User login
Bortezomib-based regimen + transplant increased progression-free survival in primary plasma cell leukemia
In a prospective study of 40 patients with primary plasma cell leukemia, upfront autotransplantation followed by allotransplant for younger patients and by consolidation/maintenance for older patients was associated with a median overall survival of 36.3 months and a median progression-free survival of 15.1 months.
Patients with this aggressive form of multiple myeloma received a regimen that combined standard chemotherapy, a proteasome inhibitor, high-dose melphalan followed by autologous stem cell transplantation, and allogeneic transplantation or immunomodulatory drugs, reported Dr. Bruno Royer of University Hospital in Amiens, France, and his associates.
Induction therapy consisted of four 21-day cycles: Cycles 1 and 3 included subcutaneous bortezomib, intravenous pegylated doxorubicin, and oral dexamethasone; cycles 2 and 4 included subcutaneous bortezomib, oral cyclophosphamide, and oral dexamethasone. Of 39 patients – one patient died 24 hours after study inclusion – 35 completed the four cycles. The overall response rate to induction was 69%: 10% of patients had a complete response and 26% had a very good partial response. Of 27 responding patients, 25 underwent high-dose melphalan followed by autologous stem cell transplantation.
The high response rates allowed 16 patients who were younger than 66 years and had an HLA-matched donor to then receive high-dose melphalan followed by autologous stem cell transplantation followed by consolidation with either an reduced-intensity conditioning allograft or a second high-dose melphalan followed by autologous stem cell transplantation and subsequent maintenance with lenalidomide, bortezomib, and dexamethasone for 1 year, the researchers said (J Clin Oncol. 2016 Apr 25. doi: 10.1200/JCO.2015.63.1929).
A total of 20% of patients had a complete response to the entire treatment protocol, 13% had a stringent complete response, 26% had a very good partial response, 5% had stable disease, and 5% had progressive disease. Thirteen patients died of progressive disease and four died of infections, including three that occurred during induction or after allograft.
This is only the second prospective trial in patients with primary plasma cell leukemia, an aggressive form of multiple myeloma that accounts for 2%-4% of cases, the researchers said. Future prospective trials should seek to optimize induction with newer combinations, such as carfilzomib, lenalidomide, dexamethasone, or monoclonal anti-CD38 antibodies. Also, optimizing the stem cell conditioning procedure and the postallograft immunomodulation may further benefit younger patients.
Dr. Royer reported receiving honoraria from Amgen and having served as a consultant or advisor for Octapharma Plasma. Fifteen coinvestigators also reported financial relationships with a number of pharmaceutical companies.
The study by Dr. Royer and associates is the first prospective trial to confirm that bortezomib-based regimens combined with a transplantation program may be effective and feasible in a significant proportion of patients with primary plasma cell leukemia. Response to induction therapy, however, was not remarkable; thus, although both cyclophosphamide and doxorubicin have demonstrated efficacy in primary plasma cell leukemia, the introduction of lenalidomide and/or incorporation of newer agents such as pomalidomide, carfilzomib, or daratumumab could hopefully optimize the induction phase and increase the rate and quality of response in future studies.
Hopefully, sequential phases of induction therapy, multiple transplantations (if applicable), further consolidation, and maintenance should ensure rapid disease control and reduction of early deaths from initial complications, a contrasting of clonal evolution that may induce drug resistance, and activity on residual disease by decreasing the risk of relapse. Feasibility of these approaches, however, may be limited, especially for older and frail patients who are unable to tolerate intensive induction or prolonged treatments. Personalized therapies with acceptable toxicities should be considered for these patients.
Dr. Pellegrino Musto is at Referral Cancer Center of Basilicata, Rionero in Vulture, Italy. He reported receiving honoraria from Celgene, Janssen-Cilag, Novartis, Sanofi, and Bristol-Myers Squibb. These comments are from an editorial (J Clin Oncol. 2016 Apr 25. doi: 10.1200/JCO.2016.66.6115) that accompanied the published study.
The study by Dr. Royer and associates is the first prospective trial to confirm that bortezomib-based regimens combined with a transplantation program may be effective and feasible in a significant proportion of patients with primary plasma cell leukemia. Response to induction therapy, however, was not remarkable; thus, although both cyclophosphamide and doxorubicin have demonstrated efficacy in primary plasma cell leukemia, the introduction of lenalidomide and/or incorporation of newer agents such as pomalidomide, carfilzomib, or daratumumab could hopefully optimize the induction phase and increase the rate and quality of response in future studies.
Hopefully, sequential phases of induction therapy, multiple transplantations (if applicable), further consolidation, and maintenance should ensure rapid disease control and reduction of early deaths from initial complications, a contrasting of clonal evolution that may induce drug resistance, and activity on residual disease by decreasing the risk of relapse. Feasibility of these approaches, however, may be limited, especially for older and frail patients who are unable to tolerate intensive induction or prolonged treatments. Personalized therapies with acceptable toxicities should be considered for these patients.
Dr. Pellegrino Musto is at Referral Cancer Center of Basilicata, Rionero in Vulture, Italy. He reported receiving honoraria from Celgene, Janssen-Cilag, Novartis, Sanofi, and Bristol-Myers Squibb. These comments are from an editorial (J Clin Oncol. 2016 Apr 25. doi: 10.1200/JCO.2016.66.6115) that accompanied the published study.
The study by Dr. Royer and associates is the first prospective trial to confirm that bortezomib-based regimens combined with a transplantation program may be effective and feasible in a significant proportion of patients with primary plasma cell leukemia. Response to induction therapy, however, was not remarkable; thus, although both cyclophosphamide and doxorubicin have demonstrated efficacy in primary plasma cell leukemia, the introduction of lenalidomide and/or incorporation of newer agents such as pomalidomide, carfilzomib, or daratumumab could hopefully optimize the induction phase and increase the rate and quality of response in future studies.
Hopefully, sequential phases of induction therapy, multiple transplantations (if applicable), further consolidation, and maintenance should ensure rapid disease control and reduction of early deaths from initial complications, a contrasting of clonal evolution that may induce drug resistance, and activity on residual disease by decreasing the risk of relapse. Feasibility of these approaches, however, may be limited, especially for older and frail patients who are unable to tolerate intensive induction or prolonged treatments. Personalized therapies with acceptable toxicities should be considered for these patients.
Dr. Pellegrino Musto is at Referral Cancer Center of Basilicata, Rionero in Vulture, Italy. He reported receiving honoraria from Celgene, Janssen-Cilag, Novartis, Sanofi, and Bristol-Myers Squibb. These comments are from an editorial (J Clin Oncol. 2016 Apr 25. doi: 10.1200/JCO.2016.66.6115) that accompanied the published study.
In a prospective study of 40 patients with primary plasma cell leukemia, upfront autotransplantation followed by allotransplant for younger patients and by consolidation/maintenance for older patients was associated with a median overall survival of 36.3 months and a median progression-free survival of 15.1 months.
Patients with this aggressive form of multiple myeloma received a regimen that combined standard chemotherapy, a proteasome inhibitor, high-dose melphalan followed by autologous stem cell transplantation, and allogeneic transplantation or immunomodulatory drugs, reported Dr. Bruno Royer of University Hospital in Amiens, France, and his associates.
Induction therapy consisted of four 21-day cycles: Cycles 1 and 3 included subcutaneous bortezomib, intravenous pegylated doxorubicin, and oral dexamethasone; cycles 2 and 4 included subcutaneous bortezomib, oral cyclophosphamide, and oral dexamethasone. Of 39 patients – one patient died 24 hours after study inclusion – 35 completed the four cycles. The overall response rate to induction was 69%: 10% of patients had a complete response and 26% had a very good partial response. Of 27 responding patients, 25 underwent high-dose melphalan followed by autologous stem cell transplantation.
The high response rates allowed 16 patients who were younger than 66 years and had an HLA-matched donor to then receive high-dose melphalan followed by autologous stem cell transplantation followed by consolidation with either an reduced-intensity conditioning allograft or a second high-dose melphalan followed by autologous stem cell transplantation and subsequent maintenance with lenalidomide, bortezomib, and dexamethasone for 1 year, the researchers said (J Clin Oncol. 2016 Apr 25. doi: 10.1200/JCO.2015.63.1929).
A total of 20% of patients had a complete response to the entire treatment protocol, 13% had a stringent complete response, 26% had a very good partial response, 5% had stable disease, and 5% had progressive disease. Thirteen patients died of progressive disease and four died of infections, including three that occurred during induction or after allograft.
This is only the second prospective trial in patients with primary plasma cell leukemia, an aggressive form of multiple myeloma that accounts for 2%-4% of cases, the researchers said. Future prospective trials should seek to optimize induction with newer combinations, such as carfilzomib, lenalidomide, dexamethasone, or monoclonal anti-CD38 antibodies. Also, optimizing the stem cell conditioning procedure and the postallograft immunomodulation may further benefit younger patients.
Dr. Royer reported receiving honoraria from Amgen and having served as a consultant or advisor for Octapharma Plasma. Fifteen coinvestigators also reported financial relationships with a number of pharmaceutical companies.
In a prospective study of 40 patients with primary plasma cell leukemia, upfront autotransplantation followed by allotransplant for younger patients and by consolidation/maintenance for older patients was associated with a median overall survival of 36.3 months and a median progression-free survival of 15.1 months.
Patients with this aggressive form of multiple myeloma received a regimen that combined standard chemotherapy, a proteasome inhibitor, high-dose melphalan followed by autologous stem cell transplantation, and allogeneic transplantation or immunomodulatory drugs, reported Dr. Bruno Royer of University Hospital in Amiens, France, and his associates.
Induction therapy consisted of four 21-day cycles: Cycles 1 and 3 included subcutaneous bortezomib, intravenous pegylated doxorubicin, and oral dexamethasone; cycles 2 and 4 included subcutaneous bortezomib, oral cyclophosphamide, and oral dexamethasone. Of 39 patients – one patient died 24 hours after study inclusion – 35 completed the four cycles. The overall response rate to induction was 69%: 10% of patients had a complete response and 26% had a very good partial response. Of 27 responding patients, 25 underwent high-dose melphalan followed by autologous stem cell transplantation.
The high response rates allowed 16 patients who were younger than 66 years and had an HLA-matched donor to then receive high-dose melphalan followed by autologous stem cell transplantation followed by consolidation with either an reduced-intensity conditioning allograft or a second high-dose melphalan followed by autologous stem cell transplantation and subsequent maintenance with lenalidomide, bortezomib, and dexamethasone for 1 year, the researchers said (J Clin Oncol. 2016 Apr 25. doi: 10.1200/JCO.2015.63.1929).
A total of 20% of patients had a complete response to the entire treatment protocol, 13% had a stringent complete response, 26% had a very good partial response, 5% had stable disease, and 5% had progressive disease. Thirteen patients died of progressive disease and four died of infections, including three that occurred during induction or after allograft.
This is only the second prospective trial in patients with primary plasma cell leukemia, an aggressive form of multiple myeloma that accounts for 2%-4% of cases, the researchers said. Future prospective trials should seek to optimize induction with newer combinations, such as carfilzomib, lenalidomide, dexamethasone, or monoclonal anti-CD38 antibodies. Also, optimizing the stem cell conditioning procedure and the postallograft immunomodulation may further benefit younger patients.
Dr. Royer reported receiving honoraria from Amgen and having served as a consultant or advisor for Octapharma Plasma. Fifteen coinvestigators also reported financial relationships with a number of pharmaceutical companies.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Progression-free survival was improved in patients who had primary plasma cell leukemia and underwent four cycles of induction; high-dose melphalan followed by autologous stem cell transplantation; consolidation with either a reduced-intensity conditioning allograft or a second high-dose melphalan followed by autologous stem cell transplantation; and subsequent maintenance with lenalidomide, bortezomib, and dexamethasone for 1 year.
Major finding: Median overall survival was 36.3 months and median progression-free survival was 15.1 months.
Data source: A prospective phase II study of 40 adults with newly diagnosed primary plasma cell leukemia.
Disclosures: Dr. Royer reported receiving honoraria from Amgen and having served as a consultant or advisor for Octapharma Plasma. Fifteen coinvestigators also reported financial relationships with various drug companies.
Combo may be active in refractory MM

Photo courtesy of the CDC
The combination of vorinostat and bortezomib, with or without dexamethasone, can be active in patients with multiple myeloma (MM) that is refractory to novel treatments, according to researchers.
In a phase 2 trial, the combination produced an overall response rate of 11% among MM patients who were refractory to bortezomib and were either refractory to or could not receive treatment with an immunomodulatory agent (IMiD).
About 92% of patients had drug-related adverse events (AEs), and 20% had serious drug-related AEs.
These results were published in Clinical Lymphoma, Myeloma & Leukemia. The study was funded by Merck & Co., Inc., which markets vorinostat as Zolinza.
This trial (VANTAGE 095) enrolled 143 MM patients from 41 centers in 12 countries. The patients had a median age of 63 (range, 37-81) and had received a median of 4 prior lines of therapy (range, 2-17).
All 143 patients were considered refractory to previous bortezomib, which was defined as less than 25% response on therapy or progression during/less than 60 days after the completion of bortezomib therapy.
All but 1 patient had been exposed to 1 or more IMiDs (99.3%). Roughly 87% of patients exposed to IMiDs were considered to have disease refractory to at least 1 IMiD and 40% to at least 2 different IMiDs. Three percent of patients were considered ineligible for further IMiD-based therapy because of previous toxicities.
For this study, patients received 21-day cycles of bortezomib (1.3 mg/m2 intravenously; days 1, 4, 8, and 11) plus oral vorinostat at 400 mg/d on days 1 to 14.
If a patient had no change as the best response after 4 cycles of treatment or progressive disease after 2 cycles of treatment, oral dexamethasone at 20 mg on the day of and day after each dose of bortezomib could be added to the treatment regimen.
Patients were treated until disease progression, unacceptable toxicities, or withdrawal from the study.
One hundred and forty-two patients were evaluable for safety and efficacy. Fifty-seven of these patients received dexamethasone per protocol.
The overall response rate (partial response or better), as assessed by an independent adjudication committee, was 11.3%.
All 16 responses were partial responses. The median time to response was 44 days (range, 22-71), and the median duration of response was 211 days (range, 64-550 days).
Eleven patients (7.7%) had a minimal response, and 87 (61.3%) had stable disease.
The median progression-free survival was 3.13 months, and the median time to progression was 3.47 months. The median overall survival was 11.2 months, with a 2-year survival rate of 32%.
All 142 patients had at least 1 AE, and 131 (92.3%) had drug-related AEs. Most AEs were grade 3 (28.9%) or 4 (50.7%). Serious AEs occurred in 64.8% of patients, and serious drug-related AEs occurred in 20.4%.
Twenty-seven patients (19%) discontinued treatment due to an AE. And 24 patients (16.9%) died from an AE, although 18 of these deaths were attributable to progression of underlying MM.
The most common AEs were thrombocytopenia (69.7%), nausea (57.0%), diarrhea (53.5%), anemia (52.1%), and fatigue (48.6%). ![]()
Photo courtesy of the CDC
The combination of vorinostat and bortezomib, with or without dexamethasone, can be active in patients with multiple myeloma (MM) that is refractory to novel treatments, according to researchers.
In a phase 2 trial, the combination produced an overall response rate of 11% among MM patients who were refractory to bortezomib and were either refractory to or could not receive treatment with an immunomodulatory agent (IMiD).
About 92% of patients had drug-related adverse events (AEs), and 20% had serious drug-related AEs.
These results were published in Clinical Lymphoma, Myeloma & Leukemia. The study was funded by Merck & Co., Inc., which markets vorinostat as Zolinza.
This trial (VANTAGE 095) enrolled 143 MM patients from 41 centers in 12 countries. The patients had a median age of 63 (range, 37-81) and had received a median of 4 prior lines of therapy (range, 2-17).
All 143 patients were considered refractory to previous bortezomib, which was defined as less than 25% response on therapy or progression during/less than 60 days after the completion of bortezomib therapy.
All but 1 patient had been exposed to 1 or more IMiDs (99.3%). Roughly 87% of patients exposed to IMiDs were considered to have disease refractory to at least 1 IMiD and 40% to at least 2 different IMiDs. Three percent of patients were considered ineligible for further IMiD-based therapy because of previous toxicities.
For this study, patients received 21-day cycles of bortezomib (1.3 mg/m2 intravenously; days 1, 4, 8, and 11) plus oral vorinostat at 400 mg/d on days 1 to 14.
If a patient had no change as the best response after 4 cycles of treatment or progressive disease after 2 cycles of treatment, oral dexamethasone at 20 mg on the day of and day after each dose of bortezomib could be added to the treatment regimen.
Patients were treated until disease progression, unacceptable toxicities, or withdrawal from the study.
One hundred and forty-two patients were evaluable for safety and efficacy. Fifty-seven of these patients received dexamethasone per protocol.
The overall response rate (partial response or better), as assessed by an independent adjudication committee, was 11.3%.
All 16 responses were partial responses. The median time to response was 44 days (range, 22-71), and the median duration of response was 211 days (range, 64-550 days).
Eleven patients (7.7%) had a minimal response, and 87 (61.3%) had stable disease.
The median progression-free survival was 3.13 months, and the median time to progression was 3.47 months. The median overall survival was 11.2 months, with a 2-year survival rate of 32%.
All 142 patients had at least 1 AE, and 131 (92.3%) had drug-related AEs. Most AEs were grade 3 (28.9%) or 4 (50.7%). Serious AEs occurred in 64.8% of patients, and serious drug-related AEs occurred in 20.4%.
Twenty-seven patients (19%) discontinued treatment due to an AE. And 24 patients (16.9%) died from an AE, although 18 of these deaths were attributable to progression of underlying MM.
The most common AEs were thrombocytopenia (69.7%), nausea (57.0%), diarrhea (53.5%), anemia (52.1%), and fatigue (48.6%). ![]()
Photo courtesy of the CDC
The combination of vorinostat and bortezomib, with or without dexamethasone, can be active in patients with multiple myeloma (MM) that is refractory to novel treatments, according to researchers.
In a phase 2 trial, the combination produced an overall response rate of 11% among MM patients who were refractory to bortezomib and were either refractory to or could not receive treatment with an immunomodulatory agent (IMiD).
About 92% of patients had drug-related adverse events (AEs), and 20% had serious drug-related AEs.
These results were published in Clinical Lymphoma, Myeloma & Leukemia. The study was funded by Merck & Co., Inc., which markets vorinostat as Zolinza.
This trial (VANTAGE 095) enrolled 143 MM patients from 41 centers in 12 countries. The patients had a median age of 63 (range, 37-81) and had received a median of 4 prior lines of therapy (range, 2-17).
All 143 patients were considered refractory to previous bortezomib, which was defined as less than 25% response on therapy or progression during/less than 60 days after the completion of bortezomib therapy.
All but 1 patient had been exposed to 1 or more IMiDs (99.3%). Roughly 87% of patients exposed to IMiDs were considered to have disease refractory to at least 1 IMiD and 40% to at least 2 different IMiDs. Three percent of patients were considered ineligible for further IMiD-based therapy because of previous toxicities.
For this study, patients received 21-day cycles of bortezomib (1.3 mg/m2 intravenously; days 1, 4, 8, and 11) plus oral vorinostat at 400 mg/d on days 1 to 14.
If a patient had no change as the best response after 4 cycles of treatment or progressive disease after 2 cycles of treatment, oral dexamethasone at 20 mg on the day of and day after each dose of bortezomib could be added to the treatment regimen.
Patients were treated until disease progression, unacceptable toxicities, or withdrawal from the study.
One hundred and forty-two patients were evaluable for safety and efficacy. Fifty-seven of these patients received dexamethasone per protocol.
The overall response rate (partial response or better), as assessed by an independent adjudication committee, was 11.3%.
All 16 responses were partial responses. The median time to response was 44 days (range, 22-71), and the median duration of response was 211 days (range, 64-550 days).
Eleven patients (7.7%) had a minimal response, and 87 (61.3%) had stable disease.
The median progression-free survival was 3.13 months, and the median time to progression was 3.47 months. The median overall survival was 11.2 months, with a 2-year survival rate of 32%.
All 142 patients had at least 1 AE, and 131 (92.3%) had drug-related AEs. Most AEs were grade 3 (28.9%) or 4 (50.7%). Serious AEs occurred in 64.8% of patients, and serious drug-related AEs occurred in 20.4%.
Twenty-seven patients (19%) discontinued treatment due to an AE. And 24 patients (16.9%) died from an AE, although 18 of these deaths were attributable to progression of underlying MM.
The most common AEs were thrombocytopenia (69.7%), nausea (57.0%), diarrhea (53.5%), anemia (52.1%), and fatigue (48.6%). ![]()
Combo could improve treatment of MM, team says

multiple myeloma
Combining a calcineurin inhibitor and a histone deacetylase (HDAC) inhibitor could improve the treatment of multiple myeloma (MM), according to researchers.
The team found that MM cells express high levels of the protein phosphatase PPP3CA, a subunit of calcineurin.
And combining the calcineurin inhibitor FK506 with the HDAC inhibitor panobinostat suppressed MM cell growth in vitro and decreased tumor growth in mouse models of MM.
Yoichi Imai, MD, PhD, of Tokyo Women’s Medical University in Japan, and colleagues conducted this research and reported the results in JCI Insight.
First, the team observed increased PPP3CA expression in MM cell lines and MM cells isolated from patients with advanced disease.
Then, the researchers found that panobinostat reduced PPP3CA expression in MM cell lines. And further investigation revealed that the drug induced degradation of PPP3CA through HSP90 inhibition.
When the team knocked down PPP3CA in MM cells, they observed a reduction in cell growth. And when they overexpressed PPP3CA, they observed enhanced MM cell growth.
The researchers noted that FK506 inhibits the association between PPP3CA and calcineurin B. Unfortunately, FK506 alone did not suppress the growth of MM cells in vitro.
However, when FK506 was given with panobinostat or the HDAC inhibitor ACY-1215, the researchers observed a greater reduction in MM cell growth than with either HDAC inhibitor alone.
Panobinostat and FK506 reduced the growth of MM cells that were t(4;14)-positive (KMS-11, KMS-18, and KMS-26) and t(4;14)-negative (U266 and KMS-12PE) more effectively than panobinostat alone.
In mice with MM, those treated with FK506 alone had tumor sizes similar to control mice. However, mice treated with panobinostat saw a decrease in tumor size. And this effect was enhanced by the addition of FK506.
The researchers observed reduced PPP3CA expression, enhanced histone H3 acetylation, and cleavage of caspase-3 in samples from panobinostat-treated mice. And FK506 augmented panobinostat-induced apoptosis.
The team said these results suggest that FK506 enhances the antimyeloma effect of panobinostat through PPP3CA reduction, which supports the importance of calcineurin in the pathogenesis of MM. ![]()
multiple myeloma
Combining a calcineurin inhibitor and a histone deacetylase (HDAC) inhibitor could improve the treatment of multiple myeloma (MM), according to researchers.
The team found that MM cells express high levels of the protein phosphatase PPP3CA, a subunit of calcineurin.
And combining the calcineurin inhibitor FK506 with the HDAC inhibitor panobinostat suppressed MM cell growth in vitro and decreased tumor growth in mouse models of MM.
Yoichi Imai, MD, PhD, of Tokyo Women’s Medical University in Japan, and colleagues conducted this research and reported the results in JCI Insight.
First, the team observed increased PPP3CA expression in MM cell lines and MM cells isolated from patients with advanced disease.
Then, the researchers found that panobinostat reduced PPP3CA expression in MM cell lines. And further investigation revealed that the drug induced degradation of PPP3CA through HSP90 inhibition.
When the team knocked down PPP3CA in MM cells, they observed a reduction in cell growth. And when they overexpressed PPP3CA, they observed enhanced MM cell growth.
The researchers noted that FK506 inhibits the association between PPP3CA and calcineurin B. Unfortunately, FK506 alone did not suppress the growth of MM cells in vitro.
However, when FK506 was given with panobinostat or the HDAC inhibitor ACY-1215, the researchers observed a greater reduction in MM cell growth than with either HDAC inhibitor alone.
Panobinostat and FK506 reduced the growth of MM cells that were t(4;14)-positive (KMS-11, KMS-18, and KMS-26) and t(4;14)-negative (U266 and KMS-12PE) more effectively than panobinostat alone.
In mice with MM, those treated with FK506 alone had tumor sizes similar to control mice. However, mice treated with panobinostat saw a decrease in tumor size. And this effect was enhanced by the addition of FK506.
The researchers observed reduced PPP3CA expression, enhanced histone H3 acetylation, and cleavage of caspase-3 in samples from panobinostat-treated mice. And FK506 augmented panobinostat-induced apoptosis.
The team said these results suggest that FK506 enhances the antimyeloma effect of panobinostat through PPP3CA reduction, which supports the importance of calcineurin in the pathogenesis of MM. ![]()
multiple myeloma
Combining a calcineurin inhibitor and a histone deacetylase (HDAC) inhibitor could improve the treatment of multiple myeloma (MM), according to researchers.
The team found that MM cells express high levels of the protein phosphatase PPP3CA, a subunit of calcineurin.
And combining the calcineurin inhibitor FK506 with the HDAC inhibitor panobinostat suppressed MM cell growth in vitro and decreased tumor growth in mouse models of MM.
Yoichi Imai, MD, PhD, of Tokyo Women’s Medical University in Japan, and colleagues conducted this research and reported the results in JCI Insight.
First, the team observed increased PPP3CA expression in MM cell lines and MM cells isolated from patients with advanced disease.
Then, the researchers found that panobinostat reduced PPP3CA expression in MM cell lines. And further investigation revealed that the drug induced degradation of PPP3CA through HSP90 inhibition.
When the team knocked down PPP3CA in MM cells, they observed a reduction in cell growth. And when they overexpressed PPP3CA, they observed enhanced MM cell growth.
The researchers noted that FK506 inhibits the association between PPP3CA and calcineurin B. Unfortunately, FK506 alone did not suppress the growth of MM cells in vitro.
However, when FK506 was given with panobinostat or the HDAC inhibitor ACY-1215, the researchers observed a greater reduction in MM cell growth than with either HDAC inhibitor alone.
Panobinostat and FK506 reduced the growth of MM cells that were t(4;14)-positive (KMS-11, KMS-18, and KMS-26) and t(4;14)-negative (U266 and KMS-12PE) more effectively than panobinostat alone.
In mice with MM, those treated with FK506 alone had tumor sizes similar to control mice. However, mice treated with panobinostat saw a decrease in tumor size. And this effect was enhanced by the addition of FK506.
The researchers observed reduced PPP3CA expression, enhanced histone H3 acetylation, and cleavage of caspase-3 in samples from panobinostat-treated mice. And FK506 augmented panobinostat-induced apoptosis.
The team said these results suggest that FK506 enhances the antimyeloma effect of panobinostat through PPP3CA reduction, which supports the importance of calcineurin in the pathogenesis of MM. ![]()
Daratumumab well tolerated, effective in heavily treated multiple myeloma
Daratumumab monotherapy was associated with an overall response rate of 29.2% and was well tolerated in 106 heavily treated patients with multiple myeloma, based on results from the SIRIUS trial.
Of 106 patients who received daratumumab at 16 mg/kg, 3% achieved a stringent complete response, 9% had a very good partial response, and 17% had a partial response. The median progression-free survival was 3.7 months and median duration of response was 7.4 months. The 12-month overall survival was 64·8%, and, at a subsequent cutoff, median overall survival was 17.5 months.
All of the study patients had been treated with proteasome inhibitors and immunomodulatory drugs, with a median of five previous therapies. Most patients (80%) had received autologous stem cell transplantation, and 97% were refractory to the last line of therapy before study enrollment.
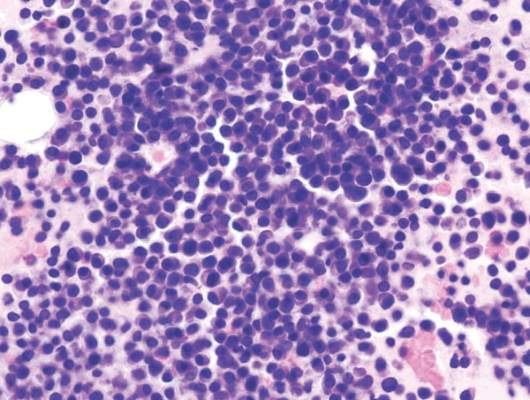
“Resistance to any previous therapy had no effect on the activity of daratumumab, lending support to a novel mechanism of action, but these findings need to be confirmed in larger studies,” wrote Dr. Sagar Lonial of Emory University, Atlanta, and colleagues (Lancet 2016;387:1551-60). Response rates were similar for patients with moderate renal impairment, those over age 75, and those with extramedullary disease or high-risk baseline cytogenetic characteristics.
Daratumumab was well tolerated, and none of the patients discontinued treatment because of treatment-related adverse events. The most common adverse events of any grade were anemia (33%), thrombocytopenia (25%), and neutropenia (23%). Additional supportive care in the form of red blood cell transfusions was received by 38% of patients, platelet transfusions by 13%, and granulocyte colony-stimulating factor by 8%. Fatigue (40%) and nausea (29%) were the most common nonhematologic adverse events. Serious adverse events were observed in 30% of patients.
Daratumumab compares favorably with other regimens such as pomalidomide alone or with dexamethasone or carfilzomib monotherapy, according to the investigators.
The favorable safety profile of daratumumab makes it an attractive candidate for combination regimens, the authors noted, and daratumumab combined with other backbone agents are currently under investigation.
This study was sponsored by Janssen, maker of daratumumab (Darzalex). Dr. Lonial reported consulting or advisory roles with Janssen and several other drug companies.
With its novel mechanism of action, single-agent activity, absence of crossresistance, and tolerability, daratumumab may prove to be a transformative new treatment for multiple myeloma.

|
| Patrice Wendling/Frontline Medical News Dr. S. Vincent Rajkumar |
The single-agent activity of daratumumab (29%) exceeds that of bortezomib (27%), lenalidomide (26%), carfilzomib (24%), or pomalidomide (18%), even in a heavily pretreated population.
The safety profile is outstanding, and therein lies the reason for enthusiasm: Daratumumab can probably be combined with currently used triplet combinations in multiple myeloma, and can potentially take these highly active regimens to new heights.
Similar to rituximab, daratumumab will probably be added to many active triplet combinations. Daratumumab will likely move rapidly to a front-line setting in clinical trials for treatment of newly diagnosed multiple myeloma, maintenance therapy, and even smoldering multiple myeloma.
However, the data are insufficient to determine the cytogenetic subtypes that respond best to daratumumab. That information will be necessary in order to best sequence drugs according to the subtype of myeloma.
It will be important also to understand how daratumumab, an anti-CD38 drug, can work optimally with elotuzumab, another newly approved monoclonal antibody that targets SLAMF7.
Dr. S. Vincent Rajkumar is with the Mayo Clinic, Rochester, Minn. These remarks were part of an editorial (Lancet 2016; 387:1490-91) accompanying the study in the Lancet.
With its novel mechanism of action, single-agent activity, absence of crossresistance, and tolerability, daratumumab may prove to be a transformative new treatment for multiple myeloma.
|
|
| Patrice Wendling/Frontline Medical News Dr. S. Vincent Rajkumar |
The single-agent activity of daratumumab (29%) exceeds that of bortezomib (27%), lenalidomide (26%), carfilzomib (24%), or pomalidomide (18%), even in a heavily pretreated population.
The safety profile is outstanding, and therein lies the reason for enthusiasm: Daratumumab can probably be combined with currently used triplet combinations in multiple myeloma, and can potentially take these highly active regimens to new heights.
Similar to rituximab, daratumumab will probably be added to many active triplet combinations. Daratumumab will likely move rapidly to a front-line setting in clinical trials for treatment of newly diagnosed multiple myeloma, maintenance therapy, and even smoldering multiple myeloma.
However, the data are insufficient to determine the cytogenetic subtypes that respond best to daratumumab. That information will be necessary in order to best sequence drugs according to the subtype of myeloma.
It will be important also to understand how daratumumab, an anti-CD38 drug, can work optimally with elotuzumab, another newly approved monoclonal antibody that targets SLAMF7.
Dr. S. Vincent Rajkumar is with the Mayo Clinic, Rochester, Minn. These remarks were part of an editorial (Lancet 2016; 387:1490-91) accompanying the study in the Lancet.
With its novel mechanism of action, single-agent activity, absence of crossresistance, and tolerability, daratumumab may prove to be a transformative new treatment for multiple myeloma.
|
|
| Patrice Wendling/Frontline Medical News Dr. S. Vincent Rajkumar |
The single-agent activity of daratumumab (29%) exceeds that of bortezomib (27%), lenalidomide (26%), carfilzomib (24%), or pomalidomide (18%), even in a heavily pretreated population.
The safety profile is outstanding, and therein lies the reason for enthusiasm: Daratumumab can probably be combined with currently used triplet combinations in multiple myeloma, and can potentially take these highly active regimens to new heights.
Similar to rituximab, daratumumab will probably be added to many active triplet combinations. Daratumumab will likely move rapidly to a front-line setting in clinical trials for treatment of newly diagnosed multiple myeloma, maintenance therapy, and even smoldering multiple myeloma.
However, the data are insufficient to determine the cytogenetic subtypes that respond best to daratumumab. That information will be necessary in order to best sequence drugs according to the subtype of myeloma.
It will be important also to understand how daratumumab, an anti-CD38 drug, can work optimally with elotuzumab, another newly approved monoclonal antibody that targets SLAMF7.
Dr. S. Vincent Rajkumar is with the Mayo Clinic, Rochester, Minn. These remarks were part of an editorial (Lancet 2016; 387:1490-91) accompanying the study in the Lancet.
Daratumumab monotherapy was associated with an overall response rate of 29.2% and was well tolerated in 106 heavily treated patients with multiple myeloma, based on results from the SIRIUS trial.
Of 106 patients who received daratumumab at 16 mg/kg, 3% achieved a stringent complete response, 9% had a very good partial response, and 17% had a partial response. The median progression-free survival was 3.7 months and median duration of response was 7.4 months. The 12-month overall survival was 64·8%, and, at a subsequent cutoff, median overall survival was 17.5 months.
All of the study patients had been treated with proteasome inhibitors and immunomodulatory drugs, with a median of five previous therapies. Most patients (80%) had received autologous stem cell transplantation, and 97% were refractory to the last line of therapy before study enrollment.
“Resistance to any previous therapy had no effect on the activity of daratumumab, lending support to a novel mechanism of action, but these findings need to be confirmed in larger studies,” wrote Dr. Sagar Lonial of Emory University, Atlanta, and colleagues (Lancet 2016;387:1551-60). Response rates were similar for patients with moderate renal impairment, those over age 75, and those with extramedullary disease or high-risk baseline cytogenetic characteristics.
Daratumumab was well tolerated, and none of the patients discontinued treatment because of treatment-related adverse events. The most common adverse events of any grade were anemia (33%), thrombocytopenia (25%), and neutropenia (23%). Additional supportive care in the form of red blood cell transfusions was received by 38% of patients, platelet transfusions by 13%, and granulocyte colony-stimulating factor by 8%. Fatigue (40%) and nausea (29%) were the most common nonhematologic adverse events. Serious adverse events were observed in 30% of patients.
Daratumumab compares favorably with other regimens such as pomalidomide alone or with dexamethasone or carfilzomib monotherapy, according to the investigators.
The favorable safety profile of daratumumab makes it an attractive candidate for combination regimens, the authors noted, and daratumumab combined with other backbone agents are currently under investigation.
This study was sponsored by Janssen, maker of daratumumab (Darzalex). Dr. Lonial reported consulting or advisory roles with Janssen and several other drug companies.
Daratumumab monotherapy was associated with an overall response rate of 29.2% and was well tolerated in 106 heavily treated patients with multiple myeloma, based on results from the SIRIUS trial.
Of 106 patients who received daratumumab at 16 mg/kg, 3% achieved a stringent complete response, 9% had a very good partial response, and 17% had a partial response. The median progression-free survival was 3.7 months and median duration of response was 7.4 months. The 12-month overall survival was 64·8%, and, at a subsequent cutoff, median overall survival was 17.5 months.
All of the study patients had been treated with proteasome inhibitors and immunomodulatory drugs, with a median of five previous therapies. Most patients (80%) had received autologous stem cell transplantation, and 97% were refractory to the last line of therapy before study enrollment.
“Resistance to any previous therapy had no effect on the activity of daratumumab, lending support to a novel mechanism of action, but these findings need to be confirmed in larger studies,” wrote Dr. Sagar Lonial of Emory University, Atlanta, and colleagues (Lancet 2016;387:1551-60). Response rates were similar for patients with moderate renal impairment, those over age 75, and those with extramedullary disease or high-risk baseline cytogenetic characteristics.
Daratumumab was well tolerated, and none of the patients discontinued treatment because of treatment-related adverse events. The most common adverse events of any grade were anemia (33%), thrombocytopenia (25%), and neutropenia (23%). Additional supportive care in the form of red blood cell transfusions was received by 38% of patients, platelet transfusions by 13%, and granulocyte colony-stimulating factor by 8%. Fatigue (40%) and nausea (29%) were the most common nonhematologic adverse events. Serious adverse events were observed in 30% of patients.
Daratumumab compares favorably with other regimens such as pomalidomide alone or with dexamethasone or carfilzomib monotherapy, according to the investigators.
The favorable safety profile of daratumumab makes it an attractive candidate for combination regimens, the authors noted, and daratumumab combined with other backbone agents are currently under investigation.
This study was sponsored by Janssen, maker of daratumumab (Darzalex). Dr. Lonial reported consulting or advisory roles with Janssen and several other drug companies.
FROM THE LANCET
Key clinical point: Daratumumab was well tolerated and showed encouraging activity in heavily treated patients with multiple myeloma.
Major finding: In 106 patients previously treated with a median of five lines of therapy, the overall response rate to daratumumab at 16 mg/kg was 29.2%; 3% achieved a stringent complete response, 9% a very good partial response, and 17% a partial response.
Data sources: Data from the SIRIUS trial for 106 patients in the 16-mg/kg group.
Disclosures: This study was sponsored by Janssen, maker of daratumumab (Darzalex). Dr. Lonial reported consulting or advisory roles with Janssen and several other drug companies.
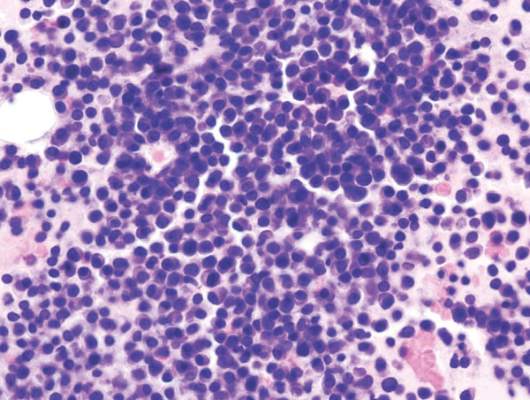
mAb can reduce CSCs in newly diagnosed MM

Photo by Linda Bartlett
NEW ORLEANS—A small study suggests that treatment with lenalidomide and dexamethasone (len-dex) prompts an increase in cancer stem cells (CSCs) for patients with newly diagnosed multiple myeloma (MM).
However, adding an anti-CD19 monoclonal antibody (mAb) to the regimen can reduce CSCs.
Most patients who received the mAb, MEDI-551, experienced a decrease in CSCs, but the cells rebounded after the patients stopped receiving MEDI-551.
And those patients who did not see a decrease in CSCs progressed. However, some patients are still in response and remain on treatment with len-dex.
The investigators believe these early results suggest prolonged treatment with len-dex and MED-551 may be safe and clinically beneficial for MM patients.
The results were presented at the 2016 AACR Annual Meeting (abstract CT102).
The study included 17 patients with newly diagnosed MM. They had a median age of 65 (range, 34-73). Most had ISS stage I (n=11), 2 had stage II, and 4 had stage III. Seven patients had t(4;14).
“We chose to carry out this clinical trial in newly diagnosed patients because our original data showed that CD19 was almost always expressed by myeloma stem cells in these patients, whereas we don’t know if that is the case in more advanced patients,” said investigator William Matsui, MD, of Johns Hopkins University School of Medicine in Baltimore, Maryland.
The patients received 28-day cycles of len-dex (len at 25 mg PO, days 1-21, and dex at 40 mg PO, weekly). Patients received MEDI-551 (at 4 mg/kg IV) in cycle 3 (day 1, 8) and cycle 4 (day 1). Responding patients continued on len-dex.
The investigators measured MM CSCs by quantifying the growth of MM colonies (CFU-MM) from marrow aspirates at baseline and at the end of cycles 2 and 4.
The team quantified peripheral blood CSCs by flow cytometry (CD19+CD27+ALDH+) at baseline and the end of cycles 2, 4, 5, and 7.
“We wanted to see if these 2 assays gave similar results, and, in this clinical trial, they were almost identical,” said investigator Carol Ann Huff, MD, also of Johns Hopkins.
“Since it is much easier to draw blood than bone marrow from our patients, we think that we can primarily use blood to track multiple myeloma stem cells in the future.”
Response
Two patients did not receive MEDI-551 due to progressive disease and noncompliance. So the investigators assessed responses in 15 patients.
After cycle 2 (len-dex alone), there were 3 very good partial responses (VGPRs), 10 partial responses (PRs), 1 molecular response, and 1 case of stable disease.
After cycle 4 (len-dex plus MEDI-551), there were 6 VGPRs, 8 PRs, and 1 molecular response.
Ten patients who completed treatment with MED-551 remain on len-dex. At the end of cycle 7, there was 1 complete response, 8 VGPRs, and 1 PR.
CFU-MM
When compared to baseline, bone-marrow-derived CFU-MM increased a median of 2.5-fold (range, 0.4-7.4) after cycle 2 but decreased a median of 0.48-fold (range, 0.14-0.85) in 14 patients after cycle 4.
The investigators compared these results to 5 newly diagnosed MM patients who only received standard treatment with len-dex.
In these patients, CFU-MM increased a median of 9.3-fold (range, 4-14) at a median of 4 months (range, 2-4). This is in spite of the fact that all of these patients had a PR or better.
Circulating CSCs
Compared to baseline, circulating MM CSCs increased a median of 1.6-fold (range, 0.4-8.6) in 14 patients after cycle 2 but decreased a median of 0.6-fold (range, 0.01-7.4) in 13 patients after cycle 4.
At the end of cycle 5, MM CSCs had increased in 4 of the 10 patients who were still on len-dex. By the end of cycle 7, MM CSCs had increased in 8 of the patients.
Circulating MM CSCs increased by the end of cycle 4 in 2 patients, and both had progressed by end of cycle 7.
Safety and next steps
The investigators said there were no serious adverse events in this trial, but 2 patients experienced grade 2 infusion reactions after the first MEDI-551 dose.
The team plans to conduct further studies to assess the long-term impact of MED-551 in MM patients and determine how the mAb might work in combination with other treatments, particularly transplant.
“In other studies at Johns Hopkins, we have found that antibody therapies can work much better after a bone marrow transplant, especially allogeneic transplants,” Dr Matsui said.
Funding and drugs for this study were provided by MedImmune Inc., the developers of MEDI-551. Drs Huff and Matsui served as a paid scientific advisory board member and a consultant to MedImmune Inc., respectively. ![]()
Photo by Linda Bartlett
NEW ORLEANS—A small study suggests that treatment with lenalidomide and dexamethasone (len-dex) prompts an increase in cancer stem cells (CSCs) for patients with newly diagnosed multiple myeloma (MM).
However, adding an anti-CD19 monoclonal antibody (mAb) to the regimen can reduce CSCs.
Most patients who received the mAb, MEDI-551, experienced a decrease in CSCs, but the cells rebounded after the patients stopped receiving MEDI-551.
And those patients who did not see a decrease in CSCs progressed. However, some patients are still in response and remain on treatment with len-dex.
The investigators believe these early results suggest prolonged treatment with len-dex and MED-551 may be safe and clinically beneficial for MM patients.
The results were presented at the 2016 AACR Annual Meeting (abstract CT102).
The study included 17 patients with newly diagnosed MM. They had a median age of 65 (range, 34-73). Most had ISS stage I (n=11), 2 had stage II, and 4 had stage III. Seven patients had t(4;14).
“We chose to carry out this clinical trial in newly diagnosed patients because our original data showed that CD19 was almost always expressed by myeloma stem cells in these patients, whereas we don’t know if that is the case in more advanced patients,” said investigator William Matsui, MD, of Johns Hopkins University School of Medicine in Baltimore, Maryland.
The patients received 28-day cycles of len-dex (len at 25 mg PO, days 1-21, and dex at 40 mg PO, weekly). Patients received MEDI-551 (at 4 mg/kg IV) in cycle 3 (day 1, 8) and cycle 4 (day 1). Responding patients continued on len-dex.
The investigators measured MM CSCs by quantifying the growth of MM colonies (CFU-MM) from marrow aspirates at baseline and at the end of cycles 2 and 4.
The team quantified peripheral blood CSCs by flow cytometry (CD19+CD27+ALDH+) at baseline and the end of cycles 2, 4, 5, and 7.
“We wanted to see if these 2 assays gave similar results, and, in this clinical trial, they were almost identical,” said investigator Carol Ann Huff, MD, also of Johns Hopkins.
“Since it is much easier to draw blood than bone marrow from our patients, we think that we can primarily use blood to track multiple myeloma stem cells in the future.”
Response
Two patients did not receive MEDI-551 due to progressive disease and noncompliance. So the investigators assessed responses in 15 patients.
After cycle 2 (len-dex alone), there were 3 very good partial responses (VGPRs), 10 partial responses (PRs), 1 molecular response, and 1 case of stable disease.
After cycle 4 (len-dex plus MEDI-551), there were 6 VGPRs, 8 PRs, and 1 molecular response.
Ten patients who completed treatment with MED-551 remain on len-dex. At the end of cycle 7, there was 1 complete response, 8 VGPRs, and 1 PR.
CFU-MM
When compared to baseline, bone-marrow-derived CFU-MM increased a median of 2.5-fold (range, 0.4-7.4) after cycle 2 but decreased a median of 0.48-fold (range, 0.14-0.85) in 14 patients after cycle 4.
The investigators compared these results to 5 newly diagnosed MM patients who only received standard treatment with len-dex.
In these patients, CFU-MM increased a median of 9.3-fold (range, 4-14) at a median of 4 months (range, 2-4). This is in spite of the fact that all of these patients had a PR or better.
Circulating CSCs
Compared to baseline, circulating MM CSCs increased a median of 1.6-fold (range, 0.4-8.6) in 14 patients after cycle 2 but decreased a median of 0.6-fold (range, 0.01-7.4) in 13 patients after cycle 4.
At the end of cycle 5, MM CSCs had increased in 4 of the 10 patients who were still on len-dex. By the end of cycle 7, MM CSCs had increased in 8 of the patients.
Circulating MM CSCs increased by the end of cycle 4 in 2 patients, and both had progressed by end of cycle 7.
Safety and next steps
The investigators said there were no serious adverse events in this trial, but 2 patients experienced grade 2 infusion reactions after the first MEDI-551 dose.
The team plans to conduct further studies to assess the long-term impact of MED-551 in MM patients and determine how the mAb might work in combination with other treatments, particularly transplant.
“In other studies at Johns Hopkins, we have found that antibody therapies can work much better after a bone marrow transplant, especially allogeneic transplants,” Dr Matsui said.
Funding and drugs for this study were provided by MedImmune Inc., the developers of MEDI-551. Drs Huff and Matsui served as a paid scientific advisory board member and a consultant to MedImmune Inc., respectively. ![]()
Photo by Linda Bartlett
NEW ORLEANS—A small study suggests that treatment with lenalidomide and dexamethasone (len-dex) prompts an increase in cancer stem cells (CSCs) for patients with newly diagnosed multiple myeloma (MM).
However, adding an anti-CD19 monoclonal antibody (mAb) to the regimen can reduce CSCs.
Most patients who received the mAb, MEDI-551, experienced a decrease in CSCs, but the cells rebounded after the patients stopped receiving MEDI-551.
And those patients who did not see a decrease in CSCs progressed. However, some patients are still in response and remain on treatment with len-dex.
The investigators believe these early results suggest prolonged treatment with len-dex and MED-551 may be safe and clinically beneficial for MM patients.
The results were presented at the 2016 AACR Annual Meeting (abstract CT102).
The study included 17 patients with newly diagnosed MM. They had a median age of 65 (range, 34-73). Most had ISS stage I (n=11), 2 had stage II, and 4 had stage III. Seven patients had t(4;14).
“We chose to carry out this clinical trial in newly diagnosed patients because our original data showed that CD19 was almost always expressed by myeloma stem cells in these patients, whereas we don’t know if that is the case in more advanced patients,” said investigator William Matsui, MD, of Johns Hopkins University School of Medicine in Baltimore, Maryland.
The patients received 28-day cycles of len-dex (len at 25 mg PO, days 1-21, and dex at 40 mg PO, weekly). Patients received MEDI-551 (at 4 mg/kg IV) in cycle 3 (day 1, 8) and cycle 4 (day 1). Responding patients continued on len-dex.
The investigators measured MM CSCs by quantifying the growth of MM colonies (CFU-MM) from marrow aspirates at baseline and at the end of cycles 2 and 4.
The team quantified peripheral blood CSCs by flow cytometry (CD19+CD27+ALDH+) at baseline and the end of cycles 2, 4, 5, and 7.
“We wanted to see if these 2 assays gave similar results, and, in this clinical trial, they were almost identical,” said investigator Carol Ann Huff, MD, also of Johns Hopkins.
“Since it is much easier to draw blood than bone marrow from our patients, we think that we can primarily use blood to track multiple myeloma stem cells in the future.”
Response
Two patients did not receive MEDI-551 due to progressive disease and noncompliance. So the investigators assessed responses in 15 patients.
After cycle 2 (len-dex alone), there were 3 very good partial responses (VGPRs), 10 partial responses (PRs), 1 molecular response, and 1 case of stable disease.
After cycle 4 (len-dex plus MEDI-551), there were 6 VGPRs, 8 PRs, and 1 molecular response.
Ten patients who completed treatment with MED-551 remain on len-dex. At the end of cycle 7, there was 1 complete response, 8 VGPRs, and 1 PR.
CFU-MM
When compared to baseline, bone-marrow-derived CFU-MM increased a median of 2.5-fold (range, 0.4-7.4) after cycle 2 but decreased a median of 0.48-fold (range, 0.14-0.85) in 14 patients after cycle 4.
The investigators compared these results to 5 newly diagnosed MM patients who only received standard treatment with len-dex.
In these patients, CFU-MM increased a median of 9.3-fold (range, 4-14) at a median of 4 months (range, 2-4). This is in spite of the fact that all of these patients had a PR or better.
Circulating CSCs
Compared to baseline, circulating MM CSCs increased a median of 1.6-fold (range, 0.4-8.6) in 14 patients after cycle 2 but decreased a median of 0.6-fold (range, 0.01-7.4) in 13 patients after cycle 4.
At the end of cycle 5, MM CSCs had increased in 4 of the 10 patients who were still on len-dex. By the end of cycle 7, MM CSCs had increased in 8 of the patients.
Circulating MM CSCs increased by the end of cycle 4 in 2 patients, and both had progressed by end of cycle 7.
Safety and next steps
The investigators said there were no serious adverse events in this trial, but 2 patients experienced grade 2 infusion reactions after the first MEDI-551 dose.
The team plans to conduct further studies to assess the long-term impact of MED-551 in MM patients and determine how the mAb might work in combination with other treatments, particularly transplant.
“In other studies at Johns Hopkins, we have found that antibody therapies can work much better after a bone marrow transplant, especially allogeneic transplants,” Dr Matsui said.
Funding and drugs for this study were provided by MedImmune Inc., the developers of MEDI-551. Drs Huff and Matsui served as a paid scientific advisory board member and a consultant to MedImmune Inc., respectively. ![]()
Targeted corticosteroids cut GVHD incidence
Short-term low-dose corticosteroid prophylaxis reduces the incidence of graft-vs.-host disease in patients who undergo allogeneic haploidentical stem-cell transplantation to treat hematologic neoplasms, according to a report published online April 18 in the Journal of Clinical Oncology.
The key to selecting patients most likely to benefit from the corticosteroid therapy is to identify those at high risk for graft-vs.-host disease (GVHD) using two biomarkers: high levels of CD56bright natural killer cells in allogeneic grafts or high CD4:CD8 ratios in bone marrow grafts, according to Dr. Ying-Jun Chang of Peking University People’s Hospital, Beijing, and associates.
The investigators performed an open-label trial involving 228 patients aged 15-60 years treated at a single medical center during an 18-month period for acute myeloid leukemia, acute lymphoblastic leukemia, chronic myeloid leukemia, myelodysplastic syndrome, or other hematologic neoplasms. Using the two biomarkers, the patients were categorized as either high or low risk for developing GVHD. They were randomly assigned to three study groups: 72 high-risk patients who received short-term low-dose corticosteroids, 73 high-risk patients who received usual care, and 83 low-risk patients who received usual care.
The cumulative 100-day incidence of acute grade-II to grade-IV GVHD was significantly lower in the high-risk patients who received prophylaxis (21%) than in the high-risk patients who did not receive prophylaxis (48%). In fact, corticosteroids decreased the rate of GVHD so that it was comparable with that in the low-risk patients (26%), Dr. Chang and associates said (J Clin Oncol. 2016 Apr 18. doi: 10.1200/JCO.2015.63l.8817).
Moreover, in the high-risk patients the median interval until GVHD developed was 25 days for those who took corticosteroids, compared with only 15 days for those who did not. Median times to myeloid recovery and platelet recovery were significantly shorter for high-risk patients who received corticosteroids than for either of the other study groups. However, 3-year overall survival and leukemia-free survival were comparable among the three study groups.
The short-term low-dose regimen of corticosteroids did not raise the rate of adverse events, including infection, which suggests that it is preferable to standard corticosteroid regimens in this patient population. The incidences of cytomegalovirus or Epstein-Barr virus reactivation, post-transplantation lymphoproliferative disorder, hemorrhagic cystitis, bacteremia, and invasive fungal infections were comparable among the three study groups. Of note, the incidences of osteonecrosis of the femoral head and secondary hypertension were significantly lower among high-risk patients who received corticosteroid prophylaxis than among those who did not.
“These results provide the first test, to our knowledge, of a novel risk-stratification-directed prophylaxis strategy that effectively prevented acute GVHD among patients who were at high risk for GVHD, without unnecessarily exposing patients who were at low risk to excessive toxicity from additional immunosuppressive agents,” Dr. Chang and associates said.
Despite the encouraging results of Chang et al, it would be premature to routinely use corticosteroid prophylaxis to prevent GVHD until further studies are completed.
This study wasn’t sufficiently powered to determine whether corticosteroids reduced treatment-specific mortality or improved overall survival. Future studies must examine these end points, as well as relapse rates, before this method of prophylaxis is widely adopted.
Dr. Edwin P. Alyea is at Dana-Farber Cancer Institute, Boston. He reported having no relevant financial disclosures. Dr. Alyea made these remarks in an editorial accompanying Dr. Chang’s report (J Clin Oncol. 2016 Apr 18. doi: 10.1200/JCO.2015.66.0902).
Despite the encouraging results of Chang et al, it would be premature to routinely use corticosteroid prophylaxis to prevent GVHD until further studies are completed.
This study wasn’t sufficiently powered to determine whether corticosteroids reduced treatment-specific mortality or improved overall survival. Future studies must examine these end points, as well as relapse rates, before this method of prophylaxis is widely adopted.
Dr. Edwin P. Alyea is at Dana-Farber Cancer Institute, Boston. He reported having no relevant financial disclosures. Dr. Alyea made these remarks in an editorial accompanying Dr. Chang’s report (J Clin Oncol. 2016 Apr 18. doi: 10.1200/JCO.2015.66.0902).
Despite the encouraging results of Chang et al, it would be premature to routinely use corticosteroid prophylaxis to prevent GVHD until further studies are completed.
This study wasn’t sufficiently powered to determine whether corticosteroids reduced treatment-specific mortality or improved overall survival. Future studies must examine these end points, as well as relapse rates, before this method of prophylaxis is widely adopted.
Dr. Edwin P. Alyea is at Dana-Farber Cancer Institute, Boston. He reported having no relevant financial disclosures. Dr. Alyea made these remarks in an editorial accompanying Dr. Chang’s report (J Clin Oncol. 2016 Apr 18. doi: 10.1200/JCO.2015.66.0902).
Short-term low-dose corticosteroid prophylaxis reduces the incidence of graft-vs.-host disease in patients who undergo allogeneic haploidentical stem-cell transplantation to treat hematologic neoplasms, according to a report published online April 18 in the Journal of Clinical Oncology.
The key to selecting patients most likely to benefit from the corticosteroid therapy is to identify those at high risk for graft-vs.-host disease (GVHD) using two biomarkers: high levels of CD56bright natural killer cells in allogeneic grafts or high CD4:CD8 ratios in bone marrow grafts, according to Dr. Ying-Jun Chang of Peking University People’s Hospital, Beijing, and associates.
The investigators performed an open-label trial involving 228 patients aged 15-60 years treated at a single medical center during an 18-month period for acute myeloid leukemia, acute lymphoblastic leukemia, chronic myeloid leukemia, myelodysplastic syndrome, or other hematologic neoplasms. Using the two biomarkers, the patients were categorized as either high or low risk for developing GVHD. They were randomly assigned to three study groups: 72 high-risk patients who received short-term low-dose corticosteroids, 73 high-risk patients who received usual care, and 83 low-risk patients who received usual care.
The cumulative 100-day incidence of acute grade-II to grade-IV GVHD was significantly lower in the high-risk patients who received prophylaxis (21%) than in the high-risk patients who did not receive prophylaxis (48%). In fact, corticosteroids decreased the rate of GVHD so that it was comparable with that in the low-risk patients (26%), Dr. Chang and associates said (J Clin Oncol. 2016 Apr 18. doi: 10.1200/JCO.2015.63l.8817).
Moreover, in the high-risk patients the median interval until GVHD developed was 25 days for those who took corticosteroids, compared with only 15 days for those who did not. Median times to myeloid recovery and platelet recovery were significantly shorter for high-risk patients who received corticosteroids than for either of the other study groups. However, 3-year overall survival and leukemia-free survival were comparable among the three study groups.
The short-term low-dose regimen of corticosteroids did not raise the rate of adverse events, including infection, which suggests that it is preferable to standard corticosteroid regimens in this patient population. The incidences of cytomegalovirus or Epstein-Barr virus reactivation, post-transplantation lymphoproliferative disorder, hemorrhagic cystitis, bacteremia, and invasive fungal infections were comparable among the three study groups. Of note, the incidences of osteonecrosis of the femoral head and secondary hypertension were significantly lower among high-risk patients who received corticosteroid prophylaxis than among those who did not.
“These results provide the first test, to our knowledge, of a novel risk-stratification-directed prophylaxis strategy that effectively prevented acute GVHD among patients who were at high risk for GVHD, without unnecessarily exposing patients who were at low risk to excessive toxicity from additional immunosuppressive agents,” Dr. Chang and associates said.
Short-term low-dose corticosteroid prophylaxis reduces the incidence of graft-vs.-host disease in patients who undergo allogeneic haploidentical stem-cell transplantation to treat hematologic neoplasms, according to a report published online April 18 in the Journal of Clinical Oncology.
The key to selecting patients most likely to benefit from the corticosteroid therapy is to identify those at high risk for graft-vs.-host disease (GVHD) using two biomarkers: high levels of CD56bright natural killer cells in allogeneic grafts or high CD4:CD8 ratios in bone marrow grafts, according to Dr. Ying-Jun Chang of Peking University People’s Hospital, Beijing, and associates.
The investigators performed an open-label trial involving 228 patients aged 15-60 years treated at a single medical center during an 18-month period for acute myeloid leukemia, acute lymphoblastic leukemia, chronic myeloid leukemia, myelodysplastic syndrome, or other hematologic neoplasms. Using the two biomarkers, the patients were categorized as either high or low risk for developing GVHD. They were randomly assigned to three study groups: 72 high-risk patients who received short-term low-dose corticosteroids, 73 high-risk patients who received usual care, and 83 low-risk patients who received usual care.
The cumulative 100-day incidence of acute grade-II to grade-IV GVHD was significantly lower in the high-risk patients who received prophylaxis (21%) than in the high-risk patients who did not receive prophylaxis (48%). In fact, corticosteroids decreased the rate of GVHD so that it was comparable with that in the low-risk patients (26%), Dr. Chang and associates said (J Clin Oncol. 2016 Apr 18. doi: 10.1200/JCO.2015.63l.8817).
Moreover, in the high-risk patients the median interval until GVHD developed was 25 days for those who took corticosteroids, compared with only 15 days for those who did not. Median times to myeloid recovery and platelet recovery were significantly shorter for high-risk patients who received corticosteroids than for either of the other study groups. However, 3-year overall survival and leukemia-free survival were comparable among the three study groups.
The short-term low-dose regimen of corticosteroids did not raise the rate of adverse events, including infection, which suggests that it is preferable to standard corticosteroid regimens in this patient population. The incidences of cytomegalovirus or Epstein-Barr virus reactivation, post-transplantation lymphoproliferative disorder, hemorrhagic cystitis, bacteremia, and invasive fungal infections were comparable among the three study groups. Of note, the incidences of osteonecrosis of the femoral head and secondary hypertension were significantly lower among high-risk patients who received corticosteroid prophylaxis than among those who did not.
“These results provide the first test, to our knowledge, of a novel risk-stratification-directed prophylaxis strategy that effectively prevented acute GVHD among patients who were at high risk for GVHD, without unnecessarily exposing patients who were at low risk to excessive toxicity from additional immunosuppressive agents,” Dr. Chang and associates said.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Short-term low-dose corticosteroid prophylaxis reduces the incidence of the GVHD in patients who undergo haploidentical stem-cell transplantation to treat hematologic neoplasms.
Major finding: The 100-day incidence of acute GVHD was significantly lower in the high-risk patients who received corticosteroid prophylaxis (21%) than in the high-risk patients who did not (48%).
Data source: An open-label randomized controlled trial involving 228 Chinese patients who underwent stem-cell transplantation.
Disclosures: This study was supported by the Beijing Committee of Science and Technology, the National High Technology Research and Development Program of China, and the National Natural Science Foundation of China. Dr. Chang and associates reported having no relevant financial disclosures.
OTX015 dose for lymphoma narrowed in phase 1 study
As a single agent for use in patients with lymphoma, an acceptable once-daily dose of OTX015 appears to be 80 mg on a 14 days on, 7 days off schedule, the results of a phase 1 study indicate.
The small-molecule inhibitor, which inhibits binding of bromodomain and exterminal proteins to acetylated histones, was associated with acceptable toxicity and efficacy in this regimen. The investigational drug is now being tested in expansion cohorts on a schedule of 14 days every 3 weeks, a regimen projected to allow for recovery from the drug’s toxic effects, Dr. Sandy Amorin of Hôpital Saint Louis, Paris, and associates reported.
The drug also is being evaluated in patients with acute leukemias.
Adults with nonleukemia hematologic malignancies that progressed on standard therapies participated in the open-label study, which was conducted at seven university hospital centers in Europe. Oral OTX015 was given once a day at one of five doses (10 mg, 20 mg, 40 mg, 80 mg, and 120 mg). The 3 + 3 study design permitted evaluation of alternative administration schedules. The primary endpoint was dose-limiting toxicity in the first treatment cycle (21 days). Secondary objectives were to evaluate safety, pharmacokinetics, and preliminary clinical activity of OTX015. The study is ongoing and is registered with ClinicalTrials.gov, number NCT01713582.
The study included 33 patients with lymphoma and 12 with myeloma; patients’ median age was 66 years, and they had received a median of four lines of prior therapy. No dose-limiting toxicities were seen in three patients given doses as high as 80 mg once a day. However, grade 4 thrombocytopenia occurred in five of six patients on a 21-day schedule of 40 mg twice a day. No patient tolerated various schedules of 120 mg once a day (Lancet Haematol. 2016;3[4]:e196-204).
The researchers then examined the 80 mg once a day dose on a continuous basis in four patients, two of whom developed grade 4 thrombocytopenia. In light of these and other toxicities, a regimen was proposed of 80 mg once a day on a schedule of 14 days on, 7 days off.
Thrombocytopenia affected 43 of 45 patients, and 26 of them had grade 3-4 events. Other grade 3-4 events were infrequent. Anemia was seen in 41, and neutropenia in 23.
Of three patients with diffuse large B-cell lymphoma, two had complete responses at 120 mg once a day, and one had a partial response at 80 mg once a day. Six additional patients, two with diffuse large B-cell lymphoma and four with indolent lymphomas, had evidence of clinical activity, but did not meet the criteria for an objective response.
The study was funded by the developers of OTX015, Oncoethix GmbH, a wholly owned subsidiary of Merck Sharp & Dohme.
On Twitter @maryjodales
As a single agent for use in patients with lymphoma, an acceptable once-daily dose of OTX015 appears to be 80 mg on a 14 days on, 7 days off schedule, the results of a phase 1 study indicate.
The small-molecule inhibitor, which inhibits binding of bromodomain and exterminal proteins to acetylated histones, was associated with acceptable toxicity and efficacy in this regimen. The investigational drug is now being tested in expansion cohorts on a schedule of 14 days every 3 weeks, a regimen projected to allow for recovery from the drug’s toxic effects, Dr. Sandy Amorin of Hôpital Saint Louis, Paris, and associates reported.
The drug also is being evaluated in patients with acute leukemias.
Adults with nonleukemia hematologic malignancies that progressed on standard therapies participated in the open-label study, which was conducted at seven university hospital centers in Europe. Oral OTX015 was given once a day at one of five doses (10 mg, 20 mg, 40 mg, 80 mg, and 120 mg). The 3 + 3 study design permitted evaluation of alternative administration schedules. The primary endpoint was dose-limiting toxicity in the first treatment cycle (21 days). Secondary objectives were to evaluate safety, pharmacokinetics, and preliminary clinical activity of OTX015. The study is ongoing and is registered with ClinicalTrials.gov, number NCT01713582.
The study included 33 patients with lymphoma and 12 with myeloma; patients’ median age was 66 years, and they had received a median of four lines of prior therapy. No dose-limiting toxicities were seen in three patients given doses as high as 80 mg once a day. However, grade 4 thrombocytopenia occurred in five of six patients on a 21-day schedule of 40 mg twice a day. No patient tolerated various schedules of 120 mg once a day (Lancet Haematol. 2016;3[4]:e196-204).
The researchers then examined the 80 mg once a day dose on a continuous basis in four patients, two of whom developed grade 4 thrombocytopenia. In light of these and other toxicities, a regimen was proposed of 80 mg once a day on a schedule of 14 days on, 7 days off.
Thrombocytopenia affected 43 of 45 patients, and 26 of them had grade 3-4 events. Other grade 3-4 events were infrequent. Anemia was seen in 41, and neutropenia in 23.
Of three patients with diffuse large B-cell lymphoma, two had complete responses at 120 mg once a day, and one had a partial response at 80 mg once a day. Six additional patients, two with diffuse large B-cell lymphoma and four with indolent lymphomas, had evidence of clinical activity, but did not meet the criteria for an objective response.
The study was funded by the developers of OTX015, Oncoethix GmbH, a wholly owned subsidiary of Merck Sharp & Dohme.
On Twitter @maryjodales
As a single agent for use in patients with lymphoma, an acceptable once-daily dose of OTX015 appears to be 80 mg on a 14 days on, 7 days off schedule, the results of a phase 1 study indicate.
The small-molecule inhibitor, which inhibits binding of bromodomain and exterminal proteins to acetylated histones, was associated with acceptable toxicity and efficacy in this regimen. The investigational drug is now being tested in expansion cohorts on a schedule of 14 days every 3 weeks, a regimen projected to allow for recovery from the drug’s toxic effects, Dr. Sandy Amorin of Hôpital Saint Louis, Paris, and associates reported.
The drug also is being evaluated in patients with acute leukemias.
Adults with nonleukemia hematologic malignancies that progressed on standard therapies participated in the open-label study, which was conducted at seven university hospital centers in Europe. Oral OTX015 was given once a day at one of five doses (10 mg, 20 mg, 40 mg, 80 mg, and 120 mg). The 3 + 3 study design permitted evaluation of alternative administration schedules. The primary endpoint was dose-limiting toxicity in the first treatment cycle (21 days). Secondary objectives were to evaluate safety, pharmacokinetics, and preliminary clinical activity of OTX015. The study is ongoing and is registered with ClinicalTrials.gov, number NCT01713582.
The study included 33 patients with lymphoma and 12 with myeloma; patients’ median age was 66 years, and they had received a median of four lines of prior therapy. No dose-limiting toxicities were seen in three patients given doses as high as 80 mg once a day. However, grade 4 thrombocytopenia occurred in five of six patients on a 21-day schedule of 40 mg twice a day. No patient tolerated various schedules of 120 mg once a day (Lancet Haematol. 2016;3[4]:e196-204).
The researchers then examined the 80 mg once a day dose on a continuous basis in four patients, two of whom developed grade 4 thrombocytopenia. In light of these and other toxicities, a regimen was proposed of 80 mg once a day on a schedule of 14 days on, 7 days off.
Thrombocytopenia affected 43 of 45 patients, and 26 of them had grade 3-4 events. Other grade 3-4 events were infrequent. Anemia was seen in 41, and neutropenia in 23.
Of three patients with diffuse large B-cell lymphoma, two had complete responses at 120 mg once a day, and one had a partial response at 80 mg once a day. Six additional patients, two with diffuse large B-cell lymphoma and four with indolent lymphomas, had evidence of clinical activity, but did not meet the criteria for an objective response.
The study was funded by the developers of OTX015, Oncoethix GmbH, a wholly owned subsidiary of Merck Sharp & Dohme.
On Twitter @maryjodales
FROM THE LANCET HAEMATOLOGY
Key clinical point: For lymphoma patients, a regimen has been determined for the small-molecule inhibitor OTX015 that was associated with acceptable toxicity and efficacy.
Major finding: On a regimen of 80 mg once a day on a schedule of 14 days on, 7 days off, thrombocytopenia affected 43 of 45 patients, and 26 of them had grade 3-4 events. However, other grade 3-4 events were infrequent.
Data source: The open-label study NCT01713582 was conducted at seven university hospital centers in Europe.
Disclosures: The study was funded by the developers of OTX015, Oncoethix GmbH, a wholly owned subsidiary of Merck Sharp & Dohme.
Feds advance cancer moonshot with expert panel, outline of goals
Federal officials took the next step in their moonshot to end cancer by announcing on April 4 a blue ribbon panel to guide the effort.
A total of 28 leading researchers, clinicians, and patient advocates have been named to the panel charged with informing the scientific direction and goals of the National Cancer Moonshot Initiative, led by Vice President Joe Biden.

“This Blue Ribbon Panel will ensure that, as [the National Institutes of Health] allocates new resources through the Moonshot, decisions will be grounded in the best science,” Vice President Biden said in a statement. “I look forward to working with this panel and many others involved with the Moonshot to make unprecedented improvements in prevention, diagnosis, and treatment of cancer.”
The key goals of the initiative were set out simultaneously in a perspective from Dr. Francis S. Collins, NIH director, and Dr. Douglas R. Lowy, director of the National Cancer Institute. The editorial was published in the New England Journal of Medicine.
“Fueled by an additional $680 million in the proposed fiscal year 2017 budget for the NIH, plus additional resources for the Food and Drug Administration, the initiative will aim to accelerate progress toward the next generation of interventions that we hope will substantially reduce cancer incidence and dramatically improve patient outcomes,” Dr. Collins and Dr. Lowy wrote. “The NIH’s most compelling opportunities for progress will be set forth by late summer 2016 in a research plan informed by the deliberations of a blue-ribbon panel of experts, which will provide scientific input to the National Cancer Advisory Board. Some possible opportunities include vaccine development, early-detection technology, single-cell genomic analysis, immunotherapy, a focus on pediatric cancer, and enhanced data sharing.”
To read the full editorial, click here.
On Twitter @denisefulton
Federal officials took the next step in their moonshot to end cancer by announcing on April 4 a blue ribbon panel to guide the effort.
A total of 28 leading researchers, clinicians, and patient advocates have been named to the panel charged with informing the scientific direction and goals of the National Cancer Moonshot Initiative, led by Vice President Joe Biden.
“This Blue Ribbon Panel will ensure that, as [the National Institutes of Health] allocates new resources through the Moonshot, decisions will be grounded in the best science,” Vice President Biden said in a statement. “I look forward to working with this panel and many others involved with the Moonshot to make unprecedented improvements in prevention, diagnosis, and treatment of cancer.”
The key goals of the initiative were set out simultaneously in a perspective from Dr. Francis S. Collins, NIH director, and Dr. Douglas R. Lowy, director of the National Cancer Institute. The editorial was published in the New England Journal of Medicine.
“Fueled by an additional $680 million in the proposed fiscal year 2017 budget for the NIH, plus additional resources for the Food and Drug Administration, the initiative will aim to accelerate progress toward the next generation of interventions that we hope will substantially reduce cancer incidence and dramatically improve patient outcomes,” Dr. Collins and Dr. Lowy wrote. “The NIH’s most compelling opportunities for progress will be set forth by late summer 2016 in a research plan informed by the deliberations of a blue-ribbon panel of experts, which will provide scientific input to the National Cancer Advisory Board. Some possible opportunities include vaccine development, early-detection technology, single-cell genomic analysis, immunotherapy, a focus on pediatric cancer, and enhanced data sharing.”
To read the full editorial, click here.
On Twitter @denisefulton
Federal officials took the next step in their moonshot to end cancer by announcing on April 4 a blue ribbon panel to guide the effort.
A total of 28 leading researchers, clinicians, and patient advocates have been named to the panel charged with informing the scientific direction and goals of the National Cancer Moonshot Initiative, led by Vice President Joe Biden.
“This Blue Ribbon Panel will ensure that, as [the National Institutes of Health] allocates new resources through the Moonshot, decisions will be grounded in the best science,” Vice President Biden said in a statement. “I look forward to working with this panel and many others involved with the Moonshot to make unprecedented improvements in prevention, diagnosis, and treatment of cancer.”
The key goals of the initiative were set out simultaneously in a perspective from Dr. Francis S. Collins, NIH director, and Dr. Douglas R. Lowy, director of the National Cancer Institute. The editorial was published in the New England Journal of Medicine.
“Fueled by an additional $680 million in the proposed fiscal year 2017 budget for the NIH, plus additional resources for the Food and Drug Administration, the initiative will aim to accelerate progress toward the next generation of interventions that we hope will substantially reduce cancer incidence and dramatically improve patient outcomes,” Dr. Collins and Dr. Lowy wrote. “The NIH’s most compelling opportunities for progress will be set forth by late summer 2016 in a research plan informed by the deliberations of a blue-ribbon panel of experts, which will provide scientific input to the National Cancer Advisory Board. Some possible opportunities include vaccine development, early-detection technology, single-cell genomic analysis, immunotherapy, a focus on pediatric cancer, and enhanced data sharing.”
To read the full editorial, click here.
On Twitter @denisefulton
FROM NEJM

Guidelines emphasize testing early and often for renal impairment in multiple myeloma
Renal status should be evaluated at diagnosis and follow-up in all myeloma patients, according to new guidance from the International Myeloma Working Group published online in the Journal of Clinical Oncology.
Renal impairment (RI) affects up to half of patients with multiple myeloma, and severe RI predicts early death, noted Dr. Meletios Dimopoulos of National and Kapodistrian University of Athens and his associates. Novel therapies have substantially increased survival for myeloma with less severe kidney disease, underscoring the importance of early treatment. To develop the guidelines, the authors reviewed all evidence from randomized trials, systematic reviews, meta-analyses, and prospective and observational studies published through December 2015 (J Clin Oncol. 2016 Mar 14. doi: 10.1200/JCO.2015.65.0044).
Grade A recommendations (evidence obtained from meta-analysis of multiple well-designed, randomized controlled trials) include the following:
Evaluate serum creatinine, estimated glomerular filtration rate, and electrolytes at initial diagnosis and follow-up assessments. Also perform the serum free light chain test, if available, and electrophoresis of a 24-hour urine specimen.
In patients with stabilized serum creatinine, evaluate GFR using the Chronic Kidney Disease Epidemiology Collaboration (preferred) or the Modification of Diet in Renal Disease formulas.
Based on GFR, determine stage of chronic kidney disease (ranging from 1, kidney damage with normal or elevated GFR, to 5, renal failure).
Bortezomib remains the foundational treatment for myeloma-related renal impairment, the authors emphasized. This 26S proteasome inhibitor should be started at the standard dose of 1.3 mg/m2 on days 1, 4, 8, and 11 of a 3-week cycle.
Another option for patients with creatinine clearance above 15 mL/min is carfilzomib, which needs no dose modification and yields similar results regardless of RI status. However, more data are needed on its renal safety, the authors said.
Patients with creatinine clearance above 30 mL/min can safely receive ixazomib in combination with lenalidomide and dexamethasone.
Those with creatinine clearance above 45 mL/min should receive pomalidomide at a dose of 4 mg/day; it is not yet clear whether the dose should be cut for more severe renal impairment.
Hypercalcemia can be treated with bisphosphonates, but patients with creatinine clearance below 30 mL/min should not receive pamidronate or zoledronic acid.
Avoid nephrotoxic agents, such as as aminoglycosides, furosemide, and contrast agents, in all patients with multiple myeloma and RI.
Dr. Dimopoulos reported receiving honoraria or financial support related to travel, accommodations, or expenses from Amgen, Celgene, Onyx Pharmaceuticals, Janssen-Cilag, Bristol-Myers Squibb, Novartis, and Genesis Pharmaceuticals. Seventeen coauthors also reported financial relationships with a number of pharmaceutical companies. The remaining three coauthors had no disclosures.
Renal status should be evaluated at diagnosis and follow-up in all myeloma patients, according to new guidance from the International Myeloma Working Group published online in the Journal of Clinical Oncology.
Renal impairment (RI) affects up to half of patients with multiple myeloma, and severe RI predicts early death, noted Dr. Meletios Dimopoulos of National and Kapodistrian University of Athens and his associates. Novel therapies have substantially increased survival for myeloma with less severe kidney disease, underscoring the importance of early treatment. To develop the guidelines, the authors reviewed all evidence from randomized trials, systematic reviews, meta-analyses, and prospective and observational studies published through December 2015 (J Clin Oncol. 2016 Mar 14. doi: 10.1200/JCO.2015.65.0044).
Grade A recommendations (evidence obtained from meta-analysis of multiple well-designed, randomized controlled trials) include the following:
Evaluate serum creatinine, estimated glomerular filtration rate, and electrolytes at initial diagnosis and follow-up assessments. Also perform the serum free light chain test, if available, and electrophoresis of a 24-hour urine specimen.
In patients with stabilized serum creatinine, evaluate GFR using the Chronic Kidney Disease Epidemiology Collaboration (preferred) or the Modification of Diet in Renal Disease formulas.
Based on GFR, determine stage of chronic kidney disease (ranging from 1, kidney damage with normal or elevated GFR, to 5, renal failure).
Bortezomib remains the foundational treatment for myeloma-related renal impairment, the authors emphasized. This 26S proteasome inhibitor should be started at the standard dose of 1.3 mg/m2 on days 1, 4, 8, and 11 of a 3-week cycle.
Another option for patients with creatinine clearance above 15 mL/min is carfilzomib, which needs no dose modification and yields similar results regardless of RI status. However, more data are needed on its renal safety, the authors said.
Patients with creatinine clearance above 30 mL/min can safely receive ixazomib in combination with lenalidomide and dexamethasone.
Those with creatinine clearance above 45 mL/min should receive pomalidomide at a dose of 4 mg/day; it is not yet clear whether the dose should be cut for more severe renal impairment.
Hypercalcemia can be treated with bisphosphonates, but patients with creatinine clearance below 30 mL/min should not receive pamidronate or zoledronic acid.
Avoid nephrotoxic agents, such as as aminoglycosides, furosemide, and contrast agents, in all patients with multiple myeloma and RI.
Dr. Dimopoulos reported receiving honoraria or financial support related to travel, accommodations, or expenses from Amgen, Celgene, Onyx Pharmaceuticals, Janssen-Cilag, Bristol-Myers Squibb, Novartis, and Genesis Pharmaceuticals. Seventeen coauthors also reported financial relationships with a number of pharmaceutical companies. The remaining three coauthors had no disclosures.
Renal status should be evaluated at diagnosis and follow-up in all myeloma patients, according to new guidance from the International Myeloma Working Group published online in the Journal of Clinical Oncology.
Renal impairment (RI) affects up to half of patients with multiple myeloma, and severe RI predicts early death, noted Dr. Meletios Dimopoulos of National and Kapodistrian University of Athens and his associates. Novel therapies have substantially increased survival for myeloma with less severe kidney disease, underscoring the importance of early treatment. To develop the guidelines, the authors reviewed all evidence from randomized trials, systematic reviews, meta-analyses, and prospective and observational studies published through December 2015 (J Clin Oncol. 2016 Mar 14. doi: 10.1200/JCO.2015.65.0044).
Grade A recommendations (evidence obtained from meta-analysis of multiple well-designed, randomized controlled trials) include the following:
Evaluate serum creatinine, estimated glomerular filtration rate, and electrolytes at initial diagnosis and follow-up assessments. Also perform the serum free light chain test, if available, and electrophoresis of a 24-hour urine specimen.
In patients with stabilized serum creatinine, evaluate GFR using the Chronic Kidney Disease Epidemiology Collaboration (preferred) or the Modification of Diet in Renal Disease formulas.
Based on GFR, determine stage of chronic kidney disease (ranging from 1, kidney damage with normal or elevated GFR, to 5, renal failure).
Bortezomib remains the foundational treatment for myeloma-related renal impairment, the authors emphasized. This 26S proteasome inhibitor should be started at the standard dose of 1.3 mg/m2 on days 1, 4, 8, and 11 of a 3-week cycle.
Another option for patients with creatinine clearance above 15 mL/min is carfilzomib, which needs no dose modification and yields similar results regardless of RI status. However, more data are needed on its renal safety, the authors said.
Patients with creatinine clearance above 30 mL/min can safely receive ixazomib in combination with lenalidomide and dexamethasone.
Those with creatinine clearance above 45 mL/min should receive pomalidomide at a dose of 4 mg/day; it is not yet clear whether the dose should be cut for more severe renal impairment.
Hypercalcemia can be treated with bisphosphonates, but patients with creatinine clearance below 30 mL/min should not receive pamidronate or zoledronic acid.
Avoid nephrotoxic agents, such as as aminoglycosides, furosemide, and contrast agents, in all patients with multiple myeloma and RI.
Dr. Dimopoulos reported receiving honoraria or financial support related to travel, accommodations, or expenses from Amgen, Celgene, Onyx Pharmaceuticals, Janssen-Cilag, Bristol-Myers Squibb, Novartis, and Genesis Pharmaceuticals. Seventeen coauthors also reported financial relationships with a number of pharmaceutical companies. The remaining three coauthors had no disclosures.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Dual inhibitor shows early promise for DLBCL

The small-molecule inhibitor CUDC-907 can provide disease control in patients with relapsed or refractory lymphoma and multiple myeloma (MM), according to researchers.
In a phase 1 trial, CUDC-907 produced responses in a small number of patients with diffuse large B-cell lymphoma (DLBCL).
And more than half of patients had stable disease while on CUDC-907, including those with MM, DLBCL, Hodgkin lymphoma (HL), and other lymphomas.
However, a majority of patients in this trial—84%—discontinued treatment due to confirmed progressive disease or signs of progression.
These results were published in The Lancet. The trial was sponsored by Curis, Inc., the company developing CUDC-907, and the Leukemia and Lymphoma Society.
“The data from the phase 1 monotherapy trial for CUDC-907, especially in heavily pretreated patients with relapsed/refractory DLBCL are very encouraging, and we look forward to data emerging from the current phase 2 trial in patients with MYC-altered DLBCL,” said study author Anas Younes, MD, of the Memorial Sloan Kettering Cancer Center in New York, New York.
CUDC-907 is an oral, dual inhibitor of class I and II histone deacetylases (HDACs), as well as class I PI3K enzymes. Specifically, CUDC-907 is designed to inhibit HDACs 1, 2, 3, 6, and 10 and PI3K-alpha, delta, and beta isoforms.
Between Jan 23, 2013, and July 27, 2015, the phase 1 trial of CUDC-907 enrolled 44 patients who were refractory to or had relapsed after 2 or more previous regimens. The patients’ median age was 63 (range, 22-83), and they had received a median of 5 prior treatments (range, 2-10).
Four patients had MM, 12 had HL, and 12 had DLBCL. The remaining 16 patients had other types of lymphoma, including lymphoplasmacytic lymphoma (n=3), small lymphocytic lymphoma (n=3), mantle cell lymphoma (n=3), follicular lymphoma (n=2), T-cell lymphoma (n=2), marginal zone lymphoma (n=1), Burkitt lymphoma (n=1), and gray zone lymphoma (n=1).
Treatment
CUDC-907 was given in a standard 3 + 3 dose-escalation design at 4 different dosing schedules—once daily, twice weekly, 3 times weekly, and daily for 5 days followed by a 2-day break (5/2)—in 21-day cycles.
Patients continued to receive CUDC-907 until disease progression or other treatment discontinuation criteria were met. The primary objective was to determine the maximum tolerated dose (MTD) and recommended phase 2 dose.
Ten patients were sequentially assigned to CUDC-907 once-daily (MTD 60 mg), 12 to twice-weekly (MTD 150 mg), 15 to 3-times-weekly (MTD 150 mg), and 7 to the 5/2 dosing schedule (MTD 60 mg).
Safety
Four dose-limiting toxicities (DLTs) occurred in 3 of 40 DLT-evaluable patients. The DLTs were diarrhea and hyperglycemia in 1 patient on 60 mg once daily, hyperglycemia in 1 patient on 150 mg twice weekly, and diarrhea in 1 patient on 150 mg 3 times weekly. There were no DLTs in patients on the 5/2 schedule.
The incidence of grade 3 or higher adverse events (AEs) was 43% (19/44). The most common of these AEs were thrombocytopenia (20%, n=9), neutropenia (7%, n=3), and hyperglycemia (7%, n=3).
Twenty-five percent of patients (11/44) had serious AEs. Three of these events were considered treatment-related. They were epistaxis and the DLTs of diarrhea and hyperglycemia.
AEs led to dose reductions in 6 patients (14%) and treatment discontinuation in 7 patients (16%).
Efficacy
Thirty-seven patients were evaluable for response, and 5 of these patients responded (14%). All responses—2 complete and 3 partial responses—occurred in patients with DLBCL.
Twenty-one of the response-evaluable patients (57%) had stable disease. This included 1 patient with DLBCL, 2 with MM, 8 with HL, and 10 with the “other” types of lymphoma.
The remaining 11 patients progressed (30%)—3 with DLBCL, 2 with MM, 2 with HL, and 4 with other lymphomas.
Thirty-seven patients (84%) discontinued CUDC-907 because of progressive disease or clinical signs of progressive disease at the data cutoff.
Based on the clinical activity of CUDC-907 in patients with relapsed/refractory DLBCL, particularly those with MYC alterations, Curis has initiated a phase 2 trial of the drug in these patients. The recommended phase 2 dose is 60 mg on the 5/2 dosing schedule. ![]()
The small-molecule inhibitor CUDC-907 can provide disease control in patients with relapsed or refractory lymphoma and multiple myeloma (MM), according to researchers.
In a phase 1 trial, CUDC-907 produced responses in a small number of patients with diffuse large B-cell lymphoma (DLBCL).
And more than half of patients had stable disease while on CUDC-907, including those with MM, DLBCL, Hodgkin lymphoma (HL), and other lymphomas.
However, a majority of patients in this trial—84%—discontinued treatment due to confirmed progressive disease or signs of progression.
These results were published in The Lancet. The trial was sponsored by Curis, Inc., the company developing CUDC-907, and the Leukemia and Lymphoma Society.
“The data from the phase 1 monotherapy trial for CUDC-907, especially in heavily pretreated patients with relapsed/refractory DLBCL are very encouraging, and we look forward to data emerging from the current phase 2 trial in patients with MYC-altered DLBCL,” said study author Anas Younes, MD, of the Memorial Sloan Kettering Cancer Center in New York, New York.
CUDC-907 is an oral, dual inhibitor of class I and II histone deacetylases (HDACs), as well as class I PI3K enzymes. Specifically, CUDC-907 is designed to inhibit HDACs 1, 2, 3, 6, and 10 and PI3K-alpha, delta, and beta isoforms.
Between Jan 23, 2013, and July 27, 2015, the phase 1 trial of CUDC-907 enrolled 44 patients who were refractory to or had relapsed after 2 or more previous regimens. The patients’ median age was 63 (range, 22-83), and they had received a median of 5 prior treatments (range, 2-10).
Four patients had MM, 12 had HL, and 12 had DLBCL. The remaining 16 patients had other types of lymphoma, including lymphoplasmacytic lymphoma (n=3), small lymphocytic lymphoma (n=3), mantle cell lymphoma (n=3), follicular lymphoma (n=2), T-cell lymphoma (n=2), marginal zone lymphoma (n=1), Burkitt lymphoma (n=1), and gray zone lymphoma (n=1).
Treatment
CUDC-907 was given in a standard 3 + 3 dose-escalation design at 4 different dosing schedules—once daily, twice weekly, 3 times weekly, and daily for 5 days followed by a 2-day break (5/2)—in 21-day cycles.
Patients continued to receive CUDC-907 until disease progression or other treatment discontinuation criteria were met. The primary objective was to determine the maximum tolerated dose (MTD) and recommended phase 2 dose.
Ten patients were sequentially assigned to CUDC-907 once-daily (MTD 60 mg), 12 to twice-weekly (MTD 150 mg), 15 to 3-times-weekly (MTD 150 mg), and 7 to the 5/2 dosing schedule (MTD 60 mg).
Safety
Four dose-limiting toxicities (DLTs) occurred in 3 of 40 DLT-evaluable patients. The DLTs were diarrhea and hyperglycemia in 1 patient on 60 mg once daily, hyperglycemia in 1 patient on 150 mg twice weekly, and diarrhea in 1 patient on 150 mg 3 times weekly. There were no DLTs in patients on the 5/2 schedule.
The incidence of grade 3 or higher adverse events (AEs) was 43% (19/44). The most common of these AEs were thrombocytopenia (20%, n=9), neutropenia (7%, n=3), and hyperglycemia (7%, n=3).
Twenty-five percent of patients (11/44) had serious AEs. Three of these events were considered treatment-related. They were epistaxis and the DLTs of diarrhea and hyperglycemia.
AEs led to dose reductions in 6 patients (14%) and treatment discontinuation in 7 patients (16%).
Efficacy
Thirty-seven patients were evaluable for response, and 5 of these patients responded (14%). All responses—2 complete and 3 partial responses—occurred in patients with DLBCL.
Twenty-one of the response-evaluable patients (57%) had stable disease. This included 1 patient with DLBCL, 2 with MM, 8 with HL, and 10 with the “other” types of lymphoma.
The remaining 11 patients progressed (30%)—3 with DLBCL, 2 with MM, 2 with HL, and 4 with other lymphomas.
Thirty-seven patients (84%) discontinued CUDC-907 because of progressive disease or clinical signs of progressive disease at the data cutoff.
Based on the clinical activity of CUDC-907 in patients with relapsed/refractory DLBCL, particularly those with MYC alterations, Curis has initiated a phase 2 trial of the drug in these patients. The recommended phase 2 dose is 60 mg on the 5/2 dosing schedule. ![]()
The small-molecule inhibitor CUDC-907 can provide disease control in patients with relapsed or refractory lymphoma and multiple myeloma (MM), according to researchers.
In a phase 1 trial, CUDC-907 produced responses in a small number of patients with diffuse large B-cell lymphoma (DLBCL).
And more than half of patients had stable disease while on CUDC-907, including those with MM, DLBCL, Hodgkin lymphoma (HL), and other lymphomas.
However, a majority of patients in this trial—84%—discontinued treatment due to confirmed progressive disease or signs of progression.
These results were published in The Lancet. The trial was sponsored by Curis, Inc., the company developing CUDC-907, and the Leukemia and Lymphoma Society.
“The data from the phase 1 monotherapy trial for CUDC-907, especially in heavily pretreated patients with relapsed/refractory DLBCL are very encouraging, and we look forward to data emerging from the current phase 2 trial in patients with MYC-altered DLBCL,” said study author Anas Younes, MD, of the Memorial Sloan Kettering Cancer Center in New York, New York.
CUDC-907 is an oral, dual inhibitor of class I and II histone deacetylases (HDACs), as well as class I PI3K enzymes. Specifically, CUDC-907 is designed to inhibit HDACs 1, 2, 3, 6, and 10 and PI3K-alpha, delta, and beta isoforms.
Between Jan 23, 2013, and July 27, 2015, the phase 1 trial of CUDC-907 enrolled 44 patients who were refractory to or had relapsed after 2 or more previous regimens. The patients’ median age was 63 (range, 22-83), and they had received a median of 5 prior treatments (range, 2-10).
Four patients had MM, 12 had HL, and 12 had DLBCL. The remaining 16 patients had other types of lymphoma, including lymphoplasmacytic lymphoma (n=3), small lymphocytic lymphoma (n=3), mantle cell lymphoma (n=3), follicular lymphoma (n=2), T-cell lymphoma (n=2), marginal zone lymphoma (n=1), Burkitt lymphoma (n=1), and gray zone lymphoma (n=1).
Treatment
CUDC-907 was given in a standard 3 + 3 dose-escalation design at 4 different dosing schedules—once daily, twice weekly, 3 times weekly, and daily for 5 days followed by a 2-day break (5/2)—in 21-day cycles.
Patients continued to receive CUDC-907 until disease progression or other treatment discontinuation criteria were met. The primary objective was to determine the maximum tolerated dose (MTD) and recommended phase 2 dose.
Ten patients were sequentially assigned to CUDC-907 once-daily (MTD 60 mg), 12 to twice-weekly (MTD 150 mg), 15 to 3-times-weekly (MTD 150 mg), and 7 to the 5/2 dosing schedule (MTD 60 mg).
Safety
Four dose-limiting toxicities (DLTs) occurred in 3 of 40 DLT-evaluable patients. The DLTs were diarrhea and hyperglycemia in 1 patient on 60 mg once daily, hyperglycemia in 1 patient on 150 mg twice weekly, and diarrhea in 1 patient on 150 mg 3 times weekly. There were no DLTs in patients on the 5/2 schedule.
The incidence of grade 3 or higher adverse events (AEs) was 43% (19/44). The most common of these AEs were thrombocytopenia (20%, n=9), neutropenia (7%, n=3), and hyperglycemia (7%, n=3).
Twenty-five percent of patients (11/44) had serious AEs. Three of these events were considered treatment-related. They were epistaxis and the DLTs of diarrhea and hyperglycemia.
AEs led to dose reductions in 6 patients (14%) and treatment discontinuation in 7 patients (16%).
Efficacy
Thirty-seven patients were evaluable for response, and 5 of these patients responded (14%). All responses—2 complete and 3 partial responses—occurred in patients with DLBCL.
Twenty-one of the response-evaluable patients (57%) had stable disease. This included 1 patient with DLBCL, 2 with MM, 8 with HL, and 10 with the “other” types of lymphoma.
The remaining 11 patients progressed (30%)—3 with DLBCL, 2 with MM, 2 with HL, and 4 with other lymphomas.
Thirty-seven patients (84%) discontinued CUDC-907 because of progressive disease or clinical signs of progressive disease at the data cutoff.
Based on the clinical activity of CUDC-907 in patients with relapsed/refractory DLBCL, particularly those with MYC alterations, Curis has initiated a phase 2 trial of the drug in these patients. The recommended phase 2 dose is 60 mg on the 5/2 dosing schedule. ![]()