diagnosis can be helpful for genetic counseling of the patient and family, for mitigating risk factors, and for precluding the use of unwarranted immunosuppressive agents,” she said.
Dr. Christopher-Stine reported having intellectual property interest in a novel Inova Diagnostics autoantibody assay detection for anti-HMGCR. She was also the safety officer for the JBT-101 Trial sponsored by Corbus and funded by the National Institutes of Health.
EXPERT ANALYSIS FROM THE WINTER RHEUMATOLOGY SYMPOSIUM
Amyloid PET may help facilitate diagnosis of inclusion body myositis
Amyloid PET imaging may help to accurately identify inclusion body myositis and distinguish it from polymyositis, which could potentially avoid misdiagnosis and unnecessary immunosuppressive medication, according to findings from a small prospective cohort study.
The study, which found significantly greater uptake of the imaging agent [18F]florbetapir (Amyvid) in muscle from patients with inclusion body myositis (IBM) than in those with polymyositis (PM), builds on previous research showing that the intramuscular beta-amyloid seen in histopathologic analysis of IBM can help distinguish it from PM, but this approach has a low sensitivity along with high diagnostic specificity. The latest diagnostic criteria for IBM have also shifted away from strict histopathologic analysis toward identifying its characteristic clinical pattern of muscle weakness, first author James B. Lilleker, PhD, of the Centre for Musculoskeletal Research at the University of Manchester (England) and his colleagues wrote in Annals of the Rheumatic Diseases.
“While this has improved sensitivity, clinically detectable weakness implies that significant and irreversible muscle damage has occurred, reducing the likelihood that novel treatments will be effective,” the researchers wrote.

Thomas E. Lloyd II, MD, PhD, codirector of the myositis center at Johns Hopkins University, Baltimore, said in an interview that IBM is difficult to diagnose because of a lack of awareness of the early clinical signs and symptoms of the disease among primary care physicians, neurologists, and rheumatologists.
“Typically, a myositis specialist can make the diagnosis based on a combination of history and exam [slowly progressive weakness affecting distal finger flexors and knee extensors] and muscle biopsy features [endomysial inflammation with rimmed vacuoles and protein aggregates],” said Dr. Lloyd, who was not involved in the current study. “However, early in the course of disease, some patients may be misdiagnosed with polymyositis due to having atypical clinical or pathological features, especially if the muscle biopsy lacks rimmed vacuoles or the patient lacks obvious finger flexor weakness.”
But amyloid PET is promising and needs further analysis comparing it with expert diagnosis, Dr. Lloyd said. “I think this imaging method has greatest potential utility to be helpful diagnostically early in the course of disease, when diagnosis of IBM can be most challenging. This approach may also have potential to identify presymptomatic patients, especially if amyloid imaging becomes used for screening for Alzheimer’s disease in the future.”
Dr. Lilleker and colleagues set out to determine whether amyloid PET with [18F]florbetapir could distinguish between IBM and PM. They identified 10 patients with IBM and 6 patients with PM and scanned each patient from shoulders to ankles with CT and PET, followed by a same-day, whole-body MRI scan. Overall, regions of interest included the left arm, right and left forearms, right and left thighs, and right and left calves. The researchers calculated the [18F]florbetapir standard uptake values (SUVs) for each region of interest while SUV ratios (SUVRs) were calculated using the lumbar fat pad reference region.
The IBM patients (9 men, 1 woman) had a mean age of 68.3 years at the time of the scan and a mean disease duration of 4 years, and they were not currently taking immunosuppressive treatments; however, patients had previously taken prednisolone (3 of 10 patients), azathioprine (1 of 10 patients), and mycophenolate (1 of 10 patients). In the PM group (4 men, 2 women), patients’ mean age was 59.7 years, with a mean disease duration of 1.5 years.
The researchers found significantly increased overall [18F]florbetapir SUVRs among all regions of interest for IBM patients (median total SUVR, 1.45; interquartile range, 1.28-2.05), compared with PM patients (total SUVR, 1.01; IQR, 0.80-1.22; P = .005). In addition, when total [18F]florbetapir SUVR was 1.28 or greater, the diagnostic sensitivity for IBM was 80% and specificity was 100%, with an area under the curve of 0.93. There were also no significant associations between total [18F]florbetapir SUVR and age at the time of the scan, disease duration, or clinical outcome measurements such as manual muscle testing of 26 muscles (MMT26), Health Assessment Questionnaire disability index, and IBM Functional Rating Scale.
Dr. Lilleker and his colleagues noted the small study size as a potential limitation, but said other factors such as age and disease severity, which differed between groups, were unlikely to affect the [18F]florbetapir SUVR.
The rarity of PM may make it difficult to determine amyloid PET imaging’s effectiveness in diagnosing IBM when compared with more traditional methods, Dr. Lloyd noted.
“Whether PET imaging is more sensitive than expert diagnosis using traditional methods [careful physical exam of individual distal interphalangeal finger flexor muscles, MRI imaging of thighs, detailed pathological analysis of muscle biopsy including immunostaining for p62/SQSTM1, detailed serum autoantibody testing, etc.] remains to be determined,” he said.
This study was supported by grants from the National Institute for Health Research Manchester Musculoskeletal Biomedical Research Centre, the Medical Research Council, and an award from the Centre for Imaging Sciences at the University of Manchester. The authors reported no conflicts of interest. Dr. Lloyd reported being a consultant for Acceleron and principal investigator for IBM clinical trials sponsored by Orphazyme and Regeneron.
SOURCE: Lilleker JB et al. Ann Rheum Dis. 2019 Feb 13. doi: 10.1136/annrheumdis-2018-214644.
Amyloid PET imaging may help to accurately identify inclusion body myositis and distinguish it from polymyositis, which could potentially avoid misdiagnosis and unnecessary immunosuppressive medication, according to findings from a small prospective cohort study.
The study, which found significantly greater uptake of the imaging agent [18F]florbetapir (Amyvid) in muscle from patients with inclusion body myositis (IBM) than in those with polymyositis (PM), builds on previous research showing that the intramuscular beta-amyloid seen in histopathologic analysis of IBM can help distinguish it from PM, but this approach has a low sensitivity along with high diagnostic specificity. The latest diagnostic criteria for IBM have also shifted away from strict histopathologic analysis toward identifying its characteristic clinical pattern of muscle weakness, first author James B. Lilleker, PhD, of the Centre for Musculoskeletal Research at the University of Manchester (England) and his colleagues wrote in Annals of the Rheumatic Diseases.
“While this has improved sensitivity, clinically detectable weakness implies that significant and irreversible muscle damage has occurred, reducing the likelihood that novel treatments will be effective,” the researchers wrote.
Thomas E. Lloyd II, MD, PhD, codirector of the myositis center at Johns Hopkins University, Baltimore, said in an interview that IBM is difficult to diagnose because of a lack of awareness of the early clinical signs and symptoms of the disease among primary care physicians, neurologists, and rheumatologists.
“Typically, a myositis specialist can make the diagnosis based on a combination of history and exam [slowly progressive weakness affecting distal finger flexors and knee extensors] and muscle biopsy features [endomysial inflammation with rimmed vacuoles and protein aggregates],” said Dr. Lloyd, who was not involved in the current study. “However, early in the course of disease, some patients may be misdiagnosed with polymyositis due to having atypical clinical or pathological features, especially if the muscle biopsy lacks rimmed vacuoles or the patient lacks obvious finger flexor weakness.”
But amyloid PET is promising and needs further analysis comparing it with expert diagnosis, Dr. Lloyd said. “I think this imaging method has greatest potential utility to be helpful diagnostically early in the course of disease, when diagnosis of IBM can be most challenging. This approach may also have potential to identify presymptomatic patients, especially if amyloid imaging becomes used for screening for Alzheimer’s disease in the future.”
Dr. Lilleker and colleagues set out to determine whether amyloid PET with [18F]florbetapir could distinguish between IBM and PM. They identified 10 patients with IBM and 6 patients with PM and scanned each patient from shoulders to ankles with CT and PET, followed by a same-day, whole-body MRI scan. Overall, regions of interest included the left arm, right and left forearms, right and left thighs, and right and left calves. The researchers calculated the [18F]florbetapir standard uptake values (SUVs) for each region of interest while SUV ratios (SUVRs) were calculated using the lumbar fat pad reference region.
The IBM patients (9 men, 1 woman) had a mean age of 68.3 years at the time of the scan and a mean disease duration of 4 years, and they were not currently taking immunosuppressive treatments; however, patients had previously taken prednisolone (3 of 10 patients), azathioprine (1 of 10 patients), and mycophenolate (1 of 10 patients). In the PM group (4 men, 2 women), patients’ mean age was 59.7 years, with a mean disease duration of 1.5 years.
The researchers found significantly increased overall [18F]florbetapir SUVRs among all regions of interest for IBM patients (median total SUVR, 1.45; interquartile range, 1.28-2.05), compared with PM patients (total SUVR, 1.01; IQR, 0.80-1.22; P = .005). In addition, when total [18F]florbetapir SUVR was 1.28 or greater, the diagnostic sensitivity for IBM was 80% and specificity was 100%, with an area under the curve of 0.93. There were also no significant associations between total [18F]florbetapir SUVR and age at the time of the scan, disease duration, or clinical outcome measurements such as manual muscle testing of 26 muscles (MMT26), Health Assessment Questionnaire disability index, and IBM Functional Rating Scale.
Dr. Lilleker and his colleagues noted the small study size as a potential limitation, but said other factors such as age and disease severity, which differed between groups, were unlikely to affect the [18F]florbetapir SUVR.
The rarity of PM may make it difficult to determine amyloid PET imaging’s effectiveness in diagnosing IBM when compared with more traditional methods, Dr. Lloyd noted.
“Whether PET imaging is more sensitive than expert diagnosis using traditional methods [careful physical exam of individual distal interphalangeal finger flexor muscles, MRI imaging of thighs, detailed pathological analysis of muscle biopsy including immunostaining for p62/SQSTM1, detailed serum autoantibody testing, etc.] remains to be determined,” he said.
This study was supported by grants from the National Institute for Health Research Manchester Musculoskeletal Biomedical Research Centre, the Medical Research Council, and an award from the Centre for Imaging Sciences at the University of Manchester. The authors reported no conflicts of interest. Dr. Lloyd reported being a consultant for Acceleron and principal investigator for IBM clinical trials sponsored by Orphazyme and Regeneron.
SOURCE: Lilleker JB et al. Ann Rheum Dis. 2019 Feb 13. doi: 10.1136/annrheumdis-2018-214644.
Amyloid PET imaging may help to accurately identify inclusion body myositis and distinguish it from polymyositis, which could potentially avoid misdiagnosis and unnecessary immunosuppressive medication, according to findings from a small prospective cohort study.
The study, which found significantly greater uptake of the imaging agent [18F]florbetapir (Amyvid) in muscle from patients with inclusion body myositis (IBM) than in those with polymyositis (PM), builds on previous research showing that the intramuscular beta-amyloid seen in histopathologic analysis of IBM can help distinguish it from PM, but this approach has a low sensitivity along with high diagnostic specificity. The latest diagnostic criteria for IBM have also shifted away from strict histopathologic analysis toward identifying its characteristic clinical pattern of muscle weakness, first author James B. Lilleker, PhD, of the Centre for Musculoskeletal Research at the University of Manchester (England) and his colleagues wrote in Annals of the Rheumatic Diseases.
“While this has improved sensitivity, clinically detectable weakness implies that significant and irreversible muscle damage has occurred, reducing the likelihood that novel treatments will be effective,” the researchers wrote.
Thomas E. Lloyd II, MD, PhD, codirector of the myositis center at Johns Hopkins University, Baltimore, said in an interview that IBM is difficult to diagnose because of a lack of awareness of the early clinical signs and symptoms of the disease among primary care physicians, neurologists, and rheumatologists.
“Typically, a myositis specialist can make the diagnosis based on a combination of history and exam [slowly progressive weakness affecting distal finger flexors and knee extensors] and muscle biopsy features [endomysial inflammation with rimmed vacuoles and protein aggregates],” said Dr. Lloyd, who was not involved in the current study. “However, early in the course of disease, some patients may be misdiagnosed with polymyositis due to having atypical clinical or pathological features, especially if the muscle biopsy lacks rimmed vacuoles or the patient lacks obvious finger flexor weakness.”
But amyloid PET is promising and needs further analysis comparing it with expert diagnosis, Dr. Lloyd said. “I think this imaging method has greatest potential utility to be helpful diagnostically early in the course of disease, when diagnosis of IBM can be most challenging. This approach may also have potential to identify presymptomatic patients, especially if amyloid imaging becomes used for screening for Alzheimer’s disease in the future.”
Dr. Lilleker and colleagues set out to determine whether amyloid PET with [18F]florbetapir could distinguish between IBM and PM. They identified 10 patients with IBM and 6 patients with PM and scanned each patient from shoulders to ankles with CT and PET, followed by a same-day, whole-body MRI scan. Overall, regions of interest included the left arm, right and left forearms, right and left thighs, and right and left calves. The researchers calculated the [18F]florbetapir standard uptake values (SUVs) for each region of interest while SUV ratios (SUVRs) were calculated using the lumbar fat pad reference region.
The IBM patients (9 men, 1 woman) had a mean age of 68.3 years at the time of the scan and a mean disease duration of 4 years, and they were not currently taking immunosuppressive treatments; however, patients had previously taken prednisolone (3 of 10 patients), azathioprine (1 of 10 patients), and mycophenolate (1 of 10 patients). In the PM group (4 men, 2 women), patients’ mean age was 59.7 years, with a mean disease duration of 1.5 years.
The researchers found significantly increased overall [18F]florbetapir SUVRs among all regions of interest for IBM patients (median total SUVR, 1.45; interquartile range, 1.28-2.05), compared with PM patients (total SUVR, 1.01; IQR, 0.80-1.22; P = .005). In addition, when total [18F]florbetapir SUVR was 1.28 or greater, the diagnostic sensitivity for IBM was 80% and specificity was 100%, with an area under the curve of 0.93. There were also no significant associations between total [18F]florbetapir SUVR and age at the time of the scan, disease duration, or clinical outcome measurements such as manual muscle testing of 26 muscles (MMT26), Health Assessment Questionnaire disability index, and IBM Functional Rating Scale.
Dr. Lilleker and his colleagues noted the small study size as a potential limitation, but said other factors such as age and disease severity, which differed between groups, were unlikely to affect the [18F]florbetapir SUVR.
The rarity of PM may make it difficult to determine amyloid PET imaging’s effectiveness in diagnosing IBM when compared with more traditional methods, Dr. Lloyd noted.
“Whether PET imaging is more sensitive than expert diagnosis using traditional methods [careful physical exam of individual distal interphalangeal finger flexor muscles, MRI imaging of thighs, detailed pathological analysis of muscle biopsy including immunostaining for p62/SQSTM1, detailed serum autoantibody testing, etc.] remains to be determined,” he said.
This study was supported by grants from the National Institute for Health Research Manchester Musculoskeletal Biomedical Research Centre, the Medical Research Council, and an award from the Centre for Imaging Sciences at the University of Manchester. The authors reported no conflicts of interest. Dr. Lloyd reported being a consultant for Acceleron and principal investigator for IBM clinical trials sponsored by Orphazyme and Regeneron.
SOURCE: Lilleker JB et al. Ann Rheum Dis. 2019 Feb 13. doi: 10.1136/annrheumdis-2018-214644.
FROM ANNALS OF THE RHEUMATIC DISEASES
Dr. Lisa Christopher-Stine: Polymyositis? It’s more likely something else
“When someone refers you [a patient with suspected] polymyositis, I want you to do a checklist in your head and say, ‘Have I thought about these five things?’ ” Dr. Christopher-Stine, director of the Johns Hopkins Myositis Center, Baltimore, said at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
The five most common diagnoses in patients labeled as having polymyositis are immune-mediated necrotizing myopathy (IMNM), overlap with other rheumatologic conditions, antisynthetase syndrome, inclusion body myositis (IBM), and muscular dystrophy, she explained.
“You may say, ‘look, it’s all what you call it,’ but I think we need to be a little bit more careful in what we call it,” she said.
IMNM
Patients with IMNM present with clinical symptoms similar to those seen in polymyositis and dermatomyositis – mainly proximal muscle weakness.
However, there are some important differences, both clinically and histologically, Dr. Christopher-Stine said.
“Look for higher [creatine kinase (CK)] levels,” she said. “In the thousands, usually multiple thousands ... like 5,000, 10,000, 2,000 ... that’s when you’re thinking about a necrotizing phenotype before you even look at the biopsy.”
CK levels will usually be under 30,000 U/L in IMNM, she noted, adding that data increasingly suggest that the extensive muscle necrosis in IMNM explains the elevated CK levels versus those seen in other myopathies.
Myalgias also tend to be more prominent in IMNM than in polymyositis.
“These folks hurt,” she said, noting that IMNM patients tend to have more extensive muscle atrophy and functional disability. “Many will be wheelchair bound within 9 months of diagnosis; it’s not subtle.”
The most important tool for making an IMNM diagnosis is muscle biopsy; look for prominent myocyte necrosis and a relative paucity of lymphocytes, she advised.
Overlap
Sometimes patients with polymyositis also have other rheumatologic conditions that shouldn’t be overlooked, therefore “overlap is its own category,” she said.
“In our experience, the most common overlap is scleroderma,” she noted, adding that the scleroderma is often, but not always, subtle, and that there may be overlapping autoantibodies.
Overt sclerodactyly is rarely seen, although a small amount may be present, but significant Raynaud’s phenomenon is common in these patients, and tiny telangiectasias across the neck are a tell-tale sign.
“Why does that matter? It’s not an esoteric argument; those are the folks that go on to have pulmonary hypertension,” she said. “They can have the same [interstitial lung disease] and all of the other internal scleroderma manifestations.”
Think about overlap and “look close phenotypically and with antibodies,” she advised.
There is also “the typical RA seropositive overlap,” she said, but lupus only rarely overlaps with myositis.
“However, the next diagnosis on the list – antisynthetase syndrome – can be a forme fruste where you first see a seronegative RA-like picture, and it’s important to think about that as well,” she said.
Antisynthetase syndrome
In patients referred for polymyositis, it’s also important to evaluate for antisynthetase syndrome, Dr. Christopher-Stine said.
The arthritis seen in the extramuscular phenotype of the syndrome is rarely deforming, but despite what many physicians were taught, “it absolutely can be erosive,” she said.
In fact, 40% of people with this syndrome present with an isolated forme fruste seronegative rheumatoid arthritis, she said.
Roughening and desquamation of the skin on the radial surface of fingers or palms – a sign known as mechanic’s hands – that doesn’t have another identifiable cause suggests this diagnosis in patients with this type of arthritis, as does interstitial lung disease and Raynaud’s phenomenon.
The Raynaud’s can be “fairly significant in the sense that it is bothersome,” but it usually doesn’t lead to ulceration or digital necrosis.
This is different from what is seen with the scleroderma phenotype, she said, adding that “if you’re starting to see gangrene and digital loss, think of something else.”
IBM
IBM is “probably the No. 1 most-missed diagnosis” among patients referred for what is initially believed to be polymyositis, Dr. Christopher-Stine said.
“I used to think that this was missed at entry, that everybody [with IBM] had all of these criteria and that rheumatologists really didn’t understand this phenotype ... but some people morph into this,” she said, explaining that they often start out looking like they have polymyositis with proximal muscle weakness.
“They may even initially respond to steroids. And then they get this phenotype,” she said.
Older men are more likely to present with the phenotype from the beginning; women, in her experience, tend to present with what appears to be polymyositis, and then develop the phenotype over time, she noted.
An IBM diagnosis requires age over 30 years, but most patients are over 50, she said.
“This is the only one of the myopathies that is preferential to men,” she added, noting that it affects men twice as often as it does women.
The syndrome is characterized by proximal strength loss and muscle atrophy. Also, a finding that a patient’s knee extensors are weaker than their hip flexors is “a fantastic bedside sign” differentiating IBM from polymyositis, she said.
That’s not to say IBM patients don’t have hip flexor weakness, but their knee extensors usually are “considerably weaker by a grade strength or more” versus their hip flexors, she explained.
“It’s a very easy bedside test. In typical other myopathies we have this, but the knee extensors aren’t that weak in general, or they’re not as weak as the hip flexors,” she added.
Another sign is distal strength loss, particularly in the forearm and finger flexors.
“I was taught to have them make a fist; don’t have them make a fist,” she said, explaining that this recruits intrinsic muscles which basically allows cheating that may mask weakness.
Instead, ask them to flex just their distal interphalangeal joints by making a claw and using the fingers to pull against your fingers, she suggested.
Mixed myopathic and neuropathic features on electromyography also indicate IBM, she said.
Muscle biopsy may be helpful, but inclusions are seen in less than one-third of IBM patients.
“At times, we have had to biopsy three times to see them at all, and some people never show them, so you have to rely on your clinical acumen if you don’t see them,” she said.
Also, keep in mind that these patients are often labeled as having treatment-resistant polymyositis.
“Please, when somebody refers to you somebody that’s treatment resistant, that may be the case, but I want you to think maybe they’re treatment resistant because they don’t have that disease.”
Muscular dystrophy
Some cases of myositis mimic certain types of muscular dystrophy, Dr. Christopher-Stine said, providing a checklist of muscular dystrophies that can look “clinically completely indistinguishable from a typical inflammatory myopathy,” and should therefore be considered in these patients.
The checklist includes Duchenne’s manifesting carrier, limb girdle muscular dystrophy type 2b, myotonic dystrophy (usually type 2), and facioscapulohumeral muscular dystrophy.
Dr. Christopher-Stine reported having intellectual property interest in a novel Inova Diagnostics autoantibody assay detection for anti-HMGCR. She was also the safety officer for the JBT-101 Trial sponsored by Corbus and funded by the National Institutes of Health.
“When someone refers you [a patient with suspected] polymyositis, I want you to do a checklist in your head and say, ‘Have I thought about these five things?’ ” Dr. Christopher-Stine, director of the Johns Hopkins Myositis Center, Baltimore, said at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
The five most common diagnoses in patients labeled as having polymyositis are immune-mediated necrotizing myopathy (IMNM), overlap with other rheumatologic conditions, antisynthetase syndrome, inclusion body myositis (IBM), and muscular dystrophy, she explained.
“You may say, ‘look, it’s all what you call it,’ but I think we need to be a little bit more careful in what we call it,” she said.
IMNM
Patients with IMNM present with clinical symptoms similar to those seen in polymyositis and dermatomyositis – mainly proximal muscle weakness.
However, there are some important differences, both clinically and histologically, Dr. Christopher-Stine said.
“Look for higher [creatine kinase (CK)] levels,” she said. “In the thousands, usually multiple thousands ... like 5,000, 10,000, 2,000 ... that’s when you’re thinking about a necrotizing phenotype before you even look at the biopsy.”
CK levels will usually be under 30,000 U/L in IMNM, she noted, adding that data increasingly suggest that the extensive muscle necrosis in IMNM explains the elevated CK levels versus those seen in other myopathies.
Myalgias also tend to be more prominent in IMNM than in polymyositis.
“These folks hurt,” she said, noting that IMNM patients tend to have more extensive muscle atrophy and functional disability. “Many will be wheelchair bound within 9 months of diagnosis; it’s not subtle.”
The most important tool for making an IMNM diagnosis is muscle biopsy; look for prominent myocyte necrosis and a relative paucity of lymphocytes, she advised.
Overlap
Sometimes patients with polymyositis also have other rheumatologic conditions that shouldn’t be overlooked, therefore “overlap is its own category,” she said.
“In our experience, the most common overlap is scleroderma,” she noted, adding that the scleroderma is often, but not always, subtle, and that there may be overlapping autoantibodies.
Overt sclerodactyly is rarely seen, although a small amount may be present, but significant Raynaud’s phenomenon is common in these patients, and tiny telangiectasias across the neck are a tell-tale sign.
“Why does that matter? It’s not an esoteric argument; those are the folks that go on to have pulmonary hypertension,” she said. “They can have the same [interstitial lung disease] and all of the other internal scleroderma manifestations.”
Think about overlap and “look close phenotypically and with antibodies,” she advised.
There is also “the typical RA seropositive overlap,” she said, but lupus only rarely overlaps with myositis.
“However, the next diagnosis on the list – antisynthetase syndrome – can be a forme fruste where you first see a seronegative RA-like picture, and it’s important to think about that as well,” she said.
Antisynthetase syndrome
In patients referred for polymyositis, it’s also important to evaluate for antisynthetase syndrome, Dr. Christopher-Stine said.
The arthritis seen in the extramuscular phenotype of the syndrome is rarely deforming, but despite what many physicians were taught, “it absolutely can be erosive,” she said.
In fact, 40% of people with this syndrome present with an isolated forme fruste seronegative rheumatoid arthritis, she said.
Roughening and desquamation of the skin on the radial surface of fingers or palms – a sign known as mechanic’s hands – that doesn’t have another identifiable cause suggests this diagnosis in patients with this type of arthritis, as does interstitial lung disease and Raynaud’s phenomenon.
The Raynaud’s can be “fairly significant in the sense that it is bothersome,” but it usually doesn’t lead to ulceration or digital necrosis.
This is different from what is seen with the scleroderma phenotype, she said, adding that “if you’re starting to see gangrene and digital loss, think of something else.”
IBM
IBM is “probably the No. 1 most-missed diagnosis” among patients referred for what is initially believed to be polymyositis, Dr. Christopher-Stine said.
“I used to think that this was missed at entry, that everybody [with IBM] had all of these criteria and that rheumatologists really didn’t understand this phenotype ... but some people morph into this,” she said, explaining that they often start out looking like they have polymyositis with proximal muscle weakness.
“They may even initially respond to steroids. And then they get this phenotype,” she said.
Older men are more likely to present with the phenotype from the beginning; women, in her experience, tend to present with what appears to be polymyositis, and then develop the phenotype over time, she noted.
An IBM diagnosis requires age over 30 years, but most patients are over 50, she said.
“This is the only one of the myopathies that is preferential to men,” she added, noting that it affects men twice as often as it does women.
The syndrome is characterized by proximal strength loss and muscle atrophy. Also, a finding that a patient’s knee extensors are weaker than their hip flexors is “a fantastic bedside sign” differentiating IBM from polymyositis, she said.
That’s not to say IBM patients don’t have hip flexor weakness, but their knee extensors usually are “considerably weaker by a grade strength or more” versus their hip flexors, she explained.
“It’s a very easy bedside test. In typical other myopathies we have this, but the knee extensors aren’t that weak in general, or they’re not as weak as the hip flexors,” she added.
Another sign is distal strength loss, particularly in the forearm and finger flexors.
“I was taught to have them make a fist; don’t have them make a fist,” she said, explaining that this recruits intrinsic muscles which basically allows cheating that may mask weakness.
Instead, ask them to flex just their distal interphalangeal joints by making a claw and using the fingers to pull against your fingers, she suggested.
Mixed myopathic and neuropathic features on electromyography also indicate IBM, she said.
Muscle biopsy may be helpful, but inclusions are seen in less than one-third of IBM patients.
“At times, we have had to biopsy three times to see them at all, and some people never show them, so you have to rely on your clinical acumen if you don’t see them,” she said.
Also, keep in mind that these patients are often labeled as having treatment-resistant polymyositis.
“Please, when somebody refers to you somebody that’s treatment resistant, that may be the case, but I want you to think maybe they’re treatment resistant because they don’t have that disease.”
Muscular dystrophy
Some cases of myositis mimic certain types of muscular dystrophy, Dr. Christopher-Stine said, providing a checklist of muscular dystrophies that can look “clinically completely indistinguishable from a typical inflammatory myopathy,” and should therefore be considered in these patients.
The checklist includes Duchenne’s manifesting carrier, limb girdle muscular dystrophy type 2b, myotonic dystrophy (usually type 2), and facioscapulohumeral muscular dystrophy.
Dr. Christopher-Stine reported having intellectual property interest in a novel Inova Diagnostics autoantibody assay detection for anti-HMGCR. She was also the safety officer for the JBT-101 Trial sponsored by Corbus and funded by the National Institutes of Health.
“When someone refers you [a patient with suspected] polymyositis, I want you to do a checklist in your head and say, ‘Have I thought about these five things?’ ” Dr. Christopher-Stine, director of the Johns Hopkins Myositis Center, Baltimore, said at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
The five most common diagnoses in patients labeled as having polymyositis are immune-mediated necrotizing myopathy (IMNM), overlap with other rheumatologic conditions, antisynthetase syndrome, inclusion body myositis (IBM), and muscular dystrophy, she explained.
“You may say, ‘look, it’s all what you call it,’ but I think we need to be a little bit more careful in what we call it,” she said.
IMNM
Patients with IMNM present with clinical symptoms similar to those seen in polymyositis and dermatomyositis – mainly proximal muscle weakness.
However, there are some important differences, both clinically and histologically, Dr. Christopher-Stine said.
“Look for higher [creatine kinase (CK)] levels,” she said. “In the thousands, usually multiple thousands ... like 5,000, 10,000, 2,000 ... that’s when you’re thinking about a necrotizing phenotype before you even look at the biopsy.”
CK levels will usually be under 30,000 U/L in IMNM, she noted, adding that data increasingly suggest that the extensive muscle necrosis in IMNM explains the elevated CK levels versus those seen in other myopathies.
Myalgias also tend to be more prominent in IMNM than in polymyositis.
“These folks hurt,” she said, noting that IMNM patients tend to have more extensive muscle atrophy and functional disability. “Many will be wheelchair bound within 9 months of diagnosis; it’s not subtle.”
The most important tool for making an IMNM diagnosis is muscle biopsy; look for prominent myocyte necrosis and a relative paucity of lymphocytes, she advised.
Overlap
Sometimes patients with polymyositis also have other rheumatologic conditions that shouldn’t be overlooked, therefore “overlap is its own category,” she said.
“In our experience, the most common overlap is scleroderma,” she noted, adding that the scleroderma is often, but not always, subtle, and that there may be overlapping autoantibodies.
Overt sclerodactyly is rarely seen, although a small amount may be present, but significant Raynaud’s phenomenon is common in these patients, and tiny telangiectasias across the neck are a tell-tale sign.
“Why does that matter? It’s not an esoteric argument; those are the folks that go on to have pulmonary hypertension,” she said. “They can have the same [interstitial lung disease] and all of the other internal scleroderma manifestations.”
Think about overlap and “look close phenotypically and with antibodies,” she advised.
There is also “the typical RA seropositive overlap,” she said, but lupus only rarely overlaps with myositis.
“However, the next diagnosis on the list – antisynthetase syndrome – can be a forme fruste where you first see a seronegative RA-like picture, and it’s important to think about that as well,” she said.
Antisynthetase syndrome
In patients referred for polymyositis, it’s also important to evaluate for antisynthetase syndrome, Dr. Christopher-Stine said.
The arthritis seen in the extramuscular phenotype of the syndrome is rarely deforming, but despite what many physicians were taught, “it absolutely can be erosive,” she said.
In fact, 40% of people with this syndrome present with an isolated forme fruste seronegative rheumatoid arthritis, she said.
Roughening and desquamation of the skin on the radial surface of fingers or palms – a sign known as mechanic’s hands – that doesn’t have another identifiable cause suggests this diagnosis in patients with this type of arthritis, as does interstitial lung disease and Raynaud’s phenomenon.
The Raynaud’s can be “fairly significant in the sense that it is bothersome,” but it usually doesn’t lead to ulceration or digital necrosis.
This is different from what is seen with the scleroderma phenotype, she said, adding that “if you’re starting to see gangrene and digital loss, think of something else.”
IBM
IBM is “probably the No. 1 most-missed diagnosis” among patients referred for what is initially believed to be polymyositis, Dr. Christopher-Stine said.
“I used to think that this was missed at entry, that everybody [with IBM] had all of these criteria and that rheumatologists really didn’t understand this phenotype ... but some people morph into this,” she said, explaining that they often start out looking like they have polymyositis with proximal muscle weakness.
“They may even initially respond to steroids. And then they get this phenotype,” she said.
Older men are more likely to present with the phenotype from the beginning; women, in her experience, tend to present with what appears to be polymyositis, and then develop the phenotype over time, she noted.
An IBM diagnosis requires age over 30 years, but most patients are over 50, she said.
“This is the only one of the myopathies that is preferential to men,” she added, noting that it affects men twice as often as it does women.
The syndrome is characterized by proximal strength loss and muscle atrophy. Also, a finding that a patient’s knee extensors are weaker than their hip flexors is “a fantastic bedside sign” differentiating IBM from polymyositis, she said.
That’s not to say IBM patients don’t have hip flexor weakness, but their knee extensors usually are “considerably weaker by a grade strength or more” versus their hip flexors, she explained.
“It’s a very easy bedside test. In typical other myopathies we have this, but the knee extensors aren’t that weak in general, or they’re not as weak as the hip flexors,” she added.
Another sign is distal strength loss, particularly in the forearm and finger flexors.
“I was taught to have them make a fist; don’t have them make a fist,” she said, explaining that this recruits intrinsic muscles which basically allows cheating that may mask weakness.
Instead, ask them to flex just their distal interphalangeal joints by making a claw and using the fingers to pull against your fingers, she suggested.
Mixed myopathic and neuropathic features on electromyography also indicate IBM, she said.
Muscle biopsy may be helpful, but inclusions are seen in less than one-third of IBM patients.
“At times, we have had to biopsy three times to see them at all, and some people never show them, so you have to rely on your clinical acumen if you don’t see them,” she said.
Also, keep in mind that these patients are often labeled as having treatment-resistant polymyositis.
“Please, when somebody refers to you somebody that’s treatment resistant, that may be the case, but I want you to think maybe they’re treatment resistant because they don’t have that disease.”
Muscular dystrophy
Some cases of myositis mimic certain types of muscular dystrophy, Dr. Christopher-Stine said, providing a checklist of muscular dystrophies that can look “clinically completely indistinguishable from a typical inflammatory myopathy,” and should therefore be considered in these patients.
The checklist includes Duchenne’s manifesting carrier, limb girdle muscular dystrophy type 2b, myotonic dystrophy (usually type 2), and facioscapulohumeral muscular dystrophy.
Dr. Christopher-Stine reported having intellectual property interest in a novel Inova Diagnostics autoantibody assay detection for anti-HMGCR. She was also the safety officer for the JBT-101 Trial sponsored by Corbus and funded by the National Institutes of Health.
EXPERT ANALYSIS FROM THE WINTER RHEUMATOLOGY SYMPOSIUM
List of medications linked to drug-induced lupus expands
leaving the overall number now standing at 118.
Among the 118 suspected drugs found in VigiBase, the WHO’s global deduplicated individual case safety reports (ICSR) database, 42 had not been previously reported in association with drug-induced lupus (DIL) and 76 had been previously reported in association with DIL in Medline. DIL was reported as a serious adverse event in 55.4% of cases, according to French researchers led by Laurent Arnaud, MD, PhD, of the department of rheumatology at Hôpitaux Universitaires de Strasbourg and Centre National de Références des Maladies Systémiques Rares, Strasbourg, France.
Dr. Arnaud and his colleagues conducted a case-noncase analysis for each drug associated with DIL in order to compare the proportion of specific adverse drug reactions (ADRs) reported for a single drug with the proportion of the same ADR for all other treatments in VigiBase, which receives reports from more than 130 country members of the WHO Programme for International Drug Monitoring and contains over 16 million deduplicated ICSRs recorded by pharmacovigilance centers since 1967. They searched for cases classified as systemic lupus erythematosus (SLE) and identified 12,166 ICSRs of DIL; from these they found 118 suspected drugs with significant pharmacovigilance signal from 8,163 ICSRs that mostly originated from the Americas (65%) and Europe (23%).
In line with what the study authors expected, the drugs associated with the highest number of DIL cases were the antitumor necrosis factor agents infliximab, adalimumab, and etanercept, and the drugs associated with the highest disproportional reporting of DIL were procainamide and hydralazine.
“This is an important finding because these are the two drugs associated with the highest risk of DIL in the literature, therefore confirming the reliability of our approach using a large pharmacovigilance database,” the researchers wrote in Annals of the Rheumatic Diseases.
Overall, DIL was considered definite for 9 drugs (procainamide, hydralazine, minocycline, quinidine, isoniazid, terbinafine, methyldopa, dihydralazine, and chlorpromazine), probable for 19 drugs, and possible for 45 drugs.
The median age of DIL onset was 49 years, which the authors noted was about 2 decades older than that of spontaneous SLE.
They also observed a marked predominance in females (female to male sex ratio, 4.3), a finding that contrasted with previous studies reporting a female to male sex ratio closer to 1:1.
Dr. Arnaud and his colleagues stated that their finding of a median delay between the reported start of suspected treatment and DIL occurrence of 172 days (interquartile range, 35-610 days) suggested that DIL mostly appears after a few months and usually within the first 2 years of treatment with the suspected drug.
“The analysis of the median reporting years for each suspected drug shows a clear evolution of suspected drugs during the past decades. This further underlines that the constantly changing spectrum of DIL should be monitored continuously, and further validates the interest of our approach using the WHO international pharmacovigilance database, the biggest database of this kind with over 16 million deduplicated ICSRs,” they wrote.
The researchers added that distinguishing DIL from SLE is important because its prognosis is usually good when the drug is withdrawn, but the spectrum of DIL is constantly evolving, with drugs once described as strongly linked to DIL now prescribed less frequently.
“The first case of DIL was reported in 1945 with sulfadiazine, while hydralazine DIL was first reported in 1953. Since then, pharmacopoeia has strongly evolved, and one could hypothesize that so has the spectrum of drugs that can induce DIL,” they wrote.
“The detailed list of suspected drugs may prove useful to physicians when confronted with potential DIL cases. Altogether, these findings may help in improving the identification of this constantly evolving disease,” they concluded.
The current study was limited by the lack of a uniform set of criteria for the diagnosis of DIL and by the level of reported details available in VigiBase.
The authors had no outside funding for the study and reported having no conflicts of interest.
SOURCE: Arnaud L et al. Ann Rheum Dis. 2019 Feb 4. doi: 10.1136/annrheumdis-2018-214598.
This new and updated list of possible lupus-inducing drugs includes a growing range of treatment categories, chemical structures, and pharmacologic actions. Yet it is still unclear what the common denominator is that links them.
Drug-induced lupus (DIL) is a peculiar adverse drug reaction that appears to be unrelated to any known property of the inducing agent, although cytokine modulating biologics are a possible exception. Nevertheless, the in vivo metabolism of dissimilar drugs to products with a common, reactive property may go some way to explaining how compounds with different pharmacologic and chemical structures could induce similar adverse reactions.
The findings by Arnaud et al. need better documentation than just positive pharmacovigilance signals. For example, a drug with a relatively high signal does not necessarily translate to a high propensity for causing lupus-like symptoms. It may be a reflection of high drug usage or an awareness of the report contributors for detecting new-onset systemic lupus erythematosus.
Regardless, this research serves to help and inform the medical community to increase the vigilance of previously unreported DIL and perhaps motivate the publication of novel, convincing case reports.
Robert L. Rubin, PhD, is with the University of New Mexico, Albuquerque. His comments are adapted from an editorial accompanying the report by Arnaud et al. (Ann Rheum Dis. 2019 Feb 13. doi: annrheumdis-2018-214785). He reported having no relevant disclosures.
This new and updated list of possible lupus-inducing drugs includes a growing range of treatment categories, chemical structures, and pharmacologic actions. Yet it is still unclear what the common denominator is that links them.
Drug-induced lupus (DIL) is a peculiar adverse drug reaction that appears to be unrelated to any known property of the inducing agent, although cytokine modulating biologics are a possible exception. Nevertheless, the in vivo metabolism of dissimilar drugs to products with a common, reactive property may go some way to explaining how compounds with different pharmacologic and chemical structures could induce similar adverse reactions.
The findings by Arnaud et al. need better documentation than just positive pharmacovigilance signals. For example, a drug with a relatively high signal does not necessarily translate to a high propensity for causing lupus-like symptoms. It may be a reflection of high drug usage or an awareness of the report contributors for detecting new-onset systemic lupus erythematosus.
Regardless, this research serves to help and inform the medical community to increase the vigilance of previously unreported DIL and perhaps motivate the publication of novel, convincing case reports.
Robert L. Rubin, PhD, is with the University of New Mexico, Albuquerque. His comments are adapted from an editorial accompanying the report by Arnaud et al. (Ann Rheum Dis. 2019 Feb 13. doi: annrheumdis-2018-214785). He reported having no relevant disclosures.
This new and updated list of possible lupus-inducing drugs includes a growing range of treatment categories, chemical structures, and pharmacologic actions. Yet it is still unclear what the common denominator is that links them.
Drug-induced lupus (DIL) is a peculiar adverse drug reaction that appears to be unrelated to any known property of the inducing agent, although cytokine modulating biologics are a possible exception. Nevertheless, the in vivo metabolism of dissimilar drugs to products with a common, reactive property may go some way to explaining how compounds with different pharmacologic and chemical structures could induce similar adverse reactions.
The findings by Arnaud et al. need better documentation than just positive pharmacovigilance signals. For example, a drug with a relatively high signal does not necessarily translate to a high propensity for causing lupus-like symptoms. It may be a reflection of high drug usage or an awareness of the report contributors for detecting new-onset systemic lupus erythematosus.
Regardless, this research serves to help and inform the medical community to increase the vigilance of previously unreported DIL and perhaps motivate the publication of novel, convincing case reports.
Robert L. Rubin, PhD, is with the University of New Mexico, Albuquerque. His comments are adapted from an editorial accompanying the report by Arnaud et al. (Ann Rheum Dis. 2019 Feb 13. doi: annrheumdis-2018-214785). He reported having no relevant disclosures.
leaving the overall number now standing at 118.
Among the 118 suspected drugs found in VigiBase, the WHO’s global deduplicated individual case safety reports (ICSR) database, 42 had not been previously reported in association with drug-induced lupus (DIL) and 76 had been previously reported in association with DIL in Medline. DIL was reported as a serious adverse event in 55.4% of cases, according to French researchers led by Laurent Arnaud, MD, PhD, of the department of rheumatology at Hôpitaux Universitaires de Strasbourg and Centre National de Références des Maladies Systémiques Rares, Strasbourg, France.
Dr. Arnaud and his colleagues conducted a case-noncase analysis for each drug associated with DIL in order to compare the proportion of specific adverse drug reactions (ADRs) reported for a single drug with the proportion of the same ADR for all other treatments in VigiBase, which receives reports from more than 130 country members of the WHO Programme for International Drug Monitoring and contains over 16 million deduplicated ICSRs recorded by pharmacovigilance centers since 1967. They searched for cases classified as systemic lupus erythematosus (SLE) and identified 12,166 ICSRs of DIL; from these they found 118 suspected drugs with significant pharmacovigilance signal from 8,163 ICSRs that mostly originated from the Americas (65%) and Europe (23%).
In line with what the study authors expected, the drugs associated with the highest number of DIL cases were the antitumor necrosis factor agents infliximab, adalimumab, and etanercept, and the drugs associated with the highest disproportional reporting of DIL were procainamide and hydralazine.
“This is an important finding because these are the two drugs associated with the highest risk of DIL in the literature, therefore confirming the reliability of our approach using a large pharmacovigilance database,” the researchers wrote in Annals of the Rheumatic Diseases.
Overall, DIL was considered definite for 9 drugs (procainamide, hydralazine, minocycline, quinidine, isoniazid, terbinafine, methyldopa, dihydralazine, and chlorpromazine), probable for 19 drugs, and possible for 45 drugs.
The median age of DIL onset was 49 years, which the authors noted was about 2 decades older than that of spontaneous SLE.
They also observed a marked predominance in females (female to male sex ratio, 4.3), a finding that contrasted with previous studies reporting a female to male sex ratio closer to 1:1.
Dr. Arnaud and his colleagues stated that their finding of a median delay between the reported start of suspected treatment and DIL occurrence of 172 days (interquartile range, 35-610 days) suggested that DIL mostly appears after a few months and usually within the first 2 years of treatment with the suspected drug.
“The analysis of the median reporting years for each suspected drug shows a clear evolution of suspected drugs during the past decades. This further underlines that the constantly changing spectrum of DIL should be monitored continuously, and further validates the interest of our approach using the WHO international pharmacovigilance database, the biggest database of this kind with over 16 million deduplicated ICSRs,” they wrote.
The researchers added that distinguishing DIL from SLE is important because its prognosis is usually good when the drug is withdrawn, but the spectrum of DIL is constantly evolving, with drugs once described as strongly linked to DIL now prescribed less frequently.
“The first case of DIL was reported in 1945 with sulfadiazine, while hydralazine DIL was first reported in 1953. Since then, pharmacopoeia has strongly evolved, and one could hypothesize that so has the spectrum of drugs that can induce DIL,” they wrote.
“The detailed list of suspected drugs may prove useful to physicians when confronted with potential DIL cases. Altogether, these findings may help in improving the identification of this constantly evolving disease,” they concluded.
The current study was limited by the lack of a uniform set of criteria for the diagnosis of DIL and by the level of reported details available in VigiBase.
The authors had no outside funding for the study and reported having no conflicts of interest.
SOURCE: Arnaud L et al. Ann Rheum Dis. 2019 Feb 4. doi: 10.1136/annrheumdis-2018-214598.
leaving the overall number now standing at 118.
Among the 118 suspected drugs found in VigiBase, the WHO’s global deduplicated individual case safety reports (ICSR) database, 42 had not been previously reported in association with drug-induced lupus (DIL) and 76 had been previously reported in association with DIL in Medline. DIL was reported as a serious adverse event in 55.4% of cases, according to French researchers led by Laurent Arnaud, MD, PhD, of the department of rheumatology at Hôpitaux Universitaires de Strasbourg and Centre National de Références des Maladies Systémiques Rares, Strasbourg, France.
Dr. Arnaud and his colleagues conducted a case-noncase analysis for each drug associated with DIL in order to compare the proportion of specific adverse drug reactions (ADRs) reported for a single drug with the proportion of the same ADR for all other treatments in VigiBase, which receives reports from more than 130 country members of the WHO Programme for International Drug Monitoring and contains over 16 million deduplicated ICSRs recorded by pharmacovigilance centers since 1967. They searched for cases classified as systemic lupus erythematosus (SLE) and identified 12,166 ICSRs of DIL; from these they found 118 suspected drugs with significant pharmacovigilance signal from 8,163 ICSRs that mostly originated from the Americas (65%) and Europe (23%).
In line with what the study authors expected, the drugs associated with the highest number of DIL cases were the antitumor necrosis factor agents infliximab, adalimumab, and etanercept, and the drugs associated with the highest disproportional reporting of DIL were procainamide and hydralazine.
“This is an important finding because these are the two drugs associated with the highest risk of DIL in the literature, therefore confirming the reliability of our approach using a large pharmacovigilance database,” the researchers wrote in Annals of the Rheumatic Diseases.
Overall, DIL was considered definite for 9 drugs (procainamide, hydralazine, minocycline, quinidine, isoniazid, terbinafine, methyldopa, dihydralazine, and chlorpromazine), probable for 19 drugs, and possible for 45 drugs.
The median age of DIL onset was 49 years, which the authors noted was about 2 decades older than that of spontaneous SLE.
They also observed a marked predominance in females (female to male sex ratio, 4.3), a finding that contrasted with previous studies reporting a female to male sex ratio closer to 1:1.
Dr. Arnaud and his colleagues stated that their finding of a median delay between the reported start of suspected treatment and DIL occurrence of 172 days (interquartile range, 35-610 days) suggested that DIL mostly appears after a few months and usually within the first 2 years of treatment with the suspected drug.
“The analysis of the median reporting years for each suspected drug shows a clear evolution of suspected drugs during the past decades. This further underlines that the constantly changing spectrum of DIL should be monitored continuously, and further validates the interest of our approach using the WHO international pharmacovigilance database, the biggest database of this kind with over 16 million deduplicated ICSRs,” they wrote.
The researchers added that distinguishing DIL from SLE is important because its prognosis is usually good when the drug is withdrawn, but the spectrum of DIL is constantly evolving, with drugs once described as strongly linked to DIL now prescribed less frequently.
“The first case of DIL was reported in 1945 with sulfadiazine, while hydralazine DIL was first reported in 1953. Since then, pharmacopoeia has strongly evolved, and one could hypothesize that so has the spectrum of drugs that can induce DIL,” they wrote.
“The detailed list of suspected drugs may prove useful to physicians when confronted with potential DIL cases. Altogether, these findings may help in improving the identification of this constantly evolving disease,” they concluded.
The current study was limited by the lack of a uniform set of criteria for the diagnosis of DIL and by the level of reported details available in VigiBase.
The authors had no outside funding for the study and reported having no conflicts of interest.
SOURCE: Arnaud L et al. Ann Rheum Dis. 2019 Feb 4. doi: 10.1136/annrheumdis-2018-214598.
FROM ANNALS OF THE RHEUMATIC DISEASES
Ehlers-Danlos syndrome: Increased IUGR risk reported
LAS VEGAS – Women with Ehlers-Danlos syndrome who became pregnant were more likely to experience antepartum hemorrhage, placenta previa, cervical incompetence, and preterm birth, according to a retrospective cohort study of national birth data. Long hospital stays also were more likely among these women.

Infants born to women with Ehlers-Danlos syndrome (EDS) were significantly more likely to have intrauterine growth retardation (IUGR) as well, an unexpected and as-yet unexplained finding, said the study’s first author, Laura Nicholls-Dempsey, MD, speaking at a poster session at the meeting sponsored by the Society for Maternal-Fetal Medicine.
Complications were infrequent overall, with a very low rate of intrauterine demise and no maternal mortality seen in the 910 women with EDS who were studied, said Dr. Nicholls-Dempsey, an ob.gyn. resident at McGill University, Montreal.
In counseling women with EDS, Dr. Nicholls-Dempsey said that she would advise them that “these are the types of things we’re going to watch out for, and we’ll see how the pregnancy goes. But we have to be careful about these: preterm birth, antepartum bleeding, placenta previa. We’ll watch the growth of the baby; we just have to be more careful about these specific things.”
Compared with women without EDS, those with the inherited connective tissue disorder had adjusted odds ratios (AORs) of 3.2 for cervical incompetence (95% confidence interval, 2.0-5.1) and 2.2 for placenta previa (95% CI, 1.3-3.9; P less than .01 for both). Absolute rates for these complications were 0.8% and 0.7% for women without EDS and 2.1% and 1.4% for women with EDS, respectively.
Women with EDS also had AORs of 1.8 for antepartum hemorrhage (2.8% versus 1.6%; 95% CI, 1.2-2.7; P less than .01). Cesarean delivery was more likely in women with EDS, with an AOR of 1.6 (37.4% versus 26.9%; 95% CI, 2.0-5.1); conversely, instrumental vaginal delivery was less likely in women with EDS (AOR = 0.5; 95% CI, 0.4-0.7; P less than .01 for both), meaning that spontaneous vaginal delivery was less likely in the EDS cohort.
The higher frequency of Cesarean deliveries may be attributable to anticipatory management by physicians seeking to avoid such complications as antepartum hemorrhage, as well as to the increased rate of placenta previa seen among the EDS cohort, Dr. Nicholls-Dempsey said.
After statistical adjustment, women in the EDS cohort were more than three times as likely to have hospital stays of both more than 7 days and 14 days (5.7% versus 2.1%, AOR = 3.1 for 7 days; 2.3% versus 0.7%, AOR = 3.8 for 14 days; P less than .01 for both).
Rates of some other maternal complications, such as pre-eclampsia, eclampsia, and gestational hypertension, were not elevated in the EDS cohort. Rates of premature rupture of membranes, chorioamnionitis, uterine rupture, postpartum hemorrhage, perineal laceration, and venous thromboembolism were also similar between groups.
However, not only was the AOR for preterm birth 1.5 for infants of women with EDS, but IUGR was more common in these neonates as well (AOR = 1.7, P less than .01 for both). The latter finding was unexpected, and Dr. Nicholls-Dempsey and her colleagues currently don’t have a mechanistic explanation for the higher IUGR rate.
Dr. Nicholls-Dempsey explained that she and her colleagues used data from the United States’ Health Care Cost and Utilization Project’s Nationwide Inpatient Sample (HCUP-NIS) to compare outcomes of women with EDS with the national sample as a whole.
Between 1999 and 2013, 13,881,592 births occurred in the HCUP-NIS cohort, with 910 deliveries to women who had EDS. These women were identified by ICD-9 codes, she said.
Comparing women with EDS to the non-EDS cohort, women with EDS were more likely to be Caucasian, have a higher income, and to be smokers; the cohorts were otherwise similar.
Ehlers-Danlos syndrome is a heterogeneous disorder involving abnormalities of collagen synthesis, with 13 known subtypes not captured in the HCUP-NIS data, Dr. Nicholls-Dempsey acknowledged. She characterized this as both a limitation but also a potential strength of the study.
“I really like this study because ... we know there’s 13 types of EDS that are genetically different ... They have their overlapping symptoms, but each one is different,” she said. “In an ideal world, we would have each subtype, and we would run this type of analysis on each subtype, to really be able to say to a patient, ‘You have this mutation, and this complication is going to be a big problem for you.’” The numbers of each subtype are so small that this is infeasible, she noted.
Still, the national sample acquired over many years offers real-world outcomes that clinicians can use in shared decision-making with EDS patients who are contemplating pregnancy or are already pregnant. Also, knowing which complications are more likely in patients with EDS can help plan optimal management of labor and delivery, Dr. Nicholls-Dempsey said.
Over the study’s 14-year span, the overall arc of EDS pregnancy outcomes is well captured regardless of mutation type. “It’s very applicable to the general population” of individuals with EDS, she noted. “Because it’s not type-specific, it’s really a good overview of what you can expect in EDS patients, regardless of the type.”
Dr. Nicholls-Dempsey reported no conflicts of interest and no outside sources of funding.
SOURCE: Nicholls-Dempsey L et al. Am J Obstet Gynecol. 2019 Jan;220(1):S381-382. Abstract 574
LAS VEGAS – Women with Ehlers-Danlos syndrome who became pregnant were more likely to experience antepartum hemorrhage, placenta previa, cervical incompetence, and preterm birth, according to a retrospective cohort study of national birth data. Long hospital stays also were more likely among these women.
Infants born to women with Ehlers-Danlos syndrome (EDS) were significantly more likely to have intrauterine growth retardation (IUGR) as well, an unexpected and as-yet unexplained finding, said the study’s first author, Laura Nicholls-Dempsey, MD, speaking at a poster session at the meeting sponsored by the Society for Maternal-Fetal Medicine.
Complications were infrequent overall, with a very low rate of intrauterine demise and no maternal mortality seen in the 910 women with EDS who were studied, said Dr. Nicholls-Dempsey, an ob.gyn. resident at McGill University, Montreal.
In counseling women with EDS, Dr. Nicholls-Dempsey said that she would advise them that “these are the types of things we’re going to watch out for, and we’ll see how the pregnancy goes. But we have to be careful about these: preterm birth, antepartum bleeding, placenta previa. We’ll watch the growth of the baby; we just have to be more careful about these specific things.”
Compared with women without EDS, those with the inherited connective tissue disorder had adjusted odds ratios (AORs) of 3.2 for cervical incompetence (95% confidence interval, 2.0-5.1) and 2.2 for placenta previa (95% CI, 1.3-3.9; P less than .01 for both). Absolute rates for these complications were 0.8% and 0.7% for women without EDS and 2.1% and 1.4% for women with EDS, respectively.
Women with EDS also had AORs of 1.8 for antepartum hemorrhage (2.8% versus 1.6%; 95% CI, 1.2-2.7; P less than .01). Cesarean delivery was more likely in women with EDS, with an AOR of 1.6 (37.4% versus 26.9%; 95% CI, 2.0-5.1); conversely, instrumental vaginal delivery was less likely in women with EDS (AOR = 0.5; 95% CI, 0.4-0.7; P less than .01 for both), meaning that spontaneous vaginal delivery was less likely in the EDS cohort.
The higher frequency of Cesarean deliveries may be attributable to anticipatory management by physicians seeking to avoid such complications as antepartum hemorrhage, as well as to the increased rate of placenta previa seen among the EDS cohort, Dr. Nicholls-Dempsey said.
After statistical adjustment, women in the EDS cohort were more than three times as likely to have hospital stays of both more than 7 days and 14 days (5.7% versus 2.1%, AOR = 3.1 for 7 days; 2.3% versus 0.7%, AOR = 3.8 for 14 days; P less than .01 for both).
Rates of some other maternal complications, such as pre-eclampsia, eclampsia, and gestational hypertension, were not elevated in the EDS cohort. Rates of premature rupture of membranes, chorioamnionitis, uterine rupture, postpartum hemorrhage, perineal laceration, and venous thromboembolism were also similar between groups.
However, not only was the AOR for preterm birth 1.5 for infants of women with EDS, but IUGR was more common in these neonates as well (AOR = 1.7, P less than .01 for both). The latter finding was unexpected, and Dr. Nicholls-Dempsey and her colleagues currently don’t have a mechanistic explanation for the higher IUGR rate.
Dr. Nicholls-Dempsey explained that she and her colleagues used data from the United States’ Health Care Cost and Utilization Project’s Nationwide Inpatient Sample (HCUP-NIS) to compare outcomes of women with EDS with the national sample as a whole.
Between 1999 and 2013, 13,881,592 births occurred in the HCUP-NIS cohort, with 910 deliveries to women who had EDS. These women were identified by ICD-9 codes, she said.
Comparing women with EDS to the non-EDS cohort, women with EDS were more likely to be Caucasian, have a higher income, and to be smokers; the cohorts were otherwise similar.
Ehlers-Danlos syndrome is a heterogeneous disorder involving abnormalities of collagen synthesis, with 13 known subtypes not captured in the HCUP-NIS data, Dr. Nicholls-Dempsey acknowledged. She characterized this as both a limitation but also a potential strength of the study.
“I really like this study because ... we know there’s 13 types of EDS that are genetically different ... They have their overlapping symptoms, but each one is different,” she said. “In an ideal world, we would have each subtype, and we would run this type of analysis on each subtype, to really be able to say to a patient, ‘You have this mutation, and this complication is going to be a big problem for you.’” The numbers of each subtype are so small that this is infeasible, she noted.
Still, the national sample acquired over many years offers real-world outcomes that clinicians can use in shared decision-making with EDS patients who are contemplating pregnancy or are already pregnant. Also, knowing which complications are more likely in patients with EDS can help plan optimal management of labor and delivery, Dr. Nicholls-Dempsey said.
Over the study’s 14-year span, the overall arc of EDS pregnancy outcomes is well captured regardless of mutation type. “It’s very applicable to the general population” of individuals with EDS, she noted. “Because it’s not type-specific, it’s really a good overview of what you can expect in EDS patients, regardless of the type.”
Dr. Nicholls-Dempsey reported no conflicts of interest and no outside sources of funding.
SOURCE: Nicholls-Dempsey L et al. Am J Obstet Gynecol. 2019 Jan;220(1):S381-382. Abstract 574
LAS VEGAS – Women with Ehlers-Danlos syndrome who became pregnant were more likely to experience antepartum hemorrhage, placenta previa, cervical incompetence, and preterm birth, according to a retrospective cohort study of national birth data. Long hospital stays also were more likely among these women.
Infants born to women with Ehlers-Danlos syndrome (EDS) were significantly more likely to have intrauterine growth retardation (IUGR) as well, an unexpected and as-yet unexplained finding, said the study’s first author, Laura Nicholls-Dempsey, MD, speaking at a poster session at the meeting sponsored by the Society for Maternal-Fetal Medicine.
Complications were infrequent overall, with a very low rate of intrauterine demise and no maternal mortality seen in the 910 women with EDS who were studied, said Dr. Nicholls-Dempsey, an ob.gyn. resident at McGill University, Montreal.
In counseling women with EDS, Dr. Nicholls-Dempsey said that she would advise them that “these are the types of things we’re going to watch out for, and we’ll see how the pregnancy goes. But we have to be careful about these: preterm birth, antepartum bleeding, placenta previa. We’ll watch the growth of the baby; we just have to be more careful about these specific things.”
Compared with women without EDS, those with the inherited connective tissue disorder had adjusted odds ratios (AORs) of 3.2 for cervical incompetence (95% confidence interval, 2.0-5.1) and 2.2 for placenta previa (95% CI, 1.3-3.9; P less than .01 for both). Absolute rates for these complications were 0.8% and 0.7% for women without EDS and 2.1% and 1.4% for women with EDS, respectively.
Women with EDS also had AORs of 1.8 for antepartum hemorrhage (2.8% versus 1.6%; 95% CI, 1.2-2.7; P less than .01). Cesarean delivery was more likely in women with EDS, with an AOR of 1.6 (37.4% versus 26.9%; 95% CI, 2.0-5.1); conversely, instrumental vaginal delivery was less likely in women with EDS (AOR = 0.5; 95% CI, 0.4-0.7; P less than .01 for both), meaning that spontaneous vaginal delivery was less likely in the EDS cohort.
The higher frequency of Cesarean deliveries may be attributable to anticipatory management by physicians seeking to avoid such complications as antepartum hemorrhage, as well as to the increased rate of placenta previa seen among the EDS cohort, Dr. Nicholls-Dempsey said.
After statistical adjustment, women in the EDS cohort were more than three times as likely to have hospital stays of both more than 7 days and 14 days (5.7% versus 2.1%, AOR = 3.1 for 7 days; 2.3% versus 0.7%, AOR = 3.8 for 14 days; P less than .01 for both).
Rates of some other maternal complications, such as pre-eclampsia, eclampsia, and gestational hypertension, were not elevated in the EDS cohort. Rates of premature rupture of membranes, chorioamnionitis, uterine rupture, postpartum hemorrhage, perineal laceration, and venous thromboembolism were also similar between groups.
However, not only was the AOR for preterm birth 1.5 for infants of women with EDS, but IUGR was more common in these neonates as well (AOR = 1.7, P less than .01 for both). The latter finding was unexpected, and Dr. Nicholls-Dempsey and her colleagues currently don’t have a mechanistic explanation for the higher IUGR rate.
Dr. Nicholls-Dempsey explained that she and her colleagues used data from the United States’ Health Care Cost and Utilization Project’s Nationwide Inpatient Sample (HCUP-NIS) to compare outcomes of women with EDS with the national sample as a whole.
Between 1999 and 2013, 13,881,592 births occurred in the HCUP-NIS cohort, with 910 deliveries to women who had EDS. These women were identified by ICD-9 codes, she said.
Comparing women with EDS to the non-EDS cohort, women with EDS were more likely to be Caucasian, have a higher income, and to be smokers; the cohorts were otherwise similar.
Ehlers-Danlos syndrome is a heterogeneous disorder involving abnormalities of collagen synthesis, with 13 known subtypes not captured in the HCUP-NIS data, Dr. Nicholls-Dempsey acknowledged. She characterized this as both a limitation but also a potential strength of the study.
“I really like this study because ... we know there’s 13 types of EDS that are genetically different ... They have their overlapping symptoms, but each one is different,” she said. “In an ideal world, we would have each subtype, and we would run this type of analysis on each subtype, to really be able to say to a patient, ‘You have this mutation, and this complication is going to be a big problem for you.’” The numbers of each subtype are so small that this is infeasible, she noted.
Still, the national sample acquired over many years offers real-world outcomes that clinicians can use in shared decision-making with EDS patients who are contemplating pregnancy or are already pregnant. Also, knowing which complications are more likely in patients with EDS can help plan optimal management of labor and delivery, Dr. Nicholls-Dempsey said.
Over the study’s 14-year span, the overall arc of EDS pregnancy outcomes is well captured regardless of mutation type. “It’s very applicable to the general population” of individuals with EDS, she noted. “Because it’s not type-specific, it’s really a good overview of what you can expect in EDS patients, regardless of the type.”
Dr. Nicholls-Dempsey reported no conflicts of interest and no outside sources of funding.
SOURCE: Nicholls-Dempsey L et al. Am J Obstet Gynecol. 2019 Jan;220(1):S381-382. Abstract 574
REPORTING FROM THE PREGNANCY MEETING