User login
Widespread Skin Thickening
The Diagnosis: Scleromyxedema
Scleromyxedema is a rare skin disorder characterized by a diffuse eruption of small waxy papules that are linearly arranged and closely spaced together. As the papular lesions coalesce, the skin thickens. Firm induration of the skin is widespread and--unlike the distribution in scleroderma and scleredema--amplified over the facial convexities, especially the glabella and ears. Histopathology reveals the classic triad of mucin accumulation, proliferation of fibroblasts, and collagen deposition with associated fibrotic changes.1
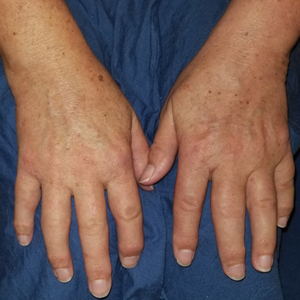
In this case, the patient exhibited the characteristic doughnut sign over the second interphalangeal joint of the right hand due to thickening and dimpling of the skin (quiz image). Histopathology from the right second finger showed the dermis with a spindled histiocytic infiltrate, fibroblasts, fibrosis, and increased interstitial mucin deposition highlighted with colloidal iron stain (Figures 1 and 2). Multinucleated giant cells also were seen in the dermis (Figure 3), and a Shikata orcein stain illustrated decreased elastic fibers (stained black) in the superficial dermis within areas of increased collagen deposition (Figure 4).
Scleromyxedema is strongly associated with a monoclonal gammopathy, which is generally mild and, in almost 80% of cases, related to λ IgG. However, the clinical implications are unclear.2,3 Scleromyxedema also is accompanied by systemic features, most commonly dysphagia, myalgias, and arthralgias, which distinguishes it from other cutaneous mucinoses. Notable morbidity and mortality are associated with involvement of the central nervous system, which can manifest as encephalopathy, seizures, or coma. Mucin deposition may be found in the myocardium and the coronary and pulmonary vasculature, resulting in rare cases of cardiac and pulmonary disease.4,5
Clinically, scleroderma also demonstrates progressive generalized skin tightening. Histopathology reveals hyalinization and thickening of the connective tissue of the deep dermis, subcutaneous fat, and muscular fascia. These changes are accompanied by a perivascular and focal interstitial lymphocytic and plasma cell infiltrate with prominent myofibroblast proliferation. In this case, the normal nail fold capillaries and absence of Raynaud phenomenon help to distinguish the diagnosis from scleroderma. In limited scleroderma, common findings include calcinosis, esophageal dysmotility, telangiectasia, and pulmonary hypertension, while systemic sclerosis can involve the internal organs, with the greatest morbidity stemming from interstitial lung disease.3,6,7
Nephrogenic systemic fibrosis is another disorder of skin thickening that involves the extremities and trunk. It is a rare complication of exposure to gadolinium contrast media in patients with advanced renal disease and those undergoing dialysis; use of gadolinium is now strictly avoided in patients with at least moderately impaired glomerular filtration rate.8 Although nephrogenic systemic fibrosis histologically looks similar to scleromyxedema, it involves deeper tissues. There is fibrosis with haphazard collagen bundle deposition in the deep dermis and subcutaneous septa. Fibroblasts and histiocytes are increased between the collagen bundles, with surrounding edema and mucin buildup. Multinucleated giant cells may or may not be present.9
Patients with scleredema present with nonpitting edema and dermal hardening over the neck, face, upper trunk, and arms. Lesional skin appears shiny and indurated. The skin is characteristically difficult to wrinkle, and the face may appear expressionless. Skin biopsy will show a thickened reticular dermis with mucin deposition and eccrine glands appearing in the upper third or mid dermis. Fibroblasts are normal in number. Scleredema generally is associated with poorly controlled diabetes, a monoclonal gammopathy, or as the aftermath of an acute infection, especially streptococcal pharyngitis. The condition may develop at any age, though nearly 50% of cases occur in children and adolescents.3,10
Interstitial granuloma annulare (GA) is a common, benign, and self-limiting dermatosis. Distinct from other skin-thickening disorders, GA is composed of smooth annular papules and plaques that do not result in skin hardening. Although GA may manifest in a generalized distribution, it is more frequently localized to the distal extremities.11 The presence of an inflammatory infiltrate surrounded by abundant mucin and collagen resembles scleromyxedema. However, GA is distinguished by palisading granulomas of histiocytes, fibroblasts, and lymphocytes lining a necrobiotic center of collagen, mucin, and fibrin.12
This patient's skin showed an immediate and marked response to intravenous immunoglobulin with dramatic softening. Such a response is well reported in the literature, and intravenous immunoglobulin is considered first-line therapy for scleromyxedema, typically in conjunction with steroids.5 Other immunosuppressive agents have been employed with reported efficacy, as have several treatments used for multiple myeloma, including thalidomide and lenalidomide.
Although spontaneous improvement infrequently occurs, a chronic progressive course is far more common with scleromyxedema. Studies are ongoing to elucidate the etiopathogenesis of scleromyxedema, scleroderma, and similar disorders to uncover the triggers that provoke the underlying dysregulation of dermal fibroblast activation and proliferation, which could offer a more precise target for effective treatments.5,13-15
- Crowe DR. Scleromyxedema. In: Crowe DR, Morgan M, Somach S, Trapp K, eds. Deadly Dermatologic Diseases: Clinicopathologic Atlas and Text. Cham, Switzerland: Springer International Publishing; 2016:139-142.
- Farmer ER, Hambrick GW Jr, Shulman LE. Papular mucinosis: a clinicopathologic study of four patients. Arch Dermatol. 1982;118:9-13.
- Rongioletti F. Mucinoses. In: Smoller BR, Rongioletti F, eds. Clinical and Pathological Aspects of Skin Diseases in Endocrine, Metabolic, Nutritional and Deposition Disease. New York, NY: Springer New York; 2010:139-152.
- Gabriel PH, Oleson GB, Bowles GA. Scleromyxoedema: a scleroderma-like disorder with systemic manifestations. Medicine. 1988;67:58-65.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Hamodat M. Scleroderma. PathologyOutlines.com. http://www.pathologyoutlines.com/topic/skinnontumorscleroderma.html. Published August 1, 2011. Updated March 29, 2019. Accessed December 11, 2019.
- Van Praet JT, Smith V, Haspeslagh M, et al. Histopathological cutaneous alterations in systemic sclerosis: a clinicopathological study. Arthritis Res Ther. 2011;13:R35.
- Swartz RD, Crofford LJ, Phan SH, et al. Nephrogenic fibrosing dermopathy: a novel cutaneous fibrosing disorder in patients with renal failure. Am J Med. 2003;114:563-572.
- Cowper SE, Rabach M, Girardi M. Clinical and histological findings in nephrogenic systemic fibrosis. Eur J Radiol. 2008;66:191-199.
- Boin F, Hummers LK. Scleroderma-like fibrosing disorders. Rheum Dis Clin North Am. 2008;34:199-ix.
- Plaza JA, Prieto VG. Inflammatory Skin Conditions. In: Modern Surgical Pathology. 2nd ed. Philadelphia, PA: Saunders; 2009:1843-1889.
- Muhlbauer JE. Granuloma annulare. J Am Acad Dermatol. 1980;3:217-230.
- Blum M, Wigley FM, Hummers LK. Scleromyxedema: a case series highlighting long-term outcomes of treatment with intravenous immunoglobulin (IVIG). Medicine. 2008;87:10-20.
- Caradonna S, Jacobe H. Thalidomide as a potential treatment for scleromyxedema. Arch Dermatol. 2004;140:277-280.
- Yeung CK, Loong F, Kwong YL. Scleromyxoedema due to a plasma cell neoplasm: rapid remission with bortezomib, thalidomide and dexamethasone. Br J Haematol. 2012;157:411.
The Diagnosis: Scleromyxedema
Scleromyxedema is a rare skin disorder characterized by a diffuse eruption of small waxy papules that are linearly arranged and closely spaced together. As the papular lesions coalesce, the skin thickens. Firm induration of the skin is widespread and--unlike the distribution in scleroderma and scleredema--amplified over the facial convexities, especially the glabella and ears. Histopathology reveals the classic triad of mucin accumulation, proliferation of fibroblasts, and collagen deposition with associated fibrotic changes.1
In this case, the patient exhibited the characteristic doughnut sign over the second interphalangeal joint of the right hand due to thickening and dimpling of the skin (quiz image). Histopathology from the right second finger showed the dermis with a spindled histiocytic infiltrate, fibroblasts, fibrosis, and increased interstitial mucin deposition highlighted with colloidal iron stain (Figures 1 and 2). Multinucleated giant cells also were seen in the dermis (Figure 3), and a Shikata orcein stain illustrated decreased elastic fibers (stained black) in the superficial dermis within areas of increased collagen deposition (Figure 4).
Scleromyxedema is strongly associated with a monoclonal gammopathy, which is generally mild and, in almost 80% of cases, related to λ IgG. However, the clinical implications are unclear.2,3 Scleromyxedema also is accompanied by systemic features, most commonly dysphagia, myalgias, and arthralgias, which distinguishes it from other cutaneous mucinoses. Notable morbidity and mortality are associated with involvement of the central nervous system, which can manifest as encephalopathy, seizures, or coma. Mucin deposition may be found in the myocardium and the coronary and pulmonary vasculature, resulting in rare cases of cardiac and pulmonary disease.4,5
Clinically, scleroderma also demonstrates progressive generalized skin tightening. Histopathology reveals hyalinization and thickening of the connective tissue of the deep dermis, subcutaneous fat, and muscular fascia. These changes are accompanied by a perivascular and focal interstitial lymphocytic and plasma cell infiltrate with prominent myofibroblast proliferation. In this case, the normal nail fold capillaries and absence of Raynaud phenomenon help to distinguish the diagnosis from scleroderma. In limited scleroderma, common findings include calcinosis, esophageal dysmotility, telangiectasia, and pulmonary hypertension, while systemic sclerosis can involve the internal organs, with the greatest morbidity stemming from interstitial lung disease.3,6,7
Nephrogenic systemic fibrosis is another disorder of skin thickening that involves the extremities and trunk. It is a rare complication of exposure to gadolinium contrast media in patients with advanced renal disease and those undergoing dialysis; use of gadolinium is now strictly avoided in patients with at least moderately impaired glomerular filtration rate.8 Although nephrogenic systemic fibrosis histologically looks similar to scleromyxedema, it involves deeper tissues. There is fibrosis with haphazard collagen bundle deposition in the deep dermis and subcutaneous septa. Fibroblasts and histiocytes are increased between the collagen bundles, with surrounding edema and mucin buildup. Multinucleated giant cells may or may not be present.9
Patients with scleredema present with nonpitting edema and dermal hardening over the neck, face, upper trunk, and arms. Lesional skin appears shiny and indurated. The skin is characteristically difficult to wrinkle, and the face may appear expressionless. Skin biopsy will show a thickened reticular dermis with mucin deposition and eccrine glands appearing in the upper third or mid dermis. Fibroblasts are normal in number. Scleredema generally is associated with poorly controlled diabetes, a monoclonal gammopathy, or as the aftermath of an acute infection, especially streptococcal pharyngitis. The condition may develop at any age, though nearly 50% of cases occur in children and adolescents.3,10
Interstitial granuloma annulare (GA) is a common, benign, and self-limiting dermatosis. Distinct from other skin-thickening disorders, GA is composed of smooth annular papules and plaques that do not result in skin hardening. Although GA may manifest in a generalized distribution, it is more frequently localized to the distal extremities.11 The presence of an inflammatory infiltrate surrounded by abundant mucin and collagen resembles scleromyxedema. However, GA is distinguished by palisading granulomas of histiocytes, fibroblasts, and lymphocytes lining a necrobiotic center of collagen, mucin, and fibrin.12
This patient's skin showed an immediate and marked response to intravenous immunoglobulin with dramatic softening. Such a response is well reported in the literature, and intravenous immunoglobulin is considered first-line therapy for scleromyxedema, typically in conjunction with steroids.5 Other immunosuppressive agents have been employed with reported efficacy, as have several treatments used for multiple myeloma, including thalidomide and lenalidomide.
Although spontaneous improvement infrequently occurs, a chronic progressive course is far more common with scleromyxedema. Studies are ongoing to elucidate the etiopathogenesis of scleromyxedema, scleroderma, and similar disorders to uncover the triggers that provoke the underlying dysregulation of dermal fibroblast activation and proliferation, which could offer a more precise target for effective treatments.5,13-15
The Diagnosis: Scleromyxedema
Scleromyxedema is a rare skin disorder characterized by a diffuse eruption of small waxy papules that are linearly arranged and closely spaced together. As the papular lesions coalesce, the skin thickens. Firm induration of the skin is widespread and--unlike the distribution in scleroderma and scleredema--amplified over the facial convexities, especially the glabella and ears. Histopathology reveals the classic triad of mucin accumulation, proliferation of fibroblasts, and collagen deposition with associated fibrotic changes.1
In this case, the patient exhibited the characteristic doughnut sign over the second interphalangeal joint of the right hand due to thickening and dimpling of the skin (quiz image). Histopathology from the right second finger showed the dermis with a spindled histiocytic infiltrate, fibroblasts, fibrosis, and increased interstitial mucin deposition highlighted with colloidal iron stain (Figures 1 and 2). Multinucleated giant cells also were seen in the dermis (Figure 3), and a Shikata orcein stain illustrated decreased elastic fibers (stained black) in the superficial dermis within areas of increased collagen deposition (Figure 4).
Scleromyxedema is strongly associated with a monoclonal gammopathy, which is generally mild and, in almost 80% of cases, related to λ IgG. However, the clinical implications are unclear.2,3 Scleromyxedema also is accompanied by systemic features, most commonly dysphagia, myalgias, and arthralgias, which distinguishes it from other cutaneous mucinoses. Notable morbidity and mortality are associated with involvement of the central nervous system, which can manifest as encephalopathy, seizures, or coma. Mucin deposition may be found in the myocardium and the coronary and pulmonary vasculature, resulting in rare cases of cardiac and pulmonary disease.4,5
Clinically, scleroderma also demonstrates progressive generalized skin tightening. Histopathology reveals hyalinization and thickening of the connective tissue of the deep dermis, subcutaneous fat, and muscular fascia. These changes are accompanied by a perivascular and focal interstitial lymphocytic and plasma cell infiltrate with prominent myofibroblast proliferation. In this case, the normal nail fold capillaries and absence of Raynaud phenomenon help to distinguish the diagnosis from scleroderma. In limited scleroderma, common findings include calcinosis, esophageal dysmotility, telangiectasia, and pulmonary hypertension, while systemic sclerosis can involve the internal organs, with the greatest morbidity stemming from interstitial lung disease.3,6,7
Nephrogenic systemic fibrosis is another disorder of skin thickening that involves the extremities and trunk. It is a rare complication of exposure to gadolinium contrast media in patients with advanced renal disease and those undergoing dialysis; use of gadolinium is now strictly avoided in patients with at least moderately impaired glomerular filtration rate.8 Although nephrogenic systemic fibrosis histologically looks similar to scleromyxedema, it involves deeper tissues. There is fibrosis with haphazard collagen bundle deposition in the deep dermis and subcutaneous septa. Fibroblasts and histiocytes are increased between the collagen bundles, with surrounding edema and mucin buildup. Multinucleated giant cells may or may not be present.9
Patients with scleredema present with nonpitting edema and dermal hardening over the neck, face, upper trunk, and arms. Lesional skin appears shiny and indurated. The skin is characteristically difficult to wrinkle, and the face may appear expressionless. Skin biopsy will show a thickened reticular dermis with mucin deposition and eccrine glands appearing in the upper third or mid dermis. Fibroblasts are normal in number. Scleredema generally is associated with poorly controlled diabetes, a monoclonal gammopathy, or as the aftermath of an acute infection, especially streptococcal pharyngitis. The condition may develop at any age, though nearly 50% of cases occur in children and adolescents.3,10
Interstitial granuloma annulare (GA) is a common, benign, and self-limiting dermatosis. Distinct from other skin-thickening disorders, GA is composed of smooth annular papules and plaques that do not result in skin hardening. Although GA may manifest in a generalized distribution, it is more frequently localized to the distal extremities.11 The presence of an inflammatory infiltrate surrounded by abundant mucin and collagen resembles scleromyxedema. However, GA is distinguished by palisading granulomas of histiocytes, fibroblasts, and lymphocytes lining a necrobiotic center of collagen, mucin, and fibrin.12
This patient's skin showed an immediate and marked response to intravenous immunoglobulin with dramatic softening. Such a response is well reported in the literature, and intravenous immunoglobulin is considered first-line therapy for scleromyxedema, typically in conjunction with steroids.5 Other immunosuppressive agents have been employed with reported efficacy, as have several treatments used for multiple myeloma, including thalidomide and lenalidomide.
Although spontaneous improvement infrequently occurs, a chronic progressive course is far more common with scleromyxedema. Studies are ongoing to elucidate the etiopathogenesis of scleromyxedema, scleroderma, and similar disorders to uncover the triggers that provoke the underlying dysregulation of dermal fibroblast activation and proliferation, which could offer a more precise target for effective treatments.5,13-15
- Crowe DR. Scleromyxedema. In: Crowe DR, Morgan M, Somach S, Trapp K, eds. Deadly Dermatologic Diseases: Clinicopathologic Atlas and Text. Cham, Switzerland: Springer International Publishing; 2016:139-142.
- Farmer ER, Hambrick GW Jr, Shulman LE. Papular mucinosis: a clinicopathologic study of four patients. Arch Dermatol. 1982;118:9-13.
- Rongioletti F. Mucinoses. In: Smoller BR, Rongioletti F, eds. Clinical and Pathological Aspects of Skin Diseases in Endocrine, Metabolic, Nutritional and Deposition Disease. New York, NY: Springer New York; 2010:139-152.
- Gabriel PH, Oleson GB, Bowles GA. Scleromyxoedema: a scleroderma-like disorder with systemic manifestations. Medicine. 1988;67:58-65.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Hamodat M. Scleroderma. PathologyOutlines.com. http://www.pathologyoutlines.com/topic/skinnontumorscleroderma.html. Published August 1, 2011. Updated March 29, 2019. Accessed December 11, 2019.
- Van Praet JT, Smith V, Haspeslagh M, et al. Histopathological cutaneous alterations in systemic sclerosis: a clinicopathological study. Arthritis Res Ther. 2011;13:R35.
- Swartz RD, Crofford LJ, Phan SH, et al. Nephrogenic fibrosing dermopathy: a novel cutaneous fibrosing disorder in patients with renal failure. Am J Med. 2003;114:563-572.
- Cowper SE, Rabach M, Girardi M. Clinical and histological findings in nephrogenic systemic fibrosis. Eur J Radiol. 2008;66:191-199.
- Boin F, Hummers LK. Scleroderma-like fibrosing disorders. Rheum Dis Clin North Am. 2008;34:199-ix.
- Plaza JA, Prieto VG. Inflammatory Skin Conditions. In: Modern Surgical Pathology. 2nd ed. Philadelphia, PA: Saunders; 2009:1843-1889.
- Muhlbauer JE. Granuloma annulare. J Am Acad Dermatol. 1980;3:217-230.
- Blum M, Wigley FM, Hummers LK. Scleromyxedema: a case series highlighting long-term outcomes of treatment with intravenous immunoglobulin (IVIG). Medicine. 2008;87:10-20.
- Caradonna S, Jacobe H. Thalidomide as a potential treatment for scleromyxedema. Arch Dermatol. 2004;140:277-280.
- Yeung CK, Loong F, Kwong YL. Scleromyxoedema due to a plasma cell neoplasm: rapid remission with bortezomib, thalidomide and dexamethasone. Br J Haematol. 2012;157:411.
- Crowe DR. Scleromyxedema. In: Crowe DR, Morgan M, Somach S, Trapp K, eds. Deadly Dermatologic Diseases: Clinicopathologic Atlas and Text. Cham, Switzerland: Springer International Publishing; 2016:139-142.
- Farmer ER, Hambrick GW Jr, Shulman LE. Papular mucinosis: a clinicopathologic study of four patients. Arch Dermatol. 1982;118:9-13.
- Rongioletti F. Mucinoses. In: Smoller BR, Rongioletti F, eds. Clinical and Pathological Aspects of Skin Diseases in Endocrine, Metabolic, Nutritional and Deposition Disease. New York, NY: Springer New York; 2010:139-152.
- Gabriel PH, Oleson GB, Bowles GA. Scleromyxoedema: a scleroderma-like disorder with systemic manifestations. Medicine. 1988;67:58-65.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Hamodat M. Scleroderma. PathologyOutlines.com. http://www.pathologyoutlines.com/topic/skinnontumorscleroderma.html. Published August 1, 2011. Updated March 29, 2019. Accessed December 11, 2019.
- Van Praet JT, Smith V, Haspeslagh M, et al. Histopathological cutaneous alterations in systemic sclerosis: a clinicopathological study. Arthritis Res Ther. 2011;13:R35.
- Swartz RD, Crofford LJ, Phan SH, et al. Nephrogenic fibrosing dermopathy: a novel cutaneous fibrosing disorder in patients with renal failure. Am J Med. 2003;114:563-572.
- Cowper SE, Rabach M, Girardi M. Clinical and histological findings in nephrogenic systemic fibrosis. Eur J Radiol. 2008;66:191-199.
- Boin F, Hummers LK. Scleroderma-like fibrosing disorders. Rheum Dis Clin North Am. 2008;34:199-ix.
- Plaza JA, Prieto VG. Inflammatory Skin Conditions. In: Modern Surgical Pathology. 2nd ed. Philadelphia, PA: Saunders; 2009:1843-1889.
- Muhlbauer JE. Granuloma annulare. J Am Acad Dermatol. 1980;3:217-230.
- Blum M, Wigley FM, Hummers LK. Scleromyxedema: a case series highlighting long-term outcomes of treatment with intravenous immunoglobulin (IVIG). Medicine. 2008;87:10-20.
- Caradonna S, Jacobe H. Thalidomide as a potential treatment for scleromyxedema. Arch Dermatol. 2004;140:277-280.
- Yeung CK, Loong F, Kwong YL. Scleromyxoedema due to a plasma cell neoplasm: rapid remission with bortezomib, thalidomide and dexamethasone. Br J Haematol. 2012;157:411.
![]()
A 62-year-old woman presented with widespread skin thickening and tightness that progressed over 2 months. Physical examination showed generalized, nonpruritic, nonpainful, flesh-colored papules along the fingers, bilateral arms and legs, chest, neck, forehead, chin, and ears. She reported mild acid reflux on review of systems. She denied a history of Raynaud phenomenon and had normal-appearing nail beds on capillaroscopy. Laboratory studies including autoimmune serologies were normal, aside from a mildly elevated monoclonal IgG spike.