User login
Flashing lights, floaters, and reduced vision
A 62-year-old woman has had flashing lights and floaters in her left eye with progressive loss of vision over the past month. She has not had recent trauma. She does not smoke.
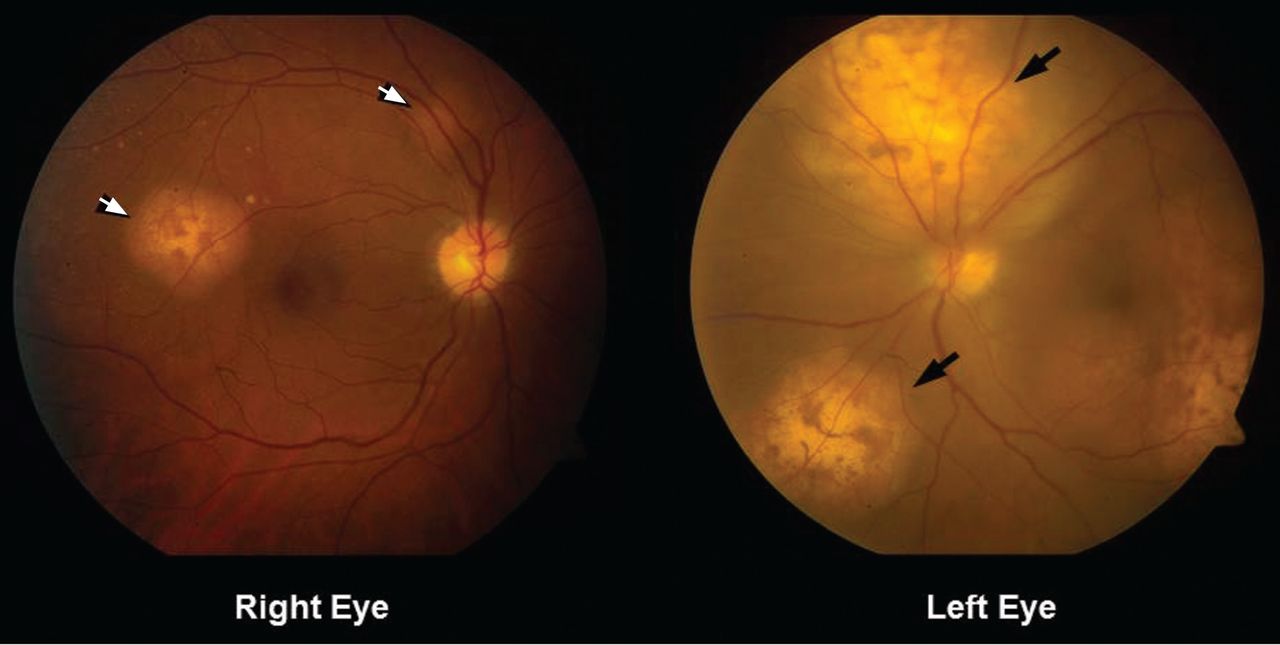
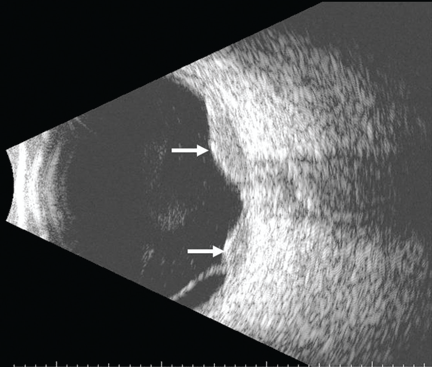
She was referred for an ophthalmologic evaluation. Her visual acuity was 20/20 in the right eye, but she could only count fingers with the left. The anterior segment appeared normal in both eyes. Funduscopic examination of the left eye revealed numerous lobulated, yellowish, choroidal lesions in the posterior pole with overlying subretinal fluid. The lesions involved the fovea, accounting for the poor visual acuity. There were two similar but smaller lesions in the right eye (Figure 1). Ultrasonography confirmed the choroidal location of the lesions (Figure 2).
Q: Which is the most likely diagnosis?
- Retinal detachment
- Choroidal melanoma
- Uveitis
- Uveal metastatic tumor
A: Uveal metastatic tumor is the correct diagnosis. Funduscopic findings of bilateral yellow choroidal lesions are consistent with metastatic cancer.
The patient was admitted to the hospital for a thorough evaluation. Computed tomography of the chest showed a 2.1-by-4.5-cm mass in the lower lobe of the left lung, highly suspicious for malignancy and associated with left hilar lymphadenopathy and right acute pulmonary embolism. Bronchoscopy showed an endobronchial tumor completely occluding the left lower lobe and the lingular orifices.
Pathologic specimens from the endobronchial tumor confirmed adenocarcinoma, consistent with a primary lung cancer.
THE OTHER DIAGNOSTIC CHOICES
Detachment or separation of the retina from the underlying pigment epithelium is one of the most commonly encountered eye emergencies.1 It requires urgent attention, since delay in treatment can cause permanent vision loss.
Retinal detachment differs from uveal metastatic tumor in that it presents and progresses rapidly. The common signs and symptoms are floaters in the center of the visual axis, a sensation of flashing lights (related to retinal traction), and, eventually, loss of vision. The detachment most often represents a break or tear (rhegmatogenous retinal detachment), but it is also a common sequela of neglected diabetic retinopathy. Exudative retinal detachment is usually secondary to uveal inflammation or a uveal tumor.
Choroidal melanoma, the most common primary intraocular malignancy, arises from melanocytes within the choroid. In most cases, it develops from preexisting melanocytic nevi.2 It may present as blurred vision, a paracentral scotoma, painless and progressive visual field loss, and floaters. Choroidal melanoma is usually pigmented (dark brown) and is invariably unilateral.
Uveitis is an inflammation of the uveal tract, which includes the iris, ciliary body, and choroid. It is classified as anterior, intermediate, or posterior uveitis or as panuveitis.3
Although flashing lights, floaters, and reduced vision can occur in uveitis, its other important presenting symptoms (ie, pain, redness, and photophobia) were absent in this patient. The absence of anterior chamber cells and corneal inflammatory deposits (keratic precipitates) also made uveitis less likely.4 However, granulomatous uveitis such as sarcoidosis can present as nodular thickening of the uvea, mimicking an intraocular tumor.5
THE MOST COMMON INTRAOCULAR MALIGNANCY
Uveal metastasis is the most common intraocular malignancy6 and is found on autopsy in up to 12% of people who die of cancer; it involves both eyes in 4.4% of cases. Multiple metastases are seen in one eye in up to 20% of cases.7
The tumors are most often in the choroid, probably because of its extensive blood supply. Breast cancer (in women) and lung cancer (in men) are the most common cancers with uveal metastasis.8 Uveal metastasis from cancers of the prostate, kidney, thyroid, and gastrointestinal tract and from lymphoma and leukemia is less common.8
Patients with choroidal metastasis can see flashing lights, floating spots, and distortion of their vision. In such patients, a careful history and physical examination can uncover signs and symptoms of the hidden cancer, especially of lung cancer.9
Once uveal metastasis is suspected, both eyes and orbits and the central nervous system should be examined, as this disease tends to present bilaterally and to involve the central nervous system.10 Uveal metastases respond to chemotherapy and radiotherapy, depending on the nature of the primary tumor. In general, treatment is based on the extent of the metastasis, prior treatments, and the patient’s overall functional status.
- Hatten B, Browne V. Retinal detachment. Emerg Med J 2011; 28:83.
- Factors predictive of growth and treatment of small choroidal melanoma: COMS Report No. 5. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol 1997; 115:1537–1544.
- Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveitis Nomenclature (SUN) Working Group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol 2005; 140:509–516.
- Wertheim MS, Mathers WD, Planck SJ, et al. In vivo confocal microscopy of keratic precipitates. Arch Ophthalmol 2004; 122:1773–1781.
- Desai UR, Tawansy KA, Joondeph BC, Schiffman RM. Choroidal granulomas in systemic sarcoidosis. Retina 2001; 21:40–47.
- Singh AD, Damato BE, Pe’er J, Murphree AL, Perry JD, eds. Uveal metastatic tumors. In: Clinical Ophthalmic Oncology. Philadelphia, PA: Saunders-Elsevier; 2007:322–327.
- Eliassi-Rad B, Albert DM, Green WR. Frequency of ocular metastases in patients dying of cancer in eye bank populations. Br J Ophthalmol 1996; 80:125–128.
- Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology 1997; 104:1265–1276.
- Herrag M, Lahmiti S, Yazidi AA, Le Lez ML, Diot P. Choroidal metastasis revealing a lung adenocarcinoma. Ann Thorac Surg 2010; 89:1013–1014.
- Kanthan GL, Jayamohan J, Yip D, Conway RM. Management of metastatic carcinoma of the uveal tract: an evidence-based analysis. Clin Exp Ophthalmol 2007; 35:553–565.
A 62-year-old woman has had flashing lights and floaters in her left eye with progressive loss of vision over the past month. She has not had recent trauma. She does not smoke.
She was referred for an ophthalmologic evaluation. Her visual acuity was 20/20 in the right eye, but she could only count fingers with the left. The anterior segment appeared normal in both eyes. Funduscopic examination of the left eye revealed numerous lobulated, yellowish, choroidal lesions in the posterior pole with overlying subretinal fluid. The lesions involved the fovea, accounting for the poor visual acuity. There were two similar but smaller lesions in the right eye (Figure 1). Ultrasonography confirmed the choroidal location of the lesions (Figure 2).
Q: Which is the most likely diagnosis?
- Retinal detachment
- Choroidal melanoma
- Uveitis
- Uveal metastatic tumor
A: Uveal metastatic tumor is the correct diagnosis. Funduscopic findings of bilateral yellow choroidal lesions are consistent with metastatic cancer.
The patient was admitted to the hospital for a thorough evaluation. Computed tomography of the chest showed a 2.1-by-4.5-cm mass in the lower lobe of the left lung, highly suspicious for malignancy and associated with left hilar lymphadenopathy and right acute pulmonary embolism. Bronchoscopy showed an endobronchial tumor completely occluding the left lower lobe and the lingular orifices.
Pathologic specimens from the endobronchial tumor confirmed adenocarcinoma, consistent with a primary lung cancer.
THE OTHER DIAGNOSTIC CHOICES
Detachment or separation of the retina from the underlying pigment epithelium is one of the most commonly encountered eye emergencies.1 It requires urgent attention, since delay in treatment can cause permanent vision loss.
Retinal detachment differs from uveal metastatic tumor in that it presents and progresses rapidly. The common signs and symptoms are floaters in the center of the visual axis, a sensation of flashing lights (related to retinal traction), and, eventually, loss of vision. The detachment most often represents a break or tear (rhegmatogenous retinal detachment), but it is also a common sequela of neglected diabetic retinopathy. Exudative retinal detachment is usually secondary to uveal inflammation or a uveal tumor.
Choroidal melanoma, the most common primary intraocular malignancy, arises from melanocytes within the choroid. In most cases, it develops from preexisting melanocytic nevi.2 It may present as blurred vision, a paracentral scotoma, painless and progressive visual field loss, and floaters. Choroidal melanoma is usually pigmented (dark brown) and is invariably unilateral.
Uveitis is an inflammation of the uveal tract, which includes the iris, ciliary body, and choroid. It is classified as anterior, intermediate, or posterior uveitis or as panuveitis.3
Although flashing lights, floaters, and reduced vision can occur in uveitis, its other important presenting symptoms (ie, pain, redness, and photophobia) were absent in this patient. The absence of anterior chamber cells and corneal inflammatory deposits (keratic precipitates) also made uveitis less likely.4 However, granulomatous uveitis such as sarcoidosis can present as nodular thickening of the uvea, mimicking an intraocular tumor.5
THE MOST COMMON INTRAOCULAR MALIGNANCY
Uveal metastasis is the most common intraocular malignancy6 and is found on autopsy in up to 12% of people who die of cancer; it involves both eyes in 4.4% of cases. Multiple metastases are seen in one eye in up to 20% of cases.7
The tumors are most often in the choroid, probably because of its extensive blood supply. Breast cancer (in women) and lung cancer (in men) are the most common cancers with uveal metastasis.8 Uveal metastasis from cancers of the prostate, kidney, thyroid, and gastrointestinal tract and from lymphoma and leukemia is less common.8
Patients with choroidal metastasis can see flashing lights, floating spots, and distortion of their vision. In such patients, a careful history and physical examination can uncover signs and symptoms of the hidden cancer, especially of lung cancer.9
Once uveal metastasis is suspected, both eyes and orbits and the central nervous system should be examined, as this disease tends to present bilaterally and to involve the central nervous system.10 Uveal metastases respond to chemotherapy and radiotherapy, depending on the nature of the primary tumor. In general, treatment is based on the extent of the metastasis, prior treatments, and the patient’s overall functional status.
A 62-year-old woman has had flashing lights and floaters in her left eye with progressive loss of vision over the past month. She has not had recent trauma. She does not smoke.
She was referred for an ophthalmologic evaluation. Her visual acuity was 20/20 in the right eye, but she could only count fingers with the left. The anterior segment appeared normal in both eyes. Funduscopic examination of the left eye revealed numerous lobulated, yellowish, choroidal lesions in the posterior pole with overlying subretinal fluid. The lesions involved the fovea, accounting for the poor visual acuity. There were two similar but smaller lesions in the right eye (Figure 1). Ultrasonography confirmed the choroidal location of the lesions (Figure 2).
Q: Which is the most likely diagnosis?
- Retinal detachment
- Choroidal melanoma
- Uveitis
- Uveal metastatic tumor
A: Uveal metastatic tumor is the correct diagnosis. Funduscopic findings of bilateral yellow choroidal lesions are consistent with metastatic cancer.
The patient was admitted to the hospital for a thorough evaluation. Computed tomography of the chest showed a 2.1-by-4.5-cm mass in the lower lobe of the left lung, highly suspicious for malignancy and associated with left hilar lymphadenopathy and right acute pulmonary embolism. Bronchoscopy showed an endobronchial tumor completely occluding the left lower lobe and the lingular orifices.
Pathologic specimens from the endobronchial tumor confirmed adenocarcinoma, consistent with a primary lung cancer.
THE OTHER DIAGNOSTIC CHOICES
Detachment or separation of the retina from the underlying pigment epithelium is one of the most commonly encountered eye emergencies.1 It requires urgent attention, since delay in treatment can cause permanent vision loss.
Retinal detachment differs from uveal metastatic tumor in that it presents and progresses rapidly. The common signs and symptoms are floaters in the center of the visual axis, a sensation of flashing lights (related to retinal traction), and, eventually, loss of vision. The detachment most often represents a break or tear (rhegmatogenous retinal detachment), but it is also a common sequela of neglected diabetic retinopathy. Exudative retinal detachment is usually secondary to uveal inflammation or a uveal tumor.
Choroidal melanoma, the most common primary intraocular malignancy, arises from melanocytes within the choroid. In most cases, it develops from preexisting melanocytic nevi.2 It may present as blurred vision, a paracentral scotoma, painless and progressive visual field loss, and floaters. Choroidal melanoma is usually pigmented (dark brown) and is invariably unilateral.
Uveitis is an inflammation of the uveal tract, which includes the iris, ciliary body, and choroid. It is classified as anterior, intermediate, or posterior uveitis or as panuveitis.3
Although flashing lights, floaters, and reduced vision can occur in uveitis, its other important presenting symptoms (ie, pain, redness, and photophobia) were absent in this patient. The absence of anterior chamber cells and corneal inflammatory deposits (keratic precipitates) also made uveitis less likely.4 However, granulomatous uveitis such as sarcoidosis can present as nodular thickening of the uvea, mimicking an intraocular tumor.5
THE MOST COMMON INTRAOCULAR MALIGNANCY
Uveal metastasis is the most common intraocular malignancy6 and is found on autopsy in up to 12% of people who die of cancer; it involves both eyes in 4.4% of cases. Multiple metastases are seen in one eye in up to 20% of cases.7
The tumors are most often in the choroid, probably because of its extensive blood supply. Breast cancer (in women) and lung cancer (in men) are the most common cancers with uveal metastasis.8 Uveal metastasis from cancers of the prostate, kidney, thyroid, and gastrointestinal tract and from lymphoma and leukemia is less common.8
Patients with choroidal metastasis can see flashing lights, floating spots, and distortion of their vision. In such patients, a careful history and physical examination can uncover signs and symptoms of the hidden cancer, especially of lung cancer.9
Once uveal metastasis is suspected, both eyes and orbits and the central nervous system should be examined, as this disease tends to present bilaterally and to involve the central nervous system.10 Uveal metastases respond to chemotherapy and radiotherapy, depending on the nature of the primary tumor. In general, treatment is based on the extent of the metastasis, prior treatments, and the patient’s overall functional status.
- Hatten B, Browne V. Retinal detachment. Emerg Med J 2011; 28:83.
- Factors predictive of growth and treatment of small choroidal melanoma: COMS Report No. 5. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol 1997; 115:1537–1544.
- Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveitis Nomenclature (SUN) Working Group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol 2005; 140:509–516.
- Wertheim MS, Mathers WD, Planck SJ, et al. In vivo confocal microscopy of keratic precipitates. Arch Ophthalmol 2004; 122:1773–1781.
- Desai UR, Tawansy KA, Joondeph BC, Schiffman RM. Choroidal granulomas in systemic sarcoidosis. Retina 2001; 21:40–47.
- Singh AD, Damato BE, Pe’er J, Murphree AL, Perry JD, eds. Uveal metastatic tumors. In: Clinical Ophthalmic Oncology. Philadelphia, PA: Saunders-Elsevier; 2007:322–327.
- Eliassi-Rad B, Albert DM, Green WR. Frequency of ocular metastases in patients dying of cancer in eye bank populations. Br J Ophthalmol 1996; 80:125–128.
- Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology 1997; 104:1265–1276.
- Herrag M, Lahmiti S, Yazidi AA, Le Lez ML, Diot P. Choroidal metastasis revealing a lung adenocarcinoma. Ann Thorac Surg 2010; 89:1013–1014.
- Kanthan GL, Jayamohan J, Yip D, Conway RM. Management of metastatic carcinoma of the uveal tract: an evidence-based analysis. Clin Exp Ophthalmol 2007; 35:553–565.
- Hatten B, Browne V. Retinal detachment. Emerg Med J 2011; 28:83.
- Factors predictive of growth and treatment of small choroidal melanoma: COMS Report No. 5. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol 1997; 115:1537–1544.
- Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveitis Nomenclature (SUN) Working Group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol 2005; 140:509–516.
- Wertheim MS, Mathers WD, Planck SJ, et al. In vivo confocal microscopy of keratic precipitates. Arch Ophthalmol 2004; 122:1773–1781.
- Desai UR, Tawansy KA, Joondeph BC, Schiffman RM. Choroidal granulomas in systemic sarcoidosis. Retina 2001; 21:40–47.
- Singh AD, Damato BE, Pe’er J, Murphree AL, Perry JD, eds. Uveal metastatic tumors. In: Clinical Ophthalmic Oncology. Philadelphia, PA: Saunders-Elsevier; 2007:322–327.
- Eliassi-Rad B, Albert DM, Green WR. Frequency of ocular metastases in patients dying of cancer in eye bank populations. Br J Ophthalmol 1996; 80:125–128.
- Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology 1997; 104:1265–1276.
- Herrag M, Lahmiti S, Yazidi AA, Le Lez ML, Diot P. Choroidal metastasis revealing a lung adenocarcinoma. Ann Thorac Surg 2010; 89:1013–1014.
- Kanthan GL, Jayamohan J, Yip D, Conway RM. Management of metastatic carcinoma of the uveal tract: an evidence-based analysis. Clin Exp Ophthalmol 2007; 35:553–565.
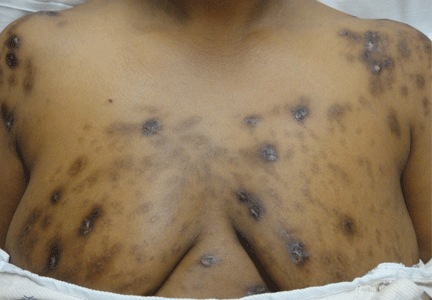
Hyperpigmentation and hypotension
A 65-year-old man presents with a 2-month history of generalized weakness, dizziness, and blurred vision. His symptoms began gradually and have been progressing over the last few weeks, so that they now affect his ability to perform normal daily activities.
He has lost 20 lb and has become anorectic. He has no fever, night sweats, headache, cough, hemoptysis, or dyspnea. He has no history of abdominal pain, changes in bowel habits, nausea, vomiting, or urinary symptoms. He was admitted 6 weeks ago for the same symptoms; he was treated for hypotension and received intravenous (IV) fluids and electrolyte supplements for dehydration.
He has a history of hypertension, stroke, vascular dementia, and atrial fibrillation. He is taking warfarin (Coumadin), extended-release diltiazem (Cardizem), simvastatin (Zocor), and donepezil (Aricept). He underwent right hemicolectomy 5 years ago for a large tubular adenoma with high-grade dysplasia in the cecum.
Initial laboratory values are as follows:
- White blood cell count 7.4 × 109/L (reference range 4.5–11.0), with a normal differential
- Mild anemia, with a hemoglobin of 116 g/L (140–175)
- Activated partial thromboplastin time 59.9 sec (23.0–32.4)
- Serum sodium 135 mmol/L (136–142)
- Serum potassium 4.6 mmol/L (3.5–5.0)
- Aspartate aminotransferase 58 U/L (10–30)
- Alanine aminotransferase 16 U/L (10–40)
- Alkaline phosphatase 328 U/L (30–120)
- Urea, creatinine, and corrected calcium are normal.
Electrocardiography shows atrial fibrillation with low-voltage QRS complexes. Chest radiography is normal. A stool test is negative for occult blood. A workup for sepsis is negative.
Q: Which is the appropriate test at this point to determine the cause of the hypotension?
- Serum parathyroid-hormone-related protein
- Baseline serum cortisol, plasma adrenocorticotropic hormone (ACTH) levels, and an ACTH stimulation test with cosyntropin (Cortrosyn)
- Serum thyrotropin level
- Aspiration biopsy of subcutaneous fat with Congo red and immunostaining
- Late-night salivary cortisol
A: The correct next step is to measure baseline serum cortisol, to test ACTH levels, and to order an ACTH stimulation test with cosyntropin.
Primary adrenocortical insufficiency should be considered in patients with metastatic malignancy who present with peripheral vascular collapse, particularly when it is associated with cutaneous hyperpigmentation, chronic malaise, fatigue, weakness, anorexia, weight loss, hypoglycemia, and electrolyte disturbances such as hyponatremia and hyperkalemia.
Checking the baseline serum cortisol and ACTH levels and cosyntropin stimulation testing are vital steps in making an early diagnosis of primary adrenocortical insufficiency. Inappropriately low serum cortisol is highly suggestive of primary adrenal insufficiency, especially if accompanied by simultaneous elevation of the plasma ACTH level. The result of the ACTH stimulation test with cosyntropin is often confirmatory.
Measuring the serum parathyroid-hormone-related protein level is not indicated, since the patient has a normal corrected calcium. Patients with ectopic Cushing syndrome may present with weight loss due to underlying malignancy, but the presence of hypotension and a lack of hypokalemia makes such a diagnosis unlikely, and, therefore, measurement of late-night salivary cortisol is not the best answer. Amyloidosis, hypothyroidism, or hyperthyroidism are unlikely to have this patient’s presentation.
RESULTS OF FURTHER EVALUATION
Our patient’s ACTH serum level was elevated, and an ACTH stimulation test with cosyntropin confirmed the diagnosis of primary adrenal insufficiency.
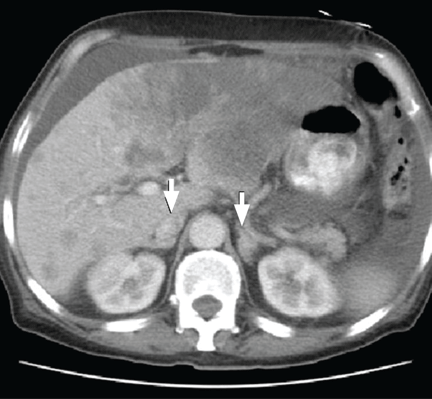
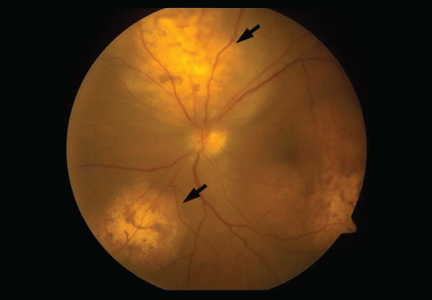
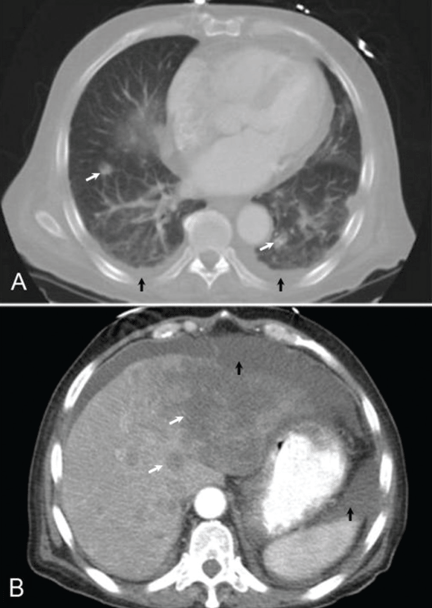
CT of the abdomen failed to demonstrate primary tumors, but both adrenal glands were enlarged, likely from metastasis (Figure 4). His hypotension responded to treatment with hydrocortisone and fludrocortisone, and his symptoms resolved. No further testing or therapy was directed to the primary occult malignancy, as it was considered advanced. The prognosis was discussed with the patient, and he deferred any further management and was discharged to hospice care. He died a few months later.
PRIMARY ADRENOCORTICAL INSUFFICIENCY
Primary adrenocortical insufficiency is an uncommon disorder caused by destruction or dysfunction of the adrenal cortices. It is characterized by chronic deficiency of cortisol, aldosterone, and adrenal androgens. In the United States, nearly 6 million people are considered to have undiagnosed adrenal insufficiency, which is clinically significant only during times of physiologic stress.1
Primary adrenocortical insufficiency affects men and women equally. However, the idiopathic autoimmune form of adrenal insufficiency (Addison disease) is two to three times more common in women than in men.
If the condition is undiagnosed or ineffectively treated, the risk of significant morbidity and death is high. Symptoms and signs are nonspecific, and the onset is insidious.
Almost all patients with primary adrenal insufficiency have malaise, fatigue, anorexia, and weight loss. Vomiting, abdominal pain, and fever are more common during an adrenal crisis, when a patient with subclinical disease is subjected to major stress. Postural dizziness or syncope is a common result of volume depletion and hypotension.2–4 It is commonly accompanied by hyponatremia and hyperkalemia.
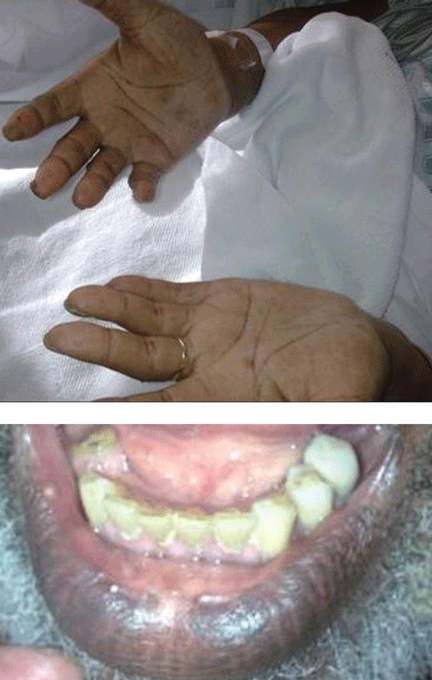
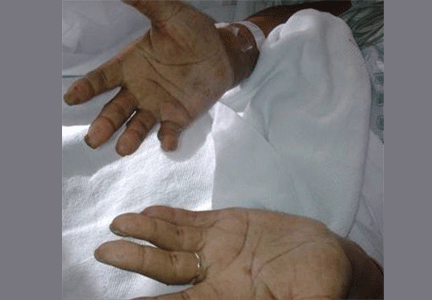
Hyperpigmentation is the most characteristic physical finding and is caused by an ACTH-mediated increase in melanin content in the skin.2,4,5 The resulting brown hyperpigmentation is most obvious in areas exposed to sunlight (face, neck, backs of hands), and in areas exposed to chronic friction or pressure, such as the elbows, knees, knuckles, waist, and shoulders (brassiere straps).4 Pigmentation is also prominent in the palmar creases, areolae, axillae, perineum, surgical scars, and umbilicus. Other patterns of hyperpigmentation are patchy pigmentation on the inner surface of lips, the buccal mucosa, under the tongue, and on the hard palate.3,5 The hyperpigmentation begins to fade within several days and largely disappears after a few months of adequate glucocorticoid therapy.4
In the United States, 80% of cases of primary adrenocortical insufficiency are caused by autoimmune adrenal destruction. The remainder are caused by infectious diseases (eg, tuberculosis, fungal infection, cytomegalovirus infection, and Mycobacterium aviumintracellulare infection in the context of human immunodeficiency virus infection), by infiltration of the adrenal glands by metastatic cancer, by adrenal hemorrhage, or by drugs such as ketoconazole, fluconazole (Diflucan), metyrapone (Metopirone), mitotane (Lysodren), and etomidate (Amidate).4,6
Adrenal metastatic disease
Infiltration of the adrenal glands by metastatic cancer is not uncommon, probably because of their rich sinusoidal blood supply, and the adrenals are the fourth most common site of metastasis. Common primary tumors are lung, breast, melanoma, gastric, esophageal, and colorectal cancers, while metastasis due to an undetermined primary tumor is the least common.7
Clinically evident adrenal insufficiency produced by metastatic carcinoma is uncommon because most of the adrenal cortex must be destroyed before hypofunction becomes evident.7–9
Malignancy rarely presents first as adrenal insufficiency caused by metastatic infiltration.10
Hormonal therapy may significantly improve symptoms and quality of life in patients with metastatic adrenal insufficiency.8,11
DIAGNOSIS AND MANAGEMENT
Once primary adrenal insufficiency is suspected, prompt diagnosis and treatment are essential. A low plasma cortisol level (< 3 μg/dL) at 8 am is highly suggestive of adrenal insufficiency if exposure to exogenous glucocorticoids has been excluded (including oral, inhaled, and injected),12,13 especially if accompanied by simultaneous elevation of the plasma ACTH level (usually > 200 pg/mL). An 8 am cortisol concentration above 15 μg/dL makes adrenal insufficiency highly unlikely, but levels between 3 and 15 μg/dL are nondiagnostic and need to be further evaluated by an ACTH stimulation test with cosyntropin.4,7
Imaging in primary adrenal insufficiency may be considered when the condition is not clearly autoimmune.14 Abdominal CT is the ideal imaging test for detecting abnormal adrenal glands. CT shows small, noncalcified adrenals in autoimmune Addison disease. It demonstrates enlarged adrenals in about 85% of cases caused by metastatic or granulomatous disease; and calcification is noted in cases of tuberculous adrenal disease.4
Management involves treating the underlying cause and starting hormone replacement therapy. Hormonal therapy consists of corticosteroids and mineralocorticoids; hydrocortisone is the drug of choice and is usually given with fludrocortisone acetate, which has a potent sodium-retaining effect. In the presence of a stressor (fever, surgery, severe illness), the dose of hydrocortisone should be doubled (> 50 mg hydrocortisone per day) for at least 3 to 5 days.2,4
- Erichsen MM, Løvås K, Fougner KJ, et al. Normal overall mortality rate in Addison’s disease, but young patients are at risk of premature death. Eur J Endocrinol 2009; 160:233–237.
- Oelkers W. Adrenal insufficiency. N Engl J Med 1996; 335:1206–1212.
- Redman BG, Pazdur R, Zingas AP, Loredo R. Prospective evaluation of adrenal insufficiency in patients with adrenal metastasis. Cancer 1987; 60:103–107.
- Berger M., Hypofunction of the adrenal cortex in infancy. Manit Med Rev 1949; 29:132.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, café au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Zelissen PM, Bast EJ, Croughs RJ. Associated autoimmunity in Addison’s disease. J Autoimmun 1995; 8:121–130.
- Lutz A, Stojkovic M, Schmidt M, Arlt W, Allolio B, Reincke M. Adrenocortical function in patients with macrometastases of the adrenal gland. Eur J Endocrinol 2000; 143:91–97.
- Kung AW, Pun KK, Lam K, Wang C, Leung CY. Addisonian crisis as presenting feature in malignancies. Cancer 1990; 65:177–179.
- Cedermark BJ, Sjöberg HE. The clinical significance of metastases to the adrenal glands. Surg Gynecol Obstet 1981; 152:607–610.
- Rosenthal FD, Davies MK, Burden AC. Malignant disease presenting as Addison’s disease. Br Med J 1978; 1:1591–1592.
- Seidenwurm DJ, Elmer EB, Kaplan LM, Williams EK, Morris DG, Hoffman AR. Metastases to the adrenal glands and the development of Addison’s disease. Cancer 1984; 54:552–557.
- Santiago AH, Ratzan S. Acute adrenal crisis in an asthmatic child treated with inhaled fluticasone proprionate. Int J Pediatr Endocrinol 2010; 2010. pii:749239.
- Holme J, Tomlinson JW, Stockley RA, Stewart PM, Barlow N, Sullivan AL. Adrenal suppression in bronchiectasis and the impact of inhaled corticosteroids. Eur Respir J 2008; 32:1047–1052.
- Mohammad K, Sadikot RT. Adrenal insufficiency as a presenting manifestation of nonsmall cell lung cancer. South Med J 2009; 102:665–667.
A 65-year-old man presents with a 2-month history of generalized weakness, dizziness, and blurred vision. His symptoms began gradually and have been progressing over the last few weeks, so that they now affect his ability to perform normal daily activities.
He has lost 20 lb and has become anorectic. He has no fever, night sweats, headache, cough, hemoptysis, or dyspnea. He has no history of abdominal pain, changes in bowel habits, nausea, vomiting, or urinary symptoms. He was admitted 6 weeks ago for the same symptoms; he was treated for hypotension and received intravenous (IV) fluids and electrolyte supplements for dehydration.
He has a history of hypertension, stroke, vascular dementia, and atrial fibrillation. He is taking warfarin (Coumadin), extended-release diltiazem (Cardizem), simvastatin (Zocor), and donepezil (Aricept). He underwent right hemicolectomy 5 years ago for a large tubular adenoma with high-grade dysplasia in the cecum.
Initial laboratory values are as follows:
- White blood cell count 7.4 × 109/L (reference range 4.5–11.0), with a normal differential
- Mild anemia, with a hemoglobin of 116 g/L (140–175)
- Activated partial thromboplastin time 59.9 sec (23.0–32.4)
- Serum sodium 135 mmol/L (136–142)
- Serum potassium 4.6 mmol/L (3.5–5.0)
- Aspartate aminotransferase 58 U/L (10–30)
- Alanine aminotransferase 16 U/L (10–40)
- Alkaline phosphatase 328 U/L (30–120)
- Urea, creatinine, and corrected calcium are normal.
Electrocardiography shows atrial fibrillation with low-voltage QRS complexes. Chest radiography is normal. A stool test is negative for occult blood. A workup for sepsis is negative.
Q: Which is the appropriate test at this point to determine the cause of the hypotension?
- Serum parathyroid-hormone-related protein
- Baseline serum cortisol, plasma adrenocorticotropic hormone (ACTH) levels, and an ACTH stimulation test with cosyntropin (Cortrosyn)
- Serum thyrotropin level
- Aspiration biopsy of subcutaneous fat with Congo red and immunostaining
- Late-night salivary cortisol
A: The correct next step is to measure baseline serum cortisol, to test ACTH levels, and to order an ACTH stimulation test with cosyntropin.
Primary adrenocortical insufficiency should be considered in patients with metastatic malignancy who present with peripheral vascular collapse, particularly when it is associated with cutaneous hyperpigmentation, chronic malaise, fatigue, weakness, anorexia, weight loss, hypoglycemia, and electrolyte disturbances such as hyponatremia and hyperkalemia.
Checking the baseline serum cortisol and ACTH levels and cosyntropin stimulation testing are vital steps in making an early diagnosis of primary adrenocortical insufficiency. Inappropriately low serum cortisol is highly suggestive of primary adrenal insufficiency, especially if accompanied by simultaneous elevation of the plasma ACTH level. The result of the ACTH stimulation test with cosyntropin is often confirmatory.
Measuring the serum parathyroid-hormone-related protein level is not indicated, since the patient has a normal corrected calcium. Patients with ectopic Cushing syndrome may present with weight loss due to underlying malignancy, but the presence of hypotension and a lack of hypokalemia makes such a diagnosis unlikely, and, therefore, measurement of late-night salivary cortisol is not the best answer. Amyloidosis, hypothyroidism, or hyperthyroidism are unlikely to have this patient’s presentation.
RESULTS OF FURTHER EVALUATION
Our patient’s ACTH serum level was elevated, and an ACTH stimulation test with cosyntropin confirmed the diagnosis of primary adrenal insufficiency.
CT of the abdomen failed to demonstrate primary tumors, but both adrenal glands were enlarged, likely from metastasis (Figure 4). His hypotension responded to treatment with hydrocortisone and fludrocortisone, and his symptoms resolved. No further testing or therapy was directed to the primary occult malignancy, as it was considered advanced. The prognosis was discussed with the patient, and he deferred any further management and was discharged to hospice care. He died a few months later.
PRIMARY ADRENOCORTICAL INSUFFICIENCY
Primary adrenocortical insufficiency is an uncommon disorder caused by destruction or dysfunction of the adrenal cortices. It is characterized by chronic deficiency of cortisol, aldosterone, and adrenal androgens. In the United States, nearly 6 million people are considered to have undiagnosed adrenal insufficiency, which is clinically significant only during times of physiologic stress.1
Primary adrenocortical insufficiency affects men and women equally. However, the idiopathic autoimmune form of adrenal insufficiency (Addison disease) is two to three times more common in women than in men.
If the condition is undiagnosed or ineffectively treated, the risk of significant morbidity and death is high. Symptoms and signs are nonspecific, and the onset is insidious.
Almost all patients with primary adrenal insufficiency have malaise, fatigue, anorexia, and weight loss. Vomiting, abdominal pain, and fever are more common during an adrenal crisis, when a patient with subclinical disease is subjected to major stress. Postural dizziness or syncope is a common result of volume depletion and hypotension.2–4 It is commonly accompanied by hyponatremia and hyperkalemia.
Hyperpigmentation is the most characteristic physical finding and is caused by an ACTH-mediated increase in melanin content in the skin.2,4,5 The resulting brown hyperpigmentation is most obvious in areas exposed to sunlight (face, neck, backs of hands), and in areas exposed to chronic friction or pressure, such as the elbows, knees, knuckles, waist, and shoulders (brassiere straps).4 Pigmentation is also prominent in the palmar creases, areolae, axillae, perineum, surgical scars, and umbilicus. Other patterns of hyperpigmentation are patchy pigmentation on the inner surface of lips, the buccal mucosa, under the tongue, and on the hard palate.3,5 The hyperpigmentation begins to fade within several days and largely disappears after a few months of adequate glucocorticoid therapy.4
In the United States, 80% of cases of primary adrenocortical insufficiency are caused by autoimmune adrenal destruction. The remainder are caused by infectious diseases (eg, tuberculosis, fungal infection, cytomegalovirus infection, and Mycobacterium aviumintracellulare infection in the context of human immunodeficiency virus infection), by infiltration of the adrenal glands by metastatic cancer, by adrenal hemorrhage, or by drugs such as ketoconazole, fluconazole (Diflucan), metyrapone (Metopirone), mitotane (Lysodren), and etomidate (Amidate).4,6
Adrenal metastatic disease
Infiltration of the adrenal glands by metastatic cancer is not uncommon, probably because of their rich sinusoidal blood supply, and the adrenals are the fourth most common site of metastasis. Common primary tumors are lung, breast, melanoma, gastric, esophageal, and colorectal cancers, while metastasis due to an undetermined primary tumor is the least common.7
Clinically evident adrenal insufficiency produced by metastatic carcinoma is uncommon because most of the adrenal cortex must be destroyed before hypofunction becomes evident.7–9
Malignancy rarely presents first as adrenal insufficiency caused by metastatic infiltration.10
Hormonal therapy may significantly improve symptoms and quality of life in patients with metastatic adrenal insufficiency.8,11
DIAGNOSIS AND MANAGEMENT
Once primary adrenal insufficiency is suspected, prompt diagnosis and treatment are essential. A low plasma cortisol level (< 3 μg/dL) at 8 am is highly suggestive of adrenal insufficiency if exposure to exogenous glucocorticoids has been excluded (including oral, inhaled, and injected),12,13 especially if accompanied by simultaneous elevation of the plasma ACTH level (usually > 200 pg/mL). An 8 am cortisol concentration above 15 μg/dL makes adrenal insufficiency highly unlikely, but levels between 3 and 15 μg/dL are nondiagnostic and need to be further evaluated by an ACTH stimulation test with cosyntropin.4,7
Imaging in primary adrenal insufficiency may be considered when the condition is not clearly autoimmune.14 Abdominal CT is the ideal imaging test for detecting abnormal adrenal glands. CT shows small, noncalcified adrenals in autoimmune Addison disease. It demonstrates enlarged adrenals in about 85% of cases caused by metastatic or granulomatous disease; and calcification is noted in cases of tuberculous adrenal disease.4
Management involves treating the underlying cause and starting hormone replacement therapy. Hormonal therapy consists of corticosteroids and mineralocorticoids; hydrocortisone is the drug of choice and is usually given with fludrocortisone acetate, which has a potent sodium-retaining effect. In the presence of a stressor (fever, surgery, severe illness), the dose of hydrocortisone should be doubled (> 50 mg hydrocortisone per day) for at least 3 to 5 days.2,4
A 65-year-old man presents with a 2-month history of generalized weakness, dizziness, and blurred vision. His symptoms began gradually and have been progressing over the last few weeks, so that they now affect his ability to perform normal daily activities.
He has lost 20 lb and has become anorectic. He has no fever, night sweats, headache, cough, hemoptysis, or dyspnea. He has no history of abdominal pain, changes in bowel habits, nausea, vomiting, or urinary symptoms. He was admitted 6 weeks ago for the same symptoms; he was treated for hypotension and received intravenous (IV) fluids and electrolyte supplements for dehydration.
He has a history of hypertension, stroke, vascular dementia, and atrial fibrillation. He is taking warfarin (Coumadin), extended-release diltiazem (Cardizem), simvastatin (Zocor), and donepezil (Aricept). He underwent right hemicolectomy 5 years ago for a large tubular adenoma with high-grade dysplasia in the cecum.
Initial laboratory values are as follows:
- White blood cell count 7.4 × 109/L (reference range 4.5–11.0), with a normal differential
- Mild anemia, with a hemoglobin of 116 g/L (140–175)
- Activated partial thromboplastin time 59.9 sec (23.0–32.4)
- Serum sodium 135 mmol/L (136–142)
- Serum potassium 4.6 mmol/L (3.5–5.0)
- Aspartate aminotransferase 58 U/L (10–30)
- Alanine aminotransferase 16 U/L (10–40)
- Alkaline phosphatase 328 U/L (30–120)
- Urea, creatinine, and corrected calcium are normal.
Electrocardiography shows atrial fibrillation with low-voltage QRS complexes. Chest radiography is normal. A stool test is negative for occult blood. A workup for sepsis is negative.
Q: Which is the appropriate test at this point to determine the cause of the hypotension?
- Serum parathyroid-hormone-related protein
- Baseline serum cortisol, plasma adrenocorticotropic hormone (ACTH) levels, and an ACTH stimulation test with cosyntropin (Cortrosyn)
- Serum thyrotropin level
- Aspiration biopsy of subcutaneous fat with Congo red and immunostaining
- Late-night salivary cortisol
A: The correct next step is to measure baseline serum cortisol, to test ACTH levels, and to order an ACTH stimulation test with cosyntropin.
Primary adrenocortical insufficiency should be considered in patients with metastatic malignancy who present with peripheral vascular collapse, particularly when it is associated with cutaneous hyperpigmentation, chronic malaise, fatigue, weakness, anorexia, weight loss, hypoglycemia, and electrolyte disturbances such as hyponatremia and hyperkalemia.
Checking the baseline serum cortisol and ACTH levels and cosyntropin stimulation testing are vital steps in making an early diagnosis of primary adrenocortical insufficiency. Inappropriately low serum cortisol is highly suggestive of primary adrenal insufficiency, especially if accompanied by simultaneous elevation of the plasma ACTH level. The result of the ACTH stimulation test with cosyntropin is often confirmatory.
Measuring the serum parathyroid-hormone-related protein level is not indicated, since the patient has a normal corrected calcium. Patients with ectopic Cushing syndrome may present with weight loss due to underlying malignancy, but the presence of hypotension and a lack of hypokalemia makes such a diagnosis unlikely, and, therefore, measurement of late-night salivary cortisol is not the best answer. Amyloidosis, hypothyroidism, or hyperthyroidism are unlikely to have this patient’s presentation.
RESULTS OF FURTHER EVALUATION
Our patient’s ACTH serum level was elevated, and an ACTH stimulation test with cosyntropin confirmed the diagnosis of primary adrenal insufficiency.
CT of the abdomen failed to demonstrate primary tumors, but both adrenal glands were enlarged, likely from metastasis (Figure 4). His hypotension responded to treatment with hydrocortisone and fludrocortisone, and his symptoms resolved. No further testing or therapy was directed to the primary occult malignancy, as it was considered advanced. The prognosis was discussed with the patient, and he deferred any further management and was discharged to hospice care. He died a few months later.
PRIMARY ADRENOCORTICAL INSUFFICIENCY
Primary adrenocortical insufficiency is an uncommon disorder caused by destruction or dysfunction of the adrenal cortices. It is characterized by chronic deficiency of cortisol, aldosterone, and adrenal androgens. In the United States, nearly 6 million people are considered to have undiagnosed adrenal insufficiency, which is clinically significant only during times of physiologic stress.1
Primary adrenocortical insufficiency affects men and women equally. However, the idiopathic autoimmune form of adrenal insufficiency (Addison disease) is two to three times more common in women than in men.
If the condition is undiagnosed or ineffectively treated, the risk of significant morbidity and death is high. Symptoms and signs are nonspecific, and the onset is insidious.
Almost all patients with primary adrenal insufficiency have malaise, fatigue, anorexia, and weight loss. Vomiting, abdominal pain, and fever are more common during an adrenal crisis, when a patient with subclinical disease is subjected to major stress. Postural dizziness or syncope is a common result of volume depletion and hypotension.2–4 It is commonly accompanied by hyponatremia and hyperkalemia.
Hyperpigmentation is the most characteristic physical finding and is caused by an ACTH-mediated increase in melanin content in the skin.2,4,5 The resulting brown hyperpigmentation is most obvious in areas exposed to sunlight (face, neck, backs of hands), and in areas exposed to chronic friction or pressure, such as the elbows, knees, knuckles, waist, and shoulders (brassiere straps).4 Pigmentation is also prominent in the palmar creases, areolae, axillae, perineum, surgical scars, and umbilicus. Other patterns of hyperpigmentation are patchy pigmentation on the inner surface of lips, the buccal mucosa, under the tongue, and on the hard palate.3,5 The hyperpigmentation begins to fade within several days and largely disappears after a few months of adequate glucocorticoid therapy.4
In the United States, 80% of cases of primary adrenocortical insufficiency are caused by autoimmune adrenal destruction. The remainder are caused by infectious diseases (eg, tuberculosis, fungal infection, cytomegalovirus infection, and Mycobacterium aviumintracellulare infection in the context of human immunodeficiency virus infection), by infiltration of the adrenal glands by metastatic cancer, by adrenal hemorrhage, or by drugs such as ketoconazole, fluconazole (Diflucan), metyrapone (Metopirone), mitotane (Lysodren), and etomidate (Amidate).4,6
Adrenal metastatic disease
Infiltration of the adrenal glands by metastatic cancer is not uncommon, probably because of their rich sinusoidal blood supply, and the adrenals are the fourth most common site of metastasis. Common primary tumors are lung, breast, melanoma, gastric, esophageal, and colorectal cancers, while metastasis due to an undetermined primary tumor is the least common.7
Clinically evident adrenal insufficiency produced by metastatic carcinoma is uncommon because most of the adrenal cortex must be destroyed before hypofunction becomes evident.7–9
Malignancy rarely presents first as adrenal insufficiency caused by metastatic infiltration.10
Hormonal therapy may significantly improve symptoms and quality of life in patients with metastatic adrenal insufficiency.8,11
DIAGNOSIS AND MANAGEMENT
Once primary adrenal insufficiency is suspected, prompt diagnosis and treatment are essential. A low plasma cortisol level (< 3 μg/dL) at 8 am is highly suggestive of adrenal insufficiency if exposure to exogenous glucocorticoids has been excluded (including oral, inhaled, and injected),12,13 especially if accompanied by simultaneous elevation of the plasma ACTH level (usually > 200 pg/mL). An 8 am cortisol concentration above 15 μg/dL makes adrenal insufficiency highly unlikely, but levels between 3 and 15 μg/dL are nondiagnostic and need to be further evaluated by an ACTH stimulation test with cosyntropin.4,7
Imaging in primary adrenal insufficiency may be considered when the condition is not clearly autoimmune.14 Abdominal CT is the ideal imaging test for detecting abnormal adrenal glands. CT shows small, noncalcified adrenals in autoimmune Addison disease. It demonstrates enlarged adrenals in about 85% of cases caused by metastatic or granulomatous disease; and calcification is noted in cases of tuberculous adrenal disease.4
Management involves treating the underlying cause and starting hormone replacement therapy. Hormonal therapy consists of corticosteroids and mineralocorticoids; hydrocortisone is the drug of choice and is usually given with fludrocortisone acetate, which has a potent sodium-retaining effect. In the presence of a stressor (fever, surgery, severe illness), the dose of hydrocortisone should be doubled (> 50 mg hydrocortisone per day) for at least 3 to 5 days.2,4
- Erichsen MM, Løvås K, Fougner KJ, et al. Normal overall mortality rate in Addison’s disease, but young patients are at risk of premature death. Eur J Endocrinol 2009; 160:233–237.
- Oelkers W. Adrenal insufficiency. N Engl J Med 1996; 335:1206–1212.
- Redman BG, Pazdur R, Zingas AP, Loredo R. Prospective evaluation of adrenal insufficiency in patients with adrenal metastasis. Cancer 1987; 60:103–107.
- Berger M., Hypofunction of the adrenal cortex in infancy. Manit Med Rev 1949; 29:132.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, café au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Zelissen PM, Bast EJ, Croughs RJ. Associated autoimmunity in Addison’s disease. J Autoimmun 1995; 8:121–130.
- Lutz A, Stojkovic M, Schmidt M, Arlt W, Allolio B, Reincke M. Adrenocortical function in patients with macrometastases of the adrenal gland. Eur J Endocrinol 2000; 143:91–97.
- Kung AW, Pun KK, Lam K, Wang C, Leung CY. Addisonian crisis as presenting feature in malignancies. Cancer 1990; 65:177–179.
- Cedermark BJ, Sjöberg HE. The clinical significance of metastases to the adrenal glands. Surg Gynecol Obstet 1981; 152:607–610.
- Rosenthal FD, Davies MK, Burden AC. Malignant disease presenting as Addison’s disease. Br Med J 1978; 1:1591–1592.
- Seidenwurm DJ, Elmer EB, Kaplan LM, Williams EK, Morris DG, Hoffman AR. Metastases to the adrenal glands and the development of Addison’s disease. Cancer 1984; 54:552–557.
- Santiago AH, Ratzan S. Acute adrenal crisis in an asthmatic child treated with inhaled fluticasone proprionate. Int J Pediatr Endocrinol 2010; 2010. pii:749239.
- Holme J, Tomlinson JW, Stockley RA, Stewart PM, Barlow N, Sullivan AL. Adrenal suppression in bronchiectasis and the impact of inhaled corticosteroids. Eur Respir J 2008; 32:1047–1052.
- Mohammad K, Sadikot RT. Adrenal insufficiency as a presenting manifestation of nonsmall cell lung cancer. South Med J 2009; 102:665–667.
- Erichsen MM, Løvås K, Fougner KJ, et al. Normal overall mortality rate in Addison’s disease, but young patients are at risk of premature death. Eur J Endocrinol 2009; 160:233–237.
- Oelkers W. Adrenal insufficiency. N Engl J Med 1996; 335:1206–1212.
- Redman BG, Pazdur R, Zingas AP, Loredo R. Prospective evaluation of adrenal insufficiency in patients with adrenal metastasis. Cancer 1987; 60:103–107.
- Berger M., Hypofunction of the adrenal cortex in infancy. Manit Med Rev 1949; 29:132.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, café au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Zelissen PM, Bast EJ, Croughs RJ. Associated autoimmunity in Addison’s disease. J Autoimmun 1995; 8:121–130.
- Lutz A, Stojkovic M, Schmidt M, Arlt W, Allolio B, Reincke M. Adrenocortical function in patients with macrometastases of the adrenal gland. Eur J Endocrinol 2000; 143:91–97.
- Kung AW, Pun KK, Lam K, Wang C, Leung CY. Addisonian crisis as presenting feature in malignancies. Cancer 1990; 65:177–179.
- Cedermark BJ, Sjöberg HE. The clinical significance of metastases to the adrenal glands. Surg Gynecol Obstet 1981; 152:607–610.
- Rosenthal FD, Davies MK, Burden AC. Malignant disease presenting as Addison’s disease. Br Med J 1978; 1:1591–1592.
- Seidenwurm DJ, Elmer EB, Kaplan LM, Williams EK, Morris DG, Hoffman AR. Metastases to the adrenal glands and the development of Addison’s disease. Cancer 1984; 54:552–557.
- Santiago AH, Ratzan S. Acute adrenal crisis in an asthmatic child treated with inhaled fluticasone proprionate. Int J Pediatr Endocrinol 2010; 2010. pii:749239.
- Holme J, Tomlinson JW, Stockley RA, Stewart PM, Barlow N, Sullivan AL. Adrenal suppression in bronchiectasis and the impact of inhaled corticosteroids. Eur Respir J 2008; 32:1047–1052.
- Mohammad K, Sadikot RT. Adrenal insufficiency as a presenting manifestation of nonsmall cell lung cancer. South Med J 2009; 102:665–667.
Factor V Leiden: How great is the risk of venous thromboembolism?
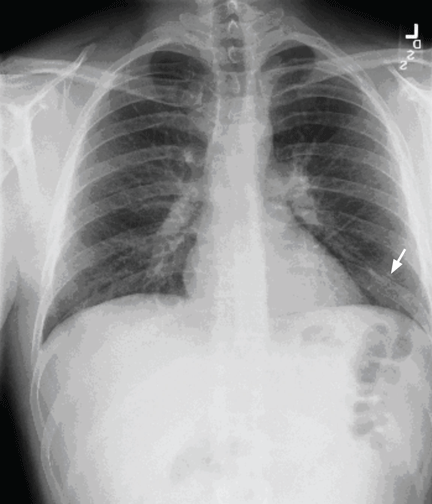
A 29-year-old white man with no chronic medical problems presents to the emergency department with shortness of breath, left-sided pleuritic chest pain, cough, and hemoptysis. These symptoms began abruptly 1 day ago and have persisted. He also has mild pain and swelling in both calves. He denies having any fever, night sweats, or chills. On further questioning, he reports having taken a long, nonstop driving trip that lasted 8 hours 1 week ago.
His medical history is negative, and he specifically reports no history of deep venous thrombosis or pulmonary embolism. He underwent appendectomy 10 years ago but has had no other operations. He does not take any medications. His family history is noncontributory and is negative for venous thromboembolism. He smokes and uses alcohol occasionally but not illicit drugs.
Examination. He appears to be in considerable distress because of his chest pain. His temperature is 100.4°F (38.0°C), blood pressure 125/70 mm Hg, heart rate 125 beats per minute, respiratory rate 26 breaths per minute, oxygen saturation 92% on room air, and body mass index 19 kg/m2.
Chest examination reveals diminished vesicular breathing in the left base, which is normal to percussion without added sounds. Both calves are swollen and tender to palpation without skin discoloration. The rest of his examination is normal.
Laboratory values:
- White blood cell count 9.3 × 109/L (reference range 4.5–11.0)
- Hemoglobin 15.9 g/dL (14.0–17.5)
- Platelets 205 × 109/L (150–350)
- Sodium 140 mEq/L (136–142)
- Potassium 3.9 mEq/L (3.5–5.0)
- Chloride 108 mEq/L (96–106)
- Bicarbonate 23 mEq/L (21–28)
- Blood urea nitrogen 14 mg/dL (8–23)
- Creatinine 0.9 mg/dL (0.6–1.2)
- Glucose 95 mg/dL (70–110)
- International normalized ratio (INR) 0.90 (0.00–1.2)
- Partial thromboplastin time 27.5 seconds (24.6–31.8)
- Creatine phosphokinase 205 U/L (39–308)
- Troponin T < 0.015 ng/mL (0.01–0.045).
Pulmonary embolism is diagnosed
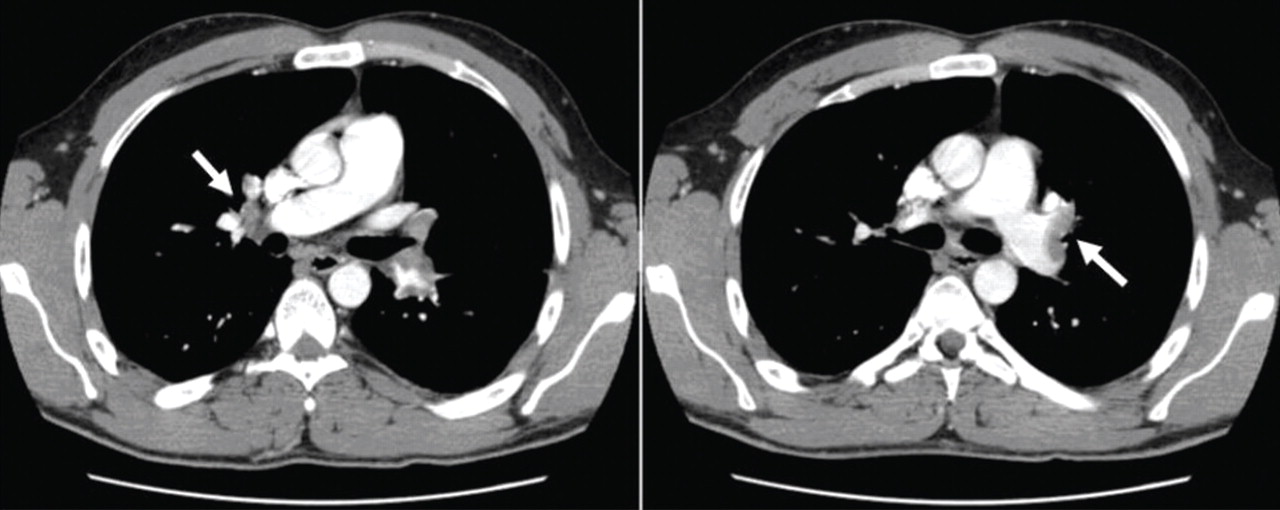
Factor V Leiden is diagnosed, and the patient recovers with treatment
Anticoagulation is started in the emergency department.
Given this patient’s young age and clot burden, a hypercoagulable state is suspected. Thrombophilia screening is performed, with tests for the factor V Leiden mutation, the prothrombin G20210A mutation, and antiphospholipid and lupus anticoagulant antibodies. The rest of the thrombophilia panel, including antithrombin III, factor VIII, protein C, and protein S, is deferred because the levels of these substances would be expected to change during the acute thrombosis.
The direct test for factor V Leiden mutation is positive for the heterozygous type. The test for the prothrombin G20210A mutation is negative, and his antiphospholipid antibody levels, including the lupus anticoagulant titer, are within normal limits.
The patient is kept on a standard regimen of unfractionated heparin, overlapped with warfarin (Coumadin) until his INR is 2.0 to 3.0 on 2 consecutive days. His hospital course is uneventful and his condition gradually improves.
He is discharged home to continue on oral anticoagulation for 6 months with a target INR of 2.0 to 3.0. Two weeks after completing his anticoagulation therapy, his levels of antithrombin III, factor VIII, protein C, and protein S are all within normal limits.
FACTOR V LEIDEN IS COMMON
Factor V Leiden is the most common inherited thrombophilia, with a prevalence of 3% to 7% in the general US population,1 approximately 5% in whites, 2.2% in Hispanics, and 1.2% in blacks.2 Its prevalence in patients with venous thromboembolism, however, is 50%.1,3 The annual incidence of venous thromboembolism in patients with factor V Leiden is 0.5%.4,5
MORE COAGULATION, LESS ANTICOAGULATION
Factor V has a critical position in both the coagulant and anticoagulant pathways. Factor V Leiden results in a hypercoagulable state by both increasing coagulation and decreasing anticoagulation.
This mutation causes factor V to be resistant to being cleaved and inactivated by activated protein C, a condition known as APC resistance. As a result, more factor Va is available within the prothrombinase complex, increasing coagulation by increased generation of thrombin.6–8
Furthermore, a cofactor formed by cleavage of factor V at position 506 is thought to support activated protein C in degrading factor VIIIa (in the tenase complex), along with protein S. People with factor V Leiden lack this cleavage product and thus have less anticoagulant activity from activated protein C. The increased coagulation and decreased anticoagulation appear to contribute equally to the hypercoagulable state in factor V Leiden-associated APC resistance.9–11
Heterozygosity for the factor V Leiden mutation accounts for 90% to 95% of cases of APC resistance. A much smaller number of people are homozygous for it.1
People who are homozygous for factor V Leiden are at higher risk of venous thromboembolism than those who are heterozygous for it, since the latter group’s blood contains both factor V Leiden and normal factor V. The normal factor V allows anticoagulation via the second pathway of inactivation of factor VIIIa by activated protein C, giving some protection against thrombosis. In people who are homozygous for factor V Leiden, the lack of normal factor V acting as an anticoagulant protein results in a higher thrombotic risk.9–11
Other factor V mutations may also cause APC resistance
Although factor V Leiden is the only genetic defect for which a causal relationship with APC resistance has been clearly determined, other, rarer hereditary factor V mutations or polymorphisms have been described, such as factor V Cambridge (Arg306Thr)12 and factor V Hong Kong (Arg306Gly).13 These mutations may result in APC resistance, but their clinical association with thrombosis is less clear.14 Factor V Liverpool (Ile359Thr) is associated with a higher risk of thrombosis, apparently because of reduced APC-mediated inactivation of factor Va and because it is a poor cofactor with activated protein C for the inactivation of factor VIIIa.15
An R2 haplotype has also been described in association with APC resistance.16,17 The phenomenon may be due to a reduction in activated protein C cofactor activity.9 However, not all studies have been convincing regarding the role of this haplotype in clinical disease.18 Coinheritance of this haplotype with factor V Leiden may increase the risk of venous thromboembolism above that associated with factor V Leiden alone.19
Although factor V Leiden is the most common cause of inherited APC resistance, other changes in hemostasis cause acquired APC resistance and may contribute to the thrombotic tendency in these patients.20–22 The most common causes of acquired APC resistance include elevated factor VIII levels,23–25 pregnancy,26–28 use of oral contraceptives,29,30 and antiphospholipid antibodies.31
USUALLY MANIFESTS AS DEEP VEIN THROMBOSIS
Factor V Leiden usually manifests as deep vein thrombosis with or without pulmonary embolism, but thrombosis in unusual locations also occurs.32
The risk of a first episode of venous thromboembolism is two to five times higher with heterozygous factor V Leiden. However, even though the relative risk is high, the absolute risk is low. Furthermore, despite the higher risk of venous thrombosis, there is no evidence that heterozygosity for factor V Leiden increases the overall mortality rate.4,33–36
In people with homozygous factor V Leiden or with combined inherited thrombophilias, the risk of venous thromboembolism is increased to a greater degree: it is 20 to 50 times higher.7,8,37–39 However, whether the risk of death is higher is not clear.
VENOUS THROMBOEMBOLISM IS MULTIFACTORIAL
The pathogenesis of venous thromboembolism is multifactorial and involves an interaction between inherited and acquired factors. Very often, people with factor V Leiden have additional risk factors that contribute to the development of venous clots, and it is very unusual for them to have thrombosis in the absence of these additional factors.
These factors include older age, surgery, obesity, prolonged travel, immobility, hospitalization, oral contraceptive use, hormonal replacement therapy, pregnancy, and malignancy. They increase the risk of venous thrombosis in normal individuals as well, but more so in people with factor V Leiden.40–43
Testing for other known causes of thrombophilia may also be pursued. These include elevated homocysteine levels, the factor II (prothrombin) G20210A mutation, anticardiolipin antibody, lupus anticoagulant, and deficiencies of antithrombin III, protein C, and protein S.
Factor V Leiden by itself does not appear to increase the risk of arterial thrombosis, ie, heart attack and stroke.33,38,44–46
Family history: A risk indicator for venous thrombosis
Family history is an important indicator of risk for a first venous thromboembolic event, regardless of other risk factors identified. The risk of a first event is two to three times higher in people with a family history of thrombosis in a first-degree relative. The risk is four times higher when multiple family members are affected, at least one of them before age 50.47
In people with genetic thrombophilia, the risk of thrombosis (especially unprovoked thrombosis at a young age) is also higher in those with a strong family history than in those without a family history. In those with factor V Leiden, the risk of venous thromboembolism is three to four times higher if there is a positive family history. The risk is five times higher in carriers of factor V Leiden with a family history of venous thromboembolism before age 50, and 13 times higher in those with more than one affected family member.47
Possible shared environmental factors or coinheritance of other unidentified genetic factors may also contribute to the higher susceptibility in thrombosis-prone families.
TESTING FOR APC RESISTANCE AND FACTOR V LEIDEN
The factor V Leiden mutation can be detected directly by genetic testing of peripheral blood mononuclear cells. This method is relatively time-consuming and expensive, however.
At present, the most cost-effective approach is to test first for APC resistance using a second-generation coagulation assay—the modified APC sensitivity test. In this clot-based method, the patient’s sample is prediluted with factor V-deficient plasma to eliminate the effect of lupus anticoagulants and factor deficiencies that could prolong the baseline clotting time, and heparin is inactivated by polybrene. Then either an augmented partial-thromboplastin-time-based assay or a tissue-factor-dependent factor V assay is performed.
This test is nearly 100% sensitive and specific for factor V Leiden, in contrast to the first-generation, or classic, APC sensitivity test, which lacked specificity and sensitivity for it.9–11,48–60 This modification also permits testing of patients receiving anticoagulants or who have abnormal augmented partial thromboplastin times due to coagulation factor deficiencies.
A positive result on the modified APC sensitivity test should be confirmed by a direct genetic test for the factor V Leiden mutation. An APC resistance assay is unnecessary if a direct genetic test is used initially.
HOW LONG TO GIVE ANTICOAGULATION AFTER VENOUS THROMBOEMBOLISM?
Patients who have had an episode of venous thromboembolism have to be treated with anticoagulants.
In general, the initial management of venous thromboembolism in patients with heritable thrombophilias is no different from that in any other patient with a clot. Anticoagulants such as warfarin are given at a target INR of 2.5 (range 2.0–3.0).32 The duration of treatment is based on the risk factors that resulted in the thrombotic event.
After a first event, some authorities recommend anticoagulant therapy for 6 months.32 A shorter period (3 months) is recommended if there is a transient risk factor (eg, surgery, oral contraceptive use, travel, pregnancy, the puerperium) and the thrombosis is confined to distal veins (eg, the calf veins).32
Factor V Leiden does not necessarily increase the risk of recurrent events in patients who have a transient risk factor. Therefore, people who are heterozygous for this mutation do not usually need to be treated lifelong with anticoagulants if they have had only one episode of deep vein thrombosis or pulmonary embolism, given the risk of bleeding associated with anticoagulation, unless they have additional risk factors.
Conditions in which indefinite anticoagulation may be required after careful consideration of the risks and benefits are:
- Life-threatening events such as near-fatal pulmonary embolism
- Cerebral or visceral vein thrombosis
- Recurrent thrombotic events
- Additional persistent risk factors (eg, active malignant neoplasm, extremity paresis, and antiphospholipid antibodies)
- Combined thrombophilias (eg, combined heterozygosity for factor V Leiden and the prothrombin G20210A mutation)
- Homozygosity for factor V Leiden.32,46,48
Factor V Leiden by itself or combined with other thrombophilic abnormalities is not associated with a higher risk of recurrent venous thromboembolism during warfarin therapy (a possible exception is the combination of factor V Leiden plus antiphospholipid antibodies).32,34 Furthermore, current evidence suggests that no thrombophilic defect is a clinically important risk factor for recurrent venous thromboembolism after anticoagulant therapy is stopped. All these facts indicate that clinical factors are probably more important than laboratory abnormalities in determining the duration of anticoagulation therapy.32,35,36,61–63
PRIMARY PROPHYLAXIS IN PATIENTS WITH FACTOR V LEIDEN
Factor V Leiden is only one of many risk factors for deep vein thrombosis or pulmonary embolism. If carriers of factor V Leiden have never had a blood clot, then they are not routinely treated with an anticoagulant. Rather, they should be counseled about reducing or eliminating other factors that may add to their risk of developing a clot in the future.
Usually, the effect of risk factors is additive: the more risk factors present, the higher the risk.46,50 Sometimes, however, the effect of multiple risk factors is more than additive.
Some risk factors, such as genetics or age, are not alterable, but many can be controlled by medications or lifestyle modifications. Therefore, general measures and precautions are recommended to minimize the risk of thrombosis. For example:
Losing weight (if the patient is overweight) is an important intervention for risk reduction, since obesity is probably the most common modifiable risk factor for developing blood clots.
Avoiding long periods of immobility is recommended. For example, if the patient is taking a long car ride (more than 2 hours), then stopping every few hours and walking around for a few minutes is a good way to keep the blood circulating. If the patient has a desk job, getting up and walking around the office periodically is advised. On long airplane trips, a walk in the aisle every so often and preventing dehydration by drinking plenty of fluids and avoiding alcohol are recommended.
Wearing elastic stockings with a graduated elastic pressure may prevent deep venous thrombosis from developing on long flights.63–65
Staying active and getting regular exercise through such activities as walking, bicycling, or swimming are helpful.
Avoiding smoking is critical.50,63
Thromboprophylaxis is recommended for most acutely ill hospitalized patients, especially those confined to bed with additional risk factors. Guidelines for prophylaxis are based on an individualized risk assessment and not on thrombophilia status. Prophylactic anticoagulation is routinely recommended for patients undergoing major high-risk surgery, such as an orthopedic, urologic, gynecologic, or bariatric procedure. Any excess thrombotic risk conferred by thrombophilia is likely small compared with the risk of surgery, and recommendations on the duration and intensity of thromboprophylaxis are not based on thrombophilic status.46,48
Education. Pain, swelling, redness of a limb, unexplained shortness of breath, and chest pain are the most common symptoms of deep vein thrombosis and pulmonary embolism.46,50 It is crucial to teach patients with factor V Leiden to recognize these symptoms and to seek early medical attention in case they experience any of them.
SCREENING FAMILY MEMBERS FOR THE FACTOR V LEIDEN MUTATION
Factor V Leiden by itself is a relatively mild thrombophilic defect that does not cause thrombosis in all carriers, and there is no evidence that early diagnosis reduces rates of morbidity or mortality. Therefore, routine screening of all asymptomatic relatives of affected patients with venous thrombosis is not recommended. Rather, the decision to screen should be made on an individual basis.50,66
Screening may be beneficial in selected cases, especially when patients have a strong family history of recurrent venous thrombosis at a young age (younger than 50 years) and the family member has additional risk factors for venous thromboembolism such as oral contraception or is planning for pregnancy.32,48,49,66
- Rees DC, Cox M, Clegg JB. World distribution of factor V Leiden. Lancet 1995; 346:1133–1134.
- Ridker PM, Miletich JP, Hennekens CH, Buring JE. Ethnic distribution of factor V Leiden in 4047 men and women. Implications for venous thromboembolism screening. JAMA 1997; 277:1305–1307.
- Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood 1995; 85:1504–1508.
- Stolz E, Kemkes-Matthes B, Pötzsch B, et al. Screening for thrombophilic risk factors among 25 German patients with cerebral venous thrombosis. Acta Neurol Scand 2000; 102:31–36.
- Langlois NJ, Wells PS. Risk of venous thromboembolism in relatives of symptomatic probands with thrombophilia: a systematic review. Thromb Haemost 2003; 90:17–26.
- Juul K, Tybjaerg-Hansen A, Mortensen J, Lange P, Vestbo J, Nordestgaard BG. Factor V leiden homozygosity, dyspnea, and reduced pulmonary function. Arch Intern Med 2005; 165:2032–2036.
- Bertina RM, Koeleman BP, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994; 369:64–67.
- Dahlbäck B. New molecular insights into the genetics of thrombophilia. Resistance to activated protein C caused by Arg506 to Gln mutation in factor V as a pathogenic risk factor for venous thrombosis. Thromb Haemost 1995; 74:139–148.
- Castoldi E, Brugge JM, Nicolaes GA, Girelli D, Tans G, Rosing J. Impaired APC cofactor activity of factor V plays a major role in the APC resistance associated with the factor V Leiden (R506Q) and R2 (H1299R) mutations. Blood 2004; 103:4173–4179.
- Dahlback B. Anticoagulant factor V and thrombosis risk (editorial). Blood 2004; 103:3995.
- Simioni P, Castoldi E, Lunghi B, Tormene D, Rosing J, Bernardi F. An underestimated combination of opposites resulting in enhanced thrombotic tendency. Blood 2005; 106:2363–2365.
- Williamson D, Brown K, Luddington R, Baglin C, Baglin T. Factor V Cambridge: a new mutation (Arg306-->Thr) associated with resistance to activated protein C. Blood 1998; 91:1140–1144.
- Chan WP, Lee CK, Kwong YL, Lam CK, Liang R. A novel mutation of Arg306 of factor V gene in Hong Kong Chinese. Blood 1998; 91:1135–1139.
- Liang R, Lee CK, Wat MS, Kwong YL, Lam CK, Liu HW. Clinical significance of Arg306 mutations of factor V gene. Blood 1998; 92:2599–2600.
- Steen M, Norstrøm EA, Tholander AL, et al. Functional characterization of factor V-Ile359Thr: a novel mutation associated with thrombosis. Blood 2004; 103:3381–3387.
- Bernardi F, Faioni EM, Castoldi E, et al. A factor V genetic component differing from factor V R506Q contributes to the activated protein C resistance phenotype. Blood 1997; 90:1552–1557.
- Lunghi B, Castoldi E, Mingozzi F, Bernardi F. A new factor V gene polymorphism (His 1254 Arg) present in subjects of African origin mimics the R2 polymorphism (His 1299 Arg). Blood 1998; 91:364–365.
- Luddington R, Jackson A, Pannerselvam S, Brown K, Baglin T. The factor V R2 allele: risk of venous thromboembolism, factor V levels and resistance to activated protein C. Thromb Haemost 2000; 83:204–208.
- Faioni EM, Franchi F, Bucciarelli P, et al. Coinheritance of the HR2 haplotype in the factor V gene confers an increased risk of venous thromboembolism to carriers of factor V R506Q (factor V Leiden). Blood 1999; 94:3062–3066.
- Clark P, Walker ID. The phenomenon known as acquired activated protein C resistance. Br J Haematol 2001; 115:767–773.
- Tosetto A, Simioni M, Madeo D, Rodeghiero F. Intraindividual consistency of the activated protein C resistance phenotype. Br J Haematol 2004; 126:405–409.
- de Visser MC, Rosendaal FR, Bertina RM. A reduced sensitivity for activated protein C in the absence of factor V Leiden increases the risk of venous thrombosis. Blood 1999; 93:1271–1276.
- Kraaijenhagen RA, in’t Anker PS, Koopman MM, et al. High plasma concentration of factor VIIIc is a major risk factor for venous thromboembolism. Thromb Haemost 2000; 83:5–9.
- Kamphuisen PW, Eikenboom JC, Bertina RM. Elevated factor VIII levels and the risk of thrombosis. Arterioscler Thromb Vasc Biol 2001; 21:731–738.
- Koster T, Blann AD, Briët E, Vandenbroucke JP, Rosendaal FR. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep-vein thrombosis. Lancet 1995; 345:152–155.
- Clark P, Brennand J, Conkie JA, McCall F, Greer IA, Walker ID. Activated protein C sensitivity, protein C, protein S and coagulation in normal pregnancy. Thromb Haemost 1998; 79:1166–1170.
- Cumming AM, Tait RC, Fildes S, Yoong A, Keeney S, Hay CR. Development of resistance to activated protein C during pregnancy. Br J Haematol 1995; 90:725–727.
- Mathonnet F, de Mazancourt P, Bastenaire B, et al. Activated protein C sensitivity ratio in pregnant women at delivery. Br J Haematol 1996; 92:244–246.
- Post MS, Rosing J, Van Der Mooren MJ, et al; Ageing Women’ and the Institute for Cardiovascular Research-Vrije Universiteit (ICaRVU). Increased resistance to activated protein C after short-term oral hormone replacement therapy in healthy post-menopausal women. Br J Haematol 2002; 119:1017–1023.
- Olivieri O, Friso S, Manzato F, et al. Resistance to activated protein C in healthy women taking oral contraceptives. Br J Haematol 1995; 91:465–470.
- Bokarewa MI, Blombäck M, Egberg N, Rosén S. A new variant of interaction between phospholipid antibodies and the protein C system. Blood Coagul Fibrinolysis 1994; 5:37–41.
- Baglin T, Gray E, Greaves M, et al; British Committee for Standards in Haematology. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010; 149:209–220.
- van Stralen KJ, Doggen CJ, Bezemer ID, Pomp ER, Lisman T, Rosendaal FR. Mechanisms of the factor V Leiden paradox. Arterioscler Thromb Vasc Biol 2008; 28:1872–1877.
- Agaoglu N, Mustafa NA, Turkyilmaz S. Prothrombotic disorders in patients with mesenteric vein thrombosis. J Invest Surg 2003; 16:299–304.
- El-Karaksy H, El-Koofy N, El-Hawary M, et al. Prevalence of factor V Leiden mutation and other hereditary thrombophilic factors in Egyptian children with portal vein thrombosis: results of a single-center case-control study. Ann Hematol 2004; 83:712–715.
- Heijmans BT, Westendorp RG, Knook DL, Kluft C, Slagboom PE. The risk of mortality and the factor V Leiden mutation in a population-based cohort. Thromb Haemost 1998; 80:607–609.
- Turkstra F, Karemaker R, Kuijer PM, Prins MH, Büller HR. Is the prevalence of the factor V Leiden mutation in patients with pulmonary embolism and deep vein thrombosis really different? Thromb Haemost 1999; 81:345–348.
- Ridker PM, Hennekens CH, Lindpaintner K, Stampfer MJ, Eisenberg PR, Miletich JP. Mutation in the gene coding for coagulation factor V and the risk of myocardial infarction, stroke, and venous thrombosis in apparently healthy men. N Engl J Med 1995; 332:912–917.
- Manten B, Westendorp RG, Koster T, Reitsma PH, Rosendaal FR. Risk factor profiles in patients with different clinical manifestations of venous thromboembolism: a focus on the factor V Leiden mutation. Thromb Haemost 1996; 76:510–513.
- Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 2005; 293:715–722.
- Bloemenkamp KW, Rosendaal FR, Helmerhorst FM, Büller HR, Vandenbroucke JP. Enhancement by factor V Leiden mutation of risk of deep-vein thrombosis associated with oral contraceptives containing a third-generation progestagen. Lancet 1995; 346:1593–1596.
- Murphy PT. Factor V Leiden and venous thromboembolism. Ann Intern Med 2004; 141:483–484.
- Nizankowska-Mogilnicka E, Adamek L, Grzanka P, et al. Genetic polymorphisms associated with acute pulmonary embolism and deep venous thrombosis. Eur Respir J 2003; 21:25–30.
- Arsov T, Miladinova D, Spiroski M. Factor V Leiden is associated with higher risk of deep venous thrombosis of large blood vessels. Croat Med J 2006; 47:433–439.
- Simioni P, Prandoni P, Lensing AW, et al. Risk for subsequent venous thromboembolic complications in carriers of the prothrombin or the factor V gene mutation with a first episode of deep-vein thrombosis. Blood 2000; 96:3329–3333.
- Ornstein DL, Cushman M. Cardiology patient page. Factor V Leiden. Circulation 2003; 107:e94–e97.
- Bezemer ID, van der Meer FJ, Eikenboom JC, Rosendaal FR, Doggen CJ. The value of family history as a risk indicator for venous thrombosis. Arch Intern Med 2009; 169:610–615.
- Press RD, Bauer KA, Kujovich JL, Heit JA. Clinical utility of factor V leiden (R506Q) testing for the diagnosis and management of thromboembolic disorders. Arch Pathol Lab Med 2002; 126:1304–1318.
- Gadelha T, Roldán V, Lecumberri R, et al; RIETE Investigators. Clinical characteristics of patients with factor V Leiden or prothrombin G20210A and a first episode of venous thromboembolism. Findings from the RIETE Registry. Thromb Res 2010; 126:283–286.
- Severinsen MT, Overvad K, Johnsen SP, et al. Genetic susceptibility, smoking, obesity and risk of venous thromboembolism. Br J Haematol 2010; 149:273–279.
- Kujovich JL. Factor V Leiden thrombophilia. Genet Med 2011; 13:1–16.
- Lijfering WM, Brouwer JL, Veeger NJ, et al. Selective testing for thrombophilia in patients with first venous thrombosis: results from a retrospective family cohort study on absolute thrombotic risk for currently known thrombophilic defects in 2479 relatives. Blood 2009; 113:5314–5322.
- Kearon C, Julian JA, Kovacs MJ, et al; ELATE Investigators. Influence of thrombophilia on risk of recurrent venous thromboembolism while on warfarin: results from a randomized trial. Blood 2008; 112:4432–4436.
- Ho WK, Hankey GJ, Quinlan DJ, Eikelboom JW. Risk of recurrent venous thromboembolism in patients with common thrombophilia: a systematic review. Arch Intern Med 2006; 166:729–736.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA 2005; 293:2352–2361.
- Strobl FJ, Hoffman S, Huber S, Williams EC, Voelkerding KV. Activated protein C resistance assay performance: improvement by sample dilution with factor V-deficient plasma. Arch Pathol Lab Med 1998; 122:430–433.
- Legnani C, Palareti G, Biagi R, et al. Activated protein C resistance: a comparison between two clotting assays and their relationship to the presence of the factor V Leiden mutation. Br J Haematol 1996; 93:694–699.
- Gouault-Heilmann M, Leroy-Matheron C. Factor V Leiden-dependent APC resistance: improved sensitivity and specificity of the APC resistance test by plasma dilution in factor V-depleted plasma. Thromb Res 1996; 82:281–283.
- Svensson PJ, Zöller B, Dahlbäck B. Evaluation of original and modified APC-resistance tests in unselected outpatients with clinically suspected thrombosis and in healthy controls. Thromb Haemost 1997; 77:332–335.
- Tripodi A, Negri B, Bertina RM, Mannucci PM. Screening for the FV:Q506 mutation—evaluation of thirteen plasma-based methods for their diagnostic efficacy in comparison with DNA analysis. Thromb Haemost 1997; 77:436–439.
- Wåhlander K, Larson G, Lindahl TL, et al. Factor V Leiden (G1691A) and prothrombin gene G20210A mutations as potential risk factors for venous thromboembolism after total hip or total knee replacement surgery. Thromb Haemost 2002; 87:580–585.
- Joseph JE, Low J, Courtenay B, Neil MJ, McGrath M, Ma D. A single-centre prospective study of clinical and haemostatic risk factors for venous thromboembolism following lower limb arthroplasty. Br J Haematol 2005; 129:87–92.
- Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):381S–453S.
- Brenner B. Prophylaxis for travel-related thrombosis? Yes. J Thromb Haemost 2004; 2:2089–2091.
- Gavish I, Brenner B. Air travel and the risk of thromboembolism. Intern Emerg Med 2011; 6:113–116.
- Grody WW, Griffin JH, Taylor AK, Korf BR, Heit JA; ACMG Factor V Leiden Working Group. American College of Medical Genetics consensus statement on factor V Leiden mutation testing. Genet Med 2001; 3:139–148.
A 29-year-old white man with no chronic medical problems presents to the emergency department with shortness of breath, left-sided pleuritic chest pain, cough, and hemoptysis. These symptoms began abruptly 1 day ago and have persisted. He also has mild pain and swelling in both calves. He denies having any fever, night sweats, or chills. On further questioning, he reports having taken a long, nonstop driving trip that lasted 8 hours 1 week ago.
His medical history is negative, and he specifically reports no history of deep venous thrombosis or pulmonary embolism. He underwent appendectomy 10 years ago but has had no other operations. He does not take any medications. His family history is noncontributory and is negative for venous thromboembolism. He smokes and uses alcohol occasionally but not illicit drugs.
Examination. He appears to be in considerable distress because of his chest pain. His temperature is 100.4°F (38.0°C), blood pressure 125/70 mm Hg, heart rate 125 beats per minute, respiratory rate 26 breaths per minute, oxygen saturation 92% on room air, and body mass index 19 kg/m2.
Chest examination reveals diminished vesicular breathing in the left base, which is normal to percussion without added sounds. Both calves are swollen and tender to palpation without skin discoloration. The rest of his examination is normal.
Laboratory values:
- White blood cell count 9.3 × 109/L (reference range 4.5–11.0)
- Hemoglobin 15.9 g/dL (14.0–17.5)
- Platelets 205 × 109/L (150–350)
- Sodium 140 mEq/L (136–142)
- Potassium 3.9 mEq/L (3.5–5.0)
- Chloride 108 mEq/L (96–106)
- Bicarbonate 23 mEq/L (21–28)
- Blood urea nitrogen 14 mg/dL (8–23)
- Creatinine 0.9 mg/dL (0.6–1.2)
- Glucose 95 mg/dL (70–110)
- International normalized ratio (INR) 0.90 (0.00–1.2)
- Partial thromboplastin time 27.5 seconds (24.6–31.8)
- Creatine phosphokinase 205 U/L (39–308)
- Troponin T < 0.015 ng/mL (0.01–0.045).
Pulmonary embolism is diagnosed
Factor V Leiden is diagnosed, and the patient recovers with treatment
Anticoagulation is started in the emergency department.
Given this patient’s young age and clot burden, a hypercoagulable state is suspected. Thrombophilia screening is performed, with tests for the factor V Leiden mutation, the prothrombin G20210A mutation, and antiphospholipid and lupus anticoagulant antibodies. The rest of the thrombophilia panel, including antithrombin III, factor VIII, protein C, and protein S, is deferred because the levels of these substances would be expected to change during the acute thrombosis.
The direct test for factor V Leiden mutation is positive for the heterozygous type. The test for the prothrombin G20210A mutation is negative, and his antiphospholipid antibody levels, including the lupus anticoagulant titer, are within normal limits.
The patient is kept on a standard regimen of unfractionated heparin, overlapped with warfarin (Coumadin) until his INR is 2.0 to 3.0 on 2 consecutive days. His hospital course is uneventful and his condition gradually improves.
He is discharged home to continue on oral anticoagulation for 6 months with a target INR of 2.0 to 3.0. Two weeks after completing his anticoagulation therapy, his levels of antithrombin III, factor VIII, protein C, and protein S are all within normal limits.
FACTOR V LEIDEN IS COMMON
Factor V Leiden is the most common inherited thrombophilia, with a prevalence of 3% to 7% in the general US population,1 approximately 5% in whites, 2.2% in Hispanics, and 1.2% in blacks.2 Its prevalence in patients with venous thromboembolism, however, is 50%.1,3 The annual incidence of venous thromboembolism in patients with factor V Leiden is 0.5%.4,5
MORE COAGULATION, LESS ANTICOAGULATION
Factor V has a critical position in both the coagulant and anticoagulant pathways. Factor V Leiden results in a hypercoagulable state by both increasing coagulation and decreasing anticoagulation.
This mutation causes factor V to be resistant to being cleaved and inactivated by activated protein C, a condition known as APC resistance. As a result, more factor Va is available within the prothrombinase complex, increasing coagulation by increased generation of thrombin.6–8
Furthermore, a cofactor formed by cleavage of factor V at position 506 is thought to support activated protein C in degrading factor VIIIa (in the tenase complex), along with protein S. People with factor V Leiden lack this cleavage product and thus have less anticoagulant activity from activated protein C. The increased coagulation and decreased anticoagulation appear to contribute equally to the hypercoagulable state in factor V Leiden-associated APC resistance.9–11
Heterozygosity for the factor V Leiden mutation accounts for 90% to 95% of cases of APC resistance. A much smaller number of people are homozygous for it.1
People who are homozygous for factor V Leiden are at higher risk of venous thromboembolism than those who are heterozygous for it, since the latter group’s blood contains both factor V Leiden and normal factor V. The normal factor V allows anticoagulation via the second pathway of inactivation of factor VIIIa by activated protein C, giving some protection against thrombosis. In people who are homozygous for factor V Leiden, the lack of normal factor V acting as an anticoagulant protein results in a higher thrombotic risk.9–11
Other factor V mutations may also cause APC resistance
Although factor V Leiden is the only genetic defect for which a causal relationship with APC resistance has been clearly determined, other, rarer hereditary factor V mutations or polymorphisms have been described, such as factor V Cambridge (Arg306Thr)12 and factor V Hong Kong (Arg306Gly).13 These mutations may result in APC resistance, but their clinical association with thrombosis is less clear.14 Factor V Liverpool (Ile359Thr) is associated with a higher risk of thrombosis, apparently because of reduced APC-mediated inactivation of factor Va and because it is a poor cofactor with activated protein C for the inactivation of factor VIIIa.15
An R2 haplotype has also been described in association with APC resistance.16,17 The phenomenon may be due to a reduction in activated protein C cofactor activity.9 However, not all studies have been convincing regarding the role of this haplotype in clinical disease.18 Coinheritance of this haplotype with factor V Leiden may increase the risk of venous thromboembolism above that associated with factor V Leiden alone.19
Although factor V Leiden is the most common cause of inherited APC resistance, other changes in hemostasis cause acquired APC resistance and may contribute to the thrombotic tendency in these patients.20–22 The most common causes of acquired APC resistance include elevated factor VIII levels,23–25 pregnancy,26–28 use of oral contraceptives,29,30 and antiphospholipid antibodies.31
USUALLY MANIFESTS AS DEEP VEIN THROMBOSIS
Factor V Leiden usually manifests as deep vein thrombosis with or without pulmonary embolism, but thrombosis in unusual locations also occurs.32
The risk of a first episode of venous thromboembolism is two to five times higher with heterozygous factor V Leiden. However, even though the relative risk is high, the absolute risk is low. Furthermore, despite the higher risk of venous thrombosis, there is no evidence that heterozygosity for factor V Leiden increases the overall mortality rate.4,33–36
In people with homozygous factor V Leiden or with combined inherited thrombophilias, the risk of venous thromboembolism is increased to a greater degree: it is 20 to 50 times higher.7,8,37–39 However, whether the risk of death is higher is not clear.
VENOUS THROMBOEMBOLISM IS MULTIFACTORIAL
The pathogenesis of venous thromboembolism is multifactorial and involves an interaction between inherited and acquired factors. Very often, people with factor V Leiden have additional risk factors that contribute to the development of venous clots, and it is very unusual for them to have thrombosis in the absence of these additional factors.
These factors include older age, surgery, obesity, prolonged travel, immobility, hospitalization, oral contraceptive use, hormonal replacement therapy, pregnancy, and malignancy. They increase the risk of venous thrombosis in normal individuals as well, but more so in people with factor V Leiden.40–43
Testing for other known causes of thrombophilia may also be pursued. These include elevated homocysteine levels, the factor II (prothrombin) G20210A mutation, anticardiolipin antibody, lupus anticoagulant, and deficiencies of antithrombin III, protein C, and protein S.
Factor V Leiden by itself does not appear to increase the risk of arterial thrombosis, ie, heart attack and stroke.33,38,44–46
Family history: A risk indicator for venous thrombosis
Family history is an important indicator of risk for a first venous thromboembolic event, regardless of other risk factors identified. The risk of a first event is two to three times higher in people with a family history of thrombosis in a first-degree relative. The risk is four times higher when multiple family members are affected, at least one of them before age 50.47
In people with genetic thrombophilia, the risk of thrombosis (especially unprovoked thrombosis at a young age) is also higher in those with a strong family history than in those without a family history. In those with factor V Leiden, the risk of venous thromboembolism is three to four times higher if there is a positive family history. The risk is five times higher in carriers of factor V Leiden with a family history of venous thromboembolism before age 50, and 13 times higher in those with more than one affected family member.47
Possible shared environmental factors or coinheritance of other unidentified genetic factors may also contribute to the higher susceptibility in thrombosis-prone families.
TESTING FOR APC RESISTANCE AND FACTOR V LEIDEN
The factor V Leiden mutation can be detected directly by genetic testing of peripheral blood mononuclear cells. This method is relatively time-consuming and expensive, however.
At present, the most cost-effective approach is to test first for APC resistance using a second-generation coagulation assay—the modified APC sensitivity test. In this clot-based method, the patient’s sample is prediluted with factor V-deficient plasma to eliminate the effect of lupus anticoagulants and factor deficiencies that could prolong the baseline clotting time, and heparin is inactivated by polybrene. Then either an augmented partial-thromboplastin-time-based assay or a tissue-factor-dependent factor V assay is performed.
This test is nearly 100% sensitive and specific for factor V Leiden, in contrast to the first-generation, or classic, APC sensitivity test, which lacked specificity and sensitivity for it.9–11,48–60 This modification also permits testing of patients receiving anticoagulants or who have abnormal augmented partial thromboplastin times due to coagulation factor deficiencies.
A positive result on the modified APC sensitivity test should be confirmed by a direct genetic test for the factor V Leiden mutation. An APC resistance assay is unnecessary if a direct genetic test is used initially.
HOW LONG TO GIVE ANTICOAGULATION AFTER VENOUS THROMBOEMBOLISM?
Patients who have had an episode of venous thromboembolism have to be treated with anticoagulants.
In general, the initial management of venous thromboembolism in patients with heritable thrombophilias is no different from that in any other patient with a clot. Anticoagulants such as warfarin are given at a target INR of 2.5 (range 2.0–3.0).32 The duration of treatment is based on the risk factors that resulted in the thrombotic event.
After a first event, some authorities recommend anticoagulant therapy for 6 months.32 A shorter period (3 months) is recommended if there is a transient risk factor (eg, surgery, oral contraceptive use, travel, pregnancy, the puerperium) and the thrombosis is confined to distal veins (eg, the calf veins).32
Factor V Leiden does not necessarily increase the risk of recurrent events in patients who have a transient risk factor. Therefore, people who are heterozygous for this mutation do not usually need to be treated lifelong with anticoagulants if they have had only one episode of deep vein thrombosis or pulmonary embolism, given the risk of bleeding associated with anticoagulation, unless they have additional risk factors.
Conditions in which indefinite anticoagulation may be required after careful consideration of the risks and benefits are:
- Life-threatening events such as near-fatal pulmonary embolism
- Cerebral or visceral vein thrombosis
- Recurrent thrombotic events
- Additional persistent risk factors (eg, active malignant neoplasm, extremity paresis, and antiphospholipid antibodies)
- Combined thrombophilias (eg, combined heterozygosity for factor V Leiden and the prothrombin G20210A mutation)
- Homozygosity for factor V Leiden.32,46,48
Factor V Leiden by itself or combined with other thrombophilic abnormalities is not associated with a higher risk of recurrent venous thromboembolism during warfarin therapy (a possible exception is the combination of factor V Leiden plus antiphospholipid antibodies).32,34 Furthermore, current evidence suggests that no thrombophilic defect is a clinically important risk factor for recurrent venous thromboembolism after anticoagulant therapy is stopped. All these facts indicate that clinical factors are probably more important than laboratory abnormalities in determining the duration of anticoagulation therapy.32,35,36,61–63
PRIMARY PROPHYLAXIS IN PATIENTS WITH FACTOR V LEIDEN
Factor V Leiden is only one of many risk factors for deep vein thrombosis or pulmonary embolism. If carriers of factor V Leiden have never had a blood clot, then they are not routinely treated with an anticoagulant. Rather, they should be counseled about reducing or eliminating other factors that may add to their risk of developing a clot in the future.
Usually, the effect of risk factors is additive: the more risk factors present, the higher the risk.46,50 Sometimes, however, the effect of multiple risk factors is more than additive.
Some risk factors, such as genetics or age, are not alterable, but many can be controlled by medications or lifestyle modifications. Therefore, general measures and precautions are recommended to minimize the risk of thrombosis. For example:
Losing weight (if the patient is overweight) is an important intervention for risk reduction, since obesity is probably the most common modifiable risk factor for developing blood clots.
Avoiding long periods of immobility is recommended. For example, if the patient is taking a long car ride (more than 2 hours), then stopping every few hours and walking around for a few minutes is a good way to keep the blood circulating. If the patient has a desk job, getting up and walking around the office periodically is advised. On long airplane trips, a walk in the aisle every so often and preventing dehydration by drinking plenty of fluids and avoiding alcohol are recommended.
Wearing elastic stockings with a graduated elastic pressure may prevent deep venous thrombosis from developing on long flights.63–65
Staying active and getting regular exercise through such activities as walking, bicycling, or swimming are helpful.
Avoiding smoking is critical.50,63
Thromboprophylaxis is recommended for most acutely ill hospitalized patients, especially those confined to bed with additional risk factors. Guidelines for prophylaxis are based on an individualized risk assessment and not on thrombophilia status. Prophylactic anticoagulation is routinely recommended for patients undergoing major high-risk surgery, such as an orthopedic, urologic, gynecologic, or bariatric procedure. Any excess thrombotic risk conferred by thrombophilia is likely small compared with the risk of surgery, and recommendations on the duration and intensity of thromboprophylaxis are not based on thrombophilic status.46,48
Education. Pain, swelling, redness of a limb, unexplained shortness of breath, and chest pain are the most common symptoms of deep vein thrombosis and pulmonary embolism.46,50 It is crucial to teach patients with factor V Leiden to recognize these symptoms and to seek early medical attention in case they experience any of them.
SCREENING FAMILY MEMBERS FOR THE FACTOR V LEIDEN MUTATION
Factor V Leiden by itself is a relatively mild thrombophilic defect that does not cause thrombosis in all carriers, and there is no evidence that early diagnosis reduces rates of morbidity or mortality. Therefore, routine screening of all asymptomatic relatives of affected patients with venous thrombosis is not recommended. Rather, the decision to screen should be made on an individual basis.50,66
Screening may be beneficial in selected cases, especially when patients have a strong family history of recurrent venous thrombosis at a young age (younger than 50 years) and the family member has additional risk factors for venous thromboembolism such as oral contraception or is planning for pregnancy.32,48,49,66
A 29-year-old white man with no chronic medical problems presents to the emergency department with shortness of breath, left-sided pleuritic chest pain, cough, and hemoptysis. These symptoms began abruptly 1 day ago and have persisted. He also has mild pain and swelling in both calves. He denies having any fever, night sweats, or chills. On further questioning, he reports having taken a long, nonstop driving trip that lasted 8 hours 1 week ago.
His medical history is negative, and he specifically reports no history of deep venous thrombosis or pulmonary embolism. He underwent appendectomy 10 years ago but has had no other operations. He does not take any medications. His family history is noncontributory and is negative for venous thromboembolism. He smokes and uses alcohol occasionally but not illicit drugs.
Examination. He appears to be in considerable distress because of his chest pain. His temperature is 100.4°F (38.0°C), blood pressure 125/70 mm Hg, heart rate 125 beats per minute, respiratory rate 26 breaths per minute, oxygen saturation 92% on room air, and body mass index 19 kg/m2.
Chest examination reveals diminished vesicular breathing in the left base, which is normal to percussion without added sounds. Both calves are swollen and tender to palpation without skin discoloration. The rest of his examination is normal.
Laboratory values:
- White blood cell count 9.3 × 109/L (reference range 4.5–11.0)
- Hemoglobin 15.9 g/dL (14.0–17.5)
- Platelets 205 × 109/L (150–350)
- Sodium 140 mEq/L (136–142)
- Potassium 3.9 mEq/L (3.5–5.0)
- Chloride 108 mEq/L (96–106)
- Bicarbonate 23 mEq/L (21–28)
- Blood urea nitrogen 14 mg/dL (8–23)
- Creatinine 0.9 mg/dL (0.6–1.2)
- Glucose 95 mg/dL (70–110)
- International normalized ratio (INR) 0.90 (0.00–1.2)
- Partial thromboplastin time 27.5 seconds (24.6–31.8)
- Creatine phosphokinase 205 U/L (39–308)
- Troponin T < 0.015 ng/mL (0.01–0.045).
Pulmonary embolism is diagnosed
Factor V Leiden is diagnosed, and the patient recovers with treatment
Anticoagulation is started in the emergency department.
Given this patient’s young age and clot burden, a hypercoagulable state is suspected. Thrombophilia screening is performed, with tests for the factor V Leiden mutation, the prothrombin G20210A mutation, and antiphospholipid and lupus anticoagulant antibodies. The rest of the thrombophilia panel, including antithrombin III, factor VIII, protein C, and protein S, is deferred because the levels of these substances would be expected to change during the acute thrombosis.
The direct test for factor V Leiden mutation is positive for the heterozygous type. The test for the prothrombin G20210A mutation is negative, and his antiphospholipid antibody levels, including the lupus anticoagulant titer, are within normal limits.
The patient is kept on a standard regimen of unfractionated heparin, overlapped with warfarin (Coumadin) until his INR is 2.0 to 3.0 on 2 consecutive days. His hospital course is uneventful and his condition gradually improves.
He is discharged home to continue on oral anticoagulation for 6 months with a target INR of 2.0 to 3.0. Two weeks after completing his anticoagulation therapy, his levels of antithrombin III, factor VIII, protein C, and protein S are all within normal limits.
FACTOR V LEIDEN IS COMMON
Factor V Leiden is the most common inherited thrombophilia, with a prevalence of 3% to 7% in the general US population,1 approximately 5% in whites, 2.2% in Hispanics, and 1.2% in blacks.2 Its prevalence in patients with venous thromboembolism, however, is 50%.1,3 The annual incidence of venous thromboembolism in patients with factor V Leiden is 0.5%.4,5
MORE COAGULATION, LESS ANTICOAGULATION
Factor V has a critical position in both the coagulant and anticoagulant pathways. Factor V Leiden results in a hypercoagulable state by both increasing coagulation and decreasing anticoagulation.
This mutation causes factor V to be resistant to being cleaved and inactivated by activated protein C, a condition known as APC resistance. As a result, more factor Va is available within the prothrombinase complex, increasing coagulation by increased generation of thrombin.6–8
Furthermore, a cofactor formed by cleavage of factor V at position 506 is thought to support activated protein C in degrading factor VIIIa (in the tenase complex), along with protein S. People with factor V Leiden lack this cleavage product and thus have less anticoagulant activity from activated protein C. The increased coagulation and decreased anticoagulation appear to contribute equally to the hypercoagulable state in factor V Leiden-associated APC resistance.9–11
Heterozygosity for the factor V Leiden mutation accounts for 90% to 95% of cases of APC resistance. A much smaller number of people are homozygous for it.1
People who are homozygous for factor V Leiden are at higher risk of venous thromboembolism than those who are heterozygous for it, since the latter group’s blood contains both factor V Leiden and normal factor V. The normal factor V allows anticoagulation via the second pathway of inactivation of factor VIIIa by activated protein C, giving some protection against thrombosis. In people who are homozygous for factor V Leiden, the lack of normal factor V acting as an anticoagulant protein results in a higher thrombotic risk.9–11
Other factor V mutations may also cause APC resistance
Although factor V Leiden is the only genetic defect for which a causal relationship with APC resistance has been clearly determined, other, rarer hereditary factor V mutations or polymorphisms have been described, such as factor V Cambridge (Arg306Thr)12 and factor V Hong Kong (Arg306Gly).13 These mutations may result in APC resistance, but their clinical association with thrombosis is less clear.14 Factor V Liverpool (Ile359Thr) is associated with a higher risk of thrombosis, apparently because of reduced APC-mediated inactivation of factor Va and because it is a poor cofactor with activated protein C for the inactivation of factor VIIIa.15
An R2 haplotype has also been described in association with APC resistance.16,17 The phenomenon may be due to a reduction in activated protein C cofactor activity.9 However, not all studies have been convincing regarding the role of this haplotype in clinical disease.18 Coinheritance of this haplotype with factor V Leiden may increase the risk of venous thromboembolism above that associated with factor V Leiden alone.19
Although factor V Leiden is the most common cause of inherited APC resistance, other changes in hemostasis cause acquired APC resistance and may contribute to the thrombotic tendency in these patients.20–22 The most common causes of acquired APC resistance include elevated factor VIII levels,23–25 pregnancy,26–28 use of oral contraceptives,29,30 and antiphospholipid antibodies.31
USUALLY MANIFESTS AS DEEP VEIN THROMBOSIS
Factor V Leiden usually manifests as deep vein thrombosis with or without pulmonary embolism, but thrombosis in unusual locations also occurs.32
The risk of a first episode of venous thromboembolism is two to five times higher with heterozygous factor V Leiden. However, even though the relative risk is high, the absolute risk is low. Furthermore, despite the higher risk of venous thrombosis, there is no evidence that heterozygosity for factor V Leiden increases the overall mortality rate.4,33–36
In people with homozygous factor V Leiden or with combined inherited thrombophilias, the risk of venous thromboembolism is increased to a greater degree: it is 20 to 50 times higher.7,8,37–39 However, whether the risk of death is higher is not clear.
VENOUS THROMBOEMBOLISM IS MULTIFACTORIAL
The pathogenesis of venous thromboembolism is multifactorial and involves an interaction between inherited and acquired factors. Very often, people with factor V Leiden have additional risk factors that contribute to the development of venous clots, and it is very unusual for them to have thrombosis in the absence of these additional factors.
These factors include older age, surgery, obesity, prolonged travel, immobility, hospitalization, oral contraceptive use, hormonal replacement therapy, pregnancy, and malignancy. They increase the risk of venous thrombosis in normal individuals as well, but more so in people with factor V Leiden.40–43
Testing for other known causes of thrombophilia may also be pursued. These include elevated homocysteine levels, the factor II (prothrombin) G20210A mutation, anticardiolipin antibody, lupus anticoagulant, and deficiencies of antithrombin III, protein C, and protein S.
Factor V Leiden by itself does not appear to increase the risk of arterial thrombosis, ie, heart attack and stroke.33,38,44–46
Family history: A risk indicator for venous thrombosis
Family history is an important indicator of risk for a first venous thromboembolic event, regardless of other risk factors identified. The risk of a first event is two to three times higher in people with a family history of thrombosis in a first-degree relative. The risk is four times higher when multiple family members are affected, at least one of them before age 50.47
In people with genetic thrombophilia, the risk of thrombosis (especially unprovoked thrombosis at a young age) is also higher in those with a strong family history than in those without a family history. In those with factor V Leiden, the risk of venous thromboembolism is three to four times higher if there is a positive family history. The risk is five times higher in carriers of factor V Leiden with a family history of venous thromboembolism before age 50, and 13 times higher in those with more than one affected family member.47
Possible shared environmental factors or coinheritance of other unidentified genetic factors may also contribute to the higher susceptibility in thrombosis-prone families.
TESTING FOR APC RESISTANCE AND FACTOR V LEIDEN
The factor V Leiden mutation can be detected directly by genetic testing of peripheral blood mononuclear cells. This method is relatively time-consuming and expensive, however.
At present, the most cost-effective approach is to test first for APC resistance using a second-generation coagulation assay—the modified APC sensitivity test. In this clot-based method, the patient’s sample is prediluted with factor V-deficient plasma to eliminate the effect of lupus anticoagulants and factor deficiencies that could prolong the baseline clotting time, and heparin is inactivated by polybrene. Then either an augmented partial-thromboplastin-time-based assay or a tissue-factor-dependent factor V assay is performed.
This test is nearly 100% sensitive and specific for factor V Leiden, in contrast to the first-generation, or classic, APC sensitivity test, which lacked specificity and sensitivity for it.9–11,48–60 This modification also permits testing of patients receiving anticoagulants or who have abnormal augmented partial thromboplastin times due to coagulation factor deficiencies.
A positive result on the modified APC sensitivity test should be confirmed by a direct genetic test for the factor V Leiden mutation. An APC resistance assay is unnecessary if a direct genetic test is used initially.
HOW LONG TO GIVE ANTICOAGULATION AFTER VENOUS THROMBOEMBOLISM?
Patients who have had an episode of venous thromboembolism have to be treated with anticoagulants.
In general, the initial management of venous thromboembolism in patients with heritable thrombophilias is no different from that in any other patient with a clot. Anticoagulants such as warfarin are given at a target INR of 2.5 (range 2.0–3.0).32 The duration of treatment is based on the risk factors that resulted in the thrombotic event.
After a first event, some authorities recommend anticoagulant therapy for 6 months.32 A shorter period (3 months) is recommended if there is a transient risk factor (eg, surgery, oral contraceptive use, travel, pregnancy, the puerperium) and the thrombosis is confined to distal veins (eg, the calf veins).32
Factor V Leiden does not necessarily increase the risk of recurrent events in patients who have a transient risk factor. Therefore, people who are heterozygous for this mutation do not usually need to be treated lifelong with anticoagulants if they have had only one episode of deep vein thrombosis or pulmonary embolism, given the risk of bleeding associated with anticoagulation, unless they have additional risk factors.
Conditions in which indefinite anticoagulation may be required after careful consideration of the risks and benefits are:
- Life-threatening events such as near-fatal pulmonary embolism
- Cerebral or visceral vein thrombosis
- Recurrent thrombotic events
- Additional persistent risk factors (eg, active malignant neoplasm, extremity paresis, and antiphospholipid antibodies)
- Combined thrombophilias (eg, combined heterozygosity for factor V Leiden and the prothrombin G20210A mutation)
- Homozygosity for factor V Leiden.32,46,48
Factor V Leiden by itself or combined with other thrombophilic abnormalities is not associated with a higher risk of recurrent venous thromboembolism during warfarin therapy (a possible exception is the combination of factor V Leiden plus antiphospholipid antibodies).32,34 Furthermore, current evidence suggests that no thrombophilic defect is a clinically important risk factor for recurrent venous thromboembolism after anticoagulant therapy is stopped. All these facts indicate that clinical factors are probably more important than laboratory abnormalities in determining the duration of anticoagulation therapy.32,35,36,61–63
PRIMARY PROPHYLAXIS IN PATIENTS WITH FACTOR V LEIDEN
Factor V Leiden is only one of many risk factors for deep vein thrombosis or pulmonary embolism. If carriers of factor V Leiden have never had a blood clot, then they are not routinely treated with an anticoagulant. Rather, they should be counseled about reducing or eliminating other factors that may add to their risk of developing a clot in the future.
Usually, the effect of risk factors is additive: the more risk factors present, the higher the risk.46,50 Sometimes, however, the effect of multiple risk factors is more than additive.
Some risk factors, such as genetics or age, are not alterable, but many can be controlled by medications or lifestyle modifications. Therefore, general measures and precautions are recommended to minimize the risk of thrombosis. For example:
Losing weight (if the patient is overweight) is an important intervention for risk reduction, since obesity is probably the most common modifiable risk factor for developing blood clots.
Avoiding long periods of immobility is recommended. For example, if the patient is taking a long car ride (more than 2 hours), then stopping every few hours and walking around for a few minutes is a good way to keep the blood circulating. If the patient has a desk job, getting up and walking around the office periodically is advised. On long airplane trips, a walk in the aisle every so often and preventing dehydration by drinking plenty of fluids and avoiding alcohol are recommended.
Wearing elastic stockings with a graduated elastic pressure may prevent deep venous thrombosis from developing on long flights.63–65
Staying active and getting regular exercise through such activities as walking, bicycling, or swimming are helpful.
Avoiding smoking is critical.50,63
Thromboprophylaxis is recommended for most acutely ill hospitalized patients, especially those confined to bed with additional risk factors. Guidelines for prophylaxis are based on an individualized risk assessment and not on thrombophilia status. Prophylactic anticoagulation is routinely recommended for patients undergoing major high-risk surgery, such as an orthopedic, urologic, gynecologic, or bariatric procedure. Any excess thrombotic risk conferred by thrombophilia is likely small compared with the risk of surgery, and recommendations on the duration and intensity of thromboprophylaxis are not based on thrombophilic status.46,48
Education. Pain, swelling, redness of a limb, unexplained shortness of breath, and chest pain are the most common symptoms of deep vein thrombosis and pulmonary embolism.46,50 It is crucial to teach patients with factor V Leiden to recognize these symptoms and to seek early medical attention in case they experience any of them.
SCREENING FAMILY MEMBERS FOR THE FACTOR V LEIDEN MUTATION
Factor V Leiden by itself is a relatively mild thrombophilic defect that does not cause thrombosis in all carriers, and there is no evidence that early diagnosis reduces rates of morbidity or mortality. Therefore, routine screening of all asymptomatic relatives of affected patients with venous thrombosis is not recommended. Rather, the decision to screen should be made on an individual basis.50,66
Screening may be beneficial in selected cases, especially when patients have a strong family history of recurrent venous thrombosis at a young age (younger than 50 years) and the family member has additional risk factors for venous thromboembolism such as oral contraception or is planning for pregnancy.32,48,49,66
- Rees DC, Cox M, Clegg JB. World distribution of factor V Leiden. Lancet 1995; 346:1133–1134.
- Ridker PM, Miletich JP, Hennekens CH, Buring JE. Ethnic distribution of factor V Leiden in 4047 men and women. Implications for venous thromboembolism screening. JAMA 1997; 277:1305–1307.
- Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood 1995; 85:1504–1508.
- Stolz E, Kemkes-Matthes B, Pötzsch B, et al. Screening for thrombophilic risk factors among 25 German patients with cerebral venous thrombosis. Acta Neurol Scand 2000; 102:31–36.
- Langlois NJ, Wells PS. Risk of venous thromboembolism in relatives of symptomatic probands with thrombophilia: a systematic review. Thromb Haemost 2003; 90:17–26.
- Juul K, Tybjaerg-Hansen A, Mortensen J, Lange P, Vestbo J, Nordestgaard BG. Factor V leiden homozygosity, dyspnea, and reduced pulmonary function. Arch Intern Med 2005; 165:2032–2036.
- Bertina RM, Koeleman BP, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994; 369:64–67.
- Dahlbäck B. New molecular insights into the genetics of thrombophilia. Resistance to activated protein C caused by Arg506 to Gln mutation in factor V as a pathogenic risk factor for venous thrombosis. Thromb Haemost 1995; 74:139–148.
- Castoldi E, Brugge JM, Nicolaes GA, Girelli D, Tans G, Rosing J. Impaired APC cofactor activity of factor V plays a major role in the APC resistance associated with the factor V Leiden (R506Q) and R2 (H1299R) mutations. Blood 2004; 103:4173–4179.
- Dahlback B. Anticoagulant factor V and thrombosis risk (editorial). Blood 2004; 103:3995.
- Simioni P, Castoldi E, Lunghi B, Tormene D, Rosing J, Bernardi F. An underestimated combination of opposites resulting in enhanced thrombotic tendency. Blood 2005; 106:2363–2365.
- Williamson D, Brown K, Luddington R, Baglin C, Baglin T. Factor V Cambridge: a new mutation (Arg306-->Thr) associated with resistance to activated protein C. Blood 1998; 91:1140–1144.
- Chan WP, Lee CK, Kwong YL, Lam CK, Liang R. A novel mutation of Arg306 of factor V gene in Hong Kong Chinese. Blood 1998; 91:1135–1139.
- Liang R, Lee CK, Wat MS, Kwong YL, Lam CK, Liu HW. Clinical significance of Arg306 mutations of factor V gene. Blood 1998; 92:2599–2600.
- Steen M, Norstrøm EA, Tholander AL, et al. Functional characterization of factor V-Ile359Thr: a novel mutation associated with thrombosis. Blood 2004; 103:3381–3387.
- Bernardi F, Faioni EM, Castoldi E, et al. A factor V genetic component differing from factor V R506Q contributes to the activated protein C resistance phenotype. Blood 1997; 90:1552–1557.
- Lunghi B, Castoldi E, Mingozzi F, Bernardi F. A new factor V gene polymorphism (His 1254 Arg) present in subjects of African origin mimics the R2 polymorphism (His 1299 Arg). Blood 1998; 91:364–365.
- Luddington R, Jackson A, Pannerselvam S, Brown K, Baglin T. The factor V R2 allele: risk of venous thromboembolism, factor V levels and resistance to activated protein C. Thromb Haemost 2000; 83:204–208.
- Faioni EM, Franchi F, Bucciarelli P, et al. Coinheritance of the HR2 haplotype in the factor V gene confers an increased risk of venous thromboembolism to carriers of factor V R506Q (factor V Leiden). Blood 1999; 94:3062–3066.
- Clark P, Walker ID. The phenomenon known as acquired activated protein C resistance. Br J Haematol 2001; 115:767–773.
- Tosetto A, Simioni M, Madeo D, Rodeghiero F. Intraindividual consistency of the activated protein C resistance phenotype. Br J Haematol 2004; 126:405–409.
- de Visser MC, Rosendaal FR, Bertina RM. A reduced sensitivity for activated protein C in the absence of factor V Leiden increases the risk of venous thrombosis. Blood 1999; 93:1271–1276.
- Kraaijenhagen RA, in’t Anker PS, Koopman MM, et al. High plasma concentration of factor VIIIc is a major risk factor for venous thromboembolism. Thromb Haemost 2000; 83:5–9.
- Kamphuisen PW, Eikenboom JC, Bertina RM. Elevated factor VIII levels and the risk of thrombosis. Arterioscler Thromb Vasc Biol 2001; 21:731–738.
- Koster T, Blann AD, Briët E, Vandenbroucke JP, Rosendaal FR. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep-vein thrombosis. Lancet 1995; 345:152–155.
- Clark P, Brennand J, Conkie JA, McCall F, Greer IA, Walker ID. Activated protein C sensitivity, protein C, protein S and coagulation in normal pregnancy. Thromb Haemost 1998; 79:1166–1170.
- Cumming AM, Tait RC, Fildes S, Yoong A, Keeney S, Hay CR. Development of resistance to activated protein C during pregnancy. Br J Haematol 1995; 90:725–727.
- Mathonnet F, de Mazancourt P, Bastenaire B, et al. Activated protein C sensitivity ratio in pregnant women at delivery. Br J Haematol 1996; 92:244–246.
- Post MS, Rosing J, Van Der Mooren MJ, et al; Ageing Women’ and the Institute for Cardiovascular Research-Vrije Universiteit (ICaRVU). Increased resistance to activated protein C after short-term oral hormone replacement therapy in healthy post-menopausal women. Br J Haematol 2002; 119:1017–1023.
- Olivieri O, Friso S, Manzato F, et al. Resistance to activated protein C in healthy women taking oral contraceptives. Br J Haematol 1995; 91:465–470.
- Bokarewa MI, Blombäck M, Egberg N, Rosén S. A new variant of interaction between phospholipid antibodies and the protein C system. Blood Coagul Fibrinolysis 1994; 5:37–41.
- Baglin T, Gray E, Greaves M, et al; British Committee for Standards in Haematology. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010; 149:209–220.
- van Stralen KJ, Doggen CJ, Bezemer ID, Pomp ER, Lisman T, Rosendaal FR. Mechanisms of the factor V Leiden paradox. Arterioscler Thromb Vasc Biol 2008; 28:1872–1877.
- Agaoglu N, Mustafa NA, Turkyilmaz S. Prothrombotic disorders in patients with mesenteric vein thrombosis. J Invest Surg 2003; 16:299–304.
- El-Karaksy H, El-Koofy N, El-Hawary M, et al. Prevalence of factor V Leiden mutation and other hereditary thrombophilic factors in Egyptian children with portal vein thrombosis: results of a single-center case-control study. Ann Hematol 2004; 83:712–715.
- Heijmans BT, Westendorp RG, Knook DL, Kluft C, Slagboom PE. The risk of mortality and the factor V Leiden mutation in a population-based cohort. Thromb Haemost 1998; 80:607–609.
- Turkstra F, Karemaker R, Kuijer PM, Prins MH, Büller HR. Is the prevalence of the factor V Leiden mutation in patients with pulmonary embolism and deep vein thrombosis really different? Thromb Haemost 1999; 81:345–348.
- Ridker PM, Hennekens CH, Lindpaintner K, Stampfer MJ, Eisenberg PR, Miletich JP. Mutation in the gene coding for coagulation factor V and the risk of myocardial infarction, stroke, and venous thrombosis in apparently healthy men. N Engl J Med 1995; 332:912–917.
- Manten B, Westendorp RG, Koster T, Reitsma PH, Rosendaal FR. Risk factor profiles in patients with different clinical manifestations of venous thromboembolism: a focus on the factor V Leiden mutation. Thromb Haemost 1996; 76:510–513.
- Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 2005; 293:715–722.
- Bloemenkamp KW, Rosendaal FR, Helmerhorst FM, Büller HR, Vandenbroucke JP. Enhancement by factor V Leiden mutation of risk of deep-vein thrombosis associated with oral contraceptives containing a third-generation progestagen. Lancet 1995; 346:1593–1596.
- Murphy PT. Factor V Leiden and venous thromboembolism. Ann Intern Med 2004; 141:483–484.
- Nizankowska-Mogilnicka E, Adamek L, Grzanka P, et al. Genetic polymorphisms associated with acute pulmonary embolism and deep venous thrombosis. Eur Respir J 2003; 21:25–30.
- Arsov T, Miladinova D, Spiroski M. Factor V Leiden is associated with higher risk of deep venous thrombosis of large blood vessels. Croat Med J 2006; 47:433–439.
- Simioni P, Prandoni P, Lensing AW, et al. Risk for subsequent venous thromboembolic complications in carriers of the prothrombin or the factor V gene mutation with a first episode of deep-vein thrombosis. Blood 2000; 96:3329–3333.
- Ornstein DL, Cushman M. Cardiology patient page. Factor V Leiden. Circulation 2003; 107:e94–e97.
- Bezemer ID, van der Meer FJ, Eikenboom JC, Rosendaal FR, Doggen CJ. The value of family history as a risk indicator for venous thrombosis. Arch Intern Med 2009; 169:610–615.
- Press RD, Bauer KA, Kujovich JL, Heit JA. Clinical utility of factor V leiden (R506Q) testing for the diagnosis and management of thromboembolic disorders. Arch Pathol Lab Med 2002; 126:1304–1318.
- Gadelha T, Roldán V, Lecumberri R, et al; RIETE Investigators. Clinical characteristics of patients with factor V Leiden or prothrombin G20210A and a first episode of venous thromboembolism. Findings from the RIETE Registry. Thromb Res 2010; 126:283–286.
- Severinsen MT, Overvad K, Johnsen SP, et al. Genetic susceptibility, smoking, obesity and risk of venous thromboembolism. Br J Haematol 2010; 149:273–279.
- Kujovich JL. Factor V Leiden thrombophilia. Genet Med 2011; 13:1–16.
- Lijfering WM, Brouwer JL, Veeger NJ, et al. Selective testing for thrombophilia in patients with first venous thrombosis: results from a retrospective family cohort study on absolute thrombotic risk for currently known thrombophilic defects in 2479 relatives. Blood 2009; 113:5314–5322.
- Kearon C, Julian JA, Kovacs MJ, et al; ELATE Investigators. Influence of thrombophilia on risk of recurrent venous thromboembolism while on warfarin: results from a randomized trial. Blood 2008; 112:4432–4436.
- Ho WK, Hankey GJ, Quinlan DJ, Eikelboom JW. Risk of recurrent venous thromboembolism in patients with common thrombophilia: a systematic review. Arch Intern Med 2006; 166:729–736.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA 2005; 293:2352–2361.
- Strobl FJ, Hoffman S, Huber S, Williams EC, Voelkerding KV. Activated protein C resistance assay performance: improvement by sample dilution with factor V-deficient plasma. Arch Pathol Lab Med 1998; 122:430–433.
- Legnani C, Palareti G, Biagi R, et al. Activated protein C resistance: a comparison between two clotting assays and their relationship to the presence of the factor V Leiden mutation. Br J Haematol 1996; 93:694–699.
- Gouault-Heilmann M, Leroy-Matheron C. Factor V Leiden-dependent APC resistance: improved sensitivity and specificity of the APC resistance test by plasma dilution in factor V-depleted plasma. Thromb Res 1996; 82:281–283.
- Svensson PJ, Zöller B, Dahlbäck B. Evaluation of original and modified APC-resistance tests in unselected outpatients with clinically suspected thrombosis and in healthy controls. Thromb Haemost 1997; 77:332–335.
- Tripodi A, Negri B, Bertina RM, Mannucci PM. Screening for the FV:Q506 mutation—evaluation of thirteen plasma-based methods for their diagnostic efficacy in comparison with DNA analysis. Thromb Haemost 1997; 77:436–439.
- Wåhlander K, Larson G, Lindahl TL, et al. Factor V Leiden (G1691A) and prothrombin gene G20210A mutations as potential risk factors for venous thromboembolism after total hip or total knee replacement surgery. Thromb Haemost 2002; 87:580–585.
- Joseph JE, Low J, Courtenay B, Neil MJ, McGrath M, Ma D. A single-centre prospective study of clinical and haemostatic risk factors for venous thromboembolism following lower limb arthroplasty. Br J Haematol 2005; 129:87–92.
- Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):381S–453S.
- Brenner B. Prophylaxis for travel-related thrombosis? Yes. J Thromb Haemost 2004; 2:2089–2091.
- Gavish I, Brenner B. Air travel and the risk of thromboembolism. Intern Emerg Med 2011; 6:113–116.
- Grody WW, Griffin JH, Taylor AK, Korf BR, Heit JA; ACMG Factor V Leiden Working Group. American College of Medical Genetics consensus statement on factor V Leiden mutation testing. Genet Med 2001; 3:139–148.
- Rees DC, Cox M, Clegg JB. World distribution of factor V Leiden. Lancet 1995; 346:1133–1134.
- Ridker PM, Miletich JP, Hennekens CH, Buring JE. Ethnic distribution of factor V Leiden in 4047 men and women. Implications for venous thromboembolism screening. JAMA 1997; 277:1305–1307.
- Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood 1995; 85:1504–1508.
- Stolz E, Kemkes-Matthes B, Pötzsch B, et al. Screening for thrombophilic risk factors among 25 German patients with cerebral venous thrombosis. Acta Neurol Scand 2000; 102:31–36.
- Langlois NJ, Wells PS. Risk of venous thromboembolism in relatives of symptomatic probands with thrombophilia: a systematic review. Thromb Haemost 2003; 90:17–26.
- Juul K, Tybjaerg-Hansen A, Mortensen J, Lange P, Vestbo J, Nordestgaard BG. Factor V leiden homozygosity, dyspnea, and reduced pulmonary function. Arch Intern Med 2005; 165:2032–2036.
- Bertina RM, Koeleman BP, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994; 369:64–67.
- Dahlbäck B. New molecular insights into the genetics of thrombophilia. Resistance to activated protein C caused by Arg506 to Gln mutation in factor V as a pathogenic risk factor for venous thrombosis. Thromb Haemost 1995; 74:139–148.
- Castoldi E, Brugge JM, Nicolaes GA, Girelli D, Tans G, Rosing J. Impaired APC cofactor activity of factor V plays a major role in the APC resistance associated with the factor V Leiden (R506Q) and R2 (H1299R) mutations. Blood 2004; 103:4173–4179.
- Dahlback B. Anticoagulant factor V and thrombosis risk (editorial). Blood 2004; 103:3995.
- Simioni P, Castoldi E, Lunghi B, Tormene D, Rosing J, Bernardi F. An underestimated combination of opposites resulting in enhanced thrombotic tendency. Blood 2005; 106:2363–2365.
- Williamson D, Brown K, Luddington R, Baglin C, Baglin T. Factor V Cambridge: a new mutation (Arg306-->Thr) associated with resistance to activated protein C. Blood 1998; 91:1140–1144.
- Chan WP, Lee CK, Kwong YL, Lam CK, Liang R. A novel mutation of Arg306 of factor V gene in Hong Kong Chinese. Blood 1998; 91:1135–1139.
- Liang R, Lee CK, Wat MS, Kwong YL, Lam CK, Liu HW. Clinical significance of Arg306 mutations of factor V gene. Blood 1998; 92:2599–2600.
- Steen M, Norstrøm EA, Tholander AL, et al. Functional characterization of factor V-Ile359Thr: a novel mutation associated with thrombosis. Blood 2004; 103:3381–3387.
- Bernardi F, Faioni EM, Castoldi E, et al. A factor V genetic component differing from factor V R506Q contributes to the activated protein C resistance phenotype. Blood 1997; 90:1552–1557.
- Lunghi B, Castoldi E, Mingozzi F, Bernardi F. A new factor V gene polymorphism (His 1254 Arg) present in subjects of African origin mimics the R2 polymorphism (His 1299 Arg). Blood 1998; 91:364–365.
- Luddington R, Jackson A, Pannerselvam S, Brown K, Baglin T. The factor V R2 allele: risk of venous thromboembolism, factor V levels and resistance to activated protein C. Thromb Haemost 2000; 83:204–208.
- Faioni EM, Franchi F, Bucciarelli P, et al. Coinheritance of the HR2 haplotype in the factor V gene confers an increased risk of venous thromboembolism to carriers of factor V R506Q (factor V Leiden). Blood 1999; 94:3062–3066.
- Clark P, Walker ID. The phenomenon known as acquired activated protein C resistance. Br J Haematol 2001; 115:767–773.
- Tosetto A, Simioni M, Madeo D, Rodeghiero F. Intraindividual consistency of the activated protein C resistance phenotype. Br J Haematol 2004; 126:405–409.
- de Visser MC, Rosendaal FR, Bertina RM. A reduced sensitivity for activated protein C in the absence of factor V Leiden increases the risk of venous thrombosis. Blood 1999; 93:1271–1276.
- Kraaijenhagen RA, in’t Anker PS, Koopman MM, et al. High plasma concentration of factor VIIIc is a major risk factor for venous thromboembolism. Thromb Haemost 2000; 83:5–9.
- Kamphuisen PW, Eikenboom JC, Bertina RM. Elevated factor VIII levels and the risk of thrombosis. Arterioscler Thromb Vasc Biol 2001; 21:731–738.
- Koster T, Blann AD, Briët E, Vandenbroucke JP, Rosendaal FR. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep-vein thrombosis. Lancet 1995; 345:152–155.
- Clark P, Brennand J, Conkie JA, McCall F, Greer IA, Walker ID. Activated protein C sensitivity, protein C, protein S and coagulation in normal pregnancy. Thromb Haemost 1998; 79:1166–1170.
- Cumming AM, Tait RC, Fildes S, Yoong A, Keeney S, Hay CR. Development of resistance to activated protein C during pregnancy. Br J Haematol 1995; 90:725–727.
- Mathonnet F, de Mazancourt P, Bastenaire B, et al. Activated protein C sensitivity ratio in pregnant women at delivery. Br J Haematol 1996; 92:244–246.
- Post MS, Rosing J, Van Der Mooren MJ, et al; Ageing Women’ and the Institute for Cardiovascular Research-Vrije Universiteit (ICaRVU). Increased resistance to activated protein C after short-term oral hormone replacement therapy in healthy post-menopausal women. Br J Haematol 2002; 119:1017–1023.
- Olivieri O, Friso S, Manzato F, et al. Resistance to activated protein C in healthy women taking oral contraceptives. Br J Haematol 1995; 91:465–470.
- Bokarewa MI, Blombäck M, Egberg N, Rosén S. A new variant of interaction between phospholipid antibodies and the protein C system. Blood Coagul Fibrinolysis 1994; 5:37–41.
- Baglin T, Gray E, Greaves M, et al; British Committee for Standards in Haematology. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010; 149:209–220.
- van Stralen KJ, Doggen CJ, Bezemer ID, Pomp ER, Lisman T, Rosendaal FR. Mechanisms of the factor V Leiden paradox. Arterioscler Thromb Vasc Biol 2008; 28:1872–1877.
- Agaoglu N, Mustafa NA, Turkyilmaz S. Prothrombotic disorders in patients with mesenteric vein thrombosis. J Invest Surg 2003; 16:299–304.
- El-Karaksy H, El-Koofy N, El-Hawary M, et al. Prevalence of factor V Leiden mutation and other hereditary thrombophilic factors in Egyptian children with portal vein thrombosis: results of a single-center case-control study. Ann Hematol 2004; 83:712–715.
- Heijmans BT, Westendorp RG, Knook DL, Kluft C, Slagboom PE. The risk of mortality and the factor V Leiden mutation in a population-based cohort. Thromb Haemost 1998; 80:607–609.
- Turkstra F, Karemaker R, Kuijer PM, Prins MH, Büller HR. Is the prevalence of the factor V Leiden mutation in patients with pulmonary embolism and deep vein thrombosis really different? Thromb Haemost 1999; 81:345–348.
- Ridker PM, Hennekens CH, Lindpaintner K, Stampfer MJ, Eisenberg PR, Miletich JP. Mutation in the gene coding for coagulation factor V and the risk of myocardial infarction, stroke, and venous thrombosis in apparently healthy men. N Engl J Med 1995; 332:912–917.
- Manten B, Westendorp RG, Koster T, Reitsma PH, Rosendaal FR. Risk factor profiles in patients with different clinical manifestations of venous thromboembolism: a focus on the factor V Leiden mutation. Thromb Haemost 1996; 76:510–513.
- Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 2005; 293:715–722.
- Bloemenkamp KW, Rosendaal FR, Helmerhorst FM, Büller HR, Vandenbroucke JP. Enhancement by factor V Leiden mutation of risk of deep-vein thrombosis associated with oral contraceptives containing a third-generation progestagen. Lancet 1995; 346:1593–1596.
- Murphy PT. Factor V Leiden and venous thromboembolism. Ann Intern Med 2004; 141:483–484.
- Nizankowska-Mogilnicka E, Adamek L, Grzanka P, et al. Genetic polymorphisms associated with acute pulmonary embolism and deep venous thrombosis. Eur Respir J 2003; 21:25–30.
- Arsov T, Miladinova D, Spiroski M. Factor V Leiden is associated with higher risk of deep venous thrombosis of large blood vessels. Croat Med J 2006; 47:433–439.
- Simioni P, Prandoni P, Lensing AW, et al. Risk for subsequent venous thromboembolic complications in carriers of the prothrombin or the factor V gene mutation with a first episode of deep-vein thrombosis. Blood 2000; 96:3329–3333.
- Ornstein DL, Cushman M. Cardiology patient page. Factor V Leiden. Circulation 2003; 107:e94–e97.
- Bezemer ID, van der Meer FJ, Eikenboom JC, Rosendaal FR, Doggen CJ. The value of family history as a risk indicator for venous thrombosis. Arch Intern Med 2009; 169:610–615.
- Press RD, Bauer KA, Kujovich JL, Heit JA. Clinical utility of factor V leiden (R506Q) testing for the diagnosis and management of thromboembolic disorders. Arch Pathol Lab Med 2002; 126:1304–1318.
- Gadelha T, Roldán V, Lecumberri R, et al; RIETE Investigators. Clinical characteristics of patients with factor V Leiden or prothrombin G20210A and a first episode of venous thromboembolism. Findings from the RIETE Registry. Thromb Res 2010; 126:283–286.
- Severinsen MT, Overvad K, Johnsen SP, et al. Genetic susceptibility, smoking, obesity and risk of venous thromboembolism. Br J Haematol 2010; 149:273–279.
- Kujovich JL. Factor V Leiden thrombophilia. Genet Med 2011; 13:1–16.
- Lijfering WM, Brouwer JL, Veeger NJ, et al. Selective testing for thrombophilia in patients with first venous thrombosis: results from a retrospective family cohort study on absolute thrombotic risk for currently known thrombophilic defects in 2479 relatives. Blood 2009; 113:5314–5322.
- Kearon C, Julian JA, Kovacs MJ, et al; ELATE Investigators. Influence of thrombophilia on risk of recurrent venous thromboembolism while on warfarin: results from a randomized trial. Blood 2008; 112:4432–4436.
- Ho WK, Hankey GJ, Quinlan DJ, Eikelboom JW. Risk of recurrent venous thromboembolism in patients with common thrombophilia: a systematic review. Arch Intern Med 2006; 166:729–736.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA 2005; 293:2352–2361.
- Strobl FJ, Hoffman S, Huber S, Williams EC, Voelkerding KV. Activated protein C resistance assay performance: improvement by sample dilution with factor V-deficient plasma. Arch Pathol Lab Med 1998; 122:430–433.
- Legnani C, Palareti G, Biagi R, et al. Activated protein C resistance: a comparison between two clotting assays and their relationship to the presence of the factor V Leiden mutation. Br J Haematol 1996; 93:694–699.
- Gouault-Heilmann M, Leroy-Matheron C. Factor V Leiden-dependent APC resistance: improved sensitivity and specificity of the APC resistance test by plasma dilution in factor V-depleted plasma. Thromb Res 1996; 82:281–283.
- Svensson PJ, Zöller B, Dahlbäck B. Evaluation of original and modified APC-resistance tests in unselected outpatients with clinically suspected thrombosis and in healthy controls. Thromb Haemost 1997; 77:332–335.
- Tripodi A, Negri B, Bertina RM, Mannucci PM. Screening for the FV:Q506 mutation—evaluation of thirteen plasma-based methods for their diagnostic efficacy in comparison with DNA analysis. Thromb Haemost 1997; 77:436–439.
- Wåhlander K, Larson G, Lindahl TL, et al. Factor V Leiden (G1691A) and prothrombin gene G20210A mutations as potential risk factors for venous thromboembolism after total hip or total knee replacement surgery. Thromb Haemost 2002; 87:580–585.
- Joseph JE, Low J, Courtenay B, Neil MJ, McGrath M, Ma D. A single-centre prospective study of clinical and haemostatic risk factors for venous thromboembolism following lower limb arthroplasty. Br J Haematol 2005; 129:87–92.
- Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):381S–453S.
- Brenner B. Prophylaxis for travel-related thrombosis? Yes. J Thromb Haemost 2004; 2:2089–2091.
- Gavish I, Brenner B. Air travel and the risk of thromboembolism. Intern Emerg Med 2011; 6:113–116.
- Grody WW, Griffin JH, Taylor AK, Korf BR, Heit JA; ACMG Factor V Leiden Working Group. American College of Medical Genetics consensus statement on factor V Leiden mutation testing. Genet Med 2001; 3:139–148.
KEY POINTS
- The pathogenesis of venous thromboembolism is complex and multifactorial, often reflecting the interplay between environmental, clinical, and genetic factors.
- Factor V Leiden increases the risk of venous thromboembolism but by itself does not appear to increase the risk of arterial thrombosis.
- Often, people with factor V Leiden may have additional risk factors that increase the rate of venous clots, such as older age, surgery, obesity, immobility, prolonged travel, hospitalization, oral contraceptive use, hormonal replacement therapy, pregnancy, and malignancy.
- General measures and precautions are needed to minimize the risk of venous thromboembolism in people with the factor V Leiden mutation, especially when modifiable factors are present, such as obesity and long periods of immobilization.
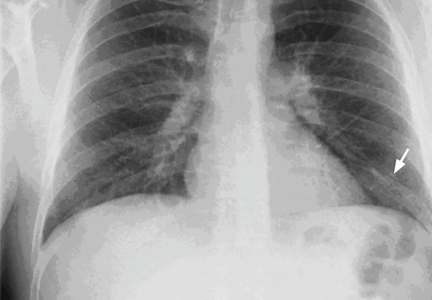
Unmasking gastric cancer
A 50-year-old male Japanese immigrant with a history of smoking and occasional untreated heartburn presented with the recent onset of flank pain, weight loss, headache, syncope, and blurred vision.
Previously healthy, he began feeling moderate pain in his left flank 1 month ago; it was diagnosed as kidney stones and was treated conservatively. Two weeks later he had an episode of syncope and soon after developed blurred vision, mainly in his left eye, along with severe bifrontal headache. An eye examination and magnetic resonance imaging of the brain indicated optic neuritis, for which he was given glucocorticoids intravenously for 3 days, with moderate improvement.
As his symptoms continued over the next 2 weeks, he lost 20 lb (9.1 kg) due to the pain, loss of appetite, nausea, and occasional vomiting.
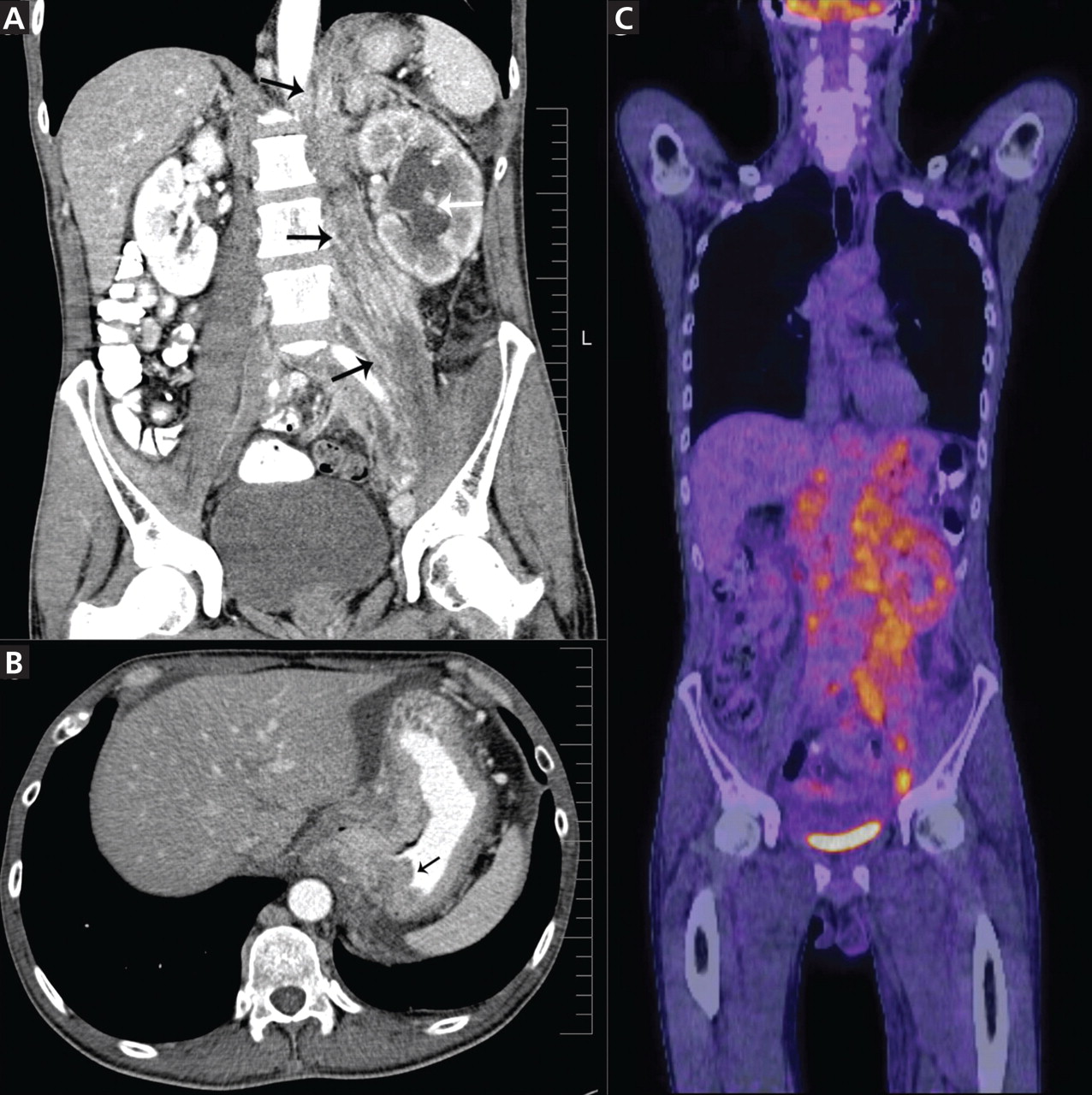
Positron emission tomography showed the retroperitoneal infiltrative process and the thickened gastric cardia to be hypermetabolic (Figure 1C).
The area of retroperitoneal infiltration was biopsied under CT guidance, and pathologic study showed poorly differentiated carcinoma with signet-ring cells, a feature of gastric cancer.
The patient underwent lumbar puncture. His cerebrospinal fluid had 206 white blood cells/μL (reference range 0–5) and large numbers of poorly differentiated malignant cells, most consistent with adenocarcinoma on cytologic study.
duodenoscopy (EGD) revealed a large, ulcerated, submucosal, nodular mass in the cardia of the stomach extending to the gastroesophageal junction (Figure 2A). Biopsy of the mass again revealed poorly differentiated adenocarcinoma with scattered signet-ring cells undermining the gastric mucosa, favoring a gastric origin (Figure 2B).
THREE SUBTYPES OF GASTRIC CANCER
Worldwide, gastric cancer is the third most common type of cancer and the second most common cause of cancer-related deaths.1 In the United States, blacks and people of Asian ancestry have almost twice the risk of death, with the highest incidence and mortality rates.2,3
Most cases of gastric adenocarcinoma can be categorized as either intestinal or diffuse, but a new proximal subtype is emerging.4
Intestinal-type gastric adenocarcinoma is the most common subtype and accounts for almost all the ethnic and geographic variation in incidence.2 The lesions are often ulcerative and distal; the pathogenesis is stepwise and is initiated by chronic inflammation. Risk factors include old age, Helicobacter pylori infection, tobacco smoking, family history, and high salt intake, with an observed risk-reduction with the use of nonsteroidal anti-inflammatory drugs and with a high intake of fruits and vegetables.3
Diffuse gastric adenocarcinoma, on the other hand, has a uniform distribution worldwide, and its incidence is increasing. It typically carries a poor prognosis. Evidence thus far has shown its pathogenesis to be independent of chronic inflammation, but it has a strong tendency to be hereditary.3
Proximal gastric adenocarcinoma is observed in the gastric cardia and near the gastroesophageal junction. It is often grouped with the distal esophageal adenocarcinomas and has similar risk factors, including reflux disease, obesity, alcohol abuse, and tobacco smoking. Interestingly, however, H pylori infection does not contribute to the pathogenesis of this type, and it may even have a protective role.3
DIFFICULT TO DETECT EARLY
Gastric cancer is difficult to detect early enough in its course to be cured. Understanding its risk factors, recognizing its common symptoms, and regarding its uncommon symptoms with suspicion may lead to earlier diagnosis and more effective treatment.
Our patient’s proximal gastric cancer was diagnosed late even though he had several risk factors for it (he was Japanese, he was a smoker, and he had gastroesophageal reflux disease) because of a late and atypical presentation with misleading paraneoplastic symptoms.
Early diagnosis is difficult because most patients have no symptoms in the early stage; weight loss and abdominal pain are often late signs of tumor progression.
Screening may be justified in high-risk groups in the United States, although the issue is debatable. Diagnostic imaging is the only effective method for screening,5 with EGD considered the first-line targeted evaluation should there be suspicion of gastric cancer either from the clinical presentation or from barium swallow.6 Candidates for screening may include elderly patients with atrophic gastritis or pernicious anemia, immigrants from countries with high rates of gastric carcinoma, and people with a family history of gastrointestinal cancer.7
- Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55:74–108.
- Crew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol 2006; 12:354–362.
- Shah MA, Kelsen DP. Gastric cancer: a primer on the epidemiology and biology of the disease and an overview of the medical management of advanced disease. J Natl Compr Canc Netw 2010; 8:437–447.
- Fine G, Chan K. Alimentary tract. In:Kissane JM, editor. Anderson’s Pathology. 8th ed. Saint Louis, MO: Mosby; 1985:1055–1095.
- Kunisaki C, Ishino J, Nakajima S, et al. Outcomes of mass screening for gastric carcinoma. Ann Surg Oncol 2006; 13:221–228.
- Cappell MS, Friedel D. The role of esophagogastroduodenoscopy in the diagnosis and management of upper gastrointestinal disorders. Med Clin North Am 2002; 86:1165–1216.
- Hisamuchi S, Fukao P, Sugawara N, et al. Evaluation of mass screening programme for stomach cancer in Japan. In:Miller AB, Chamberlain J, Day NE, et al, editors. Cancer Screening. Cambridge, UK: Cambridge University Press; 1991:357–372.
A 50-year-old male Japanese immigrant with a history of smoking and occasional untreated heartburn presented with the recent onset of flank pain, weight loss, headache, syncope, and blurred vision.
Previously healthy, he began feeling moderate pain in his left flank 1 month ago; it was diagnosed as kidney stones and was treated conservatively. Two weeks later he had an episode of syncope and soon after developed blurred vision, mainly in his left eye, along with severe bifrontal headache. An eye examination and magnetic resonance imaging of the brain indicated optic neuritis, for which he was given glucocorticoids intravenously for 3 days, with moderate improvement.
As his symptoms continued over the next 2 weeks, he lost 20 lb (9.1 kg) due to the pain, loss of appetite, nausea, and occasional vomiting.
Positron emission tomography showed the retroperitoneal infiltrative process and the thickened gastric cardia to be hypermetabolic (Figure 1C).
The area of retroperitoneal infiltration was biopsied under CT guidance, and pathologic study showed poorly differentiated carcinoma with signet-ring cells, a feature of gastric cancer.
The patient underwent lumbar puncture. His cerebrospinal fluid had 206 white blood cells/μL (reference range 0–5) and large numbers of poorly differentiated malignant cells, most consistent with adenocarcinoma on cytologic study.
duodenoscopy (EGD) revealed a large, ulcerated, submucosal, nodular mass in the cardia of the stomach extending to the gastroesophageal junction (Figure 2A). Biopsy of the mass again revealed poorly differentiated adenocarcinoma with scattered signet-ring cells undermining the gastric mucosa, favoring a gastric origin (Figure 2B).
THREE SUBTYPES OF GASTRIC CANCER
Worldwide, gastric cancer is the third most common type of cancer and the second most common cause of cancer-related deaths.1 In the United States, blacks and people of Asian ancestry have almost twice the risk of death, with the highest incidence and mortality rates.2,3
Most cases of gastric adenocarcinoma can be categorized as either intestinal or diffuse, but a new proximal subtype is emerging.4
Intestinal-type gastric adenocarcinoma is the most common subtype and accounts for almost all the ethnic and geographic variation in incidence.2 The lesions are often ulcerative and distal; the pathogenesis is stepwise and is initiated by chronic inflammation. Risk factors include old age, Helicobacter pylori infection, tobacco smoking, family history, and high salt intake, with an observed risk-reduction with the use of nonsteroidal anti-inflammatory drugs and with a high intake of fruits and vegetables.3
Diffuse gastric adenocarcinoma, on the other hand, has a uniform distribution worldwide, and its incidence is increasing. It typically carries a poor prognosis. Evidence thus far has shown its pathogenesis to be independent of chronic inflammation, but it has a strong tendency to be hereditary.3
Proximal gastric adenocarcinoma is observed in the gastric cardia and near the gastroesophageal junction. It is often grouped with the distal esophageal adenocarcinomas and has similar risk factors, including reflux disease, obesity, alcohol abuse, and tobacco smoking. Interestingly, however, H pylori infection does not contribute to the pathogenesis of this type, and it may even have a protective role.3
DIFFICULT TO DETECT EARLY
Gastric cancer is difficult to detect early enough in its course to be cured. Understanding its risk factors, recognizing its common symptoms, and regarding its uncommon symptoms with suspicion may lead to earlier diagnosis and more effective treatment.
Our patient’s proximal gastric cancer was diagnosed late even though he had several risk factors for it (he was Japanese, he was a smoker, and he had gastroesophageal reflux disease) because of a late and atypical presentation with misleading paraneoplastic symptoms.
Early diagnosis is difficult because most patients have no symptoms in the early stage; weight loss and abdominal pain are often late signs of tumor progression.
Screening may be justified in high-risk groups in the United States, although the issue is debatable. Diagnostic imaging is the only effective method for screening,5 with EGD considered the first-line targeted evaluation should there be suspicion of gastric cancer either from the clinical presentation or from barium swallow.6 Candidates for screening may include elderly patients with atrophic gastritis or pernicious anemia, immigrants from countries with high rates of gastric carcinoma, and people with a family history of gastrointestinal cancer.7
A 50-year-old male Japanese immigrant with a history of smoking and occasional untreated heartburn presented with the recent onset of flank pain, weight loss, headache, syncope, and blurred vision.
Previously healthy, he began feeling moderate pain in his left flank 1 month ago; it was diagnosed as kidney stones and was treated conservatively. Two weeks later he had an episode of syncope and soon after developed blurred vision, mainly in his left eye, along with severe bifrontal headache. An eye examination and magnetic resonance imaging of the brain indicated optic neuritis, for which he was given glucocorticoids intravenously for 3 days, with moderate improvement.
As his symptoms continued over the next 2 weeks, he lost 20 lb (9.1 kg) due to the pain, loss of appetite, nausea, and occasional vomiting.
Positron emission tomography showed the retroperitoneal infiltrative process and the thickened gastric cardia to be hypermetabolic (Figure 1C).
The area of retroperitoneal infiltration was biopsied under CT guidance, and pathologic study showed poorly differentiated carcinoma with signet-ring cells, a feature of gastric cancer.
The patient underwent lumbar puncture. His cerebrospinal fluid had 206 white blood cells/μL (reference range 0–5) and large numbers of poorly differentiated malignant cells, most consistent with adenocarcinoma on cytologic study.
duodenoscopy (EGD) revealed a large, ulcerated, submucosal, nodular mass in the cardia of the stomach extending to the gastroesophageal junction (Figure 2A). Biopsy of the mass again revealed poorly differentiated adenocarcinoma with scattered signet-ring cells undermining the gastric mucosa, favoring a gastric origin (Figure 2B).
THREE SUBTYPES OF GASTRIC CANCER
Worldwide, gastric cancer is the third most common type of cancer and the second most common cause of cancer-related deaths.1 In the United States, blacks and people of Asian ancestry have almost twice the risk of death, with the highest incidence and mortality rates.2,3
Most cases of gastric adenocarcinoma can be categorized as either intestinal or diffuse, but a new proximal subtype is emerging.4
Intestinal-type gastric adenocarcinoma is the most common subtype and accounts for almost all the ethnic and geographic variation in incidence.2 The lesions are often ulcerative and distal; the pathogenesis is stepwise and is initiated by chronic inflammation. Risk factors include old age, Helicobacter pylori infection, tobacco smoking, family history, and high salt intake, with an observed risk-reduction with the use of nonsteroidal anti-inflammatory drugs and with a high intake of fruits and vegetables.3
Diffuse gastric adenocarcinoma, on the other hand, has a uniform distribution worldwide, and its incidence is increasing. It typically carries a poor prognosis. Evidence thus far has shown its pathogenesis to be independent of chronic inflammation, but it has a strong tendency to be hereditary.3
Proximal gastric adenocarcinoma is observed in the gastric cardia and near the gastroesophageal junction. It is often grouped with the distal esophageal adenocarcinomas and has similar risk factors, including reflux disease, obesity, alcohol abuse, and tobacco smoking. Interestingly, however, H pylori infection does not contribute to the pathogenesis of this type, and it may even have a protective role.3
DIFFICULT TO DETECT EARLY
Gastric cancer is difficult to detect early enough in its course to be cured. Understanding its risk factors, recognizing its common symptoms, and regarding its uncommon symptoms with suspicion may lead to earlier diagnosis and more effective treatment.
Our patient’s proximal gastric cancer was diagnosed late even though he had several risk factors for it (he was Japanese, he was a smoker, and he had gastroesophageal reflux disease) because of a late and atypical presentation with misleading paraneoplastic symptoms.
Early diagnosis is difficult because most patients have no symptoms in the early stage; weight loss and abdominal pain are often late signs of tumor progression.
Screening may be justified in high-risk groups in the United States, although the issue is debatable. Diagnostic imaging is the only effective method for screening,5 with EGD considered the first-line targeted evaluation should there be suspicion of gastric cancer either from the clinical presentation or from barium swallow.6 Candidates for screening may include elderly patients with atrophic gastritis or pernicious anemia, immigrants from countries with high rates of gastric carcinoma, and people with a family history of gastrointestinal cancer.7
- Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55:74–108.
- Crew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol 2006; 12:354–362.
- Shah MA, Kelsen DP. Gastric cancer: a primer on the epidemiology and biology of the disease and an overview of the medical management of advanced disease. J Natl Compr Canc Netw 2010; 8:437–447.
- Fine G, Chan K. Alimentary tract. In:Kissane JM, editor. Anderson’s Pathology. 8th ed. Saint Louis, MO: Mosby; 1985:1055–1095.
- Kunisaki C, Ishino J, Nakajima S, et al. Outcomes of mass screening for gastric carcinoma. Ann Surg Oncol 2006; 13:221–228.
- Cappell MS, Friedel D. The role of esophagogastroduodenoscopy in the diagnosis and management of upper gastrointestinal disorders. Med Clin North Am 2002; 86:1165–1216.
- Hisamuchi S, Fukao P, Sugawara N, et al. Evaluation of mass screening programme for stomach cancer in Japan. In:Miller AB, Chamberlain J, Day NE, et al, editors. Cancer Screening. Cambridge, UK: Cambridge University Press; 1991:357–372.
- Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55:74–108.
- Crew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol 2006; 12:354–362.
- Shah MA, Kelsen DP. Gastric cancer: a primer on the epidemiology and biology of the disease and an overview of the medical management of advanced disease. J Natl Compr Canc Netw 2010; 8:437–447.
- Fine G, Chan K. Alimentary tract. In:Kissane JM, editor. Anderson’s Pathology. 8th ed. Saint Louis, MO: Mosby; 1985:1055–1095.
- Kunisaki C, Ishino J, Nakajima S, et al. Outcomes of mass screening for gastric carcinoma. Ann Surg Oncol 2006; 13:221–228.
- Cappell MS, Friedel D. The role of esophagogastroduodenoscopy in the diagnosis and management of upper gastrointestinal disorders. Med Clin North Am 2002; 86:1165–1216.
- Hisamuchi S, Fukao P, Sugawara N, et al. Evaluation of mass screening programme for stomach cancer in Japan. In:Miller AB, Chamberlain J, Day NE, et al, editors. Cancer Screening. Cambridge, UK: Cambridge University Press; 1991:357–372.
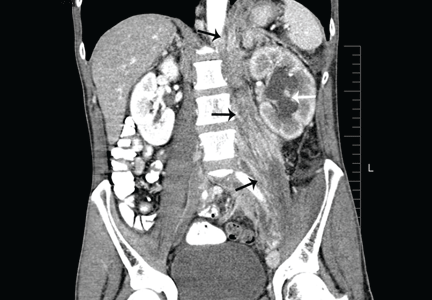
A 40-year-old woman with excoriated skin lesions
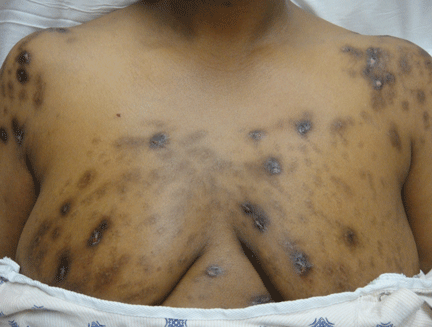
She denies any history of itching, insect bites, exposure to new medications or jewelry, allergies, recent change in medications, travel, or intravenous drug abuse.
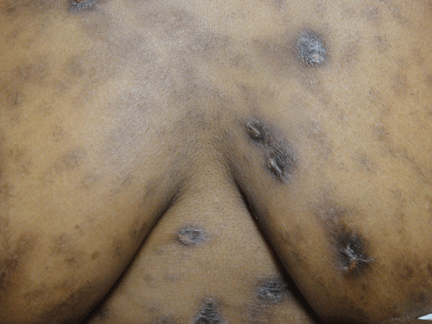
Q: Which is the most likely diagnosis?
- Allergic contact dermatitis
- Xerosis
- Dermatotillomania
- Folliculitis
- Infestation (scabies)
A: Dermatotillomania, ie, pathologic skin picking, is the correct diagnosis. On further questioning, the patient reveals that the wounds have been self-inflicted over many years, starting in her adolescence. The wounds are located only in areas she can reach. She admits that social and emotional stressors had made the condition significantly worse and that lately she had lost control of her skin-picking. She denies nail-biting, trichotillomania, or obsessive-compulsive behavior.
As for the other possible diagnoses:
Allergic contact dermatitis occurs when contact with a particular substance elicits a hypersensitivity reaction. This reaction is of the delayed type (type IV). The affected individual can develop skin erythema and swelling with vesicles that are intensely pruritic at the contact site. The erythema may become evident hours after exposure, or not until weeks later, which can make the diagnosis difficult at times.
Our patient’s lesions were not pruritic, and she denied recent exposure to allergens.
Xerosis. Xerotic (dry) skin is usually rough, with fine scales and fissures. Xerosis can affect people of all ages and is often more intense during the winter. It affects mainly the arms, legs, and hands. Patients note pruritus, which can be treated with liberal use of emollients and tepid water baths.
Our patient’s lesions were scarred, hyperpigmented, and nonpruritic.
Folliculitis is a superficial infection of the hair follicle that presents as an erythematous pustule on the extremities, buttocks, or scalp. The pustule can be tender to palpation and can progress to an abscess that requires incision and drainage and intravenous antibiotics. A moist environment and poor hygiene are predisposing factors. Staphylococcus aureus is the culprit in most cases.
Our patient’s lesions were on the chest and upper back, where hair follicles were sparse or absent, and there was no erythema or tenderness.
Scabies is a skin infestation with Sarcoptes scabiei mites, which burrow in the skin and cause intense pruritus, especially at night. Scabies usually affects the sides and webs of the fingers and skin folds. Sexual contact is a common way of transmission; however, transmission can also occur by sharing beds and towels.
Patients with dermatotillomania lack intense pruritus, and skin-picking occurs during the day, while the patient is awake.
SELF-INFLICTED WOUNDS
Pathologic skin-picking, neurotic excoriation, excoriated acne, or dermatotillomania results from scratching, picking, gouging, or squeezing of one’s skin via teeth, fingernails, tweezers, or other objects.1–3 Lesions are usually found on skin that the patient can easily reach, such as the face, chest, upper and lower extremities, and upper back.4
The prevalence of pathologic skin-picking is estimated at 2% in dermatology patients.5 The overall prevalence of psychiatric disorders in all dermatology outpatients is estimated at 30% to 40%. Women outnumber men with this disorder.6
Dermatotillomania is thought to be on the spectrum of obsessive-compulsive disorder, in which patients exhibit impulses and compulsions.5 It starts in childhood or early adulthood, with an average lifetime duration of 21 years.7 It is usually associated with anxiety, depression, obsessive-compulsive traits, eating disorders, body dysmorphic disorders, or hypochondriasis. Psychosocial stress is the main trigger. Patients report feelings of tension and stress before picking and relief while picking; there is no suicidal ideation.8
Treatments are both pharmacologic and behavioral.9 Cognitive behavioral therapy and habit reversal therapy have each been successful when used alone.8 In addition, several case reports10 and double-blind studies11,12 have shown that treatment with a selective serotonin-reuptake inhibitor (SSRI) can reduce pathologic skin-picking.13,14 However, SSRIs have also been reported to induce or aggravate this behavior in patients with underlying mild skin-picking and a family history of skin-picking.15 Thus, it is pertinent to extract a detailed history from the patient before prescribing an SSRI.
We referred our patient for behavioral therapy and prescribed fluoxetine (Prozac) 20 mg daily. She showed improvement in symptoms in 4 weeks and has since stopped skin-picking completely.
- Arnold LM. Phenomenology and therapeutic options for dermatotillomania. Expert Rev Neurother 2002; 2:725–730.
- Bohne A, Keuthen N, Wilhelm S. Pathologic hairpulling, skin picking, and nail biting. Ann Clin Psychiatry 2005; 17:227–232.
- Gattu S, Rashid RM, Khachemoune A. Self-induced skin lesions: a review of dermatitis artefacta. Cutis 2009; 84:247–251.
- Keuthen NJ, Deckersbach T, Wilhelm S, et al. Repetitive skin-picking in a student population and comparison with a sample of self-injurious skin-pickers. Psychosomatics 2000; 41:210–215.
- Arnold LM, Auchenbach MB, McElroy SL. Psychogenic excoriation. Clinical features, proposed diagnostic criteria, epidemiology and approaches to treatment. CNS Drugs 2001; 15:351–359.
- Wilhelm S, Keuthen NJ, Deckersbach T, et al. Self-injurious skin picking: clinical characteristics and comorbidity. J Clin Psychiatry 1999; 60:454–459.
- Gupta MA, Gupta AK, Haberman HF. Neurotic excoriations: a review and some new perspectives. Compr Psychiatry 1986; 27:381–386.
- Rosenbaum MS, Ayllon T. The behavioral treatment of neurodermatitis through habit-reversal. Behav Res Ther 1981; 19:313–318.
- Deckersbach T, Wilhelm S, Keuthen N. Self-injurious skin picking: clinical characteristics, assessment methods, and treatment modalities. Brief Treatment and Crisis Intervention 2003; 3:249–260.
- Sharma H. Psychogenic excoriation responding to fluoxetine: a case report. J Indian Med Assoc 2008; 106:245,262.
- Bloch MR, Elliott M, Thompson H, Koran LM. Fluoxetine in pathologic skin-picking: open-label and double-blind results. Psychosomatics 2001; 42:314–319.
- Simeon D, Stein DJ, Gross S, Islam N, Schmeidler J, Hollander E. A double-blind trial of fluoxetine in pathologic skin picking. J Clin Psychiatry 1997; 58:341–347.
- Gupta MA, Gupta AK. The use of antidepressant drugs in dermatology. J Eur Acad Dermatol Venereol 2001; 15:512–518.
- Keuthen NJ, Jameson M, Loh R, Deckersbach T, Wilhelm S, Dougherty DD. Open-label escitalopram treatment for pathological skin picking. Int Clin Psychopharmacol 2007; 22:268–274.
- Denys D, van Megen HJ, Westenberg HG. Emerging skin-picking behaviour after serotonin reuptake inhibitor-treatment in patients with obsessive-compulsive disorder: possible mechanisms and implications for clinical care. J Psychopharmacol 2003; 17:127–129.
She denies any history of itching, insect bites, exposure to new medications or jewelry, allergies, recent change in medications, travel, or intravenous drug abuse.
Q: Which is the most likely diagnosis?
- Allergic contact dermatitis
- Xerosis
- Dermatotillomania
- Folliculitis
- Infestation (scabies)
A: Dermatotillomania, ie, pathologic skin picking, is the correct diagnosis. On further questioning, the patient reveals that the wounds have been self-inflicted over many years, starting in her adolescence. The wounds are located only in areas she can reach. She admits that social and emotional stressors had made the condition significantly worse and that lately she had lost control of her skin-picking. She denies nail-biting, trichotillomania, or obsessive-compulsive behavior.
As for the other possible diagnoses:
Allergic contact dermatitis occurs when contact with a particular substance elicits a hypersensitivity reaction. This reaction is of the delayed type (type IV). The affected individual can develop skin erythema and swelling with vesicles that are intensely pruritic at the contact site. The erythema may become evident hours after exposure, or not until weeks later, which can make the diagnosis difficult at times.
Our patient’s lesions were not pruritic, and she denied recent exposure to allergens.
Xerosis. Xerotic (dry) skin is usually rough, with fine scales and fissures. Xerosis can affect people of all ages and is often more intense during the winter. It affects mainly the arms, legs, and hands. Patients note pruritus, which can be treated with liberal use of emollients and tepid water baths.
Our patient’s lesions were scarred, hyperpigmented, and nonpruritic.
Folliculitis is a superficial infection of the hair follicle that presents as an erythematous pustule on the extremities, buttocks, or scalp. The pustule can be tender to palpation and can progress to an abscess that requires incision and drainage and intravenous antibiotics. A moist environment and poor hygiene are predisposing factors. Staphylococcus aureus is the culprit in most cases.
Our patient’s lesions were on the chest and upper back, where hair follicles were sparse or absent, and there was no erythema or tenderness.
Scabies is a skin infestation with Sarcoptes scabiei mites, which burrow in the skin and cause intense pruritus, especially at night. Scabies usually affects the sides and webs of the fingers and skin folds. Sexual contact is a common way of transmission; however, transmission can also occur by sharing beds and towels.
Patients with dermatotillomania lack intense pruritus, and skin-picking occurs during the day, while the patient is awake.
SELF-INFLICTED WOUNDS
Pathologic skin-picking, neurotic excoriation, excoriated acne, or dermatotillomania results from scratching, picking, gouging, or squeezing of one’s skin via teeth, fingernails, tweezers, or other objects.1–3 Lesions are usually found on skin that the patient can easily reach, such as the face, chest, upper and lower extremities, and upper back.4
The prevalence of pathologic skin-picking is estimated at 2% in dermatology patients.5 The overall prevalence of psychiatric disorders in all dermatology outpatients is estimated at 30% to 40%. Women outnumber men with this disorder.6
Dermatotillomania is thought to be on the spectrum of obsessive-compulsive disorder, in which patients exhibit impulses and compulsions.5 It starts in childhood or early adulthood, with an average lifetime duration of 21 years.7 It is usually associated with anxiety, depression, obsessive-compulsive traits, eating disorders, body dysmorphic disorders, or hypochondriasis. Psychosocial stress is the main trigger. Patients report feelings of tension and stress before picking and relief while picking; there is no suicidal ideation.8
Treatments are both pharmacologic and behavioral.9 Cognitive behavioral therapy and habit reversal therapy have each been successful when used alone.8 In addition, several case reports10 and double-blind studies11,12 have shown that treatment with a selective serotonin-reuptake inhibitor (SSRI) can reduce pathologic skin-picking.13,14 However, SSRIs have also been reported to induce or aggravate this behavior in patients with underlying mild skin-picking and a family history of skin-picking.15 Thus, it is pertinent to extract a detailed history from the patient before prescribing an SSRI.
We referred our patient for behavioral therapy and prescribed fluoxetine (Prozac) 20 mg daily. She showed improvement in symptoms in 4 weeks and has since stopped skin-picking completely.
She denies any history of itching, insect bites, exposure to new medications or jewelry, allergies, recent change in medications, travel, or intravenous drug abuse.
Q: Which is the most likely diagnosis?
- Allergic contact dermatitis
- Xerosis
- Dermatotillomania
- Folliculitis
- Infestation (scabies)
A: Dermatotillomania, ie, pathologic skin picking, is the correct diagnosis. On further questioning, the patient reveals that the wounds have been self-inflicted over many years, starting in her adolescence. The wounds are located only in areas she can reach. She admits that social and emotional stressors had made the condition significantly worse and that lately she had lost control of her skin-picking. She denies nail-biting, trichotillomania, or obsessive-compulsive behavior.
As for the other possible diagnoses:
Allergic contact dermatitis occurs when contact with a particular substance elicits a hypersensitivity reaction. This reaction is of the delayed type (type IV). The affected individual can develop skin erythema and swelling with vesicles that are intensely pruritic at the contact site. The erythema may become evident hours after exposure, or not until weeks later, which can make the diagnosis difficult at times.
Our patient’s lesions were not pruritic, and she denied recent exposure to allergens.
Xerosis. Xerotic (dry) skin is usually rough, with fine scales and fissures. Xerosis can affect people of all ages and is often more intense during the winter. It affects mainly the arms, legs, and hands. Patients note pruritus, which can be treated with liberal use of emollients and tepid water baths.
Our patient’s lesions were scarred, hyperpigmented, and nonpruritic.
Folliculitis is a superficial infection of the hair follicle that presents as an erythematous pustule on the extremities, buttocks, or scalp. The pustule can be tender to palpation and can progress to an abscess that requires incision and drainage and intravenous antibiotics. A moist environment and poor hygiene are predisposing factors. Staphylococcus aureus is the culprit in most cases.
Our patient’s lesions were on the chest and upper back, where hair follicles were sparse or absent, and there was no erythema or tenderness.
Scabies is a skin infestation with Sarcoptes scabiei mites, which burrow in the skin and cause intense pruritus, especially at night. Scabies usually affects the sides and webs of the fingers and skin folds. Sexual contact is a common way of transmission; however, transmission can also occur by sharing beds and towels.
Patients with dermatotillomania lack intense pruritus, and skin-picking occurs during the day, while the patient is awake.
SELF-INFLICTED WOUNDS
Pathologic skin-picking, neurotic excoriation, excoriated acne, or dermatotillomania results from scratching, picking, gouging, or squeezing of one’s skin via teeth, fingernails, tweezers, or other objects.1–3 Lesions are usually found on skin that the patient can easily reach, such as the face, chest, upper and lower extremities, and upper back.4
The prevalence of pathologic skin-picking is estimated at 2% in dermatology patients.5 The overall prevalence of psychiatric disorders in all dermatology outpatients is estimated at 30% to 40%. Women outnumber men with this disorder.6
Dermatotillomania is thought to be on the spectrum of obsessive-compulsive disorder, in which patients exhibit impulses and compulsions.5 It starts in childhood or early adulthood, with an average lifetime duration of 21 years.7 It is usually associated with anxiety, depression, obsessive-compulsive traits, eating disorders, body dysmorphic disorders, or hypochondriasis. Psychosocial stress is the main trigger. Patients report feelings of tension and stress before picking and relief while picking; there is no suicidal ideation.8
Treatments are both pharmacologic and behavioral.9 Cognitive behavioral therapy and habit reversal therapy have each been successful when used alone.8 In addition, several case reports10 and double-blind studies11,12 have shown that treatment with a selective serotonin-reuptake inhibitor (SSRI) can reduce pathologic skin-picking.13,14 However, SSRIs have also been reported to induce or aggravate this behavior in patients with underlying mild skin-picking and a family history of skin-picking.15 Thus, it is pertinent to extract a detailed history from the patient before prescribing an SSRI.
We referred our patient for behavioral therapy and prescribed fluoxetine (Prozac) 20 mg daily. She showed improvement in symptoms in 4 weeks and has since stopped skin-picking completely.
- Arnold LM. Phenomenology and therapeutic options for dermatotillomania. Expert Rev Neurother 2002; 2:725–730.
- Bohne A, Keuthen N, Wilhelm S. Pathologic hairpulling, skin picking, and nail biting. Ann Clin Psychiatry 2005; 17:227–232.
- Gattu S, Rashid RM, Khachemoune A. Self-induced skin lesions: a review of dermatitis artefacta. Cutis 2009; 84:247–251.
- Keuthen NJ, Deckersbach T, Wilhelm S, et al. Repetitive skin-picking in a student population and comparison with a sample of self-injurious skin-pickers. Psychosomatics 2000; 41:210–215.
- Arnold LM, Auchenbach MB, McElroy SL. Psychogenic excoriation. Clinical features, proposed diagnostic criteria, epidemiology and approaches to treatment. CNS Drugs 2001; 15:351–359.
- Wilhelm S, Keuthen NJ, Deckersbach T, et al. Self-injurious skin picking: clinical characteristics and comorbidity. J Clin Psychiatry 1999; 60:454–459.
- Gupta MA, Gupta AK, Haberman HF. Neurotic excoriations: a review and some new perspectives. Compr Psychiatry 1986; 27:381–386.
- Rosenbaum MS, Ayllon T. The behavioral treatment of neurodermatitis through habit-reversal. Behav Res Ther 1981; 19:313–318.
- Deckersbach T, Wilhelm S, Keuthen N. Self-injurious skin picking: clinical characteristics, assessment methods, and treatment modalities. Brief Treatment and Crisis Intervention 2003; 3:249–260.
- Sharma H. Psychogenic excoriation responding to fluoxetine: a case report. J Indian Med Assoc 2008; 106:245,262.
- Bloch MR, Elliott M, Thompson H, Koran LM. Fluoxetine in pathologic skin-picking: open-label and double-blind results. Psychosomatics 2001; 42:314–319.
- Simeon D, Stein DJ, Gross S, Islam N, Schmeidler J, Hollander E. A double-blind trial of fluoxetine in pathologic skin picking. J Clin Psychiatry 1997; 58:341–347.
- Gupta MA, Gupta AK. The use of antidepressant drugs in dermatology. J Eur Acad Dermatol Venereol 2001; 15:512–518.
- Keuthen NJ, Jameson M, Loh R, Deckersbach T, Wilhelm S, Dougherty DD. Open-label escitalopram treatment for pathological skin picking. Int Clin Psychopharmacol 2007; 22:268–274.
- Denys D, van Megen HJ, Westenberg HG. Emerging skin-picking behaviour after serotonin reuptake inhibitor-treatment in patients with obsessive-compulsive disorder: possible mechanisms and implications for clinical care. J Psychopharmacol 2003; 17:127–129.
- Arnold LM. Phenomenology and therapeutic options for dermatotillomania. Expert Rev Neurother 2002; 2:725–730.
- Bohne A, Keuthen N, Wilhelm S. Pathologic hairpulling, skin picking, and nail biting. Ann Clin Psychiatry 2005; 17:227–232.
- Gattu S, Rashid RM, Khachemoune A. Self-induced skin lesions: a review of dermatitis artefacta. Cutis 2009; 84:247–251.
- Keuthen NJ, Deckersbach T, Wilhelm S, et al. Repetitive skin-picking in a student population and comparison with a sample of self-injurious skin-pickers. Psychosomatics 2000; 41:210–215.
- Arnold LM, Auchenbach MB, McElroy SL. Psychogenic excoriation. Clinical features, proposed diagnostic criteria, epidemiology and approaches to treatment. CNS Drugs 2001; 15:351–359.
- Wilhelm S, Keuthen NJ, Deckersbach T, et al. Self-injurious skin picking: clinical characteristics and comorbidity. J Clin Psychiatry 1999; 60:454–459.
- Gupta MA, Gupta AK, Haberman HF. Neurotic excoriations: a review and some new perspectives. Compr Psychiatry 1986; 27:381–386.
- Rosenbaum MS, Ayllon T. The behavioral treatment of neurodermatitis through habit-reversal. Behav Res Ther 1981; 19:313–318.
- Deckersbach T, Wilhelm S, Keuthen N. Self-injurious skin picking: clinical characteristics, assessment methods, and treatment modalities. Brief Treatment and Crisis Intervention 2003; 3:249–260.
- Sharma H. Psychogenic excoriation responding to fluoxetine: a case report. J Indian Med Assoc 2008; 106:245,262.
- Bloch MR, Elliott M, Thompson H, Koran LM. Fluoxetine in pathologic skin-picking: open-label and double-blind results. Psychosomatics 2001; 42:314–319.
- Simeon D, Stein DJ, Gross S, Islam N, Schmeidler J, Hollander E. A double-blind trial of fluoxetine in pathologic skin picking. J Clin Psychiatry 1997; 58:341–347.
- Gupta MA, Gupta AK. The use of antidepressant drugs in dermatology. J Eur Acad Dermatol Venereol 2001; 15:512–518.
- Keuthen NJ, Jameson M, Loh R, Deckersbach T, Wilhelm S, Dougherty DD. Open-label escitalopram treatment for pathological skin picking. Int Clin Psychopharmacol 2007; 22:268–274.
- Denys D, van Megen HJ, Westenberg HG. Emerging skin-picking behaviour after serotonin reuptake inhibitor-treatment in patients with obsessive-compulsive disorder: possible mechanisms and implications for clinical care. J Psychopharmacol 2003; 17:127–129.