User login
Study results support screening of childhood HL survivors
A new study indicates that early screening can reduce the risk of death from breast cancer among female survivors of childhood Hodgkin lymphoma (HL) who received chest radiation.
Researchers found evidence to suggest that starting mammograms at age 25 can reduce the risk of breast cancer death among these patients, but using MRI can reduce the risk further.
Unfortunately, both methods come with a risk of false-positive results.
David Hodgson, MD, of the University of Toronto in Canada, and his colleagues reported these findings in the Journal of the National Cancer Institute.
Dr Hodgson estimates there are thousands of HL survivors in North America treated throughout the 1990s and later who received chest radiation and are unaware they are at risk of breast cancer and eligible for early screening.
“Many of these are women who received radiotherapy to more normal tissue or at higher doses than are used currently,” he said. “But even for more recently treated patients, screening should reduce the risk of breast cancer death.”
For this study, Dr Hodgson and his colleagues gathered published information from dozens of studies about the risk of developing breast cancer in childhood HL survivors, the accuracy of different forms of breast cancer screening, and the rates at which women agree to be screened when asked.
Using mathematical models, the researchers used these data to quantify the effectiveness of starting screening early—at age 25.
The team found that, using mammography, about 260 survivors of childhood HL would need to be invited to have early breast cancer screening to prevent 1 breast cancer death.
However, the use of MRI for screening improved the effectiveness considerably. It reduced the number of women needing screening to prevent 1 breast cancer death to less than 80.
For HL survivors treated at age 15, the absolute risk of breast cancer mortality by age 75 was predicted to decrease from 16.65% with no early screening to 16.28% with annual mammography, 15.40% with annual MRI, 15.38% with same-day annual mammography and MRI, and 15.37% with alternating mammography and MRI every 6 months.
Dr Hodgson cautioned that there is a risk of false-positive screening results, particuarly with MRI, given that the method can detect many changes in breast tissue, most of which are not cancer.
The data suggested that, from age 25 to 75, at least one false-positive result would occur in 48% of women screened with mammography, 74% screened with MRI alone, and 79% screened with both methods.
The number of false-positives per 1000 screens would be 29.98 with mammography, 71.71 with MRI, and 99.52 with either same-day mammography and MRI or alternating mammography and MRI.
“So this is important for patients to know and for physicians to counsel patients about because it’s stressful for a patient to be called back about suspicious findings,” Dr Hodgson said. ![]()
A new study indicates that early screening can reduce the risk of death from breast cancer among female survivors of childhood Hodgkin lymphoma (HL) who received chest radiation.
Researchers found evidence to suggest that starting mammograms at age 25 can reduce the risk of breast cancer death among these patients, but using MRI can reduce the risk further.
Unfortunately, both methods come with a risk of false-positive results.
David Hodgson, MD, of the University of Toronto in Canada, and his colleagues reported these findings in the Journal of the National Cancer Institute.
Dr Hodgson estimates there are thousands of HL survivors in North America treated throughout the 1990s and later who received chest radiation and are unaware they are at risk of breast cancer and eligible for early screening.
“Many of these are women who received radiotherapy to more normal tissue or at higher doses than are used currently,” he said. “But even for more recently treated patients, screening should reduce the risk of breast cancer death.”
For this study, Dr Hodgson and his colleagues gathered published information from dozens of studies about the risk of developing breast cancer in childhood HL survivors, the accuracy of different forms of breast cancer screening, and the rates at which women agree to be screened when asked.
Using mathematical models, the researchers used these data to quantify the effectiveness of starting screening early—at age 25.
The team found that, using mammography, about 260 survivors of childhood HL would need to be invited to have early breast cancer screening to prevent 1 breast cancer death.
However, the use of MRI for screening improved the effectiveness considerably. It reduced the number of women needing screening to prevent 1 breast cancer death to less than 80.
For HL survivors treated at age 15, the absolute risk of breast cancer mortality by age 75 was predicted to decrease from 16.65% with no early screening to 16.28% with annual mammography, 15.40% with annual MRI, 15.38% with same-day annual mammography and MRI, and 15.37% with alternating mammography and MRI every 6 months.
Dr Hodgson cautioned that there is a risk of false-positive screening results, particuarly with MRI, given that the method can detect many changes in breast tissue, most of which are not cancer.
The data suggested that, from age 25 to 75, at least one false-positive result would occur in 48% of women screened with mammography, 74% screened with MRI alone, and 79% screened with both methods.
The number of false-positives per 1000 screens would be 29.98 with mammography, 71.71 with MRI, and 99.52 with either same-day mammography and MRI or alternating mammography and MRI.
“So this is important for patients to know and for physicians to counsel patients about because it’s stressful for a patient to be called back about suspicious findings,” Dr Hodgson said. ![]()
A new study indicates that early screening can reduce the risk of death from breast cancer among female survivors of childhood Hodgkin lymphoma (HL) who received chest radiation.
Researchers found evidence to suggest that starting mammograms at age 25 can reduce the risk of breast cancer death among these patients, but using MRI can reduce the risk further.
Unfortunately, both methods come with a risk of false-positive results.
David Hodgson, MD, of the University of Toronto in Canada, and his colleagues reported these findings in the Journal of the National Cancer Institute.
Dr Hodgson estimates there are thousands of HL survivors in North America treated throughout the 1990s and later who received chest radiation and are unaware they are at risk of breast cancer and eligible for early screening.
“Many of these are women who received radiotherapy to more normal tissue or at higher doses than are used currently,” he said. “But even for more recently treated patients, screening should reduce the risk of breast cancer death.”
For this study, Dr Hodgson and his colleagues gathered published information from dozens of studies about the risk of developing breast cancer in childhood HL survivors, the accuracy of different forms of breast cancer screening, and the rates at which women agree to be screened when asked.
Using mathematical models, the researchers used these data to quantify the effectiveness of starting screening early—at age 25.
The team found that, using mammography, about 260 survivors of childhood HL would need to be invited to have early breast cancer screening to prevent 1 breast cancer death.
However, the use of MRI for screening improved the effectiveness considerably. It reduced the number of women needing screening to prevent 1 breast cancer death to less than 80.
For HL survivors treated at age 15, the absolute risk of breast cancer mortality by age 75 was predicted to decrease from 16.65% with no early screening to 16.28% with annual mammography, 15.40% with annual MRI, 15.38% with same-day annual mammography and MRI, and 15.37% with alternating mammography and MRI every 6 months.
Dr Hodgson cautioned that there is a risk of false-positive screening results, particuarly with MRI, given that the method can detect many changes in breast tissue, most of which are not cancer.
The data suggested that, from age 25 to 75, at least one false-positive result would occur in 48% of women screened with mammography, 74% screened with MRI alone, and 79% screened with both methods.
The number of false-positives per 1000 screens would be 29.98 with mammography, 71.71 with MRI, and 99.52 with either same-day mammography and MRI or alternating mammography and MRI.
“So this is important for patients to know and for physicians to counsel patients about because it’s stressful for a patient to be called back about suspicious findings,” Dr Hodgson said. ![]()
NICE recommends device for managing SCD

Image courtesy of Terumo BCT
The National Institute for Health and Care Excellence (NICE) has issued a guidance recommending a new device for managing sickle cell disease (SCD).
The device is the Spectra Optia Apheresis System for automated red blood cell (RBC) exchange in patients with SCD who need regular blood transfusions.
The Spectra Optia Apheresis System automatically replaces sickled RBCs with healthy RBCs.
The system is made up of 3 components: an apheresis machine, embedded software, and a single-use, disposable blood tubing set.
NICE said the Spectra Optia system is faster than manual RBC exchange, and patients need RBC exchange less often with this device. In addition, the system could provide considerable savings to the National Health Service (NHS) in England.
“The device could save the NHS in England an estimated £13 million each year—around £18,000 per patient—with the size of the saving depending on the patient’s condition and the equipment already owned by the NHS,” said Carole Longson, director of the NICE Centre for Health Technology Evaluation.
“We also recommend that specialists collaborate to collect and publish data on some outcomes of treatment with Spectra Optia to provide further clinical evidence. It would be particularly helpful to have long-term data on how automated and manual exchange affects the amount of iron in the body and the need to treat this complication.”
Treatment with the Spectra Optia system is intended to be iron-neutral, meaning that patients who are already iron-overloaded can have their condition managed effectively.
The Spectra Optia Apheresis System is manufactured by Terumo BCT.
Costs and savings
The list prices (excluding tax) for the components of the Spectra Optia Apheresis System are as follows:
- Spectra Optia device: £45,350
- RBC exchange software: £6700
- Spectra Optia exchange set: £1007 per 6
- Astotube with injection port: £218 per 50
- ACD-A anticoagulant (750 ml): £57 per 12
- Service charge: £4572 per year.
Bulk order discounts are available.
Based on current evidence and expert advice on the anticipated benefits of the technology when used in patients with iron overload, cost modelling shows that, in most cases, using Spectra Optia is cost-saving compared with manual RBC exchange or top-up transfusion.
The savings depend on the iron overload status of the patient and are more likely to be achieved if devices already owned by the NHS can be used to treat SCD.
The estimated cost saving for adopting Spectra Optia is £18,100 per patient per year, which has the potential to save the NHS £12.9 million each year. ![]()
Image courtesy of Terumo BCT
The National Institute for Health and Care Excellence (NICE) has issued a guidance recommending a new device for managing sickle cell disease (SCD).
The device is the Spectra Optia Apheresis System for automated red blood cell (RBC) exchange in patients with SCD who need regular blood transfusions.
The Spectra Optia Apheresis System automatically replaces sickled RBCs with healthy RBCs.
The system is made up of 3 components: an apheresis machine, embedded software, and a single-use, disposable blood tubing set.
NICE said the Spectra Optia system is faster than manual RBC exchange, and patients need RBC exchange less often with this device. In addition, the system could provide considerable savings to the National Health Service (NHS) in England.
“The device could save the NHS in England an estimated £13 million each year—around £18,000 per patient—with the size of the saving depending on the patient’s condition and the equipment already owned by the NHS,” said Carole Longson, director of the NICE Centre for Health Technology Evaluation.
“We also recommend that specialists collaborate to collect and publish data on some outcomes of treatment with Spectra Optia to provide further clinical evidence. It would be particularly helpful to have long-term data on how automated and manual exchange affects the amount of iron in the body and the need to treat this complication.”
Treatment with the Spectra Optia system is intended to be iron-neutral, meaning that patients who are already iron-overloaded can have their condition managed effectively.
The Spectra Optia Apheresis System is manufactured by Terumo BCT.
Costs and savings
The list prices (excluding tax) for the components of the Spectra Optia Apheresis System are as follows:
- Spectra Optia device: £45,350
- RBC exchange software: £6700
- Spectra Optia exchange set: £1007 per 6
- Astotube with injection port: £218 per 50
- ACD-A anticoagulant (750 ml): £57 per 12
- Service charge: £4572 per year.
Bulk order discounts are available.
Based on current evidence and expert advice on the anticipated benefits of the technology when used in patients with iron overload, cost modelling shows that, in most cases, using Spectra Optia is cost-saving compared with manual RBC exchange or top-up transfusion.
The savings depend on the iron overload status of the patient and are more likely to be achieved if devices already owned by the NHS can be used to treat SCD.
The estimated cost saving for adopting Spectra Optia is £18,100 per patient per year, which has the potential to save the NHS £12.9 million each year. ![]()
Image courtesy of Terumo BCT
The National Institute for Health and Care Excellence (NICE) has issued a guidance recommending a new device for managing sickle cell disease (SCD).
The device is the Spectra Optia Apheresis System for automated red blood cell (RBC) exchange in patients with SCD who need regular blood transfusions.
The Spectra Optia Apheresis System automatically replaces sickled RBCs with healthy RBCs.
The system is made up of 3 components: an apheresis machine, embedded software, and a single-use, disposable blood tubing set.
NICE said the Spectra Optia system is faster than manual RBC exchange, and patients need RBC exchange less often with this device. In addition, the system could provide considerable savings to the National Health Service (NHS) in England.
“The device could save the NHS in England an estimated £13 million each year—around £18,000 per patient—with the size of the saving depending on the patient’s condition and the equipment already owned by the NHS,” said Carole Longson, director of the NICE Centre for Health Technology Evaluation.
“We also recommend that specialists collaborate to collect and publish data on some outcomes of treatment with Spectra Optia to provide further clinical evidence. It would be particularly helpful to have long-term data on how automated and manual exchange affects the amount of iron in the body and the need to treat this complication.”
Treatment with the Spectra Optia system is intended to be iron-neutral, meaning that patients who are already iron-overloaded can have their condition managed effectively.
The Spectra Optia Apheresis System is manufactured by Terumo BCT.
Costs and savings
The list prices (excluding tax) for the components of the Spectra Optia Apheresis System are as follows:
- Spectra Optia device: £45,350
- RBC exchange software: £6700
- Spectra Optia exchange set: £1007 per 6
- Astotube with injection port: £218 per 50
- ACD-A anticoagulant (750 ml): £57 per 12
- Service charge: £4572 per year.
Bulk order discounts are available.
Based on current evidence and expert advice on the anticipated benefits of the technology when used in patients with iron overload, cost modelling shows that, in most cases, using Spectra Optia is cost-saving compared with manual RBC exchange or top-up transfusion.
The savings depend on the iron overload status of the patient and are more likely to be achieved if devices already owned by the NHS can be used to treat SCD.
The estimated cost saving for adopting Spectra Optia is £18,100 per patient per year, which has the potential to save the NHS £12.9 million each year. ![]()
Transfusion doesn’t cause NEC, study suggests
Photo by Daniel Gay
Red blood cell (RBC) transfusions do not increase the risk of a serious intestinal disorder in very low-birth-weight (VLBW) infants, according to a study published in JAMA.
Past research has suggested RBC transfusions increase the risk of necrotizing enterocolitis (NEC) among VLBW infants.
But other studies have shown no association between transfusions and NEC or suggested transfusions actually have a protective effect.
So researchers set out to determine whether RBC transfusions or severe anemia were associated with the rate of NEC among VLBW infants. The results suggested a significant association for severe anemia but not RBC transfusion.
To conduct this study, Ravi M. Patel, MD, of the Emory University School of Medicine in Atlanta, Georgia, and his colleagues assessed 598 VLBW infants from 3 neonatal intensive care units in Atlanta.
The team followed the infants for 90 days or until they were discharged from the hospital, transferred to a non-study-affiliated hospital, or died (whichever came first).
Forty-four (7.4%) infants developed NEC, and 32 (5.4%) died (of any cause). Roughly half of the infants (n=319, 53%) received RBC transfusions (n=1430).
The unadjusted cumulative incidence of NEC at week 8 was 9.9% in infants who received transfusions and 4.6% in those who did not.
However, in multivariable analysis, exposure to RBC transfusion in a given week was not significantly related to the rate of NEC. The hazard ratio was 0.44 (P=0.09).
On the other hand, the rate of NEC was significantly higher among infants with severe anemia in a given week than in those without severe anemia. The hazard ratio was 5.99 (P=0.001).
The researchers said these results suggest preventing severe anemia may be more clinically important than minimizing the use of RBC transfusion as a strategy to decrease the risk of NEC in VLBW infants.
However, such a strategy might impact other important neonatal outcomes, so further study is needed. ![]()
Photo by Daniel Gay
Red blood cell (RBC) transfusions do not increase the risk of a serious intestinal disorder in very low-birth-weight (VLBW) infants, according to a study published in JAMA.
Past research has suggested RBC transfusions increase the risk of necrotizing enterocolitis (NEC) among VLBW infants.
But other studies have shown no association between transfusions and NEC or suggested transfusions actually have a protective effect.
So researchers set out to determine whether RBC transfusions or severe anemia were associated with the rate of NEC among VLBW infants. The results suggested a significant association for severe anemia but not RBC transfusion.
To conduct this study, Ravi M. Patel, MD, of the Emory University School of Medicine in Atlanta, Georgia, and his colleagues assessed 598 VLBW infants from 3 neonatal intensive care units in Atlanta.
The team followed the infants for 90 days or until they were discharged from the hospital, transferred to a non-study-affiliated hospital, or died (whichever came first).
Forty-four (7.4%) infants developed NEC, and 32 (5.4%) died (of any cause). Roughly half of the infants (n=319, 53%) received RBC transfusions (n=1430).
The unadjusted cumulative incidence of NEC at week 8 was 9.9% in infants who received transfusions and 4.6% in those who did not.
However, in multivariable analysis, exposure to RBC transfusion in a given week was not significantly related to the rate of NEC. The hazard ratio was 0.44 (P=0.09).
On the other hand, the rate of NEC was significantly higher among infants with severe anemia in a given week than in those without severe anemia. The hazard ratio was 5.99 (P=0.001).
The researchers said these results suggest preventing severe anemia may be more clinically important than minimizing the use of RBC transfusion as a strategy to decrease the risk of NEC in VLBW infants.
However, such a strategy might impact other important neonatal outcomes, so further study is needed. ![]()
Photo by Daniel Gay
Red blood cell (RBC) transfusions do not increase the risk of a serious intestinal disorder in very low-birth-weight (VLBW) infants, according to a study published in JAMA.
Past research has suggested RBC transfusions increase the risk of necrotizing enterocolitis (NEC) among VLBW infants.
But other studies have shown no association between transfusions and NEC or suggested transfusions actually have a protective effect.
So researchers set out to determine whether RBC transfusions or severe anemia were associated with the rate of NEC among VLBW infants. The results suggested a significant association for severe anemia but not RBC transfusion.
To conduct this study, Ravi M. Patel, MD, of the Emory University School of Medicine in Atlanta, Georgia, and his colleagues assessed 598 VLBW infants from 3 neonatal intensive care units in Atlanta.
The team followed the infants for 90 days or until they were discharged from the hospital, transferred to a non-study-affiliated hospital, or died (whichever came first).
Forty-four (7.4%) infants developed NEC, and 32 (5.4%) died (of any cause). Roughly half of the infants (n=319, 53%) received RBC transfusions (n=1430).
The unadjusted cumulative incidence of NEC at week 8 was 9.9% in infants who received transfusions and 4.6% in those who did not.
However, in multivariable analysis, exposure to RBC transfusion in a given week was not significantly related to the rate of NEC. The hazard ratio was 0.44 (P=0.09).
On the other hand, the rate of NEC was significantly higher among infants with severe anemia in a given week than in those without severe anemia. The hazard ratio was 5.99 (P=0.001).
The researchers said these results suggest preventing severe anemia may be more clinically important than minimizing the use of RBC transfusion as a strategy to decrease the risk of NEC in VLBW infants.
However, such a strategy might impact other important neonatal outcomes, so further study is needed. ![]()
Team identifies potential target for aggressive AML

Photo by Rhoda Baer
Research published in The Journal of Clinical Investigation has revealed a potential therapeutic target for an aggressive form of acute myeloid leukemia (AML).
Investigators studied AML characterized by overexpression of the gene meningioma-1 (MN1), which is not a druggable target.
The team found that MN1 overexpression induces aggressive AML that is dependent on a gene expression program controlled by 2 histone methyltransferases.
And 1 of these histone methyltransferases can be targeted by drugs currently in clinical development.
To make these discoveries, the investigators forced expression of MN1 in mice, which induced AML, and looked for changes in other genes. They found that MN1 overexpression prompted the activation of genes already linked to AML development—HoxA9 and Meis1.
HoxA9 and Meis1 are key targets of the histone methyltransferases Mll1 and Dot1l. It turned out that Mll1 and Dotl1 are essential for creating the environment MN1 needs to cause AML.
“In mice, we put MN1 in first, leading to AML,” explained study author Kathrin Bernt, MD, of the University of Colorado Anschutz Medical Campus.
“Then, we knocked out these chromatin-regulating molecules, Mll1 or Dot1l. When we did that, the leukemia collapsed.”
The investigators also studied samples from AML patients and found that samples with overexpression of MN1 and HOXA9 were sensitive to the DOT1L inhibitor EPZ004777. The drug induced dose-dependent decreases in cell growth and the fraction of cycling cells, as well as an increase in apoptosis.
Anticancer agents targeting DOT1L are already in clinical trials. One such inhibitor, EPZ-5676, is being tested in a phase 1 trial of pediatric patients with aggressive leukemias (NCT02141828).
“The existing trial targets patients with rearrangements in the gene MLL1,” Dr Bernt noted. “Our study shows another subset of patients that may benefit from this or other therapies aimed at DOT1L inhibition—namely, patients with MN1 overexpression.”
Dr Bernt added, however, that the investigators must still determine the cutoff of MN1 overexpression at which AML is susceptible to DOT1L inhibition.
“Overexpression exists along a spectrum,” she explained. “At what degree of MN1 overexpression does it become clinically significant?” ![]()
Photo by Rhoda Baer
Research published in The Journal of Clinical Investigation has revealed a potential therapeutic target for an aggressive form of acute myeloid leukemia (AML).
Investigators studied AML characterized by overexpression of the gene meningioma-1 (MN1), which is not a druggable target.
The team found that MN1 overexpression induces aggressive AML that is dependent on a gene expression program controlled by 2 histone methyltransferases.
And 1 of these histone methyltransferases can be targeted by drugs currently in clinical development.
To make these discoveries, the investigators forced expression of MN1 in mice, which induced AML, and looked for changes in other genes. They found that MN1 overexpression prompted the activation of genes already linked to AML development—HoxA9 and Meis1.
HoxA9 and Meis1 are key targets of the histone methyltransferases Mll1 and Dot1l. It turned out that Mll1 and Dotl1 are essential for creating the environment MN1 needs to cause AML.
“In mice, we put MN1 in first, leading to AML,” explained study author Kathrin Bernt, MD, of the University of Colorado Anschutz Medical Campus.
“Then, we knocked out these chromatin-regulating molecules, Mll1 or Dot1l. When we did that, the leukemia collapsed.”
The investigators also studied samples from AML patients and found that samples with overexpression of MN1 and HOXA9 were sensitive to the DOT1L inhibitor EPZ004777. The drug induced dose-dependent decreases in cell growth and the fraction of cycling cells, as well as an increase in apoptosis.
Anticancer agents targeting DOT1L are already in clinical trials. One such inhibitor, EPZ-5676, is being tested in a phase 1 trial of pediatric patients with aggressive leukemias (NCT02141828).
“The existing trial targets patients with rearrangements in the gene MLL1,” Dr Bernt noted. “Our study shows another subset of patients that may benefit from this or other therapies aimed at DOT1L inhibition—namely, patients with MN1 overexpression.”
Dr Bernt added, however, that the investigators must still determine the cutoff of MN1 overexpression at which AML is susceptible to DOT1L inhibition.
“Overexpression exists along a spectrum,” she explained. “At what degree of MN1 overexpression does it become clinically significant?” ![]()
Photo by Rhoda Baer
Research published in The Journal of Clinical Investigation has revealed a potential therapeutic target for an aggressive form of acute myeloid leukemia (AML).
Investigators studied AML characterized by overexpression of the gene meningioma-1 (MN1), which is not a druggable target.
The team found that MN1 overexpression induces aggressive AML that is dependent on a gene expression program controlled by 2 histone methyltransferases.
And 1 of these histone methyltransferases can be targeted by drugs currently in clinical development.
To make these discoveries, the investigators forced expression of MN1 in mice, which induced AML, and looked for changes in other genes. They found that MN1 overexpression prompted the activation of genes already linked to AML development—HoxA9 and Meis1.
HoxA9 and Meis1 are key targets of the histone methyltransferases Mll1 and Dot1l. It turned out that Mll1 and Dotl1 are essential for creating the environment MN1 needs to cause AML.
“In mice, we put MN1 in first, leading to AML,” explained study author Kathrin Bernt, MD, of the University of Colorado Anschutz Medical Campus.
“Then, we knocked out these chromatin-regulating molecules, Mll1 or Dot1l. When we did that, the leukemia collapsed.”
The investigators also studied samples from AML patients and found that samples with overexpression of MN1 and HOXA9 were sensitive to the DOT1L inhibitor EPZ004777. The drug induced dose-dependent decreases in cell growth and the fraction of cycling cells, as well as an increase in apoptosis.
Anticancer agents targeting DOT1L are already in clinical trials. One such inhibitor, EPZ-5676, is being tested in a phase 1 trial of pediatric patients with aggressive leukemias (NCT02141828).
“The existing trial targets patients with rearrangements in the gene MLL1,” Dr Bernt noted. “Our study shows another subset of patients that may benefit from this or other therapies aimed at DOT1L inhibition—namely, patients with MN1 overexpression.”
Dr Bernt added, however, that the investigators must still determine the cutoff of MN1 overexpression at which AML is susceptible to DOT1L inhibition.
“Overexpression exists along a spectrum,” she explained. “At what degree of MN1 overexpression does it become clinically significant?” ![]()
EC approves drug for iron-deficiency anemia in IBD

The European Commission (EC) has granted marketing authorization for ferric maltol (Feraccru) to treat iron-deficiency anemia in adults with inflammatory bowel disease (IBD).
The product can now be marketed for this indication in the 28 member countries of the European Union, as well as Iceland, Liechtenstein, and Norway.
The company developing the drug, Shield Therapeutics, said it will begin a roll-out of commercialization in the coming months.
Feraccru contains iron in a stable ferric state as a complex with a trimaltol ligand—ferric maltol. It is formulated as 30 mg of ferric iron in a hard gelatin capsule.
The complex is designed to provide iron for uptake across the intestinal wall and transfer to the iron transport and storage proteins—transferrin and ferritin, respectively. Feraccru dissociates on uptake from the gastrointestinal tract, and ferric maltol itself does not appear to enter the systemic circulation.
Phase 3 trials
The EC’s approval of ferric maltol is based on results of 2 phase 3 studies—AEGIS 1 and AEGIS 2. The results of these studies were published in Inflammatory Bowel Diseases in March 2015.
Together, the trials included 128 adult patients with IBD (ulcerative colitis and Crohn’s disease), iron-deficiency anemia, and recorded intolerance of ferrous sulphate. They were randomized to receive either 30 mg of ferric maltol twice a day or a matched placebo capsule for 12 weeks.
The primary efficacy endpoint was the change in hemoglobin (Hb) levels from baseline to week 12. The mean improvement in Hb in the ferric maltol group compared to the placebo group was 2.25 g/dL (P<0.0001).
The absolute mean Hb concentration improved from 11.00 g/dL at baseline to 13.20 g/dL at week 12 in the ferric maltol group. In the placebo group, the mean Hb values were similar at baseline and week 12—11.10 g/dL and 11.20 g/dL, respectively.
The incidence of treatment-emergent adverse events (AEs) was 58% in the ferric maltol group and 72% in the placebo group. However, not all of these events were considered treatment-related.
AEs that were considered treatment-related occurred in 25% of ferric maltol-treated patients and 11.7% of placebo-treated patients. The most common treatment-related AEs in the ferric maltol group were abdominal pain, constipation, and flatulence—each occurring in 6.7% of patients.
“The phase 3 clinical studies clearly demonstrated Feraccru’s effectiveness,” said Andreas Stallmach, MD, of University Clinic Jena in Germany.
“[T]his pan-European marketing authorization gives treating physicians like myself the opportunity to fulfill an important and currently unmet need for patients who are unable to tolerate other oral products, as Feraccru could provide an oral alternative to intravenous iron infusion.’’ ![]()
The European Commission (EC) has granted marketing authorization for ferric maltol (Feraccru) to treat iron-deficiency anemia in adults with inflammatory bowel disease (IBD).
The product can now be marketed for this indication in the 28 member countries of the European Union, as well as Iceland, Liechtenstein, and Norway.
The company developing the drug, Shield Therapeutics, said it will begin a roll-out of commercialization in the coming months.
Feraccru contains iron in a stable ferric state as a complex with a trimaltol ligand—ferric maltol. It is formulated as 30 mg of ferric iron in a hard gelatin capsule.
The complex is designed to provide iron for uptake across the intestinal wall and transfer to the iron transport and storage proteins—transferrin and ferritin, respectively. Feraccru dissociates on uptake from the gastrointestinal tract, and ferric maltol itself does not appear to enter the systemic circulation.
Phase 3 trials
The EC’s approval of ferric maltol is based on results of 2 phase 3 studies—AEGIS 1 and AEGIS 2. The results of these studies were published in Inflammatory Bowel Diseases in March 2015.
Together, the trials included 128 adult patients with IBD (ulcerative colitis and Crohn’s disease), iron-deficiency anemia, and recorded intolerance of ferrous sulphate. They were randomized to receive either 30 mg of ferric maltol twice a day or a matched placebo capsule for 12 weeks.
The primary efficacy endpoint was the change in hemoglobin (Hb) levels from baseline to week 12. The mean improvement in Hb in the ferric maltol group compared to the placebo group was 2.25 g/dL (P<0.0001).
The absolute mean Hb concentration improved from 11.00 g/dL at baseline to 13.20 g/dL at week 12 in the ferric maltol group. In the placebo group, the mean Hb values were similar at baseline and week 12—11.10 g/dL and 11.20 g/dL, respectively.
The incidence of treatment-emergent adverse events (AEs) was 58% in the ferric maltol group and 72% in the placebo group. However, not all of these events were considered treatment-related.
AEs that were considered treatment-related occurred in 25% of ferric maltol-treated patients and 11.7% of placebo-treated patients. The most common treatment-related AEs in the ferric maltol group were abdominal pain, constipation, and flatulence—each occurring in 6.7% of patients.
“The phase 3 clinical studies clearly demonstrated Feraccru’s effectiveness,” said Andreas Stallmach, MD, of University Clinic Jena in Germany.
“[T]his pan-European marketing authorization gives treating physicians like myself the opportunity to fulfill an important and currently unmet need for patients who are unable to tolerate other oral products, as Feraccru could provide an oral alternative to intravenous iron infusion.’’ ![]()
The European Commission (EC) has granted marketing authorization for ferric maltol (Feraccru) to treat iron-deficiency anemia in adults with inflammatory bowel disease (IBD).
The product can now be marketed for this indication in the 28 member countries of the European Union, as well as Iceland, Liechtenstein, and Norway.
The company developing the drug, Shield Therapeutics, said it will begin a roll-out of commercialization in the coming months.
Feraccru contains iron in a stable ferric state as a complex with a trimaltol ligand—ferric maltol. It is formulated as 30 mg of ferric iron in a hard gelatin capsule.
The complex is designed to provide iron for uptake across the intestinal wall and transfer to the iron transport and storage proteins—transferrin and ferritin, respectively. Feraccru dissociates on uptake from the gastrointestinal tract, and ferric maltol itself does not appear to enter the systemic circulation.
Phase 3 trials
The EC’s approval of ferric maltol is based on results of 2 phase 3 studies—AEGIS 1 and AEGIS 2. The results of these studies were published in Inflammatory Bowel Diseases in March 2015.
Together, the trials included 128 adult patients with IBD (ulcerative colitis and Crohn’s disease), iron-deficiency anemia, and recorded intolerance of ferrous sulphate. They were randomized to receive either 30 mg of ferric maltol twice a day or a matched placebo capsule for 12 weeks.
The primary efficacy endpoint was the change in hemoglobin (Hb) levels from baseline to week 12. The mean improvement in Hb in the ferric maltol group compared to the placebo group was 2.25 g/dL (P<0.0001).
The absolute mean Hb concentration improved from 11.00 g/dL at baseline to 13.20 g/dL at week 12 in the ferric maltol group. In the placebo group, the mean Hb values were similar at baseline and week 12—11.10 g/dL and 11.20 g/dL, respectively.
The incidence of treatment-emergent adverse events (AEs) was 58% in the ferric maltol group and 72% in the placebo group. However, not all of these events were considered treatment-related.
AEs that were considered treatment-related occurred in 25% of ferric maltol-treated patients and 11.7% of placebo-treated patients. The most common treatment-related AEs in the ferric maltol group were abdominal pain, constipation, and flatulence—each occurring in 6.7% of patients.
“The phase 3 clinical studies clearly demonstrated Feraccru’s effectiveness,” said Andreas Stallmach, MD, of University Clinic Jena in Germany.
“[T]his pan-European marketing authorization gives treating physicians like myself the opportunity to fulfill an important and currently unmet need for patients who are unable to tolerate other oral products, as Feraccru could provide an oral alternative to intravenous iron infusion.’’ ![]()
Vaccine candidate fails to prevent malaria in humans
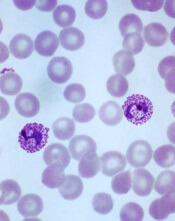
Plasmodium vivax
Image by Mae Melvin
Results of a phase 1 trial suggest a vaccine candidate does not prevent Plasmodium vivax malaria.
The vaccine, VMP001/AS01B, did not prevent malaria in any of the 30 subjects tested, although it did significantly delay parasitemia in 59% of subjects.
Lieutenant Colonel Jason W. Bennett, of the Walter Reed Army Institute
of Research (WRAIR) in Silver Spring, Maryland, and his colleagues reported these results in PLOS Neglected Tropical Diseases.
The vaccine antigen VMP001 is an Escherichia coli-produced, synthetic, chimeric, recombinant protein that incorporates the 3 major domains of circumsporozoite protein but is distinct from the native molecule. The VMP001 antigen was adjuvanted with AS01B, a proprietary liposome-based adjuvant system from GlaxoSmithKline.
For this phase 1 study, the investigators tested VMP001/AS01B in 30 healthy volunteers. The subjects received 3 intramuscular injections of VMP001 at 15 μg (n=10), 30 μg (n=10), or 60 μg (n=10), respectively, all formulated in 500 μL of AS01B at each immunization.
Fourteen days after the third immunization, the subjects were exposed to mosquitoes carrying P vivax. Six non-vaccinated control subjects were exposed to the mosquitoes as well.
The investigators said VMP001/AS01B was well tolerated and generated robust immune responses. However, it did not protect the subjects from malaria infection.
Still, the vaccine induced a “small but significant” delay in time to parasitemia in 59% of vaccinated subjects, when compared to controls.
Colonel Robert Paris, director of the US Military Malaria Research Program at WRAIR, believes the investigators can use the results of this study to design a better vaccine for P vivax malaria.
“Findings from the analysis of the immune response of vaccinated subjects have given us clues to improve vaccine candidates,” he said. “[S]tudies are now underway at WRAIR to develop next-generation vivax vaccines.” ![]()
Plasmodium vivax
Image by Mae Melvin
Results of a phase 1 trial suggest a vaccine candidate does not prevent Plasmodium vivax malaria.
The vaccine, VMP001/AS01B, did not prevent malaria in any of the 30 subjects tested, although it did significantly delay parasitemia in 59% of subjects.
Lieutenant Colonel Jason W. Bennett, of the Walter Reed Army Institute
of Research (WRAIR) in Silver Spring, Maryland, and his colleagues reported these results in PLOS Neglected Tropical Diseases.
The vaccine antigen VMP001 is an Escherichia coli-produced, synthetic, chimeric, recombinant protein that incorporates the 3 major domains of circumsporozoite protein but is distinct from the native molecule. The VMP001 antigen was adjuvanted with AS01B, a proprietary liposome-based adjuvant system from GlaxoSmithKline.
For this phase 1 study, the investigators tested VMP001/AS01B in 30 healthy volunteers. The subjects received 3 intramuscular injections of VMP001 at 15 μg (n=10), 30 μg (n=10), or 60 μg (n=10), respectively, all formulated in 500 μL of AS01B at each immunization.
Fourteen days after the third immunization, the subjects were exposed to mosquitoes carrying P vivax. Six non-vaccinated control subjects were exposed to the mosquitoes as well.
The investigators said VMP001/AS01B was well tolerated and generated robust immune responses. However, it did not protect the subjects from malaria infection.
Still, the vaccine induced a “small but significant” delay in time to parasitemia in 59% of vaccinated subjects, when compared to controls.
Colonel Robert Paris, director of the US Military Malaria Research Program at WRAIR, believes the investigators can use the results of this study to design a better vaccine for P vivax malaria.
“Findings from the analysis of the immune response of vaccinated subjects have given us clues to improve vaccine candidates,” he said. “[S]tudies are now underway at WRAIR to develop next-generation vivax vaccines.” ![]()
Plasmodium vivax
Image by Mae Melvin
Results of a phase 1 trial suggest a vaccine candidate does not prevent Plasmodium vivax malaria.
The vaccine, VMP001/AS01B, did not prevent malaria in any of the 30 subjects tested, although it did significantly delay parasitemia in 59% of subjects.
Lieutenant Colonel Jason W. Bennett, of the Walter Reed Army Institute
of Research (WRAIR) in Silver Spring, Maryland, and his colleagues reported these results in PLOS Neglected Tropical Diseases.
The vaccine antigen VMP001 is an Escherichia coli-produced, synthetic, chimeric, recombinant protein that incorporates the 3 major domains of circumsporozoite protein but is distinct from the native molecule. The VMP001 antigen was adjuvanted with AS01B, a proprietary liposome-based adjuvant system from GlaxoSmithKline.
For this phase 1 study, the investigators tested VMP001/AS01B in 30 healthy volunteers. The subjects received 3 intramuscular injections of VMP001 at 15 μg (n=10), 30 μg (n=10), or 60 μg (n=10), respectively, all formulated in 500 μL of AS01B at each immunization.
Fourteen days after the third immunization, the subjects were exposed to mosquitoes carrying P vivax. Six non-vaccinated control subjects were exposed to the mosquitoes as well.
The investigators said VMP001/AS01B was well tolerated and generated robust immune responses. However, it did not protect the subjects from malaria infection.
Still, the vaccine induced a “small but significant” delay in time to parasitemia in 59% of vaccinated subjects, when compared to controls.
Colonel Robert Paris, director of the US Military Malaria Research Program at WRAIR, believes the investigators can use the results of this study to design a better vaccine for P vivax malaria.
“Findings from the analysis of the immune response of vaccinated subjects have given us clues to improve vaccine candidates,” he said. “[S]tudies are now underway at WRAIR to develop next-generation vivax vaccines.” ![]()
Combining inhibitors to treat AML

Preclinical research has revealed a treatment approach that could prove effective against acute myeloid leukemia (AML).
Researchers tested the IAP inhibitor birinapant in combination with p38 inhibitors and observed antileukemic activity in mouse models of AML and samples from patients with the disease.
Combination treatment proved more effective than either agent alone, and the combination was less toxic than single-agent chemotherapy.
Najoua Lalaoui, PhD, of the Walter and Eliza Hall Institute of Medical Research in Parkville, Victoria, Australia, and her colleagues conducted this research and relayed the results in an article published in Cancer Cell.
The researchers generated several mouse models of AML—MLL-ENL ± NRasG12D, MLL-AF9 ± NrasG12D, AML1-ETO9a + NrasG12D, CBFβ-MYH11 + NrasG12D, NUP98-HoxA9, and HoxA9/Meis1.
In these models, the team tested birinapant with 1 of 2 p38 inhibitors—LY2228820 or SCIO-469—or with the MK2 inhibitor PF-3644022. They said each combination “dramatically” increased cell death, when compared to birinapant alone, in most models. The exceptions were AML1-ETO9a + NrasG12D and CBFβ-MYH11 + NrasG12D.
Next, the researchers tested LY2228820 plus birinapant in samples from 8 AML patients. The samples had FLT3-ITD mutations (patients 1, 2, 4, 6, and 7), a FLT3 D835 missense mutation (patient 4), nucleophosmin exon-12 mutations (patients 2 and 4), an MLL translocation (patient 3), inv(3) (patient 1), and inv(16) (patient 8).
All 8 samples were sensitive to birinapant alone. And although LY2228820 alone did not induce cell death in any of the samples, the drug had a synergistic effect with birinapant in 4 of the samples (patients 2, 3, 4, and 7).
The researchers also found that peripheral blood mononuclear cells from healthy donors proved more resistant to combination LY2228820 (at 10 µM) and birinapant (at 500 nM) than to cytarabine (10 µM), daunorubicin (at 0.4 µM), or idarubicin (at 0.4 µM).
In addition, 4 weeks of treatment with birinapant and LY2228820 was well-tolerated in mice without tumors.
Finally, the researchers tested birinapant and LY2228820, either alone or in combination, in mouse models of MLL-ENL, MLL-AF9, and NRasG12D mutant/MLL-AF9/Luc AML.
Combination treatment prolonged survival in all 3 models, when compared with mice that received single agents or no treatment. However, unlike in the MLL-ENL and MLL-AF9 models, the combination was unable to cure NRasG12D mutant/MLL-AF9/Luc mice of their leukemia.
“Our findings have made us hopeful that a combination of birinapant and a p38 inhibitor may be more effective in treating AML than current therapies and also have less toxicity for patients,” Dr Lalaoui said.
“We tested forms of AML that are highly resistant to chemotherapy and found that birinapant and p38 inhibitors could even kill these cancer cells, which is great news.”
Birinapant is being developed by TetraLogic Pharmaceuticals Corporation, and some of the researchers involved in this work reported relationships with the company. ![]()
Preclinical research has revealed a treatment approach that could prove effective against acute myeloid leukemia (AML).
Researchers tested the IAP inhibitor birinapant in combination with p38 inhibitors and observed antileukemic activity in mouse models of AML and samples from patients with the disease.
Combination treatment proved more effective than either agent alone, and the combination was less toxic than single-agent chemotherapy.
Najoua Lalaoui, PhD, of the Walter and Eliza Hall Institute of Medical Research in Parkville, Victoria, Australia, and her colleagues conducted this research and relayed the results in an article published in Cancer Cell.
The researchers generated several mouse models of AML—MLL-ENL ± NRasG12D, MLL-AF9 ± NrasG12D, AML1-ETO9a + NrasG12D, CBFβ-MYH11 + NrasG12D, NUP98-HoxA9, and HoxA9/Meis1.
In these models, the team tested birinapant with 1 of 2 p38 inhibitors—LY2228820 or SCIO-469—or with the MK2 inhibitor PF-3644022. They said each combination “dramatically” increased cell death, when compared to birinapant alone, in most models. The exceptions were AML1-ETO9a + NrasG12D and CBFβ-MYH11 + NrasG12D.
Next, the researchers tested LY2228820 plus birinapant in samples from 8 AML patients. The samples had FLT3-ITD mutations (patients 1, 2, 4, 6, and 7), a FLT3 D835 missense mutation (patient 4), nucleophosmin exon-12 mutations (patients 2 and 4), an MLL translocation (patient 3), inv(3) (patient 1), and inv(16) (patient 8).
All 8 samples were sensitive to birinapant alone. And although LY2228820 alone did not induce cell death in any of the samples, the drug had a synergistic effect with birinapant in 4 of the samples (patients 2, 3, 4, and 7).
The researchers also found that peripheral blood mononuclear cells from healthy donors proved more resistant to combination LY2228820 (at 10 µM) and birinapant (at 500 nM) than to cytarabine (10 µM), daunorubicin (at 0.4 µM), or idarubicin (at 0.4 µM).
In addition, 4 weeks of treatment with birinapant and LY2228820 was well-tolerated in mice without tumors.
Finally, the researchers tested birinapant and LY2228820, either alone or in combination, in mouse models of MLL-ENL, MLL-AF9, and NRasG12D mutant/MLL-AF9/Luc AML.
Combination treatment prolonged survival in all 3 models, when compared with mice that received single agents or no treatment. However, unlike in the MLL-ENL and MLL-AF9 models, the combination was unable to cure NRasG12D mutant/MLL-AF9/Luc mice of their leukemia.
“Our findings have made us hopeful that a combination of birinapant and a p38 inhibitor may be more effective in treating AML than current therapies and also have less toxicity for patients,” Dr Lalaoui said.
“We tested forms of AML that are highly resistant to chemotherapy and found that birinapant and p38 inhibitors could even kill these cancer cells, which is great news.”
Birinapant is being developed by TetraLogic Pharmaceuticals Corporation, and some of the researchers involved in this work reported relationships with the company. ![]()
Preclinical research has revealed a treatment approach that could prove effective against acute myeloid leukemia (AML).
Researchers tested the IAP inhibitor birinapant in combination with p38 inhibitors and observed antileukemic activity in mouse models of AML and samples from patients with the disease.
Combination treatment proved more effective than either agent alone, and the combination was less toxic than single-agent chemotherapy.
Najoua Lalaoui, PhD, of the Walter and Eliza Hall Institute of Medical Research in Parkville, Victoria, Australia, and her colleagues conducted this research and relayed the results in an article published in Cancer Cell.
The researchers generated several mouse models of AML—MLL-ENL ± NRasG12D, MLL-AF9 ± NrasG12D, AML1-ETO9a + NrasG12D, CBFβ-MYH11 + NrasG12D, NUP98-HoxA9, and HoxA9/Meis1.
In these models, the team tested birinapant with 1 of 2 p38 inhibitors—LY2228820 or SCIO-469—or with the MK2 inhibitor PF-3644022. They said each combination “dramatically” increased cell death, when compared to birinapant alone, in most models. The exceptions were AML1-ETO9a + NrasG12D and CBFβ-MYH11 + NrasG12D.
Next, the researchers tested LY2228820 plus birinapant in samples from 8 AML patients. The samples had FLT3-ITD mutations (patients 1, 2, 4, 6, and 7), a FLT3 D835 missense mutation (patient 4), nucleophosmin exon-12 mutations (patients 2 and 4), an MLL translocation (patient 3), inv(3) (patient 1), and inv(16) (patient 8).
All 8 samples were sensitive to birinapant alone. And although LY2228820 alone did not induce cell death in any of the samples, the drug had a synergistic effect with birinapant in 4 of the samples (patients 2, 3, 4, and 7).
The researchers also found that peripheral blood mononuclear cells from healthy donors proved more resistant to combination LY2228820 (at 10 µM) and birinapant (at 500 nM) than to cytarabine (10 µM), daunorubicin (at 0.4 µM), or idarubicin (at 0.4 µM).
In addition, 4 weeks of treatment with birinapant and LY2228820 was well-tolerated in mice without tumors.
Finally, the researchers tested birinapant and LY2228820, either alone or in combination, in mouse models of MLL-ENL, MLL-AF9, and NRasG12D mutant/MLL-AF9/Luc AML.
Combination treatment prolonged survival in all 3 models, when compared with mice that received single agents or no treatment. However, unlike in the MLL-ENL and MLL-AF9 models, the combination was unable to cure NRasG12D mutant/MLL-AF9/Luc mice of their leukemia.
“Our findings have made us hopeful that a combination of birinapant and a p38 inhibitor may be more effective in treating AML than current therapies and also have less toxicity for patients,” Dr Lalaoui said.
“We tested forms of AML that are highly resistant to chemotherapy and found that birinapant and p38 inhibitors could even kill these cancer cells, which is great news.”
Birinapant is being developed by TetraLogic Pharmaceuticals Corporation, and some of the researchers involved in this work reported relationships with the company.
Drug granted orphan designation for hemolytic anemia

The European Commission (EC) has granted orphan drug designation for TNT009 to treat autoimmune hemolytic anemia, including cold agglutinin disease.
TNT009 is a monoclonal antibody that selectively inhibits the classical complement pathway by targeting C1s, a serine protease within the C1-complex in the complement pathway.
The drug thereby prevents downstream disease processes involving phagocytosis, inflammation, and cell lysis.
TNT009 is being developed by True North Therapeutics.
The drug is currently in development for the treatment of autoimmune hemolytic anemia, which is characterized by the premature destruction of healthy red blood cells by autoantibodies.
In cold agglutinin disease, this destruction of red blood cells results in anemia, fatigue, and potentially fatal thrombosis.
TNT009 is also being evaluated in patients with bullous pemphigoid and end-stage renal disease.
Top-line results from a phase 1b trial of TNT009 are expected in mid-2016.
About orphan designation
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the European Union. The product must provide significant benefit to those affected by the condition.
Orphan drug designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is on the market.
The European Commission (EC) has granted orphan drug designation for TNT009 to treat autoimmune hemolytic anemia, including cold agglutinin disease.
TNT009 is a monoclonal antibody that selectively inhibits the classical complement pathway by targeting C1s, a serine protease within the C1-complex in the complement pathway.
The drug thereby prevents downstream disease processes involving phagocytosis, inflammation, and cell lysis.
TNT009 is being developed by True North Therapeutics.
The drug is currently in development for the treatment of autoimmune hemolytic anemia, which is characterized by the premature destruction of healthy red blood cells by autoantibodies.
In cold agglutinin disease, this destruction of red blood cells results in anemia, fatigue, and potentially fatal thrombosis.
TNT009 is also being evaluated in patients with bullous pemphigoid and end-stage renal disease.
Top-line results from a phase 1b trial of TNT009 are expected in mid-2016.
About orphan designation
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the European Union. The product must provide significant benefit to those affected by the condition.
Orphan drug designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is on the market.
The European Commission (EC) has granted orphan drug designation for TNT009 to treat autoimmune hemolytic anemia, including cold agglutinin disease.
TNT009 is a monoclonal antibody that selectively inhibits the classical complement pathway by targeting C1s, a serine protease within the C1-complex in the complement pathway.
The drug thereby prevents downstream disease processes involving phagocytosis, inflammation, and cell lysis.
TNT009 is being developed by True North Therapeutics.
The drug is currently in development for the treatment of autoimmune hemolytic anemia, which is characterized by the premature destruction of healthy red blood cells by autoantibodies.
In cold agglutinin disease, this destruction of red blood cells results in anemia, fatigue, and potentially fatal thrombosis.
TNT009 is also being evaluated in patients with bullous pemphigoid and end-stage renal disease.
Top-line results from a phase 1b trial of TNT009 are expected in mid-2016.
About orphan designation
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the European Union. The product must provide significant benefit to those affected by the condition.
Orphan drug designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is on the market.
EC grants venetoclax orphan designation for AML

The European Commission has granted orphan drug designation for the oral BCL-2 inhibitor venetoclax to treat acute myeloid leukemia (AML).
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the
European Union. The product must provide significant benefit to those affected by the condition.
Orphan drug designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is on the market.
Venetoclax is being developed by AbbVie in partnership with Genentech and Roche.
Phase 2 study
Results from a phase 2 study of venetoclax in AML were presented at ASH 2014. At that time, the trial had enrolled 32 patients, 30 of whom had relapsed or refractory disease. Patients had a median age of 71 (range, 19 to 84), and half were male.
The overall response rate was 15.5%, with 1 patient achieving a complete response (CR) and 4 achieving a CR with incomplete count recovery (CRi).
The researchers noted that 3 of the patients who had a CR/CRi had IDH mutations. Two of these patients also achieved minimal residual disease negativity.
The median bone marrow blast count in evaluable patients decreased 36% after treatment, and 6 patients (19%) had at least a 50% reduction in bone marrow blasts.
Common adverse events following treatment (occurring in at least 25% of patients) included nausea, diarrhea, fatigue, neutropenia, and vomiting.
Grade 3/4 adverse events (occurring in 3 or more patients) included febrile neutropenia, anemia, and pneumonia. No patient died as a result of treatment-related adverse events.
The European Commission has granted orphan drug designation for the oral BCL-2 inhibitor venetoclax to treat acute myeloid leukemia (AML).
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the
European Union. The product must provide significant benefit to those affected by the condition.
Orphan drug designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is on the market.
Venetoclax is being developed by AbbVie in partnership with Genentech and Roche.
Phase 2 study
Results from a phase 2 study of venetoclax in AML were presented at ASH 2014. At that time, the trial had enrolled 32 patients, 30 of whom had relapsed or refractory disease. Patients had a median age of 71 (range, 19 to 84), and half were male.
The overall response rate was 15.5%, with 1 patient achieving a complete response (CR) and 4 achieving a CR with incomplete count recovery (CRi).
The researchers noted that 3 of the patients who had a CR/CRi had IDH mutations. Two of these patients also achieved minimal residual disease negativity.
The median bone marrow blast count in evaluable patients decreased 36% after treatment, and 6 patients (19%) had at least a 50% reduction in bone marrow blasts.
Common adverse events following treatment (occurring in at least 25% of patients) included nausea, diarrhea, fatigue, neutropenia, and vomiting.
Grade 3/4 adverse events (occurring in 3 or more patients) included febrile neutropenia, anemia, and pneumonia. No patient died as a result of treatment-related adverse events.
The European Commission has granted orphan drug designation for the oral BCL-2 inhibitor venetoclax to treat acute myeloid leukemia (AML).
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the
European Union. The product must provide significant benefit to those affected by the condition.
Orphan drug designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is on the market.
Venetoclax is being developed by AbbVie in partnership with Genentech and Roche.
Phase 2 study
Results from a phase 2 study of venetoclax in AML were presented at ASH 2014. At that time, the trial had enrolled 32 patients, 30 of whom had relapsed or refractory disease. Patients had a median age of 71 (range, 19 to 84), and half were male.
The overall response rate was 15.5%, with 1 patient achieving a complete response (CR) and 4 achieving a CR with incomplete count recovery (CRi).
The researchers noted that 3 of the patients who had a CR/CRi had IDH mutations. Two of these patients also achieved minimal residual disease negativity.
The median bone marrow blast count in evaluable patients decreased 36% after treatment, and 6 patients (19%) had at least a 50% reduction in bone marrow blasts.
Common adverse events following treatment (occurring in at least 25% of patients) included nausea, diarrhea, fatigue, neutropenia, and vomiting.
Grade 3/4 adverse events (occurring in 3 or more patients) included febrile neutropenia, anemia, and pneumonia. No patient died as a result of treatment-related adverse events.
CHMP recommends fusion protein for hemophilia B

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that albutrepenonacog alfa (Idelvion) receive marketing authorization to treat patients with hemophilia B.
Albutrepenonacog alfa is a recombinant fusion protein linking coagulation factor IX with albumin.
The CHMP’s recommendation will be reviewed by the European Commission, which usually follows the CHMP’s advice.
In 2010, the European Commission granted albutrepenonacog alfa orphan designation as a treatment for hemophilia B.
Albutrepenonacog alfa is being developed by CSL Behring. The product is approved for use in Canada. Regulatory agencies in the US, Australia, Switzerland, and Japan are reviewing applications for the drug.
Phase 3 trial
The CHMP’s recommendation to approve albutrepenonacog alfa is based on the PROLONG-9FP clinical development program. PROLONG-9FP includes phase 1, 2, and 3 studies evaluating the safety and efficacy of albutrepenonacog alfa in adults and children (ages 1 to 61) with hemophilia B.
Data from the phase 3 study were recently published in Blood. This study included 63 previously treated male patients with severe hemophilia B. They had a mean age of 33 (range, 12 to 61).
The patients were divided into 2 groups. Group 1 (n=40) received routine prophylaxis with albutrepenonacog alfa once every 7 days for 26 weeks, followed by a 7-, 10- or 14-day prophylaxis regimen for a mean of 50, 38, or 51 weeks, respectively.
Group 2 received on-demand treatment with albutrepenonacog alfa for bleeding episodes for 26 weeks (n=23) and then switched to a 7-day prophylaxis regimen for a mean of 45 weeks (n=19).
The median annualized bleeding rate (ABR) was 2.0 in the prophylaxis arm (group 1) and 23.5 in the on-demand treatment arm (group 2). The median spontaneous ABRs were 0.0 and 17.0, respectively.
For patients in group 2, there was a significant reduction in median ABRs when patients switched from on-demand treatment to prophylaxis—19.22 and 1.58, respectively (P<0.0001). And there was a significant reduction in median spontaneous ABRs—15.43 and 0.00, respectively (P<0.0001).
Overall, 98.6% of bleeding episodes were treated successfully, including 93.6% that were treated with a single injection of albutrepenonacog alfa.
None of the patients developed inhibitors or experienced thromboembolic events, anaphylaxis, or life-threatening adverse events (AEs).
There were 347 treatment-emergent AEs reported in 54 (85.7%) patients. The most common were nasopharyngitis (25.4%), headache (23.8%), arthralgia (4.3%), and influenza (11.1%).
Eleven mild/moderate AEs in 5 patients (7.9%) were considered possibly related to albutrepenonacog alfa. Two patients discontinued treatment due to AEs—1 with hypersensitivity and 1 with headache.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that albutrepenonacog alfa (Idelvion) receive marketing authorization to treat patients with hemophilia B.
Albutrepenonacog alfa is a recombinant fusion protein linking coagulation factor IX with albumin.
The CHMP’s recommendation will be reviewed by the European Commission, which usually follows the CHMP’s advice.
In 2010, the European Commission granted albutrepenonacog alfa orphan designation as a treatment for hemophilia B.
Albutrepenonacog alfa is being developed by CSL Behring. The product is approved for use in Canada. Regulatory agencies in the US, Australia, Switzerland, and Japan are reviewing applications for the drug.
Phase 3 trial
The CHMP’s recommendation to approve albutrepenonacog alfa is based on the PROLONG-9FP clinical development program. PROLONG-9FP includes phase 1, 2, and 3 studies evaluating the safety and efficacy of albutrepenonacog alfa in adults and children (ages 1 to 61) with hemophilia B.
Data from the phase 3 study were recently published in Blood. This study included 63 previously treated male patients with severe hemophilia B. They had a mean age of 33 (range, 12 to 61).
The patients were divided into 2 groups. Group 1 (n=40) received routine prophylaxis with albutrepenonacog alfa once every 7 days for 26 weeks, followed by a 7-, 10- or 14-day prophylaxis regimen for a mean of 50, 38, or 51 weeks, respectively.
Group 2 received on-demand treatment with albutrepenonacog alfa for bleeding episodes for 26 weeks (n=23) and then switched to a 7-day prophylaxis regimen for a mean of 45 weeks (n=19).
The median annualized bleeding rate (ABR) was 2.0 in the prophylaxis arm (group 1) and 23.5 in the on-demand treatment arm (group 2). The median spontaneous ABRs were 0.0 and 17.0, respectively.
For patients in group 2, there was a significant reduction in median ABRs when patients switched from on-demand treatment to prophylaxis—19.22 and 1.58, respectively (P<0.0001). And there was a significant reduction in median spontaneous ABRs—15.43 and 0.00, respectively (P<0.0001).
Overall, 98.6% of bleeding episodes were treated successfully, including 93.6% that were treated with a single injection of albutrepenonacog alfa.
None of the patients developed inhibitors or experienced thromboembolic events, anaphylaxis, or life-threatening adverse events (AEs).
There were 347 treatment-emergent AEs reported in 54 (85.7%) patients. The most common were nasopharyngitis (25.4%), headache (23.8%), arthralgia (4.3%), and influenza (11.1%).
Eleven mild/moderate AEs in 5 patients (7.9%) were considered possibly related to albutrepenonacog alfa. Two patients discontinued treatment due to AEs—1 with hypersensitivity and 1 with headache.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that albutrepenonacog alfa (Idelvion) receive marketing authorization to treat patients with hemophilia B.
Albutrepenonacog alfa is a recombinant fusion protein linking coagulation factor IX with albumin.
The CHMP’s recommendation will be reviewed by the European Commission, which usually follows the CHMP’s advice.
In 2010, the European Commission granted albutrepenonacog alfa orphan designation as a treatment for hemophilia B.
Albutrepenonacog alfa is being developed by CSL Behring. The product is approved for use in Canada. Regulatory agencies in the US, Australia, Switzerland, and Japan are reviewing applications for the drug.
Phase 3 trial
The CHMP’s recommendation to approve albutrepenonacog alfa is based on the PROLONG-9FP clinical development program. PROLONG-9FP includes phase 1, 2, and 3 studies evaluating the safety and efficacy of albutrepenonacog alfa in adults and children (ages 1 to 61) with hemophilia B.
Data from the phase 3 study were recently published in Blood. This study included 63 previously treated male patients with severe hemophilia B. They had a mean age of 33 (range, 12 to 61).
The patients were divided into 2 groups. Group 1 (n=40) received routine prophylaxis with albutrepenonacog alfa once every 7 days for 26 weeks, followed by a 7-, 10- or 14-day prophylaxis regimen for a mean of 50, 38, or 51 weeks, respectively.
Group 2 received on-demand treatment with albutrepenonacog alfa for bleeding episodes for 26 weeks (n=23) and then switched to a 7-day prophylaxis regimen for a mean of 45 weeks (n=19).
The median annualized bleeding rate (ABR) was 2.0 in the prophylaxis arm (group 1) and 23.5 in the on-demand treatment arm (group 2). The median spontaneous ABRs were 0.0 and 17.0, respectively.
For patients in group 2, there was a significant reduction in median ABRs when patients switched from on-demand treatment to prophylaxis—19.22 and 1.58, respectively (P<0.0001). And there was a significant reduction in median spontaneous ABRs—15.43 and 0.00, respectively (P<0.0001).
Overall, 98.6% of bleeding episodes were treated successfully, including 93.6% that were treated with a single injection of albutrepenonacog alfa.
None of the patients developed inhibitors or experienced thromboembolic events, anaphylaxis, or life-threatening adverse events (AEs).
There were 347 treatment-emergent AEs reported in 54 (85.7%) patients. The most common were nasopharyngitis (25.4%), headache (23.8%), arthralgia (4.3%), and influenza (11.1%).
Eleven mild/moderate AEs in 5 patients (7.9%) were considered possibly related to albutrepenonacog alfa. Two patients discontinued treatment due to AEs—1 with hypersensitivity and 1 with headache.