User login
FDA addressing risk of cyberattacks on medical devices
The Food and Drug Administration is encouraging health care professionals –and patients – to report adverse events associated with medical devices, as part of an effort to address the potential for cyberattacks on medical equipment and medical devices, including implanted medical devices.
The agency has become aware of "cybersecurity vulnerabilities and incidents that could directly affect medical devices or hospital network operations," according to a statement on the FDA’s MedWatch site.
The statement adds, however, that the FDA "is not aware of any patient injuries or deaths associated with these incidents nor do we have any indication that any specific devices or systems in clinical use have been purposely targeted at this time."
The FDA is working with manufacturers and other federal agencies to address this issue. The agency has recommended that manufacturers and health care facilities institute safeguards to reduce the risk of medical device failures caused by a cyberattack, including evaluating network security at hospitals and health care facilities. Efforts include draft guidance for industry and FDA staff.
Adverse events associated with devices should be reported to the MedWatch Safety Information and Adverse Event Reporting Program or by calling 800-332-1081.
The Food and Drug Administration is encouraging health care professionals –and patients – to report adverse events associated with medical devices, as part of an effort to address the potential for cyberattacks on medical equipment and medical devices, including implanted medical devices.
The agency has become aware of "cybersecurity vulnerabilities and incidents that could directly affect medical devices or hospital network operations," according to a statement on the FDA’s MedWatch site.
The statement adds, however, that the FDA "is not aware of any patient injuries or deaths associated with these incidents nor do we have any indication that any specific devices or systems in clinical use have been purposely targeted at this time."
The FDA is working with manufacturers and other federal agencies to address this issue. The agency has recommended that manufacturers and health care facilities institute safeguards to reduce the risk of medical device failures caused by a cyberattack, including evaluating network security at hospitals and health care facilities. Efforts include draft guidance for industry and FDA staff.
Adverse events associated with devices should be reported to the MedWatch Safety Information and Adverse Event Reporting Program or by calling 800-332-1081.
The Food and Drug Administration is encouraging health care professionals –and patients – to report adverse events associated with medical devices, as part of an effort to address the potential for cyberattacks on medical equipment and medical devices, including implanted medical devices.
The agency has become aware of "cybersecurity vulnerabilities and incidents that could directly affect medical devices or hospital network operations," according to a statement on the FDA’s MedWatch site.
The statement adds, however, that the FDA "is not aware of any patient injuries or deaths associated with these incidents nor do we have any indication that any specific devices or systems in clinical use have been purposely targeted at this time."
The FDA is working with manufacturers and other federal agencies to address this issue. The agency has recommended that manufacturers and health care facilities institute safeguards to reduce the risk of medical device failures caused by a cyberattack, including evaluating network security at hospitals and health care facilities. Efforts include draft guidance for industry and FDA staff.
Adverse events associated with devices should be reported to the MedWatch Safety Information and Adverse Event Reporting Program or by calling 800-332-1081.
H. pylori test OK for people on PPIs, with caution
The Helicobacter pylori breath test can now be used in patients who have taken proton pump inhibitors within the previous 2 weeks, according to the manufacturer, Otsuka America Pharmaceutical Inc.
In a June 4 statement, the company said that the labeling of the BreathTek UBT for H. pylori kit has been updated to include this information, which is based on studies that evaluated the effects of PPI use in people taking the test. However, in this scenario, "the test result needs to be interpreted with caution," and the recommendation that patients not take antimicrobials, PPIs, and bismuth preparations within 2 weeks of the test still stands, the statement adds.
The label change makes it possible to test patients on a PPI for an initial diagnosis of H. pylori infection without stopping the PPI, providing "an opportunity of earlier detection and start of eradication therapy for H. pylori infection," according to Otsuka. Ingestion of PPIs within 2 weeks of the test can cause a false negative result, and the test should be repeated 2 weeks after the PPI therapy is stopped, if a patient has a negative test while taking a PPI, according to the warnings and precautions for the product.
Ingestion of bismuth preparations or antimicrobials can also cause a false negative result, if taken within 2 weeks of the test.
The Helicobacter pylori breath test can now be used in patients who have taken proton pump inhibitors within the previous 2 weeks, according to the manufacturer, Otsuka America Pharmaceutical Inc.
In a June 4 statement, the company said that the labeling of the BreathTek UBT for H. pylori kit has been updated to include this information, which is based on studies that evaluated the effects of PPI use in people taking the test. However, in this scenario, "the test result needs to be interpreted with caution," and the recommendation that patients not take antimicrobials, PPIs, and bismuth preparations within 2 weeks of the test still stands, the statement adds.
The label change makes it possible to test patients on a PPI for an initial diagnosis of H. pylori infection without stopping the PPI, providing "an opportunity of earlier detection and start of eradication therapy for H. pylori infection," according to Otsuka. Ingestion of PPIs within 2 weeks of the test can cause a false negative result, and the test should be repeated 2 weeks after the PPI therapy is stopped, if a patient has a negative test while taking a PPI, according to the warnings and precautions for the product.
Ingestion of bismuth preparations or antimicrobials can also cause a false negative result, if taken within 2 weeks of the test.
The Helicobacter pylori breath test can now be used in patients who have taken proton pump inhibitors within the previous 2 weeks, according to the manufacturer, Otsuka America Pharmaceutical Inc.
In a June 4 statement, the company said that the labeling of the BreathTek UBT for H. pylori kit has been updated to include this information, which is based on studies that evaluated the effects of PPI use in people taking the test. However, in this scenario, "the test result needs to be interpreted with caution," and the recommendation that patients not take antimicrobials, PPIs, and bismuth preparations within 2 weeks of the test still stands, the statement adds.
The label change makes it possible to test patients on a PPI for an initial diagnosis of H. pylori infection without stopping the PPI, providing "an opportunity of earlier detection and start of eradication therapy for H. pylori infection," according to Otsuka. Ingestion of PPIs within 2 weeks of the test can cause a false negative result, and the test should be repeated 2 weeks after the PPI therapy is stopped, if a patient has a negative test while taking a PPI, according to the warnings and precautions for the product.
Ingestion of bismuth preparations or antimicrobials can also cause a false negative result, if taken within 2 weeks of the test.
HIV prevention recommendations for IV drug users updated
The daily use of tenofovir reduced the risk of new HIV infections by almost 50% in a Thai study of intravenous drug users, results that are the basis of a recommendation to consider this approach in the United States, according to the Centers for Disease Control and Prevention.
"Based on these findings, CDC recommends that preexposure prophylaxis (PrEP) be considered as one of several prevention options for persons at very high risk for HIV acquisition through the injection of illicit drugs," the CDC concluded in an update on the interim guidance for PrEP for preventing HIV infection. The report will appear in the June 14 issue of the Morbidity and Mortality Weekly Report (MMWR 2013;62:463-5).
The CDC previously issued interim guidelines on the use of PrEP for men who have sex with men and heterosexually active adults.
Providing PrEP to IV drug users "at very high risk for HIV acquisition could contribute to the reduction of HIV incidence in the United States," the report stated. In 2010, 8% of the new HIV infections in the United States were due to intravenous drug use and every year, about 50,000 people in the United State become infected with HIV, according to the CDC.
Although tenofovir was the only oral antiretroviral drug in the study, the CDC is recommending the fixed-dose combination of 300 mg of tenofovir with 200 mg of emtricitabine, marketed as Truvada, as the "preferred PrEP regimen" for IV drug users.
Truvada was approved by the FDA in 2012 for preexposure prophylaxis of sexually acquired HIV in men who have sex with men and heterosexually active men and women, based on studies in these two populations, but not in IV drug users. And the addition of emtricitabine is not associated with increased side effects. Both drugs are nucleoside analog HIV-1 reverse transcriptase inhibitors.
In the Bangkok Tenofovir Study, a randomized-double blind study of 2,413 IV drug users aged 20-60 years, participants received a daily dose of 300 mg of tenofovir or placebo. Over a mean follow-up of 4.6 years, 17 people in the tenofovir group and 33 people in the placebo group became infected with HIV. The HIV incidence was 0.35 per 100 person-years among those on tenofovir vs. 0.68 per 100 person-years among those on placebo-a 48.9% reduction in HIV incidence. The drug was taken for an average of 84% of days.
There were no significant differences in the rates of adverse events, serious adverse events, mortality, grade 3-4 laboratory abnormalities, or elevated serum creatine in the two groups. Those on tenofovir experienced more nausea and vomiting in the first 2 months, but not after that time. The results of the study were published by the Thailand Ministry of Health online in the Lancet [doi: 10.1016/S0140-6736(13)61127-7]).
The CDC is working with other federal agencies to prepare guidelines on the use of PrEP in men who have sex with men, heterosexually active men and women, and IV drug users, which are expected to be released this year.
The daily use of tenofovir reduced the risk of new HIV infections by almost 50% in a Thai study of intravenous drug users, results that are the basis of a recommendation to consider this approach in the United States, according to the Centers for Disease Control and Prevention.
"Based on these findings, CDC recommends that preexposure prophylaxis (PrEP) be considered as one of several prevention options for persons at very high risk for HIV acquisition through the injection of illicit drugs," the CDC concluded in an update on the interim guidance for PrEP for preventing HIV infection. The report will appear in the June 14 issue of the Morbidity and Mortality Weekly Report (MMWR 2013;62:463-5).
The CDC previously issued interim guidelines on the use of PrEP for men who have sex with men and heterosexually active adults.
Providing PrEP to IV drug users "at very high risk for HIV acquisition could contribute to the reduction of HIV incidence in the United States," the report stated. In 2010, 8% of the new HIV infections in the United States were due to intravenous drug use and every year, about 50,000 people in the United State become infected with HIV, according to the CDC.
Although tenofovir was the only oral antiretroviral drug in the study, the CDC is recommending the fixed-dose combination of 300 mg of tenofovir with 200 mg of emtricitabine, marketed as Truvada, as the "preferred PrEP regimen" for IV drug users.
Truvada was approved by the FDA in 2012 for preexposure prophylaxis of sexually acquired HIV in men who have sex with men and heterosexually active men and women, based on studies in these two populations, but not in IV drug users. And the addition of emtricitabine is not associated with increased side effects. Both drugs are nucleoside analog HIV-1 reverse transcriptase inhibitors.
In the Bangkok Tenofovir Study, a randomized-double blind study of 2,413 IV drug users aged 20-60 years, participants received a daily dose of 300 mg of tenofovir or placebo. Over a mean follow-up of 4.6 years, 17 people in the tenofovir group and 33 people in the placebo group became infected with HIV. The HIV incidence was 0.35 per 100 person-years among those on tenofovir vs. 0.68 per 100 person-years among those on placebo-a 48.9% reduction in HIV incidence. The drug was taken for an average of 84% of days.
There were no significant differences in the rates of adverse events, serious adverse events, mortality, grade 3-4 laboratory abnormalities, or elevated serum creatine in the two groups. Those on tenofovir experienced more nausea and vomiting in the first 2 months, but not after that time. The results of the study were published by the Thailand Ministry of Health online in the Lancet [doi: 10.1016/S0140-6736(13)61127-7]).
The CDC is working with other federal agencies to prepare guidelines on the use of PrEP in men who have sex with men, heterosexually active men and women, and IV drug users, which are expected to be released this year.
The daily use of tenofovir reduced the risk of new HIV infections by almost 50% in a Thai study of intravenous drug users, results that are the basis of a recommendation to consider this approach in the United States, according to the Centers for Disease Control and Prevention.
"Based on these findings, CDC recommends that preexposure prophylaxis (PrEP) be considered as one of several prevention options for persons at very high risk for HIV acquisition through the injection of illicit drugs," the CDC concluded in an update on the interim guidance for PrEP for preventing HIV infection. The report will appear in the June 14 issue of the Morbidity and Mortality Weekly Report (MMWR 2013;62:463-5).
The CDC previously issued interim guidelines on the use of PrEP for men who have sex with men and heterosexually active adults.
Providing PrEP to IV drug users "at very high risk for HIV acquisition could contribute to the reduction of HIV incidence in the United States," the report stated. In 2010, 8% of the new HIV infections in the United States were due to intravenous drug use and every year, about 50,000 people in the United State become infected with HIV, according to the CDC.
Although tenofovir was the only oral antiretroviral drug in the study, the CDC is recommending the fixed-dose combination of 300 mg of tenofovir with 200 mg of emtricitabine, marketed as Truvada, as the "preferred PrEP regimen" for IV drug users.
Truvada was approved by the FDA in 2012 for preexposure prophylaxis of sexually acquired HIV in men who have sex with men and heterosexually active men and women, based on studies in these two populations, but not in IV drug users. And the addition of emtricitabine is not associated with increased side effects. Both drugs are nucleoside analog HIV-1 reverse transcriptase inhibitors.
In the Bangkok Tenofovir Study, a randomized-double blind study of 2,413 IV drug users aged 20-60 years, participants received a daily dose of 300 mg of tenofovir or placebo. Over a mean follow-up of 4.6 years, 17 people in the tenofovir group and 33 people in the placebo group became infected with HIV. The HIV incidence was 0.35 per 100 person-years among those on tenofovir vs. 0.68 per 100 person-years among those on placebo-a 48.9% reduction in HIV incidence. The drug was taken for an average of 84% of days.
There were no significant differences in the rates of adverse events, serious adverse events, mortality, grade 3-4 laboratory abnormalities, or elevated serum creatine in the two groups. Those on tenofovir experienced more nausea and vomiting in the first 2 months, but not after that time. The results of the study were published by the Thailand Ministry of Health online in the Lancet [doi: 10.1016/S0140-6736(13)61127-7]).
The CDC is working with other federal agencies to prepare guidelines on the use of PrEP in men who have sex with men, heterosexually active men and women, and IV drug users, which are expected to be released this year.
FROM MMWR
FDA panels revisit rosiglitazone's cardiovascular safety
The majority of two FDA advisory panels voted to modify or remove the prescribing restrictions on the type 2 diabetes drug rosiglitazone, based on the readjudicated results of a cardiovascular outcomes study, the Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycemia in Diabetes (RECORD) trial. Meeting participants spoke with IMNG Medical Media about the panels' recommendations.
The majority of two FDA advisory panels voted to modify or remove the prescribing restrictions on the type 2 diabetes drug rosiglitazone, based on the readjudicated results of a cardiovascular outcomes study, the Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycemia in Diabetes (RECORD) trial. Meeting participants spoke with IMNG Medical Media about the panels' recommendations.
The majority of two FDA advisory panels voted to modify or remove the prescribing restrictions on the type 2 diabetes drug rosiglitazone, based on the readjudicated results of a cardiovascular outcomes study, the Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycemia in Diabetes (RECORD) trial. Meeting participants spoke with IMNG Medical Media about the panels' recommendations.

Exercise program improved strength, walking in AS
Patients with ankylosing spondylitis who participated in a progressive muscle strengthening program gained significant improvements in muscle strength and walking performance after 4 months, compared with those who did not participate, Dr. Fabio Jennings reported at the annual European Congress of Rheumatology.
The exercise program, which involved resistance training with the Swiss ball, was also well tolerated and was not associated with negative effects on disease activity, said Dr. Jennings of the rheumatology division at the Federal University of Sao Paulo, Brazil.
Dr. Jennings and his colleagues performed a randomized, controlled, single-blind, prospective trial of 60 patients with ankylosing spondylitis (AS).
Exercise is recommended for people with AS, but the benefits of a specific exercise program have not been well defined, Dr. Jennings noted.
In the study, 30 patients were randomized to the supervised exercise program, which entailed eight resistance exercises using free weights on a Swiss ball twice a week for 16 weeks, with increases in load every 4 weeks. The 30 patients in the control group continued regular treatment with medications, with no exercise. Demographics, clinical features, and medications were similar in the two groups at baseline.
The impact of the exercise on functional capacity, quality of life, muscle strength, and mobility was evaluated using the BASFI (Bath Ankylosing Spondylitis Functional Index), HAQ-S (Health Assessment Questionnaire for Spondyloarthropathies), the 6-minute walk test, and other assessment tools, every 4 weeks. Disease activity was measured with the BASDAI (Bath Ankylosing Spondylitis Disease Activity Index), erythrocyte sedimentation rate (ESR), and C-reactive protein levels.
After 4 months, there were statistically significant differences between the two groups in the strengths of the muscles used in performing the exercises (abdominal, squat, triceps, reverse fly, and rowing exercises), favoring the intervention group. These patients also had significant improvements in the 6-minute walk test; and they were satisfied with the treatment, as reflected by significant differences between the two groups in the Likert scale used to assess patient satisfaction at all time points measured.
In addition, disease activity – as measured by BASDAI, ESR, and C-reactive protein – did not worsen among those who participated in the exercise program.
The study showed that this type of exercise program, using a Swiss ball to improve muscle strength, "is a beneficial and safe intervention" in people with AS, lead author Mr. Marcelo Cardoso de Souza, a physiotherapist and member of a multidisciplinary rehabilitation team at the university, said in an interview. For these patients, improvement in muscular performance and functional capacity is important, he said, noting that before clinicians refer patients to such a program, patients should undergo a medical evaluation, and the program should be provided by an experienced professional.
None of the investigators had relevant financial conflicts of interest.
Patients with ankylosing spondylitis who participated in a progressive muscle strengthening program gained significant improvements in muscle strength and walking performance after 4 months, compared with those who did not participate, Dr. Fabio Jennings reported at the annual European Congress of Rheumatology.
The exercise program, which involved resistance training with the Swiss ball, was also well tolerated and was not associated with negative effects on disease activity, said Dr. Jennings of the rheumatology division at the Federal University of Sao Paulo, Brazil.
Dr. Jennings and his colleagues performed a randomized, controlled, single-blind, prospective trial of 60 patients with ankylosing spondylitis (AS).
Exercise is recommended for people with AS, but the benefits of a specific exercise program have not been well defined, Dr. Jennings noted.
In the study, 30 patients were randomized to the supervised exercise program, which entailed eight resistance exercises using free weights on a Swiss ball twice a week for 16 weeks, with increases in load every 4 weeks. The 30 patients in the control group continued regular treatment with medications, with no exercise. Demographics, clinical features, and medications were similar in the two groups at baseline.
The impact of the exercise on functional capacity, quality of life, muscle strength, and mobility was evaluated using the BASFI (Bath Ankylosing Spondylitis Functional Index), HAQ-S (Health Assessment Questionnaire for Spondyloarthropathies), the 6-minute walk test, and other assessment tools, every 4 weeks. Disease activity was measured with the BASDAI (Bath Ankylosing Spondylitis Disease Activity Index), erythrocyte sedimentation rate (ESR), and C-reactive protein levels.
After 4 months, there were statistically significant differences between the two groups in the strengths of the muscles used in performing the exercises (abdominal, squat, triceps, reverse fly, and rowing exercises), favoring the intervention group. These patients also had significant improvements in the 6-minute walk test; and they were satisfied with the treatment, as reflected by significant differences between the two groups in the Likert scale used to assess patient satisfaction at all time points measured.
In addition, disease activity – as measured by BASDAI, ESR, and C-reactive protein – did not worsen among those who participated in the exercise program.
The study showed that this type of exercise program, using a Swiss ball to improve muscle strength, "is a beneficial and safe intervention" in people with AS, lead author Mr. Marcelo Cardoso de Souza, a physiotherapist and member of a multidisciplinary rehabilitation team at the university, said in an interview. For these patients, improvement in muscular performance and functional capacity is important, he said, noting that before clinicians refer patients to such a program, patients should undergo a medical evaluation, and the program should be provided by an experienced professional.
None of the investigators had relevant financial conflicts of interest.
Patients with ankylosing spondylitis who participated in a progressive muscle strengthening program gained significant improvements in muscle strength and walking performance after 4 months, compared with those who did not participate, Dr. Fabio Jennings reported at the annual European Congress of Rheumatology.
The exercise program, which involved resistance training with the Swiss ball, was also well tolerated and was not associated with negative effects on disease activity, said Dr. Jennings of the rheumatology division at the Federal University of Sao Paulo, Brazil.
Dr. Jennings and his colleagues performed a randomized, controlled, single-blind, prospective trial of 60 patients with ankylosing spondylitis (AS).
Exercise is recommended for people with AS, but the benefits of a specific exercise program have not been well defined, Dr. Jennings noted.
In the study, 30 patients were randomized to the supervised exercise program, which entailed eight resistance exercises using free weights on a Swiss ball twice a week for 16 weeks, with increases in load every 4 weeks. The 30 patients in the control group continued regular treatment with medications, with no exercise. Demographics, clinical features, and medications were similar in the two groups at baseline.
The impact of the exercise on functional capacity, quality of life, muscle strength, and mobility was evaluated using the BASFI (Bath Ankylosing Spondylitis Functional Index), HAQ-S (Health Assessment Questionnaire for Spondyloarthropathies), the 6-minute walk test, and other assessment tools, every 4 weeks. Disease activity was measured with the BASDAI (Bath Ankylosing Spondylitis Disease Activity Index), erythrocyte sedimentation rate (ESR), and C-reactive protein levels.
After 4 months, there were statistically significant differences between the two groups in the strengths of the muscles used in performing the exercises (abdominal, squat, triceps, reverse fly, and rowing exercises), favoring the intervention group. These patients also had significant improvements in the 6-minute walk test; and they were satisfied with the treatment, as reflected by significant differences between the two groups in the Likert scale used to assess patient satisfaction at all time points measured.
In addition, disease activity – as measured by BASDAI, ESR, and C-reactive protein – did not worsen among those who participated in the exercise program.
The study showed that this type of exercise program, using a Swiss ball to improve muscle strength, "is a beneficial and safe intervention" in people with AS, lead author Mr. Marcelo Cardoso de Souza, a physiotherapist and member of a multidisciplinary rehabilitation team at the university, said in an interview. For these patients, improvement in muscular performance and functional capacity is important, he said, noting that before clinicians refer patients to such a program, patients should undergo a medical evaluation, and the program should be provided by an experienced professional.
None of the investigators had relevant financial conflicts of interest.
FROM THE EULAR CONGRESS 2013
MRI score of joint narrowing has research promise
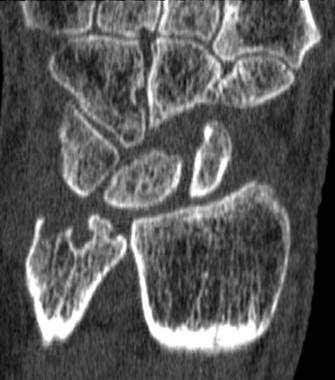
A magnetic resonance imaging scoring system of joint-space narrowing in rheumatoid arthritis showed "a very high" agreement with computed tomography scores and may become a useful tool in rheumatoid arthritis clinical trials after further validation, judging from data presented by Dr. Uffe Møller Døhn.
In a small study, which was conducted to validate the OMERACT-RAMRIS MRI JSN scoring system in the wrists and metacarpophalangeal (MCP) joints, there was a very high agreement between the joint-space narrowing scores on MRI and CT and moderate agreement between scores on MRI and x-ray, said Dr. Møller Døhn of Copenhagen University Hospital at Glostrup at the annual European Congress of Rheumatology. In addition, there was "high to very high" inter- and intrareader reliability, particularly for the wrist joints.
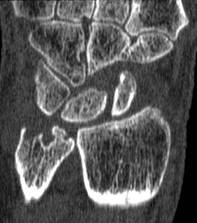
An OMERACT (Outcome Measures in Rheumatology) initiative, this scoring system is being developed to provide a more precise and sensitive method of measuring joint space damage in patients with rheumatoid arthritis (RA), but it needs to be validated through comparisons to other imaging methods.
To evaluate the degree of agreement with CT and x-ray scores, this study assessed MRI and CT images of the wrist and the second to fifth metacarpophalangeal (MCP 2-5) joints of 14 people with RA and one healthy control, who were from a clinical trial. Three readers assessed the images twice, and a single reader scored x-rays using the Sharp-Van der Heidje method, said Dr. Møller Døhn, who is in the center for rheumatology and spine diseases at the hospital.
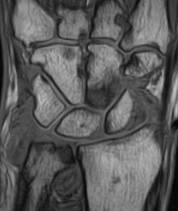
The MRI scores of joint space narrowing "were very highly correlated" with CT scores, when comparing the wrist and MCP scores both separately and combined: Using intraclass correlation coefficients (ICCs) as a measure of agreement between scores and scorers, the MRI and CT scores for joint space narrowing were 0.94 for the MCP joints, 0.92 for the wrist, and 0.92 for the wrist and MCP joints combined. But the ICCs for the x-ray joint space narrowing scores were lower: With MRI scores, the ICCs were 0.49 for the MCP 2-5 joints and 0.55 for the wrist. With CT scores, the ICCs were 0.56 for the MCP 2-5 joints and 0.43 for the wrist.
"The most important next step is to test the scoring system in a longitudinal setting, in order to investigate the sensitivity to change," Dr. Møller Døhn said in an interview before the congress. "Before the system can be implemented as an outcome measure in clinical trials, we need to know if it is more sensitive than other methods that are already available. If it turns out that [joint space narrowing] assessment of several joints on x-ray is just as good as - or better than - MRI, then it does not add information to what we already use today."
Dr. Møller Døhn reported that he had no relevant financial disclosures.
A magnetic resonance imaging scoring system of joint-space narrowing in rheumatoid arthritis showed "a very high" agreement with computed tomography scores and may become a useful tool in rheumatoid arthritis clinical trials after further validation, judging from data presented by Dr. Uffe Møller Døhn.
In a small study, which was conducted to validate the OMERACT-RAMRIS MRI JSN scoring system in the wrists and metacarpophalangeal (MCP) joints, there was a very high agreement between the joint-space narrowing scores on MRI and CT and moderate agreement between scores on MRI and x-ray, said Dr. Møller Døhn of Copenhagen University Hospital at Glostrup at the annual European Congress of Rheumatology. In addition, there was "high to very high" inter- and intrareader reliability, particularly for the wrist joints.
An OMERACT (Outcome Measures in Rheumatology) initiative, this scoring system is being developed to provide a more precise and sensitive method of measuring joint space damage in patients with rheumatoid arthritis (RA), but it needs to be validated through comparisons to other imaging methods.
To evaluate the degree of agreement with CT and x-ray scores, this study assessed MRI and CT images of the wrist and the second to fifth metacarpophalangeal (MCP 2-5) joints of 14 people with RA and one healthy control, who were from a clinical trial. Three readers assessed the images twice, and a single reader scored x-rays using the Sharp-Van der Heidje method, said Dr. Møller Døhn, who is in the center for rheumatology and spine diseases at the hospital.
The MRI scores of joint space narrowing "were very highly correlated" with CT scores, when comparing the wrist and MCP scores both separately and combined: Using intraclass correlation coefficients (ICCs) as a measure of agreement between scores and scorers, the MRI and CT scores for joint space narrowing were 0.94 for the MCP joints, 0.92 for the wrist, and 0.92 for the wrist and MCP joints combined. But the ICCs for the x-ray joint space narrowing scores were lower: With MRI scores, the ICCs were 0.49 for the MCP 2-5 joints and 0.55 for the wrist. With CT scores, the ICCs were 0.56 for the MCP 2-5 joints and 0.43 for the wrist.
"The most important next step is to test the scoring system in a longitudinal setting, in order to investigate the sensitivity to change," Dr. Møller Døhn said in an interview before the congress. "Before the system can be implemented as an outcome measure in clinical trials, we need to know if it is more sensitive than other methods that are already available. If it turns out that [joint space narrowing] assessment of several joints on x-ray is just as good as - or better than - MRI, then it does not add information to what we already use today."
Dr. Møller Døhn reported that he had no relevant financial disclosures.
A magnetic resonance imaging scoring system of joint-space narrowing in rheumatoid arthritis showed "a very high" agreement with computed tomography scores and may become a useful tool in rheumatoid arthritis clinical trials after further validation, judging from data presented by Dr. Uffe Møller Døhn.
In a small study, which was conducted to validate the OMERACT-RAMRIS MRI JSN scoring system in the wrists and metacarpophalangeal (MCP) joints, there was a very high agreement between the joint-space narrowing scores on MRI and CT and moderate agreement between scores on MRI and x-ray, said Dr. Møller Døhn of Copenhagen University Hospital at Glostrup at the annual European Congress of Rheumatology. In addition, there was "high to very high" inter- and intrareader reliability, particularly for the wrist joints.
An OMERACT (Outcome Measures in Rheumatology) initiative, this scoring system is being developed to provide a more precise and sensitive method of measuring joint space damage in patients with rheumatoid arthritis (RA), but it needs to be validated through comparisons to other imaging methods.
To evaluate the degree of agreement with CT and x-ray scores, this study assessed MRI and CT images of the wrist and the second to fifth metacarpophalangeal (MCP 2-5) joints of 14 people with RA and one healthy control, who were from a clinical trial. Three readers assessed the images twice, and a single reader scored x-rays using the Sharp-Van der Heidje method, said Dr. Møller Døhn, who is in the center for rheumatology and spine diseases at the hospital.
The MRI scores of joint space narrowing "were very highly correlated" with CT scores, when comparing the wrist and MCP scores both separately and combined: Using intraclass correlation coefficients (ICCs) as a measure of agreement between scores and scorers, the MRI and CT scores for joint space narrowing were 0.94 for the MCP joints, 0.92 for the wrist, and 0.92 for the wrist and MCP joints combined. But the ICCs for the x-ray joint space narrowing scores were lower: With MRI scores, the ICCs were 0.49 for the MCP 2-5 joints and 0.55 for the wrist. With CT scores, the ICCs were 0.56 for the MCP 2-5 joints and 0.43 for the wrist.
"The most important next step is to test the scoring system in a longitudinal setting, in order to investigate the sensitivity to change," Dr. Møller Døhn said in an interview before the congress. "Before the system can be implemented as an outcome measure in clinical trials, we need to know if it is more sensitive than other methods that are already available. If it turns out that [joint space narrowing] assessment of several joints on x-ray is just as good as - or better than - MRI, then it does not add information to what we already use today."
Dr. Møller Døhn reported that he had no relevant financial disclosures.
FROM THE EULAR CONGRESS 2013
Ustekinumab benefits in PsA sustained through 1 year
MADRID – The lessening of the signs and symptoms of psoriatic arthritis that occurs during the first 6 months of ustekinumab treatment persisted and improved further at the end of 1 year, with a favorable safety profile, according to 52-week data from the PSUMMIT II trial.
The sustained benefits in American College of Rheumatology (ACR) 20 responses and other efficacy endpoints were evident even in patients who had been treated previously with anti-tumor necrosis factor (anti-TNF) agents and among those who were anti-TNF naïve, although the benefits were greater in the latter group patients, according to Dr. Christopher T. Ritchlin, a professor in the department of medicine, allergy/immunology, and rheumatology at the University of Rochester (N.Y.). This includes beneficial effects on skin and enthesitis, Dr. Ritchlin said at the annual European Congress of Rheumatology.

The PSUMMIT II study is a follow-up to the PSUMMIT I study, the findings of which showed that ustekinumab, a human interleukin (IL)-12 and IL-23 antagonist, showed significant effectiveness in patients with psoriatic arthritis (PsA) who had not been exposed to anti-TNF drugs.
Ustekinumab is currently approved for treating moderate to severe plaque psoriasis in adults who are candidates for phototherapy or systemic therapy in the United States, or those who have failed to respond to, have a contraindication to, or are intolerant of other systemic therapies in Europe. In December 2012, the manufacturer, Janssen, announced that it had filed for further approval for ustekinumab in both the United States and Europe for the treatment of active disease.
The PSUMMIT II study enrolled 312 patients with active PsA who had five or more tender and five or more swollen joints, and a C-reactive protein level of 0.3 mg/dL or higher. Patients who had been treated previously with anti-TNF therapy (n = 180) and those naive to anti-TNF therapy (n = 132) were included and randomized to one of two doses of ustekinumab (45 mg or 90 mg) or placebo administered at week 0, 4, and 12. At 16 weeks, patients with less than a 5% improvement in tender and swollen joint counts on placebo were switched to active treatment, those on 45 mg ustekinumab had their dose upped to 90 mg, and those on 90 mg remained on that dose.
At 6 months, significantly more patients treated with ustekinumab than placebo achieved the primary endpoint of an ACR 20, and more patients on active treatment had an ACR 50, and at least a 75% improvement in the Psoriasis Area and Severity Index (PASI 75).
These results were sustained at 1 year, with 47%-48% of those on 45 mg and 90 mg, and 56% of those who switched from placebo to the 45-mg dose, achieving an ACR 20. In addition, 26%-29% achieved an ACR 50 (29% for those switched from placebo), and 13%-18% achieved an ACR 70 (15% for placebo). There were also improvements associated with treatment in HAQ-DI (Health Assessment Questionnaire-Disability Index) scores at week 52, according to Dr. Ritchlin. The mean change in HAQ-DI scores from baseline to week 52 were -0.21 for placebo, -0.20 for the 45-mg dose of ustekinumab, and -0.28 for the 90-mg dose.
Among those who had not been treated before with an anti-TNF agent, 59%-60% of those on ustekinumab (73% for those switched from placebo) achieved an ACR 20 at week 52, compared with 37%-41% of those who had taken an anti-TNF agent previously before being treated with ustekinumab (30% for placebo). Although responses among anti-TNF naive patients were superior, the responses among those who had been treated with these agents previously were still significantly improved, an indication that ustekinumab "offers an alternative for patients who cannot take or fail anti-TNF agents," Dr. Ritchlin said in the interview.
Treatment was "very effective" for skin symptoms and for enthesitis, he noted. Compared with baseline, dactylitis was improved by a median of 95% among those on the 45-mg dose, 91% among those on the 90-mg dose, and 100% in those who switched from placebo to the 45-mg dose of ustekinumab. Similar improvements in enthesitis were seen, with the highest improvement (60%) seen with the highest dose of ustekinumab. PASI scores at baseline ranged from 11 to 13 and improved by 56%-64% by follow-up at week 52.
In general, ustekinumab was well tolerated, with no deaths or cases of tuberculosis reported and with similar rates of adverse events and serious adverse events between the two doses (just under 6%). There were two malignancies: one breast cancer and one squamous cell carcinoma in two patients taking the 90-mg dose of ustekinumab, who had both been treated with anti-TNFs previously. The rate of serious infections was less than 1% among those treated with ustekinumab. Through 60 weeks of treatment, there were three major adverse cardiovascular events, all myocardial infarctions, in patients treated with ustekinumab. These patients all had multiple cardiovascular risk factors, Dr. Ritchlin said. They had also been exposed previously to anti-TNF treatment.
Radiographic data from the trial are expected and likely to be available by the end of the year for presentation at the annual American College of Rheumatology meeting.
Dr. Ritchlin disclosed having received grant and research support from Janssen. Four of the nine remaining authors are Janssen employees and shareholders of Johnson & Johnson, Janssen’s parent company. Ustekinumab is marketed as Stelara in the United States.
Sara Freeman contributed to this report.
MADRID – The lessening of the signs and symptoms of psoriatic arthritis that occurs during the first 6 months of ustekinumab treatment persisted and improved further at the end of 1 year, with a favorable safety profile, according to 52-week data from the PSUMMIT II trial.
The sustained benefits in American College of Rheumatology (ACR) 20 responses and other efficacy endpoints were evident even in patients who had been treated previously with anti-tumor necrosis factor (anti-TNF) agents and among those who were anti-TNF naïve, although the benefits were greater in the latter group patients, according to Dr. Christopher T. Ritchlin, a professor in the department of medicine, allergy/immunology, and rheumatology at the University of Rochester (N.Y.). This includes beneficial effects on skin and enthesitis, Dr. Ritchlin said at the annual European Congress of Rheumatology.
The PSUMMIT II study is a follow-up to the PSUMMIT I study, the findings of which showed that ustekinumab, a human interleukin (IL)-12 and IL-23 antagonist, showed significant effectiveness in patients with psoriatic arthritis (PsA) who had not been exposed to anti-TNF drugs.
Ustekinumab is currently approved for treating moderate to severe plaque psoriasis in adults who are candidates for phototherapy or systemic therapy in the United States, or those who have failed to respond to, have a contraindication to, or are intolerant of other systemic therapies in Europe. In December 2012, the manufacturer, Janssen, announced that it had filed for further approval for ustekinumab in both the United States and Europe for the treatment of active disease.
The PSUMMIT II study enrolled 312 patients with active PsA who had five or more tender and five or more swollen joints, and a C-reactive protein level of 0.3 mg/dL or higher. Patients who had been treated previously with anti-TNF therapy (n = 180) and those naive to anti-TNF therapy (n = 132) were included and randomized to one of two doses of ustekinumab (45 mg or 90 mg) or placebo administered at week 0, 4, and 12. At 16 weeks, patients with less than a 5% improvement in tender and swollen joint counts on placebo were switched to active treatment, those on 45 mg ustekinumab had their dose upped to 90 mg, and those on 90 mg remained on that dose.
At 6 months, significantly more patients treated with ustekinumab than placebo achieved the primary endpoint of an ACR 20, and more patients on active treatment had an ACR 50, and at least a 75% improvement in the Psoriasis Area and Severity Index (PASI 75).
These results were sustained at 1 year, with 47%-48% of those on 45 mg and 90 mg, and 56% of those who switched from placebo to the 45-mg dose, achieving an ACR 20. In addition, 26%-29% achieved an ACR 50 (29% for those switched from placebo), and 13%-18% achieved an ACR 70 (15% for placebo). There were also improvements associated with treatment in HAQ-DI (Health Assessment Questionnaire-Disability Index) scores at week 52, according to Dr. Ritchlin. The mean change in HAQ-DI scores from baseline to week 52 were -0.21 for placebo, -0.20 for the 45-mg dose of ustekinumab, and -0.28 for the 90-mg dose.
Among those who had not been treated before with an anti-TNF agent, 59%-60% of those on ustekinumab (73% for those switched from placebo) achieved an ACR 20 at week 52, compared with 37%-41% of those who had taken an anti-TNF agent previously before being treated with ustekinumab (30% for placebo). Although responses among anti-TNF naive patients were superior, the responses among those who had been treated with these agents previously were still significantly improved, an indication that ustekinumab "offers an alternative for patients who cannot take or fail anti-TNF agents," Dr. Ritchlin said in the interview.
Treatment was "very effective" for skin symptoms and for enthesitis, he noted. Compared with baseline, dactylitis was improved by a median of 95% among those on the 45-mg dose, 91% among those on the 90-mg dose, and 100% in those who switched from placebo to the 45-mg dose of ustekinumab. Similar improvements in enthesitis were seen, with the highest improvement (60%) seen with the highest dose of ustekinumab. PASI scores at baseline ranged from 11 to 13 and improved by 56%-64% by follow-up at week 52.
In general, ustekinumab was well tolerated, with no deaths or cases of tuberculosis reported and with similar rates of adverse events and serious adverse events between the two doses (just under 6%). There were two malignancies: one breast cancer and one squamous cell carcinoma in two patients taking the 90-mg dose of ustekinumab, who had both been treated with anti-TNFs previously. The rate of serious infections was less than 1% among those treated with ustekinumab. Through 60 weeks of treatment, there were three major adverse cardiovascular events, all myocardial infarctions, in patients treated with ustekinumab. These patients all had multiple cardiovascular risk factors, Dr. Ritchlin said. They had also been exposed previously to anti-TNF treatment.
Radiographic data from the trial are expected and likely to be available by the end of the year for presentation at the annual American College of Rheumatology meeting.
Dr. Ritchlin disclosed having received grant and research support from Janssen. Four of the nine remaining authors are Janssen employees and shareholders of Johnson & Johnson, Janssen’s parent company. Ustekinumab is marketed as Stelara in the United States.
Sara Freeman contributed to this report.
MADRID – The lessening of the signs and symptoms of psoriatic arthritis that occurs during the first 6 months of ustekinumab treatment persisted and improved further at the end of 1 year, with a favorable safety profile, according to 52-week data from the PSUMMIT II trial.
The sustained benefits in American College of Rheumatology (ACR) 20 responses and other efficacy endpoints were evident even in patients who had been treated previously with anti-tumor necrosis factor (anti-TNF) agents and among those who were anti-TNF naïve, although the benefits were greater in the latter group patients, according to Dr. Christopher T. Ritchlin, a professor in the department of medicine, allergy/immunology, and rheumatology at the University of Rochester (N.Y.). This includes beneficial effects on skin and enthesitis, Dr. Ritchlin said at the annual European Congress of Rheumatology.
The PSUMMIT II study is a follow-up to the PSUMMIT I study, the findings of which showed that ustekinumab, a human interleukin (IL)-12 and IL-23 antagonist, showed significant effectiveness in patients with psoriatic arthritis (PsA) who had not been exposed to anti-TNF drugs.
Ustekinumab is currently approved for treating moderate to severe plaque psoriasis in adults who are candidates for phototherapy or systemic therapy in the United States, or those who have failed to respond to, have a contraindication to, or are intolerant of other systemic therapies in Europe. In December 2012, the manufacturer, Janssen, announced that it had filed for further approval for ustekinumab in both the United States and Europe for the treatment of active disease.
The PSUMMIT II study enrolled 312 patients with active PsA who had five or more tender and five or more swollen joints, and a C-reactive protein level of 0.3 mg/dL or higher. Patients who had been treated previously with anti-TNF therapy (n = 180) and those naive to anti-TNF therapy (n = 132) were included and randomized to one of two doses of ustekinumab (45 mg or 90 mg) or placebo administered at week 0, 4, and 12. At 16 weeks, patients with less than a 5% improvement in tender and swollen joint counts on placebo were switched to active treatment, those on 45 mg ustekinumab had their dose upped to 90 mg, and those on 90 mg remained on that dose.
At 6 months, significantly more patients treated with ustekinumab than placebo achieved the primary endpoint of an ACR 20, and more patients on active treatment had an ACR 50, and at least a 75% improvement in the Psoriasis Area and Severity Index (PASI 75).
These results were sustained at 1 year, with 47%-48% of those on 45 mg and 90 mg, and 56% of those who switched from placebo to the 45-mg dose, achieving an ACR 20. In addition, 26%-29% achieved an ACR 50 (29% for those switched from placebo), and 13%-18% achieved an ACR 70 (15% for placebo). There were also improvements associated with treatment in HAQ-DI (Health Assessment Questionnaire-Disability Index) scores at week 52, according to Dr. Ritchlin. The mean change in HAQ-DI scores from baseline to week 52 were -0.21 for placebo, -0.20 for the 45-mg dose of ustekinumab, and -0.28 for the 90-mg dose.
Among those who had not been treated before with an anti-TNF agent, 59%-60% of those on ustekinumab (73% for those switched from placebo) achieved an ACR 20 at week 52, compared with 37%-41% of those who had taken an anti-TNF agent previously before being treated with ustekinumab (30% for placebo). Although responses among anti-TNF naive patients were superior, the responses among those who had been treated with these agents previously were still significantly improved, an indication that ustekinumab "offers an alternative for patients who cannot take or fail anti-TNF agents," Dr. Ritchlin said in the interview.
Treatment was "very effective" for skin symptoms and for enthesitis, he noted. Compared with baseline, dactylitis was improved by a median of 95% among those on the 45-mg dose, 91% among those on the 90-mg dose, and 100% in those who switched from placebo to the 45-mg dose of ustekinumab. Similar improvements in enthesitis were seen, with the highest improvement (60%) seen with the highest dose of ustekinumab. PASI scores at baseline ranged from 11 to 13 and improved by 56%-64% by follow-up at week 52.
In general, ustekinumab was well tolerated, with no deaths or cases of tuberculosis reported and with similar rates of adverse events and serious adverse events between the two doses (just under 6%). There were two malignancies: one breast cancer and one squamous cell carcinoma in two patients taking the 90-mg dose of ustekinumab, who had both been treated with anti-TNFs previously. The rate of serious infections was less than 1% among those treated with ustekinumab. Through 60 weeks of treatment, there were three major adverse cardiovascular events, all myocardial infarctions, in patients treated with ustekinumab. These patients all had multiple cardiovascular risk factors, Dr. Ritchlin said. They had also been exposed previously to anti-TNF treatment.
Radiographic data from the trial are expected and likely to be available by the end of the year for presentation at the annual American College of Rheumatology meeting.
Dr. Ritchlin disclosed having received grant and research support from Janssen. Four of the nine remaining authors are Janssen employees and shareholders of Johnson & Johnson, Janssen’s parent company. Ustekinumab is marketed as Stelara in the United States.
Sara Freeman contributed to this report.
AT THE EULAR CONGRESS 2013

FDA panel supports easing rosiglitazone restrictions
SILVER SPRING, MD. – Reassured by the results of a reanalysis of data from a cardiovascular safety trial of rosiglitazone, the majority of a Food and Drug Administration advisory panel recommended either loosening or lifting the restrictions placed on prescribing of the type 2 diabetes drug in 2010.
At a June 6 meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee, 13 panel members voted to modify the Risk Evaluation and Mitigation Strategy (REMS) required for the drug and seven recommended that it be removed altogether. Of the remaining six panelists, five recommended that the REMS be continued as it currently exists and one panelist, the consumer representative, voted to withdraw the drug from the market.

The FDA convened the 2-day meeting to review the results of the readjudication of data from the RECORD (Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycemia in Diabetes) trial, conducted by the Duke Clinical Research Institute (DCRI), Durham, N.C., which was commissioned to do the reanalysis by rosiglitazone manufacturer GlaxoSmithKline, which was directed to do so by the FDA.* The DCRI reanalysis found no significant differences in the composite of CV death, MI, or stroke; numerically fewer CV deaths and numerically fewer strokes in the rosiglitazone arm; and a lower risk of all cause death. There were more MIs in the rosiglitazone arm, but the difference was not significant.
On the basis of those results, the panel did not conclude that the data showed there was no increased cardiovascular risk, but generally felt reassured by the results. One of the cardiologists on the panel, Dr. Marvin Konstam, professor of medicine, Tufts University, Boston, said that the readjudicated results of RECORD "do not remove my concern about the cardiovascular safety of rosiglitazone, but in my mind, they move the needle in a direction that seems to me is appropriate to shift the burden of decision making for prescribing this drug to physicians in terms of interpreting the entirety of the data."Dr. Konstam voted for modification of the REMS.
"I still think there’s unresolved concern about an adverse cardiovascular effect or effects, but I don’t wish to deprive qualified patients rosiglitazone as an option, even in this context," said Dr. Robert Smith, an endocrinologist who is professor of medicine at Brown University, Providence, R.I.., who voted that the REMS should stay in place.*
Endocrinologist Dr. Ellen Seely, professor of medicine at Harvard Medical School, Boston, was among those voting to remove the REMS. Although she was not convinced that there was no such risk associated with rosiglitazone, "I don’t think we have data to support there is a cardiovascular risk," she said. There is a great need for drugs that type 2 diabetes that do not cause hypoglycemia, and it is helpful to have options for those patients who have exhausted most treatment options, who may not want to take pioglitazone because of the association with bladder cancer, or who are resistant to taking insulin, she added.
Under the REMS, which was fully implemented in May 2011, prescribers of rosiglitazone must be certified, and the drug can only be dispensed from one of four speciality mail-order pharmacies to patients with documentation to prove that the prescriber has discussed with them the potential increased risk of MI associated with the drug – and that they are either already taking rosiglitazone, or are unable to achieve glycemic control on other medications, and "in consultation with their health care provider, have decided not to take pioglitazone for medical reasons."
The FDA announced the REMS would be required in September 2010, several months after the same advisory panels met to discuss data from meta-analyses and other studies evaluating the CV safety of rosiglitazone, and the majority recommended restrictions on prescribing and a significant proportion recommending that it be taken off the market. The FDA also requested that GSK commission an independent blinded readjudication of RECORD, an open-label CV safety trial conducted outside of the United States, to address concerns of the panelists at the July 2010 meeting about the integrity and reliability of the data because of missing data and other issues.
The panel was satisfied with the results and largely agreed that these concerns were allayed and that the RECORD results could be considered along with the other studies that provided safety data on rosiglitazone, although none were ideal.
One of the panelists who voted to keep the REMS as it exists, Dr. Maria Suarez-Almazor of University of Texas MD Anderson Cancer Center, Houston, said she voted for no change because "there were consistent signals for MI in the observational data and also in the RECORD trial," and although they were not statistically significant, she did not see a clear benefit of using this drug over pioglitazone, other than the bladder cancer issue.
In 2011, 59% of all available rosiglitazone products, including combination products that contain the drug, were in retail pharmacies, and 34% were available through mail-order pharmacies. In 2012, 97% were in mail-order pharmacies and less than 0.1% were in retail pharmacies, according to the FDA. Between July 2007 and July 2010, 1.6 million patients received prescriptions for rosiglitazone, dropping to about 4,600 patients in 2012. Based on national prescription data, between May 2011 and December 2012, after the REMS was put in place, rosiglitazone was most commonly prescribed by physicians in family or general practice (about 52% of prescribers) and internists (about 32%), followed by endocrinologists (almost 4%), according to the FDA. These proportions are similar to the period from 2007, up until the REMS.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
In a statement released by GSK shortly after the panel meeting ended, Dr. James Shannon, GSK’s chief medical officer, said that the company "will continue to work with the FDA as it considers the recommendation of the committee." Rosiglitazone was approved by the FDA in 1999. Earlier, on the first day of the meeting, GSK indicated that it had no plans to market rosiglitazone or any products containing it.
Correction, 06/07/2013: An earlier version of this story misstated the organization that commissioned the reanalysis of the data.
Correction, 06/12/2013: An earlier version of Dr. Nissen's video interview incorrectly stated that the rest of the world had banned rosiglitazone. The drug remains available in some countries, in addition to the United States.
Correction, 06/12/2013: An earlier version of this story misstated Dr. Smith's vote.
SILVER SPRING, MD. – Reassured by the results of a reanalysis of data from a cardiovascular safety trial of rosiglitazone, the majority of a Food and Drug Administration advisory panel recommended either loosening or lifting the restrictions placed on prescribing of the type 2 diabetes drug in 2010.
At a June 6 meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee, 13 panel members voted to modify the Risk Evaluation and Mitigation Strategy (REMS) required for the drug and seven recommended that it be removed altogether. Of the remaining six panelists, five recommended that the REMS be continued as it currently exists and one panelist, the consumer representative, voted to withdraw the drug from the market.
The FDA convened the 2-day meeting to review the results of the readjudication of data from the RECORD (Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycemia in Diabetes) trial, conducted by the Duke Clinical Research Institute (DCRI), Durham, N.C., which was commissioned to do the reanalysis by rosiglitazone manufacturer GlaxoSmithKline, which was directed to do so by the FDA.* The DCRI reanalysis found no significant differences in the composite of CV death, MI, or stroke; numerically fewer CV deaths and numerically fewer strokes in the rosiglitazone arm; and a lower risk of all cause death. There were more MIs in the rosiglitazone arm, but the difference was not significant.
On the basis of those results, the panel did not conclude that the data showed there was no increased cardiovascular risk, but generally felt reassured by the results. One of the cardiologists on the panel, Dr. Marvin Konstam, professor of medicine, Tufts University, Boston, said that the readjudicated results of RECORD "do not remove my concern about the cardiovascular safety of rosiglitazone, but in my mind, they move the needle in a direction that seems to me is appropriate to shift the burden of decision making for prescribing this drug to physicians in terms of interpreting the entirety of the data."Dr. Konstam voted for modification of the REMS.
"I still think there’s unresolved concern about an adverse cardiovascular effect or effects, but I don’t wish to deprive qualified patients rosiglitazone as an option, even in this context," said Dr. Robert Smith, an endocrinologist who is professor of medicine at Brown University, Providence, R.I.., who voted that the REMS should stay in place.*
Endocrinologist Dr. Ellen Seely, professor of medicine at Harvard Medical School, Boston, was among those voting to remove the REMS. Although she was not convinced that there was no such risk associated with rosiglitazone, "I don’t think we have data to support there is a cardiovascular risk," she said. There is a great need for drugs that type 2 diabetes that do not cause hypoglycemia, and it is helpful to have options for those patients who have exhausted most treatment options, who may not want to take pioglitazone because of the association with bladder cancer, or who are resistant to taking insulin, she added.
Under the REMS, which was fully implemented in May 2011, prescribers of rosiglitazone must be certified, and the drug can only be dispensed from one of four speciality mail-order pharmacies to patients with documentation to prove that the prescriber has discussed with them the potential increased risk of MI associated with the drug – and that they are either already taking rosiglitazone, or are unable to achieve glycemic control on other medications, and "in consultation with their health care provider, have decided not to take pioglitazone for medical reasons."
The FDA announced the REMS would be required in September 2010, several months after the same advisory panels met to discuss data from meta-analyses and other studies evaluating the CV safety of rosiglitazone, and the majority recommended restrictions on prescribing and a significant proportion recommending that it be taken off the market. The FDA also requested that GSK commission an independent blinded readjudication of RECORD, an open-label CV safety trial conducted outside of the United States, to address concerns of the panelists at the July 2010 meeting about the integrity and reliability of the data because of missing data and other issues.
The panel was satisfied with the results and largely agreed that these concerns were allayed and that the RECORD results could be considered along with the other studies that provided safety data on rosiglitazone, although none were ideal.
One of the panelists who voted to keep the REMS as it exists, Dr. Maria Suarez-Almazor of University of Texas MD Anderson Cancer Center, Houston, said she voted for no change because "there were consistent signals for MI in the observational data and also in the RECORD trial," and although they were not statistically significant, she did not see a clear benefit of using this drug over pioglitazone, other than the bladder cancer issue.
In 2011, 59% of all available rosiglitazone products, including combination products that contain the drug, were in retail pharmacies, and 34% were available through mail-order pharmacies. In 2012, 97% were in mail-order pharmacies and less than 0.1% were in retail pharmacies, according to the FDA. Between July 2007 and July 2010, 1.6 million patients received prescriptions for rosiglitazone, dropping to about 4,600 patients in 2012. Based on national prescription data, between May 2011 and December 2012, after the REMS was put in place, rosiglitazone was most commonly prescribed by physicians in family or general practice (about 52% of prescribers) and internists (about 32%), followed by endocrinologists (almost 4%), according to the FDA. These proportions are similar to the period from 2007, up until the REMS.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
In a statement released by GSK shortly after the panel meeting ended, Dr. James Shannon, GSK’s chief medical officer, said that the company "will continue to work with the FDA as it considers the recommendation of the committee." Rosiglitazone was approved by the FDA in 1999. Earlier, on the first day of the meeting, GSK indicated that it had no plans to market rosiglitazone or any products containing it.
Correction, 06/07/2013: An earlier version of this story misstated the organization that commissioned the reanalysis of the data.
Correction, 06/12/2013: An earlier version of Dr. Nissen's video interview incorrectly stated that the rest of the world had banned rosiglitazone. The drug remains available in some countries, in addition to the United States.
Correction, 06/12/2013: An earlier version of this story misstated Dr. Smith's vote.
SILVER SPRING, MD. – Reassured by the results of a reanalysis of data from a cardiovascular safety trial of rosiglitazone, the majority of a Food and Drug Administration advisory panel recommended either loosening or lifting the restrictions placed on prescribing of the type 2 diabetes drug in 2010.
At a June 6 meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee, 13 panel members voted to modify the Risk Evaluation and Mitigation Strategy (REMS) required for the drug and seven recommended that it be removed altogether. Of the remaining six panelists, five recommended that the REMS be continued as it currently exists and one panelist, the consumer representative, voted to withdraw the drug from the market.
The FDA convened the 2-day meeting to review the results of the readjudication of data from the RECORD (Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycemia in Diabetes) trial, conducted by the Duke Clinical Research Institute (DCRI), Durham, N.C., which was commissioned to do the reanalysis by rosiglitazone manufacturer GlaxoSmithKline, which was directed to do so by the FDA.* The DCRI reanalysis found no significant differences in the composite of CV death, MI, or stroke; numerically fewer CV deaths and numerically fewer strokes in the rosiglitazone arm; and a lower risk of all cause death. There were more MIs in the rosiglitazone arm, but the difference was not significant.
On the basis of those results, the panel did not conclude that the data showed there was no increased cardiovascular risk, but generally felt reassured by the results. One of the cardiologists on the panel, Dr. Marvin Konstam, professor of medicine, Tufts University, Boston, said that the readjudicated results of RECORD "do not remove my concern about the cardiovascular safety of rosiglitazone, but in my mind, they move the needle in a direction that seems to me is appropriate to shift the burden of decision making for prescribing this drug to physicians in terms of interpreting the entirety of the data."Dr. Konstam voted for modification of the REMS.
"I still think there’s unresolved concern about an adverse cardiovascular effect or effects, but I don’t wish to deprive qualified patients rosiglitazone as an option, even in this context," said Dr. Robert Smith, an endocrinologist who is professor of medicine at Brown University, Providence, R.I.., who voted that the REMS should stay in place.*
Endocrinologist Dr. Ellen Seely, professor of medicine at Harvard Medical School, Boston, was among those voting to remove the REMS. Although she was not convinced that there was no such risk associated with rosiglitazone, "I don’t think we have data to support there is a cardiovascular risk," she said. There is a great need for drugs that type 2 diabetes that do not cause hypoglycemia, and it is helpful to have options for those patients who have exhausted most treatment options, who may not want to take pioglitazone because of the association with bladder cancer, or who are resistant to taking insulin, she added.
Under the REMS, which was fully implemented in May 2011, prescribers of rosiglitazone must be certified, and the drug can only be dispensed from one of four speciality mail-order pharmacies to patients with documentation to prove that the prescriber has discussed with them the potential increased risk of MI associated with the drug – and that they are either already taking rosiglitazone, or are unable to achieve glycemic control on other medications, and "in consultation with their health care provider, have decided not to take pioglitazone for medical reasons."
The FDA announced the REMS would be required in September 2010, several months after the same advisory panels met to discuss data from meta-analyses and other studies evaluating the CV safety of rosiglitazone, and the majority recommended restrictions on prescribing and a significant proportion recommending that it be taken off the market. The FDA also requested that GSK commission an independent blinded readjudication of RECORD, an open-label CV safety trial conducted outside of the United States, to address concerns of the panelists at the July 2010 meeting about the integrity and reliability of the data because of missing data and other issues.
The panel was satisfied with the results and largely agreed that these concerns were allayed and that the RECORD results could be considered along with the other studies that provided safety data on rosiglitazone, although none were ideal.
One of the panelists who voted to keep the REMS as it exists, Dr. Maria Suarez-Almazor of University of Texas MD Anderson Cancer Center, Houston, said she voted for no change because "there were consistent signals for MI in the observational data and also in the RECORD trial," and although they were not statistically significant, she did not see a clear benefit of using this drug over pioglitazone, other than the bladder cancer issue.
In 2011, 59% of all available rosiglitazone products, including combination products that contain the drug, were in retail pharmacies, and 34% were available through mail-order pharmacies. In 2012, 97% were in mail-order pharmacies and less than 0.1% were in retail pharmacies, according to the FDA. Between July 2007 and July 2010, 1.6 million patients received prescriptions for rosiglitazone, dropping to about 4,600 patients in 2012. Based on national prescription data, between May 2011 and December 2012, after the REMS was put in place, rosiglitazone was most commonly prescribed by physicians in family or general practice (about 52% of prescribers) and internists (about 32%), followed by endocrinologists (almost 4%), according to the FDA. These proportions are similar to the period from 2007, up until the REMS.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
In a statement released by GSK shortly after the panel meeting ended, Dr. James Shannon, GSK’s chief medical officer, said that the company "will continue to work with the FDA as it considers the recommendation of the committee." Rosiglitazone was approved by the FDA in 1999. Earlier, on the first day of the meeting, GSK indicated that it had no plans to market rosiglitazone or any products containing it.
Correction, 06/07/2013: An earlier version of this story misstated the organization that commissioned the reanalysis of the data.
Correction, 06/12/2013: An earlier version of Dr. Nissen's video interview incorrectly stated that the rest of the world had banned rosiglitazone. The drug remains available in some countries, in addition to the United States.
Correction, 06/12/2013: An earlier version of this story misstated Dr. Smith's vote.
AT AN FDA ADVISORY PANEL MEETING

FDA panel supports easing rosiglitazone restrictions
SILVER SPRING, MD. – Reassured by the results of a reanalysis of data from a cardiovascular safety trial of rosiglitazone, the majority of a Food and Drug Administration advisory panel recommended either loosening or lifting the restrictions placed on prescribing of the type 2 diabetes drug in 2010.
At a June 6 meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee, 13 panel members voted to modify the Risk Evaluation and Mitigation Strategy (REMS) required for the drug and seven recommended that it be removed altogether. Of the remaining six panelists, five recommended that the REMS be continued as it currently exists and one panelist, the consumer representative, voted to withdraw the drug from the market.
The FDA convened the 2-day meeting to review the results of the readjudication of data from the RECORD (Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycemia in Diabetes) trial, conducted by the Duke Clinical Research Institute (DCRI), Durham, N.C., which was commissioned to do the reanalysis by rosiglitazone manufacturer GlaxoSmithKline, which was directed to do so by the FDA.* The DCRI reanalysis found no significant differences in the composite of CV death, MI, or stroke; numerically fewer CV deaths and numerically fewer strokes in the rosiglitazone arm; and a lower risk of all cause death. There were more MIs in the rosiglitazone arm, but the difference was not significant.
On the basis of those results, the panel did not conclude that the data showed there was no increased cardiovascular risk, but generally felt reassured by the results. One of the cardiologists on the panel, Dr. Marvin Konstam, professor of medicine, Tufts University, Boston, said that the readjudicated results of RECORD "do not remove my concern about the cardiovascular safety of rosiglitazone, but in my mind, they move the needle in a direction that seems to me is appropriate to shift the burden of decision making for prescribing this drug to physicians in terms of interpreting the entirety of the data."Dr. Konstam voted for modification of the REMS.
"I still think there’s unresolved concern about an adverse cardiovascular effect or effects, but I don’t wish to deprive qualified patients rosiglitazone as an option, even in this context," said Dr. Robert Smith, an endocrinologist who is professor of medicine at Brown University, Providence, R.I.., who voted that the REMS should stay in place.*
Endocrinologist Dr. Ellen Seely, professor of medicine at Harvard Medical School, Boston, was among those voting to remove the REMS. Although she was not convinced that there was no such risk associated with rosiglitazone, "I don’t think we have data to support there is a cardiovascular risk," she said. There is a great need for drugs that type 2 diabetes that do not cause hypoglycemia, and it is helpful to have options for those patients who have exhausted most treatment options, who may not want to take pioglitazone because of the association with bladder cancer, or who are resistant to taking insulin, she added.
Under the REMS, which was fully implemented in May 2011, prescribers of rosiglitazone must be certified, and the drug can only be dispensed from one of four speciality mail-order pharmacies to patients with documentation to prove that the prescriber has discussed with them the potential increased risk of MI associated with the drug – and that they are either already taking rosiglitazone, or are unable to achieve glycemic control on other medications, and "in consultation with their health care provider, have decided not to take pioglitazone for medical reasons."
The FDA announced the REMS would be required in September 2010, several months after the same advisory panels met to discuss data from meta-analyses and other studies evaluating the CV safety of rosiglitazone, and the majority recommended restrictions on prescribing and a significant proportion recommending that it be taken off the market. The FDA also requested that GSK commission an independent blinded readjudication of RECORD, an open-label CV safety trial conducted outside of the United States, to address concerns of the panelists at the July 2010 meeting about the integrity and reliability of the data because of missing data and other issues.
The panel was satisfied with the results and largely agreed that these concerns were allayed and that the RECORD results could be considered along with the other studies that provided safety data on rosiglitazone, although none were ideal.
One of the panelists who voted to keep the REMS as it exists, Dr. Maria Suarez-Almazor of University of Texas MD Anderson Cancer Center, Houston, said she voted for no change because "there were consistent signals for MI in the observational data and also in the RECORD trial," and although they were not statistically significant, she did not see a clear benefit of using this drug over pioglitazone, other than the bladder cancer issue.
In 2011, 59% of all available rosiglitazone products, including combination products that contain the drug, were in retail pharmacies, and 34% were available through mail-order pharmacies. In 2012, 97% were in mail-order pharmacies and less than 0.1% were in retail pharmacies, according to the FDA. Between July 2007 and July 2010, 1.6 million patients received prescriptions for rosiglitazone, dropping to about 4,600 patients in 2012. Based on national prescription data, between May 2011 and December 2012, after the REMS was put in place, rosiglitazone was most commonly prescribed by physicians in family or general practice (about 52% of prescribers) and internists (about 32%), followed by endocrinologists (almost 4%), according to the FDA. These proportions are similar to the period from 2007, up until the REMS.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
In a statement released by GSK shortly after the panel meeting ended, Dr. James Shannon, GSK’s chief medical officer, said that the company "will continue to work with the FDA as it considers the recommendation of the committee." Rosiglitazone was approved by the FDA in 1999. Earlier, on the first day of the meeting, GSK indicated that it had no plans to market rosiglitazone or any products containing it.
Correction, 06/07/2013: An earlier version of this story misstated the organization that commissioned the reanalysis of the data.
Correction, 06/12/2013: An earlier version of Dr. Nissen's video interview incorrectly stated that the rest of the world had banned rosiglitazone. The drug remains available in some countries, in addition to the United States.
Correction, 06/12/2013: An earlier version of this story misstated Dr. Smith's vote.
SILVER SPRING, MD. – Reassured by the results of a reanalysis of data from a cardiovascular safety trial of rosiglitazone, the majority of a Food and Drug Administration advisory panel recommended either loosening or lifting the restrictions placed on prescribing of the type 2 diabetes drug in 2010.
At a June 6 meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee, 13 panel members voted to modify the Risk Evaluation and Mitigation Strategy (REMS) required for the drug and seven recommended that it be removed altogether. Of the remaining six panelists, five recommended that the REMS be continued as it currently exists and one panelist, the consumer representative, voted to withdraw the drug from the market.
The FDA convened the 2-day meeting to review the results of the readjudication of data from the RECORD (Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycemia in Diabetes) trial, conducted by the Duke Clinical Research Institute (DCRI), Durham, N.C., which was commissioned to do the reanalysis by rosiglitazone manufacturer GlaxoSmithKline, which was directed to do so by the FDA.* The DCRI reanalysis found no significant differences in the composite of CV death, MI, or stroke; numerically fewer CV deaths and numerically fewer strokes in the rosiglitazone arm; and a lower risk of all cause death. There were more MIs in the rosiglitazone arm, but the difference was not significant.
On the basis of those results, the panel did not conclude that the data showed there was no increased cardiovascular risk, but generally felt reassured by the results. One of the cardiologists on the panel, Dr. Marvin Konstam, professor of medicine, Tufts University, Boston, said that the readjudicated results of RECORD "do not remove my concern about the cardiovascular safety of rosiglitazone, but in my mind, they move the needle in a direction that seems to me is appropriate to shift the burden of decision making for prescribing this drug to physicians in terms of interpreting the entirety of the data."Dr. Konstam voted for modification of the REMS.
"I still think there’s unresolved concern about an adverse cardiovascular effect or effects, but I don’t wish to deprive qualified patients rosiglitazone as an option, even in this context," said Dr. Robert Smith, an endocrinologist who is professor of medicine at Brown University, Providence, R.I.., who voted that the REMS should stay in place.*
Endocrinologist Dr. Ellen Seely, professor of medicine at Harvard Medical School, Boston, was among those voting to remove the REMS. Although she was not convinced that there was no such risk associated with rosiglitazone, "I don’t think we have data to support there is a cardiovascular risk," she said. There is a great need for drugs that type 2 diabetes that do not cause hypoglycemia, and it is helpful to have options for those patients who have exhausted most treatment options, who may not want to take pioglitazone because of the association with bladder cancer, or who are resistant to taking insulin, she added.
Under the REMS, which was fully implemented in May 2011, prescribers of rosiglitazone must be certified, and the drug can only be dispensed from one of four speciality mail-order pharmacies to patients with documentation to prove that the prescriber has discussed with them the potential increased risk of MI associated with the drug – and that they are either already taking rosiglitazone, or are unable to achieve glycemic control on other medications, and "in consultation with their health care provider, have decided not to take pioglitazone for medical reasons."
The FDA announced the REMS would be required in September 2010, several months after the same advisory panels met to discuss data from meta-analyses and other studies evaluating the CV safety of rosiglitazone, and the majority recommended restrictions on prescribing and a significant proportion recommending that it be taken off the market. The FDA also requested that GSK commission an independent blinded readjudication of RECORD, an open-label CV safety trial conducted outside of the United States, to address concerns of the panelists at the July 2010 meeting about the integrity and reliability of the data because of missing data and other issues.
The panel was satisfied with the results and largely agreed that these concerns were allayed and that the RECORD results could be considered along with the other studies that provided safety data on rosiglitazone, although none were ideal.
One of the panelists who voted to keep the REMS as it exists, Dr. Maria Suarez-Almazor of University of Texas MD Anderson Cancer Center, Houston, said she voted for no change because "there were consistent signals for MI in the observational data and also in the RECORD trial," and although they were not statistically significant, she did not see a clear benefit of using this drug over pioglitazone, other than the bladder cancer issue.
In 2011, 59% of all available rosiglitazone products, including combination products that contain the drug, were in retail pharmacies, and 34% were available through mail-order pharmacies. In 2012, 97% were in mail-order pharmacies and less than 0.1% were in retail pharmacies, according to the FDA. Between July 2007 and July 2010, 1.6 million patients received prescriptions for rosiglitazone, dropping to about 4,600 patients in 2012. Based on national prescription data, between May 2011 and December 2012, after the REMS was put in place, rosiglitazone was most commonly prescribed by physicians in family or general practice (about 52% of prescribers) and internists (about 32%), followed by endocrinologists (almost 4%), according to the FDA. These proportions are similar to the period from 2007, up until the REMS.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
In a statement released by GSK shortly after the panel meeting ended, Dr. James Shannon, GSK’s chief medical officer, said that the company "will continue to work with the FDA as it considers the recommendation of the committee." Rosiglitazone was approved by the FDA in 1999. Earlier, on the first day of the meeting, GSK indicated that it had no plans to market rosiglitazone or any products containing it.
Correction, 06/07/2013: An earlier version of this story misstated the organization that commissioned the reanalysis of the data.
Correction, 06/12/2013: An earlier version of Dr. Nissen's video interview incorrectly stated that the rest of the world had banned rosiglitazone. The drug remains available in some countries, in addition to the United States.
Correction, 06/12/2013: An earlier version of this story misstated Dr. Smith's vote.
SILVER SPRING, MD. – Reassured by the results of a reanalysis of data from a cardiovascular safety trial of rosiglitazone, the majority of a Food and Drug Administration advisory panel recommended either loosening or lifting the restrictions placed on prescribing of the type 2 diabetes drug in 2010.
At a June 6 meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee, 13 panel members voted to modify the Risk Evaluation and Mitigation Strategy (REMS) required for the drug and seven recommended that it be removed altogether. Of the remaining six panelists, five recommended that the REMS be continued as it currently exists and one panelist, the consumer representative, voted to withdraw the drug from the market.
The FDA convened the 2-day meeting to review the results of the readjudication of data from the RECORD (Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycemia in Diabetes) trial, conducted by the Duke Clinical Research Institute (DCRI), Durham, N.C., which was commissioned to do the reanalysis by rosiglitazone manufacturer GlaxoSmithKline, which was directed to do so by the FDA.* The DCRI reanalysis found no significant differences in the composite of CV death, MI, or stroke; numerically fewer CV deaths and numerically fewer strokes in the rosiglitazone arm; and a lower risk of all cause death. There were more MIs in the rosiglitazone arm, but the difference was not significant.
On the basis of those results, the panel did not conclude that the data showed there was no increased cardiovascular risk, but generally felt reassured by the results. One of the cardiologists on the panel, Dr. Marvin Konstam, professor of medicine, Tufts University, Boston, said that the readjudicated results of RECORD "do not remove my concern about the cardiovascular safety of rosiglitazone, but in my mind, they move the needle in a direction that seems to me is appropriate to shift the burden of decision making for prescribing this drug to physicians in terms of interpreting the entirety of the data."Dr. Konstam voted for modification of the REMS.
"I still think there’s unresolved concern about an adverse cardiovascular effect or effects, but I don’t wish to deprive qualified patients rosiglitazone as an option, even in this context," said Dr. Robert Smith, an endocrinologist who is professor of medicine at Brown University, Providence, R.I.., who voted that the REMS should stay in place.*
Endocrinologist Dr. Ellen Seely, professor of medicine at Harvard Medical School, Boston, was among those voting to remove the REMS. Although she was not convinced that there was no such risk associated with rosiglitazone, "I don’t think we have data to support there is a cardiovascular risk," she said. There is a great need for drugs that type 2 diabetes that do not cause hypoglycemia, and it is helpful to have options for those patients who have exhausted most treatment options, who may not want to take pioglitazone because of the association with bladder cancer, or who are resistant to taking insulin, she added.
Under the REMS, which was fully implemented in May 2011, prescribers of rosiglitazone must be certified, and the drug can only be dispensed from one of four speciality mail-order pharmacies to patients with documentation to prove that the prescriber has discussed with them the potential increased risk of MI associated with the drug – and that they are either already taking rosiglitazone, or are unable to achieve glycemic control on other medications, and "in consultation with their health care provider, have decided not to take pioglitazone for medical reasons."
The FDA announced the REMS would be required in September 2010, several months after the same advisory panels met to discuss data from meta-analyses and other studies evaluating the CV safety of rosiglitazone, and the majority recommended restrictions on prescribing and a significant proportion recommending that it be taken off the market. The FDA also requested that GSK commission an independent blinded readjudication of RECORD, an open-label CV safety trial conducted outside of the United States, to address concerns of the panelists at the July 2010 meeting about the integrity and reliability of the data because of missing data and other issues.
The panel was satisfied with the results and largely agreed that these concerns were allayed and that the RECORD results could be considered along with the other studies that provided safety data on rosiglitazone, although none were ideal.
One of the panelists who voted to keep the REMS as it exists, Dr. Maria Suarez-Almazor of University of Texas MD Anderson Cancer Center, Houston, said she voted for no change because "there were consistent signals for MI in the observational data and also in the RECORD trial," and although they were not statistically significant, she did not see a clear benefit of using this drug over pioglitazone, other than the bladder cancer issue.
In 2011, 59% of all available rosiglitazone products, including combination products that contain the drug, were in retail pharmacies, and 34% were available through mail-order pharmacies. In 2012, 97% were in mail-order pharmacies and less than 0.1% were in retail pharmacies, according to the FDA. Between July 2007 and July 2010, 1.6 million patients received prescriptions for rosiglitazone, dropping to about 4,600 patients in 2012. Based on national prescription data, between May 2011 and December 2012, after the REMS was put in place, rosiglitazone was most commonly prescribed by physicians in family or general practice (about 52% of prescribers) and internists (about 32%), followed by endocrinologists (almost 4%), according to the FDA. These proportions are similar to the period from 2007, up until the REMS.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
In a statement released by GSK shortly after the panel meeting ended, Dr. James Shannon, GSK’s chief medical officer, said that the company "will continue to work with the FDA as it considers the recommendation of the committee." Rosiglitazone was approved by the FDA in 1999. Earlier, on the first day of the meeting, GSK indicated that it had no plans to market rosiglitazone or any products containing it.
Correction, 06/07/2013: An earlier version of this story misstated the organization that commissioned the reanalysis of the data.
Correction, 06/12/2013: An earlier version of Dr. Nissen's video interview incorrectly stated that the rest of the world had banned rosiglitazone. The drug remains available in some countries, in addition to the United States.
Correction, 06/12/2013: An earlier version of this story misstated Dr. Smith's vote.
FDA panel starts review of rosiglitazone CV data
SILVER SPRING, MD. – A reanalysis of data from a controversial cardiovascular safety study of rosiglitazone was well-conducted and provided results that were "highly similar" to the original results of the study, according to Food and Drug Administration officials who spoke during the first day of a 2-day advisory panel meeting focusing on the results of the reanalysis.
The meeting – of the FDA’s Endocrinologic Drugs and Drug Safety and Risk Management advisory committees – is being held to discuss the results of the independent readjudication of the results of the RECORD (Rosiglitazone Evaluated for Cardiovascular Outcomes in Oral Agent Combination Therapy for Type 2 Diabetes) trial, a prospective, open-label, noninferiority study conducted by rosiglitazone manufacturer GlaxoSmithKline in response to a request by European drug regulators to address concerns over possible CV risks of the drug.
RECORD is the only large randomized controlled CV outcomes study of rosiglitazone, and overall, the readjudication of the study "appears to support the previous observation that, in this trial, rosiglitazone was not associated with an increased risk of death or major adverse cardiovascular events," Dr. Karen Mahoney, diabetes team leader in the FDA’s division of metabolism and endocrinology products said at the meeting.
How the panel interprets these data could impact the availability of rosiglitazone, which is used to treat about 3,400 patients in the United States, down from about 117,000 patients the year before the FDA instituted a risk evaluation and mitigation strategy (REMS) to limit its use because of cardiovascular safety concerns. On the second day of the meeting, June 6, the panel will discuss these data and will vote on whether the drug should remain on the market with the same restrictions, whether the REMs and other restrictions should be lifted or modified, or whether it should be taken off the market altogether based on the readjudicated results and other available data. In Europe, rosiglitazone is no longer marketed because of concerns over CV risks.
There have been longstanding concerns that rosiglitazone may be associated with an increased risk of ischemic CV events, but the data have been mixed and limited by study designs and other issues.
But in 2007, a meta-analysis of 42 studies showed that rosiglitazone increased the risk of MI by up to 43% and the risk of CV death by up to 64% (N. Engl. J. Med. 2007;356:2457-71); the lead author was Dr. Steven Nissen, a vocal advocate for removing rosiglitazone from the market.
Considering these and other data, at a meeting of the same panels in July 2010, the majority of the panelists agreed that the data raised significant safety concerns regarding the risk of rosiglitazone and most recommended that the use of the drug be limited, but a significant proportion recommended that it be taken off the U.S. market. In September 2010, the FDA announced that rosiglitazone would remain available, with the REMS, limiting the drug to patients who were already on rosiglitazone, or for patients whose blood glucose could not be managed adequately on other medications and, could not take the only other approved thiazolidinedione (TZD) pioglitazone, "for medical reasons."
At that time, the FDA asked GSK to commission an independent readjudication of the RECORD data, which found a nonsignificant increase in MI in the rosiglitazone arm, along with a nonsignificant reduction in the risk of stroke and all-cause mortality. But the reliability of the study has been questioned because of the study design and other issues such as missing data, and in September 2010, when the FDA announced that rosiglitazone would be marketed under a risk evaluation and mitigation strategy, the agency requested that the company commission an independent readjudication of the data to determine if the results could reliably be used to evaluate the drug’s CV safety.
RECORD enrolled about 4,500 patients with type 2 diabetes in 364 centers in 25 countries outside of the United States, comparing CV outcomes among those on rosiglitazone with metformin or a sulfonylurea and metformin. Because of limitations of the study, the reliability of these results has been questioned by experts within and outside the FDA. GSK commissioned the Duke Clinical Research Institute (DCRI) to conduct the reanalysis, which involved blinded adjudication of more than 2,200 deaths, MIs, and strokes. The results, presented at the meeting on June 5, were consistent with the original RECORD results, with no significant differences in the composite of CV death, MI, or stroke; numerically fewer CV deaths and numerically fewer strokes in the rosiglitazone arm; and a lower risk of all cause death. There were more MIs in the rosiglitazone arm, but the difference was not significant.
Dr. Preston Dunnmon, a medical reviewer in the FDA’s division of cardiovascular and renal drug products, said that the adjudication was "well conceived, well executed, and comprehensive" and that the small number of additional MACE events identified during adjudication did not change the original results reported for RECORD. The conclusions do not consider the impact of the open-label design of the study, but he said there was no convincing evidence of any manipulation, intentional or otherwise, of the safety outcomes in RECORD.
However, one FDA reviewer adamantly disagreed. Dr. Thomas Marciniak, medical team leader in the FDA’s division of cardiovascular and renal products, presented what he said were his own professional opinions, not the FDA’s official views. He said that RECORD was inadequately designed and not reliable, and "confirms and extends the recognized concerns regarding increased heart failure and heart failure deaths with rosiglitazone," and suggests that rosiglitazone increases the risk of MI.
He said that the readjudication process was not truly independent, did not collect much additional information, and that it did not overcome the flaws in the study’s design and what he said was mishandling of data.
Several panelists remarked that the unblinded design of the study was a limitation.
At the July 2010 meeting, 12 of the panelists voted to take the drug off the market, and 17 voted to continue marketing the drug, but with revisions. Compared to the year before the REMS program was instituted, the drug was available in about 55,000 pharmacies and was prescribed by almost 109,000 clinicians, which has dropped to 4 pharmacies and about 2,700 prescribers, according to GSK.
SILVER SPRING, MD. – A reanalysis of data from a controversial cardiovascular safety study of rosiglitazone was well-conducted and provided results that were "highly similar" to the original results of the study, according to Food and Drug Administration officials who spoke during the first day of a 2-day advisory panel meeting focusing on the results of the reanalysis.
The meeting – of the FDA’s Endocrinologic Drugs and Drug Safety and Risk Management advisory committees – is being held to discuss the results of the independent readjudication of the results of the RECORD (Rosiglitazone Evaluated for Cardiovascular Outcomes in Oral Agent Combination Therapy for Type 2 Diabetes) trial, a prospective, open-label, noninferiority study conducted by rosiglitazone manufacturer GlaxoSmithKline in response to a request by European drug regulators to address concerns over possible CV risks of the drug.
RECORD is the only large randomized controlled CV outcomes study of rosiglitazone, and overall, the readjudication of the study "appears to support the previous observation that, in this trial, rosiglitazone was not associated with an increased risk of death or major adverse cardiovascular events," Dr. Karen Mahoney, diabetes team leader in the FDA’s division of metabolism and endocrinology products said at the meeting.
How the panel interprets these data could impact the availability of rosiglitazone, which is used to treat about 3,400 patients in the United States, down from about 117,000 patients the year before the FDA instituted a risk evaluation and mitigation strategy (REMS) to limit its use because of cardiovascular safety concerns. On the second day of the meeting, June 6, the panel will discuss these data and will vote on whether the drug should remain on the market with the same restrictions, whether the REMs and other restrictions should be lifted or modified, or whether it should be taken off the market altogether based on the readjudicated results and other available data. In Europe, rosiglitazone is no longer marketed because of concerns over CV risks.
There have been longstanding concerns that rosiglitazone may be associated with an increased risk of ischemic CV events, but the data have been mixed and limited by study designs and other issues.
But in 2007, a meta-analysis of 42 studies showed that rosiglitazone increased the risk of MI by up to 43% and the risk of CV death by up to 64% (N. Engl. J. Med. 2007;356:2457-71); the lead author was Dr. Steven Nissen, a vocal advocate for removing rosiglitazone from the market.
Considering these and other data, at a meeting of the same panels in July 2010, the majority of the panelists agreed that the data raised significant safety concerns regarding the risk of rosiglitazone and most recommended that the use of the drug be limited, but a significant proportion recommended that it be taken off the U.S. market. In September 2010, the FDA announced that rosiglitazone would remain available, with the REMS, limiting the drug to patients who were already on rosiglitazone, or for patients whose blood glucose could not be managed adequately on other medications and, could not take the only other approved thiazolidinedione (TZD) pioglitazone, "for medical reasons."
At that time, the FDA asked GSK to commission an independent readjudication of the RECORD data, which found a nonsignificant increase in MI in the rosiglitazone arm, along with a nonsignificant reduction in the risk of stroke and all-cause mortality. But the reliability of the study has been questioned because of the study design and other issues such as missing data, and in September 2010, when the FDA announced that rosiglitazone would be marketed under a risk evaluation and mitigation strategy, the agency requested that the company commission an independent readjudication of the data to determine if the results could reliably be used to evaluate the drug’s CV safety.
RECORD enrolled about 4,500 patients with type 2 diabetes in 364 centers in 25 countries outside of the United States, comparing CV outcomes among those on rosiglitazone with metformin or a sulfonylurea and metformin. Because of limitations of the study, the reliability of these results has been questioned by experts within and outside the FDA. GSK commissioned the Duke Clinical Research Institute (DCRI) to conduct the reanalysis, which involved blinded adjudication of more than 2,200 deaths, MIs, and strokes. The results, presented at the meeting on June 5, were consistent with the original RECORD results, with no significant differences in the composite of CV death, MI, or stroke; numerically fewer CV deaths and numerically fewer strokes in the rosiglitazone arm; and a lower risk of all cause death. There were more MIs in the rosiglitazone arm, but the difference was not significant.
Dr. Preston Dunnmon, a medical reviewer in the FDA’s division of cardiovascular and renal drug products, said that the adjudication was "well conceived, well executed, and comprehensive" and that the small number of additional MACE events identified during adjudication did not change the original results reported for RECORD. The conclusions do not consider the impact of the open-label design of the study, but he said there was no convincing evidence of any manipulation, intentional or otherwise, of the safety outcomes in RECORD.
However, one FDA reviewer adamantly disagreed. Dr. Thomas Marciniak, medical team leader in the FDA’s division of cardiovascular and renal products, presented what he said were his own professional opinions, not the FDA’s official views. He said that RECORD was inadequately designed and not reliable, and "confirms and extends the recognized concerns regarding increased heart failure and heart failure deaths with rosiglitazone," and suggests that rosiglitazone increases the risk of MI.
He said that the readjudication process was not truly independent, did not collect much additional information, and that it did not overcome the flaws in the study’s design and what he said was mishandling of data.
Several panelists remarked that the unblinded design of the study was a limitation.
At the July 2010 meeting, 12 of the panelists voted to take the drug off the market, and 17 voted to continue marketing the drug, but with revisions. Compared to the year before the REMS program was instituted, the drug was available in about 55,000 pharmacies and was prescribed by almost 109,000 clinicians, which has dropped to 4 pharmacies and about 2,700 prescribers, according to GSK.
SILVER SPRING, MD. – A reanalysis of data from a controversial cardiovascular safety study of rosiglitazone was well-conducted and provided results that were "highly similar" to the original results of the study, according to Food and Drug Administration officials who spoke during the first day of a 2-day advisory panel meeting focusing on the results of the reanalysis.
The meeting – of the FDA’s Endocrinologic Drugs and Drug Safety and Risk Management advisory committees – is being held to discuss the results of the independent readjudication of the results of the RECORD (Rosiglitazone Evaluated for Cardiovascular Outcomes in Oral Agent Combination Therapy for Type 2 Diabetes) trial, a prospective, open-label, noninferiority study conducted by rosiglitazone manufacturer GlaxoSmithKline in response to a request by European drug regulators to address concerns over possible CV risks of the drug.
RECORD is the only large randomized controlled CV outcomes study of rosiglitazone, and overall, the readjudication of the study "appears to support the previous observation that, in this trial, rosiglitazone was not associated with an increased risk of death or major adverse cardiovascular events," Dr. Karen Mahoney, diabetes team leader in the FDA’s division of metabolism and endocrinology products said at the meeting.
How the panel interprets these data could impact the availability of rosiglitazone, which is used to treat about 3,400 patients in the United States, down from about 117,000 patients the year before the FDA instituted a risk evaluation and mitigation strategy (REMS) to limit its use because of cardiovascular safety concerns. On the second day of the meeting, June 6, the panel will discuss these data and will vote on whether the drug should remain on the market with the same restrictions, whether the REMs and other restrictions should be lifted or modified, or whether it should be taken off the market altogether based on the readjudicated results and other available data. In Europe, rosiglitazone is no longer marketed because of concerns over CV risks.
There have been longstanding concerns that rosiglitazone may be associated with an increased risk of ischemic CV events, but the data have been mixed and limited by study designs and other issues.
But in 2007, a meta-analysis of 42 studies showed that rosiglitazone increased the risk of MI by up to 43% and the risk of CV death by up to 64% (N. Engl. J. Med. 2007;356:2457-71); the lead author was Dr. Steven Nissen, a vocal advocate for removing rosiglitazone from the market.
Considering these and other data, at a meeting of the same panels in July 2010, the majority of the panelists agreed that the data raised significant safety concerns regarding the risk of rosiglitazone and most recommended that the use of the drug be limited, but a significant proportion recommended that it be taken off the U.S. market. In September 2010, the FDA announced that rosiglitazone would remain available, with the REMS, limiting the drug to patients who were already on rosiglitazone, or for patients whose blood glucose could not be managed adequately on other medications and, could not take the only other approved thiazolidinedione (TZD) pioglitazone, "for medical reasons."
At that time, the FDA asked GSK to commission an independent readjudication of the RECORD data, which found a nonsignificant increase in MI in the rosiglitazone arm, along with a nonsignificant reduction in the risk of stroke and all-cause mortality. But the reliability of the study has been questioned because of the study design and other issues such as missing data, and in September 2010, when the FDA announced that rosiglitazone would be marketed under a risk evaluation and mitigation strategy, the agency requested that the company commission an independent readjudication of the data to determine if the results could reliably be used to evaluate the drug’s CV safety.
RECORD enrolled about 4,500 patients with type 2 diabetes in 364 centers in 25 countries outside of the United States, comparing CV outcomes among those on rosiglitazone with metformin or a sulfonylurea and metformin. Because of limitations of the study, the reliability of these results has been questioned by experts within and outside the FDA. GSK commissioned the Duke Clinical Research Institute (DCRI) to conduct the reanalysis, which involved blinded adjudication of more than 2,200 deaths, MIs, and strokes. The results, presented at the meeting on June 5, were consistent with the original RECORD results, with no significant differences in the composite of CV death, MI, or stroke; numerically fewer CV deaths and numerically fewer strokes in the rosiglitazone arm; and a lower risk of all cause death. There were more MIs in the rosiglitazone arm, but the difference was not significant.
Dr. Preston Dunnmon, a medical reviewer in the FDA’s division of cardiovascular and renal drug products, said that the adjudication was "well conceived, well executed, and comprehensive" and that the small number of additional MACE events identified during adjudication did not change the original results reported for RECORD. The conclusions do not consider the impact of the open-label design of the study, but he said there was no convincing evidence of any manipulation, intentional or otherwise, of the safety outcomes in RECORD.
However, one FDA reviewer adamantly disagreed. Dr. Thomas Marciniak, medical team leader in the FDA’s division of cardiovascular and renal products, presented what he said were his own professional opinions, not the FDA’s official views. He said that RECORD was inadequately designed and not reliable, and "confirms and extends the recognized concerns regarding increased heart failure and heart failure deaths with rosiglitazone," and suggests that rosiglitazone increases the risk of MI.
He said that the readjudication process was not truly independent, did not collect much additional information, and that it did not overcome the flaws in the study’s design and what he said was mishandling of data.
Several panelists remarked that the unblinded design of the study was a limitation.
At the July 2010 meeting, 12 of the panelists voted to take the drug off the market, and 17 voted to continue marketing the drug, but with revisions. Compared to the year before the REMS program was instituted, the drug was available in about 55,000 pharmacies and was prescribed by almost 109,000 clinicians, which has dropped to 4 pharmacies and about 2,700 prescribers, according to GSK.
AT AN FDA ADVISORY COMMITTEE MEETING