User login
Drug appears safe, effective for ocular GVHD

Photo courtesy of
Massachusetts Eye and Ear
A topical immunosuppressant may be a feasible treatment option for ocular graft-versus-host-disease (GVHD), according to a phase 1/2 study.
The immunosuppressant, tacrolimus, proved equally as effective as the steroid methylprednisolone.
And although tacrolimus was significantly more likely to produce a burning sensation, the drug did not significantly increase intraocular pressure (IOP) the way methylprednisolone did.
These results were published in Ophthalmology. The study was supported by the National Eye Institute, National Institutes of Health, and Research to Prevent Blindness.
“We found tacrolimus to be very effective—just as good as the steroid, in the reduction of ocular symptoms of GVHD,” said study author Reza Dana, MD, of Massachusetts Eye and Ear Infirmary in Boston.
“We saw this improvement without any of the negative effects, such as a rise in pressure in the eye, as we saw with the steroid.”
The trial included 40 patients with ocular GVHD. They had received a hematopoietic stem cell transplant to treat leukemias, lymphomas, multiple myeloma, myelodysplastic syndromes, and thrombocytopenia.
The patients were randomized to receive topical tacrolimus 0.05% (n=24) or topical methylprednisolone 0.5% (n=16). Their median ages were 54±12 and 58±11, respectively. And the mean baseline ocular GVHD duration was 27±34 months and 28±33 months, respectively.
The patients received treatment for 10 weeks. Three patients were lost to follow-up in the tacrolimus group, 3 withdrew from the study because of a burning sensation when applying tacrolimus, and 1 patient from the methylprednisolone group was withdrawn due to a geographic corneal epithelial defect.
Safety/tolerability
There were no major adverse events in either treatment group, and there was no significant difference in the composite tolerability scores between the groups (P=0.06).
Tolerability scores were calculated based on patients’ reports of burning sensation, discharge, redness, itchiness, and foreign body sensation (scale, 0-4 for each variable).
The burning sensation score was higher in the tacrolimus group than the methylprednisolone group. At week 5, the scores were 3.6 and 1.6, respectively (P<0.001). At week 10, they were 3.5 and 2.2, respectively (P=0.002).
There was no significant change from baseline in the mean IOP in the tacrolimus group at week 5 (P=0.50) or week 10 (P=0.20). But there was a significant change in the methylprednisolone group at both time points (P=0.04 for both).
Still, this did not amount to a significant difference between the treatment groups. In the tacrolimus group, the mean IOP was 15.6 mmHg at baseline, 16 mmHg at week 5, and 16.5 at week 10. In the methylprednisolone group the mean IOPs were 15.3 mmHg, 17.5 mmHg, and 17 mmHg, respectively.
Efficacy
The main efficacy endpoints were corneal fluorescein staining (CFS), tear film break-up time (TBUT), Schirmer test results, and expression of the ocular surface inflammatory markers human leukocyte antigen-DR (HLA-DR) and intercellular adhesion molecule-1 (ICAM-1).
Topical tacrolimus was more effective than methylprednisolone in reducing the CFS score from baseline to week 10. CFS scores were reduced by 55% and 23%, respectively (P=0.01).
There was a significant increase in TBUT from baseline to week 10 in the tacrolimus group (0.7-2.6 seconds, P=0.003) but not in the methylprednisolone group (0.6-1.0 seconds, P=0.42). However, there was no significant difference in TBUT changes between the treatment groups (P=0.06).
Schirmer test scores did not change significantly in either treatment group.
ICAM-1 expression decreased significantly from baseline in both the tacrolimus group (39% reduction, P=0.003) and the methylprednisolone group (40% reduction, P=0.008).
HLA-DR expression decreased significantly in the tacrolimus group (46% reduction, P=0.03) but not the methylprednisolone group (24% reduction, P=0.09).
“The problem with steroid treatment for ocular GVHD is that it can cause the pressure in the eye to rise, and it can also cause cataracts,” Dr Dana noted.
“The results of this trial give us reassurance that [tacrolimus] is another effective treatment for GVHD, without the negative side effects of steroids. This is a game-changer in terms of managing their care.” ![]()
Photo courtesy of
Massachusetts Eye and Ear
A topical immunosuppressant may be a feasible treatment option for ocular graft-versus-host-disease (GVHD), according to a phase 1/2 study.
The immunosuppressant, tacrolimus, proved equally as effective as the steroid methylprednisolone.
And although tacrolimus was significantly more likely to produce a burning sensation, the drug did not significantly increase intraocular pressure (IOP) the way methylprednisolone did.
These results were published in Ophthalmology. The study was supported by the National Eye Institute, National Institutes of Health, and Research to Prevent Blindness.
“We found tacrolimus to be very effective—just as good as the steroid, in the reduction of ocular symptoms of GVHD,” said study author Reza Dana, MD, of Massachusetts Eye and Ear Infirmary in Boston.
“We saw this improvement without any of the negative effects, such as a rise in pressure in the eye, as we saw with the steroid.”
The trial included 40 patients with ocular GVHD. They had received a hematopoietic stem cell transplant to treat leukemias, lymphomas, multiple myeloma, myelodysplastic syndromes, and thrombocytopenia.
The patients were randomized to receive topical tacrolimus 0.05% (n=24) or topical methylprednisolone 0.5% (n=16). Their median ages were 54±12 and 58±11, respectively. And the mean baseline ocular GVHD duration was 27±34 months and 28±33 months, respectively.
The patients received treatment for 10 weeks. Three patients were lost to follow-up in the tacrolimus group, 3 withdrew from the study because of a burning sensation when applying tacrolimus, and 1 patient from the methylprednisolone group was withdrawn due to a geographic corneal epithelial defect.
Safety/tolerability
There were no major adverse events in either treatment group, and there was no significant difference in the composite tolerability scores between the groups (P=0.06).
Tolerability scores were calculated based on patients’ reports of burning sensation, discharge, redness, itchiness, and foreign body sensation (scale, 0-4 for each variable).
The burning sensation score was higher in the tacrolimus group than the methylprednisolone group. At week 5, the scores were 3.6 and 1.6, respectively (P<0.001). At week 10, they were 3.5 and 2.2, respectively (P=0.002).
There was no significant change from baseline in the mean IOP in the tacrolimus group at week 5 (P=0.50) or week 10 (P=0.20). But there was a significant change in the methylprednisolone group at both time points (P=0.04 for both).
Still, this did not amount to a significant difference between the treatment groups. In the tacrolimus group, the mean IOP was 15.6 mmHg at baseline, 16 mmHg at week 5, and 16.5 at week 10. In the methylprednisolone group the mean IOPs were 15.3 mmHg, 17.5 mmHg, and 17 mmHg, respectively.
Efficacy
The main efficacy endpoints were corneal fluorescein staining (CFS), tear film break-up time (TBUT), Schirmer test results, and expression of the ocular surface inflammatory markers human leukocyte antigen-DR (HLA-DR) and intercellular adhesion molecule-1 (ICAM-1).
Topical tacrolimus was more effective than methylprednisolone in reducing the CFS score from baseline to week 10. CFS scores were reduced by 55% and 23%, respectively (P=0.01).
There was a significant increase in TBUT from baseline to week 10 in the tacrolimus group (0.7-2.6 seconds, P=0.003) but not in the methylprednisolone group (0.6-1.0 seconds, P=0.42). However, there was no significant difference in TBUT changes between the treatment groups (P=0.06).
Schirmer test scores did not change significantly in either treatment group.
ICAM-1 expression decreased significantly from baseline in both the tacrolimus group (39% reduction, P=0.003) and the methylprednisolone group (40% reduction, P=0.008).
HLA-DR expression decreased significantly in the tacrolimus group (46% reduction, P=0.03) but not the methylprednisolone group (24% reduction, P=0.09).
“The problem with steroid treatment for ocular GVHD is that it can cause the pressure in the eye to rise, and it can also cause cataracts,” Dr Dana noted.
“The results of this trial give us reassurance that [tacrolimus] is another effective treatment for GVHD, without the negative side effects of steroids. This is a game-changer in terms of managing their care.” ![]()
Photo courtesy of
Massachusetts Eye and Ear
A topical immunosuppressant may be a feasible treatment option for ocular graft-versus-host-disease (GVHD), according to a phase 1/2 study.
The immunosuppressant, tacrolimus, proved equally as effective as the steroid methylprednisolone.
And although tacrolimus was significantly more likely to produce a burning sensation, the drug did not significantly increase intraocular pressure (IOP) the way methylprednisolone did.
These results were published in Ophthalmology. The study was supported by the National Eye Institute, National Institutes of Health, and Research to Prevent Blindness.
“We found tacrolimus to be very effective—just as good as the steroid, in the reduction of ocular symptoms of GVHD,” said study author Reza Dana, MD, of Massachusetts Eye and Ear Infirmary in Boston.
“We saw this improvement without any of the negative effects, such as a rise in pressure in the eye, as we saw with the steroid.”
The trial included 40 patients with ocular GVHD. They had received a hematopoietic stem cell transplant to treat leukemias, lymphomas, multiple myeloma, myelodysplastic syndromes, and thrombocytopenia.
The patients were randomized to receive topical tacrolimus 0.05% (n=24) or topical methylprednisolone 0.5% (n=16). Their median ages were 54±12 and 58±11, respectively. And the mean baseline ocular GVHD duration was 27±34 months and 28±33 months, respectively.
The patients received treatment for 10 weeks. Three patients were lost to follow-up in the tacrolimus group, 3 withdrew from the study because of a burning sensation when applying tacrolimus, and 1 patient from the methylprednisolone group was withdrawn due to a geographic corneal epithelial defect.
Safety/tolerability
There were no major adverse events in either treatment group, and there was no significant difference in the composite tolerability scores between the groups (P=0.06).
Tolerability scores were calculated based on patients’ reports of burning sensation, discharge, redness, itchiness, and foreign body sensation (scale, 0-4 for each variable).
The burning sensation score was higher in the tacrolimus group than the methylprednisolone group. At week 5, the scores were 3.6 and 1.6, respectively (P<0.001). At week 10, they were 3.5 and 2.2, respectively (P=0.002).
There was no significant change from baseline in the mean IOP in the tacrolimus group at week 5 (P=0.50) or week 10 (P=0.20). But there was a significant change in the methylprednisolone group at both time points (P=0.04 for both).
Still, this did not amount to a significant difference between the treatment groups. In the tacrolimus group, the mean IOP was 15.6 mmHg at baseline, 16 mmHg at week 5, and 16.5 at week 10. In the methylprednisolone group the mean IOPs were 15.3 mmHg, 17.5 mmHg, and 17 mmHg, respectively.
Efficacy
The main efficacy endpoints were corneal fluorescein staining (CFS), tear film break-up time (TBUT), Schirmer test results, and expression of the ocular surface inflammatory markers human leukocyte antigen-DR (HLA-DR) and intercellular adhesion molecule-1 (ICAM-1).
Topical tacrolimus was more effective than methylprednisolone in reducing the CFS score from baseline to week 10. CFS scores were reduced by 55% and 23%, respectively (P=0.01).
There was a significant increase in TBUT from baseline to week 10 in the tacrolimus group (0.7-2.6 seconds, P=0.003) but not in the methylprednisolone group (0.6-1.0 seconds, P=0.42). However, there was no significant difference in TBUT changes between the treatment groups (P=0.06).
Schirmer test scores did not change significantly in either treatment group.
ICAM-1 expression decreased significantly from baseline in both the tacrolimus group (39% reduction, P=0.003) and the methylprednisolone group (40% reduction, P=0.008).
HLA-DR expression decreased significantly in the tacrolimus group (46% reduction, P=0.03) but not the methylprednisolone group (24% reduction, P=0.09).
“The problem with steroid treatment for ocular GVHD is that it can cause the pressure in the eye to rise, and it can also cause cataracts,” Dr Dana noted.
“The results of this trial give us reassurance that [tacrolimus] is another effective treatment for GVHD, without the negative side effects of steroids. This is a game-changer in terms of managing their care.” ![]()
Alternative splicing regulates activity of MALT1

Photo courtesy of
Helmholtz Zentrum München
Researchers say they have gained new insights into the workings of MALT1, a protein that controls the activation of lymphocytes.
The team found that, through alternative splicing, two variants of MALT1 may arise, and one has a stronger effect than the other.
The researchers say understanding this process is important because previous research has suggested MALT1 may be a therapeutic
target for lymphomas and other diseases.
Isabel Meininger, a doctoral student at Helmholtz Zentrum München in Neuherberg, Germany, and her colleagues described their MALT1-related findings in Nature Communications.
“To our surprise, we showed that MALT1 is regulated by post-transcriptional splicing,” Meininger said. “Depending on which MALT1 variant is expressed, the immune system is activated more or less.”
The researchers explained that most pre-messenger RNAs in mammals are prone to alternative splicing, which results in the generation of multiple transcripts and proteins with diverse functions.
In the case of MALT1, the variants MALT1A and MALT1B differ only through the presence of exon 7, a short sequence that encodes 11 amino acids.
If exon 7 is missing, as in the case of MALT1B, the protein’s ability to stimulate T cells is impaired. So MALT1A has a stronger effect on T cells than MALT1B.
The researchers also found that a molecule called hnRNP U (heterogeneous nuclear ribonucleoprotein U) regulates which of the two variants is preferably expressed.
If hnRNP U is present in small amounts, higher levels of MALT1A are expressed, resulting in stronger activation of T cells. With higher levels of hnRNP U, higher levels of MALT1B are expressed, and the response of the T cells is weaker.
“Our findings contribute to a better understanding of the function of MALT1 and enable us to better assess the potential impact of a pharmacological effect on this promising drug candidate,” said study author Daniel Krappmann, PhD, of Helmholtz Zentrum München.
In a previous study, Dr Krappmann and his team identified small molecules that can inhibit MALT1 to treat diffuse large B-cell lymphoma.
In future studies, the researchers want to confirm, in a preclinical model, the effects of MALT1 splicing on the immune system and disease development. ![]()
Photo courtesy of
Helmholtz Zentrum München
Researchers say they have gained new insights into the workings of MALT1, a protein that controls the activation of lymphocytes.
The team found that, through alternative splicing, two variants of MALT1 may arise, and one has a stronger effect than the other.
The researchers say understanding this process is important because previous research has suggested MALT1 may be a therapeutic
target for lymphomas and other diseases.
Isabel Meininger, a doctoral student at Helmholtz Zentrum München in Neuherberg, Germany, and her colleagues described their MALT1-related findings in Nature Communications.
“To our surprise, we showed that MALT1 is regulated by post-transcriptional splicing,” Meininger said. “Depending on which MALT1 variant is expressed, the immune system is activated more or less.”
The researchers explained that most pre-messenger RNAs in mammals are prone to alternative splicing, which results in the generation of multiple transcripts and proteins with diverse functions.
In the case of MALT1, the variants MALT1A and MALT1B differ only through the presence of exon 7, a short sequence that encodes 11 amino acids.
If exon 7 is missing, as in the case of MALT1B, the protein’s ability to stimulate T cells is impaired. So MALT1A has a stronger effect on T cells than MALT1B.
The researchers also found that a molecule called hnRNP U (heterogeneous nuclear ribonucleoprotein U) regulates which of the two variants is preferably expressed.
If hnRNP U is present in small amounts, higher levels of MALT1A are expressed, resulting in stronger activation of T cells. With higher levels of hnRNP U, higher levels of MALT1B are expressed, and the response of the T cells is weaker.
“Our findings contribute to a better understanding of the function of MALT1 and enable us to better assess the potential impact of a pharmacological effect on this promising drug candidate,” said study author Daniel Krappmann, PhD, of Helmholtz Zentrum München.
In a previous study, Dr Krappmann and his team identified small molecules that can inhibit MALT1 to treat diffuse large B-cell lymphoma.
In future studies, the researchers want to confirm, in a preclinical model, the effects of MALT1 splicing on the immune system and disease development. ![]()
Photo courtesy of
Helmholtz Zentrum München
Researchers say they have gained new insights into the workings of MALT1, a protein that controls the activation of lymphocytes.
The team found that, through alternative splicing, two variants of MALT1 may arise, and one has a stronger effect than the other.
The researchers say understanding this process is important because previous research has suggested MALT1 may be a therapeutic
target for lymphomas and other diseases.
Isabel Meininger, a doctoral student at Helmholtz Zentrum München in Neuherberg, Germany, and her colleagues described their MALT1-related findings in Nature Communications.
“To our surprise, we showed that MALT1 is regulated by post-transcriptional splicing,” Meininger said. “Depending on which MALT1 variant is expressed, the immune system is activated more or less.”
The researchers explained that most pre-messenger RNAs in mammals are prone to alternative splicing, which results in the generation of multiple transcripts and proteins with diverse functions.
In the case of MALT1, the variants MALT1A and MALT1B differ only through the presence of exon 7, a short sequence that encodes 11 amino acids.
If exon 7 is missing, as in the case of MALT1B, the protein’s ability to stimulate T cells is impaired. So MALT1A has a stronger effect on T cells than MALT1B.
The researchers also found that a molecule called hnRNP U (heterogeneous nuclear ribonucleoprotein U) regulates which of the two variants is preferably expressed.
If hnRNP U is present in small amounts, higher levels of MALT1A are expressed, resulting in stronger activation of T cells. With higher levels of hnRNP U, higher levels of MALT1B are expressed, and the response of the T cells is weaker.
“Our findings contribute to a better understanding of the function of MALT1 and enable us to better assess the potential impact of a pharmacological effect on this promising drug candidate,” said study author Daniel Krappmann, PhD, of Helmholtz Zentrum München.
In a previous study, Dr Krappmann and his team identified small molecules that can inhibit MALT1 to treat diffuse large B-cell lymphoma.
In future studies, the researchers want to confirm, in a preclinical model, the effects of MALT1 splicing on the immune system and disease development. ![]()
TCD screening underused in sickle cell patients

with sickle cell anemia
Photo courtesy of St. Jude
Results of a large, retrospective study suggest the use of transcranial Doppler (TCD) screening is on the rise in US children and adolescents with sickle cell anemia.
However, the rate of TCD screening in these patients falls well below national recommendations.
In addition, TCD screening rates vary greatly by state, and the use of screening tends to decrease as patients grow older.
Sarah L. Reeves, PhD, of University of Michigan, Ann Arbor, and her colleagues reported these findings in JAMA Pediatrics.
The researchers noted that guidelines from the National Heart, Lung, and Blood Institute recommend that patients with sickle cell anemia receive annual TCD screenings from age 2 to 16 to identify those patients at the highest risk of stroke.
Dr Reeves and her colleagues wanted to determine if this recommendation is being followed. So they analyzed Medicaid claims data from 2005 through 2010 for Florida, Illinois, Louisiana, Michigan, South Carolina, and Texas.
The data included 4775 patients, ages 2 to 16, with sickle cell anemia. For these patients, TCD screening rates increased from 22% in 2005 to 44% in 2010 (P<0.001).
The researchers found that TCD screening rates varied significantly by state (P=0.004), and Texas had the lowest screening rate at any time point (7% in 2005).
The team also analyzed a subset of 2388 patients who were enrolled for 2 or more consecutive years to examine potential predictors of TCD screening.
This analysis revealed that, with each year of increasing age, a patient’s odds of receiving TCD screening decreased (odds ratio=0.97, P=0.002).
On the other hand, an increasing number of well-child visits was associated with higher odds of receiving TCD screening (odds ratio=1.10, P=0.007).
And the odds of receiving TCD screening were higher for patients who previously underwent TCD screening (odds ratio=2.44, P<0.001).
The researchers said these results suggest that, despite national recommendations, TCD screening rates remain low in young patients with sickle cell anemia in the US. ![]()
with sickle cell anemia
Photo courtesy of St. Jude
Results of a large, retrospective study suggest the use of transcranial Doppler (TCD) screening is on the rise in US children and adolescents with sickle cell anemia.
However, the rate of TCD screening in these patients falls well below national recommendations.
In addition, TCD screening rates vary greatly by state, and the use of screening tends to decrease as patients grow older.
Sarah L. Reeves, PhD, of University of Michigan, Ann Arbor, and her colleagues reported these findings in JAMA Pediatrics.
The researchers noted that guidelines from the National Heart, Lung, and Blood Institute recommend that patients with sickle cell anemia receive annual TCD screenings from age 2 to 16 to identify those patients at the highest risk of stroke.
Dr Reeves and her colleagues wanted to determine if this recommendation is being followed. So they analyzed Medicaid claims data from 2005 through 2010 for Florida, Illinois, Louisiana, Michigan, South Carolina, and Texas.
The data included 4775 patients, ages 2 to 16, with sickle cell anemia. For these patients, TCD screening rates increased from 22% in 2005 to 44% in 2010 (P<0.001).
The researchers found that TCD screening rates varied significantly by state (P=0.004), and Texas had the lowest screening rate at any time point (7% in 2005).
The team also analyzed a subset of 2388 patients who were enrolled for 2 or more consecutive years to examine potential predictors of TCD screening.
This analysis revealed that, with each year of increasing age, a patient’s odds of receiving TCD screening decreased (odds ratio=0.97, P=0.002).
On the other hand, an increasing number of well-child visits was associated with higher odds of receiving TCD screening (odds ratio=1.10, P=0.007).
And the odds of receiving TCD screening were higher for patients who previously underwent TCD screening (odds ratio=2.44, P<0.001).
The researchers said these results suggest that, despite national recommendations, TCD screening rates remain low in young patients with sickle cell anemia in the US. ![]()
with sickle cell anemia
Photo courtesy of St. Jude
Results of a large, retrospective study suggest the use of transcranial Doppler (TCD) screening is on the rise in US children and adolescents with sickle cell anemia.
However, the rate of TCD screening in these patients falls well below national recommendations.
In addition, TCD screening rates vary greatly by state, and the use of screening tends to decrease as patients grow older.
Sarah L. Reeves, PhD, of University of Michigan, Ann Arbor, and her colleagues reported these findings in JAMA Pediatrics.
The researchers noted that guidelines from the National Heart, Lung, and Blood Institute recommend that patients with sickle cell anemia receive annual TCD screenings from age 2 to 16 to identify those patients at the highest risk of stroke.
Dr Reeves and her colleagues wanted to determine if this recommendation is being followed. So they analyzed Medicaid claims data from 2005 through 2010 for Florida, Illinois, Louisiana, Michigan, South Carolina, and Texas.
The data included 4775 patients, ages 2 to 16, with sickle cell anemia. For these patients, TCD screening rates increased from 22% in 2005 to 44% in 2010 (P<0.001).
The researchers found that TCD screening rates varied significantly by state (P=0.004), and Texas had the lowest screening rate at any time point (7% in 2005).
The team also analyzed a subset of 2388 patients who were enrolled for 2 or more consecutive years to examine potential predictors of TCD screening.
This analysis revealed that, with each year of increasing age, a patient’s odds of receiving TCD screening decreased (odds ratio=0.97, P=0.002).
On the other hand, an increasing number of well-child visits was associated with higher odds of receiving TCD screening (odds ratio=1.10, P=0.007).
And the odds of receiving TCD screening were higher for patients who previously underwent TCD screening (odds ratio=2.44, P<0.001).
The researchers said these results suggest that, despite national recommendations, TCD screening rates remain low in young patients with sickle cell anemia in the US. ![]()
Company recalls hemostasis valves

Photo courtesy of
Vascular Solutions, Inc.
Vascular Solutions, Inc. has issued a US-wide recall of Guardian II hemostasis valves used in catheterization procedures.
Specific lots of the products have been recalled because they pose an increased risk of air leakage that may lead to an air embolism, which could result in serious injury or death.
This recall only affects the Guardian II hemostasis valves and does not include the Guardian II NC hemostasis valves.
No injuries have been reported in association with this issue to date.
Healthcare facilities that have the affected Guardian II hemostasis valves should remove the products from their inventory and return them to Vascular Solutions.
The recalled products were manufactured from March 2015 to February 2016 and distributed from April 2015 to February 2016.
The recalled products are specific lots of Model Numbers 8210 and 8211. A listing of the recalled lots is available from Vascular Solutions and has been provided to each facility that purchased the affected products.
A total of 26,550 devices have been manufactured, with 5283 distributed in the US. The condition that led to the recall may affect approximately 2.4% of recalled devices.
Vascular Solutions voluntarily initiated the recall on March 3, 2016, through an Urgent Medical Device Recall notification distributed to purchasers of the affected products. The notification identified the specific lots subject to the recall and included instructions on how to return the affected products.
The US Food and Drug Administration (FDA) classified this as a Class I recall. The FDA defines Class I recalls as “a situation in which there is a reasonable probability that the use of, or exposure to, a violative product will cause serious adverse health consequences or death.”
Consumers with questions may contact the company by phone at 1-888-240-6001, Monday through Friday, between the hours of 8:00 am and 5:00 pm Central Time or by email at [email protected].
Adverse reactions or quality problems associated with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
Photo courtesy of
Vascular Solutions, Inc.
Vascular Solutions, Inc. has issued a US-wide recall of Guardian II hemostasis valves used in catheterization procedures.
Specific lots of the products have been recalled because they pose an increased risk of air leakage that may lead to an air embolism, which could result in serious injury or death.
This recall only affects the Guardian II hemostasis valves and does not include the Guardian II NC hemostasis valves.
No injuries have been reported in association with this issue to date.
Healthcare facilities that have the affected Guardian II hemostasis valves should remove the products from their inventory and return them to Vascular Solutions.
The recalled products were manufactured from March 2015 to February 2016 and distributed from April 2015 to February 2016.
The recalled products are specific lots of Model Numbers 8210 and 8211. A listing of the recalled lots is available from Vascular Solutions and has been provided to each facility that purchased the affected products.
A total of 26,550 devices have been manufactured, with 5283 distributed in the US. The condition that led to the recall may affect approximately 2.4% of recalled devices.
Vascular Solutions voluntarily initiated the recall on March 3, 2016, through an Urgent Medical Device Recall notification distributed to purchasers of the affected products. The notification identified the specific lots subject to the recall and included instructions on how to return the affected products.
The US Food and Drug Administration (FDA) classified this as a Class I recall. The FDA defines Class I recalls as “a situation in which there is a reasonable probability that the use of, or exposure to, a violative product will cause serious adverse health consequences or death.”
Consumers with questions may contact the company by phone at 1-888-240-6001, Monday through Friday, between the hours of 8:00 am and 5:00 pm Central Time or by email at [email protected].
Adverse reactions or quality problems associated with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
Photo courtesy of
Vascular Solutions, Inc.
Vascular Solutions, Inc. has issued a US-wide recall of Guardian II hemostasis valves used in catheterization procedures.
Specific lots of the products have been recalled because they pose an increased risk of air leakage that may lead to an air embolism, which could result in serious injury or death.
This recall only affects the Guardian II hemostasis valves and does not include the Guardian II NC hemostasis valves.
No injuries have been reported in association with this issue to date.
Healthcare facilities that have the affected Guardian II hemostasis valves should remove the products from their inventory and return them to Vascular Solutions.
The recalled products were manufactured from March 2015 to February 2016 and distributed from April 2015 to February 2016.
The recalled products are specific lots of Model Numbers 8210 and 8211. A listing of the recalled lots is available from Vascular Solutions and has been provided to each facility that purchased the affected products.
A total of 26,550 devices have been manufactured, with 5283 distributed in the US. The condition that led to the recall may affect approximately 2.4% of recalled devices.
Vascular Solutions voluntarily initiated the recall on March 3, 2016, through an Urgent Medical Device Recall notification distributed to purchasers of the affected products. The notification identified the specific lots subject to the recall and included instructions on how to return the affected products.
The US Food and Drug Administration (FDA) classified this as a Class I recall. The FDA defines Class I recalls as “a situation in which there is a reasonable probability that the use of, or exposure to, a violative product will cause serious adverse health consequences or death.”
Consumers with questions may contact the company by phone at 1-888-240-6001, Monday through Friday, between the hours of 8:00 am and 5:00 pm Central Time or by email at [email protected].
Adverse reactions or quality problems associated with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
Survey says docs don’t know FDA requirements

A survey of nearly 700 US physicians revealed that many do not know the basic requirements for a drug to receive approval from the US Food and
Drug Administration (FDA).
The results also suggested that most physicians don’t understand the FDA’s “breakthrough therapy” designation.
Since 2012, the FDA has been able to designate a drug as a breakthrough therapy if preliminary clinical evidence suggests it provides an advantage
over existing options.
Aaron S. Kesselheim, MD, of Brigham and Women’s Hospital in Boston, Massachusetts, and his colleagues wanted to determine how much physicians understood about this designation and if they had a basic understanding of the FDA’s requirements for drug approval.
So the researchers surveyed internists and specialists from the American Board of Internal Medicine’s diplomate list and reported the results of this survey in JAMA.
Of the 1148 physicians contacted, 692 (60%) responded. Participants were asked 3 questions about FDA approval and 5 about breakthrough therapies.
FDA approval
Seventy-three percent of respondents incorrectly believed that, for a drug to gain FDA approval, it must be as effective as drugs that are already approved.
However, 85% of respondents answered correctly that FDA-approved drugs typically have a favorable benefit/harm ratio.
Seventy percent of respondents incorrectly believed that FDA approval requires a drug to have both a statistically significant and a clinically important effect.
Only 6% of respondents knew the correct answer—that neither is required.
Breakthrough designation
Twenty percent of respondents said they were “familiar” (17%) or “very familiar” (3%) with breakthrough therapy designation, while 37% said they were “a little familiar,” and 42% said they were “not familiar at all.”
Fifty-eight percent of respondents said they were “fairly certain” that an FDA-approved breakthrough drug represents a major advance over currently approved treatments for its indication. Thirty-one percent said they were “fairly uncertain,” 5% said they were “very uncertain,” and 6% said they were “very certain.”
Fifty-two percent of respondents incorrectly believed that strong evidence (ie, randomized trials) is needed to earn the breakthrough designation.
However, 45% correctly answered that only preliminary evidence (eg, uncontrolled studies or studies testing surrogate outcomes) is needed. Four percent said very preliminary evidence (eg, in vitro laboratory or animal studies) is needed.
Seventy-seven percent of respondents incorrectly believed that, when the FDA grants breakthrough designation, there is high-quality evidence that the drug is more effective than currently approved treatments.
But 74% of respondents answered correctly that breakthrough designation does not mean there is high-quality evidence that the drug is safer than currently approved treatments.
Dr Kesselheim and his colleagues said the misconceptions identified in this survey may lead physicians to overprescribe newly approved drugs—particularly breakthrough therapies—and inadequately communicate how well these drugs work to patients. ![]()
A survey of nearly 700 US physicians revealed that many do not know the basic requirements for a drug to receive approval from the US Food and
Drug Administration (FDA).
The results also suggested that most physicians don’t understand the FDA’s “breakthrough therapy” designation.
Since 2012, the FDA has been able to designate a drug as a breakthrough therapy if preliminary clinical evidence suggests it provides an advantage
over existing options.
Aaron S. Kesselheim, MD, of Brigham and Women’s Hospital in Boston, Massachusetts, and his colleagues wanted to determine how much physicians understood about this designation and if they had a basic understanding of the FDA’s requirements for drug approval.
So the researchers surveyed internists and specialists from the American Board of Internal Medicine’s diplomate list and reported the results of this survey in JAMA.
Of the 1148 physicians contacted, 692 (60%) responded. Participants were asked 3 questions about FDA approval and 5 about breakthrough therapies.
FDA approval
Seventy-three percent of respondents incorrectly believed that, for a drug to gain FDA approval, it must be as effective as drugs that are already approved.
However, 85% of respondents answered correctly that FDA-approved drugs typically have a favorable benefit/harm ratio.
Seventy percent of respondents incorrectly believed that FDA approval requires a drug to have both a statistically significant and a clinically important effect.
Only 6% of respondents knew the correct answer—that neither is required.
Breakthrough designation
Twenty percent of respondents said they were “familiar” (17%) or “very familiar” (3%) with breakthrough therapy designation, while 37% said they were “a little familiar,” and 42% said they were “not familiar at all.”
Fifty-eight percent of respondents said they were “fairly certain” that an FDA-approved breakthrough drug represents a major advance over currently approved treatments for its indication. Thirty-one percent said they were “fairly uncertain,” 5% said they were “very uncertain,” and 6% said they were “very certain.”
Fifty-two percent of respondents incorrectly believed that strong evidence (ie, randomized trials) is needed to earn the breakthrough designation.
However, 45% correctly answered that only preliminary evidence (eg, uncontrolled studies or studies testing surrogate outcomes) is needed. Four percent said very preliminary evidence (eg, in vitro laboratory or animal studies) is needed.
Seventy-seven percent of respondents incorrectly believed that, when the FDA grants breakthrough designation, there is high-quality evidence that the drug is more effective than currently approved treatments.
But 74% of respondents answered correctly that breakthrough designation does not mean there is high-quality evidence that the drug is safer than currently approved treatments.
Dr Kesselheim and his colleagues said the misconceptions identified in this survey may lead physicians to overprescribe newly approved drugs—particularly breakthrough therapies—and inadequately communicate how well these drugs work to patients. ![]()
A survey of nearly 700 US physicians revealed that many do not know the basic requirements for a drug to receive approval from the US Food and
Drug Administration (FDA).
The results also suggested that most physicians don’t understand the FDA’s “breakthrough therapy” designation.
Since 2012, the FDA has been able to designate a drug as a breakthrough therapy if preliminary clinical evidence suggests it provides an advantage
over existing options.
Aaron S. Kesselheim, MD, of Brigham and Women’s Hospital in Boston, Massachusetts, and his colleagues wanted to determine how much physicians understood about this designation and if they had a basic understanding of the FDA’s requirements for drug approval.
So the researchers surveyed internists and specialists from the American Board of Internal Medicine’s diplomate list and reported the results of this survey in JAMA.
Of the 1148 physicians contacted, 692 (60%) responded. Participants were asked 3 questions about FDA approval and 5 about breakthrough therapies.
FDA approval
Seventy-three percent of respondents incorrectly believed that, for a drug to gain FDA approval, it must be as effective as drugs that are already approved.
However, 85% of respondents answered correctly that FDA-approved drugs typically have a favorable benefit/harm ratio.
Seventy percent of respondents incorrectly believed that FDA approval requires a drug to have both a statistically significant and a clinically important effect.
Only 6% of respondents knew the correct answer—that neither is required.
Breakthrough designation
Twenty percent of respondents said they were “familiar” (17%) or “very familiar” (3%) with breakthrough therapy designation, while 37% said they were “a little familiar,” and 42% said they were “not familiar at all.”
Fifty-eight percent of respondents said they were “fairly certain” that an FDA-approved breakthrough drug represents a major advance over currently approved treatments for its indication. Thirty-one percent said they were “fairly uncertain,” 5% said they were “very uncertain,” and 6% said they were “very certain.”
Fifty-two percent of respondents incorrectly believed that strong evidence (ie, randomized trials) is needed to earn the breakthrough designation.
However, 45% correctly answered that only preliminary evidence (eg, uncontrolled studies or studies testing surrogate outcomes) is needed. Four percent said very preliminary evidence (eg, in vitro laboratory or animal studies) is needed.
Seventy-seven percent of respondents incorrectly believed that, when the FDA grants breakthrough designation, there is high-quality evidence that the drug is more effective than currently approved treatments.
But 74% of respondents answered correctly that breakthrough designation does not mean there is high-quality evidence that the drug is safer than currently approved treatments.
Dr Kesselheim and his colleagues said the misconceptions identified in this survey may lead physicians to overprescribe newly approved drugs—particularly breakthrough therapies—and inadequately communicate how well these drugs work to patients. ![]()
Manufacturing methods can damage red cells

Photo by Elise Amendola
Certain methods of manufacturing red cell concentrates (RCCs) may be less damaging than others, according to research published in Vox Sanguinis.
The study showed that damage-associated molecular patterns (DAMPs) in stored blood differ according to the process and materials used to collect or prepare RCCs for transfusion.
Researchers believe this discovery could help reduce adverse reactions in transfusion recipients and potentially impact how blood is collected around the world.
To conduct this study, the researchers compared RCCs collected at blood donation centers in the US and Canada. The team examined the influence of manufacturing methods on the levels of mitochondrial (mt) DNA and extracellular vesicle (EV) DAMPs in RCCs.
“Working with the American team at Blood Systems Research Institute was key to this research because of the wide variations in blood manufacturing processes present in the US,” said Jason Acker, PhD, of Canadian Blood Services’ Centre for Innovation in Edmonton, Alberta.
“In countries like Canada, where there is a national blood service, manufacturing methods are largely standardized, so it is difficult to compare various methods. But blood collection in the US is characterized by dozens of independent blood centers that use a variety of available manufacturing processes. The Americans provided the variations we needed to measure red cell damage and to ascertain whether it can be attributed to different manufacturing methods.”
Manufacturing methods
The researchers evaluated 87 RCCs prepared using 9 different methods, outlined in the following table.
| Name | Processing method | Collection/
manufacturing method |
Anticoagulant/
additive solution (AS) |
Leuko-reduction (LR) method | LR temperature and timing |
| FenBC (n=6) | Semi-automated whole blood (WB) processing
following overnight (O/N) hold at 18–24 °C: Buffy coat (BC) method |
Collection set: Fenwal CGR6494B, Quad OptiPure RC 9SBT WB 500 ml Component processing: Compomat G4 | Citrate phosphate dextrose (CPD)/saline adenine glucose mannitol (SAGM) | Filtration of RCC | 20–24°C within 24 h of the stop bleed time |
| MacoBC
(n=6) |
Semi-automated WB processing
following O/N hold at 18–24°C: BC method |
Collection set: Macopharma LQT 7291
LX Leucoflex LCR-Diamond Quadruple Bottom and Top System, WB 500 ml Component processing: Compomat G4 |
CPD/SAGM | Filtration of RCC | 20–24°C within 24 h of the stop bleed time |
| FenWBF
(n=6) |
Semi-automated WB processing:
WB filtration method |
Collection set: Fenwal CGR8441B, Quad
PackPure WB 500 ml Component processing: Compomat G4 |
CPD/SAGM | Filtration of WB | 1–6°C within 72 h of the stop bleed time |
| MacoWBF
(n=6) |
Semi-automated WB processing:
WB filtration method |
Collection set: Macopharma Leucoflex MTL1 Quadruple Top and Top System, WB 500 ml
Component processing: Compomat G4 |
CPD/SAGM | Filtration of WB | 1–6°C within 72 h of the stop bleed time |
| FenMAN
(n=12) |
Manual WB processing | Fenwal 4R1582 Double Blood-Pack Unit
500 ml, with Flex-Excel Red Cell Filter |
CPD/AS-1 | Filtration of RCC | Room temperature (RT) within 8 h of stop bleed time |
| FenMAN-non-LR
(n=12) |
Manual WB processing | Fenwal 4R1587P Triple Blood-Pack Unit 500 ml | CPD/AS-1 | Not applicable | Not applicable |
| Alyx (n=15) | Apheresis | Fenwal Software: 3.0; 4R5720 Alyx 2RBC-LR Kit | Acid citrate dextrose A (ACD-A)/AS-1 | Filtration of RCC | RT post-
collection |
| MCS+ (n=12) | Apheresis | Haemonetics; Software Rev H or L; 0832F-00, 2RBC filtered | CP2D/AS-3 | Filtration of RCC | RT if <8 h/
cold if >8 h post- collection |
| Trima (n=12) | Apheresis | Terumo BCT; Software 6.0.6; Trima Accel 80500 kit | ACD-A/AS-3 | Filtration of RCC | RT if <8 h/ cold if >8 h post-
collection |
Results
For all RCCs, the researchers assessed the levels of mtDNA and the number and cell of origin of EVs on storage days 5 and 42.
They observed a 100-fold difference in mtDNA levels between the different methods.
The highest mtDNA levels were in the non-leukoreduced RCCs, followed by the MCS+ and Trima apheresis RCCs. The mean levels were 5.3 x 105 copies/µL, 1.3 x 105 copies/µL, and 1.2 x 105 copies/µL, respectively.
The lowest mtDNA levels were seen with the semi-automated methods. The mean levels ranged from 3.8 x 103 copies/µL to 5.9 x 103 copies/µL.
The researchers also saw a 10-fold difference in EV levels between the different methods.
The team detected red blood cell-derived CD235a+ EVs in fresh RCCs, which increased in most RCCs over the storage period (but not for FenBC and MCS+).
Platelet-derived CD41a+ EVs were highest in non-leukoreduced and Trima RCCs and did not change significantly during storage.
White blood cell-derived CD11b+ and CD66b+ EVs were low in most RCCs (though not in Trima and FenMAN-non-LR RCCs), and their levels did not significantly change during storage.
White blood cell-derived CD14+ EVs were negligible in fresh RCCs but increased in several RCCs during storage (FenMAN, Alyx, MCS+, and Trima).
Next steps
“There must be more testing of the apheresis collections equipment, blood bags, leukoreduction filters, and other variations in manufacturing methods to determine what single element or combination of elements in the various red blood cell manufacturing processes result in high levels of DAMPs and why,” said Michael Busch, MD, PhD, of Blood Systems Research Institute in San Francisco, California.
“We also need to understand how mitochondrial DAMPs are involved in adverse reactions to red blood cell transfusions,” added Sonia Bakkour, PhD, also from Blood Systems Research Institute.
“Some recently published studies on platelet components link high levels of mitochondrial DAMPs to adverse transfusion reactions. We need to see if DAMPs have similar adverse effects on recipients of red blood cell transfusions.”
“We think that our research could lead to finding the best way to manufacture red blood cells,” Dr Acker noted.
“It’s clear now that manufacturing methods matter. We . . . are keen to explore what’s in the blood bag or in the filters or in the tubing, for example, that can be minimized or eliminated, improving the outcome in patients who receive blood transfusions.” ![]()
Photo by Elise Amendola
Certain methods of manufacturing red cell concentrates (RCCs) may be less damaging than others, according to research published in Vox Sanguinis.
The study showed that damage-associated molecular patterns (DAMPs) in stored blood differ according to the process and materials used to collect or prepare RCCs for transfusion.
Researchers believe this discovery could help reduce adverse reactions in transfusion recipients and potentially impact how blood is collected around the world.
To conduct this study, the researchers compared RCCs collected at blood donation centers in the US and Canada. The team examined the influence of manufacturing methods on the levels of mitochondrial (mt) DNA and extracellular vesicle (EV) DAMPs in RCCs.
“Working with the American team at Blood Systems Research Institute was key to this research because of the wide variations in blood manufacturing processes present in the US,” said Jason Acker, PhD, of Canadian Blood Services’ Centre for Innovation in Edmonton, Alberta.
“In countries like Canada, where there is a national blood service, manufacturing methods are largely standardized, so it is difficult to compare various methods. But blood collection in the US is characterized by dozens of independent blood centers that use a variety of available manufacturing processes. The Americans provided the variations we needed to measure red cell damage and to ascertain whether it can be attributed to different manufacturing methods.”
Manufacturing methods
The researchers evaluated 87 RCCs prepared using 9 different methods, outlined in the following table.
| Name | Processing method | Collection/
manufacturing method |
Anticoagulant/
additive solution (AS) |
Leuko-reduction (LR) method | LR temperature and timing |
| FenBC (n=6) | Semi-automated whole blood (WB) processing
following overnight (O/N) hold at 18–24 °C: Buffy coat (BC) method |
Collection set: Fenwal CGR6494B, Quad OptiPure RC 9SBT WB 500 ml Component processing: Compomat G4 | Citrate phosphate dextrose (CPD)/saline adenine glucose mannitol (SAGM) | Filtration of RCC | 20–24°C within 24 h of the stop bleed time |
| MacoBC
(n=6) |
Semi-automated WB processing
following O/N hold at 18–24°C: BC method |
Collection set: Macopharma LQT 7291
LX Leucoflex LCR-Diamond Quadruple Bottom and Top System, WB 500 ml Component processing: Compomat G4 |
CPD/SAGM | Filtration of RCC | 20–24°C within 24 h of the stop bleed time |
| FenWBF
(n=6) |
Semi-automated WB processing:
WB filtration method |
Collection set: Fenwal CGR8441B, Quad
PackPure WB 500 ml Component processing: Compomat G4 |
CPD/SAGM | Filtration of WB | 1–6°C within 72 h of the stop bleed time |
| MacoWBF
(n=6) |
Semi-automated WB processing:
WB filtration method |
Collection set: Macopharma Leucoflex MTL1 Quadruple Top and Top System, WB 500 ml
Component processing: Compomat G4 |
CPD/SAGM | Filtration of WB | 1–6°C within 72 h of the stop bleed time |
| FenMAN
(n=12) |
Manual WB processing | Fenwal 4R1582 Double Blood-Pack Unit
500 ml, with Flex-Excel Red Cell Filter |
CPD/AS-1 | Filtration of RCC | Room temperature (RT) within 8 h of stop bleed time |
| FenMAN-non-LR
(n=12) |
Manual WB processing | Fenwal 4R1587P Triple Blood-Pack Unit 500 ml | CPD/AS-1 | Not applicable | Not applicable |
| Alyx (n=15) | Apheresis | Fenwal Software: 3.0; 4R5720 Alyx 2RBC-LR Kit | Acid citrate dextrose A (ACD-A)/AS-1 | Filtration of RCC | RT post-
collection |
| MCS+ (n=12) | Apheresis | Haemonetics; Software Rev H or L; 0832F-00, 2RBC filtered | CP2D/AS-3 | Filtration of RCC | RT if <8 h/
cold if >8 h post- collection |
| Trima (n=12) | Apheresis | Terumo BCT; Software 6.0.6; Trima Accel 80500 kit | ACD-A/AS-3 | Filtration of RCC | RT if <8 h/ cold if >8 h post-
collection |
Results
For all RCCs, the researchers assessed the levels of mtDNA and the number and cell of origin of EVs on storage days 5 and 42.
They observed a 100-fold difference in mtDNA levels between the different methods.
The highest mtDNA levels were in the non-leukoreduced RCCs, followed by the MCS+ and Trima apheresis RCCs. The mean levels were 5.3 x 105 copies/µL, 1.3 x 105 copies/µL, and 1.2 x 105 copies/µL, respectively.
The lowest mtDNA levels were seen with the semi-automated methods. The mean levels ranged from 3.8 x 103 copies/µL to 5.9 x 103 copies/µL.
The researchers also saw a 10-fold difference in EV levels between the different methods.
The team detected red blood cell-derived CD235a+ EVs in fresh RCCs, which increased in most RCCs over the storage period (but not for FenBC and MCS+).
Platelet-derived CD41a+ EVs were highest in non-leukoreduced and Trima RCCs and did not change significantly during storage.
White blood cell-derived CD11b+ and CD66b+ EVs were low in most RCCs (though not in Trima and FenMAN-non-LR RCCs), and their levels did not significantly change during storage.
White blood cell-derived CD14+ EVs were negligible in fresh RCCs but increased in several RCCs during storage (FenMAN, Alyx, MCS+, and Trima).
Next steps
“There must be more testing of the apheresis collections equipment, blood bags, leukoreduction filters, and other variations in manufacturing methods to determine what single element or combination of elements in the various red blood cell manufacturing processes result in high levels of DAMPs and why,” said Michael Busch, MD, PhD, of Blood Systems Research Institute in San Francisco, California.
“We also need to understand how mitochondrial DAMPs are involved in adverse reactions to red blood cell transfusions,” added Sonia Bakkour, PhD, also from Blood Systems Research Institute.
“Some recently published studies on platelet components link high levels of mitochondrial DAMPs to adverse transfusion reactions. We need to see if DAMPs have similar adverse effects on recipients of red blood cell transfusions.”
“We think that our research could lead to finding the best way to manufacture red blood cells,” Dr Acker noted.
“It’s clear now that manufacturing methods matter. We . . . are keen to explore what’s in the blood bag or in the filters or in the tubing, for example, that can be minimized or eliminated, improving the outcome in patients who receive blood transfusions.” ![]()
Photo by Elise Amendola
Certain methods of manufacturing red cell concentrates (RCCs) may be less damaging than others, according to research published in Vox Sanguinis.
The study showed that damage-associated molecular patterns (DAMPs) in stored blood differ according to the process and materials used to collect or prepare RCCs for transfusion.
Researchers believe this discovery could help reduce adverse reactions in transfusion recipients and potentially impact how blood is collected around the world.
To conduct this study, the researchers compared RCCs collected at blood donation centers in the US and Canada. The team examined the influence of manufacturing methods on the levels of mitochondrial (mt) DNA and extracellular vesicle (EV) DAMPs in RCCs.
“Working with the American team at Blood Systems Research Institute was key to this research because of the wide variations in blood manufacturing processes present in the US,” said Jason Acker, PhD, of Canadian Blood Services’ Centre for Innovation in Edmonton, Alberta.
“In countries like Canada, where there is a national blood service, manufacturing methods are largely standardized, so it is difficult to compare various methods. But blood collection in the US is characterized by dozens of independent blood centers that use a variety of available manufacturing processes. The Americans provided the variations we needed to measure red cell damage and to ascertain whether it can be attributed to different manufacturing methods.”
Manufacturing methods
The researchers evaluated 87 RCCs prepared using 9 different methods, outlined in the following table.
| Name | Processing method | Collection/
manufacturing method |
Anticoagulant/
additive solution (AS) |
Leuko-reduction (LR) method | LR temperature and timing |
| FenBC (n=6) | Semi-automated whole blood (WB) processing
following overnight (O/N) hold at 18–24 °C: Buffy coat (BC) method |
Collection set: Fenwal CGR6494B, Quad OptiPure RC 9SBT WB 500 ml Component processing: Compomat G4 | Citrate phosphate dextrose (CPD)/saline adenine glucose mannitol (SAGM) | Filtration of RCC | 20–24°C within 24 h of the stop bleed time |
| MacoBC
(n=6) |
Semi-automated WB processing
following O/N hold at 18–24°C: BC method |
Collection set: Macopharma LQT 7291
LX Leucoflex LCR-Diamond Quadruple Bottom and Top System, WB 500 ml Component processing: Compomat G4 |
CPD/SAGM | Filtration of RCC | 20–24°C within 24 h of the stop bleed time |
| FenWBF
(n=6) |
Semi-automated WB processing:
WB filtration method |
Collection set: Fenwal CGR8441B, Quad
PackPure WB 500 ml Component processing: Compomat G4 |
CPD/SAGM | Filtration of WB | 1–6°C within 72 h of the stop bleed time |
| MacoWBF
(n=6) |
Semi-automated WB processing:
WB filtration method |
Collection set: Macopharma Leucoflex MTL1 Quadruple Top and Top System, WB 500 ml
Component processing: Compomat G4 |
CPD/SAGM | Filtration of WB | 1–6°C within 72 h of the stop bleed time |
| FenMAN
(n=12) |
Manual WB processing | Fenwal 4R1582 Double Blood-Pack Unit
500 ml, with Flex-Excel Red Cell Filter |
CPD/AS-1 | Filtration of RCC | Room temperature (RT) within 8 h of stop bleed time |
| FenMAN-non-LR
(n=12) |
Manual WB processing | Fenwal 4R1587P Triple Blood-Pack Unit 500 ml | CPD/AS-1 | Not applicable | Not applicable |
| Alyx (n=15) | Apheresis | Fenwal Software: 3.0; 4R5720 Alyx 2RBC-LR Kit | Acid citrate dextrose A (ACD-A)/AS-1 | Filtration of RCC | RT post-
collection |
| MCS+ (n=12) | Apheresis | Haemonetics; Software Rev H or L; 0832F-00, 2RBC filtered | CP2D/AS-3 | Filtration of RCC | RT if <8 h/
cold if >8 h post- collection |
| Trima (n=12) | Apheresis | Terumo BCT; Software 6.0.6; Trima Accel 80500 kit | ACD-A/AS-3 | Filtration of RCC | RT if <8 h/ cold if >8 h post-
collection |
Results
For all RCCs, the researchers assessed the levels of mtDNA and the number and cell of origin of EVs on storage days 5 and 42.
They observed a 100-fold difference in mtDNA levels between the different methods.
The highest mtDNA levels were in the non-leukoreduced RCCs, followed by the MCS+ and Trima apheresis RCCs. The mean levels were 5.3 x 105 copies/µL, 1.3 x 105 copies/µL, and 1.2 x 105 copies/µL, respectively.
The lowest mtDNA levels were seen with the semi-automated methods. The mean levels ranged from 3.8 x 103 copies/µL to 5.9 x 103 copies/µL.
The researchers also saw a 10-fold difference in EV levels between the different methods.
The team detected red blood cell-derived CD235a+ EVs in fresh RCCs, which increased in most RCCs over the storage period (but not for FenBC and MCS+).
Platelet-derived CD41a+ EVs were highest in non-leukoreduced and Trima RCCs and did not change significantly during storage.
White blood cell-derived CD11b+ and CD66b+ EVs were low in most RCCs (though not in Trima and FenMAN-non-LR RCCs), and their levels did not significantly change during storage.
White blood cell-derived CD14+ EVs were negligible in fresh RCCs but increased in several RCCs during storage (FenMAN, Alyx, MCS+, and Trima).
Next steps
“There must be more testing of the apheresis collections equipment, blood bags, leukoreduction filters, and other variations in manufacturing methods to determine what single element or combination of elements in the various red blood cell manufacturing processes result in high levels of DAMPs and why,” said Michael Busch, MD, PhD, of Blood Systems Research Institute in San Francisco, California.
“We also need to understand how mitochondrial DAMPs are involved in adverse reactions to red blood cell transfusions,” added Sonia Bakkour, PhD, also from Blood Systems Research Institute.
“Some recently published studies on platelet components link high levels of mitochondrial DAMPs to adverse transfusion reactions. We need to see if DAMPs have similar adverse effects on recipients of red blood cell transfusions.”
“We think that our research could lead to finding the best way to manufacture red blood cells,” Dr Acker noted.
“It’s clear now that manufacturing methods matter. We . . . are keen to explore what’s in the blood bag or in the filters or in the tubing, for example, that can be minimized or eliminated, improving the outcome in patients who receive blood transfusions.” ![]()
FDA grants drug accelerated approval for CLL

Photo courtesy of the CDC
The US Food and Drug Administration (FDA) has granted accelerated approval for venetoclax (Venclexta) to treat patients with chronic lymphocytic leukemia (CLL) who have 17p deletion and have received at least one prior therapy.
Venetoclax will be available in the US within about a week, according to the companies developing the drug, AbbVie and Genentech (a member of the
Roche Group).
The companies said they plan to offer patient assistance programs for qualifying patients who wish to receive venetoclax.
Venetoclax (formerly ABT-199) is the first FDA-approved treatment that targets the BCL-2 protein, which is overexpressed in many patients with CLL.
The drug is indicated for daily use after 17p deletion is confirmed via the FDA-approved companion diagnostic Vysis CLL FISH probe kit, which is manufactured by Abbott Molecular.
The FDA granted venetoclax accelerated approval rather than traditional approval because the drug has not yet shown a clinical benefit. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of venetoclax for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted venetoclax breakthrough therapy designation, priority review status, and orphan drug designation.
Phase 2 trial
Results from the pivotal phase 2 trial of venetoclax (M13-982, NCT01889186) were presented at the 2015 ASH Annual Meeting. According to those data, the trial enrolled 107 patients with relapsed or refractory CLL and 17p deletion.
Patients received venetoclax at 400 mg once daily following a weekly ramp-up schedule for the first 5 weeks. The primary endpoint was overall response rate, as determined by an independent review committee.
Eighty-five patients responded to treatment, for an overall response rate of 79.4%. Eight patients (7.5%) achieved a complete response or complete response with incomplete count recovery, 3 (2.8%) had a near partial response, and 74 (69.2%) had a partial response. Twenty-two patients (20.6%) did not respond.
As of the ASH presentation, the median duration of response had not been reached. The same was true for progression-free survival and overall survival. The progression-free survival estimate for 12 months was 72.0%, and the overall survival estimate was 86.7%.
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%). (About 22% of patients had neutropenia at baseline.)
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%). Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher infections.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
Laboratory tumor lysis syndrome (TLS) occurred in 5 patients during the ramp-up period only. Two patients required a dose interruption of 1 day each. There were no clinical TLS events.
In the past, TLS has caused deaths in patients receiving venetoclax. In response, AbbVie stopped dose-escalation in patients receiving the drug and suspended enrollment in phase 1 trials.
However, researchers subsequently found that a modified dosing schedule, prophylaxis, and patient monitoring can reduce the risk of TLS.
Venetoclax is currently being evaluated in phase 3 trials for the treatment of relapsed, refractory, and previously untreated CLL. ![]()
Photo courtesy of the CDC
The US Food and Drug Administration (FDA) has granted accelerated approval for venetoclax (Venclexta) to treat patients with chronic lymphocytic leukemia (CLL) who have 17p deletion and have received at least one prior therapy.
Venetoclax will be available in the US within about a week, according to the companies developing the drug, AbbVie and Genentech (a member of the
Roche Group).
The companies said they plan to offer patient assistance programs for qualifying patients who wish to receive venetoclax.
Venetoclax (formerly ABT-199) is the first FDA-approved treatment that targets the BCL-2 protein, which is overexpressed in many patients with CLL.
The drug is indicated for daily use after 17p deletion is confirmed via the FDA-approved companion diagnostic Vysis CLL FISH probe kit, which is manufactured by Abbott Molecular.
The FDA granted venetoclax accelerated approval rather than traditional approval because the drug has not yet shown a clinical benefit. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of venetoclax for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted venetoclax breakthrough therapy designation, priority review status, and orphan drug designation.
Phase 2 trial
Results from the pivotal phase 2 trial of venetoclax (M13-982, NCT01889186) were presented at the 2015 ASH Annual Meeting. According to those data, the trial enrolled 107 patients with relapsed or refractory CLL and 17p deletion.
Patients received venetoclax at 400 mg once daily following a weekly ramp-up schedule for the first 5 weeks. The primary endpoint was overall response rate, as determined by an independent review committee.
Eighty-five patients responded to treatment, for an overall response rate of 79.4%. Eight patients (7.5%) achieved a complete response or complete response with incomplete count recovery, 3 (2.8%) had a near partial response, and 74 (69.2%) had a partial response. Twenty-two patients (20.6%) did not respond.
As of the ASH presentation, the median duration of response had not been reached. The same was true for progression-free survival and overall survival. The progression-free survival estimate for 12 months was 72.0%, and the overall survival estimate was 86.7%.
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%). (About 22% of patients had neutropenia at baseline.)
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%). Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher infections.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
Laboratory tumor lysis syndrome (TLS) occurred in 5 patients during the ramp-up period only. Two patients required a dose interruption of 1 day each. There were no clinical TLS events.
In the past, TLS has caused deaths in patients receiving venetoclax. In response, AbbVie stopped dose-escalation in patients receiving the drug and suspended enrollment in phase 1 trials.
However, researchers subsequently found that a modified dosing schedule, prophylaxis, and patient monitoring can reduce the risk of TLS.
Venetoclax is currently being evaluated in phase 3 trials for the treatment of relapsed, refractory, and previously untreated CLL. ![]()
Photo courtesy of the CDC
The US Food and Drug Administration (FDA) has granted accelerated approval for venetoclax (Venclexta) to treat patients with chronic lymphocytic leukemia (CLL) who have 17p deletion and have received at least one prior therapy.
Venetoclax will be available in the US within about a week, according to the companies developing the drug, AbbVie and Genentech (a member of the
Roche Group).
The companies said they plan to offer patient assistance programs for qualifying patients who wish to receive venetoclax.
Venetoclax (formerly ABT-199) is the first FDA-approved treatment that targets the BCL-2 protein, which is overexpressed in many patients with CLL.
The drug is indicated for daily use after 17p deletion is confirmed via the FDA-approved companion diagnostic Vysis CLL FISH probe kit, which is manufactured by Abbott Molecular.
The FDA granted venetoclax accelerated approval rather than traditional approval because the drug has not yet shown a clinical benefit. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of venetoclax for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted venetoclax breakthrough therapy designation, priority review status, and orphan drug designation.
Phase 2 trial
Results from the pivotal phase 2 trial of venetoclax (M13-982, NCT01889186) were presented at the 2015 ASH Annual Meeting. According to those data, the trial enrolled 107 patients with relapsed or refractory CLL and 17p deletion.
Patients received venetoclax at 400 mg once daily following a weekly ramp-up schedule for the first 5 weeks. The primary endpoint was overall response rate, as determined by an independent review committee.
Eighty-five patients responded to treatment, for an overall response rate of 79.4%. Eight patients (7.5%) achieved a complete response or complete response with incomplete count recovery, 3 (2.8%) had a near partial response, and 74 (69.2%) had a partial response. Twenty-two patients (20.6%) did not respond.
As of the ASH presentation, the median duration of response had not been reached. The same was true for progression-free survival and overall survival. The progression-free survival estimate for 12 months was 72.0%, and the overall survival estimate was 86.7%.
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%). (About 22% of patients had neutropenia at baseline.)
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%). Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher infections.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
Laboratory tumor lysis syndrome (TLS) occurred in 5 patients during the ramp-up period only. Two patients required a dose interruption of 1 day each. There were no clinical TLS events.
In the past, TLS has caused deaths in patients receiving venetoclax. In response, AbbVie stopped dose-escalation in patients receiving the drug and suspended enrollment in phase 1 trials.
However, researchers subsequently found that a modified dosing schedule, prophylaxis, and patient monitoring can reduce the risk of TLS.
Venetoclax is currently being evaluated in phase 3 trials for the treatment of relapsed, refractory, and previously untreated CLL.
Protein distribution impacts T cells’ fate
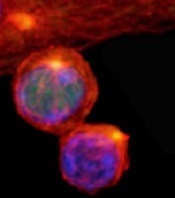
(with c-Myc in green)
Image courtesy of
Katherine Verbist and St. Jude
New research published in Nature helps explain how 2 types of cells arise from activated T cells.
Investigators found that distribution of the regulatory protein c-Myc during asymmetric cell division impacts an activated T cell’s fate, determining whether it will become an effector T cell or a memory T cell.
The team therefore believes that manipulating c-Myc levels could make vaccines more effective or advance immunotherapies for cancer treatment.
“Our work suggests that it may be possible to manipulate the immune response by nudging production of c-Myc in one direction or the other,” said study author Douglas Green, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Potentially, that could mean more effective vaccines or help to advance T-cell immune therapy for cancer treatment.”
Through a series of experiments, Dr Green and his colleagues found that, during asymmetric cell division of activated T cells, high levels of c-Myc accumulated in one daughter cell.
There, c-Myc launched and sustained the rapid proliferation of effector T cells, including those in mice infected with the influenza virus.
In contrast, daughter cells with low levels of c-Myc functioned like memory T cells, proliferating to mount an immune response a month later when mice were again exposed to the virus.
The investigators also identified the metabolic and signaling pathways that serve as a positive feedback loop to sustain the high levels of c-Myc that effector T cells require to maintain their identities and function.
The team showed that disrupting certain components of the system disturbed c-Myc production, which altered the fate of T cells and caused effector T cells to operate like memory T cells.
“While daughter cells of activated T cells seem to have very different fates, we showed their behavior could be altered by manipulating these metabolic and regulatory pathways to increase or decrease c-Myc levels,” Dr Green said.
(with c-Myc in green)
Image courtesy of
Katherine Verbist and St. Jude
New research published in Nature helps explain how 2 types of cells arise from activated T cells.
Investigators found that distribution of the regulatory protein c-Myc during asymmetric cell division impacts an activated T cell’s fate, determining whether it will become an effector T cell or a memory T cell.
The team therefore believes that manipulating c-Myc levels could make vaccines more effective or advance immunotherapies for cancer treatment.
“Our work suggests that it may be possible to manipulate the immune response by nudging production of c-Myc in one direction or the other,” said study author Douglas Green, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Potentially, that could mean more effective vaccines or help to advance T-cell immune therapy for cancer treatment.”
Through a series of experiments, Dr Green and his colleagues found that, during asymmetric cell division of activated T cells, high levels of c-Myc accumulated in one daughter cell.
There, c-Myc launched and sustained the rapid proliferation of effector T cells, including those in mice infected with the influenza virus.
In contrast, daughter cells with low levels of c-Myc functioned like memory T cells, proliferating to mount an immune response a month later when mice were again exposed to the virus.
The investigators also identified the metabolic and signaling pathways that serve as a positive feedback loop to sustain the high levels of c-Myc that effector T cells require to maintain their identities and function.
The team showed that disrupting certain components of the system disturbed c-Myc production, which altered the fate of T cells and caused effector T cells to operate like memory T cells.
“While daughter cells of activated T cells seem to have very different fates, we showed their behavior could be altered by manipulating these metabolic and regulatory pathways to increase or decrease c-Myc levels,” Dr Green said.
(with c-Myc in green)
Image courtesy of
Katherine Verbist and St. Jude
New research published in Nature helps explain how 2 types of cells arise from activated T cells.
Investigators found that distribution of the regulatory protein c-Myc during asymmetric cell division impacts an activated T cell’s fate, determining whether it will become an effector T cell or a memory T cell.
The team therefore believes that manipulating c-Myc levels could make vaccines more effective or advance immunotherapies for cancer treatment.
“Our work suggests that it may be possible to manipulate the immune response by nudging production of c-Myc in one direction or the other,” said study author Douglas Green, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Potentially, that could mean more effective vaccines or help to advance T-cell immune therapy for cancer treatment.”
Through a series of experiments, Dr Green and his colleagues found that, during asymmetric cell division of activated T cells, high levels of c-Myc accumulated in one daughter cell.
There, c-Myc launched and sustained the rapid proliferation of effector T cells, including those in mice infected with the influenza virus.
In contrast, daughter cells with low levels of c-Myc functioned like memory T cells, proliferating to mount an immune response a month later when mice were again exposed to the virus.
The investigators also identified the metabolic and signaling pathways that serve as a positive feedback loop to sustain the high levels of c-Myc that effector T cells require to maintain their identities and function.
The team showed that disrupting certain components of the system disturbed c-Myc production, which altered the fate of T cells and caused effector T cells to operate like memory T cells.
“While daughter cells of activated T cells seem to have very different fates, we showed their behavior could be altered by manipulating these metabolic and regulatory pathways to increase or decrease c-Myc levels,” Dr Green said.
Team creates public repository of xenografts

Photo by Rhoda Baer
Researchers have established a public repository of leukemia and lymphoma xenografts, according to a report in Cancer Cell.
The repository, known as the Public Repository of Xenografts (PRoXe), contains material from bone marrow, peripheral blood, and lymph nodes of mice.
It also contains information on the patients from whom the cancer tissues were derived and details on the characteristics of the tumors themselves.
PRoXe is based at Dana-Farber Cancer Institute in Boston, Massachusetts, but it has a web portal that can be accessed by researchers around the world.
Those who register with PRoxE can access the repository and search for cells from patients with specific hematologic malignancies.
Researchers can then have frozen cells shipped to them and transplant the cells into mice to create patient-derived xenograft (PDX) models for testing drugs.
“About 90% of compounds that show anticancer activity in preclinical tests don’t work when given to patients,” said David Weinstock, MD, of the Dana-Farber Cancer Institute.
“By trying drugs in PDX models, we can ‘mimic’ large and expensive human clinical trials and get answers about efficacy more quickly, less expensively, and without the need for patients to get investigational drugs that won’t work.”
To demonstrate how PRoXe can be used, Dr Weinstock and his colleagues tested the MDM2 inhibitor CGM097 against B-cell acute lymphoblastic leukemia (B-ALL) in 2 groups of mice. One group had a mutation in the TP53 tumor suppressor gene, and the other did not.
The researchers observed superior survival in the mice with wild-type TP53. This result corresponds with the results of previous research, which showed that inhibitors that disrupt the MDM2-p53 interaction can be effective in tumor models and patient tumors with wild-type TP53.
Dr Weinstock and his colleagues said their results provide strong preclinical evidence for testing CGM097 in patients who have been extensively treated for B-ALL and have wild-type TP53.
Dr Weinstock also noted that the leaders of ProXe are negotiating with a number of academic centers in an attempt to incorporate their PDX models into the repository.
The Cancer Cell manuscript had 95 authors from 14 different centers that contributed samples, models, and/or effort to the project.
Photo by Rhoda Baer
Researchers have established a public repository of leukemia and lymphoma xenografts, according to a report in Cancer Cell.
The repository, known as the Public Repository of Xenografts (PRoXe), contains material from bone marrow, peripheral blood, and lymph nodes of mice.
It also contains information on the patients from whom the cancer tissues were derived and details on the characteristics of the tumors themselves.
PRoXe is based at Dana-Farber Cancer Institute in Boston, Massachusetts, but it has a web portal that can be accessed by researchers around the world.
Those who register with PRoxE can access the repository and search for cells from patients with specific hematologic malignancies.
Researchers can then have frozen cells shipped to them and transplant the cells into mice to create patient-derived xenograft (PDX) models for testing drugs.
“About 90% of compounds that show anticancer activity in preclinical tests don’t work when given to patients,” said David Weinstock, MD, of the Dana-Farber Cancer Institute.
“By trying drugs in PDX models, we can ‘mimic’ large and expensive human clinical trials and get answers about efficacy more quickly, less expensively, and without the need for patients to get investigational drugs that won’t work.”
To demonstrate how PRoXe can be used, Dr Weinstock and his colleagues tested the MDM2 inhibitor CGM097 against B-cell acute lymphoblastic leukemia (B-ALL) in 2 groups of mice. One group had a mutation in the TP53 tumor suppressor gene, and the other did not.
The researchers observed superior survival in the mice with wild-type TP53. This result corresponds with the results of previous research, which showed that inhibitors that disrupt the MDM2-p53 interaction can be effective in tumor models and patient tumors with wild-type TP53.
Dr Weinstock and his colleagues said their results provide strong preclinical evidence for testing CGM097 in patients who have been extensively treated for B-ALL and have wild-type TP53.
Dr Weinstock also noted that the leaders of ProXe are negotiating with a number of academic centers in an attempt to incorporate their PDX models into the repository.
The Cancer Cell manuscript had 95 authors from 14 different centers that contributed samples, models, and/or effort to the project.
Photo by Rhoda Baer
Researchers have established a public repository of leukemia and lymphoma xenografts, according to a report in Cancer Cell.
The repository, known as the Public Repository of Xenografts (PRoXe), contains material from bone marrow, peripheral blood, and lymph nodes of mice.
It also contains information on the patients from whom the cancer tissues were derived and details on the characteristics of the tumors themselves.
PRoXe is based at Dana-Farber Cancer Institute in Boston, Massachusetts, but it has a web portal that can be accessed by researchers around the world.
Those who register with PRoxE can access the repository and search for cells from patients with specific hematologic malignancies.
Researchers can then have frozen cells shipped to them and transplant the cells into mice to create patient-derived xenograft (PDX) models for testing drugs.
“About 90% of compounds that show anticancer activity in preclinical tests don’t work when given to patients,” said David Weinstock, MD, of the Dana-Farber Cancer Institute.
“By trying drugs in PDX models, we can ‘mimic’ large and expensive human clinical trials and get answers about efficacy more quickly, less expensively, and without the need for patients to get investigational drugs that won’t work.”
To demonstrate how PRoXe can be used, Dr Weinstock and his colleagues tested the MDM2 inhibitor CGM097 against B-cell acute lymphoblastic leukemia (B-ALL) in 2 groups of mice. One group had a mutation in the TP53 tumor suppressor gene, and the other did not.
The researchers observed superior survival in the mice with wild-type TP53. This result corresponds with the results of previous research, which showed that inhibitors that disrupt the MDM2-p53 interaction can be effective in tumor models and patient tumors with wild-type TP53.
Dr Weinstock and his colleagues said their results provide strong preclinical evidence for testing CGM097 in patients who have been extensively treated for B-ALL and have wild-type TP53.
Dr Weinstock also noted that the leaders of ProXe are negotiating with a number of academic centers in an attempt to incorporate their PDX models into the repository.
The Cancer Cell manuscript had 95 authors from 14 different centers that contributed samples, models, and/or effort to the project.
PET-guided chemo improves PFS in advanced HL
Image by Jens Langner
Using PET imaging to guide chemotherapy can improve outcomes for patients with advanced Hodgkin lymphoma (HL), according to research published in the Journal of Clinical Oncology.
The study indicated that PET-guided treatment can improve progression-free survival (PFS) among patients who do not achieve remission with 2 cycles of ABVD.
The results also suggested the approach can spare some patients unnecessary toxicity.
“The goal of cancer treatment is to cure as many people as possible with as little toxicity as possible,” said study author Oliver Press, MD, PhD, of the Fred Hutchinson Cancer Research Center in Seattle, Washington.
“We found a promising way to do that by tailoring treatment to Hodgkin patients, an approach which could lead to a new standard of care.”
Dr Press and his colleagues began this study with 358 HIV-negative patients with advanced HL, but only 336 of them were eligible and evaluable at baseline.
The patients’ median age was 32 (range, 18 to 60). Fifty-two percent had stage III disease, 48% had stage IV, 49% had an International Prognostic Score of 0 to 2, and 51% had a score of 3 to 7.
All of the patients received ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) and underwent a PET scan to gauge their response after 2 cycles (PET2).
If the scan was negative, patients received a final 4 cycles of ABVD. If the scan was positive, patients received 6 cycles of eBEACOPP (escalated bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisolone).
Central review of the PET2 scan was performed in 331 patients. Most (82%, n=271) had a negative scan.
Forty-nine of the 60 PET2-positive patients went on to receive eBEACOPP as planned, but 11 patients declined.
The median follow-up was 39.7 months. For the entire study cohort, the estimated 2-year overall survival was 98%, and the estimated 2-year PFS was 79%.
The 2-year estimated PFS was 82% for PET2-negative patients and 64% for PET2-positive patients. The researchers noted that, typically, if HL patients have a positive PET scan after 2 rounds of ABVD, their expected 2-year PFS ranges from about 15% to 30%.
The researchers also pointed out that eBEACOPP was significantly more toxic than ABVD. The incidence of grade 4/5 toxicities was 85.7% and 36.7%, respectively (P<0.001).
There were 3 treatment-related deaths—1 in the ABVD group and 2 in the eBEACOPP group. And 6 patients developed secondary malignancies—3 in each group—including 2 non-Hodgkin lymphomas, 2 kidney cancers, 1 melanoma, and 1 skin cancer.
“[O]nly 20% of the patients in our trial were exposed to eBEACOPP, which means [most patients] weren’t exposed to its bad effects,” said Jonathan Friedberg, MD, of the University of Rochester Medical Center in Rochester, New York.
“That’s important because many people diagnosed with Hodgkin lymphoma are in their 20s and 30s and want to have children. This response-adapted therapy would ensure that the people who need the more toxic drugs receive them and would spare others from infertility and serious toxicities.”
Image by Jens Langner
Using PET imaging to guide chemotherapy can improve outcomes for patients with advanced Hodgkin lymphoma (HL), according to research published in the Journal of Clinical Oncology.
The study indicated that PET-guided treatment can improve progression-free survival (PFS) among patients who do not achieve remission with 2 cycles of ABVD.
The results also suggested the approach can spare some patients unnecessary toxicity.
“The goal of cancer treatment is to cure as many people as possible with as little toxicity as possible,” said study author Oliver Press, MD, PhD, of the Fred Hutchinson Cancer Research Center in Seattle, Washington.
“We found a promising way to do that by tailoring treatment to Hodgkin patients, an approach which could lead to a new standard of care.”
Dr Press and his colleagues began this study with 358 HIV-negative patients with advanced HL, but only 336 of them were eligible and evaluable at baseline.
The patients’ median age was 32 (range, 18 to 60). Fifty-two percent had stage III disease, 48% had stage IV, 49% had an International Prognostic Score of 0 to 2, and 51% had a score of 3 to 7.
All of the patients received ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) and underwent a PET scan to gauge their response after 2 cycles (PET2).
If the scan was negative, patients received a final 4 cycles of ABVD. If the scan was positive, patients received 6 cycles of eBEACOPP (escalated bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisolone).
Central review of the PET2 scan was performed in 331 patients. Most (82%, n=271) had a negative scan.
Forty-nine of the 60 PET2-positive patients went on to receive eBEACOPP as planned, but 11 patients declined.
The median follow-up was 39.7 months. For the entire study cohort, the estimated 2-year overall survival was 98%, and the estimated 2-year PFS was 79%.
The 2-year estimated PFS was 82% for PET2-negative patients and 64% for PET2-positive patients. The researchers noted that, typically, if HL patients have a positive PET scan after 2 rounds of ABVD, their expected 2-year PFS ranges from about 15% to 30%.
The researchers also pointed out that eBEACOPP was significantly more toxic than ABVD. The incidence of grade 4/5 toxicities was 85.7% and 36.7%, respectively (P<0.001).
There were 3 treatment-related deaths—1 in the ABVD group and 2 in the eBEACOPP group. And 6 patients developed secondary malignancies—3 in each group—including 2 non-Hodgkin lymphomas, 2 kidney cancers, 1 melanoma, and 1 skin cancer.
“[O]nly 20% of the patients in our trial were exposed to eBEACOPP, which means [most patients] weren’t exposed to its bad effects,” said Jonathan Friedberg, MD, of the University of Rochester Medical Center in Rochester, New York.
“That’s important because many people diagnosed with Hodgkin lymphoma are in their 20s and 30s and want to have children. This response-adapted therapy would ensure that the people who need the more toxic drugs receive them and would spare others from infertility and serious toxicities.”
Image by Jens Langner
Using PET imaging to guide chemotherapy can improve outcomes for patients with advanced Hodgkin lymphoma (HL), according to research published in the Journal of Clinical Oncology.
The study indicated that PET-guided treatment can improve progression-free survival (PFS) among patients who do not achieve remission with 2 cycles of ABVD.
The results also suggested the approach can spare some patients unnecessary toxicity.
“The goal of cancer treatment is to cure as many people as possible with as little toxicity as possible,” said study author Oliver Press, MD, PhD, of the Fred Hutchinson Cancer Research Center in Seattle, Washington.
“We found a promising way to do that by tailoring treatment to Hodgkin patients, an approach which could lead to a new standard of care.”
Dr Press and his colleagues began this study with 358 HIV-negative patients with advanced HL, but only 336 of them were eligible and evaluable at baseline.
The patients’ median age was 32 (range, 18 to 60). Fifty-two percent had stage III disease, 48% had stage IV, 49% had an International Prognostic Score of 0 to 2, and 51% had a score of 3 to 7.
All of the patients received ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) and underwent a PET scan to gauge their response after 2 cycles (PET2).
If the scan was negative, patients received a final 4 cycles of ABVD. If the scan was positive, patients received 6 cycles of eBEACOPP (escalated bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisolone).
Central review of the PET2 scan was performed in 331 patients. Most (82%, n=271) had a negative scan.
Forty-nine of the 60 PET2-positive patients went on to receive eBEACOPP as planned, but 11 patients declined.
The median follow-up was 39.7 months. For the entire study cohort, the estimated 2-year overall survival was 98%, and the estimated 2-year PFS was 79%.
The 2-year estimated PFS was 82% for PET2-negative patients and 64% for PET2-positive patients. The researchers noted that, typically, if HL patients have a positive PET scan after 2 rounds of ABVD, their expected 2-year PFS ranges from about 15% to 30%.
The researchers also pointed out that eBEACOPP was significantly more toxic than ABVD. The incidence of grade 4/5 toxicities was 85.7% and 36.7%, respectively (P<0.001).
There were 3 treatment-related deaths—1 in the ABVD group and 2 in the eBEACOPP group. And 6 patients developed secondary malignancies—3 in each group—including 2 non-Hodgkin lymphomas, 2 kidney cancers, 1 melanoma, and 1 skin cancer.
“[O]nly 20% of the patients in our trial were exposed to eBEACOPP, which means [most patients] weren’t exposed to its bad effects,” said Jonathan Friedberg, MD, of the University of Rochester Medical Center in Rochester, New York.
“That’s important because many people diagnosed with Hodgkin lymphoma are in their 20s and 30s and want to have children. This response-adapted therapy would ensure that the people who need the more toxic drugs receive them and would spare others from infertility and serious toxicities.”