User login
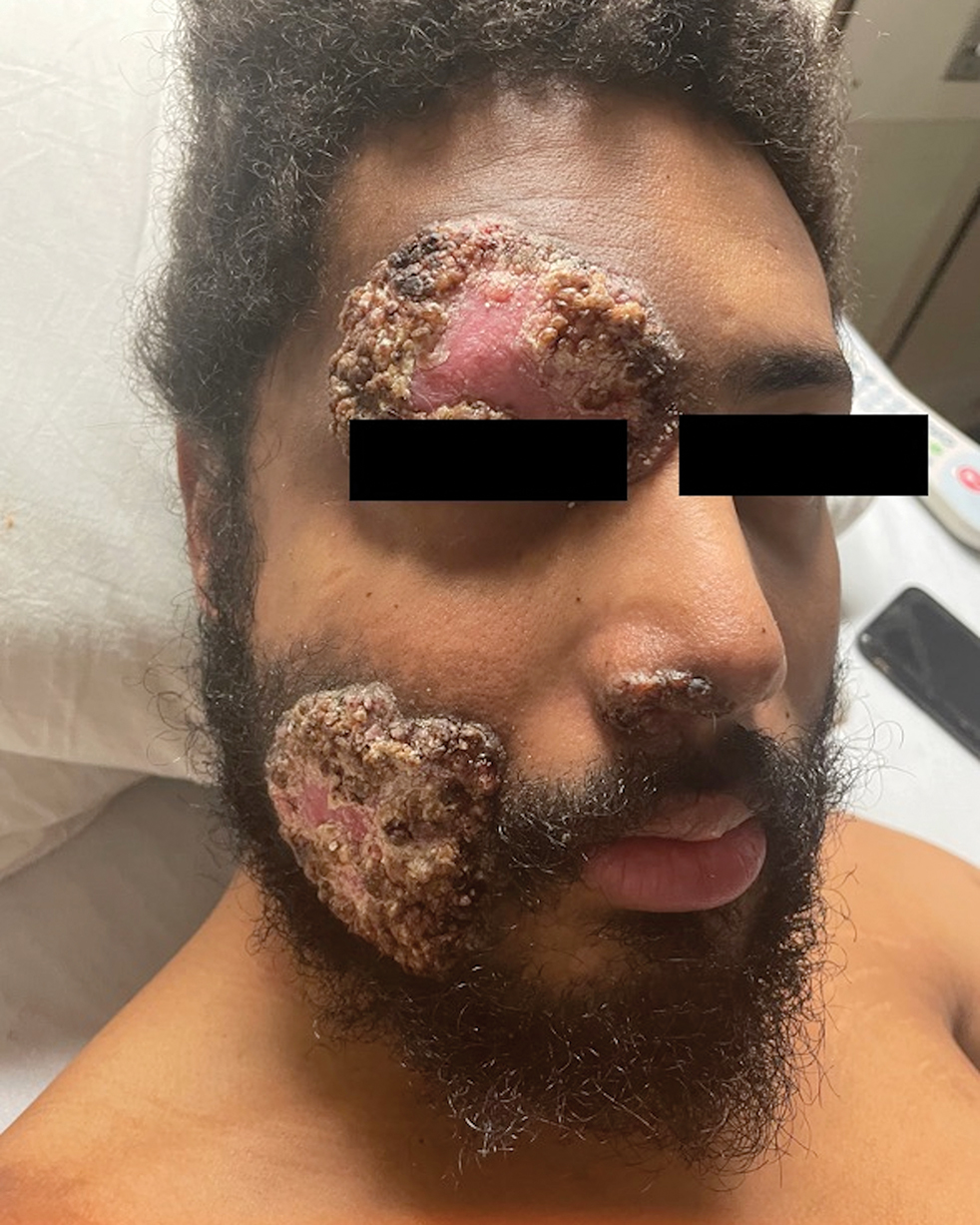
Multiple Fungating Plaques on the Face, Arms, and Legs
Multiple Fungating Plaques on the Face, Arms, and Legs
THE DIAGNOSIS: Mpox
Histologic examination demonstrated dense aggregates of necrotic cellular debris composed of karyorrhectic nuclear fragments intermixed with neutrophils, lymphocytes, and histiocytes. Eosinophilic intracytoplasmic inclusions also were observed (Figure 1). The bacterial, fungal, and mycobacterial histologic special stains and cultures were negative. Three weeks after the initial visit with dermatology, the patient was admitted to the hospital for worsening symptoms of fever, chills, and painful erythema surrounding the skin lesions. Serology and viral workup revealed a positive mpox polymerase chain reaction test, suggesting a diagnosis of mpox. Following the Centers for Disease Control and Prevention protocol, the patient was started on oral tecovirimat 200 mg twice daily for 3 weeks and intravenous infusions of cidofovir 345 mg once weekly for 2 weeks. After treatment was initiated, the skin lesions showed rapid improvement (Figure 2), and he was discharged from the hospital after finishing the second dose of cidofovir. Four months after the initial dermatology consultation, the lesions had resolved completely with residual scarring. At that time, the patient had full movement of the right eye.
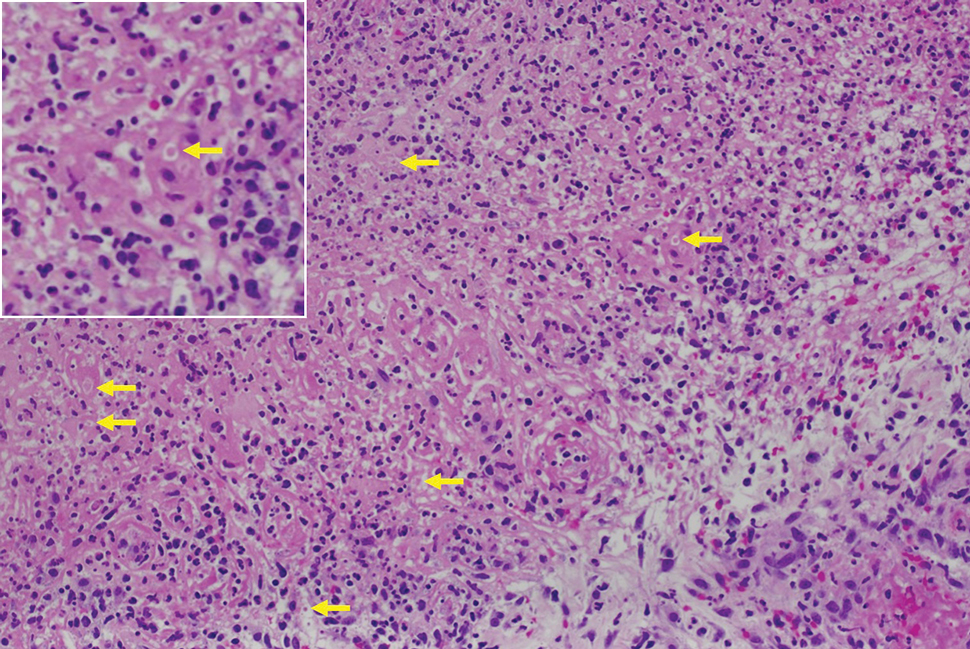
shows higher digital magnification of eosinophilic inclusions observed throughout the biopsy specimen (original magnification ×400).
Mpox virus is a member of the Poxviridae family of zoonotic viruses, which are transmitted from animals to humans. The mpox virus is brick-shaped (rectangular) and has a genome of linear double-stranded DNA encoding 180 proteins.1 Primates and rodents are the typical host reservoirs for viral circulation of mpox.2 Animal-to-human transmission occurs through direct contact with mucous membranes, bodily fluids, or tissues of an infected animal. Human-to-human transmission occurs through direct contact with infected mucous membranes, bodily fluids, respiratory droplets, and contaminated fomites.2
Symptoms typically occur within 1 week of exposure to the mpox virus. Prodromal symptoms of fever, sore throat, body aches, and headaches last for 3 days.1 Many patients experience a facial rash that spreads to the arms and legs over a period of 2 to 4 weeks. The rash initially manifests as small papules that progress to painful pustules and vesicles measuring 0.5 to 1.0 cm in diameter.3 The mpox virus is transmitted through these skin lesions until they crust over and re-epithelialize.1 The case fatality rate for mpox infection remains low (0.18%).4
Mpox outbreaks mainly were limited to central and western Africa prior to 2022. From May 17, 2022, through October 6, 2022, 26,384 cases of mpox were reported in the United States.5 During this outbreak, immunocompromised patients diagnosed with HIV and men who have sex with men were disproportionately affected.5
Due to the similarities between the smallpox virus and other orthopoxviruses, certain smallpox vaccines have been indicated for pre-exposure prophylaxis.6 The efficacy of prophylactic vaccination is believed to stem from the production of neutralizing antibodies that are cross-protective against other orthopoxviruses, including mpox.7 The 2 vaccines approved in the United States for mpox prophylaxis are JYNNEOS and ACAM2000, which are both live attenuated vaccines. Pre-exposure prophylaxis is indicated for patients at risk for severe disease, including men who have sex with men, individuals diagnosed with HIV or other immunosuppressive disorders, and individuals with recent diagnoses of one or more sexually transmitted diseases.8
Most mpox cases resolve within 2 to 4 weeks and only require supportive care (eg, nonsteroidal anti-inflammatory drugs, topical steroids, topical anesthetics) to treat pain.8 For patients at risk for severe disease, antiviral medications are warranted. Tecovirimat, brincidofovir, and cidofovir are antiviral medications used to treat smallpox that are thought to be effective against mpox.8,9 Tecovirimat and cidofovir have been shown to be effective against mpox in animal trials, but randomized or nonrandomized trials have not been performed in humans.9-11 Tecovirimat currently is available for the treatment of severe mpox in patients who meet the Centers for Disease Control and Prevention’s Investigational New Drug protocol; for these patients, a 200-mg course is administered orally or intravenously every 12 hours for 2 weeks.8
- Lu J, Xing H, Wang C, et al. Mpox (formerly monkeypox): pathogenesis, prevention, and treatment. Signal Transduct Target Ther. 2023;8:458. doi:10.1038/s41392-023-01675-
- Lim CK, Roberts J, Moso M, et al. Mpox diagnostics: review of current and emerging technologies. J Med Virol. 2023;95:e28429. doi:10.1002/jmv.28429
- Brown K, Leggat PA. Human monkeypox: current state of knowledge and implications for the future. Trop Med Infect Dis. 2016;1:8. doi:10.3390/tropicalmed1010008
- World Health Organization. Mpox (monkeypox) World Health Organization. Published April 18, 2023. Accessed May 28, 2025. https://www.who.int/news-room/fact-sheets/detail/monkeypox
- Kava CM, Rohraff DM, Wallace B, et al. Epidemiologic features of the monkeypox outbreak and the public health response—United States, May 17–October 6, 2022. 2022:1449-1456. https://www.cdc.gov/mmwr/volumes/71/wr/mm7145a4.htm?s_cid=mm7145a4_w
- Rizk JG, Lippi G, Henry BM, et al. Prevention and treatment of monkeypox. Drugs. 2022;82:957-963. doi:10.1007/s40265-022-01742-y
- Edghill-Smith Y, Golding H, Manischewitz J, et al. Smallpox vaccine-induced antibodies are necessary and sufficient for protection against monkeypox virus. Nat Med. 2005;11:740-747. doi:10.1038 /nm1261
- Centers for Disease Control and Prevention. Mpox treatment information for healthcare professionals. Updated June 18, 2024. Accessed May 28, 2025. https://www.cdc.gov/mpox/hcp/clinical-care/?CDC_AAref_Val=https://www.cdc.gov/poxvirus/mpox/clinicians/treatment.html
- Mitja O, Ogoina D, Titanji BK, et al. Monkeypox. Lancet. 2023;401:60-74. doi:10.1016/S0140-6736(22)02075-X
- Huggins J, Goff A, Hensley L, et al. Nonhuman primates are protected from smallpox virus or monkeypox virus challenges by the antiviral drug ST-246. Antimicrob Agents Chemother. 2009;53:2620-2625. doi:10.1128/aac.00021-09
- Grosenbach DW, Honeychurch K, Rose EA, et al. Oral tecovirimat for the treatment of smallpox. N Engl J Med. 2018;379:44-53. doi:10.1056 /nejmoa1705688
THE DIAGNOSIS: Mpox
Histologic examination demonstrated dense aggregates of necrotic cellular debris composed of karyorrhectic nuclear fragments intermixed with neutrophils, lymphocytes, and histiocytes. Eosinophilic intracytoplasmic inclusions also were observed (Figure 1). The bacterial, fungal, and mycobacterial histologic special stains and cultures were negative. Three weeks after the initial visit with dermatology, the patient was admitted to the hospital for worsening symptoms of fever, chills, and painful erythema surrounding the skin lesions. Serology and viral workup revealed a positive mpox polymerase chain reaction test, suggesting a diagnosis of mpox. Following the Centers for Disease Control and Prevention protocol, the patient was started on oral tecovirimat 200 mg twice daily for 3 weeks and intravenous infusions of cidofovir 345 mg once weekly for 2 weeks. After treatment was initiated, the skin lesions showed rapid improvement (Figure 2), and he was discharged from the hospital after finishing the second dose of cidofovir. Four months after the initial dermatology consultation, the lesions had resolved completely with residual scarring. At that time, the patient had full movement of the right eye.
shows higher digital magnification of eosinophilic inclusions observed throughout the biopsy specimen (original magnification ×400).
Mpox virus is a member of the Poxviridae family of zoonotic viruses, which are transmitted from animals to humans. The mpox virus is brick-shaped (rectangular) and has a genome of linear double-stranded DNA encoding 180 proteins.1 Primates and rodents are the typical host reservoirs for viral circulation of mpox.2 Animal-to-human transmission occurs through direct contact with mucous membranes, bodily fluids, or tissues of an infected animal. Human-to-human transmission occurs through direct contact with infected mucous membranes, bodily fluids, respiratory droplets, and contaminated fomites.2
Symptoms typically occur within 1 week of exposure to the mpox virus. Prodromal symptoms of fever, sore throat, body aches, and headaches last for 3 days.1 Many patients experience a facial rash that spreads to the arms and legs over a period of 2 to 4 weeks. The rash initially manifests as small papules that progress to painful pustules and vesicles measuring 0.5 to 1.0 cm in diameter.3 The mpox virus is transmitted through these skin lesions until they crust over and re-epithelialize.1 The case fatality rate for mpox infection remains low (0.18%).4
Mpox outbreaks mainly were limited to central and western Africa prior to 2022. From May 17, 2022, through October 6, 2022, 26,384 cases of mpox were reported in the United States.5 During this outbreak, immunocompromised patients diagnosed with HIV and men who have sex with men were disproportionately affected.5
Due to the similarities between the smallpox virus and other orthopoxviruses, certain smallpox vaccines have been indicated for pre-exposure prophylaxis.6 The efficacy of prophylactic vaccination is believed to stem from the production of neutralizing antibodies that are cross-protective against other orthopoxviruses, including mpox.7 The 2 vaccines approved in the United States for mpox prophylaxis are JYNNEOS and ACAM2000, which are both live attenuated vaccines. Pre-exposure prophylaxis is indicated for patients at risk for severe disease, including men who have sex with men, individuals diagnosed with HIV or other immunosuppressive disorders, and individuals with recent diagnoses of one or more sexually transmitted diseases.8
Most mpox cases resolve within 2 to 4 weeks and only require supportive care (eg, nonsteroidal anti-inflammatory drugs, topical steroids, topical anesthetics) to treat pain.8 For patients at risk for severe disease, antiviral medications are warranted. Tecovirimat, brincidofovir, and cidofovir are antiviral medications used to treat smallpox that are thought to be effective against mpox.8,9 Tecovirimat and cidofovir have been shown to be effective against mpox in animal trials, but randomized or nonrandomized trials have not been performed in humans.9-11 Tecovirimat currently is available for the treatment of severe mpox in patients who meet the Centers for Disease Control and Prevention’s Investigational New Drug protocol; for these patients, a 200-mg course is administered orally or intravenously every 12 hours for 2 weeks.8
THE DIAGNOSIS: Mpox
Histologic examination demonstrated dense aggregates of necrotic cellular debris composed of karyorrhectic nuclear fragments intermixed with neutrophils, lymphocytes, and histiocytes. Eosinophilic intracytoplasmic inclusions also were observed (Figure 1). The bacterial, fungal, and mycobacterial histologic special stains and cultures were negative. Three weeks after the initial visit with dermatology, the patient was admitted to the hospital for worsening symptoms of fever, chills, and painful erythema surrounding the skin lesions. Serology and viral workup revealed a positive mpox polymerase chain reaction test, suggesting a diagnosis of mpox. Following the Centers for Disease Control and Prevention protocol, the patient was started on oral tecovirimat 200 mg twice daily for 3 weeks and intravenous infusions of cidofovir 345 mg once weekly for 2 weeks. After treatment was initiated, the skin lesions showed rapid improvement (Figure 2), and he was discharged from the hospital after finishing the second dose of cidofovir. Four months after the initial dermatology consultation, the lesions had resolved completely with residual scarring. At that time, the patient had full movement of the right eye.
shows higher digital magnification of eosinophilic inclusions observed throughout the biopsy specimen (original magnification ×400).
Mpox virus is a member of the Poxviridae family of zoonotic viruses, which are transmitted from animals to humans. The mpox virus is brick-shaped (rectangular) and has a genome of linear double-stranded DNA encoding 180 proteins.1 Primates and rodents are the typical host reservoirs for viral circulation of mpox.2 Animal-to-human transmission occurs through direct contact with mucous membranes, bodily fluids, or tissues of an infected animal. Human-to-human transmission occurs through direct contact with infected mucous membranes, bodily fluids, respiratory droplets, and contaminated fomites.2
Symptoms typically occur within 1 week of exposure to the mpox virus. Prodromal symptoms of fever, sore throat, body aches, and headaches last for 3 days.1 Many patients experience a facial rash that spreads to the arms and legs over a period of 2 to 4 weeks. The rash initially manifests as small papules that progress to painful pustules and vesicles measuring 0.5 to 1.0 cm in diameter.3 The mpox virus is transmitted through these skin lesions until they crust over and re-epithelialize.1 The case fatality rate for mpox infection remains low (0.18%).4
Mpox outbreaks mainly were limited to central and western Africa prior to 2022. From May 17, 2022, through October 6, 2022, 26,384 cases of mpox were reported in the United States.5 During this outbreak, immunocompromised patients diagnosed with HIV and men who have sex with men were disproportionately affected.5
Due to the similarities between the smallpox virus and other orthopoxviruses, certain smallpox vaccines have been indicated for pre-exposure prophylaxis.6 The efficacy of prophylactic vaccination is believed to stem from the production of neutralizing antibodies that are cross-protective against other orthopoxviruses, including mpox.7 The 2 vaccines approved in the United States for mpox prophylaxis are JYNNEOS and ACAM2000, which are both live attenuated vaccines. Pre-exposure prophylaxis is indicated for patients at risk for severe disease, including men who have sex with men, individuals diagnosed with HIV or other immunosuppressive disorders, and individuals with recent diagnoses of one or more sexually transmitted diseases.8
Most mpox cases resolve within 2 to 4 weeks and only require supportive care (eg, nonsteroidal anti-inflammatory drugs, topical steroids, topical anesthetics) to treat pain.8 For patients at risk for severe disease, antiviral medications are warranted. Tecovirimat, brincidofovir, and cidofovir are antiviral medications used to treat smallpox that are thought to be effective against mpox.8,9 Tecovirimat and cidofovir have been shown to be effective against mpox in animal trials, but randomized or nonrandomized trials have not been performed in humans.9-11 Tecovirimat currently is available for the treatment of severe mpox in patients who meet the Centers for Disease Control and Prevention’s Investigational New Drug protocol; for these patients, a 200-mg course is administered orally or intravenously every 12 hours for 2 weeks.8
- Lu J, Xing H, Wang C, et al. Mpox (formerly monkeypox): pathogenesis, prevention, and treatment. Signal Transduct Target Ther. 2023;8:458. doi:10.1038/s41392-023-01675-
- Lim CK, Roberts J, Moso M, et al. Mpox diagnostics: review of current and emerging technologies. J Med Virol. 2023;95:e28429. doi:10.1002/jmv.28429
- Brown K, Leggat PA. Human monkeypox: current state of knowledge and implications for the future. Trop Med Infect Dis. 2016;1:8. doi:10.3390/tropicalmed1010008
- World Health Organization. Mpox (monkeypox) World Health Organization. Published April 18, 2023. Accessed May 28, 2025. https://www.who.int/news-room/fact-sheets/detail/monkeypox
- Kava CM, Rohraff DM, Wallace B, et al. Epidemiologic features of the monkeypox outbreak and the public health response—United States, May 17–October 6, 2022. 2022:1449-1456. https://www.cdc.gov/mmwr/volumes/71/wr/mm7145a4.htm?s_cid=mm7145a4_w
- Rizk JG, Lippi G, Henry BM, et al. Prevention and treatment of monkeypox. Drugs. 2022;82:957-963. doi:10.1007/s40265-022-01742-y
- Edghill-Smith Y, Golding H, Manischewitz J, et al. Smallpox vaccine-induced antibodies are necessary and sufficient for protection against monkeypox virus. Nat Med. 2005;11:740-747. doi:10.1038 /nm1261
- Centers for Disease Control and Prevention. Mpox treatment information for healthcare professionals. Updated June 18, 2024. Accessed May 28, 2025. https://www.cdc.gov/mpox/hcp/clinical-care/?CDC_AAref_Val=https://www.cdc.gov/poxvirus/mpox/clinicians/treatment.html
- Mitja O, Ogoina D, Titanji BK, et al. Monkeypox. Lancet. 2023;401:60-74. doi:10.1016/S0140-6736(22)02075-X
- Huggins J, Goff A, Hensley L, et al. Nonhuman primates are protected from smallpox virus or monkeypox virus challenges by the antiviral drug ST-246. Antimicrob Agents Chemother. 2009;53:2620-2625. doi:10.1128/aac.00021-09
- Grosenbach DW, Honeychurch K, Rose EA, et al. Oral tecovirimat for the treatment of smallpox. N Engl J Med. 2018;379:44-53. doi:10.1056 /nejmoa1705688
- Lu J, Xing H, Wang C, et al. Mpox (formerly monkeypox): pathogenesis, prevention, and treatment. Signal Transduct Target Ther. 2023;8:458. doi:10.1038/s41392-023-01675-
- Lim CK, Roberts J, Moso M, et al. Mpox diagnostics: review of current and emerging technologies. J Med Virol. 2023;95:e28429. doi:10.1002/jmv.28429
- Brown K, Leggat PA. Human monkeypox: current state of knowledge and implications for the future. Trop Med Infect Dis. 2016;1:8. doi:10.3390/tropicalmed1010008
- World Health Organization. Mpox (monkeypox) World Health Organization. Published April 18, 2023. Accessed May 28, 2025. https://www.who.int/news-room/fact-sheets/detail/monkeypox
- Kava CM, Rohraff DM, Wallace B, et al. Epidemiologic features of the monkeypox outbreak and the public health response—United States, May 17–October 6, 2022. 2022:1449-1456. https://www.cdc.gov/mmwr/volumes/71/wr/mm7145a4.htm?s_cid=mm7145a4_w
- Rizk JG, Lippi G, Henry BM, et al. Prevention and treatment of monkeypox. Drugs. 2022;82:957-963. doi:10.1007/s40265-022-01742-y
- Edghill-Smith Y, Golding H, Manischewitz J, et al. Smallpox vaccine-induced antibodies are necessary and sufficient for protection against monkeypox virus. Nat Med. 2005;11:740-747. doi:10.1038 /nm1261
- Centers for Disease Control and Prevention. Mpox treatment information for healthcare professionals. Updated June 18, 2024. Accessed May 28, 2025. https://www.cdc.gov/mpox/hcp/clinical-care/?CDC_AAref_Val=https://www.cdc.gov/poxvirus/mpox/clinicians/treatment.html
- Mitja O, Ogoina D, Titanji BK, et al. Monkeypox. Lancet. 2023;401:60-74. doi:10.1016/S0140-6736(22)02075-X
- Huggins J, Goff A, Hensley L, et al. Nonhuman primates are protected from smallpox virus or monkeypox virus challenges by the antiviral drug ST-246. Antimicrob Agents Chemother. 2009;53:2620-2625. doi:10.1128/aac.00021-09
- Grosenbach DW, Honeychurch K, Rose EA, et al. Oral tecovirimat for the treatment of smallpox. N Engl J Med. 2018;379:44-53. doi:10.1056 /nejmoa1705688
Multiple Fungating Plaques on the Face, Arms, and Legs
Multiple Fungating Plaques on the Face, Arms, and Legs
A 27-year-old man presented to his primary care physician after he was struck in the head by a tree branch while working outside. The next day, ulcerating lesions emerged on the right supraorbital ridge, along with subjective fevers, chills, fatigue, and shortness of breath. The patient reported a history of unprotected sexual intercourse with a male partner who was HIV positive. His medical history included syphilis status posttreatment with a course of 5 penicillin injections, hepatitis C, and HIV diagnosed one month prior to presentation (CD4 count, 169 cells/mm3 [reference range, 500-1500 cells/mm3]). A punch biopsy performed by the primary care physician revealed suppurative granulomatous inflammation, and the patient was prescribed antibiotics with mild improvement. He then was referred to dermatology for further evaluation of the ulcerating lesions.
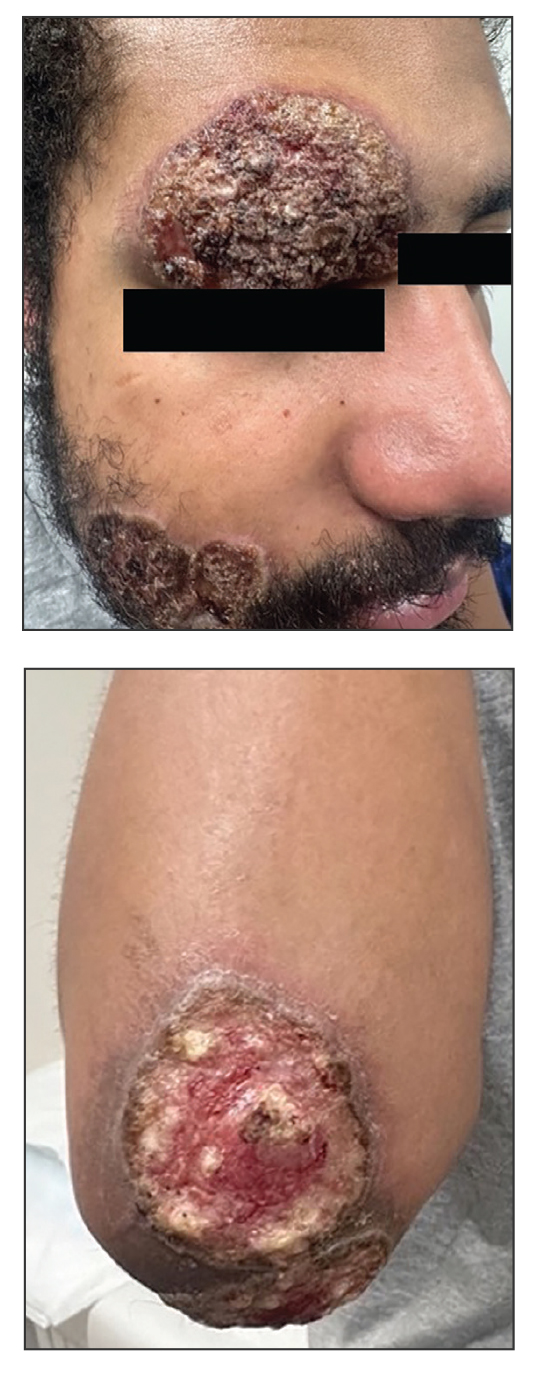
Three months after the initial trauma, the patient presented to the dermatology clinic for evaluation of multiple large fungating plaques affecting multiple sites on the face (top), arms (bottom), and legs. Physical examination revealed large circinate verrucous plaques involving the right supraorbital ridge and eyelid. The patient was unable to fully open the right eye. Similar plaques also were observed on the right malar cheek, arms, and feet. Four 5-mm punch biopsies from lesions on the right elbow and left ankle were obtained with fungal and bacterial cultures.

Hyperpigmented Flexural Plaques, Hypohidrosis, and Hypotrichosis
The Diagnosis: Lelis Syndrome
Histopathology revealed spongiotic dermatitis with marked acanthosis and hyperkeratosis (Figure, A) with fungal colonization of the stratum corneum (Figure, B). Our patient was diagnosed with Lelis syndrome (also referred to as ectodermal dysplasia with acanthosis nigricans syndrome), a rare condition with hypotrichosis and hypohidrosis resulting from ectodermal dysplasia.1,2 The pruritic rash was diagnosed as chronic dermatitis due to fungal colonization in the setting of acanthosis nigricans. The fungal infection was treated with a 4-week course of oral fluconazole 200 mg/wk, ketoconazole cream 2% twice daily, and discontinuation of topical steroids, resulting in the thinning of the plaques on the neck and antecubital fossae as well as resolution of the pruritus. Following antifungal treatment, our patient was started on tazarotene cream 0.1% for acanthosis nigricans.
. B, Grocott-Gomori methenamine-silver staining showed numerous fungal elements")
Ectodermal dysplasias are inherited disorders with abnormalities of the skin, hair, sweat glands, nails, teeth, and sometimes internal organs.3 Patients with Lelis syndrome may have other manifestations of ectodermal dysplasia in addition to hypohidrosis and hypotrichosis, including deafness and abnormal dentition,1,3 as seen in our patient. Intellectual disability has been described in many types of ectodermal dysplasia, including Lelis syndrome, but the association may be obscured by neurologic damage after repeat episodes of hyperthermia in infancy due to anhidrosis or hypohidrosis.4
When evaluating the differential diagnoses, the presence of hypotrichosis and hypohidrosis indicating ectodermal dysplasia is key. Confluent and reticulated papillomatosis presents with hyperkeratosis, papillomatosis, and focal acanthosis on histopathology. It can present on the neck and antecubital fossae; however, it is not associated with hypohidrosis and hypotrichosis.5 Although activating fibroblast growth factor receptor, FGFR, mutations have been implicated in the development of acanthosis nigricans in a variety of syndromes, these diagnoses are associated with abnormalities in skeletal development such as craniosynostosis and short stature; hypotrichosis and hypohidrosis are not seen.6,7 HAIR-AN (hyperandrogenism, insulin resistance, and acanthosis nigricans) syndrome typically presents in the prepubertal period with obesity and insulin resistance; acanthosis nigricans and alopecia can occur due to insulin resistance and hyperandrogenism, but concurrent clitoromegaly and hirsutism are common.6 Sudden onset of extensive acanthosis nigricans also is among the paraneoplastic dermatoses; it has been associated with multiple malignancies, but in these cases, hypotrichosis and hypohidrosis are not observed. Adenocarcinomas are the most common neoplasms associated with paraneoplastic acanthosis nigricans, which occurs through growth factor secretion by tumor cells stimulating hyperkeratosis and papillomatosis.6
Lelis syndrome is rare, and our case is unique because the patient had severe manifestations of acanthosis nigricans and hypotrichosis. Because the inheritance pattern and specific genetics of the condition have not been fully elucidated, the diagnosis primarily is clinical.1,8 Diagnosis may be complicated by the variety of other signs that can accompany acanthosis nigricans, hypohidrosis, and hypotrichosis.1,2 The condition also may alter or obscure presentation of other dermatologic conditions, as in our case.
Although there is no cure for Lelis syndrome, one case report described treatment with acitretin that resulted in marked improvement of the patient’s hyperkeratosis and acanthosis nigricans.9 Due to lack of health insurance coverage of acitretin, our patient was started on tazarotene cream 0.1% for acanthosis nigricans. General treatment of ectodermal dysplasia primarily consists of multidisciplinary symptom management, including careful monitoring of temperature and heat intolerance as well as provision of dental prosthetics.4,10 For ectodermal dysplasias caused by identified genetic mutations, prenatal interventions targeting gene pathways offer potentially curative treatment.10 However, for Lelis syndrome, along with many other disorders of ectodermal dysplasia, mitigation of signs and symptoms remains the primary treatment objective. Despite its rarity, increased awareness of Lelis syndrome is important to increase knowledge of ectodermal dysplasia syndromes and allow for the investigation of potential treatment options.
- Steiner CE, Cintra ML, Marques-de-Faria AP. Ectodermal dysplasia with acanthosis nigricans (Lelis syndrome). Am J Med Genet. 2002;113:381-384. doi:10.1002/ajmg.b.10787
- Lelis J. Autosomal recessive ectodermal dysplasia. Cutis. 1992; 49:435-437.
- Itin PH, Fistarol SK. Ectodermal dysplasias. Am J Med Genet C Semin Med Genet. 2004;131C:45-51. doi:10.1002/ajmg.c.30033
- Blüschke G, Nüsken KD, Schneider H. Prevalence and prevention of severe complications of hypohidrotic ectodermal dysplasia in infancy. Early Hum Dev. 2010;86:397-399. doi:10.1016/j .earlhumdev.2010.04.008
- Le C, Bedocs PM. Confluent and reticulated papillomatosis. StatPearls. StatPearls Publishing; 2022. http://www.ncbi.nlm.nih.gov/books/NBK459130/
- Das A, Datta D, Kassir M, et al. Acanthosis nigricans: a review. J Cosmet Dermatol. 2020;19:1857-1865. doi:10.1111/jocd.13544
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101. doi:10 .1046/j.1365-2133.2002.05150.x
- van Steensel MAM, van der Hout AH. Lelis syndrome may be a manifestation of hypohidrotic ectodermal dysplasia. Am J Med Genet A. 2009;149A:1612-1613. doi:10.1002/ajmg.a.32945
- Yoshimura AM, Neves Ferreira Velho PE, Ferreira Magalhães R, et al. Lelis’ syndrome: treatment with acitretin. Int J Dermatol. 2008;47: 1330-1331. doi:10.1111/j.1365-4632.2008.03874.x
- Schneider H. Ectodermal dysplasias: new perspectives on the treatment of so far immedicable genetic disorders. Front Genet. 2022;13:1000744. doi:10.3389/fgene.2022.1000744
The Diagnosis: Lelis Syndrome
Histopathology revealed spongiotic dermatitis with marked acanthosis and hyperkeratosis (Figure, A) with fungal colonization of the stratum corneum (Figure, B). Our patient was diagnosed with Lelis syndrome (also referred to as ectodermal dysplasia with acanthosis nigricans syndrome), a rare condition with hypotrichosis and hypohidrosis resulting from ectodermal dysplasia.1,2 The pruritic rash was diagnosed as chronic dermatitis due to fungal colonization in the setting of acanthosis nigricans. The fungal infection was treated with a 4-week course of oral fluconazole 200 mg/wk, ketoconazole cream 2% twice daily, and discontinuation of topical steroids, resulting in the thinning of the plaques on the neck and antecubital fossae as well as resolution of the pruritus. Following antifungal treatment, our patient was started on tazarotene cream 0.1% for acanthosis nigricans.
Ectodermal dysplasias are inherited disorders with abnormalities of the skin, hair, sweat glands, nails, teeth, and sometimes internal organs.3 Patients with Lelis syndrome may have other manifestations of ectodermal dysplasia in addition to hypohidrosis and hypotrichosis, including deafness and abnormal dentition,1,3 as seen in our patient. Intellectual disability has been described in many types of ectodermal dysplasia, including Lelis syndrome, but the association may be obscured by neurologic damage after repeat episodes of hyperthermia in infancy due to anhidrosis or hypohidrosis.4
When evaluating the differential diagnoses, the presence of hypotrichosis and hypohidrosis indicating ectodermal dysplasia is key. Confluent and reticulated papillomatosis presents with hyperkeratosis, papillomatosis, and focal acanthosis on histopathology. It can present on the neck and antecubital fossae; however, it is not associated with hypohidrosis and hypotrichosis.5 Although activating fibroblast growth factor receptor, FGFR, mutations have been implicated in the development of acanthosis nigricans in a variety of syndromes, these diagnoses are associated with abnormalities in skeletal development such as craniosynostosis and short stature; hypotrichosis and hypohidrosis are not seen.6,7 HAIR-AN (hyperandrogenism, insulin resistance, and acanthosis nigricans) syndrome typically presents in the prepubertal period with obesity and insulin resistance; acanthosis nigricans and alopecia can occur due to insulin resistance and hyperandrogenism, but concurrent clitoromegaly and hirsutism are common.6 Sudden onset of extensive acanthosis nigricans also is among the paraneoplastic dermatoses; it has been associated with multiple malignancies, but in these cases, hypotrichosis and hypohidrosis are not observed. Adenocarcinomas are the most common neoplasms associated with paraneoplastic acanthosis nigricans, which occurs through growth factor secretion by tumor cells stimulating hyperkeratosis and papillomatosis.6
Lelis syndrome is rare, and our case is unique because the patient had severe manifestations of acanthosis nigricans and hypotrichosis. Because the inheritance pattern and specific genetics of the condition have not been fully elucidated, the diagnosis primarily is clinical.1,8 Diagnosis may be complicated by the variety of other signs that can accompany acanthosis nigricans, hypohidrosis, and hypotrichosis.1,2 The condition also may alter or obscure presentation of other dermatologic conditions, as in our case.
Although there is no cure for Lelis syndrome, one case report described treatment with acitretin that resulted in marked improvement of the patient’s hyperkeratosis and acanthosis nigricans.9 Due to lack of health insurance coverage of acitretin, our patient was started on tazarotene cream 0.1% for acanthosis nigricans. General treatment of ectodermal dysplasia primarily consists of multidisciplinary symptom management, including careful monitoring of temperature and heat intolerance as well as provision of dental prosthetics.4,10 For ectodermal dysplasias caused by identified genetic mutations, prenatal interventions targeting gene pathways offer potentially curative treatment.10 However, for Lelis syndrome, along with many other disorders of ectodermal dysplasia, mitigation of signs and symptoms remains the primary treatment objective. Despite its rarity, increased awareness of Lelis syndrome is important to increase knowledge of ectodermal dysplasia syndromes and allow for the investigation of potential treatment options.
The Diagnosis: Lelis Syndrome
Histopathology revealed spongiotic dermatitis with marked acanthosis and hyperkeratosis (Figure, A) with fungal colonization of the stratum corneum (Figure, B). Our patient was diagnosed with Lelis syndrome (also referred to as ectodermal dysplasia with acanthosis nigricans syndrome), a rare condition with hypotrichosis and hypohidrosis resulting from ectodermal dysplasia.1,2 The pruritic rash was diagnosed as chronic dermatitis due to fungal colonization in the setting of acanthosis nigricans. The fungal infection was treated with a 4-week course of oral fluconazole 200 mg/wk, ketoconazole cream 2% twice daily, and discontinuation of topical steroids, resulting in the thinning of the plaques on the neck and antecubital fossae as well as resolution of the pruritus. Following antifungal treatment, our patient was started on tazarotene cream 0.1% for acanthosis nigricans.
Ectodermal dysplasias are inherited disorders with abnormalities of the skin, hair, sweat glands, nails, teeth, and sometimes internal organs.3 Patients with Lelis syndrome may have other manifestations of ectodermal dysplasia in addition to hypohidrosis and hypotrichosis, including deafness and abnormal dentition,1,3 as seen in our patient. Intellectual disability has been described in many types of ectodermal dysplasia, including Lelis syndrome, but the association may be obscured by neurologic damage after repeat episodes of hyperthermia in infancy due to anhidrosis or hypohidrosis.4
When evaluating the differential diagnoses, the presence of hypotrichosis and hypohidrosis indicating ectodermal dysplasia is key. Confluent and reticulated papillomatosis presents with hyperkeratosis, papillomatosis, and focal acanthosis on histopathology. It can present on the neck and antecubital fossae; however, it is not associated with hypohidrosis and hypotrichosis.5 Although activating fibroblast growth factor receptor, FGFR, mutations have been implicated in the development of acanthosis nigricans in a variety of syndromes, these diagnoses are associated with abnormalities in skeletal development such as craniosynostosis and short stature; hypotrichosis and hypohidrosis are not seen.6,7 HAIR-AN (hyperandrogenism, insulin resistance, and acanthosis nigricans) syndrome typically presents in the prepubertal period with obesity and insulin resistance; acanthosis nigricans and alopecia can occur due to insulin resistance and hyperandrogenism, but concurrent clitoromegaly and hirsutism are common.6 Sudden onset of extensive acanthosis nigricans also is among the paraneoplastic dermatoses; it has been associated with multiple malignancies, but in these cases, hypotrichosis and hypohidrosis are not observed. Adenocarcinomas are the most common neoplasms associated with paraneoplastic acanthosis nigricans, which occurs through growth factor secretion by tumor cells stimulating hyperkeratosis and papillomatosis.6
Lelis syndrome is rare, and our case is unique because the patient had severe manifestations of acanthosis nigricans and hypotrichosis. Because the inheritance pattern and specific genetics of the condition have not been fully elucidated, the diagnosis primarily is clinical.1,8 Diagnosis may be complicated by the variety of other signs that can accompany acanthosis nigricans, hypohidrosis, and hypotrichosis.1,2 The condition also may alter or obscure presentation of other dermatologic conditions, as in our case.
Although there is no cure for Lelis syndrome, one case report described treatment with acitretin that resulted in marked improvement of the patient’s hyperkeratosis and acanthosis nigricans.9 Due to lack of health insurance coverage of acitretin, our patient was started on tazarotene cream 0.1% for acanthosis nigricans. General treatment of ectodermal dysplasia primarily consists of multidisciplinary symptom management, including careful monitoring of temperature and heat intolerance as well as provision of dental prosthetics.4,10 For ectodermal dysplasias caused by identified genetic mutations, prenatal interventions targeting gene pathways offer potentially curative treatment.10 However, for Lelis syndrome, along with many other disorders of ectodermal dysplasia, mitigation of signs and symptoms remains the primary treatment objective. Despite its rarity, increased awareness of Lelis syndrome is important to increase knowledge of ectodermal dysplasia syndromes and allow for the investigation of potential treatment options.
- Steiner CE, Cintra ML, Marques-de-Faria AP. Ectodermal dysplasia with acanthosis nigricans (Lelis syndrome). Am J Med Genet. 2002;113:381-384. doi:10.1002/ajmg.b.10787
- Lelis J. Autosomal recessive ectodermal dysplasia. Cutis. 1992; 49:435-437.
- Itin PH, Fistarol SK. Ectodermal dysplasias. Am J Med Genet C Semin Med Genet. 2004;131C:45-51. doi:10.1002/ajmg.c.30033
- Blüschke G, Nüsken KD, Schneider H. Prevalence and prevention of severe complications of hypohidrotic ectodermal dysplasia in infancy. Early Hum Dev. 2010;86:397-399. doi:10.1016/j .earlhumdev.2010.04.008
- Le C, Bedocs PM. Confluent and reticulated papillomatosis. StatPearls. StatPearls Publishing; 2022. http://www.ncbi.nlm.nih.gov/books/NBK459130/
- Das A, Datta D, Kassir M, et al. Acanthosis nigricans: a review. J Cosmet Dermatol. 2020;19:1857-1865. doi:10.1111/jocd.13544
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101. doi:10 .1046/j.1365-2133.2002.05150.x
- van Steensel MAM, van der Hout AH. Lelis syndrome may be a manifestation of hypohidrotic ectodermal dysplasia. Am J Med Genet A. 2009;149A:1612-1613. doi:10.1002/ajmg.a.32945
- Yoshimura AM, Neves Ferreira Velho PE, Ferreira Magalhães R, et al. Lelis’ syndrome: treatment with acitretin. Int J Dermatol. 2008;47: 1330-1331. doi:10.1111/j.1365-4632.2008.03874.x
- Schneider H. Ectodermal dysplasias: new perspectives on the treatment of so far immedicable genetic disorders. Front Genet. 2022;13:1000744. doi:10.3389/fgene.2022.1000744
- Steiner CE, Cintra ML, Marques-de-Faria AP. Ectodermal dysplasia with acanthosis nigricans (Lelis syndrome). Am J Med Genet. 2002;113:381-384. doi:10.1002/ajmg.b.10787
- Lelis J. Autosomal recessive ectodermal dysplasia. Cutis. 1992; 49:435-437.
- Itin PH, Fistarol SK. Ectodermal dysplasias. Am J Med Genet C Semin Med Genet. 2004;131C:45-51. doi:10.1002/ajmg.c.30033
- Blüschke G, Nüsken KD, Schneider H. Prevalence and prevention of severe complications of hypohidrotic ectodermal dysplasia in infancy. Early Hum Dev. 2010;86:397-399. doi:10.1016/j .earlhumdev.2010.04.008
- Le C, Bedocs PM. Confluent and reticulated papillomatosis. StatPearls. StatPearls Publishing; 2022. http://www.ncbi.nlm.nih.gov/books/NBK459130/
- Das A, Datta D, Kassir M, et al. Acanthosis nigricans: a review. J Cosmet Dermatol. 2020;19:1857-1865. doi:10.1111/jocd.13544
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101. doi:10 .1046/j.1365-2133.2002.05150.x
- van Steensel MAM, van der Hout AH. Lelis syndrome may be a manifestation of hypohidrotic ectodermal dysplasia. Am J Med Genet A. 2009;149A:1612-1613. doi:10.1002/ajmg.a.32945
- Yoshimura AM, Neves Ferreira Velho PE, Ferreira Magalhães R, et al. Lelis’ syndrome: treatment with acitretin. Int J Dermatol. 2008;47: 1330-1331. doi:10.1111/j.1365-4632.2008.03874.x
- Schneider H. Ectodermal dysplasias: new perspectives on the treatment of so far immedicable genetic disorders. Front Genet. 2022;13:1000744. doi:10.3389/fgene.2022.1000744
A 61-year-old woman with a history of hypohidrosis and deafness presented with a pruritic rash on the neck and antecubital fossae of several years’ duration. Prior treatment with topical corticosteroids failed to resolve the rash. Physical examination revealed thick, velvety, hyperpigmented plaques on the inframammary folds, axillae, groin, posterior neck, and antecubital fossae with lichenification of the latter 2 areas. Many pedunculated papules were seen on the face, chest, shoulders, and trunk, as well as diffuse hair thinning, particularly of the frontal and vertex scalp. Eyebrows, eyelashes, and axillary hair were absent. Two 5-mm punch biopsies of the antecubital fossa and inframammary fold were obtained for histopathologic analysis.


Verruciform Plaques Within a Tattoo of an HIV-Positive Patient
The Diagnosis: Lichenoid Reaction With Pseudoepitheliomatous Hyperplasia
A shave biopsy of the left ankle and a punch biopsy of the left medial calf were performed and sent for histologic examination and acid-fast stain. Bacterial, fungal, and mycobacterial tissue cultures also were sent for testing. The findings from direct examination were negative, and tissue cultures exhibited no growth. The shave and punch biopsies and histology revealed pseudoepitheliomatous hyperplasia (PEH) with keratinocyte necrosis, satellitosis, and areas of acute folliculitis (Figure 1). A lichenoid hypersensitivity mixed infiltrate that included histiocytes admixed with anthracotic and red-orange pigment, lymphocytes, plasma cells, neutrophils, and rare eosinophils was noted in the dermis. Given these clinical and histopathologic findings, the patient was diagnosed with red-pigment tattoo lichenoid reaction with PEH.
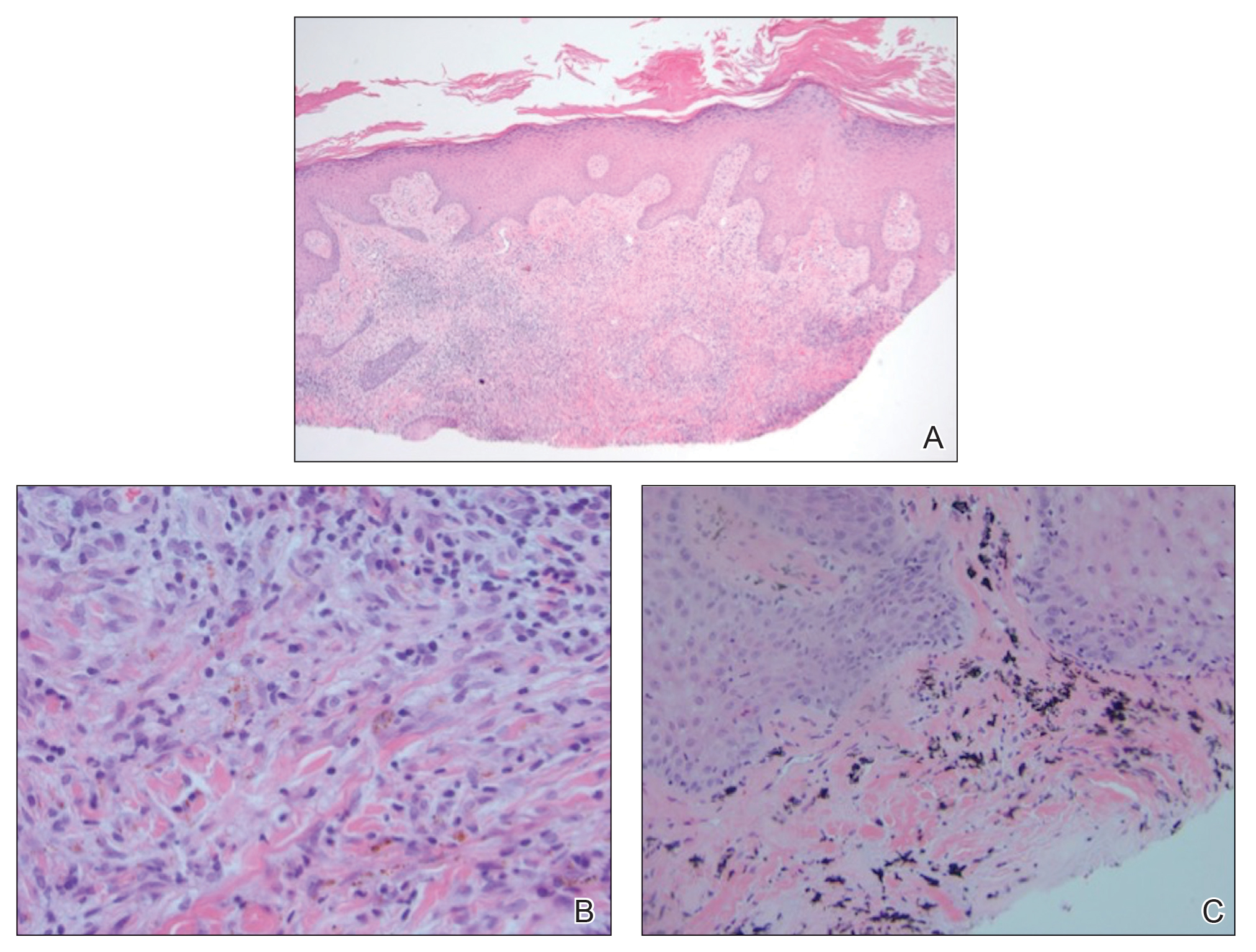
Tattoo-related inflammatory reactions can manifest clinically as allergic contact dermatitis, photodermatitis, infection, malignancy, foreign body granulomas, and delayed hypersensitivity reactions with myriad associated histopathologic patterns including spongiotic, psoriasiform, granulomatous, and lichenoid (as seen in our patient). Lichenoid tattoo reactions are the most common histopathologic variants of delayed hypersensitivity seen, mostly with cinnabar or red dye.1 However, there is a paucity of cases in the literature of PEH following tattooing with red dye. Interestingly, lichenoid tissue reaction accompanies PEH in all reported cases.2
Pseudoepitheliomatous hyperplasia can mimic squamous cell carcinoma and keratoacanthoma (KA) both clinically and histologically. All 3 conditions may exhibit epithelial hyperplasia with prominent dilated hyperplastic infundibula. In a case series of 11 presumed KAs within tattoos, Fraga and Prossick2 reported 82% (9/11) of the lesions were located strictly in areas with red pigment, and many were associated with a lichenoid tissue reaction. Kazlouskaya and Junkins-Hopkins3 previously described cases of KAs in tattoos that may represent PEH.
When treating lesions with this histologic appearance, consider the clinical and histologic overlap between KAs and PEH. Our patient was managed with clobetasol ointment 0.05% under occlusion followed by intralesional triamcinolone acetonide with notable improvement of the verrucous plaques on the left lateral malleolus (Figure 2). He also noted near resolution of the papules on the pretibial shin and complete resolution of all associated pruritus and burning. Calcineurin inhibitors, photochemotherapy, CO2 laser, excimer laser, and surgical removal with interval grafting also were considered.

It is important to recognize PEH in the differential of eruptions occurring within tattoos to avoid unnecessary invasive surgical procedures such as complete surgical excision of a KA to avoid malignant transformation.
- Mortimer NJ, Chave TA, Johnston GA. Red tattoo reactions. Clin Exp Dermatol. 2003;28:508-510.
- Fraga GR, Prossick TA. Tattoo-associated keratoacanthomas: a series of 8 patients with 11 keratoacanthomas. J Cutan Pathol. 2010;37:85-90.
- Kazlouskaya V, Junkins-Hopkins JM. Pseudoepitheliomatous hyperplasia in a red pigment tattoo: a separate entity or hypertrophic lichen planus-like reaction? J Clin Aesthet Dermatol. 2015;8:48-52.
The Diagnosis: Lichenoid Reaction With Pseudoepitheliomatous Hyperplasia
A shave biopsy of the left ankle and a punch biopsy of the left medial calf were performed and sent for histologic examination and acid-fast stain. Bacterial, fungal, and mycobacterial tissue cultures also were sent for testing. The findings from direct examination were negative, and tissue cultures exhibited no growth. The shave and punch biopsies and histology revealed pseudoepitheliomatous hyperplasia (PEH) with keratinocyte necrosis, satellitosis, and areas of acute folliculitis (Figure 1). A lichenoid hypersensitivity mixed infiltrate that included histiocytes admixed with anthracotic and red-orange pigment, lymphocytes, plasma cells, neutrophils, and rare eosinophils was noted in the dermis. Given these clinical and histopathologic findings, the patient was diagnosed with red-pigment tattoo lichenoid reaction with PEH.
Tattoo-related inflammatory reactions can manifest clinically as allergic contact dermatitis, photodermatitis, infection, malignancy, foreign body granulomas, and delayed hypersensitivity reactions with myriad associated histopathologic patterns including spongiotic, psoriasiform, granulomatous, and lichenoid (as seen in our patient). Lichenoid tattoo reactions are the most common histopathologic variants of delayed hypersensitivity seen, mostly with cinnabar or red dye.1 However, there is a paucity of cases in the literature of PEH following tattooing with red dye. Interestingly, lichenoid tissue reaction accompanies PEH in all reported cases.2
Pseudoepitheliomatous hyperplasia can mimic squamous cell carcinoma and keratoacanthoma (KA) both clinically and histologically. All 3 conditions may exhibit epithelial hyperplasia with prominent dilated hyperplastic infundibula. In a case series of 11 presumed KAs within tattoos, Fraga and Prossick2 reported 82% (9/11) of the lesions were located strictly in areas with red pigment, and many were associated with a lichenoid tissue reaction. Kazlouskaya and Junkins-Hopkins3 previously described cases of KAs in tattoos that may represent PEH.
When treating lesions with this histologic appearance, consider the clinical and histologic overlap between KAs and PEH. Our patient was managed with clobetasol ointment 0.05% under occlusion followed by intralesional triamcinolone acetonide with notable improvement of the verrucous plaques on the left lateral malleolus (Figure 2). He also noted near resolution of the papules on the pretibial shin and complete resolution of all associated pruritus and burning. Calcineurin inhibitors, photochemotherapy, CO2 laser, excimer laser, and surgical removal with interval grafting also were considered.
It is important to recognize PEH in the differential of eruptions occurring within tattoos to avoid unnecessary invasive surgical procedures such as complete surgical excision of a KA to avoid malignant transformation.
The Diagnosis: Lichenoid Reaction With Pseudoepitheliomatous Hyperplasia
A shave biopsy of the left ankle and a punch biopsy of the left medial calf were performed and sent for histologic examination and acid-fast stain. Bacterial, fungal, and mycobacterial tissue cultures also were sent for testing. The findings from direct examination were negative, and tissue cultures exhibited no growth. The shave and punch biopsies and histology revealed pseudoepitheliomatous hyperplasia (PEH) with keratinocyte necrosis, satellitosis, and areas of acute folliculitis (Figure 1). A lichenoid hypersensitivity mixed infiltrate that included histiocytes admixed with anthracotic and red-orange pigment, lymphocytes, plasma cells, neutrophils, and rare eosinophils was noted in the dermis. Given these clinical and histopathologic findings, the patient was diagnosed with red-pigment tattoo lichenoid reaction with PEH.
Tattoo-related inflammatory reactions can manifest clinically as allergic contact dermatitis, photodermatitis, infection, malignancy, foreign body granulomas, and delayed hypersensitivity reactions with myriad associated histopathologic patterns including spongiotic, psoriasiform, granulomatous, and lichenoid (as seen in our patient). Lichenoid tattoo reactions are the most common histopathologic variants of delayed hypersensitivity seen, mostly with cinnabar or red dye.1 However, there is a paucity of cases in the literature of PEH following tattooing with red dye. Interestingly, lichenoid tissue reaction accompanies PEH in all reported cases.2
Pseudoepitheliomatous hyperplasia can mimic squamous cell carcinoma and keratoacanthoma (KA) both clinically and histologically. All 3 conditions may exhibit epithelial hyperplasia with prominent dilated hyperplastic infundibula. In a case series of 11 presumed KAs within tattoos, Fraga and Prossick2 reported 82% (9/11) of the lesions were located strictly in areas with red pigment, and many were associated with a lichenoid tissue reaction. Kazlouskaya and Junkins-Hopkins3 previously described cases of KAs in tattoos that may represent PEH.
When treating lesions with this histologic appearance, consider the clinical and histologic overlap between KAs and PEH. Our patient was managed with clobetasol ointment 0.05% under occlusion followed by intralesional triamcinolone acetonide with notable improvement of the verrucous plaques on the left lateral malleolus (Figure 2). He also noted near resolution of the papules on the pretibial shin and complete resolution of all associated pruritus and burning. Calcineurin inhibitors, photochemotherapy, CO2 laser, excimer laser, and surgical removal with interval grafting also were considered.
It is important to recognize PEH in the differential of eruptions occurring within tattoos to avoid unnecessary invasive surgical procedures such as complete surgical excision of a KA to avoid malignant transformation.
- Mortimer NJ, Chave TA, Johnston GA. Red tattoo reactions. Clin Exp Dermatol. 2003;28:508-510.
- Fraga GR, Prossick TA. Tattoo-associated keratoacanthomas: a series of 8 patients with 11 keratoacanthomas. J Cutan Pathol. 2010;37:85-90.
- Kazlouskaya V, Junkins-Hopkins JM. Pseudoepitheliomatous hyperplasia in a red pigment tattoo: a separate entity or hypertrophic lichen planus-like reaction? J Clin Aesthet Dermatol. 2015;8:48-52.
- Mortimer NJ, Chave TA, Johnston GA. Red tattoo reactions. Clin Exp Dermatol. 2003;28:508-510.
- Fraga GR, Prossick TA. Tattoo-associated keratoacanthomas: a series of 8 patients with 11 keratoacanthomas. J Cutan Pathol. 2010;37:85-90.
- Kazlouskaya V, Junkins-Hopkins JM. Pseudoepitheliomatous hyperplasia in a red pigment tattoo: a separate entity or hypertrophic lichen planus-like reaction? J Clin Aesthet Dermatol. 2015;8:48-52.
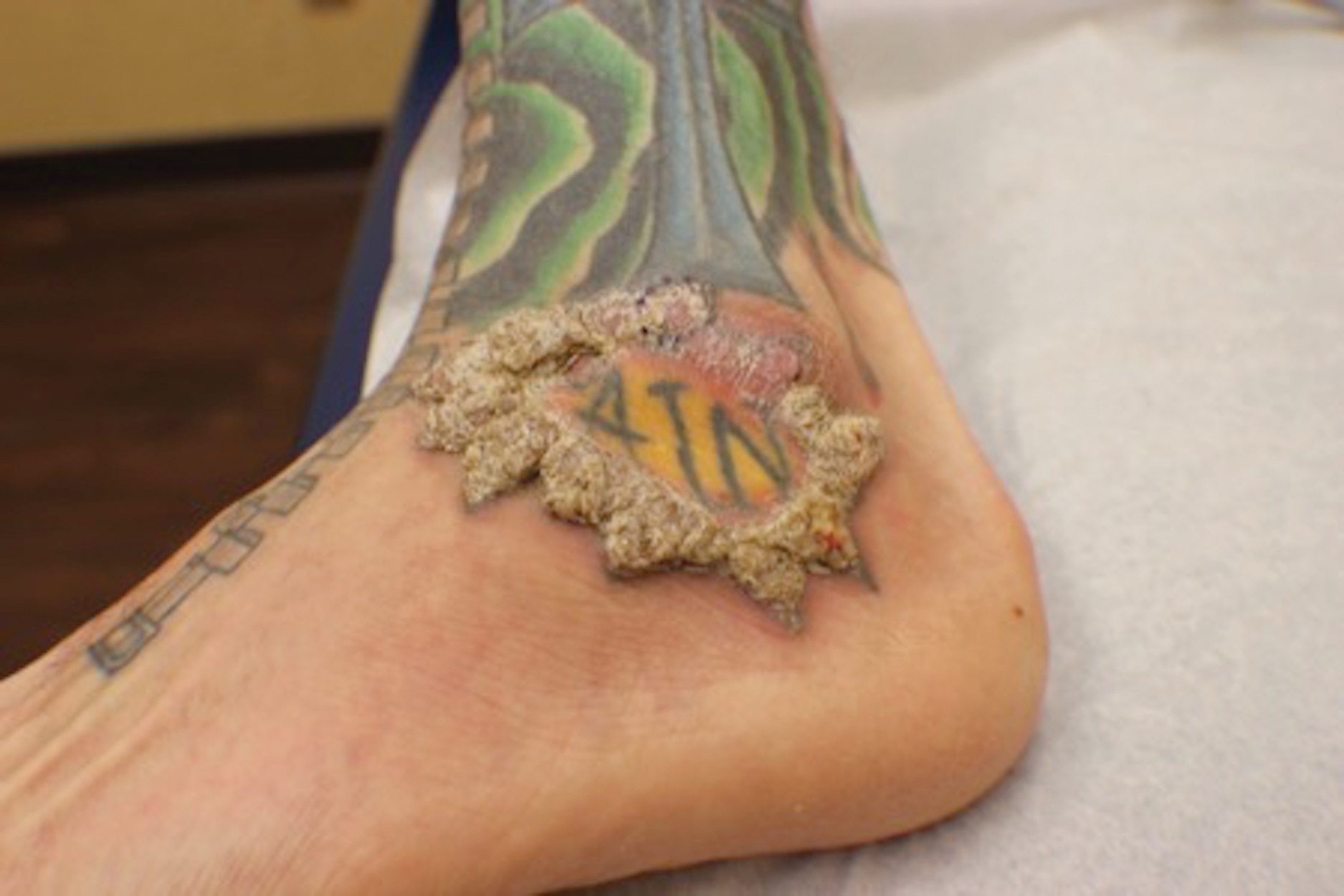
A 40-year-old man with a medical history of human immunodeficiency virus infection managed with highly active antiretroviral therapy (CD4 count, 888 cells/mm3 and an undetectable viral load), psoriasis, and recurrent condyloma acuminatum presented with exophytic, annular, hyperkeratotic, verrucous plaques on the left lateral malleolus with multiple erythematous hyperkeratotic papules on the pretibial shin of the left leg of 6 months' duration. These plaques and papules were localized to areas where red dye was used in a tattoo the patient had received 2 years prior to presentation. There was no associated fluctuance or drainage. The patient reported paroxysmal pruritus and burning pain.
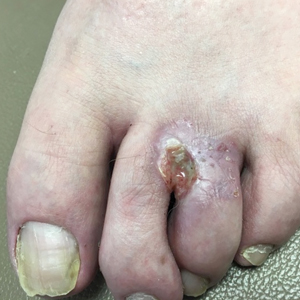
Nonhealing Eroded Plaque on an Interdigital Web Space of the Foot
The Diagnosis: Basal Cell Nevus Syndrome
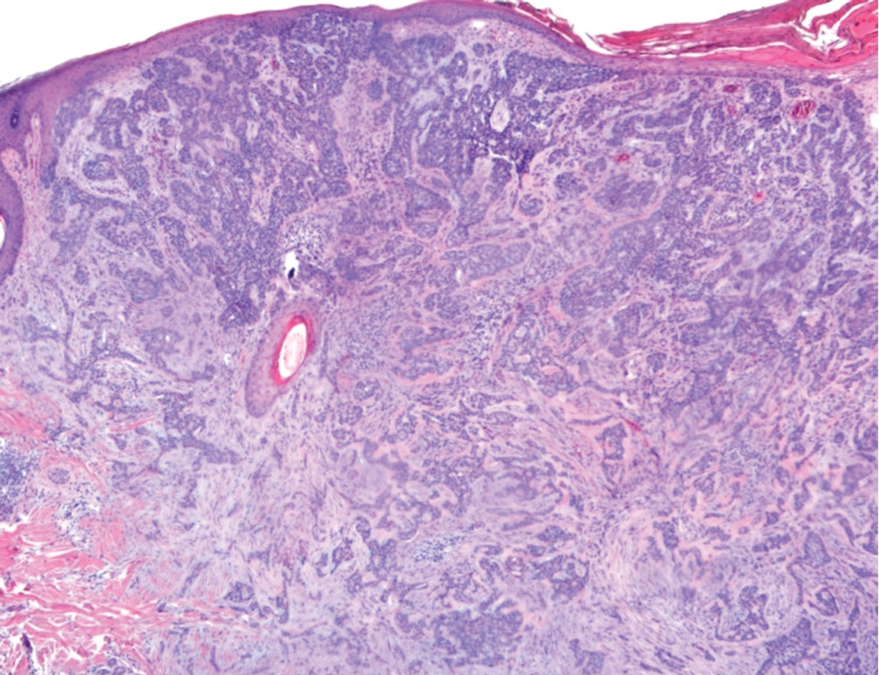
Given the patient’s history of numerous basal cell carcinomas (BCCs), odontogenic keratocysts, palmar pits, and a nonhealing ulcer, the clinical presentation was highly suggestive of interdigital BCC in the setting of basal cell nevus syndrome (BCNS). A shave biopsy was performed revealing islands of basaloid cells with peripheral palisading and a retraction artifact surrounded by fibromyxoid stroma, consistent with nodular and infiltrative BCC (Figure 1).
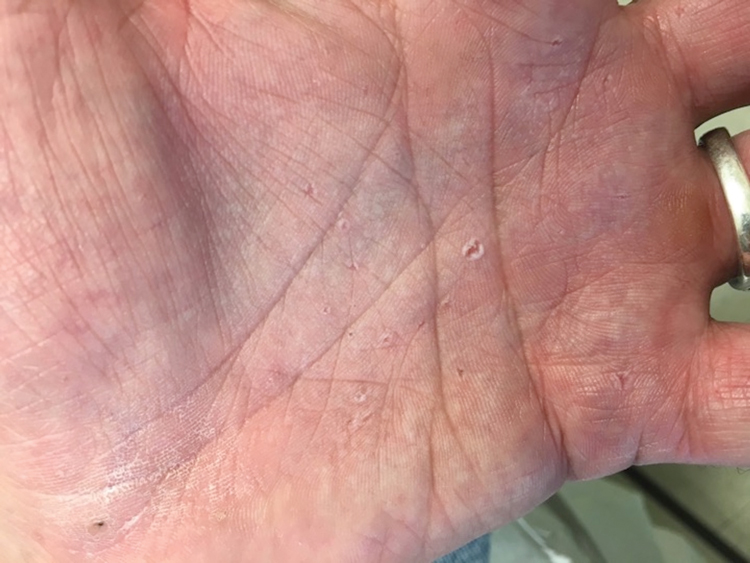
Basal cell nevus syndrome (also known as Gorlin syndrome) is a rare neurocutaneous syndrome that manifests with multiple BCCs; palmar and plantar pits (Figure 2); central nervous system tumors; and skeletal anomalies including jaw cysts, macrocephaly, frontal bossing, and bifid ribs.1 It is an autosomal-dominant condition caused by mutations in the PTCH1 gene, a tumor suppressor gene involved in the Hedgehog signaling pathway.2 Basal cell carcinoma is the most distinctive feature of BCNS, causing notable morbidity. Tumors typically present between puberty and 35 years of age, and patients can have anywhere from a few to thousands of tumors. They rarely become locally aggressive; however, with radiation therapy, proliferation and local invasion may occur within a few years. Therefore, radiotherapy should be avoided in these patients.1
Although the most common sites for BCCs in BCNS are the head, neck, and back, there is a higher rate of occurrence on sun-protected areas in BCNS compared to the general population.3 Our patient presented with interdigital BCC of the foot, which is an extremely rare occurrence. PubMed and Ovid searches using the terms basal cell carcinoma, BCC, foot, interdigital, and nonmelanoma skin cancer revealed only 3 cases of interdigital BCC of the foot. One case was associated with prior surgical trauma, the second presented as a junctional nevus, and the third did not appear to have any associated inciting factors.4-6 Dermatologists need to have a low threshold for biopsy for any unusual nonhealing lesions, especially in the setting of BCNS. Basal cell carcinomas in BCNS cannot be histologically differentiated from sporadic BCCs, and management largely depends on the size, location, recurrence, and number of lesions. Treatment methods range from topical agents to Mohs micrographic surgery.1
Nonhealing lesions of the foot may give an initial clinical impression of infection overlying peripheral vascular disease or diabetes mellitus with the possibility of associated osteomyelitis. Our patient had no clinical history to suggest peripheral vascular disease or diabetes mellitus, and he had palpable dorsalis pedis pulses as well as a normal neurologic examination. Clinicians also may consider fungal infection in the differential diagnosis. Erosio interdigitalis blastomycetica is a superficial yeast infection described as a well-defined, red, shiny plaque found in chronically wet areas, usually affecting the third or fourth interdigital spaces of the fingers.7 However, the lack of improvement with antibiotics and antifungals argued against bacterial or fungal infection in our patient. Although BCC also is a common feature of Bazex Dupré-Christol syndrome, it also is characterized by follicular atrophoderma, milia, hypohidrosis, and hypotrichosis,8 which were not evident in our patient. Pseudomonas hot foot syndrome is characterized by painful, plantar, erythematous nodules after exposure to ontaminated water that typically is self-limited but does respond to antibiotics for Pseudomonas.9
Our patient underwent Mohs micrographic surgery with a complex repair utilizing a full-thickness skin graft. There were no signs of recurrence at 3-month follow-up, and he was counseled on the importance of sun-protective behaviors along with regular dermatologic follow-up.
1. Gorlin RJ. Nevoid basal cell (Gorlin) syndrome. Genet Med. 2004; 6:530-539.
2. Bale A. The nevoid basal cell carcinoma syndrome: genetics and mechanism of carcinogenesis. Cancer Invest. 1997;15:180-186.
3. Goldstein AM, Bale SJ, Peck GL, et al. Sun exposure and basal cell carcinomas in nevoid basal cell carcinoma syndrome. J Am Acad Dermatol. 1993;29:34-41.
4. Silvers SH. Interdigital pedal basal cell carcinoma. Cutis. 1983;31:199-200.
5. Weitzner S. Basal cell carcinoma of toeweb presenting as a junctional nevus. Southwest Med. 1968;49:175.
6. Niwa A, Pimentel E. Basal cell carcinoma in unusual locations. An Bras Dermatol. 2006;81:281-284.
7. Mitchell JH. Erosio interdigitalis blastomycetica. Arch Derm Syphilol. 1922;6:675-679.
8. Kidd A, Carson L, Gregory DW, et al. A Scottish family with Bazex-Dupré-Christol syndrome: follicular atrophoderma, congenital hypotrichosis, and basal cell carcinoma. J Med Genet. 1996;33:493-497.
9. Yu Y, Cheng AS, Wang L, et al. Hot tub folliculitis or hot hand-foot syndrome caused by Pseudomonas aeruginosa. J Am Acad Dermatol. 2007;57:596-600.
The Diagnosis: Basal Cell Nevus Syndrome
Given the patient’s history of numerous basal cell carcinomas (BCCs), odontogenic keratocysts, palmar pits, and a nonhealing ulcer, the clinical presentation was highly suggestive of interdigital BCC in the setting of basal cell nevus syndrome (BCNS). A shave biopsy was performed revealing islands of basaloid cells with peripheral palisading and a retraction artifact surrounded by fibromyxoid stroma, consistent with nodular and infiltrative BCC (Figure 1).
Basal cell nevus syndrome (also known as Gorlin syndrome) is a rare neurocutaneous syndrome that manifests with multiple BCCs; palmar and plantar pits (Figure 2); central nervous system tumors; and skeletal anomalies including jaw cysts, macrocephaly, frontal bossing, and bifid ribs.1 It is an autosomal-dominant condition caused by mutations in the PTCH1 gene, a tumor suppressor gene involved in the Hedgehog signaling pathway.2 Basal cell carcinoma is the most distinctive feature of BCNS, causing notable morbidity. Tumors typically present between puberty and 35 years of age, and patients can have anywhere from a few to thousands of tumors. They rarely become locally aggressive; however, with radiation therapy, proliferation and local invasion may occur within a few years. Therefore, radiotherapy should be avoided in these patients.1
Although the most common sites for BCCs in BCNS are the head, neck, and back, there is a higher rate of occurrence on sun-protected areas in BCNS compared to the general population.3 Our patient presented with interdigital BCC of the foot, which is an extremely rare occurrence. PubMed and Ovid searches using the terms basal cell carcinoma, BCC, foot, interdigital, and nonmelanoma skin cancer revealed only 3 cases of interdigital BCC of the foot. One case was associated with prior surgical trauma, the second presented as a junctional nevus, and the third did not appear to have any associated inciting factors.4-6 Dermatologists need to have a low threshold for biopsy for any unusual nonhealing lesions, especially in the setting of BCNS. Basal cell carcinomas in BCNS cannot be histologically differentiated from sporadic BCCs, and management largely depends on the size, location, recurrence, and number of lesions. Treatment methods range from topical agents to Mohs micrographic surgery.1
Nonhealing lesions of the foot may give an initial clinical impression of infection overlying peripheral vascular disease or diabetes mellitus with the possibility of associated osteomyelitis. Our patient had no clinical history to suggest peripheral vascular disease or diabetes mellitus, and he had palpable dorsalis pedis pulses as well as a normal neurologic examination. Clinicians also may consider fungal infection in the differential diagnosis. Erosio interdigitalis blastomycetica is a superficial yeast infection described as a well-defined, red, shiny plaque found in chronically wet areas, usually affecting the third or fourth interdigital spaces of the fingers.7 However, the lack of improvement with antibiotics and antifungals argued against bacterial or fungal infection in our patient. Although BCC also is a common feature of Bazex Dupré-Christol syndrome, it also is characterized by follicular atrophoderma, milia, hypohidrosis, and hypotrichosis,8 which were not evident in our patient. Pseudomonas hot foot syndrome is characterized by painful, plantar, erythematous nodules after exposure to ontaminated water that typically is self-limited but does respond to antibiotics for Pseudomonas.9
Our patient underwent Mohs micrographic surgery with a complex repair utilizing a full-thickness skin graft. There were no signs of recurrence at 3-month follow-up, and he was counseled on the importance of sun-protective behaviors along with regular dermatologic follow-up.
The Diagnosis: Basal Cell Nevus Syndrome
Given the patient’s history of numerous basal cell carcinomas (BCCs), odontogenic keratocysts, palmar pits, and a nonhealing ulcer, the clinical presentation was highly suggestive of interdigital BCC in the setting of basal cell nevus syndrome (BCNS). A shave biopsy was performed revealing islands of basaloid cells with peripheral palisading and a retraction artifact surrounded by fibromyxoid stroma, consistent with nodular and infiltrative BCC (Figure 1).
Basal cell nevus syndrome (also known as Gorlin syndrome) is a rare neurocutaneous syndrome that manifests with multiple BCCs; palmar and plantar pits (Figure 2); central nervous system tumors; and skeletal anomalies including jaw cysts, macrocephaly, frontal bossing, and bifid ribs.1 It is an autosomal-dominant condition caused by mutations in the PTCH1 gene, a tumor suppressor gene involved in the Hedgehog signaling pathway.2 Basal cell carcinoma is the most distinctive feature of BCNS, causing notable morbidity. Tumors typically present between puberty and 35 years of age, and patients can have anywhere from a few to thousands of tumors. They rarely become locally aggressive; however, with radiation therapy, proliferation and local invasion may occur within a few years. Therefore, radiotherapy should be avoided in these patients.1
Although the most common sites for BCCs in BCNS are the head, neck, and back, there is a higher rate of occurrence on sun-protected areas in BCNS compared to the general population.3 Our patient presented with interdigital BCC of the foot, which is an extremely rare occurrence. PubMed and Ovid searches using the terms basal cell carcinoma, BCC, foot, interdigital, and nonmelanoma skin cancer revealed only 3 cases of interdigital BCC of the foot. One case was associated with prior surgical trauma, the second presented as a junctional nevus, and the third did not appear to have any associated inciting factors.4-6 Dermatologists need to have a low threshold for biopsy for any unusual nonhealing lesions, especially in the setting of BCNS. Basal cell carcinomas in BCNS cannot be histologically differentiated from sporadic BCCs, and management largely depends on the size, location, recurrence, and number of lesions. Treatment methods range from topical agents to Mohs micrographic surgery.1
Nonhealing lesions of the foot may give an initial clinical impression of infection overlying peripheral vascular disease or diabetes mellitus with the possibility of associated osteomyelitis. Our patient had no clinical history to suggest peripheral vascular disease or diabetes mellitus, and he had palpable dorsalis pedis pulses as well as a normal neurologic examination. Clinicians also may consider fungal infection in the differential diagnosis. Erosio interdigitalis blastomycetica is a superficial yeast infection described as a well-defined, red, shiny plaque found in chronically wet areas, usually affecting the third or fourth interdigital spaces of the fingers.7 However, the lack of improvement with antibiotics and antifungals argued against bacterial or fungal infection in our patient. Although BCC also is a common feature of Bazex Dupré-Christol syndrome, it also is characterized by follicular atrophoderma, milia, hypohidrosis, and hypotrichosis,8 which were not evident in our patient. Pseudomonas hot foot syndrome is characterized by painful, plantar, erythematous nodules after exposure to ontaminated water that typically is self-limited but does respond to antibiotics for Pseudomonas.9
Our patient underwent Mohs micrographic surgery with a complex repair utilizing a full-thickness skin graft. There were no signs of recurrence at 3-month follow-up, and he was counseled on the importance of sun-protective behaviors along with regular dermatologic follow-up.
1. Gorlin RJ. Nevoid basal cell (Gorlin) syndrome. Genet Med. 2004; 6:530-539.
2. Bale A. The nevoid basal cell carcinoma syndrome: genetics and mechanism of carcinogenesis. Cancer Invest. 1997;15:180-186.
3. Goldstein AM, Bale SJ, Peck GL, et al. Sun exposure and basal cell carcinomas in nevoid basal cell carcinoma syndrome. J Am Acad Dermatol. 1993;29:34-41.
4. Silvers SH. Interdigital pedal basal cell carcinoma. Cutis. 1983;31:199-200.
5. Weitzner S. Basal cell carcinoma of toeweb presenting as a junctional nevus. Southwest Med. 1968;49:175.
6. Niwa A, Pimentel E. Basal cell carcinoma in unusual locations. An Bras Dermatol. 2006;81:281-284.
7. Mitchell JH. Erosio interdigitalis blastomycetica. Arch Derm Syphilol. 1922;6:675-679.
8. Kidd A, Carson L, Gregory DW, et al. A Scottish family with Bazex-Dupré-Christol syndrome: follicular atrophoderma, congenital hypotrichosis, and basal cell carcinoma. J Med Genet. 1996;33:493-497.
9. Yu Y, Cheng AS, Wang L, et al. Hot tub folliculitis or hot hand-foot syndrome caused by Pseudomonas aeruginosa. J Am Acad Dermatol. 2007;57:596-600.
1. Gorlin RJ. Nevoid basal cell (Gorlin) syndrome. Genet Med. 2004; 6:530-539.
2. Bale A. The nevoid basal cell carcinoma syndrome: genetics and mechanism of carcinogenesis. Cancer Invest. 1997;15:180-186.
3. Goldstein AM, Bale SJ, Peck GL, et al. Sun exposure and basal cell carcinomas in nevoid basal cell carcinoma syndrome. J Am Acad Dermatol. 1993;29:34-41.
4. Silvers SH. Interdigital pedal basal cell carcinoma. Cutis. 1983;31:199-200.
5. Weitzner S. Basal cell carcinoma of toeweb presenting as a junctional nevus. Southwest Med. 1968;49:175.
6. Niwa A, Pimentel E. Basal cell carcinoma in unusual locations. An Bras Dermatol. 2006;81:281-284.
7. Mitchell JH. Erosio interdigitalis blastomycetica. Arch Derm Syphilol. 1922;6:675-679.
8. Kidd A, Carson L, Gregory DW, et al. A Scottish family with Bazex-Dupré-Christol syndrome: follicular atrophoderma, congenital hypotrichosis, and basal cell carcinoma. J Med Genet. 1996;33:493-497.
9. Yu Y, Cheng AS, Wang L, et al. Hot tub folliculitis or hot hand-foot syndrome caused by Pseudomonas aeruginosa. J Am Acad Dermatol. 2007;57:596-600.
A 53-year-old man with a history of numerous basal cell carcinomas and odontogenic keratocysts presented with a nonhealing erosion between the left second and third toes of several months’ duration. He was treated empirically with multiple courses of topical and systemic antibiotics as well as antifungals with minimal improvement. Physical examination revealed a 1.2×0.6-cm eroded plaque with rolled borders on the left second toe web; bilateral palmar pits; diffuse actinic damage; and several well-healed surgical scars on the head, neck, and back. Neurologic examination was normal, and dorsalis pedis pulses were equal and palpable bilaterally.