User login
Two new tests can detect CJD

Credit: Elise Amendola
Two groups of scientists have developed new tests to diagnose Creutzfeldt-Jakob disease (CJD).
One test uses samples collected from nasal passages to detect sporadic CJD, and the other uses urine samples to identify variant CJD.
The researchers said these tests provide simple methods for differentiating CJD from other diseases and could help prevent the transmission of CJD via blood
transfusions, transplants, or contaminated surgical instruments.
Both tests are described in The New England Journal of Medicine.
Nasal test for sporadic CJD
In one NEJM article, Byron Caughey, PhD, of the National Institute of Allergy and Infectious Diseases in Rockville, Maryland, and his colleagues detailed their results with the nasal test.
The researchers collected 31 nasal samples from patients with sporadic CJD and 43 samples from patients who had other neurologic diseases or no neurologic disease. The team brushed the inside of a subject’s nose to collect olfactory neurons connected to the brain.
Testing these samples allowed the researchers to correctly identify 30 of the 31 sporadic CJD patients (97% sensitivity). The tests also correctly showed negative results for all 43 of the non-CJD patients (100% specificity).
By comparison, tests using cerebral spinal fluid, which is currently used to detect sporadic CJD, were 77% sensitive and 100% specific. And these results took twice as long to obtain.
While continuing to validate the new testing method in CJD patients, the scientists are looking to expand their research to diagnose forms of prion diseases in sheep, cattle, and wildlife. The team also hopes to replace the nasal brush with an even simpler swabbing approach.
Urine test for variant CJD
In another NEJM article, Fabio Moda, PhD, of the University of Texas Medical School at Houston, and his colleagues described results observed with their urine test.
The team noted that the infectious agent in transmissible spongiform encephalopathies appears to be composed exclusively of the misfolded form of the prion protein, PrPSc. So they set out to determine if they could detect PrPSc in the urine of patients with CJD.
The researchers analyzed urine samples from healthy individuals (n=52) and patients with variant CJD (n=68), sporadic CJD (n=14), genetic forms of prion disease (n=4), other neurodegenerative disorders (n=50), and nondegenerative neurologic disorders (n=50).
The group found they could only detect PrPSc in samples from patients with variant CJD. They found “minute quantities” of PrPSc in 13 of the 14 urine samples from variant CJD patients, but PrPSc was not present in any of the samples from the other patients or the healthy individuals.
This suggests the test has a sensitivity of 92.9% and a specificity of 100%. ![]()
Credit: Elise Amendola
Two groups of scientists have developed new tests to diagnose Creutzfeldt-Jakob disease (CJD).
One test uses samples collected from nasal passages to detect sporadic CJD, and the other uses urine samples to identify variant CJD.
The researchers said these tests provide simple methods for differentiating CJD from other diseases and could help prevent the transmission of CJD via blood
transfusions, transplants, or contaminated surgical instruments.
Both tests are described in The New England Journal of Medicine.
Nasal test for sporadic CJD
In one NEJM article, Byron Caughey, PhD, of the National Institute of Allergy and Infectious Diseases in Rockville, Maryland, and his colleagues detailed their results with the nasal test.
The researchers collected 31 nasal samples from patients with sporadic CJD and 43 samples from patients who had other neurologic diseases or no neurologic disease. The team brushed the inside of a subject’s nose to collect olfactory neurons connected to the brain.
Testing these samples allowed the researchers to correctly identify 30 of the 31 sporadic CJD patients (97% sensitivity). The tests also correctly showed negative results for all 43 of the non-CJD patients (100% specificity).
By comparison, tests using cerebral spinal fluid, which is currently used to detect sporadic CJD, were 77% sensitive and 100% specific. And these results took twice as long to obtain.
While continuing to validate the new testing method in CJD patients, the scientists are looking to expand their research to diagnose forms of prion diseases in sheep, cattle, and wildlife. The team also hopes to replace the nasal brush with an even simpler swabbing approach.
Urine test for variant CJD
In another NEJM article, Fabio Moda, PhD, of the University of Texas Medical School at Houston, and his colleagues described results observed with their urine test.
The team noted that the infectious agent in transmissible spongiform encephalopathies appears to be composed exclusively of the misfolded form of the prion protein, PrPSc. So they set out to determine if they could detect PrPSc in the urine of patients with CJD.
The researchers analyzed urine samples from healthy individuals (n=52) and patients with variant CJD (n=68), sporadic CJD (n=14), genetic forms of prion disease (n=4), other neurodegenerative disorders (n=50), and nondegenerative neurologic disorders (n=50).
The group found they could only detect PrPSc in samples from patients with variant CJD. They found “minute quantities” of PrPSc in 13 of the 14 urine samples from variant CJD patients, but PrPSc was not present in any of the samples from the other patients or the healthy individuals.
This suggests the test has a sensitivity of 92.9% and a specificity of 100%. ![]()
Credit: Elise Amendola
Two groups of scientists have developed new tests to diagnose Creutzfeldt-Jakob disease (CJD).
One test uses samples collected from nasal passages to detect sporadic CJD, and the other uses urine samples to identify variant CJD.
The researchers said these tests provide simple methods for differentiating CJD from other diseases and could help prevent the transmission of CJD via blood
transfusions, transplants, or contaminated surgical instruments.
Both tests are described in The New England Journal of Medicine.
Nasal test for sporadic CJD
In one NEJM article, Byron Caughey, PhD, of the National Institute of Allergy and Infectious Diseases in Rockville, Maryland, and his colleagues detailed their results with the nasal test.
The researchers collected 31 nasal samples from patients with sporadic CJD and 43 samples from patients who had other neurologic diseases or no neurologic disease. The team brushed the inside of a subject’s nose to collect olfactory neurons connected to the brain.
Testing these samples allowed the researchers to correctly identify 30 of the 31 sporadic CJD patients (97% sensitivity). The tests also correctly showed negative results for all 43 of the non-CJD patients (100% specificity).
By comparison, tests using cerebral spinal fluid, which is currently used to detect sporadic CJD, were 77% sensitive and 100% specific. And these results took twice as long to obtain.
While continuing to validate the new testing method in CJD patients, the scientists are looking to expand their research to diagnose forms of prion diseases in sheep, cattle, and wildlife. The team also hopes to replace the nasal brush with an even simpler swabbing approach.
Urine test for variant CJD
In another NEJM article, Fabio Moda, PhD, of the University of Texas Medical School at Houston, and his colleagues described results observed with their urine test.
The team noted that the infectious agent in transmissible spongiform encephalopathies appears to be composed exclusively of the misfolded form of the prion protein, PrPSc. So they set out to determine if they could detect PrPSc in the urine of patients with CJD.
The researchers analyzed urine samples from healthy individuals (n=52) and patients with variant CJD (n=68), sporadic CJD (n=14), genetic forms of prion disease (n=4), other neurodegenerative disorders (n=50), and nondegenerative neurologic disorders (n=50).
The group found they could only detect PrPSc in samples from patients with variant CJD. They found “minute quantities” of PrPSc in 13 of the 14 urine samples from variant CJD patients, but PrPSc was not present in any of the samples from the other patients or the healthy individuals.
This suggests the test has a sensitivity of 92.9% and a specificity of 100%. ![]()
CHMP recommends antifungal agent
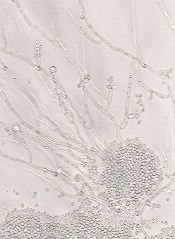
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for intravenous (IV) posaconazole (Noxafil), an antifungal agent.
If the European Commission affirms the CHMP opinion, IV posaconazole will be authorized for use in the European Union, Iceland, Liechtenstein, and Norway.
The commission previously granted marketing authorization for posaconazole delayed-release tablets and oral suspension.
Posaconazole is used to prevent invasive fungal infections in severely immunocompromised patients, such as hematopoietic stem cell transplant recipients with graft-vs-host disease or patients with hematologic malignancies and prolonged neutropenia from chemotherapy.
The drug is also used to treat fungal diseases—invasive aspergillosis, fusariosis, chromoblastomycosis, mycetoma, and coccidioidomycosis—when other antifungal agents—amphotericin B, itraconazole, or fluconazole—cannot be tolerated or have failed.
And posaconazole oral suspension is used as a first-line treatment for thrush, a fungal infection of the mouth and throat due to Candida.
Posaconazole injection is administered with a loading dose of 300 mg twice a day on the first day of therapy, then 300 mg once a day thereafter. It is given through a central venous line by IV infusion over approximately 90 minutes.
Once combined with a mixture of IV solution (150 mL of 5% dextrose in water or sodium chloride 0.9%), posaconazole should be administered immediately. If not used immediately, the solution can be stored up to 24 hours if refrigerated at 2°-8° C (36°-46° F).
The safety and effectiveness of IV posaconazole in patients younger than 18 years has not been established. IV posaconazole should not be used in pediatric patients because of non-clinical safety concerns.
Co-administration of drugs that can decrease the plasma concentration of posaconazole should be avoided unless the benefit outweighs the risk. If such drugs are necessary, patients should be monitored closely for breakthrough fungal infections.
Patients with known hypersensitivity to posaconazole or other azole antifungal medicines should not receive posaconazole. The drug should not be given with sirolimus, pimozide, quinidine, atorvastatin, lovastatin, simvastatin, or ergot alkaloids.
Drugs such as cyclosporine and tacrolimus require dose adjustments and frequent blood monitoring when administered with posaconazole. Serious side effects, including nephrotoxicity, leukoencephalopathy, and death, have been reported in patients with increased cyclosporine or tacrolimus blood levels.
Healthcare professionals should use caution when administering posaconazole to patients at risk of developing an irregular heart rhythm, as the drug has been shown to prolong the QT interval, and cases of potentially fatal irregular heart rhythm (torsades de pointes) have been reported in patients taking posaconazole.
Hepatic reactions have been reported as well. This includes mild to moderate elevations in ALT, AST, alkaline phosphatase, total bilirubin, and/or clinical hepatitis. Monitoring or discontinuation may be necessary in patients with hepatic reactions to posaconazole.
IV posaconazole should be avoided in patients with moderate or severe renal impairment (estimated glomerular filtration rate <50 mL/min), unless an assessment of the benefit/risk to the patient justifies the use of posaconazole.
In clinical trials, the adverse events associated with IV posaconazole were generally similar to those in trials of posaconazole oral suspension. The most frequently reported events were diarrhea (32%), hypokalemia (22%), fever (21%), and nausea (19%).
IV posaconazole is under development by MSD (known as Merck in the US and Canada). ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for intravenous (IV) posaconazole (Noxafil), an antifungal agent.
If the European Commission affirms the CHMP opinion, IV posaconazole will be authorized for use in the European Union, Iceland, Liechtenstein, and Norway.
The commission previously granted marketing authorization for posaconazole delayed-release tablets and oral suspension.
Posaconazole is used to prevent invasive fungal infections in severely immunocompromised patients, such as hematopoietic stem cell transplant recipients with graft-vs-host disease or patients with hematologic malignancies and prolonged neutropenia from chemotherapy.
The drug is also used to treat fungal diseases—invasive aspergillosis, fusariosis, chromoblastomycosis, mycetoma, and coccidioidomycosis—when other antifungal agents—amphotericin B, itraconazole, or fluconazole—cannot be tolerated or have failed.
And posaconazole oral suspension is used as a first-line treatment for thrush, a fungal infection of the mouth and throat due to Candida.
Posaconazole injection is administered with a loading dose of 300 mg twice a day on the first day of therapy, then 300 mg once a day thereafter. It is given through a central venous line by IV infusion over approximately 90 minutes.
Once combined with a mixture of IV solution (150 mL of 5% dextrose in water or sodium chloride 0.9%), posaconazole should be administered immediately. If not used immediately, the solution can be stored up to 24 hours if refrigerated at 2°-8° C (36°-46° F).
The safety and effectiveness of IV posaconazole in patients younger than 18 years has not been established. IV posaconazole should not be used in pediatric patients because of non-clinical safety concerns.
Co-administration of drugs that can decrease the plasma concentration of posaconazole should be avoided unless the benefit outweighs the risk. If such drugs are necessary, patients should be monitored closely for breakthrough fungal infections.
Patients with known hypersensitivity to posaconazole or other azole antifungal medicines should not receive posaconazole. The drug should not be given with sirolimus, pimozide, quinidine, atorvastatin, lovastatin, simvastatin, or ergot alkaloids.
Drugs such as cyclosporine and tacrolimus require dose adjustments and frequent blood monitoring when administered with posaconazole. Serious side effects, including nephrotoxicity, leukoencephalopathy, and death, have been reported in patients with increased cyclosporine or tacrolimus blood levels.
Healthcare professionals should use caution when administering posaconazole to patients at risk of developing an irregular heart rhythm, as the drug has been shown to prolong the QT interval, and cases of potentially fatal irregular heart rhythm (torsades de pointes) have been reported in patients taking posaconazole.
Hepatic reactions have been reported as well. This includes mild to moderate elevations in ALT, AST, alkaline phosphatase, total bilirubin, and/or clinical hepatitis. Monitoring or discontinuation may be necessary in patients with hepatic reactions to posaconazole.
IV posaconazole should be avoided in patients with moderate or severe renal impairment (estimated glomerular filtration rate <50 mL/min), unless an assessment of the benefit/risk to the patient justifies the use of posaconazole.
In clinical trials, the adverse events associated with IV posaconazole were generally similar to those in trials of posaconazole oral suspension. The most frequently reported events were diarrhea (32%), hypokalemia (22%), fever (21%), and nausea (19%).
IV posaconazole is under development by MSD (known as Merck in the US and Canada). ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for intravenous (IV) posaconazole (Noxafil), an antifungal agent.
If the European Commission affirms the CHMP opinion, IV posaconazole will be authorized for use in the European Union, Iceland, Liechtenstein, and Norway.
The commission previously granted marketing authorization for posaconazole delayed-release tablets and oral suspension.
Posaconazole is used to prevent invasive fungal infections in severely immunocompromised patients, such as hematopoietic stem cell transplant recipients with graft-vs-host disease or patients with hematologic malignancies and prolonged neutropenia from chemotherapy.
The drug is also used to treat fungal diseases—invasive aspergillosis, fusariosis, chromoblastomycosis, mycetoma, and coccidioidomycosis—when other antifungal agents—amphotericin B, itraconazole, or fluconazole—cannot be tolerated or have failed.
And posaconazole oral suspension is used as a first-line treatment for thrush, a fungal infection of the mouth and throat due to Candida.
Posaconazole injection is administered with a loading dose of 300 mg twice a day on the first day of therapy, then 300 mg once a day thereafter. It is given through a central venous line by IV infusion over approximately 90 minutes.
Once combined with a mixture of IV solution (150 mL of 5% dextrose in water or sodium chloride 0.9%), posaconazole should be administered immediately. If not used immediately, the solution can be stored up to 24 hours if refrigerated at 2°-8° C (36°-46° F).
The safety and effectiveness of IV posaconazole in patients younger than 18 years has not been established. IV posaconazole should not be used in pediatric patients because of non-clinical safety concerns.
Co-administration of drugs that can decrease the plasma concentration of posaconazole should be avoided unless the benefit outweighs the risk. If such drugs are necessary, patients should be monitored closely for breakthrough fungal infections.
Patients with known hypersensitivity to posaconazole or other azole antifungal medicines should not receive posaconazole. The drug should not be given with sirolimus, pimozide, quinidine, atorvastatin, lovastatin, simvastatin, or ergot alkaloids.
Drugs such as cyclosporine and tacrolimus require dose adjustments and frequent blood monitoring when administered with posaconazole. Serious side effects, including nephrotoxicity, leukoencephalopathy, and death, have been reported in patients with increased cyclosporine or tacrolimus blood levels.
Healthcare professionals should use caution when administering posaconazole to patients at risk of developing an irregular heart rhythm, as the drug has been shown to prolong the QT interval, and cases of potentially fatal irregular heart rhythm (torsades de pointes) have been reported in patients taking posaconazole.
Hepatic reactions have been reported as well. This includes mild to moderate elevations in ALT, AST, alkaline phosphatase, total bilirubin, and/or clinical hepatitis. Monitoring or discontinuation may be necessary in patients with hepatic reactions to posaconazole.
IV posaconazole should be avoided in patients with moderate or severe renal impairment (estimated glomerular filtration rate <50 mL/min), unless an assessment of the benefit/risk to the patient justifies the use of posaconazole.
In clinical trials, the adverse events associated with IV posaconazole were generally similar to those in trials of posaconazole oral suspension. The most frequently reported events were diarrhea (32%), hypokalemia (22%), fever (21%), and nausea (19%).
IV posaconazole is under development by MSD (known as Merck in the US and Canada). ![]()
STAP cell researcher commits suicide

Credit: NIH
An author of the retracted Nature papers on STAP cells (stimulus-triggered acquisition of pluripotency cells) has committed suicide at the age of 52.
Yoshiki Sasai, MD, PhD, was found dead at the RIKEN Center for Developmental Biology in Kobe, Japan, where he was deputy director.
Dr Sasai reportedly hanged himself and left several suicide notes.
Members of the scientific community have expressed shock and sadness upon learning of Dr Sasai’s death.
RIKEN President Ryoji Noyori, PhD, said he was “overcome with grief” when he heard the unfortunate news.
“The scientific world has lost a talented and dedicated researcher, who earned our deep respect for the advanced research he carried out over many years,” Dr Noyori said.
Nature’s editor-in-chief, Phil Campbell, PhD, echoed that sentiment, saying, “Yoshiki Sasai was an exceptional scientist, and he has left an extraordinary legacy of pioneering work across many fields within stem cell and developmental biology.”
Dr Sasai was a respected expert on embryonic stem cells, but the STAP cell scandal damaged his reputation and reportedly took a toll on his health. According to a spokesperson at RIKEN, Dr Sasai was hospitalized for stress and required counseling in the wake of the scandal.
Dr Sasai had worked closely with the lead author of the STAP cell papers, Haruko Obokata, PhD, although he said his main duty was editing the papers.
The papers, an article and a letter, were published in Nature in January. They recounted the creation of STAP cells—inducing pluripotency in somatic cells by exposing them to a low-pH environment.
Not long after the papers were published, members of the scientific community began to question the validity of the research. They voiced concerns about published images, possible plagiarism, and an inability to replicate the experiments described.
So RIKEN launched an investigation. In April, the investigative committee concluded that Dr Obokata was guilty of research misconduct, while Dr Sasai and another author from RIKEN, Teruhiko Wakayama, PhD, were guilty of negligence.
RIKEN also said the researchers would be disciplined, although details were not released.
At a subsequent news conference, Dr Sasai said the Nature papers should be retracted because of the errors and inconsistencies, but the data do indicate the STAP cell phenomenon is real.
Likewise, Dr Obokata insisted the phenomenon is real and appealed the findings of RIKEN’s investigation. But RIKEN said another investigation was not warranted and called for a retraction of the papers. In July, Nature published retractions.
A RIKEN group is still attempting to recreate the STAP cell phenomenon, with Dr Obokata’s help. RIKEN plans to release an interim report on this attempt soon.
Other researchers said they have tried and failed to replicate the STAP cell experiments. One group reported their failed attempt in F1000Research. ![]()
Credit: NIH
An author of the retracted Nature papers on STAP cells (stimulus-triggered acquisition of pluripotency cells) has committed suicide at the age of 52.
Yoshiki Sasai, MD, PhD, was found dead at the RIKEN Center for Developmental Biology in Kobe, Japan, where he was deputy director.
Dr Sasai reportedly hanged himself and left several suicide notes.
Members of the scientific community have expressed shock and sadness upon learning of Dr Sasai’s death.
RIKEN President Ryoji Noyori, PhD, said he was “overcome with grief” when he heard the unfortunate news.
“The scientific world has lost a talented and dedicated researcher, who earned our deep respect for the advanced research he carried out over many years,” Dr Noyori said.
Nature’s editor-in-chief, Phil Campbell, PhD, echoed that sentiment, saying, “Yoshiki Sasai was an exceptional scientist, and he has left an extraordinary legacy of pioneering work across many fields within stem cell and developmental biology.”
Dr Sasai was a respected expert on embryonic stem cells, but the STAP cell scandal damaged his reputation and reportedly took a toll on his health. According to a spokesperson at RIKEN, Dr Sasai was hospitalized for stress and required counseling in the wake of the scandal.
Dr Sasai had worked closely with the lead author of the STAP cell papers, Haruko Obokata, PhD, although he said his main duty was editing the papers.
The papers, an article and a letter, were published in Nature in January. They recounted the creation of STAP cells—inducing pluripotency in somatic cells by exposing them to a low-pH environment.
Not long after the papers were published, members of the scientific community began to question the validity of the research. They voiced concerns about published images, possible plagiarism, and an inability to replicate the experiments described.
So RIKEN launched an investigation. In April, the investigative committee concluded that Dr Obokata was guilty of research misconduct, while Dr Sasai and another author from RIKEN, Teruhiko Wakayama, PhD, were guilty of negligence.
RIKEN also said the researchers would be disciplined, although details were not released.
At a subsequent news conference, Dr Sasai said the Nature papers should be retracted because of the errors and inconsistencies, but the data do indicate the STAP cell phenomenon is real.
Likewise, Dr Obokata insisted the phenomenon is real and appealed the findings of RIKEN’s investigation. But RIKEN said another investigation was not warranted and called for a retraction of the papers. In July, Nature published retractions.
A RIKEN group is still attempting to recreate the STAP cell phenomenon, with Dr Obokata’s help. RIKEN plans to release an interim report on this attempt soon.
Other researchers said they have tried and failed to replicate the STAP cell experiments. One group reported their failed attempt in F1000Research. ![]()
Credit: NIH
An author of the retracted Nature papers on STAP cells (stimulus-triggered acquisition of pluripotency cells) has committed suicide at the age of 52.
Yoshiki Sasai, MD, PhD, was found dead at the RIKEN Center for Developmental Biology in Kobe, Japan, where he was deputy director.
Dr Sasai reportedly hanged himself and left several suicide notes.
Members of the scientific community have expressed shock and sadness upon learning of Dr Sasai’s death.
RIKEN President Ryoji Noyori, PhD, said he was “overcome with grief” when he heard the unfortunate news.
“The scientific world has lost a talented and dedicated researcher, who earned our deep respect for the advanced research he carried out over many years,” Dr Noyori said.
Nature’s editor-in-chief, Phil Campbell, PhD, echoed that sentiment, saying, “Yoshiki Sasai was an exceptional scientist, and he has left an extraordinary legacy of pioneering work across many fields within stem cell and developmental biology.”
Dr Sasai was a respected expert on embryonic stem cells, but the STAP cell scandal damaged his reputation and reportedly took a toll on his health. According to a spokesperson at RIKEN, Dr Sasai was hospitalized for stress and required counseling in the wake of the scandal.
Dr Sasai had worked closely with the lead author of the STAP cell papers, Haruko Obokata, PhD, although he said his main duty was editing the papers.
The papers, an article and a letter, were published in Nature in January. They recounted the creation of STAP cells—inducing pluripotency in somatic cells by exposing them to a low-pH environment.
Not long after the papers were published, members of the scientific community began to question the validity of the research. They voiced concerns about published images, possible plagiarism, and an inability to replicate the experiments described.
So RIKEN launched an investigation. In April, the investigative committee concluded that Dr Obokata was guilty of research misconduct, while Dr Sasai and another author from RIKEN, Teruhiko Wakayama, PhD, were guilty of negligence.
RIKEN also said the researchers would be disciplined, although details were not released.
At a subsequent news conference, Dr Sasai said the Nature papers should be retracted because of the errors and inconsistencies, but the data do indicate the STAP cell phenomenon is real.
Likewise, Dr Obokata insisted the phenomenon is real and appealed the findings of RIKEN’s investigation. But RIKEN said another investigation was not warranted and called for a retraction of the papers. In July, Nature published retractions.
A RIKEN group is still attempting to recreate the STAP cell phenomenon, with Dr Obokata’s help. RIKEN plans to release an interim report on this attempt soon.
Other researchers said they have tried and failed to replicate the STAP cell experiments. One group reported their failed attempt in F1000Research. ![]()
Study reveals why HSCs falter with age

in the bone marrow
A new study helps explain how blood production declines with age and why older individuals are not suitable donors for hematopoietic stem cell (HSC) transplant.
The research also reveals a potential approach for mitigating
the negative effects of aging on the blood, which can lead to anemia,
bone marrow failure, and myeloid malignancies.
The study, conducted in mice, suggests HSCs falter with age because they lose the ability to replicate their DNA accurately and efficiently during cell division.
Emmanuelle Passegué, PhD, of the University of California San Francisco, and her colleagues reported this discovery in Nature.
The researchers analyzed old HSCs in mice and found a scarcity of protein components needed to form the mini-chromosome maintenance helicase. This molecular machine unwinds double-stranded DNA so the cell’s genetic material can be duplicated and allocated to daughter cells later in cell division.
The HSCs were stressed by the loss of this machine’s activity. As a result, they had an increased risk for DNA damage and death when forced to divide.
On the other hand, the cells tended to survive unless they were confronted with a “strong replication challenge” like transplantation.
The researchers also discovered that even after the stress associated with DNA replication, old HSCs retained molecular tags on histones, a feature often associated with DNA damage.
However, these old survivors could repair induced DNA damage as efficiently as young stem cells.
“Old stem cells are not just sitting there with damaged DNA ready to develop cancer, as it has long been postulated,” Dr Passegué said.
Of course, not all was well in the old, surviving HSCs. The molecular tags accumulated on genes needed to make ribosomes.
Dr Passegué said she will further explore the consequences of reduced protein production as part of her ongoing research. She hopes it might be possible to prevent declining stem cell populations by developing a drug to prevent the loss of the helicase components needed to unwind and replicate DNA, thereby avoiding immune system failure. ![]()
in the bone marrow
A new study helps explain how blood production declines with age and why older individuals are not suitable donors for hematopoietic stem cell (HSC) transplant.
The research also reveals a potential approach for mitigating
the negative effects of aging on the blood, which can lead to anemia,
bone marrow failure, and myeloid malignancies.
The study, conducted in mice, suggests HSCs falter with age because they lose the ability to replicate their DNA accurately and efficiently during cell division.
Emmanuelle Passegué, PhD, of the University of California San Francisco, and her colleagues reported this discovery in Nature.
The researchers analyzed old HSCs in mice and found a scarcity of protein components needed to form the mini-chromosome maintenance helicase. This molecular machine unwinds double-stranded DNA so the cell’s genetic material can be duplicated and allocated to daughter cells later in cell division.
The HSCs were stressed by the loss of this machine’s activity. As a result, they had an increased risk for DNA damage and death when forced to divide.
On the other hand, the cells tended to survive unless they were confronted with a “strong replication challenge” like transplantation.
The researchers also discovered that even after the stress associated with DNA replication, old HSCs retained molecular tags on histones, a feature often associated with DNA damage.
However, these old survivors could repair induced DNA damage as efficiently as young stem cells.
“Old stem cells are not just sitting there with damaged DNA ready to develop cancer, as it has long been postulated,” Dr Passegué said.
Of course, not all was well in the old, surviving HSCs. The molecular tags accumulated on genes needed to make ribosomes.
Dr Passegué said she will further explore the consequences of reduced protein production as part of her ongoing research. She hopes it might be possible to prevent declining stem cell populations by developing a drug to prevent the loss of the helicase components needed to unwind and replicate DNA, thereby avoiding immune system failure. ![]()
in the bone marrow
A new study helps explain how blood production declines with age and why older individuals are not suitable donors for hematopoietic stem cell (HSC) transplant.
The research also reveals a potential approach for mitigating
the negative effects of aging on the blood, which can lead to anemia,
bone marrow failure, and myeloid malignancies.
The study, conducted in mice, suggests HSCs falter with age because they lose the ability to replicate their DNA accurately and efficiently during cell division.
Emmanuelle Passegué, PhD, of the University of California San Francisco, and her colleagues reported this discovery in Nature.
The researchers analyzed old HSCs in mice and found a scarcity of protein components needed to form the mini-chromosome maintenance helicase. This molecular machine unwinds double-stranded DNA so the cell’s genetic material can be duplicated and allocated to daughter cells later in cell division.
The HSCs were stressed by the loss of this machine’s activity. As a result, they had an increased risk for DNA damage and death when forced to divide.
On the other hand, the cells tended to survive unless they were confronted with a “strong replication challenge” like transplantation.
The researchers also discovered that even after the stress associated with DNA replication, old HSCs retained molecular tags on histones, a feature often associated with DNA damage.
However, these old survivors could repair induced DNA damage as efficiently as young stem cells.
“Old stem cells are not just sitting there with damaged DNA ready to develop cancer, as it has long been postulated,” Dr Passegué said.
Of course, not all was well in the old, surviving HSCs. The molecular tags accumulated on genes needed to make ribosomes.
Dr Passegué said she will further explore the consequences of reduced protein production as part of her ongoing research. She hopes it might be possible to prevent declining stem cell populations by developing a drug to prevent the loss of the helicase components needed to unwind and replicate DNA, thereby avoiding immune system failure. ![]()
Early HSCT best for infants with SCID, study shows
![]()
Credit: Chad McNeeley
Children with severe combined immune deficiency (SCID) have a good chance of survival if they undergo hematopoietic stem cell transplant (HSCT) within 3.5 months of birth, a new analysis suggests.
The risk of death is also lower if patients are free of infection at transplant and have a matched sibling donor.
“Survival is much, much better if infants undergo transplant before they turn 3.5 months old and before they contract any SCID-related infections,” said Sung-Yun Pai, MD, of the Dana-Farber Cancer Institute in Boston.
This underlines the importance of screening newborns for SCID, she added.
Dr Pai and her colleagues expressed this viewpoint, and detailed the research to support it, in The New England Journal of Medicine.
The team analyzed data on 240 children with SCID who were transplanted at 25 centers across North America between January 1, 2000, and December 31, 2009, (before the US Department of Health and Human Services recommended newborn screening for SCID in 2010).
The researchers assessed the patients’ outcomes according to age, infection status, donor source, and conditioning regimen.
Results revealed that children who underwent transplant before 3.5 months of age had excellent survival, regardless of donor source or infection status, as did patients with a matched sibling donor.
Children transplanted after 3.5 months also had a high survival rate regardless of donor source, as long as they did not have an active infection at the time of transplant.
Overall, 74% of patients survived at least 5 years. The 5-year survival rate was 97% in patients with a matched sibling donor, 94% among patients transplanted within 3.5 months of birth, 90% among patients who never had an infection, and 82% in patients whose infection resolved before transplant.
Survival was low—50%—among patients who were older than 3.5 months and had active infections at the time of transplant.
Actively infected infants who did not have a matched sibling donor and received immunosuppressive therapy or chemotherapy before transplant had particularly poor survival as well, ranging from 39% to 53%.
“This study accomplishes several things,” Dr Pai said. “First, it creates a baseline with which to compare patient outcomes since the advent of newborn screening for SCID. Second, it provides guidance for clinicians regarding the use of chemotherapy conditioning before transplantation.”
“Third, it highlights the relative impacts of infection status and patient age on transplant success. Lastly, it establishes the importance of early detection and transplantation, which points to the benefit of expanding newborn screening for SCID as broadly as possible.” ![]()
![]()
Credit: Chad McNeeley
Children with severe combined immune deficiency (SCID) have a good chance of survival if they undergo hematopoietic stem cell transplant (HSCT) within 3.5 months of birth, a new analysis suggests.
The risk of death is also lower if patients are free of infection at transplant and have a matched sibling donor.
“Survival is much, much better if infants undergo transplant before they turn 3.5 months old and before they contract any SCID-related infections,” said Sung-Yun Pai, MD, of the Dana-Farber Cancer Institute in Boston.
This underlines the importance of screening newborns for SCID, she added.
Dr Pai and her colleagues expressed this viewpoint, and detailed the research to support it, in The New England Journal of Medicine.
The team analyzed data on 240 children with SCID who were transplanted at 25 centers across North America between January 1, 2000, and December 31, 2009, (before the US Department of Health and Human Services recommended newborn screening for SCID in 2010).
The researchers assessed the patients’ outcomes according to age, infection status, donor source, and conditioning regimen.
Results revealed that children who underwent transplant before 3.5 months of age had excellent survival, regardless of donor source or infection status, as did patients with a matched sibling donor.
Children transplanted after 3.5 months also had a high survival rate regardless of donor source, as long as they did not have an active infection at the time of transplant.
Overall, 74% of patients survived at least 5 years. The 5-year survival rate was 97% in patients with a matched sibling donor, 94% among patients transplanted within 3.5 months of birth, 90% among patients who never had an infection, and 82% in patients whose infection resolved before transplant.
Survival was low—50%—among patients who were older than 3.5 months and had active infections at the time of transplant.
Actively infected infants who did not have a matched sibling donor and received immunosuppressive therapy or chemotherapy before transplant had particularly poor survival as well, ranging from 39% to 53%.
“This study accomplishes several things,” Dr Pai said. “First, it creates a baseline with which to compare patient outcomes since the advent of newborn screening for SCID. Second, it provides guidance for clinicians regarding the use of chemotherapy conditioning before transplantation.”
“Third, it highlights the relative impacts of infection status and patient age on transplant success. Lastly, it establishes the importance of early detection and transplantation, which points to the benefit of expanding newborn screening for SCID as broadly as possible.” ![]()
![]()
Credit: Chad McNeeley
Children with severe combined immune deficiency (SCID) have a good chance of survival if they undergo hematopoietic stem cell transplant (HSCT) within 3.5 months of birth, a new analysis suggests.
The risk of death is also lower if patients are free of infection at transplant and have a matched sibling donor.
“Survival is much, much better if infants undergo transplant before they turn 3.5 months old and before they contract any SCID-related infections,” said Sung-Yun Pai, MD, of the Dana-Farber Cancer Institute in Boston.
This underlines the importance of screening newborns for SCID, she added.
Dr Pai and her colleagues expressed this viewpoint, and detailed the research to support it, in The New England Journal of Medicine.
The team analyzed data on 240 children with SCID who were transplanted at 25 centers across North America between January 1, 2000, and December 31, 2009, (before the US Department of Health and Human Services recommended newborn screening for SCID in 2010).
The researchers assessed the patients’ outcomes according to age, infection status, donor source, and conditioning regimen.
Results revealed that children who underwent transplant before 3.5 months of age had excellent survival, regardless of donor source or infection status, as did patients with a matched sibling donor.
Children transplanted after 3.5 months also had a high survival rate regardless of donor source, as long as they did not have an active infection at the time of transplant.
Overall, 74% of patients survived at least 5 years. The 5-year survival rate was 97% in patients with a matched sibling donor, 94% among patients transplanted within 3.5 months of birth, 90% among patients who never had an infection, and 82% in patients whose infection resolved before transplant.
Survival was low—50%—among patients who were older than 3.5 months and had active infections at the time of transplant.
Actively infected infants who did not have a matched sibling donor and received immunosuppressive therapy or chemotherapy before transplant had particularly poor survival as well, ranging from 39% to 53%.
“This study accomplishes several things,” Dr Pai said. “First, it creates a baseline with which to compare patient outcomes since the advent of newborn screening for SCID. Second, it provides guidance for clinicians regarding the use of chemotherapy conditioning before transplantation.”
“Third, it highlights the relative impacts of infection status and patient age on transplant success. Lastly, it establishes the importance of early detection and transplantation, which points to the benefit of expanding newborn screening for SCID as broadly as possible.” ![]()
Optimal HSCT donors often elusive, study suggests
![]()
Credit: Chad McNeeley
New research suggests a majority of US patients who need unrelated hematopoietic stem cell transplants can find a suitable donor on the Be The Match Registry.
However, the likelihood of finding an 8/8 HLA-matched adult donor is often low, particularly for patients of diverse ethnic or racial backgrounds.
And finding a 6/6 HLA-matched cord blood donor is a long shot regardless of race or ethnicity, although patients younger than 20 years of age have better odds.
These findings appear in NEJM.
“This research confirms that physicians should identify the best available donor with minimal delay,” said study author Dennis Confer, MD, chief medical officer at National Marrow Donor Program/Be the Match in Minneapolis.
“Transplant should not be postponed in anticipation of finding a perfect match. Using a suitable match reflects current clinical practice.”
Dr Confer and his colleagues built population-based genetic models for 21 racial and ethnic groups to predict the likelihood of identifying a suitable adult or cord blood donor for each group.
The researchers used data on HLA genotypes and cord blood unit cell doses from the National Marrow Donor Program’s Be the Match registry, which included 10,759,087 adult donors and 186,166 cord blood units at the end of 2012.
The team found the likelihood of identifying an 8/8 HLA-matched donor is highest for white patients of European descent, at 75%, but it’s only 46% for white patients of Middle Eastern or North African descent.
For black Americans of all ethnic backgrounds, the probability of finding an 8/8 matched donor ranges from 16% (the lowest figure) to 19%.
And figures range from 27% to 57% for Hispanics, Asians, Pacific Islanders, and Native Americans (which includes individuals from the Caribbean and North, Central, and South America).
The likelihood of identifying a 7/8 matched donor is, again, highest for white patients of European descent, at 97%. And it’s 90% for white patients of Middle Eastern or North African descent.
For black Americans of all ethnic backgrounds, the likelihood of finding a 7/8 matched donor ranges from 66% (the lowest figure) to 76%. And it ranges from 72% to 91% for Hispanics, Asians, Pacific Islanders, and Native Americans.
The probability of identifying a 6/6 cord blood match is low for all racial/ethnic groups, but age plays a role. For patients age 20 and older, figures range from 1%—for both African and black Caribbean patients—to 17% for white Europeans. For patients younger than 20, figures range from 6% to 38% for the same groups.
For patients 20 and older, the likelihood of finding a 5/6 cord blood match ranges from 23% for African patients to 66% for white Europeans. And for the younger age group, the figures range from 56% to 87% for the same groups.
“We cannot yet find a suitably matched and available donor for every patient,” Dr Confer noted. “So we cannot slow down our efforts to expand the registry and fund more research to overcome these challenges.”
“To find a match for all patients, it is critical that those who join the registry remain committed to donate when called, and that we continue to add people to the Be The Match Registry for racial and ethnic groups of highest need.” ![]()
![]()
Credit: Chad McNeeley
New research suggests a majority of US patients who need unrelated hematopoietic stem cell transplants can find a suitable donor on the Be The Match Registry.
However, the likelihood of finding an 8/8 HLA-matched adult donor is often low, particularly for patients of diverse ethnic or racial backgrounds.
And finding a 6/6 HLA-matched cord blood donor is a long shot regardless of race or ethnicity, although patients younger than 20 years of age have better odds.
These findings appear in NEJM.
“This research confirms that physicians should identify the best available donor with minimal delay,” said study author Dennis Confer, MD, chief medical officer at National Marrow Donor Program/Be the Match in Minneapolis.
“Transplant should not be postponed in anticipation of finding a perfect match. Using a suitable match reflects current clinical practice.”
Dr Confer and his colleagues built population-based genetic models for 21 racial and ethnic groups to predict the likelihood of identifying a suitable adult or cord blood donor for each group.
The researchers used data on HLA genotypes and cord blood unit cell doses from the National Marrow Donor Program’s Be the Match registry, which included 10,759,087 adult donors and 186,166 cord blood units at the end of 2012.
The team found the likelihood of identifying an 8/8 HLA-matched donor is highest for white patients of European descent, at 75%, but it’s only 46% for white patients of Middle Eastern or North African descent.
For black Americans of all ethnic backgrounds, the probability of finding an 8/8 matched donor ranges from 16% (the lowest figure) to 19%.
And figures range from 27% to 57% for Hispanics, Asians, Pacific Islanders, and Native Americans (which includes individuals from the Caribbean and North, Central, and South America).
The likelihood of identifying a 7/8 matched donor is, again, highest for white patients of European descent, at 97%. And it’s 90% for white patients of Middle Eastern or North African descent.
For black Americans of all ethnic backgrounds, the likelihood of finding a 7/8 matched donor ranges from 66% (the lowest figure) to 76%. And it ranges from 72% to 91% for Hispanics, Asians, Pacific Islanders, and Native Americans.
The probability of identifying a 6/6 cord blood match is low for all racial/ethnic groups, but age plays a role. For patients age 20 and older, figures range from 1%—for both African and black Caribbean patients—to 17% for white Europeans. For patients younger than 20, figures range from 6% to 38% for the same groups.
For patients 20 and older, the likelihood of finding a 5/6 cord blood match ranges from 23% for African patients to 66% for white Europeans. And for the younger age group, the figures range from 56% to 87% for the same groups.
“We cannot yet find a suitably matched and available donor for every patient,” Dr Confer noted. “So we cannot slow down our efforts to expand the registry and fund more research to overcome these challenges.”
“To find a match for all patients, it is critical that those who join the registry remain committed to donate when called, and that we continue to add people to the Be The Match Registry for racial and ethnic groups of highest need.” ![]()
![]()
Credit: Chad McNeeley
New research suggests a majority of US patients who need unrelated hematopoietic stem cell transplants can find a suitable donor on the Be The Match Registry.
However, the likelihood of finding an 8/8 HLA-matched adult donor is often low, particularly for patients of diverse ethnic or racial backgrounds.
And finding a 6/6 HLA-matched cord blood donor is a long shot regardless of race or ethnicity, although patients younger than 20 years of age have better odds.
These findings appear in NEJM.
“This research confirms that physicians should identify the best available donor with minimal delay,” said study author Dennis Confer, MD, chief medical officer at National Marrow Donor Program/Be the Match in Minneapolis.
“Transplant should not be postponed in anticipation of finding a perfect match. Using a suitable match reflects current clinical practice.”
Dr Confer and his colleagues built population-based genetic models for 21 racial and ethnic groups to predict the likelihood of identifying a suitable adult or cord blood donor for each group.
The researchers used data on HLA genotypes and cord blood unit cell doses from the National Marrow Donor Program’s Be the Match registry, which included 10,759,087 adult donors and 186,166 cord blood units at the end of 2012.
The team found the likelihood of identifying an 8/8 HLA-matched donor is highest for white patients of European descent, at 75%, but it’s only 46% for white patients of Middle Eastern or North African descent.
For black Americans of all ethnic backgrounds, the probability of finding an 8/8 matched donor ranges from 16% (the lowest figure) to 19%.
And figures range from 27% to 57% for Hispanics, Asians, Pacific Islanders, and Native Americans (which includes individuals from the Caribbean and North, Central, and South America).
The likelihood of identifying a 7/8 matched donor is, again, highest for white patients of European descent, at 97%. And it’s 90% for white patients of Middle Eastern or North African descent.
For black Americans of all ethnic backgrounds, the likelihood of finding a 7/8 matched donor ranges from 66% (the lowest figure) to 76%. And it ranges from 72% to 91% for Hispanics, Asians, Pacific Islanders, and Native Americans.
The probability of identifying a 6/6 cord blood match is low for all racial/ethnic groups, but age plays a role. For patients age 20 and older, figures range from 1%—for both African and black Caribbean patients—to 17% for white Europeans. For patients younger than 20, figures range from 6% to 38% for the same groups.
For patients 20 and older, the likelihood of finding a 5/6 cord blood match ranges from 23% for African patients to 66% for white Europeans. And for the younger age group, the figures range from 56% to 87% for the same groups.
“We cannot yet find a suitably matched and available donor for every patient,” Dr Confer noted. “So we cannot slow down our efforts to expand the registry and fund more research to overcome these challenges.”
“To find a match for all patients, it is critical that those who join the registry remain committed to donate when called, and that we continue to add people to the Be The Match Registry for racial and ethnic groups of highest need.” ![]()
Two blood cancer patients cleared of HIV

Credit: CDC
MELBOURNE—Two men with hematologic malignancies who were also HIV-positive appear to be free of the virus after receiving stem cell transplants.
The patients have undetectable levels of HIV and remain free of their cancers—acute myeloid leukemia and non-Hodgkin lymphoma—more than 3 years after their transplants.
Importantly, the patients’ stem cell donors were not homozygous for CCR5-delta 32, a mutation that affords protection against HIV.
The researchers said these results herald a new direction in HIV research and provide hope for HIV-positive patients with leukemia and lymphoma.
The work was presented at the “Towards an HIV Cure Symposium,” which is part of the 20th International AIDS Conference.
Both patients were treated at St Vincent’s Hospital in partnership with the University of New South Wales’s Kirby Institute in Sydney, Australia.
One patient underwent a transplant in 2010 to treat his non-Hodgkin lymphoma, and his donor had 1 copy of CCR5-delta 32.
The second patient underwent a similar procedure for acute myeloid leukemia in 2011, and his donor did not have any copies of CCR5-delta 32.
Nevertheless, both patients were successfully cleared of HIV, although they remain on antiretroviral therapy as a protective measure.
“We’re so pleased that both patients are doing reasonably well years after the treatment for their cancers and remain free of both the original cancer and the HIV virus,” said David Cooper, MBBS, MD, DSc, of the Kirby Institute and St Vincent’s Hospital.
Until now, the only person considered to have cleared HIV is an American man, Timothy Ray Brown, who underwent 2 stem cell transplants in Berlin (in 2007 and 2008).
The cells in his second transplant included both copies of CCR5-delta 32, which affords protection against HIV and is found in less than 1% of the population. The man is no longer on antiretroviral therapy and remains free of HIV.
In Boston, 2 other patients underwent similar transplants in 2012, but the donor cells did not contain CCR5-delta 32. In both cases, HIV returned after antiretroviral treatment was stopped.
“It is very difficult to find a match for bone marrow donors and even more so to find one that affords protective immunity against HIV,” Dr Cooper said.
While his group’s results are a significant development, the researchers stressed that transplants are not a general functional “cure” for the up to 38.8 million people infected with HIV worldwide.
“This is a terrific, unexpected result for people with malignancy and HIV,” said Sam Milliken, MBBS, of St Vincent’s Hospital. “It may well give us a whole new insight into HIV, using the principles of stem cell transplantation.”
“It is important to caution that, at this stage, this form of treatment is far too dangerous for treating patients with HIV alone, but there may be potential for using transplants as an effective treatment modality for HIV down the track.”
The researchers said the 2 Sydney patients will be the subject of investigations to determine where any residual virus might be hiding and how it can be controlled. And the patients’ results point to a new direction for HIV research.
“We still don’t know why these patients have undetectable viral loads,” said Kersten Koelsch, MD, of the Kirby Institute. “One theory is that the induction therapy helps to destroy the cells in which the virus is hiding and that any remaining infected cells are destroyed by the patient’s new immune system.”
“We need more research to establish why and how bone marrow transplantation clears the virus. We also want to explore the predictors of sustained viral clearance and how this might be able to be exploited without the need for bone marrow transplantation.”
For the time being, the results mean that more patients who are eligible for transplant might be able to participate in clinical trials to determine the value of this procedure in HIV. ![]()
Credit: CDC
MELBOURNE—Two men with hematologic malignancies who were also HIV-positive appear to be free of the virus after receiving stem cell transplants.
The patients have undetectable levels of HIV and remain free of their cancers—acute myeloid leukemia and non-Hodgkin lymphoma—more than 3 years after their transplants.
Importantly, the patients’ stem cell donors were not homozygous for CCR5-delta 32, a mutation that affords protection against HIV.
The researchers said these results herald a new direction in HIV research and provide hope for HIV-positive patients with leukemia and lymphoma.
The work was presented at the “Towards an HIV Cure Symposium,” which is part of the 20th International AIDS Conference.
Both patients were treated at St Vincent’s Hospital in partnership with the University of New South Wales’s Kirby Institute in Sydney, Australia.
One patient underwent a transplant in 2010 to treat his non-Hodgkin lymphoma, and his donor had 1 copy of CCR5-delta 32.
The second patient underwent a similar procedure for acute myeloid leukemia in 2011, and his donor did not have any copies of CCR5-delta 32.
Nevertheless, both patients were successfully cleared of HIV, although they remain on antiretroviral therapy as a protective measure.
“We’re so pleased that both patients are doing reasonably well years after the treatment for their cancers and remain free of both the original cancer and the HIV virus,” said David Cooper, MBBS, MD, DSc, of the Kirby Institute and St Vincent’s Hospital.
Until now, the only person considered to have cleared HIV is an American man, Timothy Ray Brown, who underwent 2 stem cell transplants in Berlin (in 2007 and 2008).
The cells in his second transplant included both copies of CCR5-delta 32, which affords protection against HIV and is found in less than 1% of the population. The man is no longer on antiretroviral therapy and remains free of HIV.
In Boston, 2 other patients underwent similar transplants in 2012, but the donor cells did not contain CCR5-delta 32. In both cases, HIV returned after antiretroviral treatment was stopped.
“It is very difficult to find a match for bone marrow donors and even more so to find one that affords protective immunity against HIV,” Dr Cooper said.
While his group’s results are a significant development, the researchers stressed that transplants are not a general functional “cure” for the up to 38.8 million people infected with HIV worldwide.
“This is a terrific, unexpected result for people with malignancy and HIV,” said Sam Milliken, MBBS, of St Vincent’s Hospital. “It may well give us a whole new insight into HIV, using the principles of stem cell transplantation.”
“It is important to caution that, at this stage, this form of treatment is far too dangerous for treating patients with HIV alone, but there may be potential for using transplants as an effective treatment modality for HIV down the track.”
The researchers said the 2 Sydney patients will be the subject of investigations to determine where any residual virus might be hiding and how it can be controlled. And the patients’ results point to a new direction for HIV research.
“We still don’t know why these patients have undetectable viral loads,” said Kersten Koelsch, MD, of the Kirby Institute. “One theory is that the induction therapy helps to destroy the cells in which the virus is hiding and that any remaining infected cells are destroyed by the patient’s new immune system.”
“We need more research to establish why and how bone marrow transplantation clears the virus. We also want to explore the predictors of sustained viral clearance and how this might be able to be exploited without the need for bone marrow transplantation.”
For the time being, the results mean that more patients who are eligible for transplant might be able to participate in clinical trials to determine the value of this procedure in HIV. ![]()
Credit: CDC
MELBOURNE—Two men with hematologic malignancies who were also HIV-positive appear to be free of the virus after receiving stem cell transplants.
The patients have undetectable levels of HIV and remain free of their cancers—acute myeloid leukemia and non-Hodgkin lymphoma—more than 3 years after their transplants.
Importantly, the patients’ stem cell donors were not homozygous for CCR5-delta 32, a mutation that affords protection against HIV.
The researchers said these results herald a new direction in HIV research and provide hope for HIV-positive patients with leukemia and lymphoma.
The work was presented at the “Towards an HIV Cure Symposium,” which is part of the 20th International AIDS Conference.
Both patients were treated at St Vincent’s Hospital in partnership with the University of New South Wales’s Kirby Institute in Sydney, Australia.
One patient underwent a transplant in 2010 to treat his non-Hodgkin lymphoma, and his donor had 1 copy of CCR5-delta 32.
The second patient underwent a similar procedure for acute myeloid leukemia in 2011, and his donor did not have any copies of CCR5-delta 32.
Nevertheless, both patients were successfully cleared of HIV, although they remain on antiretroviral therapy as a protective measure.
“We’re so pleased that both patients are doing reasonably well years after the treatment for their cancers and remain free of both the original cancer and the HIV virus,” said David Cooper, MBBS, MD, DSc, of the Kirby Institute and St Vincent’s Hospital.
Until now, the only person considered to have cleared HIV is an American man, Timothy Ray Brown, who underwent 2 stem cell transplants in Berlin (in 2007 and 2008).
The cells in his second transplant included both copies of CCR5-delta 32, which affords protection against HIV and is found in less than 1% of the population. The man is no longer on antiretroviral therapy and remains free of HIV.
In Boston, 2 other patients underwent similar transplants in 2012, but the donor cells did not contain CCR5-delta 32. In both cases, HIV returned after antiretroviral treatment was stopped.
“It is very difficult to find a match for bone marrow donors and even more so to find one that affords protective immunity against HIV,” Dr Cooper said.
While his group’s results are a significant development, the researchers stressed that transplants are not a general functional “cure” for the up to 38.8 million people infected with HIV worldwide.
“This is a terrific, unexpected result for people with malignancy and HIV,” said Sam Milliken, MBBS, of St Vincent’s Hospital. “It may well give us a whole new insight into HIV, using the principles of stem cell transplantation.”
“It is important to caution that, at this stage, this form of treatment is far too dangerous for treating patients with HIV alone, but there may be potential for using transplants as an effective treatment modality for HIV down the track.”
The researchers said the 2 Sydney patients will be the subject of investigations to determine where any residual virus might be hiding and how it can be controlled. And the patients’ results point to a new direction for HIV research.
“We still don’t know why these patients have undetectable viral loads,” said Kersten Koelsch, MD, of the Kirby Institute. “One theory is that the induction therapy helps to destroy the cells in which the virus is hiding and that any remaining infected cells are destroyed by the patient’s new immune system.”
“We need more research to establish why and how bone marrow transplantation clears the virus. We also want to explore the predictors of sustained viral clearance and how this might be able to be exploited without the need for bone marrow transplantation.”
For the time being, the results mean that more patients who are eligible for transplant might be able to participate in clinical trials to determine the value of this procedure in HIV.
HSC engraftment across the species barrier

Scientists say they’ve generated a mouse model that supports the transplantation of human hematopoietic stem cells (HSCs), despite the species barrier and without the need for irradiation.
The group used a mutation of the Kit receptor in the mouse stem cells to facilitate the engraftment of human cells.
In this model, human HSCs can expand and differentiate into all blood cell types without any additional treatment.
Even cells of the innate immune system that are not typically found in “humanized” mice were efficiently generated in this mouse.
Furthermore, the stem cells can be maintained in the mouse over a longer period of time.
The researchers reported these results in Cell Stem Cell.
“Our goal was to develop an optimal model for the transplantation and study of human blood stem cells,” said study author Claudia Waskow, PhD, of Technische Universität Dresden in Germany.
To achieve optimal stem cell engraftment, she and her colleagues introduced a naturally occurring mutation of the Kit receptor into mice lacking a functional immune system.
In this way, the team circumvented the 2 major obstacles of HSC transplantation: the rejection by the recipient’s immune system and the absence of free niche space for the incoming donor stem cells in the recipient’s bone marrow.
The Kit mutation in the new mouse model impairs the recipient’s stem cell compartment in such a way that the endogenous HSCs can be easily replaced by human donor stem cells with a functional Kit receptor.
The researchers said this replacement works so efficiently that irradiation can be completely omitted, allowing the study of human blood development in a physiological setting. The model can now be used to study diseases of the human blood and immune system or to test new treatment options.
The results of this research also show that the Kit receptor is important for the function of human HSCs, notably in a transplant setting. The researchers said future studies will focus on using this knowledge to improve conditioning therapy for patients undergoing HSC transplant.
Scientists say they’ve generated a mouse model that supports the transplantation of human hematopoietic stem cells (HSCs), despite the species barrier and without the need for irradiation.
The group used a mutation of the Kit receptor in the mouse stem cells to facilitate the engraftment of human cells.
In this model, human HSCs can expand and differentiate into all blood cell types without any additional treatment.
Even cells of the innate immune system that are not typically found in “humanized” mice were efficiently generated in this mouse.
Furthermore, the stem cells can be maintained in the mouse over a longer period of time.
The researchers reported these results in Cell Stem Cell.
“Our goal was to develop an optimal model for the transplantation and study of human blood stem cells,” said study author Claudia Waskow, PhD, of Technische Universität Dresden in Germany.
To achieve optimal stem cell engraftment, she and her colleagues introduced a naturally occurring mutation of the Kit receptor into mice lacking a functional immune system.
In this way, the team circumvented the 2 major obstacles of HSC transplantation: the rejection by the recipient’s immune system and the absence of free niche space for the incoming donor stem cells in the recipient’s bone marrow.
The Kit mutation in the new mouse model impairs the recipient’s stem cell compartment in such a way that the endogenous HSCs can be easily replaced by human donor stem cells with a functional Kit receptor.
The researchers said this replacement works so efficiently that irradiation can be completely omitted, allowing the study of human blood development in a physiological setting. The model can now be used to study diseases of the human blood and immune system or to test new treatment options.
The results of this research also show that the Kit receptor is important for the function of human HSCs, notably in a transplant setting. The researchers said future studies will focus on using this knowledge to improve conditioning therapy for patients undergoing HSC transplant.
Scientists say they’ve generated a mouse model that supports the transplantation of human hematopoietic stem cells (HSCs), despite the species barrier and without the need for irradiation.
The group used a mutation of the Kit receptor in the mouse stem cells to facilitate the engraftment of human cells.
In this model, human HSCs can expand and differentiate into all blood cell types without any additional treatment.
Even cells of the innate immune system that are not typically found in “humanized” mice were efficiently generated in this mouse.
Furthermore, the stem cells can be maintained in the mouse over a longer period of time.
The researchers reported these results in Cell Stem Cell.
“Our goal was to develop an optimal model for the transplantation and study of human blood stem cells,” said study author Claudia Waskow, PhD, of Technische Universität Dresden in Germany.
To achieve optimal stem cell engraftment, she and her colleagues introduced a naturally occurring mutation of the Kit receptor into mice lacking a functional immune system.
In this way, the team circumvented the 2 major obstacles of HSC transplantation: the rejection by the recipient’s immune system and the absence of free niche space for the incoming donor stem cells in the recipient’s bone marrow.
The Kit mutation in the new mouse model impairs the recipient’s stem cell compartment in such a way that the endogenous HSCs can be easily replaced by human donor stem cells with a functional Kit receptor.
The researchers said this replacement works so efficiently that irradiation can be completely omitted, allowing the study of human blood development in a physiological setting. The model can now be used to study diseases of the human blood and immune system or to test new treatment options.
The results of this research also show that the Kit receptor is important for the function of human HSCs, notably in a transplant setting. The researchers said future studies will focus on using this knowledge to improve conditioning therapy for patients undergoing HSC transplant.
Gene editing doesn’t increase mutations in iPSCs
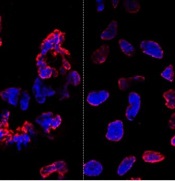
misshapen nuclear envelopes
(red) from iPSCs (DNA in blue).
The right panel shows
gene-edited iPSCs.
Credit: Salk Institute
Results of new research may ease previous concerns that gene-editing techniques could add unwanted mutations to stem cells.
Researchers compared gene editing techniques in lines of induced pluripotent stem cells (iPSCs) derived from a patient with sickle cell disease (SCD).
And they found that neither viral nor nuclease-based gene-editing methods increased the frequency of mutations in the iPSCs.
The team reported these results in Cell Stem Cell.
“The ability to precisely modify the DNA of stem cells has greatly accelerated research on human diseases and cell therapy,” said senior study author Juan Carlos Izpisua Belmonte, PhD, of the Salk Institute for Biological Studies in La Jolla, California.
“To successfully translate this technology into the clinic, we first need to scrutinize the safety of these modified stem cells, such as their genome stability and mutational load.”
Previously, Dr Belmonte’s lab pioneered the use of modified viruses, called helper-dependent adenoviral vectors (HDAdVs), to correct the genetic mutation that causes SCD.
He and his colleagues used HDAdVs to replace the mutated gene in a line of iPSCs with a mutant-free version, creating stem cells that could, theoretically, be infused into patients’ bone marrow and help create healthy blood cells.
Before such technologies are applied to humans, though, Dr Belmonte and his colleagues wanted to know whether there were risks related to editing the genes in iPSCs.
“As cells are being reprogrammed into stem cells, they tend to accumulate many mutations,” said Mo Li, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“So people naturally worry that any process you perform with these cells in vitro—including gene editing—might generate even more mutations.”
To find out whether this was the case, the researchers conducted tests in a line of SCD-derived iPSCs.
They edited the genes of some cells using 1 of 2 HDAdV designs. And they edited others using 1 of 2 transcription activator-like effector nuclease (TALEN) proteins.
They kept the rest of the SCD iPSCs in culture without editing them. Then, the team sequenced the entire genome of each cell from the 4 edits and control experiment.
While all of the cells gained a low level of random gene mutations during the experiments, the cells that had undergone gene-editing—whether through HDAdV- or TALEN-based approaches—had no more mutations than the cells kept in culture.
“We were pleasantly surprised by the results,” said Keiichiro Suzuki, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“People have found thousands of mutations introduced during iPSC reprogramming. We found less than a hundred single nucleotide variants in all cases.”
The researchers noted that this finding doesn’t necessarily mean there are no inherent risks to using stem cells with edited genes. However, it does suggest the editing process doesn’t make iPSCs any less safe.
“We concluded that the risk of mutation isn’t inherently connected to gene editing,” Dr Li said. “These cells present the same risks as using any other cells manipulated for cell or gene therapy.”
The Belmonte group is now planning more studies to address whether gene-repair in other cell types, using other approaches, or targeting other genes could be more or less likely to cause unwanted mutations.
For now, they hope their findings encourage those in the field to keep pursuing gene-editing techniques as a potential way to treat genetic diseases in the future. ![]()
misshapen nuclear envelopes
(red) from iPSCs (DNA in blue).
The right panel shows
gene-edited iPSCs.
Credit: Salk Institute
Results of new research may ease previous concerns that gene-editing techniques could add unwanted mutations to stem cells.
Researchers compared gene editing techniques in lines of induced pluripotent stem cells (iPSCs) derived from a patient with sickle cell disease (SCD).
And they found that neither viral nor nuclease-based gene-editing methods increased the frequency of mutations in the iPSCs.
The team reported these results in Cell Stem Cell.
“The ability to precisely modify the DNA of stem cells has greatly accelerated research on human diseases and cell therapy,” said senior study author Juan Carlos Izpisua Belmonte, PhD, of the Salk Institute for Biological Studies in La Jolla, California.
“To successfully translate this technology into the clinic, we first need to scrutinize the safety of these modified stem cells, such as their genome stability and mutational load.”
Previously, Dr Belmonte’s lab pioneered the use of modified viruses, called helper-dependent adenoviral vectors (HDAdVs), to correct the genetic mutation that causes SCD.
He and his colleagues used HDAdVs to replace the mutated gene in a line of iPSCs with a mutant-free version, creating stem cells that could, theoretically, be infused into patients’ bone marrow and help create healthy blood cells.
Before such technologies are applied to humans, though, Dr Belmonte and his colleagues wanted to know whether there were risks related to editing the genes in iPSCs.
“As cells are being reprogrammed into stem cells, they tend to accumulate many mutations,” said Mo Li, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“So people naturally worry that any process you perform with these cells in vitro—including gene editing—might generate even more mutations.”
To find out whether this was the case, the researchers conducted tests in a line of SCD-derived iPSCs.
They edited the genes of some cells using 1 of 2 HDAdV designs. And they edited others using 1 of 2 transcription activator-like effector nuclease (TALEN) proteins.
They kept the rest of the SCD iPSCs in culture without editing them. Then, the team sequenced the entire genome of each cell from the 4 edits and control experiment.
While all of the cells gained a low level of random gene mutations during the experiments, the cells that had undergone gene-editing—whether through HDAdV- or TALEN-based approaches—had no more mutations than the cells kept in culture.
“We were pleasantly surprised by the results,” said Keiichiro Suzuki, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“People have found thousands of mutations introduced during iPSC reprogramming. We found less than a hundred single nucleotide variants in all cases.”
The researchers noted that this finding doesn’t necessarily mean there are no inherent risks to using stem cells with edited genes. However, it does suggest the editing process doesn’t make iPSCs any less safe.
“We concluded that the risk of mutation isn’t inherently connected to gene editing,” Dr Li said. “These cells present the same risks as using any other cells manipulated for cell or gene therapy.”
The Belmonte group is now planning more studies to address whether gene-repair in other cell types, using other approaches, or targeting other genes could be more or less likely to cause unwanted mutations.
For now, they hope their findings encourage those in the field to keep pursuing gene-editing techniques as a potential way to treat genetic diseases in the future. ![]()
misshapen nuclear envelopes
(red) from iPSCs (DNA in blue).
The right panel shows
gene-edited iPSCs.
Credit: Salk Institute
Results of new research may ease previous concerns that gene-editing techniques could add unwanted mutations to stem cells.
Researchers compared gene editing techniques in lines of induced pluripotent stem cells (iPSCs) derived from a patient with sickle cell disease (SCD).
And they found that neither viral nor nuclease-based gene-editing methods increased the frequency of mutations in the iPSCs.
The team reported these results in Cell Stem Cell.
“The ability to precisely modify the DNA of stem cells has greatly accelerated research on human diseases and cell therapy,” said senior study author Juan Carlos Izpisua Belmonte, PhD, of the Salk Institute for Biological Studies in La Jolla, California.
“To successfully translate this technology into the clinic, we first need to scrutinize the safety of these modified stem cells, such as their genome stability and mutational load.”
Previously, Dr Belmonte’s lab pioneered the use of modified viruses, called helper-dependent adenoviral vectors (HDAdVs), to correct the genetic mutation that causes SCD.
He and his colleagues used HDAdVs to replace the mutated gene in a line of iPSCs with a mutant-free version, creating stem cells that could, theoretically, be infused into patients’ bone marrow and help create healthy blood cells.
Before such technologies are applied to humans, though, Dr Belmonte and his colleagues wanted to know whether there were risks related to editing the genes in iPSCs.
“As cells are being reprogrammed into stem cells, they tend to accumulate many mutations,” said Mo Li, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“So people naturally worry that any process you perform with these cells in vitro—including gene editing—might generate even more mutations.”
To find out whether this was the case, the researchers conducted tests in a line of SCD-derived iPSCs.
They edited the genes of some cells using 1 of 2 HDAdV designs. And they edited others using 1 of 2 transcription activator-like effector nuclease (TALEN) proteins.
They kept the rest of the SCD iPSCs in culture without editing them. Then, the team sequenced the entire genome of each cell from the 4 edits and control experiment.
While all of the cells gained a low level of random gene mutations during the experiments, the cells that had undergone gene-editing—whether through HDAdV- or TALEN-based approaches—had no more mutations than the cells kept in culture.
“We were pleasantly surprised by the results,” said Keiichiro Suzuki, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“People have found thousands of mutations introduced during iPSC reprogramming. We found less than a hundred single nucleotide variants in all cases.”
The researchers noted that this finding doesn’t necessarily mean there are no inherent risks to using stem cells with edited genes. However, it does suggest the editing process doesn’t make iPSCs any less safe.
“We concluded that the risk of mutation isn’t inherently connected to gene editing,” Dr Li said. “These cells present the same risks as using any other cells manipulated for cell or gene therapy.”
The Belmonte group is now planning more studies to address whether gene-repair in other cell types, using other approaches, or targeting other genes could be more or less likely to cause unwanted mutations.
For now, they hope their findings encourage those in the field to keep pursuing gene-editing techniques as a potential way to treat genetic diseases in the future. ![]()
CAR T cells may fight fungal infections
T cells modified using the Sleeping Beauty gene transfer system may help fight infections caused by invasive Aspergillus fungus.
Sleeping Beauty is already being used to create chimeric antigen receptor (CAR) T cells to treat leukemias and lymphomas.
And now, researchers have found the system may also be effective for combatting fungal infections that can be deadly for immunosuppressed patients, such as those receiving transplants to treat hematologic cancers.
“We demonstrated a new approach for Aspergillus immunotherapy based on redirecting T-cell specificity through a CAR that recognizes carbohydrate antigen on the fungal cell wall,” said study author Laurence Cooper, MD, PhD, of MD Anderson Cancer Center in Houston, Texas.
He and his colleagues described this approach in the Proceedings of the National Academy of Sciences.
Dr Cooper originally learned about Sleeping Beauty gene transfer from a study published by Perry Hackett, PhD, a professor at the University of Minnesota who created the process.
The system is named Sleeping Beauty because Dr Hackett was able to “awaken” an extinct transposon—DNA that can replicate itself and insert the copy back into the genome—and package it with a gene he wants to transfer into a plasmid. An associated transposase enzyme binds to the plasmid, cuts the transposon and gene out of the plasmid, and pastes it into the target DNA sequence.
Dr Cooper and his colleagues have found they can use this process to engineer T cells that target sugar molecules in the Aspergillus cell walls, thereby killing the fungus.
Specifically, the team adapted the pattern-recognition receptor Dectin-1 to activate T cells via chimeric CD28 and CD3-ζ (D-CAR) upon binding with carbohydrate in the cell wall of Aspergillus germlings. They used Sleeping Beauty to modify the T cells to express D-CAR.
These D-CAR+ T cells exhibited specificity for β-glucan, and this inhibited Aspergillus growth both in vitro and in vivo. Furthermore, the researchers found that treating D-CAR+ T cells with steroids did not significantly compromise antifungal activity.
“The [D-CAR+ T cells] can be manipulated in a manner suitable for human application,” Dr Cooper said, “enabling this immunology to be translated into immunotherapy.”
T cells modified using the Sleeping Beauty gene transfer system may help fight infections caused by invasive Aspergillus fungus.
Sleeping Beauty is already being used to create chimeric antigen receptor (CAR) T cells to treat leukemias and lymphomas.
And now, researchers have found the system may also be effective for combatting fungal infections that can be deadly for immunosuppressed patients, such as those receiving transplants to treat hematologic cancers.
“We demonstrated a new approach for Aspergillus immunotherapy based on redirecting T-cell specificity through a CAR that recognizes carbohydrate antigen on the fungal cell wall,” said study author Laurence Cooper, MD, PhD, of MD Anderson Cancer Center in Houston, Texas.
He and his colleagues described this approach in the Proceedings of the National Academy of Sciences.
Dr Cooper originally learned about Sleeping Beauty gene transfer from a study published by Perry Hackett, PhD, a professor at the University of Minnesota who created the process.
The system is named Sleeping Beauty because Dr Hackett was able to “awaken” an extinct transposon—DNA that can replicate itself and insert the copy back into the genome—and package it with a gene he wants to transfer into a plasmid. An associated transposase enzyme binds to the plasmid, cuts the transposon and gene out of the plasmid, and pastes it into the target DNA sequence.
Dr Cooper and his colleagues have found they can use this process to engineer T cells that target sugar molecules in the Aspergillus cell walls, thereby killing the fungus.
Specifically, the team adapted the pattern-recognition receptor Dectin-1 to activate T cells via chimeric CD28 and CD3-ζ (D-CAR) upon binding with carbohydrate in the cell wall of Aspergillus germlings. They used Sleeping Beauty to modify the T cells to express D-CAR.
These D-CAR+ T cells exhibited specificity for β-glucan, and this inhibited Aspergillus growth both in vitro and in vivo. Furthermore, the researchers found that treating D-CAR+ T cells with steroids did not significantly compromise antifungal activity.
“The [D-CAR+ T cells] can be manipulated in a manner suitable for human application,” Dr Cooper said, “enabling this immunology to be translated into immunotherapy.”
T cells modified using the Sleeping Beauty gene transfer system may help fight infections caused by invasive Aspergillus fungus.
Sleeping Beauty is already being used to create chimeric antigen receptor (CAR) T cells to treat leukemias and lymphomas.
And now, researchers have found the system may also be effective for combatting fungal infections that can be deadly for immunosuppressed patients, such as those receiving transplants to treat hematologic cancers.
“We demonstrated a new approach for Aspergillus immunotherapy based on redirecting T-cell specificity through a CAR that recognizes carbohydrate antigen on the fungal cell wall,” said study author Laurence Cooper, MD, PhD, of MD Anderson Cancer Center in Houston, Texas.
He and his colleagues described this approach in the Proceedings of the National Academy of Sciences.
Dr Cooper originally learned about Sleeping Beauty gene transfer from a study published by Perry Hackett, PhD, a professor at the University of Minnesota who created the process.
The system is named Sleeping Beauty because Dr Hackett was able to “awaken” an extinct transposon—DNA that can replicate itself and insert the copy back into the genome—and package it with a gene he wants to transfer into a plasmid. An associated transposase enzyme binds to the plasmid, cuts the transposon and gene out of the plasmid, and pastes it into the target DNA sequence.
Dr Cooper and his colleagues have found they can use this process to engineer T cells that target sugar molecules in the Aspergillus cell walls, thereby killing the fungus.
Specifically, the team adapted the pattern-recognition receptor Dectin-1 to activate T cells via chimeric CD28 and CD3-ζ (D-CAR) upon binding with carbohydrate in the cell wall of Aspergillus germlings. They used Sleeping Beauty to modify the T cells to express D-CAR.
These D-CAR+ T cells exhibited specificity for β-glucan, and this inhibited Aspergillus growth both in vitro and in vivo. Furthermore, the researchers found that treating D-CAR+ T cells with steroids did not significantly compromise antifungal activity.
“The [D-CAR+ T cells] can be manipulated in a manner suitable for human application,” Dr Cooper said, “enabling this immunology to be translated into immunotherapy.”