User login
Jeff Evans has been editor of Rheumatology News/MDedge Rheumatology and the EULAR Congress News since 2013. He started at Frontline Medical Communications in 2001 and was a reporter for 8 years before serving as editor of Clinical Neurology News and World Neurology, and briefly as editor of GI & Hepatology News. He graduated cum laude from Cornell University (New York) with a BA in biological sciences, concentrating in neurobiology and behavior.
Stroke History Did Not Alter Dabigatran's Safety, Efficacy
Patients with atrial fibrillation who were taking the anticoagulant dabigatran for secondary stroke prevention suffered an ischemic stroke or systemic embolism at a rate similar to patients taking warfarin in a prespecified subgroup analysis of patients from the 2-year RE-LY trial.
This analysis of 3,623 patients was consistent with the overall results found in the RE-LY (Randomized Evaluation of Long-Term Anticoagulation Therapy) trial cohort of 18,113 patients. Significant differences in the rates of intracranial bleeding between patients treated with dabigatran and those taking warfarin that had been observed in the overall results of the trial also were seen among those with a history of ischemic stroke or TIA.
“Although the subgroup analyses were not powered to detect whether the effects of dabigatran compared with warfarin varied by subgroup, the overlapping 95% confidence intervals suggest that major variations in the relative effects of the drugs between the patients with or without previous stroke or transient ischemic attack are unlikely,” Dr. Hans-Christoph Diener of University Hospital Essen (Germany) and his colleagues wrote (Lancet Neurol. 2010 Nov. 8 [doi:10.1016/S1474-4422(10)70274-X]).
The Food and Drug Administration approved the drug in October at doses of 150 mg and 75 mg for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. The approval was based on the overall results of the open-label RE-LY trial, which randomized patients with atrial fibrillation to 110 mg or 150 mg dabigatran twice daily or warfarin adjusted to an international normalized ratio of 2.0-3.0.
In the overall trial cohort, a stroke or systemic embolism occurred significantly more often among patients with a previous stroke or TIA (2.38% per year) than in those without such history (1.22% per year).
The primary outcome of stroke or systemic embolism occurred at similar rates between patients with a previous stroke or TIA who took warfarin (2.78% per year), 110 mg dabigatran (2.32% per year), and 150 mg dabigatran (2.07% per year). In the overall study population, the rate of stroke or systemic embolism did not differ among groups, occurring at 1.71% per year in patients on warfarin, 1.54% per year in patients on 110 mg dabigatran, and 1.11% per year in those on 150 mg dabigatran.
In the subgroup, intracranial bleeding occurred at a significantly lower rate in patients who took 110 mg dabigatran, compared with those who took warfarin (0.25% vs. 1.28% per year).
Patients with a history of stroke or TIA who took the 110-mg dose of dabigatran had a significantly lower rate of vascular death and all-cause mortality than did patients who received warfarin, but this effect was not seen in the 150-mg group.
Based on the results in patients with a previous stroke or TIA, the investigators suggested that “150 mg dabigatran might provide better protection against stroke than warfarin, whereas 110 mg dabigatran is as efficacious as warfarin and reduces adverse events (bleeding complications and mortality).” And indeed, the FDA's Cardiovascular and Renal Drugs Committee that evaluated dabigatran in September came to a similar conclusion, although no superiority claim over warfarin could be made. Additionally, the FDA did not include the 110-mg dosage that established noninferiority in its approved dosages, recommending the regimen of 150 mg twice daily, except in patients with impaired renal function, who would take 75 mg twice daily.
Boehringer Ingelheim GmbH funded the study and is marketing dabigatran as Pradaxa. Dr. Diener and some of his authors disclosed financial relationships with this company and others that manufacture or market drugs for the prevention or treatment of stroke. One author is an employee of Boehringer Ingelheim.
View on the News
Subgroup Analysis Offers Guidance
This subgroup analysis begins to fill the void of data on the benefit of oral coagulation for secondary stroke prevention and the safety of oral coagulation in patients with a previous ischemic stroke or TIA, according to Dr. Deidre A. Lane and Dr. Gregory Y.H. Lip of the University of Birmingham (England).
The analysis offers some guidance to physicians when deciding which dose of dabigatran to prescribe after going through an individualized stroke and bleeding risk assessment.
“Because of the necessary trade-off between stroke prevention and bleeding with both doses of dabigatran, consultation with patients regarding their preferences for treatment dose will be even more important to ascertain their threshold for stroke prevention over increased bleeding risk or vice versa,” they wrote.
Editor's Note: The approved dosages and indications differ between the countries in which dabigatran was approved.
DR. LANE AND DR. LIP wrote their comments in an editorial accompanying the paper (Lancet Neurol. 2010 Nov. 8 [doi:10.1016/S1474-4422(10)70275-1]). Both report having received funding for research and lecturing from manufacturers of drugs used to treat atrial fibrillation, including Boehringer Ingelheim.
Patients with atrial fibrillation who were taking the anticoagulant dabigatran for secondary stroke prevention suffered an ischemic stroke or systemic embolism at a rate similar to patients taking warfarin in a prespecified subgroup analysis of patients from the 2-year RE-LY trial.
This analysis of 3,623 patients was consistent with the overall results found in the RE-LY (Randomized Evaluation of Long-Term Anticoagulation Therapy) trial cohort of 18,113 patients. Significant differences in the rates of intracranial bleeding between patients treated with dabigatran and those taking warfarin that had been observed in the overall results of the trial also were seen among those with a history of ischemic stroke or TIA.
“Although the subgroup analyses were not powered to detect whether the effects of dabigatran compared with warfarin varied by subgroup, the overlapping 95% confidence intervals suggest that major variations in the relative effects of the drugs between the patients with or without previous stroke or transient ischemic attack are unlikely,” Dr. Hans-Christoph Diener of University Hospital Essen (Germany) and his colleagues wrote (Lancet Neurol. 2010 Nov. 8 [doi:10.1016/S1474-4422(10)70274-X]).
The Food and Drug Administration approved the drug in October at doses of 150 mg and 75 mg for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. The approval was based on the overall results of the open-label RE-LY trial, which randomized patients with atrial fibrillation to 110 mg or 150 mg dabigatran twice daily or warfarin adjusted to an international normalized ratio of 2.0-3.0.
In the overall trial cohort, a stroke or systemic embolism occurred significantly more often among patients with a previous stroke or TIA (2.38% per year) than in those without such history (1.22% per year).
The primary outcome of stroke or systemic embolism occurred at similar rates between patients with a previous stroke or TIA who took warfarin (2.78% per year), 110 mg dabigatran (2.32% per year), and 150 mg dabigatran (2.07% per year). In the overall study population, the rate of stroke or systemic embolism did not differ among groups, occurring at 1.71% per year in patients on warfarin, 1.54% per year in patients on 110 mg dabigatran, and 1.11% per year in those on 150 mg dabigatran.
In the subgroup, intracranial bleeding occurred at a significantly lower rate in patients who took 110 mg dabigatran, compared with those who took warfarin (0.25% vs. 1.28% per year).
Patients with a history of stroke or TIA who took the 110-mg dose of dabigatran had a significantly lower rate of vascular death and all-cause mortality than did patients who received warfarin, but this effect was not seen in the 150-mg group.
Based on the results in patients with a previous stroke or TIA, the investigators suggested that “150 mg dabigatran might provide better protection against stroke than warfarin, whereas 110 mg dabigatran is as efficacious as warfarin and reduces adverse events (bleeding complications and mortality).” And indeed, the FDA's Cardiovascular and Renal Drugs Committee that evaluated dabigatran in September came to a similar conclusion, although no superiority claim over warfarin could be made. Additionally, the FDA did not include the 110-mg dosage that established noninferiority in its approved dosages, recommending the regimen of 150 mg twice daily, except in patients with impaired renal function, who would take 75 mg twice daily.
Boehringer Ingelheim GmbH funded the study and is marketing dabigatran as Pradaxa. Dr. Diener and some of his authors disclosed financial relationships with this company and others that manufacture or market drugs for the prevention or treatment of stroke. One author is an employee of Boehringer Ingelheim.
View on the News
Subgroup Analysis Offers Guidance
This subgroup analysis begins to fill the void of data on the benefit of oral coagulation for secondary stroke prevention and the safety of oral coagulation in patients with a previous ischemic stroke or TIA, according to Dr. Deidre A. Lane and Dr. Gregory Y.H. Lip of the University of Birmingham (England).
The analysis offers some guidance to physicians when deciding which dose of dabigatran to prescribe after going through an individualized stroke and bleeding risk assessment.
“Because of the necessary trade-off between stroke prevention and bleeding with both doses of dabigatran, consultation with patients regarding their preferences for treatment dose will be even more important to ascertain their threshold for stroke prevention over increased bleeding risk or vice versa,” they wrote.
Editor's Note: The approved dosages and indications differ between the countries in which dabigatran was approved.
DR. LANE AND DR. LIP wrote their comments in an editorial accompanying the paper (Lancet Neurol. 2010 Nov. 8 [doi:10.1016/S1474-4422(10)70275-1]). Both report having received funding for research and lecturing from manufacturers of drugs used to treat atrial fibrillation, including Boehringer Ingelheim.
Patients with atrial fibrillation who were taking the anticoagulant dabigatran for secondary stroke prevention suffered an ischemic stroke or systemic embolism at a rate similar to patients taking warfarin in a prespecified subgroup analysis of patients from the 2-year RE-LY trial.
This analysis of 3,623 patients was consistent with the overall results found in the RE-LY (Randomized Evaluation of Long-Term Anticoagulation Therapy) trial cohort of 18,113 patients. Significant differences in the rates of intracranial bleeding between patients treated with dabigatran and those taking warfarin that had been observed in the overall results of the trial also were seen among those with a history of ischemic stroke or TIA.
“Although the subgroup analyses were not powered to detect whether the effects of dabigatran compared with warfarin varied by subgroup, the overlapping 95% confidence intervals suggest that major variations in the relative effects of the drugs between the patients with or without previous stroke or transient ischemic attack are unlikely,” Dr. Hans-Christoph Diener of University Hospital Essen (Germany) and his colleagues wrote (Lancet Neurol. 2010 Nov. 8 [doi:10.1016/S1474-4422(10)70274-X]).
The Food and Drug Administration approved the drug in October at doses of 150 mg and 75 mg for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. The approval was based on the overall results of the open-label RE-LY trial, which randomized patients with atrial fibrillation to 110 mg or 150 mg dabigatran twice daily or warfarin adjusted to an international normalized ratio of 2.0-3.0.
In the overall trial cohort, a stroke or systemic embolism occurred significantly more often among patients with a previous stroke or TIA (2.38% per year) than in those without such history (1.22% per year).
The primary outcome of stroke or systemic embolism occurred at similar rates between patients with a previous stroke or TIA who took warfarin (2.78% per year), 110 mg dabigatran (2.32% per year), and 150 mg dabigatran (2.07% per year). In the overall study population, the rate of stroke or systemic embolism did not differ among groups, occurring at 1.71% per year in patients on warfarin, 1.54% per year in patients on 110 mg dabigatran, and 1.11% per year in those on 150 mg dabigatran.
In the subgroup, intracranial bleeding occurred at a significantly lower rate in patients who took 110 mg dabigatran, compared with those who took warfarin (0.25% vs. 1.28% per year).
Patients with a history of stroke or TIA who took the 110-mg dose of dabigatran had a significantly lower rate of vascular death and all-cause mortality than did patients who received warfarin, but this effect was not seen in the 150-mg group.
Based on the results in patients with a previous stroke or TIA, the investigators suggested that “150 mg dabigatran might provide better protection against stroke than warfarin, whereas 110 mg dabigatran is as efficacious as warfarin and reduces adverse events (bleeding complications and mortality).” And indeed, the FDA's Cardiovascular and Renal Drugs Committee that evaluated dabigatran in September came to a similar conclusion, although no superiority claim over warfarin could be made. Additionally, the FDA did not include the 110-mg dosage that established noninferiority in its approved dosages, recommending the regimen of 150 mg twice daily, except in patients with impaired renal function, who would take 75 mg twice daily.
Boehringer Ingelheim GmbH funded the study and is marketing dabigatran as Pradaxa. Dr. Diener and some of his authors disclosed financial relationships with this company and others that manufacture or market drugs for the prevention or treatment of stroke. One author is an employee of Boehringer Ingelheim.
View on the News
Subgroup Analysis Offers Guidance
This subgroup analysis begins to fill the void of data on the benefit of oral coagulation for secondary stroke prevention and the safety of oral coagulation in patients with a previous ischemic stroke or TIA, according to Dr. Deidre A. Lane and Dr. Gregory Y.H. Lip of the University of Birmingham (England).
The analysis offers some guidance to physicians when deciding which dose of dabigatran to prescribe after going through an individualized stroke and bleeding risk assessment.
“Because of the necessary trade-off between stroke prevention and bleeding with both doses of dabigatran, consultation with patients regarding their preferences for treatment dose will be even more important to ascertain their threshold for stroke prevention over increased bleeding risk or vice versa,” they wrote.
Editor's Note: The approved dosages and indications differ between the countries in which dabigatran was approved.
DR. LANE AND DR. LIP wrote their comments in an editorial accompanying the paper (Lancet Neurol. 2010 Nov. 8 [doi:10.1016/S1474-4422(10)70275-1]). Both report having received funding for research and lecturing from manufacturers of drugs used to treat atrial fibrillation, including Boehringer Ingelheim.
New Multiple Sclerosis Lesions Accrue Seasonally
Major Finding: The point estimates for the rate of new T2 lesion accrual per day in MS were higher in the spring (0.024) and summer (0.030) than in the fall (0.010) or winter (0.016).
Data Source: A retrospective, observational study of brain MRI scans in 44 MS patients during 1991-1993.
Disease activity on MRI in multiple sclerosis patients is most likely to occur and is most intense in the spring and summer, according to a retrospective, observational study of a 3-year period in Boston.
Although the rates of clinical attacks and new contrast-enhancing lesions were not associated with significant seasonal differences, new T2 lesions developed in the spring and summer at nearly twice the rate as in the fall and winter. This finding “may raise concerns for design and analysis of clinical trials with MRI outcome measures. If left unaccounted this effect could bias longitudinal assessment both at the individual as well as group level,” wrote Dominik S. Meier, Ph.D., of Brigham and Women's Hospital, Boston, and colleagues.
The findings agreed with previous studies that measured the seasonality of clinical markers in Japan, Sweden, and the United States (Ohio and Arizona). Another three studies that examined MRI markers across the seasons had biased inclusion criteria or poor longitudinal follow-up, according to the investigators.
They matched meteorological data with clinical data from 44 patients who underwent 939 brain MRI scans during 1991-1993. The cohort included 13 patients with chronic progressive MS and 31 with relapsing-remitting MS. They had a mean age of 38 years, a mean disease duration of 8 years, and a mean Expanded Disability Status Scale score of 3.9.
Each patient had eight weekly scans, followed by eight scans every other week and six monthly examinations. No patient received disease-modifying therapy (Neurology 2010;75:799-806).
In the study, 31 patients developed 310 new T2 lesions, whereas 13 patients had no new lesions. In 42 patients, imaging detected a mean of 22 new contrast-enhancing lesions per patient. Clinical attacks during this period were recorded on 51 occasions in 24 patients, with a mean of 2.1 per patient.
The distribution of disease activity across the seasons was distinctly higher in the spring and summer even after applying several different methods of correcting for individual disease severity.
Point estimates for the rate of new T2 lesion accrual per day were higher in the spring (0.024) and summer (0.030) than in the fall (0.010) or winter (0.016).
Disease activity also was strongly correlated with warmer temperature and greater solar radiation, but not precipitation.
Patients with chronic, progressive MS tended to have an earlier and more pronounced high-activity period but lacked the peak of activity in August found in relapsing-remitting patients.
The findings did not change significantly in a separate analysis that excluded 18 patients who had been treated with brief bouts of steroids.
The findings could have an impact on MS clinical trials. The magnitude of an effect of the spring and summer on disease activity is likely affected by factors such as genetic affinity, disease phenotype, and geographic location, which “will have particular implications for multicenter trials that pool data from geographically distant locations,” the investigators wrote.
They also noted that biases may arise in studies that use prescreening MRI or in trials with crossover arms, depending on the timing of the trial arms.
Many of the investigators involved in this study disclosed that they had received research support from the National Institutes of Health and the National Multiple Sclerosis Society, as well as research support, speaker honoraria, or served on scientific advisory boards from MS drug manufacturers, including Biogen Idec, Genentech, EMD Serono and Teva Pharmaceutical Industries.
View on the news
MRI Variation Is a Concern
Evidence for environmental factors in the pathogenesis of multiple sclerosis has accumulated ever since Dr. John F. Kurtzke's pioneering epidemiological work in the 1960s. Epstein-Barr virus infection, smoking, and vitamin D status have all been shown to exert effects on MS risk. There also have been reports of seasonal variations in disease activity measured as relapse rate and occurrence of optic neuritis, with higher incidence of both in spring and summer. Several studies have shown a correlation between month of birth and MS risk, again with the highest risk in spring and summer.
Dr. Meier and colleagues report further evidence for a seasonal effect on disease activity and MS using serial MRI examinations. They elegantly show that disease activity, measured as new T2 lesions over time, varies over the year with a peak in spring and summer. They also show strong associations with solar radiation and daily temperature, but not precipitation. The levels of vitamin D in serum might be the causal link between season and disease activity, but this assumption remains to be proven.
What is of great concern, however, is that MRI variables, a common measure of disease activity and thus treatment efficacy, seem to be influenced by season. This could bias trials aimed at assessing the effect of drugs on disease activity and need to be considered when designing future studies.
JONATAN SALZER, M.D., is a doctoral student in the department of pharmacology and clinical neuroscience at Umeå (Sweden) University. He has no relevant disclosures.
{kind=link}
Vitals
Major Finding: The point estimates for the rate of new T2 lesion accrual per day in MS were higher in the spring (0.024) and summer (0.030) than in the fall (0.010) or winter (0.016).
Data Source: A retrospective, observational study of brain MRI scans in 44 MS patients during 1991-1993.
Disease activity on MRI in multiple sclerosis patients is most likely to occur and is most intense in the spring and summer, according to a retrospective, observational study of a 3-year period in Boston.
Although the rates of clinical attacks and new contrast-enhancing lesions were not associated with significant seasonal differences, new T2 lesions developed in the spring and summer at nearly twice the rate as in the fall and winter. This finding “may raise concerns for design and analysis of clinical trials with MRI outcome measures. If left unaccounted this effect could bias longitudinal assessment both at the individual as well as group level,” wrote Dominik S. Meier, Ph.D., of Brigham and Women's Hospital, Boston, and colleagues.
The findings agreed with previous studies that measured the seasonality of clinical markers in Japan, Sweden, and the United States (Ohio and Arizona). Another three studies that examined MRI markers across the seasons had biased inclusion criteria or poor longitudinal follow-up, according to the investigators.
They matched meteorological data with clinical data from 44 patients who underwent 939 brain MRI scans during 1991-1993. The cohort included 13 patients with chronic progressive MS and 31 with relapsing-remitting MS. They had a mean age of 38 years, a mean disease duration of 8 years, and a mean Expanded Disability Status Scale score of 3.9.
Each patient had eight weekly scans, followed by eight scans every other week and six monthly examinations. No patient received disease-modifying therapy (Neurology 2010;75:799-806).
In the study, 31 patients developed 310 new T2 lesions, whereas 13 patients had no new lesions. In 42 patients, imaging detected a mean of 22 new contrast-enhancing lesions per patient. Clinical attacks during this period were recorded on 51 occasions in 24 patients, with a mean of 2.1 per patient.
The distribution of disease activity across the seasons was distinctly higher in the spring and summer even after applying several different methods of correcting for individual disease severity.
Point estimates for the rate of new T2 lesion accrual per day were higher in the spring (0.024) and summer (0.030) than in the fall (0.010) or winter (0.016).
Disease activity also was strongly correlated with warmer temperature and greater solar radiation, but not precipitation.
Patients with chronic, progressive MS tended to have an earlier and more pronounced high-activity period but lacked the peak of activity in August found in relapsing-remitting patients.
The findings did not change significantly in a separate analysis that excluded 18 patients who had been treated with brief bouts of steroids.
The findings could have an impact on MS clinical trials. The magnitude of an effect of the spring and summer on disease activity is likely affected by factors such as genetic affinity, disease phenotype, and geographic location, which “will have particular implications for multicenter trials that pool data from geographically distant locations,” the investigators wrote.
They also noted that biases may arise in studies that use prescreening MRI or in trials with crossover arms, depending on the timing of the trial arms.
Many of the investigators involved in this study disclosed that they had received research support from the National Institutes of Health and the National Multiple Sclerosis Society, as well as research support, speaker honoraria, or served on scientific advisory boards from MS drug manufacturers, including Biogen Idec, Genentech, EMD Serono and Teva Pharmaceutical Industries.
View on the news
MRI Variation Is a Concern
Evidence for environmental factors in the pathogenesis of multiple sclerosis has accumulated ever since Dr. John F. Kurtzke's pioneering epidemiological work in the 1960s. Epstein-Barr virus infection, smoking, and vitamin D status have all been shown to exert effects on MS risk. There also have been reports of seasonal variations in disease activity measured as relapse rate and occurrence of optic neuritis, with higher incidence of both in spring and summer. Several studies have shown a correlation between month of birth and MS risk, again with the highest risk in spring and summer.
Dr. Meier and colleagues report further evidence for a seasonal effect on disease activity and MS using serial MRI examinations. They elegantly show that disease activity, measured as new T2 lesions over time, varies over the year with a peak in spring and summer. They also show strong associations with solar radiation and daily temperature, but not precipitation. The levels of vitamin D in serum might be the causal link between season and disease activity, but this assumption remains to be proven.
What is of great concern, however, is that MRI variables, a common measure of disease activity and thus treatment efficacy, seem to be influenced by season. This could bias trials aimed at assessing the effect of drugs on disease activity and need to be considered when designing future studies.
JONATAN SALZER, M.D., is a doctoral student in the department of pharmacology and clinical neuroscience at Umeå (Sweden) University. He has no relevant disclosures.
Vitals
Major Finding: The point estimates for the rate of new T2 lesion accrual per day in MS were higher in the spring (0.024) and summer (0.030) than in the fall (0.010) or winter (0.016).
Data Source: A retrospective, observational study of brain MRI scans in 44 MS patients during 1991-1993.
Disease activity on MRI in multiple sclerosis patients is most likely to occur and is most intense in the spring and summer, according to a retrospective, observational study of a 3-year period in Boston.
Although the rates of clinical attacks and new contrast-enhancing lesions were not associated with significant seasonal differences, new T2 lesions developed in the spring and summer at nearly twice the rate as in the fall and winter. This finding “may raise concerns for design and analysis of clinical trials with MRI outcome measures. If left unaccounted this effect could bias longitudinal assessment both at the individual as well as group level,” wrote Dominik S. Meier, Ph.D., of Brigham and Women's Hospital, Boston, and colleagues.
The findings agreed with previous studies that measured the seasonality of clinical markers in Japan, Sweden, and the United States (Ohio and Arizona). Another three studies that examined MRI markers across the seasons had biased inclusion criteria or poor longitudinal follow-up, according to the investigators.
They matched meteorological data with clinical data from 44 patients who underwent 939 brain MRI scans during 1991-1993. The cohort included 13 patients with chronic progressive MS and 31 with relapsing-remitting MS. They had a mean age of 38 years, a mean disease duration of 8 years, and a mean Expanded Disability Status Scale score of 3.9.
Each patient had eight weekly scans, followed by eight scans every other week and six monthly examinations. No patient received disease-modifying therapy (Neurology 2010;75:799-806).
In the study, 31 patients developed 310 new T2 lesions, whereas 13 patients had no new lesions. In 42 patients, imaging detected a mean of 22 new contrast-enhancing lesions per patient. Clinical attacks during this period were recorded on 51 occasions in 24 patients, with a mean of 2.1 per patient.
The distribution of disease activity across the seasons was distinctly higher in the spring and summer even after applying several different methods of correcting for individual disease severity.
Point estimates for the rate of new T2 lesion accrual per day were higher in the spring (0.024) and summer (0.030) than in the fall (0.010) or winter (0.016).
Disease activity also was strongly correlated with warmer temperature and greater solar radiation, but not precipitation.
Patients with chronic, progressive MS tended to have an earlier and more pronounced high-activity period but lacked the peak of activity in August found in relapsing-remitting patients.
The findings did not change significantly in a separate analysis that excluded 18 patients who had been treated with brief bouts of steroids.
The findings could have an impact on MS clinical trials. The magnitude of an effect of the spring and summer on disease activity is likely affected by factors such as genetic affinity, disease phenotype, and geographic location, which “will have particular implications for multicenter trials that pool data from geographically distant locations,” the investigators wrote.
They also noted that biases may arise in studies that use prescreening MRI or in trials with crossover arms, depending on the timing of the trial arms.
Many of the investigators involved in this study disclosed that they had received research support from the National Institutes of Health and the National Multiple Sclerosis Society, as well as research support, speaker honoraria, or served on scientific advisory boards from MS drug manufacturers, including Biogen Idec, Genentech, EMD Serono and Teva Pharmaceutical Industries.
View on the news
MRI Variation Is a Concern
Evidence for environmental factors in the pathogenesis of multiple sclerosis has accumulated ever since Dr. John F. Kurtzke's pioneering epidemiological work in the 1960s. Epstein-Barr virus infection, smoking, and vitamin D status have all been shown to exert effects on MS risk. There also have been reports of seasonal variations in disease activity measured as relapse rate and occurrence of optic neuritis, with higher incidence of both in spring and summer. Several studies have shown a correlation between month of birth and MS risk, again with the highest risk in spring and summer.
Dr. Meier and colleagues report further evidence for a seasonal effect on disease activity and MS using serial MRI examinations. They elegantly show that disease activity, measured as new T2 lesions over time, varies over the year with a peak in spring and summer. They also show strong associations with solar radiation and daily temperature, but not precipitation. The levels of vitamin D in serum might be the causal link between season and disease activity, but this assumption remains to be proven.
What is of great concern, however, is that MRI variables, a common measure of disease activity and thus treatment efficacy, seem to be influenced by season. This could bias trials aimed at assessing the effect of drugs on disease activity and need to be considered when designing future studies.
JONATAN SALZER, M.D., is a doctoral student in the department of pharmacology and clinical neuroscience at Umeå (Sweden) University. He has no relevant disclosures.
Vitals
Video Analysis Prompts Shift In Thinking on Causes of Falls
WASHINGTON – More often than not, elderly patients who fall in long-term care facilities do not trip or stumble while walking, but are instead transitioning from standing still or initiating a new activity at the time of their fall, according to an analysis of video-recorded falls.
“These results challenge traditional assumptions regarding the cause and circumstance of falls in older adults living in long-term care,” Stephen N. Robinovitch, Ph.D., said at the meeting.
About half of older adults living in long-term care facilities fall each year, whereas the annual incidence is about 30% among older adults living in the community, said Dr. Robinovitch of the department of biomedical physiology and kinesiology at Simon Fraser University, Burnaby, B.C.
Studies of self-reported falls have suggested that about half of all falls result from slips and trips, while the rest are ascribed to losing balance, changing posture, or a leg giving way. In these studies, the most common activities at the time of a fall were walking, turning, transferring, and reaching.
As part of the ongoing Vancouver Fall Mechanisms Study, Dr. Robinovitch and his colleagues are working with two long-term care facilities in British Columbia to develop “real-life laboratories” where they can witness activity before and during falls instead of relying on self-reports.
In common areas throughout the two facilities (each with about 230 beds), the investigators used 270 digital video cameras to record 184 falls by 124 residents during a 2-year period. Three expert reviewers classified the key characteristics of each fall. “A lot of what our data are suggesting is that falls among this population are highly variable,” Dr. Robinovitch said in an interview.
Unlike previous studies of falls, the videos indicated that an incorrect transfer of weight caused most falls (51%). Trips were estimated to account for 22% of falls, and slips for only 4%. Hitting or bumping something caused 21% of falls, collapsing was to blame in 10% of falls, and losing support from an external object was the cause in 13%. Each fall could have multiple causes.
At the time of a fall, four activities were significantly more common than others: walking forward (26%), standing quietly (22%), sitting down or lowering (16%), and initiating walking (16%). “In clinical evaluation, you have to consider…all four of these activities as equally important,” Dr. Robinovitch said.
Dr. Robinovitch noted that many older adults, especially older women, are unable to react quickly enough to take a corrective step or can't break a fall with their hands. In the video study, residents hit their heads in 30% of falls, their hip in 46%, and their hands in 54%.
Impact to the hands did not affect the probability of impact to the head. This suggests that even though older adults appear to maintain the protective response of moving their hands to arrest a fall, strengthening exercises are warranted to improve the effect of this response, he said.
WASHINGTON – More often than not, elderly patients who fall in long-term care facilities do not trip or stumble while walking, but are instead transitioning from standing still or initiating a new activity at the time of their fall, according to an analysis of video-recorded falls.
“These results challenge traditional assumptions regarding the cause and circumstance of falls in older adults living in long-term care,” Stephen N. Robinovitch, Ph.D., said at the meeting.
About half of older adults living in long-term care facilities fall each year, whereas the annual incidence is about 30% among older adults living in the community, said Dr. Robinovitch of the department of biomedical physiology and kinesiology at Simon Fraser University, Burnaby, B.C.
Studies of self-reported falls have suggested that about half of all falls result from slips and trips, while the rest are ascribed to losing balance, changing posture, or a leg giving way. In these studies, the most common activities at the time of a fall were walking, turning, transferring, and reaching.
As part of the ongoing Vancouver Fall Mechanisms Study, Dr. Robinovitch and his colleagues are working with two long-term care facilities in British Columbia to develop “real-life laboratories” where they can witness activity before and during falls instead of relying on self-reports.
In common areas throughout the two facilities (each with about 230 beds), the investigators used 270 digital video cameras to record 184 falls by 124 residents during a 2-year period. Three expert reviewers classified the key characteristics of each fall. “A lot of what our data are suggesting is that falls among this population are highly variable,” Dr. Robinovitch said in an interview.
Unlike previous studies of falls, the videos indicated that an incorrect transfer of weight caused most falls (51%). Trips were estimated to account for 22% of falls, and slips for only 4%. Hitting or bumping something caused 21% of falls, collapsing was to blame in 10% of falls, and losing support from an external object was the cause in 13%. Each fall could have multiple causes.
At the time of a fall, four activities were significantly more common than others: walking forward (26%), standing quietly (22%), sitting down or lowering (16%), and initiating walking (16%). “In clinical evaluation, you have to consider…all four of these activities as equally important,” Dr. Robinovitch said.
Dr. Robinovitch noted that many older adults, especially older women, are unable to react quickly enough to take a corrective step or can't break a fall with their hands. In the video study, residents hit their heads in 30% of falls, their hip in 46%, and their hands in 54%.
Impact to the hands did not affect the probability of impact to the head. This suggests that even though older adults appear to maintain the protective response of moving their hands to arrest a fall, strengthening exercises are warranted to improve the effect of this response, he said.
WASHINGTON – More often than not, elderly patients who fall in long-term care facilities do not trip or stumble while walking, but are instead transitioning from standing still or initiating a new activity at the time of their fall, according to an analysis of video-recorded falls.
“These results challenge traditional assumptions regarding the cause and circumstance of falls in older adults living in long-term care,” Stephen N. Robinovitch, Ph.D., said at the meeting.
About half of older adults living in long-term care facilities fall each year, whereas the annual incidence is about 30% among older adults living in the community, said Dr. Robinovitch of the department of biomedical physiology and kinesiology at Simon Fraser University, Burnaby, B.C.
Studies of self-reported falls have suggested that about half of all falls result from slips and trips, while the rest are ascribed to losing balance, changing posture, or a leg giving way. In these studies, the most common activities at the time of a fall were walking, turning, transferring, and reaching.
As part of the ongoing Vancouver Fall Mechanisms Study, Dr. Robinovitch and his colleagues are working with two long-term care facilities in British Columbia to develop “real-life laboratories” where they can witness activity before and during falls instead of relying on self-reports.
In common areas throughout the two facilities (each with about 230 beds), the investigators used 270 digital video cameras to record 184 falls by 124 residents during a 2-year period. Three expert reviewers classified the key characteristics of each fall. “A lot of what our data are suggesting is that falls among this population are highly variable,” Dr. Robinovitch said in an interview.
Unlike previous studies of falls, the videos indicated that an incorrect transfer of weight caused most falls (51%). Trips were estimated to account for 22% of falls, and slips for only 4%. Hitting or bumping something caused 21% of falls, collapsing was to blame in 10% of falls, and losing support from an external object was the cause in 13%. Each fall could have multiple causes.
At the time of a fall, four activities were significantly more common than others: walking forward (26%), standing quietly (22%), sitting down or lowering (16%), and initiating walking (16%). “In clinical evaluation, you have to consider…all four of these activities as equally important,” Dr. Robinovitch said.
Dr. Robinovitch noted that many older adults, especially older women, are unable to react quickly enough to take a corrective step or can't break a fall with their hands. In the video study, residents hit their heads in 30% of falls, their hip in 46%, and their hands in 54%.
Impact to the hands did not affect the probability of impact to the head. This suggests that even though older adults appear to maintain the protective response of moving their hands to arrest a fall, strengthening exercises are warranted to improve the effect of this response, he said.
FROM THE INTERNATIONAL CONGRESS ON GAIT AND MENTAL FUNCTION
Stroke History Did Not Change Safety, Effectiveness of Dabigatran
Patients with atrial fibrillation who were taking the anticoagulant dabigatran for secondary stroke prevention suffered an ischemic stroke or systemic embolism at a rate similar to patients taking warfarin in a prespecified subgroup analysis of patients from the 2-year RE-LY trial.
This analysis of 3,623 patients, published online Nov. 8 in the Lancet Neurology, was consistent with the overall results found in the RE-LY (Randomized Evaluation of Long-Term Anticoagulation Therapy) trial cohort of 18,113 patients. Significant differences in the rates of intracranial bleeding between patients treated with dabigatran and those taking warfarin that had been observed in the overall results of the trial also were seen among those with a history of ischemic stroke or TIA.
"Although the subgroup analyses were not powered to detect whether the effects of dabigatran compared with warfarin varied by subgroup, the overlapping 95% confidence intervals suggest that major variations in the relative effects of the drugs between the patients with or without previous stroke or transient ischemic attack are unlikely," Dr. Hans-Christoph Diener of University Hospital Essen (Germany) and his colleagues wrote (Lancet Neurol. 2010 Nov. 8 [doi:10.1016/S1474-4422(10)70274-X]).
The Food and Drug Administration approved the drug last month at doses of 150 mg and 75 mg for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. The approval was based on the overall results of the open-label RE-LY trial, which randomized patients with atrial fibrillation to 110 mg or 150 mg dabigatran twice daily or warfarin adjusted to an international normalized ratio of 2.0-3.0.
The drug was approved in 2008 in the European Union, Canada, and other countries for a shorter term indication, primary prevention of venous thromboembolic events in adults after elective total hip or knee replacement surgery. Canada added the indication for stroke prevention in atrial fibrillation in October.
In the overall trial cohort, a stroke or systemic embolism occurred significantly more often among patients with a previous stroke or TIA (2.38% per year) than in those without such history (1.22% per year).
The primary outcome of stroke or systemic embolism occurred at similar rates between patients with a previous stroke or TIA who took warfarin (2.78% per year), 110 mg dabigatran (2.32% per year), and 150 mg dabigatran (2.07% per year). In the overall study population, the rate of stroke or systemic embolism did not differ among groups, occurring at 1.71% per year in patients on warfarin, 1.54% per year in patients on 110 mg dabigatran, and 1.11% per year in those on 150 mg dabigatran.
In the subgroup, intracranial bleeding occurred at a significantly lower rate in patients who took 110 mg dabigatran, compared with those who took warfarin (0.25% vs. 1.28% per year).
Patients with a history of stroke or TIA who took the 110-mg dose of dabigatran had a significantly lower rate of vascular death and all-cause mortality than did patients who received warfarin, but this effect was not seen in the 150-mg group. In this subgroup, major bleeding also occurred at a significantly lower rate among only those who received 110 mg dabigatran.
The use of antiplatelet agents or nonsteroidal anti-inflammatory drugs was balanced among the subgroups across the three treatment groups.
Based on the results in patients with a previous stroke or TIA, the investigators suggested that "150 mg dabigatran might provide better protection against stroke than warfarin, whereas 110 mg dabigatran is as efficacious as warfarin and reduces adverse events (bleeding complications and mortality)." And indeed, the FDA’s Cardiovascular and Renal Drugs Committee that evaluated dabigatran in September came to a similar conclusion, although no superiority claim over warfarin could be made. Additionally, the FDA did not include the 110-mg dosage that established noninferiority in its approved dosages, recommending the regimen of 150 mg twice daily, except in patients with impaired renal function, who would take 75 mg twice daily.
They noted that because the RE-LY trial excluded all patients with ischemic stroke or TIA within the past 2 weeks before enrollment, it "cannot provide information on the efficacy of dabigatran in the early phase after transient ischemic attack or stroke."
How dabigatran might achieve a reduction in intracranial bleeding beyond a more stable anticoagulation "is not yet known," but Dr. Diener and his associates said that it might result from an inability to cross the blood-brain barrier.
In an editorial accompanying the paper, Dr. Deidre A. Lane and Dr. Gregory Y.H. Lip wrote that this subgroup analysis of the RE-LY trial is important because it begins to fill the void of data on the benefit of oral coagulation for secondary stroke prevention and the safety of oral coagulation in patients with a previous ischemic stroke or TIA (Lancet Neurol. 2010 Nov. 8 [doi:10.1016/S1474-4422(10)70275-1]).
The analysis offers some guidance to physicians when deciding which dose of dabigatran to prescribe after going through an individualized stroke and bleeding risk assessment.
"Because of the necessary trade-off between stroke prevention and bleeding with both doses of dabigatran, consultation with patients regarding their preferences for treatment dose will be even more important to ascertain their threshold for stroke prevention over increased bleeding risk or vice versa," Dr. Lane and Dr. Lip of the University of Birmingham (England) wrote.
Boehringer Ingelheim GmbH funded the study and is marketing dabigatran as Pradaxa. Dr. Diener and some of his authors disclosed financial relationships with this company and others that manufacture or market drugs for the prevention or treatment of stroke. One author is an employee of Boehringer Ingelheim.
Dr. Lane and Dr. Lip both reported having received funding for research and lecturing from different manufacturers of drugs used for the treatment of atrial fibrillation, including Boehringer Ingelheim.
This subgroup analysis of the RE-LY trial is important because it begins to fill the void of data on the benefit of oral coagulation for secondary stroke prevention and the safety of oral coagulation in patients with a previous ischemic stroke or TIA, according to Dr. Deidre A. Lane and Dr. Gregory Y.H. Lip of the University of Birmingham (England).
The analysis offers some guidance to physicians when deciding which dose of dabigatran to prescribe after going through an individualized stroke and bleeding risk assessment.
"Because of the necessary trade-off between stroke prevention and bleeding with both doses of dabigatran, consultation with patients regarding their preferences for treatment dose will be even more important to ascertain their threshold for stroke prevention over increased bleeding risk or vice versa," the doctors wrote.
Editor’s Note: The approved dosages and indications differ between the countries in which dabigatran was approved.
Dr. Lane and Dr. Lip wrote their comments in an editorial accompanying the paper (Lancet Neurol. 2010 Nov. 8 [doi:10.1016/S1474-4422(10)70275-1]). Both report having received funding for research and lecturing from different manufacturers of drugs used for the treatment of atrial fibrillation, including Boehringer Ingelheim.
This subgroup analysis of the RE-LY trial is important because it begins to fill the void of data on the benefit of oral coagulation for secondary stroke prevention and the safety of oral coagulation in patients with a previous ischemic stroke or TIA, according to Dr. Deidre A. Lane and Dr. Gregory Y.H. Lip of the University of Birmingham (England).
The analysis offers some guidance to physicians when deciding which dose of dabigatran to prescribe after going through an individualized stroke and bleeding risk assessment.
"Because of the necessary trade-off between stroke prevention and bleeding with both doses of dabigatran, consultation with patients regarding their preferences for treatment dose will be even more important to ascertain their threshold for stroke prevention over increased bleeding risk or vice versa," the doctors wrote.
Editor’s Note: The approved dosages and indications differ between the countries in which dabigatran was approved.
Dr. Lane and Dr. Lip wrote their comments in an editorial accompanying the paper (Lancet Neurol. 2010 Nov. 8 [doi:10.1016/S1474-4422(10)70275-1]). Both report having received funding for research and lecturing from different manufacturers of drugs used for the treatment of atrial fibrillation, including Boehringer Ingelheim.
This subgroup analysis of the RE-LY trial is important because it begins to fill the void of data on the benefit of oral coagulation for secondary stroke prevention and the safety of oral coagulation in patients with a previous ischemic stroke or TIA, according to Dr. Deidre A. Lane and Dr. Gregory Y.H. Lip of the University of Birmingham (England).
The analysis offers some guidance to physicians when deciding which dose of dabigatran to prescribe after going through an individualized stroke and bleeding risk assessment.
"Because of the necessary trade-off between stroke prevention and bleeding with both doses of dabigatran, consultation with patients regarding their preferences for treatment dose will be even more important to ascertain their threshold for stroke prevention over increased bleeding risk or vice versa," the doctors wrote.
Editor’s Note: The approved dosages and indications differ between the countries in which dabigatran was approved.
Dr. Lane and Dr. Lip wrote their comments in an editorial accompanying the paper (Lancet Neurol. 2010 Nov. 8 [doi:10.1016/S1474-4422(10)70275-1]). Both report having received funding for research and lecturing from different manufacturers of drugs used for the treatment of atrial fibrillation, including Boehringer Ingelheim.
Patients with atrial fibrillation who were taking the anticoagulant dabigatran for secondary stroke prevention suffered an ischemic stroke or systemic embolism at a rate similar to patients taking warfarin in a prespecified subgroup analysis of patients from the 2-year RE-LY trial.
This analysis of 3,623 patients, published online Nov. 8 in the Lancet Neurology, was consistent with the overall results found in the RE-LY (Randomized Evaluation of Long-Term Anticoagulation Therapy) trial cohort of 18,113 patients. Significant differences in the rates of intracranial bleeding between patients treated with dabigatran and those taking warfarin that had been observed in the overall results of the trial also were seen among those with a history of ischemic stroke or TIA.
"Although the subgroup analyses were not powered to detect whether the effects of dabigatran compared with warfarin varied by subgroup, the overlapping 95% confidence intervals suggest that major variations in the relative effects of the drugs between the patients with or without previous stroke or transient ischemic attack are unlikely," Dr. Hans-Christoph Diener of University Hospital Essen (Germany) and his colleagues wrote (Lancet Neurol. 2010 Nov. 8 [doi:10.1016/S1474-4422(10)70274-X]).
The Food and Drug Administration approved the drug last month at doses of 150 mg and 75 mg for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. The approval was based on the overall results of the open-label RE-LY trial, which randomized patients with atrial fibrillation to 110 mg or 150 mg dabigatran twice daily or warfarin adjusted to an international normalized ratio of 2.0-3.0.
The drug was approved in 2008 in the European Union, Canada, and other countries for a shorter term indication, primary prevention of venous thromboembolic events in adults after elective total hip or knee replacement surgery. Canada added the indication for stroke prevention in atrial fibrillation in October.
In the overall trial cohort, a stroke or systemic embolism occurred significantly more often among patients with a previous stroke or TIA (2.38% per year) than in those without such history (1.22% per year).
The primary outcome of stroke or systemic embolism occurred at similar rates between patients with a previous stroke or TIA who took warfarin (2.78% per year), 110 mg dabigatran (2.32% per year), and 150 mg dabigatran (2.07% per year). In the overall study population, the rate of stroke or systemic embolism did not differ among groups, occurring at 1.71% per year in patients on warfarin, 1.54% per year in patients on 110 mg dabigatran, and 1.11% per year in those on 150 mg dabigatran.
In the subgroup, intracranial bleeding occurred at a significantly lower rate in patients who took 110 mg dabigatran, compared with those who took warfarin (0.25% vs. 1.28% per year).
Patients with a history of stroke or TIA who took the 110-mg dose of dabigatran had a significantly lower rate of vascular death and all-cause mortality than did patients who received warfarin, but this effect was not seen in the 150-mg group. In this subgroup, major bleeding also occurred at a significantly lower rate among only those who received 110 mg dabigatran.
The use of antiplatelet agents or nonsteroidal anti-inflammatory drugs was balanced among the subgroups across the three treatment groups.
Based on the results in patients with a previous stroke or TIA, the investigators suggested that "150 mg dabigatran might provide better protection against stroke than warfarin, whereas 110 mg dabigatran is as efficacious as warfarin and reduces adverse events (bleeding complications and mortality)." And indeed, the FDA’s Cardiovascular and Renal Drugs Committee that evaluated dabigatran in September came to a similar conclusion, although no superiority claim over warfarin could be made. Additionally, the FDA did not include the 110-mg dosage that established noninferiority in its approved dosages, recommending the regimen of 150 mg twice daily, except in patients with impaired renal function, who would take 75 mg twice daily.
They noted that because the RE-LY trial excluded all patients with ischemic stroke or TIA within the past 2 weeks before enrollment, it "cannot provide information on the efficacy of dabigatran in the early phase after transient ischemic attack or stroke."
How dabigatran might achieve a reduction in intracranial bleeding beyond a more stable anticoagulation "is not yet known," but Dr. Diener and his associates said that it might result from an inability to cross the blood-brain barrier.
In an editorial accompanying the paper, Dr. Deidre A. Lane and Dr. Gregory Y.H. Lip wrote that this subgroup analysis of the RE-LY trial is important because it begins to fill the void of data on the benefit of oral coagulation for secondary stroke prevention and the safety of oral coagulation in patients with a previous ischemic stroke or TIA (Lancet Neurol. 2010 Nov. 8 [doi:10.1016/S1474-4422(10)70275-1]).
The analysis offers some guidance to physicians when deciding which dose of dabigatran to prescribe after going through an individualized stroke and bleeding risk assessment.
"Because of the necessary trade-off between stroke prevention and bleeding with both doses of dabigatran, consultation with patients regarding their preferences for treatment dose will be even more important to ascertain their threshold for stroke prevention over increased bleeding risk or vice versa," Dr. Lane and Dr. Lip of the University of Birmingham (England) wrote.
Boehringer Ingelheim GmbH funded the study and is marketing dabigatran as Pradaxa. Dr. Diener and some of his authors disclosed financial relationships with this company and others that manufacture or market drugs for the prevention or treatment of stroke. One author is an employee of Boehringer Ingelheim.
Dr. Lane and Dr. Lip both reported having received funding for research and lecturing from different manufacturers of drugs used for the treatment of atrial fibrillation, including Boehringer Ingelheim.
Patients with atrial fibrillation who were taking the anticoagulant dabigatran for secondary stroke prevention suffered an ischemic stroke or systemic embolism at a rate similar to patients taking warfarin in a prespecified subgroup analysis of patients from the 2-year RE-LY trial.
This analysis of 3,623 patients, published online Nov. 8 in the Lancet Neurology, was consistent with the overall results found in the RE-LY (Randomized Evaluation of Long-Term Anticoagulation Therapy) trial cohort of 18,113 patients. Significant differences in the rates of intracranial bleeding between patients treated with dabigatran and those taking warfarin that had been observed in the overall results of the trial also were seen among those with a history of ischemic stroke or TIA.
"Although the subgroup analyses were not powered to detect whether the effects of dabigatran compared with warfarin varied by subgroup, the overlapping 95% confidence intervals suggest that major variations in the relative effects of the drugs between the patients with or without previous stroke or transient ischemic attack are unlikely," Dr. Hans-Christoph Diener of University Hospital Essen (Germany) and his colleagues wrote (Lancet Neurol. 2010 Nov. 8 [doi:10.1016/S1474-4422(10)70274-X]).
The Food and Drug Administration approved the drug last month at doses of 150 mg and 75 mg for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. The approval was based on the overall results of the open-label RE-LY trial, which randomized patients with atrial fibrillation to 110 mg or 150 mg dabigatran twice daily or warfarin adjusted to an international normalized ratio of 2.0-3.0.
The drug was approved in 2008 in the European Union, Canada, and other countries for a shorter term indication, primary prevention of venous thromboembolic events in adults after elective total hip or knee replacement surgery. Canada added the indication for stroke prevention in atrial fibrillation in October.
In the overall trial cohort, a stroke or systemic embolism occurred significantly more often among patients with a previous stroke or TIA (2.38% per year) than in those without such history (1.22% per year).
The primary outcome of stroke or systemic embolism occurred at similar rates between patients with a previous stroke or TIA who took warfarin (2.78% per year), 110 mg dabigatran (2.32% per year), and 150 mg dabigatran (2.07% per year). In the overall study population, the rate of stroke or systemic embolism did not differ among groups, occurring at 1.71% per year in patients on warfarin, 1.54% per year in patients on 110 mg dabigatran, and 1.11% per year in those on 150 mg dabigatran.
In the subgroup, intracranial bleeding occurred at a significantly lower rate in patients who took 110 mg dabigatran, compared with those who took warfarin (0.25% vs. 1.28% per year).
Patients with a history of stroke or TIA who took the 110-mg dose of dabigatran had a significantly lower rate of vascular death and all-cause mortality than did patients who received warfarin, but this effect was not seen in the 150-mg group. In this subgroup, major bleeding also occurred at a significantly lower rate among only those who received 110 mg dabigatran.
The use of antiplatelet agents or nonsteroidal anti-inflammatory drugs was balanced among the subgroups across the three treatment groups.
Based on the results in patients with a previous stroke or TIA, the investigators suggested that "150 mg dabigatran might provide better protection against stroke than warfarin, whereas 110 mg dabigatran is as efficacious as warfarin and reduces adverse events (bleeding complications and mortality)." And indeed, the FDA’s Cardiovascular and Renal Drugs Committee that evaluated dabigatran in September came to a similar conclusion, although no superiority claim over warfarin could be made. Additionally, the FDA did not include the 110-mg dosage that established noninferiority in its approved dosages, recommending the regimen of 150 mg twice daily, except in patients with impaired renal function, who would take 75 mg twice daily.
They noted that because the RE-LY trial excluded all patients with ischemic stroke or TIA within the past 2 weeks before enrollment, it "cannot provide information on the efficacy of dabigatran in the early phase after transient ischemic attack or stroke."
How dabigatran might achieve a reduction in intracranial bleeding beyond a more stable anticoagulation "is not yet known," but Dr. Diener and his associates said that it might result from an inability to cross the blood-brain barrier.
In an editorial accompanying the paper, Dr. Deidre A. Lane and Dr. Gregory Y.H. Lip wrote that this subgroup analysis of the RE-LY trial is important because it begins to fill the void of data on the benefit of oral coagulation for secondary stroke prevention and the safety of oral coagulation in patients with a previous ischemic stroke or TIA (Lancet Neurol. 2010 Nov. 8 [doi:10.1016/S1474-4422(10)70275-1]).
The analysis offers some guidance to physicians when deciding which dose of dabigatran to prescribe after going through an individualized stroke and bleeding risk assessment.
"Because of the necessary trade-off between stroke prevention and bleeding with both doses of dabigatran, consultation with patients regarding their preferences for treatment dose will be even more important to ascertain their threshold for stroke prevention over increased bleeding risk or vice versa," Dr. Lane and Dr. Lip of the University of Birmingham (England) wrote.
Boehringer Ingelheim GmbH funded the study and is marketing dabigatran as Pradaxa. Dr. Diener and some of his authors disclosed financial relationships with this company and others that manufacture or market drugs for the prevention or treatment of stroke. One author is an employee of Boehringer Ingelheim.
Dr. Lane and Dr. Lip both reported having received funding for research and lecturing from different manufacturers of drugs used for the treatment of atrial fibrillation, including Boehringer Ingelheim.
FROM THE LANCET NEUROLOGY
Major Finding: The primary outcome of stroke or systemic embolism occurred at similar rates among patients with a previous stroke or TIA taking warfarin (2.78% per year), 110 mg dabigatran (2.32% per year), or 150 mg dabigatran (2.07% per year).
Data Source: A subgroup analysis of 3,623 patients with atrial fibrillation from the RE-LY trial who had a history of TIA or ischemic stroke.
Disclosures: Boehringer Ingelheim funded the study. Dr. Diener and some of his coauthors disclosed financial relationships with this company and others that manufacture or market drugs for the prevention or treatment of stroke. One author is an employee of Boehringer Ingelheim.
Alzheimer's Trial Dims Outlook for Dimebon
The investigational drug dimebon failed to show any benefit over placebo for patients with mild to moderate Alzheimer's disease on any of the efficacy end points in a 6-month, phase III trial, drug manufacturers Medivation Inc. and Pfizer Inc. announced.
Although the tolerability of the drug was confirmed in the efficacy study, called CONNECTION, and in a separate phase III safety and tolerability study, the results put the future of the drug in doubt.
Dr. Marwan N. Sabbagh said that he and other Alzheimer's disease (AD) investigators “were extremely disappointed with the results, and frankly, surprised.”
The disappointing efficacy results in the CONNECTION trial came as a surprise because dimebon showed strong signs of efficacy in an earlier phase II trial of 183 patients in Russia (Lancet 2008;372:207-15).
Four other phase III trials of dimebon (proposed generic name latrepirdine) are currently enrolling patients. In a 12-month trial called CONCERT, the drug is being testing in combination with donepezil (Aricept) in patients with mild to moderate AD.
Two other trials – CONTACT and CONSTELLATION – are testing dimebon in combination with donepezil or memantine (Namenda), respectively, for moderate to severe AD.
The fourth study, the HORIZON trial, is enrolling patients with Huntington's disease after dimebon was well tolerated and showed some signs of improving cognition in a phase II trial.
The remaining trials in AD patients will help to determine whether dimebon has a synergistic effect with donepezil or memantine, Dr. Sabbagh said. “If it doesn't show any shred of evidence in those two studies, I think the future of dimebon is seriously in doubt, unless it shows a benefit for Huntington's.”
Dr. Sabbagh is the medical and scientific director of the Cleo Roberts Center of Clinical Research at the Banner Sun Health Research Institute, Sun City, Ariz. His center was involved in a phase I study of dimebon and is participating in the CONCERT trial. He said that he has no other relevant disclosures.
Investigators believed that dimebon blocked the induction of the mitochondrial membrane permeability transition pore, which when open may lead to a loss of energy production and intake of small molecules that contribute to cell death (Ann. N.Y. Acad. Sci. 2003;993:334-44). Other studies have shown it increases neurite outgrowth and can raise amyloid-beta levels in interstitial brain fluid of transgenic mouse models of AD (“Dimebon's Effect May Challenge Amyloid Theory,” September/October 2009, p. 11).
The rise and apparent fall of dimebon in thecclinical drug development processhairrorsed the recent history of tramiprosate and tarenflurbil for AD, both of whichihad positive results in phase II trials that were not replicated in phase III trialss.
It could be near the end of 2011 before another drug for AD comes through the Food and Drug Administration's review process for potential approval. The candidates, that will probably be reviewed first arenoemagacestat, a gamma-secretase inhibitor, and bapineuzumab, a monoclonal antibody against amyloid-beta, Dr. Sabbagh said.
The CONNECTION study enrolled 598 patients with mild to moderate AD at 63 sites in North America, Europe, and South America. Patients were randomized to dimebon 20 mg, dimebon 5 mg, or placebo three times daily for 6 months.
Jessica Merrill of “The Pink Sheet” contributed to this report.
{kind=link}
Alzheimer's researchers 'were extremely disappointed with the results, and frankly, surprised.'
Source DR. SABBAGH
The investigational drug dimebon failed to show any benefit over placebo for patients with mild to moderate Alzheimer's disease on any of the efficacy end points in a 6-month, phase III trial, drug manufacturers Medivation Inc. and Pfizer Inc. announced.
Although the tolerability of the drug was confirmed in the efficacy study, called CONNECTION, and in a separate phase III safety and tolerability study, the results put the future of the drug in doubt.
Dr. Marwan N. Sabbagh said that he and other Alzheimer's disease (AD) investigators “were extremely disappointed with the results, and frankly, surprised.”
The disappointing efficacy results in the CONNECTION trial came as a surprise because dimebon showed strong signs of efficacy in an earlier phase II trial of 183 patients in Russia (Lancet 2008;372:207-15).
Four other phase III trials of dimebon (proposed generic name latrepirdine) are currently enrolling patients. In a 12-month trial called CONCERT, the drug is being testing in combination with donepezil (Aricept) in patients with mild to moderate AD.
Two other trials – CONTACT and CONSTELLATION – are testing dimebon in combination with donepezil or memantine (Namenda), respectively, for moderate to severe AD.
The fourth study, the HORIZON trial, is enrolling patients with Huntington's disease after dimebon was well tolerated and showed some signs of improving cognition in a phase II trial.
The remaining trials in AD patients will help to determine whether dimebon has a synergistic effect with donepezil or memantine, Dr. Sabbagh said. “If it doesn't show any shred of evidence in those two studies, I think the future of dimebon is seriously in doubt, unless it shows a benefit for Huntington's.”
Dr. Sabbagh is the medical and scientific director of the Cleo Roberts Center of Clinical Research at the Banner Sun Health Research Institute, Sun City, Ariz. His center was involved in a phase I study of dimebon and is participating in the CONCERT trial. He said that he has no other relevant disclosures.
Investigators believed that dimebon blocked the induction of the mitochondrial membrane permeability transition pore, which when open may lead to a loss of energy production and intake of small molecules that contribute to cell death (Ann. N.Y. Acad. Sci. 2003;993:334-44). Other studies have shown it increases neurite outgrowth and can raise amyloid-beta levels in interstitial brain fluid of transgenic mouse models of AD (“Dimebon's Effect May Challenge Amyloid Theory,” September/October 2009, p. 11).
The rise and apparent fall of dimebon in thecclinical drug development processhairrorsed the recent history of tramiprosate and tarenflurbil for AD, both of whichihad positive results in phase II trials that were not replicated in phase III trialss.
It could be near the end of 2011 before another drug for AD comes through the Food and Drug Administration's review process for potential approval. The candidates, that will probably be reviewed first arenoemagacestat, a gamma-secretase inhibitor, and bapineuzumab, a monoclonal antibody against amyloid-beta, Dr. Sabbagh said.
The CONNECTION study enrolled 598 patients with mild to moderate AD at 63 sites in North America, Europe, and South America. Patients were randomized to dimebon 20 mg, dimebon 5 mg, or placebo three times daily for 6 months.
Jessica Merrill of “The Pink Sheet” contributed to this report.
Alzheimer's researchers 'were extremely disappointed with the results, and frankly, surprised.'
Source DR. SABBAGH
The investigational drug dimebon failed to show any benefit over placebo for patients with mild to moderate Alzheimer's disease on any of the efficacy end points in a 6-month, phase III trial, drug manufacturers Medivation Inc. and Pfizer Inc. announced.
Although the tolerability of the drug was confirmed in the efficacy study, called CONNECTION, and in a separate phase III safety and tolerability study, the results put the future of the drug in doubt.
Dr. Marwan N. Sabbagh said that he and other Alzheimer's disease (AD) investigators “were extremely disappointed with the results, and frankly, surprised.”
The disappointing efficacy results in the CONNECTION trial came as a surprise because dimebon showed strong signs of efficacy in an earlier phase II trial of 183 patients in Russia (Lancet 2008;372:207-15).
Four other phase III trials of dimebon (proposed generic name latrepirdine) are currently enrolling patients. In a 12-month trial called CONCERT, the drug is being testing in combination with donepezil (Aricept) in patients with mild to moderate AD.
Two other trials – CONTACT and CONSTELLATION – are testing dimebon in combination with donepezil or memantine (Namenda), respectively, for moderate to severe AD.
The fourth study, the HORIZON trial, is enrolling patients with Huntington's disease after dimebon was well tolerated and showed some signs of improving cognition in a phase II trial.
The remaining trials in AD patients will help to determine whether dimebon has a synergistic effect with donepezil or memantine, Dr. Sabbagh said. “If it doesn't show any shred of evidence in those two studies, I think the future of dimebon is seriously in doubt, unless it shows a benefit for Huntington's.”
Dr. Sabbagh is the medical and scientific director of the Cleo Roberts Center of Clinical Research at the Banner Sun Health Research Institute, Sun City, Ariz. His center was involved in a phase I study of dimebon and is participating in the CONCERT trial. He said that he has no other relevant disclosures.
Investigators believed that dimebon blocked the induction of the mitochondrial membrane permeability transition pore, which when open may lead to a loss of energy production and intake of small molecules that contribute to cell death (Ann. N.Y. Acad. Sci. 2003;993:334-44). Other studies have shown it increases neurite outgrowth and can raise amyloid-beta levels in interstitial brain fluid of transgenic mouse models of AD (“Dimebon's Effect May Challenge Amyloid Theory,” September/October 2009, p. 11).
The rise and apparent fall of dimebon in thecclinical drug development processhairrorsed the recent history of tramiprosate and tarenflurbil for AD, both of whichihad positive results in phase II trials that were not replicated in phase III trialss.
It could be near the end of 2011 before another drug for AD comes through the Food and Drug Administration's review process for potential approval. The candidates, that will probably be reviewed first arenoemagacestat, a gamma-secretase inhibitor, and bapineuzumab, a monoclonal antibody against amyloid-beta, Dr. Sabbagh said.
The CONNECTION study enrolled 598 patients with mild to moderate AD at 63 sites in North America, Europe, and South America. Patients were randomized to dimebon 20 mg, dimebon 5 mg, or placebo three times daily for 6 months.
Jessica Merrill of “The Pink Sheet” contributed to this report.
Alzheimer's researchers 'were extremely disappointed with the results, and frankly, surprised.'
Source DR. SABBAGH
Study Directly Links Mechanism of Aura to Migraine Pain
Frustration has long plagued researchers who have sought to link the visual auras experienced by some migraineurs with the later onset of headache pain. But now, direct evidence from a new study in rats suggests that auras – presumed to be caused by waves of depression of spontaneous electrical activity that propagate slowly through the occipital lobe of the cortex – can trigger the activation of meningeal nociceptors.
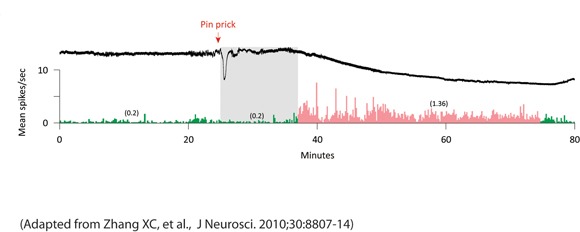
XiChun Zhang, Ph.D., and colleagues at Beth Israel Deaconess Medical Center, Boston, reported that those nociceptors in the trigeminal ganglion of rats became activated either immediately or after an average delay of 14 minutes following the administration of a pin prick, electrical pulses, or potassium chloride to the rats’ visual cortex to induce waves of cortical spreading depression (CSD).
The investigators conducted 83 trials of the three methods of cortical stimulation in 53 rats. Of 64 trials that induced waves of CSD, 31 resulted in increases in neuronal firing rates (25% or greater than baseline) that lasted at least 11 minutes. Sustained neuronal activation was not seen in all but 1 of the 19 trials that failed to produce CSD (J. Neurosci. 2010;30:8807-14).
Long-lasting neuronal activation coincided with the wave of CSD in 10 of the 31 trials in which both waves of CSD and increased neuronal firing were observed.
“The immediate activation of meningeal nociceptors may be clinically relevant to uncommon cases where migraine aura appears together with the headache,” Dr. Zhang and associates wrote.
In the other 21 trials that recorded CSD and increased neuronal firing, long-lasting neuronal activation began a mean of 14 minutes after the CSD waves.
The delayed neuronal activation observed in those 21 trials “may be relevant to the typical delay between the onset of aura and the onset of migraine headache, though the underlying mechanisms remain unknown,” wrote Dr. Zhang and coauthors. They noted that this observation may be the “most clinically promising” because “intervention during the aura phase with drugs that would block the delayed induction of neuronal activation could potentially preempt the onset of migraine headache.”
There were no differences in the pattern of neuronal activation between C- and A-delta-nociceptors or between the methods of cortical stimulation. Responses to cortical stimulation in rats that had their ipsilateral sphenopalatine ganglion excised were no different from those in other rats, which indicated that the parasympathetic innervation of the dura does not contribute to the long-lasting activation of meningeal nociceptors.
The investigators proposed that the sustained activation of meningeal nociceptors could be the result of either a “short-lasting release of algesic molecules” during CSD that promotes an acute activation of the nociceptor and gives rise to an ongoing sensitization that typically outlasts the stimulus by 30-60 minutes, or an ongoing release of algesic molecules for up to 1 hour during CSD.
However, because this period of sustained activation of the meningeal nociceptors may not be sufficient “in and of itself” to explain the 4- to 72-hour duration of the headache phase of migraine, Dr. Zhang and associates proposed that the “duration of nociceptor activation may be sufficient to promote ongoing activity of central trigeminovascular neurons that eventually becomes independent of incoming signals from the nociceptors and can last many hours.”
Dr. Zhang’s research was funded by grants from the National Institutes of Health. Dr. Black has no relevant disclosures.
The relationship between CSD and migraine aura has a pathophysiological overlap that has led to the widely accepted theory that CSD is the electrophysiological substrate underlying the migraine aura. The question as to whether CSD is the brain mechanism causing the migraine headache is more controversial. Although circumstantial evidence and indirect data, including neuroimaging studies, provide strong evidence that CSD does occur in migraine, there is still no definitive demonstration in a migraine patient that CSD causes the proposed activation of nociceptors in the cranial pain-sensitive structures, specifically the meninges, large blood vessels, and large venous sinuses.
The fact that the majority of migraine attacks are not preceded by aura is explained by the theory that CSD events may have variations in expression and may occur in subcortical tissue such as the hippocampus, cerebellum, and striatum. In this situation, an intense and steady depolarization propagates centrifugally in grey matter and occurs within regions of the brain that remain clinically silent. The depolarization does not follow functional divisions or arterial territories, and quickly depresses brain function by transiently abolishing all spontaneous and evoked synaptic activity. Although CSD can be evoked experimentally in subcortical tissue, if this were to happen in a migraineur, one could postulate that the patient may not experience an aura but would still develop headache as a consequence of trigeminal activation. Thus, while CSD has been implicated in both aura and headache, it is less clear as to whether this pandepolarization actually initiates the migraine pain in humans.
In 1941, Karl Spencer Lashley, Ph.D., an eminent psychologist at Harvard University, published a classic paper, “Patterns of cerebral integration indicated by scatomas of migraine,” in which he described his own visual aura (Arch. Neurol. Psychiatry 1941;46:331-9). He meticulously mapped his aura and postulated that if his aura migrated, so did the underlying CNS process. Using those sketches, he was able to calculate that his migraine aura resulted from a wave of intense excitation of the visual cortex followed by complete inhibition of activity. He concluded that the cortical process progressed at a speed of 3 mm/min across the visual cortex. The migraine scotomata he drew have become known as “Lashley’s aura.”
Coincidentally, Aristides Le?o, a researcher from Brazil, was also at Harvard working toward his PhD in physiology. Early in his studies, he noted an interesting cortical response elicited by stimulation of the cortex in rabbits. The distinctive feature of the response was a spread of electrical activity, with a recovery of the initial pattern of spontaneous activity occurring after 5-10 minutes. The speed of the spread was in the range of 3 mm/min, exactly the rate reported by Lashley. Despite being totally unaware of Lashley’s publication 3 years earlier, Le?o noticed the similarity of this spreading depression to the migraine aura. In 1944, he published his research paper, “Spreading depression of activity in the cerebral cortex” (J. Neurophysiol. 1944;7:359-90). This original paper on CSD is still considered one of the classics in the field of neurophysiology. Dr. Lashley’s and Dr. Le?o’s observations changed the way migraine pathophysiology is scientifically conceived.
A review of the early contributors who set the foundation of our current understanding of headache would not be complete without reference to Dr. Harold G. Wolff. His enormous contribution to neurology included landmark studies that led to the modernization of the science of migraine and the anatomy of headache. One such contribution was the discovery that the brain itself is largely insensate. In a 1940 paper coauthored with neurosurgeon Dr. Bronson Ray, Dr. Wolff reported that mechanical stimulation of the brain parenchyma did not cause pain in patients during craniotomy, but that the crucial structures that produced pain were the dura mater, large intracranial blood vessels, and the venous sinuses (Arch. Surg. 1940;41:813-56). The innervation of the pain-sensitive intracranial structures is largely supplied by branches of the first division of the trigeminal nerve. Because the dura mater and large intracranial vessels are pain producing, they have been used in research to model trigeminovascular nociception.
It should also be noted that Dr. Wolff wrote the first textbook on headaches, entitled “Wolff’s Headache and Other Head Pain.” He wrote the first (1948) and second editions (1963) entirely by himself.
The recent study by Dr. Zhang and colleagues at Beth Israel Deaconess Medical Center, Boston, is an extension of earlier work by Dr. Lashley, Dr. Le?o, Dr. Wolff, and others. The purpose of this elegant study was to determine whether CSD can give rise to activation of nociceptors that innervate the meninges. The discovery that the induction of CSD by focal stimulation of the rat visual cortex can lead to long-lasting activation of meningeal receptors is extremely important, especially if it is assumed that the headache phase of migraine is driven by ongoing neuronal activity along the trigeminovascular pathway. It is important to recognize that we have no definitive demonstration to document that what occurs in the animal model directly translates into what takes place in the brain of a migraineur. However, it is this type of sophisticated research that allows us to work toward a better understanding of the mechanisms of migraine with and without aura. The recognition that CSD can trigger activation of nociceptive meningeal receptors in an animal model that has blood vessels and anatomy similar to humans provides important scientific data that could lead to beneficial clinical application.
Stuart B. Black, M.D., is chief of neurology and co-medical director of the neuroscience center at Baylor University Medical Center in Dallas. He has no relevant disclosures.
The relationship between CSD and migraine aura has a pathophysiological overlap that has led to the widely accepted theory that CSD is the electrophysiological substrate underlying the migraine aura. The question as to whether CSD is the brain mechanism causing the migraine headache is more controversial. Although circumstantial evidence and indirect data, including neuroimaging studies, provide strong evidence that CSD does occur in migraine, there is still no definitive demonstration in a migraine patient that CSD causes the proposed activation of nociceptors in the cranial pain-sensitive structures, specifically the meninges, large blood vessels, and large venous sinuses.
The fact that the majority of migraine attacks are not preceded by aura is explained by the theory that CSD events may have variations in expression and may occur in subcortical tissue such as the hippocampus, cerebellum, and striatum. In this situation, an intense and steady depolarization propagates centrifugally in grey matter and occurs within regions of the brain that remain clinically silent. The depolarization does not follow functional divisions or arterial territories, and quickly depresses brain function by transiently abolishing all spontaneous and evoked synaptic activity. Although CSD can be evoked experimentally in subcortical tissue, if this were to happen in a migraineur, one could postulate that the patient may not experience an aura but would still develop headache as a consequence of trigeminal activation. Thus, while CSD has been implicated in both aura and headache, it is less clear as to whether this pandepolarization actually initiates the migraine pain in humans.
In 1941, Karl Spencer Lashley, Ph.D., an eminent psychologist at Harvard University, published a classic paper, “Patterns of cerebral integration indicated by scatomas of migraine,” in which he described his own visual aura (Arch. Neurol. Psychiatry 1941;46:331-9). He meticulously mapped his aura and postulated that if his aura migrated, so did the underlying CNS process. Using those sketches, he was able to calculate that his migraine aura resulted from a wave of intense excitation of the visual cortex followed by complete inhibition of activity. He concluded that the cortical process progressed at a speed of 3 mm/min across the visual cortex. The migraine scotomata he drew have become known as “Lashley’s aura.”
Coincidentally, Aristides Le?o, a researcher from Brazil, was also at Harvard working toward his PhD in physiology. Early in his studies, he noted an interesting cortical response elicited by stimulation of the cortex in rabbits. The distinctive feature of the response was a spread of electrical activity, with a recovery of the initial pattern of spontaneous activity occurring after 5-10 minutes. The speed of the spread was in the range of 3 mm/min, exactly the rate reported by Lashley. Despite being totally unaware of Lashley’s publication 3 years earlier, Le?o noticed the similarity of this spreading depression to the migraine aura. In 1944, he published his research paper, “Spreading depression of activity in the cerebral cortex” (J. Neurophysiol. 1944;7:359-90). This original paper on CSD is still considered one of the classics in the field of neurophysiology. Dr. Lashley’s and Dr. Le?o’s observations changed the way migraine pathophysiology is scientifically conceived.
A review of the early contributors who set the foundation of our current understanding of headache would not be complete without reference to Dr. Harold G. Wolff. His enormous contribution to neurology included landmark studies that led to the modernization of the science of migraine and the anatomy of headache. One such contribution was the discovery that the brain itself is largely insensate. In a 1940 paper coauthored with neurosurgeon Dr. Bronson Ray, Dr. Wolff reported that mechanical stimulation of the brain parenchyma did not cause pain in patients during craniotomy, but that the crucial structures that produced pain were the dura mater, large intracranial blood vessels, and the venous sinuses (Arch. Surg. 1940;41:813-56). The innervation of the pain-sensitive intracranial structures is largely supplied by branches of the first division of the trigeminal nerve. Because the dura mater and large intracranial vessels are pain producing, they have been used in research to model trigeminovascular nociception.
It should also be noted that Dr. Wolff wrote the first textbook on headaches, entitled “Wolff’s Headache and Other Head Pain.” He wrote the first (1948) and second editions (1963) entirely by himself.
The recent study by Dr. Zhang and colleagues at Beth Israel Deaconess Medical Center, Boston, is an extension of earlier work by Dr. Lashley, Dr. Le?o, Dr. Wolff, and others. The purpose of this elegant study was to determine whether CSD can give rise to activation of nociceptors that innervate the meninges. The discovery that the induction of CSD by focal stimulation of the rat visual cortex can lead to long-lasting activation of meningeal receptors is extremely important, especially if it is assumed that the headache phase of migraine is driven by ongoing neuronal activity along the trigeminovascular pathway. It is important to recognize that we have no definitive demonstration to document that what occurs in the animal model directly translates into what takes place in the brain of a migraineur. However, it is this type of sophisticated research that allows us to work toward a better understanding of the mechanisms of migraine with and without aura. The recognition that CSD can trigger activation of nociceptive meningeal receptors in an animal model that has blood vessels and anatomy similar to humans provides important scientific data that could lead to beneficial clinical application.
Stuart B. Black, M.D., is chief of neurology and co-medical director of the neuroscience center at Baylor University Medical Center in Dallas. He has no relevant disclosures.
The relationship between CSD and migraine aura has a pathophysiological overlap that has led to the widely accepted theory that CSD is the electrophysiological substrate underlying the migraine aura. The question as to whether CSD is the brain mechanism causing the migraine headache is more controversial. Although circumstantial evidence and indirect data, including neuroimaging studies, provide strong evidence that CSD does occur in migraine, there is still no definitive demonstration in a migraine patient that CSD causes the proposed activation of nociceptors in the cranial pain-sensitive structures, specifically the meninges, large blood vessels, and large venous sinuses.
The fact that the majority of migraine attacks are not preceded by aura is explained by the theory that CSD events may have variations in expression and may occur in subcortical tissue such as the hippocampus, cerebellum, and striatum. In this situation, an intense and steady depolarization propagates centrifugally in grey matter and occurs within regions of the brain that remain clinically silent. The depolarization does not follow functional divisions or arterial territories, and quickly depresses brain function by transiently abolishing all spontaneous and evoked synaptic activity. Although CSD can be evoked experimentally in subcortical tissue, if this were to happen in a migraineur, one could postulate that the patient may not experience an aura but would still develop headache as a consequence of trigeminal activation. Thus, while CSD has been implicated in both aura and headache, it is less clear as to whether this pandepolarization actually initiates the migraine pain in humans.
In 1941, Karl Spencer Lashley, Ph.D., an eminent psychologist at Harvard University, published a classic paper, “Patterns of cerebral integration indicated by scatomas of migraine,” in which he described his own visual aura (Arch. Neurol. Psychiatry 1941;46:331-9). He meticulously mapped his aura and postulated that if his aura migrated, so did the underlying CNS process. Using those sketches, he was able to calculate that his migraine aura resulted from a wave of intense excitation of the visual cortex followed by complete inhibition of activity. He concluded that the cortical process progressed at a speed of 3 mm/min across the visual cortex. The migraine scotomata he drew have become known as “Lashley’s aura.”
Coincidentally, Aristides Le?o, a researcher from Brazil, was also at Harvard working toward his PhD in physiology. Early in his studies, he noted an interesting cortical response elicited by stimulation of the cortex in rabbits. The distinctive feature of the response was a spread of electrical activity, with a recovery of the initial pattern of spontaneous activity occurring after 5-10 minutes. The speed of the spread was in the range of 3 mm/min, exactly the rate reported by Lashley. Despite being totally unaware of Lashley’s publication 3 years earlier, Le?o noticed the similarity of this spreading depression to the migraine aura. In 1944, he published his research paper, “Spreading depression of activity in the cerebral cortex” (J. Neurophysiol. 1944;7:359-90). This original paper on CSD is still considered one of the classics in the field of neurophysiology. Dr. Lashley’s and Dr. Le?o’s observations changed the way migraine pathophysiology is scientifically conceived.
A review of the early contributors who set the foundation of our current understanding of headache would not be complete without reference to Dr. Harold G. Wolff. His enormous contribution to neurology included landmark studies that led to the modernization of the science of migraine and the anatomy of headache. One such contribution was the discovery that the brain itself is largely insensate. In a 1940 paper coauthored with neurosurgeon Dr. Bronson Ray, Dr. Wolff reported that mechanical stimulation of the brain parenchyma did not cause pain in patients during craniotomy, but that the crucial structures that produced pain were the dura mater, large intracranial blood vessels, and the venous sinuses (Arch. Surg. 1940;41:813-56). The innervation of the pain-sensitive intracranial structures is largely supplied by branches of the first division of the trigeminal nerve. Because the dura mater and large intracranial vessels are pain producing, they have been used in research to model trigeminovascular nociception.
It should also be noted that Dr. Wolff wrote the first textbook on headaches, entitled “Wolff’s Headache and Other Head Pain.” He wrote the first (1948) and second editions (1963) entirely by himself.
The recent study by Dr. Zhang and colleagues at Beth Israel Deaconess Medical Center, Boston, is an extension of earlier work by Dr. Lashley, Dr. Le?o, Dr. Wolff, and others. The purpose of this elegant study was to determine whether CSD can give rise to activation of nociceptors that innervate the meninges. The discovery that the induction of CSD by focal stimulation of the rat visual cortex can lead to long-lasting activation of meningeal receptors is extremely important, especially if it is assumed that the headache phase of migraine is driven by ongoing neuronal activity along the trigeminovascular pathway. It is important to recognize that we have no definitive demonstration to document that what occurs in the animal model directly translates into what takes place in the brain of a migraineur. However, it is this type of sophisticated research that allows us to work toward a better understanding of the mechanisms of migraine with and without aura. The recognition that CSD can trigger activation of nociceptive meningeal receptors in an animal model that has blood vessels and anatomy similar to humans provides important scientific data that could lead to beneficial clinical application.
Stuart B. Black, M.D., is chief of neurology and co-medical director of the neuroscience center at Baylor University Medical Center in Dallas. He has no relevant disclosures.
Frustration has long plagued researchers who have sought to link the visual auras experienced by some migraineurs with the later onset of headache pain. But now, direct evidence from a new study in rats suggests that auras – presumed to be caused by waves of depression of spontaneous electrical activity that propagate slowly through the occipital lobe of the cortex – can trigger the activation of meningeal nociceptors.
XiChun Zhang, Ph.D., and colleagues at Beth Israel Deaconess Medical Center, Boston, reported that those nociceptors in the trigeminal ganglion of rats became activated either immediately or after an average delay of 14 minutes following the administration of a pin prick, electrical pulses, or potassium chloride to the rats’ visual cortex to induce waves of cortical spreading depression (CSD).
The investigators conducted 83 trials of the three methods of cortical stimulation in 53 rats. Of 64 trials that induced waves of CSD, 31 resulted in increases in neuronal firing rates (25% or greater than baseline) that lasted at least 11 minutes. Sustained neuronal activation was not seen in all but 1 of the 19 trials that failed to produce CSD (J. Neurosci. 2010;30:8807-14).
Long-lasting neuronal activation coincided with the wave of CSD in 10 of the 31 trials in which both waves of CSD and increased neuronal firing were observed.
“The immediate activation of meningeal nociceptors may be clinically relevant to uncommon cases where migraine aura appears together with the headache,” Dr. Zhang and associates wrote.
In the other 21 trials that recorded CSD and increased neuronal firing, long-lasting neuronal activation began a mean of 14 minutes after the CSD waves.
The delayed neuronal activation observed in those 21 trials “may be relevant to the typical delay between the onset of aura and the onset of migraine headache, though the underlying mechanisms remain unknown,” wrote Dr. Zhang and coauthors. They noted that this observation may be the “most clinically promising” because “intervention during the aura phase with drugs that would block the delayed induction of neuronal activation could potentially preempt the onset of migraine headache.”
There were no differences in the pattern of neuronal activation between C- and A-delta-nociceptors or between the methods of cortical stimulation. Responses to cortical stimulation in rats that had their ipsilateral sphenopalatine ganglion excised were no different from those in other rats, which indicated that the parasympathetic innervation of the dura does not contribute to the long-lasting activation of meningeal nociceptors.
The investigators proposed that the sustained activation of meningeal nociceptors could be the result of either a “short-lasting release of algesic molecules” during CSD that promotes an acute activation of the nociceptor and gives rise to an ongoing sensitization that typically outlasts the stimulus by 30-60 minutes, or an ongoing release of algesic molecules for up to 1 hour during CSD.
However, because this period of sustained activation of the meningeal nociceptors may not be sufficient “in and of itself” to explain the 4- to 72-hour duration of the headache phase of migraine, Dr. Zhang and associates proposed that the “duration of nociceptor activation may be sufficient to promote ongoing activity of central trigeminovascular neurons that eventually becomes independent of incoming signals from the nociceptors and can last many hours.”
Dr. Zhang’s research was funded by grants from the National Institutes of Health. Dr. Black has no relevant disclosures.
Frustration has long plagued researchers who have sought to link the visual auras experienced by some migraineurs with the later onset of headache pain. But now, direct evidence from a new study in rats suggests that auras – presumed to be caused by waves of depression of spontaneous electrical activity that propagate slowly through the occipital lobe of the cortex – can trigger the activation of meningeal nociceptors.
XiChun Zhang, Ph.D., and colleagues at Beth Israel Deaconess Medical Center, Boston, reported that those nociceptors in the trigeminal ganglion of rats became activated either immediately or after an average delay of 14 minutes following the administration of a pin prick, electrical pulses, or potassium chloride to the rats’ visual cortex to induce waves of cortical spreading depression (CSD).
The investigators conducted 83 trials of the three methods of cortical stimulation in 53 rats. Of 64 trials that induced waves of CSD, 31 resulted in increases in neuronal firing rates (25% or greater than baseline) that lasted at least 11 minutes. Sustained neuronal activation was not seen in all but 1 of the 19 trials that failed to produce CSD (J. Neurosci. 2010;30:8807-14).
Long-lasting neuronal activation coincided with the wave of CSD in 10 of the 31 trials in which both waves of CSD and increased neuronal firing were observed.
“The immediate activation of meningeal nociceptors may be clinically relevant to uncommon cases where migraine aura appears together with the headache,” Dr. Zhang and associates wrote.
In the other 21 trials that recorded CSD and increased neuronal firing, long-lasting neuronal activation began a mean of 14 minutes after the CSD waves.
The delayed neuronal activation observed in those 21 trials “may be relevant to the typical delay between the onset of aura and the onset of migraine headache, though the underlying mechanisms remain unknown,” wrote Dr. Zhang and coauthors. They noted that this observation may be the “most clinically promising” because “intervention during the aura phase with drugs that would block the delayed induction of neuronal activation could potentially preempt the onset of migraine headache.”
There were no differences in the pattern of neuronal activation between C- and A-delta-nociceptors or between the methods of cortical stimulation. Responses to cortical stimulation in rats that had their ipsilateral sphenopalatine ganglion excised were no different from those in other rats, which indicated that the parasympathetic innervation of the dura does not contribute to the long-lasting activation of meningeal nociceptors.
The investigators proposed that the sustained activation of meningeal nociceptors could be the result of either a “short-lasting release of algesic molecules” during CSD that promotes an acute activation of the nociceptor and gives rise to an ongoing sensitization that typically outlasts the stimulus by 30-60 minutes, or an ongoing release of algesic molecules for up to 1 hour during CSD.
However, because this period of sustained activation of the meningeal nociceptors may not be sufficient “in and of itself” to explain the 4- to 72-hour duration of the headache phase of migraine, Dr. Zhang and associates proposed that the “duration of nociceptor activation may be sufficient to promote ongoing activity of central trigeminovascular neurons that eventually becomes independent of incoming signals from the nociceptors and can last many hours.”
Dr. Zhang’s research was funded by grants from the National Institutes of Health. Dr. Black has no relevant disclosures.
Drug Combo May Prevent Glioblastoma Recurrence
Gamma-secretase inhibitors could play an important role in augmenting the effectiveness of temozolomide chemotherapy for glioblastoma multiforme if the results obtained in recent in vitro, ex vivo, and in vivo experiments are supported in future studies.

Although temozolomide (TMZ) has increased the 2-year survival rate of patients with glioblastoma multiforme (GBM) when it is used in combination with surgical resection and radiotherapy, some cells still escape treatment in most patients and contribute to local tumor recurrence. Gamma-secretase inhibitors (GSIs) are an attractive therapeutic option to bring to the clinic because they have been found in previous studies to stop both glioblastoma cell growth and the formation of glioblastoma neurospheres by blocking the Notch signaling pathway, which is commonly overexpressed in glioblastoma cells, according to Candace A. Gilbert and her colleagues at the University of Massachusetts, Worcester.
Before Ms. Gilbert and her associates tested the combination of TMZ and a GSI in vivo, they tested TMZ alone, a GSI alone, and both together on neurosphere cultures derived from patients’ GBMs. Although GSI treatment alone decreased Notch pathway signaling and reduced neurosphere formation, it could not stop the proliferation of GBM cells and the formation of secondary neurospheres. Although treatment with TMZ alone and combined treatment with TMZ and a GSI yielded similar decreases in initial neurosphere formation, cultures that were treated with the combination recovered to a smaller size, and there were fewer of them than was the case for those that were treated with TMZ alone. When these neurosphere cultures were dissociated to single cells and replated, the cells that underwent combination treatment formed far fewer secondary neurospheres than did those that had been treated with only TMZ. Treatment with both drugs also led to significantly fewer cells in each neurosphere that were capable of self-renewal.
Further in vitro experiments of the combination of drugs showed that a single dose of a GSI could reduce neurosphere recovery and the formation of secondary neurospheres only when the GSI was administered 24 hours after TMZ, in comparison with TMZ alone.
When the tumor cells were treated and then injected subcutaneously into immunocompromised mice, the researchers observed palpable tumor growth in very few mice that received cells treated with TMZ and a GSI (tumor latencies, 43-96 days), compared with tumor growth in all mice that received cells treated with a GSI alone (latencies, 3-16 days) and growth in nearly all mice that received cells treated with TMZ alone (latencies, 25-43 days).
In another group of immunocompromised mice, tumor cells that were injected subcutaneously were allowed to grow to 150 mm3. Ms. Gilbert and her associates found that the xenografts were completely eliminated in half of immunocompromised mice by an intraperitoneal injection of TMZ followed by ingestion of a GSI mixed into their food supply. The mice also survived free of a palpable tumor until they were euthanized at 150 days. The remaining half of the mice that received the combination treatment showed tumor progression at a mean of 26 days.
All tumor masses progressed (doubled in size) in mice that received only TMZ.
“Because Notch activity is associated with GBM stem cell function and survival, and the cells that survive TMZ-only treatment are capable of self-renewal and tumor initiation, it is probable that the cells targeted by TMZ plus GSI treatment possess a cancer stem cell phenotype,” the researchers wrote (Cancer Res. 2010 Aug. 10 [doi:10.1158/0008-5472.CAN-10-1378]).
They suggested that the variability of response to combined treatment in the in vivo studies could have been the result of a TMZ concentration that was not “high enough to induce a cell cycle arrest in all of the cells capable of recovery, which could hinder GSI enhancement,” or a “slight variability” in food consumption.
The need for only a single dose of a GSI to enhance TMZ therapy is beneficial, the researchers noted, because GSIs can cause cytotoxicity in the gastrointestinal tract. They found no change in the weight of the mice during combined treatment.
“These studies suggest a role for TMZ plus GSI therapy to reduce recurrences in patients with low tumor burden after surgical resection of the bulk tumor,” wrote the investigators, who acknowledged that they ultimately need to include radiation in their treatment schedule to see how it contributes to combination therapy with TMZ and GSIs.
In comments on this study, Dr. Alyx B. Porter of the Mayo Clinic in Arizona said that it “certainly provides a springboard for considering future directions in the use of GSIs, and may indeed provide further treatment options for this patient population in whom options are greatly needed.”
Dr. Porter noted that since 2005, the treatment standard for GBM has been concomitant TMZ with radiotherapy followed by adjuvant TMZ. This resulted in a 2-year survival rate of 26.5%, which was higher than any prior treatment regimen had shown. The time to progression on this regimen is about 6 months, she said, which points to the refractory and aggressive nature of this tumor.
Thus far, she added, bevacizumab has been the only agent approved by the Food and Drug Administration for use in the setting of recurrent GBM. Given the dismal prognosis for this patient population, novel agents are needed not only to augment up-front therapy to prevent recurrence but also to provide further treatment options in the recurrent setting. Dr. Porter described the study of Ms. Gilbert and colleagues as “eloquent,” and noted “the remarkable in vitro and in vivo data” suggesting that GSI and TMZ act together to halt neurosphere replication, and the finding that administering a GSI after TMZ may have the maximum impact in affecting neurosphere repopulation. “These data suggest that GSIs may indeed have an impact on glioblastoma recurrence and time to progression.”
Dr. Porter agreed with the authors’ observation that future studies to assess the total impact of the GSI in the GBM population will need to incorporate irradiation in addition to TMZ to reflect a more accurate sense of the full effect and toxicity of the GSI.
The National Institutes of Health and the CVIP Technology Development Fund funded the research. The investigators had no conflicts of interest to disclose.
Gamma-secretase inhibitors could play an important role in augmenting the effectiveness of temozolomide chemotherapy for glioblastoma multiforme if the results obtained in recent in vitro, ex vivo, and in vivo experiments are supported in future studies.
Although temozolomide (TMZ) has increased the 2-year survival rate of patients with glioblastoma multiforme (GBM) when it is used in combination with surgical resection and radiotherapy, some cells still escape treatment in most patients and contribute to local tumor recurrence. Gamma-secretase inhibitors (GSIs) are an attractive therapeutic option to bring to the clinic because they have been found in previous studies to stop both glioblastoma cell growth and the formation of glioblastoma neurospheres by blocking the Notch signaling pathway, which is commonly overexpressed in glioblastoma cells, according to Candace A. Gilbert and her colleagues at the University of Massachusetts, Worcester.
Before Ms. Gilbert and her associates tested the combination of TMZ and a GSI in vivo, they tested TMZ alone, a GSI alone, and both together on neurosphere cultures derived from patients’ GBMs. Although GSI treatment alone decreased Notch pathway signaling and reduced neurosphere formation, it could not stop the proliferation of GBM cells and the formation of secondary neurospheres. Although treatment with TMZ alone and combined treatment with TMZ and a GSI yielded similar decreases in initial neurosphere formation, cultures that were treated with the combination recovered to a smaller size, and there were fewer of them than was the case for those that were treated with TMZ alone. When these neurosphere cultures were dissociated to single cells and replated, the cells that underwent combination treatment formed far fewer secondary neurospheres than did those that had been treated with only TMZ. Treatment with both drugs also led to significantly fewer cells in each neurosphere that were capable of self-renewal.
Further in vitro experiments of the combination of drugs showed that a single dose of a GSI could reduce neurosphere recovery and the formation of secondary neurospheres only when the GSI was administered 24 hours after TMZ, in comparison with TMZ alone.
When the tumor cells were treated and then injected subcutaneously into immunocompromised mice, the researchers observed palpable tumor growth in very few mice that received cells treated with TMZ and a GSI (tumor latencies, 43-96 days), compared with tumor growth in all mice that received cells treated with a GSI alone (latencies, 3-16 days) and growth in nearly all mice that received cells treated with TMZ alone (latencies, 25-43 days).
In another group of immunocompromised mice, tumor cells that were injected subcutaneously were allowed to grow to 150 mm3. Ms. Gilbert and her associates found that the xenografts were completely eliminated in half of immunocompromised mice by an intraperitoneal injection of TMZ followed by ingestion of a GSI mixed into their food supply. The mice also survived free of a palpable tumor until they were euthanized at 150 days. The remaining half of the mice that received the combination treatment showed tumor progression at a mean of 26 days.
All tumor masses progressed (doubled in size) in mice that received only TMZ.
“Because Notch activity is associated with GBM stem cell function and survival, and the cells that survive TMZ-only treatment are capable of self-renewal and tumor initiation, it is probable that the cells targeted by TMZ plus GSI treatment possess a cancer stem cell phenotype,” the researchers wrote (Cancer Res. 2010 Aug. 10 [doi:10.1158/0008-5472.CAN-10-1378]).
They suggested that the variability of response to combined treatment in the in vivo studies could have been the result of a TMZ concentration that was not “high enough to induce a cell cycle arrest in all of the cells capable of recovery, which could hinder GSI enhancement,” or a “slight variability” in food consumption.
The need for only a single dose of a GSI to enhance TMZ therapy is beneficial, the researchers noted, because GSIs can cause cytotoxicity in the gastrointestinal tract. They found no change in the weight of the mice during combined treatment.
“These studies suggest a role for TMZ plus GSI therapy to reduce recurrences in patients with low tumor burden after surgical resection of the bulk tumor,” wrote the investigators, who acknowledged that they ultimately need to include radiation in their treatment schedule to see how it contributes to combination therapy with TMZ and GSIs.
In comments on this study, Dr. Alyx B. Porter of the Mayo Clinic in Arizona said that it “certainly provides a springboard for considering future directions in the use of GSIs, and may indeed provide further treatment options for this patient population in whom options are greatly needed.”
Dr. Porter noted that since 2005, the treatment standard for GBM has been concomitant TMZ with radiotherapy followed by adjuvant TMZ. This resulted in a 2-year survival rate of 26.5%, which was higher than any prior treatment regimen had shown. The time to progression on this regimen is about 6 months, she said, which points to the refractory and aggressive nature of this tumor.
Thus far, she added, bevacizumab has been the only agent approved by the Food and Drug Administration for use in the setting of recurrent GBM. Given the dismal prognosis for this patient population, novel agents are needed not only to augment up-front therapy to prevent recurrence but also to provide further treatment options in the recurrent setting. Dr. Porter described the study of Ms. Gilbert and colleagues as “eloquent,” and noted “the remarkable in vitro and in vivo data” suggesting that GSI and TMZ act together to halt neurosphere replication, and the finding that administering a GSI after TMZ may have the maximum impact in affecting neurosphere repopulation. “These data suggest that GSIs may indeed have an impact on glioblastoma recurrence and time to progression.”
Dr. Porter agreed with the authors’ observation that future studies to assess the total impact of the GSI in the GBM population will need to incorporate irradiation in addition to TMZ to reflect a more accurate sense of the full effect and toxicity of the GSI.
The National Institutes of Health and the CVIP Technology Development Fund funded the research. The investigators had no conflicts of interest to disclose.
Gamma-secretase inhibitors could play an important role in augmenting the effectiveness of temozolomide chemotherapy for glioblastoma multiforme if the results obtained in recent in vitro, ex vivo, and in vivo experiments are supported in future studies.
Although temozolomide (TMZ) has increased the 2-year survival rate of patients with glioblastoma multiforme (GBM) when it is used in combination with surgical resection and radiotherapy, some cells still escape treatment in most patients and contribute to local tumor recurrence. Gamma-secretase inhibitors (GSIs) are an attractive therapeutic option to bring to the clinic because they have been found in previous studies to stop both glioblastoma cell growth and the formation of glioblastoma neurospheres by blocking the Notch signaling pathway, which is commonly overexpressed in glioblastoma cells, according to Candace A. Gilbert and her colleagues at the University of Massachusetts, Worcester.
Before Ms. Gilbert and her associates tested the combination of TMZ and a GSI in vivo, they tested TMZ alone, a GSI alone, and both together on neurosphere cultures derived from patients’ GBMs. Although GSI treatment alone decreased Notch pathway signaling and reduced neurosphere formation, it could not stop the proliferation of GBM cells and the formation of secondary neurospheres. Although treatment with TMZ alone and combined treatment with TMZ and a GSI yielded similar decreases in initial neurosphere formation, cultures that were treated with the combination recovered to a smaller size, and there were fewer of them than was the case for those that were treated with TMZ alone. When these neurosphere cultures were dissociated to single cells and replated, the cells that underwent combination treatment formed far fewer secondary neurospheres than did those that had been treated with only TMZ. Treatment with both drugs also led to significantly fewer cells in each neurosphere that were capable of self-renewal.
Further in vitro experiments of the combination of drugs showed that a single dose of a GSI could reduce neurosphere recovery and the formation of secondary neurospheres only when the GSI was administered 24 hours after TMZ, in comparison with TMZ alone.
When the tumor cells were treated and then injected subcutaneously into immunocompromised mice, the researchers observed palpable tumor growth in very few mice that received cells treated with TMZ and a GSI (tumor latencies, 43-96 days), compared with tumor growth in all mice that received cells treated with a GSI alone (latencies, 3-16 days) and growth in nearly all mice that received cells treated with TMZ alone (latencies, 25-43 days).
In another group of immunocompromised mice, tumor cells that were injected subcutaneously were allowed to grow to 150 mm3. Ms. Gilbert and her associates found that the xenografts were completely eliminated in half of immunocompromised mice by an intraperitoneal injection of TMZ followed by ingestion of a GSI mixed into their food supply. The mice also survived free of a palpable tumor until they were euthanized at 150 days. The remaining half of the mice that received the combination treatment showed tumor progression at a mean of 26 days.
All tumor masses progressed (doubled in size) in mice that received only TMZ.
“Because Notch activity is associated with GBM stem cell function and survival, and the cells that survive TMZ-only treatment are capable of self-renewal and tumor initiation, it is probable that the cells targeted by TMZ plus GSI treatment possess a cancer stem cell phenotype,” the researchers wrote (Cancer Res. 2010 Aug. 10 [doi:10.1158/0008-5472.CAN-10-1378]).
They suggested that the variability of response to combined treatment in the in vivo studies could have been the result of a TMZ concentration that was not “high enough to induce a cell cycle arrest in all of the cells capable of recovery, which could hinder GSI enhancement,” or a “slight variability” in food consumption.
The need for only a single dose of a GSI to enhance TMZ therapy is beneficial, the researchers noted, because GSIs can cause cytotoxicity in the gastrointestinal tract. They found no change in the weight of the mice during combined treatment.
“These studies suggest a role for TMZ plus GSI therapy to reduce recurrences in patients with low tumor burden after surgical resection of the bulk tumor,” wrote the investigators, who acknowledged that they ultimately need to include radiation in their treatment schedule to see how it contributes to combination therapy with TMZ and GSIs.
In comments on this study, Dr. Alyx B. Porter of the Mayo Clinic in Arizona said that it “certainly provides a springboard for considering future directions in the use of GSIs, and may indeed provide further treatment options for this patient population in whom options are greatly needed.”
Dr. Porter noted that since 2005, the treatment standard for GBM has been concomitant TMZ with radiotherapy followed by adjuvant TMZ. This resulted in a 2-year survival rate of 26.5%, which was higher than any prior treatment regimen had shown. The time to progression on this regimen is about 6 months, she said, which points to the refractory and aggressive nature of this tumor.
Thus far, she added, bevacizumab has been the only agent approved by the Food and Drug Administration for use in the setting of recurrent GBM. Given the dismal prognosis for this patient population, novel agents are needed not only to augment up-front therapy to prevent recurrence but also to provide further treatment options in the recurrent setting. Dr. Porter described the study of Ms. Gilbert and colleagues as “eloquent,” and noted “the remarkable in vitro and in vivo data” suggesting that GSI and TMZ act together to halt neurosphere replication, and the finding that administering a GSI after TMZ may have the maximum impact in affecting neurosphere repopulation. “These data suggest that GSIs may indeed have an impact on glioblastoma recurrence and time to progression.”
Dr. Porter agreed with the authors’ observation that future studies to assess the total impact of the GSI in the GBM population will need to incorporate irradiation in addition to TMZ to reflect a more accurate sense of the full effect and toxicity of the GSI.
The National Institutes of Health and the CVIP Technology Development Fund funded the research. The investigators had no conflicts of interest to disclose.
Report Highlights Gaps in Alzheimer's Research
BETHESDA, MD. – Current knowledge about the epidemiology of Alzheimer's disease and cognitive decline has not provided enough evidence to recommend specific, preventive interventions, according to a draft “state-of-the-science” report issued by a panel of experts assembled by the National Institutes of Health.
The 15-member panel found that there is not enough evidence to support the use of pharmaceutical agents or dietary supplements to prevent cognitive decline or Alzheimer's disease, but ongoing additional studies of antihypertensive medications, omega-3 fatty acids, physical activity, and cognitive engagement “may provide new insight into the prevention of delay of cognitive decline or Alzheimer's disease.
“We're hoping that our report is going to supply physicians with accurate information that they can give to their patients” to clarify what interventions may be worth continuing or pursuing and which should be discontinued, said panelist Dr. Carl C. Bell, director of public and community psychiatry at the University of Illinois at Chicago, said at a press telebriefing.
A wide range of modifiable factors has been reported to be associated with risk for Alzheimer's disease, such as diabetes diet, medication, or lifestyle, but the overall quality of evidence from these studies is low, the panel said.
Panel member Arnold L. Potosky, Ph.D., of Georgetown University in Washington said that it is important for physicians to discuss participation in clinical studies with their patients.
The panel recommended that further research should include:
▸ The development and use of rigorous, consensus-based diagnostic criteria for Alzheimer's disease and mild cognitive impairment.
▸ The development and use of a standardized, well-validated, and culturally sensitive battery of outcome measures across research studies.
▸ The collection of data from caregivers of people with mild cognitive impairment or early Alzheimer's disease in a systematic manner in observational studies and randomized, controlled trials.
▸ The conduct of large-scale, long-term population-based studies with well-validated exposure and outcome measures in people followed from middle to old age.
▸ The leveraging of alternative research resources and platforms that facilitate long-term longitudinal assessments, such as a multicenter Alzheimer's disease registry or observational studies within large health care delivery systems with defined populations and well-developed electronic health records.
▸ The creation of a simple, inexpensive, quantitative instrument that can be administered by a trained nonexpert to assess change in cognitive status over time.
The panel based its draft statement on an evidence report from the Evidence-Based Practice Center at Duke University's Clinical Research Institute, which was commissioned by the Agency for Healthcare Research and Quality.
{kind=link}
The report aims 'to clarify what interventions may be worth continuing … and which should be discontinued.'
Source DR. BELL
BETHESDA, MD. – Current knowledge about the epidemiology of Alzheimer's disease and cognitive decline has not provided enough evidence to recommend specific, preventive interventions, according to a draft “state-of-the-science” report issued by a panel of experts assembled by the National Institutes of Health.
The 15-member panel found that there is not enough evidence to support the use of pharmaceutical agents or dietary supplements to prevent cognitive decline or Alzheimer's disease, but ongoing additional studies of antihypertensive medications, omega-3 fatty acids, physical activity, and cognitive engagement “may provide new insight into the prevention of delay of cognitive decline or Alzheimer's disease.
“We're hoping that our report is going to supply physicians with accurate information that they can give to their patients” to clarify what interventions may be worth continuing or pursuing and which should be discontinued, said panelist Dr. Carl C. Bell, director of public and community psychiatry at the University of Illinois at Chicago, said at a press telebriefing.
A wide range of modifiable factors has been reported to be associated with risk for Alzheimer's disease, such as diabetes diet, medication, or lifestyle, but the overall quality of evidence from these studies is low, the panel said.
Panel member Arnold L. Potosky, Ph.D., of Georgetown University in Washington said that it is important for physicians to discuss participation in clinical studies with their patients.
The panel recommended that further research should include:
▸ The development and use of rigorous, consensus-based diagnostic criteria for Alzheimer's disease and mild cognitive impairment.
▸ The development and use of a standardized, well-validated, and culturally sensitive battery of outcome measures across research studies.
▸ The collection of data from caregivers of people with mild cognitive impairment or early Alzheimer's disease in a systematic manner in observational studies and randomized, controlled trials.
▸ The conduct of large-scale, long-term population-based studies with well-validated exposure and outcome measures in people followed from middle to old age.
▸ The leveraging of alternative research resources and platforms that facilitate long-term longitudinal assessments, such as a multicenter Alzheimer's disease registry or observational studies within large health care delivery systems with defined populations and well-developed electronic health records.
▸ The creation of a simple, inexpensive, quantitative instrument that can be administered by a trained nonexpert to assess change in cognitive status over time.
The panel based its draft statement on an evidence report from the Evidence-Based Practice Center at Duke University's Clinical Research Institute, which was commissioned by the Agency for Healthcare Research and Quality.
The report aims 'to clarify what interventions may be worth continuing … and which should be discontinued.'
Source DR. BELL
BETHESDA, MD. – Current knowledge about the epidemiology of Alzheimer's disease and cognitive decline has not provided enough evidence to recommend specific, preventive interventions, according to a draft “state-of-the-science” report issued by a panel of experts assembled by the National Institutes of Health.
The 15-member panel found that there is not enough evidence to support the use of pharmaceutical agents or dietary supplements to prevent cognitive decline or Alzheimer's disease, but ongoing additional studies of antihypertensive medications, omega-3 fatty acids, physical activity, and cognitive engagement “may provide new insight into the prevention of delay of cognitive decline or Alzheimer's disease.
“We're hoping that our report is going to supply physicians with accurate information that they can give to their patients” to clarify what interventions may be worth continuing or pursuing and which should be discontinued, said panelist Dr. Carl C. Bell, director of public and community psychiatry at the University of Illinois at Chicago, said at a press telebriefing.
A wide range of modifiable factors has been reported to be associated with risk for Alzheimer's disease, such as diabetes diet, medication, or lifestyle, but the overall quality of evidence from these studies is low, the panel said.
Panel member Arnold L. Potosky, Ph.D., of Georgetown University in Washington said that it is important for physicians to discuss participation in clinical studies with their patients.
The panel recommended that further research should include:
▸ The development and use of rigorous, consensus-based diagnostic criteria for Alzheimer's disease and mild cognitive impairment.
▸ The development and use of a standardized, well-validated, and culturally sensitive battery of outcome measures across research studies.
▸ The collection of data from caregivers of people with mild cognitive impairment or early Alzheimer's disease in a systematic manner in observational studies and randomized, controlled trials.
▸ The conduct of large-scale, long-term population-based studies with well-validated exposure and outcome measures in people followed from middle to old age.
▸ The leveraging of alternative research resources and platforms that facilitate long-term longitudinal assessments, such as a multicenter Alzheimer's disease registry or observational studies within large health care delivery systems with defined populations and well-developed electronic health records.
▸ The creation of a simple, inexpensive, quantitative instrument that can be administered by a trained nonexpert to assess change in cognitive status over time.
The panel based its draft statement on an evidence report from the Evidence-Based Practice Center at Duke University's Clinical Research Institute, which was commissioned by the Agency for Healthcare Research and Quality.
The report aims 'to clarify what interventions may be worth continuing … and which should be discontinued.'
Source DR. BELL
IVIG Reduced Brain Atrophy in Alzheimer's
Major Finding: Treatment with a range of doses of IVIG for 18 months resulted in a mean increase of 6.7% in lateral ventricular volume, which was significantly lower than the 12.3% increase observed with placebo.
Data Source: A double-blind, randomized, placebo-controlled phase II trial of 24 patients with mild to moderate Alzheimer's disease.
Disclosures: Baxter Healthcare sponsored the study of IVIG, with additional support from the Citigroup Foundation and the National Institutes of Health. Dr. Relkin reporting no relevant disclosures besides receiving a research grant from Baxter Healthcare to study IVIG.
TORONTO – Intravenous immunoglobulin therapy reduced brain atrophy in patients with mild to moderate Alzheimer's disease in a small phase II trial. The finding suggests that specific IgG antibody components found in the blood product might be treatment candidates for the disease.
“Relative to what we have available right now [to treat Alzheimer's disease], this is a very promising outcome, and it's associated with a reduction in the rate of brain atrophy comparable with age-matched normals,” Dr. Norman Relkin said during a poster presentation.
Enlargement of the cerebral lateral ventricles is known to occur as a consequence of brain atrophy in Alzheimer's disease (AD). This increase in ventricular volume is correlated with cognitive decline and increases in Alzheimer's disease neuropathology.
Dr. Relkin and his colleagues compared intravenous immunoglobulin (IVIG) therapy against placebo in a 6-month, double-blind, randomized study of 24 patients with mild to moderate AD. In a 12-month extension phase of the study, 16 patients who originally were randomized to IVIG continued to receive the same doses of IVIG, whereas 8 placebo-treated patients were re-randomized to one of four doses of IVIG. The investigators used an IVIG product produced by Baxter Healthcare called Gammagard.
IVIG exhibited a dose-dependent effect on brain atrophy in which higher doses resulted in less atrophy. Among 14 IVIG-treated patients who underwent volumetric MRI at baseline and after 18 months, the yearly increase in lateral ventricle volume measured with volumetric MRI was lowest in patients treated with 0.4 mg/kg every 2 weeks (2.4%) and highest in those treated with 0.2 mg/kg every 2 weeks (11.2%). The doses of IVIG given to patients ranged from 0.2 mg/kg every 2 weeks to 0.8 mg/kg every 4 weeks.
The volume of the lateral ventricles increased by a mean of 6.7% per year during treatment with IVIG (all doses combined), which was significantly lower than the 12.3% annual rate of increase observed in six placebo-treated patients. Only the 0.4 mg/kg dose of IVIG given every 2 weeks resulted in significantly less change in total brain volume than did treatment with placebo (−0.62% vs. −2.24%, respectively).
“In addition to the brain imaging, we have previously shown changes in cerebrospinal fluid and plasma amyloid levels … and levels of cerebral metabolism changing in response to treatment,” said Dr. Relkin, director of the Memory Disorders Program at New York–Presbyterian Hospital/Weill Cornell Medical Center.
The reduction in brain atrophy was significantly correlated with improvement in clinical outcomes at 18 months on the Clinical Global Impression of Change and the cognitive subscale of the Alzheimer's Disease Assessment Scale. Patients' baseline characteristics were not correlated with volumetric MRI outcomes.
“This is a 'kitchen sink' approach, so the next step is to find what is in [IVIG] that is causing the therapeutic effect. … We know that it has a fairly good complement of antiamyloid antibodies. Those are prime candidates, but we don't know for sure yet that those are ones responsible for a therapeutic effect,” Dr. Relkin said in an interview.
In addition to an ongoing, multicenter, phase III study of IVIG in 360 patients with mild to moderate AD, Dr. Relkin and his colleagues are testing subsets of antibodies within IVIG in cell culture–based studies and preclinical animal models to see which components are therapeutically relevant. “We are not encouraging people to use [IVIG] off-label for Alzheimer's disease, even though it has been safe and well tolerated in these small studies,” he said. “It has never been studied in the Alzheimer's population before.”
Baxter Healthcare sponsored the phase I and II studies of IVIG, with additional support from the Citigroup Foundation and the National Institutes of Health. The phase III trial is cosponsored by Baxter and the NIH. Dr. Relkin reported no relevant disclosures besides receiving a research grant from Baxter Healthcare to study IVIG.
{kind=link}
The outcome is associated with a reduction in the brain atrophy rate, comparable with age-matched normals.
Source DR. RELKIN
{kind=link}
After 18 months, the ventricular enlargement rate was greater with placebo (left) than it was with IVIG (right).
Source Courtesy Dr. Dana Moore and Dr. Norman Relkin
Major Finding: Treatment with a range of doses of IVIG for 18 months resulted in a mean increase of 6.7% in lateral ventricular volume, which was significantly lower than the 12.3% increase observed with placebo.
Data Source: A double-blind, randomized, placebo-controlled phase II trial of 24 patients with mild to moderate Alzheimer's disease.
Disclosures: Baxter Healthcare sponsored the study of IVIG, with additional support from the Citigroup Foundation and the National Institutes of Health. Dr. Relkin reporting no relevant disclosures besides receiving a research grant from Baxter Healthcare to study IVIG.
TORONTO – Intravenous immunoglobulin therapy reduced brain atrophy in patients with mild to moderate Alzheimer's disease in a small phase II trial. The finding suggests that specific IgG antibody components found in the blood product might be treatment candidates for the disease.
“Relative to what we have available right now [to treat Alzheimer's disease], this is a very promising outcome, and it's associated with a reduction in the rate of brain atrophy comparable with age-matched normals,” Dr. Norman Relkin said during a poster presentation.
Enlargement of the cerebral lateral ventricles is known to occur as a consequence of brain atrophy in Alzheimer's disease (AD). This increase in ventricular volume is correlated with cognitive decline and increases in Alzheimer's disease neuropathology.
Dr. Relkin and his colleagues compared intravenous immunoglobulin (IVIG) therapy against placebo in a 6-month, double-blind, randomized study of 24 patients with mild to moderate AD. In a 12-month extension phase of the study, 16 patients who originally were randomized to IVIG continued to receive the same doses of IVIG, whereas 8 placebo-treated patients were re-randomized to one of four doses of IVIG. The investigators used an IVIG product produced by Baxter Healthcare called Gammagard.
IVIG exhibited a dose-dependent effect on brain atrophy in which higher doses resulted in less atrophy. Among 14 IVIG-treated patients who underwent volumetric MRI at baseline and after 18 months, the yearly increase in lateral ventricle volume measured with volumetric MRI was lowest in patients treated with 0.4 mg/kg every 2 weeks (2.4%) and highest in those treated with 0.2 mg/kg every 2 weeks (11.2%). The doses of IVIG given to patients ranged from 0.2 mg/kg every 2 weeks to 0.8 mg/kg every 4 weeks.
The volume of the lateral ventricles increased by a mean of 6.7% per year during treatment with IVIG (all doses combined), which was significantly lower than the 12.3% annual rate of increase observed in six placebo-treated patients. Only the 0.4 mg/kg dose of IVIG given every 2 weeks resulted in significantly less change in total brain volume than did treatment with placebo (−0.62% vs. −2.24%, respectively).
“In addition to the brain imaging, we have previously shown changes in cerebrospinal fluid and plasma amyloid levels … and levels of cerebral metabolism changing in response to treatment,” said Dr. Relkin, director of the Memory Disorders Program at New York–Presbyterian Hospital/Weill Cornell Medical Center.
The reduction in brain atrophy was significantly correlated with improvement in clinical outcomes at 18 months on the Clinical Global Impression of Change and the cognitive subscale of the Alzheimer's Disease Assessment Scale. Patients' baseline characteristics were not correlated with volumetric MRI outcomes.
“This is a 'kitchen sink' approach, so the next step is to find what is in [IVIG] that is causing the therapeutic effect. … We know that it has a fairly good complement of antiamyloid antibodies. Those are prime candidates, but we don't know for sure yet that those are ones responsible for a therapeutic effect,” Dr. Relkin said in an interview.
In addition to an ongoing, multicenter, phase III study of IVIG in 360 patients with mild to moderate AD, Dr. Relkin and his colleagues are testing subsets of antibodies within IVIG in cell culture–based studies and preclinical animal models to see which components are therapeutically relevant. “We are not encouraging people to use [IVIG] off-label for Alzheimer's disease, even though it has been safe and well tolerated in these small studies,” he said. “It has never been studied in the Alzheimer's population before.”
Baxter Healthcare sponsored the phase I and II studies of IVIG, with additional support from the Citigroup Foundation and the National Institutes of Health. The phase III trial is cosponsored by Baxter and the NIH. Dr. Relkin reported no relevant disclosures besides receiving a research grant from Baxter Healthcare to study IVIG.
The outcome is associated with a reduction in the brain atrophy rate, comparable with age-matched normals.
Source DR. RELKIN
After 18 months, the ventricular enlargement rate was greater with placebo (left) than it was with IVIG (right).
Source Courtesy Dr. Dana Moore and Dr. Norman Relkin
Major Finding: Treatment with a range of doses of IVIG for 18 months resulted in a mean increase of 6.7% in lateral ventricular volume, which was significantly lower than the 12.3% increase observed with placebo.
Data Source: A double-blind, randomized, placebo-controlled phase II trial of 24 patients with mild to moderate Alzheimer's disease.
Disclosures: Baxter Healthcare sponsored the study of IVIG, with additional support from the Citigroup Foundation and the National Institutes of Health. Dr. Relkin reporting no relevant disclosures besides receiving a research grant from Baxter Healthcare to study IVIG.
TORONTO – Intravenous immunoglobulin therapy reduced brain atrophy in patients with mild to moderate Alzheimer's disease in a small phase II trial. The finding suggests that specific IgG antibody components found in the blood product might be treatment candidates for the disease.
“Relative to what we have available right now [to treat Alzheimer's disease], this is a very promising outcome, and it's associated with a reduction in the rate of brain atrophy comparable with age-matched normals,” Dr. Norman Relkin said during a poster presentation.
Enlargement of the cerebral lateral ventricles is known to occur as a consequence of brain atrophy in Alzheimer's disease (AD). This increase in ventricular volume is correlated with cognitive decline and increases in Alzheimer's disease neuropathology.
Dr. Relkin and his colleagues compared intravenous immunoglobulin (IVIG) therapy against placebo in a 6-month, double-blind, randomized study of 24 patients with mild to moderate AD. In a 12-month extension phase of the study, 16 patients who originally were randomized to IVIG continued to receive the same doses of IVIG, whereas 8 placebo-treated patients were re-randomized to one of four doses of IVIG. The investigators used an IVIG product produced by Baxter Healthcare called Gammagard.
IVIG exhibited a dose-dependent effect on brain atrophy in which higher doses resulted in less atrophy. Among 14 IVIG-treated patients who underwent volumetric MRI at baseline and after 18 months, the yearly increase in lateral ventricle volume measured with volumetric MRI was lowest in patients treated with 0.4 mg/kg every 2 weeks (2.4%) and highest in those treated with 0.2 mg/kg every 2 weeks (11.2%). The doses of IVIG given to patients ranged from 0.2 mg/kg every 2 weeks to 0.8 mg/kg every 4 weeks.
The volume of the lateral ventricles increased by a mean of 6.7% per year during treatment with IVIG (all doses combined), which was significantly lower than the 12.3% annual rate of increase observed in six placebo-treated patients. Only the 0.4 mg/kg dose of IVIG given every 2 weeks resulted in significantly less change in total brain volume than did treatment with placebo (−0.62% vs. −2.24%, respectively).
“In addition to the brain imaging, we have previously shown changes in cerebrospinal fluid and plasma amyloid levels … and levels of cerebral metabolism changing in response to treatment,” said Dr. Relkin, director of the Memory Disorders Program at New York–Presbyterian Hospital/Weill Cornell Medical Center.
The reduction in brain atrophy was significantly correlated with improvement in clinical outcomes at 18 months on the Clinical Global Impression of Change and the cognitive subscale of the Alzheimer's Disease Assessment Scale. Patients' baseline characteristics were not correlated with volumetric MRI outcomes.
“This is a 'kitchen sink' approach, so the next step is to find what is in [IVIG] that is causing the therapeutic effect. … We know that it has a fairly good complement of antiamyloid antibodies. Those are prime candidates, but we don't know for sure yet that those are ones responsible for a therapeutic effect,” Dr. Relkin said in an interview.
In addition to an ongoing, multicenter, phase III study of IVIG in 360 patients with mild to moderate AD, Dr. Relkin and his colleagues are testing subsets of antibodies within IVIG in cell culture–based studies and preclinical animal models to see which components are therapeutically relevant. “We are not encouraging people to use [IVIG] off-label for Alzheimer's disease, even though it has been safe and well tolerated in these small studies,” he said. “It has never been studied in the Alzheimer's population before.”
Baxter Healthcare sponsored the phase I and II studies of IVIG, with additional support from the Citigroup Foundation and the National Institutes of Health. The phase III trial is cosponsored by Baxter and the NIH. Dr. Relkin reported no relevant disclosures besides receiving a research grant from Baxter Healthcare to study IVIG.
The outcome is associated with a reduction in the brain atrophy rate, comparable with age-matched normals.
Source DR. RELKIN
After 18 months, the ventricular enlargement rate was greater with placebo (left) than it was with IVIG (right).
Source Courtesy Dr. Dana Moore and Dr. Norman Relkin
Generic and Brand-Name AEDs Bioequivalent
TORONTO — Most generic formulations of antiepileptic drugs have pharmacokinetics that closely match their brand-name reference, according to an analysis of bioequivalence studies submitted to the Food and Drug Administration.
These results suggest that most switches from brand-name to generic formulations of antiepileptic drugs (AEDs) are safe and do not lead to clinically significant changes in blood concentrations, Dr. Gregory L. Krauss said.
However, he cautioned that generic-to-generic switches of AED formulations should be minimized because simulations of these switches in his study resulted in a wide variability of blood concentrations, particularly for AEDs with low solubility.
“This is an unaddressed area in U.S. regulations, but there are over 500 potential switches between different pairs of generic AEDs at the same dose,” said Dr. Krauss, professor of neurology at Johns Hopkins University, Baltimore. “Switches between generic formulations may cause undesirable shifts in AED concentrations. These sorts of patterns should be examined in clinical studies, particularly ones that would enroll patients who are intolerant to AEDs, elderly, or taking polytherapy.”
After several years of sending Freedom of Information Act data requests to the FDA, Dr. Krauss and his associates at Johns Hopkins were eventually able to collaborate on the study with officials at the agency.
Bioequivalence is determined in randomized, crossover, pharmacokinetics studies with a small number of healthy volunteers who receive single doses of the generic and references drugs.
In these studies, the FDA defines a test product to be bioequivalent to a reference product when the 90% confidence intervals for test-to-reference ratios of the area under the plasma concentration time curve (AUC) and the maximum plasma concentration (Cmax) are within an acceptance range of 80%-125%. AUC measures how much drug is absorbed in a given time, whereas Cmax measures the maximum plasma concentration of a drug.
The investigators examined 147 AED formulations, excluding extended-release products, in 251 bioequivalence studies. All 7,125 participants in these studies were adults (mean age, 32 years; 79% male), but only 44 were older than 65 years.
Overall, 54% of the participants were white, 26% were Asian, 10% were black, 3% were Hispanic, and 7% were other race/ethnicity.
In 99% of the studies, the AUC for both reference and generic formulations varied by less than 15%. In comparison, 89% of Cmax studies found that measurements between reference and generic formulations varied by less than 15%. The remaining bioequivalence studies evaluated formulations with AUC and Cmax measurements that varied 15%-25%.
For example, divalproex generic products were largely similar to Depakote in terms of AUC and Cmax. But some products did not perform as well as others and had very broad 90% confidence intervals for both AUC and Cmax.
Some generic AEDs had confidence intervals for AUC or Cmax ratios that were much less or much greater than a ratio of 1, meaning that for some switches one would expect slightly lower blood concentrations of the active ingredient and for other switches one would expect slightly higher blood concentrations.
But when a switch is made from a generic formulation of a drug with a confidence interval completely below 1 to a generic formulation with a confidence interval completely above 1, Dr. Krauss noted that there is likely to be a bigger change in blood concentration than with brand-name to generic switches.
The investigators found generally greater differences in Cmax between generic and reference formulations than they did for AUC. One of the greatest differences in Cmax was found in carbamazepine formulations. For instance, only 9% of generic formulations of carbamazepine were within 5% of the reference product, whereas 64% of formulations were within 5%-10% of the reference, 18% were within 10%-15%, and 9% were within 15%-25%.
Reference drugs did not provide more stable delivery of active ingredients to individuals, compared with generic formulations. The standard deviations between the generic formulations and a reference drug were nearly identical for most drugs. In terms of intersubject variability, “there's really no difference,” Dr. Krauss said.
Disclosures: Dr. Krauss said neither he nor his colleagues had relevant conflicts.
{kind=link}
In terms of intersubject variability, “there's really no difference,” Dr. Gregory L. Krauss said.
Source Courtesy Willette Kearney-Horne
My Take
Study Did Not Examine the Real At-Risk Population
The data presented by Dr. Krauss give us a deeper understanding of the variability among generic AED products. It is important to note that this study is based on data generated from people who will never take an AED. These normal subjects received only a single dose of the drug and were not taking any concomitant medications. There are large potential differences between this population and patients with epilepsy who are taking two or three other AEDs or non-AEDs and who might be older have taken an AED daily for many years. Those are the people in whom I'm most concerned about therapeutic equivalence.
There may be subsets of individuals who are at increased risk for seizures with small changes in bioequivalence, such as those who have had life-threatening status epilepticus in the past, pregnant women, people with epilepsy who have been seizure free for many years, and people with other serious medical conditions.
We don't really know what percentage change in AUC or Cmax between products is actually safe—that is, which ranges of bioequivalence translate to therapeutic equivalence and which do not. In his study, Dr. Krauss is suggesting that certain ranges of difference between products should be safe and others perhaps not so safe. Unfortunately, we have no data to support that inference. There are no data providing evidence that 90% confidence intervals in the 80%-125% range, which are the current FDA standard, translate to therapeutic equivalence. The FDA created this range based on expert opinion.
A recent FDA advisory committee indicated that the range for generic AED confidence intervals may not be optimal for patients with epilepsy, but the committee did not agree upon any specific recommendations.
The FDA states that all brand name–to-generic or generic-to-generic switches are safe for all people with epilepsy. I believe the only way to test this is to perform a prospective, randomized study of people with epilepsy.
MICHAEL PRIVITERA, M.D., is a professor of neurology at the University of Cincinnati and is director of the Cincinnati Epilepsy Center. He has received research funding and honoraria for speaking or consulting from UCB, Johnson & Johnson, Pfizer, Eisai, the National Institutes of Health, and the American Epilepsy Society.
{kind=link}
TORONTO — Most generic formulations of antiepileptic drugs have pharmacokinetics that closely match their brand-name reference, according to an analysis of bioequivalence studies submitted to the Food and Drug Administration.
These results suggest that most switches from brand-name to generic formulations of antiepileptic drugs (AEDs) are safe and do not lead to clinically significant changes in blood concentrations, Dr. Gregory L. Krauss said.
However, he cautioned that generic-to-generic switches of AED formulations should be minimized because simulations of these switches in his study resulted in a wide variability of blood concentrations, particularly for AEDs with low solubility.
“This is an unaddressed area in U.S. regulations, but there are over 500 potential switches between different pairs of generic AEDs at the same dose,” said Dr. Krauss, professor of neurology at Johns Hopkins University, Baltimore. “Switches between generic formulations may cause undesirable shifts in AED concentrations. These sorts of patterns should be examined in clinical studies, particularly ones that would enroll patients who are intolerant to AEDs, elderly, or taking polytherapy.”
After several years of sending Freedom of Information Act data requests to the FDA, Dr. Krauss and his associates at Johns Hopkins were eventually able to collaborate on the study with officials at the agency.
Bioequivalence is determined in randomized, crossover, pharmacokinetics studies with a small number of healthy volunteers who receive single doses of the generic and references drugs.
In these studies, the FDA defines a test product to be bioequivalent to a reference product when the 90% confidence intervals for test-to-reference ratios of the area under the plasma concentration time curve (AUC) and the maximum plasma concentration (Cmax) are within an acceptance range of 80%-125%. AUC measures how much drug is absorbed in a given time, whereas Cmax measures the maximum plasma concentration of a drug.
The investigators examined 147 AED formulations, excluding extended-release products, in 251 bioequivalence studies. All 7,125 participants in these studies were adults (mean age, 32 years; 79% male), but only 44 were older than 65 years.
Overall, 54% of the participants were white, 26% were Asian, 10% were black, 3% were Hispanic, and 7% were other race/ethnicity.
In 99% of the studies, the AUC for both reference and generic formulations varied by less than 15%. In comparison, 89% of Cmax studies found that measurements between reference and generic formulations varied by less than 15%. The remaining bioequivalence studies evaluated formulations with AUC and Cmax measurements that varied 15%-25%.
For example, divalproex generic products were largely similar to Depakote in terms of AUC and Cmax. But some products did not perform as well as others and had very broad 90% confidence intervals for both AUC and Cmax.
Some generic AEDs had confidence intervals for AUC or Cmax ratios that were much less or much greater than a ratio of 1, meaning that for some switches one would expect slightly lower blood concentrations of the active ingredient and for other switches one would expect slightly higher blood concentrations.
But when a switch is made from a generic formulation of a drug with a confidence interval completely below 1 to a generic formulation with a confidence interval completely above 1, Dr. Krauss noted that there is likely to be a bigger change in blood concentration than with brand-name to generic switches.
The investigators found generally greater differences in Cmax between generic and reference formulations than they did for AUC. One of the greatest differences in Cmax was found in carbamazepine formulations. For instance, only 9% of generic formulations of carbamazepine were within 5% of the reference product, whereas 64% of formulations were within 5%-10% of the reference, 18% were within 10%-15%, and 9% were within 15%-25%.
Reference drugs did not provide more stable delivery of active ingredients to individuals, compared with generic formulations. The standard deviations between the generic formulations and a reference drug were nearly identical for most drugs. In terms of intersubject variability, “there's really no difference,” Dr. Krauss said.
Disclosures: Dr. Krauss said neither he nor his colleagues had relevant conflicts.
In terms of intersubject variability, “there's really no difference,” Dr. Gregory L. Krauss said.
Source Courtesy Willette Kearney-Horne
My Take
Study Did Not Examine the Real At-Risk Population
The data presented by Dr. Krauss give us a deeper understanding of the variability among generic AED products. It is important to note that this study is based on data generated from people who will never take an AED. These normal subjects received only a single dose of the drug and were not taking any concomitant medications. There are large potential differences between this population and patients with epilepsy who are taking two or three other AEDs or non-AEDs and who might be older have taken an AED daily for many years. Those are the people in whom I'm most concerned about therapeutic equivalence.
There may be subsets of individuals who are at increased risk for seizures with small changes in bioequivalence, such as those who have had life-threatening status epilepticus in the past, pregnant women, people with epilepsy who have been seizure free for many years, and people with other serious medical conditions.
We don't really know what percentage change in AUC or Cmax between products is actually safe—that is, which ranges of bioequivalence translate to therapeutic equivalence and which do not. In his study, Dr. Krauss is suggesting that certain ranges of difference between products should be safe and others perhaps not so safe. Unfortunately, we have no data to support that inference. There are no data providing evidence that 90% confidence intervals in the 80%-125% range, which are the current FDA standard, translate to therapeutic equivalence. The FDA created this range based on expert opinion.
A recent FDA advisory committee indicated that the range for generic AED confidence intervals may not be optimal for patients with epilepsy, but the committee did not agree upon any specific recommendations.
The FDA states that all brand name–to-generic or generic-to-generic switches are safe for all people with epilepsy. I believe the only way to test this is to perform a prospective, randomized study of people with epilepsy.
MICHAEL PRIVITERA, M.D., is a professor of neurology at the University of Cincinnati and is director of the Cincinnati Epilepsy Center. He has received research funding and honoraria for speaking or consulting from UCB, Johnson & Johnson, Pfizer, Eisai, the National Institutes of Health, and the American Epilepsy Society.
TORONTO — Most generic formulations of antiepileptic drugs have pharmacokinetics that closely match their brand-name reference, according to an analysis of bioequivalence studies submitted to the Food and Drug Administration.
These results suggest that most switches from brand-name to generic formulations of antiepileptic drugs (AEDs) are safe and do not lead to clinically significant changes in blood concentrations, Dr. Gregory L. Krauss said.
However, he cautioned that generic-to-generic switches of AED formulations should be minimized because simulations of these switches in his study resulted in a wide variability of blood concentrations, particularly for AEDs with low solubility.
“This is an unaddressed area in U.S. regulations, but there are over 500 potential switches between different pairs of generic AEDs at the same dose,” said Dr. Krauss, professor of neurology at Johns Hopkins University, Baltimore. “Switches between generic formulations may cause undesirable shifts in AED concentrations. These sorts of patterns should be examined in clinical studies, particularly ones that would enroll patients who are intolerant to AEDs, elderly, or taking polytherapy.”
After several years of sending Freedom of Information Act data requests to the FDA, Dr. Krauss and his associates at Johns Hopkins were eventually able to collaborate on the study with officials at the agency.
Bioequivalence is determined in randomized, crossover, pharmacokinetics studies with a small number of healthy volunteers who receive single doses of the generic and references drugs.
In these studies, the FDA defines a test product to be bioequivalent to a reference product when the 90% confidence intervals for test-to-reference ratios of the area under the plasma concentration time curve (AUC) and the maximum plasma concentration (Cmax) are within an acceptance range of 80%-125%. AUC measures how much drug is absorbed in a given time, whereas Cmax measures the maximum plasma concentration of a drug.
The investigators examined 147 AED formulations, excluding extended-release products, in 251 bioequivalence studies. All 7,125 participants in these studies were adults (mean age, 32 years; 79% male), but only 44 were older than 65 years.
Overall, 54% of the participants were white, 26% were Asian, 10% were black, 3% were Hispanic, and 7% were other race/ethnicity.
In 99% of the studies, the AUC for both reference and generic formulations varied by less than 15%. In comparison, 89% of Cmax studies found that measurements between reference and generic formulations varied by less than 15%. The remaining bioequivalence studies evaluated formulations with AUC and Cmax measurements that varied 15%-25%.
For example, divalproex generic products were largely similar to Depakote in terms of AUC and Cmax. But some products did not perform as well as others and had very broad 90% confidence intervals for both AUC and Cmax.
Some generic AEDs had confidence intervals for AUC or Cmax ratios that were much less or much greater than a ratio of 1, meaning that for some switches one would expect slightly lower blood concentrations of the active ingredient and for other switches one would expect slightly higher blood concentrations.
But when a switch is made from a generic formulation of a drug with a confidence interval completely below 1 to a generic formulation with a confidence interval completely above 1, Dr. Krauss noted that there is likely to be a bigger change in blood concentration than with brand-name to generic switches.
The investigators found generally greater differences in Cmax between generic and reference formulations than they did for AUC. One of the greatest differences in Cmax was found in carbamazepine formulations. For instance, only 9% of generic formulations of carbamazepine were within 5% of the reference product, whereas 64% of formulations were within 5%-10% of the reference, 18% were within 10%-15%, and 9% were within 15%-25%.
Reference drugs did not provide more stable delivery of active ingredients to individuals, compared with generic formulations. The standard deviations between the generic formulations and a reference drug were nearly identical for most drugs. In terms of intersubject variability, “there's really no difference,” Dr. Krauss said.
Disclosures: Dr. Krauss said neither he nor his colleagues had relevant conflicts.
In terms of intersubject variability, “there's really no difference,” Dr. Gregory L. Krauss said.
Source Courtesy Willette Kearney-Horne
My Take
Study Did Not Examine the Real At-Risk Population
The data presented by Dr. Krauss give us a deeper understanding of the variability among generic AED products. It is important to note that this study is based on data generated from people who will never take an AED. These normal subjects received only a single dose of the drug and were not taking any concomitant medications. There are large potential differences between this population and patients with epilepsy who are taking two or three other AEDs or non-AEDs and who might be older have taken an AED daily for many years. Those are the people in whom I'm most concerned about therapeutic equivalence.
There may be subsets of individuals who are at increased risk for seizures with small changes in bioequivalence, such as those who have had life-threatening status epilepticus in the past, pregnant women, people with epilepsy who have been seizure free for many years, and people with other serious medical conditions.
We don't really know what percentage change in AUC or Cmax between products is actually safe—that is, which ranges of bioequivalence translate to therapeutic equivalence and which do not. In his study, Dr. Krauss is suggesting that certain ranges of difference between products should be safe and others perhaps not so safe. Unfortunately, we have no data to support that inference. There are no data providing evidence that 90% confidence intervals in the 80%-125% range, which are the current FDA standard, translate to therapeutic equivalence. The FDA created this range based on expert opinion.
A recent FDA advisory committee indicated that the range for generic AED confidence intervals may not be optimal for patients with epilepsy, but the committee did not agree upon any specific recommendations.
The FDA states that all brand name–to-generic or generic-to-generic switches are safe for all people with epilepsy. I believe the only way to test this is to perform a prospective, randomized study of people with epilepsy.
MICHAEL PRIVITERA, M.D., is a professor of neurology at the University of Cincinnati and is director of the Cincinnati Epilepsy Center. He has received research funding and honoraria for speaking or consulting from UCB, Johnson & Johnson, Pfizer, Eisai, the National Institutes of Health, and the American Epilepsy Society.