User login
Emotional Distress, Barriers to Care, and Health-Related Quality of Life in Sickle Cell Disease
From the UCSF Benioff Children’s Hospital Oakland, Oakland, CA
Abstract
- Objective: Emotional distress may adversely affect the course and complicate treatment for individuals with sickle cell disease (SCD). We evaluated variables associated with physical and mental components of health-related quality of life (HRQL) in SCD in the context of a biobehavioral model.
- Methods: We conducted a cross-sectional cohort study of 77 adults with SCD (18–69 years; 60% female; 73% Hgb SS) attending an urban, academic medical center. We measured emotional distress (Patient Health Questionnaire–9, Generalized Anxiety Disorder 7-item scale), clinical complications and utilization, barriers to health care, sociodemo-graphics and HRQL (SF-36 Health Survey). We developed models predictive of physical and mental HRQL by conducting stepwise regression analyses.
- Results: Sample prevalence of moderate to severe depression and anxiety symptoms was 33% and 36%, respectively; prevalence of impaired physical and mental HRQL was 17% and 16%, respectively. Increased symptoms of depression, older age, and ≥ 3 emergency department visits in the previous 12 months were independently associated with lower ratings of physical HRQL, controlling for anxiety and sex. Increased symptoms of depression were independently associated with lower ratings of mental HRQL, controlling for barriers to care, insurance status, lifetime complications of SCD, and sex.
- Conclusion: Emotional distress is an important contributor to both physical and mental HRQL for adults with SCD, although sociodemographic variables and barriers to care must also be considered. Innovative approaches that integrate mental health interventions with SCD clinical care are needed.
Emotional distress, including symptoms of depression and anxiety, may adversely affect the course and complicate the treatment of chronic physical conditions [1]. For patients with sickle cell disease (SCD), a group of inherited red blood cell conditions, symptoms of depression and anxiety are more prevalent compared with rates found in the general population [2–8]. The most common symptom of SCD is acute pain events, and other complications range from mild to life-threatening, including anemia, increased risk of infection, acute chest syndrome, stroke, skin ulcers, and pulmonary hypertension [9]. Depression in adults with SCD has been associated with increased sickle cell vaso-occlusive pain events, poor pain control, multiple blood transfusions, and prescription of the disease-modifying therapy hydroxyurea [4]. Adults with SCD and comorbid depression and anxiety had more daily pain and greater distress and interference from pain compared with those who did not have comorbid depression or anxiety [10]. Patients have linked emotional distress and episodes of illness [11], and research has found a relation between pain episodes and depression [12]. In a diary study, negative mood was significantly higher on pain days compared with non-pain days [13].
Studies examining the consequences of emotional distress on health-related quality of life (HRQL) for patients with SCD are emerging. Depressed adults with SCD rated their quality of life on the SF-36 Health Survey [14] as significantly poorer in all areas compared with non-depressed adults with SCD [15]. In regression models, depression was a stronger predictor of SF-36 scores than demographics, hemoglobin type, and pain measures. In a multi-site study [16], 1046 adults with SCD completed the SF-36. Increasing age was associated with significantly lower scores on all subscales except mental health, while female sex additionally contributed to diminished physical function and vitality scale scores in multivariate models [16]. The presence of a mood disorder was associated with bodily pain, and diminished vitality, social functioning, emotional role, and the mental component of HRQL. Medical complications other than pain were not associated with impaired HRQL. Anie and colleagues [17,18] have highlighted the contributions of sickle cell–related pain to diminished mood and HRQL, both in the acute hospital phase and 1 week post discharge.
A comprehensive literature review of patient-reported outcomes for adults with SCD revealed broad categories of the impact of SCD and its treatment on the lives of adults [19]. Categories included pain and pain management, emotional distress, poor social role functioning, diminished overall quality of life, and poor quality of care. Follow-up individual and group interviews with adults with SCD (n = 122) as well as individual interviews with their providers (n = 15) revealed findings consistent with the literature review on the major effects of pain on the lives of adults with SCD, interwoven with emotional distress, poor quality of care, and stigmatization [19].
In the present study, our goal was to describe variables associated with physical and mental HRQL in SCD within the context of the recently published comprehensive conceptual model of broad clinical and life effects associated with SCD [19]. The present analysis uses an existing clinical database and evaluates the effects of the relations between clinical complications of SCD, emotional distress, health care utilization, and HRQL. Our model includes barriers to health care that might prevent vulnerable patients from accessing needed health care services. Sociodemographic variables including ethnic and racial minority status and lower socioeconomic status and educational attainment may create barriers to health care for patients with SCD, as they do for individuals with other chronic conditions [20–23]. Over 60% of patients with SCD are on public insurance [24] and can have difficulties with accessing quality health care [25]. Negative provider attitudes and stigmatization when patients are seeking care for acute pain episodes have been highlighted by patients as major barriers to seeking health care [19,26–28]. In a qualitative study, 45 youth with SCD reported that competing school or peer-group activities, “feeling good,” poor patient-provider relationships, adverse clinic experiences, and forgetting were barriers to clinic attendance [29]. Limited research suggests that barriers to accessing health care are associated with poorer HRQL [30,31]; however no studies were identified that directly evaluated the relation between barriers to care and HRQL for populations with SCD.
We hypothesized that clinical complications of SCD, including pain, and barriers to accessing health care would be independently associated with the physical component of HRQL for adult patients with SCD, controlling for demographic variables. Further, we hypothesized that emotional distress, clinical complications of SCD, and barriers to accessing health care would be independently associated with the mental component of HRQL for adult patients with SCD, controlling for demographic variables.
Methods
Patient Recruitment
Participants were 18 years and older and were a subgroup selected from a larger prospective cohort enrolled in the Sickle Cell Disease Treatment Demonstration Program (SCDTDP) funded by the Health Resources and Services Administration (HRSA). As 1 of 7 SCDTDP grantees, our network collected common demographic, disease-related, and HRQL data as the other grantees to examine sickle cell health and health care [32]. Enrollment at our site was n = 115 from birth through adult, with data collection occurring at baseline in 2010 and annually through 2014. Participants were eligible for enrollment if they had any confirmed diagnosis of SCD and if they were seen at any facility treating SCD in the San Francisco Bay Area region. Interpreter services were available where English was a second language; however, no participant requested those services. The data collection site was an urban comprehensive sickle cell center. Participants were recruited through mailings, posted flyers, or were introduced to the project by their clinical providers. The institutional review boards of the sponsoring hospitals approved all procedures. This report describes analyses from the baseline data collected in 2010 and excludes pediatric patients under the age of 18 years, as we developed our conceptual model based on the adult SCD literature.
Procedures
Patients directly contacted the project coordinator or were introduced by their health care provider. The project coordinator explained the study in more detail, and if the patient agreed to participate, the project coordinator obtained thier informed consent. Participants completed the study materials in a private space in the clinic immediately after or were scheduled for a separate visit at a convenient time and location. Participants with known or observed difficulties with reading completed the questionnaires as an interview. We allowed participants who were unable to complete the forms in one visit to take them home or schedule a follow-up visit to complete them. We asked participants who took the questionnaires home to return them within 2 business days and provided them with a stamped addressed envelope. Participants were compensated with gift cards for their involvement.
Measures
Demographics and Clinical Characteristics
Participants completed an Individual Utilization Questionnaire created for the SCDTDP grantees [32], either as an interview or in paper and pencil format. Participants indicated their age, race and ethnicity, education level, type of insurance, and annual household income. They indicated the type of SCD, number of hospital days and emergency department (ED) visits in the previous 12 months, disease-modifying therapies including hydroxyurea or transfusions, and lifetime incidence of sickle cell–related complications. Complications included pain, acute chest syndrome, fever, severe infection, stroke, kidney damage, gallbladder attack, spleen problems and priapism. Medical data was verified by reviewing medical records when possible; the clinical databases in the hematology/oncology department at the sponsoring hospital are maintained using Microsoft SQL Server, a relational database management system designed for the enterprise environment. However, not all of the participating institutions were linked via this common clinical database or by an electronic health record at the time the study was conducted.
Barriers to Care
We modified a checklist of barriers to accessing health care for patients with a range of chronic conditions [33] to create a SCD-specific checklist [34]. The final checklist consists of 53 items organized into 8 categories including insurance, transportation, accommodations and accessibility, provider knowledge and attitudes, social support, individual barriers such as forgetting or difficulties understanding instructions, emotional barriers such as fear or anger, and barriers posed by SCD itself (eg, pain, fatigue). Participants check off any applicable barrier, yielding a total score ranging from 0 to 53. The checklist overall has demonstrated face validity and test-retest reliability (Pearson r = 0.74, P < 0.05).
Depressive Symptoms
Adults with SCD completed the PHQ-9, the 9-item depression scale of the Patient Health Questionnaire [35]. The PHQ-9 is a tool for assisting primary care clinicians in assessing symptoms of depression, based on criteria from the Diagnostic and Statistical Manual 4th edition (DSM-IV [36]). The PHQ-9 asks about such symptoms as sleep disturbance and difficulty concentrating over the past 2 weeks with scores ranging from 0 (Not at all) to 3 (Every day). The total symptom count is based on the number of items in which the respondent answered as “more than half of days” or greater, and scores are categorized as reflecting no (< 10), mild (10–14), moderate (15–19) or severe (≥ 20) symptoms of depression. Respondents indicate how difficult the symptoms make it for them to engage in daily activities from 0 (Not difficult at all) to 3 (Extremely difficult). The sensitivity and diagnostic and criterion validity of the PHQ-9 have been established [37]. The internal consistency of the PHQ-9 is high, with α > 0.85 in several studies and 48-hour test-retest reliability of 0.84. The PHQ has been used widely, including with African-American and Hispanic populations, and with individuals with chronic conditions [38].
Symptoms of Anxiety
Participants completed the Generalized Anxiety Disorder 7-item (GAD-7) questionnaire for screening and measuring severity of generalized anxiety disorder [39]. The GAD-7 asks about such symptoms as feeling nervous, anxious, or on edge over the past two weeks. Scores from all 7 items are added to obtain a total score [40]. Cut-points of 5, 10, and 15 represent mild, moderate, and severe levels of anxiety symptoms. Respondents indicate how difficult the symptoms make it for them to engage in daily activities from 0 (Not difficult at all) to 3 (Extremely difficult). The internal consistency of the GAD-7 is excellent (α = 0.92). Test-retest reliability is also good (Pearson r = 0.83) as is procedural validity (intraclass correlation = 0.83). The GAD-7 has excellent sensitivity and specificity to identify generalized anxiety disorder [41].
Health-Related Quality of Life
Participants completed the SF-36, which asks about the patient’s health status in the past week [14]. Eight subscales include physical functioning, role-physical, bodily pain, general health, vitality, social functioning, role-emotional and mental health. Two summary measures, the Physical Component Summary and the Mental Component Summary, are calculated from 4 scales each. Use of the summary measures has been shown to increase the reliability of scores and improve the validity of scores in discriminating between physical and psychosocial outcomes [14]. Higher scores represent better HRQL, with a mean score of 50 (SD = 50) for the general population. Internal consistency estimates for the component summary scores are α > 0.89, item discriminant validity estimates are greater than 92.5% and 2-week test-retest reliability was excellent. Scores on the SF-36 have been divided into categories of HRQL functioning [42,43]. Participants in the impaired to very impaired category have scores ≤ mean – 1 SD while participants with average to above average functioning have scores > mean – 1 SD.
The SF-36 has been used extensively in observational and randomized studies for a range of illness conditions. In SCD, some aspects of HRQL as measured by the SF-36 improved for adult patients who responded to hydroxyurea [44]. Participants in the Pain in Sickle Cell Epidemiology Study scored lower than national norms on all SF-36 subscales except psychosocial functioning [45]. HRQL decreased significantly as daily pain intensity increased [45]. Further, women reported worse bodily pain compared with men [46].
Data Analyses
All biostatistical analyses were conducted using Stata 13 [47]. Continuous variables were examined for normality with measures of skewness and peakedness. All variables satisfied the assumptions of normality with the exception of barriers to health care and ED utilization. The variable barriers to health care was transformed using a square root transformation, resulting in a more normally distributed variable. ED utilization was dichotomized as 0–2 versus 3 or more ED visits in the previous 12 months, based on the distribution of utilization in the sample. The cutpoint of ≥ 3 annual ED visits is consistent with other literature on SCD clinical severity [48].
Descriptive statistics were computed to include means, standard deviations and frequencies. Sociodemographic variables (age, sex, insurance status [public or private] and income) were examined as potential covariates using Pearson correlations and t tests. Associations among emotional distress (anxiety and depression symptoms), clinical complications and ED utilization, barriers to health care, and the outcomes of the Physical and Mental Component Summary scores from the SF-36 were examined using Pearson correlations. We conducted stepwise regression with forward selection to determine models predictive of physical and mental HRQL. We tested the addition of each chosen variable (anxiety symptoms, depression symptoms, clinical complications, ED utilization, barriers to health care, age, sex, insurance status, and income), adding the variables (if any) that were most correlated with the outcome, and repeated the process until the model was not improved. A significance level of 0.05 was used for all statistical tests.
Results
Demographic and Clinical Characteristics
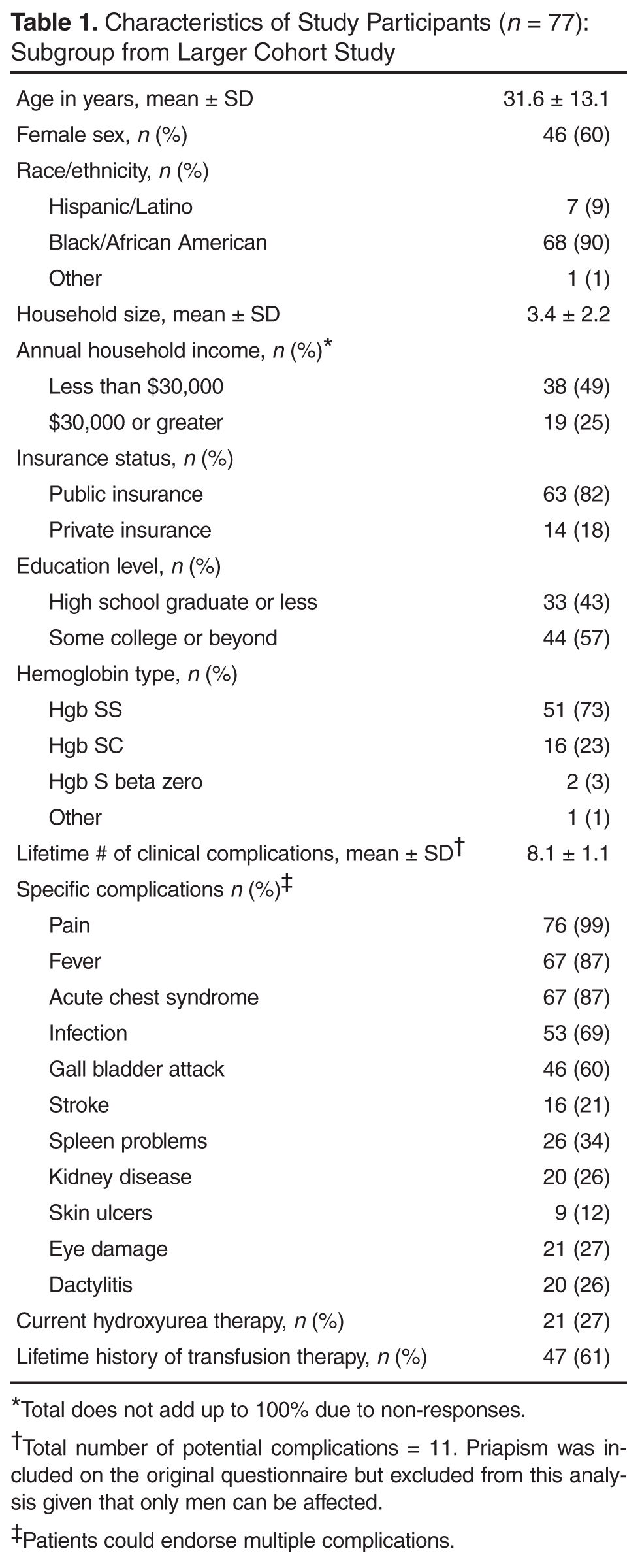
The majority of patients (73%) were diagnosed with Hgb SS disease and the most common lifetime complication was pain, reported by almost all of participants (Table 1). The next most common complication was fever, followed by acute chest syndrome. Twenty-seven percent of participants were currently on the disease-modifying therapy hydroxyurea, while 61% had a lifetime history of transfusion therapy. These data were verified with information from the clinical database for 73 participants (95%).
The median number of ED visits in the previous year was 1 (range, 0–50), with 19 patients (25%) with zero visits. The median number of hospital days in the previous year was 13 (range, 0–81). Twenty-nine patients (38%) had no hospital days in the previous year. These data were verified with information from the clinical database for 53 participants (69%), since hospital and ED visits occurred at institutions not always linked with the clinical databases at the sponsoring hospitals.
Emotional Distress, Barriers to Care, and Health-Related Quality of Life
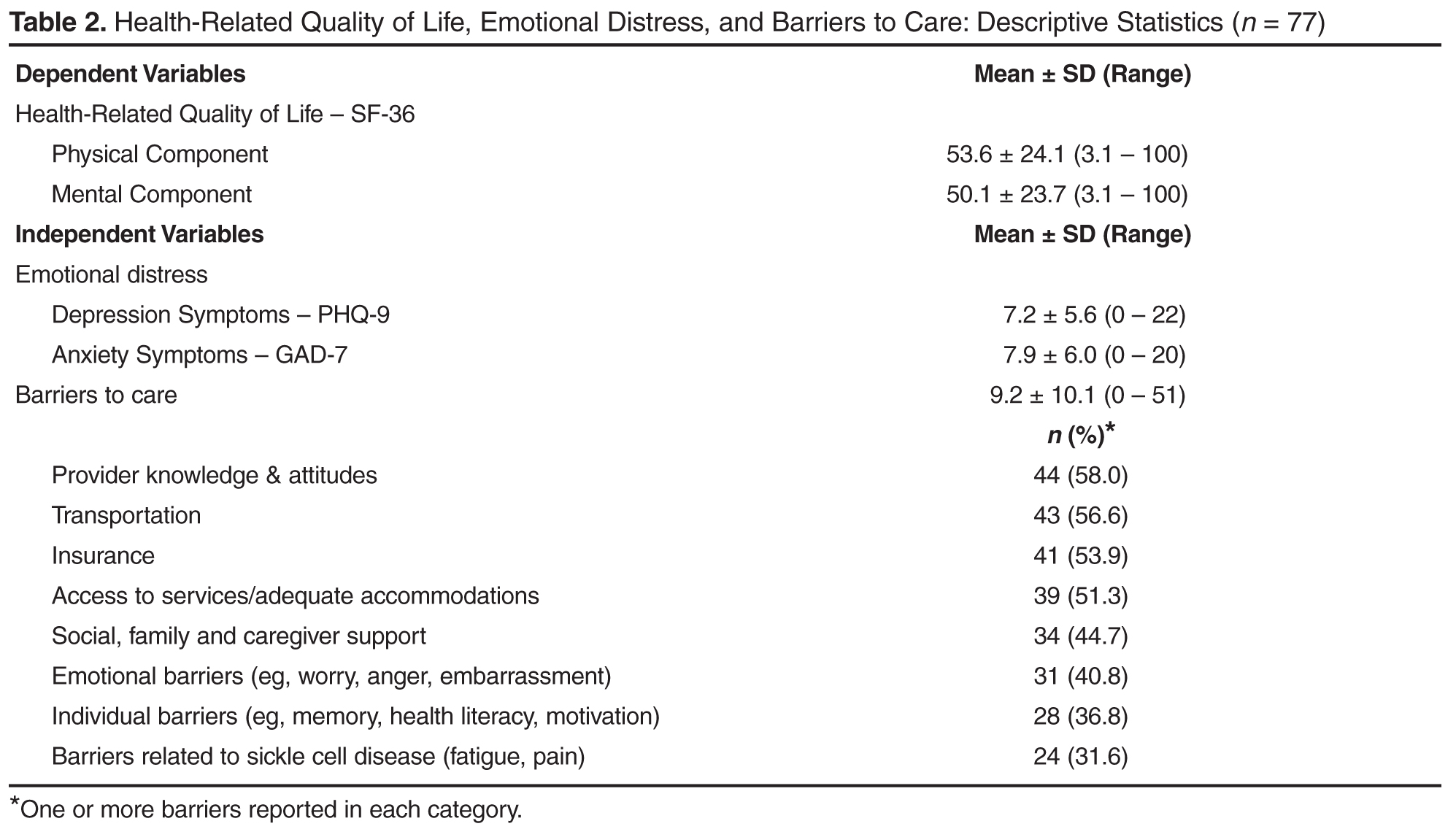
The mean score on the GAD-7 was 7.9 (SD = 6.0, α = 0.90, Table 2). The prevalence of moderate to severe symptoms of anxiety (scores ≥ 10) was 36.4% (n = 28). Fourteen patients with moderate to severe symptoms (50%) reported that anxiety symptoms created some difficulty in work, daily activities, or relationships. Twelve patients (43%) reported that symptoms created very much to extreme difficulty in work, daily activities, or relationships. Fifteen patients (29%) with moderate to severe symptoms of anxiety or depression exhibited comorbid anxiety and depression.
The mean Physical Component Summary score on the SF-36 was 53.6 (SD = 24.1, α = 0.94, Table 2). The prevalence of impaired to very impaired HRQL in the physical domain was 17% (n = 13). The mean Mental Component Summary score on the SF-36 for the sample was 50.1 (SD = 23.7, α = 0.93), with a prevalence of 16% (n = 12) in the impaired to very impaired range for HRQL in the mental domain.
The mean number of barriers from the barriers checklist was 9.2 (SD = 10.1) out of 53 possible. Sixty-five participants (86%) reported at least 1 barrier to accessing health care (Table 2). The most frequently cited barriers to care were provider knowledge and attitudes, followed by transportation, insurance, and access to services (eg, hours and location of services). Less frequently cited barriers to care were individual barriers, including memory, health literacy and motivation, as well as those related to SCD itself, ie, fatigue and pain.
Sociodemographic Variables, Emotional Distress, and Health-Related Quality of Life
Symptoms of anxiety and depression were highly correlated with one another, as would be expected (r = 0.75, P < 0.001). Physical and mental HRQL were significantly correlated with symptoms of depression (r = –0.67, P < 0.001 for physical HRQL component and r = –0.70 for mental HRQL component, P < 0.001), with impaired HRQL in both domains correlated with greater symptoms of depression. Physical and Mental Component Summary scores were significantly correlated with symptoms of anxiety (r = –0.58, P < 0.001 for the physical component and r = –0.62 for the mental component, P < 0.001), with impaired HRQL in both domains correlated with greater symptoms of anxiety. Ratings of difficulty with daily functioning from depressive symptoms were correlated with impaired HRQL in the physical (r = –0.46, P < 0.01) and mental domains (r = –0.52, P < 0.001). Ratings of difficulty with daily functioning from anxiety symptoms were also correlated with impaired HRQL in the physical (r = –0.58, P < 0.001) and mental domains (r = –0.63, P < 0.001). Reports of more barriers to health care were significantly correlated with reports of more depressive and anxiety symptoms (r = 0.53, P < 0.001 and r = 0.48, P < 0.001), with lower Mental Component Summary scores (r = –0.43, P < 0.05), and with more ED visits in the past year (r = 0.43, P < 0.05).
Relations Between Independent Variables and Outcomes
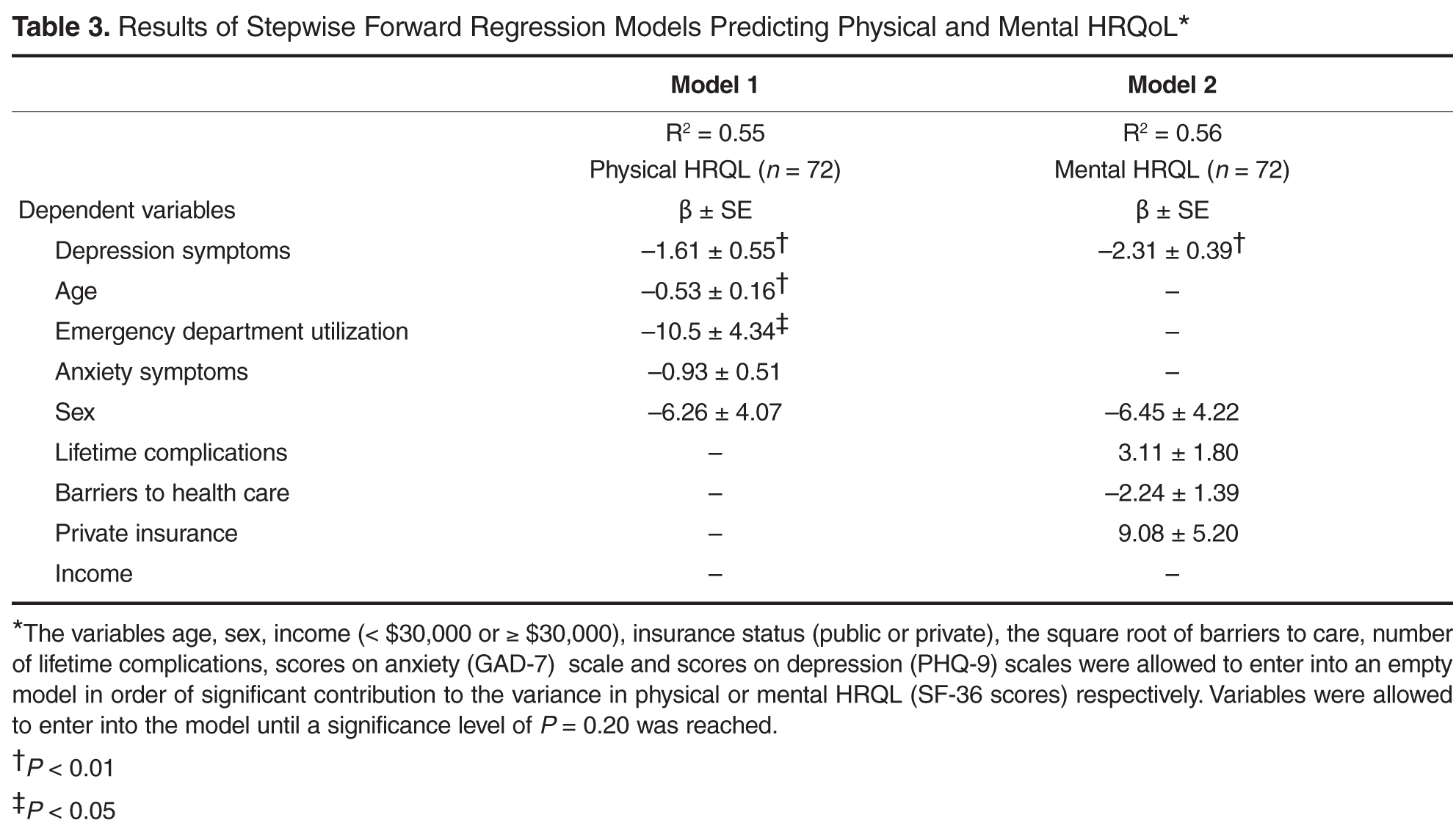
Discussion
Results of this study showed that as expected, symptoms of depression were independently associated with the mental component of HRQL, controlling for other variables. Symptoms of depression were also independently associated with the physical component of HRQL. The effect size for both models was moderate but comparable to effect sizes of other studies of predictive models of physical and mental HRQL in SCD [49]. Our findings were consistent with previous literature, with older age and increased ED utilization independently associated with lower ratings of physical HRQL, with sex and anxiety symptoms entering into the predictive model [15–18,44,45]. Contrary to our hypotheses, barriers to accessing health care were not independently associated with physical or mental HRQL but did contribute to the model for mental HRQL, as did clinical complications and private insurance status.
While our sample was similar to previous samples in mean age and percentage of women participants, our patients reported significantly higher physical HRQL scores, and a wider range of HRQL scores (eg, 53.6,
SD = 24.1 compared with 39.6, SD = 10.0 [16]). The mean Physical Component Summary score was in fact similar to the general population mean of 50. This may reflect improvements in quality of care and subsequent overall improved patient health and HRQL given that these data were collected in year 2 of the HRSA SCDTDP. As an SCDTDP grantee, we implemented goals to improve coordination of service delivery and to increase access to care. However, it should also be considered that there was a selection bias in our study, in favor of those with better HRQL. Nevertheless, as already noted, our findings are consistent with previous literature with regard to inter-relations between variables, ie, associations between lower physical HRQL ratings and symptoms of depression, older age, and increased ED utilization [15]. Future studies in SCD that directly evaluate reported access to a medical home in relation to HRQL are needed to assess the impact of access to care and care coordination on HRQL ratings.
Our use of a data collection tool that focused on lifetime rather than acute history of complications may have contributed to our failure to find a relation between clinical manifestations and physical HRQL. Further, we were not able to assess the effects of pain separately from other complications, since almost every participant reported a lifetime history of pain. However, our findings were consistent with those of researchers who have found psychosocial and sociodemographic factors, versus clinical manifestations, to be major influences on both physical and mental HRQL for individuals with SCD and other chronic and life-threatening conditions [15, 16, 50]. Our confidence is increased in this finding, given that we were able to verify self-reports of clinical manifestations with our clinical database. Our results contribute to the developing body of knowledge that emphasizes the importance of understanding the broad impact on the lives of adults of living with SCD, not just the physical symptomatology.
There has been limited research on barriers to accessing health care as associated with HRQL for SCD populations. Health care barriers have been identified for ethnic minorities, even within patient-centered medical homes, with minority status moderating the effect of barriers to care on HRQL [30]. Our findings that barriers to health care were correlated with depression and anxiety symptoms, mental HRQL, and greater ED utilization support the need to view SCD care within a biobehavioral framework. Health care provider negative attitudes and lack of knowledge were the most frequently cited barriers for adults in our study, particularly in the context of ED and inpatient care. These findings are similar to other studies that have highlighted the impact of these provider variables on quality of care [26,51]. We were not able to separate out effects of ethnic minority status, given that our patients were predominantly African American.
Contributors to poor HRQL that have been identified in SCD are poverty [42] and public insurance status [49]. While over half of our participants had family incomes of less than $30,000, despite a mean household size of 3 members, we did not find that income contributed to either of our models predicting physical or mental HRQL. Over half of our patients were well educated, which could have moderated the effect of their low incomes, but we did not measure other potential moderators such as active coping and supportive relationships [19]. These analyses were beyond the scope of our existing database, but future studies are needed on such resilience factors and processes. Our adults were predominantly on public insurance and we did find that private insurance status was positively associated with higher ratings of mental HRQL, consistent with other SCD research [49]. Taken together, our findings underscore the importance of considering the interplay between emotional distress, sociodemographic and clinical factors and quality of care in order to address risk factors for poor patient-reported outcomes [52,53].
There have not been previous reports of symptoms of emotional distress in SCD using the PHQ-9 and GAD-7, but both measures have been used widely for depression and anxiety screening, including with African-American populations. We selected these over other measures for their brevity, free availability, and psychometric properties. Our prevalence of moderate to severe depression and anxiety symptoms in the present study was similar to what has been found using other tools [2–8]. The PHQ-9 and GAD-7 also provide ratings of symptom interference on daily functioning, and we found that these ratings were associated with impaired physical and mental HRQL. Given that there generally are limited mental health resources in the communities where individuals with SCD reside and are treated, ratings of emotional distress and HRQL can be taken together to stratify those patients with the most immediate need for interventions. Further, screening can be used for early detection with the goal to intervene and prevent the progression of symptoms of emotional distress to long-term, disabling mental health disorders [54]. There is a need for innovative and cost-effective strategies for assessment and treatment of mental health symptoms and disorders for patients with SCD. One model for evidence-based practice in the management of emotional distress for patients with in SCD is the collaborative care model.
The collaborative care model integrates physical and mental health care in the patient-centered medical home and focuses on treating the whole person and family [55]. In this model, a care management staff (eg, nurse, social worker, psychologist) is integrated with the primary care team. The care management staff, in consultation with a psychiatrist, provides evidence-based care coordination, brief behavioral interventions, and support for other treatments, including medications. The effectiveness of collaborative care programs has been demonstrated for ethnic minority and safety net populations such as the SCD population, which is disproportionately low-income and on public insurance [56, 57]. Future research with SCD populations should investigate such interventions as the collaborative care model that addresses both emotional distress and barriers to care.
Limitations
Our results need to be interpreted with caution given the small sample size and the potential bias introduced by non-random sampling. In addition, as our patients are from an urban setting, findings might not generalize to rural populations. This study was cross sectional so no inferences can be made with regard to causality and temporal relations between anxiety symptoms, barriers to care, and HRQL. Our strategy for measuring total clinical complications and barriers to care conserved power but it was not possible to evaluate if specific complications or barriers may have exerted a greater impact on HRQL compared with others. Similarly, other studies have examined specific domains of HRQL, while we limited our analysis to the Physical and Mental Component Summary scores. The utilization questionnaire was designed to assess only lifetime complications, not complications more proximal to the HRQL ratings.
Patient-reported outcomes, now widely accepted as outcome measures, elicit patients’ descriptions of the impact of their condition on their day-to-day lives [34, 58–60]. However, measures of mental health symptoms and HRQL may be subject to recall bias, measurement error, and confounding [61,62]. Nevertheless, a range of studies support the idea that mental health symptoms and HRQL are distinct constructs, and that patients with physical and mental health symptoms are vulnerable to lower ratings of HRQL [63,64]. Disease-modifying therapies such as hydroxyurea can contribute to improved ratings of HRQL [44,65], but we were not able to evaluate the contribution of hydroxyurea to HRQL as it appears to have been underutilized in our sample.
Conclusion
We evaluated emotional distress and other variables in the context of a biobehavioral model of HRQL outcomes for adults with SCD. Integrating the patient's perspective of the impact of the disease and its treatment with assessment of clinical indications is critical to implementing and evaluating effective therapies [25]. However, there are conceptual challenges in determining what actually contributes to HRQL from the patient’s perspective in the context of genetic disorders such as SCD [50]. Our findings highlight the importance of incorporating comprehensive psychosocial screening in order to support optimal HRQL in SCD. Providers may be reluctant to include such screening if, as is often the case, mental health services are difficult to access. Models such as the collaborative care model, which include mental health interventions within the sickle cell center or primary care provider’s office, should be implented. Barriers to care and HRQL should also be routinely evaluated for patients with SCD. Use of disease-specific tools, such as the Adult Sickle Cell Quality of Life measurement system [66], may increase the specificity needed to detect differences within adults with SCD and improvements related to interventions, whether medical or psychosocial. Contributors to HRQL in SCD go beyond clinical manifestations to include psychological and social factors, as well as provider and health system variables. Research conducted within the framework of a comprehensive conceptual model of broad clinical and life effects associated with SCD can inform clinical applications that ultimately enhance HRQL for patients with SCD.
Acknowledgment: The authors wish to thank San Keller, PhD, for her helpful comments on a previous version of this manuscript.
Corresponding author: Marsha J. Treadwell, PhD, Hematology/Oncology Dept., UCSF Benioff Children’s Hospital Oakland, 747 52nd St., Oakland, CA 94609, [email protected].
Funding/support: This research was conducted as part of the National Initiative for Children’s Healthcare Quality (NICHQ) Working to Improve Sickle Cell Healthcare (WISCH) project. Further support came from a grant from the Health Resources and Services Administration (HRSA) Sickle Cell Disease Treatment Demonstration Project Grant No. U1EMC16492 and from the National Institutes of Health (NIH) Clinical and Translational Science Award UL1 RR024131. The views expressed in this publication do not necessarily reflect the views of WISCH, NICHQ, HRSA or NIH.
Financial disclosures: None.
Author contributions: conception and design, MJT; analysis and interpretation of data, MJT, GG; drafting of article, MJT, GG; critical revision of the article, MJT, KK, FB; statistical expertise, GG; obtaining of funding, MJT; administrative or technical support, KK, FB; collection and assembly of data, KK, FB.
From the UCSF Benioff Children’s Hospital Oakland, Oakland, CA
Abstract
- Objective: Emotional distress may adversely affect the course and complicate treatment for individuals with sickle cell disease (SCD). We evaluated variables associated with physical and mental components of health-related quality of life (HRQL) in SCD in the context of a biobehavioral model.
- Methods: We conducted a cross-sectional cohort study of 77 adults with SCD (18–69 years; 60% female; 73% Hgb SS) attending an urban, academic medical center. We measured emotional distress (Patient Health Questionnaire–9, Generalized Anxiety Disorder 7-item scale), clinical complications and utilization, barriers to health care, sociodemo-graphics and HRQL (SF-36 Health Survey). We developed models predictive of physical and mental HRQL by conducting stepwise regression analyses.
- Results: Sample prevalence of moderate to severe depression and anxiety symptoms was 33% and 36%, respectively; prevalence of impaired physical and mental HRQL was 17% and 16%, respectively. Increased symptoms of depression, older age, and ≥ 3 emergency department visits in the previous 12 months were independently associated with lower ratings of physical HRQL, controlling for anxiety and sex. Increased symptoms of depression were independently associated with lower ratings of mental HRQL, controlling for barriers to care, insurance status, lifetime complications of SCD, and sex.
- Conclusion: Emotional distress is an important contributor to both physical and mental HRQL for adults with SCD, although sociodemographic variables and barriers to care must also be considered. Innovative approaches that integrate mental health interventions with SCD clinical care are needed.
Emotional distress, including symptoms of depression and anxiety, may adversely affect the course and complicate the treatment of chronic physical conditions [1]. For patients with sickle cell disease (SCD), a group of inherited red blood cell conditions, symptoms of depression and anxiety are more prevalent compared with rates found in the general population [2–8]. The most common symptom of SCD is acute pain events, and other complications range from mild to life-threatening, including anemia, increased risk of infection, acute chest syndrome, stroke, skin ulcers, and pulmonary hypertension [9]. Depression in adults with SCD has been associated with increased sickle cell vaso-occlusive pain events, poor pain control, multiple blood transfusions, and prescription of the disease-modifying therapy hydroxyurea [4]. Adults with SCD and comorbid depression and anxiety had more daily pain and greater distress and interference from pain compared with those who did not have comorbid depression or anxiety [10]. Patients have linked emotional distress and episodes of illness [11], and research has found a relation between pain episodes and depression [12]. In a diary study, negative mood was significantly higher on pain days compared with non-pain days [13].
Studies examining the consequences of emotional distress on health-related quality of life (HRQL) for patients with SCD are emerging. Depressed adults with SCD rated their quality of life on the SF-36 Health Survey [14] as significantly poorer in all areas compared with non-depressed adults with SCD [15]. In regression models, depression was a stronger predictor of SF-36 scores than demographics, hemoglobin type, and pain measures. In a multi-site study [16], 1046 adults with SCD completed the SF-36. Increasing age was associated with significantly lower scores on all subscales except mental health, while female sex additionally contributed to diminished physical function and vitality scale scores in multivariate models [16]. The presence of a mood disorder was associated with bodily pain, and diminished vitality, social functioning, emotional role, and the mental component of HRQL. Medical complications other than pain were not associated with impaired HRQL. Anie and colleagues [17,18] have highlighted the contributions of sickle cell–related pain to diminished mood and HRQL, both in the acute hospital phase and 1 week post discharge.
A comprehensive literature review of patient-reported outcomes for adults with SCD revealed broad categories of the impact of SCD and its treatment on the lives of adults [19]. Categories included pain and pain management, emotional distress, poor social role functioning, diminished overall quality of life, and poor quality of care. Follow-up individual and group interviews with adults with SCD (n = 122) as well as individual interviews with their providers (n = 15) revealed findings consistent with the literature review on the major effects of pain on the lives of adults with SCD, interwoven with emotional distress, poor quality of care, and stigmatization [19].
In the present study, our goal was to describe variables associated with physical and mental HRQL in SCD within the context of the recently published comprehensive conceptual model of broad clinical and life effects associated with SCD [19]. The present analysis uses an existing clinical database and evaluates the effects of the relations between clinical complications of SCD, emotional distress, health care utilization, and HRQL. Our model includes barriers to health care that might prevent vulnerable patients from accessing needed health care services. Sociodemographic variables including ethnic and racial minority status and lower socioeconomic status and educational attainment may create barriers to health care for patients with SCD, as they do for individuals with other chronic conditions [20–23]. Over 60% of patients with SCD are on public insurance [24] and can have difficulties with accessing quality health care [25]. Negative provider attitudes and stigmatization when patients are seeking care for acute pain episodes have been highlighted by patients as major barriers to seeking health care [19,26–28]. In a qualitative study, 45 youth with SCD reported that competing school or peer-group activities, “feeling good,” poor patient-provider relationships, adverse clinic experiences, and forgetting were barriers to clinic attendance [29]. Limited research suggests that barriers to accessing health care are associated with poorer HRQL [30,31]; however no studies were identified that directly evaluated the relation between barriers to care and HRQL for populations with SCD.
We hypothesized that clinical complications of SCD, including pain, and barriers to accessing health care would be independently associated with the physical component of HRQL for adult patients with SCD, controlling for demographic variables. Further, we hypothesized that emotional distress, clinical complications of SCD, and barriers to accessing health care would be independently associated with the mental component of HRQL for adult patients with SCD, controlling for demographic variables.
Methods
Patient Recruitment
Participants were 18 years and older and were a subgroup selected from a larger prospective cohort enrolled in the Sickle Cell Disease Treatment Demonstration Program (SCDTDP) funded by the Health Resources and Services Administration (HRSA). As 1 of 7 SCDTDP grantees, our network collected common demographic, disease-related, and HRQL data as the other grantees to examine sickle cell health and health care [32]. Enrollment at our site was n = 115 from birth through adult, with data collection occurring at baseline in 2010 and annually through 2014. Participants were eligible for enrollment if they had any confirmed diagnosis of SCD and if they were seen at any facility treating SCD in the San Francisco Bay Area region. Interpreter services were available where English was a second language; however, no participant requested those services. The data collection site was an urban comprehensive sickle cell center. Participants were recruited through mailings, posted flyers, or were introduced to the project by their clinical providers. The institutional review boards of the sponsoring hospitals approved all procedures. This report describes analyses from the baseline data collected in 2010 and excludes pediatric patients under the age of 18 years, as we developed our conceptual model based on the adult SCD literature.
Procedures
Patients directly contacted the project coordinator or were introduced by their health care provider. The project coordinator explained the study in more detail, and if the patient agreed to participate, the project coordinator obtained thier informed consent. Participants completed the study materials in a private space in the clinic immediately after or were scheduled for a separate visit at a convenient time and location. Participants with known or observed difficulties with reading completed the questionnaires as an interview. We allowed participants who were unable to complete the forms in one visit to take them home or schedule a follow-up visit to complete them. We asked participants who took the questionnaires home to return them within 2 business days and provided them with a stamped addressed envelope. Participants were compensated with gift cards for their involvement.
Measures
Demographics and Clinical Characteristics
Participants completed an Individual Utilization Questionnaire created for the SCDTDP grantees [32], either as an interview or in paper and pencil format. Participants indicated their age, race and ethnicity, education level, type of insurance, and annual household income. They indicated the type of SCD, number of hospital days and emergency department (ED) visits in the previous 12 months, disease-modifying therapies including hydroxyurea or transfusions, and lifetime incidence of sickle cell–related complications. Complications included pain, acute chest syndrome, fever, severe infection, stroke, kidney damage, gallbladder attack, spleen problems and priapism. Medical data was verified by reviewing medical records when possible; the clinical databases in the hematology/oncology department at the sponsoring hospital are maintained using Microsoft SQL Server, a relational database management system designed for the enterprise environment. However, not all of the participating institutions were linked via this common clinical database or by an electronic health record at the time the study was conducted.
Barriers to Care
We modified a checklist of barriers to accessing health care for patients with a range of chronic conditions [33] to create a SCD-specific checklist [34]. The final checklist consists of 53 items organized into 8 categories including insurance, transportation, accommodations and accessibility, provider knowledge and attitudes, social support, individual barriers such as forgetting or difficulties understanding instructions, emotional barriers such as fear or anger, and barriers posed by SCD itself (eg, pain, fatigue). Participants check off any applicable barrier, yielding a total score ranging from 0 to 53. The checklist overall has demonstrated face validity and test-retest reliability (Pearson r = 0.74, P < 0.05).
Depressive Symptoms
Adults with SCD completed the PHQ-9, the 9-item depression scale of the Patient Health Questionnaire [35]. The PHQ-9 is a tool for assisting primary care clinicians in assessing symptoms of depression, based on criteria from the Diagnostic and Statistical Manual 4th edition (DSM-IV [36]). The PHQ-9 asks about such symptoms as sleep disturbance and difficulty concentrating over the past 2 weeks with scores ranging from 0 (Not at all) to 3 (Every day). The total symptom count is based on the number of items in which the respondent answered as “more than half of days” or greater, and scores are categorized as reflecting no (< 10), mild (10–14), moderate (15–19) or severe (≥ 20) symptoms of depression. Respondents indicate how difficult the symptoms make it for them to engage in daily activities from 0 (Not difficult at all) to 3 (Extremely difficult). The sensitivity and diagnostic and criterion validity of the PHQ-9 have been established [37]. The internal consistency of the PHQ-9 is high, with α > 0.85 in several studies and 48-hour test-retest reliability of 0.84. The PHQ has been used widely, including with African-American and Hispanic populations, and with individuals with chronic conditions [38].
Symptoms of Anxiety
Participants completed the Generalized Anxiety Disorder 7-item (GAD-7) questionnaire for screening and measuring severity of generalized anxiety disorder [39]. The GAD-7 asks about such symptoms as feeling nervous, anxious, or on edge over the past two weeks. Scores from all 7 items are added to obtain a total score [40]. Cut-points of 5, 10, and 15 represent mild, moderate, and severe levels of anxiety symptoms. Respondents indicate how difficult the symptoms make it for them to engage in daily activities from 0 (Not difficult at all) to 3 (Extremely difficult). The internal consistency of the GAD-7 is excellent (α = 0.92). Test-retest reliability is also good (Pearson r = 0.83) as is procedural validity (intraclass correlation = 0.83). The GAD-7 has excellent sensitivity and specificity to identify generalized anxiety disorder [41].
Health-Related Quality of Life
Participants completed the SF-36, which asks about the patient’s health status in the past week [14]. Eight subscales include physical functioning, role-physical, bodily pain, general health, vitality, social functioning, role-emotional and mental health. Two summary measures, the Physical Component Summary and the Mental Component Summary, are calculated from 4 scales each. Use of the summary measures has been shown to increase the reliability of scores and improve the validity of scores in discriminating between physical and psychosocial outcomes [14]. Higher scores represent better HRQL, with a mean score of 50 (SD = 50) for the general population. Internal consistency estimates for the component summary scores are α > 0.89, item discriminant validity estimates are greater than 92.5% and 2-week test-retest reliability was excellent. Scores on the SF-36 have been divided into categories of HRQL functioning [42,43]. Participants in the impaired to very impaired category have scores ≤ mean – 1 SD while participants with average to above average functioning have scores > mean – 1 SD.
The SF-36 has been used extensively in observational and randomized studies for a range of illness conditions. In SCD, some aspects of HRQL as measured by the SF-36 improved for adult patients who responded to hydroxyurea [44]. Participants in the Pain in Sickle Cell Epidemiology Study scored lower than national norms on all SF-36 subscales except psychosocial functioning [45]. HRQL decreased significantly as daily pain intensity increased [45]. Further, women reported worse bodily pain compared with men [46].
Data Analyses
All biostatistical analyses were conducted using Stata 13 [47]. Continuous variables were examined for normality with measures of skewness and peakedness. All variables satisfied the assumptions of normality with the exception of barriers to health care and ED utilization. The variable barriers to health care was transformed using a square root transformation, resulting in a more normally distributed variable. ED utilization was dichotomized as 0–2 versus 3 or more ED visits in the previous 12 months, based on the distribution of utilization in the sample. The cutpoint of ≥ 3 annual ED visits is consistent with other literature on SCD clinical severity [48].
Descriptive statistics were computed to include means, standard deviations and frequencies. Sociodemographic variables (age, sex, insurance status [public or private] and income) were examined as potential covariates using Pearson correlations and t tests. Associations among emotional distress (anxiety and depression symptoms), clinical complications and ED utilization, barriers to health care, and the outcomes of the Physical and Mental Component Summary scores from the SF-36 were examined using Pearson correlations. We conducted stepwise regression with forward selection to determine models predictive of physical and mental HRQL. We tested the addition of each chosen variable (anxiety symptoms, depression symptoms, clinical complications, ED utilization, barriers to health care, age, sex, insurance status, and income), adding the variables (if any) that were most correlated with the outcome, and repeated the process until the model was not improved. A significance level of 0.05 was used for all statistical tests.
Results
Demographic and Clinical Characteristics
The majority of patients (73%) were diagnosed with Hgb SS disease and the most common lifetime complication was pain, reported by almost all of participants (Table 1). The next most common complication was fever, followed by acute chest syndrome. Twenty-seven percent of participants were currently on the disease-modifying therapy hydroxyurea, while 61% had a lifetime history of transfusion therapy. These data were verified with information from the clinical database for 73 participants (95%).
The median number of ED visits in the previous year was 1 (range, 0–50), with 19 patients (25%) with zero visits. The median number of hospital days in the previous year was 13 (range, 0–81). Twenty-nine patients (38%) had no hospital days in the previous year. These data were verified with information from the clinical database for 53 participants (69%), since hospital and ED visits occurred at institutions not always linked with the clinical databases at the sponsoring hospitals.
Emotional Distress, Barriers to Care, and Health-Related Quality of Life
The mean score on the GAD-7 was 7.9 (SD = 6.0, α = 0.90, Table 2). The prevalence of moderate to severe symptoms of anxiety (scores ≥ 10) was 36.4% (n = 28). Fourteen patients with moderate to severe symptoms (50%) reported that anxiety symptoms created some difficulty in work, daily activities, or relationships. Twelve patients (43%) reported that symptoms created very much to extreme difficulty in work, daily activities, or relationships. Fifteen patients (29%) with moderate to severe symptoms of anxiety or depression exhibited comorbid anxiety and depression.
The mean Physical Component Summary score on the SF-36 was 53.6 (SD = 24.1, α = 0.94, Table 2). The prevalence of impaired to very impaired HRQL in the physical domain was 17% (n = 13). The mean Mental Component Summary score on the SF-36 for the sample was 50.1 (SD = 23.7, α = 0.93), with a prevalence of 16% (n = 12) in the impaired to very impaired range for HRQL in the mental domain.
The mean number of barriers from the barriers checklist was 9.2 (SD = 10.1) out of 53 possible. Sixty-five participants (86%) reported at least 1 barrier to accessing health care (Table 2). The most frequently cited barriers to care were provider knowledge and attitudes, followed by transportation, insurance, and access to services (eg, hours and location of services). Less frequently cited barriers to care were individual barriers, including memory, health literacy and motivation, as well as those related to SCD itself, ie, fatigue and pain.
Sociodemographic Variables, Emotional Distress, and Health-Related Quality of Life
Symptoms of anxiety and depression were highly correlated with one another, as would be expected (r = 0.75, P < 0.001). Physical and mental HRQL were significantly correlated with symptoms of depression (r = –0.67, P < 0.001 for physical HRQL component and r = –0.70 for mental HRQL component, P < 0.001), with impaired HRQL in both domains correlated with greater symptoms of depression. Physical and Mental Component Summary scores were significantly correlated with symptoms of anxiety (r = –0.58, P < 0.001 for the physical component and r = –0.62 for the mental component, P < 0.001), with impaired HRQL in both domains correlated with greater symptoms of anxiety. Ratings of difficulty with daily functioning from depressive symptoms were correlated with impaired HRQL in the physical (r = –0.46, P < 0.01) and mental domains (r = –0.52, P < 0.001). Ratings of difficulty with daily functioning from anxiety symptoms were also correlated with impaired HRQL in the physical (r = –0.58, P < 0.001) and mental domains (r = –0.63, P < 0.001). Reports of more barriers to health care were significantly correlated with reports of more depressive and anxiety symptoms (r = 0.53, P < 0.001 and r = 0.48, P < 0.001), with lower Mental Component Summary scores (r = –0.43, P < 0.05), and with more ED visits in the past year (r = 0.43, P < 0.05).
Relations Between Independent Variables and Outcomes
Discussion
Results of this study showed that as expected, symptoms of depression were independently associated with the mental component of HRQL, controlling for other variables. Symptoms of depression were also independently associated with the physical component of HRQL. The effect size for both models was moderate but comparable to effect sizes of other studies of predictive models of physical and mental HRQL in SCD [49]. Our findings were consistent with previous literature, with older age and increased ED utilization independently associated with lower ratings of physical HRQL, with sex and anxiety symptoms entering into the predictive model [15–18,44,45]. Contrary to our hypotheses, barriers to accessing health care were not independently associated with physical or mental HRQL but did contribute to the model for mental HRQL, as did clinical complications and private insurance status.
While our sample was similar to previous samples in mean age and percentage of women participants, our patients reported significantly higher physical HRQL scores, and a wider range of HRQL scores (eg, 53.6,
SD = 24.1 compared with 39.6, SD = 10.0 [16]). The mean Physical Component Summary score was in fact similar to the general population mean of 50. This may reflect improvements in quality of care and subsequent overall improved patient health and HRQL given that these data were collected in year 2 of the HRSA SCDTDP. As an SCDTDP grantee, we implemented goals to improve coordination of service delivery and to increase access to care. However, it should also be considered that there was a selection bias in our study, in favor of those with better HRQL. Nevertheless, as already noted, our findings are consistent with previous literature with regard to inter-relations between variables, ie, associations between lower physical HRQL ratings and symptoms of depression, older age, and increased ED utilization [15]. Future studies in SCD that directly evaluate reported access to a medical home in relation to HRQL are needed to assess the impact of access to care and care coordination on HRQL ratings.
Our use of a data collection tool that focused on lifetime rather than acute history of complications may have contributed to our failure to find a relation between clinical manifestations and physical HRQL. Further, we were not able to assess the effects of pain separately from other complications, since almost every participant reported a lifetime history of pain. However, our findings were consistent with those of researchers who have found psychosocial and sociodemographic factors, versus clinical manifestations, to be major influences on both physical and mental HRQL for individuals with SCD and other chronic and life-threatening conditions [15, 16, 50]. Our confidence is increased in this finding, given that we were able to verify self-reports of clinical manifestations with our clinical database. Our results contribute to the developing body of knowledge that emphasizes the importance of understanding the broad impact on the lives of adults of living with SCD, not just the physical symptomatology.
There has been limited research on barriers to accessing health care as associated with HRQL for SCD populations. Health care barriers have been identified for ethnic minorities, even within patient-centered medical homes, with minority status moderating the effect of barriers to care on HRQL [30]. Our findings that barriers to health care were correlated with depression and anxiety symptoms, mental HRQL, and greater ED utilization support the need to view SCD care within a biobehavioral framework. Health care provider negative attitudes and lack of knowledge were the most frequently cited barriers for adults in our study, particularly in the context of ED and inpatient care. These findings are similar to other studies that have highlighted the impact of these provider variables on quality of care [26,51]. We were not able to separate out effects of ethnic minority status, given that our patients were predominantly African American.
Contributors to poor HRQL that have been identified in SCD are poverty [42] and public insurance status [49]. While over half of our participants had family incomes of less than $30,000, despite a mean household size of 3 members, we did not find that income contributed to either of our models predicting physical or mental HRQL. Over half of our patients were well educated, which could have moderated the effect of their low incomes, but we did not measure other potential moderators such as active coping and supportive relationships [19]. These analyses were beyond the scope of our existing database, but future studies are needed on such resilience factors and processes. Our adults were predominantly on public insurance and we did find that private insurance status was positively associated with higher ratings of mental HRQL, consistent with other SCD research [49]. Taken together, our findings underscore the importance of considering the interplay between emotional distress, sociodemographic and clinical factors and quality of care in order to address risk factors for poor patient-reported outcomes [52,53].
There have not been previous reports of symptoms of emotional distress in SCD using the PHQ-9 and GAD-7, but both measures have been used widely for depression and anxiety screening, including with African-American populations. We selected these over other measures for their brevity, free availability, and psychometric properties. Our prevalence of moderate to severe depression and anxiety symptoms in the present study was similar to what has been found using other tools [2–8]. The PHQ-9 and GAD-7 also provide ratings of symptom interference on daily functioning, and we found that these ratings were associated with impaired physical and mental HRQL. Given that there generally are limited mental health resources in the communities where individuals with SCD reside and are treated, ratings of emotional distress and HRQL can be taken together to stratify those patients with the most immediate need for interventions. Further, screening can be used for early detection with the goal to intervene and prevent the progression of symptoms of emotional distress to long-term, disabling mental health disorders [54]. There is a need for innovative and cost-effective strategies for assessment and treatment of mental health symptoms and disorders for patients with SCD. One model for evidence-based practice in the management of emotional distress for patients with in SCD is the collaborative care model.
The collaborative care model integrates physical and mental health care in the patient-centered medical home and focuses on treating the whole person and family [55]. In this model, a care management staff (eg, nurse, social worker, psychologist) is integrated with the primary care team. The care management staff, in consultation with a psychiatrist, provides evidence-based care coordination, brief behavioral interventions, and support for other treatments, including medications. The effectiveness of collaborative care programs has been demonstrated for ethnic minority and safety net populations such as the SCD population, which is disproportionately low-income and on public insurance [56, 57]. Future research with SCD populations should investigate such interventions as the collaborative care model that addresses both emotional distress and barriers to care.
Limitations
Our results need to be interpreted with caution given the small sample size and the potential bias introduced by non-random sampling. In addition, as our patients are from an urban setting, findings might not generalize to rural populations. This study was cross sectional so no inferences can be made with regard to causality and temporal relations between anxiety symptoms, barriers to care, and HRQL. Our strategy for measuring total clinical complications and barriers to care conserved power but it was not possible to evaluate if specific complications or barriers may have exerted a greater impact on HRQL compared with others. Similarly, other studies have examined specific domains of HRQL, while we limited our analysis to the Physical and Mental Component Summary scores. The utilization questionnaire was designed to assess only lifetime complications, not complications more proximal to the HRQL ratings.
Patient-reported outcomes, now widely accepted as outcome measures, elicit patients’ descriptions of the impact of their condition on their day-to-day lives [34, 58–60]. However, measures of mental health symptoms and HRQL may be subject to recall bias, measurement error, and confounding [61,62]. Nevertheless, a range of studies support the idea that mental health symptoms and HRQL are distinct constructs, and that patients with physical and mental health symptoms are vulnerable to lower ratings of HRQL [63,64]. Disease-modifying therapies such as hydroxyurea can contribute to improved ratings of HRQL [44,65], but we were not able to evaluate the contribution of hydroxyurea to HRQL as it appears to have been underutilized in our sample.
Conclusion
We evaluated emotional distress and other variables in the context of a biobehavioral model of HRQL outcomes for adults with SCD. Integrating the patient's perspective of the impact of the disease and its treatment with assessment of clinical indications is critical to implementing and evaluating effective therapies [25]. However, there are conceptual challenges in determining what actually contributes to HRQL from the patient’s perspective in the context of genetic disorders such as SCD [50]. Our findings highlight the importance of incorporating comprehensive psychosocial screening in order to support optimal HRQL in SCD. Providers may be reluctant to include such screening if, as is often the case, mental health services are difficult to access. Models such as the collaborative care model, which include mental health interventions within the sickle cell center or primary care provider’s office, should be implented. Barriers to care and HRQL should also be routinely evaluated for patients with SCD. Use of disease-specific tools, such as the Adult Sickle Cell Quality of Life measurement system [66], may increase the specificity needed to detect differences within adults with SCD and improvements related to interventions, whether medical or psychosocial. Contributors to HRQL in SCD go beyond clinical manifestations to include psychological and social factors, as well as provider and health system variables. Research conducted within the framework of a comprehensive conceptual model of broad clinical and life effects associated with SCD can inform clinical applications that ultimately enhance HRQL for patients with SCD.
Acknowledgment: The authors wish to thank San Keller, PhD, for her helpful comments on a previous version of this manuscript.
Corresponding author: Marsha J. Treadwell, PhD, Hematology/Oncology Dept., UCSF Benioff Children’s Hospital Oakland, 747 52nd St., Oakland, CA 94609, [email protected].
Funding/support: This research was conducted as part of the National Initiative for Children’s Healthcare Quality (NICHQ) Working to Improve Sickle Cell Healthcare (WISCH) project. Further support came from a grant from the Health Resources and Services Administration (HRSA) Sickle Cell Disease Treatment Demonstration Project Grant No. U1EMC16492 and from the National Institutes of Health (NIH) Clinical and Translational Science Award UL1 RR024131. The views expressed in this publication do not necessarily reflect the views of WISCH, NICHQ, HRSA or NIH.
Financial disclosures: None.
Author contributions: conception and design, MJT; analysis and interpretation of data, MJT, GG; drafting of article, MJT, GG; critical revision of the article, MJT, KK, FB; statistical expertise, GG; obtaining of funding, MJT; administrative or technical support, KK, FB; collection and assembly of data, KK, FB.
From the UCSF Benioff Children’s Hospital Oakland, Oakland, CA
Abstract
- Objective: Emotional distress may adversely affect the course and complicate treatment for individuals with sickle cell disease (SCD). We evaluated variables associated with physical and mental components of health-related quality of life (HRQL) in SCD in the context of a biobehavioral model.
- Methods: We conducted a cross-sectional cohort study of 77 adults with SCD (18–69 years; 60% female; 73% Hgb SS) attending an urban, academic medical center. We measured emotional distress (Patient Health Questionnaire–9, Generalized Anxiety Disorder 7-item scale), clinical complications and utilization, barriers to health care, sociodemo-graphics and HRQL (SF-36 Health Survey). We developed models predictive of physical and mental HRQL by conducting stepwise regression analyses.
- Results: Sample prevalence of moderate to severe depression and anxiety symptoms was 33% and 36%, respectively; prevalence of impaired physical and mental HRQL was 17% and 16%, respectively. Increased symptoms of depression, older age, and ≥ 3 emergency department visits in the previous 12 months were independently associated with lower ratings of physical HRQL, controlling for anxiety and sex. Increased symptoms of depression were independently associated with lower ratings of mental HRQL, controlling for barriers to care, insurance status, lifetime complications of SCD, and sex.
- Conclusion: Emotional distress is an important contributor to both physical and mental HRQL for adults with SCD, although sociodemographic variables and barriers to care must also be considered. Innovative approaches that integrate mental health interventions with SCD clinical care are needed.
Emotional distress, including symptoms of depression and anxiety, may adversely affect the course and complicate the treatment of chronic physical conditions [1]. For patients with sickle cell disease (SCD), a group of inherited red blood cell conditions, symptoms of depression and anxiety are more prevalent compared with rates found in the general population [2–8]. The most common symptom of SCD is acute pain events, and other complications range from mild to life-threatening, including anemia, increased risk of infection, acute chest syndrome, stroke, skin ulcers, and pulmonary hypertension [9]. Depression in adults with SCD has been associated with increased sickle cell vaso-occlusive pain events, poor pain control, multiple blood transfusions, and prescription of the disease-modifying therapy hydroxyurea [4]. Adults with SCD and comorbid depression and anxiety had more daily pain and greater distress and interference from pain compared with those who did not have comorbid depression or anxiety [10]. Patients have linked emotional distress and episodes of illness [11], and research has found a relation between pain episodes and depression [12]. In a diary study, negative mood was significantly higher on pain days compared with non-pain days [13].
Studies examining the consequences of emotional distress on health-related quality of life (HRQL) for patients with SCD are emerging. Depressed adults with SCD rated their quality of life on the SF-36 Health Survey [14] as significantly poorer in all areas compared with non-depressed adults with SCD [15]. In regression models, depression was a stronger predictor of SF-36 scores than demographics, hemoglobin type, and pain measures. In a multi-site study [16], 1046 adults with SCD completed the SF-36. Increasing age was associated with significantly lower scores on all subscales except mental health, while female sex additionally contributed to diminished physical function and vitality scale scores in multivariate models [16]. The presence of a mood disorder was associated with bodily pain, and diminished vitality, social functioning, emotional role, and the mental component of HRQL. Medical complications other than pain were not associated with impaired HRQL. Anie and colleagues [17,18] have highlighted the contributions of sickle cell–related pain to diminished mood and HRQL, both in the acute hospital phase and 1 week post discharge.
A comprehensive literature review of patient-reported outcomes for adults with SCD revealed broad categories of the impact of SCD and its treatment on the lives of adults [19]. Categories included pain and pain management, emotional distress, poor social role functioning, diminished overall quality of life, and poor quality of care. Follow-up individual and group interviews with adults with SCD (n = 122) as well as individual interviews with their providers (n = 15) revealed findings consistent with the literature review on the major effects of pain on the lives of adults with SCD, interwoven with emotional distress, poor quality of care, and stigmatization [19].
In the present study, our goal was to describe variables associated with physical and mental HRQL in SCD within the context of the recently published comprehensive conceptual model of broad clinical and life effects associated with SCD [19]. The present analysis uses an existing clinical database and evaluates the effects of the relations between clinical complications of SCD, emotional distress, health care utilization, and HRQL. Our model includes barriers to health care that might prevent vulnerable patients from accessing needed health care services. Sociodemographic variables including ethnic and racial minority status and lower socioeconomic status and educational attainment may create barriers to health care for patients with SCD, as they do for individuals with other chronic conditions [20–23]. Over 60% of patients with SCD are on public insurance [24] and can have difficulties with accessing quality health care [25]. Negative provider attitudes and stigmatization when patients are seeking care for acute pain episodes have been highlighted by patients as major barriers to seeking health care [19,26–28]. In a qualitative study, 45 youth with SCD reported that competing school or peer-group activities, “feeling good,” poor patient-provider relationships, adverse clinic experiences, and forgetting were barriers to clinic attendance [29]. Limited research suggests that barriers to accessing health care are associated with poorer HRQL [30,31]; however no studies were identified that directly evaluated the relation between barriers to care and HRQL for populations with SCD.
We hypothesized that clinical complications of SCD, including pain, and barriers to accessing health care would be independently associated with the physical component of HRQL for adult patients with SCD, controlling for demographic variables. Further, we hypothesized that emotional distress, clinical complications of SCD, and barriers to accessing health care would be independently associated with the mental component of HRQL for adult patients with SCD, controlling for demographic variables.
Methods
Patient Recruitment
Participants were 18 years and older and were a subgroup selected from a larger prospective cohort enrolled in the Sickle Cell Disease Treatment Demonstration Program (SCDTDP) funded by the Health Resources and Services Administration (HRSA). As 1 of 7 SCDTDP grantees, our network collected common demographic, disease-related, and HRQL data as the other grantees to examine sickle cell health and health care [32]. Enrollment at our site was n = 115 from birth through adult, with data collection occurring at baseline in 2010 and annually through 2014. Participants were eligible for enrollment if they had any confirmed diagnosis of SCD and if they were seen at any facility treating SCD in the San Francisco Bay Area region. Interpreter services were available where English was a second language; however, no participant requested those services. The data collection site was an urban comprehensive sickle cell center. Participants were recruited through mailings, posted flyers, or were introduced to the project by their clinical providers. The institutional review boards of the sponsoring hospitals approved all procedures. This report describes analyses from the baseline data collected in 2010 and excludes pediatric patients under the age of 18 years, as we developed our conceptual model based on the adult SCD literature.
Procedures
Patients directly contacted the project coordinator or were introduced by their health care provider. The project coordinator explained the study in more detail, and if the patient agreed to participate, the project coordinator obtained thier informed consent. Participants completed the study materials in a private space in the clinic immediately after or were scheduled for a separate visit at a convenient time and location. Participants with known or observed difficulties with reading completed the questionnaires as an interview. We allowed participants who were unable to complete the forms in one visit to take them home or schedule a follow-up visit to complete them. We asked participants who took the questionnaires home to return them within 2 business days and provided them with a stamped addressed envelope. Participants were compensated with gift cards for their involvement.
Measures
Demographics and Clinical Characteristics
Participants completed an Individual Utilization Questionnaire created for the SCDTDP grantees [32], either as an interview or in paper and pencil format. Participants indicated their age, race and ethnicity, education level, type of insurance, and annual household income. They indicated the type of SCD, number of hospital days and emergency department (ED) visits in the previous 12 months, disease-modifying therapies including hydroxyurea or transfusions, and lifetime incidence of sickle cell–related complications. Complications included pain, acute chest syndrome, fever, severe infection, stroke, kidney damage, gallbladder attack, spleen problems and priapism. Medical data was verified by reviewing medical records when possible; the clinical databases in the hematology/oncology department at the sponsoring hospital are maintained using Microsoft SQL Server, a relational database management system designed for the enterprise environment. However, not all of the participating institutions were linked via this common clinical database or by an electronic health record at the time the study was conducted.
Barriers to Care
We modified a checklist of barriers to accessing health care for patients with a range of chronic conditions [33] to create a SCD-specific checklist [34]. The final checklist consists of 53 items organized into 8 categories including insurance, transportation, accommodations and accessibility, provider knowledge and attitudes, social support, individual barriers such as forgetting or difficulties understanding instructions, emotional barriers such as fear or anger, and barriers posed by SCD itself (eg, pain, fatigue). Participants check off any applicable barrier, yielding a total score ranging from 0 to 53. The checklist overall has demonstrated face validity and test-retest reliability (Pearson r = 0.74, P < 0.05).
Depressive Symptoms
Adults with SCD completed the PHQ-9, the 9-item depression scale of the Patient Health Questionnaire [35]. The PHQ-9 is a tool for assisting primary care clinicians in assessing symptoms of depression, based on criteria from the Diagnostic and Statistical Manual 4th edition (DSM-IV [36]). The PHQ-9 asks about such symptoms as sleep disturbance and difficulty concentrating over the past 2 weeks with scores ranging from 0 (Not at all) to 3 (Every day). The total symptom count is based on the number of items in which the respondent answered as “more than half of days” or greater, and scores are categorized as reflecting no (< 10), mild (10–14), moderate (15–19) or severe (≥ 20) symptoms of depression. Respondents indicate how difficult the symptoms make it for them to engage in daily activities from 0 (Not difficult at all) to 3 (Extremely difficult). The sensitivity and diagnostic and criterion validity of the PHQ-9 have been established [37]. The internal consistency of the PHQ-9 is high, with α > 0.85 in several studies and 48-hour test-retest reliability of 0.84. The PHQ has been used widely, including with African-American and Hispanic populations, and with individuals with chronic conditions [38].
Symptoms of Anxiety
Participants completed the Generalized Anxiety Disorder 7-item (GAD-7) questionnaire for screening and measuring severity of generalized anxiety disorder [39]. The GAD-7 asks about such symptoms as feeling nervous, anxious, or on edge over the past two weeks. Scores from all 7 items are added to obtain a total score [40]. Cut-points of 5, 10, and 15 represent mild, moderate, and severe levels of anxiety symptoms. Respondents indicate how difficult the symptoms make it for them to engage in daily activities from 0 (Not difficult at all) to 3 (Extremely difficult). The internal consistency of the GAD-7 is excellent (α = 0.92). Test-retest reliability is also good (Pearson r = 0.83) as is procedural validity (intraclass correlation = 0.83). The GAD-7 has excellent sensitivity and specificity to identify generalized anxiety disorder [41].
Health-Related Quality of Life
Participants completed the SF-36, which asks about the patient’s health status in the past week [14]. Eight subscales include physical functioning, role-physical, bodily pain, general health, vitality, social functioning, role-emotional and mental health. Two summary measures, the Physical Component Summary and the Mental Component Summary, are calculated from 4 scales each. Use of the summary measures has been shown to increase the reliability of scores and improve the validity of scores in discriminating between physical and psychosocial outcomes [14]. Higher scores represent better HRQL, with a mean score of 50 (SD = 50) for the general population. Internal consistency estimates for the component summary scores are α > 0.89, item discriminant validity estimates are greater than 92.5% and 2-week test-retest reliability was excellent. Scores on the SF-36 have been divided into categories of HRQL functioning [42,43]. Participants in the impaired to very impaired category have scores ≤ mean – 1 SD while participants with average to above average functioning have scores > mean – 1 SD.
The SF-36 has been used extensively in observational and randomized studies for a range of illness conditions. In SCD, some aspects of HRQL as measured by the SF-36 improved for adult patients who responded to hydroxyurea [44]. Participants in the Pain in Sickle Cell Epidemiology Study scored lower than national norms on all SF-36 subscales except psychosocial functioning [45]. HRQL decreased significantly as daily pain intensity increased [45]. Further, women reported worse bodily pain compared with men [46].
Data Analyses
All biostatistical analyses were conducted using Stata 13 [47]. Continuous variables were examined for normality with measures of skewness and peakedness. All variables satisfied the assumptions of normality with the exception of barriers to health care and ED utilization. The variable barriers to health care was transformed using a square root transformation, resulting in a more normally distributed variable. ED utilization was dichotomized as 0–2 versus 3 or more ED visits in the previous 12 months, based on the distribution of utilization in the sample. The cutpoint of ≥ 3 annual ED visits is consistent with other literature on SCD clinical severity [48].
Descriptive statistics were computed to include means, standard deviations and frequencies. Sociodemographic variables (age, sex, insurance status [public or private] and income) were examined as potential covariates using Pearson correlations and t tests. Associations among emotional distress (anxiety and depression symptoms), clinical complications and ED utilization, barriers to health care, and the outcomes of the Physical and Mental Component Summary scores from the SF-36 were examined using Pearson correlations. We conducted stepwise regression with forward selection to determine models predictive of physical and mental HRQL. We tested the addition of each chosen variable (anxiety symptoms, depression symptoms, clinical complications, ED utilization, barriers to health care, age, sex, insurance status, and income), adding the variables (if any) that were most correlated with the outcome, and repeated the process until the model was not improved. A significance level of 0.05 was used for all statistical tests.
Results
Demographic and Clinical Characteristics
The majority of patients (73%) were diagnosed with Hgb SS disease and the most common lifetime complication was pain, reported by almost all of participants (Table 1). The next most common complication was fever, followed by acute chest syndrome. Twenty-seven percent of participants were currently on the disease-modifying therapy hydroxyurea, while 61% had a lifetime history of transfusion therapy. These data were verified with information from the clinical database for 73 participants (95%).
The median number of ED visits in the previous year was 1 (range, 0–50), with 19 patients (25%) with zero visits. The median number of hospital days in the previous year was 13 (range, 0–81). Twenty-nine patients (38%) had no hospital days in the previous year. These data were verified with information from the clinical database for 53 participants (69%), since hospital and ED visits occurred at institutions not always linked with the clinical databases at the sponsoring hospitals.
Emotional Distress, Barriers to Care, and Health-Related Quality of Life
The mean score on the GAD-7 was 7.9 (SD = 6.0, α = 0.90, Table 2). The prevalence of moderate to severe symptoms of anxiety (scores ≥ 10) was 36.4% (n = 28). Fourteen patients with moderate to severe symptoms (50%) reported that anxiety symptoms created some difficulty in work, daily activities, or relationships. Twelve patients (43%) reported that symptoms created very much to extreme difficulty in work, daily activities, or relationships. Fifteen patients (29%) with moderate to severe symptoms of anxiety or depression exhibited comorbid anxiety and depression.
The mean Physical Component Summary score on the SF-36 was 53.6 (SD = 24.1, α = 0.94, Table 2). The prevalence of impaired to very impaired HRQL in the physical domain was 17% (n = 13). The mean Mental Component Summary score on the SF-36 for the sample was 50.1 (SD = 23.7, α = 0.93), with a prevalence of 16% (n = 12) in the impaired to very impaired range for HRQL in the mental domain.
The mean number of barriers from the barriers checklist was 9.2 (SD = 10.1) out of 53 possible. Sixty-five participants (86%) reported at least 1 barrier to accessing health care (Table 2). The most frequently cited barriers to care were provider knowledge and attitudes, followed by transportation, insurance, and access to services (eg, hours and location of services). Less frequently cited barriers to care were individual barriers, including memory, health literacy and motivation, as well as those related to SCD itself, ie, fatigue and pain.
Sociodemographic Variables, Emotional Distress, and Health-Related Quality of Life
Symptoms of anxiety and depression were highly correlated with one another, as would be expected (r = 0.75, P < 0.001). Physical and mental HRQL were significantly correlated with symptoms of depression (r = –0.67, P < 0.001 for physical HRQL component and r = –0.70 for mental HRQL component, P < 0.001), with impaired HRQL in both domains correlated with greater symptoms of depression. Physical and Mental Component Summary scores were significantly correlated with symptoms of anxiety (r = –0.58, P < 0.001 for the physical component and r = –0.62 for the mental component, P < 0.001), with impaired HRQL in both domains correlated with greater symptoms of anxiety. Ratings of difficulty with daily functioning from depressive symptoms were correlated with impaired HRQL in the physical (r = –0.46, P < 0.01) and mental domains (r = –0.52, P < 0.001). Ratings of difficulty with daily functioning from anxiety symptoms were also correlated with impaired HRQL in the physical (r = –0.58, P < 0.001) and mental domains (r = –0.63, P < 0.001). Reports of more barriers to health care were significantly correlated with reports of more depressive and anxiety symptoms (r = 0.53, P < 0.001 and r = 0.48, P < 0.001), with lower Mental Component Summary scores (r = –0.43, P < 0.05), and with more ED visits in the past year (r = 0.43, P < 0.05).
Relations Between Independent Variables and Outcomes
Discussion
Results of this study showed that as expected, symptoms of depression were independently associated with the mental component of HRQL, controlling for other variables. Symptoms of depression were also independently associated with the physical component of HRQL. The effect size for both models was moderate but comparable to effect sizes of other studies of predictive models of physical and mental HRQL in SCD [49]. Our findings were consistent with previous literature, with older age and increased ED utilization independently associated with lower ratings of physical HRQL, with sex and anxiety symptoms entering into the predictive model [15–18,44,45]. Contrary to our hypotheses, barriers to accessing health care were not independently associated with physical or mental HRQL but did contribute to the model for mental HRQL, as did clinical complications and private insurance status.
While our sample was similar to previous samples in mean age and percentage of women participants, our patients reported significantly higher physical HRQL scores, and a wider range of HRQL scores (eg, 53.6,
SD = 24.1 compared with 39.6, SD = 10.0 [16]). The mean Physical Component Summary score was in fact similar to the general population mean of 50. This may reflect improvements in quality of care and subsequent overall improved patient health and HRQL given that these data were collected in year 2 of the HRSA SCDTDP. As an SCDTDP grantee, we implemented goals to improve coordination of service delivery and to increase access to care. However, it should also be considered that there was a selection bias in our study, in favor of those with better HRQL. Nevertheless, as already noted, our findings are consistent with previous literature with regard to inter-relations between variables, ie, associations between lower physical HRQL ratings and symptoms of depression, older age, and increased ED utilization [15]. Future studies in SCD that directly evaluate reported access to a medical home in relation to HRQL are needed to assess the impact of access to care and care coordination on HRQL ratings.
Our use of a data collection tool that focused on lifetime rather than acute history of complications may have contributed to our failure to find a relation between clinical manifestations and physical HRQL. Further, we were not able to assess the effects of pain separately from other complications, since almost every participant reported a lifetime history of pain. However, our findings were consistent with those of researchers who have found psychosocial and sociodemographic factors, versus clinical manifestations, to be major influences on both physical and mental HRQL for individuals with SCD and other chronic and life-threatening conditions [15, 16, 50]. Our confidence is increased in this finding, given that we were able to verify self-reports of clinical manifestations with our clinical database. Our results contribute to the developing body of knowledge that emphasizes the importance of understanding the broad impact on the lives of adults of living with SCD, not just the physical symptomatology.
There has been limited research on barriers to accessing health care as associated with HRQL for SCD populations. Health care barriers have been identified for ethnic minorities, even within patient-centered medical homes, with minority status moderating the effect of barriers to care on HRQL [30]. Our findings that barriers to health care were correlated with depression and anxiety symptoms, mental HRQL, and greater ED utilization support the need to view SCD care within a biobehavioral framework. Health care provider negative attitudes and lack of knowledge were the most frequently cited barriers for adults in our study, particularly in the context of ED and inpatient care. These findings are similar to other studies that have highlighted the impact of these provider variables on quality of care [26,51]. We were not able to separate out effects of ethnic minority status, given that our patients were predominantly African American.
Contributors to poor HRQL that have been identified in SCD are poverty [42] and public insurance status [49]. While over half of our participants had family incomes of less than $30,000, despite a mean household size of 3 members, we did not find that income contributed to either of our models predicting physical or mental HRQL. Over half of our patients were well educated, which could have moderated the effect of their low incomes, but we did not measure other potential moderators such as active coping and supportive relationships [19]. These analyses were beyond the scope of our existing database, but future studies are needed on such resilience factors and processes. Our adults were predominantly on public insurance and we did find that private insurance status was positively associated with higher ratings of mental HRQL, consistent with other SCD research [49]. Taken together, our findings underscore the importance of considering the interplay between emotional distress, sociodemographic and clinical factors and quality of care in order to address risk factors for poor patient-reported outcomes [52,53].
There have not been previous reports of symptoms of emotional distress in SCD using the PHQ-9 and GAD-7, but both measures have been used widely for depression and anxiety screening, including with African-American populations. We selected these over other measures for their brevity, free availability, and psychometric properties. Our prevalence of moderate to severe depression and anxiety symptoms in the present study was similar to what has been found using other tools [2–8]. The PHQ-9 and GAD-7 also provide ratings of symptom interference on daily functioning, and we found that these ratings were associated with impaired physical and mental HRQL. Given that there generally are limited mental health resources in the communities where individuals with SCD reside and are treated, ratings of emotional distress and HRQL can be taken together to stratify those patients with the most immediate need for interventions. Further, screening can be used for early detection with the goal to intervene and prevent the progression of symptoms of emotional distress to long-term, disabling mental health disorders [54]. There is a need for innovative and cost-effective strategies for assessment and treatment of mental health symptoms and disorders for patients with SCD. One model for evidence-based practice in the management of emotional distress for patients with in SCD is the collaborative care model.
The collaborative care model integrates physical and mental health care in the patient-centered medical home and focuses on treating the whole person and family [55]. In this model, a care management staff (eg, nurse, social worker, psychologist) is integrated with the primary care team. The care management staff, in consultation with a psychiatrist, provides evidence-based care coordination, brief behavioral interventions, and support for other treatments, including medications. The effectiveness of collaborative care programs has been demonstrated for ethnic minority and safety net populations such as the SCD population, which is disproportionately low-income and on public insurance [56, 57]. Future research with SCD populations should investigate such interventions as the collaborative care model that addresses both emotional distress and barriers to care.
Limitations
Our results need to be interpreted with caution given the small sample size and the potential bias introduced by non-random sampling. In addition, as our patients are from an urban setting, findings might not generalize to rural populations. This study was cross sectional so no inferences can be made with regard to causality and temporal relations between anxiety symptoms, barriers to care, and HRQL. Our strategy for measuring total clinical complications and barriers to care conserved power but it was not possible to evaluate if specific complications or barriers may have exerted a greater impact on HRQL compared with others. Similarly, other studies have examined specific domains of HRQL, while we limited our analysis to the Physical and Mental Component Summary scores. The utilization questionnaire was designed to assess only lifetime complications, not complications more proximal to the HRQL ratings.
Patient-reported outcomes, now widely accepted as outcome measures, elicit patients’ descriptions of the impact of their condition on their day-to-day lives [34, 58–60]. However, measures of mental health symptoms and HRQL may be subject to recall bias, measurement error, and confounding [61,62]. Nevertheless, a range of studies support the idea that mental health symptoms and HRQL are distinct constructs, and that patients with physical and mental health symptoms are vulnerable to lower ratings of HRQL [63,64]. Disease-modifying therapies such as hydroxyurea can contribute to improved ratings of HRQL [44,65], but we were not able to evaluate the contribution of hydroxyurea to HRQL as it appears to have been underutilized in our sample.
Conclusion
We evaluated emotional distress and other variables in the context of a biobehavioral model of HRQL outcomes for adults with SCD. Integrating the patient's perspective of the impact of the disease and its treatment with assessment of clinical indications is critical to implementing and evaluating effective therapies [25]. However, there are conceptual challenges in determining what actually contributes to HRQL from the patient’s perspective in the context of genetic disorders such as SCD [50]. Our findings highlight the importance of incorporating comprehensive psychosocial screening in order to support optimal HRQL in SCD. Providers may be reluctant to include such screening if, as is often the case, mental health services are difficult to access. Models such as the collaborative care model, which include mental health interventions within the sickle cell center or primary care provider’s office, should be implented. Barriers to care and HRQL should also be routinely evaluated for patients with SCD. Use of disease-specific tools, such as the Adult Sickle Cell Quality of Life measurement system [66], may increase the specificity needed to detect differences within adults with SCD and improvements related to interventions, whether medical or psychosocial. Contributors to HRQL in SCD go beyond clinical manifestations to include psychological and social factors, as well as provider and health system variables. Research conducted within the framework of a comprehensive conceptual model of broad clinical and life effects associated with SCD can inform clinical applications that ultimately enhance HRQL for patients with SCD.
Acknowledgment: The authors wish to thank San Keller, PhD, for her helpful comments on a previous version of this manuscript.
Corresponding author: Marsha J. Treadwell, PhD, Hematology/Oncology Dept., UCSF Benioff Children’s Hospital Oakland, 747 52nd St., Oakland, CA 94609, [email protected].
Funding/support: This research was conducted as part of the National Initiative for Children’s Healthcare Quality (NICHQ) Working to Improve Sickle Cell Healthcare (WISCH) project. Further support came from a grant from the Health Resources and Services Administration (HRSA) Sickle Cell Disease Treatment Demonstration Project Grant No. U1EMC16492 and from the National Institutes of Health (NIH) Clinical and Translational Science Award UL1 RR024131. The views expressed in this publication do not necessarily reflect the views of WISCH, NICHQ, HRSA or NIH.
Financial disclosures: None.
Author contributions: conception and design, MJT; analysis and interpretation of data, MJT, GG; drafting of article, MJT, GG; critical revision of the article, MJT, KK, FB; statistical expertise, GG; obtaining of funding, MJT; administrative or technical support, KK, FB; collection and assembly of data, KK, FB.
A Quality Improvement Initiative to Improve Emergency Department Care for Pediatric Patients with Sickle Cell Disease
From the Children’s Hospital & Research Center Oakland, Oakland, CA.
Abstract
- Objective: To determine whether a quality improvement (QI) initiative would result in more timely assessment and treatment of acute sickle cell–related pain for pediatric patients with sickle cell disease (SCD) treated in the emergency department (ED).
- Methods: We created and implemented a protocol for SCD pain management in the ED with the goals of improving (1) mean time from triage to first analgesic dose; (2) percentage of patients that received their first analgesic dose within 30 minutes of triage, and (3) percentage of patients who had pain assessment performed within 30 minutes of triage and who were re-assessed within 30 minutes after the first analgesic dose.
- Results: Significant improvements were achieved between baseline (55 patient visits) and post order set implementation (165 visits) in time from triage to administration of first analgesic (decreased from 89.9 ± 50.5 to 35.2 ± 22.8 minutes, P < 0.001); percentage of patient visits receiving pain medications within 30 minutes of triage (from 7% to 53%, P < 0.001); percentage of patient visits assessed within 30 minutes of triage (from 64% to 99.4%, P < 0.001); and percentage of patient visits re-assessed within 30 minutes of initial analgesic (from 54% to 86%, P < 0.001).
- Conclusions: Implementation of a QI initiative in the ED led to expeditious care for pediatric patients with SCD presenting with pain. A QI framework provided us with unique challenges but also invaluable lessons as we address our objective of decreasing the quality gap in SCD medical care.
Pain is the leading cause of emergency department (ED) visits for patients with sickle cell disease (SCD) [1]. In the United States, 78% of the nearly 200,000 annual ED visits for SCD are for a complaint of pain [1]. Guidelines for the management of sickle cell vaso-occlusive pain episodes (VOE) suggest prompt initiation of parenteral opioids, use of effective opioid doses, and repeat opioid doses at frequent intervals [2–4]. Adherence to guidelines is poor. Both pediatric and adult patients with SCD experience delays in the initiation of analgesics and are routinely undertreated with respect to opioid dosing [5–8]. Even after controlling for race, the delays in time to analgesic administration experienced by patients with SCD exceed the delays encountered by patients who present to the ED with other types of pain [5,9]. These disparities warrant efforts designed to improve the delivery of quality care to patients with SCD.
Barriers to rapid and appropriate care of VOE in the ED are multifactorial and include systems-based limitations, such as acuity of the ED census, staffing limitations (eg, nurse-to-patient ratios), and facility limitations (eg, room availability) [6]. Provider-based limitations may include lack of awareness of available guidelines [10]. Biases and misunderstandings amongst providers about sickle cell pain and adequate medication dosing may also play a role [11–13]. These provider biases often lead to undertreatment of the pain, which in turn can lead to pseudoaddiction (drug-seeking behavior due to inadequate treatment) and a cycle of increased ED and inpatient utilization [14,15].
Patient-specific barriers to effective ED management of pain are equally complex. Previous negative experiences in the ED can lead patients and families to delay seeking care or avoid the ED altogether despite severe VOE pain [16]. Patients report frustration with the lack of consideration that they receive for their reports of pain, perceived insensitivity of hospital staff, inadequate analgesic administration, staff preoccupation with concerns of drug addiction, and an overall lack of respect and trust [17–19]. Patients also perceive a lack of knowledge of SCD and its treatments on the part of ED staff [7]. Other barriers to effective management are technical in nature, such as difficulty in establishing timely intravenous (IV) access.
Gaps and variations in quality of care contribute to poor outcomes for patients with SCD [20,21]. To help address these inequities, the Working to Improve Sickle Cell Healthcare (WISCH) project began in 2010 to improve care and outcomes for patients with SCD. WISCH is a collaborative quality improvement (QI) project funded by the Health Resources and Services Administration (HRSA) that has the goal to use improvement science to improve outcomes for patients with SCD across the life course (Ed note: see Editorial by Oyeku et al in this issue). As one of the HRSA-WISCH grantee networks, we undertook a QI project designed to decrease the quality gap in SCD medical care by creating and implementing a protocol for ED pain management for pediatric patients. Goals of the project were to improve the timely and appropriate assessment and treatment of acute VOE in the ED.
Methods
Setting
This ED QI initiative was implemented at Children’s Hospital & Research Center Oakland, an urban free-standing pediatric hospital that serves a demographically diverse population. The hospital ED sees over 45,000 visits per year, with 250 visits per year for VOE. Residents in pediatrics, family medicine, and emergency medicine staff the ED. All attending physicians are subspecialists in pediatric emergency medicine. Study procedures were approved by the hospital’s institutional review board.
Intervention
A multidisciplinary team consisting of ED staff and sickle cell center staff drafted a nursing-driven protocol for the assessment and management of acute pain associated with VOE, incorporating elements from a protocol in use by another WISCH collaborative member. The protocol called for the immediate triage and assessment of all patients with SCD who presented with moderate to severe pain suggestive of VOE. Moderate to severe pain was defined as a pain score of ≥ 5 on a numeric scale of 0 to 10, where 0 = no pain and 10 = the worst pain imaginable. Exclusion criteria included a chief complaint of pain not considered secondary to VOE (eg, trauma, fracture). Patients were also excluded if they had been transferred from another facility. The protocol called for IV pain medication to be administered within 10 minutes of the patient being roomed, with re-evaluation at 20-minute intervals and re-dosing of pain medication based on the patient’s subsequent pain rating.
Measures
We selected performance measures from the bank developed by the WISCH team to track improvement and evaluate progress. These performance measures included (1) mean time from triage to first analgesic dose, (2) percentage of patients that received their first dose of analgesic within 30 minutes of triage, (3) percentage of patients who had a pain assessment performed within 30 minutes of triage, and (4) percentage of patients re-assessed within 30 minutes after the first dose of analgesic had been administered. Our aims were to have 80% of patients assessed and given pain medications within 30 minutes of triage, and to have 80% of patients re-assessed within 30 minutes after having received their first dose of an analgesic, within 12 months of implementing our intervention.
Data Collection and Analysis
The WISCH project coordinator reviewed records of visits to the ED for a baseline period of 6 months and post-order set implementaton. Demographic data (age, gender), clinical data (hemoglobin type), pain scores, utilization data (number of ED visits during the study period), and data pertaining to the metrics chosen from the WISCH measurement bank were extracted from each eligible patient’s ED chart after the visit was completed. If patients were admitted, their length of hospitalization was extracted from their inpatient medical record.
All biostatistical analyses were conducted using Stata 9.2 (StataCorp, College Station, TX). Descriptive statistics computed at 2 time-points (pre and post order set implementation) were utilized to examine means, standard deviations and percentages. The 2 time-points were initially compared at the visit level of measurement, using Student’s t tests corrected for unequal variances where necessary for continuous variables and chi-square analyses for categorical variables, to evaluate if there was an improvement in timely triage, assessment, and treatment of acute VOE pain for all ED visits pre and post order set implementation. To account for trends and possible correlations across the months post order set implementation, we ran a mixed linear model with repeated measures over time to compare visits during all months post order set implementation with the baseline months, for metric 1, time from triage to first pain medication. If significant differences were found, we used Dunnett’s method of multiple comparisons to determine which months differed from baseline. For metrics 2 through 4, we ran linear models with a binary outcome, a logit link function and using general estimating equations to determine trends and to account for correlations over time.
Secondary analyses were conducted to evaluate whether mean pain scores were significantly different over the course of the ED visit for the 78 unique patients seen post order set implementation. A multivariable mixed linear model, for the outcome of the third pain score, was used to assess the associations with prior scores and to control for potential covariates (age, gender, number of ED visits, hemoglobin type) that were determined in advance. A statistical significance level of 0.05 was used for all tests.
Results
Baseline data were collected from December 2011 to May 2012. The protocol was implemented in July 2012 and was utilized during 165 ED visits (91% of eligible visits) through April 2013. There were no statistically significant differences in demographic or clinical characteristics between the 55 patients whose charts were reviewed prior to implementing the order set and the 78 unique patients treated thereafter. Pre order set implementation, the mean age was 14.6 ± 6.4 years; 60% were female and the primary diagnosis was HgbSS disease (61.8% of diagnoses). Post order set implementation, the mean age was 16.0 ± 8.0 years; 51.3% were female and the primary diagnosis was HgbSS disease (61.5% of diagnoses). The mean number of visits was 1.5 visits per patient with a range of 1–8 visits, both pre and post order set implementation. Thirty-one patients had ED visits at both time periods.
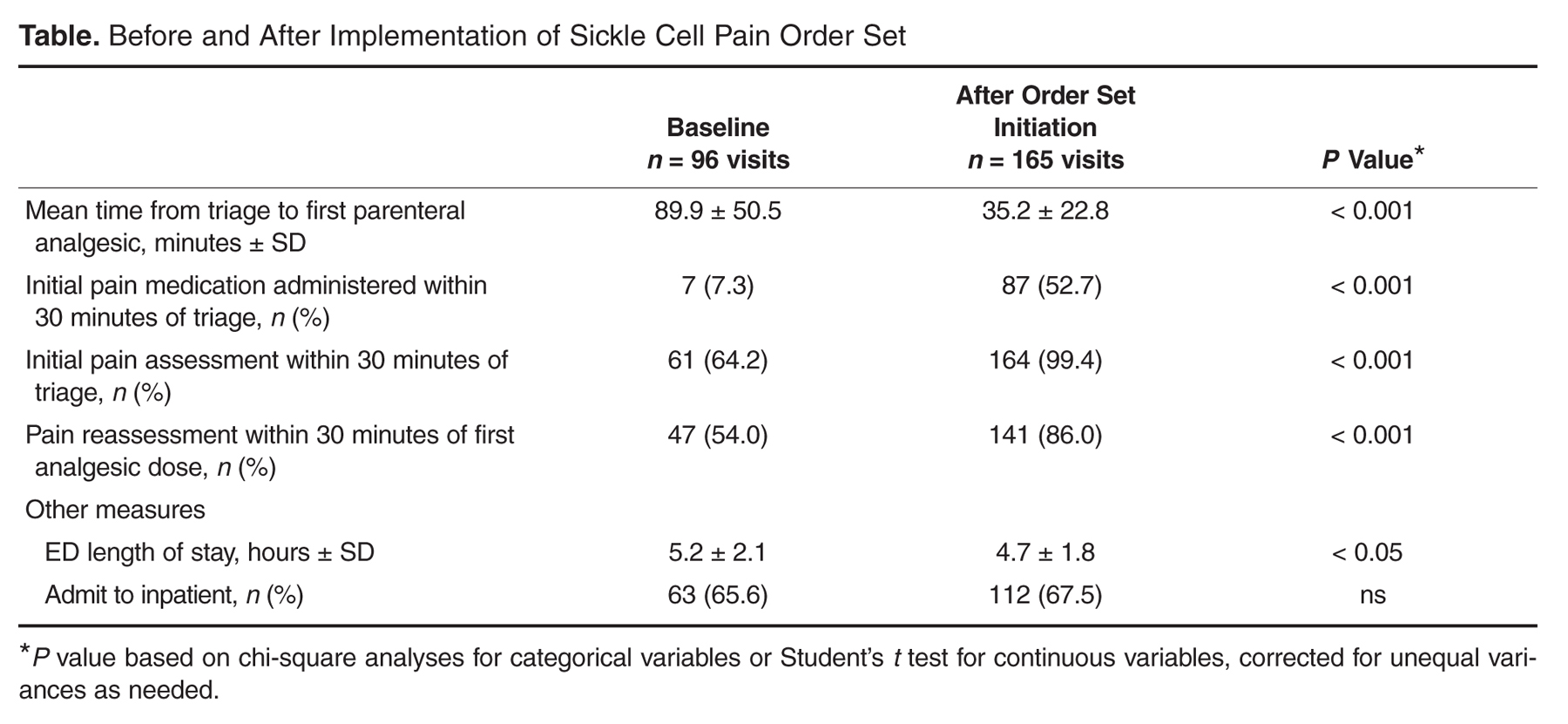
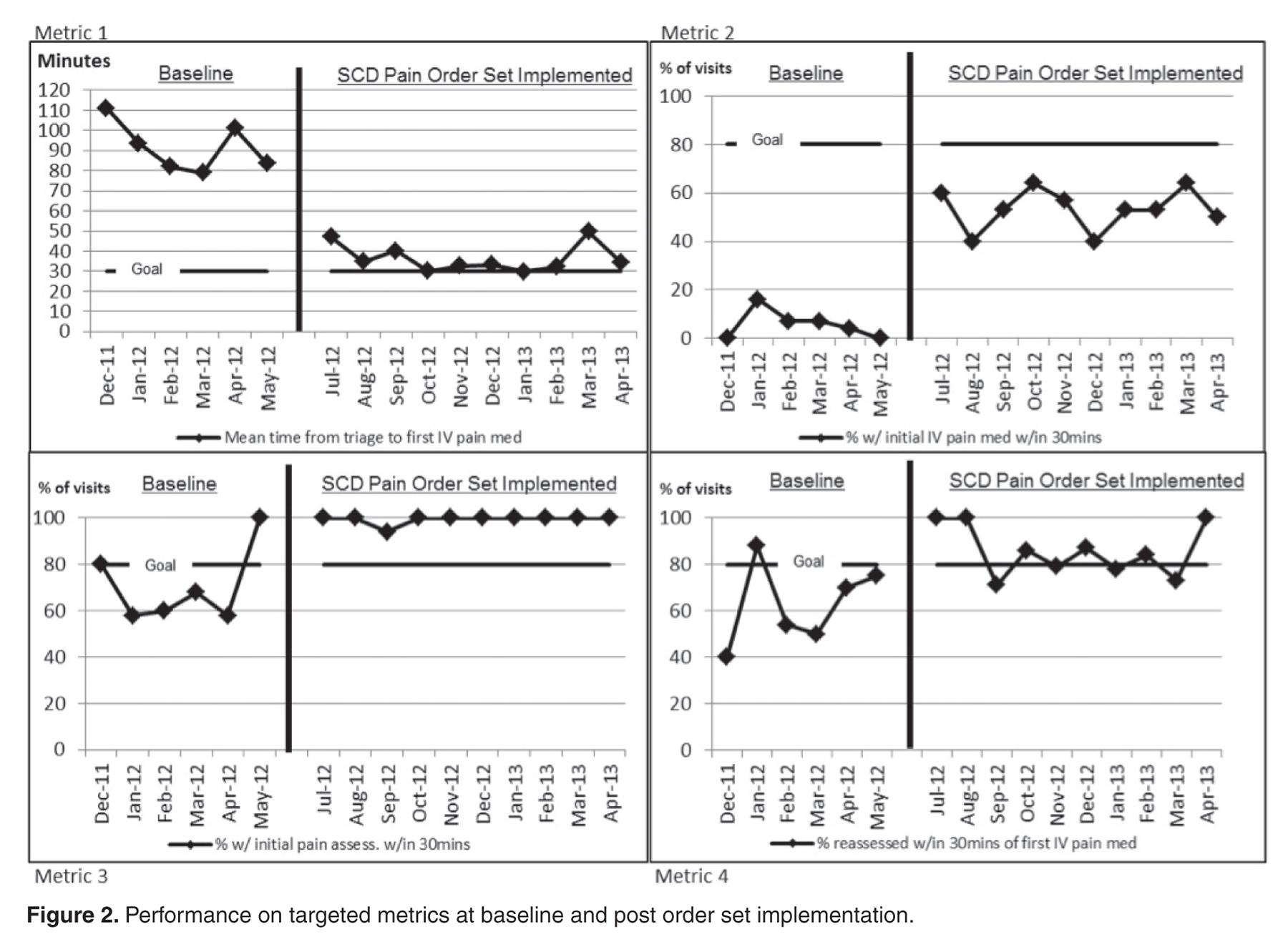
It can be seen in Figure 2 that staff performance on 3 of the 4 metrics (with the exception of initial analgesic within 30 minutes of triage) began to improve prior to implementing the order set. The mean length of ED stays decreased by 30 minutes, from a mean of 5.2 hours down to 4.7 hours (P < 0.05, Table). There was no significant change in the percentage of patients admitted to the inpatient unit.
We performed secondary analyses to determine if performance on our first metric, mean time from triage to first analgesic dose, was associated with any improvement on the third pain assessment for the patients enrolled post order set implementation. Looking at the first ED visit during the study period for the 78 unique patients, we found significant decreases in mean pain scores from the first to the second, from the second to the third, and from the first to the third assessment (P < 0.01). The mean pain scores were 8.3 ± 1.8, 5.9 ± 2.8, and 5.1 ± 3.0 on initial, second and third assessments, respectively. A multivariable model controlling for gender, hemoglobin type, number of ED visits and time to first pain medication showed that only the score at the second pain assessment (β = 0.88 ± 0.08, P < 0.001) was a significant predictor of the score at the third pain assessment.
Discussion
We demonstrated that a QI initiative to improve acute pain management resulted in more timely assessment and treatment of pain in pediatric patients with SCD. Significant improvements from baseline were achieved and sustained over a 10-month period in all 4 targeted metrics. We consistently exceeded our goal of having 80% of patients assessed within 30 minutes of triage, and our mean time to first pain medication (35.2 ± 22.8 minutes) came close to our goal of 30 minutes from triage. While we also achieved our goal to have 80% of patients re-assessed within 30 minutes after having received their first dose of an analgesic, we fell short in the percent who received their initial pain medication within 30 minutes of triage (52.7% versus goal of 80%). Although the length of stay in the ED decreased, no change was observed in the percentage of patients who required admission to the inpatient unit. A secondary analysis showed that mean pain scores significantly decreased over the course of the ED visit, from severe to moderate intensity.
The improvements that we observed began prior to implementation of the order set. We recognize that simply raising awareness and educating staff about the importance of timely and appropriate assessment and treatment of acute sickle cell related pain in the ED might be a potential confounder of our results. However, changes were sustained for 10 months post order set implementation and beyond, with no evidence that the performance on the target metrics is drifting back to baseline levels. Education and awareness-raising alone rarely result in sustained application of clinical practice guidelines [22]. We collaborated with NICHQ and other HRSA-WISCH grantees to systematically implement improvement science to ensure that the changes that we observed were indeed improvements and would be sustained [23] by first changing the system of care in the ED by introducing a standard order set [24,25]. We put a system into place to track use of the order set and to work with providers almost immediately if deviations were observed, to understand and overcome any barriers to the order set implementation. Systems in the ED and in the sickle cell center were aligned with the hospital’s QI initiatives [23].
Another strategy that we used to insure that the changes we observed would be sustained was to create a multidisciplinary team to build knowledge, skills, and new practices, including learning from other WISCH grantees and the NICHQ coordinating center [23]. We modified and adapted the intervention to our specific context [25]; although the outline of the order set was influenced by our WISCH colleagues, the final order set was structured to be consistent with other protocols within our institution. Finally, we included consumer input in the design of the project from the outset.
A previous study of a multi-institutional QI initiative aimed at improving acute SCD pain management for adult patients in the ED was unable to demonstrate an improvement in time to administration of initial analgesic [26]. Our study with pediatric patients was able to demonstrate a clinically meaningful decrease in the time to administration of first parenteral analgesic. The factors that account for the discrepant findings between these studies are likely multifactorial. Age (ie, pediatric vs. adult patients) may have played a role given that IV access may become increasingly difficult as patients with SCD age [26]. Education for providers should include the importance of alternative methods of administration of opioids, including subcutaneous and intranasal routes, to avoid delays when IV access is difficult. It is possible that negative provider attitudes converge with the documented increase in patient visits during the young adult years [27]. This may set up a challenging feedback loop wherein these vulnerable young adults are faced with greater stigma and consequently receive lower quality care, even when there is an attempt to carry out a standardized protocol.
We did not find that the QI intervention resulted in decreased admissions to the inpatient unit, with 68% of visits resulting in admission. In a recent pediatric SCD study, hospital admissions for pain control accounted for 78% of all admissions and 70% of readmissions within 30 days [28]. The investigators found that use of a SCD analgesic protocol including patient-controlled analgesia (PCA) improved quality of care as well as hospital readmission rates within 30 days (from 28% to 11%). Our ED QI protocol focused on only the first 90 minutes of the visit for pain. Our team has discussed the potential for starting the PCA in the ED and we should build on our success to focus on specific care that patients receive beyond their initial presentation. Further, we introduced pain action planning into outpatient care and need to continue to improve positive patient self-management strategies to ensure more seamless transition of pain management between home, ED, and inpatient settings.
Several valuable lessons were learned over the course of the ED QI initiative. Previous researchers [28] have emphasized the importance of coupling provider education with standardized order sets in efforts to improve the care of patients with SCD. Although we did not offer monthly formal education to our providers, the immediate follow-up when there were protocol deviations most likely served as teaching moments. These teaching moments also surfaced when some ED and hematology providers expressed concerns about the risk for oversedation with the rapid reassessment of pain and re-dosing of pain medications. Although rare, some parents also expressed that their child was being treated too vigorously with opioids. Our project highlighted the element of stigma that still accompanies the use of opioids for SCD pain management.
The project could not have been undertaken were it not for a small but determined multidisciplinary team of individuals who were personally invested in seeing the project come to fruition. The identification of physician and nurse champions who were enthusiastic about the project, invested in its conduct, and committed to its success was a cornerstone of the project’s success. These champions played an essential role in engaging staff interest in the project and oversaw the practicalities of implementing a new protocol in the ED. A spirit of collaboration, teamwork, and good communication between all involved parties was also critical. At the same time, we incorporated input from the treating ED and hematology clinicians using PDSA cycles as we were refining our protocol. We believe that our process enhanced buy-in from participating providers and clarified any issues that needed to be addressed in our setting, resulting in accelerated and sustained quality improvement.
Limitations
Although protocol-driven interventions are designed to provide a certain degree of uniformity of care, the protocol was not designed nor utilized in such a way that it superseded the best medical judgment of the treating clinicians. Deviations from the protocol were permissible when they were felt to be in the patient’s best interest. The study did not control for confounding variables such as disease severity, how long the patient had been in pain prior to coming to the ED, nor did we assess therapeutic interventions the patient had utilized at home prior to seeking out care in the ED. All of these factors could affect how well a patient might respond to treatment. We believe that sharing baseline data and monthly progress via run charts (graphs of data over time) with ED and sickle cell center staff and with consumer representatives enhanced the pace and focus of the project [23]. We had a dedicated person managing our data in real time through our HRSA funding, thus the project might not be generalizable to other institutions that do not have such staffing or access to the technology to allow project progress to be closely monitored by stakeholders.
Future Directions
With the goal of further reducing the time to administration of first analgesic dose in the ED setting, intranasal fentanyl will be utilized in our ED as the initial drug of choice for patients who do not object to or have a contraindication to its use. Collection of data from patients and family members is being undertaken to assess consumer satisfaction with the ED QI initiative. Recognizing that the ED management of acute pain addresses only one aspect of sickle cell pain, we are looking at ways to more comprehensively address pain. Individualized outpatient pain management plans are being created and patients and families are being encouraged and empowered to become active partners with their sickle cell providers in their own care. Although our initial efforts have focused on our pediatric patients, an additional aim of our project is to broaden the scope of our ED QI initiative to include community hospitals in the region that serve adult patients with SCD.
Conclusion
Implementation of a QI initiative in the ED has led to expeditious care for pediatric patients with SCD presenting with VOE. A multidisciplinary approach, ongoing staff education, and commitment to the initiative have been necessary to sustain the improvements. Our success can provide a template for other QI initiatives in the ED that translate to improved patient care for other diseases. A QI framework provided us with unique challenges but also invaluable lessons as we addressed our objective to improve outcomes for patients with SCD across the life course.
Acknowledgments: The authors wish to thank Theresa Freitas, RN, Lisa Hale, PNP, Carolyn Hoppe, MD, Ileana Mendez, RN, Helen Mitchell, Mary Rutherford, MD, Augusta Saulys, MD and the Children’s Hospital & Research Center Oakland Emergency Medicine Department and Sickle Cell Center for their support.
Corresponding author: Marsha Treadwell, PhD, Children’s Hospital & Research Center Oakland, 747 52nd St, Oakland, CA 94609, [email protected].
Funding/support: This research was conducted as part of the National Initiative for Children’s Healthcare Quality (NICHQ) Working to Improve Sickle Cell Healthcare (WISCH) project. Further support came from a grant from the Health Resources and Services Administration Sickle Cell Disease Treatment Demonstration Project Grant No. U1EMC16492 and from NIH CTSA grant UL1 RR024131. The views expressed in this publication do not necessarily reflect the views of WISCH, NICHQ, or HRSA.
1. Yusuf HR, Atrash HK, Grosse SD, et al. Emergency department visits made by patients with sickle cell disease: a descriptive study, 1999-2007. Am J Preventive Med 2010;38 (4 Suppl):S536–41.
2. Benjamin L, Dampier C, Jacox A, et al. Guideline for the management of acute and chronic pain in sickle cell disease. American Pain Society; 1999.
3. Rees DC, Olujohungbe AD, Parker NE, et al. Guidelines for the management of the acute painful crisis in sickle cell disease. Br J Haematology 2003;120:744–52.
4. Solomon LR. Pain management in adults with sickle cell disease in a medical center emergency department. J Nat Med Assoc 2010;102:1025–32.
5. Lazio MP, Costello HH, Courtney DM, et al. A comparison of analgesic management for emergency department patients with sickle cell disease and renal colic. Clin J Pain 2010;26:199–205.
6. Shenoi R, Ma L, Syblik D, Yusuf S. Emergency department crowding and analgesic delay in pediatric sickle cell pain crises. Ped Emerg Care 2011;27:911–7.
7. Tanabe P, Artz N, Mark Courtney D, et al. Adult emergency department patients with sickle cell pain crisis: a learning collaborative model to improve analgesic management. Acad Emerg Med 2010;17:399–407.
8. Zempsky WT. Evaluation and treatment of sickle cell pain in the emergency department: paths to a better future. Clin Ped Emerg Med 2010;11:265–73.
9. Haywood C Jr, Tanabe P, Naik R, et al. The impact of race and disease on sickle cell patient wait times in the emergency department. Am J Emerg Med 2013;31:651–6.
10. Solomon LR. Treatment and prevention of pain due to vaso-occlusive crises in adults with sickle cell disease: an educational void. Blood 2008;111:997–1003.
11. Ballas SK. New era dawns on sickle cell pain. Blood 2010;116:311–2.
12. Haywood C Jr, Lanzkron S, Ratanawongsa N, et al. The association of provider communication with trust among adults with sickle cell disease. J Gen Intern Med 2010;25:543–8.
13. Zempsky WT. Treatment of sickle cell pain: fostering trust and justice. JAMA 2009;302:2479–80.
14. Elander J, Lusher J, Bevan D, Telfer P. Pain management and symptoms of substance dependence among patients with sickle cell disease. Soc Sci Med 2003;57:1683–96.
15. Elander J, Lusher J, Bevan D, et al. Understanding the causes of problematic pain management in sickle cell disease: evidence that pseudoaddiction plays a more important role than genuine analgesic dependence. J Pain Sympt Manag 2004;27:156–69.
16. Smith WR, Penberthy LT, Bovbjerg VE, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med 2008;148:94–101.
17. Harris A, Parker N, Baker C. Adults with sickle cell. Psychol Health Med 1998;3:171–9.
18. Jenerette CM, Brewer C. Health-related stigma in young adults with sickle cell disease. J Nat Med Assoc 2010;102:1050–5.
19. Maxwell K, Streetly A, Bevan D. Experiences of hospital care and treatment seeking for pain from sickle cell disease: qualitative study. BMJ 1999;318:1585–90.
20. Oyeku SO, Wang CJ, Scoville R, et al. Hemoglobinopathy Learning Collaborative: using quality improvement (QI) to achieve equity in health care quality, coordination, and outcomes for sickle cell disease. J Health Care Poor Underserved 2012;23(3 Suppl):34–48.
21. Wang CJ, Kavanagh PL, Little AA, et al. Quality-of-care indicators for children with sickle cell disease. Pediatrics 2011;128:484–93.
22. Mansouri M, Lockyer J. A meta-analysis of continuing medical education effectiveness. J Contin Ed Health Prof 2007;27:6–15.
23. The breakthrough series: IHI’s collaborative model for achieving breakthrough improvement. Boston: Institute for Healthcare Improvement; 2003.
24. Berwick DM. Improvement, trust, and the healthcare workforce. Qual Safety Health Care 2003;12:448–52.
25. Hovlid E, Bukve O, Haug K, et al. Sustainability of healthcare improvement: what can we learn from learning theory? BMC Health Serv Res 2012;12:235.
26. Tanabe P, Hafner JW, Martinovich Z, Artz N. Adult emergency department patients with sickle cell pain crisis: results from a quality improvement learning collaborative model to improve analgesic management. Acad Emerg Med 2012;19:430–8.
27. Brousseau DC, Owens PL, Mosso AL, et al. Acute care utilization and rehospitalizations for sickle cell disease. JAMA 2010;303:1288–94.
28. Frei-Jones MJ, Field JJ, DeBaun MR. Multi-modal intervention and prospective implementation of standardized sickle cell pain admission orders reduces 30-day readmission rate. Pediatr Blood Cancer 2009;53:401–5.
From the Children’s Hospital & Research Center Oakland, Oakland, CA.
Abstract
- Objective: To determine whether a quality improvement (QI) initiative would result in more timely assessment and treatment of acute sickle cell–related pain for pediatric patients with sickle cell disease (SCD) treated in the emergency department (ED).
- Methods: We created and implemented a protocol for SCD pain management in the ED with the goals of improving (1) mean time from triage to first analgesic dose; (2) percentage of patients that received their first analgesic dose within 30 minutes of triage, and (3) percentage of patients who had pain assessment performed within 30 minutes of triage and who were re-assessed within 30 minutes after the first analgesic dose.
- Results: Significant improvements were achieved between baseline (55 patient visits) and post order set implementation (165 visits) in time from triage to administration of first analgesic (decreased from 89.9 ± 50.5 to 35.2 ± 22.8 minutes, P < 0.001); percentage of patient visits receiving pain medications within 30 minutes of triage (from 7% to 53%, P < 0.001); percentage of patient visits assessed within 30 minutes of triage (from 64% to 99.4%, P < 0.001); and percentage of patient visits re-assessed within 30 minutes of initial analgesic (from 54% to 86%, P < 0.001).
- Conclusions: Implementation of a QI initiative in the ED led to expeditious care for pediatric patients with SCD presenting with pain. A QI framework provided us with unique challenges but also invaluable lessons as we address our objective of decreasing the quality gap in SCD medical care.
Pain is the leading cause of emergency department (ED) visits for patients with sickle cell disease (SCD) [1]. In the United States, 78% of the nearly 200,000 annual ED visits for SCD are for a complaint of pain [1]. Guidelines for the management of sickle cell vaso-occlusive pain episodes (VOE) suggest prompt initiation of parenteral opioids, use of effective opioid doses, and repeat opioid doses at frequent intervals [2–4]. Adherence to guidelines is poor. Both pediatric and adult patients with SCD experience delays in the initiation of analgesics and are routinely undertreated with respect to opioid dosing [5–8]. Even after controlling for race, the delays in time to analgesic administration experienced by patients with SCD exceed the delays encountered by patients who present to the ED with other types of pain [5,9]. These disparities warrant efforts designed to improve the delivery of quality care to patients with SCD.
Barriers to rapid and appropriate care of VOE in the ED are multifactorial and include systems-based limitations, such as acuity of the ED census, staffing limitations (eg, nurse-to-patient ratios), and facility limitations (eg, room availability) [6]. Provider-based limitations may include lack of awareness of available guidelines [10]. Biases and misunderstandings amongst providers about sickle cell pain and adequate medication dosing may also play a role [11–13]. These provider biases often lead to undertreatment of the pain, which in turn can lead to pseudoaddiction (drug-seeking behavior due to inadequate treatment) and a cycle of increased ED and inpatient utilization [14,15].
Patient-specific barriers to effective ED management of pain are equally complex. Previous negative experiences in the ED can lead patients and families to delay seeking care or avoid the ED altogether despite severe VOE pain [16]. Patients report frustration with the lack of consideration that they receive for their reports of pain, perceived insensitivity of hospital staff, inadequate analgesic administration, staff preoccupation with concerns of drug addiction, and an overall lack of respect and trust [17–19]. Patients also perceive a lack of knowledge of SCD and its treatments on the part of ED staff [7]. Other barriers to effective management are technical in nature, such as difficulty in establishing timely intravenous (IV) access.
Gaps and variations in quality of care contribute to poor outcomes for patients with SCD [20,21]. To help address these inequities, the Working to Improve Sickle Cell Healthcare (WISCH) project began in 2010 to improve care and outcomes for patients with SCD. WISCH is a collaborative quality improvement (QI) project funded by the Health Resources and Services Administration (HRSA) that has the goal to use improvement science to improve outcomes for patients with SCD across the life course (Ed note: see Editorial by Oyeku et al in this issue). As one of the HRSA-WISCH grantee networks, we undertook a QI project designed to decrease the quality gap in SCD medical care by creating and implementing a protocol for ED pain management for pediatric patients. Goals of the project were to improve the timely and appropriate assessment and treatment of acute VOE in the ED.
Methods
Setting
This ED QI initiative was implemented at Children’s Hospital & Research Center Oakland, an urban free-standing pediatric hospital that serves a demographically diverse population. The hospital ED sees over 45,000 visits per year, with 250 visits per year for VOE. Residents in pediatrics, family medicine, and emergency medicine staff the ED. All attending physicians are subspecialists in pediatric emergency medicine. Study procedures were approved by the hospital’s institutional review board.
Intervention
A multidisciplinary team consisting of ED staff and sickle cell center staff drafted a nursing-driven protocol for the assessment and management of acute pain associated with VOE, incorporating elements from a protocol in use by another WISCH collaborative member. The protocol called for the immediate triage and assessment of all patients with SCD who presented with moderate to severe pain suggestive of VOE. Moderate to severe pain was defined as a pain score of ≥ 5 on a numeric scale of 0 to 10, where 0 = no pain and 10 = the worst pain imaginable. Exclusion criteria included a chief complaint of pain not considered secondary to VOE (eg, trauma, fracture). Patients were also excluded if they had been transferred from another facility. The protocol called for IV pain medication to be administered within 10 minutes of the patient being roomed, with re-evaluation at 20-minute intervals and re-dosing of pain medication based on the patient’s subsequent pain rating.
Measures
We selected performance measures from the bank developed by the WISCH team to track improvement and evaluate progress. These performance measures included (1) mean time from triage to first analgesic dose, (2) percentage of patients that received their first dose of analgesic within 30 minutes of triage, (3) percentage of patients who had a pain assessment performed within 30 minutes of triage, and (4) percentage of patients re-assessed within 30 minutes after the first dose of analgesic had been administered. Our aims were to have 80% of patients assessed and given pain medications within 30 minutes of triage, and to have 80% of patients re-assessed within 30 minutes after having received their first dose of an analgesic, within 12 months of implementing our intervention.
Data Collection and Analysis
The WISCH project coordinator reviewed records of visits to the ED for a baseline period of 6 months and post-order set implementaton. Demographic data (age, gender), clinical data (hemoglobin type), pain scores, utilization data (number of ED visits during the study period), and data pertaining to the metrics chosen from the WISCH measurement bank were extracted from each eligible patient’s ED chart after the visit was completed. If patients were admitted, their length of hospitalization was extracted from their inpatient medical record.
All biostatistical analyses were conducted using Stata 9.2 (StataCorp, College Station, TX). Descriptive statistics computed at 2 time-points (pre and post order set implementation) were utilized to examine means, standard deviations and percentages. The 2 time-points were initially compared at the visit level of measurement, using Student’s t tests corrected for unequal variances where necessary for continuous variables and chi-square analyses for categorical variables, to evaluate if there was an improvement in timely triage, assessment, and treatment of acute VOE pain for all ED visits pre and post order set implementation. To account for trends and possible correlations across the months post order set implementation, we ran a mixed linear model with repeated measures over time to compare visits during all months post order set implementation with the baseline months, for metric 1, time from triage to first pain medication. If significant differences were found, we used Dunnett’s method of multiple comparisons to determine which months differed from baseline. For metrics 2 through 4, we ran linear models with a binary outcome, a logit link function and using general estimating equations to determine trends and to account for correlations over time.
Secondary analyses were conducted to evaluate whether mean pain scores were significantly different over the course of the ED visit for the 78 unique patients seen post order set implementation. A multivariable mixed linear model, for the outcome of the third pain score, was used to assess the associations with prior scores and to control for potential covariates (age, gender, number of ED visits, hemoglobin type) that were determined in advance. A statistical significance level of 0.05 was used for all tests.
Results
Baseline data were collected from December 2011 to May 2012. The protocol was implemented in July 2012 and was utilized during 165 ED visits (91% of eligible visits) through April 2013. There were no statistically significant differences in demographic or clinical characteristics between the 55 patients whose charts were reviewed prior to implementing the order set and the 78 unique patients treated thereafter. Pre order set implementation, the mean age was 14.6 ± 6.4 years; 60% were female and the primary diagnosis was HgbSS disease (61.8% of diagnoses). Post order set implementation, the mean age was 16.0 ± 8.0 years; 51.3% were female and the primary diagnosis was HgbSS disease (61.5% of diagnoses). The mean number of visits was 1.5 visits per patient with a range of 1–8 visits, both pre and post order set implementation. Thirty-one patients had ED visits at both time periods.
It can be seen in Figure 2 that staff performance on 3 of the 4 metrics (with the exception of initial analgesic within 30 minutes of triage) began to improve prior to implementing the order set. The mean length of ED stays decreased by 30 minutes, from a mean of 5.2 hours down to 4.7 hours (P < 0.05, Table). There was no significant change in the percentage of patients admitted to the inpatient unit.
We performed secondary analyses to determine if performance on our first metric, mean time from triage to first analgesic dose, was associated with any improvement on the third pain assessment for the patients enrolled post order set implementation. Looking at the first ED visit during the study period for the 78 unique patients, we found significant decreases in mean pain scores from the first to the second, from the second to the third, and from the first to the third assessment (P < 0.01). The mean pain scores were 8.3 ± 1.8, 5.9 ± 2.8, and 5.1 ± 3.0 on initial, second and third assessments, respectively. A multivariable model controlling for gender, hemoglobin type, number of ED visits and time to first pain medication showed that only the score at the second pain assessment (β = 0.88 ± 0.08, P < 0.001) was a significant predictor of the score at the third pain assessment.
Discussion
We demonstrated that a QI initiative to improve acute pain management resulted in more timely assessment and treatment of pain in pediatric patients with SCD. Significant improvements from baseline were achieved and sustained over a 10-month period in all 4 targeted metrics. We consistently exceeded our goal of having 80% of patients assessed within 30 minutes of triage, and our mean time to first pain medication (35.2 ± 22.8 minutes) came close to our goal of 30 minutes from triage. While we also achieved our goal to have 80% of patients re-assessed within 30 minutes after having received their first dose of an analgesic, we fell short in the percent who received their initial pain medication within 30 minutes of triage (52.7% versus goal of 80%). Although the length of stay in the ED decreased, no change was observed in the percentage of patients who required admission to the inpatient unit. A secondary analysis showed that mean pain scores significantly decreased over the course of the ED visit, from severe to moderate intensity.
The improvements that we observed began prior to implementation of the order set. We recognize that simply raising awareness and educating staff about the importance of timely and appropriate assessment and treatment of acute sickle cell related pain in the ED might be a potential confounder of our results. However, changes were sustained for 10 months post order set implementation and beyond, with no evidence that the performance on the target metrics is drifting back to baseline levels. Education and awareness-raising alone rarely result in sustained application of clinical practice guidelines [22]. We collaborated with NICHQ and other HRSA-WISCH grantees to systematically implement improvement science to ensure that the changes that we observed were indeed improvements and would be sustained [23] by first changing the system of care in the ED by introducing a standard order set [24,25]. We put a system into place to track use of the order set and to work with providers almost immediately if deviations were observed, to understand and overcome any barriers to the order set implementation. Systems in the ED and in the sickle cell center were aligned with the hospital’s QI initiatives [23].
Another strategy that we used to insure that the changes we observed would be sustained was to create a multidisciplinary team to build knowledge, skills, and new practices, including learning from other WISCH grantees and the NICHQ coordinating center [23]. We modified and adapted the intervention to our specific context [25]; although the outline of the order set was influenced by our WISCH colleagues, the final order set was structured to be consistent with other protocols within our institution. Finally, we included consumer input in the design of the project from the outset.
A previous study of a multi-institutional QI initiative aimed at improving acute SCD pain management for adult patients in the ED was unable to demonstrate an improvement in time to administration of initial analgesic [26]. Our study with pediatric patients was able to demonstrate a clinically meaningful decrease in the time to administration of first parenteral analgesic. The factors that account for the discrepant findings between these studies are likely multifactorial. Age (ie, pediatric vs. adult patients) may have played a role given that IV access may become increasingly difficult as patients with SCD age [26]. Education for providers should include the importance of alternative methods of administration of opioids, including subcutaneous and intranasal routes, to avoid delays when IV access is difficult. It is possible that negative provider attitudes converge with the documented increase in patient visits during the young adult years [27]. This may set up a challenging feedback loop wherein these vulnerable young adults are faced with greater stigma and consequently receive lower quality care, even when there is an attempt to carry out a standardized protocol.
We did not find that the QI intervention resulted in decreased admissions to the inpatient unit, with 68% of visits resulting in admission. In a recent pediatric SCD study, hospital admissions for pain control accounted for 78% of all admissions and 70% of readmissions within 30 days [28]. The investigators found that use of a SCD analgesic protocol including patient-controlled analgesia (PCA) improved quality of care as well as hospital readmission rates within 30 days (from 28% to 11%). Our ED QI protocol focused on only the first 90 minutes of the visit for pain. Our team has discussed the potential for starting the PCA in the ED and we should build on our success to focus on specific care that patients receive beyond their initial presentation. Further, we introduced pain action planning into outpatient care and need to continue to improve positive patient self-management strategies to ensure more seamless transition of pain management between home, ED, and inpatient settings.
Several valuable lessons were learned over the course of the ED QI initiative. Previous researchers [28] have emphasized the importance of coupling provider education with standardized order sets in efforts to improve the care of patients with SCD. Although we did not offer monthly formal education to our providers, the immediate follow-up when there were protocol deviations most likely served as teaching moments. These teaching moments also surfaced when some ED and hematology providers expressed concerns about the risk for oversedation with the rapid reassessment of pain and re-dosing of pain medications. Although rare, some parents also expressed that their child was being treated too vigorously with opioids. Our project highlighted the element of stigma that still accompanies the use of opioids for SCD pain management.
The project could not have been undertaken were it not for a small but determined multidisciplinary team of individuals who were personally invested in seeing the project come to fruition. The identification of physician and nurse champions who were enthusiastic about the project, invested in its conduct, and committed to its success was a cornerstone of the project’s success. These champions played an essential role in engaging staff interest in the project and oversaw the practicalities of implementing a new protocol in the ED. A spirit of collaboration, teamwork, and good communication between all involved parties was also critical. At the same time, we incorporated input from the treating ED and hematology clinicians using PDSA cycles as we were refining our protocol. We believe that our process enhanced buy-in from participating providers and clarified any issues that needed to be addressed in our setting, resulting in accelerated and sustained quality improvement.
Limitations
Although protocol-driven interventions are designed to provide a certain degree of uniformity of care, the protocol was not designed nor utilized in such a way that it superseded the best medical judgment of the treating clinicians. Deviations from the protocol were permissible when they were felt to be in the patient’s best interest. The study did not control for confounding variables such as disease severity, how long the patient had been in pain prior to coming to the ED, nor did we assess therapeutic interventions the patient had utilized at home prior to seeking out care in the ED. All of these factors could affect how well a patient might respond to treatment. We believe that sharing baseline data and monthly progress via run charts (graphs of data over time) with ED and sickle cell center staff and with consumer representatives enhanced the pace and focus of the project [23]. We had a dedicated person managing our data in real time through our HRSA funding, thus the project might not be generalizable to other institutions that do not have such staffing or access to the technology to allow project progress to be closely monitored by stakeholders.
Future Directions
With the goal of further reducing the time to administration of first analgesic dose in the ED setting, intranasal fentanyl will be utilized in our ED as the initial drug of choice for patients who do not object to or have a contraindication to its use. Collection of data from patients and family members is being undertaken to assess consumer satisfaction with the ED QI initiative. Recognizing that the ED management of acute pain addresses only one aspect of sickle cell pain, we are looking at ways to more comprehensively address pain. Individualized outpatient pain management plans are being created and patients and families are being encouraged and empowered to become active partners with their sickle cell providers in their own care. Although our initial efforts have focused on our pediatric patients, an additional aim of our project is to broaden the scope of our ED QI initiative to include community hospitals in the region that serve adult patients with SCD.
Conclusion
Implementation of a QI initiative in the ED has led to expeditious care for pediatric patients with SCD presenting with VOE. A multidisciplinary approach, ongoing staff education, and commitment to the initiative have been necessary to sustain the improvements. Our success can provide a template for other QI initiatives in the ED that translate to improved patient care for other diseases. A QI framework provided us with unique challenges but also invaluable lessons as we addressed our objective to improve outcomes for patients with SCD across the life course.
Acknowledgments: The authors wish to thank Theresa Freitas, RN, Lisa Hale, PNP, Carolyn Hoppe, MD, Ileana Mendez, RN, Helen Mitchell, Mary Rutherford, MD, Augusta Saulys, MD and the Children’s Hospital & Research Center Oakland Emergency Medicine Department and Sickle Cell Center for their support.
Corresponding author: Marsha Treadwell, PhD, Children’s Hospital & Research Center Oakland, 747 52nd St, Oakland, CA 94609, [email protected].
Funding/support: This research was conducted as part of the National Initiative for Children’s Healthcare Quality (NICHQ) Working to Improve Sickle Cell Healthcare (WISCH) project. Further support came from a grant from the Health Resources and Services Administration Sickle Cell Disease Treatment Demonstration Project Grant No. U1EMC16492 and from NIH CTSA grant UL1 RR024131. The views expressed in this publication do not necessarily reflect the views of WISCH, NICHQ, or HRSA.
From the Children’s Hospital & Research Center Oakland, Oakland, CA.
Abstract
- Objective: To determine whether a quality improvement (QI) initiative would result in more timely assessment and treatment of acute sickle cell–related pain for pediatric patients with sickle cell disease (SCD) treated in the emergency department (ED).
- Methods: We created and implemented a protocol for SCD pain management in the ED with the goals of improving (1) mean time from triage to first analgesic dose; (2) percentage of patients that received their first analgesic dose within 30 minutes of triage, and (3) percentage of patients who had pain assessment performed within 30 minutes of triage and who were re-assessed within 30 minutes after the first analgesic dose.
- Results: Significant improvements were achieved between baseline (55 patient visits) and post order set implementation (165 visits) in time from triage to administration of first analgesic (decreased from 89.9 ± 50.5 to 35.2 ± 22.8 minutes, P < 0.001); percentage of patient visits receiving pain medications within 30 minutes of triage (from 7% to 53%, P < 0.001); percentage of patient visits assessed within 30 minutes of triage (from 64% to 99.4%, P < 0.001); and percentage of patient visits re-assessed within 30 minutes of initial analgesic (from 54% to 86%, P < 0.001).
- Conclusions: Implementation of a QI initiative in the ED led to expeditious care for pediatric patients with SCD presenting with pain. A QI framework provided us with unique challenges but also invaluable lessons as we address our objective of decreasing the quality gap in SCD medical care.
Pain is the leading cause of emergency department (ED) visits for patients with sickle cell disease (SCD) [1]. In the United States, 78% of the nearly 200,000 annual ED visits for SCD are for a complaint of pain [1]. Guidelines for the management of sickle cell vaso-occlusive pain episodes (VOE) suggest prompt initiation of parenteral opioids, use of effective opioid doses, and repeat opioid doses at frequent intervals [2–4]. Adherence to guidelines is poor. Both pediatric and adult patients with SCD experience delays in the initiation of analgesics and are routinely undertreated with respect to opioid dosing [5–8]. Even after controlling for race, the delays in time to analgesic administration experienced by patients with SCD exceed the delays encountered by patients who present to the ED with other types of pain [5,9]. These disparities warrant efforts designed to improve the delivery of quality care to patients with SCD.
Barriers to rapid and appropriate care of VOE in the ED are multifactorial and include systems-based limitations, such as acuity of the ED census, staffing limitations (eg, nurse-to-patient ratios), and facility limitations (eg, room availability) [6]. Provider-based limitations may include lack of awareness of available guidelines [10]. Biases and misunderstandings amongst providers about sickle cell pain and adequate medication dosing may also play a role [11–13]. These provider biases often lead to undertreatment of the pain, which in turn can lead to pseudoaddiction (drug-seeking behavior due to inadequate treatment) and a cycle of increased ED and inpatient utilization [14,15].
Patient-specific barriers to effective ED management of pain are equally complex. Previous negative experiences in the ED can lead patients and families to delay seeking care or avoid the ED altogether despite severe VOE pain [16]. Patients report frustration with the lack of consideration that they receive for their reports of pain, perceived insensitivity of hospital staff, inadequate analgesic administration, staff preoccupation with concerns of drug addiction, and an overall lack of respect and trust [17–19]. Patients also perceive a lack of knowledge of SCD and its treatments on the part of ED staff [7]. Other barriers to effective management are technical in nature, such as difficulty in establishing timely intravenous (IV) access.
Gaps and variations in quality of care contribute to poor outcomes for patients with SCD [20,21]. To help address these inequities, the Working to Improve Sickle Cell Healthcare (WISCH) project began in 2010 to improve care and outcomes for patients with SCD. WISCH is a collaborative quality improvement (QI) project funded by the Health Resources and Services Administration (HRSA) that has the goal to use improvement science to improve outcomes for patients with SCD across the life course (Ed note: see Editorial by Oyeku et al in this issue). As one of the HRSA-WISCH grantee networks, we undertook a QI project designed to decrease the quality gap in SCD medical care by creating and implementing a protocol for ED pain management for pediatric patients. Goals of the project were to improve the timely and appropriate assessment and treatment of acute VOE in the ED.
Methods
Setting
This ED QI initiative was implemented at Children’s Hospital & Research Center Oakland, an urban free-standing pediatric hospital that serves a demographically diverse population. The hospital ED sees over 45,000 visits per year, with 250 visits per year for VOE. Residents in pediatrics, family medicine, and emergency medicine staff the ED. All attending physicians are subspecialists in pediatric emergency medicine. Study procedures were approved by the hospital’s institutional review board.
Intervention
A multidisciplinary team consisting of ED staff and sickle cell center staff drafted a nursing-driven protocol for the assessment and management of acute pain associated with VOE, incorporating elements from a protocol in use by another WISCH collaborative member. The protocol called for the immediate triage and assessment of all patients with SCD who presented with moderate to severe pain suggestive of VOE. Moderate to severe pain was defined as a pain score of ≥ 5 on a numeric scale of 0 to 10, where 0 = no pain and 10 = the worst pain imaginable. Exclusion criteria included a chief complaint of pain not considered secondary to VOE (eg, trauma, fracture). Patients were also excluded if they had been transferred from another facility. The protocol called for IV pain medication to be administered within 10 minutes of the patient being roomed, with re-evaluation at 20-minute intervals and re-dosing of pain medication based on the patient’s subsequent pain rating.
Measures
We selected performance measures from the bank developed by the WISCH team to track improvement and evaluate progress. These performance measures included (1) mean time from triage to first analgesic dose, (2) percentage of patients that received their first dose of analgesic within 30 minutes of triage, (3) percentage of patients who had a pain assessment performed within 30 minutes of triage, and (4) percentage of patients re-assessed within 30 minutes after the first dose of analgesic had been administered. Our aims were to have 80% of patients assessed and given pain medications within 30 minutes of triage, and to have 80% of patients re-assessed within 30 minutes after having received their first dose of an analgesic, within 12 months of implementing our intervention.
Data Collection and Analysis
The WISCH project coordinator reviewed records of visits to the ED for a baseline period of 6 months and post-order set implementaton. Demographic data (age, gender), clinical data (hemoglobin type), pain scores, utilization data (number of ED visits during the study period), and data pertaining to the metrics chosen from the WISCH measurement bank were extracted from each eligible patient’s ED chart after the visit was completed. If patients were admitted, their length of hospitalization was extracted from their inpatient medical record.
All biostatistical analyses were conducted using Stata 9.2 (StataCorp, College Station, TX). Descriptive statistics computed at 2 time-points (pre and post order set implementation) were utilized to examine means, standard deviations and percentages. The 2 time-points were initially compared at the visit level of measurement, using Student’s t tests corrected for unequal variances where necessary for continuous variables and chi-square analyses for categorical variables, to evaluate if there was an improvement in timely triage, assessment, and treatment of acute VOE pain for all ED visits pre and post order set implementation. To account for trends and possible correlations across the months post order set implementation, we ran a mixed linear model with repeated measures over time to compare visits during all months post order set implementation with the baseline months, for metric 1, time from triage to first pain medication. If significant differences were found, we used Dunnett’s method of multiple comparisons to determine which months differed from baseline. For metrics 2 through 4, we ran linear models with a binary outcome, a logit link function and using general estimating equations to determine trends and to account for correlations over time.
Secondary analyses were conducted to evaluate whether mean pain scores were significantly different over the course of the ED visit for the 78 unique patients seen post order set implementation. A multivariable mixed linear model, for the outcome of the third pain score, was used to assess the associations with prior scores and to control for potential covariates (age, gender, number of ED visits, hemoglobin type) that were determined in advance. A statistical significance level of 0.05 was used for all tests.
Results
Baseline data were collected from December 2011 to May 2012. The protocol was implemented in July 2012 and was utilized during 165 ED visits (91% of eligible visits) through April 2013. There were no statistically significant differences in demographic or clinical characteristics between the 55 patients whose charts were reviewed prior to implementing the order set and the 78 unique patients treated thereafter. Pre order set implementation, the mean age was 14.6 ± 6.4 years; 60% were female and the primary diagnosis was HgbSS disease (61.8% of diagnoses). Post order set implementation, the mean age was 16.0 ± 8.0 years; 51.3% were female and the primary diagnosis was HgbSS disease (61.5% of diagnoses). The mean number of visits was 1.5 visits per patient with a range of 1–8 visits, both pre and post order set implementation. Thirty-one patients had ED visits at both time periods.
It can be seen in Figure 2 that staff performance on 3 of the 4 metrics (with the exception of initial analgesic within 30 minutes of triage) began to improve prior to implementing the order set. The mean length of ED stays decreased by 30 minutes, from a mean of 5.2 hours down to 4.7 hours (P < 0.05, Table). There was no significant change in the percentage of patients admitted to the inpatient unit.
We performed secondary analyses to determine if performance on our first metric, mean time from triage to first analgesic dose, was associated with any improvement on the third pain assessment for the patients enrolled post order set implementation. Looking at the first ED visit during the study period for the 78 unique patients, we found significant decreases in mean pain scores from the first to the second, from the second to the third, and from the first to the third assessment (P < 0.01). The mean pain scores were 8.3 ± 1.8, 5.9 ± 2.8, and 5.1 ± 3.0 on initial, second and third assessments, respectively. A multivariable model controlling for gender, hemoglobin type, number of ED visits and time to first pain medication showed that only the score at the second pain assessment (β = 0.88 ± 0.08, P < 0.001) was a significant predictor of the score at the third pain assessment.
Discussion
We demonstrated that a QI initiative to improve acute pain management resulted in more timely assessment and treatment of pain in pediatric patients with SCD. Significant improvements from baseline were achieved and sustained over a 10-month period in all 4 targeted metrics. We consistently exceeded our goal of having 80% of patients assessed within 30 minutes of triage, and our mean time to first pain medication (35.2 ± 22.8 minutes) came close to our goal of 30 minutes from triage. While we also achieved our goal to have 80% of patients re-assessed within 30 minutes after having received their first dose of an analgesic, we fell short in the percent who received their initial pain medication within 30 minutes of triage (52.7% versus goal of 80%). Although the length of stay in the ED decreased, no change was observed in the percentage of patients who required admission to the inpatient unit. A secondary analysis showed that mean pain scores significantly decreased over the course of the ED visit, from severe to moderate intensity.
The improvements that we observed began prior to implementation of the order set. We recognize that simply raising awareness and educating staff about the importance of timely and appropriate assessment and treatment of acute sickle cell related pain in the ED might be a potential confounder of our results. However, changes were sustained for 10 months post order set implementation and beyond, with no evidence that the performance on the target metrics is drifting back to baseline levels. Education and awareness-raising alone rarely result in sustained application of clinical practice guidelines [22]. We collaborated with NICHQ and other HRSA-WISCH grantees to systematically implement improvement science to ensure that the changes that we observed were indeed improvements and would be sustained [23] by first changing the system of care in the ED by introducing a standard order set [24,25]. We put a system into place to track use of the order set and to work with providers almost immediately if deviations were observed, to understand and overcome any barriers to the order set implementation. Systems in the ED and in the sickle cell center were aligned with the hospital’s QI initiatives [23].
Another strategy that we used to insure that the changes we observed would be sustained was to create a multidisciplinary team to build knowledge, skills, and new practices, including learning from other WISCH grantees and the NICHQ coordinating center [23]. We modified and adapted the intervention to our specific context [25]; although the outline of the order set was influenced by our WISCH colleagues, the final order set was structured to be consistent with other protocols within our institution. Finally, we included consumer input in the design of the project from the outset.
A previous study of a multi-institutional QI initiative aimed at improving acute SCD pain management for adult patients in the ED was unable to demonstrate an improvement in time to administration of initial analgesic [26]. Our study with pediatric patients was able to demonstrate a clinically meaningful decrease in the time to administration of first parenteral analgesic. The factors that account for the discrepant findings between these studies are likely multifactorial. Age (ie, pediatric vs. adult patients) may have played a role given that IV access may become increasingly difficult as patients with SCD age [26]. Education for providers should include the importance of alternative methods of administration of opioids, including subcutaneous and intranasal routes, to avoid delays when IV access is difficult. It is possible that negative provider attitudes converge with the documented increase in patient visits during the young adult years [27]. This may set up a challenging feedback loop wherein these vulnerable young adults are faced with greater stigma and consequently receive lower quality care, even when there is an attempt to carry out a standardized protocol.
We did not find that the QI intervention resulted in decreased admissions to the inpatient unit, with 68% of visits resulting in admission. In a recent pediatric SCD study, hospital admissions for pain control accounted for 78% of all admissions and 70% of readmissions within 30 days [28]. The investigators found that use of a SCD analgesic protocol including patient-controlled analgesia (PCA) improved quality of care as well as hospital readmission rates within 30 days (from 28% to 11%). Our ED QI protocol focused on only the first 90 minutes of the visit for pain. Our team has discussed the potential for starting the PCA in the ED and we should build on our success to focus on specific care that patients receive beyond their initial presentation. Further, we introduced pain action planning into outpatient care and need to continue to improve positive patient self-management strategies to ensure more seamless transition of pain management between home, ED, and inpatient settings.
Several valuable lessons were learned over the course of the ED QI initiative. Previous researchers [28] have emphasized the importance of coupling provider education with standardized order sets in efforts to improve the care of patients with SCD. Although we did not offer monthly formal education to our providers, the immediate follow-up when there were protocol deviations most likely served as teaching moments. These teaching moments also surfaced when some ED and hematology providers expressed concerns about the risk for oversedation with the rapid reassessment of pain and re-dosing of pain medications. Although rare, some parents also expressed that their child was being treated too vigorously with opioids. Our project highlighted the element of stigma that still accompanies the use of opioids for SCD pain management.
The project could not have been undertaken were it not for a small but determined multidisciplinary team of individuals who were personally invested in seeing the project come to fruition. The identification of physician and nurse champions who were enthusiastic about the project, invested in its conduct, and committed to its success was a cornerstone of the project’s success. These champions played an essential role in engaging staff interest in the project and oversaw the practicalities of implementing a new protocol in the ED. A spirit of collaboration, teamwork, and good communication between all involved parties was also critical. At the same time, we incorporated input from the treating ED and hematology clinicians using PDSA cycles as we were refining our protocol. We believe that our process enhanced buy-in from participating providers and clarified any issues that needed to be addressed in our setting, resulting in accelerated and sustained quality improvement.
Limitations
Although protocol-driven interventions are designed to provide a certain degree of uniformity of care, the protocol was not designed nor utilized in such a way that it superseded the best medical judgment of the treating clinicians. Deviations from the protocol were permissible when they were felt to be in the patient’s best interest. The study did not control for confounding variables such as disease severity, how long the patient had been in pain prior to coming to the ED, nor did we assess therapeutic interventions the patient had utilized at home prior to seeking out care in the ED. All of these factors could affect how well a patient might respond to treatment. We believe that sharing baseline data and monthly progress via run charts (graphs of data over time) with ED and sickle cell center staff and with consumer representatives enhanced the pace and focus of the project [23]. We had a dedicated person managing our data in real time through our HRSA funding, thus the project might not be generalizable to other institutions that do not have such staffing or access to the technology to allow project progress to be closely monitored by stakeholders.
Future Directions
With the goal of further reducing the time to administration of first analgesic dose in the ED setting, intranasal fentanyl will be utilized in our ED as the initial drug of choice for patients who do not object to or have a contraindication to its use. Collection of data from patients and family members is being undertaken to assess consumer satisfaction with the ED QI initiative. Recognizing that the ED management of acute pain addresses only one aspect of sickle cell pain, we are looking at ways to more comprehensively address pain. Individualized outpatient pain management plans are being created and patients and families are being encouraged and empowered to become active partners with their sickle cell providers in their own care. Although our initial efforts have focused on our pediatric patients, an additional aim of our project is to broaden the scope of our ED QI initiative to include community hospitals in the region that serve adult patients with SCD.
Conclusion
Implementation of a QI initiative in the ED has led to expeditious care for pediatric patients with SCD presenting with VOE. A multidisciplinary approach, ongoing staff education, and commitment to the initiative have been necessary to sustain the improvements. Our success can provide a template for other QI initiatives in the ED that translate to improved patient care for other diseases. A QI framework provided us with unique challenges but also invaluable lessons as we addressed our objective to improve outcomes for patients with SCD across the life course.
Acknowledgments: The authors wish to thank Theresa Freitas, RN, Lisa Hale, PNP, Carolyn Hoppe, MD, Ileana Mendez, RN, Helen Mitchell, Mary Rutherford, MD, Augusta Saulys, MD and the Children’s Hospital & Research Center Oakland Emergency Medicine Department and Sickle Cell Center for their support.
Corresponding author: Marsha Treadwell, PhD, Children’s Hospital & Research Center Oakland, 747 52nd St, Oakland, CA 94609, [email protected].
Funding/support: This research was conducted as part of the National Initiative for Children’s Healthcare Quality (NICHQ) Working to Improve Sickle Cell Healthcare (WISCH) project. Further support came from a grant from the Health Resources and Services Administration Sickle Cell Disease Treatment Demonstration Project Grant No. U1EMC16492 and from NIH CTSA grant UL1 RR024131. The views expressed in this publication do not necessarily reflect the views of WISCH, NICHQ, or HRSA.
1. Yusuf HR, Atrash HK, Grosse SD, et al. Emergency department visits made by patients with sickle cell disease: a descriptive study, 1999-2007. Am J Preventive Med 2010;38 (4 Suppl):S536–41.
2. Benjamin L, Dampier C, Jacox A, et al. Guideline for the management of acute and chronic pain in sickle cell disease. American Pain Society; 1999.
3. Rees DC, Olujohungbe AD, Parker NE, et al. Guidelines for the management of the acute painful crisis in sickle cell disease. Br J Haematology 2003;120:744–52.
4. Solomon LR. Pain management in adults with sickle cell disease in a medical center emergency department. J Nat Med Assoc 2010;102:1025–32.
5. Lazio MP, Costello HH, Courtney DM, et al. A comparison of analgesic management for emergency department patients with sickle cell disease and renal colic. Clin J Pain 2010;26:199–205.
6. Shenoi R, Ma L, Syblik D, Yusuf S. Emergency department crowding and analgesic delay in pediatric sickle cell pain crises. Ped Emerg Care 2011;27:911–7.
7. Tanabe P, Artz N, Mark Courtney D, et al. Adult emergency department patients with sickle cell pain crisis: a learning collaborative model to improve analgesic management. Acad Emerg Med 2010;17:399–407.
8. Zempsky WT. Evaluation and treatment of sickle cell pain in the emergency department: paths to a better future. Clin Ped Emerg Med 2010;11:265–73.
9. Haywood C Jr, Tanabe P, Naik R, et al. The impact of race and disease on sickle cell patient wait times in the emergency department. Am J Emerg Med 2013;31:651–6.
10. Solomon LR. Treatment and prevention of pain due to vaso-occlusive crises in adults with sickle cell disease: an educational void. Blood 2008;111:997–1003.
11. Ballas SK. New era dawns on sickle cell pain. Blood 2010;116:311–2.
12. Haywood C Jr, Lanzkron S, Ratanawongsa N, et al. The association of provider communication with trust among adults with sickle cell disease. J Gen Intern Med 2010;25:543–8.
13. Zempsky WT. Treatment of sickle cell pain: fostering trust and justice. JAMA 2009;302:2479–80.
14. Elander J, Lusher J, Bevan D, Telfer P. Pain management and symptoms of substance dependence among patients with sickle cell disease. Soc Sci Med 2003;57:1683–96.
15. Elander J, Lusher J, Bevan D, et al. Understanding the causes of problematic pain management in sickle cell disease: evidence that pseudoaddiction plays a more important role than genuine analgesic dependence. J Pain Sympt Manag 2004;27:156–69.
16. Smith WR, Penberthy LT, Bovbjerg VE, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med 2008;148:94–101.
17. Harris A, Parker N, Baker C. Adults with sickle cell. Psychol Health Med 1998;3:171–9.
18. Jenerette CM, Brewer C. Health-related stigma in young adults with sickle cell disease. J Nat Med Assoc 2010;102:1050–5.
19. Maxwell K, Streetly A, Bevan D. Experiences of hospital care and treatment seeking for pain from sickle cell disease: qualitative study. BMJ 1999;318:1585–90.
20. Oyeku SO, Wang CJ, Scoville R, et al. Hemoglobinopathy Learning Collaborative: using quality improvement (QI) to achieve equity in health care quality, coordination, and outcomes for sickle cell disease. J Health Care Poor Underserved 2012;23(3 Suppl):34–48.
21. Wang CJ, Kavanagh PL, Little AA, et al. Quality-of-care indicators for children with sickle cell disease. Pediatrics 2011;128:484–93.
22. Mansouri M, Lockyer J. A meta-analysis of continuing medical education effectiveness. J Contin Ed Health Prof 2007;27:6–15.
23. The breakthrough series: IHI’s collaborative model for achieving breakthrough improvement. Boston: Institute for Healthcare Improvement; 2003.
24. Berwick DM. Improvement, trust, and the healthcare workforce. Qual Safety Health Care 2003;12:448–52.
25. Hovlid E, Bukve O, Haug K, et al. Sustainability of healthcare improvement: what can we learn from learning theory? BMC Health Serv Res 2012;12:235.
26. Tanabe P, Hafner JW, Martinovich Z, Artz N. Adult emergency department patients with sickle cell pain crisis: results from a quality improvement learning collaborative model to improve analgesic management. Acad Emerg Med 2012;19:430–8.
27. Brousseau DC, Owens PL, Mosso AL, et al. Acute care utilization and rehospitalizations for sickle cell disease. JAMA 2010;303:1288–94.
28. Frei-Jones MJ, Field JJ, DeBaun MR. Multi-modal intervention and prospective implementation of standardized sickle cell pain admission orders reduces 30-day readmission rate. Pediatr Blood Cancer 2009;53:401–5.
1. Yusuf HR, Atrash HK, Grosse SD, et al. Emergency department visits made by patients with sickle cell disease: a descriptive study, 1999-2007. Am J Preventive Med 2010;38 (4 Suppl):S536–41.
2. Benjamin L, Dampier C, Jacox A, et al. Guideline for the management of acute and chronic pain in sickle cell disease. American Pain Society; 1999.
3. Rees DC, Olujohungbe AD, Parker NE, et al. Guidelines for the management of the acute painful crisis in sickle cell disease. Br J Haematology 2003;120:744–52.
4. Solomon LR. Pain management in adults with sickle cell disease in a medical center emergency department. J Nat Med Assoc 2010;102:1025–32.
5. Lazio MP, Costello HH, Courtney DM, et al. A comparison of analgesic management for emergency department patients with sickle cell disease and renal colic. Clin J Pain 2010;26:199–205.
6. Shenoi R, Ma L, Syblik D, Yusuf S. Emergency department crowding and analgesic delay in pediatric sickle cell pain crises. Ped Emerg Care 2011;27:911–7.
7. Tanabe P, Artz N, Mark Courtney D, et al. Adult emergency department patients with sickle cell pain crisis: a learning collaborative model to improve analgesic management. Acad Emerg Med 2010;17:399–407.
8. Zempsky WT. Evaluation and treatment of sickle cell pain in the emergency department: paths to a better future. Clin Ped Emerg Med 2010;11:265–73.
9. Haywood C Jr, Tanabe P, Naik R, et al. The impact of race and disease on sickle cell patient wait times in the emergency department. Am J Emerg Med 2013;31:651–6.
10. Solomon LR. Treatment and prevention of pain due to vaso-occlusive crises in adults with sickle cell disease: an educational void. Blood 2008;111:997–1003.
11. Ballas SK. New era dawns on sickle cell pain. Blood 2010;116:311–2.
12. Haywood C Jr, Lanzkron S, Ratanawongsa N, et al. The association of provider communication with trust among adults with sickle cell disease. J Gen Intern Med 2010;25:543–8.
13. Zempsky WT. Treatment of sickle cell pain: fostering trust and justice. JAMA 2009;302:2479–80.
14. Elander J, Lusher J, Bevan D, Telfer P. Pain management and symptoms of substance dependence among patients with sickle cell disease. Soc Sci Med 2003;57:1683–96.
15. Elander J, Lusher J, Bevan D, et al. Understanding the causes of problematic pain management in sickle cell disease: evidence that pseudoaddiction plays a more important role than genuine analgesic dependence. J Pain Sympt Manag 2004;27:156–69.
16. Smith WR, Penberthy LT, Bovbjerg VE, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med 2008;148:94–101.
17. Harris A, Parker N, Baker C. Adults with sickle cell. Psychol Health Med 1998;3:171–9.
18. Jenerette CM, Brewer C. Health-related stigma in young adults with sickle cell disease. J Nat Med Assoc 2010;102:1050–5.
19. Maxwell K, Streetly A, Bevan D. Experiences of hospital care and treatment seeking for pain from sickle cell disease: qualitative study. BMJ 1999;318:1585–90.
20. Oyeku SO, Wang CJ, Scoville R, et al. Hemoglobinopathy Learning Collaborative: using quality improvement (QI) to achieve equity in health care quality, coordination, and outcomes for sickle cell disease. J Health Care Poor Underserved 2012;23(3 Suppl):34–48.
21. Wang CJ, Kavanagh PL, Little AA, et al. Quality-of-care indicators for children with sickle cell disease. Pediatrics 2011;128:484–93.
22. Mansouri M, Lockyer J. A meta-analysis of continuing medical education effectiveness. J Contin Ed Health Prof 2007;27:6–15.
23. The breakthrough series: IHI’s collaborative model for achieving breakthrough improvement. Boston: Institute for Healthcare Improvement; 2003.
24. Berwick DM. Improvement, trust, and the healthcare workforce. Qual Safety Health Care 2003;12:448–52.
25. Hovlid E, Bukve O, Haug K, et al. Sustainability of healthcare improvement: what can we learn from learning theory? BMC Health Serv Res 2012;12:235.
26. Tanabe P, Hafner JW, Martinovich Z, Artz N. Adult emergency department patients with sickle cell pain crisis: results from a quality improvement learning collaborative model to improve analgesic management. Acad Emerg Med 2012;19:430–8.
27. Brousseau DC, Owens PL, Mosso AL, et al. Acute care utilization and rehospitalizations for sickle cell disease. JAMA 2010;303:1288–94.
28. Frei-Jones MJ, Field JJ, DeBaun MR. Multi-modal intervention and prospective implementation of standardized sickle cell pain admission orders reduces 30-day readmission rate. Pediatr Blood Cancer 2009;53:401–5.