User login
Combo disappoints in newly diagnosed MM

Top-line results from the phase 3 CLARION trial suggest that treatment with carfilzomib, melphalan, and prednisone (KMP) is not superior to treatment with bortezomib, melphalan, and prednisone (VMP).
The trial was designed to compare KMP and VMP in patients with newly diagnosed multiple myeloma (MM) who were ineligible for hematopoietic stem cell transplant.
The results showed that progression-free survival (PFS) rates were similar with the 2 regimens.
And although overall survival data are not yet mature, there seems to be a trend favoring the VMP regimen.
Amgen, the company developing carfilzomib, released these results yesterday.
“The CLARION results, generated in the context of a melphalan-containing regimen, are disappointing, especially given the robust data we’ve seen in the second-line setting,” said Sean E. Harper, MD, executive vice president of Research and Development at Amgen.
“However, the myeloma landscape has changed dramatically since the design of the CLARION study, with very few newly diagnosed patients treated with
melphalan-based regimens, particularly in the US. We remain committed to exploring Kyprolis in combination with other agents to advance the treatment of multiple myeloma.”
Dr Harper said he could not comment on whether the CLARION trial will continue, as Amgen hopes to present data from the trial at the 2016 ASH Annual Meeting.
The CLARION trial is a head-to-head, randomized study in transplant-ineligible patients with newly diagnosed MM. A total of 955 patients were randomized 1:1 to receive KMP or VMP for 54 weeks. The median patient age was 72.
The trial did not meet the primary endpoint of superiority in PFS. The median PFS was 22.3 months in the KMP arm and 22.1 months in the VMP arm. The hazard ratio was 0.91 (95% CI, 0.75-1.10), and the difference between the arms was not statistically significant.
The data for overall survival, a secondary endpoint, are not yet mature. But the observed hazard ratio was 1.21 (95% CI, 0.90-1.64), and there was no significant difference between the treatment arms.
The incidence of grade 3 or higher adverse events was 74.7% in the KMP arm and 76.2% in the VMP arm.
The incidence of grade 2 or higher peripheral neuropathy, a secondary endpoint, was 2.5% in the KMP arm and 35.1% in the VMP arm.
Fatal treatment-emergent adverse events occurred in 6.5% of patients in the KMP arm and 4.3% of those in the VMP arm. ![]()
Top-line results from the phase 3 CLARION trial suggest that treatment with carfilzomib, melphalan, and prednisone (KMP) is not superior to treatment with bortezomib, melphalan, and prednisone (VMP).
The trial was designed to compare KMP and VMP in patients with newly diagnosed multiple myeloma (MM) who were ineligible for hematopoietic stem cell transplant.
The results showed that progression-free survival (PFS) rates were similar with the 2 regimens.
And although overall survival data are not yet mature, there seems to be a trend favoring the VMP regimen.
Amgen, the company developing carfilzomib, released these results yesterday.
“The CLARION results, generated in the context of a melphalan-containing regimen, are disappointing, especially given the robust data we’ve seen in the second-line setting,” said Sean E. Harper, MD, executive vice president of Research and Development at Amgen.
“However, the myeloma landscape has changed dramatically since the design of the CLARION study, with very few newly diagnosed patients treated with
melphalan-based regimens, particularly in the US. We remain committed to exploring Kyprolis in combination with other agents to advance the treatment of multiple myeloma.”
Dr Harper said he could not comment on whether the CLARION trial will continue, as Amgen hopes to present data from the trial at the 2016 ASH Annual Meeting.
The CLARION trial is a head-to-head, randomized study in transplant-ineligible patients with newly diagnosed MM. A total of 955 patients were randomized 1:1 to receive KMP or VMP for 54 weeks. The median patient age was 72.
The trial did not meet the primary endpoint of superiority in PFS. The median PFS was 22.3 months in the KMP arm and 22.1 months in the VMP arm. The hazard ratio was 0.91 (95% CI, 0.75-1.10), and the difference between the arms was not statistically significant.
The data for overall survival, a secondary endpoint, are not yet mature. But the observed hazard ratio was 1.21 (95% CI, 0.90-1.64), and there was no significant difference between the treatment arms.
The incidence of grade 3 or higher adverse events was 74.7% in the KMP arm and 76.2% in the VMP arm.
The incidence of grade 2 or higher peripheral neuropathy, a secondary endpoint, was 2.5% in the KMP arm and 35.1% in the VMP arm.
Fatal treatment-emergent adverse events occurred in 6.5% of patients in the KMP arm and 4.3% of those in the VMP arm. ![]()
Top-line results from the phase 3 CLARION trial suggest that treatment with carfilzomib, melphalan, and prednisone (KMP) is not superior to treatment with bortezomib, melphalan, and prednisone (VMP).
The trial was designed to compare KMP and VMP in patients with newly diagnosed multiple myeloma (MM) who were ineligible for hematopoietic stem cell transplant.
The results showed that progression-free survival (PFS) rates were similar with the 2 regimens.
And although overall survival data are not yet mature, there seems to be a trend favoring the VMP regimen.
Amgen, the company developing carfilzomib, released these results yesterday.
“The CLARION results, generated in the context of a melphalan-containing regimen, are disappointing, especially given the robust data we’ve seen in the second-line setting,” said Sean E. Harper, MD, executive vice president of Research and Development at Amgen.
“However, the myeloma landscape has changed dramatically since the design of the CLARION study, with very few newly diagnosed patients treated with
melphalan-based regimens, particularly in the US. We remain committed to exploring Kyprolis in combination with other agents to advance the treatment of multiple myeloma.”
Dr Harper said he could not comment on whether the CLARION trial will continue, as Amgen hopes to present data from the trial at the 2016 ASH Annual Meeting.
The CLARION trial is a head-to-head, randomized study in transplant-ineligible patients with newly diagnosed MM. A total of 955 patients were randomized 1:1 to receive KMP or VMP for 54 weeks. The median patient age was 72.
The trial did not meet the primary endpoint of superiority in PFS. The median PFS was 22.3 months in the KMP arm and 22.1 months in the VMP arm. The hazard ratio was 0.91 (95% CI, 0.75-1.10), and the difference between the arms was not statistically significant.
The data for overall survival, a secondary endpoint, are not yet mature. But the observed hazard ratio was 1.21 (95% CI, 0.90-1.64), and there was no significant difference between the treatment arms.
The incidence of grade 3 or higher adverse events was 74.7% in the KMP arm and 76.2% in the VMP arm.
The incidence of grade 2 or higher peripheral neuropathy, a secondary endpoint, was 2.5% in the KMP arm and 35.1% in the VMP arm.
Fatal treatment-emergent adverse events occurred in 6.5% of patients in the KMP arm and 4.3% of those in the VMP arm. ![]()
Malaria vaccine receives fast track designation

Image by Ute Frevert
and Margaret Shear
The US Food and Drug Administration (FDA) has granted fast track designation to an investigational malaria vaccine.
Sanaria® PfSPZ Vaccine is composed of live but weakened Plasmodium falciparum sporozoites and is being developed by Sanaria, Inc.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the FDA’s fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the biologic license application or new drug application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA.
Trials of Sanaria® PfSPZ Vaccine
In a phase 1 study published in Science in 2013, 80% of patients who received higher doses of Sanaria® PfSPZ Vaccine were protected from malaria at 3 weeks after vaccination.
In another phase 1 study published in Nature Medicine in 2016, 4 doses of the vaccine protected 73% of subjects from malaria at 3 weeks and 55% of subjects at 21 weeks.
Five of the subjects without parasitemia at 21 weeks were exposed to malaria-carrying mosquitoes again at 59 weeks, and none developed parasitemia.
To date, 1165 volunteers have received Sanaria’s PfSPZ-based products in more than 2 dozen clinical trials in the US, Europe, and Africa.
Clinical trials are in progress in Tanzania, Kenya, Mali, Burkina Faso, Germany, and the US, and are intended to begin soon in Equatorial Guinea. ![]()
Image by Ute Frevert
and Margaret Shear
The US Food and Drug Administration (FDA) has granted fast track designation to an investigational malaria vaccine.
Sanaria® PfSPZ Vaccine is composed of live but weakened Plasmodium falciparum sporozoites and is being developed by Sanaria, Inc.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the FDA’s fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the biologic license application or new drug application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA.
Trials of Sanaria® PfSPZ Vaccine
In a phase 1 study published in Science in 2013, 80% of patients who received higher doses of Sanaria® PfSPZ Vaccine were protected from malaria at 3 weeks after vaccination.
In another phase 1 study published in Nature Medicine in 2016, 4 doses of the vaccine protected 73% of subjects from malaria at 3 weeks and 55% of subjects at 21 weeks.
Five of the subjects without parasitemia at 21 weeks were exposed to malaria-carrying mosquitoes again at 59 weeks, and none developed parasitemia.
To date, 1165 volunteers have received Sanaria’s PfSPZ-based products in more than 2 dozen clinical trials in the US, Europe, and Africa.
Clinical trials are in progress in Tanzania, Kenya, Mali, Burkina Faso, Germany, and the US, and are intended to begin soon in Equatorial Guinea. ![]()
Image by Ute Frevert
and Margaret Shear
The US Food and Drug Administration (FDA) has granted fast track designation to an investigational malaria vaccine.
Sanaria® PfSPZ Vaccine is composed of live but weakened Plasmodium falciparum sporozoites and is being developed by Sanaria, Inc.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the FDA’s fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the biologic license application or new drug application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA.
Trials of Sanaria® PfSPZ Vaccine
In a phase 1 study published in Science in 2013, 80% of patients who received higher doses of Sanaria® PfSPZ Vaccine were protected from malaria at 3 weeks after vaccination.
In another phase 1 study published in Nature Medicine in 2016, 4 doses of the vaccine protected 73% of subjects from malaria at 3 weeks and 55% of subjects at 21 weeks.
Five of the subjects without parasitemia at 21 weeks were exposed to malaria-carrying mosquitoes again at 59 weeks, and none developed parasitemia.
To date, 1165 volunteers have received Sanaria’s PfSPZ-based products in more than 2 dozen clinical trials in the US, Europe, and Africa.
Clinical trials are in progress in Tanzania, Kenya, Mali, Burkina Faso, Germany, and the US, and are intended to begin soon in Equatorial Guinea. ![]()
EMA says plasma/urine-derived meds are safe from Zika

Photo by Cristina Granados
Patients who take plasma-derived or urine-derived medicines do not have to worry about these products being contaminated with Zika virus, according to the European Medicines Agency (EMA).
The agency said assessments have confirmed that manufacturing processes for these medicines—which include coagulation factors, immunoglobulins, and urokinase products—successfully inactivate or remove the Zika virus.
These medicines are produced from body fluids that might be sourced in parts of the world where the Zika virus is prevalent. So regulators in the European Union (EU) sought reassurance that there is no risk of the virus contaminating the final product and thus affecting the patients taking these medicines.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) investigated the potential risk with plasma-derived medicinal products.
And the Co-ordination Group for Mutual Recognition and Decentralised Procedures—Human (CMDh) has coordinated the assessment by EU member states on the potential risk with urine-derived medicinal products.
The CHMP concluded at its meeting last week that the manufacturing processes used for plasma-derived products—including, for example, the solvent/detergent method to inactivate viruses, pasteurization, and virus filtration—inactivate or remove the Zika virus from the finished product.
The CHMP therefore concluded that no additional safety measures, such as the testing or exclusion of certain plasma donors, were necessary.
The CMDh, following an assessment of data, concluded that the manufacturing processes for urine-derived products contain complementary steps with inactivation/removal capacity for enveloped viruses, which are considered sufficient for eliminating Zika virus.
Additional safety measures, such as screening urine donors/donations or deferring donors returning from Zika-affected areas, are not considered necessary.
The findings from these assessments are available in a report from the CHMP’s Biologics Working Party.
The Biologics Working Party recommendation on plasma-derived products is in line with the guidance published in July 2016 by the European Centre for Disease Prevention and Control. ![]()
Photo by Cristina Granados
Patients who take plasma-derived or urine-derived medicines do not have to worry about these products being contaminated with Zika virus, according to the European Medicines Agency (EMA).
The agency said assessments have confirmed that manufacturing processes for these medicines—which include coagulation factors, immunoglobulins, and urokinase products—successfully inactivate or remove the Zika virus.
These medicines are produced from body fluids that might be sourced in parts of the world where the Zika virus is prevalent. So regulators in the European Union (EU) sought reassurance that there is no risk of the virus contaminating the final product and thus affecting the patients taking these medicines.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) investigated the potential risk with plasma-derived medicinal products.
And the Co-ordination Group for Mutual Recognition and Decentralised Procedures—Human (CMDh) has coordinated the assessment by EU member states on the potential risk with urine-derived medicinal products.
The CHMP concluded at its meeting last week that the manufacturing processes used for plasma-derived products—including, for example, the solvent/detergent method to inactivate viruses, pasteurization, and virus filtration—inactivate or remove the Zika virus from the finished product.
The CHMP therefore concluded that no additional safety measures, such as the testing or exclusion of certain plasma donors, were necessary.
The CMDh, following an assessment of data, concluded that the manufacturing processes for urine-derived products contain complementary steps with inactivation/removal capacity for enveloped viruses, which are considered sufficient for eliminating Zika virus.
Additional safety measures, such as screening urine donors/donations or deferring donors returning from Zika-affected areas, are not considered necessary.
The findings from these assessments are available in a report from the CHMP’s Biologics Working Party.
The Biologics Working Party recommendation on plasma-derived products is in line with the guidance published in July 2016 by the European Centre for Disease Prevention and Control. ![]()
Photo by Cristina Granados
Patients who take plasma-derived or urine-derived medicines do not have to worry about these products being contaminated with Zika virus, according to the European Medicines Agency (EMA).
The agency said assessments have confirmed that manufacturing processes for these medicines—which include coagulation factors, immunoglobulins, and urokinase products—successfully inactivate or remove the Zika virus.
These medicines are produced from body fluids that might be sourced in parts of the world where the Zika virus is prevalent. So regulators in the European Union (EU) sought reassurance that there is no risk of the virus contaminating the final product and thus affecting the patients taking these medicines.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) investigated the potential risk with plasma-derived medicinal products.
And the Co-ordination Group for Mutual Recognition and Decentralised Procedures—Human (CMDh) has coordinated the assessment by EU member states on the potential risk with urine-derived medicinal products.
The CHMP concluded at its meeting last week that the manufacturing processes used for plasma-derived products—including, for example, the solvent/detergent method to inactivate viruses, pasteurization, and virus filtration—inactivate or remove the Zika virus from the finished product.
The CHMP therefore concluded that no additional safety measures, such as the testing or exclusion of certain plasma donors, were necessary.
The CMDh, following an assessment of data, concluded that the manufacturing processes for urine-derived products contain complementary steps with inactivation/removal capacity for enveloped viruses, which are considered sufficient for eliminating Zika virus.
Additional safety measures, such as screening urine donors/donations or deferring donors returning from Zika-affected areas, are not considered necessary.
The findings from these assessments are available in a report from the CHMP’s Biologics Working Party.
The Biologics Working Party recommendation on plasma-derived products is in line with the guidance published in July 2016 by the European Centre for Disease Prevention and Control. ![]()
Team creates method for predicting drug toxicity

for a clinical trial
Photo by Esther Dyson
Researchers have devised a method for predicting whether experimental drugs will fail clinical trials due to excessive toxicity.
They say the method, known as PrOCTOR, upends conventional wisdom about the criteria on which to evaluate new drugs’ safety.
Rather than exclusively examining molecular structure to determine viability, PrOCTOR combines a host of structural features and features related to how the drug binds to molecules in the body.
“We looked more broadly at drug molecule features that drug developers thought were unimportant in predicting drug safety in the past. Then, we let the data speak for itself,” said study author Olivier Elemento, PhD, of Weill Cornell Medicine in New York, New York.
Dr Elemento and his colleagues described PrOCTOR in Cell Chemical Biology.
PrOCTOR was inspired by an approach that baseball statisticians adopted to better predict which players would be successful offensively. Instead of relying on collective wisdom from baseball insiders, statisticians decided to use an objective numbers analysis to measure in-game productivity, a strategy known as “Moneyball.”
Similarly, Dr Elemento and his colleagues developed a computational method that analyzes data from 48 different features of a drug—from molecular weight to details about its target—to determine whether it would be safe for clinical use.
Using machine learning, the researchers trained PrOCTOR on drugs approved by the US Food and Drug Administration (FDA) as well as drugs that failed clinical trials due to toxicity problems.
Based on this information, the researchers created “PrOCTOR scores” that could help distinguish drugs approved by the FDA from those that failed for toxicity.
They tested PrOCTOR on hundreds of additional drugs approved in Europe and Japan and using side effect data on approved drugs collected by the FDA.
The researchers said PrOCTOR was able to accurately recognize toxic side effects that were a consequence of a drug’s chemical features and its target. Records revealing that many of these drugs had failed clinical trials supported PrOCTOR’s accuracy.
“We were able to find several features that led to a very predictive model,” Dr Elemento said. “Hopefully, this approach could be used to determine whether it’s worth pursuing a drug prior to starting human trials.” ![]()
for a clinical trial
Photo by Esther Dyson
Researchers have devised a method for predicting whether experimental drugs will fail clinical trials due to excessive toxicity.
They say the method, known as PrOCTOR, upends conventional wisdom about the criteria on which to evaluate new drugs’ safety.
Rather than exclusively examining molecular structure to determine viability, PrOCTOR combines a host of structural features and features related to how the drug binds to molecules in the body.
“We looked more broadly at drug molecule features that drug developers thought were unimportant in predicting drug safety in the past. Then, we let the data speak for itself,” said study author Olivier Elemento, PhD, of Weill Cornell Medicine in New York, New York.
Dr Elemento and his colleagues described PrOCTOR in Cell Chemical Biology.
PrOCTOR was inspired by an approach that baseball statisticians adopted to better predict which players would be successful offensively. Instead of relying on collective wisdom from baseball insiders, statisticians decided to use an objective numbers analysis to measure in-game productivity, a strategy known as “Moneyball.”
Similarly, Dr Elemento and his colleagues developed a computational method that analyzes data from 48 different features of a drug—from molecular weight to details about its target—to determine whether it would be safe for clinical use.
Using machine learning, the researchers trained PrOCTOR on drugs approved by the US Food and Drug Administration (FDA) as well as drugs that failed clinical trials due to toxicity problems.
Based on this information, the researchers created “PrOCTOR scores” that could help distinguish drugs approved by the FDA from those that failed for toxicity.
They tested PrOCTOR on hundreds of additional drugs approved in Europe and Japan and using side effect data on approved drugs collected by the FDA.
The researchers said PrOCTOR was able to accurately recognize toxic side effects that were a consequence of a drug’s chemical features and its target. Records revealing that many of these drugs had failed clinical trials supported PrOCTOR’s accuracy.
“We were able to find several features that led to a very predictive model,” Dr Elemento said. “Hopefully, this approach could be used to determine whether it’s worth pursuing a drug prior to starting human trials.” ![]()
for a clinical trial
Photo by Esther Dyson
Researchers have devised a method for predicting whether experimental drugs will fail clinical trials due to excessive toxicity.
They say the method, known as PrOCTOR, upends conventional wisdom about the criteria on which to evaluate new drugs’ safety.
Rather than exclusively examining molecular structure to determine viability, PrOCTOR combines a host of structural features and features related to how the drug binds to molecules in the body.
“We looked more broadly at drug molecule features that drug developers thought were unimportant in predicting drug safety in the past. Then, we let the data speak for itself,” said study author Olivier Elemento, PhD, of Weill Cornell Medicine in New York, New York.
Dr Elemento and his colleagues described PrOCTOR in Cell Chemical Biology.
PrOCTOR was inspired by an approach that baseball statisticians adopted to better predict which players would be successful offensively. Instead of relying on collective wisdom from baseball insiders, statisticians decided to use an objective numbers analysis to measure in-game productivity, a strategy known as “Moneyball.”
Similarly, Dr Elemento and his colleagues developed a computational method that analyzes data from 48 different features of a drug—from molecular weight to details about its target—to determine whether it would be safe for clinical use.
Using machine learning, the researchers trained PrOCTOR on drugs approved by the US Food and Drug Administration (FDA) as well as drugs that failed clinical trials due to toxicity problems.
Based on this information, the researchers created “PrOCTOR scores” that could help distinguish drugs approved by the FDA from those that failed for toxicity.
They tested PrOCTOR on hundreds of additional drugs approved in Europe and Japan and using side effect data on approved drugs collected by the FDA.
The researchers said PrOCTOR was able to accurately recognize toxic side effects that were a consequence of a drug’s chemical features and its target. Records revealing that many of these drugs had failed clinical trials supported PrOCTOR’s accuracy.
“We were able to find several features that led to a very predictive model,” Dr Elemento said. “Hopefully, this approach could be used to determine whether it’s worth pursuing a drug prior to starting human trials.” ![]()
CHMP recommends approval of ixazomib for MM
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that ixazomib (NinlaroTM) receive conditional marketing authorization.
The recommendation is for ixazomib in combination with lenalidomide and dexamethasone as a treatment for adults with multiple myeloma (MM) who have received at least 1 prior therapy.
The CHMP’s recommendation will be reviewed by the European Commission (EC).
If the EC grants the authorization, ixazomib will be the first oral proteasome inhibitor approved for use across the European Economic Area.
Ixazomib is being developed by Takeda Pharmaceutical Company Limited.
Phase 3 trial
The CHMP’s positive opinion of ixazomib is based on results from the phase 3 TOURMALINE-MM1 trial, which were presented at the 2015 ASH Annual Meeting.
The trial included 722 patients with relapsed or refractory MM. The patients were randomized to receive ixazomib, lenalidomide, and dexamethasone (IRd, n=360) or placebo, lenalidomide, and dexamethasone (Rd, n=362).
Baseline patient characteristics were similar between the treatment arms. Fifty-nine percent of patients in both arms had received 1 prior line of therapy, and 41% in both arms had 2 or 3 prior lines of therapy.
Seventy-eight percent of patients responded to IRd, and 72% responded to Rd (P=0.035). The rates of complete response were 12% and 7%, respectively (P=0.019).
At a median follow-up of about 15 months, the median progression-free survival was 20.6 months in the IRd arm and 14.7 months in the Rd arm. The hazard ratio was 0.742 (P=0.012).
At a median follow-up of about 23 months, the median overall survival had not been reached in either treatment arm.
The incidence of adverse events (AEs) was 98% in the IRd arm and 99% in the Rd arm. The incidence of grade 3 or higher AEs was 74% and 69%, respectively. The incidence of serious AEs was 47% and 49%, respectively.
Common AEs in the IRd and Rd arms, respectively, were diarrhea (45% vs 39%), constipation (35% vs 26%), nausea (29% vs 22%), vomiting (23% vs 12%), rash (36% vs 23%), back pain (24% vs 17%), upper respiratory tract infection (23% vs 19%), thrombocytopenia (31% vs 16%), peripheral neuropathy (27% vs 22%), peripheral edema (28% vs 20%), thromboembolism (8% vs 11%), and neutropenia (33% vs 31%).
About conditional authorization
Conditional marketing authorization represents an expedited path for approval. The EC grants this type of authorization before pivotal registration studies are completed.
Conditional marketing authorization is granted to products whose benefits are thought to outweigh their risks, products that address unmet needs, and products that are expected to provide a significant public health benefit.
If ixazomib receives conditional marketing authorization, Takeda will be required to provide post-approval updates on safety and efficacy analyses for TOURMALINE-MM1 and some other ongoing studies to demonstrate the treatment’s long-term effects. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that ixazomib (NinlaroTM) receive conditional marketing authorization.
The recommendation is for ixazomib in combination with lenalidomide and dexamethasone as a treatment for adults with multiple myeloma (MM) who have received at least 1 prior therapy.
The CHMP’s recommendation will be reviewed by the European Commission (EC).
If the EC grants the authorization, ixazomib will be the first oral proteasome inhibitor approved for use across the European Economic Area.
Ixazomib is being developed by Takeda Pharmaceutical Company Limited.
Phase 3 trial
The CHMP’s positive opinion of ixazomib is based on results from the phase 3 TOURMALINE-MM1 trial, which were presented at the 2015 ASH Annual Meeting.
The trial included 722 patients with relapsed or refractory MM. The patients were randomized to receive ixazomib, lenalidomide, and dexamethasone (IRd, n=360) or placebo, lenalidomide, and dexamethasone (Rd, n=362).
Baseline patient characteristics were similar between the treatment arms. Fifty-nine percent of patients in both arms had received 1 prior line of therapy, and 41% in both arms had 2 or 3 prior lines of therapy.
Seventy-eight percent of patients responded to IRd, and 72% responded to Rd (P=0.035). The rates of complete response were 12% and 7%, respectively (P=0.019).
At a median follow-up of about 15 months, the median progression-free survival was 20.6 months in the IRd arm and 14.7 months in the Rd arm. The hazard ratio was 0.742 (P=0.012).
At a median follow-up of about 23 months, the median overall survival had not been reached in either treatment arm.
The incidence of adverse events (AEs) was 98% in the IRd arm and 99% in the Rd arm. The incidence of grade 3 or higher AEs was 74% and 69%, respectively. The incidence of serious AEs was 47% and 49%, respectively.
Common AEs in the IRd and Rd arms, respectively, were diarrhea (45% vs 39%), constipation (35% vs 26%), nausea (29% vs 22%), vomiting (23% vs 12%), rash (36% vs 23%), back pain (24% vs 17%), upper respiratory tract infection (23% vs 19%), thrombocytopenia (31% vs 16%), peripheral neuropathy (27% vs 22%), peripheral edema (28% vs 20%), thromboembolism (8% vs 11%), and neutropenia (33% vs 31%).
About conditional authorization
Conditional marketing authorization represents an expedited path for approval. The EC grants this type of authorization before pivotal registration studies are completed.
Conditional marketing authorization is granted to products whose benefits are thought to outweigh their risks, products that address unmet needs, and products that are expected to provide a significant public health benefit.
If ixazomib receives conditional marketing authorization, Takeda will be required to provide post-approval updates on safety and efficacy analyses for TOURMALINE-MM1 and some other ongoing studies to demonstrate the treatment’s long-term effects. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that ixazomib (NinlaroTM) receive conditional marketing authorization.
The recommendation is for ixazomib in combination with lenalidomide and dexamethasone as a treatment for adults with multiple myeloma (MM) who have received at least 1 prior therapy.
The CHMP’s recommendation will be reviewed by the European Commission (EC).
If the EC grants the authorization, ixazomib will be the first oral proteasome inhibitor approved for use across the European Economic Area.
Ixazomib is being developed by Takeda Pharmaceutical Company Limited.
Phase 3 trial
The CHMP’s positive opinion of ixazomib is based on results from the phase 3 TOURMALINE-MM1 trial, which were presented at the 2015 ASH Annual Meeting.
The trial included 722 patients with relapsed or refractory MM. The patients were randomized to receive ixazomib, lenalidomide, and dexamethasone (IRd, n=360) or placebo, lenalidomide, and dexamethasone (Rd, n=362).
Baseline patient characteristics were similar between the treatment arms. Fifty-nine percent of patients in both arms had received 1 prior line of therapy, and 41% in both arms had 2 or 3 prior lines of therapy.
Seventy-eight percent of patients responded to IRd, and 72% responded to Rd (P=0.035). The rates of complete response were 12% and 7%, respectively (P=0.019).
At a median follow-up of about 15 months, the median progression-free survival was 20.6 months in the IRd arm and 14.7 months in the Rd arm. The hazard ratio was 0.742 (P=0.012).
At a median follow-up of about 23 months, the median overall survival had not been reached in either treatment arm.
The incidence of adverse events (AEs) was 98% in the IRd arm and 99% in the Rd arm. The incidence of grade 3 or higher AEs was 74% and 69%, respectively. The incidence of serious AEs was 47% and 49%, respectively.
Common AEs in the IRd and Rd arms, respectively, were diarrhea (45% vs 39%), constipation (35% vs 26%), nausea (29% vs 22%), vomiting (23% vs 12%), rash (36% vs 23%), back pain (24% vs 17%), upper respiratory tract infection (23% vs 19%), thrombocytopenia (31% vs 16%), peripheral neuropathy (27% vs 22%), peripheral edema (28% vs 20%), thromboembolism (8% vs 11%), and neutropenia (33% vs 31%).
About conditional authorization
Conditional marketing authorization represents an expedited path for approval. The EC grants this type of authorization before pivotal registration studies are completed.
Conditional marketing authorization is granted to products whose benefits are thought to outweigh their risks, products that address unmet needs, and products that are expected to provide a significant public health benefit.
If ixazomib receives conditional marketing authorization, Takeda will be required to provide post-approval updates on safety and efficacy analyses for TOURMALINE-MM1 and some other ongoing studies to demonstrate the treatment’s long-term effects. ![]()
Drug granted fast track designation for MF

mycosis fungoides
The US Food and Drug Administration (FDA) has granted fast track designation to Resimmune® (A-dmDT390-bisFv[UCHT1]) for the treatment of mycosis fungoides (MF).
Resimmune is an anti-CD3 immunotoxin that targets and transiently depletes a high percentage of malignant and normal T cells.
Researchers believe this high rate of T-cell depletion may “reset” a patient’s immune system, leading to immunomodulation. However, the safety and efficacy of Resimmune has not been established.
Resimmune is being developed by Angimmune LLC.
Resimmune research
The FDA’s fast track designation for Resimmune is based on results from a phase 1 trial, which were published in haematologica last year.
The trial included 25 patients with cutaneous T-cell lymphoma (CTCL)—17 with stage IB-IIB disease and 8 with stage III-IV disease. The patients received Resimmune at varying doses—between 2.5 µg/kg and 11.25 µg/kg.
Safety
Drug-related adverse events for the entire study cohort (n=30) included:
- Grade 2-3 AST/ALT elevations (n=6)
- Grade 2 chills (n=10)
- Grade 5 EBV infection (n=1)
- Grade 3 EBV/CMV infection (n=5)
- Grade 2 fever (n=6)
- Grade 5 heart failure (n=2)
- Grade 2-3 hypoalbuminemia (n=11)
- Grade 2 hypomagnesemia (n=1)
- Grade 3 hypophosphatemia (n=3)
- Grade 2-4 hypotension (n=3)
- Grade 4 hypoxia (n=1)
- Grade 4 liver failure (n=1)
- Grade 4 metabolic acidosis (n=1)
- Grade 3 supraventricular tachycardia (n=2)
- Grade 3-4 uremia (n=3)
- Grade 2-4 vascular leak syndrome (n=5).
Efficacy and phase 2
Nine of the CTCL patients (36%) responded to Resimmune, and 4 (16%) had complete responses (CRs). The duration of CR was more than 38 months for 1 patient, more than 60 months for another, and more than 72 months for 2 patients.
Of the 5 partial responses, 2 lasted 3 months, 1 lasted more than 3 months, 1 lasted more than 6 months, and the longest lasted 14 months.
The researchers said this trial revealed a subgroup of CTCL patients with a very high response rate.
When they excluded patients whose mSWAT scores never exceeded 50 and who never had lymph node involvement or stage III disease, the researchers were left with 9 patients. This subgroup had an overall response rate of 89% and a CR rate of 50% (at 6 months of follow-up).
“Results from our phase 1 trial demonstrated a particularly robust response among certain patients with early stage disease, and the randomized phase 2 trial is designed to further explore this potential therapeutic benefit,” said David Neville, MD, president and chief scientific officer of Angimmune.
The objective of this phase 2 trial is to document the incidence of CRs with Resimmune compared to oral vorinostat after a maximum of 12 months of treatment. The trial will enroll patients with stage IB/IIB MF with mSWAT < 50 who have never had lymphoid disease or a prior hematopoietic stem cell transplant.
About fast track designation
The FDA’s fast track program is designed to facilitate and expedite the development and review of new drugs intended to treat serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the drug may be allowed to submit sections of the biologic license application or new drug application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings with the FDA to discuss the drug’s development plan and ensure collection of the appropriate data needed to support drug approval. And the designation allows for more frequent written communication from the FDA about things such as the design of proposed clinical trials and the use of biomarkers. ![]()
mycosis fungoides
The US Food and Drug Administration (FDA) has granted fast track designation to Resimmune® (A-dmDT390-bisFv[UCHT1]) for the treatment of mycosis fungoides (MF).
Resimmune is an anti-CD3 immunotoxin that targets and transiently depletes a high percentage of malignant and normal T cells.
Researchers believe this high rate of T-cell depletion may “reset” a patient’s immune system, leading to immunomodulation. However, the safety and efficacy of Resimmune has not been established.
Resimmune is being developed by Angimmune LLC.
Resimmune research
The FDA’s fast track designation for Resimmune is based on results from a phase 1 trial, which were published in haematologica last year.
The trial included 25 patients with cutaneous T-cell lymphoma (CTCL)—17 with stage IB-IIB disease and 8 with stage III-IV disease. The patients received Resimmune at varying doses—between 2.5 µg/kg and 11.25 µg/kg.
Safety
Drug-related adverse events for the entire study cohort (n=30) included:
- Grade 2-3 AST/ALT elevations (n=6)
- Grade 2 chills (n=10)
- Grade 5 EBV infection (n=1)
- Grade 3 EBV/CMV infection (n=5)
- Grade 2 fever (n=6)
- Grade 5 heart failure (n=2)
- Grade 2-3 hypoalbuminemia (n=11)
- Grade 2 hypomagnesemia (n=1)
- Grade 3 hypophosphatemia (n=3)
- Grade 2-4 hypotension (n=3)
- Grade 4 hypoxia (n=1)
- Grade 4 liver failure (n=1)
- Grade 4 metabolic acidosis (n=1)
- Grade 3 supraventricular tachycardia (n=2)
- Grade 3-4 uremia (n=3)
- Grade 2-4 vascular leak syndrome (n=5).
Efficacy and phase 2
Nine of the CTCL patients (36%) responded to Resimmune, and 4 (16%) had complete responses (CRs). The duration of CR was more than 38 months for 1 patient, more than 60 months for another, and more than 72 months for 2 patients.
Of the 5 partial responses, 2 lasted 3 months, 1 lasted more than 3 months, 1 lasted more than 6 months, and the longest lasted 14 months.
The researchers said this trial revealed a subgroup of CTCL patients with a very high response rate.
When they excluded patients whose mSWAT scores never exceeded 50 and who never had lymph node involvement or stage III disease, the researchers were left with 9 patients. This subgroup had an overall response rate of 89% and a CR rate of 50% (at 6 months of follow-up).
“Results from our phase 1 trial demonstrated a particularly robust response among certain patients with early stage disease, and the randomized phase 2 trial is designed to further explore this potential therapeutic benefit,” said David Neville, MD, president and chief scientific officer of Angimmune.
The objective of this phase 2 trial is to document the incidence of CRs with Resimmune compared to oral vorinostat after a maximum of 12 months of treatment. The trial will enroll patients with stage IB/IIB MF with mSWAT < 50 who have never had lymphoid disease or a prior hematopoietic stem cell transplant.
About fast track designation
The FDA’s fast track program is designed to facilitate and expedite the development and review of new drugs intended to treat serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the drug may be allowed to submit sections of the biologic license application or new drug application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings with the FDA to discuss the drug’s development plan and ensure collection of the appropriate data needed to support drug approval. And the designation allows for more frequent written communication from the FDA about things such as the design of proposed clinical trials and the use of biomarkers. ![]()
mycosis fungoides
The US Food and Drug Administration (FDA) has granted fast track designation to Resimmune® (A-dmDT390-bisFv[UCHT1]) for the treatment of mycosis fungoides (MF).
Resimmune is an anti-CD3 immunotoxin that targets and transiently depletes a high percentage of malignant and normal T cells.
Researchers believe this high rate of T-cell depletion may “reset” a patient’s immune system, leading to immunomodulation. However, the safety and efficacy of Resimmune has not been established.
Resimmune is being developed by Angimmune LLC.
Resimmune research
The FDA’s fast track designation for Resimmune is based on results from a phase 1 trial, which were published in haematologica last year.
The trial included 25 patients with cutaneous T-cell lymphoma (CTCL)—17 with stage IB-IIB disease and 8 with stage III-IV disease. The patients received Resimmune at varying doses—between 2.5 µg/kg and 11.25 µg/kg.
Safety
Drug-related adverse events for the entire study cohort (n=30) included:
- Grade 2-3 AST/ALT elevations (n=6)
- Grade 2 chills (n=10)
- Grade 5 EBV infection (n=1)
- Grade 3 EBV/CMV infection (n=5)
- Grade 2 fever (n=6)
- Grade 5 heart failure (n=2)
- Grade 2-3 hypoalbuminemia (n=11)
- Grade 2 hypomagnesemia (n=1)
- Grade 3 hypophosphatemia (n=3)
- Grade 2-4 hypotension (n=3)
- Grade 4 hypoxia (n=1)
- Grade 4 liver failure (n=1)
- Grade 4 metabolic acidosis (n=1)
- Grade 3 supraventricular tachycardia (n=2)
- Grade 3-4 uremia (n=3)
- Grade 2-4 vascular leak syndrome (n=5).
Efficacy and phase 2
Nine of the CTCL patients (36%) responded to Resimmune, and 4 (16%) had complete responses (CRs). The duration of CR was more than 38 months for 1 patient, more than 60 months for another, and more than 72 months for 2 patients.
Of the 5 partial responses, 2 lasted 3 months, 1 lasted more than 3 months, 1 lasted more than 6 months, and the longest lasted 14 months.
The researchers said this trial revealed a subgroup of CTCL patients with a very high response rate.
When they excluded patients whose mSWAT scores never exceeded 50 and who never had lymph node involvement or stage III disease, the researchers were left with 9 patients. This subgroup had an overall response rate of 89% and a CR rate of 50% (at 6 months of follow-up).
“Results from our phase 1 trial demonstrated a particularly robust response among certain patients with early stage disease, and the randomized phase 2 trial is designed to further explore this potential therapeutic benefit,” said David Neville, MD, president and chief scientific officer of Angimmune.
The objective of this phase 2 trial is to document the incidence of CRs with Resimmune compared to oral vorinostat after a maximum of 12 months of treatment. The trial will enroll patients with stage IB/IIB MF with mSWAT < 50 who have never had lymphoid disease or a prior hematopoietic stem cell transplant.
About fast track designation
The FDA’s fast track program is designed to facilitate and expedite the development and review of new drugs intended to treat serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the drug may be allowed to submit sections of the biologic license application or new drug application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings with the FDA to discuss the drug’s development plan and ensure collection of the appropriate data needed to support drug approval. And the designation allows for more frequent written communication from the FDA about things such as the design of proposed clinical trials and the use of biomarkers. ![]()
FDA grants drug orphan designation for PNH

The US Food and Drug Administration (FDA) has granted orphan drug designation to Coversin as a treatment for paroxysmal nocturnal hemoglobinuria (PNH).
Coversin is a recombinant small protein (16,740 Da) derived from a native protein found in the saliva of the Ornithodoros moubata tick.
The drug is a second-generation complement inhibitor that acts on complement component C5, preventing release of C5a and formation of C5b-9 (also known as the membrane attack complex).
Coversin is being developed by Akari Therapeutics.
In vitro experiments have shown that Coversin inhibits red blood cell lysis in PNH, and Coversin can achieve full complement inhibition in the blood of PNH patients who are resistant to eculizumab.
In a phase 1a trial of healthy volunteers, Coversin completely inhibited complement C5 activity within 12 hours of administration.
Akari Therapeutics is currently conducting a phase 1b study of Coversin in healthy volunteers and is enrolling patients with eculizumab-resistant PNH in a phase 2 trial.
The company has also been administering Coversin to a patient with eculizumab-resistant PNH. Thus far, Coversin has prevented hemolytic episodes and improved disease symptoms in this patient. The only drug-related adverse event has been occasional local and transient irritation at the injection site.
“We have continued to see complete complement inhibition and symptom control in a PNH patient with resistance to eculizumab, who has been self-administering subcutaneous Coversin for over 7 months,” said Gur Roshwalb, MD, CEO of Akari Therapeutics.
“We believe that Coversin, when approved, could provide important benefits for all patients with PNH.”
Coversin is also being studied in atypical hemolytic uremic syndrome and Guillain Barré syndrome.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to Coversin as a treatment for paroxysmal nocturnal hemoglobinuria (PNH).
Coversin is a recombinant small protein (16,740 Da) derived from a native protein found in the saliva of the Ornithodoros moubata tick.
The drug is a second-generation complement inhibitor that acts on complement component C5, preventing release of C5a and formation of C5b-9 (also known as the membrane attack complex).
Coversin is being developed by Akari Therapeutics.
In vitro experiments have shown that Coversin inhibits red blood cell lysis in PNH, and Coversin can achieve full complement inhibition in the blood of PNH patients who are resistant to eculizumab.
In a phase 1a trial of healthy volunteers, Coversin completely inhibited complement C5 activity within 12 hours of administration.
Akari Therapeutics is currently conducting a phase 1b study of Coversin in healthy volunteers and is enrolling patients with eculizumab-resistant PNH in a phase 2 trial.
The company has also been administering Coversin to a patient with eculizumab-resistant PNH. Thus far, Coversin has prevented hemolytic episodes and improved disease symptoms in this patient. The only drug-related adverse event has been occasional local and transient irritation at the injection site.
“We have continued to see complete complement inhibition and symptom control in a PNH patient with resistance to eculizumab, who has been self-administering subcutaneous Coversin for over 7 months,” said Gur Roshwalb, MD, CEO of Akari Therapeutics.
“We believe that Coversin, when approved, could provide important benefits for all patients with PNH.”
Coversin is also being studied in atypical hemolytic uremic syndrome and Guillain Barré syndrome.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to Coversin as a treatment for paroxysmal nocturnal hemoglobinuria (PNH).
Coversin is a recombinant small protein (16,740 Da) derived from a native protein found in the saliva of the Ornithodoros moubata tick.
The drug is a second-generation complement inhibitor that acts on complement component C5, preventing release of C5a and formation of C5b-9 (also known as the membrane attack complex).
Coversin is being developed by Akari Therapeutics.
In vitro experiments have shown that Coversin inhibits red blood cell lysis in PNH, and Coversin can achieve full complement inhibition in the blood of PNH patients who are resistant to eculizumab.
In a phase 1a trial of healthy volunteers, Coversin completely inhibited complement C5 activity within 12 hours of administration.
Akari Therapeutics is currently conducting a phase 1b study of Coversin in healthy volunteers and is enrolling patients with eculizumab-resistant PNH in a phase 2 trial.
The company has also been administering Coversin to a patient with eculizumab-resistant PNH. Thus far, Coversin has prevented hemolytic episodes and improved disease symptoms in this patient. The only drug-related adverse event has been occasional local and transient irritation at the injection site.
“We have continued to see complete complement inhibition and symptom control in a PNH patient with resistance to eculizumab, who has been self-administering subcutaneous Coversin for over 7 months,” said Gur Roshwalb, MD, CEO of Akari Therapeutics.
“We believe that Coversin, when approved, could provide important benefits for all patients with PNH.”
Coversin is also being studied in atypical hemolytic uremic syndrome and Guillain Barré syndrome.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
FDA expands approval of blinatumomab

and solution for infusion
Photo courtesy of Amgen
The US Food and Drug Administration (FDA) has granted accelerated approval for blinatumomab (Blincyto®) to treat pediatric patients with Philadelphia chromosome-negative (Ph-) relapsed or refractory B-cell precursor acute lymphoblastic leukemia (BCP-ALL).
The approval is based on results from a phase 1/2 study.
Continued approval of blinatumomab for this indication may be contingent upon verification of clinical benefit in subsequent trials.
Blinatumomab is a bispecific, CD19-directed, CD3 T-cell engager (BiTE®) antibody construct that binds specifically to CD19 expressed on the surface of cells of B-lineage origin and CD3 expressed on the surface of T cells.
In 2014, blinatumomab received accelerated approval from the FDA to treat adults with Ph- relapsed or refractory BCP-ALL. The FDA has also granted the drug priority review, breakthrough therapy designation, and orphan drug designation.
Blinatumomab is being developed by Amgen. Full prescribing information is available at www.blincyto.com.
‘205 trial
The latest accelerated approval of blinatumomab is based on results from the phase 1/2 ‘205 trial, in which researchers evaluated blinatumomab in 93 pediatric patients with relapsed or refractory BCP-ALL.
Amgen said treatment in this study has been completed, subjects are being monitored for long-term efficacy, and the data will be submitted for publication.
Initial results from this study were presented at ASH 2014. The abstract included data on 39 patients with relapsed/refractory BCP-ALL and a median age of 9 (range, 2-16).
The patients received blinatumomab at a dose of 5-15 µg/m²/day. Nineteen patients completed 1 cycle of blinatumomab, 4 completed 2 cycles, and 2 completed 3 cycles.
During the first 2 treatment cycles, 12 patients achieved a complete response, 5 of whom were negative for minimal residual disease.
Six of the complete responders went on to transplant. The median relapse-free survival for complete responders was 5.6 months.
At 6 months of follow-up, the median overall survival for all 39 patients was 4.3 months.
All of the patients experienced adverse events. The most common were pyrexia (74%), anemia (33%), nausea (31%), headache (28%), hypertension (26%), increased alanine aminotransferase (23%), and cough (21%).
The most common grade 3 or higher events were anemia (26%), pyrexia (21%), increased alanine aminotransferase (18%), increased aspartate aminotransferase (18%), and febrile neutropenia (15%).
Three patients developed cytokine release syndrome (2 grade 3).
and solution for infusion
Photo courtesy of Amgen
The US Food and Drug Administration (FDA) has granted accelerated approval for blinatumomab (Blincyto®) to treat pediatric patients with Philadelphia chromosome-negative (Ph-) relapsed or refractory B-cell precursor acute lymphoblastic leukemia (BCP-ALL).
The approval is based on results from a phase 1/2 study.
Continued approval of blinatumomab for this indication may be contingent upon verification of clinical benefit in subsequent trials.
Blinatumomab is a bispecific, CD19-directed, CD3 T-cell engager (BiTE®) antibody construct that binds specifically to CD19 expressed on the surface of cells of B-lineage origin and CD3 expressed on the surface of T cells.
In 2014, blinatumomab received accelerated approval from the FDA to treat adults with Ph- relapsed or refractory BCP-ALL. The FDA has also granted the drug priority review, breakthrough therapy designation, and orphan drug designation.
Blinatumomab is being developed by Amgen. Full prescribing information is available at www.blincyto.com.
‘205 trial
The latest accelerated approval of blinatumomab is based on results from the phase 1/2 ‘205 trial, in which researchers evaluated blinatumomab in 93 pediatric patients with relapsed or refractory BCP-ALL.
Amgen said treatment in this study has been completed, subjects are being monitored for long-term efficacy, and the data will be submitted for publication.
Initial results from this study were presented at ASH 2014. The abstract included data on 39 patients with relapsed/refractory BCP-ALL and a median age of 9 (range, 2-16).
The patients received blinatumomab at a dose of 5-15 µg/m²/day. Nineteen patients completed 1 cycle of blinatumomab, 4 completed 2 cycles, and 2 completed 3 cycles.
During the first 2 treatment cycles, 12 patients achieved a complete response, 5 of whom were negative for minimal residual disease.
Six of the complete responders went on to transplant. The median relapse-free survival for complete responders was 5.6 months.
At 6 months of follow-up, the median overall survival for all 39 patients was 4.3 months.
All of the patients experienced adverse events. The most common were pyrexia (74%), anemia (33%), nausea (31%), headache (28%), hypertension (26%), increased alanine aminotransferase (23%), and cough (21%).
The most common grade 3 or higher events were anemia (26%), pyrexia (21%), increased alanine aminotransferase (18%), increased aspartate aminotransferase (18%), and febrile neutropenia (15%).
Three patients developed cytokine release syndrome (2 grade 3).
and solution for infusion
Photo courtesy of Amgen
The US Food and Drug Administration (FDA) has granted accelerated approval for blinatumomab (Blincyto®) to treat pediatric patients with Philadelphia chromosome-negative (Ph-) relapsed or refractory B-cell precursor acute lymphoblastic leukemia (BCP-ALL).
The approval is based on results from a phase 1/2 study.
Continued approval of blinatumomab for this indication may be contingent upon verification of clinical benefit in subsequent trials.
Blinatumomab is a bispecific, CD19-directed, CD3 T-cell engager (BiTE®) antibody construct that binds specifically to CD19 expressed on the surface of cells of B-lineage origin and CD3 expressed on the surface of T cells.
In 2014, blinatumomab received accelerated approval from the FDA to treat adults with Ph- relapsed or refractory BCP-ALL. The FDA has also granted the drug priority review, breakthrough therapy designation, and orphan drug designation.
Blinatumomab is being developed by Amgen. Full prescribing information is available at www.blincyto.com.
‘205 trial
The latest accelerated approval of blinatumomab is based on results from the phase 1/2 ‘205 trial, in which researchers evaluated blinatumomab in 93 pediatric patients with relapsed or refractory BCP-ALL.
Amgen said treatment in this study has been completed, subjects are being monitored for long-term efficacy, and the data will be submitted for publication.
Initial results from this study were presented at ASH 2014. The abstract included data on 39 patients with relapsed/refractory BCP-ALL and a median age of 9 (range, 2-16).
The patients received blinatumomab at a dose of 5-15 µg/m²/day. Nineteen patients completed 1 cycle of blinatumomab, 4 completed 2 cycles, and 2 completed 3 cycles.
During the first 2 treatment cycles, 12 patients achieved a complete response, 5 of whom were negative for minimal residual disease.
Six of the complete responders went on to transplant. The median relapse-free survival for complete responders was 5.6 months.
At 6 months of follow-up, the median overall survival for all 39 patients was 4.3 months.
All of the patients experienced adverse events. The most common were pyrexia (74%), anemia (33%), nausea (31%), headache (28%), hypertension (26%), increased alanine aminotransferase (23%), and cough (21%).
The most common grade 3 or higher events were anemia (26%), pyrexia (21%), increased alanine aminotransferase (18%), increased aspartate aminotransferase (18%), and febrile neutropenia (15%).
Three patients developed cytokine release syndrome (2 grade 3).
FDA approves new indication for ofatumumab in CLL

Photo courtesy of GSK
The US Food and Drug Administration (FDA) has approved the use of ofatumumab (Arzerra®) in combination with fludarabine and cyclophosphamide to treat patients with relapsed chronic lymphocytic leukemia (CLL).
Ofatumumab was previously approved by the FDA for use in combination with chlorambucil to treat previously untreated CLL patients who cannot receive fludarabine-based therapy, as monotherapy for CLL that is refractory to fludarabine and alemtuzumab, and as maintenance therapy for patients who are in complete or partial response after receiving at least 2 lines of therapy for recurrent or progressive CLL.
Ofatumumab is a monoclonal antibody designed to target CD20.
The drug’s prescribing information includes a boxed warning noting that hepatitis B virus reactivation can occur in patients receiving CD20-directed cytolytic antibodies, including ofatumumab. In some cases, this results in fulminant hepatitis, hepatic failure, and death.
The boxed warning also states that progressive multifocal leukoencephalopathy, resulting in death, can occur in patients receiving CD20-directed cytolytic antibodies, including ofatumumab.
Ofatumumab is marketed under a collaboration agreement between Genmab and Novartis.
COMPLEMENT 2 trial
The FDA’s latest approval for ofatumumab is based on results of the phase 3 COMPLEMENT 2 trial. Novartis reported top-line results from this study in April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed.
Photo courtesy of GSK
The US Food and Drug Administration (FDA) has approved the use of ofatumumab (Arzerra®) in combination with fludarabine and cyclophosphamide to treat patients with relapsed chronic lymphocytic leukemia (CLL).
Ofatumumab was previously approved by the FDA for use in combination with chlorambucil to treat previously untreated CLL patients who cannot receive fludarabine-based therapy, as monotherapy for CLL that is refractory to fludarabine and alemtuzumab, and as maintenance therapy for patients who are in complete or partial response after receiving at least 2 lines of therapy for recurrent or progressive CLL.
Ofatumumab is a monoclonal antibody designed to target CD20.
The drug’s prescribing information includes a boxed warning noting that hepatitis B virus reactivation can occur in patients receiving CD20-directed cytolytic antibodies, including ofatumumab. In some cases, this results in fulminant hepatitis, hepatic failure, and death.
The boxed warning also states that progressive multifocal leukoencephalopathy, resulting in death, can occur in patients receiving CD20-directed cytolytic antibodies, including ofatumumab.
Ofatumumab is marketed under a collaboration agreement between Genmab and Novartis.
COMPLEMENT 2 trial
The FDA’s latest approval for ofatumumab is based on results of the phase 3 COMPLEMENT 2 trial. Novartis reported top-line results from this study in April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed.
Photo courtesy of GSK
The US Food and Drug Administration (FDA) has approved the use of ofatumumab (Arzerra®) in combination with fludarabine and cyclophosphamide to treat patients with relapsed chronic lymphocytic leukemia (CLL).
Ofatumumab was previously approved by the FDA for use in combination with chlorambucil to treat previously untreated CLL patients who cannot receive fludarabine-based therapy, as monotherapy for CLL that is refractory to fludarabine and alemtuzumab, and as maintenance therapy for patients who are in complete or partial response after receiving at least 2 lines of therapy for recurrent or progressive CLL.
Ofatumumab is a monoclonal antibody designed to target CD20.
The drug’s prescribing information includes a boxed warning noting that hepatitis B virus reactivation can occur in patients receiving CD20-directed cytolytic antibodies, including ofatumumab. In some cases, this results in fulminant hepatitis, hepatic failure, and death.
The boxed warning also states that progressive multifocal leukoencephalopathy, resulting in death, can occur in patients receiving CD20-directed cytolytic antibodies, including ofatumumab.
Ofatumumab is marketed under a collaboration agreement between Genmab and Novartis.
COMPLEMENT 2 trial
The FDA’s latest approval for ofatumumab is based on results of the phase 3 COMPLEMENT 2 trial. Novartis reported top-line results from this study in April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed.
NICE approves bosutinib for routine NHS use
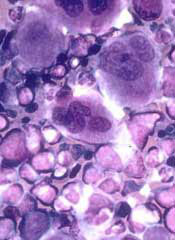
The National Institute for Health and Care Excellence (NICE) has issued a final guidance recommending that bosutinib (Bosulif), a tyrosine kinase inhibitor used to treat certain patients with chronic myeloid leukemia (CML), be made available through the National Health Service (NHS).
This means patients will no longer have to apply to the Cancer Drugs Fund (CDF) to obtain bosutinib.
The CDF is money the government sets aside to pay for cancer drugs that haven’t been approved by NICE and aren’t available within the NHS in England.
Following the decision to reform the CDF earlier this year, NICE began to reappraise all drugs currently in the CDF in April. Bosutinib is the first drug to be looked at through this reconsideration process.
NICE previously considered making bosutinib available through the NHS in 2013 but decided the drug was not cost-effective. So bosutinib was made available to patients via the CDF.
As part of the reappraisal process, Pfizer offered a discount for bosutinib. Taking this discount into consideration, as well as the limited treatment options for CML patients, NICE decided bosutinib is cost-effective.
Bosutinib has conditional approval from the European Commission to treat adults with Philadelphia-chromosome-positive CML in chronic phase, accelerated phase, or blast phase, but only if those patients have previously received one or more tyrosine kinase inhibitors and are not considered eligible for treatment with imatinib, nilotinib, or dasatinib.
The National Institute for Health and Care Excellence (NICE) has issued a final guidance recommending that bosutinib (Bosulif), a tyrosine kinase inhibitor used to treat certain patients with chronic myeloid leukemia (CML), be made available through the National Health Service (NHS).
This means patients will no longer have to apply to the Cancer Drugs Fund (CDF) to obtain bosutinib.
The CDF is money the government sets aside to pay for cancer drugs that haven’t been approved by NICE and aren’t available within the NHS in England.
Following the decision to reform the CDF earlier this year, NICE began to reappraise all drugs currently in the CDF in April. Bosutinib is the first drug to be looked at through this reconsideration process.
NICE previously considered making bosutinib available through the NHS in 2013 but decided the drug was not cost-effective. So bosutinib was made available to patients via the CDF.
As part of the reappraisal process, Pfizer offered a discount for bosutinib. Taking this discount into consideration, as well as the limited treatment options for CML patients, NICE decided bosutinib is cost-effective.
Bosutinib has conditional approval from the European Commission to treat adults with Philadelphia-chromosome-positive CML in chronic phase, accelerated phase, or blast phase, but only if those patients have previously received one or more tyrosine kinase inhibitors and are not considered eligible for treatment with imatinib, nilotinib, or dasatinib.
The National Institute for Health and Care Excellence (NICE) has issued a final guidance recommending that bosutinib (Bosulif), a tyrosine kinase inhibitor used to treat certain patients with chronic myeloid leukemia (CML), be made available through the National Health Service (NHS).
This means patients will no longer have to apply to the Cancer Drugs Fund (CDF) to obtain bosutinib.
The CDF is money the government sets aside to pay for cancer drugs that haven’t been approved by NICE and aren’t available within the NHS in England.
Following the decision to reform the CDF earlier this year, NICE began to reappraise all drugs currently in the CDF in April. Bosutinib is the first drug to be looked at through this reconsideration process.
NICE previously considered making bosutinib available through the NHS in 2013 but decided the drug was not cost-effective. So bosutinib was made available to patients via the CDF.
As part of the reappraisal process, Pfizer offered a discount for bosutinib. Taking this discount into consideration, as well as the limited treatment options for CML patients, NICE decided bosutinib is cost-effective.
Bosutinib has conditional approval from the European Commission to treat adults with Philadelphia-chromosome-positive CML in chronic phase, accelerated phase, or blast phase, but only if those patients have previously received one or more tyrosine kinase inhibitors and are not considered eligible for treatment with imatinib, nilotinib, or dasatinib.