User login
In reply: The role of sentinel lymph node biopsy after excision of melanomas
In Reply: Thank you for your important question. Examination of the excision specimen of the patient’s primary cutaneous melanoma lesion demonstrated a Breslow depth of 1.92 mm. He did indeed undergo sentinel lymph node biopsy at the time of excision. Histologic examination of the biopsy specimen was negative for evidence of metastatic melanoma. Despite this, he obviously developed metastatic disease several years later.
As you allude to, sentinel lymph node biopsy is an important minimally invasive procedure in patients with melanoma. Morton et al1 compared it with nodal observation and found that in patients with at least intermediate-thickness cutaneous melanoma, sentinel node biopsy significantly prolonged disease-free survival for all patients and improved melanoma-specific survival rates for patients with nodal metastases from intermediate-thickness melanomas (1.2–3.5 mm).1 However, it remains an imperfect procedure, and a percentage of patients develop recurrence or metastasis despite a negative biopsy. In a recent study by Jones et al,2 16% of melanoma patients in a cohort with a negative sentinel node biopsy developed recurrence.2 In these unfortunate patients, medications such as CTLA-4 inhibitors and PD-1 inhibitors now offer hope for prolonged survival.
- Morton DL, Thompson JF, Cochran AJ, et al, for the MSLT Group. Final trial report of sentinel-node biopsy versus nodal observation in melanoma. N Engl J Med 2014; 370:599–609.
- Jones EL, Jones TS, Nathan Pearlman NW, et al. Long-term follow-up and survival of patients following a recurrence of melanoma after a negative sentinel lymph node biopsy result. JAMA Surg 2013; 148:456–461.
In Reply: Thank you for your important question. Examination of the excision specimen of the patient’s primary cutaneous melanoma lesion demonstrated a Breslow depth of 1.92 mm. He did indeed undergo sentinel lymph node biopsy at the time of excision. Histologic examination of the biopsy specimen was negative for evidence of metastatic melanoma. Despite this, he obviously developed metastatic disease several years later.
As you allude to, sentinel lymph node biopsy is an important minimally invasive procedure in patients with melanoma. Morton et al1 compared it with nodal observation and found that in patients with at least intermediate-thickness cutaneous melanoma, sentinel node biopsy significantly prolonged disease-free survival for all patients and improved melanoma-specific survival rates for patients with nodal metastases from intermediate-thickness melanomas (1.2–3.5 mm).1 However, it remains an imperfect procedure, and a percentage of patients develop recurrence or metastasis despite a negative biopsy. In a recent study by Jones et al,2 16% of melanoma patients in a cohort with a negative sentinel node biopsy developed recurrence.2 In these unfortunate patients, medications such as CTLA-4 inhibitors and PD-1 inhibitors now offer hope for prolonged survival.
In Reply: Thank you for your important question. Examination of the excision specimen of the patient’s primary cutaneous melanoma lesion demonstrated a Breslow depth of 1.92 mm. He did indeed undergo sentinel lymph node biopsy at the time of excision. Histologic examination of the biopsy specimen was negative for evidence of metastatic melanoma. Despite this, he obviously developed metastatic disease several years later.
As you allude to, sentinel lymph node biopsy is an important minimally invasive procedure in patients with melanoma. Morton et al1 compared it with nodal observation and found that in patients with at least intermediate-thickness cutaneous melanoma, sentinel node biopsy significantly prolonged disease-free survival for all patients and improved melanoma-specific survival rates for patients with nodal metastases from intermediate-thickness melanomas (1.2–3.5 mm).1 However, it remains an imperfect procedure, and a percentage of patients develop recurrence or metastasis despite a negative biopsy. In a recent study by Jones et al,2 16% of melanoma patients in a cohort with a negative sentinel node biopsy developed recurrence.2 In these unfortunate patients, medications such as CTLA-4 inhibitors and PD-1 inhibitors now offer hope for prolonged survival.
- Morton DL, Thompson JF, Cochran AJ, et al, for the MSLT Group. Final trial report of sentinel-node biopsy versus nodal observation in melanoma. N Engl J Med 2014; 370:599–609.
- Jones EL, Jones TS, Nathan Pearlman NW, et al. Long-term follow-up and survival of patients following a recurrence of melanoma after a negative sentinel lymph node biopsy result. JAMA Surg 2013; 148:456–461.
- Morton DL, Thompson JF, Cochran AJ, et al, for the MSLT Group. Final trial report of sentinel-node biopsy versus nodal observation in melanoma. N Engl J Med 2014; 370:599–609.
- Jones EL, Jones TS, Nathan Pearlman NW, et al. Long-term follow-up and survival of patients following a recurrence of melanoma after a negative sentinel lymph node biopsy result. JAMA Surg 2013; 148:456–461.
Parvovirus mimicking acute HIV infection
A 25-year-old Jamaican man presented to the emergency department for evaluation of a rash on his face, back, and hands (Figure 1). He recalled a puncture injury after handling garbage at work. He denied recent travel, blood transfusions, or sick contacts. He was not aware of any recent arthropod bites. All standard vaccines including a tetanus booster were up to date.
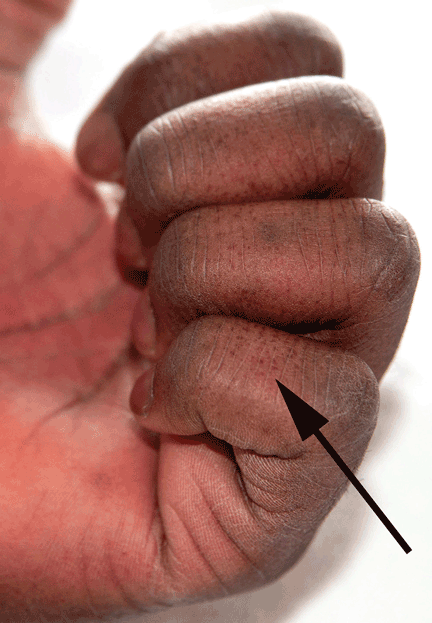
Examination revealed edema of the hands and uvula. He was discharged with diphenhydramine and a short course of a systemic corticosteroid for presumed contact dermatitis.
He returned 4 days later with new symptoms, including sore throat, fever with a temperature of 103°F (39.4°C), oropharyngeal pain, dysphagia, dysuria, and purpura on the hands, abdomen, and legs. He was admitted to the hospital.

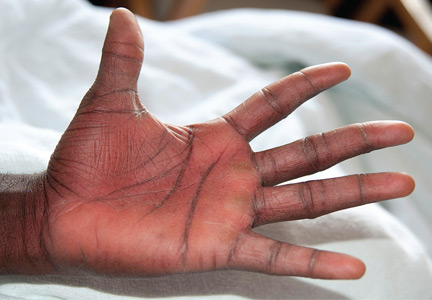
Examination revealed bright red confluent erythema of both legs extending to the lower abdomen, with petechiae on the palms (Figure 2), soles, toes, and fingers. Several small scrotal ulcers with well-defined borders were noted. Oral examination revealed white-yellow adherent plaques on the tongue and similar small ulcers on the lower lip and soft and hard palates.
A complete blood cell count revealed absolute lymphopenia, with a white blood cell count of 0.64 × 109/L (reference range 1.0–4.8) and a neutrophil percentage of 78.9% (39%–68%). Other values were within normal limits, with a red blood cell count of 5.3 × 1012/L (3.9–5.5) and a platelet count of 161 × 109/L (150–350). Serum liver enzymes were also within normal limits.
Treatment with intravenous fluids and intramuscular penicillin G was started empirically, pending a workup for infectious disease. Tests for syphilis immunoglobulin G (IgG), streptococci, anti-streptolysin O, Epstein-Barr virus, and human immunodeficiency virus (HIV) 1 and 2 were negative. The scrotal ulcers were swabbed, and culture and direct fluorescent antibody testing for cytomegalovirus and herpes simplex virus were negative. Urine testing for gonococcal and chlamydial infection was negative.
On the fifth day of hospitalization, the patient’s condition was improving, but there was still no definitive diagnosis. Consultation with the inpatient dermatology team prompted testing for parvovirus B19 infection, based on the gloves-and-socks distribution of the purpura. Testing revealed a slightly elevated parvovirus B19 IgG titer (2.61) and a significantly elevated parvovirus B19 IgM titer (12.74), which confirmed acute parvovirus infection.
The patient’s condition improved over several days with fluid administration, and he was discharged in good condition. He returned 1 week later for a follow-up appointment, at which time only superficial desquamation was noted in the areas previously affected by purpura.
PARVOVIRUS B19: NOT ONLY IN CHILDREN
Parvovirus B19 is responsible for the common childhood viral exanthem known as fifth disease.1 However, although much less common, the virus can also affect young adults, precipitating a dermatosis referred to as gloves-and-socks syndrome characterized by purpura on the hands and feet,2,3 and with a higher incidence in the spring and summer.1
Although papular-purpuric gloves-and- socks syndrome is characterized by purpura on the hands and feet, the cheeks, oral mucosa, inner thighs, buttocks, and genitalia are affected in about 50% of patients.2 In one report, in two-thirds of adult patients the presentation was caused by parvovirus B19 infection,4 but the syndrome has also been associated with Epstein-Barr virus, cytomegalovirus, human herpesvirus types 6 and 7, hepatitis B virus, rubella virus, and varicella zoster virus.4
Parvovirus B19 infection is commonly associated with systemic manifestations such as fever, fatigue, and lymphadenopathy, as well as swelling of the lips, cutaneous and mucosal ulcerations, polyarthritis, and petechiae involving the hard palate, the soft palate, or both.1
The syndrome is self-limited and resolves within 1 to 2 weeks.1
THE DIAGNOSTIC CHALLENGE
The differential diagnosis of the syndrome’s gloves-and-socks presentation includes hand-foot-mouth disease, erythema multiforme, Henoch-Schönlein purpura, and Kawasaki disease,4 in addition to viral exanthems and sexually transmitted diseases. Our patient’s fever, rash, and absolute lymphopenia focused attention on possible HIV infection, which caused the patient significant anxiety while awaiting the results of HIV testing. Heightened awareness of the cutaneous presentation of parvovirus B19 infection can help avoid unnecessary hospitalization and patient anxiety.
- Smith PT, Landry ML, Carey H, Krasnoff J, Cooney E. Papular-purpuric gloves and socks syndrome associated with acute parvovirus B19 infection: case report and review. Clin Infect Dis 1998; 27:164–168.
- Harms M, Feldmann R, Saurat JH. Papular-purpuric “gloves and socks” syndrome. J Am Acad Dermatol 1990; 23:850–854.
- Bagot M, Revuz J. Papular-purpuric “gloves and socks” syndrome: primary infection with parvovirus B19? J Am Acad Dermatol 1991; 25:341–342.
- Gutermuth J, Nadas K, Zirbs M, et al. Papular-purpuric gloves and socks syndrome. Lancet 2011; 378:198.
A 25-year-old Jamaican man presented to the emergency department for evaluation of a rash on his face, back, and hands (Figure 1). He recalled a puncture injury after handling garbage at work. He denied recent travel, blood transfusions, or sick contacts. He was not aware of any recent arthropod bites. All standard vaccines including a tetanus booster were up to date.
Examination revealed edema of the hands and uvula. He was discharged with diphenhydramine and a short course of a systemic corticosteroid for presumed contact dermatitis.
He returned 4 days later with new symptoms, including sore throat, fever with a temperature of 103°F (39.4°C), oropharyngeal pain, dysphagia, dysuria, and purpura on the hands, abdomen, and legs. He was admitted to the hospital.
Examination revealed bright red confluent erythema of both legs extending to the lower abdomen, with petechiae on the palms (Figure 2), soles, toes, and fingers. Several small scrotal ulcers with well-defined borders were noted. Oral examination revealed white-yellow adherent plaques on the tongue and similar small ulcers on the lower lip and soft and hard palates.
A complete blood cell count revealed absolute lymphopenia, with a white blood cell count of 0.64 × 109/L (reference range 1.0–4.8) and a neutrophil percentage of 78.9% (39%–68%). Other values were within normal limits, with a red blood cell count of 5.3 × 1012/L (3.9–5.5) and a platelet count of 161 × 109/L (150–350). Serum liver enzymes were also within normal limits.
Treatment with intravenous fluids and intramuscular penicillin G was started empirically, pending a workup for infectious disease. Tests for syphilis immunoglobulin G (IgG), streptococci, anti-streptolysin O, Epstein-Barr virus, and human immunodeficiency virus (HIV) 1 and 2 were negative. The scrotal ulcers were swabbed, and culture and direct fluorescent antibody testing for cytomegalovirus and herpes simplex virus were negative. Urine testing for gonococcal and chlamydial infection was negative.
On the fifth day of hospitalization, the patient’s condition was improving, but there was still no definitive diagnosis. Consultation with the inpatient dermatology team prompted testing for parvovirus B19 infection, based on the gloves-and-socks distribution of the purpura. Testing revealed a slightly elevated parvovirus B19 IgG titer (2.61) and a significantly elevated parvovirus B19 IgM titer (12.74), which confirmed acute parvovirus infection.
The patient’s condition improved over several days with fluid administration, and he was discharged in good condition. He returned 1 week later for a follow-up appointment, at which time only superficial desquamation was noted in the areas previously affected by purpura.
PARVOVIRUS B19: NOT ONLY IN CHILDREN
Parvovirus B19 is responsible for the common childhood viral exanthem known as fifth disease.1 However, although much less common, the virus can also affect young adults, precipitating a dermatosis referred to as gloves-and-socks syndrome characterized by purpura on the hands and feet,2,3 and with a higher incidence in the spring and summer.1
Although papular-purpuric gloves-and- socks syndrome is characterized by purpura on the hands and feet, the cheeks, oral mucosa, inner thighs, buttocks, and genitalia are affected in about 50% of patients.2 In one report, in two-thirds of adult patients the presentation was caused by parvovirus B19 infection,4 but the syndrome has also been associated with Epstein-Barr virus, cytomegalovirus, human herpesvirus types 6 and 7, hepatitis B virus, rubella virus, and varicella zoster virus.4
Parvovirus B19 infection is commonly associated with systemic manifestations such as fever, fatigue, and lymphadenopathy, as well as swelling of the lips, cutaneous and mucosal ulcerations, polyarthritis, and petechiae involving the hard palate, the soft palate, or both.1
The syndrome is self-limited and resolves within 1 to 2 weeks.1
THE DIAGNOSTIC CHALLENGE
The differential diagnosis of the syndrome’s gloves-and-socks presentation includes hand-foot-mouth disease, erythema multiforme, Henoch-Schönlein purpura, and Kawasaki disease,4 in addition to viral exanthems and sexually transmitted diseases. Our patient’s fever, rash, and absolute lymphopenia focused attention on possible HIV infection, which caused the patient significant anxiety while awaiting the results of HIV testing. Heightened awareness of the cutaneous presentation of parvovirus B19 infection can help avoid unnecessary hospitalization and patient anxiety.
A 25-year-old Jamaican man presented to the emergency department for evaluation of a rash on his face, back, and hands (Figure 1). He recalled a puncture injury after handling garbage at work. He denied recent travel, blood transfusions, or sick contacts. He was not aware of any recent arthropod bites. All standard vaccines including a tetanus booster were up to date.
Examination revealed edema of the hands and uvula. He was discharged with diphenhydramine and a short course of a systemic corticosteroid for presumed contact dermatitis.
He returned 4 days later with new symptoms, including sore throat, fever with a temperature of 103°F (39.4°C), oropharyngeal pain, dysphagia, dysuria, and purpura on the hands, abdomen, and legs. He was admitted to the hospital.
Examination revealed bright red confluent erythema of both legs extending to the lower abdomen, with petechiae on the palms (Figure 2), soles, toes, and fingers. Several small scrotal ulcers with well-defined borders were noted. Oral examination revealed white-yellow adherent plaques on the tongue and similar small ulcers on the lower lip and soft and hard palates.
A complete blood cell count revealed absolute lymphopenia, with a white blood cell count of 0.64 × 109/L (reference range 1.0–4.8) and a neutrophil percentage of 78.9% (39%–68%). Other values were within normal limits, with a red blood cell count of 5.3 × 1012/L (3.9–5.5) and a platelet count of 161 × 109/L (150–350). Serum liver enzymes were also within normal limits.
Treatment with intravenous fluids and intramuscular penicillin G was started empirically, pending a workup for infectious disease. Tests for syphilis immunoglobulin G (IgG), streptococci, anti-streptolysin O, Epstein-Barr virus, and human immunodeficiency virus (HIV) 1 and 2 were negative. The scrotal ulcers were swabbed, and culture and direct fluorescent antibody testing for cytomegalovirus and herpes simplex virus were negative. Urine testing for gonococcal and chlamydial infection was negative.
On the fifth day of hospitalization, the patient’s condition was improving, but there was still no definitive diagnosis. Consultation with the inpatient dermatology team prompted testing for parvovirus B19 infection, based on the gloves-and-socks distribution of the purpura. Testing revealed a slightly elevated parvovirus B19 IgG titer (2.61) and a significantly elevated parvovirus B19 IgM titer (12.74), which confirmed acute parvovirus infection.
The patient’s condition improved over several days with fluid administration, and he was discharged in good condition. He returned 1 week later for a follow-up appointment, at which time only superficial desquamation was noted in the areas previously affected by purpura.
PARVOVIRUS B19: NOT ONLY IN CHILDREN
Parvovirus B19 is responsible for the common childhood viral exanthem known as fifth disease.1 However, although much less common, the virus can also affect young adults, precipitating a dermatosis referred to as gloves-and-socks syndrome characterized by purpura on the hands and feet,2,3 and with a higher incidence in the spring and summer.1
Although papular-purpuric gloves-and- socks syndrome is characterized by purpura on the hands and feet, the cheeks, oral mucosa, inner thighs, buttocks, and genitalia are affected in about 50% of patients.2 In one report, in two-thirds of adult patients the presentation was caused by parvovirus B19 infection,4 but the syndrome has also been associated with Epstein-Barr virus, cytomegalovirus, human herpesvirus types 6 and 7, hepatitis B virus, rubella virus, and varicella zoster virus.4
Parvovirus B19 infection is commonly associated with systemic manifestations such as fever, fatigue, and lymphadenopathy, as well as swelling of the lips, cutaneous and mucosal ulcerations, polyarthritis, and petechiae involving the hard palate, the soft palate, or both.1
The syndrome is self-limited and resolves within 1 to 2 weeks.1
THE DIAGNOSTIC CHALLENGE
The differential diagnosis of the syndrome’s gloves-and-socks presentation includes hand-foot-mouth disease, erythema multiforme, Henoch-Schönlein purpura, and Kawasaki disease,4 in addition to viral exanthems and sexually transmitted diseases. Our patient’s fever, rash, and absolute lymphopenia focused attention on possible HIV infection, which caused the patient significant anxiety while awaiting the results of HIV testing. Heightened awareness of the cutaneous presentation of parvovirus B19 infection can help avoid unnecessary hospitalization and patient anxiety.
- Smith PT, Landry ML, Carey H, Krasnoff J, Cooney E. Papular-purpuric gloves and socks syndrome associated with acute parvovirus B19 infection: case report and review. Clin Infect Dis 1998; 27:164–168.
- Harms M, Feldmann R, Saurat JH. Papular-purpuric “gloves and socks” syndrome. J Am Acad Dermatol 1990; 23:850–854.
- Bagot M, Revuz J. Papular-purpuric “gloves and socks” syndrome: primary infection with parvovirus B19? J Am Acad Dermatol 1991; 25:341–342.
- Gutermuth J, Nadas K, Zirbs M, et al. Papular-purpuric gloves and socks syndrome. Lancet 2011; 378:198.
- Smith PT, Landry ML, Carey H, Krasnoff J, Cooney E. Papular-purpuric gloves and socks syndrome associated with acute parvovirus B19 infection: case report and review. Clin Infect Dis 1998; 27:164–168.
- Harms M, Feldmann R, Saurat JH. Papular-purpuric “gloves and socks” syndrome. J Am Acad Dermatol 1990; 23:850–854.
- Bagot M, Revuz J. Papular-purpuric “gloves and socks” syndrome: primary infection with parvovirus B19? J Am Acad Dermatol 1991; 25:341–342.
- Gutermuth J, Nadas K, Zirbs M, et al. Papular-purpuric gloves and socks syndrome. Lancet 2011; 378:198.
Dermatology update: The dawn of targeted treatment
New targeted therapies are changing the way patients with advanced dermatologic diseases are treated. Innovative molecular biology techniques developed as far back as the 1970s have engendered tremendous insight into the cellular and molecular pathogenesis of numerous diseases. Novel medications based on these insights are now bearing fruit, as directed biologic therapies that are revolutionizing clinical practice are increasingly becoming available.
This article reviews advances in targeted therapies for advanced basal cell carcinoma, psoriasis, and metastatic melanoma.
TARGETED THERAPY FOR BASAL CELL CARCINOMA
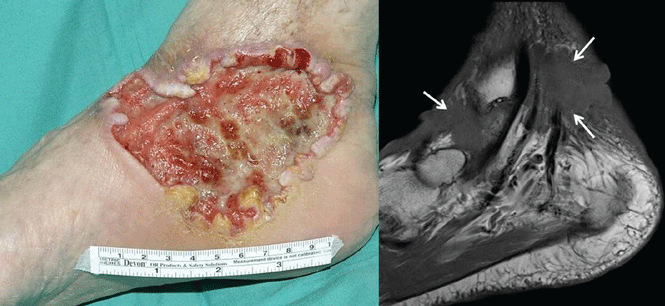
Case 1. A 56-year-old man presents with a progressively enlarging leg ulcer. Although it has been treated empirically for years as a venous stasis ulcer, biopsy reveals that it is basal cell carcinoma. Imaging shows muscle and tendon invasion, making surgical intervention short of amputation challenging (Figure 1). What are his options?
Basal cell carcinoma is the most common cancer in humans, accounting for 25% of all cancers and more than 2 million cases in the United States every year. In most cases, surgical excision is curative, but a subset of patients have inoperable, locally advanced, or metastatic disease that drastically limits treatment options. The median survival in metastatic basal cell carcinoma is 24 months, and conventional chemotherapy has not been shown to improve the prognosis.1,2
In addition to the burden of sporadic basal cell carcinoma, patients with the rare autosomal-dominant genetic disorder basal cell nevus syndrome (Gorlin syndrome) develop multiple basal cell lesions over their lifetime. The syndrome may also involve abnormalities of the skeletal system, genitourinary tract, and central nervous system, including development of medulloblastoma.
In Gorlin syndrome, basal cell carcinomas occur often and early; about half of white patients with the syndrome develop their first lesions by age 21, and 90% by age 35. The lesions occur in multiple numbers and can develop anywhere on the body, including on non–sun-exposed areas. Patients who have Gorlin syndrome need meticulous monitoring every 2 to 3 months so that basal cell lesions can be recognized early and treated before they become locally advanced. Keeping up with the numerous medical appointments and invasive treatments can be physically and mentally taxing for patients.
Specific pathway and mutations identified
In 1996, Gorlin syndrome was found to be caused by mutations of the human homolog of the PATCHED gene, which codes for a receptor in the “hedgehog” pathway.3 Two years later, the same mutations were found to be involved in many sporadic basal cell carcinomas, and we now believe that at least 85% of basal cell carcinomas involve abnormal activation of hedgehog pathway signaling.4,5
Vismodegib developed as targeted therapy
In 2009, Robarge et al6 described a potent inhibitor of the hedgehog pathway that was later optimized for potency and desirable pharmacologic traits, resulting in the drug vismodegib.7,8
Two phase 2 multicenter clinical trials9,10 of vismodegib were published in 2012. In the first, which was not randomized,9 33 patients with metastatic basal cell carcinoma and 63 patients with locally advanced disease were treated with vismodegib. Of those with metastatic disease, 30% achieved an objective response. Of those with locally advanced disease, 43% achieved an objective response and 21% achieved a complete response.
In the second trial,10 patients with Gorlin syndrome were randomized to either vismodegib (26 patients) or placebo (16 patients). After 8 months, the vismodegib group had developed significantly fewer new surgically eligible tumors (2 vs 29 per year), their tumors were smaller (change from baseline of the sum of the longest diameters –65% vs –11%), and they needed fewer surgeries (mean 0.31 vs 4.4 per patient). No tumors progressed in the treatment group. Results in some patients were dramatic, with complete healing of large ulcerative tumors. The trial was ended early in view of significant efficacy in the treatment group.
Based on these trials, the US Food and Drug Administration (FDA) approved vismodegib for treating metastatic and locally advanced basal cell carcinoma.
Resistance and adverse effects common
Unfortunately, vismodegib has significant drawbacks. About 20% of patients develop resistance, with tumors recurring after several months of therapy.11 Adverse effects most commonly reported were muscle spasms (68%), alopecia (63%), taste distortion (51%), weight loss (46%), and fatigue (36%). Although these effects were considered mild or moderate, they tended to persist, and almost every patient developed at least one. In the nonrandomized trial,9 more than 25% of patients discontinued treatment because of adverse effects, and more than half of patients did the same in the basal cell nevus syndrome trial.10
New uses may reduce shortcomings
Studies are under way to determine how best to use vismodegib.
One possibility is to use the drug for a few months to shrink tumors to the point that they become eligible for surgery. This is especially important for high-risk tumors, such as those near the eye or other vital structures. In 11 patients, Ally et al12 found that the surgical defect area was reduced by 27% from baseline after 4 months of treatment with vismodegib, allowing for curative surgery in some.
Another option is to combine vismodegib with other agents—either new ones on the horizon or existing nonspecific medications. For example, the antifungal itraconazole has been shown to inhibit hedgehog signaling and perhaps could be combined with vismodegib to increase response and reduce resistance.
Finally, a topical or intralesional form of vismodegib would be useful not only to reduce systemic toxicity, but also to increase efficacy when combined with other topical or systemic medications.
TARGETED THERAPY FOR PSORIASIS VULGARIS
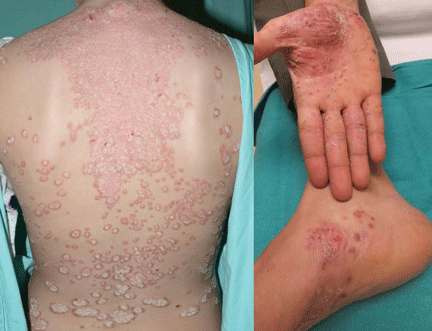
Case 2. A 28-year-old woman presents with worsening psoriasis. About 35% of her body surface is involved, including the palms and soles, making it difficult for her to perform activities of daily living (Figure 2). What are her options?
Psoriasis is a chronic immune-mediated disease that affects up to 3% of people worldwide. In its moderate to severe forms, we recognize psoriasis as a systemic inflammatory disease that may adversely affect organ systems other than the skin. Commonly associated comorbid diseases include inflammatory (psoriatic) arthritis, cardiovascular disease, malignancies (eg, lymphoma), and inflammatory bowel disease. In addition, patients are well known to have significantly impaired quality of life because of low self-esteem, stigmatization affecting their social and work relationships, and, in up to 60%, clinical depression.13,14 The onset of psoriatic arthritis, particularly of erosive disease, is an important decision point for starting aggressive treatment, as joint destruction is irreversible.
Early targeted therapy aimed at TNF alpha, IL-12, and IL-23
Histologically, psoriasis involves thickening of the epidermis caused by hyperproliferation of keratinocytes. Based on this, prior to the 1980s, the dominant hypothesis concerning its pathogenesis was that it was caused by an inherent defect of keratinocytes. In the 1980s and 1990s, however, molecular research revealed that psoriasis was an immune-mediated disease caused by immunologic dysregulation predominantly involving T-helper 1 (Th-1) cells, with the inflammatory cytokines tumor necrosis factor (TNF) alpha, interferon gamma, interleukin (IL) 12, and IL-23 playing prominent roles.15 These findings led to the development and FDA approval of the first effective, targeted, psoriasis treatments, TNF-alpha inhibitors and the IL-12/23 inhibitor ustekinumab.
Etanercept, the first TNF-alpha inhibitor to become available, was approved in 2004 for moderate to severe psoriasis. In 2008, the IL-12/23 inhibitor ustekinumab was approved for the same indication. These drugs are efficacious, are generally safe, and have revolutionized the treatment of psoriasis and psoriatic arthritis, and they are now prescribed on a daily basis.16,17
In the clinical trials that led to approval of these drugs, the main outcome measure was the Psoriasis Area and Severity Index (PASI), a clinical scoring tool that assesses clinical aspects of psoriatic disease including body surface area involvement, degree of thickness, erythema, and scaling of psoriatic plaques. PASI scores range from 0 (no psoriasis) to 72 (most severe psoriasis). Achieving “PASI 75” indicates at least 75% improvement from the baseline score and represents the most common primary outcome measure in clinical trials assessing efficacy of new treatments. Up to 80% of patients who received currently available TNF-alpha inhibitors and ustekinumab in pivotal clinical trials achieved PASI 75 when assessed at 12 to 16 weeks after starting treatment. A moderate percentage of patients (19%–57%, depending on the trial) achieved 90% improvement (PASI 90), and a minority (up to 18%) achieved PASI 100, indicating complete clearing of their psoriasis.18–22
Newly developed therapies target IL-17A
In the mid-2000s, Th-17 cells were discovered, a new lineage of T cells distinct from Th-1 and Th-2 cells. Th-17 cells are characterized by their production of IL-17, a pro-inflammatory cytokine with six family members (IL-17A through IL-17F). Over the next few years, experiments revealed that Th-17 cells and IL-17A play key roles in psoriasis immunologic dysregulation.15 These findings led to a paradigm shift in hypotheses concerning psoriasis pathogenesis, with Th-17 cells and IL-17 replacing Th-1 cells and associated cytokines as dominant mediators of tissue damage.
Additionally, these findings led to new ideas for treatment. Three monoclonal antibodies that target IL-17 inhibition are currently under investigation. Secukinumab and ixekizumab bind to IL-17A and inhibit it from downstream signaling, whereas brodalumab binds to the IL-17A receptor, blocking all six IL-17 cytokines (IL-17A to IL-17F).23
Clinical trials of IL-17 inhibitors show excellent skin improvement
Secukinumab. In 2014, the results of two phase 3 trials of secukinumab were published.24
In the Efficacy and Safety of Subcutaneous Secukinumab for Moderate to Severe Chronic Plaque-type Psoriasis for up to 1 Year trial,24 patients were given either secukinumab 300 mg or 150 mg subcutaneously at defined time points; 82% and 72%, respectively, attained PASI 75 at 12 weeks.
Similar results were seen in the Safety and Efficacy of Secukinumab Compared to Etanercept in Subjects With Moderate to Severe, Chronic Plaque-Type Psoriasis study,24 in which PASI 75 was achieved by 77% of patients receiving secukinumab 300 mg, 67% of those receiving secukinumab 150 mg, and only 44% of those receiving etanercept 50 mg twice weekly at 12 weeks. Rates of infection with secukinumab and etanercept were similar.
The most striking results of these trials were that more than half of patients receiving the 300-mg dose achieved at least 90% improvement in their PASI score (PASI 90) by week 12, and in more than a quarter of patients the psoriasis completely cleared (PASI 100). These results were dramatically better than for etanercept (PASI 90 21%; PASI 100 4%).
Additionally, secukinumab worked fast. The median time to PASI 50 with secukinumab 300 mg was less than half that seen with etanercept (3 weeks vs 7 weeks).
Ixekizumab. In 2012, a phase 2 trial evaluated subcutaneous injections of ixekizumab in dosages ranging from 10 to 150 mg at defined intervals for 16 weeks.25 Of those receiving the highest dosage, 82% attained PASI 75 at 12 weeks, on par with what is noted in patients receiving TNF-alpha inhibitors and IL-12/23 inhibitors. Remarkably, however, almost three-quarters of patients (71%) achieved PASI 90, and 39% achieved PASI 100. Improvement in psoriasis was apparent as early as 1 week after injection.
Brodalumab. A 2012 phase 2 trial of various dosages of the IL-17 receptor inhibitor brodalumab26 also showed excellent PASI 75 achievement with the highest dosage (82%). Astonishingly, though, PASI 90 was achieved by 75% of patients, and PASI 100 by 62%.
Overall, although the percentages of patients achieving PASI 75 with the new IL-17 inhibitor drugs are comparable to those seen with TNF-alpha inhibitors and IL-12/23 inhibitors, the extraordinarily high percentages of patients who achieved PASI 90 and PASI 100 are unprecedented.18–22
Arthritis improvement not shown
Where the IL-17 inhibitors eventually settle within algorithms of psoriasis treatment largely depends on their efficacy in treating psoriatic arthritis compared with TNF-alpha inhibitors and IL-12/23 inhibitors. Joint inflammation is typically evaluated with the American College of Rheumatology (ACR) scoring tool, which in simple terms can be thought of as analogous to the PASI scoring tool for the skin. Although the ACR scoring tool was developed to assess joint inflammation in clinical trials for patients with rheumatoid arthritis, it is commonly used to assess improvement of psoriatic arthritis in clinical trials. The ACR tool involves assessing and scoring the number of swollen and tender joints, but also incorporates serologic assessment of acute-phase reactants (erythrocyte sedimentation rate or C-reactive protein level), patient and physician global assessment, pain, and function. ACR 20 implies roughly a 20% improvement in these criteria, whereas ACR 50 indicates 50% improvement, and so on.
Two phase 2 trials of IL-17 inhibitors for psoriatic arthritis have been published, one with secukinumab27 and one with brodalumab.28 Neither had impressive improvement in the ACR score vs TNF inhibitors—39% for ACR 20 at week 12 and less than 10% for ACR 70. Clinical trial design may have played a role, and phase 3 trials are under way for all three IL-17 inhibitors.
Adverse effects of IL-17 inhibitors
For the most part, adverse effects reported with the IL-17 inhibitors have been mild and similar to those reported with available biologic treatments for psoriasis. Adverse effects most commonly reported have been nasopharyngitis, upper respiratory infection, arthralgia, and mild injection-site reactions. In the future, attention will be paid to the rate of infections known to be associated with IL-17, mainly localized infections with Staphylococcus aureus and Candida species. Some patients have developed Candida esophagitis, but this appears to resolve with discontinuation of the drugs. Neutropenia has occurred, but very few patients have developed grade 3 (500–1,000 cells/mm3) or worse. All adverse effects were reversible with discontinuation of treatment.
Approval of secukinumab, and current studies of IL-17 inhibitors
On January 21, 2015, secukinumab was approved by the FDA for treatment of moderate to severe psoriasis vulgaris in adult patients and is now available by prescription.
More trials of IL-17 inhibitors for the treatment of psoriasis and psoriatic arthritis are under way and are at various phases at the time of this writing.23
TARGETED THERAPY FOR ADVANCED MELANOMA
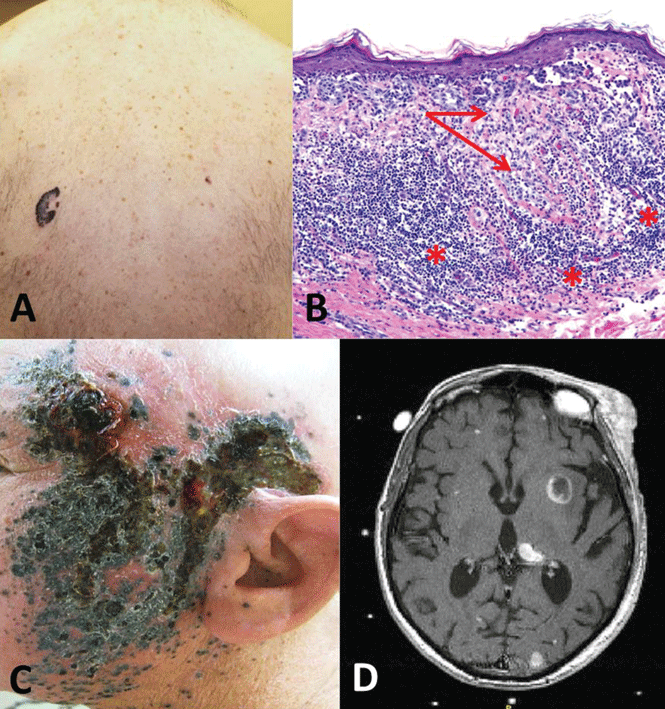
Case 3. A 58-year-old man presents with an irregular pigmented lesion on his back. Biopsy shows malignant melanoma with an intense, chronic inflammatory infiltrate surrounding the tumor (Figure 3). The tumor was surgically excised with standard margins. Two years later, the patient developed multiple pigmented lesions on the face and complained of headache. Magnetic resonance imaging of the brain revealed multiple enhancing lesions consistent with metastatic melanoma (Figure 3). What are this patient’s options?
Melanoma is the fifth most common cancer in humans, with about 132,000 new cases diagnosed worldwide each year and 48,000 deaths from advanced disease. Its incidence has risen rapidly over the last few decades. Advanced disease has a poor prognosis, with the median overall survival less than 1 year and 5-year survival less than 10%.
Despite decades of research, a paucity of FDA-approved medications were available to treat advanced melanoma until recently. The alkylating agent dacarbazine was approved in 1975, interferon alpha in 1995, and high-dose IL-2 in 1998. Although some patients respond, studies have not shown significant improvement in survival with any of these medications.29–31
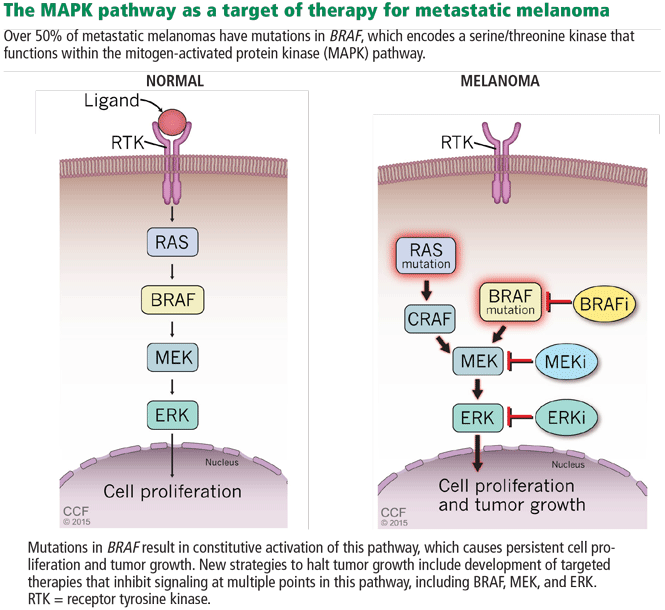
In 2002, Davies et al32 found that 50% to 65% of metastatic cutaneous melanomas have a mutation in the BRAF gene. Interestingly, 80% of these patients share a single specific mutation: substitution of glutamic acid for valine in codon 600 (BRAF V600E). The second most common mutation is a single substitution of a lysine for that same valine (BRAF V600K). Additionally, NRAS is mutated in about 20% of melanomas. These discoveries implicated a mitogen-activated protein kinase (MAPK) pathway (Figure 4) as playing a critical role in metastatic melanoma for a large percentage of patients.29
Based on this knowledge, several targeted therapies for melanoma have been developed, and some have been approved.
BRAF inhibitors—first success against melanoma
Vemurafenib. In 2010, Flaherty et al33 reported on a phase 1 and phase 2 clinical trial of vemurafenib (960 mg orally twice daily), a potent inhibitor of BRAF with the V600E mutation. They demonstrated a clinical benefit in 80% of patients with stage IV BRAF-mutant melanoma, an unprecedented response that opened the door to changes in the treatment of metastatic melanoma.
The phase 3 BRAF Inhibitor in Melanoma (BRIM)-3 clinical trial,34 published in 2011, randomized 675 previously untreated patients with advanced melanoma to either vemurafenib 960 mg orally twice daily or dacarbazine, the standard of care. The trial was terminated early when an interim analysis showed a significant overall advantage for vemurafenib (median progression-free survival 5.3 months vs 1.6 months for dacarbazine). Based on these results, vemurafenib was FDA-approved in August 2011 for use in patients with BRAF-mutant melanoma.
Dabrafenib. In a phase 3 clinical trial in 2012, Hauschild et al35 randomized 250 patients with BRAF (V600E)-mutated melanoma in a 3:1 ratio to receive either dabrafenib, a more potent second-generation BRAF inhibitor, or dacarbazine. Half of patients responded to dabrafenib, with a significantly improved progression-free survival rate (5.1 vs 2.7 months respectively), leading to FDA approval for its use in BRAF-mutant melanoma in May 2013.
Adverse effects common to vemurafenib and dabrafenib include rash, fatigue, fever, and joint pain. In addition, up to 25% of patients develop multiple secondary cutaneous squamous cell carcinomas and keratoacanthomas, usually within the first few months of therapy, which are believed to be caused by paradoxical activation of the MAPK pathway.
A more important problem with these medications is the development of resistance. Tumors typically progress again after a median progression-free survival of 6 to 7 months.
MEK inhibitors—another line of defense
Inhibitors of MEK—a serine-threonine kinase that is part of the same MAPK pathway involving BRAF—have been developed as well.
Trametinib. In 2012, trametinib, an allosteric MEK inhibitor, was used in an open-label phase 3 trial in 322 patients with advanced melanoma. Progression-free survival was 4.8 months for trametinib-treated patients compared with 1.5 months for the standard chemotherapy group (dacarbazine or paclitaxel).36 These results led to FDA approval of trametinib in May 2013 for treating BRAF-mutant melanoma.29
Cobimetinib is a second MEK inhibitor being evaluated alone and in combination with other targeted treatments for advanced melanoma.
Both MEK inhibitors have adverse effects similar to those seen with the BRAF inhibitors, including diarrhea, rash, fatigue, and edema. They also tend to cause asymptomatic elevated creatine kinase and transient retinopathy, reduced ejection fraction, and ventricular dysfunction. Unlike BRAF inhibitors, they are not associated with development of secondary cutaneous squamous cell carcinomas or keratoacanthomas. However, as with BRAF inhibitors, resistance is a problem with MEK inhibitors, with most patients relapsing less than a year after starting therapy.
Combination therapy improves outcomes
Possible mechanisms underlying resistance to these medications are being studied. A number of important factors appear to drive resistance, including expression of truncated BRAF proteins that do not bind the BRAF inhibitors and still activate downstream signaling, and amplification of BRAF to such a degree that it overwhelms the medications. This has led to the idea of combining BRAF inhibitors and MEK inhibitors to block the MAPK pathway at two points, potentially increasing the response and decreasing resistance.
Two trials have evaluated combinations of BRAF and MEK inhibitors in patients with advanced melanoma. Larkin et al37 conducted a phase 3 study evaluating combined vemurafenib (a BRAF inhibitor) and cobimetinib (a MEK inhibitor) vs combined vemurafenib and placebo. Survival with the combination therapy was 9.9 months vs 6.2 months with the single treatment.
The incidence of serious adverse effects was not significantly increased with the combination therapy, and keratoacanthomas, cutaneous squamous cell carcinomas, alopecia, and arthralgias were reduced compared with the vemurafenib and placebo group.
Another trial38 evaluating combined dabrafenib (a BRAF inhibitor) and trametinib (a MEK inhibitor) vs combined dabrafenib and placebo had similar findings: increased survival in the combined therapy group (9.3 months vs 8.8 months) and lower rates of squamous cell carcinoma (2% vs 9%).
In January 2014, the FDA approved the combination of BRAF and MEK inhibitors for the treatment of BRAF-mutant metastatic melanoma based on improved survival and generally reduced adverse effects.
IMMUNOTHERAPIES FOR NON-BRAF MELANOMA
Although BRAF and MEK inhibitors represent tremendous advances, their use is limited to the approximately 50% to 65% of patients with advanced melanoma who have BRAF V600 mutations. For others, only the traditional standard medications have been available until recently.
Two of those standard FDA-approved medications, interferon alpha-2b and IL-2, represent immunotherapies. Interferon alpha-2b up-regulates antigen presentation and increases antigen recognition by T cells. Overall, about 20% of patients in clinical trials have achieved responses.
IL-2 is a cytokine that increases T-cell proliferation and maturation into effector T cells. High-dose IL-2 has produced responses in 15% of patients, with a durable complete response in a small proportion.
Though success with these medications was modest, the fact that some patients responded to them indicates that immunotherapy could be a viable strategy for treating metastatic melanoma.30 This is underscored by the fact that some patients can mount an adaptive immune response specifically directed against antigenic proteins expressed in their tumors, resulting in expansion of cytotoxic T cells and control or even elimination of the malignancy.30
Tumors manipulate host immune checkpoints
Molecular biology has provided tremendous insight into tumor immunology over the past several decades, and we now recognize that a hallmark of cancer is escape from immune control.
Cancer cells contain a multitude of mutations that produce proteins that should be recognized by the immune system as foreign but in most individuals are not. This is because T-cell activity is down-regulated in cancer due to cancer cells’ ability to manipulate the host’s normal immunologic inhibitory pathways critical for maintaining self-tolerance.
In general, T-cell activation is initiated when an antigen-presenting cell presents an antigen to a T cell in a major histocompatibility complex-restricted manner. To prevent T cells from being activated by self-antigens and initiating autoimmunity, the interaction between antigen-presenting cells and T cells is regulated by checkpoints (Figure 5). First, for an antigen-presenting cell/T-cell interaction to result in T-cell activation, the T-cell receptor CD28 must bind CD80 on the antigen-presenting cell to drive a “positive” signal. Early in the interaction, the T-cell receptor CTLA-4 is up-regulated and competes with CD28 for binding of CD80. If CTLA-4, and not CD28, binds CD80, a “negative” signal is sent to the T cell and down-regulates it, making the interaction unproductive. Importantly, it is the CTLA-4:CD80 interaction that appears to be crucial for the ability of tumors to dampen T-cell responses to cancer cells.
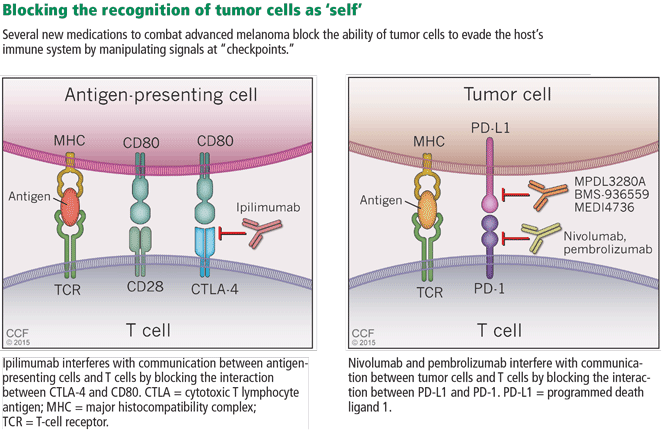
Ipilimumab is a fully humanized monoclonal antibody that binds to CTLA-4, blocking its ability to bind to CD80 and thereby enhancing T-cell activation. In a phase 3 trial, Hodi et al39 evaluated its use in treating advanced melanoma, with some enrolled patients having failed IL-2 treatment. Patients receiving ipilimumab with or without a glycoprotein-100 peptide vaccine (gp100) had an overall survival benefit of 10.1 months compared with 6.4 months for patients treated with gp100 alone. At 24 months, the survival rate with ipilimumab alone was 23.5%, almost double that of patients receiving gp100 alone.
Ipilimumab received FDA approval for treatment of metastatic melanoma in March 2011. This, and the BRAF inhibitors, were the first drugs approved by the FDA for the treatment of advanced melanoma in more than a decade.
Common adverse effects of ipilimumab include fatigue, diarrhea, rash, and pruritus. As expected, given its mechanism of action, up to about 25% of patients experience severe autoimmune-related events that may variably manifest as colitis, rash, hepatitis, neuritis, hypothyroidism, hypopituitarism, and hypophysitis. Another problem with this medication is that a subset of patients do not respond.
Cancer cells disguised as normal cells
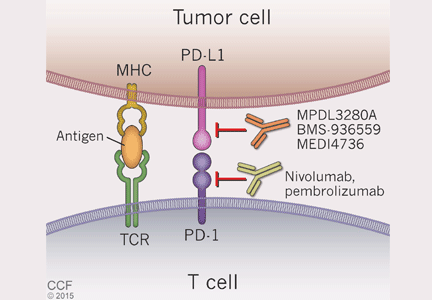
Cancer cells can also manipulate another immunologic checkpoint to evade attack by the host immune system (Figure 5). Cytotoxic T cells may recognize antigens on tumor cells and become activated and primed to directly destroy them. However, tumor cells, like normal cells express the programmed death ligands RTK-L1 and PD-L2. These ligands function to bind to the PD-1 receptor on activated T cells to indicate they are “self” and inhibit the cytotoxic T cells from destroying them.
Evasion of immune system attack by manipulating checkpoints involving CTLA-4 and PD-1 helps explain why malignancies can seemingly be associated with brisk inflammatory responses, such as the tumor in Case 3, yet progress and eventually metastasize (Figure 3).
Two medications—nivolumab and pembrolizumab—have been developed in an attempt to disrupt the ability of tumor cells to trick the immune system into accepting them as “self” by manipulating the PD-L1/PD-L2: PD-1 interaction. Both drugs are monoclonal antibodies that bind to PD-1 and, thus, effectively block the ability of PD-L1 or PD-L2 on tumor cells to bind these ligands and signal to activated T cells that they are “self.” This blocking allows T cells to then carry out their killing of tumor cells they initially recognize as foreign.
Nivolumab. In 2014, a phase 3 trial40 compared nivolumab and dacarbazine in patients with untreated advanced melanoma without a BRAF mutation. Objective response rates were 40.0% in the nivolumab group vs 13.9% in the dacarbazine group. This trial was stopped early because of significantly better survival rates in patients taking nivolumab compared with standard chemotherapy.
Interestingly, only 35% of patients who responded to nivolumab had evidence of PD-L1 expression on the surface of their tumor cells as assessed by immunohistochemical assay. Regardless of PD-L1 status, nivolumab-treated patients had improved overall survival compared with those treated with dacarbazine. The response rate with nivolumab was only slightly better in the subgroup of patients whose tumors expressed PD-L1 than in the subgroup without PD-L1.
On December 22, 2014, the FDA granted accelerated approval to nivolumab for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab treatment and, if BRAF V600 mutation-positive, a BRAF inhibitor.
Pembrolizumab. Also in 2014, an open-label, randomized, phase 1b trial of pembrolizumab treatment at two different dosage schedules was conducted in patients with advanced melanoma that had become refractory either to ipilimumab or a BRAF inhibitor.41 Treatment with pembrolizumab had an objective response rate of 26% at both doses.
In September 2014, the FDA granted accelerated approval for the use of pembrolizumab to treat patients with unresectable or metastatic melanoma and disease progression following treatment with ipilimumab or a BRAF inhibitor.
Adverse effects of PD-1 inhibitors are similar to those seen with ipilimumab, the most common (occurring in at least 20%) being fatigue, cough, nausea, pruritus, rash, decreased appetite, constipation, muscle pain, and diarrhea. Serious effects from pembrolizumab (occurring in at least 2%) were kidney failure, dyspnea, pneumonia, and cellulitis. As seen with ipilimumab, clinically significant autoimmune adverse reactions occur with PD-1 inhibitors, including pneumonitis, colitis, hypophysitis, nephritis, and hepatitis.
Combination therapy under investigation
A phase 1 trial using combination therapy with both immune checkpoint inhibitors—nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4)—in patients with treatment-resistant metastatic melanoma was published in 2013.42 More than half of patients achieved objective responses, with tumor regression of at least 80% in those who had a response. Tumor response was evident in all subgroups of patients studied—those with pretreatment elevated lactate dehydrogenase levels (one of the strongest prognostic factors in metastatic melanoma), metastases to distant sites, and bulky, multifocal tumor burden. Based on these results, a phase 3 trial is now under way looking at the combination of these two medications vs either one alone.
In summary, targeted treatments are changing the paradigm of how common dermatologic conditions associated with significant morbidity and mortality are treated. Although implementation of the above treatments into everyday clinical practice is exciting, future studies surrounding each are needed to address unanswered issues, such as the optimal dosing and treatment schedules in terms of both disease response and inhibition of resistance, optimal patient/disease characteristics for use, and optimal drug treatment combinations. In the meantime, basic research still utilizing classic molecular biology techniques to uncover pathogenic disease mechanisms in even more detail is ongoing and hopefully will lead to development of even better targeted treatments or even cures for these diseases.
- Lyons TG, O’Kane GM, Kelly CM. Efficacy and safety of vismodegib: a new therapeutic agent in the treatment of basal cell carcinoma. Expert Opin Drug Saf 2014; 13:1125–1132.
- McCusker M, Basset-Sequin N, Dummer R, et al. Metastatic basal cell carcinoma: prognosis dependent on anatomic site and spread of disease. Eur J Cancer 2014; 50:774–783.
- Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996; 85:841–851.
- Aszterbaum M, Rothman A, Johnson RL, et al. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J Invest Dermatol 1998; 110:885–888.
- Ingham PW, Placzek M. Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nat Rev Genet 2006; 7:841–850.
- Robarge KD, Brunton SA, Castanedo GM, et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg Med Chem Lett 2009; 19:5576–5581.
- Proctor AE, Thompson LA, O’Bryant CL. Vismodegib: an inhibitor of the Hedgehog signaling pathway in the treatment of basal cell carcinoma. Ann Pharmacother 2014; 48:99–106.
- Dessinioti C, Plaka M, Stratigos AJ. Vismodegib for the treatment of basal cell carcinoma: results and implications of the ERIVANCE BCC trial. Future Oncol 2014; 10:927–936.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med 2012; 366:2171–2179.
- Tang JY, Mackay-Wiggan JM, Aszterbaum M, et al. Inhibiting the hedgehog pathway in patients with basal-cell nevus syndrome. N Engl J Med 2012; 366:2180–2188.
- Brinkhuizen T, Reinders MG, van Geel M, et al. Acquired resistance to the Hedgehog pathway inhibitor vismodegib due to smoothened mutations in treatment of locally advanced basal cell carcinoma. J Am Acad Dermatol 2014; 71:1005–1008.
- Ally MS, Aasi S, Wysong A, et al. An investigator-initiated open-label clinical trial of vismodegib as a neoadjuvant to surgery for high-risk basal cell carcinoma. J Am Acad Dermatol 2014; 71:904–911.
- Rapp SR, Feldman SR, Exum ML, Fleischer AB Jr, Reboussin DM. Psoriasis causes as much disability as other major medical diseases. J Am Acad Dermatol 1999; 41:401–407.
- Gelfand JM, Niemann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. JAMA 2006; 296:1735–1741.
- Lynde CW, Poulin Y, Vender R, Bourcier M, Khalil S. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol 2014; 71:141–150.
- Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther 2008; 117:244–279.
- Nestle FL, Kaplan DH, Barker J. Psoriasis. N Engl J Med 2009; 361:496–509.
- Mentor A, Tyring SK, Gordon K, et al. Adalimumab therapy for moderate to severe psoriasis: a randomized, controlled phase III trial. J Am Acad Dermatol 2007; 58:106–115.
- Leonardi CL, Powers JL, Matheson RT, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med 2003; 349:2014–2022.
- Reich K, Nestle FO, Papp K, et al. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet 2005; 366:1367–1374.
- Leonardi CL, Kimball AB, Papp KA, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008; 371:1665–1674.
- Papp KA, Langley RG, Lebwohl M, et al; PHOENIX 2 study investigators. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet 2008; 371:1675–1684.
- Leonardi CL, Gordon KB. New and emerging therapies in psoriasis. Semin Cut Med Surg 2014; 33(suppl 2):S37–S41.
- Langley RG, Elewski BE, Lebwohl, et al for the ERASURE and FIXTURE Study Groups. Secukinumab in plaque psorisis—results of two phase 3 trials. N Engl J Med 2014; 371:326–338.
- Leonardi C, Matheson R, Zachariae C. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med 2012; 366:1190–1199.
- Papp KA, Leonardi C, Menter A, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med 2012; 366:1181–1189.
- McInnes IB, Sieper J, Braun J, et al. Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: a 24-week, randomised, double-blind, placebo-controlled, phase II proof-of-concept trial. Ann Rheum Dis 2014; 73:349–356.
- Mease PJ, Genovese MC, Greenwald MW, et al. Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. N Engl J Med 2014; 370:2295–2306.
- Girotti MR, Saturno G, Lorigan P, Marais R. No longer an untreatable disease: how targeted and immunotherapies have changed the management of melanoma patients. Molec Oncol 2014, 8:1140–1158.
- Saranga-Perry V, Ambe C, Zager JS, Kudchadkar RR. Recent developments in the medical and surgical treatment of melanoma. CA Canc J Clin 2014; 64:171–185.
- Shah DJ, Dronca RS. Latest advances in chemotherapeutic, targeted, and immune approaches in the treatment of metastatic melanoma. Mayo Clin Proc 2014; 89:504–519.
- Davies H, Ignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417:949–954.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363:809–819.
- Chapman PB, Hauschild A, Robert C. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364:2507–2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012; 380:358–365.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012; 367:107–114.
- Larkin J, Ascierto PA, Dreno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014; 371:1867–1876.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Eng J Med 2014; 371:1877–1888.
- Hodi FS, O’Day SJ, McDermott DF, Weber RW. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711–723.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015; 372:320–330.
- Robert C, Ribas A, Wolchok JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 2014; 384:1109–1117.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013; 369:122–133.
New targeted therapies are changing the way patients with advanced dermatologic diseases are treated. Innovative molecular biology techniques developed as far back as the 1970s have engendered tremendous insight into the cellular and molecular pathogenesis of numerous diseases. Novel medications based on these insights are now bearing fruit, as directed biologic therapies that are revolutionizing clinical practice are increasingly becoming available.
This article reviews advances in targeted therapies for advanced basal cell carcinoma, psoriasis, and metastatic melanoma.
TARGETED THERAPY FOR BASAL CELL CARCINOMA
Case 1. A 56-year-old man presents with a progressively enlarging leg ulcer. Although it has been treated empirically for years as a venous stasis ulcer, biopsy reveals that it is basal cell carcinoma. Imaging shows muscle and tendon invasion, making surgical intervention short of amputation challenging (Figure 1). What are his options?
Basal cell carcinoma is the most common cancer in humans, accounting for 25% of all cancers and more than 2 million cases in the United States every year. In most cases, surgical excision is curative, but a subset of patients have inoperable, locally advanced, or metastatic disease that drastically limits treatment options. The median survival in metastatic basal cell carcinoma is 24 months, and conventional chemotherapy has not been shown to improve the prognosis.1,2
In addition to the burden of sporadic basal cell carcinoma, patients with the rare autosomal-dominant genetic disorder basal cell nevus syndrome (Gorlin syndrome) develop multiple basal cell lesions over their lifetime. The syndrome may also involve abnormalities of the skeletal system, genitourinary tract, and central nervous system, including development of medulloblastoma.
In Gorlin syndrome, basal cell carcinomas occur often and early; about half of white patients with the syndrome develop their first lesions by age 21, and 90% by age 35. The lesions occur in multiple numbers and can develop anywhere on the body, including on non–sun-exposed areas. Patients who have Gorlin syndrome need meticulous monitoring every 2 to 3 months so that basal cell lesions can be recognized early and treated before they become locally advanced. Keeping up with the numerous medical appointments and invasive treatments can be physically and mentally taxing for patients.
Specific pathway and mutations identified
In 1996, Gorlin syndrome was found to be caused by mutations of the human homolog of the PATCHED gene, which codes for a receptor in the “hedgehog” pathway.3 Two years later, the same mutations were found to be involved in many sporadic basal cell carcinomas, and we now believe that at least 85% of basal cell carcinomas involve abnormal activation of hedgehog pathway signaling.4,5
Vismodegib developed as targeted therapy
In 2009, Robarge et al6 described a potent inhibitor of the hedgehog pathway that was later optimized for potency and desirable pharmacologic traits, resulting in the drug vismodegib.7,8
Two phase 2 multicenter clinical trials9,10 of vismodegib were published in 2012. In the first, which was not randomized,9 33 patients with metastatic basal cell carcinoma and 63 patients with locally advanced disease were treated with vismodegib. Of those with metastatic disease, 30% achieved an objective response. Of those with locally advanced disease, 43% achieved an objective response and 21% achieved a complete response.
In the second trial,10 patients with Gorlin syndrome were randomized to either vismodegib (26 patients) or placebo (16 patients). After 8 months, the vismodegib group had developed significantly fewer new surgically eligible tumors (2 vs 29 per year), their tumors were smaller (change from baseline of the sum of the longest diameters –65% vs –11%), and they needed fewer surgeries (mean 0.31 vs 4.4 per patient). No tumors progressed in the treatment group. Results in some patients were dramatic, with complete healing of large ulcerative tumors. The trial was ended early in view of significant efficacy in the treatment group.
Based on these trials, the US Food and Drug Administration (FDA) approved vismodegib for treating metastatic and locally advanced basal cell carcinoma.
Resistance and adverse effects common
Unfortunately, vismodegib has significant drawbacks. About 20% of patients develop resistance, with tumors recurring after several months of therapy.11 Adverse effects most commonly reported were muscle spasms (68%), alopecia (63%), taste distortion (51%), weight loss (46%), and fatigue (36%). Although these effects were considered mild or moderate, they tended to persist, and almost every patient developed at least one. In the nonrandomized trial,9 more than 25% of patients discontinued treatment because of adverse effects, and more than half of patients did the same in the basal cell nevus syndrome trial.10
New uses may reduce shortcomings
Studies are under way to determine how best to use vismodegib.
One possibility is to use the drug for a few months to shrink tumors to the point that they become eligible for surgery. This is especially important for high-risk tumors, such as those near the eye or other vital structures. In 11 patients, Ally et al12 found that the surgical defect area was reduced by 27% from baseline after 4 months of treatment with vismodegib, allowing for curative surgery in some.
Another option is to combine vismodegib with other agents—either new ones on the horizon or existing nonspecific medications. For example, the antifungal itraconazole has been shown to inhibit hedgehog signaling and perhaps could be combined with vismodegib to increase response and reduce resistance.
Finally, a topical or intralesional form of vismodegib would be useful not only to reduce systemic toxicity, but also to increase efficacy when combined with other topical or systemic medications.
TARGETED THERAPY FOR PSORIASIS VULGARIS
Case 2. A 28-year-old woman presents with worsening psoriasis. About 35% of her body surface is involved, including the palms and soles, making it difficult for her to perform activities of daily living (Figure 2). What are her options?
Psoriasis is a chronic immune-mediated disease that affects up to 3% of people worldwide. In its moderate to severe forms, we recognize psoriasis as a systemic inflammatory disease that may adversely affect organ systems other than the skin. Commonly associated comorbid diseases include inflammatory (psoriatic) arthritis, cardiovascular disease, malignancies (eg, lymphoma), and inflammatory bowel disease. In addition, patients are well known to have significantly impaired quality of life because of low self-esteem, stigmatization affecting their social and work relationships, and, in up to 60%, clinical depression.13,14 The onset of psoriatic arthritis, particularly of erosive disease, is an important decision point for starting aggressive treatment, as joint destruction is irreversible.
Early targeted therapy aimed at TNF alpha, IL-12, and IL-23
Histologically, psoriasis involves thickening of the epidermis caused by hyperproliferation of keratinocytes. Based on this, prior to the 1980s, the dominant hypothesis concerning its pathogenesis was that it was caused by an inherent defect of keratinocytes. In the 1980s and 1990s, however, molecular research revealed that psoriasis was an immune-mediated disease caused by immunologic dysregulation predominantly involving T-helper 1 (Th-1) cells, with the inflammatory cytokines tumor necrosis factor (TNF) alpha, interferon gamma, interleukin (IL) 12, and IL-23 playing prominent roles.15 These findings led to the development and FDA approval of the first effective, targeted, psoriasis treatments, TNF-alpha inhibitors and the IL-12/23 inhibitor ustekinumab.
Etanercept, the first TNF-alpha inhibitor to become available, was approved in 2004 for moderate to severe psoriasis. In 2008, the IL-12/23 inhibitor ustekinumab was approved for the same indication. These drugs are efficacious, are generally safe, and have revolutionized the treatment of psoriasis and psoriatic arthritis, and they are now prescribed on a daily basis.16,17
In the clinical trials that led to approval of these drugs, the main outcome measure was the Psoriasis Area and Severity Index (PASI), a clinical scoring tool that assesses clinical aspects of psoriatic disease including body surface area involvement, degree of thickness, erythema, and scaling of psoriatic plaques. PASI scores range from 0 (no psoriasis) to 72 (most severe psoriasis). Achieving “PASI 75” indicates at least 75% improvement from the baseline score and represents the most common primary outcome measure in clinical trials assessing efficacy of new treatments. Up to 80% of patients who received currently available TNF-alpha inhibitors and ustekinumab in pivotal clinical trials achieved PASI 75 when assessed at 12 to 16 weeks after starting treatment. A moderate percentage of patients (19%–57%, depending on the trial) achieved 90% improvement (PASI 90), and a minority (up to 18%) achieved PASI 100, indicating complete clearing of their psoriasis.18–22
Newly developed therapies target IL-17A
In the mid-2000s, Th-17 cells were discovered, a new lineage of T cells distinct from Th-1 and Th-2 cells. Th-17 cells are characterized by their production of IL-17, a pro-inflammatory cytokine with six family members (IL-17A through IL-17F). Over the next few years, experiments revealed that Th-17 cells and IL-17A play key roles in psoriasis immunologic dysregulation.15 These findings led to a paradigm shift in hypotheses concerning psoriasis pathogenesis, with Th-17 cells and IL-17 replacing Th-1 cells and associated cytokines as dominant mediators of tissue damage.
Additionally, these findings led to new ideas for treatment. Three monoclonal antibodies that target IL-17 inhibition are currently under investigation. Secukinumab and ixekizumab bind to IL-17A and inhibit it from downstream signaling, whereas brodalumab binds to the IL-17A receptor, blocking all six IL-17 cytokines (IL-17A to IL-17F).23
Clinical trials of IL-17 inhibitors show excellent skin improvement
Secukinumab. In 2014, the results of two phase 3 trials of secukinumab were published.24
In the Efficacy and Safety of Subcutaneous Secukinumab for Moderate to Severe Chronic Plaque-type Psoriasis for up to 1 Year trial,24 patients were given either secukinumab 300 mg or 150 mg subcutaneously at defined time points; 82% and 72%, respectively, attained PASI 75 at 12 weeks.
Similar results were seen in the Safety and Efficacy of Secukinumab Compared to Etanercept in Subjects With Moderate to Severe, Chronic Plaque-Type Psoriasis study,24 in which PASI 75 was achieved by 77% of patients receiving secukinumab 300 mg, 67% of those receiving secukinumab 150 mg, and only 44% of those receiving etanercept 50 mg twice weekly at 12 weeks. Rates of infection with secukinumab and etanercept were similar.
The most striking results of these trials were that more than half of patients receiving the 300-mg dose achieved at least 90% improvement in their PASI score (PASI 90) by week 12, and in more than a quarter of patients the psoriasis completely cleared (PASI 100). These results were dramatically better than for etanercept (PASI 90 21%; PASI 100 4%).
Additionally, secukinumab worked fast. The median time to PASI 50 with secukinumab 300 mg was less than half that seen with etanercept (3 weeks vs 7 weeks).
Ixekizumab. In 2012, a phase 2 trial evaluated subcutaneous injections of ixekizumab in dosages ranging from 10 to 150 mg at defined intervals for 16 weeks.25 Of those receiving the highest dosage, 82% attained PASI 75 at 12 weeks, on par with what is noted in patients receiving TNF-alpha inhibitors and IL-12/23 inhibitors. Remarkably, however, almost three-quarters of patients (71%) achieved PASI 90, and 39% achieved PASI 100. Improvement in psoriasis was apparent as early as 1 week after injection.
Brodalumab. A 2012 phase 2 trial of various dosages of the IL-17 receptor inhibitor brodalumab26 also showed excellent PASI 75 achievement with the highest dosage (82%). Astonishingly, though, PASI 90 was achieved by 75% of patients, and PASI 100 by 62%.
Overall, although the percentages of patients achieving PASI 75 with the new IL-17 inhibitor drugs are comparable to those seen with TNF-alpha inhibitors and IL-12/23 inhibitors, the extraordinarily high percentages of patients who achieved PASI 90 and PASI 100 are unprecedented.18–22
Arthritis improvement not shown
Where the IL-17 inhibitors eventually settle within algorithms of psoriasis treatment largely depends on their efficacy in treating psoriatic arthritis compared with TNF-alpha inhibitors and IL-12/23 inhibitors. Joint inflammation is typically evaluated with the American College of Rheumatology (ACR) scoring tool, which in simple terms can be thought of as analogous to the PASI scoring tool for the skin. Although the ACR scoring tool was developed to assess joint inflammation in clinical trials for patients with rheumatoid arthritis, it is commonly used to assess improvement of psoriatic arthritis in clinical trials. The ACR tool involves assessing and scoring the number of swollen and tender joints, but also incorporates serologic assessment of acute-phase reactants (erythrocyte sedimentation rate or C-reactive protein level), patient and physician global assessment, pain, and function. ACR 20 implies roughly a 20% improvement in these criteria, whereas ACR 50 indicates 50% improvement, and so on.
Two phase 2 trials of IL-17 inhibitors for psoriatic arthritis have been published, one with secukinumab27 and one with brodalumab.28 Neither had impressive improvement in the ACR score vs TNF inhibitors—39% for ACR 20 at week 12 and less than 10% for ACR 70. Clinical trial design may have played a role, and phase 3 trials are under way for all three IL-17 inhibitors.
Adverse effects of IL-17 inhibitors
For the most part, adverse effects reported with the IL-17 inhibitors have been mild and similar to those reported with available biologic treatments for psoriasis. Adverse effects most commonly reported have been nasopharyngitis, upper respiratory infection, arthralgia, and mild injection-site reactions. In the future, attention will be paid to the rate of infections known to be associated with IL-17, mainly localized infections with Staphylococcus aureus and Candida species. Some patients have developed Candida esophagitis, but this appears to resolve with discontinuation of the drugs. Neutropenia has occurred, but very few patients have developed grade 3 (500–1,000 cells/mm3) or worse. All adverse effects were reversible with discontinuation of treatment.
Approval of secukinumab, and current studies of IL-17 inhibitors
On January 21, 2015, secukinumab was approved by the FDA for treatment of moderate to severe psoriasis vulgaris in adult patients and is now available by prescription.
More trials of IL-17 inhibitors for the treatment of psoriasis and psoriatic arthritis are under way and are at various phases at the time of this writing.23
TARGETED THERAPY FOR ADVANCED MELANOMA
Case 3. A 58-year-old man presents with an irregular pigmented lesion on his back. Biopsy shows malignant melanoma with an intense, chronic inflammatory infiltrate surrounding the tumor (Figure 3). The tumor was surgically excised with standard margins. Two years later, the patient developed multiple pigmented lesions on the face and complained of headache. Magnetic resonance imaging of the brain revealed multiple enhancing lesions consistent with metastatic melanoma (Figure 3). What are this patient’s options?
Melanoma is the fifth most common cancer in humans, with about 132,000 new cases diagnosed worldwide each year and 48,000 deaths from advanced disease. Its incidence has risen rapidly over the last few decades. Advanced disease has a poor prognosis, with the median overall survival less than 1 year and 5-year survival less than 10%.
Despite decades of research, a paucity of FDA-approved medications were available to treat advanced melanoma until recently. The alkylating agent dacarbazine was approved in 1975, interferon alpha in 1995, and high-dose IL-2 in 1998. Although some patients respond, studies have not shown significant improvement in survival with any of these medications.29–31
In 2002, Davies et al32 found that 50% to 65% of metastatic cutaneous melanomas have a mutation in the BRAF gene. Interestingly, 80% of these patients share a single specific mutation: substitution of glutamic acid for valine in codon 600 (BRAF V600E). The second most common mutation is a single substitution of a lysine for that same valine (BRAF V600K). Additionally, NRAS is mutated in about 20% of melanomas. These discoveries implicated a mitogen-activated protein kinase (MAPK) pathway (Figure 4) as playing a critical role in metastatic melanoma for a large percentage of patients.29
Based on this knowledge, several targeted therapies for melanoma have been developed, and some have been approved.
BRAF inhibitors—first success against melanoma
Vemurafenib. In 2010, Flaherty et al33 reported on a phase 1 and phase 2 clinical trial of vemurafenib (960 mg orally twice daily), a potent inhibitor of BRAF with the V600E mutation. They demonstrated a clinical benefit in 80% of patients with stage IV BRAF-mutant melanoma, an unprecedented response that opened the door to changes in the treatment of metastatic melanoma.
The phase 3 BRAF Inhibitor in Melanoma (BRIM)-3 clinical trial,34 published in 2011, randomized 675 previously untreated patients with advanced melanoma to either vemurafenib 960 mg orally twice daily or dacarbazine, the standard of care. The trial was terminated early when an interim analysis showed a significant overall advantage for vemurafenib (median progression-free survival 5.3 months vs 1.6 months for dacarbazine). Based on these results, vemurafenib was FDA-approved in August 2011 for use in patients with BRAF-mutant melanoma.
Dabrafenib. In a phase 3 clinical trial in 2012, Hauschild et al35 randomized 250 patients with BRAF (V600E)-mutated melanoma in a 3:1 ratio to receive either dabrafenib, a more potent second-generation BRAF inhibitor, or dacarbazine. Half of patients responded to dabrafenib, with a significantly improved progression-free survival rate (5.1 vs 2.7 months respectively), leading to FDA approval for its use in BRAF-mutant melanoma in May 2013.
Adverse effects common to vemurafenib and dabrafenib include rash, fatigue, fever, and joint pain. In addition, up to 25% of patients develop multiple secondary cutaneous squamous cell carcinomas and keratoacanthomas, usually within the first few months of therapy, which are believed to be caused by paradoxical activation of the MAPK pathway.
A more important problem with these medications is the development of resistance. Tumors typically progress again after a median progression-free survival of 6 to 7 months.
MEK inhibitors—another line of defense
Inhibitors of MEK—a serine-threonine kinase that is part of the same MAPK pathway involving BRAF—have been developed as well.
Trametinib. In 2012, trametinib, an allosteric MEK inhibitor, was used in an open-label phase 3 trial in 322 patients with advanced melanoma. Progression-free survival was 4.8 months for trametinib-treated patients compared with 1.5 months for the standard chemotherapy group (dacarbazine or paclitaxel).36 These results led to FDA approval of trametinib in May 2013 for treating BRAF-mutant melanoma.29
Cobimetinib is a second MEK inhibitor being evaluated alone and in combination with other targeted treatments for advanced melanoma.
Both MEK inhibitors have adverse effects similar to those seen with the BRAF inhibitors, including diarrhea, rash, fatigue, and edema. They also tend to cause asymptomatic elevated creatine kinase and transient retinopathy, reduced ejection fraction, and ventricular dysfunction. Unlike BRAF inhibitors, they are not associated with development of secondary cutaneous squamous cell carcinomas or keratoacanthomas. However, as with BRAF inhibitors, resistance is a problem with MEK inhibitors, with most patients relapsing less than a year after starting therapy.
Combination therapy improves outcomes
Possible mechanisms underlying resistance to these medications are being studied. A number of important factors appear to drive resistance, including expression of truncated BRAF proteins that do not bind the BRAF inhibitors and still activate downstream signaling, and amplification of BRAF to such a degree that it overwhelms the medications. This has led to the idea of combining BRAF inhibitors and MEK inhibitors to block the MAPK pathway at two points, potentially increasing the response and decreasing resistance.
Two trials have evaluated combinations of BRAF and MEK inhibitors in patients with advanced melanoma. Larkin et al37 conducted a phase 3 study evaluating combined vemurafenib (a BRAF inhibitor) and cobimetinib (a MEK inhibitor) vs combined vemurafenib and placebo. Survival with the combination therapy was 9.9 months vs 6.2 months with the single treatment.
The incidence of serious adverse effects was not significantly increased with the combination therapy, and keratoacanthomas, cutaneous squamous cell carcinomas, alopecia, and arthralgias were reduced compared with the vemurafenib and placebo group.
Another trial38 evaluating combined dabrafenib (a BRAF inhibitor) and trametinib (a MEK inhibitor) vs combined dabrafenib and placebo had similar findings: increased survival in the combined therapy group (9.3 months vs 8.8 months) and lower rates of squamous cell carcinoma (2% vs 9%).
In January 2014, the FDA approved the combination of BRAF and MEK inhibitors for the treatment of BRAF-mutant metastatic melanoma based on improved survival and generally reduced adverse effects.
IMMUNOTHERAPIES FOR NON-BRAF MELANOMA
Although BRAF and MEK inhibitors represent tremendous advances, their use is limited to the approximately 50% to 65% of patients with advanced melanoma who have BRAF V600 mutations. For others, only the traditional standard medications have been available until recently.
Two of those standard FDA-approved medications, interferon alpha-2b and IL-2, represent immunotherapies. Interferon alpha-2b up-regulates antigen presentation and increases antigen recognition by T cells. Overall, about 20% of patients in clinical trials have achieved responses.
IL-2 is a cytokine that increases T-cell proliferation and maturation into effector T cells. High-dose IL-2 has produced responses in 15% of patients, with a durable complete response in a small proportion.
Though success with these medications was modest, the fact that some patients responded to them indicates that immunotherapy could be a viable strategy for treating metastatic melanoma.30 This is underscored by the fact that some patients can mount an adaptive immune response specifically directed against antigenic proteins expressed in their tumors, resulting in expansion of cytotoxic T cells and control or even elimination of the malignancy.30
Tumors manipulate host immune checkpoints
Molecular biology has provided tremendous insight into tumor immunology over the past several decades, and we now recognize that a hallmark of cancer is escape from immune control.
Cancer cells contain a multitude of mutations that produce proteins that should be recognized by the immune system as foreign but in most individuals are not. This is because T-cell activity is down-regulated in cancer due to cancer cells’ ability to manipulate the host’s normal immunologic inhibitory pathways critical for maintaining self-tolerance.
In general, T-cell activation is initiated when an antigen-presenting cell presents an antigen to a T cell in a major histocompatibility complex-restricted manner. To prevent T cells from being activated by self-antigens and initiating autoimmunity, the interaction between antigen-presenting cells and T cells is regulated by checkpoints (Figure 5). First, for an antigen-presenting cell/T-cell interaction to result in T-cell activation, the T-cell receptor CD28 must bind CD80 on the antigen-presenting cell to drive a “positive” signal. Early in the interaction, the T-cell receptor CTLA-4 is up-regulated and competes with CD28 for binding of CD80. If CTLA-4, and not CD28, binds CD80, a “negative” signal is sent to the T cell and down-regulates it, making the interaction unproductive. Importantly, it is the CTLA-4:CD80 interaction that appears to be crucial for the ability of tumors to dampen T-cell responses to cancer cells.
Ipilimumab is a fully humanized monoclonal antibody that binds to CTLA-4, blocking its ability to bind to CD80 and thereby enhancing T-cell activation. In a phase 3 trial, Hodi et al39 evaluated its use in treating advanced melanoma, with some enrolled patients having failed IL-2 treatment. Patients receiving ipilimumab with or without a glycoprotein-100 peptide vaccine (gp100) had an overall survival benefit of 10.1 months compared with 6.4 months for patients treated with gp100 alone. At 24 months, the survival rate with ipilimumab alone was 23.5%, almost double that of patients receiving gp100 alone.
Ipilimumab received FDA approval for treatment of metastatic melanoma in March 2011. This, and the BRAF inhibitors, were the first drugs approved by the FDA for the treatment of advanced melanoma in more than a decade.
Common adverse effects of ipilimumab include fatigue, diarrhea, rash, and pruritus. As expected, given its mechanism of action, up to about 25% of patients experience severe autoimmune-related events that may variably manifest as colitis, rash, hepatitis, neuritis, hypothyroidism, hypopituitarism, and hypophysitis. Another problem with this medication is that a subset of patients do not respond.
Cancer cells disguised as normal cells
Cancer cells can also manipulate another immunologic checkpoint to evade attack by the host immune system (Figure 5). Cytotoxic T cells may recognize antigens on tumor cells and become activated and primed to directly destroy them. However, tumor cells, like normal cells express the programmed death ligands RTK-L1 and PD-L2. These ligands function to bind to the PD-1 receptor on activated T cells to indicate they are “self” and inhibit the cytotoxic T cells from destroying them.
Evasion of immune system attack by manipulating checkpoints involving CTLA-4 and PD-1 helps explain why malignancies can seemingly be associated with brisk inflammatory responses, such as the tumor in Case 3, yet progress and eventually metastasize (Figure 3).
Two medications—nivolumab and pembrolizumab—have been developed in an attempt to disrupt the ability of tumor cells to trick the immune system into accepting them as “self” by manipulating the PD-L1/PD-L2: PD-1 interaction. Both drugs are monoclonal antibodies that bind to PD-1 and, thus, effectively block the ability of PD-L1 or PD-L2 on tumor cells to bind these ligands and signal to activated T cells that they are “self.” This blocking allows T cells to then carry out their killing of tumor cells they initially recognize as foreign.
Nivolumab. In 2014, a phase 3 trial40 compared nivolumab and dacarbazine in patients with untreated advanced melanoma without a BRAF mutation. Objective response rates were 40.0% in the nivolumab group vs 13.9% in the dacarbazine group. This trial was stopped early because of significantly better survival rates in patients taking nivolumab compared with standard chemotherapy.
Interestingly, only 35% of patients who responded to nivolumab had evidence of PD-L1 expression on the surface of their tumor cells as assessed by immunohistochemical assay. Regardless of PD-L1 status, nivolumab-treated patients had improved overall survival compared with those treated with dacarbazine. The response rate with nivolumab was only slightly better in the subgroup of patients whose tumors expressed PD-L1 than in the subgroup without PD-L1.
On December 22, 2014, the FDA granted accelerated approval to nivolumab for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab treatment and, if BRAF V600 mutation-positive, a BRAF inhibitor.
Pembrolizumab. Also in 2014, an open-label, randomized, phase 1b trial of pembrolizumab treatment at two different dosage schedules was conducted in patients with advanced melanoma that had become refractory either to ipilimumab or a BRAF inhibitor.41 Treatment with pembrolizumab had an objective response rate of 26% at both doses.
In September 2014, the FDA granted accelerated approval for the use of pembrolizumab to treat patients with unresectable or metastatic melanoma and disease progression following treatment with ipilimumab or a BRAF inhibitor.
Adverse effects of PD-1 inhibitors are similar to those seen with ipilimumab, the most common (occurring in at least 20%) being fatigue, cough, nausea, pruritus, rash, decreased appetite, constipation, muscle pain, and diarrhea. Serious effects from pembrolizumab (occurring in at least 2%) were kidney failure, dyspnea, pneumonia, and cellulitis. As seen with ipilimumab, clinically significant autoimmune adverse reactions occur with PD-1 inhibitors, including pneumonitis, colitis, hypophysitis, nephritis, and hepatitis.
Combination therapy under investigation
A phase 1 trial using combination therapy with both immune checkpoint inhibitors—nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4)—in patients with treatment-resistant metastatic melanoma was published in 2013.42 More than half of patients achieved objective responses, with tumor regression of at least 80% in those who had a response. Tumor response was evident in all subgroups of patients studied—those with pretreatment elevated lactate dehydrogenase levels (one of the strongest prognostic factors in metastatic melanoma), metastases to distant sites, and bulky, multifocal tumor burden. Based on these results, a phase 3 trial is now under way looking at the combination of these two medications vs either one alone.
In summary, targeted treatments are changing the paradigm of how common dermatologic conditions associated with significant morbidity and mortality are treated. Although implementation of the above treatments into everyday clinical practice is exciting, future studies surrounding each are needed to address unanswered issues, such as the optimal dosing and treatment schedules in terms of both disease response and inhibition of resistance, optimal patient/disease characteristics for use, and optimal drug treatment combinations. In the meantime, basic research still utilizing classic molecular biology techniques to uncover pathogenic disease mechanisms in even more detail is ongoing and hopefully will lead to development of even better targeted treatments or even cures for these diseases.
New targeted therapies are changing the way patients with advanced dermatologic diseases are treated. Innovative molecular biology techniques developed as far back as the 1970s have engendered tremendous insight into the cellular and molecular pathogenesis of numerous diseases. Novel medications based on these insights are now bearing fruit, as directed biologic therapies that are revolutionizing clinical practice are increasingly becoming available.
This article reviews advances in targeted therapies for advanced basal cell carcinoma, psoriasis, and metastatic melanoma.
TARGETED THERAPY FOR BASAL CELL CARCINOMA
Case 1. A 56-year-old man presents with a progressively enlarging leg ulcer. Although it has been treated empirically for years as a venous stasis ulcer, biopsy reveals that it is basal cell carcinoma. Imaging shows muscle and tendon invasion, making surgical intervention short of amputation challenging (Figure 1). What are his options?
Basal cell carcinoma is the most common cancer in humans, accounting for 25% of all cancers and more than 2 million cases in the United States every year. In most cases, surgical excision is curative, but a subset of patients have inoperable, locally advanced, or metastatic disease that drastically limits treatment options. The median survival in metastatic basal cell carcinoma is 24 months, and conventional chemotherapy has not been shown to improve the prognosis.1,2
In addition to the burden of sporadic basal cell carcinoma, patients with the rare autosomal-dominant genetic disorder basal cell nevus syndrome (Gorlin syndrome) develop multiple basal cell lesions over their lifetime. The syndrome may also involve abnormalities of the skeletal system, genitourinary tract, and central nervous system, including development of medulloblastoma.
In Gorlin syndrome, basal cell carcinomas occur often and early; about half of white patients with the syndrome develop their first lesions by age 21, and 90% by age 35. The lesions occur in multiple numbers and can develop anywhere on the body, including on non–sun-exposed areas. Patients who have Gorlin syndrome need meticulous monitoring every 2 to 3 months so that basal cell lesions can be recognized early and treated before they become locally advanced. Keeping up with the numerous medical appointments and invasive treatments can be physically and mentally taxing for patients.
Specific pathway and mutations identified
In 1996, Gorlin syndrome was found to be caused by mutations of the human homolog of the PATCHED gene, which codes for a receptor in the “hedgehog” pathway.3 Two years later, the same mutations were found to be involved in many sporadic basal cell carcinomas, and we now believe that at least 85% of basal cell carcinomas involve abnormal activation of hedgehog pathway signaling.4,5
Vismodegib developed as targeted therapy
In 2009, Robarge et al6 described a potent inhibitor of the hedgehog pathway that was later optimized for potency and desirable pharmacologic traits, resulting in the drug vismodegib.7,8
Two phase 2 multicenter clinical trials9,10 of vismodegib were published in 2012. In the first, which was not randomized,9 33 patients with metastatic basal cell carcinoma and 63 patients with locally advanced disease were treated with vismodegib. Of those with metastatic disease, 30% achieved an objective response. Of those with locally advanced disease, 43% achieved an objective response and 21% achieved a complete response.
In the second trial,10 patients with Gorlin syndrome were randomized to either vismodegib (26 patients) or placebo (16 patients). After 8 months, the vismodegib group had developed significantly fewer new surgically eligible tumors (2 vs 29 per year), their tumors were smaller (change from baseline of the sum of the longest diameters –65% vs –11%), and they needed fewer surgeries (mean 0.31 vs 4.4 per patient). No tumors progressed in the treatment group. Results in some patients were dramatic, with complete healing of large ulcerative tumors. The trial was ended early in view of significant efficacy in the treatment group.
Based on these trials, the US Food and Drug Administration (FDA) approved vismodegib for treating metastatic and locally advanced basal cell carcinoma.
Resistance and adverse effects common
Unfortunately, vismodegib has significant drawbacks. About 20% of patients develop resistance, with tumors recurring after several months of therapy.11 Adverse effects most commonly reported were muscle spasms (68%), alopecia (63%), taste distortion (51%), weight loss (46%), and fatigue (36%). Although these effects were considered mild or moderate, they tended to persist, and almost every patient developed at least one. In the nonrandomized trial,9 more than 25% of patients discontinued treatment because of adverse effects, and more than half of patients did the same in the basal cell nevus syndrome trial.10
New uses may reduce shortcomings
Studies are under way to determine how best to use vismodegib.
One possibility is to use the drug for a few months to shrink tumors to the point that they become eligible for surgery. This is especially important for high-risk tumors, such as those near the eye or other vital structures. In 11 patients, Ally et al12 found that the surgical defect area was reduced by 27% from baseline after 4 months of treatment with vismodegib, allowing for curative surgery in some.
Another option is to combine vismodegib with other agents—either new ones on the horizon or existing nonspecific medications. For example, the antifungal itraconazole has been shown to inhibit hedgehog signaling and perhaps could be combined with vismodegib to increase response and reduce resistance.
Finally, a topical or intralesional form of vismodegib would be useful not only to reduce systemic toxicity, but also to increase efficacy when combined with other topical or systemic medications.
TARGETED THERAPY FOR PSORIASIS VULGARIS
Case 2. A 28-year-old woman presents with worsening psoriasis. About 35% of her body surface is involved, including the palms and soles, making it difficult for her to perform activities of daily living (Figure 2). What are her options?
Psoriasis is a chronic immune-mediated disease that affects up to 3% of people worldwide. In its moderate to severe forms, we recognize psoriasis as a systemic inflammatory disease that may adversely affect organ systems other than the skin. Commonly associated comorbid diseases include inflammatory (psoriatic) arthritis, cardiovascular disease, malignancies (eg, lymphoma), and inflammatory bowel disease. In addition, patients are well known to have significantly impaired quality of life because of low self-esteem, stigmatization affecting their social and work relationships, and, in up to 60%, clinical depression.13,14 The onset of psoriatic arthritis, particularly of erosive disease, is an important decision point for starting aggressive treatment, as joint destruction is irreversible.
Early targeted therapy aimed at TNF alpha, IL-12, and IL-23
Histologically, psoriasis involves thickening of the epidermis caused by hyperproliferation of keratinocytes. Based on this, prior to the 1980s, the dominant hypothesis concerning its pathogenesis was that it was caused by an inherent defect of keratinocytes. In the 1980s and 1990s, however, molecular research revealed that psoriasis was an immune-mediated disease caused by immunologic dysregulation predominantly involving T-helper 1 (Th-1) cells, with the inflammatory cytokines tumor necrosis factor (TNF) alpha, interferon gamma, interleukin (IL) 12, and IL-23 playing prominent roles.15 These findings led to the development and FDA approval of the first effective, targeted, psoriasis treatments, TNF-alpha inhibitors and the IL-12/23 inhibitor ustekinumab.
Etanercept, the first TNF-alpha inhibitor to become available, was approved in 2004 for moderate to severe psoriasis. In 2008, the IL-12/23 inhibitor ustekinumab was approved for the same indication. These drugs are efficacious, are generally safe, and have revolutionized the treatment of psoriasis and psoriatic arthritis, and they are now prescribed on a daily basis.16,17
In the clinical trials that led to approval of these drugs, the main outcome measure was the Psoriasis Area and Severity Index (PASI), a clinical scoring tool that assesses clinical aspects of psoriatic disease including body surface area involvement, degree of thickness, erythema, and scaling of psoriatic plaques. PASI scores range from 0 (no psoriasis) to 72 (most severe psoriasis). Achieving “PASI 75” indicates at least 75% improvement from the baseline score and represents the most common primary outcome measure in clinical trials assessing efficacy of new treatments. Up to 80% of patients who received currently available TNF-alpha inhibitors and ustekinumab in pivotal clinical trials achieved PASI 75 when assessed at 12 to 16 weeks after starting treatment. A moderate percentage of patients (19%–57%, depending on the trial) achieved 90% improvement (PASI 90), and a minority (up to 18%) achieved PASI 100, indicating complete clearing of their psoriasis.18–22
Newly developed therapies target IL-17A
In the mid-2000s, Th-17 cells were discovered, a new lineage of T cells distinct from Th-1 and Th-2 cells. Th-17 cells are characterized by their production of IL-17, a pro-inflammatory cytokine with six family members (IL-17A through IL-17F). Over the next few years, experiments revealed that Th-17 cells and IL-17A play key roles in psoriasis immunologic dysregulation.15 These findings led to a paradigm shift in hypotheses concerning psoriasis pathogenesis, with Th-17 cells and IL-17 replacing Th-1 cells and associated cytokines as dominant mediators of tissue damage.
Additionally, these findings led to new ideas for treatment. Three monoclonal antibodies that target IL-17 inhibition are currently under investigation. Secukinumab and ixekizumab bind to IL-17A and inhibit it from downstream signaling, whereas brodalumab binds to the IL-17A receptor, blocking all six IL-17 cytokines (IL-17A to IL-17F).23
Clinical trials of IL-17 inhibitors show excellent skin improvement
Secukinumab. In 2014, the results of two phase 3 trials of secukinumab were published.24
In the Efficacy and Safety of Subcutaneous Secukinumab for Moderate to Severe Chronic Plaque-type Psoriasis for up to 1 Year trial,24 patients were given either secukinumab 300 mg or 150 mg subcutaneously at defined time points; 82% and 72%, respectively, attained PASI 75 at 12 weeks.
Similar results were seen in the Safety and Efficacy of Secukinumab Compared to Etanercept in Subjects With Moderate to Severe, Chronic Plaque-Type Psoriasis study,24 in which PASI 75 was achieved by 77% of patients receiving secukinumab 300 mg, 67% of those receiving secukinumab 150 mg, and only 44% of those receiving etanercept 50 mg twice weekly at 12 weeks. Rates of infection with secukinumab and etanercept were similar.
The most striking results of these trials were that more than half of patients receiving the 300-mg dose achieved at least 90% improvement in their PASI score (PASI 90) by week 12, and in more than a quarter of patients the psoriasis completely cleared (PASI 100). These results were dramatically better than for etanercept (PASI 90 21%; PASI 100 4%).
Additionally, secukinumab worked fast. The median time to PASI 50 with secukinumab 300 mg was less than half that seen with etanercept (3 weeks vs 7 weeks).
Ixekizumab. In 2012, a phase 2 trial evaluated subcutaneous injections of ixekizumab in dosages ranging from 10 to 150 mg at defined intervals for 16 weeks.25 Of those receiving the highest dosage, 82% attained PASI 75 at 12 weeks, on par with what is noted in patients receiving TNF-alpha inhibitors and IL-12/23 inhibitors. Remarkably, however, almost three-quarters of patients (71%) achieved PASI 90, and 39% achieved PASI 100. Improvement in psoriasis was apparent as early as 1 week after injection.
Brodalumab. A 2012 phase 2 trial of various dosages of the IL-17 receptor inhibitor brodalumab26 also showed excellent PASI 75 achievement with the highest dosage (82%). Astonishingly, though, PASI 90 was achieved by 75% of patients, and PASI 100 by 62%.
Overall, although the percentages of patients achieving PASI 75 with the new IL-17 inhibitor drugs are comparable to those seen with TNF-alpha inhibitors and IL-12/23 inhibitors, the extraordinarily high percentages of patients who achieved PASI 90 and PASI 100 are unprecedented.18–22
Arthritis improvement not shown
Where the IL-17 inhibitors eventually settle within algorithms of psoriasis treatment largely depends on their efficacy in treating psoriatic arthritis compared with TNF-alpha inhibitors and IL-12/23 inhibitors. Joint inflammation is typically evaluated with the American College of Rheumatology (ACR) scoring tool, which in simple terms can be thought of as analogous to the PASI scoring tool for the skin. Although the ACR scoring tool was developed to assess joint inflammation in clinical trials for patients with rheumatoid arthritis, it is commonly used to assess improvement of psoriatic arthritis in clinical trials. The ACR tool involves assessing and scoring the number of swollen and tender joints, but also incorporates serologic assessment of acute-phase reactants (erythrocyte sedimentation rate or C-reactive protein level), patient and physician global assessment, pain, and function. ACR 20 implies roughly a 20% improvement in these criteria, whereas ACR 50 indicates 50% improvement, and so on.
Two phase 2 trials of IL-17 inhibitors for psoriatic arthritis have been published, one with secukinumab27 and one with brodalumab.28 Neither had impressive improvement in the ACR score vs TNF inhibitors—39% for ACR 20 at week 12 and less than 10% for ACR 70. Clinical trial design may have played a role, and phase 3 trials are under way for all three IL-17 inhibitors.
Adverse effects of IL-17 inhibitors
For the most part, adverse effects reported with the IL-17 inhibitors have been mild and similar to those reported with available biologic treatments for psoriasis. Adverse effects most commonly reported have been nasopharyngitis, upper respiratory infection, arthralgia, and mild injection-site reactions. In the future, attention will be paid to the rate of infections known to be associated with IL-17, mainly localized infections with Staphylococcus aureus and Candida species. Some patients have developed Candida esophagitis, but this appears to resolve with discontinuation of the drugs. Neutropenia has occurred, but very few patients have developed grade 3 (500–1,000 cells/mm3) or worse. All adverse effects were reversible with discontinuation of treatment.
Approval of secukinumab, and current studies of IL-17 inhibitors
On January 21, 2015, secukinumab was approved by the FDA for treatment of moderate to severe psoriasis vulgaris in adult patients and is now available by prescription.
More trials of IL-17 inhibitors for the treatment of psoriasis and psoriatic arthritis are under way and are at various phases at the time of this writing.23
TARGETED THERAPY FOR ADVANCED MELANOMA
Case 3. A 58-year-old man presents with an irregular pigmented lesion on his back. Biopsy shows malignant melanoma with an intense, chronic inflammatory infiltrate surrounding the tumor (Figure 3). The tumor was surgically excised with standard margins. Two years later, the patient developed multiple pigmented lesions on the face and complained of headache. Magnetic resonance imaging of the brain revealed multiple enhancing lesions consistent with metastatic melanoma (Figure 3). What are this patient’s options?
Melanoma is the fifth most common cancer in humans, with about 132,000 new cases diagnosed worldwide each year and 48,000 deaths from advanced disease. Its incidence has risen rapidly over the last few decades. Advanced disease has a poor prognosis, with the median overall survival less than 1 year and 5-year survival less than 10%.
Despite decades of research, a paucity of FDA-approved medications were available to treat advanced melanoma until recently. The alkylating agent dacarbazine was approved in 1975, interferon alpha in 1995, and high-dose IL-2 in 1998. Although some patients respond, studies have not shown significant improvement in survival with any of these medications.29–31
In 2002, Davies et al32 found that 50% to 65% of metastatic cutaneous melanomas have a mutation in the BRAF gene. Interestingly, 80% of these patients share a single specific mutation: substitution of glutamic acid for valine in codon 600 (BRAF V600E). The second most common mutation is a single substitution of a lysine for that same valine (BRAF V600K). Additionally, NRAS is mutated in about 20% of melanomas. These discoveries implicated a mitogen-activated protein kinase (MAPK) pathway (Figure 4) as playing a critical role in metastatic melanoma for a large percentage of patients.29
Based on this knowledge, several targeted therapies for melanoma have been developed, and some have been approved.
BRAF inhibitors—first success against melanoma
Vemurafenib. In 2010, Flaherty et al33 reported on a phase 1 and phase 2 clinical trial of vemurafenib (960 mg orally twice daily), a potent inhibitor of BRAF with the V600E mutation. They demonstrated a clinical benefit in 80% of patients with stage IV BRAF-mutant melanoma, an unprecedented response that opened the door to changes in the treatment of metastatic melanoma.
The phase 3 BRAF Inhibitor in Melanoma (BRIM)-3 clinical trial,34 published in 2011, randomized 675 previously untreated patients with advanced melanoma to either vemurafenib 960 mg orally twice daily or dacarbazine, the standard of care. The trial was terminated early when an interim analysis showed a significant overall advantage for vemurafenib (median progression-free survival 5.3 months vs 1.6 months for dacarbazine). Based on these results, vemurafenib was FDA-approved in August 2011 for use in patients with BRAF-mutant melanoma.
Dabrafenib. In a phase 3 clinical trial in 2012, Hauschild et al35 randomized 250 patients with BRAF (V600E)-mutated melanoma in a 3:1 ratio to receive either dabrafenib, a more potent second-generation BRAF inhibitor, or dacarbazine. Half of patients responded to dabrafenib, with a significantly improved progression-free survival rate (5.1 vs 2.7 months respectively), leading to FDA approval for its use in BRAF-mutant melanoma in May 2013.
Adverse effects common to vemurafenib and dabrafenib include rash, fatigue, fever, and joint pain. In addition, up to 25% of patients develop multiple secondary cutaneous squamous cell carcinomas and keratoacanthomas, usually within the first few months of therapy, which are believed to be caused by paradoxical activation of the MAPK pathway.
A more important problem with these medications is the development of resistance. Tumors typically progress again after a median progression-free survival of 6 to 7 months.
MEK inhibitors—another line of defense
Inhibitors of MEK—a serine-threonine kinase that is part of the same MAPK pathway involving BRAF—have been developed as well.
Trametinib. In 2012, trametinib, an allosteric MEK inhibitor, was used in an open-label phase 3 trial in 322 patients with advanced melanoma. Progression-free survival was 4.8 months for trametinib-treated patients compared with 1.5 months for the standard chemotherapy group (dacarbazine or paclitaxel).36 These results led to FDA approval of trametinib in May 2013 for treating BRAF-mutant melanoma.29
Cobimetinib is a second MEK inhibitor being evaluated alone and in combination with other targeted treatments for advanced melanoma.
Both MEK inhibitors have adverse effects similar to those seen with the BRAF inhibitors, including diarrhea, rash, fatigue, and edema. They also tend to cause asymptomatic elevated creatine kinase and transient retinopathy, reduced ejection fraction, and ventricular dysfunction. Unlike BRAF inhibitors, they are not associated with development of secondary cutaneous squamous cell carcinomas or keratoacanthomas. However, as with BRAF inhibitors, resistance is a problem with MEK inhibitors, with most patients relapsing less than a year after starting therapy.
Combination therapy improves outcomes
Possible mechanisms underlying resistance to these medications are being studied. A number of important factors appear to drive resistance, including expression of truncated BRAF proteins that do not bind the BRAF inhibitors and still activate downstream signaling, and amplification of BRAF to such a degree that it overwhelms the medications. This has led to the idea of combining BRAF inhibitors and MEK inhibitors to block the MAPK pathway at two points, potentially increasing the response and decreasing resistance.
Two trials have evaluated combinations of BRAF and MEK inhibitors in patients with advanced melanoma. Larkin et al37 conducted a phase 3 study evaluating combined vemurafenib (a BRAF inhibitor) and cobimetinib (a MEK inhibitor) vs combined vemurafenib and placebo. Survival with the combination therapy was 9.9 months vs 6.2 months with the single treatment.
The incidence of serious adverse effects was not significantly increased with the combination therapy, and keratoacanthomas, cutaneous squamous cell carcinomas, alopecia, and arthralgias were reduced compared with the vemurafenib and placebo group.
Another trial38 evaluating combined dabrafenib (a BRAF inhibitor) and trametinib (a MEK inhibitor) vs combined dabrafenib and placebo had similar findings: increased survival in the combined therapy group (9.3 months vs 8.8 months) and lower rates of squamous cell carcinoma (2% vs 9%).
In January 2014, the FDA approved the combination of BRAF and MEK inhibitors for the treatment of BRAF-mutant metastatic melanoma based on improved survival and generally reduced adverse effects.
IMMUNOTHERAPIES FOR NON-BRAF MELANOMA
Although BRAF and MEK inhibitors represent tremendous advances, their use is limited to the approximately 50% to 65% of patients with advanced melanoma who have BRAF V600 mutations. For others, only the traditional standard medications have been available until recently.
Two of those standard FDA-approved medications, interferon alpha-2b and IL-2, represent immunotherapies. Interferon alpha-2b up-regulates antigen presentation and increases antigen recognition by T cells. Overall, about 20% of patients in clinical trials have achieved responses.
IL-2 is a cytokine that increases T-cell proliferation and maturation into effector T cells. High-dose IL-2 has produced responses in 15% of patients, with a durable complete response in a small proportion.
Though success with these medications was modest, the fact that some patients responded to them indicates that immunotherapy could be a viable strategy for treating metastatic melanoma.30 This is underscored by the fact that some patients can mount an adaptive immune response specifically directed against antigenic proteins expressed in their tumors, resulting in expansion of cytotoxic T cells and control or even elimination of the malignancy.30
Tumors manipulate host immune checkpoints
Molecular biology has provided tremendous insight into tumor immunology over the past several decades, and we now recognize that a hallmark of cancer is escape from immune control.
Cancer cells contain a multitude of mutations that produce proteins that should be recognized by the immune system as foreign but in most individuals are not. This is because T-cell activity is down-regulated in cancer due to cancer cells’ ability to manipulate the host’s normal immunologic inhibitory pathways critical for maintaining self-tolerance.
In general, T-cell activation is initiated when an antigen-presenting cell presents an antigen to a T cell in a major histocompatibility complex-restricted manner. To prevent T cells from being activated by self-antigens and initiating autoimmunity, the interaction between antigen-presenting cells and T cells is regulated by checkpoints (Figure 5). First, for an antigen-presenting cell/T-cell interaction to result in T-cell activation, the T-cell receptor CD28 must bind CD80 on the antigen-presenting cell to drive a “positive” signal. Early in the interaction, the T-cell receptor CTLA-4 is up-regulated and competes with CD28 for binding of CD80. If CTLA-4, and not CD28, binds CD80, a “negative” signal is sent to the T cell and down-regulates it, making the interaction unproductive. Importantly, it is the CTLA-4:CD80 interaction that appears to be crucial for the ability of tumors to dampen T-cell responses to cancer cells.
Ipilimumab is a fully humanized monoclonal antibody that binds to CTLA-4, blocking its ability to bind to CD80 and thereby enhancing T-cell activation. In a phase 3 trial, Hodi et al39 evaluated its use in treating advanced melanoma, with some enrolled patients having failed IL-2 treatment. Patients receiving ipilimumab with or without a glycoprotein-100 peptide vaccine (gp100) had an overall survival benefit of 10.1 months compared with 6.4 months for patients treated with gp100 alone. At 24 months, the survival rate with ipilimumab alone was 23.5%, almost double that of patients receiving gp100 alone.
Ipilimumab received FDA approval for treatment of metastatic melanoma in March 2011. This, and the BRAF inhibitors, were the first drugs approved by the FDA for the treatment of advanced melanoma in more than a decade.
Common adverse effects of ipilimumab include fatigue, diarrhea, rash, and pruritus. As expected, given its mechanism of action, up to about 25% of patients experience severe autoimmune-related events that may variably manifest as colitis, rash, hepatitis, neuritis, hypothyroidism, hypopituitarism, and hypophysitis. Another problem with this medication is that a subset of patients do not respond.
Cancer cells disguised as normal cells
Cancer cells can also manipulate another immunologic checkpoint to evade attack by the host immune system (Figure 5). Cytotoxic T cells may recognize antigens on tumor cells and become activated and primed to directly destroy them. However, tumor cells, like normal cells express the programmed death ligands RTK-L1 and PD-L2. These ligands function to bind to the PD-1 receptor on activated T cells to indicate they are “self” and inhibit the cytotoxic T cells from destroying them.
Evasion of immune system attack by manipulating checkpoints involving CTLA-4 and PD-1 helps explain why malignancies can seemingly be associated with brisk inflammatory responses, such as the tumor in Case 3, yet progress and eventually metastasize (Figure 3).
Two medications—nivolumab and pembrolizumab—have been developed in an attempt to disrupt the ability of tumor cells to trick the immune system into accepting them as “self” by manipulating the PD-L1/PD-L2: PD-1 interaction. Both drugs are monoclonal antibodies that bind to PD-1 and, thus, effectively block the ability of PD-L1 or PD-L2 on tumor cells to bind these ligands and signal to activated T cells that they are “self.” This blocking allows T cells to then carry out their killing of tumor cells they initially recognize as foreign.
Nivolumab. In 2014, a phase 3 trial40 compared nivolumab and dacarbazine in patients with untreated advanced melanoma without a BRAF mutation. Objective response rates were 40.0% in the nivolumab group vs 13.9% in the dacarbazine group. This trial was stopped early because of significantly better survival rates in patients taking nivolumab compared with standard chemotherapy.
Interestingly, only 35% of patients who responded to nivolumab had evidence of PD-L1 expression on the surface of their tumor cells as assessed by immunohistochemical assay. Regardless of PD-L1 status, nivolumab-treated patients had improved overall survival compared with those treated with dacarbazine. The response rate with nivolumab was only slightly better in the subgroup of patients whose tumors expressed PD-L1 than in the subgroup without PD-L1.
On December 22, 2014, the FDA granted accelerated approval to nivolumab for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab treatment and, if BRAF V600 mutation-positive, a BRAF inhibitor.
Pembrolizumab. Also in 2014, an open-label, randomized, phase 1b trial of pembrolizumab treatment at two different dosage schedules was conducted in patients with advanced melanoma that had become refractory either to ipilimumab or a BRAF inhibitor.41 Treatment with pembrolizumab had an objective response rate of 26% at both doses.
In September 2014, the FDA granted accelerated approval for the use of pembrolizumab to treat patients with unresectable or metastatic melanoma and disease progression following treatment with ipilimumab or a BRAF inhibitor.
Adverse effects of PD-1 inhibitors are similar to those seen with ipilimumab, the most common (occurring in at least 20%) being fatigue, cough, nausea, pruritus, rash, decreased appetite, constipation, muscle pain, and diarrhea. Serious effects from pembrolizumab (occurring in at least 2%) were kidney failure, dyspnea, pneumonia, and cellulitis. As seen with ipilimumab, clinically significant autoimmune adverse reactions occur with PD-1 inhibitors, including pneumonitis, colitis, hypophysitis, nephritis, and hepatitis.
Combination therapy under investigation
A phase 1 trial using combination therapy with both immune checkpoint inhibitors—nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4)—in patients with treatment-resistant metastatic melanoma was published in 2013.42 More than half of patients achieved objective responses, with tumor regression of at least 80% in those who had a response. Tumor response was evident in all subgroups of patients studied—those with pretreatment elevated lactate dehydrogenase levels (one of the strongest prognostic factors in metastatic melanoma), metastases to distant sites, and bulky, multifocal tumor burden. Based on these results, a phase 3 trial is now under way looking at the combination of these two medications vs either one alone.
In summary, targeted treatments are changing the paradigm of how common dermatologic conditions associated with significant morbidity and mortality are treated. Although implementation of the above treatments into everyday clinical practice is exciting, future studies surrounding each are needed to address unanswered issues, such as the optimal dosing and treatment schedules in terms of both disease response and inhibition of resistance, optimal patient/disease characteristics for use, and optimal drug treatment combinations. In the meantime, basic research still utilizing classic molecular biology techniques to uncover pathogenic disease mechanisms in even more detail is ongoing and hopefully will lead to development of even better targeted treatments or even cures for these diseases.
- Lyons TG, O’Kane GM, Kelly CM. Efficacy and safety of vismodegib: a new therapeutic agent in the treatment of basal cell carcinoma. Expert Opin Drug Saf 2014; 13:1125–1132.
- McCusker M, Basset-Sequin N, Dummer R, et al. Metastatic basal cell carcinoma: prognosis dependent on anatomic site and spread of disease. Eur J Cancer 2014; 50:774–783.
- Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996; 85:841–851.
- Aszterbaum M, Rothman A, Johnson RL, et al. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J Invest Dermatol 1998; 110:885–888.
- Ingham PW, Placzek M. Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nat Rev Genet 2006; 7:841–850.
- Robarge KD, Brunton SA, Castanedo GM, et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg Med Chem Lett 2009; 19:5576–5581.
- Proctor AE, Thompson LA, O’Bryant CL. Vismodegib: an inhibitor of the Hedgehog signaling pathway in the treatment of basal cell carcinoma. Ann Pharmacother 2014; 48:99–106.
- Dessinioti C, Plaka M, Stratigos AJ. Vismodegib for the treatment of basal cell carcinoma: results and implications of the ERIVANCE BCC trial. Future Oncol 2014; 10:927–936.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med 2012; 366:2171–2179.
- Tang JY, Mackay-Wiggan JM, Aszterbaum M, et al. Inhibiting the hedgehog pathway in patients with basal-cell nevus syndrome. N Engl J Med 2012; 366:2180–2188.
- Brinkhuizen T, Reinders MG, van Geel M, et al. Acquired resistance to the Hedgehog pathway inhibitor vismodegib due to smoothened mutations in treatment of locally advanced basal cell carcinoma. J Am Acad Dermatol 2014; 71:1005–1008.
- Ally MS, Aasi S, Wysong A, et al. An investigator-initiated open-label clinical trial of vismodegib as a neoadjuvant to surgery for high-risk basal cell carcinoma. J Am Acad Dermatol 2014; 71:904–911.
- Rapp SR, Feldman SR, Exum ML, Fleischer AB Jr, Reboussin DM. Psoriasis causes as much disability as other major medical diseases. J Am Acad Dermatol 1999; 41:401–407.
- Gelfand JM, Niemann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. JAMA 2006; 296:1735–1741.
- Lynde CW, Poulin Y, Vender R, Bourcier M, Khalil S. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol 2014; 71:141–150.
- Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther 2008; 117:244–279.
- Nestle FL, Kaplan DH, Barker J. Psoriasis. N Engl J Med 2009; 361:496–509.
- Mentor A, Tyring SK, Gordon K, et al. Adalimumab therapy for moderate to severe psoriasis: a randomized, controlled phase III trial. J Am Acad Dermatol 2007; 58:106–115.
- Leonardi CL, Powers JL, Matheson RT, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med 2003; 349:2014–2022.
- Reich K, Nestle FO, Papp K, et al. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet 2005; 366:1367–1374.
- Leonardi CL, Kimball AB, Papp KA, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008; 371:1665–1674.
- Papp KA, Langley RG, Lebwohl M, et al; PHOENIX 2 study investigators. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet 2008; 371:1675–1684.
- Leonardi CL, Gordon KB. New and emerging therapies in psoriasis. Semin Cut Med Surg 2014; 33(suppl 2):S37–S41.
- Langley RG, Elewski BE, Lebwohl, et al for the ERASURE and FIXTURE Study Groups. Secukinumab in plaque psorisis—results of two phase 3 trials. N Engl J Med 2014; 371:326–338.
- Leonardi C, Matheson R, Zachariae C. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med 2012; 366:1190–1199.
- Papp KA, Leonardi C, Menter A, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med 2012; 366:1181–1189.
- McInnes IB, Sieper J, Braun J, et al. Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: a 24-week, randomised, double-blind, placebo-controlled, phase II proof-of-concept trial. Ann Rheum Dis 2014; 73:349–356.
- Mease PJ, Genovese MC, Greenwald MW, et al. Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. N Engl J Med 2014; 370:2295–2306.
- Girotti MR, Saturno G, Lorigan P, Marais R. No longer an untreatable disease: how targeted and immunotherapies have changed the management of melanoma patients. Molec Oncol 2014, 8:1140–1158.
- Saranga-Perry V, Ambe C, Zager JS, Kudchadkar RR. Recent developments in the medical and surgical treatment of melanoma. CA Canc J Clin 2014; 64:171–185.
- Shah DJ, Dronca RS. Latest advances in chemotherapeutic, targeted, and immune approaches in the treatment of metastatic melanoma. Mayo Clin Proc 2014; 89:504–519.
- Davies H, Ignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417:949–954.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363:809–819.
- Chapman PB, Hauschild A, Robert C. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364:2507–2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012; 380:358–365.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012; 367:107–114.
- Larkin J, Ascierto PA, Dreno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014; 371:1867–1876.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Eng J Med 2014; 371:1877–1888.
- Hodi FS, O’Day SJ, McDermott DF, Weber RW. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711–723.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015; 372:320–330.
- Robert C, Ribas A, Wolchok JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 2014; 384:1109–1117.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013; 369:122–133.
- Lyons TG, O’Kane GM, Kelly CM. Efficacy and safety of vismodegib: a new therapeutic agent in the treatment of basal cell carcinoma. Expert Opin Drug Saf 2014; 13:1125–1132.
- McCusker M, Basset-Sequin N, Dummer R, et al. Metastatic basal cell carcinoma: prognosis dependent on anatomic site and spread of disease. Eur J Cancer 2014; 50:774–783.
- Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996; 85:841–851.
- Aszterbaum M, Rothman A, Johnson RL, et al. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J Invest Dermatol 1998; 110:885–888.
- Ingham PW, Placzek M. Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nat Rev Genet 2006; 7:841–850.
- Robarge KD, Brunton SA, Castanedo GM, et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg Med Chem Lett 2009; 19:5576–5581.
- Proctor AE, Thompson LA, O’Bryant CL. Vismodegib: an inhibitor of the Hedgehog signaling pathway in the treatment of basal cell carcinoma. Ann Pharmacother 2014; 48:99–106.
- Dessinioti C, Plaka M, Stratigos AJ. Vismodegib for the treatment of basal cell carcinoma: results and implications of the ERIVANCE BCC trial. Future Oncol 2014; 10:927–936.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med 2012; 366:2171–2179.
- Tang JY, Mackay-Wiggan JM, Aszterbaum M, et al. Inhibiting the hedgehog pathway in patients with basal-cell nevus syndrome. N Engl J Med 2012; 366:2180–2188.
- Brinkhuizen T, Reinders MG, van Geel M, et al. Acquired resistance to the Hedgehog pathway inhibitor vismodegib due to smoothened mutations in treatment of locally advanced basal cell carcinoma. J Am Acad Dermatol 2014; 71:1005–1008.
- Ally MS, Aasi S, Wysong A, et al. An investigator-initiated open-label clinical trial of vismodegib as a neoadjuvant to surgery for high-risk basal cell carcinoma. J Am Acad Dermatol 2014; 71:904–911.
- Rapp SR, Feldman SR, Exum ML, Fleischer AB Jr, Reboussin DM. Psoriasis causes as much disability as other major medical diseases. J Am Acad Dermatol 1999; 41:401–407.
- Gelfand JM, Niemann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. JAMA 2006; 296:1735–1741.
- Lynde CW, Poulin Y, Vender R, Bourcier M, Khalil S. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol 2014; 71:141–150.
- Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther 2008; 117:244–279.
- Nestle FL, Kaplan DH, Barker J. Psoriasis. N Engl J Med 2009; 361:496–509.
- Mentor A, Tyring SK, Gordon K, et al. Adalimumab therapy for moderate to severe psoriasis: a randomized, controlled phase III trial. J Am Acad Dermatol 2007; 58:106–115.
- Leonardi CL, Powers JL, Matheson RT, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med 2003; 349:2014–2022.
- Reich K, Nestle FO, Papp K, et al. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet 2005; 366:1367–1374.
- Leonardi CL, Kimball AB, Papp KA, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008; 371:1665–1674.
- Papp KA, Langley RG, Lebwohl M, et al; PHOENIX 2 study investigators. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet 2008; 371:1675–1684.
- Leonardi CL, Gordon KB. New and emerging therapies in psoriasis. Semin Cut Med Surg 2014; 33(suppl 2):S37–S41.
- Langley RG, Elewski BE, Lebwohl, et al for the ERASURE and FIXTURE Study Groups. Secukinumab in plaque psorisis—results of two phase 3 trials. N Engl J Med 2014; 371:326–338.
- Leonardi C, Matheson R, Zachariae C. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med 2012; 366:1190–1199.
- Papp KA, Leonardi C, Menter A, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med 2012; 366:1181–1189.
- McInnes IB, Sieper J, Braun J, et al. Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: a 24-week, randomised, double-blind, placebo-controlled, phase II proof-of-concept trial. Ann Rheum Dis 2014; 73:349–356.
- Mease PJ, Genovese MC, Greenwald MW, et al. Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. N Engl J Med 2014; 370:2295–2306.
- Girotti MR, Saturno G, Lorigan P, Marais R. No longer an untreatable disease: how targeted and immunotherapies have changed the management of melanoma patients. Molec Oncol 2014, 8:1140–1158.
- Saranga-Perry V, Ambe C, Zager JS, Kudchadkar RR. Recent developments in the medical and surgical treatment of melanoma. CA Canc J Clin 2014; 64:171–185.
- Shah DJ, Dronca RS. Latest advances in chemotherapeutic, targeted, and immune approaches in the treatment of metastatic melanoma. Mayo Clin Proc 2014; 89:504–519.
- Davies H, Ignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417:949–954.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363:809–819.
- Chapman PB, Hauschild A, Robert C. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364:2507–2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012; 380:358–365.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012; 367:107–114.
- Larkin J, Ascierto PA, Dreno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014; 371:1867–1876.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Eng J Med 2014; 371:1877–1888.
- Hodi FS, O’Day SJ, McDermott DF, Weber RW. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711–723.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015; 372:320–330.
- Robert C, Ribas A, Wolchok JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 2014; 384:1109–1117.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013; 369:122–133.
KEY POINTS
- Vismodegib, an inhibitor of the “hedgehog” pathway, dramatically shrinks basal cell carcinomas, but resistance and adverse effects remain troublesome. Using it to shrink tumors to operable size may be its best future role.
- Th-17 cells and interleukin 17 are now thought to play central roles in the pathogenesis of psoriasis. Clinical trials of new drugs that block interleukin 17 show striking improvement in skin manifestations with few side effects. Benefits in psoriatic arthritis have not yet been shown.
- About half of patients with melanoma harbor BRAF mutations, and new treatments that target this pathway have improved survival rates. For melanoma not involving BRAF mutations, a better understanding of how tumors evade immune control has led to improved immunotherapies. These targeted medications mark the first major advancements in metastatic melanoma treatment in decades.