User login
Hot & Bothered About Kidney Stones
The long lazy days of summer are ending: The warm evenings, the iced tea, the sounds of kids playing at the pool being overshadowed by the loud moans of the patient with kidney stones ….
Kidney stones (nephrolithiasis) are collections of crystals that coalesce into a hard ball and can lodge in any location of the urinary collecting systems. More than half a million patients seen in US emergency departments each year will receive a diagnosis of nephrolithiasis.1
But the problem is much more common in the summer, thanks to the double whammy of heat and humidity.2-4 Research indicates that it is not geographic area but instead the effects of climate that impact stone incidence.5 As climate change occurs, it is expected that the incidence of kidney stones will rise.
There is a “stone belt” that covers the southern portion of the United States (see Figure). As reported in Kidney International, this area is growing due to climate change and is expected to reach as far north as Nebraska, Illinois, Pennsylvania, and Oregon by 2095. Thus, the incidence of stone formation will increase throughout the 21st century in many parts of the US.6
Kidney stones are more common in men than in women and in white than in nonwhite persons (by three to four times). Peak incidence occurs between ages 20 and 50.1 Heat plays a greater role in the increased incidence of stone formation in men for unknown reasons.6
Stones that lodge in the ureter or the calyces of the kidney will often cause obstruction. When the flow of urine is obstructed, infection, loss of kidney function, and chronic permanent damage can result. Thus, decreasing the incidence of stones is vital at any time of the year—but most significant during the summer.
All patients with a history of stones require fluid hydration, up to 2.5 L/d, with extra intake during the heat of summer.7 Patients who travel to hot, humid regions must be encouraged to increase fluid consumption. Often, foreign travel can be problematic due to a decrease in access to clean drinking water and/or lavatory facilities. It is incumbent upon the practitioner to review risk for kidney stones with patients who plan to travel to warm areas.
As the summer season closes and school starts, this is a perfect time to review the causes, treatment, and most importantly, the methods to decrease recurrent kidney stone formation with patients. Each incident of stone formation for our patients can translate to an increased incidence of chronic kidney disease and a 50% risk for another stone during their lifetime.1
REFERENCES
1. National Kidney Foundation. Kidney stones. www.kidney.org/atoz/content/kidneystones.cfm. Accessed September 10, 2014.
2.Schade GR, Faerber GJ. Urinary tract stones. Prim Care. 2010;37(3):565-581, ix.
3. Pearle MS, Calhoun E, Curhan GC. Urolithiasis. In: Litwin MS, Saigal CS, eds. Urologic Diseases in America. National Institute of Diabetes and Digestive and Kidney Diseases. 2007:281-320. http://kidney.niddk.nih.gov/statistics/uda/Urologic_Diseases_in_America.pdf. Accessed September 10, 2014.
4. Romero V, Akpinar H, Assimos DG. Kidney stones: a global picture of prevalence, incidence and associated risk factors. Rev Urol. 2010;12(2-3):e86-e96.
5. Eisner BH, Sheth S, Herrick B, et al. The effects of ambient temperature, humidity and season of year on urine composition in patients with nephrolithiasis. BJU Int. 2012;110(11c):E1014–E1017.
6. Fakheri RJ, Goldfarb DS. Ambient temperature as a contributor to kidney stone formation: Implications of global warming. Kidney Int. 2011;79:1178–1185.
7. Lipkin ME, Preminger GM. Demystifying the medical management of nephrolithiasis. Rev Urol. 2011;13(1):34-38.
The long lazy days of summer are ending: The warm evenings, the iced tea, the sounds of kids playing at the pool being overshadowed by the loud moans of the patient with kidney stones ….
Kidney stones (nephrolithiasis) are collections of crystals that coalesce into a hard ball and can lodge in any location of the urinary collecting systems. More than half a million patients seen in US emergency departments each year will receive a diagnosis of nephrolithiasis.1
But the problem is much more common in the summer, thanks to the double whammy of heat and humidity.2-4 Research indicates that it is not geographic area but instead the effects of climate that impact stone incidence.5 As climate change occurs, it is expected that the incidence of kidney stones will rise.
There is a “stone belt” that covers the southern portion of the United States (see Figure). As reported in Kidney International, this area is growing due to climate change and is expected to reach as far north as Nebraska, Illinois, Pennsylvania, and Oregon by 2095. Thus, the incidence of stone formation will increase throughout the 21st century in many parts of the US.6
Kidney stones are more common in men than in women and in white than in nonwhite persons (by three to four times). Peak incidence occurs between ages 20 and 50.1 Heat plays a greater role in the increased incidence of stone formation in men for unknown reasons.6
Stones that lodge in the ureter or the calyces of the kidney will often cause obstruction. When the flow of urine is obstructed, infection, loss of kidney function, and chronic permanent damage can result. Thus, decreasing the incidence of stones is vital at any time of the year—but most significant during the summer.
All patients with a history of stones require fluid hydration, up to 2.5 L/d, with extra intake during the heat of summer.7 Patients who travel to hot, humid regions must be encouraged to increase fluid consumption. Often, foreign travel can be problematic due to a decrease in access to clean drinking water and/or lavatory facilities. It is incumbent upon the practitioner to review risk for kidney stones with patients who plan to travel to warm areas.
As the summer season closes and school starts, this is a perfect time to review the causes, treatment, and most importantly, the methods to decrease recurrent kidney stone formation with patients. Each incident of stone formation for our patients can translate to an increased incidence of chronic kidney disease and a 50% risk for another stone during their lifetime.1
REFERENCES
1. National Kidney Foundation. Kidney stones. www.kidney.org/atoz/content/kidneystones.cfm. Accessed September 10, 2014.
2.Schade GR, Faerber GJ. Urinary tract stones. Prim Care. 2010;37(3):565-581, ix.
3. Pearle MS, Calhoun E, Curhan GC. Urolithiasis. In: Litwin MS, Saigal CS, eds. Urologic Diseases in America. National Institute of Diabetes and Digestive and Kidney Diseases. 2007:281-320. http://kidney.niddk.nih.gov/statistics/uda/Urologic_Diseases_in_America.pdf. Accessed September 10, 2014.
4. Romero V, Akpinar H, Assimos DG. Kidney stones: a global picture of prevalence, incidence and associated risk factors. Rev Urol. 2010;12(2-3):e86-e96.
5. Eisner BH, Sheth S, Herrick B, et al. The effects of ambient temperature, humidity and season of year on urine composition in patients with nephrolithiasis. BJU Int. 2012;110(11c):E1014–E1017.
6. Fakheri RJ, Goldfarb DS. Ambient temperature as a contributor to kidney stone formation: Implications of global warming. Kidney Int. 2011;79:1178–1185.
7. Lipkin ME, Preminger GM. Demystifying the medical management of nephrolithiasis. Rev Urol. 2011;13(1):34-38.
The long lazy days of summer are ending: The warm evenings, the iced tea, the sounds of kids playing at the pool being overshadowed by the loud moans of the patient with kidney stones ….
Kidney stones (nephrolithiasis) are collections of crystals that coalesce into a hard ball and can lodge in any location of the urinary collecting systems. More than half a million patients seen in US emergency departments each year will receive a diagnosis of nephrolithiasis.1
But the problem is much more common in the summer, thanks to the double whammy of heat and humidity.2-4 Research indicates that it is not geographic area but instead the effects of climate that impact stone incidence.5 As climate change occurs, it is expected that the incidence of kidney stones will rise.
There is a “stone belt” that covers the southern portion of the United States (see Figure). As reported in Kidney International, this area is growing due to climate change and is expected to reach as far north as Nebraska, Illinois, Pennsylvania, and Oregon by 2095. Thus, the incidence of stone formation will increase throughout the 21st century in many parts of the US.6
Kidney stones are more common in men than in women and in white than in nonwhite persons (by three to four times). Peak incidence occurs between ages 20 and 50.1 Heat plays a greater role in the increased incidence of stone formation in men for unknown reasons.6
Stones that lodge in the ureter or the calyces of the kidney will often cause obstruction. When the flow of urine is obstructed, infection, loss of kidney function, and chronic permanent damage can result. Thus, decreasing the incidence of stones is vital at any time of the year—but most significant during the summer.
All patients with a history of stones require fluid hydration, up to 2.5 L/d, with extra intake during the heat of summer.7 Patients who travel to hot, humid regions must be encouraged to increase fluid consumption. Often, foreign travel can be problematic due to a decrease in access to clean drinking water and/or lavatory facilities. It is incumbent upon the practitioner to review risk for kidney stones with patients who plan to travel to warm areas.
As the summer season closes and school starts, this is a perfect time to review the causes, treatment, and most importantly, the methods to decrease recurrent kidney stone formation with patients. Each incident of stone formation for our patients can translate to an increased incidence of chronic kidney disease and a 50% risk for another stone during their lifetime.1
REFERENCES
1. National Kidney Foundation. Kidney stones. www.kidney.org/atoz/content/kidneystones.cfm. Accessed September 10, 2014.
2.Schade GR, Faerber GJ. Urinary tract stones. Prim Care. 2010;37(3):565-581, ix.
3. Pearle MS, Calhoun E, Curhan GC. Urolithiasis. In: Litwin MS, Saigal CS, eds. Urologic Diseases in America. National Institute of Diabetes and Digestive and Kidney Diseases. 2007:281-320. http://kidney.niddk.nih.gov/statistics/uda/Urologic_Diseases_in_America.pdf. Accessed September 10, 2014.
4. Romero V, Akpinar H, Assimos DG. Kidney stones: a global picture of prevalence, incidence and associated risk factors. Rev Urol. 2010;12(2-3):e86-e96.
5. Eisner BH, Sheth S, Herrick B, et al. The effects of ambient temperature, humidity and season of year on urine composition in patients with nephrolithiasis. BJU Int. 2012;110(11c):E1014–E1017.
6. Fakheri RJ, Goldfarb DS. Ambient temperature as a contributor to kidney stone formation: Implications of global warming. Kidney Int. 2011;79:1178–1185.
7. Lipkin ME, Preminger GM. Demystifying the medical management of nephrolithiasis. Rev Urol. 2011;13(1):34-38.
Statins do not worsen diabetes microvascular complications, may be protective
Contrary to expectations, statin use before the development of type II diabetes did not worsen microvascular complications such as retinopathy, neuropathy, and gangrene of the foot.
In fact, despite concerns that statins have been seen to increase glucose levels and the risk of diabetes development, they may provide a protective effect from these conditions in newly developed diabetic patients, according to an analysis of data from more than 60,000 individuals in the Danish Patient Registry.
"The cumulative incidences of diabetic retinopathy, diabetic neuropathy, and gangrene were reduced in statin users compared with non–statin users, but [the] risk of diabetic nephropathy was similar for all patients with diabetes," stated Dr. Sune F. Nielsen, Ph.D., and Dr. Børge G. Nordestgaard of the Herlev Hospital, Copenhagen University Hospital. However, they did find that statin use, as previously seen, did significantly increase the risk of developing diabetes in the first place. Their study was published online Sept. 10 in the Lancet Diabetes & Endocrinology (2014 Sept. 10 [doi: 10.1016/S2213-8587(14)70173-1]).
The researchers performed a nested matched study of all men and women living in Denmark who were diagnosed with incident diabetes during 1996-2009 at age 40 years or older, and assessed their outcomes through use of the Danish Civil Registration System, the Danish Patient Registry, and the Danish Registry of Medicinal Product Statistics. After exclusions, 62,716 patients with diabetes were randomly selected for the study: 15,679 statin users and 47,037 non–statin users. The primary outcome was the incidence of diabetic retinopathy, diabetic neuropathy, diabetic nephropathy, and gangrene of the foot. The design "captured 100% of individuals in Denmark who had ever used a statin within the time frame of the study."
Follow-up was censored at date of death for 9,560 individuals. During 215,725 person-years of follow-up, diabetic retinopathy was recorded in 2,866 patients, diabetic neuropathy in 1,406, diabetic nephropathy in 1,248, and gangrene of the foot in 2,392.
Over a median follow-up of 2.7 years, statin users were significantly less likely to be diagnosed with diabetic neuropathy (hazard ratio, 0.66; 95% confidence interval, 0.57-0.75: P less than .0001) and diabetic retinopathy (HR, 0.60; 95% CI 0.54-0.66: P less than .0001) than were those who had not received statins. However, no difference was noted in the incidence of diabetic nephropathy (HR, 0.97; 95% CI, 0.85-1.10; P = .62).
In contrast, the researchers found that statin use significantly increased the risk of developing diabetes in people who did not have the disease when the study began. When they compared a random selection of 272,994 non–statin users with 90,998 statin users, the multivariable adjusted hazard ratio for the risk of developing diabetes was 1.17 (95% CI, 1.14-1.21). These results are similar to those seen in previous randomized studies of statin use.
"In conclusion, we found no evidence that statin use is associated with an increased risk of microvascular disease; this result is important and clinically reassuring on its own. Whether or not statins are protective against some forms of microvascular disease, a possibility raised by these data, and by which mechanism, will need to be addressed in studies similar to ours, or in Mendelian randomization studies," said Dr. Nielsen and Dr. Nordestgaard. "Ideally, however, this question should be addressed in the setting of a randomized controlled trial," they added.
Dr. Nordestgaard has received consultancy fees or lecture honoraria from AstraZeneca, Pfizer, and Merck, and Dr. Nielsen declared no competing interests. The work was supported by Herlev Hospital, Copenhagen University Hospital.
Pharmacoepidemiological studies need cautious interpretation and can be regarded only as hypothesis generating; Dr. Nielsen and Dr. Nordestgaard are appropriately circumspect.
The study has many strengths, such as its size, the quality and coverage of the national registry, and external validity – i.e., statin use was associated with an increased risk of diabetes, an effect size similar to that reported in randomized trials of statins. However, important weaknesses of the study include the absence of data on important predictors of microvascular disease – e.g., hemoglobin A1c, urine albumin, and blood pressure. For now, any benefit of statins on microvascular complications remains unproven.
Dr. David Preiss, of the University of Glasgow (Scotland), is cochair of the Scottish Lipid Forum, whose annual meeting is supported by grants from pharmaceutical companies including MSD, AstraZeneca, and Sanofi. The remarks are taken from his accompanying commentary (Lancet Diabetes Endocrinol. 2014 Sept. 10 [doi: 10.1016/S2213-8587(14)70173-1]).
Pharmacoepidemiological studies need cautious interpretation and can be regarded only as hypothesis generating; Dr. Nielsen and Dr. Nordestgaard are appropriately circumspect.
The study has many strengths, such as its size, the quality and coverage of the national registry, and external validity – i.e., statin use was associated with an increased risk of diabetes, an effect size similar to that reported in randomized trials of statins. However, important weaknesses of the study include the absence of data on important predictors of microvascular disease – e.g., hemoglobin A1c, urine albumin, and blood pressure. For now, any benefit of statins on microvascular complications remains unproven.
Dr. David Preiss, of the University of Glasgow (Scotland), is cochair of the Scottish Lipid Forum, whose annual meeting is supported by grants from pharmaceutical companies including MSD, AstraZeneca, and Sanofi. The remarks are taken from his accompanying commentary (Lancet Diabetes Endocrinol. 2014 Sept. 10 [doi: 10.1016/S2213-8587(14)70173-1]).
Pharmacoepidemiological studies need cautious interpretation and can be regarded only as hypothesis generating; Dr. Nielsen and Dr. Nordestgaard are appropriately circumspect.
The study has many strengths, such as its size, the quality and coverage of the national registry, and external validity – i.e., statin use was associated with an increased risk of diabetes, an effect size similar to that reported in randomized trials of statins. However, important weaknesses of the study include the absence of data on important predictors of microvascular disease – e.g., hemoglobin A1c, urine albumin, and blood pressure. For now, any benefit of statins on microvascular complications remains unproven.
Dr. David Preiss, of the University of Glasgow (Scotland), is cochair of the Scottish Lipid Forum, whose annual meeting is supported by grants from pharmaceutical companies including MSD, AstraZeneca, and Sanofi. The remarks are taken from his accompanying commentary (Lancet Diabetes Endocrinol. 2014 Sept. 10 [doi: 10.1016/S2213-8587(14)70173-1]).
Contrary to expectations, statin use before the development of type II diabetes did not worsen microvascular complications such as retinopathy, neuropathy, and gangrene of the foot.
In fact, despite concerns that statins have been seen to increase glucose levels and the risk of diabetes development, they may provide a protective effect from these conditions in newly developed diabetic patients, according to an analysis of data from more than 60,000 individuals in the Danish Patient Registry.
"The cumulative incidences of diabetic retinopathy, diabetic neuropathy, and gangrene were reduced in statin users compared with non–statin users, but [the] risk of diabetic nephropathy was similar for all patients with diabetes," stated Dr. Sune F. Nielsen, Ph.D., and Dr. Børge G. Nordestgaard of the Herlev Hospital, Copenhagen University Hospital. However, they did find that statin use, as previously seen, did significantly increase the risk of developing diabetes in the first place. Their study was published online Sept. 10 in the Lancet Diabetes & Endocrinology (2014 Sept. 10 [doi: 10.1016/S2213-8587(14)70173-1]).
The researchers performed a nested matched study of all men and women living in Denmark who were diagnosed with incident diabetes during 1996-2009 at age 40 years or older, and assessed their outcomes through use of the Danish Civil Registration System, the Danish Patient Registry, and the Danish Registry of Medicinal Product Statistics. After exclusions, 62,716 patients with diabetes were randomly selected for the study: 15,679 statin users and 47,037 non–statin users. The primary outcome was the incidence of diabetic retinopathy, diabetic neuropathy, diabetic nephropathy, and gangrene of the foot. The design "captured 100% of individuals in Denmark who had ever used a statin within the time frame of the study."
Follow-up was censored at date of death for 9,560 individuals. During 215,725 person-years of follow-up, diabetic retinopathy was recorded in 2,866 patients, diabetic neuropathy in 1,406, diabetic nephropathy in 1,248, and gangrene of the foot in 2,392.
Over a median follow-up of 2.7 years, statin users were significantly less likely to be diagnosed with diabetic neuropathy (hazard ratio, 0.66; 95% confidence interval, 0.57-0.75: P less than .0001) and diabetic retinopathy (HR, 0.60; 95% CI 0.54-0.66: P less than .0001) than were those who had not received statins. However, no difference was noted in the incidence of diabetic nephropathy (HR, 0.97; 95% CI, 0.85-1.10; P = .62).
In contrast, the researchers found that statin use significantly increased the risk of developing diabetes in people who did not have the disease when the study began. When they compared a random selection of 272,994 non–statin users with 90,998 statin users, the multivariable adjusted hazard ratio for the risk of developing diabetes was 1.17 (95% CI, 1.14-1.21). These results are similar to those seen in previous randomized studies of statin use.
"In conclusion, we found no evidence that statin use is associated with an increased risk of microvascular disease; this result is important and clinically reassuring on its own. Whether or not statins are protective against some forms of microvascular disease, a possibility raised by these data, and by which mechanism, will need to be addressed in studies similar to ours, or in Mendelian randomization studies," said Dr. Nielsen and Dr. Nordestgaard. "Ideally, however, this question should be addressed in the setting of a randomized controlled trial," they added.
Dr. Nordestgaard has received consultancy fees or lecture honoraria from AstraZeneca, Pfizer, and Merck, and Dr. Nielsen declared no competing interests. The work was supported by Herlev Hospital, Copenhagen University Hospital.
Contrary to expectations, statin use before the development of type II diabetes did not worsen microvascular complications such as retinopathy, neuropathy, and gangrene of the foot.
In fact, despite concerns that statins have been seen to increase glucose levels and the risk of diabetes development, they may provide a protective effect from these conditions in newly developed diabetic patients, according to an analysis of data from more than 60,000 individuals in the Danish Patient Registry.
"The cumulative incidences of diabetic retinopathy, diabetic neuropathy, and gangrene were reduced in statin users compared with non–statin users, but [the] risk of diabetic nephropathy was similar for all patients with diabetes," stated Dr. Sune F. Nielsen, Ph.D., and Dr. Børge G. Nordestgaard of the Herlev Hospital, Copenhagen University Hospital. However, they did find that statin use, as previously seen, did significantly increase the risk of developing diabetes in the first place. Their study was published online Sept. 10 in the Lancet Diabetes & Endocrinology (2014 Sept. 10 [doi: 10.1016/S2213-8587(14)70173-1]).
The researchers performed a nested matched study of all men and women living in Denmark who were diagnosed with incident diabetes during 1996-2009 at age 40 years or older, and assessed their outcomes through use of the Danish Civil Registration System, the Danish Patient Registry, and the Danish Registry of Medicinal Product Statistics. After exclusions, 62,716 patients with diabetes were randomly selected for the study: 15,679 statin users and 47,037 non–statin users. The primary outcome was the incidence of diabetic retinopathy, diabetic neuropathy, diabetic nephropathy, and gangrene of the foot. The design "captured 100% of individuals in Denmark who had ever used a statin within the time frame of the study."
Follow-up was censored at date of death for 9,560 individuals. During 215,725 person-years of follow-up, diabetic retinopathy was recorded in 2,866 patients, diabetic neuropathy in 1,406, diabetic nephropathy in 1,248, and gangrene of the foot in 2,392.
Over a median follow-up of 2.7 years, statin users were significantly less likely to be diagnosed with diabetic neuropathy (hazard ratio, 0.66; 95% confidence interval, 0.57-0.75: P less than .0001) and diabetic retinopathy (HR, 0.60; 95% CI 0.54-0.66: P less than .0001) than were those who had not received statins. However, no difference was noted in the incidence of diabetic nephropathy (HR, 0.97; 95% CI, 0.85-1.10; P = .62).
In contrast, the researchers found that statin use significantly increased the risk of developing diabetes in people who did not have the disease when the study began. When they compared a random selection of 272,994 non–statin users with 90,998 statin users, the multivariable adjusted hazard ratio for the risk of developing diabetes was 1.17 (95% CI, 1.14-1.21). These results are similar to those seen in previous randomized studies of statin use.
"In conclusion, we found no evidence that statin use is associated with an increased risk of microvascular disease; this result is important and clinically reassuring on its own. Whether or not statins are protective against some forms of microvascular disease, a possibility raised by these data, and by which mechanism, will need to be addressed in studies similar to ours, or in Mendelian randomization studies," said Dr. Nielsen and Dr. Nordestgaard. "Ideally, however, this question should be addressed in the setting of a randomized controlled trial," they added.
Dr. Nordestgaard has received consultancy fees or lecture honoraria from AstraZeneca, Pfizer, and Merck, and Dr. Nielsen declared no competing interests. The work was supported by Herlev Hospital, Copenhagen University Hospital.
FROM THE LANCET DIABETES & ENDOCRINOLOGY
Key clinical point: Statins may protect against microvascular complications in diabetes patients.
Major finding: Statin users were significantly less likely to be diagnosed with diabetic neuropathy (HR, 0.66) and diabetic retinopathy (HR, 0.60) than non–statin users.
Data source: A registry study compared 62,716 patients with diabetes: 15,679 statin users and 47,037 non–statin users.
Disclosures: Dr. Nordestgaard has received consultancy fees or lecture honoraria from AstraZeneca, Pfizer, and Merck, and Dr. Nielsen declared no competing interests. The work was supported by Herlev Hospital, Copenhagen University Hospital.
High PRA is risk factor for death while on kidney transplant wait list
SAN FRANCISCO – Patients awaiting kidney transplantation are more likely to die from any cause and specifically from cardiovascular causes if they have greater immune sensitization as assessed from panel-reactive antibodies, according to a study reported at the annual meeting of the 2014 World Transplant Congress.
"We find PRAs [panel-reactive antibodies] to be a predictor of mortality in wait-listed kidney transplant candidates," commented lead investigator Dr. Ruth Sapir-Pichhadze, a research fellow at the University of Toronto.
"When looking at all-cause mortality, our findings support the sliding scale of allocation points by PRA," whereby patients are given higher priority on the waiting list, she added. "When looking at cardiovascular mortality, our findings give rise to a need to conduct further studies to corroborate our findings and investigate the mechanisms by which PRA confers added risk."
In comments provided by e-mail, one of the session’s cochairs, Dr. Jonathan Bromberg of the University of Maryland, Baltimore, said, "The results from this abstract provide a new twist to the analysis of highly sensitized patients, suggesting that a high PRA may be associated with increased cardiovascular and all-cause mortality."
It was difficult to know whether analyses had captured all potential confounders, according to Dr. Bromberg. "Nonetheless, if we take the results at face value, they suggest even more urgency for transplanting this group of patients."
"Since patients with high PRAs already have an advantage on the organ wait list, the answer for these patients lies not in giving them more advantage on the wait list, but rather expanding the living and deceased donor organ supply, and also devising new methods to prevent and treat antibody-mediated rejection, which currently are still inadequate to ensure excellent allograft function in this challenging group of recipients," he concluded.
Dr. Sapir-Pichhadze and her colleagues studied 161,308 adult patients wait-listed for a first kidney transplant between 2000 and 2009 in the Scientific Registry of Transplant Recipients.
The patients had serial measurements of PRAs, which target human leukocyte antigen and are used to assess likely compatibility with donor organs, and were followed until transplantation, death, or end of observation in 2010.
Multivariate analyses showed that when patients having a time-varying PRA of 0% were the reference group, the risk of cardiovascular mortality was elevated for peers with a PRA of 1%-19% (hazard ratio, 1.05), a PRA of 20%-79% (1.11), or a PRA of 80%-100% (1.21), Dr. Sapir-Pichhadze reported at the congress, which was sponsored by the American Society of Transplant Surgeons.
Sensitivity analyses showed that this association was especially pronounced among patients at low risk for comorbidity, defined as those who were under age 40, had been on dialysis less than a year, and did not have coronary artery disease, diabetes, or peripheral vascular disease.
The risk of all-cause mortality was similarly elevated for patients with a PRA of 20%-79% (hazard ratio, 1.11) or a PRA of 80%-100% (1.17). And sensitivity analyses again showed that this association was especially pronounced among the subset at low risk for comorbidity.
Findings were much the same when only baseline PRA was considered, according to Dr. Sapir-Pichhadze, who disclosed no conflicts of interest relevant to the research.
The other session cochair, Dr. Jon Von Visger, of the Ohio State University in Columbus, asked, "Is this association of mortality and higher PRA independent of time on wait list?"
"We addressed this issue, considering how important it is, using two methods," she replied; one was inclusion in models of the time from dialysis initiation to wait listing as a covariate, and the other was performance of a competing risk analysis. And the association persisted in both cases.
A session attendee asked, "Have you looked for a correlation between PRA and C-reactive protein?"
"This is a registry type of analysis, and CRP is not recorded there," Dr. Sapir-Pichhadze replied, while acknowledging that the question is important. "This is where prospective cohort studies potentially would account for this kind of variable, and see if CRP explains similarly [the association that] PRA would otherwise explain."
Dr. Sapir-Pichhadze disclosed no relevant conflicts of interest.
SAN FRANCISCO – Patients awaiting kidney transplantation are more likely to die from any cause and specifically from cardiovascular causes if they have greater immune sensitization as assessed from panel-reactive antibodies, according to a study reported at the annual meeting of the 2014 World Transplant Congress.
"We find PRAs [panel-reactive antibodies] to be a predictor of mortality in wait-listed kidney transplant candidates," commented lead investigator Dr. Ruth Sapir-Pichhadze, a research fellow at the University of Toronto.
"When looking at all-cause mortality, our findings support the sliding scale of allocation points by PRA," whereby patients are given higher priority on the waiting list, she added. "When looking at cardiovascular mortality, our findings give rise to a need to conduct further studies to corroborate our findings and investigate the mechanisms by which PRA confers added risk."
In comments provided by e-mail, one of the session’s cochairs, Dr. Jonathan Bromberg of the University of Maryland, Baltimore, said, "The results from this abstract provide a new twist to the analysis of highly sensitized patients, suggesting that a high PRA may be associated with increased cardiovascular and all-cause mortality."
It was difficult to know whether analyses had captured all potential confounders, according to Dr. Bromberg. "Nonetheless, if we take the results at face value, they suggest even more urgency for transplanting this group of patients."
"Since patients with high PRAs already have an advantage on the organ wait list, the answer for these patients lies not in giving them more advantage on the wait list, but rather expanding the living and deceased donor organ supply, and also devising new methods to prevent and treat antibody-mediated rejection, which currently are still inadequate to ensure excellent allograft function in this challenging group of recipients," he concluded.
Dr. Sapir-Pichhadze and her colleagues studied 161,308 adult patients wait-listed for a first kidney transplant between 2000 and 2009 in the Scientific Registry of Transplant Recipients.
The patients had serial measurements of PRAs, which target human leukocyte antigen and are used to assess likely compatibility with donor organs, and were followed until transplantation, death, or end of observation in 2010.
Multivariate analyses showed that when patients having a time-varying PRA of 0% were the reference group, the risk of cardiovascular mortality was elevated for peers with a PRA of 1%-19% (hazard ratio, 1.05), a PRA of 20%-79% (1.11), or a PRA of 80%-100% (1.21), Dr. Sapir-Pichhadze reported at the congress, which was sponsored by the American Society of Transplant Surgeons.
Sensitivity analyses showed that this association was especially pronounced among patients at low risk for comorbidity, defined as those who were under age 40, had been on dialysis less than a year, and did not have coronary artery disease, diabetes, or peripheral vascular disease.
The risk of all-cause mortality was similarly elevated for patients with a PRA of 20%-79% (hazard ratio, 1.11) or a PRA of 80%-100% (1.17). And sensitivity analyses again showed that this association was especially pronounced among the subset at low risk for comorbidity.
Findings were much the same when only baseline PRA was considered, according to Dr. Sapir-Pichhadze, who disclosed no conflicts of interest relevant to the research.
The other session cochair, Dr. Jon Von Visger, of the Ohio State University in Columbus, asked, "Is this association of mortality and higher PRA independent of time on wait list?"
"We addressed this issue, considering how important it is, using two methods," she replied; one was inclusion in models of the time from dialysis initiation to wait listing as a covariate, and the other was performance of a competing risk analysis. And the association persisted in both cases.
A session attendee asked, "Have you looked for a correlation between PRA and C-reactive protein?"
"This is a registry type of analysis, and CRP is not recorded there," Dr. Sapir-Pichhadze replied, while acknowledging that the question is important. "This is where prospective cohort studies potentially would account for this kind of variable, and see if CRP explains similarly [the association that] PRA would otherwise explain."
Dr. Sapir-Pichhadze disclosed no relevant conflicts of interest.
SAN FRANCISCO – Patients awaiting kidney transplantation are more likely to die from any cause and specifically from cardiovascular causes if they have greater immune sensitization as assessed from panel-reactive antibodies, according to a study reported at the annual meeting of the 2014 World Transplant Congress.
"We find PRAs [panel-reactive antibodies] to be a predictor of mortality in wait-listed kidney transplant candidates," commented lead investigator Dr. Ruth Sapir-Pichhadze, a research fellow at the University of Toronto.
"When looking at all-cause mortality, our findings support the sliding scale of allocation points by PRA," whereby patients are given higher priority on the waiting list, she added. "When looking at cardiovascular mortality, our findings give rise to a need to conduct further studies to corroborate our findings and investigate the mechanisms by which PRA confers added risk."
In comments provided by e-mail, one of the session’s cochairs, Dr. Jonathan Bromberg of the University of Maryland, Baltimore, said, "The results from this abstract provide a new twist to the analysis of highly sensitized patients, suggesting that a high PRA may be associated with increased cardiovascular and all-cause mortality."
It was difficult to know whether analyses had captured all potential confounders, according to Dr. Bromberg. "Nonetheless, if we take the results at face value, they suggest even more urgency for transplanting this group of patients."
"Since patients with high PRAs already have an advantage on the organ wait list, the answer for these patients lies not in giving them more advantage on the wait list, but rather expanding the living and deceased donor organ supply, and also devising new methods to prevent and treat antibody-mediated rejection, which currently are still inadequate to ensure excellent allograft function in this challenging group of recipients," he concluded.
Dr. Sapir-Pichhadze and her colleagues studied 161,308 adult patients wait-listed for a first kidney transplant between 2000 and 2009 in the Scientific Registry of Transplant Recipients.
The patients had serial measurements of PRAs, which target human leukocyte antigen and are used to assess likely compatibility with donor organs, and were followed until transplantation, death, or end of observation in 2010.
Multivariate analyses showed that when patients having a time-varying PRA of 0% were the reference group, the risk of cardiovascular mortality was elevated for peers with a PRA of 1%-19% (hazard ratio, 1.05), a PRA of 20%-79% (1.11), or a PRA of 80%-100% (1.21), Dr. Sapir-Pichhadze reported at the congress, which was sponsored by the American Society of Transplant Surgeons.
Sensitivity analyses showed that this association was especially pronounced among patients at low risk for comorbidity, defined as those who were under age 40, had been on dialysis less than a year, and did not have coronary artery disease, diabetes, or peripheral vascular disease.
The risk of all-cause mortality was similarly elevated for patients with a PRA of 20%-79% (hazard ratio, 1.11) or a PRA of 80%-100% (1.17). And sensitivity analyses again showed that this association was especially pronounced among the subset at low risk for comorbidity.
Findings were much the same when only baseline PRA was considered, according to Dr. Sapir-Pichhadze, who disclosed no conflicts of interest relevant to the research.
The other session cochair, Dr. Jon Von Visger, of the Ohio State University in Columbus, asked, "Is this association of mortality and higher PRA independent of time on wait list?"
"We addressed this issue, considering how important it is, using two methods," she replied; one was inclusion in models of the time from dialysis initiation to wait listing as a covariate, and the other was performance of a competing risk analysis. And the association persisted in both cases.
A session attendee asked, "Have you looked for a correlation between PRA and C-reactive protein?"
"This is a registry type of analysis, and CRP is not recorded there," Dr. Sapir-Pichhadze replied, while acknowledging that the question is important. "This is where prospective cohort studies potentially would account for this kind of variable, and see if CRP explains similarly [the association that] PRA would otherwise explain."
Dr. Sapir-Pichhadze disclosed no relevant conflicts of interest.
AT THE 2014 WORLD TRANSPLANT CONGRESS
Key clinical point: Wait-listed kidney transplant patients should be tested for higher PRA levels for increased risk of mortality.
Major finding: The risks of cardiovascular mortality and all-cause mortality rose with PRA category; they were 21% and 17% higher, respectively, for patients in the highest versus lowest category.
Data source: A cohort study of 161,308 adult patients wait-listed for a first kidney transplant during 2000-2009.
Disclosures: Dr. Sapir-Pichhadze disclosed no relevant conflicts of interest.
Right-sided living donor kidney transplant found safe
SAN FRANCISCO – The practice of preferentially using left instead of right kidneys in living donor kidney transplantation may no longer be justified in the era of contemporary laparoscopic surgery, suggests a national study reported at the 2014 World Transplant Congress.
"The current approach in many centers is to prefer left living donor nephrectomy due to longer vessel length...Right donor nephrectomy, at least in our center and I think in most centers, has generally been reserved for cases of multiple or complex vessels on the left or incidental anatomical abnormalities on the right like cysts or stones," commented presenting author Dr. Tim E. Taber of Indiana University in Indianapolis.
Only one in seven of the roughly 59,000 living donor kidney transplants studied was performed using a right kidney. However, most short- and long-term outcomes were statistically indistinguishable between recipients of left and right kidneys, and the differences that were significant were small, he reported at the congress sponsored by the American Society of Transplant Surgeons.
"Our [study] is the largest national analysis or most recent large data analysis done on this subject in today’s surgical era of established laparoscopic living donor nephrectomies. There may be a minor risk for slightly inferior outcomes with right versus left kidneys," Dr. Taber concluded.
"Right-donor nephrectomy continues to be performed with great reluctance," he added. Yet, "under the accepted principles of live-donor nephrectomy, with enough surgical expertise, right-donor nephrectomy can be performed successfully. Right kidneys seem to have a very small difference, if any, in outcomes as compared to left kidneys. Surgical expertise and experience should be tailored toward this aspect."
A session attendee from Brazil commented, "We [prefer] to choose the right kidney in situations where we have one artery on the right side and multiple arteries on the left side." In these cases, his group uses an approach to the vasculature adopted from pancreas transplantation. "We have identical results with the right and left side," he reported.
Dr. Lloyd E. Ratner, director of renal and pancreatic transplantation at Columbia University Medical Center in New York, who also attended the session, said, "I feel somewhat responsible for causing this problem with the right kidney because we were the ones that originally described the higher thrombosis rate with the right kidney with the laparoscopic donor nephrectomies. And I think it scared everyone off from this topic."
As several attendees noted, "there are surgical ways of getting around this," he agreed, offering two more options. "The first is that if we get a short vein, we’re not reluctant at all to put a piece of Dacron onto it, so you don’t even need to dig out the saphenous and cause additional time or morbidity to the patient. And the nice thing about the Dacron grafts is that they are corrugated and they don’t collapse. They also stretch, so you don’t need to cut them exactly precisely," he said.
"And number two is when you are stapling ... it’s often useful to be able to staple onto the cava and not get the vein in one staple byte." By using two passes in the appropriate configuration, "you actually get a cuff of cava, then you have plenty of vein," he explained.
In the study, Dr. Taber and colleagues retrospectively analyzed data from 58,599 adult living donor kidney transplants performed during 2000-2009 and captured in the United Network for Organ Sharing (UNOS) database. In 86% of cases, surgeons used the donor’s left kidney.
Recipients of left and right kidneys were statistically indistinguishable with respect to hospital length of stay, treatment for acute rejection within 6 months, acute rejection as a cause of graft failure, inadequate urine production in the first 24 hours, primary graft failure, graft thrombosis or surgical complication as a contributory cause of graft failure, and 1-year graft survival.
Those receiving a right kidney did have significant but small increases in rates of delayed graft function, as defined by the need for dialysis within 7 days of transplantation (5.7% vs. 4.2%), lack of decline in serum creatinine in the first 24 hours (19.7% vs. 16.4%), treatment for acute rejection within 1 year (12.7% vs. 11.8%), and graft thrombosis as the cause of graft failure (1.1% vs. 0.8%).
The Kaplan-Meier cumulative rate of graft survival was better for left kidneys than for right kidneys (P = .006), but "these are essentially superimposed numbers," said Dr. Taber, who disclosed no conflicts of interest related to the research.
The study had limitations, such as its retrospective design, lack of more detailed information about donor and recipient outcomes, and reliance on data as reported by centers, he acknowledged. Also, such large studies may pick up small differences that are not clinically meaningful.
"With ever-increasing demands for living donor transplantation, right-donor nephrectomies are being considered more often. Every effort should be made to leave the donor with the higher-functioning kidney, but at the same time maximizing the living donor pool," Dr. Taber concluded.
Dr. Taber disclosed no relevant conflicts of interest.
SAN FRANCISCO – The practice of preferentially using left instead of right kidneys in living donor kidney transplantation may no longer be justified in the era of contemporary laparoscopic surgery, suggests a national study reported at the 2014 World Transplant Congress.
"The current approach in many centers is to prefer left living donor nephrectomy due to longer vessel length...Right donor nephrectomy, at least in our center and I think in most centers, has generally been reserved for cases of multiple or complex vessels on the left or incidental anatomical abnormalities on the right like cysts or stones," commented presenting author Dr. Tim E. Taber of Indiana University in Indianapolis.
Only one in seven of the roughly 59,000 living donor kidney transplants studied was performed using a right kidney. However, most short- and long-term outcomes were statistically indistinguishable between recipients of left and right kidneys, and the differences that were significant were small, he reported at the congress sponsored by the American Society of Transplant Surgeons.
"Our [study] is the largest national analysis or most recent large data analysis done on this subject in today’s surgical era of established laparoscopic living donor nephrectomies. There may be a minor risk for slightly inferior outcomes with right versus left kidneys," Dr. Taber concluded.
"Right-donor nephrectomy continues to be performed with great reluctance," he added. Yet, "under the accepted principles of live-donor nephrectomy, with enough surgical expertise, right-donor nephrectomy can be performed successfully. Right kidneys seem to have a very small difference, if any, in outcomes as compared to left kidneys. Surgical expertise and experience should be tailored toward this aspect."
A session attendee from Brazil commented, "We [prefer] to choose the right kidney in situations where we have one artery on the right side and multiple arteries on the left side." In these cases, his group uses an approach to the vasculature adopted from pancreas transplantation. "We have identical results with the right and left side," he reported.
Dr. Lloyd E. Ratner, director of renal and pancreatic transplantation at Columbia University Medical Center in New York, who also attended the session, said, "I feel somewhat responsible for causing this problem with the right kidney because we were the ones that originally described the higher thrombosis rate with the right kidney with the laparoscopic donor nephrectomies. And I think it scared everyone off from this topic."
As several attendees noted, "there are surgical ways of getting around this," he agreed, offering two more options. "The first is that if we get a short vein, we’re not reluctant at all to put a piece of Dacron onto it, so you don’t even need to dig out the saphenous and cause additional time or morbidity to the patient. And the nice thing about the Dacron grafts is that they are corrugated and they don’t collapse. They also stretch, so you don’t need to cut them exactly precisely," he said.
"And number two is when you are stapling ... it’s often useful to be able to staple onto the cava and not get the vein in one staple byte." By using two passes in the appropriate configuration, "you actually get a cuff of cava, then you have plenty of vein," he explained.
In the study, Dr. Taber and colleagues retrospectively analyzed data from 58,599 adult living donor kidney transplants performed during 2000-2009 and captured in the United Network for Organ Sharing (UNOS) database. In 86% of cases, surgeons used the donor’s left kidney.
Recipients of left and right kidneys were statistically indistinguishable with respect to hospital length of stay, treatment for acute rejection within 6 months, acute rejection as a cause of graft failure, inadequate urine production in the first 24 hours, primary graft failure, graft thrombosis or surgical complication as a contributory cause of graft failure, and 1-year graft survival.
Those receiving a right kidney did have significant but small increases in rates of delayed graft function, as defined by the need for dialysis within 7 days of transplantation (5.7% vs. 4.2%), lack of decline in serum creatinine in the first 24 hours (19.7% vs. 16.4%), treatment for acute rejection within 1 year (12.7% vs. 11.8%), and graft thrombosis as the cause of graft failure (1.1% vs. 0.8%).
The Kaplan-Meier cumulative rate of graft survival was better for left kidneys than for right kidneys (P = .006), but "these are essentially superimposed numbers," said Dr. Taber, who disclosed no conflicts of interest related to the research.
The study had limitations, such as its retrospective design, lack of more detailed information about donor and recipient outcomes, and reliance on data as reported by centers, he acknowledged. Also, such large studies may pick up small differences that are not clinically meaningful.
"With ever-increasing demands for living donor transplantation, right-donor nephrectomies are being considered more often. Every effort should be made to leave the donor with the higher-functioning kidney, but at the same time maximizing the living donor pool," Dr. Taber concluded.
Dr. Taber disclosed no relevant conflicts of interest.
SAN FRANCISCO – The practice of preferentially using left instead of right kidneys in living donor kidney transplantation may no longer be justified in the era of contemporary laparoscopic surgery, suggests a national study reported at the 2014 World Transplant Congress.
"The current approach in many centers is to prefer left living donor nephrectomy due to longer vessel length...Right donor nephrectomy, at least in our center and I think in most centers, has generally been reserved for cases of multiple or complex vessels on the left or incidental anatomical abnormalities on the right like cysts or stones," commented presenting author Dr. Tim E. Taber of Indiana University in Indianapolis.
Only one in seven of the roughly 59,000 living donor kidney transplants studied was performed using a right kidney. However, most short- and long-term outcomes were statistically indistinguishable between recipients of left and right kidneys, and the differences that were significant were small, he reported at the congress sponsored by the American Society of Transplant Surgeons.
"Our [study] is the largest national analysis or most recent large data analysis done on this subject in today’s surgical era of established laparoscopic living donor nephrectomies. There may be a minor risk for slightly inferior outcomes with right versus left kidneys," Dr. Taber concluded.
"Right-donor nephrectomy continues to be performed with great reluctance," he added. Yet, "under the accepted principles of live-donor nephrectomy, with enough surgical expertise, right-donor nephrectomy can be performed successfully. Right kidneys seem to have a very small difference, if any, in outcomes as compared to left kidneys. Surgical expertise and experience should be tailored toward this aspect."
A session attendee from Brazil commented, "We [prefer] to choose the right kidney in situations where we have one artery on the right side and multiple arteries on the left side." In these cases, his group uses an approach to the vasculature adopted from pancreas transplantation. "We have identical results with the right and left side," he reported.
Dr. Lloyd E. Ratner, director of renal and pancreatic transplantation at Columbia University Medical Center in New York, who also attended the session, said, "I feel somewhat responsible for causing this problem with the right kidney because we were the ones that originally described the higher thrombosis rate with the right kidney with the laparoscopic donor nephrectomies. And I think it scared everyone off from this topic."
As several attendees noted, "there are surgical ways of getting around this," he agreed, offering two more options. "The first is that if we get a short vein, we’re not reluctant at all to put a piece of Dacron onto it, so you don’t even need to dig out the saphenous and cause additional time or morbidity to the patient. And the nice thing about the Dacron grafts is that they are corrugated and they don’t collapse. They also stretch, so you don’t need to cut them exactly precisely," he said.
"And number two is when you are stapling ... it’s often useful to be able to staple onto the cava and not get the vein in one staple byte." By using two passes in the appropriate configuration, "you actually get a cuff of cava, then you have plenty of vein," he explained.
In the study, Dr. Taber and colleagues retrospectively analyzed data from 58,599 adult living donor kidney transplants performed during 2000-2009 and captured in the United Network for Organ Sharing (UNOS) database. In 86% of cases, surgeons used the donor’s left kidney.
Recipients of left and right kidneys were statistically indistinguishable with respect to hospital length of stay, treatment for acute rejection within 6 months, acute rejection as a cause of graft failure, inadequate urine production in the first 24 hours, primary graft failure, graft thrombosis or surgical complication as a contributory cause of graft failure, and 1-year graft survival.
Those receiving a right kidney did have significant but small increases in rates of delayed graft function, as defined by the need for dialysis within 7 days of transplantation (5.7% vs. 4.2%), lack of decline in serum creatinine in the first 24 hours (19.7% vs. 16.4%), treatment for acute rejection within 1 year (12.7% vs. 11.8%), and graft thrombosis as the cause of graft failure (1.1% vs. 0.8%).
The Kaplan-Meier cumulative rate of graft survival was better for left kidneys than for right kidneys (P = .006), but "these are essentially superimposed numbers," said Dr. Taber, who disclosed no conflicts of interest related to the research.
The study had limitations, such as its retrospective design, lack of more detailed information about donor and recipient outcomes, and reliance on data as reported by centers, he acknowledged. Also, such large studies may pick up small differences that are not clinically meaningful.
"With ever-increasing demands for living donor transplantation, right-donor nephrectomies are being considered more often. Every effort should be made to leave the donor with the higher-functioning kidney, but at the same time maximizing the living donor pool," Dr. Taber concluded.
Dr. Taber disclosed no relevant conflicts of interest.
FROM THE 2014 WORLD TRANSPLANT CONGRESS
Key clinical point: Choices of kidney to transplant may not need to hinge on left or right donor organ as a deciding factor.
Major Finding: Recipients of left and right kidneys were statistically indistinguishable with respect to hospital length of stay, treatment for acute rejection within 6 months, acute rejection as a cause of graft failure, inadequate urine production in the first 24 hours, and primary graft failure for acute rejection.
Data Source: A national retrospective cohort study of 58,599 adult living donor kidney transplants done during 2000-2009
Disclosures: Dr. Taber disclosed no relevant conflicts of interest.

Polycystic kidney disease: Molecular understanding dictating management

Dr. Braun is an iconic figure in Cleveland Clinic medicine. He is the consummate internist, nephrologist, and transplantation physician, but he is also a critical thinker. He strives to understand (and explain) what underpins our clinical observations and therapeutic decisions. He asks the “why” questions. As he ticked through the manifestations of PKD and the diagnostic dilemmas that arise in taking care of these patients, and then transitioned into explaining the interesting though incomplete current molecular understanding of this relatively prevalent genetic disorder, I heard many of the same questions I had asked myself 30 years ago. But this time I was getting some answers.
How can one be certain a cyst is infected? How do these cysts form and expand without apparent communication with the tubular lumens? (Intracystic bleeding and infection may not be reflected in the urinalysis, although the organism isolated from infected cysts is frequently Escherichia coli.) If renal cysts are formed from tubular epithelial cells that are preprogrammed to self-organize into lumen-like structures, how does the same genetic defect predispose to cyst formation in organs such as the liver, or to aneurysms in blood vessels in the brain? Why does the disease take so long to express itself, and why is its expression so variable?
The patient did well during his hospital stay 30 years ago. As I recall, he had staphylococcal bacteremia with an infected cyst. We discussed the clinical scenario but had no suggestions as to how to prevent the growth of what we now know are about 60 subclinical cysts for every one that we recognize. And we certainly didn’t discuss the idea that the disease process may be partially driven by dysfunctional nonmotile cilia that should respond to urine flow by appropriately directing regeneration and proliferation of renal tubular cells.
I love getting answers to questions that I didn’t know enough to ask.
Dr. Braun is an iconic figure in Cleveland Clinic medicine. He is the consummate internist, nephrologist, and transplantation physician, but he is also a critical thinker. He strives to understand (and explain) what underpins our clinical observations and therapeutic decisions. He asks the “why” questions. As he ticked through the manifestations of PKD and the diagnostic dilemmas that arise in taking care of these patients, and then transitioned into explaining the interesting though incomplete current molecular understanding of this relatively prevalent genetic disorder, I heard many of the same questions I had asked myself 30 years ago. But this time I was getting some answers.
How can one be certain a cyst is infected? How do these cysts form and expand without apparent communication with the tubular lumens? (Intracystic bleeding and infection may not be reflected in the urinalysis, although the organism isolated from infected cysts is frequently Escherichia coli.) If renal cysts are formed from tubular epithelial cells that are preprogrammed to self-organize into lumen-like structures, how does the same genetic defect predispose to cyst formation in organs such as the liver, or to aneurysms in blood vessels in the brain? Why does the disease take so long to express itself, and why is its expression so variable?
The patient did well during his hospital stay 30 years ago. As I recall, he had staphylococcal bacteremia with an infected cyst. We discussed the clinical scenario but had no suggestions as to how to prevent the growth of what we now know are about 60 subclinical cysts for every one that we recognize. And we certainly didn’t discuss the idea that the disease process may be partially driven by dysfunctional nonmotile cilia that should respond to urine flow by appropriately directing regeneration and proliferation of renal tubular cells.
I love getting answers to questions that I didn’t know enough to ask.
Dr. Braun is an iconic figure in Cleveland Clinic medicine. He is the consummate internist, nephrologist, and transplantation physician, but he is also a critical thinker. He strives to understand (and explain) what underpins our clinical observations and therapeutic decisions. He asks the “why” questions. As he ticked through the manifestations of PKD and the diagnostic dilemmas that arise in taking care of these patients, and then transitioned into explaining the interesting though incomplete current molecular understanding of this relatively prevalent genetic disorder, I heard many of the same questions I had asked myself 30 years ago. But this time I was getting some answers.
How can one be certain a cyst is infected? How do these cysts form and expand without apparent communication with the tubular lumens? (Intracystic bleeding and infection may not be reflected in the urinalysis, although the organism isolated from infected cysts is frequently Escherichia coli.) If renal cysts are formed from tubular epithelial cells that are preprogrammed to self-organize into lumen-like structures, how does the same genetic defect predispose to cyst formation in organs such as the liver, or to aneurysms in blood vessels in the brain? Why does the disease take so long to express itself, and why is its expression so variable?
The patient did well during his hospital stay 30 years ago. As I recall, he had staphylococcal bacteremia with an infected cyst. We discussed the clinical scenario but had no suggestions as to how to prevent the growth of what we now know are about 60 subclinical cysts for every one that we recognize. And we certainly didn’t discuss the idea that the disease process may be partially driven by dysfunctional nonmotile cilia that should respond to urine flow by appropriately directing regeneration and proliferation of renal tubular cells.
I love getting answers to questions that I didn’t know enough to ask.
Advances in autosomal dominant polycystic kidney disease—2014 and beyond
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited renal disease, has an estimated prevalence of 1:400 to 1:1,000 live births in the United States, and occurs worldwide.1,2 There are about 700,000 people living with it in the United States, and about 6,000 new cases arise annually. It accounts for nearly 5% of all patients with end-stage renal disease in the United States.3
This paper will offer an overview of the pathogenesis of renal cysts, review some of the clinical aspects of ADPKD including diagnosis and management of complications, and discuss recent drug trials and current management.
TWO TYPES—PKD1 IS MORE COMMON AND PROGRESSES MORE RAPIDLY
Two major forms of ADPKD are recognized and can usually be determined by genetic testing: PKD1, accounting for about 85% of cases, and PKD2, accounting for 15%.
The gene locus for PKD1 is on the short arm of the 16th chromosome (16p13.3), and its glycoprotein gene product is polycystin 1 (PC1), a large molecule with 4,303 amino acids.2 PC1 has a long N-terminal extracellular tail that can function as a mechanosensor. Disease progression is much faster with PKD1, and end-stage renal disease usually occurs before age 56.4
In PKD2, the gene locus is on the long arm of the fourth chromosome (4q21–23), and has a smaller glycoprotein gene product, polycystin 2 (PC2), that plays a role in calcium transport. The disease course of PKD2 tends to be slower. End-stage renal disease might not develop in the patient’s lifetime, since it typically develops when the patient is more than 70 years old.4
Although the growth rate of renal cysts is similar between the two types, patients with PKD1 develop about twice as many cysts as those with PDK2, and their cyst development starts at a younger age.5
Typically, patients have a clear phenotype and a positive family history, but in about 10% of possible ADPKD cases, there is no family history of ADPKD. Genetic variations such as incompletely penetrant PKD1 alleles,6 hypomorphic alleles,7 and trans-heterozygous mutations8 account for at least some of these cases.
IMAGING CRITERIA HAVE BROADENED
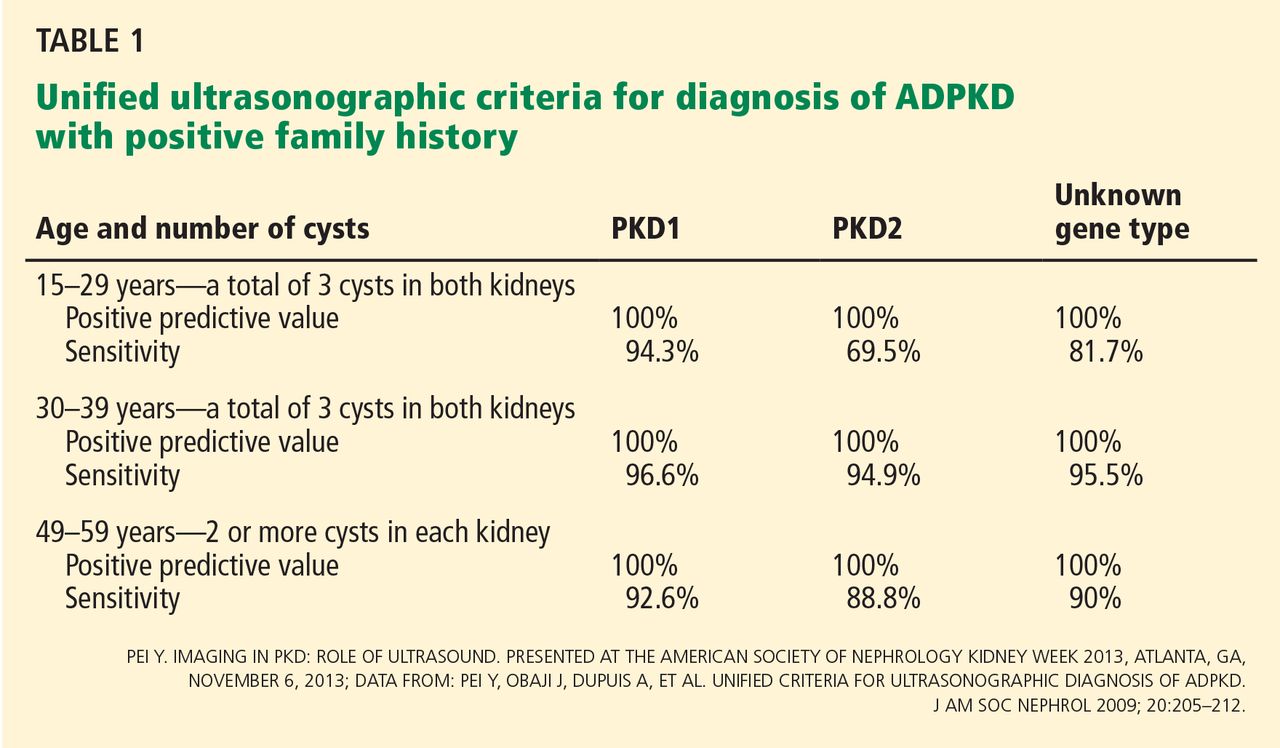
Ultrasonographic criteria for the diagnosis of ADPKD that were published in 1994 were based on patients who had a family history of PKD1.9 The criteria have since been modified (the “unified criteria”) to include patients with a family history of PKD2 who begin cyst development at a later age and with lower numbers.10 For patients ages 30 to 39, a previously difficult diagnostic group, the criterion for the minimum number of cysts visible on ultrasonography changed from four to three, improving the sensitivity of detecting disease from approximately 76% to approximately 95% (Table 1).9,10 It is important to note that these criteria apply only to patients “at risk,” ie, with a positive family history of ADPKD.
Computed tomography (CT) and magnetic resonance imaging (MRI) classically show bilaterally enlarged multicystic kidneys, though variations can be seen.
DISEASE CAN PRESENT IN MYRIAD WAYS

Although cystic kidney disease is the basic underlying problem, undiagnosed patients may present with a variety of symptoms caused by other manifestations of ADPKD (Table 2).
Hypertension is the most common presentation, occurring in about 50% of patients ages 20 to 34, and essentially 100% of those with end-stage renal disease.11 It is associated with up-regulation of the renin-angiotensin-aldosterone system.
Pain is typically located in the abdomen, flank, or back and can occur in a localized or diffuse manner. Early abdominal distress is often simply described as “fullness.” Localized pain is usually caused by bleeding into or rupture of a cyst, renal stones, or infection.12 Because renal cysts are noncommunicating, bleeding can occur into a cyst and cause pain without gross hematuria. Compression by greatly enlarged kidneys, liver, or both can cause a variety of gastrointestinal symptoms such as reflux esophagitis and varying degrees of constipation. Diffuse pain is often musculoskeletal and related to exaggerated lordosis from increasing abdominal size due to enlarging cystic kidneys and sometimes liver.12 In carefully selected cases, cyst aspiration may be helpful.11
Although renal carcinomas are rare and not more frequent than in the general population, they can occur at an earlier age and with constitutional symptoms.11
Urinary tract infections are increased in frequency. A patient may have a simple urinary tract infection that is cured with the appropriate antibiotic. However, a urinary tract infection repeatedly recurring with the same organism is a strong clue that an infected cyst is the source and requires more intensive treatment with the appropriate cyst-penetrating antibiotic. On the other hand, because cysts are noncommunicating, an infected cyst might be present despite a negative urine culture.
Identifying infected cysts can be a challenge with conventional imaging techniques, but combined positron emission tomography and CT (PET/CT) can be a valuable though expensive diagnostic tool to identify an infected kidney or liver cyst, or to identify an unsuspected source of the pain and infection.13
Jouret et al13 evaluated 27 PET/CT scans performed in 24 patients with ADPKD and suspicion of an abdominal infection. Patients were deemed to have probable cyst infection if they met all of the following criteria: temperature more than 38°C for longer than 3 days, loin or liver tenderness, plasma C-reactive protein level greater than 5 mg/dL, and no evidence of intracystic bleeding on CT. Patients with only two or three of these criteria were classified as having fever of unknown origin. Diagnosis of cyst infection was confirmed by cyst fluid analysis.
PET/CT identified a kidney or liver cyst infection in 85% of 13 infectious events in 11 patients who met all the criteria for probable cyst infection; CT alone contributed to the diagnosis in only one patient.13 In those with fever of unknown origin, PET/CT identified a source of infection in 64% of 14 events in 13 patients: two infected renal cysts, as well as one patient each with other infections that would be difficult to diagnose clinically, ie, small bowel diverticulitis, psoas abscess, diverticulitis of the right colon, pyelonephritis in a transplanted kidney, infected abdominal aortic aneurysm, prostatitis, colitis, and Helicobacter pylori gastritis. Results of PET/CT were negative in five patients with intracystic bleeding.
Kidney stones occur in 20% to 36% of patients.11,14 Uric acid stones occur at almost the same frequency as calcium oxalate stones.
Chronic kidney disease not previously diagnosed may be the presenting condition in a small percentage of patients, sometimes those in whom much earlier hypertension was not fully evaluated. ADPKD is typically not associated with significant proteinuria (eg, nephrotic range), and the presence of heavy proteinuria usually indicates the presence of a superimposed primary glomerulopathy.15
Cysts in other locations. By MRI, liver cysts are present in 58% of patients ages 15 to 24, rising to 94% in those ages 35 to 46.11 Because liver cysts are estrogen-dependent, they are more prominent in women. A small percentage of patients develop cysts in the pancreas (5%), arachnoid membranes (8%), and seminal vesicles (40% of men with ADPKD).11
Cardiovascular abnormalities occur in almost one-third of patients with ADPKD, usually as mitral and aortic valve abnormalities.16 Aneurysms of the aortic root and abdominal aorta can also occur, in addition to intracranial aneurysms (see below).17
Intracranial aneurysms are not uncommon, and size usually determines their risk.
Intracranial aneurysms are strongly influenced by family history: 16% of ADPKD patients with a family history of intracranial aneurysm also develop them, compared with 5% to 6% of patients with no family history.11 The anterior cerebral circulation is involved in about 80% of cases. A sentinel or sudden “thunderclap” headache is a classic presentation that may precede full-blown rupture in about 17% of cases.18 Patients who rupture an intracranial aneurysm have a mean age of 39, usually have normal renal function, and can be normotensive.11
For patients with no history of subarachnoid hemorrhage, the 5-year cumulative rupture rates for patients with aneurysms located in the internal carotid artery, anterior communicating or anterior cerebral artery, or middle cerebral artery were 0% for aneurysms less than 7 mm, 2.6% for those 7 to 12 mm, 14.5% for those 13 to 24 mm, and 40% for those 25 mm or larger, with higher rates for the same sizes in the posterior circulation.11
In patients without symptoms, size is correlated with risk of rupture: less than 4 mm is usually associated with very low risk, 4 to less than 7 mm with moderate risk, and 7 mm or more with increasing risk. An aneurysm larger than 10 mm is associated with roughly a 1% risk of rupture per year.19
Irazabal et al20 retrospectively studied 407 patients with ADPKD who were screened for intracranial aneurysm. Saccular aneurysms were detected in 45 patients; most were small (median diameter 3.5 mm). During cumulative imaging follow-up of 243 years, only one new intracranial aneurysm was detected (increasing from 2 to 4.4 mm over 144 months) and two previously identified aneurysms grew (one increasing 4.5 to 5.9 mm over 69 months and the other 4.7 to 6.2 mm over 184 months). No change occurred in 28 patients. Seven patients were lost to follow-up, however. During cumulative clinical follow-up of 316 years, no aneurysm ruptured. Two patients were lost to follow-up, three had surgical clipping, and five died of unrelated causes. The authors concluded that presymptomatic intracranial aneurysms are usually small, and that growth and rupture risks are no higher than for unruptured intracranial aneurysms in the general population. A 2014 study also suggests a conservative approach for managing intracranial aneurysm in the general population.21
In asymptomatic ADPKD patients, it is reasonable to reserve screening for those with a positive family history of intracranial aneurysm or subarachnoid hemorrhage, those with a previous ruptured aneurysm, those in high-risk professions (eg, pilots), and for patients prior to anticoagulant therapy or major surgery possibly associated with hemodynamic instability.11,22 Certain extremely anxious patients might also need to be studied. Screening can be performed with magnetic resonance angiography without gadolinium contrast. It is prudent to have patients with an intracranial aneurysm thoroughly evaluated by an experienced neurosurgeon with continued follow-up.
PROGRESSION OF ADPKD
The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) study23 evaluated 241 patients with ADPKD (ages 15 to 46) by measuring the annual rate of change in total kidney volume, total cyst volume, and iothalamate glomerular filtration rate (GFR) over 3 years. The annual increase in total kidney volume averaged 5.3%,23 though the reported range with various imaging techniques is from 4% to 12.8% in adults.24 This study focused on macrocystic disease, ie, cysts that are visible by MRI and measurably increase total kidney volume. Although larger total kidney volume at baseline generally predicted a more rapid decline in GFR, there were wide and overlapping variations in yearly GFR declines within and among different total-kidney-volume groups.23
SPECIAL CLINICAL PROBLEMS IN ADPKD
Case 1: A man with ADPKD develops new and increasing proteinuria
A 55-year-old man with ADPKD and stage 3 chronic kidney disease developed new and increasing proteinuria, rising to 5,500 mg per 24 hours. What is the most likely explanation?
- Rapidly progressive renal failure with increasing proteinuria in ADPKD
- Bilateral renal vein thromboses because of cyst compression
- Malignant hypertension with bilateral renal artery compression
- Superimposed primary glomerulopathy
- Multiple infected renal cysts with pyonephrosis
Answer: Superimposed primary glomerulopathy.
ADPKD (similar to uncomplicated obstructive uropathy, pyelonephritis, main renal artery disease, and often cases of interstitial nephritis without secondary glomerular changes) typically does not result in nephrotic-range proteinuria. A superimposed primary glomerulopathy, focal segmental glomerulosclerosis, was the biopsy-proved diagnosis.
At least 21 cases have been reported of AD-PKD with nephrotic-range proteinuria and a renal biopsy showing a primary glomerulopathy, including focal segmental glomerulosclerosis (5 cases), minimal-change disease (5), membranous nephropathy (3), IgA nephropathy (2), and one each of crescentic glomerulonephropathy, diabetic nephropathy, membranoproliferative glomerulonephritis, postinfectious glomerulonephropathy, amyloid glomerulopathy, and mesangioproliferative glomerulopathy.15 Treatment was directed at the primary glomerulopathy, and the outcomes corresponded to the primary diagnosis (eg, with appropriate treatment, three of the five patients with focal segmental glomerulosclerosis progressed to end-stage renal disease, all of the patients with minimal-change disease went into remission, and one of the two cases with IgA nephropathy improved).15
Case 2: A woman with ADPKD and advanced renal failure develops shortness of breath
A 47-year-old woman with very large polycystic kidneys (total kidney volume 7,500 mL; normal range for a single kidney approximately 136–295 mL, mean 196)25 and estimated GFR of 25 mL/min developed new-onset shortness of breath while climbing steps and later even when making a bed. She had no chest pain, cough, or edema. She was sent directly to the emergency department and was admitted and treated; her condition improved, and she was discharged after 6 days. What did she have?
- Presentation of rare cystic pulmonary disease in ADPKD
- Onset of pneumonia with early bacteremia
- Progressive reduction in ventilatory capacity from massive polycystic kidneys and liver elevating both sides of the diaphragm
- Pulmonary emboli from an iliac vein or inferior vena cava source
- Progressive anemia accompanying rapidly worsening stage 4 chronic kidney disease
Answer: She had pulmonary emboli from an iliac vein (right) or inferior vena cava source.
Pulmonary emboli in ADPKD can be caused by thrombi in the inferior vena cava or the iliac or femoral vein because of compression by a massive right polycystic kidney. Four cases were reported at Mayo Clinic,26 three diagnosed by MRI and one with CT. One additional case occurred at Cleveland Clinic. All patients survived after treatment with anticoagulation therapy; early nephrectomy was required in two cases.
Interestingly, following kidney transplantation, the patients at greatest risk for pulmonary emboli are those with ADPKD as their original disease.27
RENAL CYSTS RESULT FROM COMBINED MUTATIONS, INJURY
The germline ADPKD mutation that occurs in one allele of all renal tubular epithelial cells is necessary but not sufficient for cystogenesis.28 One or more additional somatic mutations of the normal allele—the “second hit”—also develop within individual tubular epithelial cells.28,29 These epithelial cells undergo clonal proliferation, resulting in tubular dilatation and cyst formation. Monoclonality of cells in cysts has been documented.
Ischemia-reperfusion injury can be viewed as a “third hit.”30 In PKD1 knockout mice, which at 5 weeks of age normally develop only mild cystic kidney disease, the superimposition of unilateral ischemia-reperfusion injury at 8 weeks caused widespread and rapid cyst formation. It is believed that acute renal injury reactivates developmental signaling pathways within 48 hours that trigger epithelial cell proliferation and then cyst development detectable by MRI 2 weeks later. Although this phenomenon has not been documented in humans, it is a cautionary tale.
CYSTOGENESIS INVOLVES MULTIPLE PATHWAYS
A comprehensive description of pathways leading to renal cyst formation is beyond the scope of this article, and the reader is referred to much more detailed and extensive reviews.2,31 Disturbances in at least three major interconnected pathways promote cystogenesis in renal tubular epithelial cells:
- Normal calcium transport into the endoplasmic reticulum is disrupted by abnormal polycystins on the surface of the primary cilium
- Vasopressin and other stimuli increase the production of cyclic adenosine monophosphate (cAMP)
- The mammalian target of rapamycin (mTOR) proliferative pathway is up-regulated.
DISRUPTION OF CALCIUM TRANSPORT IN THE PRIMARY CILIUM
Primary cilia are nonmotile cellular organelles of varying size, from about 0.25 μm up to about 1 μm.32 Each primary cilium has nine peripheral pairs of microtubules but lacks a centrally located pair that is present in motile cilia. Primary cilia are ubiquitous and have been highly conserved throughout evolution. A single cilium is present on almost all vertebral cells.33
Cilial defects have been identified in autosomal dominant as well as recessive diseases and are known as ciliopathies.33 Although rare in humans, they can affect a broad spectrum of organs other than the kidney, including the eye, liver, and brain.33
Urine flow in a renal tubule is believed to exert mechanical stimulation on the extracellular flagellum-like N-terminal tail of PC1 that extends from a primary cilium into the urinary space. PC1 in concert with PC2 opens PC2 calcium channels, allowing calcium ions to flow down the microtubules to ryanodine receptors and the basal body.32,33 This leads to local release of calcium ions that regulate cell proliferation.32,34 However, in ADPKD kidneys, PC1 and PC2 molecules are sparse or mutated, resulting in defective calcium transport, increased and unregulated tubular epithelial cell proliferation, and cyst formation.
In a totally different clinical setting, biopsies of human renal transplants that sustained acute tubular necrosis during transplantation reveal that a cilium dramatically elongates in response to injury,35 possibly as a compensatory mechanism to maintain calcium transport in the presence of meager urine flow and to restore the proliferation of tubular epithelial cells in a regulated repair process.
THE ROLE OF VASOPRESSIN AND ACTIVATION OF cAMP
In classic experiments, Wang et al36 cross-bred rats having genetically inherited polycystic kidney disease (actually, autosomal recessive polycystic kidney disease) with Brattleboro rats that completely lack vasopressin. At 10 and 20 weeks of age, the offspring had virtually complete inhibition of cystogenesis because of the absence of vasopressin. However, when vasopressin was restored by exogenous administration continuously for 8 weeks, the animals formed massive renal cysts.
Vasopressin activates cAMP, which then functions as a second messenger in cell signaling. cAMP increases the activation of the protein kinase A (PKA) pathway, which in turn increases downstream activity of the B-raf/ERK pathway. Up-regulation of cAMP and PKA appears to perpetuate activation of canonical Wnt signaling, down-regulate non-canonical Wnt/planar cell polarity signaling, and lead to loss of tubular diameter control, resulting in cyst formation.31 Normally, cAMP is degraded by phosphodiesterase. However, because of the primary cilium calcium transport defect in ADPKD, phosphodiesterase is reduced and cAMP persists.37 In conjunction with the defective primary cilial calcium transport, cAMP exerts a proliferative effect on renal tubular epithelial cells that is opposite to its effect in normal kidneys.31,32 cAMP also up-regulates the cystic fibrosis transmembrane conductance regulator (CFTR) that promotes chloride ion transport. Sodium ions follow the chloride ions, leading to fluid accumulation and cyst enlargement.31
Inhibiting vasopressin by increasing water intake
A simple key mechanism for limiting vasopressin secretion is by sufficient water ingestion. Nagao et al38 found that rats with polycystic kidney disease given water with 5% glucose (resulting in 3.5-fold increased fluid intake compared with rats given tap water) had a 68% reduction in urinary vasopressin and a urine osmolality less than 290 mOsm/kg. The high-water-intake rats had dramatically reduced cystic areas in the kidney and a 28% reduction of kidney-to-body weight ratio vs controls.
In an obvious oversimplification, these findings raised the question of whether a sufficient increase in water intake could be an effective therapy for polycystic kidney disease.39 A pilot clinical study evaluated changes in urine osmolality in eight patients with ADPKD who had normal renal function.40 At baseline, 24-hour urine osmolality was typically elevated to approximately 753 mOsm/kg compared to the plasma at 285 mOsm/kg, indicating that antidiuresis is the usual state. During the 2-week study, urine volume and osmolality were measured, and additional water intake was adjusted in order to achieve a urine osmolality goal of 285 ± 45 mOsm/kg. These adjustments resulted in water intake that appeared to be in the range of 2,400 to 3,000 mL per 24 hours. The major limitations of the study were that it was very short term, and there was no opportunity to measure changes in total kidney volume or estimated GFR.
In a recent preliminary report from Japan, high water intake (2,500–3,000 mL daily) in 18 ADPKD patients was compared over 12 months with ad libitum water intake in 14 ADPKD controls (clinicaltrials.gov NCT 01348505). There was no statistically significant change in total kidney volume or cystatin-estimated GFR in those on high water intake, but serious defects in study design (patients in the high water intake group were allowed to decrease their intake if it was causing them difficulty, and patients in the ad libitum water intake group had no measurement of their actual water intake) prevent any conclusions because there was no evidence that the groups were different from one another with respect to the key element of the study, namely, water intake.
Blocking the vasopressin receptor slows disease progression
Using another approach, Gattone et al41 inhibited the effect of vasopressin by blocking the vasopressin 2 receptor (V2R) in mouse and rat models of polycystic kidney disease, using an experimental drug, OPC31260. The drug halted disease progression and, in one situation, appeared to cause regression of established disease. As noted by Torres and Harris,31 even though both increased water intake and V2R antagonists decrease cAMP in the distal tubules and collecting ducts, circulating levels of vasopressin are decreased by increased water intake but increased by V2R antagonists.
After these remarkable results in animal models, clinical trials of the V2R antagonist tolvaptan were conducted in patients with ADPKD. In the Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes 3:4 study,42 1,445 adults (ages 18 to 50) with ADPKD in 133 centers worldwide were randomized to receive either tolvaptan or placebo for 3 years. Key inclusion criteria included good renal function (estimated GFR ≥ 60 mL/min) and total kidney volume of at least 750 mL (mean 1,700 mL) as measured by MRI. Tolvaptan was titrated to the highest tolerated twice-daily dose (average total of 95 mg/day). All patients were advised to maintain good hydration and to avoid thirst by drinking a glass of water after each urination. Unfortunately, neither water intake nor urine output was measured.
The primary end point was the annual rate of change in total kidney volume, with secondary end points of clinical progression (worsening kidney function, pain, hypertension, albuminuria), and rate of decline in kidney function as measured by the slope of the reciprocal of serum creatinine.42
Patients in the tolvaptan arm had a slower annual increase in total kidney volume than controls (2.8% vs 5.5%, respectively, P < .001) and a slower annual decline in renal function (−2.61 vs −3.81 mg/mL−1, respectively, P < .001).42 More participants in the treatment group withdrew than in the placebo group (23% vs 14%, respectively).
Adverse events occurred more frequently with tolvaptan.42 Liver enzyme elevations of greater than three times the upper limit of normal occurred in 4.4% of patients in the treatment group, leading to a drug warning issued in January 2013. As expected, side effects related to diuresis (urinary frequency, nocturia, polyuria, and thirst) were more frequent in the treatment group, occurring in up to 55% of participants.
The authors noted, “Although maintaining hydration helped ensure that the blinding in the study was maintained, the suppression of vasopressin release in the placebo group may have led to an underestimation of the beneficial effect of tolvaptan and may account for the lower rates of kidney growth observed in the placebo group.”42
In 2013, the US Food and Drug Administration (FDA) denied a new drug application for tolvaptan as a treatment for ADPKD.
THE mTOR PATHWAY IS UP-REGULATED
The mTOR pathway that plays a major role in cell growth and proliferation includes interaction of the cytoplasmic tail of polycystin 1 with tuberin.43 Activation products of mTOR, including phospho-S6K, have been found in tubular epithelial cells lining cysts of ADPKD kidneys but not in normal kidneys.43 Mutant mice with polycystic disease had phospho-S6K in tubular epithelial cells of cysts, whereas those treated with the mTOR inhibitor rapamycin did not.43 But subsequent studies have shown that only a low level of mTOR activation is present in 65% to 70% of ADPKD cysts.44
Two major studies of the treatment of ADPKD with rapamycin that were published contemporaneously in 2010 failed to demonstrate any significant benefit with mTOR inhibitor treatment.45,46
Serra et al45 conducted an 18-month, open-label trial of 100 ADPKD patients ages 18 to 40 with an estimated GFR (eGFR) of at least 70 mL/min. Patients were randomized to receive rapamycin, given as sirolimus 2 mg per day, or standard care. The primary end point was the reduction in the growth rate of total kidney volume, measured by MRI. Secondary end points were eGFR and protein excretion (albumin-creatinine ratio). No significant difference was found in total kidney volume, but a nonsignificant stabilization of eGFR was noted.
Walz et al46 in a 2-year, multicenter, double-blind trial, randomized 433 patients (mean age 44; mean eGFR 54.5 mL/min) to treatment with either the short-acting mTOR inhibitor everolimus (2.5 mg twice daily) or placebo. Although patients in the treatment group had less of an increase in total kidney volume (significant at 1 year but not at 2 years), they tended to show a decline in eGFR. But further analysis showed that the only patients who had a reduction in eGFR were males who already had impaired kidney function at baseline.47
In a pilot study, 30 patients with ADPKD (mean age 49) were randomized to one of three therapies:
- Low-dose rapamycin (trough blood level 2–5 ng/mL)
- Standard-dose rapamycin (trough blood level > 5–8 ng/mL)
- Standard care without rapamycin.48
In contrast to other studies, the primary end point was the change in iothalamate GFR at 12 months, with change in total kidney volume being a secondary end point.
At 12 months, with 26 patients completing the study, the low-dose rapamycin group (n = 9) had a significant increase in iothalamate GFR of 7.7 ± 12.5 mL/min/1.73 m2, whereas the standard-dose rapamycin group (n = 8) had a nonsignificant increase of 1.6 ± 12.1 mL/min/1.73 m2, and the no-rapamycin group (n = 9) had a fall in iothalamate GFR of 11.2 ± 9.1 mL/min/1.73 m2 (P = .005 for low-dose vs no rapamycin; P = .07 for standard-dose vs no rapamycin; P = .52 for low-dose vs standard-dose rapamycin; and P = .002 for combined low-dose and standard-dose rapamycin vs no rapamycin.).48 These differences were observed despite there being no significant change in total kidney volume in any of the groups. Patients on low-dose rapamycin had fewer adverse effects than those on standard dose and were more often able to continue therapy for the entire study. This, and the use of iothalamate GFR rather than eGFR to measure GFR, are believed to be the main reasons that low-dose effects were more pronounced than those with standard doses. One may speculate that rapamycin may have its effect on microcysts and cystogenic cells, resulting in stabilization of or improvement in renal function without detectable slowing in total kidney volume enlargement. Mechanisms for this possibility involve new concepts, as discussed below.
NEW CONCEPTS
Specialized cells also promote renal cyst formation
Specialized cells that promote cyst formation have been identified by Karihaloo et al49 in a mouse model of polycystic kidney disease. In this model, alternatively activated macrophages homed to cystic areas and promoted cyst growth. These findings suggested that interrupting the homing and proliferative signals of macrophages could be a therapeutic target for ADPKD. Although rapamycin can suppress macrophage proliferation by macrophage colony-stimulating factor and inhibit macrophage function,50 alternatively activated macrophages have not been specifically studied for rapamycin responsiveness.
More promising is evidence that CD133+ progenitor cells from human ADPKD kidneys—but not from normal human kidneys—form cysts in vitro and in severe combined immunodeficient mouse models.51 Treatment with rapamycin decreased proliferation of the de-differentiated CD133+ cells from ADPKD patients and reduced cystogenesis.51
Visible cysts are the tip of the iceberg
Using ADPKD nephrectomy specimens from eight patients, Grantham et al52 compared cyst counts by MRI and by histology and found that for every renal cyst detected by MRI, about 62 smaller cysts (< 0.9 mm) are present in the kidney. For a typical patient having an average of 587 cysts in both kidneys that are detectable by MRI, this means that more than 36,000 cysts are actually present, and MRI detects less than 2% of the total cysts present.
Although microcysts are too small to contribute much to total kidney volume, they can interfere with kidney function. Microcysts can reduce GFR in two major ways: by compressing microvasculature, tubules, and glomeruli in the cortex; or by blocking the drainage of multiple upstream nephrons when they form in or block medullary collecting ducts.52 Although the growth rates of microcysts less than 1 mm in size have not yet been measured, the adult combined growth rates of the renal cyst component is approximately 12% per year, with yearly individual cyst growth rates up to 71%, and with fetal cyst growth rates even higher for cysts larger than 7.0 mm.53 Before and during an accelerated growth period, microcysts may be susceptible to certain therapies that could first improve GFR and only later change measurable total kidney volume by slowing microcyst progression to macrocysts either directly or through specialized cells that may be sensitive to rapamycin.
CURRENT MANAGEMENT OF ADPKD
Blood pressure control is essential—but too low is not good. For adult patients with hypertension caused by ADPKD, an acceptable blood pressure range is 120–130/70–80 mm Hg. However, further information from recently published blood pressure guidelines54 and the results of the Halt Progression of Polycystic Kidney Disease (HALT-PKD) study to be reported in late 201455 may provide more precise ranges for blood pressure control in ADPKD.
Although substantial experimental evidence exists for the benefits of inhibiting the up-regulation of the renin-angiotensin-aldosterone system in ADPKD, clinical proof is not yet available to confirm that angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) are preferred therapy.55 This may be determined by results of the HALT-PKD study, due for release in late 2014.55
Controlling blood pressure should be done with caution. Patients with low GFRs whose blood pressure is too low tend to have a more rapid decline of GFR, as suggested in the Modification of Diet in Renal Disease (MDRD) study in 1995.56
Experimental evidence suggests that avoiding calcium channel blockers may be advisable. Yamaguchi et al34 found that calcium channel blockers worsen the calcium transport defect and convert tubular epithelial cells to a proliferative phenotype.34
High fluid intake (2,500–3,000 mL/day), because it suppresses vasopressin, may be useful if permitted by several factors such as the patient’s cardiopulmonary and renal and electrolyte status, other medications, and diet.31 The reader is referred to a detailed description of the precautions necessary when prescribing high water intake.31 Patients should have their fluid intake managed by a physician and their renal function and serum sodium and electrolytes monitored regularly in order to avoid hyponatremia. Severe hyponatremia has occurred in patients with ADPKD and impaired kidney function who drank excessive quantities of water. Cardiac and pulmonary complications from excessive fluid intake are also possible, especially with a low GFR and compromised cardiac function.
A low-sodium diet, if not a contributing factor in hyponatremia, can be used under physician direction in the management of hypertension as well as in the prevention of calcium oxalate kidney stones.
Caffeine should be avoided because it may interfere with the activity of the phosphodiesterase that is necessary for the catabolism of cAMP to 5′AMP.
A low-protein diet is of unproven benefit,56 but it is prudent to avoid high protein intake.57
Complications such as bleeding (into or from cysts), infection (urinary tract, kidney cysts, and liver cysts), kidney stones, and urinary tract obstruction should be treated promptly and may require hospitalization.
Regular symptom reviews and physical examinations need to be performed with nonrenal concerns also in mind, such as intracranial aneurysms and cardiac valve lesions.11,58
Formal genetic counseling and molecular testing are becoming more frequently indicated as more complex inheritance patterns arise.6–8,59
Renal replacement therapy in the form of dialysis or transplantation is usually available for the patient when end-stage renal disease occurs. In the largest study thus far, ADPKD patient survival with a kidney transplant was similar to that of patients without ADPKD (about 93% at 5 years), and from 5 years to 15 years death-censored graft survival was actually better.60 Thromboembolic events are more frequent after transplantation,27,60 but they may also occur before transplantation from a massive right kidney compressing the iliac vein or the inferior vena cava, or both, leading to thrombus formation.26
Investigational as well as standard drug studies have intensified. Results from a large randomized study in approximately 1,000 adult ADPKD patients that evaluated over 6 to 8 years the effects of ACE inhibition with or without ARB treatment of hypertension, at both usual and lower blood pressure ranges in those with good renal function, are expected in late 2014.55 Outcomes from a few small clinical studies, eg, one with long-acting somatostatin31,61 and one using low-dose rapamycin48 in adults with ADPKD, will require confirmation in large randomized placebo-controlled clinical studies. In a new 3-year randomized placebo-controlled study of 91 children and young adults (ages 8 to 22) with ADPKD and essentially normal renal function who continued treatment with lisinopril, the addition of pravastatin (20 mg or 40 mg daily based on age) resulted in a significant reduction in the number of patients (46% vs 68%, respectively, P = .03) experiencing a greater than 20% change (increase) in height-adjusted total kidney volume.62 Change in GFR was not reported,62 but an earlier 4-week study in 10 patients treated with simvastatin did show an increase in renal blood flow and GFR.63 Numerous other agents that lack human studies include some described in older experimental work (eg, amiloride,31,64 citrate31,65) and many others from a growing list of potential therapeutic targets.31,66–73 It must be emphasized that there is no FDA-approved medication specifically for the treatment of ADPKD.
Future specific treatments of ADPKD may also involve minimally toxic doses of combination or sequential therapy directed at precystic and then both micro- and macrocystic/cystic fluid expansion aspects of ADPKD.48,74 Unfortunately, at the present time there is no specific FDA-approved therapy for ADPKD.
- Torres VE, Harris PC. Mechanisms of disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol 2006; 2:40–55.
- Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int 2009; 76:149–168.
- United States Renal Data System. 2013 atlas of CKD & ESRD. Volume 2 - atlas ESRD:172. www.usrds.org/atlas.aspx. Accessed June 4, 2014.
- Barua M, Cil O, Paerson AD, et al. Family history of renal disease severity predicts the mutated gene in ADPKD. J Am Soc Nephrol 2009, 20:1833–1838.
- Harris PC, Bae KT, Rossetti S, et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2006; 17:3013–3019.
- Vujic M, Heyer CM, Ars E, et al. Incompletely penetrant PKD1 alleles mimic the renal manifestations of ARPKD. J Am Soc Nephrol 2010; 21:1097–1102.
- Harris PC. What is the role of somatic mutation in autosomal dominant polycystic kidney disease? J Am Soc Nephrol 2010; 21:1073–1076.
- Watnick T, He N, Wang K, et al. Mutations of PKD1 in ADPKD2 cysts suggest a pathogenic effect of trans-heterozygous mutations. Nat Genet 2000; 25:143–144.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994; 343:824–827.
- Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol 2009; 20:205–212.
- Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet 2007; 369:1287–1301.
- Bajwa ZH, Sial KA, Malik AB, Steinman TI. Pain patterns in patients with polycystic kidney disease. Kidney Int 2004; 66:1561–1569.
- Jouret F, Lhommel R, Beguin C, et al. Positron-emission computed tomography in cyst infection diagnosis in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2011; 6:1644–1650.
- Nishiura JL, Neves RF, Eloi SR, Cintra SM, Ajzen SA, Heilberg IP. Evaluation of nephrolithiasis in autosomal dominant polycystic kidney disease patients. Clin J Am Soc Nephrol 2009; 4:838–844.
- Hiura T, Yamazaki H, Saeki T, et al. Nephrotic syndrome and IgA nephropathy in polycystic kidney disease. Clin Exp Nephrol 2006; 10:136–139.
- Hossack KF, Leddy CL, Johnson AM, Schrier RW, Gabow PA. Echocardiographic findings in autosomal dominant polycystic kidney disease. N Engl J Med 1988; 319:907–912.
- Rossetti S, Chauveau D, Kubly V, et al. Association of mutation position in polycystic kidney disease 1 (PKD1) gene and development of a vascular phenotype. Lancet 2003; 361:2196–2201.
- Linn FH, Wijdicks EF, van der Graaf Y, Weerdesteyn-van Vliet FA, Bartelds AI, van Gijn J. Prospective study of sentinel headache in aneurismal subarachnoid haemorrhage. Lancet 1994; 344:590–593.
- Belz MM, Fick-Brosnahan GM, Hughes RL, et al. Recurrence of intracranial aneurysms in autosomal-dominant polycystic kidney disease. Kidney Int 2003; 63:1824–1830.
- Irazabal MV, Huston J, Kubly V, et al. Extended follow-up of unruptured intracranial aneurysms detected by presymptomatic screening in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2011; 6:1274–1285.
- Salman A-S, White PM, Counsell CE, et al; Scottish Audit of Intracranial Vascular Malformations Collaborators. Outcome after conservative management or intervention for unruptured brain arteriovenous malformations. JAMA 2014; 311:1661–1669.
- Vijay A, Vijay A, Pankaj P. Autosomal dominant polycystic kidney disease: a comprehensive review. Nephrourol Mon 2010; 2:172–192.
- Grantham JJ, Torres VE, Chapman AB, et al; CRISP Investigators. Volume progression in polycystic kidney disease. N Engl J Med 2006; 354:2122–2130.
- Bae KT, Grantham JJ. Imaging for the prognosis of autosomal dominant polycystic kidney disease. Nat Rev Nephrol 2010; 6:96–106.
- van den Dool SW, Wasser NM, de Fijter JW, Hoekstra J, van der Geest RJ. Functional renal volume: quantitative analysis at gadolinium-enhanced MR angiography—feasibility study in healthy potential kidney donors. Radiology 2005; 236:189–195.
- O’Sullivan DA, Torres VE, Heit JA, Liggett S, King BF. Compression of the inferior vena cava by right renal cysts: an unusual cause of IVC and/or iliofemoral thrombosis with pulmonary embolism in autosomal dominant polycystic kidney disease. Clin Nephrol 1998; 49:332–334.
- Tveit DP, Hypolite I, Bucci J, et al. Risk factors for hospitalizations resulting from pulmonary embolism after renal transplantation in the United States. J Nephrol 2001; 14:361–368.
- Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 2001; 7:151–156.
- Qian F, Germino GG. “Mistakes happen”: somatic mutation and disease. Am J Hum Genet 1997; 61:1000–1005.
- Takakura A, Contrino L, Zhou X, et al. Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum Mol Genet 2009; 18:2523–2531.
- Torres VE, Harris PC. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol 2014; 25:18–32.
- Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33:129–137.
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med 2011; 364:1533–1543.
- Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004; 279:40419–40430.
- Verghese E, Ricardo SD, Weidenfeld R, et al. Renal primary cilia lengthen after acute tubular necrosis. J Am Soc Nephrol 2009; 20:2147–2153.
- Wang X, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 2008; 19:102–108.
- Torres VE. Cyclic AMP, at the hub of the cystic cycle. Kidney Int 2004; 66:1283–1285.
- Nagao S, Nishii K, Katsuyama M, et al. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 2006; 17:2220–2227.
- Grantham JJ. Therapy for polycystic kidney disease? It’s water, stupid! J Am Soc Nephrol 2008; 19:1–7.
- Wang CJ, Creed C, Winklhofer FT, Grantham JJ. Water prescription in autosomal dominant polycystic kidney disease: a pilot study. Clin J Am Soc Nephrol 2011; 6:192–197.
- Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003; 9:1323–1326.
- Torres VE, Chapman AB, Devuyst O, et al; TEMPO 3:4 Trial Investigators. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2012; 367:2407–2418.
- Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A 2006; 103:5466–5471.
- Hartman TR, Liu D, Zilfou JT, et al. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum Mol Genet 2009; 18:161–163.
- Serra AL, Poster D, Kistler AD, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med 2010; 363:820–829.
- Walz G, Budde K, Mannaa M, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2010; 363:830–840. Errata in: N Engl J Med 2010; 363:1190 and N Engl J Med 2010; 363:1977.
- Walz G, Budde K, Eckardt K-U. mTOR inhibitors and autosomal dominant polycystic kidney disease (correspondence). N Engl J Med 2011; 364:287–288.
- Braun WE, Schold JD, Stephany BR, Spinko RA, Herfs BR. Low dose rapamycin (sirolimus) effects in autosomal dominant polycystic kidney disease: an open-label randomized control pilot study. Clin J Am Soc Nephrol 2014; 9:881–888.
- Karihaloo A, Koraishy F, Huen SC, et al. Macrophages promote cyst growth in polycystic kidney disease. J Am Soc Nephrol 2011; 22:1809–1814.
- Fox R, Nhan TQ, Law GL, Morris DR, Liles WC, Schwartz SM. PSGL-1 and mTOR regulate translation of ROCK-1 and physiological functions of macrophages. EMBO J 2007; 26:505–515. Erratum in: EMBO J 2007; 26:2605.
- Carvalhosa R, Deambrosis I, Carrera P, et al. Cystogenic potential of CD133+ progenitor cells of human polycystic kidneys. J Pathol 2011; 225:129–141.
- Grantham JJ, Mulamalla S, Grantham CJ, et al. Detected renal cysts are tips of the iceberg in adults with ADPKD. Clin J Am Soc Nephrol 2012; 7:1087–1093.
- Grantham JJ, Cook LT, Wetzel LH, Cadnapaphornchai MA, Bae KT. Evidence of extraordinary growth in the progressive enlargement of renal cysts. Clin J Am Soc Nephrol 2010; 5:889–896.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Chapman AB, Torres VE, Perrone RD, et al. The HALT polycystic kidney disease trials: design and implementation. Clin J Am Soc Nephrol 2010; 5:102–109.
- Klahr S, Breyer JA, Beck GJ, et al. Dietary protein restriction, blood pressure control, and the progression of polycystic kidney disease. Modification of Diet in Renal Disease Study Group. J Am Soc Nephrol 1995; 5:2037–2047.
- Thilly N. Low-protein diet in chronic kidney disease: from questions of effectiveness to those of feasibility. Nephrol Dial Transplant 2013; 28:2203–2205.
- Luciano RL, Dahl NK. Extra-renal manifestations of autosomal dominant polycystic kidney disease (ADPKD): considerations for routine screening and management. Nephrol Dial Transplant 2014; 29:247–254.
- Harris PC, Rossetti S. Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat Rev Nephrol 2010; 6:197–206.
- Jacquet A, Pallet N, Kessler M, et al. Outcomes of renal transplantation in patients with autosomal dominant polycystic kidney disease: a nationwide longitudinal study. Transpl Int 2011; 24:582–587.
- Ruggenenti P, Remuzzi A, Ondei P, et al. Safety and efficacy of long-acting somatostatin treatment in autosomal-dominant polycystic kidney disease. Kidney Int 2005; 68:206–216.
- Cadnapaphornchai MA, George DM, McFann K, et al. Effect of pravastatin on total kidney volume, left ventricular mass index, and microalbuminuria in pediatric autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2014; 9:889–896.
- van Dijk MA, Kamper AM, van Veen S, Souverjin JH, Blauw GJ. Effect of simvastatin on renal function in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2001; 16:2152–2157.
- Grantham JJ, Uchich M, Cragoe EL, et al. Chemical modification of cell proliferation and fluid secretion in renal cysts. Kidney Int 1989; 35:1379–1389.
- Tanner GA. Potassium citrate/citric acid intake improves renal function in rats with polycystic kidney disease. J Am Soc Nephrol 1998; 9:1242–1248.
- Belibi FA, Edelstein CL. Novel targets for the treatment of autosomal dominant polycystic kidney disease. Expert Opin Investig Drugs 2010; 19:315–328.
- Tao Y, Kim J, Yin Y, et al. VEGF receptor inhibition slows the progression of polycystic kidney disease. Kidney Int 2007; 72:1358–1366.
- Terryn S, Ho A, Beauwens R, Devuyst O. Fluid transport and cystogenesis in autosomal dominant polycystic kidney disease. Biochim Biophys Acta 2011; 1812:1314–1321.
- Thiagarajah JR, Verkman AS. CFTR inhibitors for treating diarrheal disease. Clin Pharmacol Ther 2012; 92:287–290.
- Boehn SN, Spahn S, Neudecker S, et al. Inhibition of Comt with tolcapone slows proression of polycystic kidney disease in the more severely affected PKD/Mhm (cy/+) substrain of the Hannover Sprague-Dawley rat. Nephrol Dial Transplant 2013; 28:2045–2058.
- Rees S, Kittikulsuth W, Roos K, Strait KA, Van Hoek A, Kohan DE. Adenylyl cyclase 6 deficiency ameliorates polycystic kidney disease. J Am Soc Nephrol 2014; 25:232–237.
- Buchholz B, Schley G, Faria D, et al. Hypoxia-inducible factor-1a causes renal cyst expansion through calcium-activated chloride secretion. J Am Soc Nephrol 2014; 25:465–474.
- Wallace DP, White C, Savinkova L, et al. Periostin promotes renal cyst growth and interstitial fibrosis in polycystic kidney disease. Kidney Int 2014; 85:845–854.
- Leuenroth SJ, Crews CM. Targeting cyst initiation in ADPKD. J Am Soc Nephrol 2009; 20:1–3.
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited renal disease, has an estimated prevalence of 1:400 to 1:1,000 live births in the United States, and occurs worldwide.1,2 There are about 700,000 people living with it in the United States, and about 6,000 new cases arise annually. It accounts for nearly 5% of all patients with end-stage renal disease in the United States.3
This paper will offer an overview of the pathogenesis of renal cysts, review some of the clinical aspects of ADPKD including diagnosis and management of complications, and discuss recent drug trials and current management.
TWO TYPES—PKD1 IS MORE COMMON AND PROGRESSES MORE RAPIDLY
Two major forms of ADPKD are recognized and can usually be determined by genetic testing: PKD1, accounting for about 85% of cases, and PKD2, accounting for 15%.
The gene locus for PKD1 is on the short arm of the 16th chromosome (16p13.3), and its glycoprotein gene product is polycystin 1 (PC1), a large molecule with 4,303 amino acids.2 PC1 has a long N-terminal extracellular tail that can function as a mechanosensor. Disease progression is much faster with PKD1, and end-stage renal disease usually occurs before age 56.4
In PKD2, the gene locus is on the long arm of the fourth chromosome (4q21–23), and has a smaller glycoprotein gene product, polycystin 2 (PC2), that plays a role in calcium transport. The disease course of PKD2 tends to be slower. End-stage renal disease might not develop in the patient’s lifetime, since it typically develops when the patient is more than 70 years old.4
Although the growth rate of renal cysts is similar between the two types, patients with PKD1 develop about twice as many cysts as those with PDK2, and their cyst development starts at a younger age.5
Typically, patients have a clear phenotype and a positive family history, but in about 10% of possible ADPKD cases, there is no family history of ADPKD. Genetic variations such as incompletely penetrant PKD1 alleles,6 hypomorphic alleles,7 and trans-heterozygous mutations8 account for at least some of these cases.
IMAGING CRITERIA HAVE BROADENED
Ultrasonographic criteria for the diagnosis of ADPKD that were published in 1994 were based on patients who had a family history of PKD1.9 The criteria have since been modified (the “unified criteria”) to include patients with a family history of PKD2 who begin cyst development at a later age and with lower numbers.10 For patients ages 30 to 39, a previously difficult diagnostic group, the criterion for the minimum number of cysts visible on ultrasonography changed from four to three, improving the sensitivity of detecting disease from approximately 76% to approximately 95% (Table 1).9,10 It is important to note that these criteria apply only to patients “at risk,” ie, with a positive family history of ADPKD.
Computed tomography (CT) and magnetic resonance imaging (MRI) classically show bilaterally enlarged multicystic kidneys, though variations can be seen.
DISEASE CAN PRESENT IN MYRIAD WAYS
Although cystic kidney disease is the basic underlying problem, undiagnosed patients may present with a variety of symptoms caused by other manifestations of ADPKD (Table 2).
Hypertension is the most common presentation, occurring in about 50% of patients ages 20 to 34, and essentially 100% of those with end-stage renal disease.11 It is associated with up-regulation of the renin-angiotensin-aldosterone system.
Pain is typically located in the abdomen, flank, or back and can occur in a localized or diffuse manner. Early abdominal distress is often simply described as “fullness.” Localized pain is usually caused by bleeding into or rupture of a cyst, renal stones, or infection.12 Because renal cysts are noncommunicating, bleeding can occur into a cyst and cause pain without gross hematuria. Compression by greatly enlarged kidneys, liver, or both can cause a variety of gastrointestinal symptoms such as reflux esophagitis and varying degrees of constipation. Diffuse pain is often musculoskeletal and related to exaggerated lordosis from increasing abdominal size due to enlarging cystic kidneys and sometimes liver.12 In carefully selected cases, cyst aspiration may be helpful.11
Although renal carcinomas are rare and not more frequent than in the general population, they can occur at an earlier age and with constitutional symptoms.11
Urinary tract infections are increased in frequency. A patient may have a simple urinary tract infection that is cured with the appropriate antibiotic. However, a urinary tract infection repeatedly recurring with the same organism is a strong clue that an infected cyst is the source and requires more intensive treatment with the appropriate cyst-penetrating antibiotic. On the other hand, because cysts are noncommunicating, an infected cyst might be present despite a negative urine culture.
Identifying infected cysts can be a challenge with conventional imaging techniques, but combined positron emission tomography and CT (PET/CT) can be a valuable though expensive diagnostic tool to identify an infected kidney or liver cyst, or to identify an unsuspected source of the pain and infection.13
Jouret et al13 evaluated 27 PET/CT scans performed in 24 patients with ADPKD and suspicion of an abdominal infection. Patients were deemed to have probable cyst infection if they met all of the following criteria: temperature more than 38°C for longer than 3 days, loin or liver tenderness, plasma C-reactive protein level greater than 5 mg/dL, and no evidence of intracystic bleeding on CT. Patients with only two or three of these criteria were classified as having fever of unknown origin. Diagnosis of cyst infection was confirmed by cyst fluid analysis.
PET/CT identified a kidney or liver cyst infection in 85% of 13 infectious events in 11 patients who met all the criteria for probable cyst infection; CT alone contributed to the diagnosis in only one patient.13 In those with fever of unknown origin, PET/CT identified a source of infection in 64% of 14 events in 13 patients: two infected renal cysts, as well as one patient each with other infections that would be difficult to diagnose clinically, ie, small bowel diverticulitis, psoas abscess, diverticulitis of the right colon, pyelonephritis in a transplanted kidney, infected abdominal aortic aneurysm, prostatitis, colitis, and Helicobacter pylori gastritis. Results of PET/CT were negative in five patients with intracystic bleeding.
Kidney stones occur in 20% to 36% of patients.11,14 Uric acid stones occur at almost the same frequency as calcium oxalate stones.
Chronic kidney disease not previously diagnosed may be the presenting condition in a small percentage of patients, sometimes those in whom much earlier hypertension was not fully evaluated. ADPKD is typically not associated with significant proteinuria (eg, nephrotic range), and the presence of heavy proteinuria usually indicates the presence of a superimposed primary glomerulopathy.15
Cysts in other locations. By MRI, liver cysts are present in 58% of patients ages 15 to 24, rising to 94% in those ages 35 to 46.11 Because liver cysts are estrogen-dependent, they are more prominent in women. A small percentage of patients develop cysts in the pancreas (5%), arachnoid membranes (8%), and seminal vesicles (40% of men with ADPKD).11
Cardiovascular abnormalities occur in almost one-third of patients with ADPKD, usually as mitral and aortic valve abnormalities.16 Aneurysms of the aortic root and abdominal aorta can also occur, in addition to intracranial aneurysms (see below).17
Intracranial aneurysms are not uncommon, and size usually determines their risk.
Intracranial aneurysms are strongly influenced by family history: 16% of ADPKD patients with a family history of intracranial aneurysm also develop them, compared with 5% to 6% of patients with no family history.11 The anterior cerebral circulation is involved in about 80% of cases. A sentinel or sudden “thunderclap” headache is a classic presentation that may precede full-blown rupture in about 17% of cases.18 Patients who rupture an intracranial aneurysm have a mean age of 39, usually have normal renal function, and can be normotensive.11
For patients with no history of subarachnoid hemorrhage, the 5-year cumulative rupture rates for patients with aneurysms located in the internal carotid artery, anterior communicating or anterior cerebral artery, or middle cerebral artery were 0% for aneurysms less than 7 mm, 2.6% for those 7 to 12 mm, 14.5% for those 13 to 24 mm, and 40% for those 25 mm or larger, with higher rates for the same sizes in the posterior circulation.11
In patients without symptoms, size is correlated with risk of rupture: less than 4 mm is usually associated with very low risk, 4 to less than 7 mm with moderate risk, and 7 mm or more with increasing risk. An aneurysm larger than 10 mm is associated with roughly a 1% risk of rupture per year.19
Irazabal et al20 retrospectively studied 407 patients with ADPKD who were screened for intracranial aneurysm. Saccular aneurysms were detected in 45 patients; most were small (median diameter 3.5 mm). During cumulative imaging follow-up of 243 years, only one new intracranial aneurysm was detected (increasing from 2 to 4.4 mm over 144 months) and two previously identified aneurysms grew (one increasing 4.5 to 5.9 mm over 69 months and the other 4.7 to 6.2 mm over 184 months). No change occurred in 28 patients. Seven patients were lost to follow-up, however. During cumulative clinical follow-up of 316 years, no aneurysm ruptured. Two patients were lost to follow-up, three had surgical clipping, and five died of unrelated causes. The authors concluded that presymptomatic intracranial aneurysms are usually small, and that growth and rupture risks are no higher than for unruptured intracranial aneurysms in the general population. A 2014 study also suggests a conservative approach for managing intracranial aneurysm in the general population.21
In asymptomatic ADPKD patients, it is reasonable to reserve screening for those with a positive family history of intracranial aneurysm or subarachnoid hemorrhage, those with a previous ruptured aneurysm, those in high-risk professions (eg, pilots), and for patients prior to anticoagulant therapy or major surgery possibly associated with hemodynamic instability.11,22 Certain extremely anxious patients might also need to be studied. Screening can be performed with magnetic resonance angiography without gadolinium contrast. It is prudent to have patients with an intracranial aneurysm thoroughly evaluated by an experienced neurosurgeon with continued follow-up.
PROGRESSION OF ADPKD
The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) study23 evaluated 241 patients with ADPKD (ages 15 to 46) by measuring the annual rate of change in total kidney volume, total cyst volume, and iothalamate glomerular filtration rate (GFR) over 3 years. The annual increase in total kidney volume averaged 5.3%,23 though the reported range with various imaging techniques is from 4% to 12.8% in adults.24 This study focused on macrocystic disease, ie, cysts that are visible by MRI and measurably increase total kidney volume. Although larger total kidney volume at baseline generally predicted a more rapid decline in GFR, there were wide and overlapping variations in yearly GFR declines within and among different total-kidney-volume groups.23
SPECIAL CLINICAL PROBLEMS IN ADPKD
Case 1: A man with ADPKD develops new and increasing proteinuria
A 55-year-old man with ADPKD and stage 3 chronic kidney disease developed new and increasing proteinuria, rising to 5,500 mg per 24 hours. What is the most likely explanation?
- Rapidly progressive renal failure with increasing proteinuria in ADPKD
- Bilateral renal vein thromboses because of cyst compression
- Malignant hypertension with bilateral renal artery compression
- Superimposed primary glomerulopathy
- Multiple infected renal cysts with pyonephrosis
Answer: Superimposed primary glomerulopathy.
ADPKD (similar to uncomplicated obstructive uropathy, pyelonephritis, main renal artery disease, and often cases of interstitial nephritis without secondary glomerular changes) typically does not result in nephrotic-range proteinuria. A superimposed primary glomerulopathy, focal segmental glomerulosclerosis, was the biopsy-proved diagnosis.
At least 21 cases have been reported of AD-PKD with nephrotic-range proteinuria and a renal biopsy showing a primary glomerulopathy, including focal segmental glomerulosclerosis (5 cases), minimal-change disease (5), membranous nephropathy (3), IgA nephropathy (2), and one each of crescentic glomerulonephropathy, diabetic nephropathy, membranoproliferative glomerulonephritis, postinfectious glomerulonephropathy, amyloid glomerulopathy, and mesangioproliferative glomerulopathy.15 Treatment was directed at the primary glomerulopathy, and the outcomes corresponded to the primary diagnosis (eg, with appropriate treatment, three of the five patients with focal segmental glomerulosclerosis progressed to end-stage renal disease, all of the patients with minimal-change disease went into remission, and one of the two cases with IgA nephropathy improved).15
Case 2: A woman with ADPKD and advanced renal failure develops shortness of breath
A 47-year-old woman with very large polycystic kidneys (total kidney volume 7,500 mL; normal range for a single kidney approximately 136–295 mL, mean 196)25 and estimated GFR of 25 mL/min developed new-onset shortness of breath while climbing steps and later even when making a bed. She had no chest pain, cough, or edema. She was sent directly to the emergency department and was admitted and treated; her condition improved, and she was discharged after 6 days. What did she have?
- Presentation of rare cystic pulmonary disease in ADPKD
- Onset of pneumonia with early bacteremia
- Progressive reduction in ventilatory capacity from massive polycystic kidneys and liver elevating both sides of the diaphragm
- Pulmonary emboli from an iliac vein or inferior vena cava source
- Progressive anemia accompanying rapidly worsening stage 4 chronic kidney disease
Answer: She had pulmonary emboli from an iliac vein (right) or inferior vena cava source.
Pulmonary emboli in ADPKD can be caused by thrombi in the inferior vena cava or the iliac or femoral vein because of compression by a massive right polycystic kidney. Four cases were reported at Mayo Clinic,26 three diagnosed by MRI and one with CT. One additional case occurred at Cleveland Clinic. All patients survived after treatment with anticoagulation therapy; early nephrectomy was required in two cases.
Interestingly, following kidney transplantation, the patients at greatest risk for pulmonary emboli are those with ADPKD as their original disease.27
RENAL CYSTS RESULT FROM COMBINED MUTATIONS, INJURY
The germline ADPKD mutation that occurs in one allele of all renal tubular epithelial cells is necessary but not sufficient for cystogenesis.28 One or more additional somatic mutations of the normal allele—the “second hit”—also develop within individual tubular epithelial cells.28,29 These epithelial cells undergo clonal proliferation, resulting in tubular dilatation and cyst formation. Monoclonality of cells in cysts has been documented.
Ischemia-reperfusion injury can be viewed as a “third hit.”30 In PKD1 knockout mice, which at 5 weeks of age normally develop only mild cystic kidney disease, the superimposition of unilateral ischemia-reperfusion injury at 8 weeks caused widespread and rapid cyst formation. It is believed that acute renal injury reactivates developmental signaling pathways within 48 hours that trigger epithelial cell proliferation and then cyst development detectable by MRI 2 weeks later. Although this phenomenon has not been documented in humans, it is a cautionary tale.
CYSTOGENESIS INVOLVES MULTIPLE PATHWAYS
A comprehensive description of pathways leading to renal cyst formation is beyond the scope of this article, and the reader is referred to much more detailed and extensive reviews.2,31 Disturbances in at least three major interconnected pathways promote cystogenesis in renal tubular epithelial cells:
- Normal calcium transport into the endoplasmic reticulum is disrupted by abnormal polycystins on the surface of the primary cilium
- Vasopressin and other stimuli increase the production of cyclic adenosine monophosphate (cAMP)
- The mammalian target of rapamycin (mTOR) proliferative pathway is up-regulated.
DISRUPTION OF CALCIUM TRANSPORT IN THE PRIMARY CILIUM
Primary cilia are nonmotile cellular organelles of varying size, from about 0.25 μm up to about 1 μm.32 Each primary cilium has nine peripheral pairs of microtubules but lacks a centrally located pair that is present in motile cilia. Primary cilia are ubiquitous and have been highly conserved throughout evolution. A single cilium is present on almost all vertebral cells.33
Cilial defects have been identified in autosomal dominant as well as recessive diseases and are known as ciliopathies.33 Although rare in humans, they can affect a broad spectrum of organs other than the kidney, including the eye, liver, and brain.33
Urine flow in a renal tubule is believed to exert mechanical stimulation on the extracellular flagellum-like N-terminal tail of PC1 that extends from a primary cilium into the urinary space. PC1 in concert with PC2 opens PC2 calcium channels, allowing calcium ions to flow down the microtubules to ryanodine receptors and the basal body.32,33 This leads to local release of calcium ions that regulate cell proliferation.32,34 However, in ADPKD kidneys, PC1 and PC2 molecules are sparse or mutated, resulting in defective calcium transport, increased and unregulated tubular epithelial cell proliferation, and cyst formation.
In a totally different clinical setting, biopsies of human renal transplants that sustained acute tubular necrosis during transplantation reveal that a cilium dramatically elongates in response to injury,35 possibly as a compensatory mechanism to maintain calcium transport in the presence of meager urine flow and to restore the proliferation of tubular epithelial cells in a regulated repair process.
THE ROLE OF VASOPRESSIN AND ACTIVATION OF cAMP
In classic experiments, Wang et al36 cross-bred rats having genetically inherited polycystic kidney disease (actually, autosomal recessive polycystic kidney disease) with Brattleboro rats that completely lack vasopressin. At 10 and 20 weeks of age, the offspring had virtually complete inhibition of cystogenesis because of the absence of vasopressin. However, when vasopressin was restored by exogenous administration continuously for 8 weeks, the animals formed massive renal cysts.
Vasopressin activates cAMP, which then functions as a second messenger in cell signaling. cAMP increases the activation of the protein kinase A (PKA) pathway, which in turn increases downstream activity of the B-raf/ERK pathway. Up-regulation of cAMP and PKA appears to perpetuate activation of canonical Wnt signaling, down-regulate non-canonical Wnt/planar cell polarity signaling, and lead to loss of tubular diameter control, resulting in cyst formation.31 Normally, cAMP is degraded by phosphodiesterase. However, because of the primary cilium calcium transport defect in ADPKD, phosphodiesterase is reduced and cAMP persists.37 In conjunction with the defective primary cilial calcium transport, cAMP exerts a proliferative effect on renal tubular epithelial cells that is opposite to its effect in normal kidneys.31,32 cAMP also up-regulates the cystic fibrosis transmembrane conductance regulator (CFTR) that promotes chloride ion transport. Sodium ions follow the chloride ions, leading to fluid accumulation and cyst enlargement.31
Inhibiting vasopressin by increasing water intake
A simple key mechanism for limiting vasopressin secretion is by sufficient water ingestion. Nagao et al38 found that rats with polycystic kidney disease given water with 5% glucose (resulting in 3.5-fold increased fluid intake compared with rats given tap water) had a 68% reduction in urinary vasopressin and a urine osmolality less than 290 mOsm/kg. The high-water-intake rats had dramatically reduced cystic areas in the kidney and a 28% reduction of kidney-to-body weight ratio vs controls.
In an obvious oversimplification, these findings raised the question of whether a sufficient increase in water intake could be an effective therapy for polycystic kidney disease.39 A pilot clinical study evaluated changes in urine osmolality in eight patients with ADPKD who had normal renal function.40 At baseline, 24-hour urine osmolality was typically elevated to approximately 753 mOsm/kg compared to the plasma at 285 mOsm/kg, indicating that antidiuresis is the usual state. During the 2-week study, urine volume and osmolality were measured, and additional water intake was adjusted in order to achieve a urine osmolality goal of 285 ± 45 mOsm/kg. These adjustments resulted in water intake that appeared to be in the range of 2,400 to 3,000 mL per 24 hours. The major limitations of the study were that it was very short term, and there was no opportunity to measure changes in total kidney volume or estimated GFR.
In a recent preliminary report from Japan, high water intake (2,500–3,000 mL daily) in 18 ADPKD patients was compared over 12 months with ad libitum water intake in 14 ADPKD controls (clinicaltrials.gov NCT 01348505). There was no statistically significant change in total kidney volume or cystatin-estimated GFR in those on high water intake, but serious defects in study design (patients in the high water intake group were allowed to decrease their intake if it was causing them difficulty, and patients in the ad libitum water intake group had no measurement of their actual water intake) prevent any conclusions because there was no evidence that the groups were different from one another with respect to the key element of the study, namely, water intake.
Blocking the vasopressin receptor slows disease progression
Using another approach, Gattone et al41 inhibited the effect of vasopressin by blocking the vasopressin 2 receptor (V2R) in mouse and rat models of polycystic kidney disease, using an experimental drug, OPC31260. The drug halted disease progression and, in one situation, appeared to cause regression of established disease. As noted by Torres and Harris,31 even though both increased water intake and V2R antagonists decrease cAMP in the distal tubules and collecting ducts, circulating levels of vasopressin are decreased by increased water intake but increased by V2R antagonists.
After these remarkable results in animal models, clinical trials of the V2R antagonist tolvaptan were conducted in patients with ADPKD. In the Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes 3:4 study,42 1,445 adults (ages 18 to 50) with ADPKD in 133 centers worldwide were randomized to receive either tolvaptan or placebo for 3 years. Key inclusion criteria included good renal function (estimated GFR ≥ 60 mL/min) and total kidney volume of at least 750 mL (mean 1,700 mL) as measured by MRI. Tolvaptan was titrated to the highest tolerated twice-daily dose (average total of 95 mg/day). All patients were advised to maintain good hydration and to avoid thirst by drinking a glass of water after each urination. Unfortunately, neither water intake nor urine output was measured.
The primary end point was the annual rate of change in total kidney volume, with secondary end points of clinical progression (worsening kidney function, pain, hypertension, albuminuria), and rate of decline in kidney function as measured by the slope of the reciprocal of serum creatinine.42
Patients in the tolvaptan arm had a slower annual increase in total kidney volume than controls (2.8% vs 5.5%, respectively, P < .001) and a slower annual decline in renal function (−2.61 vs −3.81 mg/mL−1, respectively, P < .001).42 More participants in the treatment group withdrew than in the placebo group (23% vs 14%, respectively).
Adverse events occurred more frequently with tolvaptan.42 Liver enzyme elevations of greater than three times the upper limit of normal occurred in 4.4% of patients in the treatment group, leading to a drug warning issued in January 2013. As expected, side effects related to diuresis (urinary frequency, nocturia, polyuria, and thirst) were more frequent in the treatment group, occurring in up to 55% of participants.
The authors noted, “Although maintaining hydration helped ensure that the blinding in the study was maintained, the suppression of vasopressin release in the placebo group may have led to an underestimation of the beneficial effect of tolvaptan and may account for the lower rates of kidney growth observed in the placebo group.”42
In 2013, the US Food and Drug Administration (FDA) denied a new drug application for tolvaptan as a treatment for ADPKD.
THE mTOR PATHWAY IS UP-REGULATED
The mTOR pathway that plays a major role in cell growth and proliferation includes interaction of the cytoplasmic tail of polycystin 1 with tuberin.43 Activation products of mTOR, including phospho-S6K, have been found in tubular epithelial cells lining cysts of ADPKD kidneys but not in normal kidneys.43 Mutant mice with polycystic disease had phospho-S6K in tubular epithelial cells of cysts, whereas those treated with the mTOR inhibitor rapamycin did not.43 But subsequent studies have shown that only a low level of mTOR activation is present in 65% to 70% of ADPKD cysts.44
Two major studies of the treatment of ADPKD with rapamycin that were published contemporaneously in 2010 failed to demonstrate any significant benefit with mTOR inhibitor treatment.45,46
Serra et al45 conducted an 18-month, open-label trial of 100 ADPKD patients ages 18 to 40 with an estimated GFR (eGFR) of at least 70 mL/min. Patients were randomized to receive rapamycin, given as sirolimus 2 mg per day, or standard care. The primary end point was the reduction in the growth rate of total kidney volume, measured by MRI. Secondary end points were eGFR and protein excretion (albumin-creatinine ratio). No significant difference was found in total kidney volume, but a nonsignificant stabilization of eGFR was noted.
Walz et al46 in a 2-year, multicenter, double-blind trial, randomized 433 patients (mean age 44; mean eGFR 54.5 mL/min) to treatment with either the short-acting mTOR inhibitor everolimus (2.5 mg twice daily) or placebo. Although patients in the treatment group had less of an increase in total kidney volume (significant at 1 year but not at 2 years), they tended to show a decline in eGFR. But further analysis showed that the only patients who had a reduction in eGFR were males who already had impaired kidney function at baseline.47
In a pilot study, 30 patients with ADPKD (mean age 49) were randomized to one of three therapies:
- Low-dose rapamycin (trough blood level 2–5 ng/mL)
- Standard-dose rapamycin (trough blood level > 5–8 ng/mL)
- Standard care without rapamycin.48
In contrast to other studies, the primary end point was the change in iothalamate GFR at 12 months, with change in total kidney volume being a secondary end point.
At 12 months, with 26 patients completing the study, the low-dose rapamycin group (n = 9) had a significant increase in iothalamate GFR of 7.7 ± 12.5 mL/min/1.73 m2, whereas the standard-dose rapamycin group (n = 8) had a nonsignificant increase of 1.6 ± 12.1 mL/min/1.73 m2, and the no-rapamycin group (n = 9) had a fall in iothalamate GFR of 11.2 ± 9.1 mL/min/1.73 m2 (P = .005 for low-dose vs no rapamycin; P = .07 for standard-dose vs no rapamycin; P = .52 for low-dose vs standard-dose rapamycin; and P = .002 for combined low-dose and standard-dose rapamycin vs no rapamycin.).48 These differences were observed despite there being no significant change in total kidney volume in any of the groups. Patients on low-dose rapamycin had fewer adverse effects than those on standard dose and were more often able to continue therapy for the entire study. This, and the use of iothalamate GFR rather than eGFR to measure GFR, are believed to be the main reasons that low-dose effects were more pronounced than those with standard doses. One may speculate that rapamycin may have its effect on microcysts and cystogenic cells, resulting in stabilization of or improvement in renal function without detectable slowing in total kidney volume enlargement. Mechanisms for this possibility involve new concepts, as discussed below.
NEW CONCEPTS
Specialized cells also promote renal cyst formation
Specialized cells that promote cyst formation have been identified by Karihaloo et al49 in a mouse model of polycystic kidney disease. In this model, alternatively activated macrophages homed to cystic areas and promoted cyst growth. These findings suggested that interrupting the homing and proliferative signals of macrophages could be a therapeutic target for ADPKD. Although rapamycin can suppress macrophage proliferation by macrophage colony-stimulating factor and inhibit macrophage function,50 alternatively activated macrophages have not been specifically studied for rapamycin responsiveness.
More promising is evidence that CD133+ progenitor cells from human ADPKD kidneys—but not from normal human kidneys—form cysts in vitro and in severe combined immunodeficient mouse models.51 Treatment with rapamycin decreased proliferation of the de-differentiated CD133+ cells from ADPKD patients and reduced cystogenesis.51
Visible cysts are the tip of the iceberg
Using ADPKD nephrectomy specimens from eight patients, Grantham et al52 compared cyst counts by MRI and by histology and found that for every renal cyst detected by MRI, about 62 smaller cysts (< 0.9 mm) are present in the kidney. For a typical patient having an average of 587 cysts in both kidneys that are detectable by MRI, this means that more than 36,000 cysts are actually present, and MRI detects less than 2% of the total cysts present.
Although microcysts are too small to contribute much to total kidney volume, they can interfere with kidney function. Microcysts can reduce GFR in two major ways: by compressing microvasculature, tubules, and glomeruli in the cortex; or by blocking the drainage of multiple upstream nephrons when they form in or block medullary collecting ducts.52 Although the growth rates of microcysts less than 1 mm in size have not yet been measured, the adult combined growth rates of the renal cyst component is approximately 12% per year, with yearly individual cyst growth rates up to 71%, and with fetal cyst growth rates even higher for cysts larger than 7.0 mm.53 Before and during an accelerated growth period, microcysts may be susceptible to certain therapies that could first improve GFR and only later change measurable total kidney volume by slowing microcyst progression to macrocysts either directly or through specialized cells that may be sensitive to rapamycin.
CURRENT MANAGEMENT OF ADPKD
Blood pressure control is essential—but too low is not good. For adult patients with hypertension caused by ADPKD, an acceptable blood pressure range is 120–130/70–80 mm Hg. However, further information from recently published blood pressure guidelines54 and the results of the Halt Progression of Polycystic Kidney Disease (HALT-PKD) study to be reported in late 201455 may provide more precise ranges for blood pressure control in ADPKD.
Although substantial experimental evidence exists for the benefits of inhibiting the up-regulation of the renin-angiotensin-aldosterone system in ADPKD, clinical proof is not yet available to confirm that angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) are preferred therapy.55 This may be determined by results of the HALT-PKD study, due for release in late 2014.55
Controlling blood pressure should be done with caution. Patients with low GFRs whose blood pressure is too low tend to have a more rapid decline of GFR, as suggested in the Modification of Diet in Renal Disease (MDRD) study in 1995.56
Experimental evidence suggests that avoiding calcium channel blockers may be advisable. Yamaguchi et al34 found that calcium channel blockers worsen the calcium transport defect and convert tubular epithelial cells to a proliferative phenotype.34
High fluid intake (2,500–3,000 mL/day), because it suppresses vasopressin, may be useful if permitted by several factors such as the patient’s cardiopulmonary and renal and electrolyte status, other medications, and diet.31 The reader is referred to a detailed description of the precautions necessary when prescribing high water intake.31 Patients should have their fluid intake managed by a physician and their renal function and serum sodium and electrolytes monitored regularly in order to avoid hyponatremia. Severe hyponatremia has occurred in patients with ADPKD and impaired kidney function who drank excessive quantities of water. Cardiac and pulmonary complications from excessive fluid intake are also possible, especially with a low GFR and compromised cardiac function.
A low-sodium diet, if not a contributing factor in hyponatremia, can be used under physician direction in the management of hypertension as well as in the prevention of calcium oxalate kidney stones.
Caffeine should be avoided because it may interfere with the activity of the phosphodiesterase that is necessary for the catabolism of cAMP to 5′AMP.
A low-protein diet is of unproven benefit,56 but it is prudent to avoid high protein intake.57
Complications such as bleeding (into or from cysts), infection (urinary tract, kidney cysts, and liver cysts), kidney stones, and urinary tract obstruction should be treated promptly and may require hospitalization.
Regular symptom reviews and physical examinations need to be performed with nonrenal concerns also in mind, such as intracranial aneurysms and cardiac valve lesions.11,58
Formal genetic counseling and molecular testing are becoming more frequently indicated as more complex inheritance patterns arise.6–8,59
Renal replacement therapy in the form of dialysis or transplantation is usually available for the patient when end-stage renal disease occurs. In the largest study thus far, ADPKD patient survival with a kidney transplant was similar to that of patients without ADPKD (about 93% at 5 years), and from 5 years to 15 years death-censored graft survival was actually better.60 Thromboembolic events are more frequent after transplantation,27,60 but they may also occur before transplantation from a massive right kidney compressing the iliac vein or the inferior vena cava, or both, leading to thrombus formation.26
Investigational as well as standard drug studies have intensified. Results from a large randomized study in approximately 1,000 adult ADPKD patients that evaluated over 6 to 8 years the effects of ACE inhibition with or without ARB treatment of hypertension, at both usual and lower blood pressure ranges in those with good renal function, are expected in late 2014.55 Outcomes from a few small clinical studies, eg, one with long-acting somatostatin31,61 and one using low-dose rapamycin48 in adults with ADPKD, will require confirmation in large randomized placebo-controlled clinical studies. In a new 3-year randomized placebo-controlled study of 91 children and young adults (ages 8 to 22) with ADPKD and essentially normal renal function who continued treatment with lisinopril, the addition of pravastatin (20 mg or 40 mg daily based on age) resulted in a significant reduction in the number of patients (46% vs 68%, respectively, P = .03) experiencing a greater than 20% change (increase) in height-adjusted total kidney volume.62 Change in GFR was not reported,62 but an earlier 4-week study in 10 patients treated with simvastatin did show an increase in renal blood flow and GFR.63 Numerous other agents that lack human studies include some described in older experimental work (eg, amiloride,31,64 citrate31,65) and many others from a growing list of potential therapeutic targets.31,66–73 It must be emphasized that there is no FDA-approved medication specifically for the treatment of ADPKD.
Future specific treatments of ADPKD may also involve minimally toxic doses of combination or sequential therapy directed at precystic and then both micro- and macrocystic/cystic fluid expansion aspects of ADPKD.48,74 Unfortunately, at the present time there is no specific FDA-approved therapy for ADPKD.
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited renal disease, has an estimated prevalence of 1:400 to 1:1,000 live births in the United States, and occurs worldwide.1,2 There are about 700,000 people living with it in the United States, and about 6,000 new cases arise annually. It accounts for nearly 5% of all patients with end-stage renal disease in the United States.3
This paper will offer an overview of the pathogenesis of renal cysts, review some of the clinical aspects of ADPKD including diagnosis and management of complications, and discuss recent drug trials and current management.
TWO TYPES—PKD1 IS MORE COMMON AND PROGRESSES MORE RAPIDLY
Two major forms of ADPKD are recognized and can usually be determined by genetic testing: PKD1, accounting for about 85% of cases, and PKD2, accounting for 15%.
The gene locus for PKD1 is on the short arm of the 16th chromosome (16p13.3), and its glycoprotein gene product is polycystin 1 (PC1), a large molecule with 4,303 amino acids.2 PC1 has a long N-terminal extracellular tail that can function as a mechanosensor. Disease progression is much faster with PKD1, and end-stage renal disease usually occurs before age 56.4
In PKD2, the gene locus is on the long arm of the fourth chromosome (4q21–23), and has a smaller glycoprotein gene product, polycystin 2 (PC2), that plays a role in calcium transport. The disease course of PKD2 tends to be slower. End-stage renal disease might not develop in the patient’s lifetime, since it typically develops when the patient is more than 70 years old.4
Although the growth rate of renal cysts is similar between the two types, patients with PKD1 develop about twice as many cysts as those with PDK2, and their cyst development starts at a younger age.5
Typically, patients have a clear phenotype and a positive family history, but in about 10% of possible ADPKD cases, there is no family history of ADPKD. Genetic variations such as incompletely penetrant PKD1 alleles,6 hypomorphic alleles,7 and trans-heterozygous mutations8 account for at least some of these cases.
IMAGING CRITERIA HAVE BROADENED
Ultrasonographic criteria for the diagnosis of ADPKD that were published in 1994 were based on patients who had a family history of PKD1.9 The criteria have since been modified (the “unified criteria”) to include patients with a family history of PKD2 who begin cyst development at a later age and with lower numbers.10 For patients ages 30 to 39, a previously difficult diagnostic group, the criterion for the minimum number of cysts visible on ultrasonography changed from four to three, improving the sensitivity of detecting disease from approximately 76% to approximately 95% (Table 1).9,10 It is important to note that these criteria apply only to patients “at risk,” ie, with a positive family history of ADPKD.
Computed tomography (CT) and magnetic resonance imaging (MRI) classically show bilaterally enlarged multicystic kidneys, though variations can be seen.
DISEASE CAN PRESENT IN MYRIAD WAYS
Although cystic kidney disease is the basic underlying problem, undiagnosed patients may present with a variety of symptoms caused by other manifestations of ADPKD (Table 2).
Hypertension is the most common presentation, occurring in about 50% of patients ages 20 to 34, and essentially 100% of those with end-stage renal disease.11 It is associated with up-regulation of the renin-angiotensin-aldosterone system.
Pain is typically located in the abdomen, flank, or back and can occur in a localized or diffuse manner. Early abdominal distress is often simply described as “fullness.” Localized pain is usually caused by bleeding into or rupture of a cyst, renal stones, or infection.12 Because renal cysts are noncommunicating, bleeding can occur into a cyst and cause pain without gross hematuria. Compression by greatly enlarged kidneys, liver, or both can cause a variety of gastrointestinal symptoms such as reflux esophagitis and varying degrees of constipation. Diffuse pain is often musculoskeletal and related to exaggerated lordosis from increasing abdominal size due to enlarging cystic kidneys and sometimes liver.12 In carefully selected cases, cyst aspiration may be helpful.11
Although renal carcinomas are rare and not more frequent than in the general population, they can occur at an earlier age and with constitutional symptoms.11
Urinary tract infections are increased in frequency. A patient may have a simple urinary tract infection that is cured with the appropriate antibiotic. However, a urinary tract infection repeatedly recurring with the same organism is a strong clue that an infected cyst is the source and requires more intensive treatment with the appropriate cyst-penetrating antibiotic. On the other hand, because cysts are noncommunicating, an infected cyst might be present despite a negative urine culture.
Identifying infected cysts can be a challenge with conventional imaging techniques, but combined positron emission tomography and CT (PET/CT) can be a valuable though expensive diagnostic tool to identify an infected kidney or liver cyst, or to identify an unsuspected source of the pain and infection.13
Jouret et al13 evaluated 27 PET/CT scans performed in 24 patients with ADPKD and suspicion of an abdominal infection. Patients were deemed to have probable cyst infection if they met all of the following criteria: temperature more than 38°C for longer than 3 days, loin or liver tenderness, plasma C-reactive protein level greater than 5 mg/dL, and no evidence of intracystic bleeding on CT. Patients with only two or three of these criteria were classified as having fever of unknown origin. Diagnosis of cyst infection was confirmed by cyst fluid analysis.
PET/CT identified a kidney or liver cyst infection in 85% of 13 infectious events in 11 patients who met all the criteria for probable cyst infection; CT alone contributed to the diagnosis in only one patient.13 In those with fever of unknown origin, PET/CT identified a source of infection in 64% of 14 events in 13 patients: two infected renal cysts, as well as one patient each with other infections that would be difficult to diagnose clinically, ie, small bowel diverticulitis, psoas abscess, diverticulitis of the right colon, pyelonephritis in a transplanted kidney, infected abdominal aortic aneurysm, prostatitis, colitis, and Helicobacter pylori gastritis. Results of PET/CT were negative in five patients with intracystic bleeding.
Kidney stones occur in 20% to 36% of patients.11,14 Uric acid stones occur at almost the same frequency as calcium oxalate stones.
Chronic kidney disease not previously diagnosed may be the presenting condition in a small percentage of patients, sometimes those in whom much earlier hypertension was not fully evaluated. ADPKD is typically not associated with significant proteinuria (eg, nephrotic range), and the presence of heavy proteinuria usually indicates the presence of a superimposed primary glomerulopathy.15
Cysts in other locations. By MRI, liver cysts are present in 58% of patients ages 15 to 24, rising to 94% in those ages 35 to 46.11 Because liver cysts are estrogen-dependent, they are more prominent in women. A small percentage of patients develop cysts in the pancreas (5%), arachnoid membranes (8%), and seminal vesicles (40% of men with ADPKD).11
Cardiovascular abnormalities occur in almost one-third of patients with ADPKD, usually as mitral and aortic valve abnormalities.16 Aneurysms of the aortic root and abdominal aorta can also occur, in addition to intracranial aneurysms (see below).17
Intracranial aneurysms are not uncommon, and size usually determines their risk.
Intracranial aneurysms are strongly influenced by family history: 16% of ADPKD patients with a family history of intracranial aneurysm also develop them, compared with 5% to 6% of patients with no family history.11 The anterior cerebral circulation is involved in about 80% of cases. A sentinel or sudden “thunderclap” headache is a classic presentation that may precede full-blown rupture in about 17% of cases.18 Patients who rupture an intracranial aneurysm have a mean age of 39, usually have normal renal function, and can be normotensive.11
For patients with no history of subarachnoid hemorrhage, the 5-year cumulative rupture rates for patients with aneurysms located in the internal carotid artery, anterior communicating or anterior cerebral artery, or middle cerebral artery were 0% for aneurysms less than 7 mm, 2.6% for those 7 to 12 mm, 14.5% for those 13 to 24 mm, and 40% for those 25 mm or larger, with higher rates for the same sizes in the posterior circulation.11
In patients without symptoms, size is correlated with risk of rupture: less than 4 mm is usually associated with very low risk, 4 to less than 7 mm with moderate risk, and 7 mm or more with increasing risk. An aneurysm larger than 10 mm is associated with roughly a 1% risk of rupture per year.19
Irazabal et al20 retrospectively studied 407 patients with ADPKD who were screened for intracranial aneurysm. Saccular aneurysms were detected in 45 patients; most were small (median diameter 3.5 mm). During cumulative imaging follow-up of 243 years, only one new intracranial aneurysm was detected (increasing from 2 to 4.4 mm over 144 months) and two previously identified aneurysms grew (one increasing 4.5 to 5.9 mm over 69 months and the other 4.7 to 6.2 mm over 184 months). No change occurred in 28 patients. Seven patients were lost to follow-up, however. During cumulative clinical follow-up of 316 years, no aneurysm ruptured. Two patients were lost to follow-up, three had surgical clipping, and five died of unrelated causes. The authors concluded that presymptomatic intracranial aneurysms are usually small, and that growth and rupture risks are no higher than for unruptured intracranial aneurysms in the general population. A 2014 study also suggests a conservative approach for managing intracranial aneurysm in the general population.21
In asymptomatic ADPKD patients, it is reasonable to reserve screening for those with a positive family history of intracranial aneurysm or subarachnoid hemorrhage, those with a previous ruptured aneurysm, those in high-risk professions (eg, pilots), and for patients prior to anticoagulant therapy or major surgery possibly associated with hemodynamic instability.11,22 Certain extremely anxious patients might also need to be studied. Screening can be performed with magnetic resonance angiography without gadolinium contrast. It is prudent to have patients with an intracranial aneurysm thoroughly evaluated by an experienced neurosurgeon with continued follow-up.
PROGRESSION OF ADPKD
The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) study23 evaluated 241 patients with ADPKD (ages 15 to 46) by measuring the annual rate of change in total kidney volume, total cyst volume, and iothalamate glomerular filtration rate (GFR) over 3 years. The annual increase in total kidney volume averaged 5.3%,23 though the reported range with various imaging techniques is from 4% to 12.8% in adults.24 This study focused on macrocystic disease, ie, cysts that are visible by MRI and measurably increase total kidney volume. Although larger total kidney volume at baseline generally predicted a more rapid decline in GFR, there were wide and overlapping variations in yearly GFR declines within and among different total-kidney-volume groups.23
SPECIAL CLINICAL PROBLEMS IN ADPKD
Case 1: A man with ADPKD develops new and increasing proteinuria
A 55-year-old man with ADPKD and stage 3 chronic kidney disease developed new and increasing proteinuria, rising to 5,500 mg per 24 hours. What is the most likely explanation?
- Rapidly progressive renal failure with increasing proteinuria in ADPKD
- Bilateral renal vein thromboses because of cyst compression
- Malignant hypertension with bilateral renal artery compression
- Superimposed primary glomerulopathy
- Multiple infected renal cysts with pyonephrosis
Answer: Superimposed primary glomerulopathy.
ADPKD (similar to uncomplicated obstructive uropathy, pyelonephritis, main renal artery disease, and often cases of interstitial nephritis without secondary glomerular changes) typically does not result in nephrotic-range proteinuria. A superimposed primary glomerulopathy, focal segmental glomerulosclerosis, was the biopsy-proved diagnosis.
At least 21 cases have been reported of AD-PKD with nephrotic-range proteinuria and a renal biopsy showing a primary glomerulopathy, including focal segmental glomerulosclerosis (5 cases), minimal-change disease (5), membranous nephropathy (3), IgA nephropathy (2), and one each of crescentic glomerulonephropathy, diabetic nephropathy, membranoproliferative glomerulonephritis, postinfectious glomerulonephropathy, amyloid glomerulopathy, and mesangioproliferative glomerulopathy.15 Treatment was directed at the primary glomerulopathy, and the outcomes corresponded to the primary diagnosis (eg, with appropriate treatment, three of the five patients with focal segmental glomerulosclerosis progressed to end-stage renal disease, all of the patients with minimal-change disease went into remission, and one of the two cases with IgA nephropathy improved).15
Case 2: A woman with ADPKD and advanced renal failure develops shortness of breath
A 47-year-old woman with very large polycystic kidneys (total kidney volume 7,500 mL; normal range for a single kidney approximately 136–295 mL, mean 196)25 and estimated GFR of 25 mL/min developed new-onset shortness of breath while climbing steps and later even when making a bed. She had no chest pain, cough, or edema. She was sent directly to the emergency department and was admitted and treated; her condition improved, and she was discharged after 6 days. What did she have?
- Presentation of rare cystic pulmonary disease in ADPKD
- Onset of pneumonia with early bacteremia
- Progressive reduction in ventilatory capacity from massive polycystic kidneys and liver elevating both sides of the diaphragm
- Pulmonary emboli from an iliac vein or inferior vena cava source
- Progressive anemia accompanying rapidly worsening stage 4 chronic kidney disease
Answer: She had pulmonary emboli from an iliac vein (right) or inferior vena cava source.
Pulmonary emboli in ADPKD can be caused by thrombi in the inferior vena cava or the iliac or femoral vein because of compression by a massive right polycystic kidney. Four cases were reported at Mayo Clinic,26 three diagnosed by MRI and one with CT. One additional case occurred at Cleveland Clinic. All patients survived after treatment with anticoagulation therapy; early nephrectomy was required in two cases.
Interestingly, following kidney transplantation, the patients at greatest risk for pulmonary emboli are those with ADPKD as their original disease.27
RENAL CYSTS RESULT FROM COMBINED MUTATIONS, INJURY
The germline ADPKD mutation that occurs in one allele of all renal tubular epithelial cells is necessary but not sufficient for cystogenesis.28 One or more additional somatic mutations of the normal allele—the “second hit”—also develop within individual tubular epithelial cells.28,29 These epithelial cells undergo clonal proliferation, resulting in tubular dilatation and cyst formation. Monoclonality of cells in cysts has been documented.
Ischemia-reperfusion injury can be viewed as a “third hit.”30 In PKD1 knockout mice, which at 5 weeks of age normally develop only mild cystic kidney disease, the superimposition of unilateral ischemia-reperfusion injury at 8 weeks caused widespread and rapid cyst formation. It is believed that acute renal injury reactivates developmental signaling pathways within 48 hours that trigger epithelial cell proliferation and then cyst development detectable by MRI 2 weeks later. Although this phenomenon has not been documented in humans, it is a cautionary tale.
CYSTOGENESIS INVOLVES MULTIPLE PATHWAYS
A comprehensive description of pathways leading to renal cyst formation is beyond the scope of this article, and the reader is referred to much more detailed and extensive reviews.2,31 Disturbances in at least three major interconnected pathways promote cystogenesis in renal tubular epithelial cells:
- Normal calcium transport into the endoplasmic reticulum is disrupted by abnormal polycystins on the surface of the primary cilium
- Vasopressin and other stimuli increase the production of cyclic adenosine monophosphate (cAMP)
- The mammalian target of rapamycin (mTOR) proliferative pathway is up-regulated.
DISRUPTION OF CALCIUM TRANSPORT IN THE PRIMARY CILIUM
Primary cilia are nonmotile cellular organelles of varying size, from about 0.25 μm up to about 1 μm.32 Each primary cilium has nine peripheral pairs of microtubules but lacks a centrally located pair that is present in motile cilia. Primary cilia are ubiquitous and have been highly conserved throughout evolution. A single cilium is present on almost all vertebral cells.33
Cilial defects have been identified in autosomal dominant as well as recessive diseases and are known as ciliopathies.33 Although rare in humans, they can affect a broad spectrum of organs other than the kidney, including the eye, liver, and brain.33
Urine flow in a renal tubule is believed to exert mechanical stimulation on the extracellular flagellum-like N-terminal tail of PC1 that extends from a primary cilium into the urinary space. PC1 in concert with PC2 opens PC2 calcium channels, allowing calcium ions to flow down the microtubules to ryanodine receptors and the basal body.32,33 This leads to local release of calcium ions that regulate cell proliferation.32,34 However, in ADPKD kidneys, PC1 and PC2 molecules are sparse or mutated, resulting in defective calcium transport, increased and unregulated tubular epithelial cell proliferation, and cyst formation.
In a totally different clinical setting, biopsies of human renal transplants that sustained acute tubular necrosis during transplantation reveal that a cilium dramatically elongates in response to injury,35 possibly as a compensatory mechanism to maintain calcium transport in the presence of meager urine flow and to restore the proliferation of tubular epithelial cells in a regulated repair process.
THE ROLE OF VASOPRESSIN AND ACTIVATION OF cAMP
In classic experiments, Wang et al36 cross-bred rats having genetically inherited polycystic kidney disease (actually, autosomal recessive polycystic kidney disease) with Brattleboro rats that completely lack vasopressin. At 10 and 20 weeks of age, the offspring had virtually complete inhibition of cystogenesis because of the absence of vasopressin. However, when vasopressin was restored by exogenous administration continuously for 8 weeks, the animals formed massive renal cysts.
Vasopressin activates cAMP, which then functions as a second messenger in cell signaling. cAMP increases the activation of the protein kinase A (PKA) pathway, which in turn increases downstream activity of the B-raf/ERK pathway. Up-regulation of cAMP and PKA appears to perpetuate activation of canonical Wnt signaling, down-regulate non-canonical Wnt/planar cell polarity signaling, and lead to loss of tubular diameter control, resulting in cyst formation.31 Normally, cAMP is degraded by phosphodiesterase. However, because of the primary cilium calcium transport defect in ADPKD, phosphodiesterase is reduced and cAMP persists.37 In conjunction with the defective primary cilial calcium transport, cAMP exerts a proliferative effect on renal tubular epithelial cells that is opposite to its effect in normal kidneys.31,32 cAMP also up-regulates the cystic fibrosis transmembrane conductance regulator (CFTR) that promotes chloride ion transport. Sodium ions follow the chloride ions, leading to fluid accumulation and cyst enlargement.31
Inhibiting vasopressin by increasing water intake
A simple key mechanism for limiting vasopressin secretion is by sufficient water ingestion. Nagao et al38 found that rats with polycystic kidney disease given water with 5% glucose (resulting in 3.5-fold increased fluid intake compared with rats given tap water) had a 68% reduction in urinary vasopressin and a urine osmolality less than 290 mOsm/kg. The high-water-intake rats had dramatically reduced cystic areas in the kidney and a 28% reduction of kidney-to-body weight ratio vs controls.
In an obvious oversimplification, these findings raised the question of whether a sufficient increase in water intake could be an effective therapy for polycystic kidney disease.39 A pilot clinical study evaluated changes in urine osmolality in eight patients with ADPKD who had normal renal function.40 At baseline, 24-hour urine osmolality was typically elevated to approximately 753 mOsm/kg compared to the plasma at 285 mOsm/kg, indicating that antidiuresis is the usual state. During the 2-week study, urine volume and osmolality were measured, and additional water intake was adjusted in order to achieve a urine osmolality goal of 285 ± 45 mOsm/kg. These adjustments resulted in water intake that appeared to be in the range of 2,400 to 3,000 mL per 24 hours. The major limitations of the study were that it was very short term, and there was no opportunity to measure changes in total kidney volume or estimated GFR.
In a recent preliminary report from Japan, high water intake (2,500–3,000 mL daily) in 18 ADPKD patients was compared over 12 months with ad libitum water intake in 14 ADPKD controls (clinicaltrials.gov NCT 01348505). There was no statistically significant change in total kidney volume or cystatin-estimated GFR in those on high water intake, but serious defects in study design (patients in the high water intake group were allowed to decrease their intake if it was causing them difficulty, and patients in the ad libitum water intake group had no measurement of their actual water intake) prevent any conclusions because there was no evidence that the groups were different from one another with respect to the key element of the study, namely, water intake.
Blocking the vasopressin receptor slows disease progression
Using another approach, Gattone et al41 inhibited the effect of vasopressin by blocking the vasopressin 2 receptor (V2R) in mouse and rat models of polycystic kidney disease, using an experimental drug, OPC31260. The drug halted disease progression and, in one situation, appeared to cause regression of established disease. As noted by Torres and Harris,31 even though both increased water intake and V2R antagonists decrease cAMP in the distal tubules and collecting ducts, circulating levels of vasopressin are decreased by increased water intake but increased by V2R antagonists.
After these remarkable results in animal models, clinical trials of the V2R antagonist tolvaptan were conducted in patients with ADPKD. In the Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes 3:4 study,42 1,445 adults (ages 18 to 50) with ADPKD in 133 centers worldwide were randomized to receive either tolvaptan or placebo for 3 years. Key inclusion criteria included good renal function (estimated GFR ≥ 60 mL/min) and total kidney volume of at least 750 mL (mean 1,700 mL) as measured by MRI. Tolvaptan was titrated to the highest tolerated twice-daily dose (average total of 95 mg/day). All patients were advised to maintain good hydration and to avoid thirst by drinking a glass of water after each urination. Unfortunately, neither water intake nor urine output was measured.
The primary end point was the annual rate of change in total kidney volume, with secondary end points of clinical progression (worsening kidney function, pain, hypertension, albuminuria), and rate of decline in kidney function as measured by the slope of the reciprocal of serum creatinine.42
Patients in the tolvaptan arm had a slower annual increase in total kidney volume than controls (2.8% vs 5.5%, respectively, P < .001) and a slower annual decline in renal function (−2.61 vs −3.81 mg/mL−1, respectively, P < .001).42 More participants in the treatment group withdrew than in the placebo group (23% vs 14%, respectively).
Adverse events occurred more frequently with tolvaptan.42 Liver enzyme elevations of greater than three times the upper limit of normal occurred in 4.4% of patients in the treatment group, leading to a drug warning issued in January 2013. As expected, side effects related to diuresis (urinary frequency, nocturia, polyuria, and thirst) were more frequent in the treatment group, occurring in up to 55% of participants.
The authors noted, “Although maintaining hydration helped ensure that the blinding in the study was maintained, the suppression of vasopressin release in the placebo group may have led to an underestimation of the beneficial effect of tolvaptan and may account for the lower rates of kidney growth observed in the placebo group.”42
In 2013, the US Food and Drug Administration (FDA) denied a new drug application for tolvaptan as a treatment for ADPKD.
THE mTOR PATHWAY IS UP-REGULATED
The mTOR pathway that plays a major role in cell growth and proliferation includes interaction of the cytoplasmic tail of polycystin 1 with tuberin.43 Activation products of mTOR, including phospho-S6K, have been found in tubular epithelial cells lining cysts of ADPKD kidneys but not in normal kidneys.43 Mutant mice with polycystic disease had phospho-S6K in tubular epithelial cells of cysts, whereas those treated with the mTOR inhibitor rapamycin did not.43 But subsequent studies have shown that only a low level of mTOR activation is present in 65% to 70% of ADPKD cysts.44
Two major studies of the treatment of ADPKD with rapamycin that were published contemporaneously in 2010 failed to demonstrate any significant benefit with mTOR inhibitor treatment.45,46
Serra et al45 conducted an 18-month, open-label trial of 100 ADPKD patients ages 18 to 40 with an estimated GFR (eGFR) of at least 70 mL/min. Patients were randomized to receive rapamycin, given as sirolimus 2 mg per day, or standard care. The primary end point was the reduction in the growth rate of total kidney volume, measured by MRI. Secondary end points were eGFR and protein excretion (albumin-creatinine ratio). No significant difference was found in total kidney volume, but a nonsignificant stabilization of eGFR was noted.
Walz et al46 in a 2-year, multicenter, double-blind trial, randomized 433 patients (mean age 44; mean eGFR 54.5 mL/min) to treatment with either the short-acting mTOR inhibitor everolimus (2.5 mg twice daily) or placebo. Although patients in the treatment group had less of an increase in total kidney volume (significant at 1 year but not at 2 years), they tended to show a decline in eGFR. But further analysis showed that the only patients who had a reduction in eGFR were males who already had impaired kidney function at baseline.47
In a pilot study, 30 patients with ADPKD (mean age 49) were randomized to one of three therapies:
- Low-dose rapamycin (trough blood level 2–5 ng/mL)
- Standard-dose rapamycin (trough blood level > 5–8 ng/mL)
- Standard care without rapamycin.48
In contrast to other studies, the primary end point was the change in iothalamate GFR at 12 months, with change in total kidney volume being a secondary end point.
At 12 months, with 26 patients completing the study, the low-dose rapamycin group (n = 9) had a significant increase in iothalamate GFR of 7.7 ± 12.5 mL/min/1.73 m2, whereas the standard-dose rapamycin group (n = 8) had a nonsignificant increase of 1.6 ± 12.1 mL/min/1.73 m2, and the no-rapamycin group (n = 9) had a fall in iothalamate GFR of 11.2 ± 9.1 mL/min/1.73 m2 (P = .005 for low-dose vs no rapamycin; P = .07 for standard-dose vs no rapamycin; P = .52 for low-dose vs standard-dose rapamycin; and P = .002 for combined low-dose and standard-dose rapamycin vs no rapamycin.).48 These differences were observed despite there being no significant change in total kidney volume in any of the groups. Patients on low-dose rapamycin had fewer adverse effects than those on standard dose and were more often able to continue therapy for the entire study. This, and the use of iothalamate GFR rather than eGFR to measure GFR, are believed to be the main reasons that low-dose effects were more pronounced than those with standard doses. One may speculate that rapamycin may have its effect on microcysts and cystogenic cells, resulting in stabilization of or improvement in renal function without detectable slowing in total kidney volume enlargement. Mechanisms for this possibility involve new concepts, as discussed below.
NEW CONCEPTS
Specialized cells also promote renal cyst formation
Specialized cells that promote cyst formation have been identified by Karihaloo et al49 in a mouse model of polycystic kidney disease. In this model, alternatively activated macrophages homed to cystic areas and promoted cyst growth. These findings suggested that interrupting the homing and proliferative signals of macrophages could be a therapeutic target for ADPKD. Although rapamycin can suppress macrophage proliferation by macrophage colony-stimulating factor and inhibit macrophage function,50 alternatively activated macrophages have not been specifically studied for rapamycin responsiveness.
More promising is evidence that CD133+ progenitor cells from human ADPKD kidneys—but not from normal human kidneys—form cysts in vitro and in severe combined immunodeficient mouse models.51 Treatment with rapamycin decreased proliferation of the de-differentiated CD133+ cells from ADPKD patients and reduced cystogenesis.51
Visible cysts are the tip of the iceberg
Using ADPKD nephrectomy specimens from eight patients, Grantham et al52 compared cyst counts by MRI and by histology and found that for every renal cyst detected by MRI, about 62 smaller cysts (< 0.9 mm) are present in the kidney. For a typical patient having an average of 587 cysts in both kidneys that are detectable by MRI, this means that more than 36,000 cysts are actually present, and MRI detects less than 2% of the total cysts present.
Although microcysts are too small to contribute much to total kidney volume, they can interfere with kidney function. Microcysts can reduce GFR in two major ways: by compressing microvasculature, tubules, and glomeruli in the cortex; or by blocking the drainage of multiple upstream nephrons when they form in or block medullary collecting ducts.52 Although the growth rates of microcysts less than 1 mm in size have not yet been measured, the adult combined growth rates of the renal cyst component is approximately 12% per year, with yearly individual cyst growth rates up to 71%, and with fetal cyst growth rates even higher for cysts larger than 7.0 mm.53 Before and during an accelerated growth period, microcysts may be susceptible to certain therapies that could first improve GFR and only later change measurable total kidney volume by slowing microcyst progression to macrocysts either directly or through specialized cells that may be sensitive to rapamycin.
CURRENT MANAGEMENT OF ADPKD
Blood pressure control is essential—but too low is not good. For adult patients with hypertension caused by ADPKD, an acceptable blood pressure range is 120–130/70–80 mm Hg. However, further information from recently published blood pressure guidelines54 and the results of the Halt Progression of Polycystic Kidney Disease (HALT-PKD) study to be reported in late 201455 may provide more precise ranges for blood pressure control in ADPKD.
Although substantial experimental evidence exists for the benefits of inhibiting the up-regulation of the renin-angiotensin-aldosterone system in ADPKD, clinical proof is not yet available to confirm that angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) are preferred therapy.55 This may be determined by results of the HALT-PKD study, due for release in late 2014.55
Controlling blood pressure should be done with caution. Patients with low GFRs whose blood pressure is too low tend to have a more rapid decline of GFR, as suggested in the Modification of Diet in Renal Disease (MDRD) study in 1995.56
Experimental evidence suggests that avoiding calcium channel blockers may be advisable. Yamaguchi et al34 found that calcium channel blockers worsen the calcium transport defect and convert tubular epithelial cells to a proliferative phenotype.34
High fluid intake (2,500–3,000 mL/day), because it suppresses vasopressin, may be useful if permitted by several factors such as the patient’s cardiopulmonary and renal and electrolyte status, other medications, and diet.31 The reader is referred to a detailed description of the precautions necessary when prescribing high water intake.31 Patients should have their fluid intake managed by a physician and their renal function and serum sodium and electrolytes monitored regularly in order to avoid hyponatremia. Severe hyponatremia has occurred in patients with ADPKD and impaired kidney function who drank excessive quantities of water. Cardiac and pulmonary complications from excessive fluid intake are also possible, especially with a low GFR and compromised cardiac function.
A low-sodium diet, if not a contributing factor in hyponatremia, can be used under physician direction in the management of hypertension as well as in the prevention of calcium oxalate kidney stones.
Caffeine should be avoided because it may interfere with the activity of the phosphodiesterase that is necessary for the catabolism of cAMP to 5′AMP.
A low-protein diet is of unproven benefit,56 but it is prudent to avoid high protein intake.57
Complications such as bleeding (into or from cysts), infection (urinary tract, kidney cysts, and liver cysts), kidney stones, and urinary tract obstruction should be treated promptly and may require hospitalization.
Regular symptom reviews and physical examinations need to be performed with nonrenal concerns also in mind, such as intracranial aneurysms and cardiac valve lesions.11,58
Formal genetic counseling and molecular testing are becoming more frequently indicated as more complex inheritance patterns arise.6–8,59
Renal replacement therapy in the form of dialysis or transplantation is usually available for the patient when end-stage renal disease occurs. In the largest study thus far, ADPKD patient survival with a kidney transplant was similar to that of patients without ADPKD (about 93% at 5 years), and from 5 years to 15 years death-censored graft survival was actually better.60 Thromboembolic events are more frequent after transplantation,27,60 but they may also occur before transplantation from a massive right kidney compressing the iliac vein or the inferior vena cava, or both, leading to thrombus formation.26
Investigational as well as standard drug studies have intensified. Results from a large randomized study in approximately 1,000 adult ADPKD patients that evaluated over 6 to 8 years the effects of ACE inhibition with or without ARB treatment of hypertension, at both usual and lower blood pressure ranges in those with good renal function, are expected in late 2014.55 Outcomes from a few small clinical studies, eg, one with long-acting somatostatin31,61 and one using low-dose rapamycin48 in adults with ADPKD, will require confirmation in large randomized placebo-controlled clinical studies. In a new 3-year randomized placebo-controlled study of 91 children and young adults (ages 8 to 22) with ADPKD and essentially normal renal function who continued treatment with lisinopril, the addition of pravastatin (20 mg or 40 mg daily based on age) resulted in a significant reduction in the number of patients (46% vs 68%, respectively, P = .03) experiencing a greater than 20% change (increase) in height-adjusted total kidney volume.62 Change in GFR was not reported,62 but an earlier 4-week study in 10 patients treated with simvastatin did show an increase in renal blood flow and GFR.63 Numerous other agents that lack human studies include some described in older experimental work (eg, amiloride,31,64 citrate31,65) and many others from a growing list of potential therapeutic targets.31,66–73 It must be emphasized that there is no FDA-approved medication specifically for the treatment of ADPKD.
Future specific treatments of ADPKD may also involve minimally toxic doses of combination or sequential therapy directed at precystic and then both micro- and macrocystic/cystic fluid expansion aspects of ADPKD.48,74 Unfortunately, at the present time there is no specific FDA-approved therapy for ADPKD.
- Torres VE, Harris PC. Mechanisms of disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol 2006; 2:40–55.
- Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int 2009; 76:149–168.
- United States Renal Data System. 2013 atlas of CKD & ESRD. Volume 2 - atlas ESRD:172. www.usrds.org/atlas.aspx. Accessed June 4, 2014.
- Barua M, Cil O, Paerson AD, et al. Family history of renal disease severity predicts the mutated gene in ADPKD. J Am Soc Nephrol 2009, 20:1833–1838.
- Harris PC, Bae KT, Rossetti S, et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2006; 17:3013–3019.
- Vujic M, Heyer CM, Ars E, et al. Incompletely penetrant PKD1 alleles mimic the renal manifestations of ARPKD. J Am Soc Nephrol 2010; 21:1097–1102.
- Harris PC. What is the role of somatic mutation in autosomal dominant polycystic kidney disease? J Am Soc Nephrol 2010; 21:1073–1076.
- Watnick T, He N, Wang K, et al. Mutations of PKD1 in ADPKD2 cysts suggest a pathogenic effect of trans-heterozygous mutations. Nat Genet 2000; 25:143–144.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994; 343:824–827.
- Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol 2009; 20:205–212.
- Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet 2007; 369:1287–1301.
- Bajwa ZH, Sial KA, Malik AB, Steinman TI. Pain patterns in patients with polycystic kidney disease. Kidney Int 2004; 66:1561–1569.
- Jouret F, Lhommel R, Beguin C, et al. Positron-emission computed tomography in cyst infection diagnosis in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2011; 6:1644–1650.
- Nishiura JL, Neves RF, Eloi SR, Cintra SM, Ajzen SA, Heilberg IP. Evaluation of nephrolithiasis in autosomal dominant polycystic kidney disease patients. Clin J Am Soc Nephrol 2009; 4:838–844.
- Hiura T, Yamazaki H, Saeki T, et al. Nephrotic syndrome and IgA nephropathy in polycystic kidney disease. Clin Exp Nephrol 2006; 10:136–139.
- Hossack KF, Leddy CL, Johnson AM, Schrier RW, Gabow PA. Echocardiographic findings in autosomal dominant polycystic kidney disease. N Engl J Med 1988; 319:907–912.
- Rossetti S, Chauveau D, Kubly V, et al. Association of mutation position in polycystic kidney disease 1 (PKD1) gene and development of a vascular phenotype. Lancet 2003; 361:2196–2201.
- Linn FH, Wijdicks EF, van der Graaf Y, Weerdesteyn-van Vliet FA, Bartelds AI, van Gijn J. Prospective study of sentinel headache in aneurismal subarachnoid haemorrhage. Lancet 1994; 344:590–593.
- Belz MM, Fick-Brosnahan GM, Hughes RL, et al. Recurrence of intracranial aneurysms in autosomal-dominant polycystic kidney disease. Kidney Int 2003; 63:1824–1830.
- Irazabal MV, Huston J, Kubly V, et al. Extended follow-up of unruptured intracranial aneurysms detected by presymptomatic screening in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2011; 6:1274–1285.
- Salman A-S, White PM, Counsell CE, et al; Scottish Audit of Intracranial Vascular Malformations Collaborators. Outcome after conservative management or intervention for unruptured brain arteriovenous malformations. JAMA 2014; 311:1661–1669.
- Vijay A, Vijay A, Pankaj P. Autosomal dominant polycystic kidney disease: a comprehensive review. Nephrourol Mon 2010; 2:172–192.
- Grantham JJ, Torres VE, Chapman AB, et al; CRISP Investigators. Volume progression in polycystic kidney disease. N Engl J Med 2006; 354:2122–2130.
- Bae KT, Grantham JJ. Imaging for the prognosis of autosomal dominant polycystic kidney disease. Nat Rev Nephrol 2010; 6:96–106.
- van den Dool SW, Wasser NM, de Fijter JW, Hoekstra J, van der Geest RJ. Functional renal volume: quantitative analysis at gadolinium-enhanced MR angiography—feasibility study in healthy potential kidney donors. Radiology 2005; 236:189–195.
- O’Sullivan DA, Torres VE, Heit JA, Liggett S, King BF. Compression of the inferior vena cava by right renal cysts: an unusual cause of IVC and/or iliofemoral thrombosis with pulmonary embolism in autosomal dominant polycystic kidney disease. Clin Nephrol 1998; 49:332–334.
- Tveit DP, Hypolite I, Bucci J, et al. Risk factors for hospitalizations resulting from pulmonary embolism after renal transplantation in the United States. J Nephrol 2001; 14:361–368.
- Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 2001; 7:151–156.
- Qian F, Germino GG. “Mistakes happen”: somatic mutation and disease. Am J Hum Genet 1997; 61:1000–1005.
- Takakura A, Contrino L, Zhou X, et al. Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum Mol Genet 2009; 18:2523–2531.
- Torres VE, Harris PC. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol 2014; 25:18–32.
- Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33:129–137.
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med 2011; 364:1533–1543.
- Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004; 279:40419–40430.
- Verghese E, Ricardo SD, Weidenfeld R, et al. Renal primary cilia lengthen after acute tubular necrosis. J Am Soc Nephrol 2009; 20:2147–2153.
- Wang X, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 2008; 19:102–108.
- Torres VE. Cyclic AMP, at the hub of the cystic cycle. Kidney Int 2004; 66:1283–1285.
- Nagao S, Nishii K, Katsuyama M, et al. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 2006; 17:2220–2227.
- Grantham JJ. Therapy for polycystic kidney disease? It’s water, stupid! J Am Soc Nephrol 2008; 19:1–7.
- Wang CJ, Creed C, Winklhofer FT, Grantham JJ. Water prescription in autosomal dominant polycystic kidney disease: a pilot study. Clin J Am Soc Nephrol 2011; 6:192–197.
- Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003; 9:1323–1326.
- Torres VE, Chapman AB, Devuyst O, et al; TEMPO 3:4 Trial Investigators. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2012; 367:2407–2418.
- Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A 2006; 103:5466–5471.
- Hartman TR, Liu D, Zilfou JT, et al. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum Mol Genet 2009; 18:161–163.
- Serra AL, Poster D, Kistler AD, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med 2010; 363:820–829.
- Walz G, Budde K, Mannaa M, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2010; 363:830–840. Errata in: N Engl J Med 2010; 363:1190 and N Engl J Med 2010; 363:1977.
- Walz G, Budde K, Eckardt K-U. mTOR inhibitors and autosomal dominant polycystic kidney disease (correspondence). N Engl J Med 2011; 364:287–288.
- Braun WE, Schold JD, Stephany BR, Spinko RA, Herfs BR. Low dose rapamycin (sirolimus) effects in autosomal dominant polycystic kidney disease: an open-label randomized control pilot study. Clin J Am Soc Nephrol 2014; 9:881–888.
- Karihaloo A, Koraishy F, Huen SC, et al. Macrophages promote cyst growth in polycystic kidney disease. J Am Soc Nephrol 2011; 22:1809–1814.
- Fox R, Nhan TQ, Law GL, Morris DR, Liles WC, Schwartz SM. PSGL-1 and mTOR regulate translation of ROCK-1 and physiological functions of macrophages. EMBO J 2007; 26:505–515. Erratum in: EMBO J 2007; 26:2605.
- Carvalhosa R, Deambrosis I, Carrera P, et al. Cystogenic potential of CD133+ progenitor cells of human polycystic kidneys. J Pathol 2011; 225:129–141.
- Grantham JJ, Mulamalla S, Grantham CJ, et al. Detected renal cysts are tips of the iceberg in adults with ADPKD. Clin J Am Soc Nephrol 2012; 7:1087–1093.
- Grantham JJ, Cook LT, Wetzel LH, Cadnapaphornchai MA, Bae KT. Evidence of extraordinary growth in the progressive enlargement of renal cysts. Clin J Am Soc Nephrol 2010; 5:889–896.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Chapman AB, Torres VE, Perrone RD, et al. The HALT polycystic kidney disease trials: design and implementation. Clin J Am Soc Nephrol 2010; 5:102–109.
- Klahr S, Breyer JA, Beck GJ, et al. Dietary protein restriction, blood pressure control, and the progression of polycystic kidney disease. Modification of Diet in Renal Disease Study Group. J Am Soc Nephrol 1995; 5:2037–2047.
- Thilly N. Low-protein diet in chronic kidney disease: from questions of effectiveness to those of feasibility. Nephrol Dial Transplant 2013; 28:2203–2205.
- Luciano RL, Dahl NK. Extra-renal manifestations of autosomal dominant polycystic kidney disease (ADPKD): considerations for routine screening and management. Nephrol Dial Transplant 2014; 29:247–254.
- Harris PC, Rossetti S. Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat Rev Nephrol 2010; 6:197–206.
- Jacquet A, Pallet N, Kessler M, et al. Outcomes of renal transplantation in patients with autosomal dominant polycystic kidney disease: a nationwide longitudinal study. Transpl Int 2011; 24:582–587.
- Ruggenenti P, Remuzzi A, Ondei P, et al. Safety and efficacy of long-acting somatostatin treatment in autosomal-dominant polycystic kidney disease. Kidney Int 2005; 68:206–216.
- Cadnapaphornchai MA, George DM, McFann K, et al. Effect of pravastatin on total kidney volume, left ventricular mass index, and microalbuminuria in pediatric autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2014; 9:889–896.
- van Dijk MA, Kamper AM, van Veen S, Souverjin JH, Blauw GJ. Effect of simvastatin on renal function in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2001; 16:2152–2157.
- Grantham JJ, Uchich M, Cragoe EL, et al. Chemical modification of cell proliferation and fluid secretion in renal cysts. Kidney Int 1989; 35:1379–1389.
- Tanner GA. Potassium citrate/citric acid intake improves renal function in rats with polycystic kidney disease. J Am Soc Nephrol 1998; 9:1242–1248.
- Belibi FA, Edelstein CL. Novel targets for the treatment of autosomal dominant polycystic kidney disease. Expert Opin Investig Drugs 2010; 19:315–328.
- Tao Y, Kim J, Yin Y, et al. VEGF receptor inhibition slows the progression of polycystic kidney disease. Kidney Int 2007; 72:1358–1366.
- Terryn S, Ho A, Beauwens R, Devuyst O. Fluid transport and cystogenesis in autosomal dominant polycystic kidney disease. Biochim Biophys Acta 2011; 1812:1314–1321.
- Thiagarajah JR, Verkman AS. CFTR inhibitors for treating diarrheal disease. Clin Pharmacol Ther 2012; 92:287–290.
- Boehn SN, Spahn S, Neudecker S, et al. Inhibition of Comt with tolcapone slows proression of polycystic kidney disease in the more severely affected PKD/Mhm (cy/+) substrain of the Hannover Sprague-Dawley rat. Nephrol Dial Transplant 2013; 28:2045–2058.
- Rees S, Kittikulsuth W, Roos K, Strait KA, Van Hoek A, Kohan DE. Adenylyl cyclase 6 deficiency ameliorates polycystic kidney disease. J Am Soc Nephrol 2014; 25:232–237.
- Buchholz B, Schley G, Faria D, et al. Hypoxia-inducible factor-1a causes renal cyst expansion through calcium-activated chloride secretion. J Am Soc Nephrol 2014; 25:465–474.
- Wallace DP, White C, Savinkova L, et al. Periostin promotes renal cyst growth and interstitial fibrosis in polycystic kidney disease. Kidney Int 2014; 85:845–854.
- Leuenroth SJ, Crews CM. Targeting cyst initiation in ADPKD. J Am Soc Nephrol 2009; 20:1–3.
- Torres VE, Harris PC. Mechanisms of disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol 2006; 2:40–55.
- Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int 2009; 76:149–168.
- United States Renal Data System. 2013 atlas of CKD & ESRD. Volume 2 - atlas ESRD:172. www.usrds.org/atlas.aspx. Accessed June 4, 2014.
- Barua M, Cil O, Paerson AD, et al. Family history of renal disease severity predicts the mutated gene in ADPKD. J Am Soc Nephrol 2009, 20:1833–1838.
- Harris PC, Bae KT, Rossetti S, et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2006; 17:3013–3019.
- Vujic M, Heyer CM, Ars E, et al. Incompletely penetrant PKD1 alleles mimic the renal manifestations of ARPKD. J Am Soc Nephrol 2010; 21:1097–1102.
- Harris PC. What is the role of somatic mutation in autosomal dominant polycystic kidney disease? J Am Soc Nephrol 2010; 21:1073–1076.
- Watnick T, He N, Wang K, et al. Mutations of PKD1 in ADPKD2 cysts suggest a pathogenic effect of trans-heterozygous mutations. Nat Genet 2000; 25:143–144.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994; 343:824–827.
- Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol 2009; 20:205–212.
- Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet 2007; 369:1287–1301.
- Bajwa ZH, Sial KA, Malik AB, Steinman TI. Pain patterns in patients with polycystic kidney disease. Kidney Int 2004; 66:1561–1569.
- Jouret F, Lhommel R, Beguin C, et al. Positron-emission computed tomography in cyst infection diagnosis in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2011; 6:1644–1650.
- Nishiura JL, Neves RF, Eloi SR, Cintra SM, Ajzen SA, Heilberg IP. Evaluation of nephrolithiasis in autosomal dominant polycystic kidney disease patients. Clin J Am Soc Nephrol 2009; 4:838–844.
- Hiura T, Yamazaki H, Saeki T, et al. Nephrotic syndrome and IgA nephropathy in polycystic kidney disease. Clin Exp Nephrol 2006; 10:136–139.
- Hossack KF, Leddy CL, Johnson AM, Schrier RW, Gabow PA. Echocardiographic findings in autosomal dominant polycystic kidney disease. N Engl J Med 1988; 319:907–912.
- Rossetti S, Chauveau D, Kubly V, et al. Association of mutation position in polycystic kidney disease 1 (PKD1) gene and development of a vascular phenotype. Lancet 2003; 361:2196–2201.
- Linn FH, Wijdicks EF, van der Graaf Y, Weerdesteyn-van Vliet FA, Bartelds AI, van Gijn J. Prospective study of sentinel headache in aneurismal subarachnoid haemorrhage. Lancet 1994; 344:590–593.
- Belz MM, Fick-Brosnahan GM, Hughes RL, et al. Recurrence of intracranial aneurysms in autosomal-dominant polycystic kidney disease. Kidney Int 2003; 63:1824–1830.
- Irazabal MV, Huston J, Kubly V, et al. Extended follow-up of unruptured intracranial aneurysms detected by presymptomatic screening in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2011; 6:1274–1285.
- Salman A-S, White PM, Counsell CE, et al; Scottish Audit of Intracranial Vascular Malformations Collaborators. Outcome after conservative management or intervention for unruptured brain arteriovenous malformations. JAMA 2014; 311:1661–1669.
- Vijay A, Vijay A, Pankaj P. Autosomal dominant polycystic kidney disease: a comprehensive review. Nephrourol Mon 2010; 2:172–192.
- Grantham JJ, Torres VE, Chapman AB, et al; CRISP Investigators. Volume progression in polycystic kidney disease. N Engl J Med 2006; 354:2122–2130.
- Bae KT, Grantham JJ. Imaging for the prognosis of autosomal dominant polycystic kidney disease. Nat Rev Nephrol 2010; 6:96–106.
- van den Dool SW, Wasser NM, de Fijter JW, Hoekstra J, van der Geest RJ. Functional renal volume: quantitative analysis at gadolinium-enhanced MR angiography—feasibility study in healthy potential kidney donors. Radiology 2005; 236:189–195.
- O’Sullivan DA, Torres VE, Heit JA, Liggett S, King BF. Compression of the inferior vena cava by right renal cysts: an unusual cause of IVC and/or iliofemoral thrombosis with pulmonary embolism in autosomal dominant polycystic kidney disease. Clin Nephrol 1998; 49:332–334.
- Tveit DP, Hypolite I, Bucci J, et al. Risk factors for hospitalizations resulting from pulmonary embolism after renal transplantation in the United States. J Nephrol 2001; 14:361–368.
- Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 2001; 7:151–156.
- Qian F, Germino GG. “Mistakes happen”: somatic mutation and disease. Am J Hum Genet 1997; 61:1000–1005.
- Takakura A, Contrino L, Zhou X, et al. Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum Mol Genet 2009; 18:2523–2531.
- Torres VE, Harris PC. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol 2014; 25:18–32.
- Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33:129–137.
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med 2011; 364:1533–1543.
- Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004; 279:40419–40430.
- Verghese E, Ricardo SD, Weidenfeld R, et al. Renal primary cilia lengthen after acute tubular necrosis. J Am Soc Nephrol 2009; 20:2147–2153.
- Wang X, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 2008; 19:102–108.
- Torres VE. Cyclic AMP, at the hub of the cystic cycle. Kidney Int 2004; 66:1283–1285.
- Nagao S, Nishii K, Katsuyama M, et al. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 2006; 17:2220–2227.
- Grantham JJ. Therapy for polycystic kidney disease? It’s water, stupid! J Am Soc Nephrol 2008; 19:1–7.
- Wang CJ, Creed C, Winklhofer FT, Grantham JJ. Water prescription in autosomal dominant polycystic kidney disease: a pilot study. Clin J Am Soc Nephrol 2011; 6:192–197.
- Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003; 9:1323–1326.
- Torres VE, Chapman AB, Devuyst O, et al; TEMPO 3:4 Trial Investigators. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2012; 367:2407–2418.
- Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A 2006; 103:5466–5471.
- Hartman TR, Liu D, Zilfou JT, et al. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum Mol Genet 2009; 18:161–163.
- Serra AL, Poster D, Kistler AD, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med 2010; 363:820–829.
- Walz G, Budde K, Mannaa M, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2010; 363:830–840. Errata in: N Engl J Med 2010; 363:1190 and N Engl J Med 2010; 363:1977.
- Walz G, Budde K, Eckardt K-U. mTOR inhibitors and autosomal dominant polycystic kidney disease (correspondence). N Engl J Med 2011; 364:287–288.
- Braun WE, Schold JD, Stephany BR, Spinko RA, Herfs BR. Low dose rapamycin (sirolimus) effects in autosomal dominant polycystic kidney disease: an open-label randomized control pilot study. Clin J Am Soc Nephrol 2014; 9:881–888.
- Karihaloo A, Koraishy F, Huen SC, et al. Macrophages promote cyst growth in polycystic kidney disease. J Am Soc Nephrol 2011; 22:1809–1814.
- Fox R, Nhan TQ, Law GL, Morris DR, Liles WC, Schwartz SM. PSGL-1 and mTOR regulate translation of ROCK-1 and physiological functions of macrophages. EMBO J 2007; 26:505–515. Erratum in: EMBO J 2007; 26:2605.
- Carvalhosa R, Deambrosis I, Carrera P, et al. Cystogenic potential of CD133+ progenitor cells of human polycystic kidneys. J Pathol 2011; 225:129–141.
- Grantham JJ, Mulamalla S, Grantham CJ, et al. Detected renal cysts are tips of the iceberg in adults with ADPKD. Clin J Am Soc Nephrol 2012; 7:1087–1093.
- Grantham JJ, Cook LT, Wetzel LH, Cadnapaphornchai MA, Bae KT. Evidence of extraordinary growth in the progressive enlargement of renal cysts. Clin J Am Soc Nephrol 2010; 5:889–896.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Chapman AB, Torres VE, Perrone RD, et al. The HALT polycystic kidney disease trials: design and implementation. Clin J Am Soc Nephrol 2010; 5:102–109.
- Klahr S, Breyer JA, Beck GJ, et al. Dietary protein restriction, blood pressure control, and the progression of polycystic kidney disease. Modification of Diet in Renal Disease Study Group. J Am Soc Nephrol 1995; 5:2037–2047.
- Thilly N. Low-protein diet in chronic kidney disease: from questions of effectiveness to those of feasibility. Nephrol Dial Transplant 2013; 28:2203–2205.
- Luciano RL, Dahl NK. Extra-renal manifestations of autosomal dominant polycystic kidney disease (ADPKD): considerations for routine screening and management. Nephrol Dial Transplant 2014; 29:247–254.
- Harris PC, Rossetti S. Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat Rev Nephrol 2010; 6:197–206.
- Jacquet A, Pallet N, Kessler M, et al. Outcomes of renal transplantation in patients with autosomal dominant polycystic kidney disease: a nationwide longitudinal study. Transpl Int 2011; 24:582–587.
- Ruggenenti P, Remuzzi A, Ondei P, et al. Safety and efficacy of long-acting somatostatin treatment in autosomal-dominant polycystic kidney disease. Kidney Int 2005; 68:206–216.
- Cadnapaphornchai MA, George DM, McFann K, et al. Effect of pravastatin on total kidney volume, left ventricular mass index, and microalbuminuria in pediatric autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2014; 9:889–896.
- van Dijk MA, Kamper AM, van Veen S, Souverjin JH, Blauw GJ. Effect of simvastatin on renal function in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2001; 16:2152–2157.
- Grantham JJ, Uchich M, Cragoe EL, et al. Chemical modification of cell proliferation and fluid secretion in renal cysts. Kidney Int 1989; 35:1379–1389.
- Tanner GA. Potassium citrate/citric acid intake improves renal function in rats with polycystic kidney disease. J Am Soc Nephrol 1998; 9:1242–1248.
- Belibi FA, Edelstein CL. Novel targets for the treatment of autosomal dominant polycystic kidney disease. Expert Opin Investig Drugs 2010; 19:315–328.
- Tao Y, Kim J, Yin Y, et al. VEGF receptor inhibition slows the progression of polycystic kidney disease. Kidney Int 2007; 72:1358–1366.
- Terryn S, Ho A, Beauwens R, Devuyst O. Fluid transport and cystogenesis in autosomal dominant polycystic kidney disease. Biochim Biophys Acta 2011; 1812:1314–1321.
- Thiagarajah JR, Verkman AS. CFTR inhibitors for treating diarrheal disease. Clin Pharmacol Ther 2012; 92:287–290.
- Boehn SN, Spahn S, Neudecker S, et al. Inhibition of Comt with tolcapone slows proression of polycystic kidney disease in the more severely affected PKD/Mhm (cy/+) substrain of the Hannover Sprague-Dawley rat. Nephrol Dial Transplant 2013; 28:2045–2058.
- Rees S, Kittikulsuth W, Roos K, Strait KA, Van Hoek A, Kohan DE. Adenylyl cyclase 6 deficiency ameliorates polycystic kidney disease. J Am Soc Nephrol 2014; 25:232–237.
- Buchholz B, Schley G, Faria D, et al. Hypoxia-inducible factor-1a causes renal cyst expansion through calcium-activated chloride secretion. J Am Soc Nephrol 2014; 25:465–474.
- Wallace DP, White C, Savinkova L, et al. Periostin promotes renal cyst growth and interstitial fibrosis in polycystic kidney disease. Kidney Int 2014; 85:845–854.
- Leuenroth SJ, Crews CM. Targeting cyst initiation in ADPKD. J Am Soc Nephrol 2009; 20:1–3.
KEY POINTS
- For at-risk patients in the previously difficult diagnostic group from 30 to 39 years of age, newer ultrasonographic criteria for diagnosing PKD1 and PKD2 now require a minimum total of three renal cysts.
- An intracranial aneurysm occurs in approximately 16% of ADPKD patients who have a family member with ADPKD plus an intracranial aneurysm or subarachnoid hemorrhage. Appropriate screening is warranted.
- Combined positron-emission and computed tomography helps identify infected renal or liver cysts and may uncover other unsuspected abdominal or pelvic infections.
- Cyst expansion and increasing total kidney volume might be slowed by increasing water intake to 2,500 to 3,000 mL per day, although formal documentation of this is not published. However, this must be done under a physician’s supervision because of possible adverse effects.
- Tolvaptan, a promising new drug for treating ADPKD, failed to receive US approval. Rapamycin is another potentially effective agent but has had mixed results in clinical trials.
Overcorrection of Hyponatremia
Q) A clinic patient of mine was recently admitted to the hospital with hyponatremia (serum sodium, 115 mEq/L). She was treated with 2 L of normal saline and discharged home 48 hours later, at her baseline mental status with a serum sodium level of 132 mEq/L. Two days later, she was readmitted for mental status changes, and MRI showed brain swelling. The neurologist stated this was a result of the initial treatment for her hyponatremia. How is this possible?
The cause-and-effect relationship between rapid correction of chronic hyponatremia and subsequent development of neurologic problems was discovered in the late 1970s. Central pontine and extrapontine myelinolysis (known as osmotic demyelination syndrome or ODS) is a neurologic condition that can occur from rapid sodium correction. It is diagnosed by MRI, which shows hyperintense lesions on T2-weighted images. Clinical signs include upper motor neuron signs, pseudobulbar palsy, spastic quadriparesis, and mental status changes ranging from mild confusion to coma.2
Treatment for hyponatremia should be guided by symptom management.2,3 If a patient is asymptomatic, a simple and effective strategy is to keep NPO for 24 hours, except for medications. Simple food and fluid restriction will likely increase the serum sodium level because of obligate solute losses and urinary electrolyte free water loss.2,4 While the first instinct is to feed these patients, as they often appear malnourished, this can cause a solute load leading to a too-rapid sodium correction. After 24 hours, if intake restriction is not effective, use 0.5% normal saline but with limited dosing orders, as usual saline dosing can cause too rapid a correction.2
For symptomatic patients (confusion, seizures, coma), the goal is to initially elevate sodium by 1 to 2 mEq/L per hour for the first two to three hours. Do not exceed 10 mEq/L in 24 hours or 18 mEq/L in 48 hours. Exceeding these limits puts patients at high risk for ODS. In fact, even when staying within these parameters, there is some risk for overcorrection. It is always better to go slowly.2,3
In the patient with hyponatremia due to low solute intake (eg, beer potomania), diuresis can start spontaneously after a period of food and fluid restriction. It can also be initiated with just a small amount of solute. For example, administering an IV antibiotic with a base solution of 100 mL of normal saline or a “banana bag” (an IV solution containing 0.5 to 1 L of normal saline with multivitamins/minerals that cause the fluid to be yellow) can produce several liters of diuresis.2 Once you open the floodgate, you can unintentionally cause too-rapid correction that could lead to ODS.
In chronic hyponatremic patients, low antidiuretic hormone (ADH) levels are often found; thus when a solute is introduced, there is little ADH in the system to protect against excessive water loss and electrolyte imbalance. At the same time, excessive water loss can translate to higher sodium levels and increase the risk for cerebral edema. If rapid diuresis occurs, an infusion of D5W (5% dextrose in water) to match the rate of urine output may prevent a rapid serum sodium level rise. Frequent monitoring of serum sodium levels is often necessary. In instances where ODS is already present, there are case studies of improved neurologic outcomes with reduction of serum sodium levels.2,3
While the treatment of hyponatremia at first glance seems straightforward—replace that which is lost—it can actually transform a seemingly simple problem into a complicated clinical course requiring intensive care, due to the need for frequent monitoring and intervention.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
Q) A clinic patient of mine was recently admitted to the hospital with hyponatremia (serum sodium, 115 mEq/L). She was treated with 2 L of normal saline and discharged home 48 hours later, at her baseline mental status with a serum sodium level of 132 mEq/L. Two days later, she was readmitted for mental status changes, and MRI showed brain swelling. The neurologist stated this was a result of the initial treatment for her hyponatremia. How is this possible?
The cause-and-effect relationship between rapid correction of chronic hyponatremia and subsequent development of neurologic problems was discovered in the late 1970s. Central pontine and extrapontine myelinolysis (known as osmotic demyelination syndrome or ODS) is a neurologic condition that can occur from rapid sodium correction. It is diagnosed by MRI, which shows hyperintense lesions on T2-weighted images. Clinical signs include upper motor neuron signs, pseudobulbar palsy, spastic quadriparesis, and mental status changes ranging from mild confusion to coma.2
Treatment for hyponatremia should be guided by symptom management.2,3 If a patient is asymptomatic, a simple and effective strategy is to keep NPO for 24 hours, except for medications. Simple food and fluid restriction will likely increase the serum sodium level because of obligate solute losses and urinary electrolyte free water loss.2,4 While the first instinct is to feed these patients, as they often appear malnourished, this can cause a solute load leading to a too-rapid sodium correction. After 24 hours, if intake restriction is not effective, use 0.5% normal saline but with limited dosing orders, as usual saline dosing can cause too rapid a correction.2
For symptomatic patients (confusion, seizures, coma), the goal is to initially elevate sodium by 1 to 2 mEq/L per hour for the first two to three hours. Do not exceed 10 mEq/L in 24 hours or 18 mEq/L in 48 hours. Exceeding these limits puts patients at high risk for ODS. In fact, even when staying within these parameters, there is some risk for overcorrection. It is always better to go slowly.2,3
In the patient with hyponatremia due to low solute intake (eg, beer potomania), diuresis can start spontaneously after a period of food and fluid restriction. It can also be initiated with just a small amount of solute. For example, administering an IV antibiotic with a base solution of 100 mL of normal saline or a “banana bag” (an IV solution containing 0.5 to 1 L of normal saline with multivitamins/minerals that cause the fluid to be yellow) can produce several liters of diuresis.2 Once you open the floodgate, you can unintentionally cause too-rapid correction that could lead to ODS.
In chronic hyponatremic patients, low antidiuretic hormone (ADH) levels are often found; thus when a solute is introduced, there is little ADH in the system to protect against excessive water loss and electrolyte imbalance. At the same time, excessive water loss can translate to higher sodium levels and increase the risk for cerebral edema. If rapid diuresis occurs, an infusion of D5W (5% dextrose in water) to match the rate of urine output may prevent a rapid serum sodium level rise. Frequent monitoring of serum sodium levels is often necessary. In instances where ODS is already present, there are case studies of improved neurologic outcomes with reduction of serum sodium levels.2,3
While the treatment of hyponatremia at first glance seems straightforward—replace that which is lost—it can actually transform a seemingly simple problem into a complicated clinical course requiring intensive care, due to the need for frequent monitoring and intervention.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.
Q) A clinic patient of mine was recently admitted to the hospital with hyponatremia (serum sodium, 115 mEq/L). She was treated with 2 L of normal saline and discharged home 48 hours later, at her baseline mental status with a serum sodium level of 132 mEq/L. Two days later, she was readmitted for mental status changes, and MRI showed brain swelling. The neurologist stated this was a result of the initial treatment for her hyponatremia. How is this possible?
The cause-and-effect relationship between rapid correction of chronic hyponatremia and subsequent development of neurologic problems was discovered in the late 1970s. Central pontine and extrapontine myelinolysis (known as osmotic demyelination syndrome or ODS) is a neurologic condition that can occur from rapid sodium correction. It is diagnosed by MRI, which shows hyperintense lesions on T2-weighted images. Clinical signs include upper motor neuron signs, pseudobulbar palsy, spastic quadriparesis, and mental status changes ranging from mild confusion to coma.2
Treatment for hyponatremia should be guided by symptom management.2,3 If a patient is asymptomatic, a simple and effective strategy is to keep NPO for 24 hours, except for medications. Simple food and fluid restriction will likely increase the serum sodium level because of obligate solute losses and urinary electrolyte free water loss.2,4 While the first instinct is to feed these patients, as they often appear malnourished, this can cause a solute load leading to a too-rapid sodium correction. After 24 hours, if intake restriction is not effective, use 0.5% normal saline but with limited dosing orders, as usual saline dosing can cause too rapid a correction.2
For symptomatic patients (confusion, seizures, coma), the goal is to initially elevate sodium by 1 to 2 mEq/L per hour for the first two to three hours. Do not exceed 10 mEq/L in 24 hours or 18 mEq/L in 48 hours. Exceeding these limits puts patients at high risk for ODS. In fact, even when staying within these parameters, there is some risk for overcorrection. It is always better to go slowly.2,3
In the patient with hyponatremia due to low solute intake (eg, beer potomania), diuresis can start spontaneously after a period of food and fluid restriction. It can also be initiated with just a small amount of solute. For example, administering an IV antibiotic with a base solution of 100 mL of normal saline or a “banana bag” (an IV solution containing 0.5 to 1 L of normal saline with multivitamins/minerals that cause the fluid to be yellow) can produce several liters of diuresis.2 Once you open the floodgate, you can unintentionally cause too-rapid correction that could lead to ODS.
In chronic hyponatremic patients, low antidiuretic hormone (ADH) levels are often found; thus when a solute is introduced, there is little ADH in the system to protect against excessive water loss and electrolyte imbalance. At the same time, excessive water loss can translate to higher sodium levels and increase the risk for cerebral edema. If rapid diuresis occurs, an infusion of D5W (5% dextrose in water) to match the rate of urine output may prevent a rapid serum sodium level rise. Frequent monitoring of serum sodium levels is often necessary. In instances where ODS is already present, there are case studies of improved neurologic outcomes with reduction of serum sodium levels.2,3
While the treatment of hyponatremia at first glance seems straightforward—replace that which is lost—it can actually transform a seemingly simple problem into a complicated clinical course requiring intensive care, due to the need for frequent monitoring and intervention.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.
Hyponatremia: Beer Potomania
Q) Recently, we had a patient admitted for hyponatremia with
a serum sodium level of 117 mEq/L. One of the hospitalists mentioned “beer potomania” in the differential. Not wanting to look dumb, I just agreed. What is beer potomania, and how is it related to low serum sodium?
Potomania is the excessive consumption of alcoholic beverages; beer potomania is used to refer to a dilutional hyponatremia caused by excessive consumption of beer.1 First recognized in 1971, this cause of hyponatremia is not the most common but should be in the differential if the patient is a heavy alcohol imbiber who presents with encephalopathy and low serum sodium.
When considering this diagnosis, keep in mind that hyponatremia is common among chronic alcoholics and can be due to conditions such as cirrhosis, congestive heart failure, syndrome of inappropriate antidiuretic hormone (SIADH) secretion, and hypovolemia. Less common but still belonging in the differential are pseudohyponatremia secondary to alcohol-induced severe hypertriglyceridemia and cerebral salt wasting syndrome.2,3
Beer potomania usually manifests as altered mental status, weakness, and gait disturbance with an average serum sodium concentration of 108 mEq/L.3 Other abnormal lab results consistent with this diagnosis include hypokalemia (mean potassium, 3 mEq/L) and low blood urea nitrogen and urine sodium levels.2,3 Another fairly consistent finding is a recent personal history of binge drinking (more than about 5 L, or 14 cans of beer, in 24 hours) and/or history of illness (vomiting, diarrhea) that predisposed the patient to a rapid drop in serum sodium levels.2
Based on the information presented thus far, you may ask, “Why haven’t I seen this diagnosed more often? There are a lot of beer bingers out there!” Good question. Let’s review the pathophysiology of beer potomania. When patients have poor protein and solute (food, electrolytes) intake, they can experience water intoxication with smaller-than-usual volumes of fluid. The kidneys need a certain amount of solute to facilitate free water clearance (the ability to clear excess fluid from the body). A lack of adequate solute results in a buildup of free water in the vascular system, leading to a dilutional hyponatremia.3
Free water clearance is dependent on both solute excretion and the ability to dilute urine. Someone consuming an average diet will excrete 600 to 900 mOsm/d of solute. This osmolar load in-cludes urea generated from protein (10 g of protein produces about 50 mOsm of urea), along with dietary sodium and potassium. The maximum capacity for urinary dilution is 50 mOsm/L. In a nutritionally sound person, a lot of fluid—about 20 L—would be required to overwhelm the body’s capacity for urinary dilution.2
However, when you don’t eat, the body starts to break down tissue to create energy to survive. This catabolism creates 100 to 150 mOsm/d of urea, allowing you to continue to appropriately excrete a moderate amount of fluid in spite of poor solute intake ... as long as you are not drinking excessive amounts of water.5
Alcoholics get a moderate amount of their calories via beer consumption and do not experience this endogenous protein breakdown or its resultant low urea/solute level. With low solute intake, dramatically lower fluid intake (about 14 cans of beer) will overwhelm the kidneys’ ability to clear excess free water in the body.2 Fortunately, most heavy beer drinkers continue to eat at least modestly, which is sufficient to avoid this rare type of hyponatremia. Chronic alcoholics who go on a drinking binge beyond their normal baseline alcohol consumption, or who develop a flulike illness that causes electrolyte depletion (via diarrhea or vomiting), are at higher risk for beer potomania.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
Q) Recently, we had a patient admitted for hyponatremia with
a serum sodium level of 117 mEq/L. One of the hospitalists mentioned “beer potomania” in the differential. Not wanting to look dumb, I just agreed. What is beer potomania, and how is it related to low serum sodium?
Potomania is the excessive consumption of alcoholic beverages; beer potomania is used to refer to a dilutional hyponatremia caused by excessive consumption of beer.1 First recognized in 1971, this cause of hyponatremia is not the most common but should be in the differential if the patient is a heavy alcohol imbiber who presents with encephalopathy and low serum sodium.
When considering this diagnosis, keep in mind that hyponatremia is common among chronic alcoholics and can be due to conditions such as cirrhosis, congestive heart failure, syndrome of inappropriate antidiuretic hormone (SIADH) secretion, and hypovolemia. Less common but still belonging in the differential are pseudohyponatremia secondary to alcohol-induced severe hypertriglyceridemia and cerebral salt wasting syndrome.2,3
Beer potomania usually manifests as altered mental status, weakness, and gait disturbance with an average serum sodium concentration of 108 mEq/L.3 Other abnormal lab results consistent with this diagnosis include hypokalemia (mean potassium, 3 mEq/L) and low blood urea nitrogen and urine sodium levels.2,3 Another fairly consistent finding is a recent personal history of binge drinking (more than about 5 L, or 14 cans of beer, in 24 hours) and/or history of illness (vomiting, diarrhea) that predisposed the patient to a rapid drop in serum sodium levels.2
Based on the information presented thus far, you may ask, “Why haven’t I seen this diagnosed more often? There are a lot of beer bingers out there!” Good question. Let’s review the pathophysiology of beer potomania. When patients have poor protein and solute (food, electrolytes) intake, they can experience water intoxication with smaller-than-usual volumes of fluid. The kidneys need a certain amount of solute to facilitate free water clearance (the ability to clear excess fluid from the body). A lack of adequate solute results in a buildup of free water in the vascular system, leading to a dilutional hyponatremia.3
Free water clearance is dependent on both solute excretion and the ability to dilute urine. Someone consuming an average diet will excrete 600 to 900 mOsm/d of solute. This osmolar load in-cludes urea generated from protein (10 g of protein produces about 50 mOsm of urea), along with dietary sodium and potassium. The maximum capacity for urinary dilution is 50 mOsm/L. In a nutritionally sound person, a lot of fluid—about 20 L—would be required to overwhelm the body’s capacity for urinary dilution.2
However, when you don’t eat, the body starts to break down tissue to create energy to survive. This catabolism creates 100 to 150 mOsm/d of urea, allowing you to continue to appropriately excrete a moderate amount of fluid in spite of poor solute intake ... as long as you are not drinking excessive amounts of water.5
Alcoholics get a moderate amount of their calories via beer consumption and do not experience this endogenous protein breakdown or its resultant low urea/solute level. With low solute intake, dramatically lower fluid intake (about 14 cans of beer) will overwhelm the kidneys’ ability to clear excess free water in the body.2 Fortunately, most heavy beer drinkers continue to eat at least modestly, which is sufficient to avoid this rare type of hyponatremia. Chronic alcoholics who go on a drinking binge beyond their normal baseline alcohol consumption, or who develop a flulike illness that causes electrolyte depletion (via diarrhea or vomiting), are at higher risk for beer potomania.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.
Q) Recently, we had a patient admitted for hyponatremia with
a serum sodium level of 117 mEq/L. One of the hospitalists mentioned “beer potomania” in the differential. Not wanting to look dumb, I just agreed. What is beer potomania, and how is it related to low serum sodium?
Potomania is the excessive consumption of alcoholic beverages; beer potomania is used to refer to a dilutional hyponatremia caused by excessive consumption of beer.1 First recognized in 1971, this cause of hyponatremia is not the most common but should be in the differential if the patient is a heavy alcohol imbiber who presents with encephalopathy and low serum sodium.
When considering this diagnosis, keep in mind that hyponatremia is common among chronic alcoholics and can be due to conditions such as cirrhosis, congestive heart failure, syndrome of inappropriate antidiuretic hormone (SIADH) secretion, and hypovolemia. Less common but still belonging in the differential are pseudohyponatremia secondary to alcohol-induced severe hypertriglyceridemia and cerebral salt wasting syndrome.2,3
Beer potomania usually manifests as altered mental status, weakness, and gait disturbance with an average serum sodium concentration of 108 mEq/L.3 Other abnormal lab results consistent with this diagnosis include hypokalemia (mean potassium, 3 mEq/L) and low blood urea nitrogen and urine sodium levels.2,3 Another fairly consistent finding is a recent personal history of binge drinking (more than about 5 L, or 14 cans of beer, in 24 hours) and/or history of illness (vomiting, diarrhea) that predisposed the patient to a rapid drop in serum sodium levels.2
Based on the information presented thus far, you may ask, “Why haven’t I seen this diagnosed more often? There are a lot of beer bingers out there!” Good question. Let’s review the pathophysiology of beer potomania. When patients have poor protein and solute (food, electrolytes) intake, they can experience water intoxication with smaller-than-usual volumes of fluid. The kidneys need a certain amount of solute to facilitate free water clearance (the ability to clear excess fluid from the body). A lack of adequate solute results in a buildup of free water in the vascular system, leading to a dilutional hyponatremia.3
Free water clearance is dependent on both solute excretion and the ability to dilute urine. Someone consuming an average diet will excrete 600 to 900 mOsm/d of solute. This osmolar load in-cludes urea generated from protein (10 g of protein produces about 50 mOsm of urea), along with dietary sodium and potassium. The maximum capacity for urinary dilution is 50 mOsm/L. In a nutritionally sound person, a lot of fluid—about 20 L—would be required to overwhelm the body’s capacity for urinary dilution.2
However, when you don’t eat, the body starts to break down tissue to create energy to survive. This catabolism creates 100 to 150 mOsm/d of urea, allowing you to continue to appropriately excrete a moderate amount of fluid in spite of poor solute intake ... as long as you are not drinking excessive amounts of water.5
Alcoholics get a moderate amount of their calories via beer consumption and do not experience this endogenous protein breakdown or its resultant low urea/solute level. With low solute intake, dramatically lower fluid intake (about 14 cans of beer) will overwhelm the kidneys’ ability to clear excess free water in the body.2 Fortunately, most heavy beer drinkers continue to eat at least modestly, which is sufficient to avoid this rare type of hyponatremia. Chronic alcoholics who go on a drinking binge beyond their normal baseline alcohol consumption, or who develop a flulike illness that causes electrolyte depletion (via diarrhea or vomiting), are at higher risk for beer potomania.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.
Early elimination of cyclosporine after heart transplant has renal benefit
SAN FRANCISCO – Use of an everolimus-containing regimen with early stopping of cyclosporine after de novo heart transplantation improves renal function and reduces cardiac allograft vasculopathy, without compromising graft outcomes, new data suggest.
These was among key findings of the randomized, open-label SCHEDULE (Scandinavian Heart Transplant Everolimus De Novo Study with Early Calcineurin Inhibitor Avoidance) reported at the 2014 World Transplant Congress.
"Renal dysfunction and cardiac allograft vasculopathy are markers for increased morbidity and mortality after heart transplantation," lead author Dr. Vilborg Sigurdardottir commented when introducing the study.

Patients in the trial were randomized evenly to a three-drug regimen containing the calcineurin inhibitor cyclosporine (Sandimmune) or to a four-drug regimen also containing the mTOR inhibitor everolimus (Zortress) with discontinuation of cyclosporine at week 7-11. Everolimus is currently approved by the Food and Drug Administration to prevent graft rejection in kidney and liver transplant recipients and, under another brand name, to treat some cancers.
Measured glomerular filtration rate (GFR) at 12 months, the trial’s primary outcome, was 30% better in the everolimus group than in the cyclosporine group (79.8 vs. 61.5 mL/min per 1.73 m2; P less than .001), according to results presented at the congress and recently published (Am. J. Transplant. 2014;14:1828-38).
The urinary albumin-creatinine ratio was higher in the everolimus group, but none of the patients had nephrotic levels of proteinuria.
Rates of adverse events were similar, with the exception that the everolimus group had a lower rate of cytomegalovirus infection (5% vs. 30%) and a higher rate of pneumonia (12% vs. 3%), Dr. Sigurdardottir reported at the congress, which was sponsored by the American Society of Transplant Surgeons.
The incidence of biopsy-proven acute rejection of at least grade 2R was greater with everolimus (40% vs. 18%, P = .01). However, at 12 months, the groups did not differ with respect to left ventricular function as assessed by echocardiography and biomarkers, and, in a cardiac reserve substudy, with respect to cardiac output and pulmonary capillary wedge pressure.
The incidence of cardiac allograft vasculopathy, defined as a mean media-intima thickness of at least 0.5 mm on intravascular ultrasound (IVUS), was lower in the everolimus group (51% vs. 65%, P less than .01), and progression assessed as the change in percent atheroma volume was slower in that group.
"Everolimus initiation and early cyclosporine elimination in de novo heart transplant recipients showed a highly significant improvement of renal function in terms of measured GFR, a reduced incidence of cytomegalovirus [a confirmatory result of previous large-scale studies], similar numbers of adverse and serious adverse events, and an increased incidence of treated acute rejection, however, without hemodynamic compromise and with preserved cardiac function and preserved cardiac reserve," concluded Dr. Sigurdardottir, who is medical director of heart transplantation at the Transplant Institute, Sahlgrenska University Hospital, Gothenburg, Sweden. "We saw also favorable coronary remodeling and less graft vasculopathy, as previously shown."
Among patients whose donor hearts had such disease, the increase in media-intima thickness and percent atheroma volume was less with everolimus than with cyclosporine, Dr. Sigurdardottir said. "Interestingly, we saw here that the total atheroma volume decreased between baseline and 12 months in the everolimus group in the patients who had preexisting donor disease."
An attendee from Norway said, "I am a nephrologist, and if I were to get a new heart, I’d rather have a GFR of 61 and no rejection than a GFR of 73 with rejection. Have you looked at the development of donor-specific antibodies in the ones who had rejection, because I’d like to live for more than a year – I’d like to live 3 years or 5 years or 10 years."
"You are absolutely right. At the time of transplantation, we would be looking at the acute problems, and we often see the kidney dysfunction, so we want to do something about that. But of course these studies need to tell us how patients fare longer term," Dr. Sigurdardottir agreed. None of the patients were found to have donor-specific antibodies, but the trial protocol did not mandate routine measurement, she said.
An attendee from Los Angeles commented, "We tried to do CNI [calcineurin inhibitor] weaning in 2006 and had hemodynamically compromised rejection. Now, I congratulate you on being innovative and having quadruple therapy from the get-go and then taking off the CNI. But the issue of increased rejection is important because ISHLT [International Society for Heart and Lung Transplantation] data show that that does lead to poorer outcome. It is countered by your improvement in renal function, but also your IVUS result, I think, is very important as well."
"Rejection is an important issue, but it is a common issue after transplantation. It was usually manageable. Since we didn’t see any hemodynamic compromise, it was up to each investigator to evaluate what to do. There were nine patients who converted to combination therapy," Dr. Sigurdardottir reported. "The future needs to tell us what the relevance of this rejection is, and we will do a follow-up at 3 and 5 years."
Dr. Sigurdardottir disclosed no relevant conflicts of interest. The trial was sponsored by Novartis, manufacturer of everolimus.
SAN FRANCISCO – Use of an everolimus-containing regimen with early stopping of cyclosporine after de novo heart transplantation improves renal function and reduces cardiac allograft vasculopathy, without compromising graft outcomes, new data suggest.
These was among key findings of the randomized, open-label SCHEDULE (Scandinavian Heart Transplant Everolimus De Novo Study with Early Calcineurin Inhibitor Avoidance) reported at the 2014 World Transplant Congress.
"Renal dysfunction and cardiac allograft vasculopathy are markers for increased morbidity and mortality after heart transplantation," lead author Dr. Vilborg Sigurdardottir commented when introducing the study.
Patients in the trial were randomized evenly to a three-drug regimen containing the calcineurin inhibitor cyclosporine (Sandimmune) or to a four-drug regimen also containing the mTOR inhibitor everolimus (Zortress) with discontinuation of cyclosporine at week 7-11. Everolimus is currently approved by the Food and Drug Administration to prevent graft rejection in kidney and liver transplant recipients and, under another brand name, to treat some cancers.
Measured glomerular filtration rate (GFR) at 12 months, the trial’s primary outcome, was 30% better in the everolimus group than in the cyclosporine group (79.8 vs. 61.5 mL/min per 1.73 m2; P less than .001), according to results presented at the congress and recently published (Am. J. Transplant. 2014;14:1828-38).
The urinary albumin-creatinine ratio was higher in the everolimus group, but none of the patients had nephrotic levels of proteinuria.
Rates of adverse events were similar, with the exception that the everolimus group had a lower rate of cytomegalovirus infection (5% vs. 30%) and a higher rate of pneumonia (12% vs. 3%), Dr. Sigurdardottir reported at the congress, which was sponsored by the American Society of Transplant Surgeons.
The incidence of biopsy-proven acute rejection of at least grade 2R was greater with everolimus (40% vs. 18%, P = .01). However, at 12 months, the groups did not differ with respect to left ventricular function as assessed by echocardiography and biomarkers, and, in a cardiac reserve substudy, with respect to cardiac output and pulmonary capillary wedge pressure.
The incidence of cardiac allograft vasculopathy, defined as a mean media-intima thickness of at least 0.5 mm on intravascular ultrasound (IVUS), was lower in the everolimus group (51% vs. 65%, P less than .01), and progression assessed as the change in percent atheroma volume was slower in that group.
"Everolimus initiation and early cyclosporine elimination in de novo heart transplant recipients showed a highly significant improvement of renal function in terms of measured GFR, a reduced incidence of cytomegalovirus [a confirmatory result of previous large-scale studies], similar numbers of adverse and serious adverse events, and an increased incidence of treated acute rejection, however, without hemodynamic compromise and with preserved cardiac function and preserved cardiac reserve," concluded Dr. Sigurdardottir, who is medical director of heart transplantation at the Transplant Institute, Sahlgrenska University Hospital, Gothenburg, Sweden. "We saw also favorable coronary remodeling and less graft vasculopathy, as previously shown."
Among patients whose donor hearts had such disease, the increase in media-intima thickness and percent atheroma volume was less with everolimus than with cyclosporine, Dr. Sigurdardottir said. "Interestingly, we saw here that the total atheroma volume decreased between baseline and 12 months in the everolimus group in the patients who had preexisting donor disease."
An attendee from Norway said, "I am a nephrologist, and if I were to get a new heart, I’d rather have a GFR of 61 and no rejection than a GFR of 73 with rejection. Have you looked at the development of donor-specific antibodies in the ones who had rejection, because I’d like to live for more than a year – I’d like to live 3 years or 5 years or 10 years."
"You are absolutely right. At the time of transplantation, we would be looking at the acute problems, and we often see the kidney dysfunction, so we want to do something about that. But of course these studies need to tell us how patients fare longer term," Dr. Sigurdardottir agreed. None of the patients were found to have donor-specific antibodies, but the trial protocol did not mandate routine measurement, she said.
An attendee from Los Angeles commented, "We tried to do CNI [calcineurin inhibitor] weaning in 2006 and had hemodynamically compromised rejection. Now, I congratulate you on being innovative and having quadruple therapy from the get-go and then taking off the CNI. But the issue of increased rejection is important because ISHLT [International Society for Heart and Lung Transplantation] data show that that does lead to poorer outcome. It is countered by your improvement in renal function, but also your IVUS result, I think, is very important as well."
"Rejection is an important issue, but it is a common issue after transplantation. It was usually manageable. Since we didn’t see any hemodynamic compromise, it was up to each investigator to evaluate what to do. There were nine patients who converted to combination therapy," Dr. Sigurdardottir reported. "The future needs to tell us what the relevance of this rejection is, and we will do a follow-up at 3 and 5 years."
Dr. Sigurdardottir disclosed no relevant conflicts of interest. The trial was sponsored by Novartis, manufacturer of everolimus.
SAN FRANCISCO – Use of an everolimus-containing regimen with early stopping of cyclosporine after de novo heart transplantation improves renal function and reduces cardiac allograft vasculopathy, without compromising graft outcomes, new data suggest.
These was among key findings of the randomized, open-label SCHEDULE (Scandinavian Heart Transplant Everolimus De Novo Study with Early Calcineurin Inhibitor Avoidance) reported at the 2014 World Transplant Congress.
"Renal dysfunction and cardiac allograft vasculopathy are markers for increased morbidity and mortality after heart transplantation," lead author Dr. Vilborg Sigurdardottir commented when introducing the study.
Patients in the trial were randomized evenly to a three-drug regimen containing the calcineurin inhibitor cyclosporine (Sandimmune) or to a four-drug regimen also containing the mTOR inhibitor everolimus (Zortress) with discontinuation of cyclosporine at week 7-11. Everolimus is currently approved by the Food and Drug Administration to prevent graft rejection in kidney and liver transplant recipients and, under another brand name, to treat some cancers.
Measured glomerular filtration rate (GFR) at 12 months, the trial’s primary outcome, was 30% better in the everolimus group than in the cyclosporine group (79.8 vs. 61.5 mL/min per 1.73 m2; P less than .001), according to results presented at the congress and recently published (Am. J. Transplant. 2014;14:1828-38).
The urinary albumin-creatinine ratio was higher in the everolimus group, but none of the patients had nephrotic levels of proteinuria.
Rates of adverse events were similar, with the exception that the everolimus group had a lower rate of cytomegalovirus infection (5% vs. 30%) and a higher rate of pneumonia (12% vs. 3%), Dr. Sigurdardottir reported at the congress, which was sponsored by the American Society of Transplant Surgeons.
The incidence of biopsy-proven acute rejection of at least grade 2R was greater with everolimus (40% vs. 18%, P = .01). However, at 12 months, the groups did not differ with respect to left ventricular function as assessed by echocardiography and biomarkers, and, in a cardiac reserve substudy, with respect to cardiac output and pulmonary capillary wedge pressure.
The incidence of cardiac allograft vasculopathy, defined as a mean media-intima thickness of at least 0.5 mm on intravascular ultrasound (IVUS), was lower in the everolimus group (51% vs. 65%, P less than .01), and progression assessed as the change in percent atheroma volume was slower in that group.
"Everolimus initiation and early cyclosporine elimination in de novo heart transplant recipients showed a highly significant improvement of renal function in terms of measured GFR, a reduced incidence of cytomegalovirus [a confirmatory result of previous large-scale studies], similar numbers of adverse and serious adverse events, and an increased incidence of treated acute rejection, however, without hemodynamic compromise and with preserved cardiac function and preserved cardiac reserve," concluded Dr. Sigurdardottir, who is medical director of heart transplantation at the Transplant Institute, Sahlgrenska University Hospital, Gothenburg, Sweden. "We saw also favorable coronary remodeling and less graft vasculopathy, as previously shown."
Among patients whose donor hearts had such disease, the increase in media-intima thickness and percent atheroma volume was less with everolimus than with cyclosporine, Dr. Sigurdardottir said. "Interestingly, we saw here that the total atheroma volume decreased between baseline and 12 months in the everolimus group in the patients who had preexisting donor disease."
An attendee from Norway said, "I am a nephrologist, and if I were to get a new heart, I’d rather have a GFR of 61 and no rejection than a GFR of 73 with rejection. Have you looked at the development of donor-specific antibodies in the ones who had rejection, because I’d like to live for more than a year – I’d like to live 3 years or 5 years or 10 years."
"You are absolutely right. At the time of transplantation, we would be looking at the acute problems, and we often see the kidney dysfunction, so we want to do something about that. But of course these studies need to tell us how patients fare longer term," Dr. Sigurdardottir agreed. None of the patients were found to have donor-specific antibodies, but the trial protocol did not mandate routine measurement, she said.
An attendee from Los Angeles commented, "We tried to do CNI [calcineurin inhibitor] weaning in 2006 and had hemodynamically compromised rejection. Now, I congratulate you on being innovative and having quadruple therapy from the get-go and then taking off the CNI. But the issue of increased rejection is important because ISHLT [International Society for Heart and Lung Transplantation] data show that that does lead to poorer outcome. It is countered by your improvement in renal function, but also your IVUS result, I think, is very important as well."
"Rejection is an important issue, but it is a common issue after transplantation. It was usually manageable. Since we didn’t see any hemodynamic compromise, it was up to each investigator to evaluate what to do. There were nine patients who converted to combination therapy," Dr. Sigurdardottir reported. "The future needs to tell us what the relevance of this rejection is, and we will do a follow-up at 3 and 5 years."
Dr. Sigurdardottir disclosed no relevant conflicts of interest. The trial was sponsored by Novartis, manufacturer of everolimus.
FROM THE 2014 WORLD TRANSPLANT CONGRESS
Key clinical point: For post–heart transplant patients, early cessation of cyclosporine when using an everolimus-containing regimen appears to be safe and did not compromise graft outcomes.
Major finding: Compared with patients continued on cyclosporine, patients taken off this agent at 7-11 weeks had a 30% better measured glomerular filtration rate at 12 months.
Data source: A randomized, open-label trial of 115 patients undergoing de novo heart transplantation
Disclosures: Dr. Sigurdardottir disclosed no relevant conflicts of interest. The trial was sponsored by Novartis, manufacturer of everolimus.

Primary Prevention of Diabetic Kidney Disease: Thumbs Up/Down
LAS VEGAS – Contrary to conventional wisdom, neither ACE inhibitors nor angiotensin receptor blockers have any role to play in primary prevention of diabetic kidney disease, according to Dr. Robert C. Stanton, chief of nephrology at the Harvard University’s Joslin Diabetes Center, Boston.
"I don’t see any unique indication for ACE inhibitors and ARBs for the primary prevention of kidney disease in diabetic patients, especially given that around 70% of diabetes patients will never develop kidney disease. They’re perfectly fine blood pressure pills. But as a magic kidney disease prevention drug, I don’t see any evidence for that. Of course, patients with proteinuria are another issue entirely. Those drugs absolutely are beneficial in that setting," he said at a meeting sponsored by the National Kidney Foundation.
When Dr. Stanton polled his audience electronically during the course of his talk, however, the majority of physicians indicated that they believe ACE inhibitors and ARBs are indeed useful for primary prevention of diabetic kidney disease. The evidence, Dr. Stanton emphasized, shows otherwise.
For example, a well-conducted, randomized, multicenter, placebo-controlled, 5-year clinical trial showed no benefit for enalapril or losartan in preventing kidney disease in patients with type 1 diabetes (N. Engl. J. Med. 2009;361:40-51). And three randomized controlled trials showed no primary preventive benefit for candesartan in more than 5,000 patients with type 1 or type 2 diabetes (Ann. Intern. Med. 2009;151:11-20).
Dr. Stanton noted that lots of other interventions have been proposed for the primary prevention of kidney disease in diabetes patients. Some are supported by solid evidence of benefit, others are not.
Here is his view of the preventive landscape:
• Intensive blood glucose control. "This is the easy one," he said. "A lot of us in the diabetes world feel that a hemoglobin A1c of 7% is the appropriate target for preventing many complications. It’s a reasonable target and should be achieved whether you’re talking about type 1 or type 2 patients."
The nephrologist noted that recent 25-year follow-up data from the landmark Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications study showed that fully 18 years after the intervention ended, patients assigned to intensive blood glucose control still showed highly impressive 50% reductions in the cumulative incidence of both microalbuminuria and end-stage renal disease compared with patients placed on less intensive control (Diabetes Care 2014;37:24-30).
• Smoking cessation. Smoking has been linked to a several-fold increased risk of diabetic kidney disease. "I think of diabetes as an endothelial cell disease, and smoking is the greatest endothelial cell poison we’ve come up with. So stopping smoking is something well worth doing," Dr. Stanton said.
• Blood pressure control. No question exists regarding its renoprotective effect. But recent guidelines are dizzyingly all over the map in terms of target pressure recommendations.
"I’m getting a major headache reading these articles right now. I can show you the data. Good luck! I personally like a target of 130/80 mm Hg or less, particularly when it’s not that hard to get there. But I’d let you decide what particular target you favor," he said.
He prefers 130/80 mm Hg as a target blood pressure for primary prevention of diabetic kidney disease in large part because of a meta-analysis showing that it was associated with a 10% reduction in the risk of developing microalbuminuria and an 11% decrease in end-stage renal disease (PloS Med 2012;9(8):e1001293).
• Weight loss. The growing bariatric surgery literature supports weight loss as a primary preventive strategy.
• Protein intake. There is no role for a low-protein diet – say, less than 0.8 g/kg per day – for primary prevention of kidney disease in diabetes patients. And Dr. Stanton believes a high-protein diet in the range of more than 1.5 or 2 g/kg per day is best avoided in patients with diabetes, although he stressed that the evidence on this score remains sketchy.
Still, "I would not go on a body-building diet or an Atkins-type diet," he cautioned.
• Targeting glomerular hyperfiltration. Studies have shown conflicting results. "For me, there’s no clear role for targeting hyperfiltration," said Dr. Stanton, who cited a comprehensive review that he finds persuasive (Diabetologia 2010;53:2093-104).
The key to developing more effective primary prevention strategies, according to Dr. Stanton, will be first to establish markers that clearly identify the 30% or so of diabetes patients who will go on to develop renal disease, then test novel interventions specifically in that high-risk group.
Promising biomarkers include circulating tumor necrosis factor alpha receptor levels, von Willebrand factor, monocyte chemoattractant factor, asymmetrical dimethylarginine, interleukin-6 and -8, and Fas receptor.
For example, one study showed that patients with type 2 diabetes in the top quartile for circulating TNF receptor 1 had a cumulative 12-year incidence of end-stage renal disease of 54%, compared to just 3% in patients in the other quartiles (J. Am. Soc. Nephrol. 2012;23:507-15).
"Lots of companies are looking at these now. These markers may be coming our way as indicators of people with diabetes who are likely to progress to kidney disease," Dr. Stanton said.
He reported serving as a consultant to Boehringer Ingelheim.
LAS VEGAS – Contrary to conventional wisdom, neither ACE inhibitors nor angiotensin receptor blockers have any role to play in primary prevention of diabetic kidney disease, according to Dr. Robert C. Stanton, chief of nephrology at the Harvard University’s Joslin Diabetes Center, Boston.
"I don’t see any unique indication for ACE inhibitors and ARBs for the primary prevention of kidney disease in diabetic patients, especially given that around 70% of diabetes patients will never develop kidney disease. They’re perfectly fine blood pressure pills. But as a magic kidney disease prevention drug, I don’t see any evidence for that. Of course, patients with proteinuria are another issue entirely. Those drugs absolutely are beneficial in that setting," he said at a meeting sponsored by the National Kidney Foundation.
When Dr. Stanton polled his audience electronically during the course of his talk, however, the majority of physicians indicated that they believe ACE inhibitors and ARBs are indeed useful for primary prevention of diabetic kidney disease. The evidence, Dr. Stanton emphasized, shows otherwise.
For example, a well-conducted, randomized, multicenter, placebo-controlled, 5-year clinical trial showed no benefit for enalapril or losartan in preventing kidney disease in patients with type 1 diabetes (N. Engl. J. Med. 2009;361:40-51). And three randomized controlled trials showed no primary preventive benefit for candesartan in more than 5,000 patients with type 1 or type 2 diabetes (Ann. Intern. Med. 2009;151:11-20).
Dr. Stanton noted that lots of other interventions have been proposed for the primary prevention of kidney disease in diabetes patients. Some are supported by solid evidence of benefit, others are not.
Here is his view of the preventive landscape:
• Intensive blood glucose control. "This is the easy one," he said. "A lot of us in the diabetes world feel that a hemoglobin A1c of 7% is the appropriate target for preventing many complications. It’s a reasonable target and should be achieved whether you’re talking about type 1 or type 2 patients."
The nephrologist noted that recent 25-year follow-up data from the landmark Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications study showed that fully 18 years after the intervention ended, patients assigned to intensive blood glucose control still showed highly impressive 50% reductions in the cumulative incidence of both microalbuminuria and end-stage renal disease compared with patients placed on less intensive control (Diabetes Care 2014;37:24-30).
• Smoking cessation. Smoking has been linked to a several-fold increased risk of diabetic kidney disease. "I think of diabetes as an endothelial cell disease, and smoking is the greatest endothelial cell poison we’ve come up with. So stopping smoking is something well worth doing," Dr. Stanton said.
• Blood pressure control. No question exists regarding its renoprotective effect. But recent guidelines are dizzyingly all over the map in terms of target pressure recommendations.
"I’m getting a major headache reading these articles right now. I can show you the data. Good luck! I personally like a target of 130/80 mm Hg or less, particularly when it’s not that hard to get there. But I’d let you decide what particular target you favor," he said.
He prefers 130/80 mm Hg as a target blood pressure for primary prevention of diabetic kidney disease in large part because of a meta-analysis showing that it was associated with a 10% reduction in the risk of developing microalbuminuria and an 11% decrease in end-stage renal disease (PloS Med 2012;9(8):e1001293).
• Weight loss. The growing bariatric surgery literature supports weight loss as a primary preventive strategy.
• Protein intake. There is no role for a low-protein diet – say, less than 0.8 g/kg per day – for primary prevention of kidney disease in diabetes patients. And Dr. Stanton believes a high-protein diet in the range of more than 1.5 or 2 g/kg per day is best avoided in patients with diabetes, although he stressed that the evidence on this score remains sketchy.
Still, "I would not go on a body-building diet or an Atkins-type diet," he cautioned.
• Targeting glomerular hyperfiltration. Studies have shown conflicting results. "For me, there’s no clear role for targeting hyperfiltration," said Dr. Stanton, who cited a comprehensive review that he finds persuasive (Diabetologia 2010;53:2093-104).
The key to developing more effective primary prevention strategies, according to Dr. Stanton, will be first to establish markers that clearly identify the 30% or so of diabetes patients who will go on to develop renal disease, then test novel interventions specifically in that high-risk group.
Promising biomarkers include circulating tumor necrosis factor alpha receptor levels, von Willebrand factor, monocyte chemoattractant factor, asymmetrical dimethylarginine, interleukin-6 and -8, and Fas receptor.
For example, one study showed that patients with type 2 diabetes in the top quartile for circulating TNF receptor 1 had a cumulative 12-year incidence of end-stage renal disease of 54%, compared to just 3% in patients in the other quartiles (J. Am. Soc. Nephrol. 2012;23:507-15).
"Lots of companies are looking at these now. These markers may be coming our way as indicators of people with diabetes who are likely to progress to kidney disease," Dr. Stanton said.
He reported serving as a consultant to Boehringer Ingelheim.
LAS VEGAS – Contrary to conventional wisdom, neither ACE inhibitors nor angiotensin receptor blockers have any role to play in primary prevention of diabetic kidney disease, according to Dr. Robert C. Stanton, chief of nephrology at the Harvard University’s Joslin Diabetes Center, Boston.
"I don’t see any unique indication for ACE inhibitors and ARBs for the primary prevention of kidney disease in diabetic patients, especially given that around 70% of diabetes patients will never develop kidney disease. They’re perfectly fine blood pressure pills. But as a magic kidney disease prevention drug, I don’t see any evidence for that. Of course, patients with proteinuria are another issue entirely. Those drugs absolutely are beneficial in that setting," he said at a meeting sponsored by the National Kidney Foundation.
When Dr. Stanton polled his audience electronically during the course of his talk, however, the majority of physicians indicated that they believe ACE inhibitors and ARBs are indeed useful for primary prevention of diabetic kidney disease. The evidence, Dr. Stanton emphasized, shows otherwise.
For example, a well-conducted, randomized, multicenter, placebo-controlled, 5-year clinical trial showed no benefit for enalapril or losartan in preventing kidney disease in patients with type 1 diabetes (N. Engl. J. Med. 2009;361:40-51). And three randomized controlled trials showed no primary preventive benefit for candesartan in more than 5,000 patients with type 1 or type 2 diabetes (Ann. Intern. Med. 2009;151:11-20).
Dr. Stanton noted that lots of other interventions have been proposed for the primary prevention of kidney disease in diabetes patients. Some are supported by solid evidence of benefit, others are not.
Here is his view of the preventive landscape:
• Intensive blood glucose control. "This is the easy one," he said. "A lot of us in the diabetes world feel that a hemoglobin A1c of 7% is the appropriate target for preventing many complications. It’s a reasonable target and should be achieved whether you’re talking about type 1 or type 2 patients."
The nephrologist noted that recent 25-year follow-up data from the landmark Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications study showed that fully 18 years after the intervention ended, patients assigned to intensive blood glucose control still showed highly impressive 50% reductions in the cumulative incidence of both microalbuminuria and end-stage renal disease compared with patients placed on less intensive control (Diabetes Care 2014;37:24-30).
• Smoking cessation. Smoking has been linked to a several-fold increased risk of diabetic kidney disease. "I think of diabetes as an endothelial cell disease, and smoking is the greatest endothelial cell poison we’ve come up with. So stopping smoking is something well worth doing," Dr. Stanton said.
• Blood pressure control. No question exists regarding its renoprotective effect. But recent guidelines are dizzyingly all over the map in terms of target pressure recommendations.
"I’m getting a major headache reading these articles right now. I can show you the data. Good luck! I personally like a target of 130/80 mm Hg or less, particularly when it’s not that hard to get there. But I’d let you decide what particular target you favor," he said.
He prefers 130/80 mm Hg as a target blood pressure for primary prevention of diabetic kidney disease in large part because of a meta-analysis showing that it was associated with a 10% reduction in the risk of developing microalbuminuria and an 11% decrease in end-stage renal disease (PloS Med 2012;9(8):e1001293).
• Weight loss. The growing bariatric surgery literature supports weight loss as a primary preventive strategy.
• Protein intake. There is no role for a low-protein diet – say, less than 0.8 g/kg per day – for primary prevention of kidney disease in diabetes patients. And Dr. Stanton believes a high-protein diet in the range of more than 1.5 or 2 g/kg per day is best avoided in patients with diabetes, although he stressed that the evidence on this score remains sketchy.
Still, "I would not go on a body-building diet or an Atkins-type diet," he cautioned.
• Targeting glomerular hyperfiltration. Studies have shown conflicting results. "For me, there’s no clear role for targeting hyperfiltration," said Dr. Stanton, who cited a comprehensive review that he finds persuasive (Diabetologia 2010;53:2093-104).
The key to developing more effective primary prevention strategies, according to Dr. Stanton, will be first to establish markers that clearly identify the 30% or so of diabetes patients who will go on to develop renal disease, then test novel interventions specifically in that high-risk group.
Promising biomarkers include circulating tumor necrosis factor alpha receptor levels, von Willebrand factor, monocyte chemoattractant factor, asymmetrical dimethylarginine, interleukin-6 and -8, and Fas receptor.
For example, one study showed that patients with type 2 diabetes in the top quartile for circulating TNF receptor 1 had a cumulative 12-year incidence of end-stage renal disease of 54%, compared to just 3% in patients in the other quartiles (J. Am. Soc. Nephrol. 2012;23:507-15).
"Lots of companies are looking at these now. These markers may be coming our way as indicators of people with diabetes who are likely to progress to kidney disease," Dr. Stanton said.
He reported serving as a consultant to Boehringer Ingelheim.
EXPERT ANALYSIS FROM SCM 14

{kind=link}