User login
Not so crazy: Pancreas transplants in type 2 diabetes rising
Simultaneous
Traditionally, recipients of pancreas transplants have been people with type 1 diabetes who also have either chronic kidney disease (CKD) or hypoglycemic unawareness. The former group could receive either a simultaneous pancreas-kidney or a pancreas after kidney transplant, while the latter – if they have normal kidney function – would be eligible for a pancreas transplant alone.
But increasingly in recent years, patients with type 2 diabetes and CKD have been receiving simultaneous pancreas-kidney transplants, with similar success rates to those of people with type 1 diabetes.
Such candidates are typically sufficiently fit, not morbidly obese, and taking insulin regardless of their C-peptide status, said Jon S. Odorico, MD, professor of surgery and director of pancreas and islet transplantation at the University of Wisconsin–Madison Transplant Program.
“One might ask: Is it a crazy idea to do a pancreas transplant for patients with type 2 diabetes? Based on the known mechanisms of hyperglycemia in these patients, it might seem so,” he said, noting that while individuals with type 2 diabetes usually have insulin resistance, many also have relative or absolute deficiency of insulin production.
“So by replacing beta-cell mass, pancreas transplantation addresses this beta-cell defect mechanism,” he explained when discussing the topic during a symposium held June 26 at the virtual American Diabetes Association (ADA) 81st Scientific Sessions.
Arguments in favor of simultaneous pancreas-kidney transplant in people with type 2 diabetes and CKD include the fact that type 2 diabetes is the leading cause of kidney disease in the United States – roughly 50-60% of candidates on the kidney transplant waiting list also have type 2 diabetes – and that kidney transplant alone tends to worsen diabetes control due to the required immunosuppression.
Moreover, due to a 2014 allocation policy change that separates simultaneous pancreas-kidney from kidney transplant–alone donor organs, waiting times are shorter for the former, and kidney quality is generally better than for kidney transplant alone, unless a living kidney donor is available.
And, Dr. Odorico added, “adding a pancreas to a kidney transplant does not appear to jeopardize patient survival or kidney graft survival in appropriately selected patients with diabetes.” However, he also noted that because type 2 diabetes is so heterogeneous, ideal candidates for simultaneous pancreas-kidney transplant are not yet clear.
Currently, people with type 2 diabetes account for about 20% of those receiving simultaneous pancreas-kidney transplants and about 50% of pancreas after kidney transplants. Few pancreas transplants alone are performed in type 2 diabetes because those individuals rarely experience severe life-threatening hypoglycemia, Dr. Odorico explained.
Criteria have shifted over time, C-peptide removed in 2019
In an interview, symposium moderator Peter G. Stock, MD, PhD, surgical director of the Kidney and Pancreas Transplant Program at the University of California, San Francisco, said he agreed that “it’s a surprising trend. It doesn’t make intuitive sense. In type 1 diabetes, it makes sense to replace the beta cells. But type 2 is due to a whole cluster of etiologies ... The view in the public domain is that it’s not due to the lack of insulin but problems with insulin resistance and obesity. So it doesn’t make a whole lot of sense to give you more insulin if it’s a receptor problem.”
But Dr. Stock noted that because in the past diabetes type wasn’t always rigorously assessed using C-peptide and antibody testing, which most centers measure today, “a number of transplants were done in people who turned out to have type 2. Our perception is that everybody who has type 2 is obese, but that’s not true anymore.”
Once it became apparent that some patients with type 2 diabetes who received pancreas transplants seemed to be doing well, the pancreas transplantation committee of the United Network for Organ Sharing (UNOS) established general criteria for the procedure in people with diabetes. They had to be taking insulin and have a C-peptide value of 2 ng/mL or below or taking insulin with a C-peptide greater than 2 ng/mL and a body mass index less than or equal to the maximum allowable BMI (28 kg/m2 at the time).
Dr. Stock, who chaired that committee from 2005 to 2007, said: “We thought it was risky to offer a scarce pool of donor pancreases to people with type 2 when we had people with type 1 who we know will benefit from it. So initially, the committee decided to limit pancreas transplantation to those with type 2 who have fairly low insulin requirements and BMIs that are more in the range of people with type 1. And lo and behold the results were comparable.”
Subsequent to Dr. Stock’s tenure as chair, the UNOS committee decided that the BMI and C-peptide criteria for simultaneous pancreas-kidney were no longer scientifically justifiable and were potentially discriminatory both to minority populations with type 2 diabetes and people with type 1 diabetes who have a high BMI, so in 2019, they removed them.
Individual transplant centers must follow UNOS rules, but they can also add their own criteria. Some don’t perform simultaneous pancreas-kidney transplants in people with type 2 diabetes at all.
At Dr. Odorico’s center, which began doing so in 2012, patients with type 2 diabetes account for nearly 40% of all simultaneous pancreas-kidney transplants. Indications there include age 20-60 years, insulin dependent with requirements less than 1 unit/kg/day, CKD stage 3-5, predialysis or on dialysis, and BMI <33 kg/m2.
“They are highly selected and a fairly fit group of patients,” Dr. Odorico noted.
Those who don’t meet all the requirements for simultaneous pancreas-kidney transplants may still be eligible for kidney transplant alone, from either a living or deceased donor, he said.
Dr. Stock’s criteria at UCSF are even more stringent for both BMI and insulin requirements.
SPK outcomes similar for type 1 and type 2 diabetes: Emerging data
Data to guide this area are accumulating slowly. Thus far, all studies have been retrospective and have used variable definitions for diabetes type and for graft failure. However, they’re fairly consistent in showing similar outcomes by diabetes type and little impact of C-peptide level on patient survival or survival of either kidney or pancreas graft, particularly after adjustment for confounding factors between the two types.
In a study from Dr. Odorico’s center of 284 type 1 and 39 type 2 diabetes patients undergoing simultaneous pancreas-kidney transplant between 2006 and 2017, pretransplant BMI and insulin requirements did not affect patient or graft survival in either type. There was a suggestion of greater risk for post-transplant diabetes with very high pretransplant insulin requirements (>75 units/day) but the numbers were too small to be definitive.
“It’s clear we will be doing more pancreas transplants in the future in this group of patients, and it’s ripe for further investigation,” Dr. Odorico concluded.
Beta cells for all?
Dr. Stock added one more aspect. While of course whole-organ transplantation is limited by the shortage of human donors, stem cell–derived beta cells could potentially produce an unlimited supply. Both Dr. Stock and Dr. Odorico are working on different approaches to this.
“We’re really close,” he said, noting, “the data we get for people with type 2 diabetes undergoing solid organ pancreas transplant could also be applied to cellular therapy ... We need to get a better understanding of which patients will benefit. The data we have so far are very promising.”
Dr. Odorico is scientific founder, stock equity holder, scientific advisory board chair, and a prior grant support recipient from Regenerative Medical Solutions. He has reported receiving clinical trial support from Veloxis Pharmaceuticals, CareDx, Natera, and Vertex Pharmaceuticals. Dr. Stock has reported being on the scientific advisory board of Encellin and receives funding from the California Institute of Regenerative Medicine and National Institutes of Health.
A version of this article first appeared on Medscape.com.
Simultaneous
Traditionally, recipients of pancreas transplants have been people with type 1 diabetes who also have either chronic kidney disease (CKD) or hypoglycemic unawareness. The former group could receive either a simultaneous pancreas-kidney or a pancreas after kidney transplant, while the latter – if they have normal kidney function – would be eligible for a pancreas transplant alone.
But increasingly in recent years, patients with type 2 diabetes and CKD have been receiving simultaneous pancreas-kidney transplants, with similar success rates to those of people with type 1 diabetes.
Such candidates are typically sufficiently fit, not morbidly obese, and taking insulin regardless of their C-peptide status, said Jon S. Odorico, MD, professor of surgery and director of pancreas and islet transplantation at the University of Wisconsin–Madison Transplant Program.
“One might ask: Is it a crazy idea to do a pancreas transplant for patients with type 2 diabetes? Based on the known mechanisms of hyperglycemia in these patients, it might seem so,” he said, noting that while individuals with type 2 diabetes usually have insulin resistance, many also have relative or absolute deficiency of insulin production.
“So by replacing beta-cell mass, pancreas transplantation addresses this beta-cell defect mechanism,” he explained when discussing the topic during a symposium held June 26 at the virtual American Diabetes Association (ADA) 81st Scientific Sessions.
Arguments in favor of simultaneous pancreas-kidney transplant in people with type 2 diabetes and CKD include the fact that type 2 diabetes is the leading cause of kidney disease in the United States – roughly 50-60% of candidates on the kidney transplant waiting list also have type 2 diabetes – and that kidney transplant alone tends to worsen diabetes control due to the required immunosuppression.
Moreover, due to a 2014 allocation policy change that separates simultaneous pancreas-kidney from kidney transplant–alone donor organs, waiting times are shorter for the former, and kidney quality is generally better than for kidney transplant alone, unless a living kidney donor is available.
And, Dr. Odorico added, “adding a pancreas to a kidney transplant does not appear to jeopardize patient survival or kidney graft survival in appropriately selected patients with diabetes.” However, he also noted that because type 2 diabetes is so heterogeneous, ideal candidates for simultaneous pancreas-kidney transplant are not yet clear.
Currently, people with type 2 diabetes account for about 20% of those receiving simultaneous pancreas-kidney transplants and about 50% of pancreas after kidney transplants. Few pancreas transplants alone are performed in type 2 diabetes because those individuals rarely experience severe life-threatening hypoglycemia, Dr. Odorico explained.
Criteria have shifted over time, C-peptide removed in 2019
In an interview, symposium moderator Peter G. Stock, MD, PhD, surgical director of the Kidney and Pancreas Transplant Program at the University of California, San Francisco, said he agreed that “it’s a surprising trend. It doesn’t make intuitive sense. In type 1 diabetes, it makes sense to replace the beta cells. But type 2 is due to a whole cluster of etiologies ... The view in the public domain is that it’s not due to the lack of insulin but problems with insulin resistance and obesity. So it doesn’t make a whole lot of sense to give you more insulin if it’s a receptor problem.”
But Dr. Stock noted that because in the past diabetes type wasn’t always rigorously assessed using C-peptide and antibody testing, which most centers measure today, “a number of transplants were done in people who turned out to have type 2. Our perception is that everybody who has type 2 is obese, but that’s not true anymore.”
Once it became apparent that some patients with type 2 diabetes who received pancreas transplants seemed to be doing well, the pancreas transplantation committee of the United Network for Organ Sharing (UNOS) established general criteria for the procedure in people with diabetes. They had to be taking insulin and have a C-peptide value of 2 ng/mL or below or taking insulin with a C-peptide greater than 2 ng/mL and a body mass index less than or equal to the maximum allowable BMI (28 kg/m2 at the time).
Dr. Stock, who chaired that committee from 2005 to 2007, said: “We thought it was risky to offer a scarce pool of donor pancreases to people with type 2 when we had people with type 1 who we know will benefit from it. So initially, the committee decided to limit pancreas transplantation to those with type 2 who have fairly low insulin requirements and BMIs that are more in the range of people with type 1. And lo and behold the results were comparable.”
Subsequent to Dr. Stock’s tenure as chair, the UNOS committee decided that the BMI and C-peptide criteria for simultaneous pancreas-kidney were no longer scientifically justifiable and were potentially discriminatory both to minority populations with type 2 diabetes and people with type 1 diabetes who have a high BMI, so in 2019, they removed them.
Individual transplant centers must follow UNOS rules, but they can also add their own criteria. Some don’t perform simultaneous pancreas-kidney transplants in people with type 2 diabetes at all.
At Dr. Odorico’s center, which began doing so in 2012, patients with type 2 diabetes account for nearly 40% of all simultaneous pancreas-kidney transplants. Indications there include age 20-60 years, insulin dependent with requirements less than 1 unit/kg/day, CKD stage 3-5, predialysis or on dialysis, and BMI <33 kg/m2.
“They are highly selected and a fairly fit group of patients,” Dr. Odorico noted.
Those who don’t meet all the requirements for simultaneous pancreas-kidney transplants may still be eligible for kidney transplant alone, from either a living or deceased donor, he said.
Dr. Stock’s criteria at UCSF are even more stringent for both BMI and insulin requirements.
SPK outcomes similar for type 1 and type 2 diabetes: Emerging data
Data to guide this area are accumulating slowly. Thus far, all studies have been retrospective and have used variable definitions for diabetes type and for graft failure. However, they’re fairly consistent in showing similar outcomes by diabetes type and little impact of C-peptide level on patient survival or survival of either kidney or pancreas graft, particularly after adjustment for confounding factors between the two types.
In a study from Dr. Odorico’s center of 284 type 1 and 39 type 2 diabetes patients undergoing simultaneous pancreas-kidney transplant between 2006 and 2017, pretransplant BMI and insulin requirements did not affect patient or graft survival in either type. There was a suggestion of greater risk for post-transplant diabetes with very high pretransplant insulin requirements (>75 units/day) but the numbers were too small to be definitive.
“It’s clear we will be doing more pancreas transplants in the future in this group of patients, and it’s ripe for further investigation,” Dr. Odorico concluded.
Beta cells for all?
Dr. Stock added one more aspect. While of course whole-organ transplantation is limited by the shortage of human donors, stem cell–derived beta cells could potentially produce an unlimited supply. Both Dr. Stock and Dr. Odorico are working on different approaches to this.
“We’re really close,” he said, noting, “the data we get for people with type 2 diabetes undergoing solid organ pancreas transplant could also be applied to cellular therapy ... We need to get a better understanding of which patients will benefit. The data we have so far are very promising.”
Dr. Odorico is scientific founder, stock equity holder, scientific advisory board chair, and a prior grant support recipient from Regenerative Medical Solutions. He has reported receiving clinical trial support from Veloxis Pharmaceuticals, CareDx, Natera, and Vertex Pharmaceuticals. Dr. Stock has reported being on the scientific advisory board of Encellin and receives funding from the California Institute of Regenerative Medicine and National Institutes of Health.
A version of this article first appeared on Medscape.com.
Simultaneous
Traditionally, recipients of pancreas transplants have been people with type 1 diabetes who also have either chronic kidney disease (CKD) or hypoglycemic unawareness. The former group could receive either a simultaneous pancreas-kidney or a pancreas after kidney transplant, while the latter – if they have normal kidney function – would be eligible for a pancreas transplant alone.
But increasingly in recent years, patients with type 2 diabetes and CKD have been receiving simultaneous pancreas-kidney transplants, with similar success rates to those of people with type 1 diabetes.
Such candidates are typically sufficiently fit, not morbidly obese, and taking insulin regardless of their C-peptide status, said Jon S. Odorico, MD, professor of surgery and director of pancreas and islet transplantation at the University of Wisconsin–Madison Transplant Program.
“One might ask: Is it a crazy idea to do a pancreas transplant for patients with type 2 diabetes? Based on the known mechanisms of hyperglycemia in these patients, it might seem so,” he said, noting that while individuals with type 2 diabetes usually have insulin resistance, many also have relative or absolute deficiency of insulin production.
“So by replacing beta-cell mass, pancreas transplantation addresses this beta-cell defect mechanism,” he explained when discussing the topic during a symposium held June 26 at the virtual American Diabetes Association (ADA) 81st Scientific Sessions.
Arguments in favor of simultaneous pancreas-kidney transplant in people with type 2 diabetes and CKD include the fact that type 2 diabetes is the leading cause of kidney disease in the United States – roughly 50-60% of candidates on the kidney transplant waiting list also have type 2 diabetes – and that kidney transplant alone tends to worsen diabetes control due to the required immunosuppression.
Moreover, due to a 2014 allocation policy change that separates simultaneous pancreas-kidney from kidney transplant–alone donor organs, waiting times are shorter for the former, and kidney quality is generally better than for kidney transplant alone, unless a living kidney donor is available.
And, Dr. Odorico added, “adding a pancreas to a kidney transplant does not appear to jeopardize patient survival or kidney graft survival in appropriately selected patients with diabetes.” However, he also noted that because type 2 diabetes is so heterogeneous, ideal candidates for simultaneous pancreas-kidney transplant are not yet clear.
Currently, people with type 2 diabetes account for about 20% of those receiving simultaneous pancreas-kidney transplants and about 50% of pancreas after kidney transplants. Few pancreas transplants alone are performed in type 2 diabetes because those individuals rarely experience severe life-threatening hypoglycemia, Dr. Odorico explained.
Criteria have shifted over time, C-peptide removed in 2019
In an interview, symposium moderator Peter G. Stock, MD, PhD, surgical director of the Kidney and Pancreas Transplant Program at the University of California, San Francisco, said he agreed that “it’s a surprising trend. It doesn’t make intuitive sense. In type 1 diabetes, it makes sense to replace the beta cells. But type 2 is due to a whole cluster of etiologies ... The view in the public domain is that it’s not due to the lack of insulin but problems with insulin resistance and obesity. So it doesn’t make a whole lot of sense to give you more insulin if it’s a receptor problem.”
But Dr. Stock noted that because in the past diabetes type wasn’t always rigorously assessed using C-peptide and antibody testing, which most centers measure today, “a number of transplants were done in people who turned out to have type 2. Our perception is that everybody who has type 2 is obese, but that’s not true anymore.”
Once it became apparent that some patients with type 2 diabetes who received pancreas transplants seemed to be doing well, the pancreas transplantation committee of the United Network for Organ Sharing (UNOS) established general criteria for the procedure in people with diabetes. They had to be taking insulin and have a C-peptide value of 2 ng/mL or below or taking insulin with a C-peptide greater than 2 ng/mL and a body mass index less than or equal to the maximum allowable BMI (28 kg/m2 at the time).
Dr. Stock, who chaired that committee from 2005 to 2007, said: “We thought it was risky to offer a scarce pool of donor pancreases to people with type 2 when we had people with type 1 who we know will benefit from it. So initially, the committee decided to limit pancreas transplantation to those with type 2 who have fairly low insulin requirements and BMIs that are more in the range of people with type 1. And lo and behold the results were comparable.”
Subsequent to Dr. Stock’s tenure as chair, the UNOS committee decided that the BMI and C-peptide criteria for simultaneous pancreas-kidney were no longer scientifically justifiable and were potentially discriminatory both to minority populations with type 2 diabetes and people with type 1 diabetes who have a high BMI, so in 2019, they removed them.
Individual transplant centers must follow UNOS rules, but they can also add their own criteria. Some don’t perform simultaneous pancreas-kidney transplants in people with type 2 diabetes at all.
At Dr. Odorico’s center, which began doing so in 2012, patients with type 2 diabetes account for nearly 40% of all simultaneous pancreas-kidney transplants. Indications there include age 20-60 years, insulin dependent with requirements less than 1 unit/kg/day, CKD stage 3-5, predialysis or on dialysis, and BMI <33 kg/m2.
“They are highly selected and a fairly fit group of patients,” Dr. Odorico noted.
Those who don’t meet all the requirements for simultaneous pancreas-kidney transplants may still be eligible for kidney transplant alone, from either a living or deceased donor, he said.
Dr. Stock’s criteria at UCSF are even more stringent for both BMI and insulin requirements.
SPK outcomes similar for type 1 and type 2 diabetes: Emerging data
Data to guide this area are accumulating slowly. Thus far, all studies have been retrospective and have used variable definitions for diabetes type and for graft failure. However, they’re fairly consistent in showing similar outcomes by diabetes type and little impact of C-peptide level on patient survival or survival of either kidney or pancreas graft, particularly after adjustment for confounding factors between the two types.
In a study from Dr. Odorico’s center of 284 type 1 and 39 type 2 diabetes patients undergoing simultaneous pancreas-kidney transplant between 2006 and 2017, pretransplant BMI and insulin requirements did not affect patient or graft survival in either type. There was a suggestion of greater risk for post-transplant diabetes with very high pretransplant insulin requirements (>75 units/day) but the numbers were too small to be definitive.
“It’s clear we will be doing more pancreas transplants in the future in this group of patients, and it’s ripe for further investigation,” Dr. Odorico concluded.
Beta cells for all?
Dr. Stock added one more aspect. While of course whole-organ transplantation is limited by the shortage of human donors, stem cell–derived beta cells could potentially produce an unlimited supply. Both Dr. Stock and Dr. Odorico are working on different approaches to this.
“We’re really close,” he said, noting, “the data we get for people with type 2 diabetes undergoing solid organ pancreas transplant could also be applied to cellular therapy ... We need to get a better understanding of which patients will benefit. The data we have so far are very promising.”
Dr. Odorico is scientific founder, stock equity holder, scientific advisory board chair, and a prior grant support recipient from Regenerative Medical Solutions. He has reported receiving clinical trial support from Veloxis Pharmaceuticals, CareDx, Natera, and Vertex Pharmaceuticals. Dr. Stock has reported being on the scientific advisory board of Encellin and receives funding from the California Institute of Regenerative Medicine and National Institutes of Health.
A version of this article first appeared on Medscape.com.
Third COVID-19 vaccine dose helped some transplant recipients

All of those with low titers before the third dose had high titers after receiving the additional shot, but only about 33% of those with negative initial responses had detectable antibodies after the third dose, according to the paper, published in Annals of Internal Medicine.
Researchers at Johns Hopkins, Baltimore, who keep a COVID-19 vaccine registry, perform antibody tests on all registry subjects and inform them of their results. Registry participants were asked to inform the research team if they received a third dose, and, the research team tracked the immune responses of those who did.
The participants in this case series had low antibody levels and received a third dose of the vaccine on their own between March 20 and May 10 of 2021.
Third dose results
In this cases series – thought to be the first to look at third vaccine shots in this type of patient group – all six of those who had low antibody titers before the third dose had high-positive titers after the third dose.
Of the 24 individuals who had negative antibody titers before the third dose, just 6 had high titers after the third dose.
Two of the participants had low-positive titers, and 16 were negative.
“Several of those boosted very nicely into ranges seen, using these assays, in healthy persons,” said William Werbel, MD, a fellow in infectious disease at Johns Hopkins Medicine, Baltimore, who helped lead the study. Those with negative levels, even if they responded, tended to have lower titers, he said.
“The benefits at least from an antibody perspective were not the same for everybody and so this is obviously something that needs to be considered when thinking about selecting patients” for a COVID-19 prevention strategy, he said.
Reactions to the vaccine were low to moderate, such as some arm pain and fatigue.
“Showing that something is safe in that special, vulnerable population is important,” Dr. Werbel said. “We’re all wanting to make sure that we’re doing no harm.”
Dr. Werbel noted that there was no pattern in the small series based on the organ transplanted or in the vaccines used. As their third shot, 15 of the patients received the Johnson & Johnson vaccine; 9 received Moderna; and 6 received Pfizer-BioNTech.
Welcome news, but larger studies needed
“To think that a third dose could confer protection for a significant number of people is of course extremely welcome news,” said Christian Larsen, MD, DPhil, professor of surgery in the transplantation division at Emory University, Atlanta, who was not involved in the study. “It’s the easiest conceivable next intervention.”
He added, “We just want studies to confirm that – larger studies.”
Dr. Werbel stressed the importance of looking at third doses in these patients in a more controlled fashion in a randomized trial, to more carefully monitor safety and how patients fare when starting with one type of vaccine and switching to another, for example.
Richard Wender, MD, chair of family medicine and community health at the University of Pennsylvania, Philadelphia, said the findings are a reminder that there is still a lot that is unknown about COVID-19 and vaccination.

“We still don’t know who will or will not benefit from a third dose,” he said. “And our knowledge is evolving. For example, a recent study suggested that people with previous infection and who are vaccinated may have better and longer protection than people with vaccination alone. We’re still learning.”
He added that specialists, not primary care clinicians, should be relied upon to respond to this emerging vaccination data. Primary care doctors are very busy in other ways – such as in getting children caught up on vaccinations and helping adults return to managing their chronic diseases, Dr. Wender noted.
“Their focus needs to be on helping to overcome hesitancy, mistrust, lack of information, or antivaccination sentiment to help more people feel comfortable being vaccinated – this is a lot of work and needs constant focus. In short, primary care clinicians need to focus chiefly on the unvaccinated,” he said.
“Monitoring immunization recommendations for unique at-risk populations should be the chief responsibility of teams providing subspecialty care, [such as for] transplant patients, people with chronic kidney disease, cancer patients, and people with other chronic illnesses. This will allow primary care clinicians to tackle their many complex jobs.”
Possible solutions for those with low antibody responses
Dr. Larsen said that those with ongoing low antibody responses might still have other immune responses, such as a T-cell response. Such patients also could consider changing their vaccine type, he said.
“At the more significant intervention level, there may be circumstances where one could change the immunosuppressive drugs in a controlled way that might allow a better response,” suggested Dr. Larsen. “That’s obviously going to be something that requires a lot more thought and careful study.”
Dr. Werbel said that other options might need to be considered for those having no response following a third dose. One possibility is trying a vaccine with an adjuvant, such as the Novavax version, which might be more widely available soon.
“If you’re given a third dose of a very immunogenic vaccine – something that should work – and you just have no antibody development, it seems relatively unlikely that doing the same thing again is going to help you from that perspective, and for all we know might expose you to more risk,” Dr. Werbel noted.
Participant details
None of the 30 patients were thought to have ever had COVID-19. On average, patients had received their transplant 4.5 years before their original vaccination. In 25 patients, maintenance immunosuppression included tacrolimus or cyclosporine along with mycophenolate. Corticosteroids were also used for 24 patients, sirolimus was used for one patient, and belatacept was used for another patient.
Fifty-seven percent of patients had received the Pfizer/BioNTech vaccine originally, and 43% the Moderna vaccine. Most of the patients were kidney recipients, with two heart, three liver, one lung, one pancreas and one kidney-pancreas.
Dr. Werbel, Dr. Wender, and Dr. Larsen reported no relevant disclosures.
All of those with low titers before the third dose had high titers after receiving the additional shot, but only about 33% of those with negative initial responses had detectable antibodies after the third dose, according to the paper, published in Annals of Internal Medicine.
Researchers at Johns Hopkins, Baltimore, who keep a COVID-19 vaccine registry, perform antibody tests on all registry subjects and inform them of their results. Registry participants were asked to inform the research team if they received a third dose, and, the research team tracked the immune responses of those who did.
The participants in this case series had low antibody levels and received a third dose of the vaccine on their own between March 20 and May 10 of 2021.
Third dose results
In this cases series – thought to be the first to look at third vaccine shots in this type of patient group – all six of those who had low antibody titers before the third dose had high-positive titers after the third dose.
Of the 24 individuals who had negative antibody titers before the third dose, just 6 had high titers after the third dose.
Two of the participants had low-positive titers, and 16 were negative.
“Several of those boosted very nicely into ranges seen, using these assays, in healthy persons,” said William Werbel, MD, a fellow in infectious disease at Johns Hopkins Medicine, Baltimore, who helped lead the study. Those with negative levels, even if they responded, tended to have lower titers, he said.
“The benefits at least from an antibody perspective were not the same for everybody and so this is obviously something that needs to be considered when thinking about selecting patients” for a COVID-19 prevention strategy, he said.
Reactions to the vaccine were low to moderate, such as some arm pain and fatigue.
“Showing that something is safe in that special, vulnerable population is important,” Dr. Werbel said. “We’re all wanting to make sure that we’re doing no harm.”
Dr. Werbel noted that there was no pattern in the small series based on the organ transplanted or in the vaccines used. As their third shot, 15 of the patients received the Johnson & Johnson vaccine; 9 received Moderna; and 6 received Pfizer-BioNTech.
Welcome news, but larger studies needed
“To think that a third dose could confer protection for a significant number of people is of course extremely welcome news,” said Christian Larsen, MD, DPhil, professor of surgery in the transplantation division at Emory University, Atlanta, who was not involved in the study. “It’s the easiest conceivable next intervention.”
He added, “We just want studies to confirm that – larger studies.”
Dr. Werbel stressed the importance of looking at third doses in these patients in a more controlled fashion in a randomized trial, to more carefully monitor safety and how patients fare when starting with one type of vaccine and switching to another, for example.
Richard Wender, MD, chair of family medicine and community health at the University of Pennsylvania, Philadelphia, said the findings are a reminder that there is still a lot that is unknown about COVID-19 and vaccination.
“We still don’t know who will or will not benefit from a third dose,” he said. “And our knowledge is evolving. For example, a recent study suggested that people with previous infection and who are vaccinated may have better and longer protection than people with vaccination alone. We’re still learning.”
He added that specialists, not primary care clinicians, should be relied upon to respond to this emerging vaccination data. Primary care doctors are very busy in other ways – such as in getting children caught up on vaccinations and helping adults return to managing their chronic diseases, Dr. Wender noted.
“Their focus needs to be on helping to overcome hesitancy, mistrust, lack of information, or antivaccination sentiment to help more people feel comfortable being vaccinated – this is a lot of work and needs constant focus. In short, primary care clinicians need to focus chiefly on the unvaccinated,” he said.
“Monitoring immunization recommendations for unique at-risk populations should be the chief responsibility of teams providing subspecialty care, [such as for] transplant patients, people with chronic kidney disease, cancer patients, and people with other chronic illnesses. This will allow primary care clinicians to tackle their many complex jobs.”
Possible solutions for those with low antibody responses
Dr. Larsen said that those with ongoing low antibody responses might still have other immune responses, such as a T-cell response. Such patients also could consider changing their vaccine type, he said.
“At the more significant intervention level, there may be circumstances where one could change the immunosuppressive drugs in a controlled way that might allow a better response,” suggested Dr. Larsen. “That’s obviously going to be something that requires a lot more thought and careful study.”
Dr. Werbel said that other options might need to be considered for those having no response following a third dose. One possibility is trying a vaccine with an adjuvant, such as the Novavax version, which might be more widely available soon.
“If you’re given a third dose of a very immunogenic vaccine – something that should work – and you just have no antibody development, it seems relatively unlikely that doing the same thing again is going to help you from that perspective, and for all we know might expose you to more risk,” Dr. Werbel noted.
Participant details
None of the 30 patients were thought to have ever had COVID-19. On average, patients had received their transplant 4.5 years before their original vaccination. In 25 patients, maintenance immunosuppression included tacrolimus or cyclosporine along with mycophenolate. Corticosteroids were also used for 24 patients, sirolimus was used for one patient, and belatacept was used for another patient.
Fifty-seven percent of patients had received the Pfizer/BioNTech vaccine originally, and 43% the Moderna vaccine. Most of the patients were kidney recipients, with two heart, three liver, one lung, one pancreas and one kidney-pancreas.
Dr. Werbel, Dr. Wender, and Dr. Larsen reported no relevant disclosures.
All of those with low titers before the third dose had high titers after receiving the additional shot, but only about 33% of those with negative initial responses had detectable antibodies after the third dose, according to the paper, published in Annals of Internal Medicine.
Researchers at Johns Hopkins, Baltimore, who keep a COVID-19 vaccine registry, perform antibody tests on all registry subjects and inform them of their results. Registry participants were asked to inform the research team if they received a third dose, and, the research team tracked the immune responses of those who did.
The participants in this case series had low antibody levels and received a third dose of the vaccine on their own between March 20 and May 10 of 2021.
Third dose results
In this cases series – thought to be the first to look at third vaccine shots in this type of patient group – all six of those who had low antibody titers before the third dose had high-positive titers after the third dose.
Of the 24 individuals who had negative antibody titers before the third dose, just 6 had high titers after the third dose.
Two of the participants had low-positive titers, and 16 were negative.
“Several of those boosted very nicely into ranges seen, using these assays, in healthy persons,” said William Werbel, MD, a fellow in infectious disease at Johns Hopkins Medicine, Baltimore, who helped lead the study. Those with negative levels, even if they responded, tended to have lower titers, he said.
“The benefits at least from an antibody perspective were not the same for everybody and so this is obviously something that needs to be considered when thinking about selecting patients” for a COVID-19 prevention strategy, he said.
Reactions to the vaccine were low to moderate, such as some arm pain and fatigue.
“Showing that something is safe in that special, vulnerable population is important,” Dr. Werbel said. “We’re all wanting to make sure that we’re doing no harm.”
Dr. Werbel noted that there was no pattern in the small series based on the organ transplanted or in the vaccines used. As their third shot, 15 of the patients received the Johnson & Johnson vaccine; 9 received Moderna; and 6 received Pfizer-BioNTech.
Welcome news, but larger studies needed
“To think that a third dose could confer protection for a significant number of people is of course extremely welcome news,” said Christian Larsen, MD, DPhil, professor of surgery in the transplantation division at Emory University, Atlanta, who was not involved in the study. “It’s the easiest conceivable next intervention.”
He added, “We just want studies to confirm that – larger studies.”
Dr. Werbel stressed the importance of looking at third doses in these patients in a more controlled fashion in a randomized trial, to more carefully monitor safety and how patients fare when starting with one type of vaccine and switching to another, for example.
Richard Wender, MD, chair of family medicine and community health at the University of Pennsylvania, Philadelphia, said the findings are a reminder that there is still a lot that is unknown about COVID-19 and vaccination.
“We still don’t know who will or will not benefit from a third dose,” he said. “And our knowledge is evolving. For example, a recent study suggested that people with previous infection and who are vaccinated may have better and longer protection than people with vaccination alone. We’re still learning.”
He added that specialists, not primary care clinicians, should be relied upon to respond to this emerging vaccination data. Primary care doctors are very busy in other ways – such as in getting children caught up on vaccinations and helping adults return to managing their chronic diseases, Dr. Wender noted.
“Their focus needs to be on helping to overcome hesitancy, mistrust, lack of information, or antivaccination sentiment to help more people feel comfortable being vaccinated – this is a lot of work and needs constant focus. In short, primary care clinicians need to focus chiefly on the unvaccinated,” he said.
“Monitoring immunization recommendations for unique at-risk populations should be the chief responsibility of teams providing subspecialty care, [such as for] transplant patients, people with chronic kidney disease, cancer patients, and people with other chronic illnesses. This will allow primary care clinicians to tackle their many complex jobs.”
Possible solutions for those with low antibody responses
Dr. Larsen said that those with ongoing low antibody responses might still have other immune responses, such as a T-cell response. Such patients also could consider changing their vaccine type, he said.
“At the more significant intervention level, there may be circumstances where one could change the immunosuppressive drugs in a controlled way that might allow a better response,” suggested Dr. Larsen. “That’s obviously going to be something that requires a lot more thought and careful study.”
Dr. Werbel said that other options might need to be considered for those having no response following a third dose. One possibility is trying a vaccine with an adjuvant, such as the Novavax version, which might be more widely available soon.
“If you’re given a third dose of a very immunogenic vaccine – something that should work – and you just have no antibody development, it seems relatively unlikely that doing the same thing again is going to help you from that perspective, and for all we know might expose you to more risk,” Dr. Werbel noted.
Participant details
None of the 30 patients were thought to have ever had COVID-19. On average, patients had received their transplant 4.5 years before their original vaccination. In 25 patients, maintenance immunosuppression included tacrolimus or cyclosporine along with mycophenolate. Corticosteroids were also used for 24 patients, sirolimus was used for one patient, and belatacept was used for another patient.
Fifty-seven percent of patients had received the Pfizer/BioNTech vaccine originally, and 43% the Moderna vaccine. Most of the patients were kidney recipients, with two heart, three liver, one lung, one pancreas and one kidney-pancreas.
Dr. Werbel, Dr. Wender, and Dr. Larsen reported no relevant disclosures.
Predicting outcomes in therapy-related AML
Therapy-related acute myeloid leukemia (t-AML) occurs as a complication of chemotherapy and/or radiotherapy for previous cancer or for nonmalignant disorders, with an estimated prevalence of 10%-15% of all AML cases, according to Ram Vasudevan Nampoothiri, MD, and colleagues at the Princess Margaret Cancer Center, University of Toronto.
Dr. Nampoothiri and colleagues performed a retrospective study of 68 patients with t-AML who underwent hematopoietic stem cell transplantation (HSCT) at their institution. They found significant predictors of reduced overall survival, including chromosomal rearrangements, induction regimens, donor type, patient performance status, and the type of graft-versus-host disease (GVHD) prophylaxis the patients received, as reported in Hematology/Oncology and Stem Cell Therapy.
Some populations benefit
Among the 68 patients studied, a total of 59.9% were women; and the median age was 56.5 years. All patients were analyzed for prior malignancy, therapy, time to diagnosis of t-AML, transplant details, relapse-free survival, overall survival, and predictors of outcomes.
At 2 years, the cumulative incidence of relapse, nonrelapse mortality, relapse-free survival, and overall survival were 17.9%, 34.5%, 47.6%, and 49.3%, respectively. Overall, acute and chronic GVHD occurred in 39 (57.4%) and 23 (33.8%) patients, respectively, according to the researchers.
The significant predictors of reduced overall survival were the presence of the 11q23 chromosomal rearrangement (hazard ratio, 3.24), use of induction regimens other than fludarabine, cytarabine, idarubicin, and granulocyte colony-stimulating factor or 7 + 3 (HR, 3.65), use of haploidentical donors (HR, 3.48), an Eastern Cooperative Oncology Group performance status of 2 or higher (HR, 5.83), and use of cyclosporine A–methotrexate as GVHD prophylaxis (HR, 2.41).
The researchers also found that a significant decrease in survival was seen with an increasing number of any of these prognostic factors.
A growing need
The incidence of t-AML is increasing because of longer life expectancy of the general population and also because of the improved survival of patients treated with chemotherapy and/or radiation for prior malignancies, according to the researchers.
They concluded that, even with this increasing prevalence and normally poor prognosis, “patients of t-AML having good-risk karyotypes, good performance status, and having HLA-matched donors have favorable outcomes after allo-HSCT.”
The authors reported that they had no competing financial interests.
Therapy-related acute myeloid leukemia (t-AML) occurs as a complication of chemotherapy and/or radiotherapy for previous cancer or for nonmalignant disorders, with an estimated prevalence of 10%-15% of all AML cases, according to Ram Vasudevan Nampoothiri, MD, and colleagues at the Princess Margaret Cancer Center, University of Toronto.
Dr. Nampoothiri and colleagues performed a retrospective study of 68 patients with t-AML who underwent hematopoietic stem cell transplantation (HSCT) at their institution. They found significant predictors of reduced overall survival, including chromosomal rearrangements, induction regimens, donor type, patient performance status, and the type of graft-versus-host disease (GVHD) prophylaxis the patients received, as reported in Hematology/Oncology and Stem Cell Therapy.
Some populations benefit
Among the 68 patients studied, a total of 59.9% were women; and the median age was 56.5 years. All patients were analyzed for prior malignancy, therapy, time to diagnosis of t-AML, transplant details, relapse-free survival, overall survival, and predictors of outcomes.
At 2 years, the cumulative incidence of relapse, nonrelapse mortality, relapse-free survival, and overall survival were 17.9%, 34.5%, 47.6%, and 49.3%, respectively. Overall, acute and chronic GVHD occurred in 39 (57.4%) and 23 (33.8%) patients, respectively, according to the researchers.
The significant predictors of reduced overall survival were the presence of the 11q23 chromosomal rearrangement (hazard ratio, 3.24), use of induction regimens other than fludarabine, cytarabine, idarubicin, and granulocyte colony-stimulating factor or 7 + 3 (HR, 3.65), use of haploidentical donors (HR, 3.48), an Eastern Cooperative Oncology Group performance status of 2 or higher (HR, 5.83), and use of cyclosporine A–methotrexate as GVHD prophylaxis (HR, 2.41).
The researchers also found that a significant decrease in survival was seen with an increasing number of any of these prognostic factors.
A growing need
The incidence of t-AML is increasing because of longer life expectancy of the general population and also because of the improved survival of patients treated with chemotherapy and/or radiation for prior malignancies, according to the researchers.
They concluded that, even with this increasing prevalence and normally poor prognosis, “patients of t-AML having good-risk karyotypes, good performance status, and having HLA-matched donors have favorable outcomes after allo-HSCT.”
The authors reported that they had no competing financial interests.
Therapy-related acute myeloid leukemia (t-AML) occurs as a complication of chemotherapy and/or radiotherapy for previous cancer or for nonmalignant disorders, with an estimated prevalence of 10%-15% of all AML cases, according to Ram Vasudevan Nampoothiri, MD, and colleagues at the Princess Margaret Cancer Center, University of Toronto.
Dr. Nampoothiri and colleagues performed a retrospective study of 68 patients with t-AML who underwent hematopoietic stem cell transplantation (HSCT) at their institution. They found significant predictors of reduced overall survival, including chromosomal rearrangements, induction regimens, donor type, patient performance status, and the type of graft-versus-host disease (GVHD) prophylaxis the patients received, as reported in Hematology/Oncology and Stem Cell Therapy.
Some populations benefit
Among the 68 patients studied, a total of 59.9% were women; and the median age was 56.5 years. All patients were analyzed for prior malignancy, therapy, time to diagnosis of t-AML, transplant details, relapse-free survival, overall survival, and predictors of outcomes.
At 2 years, the cumulative incidence of relapse, nonrelapse mortality, relapse-free survival, and overall survival were 17.9%, 34.5%, 47.6%, and 49.3%, respectively. Overall, acute and chronic GVHD occurred in 39 (57.4%) and 23 (33.8%) patients, respectively, according to the researchers.
The significant predictors of reduced overall survival were the presence of the 11q23 chromosomal rearrangement (hazard ratio, 3.24), use of induction regimens other than fludarabine, cytarabine, idarubicin, and granulocyte colony-stimulating factor or 7 + 3 (HR, 3.65), use of haploidentical donors (HR, 3.48), an Eastern Cooperative Oncology Group performance status of 2 or higher (HR, 5.83), and use of cyclosporine A–methotrexate as GVHD prophylaxis (HR, 2.41).
The researchers also found that a significant decrease in survival was seen with an increasing number of any of these prognostic factors.
A growing need
The incidence of t-AML is increasing because of longer life expectancy of the general population and also because of the improved survival of patients treated with chemotherapy and/or radiation for prior malignancies, according to the researchers.
They concluded that, even with this increasing prevalence and normally poor prognosis, “patients of t-AML having good-risk karyotypes, good performance status, and having HLA-matched donors have favorable outcomes after allo-HSCT.”
The authors reported that they had no competing financial interests.
FROM HEMATOLOGY/ONCOLOGY AND STEM CELL THERAPY
FDA panel supports islet cell treatment for type 1 diabetes
A Food and Drug Administration advisory panel has endorsed a pancreatic islet cell transplant therapy for the treatment of people with type 1 diabetes that can’t be managed with current therapies.

On April 15, the FDA’s Cellular, Tissue, and Gene Therapies Advisory Committee voted 12 to 4 in favor of approval of donislecel (Lantidra). There was one abstention. The panel regarded the drug as having “an overall favorable benefit-risk profile for some patients with type 1 diabetes.” The product consists of purified allogeneic pancreatic islets of Langerhans derived from cadaveric donors and is infused into the portal vein of the liver.
Benefits of the treatment include the potential for insulin independence and elimination of severe hypoglycemia. Risks are those associated with the surgical procedure and with long-term immunosuppression.
The therapy is manufactured by CellTrans. According to Jose Oberholzer, MD, the founder of CellTrans, the proposed indication is for adults with “brittle” type 1 diabetes who meet the American Diabetes Association’s (ADA) criteria for whole-organ pancreas-alone transplant (i.e., transplant of pancreas but not kidney).
The ADA criteria include the following: frequent, severe hypoglycemia, hyperglycemia, and/or ketoacidosis that requires medical attention; clinical or emotional problems regarding the use of exogenous insulin; and consistent failure of insulin-based management to prevent acute diabetes complications.
Success in two-thirds of patients in small studies
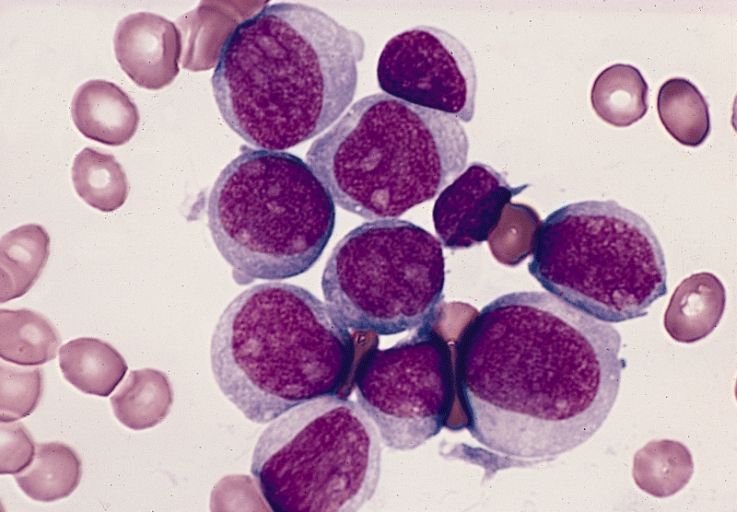
Dr. Oberholzer presented data from two single-arm open-label studies: a phase 1/2 trial initiated in 2004 with 10 patients, and a phase 3 study with 20 patients that began in 2007. The inclusion criteria differed somewhat between the two studies, but all 30 patients had hypoglycemic unawareness. Mean follow-up was 7.8 years for the phase 1/2 trial and 4.7 years for the phase 3 trial.
For all of the patients, C-peptide levels were positive after transplant. The composite endpoint for success – an A1c level of ≤ 6.5% and the absence of severe hypoglycemic episodes for 1 year – was met by 19 patients (63.3%). For five patients (16.7%), the target A1c level was not achieved, and seven patients (23.3%) experienced a severe episode of hypoglycemia.
Twenty of the 30 patients achieved insulin independence for at least 1 year.
Improvements were also seen at 1 year in mixed meal test outcomes, fasting blood glucose levels, and overall glycemic control. Graft survival 10 years post transplant was achieved by 60% of patients, Dr. Oberholzer said.
Adverse events not unexpected, but still of concern
Two patients died, one as a result of fulminant sepsis at 20 months post transplant, and the other as a result of severe dementia 9 years post transplant. Three patients experienced four serious procedure-related events, including one liver laceration and two hepatic hematomas. Elevations in portal pressure occurred in two patients.
Most adverse events were associated with immunosuppression. These included 178 infections in 26 of the 30 patients. The most common of these were herpes virus infections, Epstein-Barr virus infections, oral candidiasis, and cytomegalovirus infections. Twelve infections were severe. Renal function declined persistently in two patients (20%), and six (20%) experienced new-onset proteinuria at 1 year.
The adverse events related to the procedure and the problems associated with immunosuppression were not unexpected and were consistent with those described for patients receiving whole pancreas transplants, FDA reviewer Patricia Beaston, MD, said in her review of the CellTrans data.
Panel members support treatment for a small group of patients
During the discussion, several panel members pointed out that the target patient population for this treatment will likely be smaller today than it was when the two studies were initiated, given advances in diabetes care. Those advances include continuous glucose monitoring devices with alarms and closed-loop insulin delivery systems – the “artificial pancreas” that automatically suspends insulin delivery to prevent hypoglycemia.
Panel chair Lisa Butterfield, PhD, a surgeon and immunologist at the University of California, San Francisco, voted in favor of approval. But, she added, “I do support postapproval gathering of data to learn more about the product. ... I don’t know how many patients will really benefit, but I think it’s to be determined.”
Christopher K. Breuer, MD, a general and pediatric surgeon at the Center for Regenerative Medicine, Nationwide Children’s Hospital, Columbus, Ohio, said he supported approval for “two very small subpopulations where it would provide the only viable therapy”: those who are eligible for pancreas transplant but cannot tolerate a major operation, and those who already use the latest automated insulin delivery systems and still do not achieve acceptable glycemic control.
Temporary voting member David Harlan, MD, director of the University of Massachusetts Diabetes Center of Excellence, Worcester, Mass., voted no.
He noted that only about 100 whole pancreas-only transplants are performed annually in the United States and that such transplants are “very effective, so we’re talking about patients who aren’t pancreas transplant candidates who might get this.”
Moreover, Dr. Harlan said, “I’ve seen the awful things that can happen in posttransplant recipients. It’s really hard to get that informed consent from someone when you’re asking them to consider a future that they don’t know. When it works, it’s great. When it doesn’t work, it can be catastrophic. I just worry about opening Pandora’s box.”
The only other diabetes specialist on the panel, temporary voting member Ellen Leschek, MD, said she “reluctantly voted yes because a few people could benefit, but I think it’s a much smaller number than the company may believe.”
Dr. Leschek, of the National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, Md., said she’s concerned that “if it’s approved, too many people will get treated this way, when in fact, for a lot of those people, the risks will outweigh the benefits.”
Sandy Feng, MD, PhD, of the department of surgery at the University of California, San Francisco, pointed out that with regard to immunosuppressive therapy, “We’re concerned about the toxicity of what we currently use, but there are additional therapies being developed that might mitigate those toxicities that would be beneficial to this population.”
Dr. Feng, who voted yes, also said, “I do pancreas transplants. I can tell you that there is nothing that [patients with type 1 diabetes] like more than the freedom from dealing with the entire insulin issue. That has made a large impression on me over the last 20-plus years of clinical practice, so I do think this can help some people and will be incredibly meaningful to those people.”
FDA advisory panel members are vetted for conflicts of interest, and special waivers are granted if necessary. No such waivers were granted for this meeting.
A version of this article first appeared on Medscape.com.
A Food and Drug Administration advisory panel has endorsed a pancreatic islet cell transplant therapy for the treatment of people with type 1 diabetes that can’t be managed with current therapies.
On April 15, the FDA’s Cellular, Tissue, and Gene Therapies Advisory Committee voted 12 to 4 in favor of approval of donislecel (Lantidra). There was one abstention. The panel regarded the drug as having “an overall favorable benefit-risk profile for some patients with type 1 diabetes.” The product consists of purified allogeneic pancreatic islets of Langerhans derived from cadaveric donors and is infused into the portal vein of the liver.
Benefits of the treatment include the potential for insulin independence and elimination of severe hypoglycemia. Risks are those associated with the surgical procedure and with long-term immunosuppression.
The therapy is manufactured by CellTrans. According to Jose Oberholzer, MD, the founder of CellTrans, the proposed indication is for adults with “brittle” type 1 diabetes who meet the American Diabetes Association’s (ADA) criteria for whole-organ pancreas-alone transplant (i.e., transplant of pancreas but not kidney).
The ADA criteria include the following: frequent, severe hypoglycemia, hyperglycemia, and/or ketoacidosis that requires medical attention; clinical or emotional problems regarding the use of exogenous insulin; and consistent failure of insulin-based management to prevent acute diabetes complications.
Success in two-thirds of patients in small studies
Dr. Oberholzer presented data from two single-arm open-label studies: a phase 1/2 trial initiated in 2004 with 10 patients, and a phase 3 study with 20 patients that began in 2007. The inclusion criteria differed somewhat between the two studies, but all 30 patients had hypoglycemic unawareness. Mean follow-up was 7.8 years for the phase 1/2 trial and 4.7 years for the phase 3 trial.
For all of the patients, C-peptide levels were positive after transplant. The composite endpoint for success – an A1c level of ≤ 6.5% and the absence of severe hypoglycemic episodes for 1 year – was met by 19 patients (63.3%). For five patients (16.7%), the target A1c level was not achieved, and seven patients (23.3%) experienced a severe episode of hypoglycemia.
Twenty of the 30 patients achieved insulin independence for at least 1 year.
Improvements were also seen at 1 year in mixed meal test outcomes, fasting blood glucose levels, and overall glycemic control. Graft survival 10 years post transplant was achieved by 60% of patients, Dr. Oberholzer said.
Adverse events not unexpected, but still of concern
Two patients died, one as a result of fulminant sepsis at 20 months post transplant, and the other as a result of severe dementia 9 years post transplant. Three patients experienced four serious procedure-related events, including one liver laceration and two hepatic hematomas. Elevations in portal pressure occurred in two patients.
Most adverse events were associated with immunosuppression. These included 178 infections in 26 of the 30 patients. The most common of these were herpes virus infections, Epstein-Barr virus infections, oral candidiasis, and cytomegalovirus infections. Twelve infections were severe. Renal function declined persistently in two patients (20%), and six (20%) experienced new-onset proteinuria at 1 year.
The adverse events related to the procedure and the problems associated with immunosuppression were not unexpected and were consistent with those described for patients receiving whole pancreas transplants, FDA reviewer Patricia Beaston, MD, said in her review of the CellTrans data.
Panel members support treatment for a small group of patients
During the discussion, several panel members pointed out that the target patient population for this treatment will likely be smaller today than it was when the two studies were initiated, given advances in diabetes care. Those advances include continuous glucose monitoring devices with alarms and closed-loop insulin delivery systems – the “artificial pancreas” that automatically suspends insulin delivery to prevent hypoglycemia.
Panel chair Lisa Butterfield, PhD, a surgeon and immunologist at the University of California, San Francisco, voted in favor of approval. But, she added, “I do support postapproval gathering of data to learn more about the product. ... I don’t know how many patients will really benefit, but I think it’s to be determined.”
Christopher K. Breuer, MD, a general and pediatric surgeon at the Center for Regenerative Medicine, Nationwide Children’s Hospital, Columbus, Ohio, said he supported approval for “two very small subpopulations where it would provide the only viable therapy”: those who are eligible for pancreas transplant but cannot tolerate a major operation, and those who already use the latest automated insulin delivery systems and still do not achieve acceptable glycemic control.
Temporary voting member David Harlan, MD, director of the University of Massachusetts Diabetes Center of Excellence, Worcester, Mass., voted no.
He noted that only about 100 whole pancreas-only transplants are performed annually in the United States and that such transplants are “very effective, so we’re talking about patients who aren’t pancreas transplant candidates who might get this.”
Moreover, Dr. Harlan said, “I’ve seen the awful things that can happen in posttransplant recipients. It’s really hard to get that informed consent from someone when you’re asking them to consider a future that they don’t know. When it works, it’s great. When it doesn’t work, it can be catastrophic. I just worry about opening Pandora’s box.”
The only other diabetes specialist on the panel, temporary voting member Ellen Leschek, MD, said she “reluctantly voted yes because a few people could benefit, but I think it’s a much smaller number than the company may believe.”
Dr. Leschek, of the National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, Md., said she’s concerned that “if it’s approved, too many people will get treated this way, when in fact, for a lot of those people, the risks will outweigh the benefits.”
Sandy Feng, MD, PhD, of the department of surgery at the University of California, San Francisco, pointed out that with regard to immunosuppressive therapy, “We’re concerned about the toxicity of what we currently use, but there are additional therapies being developed that might mitigate those toxicities that would be beneficial to this population.”
Dr. Feng, who voted yes, also said, “I do pancreas transplants. I can tell you that there is nothing that [patients with type 1 diabetes] like more than the freedom from dealing with the entire insulin issue. That has made a large impression on me over the last 20-plus years of clinical practice, so I do think this can help some people and will be incredibly meaningful to those people.”
FDA advisory panel members are vetted for conflicts of interest, and special waivers are granted if necessary. No such waivers were granted for this meeting.
A version of this article first appeared on Medscape.com.
A Food and Drug Administration advisory panel has endorsed a pancreatic islet cell transplant therapy for the treatment of people with type 1 diabetes that can’t be managed with current therapies.
On April 15, the FDA’s Cellular, Tissue, and Gene Therapies Advisory Committee voted 12 to 4 in favor of approval of donislecel (Lantidra). There was one abstention. The panel regarded the drug as having “an overall favorable benefit-risk profile for some patients with type 1 diabetes.” The product consists of purified allogeneic pancreatic islets of Langerhans derived from cadaveric donors and is infused into the portal vein of the liver.
Benefits of the treatment include the potential for insulin independence and elimination of severe hypoglycemia. Risks are those associated with the surgical procedure and with long-term immunosuppression.
The therapy is manufactured by CellTrans. According to Jose Oberholzer, MD, the founder of CellTrans, the proposed indication is for adults with “brittle” type 1 diabetes who meet the American Diabetes Association’s (ADA) criteria for whole-organ pancreas-alone transplant (i.e., transplant of pancreas but not kidney).
The ADA criteria include the following: frequent, severe hypoglycemia, hyperglycemia, and/or ketoacidosis that requires medical attention; clinical or emotional problems regarding the use of exogenous insulin; and consistent failure of insulin-based management to prevent acute diabetes complications.
Success in two-thirds of patients in small studies
Dr. Oberholzer presented data from two single-arm open-label studies: a phase 1/2 trial initiated in 2004 with 10 patients, and a phase 3 study with 20 patients that began in 2007. The inclusion criteria differed somewhat between the two studies, but all 30 patients had hypoglycemic unawareness. Mean follow-up was 7.8 years for the phase 1/2 trial and 4.7 years for the phase 3 trial.
For all of the patients, C-peptide levels were positive after transplant. The composite endpoint for success – an A1c level of ≤ 6.5% and the absence of severe hypoglycemic episodes for 1 year – was met by 19 patients (63.3%). For five patients (16.7%), the target A1c level was not achieved, and seven patients (23.3%) experienced a severe episode of hypoglycemia.
Twenty of the 30 patients achieved insulin independence for at least 1 year.
Improvements were also seen at 1 year in mixed meal test outcomes, fasting blood glucose levels, and overall glycemic control. Graft survival 10 years post transplant was achieved by 60% of patients, Dr. Oberholzer said.
Adverse events not unexpected, but still of concern
Two patients died, one as a result of fulminant sepsis at 20 months post transplant, and the other as a result of severe dementia 9 years post transplant. Three patients experienced four serious procedure-related events, including one liver laceration and two hepatic hematomas. Elevations in portal pressure occurred in two patients.
Most adverse events were associated with immunosuppression. These included 178 infections in 26 of the 30 patients. The most common of these were herpes virus infections, Epstein-Barr virus infections, oral candidiasis, and cytomegalovirus infections. Twelve infections were severe. Renal function declined persistently in two patients (20%), and six (20%) experienced new-onset proteinuria at 1 year.
The adverse events related to the procedure and the problems associated with immunosuppression were not unexpected and were consistent with those described for patients receiving whole pancreas transplants, FDA reviewer Patricia Beaston, MD, said in her review of the CellTrans data.
Panel members support treatment for a small group of patients
During the discussion, several panel members pointed out that the target patient population for this treatment will likely be smaller today than it was when the two studies were initiated, given advances in diabetes care. Those advances include continuous glucose monitoring devices with alarms and closed-loop insulin delivery systems – the “artificial pancreas” that automatically suspends insulin delivery to prevent hypoglycemia.
Panel chair Lisa Butterfield, PhD, a surgeon and immunologist at the University of California, San Francisco, voted in favor of approval. But, she added, “I do support postapproval gathering of data to learn more about the product. ... I don’t know how many patients will really benefit, but I think it’s to be determined.”
Christopher K. Breuer, MD, a general and pediatric surgeon at the Center for Regenerative Medicine, Nationwide Children’s Hospital, Columbus, Ohio, said he supported approval for “two very small subpopulations where it would provide the only viable therapy”: those who are eligible for pancreas transplant but cannot tolerate a major operation, and those who already use the latest automated insulin delivery systems and still do not achieve acceptable glycemic control.
Temporary voting member David Harlan, MD, director of the University of Massachusetts Diabetes Center of Excellence, Worcester, Mass., voted no.
He noted that only about 100 whole pancreas-only transplants are performed annually in the United States and that such transplants are “very effective, so we’re talking about patients who aren’t pancreas transplant candidates who might get this.”
Moreover, Dr. Harlan said, “I’ve seen the awful things that can happen in posttransplant recipients. It’s really hard to get that informed consent from someone when you’re asking them to consider a future that they don’t know. When it works, it’s great. When it doesn’t work, it can be catastrophic. I just worry about opening Pandora’s box.”
The only other diabetes specialist on the panel, temporary voting member Ellen Leschek, MD, said she “reluctantly voted yes because a few people could benefit, but I think it’s a much smaller number than the company may believe.”
Dr. Leschek, of the National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, Md., said she’s concerned that “if it’s approved, too many people will get treated this way, when in fact, for a lot of those people, the risks will outweigh the benefits.”
Sandy Feng, MD, PhD, of the department of surgery at the University of California, San Francisco, pointed out that with regard to immunosuppressive therapy, “We’re concerned about the toxicity of what we currently use, but there are additional therapies being developed that might mitigate those toxicities that would be beneficial to this population.”
Dr. Feng, who voted yes, also said, “I do pancreas transplants. I can tell you that there is nothing that [patients with type 1 diabetes] like more than the freedom from dealing with the entire insulin issue. That has made a large impression on me over the last 20-plus years of clinical practice, so I do think this can help some people and will be incredibly meaningful to those people.”
FDA advisory panel members are vetted for conflicts of interest, and special waivers are granted if necessary. No such waivers were granted for this meeting.
A version of this article first appeared on Medscape.com.
OCS heart system earns hard-won backing of FDA panel
After more than 10 hours of intense debate, a Food and Drug Administration advisory panel gave its support to a premarket approval application (PMA) for the TransMedics Organ Care System (OCS) Heart system.
The OCS Heart is a portable extracorporeal perfusion and monitoring system designed to keep a donor heart in a normothermic, beating state. The “heart in a box” technology allows donor hearts to be transported across longer distances than is possible with standard cold storage, which can safely preserve donor hearts for about 4 hours.
The Circulatory System Devices Panel of the Medical Devices Advisory Committee voted 12 to 5, with 1 abstention, that the benefits of the OCS Heart System outweigh its risks.
The panel voted in favor of the OCS Heart being effective (10 yes, 6 no, and 2 abstaining) and safe (9 yes, 7 no, 2 abstaining) but not without mixed feelings.
James Blankenship, MD, a cardiologist at the University of New Mexico, Albuquerque, voted yes to all three questions but said: “If it had been compared to standard of care, I would have voted no to all three. But if it’s compared to getting an [left ventricular assist device] LVAD or not getting a heart at all, I would say the benefits outweigh the risks.”
Marc R. Katz, MD, chief of cardiothoracic surgery, Medical University of South Carolina, Charleston, also gave universal support, noting that the rate of heart transplantations has been flat for years. “This is a big step forward toward being able to expand that number. Now all that said, it obviously was a less-than-perfect study and I do think there needs to be some constraints put on the utilization.”
The panel reviewed data from the single-arm OCS Heart EXPAND trial and associated EXPAND Continued Access Protocol (CAP), as well the sponsor’s first OCS Heart trial, PROCEED II.
EXPAND met its effectiveness endpoint, with 88% of donor hearts successfully transplanted, an 8% incidence of severe primary graft dysfunction (PGD) 24 hours after transplantation, and 94.6% survival at 30 days.
Data from 41 patients with 30-day follow-up in the ongoing EXPAND CAP show 91% of donor hearts were utilized, a 2.4% incidence of severe PGD, and 100% 30-day survival.
The sponsor and the FDA clashed over changes made to the trial after the PMA was submitted, the appropriateness of the effectiveness outcome, and claims by the FDA that there was substantial overlap in demographic characteristics between the extended criteria donor hearts in the EXPAND trials and the standard criteria donor hearts in PROCEED II.
TransMedics previously submitted a PMA based on PROCEED II but it noted in submitted documents that it was withdrawn because of “fundamental disagreements with FDA” on the interpretation of a post hoc analysis with United Network for Organ Sharing registry data that identified increased all-cause mortality risk but comparable cardiac-related mortality in patients with OCS hearts.
During the marathon hearing, FDA officials presented several post hoc analyses, including one stratified by donor inclusion criteria, in which 30-day survival estimates were worse in recipients of single-criterion organs than for those receiving donor organs with multiple inclusion criteria (85% vs. 91.4%). In a second analysis, 2-year point estimates of survival also trended lower with donor organs having only one extended criterion.
Reported EXPAND CAP 6- and 12-month survival estimates were 100% and 93%, respectively, which was higher than EXPAND (93% and 84%), but there was substantial censoring (>50%) at 6 months and beyond, FDA officials said.
When EXPAND and CAP data were pooled, modeled survival curves shifted upward but there was a substantial site effect, with a single site contributing 46% of data, which may affect generalizability of the results, they noted.
“I voted yes for safety, no for efficacy, and no for approval and I’d just like to say I found this to be the most difficult vote in my experience on this panel,” John Hirshfeld, MD, University of Pennsylvania, Philadelphia, said. “I was very concerned that the PROCEED data suggests a possible harm, and in the absence of an interpretable comparator for the EXPAND trial, it’s really not possible to decide if there’s efficacy.”
Keith B. Allen, MD, director of surgical research at Saint Luke’s Hospital of Kansas City (Mo.), said, “I voted no on safety; I’m not going to give the company a pass. I think their animal data was sorely lacking and a lot of issues over the last 10 years could have been addressed with some key animal studies.
“For efficacy and risk/benefit, I voted yes for both,” he said. “Had this been standard of care and only PROCEED II, I would have voted no, but I do think there are a lot of hearts that go in the bucket and this is a challenging population.”
More than a dozen physicians and patients spoke at the open public hearing about the potential for the device to expand donor heart utilization, including a recipient whose own father died while waiting on the transplant list. Only about 3 out of every 10 donated hearts are used for transplant. To ensure fair access, particularly for patients in rural areas, federal changes in 2020 mandate that organs be allocated to the sickest patients first.
Data showed that the OCS Heart System was associated with shorter waiting list times, compared with U.S. averages but longer preservation times than cold static preservation.
In all, 13% of accepted donor organs were subsequently turned down after OCS heart preservation. Lactate levels were cited as the principal reason for turn-down but, FDA officials said, the validity of using lactate as a marker for transplantability is unclear.
Pathologic analysis of OCS Heart turned-down donor hearts with stable antemortem hemodynamics, normal or near-normal anatomy and normal ventricular function by echocardiography, and autopsy findings of acute diffuse or multifocal myocardial damage “suggest that in an important proportion of cases the OCS Heart system did not provide effective organ preservation or its use caused severe myocardial damage to what might have been an acceptable graft for transplant,” said Andrew Farb, MD, chief medical officer of the FDA’s Office of Cardiovascular Devices.
Proposed indication
In the present PMA, the OCS Heart System is indicated for donor hearts with one or more of the following characteristics: an expected cross-clamp or ischemic time of at least 4 hours because of donor or recipient characteristics; or an expected total cross-clamp time of at least 2 hours plus one of the following risk factors: donor age 55 or older, history of cardiac arrest and downtime of at least 20 minutes, history of alcoholism, history of diabetes, donor ejection fraction of 40%-50%,history of left ventricular hypertrophy, and donor angiogram with luminal irregularities but no significant coronary artery disease
Several members voiced concern about “indication creep” should the device be approved by the FDA, and highlighted the 2-hour cross-clamp time plus wide-ranging risk factors.

“I’m a surgeon and I voted no on all three counts,” said Murray H. Kwon, MD, Ronald Reagan University of California, Los Angeles Medical Center. “As far as risk/benefit, if it was just limited to one group – the 4-hour plus – I would say yes, but if you’re going to tell me that there’s a risk/benefit for the 2-hour with the alcoholic, I don’t know how that was proved in anything.”
Dr. Kwon was also troubled by lack of proper controls and by the one quarter of patients who ended up on mechanical circulatory support in the first 30 days after transplant. “I find that highly aberrant.”
Joaquin E. Cigarroa, MD, head of cardiovascular medicine, Oregon Health & Science University, Portland, said the unmet need for patients with refractory, end-stage heart failure is challenging and quite emotional, but also voted no across the board, citing concerns about a lack of comparator in the EXPAND trials and overall out-of-body ischemic time.
“As it relates to risk/benefit, I thought long and hard about voting yes despite all the unknowns because of this emotion, but ultimately I voted no because of the secondary 2-hours plus alcoholism, diabetes, or minor coronary disease, in which the ischemic burden and ongoing lactate production concern me,” he said.
Although the panel decision is nonbinding, there was strong support from the committee members for a randomized, postapproval trial and more complete animal studies.
A version of this article first appeared on Medscape.com.
After more than 10 hours of intense debate, a Food and Drug Administration advisory panel gave its support to a premarket approval application (PMA) for the TransMedics Organ Care System (OCS) Heart system.
The OCS Heart is a portable extracorporeal perfusion and monitoring system designed to keep a donor heart in a normothermic, beating state. The “heart in a box” technology allows donor hearts to be transported across longer distances than is possible with standard cold storage, which can safely preserve donor hearts for about 4 hours.
The Circulatory System Devices Panel of the Medical Devices Advisory Committee voted 12 to 5, with 1 abstention, that the benefits of the OCS Heart System outweigh its risks.
The panel voted in favor of the OCS Heart being effective (10 yes, 6 no, and 2 abstaining) and safe (9 yes, 7 no, 2 abstaining) but not without mixed feelings.
James Blankenship, MD, a cardiologist at the University of New Mexico, Albuquerque, voted yes to all three questions but said: “If it had been compared to standard of care, I would have voted no to all three. But if it’s compared to getting an [left ventricular assist device] LVAD or not getting a heart at all, I would say the benefits outweigh the risks.”
Marc R. Katz, MD, chief of cardiothoracic surgery, Medical University of South Carolina, Charleston, also gave universal support, noting that the rate of heart transplantations has been flat for years. “This is a big step forward toward being able to expand that number. Now all that said, it obviously was a less-than-perfect study and I do think there needs to be some constraints put on the utilization.”
The panel reviewed data from the single-arm OCS Heart EXPAND trial and associated EXPAND Continued Access Protocol (CAP), as well the sponsor’s first OCS Heart trial, PROCEED II.
EXPAND met its effectiveness endpoint, with 88% of donor hearts successfully transplanted, an 8% incidence of severe primary graft dysfunction (PGD) 24 hours after transplantation, and 94.6% survival at 30 days.
Data from 41 patients with 30-day follow-up in the ongoing EXPAND CAP show 91% of donor hearts were utilized, a 2.4% incidence of severe PGD, and 100% 30-day survival.
The sponsor and the FDA clashed over changes made to the trial after the PMA was submitted, the appropriateness of the effectiveness outcome, and claims by the FDA that there was substantial overlap in demographic characteristics between the extended criteria donor hearts in the EXPAND trials and the standard criteria donor hearts in PROCEED II.
TransMedics previously submitted a PMA based on PROCEED II but it noted in submitted documents that it was withdrawn because of “fundamental disagreements with FDA” on the interpretation of a post hoc analysis with United Network for Organ Sharing registry data that identified increased all-cause mortality risk but comparable cardiac-related mortality in patients with OCS hearts.
During the marathon hearing, FDA officials presented several post hoc analyses, including one stratified by donor inclusion criteria, in which 30-day survival estimates were worse in recipients of single-criterion organs than for those receiving donor organs with multiple inclusion criteria (85% vs. 91.4%). In a second analysis, 2-year point estimates of survival also trended lower with donor organs having only one extended criterion.
Reported EXPAND CAP 6- and 12-month survival estimates were 100% and 93%, respectively, which was higher than EXPAND (93% and 84%), but there was substantial censoring (>50%) at 6 months and beyond, FDA officials said.
When EXPAND and CAP data were pooled, modeled survival curves shifted upward but there was a substantial site effect, with a single site contributing 46% of data, which may affect generalizability of the results, they noted.
“I voted yes for safety, no for efficacy, and no for approval and I’d just like to say I found this to be the most difficult vote in my experience on this panel,” John Hirshfeld, MD, University of Pennsylvania, Philadelphia, said. “I was very concerned that the PROCEED data suggests a possible harm, and in the absence of an interpretable comparator for the EXPAND trial, it’s really not possible to decide if there’s efficacy.”
Keith B. Allen, MD, director of surgical research at Saint Luke’s Hospital of Kansas City (Mo.), said, “I voted no on safety; I’m not going to give the company a pass. I think their animal data was sorely lacking and a lot of issues over the last 10 years could have been addressed with some key animal studies.
“For efficacy and risk/benefit, I voted yes for both,” he said. “Had this been standard of care and only PROCEED II, I would have voted no, but I do think there are a lot of hearts that go in the bucket and this is a challenging population.”
More than a dozen physicians and patients spoke at the open public hearing about the potential for the device to expand donor heart utilization, including a recipient whose own father died while waiting on the transplant list. Only about 3 out of every 10 donated hearts are used for transplant. To ensure fair access, particularly for patients in rural areas, federal changes in 2020 mandate that organs be allocated to the sickest patients first.
Data showed that the OCS Heart System was associated with shorter waiting list times, compared with U.S. averages but longer preservation times than cold static preservation.
In all, 13% of accepted donor organs were subsequently turned down after OCS heart preservation. Lactate levels were cited as the principal reason for turn-down but, FDA officials said, the validity of using lactate as a marker for transplantability is unclear.
Pathologic analysis of OCS Heart turned-down donor hearts with stable antemortem hemodynamics, normal or near-normal anatomy and normal ventricular function by echocardiography, and autopsy findings of acute diffuse or multifocal myocardial damage “suggest that in an important proportion of cases the OCS Heart system did not provide effective organ preservation or its use caused severe myocardial damage to what might have been an acceptable graft for transplant,” said Andrew Farb, MD, chief medical officer of the FDA’s Office of Cardiovascular Devices.
Proposed indication
In the present PMA, the OCS Heart System is indicated for donor hearts with one or more of the following characteristics: an expected cross-clamp or ischemic time of at least 4 hours because of donor or recipient characteristics; or an expected total cross-clamp time of at least 2 hours plus one of the following risk factors: donor age 55 or older, history of cardiac arrest and downtime of at least 20 minutes, history of alcoholism, history of diabetes, donor ejection fraction of 40%-50%,history of left ventricular hypertrophy, and donor angiogram with luminal irregularities but no significant coronary artery disease
Several members voiced concern about “indication creep” should the device be approved by the FDA, and highlighted the 2-hour cross-clamp time plus wide-ranging risk factors.
“I’m a surgeon and I voted no on all three counts,” said Murray H. Kwon, MD, Ronald Reagan University of California, Los Angeles Medical Center. “As far as risk/benefit, if it was just limited to one group – the 4-hour plus – I would say yes, but if you’re going to tell me that there’s a risk/benefit for the 2-hour with the alcoholic, I don’t know how that was proved in anything.”
Dr. Kwon was also troubled by lack of proper controls and by the one quarter of patients who ended up on mechanical circulatory support in the first 30 days after transplant. “I find that highly aberrant.”
Joaquin E. Cigarroa, MD, head of cardiovascular medicine, Oregon Health & Science University, Portland, said the unmet need for patients with refractory, end-stage heart failure is challenging and quite emotional, but also voted no across the board, citing concerns about a lack of comparator in the EXPAND trials and overall out-of-body ischemic time.
“As it relates to risk/benefit, I thought long and hard about voting yes despite all the unknowns because of this emotion, but ultimately I voted no because of the secondary 2-hours plus alcoholism, diabetes, or minor coronary disease, in which the ischemic burden and ongoing lactate production concern me,” he said.
Although the panel decision is nonbinding, there was strong support from the committee members for a randomized, postapproval trial and more complete animal studies.
A version of this article first appeared on Medscape.com.
After more than 10 hours of intense debate, a Food and Drug Administration advisory panel gave its support to a premarket approval application (PMA) for the TransMedics Organ Care System (OCS) Heart system.
The OCS Heart is a portable extracorporeal perfusion and monitoring system designed to keep a donor heart in a normothermic, beating state. The “heart in a box” technology allows donor hearts to be transported across longer distances than is possible with standard cold storage, which can safely preserve donor hearts for about 4 hours.
The Circulatory System Devices Panel of the Medical Devices Advisory Committee voted 12 to 5, with 1 abstention, that the benefits of the OCS Heart System outweigh its risks.
The panel voted in favor of the OCS Heart being effective (10 yes, 6 no, and 2 abstaining) and safe (9 yes, 7 no, 2 abstaining) but not without mixed feelings.
James Blankenship, MD, a cardiologist at the University of New Mexico, Albuquerque, voted yes to all three questions but said: “If it had been compared to standard of care, I would have voted no to all three. But if it’s compared to getting an [left ventricular assist device] LVAD or not getting a heart at all, I would say the benefits outweigh the risks.”
Marc R. Katz, MD, chief of cardiothoracic surgery, Medical University of South Carolina, Charleston, also gave universal support, noting that the rate of heart transplantations has been flat for years. “This is a big step forward toward being able to expand that number. Now all that said, it obviously was a less-than-perfect study and I do think there needs to be some constraints put on the utilization.”
The panel reviewed data from the single-arm OCS Heart EXPAND trial and associated EXPAND Continued Access Protocol (CAP), as well the sponsor’s first OCS Heart trial, PROCEED II.
EXPAND met its effectiveness endpoint, with 88% of donor hearts successfully transplanted, an 8% incidence of severe primary graft dysfunction (PGD) 24 hours after transplantation, and 94.6% survival at 30 days.
Data from 41 patients with 30-day follow-up in the ongoing EXPAND CAP show 91% of donor hearts were utilized, a 2.4% incidence of severe PGD, and 100% 30-day survival.
The sponsor and the FDA clashed over changes made to the trial after the PMA was submitted, the appropriateness of the effectiveness outcome, and claims by the FDA that there was substantial overlap in demographic characteristics between the extended criteria donor hearts in the EXPAND trials and the standard criteria donor hearts in PROCEED II.
TransMedics previously submitted a PMA based on PROCEED II but it noted in submitted documents that it was withdrawn because of “fundamental disagreements with FDA” on the interpretation of a post hoc analysis with United Network for Organ Sharing registry data that identified increased all-cause mortality risk but comparable cardiac-related mortality in patients with OCS hearts.
During the marathon hearing, FDA officials presented several post hoc analyses, including one stratified by donor inclusion criteria, in which 30-day survival estimates were worse in recipients of single-criterion organs than for those receiving donor organs with multiple inclusion criteria (85% vs. 91.4%). In a second analysis, 2-year point estimates of survival also trended lower with donor organs having only one extended criterion.
Reported EXPAND CAP 6- and 12-month survival estimates were 100% and 93%, respectively, which was higher than EXPAND (93% and 84%), but there was substantial censoring (>50%) at 6 months and beyond, FDA officials said.
When EXPAND and CAP data were pooled, modeled survival curves shifted upward but there was a substantial site effect, with a single site contributing 46% of data, which may affect generalizability of the results, they noted.
“I voted yes for safety, no for efficacy, and no for approval and I’d just like to say I found this to be the most difficult vote in my experience on this panel,” John Hirshfeld, MD, University of Pennsylvania, Philadelphia, said. “I was very concerned that the PROCEED data suggests a possible harm, and in the absence of an interpretable comparator for the EXPAND trial, it’s really not possible to decide if there’s efficacy.”
Keith B. Allen, MD, director of surgical research at Saint Luke’s Hospital of Kansas City (Mo.), said, “I voted no on safety; I’m not going to give the company a pass. I think their animal data was sorely lacking and a lot of issues over the last 10 years could have been addressed with some key animal studies.
“For efficacy and risk/benefit, I voted yes for both,” he said. “Had this been standard of care and only PROCEED II, I would have voted no, but I do think there are a lot of hearts that go in the bucket and this is a challenging population.”
More than a dozen physicians and patients spoke at the open public hearing about the potential for the device to expand donor heart utilization, including a recipient whose own father died while waiting on the transplant list. Only about 3 out of every 10 donated hearts are used for transplant. To ensure fair access, particularly for patients in rural areas, federal changes in 2020 mandate that organs be allocated to the sickest patients first.
Data showed that the OCS Heart System was associated with shorter waiting list times, compared with U.S. averages but longer preservation times than cold static preservation.
In all, 13% of accepted donor organs were subsequently turned down after OCS heart preservation. Lactate levels were cited as the principal reason for turn-down but, FDA officials said, the validity of using lactate as a marker for transplantability is unclear.
Pathologic analysis of OCS Heart turned-down donor hearts with stable antemortem hemodynamics, normal or near-normal anatomy and normal ventricular function by echocardiography, and autopsy findings of acute diffuse or multifocal myocardial damage “suggest that in an important proportion of cases the OCS Heart system did not provide effective organ preservation or its use caused severe myocardial damage to what might have been an acceptable graft for transplant,” said Andrew Farb, MD, chief medical officer of the FDA’s Office of Cardiovascular Devices.
Proposed indication
In the present PMA, the OCS Heart System is indicated for donor hearts with one or more of the following characteristics: an expected cross-clamp or ischemic time of at least 4 hours because of donor or recipient characteristics; or an expected total cross-clamp time of at least 2 hours plus one of the following risk factors: donor age 55 or older, history of cardiac arrest and downtime of at least 20 minutes, history of alcoholism, history of diabetes, donor ejection fraction of 40%-50%,history of left ventricular hypertrophy, and donor angiogram with luminal irregularities but no significant coronary artery disease
Several members voiced concern about “indication creep” should the device be approved by the FDA, and highlighted the 2-hour cross-clamp time plus wide-ranging risk factors.
“I’m a surgeon and I voted no on all three counts,” said Murray H. Kwon, MD, Ronald Reagan University of California, Los Angeles Medical Center. “As far as risk/benefit, if it was just limited to one group – the 4-hour plus – I would say yes, but if you’re going to tell me that there’s a risk/benefit for the 2-hour with the alcoholic, I don’t know how that was proved in anything.”
Dr. Kwon was also troubled by lack of proper controls and by the one quarter of patients who ended up on mechanical circulatory support in the first 30 days after transplant. “I find that highly aberrant.”
Joaquin E. Cigarroa, MD, head of cardiovascular medicine, Oregon Health & Science University, Portland, said the unmet need for patients with refractory, end-stage heart failure is challenging and quite emotional, but also voted no across the board, citing concerns about a lack of comparator in the EXPAND trials and overall out-of-body ischemic time.
“As it relates to risk/benefit, I thought long and hard about voting yes despite all the unknowns because of this emotion, but ultimately I voted no because of the secondary 2-hours plus alcoholism, diabetes, or minor coronary disease, in which the ischemic burden and ongoing lactate production concern me,” he said.
Although the panel decision is nonbinding, there was strong support from the committee members for a randomized, postapproval trial and more complete animal studies.
A version of this article first appeared on Medscape.com.
Study: Good overall survival in older patients after liver transplant for HCC
Judicious organ matching for older liver transplant candidates with hepatocellular carcinoma (HCC) leads to survival outcomes similar to those in younger recipients, a case review suggests.
Overall survival (OS) rates among transplant recipients included in a prospective institutional database were 85.5% and 84% at 3 years after liver transplant in patients aged 65 years and under and those over 65 years, respectively. The 5-year survival rates were 73.9% and 77%, respectively (P = .26), Ola Ahmed, MD, of the department of abdominal organ transplantation surgery at Washington University, St. Louis, and colleagues found.
The investigators looked at 1,629 patients diagnosed with HCC between Jan. 1, 2002, and Dec. 31, 2019 of whom 700 were considered for curative surgery, including transplant in 538, and resection in 162.
The patients had a mean age of 62.8 years. Those older than 65 years were less likely to be considered or listed for transplant (27% vs. 73%, P < .01), although oncologic staging and delisting rates were similar in both groups. “This observation still holds true after controlling for other variables, including viral hepatitis and gender in the multivariable analysis (adjusted odds ratio, 0.365),” the investigators reported in the Journal of the American College of Surgeons.
The findings were also reported at the 2020 virtual Western Surgical Association 128th Scientific Session in November.
The issue of resection
Surgical intervention occurred in 597 patients, including 392 and 205 aged 65 years and younger and over 65 years, respectively.
OS was lower among patients who underwent resection, compared with the liver transplant recipients, but was similar in the older and younger age groups (3-year OS, 59% vs. 64.8% and 5-year OS, 44.8% vs. 49%; P = .13). No differences were noted in the development of local or distant metastatic disease after transplant or resection.
The two age groups had comparable ICU stays (2 days) and total hospital length of stay (6 days). There were no differences in 30- and 90-day hospital readmissions, they noted.
“On additional age analysis, 65% of transplanted patients over 65 years are currently alive and were disease free at the end of the study period, compared to only 18% of their resected counterparts (P < .01),” they wrote.
Justifying transplant
The findings are notable because despite the effectiveness of transplant as an alternative treatment for unresectable HCC, older patients are often excluded from consideration for transplant. Most studies over the past 15 years have focused on patients aged under 60 years and the ability to extrapolate results to older patients has been limited. Further, results have been conflicting in older patients, the authors explained.
“This is particularly apposite at this time with prolonged life expectancy and the growing interest in improving cancer survivorship,” they noted, adding that “there is logic in challenging existing gold standards and traditional norms with real-life medical practice.
Indeed, the current findings suggest – perhaps contrary to common perceptions – that transplant in carefully selected patients “can be justified in older age groups and provide clinically meaningful and longer survival benefits,” they said, adding that “discussions should be guided by the potential for unfair age discriminations and precise terminology of physiologic rather than actual age.
“Such insights highlight the continued need for quality improvement in the surgical management of older patients, raising questions regarding current resource utilization among different age groups and how age can influence patterns of cancer care,” they concluded.
The authors reported having no disclosures.
Judicious organ matching for older liver transplant candidates with hepatocellular carcinoma (HCC) leads to survival outcomes similar to those in younger recipients, a case review suggests.
Overall survival (OS) rates among transplant recipients included in a prospective institutional database were 85.5% and 84% at 3 years after liver transplant in patients aged 65 years and under and those over 65 years, respectively. The 5-year survival rates were 73.9% and 77%, respectively (P = .26), Ola Ahmed, MD, of the department of abdominal organ transplantation surgery at Washington University, St. Louis, and colleagues found.
The investigators looked at 1,629 patients diagnosed with HCC between Jan. 1, 2002, and Dec. 31, 2019 of whom 700 were considered for curative surgery, including transplant in 538, and resection in 162.
The patients had a mean age of 62.8 years. Those older than 65 years were less likely to be considered or listed for transplant (27% vs. 73%, P < .01), although oncologic staging and delisting rates were similar in both groups. “This observation still holds true after controlling for other variables, including viral hepatitis and gender in the multivariable analysis (adjusted odds ratio, 0.365),” the investigators reported in the Journal of the American College of Surgeons.
The findings were also reported at the 2020 virtual Western Surgical Association 128th Scientific Session in November.
The issue of resection
Surgical intervention occurred in 597 patients, including 392 and 205 aged 65 years and younger and over 65 years, respectively.
OS was lower among patients who underwent resection, compared with the liver transplant recipients, but was similar in the older and younger age groups (3-year OS, 59% vs. 64.8% and 5-year OS, 44.8% vs. 49%; P = .13). No differences were noted in the development of local or distant metastatic disease after transplant or resection.
The two age groups had comparable ICU stays (2 days) and total hospital length of stay (6 days). There were no differences in 30- and 90-day hospital readmissions, they noted.
“On additional age analysis, 65% of transplanted patients over 65 years are currently alive and were disease free at the end of the study period, compared to only 18% of their resected counterparts (P < .01),” they wrote.
Justifying transplant
The findings are notable because despite the effectiveness of transplant as an alternative treatment for unresectable HCC, older patients are often excluded from consideration for transplant. Most studies over the past 15 years have focused on patients aged under 60 years and the ability to extrapolate results to older patients has been limited. Further, results have been conflicting in older patients, the authors explained.
“This is particularly apposite at this time with prolonged life expectancy and the growing interest in improving cancer survivorship,” they noted, adding that “there is logic in challenging existing gold standards and traditional norms with real-life medical practice.
Indeed, the current findings suggest – perhaps contrary to common perceptions – that transplant in carefully selected patients “can be justified in older age groups and provide clinically meaningful and longer survival benefits,” they said, adding that “discussions should be guided by the potential for unfair age discriminations and precise terminology of physiologic rather than actual age.
“Such insights highlight the continued need for quality improvement in the surgical management of older patients, raising questions regarding current resource utilization among different age groups and how age can influence patterns of cancer care,” they concluded.
The authors reported having no disclosures.
Judicious organ matching for older liver transplant candidates with hepatocellular carcinoma (HCC) leads to survival outcomes similar to those in younger recipients, a case review suggests.
Overall survival (OS) rates among transplant recipients included in a prospective institutional database were 85.5% and 84% at 3 years after liver transplant in patients aged 65 years and under and those over 65 years, respectively. The 5-year survival rates were 73.9% and 77%, respectively (P = .26), Ola Ahmed, MD, of the department of abdominal organ transplantation surgery at Washington University, St. Louis, and colleagues found.
The investigators looked at 1,629 patients diagnosed with HCC between Jan. 1, 2002, and Dec. 31, 2019 of whom 700 were considered for curative surgery, including transplant in 538, and resection in 162.
The patients had a mean age of 62.8 years. Those older than 65 years were less likely to be considered or listed for transplant (27% vs. 73%, P < .01), although oncologic staging and delisting rates were similar in both groups. “This observation still holds true after controlling for other variables, including viral hepatitis and gender in the multivariable analysis (adjusted odds ratio, 0.365),” the investigators reported in the Journal of the American College of Surgeons.
The findings were also reported at the 2020 virtual Western Surgical Association 128th Scientific Session in November.
The issue of resection
Surgical intervention occurred in 597 patients, including 392 and 205 aged 65 years and younger and over 65 years, respectively.
OS was lower among patients who underwent resection, compared with the liver transplant recipients, but was similar in the older and younger age groups (3-year OS, 59% vs. 64.8% and 5-year OS, 44.8% vs. 49%; P = .13). No differences were noted in the development of local or distant metastatic disease after transplant or resection.
The two age groups had comparable ICU stays (2 days) and total hospital length of stay (6 days). There were no differences in 30- and 90-day hospital readmissions, they noted.
“On additional age analysis, 65% of transplanted patients over 65 years are currently alive and were disease free at the end of the study period, compared to only 18% of their resected counterparts (P < .01),” they wrote.
Justifying transplant
The findings are notable because despite the effectiveness of transplant as an alternative treatment for unresectable HCC, older patients are often excluded from consideration for transplant. Most studies over the past 15 years have focused on patients aged under 60 years and the ability to extrapolate results to older patients has been limited. Further, results have been conflicting in older patients, the authors explained.
“This is particularly apposite at this time with prolonged life expectancy and the growing interest in improving cancer survivorship,” they noted, adding that “there is logic in challenging existing gold standards and traditional norms with real-life medical practice.
Indeed, the current findings suggest – perhaps contrary to common perceptions – that transplant in carefully selected patients “can be justified in older age groups and provide clinically meaningful and longer survival benefits,” they said, adding that “discussions should be guided by the potential for unfair age discriminations and precise terminology of physiologic rather than actual age.
“Such insights highlight the continued need for quality improvement in the surgical management of older patients, raising questions regarding current resource utilization among different age groups and how age can influence patterns of cancer care,” they concluded.
The authors reported having no disclosures.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF SURGEONS
Poor survival with COVID in patients who have had HSCT
Among individuals who have received a hematopoietic stem cell transplant (HSCT), often used in the treatment of blood cancers, rates of survival are poor for those who develop COVID-19.
The probability of survival 30 days after being diagnosed with COVID-19 is only 68% for persons who have received an allogeneic HSCT and 67% for autologous HSCT recipients, according to new data from the Center for International Blood and Marrow Transplant Research.
These findings underscore the need for “stringent surveillance and aggressive treatment measures” in this population, Akshay Sharma, MBBS, of St. Jude Children’s Research Hospital, Memphis, and colleagues wrote.
The findings were published online March 1, 2021, in The Lancet Haematology.
The study is “of importance for physicians caring for HSCT recipients worldwide,” Mathieu Leclerc, MD, and Sébastien Maury, MD, Hôpital Henri Mondor, Créteil, France, commented in an accompanying editorial.
Study details
For their study, Dr. Sharma and colleagues analyzed outcomes for all HSCT recipients who developed COVID-19 and whose cases were reported to the CIBMTR. Of 318 such patients, 184 had undergone allogeneic HSCT, and 134 had undergone autologous HSCT.
Overall, about half of these patients (49%) had mild COVID-19.
Severe COVID-19 that required mechanical ventilation developed in 15% and 13% of the allogeneic and autologous HSCT recipients, respectively.
About one-fifth of patients died: 22% and 19% of allogeneic and autologous HSCT recipients, respectively.
Factors associated with greater mortality risk included age of 50 years or older (hazard ratio, 2.53), male sex (HR, 3.53), and development of COVID-19 within 12 months of undergoing HSCT (HR, 2.67).
Among autologous HSCT recipients, lymphoma was associated with higher mortality risk in comparison with a plasma cell disorder or myeloma (HR, 2.41), the authors noted.
“Two important messages can be drawn from the results reported by Sharma and colleagues,” Dr. Leclerc and Dr. Maury wrote in their editorial. “The first is the confirmation that the prognosis of COVID-19 is particularly poor in HSCT recipients, and that its prevention, in the absence of any specific curative treatment with sufficient efficacy, should be at the forefront of concerns.”
The second relates to the risk factors for death among HSCT recipients who develop COVID-19. In addition to previously known risk factors, such as age and gender, the investigators identified transplant-specific factors potentially associated with prognosis – namely, the nearly threefold increase in death among allogeneic HSCT recipients who develop COVID-19 within 12 months of transplant, they explained.
However, the findings are limited by a substantial amount of missing data, short follow-up, and the possibility of selection bias, they noted.
“Further large and well-designed studies with longer follow-up are needed to confirm and refine the results,” the editorialists wrote.
“[A] better understanding of the distinctive features of COVID-19 infection in HSCT recipients will be a necessary and essential step toward improvement of the remarkably poor prognosis observed in this setting,” they added.
The study was funded by the American Society of Hematology; the Leukemia and Lymphoma Society; the National Cancer Institute; the National Heart, Lung and Blood Institute; the National Institute of Allergy and Infectious Diseases; the National Institutes of Health; the Health Resources and Services Administration; and the Office of Naval Research. Dr. Sharma receives support for the conduct of industry-sponsored trials from Vertex Pharmaceuticals, CRISPR Therapeutics, and Novartis and consulting fees from Spotlight Therapeutics. Dr. Leclerc and Dr. Maury disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Among individuals who have received a hematopoietic stem cell transplant (HSCT), often used in the treatment of blood cancers, rates of survival are poor for those who develop COVID-19.
The probability of survival 30 days after being diagnosed with COVID-19 is only 68% for persons who have received an allogeneic HSCT and 67% for autologous HSCT recipients, according to new data from the Center for International Blood and Marrow Transplant Research.
These findings underscore the need for “stringent surveillance and aggressive treatment measures” in this population, Akshay Sharma, MBBS, of St. Jude Children’s Research Hospital, Memphis, and colleagues wrote.
The findings were published online March 1, 2021, in The Lancet Haematology.
The study is “of importance for physicians caring for HSCT recipients worldwide,” Mathieu Leclerc, MD, and Sébastien Maury, MD, Hôpital Henri Mondor, Créteil, France, commented in an accompanying editorial.
Study details
For their study, Dr. Sharma and colleagues analyzed outcomes for all HSCT recipients who developed COVID-19 and whose cases were reported to the CIBMTR. Of 318 such patients, 184 had undergone allogeneic HSCT, and 134 had undergone autologous HSCT.
Overall, about half of these patients (49%) had mild COVID-19.
Severe COVID-19 that required mechanical ventilation developed in 15% and 13% of the allogeneic and autologous HSCT recipients, respectively.
About one-fifth of patients died: 22% and 19% of allogeneic and autologous HSCT recipients, respectively.
Factors associated with greater mortality risk included age of 50 years or older (hazard ratio, 2.53), male sex (HR, 3.53), and development of COVID-19 within 12 months of undergoing HSCT (HR, 2.67).
Among autologous HSCT recipients, lymphoma was associated with higher mortality risk in comparison with a plasma cell disorder or myeloma (HR, 2.41), the authors noted.
“Two important messages can be drawn from the results reported by Sharma and colleagues,” Dr. Leclerc and Dr. Maury wrote in their editorial. “The first is the confirmation that the prognosis of COVID-19 is particularly poor in HSCT recipients, and that its prevention, in the absence of any specific curative treatment with sufficient efficacy, should be at the forefront of concerns.”
The second relates to the risk factors for death among HSCT recipients who develop COVID-19. In addition to previously known risk factors, such as age and gender, the investigators identified transplant-specific factors potentially associated with prognosis – namely, the nearly threefold increase in death among allogeneic HSCT recipients who develop COVID-19 within 12 months of transplant, they explained.
However, the findings are limited by a substantial amount of missing data, short follow-up, and the possibility of selection bias, they noted.
“Further large and well-designed studies with longer follow-up are needed to confirm and refine the results,” the editorialists wrote.
“[A] better understanding of the distinctive features of COVID-19 infection in HSCT recipients will be a necessary and essential step toward improvement of the remarkably poor prognosis observed in this setting,” they added.
The study was funded by the American Society of Hematology; the Leukemia and Lymphoma Society; the National Cancer Institute; the National Heart, Lung and Blood Institute; the National Institute of Allergy and Infectious Diseases; the National Institutes of Health; the Health Resources and Services Administration; and the Office of Naval Research. Dr. Sharma receives support for the conduct of industry-sponsored trials from Vertex Pharmaceuticals, CRISPR Therapeutics, and Novartis and consulting fees from Spotlight Therapeutics. Dr. Leclerc and Dr. Maury disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Among individuals who have received a hematopoietic stem cell transplant (HSCT), often used in the treatment of blood cancers, rates of survival are poor for those who develop COVID-19.
The probability of survival 30 days after being diagnosed with COVID-19 is only 68% for persons who have received an allogeneic HSCT and 67% for autologous HSCT recipients, according to new data from the Center for International Blood and Marrow Transplant Research.
These findings underscore the need for “stringent surveillance and aggressive treatment measures” in this population, Akshay Sharma, MBBS, of St. Jude Children’s Research Hospital, Memphis, and colleagues wrote.
The findings were published online March 1, 2021, in The Lancet Haematology.
The study is “of importance for physicians caring for HSCT recipients worldwide,” Mathieu Leclerc, MD, and Sébastien Maury, MD, Hôpital Henri Mondor, Créteil, France, commented in an accompanying editorial.
Study details
For their study, Dr. Sharma and colleagues analyzed outcomes for all HSCT recipients who developed COVID-19 and whose cases were reported to the CIBMTR. Of 318 such patients, 184 had undergone allogeneic HSCT, and 134 had undergone autologous HSCT.
Overall, about half of these patients (49%) had mild COVID-19.
Severe COVID-19 that required mechanical ventilation developed in 15% and 13% of the allogeneic and autologous HSCT recipients, respectively.
About one-fifth of patients died: 22% and 19% of allogeneic and autologous HSCT recipients, respectively.
Factors associated with greater mortality risk included age of 50 years or older (hazard ratio, 2.53), male sex (HR, 3.53), and development of COVID-19 within 12 months of undergoing HSCT (HR, 2.67).
Among autologous HSCT recipients, lymphoma was associated with higher mortality risk in comparison with a plasma cell disorder or myeloma (HR, 2.41), the authors noted.
“Two important messages can be drawn from the results reported by Sharma and colleagues,” Dr. Leclerc and Dr. Maury wrote in their editorial. “The first is the confirmation that the prognosis of COVID-19 is particularly poor in HSCT recipients, and that its prevention, in the absence of any specific curative treatment with sufficient efficacy, should be at the forefront of concerns.”
The second relates to the risk factors for death among HSCT recipients who develop COVID-19. In addition to previously known risk factors, such as age and gender, the investigators identified transplant-specific factors potentially associated with prognosis – namely, the nearly threefold increase in death among allogeneic HSCT recipients who develop COVID-19 within 12 months of transplant, they explained.
However, the findings are limited by a substantial amount of missing data, short follow-up, and the possibility of selection bias, they noted.
“Further large and well-designed studies with longer follow-up are needed to confirm and refine the results,” the editorialists wrote.
“[A] better understanding of the distinctive features of COVID-19 infection in HSCT recipients will be a necessary and essential step toward improvement of the remarkably poor prognosis observed in this setting,” they added.
The study was funded by the American Society of Hematology; the Leukemia and Lymphoma Society; the National Cancer Institute; the National Heart, Lung and Blood Institute; the National Institute of Allergy and Infectious Diseases; the National Institutes of Health; the Health Resources and Services Administration; and the Office of Naval Research. Dr. Sharma receives support for the conduct of industry-sponsored trials from Vertex Pharmaceuticals, CRISPR Therapeutics, and Novartis and consulting fees from Spotlight Therapeutics. Dr. Leclerc and Dr. Maury disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Allo-HSCT plus monoclonal antibody treatment can improve survival in patients with r/r B-ALL
The use of allogeneic hematopoietic stem cell transplantation (allo-HSCT) can improve survival in minimal residual disease (MRD)–negative remission patients with relapsed/refractory (r/r) B-cell acute lymphoblastic leukemia (B-ALL) after the start of monoclonal antibody treatment, according to the results of a landmark analysis presented at the virtual meeting of the European Society for Blood and Marrow Transplantation.
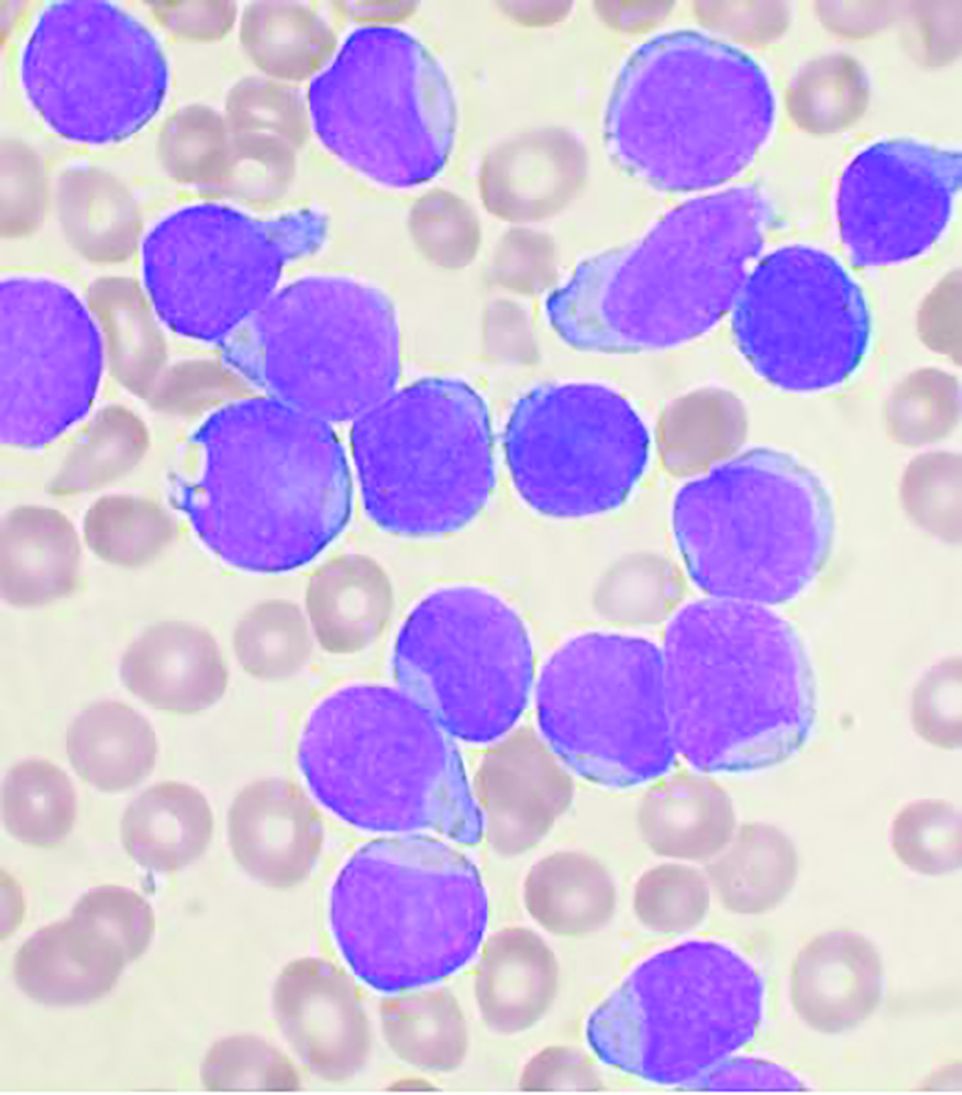
Previous studies have indicated that allo-HSCT improves the results of treatment in r/r B-ALL patients, compared with chemotherapy alone. In addition, it has been found that the monoclonal antibodies (Mab), anti-CD19-blinatumomab and anti-CD22-inotuzumab ozogamicin, induced remission in a significant proportion of such patients.
To determine if the use of allo-HSCT improves the outcome of patients in MRD-negative remission with or without Mab treatment, researchers performed a landmark analysis of 110 patients who achieved MRD-negative status after Mab treatment. The analysis examined results at 2, 4, and 6 months subsequent to the initiation of Mab treatment, according to poster presentation by Inna V. Markova, MD, and colleagues at Pavlov University, Saint Petersburg, Russian Federation.
Study details
The researchers included 110 patients who achieved MRD-negative status outside of clinical trials at a single institution in the analysis. Forty of the patients (36%) were children and 70 (64%) were adults. The median age for all patients was 23 years and the median follow up was 24 months. Fifty-seven (52%) and 53 (48%) patients received Mab for hematological relapse and persistent measurable residual disease or for molecular relapse, respectively. Therapy with Mab alone without subsequent allo-HSCT was used in 36 (31%) patients (30 received blinatumomab and 6 received inotuzumab ozogamicin). A total of 74 (69%) patients received allo-HSCT from a matched related or unrelated donor (MD-HSCT, n = 38) or haploidentical donor (Haplo-HSCT, n = 36). All patients received posttransplantation cyclophosphamide (PTCY)–based graft-versus host disease (GVHD) prophylaxis. Landmark analysis was performed at 2, 4, and 6 months after Mab therapy initiation to determine the effect of allo-HSCT on the outcome and the optimal timing of HSCT. Overall survival and disease-free survival (DFS) were used as outcomes.
Promising results
No significant differences between the MD-HSCT, Mab alone, and Haplo-HSCT groups were observed in 2-month landmark analysis (P = .4 for OS and P =.65 for DFS). However, the 4-month landmark analysis demonstrated superior overall survival and DFS in patients after MD-HSCT, but not Haplo-HSCT, compared with Mab alone: 2-year OS was 75%, 50%, and 27,7% (P = .032) and DFS was 53.5%, 51.3%, and 16.6% (P = .02) for MD-HSCT, Mab alone and Haplo-HSCT groups, respectively. In addition, 6-month analysis showed that there was no benefit from subsequent transplantation, according to the authors, with regard to overall survival (P = .11).
“Our study demonstrated that at least MD-HSCT with PTCY platform improves survival in MRD-negative remission if performed during the first 4 months after Mab initiation. Haplo-HSCT or MD-HSCT beyond 4 months are not associated with improved outcomes in this groups of patients,” the researchers concluded.
The researchers reported they had no conflicts of interest to declare.
The use of allogeneic hematopoietic stem cell transplantation (allo-HSCT) can improve survival in minimal residual disease (MRD)–negative remission patients with relapsed/refractory (r/r) B-cell acute lymphoblastic leukemia (B-ALL) after the start of monoclonal antibody treatment, according to the results of a landmark analysis presented at the virtual meeting of the European Society for Blood and Marrow Transplantation.
Previous studies have indicated that allo-HSCT improves the results of treatment in r/r B-ALL patients, compared with chemotherapy alone. In addition, it has been found that the monoclonal antibodies (Mab), anti-CD19-blinatumomab and anti-CD22-inotuzumab ozogamicin, induced remission in a significant proportion of such patients.
To determine if the use of allo-HSCT improves the outcome of patients in MRD-negative remission with or without Mab treatment, researchers performed a landmark analysis of 110 patients who achieved MRD-negative status after Mab treatment. The analysis examined results at 2, 4, and 6 months subsequent to the initiation of Mab treatment, according to poster presentation by Inna V. Markova, MD, and colleagues at Pavlov University, Saint Petersburg, Russian Federation.
Study details
The researchers included 110 patients who achieved MRD-negative status outside of clinical trials at a single institution in the analysis. Forty of the patients (36%) were children and 70 (64%) were adults. The median age for all patients was 23 years and the median follow up was 24 months. Fifty-seven (52%) and 53 (48%) patients received Mab for hematological relapse and persistent measurable residual disease or for molecular relapse, respectively. Therapy with Mab alone without subsequent allo-HSCT was used in 36 (31%) patients (30 received blinatumomab and 6 received inotuzumab ozogamicin). A total of 74 (69%) patients received allo-HSCT from a matched related or unrelated donor (MD-HSCT, n = 38) or haploidentical donor (Haplo-HSCT, n = 36). All patients received posttransplantation cyclophosphamide (PTCY)–based graft-versus host disease (GVHD) prophylaxis. Landmark analysis was performed at 2, 4, and 6 months after Mab therapy initiation to determine the effect of allo-HSCT on the outcome and the optimal timing of HSCT. Overall survival and disease-free survival (DFS) were used as outcomes.
Promising results
No significant differences between the MD-HSCT, Mab alone, and Haplo-HSCT groups were observed in 2-month landmark analysis (P = .4 for OS and P =.65 for DFS). However, the 4-month landmark analysis demonstrated superior overall survival and DFS in patients after MD-HSCT, but not Haplo-HSCT, compared with Mab alone: 2-year OS was 75%, 50%, and 27,7% (P = .032) and DFS was 53.5%, 51.3%, and 16.6% (P = .02) for MD-HSCT, Mab alone and Haplo-HSCT groups, respectively. In addition, 6-month analysis showed that there was no benefit from subsequent transplantation, according to the authors, with regard to overall survival (P = .11).
“Our study demonstrated that at least MD-HSCT with PTCY platform improves survival in MRD-negative remission if performed during the first 4 months after Mab initiation. Haplo-HSCT or MD-HSCT beyond 4 months are not associated with improved outcomes in this groups of patients,” the researchers concluded.
The researchers reported they had no conflicts of interest to declare.
The use of allogeneic hematopoietic stem cell transplantation (allo-HSCT) can improve survival in minimal residual disease (MRD)–negative remission patients with relapsed/refractory (r/r) B-cell acute lymphoblastic leukemia (B-ALL) after the start of monoclonal antibody treatment, according to the results of a landmark analysis presented at the virtual meeting of the European Society for Blood and Marrow Transplantation.
Previous studies have indicated that allo-HSCT improves the results of treatment in r/r B-ALL patients, compared with chemotherapy alone. In addition, it has been found that the monoclonal antibodies (Mab), anti-CD19-blinatumomab and anti-CD22-inotuzumab ozogamicin, induced remission in a significant proportion of such patients.
To determine if the use of allo-HSCT improves the outcome of patients in MRD-negative remission with or without Mab treatment, researchers performed a landmark analysis of 110 patients who achieved MRD-negative status after Mab treatment. The analysis examined results at 2, 4, and 6 months subsequent to the initiation of Mab treatment, according to poster presentation by Inna V. Markova, MD, and colleagues at Pavlov University, Saint Petersburg, Russian Federation.
Study details
The researchers included 110 patients who achieved MRD-negative status outside of clinical trials at a single institution in the analysis. Forty of the patients (36%) were children and 70 (64%) were adults. The median age for all patients was 23 years and the median follow up was 24 months. Fifty-seven (52%) and 53 (48%) patients received Mab for hematological relapse and persistent measurable residual disease or for molecular relapse, respectively. Therapy with Mab alone without subsequent allo-HSCT was used in 36 (31%) patients (30 received blinatumomab and 6 received inotuzumab ozogamicin). A total of 74 (69%) patients received allo-HSCT from a matched related or unrelated donor (MD-HSCT, n = 38) or haploidentical donor (Haplo-HSCT, n = 36). All patients received posttransplantation cyclophosphamide (PTCY)–based graft-versus host disease (GVHD) prophylaxis. Landmark analysis was performed at 2, 4, and 6 months after Mab therapy initiation to determine the effect of allo-HSCT on the outcome and the optimal timing of HSCT. Overall survival and disease-free survival (DFS) were used as outcomes.
Promising results
No significant differences between the MD-HSCT, Mab alone, and Haplo-HSCT groups were observed in 2-month landmark analysis (P = .4 for OS and P =.65 for DFS). However, the 4-month landmark analysis demonstrated superior overall survival and DFS in patients after MD-HSCT, but not Haplo-HSCT, compared with Mab alone: 2-year OS was 75%, 50%, and 27,7% (P = .032) and DFS was 53.5%, 51.3%, and 16.6% (P = .02) for MD-HSCT, Mab alone and Haplo-HSCT groups, respectively. In addition, 6-month analysis showed that there was no benefit from subsequent transplantation, according to the authors, with regard to overall survival (P = .11).
“Our study demonstrated that at least MD-HSCT with PTCY platform improves survival in MRD-negative remission if performed during the first 4 months after Mab initiation. Haplo-HSCT or MD-HSCT beyond 4 months are not associated with improved outcomes in this groups of patients,” the researchers concluded.
The researchers reported they had no conflicts of interest to declare.
FROM EBMT 2021
Omidubicel improves on umbilical cord blood transplants
Omidubicel, an investigational enriched umbilical cord blood product being developed by Gamida Cell for transplantation in patients with blood cancers, appears to have some advantages over standard umbilical cord blood.
The results come from a global phase 3 trial (NCT02730299) presented at the annual meeting of the European Society for Blood and Bone Marrow Transplantation.
“Transplantation with omidubicel, compared to standard cord blood transplantation, results in faster hematopoietic recovery, fewer infections, and fewer days in hospital,” said coinvestigator Guillermo F. Sanz, MD, PhD, from the Hospital Universitari i Politècnic la Fe in Valencia, Spain.
“Omidubicel should be considered as the new standard of care for patients eligible for umbilical cord blood transplantation,” Dr. Sanz concluded.
Zachariah DeFilipp, MD, from Mass General Cancer Center in Boston, a hematopoietic stem cell transplantation specialist who was not involved in the study, said in an interview that “omidubicel significantly improves the engraftment after transplant, as compared to standard cord blood transplant. For patients that lack an HLA-matched donor, this approach can help overcome the prolonged cytopenias that occur with standard cord blood transplants in adults.”
Gamida Cell plans to submit these data for approval of omidubicel by the Food and Drug Administration in the fourth quarter of 2021.
Omidubicel is also being evaluated in a phase 1/2 clinical study in patients with severe aplastic anemia (NCT03173937).
Expanding possibilities
Although umbilical cord blood stem cell grafts come from a readily available source and show greater tolerance across HLA barriers than other sources (such as bone marrow), the relatively low dose of stem cells in each unit results in delayed hematopoietic recovery, increased transplant-related morbidity and mortality, and longer hospitalizations, Dr. Sanz said.
Omidubicel consists of two cryopreserved fractions from a single cord blood unit. The product contains both noncultured CD133-negative cells, including T cells, and CD133-positive cells that are then expanded ex vivo for 21 days in the presence of nicotinamide.
“Nicotinamide increases stem and progenitor cells, inhibits differentiation and increases migration, bone marrow homing, and engraftment efficiency while preserving cellular functionality and phenotype,” Dr. Sanz explained during his presentation.
In an earlier phase 1/2 trial in 36 patients with high-risk hematologic malignancies, omidubicel was associated with hematopoietic engraftment lasting at least 10 years.
Details of phase 3 trial results
The global phase 3 trial was conducted in 125 patients (aged 13-65 years) with high-risk malignancies, including acute myeloid and lymphoblastic leukemias, myelodysplastic syndrome, chronic myeloid leukemia, lymphomas, and rare leukemias. These patients were all eligible for allogeneic stem cell transplantation but did not have matched donors.
Patients were randomly assigned to receive hematopoietic reconstitution with either omidubicel (n = 52) or standard cord blood (n = 58).
At 42 days of follow-up, the median time to neutrophil engraftment in the intention-to-treat (ITT) population, the primary endpoint, was 12 days with omidubicel versus 22 days with standard cord blood (P < .001).
In the as-treated population – the 108 patients who actually received omidubicel or standard cord blood – median time to engraftment was 10.0 versus 20.5 days, respectively (P < .001).
Rates of neutrophil engraftment at 42 days were 96% with omidubicel versus 89% with standard cord blood.
The secondary endpoint of time-to-platelet engraftment in the ITT population also favored omidubicel, with a cumulative day 42 incidence rate of 55%, compared with 35% with standard cord blood (P = .028).
In the as-treated population, median times to platelet engraftment were 37 days and 50 days, respectively (P = .023). The cumulative rates of platelet engraftment at 100 days of follow-up were 83% and 73%, respectively.
The incidence of grade 2 or 3 bacterial or invasive fungal infections by day 100 in the ITT population was 37% among patients who received omidubicel, compared with 57% for patients who received standard cord blood (P = .027). Viral infections occurred in 10% versus 26% of patients, respectively.
The incidence of acute graft versus host disease at day 100 was similar between treatment groups, and there was no significant difference at 1 year.
Relapse and nonrelapse mortality rates, as well as disease-free and overall survival rates also did not differ between groups.
In the first 100 days post transplant, patients who received omidubicel were alive and out of the hospital for a median of 60.5 days, compared with 48 days for patients who received standard cord blood (P = .005).
The study was funded by Gamida Cell. Dr. Sanz reported receiving research funding from the company and several others, and consulting fees, honoraria, speakers bureau activity, and travel expenses from other companies. Dr. DeFilipp reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Omidubicel, an investigational enriched umbilical cord blood product being developed by Gamida Cell for transplantation in patients with blood cancers, appears to have some advantages over standard umbilical cord blood.
The results come from a global phase 3 trial (NCT02730299) presented at the annual meeting of the European Society for Blood and Bone Marrow Transplantation.
“Transplantation with omidubicel, compared to standard cord blood transplantation, results in faster hematopoietic recovery, fewer infections, and fewer days in hospital,” said coinvestigator Guillermo F. Sanz, MD, PhD, from the Hospital Universitari i Politècnic la Fe in Valencia, Spain.
“Omidubicel should be considered as the new standard of care for patients eligible for umbilical cord blood transplantation,” Dr. Sanz concluded.
Zachariah DeFilipp, MD, from Mass General Cancer Center in Boston, a hematopoietic stem cell transplantation specialist who was not involved in the study, said in an interview that “omidubicel significantly improves the engraftment after transplant, as compared to standard cord blood transplant. For patients that lack an HLA-matched donor, this approach can help overcome the prolonged cytopenias that occur with standard cord blood transplants in adults.”
Gamida Cell plans to submit these data for approval of omidubicel by the Food and Drug Administration in the fourth quarter of 2021.
Omidubicel is also being evaluated in a phase 1/2 clinical study in patients with severe aplastic anemia (NCT03173937).
Expanding possibilities
Although umbilical cord blood stem cell grafts come from a readily available source and show greater tolerance across HLA barriers than other sources (such as bone marrow), the relatively low dose of stem cells in each unit results in delayed hematopoietic recovery, increased transplant-related morbidity and mortality, and longer hospitalizations, Dr. Sanz said.
Omidubicel consists of two cryopreserved fractions from a single cord blood unit. The product contains both noncultured CD133-negative cells, including T cells, and CD133-positive cells that are then expanded ex vivo for 21 days in the presence of nicotinamide.
“Nicotinamide increases stem and progenitor cells, inhibits differentiation and increases migration, bone marrow homing, and engraftment efficiency while preserving cellular functionality and phenotype,” Dr. Sanz explained during his presentation.
In an earlier phase 1/2 trial in 36 patients with high-risk hematologic malignancies, omidubicel was associated with hematopoietic engraftment lasting at least 10 years.
Details of phase 3 trial results
The global phase 3 trial was conducted in 125 patients (aged 13-65 years) with high-risk malignancies, including acute myeloid and lymphoblastic leukemias, myelodysplastic syndrome, chronic myeloid leukemia, lymphomas, and rare leukemias. These patients were all eligible for allogeneic stem cell transplantation but did not have matched donors.
Patients were randomly assigned to receive hematopoietic reconstitution with either omidubicel (n = 52) or standard cord blood (n = 58).
At 42 days of follow-up, the median time to neutrophil engraftment in the intention-to-treat (ITT) population, the primary endpoint, was 12 days with omidubicel versus 22 days with standard cord blood (P < .001).
In the as-treated population – the 108 patients who actually received omidubicel or standard cord blood – median time to engraftment was 10.0 versus 20.5 days, respectively (P < .001).
Rates of neutrophil engraftment at 42 days were 96% with omidubicel versus 89% with standard cord blood.
The secondary endpoint of time-to-platelet engraftment in the ITT population also favored omidubicel, with a cumulative day 42 incidence rate of 55%, compared with 35% with standard cord blood (P = .028).
In the as-treated population, median times to platelet engraftment were 37 days and 50 days, respectively (P = .023). The cumulative rates of platelet engraftment at 100 days of follow-up were 83% and 73%, respectively.
The incidence of grade 2 or 3 bacterial or invasive fungal infections by day 100 in the ITT population was 37% among patients who received omidubicel, compared with 57% for patients who received standard cord blood (P = .027). Viral infections occurred in 10% versus 26% of patients, respectively.
The incidence of acute graft versus host disease at day 100 was similar between treatment groups, and there was no significant difference at 1 year.
Relapse and nonrelapse mortality rates, as well as disease-free and overall survival rates also did not differ between groups.
In the first 100 days post transplant, patients who received omidubicel were alive and out of the hospital for a median of 60.5 days, compared with 48 days for patients who received standard cord blood (P = .005).
The study was funded by Gamida Cell. Dr. Sanz reported receiving research funding from the company and several others, and consulting fees, honoraria, speakers bureau activity, and travel expenses from other companies. Dr. DeFilipp reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Omidubicel, an investigational enriched umbilical cord blood product being developed by Gamida Cell for transplantation in patients with blood cancers, appears to have some advantages over standard umbilical cord blood.
The results come from a global phase 3 trial (NCT02730299) presented at the annual meeting of the European Society for Blood and Bone Marrow Transplantation.
“Transplantation with omidubicel, compared to standard cord blood transplantation, results in faster hematopoietic recovery, fewer infections, and fewer days in hospital,” said coinvestigator Guillermo F. Sanz, MD, PhD, from the Hospital Universitari i Politècnic la Fe in Valencia, Spain.
“Omidubicel should be considered as the new standard of care for patients eligible for umbilical cord blood transplantation,” Dr. Sanz concluded.
Zachariah DeFilipp, MD, from Mass General Cancer Center in Boston, a hematopoietic stem cell transplantation specialist who was not involved in the study, said in an interview that “omidubicel significantly improves the engraftment after transplant, as compared to standard cord blood transplant. For patients that lack an HLA-matched donor, this approach can help overcome the prolonged cytopenias that occur with standard cord blood transplants in adults.”
Gamida Cell plans to submit these data for approval of omidubicel by the Food and Drug Administration in the fourth quarter of 2021.
Omidubicel is also being evaluated in a phase 1/2 clinical study in patients with severe aplastic anemia (NCT03173937).
Expanding possibilities
Although umbilical cord blood stem cell grafts come from a readily available source and show greater tolerance across HLA barriers than other sources (such as bone marrow), the relatively low dose of stem cells in each unit results in delayed hematopoietic recovery, increased transplant-related morbidity and mortality, and longer hospitalizations, Dr. Sanz said.
Omidubicel consists of two cryopreserved fractions from a single cord blood unit. The product contains both noncultured CD133-negative cells, including T cells, and CD133-positive cells that are then expanded ex vivo for 21 days in the presence of nicotinamide.
“Nicotinamide increases stem and progenitor cells, inhibits differentiation and increases migration, bone marrow homing, and engraftment efficiency while preserving cellular functionality and phenotype,” Dr. Sanz explained during his presentation.
In an earlier phase 1/2 trial in 36 patients with high-risk hematologic malignancies, omidubicel was associated with hematopoietic engraftment lasting at least 10 years.
Details of phase 3 trial results
The global phase 3 trial was conducted in 125 patients (aged 13-65 years) with high-risk malignancies, including acute myeloid and lymphoblastic leukemias, myelodysplastic syndrome, chronic myeloid leukemia, lymphomas, and rare leukemias. These patients were all eligible for allogeneic stem cell transplantation but did not have matched donors.
Patients were randomly assigned to receive hematopoietic reconstitution with either omidubicel (n = 52) or standard cord blood (n = 58).
At 42 days of follow-up, the median time to neutrophil engraftment in the intention-to-treat (ITT) population, the primary endpoint, was 12 days with omidubicel versus 22 days with standard cord blood (P < .001).
In the as-treated population – the 108 patients who actually received omidubicel or standard cord blood – median time to engraftment was 10.0 versus 20.5 days, respectively (P < .001).
Rates of neutrophil engraftment at 42 days were 96% with omidubicel versus 89% with standard cord blood.
The secondary endpoint of time-to-platelet engraftment in the ITT population also favored omidubicel, with a cumulative day 42 incidence rate of 55%, compared with 35% with standard cord blood (P = .028).
In the as-treated population, median times to platelet engraftment were 37 days and 50 days, respectively (P = .023). The cumulative rates of platelet engraftment at 100 days of follow-up were 83% and 73%, respectively.
The incidence of grade 2 or 3 bacterial or invasive fungal infections by day 100 in the ITT population was 37% among patients who received omidubicel, compared with 57% for patients who received standard cord blood (P = .027). Viral infections occurred in 10% versus 26% of patients, respectively.
The incidence of acute graft versus host disease at day 100 was similar between treatment groups, and there was no significant difference at 1 year.
Relapse and nonrelapse mortality rates, as well as disease-free and overall survival rates also did not differ between groups.
In the first 100 days post transplant, patients who received omidubicel were alive and out of the hospital for a median of 60.5 days, compared with 48 days for patients who received standard cord blood (P = .005).
The study was funded by Gamida Cell. Dr. Sanz reported receiving research funding from the company and several others, and consulting fees, honoraria, speakers bureau activity, and travel expenses from other companies. Dr. DeFilipp reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
GVHD prophylaxis: Similar outcomes with PTCy and ATG
A newer regimen was no more effective than an older regimen when both were compared for graft versus host disease (GVHD) prophylaxis in patients who underwent reduced-intensity conditioning followed by a hematopoietic stem cell transplant (HSCT) from a 10/10 HLA-matched related or unrelated donor.
These results come from a multicenter randomized trial that compared posttransplant cyclophosphamide (PTCy) to antithymocyte globulin (ATG), which has been used for decades.
There were no significant differences between the two in either disease-free or overall survival, GVHD-free relapse-free survival (GRFS), or nonrelapse mortality, reported lead investigator Eolia Brissot, MD, of Hôpital Saint-Antoine, Sorbonne University, Paris.
Her presentation was judged ‘top abstract’ at the annual meeting of the European Society for Blood and Marrow Transplantation (EBMT), held virtually because of the pandemic.
ATG has been used for more than 30 years for GVHD prophylaxis in allogeneic HSCT. In contrast, PTCy is the new kid on the block, developed to facilitate haploidentical transplants using unmanipulated bone marrow cells to act as a method for selective allodepletion in vivo.
“PTCy [has proved] to be effective in preventing both acute and chronic GVHD,” Dr. Brissot said. “However, controversial outcome data remain when comparing PTCy and ATG according to the type of donors.”
Until now, she noted, there have been no prospective randomized data available for patients with donors (related or unrelated) that have 10 of 10 matched human leukocyte antigen (HLA) alleles. Hence, these were the patients studied in this latest trial, and in this population both regimens showed similar outcomes.
A bone marrow transplant specialist who was not involved in the study said that it’s a good first step.
“This is an important study to gain preliminary data to design a larger, subsequent phase 3 study,” said Zachariah DeFilipp, MD, of Mass General Cancer Center in Boston.
“The use of ATG as part of GVHD prophylaxis is common at many centers, especially in Europe, “ he explained. “The use of posttransplant cyclophosphamide is being expanded to more settings with transplant, beyond haploidentical transplant.
“Further investigations comparing the use of PTCy to ATG will help determine whether PTCy should be more broadly adopted as a standard-of-care GVHD prophylaxis approach, given currently available regimens,” he said in an interview.
Study details
The randomized phase 2b study (NCT02876679), conducted in centers in 11 cities in France, compared PTCy with ATG in patients with hematologic malignancies for whom a reduced-intensity allogeneic HSCT was indicated. This included patients aged 50 and older, and/or heavily pretreated patients who received an autologous HSCT or more than two prior lines of chemotherapy before allogeneic HSCT, as well as patients with poor performance status due to significant medical comorbidities.
Excluded from the trial were patients with creatinine clearance less than 30 mL/min; bilirubin or liver amino transferases more than three times the upper limit of normal; cardiac ejection fraction less than 40%; or pulmonary impairment with less than 50% lung carbon monoxide diffusing capacity.
Of 90 patients enrolled, 1 experienced a relapse before randomization, and the remaining 89 patients were assigned to either PTCy (experimental arm, 45 patients) or to ATG (control group, 44 patients).
Most patients had good performance status (Eastern Cooperative Oncology Group performance status 0 or 1). Diagnoses included acute myeloid and lymphoblastic leukemia, multiple myeloma, lymphomas, and myelodysplastic syndrome. The median age was 64 years, and the male to female ratio was about 2:1 in both groups.
All patients received “FB2” reduced-intensity conditioning with fludarabine30 mg/m2 per day for 4 days, and intravenous busulfan 130 mg/m2 per day for 2 days.
Patients in the experimental arm received cyclophosphamide 50 mg/kg per day on days 3 and 4 after transplant. Patients in the control group received ATG 2.5 mg/kg per day on days 3 and 2 prior to transplant.
All patients also received cyclosporine A, and those who had unrelated donors also received mycophenolate mofetil. In all, 39% of patients received cells from matched sibling donors, and 61% received cells from matched unrelated donors.
No significant differences seen
At 12 months of follow-up, there was no significant difference between the trial arms in the primary endpoint of GRFS, a composite of grade 3-4 acute GVHD, chronic GVHD requiring systemic treatment, relapse, or death. The rates of GRFS were 52.2% with PTCy vs. 45% with ATG.
Rates of disease-free survival were 68.5% with PTCy and 67.1% with ATG. The respective 12-month overall survival rates were 78.9% and 80.4%, respectively. The differences were not statistically significant.
The incidence of relapse at 1 year was 22.1% in the ATG group vs. 17.6% in the PTCy group. Respective nonrelapse mortality rates were 10.8% for the ATG group and 14% for the PTCy group. Neither difference was statistically significant.
There were also no significant between-group differences in the incidence at 12-month follow-up of either acute GVHD (34.9% PTCy arm vs. 24.3% ATG arm for grades II-IV combined, and 9.3% PTCy vs. 2.7% ATG for grades III or IV) or chronic GVHD (30.2% ATG vs. 26% PTCy).
The safety analysis showed no significant between-group differences in selected adverse events, including Epstein-Barr viral reactivation, cytomegalovirus reactivation, cardiac adverse events, or hemorrhagic cystitis.
The study was supported by Hospitals of Paris. Dr. Brissot and Dr. DeFilipp have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A newer regimen was no more effective than an older regimen when both were compared for graft versus host disease (GVHD) prophylaxis in patients who underwent reduced-intensity conditioning followed by a hematopoietic stem cell transplant (HSCT) from a 10/10 HLA-matched related or unrelated donor.
These results come from a multicenter randomized trial that compared posttransplant cyclophosphamide (PTCy) to antithymocyte globulin (ATG), which has been used for decades.
There were no significant differences between the two in either disease-free or overall survival, GVHD-free relapse-free survival (GRFS), or nonrelapse mortality, reported lead investigator Eolia Brissot, MD, of Hôpital Saint-Antoine, Sorbonne University, Paris.
Her presentation was judged ‘top abstract’ at the annual meeting of the European Society for Blood and Marrow Transplantation (EBMT), held virtually because of the pandemic.
ATG has been used for more than 30 years for GVHD prophylaxis in allogeneic HSCT. In contrast, PTCy is the new kid on the block, developed to facilitate haploidentical transplants using unmanipulated bone marrow cells to act as a method for selective allodepletion in vivo.
“PTCy [has proved] to be effective in preventing both acute and chronic GVHD,” Dr. Brissot said. “However, controversial outcome data remain when comparing PTCy and ATG according to the type of donors.”
Until now, she noted, there have been no prospective randomized data available for patients with donors (related or unrelated) that have 10 of 10 matched human leukocyte antigen (HLA) alleles. Hence, these were the patients studied in this latest trial, and in this population both regimens showed similar outcomes.
A bone marrow transplant specialist who was not involved in the study said that it’s a good first step.
“This is an important study to gain preliminary data to design a larger, subsequent phase 3 study,” said Zachariah DeFilipp, MD, of Mass General Cancer Center in Boston.
“The use of ATG as part of GVHD prophylaxis is common at many centers, especially in Europe, “ he explained. “The use of posttransplant cyclophosphamide is being expanded to more settings with transplant, beyond haploidentical transplant.
“Further investigations comparing the use of PTCy to ATG will help determine whether PTCy should be more broadly adopted as a standard-of-care GVHD prophylaxis approach, given currently available regimens,” he said in an interview.
Study details
The randomized phase 2b study (NCT02876679), conducted in centers in 11 cities in France, compared PTCy with ATG in patients with hematologic malignancies for whom a reduced-intensity allogeneic HSCT was indicated. This included patients aged 50 and older, and/or heavily pretreated patients who received an autologous HSCT or more than two prior lines of chemotherapy before allogeneic HSCT, as well as patients with poor performance status due to significant medical comorbidities.
Excluded from the trial were patients with creatinine clearance less than 30 mL/min; bilirubin or liver amino transferases more than three times the upper limit of normal; cardiac ejection fraction less than 40%; or pulmonary impairment with less than 50% lung carbon monoxide diffusing capacity.
Of 90 patients enrolled, 1 experienced a relapse before randomization, and the remaining 89 patients were assigned to either PTCy (experimental arm, 45 patients) or to ATG (control group, 44 patients).
Most patients had good performance status (Eastern Cooperative Oncology Group performance status 0 or 1). Diagnoses included acute myeloid and lymphoblastic leukemia, multiple myeloma, lymphomas, and myelodysplastic syndrome. The median age was 64 years, and the male to female ratio was about 2:1 in both groups.
All patients received “FB2” reduced-intensity conditioning with fludarabine30 mg/m2 per day for 4 days, and intravenous busulfan 130 mg/m2 per day for 2 days.
Patients in the experimental arm received cyclophosphamide 50 mg/kg per day on days 3 and 4 after transplant. Patients in the control group received ATG 2.5 mg/kg per day on days 3 and 2 prior to transplant.
All patients also received cyclosporine A, and those who had unrelated donors also received mycophenolate mofetil. In all, 39% of patients received cells from matched sibling donors, and 61% received cells from matched unrelated donors.
No significant differences seen
At 12 months of follow-up, there was no significant difference between the trial arms in the primary endpoint of GRFS, a composite of grade 3-4 acute GVHD, chronic GVHD requiring systemic treatment, relapse, or death. The rates of GRFS were 52.2% with PTCy vs. 45% with ATG.
Rates of disease-free survival were 68.5% with PTCy and 67.1% with ATG. The respective 12-month overall survival rates were 78.9% and 80.4%, respectively. The differences were not statistically significant.
The incidence of relapse at 1 year was 22.1% in the ATG group vs. 17.6% in the PTCy group. Respective nonrelapse mortality rates were 10.8% for the ATG group and 14% for the PTCy group. Neither difference was statistically significant.
There were also no significant between-group differences in the incidence at 12-month follow-up of either acute GVHD (34.9% PTCy arm vs. 24.3% ATG arm for grades II-IV combined, and 9.3% PTCy vs. 2.7% ATG for grades III or IV) or chronic GVHD (30.2% ATG vs. 26% PTCy).
The safety analysis showed no significant between-group differences in selected adverse events, including Epstein-Barr viral reactivation, cytomegalovirus reactivation, cardiac adverse events, or hemorrhagic cystitis.
The study was supported by Hospitals of Paris. Dr. Brissot and Dr. DeFilipp have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A newer regimen was no more effective than an older regimen when both were compared for graft versus host disease (GVHD) prophylaxis in patients who underwent reduced-intensity conditioning followed by a hematopoietic stem cell transplant (HSCT) from a 10/10 HLA-matched related or unrelated donor.
These results come from a multicenter randomized trial that compared posttransplant cyclophosphamide (PTCy) to antithymocyte globulin (ATG), which has been used for decades.
There were no significant differences between the two in either disease-free or overall survival, GVHD-free relapse-free survival (GRFS), or nonrelapse mortality, reported lead investigator Eolia Brissot, MD, of Hôpital Saint-Antoine, Sorbonne University, Paris.
Her presentation was judged ‘top abstract’ at the annual meeting of the European Society for Blood and Marrow Transplantation (EBMT), held virtually because of the pandemic.
ATG has been used for more than 30 years for GVHD prophylaxis in allogeneic HSCT. In contrast, PTCy is the new kid on the block, developed to facilitate haploidentical transplants using unmanipulated bone marrow cells to act as a method for selective allodepletion in vivo.
“PTCy [has proved] to be effective in preventing both acute and chronic GVHD,” Dr. Brissot said. “However, controversial outcome data remain when comparing PTCy and ATG according to the type of donors.”
Until now, she noted, there have been no prospective randomized data available for patients with donors (related or unrelated) that have 10 of 10 matched human leukocyte antigen (HLA) alleles. Hence, these were the patients studied in this latest trial, and in this population both regimens showed similar outcomes.
A bone marrow transplant specialist who was not involved in the study said that it’s a good first step.
“This is an important study to gain preliminary data to design a larger, subsequent phase 3 study,” said Zachariah DeFilipp, MD, of Mass General Cancer Center in Boston.
“The use of ATG as part of GVHD prophylaxis is common at many centers, especially in Europe, “ he explained. “The use of posttransplant cyclophosphamide is being expanded to more settings with transplant, beyond haploidentical transplant.
“Further investigations comparing the use of PTCy to ATG will help determine whether PTCy should be more broadly adopted as a standard-of-care GVHD prophylaxis approach, given currently available regimens,” he said in an interview.
Study details
The randomized phase 2b study (NCT02876679), conducted in centers in 11 cities in France, compared PTCy with ATG in patients with hematologic malignancies for whom a reduced-intensity allogeneic HSCT was indicated. This included patients aged 50 and older, and/or heavily pretreated patients who received an autologous HSCT or more than two prior lines of chemotherapy before allogeneic HSCT, as well as patients with poor performance status due to significant medical comorbidities.
Excluded from the trial were patients with creatinine clearance less than 30 mL/min; bilirubin or liver amino transferases more than three times the upper limit of normal; cardiac ejection fraction less than 40%; or pulmonary impairment with less than 50% lung carbon monoxide diffusing capacity.
Of 90 patients enrolled, 1 experienced a relapse before randomization, and the remaining 89 patients were assigned to either PTCy (experimental arm, 45 patients) or to ATG (control group, 44 patients).
Most patients had good performance status (Eastern Cooperative Oncology Group performance status 0 or 1). Diagnoses included acute myeloid and lymphoblastic leukemia, multiple myeloma, lymphomas, and myelodysplastic syndrome. The median age was 64 years, and the male to female ratio was about 2:1 in both groups.
All patients received “FB2” reduced-intensity conditioning with fludarabine30 mg/m2 per day for 4 days, and intravenous busulfan 130 mg/m2 per day for 2 days.
Patients in the experimental arm received cyclophosphamide 50 mg/kg per day on days 3 and 4 after transplant. Patients in the control group received ATG 2.5 mg/kg per day on days 3 and 2 prior to transplant.
All patients also received cyclosporine A, and those who had unrelated donors also received mycophenolate mofetil. In all, 39% of patients received cells from matched sibling donors, and 61% received cells from matched unrelated donors.
No significant differences seen
At 12 months of follow-up, there was no significant difference between the trial arms in the primary endpoint of GRFS, a composite of grade 3-4 acute GVHD, chronic GVHD requiring systemic treatment, relapse, or death. The rates of GRFS were 52.2% with PTCy vs. 45% with ATG.
Rates of disease-free survival were 68.5% with PTCy and 67.1% with ATG. The respective 12-month overall survival rates were 78.9% and 80.4%, respectively. The differences were not statistically significant.
The incidence of relapse at 1 year was 22.1% in the ATG group vs. 17.6% in the PTCy group. Respective nonrelapse mortality rates were 10.8% for the ATG group and 14% for the PTCy group. Neither difference was statistically significant.
There were also no significant between-group differences in the incidence at 12-month follow-up of either acute GVHD (34.9% PTCy arm vs. 24.3% ATG arm for grades II-IV combined, and 9.3% PTCy vs. 2.7% ATG for grades III or IV) or chronic GVHD (30.2% ATG vs. 26% PTCy).
The safety analysis showed no significant between-group differences in selected adverse events, including Epstein-Barr viral reactivation, cytomegalovirus reactivation, cardiac adverse events, or hemorrhagic cystitis.
The study was supported by Hospitals of Paris. Dr. Brissot and Dr. DeFilipp have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.