User login
Overlapping Phenotypic Features of PTEN Hamartoma Tumor Syndrome and Birt-Hogg-Dubé Syndrome
To the Editor:
PTEN hamartoma tumor syndrome (PHTS) encompasses a spectrum of disorders that most commonly are caused by autosomal-dominant germline mutations in the phosphatase and tensin homolog, PTEN, tumor suppressor gene on chromosome 10q23. We describe a patient who presented with clinical features of PHTS and Birt-Hogg-Dubé syndrome (BHDS). Because the genetic mutations associated with both PHTS and BHDS result in altered mammalian target of rapamycin (mTOR) signaling, patients may have overlapping phenotypic features.
A 51-year-old man with a history of multiple carcinomas presented for evaluation of flesh-colored papules on the cheeks, nose, tongue, and hands, in addition to numerous skin tags on the neck, axillae, and lower abdomen bilaterally. His medical history was notable for several nasal and gastrointestinal tract polyps, chromophobe renal cell carcinoma, cutaneous lipomas, atypical carcinoid syndrome of the right lung, and a multinodular thyroid. His family history was notable for small cell lung cancer in his father, breast cancer and pancreatic cancer in his maternal aunt, esophageal cancer in his maternal grandfather, and celiac disease in his daughter.

Clinical examination revealed flesh-colored, dome-shaped papules measuring 1 to 2 mm in diameter on the nose and cheeks (Figure 1). He had hyperkeratotic papules on the dorsal fingers, consistent with acral keratoses. Additionally, multiple flesh-colored papules with a cobblestonelike appearance were noted on the oral mucosa (Figure 2). Other findings included pedunculated papules on the neck, axillae, and lower abdomen bilaterally, consistent with fibroepithelial polyps, as well as hyperpigmented velvety plaques on the axillae, characteristic of acanthosis nigricans (Figure 3). A shave biopsy of a papule on the right cheek revealed a proliferation of plump stellate fibroblasts, small blood vessels, and thick collagen bundles, characteristic of a fibrous papule (Figure 4).

Differential diagnoses for our patient included BHDS and Cowden syndrome (CS). Due to the combination of extensive family history of multiorgan cancers as well as the clinical findings, he was referred to a geneticist for further evaluation. Genetic analysis was positive for a heterozygous mutation variant of uncertain significance in the PTEN gene.

The PHTS disorders include CS, Bannayan-Riley-Ruvalcaba syndrome, Lhermitte-Duclos disease, Proteus syndrome, and Proteus-like syndrome (Table).1-9 Our patient’s clinical findings were indicative of CS, a rare genodermatosis characterized by multiple hamartomas and neoplasms of ectodermal, mesodermal, and endodermal origin.1 Most CS patients develop trichilemmomas of the central face, mucocutaneous papillomatous papules, and acral and plantar keratoses by the third decade of life.1 Importantly, CS patients have an increased risk for breast, thyroid, renal, endometrial, and colorectal cancers, as well as melanoma, with estimated lifetime risks of 85%, 35%, 33%, 28%, 9%, and 6%, respectively.2,10

Regarding the pathophysiology of PHTS disorders, PTEN encodes a phosphatase that inhibits phosphoinositide 3-kinase/Akt and mTOR signaling pathways, thereby controlling cell proliferation, cell-cycle progression, and apoptosis.2,3 Loss of PTEN function, as seen in CS patients, results in an increased risk for cancer.2 Other genetic diseases, including juvenile polyposis syndrome, Proteus syndrome, tuberous sclerosis, and Peutz-Jeghers syndrome, have phenotypic similarities to PHTS.3 Specifically, loss-of-function mutations of TSC1 and TSC2, tumor suppressor genes associated with tuberous sclerosis, similarly result in dysregulation of mTOR signaling.

Our patient also had some clinical features characteristic of BHDS, such as flesh-colored facial papules, acrochordonlike lesions, and chromophobe renal cell carcinoma.11 Birt-Hogg-Dubé syndrome most often is caused by an autosomal-dominant germline mutation in FLCN, a tumor suppressor gene.11 Interestingly, FLCN interacts with AMP-activated protein kinase to help regulate mTOR signaling, which may explain phenotypic similarities seen in CS and BHDS.12
Because the PHTS disorders and BHDS result in similar functional consequences on the mTOR signaling pathway, patients can present with overlapping clinical features that may be diagnostically challenging. Management includes patient education regarding cancer risk, surveillance for early detection of malignancy, and genetic counseling for family members.2 It is important for clinicians to appreciate phenotypic similarities between PHTS and other disorders affecting mTOR signaling to prevent delays in diagnosis.
- Nosé V. Genodermatosis affecting the skin and mucosa of the head and neck: clinicopathologic, genetic, and molecular aspect—PTEN-hamartoma tumor syndrome/Cowden syndrome. Head Neck Pathol. 2016;10:131-138.
- Porto A, Roider E, Ruzicka T. Cowden syndrome: report of a case and brief review of literature. An Bras Dermatol. 2013;88(6 suppl 1):S52-S55.
- Leslie N, Longy M. Inherited PTEN mutations and the prediction of phenotype. Semin Cell Dev Biol. 2016;52:30-38.
- The National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology. genetic/familial high-risk assessment: breast and ovarian (version 1.2017). Published September 19, 2016. Accessed August 11, 2021. https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf
- Laury AR, Bongiovanni M, Tille J, et al. Thyroid pathology in PTEN-hamartoma tumor syndrome: characteristic findings of a distinct entity. Thyroid. 2011;21:135-144.
- Eng C. PTEN hamartoma tumor syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. University of Washington; 2001.
- Golden N, Tjokorda MGB, Sri M, et al. Management of unusual dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) in a developing country: case report and review of the literature. Asian J Neurosurg. 2016;11:170.
- Biesecker LG, Happle R, Mulliken JB, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84:389-395.
- Busa T, Milh M, Degardin N, et al. Clinical presentation of PTEN mutations in childhood in the absence of family history of Cowden syndrome. Eur J Paediatr Neurol. 2015;19:188-192.
- Tan MH, Mester JL, Ngeow J, et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400-407.
- Ponti G, Pellacani G, Seidenari S, et al. Cancer-associated genodermatoses: skin neoplasms as clues to hereditary tumor syndromes. Crit Rev Oncol Hematol. 2013;85:239-256.
- Baba M, Hong S, Sharma N, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006;103:15552-15557.
To the Editor:
PTEN hamartoma tumor syndrome (PHTS) encompasses a spectrum of disorders that most commonly are caused by autosomal-dominant germline mutations in the phosphatase and tensin homolog, PTEN, tumor suppressor gene on chromosome 10q23. We describe a patient who presented with clinical features of PHTS and Birt-Hogg-Dubé syndrome (BHDS). Because the genetic mutations associated with both PHTS and BHDS result in altered mammalian target of rapamycin (mTOR) signaling, patients may have overlapping phenotypic features.
A 51-year-old man with a history of multiple carcinomas presented for evaluation of flesh-colored papules on the cheeks, nose, tongue, and hands, in addition to numerous skin tags on the neck, axillae, and lower abdomen bilaterally. His medical history was notable for several nasal and gastrointestinal tract polyps, chromophobe renal cell carcinoma, cutaneous lipomas, atypical carcinoid syndrome of the right lung, and a multinodular thyroid. His family history was notable for small cell lung cancer in his father, breast cancer and pancreatic cancer in his maternal aunt, esophageal cancer in his maternal grandfather, and celiac disease in his daughter.
Clinical examination revealed flesh-colored, dome-shaped papules measuring 1 to 2 mm in diameter on the nose and cheeks (Figure 1). He had hyperkeratotic papules on the dorsal fingers, consistent with acral keratoses. Additionally, multiple flesh-colored papules with a cobblestonelike appearance were noted on the oral mucosa (Figure 2). Other findings included pedunculated papules on the neck, axillae, and lower abdomen bilaterally, consistent with fibroepithelial polyps, as well as hyperpigmented velvety plaques on the axillae, characteristic of acanthosis nigricans (Figure 3). A shave biopsy of a papule on the right cheek revealed a proliferation of plump stellate fibroblasts, small blood vessels, and thick collagen bundles, characteristic of a fibrous papule (Figure 4).
Differential diagnoses for our patient included BHDS and Cowden syndrome (CS). Due to the combination of extensive family history of multiorgan cancers as well as the clinical findings, he was referred to a geneticist for further evaluation. Genetic analysis was positive for a heterozygous mutation variant of uncertain significance in the PTEN gene.
The PHTS disorders include CS, Bannayan-Riley-Ruvalcaba syndrome, Lhermitte-Duclos disease, Proteus syndrome, and Proteus-like syndrome (Table).1-9 Our patient’s clinical findings were indicative of CS, a rare genodermatosis characterized by multiple hamartomas and neoplasms of ectodermal, mesodermal, and endodermal origin.1 Most CS patients develop trichilemmomas of the central face, mucocutaneous papillomatous papules, and acral and plantar keratoses by the third decade of life.1 Importantly, CS patients have an increased risk for breast, thyroid, renal, endometrial, and colorectal cancers, as well as melanoma, with estimated lifetime risks of 85%, 35%, 33%, 28%, 9%, and 6%, respectively.2,10
Regarding the pathophysiology of PHTS disorders, PTEN encodes a phosphatase that inhibits phosphoinositide 3-kinase/Akt and mTOR signaling pathways, thereby controlling cell proliferation, cell-cycle progression, and apoptosis.2,3 Loss of PTEN function, as seen in CS patients, results in an increased risk for cancer.2 Other genetic diseases, including juvenile polyposis syndrome, Proteus syndrome, tuberous sclerosis, and Peutz-Jeghers syndrome, have phenotypic similarities to PHTS.3 Specifically, loss-of-function mutations of TSC1 and TSC2, tumor suppressor genes associated with tuberous sclerosis, similarly result in dysregulation of mTOR signaling.
Our patient also had some clinical features characteristic of BHDS, such as flesh-colored facial papules, acrochordonlike lesions, and chromophobe renal cell carcinoma.11 Birt-Hogg-Dubé syndrome most often is caused by an autosomal-dominant germline mutation in FLCN, a tumor suppressor gene.11 Interestingly, FLCN interacts with AMP-activated protein kinase to help regulate mTOR signaling, which may explain phenotypic similarities seen in CS and BHDS.12
Because the PHTS disorders and BHDS result in similar functional consequences on the mTOR signaling pathway, patients can present with overlapping clinical features that may be diagnostically challenging. Management includes patient education regarding cancer risk, surveillance for early detection of malignancy, and genetic counseling for family members.2 It is important for clinicians to appreciate phenotypic similarities between PHTS and other disorders affecting mTOR signaling to prevent delays in diagnosis.
To the Editor:
PTEN hamartoma tumor syndrome (PHTS) encompasses a spectrum of disorders that most commonly are caused by autosomal-dominant germline mutations in the phosphatase and tensin homolog, PTEN, tumor suppressor gene on chromosome 10q23. We describe a patient who presented with clinical features of PHTS and Birt-Hogg-Dubé syndrome (BHDS). Because the genetic mutations associated with both PHTS and BHDS result in altered mammalian target of rapamycin (mTOR) signaling, patients may have overlapping phenotypic features.
A 51-year-old man with a history of multiple carcinomas presented for evaluation of flesh-colored papules on the cheeks, nose, tongue, and hands, in addition to numerous skin tags on the neck, axillae, and lower abdomen bilaterally. His medical history was notable for several nasal and gastrointestinal tract polyps, chromophobe renal cell carcinoma, cutaneous lipomas, atypical carcinoid syndrome of the right lung, and a multinodular thyroid. His family history was notable for small cell lung cancer in his father, breast cancer and pancreatic cancer in his maternal aunt, esophageal cancer in his maternal grandfather, and celiac disease in his daughter.
Clinical examination revealed flesh-colored, dome-shaped papules measuring 1 to 2 mm in diameter on the nose and cheeks (Figure 1). He had hyperkeratotic papules on the dorsal fingers, consistent with acral keratoses. Additionally, multiple flesh-colored papules with a cobblestonelike appearance were noted on the oral mucosa (Figure 2). Other findings included pedunculated papules on the neck, axillae, and lower abdomen bilaterally, consistent with fibroepithelial polyps, as well as hyperpigmented velvety plaques on the axillae, characteristic of acanthosis nigricans (Figure 3). A shave biopsy of a papule on the right cheek revealed a proliferation of plump stellate fibroblasts, small blood vessels, and thick collagen bundles, characteristic of a fibrous papule (Figure 4).
Differential diagnoses for our patient included BHDS and Cowden syndrome (CS). Due to the combination of extensive family history of multiorgan cancers as well as the clinical findings, he was referred to a geneticist for further evaluation. Genetic analysis was positive for a heterozygous mutation variant of uncertain significance in the PTEN gene.
The PHTS disorders include CS, Bannayan-Riley-Ruvalcaba syndrome, Lhermitte-Duclos disease, Proteus syndrome, and Proteus-like syndrome (Table).1-9 Our patient’s clinical findings were indicative of CS, a rare genodermatosis characterized by multiple hamartomas and neoplasms of ectodermal, mesodermal, and endodermal origin.1 Most CS patients develop trichilemmomas of the central face, mucocutaneous papillomatous papules, and acral and plantar keratoses by the third decade of life.1 Importantly, CS patients have an increased risk for breast, thyroid, renal, endometrial, and colorectal cancers, as well as melanoma, with estimated lifetime risks of 85%, 35%, 33%, 28%, 9%, and 6%, respectively.2,10
Regarding the pathophysiology of PHTS disorders, PTEN encodes a phosphatase that inhibits phosphoinositide 3-kinase/Akt and mTOR signaling pathways, thereby controlling cell proliferation, cell-cycle progression, and apoptosis.2,3 Loss of PTEN function, as seen in CS patients, results in an increased risk for cancer.2 Other genetic diseases, including juvenile polyposis syndrome, Proteus syndrome, tuberous sclerosis, and Peutz-Jeghers syndrome, have phenotypic similarities to PHTS.3 Specifically, loss-of-function mutations of TSC1 and TSC2, tumor suppressor genes associated with tuberous sclerosis, similarly result in dysregulation of mTOR signaling.
Our patient also had some clinical features characteristic of BHDS, such as flesh-colored facial papules, acrochordonlike lesions, and chromophobe renal cell carcinoma.11 Birt-Hogg-Dubé syndrome most often is caused by an autosomal-dominant germline mutation in FLCN, a tumor suppressor gene.11 Interestingly, FLCN interacts with AMP-activated protein kinase to help regulate mTOR signaling, which may explain phenotypic similarities seen in CS and BHDS.12
Because the PHTS disorders and BHDS result in similar functional consequences on the mTOR signaling pathway, patients can present with overlapping clinical features that may be diagnostically challenging. Management includes patient education regarding cancer risk, surveillance for early detection of malignancy, and genetic counseling for family members.2 It is important for clinicians to appreciate phenotypic similarities between PHTS and other disorders affecting mTOR signaling to prevent delays in diagnosis.
- Nosé V. Genodermatosis affecting the skin and mucosa of the head and neck: clinicopathologic, genetic, and molecular aspect—PTEN-hamartoma tumor syndrome/Cowden syndrome. Head Neck Pathol. 2016;10:131-138.
- Porto A, Roider E, Ruzicka T. Cowden syndrome: report of a case and brief review of literature. An Bras Dermatol. 2013;88(6 suppl 1):S52-S55.
- Leslie N, Longy M. Inherited PTEN mutations and the prediction of phenotype. Semin Cell Dev Biol. 2016;52:30-38.
- The National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology. genetic/familial high-risk assessment: breast and ovarian (version 1.2017). Published September 19, 2016. Accessed August 11, 2021. https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf
- Laury AR, Bongiovanni M, Tille J, et al. Thyroid pathology in PTEN-hamartoma tumor syndrome: characteristic findings of a distinct entity. Thyroid. 2011;21:135-144.
- Eng C. PTEN hamartoma tumor syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. University of Washington; 2001.
- Golden N, Tjokorda MGB, Sri M, et al. Management of unusual dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) in a developing country: case report and review of the literature. Asian J Neurosurg. 2016;11:170.
- Biesecker LG, Happle R, Mulliken JB, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84:389-395.
- Busa T, Milh M, Degardin N, et al. Clinical presentation of PTEN mutations in childhood in the absence of family history of Cowden syndrome. Eur J Paediatr Neurol. 2015;19:188-192.
- Tan MH, Mester JL, Ngeow J, et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400-407.
- Ponti G, Pellacani G, Seidenari S, et al. Cancer-associated genodermatoses: skin neoplasms as clues to hereditary tumor syndromes. Crit Rev Oncol Hematol. 2013;85:239-256.
- Baba M, Hong S, Sharma N, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006;103:15552-15557.
- Nosé V. Genodermatosis affecting the skin and mucosa of the head and neck: clinicopathologic, genetic, and molecular aspect—PTEN-hamartoma tumor syndrome/Cowden syndrome. Head Neck Pathol. 2016;10:131-138.
- Porto A, Roider E, Ruzicka T. Cowden syndrome: report of a case and brief review of literature. An Bras Dermatol. 2013;88(6 suppl 1):S52-S55.
- Leslie N, Longy M. Inherited PTEN mutations and the prediction of phenotype. Semin Cell Dev Biol. 2016;52:30-38.
- The National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology. genetic/familial high-risk assessment: breast and ovarian (version 1.2017). Published September 19, 2016. Accessed August 11, 2021. https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf
- Laury AR, Bongiovanni M, Tille J, et al. Thyroid pathology in PTEN-hamartoma tumor syndrome: characteristic findings of a distinct entity. Thyroid. 2011;21:135-144.
- Eng C. PTEN hamartoma tumor syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. University of Washington; 2001.
- Golden N, Tjokorda MGB, Sri M, et al. Management of unusual dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) in a developing country: case report and review of the literature. Asian J Neurosurg. 2016;11:170.
- Biesecker LG, Happle R, Mulliken JB, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84:389-395.
- Busa T, Milh M, Degardin N, et al. Clinical presentation of PTEN mutations in childhood in the absence of family history of Cowden syndrome. Eur J Paediatr Neurol. 2015;19:188-192.
- Tan MH, Mester JL, Ngeow J, et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400-407.
- Ponti G, Pellacani G, Seidenari S, et al. Cancer-associated genodermatoses: skin neoplasms as clues to hereditary tumor syndromes. Crit Rev Oncol Hematol. 2013;85:239-256.
- Baba M, Hong S, Sharma N, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006;103:15552-15557.
PRACTICE POINTS
- PTEN hamartoma tumor syndrome (PHTS) represents a spectrum of disorders caused by autosomal-dominant germline mutations in PTEN.
- Our patient presented with phenotypic features of PHTS and Birt-Hogg-Dubé syndrome. Given that both syndromes cause alterations in mammalian target of rapamycin signaling, overlapping phenotypic features may be seen.
- Recognizing overlapping phenotypic features of these syndromes will allow for timely diagnosis and surveillance for malignancy.
Pediatric-Onset Refractory Lupus Erythematosus Panniculitis Treated With Rituximab
To the Editor:
Lupus erythematosus panniculitis (LEP) is rare in the pediatric population. It can be difficult to manage, as patients may not respond to conventional treatments including hydroxychloroquine and prednisone. We report the use of rituximab in the treatment of a 20-year-old woman with LEP of the face, legs, and arms that was refractory to standard treatments. She also had a history of hemophagocytic lymphohistiocytosis (HLH). Further studies are warranted to determine the role of rituximab in the treatment of pediatric patients with LEP.
A 20-year-old woman with history of LEP and HLH initially presented with migratory violaceous nodules on the face 16 years prior to the current presentation. A skin biopsy 3 years after that initial presentation suggested a diagnosis of cutaneous lupus erythematosus. Six years later, numerous asymptomatic lesions appeared on the legs, predominantly on the calves; she was successfully treated with hydroxychloroquine and high-dose prednisone. Four years prior to the current presentation, a febrile illness prompted discontinuation of hydroxychloroquine and hospitalization, where she was first was diagnosed with HLH; she achieved remission with cyclosporine. At the current presentation, she continued to have persistent violaceous lesions on the face, lower arms, and legs with underlying nodularity (Figure 1). Skin biopsies revealed LEP and were less suggestive of HLH. She was restarted on hydroxychloroquine, which did not adequately control the disease. Rheumatologic workup was only notable for an antinuclear antibody titer of 1:80 (reference range, <1:80) in a speckled pattern.
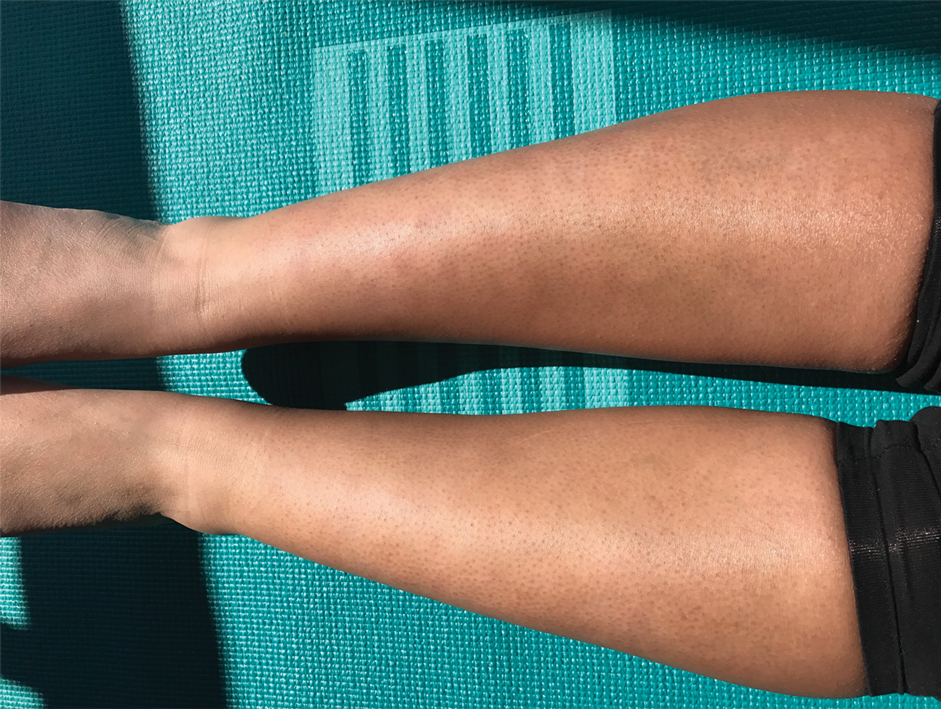
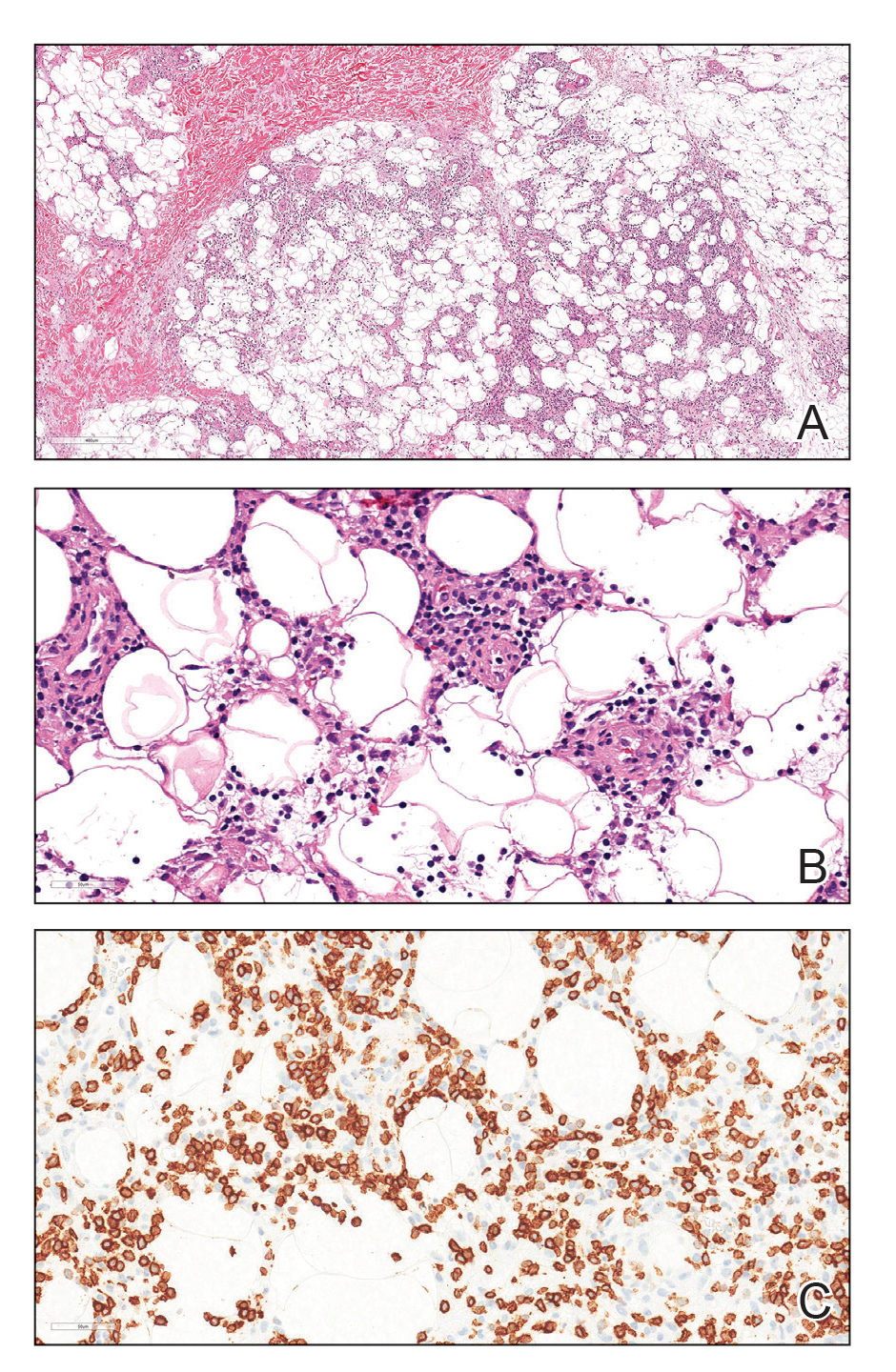
Due to the refractory nature of her condition, continued lesion development despite standard treatment, and concerns of possible scarring, we considered a trial of rituximab. Because HLH and LEP can mimic subcutaneous T-cell lymphoma, another skin biopsy was performed, which revealed a deep dermal and subcutaneous lymphohistiocytic infiltrate composed of predominantly CD3+ T cells with a mixed population of CD4+ and CD8+ cells (Figure 2). There was no evidence of transformation into lymphoma. Pathologic findings were most compatible with LEP rather than an HLH-associated panniculitis due to the lack of definitive phagocytosis. She received rituximab using body surface area–based dosing at 375 mg/m2. CD19 levels decreased to undetectable levels after the first dose. Rituximab was dosed based on clinical response; she tolerated treatment well and experienced considerable improvement in the number of lesions following completion of 4 doses at weeks 0, 1, 5, and 7 (Figure 3). She developed a flare at 7 months and improved again after another dose of rituximab.

Lupus erythematosus panniculitis is a rare variant of lupus erythematosus with an average age of presentation between 30 and 60 years.1 In children, LEP presents as recurrent subcutaneous nodules and plaques, commonly involving the face and upper arms.1,2 Long-term sequelae include local swelling and skin atrophy.3 Conventional treatment options for pediatric patients include hydroxychloroquine and corticosteroids.1 Management can be challenging due to the lack of response to conventional treatments as well as the chronic progressive nature of LEP.2 In refractory cases, cyclosporine, azathioprine, sulfones, thalidomide, mycophenolate mofetil, and cyclophosphamide are alternative treatment options.1-4
Rituximab, a chimeric monoclonal antibody targeting B-cell surface marker CD20, results in depletion of mature B cells. Use of rituximab for LEP has been described in multiple case reports involving an 8-year-old boy, 22-year-old girl, and 2 middle-aged women.2-4 In addition, a recently published case series of 4 patients with childhood-onset refractory LEP described improvement of disease activity with rituximab.5 It is important to rule out subcutaneous T-cell lymphoma before treatment with rituximab, as its histopathology can closely resemble that seen in LEP and HLH-associated cytophagic histiocytic panniculitis.1,6
Rituximab may be an effective treatment option in pediatric patients with refractory LEP. Larger studies on the use of rituximab in the pediatric population are necessary.
- Weingartner JS, Zedek DC, Burkhart CN, et al. Lupus erythematosus panniculitis in children: report of three cases and review of previously reported cases. Pediatr Dermatol. 2011;29:169-176.
- Moreno-Suárez F, Pulpillo-Ruiz Á. Rituximab for the treatment of lupus erythematosus panniculitis. Dermatol Ther. 2013;26:415-418.
- Guissa VR, Trudes G, Jesus AA, et al. Lupus erythematosus panniculitis in children and adolescents. Acta Reumatol Port. 2012;37:82-85.
- Mcardle A, Baker JF. A case of “refractory” lupus erythematosus profundus responsive to rituximab. Clin Rheumatol. 2009;28:745-746.
- Correll CK, Miller DD, Maguiness SM. Treatment of childhood-onset lupus erythematosus panniculitis with rituximab. JAMA Dermatol. 2020;156:566-569.
- Aronson IK, Worobec SM. Cytophagic histiocytic panniculitis and hemophagocytic lymphohistiocytosis: an overview. Dermatol Ther. 2010;23:389-402.
To the Editor:
Lupus erythematosus panniculitis (LEP) is rare in the pediatric population. It can be difficult to manage, as patients may not respond to conventional treatments including hydroxychloroquine and prednisone. We report the use of rituximab in the treatment of a 20-year-old woman with LEP of the face, legs, and arms that was refractory to standard treatments. She also had a history of hemophagocytic lymphohistiocytosis (HLH). Further studies are warranted to determine the role of rituximab in the treatment of pediatric patients with LEP.
A 20-year-old woman with history of LEP and HLH initially presented with migratory violaceous nodules on the face 16 years prior to the current presentation. A skin biopsy 3 years after that initial presentation suggested a diagnosis of cutaneous lupus erythematosus. Six years later, numerous asymptomatic lesions appeared on the legs, predominantly on the calves; she was successfully treated with hydroxychloroquine and high-dose prednisone. Four years prior to the current presentation, a febrile illness prompted discontinuation of hydroxychloroquine and hospitalization, where she was first was diagnosed with HLH; she achieved remission with cyclosporine. At the current presentation, she continued to have persistent violaceous lesions on the face, lower arms, and legs with underlying nodularity (Figure 1). Skin biopsies revealed LEP and were less suggestive of HLH. She was restarted on hydroxychloroquine, which did not adequately control the disease. Rheumatologic workup was only notable for an antinuclear antibody titer of 1:80 (reference range, <1:80) in a speckled pattern.
Due to the refractory nature of her condition, continued lesion development despite standard treatment, and concerns of possible scarring, we considered a trial of rituximab. Because HLH and LEP can mimic subcutaneous T-cell lymphoma, another skin biopsy was performed, which revealed a deep dermal and subcutaneous lymphohistiocytic infiltrate composed of predominantly CD3+ T cells with a mixed population of CD4+ and CD8+ cells (Figure 2). There was no evidence of transformation into lymphoma. Pathologic findings were most compatible with LEP rather than an HLH-associated panniculitis due to the lack of definitive phagocytosis. She received rituximab using body surface area–based dosing at 375 mg/m2. CD19 levels decreased to undetectable levels after the first dose. Rituximab was dosed based on clinical response; she tolerated treatment well and experienced considerable improvement in the number of lesions following completion of 4 doses at weeks 0, 1, 5, and 7 (Figure 3). She developed a flare at 7 months and improved again after another dose of rituximab.
Lupus erythematosus panniculitis is a rare variant of lupus erythematosus with an average age of presentation between 30 and 60 years.1 In children, LEP presents as recurrent subcutaneous nodules and plaques, commonly involving the face and upper arms.1,2 Long-term sequelae include local swelling and skin atrophy.3 Conventional treatment options for pediatric patients include hydroxychloroquine and corticosteroids.1 Management can be challenging due to the lack of response to conventional treatments as well as the chronic progressive nature of LEP.2 In refractory cases, cyclosporine, azathioprine, sulfones, thalidomide, mycophenolate mofetil, and cyclophosphamide are alternative treatment options.1-4
Rituximab, a chimeric monoclonal antibody targeting B-cell surface marker CD20, results in depletion of mature B cells. Use of rituximab for LEP has been described in multiple case reports involving an 8-year-old boy, 22-year-old girl, and 2 middle-aged women.2-4 In addition, a recently published case series of 4 patients with childhood-onset refractory LEP described improvement of disease activity with rituximab.5 It is important to rule out subcutaneous T-cell lymphoma before treatment with rituximab, as its histopathology can closely resemble that seen in LEP and HLH-associated cytophagic histiocytic panniculitis.1,6
Rituximab may be an effective treatment option in pediatric patients with refractory LEP. Larger studies on the use of rituximab in the pediatric population are necessary.
To the Editor:
Lupus erythematosus panniculitis (LEP) is rare in the pediatric population. It can be difficult to manage, as patients may not respond to conventional treatments including hydroxychloroquine and prednisone. We report the use of rituximab in the treatment of a 20-year-old woman with LEP of the face, legs, and arms that was refractory to standard treatments. She also had a history of hemophagocytic lymphohistiocytosis (HLH). Further studies are warranted to determine the role of rituximab in the treatment of pediatric patients with LEP.
A 20-year-old woman with history of LEP and HLH initially presented with migratory violaceous nodules on the face 16 years prior to the current presentation. A skin biopsy 3 years after that initial presentation suggested a diagnosis of cutaneous lupus erythematosus. Six years later, numerous asymptomatic lesions appeared on the legs, predominantly on the calves; she was successfully treated with hydroxychloroquine and high-dose prednisone. Four years prior to the current presentation, a febrile illness prompted discontinuation of hydroxychloroquine and hospitalization, where she was first was diagnosed with HLH; she achieved remission with cyclosporine. At the current presentation, she continued to have persistent violaceous lesions on the face, lower arms, and legs with underlying nodularity (Figure 1). Skin biopsies revealed LEP and were less suggestive of HLH. She was restarted on hydroxychloroquine, which did not adequately control the disease. Rheumatologic workup was only notable for an antinuclear antibody titer of 1:80 (reference range, <1:80) in a speckled pattern.
Due to the refractory nature of her condition, continued lesion development despite standard treatment, and concerns of possible scarring, we considered a trial of rituximab. Because HLH and LEP can mimic subcutaneous T-cell lymphoma, another skin biopsy was performed, which revealed a deep dermal and subcutaneous lymphohistiocytic infiltrate composed of predominantly CD3+ T cells with a mixed population of CD4+ and CD8+ cells (Figure 2). There was no evidence of transformation into lymphoma. Pathologic findings were most compatible with LEP rather than an HLH-associated panniculitis due to the lack of definitive phagocytosis. She received rituximab using body surface area–based dosing at 375 mg/m2. CD19 levels decreased to undetectable levels after the first dose. Rituximab was dosed based on clinical response; she tolerated treatment well and experienced considerable improvement in the number of lesions following completion of 4 doses at weeks 0, 1, 5, and 7 (Figure 3). She developed a flare at 7 months and improved again after another dose of rituximab.
Lupus erythematosus panniculitis is a rare variant of lupus erythematosus with an average age of presentation between 30 and 60 years.1 In children, LEP presents as recurrent subcutaneous nodules and plaques, commonly involving the face and upper arms.1,2 Long-term sequelae include local swelling and skin atrophy.3 Conventional treatment options for pediatric patients include hydroxychloroquine and corticosteroids.1 Management can be challenging due to the lack of response to conventional treatments as well as the chronic progressive nature of LEP.2 In refractory cases, cyclosporine, azathioprine, sulfones, thalidomide, mycophenolate mofetil, and cyclophosphamide are alternative treatment options.1-4
Rituximab, a chimeric monoclonal antibody targeting B-cell surface marker CD20, results in depletion of mature B cells. Use of rituximab for LEP has been described in multiple case reports involving an 8-year-old boy, 22-year-old girl, and 2 middle-aged women.2-4 In addition, a recently published case series of 4 patients with childhood-onset refractory LEP described improvement of disease activity with rituximab.5 It is important to rule out subcutaneous T-cell lymphoma before treatment with rituximab, as its histopathology can closely resemble that seen in LEP and HLH-associated cytophagic histiocytic panniculitis.1,6
Rituximab may be an effective treatment option in pediatric patients with refractory LEP. Larger studies on the use of rituximab in the pediatric population are necessary.
- Weingartner JS, Zedek DC, Burkhart CN, et al. Lupus erythematosus panniculitis in children: report of three cases and review of previously reported cases. Pediatr Dermatol. 2011;29:169-176.
- Moreno-Suárez F, Pulpillo-Ruiz Á. Rituximab for the treatment of lupus erythematosus panniculitis. Dermatol Ther. 2013;26:415-418.
- Guissa VR, Trudes G, Jesus AA, et al. Lupus erythematosus panniculitis in children and adolescents. Acta Reumatol Port. 2012;37:82-85.
- Mcardle A, Baker JF. A case of “refractory” lupus erythematosus profundus responsive to rituximab. Clin Rheumatol. 2009;28:745-746.
- Correll CK, Miller DD, Maguiness SM. Treatment of childhood-onset lupus erythematosus panniculitis with rituximab. JAMA Dermatol. 2020;156:566-569.
- Aronson IK, Worobec SM. Cytophagic histiocytic panniculitis and hemophagocytic lymphohistiocytosis: an overview. Dermatol Ther. 2010;23:389-402.
- Weingartner JS, Zedek DC, Burkhart CN, et al. Lupus erythematosus panniculitis in children: report of three cases and review of previously reported cases. Pediatr Dermatol. 2011;29:169-176.
- Moreno-Suárez F, Pulpillo-Ruiz Á. Rituximab for the treatment of lupus erythematosus panniculitis. Dermatol Ther. 2013;26:415-418.
- Guissa VR, Trudes G, Jesus AA, et al. Lupus erythematosus panniculitis in children and adolescents. Acta Reumatol Port. 2012;37:82-85.
- Mcardle A, Baker JF. A case of “refractory” lupus erythematosus profundus responsive to rituximab. Clin Rheumatol. 2009;28:745-746.
- Correll CK, Miller DD, Maguiness SM. Treatment of childhood-onset lupus erythematosus panniculitis with rituximab. JAMA Dermatol. 2020;156:566-569.
- Aronson IK, Worobec SM. Cytophagic histiocytic panniculitis and hemophagocytic lymphohistiocytosis: an overview. Dermatol Ther. 2010;23:389-402.
Practice Points
- Lupus erythematosus panniculitis (LEP) is rare in the pediatric population and often is difficult to treat.
- Rituximab can be an effective treatment option for refractory LEP.
- Before the initiation of rituximab, a biopsy is warranted to rule out subcutaneous T-cell lymphoma, which can mimic LEP and hemophagocytic lymphohistiocytosis–associated panniculitis.