User login
Cells might protect cancer patients from infection

receiving chemotherapy
Photo by Rhoda Baer
Researchers say they have discovered a type of macrophage that may protect against lung infections during chemotherapy.
These macrophages, found in the lungs of mice, were able to survive chemotherapy.
The macrophages could remove bacteria when activated by a vaccine, which improved survival in mice with lethal bacterial pneumonia that had received chemotherapy and were therefore depleted of neutrophils.
The researchers said these results suggest the cells—known as vaccine-induced macrophages (ViMs)— might be able to protect cancer patients from life-threatening infections.
“We have identified a new form of housekeeping macrophage in mice that may, in future, be harnessed to protect against lung infections—like bacterial pneumonia—that remain one of the greatest threats to survival of cancer patients during chemotherapy,” said Peter Murray, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Dr Murray and his colleagues detailed this discovery in PNAS.
Working in a mouse model that mimics infection in chemotherapy-treated patients, the researchers found that vaccination protected mice from Pseudomonas aeruginosa pneumonia.
The quest to understand how such protection was possible in the absence of neutrophils led the team to discover ViMs.
The researchers found that ViMs were produced in the lungs following vaccination, proliferated locally, and could persist for at least a month.
Analyses suggested ViMs are closely related to alveolar macrophages, which originate in the embryo, reside in the air-exposed surfaces of alveoli, and are self-maintained in adults.
“All lines of cellular and molecular evidence in this study point to alveolar macrophages as the source of ViMs,” Dr Murray said.
However, unlike alveolar macrophages, the population of ViMs remained stable during chemotherapy and exhibited enhanced phagocytic activity.
When ViMs were transferred to unvaccinated mice depleted of neutrophils via chemotherapy, the animals were protected from lethal Pseudomonas infections.
“We now know that increasing the number of ViMs in the tissue can compensate for the immune deficit caused by chemotherapy,” said study author Akinobu Kamei, MD, of St. Jude Children’s Research Hospital.
“In this study, we relied on vaccination prior to chemotherapy. Going forward, we will explore other, more practical methods for use at the bedside to effectively induce tissue-resident macrophages like ViMs.”
The possible approaches include using drugs or cytokines to induce protection in the immune-compromised host. ![]()
receiving chemotherapy
Photo by Rhoda Baer
Researchers say they have discovered a type of macrophage that may protect against lung infections during chemotherapy.
These macrophages, found in the lungs of mice, were able to survive chemotherapy.
The macrophages could remove bacteria when activated by a vaccine, which improved survival in mice with lethal bacterial pneumonia that had received chemotherapy and were therefore depleted of neutrophils.
The researchers said these results suggest the cells—known as vaccine-induced macrophages (ViMs)— might be able to protect cancer patients from life-threatening infections.
“We have identified a new form of housekeeping macrophage in mice that may, in future, be harnessed to protect against lung infections—like bacterial pneumonia—that remain one of the greatest threats to survival of cancer patients during chemotherapy,” said Peter Murray, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Dr Murray and his colleagues detailed this discovery in PNAS.
Working in a mouse model that mimics infection in chemotherapy-treated patients, the researchers found that vaccination protected mice from Pseudomonas aeruginosa pneumonia.
The quest to understand how such protection was possible in the absence of neutrophils led the team to discover ViMs.
The researchers found that ViMs were produced in the lungs following vaccination, proliferated locally, and could persist for at least a month.
Analyses suggested ViMs are closely related to alveolar macrophages, which originate in the embryo, reside in the air-exposed surfaces of alveoli, and are self-maintained in adults.
“All lines of cellular and molecular evidence in this study point to alveolar macrophages as the source of ViMs,” Dr Murray said.
However, unlike alveolar macrophages, the population of ViMs remained stable during chemotherapy and exhibited enhanced phagocytic activity.
When ViMs were transferred to unvaccinated mice depleted of neutrophils via chemotherapy, the animals were protected from lethal Pseudomonas infections.
“We now know that increasing the number of ViMs in the tissue can compensate for the immune deficit caused by chemotherapy,” said study author Akinobu Kamei, MD, of St. Jude Children’s Research Hospital.
“In this study, we relied on vaccination prior to chemotherapy. Going forward, we will explore other, more practical methods for use at the bedside to effectively induce tissue-resident macrophages like ViMs.”
The possible approaches include using drugs or cytokines to induce protection in the immune-compromised host. ![]()
receiving chemotherapy
Photo by Rhoda Baer
Researchers say they have discovered a type of macrophage that may protect against lung infections during chemotherapy.
These macrophages, found in the lungs of mice, were able to survive chemotherapy.
The macrophages could remove bacteria when activated by a vaccine, which improved survival in mice with lethal bacterial pneumonia that had received chemotherapy and were therefore depleted of neutrophils.
The researchers said these results suggest the cells—known as vaccine-induced macrophages (ViMs)— might be able to protect cancer patients from life-threatening infections.
“We have identified a new form of housekeeping macrophage in mice that may, in future, be harnessed to protect against lung infections—like bacterial pneumonia—that remain one of the greatest threats to survival of cancer patients during chemotherapy,” said Peter Murray, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Dr Murray and his colleagues detailed this discovery in PNAS.
Working in a mouse model that mimics infection in chemotherapy-treated patients, the researchers found that vaccination protected mice from Pseudomonas aeruginosa pneumonia.
The quest to understand how such protection was possible in the absence of neutrophils led the team to discover ViMs.
The researchers found that ViMs were produced in the lungs following vaccination, proliferated locally, and could persist for at least a month.
Analyses suggested ViMs are closely related to alveolar macrophages, which originate in the embryo, reside in the air-exposed surfaces of alveoli, and are self-maintained in adults.
“All lines of cellular and molecular evidence in this study point to alveolar macrophages as the source of ViMs,” Dr Murray said.
However, unlike alveolar macrophages, the population of ViMs remained stable during chemotherapy and exhibited enhanced phagocytic activity.
When ViMs were transferred to unvaccinated mice depleted of neutrophils via chemotherapy, the animals were protected from lethal Pseudomonas infections.
“We now know that increasing the number of ViMs in the tissue can compensate for the immune deficit caused by chemotherapy,” said study author Akinobu Kamei, MD, of St. Jude Children’s Research Hospital.
“In this study, we relied on vaccination prior to chemotherapy. Going forward, we will explore other, more practical methods for use at the bedside to effectively induce tissue-resident macrophages like ViMs.”
The possible approaches include using drugs or cytokines to induce protection in the immune-compromised host. ![]()
Preserved slides provide insight into European malaria

Photo from the Institute
of Evolutionary Biology
A new study published in PNAS has provided insights regarding the origin and spread of European malaria.
The malaria-causing parasites Plasmodium vivax and Plasmodium falciparum were eradicated in Europe in the mid-twentieth century.
Now, researchers have recovered genetic data from European samples of malaria preserved on microscope slides in the 1940s.
The team performed second-generation sequencing on DNA extracted from 3 of the slides, which generated millions of sequences of malaria-causing parasites.
The researchers were then able to reconstruct the parasites’ mitochondrial genomes and compare them with those of present-day samples worldwide.
“The European sequence of P vivax is closely related to the most common strain currently found in Central and South America,” said study author Carles Lalueza-Fox, PhD, of the Institute of Evolutionary Biology (Consejo Superior de Investigaciones Científicas-Universitat Pompeu Fabra) in Barcelona, Spain.
“This suggests that the pathogen was introduced to the Americas by European colonists after Columbus. In contrast, the European sequence of P falciparum belongs to a strain which has only been found in India. This indicates that the pathogen of the most severe form of malaria was introduced to Europe from the Indian subcontinent, probably some 2500 years ago.”
The European samples the researchers analyzed were dated between 1942 and 1944. They originate from an old antimalarial center inaugurated in 1925 in Sant Jaume d’Enveja, on the Ebro Delta, located in Spain’s north-eastern region of Tarragona.
The center’s head, Ildefonso Canicio, spent decades treating malaria sufferers who worked in the area’s rice fields and ultimately contracted the disease himself.
Following Dr Canicio’s death in 1961, some of his slides, which were used for diagnostic purposes, were saved from destruction when they were recognized by his descendants, who allowed them to be used in the current study.
“It is still possible to see malaria-carrying parasites on the slides when they are studied under the microscope,” Dr Lalueza-Fox said. “However, the quantity of the pathogen’s DNA available in a single drop of blood is very limited, and when you add to that the issue of poor preservation after 70 years, it is clear why this type of study has never been carried out.”
Still, the researchers said this study has shown that historic specimens can be an important source of insight into the genetics of extinct or eradicated pathogens.
“Analyzing the nuclear genome in these pathogens will allow us to know more about the mutations which have made current-day strains resistant to different drugs, given that the European Plasmodium which has been retrieved is older than all of these treatments,” said study author Pere Gelabert, also of the Institute of Evolutionary Biology. ![]()
Photo from the Institute
of Evolutionary Biology
A new study published in PNAS has provided insights regarding the origin and spread of European malaria.
The malaria-causing parasites Plasmodium vivax and Plasmodium falciparum were eradicated in Europe in the mid-twentieth century.
Now, researchers have recovered genetic data from European samples of malaria preserved on microscope slides in the 1940s.
The team performed second-generation sequencing on DNA extracted from 3 of the slides, which generated millions of sequences of malaria-causing parasites.
The researchers were then able to reconstruct the parasites’ mitochondrial genomes and compare them with those of present-day samples worldwide.
“The European sequence of P vivax is closely related to the most common strain currently found in Central and South America,” said study author Carles Lalueza-Fox, PhD, of the Institute of Evolutionary Biology (Consejo Superior de Investigaciones Científicas-Universitat Pompeu Fabra) in Barcelona, Spain.
“This suggests that the pathogen was introduced to the Americas by European colonists after Columbus. In contrast, the European sequence of P falciparum belongs to a strain which has only been found in India. This indicates that the pathogen of the most severe form of malaria was introduced to Europe from the Indian subcontinent, probably some 2500 years ago.”
The European samples the researchers analyzed were dated between 1942 and 1944. They originate from an old antimalarial center inaugurated in 1925 in Sant Jaume d’Enveja, on the Ebro Delta, located in Spain’s north-eastern region of Tarragona.
The center’s head, Ildefonso Canicio, spent decades treating malaria sufferers who worked in the area’s rice fields and ultimately contracted the disease himself.
Following Dr Canicio’s death in 1961, some of his slides, which were used for diagnostic purposes, were saved from destruction when they were recognized by his descendants, who allowed them to be used in the current study.
“It is still possible to see malaria-carrying parasites on the slides when they are studied under the microscope,” Dr Lalueza-Fox said. “However, the quantity of the pathogen’s DNA available in a single drop of blood is very limited, and when you add to that the issue of poor preservation after 70 years, it is clear why this type of study has never been carried out.”
Still, the researchers said this study has shown that historic specimens can be an important source of insight into the genetics of extinct or eradicated pathogens.
“Analyzing the nuclear genome in these pathogens will allow us to know more about the mutations which have made current-day strains resistant to different drugs, given that the European Plasmodium which has been retrieved is older than all of these treatments,” said study author Pere Gelabert, also of the Institute of Evolutionary Biology. ![]()
Photo from the Institute
of Evolutionary Biology
A new study published in PNAS has provided insights regarding the origin and spread of European malaria.
The malaria-causing parasites Plasmodium vivax and Plasmodium falciparum were eradicated in Europe in the mid-twentieth century.
Now, researchers have recovered genetic data from European samples of malaria preserved on microscope slides in the 1940s.
The team performed second-generation sequencing on DNA extracted from 3 of the slides, which generated millions of sequences of malaria-causing parasites.
The researchers were then able to reconstruct the parasites’ mitochondrial genomes and compare them with those of present-day samples worldwide.
“The European sequence of P vivax is closely related to the most common strain currently found in Central and South America,” said study author Carles Lalueza-Fox, PhD, of the Institute of Evolutionary Biology (Consejo Superior de Investigaciones Científicas-Universitat Pompeu Fabra) in Barcelona, Spain.
“This suggests that the pathogen was introduced to the Americas by European colonists after Columbus. In contrast, the European sequence of P falciparum belongs to a strain which has only been found in India. This indicates that the pathogen of the most severe form of malaria was introduced to Europe from the Indian subcontinent, probably some 2500 years ago.”
The European samples the researchers analyzed were dated between 1942 and 1944. They originate from an old antimalarial center inaugurated in 1925 in Sant Jaume d’Enveja, on the Ebro Delta, located in Spain’s north-eastern region of Tarragona.
The center’s head, Ildefonso Canicio, spent decades treating malaria sufferers who worked in the area’s rice fields and ultimately contracted the disease himself.
Following Dr Canicio’s death in 1961, some of his slides, which were used for diagnostic purposes, were saved from destruction when they were recognized by his descendants, who allowed them to be used in the current study.
“It is still possible to see malaria-carrying parasites on the slides when they are studied under the microscope,” Dr Lalueza-Fox said. “However, the quantity of the pathogen’s DNA available in a single drop of blood is very limited, and when you add to that the issue of poor preservation after 70 years, it is clear why this type of study has never been carried out.”
Still, the researchers said this study has shown that historic specimens can be an important source of insight into the genetics of extinct or eradicated pathogens.
“Analyzing the nuclear genome in these pathogens will allow us to know more about the mutations which have made current-day strains resistant to different drugs, given that the European Plasmodium which has been retrieved is older than all of these treatments,” said study author Pere Gelabert, also of the Institute of Evolutionary Biology. ![]()
EMA and FDA collaborate to combat rare diseases

Photo courtesy of the FDA
The European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) have set up a new working group, or “cluster,” on rare diseases.
This means the EMA and FDA will hold regular meetings via teleconference to share information on the regulation of medicines for rare diseases.
The cluster will provide a forum for the confidential exchange of draft documents, proposed policies, and detailed information supporting the scientific basis for decision-making on medicine development.
The agencies will exchange information on topics such as:
- The design of clinical trials in small populations and the use of statistical analysis methods
- The selection and validation of trial endpoints
- Preclinical evidence to support development programs
- The design of post-marketing studies—in particular, in the context of early access mechanisms such as the EMA’s conditional marketing authorization and the FDA’s accelerated approval
- Risk management strategies for long-term safety issues with medicines for rare diseases.
The first meeting of the rare diseases cluster took place on September 23, 2016. The cluster will initially meet once a month via teleconference and will be chaired jointly by the FDA and EMA.
The creation of this cluster is the latest step in the EMA’s and FDA’s wider objective to expand and reinforce international collaboration.
The clusters established by the agencies focus on areas where the parties involved could benefit from an intensified exchange of information and strengthened collaboration.
The existing EMA/FDA clusters address issues related to patient engagement, biosimilars, orphan medicines, cancer drugs, medicines for children, and pharmacovigilance, among other topics. ![]()
Photo courtesy of the FDA
The European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) have set up a new working group, or “cluster,” on rare diseases.
This means the EMA and FDA will hold regular meetings via teleconference to share information on the regulation of medicines for rare diseases.
The cluster will provide a forum for the confidential exchange of draft documents, proposed policies, and detailed information supporting the scientific basis for decision-making on medicine development.
The agencies will exchange information on topics such as:
- The design of clinical trials in small populations and the use of statistical analysis methods
- The selection and validation of trial endpoints
- Preclinical evidence to support development programs
- The design of post-marketing studies—in particular, in the context of early access mechanisms such as the EMA’s conditional marketing authorization and the FDA’s accelerated approval
- Risk management strategies for long-term safety issues with medicines for rare diseases.
The first meeting of the rare diseases cluster took place on September 23, 2016. The cluster will initially meet once a month via teleconference and will be chaired jointly by the FDA and EMA.
The creation of this cluster is the latest step in the EMA’s and FDA’s wider objective to expand and reinforce international collaboration.
The clusters established by the agencies focus on areas where the parties involved could benefit from an intensified exchange of information and strengthened collaboration.
The existing EMA/FDA clusters address issues related to patient engagement, biosimilars, orphan medicines, cancer drugs, medicines for children, and pharmacovigilance, among other topics. ![]()
Photo courtesy of the FDA
The European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) have set up a new working group, or “cluster,” on rare diseases.
This means the EMA and FDA will hold regular meetings via teleconference to share information on the regulation of medicines for rare diseases.
The cluster will provide a forum for the confidential exchange of draft documents, proposed policies, and detailed information supporting the scientific basis for decision-making on medicine development.
The agencies will exchange information on topics such as:
- The design of clinical trials in small populations and the use of statistical analysis methods
- The selection and validation of trial endpoints
- Preclinical evidence to support development programs
- The design of post-marketing studies—in particular, in the context of early access mechanisms such as the EMA’s conditional marketing authorization and the FDA’s accelerated approval
- Risk management strategies for long-term safety issues with medicines for rare diseases.
The first meeting of the rare diseases cluster took place on September 23, 2016. The cluster will initially meet once a month via teleconference and will be chaired jointly by the FDA and EMA.
The creation of this cluster is the latest step in the EMA’s and FDA’s wider objective to expand and reinforce international collaboration.
The clusters established by the agencies focus on areas where the parties involved could benefit from an intensified exchange of information and strengthened collaboration.
The existing EMA/FDA clusters address issues related to patient engagement, biosimilars, orphan medicines, cancer drugs, medicines for children, and pharmacovigilance, among other topics. ![]()
Malaria vaccine receives fast track designation

Image by Ute Frevert
and Margaret Shear
The US Food and Drug Administration (FDA) has granted fast track designation to an investigational malaria vaccine.
Sanaria® PfSPZ Vaccine is composed of live but weakened Plasmodium falciparum sporozoites and is being developed by Sanaria, Inc.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the FDA’s fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the biologic license application or new drug application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA.
Trials of Sanaria® PfSPZ Vaccine
In a phase 1 study published in Science in 2013, 80% of patients who received higher doses of Sanaria® PfSPZ Vaccine were protected from malaria at 3 weeks after vaccination.
In another phase 1 study published in Nature Medicine in 2016, 4 doses of the vaccine protected 73% of subjects from malaria at 3 weeks and 55% of subjects at 21 weeks.
Five of the subjects without parasitemia at 21 weeks were exposed to malaria-carrying mosquitoes again at 59 weeks, and none developed parasitemia.
To date, 1165 volunteers have received Sanaria’s PfSPZ-based products in more than 2 dozen clinical trials in the US, Europe, and Africa.
Clinical trials are in progress in Tanzania, Kenya, Mali, Burkina Faso, Germany, and the US, and are intended to begin soon in Equatorial Guinea. ![]()
Image by Ute Frevert
and Margaret Shear
The US Food and Drug Administration (FDA) has granted fast track designation to an investigational malaria vaccine.
Sanaria® PfSPZ Vaccine is composed of live but weakened Plasmodium falciparum sporozoites and is being developed by Sanaria, Inc.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the FDA’s fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the biologic license application or new drug application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA.
Trials of Sanaria® PfSPZ Vaccine
In a phase 1 study published in Science in 2013, 80% of patients who received higher doses of Sanaria® PfSPZ Vaccine were protected from malaria at 3 weeks after vaccination.
In another phase 1 study published in Nature Medicine in 2016, 4 doses of the vaccine protected 73% of subjects from malaria at 3 weeks and 55% of subjects at 21 weeks.
Five of the subjects without parasitemia at 21 weeks were exposed to malaria-carrying mosquitoes again at 59 weeks, and none developed parasitemia.
To date, 1165 volunteers have received Sanaria’s PfSPZ-based products in more than 2 dozen clinical trials in the US, Europe, and Africa.
Clinical trials are in progress in Tanzania, Kenya, Mali, Burkina Faso, Germany, and the US, and are intended to begin soon in Equatorial Guinea. ![]()
Image by Ute Frevert
and Margaret Shear
The US Food and Drug Administration (FDA) has granted fast track designation to an investigational malaria vaccine.
Sanaria® PfSPZ Vaccine is composed of live but weakened Plasmodium falciparum sporozoites and is being developed by Sanaria, Inc.
The FDA’s fast track program is designed to facilitate the development and expedite the review of products intended to treat or prevent serious or life-threatening conditions and address unmet medical need.
Through the FDA’s fast track program, a product may be eligible for priority review. In addition, the company developing the product may be allowed to submit sections of the biologic license application or new drug application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings and written communications with the FDA.
Trials of Sanaria® PfSPZ Vaccine
In a phase 1 study published in Science in 2013, 80% of patients who received higher doses of Sanaria® PfSPZ Vaccine were protected from malaria at 3 weeks after vaccination.
In another phase 1 study published in Nature Medicine in 2016, 4 doses of the vaccine protected 73% of subjects from malaria at 3 weeks and 55% of subjects at 21 weeks.
Five of the subjects without parasitemia at 21 weeks were exposed to malaria-carrying mosquitoes again at 59 weeks, and none developed parasitemia.
To date, 1165 volunteers have received Sanaria’s PfSPZ-based products in more than 2 dozen clinical trials in the US, Europe, and Africa.
Clinical trials are in progress in Tanzania, Kenya, Mali, Burkina Faso, Germany, and the US, and are intended to begin soon in Equatorial Guinea. ![]()
Blood pressure drugs could treat cerebral malaria

vessel with nuclei in blue,
protein skeletons in red,
and beta-catenin in green.
Image courtesy of
NYU Langone Medical Center
Drugs intended to treat high blood pressure might also be effective for treating cerebral malaria, according to preclinical research published in the Journal of Clinical Investigation.
Researchers tested these drugs—angiotensin II (Ang II) receptor modulators—in mice.
In an initial experiment, 2 of the drugs—losartan and CGP-42112A—reduced the incidence of cerebral malaria.
In another experiment, 2 other Ang II receptor modulators—irbesartan and compound 21 (C21)—were each combined with the antimalarial chloroquine.
Both combinations improved survival in mice with cerebral malaria when compared to chloroquine alone.
“About 1 in 5 patients with cerebral malaria die within 48 hours of being admitted to the hospital—the time it takes for the parasite-killing drug to take effect,” said study author Ana Rodriguez, PhD, of NYU Langone Medical Center in New York.
“If we could add a drug that stopped hemorrhages during that window, it would buy time and save lives.”
With this research, Dr Rodriguez and her colleagues showed that Ang II receptor modulators inhibit the activation of beta-catenin, a protein that plays a key role in cerebral malaria.
The team found that when Plasmodium falciparum–infected red blood cells rupture over human brain microvascular endothelial cells, this activates beta-catenin, which mediates the disruption of interendothelial junctions.
But inhibiting the activation of beta-catenin with Ang II receptor modulators protects endothelial cells from this disruption.
The researchers demonstrated this by infecting mice with P berghei ANKA, treating them with losartan or CGP-42112A, and monitoring them for neurological symptoms.
Seventy-five percent of control mice developed cerebral malaria, compared to 13.3% of mice treated with losartan and 28.5% treated with CGP-42112A.
In another experiment, the researchers treated malaria-infected mice with chloroquine, alone or in combination with irbesartan or C21, only after neurological signs were evident.
The team observed an increase in survival for mice treated with irbesartan or C21. The survival rate was 18% for the mice treated only with chloroquine, 65% for mice given irbesartan, and 73% percent for mice treated with C21.
The researchers also found that mice treated with irbesartan or C21 experienced fewer, smaller hemorrhages. And, in most cases, these mice fully recovered.
Dr Rodriguez and her colleagues said the next steps for this research will be to investigate the exact molecules coming from ruptured P falciparum–infected red blood cells that signal to beta-catenin in vessel walls, and to conduct trials that test the effects of Ang II receptor modulators in patients with cerebral malaria. ![]()
vessel with nuclei in blue,
protein skeletons in red,
and beta-catenin in green.
Image courtesy of
NYU Langone Medical Center
Drugs intended to treat high blood pressure might also be effective for treating cerebral malaria, according to preclinical research published in the Journal of Clinical Investigation.
Researchers tested these drugs—angiotensin II (Ang II) receptor modulators—in mice.
In an initial experiment, 2 of the drugs—losartan and CGP-42112A—reduced the incidence of cerebral malaria.
In another experiment, 2 other Ang II receptor modulators—irbesartan and compound 21 (C21)—were each combined with the antimalarial chloroquine.
Both combinations improved survival in mice with cerebral malaria when compared to chloroquine alone.
“About 1 in 5 patients with cerebral malaria die within 48 hours of being admitted to the hospital—the time it takes for the parasite-killing drug to take effect,” said study author Ana Rodriguez, PhD, of NYU Langone Medical Center in New York.
“If we could add a drug that stopped hemorrhages during that window, it would buy time and save lives.”
With this research, Dr Rodriguez and her colleagues showed that Ang II receptor modulators inhibit the activation of beta-catenin, a protein that plays a key role in cerebral malaria.
The team found that when Plasmodium falciparum–infected red blood cells rupture over human brain microvascular endothelial cells, this activates beta-catenin, which mediates the disruption of interendothelial junctions.
But inhibiting the activation of beta-catenin with Ang II receptor modulators protects endothelial cells from this disruption.
The researchers demonstrated this by infecting mice with P berghei ANKA, treating them with losartan or CGP-42112A, and monitoring them for neurological symptoms.
Seventy-five percent of control mice developed cerebral malaria, compared to 13.3% of mice treated with losartan and 28.5% treated with CGP-42112A.
In another experiment, the researchers treated malaria-infected mice with chloroquine, alone or in combination with irbesartan or C21, only after neurological signs were evident.
The team observed an increase in survival for mice treated with irbesartan or C21. The survival rate was 18% for the mice treated only with chloroquine, 65% for mice given irbesartan, and 73% percent for mice treated with C21.
The researchers also found that mice treated with irbesartan or C21 experienced fewer, smaller hemorrhages. And, in most cases, these mice fully recovered.
Dr Rodriguez and her colleagues said the next steps for this research will be to investigate the exact molecules coming from ruptured P falciparum–infected red blood cells that signal to beta-catenin in vessel walls, and to conduct trials that test the effects of Ang II receptor modulators in patients with cerebral malaria. ![]()
vessel with nuclei in blue,
protein skeletons in red,
and beta-catenin in green.
Image courtesy of
NYU Langone Medical Center
Drugs intended to treat high blood pressure might also be effective for treating cerebral malaria, according to preclinical research published in the Journal of Clinical Investigation.
Researchers tested these drugs—angiotensin II (Ang II) receptor modulators—in mice.
In an initial experiment, 2 of the drugs—losartan and CGP-42112A—reduced the incidence of cerebral malaria.
In another experiment, 2 other Ang II receptor modulators—irbesartan and compound 21 (C21)—were each combined with the antimalarial chloroquine.
Both combinations improved survival in mice with cerebral malaria when compared to chloroquine alone.
“About 1 in 5 patients with cerebral malaria die within 48 hours of being admitted to the hospital—the time it takes for the parasite-killing drug to take effect,” said study author Ana Rodriguez, PhD, of NYU Langone Medical Center in New York.
“If we could add a drug that stopped hemorrhages during that window, it would buy time and save lives.”
With this research, Dr Rodriguez and her colleagues showed that Ang II receptor modulators inhibit the activation of beta-catenin, a protein that plays a key role in cerebral malaria.
The team found that when Plasmodium falciparum–infected red blood cells rupture over human brain microvascular endothelial cells, this activates beta-catenin, which mediates the disruption of interendothelial junctions.
But inhibiting the activation of beta-catenin with Ang II receptor modulators protects endothelial cells from this disruption.
The researchers demonstrated this by infecting mice with P berghei ANKA, treating them with losartan or CGP-42112A, and monitoring them for neurological symptoms.
Seventy-five percent of control mice developed cerebral malaria, compared to 13.3% of mice treated with losartan and 28.5% treated with CGP-42112A.
In another experiment, the researchers treated malaria-infected mice with chloroquine, alone or in combination with irbesartan or C21, only after neurological signs were evident.
The team observed an increase in survival for mice treated with irbesartan or C21. The survival rate was 18% for the mice treated only with chloroquine, 65% for mice given irbesartan, and 73% percent for mice treated with C21.
The researchers also found that mice treated with irbesartan or C21 experienced fewer, smaller hemorrhages. And, in most cases, these mice fully recovered.
Dr Rodriguez and her colleagues said the next steps for this research will be to investigate the exact molecules coming from ruptured P falciparum–infected red blood cells that signal to beta-catenin in vessel walls, and to conduct trials that test the effects of Ang II receptor modulators in patients with cerebral malaria. ![]()
EMA says plasma/urine-derived meds are safe from Zika

Photo by Cristina Granados
Patients who take plasma-derived or urine-derived medicines do not have to worry about these products being contaminated with Zika virus, according to the European Medicines Agency (EMA).
The agency said assessments have confirmed that manufacturing processes for these medicines—which include coagulation factors, immunoglobulins, and urokinase products—successfully inactivate or remove the Zika virus.
These medicines are produced from body fluids that might be sourced in parts of the world where the Zika virus is prevalent. So regulators in the European Union (EU) sought reassurance that there is no risk of the virus contaminating the final product and thus affecting the patients taking these medicines.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) investigated the potential risk with plasma-derived medicinal products.
And the Co-ordination Group for Mutual Recognition and Decentralised Procedures—Human (CMDh) has coordinated the assessment by EU member states on the potential risk with urine-derived medicinal products.
The CHMP concluded at its meeting last week that the manufacturing processes used for plasma-derived products—including, for example, the solvent/detergent method to inactivate viruses, pasteurization, and virus filtration—inactivate or remove the Zika virus from the finished product.
The CHMP therefore concluded that no additional safety measures, such as the testing or exclusion of certain plasma donors, were necessary.
The CMDh, following an assessment of data, concluded that the manufacturing processes for urine-derived products contain complementary steps with inactivation/removal capacity for enveloped viruses, which are considered sufficient for eliminating Zika virus.
Additional safety measures, such as screening urine donors/donations or deferring donors returning from Zika-affected areas, are not considered necessary.
The findings from these assessments are available in a report from the CHMP’s Biologics Working Party.
The Biologics Working Party recommendation on plasma-derived products is in line with the guidance published in July 2016 by the European Centre for Disease Prevention and Control. ![]()
Photo by Cristina Granados
Patients who take plasma-derived or urine-derived medicines do not have to worry about these products being contaminated with Zika virus, according to the European Medicines Agency (EMA).
The agency said assessments have confirmed that manufacturing processes for these medicines—which include coagulation factors, immunoglobulins, and urokinase products—successfully inactivate or remove the Zika virus.
These medicines are produced from body fluids that might be sourced in parts of the world where the Zika virus is prevalent. So regulators in the European Union (EU) sought reassurance that there is no risk of the virus contaminating the final product and thus affecting the patients taking these medicines.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) investigated the potential risk with plasma-derived medicinal products.
And the Co-ordination Group for Mutual Recognition and Decentralised Procedures—Human (CMDh) has coordinated the assessment by EU member states on the potential risk with urine-derived medicinal products.
The CHMP concluded at its meeting last week that the manufacturing processes used for plasma-derived products—including, for example, the solvent/detergent method to inactivate viruses, pasteurization, and virus filtration—inactivate or remove the Zika virus from the finished product.
The CHMP therefore concluded that no additional safety measures, such as the testing or exclusion of certain plasma donors, were necessary.
The CMDh, following an assessment of data, concluded that the manufacturing processes for urine-derived products contain complementary steps with inactivation/removal capacity for enveloped viruses, which are considered sufficient for eliminating Zika virus.
Additional safety measures, such as screening urine donors/donations or deferring donors returning from Zika-affected areas, are not considered necessary.
The findings from these assessments are available in a report from the CHMP’s Biologics Working Party.
The Biologics Working Party recommendation on plasma-derived products is in line with the guidance published in July 2016 by the European Centre for Disease Prevention and Control. ![]()
Photo by Cristina Granados
Patients who take plasma-derived or urine-derived medicines do not have to worry about these products being contaminated with Zika virus, according to the European Medicines Agency (EMA).
The agency said assessments have confirmed that manufacturing processes for these medicines—which include coagulation factors, immunoglobulins, and urokinase products—successfully inactivate or remove the Zika virus.
These medicines are produced from body fluids that might be sourced in parts of the world where the Zika virus is prevalent. So regulators in the European Union (EU) sought reassurance that there is no risk of the virus contaminating the final product and thus affecting the patients taking these medicines.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) investigated the potential risk with plasma-derived medicinal products.
And the Co-ordination Group for Mutual Recognition and Decentralised Procedures—Human (CMDh) has coordinated the assessment by EU member states on the potential risk with urine-derived medicinal products.
The CHMP concluded at its meeting last week that the manufacturing processes used for plasma-derived products—including, for example, the solvent/detergent method to inactivate viruses, pasteurization, and virus filtration—inactivate or remove the Zika virus from the finished product.
The CHMP therefore concluded that no additional safety measures, such as the testing or exclusion of certain plasma donors, were necessary.
The CMDh, following an assessment of data, concluded that the manufacturing processes for urine-derived products contain complementary steps with inactivation/removal capacity for enveloped viruses, which are considered sufficient for eliminating Zika virus.
Additional safety measures, such as screening urine donors/donations or deferring donors returning from Zika-affected areas, are not considered necessary.
The findings from these assessments are available in a report from the CHMP’s Biologics Working Party.
The Biologics Working Party recommendation on plasma-derived products is in line with the guidance published in July 2016 by the European Centre for Disease Prevention and Control. ![]()
Compound could treat resistant malaria
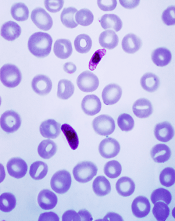
of the P falciparum parasite
Image by Mae Melvin/CDC
A novel compound could be effective against malaria infections that are resistant to currently available antimalarial drugs, according to researchers.
In a phase 2 study, the compound, KAF156, proved active against Plasmodium vivax and Plasmodium falciparum malaria.
KAF156 was able to clear both blood and liver stages of malaria parasites, including artemisinin-resistant parasites.
Most of the patients in this study had at least 1 adverse event (AE). However, most were grade 1, and there were no grade 4 or serious AEs.
Results from this study were published in NEJM. The trial was funded, in part, by Novartis, the company developing KAF156.
KAF156 is the first compound from a novel class of drugs called imidazolopiperazines. The drugs’ mechanism of action is still being characterized, but it may be related to a previously uncharacterized gene—Plasmodium falciparum cyclic amine resistance locus (Pfcarl).
From March to August 2013, researchers conducted a phase 2 trial of KAF156 at 5 centers in Thailand and Vietnam.
Twenty-one of the patients in this study had acute, uncomplicated malaria—11 with P vivax and 10 with P falciparum malaria. They received multiple doses of KAF156—400 mg once daily for 3 days.
Twenty-two of the patients studied had uncomplicated, P falciparum malaria and received a single dose of KAF156 at 800 mg.
All patients were assessed for fever and parasite clearance as well as AEs. Patients in the single-dose cohort were also followed for 28 days to assess the cure rate.
Two patients were excluded from the efficacy analysis. One was a P vivax patient receiving the 400 mg dose who turned out to have a mixed infection. And the other was a P falciparum patient who vomited repeatedly after receiving 800 mg of KAF156 (after 3 attempts at dosing).
Efficacy
In the multiple-dose cohorts, the median time to fever clearance after receiving KAF156 was 14 hours (range, 4 to 30) in patients with P vivax malaria and 6 hours (range, 4 to 24) in patients with P falciparum malaria. In the single-dose cohort, the median time to fever clearance was 4 hours (range, 4 to 66).
In the multiple-dose cohorts, the median time to parasite clearance was 24 hours (range, 16 to 36) in patients with P vivax and 45 hours (range, 36 to 66) in patients with P falciparum.
In the single-dose cohort, the median time to parasite clearance was 49 hours (range, 16 to 68). One of these patients had parasite clearance at 66 hours but an asymptomatic recurrence at 84 hours. In all of the other patients, parasitemia cleared.
During follow-up of the single-dose cohort, 1 patient had reinfection, and 7 had recrudescent infections.
Safety
Most patients had at least 1 AE, although none were serious. Seventy-two percent of patients had grade 1 AEs, 35% had grade 2 AEs, and 14% had grade 3 AEs.
Sixty percent of AEs were considered related to treatment. In the multiple-dose cohorts, 14 of the 31 AEs (45%) reported in 7 patients (1 with P vivax and 6 with P falciparum malaria) were considered drug-related. All 22 P falciparum patients in the single-dose cohort had at least 1 AE that was considered drug-related.
The most common AEs were sinus bradycardia, thrombocytopenia, hypokalemia, anemia, and hyperbilirubinemia.
Two patients experienced vomiting of grade 2 or higher. One of these patients discontinued treatment because of repeated vomiting after the 800 mg dose (mentioned above). ![]()
of the P falciparum parasite
Image by Mae Melvin/CDC
A novel compound could be effective against malaria infections that are resistant to currently available antimalarial drugs, according to researchers.
In a phase 2 study, the compound, KAF156, proved active against Plasmodium vivax and Plasmodium falciparum malaria.
KAF156 was able to clear both blood and liver stages of malaria parasites, including artemisinin-resistant parasites.
Most of the patients in this study had at least 1 adverse event (AE). However, most were grade 1, and there were no grade 4 or serious AEs.
Results from this study were published in NEJM. The trial was funded, in part, by Novartis, the company developing KAF156.
KAF156 is the first compound from a novel class of drugs called imidazolopiperazines. The drugs’ mechanism of action is still being characterized, but it may be related to a previously uncharacterized gene—Plasmodium falciparum cyclic amine resistance locus (Pfcarl).
From March to August 2013, researchers conducted a phase 2 trial of KAF156 at 5 centers in Thailand and Vietnam.
Twenty-one of the patients in this study had acute, uncomplicated malaria—11 with P vivax and 10 with P falciparum malaria. They received multiple doses of KAF156—400 mg once daily for 3 days.
Twenty-two of the patients studied had uncomplicated, P falciparum malaria and received a single dose of KAF156 at 800 mg.
All patients were assessed for fever and parasite clearance as well as AEs. Patients in the single-dose cohort were also followed for 28 days to assess the cure rate.
Two patients were excluded from the efficacy analysis. One was a P vivax patient receiving the 400 mg dose who turned out to have a mixed infection. And the other was a P falciparum patient who vomited repeatedly after receiving 800 mg of KAF156 (after 3 attempts at dosing).
Efficacy
In the multiple-dose cohorts, the median time to fever clearance after receiving KAF156 was 14 hours (range, 4 to 30) in patients with P vivax malaria and 6 hours (range, 4 to 24) in patients with P falciparum malaria. In the single-dose cohort, the median time to fever clearance was 4 hours (range, 4 to 66).
In the multiple-dose cohorts, the median time to parasite clearance was 24 hours (range, 16 to 36) in patients with P vivax and 45 hours (range, 36 to 66) in patients with P falciparum.
In the single-dose cohort, the median time to parasite clearance was 49 hours (range, 16 to 68). One of these patients had parasite clearance at 66 hours but an asymptomatic recurrence at 84 hours. In all of the other patients, parasitemia cleared.
During follow-up of the single-dose cohort, 1 patient had reinfection, and 7 had recrudescent infections.
Safety
Most patients had at least 1 AE, although none were serious. Seventy-two percent of patients had grade 1 AEs, 35% had grade 2 AEs, and 14% had grade 3 AEs.
Sixty percent of AEs were considered related to treatment. In the multiple-dose cohorts, 14 of the 31 AEs (45%) reported in 7 patients (1 with P vivax and 6 with P falciparum malaria) were considered drug-related. All 22 P falciparum patients in the single-dose cohort had at least 1 AE that was considered drug-related.
The most common AEs were sinus bradycardia, thrombocytopenia, hypokalemia, anemia, and hyperbilirubinemia.
Two patients experienced vomiting of grade 2 or higher. One of these patients discontinued treatment because of repeated vomiting after the 800 mg dose (mentioned above). ![]()
of the P falciparum parasite
Image by Mae Melvin/CDC
A novel compound could be effective against malaria infections that are resistant to currently available antimalarial drugs, according to researchers.
In a phase 2 study, the compound, KAF156, proved active against Plasmodium vivax and Plasmodium falciparum malaria.
KAF156 was able to clear both blood and liver stages of malaria parasites, including artemisinin-resistant parasites.
Most of the patients in this study had at least 1 adverse event (AE). However, most were grade 1, and there were no grade 4 or serious AEs.
Results from this study were published in NEJM. The trial was funded, in part, by Novartis, the company developing KAF156.
KAF156 is the first compound from a novel class of drugs called imidazolopiperazines. The drugs’ mechanism of action is still being characterized, but it may be related to a previously uncharacterized gene—Plasmodium falciparum cyclic amine resistance locus (Pfcarl).
From March to August 2013, researchers conducted a phase 2 trial of KAF156 at 5 centers in Thailand and Vietnam.
Twenty-one of the patients in this study had acute, uncomplicated malaria—11 with P vivax and 10 with P falciparum malaria. They received multiple doses of KAF156—400 mg once daily for 3 days.
Twenty-two of the patients studied had uncomplicated, P falciparum malaria and received a single dose of KAF156 at 800 mg.
All patients were assessed for fever and parasite clearance as well as AEs. Patients in the single-dose cohort were also followed for 28 days to assess the cure rate.
Two patients were excluded from the efficacy analysis. One was a P vivax patient receiving the 400 mg dose who turned out to have a mixed infection. And the other was a P falciparum patient who vomited repeatedly after receiving 800 mg of KAF156 (after 3 attempts at dosing).
Efficacy
In the multiple-dose cohorts, the median time to fever clearance after receiving KAF156 was 14 hours (range, 4 to 30) in patients with P vivax malaria and 6 hours (range, 4 to 24) in patients with P falciparum malaria. In the single-dose cohort, the median time to fever clearance was 4 hours (range, 4 to 66).
In the multiple-dose cohorts, the median time to parasite clearance was 24 hours (range, 16 to 36) in patients with P vivax and 45 hours (range, 36 to 66) in patients with P falciparum.
In the single-dose cohort, the median time to parasite clearance was 49 hours (range, 16 to 68). One of these patients had parasite clearance at 66 hours but an asymptomatic recurrence at 84 hours. In all of the other patients, parasitemia cleared.
During follow-up of the single-dose cohort, 1 patient had reinfection, and 7 had recrudescent infections.
Safety
Most patients had at least 1 AE, although none were serious. Seventy-two percent of patients had grade 1 AEs, 35% had grade 2 AEs, and 14% had grade 3 AEs.
Sixty percent of AEs were considered related to treatment. In the multiple-dose cohorts, 14 of the 31 AEs (45%) reported in 7 patients (1 with P vivax and 6 with P falciparum malaria) were considered drug-related. All 22 P falciparum patients in the single-dose cohort had at least 1 AE that was considered drug-related.
The most common AEs were sinus bradycardia, thrombocytopenia, hypokalemia, anemia, and hyperbilirubinemia.
Two patients experienced vomiting of grade 2 or higher. One of these patients discontinued treatment because of repeated vomiting after the 800 mg dose (mentioned above).
Cancer report details progress, predicts problems

Photo by Rhoda Baer
A new report highlights recent advances made in the fight against cancer but suggests the burden of cancer in the US is still on the rise.
The AACR Cancer Progress Report 2016 states that the number of cancer survivors in the US rose by 1 million from 2014 to 2016, reaching an estimated 15.5 million.
Meanwhile, the US Food and Drug Administration (FDA) approved new treatments for a range of cancers.
Between August 1, 2015, and July 31, 2016, the FDA approved 13 new anticancer therapies and new uses for 11 previously approved anticancer therapies.
Six of these drugs were approved to treat hematologic malignancies:
- Venetoclax for chronic lymphocytic leukemia
- Daratumumab for multiple myeloma
- Elotuzumab for multiple myeloma
- Ixazomib for multiple myeloma
- Obinutuzumab for follicular lymphoma
- Nivolumab for classical Hodgkin lymphoma.
The report notes that the use of immunotherapy, in particular, is on the rise. For example, on August 1, 2015, checkpoint inhibitors were approved for just 2 cancers—melanoma and lung cancer.
As of September 1, 2016, checkpoint inhibitors have been approved for 6 cancers—Hodgkin lymphoma, bladder cancer, head and neck cancer, kidney cancer, lung cancer, and melanoma.
“The promise of immunotherapy for cancer therapy has never been greater, and the opportunity to make significant progress in this critical area is real,” said Nancy E. Davidson, MD, president of the AACR and director of the University of Pittsburgh Cancer Institute in Pennsylvania.
“However, continued progress is going to require a sustained federal commitment to the research agenda. And in fact, if the necessary funding is provided, we will accelerate the pace of progress and, in turn, markedly reduce morbidity and mortality from cancer.”
Growing burden of cancer
The report emphasizes that although advances are being made against cancers, these diseases continue to exert an immense personal and economic toll, and the burden of cancer is expected to grow in the coming decades.
According to the report:
- More than 595,000 people in the US are projected to die from cancer in 2016
- Cancer is the number one cause of disease-related death among US children
- The number of new cancer cases in the US is predicted to rise from 1.7 million in 2015 to 2.4 million in 2035
- It is estimated that the direct medical costs of cancer care in the US in 2010 were nearly $125 billion, and these costs will rise to $156 billion in 2020.
The report states that the increasing economic and personal burden of cancer underscores the need for more research to develop new approaches to cancer prevention and treatment.
The report also highlights the recommendations of the National Cancer Moonshot Initiative Blue Ribbon Panel for accelerating the pace of progress in cancer research.
“Research has made tremendous advances against cancer,” said Margaret Foti, PhD, MD, chief executive officer of the AACR.
“However, we need to accelerate the pace of progress because it is unacceptable that 1 American will die of cancer every minute of every day this year.”
Photo by Rhoda Baer
A new report highlights recent advances made in the fight against cancer but suggests the burden of cancer in the US is still on the rise.
The AACR Cancer Progress Report 2016 states that the number of cancer survivors in the US rose by 1 million from 2014 to 2016, reaching an estimated 15.5 million.
Meanwhile, the US Food and Drug Administration (FDA) approved new treatments for a range of cancers.
Between August 1, 2015, and July 31, 2016, the FDA approved 13 new anticancer therapies and new uses for 11 previously approved anticancer therapies.
Six of these drugs were approved to treat hematologic malignancies:
- Venetoclax for chronic lymphocytic leukemia
- Daratumumab for multiple myeloma
- Elotuzumab for multiple myeloma
- Ixazomib for multiple myeloma
- Obinutuzumab for follicular lymphoma
- Nivolumab for classical Hodgkin lymphoma.
The report notes that the use of immunotherapy, in particular, is on the rise. For example, on August 1, 2015, checkpoint inhibitors were approved for just 2 cancers—melanoma and lung cancer.
As of September 1, 2016, checkpoint inhibitors have been approved for 6 cancers—Hodgkin lymphoma, bladder cancer, head and neck cancer, kidney cancer, lung cancer, and melanoma.
“The promise of immunotherapy for cancer therapy has never been greater, and the opportunity to make significant progress in this critical area is real,” said Nancy E. Davidson, MD, president of the AACR and director of the University of Pittsburgh Cancer Institute in Pennsylvania.
“However, continued progress is going to require a sustained federal commitment to the research agenda. And in fact, if the necessary funding is provided, we will accelerate the pace of progress and, in turn, markedly reduce morbidity and mortality from cancer.”
Growing burden of cancer
The report emphasizes that although advances are being made against cancers, these diseases continue to exert an immense personal and economic toll, and the burden of cancer is expected to grow in the coming decades.
According to the report:
- More than 595,000 people in the US are projected to die from cancer in 2016
- Cancer is the number one cause of disease-related death among US children
- The number of new cancer cases in the US is predicted to rise from 1.7 million in 2015 to 2.4 million in 2035
- It is estimated that the direct medical costs of cancer care in the US in 2010 were nearly $125 billion, and these costs will rise to $156 billion in 2020.
The report states that the increasing economic and personal burden of cancer underscores the need for more research to develop new approaches to cancer prevention and treatment.
The report also highlights the recommendations of the National Cancer Moonshot Initiative Blue Ribbon Panel for accelerating the pace of progress in cancer research.
“Research has made tremendous advances against cancer,” said Margaret Foti, PhD, MD, chief executive officer of the AACR.
“However, we need to accelerate the pace of progress because it is unacceptable that 1 American will die of cancer every minute of every day this year.”
Photo by Rhoda Baer
A new report highlights recent advances made in the fight against cancer but suggests the burden of cancer in the US is still on the rise.
The AACR Cancer Progress Report 2016 states that the number of cancer survivors in the US rose by 1 million from 2014 to 2016, reaching an estimated 15.5 million.
Meanwhile, the US Food and Drug Administration (FDA) approved new treatments for a range of cancers.
Between August 1, 2015, and July 31, 2016, the FDA approved 13 new anticancer therapies and new uses for 11 previously approved anticancer therapies.
Six of these drugs were approved to treat hematologic malignancies:
- Venetoclax for chronic lymphocytic leukemia
- Daratumumab for multiple myeloma
- Elotuzumab for multiple myeloma
- Ixazomib for multiple myeloma
- Obinutuzumab for follicular lymphoma
- Nivolumab for classical Hodgkin lymphoma.
The report notes that the use of immunotherapy, in particular, is on the rise. For example, on August 1, 2015, checkpoint inhibitors were approved for just 2 cancers—melanoma and lung cancer.
As of September 1, 2016, checkpoint inhibitors have been approved for 6 cancers—Hodgkin lymphoma, bladder cancer, head and neck cancer, kidney cancer, lung cancer, and melanoma.
“The promise of immunotherapy for cancer therapy has never been greater, and the opportunity to make significant progress in this critical area is real,” said Nancy E. Davidson, MD, president of the AACR and director of the University of Pittsburgh Cancer Institute in Pennsylvania.
“However, continued progress is going to require a sustained federal commitment to the research agenda. And in fact, if the necessary funding is provided, we will accelerate the pace of progress and, in turn, markedly reduce morbidity and mortality from cancer.”
Growing burden of cancer
The report emphasizes that although advances are being made against cancers, these diseases continue to exert an immense personal and economic toll, and the burden of cancer is expected to grow in the coming decades.
According to the report:
- More than 595,000 people in the US are projected to die from cancer in 2016
- Cancer is the number one cause of disease-related death among US children
- The number of new cancer cases in the US is predicted to rise from 1.7 million in 2015 to 2.4 million in 2035
- It is estimated that the direct medical costs of cancer care in the US in 2010 were nearly $125 billion, and these costs will rise to $156 billion in 2020.
The report states that the increasing economic and personal burden of cancer underscores the need for more research to develop new approaches to cancer prevention and treatment.
The report also highlights the recommendations of the National Cancer Moonshot Initiative Blue Ribbon Panel for accelerating the pace of progress in cancer research.
“Research has made tremendous advances against cancer,” said Margaret Foti, PhD, MD, chief executive officer of the AACR.
“However, we need to accelerate the pace of progress because it is unacceptable that 1 American will die of cancer every minute of every day this year.”
Dying cancer patients may be under-treated for pain

Photo courtesy of CDC
New research suggests that many patients who die of cancer do not receive strong opioid medications in their last year of life, despite the fact that these drugs are the recommended treatment for cancer-related pain.
Researchers used UK Cancer Registry Data to study more than 6000 cancer patients who died over a 7-year period.
Less than half of these patients received prescriptions for strong opioid medications in their last year of life.
Among those patients who did receive such prescriptions, many received them late.
Lucy Ziegler, PhD, of the University of Leeds in the UK, and her colleagues conducted this study and reported the results in PAIN.
The study included 6080 cancer patients who died between 2005 and 2012.
About 76% (n=4610) of these patients had received one or more prescriptions for analgesics, including 48% (n=2919) who received a strong opioid and 28% (n=1691) who received a non-opioid or weak opioid. The remaining 24% (n=1470) did not receive any prescription analgesic.
The chance of receiving strong opioids was not affected by patients’ age or sex.
However, patients who died in a hospital were 60% less likely to have a prescription for strong opioids during the last year of life, when compared with those who died in hospice.
And patients who received chemotherapy in the last year of life were 30% more likely to receive a strong opioid than patients who did not receive chemotherapy.
Timing of therapy
Among the patients who did receive strong opioids, the median time between receiving the medication and death was 9 weeks. By 6 weeks before death, just 30% of patients had been prescribed a strong opioid.
The researchers noted that these figures don’t match up with previous studies reporting that severe pain can occur “much earlier in the cancer trajectory.”
Older patients were more likely to receive their strong opioid prescription late (defined as later than 9 weeks before death). After other factors were taken into account, patients age 60 or older were about 2 to 4 times more likely to be in the late-prescribing group, compared with those age 50 or younger.
Compared to patients who died in hospice, patients who died in a hospital were 40% more likely to receive a late prescription for a strong opioid. Patients who died in their own home were 2.6 times more likely to receive a late prescription, and patients who died in a care home were 2.8 times more likely to receive a late prescription.
Patients who had surgery were 40% more likely to receive a late prescription than patients who did not undergo surgery.
But patients who received chemotherapy and/or radiotherapy were 30% more likely to have received an early prescription for a strong opioid than patients who did not receive chemo/radiotherapy.
Dr Ziegler and her colleagues noted that this study had its limitations; in particular, the lack of data on pain severity.
Still, the researchers believe their results support the hypothesis of potential under-treatment of cancer pain and suggest that many more patients with advanced cancer and pain may benefit from a strong opioid analgesic.
Photo courtesy of CDC
New research suggests that many patients who die of cancer do not receive strong opioid medications in their last year of life, despite the fact that these drugs are the recommended treatment for cancer-related pain.
Researchers used UK Cancer Registry Data to study more than 6000 cancer patients who died over a 7-year period.
Less than half of these patients received prescriptions for strong opioid medications in their last year of life.
Among those patients who did receive such prescriptions, many received them late.
Lucy Ziegler, PhD, of the University of Leeds in the UK, and her colleagues conducted this study and reported the results in PAIN.
The study included 6080 cancer patients who died between 2005 and 2012.
About 76% (n=4610) of these patients had received one or more prescriptions for analgesics, including 48% (n=2919) who received a strong opioid and 28% (n=1691) who received a non-opioid or weak opioid. The remaining 24% (n=1470) did not receive any prescription analgesic.
The chance of receiving strong opioids was not affected by patients’ age or sex.
However, patients who died in a hospital were 60% less likely to have a prescription for strong opioids during the last year of life, when compared with those who died in hospice.
And patients who received chemotherapy in the last year of life were 30% more likely to receive a strong opioid than patients who did not receive chemotherapy.
Timing of therapy
Among the patients who did receive strong opioids, the median time between receiving the medication and death was 9 weeks. By 6 weeks before death, just 30% of patients had been prescribed a strong opioid.
The researchers noted that these figures don’t match up with previous studies reporting that severe pain can occur “much earlier in the cancer trajectory.”
Older patients were more likely to receive their strong opioid prescription late (defined as later than 9 weeks before death). After other factors were taken into account, patients age 60 or older were about 2 to 4 times more likely to be in the late-prescribing group, compared with those age 50 or younger.
Compared to patients who died in hospice, patients who died in a hospital were 40% more likely to receive a late prescription for a strong opioid. Patients who died in their own home were 2.6 times more likely to receive a late prescription, and patients who died in a care home were 2.8 times more likely to receive a late prescription.
Patients who had surgery were 40% more likely to receive a late prescription than patients who did not undergo surgery.
But patients who received chemotherapy and/or radiotherapy were 30% more likely to have received an early prescription for a strong opioid than patients who did not receive chemo/radiotherapy.
Dr Ziegler and her colleagues noted that this study had its limitations; in particular, the lack of data on pain severity.
Still, the researchers believe their results support the hypothesis of potential under-treatment of cancer pain and suggest that many more patients with advanced cancer and pain may benefit from a strong opioid analgesic.
Photo courtesy of CDC
New research suggests that many patients who die of cancer do not receive strong opioid medications in their last year of life, despite the fact that these drugs are the recommended treatment for cancer-related pain.
Researchers used UK Cancer Registry Data to study more than 6000 cancer patients who died over a 7-year period.
Less than half of these patients received prescriptions for strong opioid medications in their last year of life.
Among those patients who did receive such prescriptions, many received them late.
Lucy Ziegler, PhD, of the University of Leeds in the UK, and her colleagues conducted this study and reported the results in PAIN.
The study included 6080 cancer patients who died between 2005 and 2012.
About 76% (n=4610) of these patients had received one or more prescriptions for analgesics, including 48% (n=2919) who received a strong opioid and 28% (n=1691) who received a non-opioid or weak opioid. The remaining 24% (n=1470) did not receive any prescription analgesic.
The chance of receiving strong opioids was not affected by patients’ age or sex.
However, patients who died in a hospital were 60% less likely to have a prescription for strong opioids during the last year of life, when compared with those who died in hospice.
And patients who received chemotherapy in the last year of life were 30% more likely to receive a strong opioid than patients who did not receive chemotherapy.
Timing of therapy
Among the patients who did receive strong opioids, the median time between receiving the medication and death was 9 weeks. By 6 weeks before death, just 30% of patients had been prescribed a strong opioid.
The researchers noted that these figures don’t match up with previous studies reporting that severe pain can occur “much earlier in the cancer trajectory.”
Older patients were more likely to receive their strong opioid prescription late (defined as later than 9 weeks before death). After other factors were taken into account, patients age 60 or older were about 2 to 4 times more likely to be in the late-prescribing group, compared with those age 50 or younger.
Compared to patients who died in hospice, patients who died in a hospital were 40% more likely to receive a late prescription for a strong opioid. Patients who died in their own home were 2.6 times more likely to receive a late prescription, and patients who died in a care home were 2.8 times more likely to receive a late prescription.
Patients who had surgery were 40% more likely to receive a late prescription than patients who did not undergo surgery.
But patients who received chemotherapy and/or radiotherapy were 30% more likely to have received an early prescription for a strong opioid than patients who did not receive chemo/radiotherapy.
Dr Ziegler and her colleagues noted that this study had its limitations; in particular, the lack of data on pain severity.
Still, the researchers believe their results support the hypothesis of potential under-treatment of cancer pain and suggest that many more patients with advanced cancer and pain may benefit from a strong opioid analgesic.
Team creates method for predicting drug toxicity

for a clinical trial
Photo by Esther Dyson
Researchers have devised a method for predicting whether experimental drugs will fail clinical trials due to excessive toxicity.
They say the method, known as PrOCTOR, upends conventional wisdom about the criteria on which to evaluate new drugs’ safety.
Rather than exclusively examining molecular structure to determine viability, PrOCTOR combines a host of structural features and features related to how the drug binds to molecules in the body.
“We looked more broadly at drug molecule features that drug developers thought were unimportant in predicting drug safety in the past. Then, we let the data speak for itself,” said study author Olivier Elemento, PhD, of Weill Cornell Medicine in New York, New York.
Dr Elemento and his colleagues described PrOCTOR in Cell Chemical Biology.
PrOCTOR was inspired by an approach that baseball statisticians adopted to better predict which players would be successful offensively. Instead of relying on collective wisdom from baseball insiders, statisticians decided to use an objective numbers analysis to measure in-game productivity, a strategy known as “Moneyball.”
Similarly, Dr Elemento and his colleagues developed a computational method that analyzes data from 48 different features of a drug—from molecular weight to details about its target—to determine whether it would be safe for clinical use.
Using machine learning, the researchers trained PrOCTOR on drugs approved by the US Food and Drug Administration (FDA) as well as drugs that failed clinical trials due to toxicity problems.
Based on this information, the researchers created “PrOCTOR scores” that could help distinguish drugs approved by the FDA from those that failed for toxicity.
They tested PrOCTOR on hundreds of additional drugs approved in Europe and Japan and using side effect data on approved drugs collected by the FDA.
The researchers said PrOCTOR was able to accurately recognize toxic side effects that were a consequence of a drug’s chemical features and its target. Records revealing that many of these drugs had failed clinical trials supported PrOCTOR’s accuracy.
“We were able to find several features that led to a very predictive model,” Dr Elemento said. “Hopefully, this approach could be used to determine whether it’s worth pursuing a drug prior to starting human trials.”
for a clinical trial
Photo by Esther Dyson
Researchers have devised a method for predicting whether experimental drugs will fail clinical trials due to excessive toxicity.
They say the method, known as PrOCTOR, upends conventional wisdom about the criteria on which to evaluate new drugs’ safety.
Rather than exclusively examining molecular structure to determine viability, PrOCTOR combines a host of structural features and features related to how the drug binds to molecules in the body.
“We looked more broadly at drug molecule features that drug developers thought were unimportant in predicting drug safety in the past. Then, we let the data speak for itself,” said study author Olivier Elemento, PhD, of Weill Cornell Medicine in New York, New York.
Dr Elemento and his colleagues described PrOCTOR in Cell Chemical Biology.
PrOCTOR was inspired by an approach that baseball statisticians adopted to better predict which players would be successful offensively. Instead of relying on collective wisdom from baseball insiders, statisticians decided to use an objective numbers analysis to measure in-game productivity, a strategy known as “Moneyball.”
Similarly, Dr Elemento and his colleagues developed a computational method that analyzes data from 48 different features of a drug—from molecular weight to details about its target—to determine whether it would be safe for clinical use.
Using machine learning, the researchers trained PrOCTOR on drugs approved by the US Food and Drug Administration (FDA) as well as drugs that failed clinical trials due to toxicity problems.
Based on this information, the researchers created “PrOCTOR scores” that could help distinguish drugs approved by the FDA from those that failed for toxicity.
They tested PrOCTOR on hundreds of additional drugs approved in Europe and Japan and using side effect data on approved drugs collected by the FDA.
The researchers said PrOCTOR was able to accurately recognize toxic side effects that were a consequence of a drug’s chemical features and its target. Records revealing that many of these drugs had failed clinical trials supported PrOCTOR’s accuracy.
“We were able to find several features that led to a very predictive model,” Dr Elemento said. “Hopefully, this approach could be used to determine whether it’s worth pursuing a drug prior to starting human trials.”
for a clinical trial
Photo by Esther Dyson
Researchers have devised a method for predicting whether experimental drugs will fail clinical trials due to excessive toxicity.
They say the method, known as PrOCTOR, upends conventional wisdom about the criteria on which to evaluate new drugs’ safety.
Rather than exclusively examining molecular structure to determine viability, PrOCTOR combines a host of structural features and features related to how the drug binds to molecules in the body.
“We looked more broadly at drug molecule features that drug developers thought were unimportant in predicting drug safety in the past. Then, we let the data speak for itself,” said study author Olivier Elemento, PhD, of Weill Cornell Medicine in New York, New York.
Dr Elemento and his colleagues described PrOCTOR in Cell Chemical Biology.
PrOCTOR was inspired by an approach that baseball statisticians adopted to better predict which players would be successful offensively. Instead of relying on collective wisdom from baseball insiders, statisticians decided to use an objective numbers analysis to measure in-game productivity, a strategy known as “Moneyball.”
Similarly, Dr Elemento and his colleagues developed a computational method that analyzes data from 48 different features of a drug—from molecular weight to details about its target—to determine whether it would be safe for clinical use.
Using machine learning, the researchers trained PrOCTOR on drugs approved by the US Food and Drug Administration (FDA) as well as drugs that failed clinical trials due to toxicity problems.
Based on this information, the researchers created “PrOCTOR scores” that could help distinguish drugs approved by the FDA from those that failed for toxicity.
They tested PrOCTOR on hundreds of additional drugs approved in Europe and Japan and using side effect data on approved drugs collected by the FDA.
The researchers said PrOCTOR was able to accurately recognize toxic side effects that were a consequence of a drug’s chemical features and its target. Records revealing that many of these drugs had failed clinical trials supported PrOCTOR’s accuracy.
“We were able to find several features that led to a very predictive model,” Dr Elemento said. “Hopefully, this approach could be used to determine whether it’s worth pursuing a drug prior to starting human trials.”