User login
ClinicalEdge Topic
From the journals: sarcoma around the world
EWING SARCOMA IN NEPAL: Investigators reported what they believe to be the first prospective clinical trial providing state-of-the-art chemotherapy to patients with Ewing sarcoma in Nepal. They treated 20 newly diagnosed patients with combination chemotherapy, including a course of etoposide and ifosfamide during external-beam radiotherapy. Radiotherapy was the only available treatment modality for local tumor control because advanced tumor-orthopedic services are not available in Nepal.
The 11 females and 9 males enrolled ranged in age from 6 to 37 years.
The treatment protocol—based on the Nepali-Norwegian Ewing Sarcoma Study treatment initiative— consisted of:
- Cyclophosphamide (1,200 mg/m2 as a 30-minute intravenous [IV] infusion)
- Doxorubicin (40 mg/m2/d as a 4-hour IV infusion on days 1 and 2; total dose, 80 mg/m2 in 2 days; total cumulative dose, 400 mg/m2)
- Etoposide (150 mg/m2/d as a 2-hour IV infusion; total dose, 450 mg/m2 in 3 days)
- Ifosfamide (3,000 mg/m2 over 21 to 24 hours as a 3-day continuous IV infusion; total dose, 9,000 mg/m2 in 3 days)
- Vincristine (1.5 mg/m2 IV push; maximum, 2 mg)
Patients received 5 courses of chemotherapy, then radiotherapy twice daily for 4 weeks for a total accumulated 54-Gy dose with a course of etoposide and ifosfamide, followed by 6 additional courses of chemotherapy.
Patients had primary tumors in the following sites: femur (n = 4), pubic bone (n = 1), fibula (n = 1), thoracic wall or costae (n = 4), clavicle (n = 1), craniofacial bone (n = 3), humerus (n = 3), forearm (n = 1), musculus sartorius with invasion into adjacent femur (n = 1), and uterine cervix (n = 1).
Eleven patients completed the entire treatment regimen, 6 of whom had no evidence of disease at a median follow-up of 2.3 years (range, 1.3 to 3.1 years). Four of them died of metastatic disease, and 1 experienced a recurrence 6 months later.
Three patients died due to chemotherapy- related toxicity, and 6 patients did not complete the treatment protocol, 4 of whom experienced progressive disease, were lost to follow-up, and presumed dead.
The investigators concluded that radiotherapy as the sole local treatment modality in combination with chemotherapy is feasible. They observed no fractures among the 15 patients who received radiotherapy.
SOURCE: Jha AK, Neupane P, Pradhan M, et al. Ewing sarcoma in Nepal treated with combined chemotherapy and definitive radiotherapy. J Glob Oncol. 2019;5:1-10.
PEDIATRIC SOFT TISSUE AND BONE SARCOMAS IN TANZANIA: In this retrospective review, investigators documented the epidemiologic and clinical features of pediatric sarcomas in the largest pediatric oncology center in Tanzania—Muhimbili National Hospital. Their objective in collecting the data was to compare the results with those of other countries and ultimately prioritize treatment protocols and resources for the more common pediatric sarcomas in Tanzania. Prior to this study, no data existed on the frequency and types most commonly seen in the country.
Between 2011 and 2016, the investigators collected information on 135 pediatric cases seen at the hospital. Eighty-nine cases (66%) were soft tissue sarcomas (STS) and 46 (34%) were bone sarcomas. Most patients, they reported, presented with a painless swelling.
Investigators found that, as in other countries, embryonal rhabdomyosarcoma accounted for the majority (75%) of all sarcomas seen in this study and osteosarcoma accounted for most (87%) bone sarcomas. However, unlike pediatric sarcomas in other countries, few cases of Ewing sarcoma were diagnosed during the study period.
An important disparity between Tanzania and other countries is that most patients in Tanzania present with advanced- stage disease, when the possibility of curative therapy is vastly reduced. Investigators found the lung to be the most common site of distant metastasis.
Other clinical and tumor characteristics reported in this study included:
- Slight female predominance (51%)
- Mean age, 6.3 years
- 42% of STS patients were younger than 5 years (n = 37)
- 46% of bone sarcoma patients were 10 to 15 years old (n = 21)
- Head and neck were the most common sites for STS
- Extremities were the most common sites for bone sarcomas
- Most patients presented with large tumors (>5 cm for STS and >8 cm for bone sarcomas).
The investigators believe these findings and others they reported will help them adapt treatment protocols used in Europe and America so that they will be most appropriate for their patients.
SOURCE: Siwillis EM, Dharse NJ, Scanlan T, et al. Pediatric soft tissue and bone sarcomas in Tanzania: Epidemiology and clinical features. J Glob Oncol. 2019;5:1-6.
PEDIATRIC OSTEOSARCOMA IN LEBANON: Investigators at a single institution in Lebanon reported a similar survival rate for newly diagnosed patients with pediatric osteosarcoma treated at their center as for those treated in more developed countries. In a retrospective review of the medical records of 38 patients treated at the American University of Beirut Medical Center between August 2001 and May 2012, they determined the 5-year overall survival (OS) for all patients to be 74% and the event-free survival (EFS), 62%. Patients with localized disease had a 5-year OS of 81% and an EFS of 68%. Patients with metastatic disease had OS and EFS rates of about 42%.
All patients with localized disease received chemotherapy according to the Pediatric Oncology Group 9351 protocol, which consisted of cisplatin, doxorubicin, and methotrexate. If patients had metastatic disease or tumor necrosis less than 90%, they also received ifosfamide and etoposide.
Patients were a mean age of 12.9 years at diagnosis and there were an equal number of male and female patients. Most patients (n=34) had a primary tumor site affecting the long bones around the knee.
Six patients had metastatic disease to the lungs, and 3 patients had multifocal bone disease with lung metastases.
Thirty-three patients (86.8%) underwent surgical resection after 2 courses of induction chemotherapy. Twenty-two (66.7%) of these patients had a delay in local tumor control of more than 4 weeks. And 12 patients (31.5%) had tumor necrosis of less than 90%.
The investigators analyzed the prognostic importance of age, sex, metastatic disease, tumor site, delay in local control, and degree of tumor necrosis. Bivariate analysis revealed that only the degree of tumor necrosis was a statistically significant adverse prognostic factor for EFS (P=.001) and OS (P=.002).
SOURCE: Abou Ali B, Salman M, Ghanem KM, et al. Clinical prognostic factors and outcome in pediatric osteosarcoma: Effect of delay in local control and degree of necrosis in a multidisciplinary setting in Lebanon. J Glob Oncol. 2019;5:1-8.
EWING SARCOMA IN NEPAL: Investigators reported what they believe to be the first prospective clinical trial providing state-of-the-art chemotherapy to patients with Ewing sarcoma in Nepal. They treated 20 newly diagnosed patients with combination chemotherapy, including a course of etoposide and ifosfamide during external-beam radiotherapy. Radiotherapy was the only available treatment modality for local tumor control because advanced tumor-orthopedic services are not available in Nepal.
The 11 females and 9 males enrolled ranged in age from 6 to 37 years.
The treatment protocol—based on the Nepali-Norwegian Ewing Sarcoma Study treatment initiative— consisted of:
- Cyclophosphamide (1,200 mg/m2 as a 30-minute intravenous [IV] infusion)
- Doxorubicin (40 mg/m2/d as a 4-hour IV infusion on days 1 and 2; total dose, 80 mg/m2 in 2 days; total cumulative dose, 400 mg/m2)
- Etoposide (150 mg/m2/d as a 2-hour IV infusion; total dose, 450 mg/m2 in 3 days)
- Ifosfamide (3,000 mg/m2 over 21 to 24 hours as a 3-day continuous IV infusion; total dose, 9,000 mg/m2 in 3 days)
- Vincristine (1.5 mg/m2 IV push; maximum, 2 mg)
Patients received 5 courses of chemotherapy, then radiotherapy twice daily for 4 weeks for a total accumulated 54-Gy dose with a course of etoposide and ifosfamide, followed by 6 additional courses of chemotherapy.
Patients had primary tumors in the following sites: femur (n = 4), pubic bone (n = 1), fibula (n = 1), thoracic wall or costae (n = 4), clavicle (n = 1), craniofacial bone (n = 3), humerus (n = 3), forearm (n = 1), musculus sartorius with invasion into adjacent femur (n = 1), and uterine cervix (n = 1).
Eleven patients completed the entire treatment regimen, 6 of whom had no evidence of disease at a median follow-up of 2.3 years (range, 1.3 to 3.1 years). Four of them died of metastatic disease, and 1 experienced a recurrence 6 months later.
Three patients died due to chemotherapy- related toxicity, and 6 patients did not complete the treatment protocol, 4 of whom experienced progressive disease, were lost to follow-up, and presumed dead.
The investigators concluded that radiotherapy as the sole local treatment modality in combination with chemotherapy is feasible. They observed no fractures among the 15 patients who received radiotherapy.
SOURCE: Jha AK, Neupane P, Pradhan M, et al. Ewing sarcoma in Nepal treated with combined chemotherapy and definitive radiotherapy. J Glob Oncol. 2019;5:1-10.
PEDIATRIC SOFT TISSUE AND BONE SARCOMAS IN TANZANIA: In this retrospective review, investigators documented the epidemiologic and clinical features of pediatric sarcomas in the largest pediatric oncology center in Tanzania—Muhimbili National Hospital. Their objective in collecting the data was to compare the results with those of other countries and ultimately prioritize treatment protocols and resources for the more common pediatric sarcomas in Tanzania. Prior to this study, no data existed on the frequency and types most commonly seen in the country.
Between 2011 and 2016, the investigators collected information on 135 pediatric cases seen at the hospital. Eighty-nine cases (66%) were soft tissue sarcomas (STS) and 46 (34%) were bone sarcomas. Most patients, they reported, presented with a painless swelling.
Investigators found that, as in other countries, embryonal rhabdomyosarcoma accounted for the majority (75%) of all sarcomas seen in this study and osteosarcoma accounted for most (87%) bone sarcomas. However, unlike pediatric sarcomas in other countries, few cases of Ewing sarcoma were diagnosed during the study period.
An important disparity between Tanzania and other countries is that most patients in Tanzania present with advanced- stage disease, when the possibility of curative therapy is vastly reduced. Investigators found the lung to be the most common site of distant metastasis.
Other clinical and tumor characteristics reported in this study included:
- Slight female predominance (51%)
- Mean age, 6.3 years
- 42% of STS patients were younger than 5 years (n = 37)
- 46% of bone sarcoma patients were 10 to 15 years old (n = 21)
- Head and neck were the most common sites for STS
- Extremities were the most common sites for bone sarcomas
- Most patients presented with large tumors (>5 cm for STS and >8 cm for bone sarcomas).
The investigators believe these findings and others they reported will help them adapt treatment protocols used in Europe and America so that they will be most appropriate for their patients.
SOURCE: Siwillis EM, Dharse NJ, Scanlan T, et al. Pediatric soft tissue and bone sarcomas in Tanzania: Epidemiology and clinical features. J Glob Oncol. 2019;5:1-6.
PEDIATRIC OSTEOSARCOMA IN LEBANON: Investigators at a single institution in Lebanon reported a similar survival rate for newly diagnosed patients with pediatric osteosarcoma treated at their center as for those treated in more developed countries. In a retrospective review of the medical records of 38 patients treated at the American University of Beirut Medical Center between August 2001 and May 2012, they determined the 5-year overall survival (OS) for all patients to be 74% and the event-free survival (EFS), 62%. Patients with localized disease had a 5-year OS of 81% and an EFS of 68%. Patients with metastatic disease had OS and EFS rates of about 42%.
All patients with localized disease received chemotherapy according to the Pediatric Oncology Group 9351 protocol, which consisted of cisplatin, doxorubicin, and methotrexate. If patients had metastatic disease or tumor necrosis less than 90%, they also received ifosfamide and etoposide.
Patients were a mean age of 12.9 years at diagnosis and there were an equal number of male and female patients. Most patients (n=34) had a primary tumor site affecting the long bones around the knee.
Six patients had metastatic disease to the lungs, and 3 patients had multifocal bone disease with lung metastases.
Thirty-three patients (86.8%) underwent surgical resection after 2 courses of induction chemotherapy. Twenty-two (66.7%) of these patients had a delay in local tumor control of more than 4 weeks. And 12 patients (31.5%) had tumor necrosis of less than 90%.
The investigators analyzed the prognostic importance of age, sex, metastatic disease, tumor site, delay in local control, and degree of tumor necrosis. Bivariate analysis revealed that only the degree of tumor necrosis was a statistically significant adverse prognostic factor for EFS (P=.001) and OS (P=.002).
SOURCE: Abou Ali B, Salman M, Ghanem KM, et al. Clinical prognostic factors and outcome in pediatric osteosarcoma: Effect of delay in local control and degree of necrosis in a multidisciplinary setting in Lebanon. J Glob Oncol. 2019;5:1-8.
EWING SARCOMA IN NEPAL: Investigators reported what they believe to be the first prospective clinical trial providing state-of-the-art chemotherapy to patients with Ewing sarcoma in Nepal. They treated 20 newly diagnosed patients with combination chemotherapy, including a course of etoposide and ifosfamide during external-beam radiotherapy. Radiotherapy was the only available treatment modality for local tumor control because advanced tumor-orthopedic services are not available in Nepal.
The 11 females and 9 males enrolled ranged in age from 6 to 37 years.
The treatment protocol—based on the Nepali-Norwegian Ewing Sarcoma Study treatment initiative— consisted of:
- Cyclophosphamide (1,200 mg/m2 as a 30-minute intravenous [IV] infusion)
- Doxorubicin (40 mg/m2/d as a 4-hour IV infusion on days 1 and 2; total dose, 80 mg/m2 in 2 days; total cumulative dose, 400 mg/m2)
- Etoposide (150 mg/m2/d as a 2-hour IV infusion; total dose, 450 mg/m2 in 3 days)
- Ifosfamide (3,000 mg/m2 over 21 to 24 hours as a 3-day continuous IV infusion; total dose, 9,000 mg/m2 in 3 days)
- Vincristine (1.5 mg/m2 IV push; maximum, 2 mg)
Patients received 5 courses of chemotherapy, then radiotherapy twice daily for 4 weeks for a total accumulated 54-Gy dose with a course of etoposide and ifosfamide, followed by 6 additional courses of chemotherapy.
Patients had primary tumors in the following sites: femur (n = 4), pubic bone (n = 1), fibula (n = 1), thoracic wall or costae (n = 4), clavicle (n = 1), craniofacial bone (n = 3), humerus (n = 3), forearm (n = 1), musculus sartorius with invasion into adjacent femur (n = 1), and uterine cervix (n = 1).
Eleven patients completed the entire treatment regimen, 6 of whom had no evidence of disease at a median follow-up of 2.3 years (range, 1.3 to 3.1 years). Four of them died of metastatic disease, and 1 experienced a recurrence 6 months later.
Three patients died due to chemotherapy- related toxicity, and 6 patients did not complete the treatment protocol, 4 of whom experienced progressive disease, were lost to follow-up, and presumed dead.
The investigators concluded that radiotherapy as the sole local treatment modality in combination with chemotherapy is feasible. They observed no fractures among the 15 patients who received radiotherapy.
SOURCE: Jha AK, Neupane P, Pradhan M, et al. Ewing sarcoma in Nepal treated with combined chemotherapy and definitive radiotherapy. J Glob Oncol. 2019;5:1-10.
PEDIATRIC SOFT TISSUE AND BONE SARCOMAS IN TANZANIA: In this retrospective review, investigators documented the epidemiologic and clinical features of pediatric sarcomas in the largest pediatric oncology center in Tanzania—Muhimbili National Hospital. Their objective in collecting the data was to compare the results with those of other countries and ultimately prioritize treatment protocols and resources for the more common pediatric sarcomas in Tanzania. Prior to this study, no data existed on the frequency and types most commonly seen in the country.
Between 2011 and 2016, the investigators collected information on 135 pediatric cases seen at the hospital. Eighty-nine cases (66%) were soft tissue sarcomas (STS) and 46 (34%) were bone sarcomas. Most patients, they reported, presented with a painless swelling.
Investigators found that, as in other countries, embryonal rhabdomyosarcoma accounted for the majority (75%) of all sarcomas seen in this study and osteosarcoma accounted for most (87%) bone sarcomas. However, unlike pediatric sarcomas in other countries, few cases of Ewing sarcoma were diagnosed during the study period.
An important disparity between Tanzania and other countries is that most patients in Tanzania present with advanced- stage disease, when the possibility of curative therapy is vastly reduced. Investigators found the lung to be the most common site of distant metastasis.
Other clinical and tumor characteristics reported in this study included:
- Slight female predominance (51%)
- Mean age, 6.3 years
- 42% of STS patients were younger than 5 years (n = 37)
- 46% of bone sarcoma patients were 10 to 15 years old (n = 21)
- Head and neck were the most common sites for STS
- Extremities were the most common sites for bone sarcomas
- Most patients presented with large tumors (>5 cm for STS and >8 cm for bone sarcomas).
The investigators believe these findings and others they reported will help them adapt treatment protocols used in Europe and America so that they will be most appropriate for their patients.
SOURCE: Siwillis EM, Dharse NJ, Scanlan T, et al. Pediatric soft tissue and bone sarcomas in Tanzania: Epidemiology and clinical features. J Glob Oncol. 2019;5:1-6.
PEDIATRIC OSTEOSARCOMA IN LEBANON: Investigators at a single institution in Lebanon reported a similar survival rate for newly diagnosed patients with pediatric osteosarcoma treated at their center as for those treated in more developed countries. In a retrospective review of the medical records of 38 patients treated at the American University of Beirut Medical Center between August 2001 and May 2012, they determined the 5-year overall survival (OS) for all patients to be 74% and the event-free survival (EFS), 62%. Patients with localized disease had a 5-year OS of 81% and an EFS of 68%. Patients with metastatic disease had OS and EFS rates of about 42%.
All patients with localized disease received chemotherapy according to the Pediatric Oncology Group 9351 protocol, which consisted of cisplatin, doxorubicin, and methotrexate. If patients had metastatic disease or tumor necrosis less than 90%, they also received ifosfamide and etoposide.
Patients were a mean age of 12.9 years at diagnosis and there were an equal number of male and female patients. Most patients (n=34) had a primary tumor site affecting the long bones around the knee.
Six patients had metastatic disease to the lungs, and 3 patients had multifocal bone disease with lung metastases.
Thirty-three patients (86.8%) underwent surgical resection after 2 courses of induction chemotherapy. Twenty-two (66.7%) of these patients had a delay in local tumor control of more than 4 weeks. And 12 patients (31.5%) had tumor necrosis of less than 90%.
The investigators analyzed the prognostic importance of age, sex, metastatic disease, tumor site, delay in local control, and degree of tumor necrosis. Bivariate analysis revealed that only the degree of tumor necrosis was a statistically significant adverse prognostic factor for EFS (P=.001) and OS (P=.002).
SOURCE: Abou Ali B, Salman M, Ghanem KM, et al. Clinical prognostic factors and outcome in pediatric osteosarcoma: Effect of delay in local control and degree of necrosis in a multidisciplinary setting in Lebanon. J Glob Oncol. 2019;5:1-8.
CAR T cells home in on HER2 in advanced sarcomas
ATLANTA – A novel chimeric antigen receptor (CAR) T-cell construct centered on HER2 as the target antigen was safe and showed early promise in the treatment of advanced sarcomas of bone and soft tissues in a phase I trial.

One patient, a 16-year-old girl with advanced osteosarcoma metastatic to her lungs, had a complete response to the therapy that is ongoing out to nearly 3 years, reported Shoba A. Navai, MD, from Baylor College of Medicine in Houston.
A second patient, an 8-year-old boy with rhabdomyosarcoma metastatic to bone marrow, had a complete response lasting 12 months. Upon relapse he was re-enrolled, received additional CAR T-cell infusions, and had a second complete response that has been ongoing for 17 months.
“HER2 CAR T cells can induce objective clinical responses in some patients with sarcoma, and engagement of endogenous immunity may aid in generation of tumor responses. We are currently working to validate these findings in other patients who were treated,” she said at a briefing at the annual meeting of the American Association for Cancer Research.
HER2 is a member of the human epidermal growth factor receptor family that is primarily expressed on the surface of tumor cells but is largely absent from nonmalignant tissues. HER2 can be expressed in a variety of sarcomas, including osteosarcoma, and HER2 expression in osteosarcoma correlates with worse overall survival.
Unlike HER2-positive breast cancers, however, HER2 expression levels in osteosarcoma are too low to be effectively targeted by anti-HER2 agents such as trastuzumab (Hereceptin).
But as Dr. Navai and colleagues have found, HER2 appears to be a valid target for CAR T-cell therapy in otherwise antigenically “cold” tumors – that is, tumors with few targetable antigens.
Old target, new weapon
They have developed a CAR T-cell construct using a HER2-directed antibody coupled with CD28 as the costimulatory molecule. As with other CAR T therapies, the patient’s T cells or selected T cell subsets are collected, transfected to express the antigen, and are then expanded and returned to the patient following lymphodepletion with either fludarabine alone or with cyclophosphamide.
Each patient received up to three infusions of autologous CAR T cells at a dose of 1 x 108 cells/m2, and eligible patients received up to five additional infusions without additional lymphodepletion.
Dr. Navai presented data on 10 patients treated to date, including the two mentioned before; the boy with rhabdomyosarcoma was counted as two separate patients for the purpose of the efficacy analysis.
All patients had metastatic disease, including five with osteosarcoma, three with rhabdomyosarcoma, one with Ewing sarcoma, and one with synovial sarcoma.
The lymphodepletion regimens did their job, inducing neutropenia (defined as an absolute neutrophil count less than 500 per milliliter ) for up to 14 days.
Eight patients developed grade 1 or 2 cytokine release syndrome within 24 hours of CAR T-cell infusion, and all cases completely resolved with supportive care within 5 days of onset.
In nine patients, T cells were successfully expanded, with a median peak expansion on day 7.
In all 10 patients, CAR T cells were detected by quantitative polymerase chain reaction 6 weeks after infusion.
In addition to the two patients with complete remissions already described, three patients had stable disease. The remaining patients had disease progression. At the most recent analysis, five patients were still alive, and five had died.
The infusions were safe, with no dose-limiting toxicities reported. No patient required a transfusion, and there were no opportunistic, infections, no neurotoxicities, and no lasting pulmonary or cardiac toxicities, Dr. Navai reported.
Some fare better than others
Nilofer S. Azad, MD, of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, Baltimore, who moderated the briefing, commented that the study had “very small numbers, but is still very exciting.”
She noted that the patients who benefited most from the therapy either had minimal residual disease or bone marrow disease without visceral disease; she asked Dr. Navai how this could be addressed going forward.
“The patients who seemed to have had responses both in this trial, as well as in our previous trial without lymphodepletion, tended to have less disease or more accessible disease. So we hypothesized that disease that’s in the bone marrow because it’s more accessible, or in the lungs, where also CAR T cells go after they are first infused, may be more amenable to treatment,” Dr. Navai said.
In contrast, larger tumors and more invasive disease may emit immune inhibitory signals that dampen the efficacy of CAR T cells, she added.
Development of the CAR T-cell construct is supported by the Cancer Prevention & Research Institute of Texas, Stand Up to Cancer, the St. Baldrick’s Foundation, Cookies for Kids’ Cancer, Alex’s Lemonade Stand, and a grant from the National Institutes of Health. Dr. Navai and Dr. Azad reported having no disclosures relevant to the work.
SOURCE: Navai SA et al. AACR 2019, Abstract LB-147.
ATLANTA – A novel chimeric antigen receptor (CAR) T-cell construct centered on HER2 as the target antigen was safe and showed early promise in the treatment of advanced sarcomas of bone and soft tissues in a phase I trial.
One patient, a 16-year-old girl with advanced osteosarcoma metastatic to her lungs, had a complete response to the therapy that is ongoing out to nearly 3 years, reported Shoba A. Navai, MD, from Baylor College of Medicine in Houston.
A second patient, an 8-year-old boy with rhabdomyosarcoma metastatic to bone marrow, had a complete response lasting 12 months. Upon relapse he was re-enrolled, received additional CAR T-cell infusions, and had a second complete response that has been ongoing for 17 months.
“HER2 CAR T cells can induce objective clinical responses in some patients with sarcoma, and engagement of endogenous immunity may aid in generation of tumor responses. We are currently working to validate these findings in other patients who were treated,” she said at a briefing at the annual meeting of the American Association for Cancer Research.
HER2 is a member of the human epidermal growth factor receptor family that is primarily expressed on the surface of tumor cells but is largely absent from nonmalignant tissues. HER2 can be expressed in a variety of sarcomas, including osteosarcoma, and HER2 expression in osteosarcoma correlates with worse overall survival.
Unlike HER2-positive breast cancers, however, HER2 expression levels in osteosarcoma are too low to be effectively targeted by anti-HER2 agents such as trastuzumab (Hereceptin).
But as Dr. Navai and colleagues have found, HER2 appears to be a valid target for CAR T-cell therapy in otherwise antigenically “cold” tumors – that is, tumors with few targetable antigens.
Old target, new weapon
They have developed a CAR T-cell construct using a HER2-directed antibody coupled with CD28 as the costimulatory molecule. As with other CAR T therapies, the patient’s T cells or selected T cell subsets are collected, transfected to express the antigen, and are then expanded and returned to the patient following lymphodepletion with either fludarabine alone or with cyclophosphamide.
Each patient received up to three infusions of autologous CAR T cells at a dose of 1 x 108 cells/m2, and eligible patients received up to five additional infusions without additional lymphodepletion.
Dr. Navai presented data on 10 patients treated to date, including the two mentioned before; the boy with rhabdomyosarcoma was counted as two separate patients for the purpose of the efficacy analysis.
All patients had metastatic disease, including five with osteosarcoma, three with rhabdomyosarcoma, one with Ewing sarcoma, and one with synovial sarcoma.
The lymphodepletion regimens did their job, inducing neutropenia (defined as an absolute neutrophil count less than 500 per milliliter ) for up to 14 days.
Eight patients developed grade 1 or 2 cytokine release syndrome within 24 hours of CAR T-cell infusion, and all cases completely resolved with supportive care within 5 days of onset.
In nine patients, T cells were successfully expanded, with a median peak expansion on day 7.
In all 10 patients, CAR T cells were detected by quantitative polymerase chain reaction 6 weeks after infusion.
In addition to the two patients with complete remissions already described, three patients had stable disease. The remaining patients had disease progression. At the most recent analysis, five patients were still alive, and five had died.
The infusions were safe, with no dose-limiting toxicities reported. No patient required a transfusion, and there were no opportunistic, infections, no neurotoxicities, and no lasting pulmonary or cardiac toxicities, Dr. Navai reported.
Some fare better than others
Nilofer S. Azad, MD, of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, Baltimore, who moderated the briefing, commented that the study had “very small numbers, but is still very exciting.”
She noted that the patients who benefited most from the therapy either had minimal residual disease or bone marrow disease without visceral disease; she asked Dr. Navai how this could be addressed going forward.
“The patients who seemed to have had responses both in this trial, as well as in our previous trial without lymphodepletion, tended to have less disease or more accessible disease. So we hypothesized that disease that’s in the bone marrow because it’s more accessible, or in the lungs, where also CAR T cells go after they are first infused, may be more amenable to treatment,” Dr. Navai said.
In contrast, larger tumors and more invasive disease may emit immune inhibitory signals that dampen the efficacy of CAR T cells, she added.
Development of the CAR T-cell construct is supported by the Cancer Prevention & Research Institute of Texas, Stand Up to Cancer, the St. Baldrick’s Foundation, Cookies for Kids’ Cancer, Alex’s Lemonade Stand, and a grant from the National Institutes of Health. Dr. Navai and Dr. Azad reported having no disclosures relevant to the work.
SOURCE: Navai SA et al. AACR 2019, Abstract LB-147.
ATLANTA – A novel chimeric antigen receptor (CAR) T-cell construct centered on HER2 as the target antigen was safe and showed early promise in the treatment of advanced sarcomas of bone and soft tissues in a phase I trial.
One patient, a 16-year-old girl with advanced osteosarcoma metastatic to her lungs, had a complete response to the therapy that is ongoing out to nearly 3 years, reported Shoba A. Navai, MD, from Baylor College of Medicine in Houston.
A second patient, an 8-year-old boy with rhabdomyosarcoma metastatic to bone marrow, had a complete response lasting 12 months. Upon relapse he was re-enrolled, received additional CAR T-cell infusions, and had a second complete response that has been ongoing for 17 months.
“HER2 CAR T cells can induce objective clinical responses in some patients with sarcoma, and engagement of endogenous immunity may aid in generation of tumor responses. We are currently working to validate these findings in other patients who were treated,” she said at a briefing at the annual meeting of the American Association for Cancer Research.
HER2 is a member of the human epidermal growth factor receptor family that is primarily expressed on the surface of tumor cells but is largely absent from nonmalignant tissues. HER2 can be expressed in a variety of sarcomas, including osteosarcoma, and HER2 expression in osteosarcoma correlates with worse overall survival.
Unlike HER2-positive breast cancers, however, HER2 expression levels in osteosarcoma are too low to be effectively targeted by anti-HER2 agents such as trastuzumab (Hereceptin).
But as Dr. Navai and colleagues have found, HER2 appears to be a valid target for CAR T-cell therapy in otherwise antigenically “cold” tumors – that is, tumors with few targetable antigens.
Old target, new weapon
They have developed a CAR T-cell construct using a HER2-directed antibody coupled with CD28 as the costimulatory molecule. As with other CAR T therapies, the patient’s T cells or selected T cell subsets are collected, transfected to express the antigen, and are then expanded and returned to the patient following lymphodepletion with either fludarabine alone or with cyclophosphamide.
Each patient received up to three infusions of autologous CAR T cells at a dose of 1 x 108 cells/m2, and eligible patients received up to five additional infusions without additional lymphodepletion.
Dr. Navai presented data on 10 patients treated to date, including the two mentioned before; the boy with rhabdomyosarcoma was counted as two separate patients for the purpose of the efficacy analysis.
All patients had metastatic disease, including five with osteosarcoma, three with rhabdomyosarcoma, one with Ewing sarcoma, and one with synovial sarcoma.
The lymphodepletion regimens did their job, inducing neutropenia (defined as an absolute neutrophil count less than 500 per milliliter ) for up to 14 days.
Eight patients developed grade 1 or 2 cytokine release syndrome within 24 hours of CAR T-cell infusion, and all cases completely resolved with supportive care within 5 days of onset.
In nine patients, T cells were successfully expanded, with a median peak expansion on day 7.
In all 10 patients, CAR T cells were detected by quantitative polymerase chain reaction 6 weeks after infusion.
In addition to the two patients with complete remissions already described, three patients had stable disease. The remaining patients had disease progression. At the most recent analysis, five patients were still alive, and five had died.
The infusions were safe, with no dose-limiting toxicities reported. No patient required a transfusion, and there were no opportunistic, infections, no neurotoxicities, and no lasting pulmonary or cardiac toxicities, Dr. Navai reported.
Some fare better than others
Nilofer S. Azad, MD, of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, Baltimore, who moderated the briefing, commented that the study had “very small numbers, but is still very exciting.”
She noted that the patients who benefited most from the therapy either had minimal residual disease or bone marrow disease without visceral disease; she asked Dr. Navai how this could be addressed going forward.
“The patients who seemed to have had responses both in this trial, as well as in our previous trial without lymphodepletion, tended to have less disease or more accessible disease. So we hypothesized that disease that’s in the bone marrow because it’s more accessible, or in the lungs, where also CAR T cells go after they are first infused, may be more amenable to treatment,” Dr. Navai said.
In contrast, larger tumors and more invasive disease may emit immune inhibitory signals that dampen the efficacy of CAR T cells, she added.
Development of the CAR T-cell construct is supported by the Cancer Prevention & Research Institute of Texas, Stand Up to Cancer, the St. Baldrick’s Foundation, Cookies for Kids’ Cancer, Alex’s Lemonade Stand, and a grant from the National Institutes of Health. Dr. Navai and Dr. Azad reported having no disclosures relevant to the work.
SOURCE: Navai SA et al. AACR 2019, Abstract LB-147.
REPORTING FROM AACR 2019
More Reports from the Connective Tissue Oncology Society 2018 annual meeting in Rome, November 14-17
Early Results Find Olaratumab Combo With Doxorubicin Plus Ifosfamide Safe
Initial results of the phase 1b study of olaratumab plus doxorubicin and ifosfamide have shown the combination to be safe, reported Sebastian Bauer, MD, of the West German Cancer Center, University of Duisburg-Essen, Essen, Germany, and his colleagues at CTOS 2018.
The phase 1 trial (NCT03283696) enrolled 16 patients with advanced or metastatic soft tissue sarcomas. Patients had received no prior lines of systemic therapy and had an ECOG performance status of 0-1. Adequate follow-up data were available for 10 patients.
Olaratumab (Lartruvo), which binds platelet-derived growth factor receptor alpha (PDGFRα), was given at 15 mg/kg in combination with doxorubicin (75 mg/m2 on days 1-3) and ifosfamide (10 g/m2 on days 1-4). This was followed by mandatory granulocyte-colony-stimulating factor therapy in cycles 1-6 on a 21-day cycle. Doxorubicin could be administered by continuous infusion or bolus administration and with cardiac protection. Mesna dosing was at least 60% of the ifosfamide dose.
Two of the 10 patients had dose-limiting toxicities; one had grade 4 febrile neutropenia and the other had grade 3 febrile neutropenia and grade 3 mucositis. Common related adverse events occurring in over 30% of patients included fatigue, anemia, neutropenia, thrombocytopenia, constipation, and nausea. One patient discontinued study treatment due to progressive disease, and all others were on study treatment as of the data cutoff. Among 7 patients evaluated for tumor response, 3 patients had a partial response according to RECIST and 3 other patients had stabilized disease as best overall response, for a disease control rate of 86%.
Given that 8 of 10 evaluable patients have completed the dose-limiting toxicity period without dose-limiting toxicities at the 15 mg/kg dose level of olaratumab, the study has proceeded to the next cohort. In those patients, an olaratumab loading dose of 20 mg/kg will be evaluated in cycle 1, followed by 15 mg/kg of olaratumab in subsequent cycles with the same doses of doxorubicin plus ifosfamide, the researchers wrote in their abstract.
NOTE: Since CTOS 2018, olaratumab plus doxorubicin did not meet its phase 3 endpoint of overall survival (OS) advantage in the full study population or in the leiomyosarcoma subpopulation compared to doxorubicin alone.
Anthracycline-Based Regimen Excels in FIGO-1 Uterine Leiomyosarcoma
Patients with uterine leiomyosarcomas treated with anthracycline-based regimens experienced longer disease-free survival compared to patients treated with gemcitabine and docetaxel, according to a retrospective analysis reported at CTOS 2018.
Roberta Sanfilippo, MD, of Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues reviewed all patients with FIGO stage I uterine leiomyosarcomas at two Italian centers who underwent hysterectomy with or without oophorectomy and were then treated with adjuvant chemotherapy with anthracycline-based or gemcitabine-based regimens.
Of 145 patients, 97 were treated with an anthracycline-based regimen and 48 with gemcitabine and docetaxel. The median number of cycles of anthracycline-based therapy patients received was 4 (range 2-6) and the median number of cycles with gemcitabine and docetaxel was 5 (range 3-7). Disease-free survival was 31 months in patients treated with anthracycline-based chemotherapy and 19 months in patients treated with gemcitabine and docetaxel.
These results suggest that future trials to assess the efficacy of adjuvant chemotherapy in uterine leiomyosarcoma should incorporate anthracyclines, the investigators maintain.
Trabectedin and Concurrent Low-Dose Radiotherapy Feasible
Trabectedin concurrent with lowdose radiotherapy is being examined as an option for patients with pulmonary metastatic soft tissue sarcoma (NCT02275286).
In a phase 1 study, long-lasting dimensional responses were seen in 71% of the irradiated lesions. Based on those results, trabectedin (Yondelis) at 1.5 mg/m2 will be the recommended dose for phase 2, according to Javier Martín-Broto, MD, of the Institute of Biomedicine Research (IBIS)-University Hospital Virgen del Rocio/CSIC/University of Seville, Spain, and his colleagues, reporting at CTOS 2018.
For the study, trabectedin was given along with radiotherapy (30 Gy) in 10 fractions (3 Gy/fraction). Three dose levels of trabectedin were administered: -1 (1.1 mg/m2), 1 (1.3 mg/m2), and 2 (1.5 mg/m2). Dose-limiting toxicity was defined as grade 3 or greater events excluding grade 3/4 neutropenia lasting less than 5 days, grade 3 transaminitis if it did not lead to trabectedin delay, and grade 3/4 nausea/vomiting due to inadequate prophylaxis.
Ten of the 18 patients enrolled had synovial sarcoma; 3 had undifferentiated pleomorphic sarcomas, and the other patients had either myxoid liposarcoma, dedifferentiated liposarcoma, G3 not otherwise specified sarcoma, leiomyosarcoma, or malignant peripheral nerve sheath tumor.
Patients received a median of 1 prior line of chemotherapy (range: 0-3). Twelve patients received trabectedin at dose level 1 and 6 patients at dose level 2. Grade 3/4 adverse events were neutropenia, seen in 8 patients; alanine aminotransferase (ALT) elevation, seen in 2 patients; gamma-glutamyl transferase (GGT) elevation, seen in 2 patients; anemia, seen in 2 patients; febrile neutropenia, seen in 1 patient; and pneumonitis, seen in 1 patient.
There were two dose-limiting toxicities: transient grade 4 ALT elevation at the level 1 dose and grade 4 neutropenia for more than 5 days at the level 2 dose.
Based on central radiological review of 17 evaluable patients, 2 patients achieved complete response, 3 had partial responses, 6 had stable disease, and 6 had progressive disease. The local review reported complete responses in 2 patients, partial responses in 5, stable disease in 4, and progressive disease in 6.
Of the irradiated lesions, 71% had long-lasting dimensional responses: 4 completely responded, 8 responded partially, 4 were stable, and 1 progressed.
With a median follow-up of 18 months, median progression-free survival was 2.83 months (95%CI: 2.3-3.3 months). Thirteen patients have died, with a median overall survival of 8.77 months (95%CI: 3.6-13.9) and a 12-month overall survival rate of 48%.
The investigators concluded trabectedin with concurrent radiotherapy was feasible in patients with pulmonary metastatic soft tissue sarcoma regardless of their histologic subtype.
Early Results Find Olaratumab Combo With Doxorubicin Plus Ifosfamide Safe
Initial results of the phase 1b study of olaratumab plus doxorubicin and ifosfamide have shown the combination to be safe, reported Sebastian Bauer, MD, of the West German Cancer Center, University of Duisburg-Essen, Essen, Germany, and his colleagues at CTOS 2018.
The phase 1 trial (NCT03283696) enrolled 16 patients with advanced or metastatic soft tissue sarcomas. Patients had received no prior lines of systemic therapy and had an ECOG performance status of 0-1. Adequate follow-up data were available for 10 patients.
Olaratumab (Lartruvo), which binds platelet-derived growth factor receptor alpha (PDGFRα), was given at 15 mg/kg in combination with doxorubicin (75 mg/m2 on days 1-3) and ifosfamide (10 g/m2 on days 1-4). This was followed by mandatory granulocyte-colony-stimulating factor therapy in cycles 1-6 on a 21-day cycle. Doxorubicin could be administered by continuous infusion or bolus administration and with cardiac protection. Mesna dosing was at least 60% of the ifosfamide dose.
Two of the 10 patients had dose-limiting toxicities; one had grade 4 febrile neutropenia and the other had grade 3 febrile neutropenia and grade 3 mucositis. Common related adverse events occurring in over 30% of patients included fatigue, anemia, neutropenia, thrombocytopenia, constipation, and nausea. One patient discontinued study treatment due to progressive disease, and all others were on study treatment as of the data cutoff. Among 7 patients evaluated for tumor response, 3 patients had a partial response according to RECIST and 3 other patients had stabilized disease as best overall response, for a disease control rate of 86%.
Given that 8 of 10 evaluable patients have completed the dose-limiting toxicity period without dose-limiting toxicities at the 15 mg/kg dose level of olaratumab, the study has proceeded to the next cohort. In those patients, an olaratumab loading dose of 20 mg/kg will be evaluated in cycle 1, followed by 15 mg/kg of olaratumab in subsequent cycles with the same doses of doxorubicin plus ifosfamide, the researchers wrote in their abstract.
NOTE: Since CTOS 2018, olaratumab plus doxorubicin did not meet its phase 3 endpoint of overall survival (OS) advantage in the full study population or in the leiomyosarcoma subpopulation compared to doxorubicin alone.
Anthracycline-Based Regimen Excels in FIGO-1 Uterine Leiomyosarcoma
Patients with uterine leiomyosarcomas treated with anthracycline-based regimens experienced longer disease-free survival compared to patients treated with gemcitabine and docetaxel, according to a retrospective analysis reported at CTOS 2018.
Roberta Sanfilippo, MD, of Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues reviewed all patients with FIGO stage I uterine leiomyosarcomas at two Italian centers who underwent hysterectomy with or without oophorectomy and were then treated with adjuvant chemotherapy with anthracycline-based or gemcitabine-based regimens.
Of 145 patients, 97 were treated with an anthracycline-based regimen and 48 with gemcitabine and docetaxel. The median number of cycles of anthracycline-based therapy patients received was 4 (range 2-6) and the median number of cycles with gemcitabine and docetaxel was 5 (range 3-7). Disease-free survival was 31 months in patients treated with anthracycline-based chemotherapy and 19 months in patients treated with gemcitabine and docetaxel.
These results suggest that future trials to assess the efficacy of adjuvant chemotherapy in uterine leiomyosarcoma should incorporate anthracyclines, the investigators maintain.
Trabectedin and Concurrent Low-Dose Radiotherapy Feasible
Trabectedin concurrent with lowdose radiotherapy is being examined as an option for patients with pulmonary metastatic soft tissue sarcoma (NCT02275286).
In a phase 1 study, long-lasting dimensional responses were seen in 71% of the irradiated lesions. Based on those results, trabectedin (Yondelis) at 1.5 mg/m2 will be the recommended dose for phase 2, according to Javier Martín-Broto, MD, of the Institute of Biomedicine Research (IBIS)-University Hospital Virgen del Rocio/CSIC/University of Seville, Spain, and his colleagues, reporting at CTOS 2018.
For the study, trabectedin was given along with radiotherapy (30 Gy) in 10 fractions (3 Gy/fraction). Three dose levels of trabectedin were administered: -1 (1.1 mg/m2), 1 (1.3 mg/m2), and 2 (1.5 mg/m2). Dose-limiting toxicity was defined as grade 3 or greater events excluding grade 3/4 neutropenia lasting less than 5 days, grade 3 transaminitis if it did not lead to trabectedin delay, and grade 3/4 nausea/vomiting due to inadequate prophylaxis.
Ten of the 18 patients enrolled had synovial sarcoma; 3 had undifferentiated pleomorphic sarcomas, and the other patients had either myxoid liposarcoma, dedifferentiated liposarcoma, G3 not otherwise specified sarcoma, leiomyosarcoma, or malignant peripheral nerve sheath tumor.
Patients received a median of 1 prior line of chemotherapy (range: 0-3). Twelve patients received trabectedin at dose level 1 and 6 patients at dose level 2. Grade 3/4 adverse events were neutropenia, seen in 8 patients; alanine aminotransferase (ALT) elevation, seen in 2 patients; gamma-glutamyl transferase (GGT) elevation, seen in 2 patients; anemia, seen in 2 patients; febrile neutropenia, seen in 1 patient; and pneumonitis, seen in 1 patient.
There were two dose-limiting toxicities: transient grade 4 ALT elevation at the level 1 dose and grade 4 neutropenia for more than 5 days at the level 2 dose.
Based on central radiological review of 17 evaluable patients, 2 patients achieved complete response, 3 had partial responses, 6 had stable disease, and 6 had progressive disease. The local review reported complete responses in 2 patients, partial responses in 5, stable disease in 4, and progressive disease in 6.
Of the irradiated lesions, 71% had long-lasting dimensional responses: 4 completely responded, 8 responded partially, 4 were stable, and 1 progressed.
With a median follow-up of 18 months, median progression-free survival was 2.83 months (95%CI: 2.3-3.3 months). Thirteen patients have died, with a median overall survival of 8.77 months (95%CI: 3.6-13.9) and a 12-month overall survival rate of 48%.
The investigators concluded trabectedin with concurrent radiotherapy was feasible in patients with pulmonary metastatic soft tissue sarcoma regardless of their histologic subtype.
Early Results Find Olaratumab Combo With Doxorubicin Plus Ifosfamide Safe
Initial results of the phase 1b study of olaratumab plus doxorubicin and ifosfamide have shown the combination to be safe, reported Sebastian Bauer, MD, of the West German Cancer Center, University of Duisburg-Essen, Essen, Germany, and his colleagues at CTOS 2018.
The phase 1 trial (NCT03283696) enrolled 16 patients with advanced or metastatic soft tissue sarcomas. Patients had received no prior lines of systemic therapy and had an ECOG performance status of 0-1. Adequate follow-up data were available for 10 patients.
Olaratumab (Lartruvo), which binds platelet-derived growth factor receptor alpha (PDGFRα), was given at 15 mg/kg in combination with doxorubicin (75 mg/m2 on days 1-3) and ifosfamide (10 g/m2 on days 1-4). This was followed by mandatory granulocyte-colony-stimulating factor therapy in cycles 1-6 on a 21-day cycle. Doxorubicin could be administered by continuous infusion or bolus administration and with cardiac protection. Mesna dosing was at least 60% of the ifosfamide dose.
Two of the 10 patients had dose-limiting toxicities; one had grade 4 febrile neutropenia and the other had grade 3 febrile neutropenia and grade 3 mucositis. Common related adverse events occurring in over 30% of patients included fatigue, anemia, neutropenia, thrombocytopenia, constipation, and nausea. One patient discontinued study treatment due to progressive disease, and all others were on study treatment as of the data cutoff. Among 7 patients evaluated for tumor response, 3 patients had a partial response according to RECIST and 3 other patients had stabilized disease as best overall response, for a disease control rate of 86%.
Given that 8 of 10 evaluable patients have completed the dose-limiting toxicity period without dose-limiting toxicities at the 15 mg/kg dose level of olaratumab, the study has proceeded to the next cohort. In those patients, an olaratumab loading dose of 20 mg/kg will be evaluated in cycle 1, followed by 15 mg/kg of olaratumab in subsequent cycles with the same doses of doxorubicin plus ifosfamide, the researchers wrote in their abstract.
NOTE: Since CTOS 2018, olaratumab plus doxorubicin did not meet its phase 3 endpoint of overall survival (OS) advantage in the full study population or in the leiomyosarcoma subpopulation compared to doxorubicin alone.
Anthracycline-Based Regimen Excels in FIGO-1 Uterine Leiomyosarcoma
Patients with uterine leiomyosarcomas treated with anthracycline-based regimens experienced longer disease-free survival compared to patients treated with gemcitabine and docetaxel, according to a retrospective analysis reported at CTOS 2018.
Roberta Sanfilippo, MD, of Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues reviewed all patients with FIGO stage I uterine leiomyosarcomas at two Italian centers who underwent hysterectomy with or without oophorectomy and were then treated with adjuvant chemotherapy with anthracycline-based or gemcitabine-based regimens.
Of 145 patients, 97 were treated with an anthracycline-based regimen and 48 with gemcitabine and docetaxel. The median number of cycles of anthracycline-based therapy patients received was 4 (range 2-6) and the median number of cycles with gemcitabine and docetaxel was 5 (range 3-7). Disease-free survival was 31 months in patients treated with anthracycline-based chemotherapy and 19 months in patients treated with gemcitabine and docetaxel.
These results suggest that future trials to assess the efficacy of adjuvant chemotherapy in uterine leiomyosarcoma should incorporate anthracyclines, the investigators maintain.
Trabectedin and Concurrent Low-Dose Radiotherapy Feasible
Trabectedin concurrent with lowdose radiotherapy is being examined as an option for patients with pulmonary metastatic soft tissue sarcoma (NCT02275286).
In a phase 1 study, long-lasting dimensional responses were seen in 71% of the irradiated lesions. Based on those results, trabectedin (Yondelis) at 1.5 mg/m2 will be the recommended dose for phase 2, according to Javier Martín-Broto, MD, of the Institute of Biomedicine Research (IBIS)-University Hospital Virgen del Rocio/CSIC/University of Seville, Spain, and his colleagues, reporting at CTOS 2018.
For the study, trabectedin was given along with radiotherapy (30 Gy) in 10 fractions (3 Gy/fraction). Three dose levels of trabectedin were administered: -1 (1.1 mg/m2), 1 (1.3 mg/m2), and 2 (1.5 mg/m2). Dose-limiting toxicity was defined as grade 3 or greater events excluding grade 3/4 neutropenia lasting less than 5 days, grade 3 transaminitis if it did not lead to trabectedin delay, and grade 3/4 nausea/vomiting due to inadequate prophylaxis.
Ten of the 18 patients enrolled had synovial sarcoma; 3 had undifferentiated pleomorphic sarcomas, and the other patients had either myxoid liposarcoma, dedifferentiated liposarcoma, G3 not otherwise specified sarcoma, leiomyosarcoma, or malignant peripheral nerve sheath tumor.
Patients received a median of 1 prior line of chemotherapy (range: 0-3). Twelve patients received trabectedin at dose level 1 and 6 patients at dose level 2. Grade 3/4 adverse events were neutropenia, seen in 8 patients; alanine aminotransferase (ALT) elevation, seen in 2 patients; gamma-glutamyl transferase (GGT) elevation, seen in 2 patients; anemia, seen in 2 patients; febrile neutropenia, seen in 1 patient; and pneumonitis, seen in 1 patient.
There were two dose-limiting toxicities: transient grade 4 ALT elevation at the level 1 dose and grade 4 neutropenia for more than 5 days at the level 2 dose.
Based on central radiological review of 17 evaluable patients, 2 patients achieved complete response, 3 had partial responses, 6 had stable disease, and 6 had progressive disease. The local review reported complete responses in 2 patients, partial responses in 5, stable disease in 4, and progressive disease in 6.
Of the irradiated lesions, 71% had long-lasting dimensional responses: 4 completely responded, 8 responded partially, 4 were stable, and 1 progressed.
With a median follow-up of 18 months, median progression-free survival was 2.83 months (95%CI: 2.3-3.3 months). Thirteen patients have died, with a median overall survival of 8.77 months (95%CI: 3.6-13.9) and a 12-month overall survival rate of 48%.
The investigators concluded trabectedin with concurrent radiotherapy was feasible in patients with pulmonary metastatic soft tissue sarcoma regardless of their histologic subtype.
Health care resource utilization leading to a diagnosis of soft tissue sarcoma
Introduction
Soft tissue sarcomas (STS) are a heterogeneous group of cancerous tumors, comprised of more than 50 histological subtypes that develop from soft tissues of the body (eg, fat, muscles, nerve tissue, deep skin tissue, visceral nonepithelial tissue). Due to many factors, not limited to the heterogeneity of this set of diseases and lack of screening tests, reaching a diagnosis of STS is challenging for the general practitioner as well as for the oncologist. Sarcomas may present with nonspecific and often indolent symptomology, depending on the specific histological subtype. According to the American Cancer Society, the signs and symptoms of a sarcoma include a new or growing lump, worsening abdominal pain, blood in stool or vomit, and black stools (due to abdominal bleeding).1 Unfortunately, these symptoms could be indicative of any number of other health conditions and are nonspecific to sarcoma.
As with many cancers, the early detection of disease when it may be completely resected could lead to a cure, whereas diagnosis when the disease is no longer amenable to surgery will impact patient survival. Among all forms of STS, early diagnosis when the patient has only localized disease is associated with an 80.8% five-year survival rate, which decreases to 16.4% for patients whose disease has already metastasized to other parts of the body at the time of diagnosis.2
Previous work has evaluated the relationship between duration of symptoms that may lead to a diagnosis of sarcoma and cancer outcomes. A retrospective analysis of a cohort of adults with bone or STS found no correlation between patient recall of duration of prediagnosis symptoms and survival or metastatic disease at diagnosis.3,4 Little other research was identified that examined the challenges of identifying a potential sarcoma. Despite the gap in knowledge, advocacy and patient-centered organizations emphasize the risk of delayed diagnosis and report high levels of stress and frustration among patients by the time an accurate diagnosis is obtained.5 The objective of this study was to quantify the health care experience and misdiagnoses that occurred prior to a sarcoma diagnosis compared to a cohort of matched controls.
Methods
A retrospective observational database study was conducted using detailed resource utilization and cost data from the Truven MarketScan claims database. Truven MarketScan® is a HIPAA-compliant, fully integrated patient-level database containing inpatient, outpatient, drug, lab, health risk assessment, and benefit design information from commercial and Medicare supplemental insurance plans. Additionally, the Health and Productivity Management (HPM) database, containing workplace absence, short-term disability, long-term disability, and worker’s compensation data, is linked at the individual patient level. The linkage of the claims and HPM database was used for this study.
Patients were eligible for inclusion in the cohort of a sarcoma if they had at least two ICD-9 codes of 171.x on two different days between July 1, 2004, and March 30, 2014. The date of the first eligible code was considered the index date. Patients were required to have at least 6 months of health care plan enrollment prior to the first eligible ICD-9 code to allow for prediagnosis activity to be identified in the database. Patients were also required to be 18 years of age or older on the first eligible ICD-9 code date. Patients were excluded who had evidence suggesting a diagnosis of osteosarcoma, Kaposi’s sarcoma, or gastrointestinal stromal tumors (treatment with methotrexate, ICD-9 codes of 176.x, 171.x, or 238.1), a history of any cancer before the eligible sarcoma ICD-9 code, or history of systemic anticancer therapy during the 6-month pre-index period. All patients meeting eligibility criteria were included in the matching algorithm to identify the control cohort.
The matched control cohort was required to have at least the same duration of follow-up at the case level as the matched sarcoma patient, could not have any evidence of any malignancy at any time in the database, nor could have received any systemic anticancer therapy at any time. Controls were randomly selected from the more than 100 million individual patient cases in the MarketScan database to be matched to the eligible sarcoma patient cohort exactly on age, geographic region of residence, health insurance plan type, gender, noncancer comorbid conditions (measured by Charlson Comorbidity Index items), and employment status. All factors were exact matched at the sarcoma cohort index diagnosis date. In the case of missing variables, patients were matched on missingness (eg, a case with missing employment status would be matched to a control with missing employment status).
The eligible time period for the index date of the possible sarcoma cohort and matched controls was between July 1, 2004, and March 30, 2014, which allowed for a minimum of 1-year follow-up through the end of the database available at the time of analysis.
All ICD-9 diagnostic and procedure codes present in the matched 6-month time period pre-index diagnosis were compared to explore factors that may be more likely to be present in the sarcoma cohort compared to matched controls. Univariate analysis was conducted for each prediagnosis variable. Analyses were conducted using T test for continuous variables, and Chi-square or Fisher’s exact test was used for categorical variables.
Number of physician visits, inpatient hospital stays, surgical procedures, and emergency room visits were compared between those in the sarcoma cohort and matched controls during the matched 6-month pre-index period. The post-index diagnosis employment status was also compared between groups using the HPM database. Comparisons between the sarcoma cohort and control cohort were made among the actively employed patients at baseline related to the proportion of patients who continued active employment, the proportion who permanently discontinued work, and the proportion who initially discontinued work and then returned to work at a later time. No adjustments were made for multiple comparisons.
Results
A total of 7826 controls were each matched to patients in the sarcoma cohort. The baseline characteristics of the study cohorts are provided in Table 1. 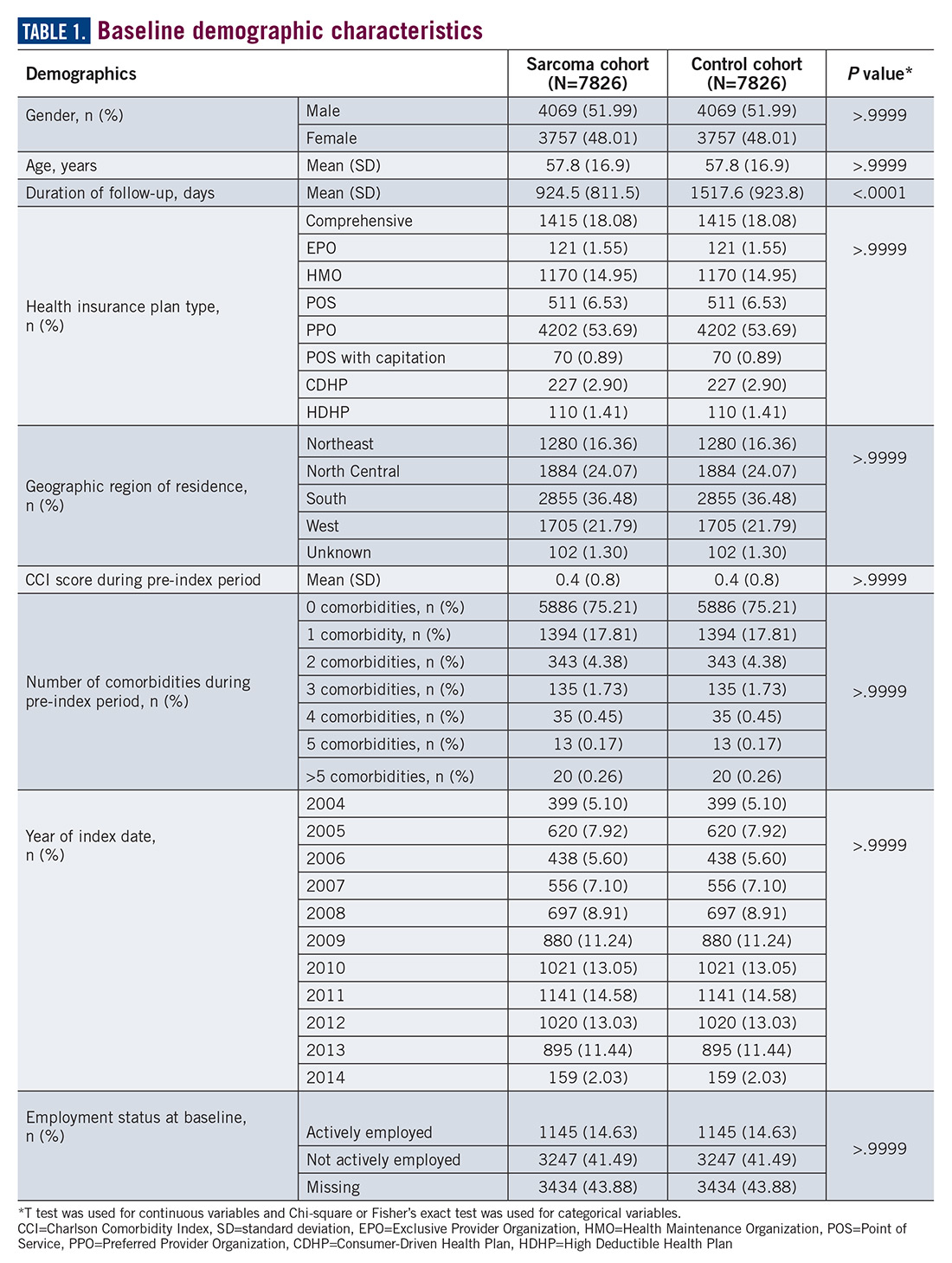
During the 6-month period before the sarcoma diagnosis (prediagnosis period), patients had significantly greater frequency of diagnoses identified than controls for uncertain neoplasms, limb pain, and hypertension (all P<.001, Table 2). 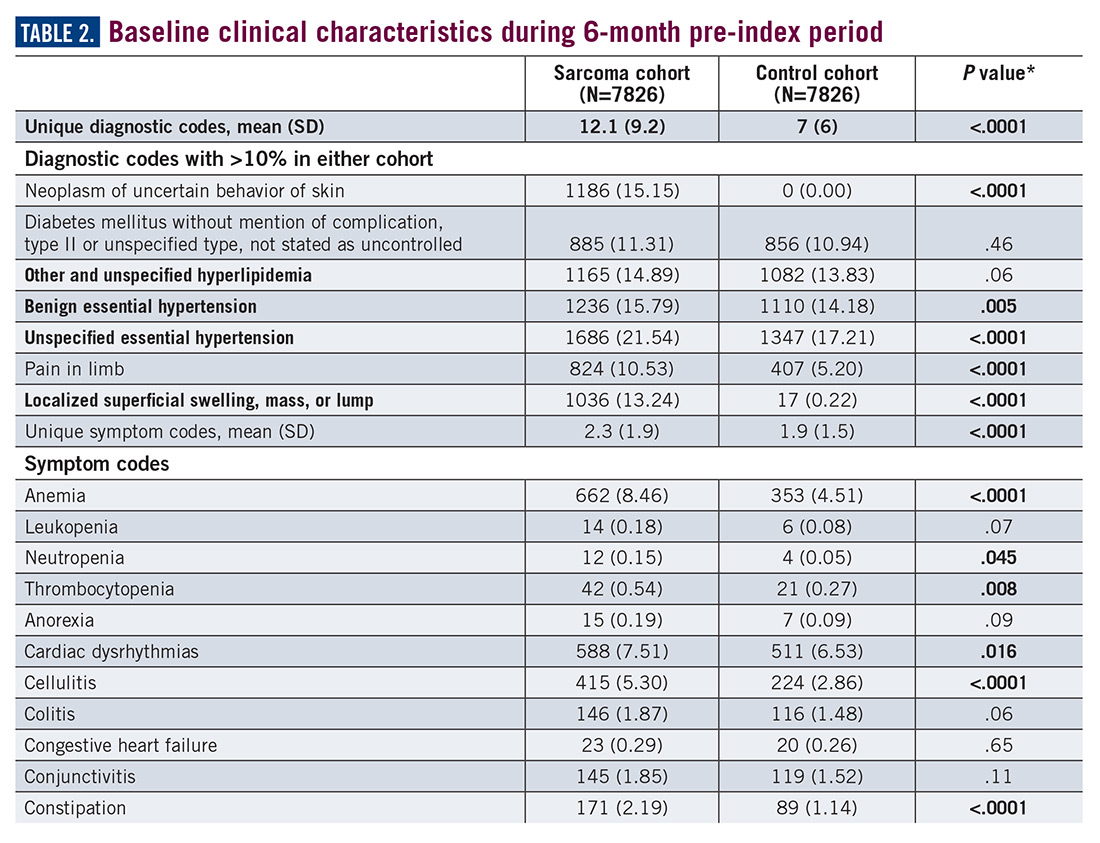
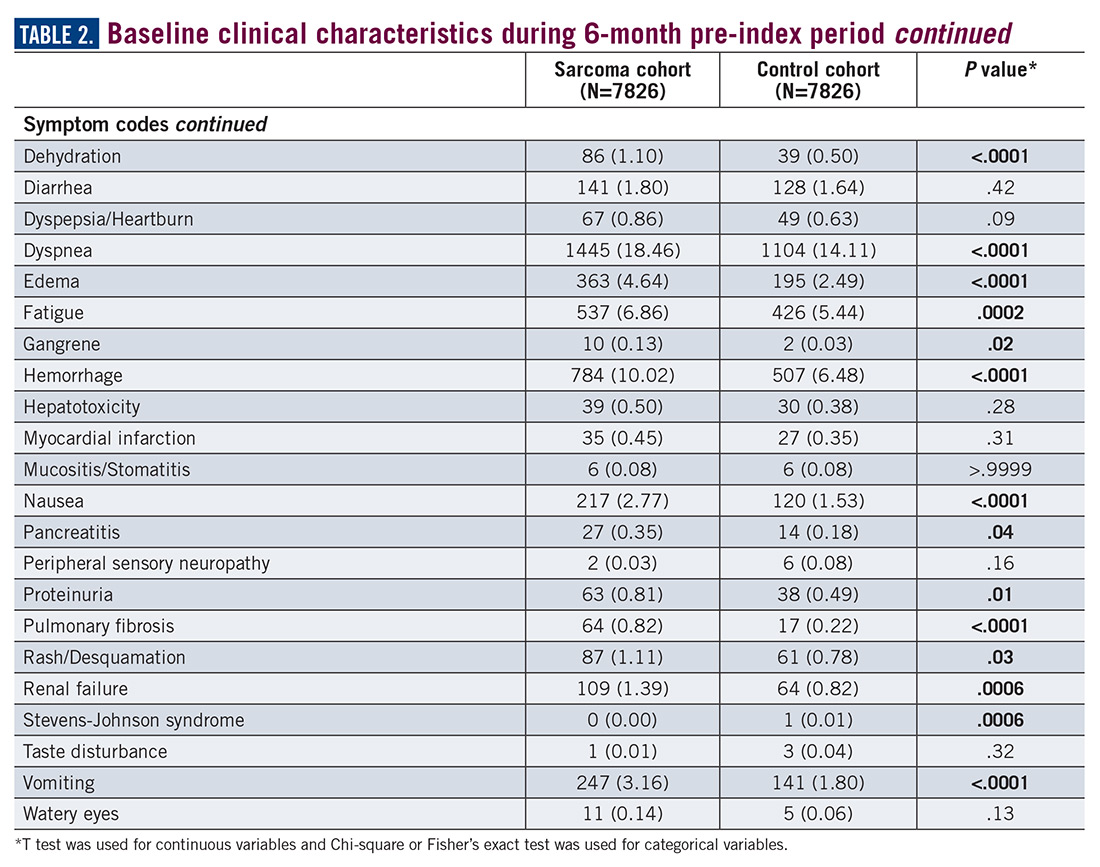
Similarly, the majority of health care resource utilization factors evaluated showed statistically higher health care use among patients later suspected of having sarcoma than matched controls (Table 3). 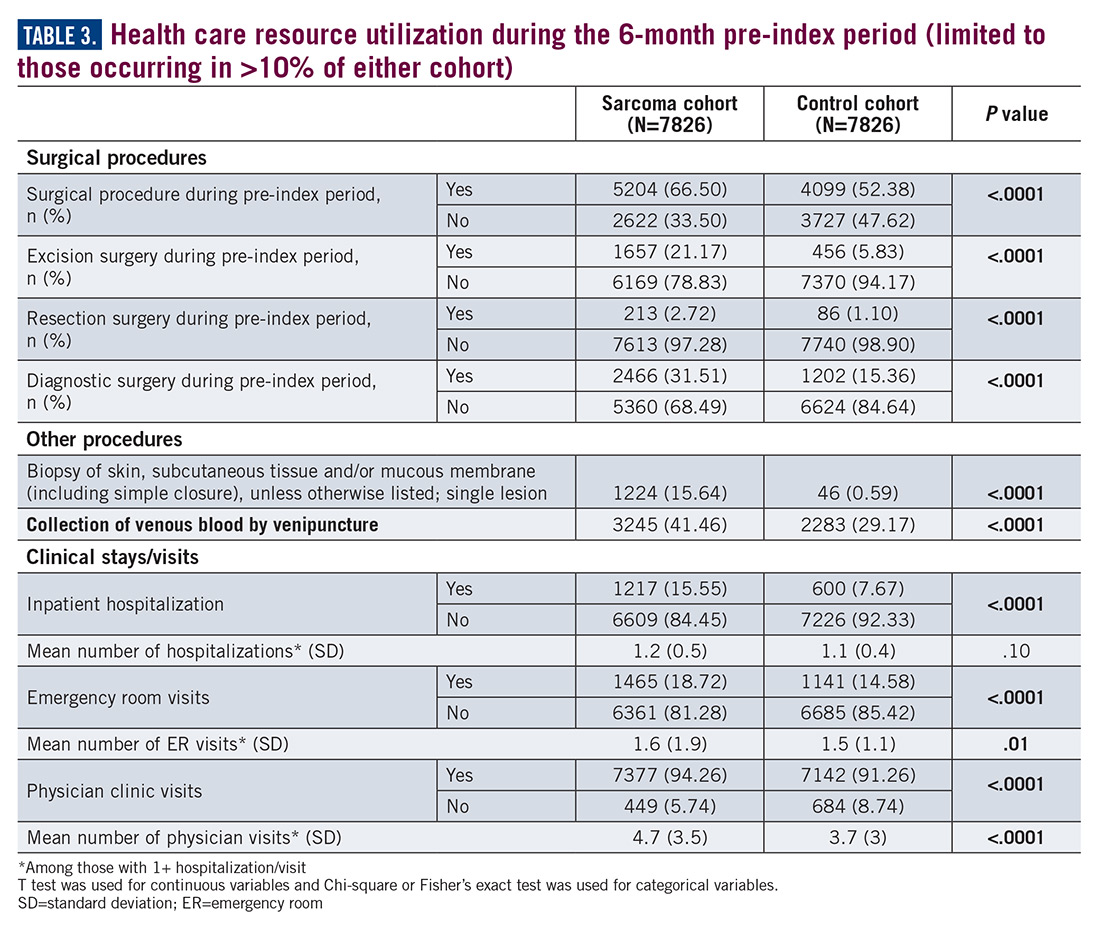
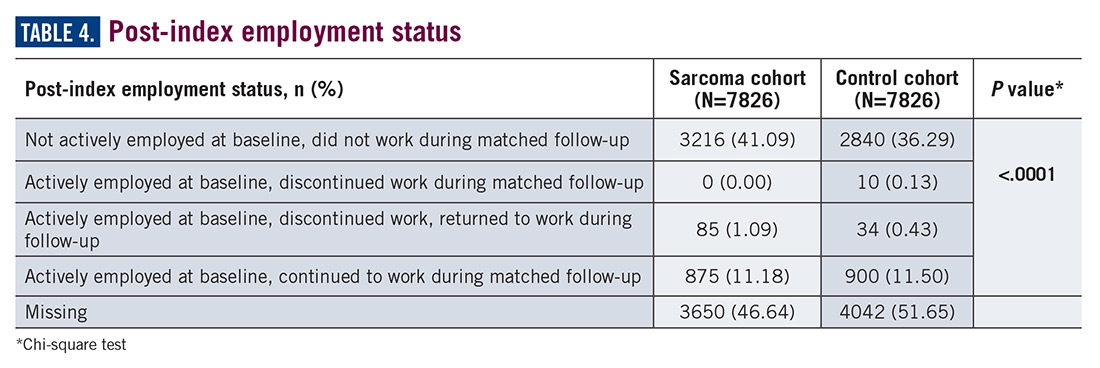
Employment status was missing for 44% of the cohort at baseline and approximately half the cohort during follow-up (Table 4).
Discussion
The symptoms experienced by patients that were recorded in claims were significantly higher across multiple categories than matched controls. However, the rates were relatively low, demonstrating the wide variability in the presentation of sarcoma. Patients had a variety of recorded problems, not limited to a lump or pain, but including hematologic, gastric, and cardiac concerns, that differed from those who had no suspected sarcoma. These factors highlight the challenges that may be facing patients who have an undetected sarcoma.
An expected finding was the difference in duration of follow-up between cohorts. This could be due to longer survival of those without a sarcoma diagnosis or due to insurance changes among those who had a sarcoma diagnosis. The absence of death data did not allow for further exploration of this finding within this study. Future research may wish to identify more comprehensive datasets to allow for the objective evaluation of the differences in time to diagnosis and stage of disease and survival, which would be the ultimate goal in order to develop potential strategies to improve patient outcomes.
This study was limited in that the sarcoma diagnosis could not be verified in a clinical record due to the de-identified nature of the claims data used for this study. Prior work has shown that the ICD coding for sarcoma is incomplete6,7; therefore it is likely there are many other patients in the claims dataset who had a suspected sarcoma but who did not have a 171.x code recorded. Hence, this study is limited to a comparison of a cohort for whom the provider specified a sarcoma code in their billing records. While there are gaps in the ability to identify the entire population of sarcoma patients, the patients with ICD codes used in this study are likely true sarcoma cases. Prior work has demonstrated that the presence of these codes accurately reflects a true sarcoma diagnosis.7 However, given the concerns with ICD coding, two sarcoma codes were required on unique days to reduce the risk of single rule-out codes or data entry error. Patients diagnosed with sarcoma demonstrate significantly greater health care resource use across variables as matched controls during the 6-month period leading to diagnosis, supporting the observations within advocacy and patient reports of the challenges faced during the process to reach an accurate diagnosis. This work may provide the initial basis for the development of strategies to more rapidly identify a potential sarcoma. Future research could also evaluate more than 6 months prior to diagnosis, to quantify the duration of time during which these differences versus controls may exist. Additionally, the cost of care may be of interest to future research to better quantify the burden of misdiagnosis on the health care system.
Acknowledgement
The authors would like to acknowledge Yun Fang, MS, for her support in the SAS coding for the analysis of this study.
Corresponding Author
Lisa M. Hess, PhD, Eli Lilly and Company. [email protected]
Disclosures
No funding was received or exchanged in the conceptualization, conduct, data collection, analysis, interpretation, or writing related to this study. This unfunded study was conducted by employees of Eli Lilly and Company.
1. ACS. Signs and Symptoms of Soft Tissue Sarcomas. 2018. https://www.cancer.org/cancer/soft-tissue-sarcoma/detection-diagnosis-staging/signs-symptoms.html. Accessed September 27, 2018.
2. SEER. Cancer Stat Facts: Soft Tissue including Heart Cancer. National Cancer Institute Surveillance, Epidemiology, and End Results Program; 2018. https://seer.cancer.gov/statfacts/html/soft.html. Accessed February 20, 2019.
3. Rougraff BT, Davis K, Lawrence J. Does length of symptoms before diagnosis of sarcoma affect patient survival? Clin Orthop Relat Res. 2007;462:181-189.
4. Rougraff BT, Lawrence J, Davis K. Length of symptoms before referral: prognostic variable for high-grade soft tissue sarcoma? Clin Orthop Relat Res. 2012;470(3):706-711.
5. LSSI. Liddy Shriver Sarcoma Initiative. Sarcoma: A diagnosis of patience. http://sarcomahelp.org/articles/patience.html. Accessed September 20, 2018.
6. Hess LM, Zhu EY, Sugihara T, Fang Y, Collins N, Nicol S. Challenges with use of the International Classification of Disease Coding (ICD-9-CM/ICD-10-CM) for soft tissue sarcoma. Perspect Health Inf Manage. 2019;16 (Spring). eCollection 2019.
7. Lyu HG, Stein LA, Saadat LV, Phicil SN, Haider A, Raut CP. Assessment of the accuracy of disease coding among patients diagnosed with sarcoma. JAMA Oncol. 2018;4(9):1293-1295.
Introduction
Soft tissue sarcomas (STS) are a heterogeneous group of cancerous tumors, comprised of more than 50 histological subtypes that develop from soft tissues of the body (eg, fat, muscles, nerve tissue, deep skin tissue, visceral nonepithelial tissue). Due to many factors, not limited to the heterogeneity of this set of diseases and lack of screening tests, reaching a diagnosis of STS is challenging for the general practitioner as well as for the oncologist. Sarcomas may present with nonspecific and often indolent symptomology, depending on the specific histological subtype. According to the American Cancer Society, the signs and symptoms of a sarcoma include a new or growing lump, worsening abdominal pain, blood in stool or vomit, and black stools (due to abdominal bleeding).1 Unfortunately, these symptoms could be indicative of any number of other health conditions and are nonspecific to sarcoma.
As with many cancers, the early detection of disease when it may be completely resected could lead to a cure, whereas diagnosis when the disease is no longer amenable to surgery will impact patient survival. Among all forms of STS, early diagnosis when the patient has only localized disease is associated with an 80.8% five-year survival rate, which decreases to 16.4% for patients whose disease has already metastasized to other parts of the body at the time of diagnosis.2
Previous work has evaluated the relationship between duration of symptoms that may lead to a diagnosis of sarcoma and cancer outcomes. A retrospective analysis of a cohort of adults with bone or STS found no correlation between patient recall of duration of prediagnosis symptoms and survival or metastatic disease at diagnosis.3,4 Little other research was identified that examined the challenges of identifying a potential sarcoma. Despite the gap in knowledge, advocacy and patient-centered organizations emphasize the risk of delayed diagnosis and report high levels of stress and frustration among patients by the time an accurate diagnosis is obtained.5 The objective of this study was to quantify the health care experience and misdiagnoses that occurred prior to a sarcoma diagnosis compared to a cohort of matched controls.
Methods
A retrospective observational database study was conducted using detailed resource utilization and cost data from the Truven MarketScan claims database. Truven MarketScan® is a HIPAA-compliant, fully integrated patient-level database containing inpatient, outpatient, drug, lab, health risk assessment, and benefit design information from commercial and Medicare supplemental insurance plans. Additionally, the Health and Productivity Management (HPM) database, containing workplace absence, short-term disability, long-term disability, and worker’s compensation data, is linked at the individual patient level. The linkage of the claims and HPM database was used for this study.
Patients were eligible for inclusion in the cohort of a sarcoma if they had at least two ICD-9 codes of 171.x on two different days between July 1, 2004, and March 30, 2014. The date of the first eligible code was considered the index date. Patients were required to have at least 6 months of health care plan enrollment prior to the first eligible ICD-9 code to allow for prediagnosis activity to be identified in the database. Patients were also required to be 18 years of age or older on the first eligible ICD-9 code date. Patients were excluded who had evidence suggesting a diagnosis of osteosarcoma, Kaposi’s sarcoma, or gastrointestinal stromal tumors (treatment with methotrexate, ICD-9 codes of 176.x, 171.x, or 238.1), a history of any cancer before the eligible sarcoma ICD-9 code, or history of systemic anticancer therapy during the 6-month pre-index period. All patients meeting eligibility criteria were included in the matching algorithm to identify the control cohort.
The matched control cohort was required to have at least the same duration of follow-up at the case level as the matched sarcoma patient, could not have any evidence of any malignancy at any time in the database, nor could have received any systemic anticancer therapy at any time. Controls were randomly selected from the more than 100 million individual patient cases in the MarketScan database to be matched to the eligible sarcoma patient cohort exactly on age, geographic region of residence, health insurance plan type, gender, noncancer comorbid conditions (measured by Charlson Comorbidity Index items), and employment status. All factors were exact matched at the sarcoma cohort index diagnosis date. In the case of missing variables, patients were matched on missingness (eg, a case with missing employment status would be matched to a control with missing employment status).
The eligible time period for the index date of the possible sarcoma cohort and matched controls was between July 1, 2004, and March 30, 2014, which allowed for a minimum of 1-year follow-up through the end of the database available at the time of analysis.
All ICD-9 diagnostic and procedure codes present in the matched 6-month time period pre-index diagnosis were compared to explore factors that may be more likely to be present in the sarcoma cohort compared to matched controls. Univariate analysis was conducted for each prediagnosis variable. Analyses were conducted using T test for continuous variables, and Chi-square or Fisher’s exact test was used for categorical variables.
Number of physician visits, inpatient hospital stays, surgical procedures, and emergency room visits were compared between those in the sarcoma cohort and matched controls during the matched 6-month pre-index period. The post-index diagnosis employment status was also compared between groups using the HPM database. Comparisons between the sarcoma cohort and control cohort were made among the actively employed patients at baseline related to the proportion of patients who continued active employment, the proportion who permanently discontinued work, and the proportion who initially discontinued work and then returned to work at a later time. No adjustments were made for multiple comparisons.
Results
A total of 7826 controls were each matched to patients in the sarcoma cohort. The baseline characteristics of the study cohorts are provided in Table 1.
During the 6-month period before the sarcoma diagnosis (prediagnosis period), patients had significantly greater frequency of diagnoses identified than controls for uncertain neoplasms, limb pain, and hypertension (all P<.001, Table 2).
Similarly, the majority of health care resource utilization factors evaluated showed statistically higher health care use among patients later suspected of having sarcoma than matched controls (Table 3).
Employment status was missing for 44% of the cohort at baseline and approximately half the cohort during follow-up (Table 4).
Discussion
The symptoms experienced by patients that were recorded in claims were significantly higher across multiple categories than matched controls. However, the rates were relatively low, demonstrating the wide variability in the presentation of sarcoma. Patients had a variety of recorded problems, not limited to a lump or pain, but including hematologic, gastric, and cardiac concerns, that differed from those who had no suspected sarcoma. These factors highlight the challenges that may be facing patients who have an undetected sarcoma.
An expected finding was the difference in duration of follow-up between cohorts. This could be due to longer survival of those without a sarcoma diagnosis or due to insurance changes among those who had a sarcoma diagnosis. The absence of death data did not allow for further exploration of this finding within this study. Future research may wish to identify more comprehensive datasets to allow for the objective evaluation of the differences in time to diagnosis and stage of disease and survival, which would be the ultimate goal in order to develop potential strategies to improve patient outcomes.
This study was limited in that the sarcoma diagnosis could not be verified in a clinical record due to the de-identified nature of the claims data used for this study. Prior work has shown that the ICD coding for sarcoma is incomplete6,7; therefore it is likely there are many other patients in the claims dataset who had a suspected sarcoma but who did not have a 171.x code recorded. Hence, this study is limited to a comparison of a cohort for whom the provider specified a sarcoma code in their billing records. While there are gaps in the ability to identify the entire population of sarcoma patients, the patients with ICD codes used in this study are likely true sarcoma cases. Prior work has demonstrated that the presence of these codes accurately reflects a true sarcoma diagnosis.7 However, given the concerns with ICD coding, two sarcoma codes were required on unique days to reduce the risk of single rule-out codes or data entry error. Patients diagnosed with sarcoma demonstrate significantly greater health care resource use across variables as matched controls during the 6-month period leading to diagnosis, supporting the observations within advocacy and patient reports of the challenges faced during the process to reach an accurate diagnosis. This work may provide the initial basis for the development of strategies to more rapidly identify a potential sarcoma. Future research could also evaluate more than 6 months prior to diagnosis, to quantify the duration of time during which these differences versus controls may exist. Additionally, the cost of care may be of interest to future research to better quantify the burden of misdiagnosis on the health care system.
Acknowledgement
The authors would like to acknowledge Yun Fang, MS, for her support in the SAS coding for the analysis of this study.
Corresponding Author
Lisa M. Hess, PhD, Eli Lilly and Company. [email protected]
Disclosures
No funding was received or exchanged in the conceptualization, conduct, data collection, analysis, interpretation, or writing related to this study. This unfunded study was conducted by employees of Eli Lilly and Company.
Introduction
Soft tissue sarcomas (STS) are a heterogeneous group of cancerous tumors, comprised of more than 50 histological subtypes that develop from soft tissues of the body (eg, fat, muscles, nerve tissue, deep skin tissue, visceral nonepithelial tissue). Due to many factors, not limited to the heterogeneity of this set of diseases and lack of screening tests, reaching a diagnosis of STS is challenging for the general practitioner as well as for the oncologist. Sarcomas may present with nonspecific and often indolent symptomology, depending on the specific histological subtype. According to the American Cancer Society, the signs and symptoms of a sarcoma include a new or growing lump, worsening abdominal pain, blood in stool or vomit, and black stools (due to abdominal bleeding).1 Unfortunately, these symptoms could be indicative of any number of other health conditions and are nonspecific to sarcoma.
As with many cancers, the early detection of disease when it may be completely resected could lead to a cure, whereas diagnosis when the disease is no longer amenable to surgery will impact patient survival. Among all forms of STS, early diagnosis when the patient has only localized disease is associated with an 80.8% five-year survival rate, which decreases to 16.4% for patients whose disease has already metastasized to other parts of the body at the time of diagnosis.2
Previous work has evaluated the relationship between duration of symptoms that may lead to a diagnosis of sarcoma and cancer outcomes. A retrospective analysis of a cohort of adults with bone or STS found no correlation between patient recall of duration of prediagnosis symptoms and survival or metastatic disease at diagnosis.3,4 Little other research was identified that examined the challenges of identifying a potential sarcoma. Despite the gap in knowledge, advocacy and patient-centered organizations emphasize the risk of delayed diagnosis and report high levels of stress and frustration among patients by the time an accurate diagnosis is obtained.5 The objective of this study was to quantify the health care experience and misdiagnoses that occurred prior to a sarcoma diagnosis compared to a cohort of matched controls.
Methods
A retrospective observational database study was conducted using detailed resource utilization and cost data from the Truven MarketScan claims database. Truven MarketScan® is a HIPAA-compliant, fully integrated patient-level database containing inpatient, outpatient, drug, lab, health risk assessment, and benefit design information from commercial and Medicare supplemental insurance plans. Additionally, the Health and Productivity Management (HPM) database, containing workplace absence, short-term disability, long-term disability, and worker’s compensation data, is linked at the individual patient level. The linkage of the claims and HPM database was used for this study.
Patients were eligible for inclusion in the cohort of a sarcoma if they had at least two ICD-9 codes of 171.x on two different days between July 1, 2004, and March 30, 2014. The date of the first eligible code was considered the index date. Patients were required to have at least 6 months of health care plan enrollment prior to the first eligible ICD-9 code to allow for prediagnosis activity to be identified in the database. Patients were also required to be 18 years of age or older on the first eligible ICD-9 code date. Patients were excluded who had evidence suggesting a diagnosis of osteosarcoma, Kaposi’s sarcoma, or gastrointestinal stromal tumors (treatment with methotrexate, ICD-9 codes of 176.x, 171.x, or 238.1), a history of any cancer before the eligible sarcoma ICD-9 code, or history of systemic anticancer therapy during the 6-month pre-index period. All patients meeting eligibility criteria were included in the matching algorithm to identify the control cohort.
The matched control cohort was required to have at least the same duration of follow-up at the case level as the matched sarcoma patient, could not have any evidence of any malignancy at any time in the database, nor could have received any systemic anticancer therapy at any time. Controls were randomly selected from the more than 100 million individual patient cases in the MarketScan database to be matched to the eligible sarcoma patient cohort exactly on age, geographic region of residence, health insurance plan type, gender, noncancer comorbid conditions (measured by Charlson Comorbidity Index items), and employment status. All factors were exact matched at the sarcoma cohort index diagnosis date. In the case of missing variables, patients were matched on missingness (eg, a case with missing employment status would be matched to a control with missing employment status).
The eligible time period for the index date of the possible sarcoma cohort and matched controls was between July 1, 2004, and March 30, 2014, which allowed for a minimum of 1-year follow-up through the end of the database available at the time of analysis.
All ICD-9 diagnostic and procedure codes present in the matched 6-month time period pre-index diagnosis were compared to explore factors that may be more likely to be present in the sarcoma cohort compared to matched controls. Univariate analysis was conducted for each prediagnosis variable. Analyses were conducted using T test for continuous variables, and Chi-square or Fisher’s exact test was used for categorical variables.
Number of physician visits, inpatient hospital stays, surgical procedures, and emergency room visits were compared between those in the sarcoma cohort and matched controls during the matched 6-month pre-index period. The post-index diagnosis employment status was also compared between groups using the HPM database. Comparisons between the sarcoma cohort and control cohort were made among the actively employed patients at baseline related to the proportion of patients who continued active employment, the proportion who permanently discontinued work, and the proportion who initially discontinued work and then returned to work at a later time. No adjustments were made for multiple comparisons.
Results
A total of 7826 controls were each matched to patients in the sarcoma cohort. The baseline characteristics of the study cohorts are provided in Table 1.
During the 6-month period before the sarcoma diagnosis (prediagnosis period), patients had significantly greater frequency of diagnoses identified than controls for uncertain neoplasms, limb pain, and hypertension (all P<.001, Table 2).
Similarly, the majority of health care resource utilization factors evaluated showed statistically higher health care use among patients later suspected of having sarcoma than matched controls (Table 3).
Employment status was missing for 44% of the cohort at baseline and approximately half the cohort during follow-up (Table 4).
Discussion
The symptoms experienced by patients that were recorded in claims were significantly higher across multiple categories than matched controls. However, the rates were relatively low, demonstrating the wide variability in the presentation of sarcoma. Patients had a variety of recorded problems, not limited to a lump or pain, but including hematologic, gastric, and cardiac concerns, that differed from those who had no suspected sarcoma. These factors highlight the challenges that may be facing patients who have an undetected sarcoma.
An expected finding was the difference in duration of follow-up between cohorts. This could be due to longer survival of those without a sarcoma diagnosis or due to insurance changes among those who had a sarcoma diagnosis. The absence of death data did not allow for further exploration of this finding within this study. Future research may wish to identify more comprehensive datasets to allow for the objective evaluation of the differences in time to diagnosis and stage of disease and survival, which would be the ultimate goal in order to develop potential strategies to improve patient outcomes.
This study was limited in that the sarcoma diagnosis could not be verified in a clinical record due to the de-identified nature of the claims data used for this study. Prior work has shown that the ICD coding for sarcoma is incomplete6,7; therefore it is likely there are many other patients in the claims dataset who had a suspected sarcoma but who did not have a 171.x code recorded. Hence, this study is limited to a comparison of a cohort for whom the provider specified a sarcoma code in their billing records. While there are gaps in the ability to identify the entire population of sarcoma patients, the patients with ICD codes used in this study are likely true sarcoma cases. Prior work has demonstrated that the presence of these codes accurately reflects a true sarcoma diagnosis.7 However, given the concerns with ICD coding, two sarcoma codes were required on unique days to reduce the risk of single rule-out codes or data entry error. Patients diagnosed with sarcoma demonstrate significantly greater health care resource use across variables as matched controls during the 6-month period leading to diagnosis, supporting the observations within advocacy and patient reports of the challenges faced during the process to reach an accurate diagnosis. This work may provide the initial basis for the development of strategies to more rapidly identify a potential sarcoma. Future research could also evaluate more than 6 months prior to diagnosis, to quantify the duration of time during which these differences versus controls may exist. Additionally, the cost of care may be of interest to future research to better quantify the burden of misdiagnosis on the health care system.
Acknowledgement
The authors would like to acknowledge Yun Fang, MS, for her support in the SAS coding for the analysis of this study.
Corresponding Author
Lisa M. Hess, PhD, Eli Lilly and Company. [email protected]
Disclosures
No funding was received or exchanged in the conceptualization, conduct, data collection, analysis, interpretation, or writing related to this study. This unfunded study was conducted by employees of Eli Lilly and Company.
1. ACS. Signs and Symptoms of Soft Tissue Sarcomas. 2018. https://www.cancer.org/cancer/soft-tissue-sarcoma/detection-diagnosis-staging/signs-symptoms.html. Accessed September 27, 2018.
2. SEER. Cancer Stat Facts: Soft Tissue including Heart Cancer. National Cancer Institute Surveillance, Epidemiology, and End Results Program; 2018. https://seer.cancer.gov/statfacts/html/soft.html. Accessed February 20, 2019.
3. Rougraff BT, Davis K, Lawrence J. Does length of symptoms before diagnosis of sarcoma affect patient survival? Clin Orthop Relat Res. 2007;462:181-189.
4. Rougraff BT, Lawrence J, Davis K. Length of symptoms before referral: prognostic variable for high-grade soft tissue sarcoma? Clin Orthop Relat Res. 2012;470(3):706-711.
5. LSSI. Liddy Shriver Sarcoma Initiative. Sarcoma: A diagnosis of patience. http://sarcomahelp.org/articles/patience.html. Accessed September 20, 2018.
6. Hess LM, Zhu EY, Sugihara T, Fang Y, Collins N, Nicol S. Challenges with use of the International Classification of Disease Coding (ICD-9-CM/ICD-10-CM) for soft tissue sarcoma. Perspect Health Inf Manage. 2019;16 (Spring). eCollection 2019.
7. Lyu HG, Stein LA, Saadat LV, Phicil SN, Haider A, Raut CP. Assessment of the accuracy of disease coding among patients diagnosed with sarcoma. JAMA Oncol. 2018;4(9):1293-1295.
1. ACS. Signs and Symptoms of Soft Tissue Sarcomas. 2018. https://www.cancer.org/cancer/soft-tissue-sarcoma/detection-diagnosis-staging/signs-symptoms.html. Accessed September 27, 2018.
2. SEER. Cancer Stat Facts: Soft Tissue including Heart Cancer. National Cancer Institute Surveillance, Epidemiology, and End Results Program; 2018. https://seer.cancer.gov/statfacts/html/soft.html. Accessed February 20, 2019.
3. Rougraff BT, Davis K, Lawrence J. Does length of symptoms before diagnosis of sarcoma affect patient survival? Clin Orthop Relat Res. 2007;462:181-189.
4. Rougraff BT, Lawrence J, Davis K. Length of symptoms before referral: prognostic variable for high-grade soft tissue sarcoma? Clin Orthop Relat Res. 2012;470(3):706-711.
5. LSSI. Liddy Shriver Sarcoma Initiative. Sarcoma: A diagnosis of patience. http://sarcomahelp.org/articles/patience.html. Accessed September 20, 2018.
6. Hess LM, Zhu EY, Sugihara T, Fang Y, Collins N, Nicol S. Challenges with use of the International Classification of Disease Coding (ICD-9-CM/ICD-10-CM) for soft tissue sarcoma. Perspect Health Inf Manage. 2019;16 (Spring). eCollection 2019.
7. Lyu HG, Stein LA, Saadat LV, Phicil SN, Haider A, Raut CP. Assessment of the accuracy of disease coding among patients diagnosed with sarcoma. JAMA Oncol. 2018;4(9):1293-1295.
Abstract
Introduction: The challenges of diagnosing soft tissue sarcoma are not well studied; however, the heterogeneity of its presentation would suggest that patients may experience a complex journey in the health care system prior to reaching an accurate diagnosis. This study was designed to evaluate the diagnoses, procedures, and health care resource utilization of patients with soft tissue sarcoma compared to a matched healthy control cohort.
Methods: Patients in the sarcoma cohort were identified in claims data by the presence of diagnosis codes for soft tissue sarcoma. Controls were matched using exact methods on demographic, employment, and insurance variables at the date of the index sarcoma diagnosis. Health care resource utilization and diagnosis and procedure codes were compared between the cohorts during the prediagnosis period (6 months prior to the index and matched date). T test was used for continuous variables and Chi-square or Fisher’s exact test was used for categorical variables.
Results: A total of 7826 sarcoma patients were matched to 7826 controls on demographic, employment, and insurance variables. Diagnoses of uncertain neoplasms, limb pain, and hypertension, as well as anemia, neutropenia, thrombocytopenia, cardiac dysrhythmia, cellulitis, constipation, dehydration, diarrhea, dyspnea, edema, fatigue, gangrene, hemorrhage, nausea, pancreatitis, proteinuria, pulmonary fibrosis, rash, renal failure, vomiting, and watery eyes were significantly greater in the sarcoma cohort versus controls (all P <.05). The majority of health care resource utilization evaluated showed statistically higher utilization in the sarcoma cohort versus matched controls.
Conclusions: Sarcoma patients had many health conditions and diagnoses that significantly differed from controls during the 6-month period prior to diagnosis. These data provide initial evidence regarding the quantity and frequency of additional health care resources used and symptoms experienced leading to the diagnosis of sarcoma.
Key words: sarcoma, diagnosis, health care resource utilization, health care economics
The Sarcoma Journal enters its second full year with renewed commitment and energy
We begin this second full year publishing The Sarcoma Journal—Official Journal of the Sarcoma Foundation of AmericaTM—with renewed energy and dedication to its founding principle of communicating authoritative and comprehensive scientific information on the diagnosis and treatment of sarcomas and sarcoma subtypes.
Despite advances in treatment and management options for our patients, the need still exists for more effective treatment strategies, new treatment paradigms, and improved understanding of disease biology. This journal, along with its online platform, plays an important role in the dissemination of reliable, peer-reviewed content to the sarcoma community and aims to bridge the gap between bench and bedside.
To this end, we have again recruited an outstanding advisory board, many of whom have long-standing affiliations with the Sarcoma Foundation of America. With their help, the editorial staff and I will continue to build the journal and make it the number one academic and practice resource for the sarcoma community.
We invite you and your colleagues to submit original research, review articles, case reports, opinion pieces, and meeting summaries for publication. In this way we will create the robust forum we all desire.

We begin this second full year publishing The Sarcoma Journal—Official Journal of the Sarcoma Foundation of AmericaTM—with renewed energy and dedication to its founding principle of communicating authoritative and comprehensive scientific information on the diagnosis and treatment of sarcomas and sarcoma subtypes.
Despite advances in treatment and management options for our patients, the need still exists for more effective treatment strategies, new treatment paradigms, and improved understanding of disease biology. This journal, along with its online platform, plays an important role in the dissemination of reliable, peer-reviewed content to the sarcoma community and aims to bridge the gap between bench and bedside.
To this end, we have again recruited an outstanding advisory board, many of whom have long-standing affiliations with the Sarcoma Foundation of America. With their help, the editorial staff and I will continue to build the journal and make it the number one academic and practice resource for the sarcoma community.
We invite you and your colleagues to submit original research, review articles, case reports, opinion pieces, and meeting summaries for publication. In this way we will create the robust forum we all desire.
We begin this second full year publishing The Sarcoma Journal—Official Journal of the Sarcoma Foundation of AmericaTM—with renewed energy and dedication to its founding principle of communicating authoritative and comprehensive scientific information on the diagnosis and treatment of sarcomas and sarcoma subtypes.
Despite advances in treatment and management options for our patients, the need still exists for more effective treatment strategies, new treatment paradigms, and improved understanding of disease biology. This journal, along with its online platform, plays an important role in the dissemination of reliable, peer-reviewed content to the sarcoma community and aims to bridge the gap between bench and bedside.
To this end, we have again recruited an outstanding advisory board, many of whom have long-standing affiliations with the Sarcoma Foundation of America. With their help, the editorial staff and I will continue to build the journal and make it the number one academic and practice resource for the sarcoma community.
We invite you and your colleagues to submit original research, review articles, case reports, opinion pieces, and meeting summaries for publication. In this way we will create the robust forum we all desire.

Sorafenib extends PFS for refractory desmoid tumors
For patients with progressive, refractory, or symptomatic desmoid tumors – also known as aggressive fibromatosis – treatment with daily sorafenib (Nexavar) was associated with durable responses and a significant improvement in progression-free survival.
After a median follow-up of 27.2 months, the 2-year progression-free survival (PFS) rate for patients randomly assigned to receive 400 mg sorafenib daily was 81%, compared with 36% for patients assigned to placebo (P less than .001), reported Mrinal M. Gounder, MD, from Memorial Sloan Kettering Cancer Center in New York City, and his colleagues.
“Other agents that are used to treat these tumors include anthracyclines [e.g., pegylated liposomal doxorubicin], vinca alkaloids, and pazopanib. On the basis of the predictable toxic-effects profile and substantial progression-free survival advantage conferred by sorafenib, the drug has antitumor activity as first-line therapy or as subsequent therapy for desmoid tumors,” they wrote in the New England Journal of Medicine.
There is no accepted standard of care for the systemic treatment for desmoid tumors, with options ranging from hormonal blockade, cytotoxic chemotherapy, and targeted agents such as tyrosine kinase inhibitors (TKIs).
Based on a retrospective study showing that the multitargeting oral TKI sorafenib was associated with a 25% response rate and acceptable safety in patients with desmoid tumors, the investigators initiated a phase 3, randomized trial to evaluate the efficacy and safety of sorafenib in this population.
They enrolled 87 patients aged 18 years or older with a histologically documented desmoid tumor that showed clinical and radiographic progression of at least 10% in maximum unidimensional measurement within the last 6 months, symptomatic disease, or recurrent or primary disease that was either inoperable or deemed to require extensive surgery.
The patients were randomized in double-blinded fashion on a 2:1 basis to receive either sorafenib 400 mg daily or placebo until progression. Crossover to sorafenib was allowed for patients assigned to placebo who experienced disease progressions.
As noted before, investigator-assessed PFS, the primary endpoint, clearly favored sorafenib.
Objective response rates before crossover were 33% in the sorafenib arm, consisting of 1 complete and 15 partial responses, and 20% in the placebo arm, consisting of 7 partial responses. The respective median times to objective response were 9.6 months versus 13.3 months. The earliest response, defined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, occurred at 2.2 months in the sorafenib arm versus 8.8 months in the placebo arm.
The authors also performed an exploratory analysis looking at MRI as a measure of response evaluation and found that “changes in T2-weighted signal intensity and volumetric measurements may be better measures of treatment effect than RECIST. This is particularly evident when the best response according to RECIST is stable disease.”
The most frequently reported adverse events among patients treated with sorafenib were grade 1 or 2 rash in 73%, fatigue in 67%, hypertension in 55%, and diarrhea in 51%. The most frequent treatment-emergent adverse events in the placebo group were rash of any kind in 42% and palmar-plantar erythrodysesthesia syndrome in 22%.
The investigators acknowledged that the mechanism of action of sorafenib in desmoid tumors is unknown, but noted that they are looking for clues in 25 sets of paired biopsy samples.
The study was supported by grants from the National Cancer Institute, Bayer, Memorial Sloan Kettering Cancer Center, the American Society of Clinical Oncology, Desmoid Tumor Research Foundation, and an Orphan Products Clinical Trials Grant from the Food and Drug Administration. Dr. Gounder reported fees for advisory board activities/consulting for Bayer, Epizyme, Karyopharm Therapeutics, Daiichi Sankyo, TRACON Pharmaceuticals, and Amgen, and travel expenses from Epizyme.
SOURCE: Gounder MM et al. N Engl J Med. 2018 Dec 19. doi: 10.1056/NEJMoa1805052.
For patients with progressive, refractory, or symptomatic desmoid tumors – also known as aggressive fibromatosis – treatment with daily sorafenib (Nexavar) was associated with durable responses and a significant improvement in progression-free survival.
After a median follow-up of 27.2 months, the 2-year progression-free survival (PFS) rate for patients randomly assigned to receive 400 mg sorafenib daily was 81%, compared with 36% for patients assigned to placebo (P less than .001), reported Mrinal M. Gounder, MD, from Memorial Sloan Kettering Cancer Center in New York City, and his colleagues.
“Other agents that are used to treat these tumors include anthracyclines [e.g., pegylated liposomal doxorubicin], vinca alkaloids, and pazopanib. On the basis of the predictable toxic-effects profile and substantial progression-free survival advantage conferred by sorafenib, the drug has antitumor activity as first-line therapy or as subsequent therapy for desmoid tumors,” they wrote in the New England Journal of Medicine.
There is no accepted standard of care for the systemic treatment for desmoid tumors, with options ranging from hormonal blockade, cytotoxic chemotherapy, and targeted agents such as tyrosine kinase inhibitors (TKIs).
Based on a retrospective study showing that the multitargeting oral TKI sorafenib was associated with a 25% response rate and acceptable safety in patients with desmoid tumors, the investigators initiated a phase 3, randomized trial to evaluate the efficacy and safety of sorafenib in this population.
They enrolled 87 patients aged 18 years or older with a histologically documented desmoid tumor that showed clinical and radiographic progression of at least 10% in maximum unidimensional measurement within the last 6 months, symptomatic disease, or recurrent or primary disease that was either inoperable or deemed to require extensive surgery.
The patients were randomized in double-blinded fashion on a 2:1 basis to receive either sorafenib 400 mg daily or placebo until progression. Crossover to sorafenib was allowed for patients assigned to placebo who experienced disease progressions.
As noted before, investigator-assessed PFS, the primary endpoint, clearly favored sorafenib.
Objective response rates before crossover were 33% in the sorafenib arm, consisting of 1 complete and 15 partial responses, and 20% in the placebo arm, consisting of 7 partial responses. The respective median times to objective response were 9.6 months versus 13.3 months. The earliest response, defined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, occurred at 2.2 months in the sorafenib arm versus 8.8 months in the placebo arm.
The authors also performed an exploratory analysis looking at MRI as a measure of response evaluation and found that “changes in T2-weighted signal intensity and volumetric measurements may be better measures of treatment effect than RECIST. This is particularly evident when the best response according to RECIST is stable disease.”
The most frequently reported adverse events among patients treated with sorafenib were grade 1 or 2 rash in 73%, fatigue in 67%, hypertension in 55%, and diarrhea in 51%. The most frequent treatment-emergent adverse events in the placebo group were rash of any kind in 42% and palmar-plantar erythrodysesthesia syndrome in 22%.
The investigators acknowledged that the mechanism of action of sorafenib in desmoid tumors is unknown, but noted that they are looking for clues in 25 sets of paired biopsy samples.
The study was supported by grants from the National Cancer Institute, Bayer, Memorial Sloan Kettering Cancer Center, the American Society of Clinical Oncology, Desmoid Tumor Research Foundation, and an Orphan Products Clinical Trials Grant from the Food and Drug Administration. Dr. Gounder reported fees for advisory board activities/consulting for Bayer, Epizyme, Karyopharm Therapeutics, Daiichi Sankyo, TRACON Pharmaceuticals, and Amgen, and travel expenses from Epizyme.
SOURCE: Gounder MM et al. N Engl J Med. 2018 Dec 19. doi: 10.1056/NEJMoa1805052.
For patients with progressive, refractory, or symptomatic desmoid tumors – also known as aggressive fibromatosis – treatment with daily sorafenib (Nexavar) was associated with durable responses and a significant improvement in progression-free survival.
After a median follow-up of 27.2 months, the 2-year progression-free survival (PFS) rate for patients randomly assigned to receive 400 mg sorafenib daily was 81%, compared with 36% for patients assigned to placebo (P less than .001), reported Mrinal M. Gounder, MD, from Memorial Sloan Kettering Cancer Center in New York City, and his colleagues.
“Other agents that are used to treat these tumors include anthracyclines [e.g., pegylated liposomal doxorubicin], vinca alkaloids, and pazopanib. On the basis of the predictable toxic-effects profile and substantial progression-free survival advantage conferred by sorafenib, the drug has antitumor activity as first-line therapy or as subsequent therapy for desmoid tumors,” they wrote in the New England Journal of Medicine.
There is no accepted standard of care for the systemic treatment for desmoid tumors, with options ranging from hormonal blockade, cytotoxic chemotherapy, and targeted agents such as tyrosine kinase inhibitors (TKIs).
Based on a retrospective study showing that the multitargeting oral TKI sorafenib was associated with a 25% response rate and acceptable safety in patients with desmoid tumors, the investigators initiated a phase 3, randomized trial to evaluate the efficacy and safety of sorafenib in this population.
They enrolled 87 patients aged 18 years or older with a histologically documented desmoid tumor that showed clinical and radiographic progression of at least 10% in maximum unidimensional measurement within the last 6 months, symptomatic disease, or recurrent or primary disease that was either inoperable or deemed to require extensive surgery.
The patients were randomized in double-blinded fashion on a 2:1 basis to receive either sorafenib 400 mg daily or placebo until progression. Crossover to sorafenib was allowed for patients assigned to placebo who experienced disease progressions.
As noted before, investigator-assessed PFS, the primary endpoint, clearly favored sorafenib.
Objective response rates before crossover were 33% in the sorafenib arm, consisting of 1 complete and 15 partial responses, and 20% in the placebo arm, consisting of 7 partial responses. The respective median times to objective response were 9.6 months versus 13.3 months. The earliest response, defined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, occurred at 2.2 months in the sorafenib arm versus 8.8 months in the placebo arm.
The authors also performed an exploratory analysis looking at MRI as a measure of response evaluation and found that “changes in T2-weighted signal intensity and volumetric measurements may be better measures of treatment effect than RECIST. This is particularly evident when the best response according to RECIST is stable disease.”
The most frequently reported adverse events among patients treated with sorafenib were grade 1 or 2 rash in 73%, fatigue in 67%, hypertension in 55%, and diarrhea in 51%. The most frequent treatment-emergent adverse events in the placebo group were rash of any kind in 42% and palmar-plantar erythrodysesthesia syndrome in 22%.
The investigators acknowledged that the mechanism of action of sorafenib in desmoid tumors is unknown, but noted that they are looking for clues in 25 sets of paired biopsy samples.
The study was supported by grants from the National Cancer Institute, Bayer, Memorial Sloan Kettering Cancer Center, the American Society of Clinical Oncology, Desmoid Tumor Research Foundation, and an Orphan Products Clinical Trials Grant from the Food and Drug Administration. Dr. Gounder reported fees for advisory board activities/consulting for Bayer, Epizyme, Karyopharm Therapeutics, Daiichi Sankyo, TRACON Pharmaceuticals, and Amgen, and travel expenses from Epizyme.
SOURCE: Gounder MM et al. N Engl J Med. 2018 Dec 19. doi: 10.1056/NEJMoa1805052.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: There is no accepted standard of systemic therapy for recurrent, refractory, or symptomatic desmoid tumors.
Major finding: Median progression-free survival with sorafenib after a median follow-up of 27.2 months was 81% versus 36% for placebo.
Study details: A double-blind, phase 3 trial with 2:1 randomization of sorafenib to placebo in 87 patients.
Disclosures: The study was supported by grants from the National Cancer Institute, Bayer, Memorial Sloan Kettering Cancer Center, the American Society of Clinical Oncology, Desmoid Tumor Research Foundation, and an Orphan Products Clinical Trials Grant from the Food and Drug Administration. Dr. Gounder reported fees for advisory board activities/consulting for Bayer, Epizyme, Karyopharm Therapeutics, Daiichi Sankyo, TRACON Pharmaceuticals, and Amgen, and travel expenses from Epizyme.
Source: Gounder MM et al. N Engl J Med. 2018 Dec 19. doi: 10.1056/NEJMoa1805052.
Soft Tissue Sarcoma Chemotherapy
Predicting response to chemotherapy
The prognostic nomogram called Sarculator was used effectively to define a high-risk subgroup of patients likely to benefit from adjuvant chemotherapy, Sandro Pasquali, MD, of the Fondazione IRCCS Istituto Nazionale dei Tumori, Milano, Italy and his colleagues reported at the meeting.
Perioperative chemotherapy was shown to afford no survival advantage over observation in the EORTC 62931 (European Organization for Research and Treatment of Cancer—62931) study of adjuvant doxorubicin plus ifosfamide (Lancet Oncol 2012;13:1045-54). However, subsequent analyses of that data attributed this finding to variations in treatment schedules and the inclusion of low-risk tumors, which may have diluted the effect of chemotherapy, the researchers said in their abstract.
Further, a recent interim report of the ISG-1001 trial showed a survival benefit for patients who received neoadjuvant epirubicin plus ifosfamide therapy for localized high-risk soft-tissue sarcoma of the extremities or trunk wall (Lancet Oncol 2017;18:812-822).
The researchers performed a retrospective analysis of individual data for 290 patients with extremity and trunk wall soft-tissue sarcomas in the EORTC-STBSG 62931 study. The Sarculator was used to calculate 10-year predicted probability of overall survival (pr-OS) for each patient.
Patients were grouped in two categories of predicted overall survival: high predicted survival (over 60%) and low predicted overall survival (60% or less). Overall survival and disease-free survival were calculated at 8 years, the study’s median follow-up.
The 8-year probability of overall survival and disease-free survival was 58% [95% confidence interval (CI): 52–63%] and 51% (95% CI: 46–57%), respectively. In the 290 patients with extremity and trunk wall soft tissue sarcomas, adjuvant chemotherapy was not associated with an overall survival benefit [Hazard ratio (HR) = 0.91, 95%CI 0.63–1.31]. The Sarcolator Nomogram detected 80 patients who were at greater risk of death compared to the 210 patients with higher predicted overall survival. The risk of death was significantly lower with adjuvant chemotherapy in the group with low predicted survival based on the Sarculator Nomogram (HR=0.50, 95%CI 0.30-0.90). Consistently, the risk of recurrence was significantly lower when adjuvant chemotherapy was used in the group with predicted overall survival of less than 60% (HR = 0.49, 95%CI 0.28-0.85) while this difference was not observed in patients with high predicted overall survival (HR = 0.95, 95%CI 0.62-1.44).
Doxorubicin plus dacarbazine deserve evaluation in prospective trials in leiomyosarcoma
Doxorubicin plus dacarbazine appeared to best the outcomes seen with doxorubicin plus ifosfamide and with doxorubicin alone in terms of overall response rate and progression free survival as first-line treatment in patients with advanced leiomyosarcomas, based on a retrospective analysis presented by Lorenzo D’Ambrosio, MD, of the Unitversity of Torino, Italy, and his associates.
As patients in the trial were not randomized to therapy, the researchers used a logistic regression model that accounted for histology, site of primary, age, gender, performance status, tumor extent, and tumor grade. Patients were then matched across the different groups by their propensity scores.The 303 patients, 216 of them women, were enrolled from 18 EORTC STBSG (European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group) sites. Doxorubicin plus dacarbazine was given to 117 patients (39%), doxorubicin plus ifosfamide was given to 71 (23%), and doxorubicin alone was given to 115 (38%). There were no significant differences among the regimens in terms of dose reductions of more than 10%, delays of greater than 72 hours, or granulocyte-colony stimulating factor use.
In the whole population, unadjusted median progression free survival was 9.4 months (95% CI 6.1-9.7 months) for those given doxorubicin plus dacarbazine, 6.8 months (4.5-9.5 months) for those given doxorubicin plus ifosfamide), and 5.4 months (3.8-6.8 months) for those given doxorubicin alone. The respective overall response rates for the three regimens were 36.8%, 21.5%, and 25.9%. When using propensity scores to adjust for lack of randomization, progression free survival was significantly longer with doxorubicin plus dacarbazine [median 9.2 months (95%CI 5.2-9.7 months) than with doxorubicin [median 4.8 months (2.3-6.0 months); HR 0.72 (0.52-0.99)]. The difference was not significant when compared to doxorubicin plus ifosfamide [8.2 months (5.2-10.1 months), HR 1.01 (0.68-1.50)]. Progression free survival did not differ significantly between doxorubicin plus ifosfamide, and doxorubicin [HR 0.71 (0.48-1.06)]. In the same matched population, overall response rates were 30.9%, 19.5%, and 25.6% for doxorubicin plus dacarbazine, doxorubicin plus ifosfamide, and doxorubicin, respectively.
Overall survival comparisons were weakened by a shorter median follow-up in the doxorubicin plus dacarbazine groups (32 months) compared to the doxorubicin plus ifosfamide group (50 months) and the doxorubicin group (46 months). With this limit, patients in the doxorubicin plus dacarbazine arm had longer overall survival [median 36.8 (27.9-47.2) months] when compared to both doxorubicin plus ifosfamide [21.9 (16.7-33.4), HR 0.65 (0.40-1.06); and doxorubicin arms 30.3 (21.0-36.3) months, HR 0.66 (0.43-0.99).
Subsequent treatments were well balanced across arms. None of the selected factors for multivariate analysis (age, sex, ECOG performance status, histotype, site of primary tumor, tumor grade, and tumor extent) significantly affected the progression free survival and overall survival associated with the treatments.
Olaratumab in combination with doxorubicin plus ifosfamide
Olaratumab at 15 mg/kg has been shown to be safe in combination with doxorubicin plus ifosfamide in a Phase 1b study (NCT03283696), reported Sebastian Bauer, MD, of the West German Cancer Center, University of Duisburg-Essen, Essen, Germany, and his colleagues.
Given that 8 of 10 evaluable patients have completed the drug-limiting toxicity period without drug-limiting toxicities at the 15 mg/kg dose level of olaratumab, the study has proceeded to the next cohort. In those patients, an olaratumab loading dose of 20 mg/kg will be evaluated in cycle 1, followed by 15 mg/kg of olaratumab in subsequent cycles with the same doses of doxorubicin plus ifosfamide, the researchers wrote in their abstract.
The phase 1 trial enrolled 16 patients with advanced or metastatic soft tissue sarcomas and no prior lines of systemic therapy and ECOG performance status 0-1. Adequate follow up data was available for 10 patients.
Olaratumab, (Lartruvo), which binds platelet-derived growth factor receptor alpha (PDGFRα), was given at 15 mg/kg in combination with doxorubicin (75 mg/m2 on days 1-3) and ifosfamide (10 g/m2 on days 1-4) followed by mandatory granulocyte-colony-stimulating factor therapy in cycles 1-6 on a 21-day cycle. Doxorubicin was allowed to be administered by continuous infusion or bolus administration and with cardiac protection. Mesna dosing was at least 60% of the ifosfamide dose.
Two of the 10 patients had dose-limiting toxicities; one had Grade 4 febrile neutropenia and the other had Grade 3 febrile neutropenia and Grade 3 mucositis. Common related adverse events occurring in over 30% of patients included fatigue, anemia, neutropenia, thrombocytopenia, constipation, and nausea. One patient discontinued study treatment due to progressive disease, and all others were on study treatment as of data cutoff. Among 7 patients evaluated for tumor response assessment, 3 patients had a partial response according to RECIST and 3 further patients had stabilized disease as best overall response for a disease control rate of 86%.
Anthracycline-based regimen excels in FIGO-1 uterine leiomyosarcoma
Future trials to assess the efficacy of adjuvant chemotherapy in uterine leiomyosarcoma should incorporate anthracyclines, according to Roberta Sanfilippo, MD, of Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues.
Disease-free survival was extended in patients with uterine leiomyosarcomas treated with anthracycline-based regimens as compared to gemcitabine and docetaxel, based on a retrospective analysis reported at the meeting by Dr. Sanfilippo.
They reviewed all patients with FIGO stage I uterine leiomyosarcomas who underwent hysterectomy with or without oophorectomy and were treated with adjuvant chemotherapy with either anthracycline-based or gemcitabine-based chemotherapy at two Italian centers.
Of 145 patients, 97 were treated with an anthracycline-based regimen and 48 with gemcitabine and docetaxel. The median number of cycles of anthracycline based regimen received was 4 (range 2-6) and with gemcitabine and docetaxel was 5 (range 3-7). Disease free survival was 31 months in patients treated with anthracycline-based chemotherapy and 19 months in patients treated with gemcitabine and docetaxel.
Trabectedin and low-dose radiotherapy
Trabectedin concurrent with low-dose radiotherapy is being examined as an option for patients with pulmonary metastatic soft tissue sarcoma (NCT02275286).
In a phase 1 study, long-lasting dimensional responses were seen in 71% of the irradiated lesions showed. Based on those results, trabectedin (Yondelis) at 1.5 mg/m 2 will be the recommended dose for phase 2, according to Javier Martín-Broto, MD, of the Institute of Biomedicine Research (IBIS)-University Hospital Virgen del Rocio/CSIC/University of Seville, Spain, and his colleagues.
For the study, trabectedin was given along with radiotherapy (30 Gy) in 10 fractions (3 Gy/fraction). Three dose levels of trabectedin were administered: -1 (1.1 mg/m 2), 1 (1.3 mg/m 2) and 2 (1.5 mg/m 2). Dose-limiting toxicity was defined as grade 3 or greater events excluding grade 3/4 neutropenia lasting less than 5 days, grade 3 transaminitis if it did not lead to trabectedin delay, and grade 3/4 nausea/vomiting due to inadequate prophylaxis.
Ten of the 18 patients enrolled had synovial sarcoma; 3 had undifferentiated pleomorphic sarcomas and the other patients had either myxoid liposarcoma, dedifferentiated liposarcoma, G3 not otherwise specified sarcoma, leiomyosarcoma, and malignant peripheral nerve sheath tumor.
Patients received a median of 1 prior line of chemotherapy (range: 0-3). Twelve patients received trabectedin at dose level 1 and 6 patients at dose level 2. Grade 3/4 adverse events were neutropenia, seen in 8 patients; alanine aminotransferase (ALT) elevation, seen in 2 patients; gamma-glutamyl transferase (GGT) elevation, seen in 2 patients; anemia, seen in 2 patients; febrile neutropenia, seen in 1 patient; and pneumonitis, seen in 1 patient.
There were two dose-limiting toxicities: transient grade 4 ALT elevation at the level 1 dose and grade 4 neutropenia for more than 5 days at the level 2 dose.
Based on central radiological review of 17 evaluable patients, 2 patients achieved complete response, 3 had partial responses, 6 had stable disease, and 6 had progressive disease. The local review reported complete responses in 2 patients, partial responses in 5, stable disease in 4, and progressive disease in 6.
On the irradiated lesions, 4 had complete responses, 8 had partial responses, 4 had stable disease, and 1 had progressive disease. With a median follow-up of 18 months, median progression-free survival was 2.83 months (95%CI: 2.3-3.3 months). Thirteen patients have died, with a median overall survival of 8.77 months (95%CI: 3.6-13.9) and a 12-month overall survival rate of 48%.
Predicting response to chemotherapy
The prognostic nomogram called Sarculator was used effectively to define a high-risk subgroup of patients likely to benefit from adjuvant chemotherapy, Sandro Pasquali, MD, of the Fondazione IRCCS Istituto Nazionale dei Tumori, Milano, Italy and his colleagues reported at the meeting.
Perioperative chemotherapy was shown to afford no survival advantage over observation in the EORTC 62931 (European Organization for Research and Treatment of Cancer—62931) study of adjuvant doxorubicin plus ifosfamide (Lancet Oncol 2012;13:1045-54). However, subsequent analyses of that data attributed this finding to variations in treatment schedules and the inclusion of low-risk tumors, which may have diluted the effect of chemotherapy, the researchers said in their abstract.
Further, a recent interim report of the ISG-1001 trial showed a survival benefit for patients who received neoadjuvant epirubicin plus ifosfamide therapy for localized high-risk soft-tissue sarcoma of the extremities or trunk wall (Lancet Oncol 2017;18:812-822).
The researchers performed a retrospective analysis of individual data for 290 patients with extremity and trunk wall soft-tissue sarcomas in the EORTC-STBSG 62931 study. The Sarculator was used to calculate 10-year predicted probability of overall survival (pr-OS) for each patient.
Patients were grouped in two categories of predicted overall survival: high predicted survival (over 60%) and low predicted overall survival (60% or less). Overall survival and disease-free survival were calculated at 8 years, the study’s median follow-up.
The 8-year probability of overall survival and disease-free survival was 58% [95% confidence interval (CI): 52–63%] and 51% (95% CI: 46–57%), respectively. In the 290 patients with extremity and trunk wall soft tissue sarcomas, adjuvant chemotherapy was not associated with an overall survival benefit [Hazard ratio (HR) = 0.91, 95%CI 0.63–1.31]. The Sarcolator Nomogram detected 80 patients who were at greater risk of death compared to the 210 patients with higher predicted overall survival. The risk of death was significantly lower with adjuvant chemotherapy in the group with low predicted survival based on the Sarculator Nomogram (HR=0.50, 95%CI 0.30-0.90). Consistently, the risk of recurrence was significantly lower when adjuvant chemotherapy was used in the group with predicted overall survival of less than 60% (HR = 0.49, 95%CI 0.28-0.85) while this difference was not observed in patients with high predicted overall survival (HR = 0.95, 95%CI 0.62-1.44).
Doxorubicin plus dacarbazine deserve evaluation in prospective trials in leiomyosarcoma
Doxorubicin plus dacarbazine appeared to best the outcomes seen with doxorubicin plus ifosfamide and with doxorubicin alone in terms of overall response rate and progression free survival as first-line treatment in patients with advanced leiomyosarcomas, based on a retrospective analysis presented by Lorenzo D’Ambrosio, MD, of the Unitversity of Torino, Italy, and his associates.
As patients in the trial were not randomized to therapy, the researchers used a logistic regression model that accounted for histology, site of primary, age, gender, performance status, tumor extent, and tumor grade. Patients were then matched across the different groups by their propensity scores.The 303 patients, 216 of them women, were enrolled from 18 EORTC STBSG (European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group) sites. Doxorubicin plus dacarbazine was given to 117 patients (39%), doxorubicin plus ifosfamide was given to 71 (23%), and doxorubicin alone was given to 115 (38%). There were no significant differences among the regimens in terms of dose reductions of more than 10%, delays of greater than 72 hours, or granulocyte-colony stimulating factor use.
In the whole population, unadjusted median progression free survival was 9.4 months (95% CI 6.1-9.7 months) for those given doxorubicin plus dacarbazine, 6.8 months (4.5-9.5 months) for those given doxorubicin plus ifosfamide), and 5.4 months (3.8-6.8 months) for those given doxorubicin alone. The respective overall response rates for the three regimens were 36.8%, 21.5%, and 25.9%. When using propensity scores to adjust for lack of randomization, progression free survival was significantly longer with doxorubicin plus dacarbazine [median 9.2 months (95%CI 5.2-9.7 months) than with doxorubicin [median 4.8 months (2.3-6.0 months); HR 0.72 (0.52-0.99)]. The difference was not significant when compared to doxorubicin plus ifosfamide [8.2 months (5.2-10.1 months), HR 1.01 (0.68-1.50)]. Progression free survival did not differ significantly between doxorubicin plus ifosfamide, and doxorubicin [HR 0.71 (0.48-1.06)]. In the same matched population, overall response rates were 30.9%, 19.5%, and 25.6% for doxorubicin plus dacarbazine, doxorubicin plus ifosfamide, and doxorubicin, respectively.
Overall survival comparisons were weakened by a shorter median follow-up in the doxorubicin plus dacarbazine groups (32 months) compared to the doxorubicin plus ifosfamide group (50 months) and the doxorubicin group (46 months). With this limit, patients in the doxorubicin plus dacarbazine arm had longer overall survival [median 36.8 (27.9-47.2) months] when compared to both doxorubicin plus ifosfamide [21.9 (16.7-33.4), HR 0.65 (0.40-1.06); and doxorubicin arms 30.3 (21.0-36.3) months, HR 0.66 (0.43-0.99).
Subsequent treatments were well balanced across arms. None of the selected factors for multivariate analysis (age, sex, ECOG performance status, histotype, site of primary tumor, tumor grade, and tumor extent) significantly affected the progression free survival and overall survival associated with the treatments.
Olaratumab in combination with doxorubicin plus ifosfamide
Olaratumab at 15 mg/kg has been shown to be safe in combination with doxorubicin plus ifosfamide in a Phase 1b study (NCT03283696), reported Sebastian Bauer, MD, of the West German Cancer Center, University of Duisburg-Essen, Essen, Germany, and his colleagues.
Given that 8 of 10 evaluable patients have completed the drug-limiting toxicity period without drug-limiting toxicities at the 15 mg/kg dose level of olaratumab, the study has proceeded to the next cohort. In those patients, an olaratumab loading dose of 20 mg/kg will be evaluated in cycle 1, followed by 15 mg/kg of olaratumab in subsequent cycles with the same doses of doxorubicin plus ifosfamide, the researchers wrote in their abstract.
The phase 1 trial enrolled 16 patients with advanced or metastatic soft tissue sarcomas and no prior lines of systemic therapy and ECOG performance status 0-1. Adequate follow up data was available for 10 patients.
Olaratumab, (Lartruvo), which binds platelet-derived growth factor receptor alpha (PDGFRα), was given at 15 mg/kg in combination with doxorubicin (75 mg/m2 on days 1-3) and ifosfamide (10 g/m2 on days 1-4) followed by mandatory granulocyte-colony-stimulating factor therapy in cycles 1-6 on a 21-day cycle. Doxorubicin was allowed to be administered by continuous infusion or bolus administration and with cardiac protection. Mesna dosing was at least 60% of the ifosfamide dose.
Two of the 10 patients had dose-limiting toxicities; one had Grade 4 febrile neutropenia and the other had Grade 3 febrile neutropenia and Grade 3 mucositis. Common related adverse events occurring in over 30% of patients included fatigue, anemia, neutropenia, thrombocytopenia, constipation, and nausea. One patient discontinued study treatment due to progressive disease, and all others were on study treatment as of data cutoff. Among 7 patients evaluated for tumor response assessment, 3 patients had a partial response according to RECIST and 3 further patients had stabilized disease as best overall response for a disease control rate of 86%.
Anthracycline-based regimen excels in FIGO-1 uterine leiomyosarcoma
Future trials to assess the efficacy of adjuvant chemotherapy in uterine leiomyosarcoma should incorporate anthracyclines, according to Roberta Sanfilippo, MD, of Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues.
Disease-free survival was extended in patients with uterine leiomyosarcomas treated with anthracycline-based regimens as compared to gemcitabine and docetaxel, based on a retrospective analysis reported at the meeting by Dr. Sanfilippo.
They reviewed all patients with FIGO stage I uterine leiomyosarcomas who underwent hysterectomy with or without oophorectomy and were treated with adjuvant chemotherapy with either anthracycline-based or gemcitabine-based chemotherapy at two Italian centers.
Of 145 patients, 97 were treated with an anthracycline-based regimen and 48 with gemcitabine and docetaxel. The median number of cycles of anthracycline based regimen received was 4 (range 2-6) and with gemcitabine and docetaxel was 5 (range 3-7). Disease free survival was 31 months in patients treated with anthracycline-based chemotherapy and 19 months in patients treated with gemcitabine and docetaxel.
Trabectedin and low-dose radiotherapy
Trabectedin concurrent with low-dose radiotherapy is being examined as an option for patients with pulmonary metastatic soft tissue sarcoma (NCT02275286).
In a phase 1 study, long-lasting dimensional responses were seen in 71% of the irradiated lesions showed. Based on those results, trabectedin (Yondelis) at 1.5 mg/m 2 will be the recommended dose for phase 2, according to Javier Martín-Broto, MD, of the Institute of Biomedicine Research (IBIS)-University Hospital Virgen del Rocio/CSIC/University of Seville, Spain, and his colleagues.
For the study, trabectedin was given along with radiotherapy (30 Gy) in 10 fractions (3 Gy/fraction). Three dose levels of trabectedin were administered: -1 (1.1 mg/m 2), 1 (1.3 mg/m 2) and 2 (1.5 mg/m 2). Dose-limiting toxicity was defined as grade 3 or greater events excluding grade 3/4 neutropenia lasting less than 5 days, grade 3 transaminitis if it did not lead to trabectedin delay, and grade 3/4 nausea/vomiting due to inadequate prophylaxis.
Ten of the 18 patients enrolled had synovial sarcoma; 3 had undifferentiated pleomorphic sarcomas and the other patients had either myxoid liposarcoma, dedifferentiated liposarcoma, G3 not otherwise specified sarcoma, leiomyosarcoma, and malignant peripheral nerve sheath tumor.
Patients received a median of 1 prior line of chemotherapy (range: 0-3). Twelve patients received trabectedin at dose level 1 and 6 patients at dose level 2. Grade 3/4 adverse events were neutropenia, seen in 8 patients; alanine aminotransferase (ALT) elevation, seen in 2 patients; gamma-glutamyl transferase (GGT) elevation, seen in 2 patients; anemia, seen in 2 patients; febrile neutropenia, seen in 1 patient; and pneumonitis, seen in 1 patient.
There were two dose-limiting toxicities: transient grade 4 ALT elevation at the level 1 dose and grade 4 neutropenia for more than 5 days at the level 2 dose.
Based on central radiological review of 17 evaluable patients, 2 patients achieved complete response, 3 had partial responses, 6 had stable disease, and 6 had progressive disease. The local review reported complete responses in 2 patients, partial responses in 5, stable disease in 4, and progressive disease in 6.
On the irradiated lesions, 4 had complete responses, 8 had partial responses, 4 had stable disease, and 1 had progressive disease. With a median follow-up of 18 months, median progression-free survival was 2.83 months (95%CI: 2.3-3.3 months). Thirteen patients have died, with a median overall survival of 8.77 months (95%CI: 3.6-13.9) and a 12-month overall survival rate of 48%.
Predicting response to chemotherapy
The prognostic nomogram called Sarculator was used effectively to define a high-risk subgroup of patients likely to benefit from adjuvant chemotherapy, Sandro Pasquali, MD, of the Fondazione IRCCS Istituto Nazionale dei Tumori, Milano, Italy and his colleagues reported at the meeting.
Perioperative chemotherapy was shown to afford no survival advantage over observation in the EORTC 62931 (European Organization for Research and Treatment of Cancer—62931) study of adjuvant doxorubicin plus ifosfamide (Lancet Oncol 2012;13:1045-54). However, subsequent analyses of that data attributed this finding to variations in treatment schedules and the inclusion of low-risk tumors, which may have diluted the effect of chemotherapy, the researchers said in their abstract.
Further, a recent interim report of the ISG-1001 trial showed a survival benefit for patients who received neoadjuvant epirubicin plus ifosfamide therapy for localized high-risk soft-tissue sarcoma of the extremities or trunk wall (Lancet Oncol 2017;18:812-822).
The researchers performed a retrospective analysis of individual data for 290 patients with extremity and trunk wall soft-tissue sarcomas in the EORTC-STBSG 62931 study. The Sarculator was used to calculate 10-year predicted probability of overall survival (pr-OS) for each patient.
Patients were grouped in two categories of predicted overall survival: high predicted survival (over 60%) and low predicted overall survival (60% or less). Overall survival and disease-free survival were calculated at 8 years, the study’s median follow-up.
The 8-year probability of overall survival and disease-free survival was 58% [95% confidence interval (CI): 52–63%] and 51% (95% CI: 46–57%), respectively. In the 290 patients with extremity and trunk wall soft tissue sarcomas, adjuvant chemotherapy was not associated with an overall survival benefit [Hazard ratio (HR) = 0.91, 95%CI 0.63–1.31]. The Sarcolator Nomogram detected 80 patients who were at greater risk of death compared to the 210 patients with higher predicted overall survival. The risk of death was significantly lower with adjuvant chemotherapy in the group with low predicted survival based on the Sarculator Nomogram (HR=0.50, 95%CI 0.30-0.90). Consistently, the risk of recurrence was significantly lower when adjuvant chemotherapy was used in the group with predicted overall survival of less than 60% (HR = 0.49, 95%CI 0.28-0.85) while this difference was not observed in patients with high predicted overall survival (HR = 0.95, 95%CI 0.62-1.44).
Doxorubicin plus dacarbazine deserve evaluation in prospective trials in leiomyosarcoma
Doxorubicin plus dacarbazine appeared to best the outcomes seen with doxorubicin plus ifosfamide and with doxorubicin alone in terms of overall response rate and progression free survival as first-line treatment in patients with advanced leiomyosarcomas, based on a retrospective analysis presented by Lorenzo D’Ambrosio, MD, of the Unitversity of Torino, Italy, and his associates.
As patients in the trial were not randomized to therapy, the researchers used a logistic regression model that accounted for histology, site of primary, age, gender, performance status, tumor extent, and tumor grade. Patients were then matched across the different groups by their propensity scores.The 303 patients, 216 of them women, were enrolled from 18 EORTC STBSG (European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group) sites. Doxorubicin plus dacarbazine was given to 117 patients (39%), doxorubicin plus ifosfamide was given to 71 (23%), and doxorubicin alone was given to 115 (38%). There were no significant differences among the regimens in terms of dose reductions of more than 10%, delays of greater than 72 hours, or granulocyte-colony stimulating factor use.
In the whole population, unadjusted median progression free survival was 9.4 months (95% CI 6.1-9.7 months) for those given doxorubicin plus dacarbazine, 6.8 months (4.5-9.5 months) for those given doxorubicin plus ifosfamide), and 5.4 months (3.8-6.8 months) for those given doxorubicin alone. The respective overall response rates for the three regimens were 36.8%, 21.5%, and 25.9%. When using propensity scores to adjust for lack of randomization, progression free survival was significantly longer with doxorubicin plus dacarbazine [median 9.2 months (95%CI 5.2-9.7 months) than with doxorubicin [median 4.8 months (2.3-6.0 months); HR 0.72 (0.52-0.99)]. The difference was not significant when compared to doxorubicin plus ifosfamide [8.2 months (5.2-10.1 months), HR 1.01 (0.68-1.50)]. Progression free survival did not differ significantly between doxorubicin plus ifosfamide, and doxorubicin [HR 0.71 (0.48-1.06)]. In the same matched population, overall response rates were 30.9%, 19.5%, and 25.6% for doxorubicin plus dacarbazine, doxorubicin plus ifosfamide, and doxorubicin, respectively.
Overall survival comparisons were weakened by a shorter median follow-up in the doxorubicin plus dacarbazine groups (32 months) compared to the doxorubicin plus ifosfamide group (50 months) and the doxorubicin group (46 months). With this limit, patients in the doxorubicin plus dacarbazine arm had longer overall survival [median 36.8 (27.9-47.2) months] when compared to both doxorubicin plus ifosfamide [21.9 (16.7-33.4), HR 0.65 (0.40-1.06); and doxorubicin arms 30.3 (21.0-36.3) months, HR 0.66 (0.43-0.99).
Subsequent treatments were well balanced across arms. None of the selected factors for multivariate analysis (age, sex, ECOG performance status, histotype, site of primary tumor, tumor grade, and tumor extent) significantly affected the progression free survival and overall survival associated with the treatments.
Olaratumab in combination with doxorubicin plus ifosfamide
Olaratumab at 15 mg/kg has been shown to be safe in combination with doxorubicin plus ifosfamide in a Phase 1b study (NCT03283696), reported Sebastian Bauer, MD, of the West German Cancer Center, University of Duisburg-Essen, Essen, Germany, and his colleagues.
Given that 8 of 10 evaluable patients have completed the drug-limiting toxicity period without drug-limiting toxicities at the 15 mg/kg dose level of olaratumab, the study has proceeded to the next cohort. In those patients, an olaratumab loading dose of 20 mg/kg will be evaluated in cycle 1, followed by 15 mg/kg of olaratumab in subsequent cycles with the same doses of doxorubicin plus ifosfamide, the researchers wrote in their abstract.
The phase 1 trial enrolled 16 patients with advanced or metastatic soft tissue sarcomas and no prior lines of systemic therapy and ECOG performance status 0-1. Adequate follow up data was available for 10 patients.
Olaratumab, (Lartruvo), which binds platelet-derived growth factor receptor alpha (PDGFRα), was given at 15 mg/kg in combination with doxorubicin (75 mg/m2 on days 1-3) and ifosfamide (10 g/m2 on days 1-4) followed by mandatory granulocyte-colony-stimulating factor therapy in cycles 1-6 on a 21-day cycle. Doxorubicin was allowed to be administered by continuous infusion or bolus administration and with cardiac protection. Mesna dosing was at least 60% of the ifosfamide dose.
Two of the 10 patients had dose-limiting toxicities; one had Grade 4 febrile neutropenia and the other had Grade 3 febrile neutropenia and Grade 3 mucositis. Common related adverse events occurring in over 30% of patients included fatigue, anemia, neutropenia, thrombocytopenia, constipation, and nausea. One patient discontinued study treatment due to progressive disease, and all others were on study treatment as of data cutoff. Among 7 patients evaluated for tumor response assessment, 3 patients had a partial response according to RECIST and 3 further patients had stabilized disease as best overall response for a disease control rate of 86%.
Anthracycline-based regimen excels in FIGO-1 uterine leiomyosarcoma
Future trials to assess the efficacy of adjuvant chemotherapy in uterine leiomyosarcoma should incorporate anthracyclines, according to Roberta Sanfilippo, MD, of Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues.
Disease-free survival was extended in patients with uterine leiomyosarcomas treated with anthracycline-based regimens as compared to gemcitabine and docetaxel, based on a retrospective analysis reported at the meeting by Dr. Sanfilippo.
They reviewed all patients with FIGO stage I uterine leiomyosarcomas who underwent hysterectomy with or without oophorectomy and were treated with adjuvant chemotherapy with either anthracycline-based or gemcitabine-based chemotherapy at two Italian centers.
Of 145 patients, 97 were treated with an anthracycline-based regimen and 48 with gemcitabine and docetaxel. The median number of cycles of anthracycline based regimen received was 4 (range 2-6) and with gemcitabine and docetaxel was 5 (range 3-7). Disease free survival was 31 months in patients treated with anthracycline-based chemotherapy and 19 months in patients treated with gemcitabine and docetaxel.
Trabectedin and low-dose radiotherapy
Trabectedin concurrent with low-dose radiotherapy is being examined as an option for patients with pulmonary metastatic soft tissue sarcoma (NCT02275286).
In a phase 1 study, long-lasting dimensional responses were seen in 71% of the irradiated lesions showed. Based on those results, trabectedin (Yondelis) at 1.5 mg/m 2 will be the recommended dose for phase 2, according to Javier Martín-Broto, MD, of the Institute of Biomedicine Research (IBIS)-University Hospital Virgen del Rocio/CSIC/University of Seville, Spain, and his colleagues.
For the study, trabectedin was given along with radiotherapy (30 Gy) in 10 fractions (3 Gy/fraction). Three dose levels of trabectedin were administered: -1 (1.1 mg/m 2), 1 (1.3 mg/m 2) and 2 (1.5 mg/m 2). Dose-limiting toxicity was defined as grade 3 or greater events excluding grade 3/4 neutropenia lasting less than 5 days, grade 3 transaminitis if it did not lead to trabectedin delay, and grade 3/4 nausea/vomiting due to inadequate prophylaxis.
Ten of the 18 patients enrolled had synovial sarcoma; 3 had undifferentiated pleomorphic sarcomas and the other patients had either myxoid liposarcoma, dedifferentiated liposarcoma, G3 not otherwise specified sarcoma, leiomyosarcoma, and malignant peripheral nerve sheath tumor.
Patients received a median of 1 prior line of chemotherapy (range: 0-3). Twelve patients received trabectedin at dose level 1 and 6 patients at dose level 2. Grade 3/4 adverse events were neutropenia, seen in 8 patients; alanine aminotransferase (ALT) elevation, seen in 2 patients; gamma-glutamyl transferase (GGT) elevation, seen in 2 patients; anemia, seen in 2 patients; febrile neutropenia, seen in 1 patient; and pneumonitis, seen in 1 patient.
There were two dose-limiting toxicities: transient grade 4 ALT elevation at the level 1 dose and grade 4 neutropenia for more than 5 days at the level 2 dose.
Based on central radiological review of 17 evaluable patients, 2 patients achieved complete response, 3 had partial responses, 6 had stable disease, and 6 had progressive disease. The local review reported complete responses in 2 patients, partial responses in 5, stable disease in 4, and progressive disease in 6.
On the irradiated lesions, 4 had complete responses, 8 had partial responses, 4 had stable disease, and 1 had progressive disease. With a median follow-up of 18 months, median progression-free survival was 2.83 months (95%CI: 2.3-3.3 months). Thirteen patients have died, with a median overall survival of 8.77 months (95%CI: 3.6-13.9) and a 12-month overall survival rate of 48%.
Reports from the annual meeting of The Connective Tissue Oncology Society held in Rome, November 14-17, 2018 Sarcoma of the Year: Intimal Sarcoma
This year’s annual meeting of The Connective Tissue Oncology Society brought new insights on intimal sarcoma. Four studies in a featured session at the meeting examined both current and novel treatments for this rare and aggressive cancer, and emphasized the need for new therapies.
Anthracycline-based regimens as preferred first-line therapies
Anthracycline-based regimens were the preferred first-line therapies used in 83 adults with intimal sarcomas in a retrospective study of data from the World Sarcoma Network, reported by Anna Maria Frezza, MD, of the, Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues. The researchers described the experience with anthracycline-based regimens as well as gemcitabine-based regimens and pazopanib among MDM2-positive patients with intimal sarcomas treated at 16 sarcoma reference centers in Europe, the United States, and Japan. Their findings speak to the need for new active drugs, which they said should target the MDM2 and CDK4 overexpression seen in patients with this rare sarcoma.Of the 83 patients studied, nearly all (76 patients) initially received an anthracycline-based regimen. Gemcitabine-based regimens were used in 29 patients and pazopanib in 10 patients; 20 of the 39 patients received more than one treatment.
Anthracycline-based regimens were associated with a 12-month progression-free survival rate of 38% in 76 patients with intimal sarcomas. All of the 76 patients received anthracycline regimens as their initial systemic therapy; 27 were treated for localized disease with a curative intent and the remaining 49 had advanced disease. The researchers also noted that anthracycline regimens were safely used in 22 patients with cardiac intimal sarcomas, as none of them died of cardiotoxicity.
Based on RECIST 1.1 measures, the overall response rate was 37% in 57 evaluable patients: 3 patients had a complete response, 18 had a partial response, 27 had stable disease, and 9 had progressive disease. For those with localized disease, the median time to progression was 14 months, and overall survival time was 51. For patients with advanced disease, the median time to progression was 8 months and overall survival was 22 months.
Outcomes were less favorable when patients were treated with gemcitabine regimens or pazopanib. In most of these cases, however, patients were either on their second (gemcitabine) or third (pazopanib) lines of therapy.
In the gemcitabine group, 2 patients were treated for localized disease with curative intent and 27 for advanced disease. Of 28 evaluable patients, best response was partial remission in 3, stable disease in 8, and progressive disease in 17. In the 27 patients with advanced disease, the median progression free survival time was 3 months and overall survival was 13 months.
All 10 patients in the pazopanib group had advanced disease and had undergone a median of two prior lines of therapy. One patient had a partial remission, 3 had stable disease, and 6 had progressive disease. The median progression free survival was 4 months and median overall survival was 12 months.
Rarest of the rare: Primary malignant sarcoma of the heart
Luke Smith, of the School of Clinical Medicine, University of Cambridge, U.K., detailed the experience of 28 patients diagnosed with sarcomas of the heart or great vessels at the university’s Royal Papworth Hospital and Addenbrooke’s Hospital between 2000 and 2018.
Based on this retrospective review, surgery offers the best chance for long-term survival for these patients, who would otherwise experience progressive heart failure and die. Adjuvant chemotherapy and radiation therapy might be able to extend their survival and improve symptomatic relief, he said, but these outcomes have not been prospectively studied.
Typically, the patients in this series, 20 with pulmonary artery sarcoma and 8 with cardiac sarcoma, presented with symptoms mimicking heart failure, pulmonary hypertension, or thromboembolic disease. Nearly all, 24 patients reported breathlessness. Eight patients had chest pain or tightness, 6 had cough, 6 had peripheral edema, 6 had constitutional symptoms, 3 had hemoptysis, and 1 had a TIA. Only 1 patient had a seriously impaired left ventricular ejection fraction of less than 30%. LVEF was normal at 55% or more in 16 patients, and moderately impaired at 30% or more in 10 patients.
Median overall survival was 17 months. The 19 patients who underwent surgical resection of their primary tumor survived much longer than the 10 patients who did not--median overall survival of 20 months vs. 9 months--but this finding may simply reflect more advanced disease in patients with inoperable disease. There were 3 perioperative deaths among the 19 patients who underwent surgery: 14 with pulmonary artery sarcomas had pulmonary endarterectomy and 4 with cardiac sarcomas underwent resection or maximal debulking of their tumors.
Based on the retrospective study, adjuvant chemotherapy and radiation were safe and may lead to better outcomes for these patients. Active chemotherapy regimens in the palliative setting included paclitaxel (angiosarcoma) and anthracycline ± ifosfamide.
Nine patients received post-surgical chemotherapy, and after completion five also had radiotherapy. The 3 cardiac sarcoma patients who had surgical resection with curative intent were treated with adjuvant ifosfamide-based chemotherapy (with close monitoring of fluid balance), and showed no evidence of disease on last follow-ups. One patient received post-operative paclitaxel following maximal debulking of a cardiac angiosarcoma.
Post-surgical anthracycline with and without ifosfamide were used in patients with pulmonary artery sarcomas with no clinical cardiotoxicity. Although the median overall survival for patients who received post-operative chemo- and radio-therapy was 28 months and the median overall survival with surgery alone was 9 months, the difference was not statistically significant.
In the palliative setting, partial responses were observed with paclitaxel and anthracycline (including liposomal doxorubicin) in patients with cardiac angiosarcoma. For pulmonary artery intimal sarcomas, partial responses were achieved with anthracycline with and without ifosfamide. Radiotherapy provided good local control.
The longest surviving pulmonary artery sarcoma patient, at 103 months, had pulmonary artery endarterectomy, followed by adjuvant epirubicin and radiotherapy. She developed lung metastases 7 years later and was treated with radiofrequency ablation. The longest surviving cardiac sarcoma patient, at 24 months, remains disease free. He had surgery to resect a high-grade undifferentiated sarcoma with involved margins, followed by adjuvant ifosfamide and radiotherapy to the right atrium.
Therapeutically exploitable genetic aberrations in intimal sarcomas
Imatinib and olaratumab might prove to be therapeutic approaches for some patients with intimal sarcomas, based on a retrospective evaluation of genetic aberrations in 11 patients with intimal sarcomas, Jason Roszik, PhD, MBA, reported at the meeting.
Dr. Roszik and his colleagues at the University of Texas MD Anderson Cancer Center, Houston, analyzed information on 11 patients with intimal sarcomas in the American Association for Cancer Research (AACR) project, Genomics Evidence Neoplasia Information Exchange (GENIE). Sampling was taken from the primary tumor in 8 patients and from the metastatic site in the other 3.
MDM2 amplifications were seen in 8 of 10 patients with available copy number alterations. Amplifications in the CDK pathway were present in 5, PDGFRA gain was seen in 4, and CDKN2A copy number loss was present in 3. Mutations that could be targeted with drugs included ALK, ATM/ATR, PTCH1 and PDGFRB, he said.
Unique genomic rearrangement events included PDE4DIP-NOTCH2 and MRPS30-ARID2 fusions. Co-occurring alterations included a NOTCH2 copy number gain in the PDE4DIP-NOTCH2 fusion tumor, and PDGFRB mutations in both fusion-positive cases.
The researchers also drew on the published findings of whole-exome sequencing and array-comparative genomic hybridization from an autopsy case of cardiac intimal sarcoma (Virchows Arch. 2017 Sep;471(3):423-428). That study identified concurrent PDGFRA amplification and PDGFRB mutation.
The researchers additionally examined clinical trial enrollments and could find no patient with intimal sarcoma among 406 sarcoma enrolled patients. Intimal sarcomas were not eligible for any clinical trial given the location of the tumors in major blood vessels.
“The somatic mutations and DNA copy number alterations in the PDGFR pathway relevant to the pathogenesis and potential targeted therapy of cardiac intimal sarcoma may be targeted by imatinib or olaratumab. Inclusion of such rare tumors in targeted therapy basket trials with a waiver for inclusion criteria is warranted,” Dr. Roszik and his colleagues concluded in the abstract of their presentation.
The promise of combination therapy
The “largest experience using multimodality therapy with proton based local therapy” for sarcomas involving the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries was reported by Yen-Lin E. Chen, MD, and her colleagues at Massachusetts General Hospital, Boston.
They examined an institutional sarcoma data repository of 13,950 patients and found 37 patients with sarcomas arising from the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries. These included 9 with unclassified pleomorphic sarcoma/malignant fibrous histiocytoma, 8 with angiosarcoma, 4 with spindle cell sarcoma, 4 with sarcoma not otherwise specified, 3 with leiomyosarcoma, 2 with osteosarcoma, 2 with Ewing sarcoma, and 1 each with chondrosarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, synovial sarcoma, and intimal sarcoma.
Two-thirds of the patients had induction chemotherapy with or without maintenance therapy. Adriamycin, ifosfamide, and taxol therapies were most common. Two-thirds received proton based radiotherapy. Of the 23 patients who underwent resection, 11 were R2 (macroscopic positive margins), 3 were R1 (microscopic positive margins), and 9 were R0 (clear margins).
The 1-year overall survival rate was 64%, which fell to 37% at 3 years and to 28% at 5 years. Median survival was 28 months, twice that typically seen in the literature, Dr. Chen said.
For patients receiving proton based radiotherapy to a median dose of 64.8 GyRBE (range 63-72 GyRBE, 3 with additional intraoperative electrons), local failure free survivals were 80%, 64%, and 52% at 1, 3, and 5 years, respectively. For patients who did not receive radiotherapy, local failure free survival rates were 13%, 10%, 10%, respectively.
Overall, the 1, 3, and 5 year metastatic free survival rates were 25%, 14%, and 14%.
Survival rate was significantly better for patients with tumors smaller than 5 cm ( P =0.036), those over 40 years old ( P =0.028), those able to have surgery ( P =0.011), and those with non-angiosarcoma histologies ( P = 0.002).
This year’s annual meeting of The Connective Tissue Oncology Society brought new insights on intimal sarcoma. Four studies in a featured session at the meeting examined both current and novel treatments for this rare and aggressive cancer, and emphasized the need for new therapies.
Anthracycline-based regimens as preferred first-line therapies
Anthracycline-based regimens were the preferred first-line therapies used in 83 adults with intimal sarcomas in a retrospective study of data from the World Sarcoma Network, reported by Anna Maria Frezza, MD, of the, Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues. The researchers described the experience with anthracycline-based regimens as well as gemcitabine-based regimens and pazopanib among MDM2-positive patients with intimal sarcomas treated at 16 sarcoma reference centers in Europe, the United States, and Japan. Their findings speak to the need for new active drugs, which they said should target the MDM2 and CDK4 overexpression seen in patients with this rare sarcoma.Of the 83 patients studied, nearly all (76 patients) initially received an anthracycline-based regimen. Gemcitabine-based regimens were used in 29 patients and pazopanib in 10 patients; 20 of the 39 patients received more than one treatment.
Anthracycline-based regimens were associated with a 12-month progression-free survival rate of 38% in 76 patients with intimal sarcomas. All of the 76 patients received anthracycline regimens as their initial systemic therapy; 27 were treated for localized disease with a curative intent and the remaining 49 had advanced disease. The researchers also noted that anthracycline regimens were safely used in 22 patients with cardiac intimal sarcomas, as none of them died of cardiotoxicity.
Based on RECIST 1.1 measures, the overall response rate was 37% in 57 evaluable patients: 3 patients had a complete response, 18 had a partial response, 27 had stable disease, and 9 had progressive disease. For those with localized disease, the median time to progression was 14 months, and overall survival time was 51. For patients with advanced disease, the median time to progression was 8 months and overall survival was 22 months.
Outcomes were less favorable when patients were treated with gemcitabine regimens or pazopanib. In most of these cases, however, patients were either on their second (gemcitabine) or third (pazopanib) lines of therapy.
In the gemcitabine group, 2 patients were treated for localized disease with curative intent and 27 for advanced disease. Of 28 evaluable patients, best response was partial remission in 3, stable disease in 8, and progressive disease in 17. In the 27 patients with advanced disease, the median progression free survival time was 3 months and overall survival was 13 months.
All 10 patients in the pazopanib group had advanced disease and had undergone a median of two prior lines of therapy. One patient had a partial remission, 3 had stable disease, and 6 had progressive disease. The median progression free survival was 4 months and median overall survival was 12 months.
Rarest of the rare: Primary malignant sarcoma of the heart
Luke Smith, of the School of Clinical Medicine, University of Cambridge, U.K., detailed the experience of 28 patients diagnosed with sarcomas of the heart or great vessels at the university’s Royal Papworth Hospital and Addenbrooke’s Hospital between 2000 and 2018.
Based on this retrospective review, surgery offers the best chance for long-term survival for these patients, who would otherwise experience progressive heart failure and die. Adjuvant chemotherapy and radiation therapy might be able to extend their survival and improve symptomatic relief, he said, but these outcomes have not been prospectively studied.
Typically, the patients in this series, 20 with pulmonary artery sarcoma and 8 with cardiac sarcoma, presented with symptoms mimicking heart failure, pulmonary hypertension, or thromboembolic disease. Nearly all, 24 patients reported breathlessness. Eight patients had chest pain or tightness, 6 had cough, 6 had peripheral edema, 6 had constitutional symptoms, 3 had hemoptysis, and 1 had a TIA. Only 1 patient had a seriously impaired left ventricular ejection fraction of less than 30%. LVEF was normal at 55% or more in 16 patients, and moderately impaired at 30% or more in 10 patients.
Median overall survival was 17 months. The 19 patients who underwent surgical resection of their primary tumor survived much longer than the 10 patients who did not--median overall survival of 20 months vs. 9 months--but this finding may simply reflect more advanced disease in patients with inoperable disease. There were 3 perioperative deaths among the 19 patients who underwent surgery: 14 with pulmonary artery sarcomas had pulmonary endarterectomy and 4 with cardiac sarcomas underwent resection or maximal debulking of their tumors.
Based on the retrospective study, adjuvant chemotherapy and radiation were safe and may lead to better outcomes for these patients. Active chemotherapy regimens in the palliative setting included paclitaxel (angiosarcoma) and anthracycline ± ifosfamide.
Nine patients received post-surgical chemotherapy, and after completion five also had radiotherapy. The 3 cardiac sarcoma patients who had surgical resection with curative intent were treated with adjuvant ifosfamide-based chemotherapy (with close monitoring of fluid balance), and showed no evidence of disease on last follow-ups. One patient received post-operative paclitaxel following maximal debulking of a cardiac angiosarcoma.
Post-surgical anthracycline with and without ifosfamide were used in patients with pulmonary artery sarcomas with no clinical cardiotoxicity. Although the median overall survival for patients who received post-operative chemo- and radio-therapy was 28 months and the median overall survival with surgery alone was 9 months, the difference was not statistically significant.
In the palliative setting, partial responses were observed with paclitaxel and anthracycline (including liposomal doxorubicin) in patients with cardiac angiosarcoma. For pulmonary artery intimal sarcomas, partial responses were achieved with anthracycline with and without ifosfamide. Radiotherapy provided good local control.
The longest surviving pulmonary artery sarcoma patient, at 103 months, had pulmonary artery endarterectomy, followed by adjuvant epirubicin and radiotherapy. She developed lung metastases 7 years later and was treated with radiofrequency ablation. The longest surviving cardiac sarcoma patient, at 24 months, remains disease free. He had surgery to resect a high-grade undifferentiated sarcoma with involved margins, followed by adjuvant ifosfamide and radiotherapy to the right atrium.
Therapeutically exploitable genetic aberrations in intimal sarcomas
Imatinib and olaratumab might prove to be therapeutic approaches for some patients with intimal sarcomas, based on a retrospective evaluation of genetic aberrations in 11 patients with intimal sarcomas, Jason Roszik, PhD, MBA, reported at the meeting.
Dr. Roszik and his colleagues at the University of Texas MD Anderson Cancer Center, Houston, analyzed information on 11 patients with intimal sarcomas in the American Association for Cancer Research (AACR) project, Genomics Evidence Neoplasia Information Exchange (GENIE). Sampling was taken from the primary tumor in 8 patients and from the metastatic site in the other 3.
MDM2 amplifications were seen in 8 of 10 patients with available copy number alterations. Amplifications in the CDK pathway were present in 5, PDGFRA gain was seen in 4, and CDKN2A copy number loss was present in 3. Mutations that could be targeted with drugs included ALK, ATM/ATR, PTCH1 and PDGFRB, he said.
Unique genomic rearrangement events included PDE4DIP-NOTCH2 and MRPS30-ARID2 fusions. Co-occurring alterations included a NOTCH2 copy number gain in the PDE4DIP-NOTCH2 fusion tumor, and PDGFRB mutations in both fusion-positive cases.
The researchers also drew on the published findings of whole-exome sequencing and array-comparative genomic hybridization from an autopsy case of cardiac intimal sarcoma (Virchows Arch. 2017 Sep;471(3):423-428). That study identified concurrent PDGFRA amplification and PDGFRB mutation.
The researchers additionally examined clinical trial enrollments and could find no patient with intimal sarcoma among 406 sarcoma enrolled patients. Intimal sarcomas were not eligible for any clinical trial given the location of the tumors in major blood vessels.
“The somatic mutations and DNA copy number alterations in the PDGFR pathway relevant to the pathogenesis and potential targeted therapy of cardiac intimal sarcoma may be targeted by imatinib or olaratumab. Inclusion of such rare tumors in targeted therapy basket trials with a waiver for inclusion criteria is warranted,” Dr. Roszik and his colleagues concluded in the abstract of their presentation.
The promise of combination therapy
The “largest experience using multimodality therapy with proton based local therapy” for sarcomas involving the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries was reported by Yen-Lin E. Chen, MD, and her colleagues at Massachusetts General Hospital, Boston.
They examined an institutional sarcoma data repository of 13,950 patients and found 37 patients with sarcomas arising from the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries. These included 9 with unclassified pleomorphic sarcoma/malignant fibrous histiocytoma, 8 with angiosarcoma, 4 with spindle cell sarcoma, 4 with sarcoma not otherwise specified, 3 with leiomyosarcoma, 2 with osteosarcoma, 2 with Ewing sarcoma, and 1 each with chondrosarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, synovial sarcoma, and intimal sarcoma.
Two-thirds of the patients had induction chemotherapy with or without maintenance therapy. Adriamycin, ifosfamide, and taxol therapies were most common. Two-thirds received proton based radiotherapy. Of the 23 patients who underwent resection, 11 were R2 (macroscopic positive margins), 3 were R1 (microscopic positive margins), and 9 were R0 (clear margins).
The 1-year overall survival rate was 64%, which fell to 37% at 3 years and to 28% at 5 years. Median survival was 28 months, twice that typically seen in the literature, Dr. Chen said.
For patients receiving proton based radiotherapy to a median dose of 64.8 GyRBE (range 63-72 GyRBE, 3 with additional intraoperative electrons), local failure free survivals were 80%, 64%, and 52% at 1, 3, and 5 years, respectively. For patients who did not receive radiotherapy, local failure free survival rates were 13%, 10%, 10%, respectively.
Overall, the 1, 3, and 5 year metastatic free survival rates were 25%, 14%, and 14%.
Survival rate was significantly better for patients with tumors smaller than 5 cm ( P =0.036), those over 40 years old ( P =0.028), those able to have surgery ( P =0.011), and those with non-angiosarcoma histologies ( P = 0.002).
This year’s annual meeting of The Connective Tissue Oncology Society brought new insights on intimal sarcoma. Four studies in a featured session at the meeting examined both current and novel treatments for this rare and aggressive cancer, and emphasized the need for new therapies.
Anthracycline-based regimens as preferred first-line therapies
Anthracycline-based regimens were the preferred first-line therapies used in 83 adults with intimal sarcomas in a retrospective study of data from the World Sarcoma Network, reported by Anna Maria Frezza, MD, of the, Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues. The researchers described the experience with anthracycline-based regimens as well as gemcitabine-based regimens and pazopanib among MDM2-positive patients with intimal sarcomas treated at 16 sarcoma reference centers in Europe, the United States, and Japan. Their findings speak to the need for new active drugs, which they said should target the MDM2 and CDK4 overexpression seen in patients with this rare sarcoma.Of the 83 patients studied, nearly all (76 patients) initially received an anthracycline-based regimen. Gemcitabine-based regimens were used in 29 patients and pazopanib in 10 patients; 20 of the 39 patients received more than one treatment.
Anthracycline-based regimens were associated with a 12-month progression-free survival rate of 38% in 76 patients with intimal sarcomas. All of the 76 patients received anthracycline regimens as their initial systemic therapy; 27 were treated for localized disease with a curative intent and the remaining 49 had advanced disease. The researchers also noted that anthracycline regimens were safely used in 22 patients with cardiac intimal sarcomas, as none of them died of cardiotoxicity.
Based on RECIST 1.1 measures, the overall response rate was 37% in 57 evaluable patients: 3 patients had a complete response, 18 had a partial response, 27 had stable disease, and 9 had progressive disease. For those with localized disease, the median time to progression was 14 months, and overall survival time was 51. For patients with advanced disease, the median time to progression was 8 months and overall survival was 22 months.
Outcomes were less favorable when patients were treated with gemcitabine regimens or pazopanib. In most of these cases, however, patients were either on their second (gemcitabine) or third (pazopanib) lines of therapy.
In the gemcitabine group, 2 patients were treated for localized disease with curative intent and 27 for advanced disease. Of 28 evaluable patients, best response was partial remission in 3, stable disease in 8, and progressive disease in 17. In the 27 patients with advanced disease, the median progression free survival time was 3 months and overall survival was 13 months.
All 10 patients in the pazopanib group had advanced disease and had undergone a median of two prior lines of therapy. One patient had a partial remission, 3 had stable disease, and 6 had progressive disease. The median progression free survival was 4 months and median overall survival was 12 months.
Rarest of the rare: Primary malignant sarcoma of the heart
Luke Smith, of the School of Clinical Medicine, University of Cambridge, U.K., detailed the experience of 28 patients diagnosed with sarcomas of the heart or great vessels at the university’s Royal Papworth Hospital and Addenbrooke’s Hospital between 2000 and 2018.
Based on this retrospective review, surgery offers the best chance for long-term survival for these patients, who would otherwise experience progressive heart failure and die. Adjuvant chemotherapy and radiation therapy might be able to extend their survival and improve symptomatic relief, he said, but these outcomes have not been prospectively studied.
Typically, the patients in this series, 20 with pulmonary artery sarcoma and 8 with cardiac sarcoma, presented with symptoms mimicking heart failure, pulmonary hypertension, or thromboembolic disease. Nearly all, 24 patients reported breathlessness. Eight patients had chest pain or tightness, 6 had cough, 6 had peripheral edema, 6 had constitutional symptoms, 3 had hemoptysis, and 1 had a TIA. Only 1 patient had a seriously impaired left ventricular ejection fraction of less than 30%. LVEF was normal at 55% or more in 16 patients, and moderately impaired at 30% or more in 10 patients.
Median overall survival was 17 months. The 19 patients who underwent surgical resection of their primary tumor survived much longer than the 10 patients who did not--median overall survival of 20 months vs. 9 months--but this finding may simply reflect more advanced disease in patients with inoperable disease. There were 3 perioperative deaths among the 19 patients who underwent surgery: 14 with pulmonary artery sarcomas had pulmonary endarterectomy and 4 with cardiac sarcomas underwent resection or maximal debulking of their tumors.
Based on the retrospective study, adjuvant chemotherapy and radiation were safe and may lead to better outcomes for these patients. Active chemotherapy regimens in the palliative setting included paclitaxel (angiosarcoma) and anthracycline ± ifosfamide.
Nine patients received post-surgical chemotherapy, and after completion five also had radiotherapy. The 3 cardiac sarcoma patients who had surgical resection with curative intent were treated with adjuvant ifosfamide-based chemotherapy (with close monitoring of fluid balance), and showed no evidence of disease on last follow-ups. One patient received post-operative paclitaxel following maximal debulking of a cardiac angiosarcoma.
Post-surgical anthracycline with and without ifosfamide were used in patients with pulmonary artery sarcomas with no clinical cardiotoxicity. Although the median overall survival for patients who received post-operative chemo- and radio-therapy was 28 months and the median overall survival with surgery alone was 9 months, the difference was not statistically significant.
In the palliative setting, partial responses were observed with paclitaxel and anthracycline (including liposomal doxorubicin) in patients with cardiac angiosarcoma. For pulmonary artery intimal sarcomas, partial responses were achieved with anthracycline with and without ifosfamide. Radiotherapy provided good local control.
The longest surviving pulmonary artery sarcoma patient, at 103 months, had pulmonary artery endarterectomy, followed by adjuvant epirubicin and radiotherapy. She developed lung metastases 7 years later and was treated with radiofrequency ablation. The longest surviving cardiac sarcoma patient, at 24 months, remains disease free. He had surgery to resect a high-grade undifferentiated sarcoma with involved margins, followed by adjuvant ifosfamide and radiotherapy to the right atrium.
Therapeutically exploitable genetic aberrations in intimal sarcomas
Imatinib and olaratumab might prove to be therapeutic approaches for some patients with intimal sarcomas, based on a retrospective evaluation of genetic aberrations in 11 patients with intimal sarcomas, Jason Roszik, PhD, MBA, reported at the meeting.
Dr. Roszik and his colleagues at the University of Texas MD Anderson Cancer Center, Houston, analyzed information on 11 patients with intimal sarcomas in the American Association for Cancer Research (AACR) project, Genomics Evidence Neoplasia Information Exchange (GENIE). Sampling was taken from the primary tumor in 8 patients and from the metastatic site in the other 3.
MDM2 amplifications were seen in 8 of 10 patients with available copy number alterations. Amplifications in the CDK pathway were present in 5, PDGFRA gain was seen in 4, and CDKN2A copy number loss was present in 3. Mutations that could be targeted with drugs included ALK, ATM/ATR, PTCH1 and PDGFRB, he said.
Unique genomic rearrangement events included PDE4DIP-NOTCH2 and MRPS30-ARID2 fusions. Co-occurring alterations included a NOTCH2 copy number gain in the PDE4DIP-NOTCH2 fusion tumor, and PDGFRB mutations in both fusion-positive cases.
The researchers also drew on the published findings of whole-exome sequencing and array-comparative genomic hybridization from an autopsy case of cardiac intimal sarcoma (Virchows Arch. 2017 Sep;471(3):423-428). That study identified concurrent PDGFRA amplification and PDGFRB mutation.
The researchers additionally examined clinical trial enrollments and could find no patient with intimal sarcoma among 406 sarcoma enrolled patients. Intimal sarcomas were not eligible for any clinical trial given the location of the tumors in major blood vessels.
“The somatic mutations and DNA copy number alterations in the PDGFR pathway relevant to the pathogenesis and potential targeted therapy of cardiac intimal sarcoma may be targeted by imatinib or olaratumab. Inclusion of such rare tumors in targeted therapy basket trials with a waiver for inclusion criteria is warranted,” Dr. Roszik and his colleagues concluded in the abstract of their presentation.
The promise of combination therapy
The “largest experience using multimodality therapy with proton based local therapy” for sarcomas involving the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries was reported by Yen-Lin E. Chen, MD, and her colleagues at Massachusetts General Hospital, Boston.
They examined an institutional sarcoma data repository of 13,950 patients and found 37 patients with sarcomas arising from the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries. These included 9 with unclassified pleomorphic sarcoma/malignant fibrous histiocytoma, 8 with angiosarcoma, 4 with spindle cell sarcoma, 4 with sarcoma not otherwise specified, 3 with leiomyosarcoma, 2 with osteosarcoma, 2 with Ewing sarcoma, and 1 each with chondrosarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, synovial sarcoma, and intimal sarcoma.
Two-thirds of the patients had induction chemotherapy with or without maintenance therapy. Adriamycin, ifosfamide, and taxol therapies were most common. Two-thirds received proton based radiotherapy. Of the 23 patients who underwent resection, 11 were R2 (macroscopic positive margins), 3 were R1 (microscopic positive margins), and 9 were R0 (clear margins).
The 1-year overall survival rate was 64%, which fell to 37% at 3 years and to 28% at 5 years. Median survival was 28 months, twice that typically seen in the literature, Dr. Chen said.
For patients receiving proton based radiotherapy to a median dose of 64.8 GyRBE (range 63-72 GyRBE, 3 with additional intraoperative electrons), local failure free survivals were 80%, 64%, and 52% at 1, 3, and 5 years, respectively. For patients who did not receive radiotherapy, local failure free survival rates were 13%, 10%, 10%, respectively.
Overall, the 1, 3, and 5 year metastatic free survival rates were 25%, 14%, and 14%.
Survival rate was significantly better for patients with tumors smaller than 5 cm ( P =0.036), those over 40 years old ( P =0.028), those able to have surgery ( P =0.011), and those with non-angiosarcoma histologies ( P = 0.002).
FDA approves larotrectinib for cancers with NTRK gene fusions
for adult and pediatric patients with solid tumors that harbor a genetic aberration known as an neurotrophic receptor tyrosine kinase (NTRK) fusion.
Specifically, the oral inhibitor of tropomyosin receptor kinase is approved for patients with solid tumors that have a NTRK gene fusion without a known acquired resistance mutation and have metastatic disease, are likely to experience severe morbidity from surgical resection, have no satisfactory alternative treatments, or have cancer that has progressed following treatment, the FDA said in a press release.
NTRK fusions are found in both children and adults with dozens of different cancer types. The genetic aberration tends to be rare in common cancers and nearly universal in certain uncommon cancers.
Approval of larotrectinib (Vitrakvi) was based on overall response rate and response duration in the first 55 patients with unresectable or metastatic solid tumors harboring an NTRK gene fusion enrolled across three multicenter, open-label, single-arm clinical trials. The ORR was 75% (95% confidence interval, 61%-85%), including 22% complete responses and 53% partial responses. At the time of database lock, median duration of response had not been reached, the FDA said.
The most common tumors were salivary gland tumors (22%), soft tissue sarcomas (20%), infantile fibrosarcomas (13%), and thyroid cancers (9%). Identification of positive NTRK gene fusion status was prospectively determined in local laboratories using next-generation sequencing or fluorescence in situ hybridization.
Results of the three trials, a phase 1 trial among 8 adult patients (LOXO-TRK-14001), a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 basket trial among 35 adult and adolescent patients (NAVIGATE), were presented at the annual meeting of the American Society of Clinical Oncology in 2017.
The safety of larotrectinib was evaluated in 176 patients enrolled across the three clinical trials, including 44 pediatric patients. The most common adverse reactions with larotrectinib were fatigue, nausea, dizziness, vomiting, increased AST, cough, increased ALT, constipation, and diarrhea, the FDA said.
The recommended larotrectinib doses are 100 mg orally twice daily for adults and 100 mg/m2 orally twice daily (maximum of 100 mg per dose) for pediatric patients.
The approval of larotrectinib follows the approval of pembrolizumab for certain solid tumors with the MSI-H biomarker, as the first approval for the treatment of cancer based on a biomarker rather than the particular body part or organ system affected by the tumor.
The FDA granted the accelerated approval of Vitrakvi to Loxo Oncology and Bayer.
for adult and pediatric patients with solid tumors that harbor a genetic aberration known as an neurotrophic receptor tyrosine kinase (NTRK) fusion.
Specifically, the oral inhibitor of tropomyosin receptor kinase is approved for patients with solid tumors that have a NTRK gene fusion without a known acquired resistance mutation and have metastatic disease, are likely to experience severe morbidity from surgical resection, have no satisfactory alternative treatments, or have cancer that has progressed following treatment, the FDA said in a press release.
NTRK fusions are found in both children and adults with dozens of different cancer types. The genetic aberration tends to be rare in common cancers and nearly universal in certain uncommon cancers.
Approval of larotrectinib (Vitrakvi) was based on overall response rate and response duration in the first 55 patients with unresectable or metastatic solid tumors harboring an NTRK gene fusion enrolled across three multicenter, open-label, single-arm clinical trials. The ORR was 75% (95% confidence interval, 61%-85%), including 22% complete responses and 53% partial responses. At the time of database lock, median duration of response had not been reached, the FDA said.
The most common tumors were salivary gland tumors (22%), soft tissue sarcomas (20%), infantile fibrosarcomas (13%), and thyroid cancers (9%). Identification of positive NTRK gene fusion status was prospectively determined in local laboratories using next-generation sequencing or fluorescence in situ hybridization.
Results of the three trials, a phase 1 trial among 8 adult patients (LOXO-TRK-14001), a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 basket trial among 35 adult and adolescent patients (NAVIGATE), were presented at the annual meeting of the American Society of Clinical Oncology in 2017.
The safety of larotrectinib was evaluated in 176 patients enrolled across the three clinical trials, including 44 pediatric patients. The most common adverse reactions with larotrectinib were fatigue, nausea, dizziness, vomiting, increased AST, cough, increased ALT, constipation, and diarrhea, the FDA said.
The recommended larotrectinib doses are 100 mg orally twice daily for adults and 100 mg/m2 orally twice daily (maximum of 100 mg per dose) for pediatric patients.
The approval of larotrectinib follows the approval of pembrolizumab for certain solid tumors with the MSI-H biomarker, as the first approval for the treatment of cancer based on a biomarker rather than the particular body part or organ system affected by the tumor.
The FDA granted the accelerated approval of Vitrakvi to Loxo Oncology and Bayer.
for adult and pediatric patients with solid tumors that harbor a genetic aberration known as an neurotrophic receptor tyrosine kinase (NTRK) fusion.
Specifically, the oral inhibitor of tropomyosin receptor kinase is approved for patients with solid tumors that have a NTRK gene fusion without a known acquired resistance mutation and have metastatic disease, are likely to experience severe morbidity from surgical resection, have no satisfactory alternative treatments, or have cancer that has progressed following treatment, the FDA said in a press release.
NTRK fusions are found in both children and adults with dozens of different cancer types. The genetic aberration tends to be rare in common cancers and nearly universal in certain uncommon cancers.
Approval of larotrectinib (Vitrakvi) was based on overall response rate and response duration in the first 55 patients with unresectable or metastatic solid tumors harboring an NTRK gene fusion enrolled across three multicenter, open-label, single-arm clinical trials. The ORR was 75% (95% confidence interval, 61%-85%), including 22% complete responses and 53% partial responses. At the time of database lock, median duration of response had not been reached, the FDA said.
The most common tumors were salivary gland tumors (22%), soft tissue sarcomas (20%), infantile fibrosarcomas (13%), and thyroid cancers (9%). Identification of positive NTRK gene fusion status was prospectively determined in local laboratories using next-generation sequencing or fluorescence in situ hybridization.
Results of the three trials, a phase 1 trial among 8 adult patients (LOXO-TRK-14001), a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 basket trial among 35 adult and adolescent patients (NAVIGATE), were presented at the annual meeting of the American Society of Clinical Oncology in 2017.
The safety of larotrectinib was evaluated in 176 patients enrolled across the three clinical trials, including 44 pediatric patients. The most common adverse reactions with larotrectinib were fatigue, nausea, dizziness, vomiting, increased AST, cough, increased ALT, constipation, and diarrhea, the FDA said.
The recommended larotrectinib doses are 100 mg orally twice daily for adults and 100 mg/m2 orally twice daily (maximum of 100 mg per dose) for pediatric patients.
The approval of larotrectinib follows the approval of pembrolizumab for certain solid tumors with the MSI-H biomarker, as the first approval for the treatment of cancer based on a biomarker rather than the particular body part or organ system affected by the tumor.
The FDA granted the accelerated approval of Vitrakvi to Loxo Oncology and Bayer.
Predicting treatment response in leiomyosarcoma, liposarcoma
Aberrations in oncogenic pathways and immune modulation influence treatment response in patients with metastatic leiomyosarcoma or liposarcoma, based on an analysis of whole-exome sequencing of tumor samples from patients in a completed phase 3 randomized trial comparing trabectedin and dacarbazine.
In that trial, trabectedin benefit was mostly seen in patients with leiomyosarcoma, as well as in patients with myxoid/round cell sarcomas, and less so in those with dedifferentiated and pleomorphic liposarcomas.
Gurpreet Kapoor, PhD, of LabConnect, Seattle, and colleagues examined aberrations in oncogenic pathways (DNA damage response, PI3K, MDM2-p53) and in immune modulation and then correlated the genomic aberrations with prospective data on clinical outcomes in the trial.
For the study, presented at the annual meeting of the American Society of Clinical Oncology in Chicago, archival tumor samples were collected from 456 of the 518 patients; 180 had uterine leiomyosarcomas, 149 had nonuterine leiomyosarcomas, 66 had dedifferentiated liposarcomas, 46 had myxoid liposarcomas, and 15 had pleomorphic liposarcomas.
Peripheral blood samples from a subset of 346 patients were also analyzed as matched normal to filter noise from nonpathogenic variants in the whole-exome sequencing.
Consistent with sarcoma data from The Cancer Genome Atlas, frequent homozygous gene deletions with relatively low mutational load were noted in these leiomyosarcoma and liposarcoma samples. TP53 and RB1 alterations were more frequent in leiomyosarcomas than in liposarcomas and were not associated with clinical outcomes. Analyses of 103 DNA damage-response genes found somatic alterations exceeded 20% across subtypes and correlated with improved progression-free survival in only uterine leiomyosarcomas (hazard ratio, 0.63; P = .03).
Genomic alterations in PI3K pathway genes were noted in 30% of myxoid liposarcomas and were associated with a worse rate of progression-free survival (HR, 3.0; P = .045).
A trend towards better overall survival was noted in dedifferentiated liposarcoma patients with MDM2 amplification as compared with normal MDM2 copy number.
Certain subtype-specific genomic aberrations in immune modulation pathways were associated with worse clinical outcomes in patients with uterine leiomyosarcoma or dedifferentiated liposarcoma. Alterations in immune suppressors were associated with improved clinical outcomes in nonuterine leiomyosarcomas and alterations in lipid metabolism were associated with improved clinical outcomes in dedifferentiated liposarcomas.
The invited discussant for the study, Mark Andrew Dickson, MD, of Memorial Sloan Kettering Cancer Center, New York, noted that “the real take-home here is that the TMBs (tumor mutation burdens) are relatively low across all of the L-type sarcomas.
“The pattern and prevalence of genomic aberrations that we’re seeing in this cohort of patients prospectively analyzed on a clinical trial are consistent with prior reports. ... including CDK4 and MDM2 in dedifferentiated liposarcoma, PI3-kinase in some myxoid/round cells, p53 in leiomyosarcoma and liposarcoma, and so on.”
Generally, tumor mutation burden is low in L-type sarcomas, and there are some intriguing associations with benefit to therapies, such as PI3-kinase pathway and potential resistance to trabectedin and high tumor mutation burden and potential sensitivity to trabectedin, that need to be explored and validated in another larger cohort, he said.
“I also am increasingly coming to terms with the fact that the tumors like leiomyosarcoma, which have low tumor mutation burden, and which so far have proven fairly immune to immunotherapy, based on all of the negative PD-1 data that we’ve seen, and that also have recurrent, relatively unactionable mutations, like p53 and Rb, remain very difficult to treat,” Dr. Dickson concluded.
SOURCE: Kapoor G et al. ASCO 2018, Abstract 11513.
Aberrations in oncogenic pathways and immune modulation influence treatment response in patients with metastatic leiomyosarcoma or liposarcoma, based on an analysis of whole-exome sequencing of tumor samples from patients in a completed phase 3 randomized trial comparing trabectedin and dacarbazine.
In that trial, trabectedin benefit was mostly seen in patients with leiomyosarcoma, as well as in patients with myxoid/round cell sarcomas, and less so in those with dedifferentiated and pleomorphic liposarcomas.
Gurpreet Kapoor, PhD, of LabConnect, Seattle, and colleagues examined aberrations in oncogenic pathways (DNA damage response, PI3K, MDM2-p53) and in immune modulation and then correlated the genomic aberrations with prospective data on clinical outcomes in the trial.
For the study, presented at the annual meeting of the American Society of Clinical Oncology in Chicago, archival tumor samples were collected from 456 of the 518 patients; 180 had uterine leiomyosarcomas, 149 had nonuterine leiomyosarcomas, 66 had dedifferentiated liposarcomas, 46 had myxoid liposarcomas, and 15 had pleomorphic liposarcomas.
Peripheral blood samples from a subset of 346 patients were also analyzed as matched normal to filter noise from nonpathogenic variants in the whole-exome sequencing.
Consistent with sarcoma data from The Cancer Genome Atlas, frequent homozygous gene deletions with relatively low mutational load were noted in these leiomyosarcoma and liposarcoma samples. TP53 and RB1 alterations were more frequent in leiomyosarcomas than in liposarcomas and were not associated with clinical outcomes. Analyses of 103 DNA damage-response genes found somatic alterations exceeded 20% across subtypes and correlated with improved progression-free survival in only uterine leiomyosarcomas (hazard ratio, 0.63; P = .03).
Genomic alterations in PI3K pathway genes were noted in 30% of myxoid liposarcomas and were associated with a worse rate of progression-free survival (HR, 3.0; P = .045).
A trend towards better overall survival was noted in dedifferentiated liposarcoma patients with MDM2 amplification as compared with normal MDM2 copy number.
Certain subtype-specific genomic aberrations in immune modulation pathways were associated with worse clinical outcomes in patients with uterine leiomyosarcoma or dedifferentiated liposarcoma. Alterations in immune suppressors were associated with improved clinical outcomes in nonuterine leiomyosarcomas and alterations in lipid metabolism were associated with improved clinical outcomes in dedifferentiated liposarcomas.
The invited discussant for the study, Mark Andrew Dickson, MD, of Memorial Sloan Kettering Cancer Center, New York, noted that “the real take-home here is that the TMBs (tumor mutation burdens) are relatively low across all of the L-type sarcomas.
“The pattern and prevalence of genomic aberrations that we’re seeing in this cohort of patients prospectively analyzed on a clinical trial are consistent with prior reports. ... including CDK4 and MDM2 in dedifferentiated liposarcoma, PI3-kinase in some myxoid/round cells, p53 in leiomyosarcoma and liposarcoma, and so on.”
Generally, tumor mutation burden is low in L-type sarcomas, and there are some intriguing associations with benefit to therapies, such as PI3-kinase pathway and potential resistance to trabectedin and high tumor mutation burden and potential sensitivity to trabectedin, that need to be explored and validated in another larger cohort, he said.
“I also am increasingly coming to terms with the fact that the tumors like leiomyosarcoma, which have low tumor mutation burden, and which so far have proven fairly immune to immunotherapy, based on all of the negative PD-1 data that we’ve seen, and that also have recurrent, relatively unactionable mutations, like p53 and Rb, remain very difficult to treat,” Dr. Dickson concluded.
SOURCE: Kapoor G et al. ASCO 2018, Abstract 11513.
Aberrations in oncogenic pathways and immune modulation influence treatment response in patients with metastatic leiomyosarcoma or liposarcoma, based on an analysis of whole-exome sequencing of tumor samples from patients in a completed phase 3 randomized trial comparing trabectedin and dacarbazine.
In that trial, trabectedin benefit was mostly seen in patients with leiomyosarcoma, as well as in patients with myxoid/round cell sarcomas, and less so in those with dedifferentiated and pleomorphic liposarcomas.
Gurpreet Kapoor, PhD, of LabConnect, Seattle, and colleagues examined aberrations in oncogenic pathways (DNA damage response, PI3K, MDM2-p53) and in immune modulation and then correlated the genomic aberrations with prospective data on clinical outcomes in the trial.
For the study, presented at the annual meeting of the American Society of Clinical Oncology in Chicago, archival tumor samples were collected from 456 of the 518 patients; 180 had uterine leiomyosarcomas, 149 had nonuterine leiomyosarcomas, 66 had dedifferentiated liposarcomas, 46 had myxoid liposarcomas, and 15 had pleomorphic liposarcomas.
Peripheral blood samples from a subset of 346 patients were also analyzed as matched normal to filter noise from nonpathogenic variants in the whole-exome sequencing.
Consistent with sarcoma data from The Cancer Genome Atlas, frequent homozygous gene deletions with relatively low mutational load were noted in these leiomyosarcoma and liposarcoma samples. TP53 and RB1 alterations were more frequent in leiomyosarcomas than in liposarcomas and were not associated with clinical outcomes. Analyses of 103 DNA damage-response genes found somatic alterations exceeded 20% across subtypes and correlated with improved progression-free survival in only uterine leiomyosarcomas (hazard ratio, 0.63; P = .03).
Genomic alterations in PI3K pathway genes were noted in 30% of myxoid liposarcomas and were associated with a worse rate of progression-free survival (HR, 3.0; P = .045).
A trend towards better overall survival was noted in dedifferentiated liposarcoma patients with MDM2 amplification as compared with normal MDM2 copy number.
Certain subtype-specific genomic aberrations in immune modulation pathways were associated with worse clinical outcomes in patients with uterine leiomyosarcoma or dedifferentiated liposarcoma. Alterations in immune suppressors were associated with improved clinical outcomes in nonuterine leiomyosarcomas and alterations in lipid metabolism were associated with improved clinical outcomes in dedifferentiated liposarcomas.
The invited discussant for the study, Mark Andrew Dickson, MD, of Memorial Sloan Kettering Cancer Center, New York, noted that “the real take-home here is that the TMBs (tumor mutation burdens) are relatively low across all of the L-type sarcomas.
“The pattern and prevalence of genomic aberrations that we’re seeing in this cohort of patients prospectively analyzed on a clinical trial are consistent with prior reports. ... including CDK4 and MDM2 in dedifferentiated liposarcoma, PI3-kinase in some myxoid/round cells, p53 in leiomyosarcoma and liposarcoma, and so on.”
Generally, tumor mutation burden is low in L-type sarcomas, and there are some intriguing associations with benefit to therapies, such as PI3-kinase pathway and potential resistance to trabectedin and high tumor mutation burden and potential sensitivity to trabectedin, that need to be explored and validated in another larger cohort, he said.
“I also am increasingly coming to terms with the fact that the tumors like leiomyosarcoma, which have low tumor mutation burden, and which so far have proven fairly immune to immunotherapy, based on all of the negative PD-1 data that we’ve seen, and that also have recurrent, relatively unactionable mutations, like p53 and Rb, remain very difficult to treat,” Dr. Dickson concluded.
SOURCE: Kapoor G et al. ASCO 2018, Abstract 11513.
REPORTING FROM ASCO 2018
Key clinical point: Aberrations in oncogenic pathways and immune modulation influence treatment response in patients with metastatic leiomyosarcoma or liposarcoma.
Major finding: Genomic alterations in PI3K pathway genes were noted in 30% of myxoid liposarcomas and were associated with a worse rate of progression-free survival (HR, 3.0; P = .045).
Study details: Archival tumor samples were collected from 456 of the 518 patients; 180 had uterine leiomyosarcomas, 149 had nonuterine leiomyosarcomas, 66 had dedifferentiated liposarcomas, 46 had myxoid liposarcomas, and 15 had pleomorphic liposarcomas in the completed phase 3 randomized trial comparing trabectedin and dacarbazine.
Disclosures: Dr. Kapoor is employed by LabConnect, Seattle. Research funding was supplied by Janssen Research & Development.
Source: Kapoor G et al. ASCO 2018, Abstract 11513.