User login
Disseminated Waxy Papules: A Sign of Systemic Disease
To the Editor:
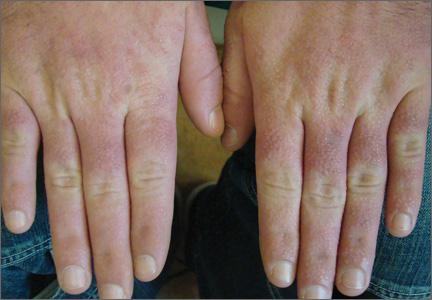
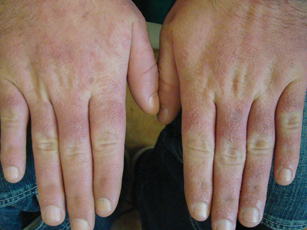
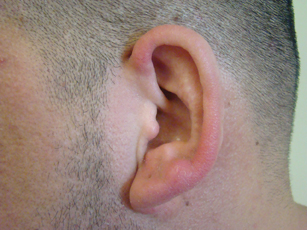
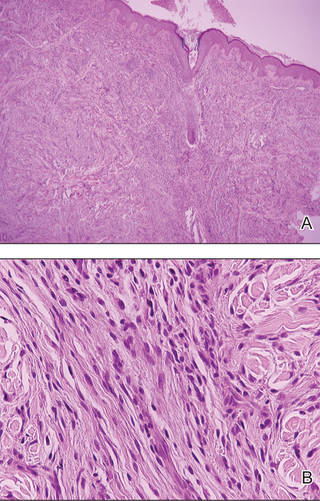
A 35-year-old man presented with asymptomatic multiple waxy firm papules on the dorsal aspect of the hands (Figure 1). On complete clinical examination, multiple similar lesions were found on the face and the lateral aspect of the neck; slightly pruritic, clustered, erythematous papules were seen in the pubic area. Deep longitudinal furrows on the glabella and swelling of the ears (Figure 2) also were present. Histology revealed a dermal infiltrate of fibroblasts (Figure 3). Abundant mucin deposition between collagen bundles stained positive with Alcian blue, confirming the diagnosis of generalized lichen myxedematosus (GLM). A thorough laboratory evaluation revealed λ light chain gammopathy. Bone marrow examination was normal. Thyroid function tests and radiography did not detect any abnormalities.
Being a musician, the patient traveled frequently and therefore did not present for follow-up. The disease ran its course. Approximately 6 months later he presented with a generalized eruption with weakness and intense bone pain. Progression to multiple myeloma was confirmed by serum protein electrophoresis and plain radiography, which revealed lytic lesions of the skull.
Generalized lichen myxedematosus, or scleromyxedema, is a form of papular mucinosis. It is a cutaneous mucinosis characterized by a generalized papular and sclerodermoid eruption, mucin deposition, increased fibroblast proliferation, fibrosis, and monoclonal gammopathy in the absence of thyroid disease. It is a rare disease that does not have a gender predilection and affects men and women aged 30 to 80 years.1,2
Clinically, GLM is characterized by a widespread symmetric eruption of small, closely spaced, waxy, firm, dome-shaped or flat-topped papules that measure 2 to 3 mm in diameter. Papules commonly form clusters on the face, neck, distal forearms, and hands. The affected skin may have a shiny sclerodermatous appearance.2
Histologically, there is a pronounced mucin deposition within the upper and mid reticular dermis, along with a proliferation of irregularly arranged fibroblasts and fibrosis.3 Collagen bundles appear pushed together into fascicles by the mucinous deposits.4
Generalized lichen myxedematosus should be distinguished from nephrogenic systemic fibrosis, granuloma annulare, amyloidosis, and Hansen disease. In nearly 83% of cases there is an accompanying paraproteinemia, most commonly an IgG λ light chain elevation.2 Monoclonal gammopathy of IgG with λ light chain has been considered as a fibroblast growth factor. Paraprotein levels do not correlate with the extent or progression of the disease.1 Nonparaprotein factors also can be responsible for excess fibroblast proliferation.2 It still is not elucidated as to how the abnormal mucin production results from the proliferation of fibroblasts.
Patients with GLM must be closely followed. The course of the disease is chronic and progressive. Extracutaneous manifestations may involve the gastrointestinal, musculoskeletal, cardiovascular, pulmonary, and central nervous systems, most likely because of mucin deposition in various organs.1,4 In these cases, the life expectancy is limited. Possibility of progression to multiple myeloma is less than 10%.1 Waldenström macroglobulinemia, Hodgkin lymphoma, and non-Hodgkin lymphomas all have been associated with GLM.2 Although extremely uncommon, spontaneous resolution has been reported after as long as a decade.5
Treatment of GLM, including melphalan, plasmapheresis, corticosteroids, cyclophosphamide, methotrexate, thalidomide, electron beam therapy, intravenous immunoglobulin therapy, extracorporeal photochemotherapy, high-dose chemotherapy with autologous stem cell rescue, and melphalan with autologous peripheral blood stem cell transplant are considered to improve the prognosis of this noncurable disease.6 As there is still no consensus on the treatment of scleromyxedema, multicenter studies on therapeutic schemes and responses are anticipated.
Generalized lichen myxedematosus is an instructive example of skin lesions being the presenting sign of a systemic disease. Progression to multiple myeloma in our patient made the prognosis less favorable.
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104.
- Cokonis Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Pomann JJ, Rudner EJ. Scleromyxedema revisited. Int J Dermatol. 2003;42:31-35.
- Dinneen AM, Dicken CH. Scleromyxedema. J Am Acad Dermatol. 1995;33:37-43.
- Heymann WR. Scleromyxedema. J Am Acad Dermatol. 2007;57:890-891.
To the Editor:
A 35-year-old man presented with asymptomatic multiple waxy firm papules on the dorsal aspect of the hands (Figure 1). On complete clinical examination, multiple similar lesions were found on the face and the lateral aspect of the neck; slightly pruritic, clustered, erythematous papules were seen in the pubic area. Deep longitudinal furrows on the glabella and swelling of the ears (Figure 2) also were present. Histology revealed a dermal infiltrate of fibroblasts (Figure 3). Abundant mucin deposition between collagen bundles stained positive with Alcian blue, confirming the diagnosis of generalized lichen myxedematosus (GLM). A thorough laboratory evaluation revealed λ light chain gammopathy. Bone marrow examination was normal. Thyroid function tests and radiography did not detect any abnormalities.
Being a musician, the patient traveled frequently and therefore did not present for follow-up. The disease ran its course. Approximately 6 months later he presented with a generalized eruption with weakness and intense bone pain. Progression to multiple myeloma was confirmed by serum protein electrophoresis and plain radiography, which revealed lytic lesions of the skull.
Generalized lichen myxedematosus, or scleromyxedema, is a form of papular mucinosis. It is a cutaneous mucinosis characterized by a generalized papular and sclerodermoid eruption, mucin deposition, increased fibroblast proliferation, fibrosis, and monoclonal gammopathy in the absence of thyroid disease. It is a rare disease that does not have a gender predilection and affects men and women aged 30 to 80 years.1,2
Clinically, GLM is characterized by a widespread symmetric eruption of small, closely spaced, waxy, firm, dome-shaped or flat-topped papules that measure 2 to 3 mm in diameter. Papules commonly form clusters on the face, neck, distal forearms, and hands. The affected skin may have a shiny sclerodermatous appearance.2
Histologically, there is a pronounced mucin deposition within the upper and mid reticular dermis, along with a proliferation of irregularly arranged fibroblasts and fibrosis.3 Collagen bundles appear pushed together into fascicles by the mucinous deposits.4
Generalized lichen myxedematosus should be distinguished from nephrogenic systemic fibrosis, granuloma annulare, amyloidosis, and Hansen disease. In nearly 83% of cases there is an accompanying paraproteinemia, most commonly an IgG λ light chain elevation.2 Monoclonal gammopathy of IgG with λ light chain has been considered as a fibroblast growth factor. Paraprotein levels do not correlate with the extent or progression of the disease.1 Nonparaprotein factors also can be responsible for excess fibroblast proliferation.2 It still is not elucidated as to how the abnormal mucin production results from the proliferation of fibroblasts.
Patients with GLM must be closely followed. The course of the disease is chronic and progressive. Extracutaneous manifestations may involve the gastrointestinal, musculoskeletal, cardiovascular, pulmonary, and central nervous systems, most likely because of mucin deposition in various organs.1,4 In these cases, the life expectancy is limited. Possibility of progression to multiple myeloma is less than 10%.1 Waldenström macroglobulinemia, Hodgkin lymphoma, and non-Hodgkin lymphomas all have been associated with GLM.2 Although extremely uncommon, spontaneous resolution has been reported after as long as a decade.5
Treatment of GLM, including melphalan, plasmapheresis, corticosteroids, cyclophosphamide, methotrexate, thalidomide, electron beam therapy, intravenous immunoglobulin therapy, extracorporeal photochemotherapy, high-dose chemotherapy with autologous stem cell rescue, and melphalan with autologous peripheral blood stem cell transplant are considered to improve the prognosis of this noncurable disease.6 As there is still no consensus on the treatment of scleromyxedema, multicenter studies on therapeutic schemes and responses are anticipated.
Generalized lichen myxedematosus is an instructive example of skin lesions being the presenting sign of a systemic disease. Progression to multiple myeloma in our patient made the prognosis less favorable.
To the Editor:
A 35-year-old man presented with asymptomatic multiple waxy firm papules on the dorsal aspect of the hands (Figure 1). On complete clinical examination, multiple similar lesions were found on the face and the lateral aspect of the neck; slightly pruritic, clustered, erythematous papules were seen in the pubic area. Deep longitudinal furrows on the glabella and swelling of the ears (Figure 2) also were present. Histology revealed a dermal infiltrate of fibroblasts (Figure 3). Abundant mucin deposition between collagen bundles stained positive with Alcian blue, confirming the diagnosis of generalized lichen myxedematosus (GLM). A thorough laboratory evaluation revealed λ light chain gammopathy. Bone marrow examination was normal. Thyroid function tests and radiography did not detect any abnormalities.
Being a musician, the patient traveled frequently and therefore did not present for follow-up. The disease ran its course. Approximately 6 months later he presented with a generalized eruption with weakness and intense bone pain. Progression to multiple myeloma was confirmed by serum protein electrophoresis and plain radiography, which revealed lytic lesions of the skull.
Generalized lichen myxedematosus, or scleromyxedema, is a form of papular mucinosis. It is a cutaneous mucinosis characterized by a generalized papular and sclerodermoid eruption, mucin deposition, increased fibroblast proliferation, fibrosis, and monoclonal gammopathy in the absence of thyroid disease. It is a rare disease that does not have a gender predilection and affects men and women aged 30 to 80 years.1,2
Clinically, GLM is characterized by a widespread symmetric eruption of small, closely spaced, waxy, firm, dome-shaped or flat-topped papules that measure 2 to 3 mm in diameter. Papules commonly form clusters on the face, neck, distal forearms, and hands. The affected skin may have a shiny sclerodermatous appearance.2
Histologically, there is a pronounced mucin deposition within the upper and mid reticular dermis, along with a proliferation of irregularly arranged fibroblasts and fibrosis.3 Collagen bundles appear pushed together into fascicles by the mucinous deposits.4
Generalized lichen myxedematosus should be distinguished from nephrogenic systemic fibrosis, granuloma annulare, amyloidosis, and Hansen disease. In nearly 83% of cases there is an accompanying paraproteinemia, most commonly an IgG λ light chain elevation.2 Monoclonal gammopathy of IgG with λ light chain has been considered as a fibroblast growth factor. Paraprotein levels do not correlate with the extent or progression of the disease.1 Nonparaprotein factors also can be responsible for excess fibroblast proliferation.2 It still is not elucidated as to how the abnormal mucin production results from the proliferation of fibroblasts.
Patients with GLM must be closely followed. The course of the disease is chronic and progressive. Extracutaneous manifestations may involve the gastrointestinal, musculoskeletal, cardiovascular, pulmonary, and central nervous systems, most likely because of mucin deposition in various organs.1,4 In these cases, the life expectancy is limited. Possibility of progression to multiple myeloma is less than 10%.1 Waldenström macroglobulinemia, Hodgkin lymphoma, and non-Hodgkin lymphomas all have been associated with GLM.2 Although extremely uncommon, spontaneous resolution has been reported after as long as a decade.5
Treatment of GLM, including melphalan, plasmapheresis, corticosteroids, cyclophosphamide, methotrexate, thalidomide, electron beam therapy, intravenous immunoglobulin therapy, extracorporeal photochemotherapy, high-dose chemotherapy with autologous stem cell rescue, and melphalan with autologous peripheral blood stem cell transplant are considered to improve the prognosis of this noncurable disease.6 As there is still no consensus on the treatment of scleromyxedema, multicenter studies on therapeutic schemes and responses are anticipated.
Generalized lichen myxedematosus is an instructive example of skin lesions being the presenting sign of a systemic disease. Progression to multiple myeloma in our patient made the prognosis less favorable.
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104.
- Cokonis Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Pomann JJ, Rudner EJ. Scleromyxedema revisited. Int J Dermatol. 2003;42:31-35.
- Dinneen AM, Dicken CH. Scleromyxedema. J Am Acad Dermatol. 1995;33:37-43.
- Heymann WR. Scleromyxedema. J Am Acad Dermatol. 2007;57:890-891.
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104.
- Cokonis Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Pomann JJ, Rudner EJ. Scleromyxedema revisited. Int J Dermatol. 2003;42:31-35.
- Dinneen AM, Dicken CH. Scleromyxedema. J Am Acad Dermatol. 1995;33:37-43.
- Heymann WR. Scleromyxedema. J Am Acad Dermatol. 2007;57:890-891.